
Charcot Marie Tooth · PDF file• First identified in 1886 by Martin Charcot, Pierre Marie...
57
Charcot-Marie-Tooth Disease By Eleanor R. Hethcox, ACNP-BC
Transcript of Charcot Marie Tooth · PDF file• First identified in 1886 by Martin Charcot, Pierre Marie...

Charcot-Marie-Tooth Disease
By Eleanor R Hethcox ACNP-BC
Charcot-Marie-Tooth Disease
bull One of the most common inherited neurological disorders
bull Prevalence is 17-40100000 world wide
bull Affecting approximately 1 in 2500 people in the US
bull Affects male and females and all cultures
bull First identified in 1886 by Martin Charcot Pierre Marie and
Howard Henry Tooth
bull AKA ndash Hereditary Motor and Sensory Neuropathy (HMSN)
or peroneal muscular atrophy
bull Affects peripheral nerves ndash specifically related to the
myelin sheath and axons
Charcot-Marie-Tooth Disease
bull Many forms of CMT exist (CMT1234X)
bull Normal life expectancy CMT is not fatal
bull Onset of symptoms is most often in adolescence or early
adulthood
bull Progression of symptoms is very gradual
bull Slow progressing weakness beginning in the distal limb
muscles generally is noted Usually starts in lower
extremities before it starts in the upper extremities
bull Severity of symptoms may vary greatly among affected
individuals
Types of Hereditary Neuropathies
Charcot-Marie-Tooth Disease
bull A clinically and genetically heterogenous group of
disorders caused by mutations in genes that affect the
normal function of the peripheral nerves
Image from Muscular Dystrophy Association of NSW
httpwwwmdnsworgauoldFactsSheetsCMThtm
Axons
Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Charcot-Marie-Tooth Disease
bull One of the most common inherited neurological disorders
bull Prevalence is 17-40100000 world wide
bull Affecting approximately 1 in 2500 people in the US
bull Affects male and females and all cultures
bull First identified in 1886 by Martin Charcot Pierre Marie and
Howard Henry Tooth
bull AKA ndash Hereditary Motor and Sensory Neuropathy (HMSN)
or peroneal muscular atrophy
bull Affects peripheral nerves ndash specifically related to the
myelin sheath and axons
Charcot-Marie-Tooth Disease
bull Many forms of CMT exist (CMT1234X)
bull Normal life expectancy CMT is not fatal
bull Onset of symptoms is most often in adolescence or early
adulthood
bull Progression of symptoms is very gradual
bull Slow progressing weakness beginning in the distal limb
muscles generally is noted Usually starts in lower
extremities before it starts in the upper extremities
bull Severity of symptoms may vary greatly among affected
individuals
Types of Hereditary Neuropathies
Charcot-Marie-Tooth Disease
bull A clinically and genetically heterogenous group of
disorders caused by mutations in genes that affect the
normal function of the peripheral nerves
Image from Muscular Dystrophy Association of NSW
httpwwwmdnsworgauoldFactsSheetsCMThtm
Axons
Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Charcot-Marie-Tooth Disease
bull Many forms of CMT exist (CMT1234X)
bull Normal life expectancy CMT is not fatal
bull Onset of symptoms is most often in adolescence or early
adulthood
bull Progression of symptoms is very gradual
bull Slow progressing weakness beginning in the distal limb
muscles generally is noted Usually starts in lower
extremities before it starts in the upper extremities
bull Severity of symptoms may vary greatly among affected
individuals
Types of Hereditary Neuropathies
Charcot-Marie-Tooth Disease
bull A clinically and genetically heterogenous group of
disorders caused by mutations in genes that affect the
normal function of the peripheral nerves
Image from Muscular Dystrophy Association of NSW
httpwwwmdnsworgauoldFactsSheetsCMThtm
Axons
Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
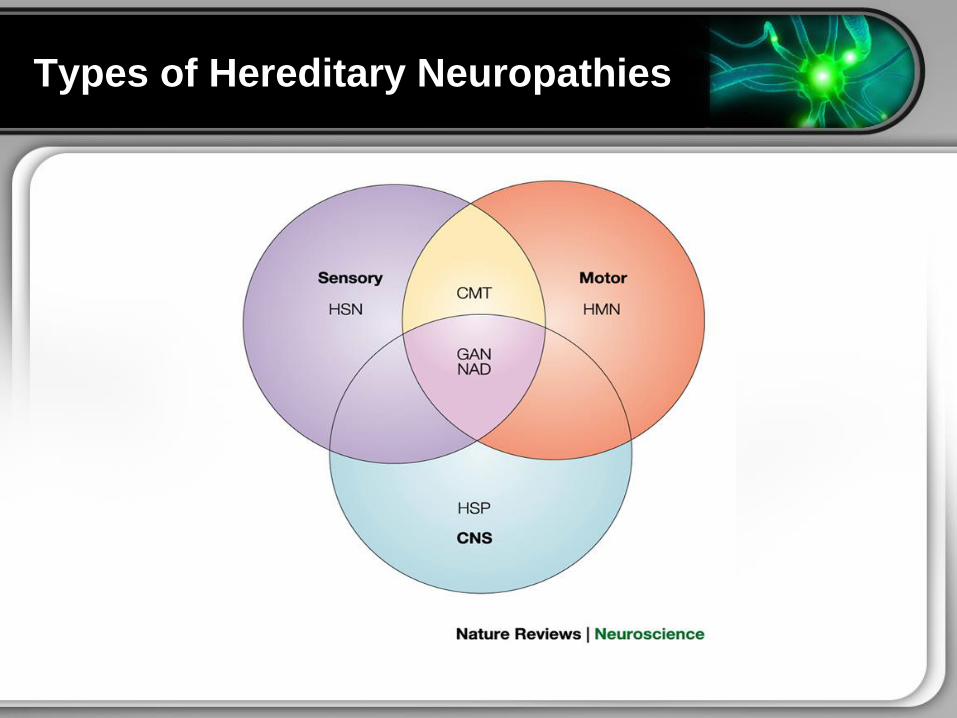
Types of Hereditary Neuropathies
Charcot-Marie-Tooth Disease
bull A clinically and genetically heterogenous group of
disorders caused by mutations in genes that affect the
normal function of the peripheral nerves
Image from Muscular Dystrophy Association of NSW
httpwwwmdnsworgauoldFactsSheetsCMThtm
Axons
Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
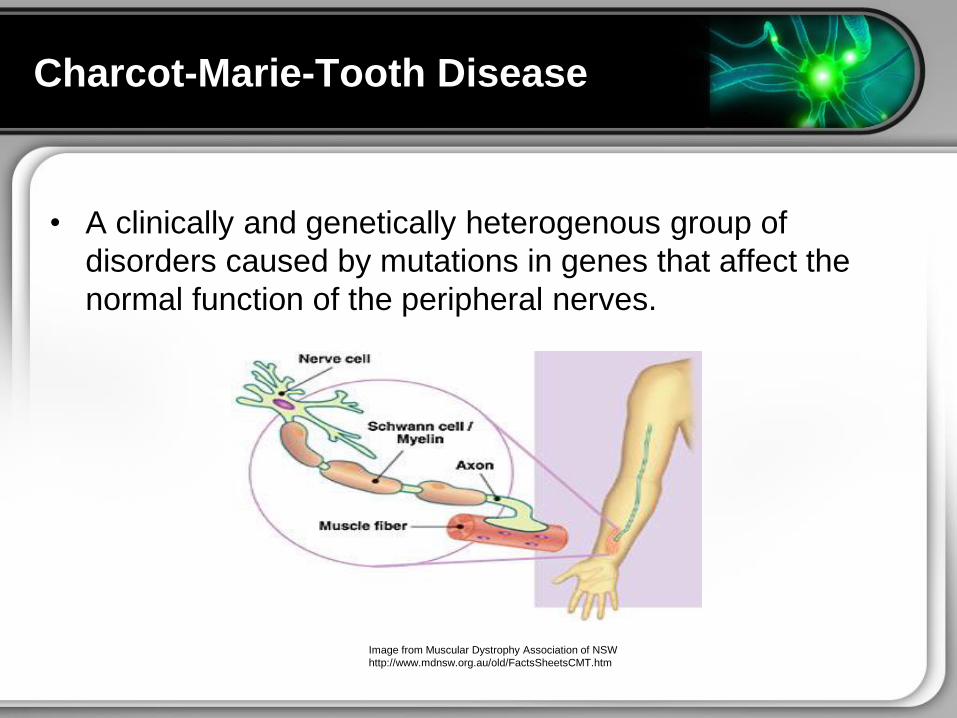
Charcot-Marie-Tooth Disease
bull A clinically and genetically heterogenous group of
disorders caused by mutations in genes that affect the
normal function of the peripheral nerves
Image from Muscular Dystrophy Association of NSW
httpwwwmdnsworgauoldFactsSheetsCMThtm
Axons
Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
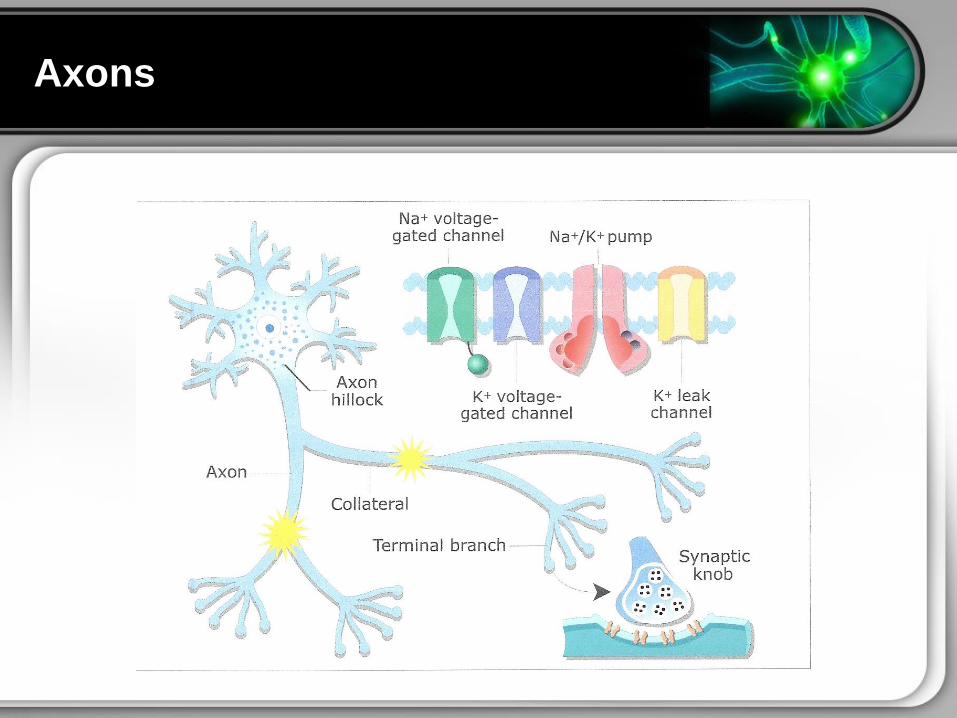
Axons
Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Functions of the Myelin Sheath
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
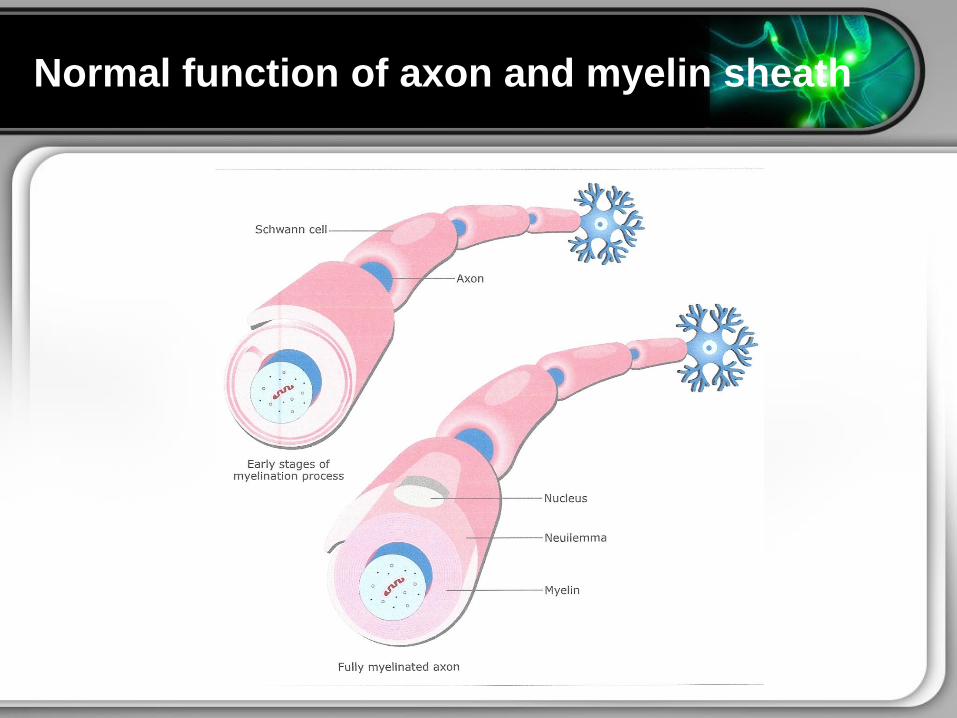
Normal function of axon and myelin sheath
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
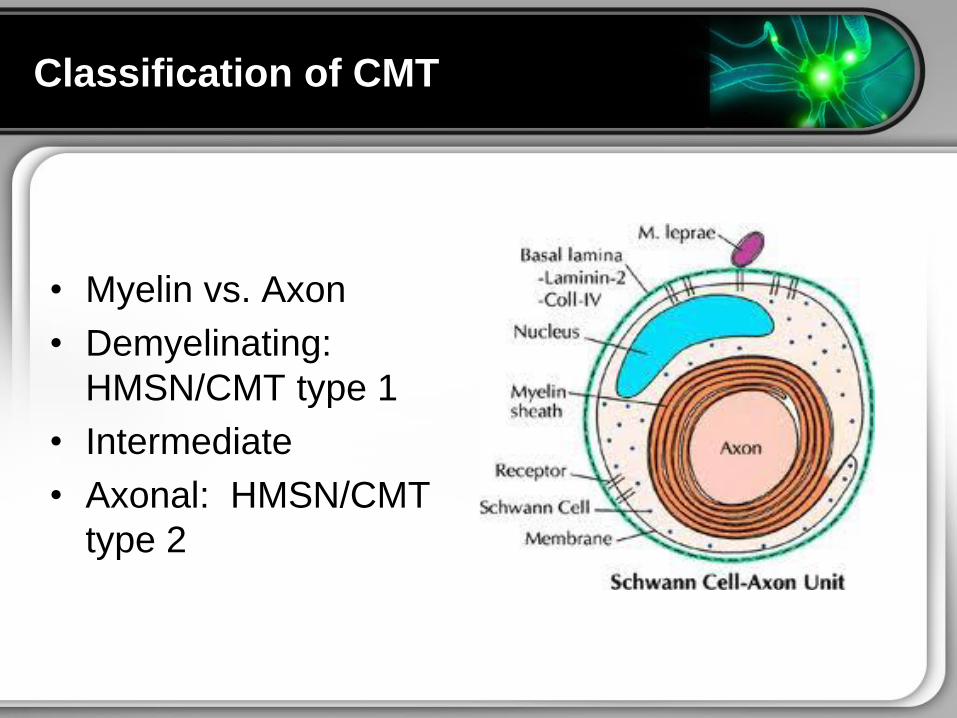
Classification of CMT
bull Myelin vs Axon
bull Demyelinating
HMSNCMT type 1
bull Intermediate
bull Axonal HMSNCMT
type 2
Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Peripheral Neuropathy
CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT ndash Clinico-electrophysiological Evaluation
CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT Inheritance Patterns
bull Extensive underling genetic heterogeneity
bull The CMT spectrum of disorders can be inherited in the following ways
1) Autosomal dominant
2) Autosomal recessive
3) X-linked inheritance (dominant and recessive)
(Spontaneous mutations have been reported)
bull More than 40 genes have been implicated as causes of
the various forms of CMT
bull Most commonly identified subtypes CMT1A CMTX1 hereditary
neuropathy with liability to pressure palsies CMT1B and CMT2A
Together these 5 subtypes account for 92 of genetically defined CMT
cases All other CMT subtypes amp assoc mutations each accounted for
lt1 of genetically defined CMT
Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Other Influences on CMT Phenotype
Even among family members with the same type of CMT
symptoms can vary widely It is therefore possible that other
genetic or environmental factors affect the development of
CMT
Comorbidities Nutritional Environment
Diabetes Mellitus
Obesity
Hypothyroidism
Exposure to Toxins
Genetic Background
Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Charcot Marie Tooth Types
CMT Type Chromosome
Inheritance
Pattern
Age of Onset Clinical Features Average
NCVs
CMT 1A
(PMP-22 dupl)
17p11AD First decade Distal Weakness 15-20 ms
CMT 1B (P0-
MPZ)
1q22AD First decade Distal Weakness lt20ms
CMT 1C CMT
(nonA nonB)
16p13AD Second
decade
Distal Weakness 26-42 ms
CMT 1D (early
growth
response
[EGR]-2)
10q21 AD First decade Distal Weakness 15-20 ms
CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT ndash Types Continued
CMT
Type
Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
CMT 1D 10q21 AD 1st decade Distal weakness 15-20 ms
CMT 1E 17p11 AD 1st decade Distal weakness 15-20 ms
CMT 1F 8p21 AD 1st decade Distal weakness 15-20 ms
CMT X Xq13 XD 2nd decade Distal weakness 25-40 ms
CMT 2A 1p36 AD 10 y Distal weakness gt 38 ms
CMT 2B 3q AD 2nd decade Distal weakness sensory loss
skin ulcers
Axon loss
normal
CMT 2C 12q23-q24 AD 1st decade Vocal cord diaphragm amp distal
weaknessgt 50 ms
CMT 2D 7p14 AD 16-30y Distal weakness upper limb
predominantly
Axon loss
normal
CMT 2E 8p21AD 10-30y Distal weakness lower limb
predominantly
Axon
lossnormal
CMT 2F 7q11-21 AD 15-25y Distal weakness Axon loss
normal
CMT 2G 12q12-q13 AD 9-76y Distal weakness Axon loss
normal
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
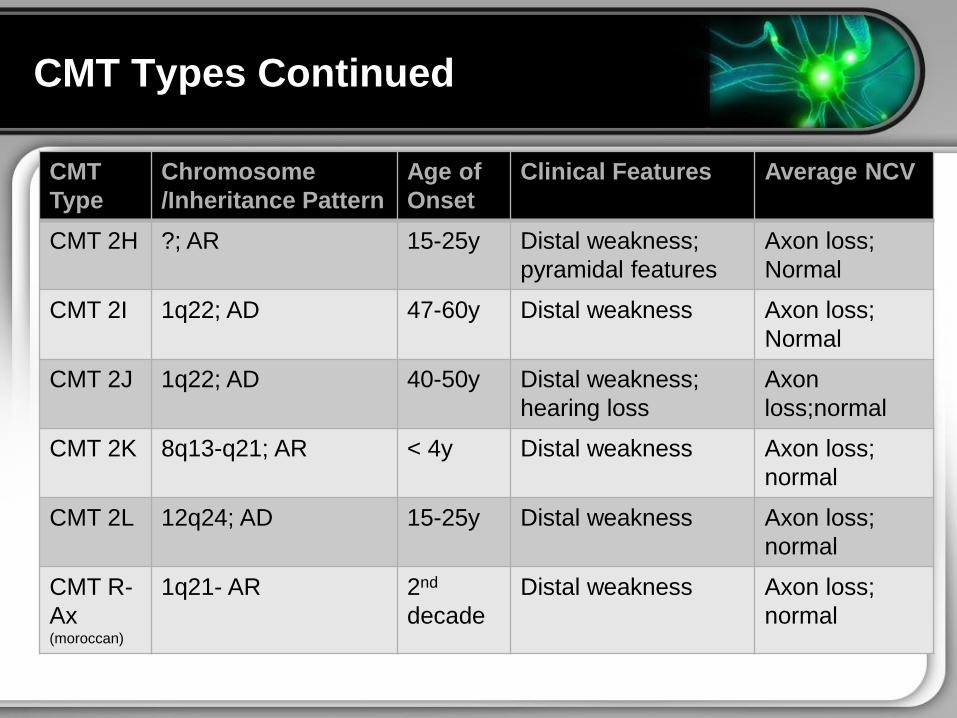
CMT Types Continued
CMT
Type
Chromosome
Inheritance Pattern
Age of
Onset
Clinical Features Average NCV
CMT 2H AR 15-25y Distal weakness
pyramidal features
Axon loss
Normal
CMT 2I 1q22 AD 47-60y Distal weakness Axon loss
Normal
CMT 2J 1q22 AD 40-50y Distal weakness
hearing loss
Axon
lossnormal
CMT 2K 8q13-q21 AR lt 4y Distal weakness Axon loss
normal
CMT 2L 12q24 AD 15-25y Distal weakness Axon loss
normal
CMT R-
Ax (moroccan)
1q21- AR 2nd
decade
Distal weakness Axon loss
normal
Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Types Continued
Type Chromosome
Inheritance
Pattern
Age of
Onset
Clinical Features Average
NCV
Cowchock
syndrome
Xq24-q26 1st
decade
Distal weakness
deafness mental
retardation
Axon loss
normal
HNPP (PMP-
22) or
tomaculous
neuropathy
17p11 AD All ages Episodic weakness and
numbness
Conduction
blocks
Dejerine-
Sottas
syndrome
(DSS) or
HMSN 3
P0 AR
PMP- 22 AD
8q23 AD
2y Severe weakness lt 10 ms
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
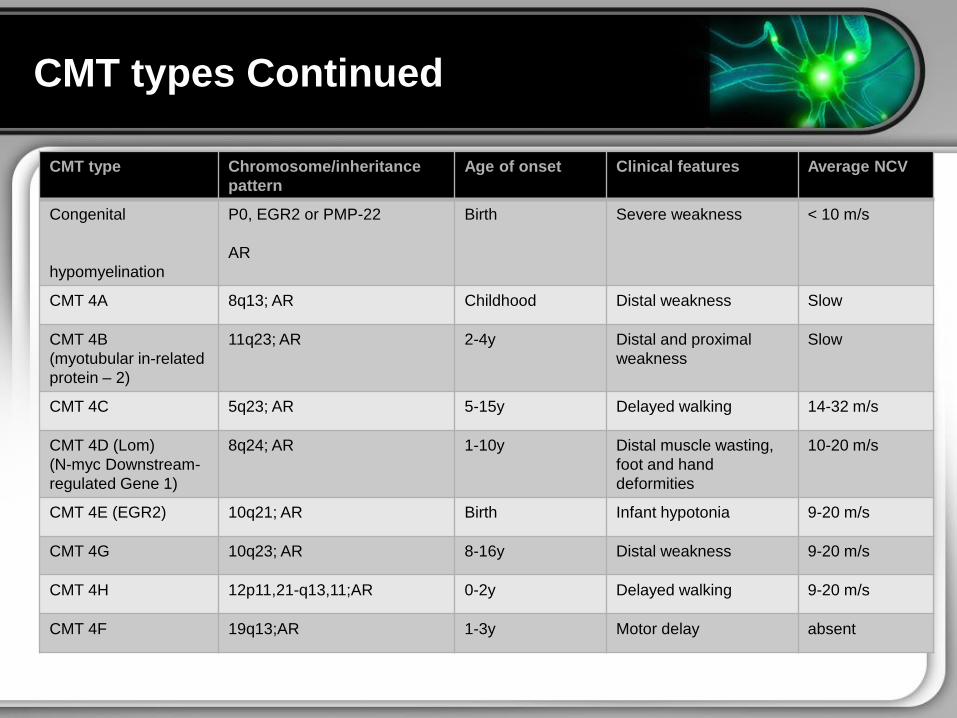
CMT types Continued
CMT type Chromosomeinheritance
pattern
Age of onset Clinical features Average NCV
Congenital
hypomyelination
P0 EGR2 or PMP-22
AR
Birth Severe weakness lt 10 ms
CMT 4A 8q13 AR Childhood Distal weakness Slow
CMT 4B
(myotubular in-related
protein ndash 2)
11q23 AR 2-4y Distal and proximal
weakness
Slow
CMT 4C 5q23 AR 5-15y Delayed walking 14-32 ms
CMT 4D (Lom)
(N-myc Downstream-
regulated Gene 1)
8q24 AR 1-10y Distal muscle wasting
foot and hand
deformities
10-20 ms
CMT 4E (EGR2) 10q21 AR Birth Infant hypotonia 9-20 ms
CMT 4G 10q23 AR 8-16y Distal weakness 9-20 ms
CMT 4H 12p1121-q1311AR 0-2y Delayed walking 9-20 ms
CMT 4F 19q13AR 1-3y Motor delay absent
CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT ndash Types (AD X-linked AR)
bull CMT 2D
bull CMT 2E
bull CMT 2F
bull CMT 2G
bull CMT 2H
bull CMT 2I
bull CMT 2J
bull CMT 2K
bull CMT 2L
bull CMT R-Ax
(Ouvrier)
bull CMT 1A
bull CMT 1B
bull CMT 1C
bull CMT 1D
bull CMT 1E
bull CMT 1F
bull CMT X
bull CMT 2A
bull CMT 2B
bull CMT 2C
bull CMT R-Ax
(Moroccan)
bull Cowchock
syndrome
bull HNPP (tomaculous
neuropathy)
bull DSS
bull Congenital
hypomyelination
(CH)
bull CMT 4A
bull CMT 4B
bull CMT 4C
bull CMT 4D
bull CMT 4E
bull CMT 4G
bull CMT 4H
bull CMT 4F
Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Most common CMT CMT linked types
bull CMT1A
bull CMT1B
bull CMT X1
bull CMT2A
bull HNPP (linked to CMT but not a type of
CMT)
These types occur most
commonly
CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT Type 1 A
PMP22
Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Genetics of CMT1 ndash demyelinating
bull Caused by
mutations in genes
that are expressed
in Schwann cells
bull Exhibit AD AR and
X-linked inheritance
bull Subdivided in types
ABCetc
CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT Type 1A Pedigree
bull Autosomal Dominant Inheritance
CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT 1A ndash Autosomal Dominant Form
bull Most common type of CMT
bull PMP22 (peripheral myelin
protein) a hydrophobic 22-
kDa glycoprotein of 160 aa
bull Largely unknown but
thought to have a role in
the initiation of myelin
spirals regulation of
growth amp differentiation of
Schwann cells and control
of thickness and stability of
myelin sheaths
CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT1A ndash PMP22 gene
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
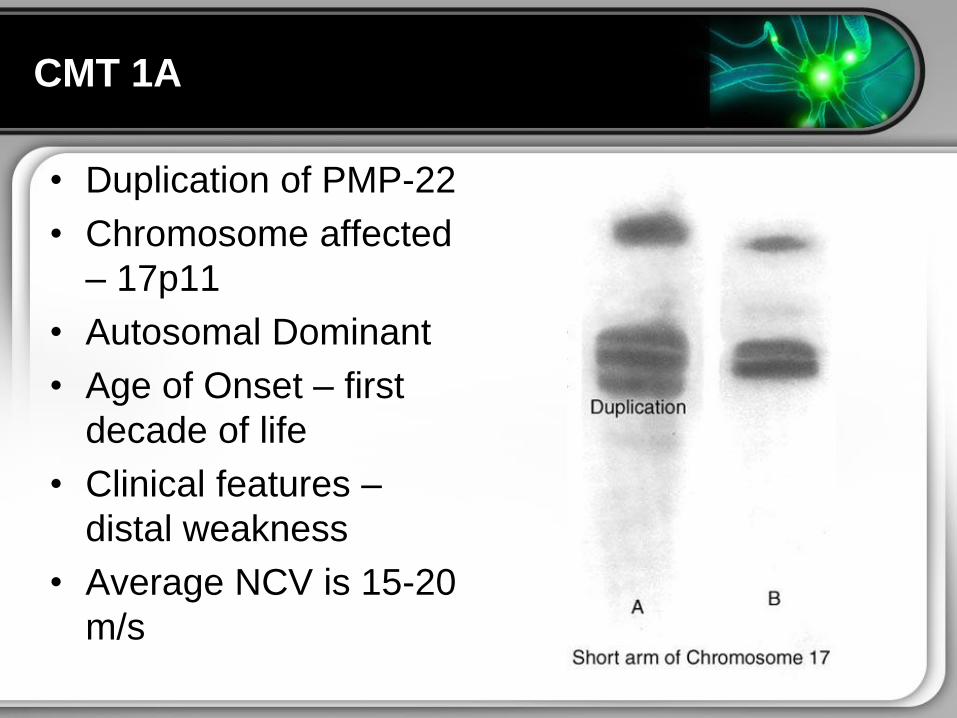
CMT 1A
bull Duplication of PMP-22
bull Chromosome affected
ndash 17p11
bull Autosomal Dominant
bull Age of Onset ndash first
decade of life
bull Clinical features ndash
distal weakness
bull Average NCV is 15-20
ms
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
CMT Type 1B
Gene ndash Myelin Protein Zero
Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Charcot Marie Tooth 1B
bull MPZ or Myelin Protein Zero
bull Normal function of MPZ is that of an adhesion molecule
bull Plays a role in myelin compaction
Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Charcot Marie Tooth 1B (CMT 1B)
bull Autosomal Dominant
bull Caused by mutations in the gene that carries the
instructions for manufacturing the myelin protein zero (P0)
bull Chromosome 1q22
bull Age of onset ndash first decade
bull Distal weakness
bull Avg NCV lt 20 ms
bull Less common than CMT1A
bull Mutations in the MPZ gene account for less than 5 of
CMT1 cases
CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT X1
Other Subtypes 23Cowchock syndrome 5
CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT X1
bull X-linked Dominant inheritance
bull Age of Onset is in the second decade of life
bull Clinical features include distal weakness
bull Average NCV ndash 25-40 ms
bull Caused by mutations in the gap junction protein beta 1 (GJB1 gene)
aka connexin 32 gene on chromosome Xq13
bull GJB1 encodes a gap junction protein that plays an important role in the
homeostasis of myelinated axons
CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT X1
bull Gene is expressed in myelinating Schwann cells but not
incorporated into the myelin sheath
bull Both sexes are affected symptoms more prominent in
boys
bull Gait problems foot deformities (pes planus or pes cavus)
bull Less common features include tremor hand weakness and
sensorineural deafness
bull Demyelination and axonal loss is observed histologically
but onion bulb formation is minimal
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
Hereditary neuropathy
with liability to pressure palsies
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
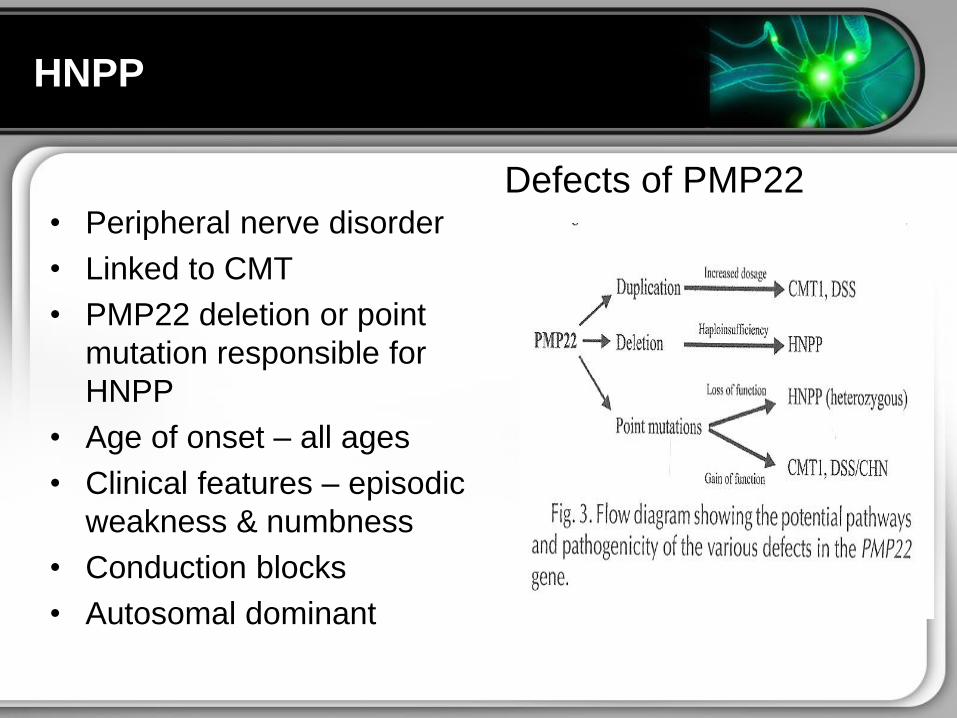
HNPP
bull Peripheral nerve disorder
bull Linked to CMT
bull PMP22 deletion or point
mutation responsible for
HNPP
bull Age of onset ndash all ages
bull Clinical features ndash episodic
weakness amp numbness
bull Conduction blocks
bull Autosomal dominant
Defects of PMP22
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
Charcot Marie Tooth
Type CMT2A
CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

CMT2A
bull Most common CMT2 Phenotype
ndash Axonal degeneration
bull Maps to chromosome 1p35-36
bull Gene most commonly
implicated is mitochondrial
fusion protein mitofusin 2
(MFN2)
bull Other genes implicated include
MT-ATP6 Dynamin 2
bull Majority of these cases are
autosomal dominant but the
genetic basis of CMT2 is
heterogenous like CMT1
bull Many other subtypes of CMT2
exist including BCDE etc
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
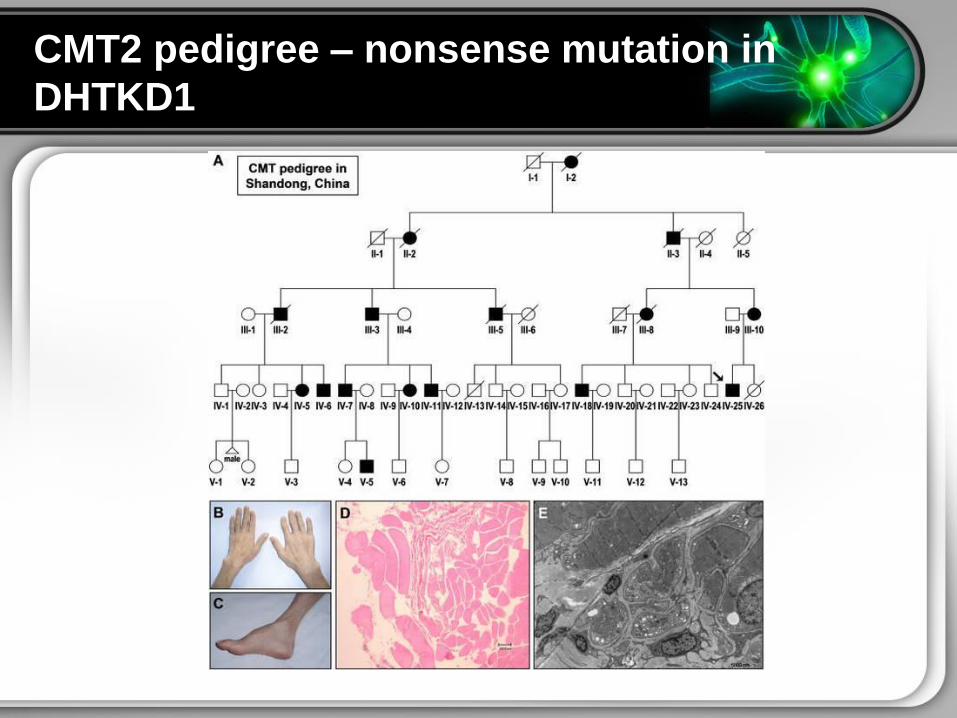
CMT2 pedigree ndash nonsense mutation in
DHTKD1
Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Charcot Marie Tooth
Work-up
Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Diagnosis of CMT Disease
bull Complete Medical
History and Physical
Examination
bull Family History very
important
bull Neurological
Examination
bull Genetic Testing
bull Nerve Conduction
Studies
bull Electromyography
bull Nerve biopsy
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
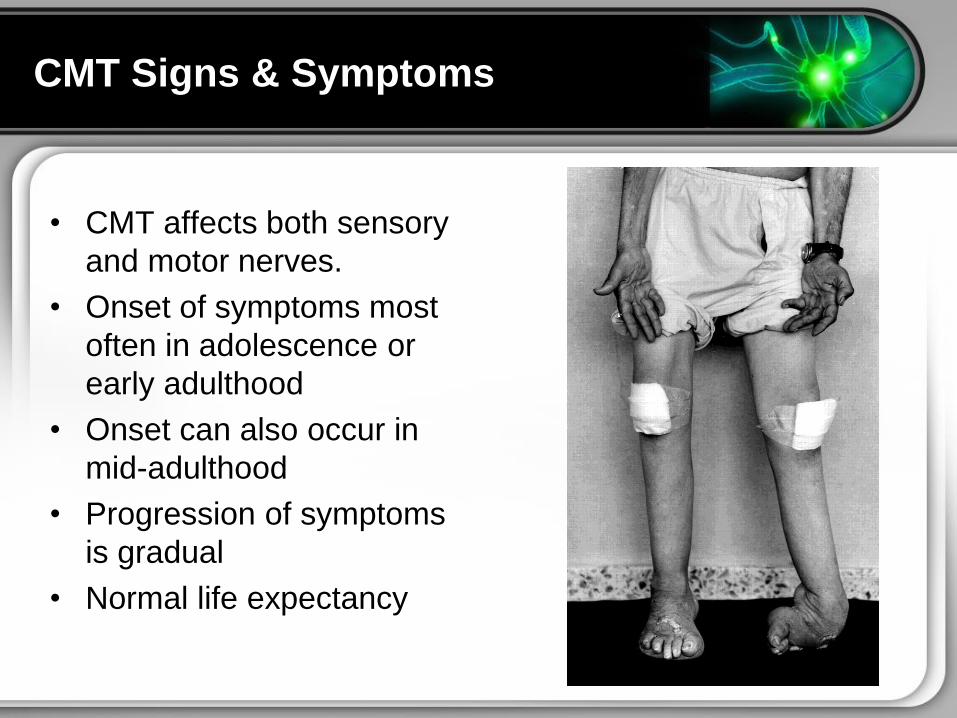
CMT Signs amp Symptoms
bull CMT affects both sensory
and motor nerves
bull Onset of symptoms most
often in adolescence or
early adulthood
bull Onset can also occur in
mid-adulthood
bull Progression of symptoms
is gradual
bull Normal life expectancy
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
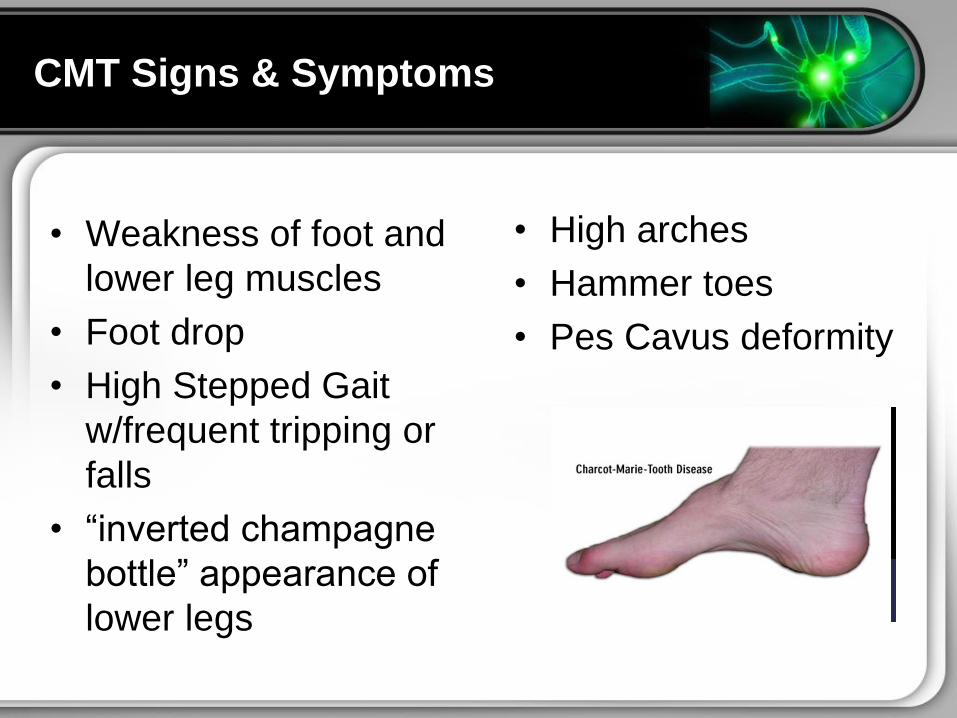
CMT Signs amp Symptoms
bull Weakness of foot and
lower leg muscles
bull Foot drop
bull High Stepped Gait
wfrequent tripping or
falls
bull ldquoinverted champagne
bottlerdquo appearance of
lower legs
bull High arches
bull Hammer toes
bull Pes Cavus deformity
Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Findings of CMT
bull Historyndash Significant family history
ndash Slowly progressing weakness
beginning in the distal limb muscles
ndash Muscle wasting weakness
ndash Onset usually occurs in the first 2
decades of life
ndash Initial co difficulty walking frequent
tripping due to foot and distal leg
weakness Frequent ankle sprains
and falls
ndash Child can be clumsy andor not
athletic
ndash Foot drop commonly occurs
ndash Steppage is common (gait)
ndash Foot deformity (pes cavus)
bull Physicalndash Distal wasting in legs ndash ldquostork leg or
inverted champagne bottle
appearancerdquo
ndash Pes cavus (high arch foot)
ndash Spinal deformities (thoracic
scoliosis) in 37-50 of patients with
CMT type 1
ndash DTRs are markedly diminished or
absent
ndash Vibration sensation and
proprioception are significantly
decreased
ndash Sensory gait ataxia and positive
Romberg test
ndash Sense of paintemp is intact
ndash Essential tremor in 30-50 of
patients
Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Genetic Testing for Charcot Marie Tooth
bull For family planning purposes
bull Natural history studies
bull Entry into clinical trials
bull Usually very expensive
bull Can be confusing for patients
bull Most general practitioners also confused due to locus
heterogeneity
bull Has implications for medical treatments
bull Several algorithms have been developed
Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Genetic Testing Algorithms
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Algorithm for Genetic Testing CMT
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
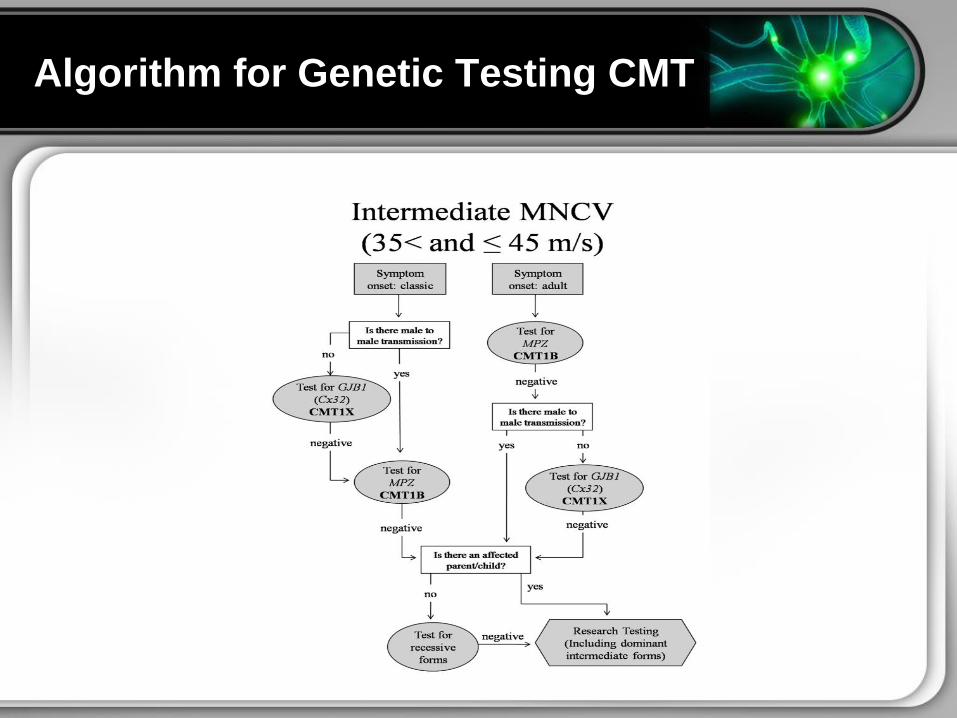
Algorithm for Genetic Testing CMT
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
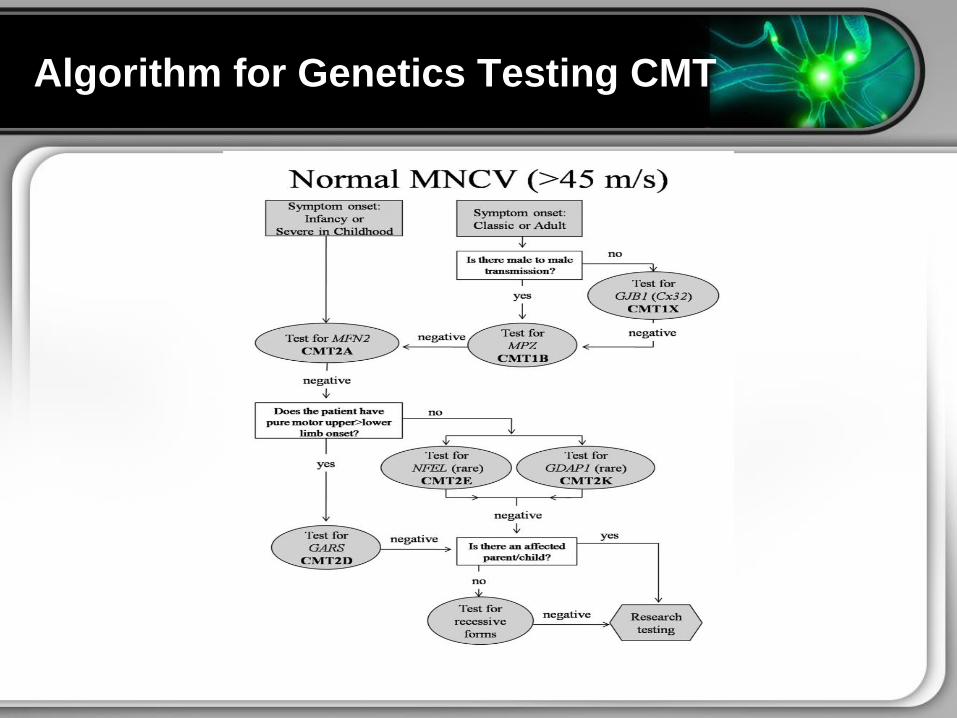
Algorithm for Genetics Testing CMT
Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Management of CMT
bull Is supportive only
bull Disease-modifying therapy IS NOT available
bull Physical Therapy
bull Occupational Therapy
bull Braces and other orthopedic devices
bull Orthopedic surgery
bull Analgesics for severe pain
bull Muscle strengthening to delay or reduce muscle atrophy
bull Avoidance of drugs known to exacerbate CMT symptoms
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
Charcot Marie Tooth
Latest Therapies
Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Promising Research for New CMT Treatments
bull Lck tyrosine kinase mediates β1-integrin signaling to
regulate Schwann cell migration and myelination
bull What we have found is that Lck is essentially the switch
that signals migration of the Schwann cells and production
of the myelin sheath Dr Tapinos said This finding sets
the stage for further research into the specific molecular
mechanisms that occur in order for this process to break
down and eventually toward developing treatments to
prevent it
Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Promising Research for New CMT Therapies
bull April 2013
bull Hitting lsquoresetrsquo in protein synthesis restores myelination suggests new
treatment for misfolded protein diseases such as CMT Alzheimerrsquos
bull Lawrence Wrabetz MD director of the institute and professor of neurology and
biochemistry in UBrsquos School of Medicine and Biomedical Sciences ldquoThe
misfolded protein diseases are an interesting and challenging group of
diseases to study CMT for example is caused by mutations in more than 40
different genesrdquo he says ldquoWhen there are so many different genes
involved and so many different mechanisms you have to find a unifying
mechanism this problem of Gadd34 turning protein synthesis on at too high a
level could be one unifying mechanism The hope is that this proof of principle
applies to more than just CMT and may lead to improved treatments for
Alzheimerrsquos Parkinsonrsquos Type 1 diabetes and the other diseases caused by
misfolded proteinsrdquo
Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Integrative Therapies for CMT
bull L-carnitine
bull TENS
bull Aconite ndash homeopathic
bull Biotin
bull Folate
(NOTE There is lack of sufficient scientific data on use of
these therapies for treatment of CMT)
Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Latest Research and Clinical Trials
bull Natural History Evaluation of Charcot Marie Tooth Disease (CMT)
Types CMT1B CMT2A CMT4A CMT4C and Others
bull Effects of Coenzyme Q10 on Charcot-Marie-Tooth Disease
bull Genetics of Charcot Marie Tooth (CMT) - Modifiers of CMT1A New
Causes of CMT2
bull Treadmill Stretching and Proprioceptive Exercise (TreSPE)
Rehabilitation Program for CharcotminusMarieminusTooth Neuropathy Type 1A
(CMT1A)
bull Development of Charcot Marie Tooth Disease (CMT) Pediatric Scale
for Children With CMT
bull Correlation Between Clinical and Electrophysiological Phenotypes in a
Population of Patients With Neuropathy Charcot-Marie-Tooth Disease
Type 1A
bull Ascorbic Acid Treatment in CMT1A Trial (AATIC)
bull Phase II Randomized Placebo-controlled Trial in Patients With
Charcot-Marie-tooth Disease Type 1A
Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

Resource Centers for CMT
National CMT Resource Center
Charcot-Marie-Tooth Association
National Society for Genetic Counselors (NSGC)
Hereditary Disease Foundation
Hereditary Neuropathy Foundation
Alliance of Genetic Support Groups
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth
Links to some CMT websitevideos
bull httpwwwnhsukvideopagescharcot-marie-tooth-
diseaseaspx
bull httpwwwmolgenuaacbeCMTMutationsHomeIPNcfm
bull httpwwwyoutubecomwatchv=Jspjvd43x7g
bull httpwwwbuffaloedunewsreleases201304056html
bull httpwwwncbinlmnihgovpmcarticlesPMC3058597
bull httpghrnlmnihgovgeneGJB1
bull httpwwwnaturecomncommsjournalv4n5fullncomms2
928htmlgenesandproteins
References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth

References
bull Cruse RP (2013) Hereditary primary motor sensory neuropathies including Charcot-Marie-Tooth disease Retrieved from
wwwuptodatecom
bull Houden H amp Reilly M (2006) Molecular Genetics of Autosomal-Dominant Demyelinating Charcot-Marie-Tooth Disease
NeuroMolecular Medicine (8 43-62)
bull Kedlaya D amp Calhoun J (2012) Charcot-Marie-Tooth Disease Clinical Presentation Retrieved from
httpemedicinemedscapecomarticle1232386-clinical
bull Martins Elisabeth N Branco Andreacute C Alvarenga Lecircnio S Uno Fausto Moraes Nilva B amp Belfort Jr Rubens (2000)
Charcot-Marie-Tooth disease and posterior scleritis a case report Arquivos Brasileiros de Oftalmologia 63(5) 413-415
Retrieved November 10 2013 from httpwwwscielobrscielophpscript=sci_arttextamppid=S0004-
27492000000500016amplng=enamptlng=en 101590S0004-27492000000500016
bull Suter U amp Scherer SS (2003) Disease mechanism in inherited neuropathies Nature Reviews Neuroscience (4 714-726)
bull Szigeti K Garcia CA amp Lupski JR (2006) Charcot-Marie-Tooth disease and related hereditary polyneuropathies Molecular
diagnostics determine aspects of medical management Genetics in Medicine (8 86-92) Retrieved from
httpwwwnaturecomgimjournalv8n2fullgim200614ahtml
bull httpstructbiovanderbiltedusandersResearch_Julia_Ver_1Researchhtml
bull Exp Neurol 2009 Aug218(2)268-73 doi 101016jexpneurol200905003 Epub 2009 May 8Role of mitofusin 2 mutations in
the physiopathology of Charcot-Marie-Tooth disease type 2ACartoni R Martinou JC
bull httpwwwclarksnutritioncomnsDisplayMonographaspStoreID=2691B1FE187D41ACB869A85CA5957A0AampDocID=condition-
charcotmarietooth


















