
DISSOCIATION OF CLATHRATE HYDRATES BELOW THE ICE POINT
10
DISSOCIATION OF CLATHRATE HYDRATES BELOW THE ICE POINT Satoshi Takeya ∗ Research Institute of Instrumentation Frontier National Institute of Advanced Industrial Science and Technology (AIST) Central 5, 1-1-1, Higashi, Tsukuba 305-8565 JAPAN John A. Ripmeester Steacie Institute for Molecular Sciences National Research Council of Canada 100 Sussex Dr., Ottawa, ON, K1A0R6 CANADA ABSTRACT The existence of enhanced preservation phenomena for structure I CH 4 hydrate in the temperature region from 240 K to 273.2 K is one remaining puzzle: this phenomenon has been termed “anomalous preservation”. So far, this has been observed only for CH 4 hydrate upon dissociation by rapid pressure-release from high pressures at which CH 4 hydrate is stable to an ambient pressure of CH 4 gas in this temperature region. Dissociation of gas hydrates by temperature- ramping depends on neither the thermodynamic stability nor the crystal structure, but the nature of the guest molecules. Additionally, anomalous preservation, the existence of gas hydrates far outside their stability zone below the melting point of ice, is shown to depend on the type of guest molecule and the morphology of the hexagonal ice grown during hydrate dissociation by rapid pressure-release. Here, the dissociation mechanism of clathrate hydrates below the ice point is discussed on the basis of the experimental results obtained so far. Keywords: self-preservation, anomalous preservation, gas hydrates, dissociation ∗ Corresponding author: Phone: +81 29 861 6006 Fax +81 28 861 4845 E-mail: [email protected] INTRODUCTION Clathrate hydrates, so called gas hydrates, are crystalline ice-like inclusion compounds consisting of water molecules hydrogen-bonded to form host cages that contain small molecules as guest. Three different crystal structural families are known, the structure depending on the nature of the guest molecules; structure I with space group Pm3n, structure II with space group Fd-3m, and structure H with space group P6/mmm.[1] Guest-host interactions play a crucial role, as hydrates are thermodynamically stable only when guest molecules occupy the host cages to a certain minimum level. Even for hydrate growth, the interaction energy and dynamics of the host and guest molecules are also important. For instance, hydrate formation starting from amorphous solid water (ASW) with enclosed guest molecules at low temperatures is an example: H 2 O molecules phase-separate from the amorphous phase to form cubic ice (Ic) first, then they form the appropriate hydrates with temperature increase.[2-4] Recently, the dissociation of clathrate hydrates has also been studied, and progress towards understanding the mechanism has been made. For instance, CH 4 hydrate can be stored at atmospheric pressure below the ice point (273 K) even though this is well outside the zone Proceedings of the 7th International Conference on Gas Hydrates (ICGH 2011), Edinburgh, Scotland, United Kingdom, July 17-21, 2011.
Transcript of DISSOCIATION OF CLATHRATE HYDRATES BELOW THE ICE POINT

DISSOCIATION OF CLATHRATE HYDRATES BELOW THE ICE POINT
Satoshi Takeya∗ Research Institute of Instrumentation Frontier
National Institute of Advanced Industrial Science and Technology (AIST) Central 5, 1-1-1, Higashi, Tsukuba 305-8565
JAPAN
John A. Ripmeester Steacie Institute for Molecular Sciences National Research Council of Canada 100 Sussex Dr., Ottawa, ON, K1A0R6
CANADA
ABSTRACT The existence of enhanced preservation phenomena for structure I CH4 hydrate in the temperature region from 240 K to 273.2 K is one remaining puzzle: this phenomenon has been termed “anomalous preservation”. So far, this has been observed only for CH4 hydrate upon dissociation by rapid pressure-release from high pressures at which CH4 hydrate is stable to an ambient pressure of CH4 gas in this temperature region. Dissociation of gas hydrates by temperature-ramping depends on neither the thermodynamic stability nor the crystal structure, but the nature of the guest molecules. Additionally, anomalous preservation, the existence of gas hydrates far outside their stability zone below the melting point of ice, is shown to depend on the type of guest molecule and the morphology of the hexagonal ice grown during hydrate dissociation by rapid pressure-release. Here, the dissociation mechanism of clathrate hydrates below the ice point is discussed on the basis of the experimental results obtained so far.
Keywords: self-preservation, anomalous preservation, gas hydrates, dissociation
∗ Corresponding author: Phone: +81 29 861 6006 Fax +81 28 861 4845 E-mail: [email protected]
INTRODUCTION Clathrate hydrates, so called gas hydrates, are crystalline ice-like inclusion compounds consisting of water molecules hydrogen-bonded to form host cages that contain small molecules as guest. Three different crystal structural families are known, the structure depending on the nature of the guest molecules; structure I with space group Pm3n, structure II with space group Fd-3m, and structure H with space group P6/mmm.[1] Guest-host interactions play a crucial role, as hydrates are thermodynamically stable only when guest molecules occupy the host cages to a certain minimum level. Even for hydrate growth, the
interaction energy and dynamics of the host and guest molecules are also important. For instance, hydrate formation starting from amorphous solid water (ASW) with enclosed guest molecules at low temperatures is an example: H2O molecules phase-separate from the amorphous phase to form cubic ice (Ic) first, then they form the appropriate hydrates with temperature increase.[2-4]
Recently, the dissociation of clathrate hydrates has also been studied, and progress towards understanding the mechanism has been made. For instance, CH4 hydrate can be stored at atmospheric pressure below the ice point (273 K) even though this is well outside the zone
Proceedings of the 7th International Conference on Gas Hydrates (ICGH 2011), Edinburgh, Scotland, United Kingdom, July 17-21, 2011.

of thermodynamic stability of the hydrate,[5] an effect that has been termed self-preservation [6] or anomalous preservation. [7] Natural gas hydrates are expected to be a new global energy source [8] and there are also concerns about this large reservoir of methane which may contribute to global warming by their dissociation. [9]
In this paper, we report on recent progress in determining the dissociation mechanism of gas hydrates below the ice point. EXPERIMENTAL METHODS In-situ observations of the dissociation process of gas hydrates have been extensively studied by measuring the surface and mass fraction changes. [10-26] However, visualization of gas hydrate coexisting with ice using conventional techniques, such as optical microscopy, confocal microscopy, X-ray absorption contrast imaging or X-ray computed tomography (CT), and magnetic resonance imaging (MRI), are difficult because both materials are composed of water molecules, even though gas hydrates include caged gas molecules. Powder diffraction techniques using X-rays or neutrons are useful as they allow us to make in-situ measurements under complex sample conditions (P, T), and these methods can give quantitative information on crystallographic transformations. It is noteworthy that even very fast scan diffraction data allow a precise analysis because of the high signal-to-noise ratio possible with the help of a high-speed detector in in-house X-ray powder diffraction studies. Neutron radiation is also a powerful diffraction tool, but the use of neutrons is limited compared to synchrotron radiation because of high costs and limited availability worldwide. Many studies involving hydrogen atoms have been executed on deuteriated materials using the neutron diffraction technique because such experiments provide unique information on the location of the deuterium atoms.
Visual observation of the actual gas hydrate rather than alternative model gas hydrates is necessary to assess the potential of gas hydrates as gas storage media, because it has been reported that self-preservation and anomalous preservation phenomena strongly depend on the type of guest molecule.[24] Cryogenic scanning electron microscopy (cryo-SEM) is used for surface observations of dissociated gas hydrates. Special resolution for the SEM is higher than for the other methods noted above and the detailed texture of
ice on the surface of dissociating gas hydrate can be obtained. It is also possible to distinguish gas hydrates and ices by means of energy dispersive X-ray spectroscopy (EDX). However, the use of these measurements is limited to only ex-situ observations under vacuum condition. Phase contrast X-ray imaging is a unique technique used to identify gas hydrates coexisting with both ice
[27] and liquid water [28], while X-ray absorption-contrast imaging requires an additive to visualize gas hydrates coexisting with water to enhance the difference in density between the hydrate and water.[29] This is because an X-ray-phase-shift cross-section is more than a hundred times larger than that of X-ray absorption.[30] The advantage of phase contrast imaging is that it is more pronounced in elements with low atomic numbers, such as carbon and oxygen. In this respect, phase contrast X-ray imaging should be viable for the probing the structure of gas hydrate coexisting with ice. However, this X-ray imaging technique is also limited only to ex-situ observations.
Now, we have many options for measuring gas hydrate dissociation, but we still need to use several experimental techniques together for comprehensive understanding of the dissociation mechanism for gas hydrates. HYDRATE DISSOCIATION The dissociation of materials is generally regarded as a spontaneous reaction, while crystallization usually consists of two events, nucleation and crystal growth. Dissociation of gas hydrates below the ice point may also consist of two events, nucleation of gas bubbles and ice particles with subsequent dissociation of the hydrate. In the case of the self-preservation phenomenon of CH4 hydrate at temperatures below 200K, dissociation is controlled by two processes, namely initial rapid dissociation followed by slow diffusion of the gas through the ice. The dissociation rate increase with temperature increase.[7] Above 230 K, the reduction in diffusion rate due to ice sheet formation, transformed from ice particles around dissociating hydrate, was suggested by Shimada et al.[18] On the other hand, Kuhs et al. reported formation of ice Ic with stacking faults, but not hexagonal ice (Ih), due to CH4 hydrate dissociation below 240 K,[15] and they suggested that transformation from ice Ic to ice Ih upon annealing hinders gas diffusion. These studies, based on crystal structure transformation, have clarified that

the dissociation of CH4 hydrate is controlled by the rate of gas diffusion through ice. The existence of unusually enhanced preservation phenomena of CH4 hydrate as reported by Stern et al. in the temperature region from 240 K to 273.2 K[7] is still one remaining puzzle, a phenomenon which has been termed anomalous preservation. The dissociation rates vary in a reproducible way with two minima at around 250 K and 268 K whereas the dissociation is fast below 240 K and appears to be thermally activated. This anomalous preservation has been observed in this temperature region only for CH4 hydrate upon dissociation by rapid pressure-release from the high pressures at which CH4 hydrate is stable, to an ambient pressure of CH4 gas. In contrast, CH4+C2H6, a mixed gas hydrate of structure II, did not show preservation behavior comparable to CH4 hydrate, whereas its dissociation pressure is lower than that of CH4 hydrate at the same temperature.[12] Additionally, anomalously stable N2, O2, CO, and Ar hydrate have also been suggested. [31] Therefore, further studies are required for developing a comprehensive understanding of the dissociation mechanism of hydrates below 273 K.
Dissociation of gas hydrates strongly depend on the sample treatment after their formation as well as initial ice content as impurity and sample size of gas hydrates. Figure 1 show two ways to take samples from synthesis conditions of elevated pressure down to atmospheric pressure prior to gas hydrate dissociation measurements: by the rapid pressure drop method and by the temperature
ramping method. Herein, self-preservation is defined as the presence of gas hydrate below 240K and anomalous preservation is defined as its existence above 240K up to the ice point. 1. Dissociation under isothermal conditions
By measuring the reaction rate for kinetic process under isothermal conditions, Arrhenius plots are often used to analyze the effect of temperature on the rates of chemical reactions. For a single rate-limited thermally activated process, an Arrhenius plot gives a straight line, from which the activation energy can be determined.
Figure 2(a) shows the changes of X-ray diffraction profiles from structure I CH4 hydrate to hexagonal ice Ih during 80 min of dissociation at 198 K. [11] The initial profile is that of CH4 hydrate at 120 K. Upon varying the experimental temperature, several diffraction peaks from ice Ih appeared and continued to increase with time,
140 160 180 200 220 240 260 280
1
10
102 Water+
CH4(g)
CH
4pr
essu
re [k
Pa]
Temperature (K)
103
104 CH4 hydrate+CH4(l)
A
B
Ice + CH4(g)
D
C
CH4 hydrate+CH4(g)
140 160 180 200 220 240 260 280
1
10
102 Water+
CH4(g)
CH
4pr
essu
re [k
Pa]
Temperature (K)
103
104 CH4 hydrate+CH4(l)
A
B
Ice + CH4(g)
D
C
CH4 hydrate+CH4(g)
Figure 1. Phase diagram for the CH4-H2O system and experimental conditions. Point A indicates the synthesis condition of sample CH4hydrate. The temperature were then reduced to point C, and quenched to B for temperature-ramping method. Otherwise, pressure was rapidly released from C to D.
15 20 25Photon energy [keV]
020
40
60
80
Tim
e [m
in]
Ih(100)Ih(002)
Ih(101)
Ih(102) Ih(110)
(222)(320) (410)(321)
(b)253K233K213K205K198K189K178K168K158K148K
10 100 1000 10000Time [min]
0
0.0
0.2
0.4
0.6
0.8
1.0
Rel
ativ
e in
tens
ity
(a)
15 20 25Photon energy [keV]
020
40
60
80
Tim
e [m
in]
Ih(100)Ih(002)
Ih(101)
Ih(102) Ih(110)
(222)(320) (410)(321)
15 20 25Photon energy [keV]
020
40
60
80
Tim
e [m
in]
Ih(100)Ih(002)
Ih(101)
Ih(102) Ih(110)
(222)(320) (410)(321)
(b)253K233K213K205K198K189K178K168K158K148K
10 100 1000 10000Time [min]
0
0.0
0.2
0.4
0.6
0.8
1.0
Rel
ativ
e in
tens
ity
253K233K213K205K198K189K178K168K158K148K
10 100 1000 10000Time [min]
0 10 100 1000 10000Time [min]
0
0.0
0.2
0.4
0.6
0.8
1.0
Rel
ativ
e in
tens
ity
(a)
Figure 2 (a) In situ, time-resolved, energy-dispersive X-ray diffraction profiles during the transformation of CH4 hydrate to ice Ih. The temperature is 198 K. Indices of the crystal planes are labeled on the diffraction peaks. (b) The integrated intensity of the (321) crystal plane of CH4 hydrate as a function of time. The intensity was scaled to initially equal one.

whereas the CH4 hydrate diffraction peak heights decreased with time. This indicates that the amount of hydrate decreased and the amount of ice increased; the dissociated CH4 hydrate likely transformed into ice Ih as it released CH4 gas. The integrated intensity of X-ray diffraction is proportional to the crystal volume; hence, their rates of change are measures of the transformation rates. Figure 2(b) shows that the initial dissociation rate of CH4 hydrate was relatively fast within the first several tens of minutes for the temperature of 189 K and above (first regime) and then became relatively slow (second regime). In the second regime, hydrate decomposition is controlled by the rate of diffusion of released CH4 gas from the inside of the particle to the outside. [11] Here, the particle is assumed to be a sphere of fixed external radius rho. The hydrate is a smaller sphere of radius rh at the center of the particle. Outside the hydrate, but inside the particle, the CH4 pressure equals the dissociation pressure of CH4 hydrate, but this gas is trapped by a shell of ice with interior radius ri and exterior radius rho. With a CH4 diffusion constant D inside the ice and a scaled radius R = rh/rho, the hydrate radius at time t is
)1(.)(6)1(2)1(30
232 t
CCCTC
rDRR
a
ad
ho
−−
=−+−
Here Cd (T) is the density of CH4 gas in the gas phase at the dissociation pressure of CH4 hydrate at temperature T, Co is the CH4 density in the hydrate, and Ca is the CH4 density in the surrounding atmosphere. Because the latent heat of the transformation is small, it was assumed that the hydrate was at the same temperature as the N2 gas. The hydrate dissociation is well described by the diffusion model at 168 - 198 K. These values correspond to an Arrhenius activation energy for diffusion of 20.1 kJ/mol. The diffusion constants are relatively high [32] and the activation energy is smaller than for other molecules for diffusion through ice Ih.[33] These results imply that the CH4 does not diffuse through a solid ice Ih layer, but instead through pores or grain boundaries. However, the hydrate decomposition at 158 K did not follow a diffusion process. Hence, the dissociation reaction or another process probably determines the dissociation rate below 158 K. The temperature range from 180K to 220 K is quite consistent with the ice
transformation temperature where ice Ic transforms to ice Ih. As reported by Kuhs et al., [15] the relative peak intensity ratios of the Ih(100) and Ih(002) reflections, which overlap with those of Ic(111), changed with time in this region due to formation of ice Ic and its transformation into ice Ih (see Fig.2(a)). At the initial stage of the hydrate dissociation, ice Ic formation was observed but ice Ic transformed into normal hexagonal ice Ih as the hydrate dissociated under isothermal condition. 2. Dissociation by temperature ramping
Warming the sample above the CH4 hydrate dissociation temperature (193 K at 0.1 MPa) by heating destabilizes the hydrate. Further warming through the ice point is then required to fully melt
160 180 200 220 240 2600.0
0.2
0.4
0.6
0.8
1.0
CH4C2H6CH3FCH2F2CHF3CF4
CO2Xe
160 180 200 220 240 2600.0
0.2
0.4
0.6
0.8
1.0
C3H8
Ar
N2
Kr
Temperature / K
O2
V/V
0
(a)
(b)
H2S
V/V
0
160 180 200 220 240 2600.0
0.2
0.4
0.6
0.8
1.0
CH4C2H6CH3FCH2F2CHF3CF4
CO2Xe
160 180 200 220 240 2600.0
0.2
0.4
0.6
0.8
1.0
C3H8
Ar
N2
Kr
Temperature / K
O2
V/V
0
(a)
(b)
H2S
V/V
0
Figure 3 The volume ratios of hydrate as a function of temperature. These volume ratios were scaled to the initial value and the solid lines indicate trends. (a) Structure I hydrate, (b) Structure II hydrate.
the ice and release any remaining gas. Differential scanning calorimetry (DSC) is a thermoanalytical technique in which the difference in the amount of heat required to increase the temperature of a sample and reference is measured as a function of temperature. When the sample undergoes a physical transformation such as phase transitions, more or less heat will need to flow to it than to the reference to maintain both at the same temperature. [5] Likewise, as the sample undergoes a phase transition, such as hydrate dissociation, the event can be detected by the change of diffraction profile by means of powder X-ray or nutron diffraction by temperarure dependent measurments. Here, the resolution of temperature for phase transitions by means of the powder diffraction method is not as high as for DSC, but the advantege of the diffraction method is that it is possible to detect crystal structure transformation by temperatur ramping.
Figure 3 shows that most of the hydrates
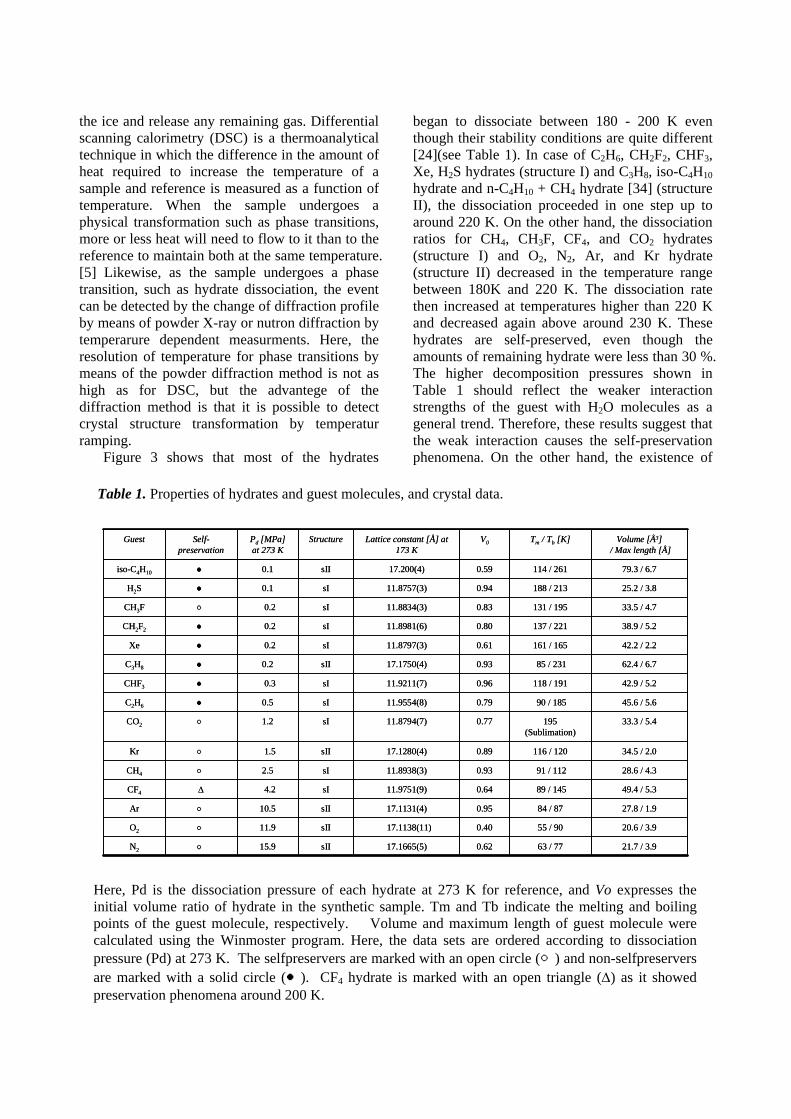
began to dissociate between 180 - 200 K even though their stability conditions are quite different [24](see Table 1). In case of C2H6, CH2F2, CHF3, Xe, H2S hydrates (structure I) and C3H8, iso-C4H10 hydrate and n-C4H10 + CH4 hydrate [34] (structure II), the dissociation proceeded in one step up to around 220 K. On the other hand, the dissociation ratios for CH4, CH3F, CF4, and CO2 hydrates (structure I) and O2, N2, Ar, and Kr hydrate (structure II) decreased in the temperature range between 180K and 220 K. The dissociation rate then increased at temperatures higher than 220 K and decreased again above around 230 K. These hydrates are self-preserved, even though the amounts of remaining hydrate were less than 30 %. The higher decomposition pressures shown in Table 1 should reflect the weaker interaction strengths of the guest with H2O molecules as a general trend. Therefore, these results suggest that the weak interaction causes the self-preservation phenomena. On the other hand, the existence of
Here, Pd is the dissociation pressure of each hydrate at 273 K for reference, and Vo expresses the initial volume ratio of hydrate in the synthetic sample. Tm and Tb indicate the melting and boiling points of the guest molecule, respectively. Volume and maximum length of guest molecule were calculated using the Winmoster program. Here, the data sets are ordered according to dissociation pressure (Pd) at 273 K. The selfpreservers are marked with an open circle (○) and non-selfpreservers are marked with a solid circle (●). CF4 hydrate is marked with an open triangle (∆) as it showed preservation phenomena around 200 K.
Table 1. Properties of hydrates and guest molecules, and crystal data.
79.3 / 6.7114 / 2610.5917.200(4)sII0.1●iso-C4H10
21.7 / 3.963 / 770.6217.1665(5)sII15.9○N2
20.6 / 3.955 / 900.4017.1138(11)sII11.9○O2
27.8 / 1.984 / 870.9517.1131(4)sII10.5○Ar
49.4 / 5.389 / 1450.6411.9751(9)sI4.2∆CF4
28.6 / 4.391 / 1120.9311.8938(3)sI2.5○CH4
34.5 / 2.0116 / 1200.8917.1280(4)sII1.5○Kr
33.3 / 5.4195 (Sublimation)
0.7711.8794(7)sI1.2○CO2
45.6 / 5.690 / 1850.7911.9554(8)sI0.5●C2H6
42.9 / 5.2118 / 1910.9611.9211(7)sI0.3●CHF3
62.4 / 6.785 / 2310.9317.1750(4)sII0.2●C3H8
42.2 / 2.2161 / 1650.6111.8797(3)sI0.2●Xe
38.9 / 5.2137 / 2210.8011.8981(6)sI0.2●CH2F2
33.5 / 4.7131 / 1950.8311.8834(3)sI0.2○CH3F
25.2 / 3.8188 / 2130.9411.8757(3)sI0.1●H2S
Volume [Å3]/ Max length [Å]
Tm / Tb [K]V0Lattice constant [Å] at 173 K
StructurePd [MPa] at 273 K
Self-preservation
Guest
79.3 / 6.7114 / 2610.5917.200(4)sII0.1●iso-C4H10
21.7 / 3.963 / 770.6217.1665(5)sII15.9○N2
20.6 / 3.955 / 900.4017.1138(11)sII11.9○O2
27.8 / 1.984 / 870.9517.1131(4)sII10.5○Ar
49.4 / 5.389 / 1450.6411.9751(9)sI4.2∆CF4
28.6 / 4.391 / 1120.9311.8938(3)sI2.5○CH4
34.5 / 2.0116 / 1200.8917.1280(4)sII1.5○Kr
33.3 / 5.4195 (Sublimation)
0.7711.8794(7)sI1.2○CO2
45.6 / 5.690 / 1850.7911.9554(8)sI0.5●C2H6
42.9 / 5.2118 / 1910.9611.9211(7)sI0.3●CHF3
62.4 / 6.785 / 2310.9317.1750(4)sII0.2●C3H8
42.2 / 2.2161 / 1650.6111.8797(3)sI0.2●Xe
38.9 / 5.2137 / 2210.8011.8981(6)sI0.2●CH2F2
33.5 / 4.7131 / 1950.8311.8834(3)sI0.2○CH3F
25.2 / 3.8188 / 2130.9411.8757(3)sI0.1●H2S
Volume [Å3]/ Max length [Å]
Tm / Tb [K]V0Lattice constant [Å] at 173 K
StructurePd [MPa] at 273 K
Self-preservation
Guest

self-preservation in a quenched high-pressure phase of structure H being stable at 460-770 MPa, is reported for the temperature range 170 K – 230 K.[35] This is consistent with the experimental results observed for normal Ar hydrate. Also, a recent study suggests the existence of self-preservation in CO2 + H2 hydrate, whereas CO2 + H2 + C3H8 hydrate dissociate in one step up to around 220 K. [36] Here, CO2 hydrate is self-preserved and H2 hydrate is expected to be self-preserved due to its very high decomposition pressure (~ 200 MPa at 273 K) [37], but C3H8 hydrate is not self-preserved. Therefore, self-preservation phenomena for hydrates, such as N2, O2, Ar, and CH4 hydrate, of these binary gas systems are also expected. On the other hand, the presence of some quantity of a second guest which does not show self-preservation, such as C2H6 or C3H8, may inhibit the onset of self-preservation phenomena. 3. Dissociation by rapid pressure-release
Anomalous behavior as obtained by applying the rapid pressure-release method may have important implications for drill cores obtained from remote natural gas hydrates or hydrate-bearing sediments, as well as for gas storage and transportation using gas hydrates [38] Figure 4 shows relative volume ratios for CH4 hydrate as a function of time in the temperature region from 257.5 to 270.5 K. In case of the measurement at 268 K, the CH4 hydrate was preserved and almost 40 % of the hydrate still remained after 12 hours even though the stability conditions (2.2 MPa of CH4 at 268 K) were quite different from those under which the sample was kept. Here, it is shown that even a thin layer of CH4 hydrate 0.3 mm in thickness can show anomalous preservation phenomena. In contrast, C2H6 hydrate, CH4+C2H6 hydrate and C3H8 hydrate dissociated just after rapid pressure-release. The dissociation pressures at 268 K for C2H6 hydrate, CH4+C2H6 hydrate (structure I), and C3H8 hydrate (structure II with space group Fd-3m) are 0.36 MPa, 0.5 MPa and 0.12 MPa, respectively, and their thermodynamic stabilities are reflected by dissociation pressures that are lower than that of CH4 hydrate. These hydrates did not show anomalous preservation, this result being consistent with the earlier macroscopic study by Stern et al.[12] Here, the experimental conditions are close to the phase equilibrium boundary for C3H8 hydrate (268 K, 0.12 MPa of C3H8), even
though the C3H8 hydrate dissociated rapidly. By the temperature ramping method, dissociation of C2H6 and C3H8 hydrate under an atmospheric pressure of N2 gas proceeded in a single step up to 220 K whereas the dissociation rate for CH4 hydrate decreased and as the sample self-preserved up to 268 K.[25] Additionally, it has been reported that CH4+C2H6 hydrate dissociates as a single entity without preferential release of either CH4 or C2H6.[39] Therefore, the preservation of gas hydrates by the rapid pressure-release method also
(a)
(b)
V/Vo
0 10 20 30 40 50 600.0
0.2
0.4
0.6
0.8
1.0
0 100 200 300 400 500 600 700 800
0.0
0.2
0.4
0.6
0.8
1.0
Time [min]
CH4 (268 K)CH4 (257.5 K)
CH4 (270.5 K) C3H8 (268 K)CH4+C2H6 (268 K)C2H6 (268 K)
V/Vo
(a)
(b)
V/Vo
0 10 20 30 40 50 600.0
0.2
0.4
0.6
0.8
1.0
0 10 20 30 40 50 600.0
0.2
0.4
0.6
0.8
1.0
0 100 200 300 400 500 600 700 800
0.0
0.2
0.4
0.6
0.8
1.0
Time [min]
CH4 (268 K)CH4 (257.5 K)
CH4 (270.5 K) C3H8 (268 K)CH4+C2H6 (268 K)C2H6 (268 K)
CH4 (268 K)CH4 (257.5 K)
CH4 (270.5 K) C3H8 (268 K)CH4+C2H6 (268 K)C2H6 (268 K)
V/Vo
Figure 4 The volume ratio V/Vo of the dissociating hydrates as a function of time. Vois the initial volume of gas hydrate. The relative volume ratios for structure I hydrate were analyzed as a function of time using the reflection for the sI(321) crystal plane at around 27.5 o in 2θ and those for structure II hydrate were analyzed using reflection sII(511) at around 26.5 o in 2θ because the integrated intensity of the x-ray diffraction profile is proportional to the crystal volume. a) Linear time scale for initial 60 min., b) Linear time scale showing only CH4 hydrates.

depends on the nature of the guest molecules, but neither on the thermodynamic stability nor the crystal structure.
The initial dissociation of the CH4 hydrate was fast and then became slower in the for both the rapid pressure release method and the temperature-ramping method. The initial growth of ice is due to nucleation and lateral growth that covers the hydrate with a coating of ice, and the slower part of the growth occurs after the surface is completely coated with ice.[10] Figure 5 shows time-dependent relative intensity ratios of the Ih(002) to Ih(100) reflections measured by PXRD. For CH4 hydrate, the relative intensity ratio at 257.5 K was consistent with the theoretical value for ice Ih, I002/I100 ~ 0.5, but the relative intensities at 268 K and 270.5 K were significantly larger. It is known that the morphology of ice Ih grown in a vapor phase of air switches from plate-like crystals (~ 271 K), which has extended (001) crystal planes, to columns (~ 268 K), to plates (~ 258 K), and predominantly to columns (< 243 K) as the temperature is decreased.[40] Because of the flow of heat, the plate-like ice likely grows in a horizontal orientation in the sample holder as the growth of ice Ih is due to an exothermic reaction. The experimental results for CH4 hydrate suggests the formation of plate-like ice Ih crystals on the surface of the hydrate layer at 268 K and 270.5 K while the formation of randomly oriented smaller size of ice Ih crystals at 257.5 K. This is supported by earlier cryo-SEM observations [12,41] that anomalously preserved hydrate is a uniformly dense material while as-synthesized hydrate has a macroscopically granular appearance. However, in the case of C2H6, CH4+C2H6 and C3H8 hydrates at 268 K, the changes in the PXRD profile with time suggested that neither anomalous preservation nor growth of plate-like ice Ih crystals took place. Here, the initial dissociation rates may depend on the type of guest molecules; C2H6 and C3H8 hydrate dissociate readily but CH4 hydrate is self-preserved during temperature-ramping as reported.[26] It is also reported that morphologies of ice change depending on the type of atmospheric gas present during growth, e.g. air, N2, O2, CO2 and H2, even though the mechanism is poorly understood.[40] The difference in ice morphology depending on guest molecules may change the dissociation rates of gas hydrates. Subsequently, the rapid dissociation of the hydrates also will give higher levels of water saturation, and this may well lead to more complex
morphologies for the ice that is formed, dendrites or needles instead of plates or columns under the isothermal condition used.[40] The former morphologies are much more likely to produce a porous ice layer than extended plates, thus further hydrate dissociation should occur readily. In fact, recent in-situ direct observations by using scanning confocal microscopy show formation of a transparent ice sheet upon dissociating CH4 hydrate while small ice particles are formed upon dissociating C2H6 hydrate by the pressure-release method.[22] Accordingly, it is concluded that the formation of the plate-like ice Ih crystal leads to the onset of anomalous preservation induced by
0 10 20 30 40 50 600.1
1
10
100
I 002
/ I 1
00
Time [min]
Ih (002)
Ih (100)sI (321)
22 24 26 2822 24 26 28
T [min]
60
0
60
0
sI (222)Ih (101)
sI (320)
2θ [degree]
Ih (100)Ih (002) sI (320)
sI (321)
Ih (101)sI (222)
2θ [degree]
CH4 (268 K)CH4 (257.5 K)
CH4 (270.5 K) C3H8 (268 K)CH4+C2H6 (268 K)C2H6 (268 K)
(a) (b)
(c)
0 10 20 30 40 50 600.1
1
10
100
I 002
/ I 1
00
Time [min]
Ih (002)
Ih (100)sI (321)
22 24 26 2822 24 26 28
T [min]
60
0
60
0
sI (222)Ih (101)
sI (320)
2θ [degree]
Ih (100)Ih (002) sI (320)
sI (321)
Ih (101)sI (222)
2θ [degree]
CH4 (268 K)CH4 (257.5 K)
CH4 (270.5 K) C3H8 (268 K)CH4+C2H6 (268 K)C2H6 (268 K)
CH4 (268 K)CH4 (257.5 K)
CH4 (270.5 K) C3H8 (268 K)CH4+C2H6 (268 K)C2H6 (268 K)
(a) (b)
(c)
Figure 5 In situ PXRD profiles during the transformation of CH4 hydrate into ice by the pressure-release method. The initial profile was measured where the hydrate was stable under high pressure. The Miller index of each reflection as derived from structure I hydrate (sI) and hexagonal ice (Ih). (a) 270.5 K, (b) 257.5 K. (c) Time dependence of the intensity ratio of the Ih(002) to Ih(100) reflections during the transformation from gas hydrates to ice. Here, initial values for the CH4 hydrate at 270.5 K are not shown because the I002/I100 ratios are at infinity. Here, mole fraction of CH4 + C2H6hydrate is CH4/C2H6= 42.5/57.5.

the rapid pressure-release method. We suggest that plate-like ice Ih crystals are more likely to inhibit the dissociation of CH4 hydrate than aggregated ice crystals of other morphologies or different crystalline phases of ice, formed by temperature-ramping above 240 K.[18] In turn, anomalous preservation is shown to depend on the morphology of the ice Ih grown which depends strongly on the thermal history of the hydrate samples as well as on the nature of the guest molecules.
It has been reported that crystals with horizontally oriented Ih (001) planes grow at the surface of supercooled liquid water at temperatures just below the ice point,[42,43] thus the existence of such a layer also supports the growth of plate-like ice crystals. At the same time, a mobile water layer may help anomalous preservation in a number of ways e.g. it may support the contact between the dissociating hydrate and ice that forms, or it may seal the plate-like crystals together to give a tight envelope around the dissociating hydrate. In the case of the diffusion-limited hydrate dissociation model,[10,11] it is applicable only in case when the hydrate forming-gas phase such as that in voids whose internal pressure corresponds to the dissociation pressure of gas hydrate exists between the ice layer and the hydrate. It suggests that the ice layer needs to be strong enough to withstand the large pressure difference between the dissociation pressure and atmospheric pressure e.g. a pressure difference corresponding to 2.3 MPa at 270.5 K for CH4 hydrate, while the fracture strength of granular polycrystalline ice decreases with increasing temperature and increasing grain size of ice. [19] Nevertheless, the thickness of the ice layer covering the dissociating CH4 hydrate at 270.5 K is estimated to be less than 0.1 mm according to the volume ratio of the preserved CH4 hydrates. It is also reported that the thickness of the ice layer covering the dissociating CO2 hydrate is estimated to be few µm by cryo-SEM observations. It is unclear that this difference of ice thickness is caused by the difference of experimental methods or a difference of guest molecules. However, these results suggest that a very thin ice layer envelops the anomalously preserved gas hydrates. On the other hand, it is known that single crystals of natural air (N2+O2) hydrate tightly enveloped in an ice crystal, as recovered from the ice sheet in Polar Regions, are preserved under atmospheric pressure for several
years. It is reported that the on-set of dissociation of the natural air hydrate occurs only after the nucleation of air bubbles at the boundary of air hydrate and the surrounding ice at 263 K.[44] This is yet more evidence for the high stability of tightly enveloped gas hydrate just below the ice point even though the mechanical strength of a thin ice layer is not strong. On the other hand, the internal texture and distribution of the ice layer in anomalously preserved CH4 hydrate above 240K remain unclear. It is important to develop a model for the ice layer that allows the highly stable CH4 hydrate to be formed on a macroscopic scale using non-destructive imaging technique such as phase contrast X-ray imaging technique noted above.
Additionally, it is suggested that the nature of the ice crystals and the interaction between guest molecules and H2O molecules [6] both affect anomalous preservation phenomena for gas hydrates. We think that the gas hydrates which show self-preservation phenomena by the temperature-ramping method in earlier studies,[24] such as N2, O2, Ar, and H2 hydrate, will also show anomalous preservation by the rapid pressure-release method. Preservation phenomena for hydrates of these binary gas systems are also expected. In addition, anomalous preservation phenomena for the CH4+C2H6 mixed gas hydrate upon dissociation by the rapid pressure-release method is also expected, depending on the exact gas composition. SUMMARY
As shown in Fig.6, ice layers grown on the dissociating gas hydrate are likely to change.
140 160 180 200 220 240 260 280
1
10
102
CH
4pr
essu
re [k
Pa]
Temperature (K)
103
104
140 160 180 200 220 240 260 280
1
10
102
CH
4pr
essu
re [k
Pa]
Temperature (K)
103
104
140 160 180 200 220 240 260 280
1
10
102
CH
4pr
essu
re [k
Pa]
Temperature (K)
103
104
Figure 6. Phase diagram for the CH4-H2O system and a sketch showing our assumed model for the transformation of CH4 hydrate to ice under 0.1 MPa.

The different texture of the ice layer may cause different dissociation rates of gas hydrates. (1) Below 200 K, dissociation has two regimes. Ice growth is relatively slow in the second regime; furthermore, between 168 K and 198 K, the hydrate decomposition follows a diffusion process that indicates that the hydrate is coated by an ice layer. The inferred diffusion coefficients for CH4 in ice approximately fit Arhennius behaviour with an activation energy of 20.1 kJ/mole for CH4 gas diffusion through pores or grain boundaries of ice. (2) It is shown that the presence of self-preservation phenomena depends on the type of guest molecule; CH4, CH3F, CF4, and CO2 hydrates (structure I) and O2, N2, Ar, and Kr hydrate (structure II) show self-preservation phenomena, and we conclude that interaction of guest molecules with H2O molecules in the ice play a crucial role in promoting this phenomenon even though there are still exceptions. We expect that mixed gas hydrates encaging molecules with weak interaction with H2O molecules such as H2 and NO should again show self-preservation phenomena even though H2 probably can diffuse out of the cages of the hydrate without decomposing the lattice, at least initially. (3) The distinct morphologies of ice Ih that appear when hydrate dissociates under different temperature-pressure profiles are shown to be correlated with the occurrence of anomalous preservation: plate-like ice crystals grew upon dissociation by rapid pressure-release, and aggregated ice crystals transformed by sintering finely powdered ice crystals grew upon dissociation by temperature-ramping. The formation of the plate-like ice crystal accompanies the onset of the anomalous preservation of gas hydrates. Then, a mobile water layer may also support the contact between the dissociating hydrate and the plate-like ice Ih. Additionally, it is suggested that the nature of the ice crystals and the interaction between guest molecules and H2O molecules both affect anomalous preservation phenomena for gas hydrates. Acknowledgements Much of the work was contributed by our colleagues at NRC: Chris Ratcliffe, Gary Enright, Dennis Klug, Konstantin Udachin, and Hailong Lu, as well as our colleague elsewhere: Tsutomu Uchida (Hokkaido Univ.), Wataru Shimada (Toyama Univ.), Akihiro Hachikubo (KIT), Ryo Ohmura (Keio Univ.), Jiro Nagao (AIST), Takao
Ebinuma (AIST), Hideo Narita (AIST) all gratefully acknowledged. REFERENCES [1] Jeffrey GA. Comprehensive Supramoloecular Chemistry, Vol. 6, Ed. Atwood JL, Davies JED, MacNicol DD, Vogtle F. Oxford: Pergamon/Elsevier, 1996. [2] Hallbrucker A. Angew. Chem. Int. Ed. 1994;33:691. [3] Tulk CA, Ba Y, Klug DD, McLaurin G, Ripmeester JA. J. Chem. Phys. 1999;110:6475. [4] Nakayama H, Klug DD, Ratcliffe CI, Ripmeester JA. Chem. Eur. J. 2003;9:2969. [5] Handa YP, J. Chem. Thermodynamics 1986; 18:891. [6] Yakushev VS, Istomin VA. Physics and Chemistry of Ice; Sapporo:Hokkaido University Press, 1992;136. [7] Stern LA, Circone S, Kirby SH, Durham WB. J. Phys. Chem. B. 2001;105:1756. [8] Makogon YF, Holditch SA, Makogon TY. J. Pet. Sci. Eng. 2007;56:14. [9] Kennedy M. Mrofka D. Borch CVD. Nature 2008;453:642. [10] Takeya S, Shimada W, Kamata Y, Ebinuma T, Uchida T, Nagao J, Narita H. J. Phys. Chem. A 2001;105:9756. [11] Takeya S, Ebinuma T, Uchida T, Nagao J, Narita H, J. Cystal Growth 2002;237-239:379. [12] Stern LA, Circone S, Kirby SH, Durham WB. Can. J. Phys. 2003;81:271. [13] Komai T, Kang S, Yoon J, Yamamoto Y, Kawamura T, Ohtake M. J. Phys. Chem. B 2004; 108:8062. [14] Circone S, Stern LA, Kirby SH. Am. Mineral. 2004;89:1192. [15] Kuhs WF, Genov G, Satykova DK, Hansen T. Phys. Chem. Chem. Phys. 2004;6:4917. [16] Giavarini C, Maccioni F. Ind. Eng. Chem. Res. 2004;43:6616. [17] Kawamura T, Yamamoto Y, Yoon JH, Sakamoto Y, Komai T, Haneda H, Ohtake M, Ohga K. Proc. 14TH ISOPE, Toulon, France, 2004;48. [18] Shimada W, Takeya S, Kamata Y, Uchida T, Nagao J, Ebinuma T, Narita H. J. Phys. Chem. B 2005;109:5802. [19] Takeya S, Uchida T, Nagao J, Ohmura R, Shimada W, Kamata Y, Ebinuma T, Narita H. Chem. Eng. Sci. 2005;60:1383. [20] Takeya K, Nango K, Sugahara T, Ohgaki K. J. Phys. Chem. B 2005;109:21086.

[21] Zhang G, Rogers RE. Chem. Eng. Sci., 2008;63:2066. [22] Nagao J, Shimomura N, Ebinuma T, Narita H. Proc. 6th Int. Conf. Gas Hydrates Vancouver, Canada, 2008. [23] Melnikov VP, Nestrov AN, Reshetnikov AM, Istomin VA, Kwon VG. Chem. Eng. Sci. 2010;65:906. [24] Takeya S, Ripmeester JA. Angew. Chem. 2008;120:1296. [25] Melnikov VP, Nestrov AN, Reshetnikov AM, Zavodovsky AG. Chem. Eng. Sci. 2009;64:1160. [26] Takeya S, Ripmeester JA. Chem. Phys. Chem. 2010;11:70. [27] Takeya S, Honda K, Yoneyama A, Hirai Y, Okuyama J, Hondoh T, Hyodo K, Takeda T. Rev. Sci. Instrum. 2006;77:053705. [28] Takeya S, Honda K, Kawamura T, Yamamoto Y, Yoneyama A, Hirai Y, Hyodo K, Takeda T. Appl. Phys. Lett. 2007;90:081920. [29] Kerkar P, Jones KW, Kleinberg R, Lindquist WB, Tomov S, Feng H, Mahajan D. Appl. Phys. Lett. 2009;95:024102. [30] Momose A, Takeda T, Itai Y. Rev. Sci. Instrum. 1995;66:1434. [31] Hallbrucker A, Mayer E. J. Chem. Soc. Faraday Trans. 1990;86:3785. [32] Satoh K, Uchida T, Hondoh T, Mae S. Proc. NIPR Symp. Polar Meteorol. Glaciol. 1996;10:73. [33] Goto K, Hondoh T, Higashi A. Jpn. J. Appl. Phys. 1986;25:351. [34] Takeya S. Sakagami H, Hachikubo H. Proc. 7th Int. Conf. Gas Hydrates Edinburgh, Scotland, 2011. [35] Ogienko AG, Kurnosov AV, Manakov AY, Larionov, EG, Ancharov AI, Sheromov MA, Nesterov AN. J. Phys. Chem. B. 2006;110:2840. [36]Kumar R, Englezos P, Moudrakovski I, Ripmeester JA. AIChE J. 2009;55:1584. [37]Lokshina KA, Zhao Y. Appl. Phys. Lett. 2006;88:131909. [38] Gudmundson J, Borrehaug A. Proc. 2nd Intnl. Conf. on Natural Gas Hydrates. Monfort JP. ed., Toulouse, France, 1996;415. [39] Rovetto LJ, Bowler KE, Stadterman LL, Dec SF, Koh CA, Sloan ED. Fluid Phase Equilib. 2007;261:407. [40] Hobbs PV. Ice Physics. Oxford: Clarendon Press, 1974. [41] Falenty A, Kuhs WF. J. Phys. Chem. B 2009;113:5975. [42] Arakawa K. J. Glaciol. 1955;2:463. [43] Fujioka T, Sekerka RF. J. Cryst. Growth
1974;24/25:84. [44] Shoji H, Langway CC, Nature 1982;298:548.


















