
Farmakologi Molecular - Reseptor Receptor theory First postulated by John Langley (1878) ◦...
28
Farmakologi Molecular - Reseptor
-
Upload
frederick-brooks -
Category
Documents
-
view
220 -
download
2
Transcript of Farmakologi Molecular - Reseptor Receptor theory First postulated by John Langley (1878) ◦...

Farmakologi Molecular - Reseptor

Receptor theoryFirst postulated by John Langley
(1878)◦ Established after his experiments using
nicotine and curare analogues on muscle contraction. Isolated muscle fibers: pilocarpine (contraction)
and atropine (inhibition). Two compounds competing for a third, but
unknown substrate.
Furthered by Paul Ehrlich (1854-1915)◦ Demonstrated that stereoselectivity was
imperative in drug-receptor signaling.

John LangleyLangley concluded that a
protoplasmic "receptive substance" must exist which the two drugs compete for directly. He further added that the effect of combination of the receptive substance with competing drugs was determined by their comparative chemical affinities for the substance and relative dose.
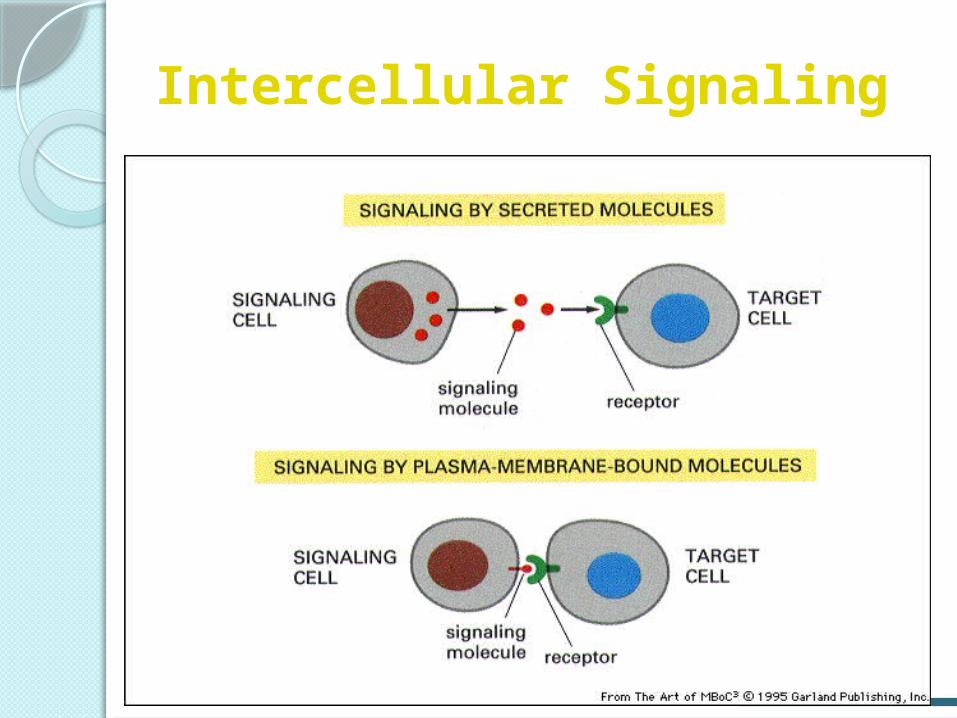
Intercellular Signaling

Classes of cell-surface receptors

• Receptor must possess structural and steric specificity for a hormone and for its close analogs as well.
• Receptors are saturable and limited (i.e. there is a finite number of binding sites).
• Hormone-receptor binding is cell specific in accordance with target organ specificity.
• Receptor must possess a high affinity for the hormone at physiological concentrations.
•Once a hormone binds to the receptor, some recognizable early chemical event must occur.
Criteria for hormone-mediated events

Affinity: The tenacity by which a drug binds to its receptor.◦ Discussion: a very lipid soluble drug may have irreversible
effects; is this high-affinity or merely a non-specific effect?
Intrinsic activity: Relative maximal effect of a drug in a particular tissue preparation when compared to the natural, endogenous ligand.◦ Full agonist – IA = 1 (*equal to the endogenous ligand)◦ Antagonist – IA = 0◦ Partial agonist – IA = 0~1 (*produces less than the
maximal response, but with maximal binding to receptors.)
Intrinsic efficacy: a drugs ability to bind a receptor and elicit a functional response◦ A measure of the formation of a drug-receptor complex.
Potency: ability of a drug to cause a measured functional change.

Receptors have two major properties:
Recognition and Transduction Recognition: The receptor protein must exist in a conformational state that allows for recognition and binding of a compound and must satisfy the following criteria:
Saturability – receptors exists in finite numbers.
Reversibility – binding must occur non-covalently due to weak intermolecular forces (H-bonding, van der Waal forces).
Stereoselectivity – receptors should recognize only one of the naturally occurring optical isomers (+ or -, d or l, or S or R).
Agonist specificity – structurally related drugs should bind well, while physically dissimilar compounds should bind poorly.
Tissue specificity – binding should occur in tissues known to be sensitive to the endogenous ligand. Binding should occur at physiologically relevant concentrations.

The failure of a drug to satisfy any of these conditions indicates non-specific binding to proteins or phospholipids in places like blood or plasma membrane components.

Receptors have two major properties:
Recognition and Transduction Transduction: The second property of a receptor is that the binding of an agonist must be transduced into some kind of functional response (biological or physiological).
Different receptor types are linked to effector systems either directly or through simple or more-complex intermediate signal amplification systems. Some examples are:
Ligand-gated ion channels – nicotinic Ach receptors Single-transmembrane receptors – RTKs like insulin or EGF receptors 7-transmembrane GPCRs – opioid receptors Soluble steroid hormones – estrogen receptor

Predicting whether a drug will cause a response in a particular tissue
Factors involving the equilibrium of a drug at a receptor. Limited diffusion Metabolism Entrapment in proteins, fat, or blood.Response depends of what the receptor is connected to. Effector type Need for any allosteric co-factors – THB on tyrosine hydroxylase. Direct receptor modification – phosphorylation

Receptor theory and receptor binding.
Must obey the Law of Mass Action and follow basic laws of thermodynamics. Primary assumption – a single ligand is binding to a homogeneous population of receptors
NH+3
COO-

kon = # of binding events/time (Rate of association) = [ligand] [receptor] kon = M-1 min-1
koff = # of dissociation events/time (Rate of dissociation) = [ligand receptor] koff = min-1
Binding occurs when ligand and receptor collide with the proper orientation and energy.
Interaction is reversible.Rate of formation [L] + [R] or dissociation [LR]
depends solely on the number of receptors, the concentration of ligand, and the rate constants kon and koff.
kon/k1
[ligand] + [receptor] [ligand receptor]
koff/k2

Receptor occupancy, activation of target cell responses, kinetics of binding
•Activation of membrane receptors and target cell responses is proportional to the degree of receptor occupancy.
•However, the hormone concentration at which half of the receptors is occupied by a ligand (Kd) is often lower than the
concentration required to elicit a half-maximal biological response (ED50)

Assumptions of the law of mass action.
All receptors are equally accessible to ligand.
No partial binding occurs; receptors are either free of ligand or bound with ligand.
Ligand is nor altered by bindingBinding is reversibleDifferent affinity states?????

Competition binding assaysAllows one to determine a rough estimate of an
unlabeled ligand’s affinity for a receptor.Competitive or non-competitive. Introduction into the incubation mixture of a
non-radioactive drug (e.g. drug B) that also binds to R will result in less of R being available for binding with D*, thus reducing the amount of [D*R] that forms. This second drug essentially competes with D* for occupation of R. Increasing concentrations of B result in decreasing amounts of [D * R] being formed.
Method:◦ Single concentration of labeled ligand◦ Multiple (log-scale) concentrations of the
unlabeled/competing ligand.

Competition binding assaysThe concentration of inhibitor which displaces 50% of
the radiolabeled ligand is known as the IC50 for that drug.
IC50 cannot be viewed as the “KD” of the inhibitor because it is just an estimate.
Ki = the equilibrium inhibitor dissociation constant.
◦ It is the concentration of the competing ligand that would bind to 50% of sites in the absence of the radioligand.
Ki can only be determined after the IC50 is known.
Uses the equation of Cheng and Prusoff.
Ki = IC50
1 + [radiolabeled ligand] Kd

Receptor antagonists.
Prevent agonist-mediated responses by preventing a drug from binding and eliciting its normal response.
Intrinsic activity = 0.No sensitivity to Na+ or GTP.Antagonists are measured by the
selectivity, affinity for their receptor, and potency.

Irreversible antagonists. Binds in an irreversible manner, usually by
covalent modification of the receptor.EEDQ (non-selective)N-ethylmalemide (NEM) or other sulfhydryl
or alkylating agents (non-selective).AntibodiesMolecular control (mutation) – EXAMPLEPrevents binding at the atomic level.Effectively and practically lowers the
number of receptors capable of binding an agonist.
Adding more agonist is uselessOnly cure: Make New Receptors by
Protein Synthesis.

Receptor subtypes
First learned for the histamine receptor.◦ histamine activation by
agonist produces smooth muscle contraction.
The residual activity in gastric secretion, even in the absence of muscle contraction, indicated the presence of histamine-sensitive receptors.
Conclusion = Receptor Subtypes.◦ Receptor subtypes are
characterized by:◦ Binding differences
(selective ligands)◦ Function◦ Molecular cloning analysis
revealing amino acid differences.
% response
100
50
0
Log [histamine].001 .01 1 10 100 1000
contraction
Gastric secretion
Contraction + antagonist

Opioid receptor subtypes
Receptor type µ-Receptorm1, m2, m3 ??
d -Receptord1, d2 ??
k -Receptork1, k2 ??
Selective agonists endomorphin-1endomorphin-2DAMGO
[D-Ala2]-deltorphin I[D-Ala2]-deltorphin IIDPDPESNC 80DSLET
enadolineU-50488U-69593
Selective antagonists CTAP naltrindoleTIPP-yICI 174864
nor-binaltorphimine

Stopping the GPCR signal
Endogenous GTPase within Ga subunit
Proteolysis of receptor-rareNT re-uptake or enzymolysisRGS proteins-regulators of GTPaseReceptor internalization/down-
regulation.


Receptor desensitizationA loss of agonist affinity, but not receptor number
after chronic agonist stimulation.◦ Best example is b2-AR.◦ Activation of PKA/GRKs◦ Phosphorylation- b-arrestin
uncoupling of receptor and G-protein results in a rightward shift of the binding curve: DESENSITIZATION.
KD of isoproterenol (1 100 nM) goes up ◦ affinity goes down◦ number of receptors does not change (Bmax does not
change). b-arrestin binds with clathrin AP-2 binding site.
◦ Complex internalizes into membrane-bound endosomes.◦ Endosomes internalizes◦ transient decrease in surface receptor number.


Adapted from Lefkowitz, 1998 (JBC, vol., 273)

Receptor down-regulationProteolytic degradation of receptor
◦ producing a net loss in total cell receptor number.
PKC involvement during endocytosisBmax can decreases (~60%); KD remains
the sameUse of endosomes and lysosomes.



















