
InnoVision Report Date Generated: Management: Jill...
30
e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved. InnoVision Report Date Generated: March 2, 2010 Owner: Amelia Medical Agent/s: John Filstein Inventor/s: William Bowman MD Management: Jill Bernard, President DNA Powered Technology Assessment Tendon Repair Device The following information resulted from the analysis of this technology by the e-Zassi Technology Assessment Software Platform. The Tendon Repair Device is a biologic and medical device combination product used in the soft tissue repair segment of the orthopedics market. It is designed as disposable, for single patient use. It is expected to be used in the physician office by a specialty practice physician. Improved clinical outcome for the patient has been identified as a benefit resulting from the use of the Tendon Repair Device. Nature of Technology – Summary Table Type of Device: Medical Device with Biologic. Learn more Device Regulatory Classification: US: Class II EU: Class III Learn more Regulatory Status: US: A regulatory submission is required, but not yet filed. EU: A regulatory filing is required but has not been submitted. Learn more Primary Intended Use: Treatment Learn more Primary and Secondary Users: Specialty practice physician,Surgeon. Learn more Key Purchasing Decision Maker: Physician in Specialty Practice. Learn more Reimbursement Outlook: Likely existing coverage applies. Learn more Intellectual Property Status: No patents are issued. Patent applications are pending. Learn more Clinical Specialty / Market: Orthopedics Learn more Market Segment / Sub-Segment: Soft tissue repair. Not Identified. Learn more Pre-Clinical / Clinical Research Status: Pre-clinical in-vitro testing is complete. Animal testing is complete. First-in-man trial is complete. Learn more Advantages and Benefits: Improved clinical outcome for the patient. Reduction in treatment time. Reduced pain. Improved quality of life. Reduced need for antibiotics. Learn more Grant Matching Results: 6 Federal/State Grant(s). Learn more Manufacturing Build Status: Three dimensional computer model(s). Functional prototype(s) for in-vitro/animal testing. Functional, clinical product. Learn more
Transcript of InnoVision Report Date Generated: Management: Jill...

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report Date Generated: March 2, 2010
Owner: Amelia MedicalAgent/s: John FilsteinInventor/s: William Bowman MDManagement: Jill Bernard, President
DNA Powered Technology Assessment
Tendon Repair DeviceThe following information resulted from the analysis of this technology by the e-Zassi Technology Assessment Software Platform. The Tendon Repair Device is a biologic and medical device combination product used in the soft tissue repair segment of the orthopedics market. It is designed as disposable, for single patient use. It is expected to be used in the physician office by a specialty practice physician. Improved clinical outcome for the patient has been identified as a benefit resulting from the use of the Tendon Repair Device.
Nature of Technology – Summary TableType of Device: Medical Device with Biologic. Learn more
Device Regulatory Classification: US: Class II EU: Class III Learn more
Regulatory Status: US: A regulatory submission is required, but not yet filed.EU: A regulatory filing is required but has not been submitted. Learn more
Primary Intended Use: Treatment Learn more
Primary and Secondary Users: Specialty practice physician,Surgeon. Learn more
Key Purchasing Decision Maker: Physician in Specialty Practice. Learn more
Reimbursement Outlook: Likely existing coverage applies. Learn more
Intellectual Property Status: No patents are issued.Patent applications are pending. Learn more
Clinical Specialty / Market: Orthopedics Learn more
Market Segment / Sub-Segment: Soft tissue repair. Not Identified. Learn more
Pre-Clinical / Clinical Research Status: Pre-clinical in-vitro testing is complete. Animal testing is complete. First-in-man trial is complete. Learn more
Advantages and Benefits: Improved clinical outcome for the patient.Reduction in treatment time.Reduced pain.Improved quality of life.Reduced need for antibiotics.
Learn more
Grant Matching Results: 6 Federal/State Grant(s). Learn more
Manufacturing Build Status: Three dimensional computer model(s).Functional prototype(s) for in-vitro/animal testing.Functional, clinical product.
Learn more

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
2
Market LandscapeThe market landscape section includes information on market segment, customers, and competitors, all of which are invaluable in devising an effective sales and marketing plan for the technology. Initially, this data covers the major geographic segments for the US and the European Union medical device market. Future versions of this report will include world-wide market data.
Market Information
Market Overview and Environmental Assessment The United States market for orthopedic devices in 2006 was the largest in the world with revenues of close to $16 billion. By 2011, it is projected the orthopedic device market to generate over $31 billion in revenues at a compound annual growth rate (CAGR) of just under 15%. The US currently accounts for 57% of worldwide orthopedic device sales and is expected to hold a 64% global market share. As in the global market, the largest share of the US market is held by joint reconstruction devices (38%), and is followed by spinal surgery devices (18%).
The fastest growing segment in the orthopedic devices market is the spinal surgery device segment with a CAGR of 19%. Joint reconstruction devices are estimated to grow by a CAGR of over 17%, and maintain a revenue leadership position with sales of over $13 billion. Other device segments, which include: accessories (bone cement and casts), orthobiologics, braces and support, trauma fixation, and diagnostics, are expected to have individual CAGRs within the range of 7-11% through 2011.
Date Generated: March 2, 2010
e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
3
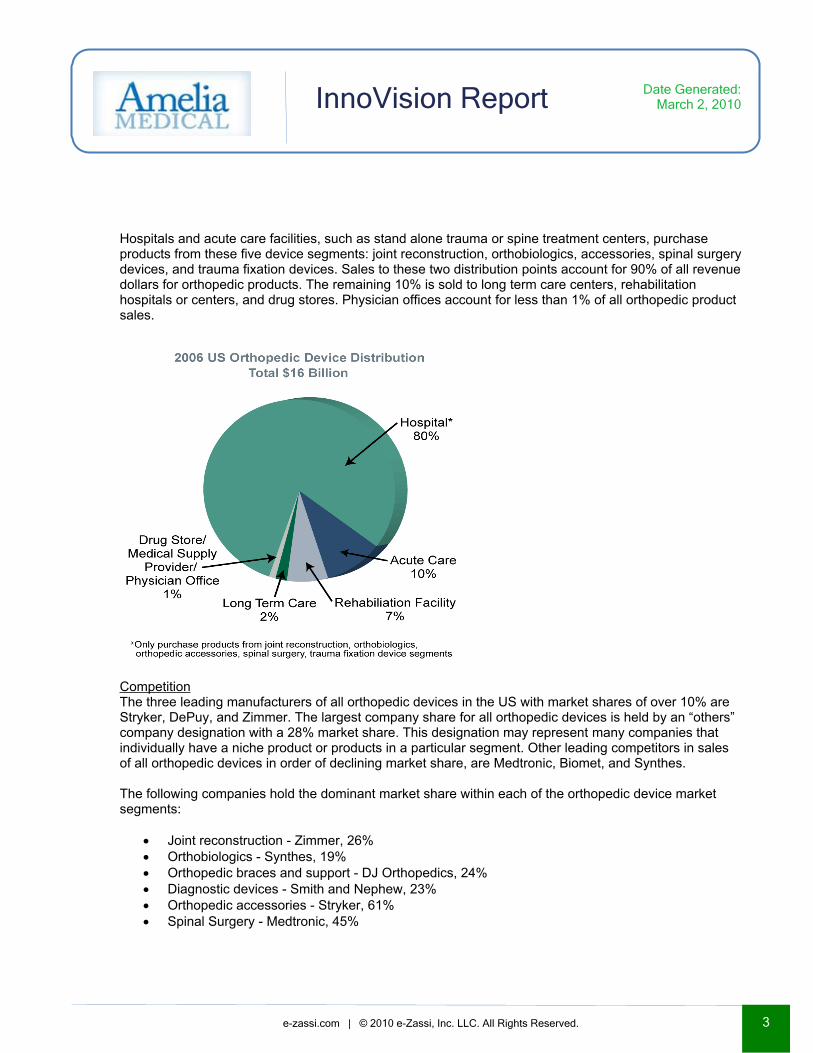
Hospitals and acute care facilities, such as stand alone trauma or spine treatment centers, purchase products from these five device segments: joint reconstruction, orthobiologics, accessories, spinal surgery devices, and trauma fixation devices. Sales to these two distribution points account for 90% of all revenue dollars for orthopedic products. The remaining 10% is sold to long term care centers, rehabilitation hospitals or centers, and drug stores. Physician offices account for less than 1% of all orthopedic product sales.
CompetitionThe three leading manufacturers of all orthopedic devices in the US with market shares of over 10% are Stryker, DePuy, and Zimmer. The largest company share for all orthopedic devices is held by an “others” company designation with a 28% market share. This designation may represent many companies that individually have a niche product or products in a particular segment. Other leading competitors in sales of all orthopedic devices in order of declining market share, are Medtronic, Biomet, and Synthes.
The following companies hold the dominant market share within each of the orthopedic device market segments:
Joint reconstruction - Zimmer, 26% Orthobiologics - Synthes, 19% Orthopedic braces and support - DJ Orthopedics, 24% Diagnostic devices - Smith and Nephew, 23% Orthopedic accessories - Stryker, 61% Spinal Surgery - Medtronic, 45%
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
4
(Global Markets Direct, Orthopedic Devices Market Profile, 2008)
US Orthopedic Soft Tissue Device Market SegmentOrthopedic soft tissue products are used to diagnose, treat, or repair injuries and disorders of the soft tissue in the joints including tendons, ligaments, and fascia. Soft tissue diagnostic and repair procedures include biopsies, excisions, carpal and tarsal tunnel surgery, capsule, ligament and tendon repairs and reconstructions.
Products in this category include:
1. Arthroscopes (to access all joints)2. Cameras and visualization systems3. Fluid management systems4. Powered shavers and drills5. Handheld instruments (for removal of bone and soft tissue)6. Radiofrequency systems 7. Metal and resorbable soft tissue repair implants (screws, anchors, tacks, etc.)
Soft Tissue ProceduresThere were more than 10 million soft tissue repair procedures performed in the US in 2007, and more than 15 million worldwide, with revenues close to $2.9 billion. Revenues for the segment increased by 13% over 2006. Primary drivers of the soft tissue market are longer and better quality of life, and a growing market preference for minimally invasive surgeries. In the United States, the expanding number of ambulatory surgery centers has also contributed to market growth.
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
5
In the United States, 3.3 million of soft tissue repair surgeries were done arthoscopically, via a scope inserted into the joint through a small opening in the skin, which is sometimes called a keyhole opening.
Arthroscopic Soft Tissue ProceduresOf all the arthroscopic soft tissue procedures done in 2007, more than 2 million were knee surgeries, half of which were meniscectomies, which are surgeries that involve surgical removal of all or part of a meniscus. Globally, the arthroscopic market is expected to continue to grow at a rate of 9% per year from $2 billion in 2006 to approximately $3.7 billion in 2011.
CompetitionThere are many companies participating in the soft tissue market, however the five largest companies together hold more than 70% of the market share. These companies are Smith & Nephew (approximately 23% share), Stryker, Johnson & Johnson, Arthrex, and ConMed Linvatec. All leading companies in this market space sell arthroscopes, visualization devices, handheld instruments for minimally invasive joint repair, suturing devices and metal and/or resorbable implants.
Knowledge Enterprises, Inc., The 2007-2008 Orthopaedic Industry Annual Report, 2008Espicom Business Intelligence, Orthopaedics Market Report, 2008
Additional United States market subsegment information is not available for the Tendon Repair Device.
European Orthopedic MarketIn 2006, the European market for orthopedic devices was $6.2 billion. By 2011, this market is projected to generate $9.0 billion in revenues. The orthopedics device market in Europe, in percentage of market, includes:
Joint reconstruction devices (66%) Orthopedics braces and supports (10%) Spinal surgery (7%) Trauma fixation (7%) Orthobiologics (4%) Orthopedic diagnostic devices (3%) Orthopedic accessories (3%)
Compound Annual Growth Rate (CAGR) forecast in percentages for the European orthopedic device market in declining order for 2006 - 2011), includes:
Spinal surgery (12%) Orthopedic accessories (8.5%) Joint reconstruction (7.5%) Orthopedic braces and supports (7.3%) Orthobiologics (6.7%) Orthopedic diagnostic devices (6.7%) Trauma fixation (5.5%)
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
6
CompetitionLeading manufacturers of orthopedic devices in Europe based on market share of revenue, in declining order are:
Stryker (16%) Zimmer (14%) Dupuy (11%) Biomet (9%) Smith and Nephew (7%) Synthes (4%) Corin (3%) Remaining 36% is distributed among other competitors with smaller shares
In Europe, orthopedic device shareholders (in percentage) by market category are:
Joint reconstruction - Zimmer (20%) Spinal surgery - Medtronic (31%) Orthobiologics - Synthes (10%) Trauma fixation - Synthes (34%) Orthopedic braces and supports - DJ Orthopedics (15%) Orthopedic diagnostic devices - Smith and Nephew (22%) Orthopedic accessories - Stryker (39%)
(Global Markets Direct, Orthopedic Devices Market Profile, 2008)
Additional European Union market subsegment information is not available for the Tendon Repair Device.
Additional information related to the minimally invasive surgery products market is located in the appendix.
Key Purchasing Decision MakerPhysician SpecialistSales representatives gain access to most specialty physicians in their private practice office or institutional clinic. Effective specialty sales representatives are strong relationship builders and communicators with deep knowledge of the product and the specialty for which it is used. A sales team with existing relationships in a particular specialty is extremely valuable in a marketing effort to this group.
A significant promotional budget may be required to increase awareness in the specialty community. Many successful new products have made large investments in peer educational initiatives that increase awareness and spread the word from doctor to doctor.
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
7
Device Primary Intended UseTreatment and Management ProductsThe intended use of the Tendon Repair Device has a direct impact on where a product is sold, the patient population, and overall marketing strategy. Planning for sufficient marketing and/or sales power to reach the potential users of the product is a critical marketing consideration.
Treatment and management products may be used in a wide variety of settings, including; acute care facilities, outpatient clinics, physician offices, and to a lesser extent, long-term care facilities, rehabilitation facilities, and/or home care. Many companies have employed successful segmentation strategies, which involve focusing marketing resources on only one segment of the potential market at a time.
Product ProfileMarketing ImplantsAn implant is a medical device that is designed to replace a missing biological structure, such as a breast or a joint. Implants are typically inserted surgically. The decision to select a specific surgical implant usually rests with the surgeon, however more and more frequently patients play a role in selecting the implant that will be used. This is particularly true with breast implants, but the patient is often consulted for other types of implants, including joint replacements and tissue implants. Patients have enough input in the decision that some companies are using direct-to –consumer advertising, including television ads, to influence their opinions. In an institutional setting, the use of a specific implant is likely to require approval by the product committee. Significant clinical and safety and efficacy data is critical to the ability to make inroads in the implant market.
RegulatoryThe medical device industry is regulated by government agencies around the world to help ensure public safety. These agencies monitor the distribution, marketing, design, and manufacturing of medical devices sold in the country of the agency. In the US, the government agency that is responsible for this regulation is the Food and Drug Administration (FDA). In the European Union, each country has its own government agency known as a Competent Authority. This section of the report includes the likely US and EU regulatory parameters and classifications for the technology.
US Regulatory
US Regulatory Status: A regulatory submission is required, but not yet filed FDA Submission No.: Not ApplicableFDA Clearance/Approval Date: Not Applicable
FDA Regulation / Device Classification / Predicate Product Code / Regulatory Pathway:
The following information resulted from the analysis of the Tendon Repair Device by the e-Zassi Technology Assessment Platform. The results shown below are potential predicate regulation and
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
8
product codes that represent similar types of legally marketed devices. This information provides insights to be shared in conversations with the FDA or regulatory consultants.
FDA Regulation No: 888.4580
FDA Regulation Name: sonic surgical instrument and accessories/attachments
Product Code Product Code Title Classification Regulatory PathwayJDX Instrument, Surgical, Sonic And
Accessory/AttachmentClass II 510(k)
LZV System, Cement Removal Extraction Class II 510(k)
The FDA assigns devices to one of three regulatory classes based on the level of control necessary to assure safety and effectiveness of the device. The Tendon Repair Device may be considered a Class II medical device because at least one of its characteristics is associated with a marginal level of risk. It is good practice to review the proposed device classification directly with the FDA. Class II devices are those for which general controls alone are insufficient to ensure safety and effectiveness. Existing methods are available to provide such assurances. In addition to complying with general controls, Class II devices are also subject to special controls. Special controls may include distinct labeling requirements, mandatory performance standards, and post market surveillance. Examples of Class II devices include powered wheelchairs, infusion pumps, and surgical drapes.The FDA will likely require some performance testing or bench testing, or both, to support the safety of the device. Confirm the Tendon Repair Device classification with the FDA by submitting a 513(g). The 513(g) request should include the following:Cover letter stating the submission is a 513(g) request for information concerning the Tendon Repair Device, including; device status, classification, and regulatory requirementsDetailed description of the Tendon Repair Device1. Concise indication(s) for Use Statement
The Tendon Repair Device proposed labeling or labeling of a marketed similar device2. If no labeling is available, note this in the cover letter3. Contact person's name, address, telephone number, and fax number on the cover letter4. Copy of the Medical Device User Fee Payment sheet (MDUFP)
Send the 513(g) request to:513(g) Coordinatorc/o Document mail Center (HFZ-401)Office of Device EvaluationCenter for Devices and Radiological Health9200 Corporate BoulevardRockville, MD 20850
The Tendon Repair Device regulatory path requires a Premarket Notification 510(k).
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
9
A 510(k) is a premarket submission made to the FDA to demonstrate that the device to be marketed is at least as safe and effective (substantially equivalent) to a legally marketed device not subject to premarket approval. The requirements for a 510(k) submission (there is no form) are described in 21 CFR 807, Subpart E. Before marketing a device, each submitter must receive an order, in the form of a letter, from the FDA which finds the device to be substantially equivalent (SE). Substantially equivalent means that the device can be marketed in the U.S. The order “clears” the device for commercial distribution. Submitters must compare their device to one or more similar legally marketed devices, make substantially equivalent claims, and then support those claims. The device’s substantial equivalent comparison to legally marketed device(s) is commonly known as the "predicate". The submitter cannot proceed to market the device in the U.S. until the SE order is received. The SE determination is based on information provided by the submitter, and is usually made within 90 days. NOTE: To evaluate potential predicate devices against which substantial equivalency claims may be drawn, visit the 510(k) Premarket Notification page on the FDA website at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm. Then, enter into the Product Code search box, a suggested product code identified by the e-Zassi US Regulatory Calculator and click the Search button on the FDA web page.A 510(k) will require accompanying regulatory fees, and additional resources to submit the substantially equivalent filing. There is a possible need to increase resources in budget, schedule, and other areas, in order to achieve regulatory approval for the Tendon Repair Device.
Classification as a Combination Product and FDA Primary Mode of Action:
The Tendon Repair Device is likely to be considered a medical device with biologic combination product. Examples of this type of combination product include:
Devices coated or impregnated with a biologic Skin substitutes with cellular components Orthopedic implants with growth factors Tissue, cellular, or gene therapy products
Combination products, such as the Tendon Repair Device require pre-clinical testing, and often clinical testing to support safety and efficacy of both the device and biologic portions of the product. It is recommended that early correspondence with the FDA be obtained for guidance on the type and amount of testing required for the product. Biologic product development and commercialization typically takes 4-6 years, with a cost of $5-7 million. The development and commercialization of a new medical device typically takes 2-4 years, with a cost of $1-2 million. The complexity of the Tendon Repair Device device/biologic combination will have a great bearing on development time and costs.
The primary mode of action (PMOA) is defined as the component of a combination product that is expected to make the greatest contribution to the overall intended therapeutic effects of the combination product. The device portion of Tendon Repair Device may hold the primary mode of action (PMOA). The FDA makes a final assignment based on information provided about the Tendon Repair Device and how the device and biologic components work together. The FDA will then assign primary jurisdiction of the Tendon Repair Device to one of its established centers for regulatory review based on the PMOA finding. If the FDA agrees that the device portion holds the PMOA, the Center for Devices and Radiological Health (CDRH) will be the lead center. CDRH personnel will review the product submission, while the
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
10
Center for Biologics Evaluation and Research (CBER) will be consulted for questions or issues regarding the Tendon Repair Device biologic component.
European Union (EU) RegulatoryEU Regulatory Status: A regulatory filing is required but has not been submitted
EU Device Classification / Regulatory Pathway:
The following information resulted from the analysis of the Tendon Repair Device by the e-Zassi Technology Assessment Platform and represents the likely EU Regulatory parameters for the Tendon Repair Device.
Classification Regulatory Pathway
Class III CE Mark / Design Dossier / Full Quality Management System
The Tendon Repair Device may be classified as a Class III device according to the European Union (EU) Medical Device Directive. Class III devices typically involve the most risk to patients; they include devices that contact the central nervous system or heart, or are absorbed by the body.
Class III device manufacturers are required to develop a full Quality Management System in compliance with ISO 13485: 2003. A Notified Body is required for certification of Class III devices. A Notified Body will ensure that the documentation and quality system that are developed are in accordance with EU Regulations. The following steps must be followed in order for the Tendon Repair Device to be shipped to Europe.
Locate a Notified Bodyo This is an outside service that will be responsible for auditing the Technical File and
Quality Management System (QMS) to ensure that they meet the requirements of the directive. This service will cost around $15,000-$30,000
o Examples of Notified Bodies include TUV, BSi, Intertek Implement a Quality Management System (QMS)
o Full QMS in compliance with ISO 13485: 2003 Prepare Design Dossier
o This provides detailed information intended to demonstrate compliance with the health and safety requirements stipulated in the directive
o This is a detailed set of documents which provides information from design and development through manufacturing
Appoint an Authorized Representative (EC REP)o There must be a physical address in Europe which is identified on the product
labeling
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
11
o There must be a person at that address that can handle regulatory issues, e.g., product complaints, recalls, Customs issues
Register with Competent Authority (if applicable)o A Competent Authority is the equivalent of the FDA in the U.S.; each country has its
own government agency that regulates medical deviceso Must be located in the same country as the Authorized Representativeo Note: Not all countries require registration
Obtain CE Certificateo This will be issued by the Notified Body after successfully passing the registration
audit and technical file reviewo Follow-up audits are required to keep CE Certificate
Prepare Declaration of Conformityo A legally binding document that certifies all of the requirements of CE Marking have
been meto Work with the Notified Body to complete this
Notified Body must certify the declaration
Clinical Research RequirementsBased on the device classification, the United States (US) approval or clearance process for medium to high risk medical devices is very different from the European Union (EU), especially in terms of the scope and size of clinical trials required for high risk devices. In general, to receive approval to market a device in the EU, the manufacturer must demonstrate that the device is safe and that it performs in a manner consistent with the manufacturer’s intended use. To receive approval or clearance to market a high-risk device (Class III and some Class II) in the US, the manufacturer must demonstrate the device to be effective as well as safe, thus requiring prospective, randomized clinical trials often involving hundreds of human subjects. Further, additional clinical data may be required for Class IIb devices in the EU. Regardless of the regulatory body (US or EU), it is important for the innovator to discuss the scope and size of clinical trials required for medium to high risk devices with the appropriate regulatory body.
The information presented within this report does not represent or intend to be an opinion, a suggestion, a comment, or a decision from the FDA, or any of its authorities, agents, employees, or third party representatives. This information is an estimate and is intended as preparation for conversations with the appropriate regulatory authority or regulatory consultant. All aspects of the technology must be evaluated in order to make an accurate classification. The regulatory authority or regulatory consultant can assist in fully assessing the technology. Ultimately, the regulatory authority reserves responsibility for classifying a new medical device.
Clinical
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
12
Pre-Clinical Research StatusIn the US and in Europe, pre-clinical trials that involve the use of animals must be implemented based on regulatory requirements, local laws, and quality standards for animal testing. In addition, in Europe, due to the concern regarding animal use in the cosmetic industry, it is also important to contact the EU Regulatory Authority to determine whether or not animal testing is required for the device under investigation.The Tendon Repair Device may benefit from animal studies used to meet biocompatibility testing requirements, establish safety and tolerance guidelines, or to provide other clinical outcomes that will support the overall product development process. In addition, animal testing data should adequately demonstrate that the device is relatively safe and that it functions as intended.
In the US and in Europe, pre-clinical trials that involve in vitro bench testing must be implemented based on regulatory requirements, local laws, and quality standards. In vitro bench testing should provide adequate data to demonstrate that the device is relatively safe and that it functions as intended.
The technology may use testing parameters, such as the following, to demonstrate optimal device operation:
Hazard analysis Temperature ranges Humidity ranges Pressure (e.g., altitude)
Table I – Overview of Pre-Clinical Research Status
In-Vitro Bench Testing Status:Pre-clinical in-vitro testing is complete
Animal Testing Status:Animal testing is complete
Human Clinical Research Status
Clinical trials that involve the participation of human subjects may be conducted anywhere in the world. However, each country or region has specific regulatory requirements for conducting clinical research and for the transporting of investigational device(s) into that country or region. The US, Europe, Canada, and Japan accept the International Conference on Harmonisation Good Clinical Practices (ICH/GCP) guidelines for designing, conducting, recording, and reporting the findings of the clinical trial. The clinical trial sponsor must ensure compliance with ICH/GCP and regulatory requirements prior to conducting clinical device trial(s) in a given country or region. The Tendon Repair Device clinical trial(s) must include qualified research professionals with the prerequisite education, training, and experience to support the quality systems requirements (QSRs) ICH/GCP, and regulatory requirements. These qualified research professionals include (but are not limited to) a medical director, a clinical monitor, a project manager, a data manager, a biostatistician, a quality assurance manager, and a regulatory affairs representative. Additionally, clinical conduct standard operating procedures (SOPs) must be either written and maintained by the medical device developer, and/or manufacturer, or transferred to a Clinical Research Organization (CRO). The developer or manufacturer, however, is ultimately responsible for compliance.
Table II – Overview of Human Clinical Research Status
ClinClinClinicalicalical
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
13
Types of Human Clinical Research Conducted:First-in-man Trial
Pilot Trial Results (US, EU):Not Identified
Clinical Trials Location:United States
Pivotal Trial Results (US, EU):Not Identified
Clinical Data Available to Support Claims:User reports the data does not support any claim at this time
Regulatory Confirmation of US/EU GCP Compliance:Yes
Clinical Research Published:No
Which regulatory body confirmed GCP Compliance?U.S. Food and Drug Administration
FDA Clinical Strategy Meeting: Met with FDA prior to clinical trials
E6 Good Clinical Practice Consolidate Guidance (ICH/GCP) Section 8, essential documents are those documents that individually and collectively permit evaluation of the trial, and the quality of the data produced. These documents serve to demonstrate the compliance of the investigator, and/or sponsor; and that the trial and data have been monitored in accordance with Good Clinical Practice (GCP) standards and all applicable regulatory requirements. Essential documents are collected before (ICH/GCP 8.2), during (ICH/GCP 8.3), and after (ICH/GCP 8.4) the clinical trial.
Table III – Overview of Essential Documents Collected for Clinical Trial(s)Type of Essential Documents Collected for Clinical Trial(s): Essential documents collected before the clinical phase of the trial started as outlined in ICH GCP Section 8.2Essential documents were collected during the clinical conduct of the trial as outlined in ICH GCP Section 8.3
Specific Essential Documents Collected During the Trial(s) (ICH GCP 8.3]*:Any revisions to protocol, informed consentsMonitoring Visit ReportsSigned informed consentsSubject screening and enrollment logs
Specific Essential Documents Collected Before the Trial(s) Began [ICH GCP 8.2]*:IRB/IEC Approval LetterInitiation Visit Monitoring Report
Specific Essential Documents Collected After the Trial(s) were completed/terminated (ICH GCP 8.4]*:Not Identified
Note: The above table illustrates only a small sample of the essential documents collected before, during, and/or after a clinical trial(s). Further review of all essential documents collected for a clinical trial(s) should be conducted to ensure compliance with ICH/GCP section 8, essential documents.
ReimbursementCoverage and payment for new, innovative devices in the global health care system is a complex process. Each country has a distinct set of criteria, coding system, and decision making process. A well designed plan is essential to success in obtaining global reimbursement for the Tendon Repair Device. Currently, e-Zassi data only includes US and German reimbursement content. Additional content for the other countries is coming soon.
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
14
US ReimbursementThe US reimbursement process requires both coverage and coding. Coverage is determining the eligible benefit and the criteria that must be met to obtain a specific covered service by various organizations involved in reimbursement for a new device, technology, or service. Coding is used to define medical conditions, to identify medical services and devices used in various clinical settings, and by who is using the new device. The following brief overview describes US codes used for identification of diseases, surgical procedures, diagnostic procedures, medical services, devices, supplies, and drugs. The following table describes the likelihood of the technology having existing reimbursement coverage in the US:
Overall Coverage: Likely existing coverage applies
International Classification of Diseases 9th Revision, Clinical Modification (ICD-9-CM) codes identify the reason for the health encounter (medical diagnosis, specific injury, or other health care reason). The codes are use to process billing and to track health statistics. They are used in-hospital, out-patient clinic, and specialty facilities. (Nursing homes and physician offices) The Center of Disease Control, National Center for Health Statistics (NCHS) manages the process for new ICD-9-CM codes. The following table describes the likelihood of an existing ICD-9-CM code being associated with the technology:
ICD-9-CM: An ICD-9-CM code is identified
Current Procedural Terminology (CPT) codes are uniform codes of descriptive terms that are used primarily to identify medical services and procedures furnished by physicians and other health care professionals. The clinical settings include a physician office, the out-patient clinic or surgical center, and other unique specialty facilities. The American Medical Association (AMA) manages the process for new CPT codes. The following table describes the likelihood of an existing CPT code being matched with the use of the technology:
CPT: A CPT code is identified
Healthcare Common Procedure Coding System (HCPCS) codes are alpha-numeric codes that describe categories of products, medical supplies, certain drugs and biologics, certain devices, and some procedures not covered by CPT codes. The HCPCS code is used for devices in a person’s home, the physician office, an out-patient clinic, or a surgery center. The Center for Medicaid and Medicaid Services HCPCS Workgroup manages the process for a new HCPCS code. The following table describes the likelihood of an existing HCPS code matching a description of the technology:
HCPCS: An HCPCS code is identified
German Reimbursement
The German health system requires that all its citizens be provided health care coverage. The majority of its population (85%) is covered under the mandatory statutory health insurance (SHI) and the remainder by a mix of private and public providers. The system is funded by the statutory health insurance (Pflegekasse) contributions, private funds, and government social security (sickness) funds. Employed
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
15
citizens must contribute, along with their employers, into the SHI and the long term care (LTC) insurance which covers community-based services. Individuals choose their mandatory health plan from one of more than 230 health insurance companies. The range of covered services and products is defined in the German Code of Social Law.
In Germany there are two different reimbursement systems I; the German Diagnostic Related Group system and the doctor’s fee scale reimbursement system.
The German Diagnostic Related Group system (G-DRG) includes codes used for treatments in hospitals. Each Federal member state (of which there are 16) establishes the base-rate for hospitals within their State, which is then calculated with the G-DRG mix-weight to determine the overall payment for a patient encounter in the hospital. The following table describes the likelihood of a G-DRG code matching the technology:
G-DRG: A G-DRG code is identified
In the doctor’s fee scale reimbursement system, EBM-Code: (Einheitlicher Bewertungsmaßstab = EBM) is the doctor’s fee scale reimbursement system for office-based physicians. Physicians are organized in Regional Physicians’ Associations (of which there are 17) with budgets set by the federal member state for management of the population base. Physicians are paid per service they provide. All insurance (SHI) fees are standardized throughout Germany. Private Insurance fees vary. The following table describes the likelihood of an EBM-code being available for the technology:
EBM-Code: An EBM-Code is identified
Other supporting codes used in the German health system include:
Operation Procedure Codes (OPS) identify surgical treatments. Operation Procedure Codes are determined by the location of treatment. In a hospital or a clinic treatments are categorized under a G-DRG, or under an EBM-Code for treatment in a doctor’s office. The following table describes the status of an OPS code associated with the technology:
OPS: An OPS code is identified
German Health System (GHS) Therapeutic Appliances List (Hilfsmittelverzeichnis) includes categories of devices used in the community for in home use. The likelihood of whether or not the technology is included is identified in the following table:
Therapeutic Appliance List:
A Therapeutic Appliances List code is identified
International Statistical Classification of Diseases (ICD) and Related Health Problems; 10th revision (ICD-10) codes track the diagnosis for utilization of services for health statistics and are used to process
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
16
payment in a hospital and other facilities. The following table describes whether or not it is likely that an ICD-10 code identifies the clinical reason for the technology:
ICD-10: An ICD-10 code was not identified
Intellectual PropertyIntellectual property considerations are a critical part of the due diligence required when determining the potential for commercializing a technology. The following information characterizes the intellectual property status of the Tendon Repair Device.
Patent StatusPatents are generally granted on a nation by nation basis. In the US, patent grants are effective for 20 years from the date of filing, and protect an invention only within the United States, and in its territories and possessions. Under US law, when a patent is granted, the grant provides the right to exclude others from making, using, offering for sale, selling, or importing the invention. When the patent is issued in the US, the patentee has the burden of enforcing the patent, not the United States Patent and Trademark Office (USPTO).
The Tendon Repair Device patent status is: No patents are issued. Patent applications are pending.
A written opinion from a patent attorney evaluating the patentability of the Tendon Repair Device was rendered. Assuming the opinion is favorable, this is a useful piece of information for potential investors or other interested parties. It should be noted that any disclosure of such an opinion is usually done only under a confidentiality disclosure agreement (CDA).
Intellectual Property Ownership and EncumbrancesOthers may have intellectual property rights to the Tendon Repair Device. Meticulous review must be conducted to ensure that all patents are properly filed, and to indicate all of the inventors involved with the Tendon Repair Device. These precautions will help ensure the parties that hold rights to the invention are properly represented during subsequent license or sale negotiations. The following individuals or organizations may have rights to the Tendon Repair Device: Co-inventor may have rights, Company or employer may have rights.
It is uncertain if encumbrances or contractual obligations for the Tendon Repair Device exist. More information is needed to determine if such obligations exist for the Tendon Repair Device.
Public Disclosure and Public Use
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
17
The protection of intellectual property is important to innovators and partners. Public disclosure and/or public use of a technology may jeopardize intellectual property protection. The following table provides information on the status of public disclosure and public use for the Tendon Repair Device.
Public Disclosure and Use Status Date of Occurrence
Public Use: No Public Use has occurred None Identified
Public Disclosure: Public Disclosure has not occurred None Identified
Trademark and Copyright StatusTrademarks and copyrights are valuable Intellectual Property (IP) components. The following table provides information on the trademark and copyright status of the Tendon Repair Device.
Trademarks and Copyrights Status Type
Trademarks: No trademark NA
Copyrights: No copyright NA
Litigation Status
It is uncertain if the Tendon Repair Device is currently involved in a litigation or legal dispute.
Technology Platform Considerations
Some medical innovations are considered technology platform innovations and may offer clinical benefit across medical specialties. When this occurs, additional intellectual property review is recommended to insure that all IP rights are protected. At this point, the Tendon Repair Device is not reported to include such a DNA technology platform. This conclusion means that the Tendon Repair Device may note only one clinical application within the patent application.
Product Development and ManufacturingSuccessful medical device development of the Tendon Repair Device requires a core development team of individuals with expertise in; Project Management, Intellectual Property, Design, Process Development, Manufacturing, Procurement, Quality, Regulatory, Reimbursement, Medical Affairs, Clinical Affairs, and Marketing. Product development requires a broad range of knowledge about the market in which the technology will participate, including current products, requirements, key features, and claims. The successful developer will also be very familiar with customers and competitors. Most importantly, the developer will have a clear understanding of user needs, the development process, and risks (patient, technical, safety, business, schedule, and cost). The following analysis includes invaluable information in devising an effective development plan for the medical device technology.
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
18
The selection of a regulatory-compliant, competent manufacturer is a key consideration for all medical product innovators. The innovator has reported the following about the Tendon Repair Device manufacturing facility:
manufacturer selected or being considered has experience with the Tendon Repair Device or similar device type has a QSR/GMP compliant quality system registered with FDA ISO certified uncertain about successful QSIT audit
Quality System Regulations (QSRs)
Global quality system regulations require that a medical device be designed, developed, and manufactured under design controls as defined by 21 CFR 820.30(h) for US and International Standards Organization (ISO) 13485 section 7.3 for International design control requirements. The quality system of a medical device company must contain procedures that define how the manufacturer will address design control elements and design transfer regulations. Both the design and manufacturing company must have a compliant quality system in place. This is true even when the design company does not plan to manufacture the medical device.
The design history file (DHF) must contain a comprehensive record of the design process as defined by the applicable design control requirements. An executive summary is created in the Phase IV Design Transfer process that contains references to specified critical required documentation. The clinical evaluation phase is only used if the clinical evaluation (clinical trials) are required or implemented.
Note: The manufacturer must have procedures in place and must maintain documentation in the design history file to demonstrate compliance with the design control requirements of §820.30 and completion of the activities identified in the design plan. The design history file must be made available for FDA inspection. The FDA will conduct routine quality system inspections of all classes of devices subject to design to evaluate the manufacturer’s compliance with design control requirements.
Medical Device Design Control Development PhaseThe innovator has reported the following for the Tendon Repair Device:
Level of planning: Business Plan, Marketing Plan
Level of documentation: Full engineering drawing package (human/clinical poduct)
Product build status: Three dimensional computer model(s), Functional prototype(s) for in-vitro/animal testing, Functional, clinical product
Design control system: In compliance to Good Manufacturing Practices
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
19
(GMP)
Development stage: Phase II Design Output
The following chart depicts the typical formal design control process phase with the current phase of development (Phase II Design Output) highlighted.
Design control deliverables status at the time of this report:
Phase I Design Input
The concept document complete and released to formal documentation control Complete
The marketing/user needs specification complete and released to formal document control Complete
The design specification complete and released to formal document control Complete
The hazard analysis complete and released to formal document control CompleteThe design failure mode and effects analysis (DFMEA) complete and released to formal document control Complete
The process failure mode and effects analysis (PFMEA) drafted Complete
The phase I design input design review conducted and approved Complete
Phase II Design Output
The design input document updated and under formal document control CompleteThe device master record (DMR) complete, updated, and under formal document control Incomplete
The risk analysis updated and under formal document control Complete
The master design verification and validation plan established Complete
The suppliers selected and qualifications in process Incomplete
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
20
The initial transfer cost of goods sold (TCOGS) drafted IncompleteThe phase II design output design review conducted and approved. The minutes from the design I input design review examined and all action items sufficiently closed out.
Incomplete
Phase III Design Verification and Validation
The design specification testing including all label performance claims complete Incomplete
The biocompatibility and accelerated aging and/or ship testing complete CompleteThe manufacturing and quality procedures under formal document control and training initiated Incomplete
The process validation (PV) protocols drafted, installation qualifications (IQs) and/or operational qualifications (OQs) complete, and the process qualifications (PQs) and product performance qualifications (PPQs) approved Incomplete
The final manufacturing plan and the transfer cost of goods sold (TCOGS) established and up to date Incomplete
The regulatory submission, clinical investigational exemption (IDE), or the institution review board (IRB) protocols and preparation fully complete Incomplete
The phase III design verification and validation design review conducted and approved. The minutes from the phase II design output design review examined and all action items sufficiently closed out.
Incomplete
Phase IV Design Transfer
The process validation (PV), installation qualification (IQ), operational qualification (OQ), process qualification (PQ), product performance qualifications (PPQs), and supplier qualifications approved
Incomplete
The device master record (DMR) complete, updated, and under formal document control Incomplete
The training and pertinent documentation updated and complete IncompleteThe design-related material review boards (MRB) closed and corrective action requests (CAR) implemented Incomplete
The bill of materials (BOM) successfully transferred to manufacturing and/or the procurement specialist. Also, ownership of tools, machines, and gauges documented
Incomplete
The initial build order submitted and the forecast updated IncompleteThe phase IV design transfer design review conducted and approved. The minutes from the phase III design verification and validation design review examined and all action items closed.
Incomplete
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
21
Material Biocompatibility Considerations and Status
All submissions for regulatory approval of medical devices must include a biocompatibility assessment of the device and the materials use therein as one of the assurances the device is safe for its intended use. The guidance documents used to determine what testing may be required are:
In Europe, refer to ISO 10993, “Biological Evaluation of Medical Devices”. In the US, refer to the FDA Blue Book Memorandum #G95-1, “Use of International Standard ISO 10993, ‘Biological Evaluation of Medical Devices’ – Part 1: Evaluation and Testing”.
The specific biocompatibility tests required for any given device are dependent upon where the device or component contacts the patient and for how long. The user provided the following information for the technology during the interview process:
Device Type: Surgical Implant
Patient Contact Type: Direct patient contact with breached or compromised surfaces, tissue, tissue fluid, bone or dentin, blood, directly (circulating) for less than 60 minutes
Sterility Requirements: Sterile when used
Biocompatibility test sample lot builds must be completed under formal design control using final manufacturing (production not prototype) processes and materials, and maximum sterilization dosage.The user has identified the following biocompatibility test requirements and status for the Tendon Repair Device:
Biocompatibility Test: Current Status:
Cytotoxicity Incomplete
Sensitization Incomplete
Irritation or Intracutaneous Reactivity Incomplete
System Toxicity (Acute) and Pyrogenicity More Information Required
Sub-chronic Toxicity (Sub-acute) Incomplete
Biocompatibility Genotoxicity Test Status Incomplete
Biocompatibility Implantation Test Status More Information Required
Haemocompatibility Incomplete
Chronic Toxicity Incomplete
Carcinogenicity Incomplete
Reproductive Developmental Not Required
Biodegradable Incomplete
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
22
Grant EligibilityThe e-Zassi grant matching engine utilizes a proprietary technology model record designed to match medical device technologies to International and US federal, state, and local grant programs. Initially, the grant database will include the National Institute of Health (NIH) current federal grant program parent announcement/omnibus solicitation PHS 2009-2 SBIR/STTR program descriptions and research topics as well as the current US state grant/loan programs available. The e-Zassi grant matching engine searches the state and NIH solicitations on a regular basis. A key feature of the e-Zassi algorithm is that it does not require confidential information to be disclosed when calculating the best NIH SBIR/STTR grant and US state grant matched to the Tendon Repair Device.The following grant opportunities have been identified as potential matches for the Tendon Repair Device.
The table below provides a brief description of each grant, as well as contact information where further information can be obtained.
Grant Name Description Contact InfoNational Institute of Health SBIR
2008 - 2009 National Heart, Lung, and Blood Institute Division of
Cardiovascular Diseases http://www.nhlbi.nih.gov
Circulatory support systems: Implantable rechargeable batteries
and alternate power sources
J. Timothy Baldwin, Ph.D. Pothur R. Srinivas, PhD 301-435-0513, 301-435-0550 [email protected];
National Institute of Health SBIR 2008 - 2009 National Institute of
Biomedical Imaging and Bioengineering
http://www.nibib.nih.gov
Biomaterials. Development of new or novel biomaterials that can be used for a broad spectrum of biomedical applications such as implantable devices; drug and gene delivery;
tissue engineering; imaging agents; and biosensors and actuators.
Research that is
Mr. Todd Merchak 301-496-8592 [email protected]
National Institute of Health SBIR 2008 - 2009 National Institute of
Biomedical Imaging and Bioengineering
http://www.nibib.nih.gov
Biomechanics and Rehabilitation Engineering. Research on
biomechanics which can be applied to a broad range of applications including implants, prosthetics,
clinical gait and posture biomechanics, traumatic injury, repair processes, rehabilitation,
sports
Mr. Todd Merchak 301-496-8592 [email protected]
National Institute of Health SBIR 2008 - 2009 National Institute of
Biomedical Imaging and Bioengineering
http://www.nibib.nih.gov
Medical Devices and Implant Science. Design, development,
evaluation and validation of medical devices and implants. This includes
exploratory research on next generation concepts for diagnostic
and therapeutic devices; development of tools for assessing
Mr. Todd Merchak 301-496-8592 [email protected]
National Institute of Health SBIR Surgical Tools and Techniques. Mr. Todd Merchak 301-496-8592
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
23
2008 - 2009 National Institute of Biomedical Imaging and
Bioengineering http://www.nibib.nih.gov
Research and development of new medical technologies to improve the outcomes of surgical interventions. Examples of relevant technologies
include: minimally invasive surgeries, energy-based
interventions such as RF ablation,
Prairie Winds Capital, LLC http://www.prairiewindscapital.com/
PWC is a RAIN® (Regional Angel Investment Network) fund member of the Rain Source Capital group,
which is itself located and headquartered in Saint Paul,
Minnesota
Dale Froehlich 605.275.2833 [email protected]
RAIN Source Capital (S/Expans) http://www.rainsourcecapital.com/
RAIN Source Capital is a multi-state network of RAIN® funds that works
with angel investors who are interested in supporting growing
companies.
Steve Mercil 651-632-2140 [email protected]
For your consideration e-Zassi is providing the following information on both federal and state grant programs.
There are certain criteria the innovator should remember when applying for a US state grant program:
The business must be owned 51% or more by a US citizen Only small to medium-sized businesses (500 employees or fewer) are eligible for US state grant
funding programs Most states require that at least 50% of business operations and/or employees be located within
the state, with the exception of angel networks or venture capital firms If a funding program includes multiple states, then it is a grant program in which the business
operation location rules may include different criteria
When the innovator prepares an SBIR proposal and submission, the following links may prove to be very helpful:
1. Tutorial and Advice on applying for NIH SBIR grants (http://www.niaid.nih.gov/ncn/sbir/pres.htm)
2. PHS-2009-2 Small Business Innovation Research Program Parent Announcement (SBIR [R43/R44]) (http://grants.nih.gov/grants/guide/pa-files/pa-09-080.html)
3. Small Business Technology Transfer Program Parent Announcement (STTR [R41/R42]) (http://grants.nih.gov/grants/guide/pa-files/pa-09-081.html)
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
24
It is important to note that if Venture Capital is listed as a source of capital in the following Innovator Objectives section then, the technology may not be eligible as an NIH SBIR grant funded project. Check with the contact source to confirm eligibility.
Innovator Objectives
Objectives: Not defined by user
Desired Role in Development/Commercialization:
CEO No day-to-day involvement with company strictly scientific/technical pursuit - contributor
Sources of Capital Acquired for the Tendon Repair Device:
Corporate partnership(s)
The following table summarizes the level of decision-making authority desired by the innovator:
Activity Make all the decisions Share responsibility for the decision
Others make the decision without me
Core Technology Decisions
√
Major Expenditures
√
Hiring of Key Employees
√
Resource Allocation
√
Strategic Partnerships
√
Selection of Board Members and Key Executives
√
Capital Formation
√
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
25
Appendix
Additional Marketing Information
Global Orthopedic Device Market Overview The global orthopedic device market includes products used to diagnose, treat, and manage disorders of the musculoskeletal system. There are more than 150 of these types of disorders, which include fractures, damage from trauma, joint destruction from diseases such as arthritis, and disabling conditions of the spine.
Segments of the global orthopedic market include:
Joint reconstruction Orthobiologics Orthopedic braces and supports Orthopedic diagnostic devices Orthopedic accessories, spinal surgery devices Trauma fixation devices Orthopedic soft tissue implants
Musculoskeletal disorders are a leading cause of disability and their treatment is very costly. “Arthritis afflicts more than 500 million people and more than 90% of people over the age of 30 will have back problems at some point in their lives.” 1
Another factor in the expansion of the orthopedic market is the growth in the population of individuals over the age of 50, which is predicted to double in the next 15 years. While the US currently represents more than 60 percent of global orthopedic sales, improved access to orthopedic surgeries in less developed markets will contribute to long-term segment growth. Other factors influencing the positive growth forecast for orthopedic devices include the availability of less invasive surgeries and the trend toward greater activity later in life.
The global orthopedic devices market is expected to increase from $30.9 billion in 2007 to $49.2 billion in 2011, at a combined average growth rate that accelerates from 10.4% in 2007 to 13.5% in 2011.
(Global Markets Direct, Orthopedic Devices Market Profile, 2008)(MedTech, InSight US Surgical Procedure Volumes, 2002)
In 2007, category revenues were as follows:
Joint reconstruction - $13.5 billion Spinal surgery - $4.6 billion
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
26
Orthobiologics - $4.2 billion Trauma fixation - $3.3 billion Orthopedic braces and supports - $2.8 billion Orthopedic diagnostic devices - $1.8 billion Orthopedic accessories - $0.7 billion, GMD
Categories projected to have the highest growth rates:
Spinal surgery - 16.1% forecasted CAGR 2006-2011 Joint reconstruction - 13.1% Orthopedic accessories - 11.7% Orthobiologics - 10.7%, GMD
Orthopedics CompetitionLeading competitors in the 2006 Global Orthopedic Device Market, in order of declining market share, include:
Stryker (15%) DePuy (12%) Zimmer (11%) Biomet (8%) Medtronic (7%) Synthese (7%) Smith and Nephew (6%) Others (34%)
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
27
In 2006, global share leaders in each of the orthopedic device categories included: Joint reconstruction - Zimmer (24%) Spinal surgery - Medtronic (38%) Orthobiologics - Synthes (14%) Trauma fixation - Synthes (38%) Orthopedic braces and supports - DJ Orthopedics (26%) Orthopedic diagnostic devices - Smith and Nephew (25%) Orthopedic accessories - Stryker (39%)
(Global Markets Direct, Orthopedic Devices Market Profile, 2008)1(MedTech, InSight US Surgical Procedure Volumes, 2002)
Minimally Invasive Surgery (MIS) Devices and Equipment OverviewMinimally invasive surgery is defined as surgery that is conducted with a minimum of trauma to the patient, without making a large incision in the body. The MIS market includes devices for a wide variety of anatomical uses and that fall into a number of different technological categories.
The following chart illustrates the Major US MIS Application Segments, 2005 and 2011 (% of Total Sales).1
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
28
Minimally invasive surgical procedures (closed or local surgery) are less invasive and traumatic to the body than open invasive surgery. These procedures involve use of laparoscopic devices and remote-control manipulation of instruments with indirect observation of the surgical field through an endoscope or similar device, and are carried out through the skin or through a body cavity or anatomical opening. This may result in shorter hospital stays, or allow outpatient treatment.
Special medical equipment may be used, such as fiber optic cables, miniature video cameras, and special surgical instruments handled via tubes inserted into the body through small openings in its surface. The images of the interior of the body are transmitted to an external video monitor and the surgeon has the possibility of making a diagnosis, visually identifying internal features, and then performing surgery based on these findings.
The following chart illustrates the US Forecast for MIS Devices and Equipment Market Sales by Application through 2011 ($ millions) 1.
The global MIS devices and instrument market is estimated at $14.9 billion in 2008, with an average annual growth rate (AAGR) of 7.5%. Revenues in 2018 are expected to top $30 billion1.
Minimally invasive devices and equipment include1: o Monitoring/visualization equipmento Robotics/computer-assisted surgeryo Electrosurgical and auxiliary equipmento Endosurgical instrumentso Surgical devices (stents, catheters, guidewires, etc.)
The following table identifies the US Forecast for MIS Equipment Types Through 2011 ($ Millions).10
Date Generated: March 2, 2010

e-zassi.com | © 2010 e-Zassi, Inc. LLC. All Rights Reserved.
InnoVision Report
29
US MIS Market OverviewIn the United States, about 4.9 million MIS procedures are performed annually. Cardiac procedures (mainly angioplasties and other heart catheterization procedures) account for the largest number, (e.g., more than 1.5 million or 31%% of all MIS procedures) Orthopedic is the fastest-growing anatomical segment, with an anticipated average annual growth rate of 8.7% through 2011, followed by cardiac surgery (7.9%) and vascular surgery (6.8%). The remaining segments all have projected average 2006 to 2011 growth rates below the US market average of 7.2%1.In the United States MIS device and equipment market, cardiac was the largest anatomical segment in 2005, with 69% of the total market, followed by orthopedic (20%), gastrointestinal (18%), and gynecology (6%). The remaining anatomical segments were all relatively small1.
The following table identifies catheterization (including angioplasty) as the largest annual number of minimally invasive cardiac procedures performed in the US annually1. This report was generated by the e-Zassi Technology Assessment software platform, a proprietary software tool available only to members of the e-Zassi medical device community, and is made available to enable collaboration on the medical technology described herein. This report reflects the input of the party requesting this report and not e-Zassi, LLC. Accordingly, e-Zassi, LLC, disclaims all liability in connection with this report. No copyright, trademark, trade name,service mark, or other proprietary notice or legend herein may be removed. Contact the requestingparty for additional information regarding this report's content.
Date Generated: March 2, 2010

30


















