![Dietary supplementation with free methionine or methionine … · 2019. 6. 27. · with MHA or DL-methionine in heat stress-exposed broilers [23, 24]. In this study, we hypothesize](https://static.fdocuments.net/doc/165x107/60e337666b3f9a31a45a96d1/dietary-supplementation-with-free-methionine-or-methionine-2019-6-27-with-mha.jpg)
Studies of Glutamate Dehydrogenase : Methionine-169: the Preferentially Carboxymethylated Residue
7
Eur. J. Biochem. 74, 379-385 (1977) Studies of Glutamate Dehydrogenase Methionine-169 : the Preferentially Carboxymethylated Residue Michael DAVID, Ihab R. RASCHED, and Horst SUND Fachbereich Biologie der Universitat Konstanz (Received July 20/December 20, 1976) In phosphate buffer at pH 7.0, 5,5'-dithio-bis(2-nitrobenzoic acid), N-ethylmaleimide or iodo- acetamide do not alter the activity of beef liver glutamate dehydrogenase. Iodoacetate, however, inactivates the enzyme irreversibly by alkylation. Combined addition of the coenzyme NADH and the substrate 2-oxoglutarate or the effector GTP protects against this inactivation. The alkylation reaction is independent of pH between pH 6 - 9 indicating that amino, imidazole or phenolic groups are probably not involved in this reaction. Titration of the thiol groups, after denaturation of the enzyme, revealed the loss of approximately one group per polypeptide chain. However, this is not due to the exclusive alkylation of a cysteine residue, since alkylation with iodo- [2-14C]aceticacid also labels a methionine residue. 50 % of the label is incorporated into methionine- 169 and only 7 % into cysteine-115, the remaining radioactivity is distributed in minor quantities (4 x) in several unidentified residues. A probable cause of the erroneous thiol groups titration is discussed. Cysteine residues have been shown to be essential for the activity of several dehydrogenases [2- 51. With respect to glutamate dehydrogenases, however, no clear evidence has been presented for a catalytic role of cysteine residues. Malcolm and Radda [6] reported that the modifi- cation with 4-iodoacetamidosalicylic acid yields after acid hydrolysis 0.93 mol S-carboxymethylcysteine/mol polypeptide chain. Rosen et al. [7] reported that the same reagent alkylates two methionine residues and two cysteine residues per polypeptide chain. They concluded that a methionyl residue is critical for the enzymic activity. Our investigations on the accessibility of the - SH groups of the enzyme to alkylation with iodoacetate demonstrated that the - SH groups under native con- ditions in phosphate but not in Tris buffer are buried and that only a large excess of the reagent partially alkylates some of them [8]. Preliminary results report- ed recently [9] suggested iodoacetate to be a specific modifier of the enzyme: 0.8 mol reagent/mol poly- peptide chain reduces the enzymic activity to 20 % while the presence of substrate and coenzyme protects the enzyme completely. Approximately one - SH group per polypeptide chain was supposed to be alkylated by the reaction, which in part contradicts our previous results [8]. -~ ~ The previous paper in this series is [I]. Enzyme. Glutamate dehydrogenase or L-glutamate:NAD(P)+ oxidoreductase (deamindting) (EC I .4.1.3). In this paper we describe in more detail the reaction of beef liver glutamate dehydrogenase with iodoace- tate. Furthermore, using iod0-[2-'~C]acetic acid, we present evidence that methionine-169 is the prefer- entially labeled residue, whereas cysteine-I 15 is alky- lated to much lesser extent. MATERIALS AND METHODS Glutamate Dehydrogenase Beef liver glutamate dehydrogenase was obtained from Boehringer Mannheim GmbH (Mannheim, Germany) as a crystalline suspension in 2 M ammo- nium sulfate. The enzyme was used after dialysis at 4 "C for 48 h against 0.067 M phosphate buffer, pH 7.6. The protein concentration was determined spec- trophotometrically at 279 nm using an A: & value of 9.7 [lo]. Absorbance measurements were made on a Zeiss spectrophotometer PMQ 3 (Zeiss, Oberkochen, Germany). The enzymic activity was measured in 0.067 M phosphate buffer, pH 7.6, using 1.15 mM NADf, 8.5 mM glutamate and 3-4 pg/ml enzyme in the for- ward reaction. Inhibition by GTP was determined under the conditions of the forward reaction, but with 50 pM GTP. Substances ADP, GTP, NADH, NAD', 2-oxoglutarate of the highest purity available, were obtained from
-
Upload
michael-david -
Category
Documents
-
view
215 -
download
3
Transcript of Studies of Glutamate Dehydrogenase : Methionine-169: the Preferentially Carboxymethylated Residue

Eur. J. Biochem. 74, 379-385 (1977)
Studies of Glutamate Dehydrogenase Methionine-169 : the Preferentially Carboxymethylated Residue
Michael DAVID, Ihab R. RASCHED, and Horst SUND
Fachbereich Biologie der Universitat Konstanz
(Received July 20/December 20, 1976)
In phosphate buffer at pH 7.0, 5,5'-dithio-bis(2-nitrobenzoic acid), N-ethylmaleimide or iodo- acetamide do not alter the activity of beef liver glutamate dehydrogenase. Iodoacetate, however, inactivates the enzyme irreversibly by alkylation. Combined addition of the coenzyme NADH and the substrate 2-oxoglutarate or the effector GTP protects against this inactivation.
The alkylation reaction is independent of pH between pH 6 - 9 indicating that amino, imidazole or phenolic groups are probably not involved in this reaction. Titration of the thiol groups, after denaturation of the enzyme, revealed the loss of approximately one group per polypeptide chain. However, this is not due to the exclusive alkylation of a cysteine residue, since alkylation with iodo- [2-14C]acetic acid also labels a methionine residue. 50 % of the label is incorporated into methionine- 169 and only 7 % into cysteine-115, the remaining radioactivity is distributed in minor quantities (4 x) in several unidentified residues. A probable cause of the erroneous thiol groups titration is discussed.
Cysteine residues have been shown to be essential for the activity of several dehydrogenases [2- 51. With respect to glutamate dehydrogenases, however, no clear evidence has been presented for a catalytic role of cysteine residues.
Malcolm and Radda [6] reported that the modifi- cation with 4-iodoacetamidosalicylic acid yields after acid hydrolysis 0.93 mol S-carboxymethylcysteine/mol polypeptide chain. Rosen et al. [7] reported that the same reagent alkylates two methionine residues and two cysteine residues per polypeptide chain. They concluded that a methionyl residue is critical for the enzymic activity.
Our investigations on the accessibility of the - SH groups of the enzyme to alkylation with iodoacetate demonstrated that the - SH groups under native con- ditions in phosphate but not in Tris buffer are buried and that only a large excess of the reagent partially alkylates some of them [8]. Preliminary results report- ed recently [9] suggested iodoacetate to be a specific modifier of the enzyme: 0.8 mol reagent/mol poly- peptide chain reduces the enzymic activity to 20 % while the presence of substrate and coenzyme protects the enzyme completely. Approximately one - SH group per polypeptide chain was supposed to be alkylated by the reaction, which in part contradicts our previous results [8]. -~ ~
The previous paper in this series is [I]. Enzyme. Glutamate dehydrogenase or L-glutamate: NAD(P)+
oxidoreductase (deamindting) (EC I .4.1.3).
In this paper we describe in more detail the reaction of beef liver glutamate dehydrogenase with iodoace- tate. Furthermore, using iod0-[2-'~C]acetic acid, we present evidence that methionine-169 is the prefer- entially labeled residue, whereas cysteine-I 15 is alky- lated to much lesser extent.
MATERIALS AND METHODS
Glutamate Dehydrogenase
Beef liver glutamate dehydrogenase was obtained from Boehringer Mannheim GmbH (Mannheim, Germany) as a crystalline suspension in 2 M ammo- nium sulfate. The enzyme was used after dialysis at 4 "C for 48 h against 0.067 M phosphate buffer, pH 7.6. The protein concentration was determined spec- trophotometrically at 279 nm using an A: & value of 9.7 [lo]. Absorbance measurements were made on a Zeiss spectrophotometer PMQ 3 (Zeiss, Oberkochen, Germany).
The enzymic activity was measured in 0.067 M phosphate buffer, pH 7.6, using 1.15 mM NADf , 8.5 mM glutamate and 3-4 pg/ml enzyme in the for- ward reaction. Inhibition by GTP was determined under the conditions of the forward reaction, but with 50 pM GTP.
Substances
ADP, GTP, NADH, NAD', 2-oxoglutarate of the highest purity available, were obtained from

380 Functional Methioninc-I69 in Glutamate Dehydrogenase
Boehringer Mannheim GmbH (Mannheim, Ger- many). Buffer substances, analytical grade, and L- glutamate were products of E. Merck (Darmstadt, Germany), guanidine hydrochloride of extreme purity was purchased from Heico (Delaware, U.S.A.), so- dium dodecylsulfate and 5,5'-dithio-bis(2-nitroben- zoic acid) from Serva (Heidelberg, Germany), iodo- acetate and iodoacetamide were obtained from Sigma Chemical Company (St Louis, U.S.A.) and iodo- [2-14C]acetate from Amersham Buchler GmbH (Braunschweig, Germany). N-Ethylmaleimide was purchased from E. Merck (Darmstadt, Germany), trypsin treated with L-1-tosylamido-2-phenylethyl chloromethyl ketone was obtained from Worthington Biochemical Corporation (Freehold, N.J., U.S.A.). All chemicals for the Edman degradation are sequenal grade and purchased from Pierce Co. (Illinois, U.S.A.).
Modification
A solution of glutamate dehydrogenase (54 pM)' was incubated with iodoacetic acid (12.5 mM) in 0.067 M phosphate buffer, pH 7.0, and allowed to react for 2 h at 20 "C in the dark. For further use samples were passed through a Sephadex G-25 column equilibrated with the same buffer or dialysed for 48 h against several changes of the buffer.
For sequence analyses and quantitative determina- tion of the degree of alkylation iod0-[2-'~C]acetic acid diluted with unlabeled iodoacetic acid to give 1.84 x lo6 counts min-' pmol-' was used for the mo- dification. The radioactivity was determined by count- ing aliquots in a Beckman LS-100 C liquid-scintilla- tion spectrophotometer. All measurements were nor- malized to a standard. Samples destinated to se- quence analysis were first separated from excess re- agent and then dialyzed against distilled water and l yophilized.
Amino Acid Analysis
The modified protein was hydrolyzed with 6 M HCl in evacuated and sealed tubes at 110 "C for 24 h. Other samples were hydrolyzed after oxidation with performic acid [ll], which converts methionine to inethionine sulfone, but not S-carboxymethylmethio- nine [12]. The pure tryptic peptides were hydrolyzed in the same way. The quantitative amino acid analyses were performed on a Biotronik Chromatography System 4010. The values in the tables are molar ratios without correction for destruction, incomplete hydro- lysis or impurities. Qualitative analyses after acid hydrolysis of the pure peptides were performed by electrophoresis at pH 1.9 and 100 V/cm. The amino
' The molecular weight used to calculate the molarity of enzyme solutions is that of the polypeptide chain (56000).
acids were visualized by staining with the ninhydrin reagent [13].
Determination of - SH Groups
Determination of - SH groups was performed both by amino acid analysis and according to Ellman [14]. Samples of modified or native enzyme (0.5 - 1 .O mg) were denatured with 0.2 o/, sodium dodecylsulfate prior to titration with 5,5'-dithio-bis(2-nitrobenzoic acid). To test the influence of iodine on the reaction a solution of the enzyme (1 mgiml) was treated with 0.2 mM iodine prior to denaturation.
Sequence and E F Z ~ Group Determination
Carboxymethylation. The carboxymethylat ion was performed at pH 8.1 in 6 M guanidine hydrochloride containing 2 mM EDTA and 0.1 M Tris-HCI at a protein concentration of 10 mg/ml [15,16]. The pro- tein solution was first reduced with dithiothreitol (40% molar excess with respect to the protein -SH groups) for 3 h at 37 "C. Iodoacetate (20% molar excess with respect to the total amount of - SH groups) was then added and the solution incubated at 25 "C for 3 h in the dark. Guanidine hydrochloride and excess reagents were removed by repeated dialysis against 1 mM HC1 at 4 "C.
Tryptic Peptides. The carboxymethylated protein (1.8 pmol with 2.8 x lo6 counts/min) was digested with L-1-tosylamido-2-phenylethyl-chloromethyl-ke- tone-treated trypsin (ratio trypsin : substrate 2 : 100, w/w) at a protein concentration of about 3 mg/ml in 0.1 M ammonium bicarbonate for 4 h at 37 "C and fol- lowed by freeze-drying. The tryptic peptides were then separated by gel filtration on a column (1.5 x 145 cm) of Sephadex G-25F, equilibrated with 5 % formic acid. 90% of the total radioactivity was recovered in two fractions, TCI (60 %) and TCII (30 %). Fraction TCI was freeze-dried and rechromatographed on the same column but filled with Sephadex G-75. 87% of the radioactivity were pooled into fraction TCIR. The fractions TCIR and TCII were further purified by high-voltage electrophoresis at 60 V/cm in cooled tanks [17] on Whatman no. 3 MM paper at pH 1.9 and 6.5 [18]. Chromatography in butan-1-ol/acetic acid/ water/pyridine (1 5/3/12/10, by vol.) was also used. In purifications involving more than one step of separa- tion on paper, the latter was stitched onto a new paper [19]. Modified peptides were detected by autoradiog- raphy, scintillation counting and staining of guide strips with a ninhydrin/cadmium reagent [13].
Sequence Analysis. End-group and sequence ana- lyses of peptides were performed by the dansyl-Edman method [20]. The dansyl-amino acids were identified by thin-layer chromatography on polyamide layers [21] in four systems [22].
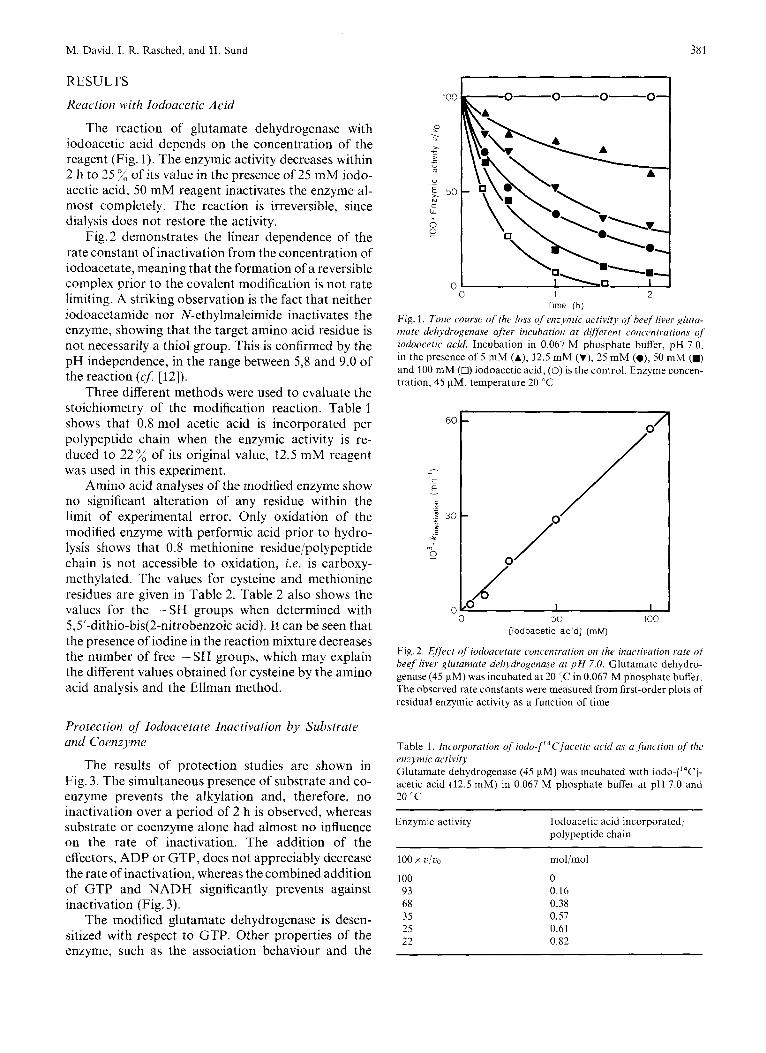
M. David, I. R. Rasched, and H. Sund 381
RESULTS
Reaction with Iodoacetic Acid
The reaction of glutamate dehydrogenase with iodoacetic acid depends on the concentration of the reagent (Fig. I ) . The enzymic activity decreases within 2 11 to 25 % of its value in the presence of 25 mM iodo- acetic acid, 50 mM reagent inactivates the enzyme al- most completely. The reaction is irreversible, since dialysis does not restore the activity.
Fig. 2 demonstrates the linear dependence of the rate constant of inactivation from the concentration of iodoacetate, meaning that the formation of a reversible complex prior to the covalent modification is not rate limiting. A striking observation is the fact that neither iodoacetamide nor N-ethylmaleimide inactivates the enzyme, showing that the target amino acid residue is not necessarily a thiol group. This is confirmed by the pH independence, in the range between 5,8 and 9.0 of the reaction (cJ [12]).
Three different methods were used to evaluate the stoichiometry of the modification reaction. Table 1 shows that 0.8 mol acetic acid is incorporated per polypeptide chain when the enzymic activity is re- duced to 22% of its original value, 12.5 mM reagent was used in this experiment.
Amino acid analyses of the modified enzyme show no significant alteration of any residue within the limit of experimental error. Only oxidation of the modified enzyme with performic acid prior to hydro- lysis shows that 0.8 methionine residue/polypeptide chain is not accessible to oxidation, i.e. is carboxy- methylated. The values for cysteine and methionine residues are given in Table 2. Table 2 also shows the values for the -SH groups when determined with 5,5'-dithio-bis(2-nitrobenzoic acid). It can be seen that the presence of iodine in the reaction mixture decreases the number of free -SH groups, which may explain the different values obtained for cysteine by the amino acid analysis and the Ellman method.
Protection of Iodoacetate Inactivation by Substrate and Coenzyme
The results of protection studies are shown in Fig. 3. The simultaneous presence of substrate and co- enzyme prevents the alkylation and, therefore, no inactivation over a period of 2 h is observed, whereas substrate or coenzyme alone had almost no influence on the rate of inactivation. The addition of the effectors, ADP or GTP, does not appreciably decrease the rate of inactivation, whereas the combined addition of GTP and NADH significantly prevents against inactivation (Fig. 3).
The modified glutamate dehydrogenase is desen- sitized with respect to GTP. Other properties of the enzyme, such as the association behaviour and the
100
P . >-
5
0
- ~
- - u 5 50 N c w 0 0
- 0 1 2
Time (h)
Fig. 1. Time course of [lie loss of en;77zic actitiity q f h w f liver glura- mate dehydrogenase after incubation at dif;erent concentrations of iodoacetic acid. Incubation in 0.067 M phosphate buffer, pH 7.0, in the presence of 5 mM (A), 12.5 mM (Y), 25 mM (O), 50 mM (M) and 100 mM (n) iodoacetic acid, (0) is the control. Enzvme concen- tration, 45 pM, temperature 20 "C
60
- c - E - L
B 3 0
* 0
0
0 0 50 100
[lodoacetic acid] (mM)
Fig. 2. Effect of iodoacetate concentration on the inactivation rate o/ beef liver glutamate delzydrogenase at p H 7.0. Glutamate dehydro- genase (45 pM) was incubated at 20 IC in 0.067 M phosphate buffer. The observed rate constants were measured from first-order plots of residual enzymic activity as a function of time
Table 1. Incorporation of iodo-["C]ucelic acid as (I Junctioii of thc enzymic activity Glutamate dehydrogenase (45 pM) was incubated with iodo-[14C]- acetic acid (12.5 mM) in 0.067 M phosphate buffer at pH 7.0 and 20 'C
Enzymic activity Iodoacetic acid incorporated/ polypeptide chain
100 x ./.o mol/mol
100 0 93 0.16 68 0.38 3s 0.57 25 0.61 22 0.82

382 Functional Methionine-169 in Glutamate Dehydrogenase
Table 2. Determination of the cysteine and methionine residues in the modif2d and unmodified glutamate dehydrogenase For experimental details see text
Enzyme Cysteine Methionine
determination amino acid amino acid according to analysis analysis Ellman [I41
mol/mol polypeptide chain
~~
~ -~
Unmodified 6.0 + 0.2 6.6 i 0.1" 13.6 i 0.2' 6.4b 14b
Modified 5.2 + 0.3 6.6 i 0.1 12.8 k 0.2'
Unmodified enzyme + 0.2 mM iodine 5.4 0.3 - -
~~
a Determined as cysteic acid.
' Determined as methioniiie sulfone Data given in [24].
0-0-0- 0
"
c W
0 e
0 I I 0 1 2
Time ( h )
Fig. 3. Time course of the loss of enzymic activity of glutamate rie- liydrogenase (45 p M ) after incubation with iodoacetate (12,s m M ) . Incubation in 0.067 M phosphate buffer, pH 7.0 and 20 "C in the presence of 10 mM 2-oxoglutarate of 10 mM glutamate (a), 1 mM NADH or 1 mM NAD' (A), 0.1 mM GTP (v), 1 mM ADP (A), 1 mM NADH + 0.1 mM GTP (V), 10 mM 2-oxoglutarate + I mM NADH (m), (0) is the control, (0) without any other addition
Fraction nurnber
Fig. 4. Gel filtration of the tryptic digest of the iodo-[2-'4C]acetic-ucid-mod$ied glutamate dehydrogenase. Conditions ; Sephadex G-25 F, 145 x 1.5 cm, in 5 "/, formic acid, flow rate 15 ml/h, 3-ml fractions were collected. Fractions marked by horizontal bar were pooled for further purification. Fractions TCI contained 60 "/:and fraction TCII 30 %of the total radioactivity. Absorbance at 280 nm (O), radioactivity (counts: min) in 50 pl (0)
overall conformation, were not altered by the modifi- cation (data are not given).
Sequence Analysis of' the Labeled Peptides
For sequence analyses radioactive labeled protein was prepared, carboxymethylated and digested with trypsin as described under Materials and Methods. After freeze-drying, the digest (100 mg containing 2.8 x lo6 counts/min) was dissolved in 1.5 ml 5 x formic acid and applied to a Sephadex G-25 F column, Fig. 4 shows the elution profile of thischromatography. Two fractions were collected containing 90 % of the
total radioactivity : fraction TCI (60 %) and fraction TCII (30 %). A further purification of fraction TCI was obtained by rechromatography on Sephadex G-75 (Fig. 5). 87 % of the radioactivity of that fraction was pooled into fraction TCIR. The fractions TCIR and TCII were further purified by paper electrophoresis at pH 1.9 and 6.5.
Fraction TCIR separated in two labeled spots: one anodic spot, containing 90% of the radioactivity of fraction TCIR (54% of the total radioactivity), was further purified by electrophoresis at pH 1.9 to yield the pure peptide TCIRP containing 52 of the total radioactivity. The second spot, proved on further
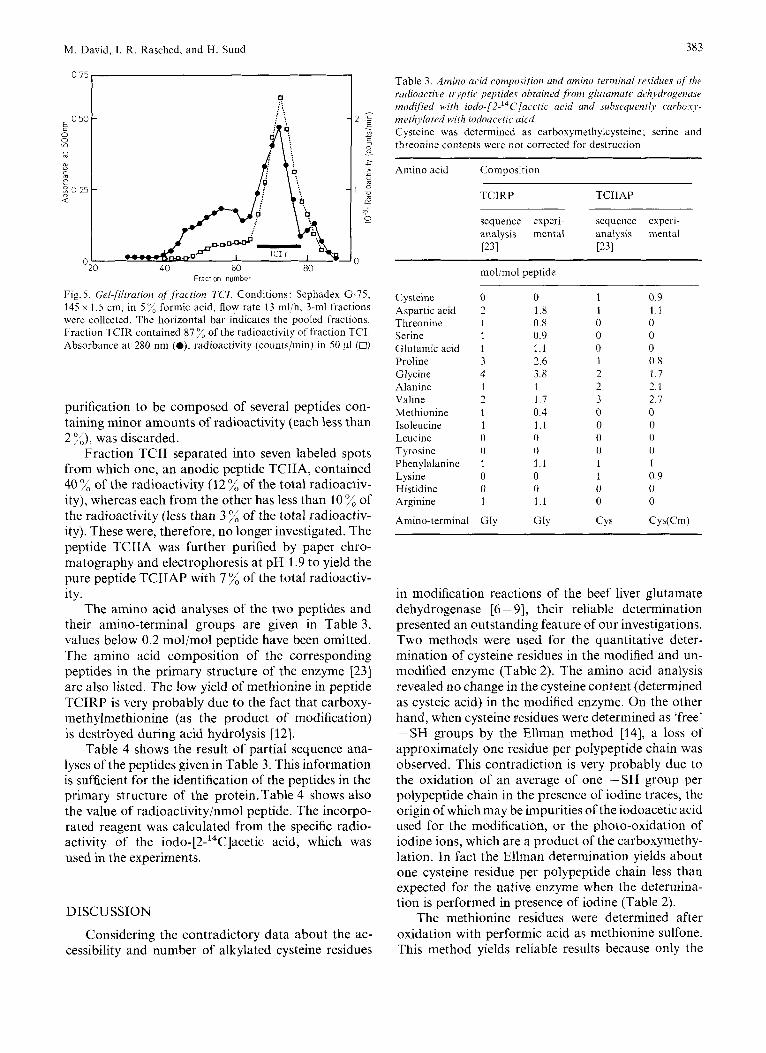
M. David, I. R. Rasched, and H. Sund 383
0 75
Fraction number
Fig. 5. Gel$ltration of Jiaction TCI. Conditions: Sephadex G-75, 145 x 1.5 cm, in 5 ”/, formic acid, flow rate 13 ml/h, 3-ml fractions were collected. The horizontal bar indicates the pooled fractions. Fraction TCIR contained 87 ”/, of the radioactivity of fraction TCI. Absorbance at 280 nin (O), radioactivity (counts/min) in 50 pl (0)
purification to be composed of several peptides con- taining minor amounts of radioactivity (each less than 2 %), was discarded.
Fraction TCII separated into seven labeled spots from which one, an anodic peptide TCIIA, contained 40 % of the radioactivity (12 % of the total radioactiv- ity), whereas each from the other has less than 10 % of the radioactivity (less than 3 % of the total radioactiv- ity). These were, therefore, no longer investigated. The peptide TCIIA was further purified by paper chro- matography and electrophoresis at pH 1.9 to yield the pure peptide TCIIAP with 7 ”/, of the total radioactiv- ity.
The amino acid analyses of the two peptides and their amino-terminal groups are given in Table 3, values below 0.2 mol/mol peptide have been omitted. The amino acid composition of the corresponding peptides in the primary structure of the enzyme 1231 are also listed. The low yield of methionine in peptide TCIRP is very probably due to the fact that carboxy- methylmethionine (as the product of modification) is destrbyed during acid hydrolysis [12].
Table 4 shows the result of partial sequence ana- lyses of the peptides given in Table 3. This information is sufficient for the identification of the peptides in the primary structure of the protein.Table 4 shows also the value of radioactivity/nmol peptide. The incorpo- rated reagent was calculated from the specific radio- activity of the iod0-[2-‘~C]acetic acid, which was used in the experiments.
DISCUSSION
Considering the contradictory data about the ac- cessibility and number of alkylated cysteine residues
Table 3 . Amino acid composition and amino terminal residues oJ iht radioactive trjptic peprides obtained from glutamate dchydrogmasu modijied with iodo-[2-14C]aci~tic acid and subsequently curhouj,- methyluted with iodoacetic uicd Cysteine was determined as carboxymethylcysteine; serine and threonine contents were not corrected for destruction
Amino acid Composition _ _ ~ ~-
TCIRP TCIIAP
sequence experi- sequence experi- analysis mental analysis mental ~ 3 1 ~ 3 1
C ysteine Aspartic acid Threonine Serine Glutamic acid Proline Glycine Alanine Valine Methionine Isoleucine Leucine Tyrosine Phenylalanine Lysine Histidine Arginine
Amino-terminal
mol/mol peptide
0 0 2 1.8 1 0.8 1 0.9 1 1.1 3 2.6 4 3.8 1 1 2 1 7 1 0.4 1 1.1 0 0 0 0 1 1.1 0 0 0 0 I 1.1
~ . _ _ _ _ _
GlY Gly
~ -~
1 0 9 1 1 1 0 0 0 0 0 0 1 0.8 2 1 7 2 2 1 3 2 7 0 0 0 0 0 0 0 0 1 1 1 0 9 0 0 0 0
CYS Cys(Cm)
in modification reactions of the beef liver glutamate dehydrogenase [6 - 91, their reliable determination presented an outstanding feature of our investigations. Two methods were used for the quantitative deter- mination of cysteine residues in the modified and un- modified enzyme (Table 2). The amino acid analysis revealed no change in the cysteine content (determined as cysteic acid) in the modified enzyme. On the other hand, when cysteine residues were determined as ‘free’ -SH groups by the Ellman method [14], a loss of approximately one residue per polypeptide chain was observed. This contradiction is very probably due to the oxidation of an average of one -SH group per polypeptide chain in the presence of iodine traces, the origin of which may be impurities of the iodoacetic acid used for the modification, or the photo-oxidation of iodine ions, which are a product of the carboxymethy- lation. In fact the Ellman determination yields about one cysteine residue per polypeptide chain less than expected for the native enzyme when the determina- tion is performed in presence of iodine (Table 2).
The methionine residues were determined after oxidation with performic acid as methionine sulfone. This method yields reliable results because only the

I 0 .- f
Functional Methionine-169 in Glutamate Dehydrogenase
‘free’ methionine residues are converted to methionine sulfone, which avoids interference with the destruction products of S-carboxymethylmethionine during the acid hydrolysis.
The beef liver glutamate dehydrogenase possesses six cysteine residues per polypeptide chain. When in- vestigated in phosphate buffer the -SH groups are almost not accessible to iodoacetic acid or 5,5’-dithio- bis(2-nitrobenzoic acid), on the other hand they be- come exposed in Tris buffer and react in the order cysteine-89, 55, 197, 115, 270 and 319 [8]. Further- more, we reported the incorporation of iodoacetic acid to the enzyme in phosphate buffer and concluded from the electrophoretic mobility of the labeled tryptic pep- tide at pH 6.5, that cysteine-115 has been alkylated. The present paper shows that methionine-169 is pre- dominantly alkylated in phosphate buffer, whereas only traces of cysteine-115 are modified (Table 4). The earlier results can be explained by the fact that both tryptic peptides containing methionine-169 and cys- teine-115 display very similar electrophoretic mobil- ities at pH 6.5 and were confounded in our conclusions.
The inactivation of glutamate dehydrogenase by alkylation with iodoacetic acid is irreversible. How- ever, the inactivation can be prevented to a great extent by the addition of NADH and the substrate or the effector GTP (Fig.3). It seems that no alteration of the native enzyme conformation occurs after car- boxymethylation as indicated by circular dichroism measurements (results shown in [26]), meaning that a loss of substrate, coenzymes or effectors binding could not simply be due to conformational shift. It can be concluded that the modified methionine-169 is not only the most reactive methionine residue in the poly- peptide chain but also essential for the enzymic activ- ity and is probably located in the active site. Rosen et ul. [7] have also reported the alkylation, using 4-iodo- acetamidosalicylic acid, of one methionine residue in glutamate dehydrogenase which is critical for the enzymic activity.
REFERENCES 1. Rasched, 1. R. , Bohn, A. & Sund, H. (1977) Eur.. J . Biochrz.
2. Harris, I. (1964) Nalurc (Lond.) 203, 30-34. 3. Woenckhaus, C., Zoltobrocki, M. & Berghauser, J . (1970)
4. Holbrook, J . J. & Stinson, R. A. (1970) Biochrm. J . 120, 289-
5. Silverstein, E. & Sulebele, G. (1970) Biochemistry, 9, 214-282. 6 . Malcolm, A. D. B. & Radda, G . K . (1970) Eur. J . Biochem. 15,
7. Rosen, N. L., Bishop, L., Burnett, J. B., Bishop, M. &Colman,
8. Hucho, F., Rasched, I . & Sund. H. (1975) Eur. J . Biochem. 52,
9. Rasched, I., Bohn, A., David, M. & Sund, H. (1975) Biocliem.
74,365 - 371.
Hoppe-Seyler’s Z . Physiol. Chem. 351, 1441 - 1448.
291.
555-561.
R. F. (1973) J . Bid. Chem. 248, 1359-7369.
221 -230.
Soc. Trans. 3, 926- 921.

M. David, I. R. Rasched, and H. Sund 385
10. Oison, J . A. & Anfinsen, C. A. (1952) J . Biol. Chem. 197, 67-
13. Hirs, C. H. W. (1967) Methods Enzyntol. I l , 197-199. 12. Gundlach, H. G., Moore, S. & Stein, W. H. (1959) J . Biol.
33. Heilmann, J., Barsolier, J. & Watzke, E. (1957) Hoppe-Seyler’s
14. Ellman, G. L. (1959) Arch. Biochem. Biophys. 82, 70-77. 15. Milstein, C. (1966) Biochem. J . 101, 352-368. 16. Jornvall, H. &Harris, J. I. (1970) Eur. J . Biochem. 13,565- 576. 17. Ryle, A. P., Sanger, F., Smith, L. F. & Kitai, R. (1955) Bio-
18. Ambler, R . P. (1963) Biochem. J . 89, 349-378. 19. Naughton, M. A. & Hagopian, H. (1962) Anal. Biochem. 3,
79
Chen?. 234, 1761 - 1764.
Z. Physiol. Chem. 309, 219-220.
chem. J . 60, 541 - 556.
276-284.
20. Grey, W. R. (1972) Methods Enzymol. 25, 333- 344. 21. Woods, K. R. & Wang, K. T. (1967) Biochim. Biophys. Acta,
22. Crowshow, K., Jessup, S. J. & Ramwell, P. W. (1967) Biochem.
23. Moon, K. & Smith, E. L. (1973) J . Biol. Chem. 248,3082- 3088. 24. Landon, M., Melomud, M. D. & Smith, E. L. (1971) J . Biol.
25. Offord, R. E. (1966) Nature (Lond.) 211, 591 - 593. 26. David, M. (1976) Dissertation, University of Konstanz, Kon-
27. Hartley, B. S. (1970) Biochem. J . 119, 805-822.
133, 369- 370.
J . 103, 79-85.
Chem. 246,2360 - 2373.
stanz.
M. David, I. R. Rasched, and H. Sund, Fachbereich Biologie der Universitat Konstanz, Postfach 77 33, D-7750 Konstanz, Federal Republic of Germany


















