
Identification of protein kinase inhibitors to reprogram ...

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1988 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 263, No. 11, Issue of April 15, pp. 5124-5131, 1988 Printed in U S A .
Aminoacridines, Potent Inhibitors of Protein Kinase C*
(Received for publication, May 6, 1987)
Yusuf A. HannunSQ and Robert M. Bell7 From Departments of $Medicine and (IBiochemistry, Duke University Medical Center, Durham, North Carolina 27710
Acridine orange, acridine yellow G, and related com- pounds potently inhibited protein kinase C (Ca2+/phos- pholipid-dependent enzyme) activity and phorbol di- butyrate binding. Inhibition was investigated in vitro using Triton X- 100 mixed micellar assays (Hannun, Y. A., Loomis, C. R., and Bell, R. M. (1986) J. Biol. Chem. 260, 10039-10043 and Hannun, Y. A., and Bell, R. M. (1986) J. Biol. Chem. 261,9341-9347). Inhibition by the acridine derivatives was subject to surface di- lution; therefore, the relevant concentration unit is mol % rather than the bulk molar concentration. Fifty per- cent inhibition of protein kinase C activity occurred at concentrations of these compounds comparable to con- centrations of sn-1,2-diacylglycerol (DAG) and phos- phatidylserine (PS) required for enzyme activation (i.e. 1-6 mol %). The mechanism of inhibition appeared to be complex: both the catalytic and regulatory sites of protein kinase C were affected. Acridine orange was a competitive inhibitor with respect to MgATP when the catalytic fragment of protein kinase C was em- ployed. Inhibition at the active site was overcome by the addition of Triton X-100 micelles or phospholipid vesicles. When the activity of intact protein kinase C was measured, inhibition was noncompetitive with re- spect to MgATP. Further kinetic analysis suggested a competitive type of inhibition with respect to PS and DAG implying an interaction of acridine compounds with the regulatory lipid cofactors or with the regula- tory domain of protein kinase C. This was further supported by demonstrating inhibition of phorbol di- butyrate binding to both protein kinase C and the lipid- binding domain generated by trypsin hydrolysis. Ac- ridine orange and acridine yellow G also inhibited thrombin-induced 40-kDa phosphorylation in human platelets and phorbol dibutyrate binding to platelets. These effects were also subject to surface dilution. These results suggest that acridine derivatives have multiple interactions with protein kinase C with the predominant effect being inhibition of activation within the regulatory domain of the enzyme. Some of the biologic effects of acridine derivatives including anti-tumor action may occur as a consequence of pro- tein kinase C inhibition.
The Ca2+- and phospholipid-dependent protein kinase, pro- tein kinase C, has emerged as a pivotal element in transducing the effects of those extracellular signals that result in phos- phatidylinositol turnover (1). The breakdown of phosphati-
* This work was supported by National Institutes of Health Grant AM20205. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ Recipient of National Institutes of Health Physician Scientist Award 1011-ES-00155.
dylinositol, especially phosphatidylinositol 4,5-bisphosphate, in response to receptor-ligand interaction results in the gen- eration of two second messengers, diacylglycerol (DAG) and inositol 1,4,5-triphosphate. Inositol 1,4,5-triphosphate mobi- lizes intracellular calcium (2), whereas DAG activates protein kinase C (3). Further interest in protein kinase C was gener- ated by the discovery that phorbol esters, potent tumor pro- moters, bind to (4) and activate the enzyme by interaction at the diacylglycerol site (5-7). Protein kinase C is implicated in the action of tumor promoters, regulation of cell differentia- tion, and oncogenesis (8). DAG levels are elevated in ras- and sis- transformed cells suggesting that oncogene products act by permanently activating this transmembrane signaling pathway (9,lO). Understanding the molecular mechanisms of regulation of protein kinase C activity has, therefore, assumed added importance.
Our laboratory has undertaken a detailed analysis of the in vitro mechanisms of regulation of protein kinase C activity by lipid cofactors using detergent/lipid mixed micellar meth- ods (11,12). Recent work has established that a 32-kDa tryptic fragment is a lipid-binding domain capable of high affinity phorbol ester binding (13-15). Thus, protein kinase C con- tains two functional domains, the lipid-binding domain and a catalytic domain (16). Knowledge of the primary structure of multiple protein kinase Cs and their respective domains emerged with the cloning of cDNAs of protein kinase C from rat (17, 18), bovine (19), and human (20) brains.
Structure-activity studies from our laboratory resulted in the identification of sphingosine (21) and the related lyso- sphingolipid compounds (22) as potent inhibitors of protein kinase C activity and of phorbol dibutyrate binding. Struc- ture-function analysis of these inhibitors, amphipathic amines, led us to discover that certain acridine derivatives were potent inhibitors of protein kinase C.
The acridine derivatives are basic compounds that exhibit fluorescence and are used as staining reagents for the detec- tion of DNA and polysaccharides in cytologic samples (23- 25). These compounds also have bacteriostatic, bacteriocidal, antiviral, and antitumor activities (25, 26). In this paper we report on the inhibition of protein kinase C activity and of phorbol ester binding by certain acridine derivatives and on the mechanism of inhibition. We also show that acridine orange and acridine yellow G inhibit the phosphorylation of a 40-kDa polypeptide in human platelets induced by stimu- lation with thrombin or dioctanoylglycerol. Finally, we discuss the relevance of this inhibition to the biologic effects of acridine compounds and the potential significance of these molecules in furthering the understanding of the molecular mechanism of protein kinase C regulation.
EXPERIMENTAL PROCEDURES
Materials Brain tissue from Charles River CD rats was used as the source of
the protein kinase C. [32P]Orthophosphate, Aquasol 11, [y3*P]ATP,
5124

Aminoacridines, Inhibitors of Protein Kinase C and [3H]PDBu' (12.5 Ci/nmol) were from Du Pont-New England Nuclear. Calf thymus histone type 1114, phospholipase C, thrombin, phenylmethylsulfonyl fluoride, bovine serum albumin, and phorbol dibutyrate were from Sigma. Ultrogel-AcA 202 was from LKB. Acri- dine orange, acridine yellow G , acriflavine HCl, 9-aminoacridine, acridine, 1-aminoanthracene, l-amino-4-bromo-2-methylanthraqui- none, and 9-acridine carboxylic acid hydrate were from Aldrich. Triton X-100 was from Research Products International Corp. 1,Z- Dioleoyl-sn-glycero-3-phosphoserine and 1,2-dioleoyl-sn-glycero-3- phosphocholine were from Avanti Polar Lipids, Inc. sn-1,2-Dioleoyl- glycerol was prepared from dioleoylphosphatidylcholine as previously described (27).
Methods
Purification of Protein Kinase C-Protein kinase C was purified to near homogeneity from rat brain according to the method of Wolf et al. (28).
Mired Micellar Assay for Protein Kinase C-Protein kinase C activity was assayed using Triton X-l00/lipid mixed micelles as previously described (11). Acridine derivatives were added from aqueous solutions, except for 1-aminoanthracene, acridine, and 1- amino-4-bromo-2-methylanthraquinone which were added from con- centrated dimethyl sulfoxide solutions.
Mixed Micellar Assay for PHIPDBU Binding to Protein Kinase C-[3H]PDBU binding was measured as previously described (29).
40-kDa Polypeptide Phosphorylation in Human Platelets-Human platelets were prepared from freshly drawn blood as described by Siess et al. (30). They were then suspended in modified Tyrode's buffer to a concentration of 2.5 X lo8 platelets/ml. Thrombin-induced 40-kDa polypeptide phosphorylation was performed as previously described (21).
The Catalytic and Lipid-binding Domains of Protein Kinase C- Protein kinase C was partially digested with trypsin as previously described to generate the catalytic and lipid-binding domains (13).
RESULTS AND DISCUSSION
Acridine Derivatives Inhibit Protein Kinase C-Acridine and some of its biologically active derivatives as well as structurally related compounds were evaluated for their ef- fects on protein kinase C activity. Acridine yellow G and acridine orange proved to be potent inhibitors of protein kinase C activity measured using the Triton X-100 mixed micellar assay (11, 12) containing 6 mol % PS and 2 mol, % diCla,. in the presence of 100 PM Ca2+ (Fig. 1). The IC6o% of acridine yellow G, the most potent inhibitor of this series, was 40 PM (1 mol %). Under the conditions of the assay, the bulk concentration of PS was 260 PM, and that of diClS1 86 PM, therefore inhibition by acridine yellow G occurred on a molar basis equivalent to half the concentration of diClel or 0.15 the concentration of PS. The importance of expressing the concentration of these inhibitors as mol % rather than bulk molar concentrastions arises from the amphipathic na- ture of these compounds which causes them to partition into the mixed micelles. As expected, the inhibition by acridine yellow G and acridine orange displayed surface dilution ki- netics (Fig. 2). The potency of these compounds was deter- mined primarily by their concentration relative to Triton X- 100 concentration, which is proportional to the number of mixed micelles present. Inhibition was dramatically reduced by increasing the concentration of Triton X-100 PS . diClS,. mixed micelles present. Inhibition remained unchanged when the concentration of acridine yellow G (or that of acridine orange) was simultaneously changed with the concentration of Triton X-100 mixed micelles to maintain a constant mole ratio of the two molecules. These results suggest two impor- tant conclusions. First, the concentrations of these molecules
The abbreviations used are: PDBu, phorbol dibutyrate; PS, phos- phatidylserine; DAG, diacylglycerol; dit,,,,, sn-1,2-dioleoylglycero~ CDTA, 1,2-cyclohexylenedinitrilotetracetic acid; ICwa, concentration at which 50% inhibition is observed.
100
80
40-
20. -b Acrldine Yellow G
5125
OO l L S 3 L - J 0.5 1.0 CONCENTRATION ( m M )
FIG. 1. Inhibition of protein kinase C activity by acridine orange and acridine yellow G . Mixed micelles of Triton X-100, 0.3% (w/v), contained 6 mol % PS and 2 mol % diCla,. 1 mol % of lipid cofactor is equivalent to 43 pM. Acridine orange (0) was added from an aqueous solution and acridine yellow G (0) was added from a concentrated 50% ethanol solution.
::I 20
75 AM Acr8dlne Oronge
1.7 mol % A c r d n e Oronge
ob 6.3 I I I I 0.6 0.9 I .2 % TX. 100 (w/v)
FIG. 2. Effects of surface dilution on inhibition by acridine orange. Mixed micelles were formed with Triton X-100 (TX.100) at 3% (w/v) containing 8 mol % PS and 2 mol % diC,,,.. . The mixed micelles were then diluted into the assay mixture to give the indicated final concentrations of Triton X-100 (w/v). Protein kinase C activity was independent of Triton X-100 concentration in the presence of fixed mol % of lipid cofactor (A). Inhibition by acridine orange was evaluated at a fixed molar concentration (0) and at a fixed mol % (with respect to Triton X-100, C'
I O 0
F 00
t- U
2 a
60 J Q
5 40 I s
E
20 crldlne Yellow G
0 0 1.0 2 .o
CONCENTRATION (mM)
FIG. 3. Inhibition of activity of the catalytic domain of pro- tein kinase C . Acridine orange was evaluated for inhibition of the catalytic fragment of protein kinase C in the presence (0) and abseme (0) of Triton X-100. Similarly, acridine yellow G was evaluated in presence (0) or absence (m) of Triton X-100.
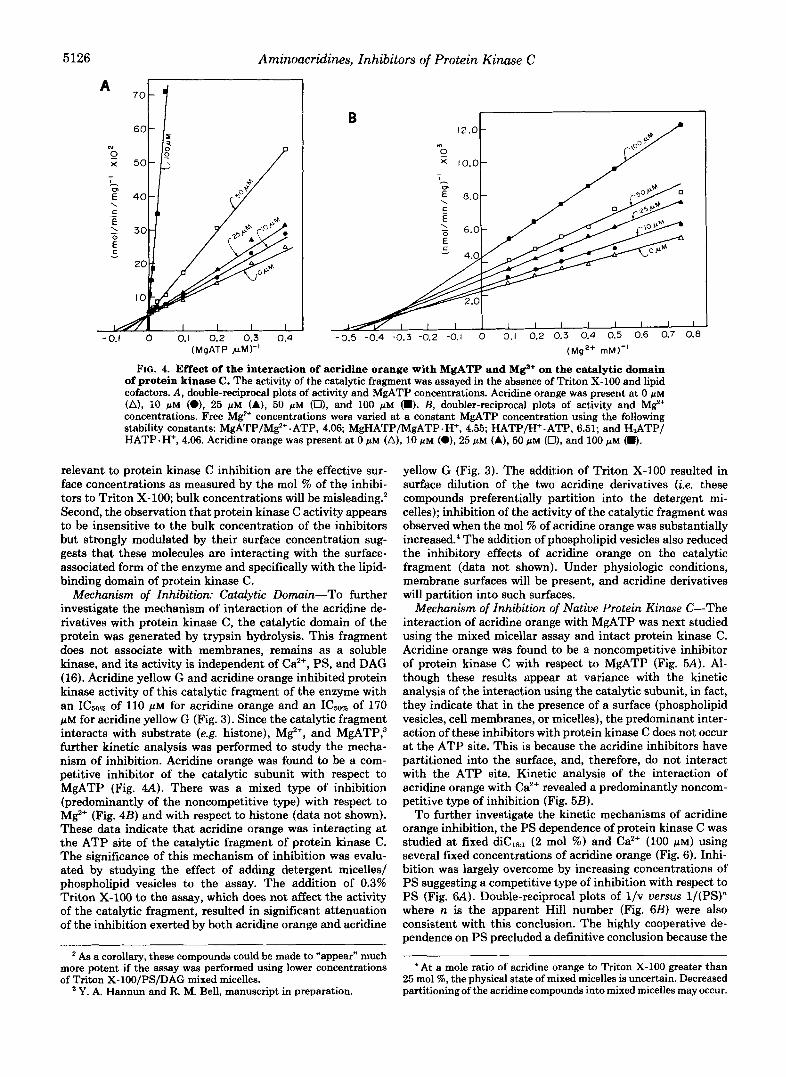
5126 Aminoacridines, Inhibitors of Protein Kinase C
-0.1 0 0.1 0.2 0.3 0.4 (MgATP ,uM)”
FIG. 4. Effect of the interaction of acridine orange with MgATP and Mg+ on the catalytic domain of protein kinase C. The activity of the catalytic fragment was assayed in the absence of Triton X-100 and lipid cofactors. A, double-reciprocal plots of activity and MgATP concentrations. Acridine orange was present at 0 p~ (A), 10 pM (O), 25 p M (A), 50 pM (Ut, and 100 pM (8). B, doubler-reciprocal plots of activity and M$+ concentrations. Free M e concentrations were varied at a constant MgATP concentration using the following stability constants: MgATP/M%+ .ATP, 4.06; MgHATP/MgATP .H+, 4.55; HATP/H+-ATP, 6.51; and H,ATP/ HATP. H+, 4.06. Acridine orange was present at 0 pM (A), 10 pM (e), 25 pM (A), 50 pM (O), and 100 NM (m).
relevant to protein kinase C inhibition are the effective sur- face concentrations as measured by the mol % of the inhibi- tors to Triton X-100; bulk concentrations will be misleading.* Second, the observation that protein kinase C activity appears to be insensitive to the bulk concentration of the inhibitors but strongly modulated by their surface concentration sug- gests that these molecules are interacting with the surface- associated form of the enzyme and specifically with the lipid- binding domain of protein kinase C.
Mechanism of Inhibition: Catalytic Domain-To further investigate the mechanism of interaction of the acridine de- rivatives with protein kinase C, the catalytic domain of the protein was generated by trypsin hydrolysis. This fragment does not associate with membranes, remains as a soluble kinase, and its activity is independent of Ca”, PS, and DAG (16). Acridine yellow G and acridine orange inhibited protein kinase activity of this catalytic fragment of the enzyme with an of 110 pM for acridine orange and an IC5,, of 170 p~ for acridine yellow G (Fig. 3). Since the catalytic fragment interacts with substrate (e.g. histone), M F , and MgATP; further kinetic analysis was performed to study the mecha- nism of inhibition. Acridine orange was found to be a com- petitive inhibitor of the catalytic subunit with respect to MgATP (Fig. 4A). There was a mixed type of inhibition (predominantly of the noncompetitive type) with respect to MgZ+ (Fig. 4B) and with respect to histone (data not shown). These data indicate that acridine orange was interacting at the ATP site of the catalytic fragment of protein kinase C . The significance of this mechanism of inhibition was evalu- ated by studying the effect of adding detergent micelles/ phospholipid vesicles to the assay. The addition of 0.3% Triton X-100 to the assay, which does not affect the activity of the catalytic fragment, resulted in significant attenuation of the inhibition exerted by both acridine orange and acridine
As a corollary, these compounds could be made to “appear” much more potent if the assay was performed using lower concentrations of Triton X-lOO/PS/DAG mixed micelles.
Y. A. Hannun and R. M. Bell, manuscript in preparation.
yellow G (Fig. 3). The addition of Triton X-100 resulted in surface dilution of the two acridine derivatives (i.e. these compounds preferentially partition into the detergent mi- celles); inhibition of the activity of the catalytic fragment was observed when the mol % of acridine orange was substantially increased: The addition of phospholipid vesicles also reduced the inhibitory effects of acridine orange on the catalytic fragment (data not shown). Under physiologic conditions, membrane surfaces will be present, and acridine derivatives will partition into such surfaces.
Mechanism of Inhibition of Native Protein Kinase C-The interaction of acridine orange with MgATP was next studied using the mixed micellar assay and intact protein kinase C. Acridine orange was found to be a noncompetitive inhibitor of protein kinase C with respect to MgATP (Fig. 5A). Al- though these results appear at variance with the kinetic analysis of the interaction using the catalytic subunit, in fact, they indicate that in the presence of a surface (phospholipid vesicles, cell membranes, or micelles), the predominant inter- action of these inhibitors with protein kinase C does not occur at the ATP site. This is because the acridine inhibitors have partitioned into the surface, and, therefore, do not interact with the ATP site. Kinetic analysis of the interaction of acridine orange with Ca2+ revealed a predominantly noncom- petitive type of inhibition (Fig. 5B).
To further investigate the kinetic mechanisms of acridine orange inhibition, the PS dependence of protein kinase C was studied at fixed diCls:l (2 mol %) and Ca2+ (100 p ~ ) using several fixed concentrations of acridine orange (Fig. 6). Inhi- bition was largely overcome by increasing concentrations of PS suggesting a competitive type of inhibition with respect to PS (Fig. 6A). Double-reciprocal plots of l /v uersus l/(PS)” where n is the apparent Hill number (Fig. 6B) were also consistent with this conclusion. The highly cooperative de- pendence on PS precluded a definitive conclusion because the
‘ At a mole ratio of acridine orange to Triton X-100 greater than 25 mol %, the physical state of mixed micelles is uncertain. Decreased partitioning of the acridine compounds into mixed micelles may occur.

Aminoacridines, Inhibitors of Protein Kinase C 5127
A
(MgATP ,AIM)" -0.04 0 0.04 0.08 0.12 0.16 0.20 0.24
(Calcium ,MI-'
FIG. 5. Effect of the interaction of acridine orange with MgATP and Caz' on protein kinase C activity. The activity of protein kinase C was assayed with Triton X-100 mixed micelles containing 6 mol % PS and 1 mol % diCIR1. A, double-reciprocal plots of activity and MgATP concentrations. Acridine orange was present at 0 pM (A), 25 pM (A), 50 pM (O), and 100 pM (m). B, double-reciprocal plots of activity and Caz+ concentrations. Acridine orange was present a t 0 pM (A), 25 pM (A), 50 pM (O), and 100 pM (m). caz+ concentrations were varied by using a CDTA . Ca" buffer.
FIG. 6. Effect of phosphatidylser- ine on acridine orange inhibition of protein kinase C. A, mixed micelles were formed with at 1 mol % and the indicated mol % of PS. Acridine or- ange was present a t 0 p M (A), 25 p M (A), 50 pM (a), and 100 pM (m). B, double- reciprocal plot of data shown in A with the concentration of PS raised to the apparent Hill number. I I I
4 6 8 IO PHOSPHATIDYLSERINE mol %
(DIOLEIN mol %)"
FIG. 7. Effect of dioleoylglycerol on acridine orange inhi- bition of protein kinase C. Double-reciprocal plots of activity and diCIR, concentration at various concentrations of acridine orange.
Hill numbers varied slightly as a function of acridine orange concentration. Finally, the dependence of protein kinase C on sn-1,2-dioleoylglycerol at fixed PS (6 mol W ) and Ca2+ (100 PM) and several concentrations of acridine orange was studied. Double-reciprocal analysis of the interaction revealed a com- petitive type of inhibition (Fig. 7). When the phorbol ester, PMA, was employed in place of diCla,. to activate protein kinase C, the double-reciprocal plots indicated a competitive
(PHOSPHATIDYLSERINE IO
type of inhibition (data not shown) similar to that seen with diCm1.
Mechanism of Inhibition of Phorbol Ester Binding-To demonstrate a direct interaction between these inhibitors and the regulatory domain of protein kinase C, the effect of acridine orange and other analogues on [3H]phorbol dibutyr- ate binding was examined. In an assay of phorbol binding using Triton X-100 mixed micelles containing 20 mol % PS (29), acridine orange was found to inhibit [3H]phorbol dibu- tyrate binding (Table I). Acridine yellow G and other acridine derivatives were also found to inhibit phorbol binding. These results demonstrate that these inhibitors interact with the regulatory domain of protein kinase C. To confirm this con- clusion, the regulatory fragment was separated from the cat- alytic fragment after limited hydrolysis with trypsin. This 32- kDa fragment binds [3H]phorbol &butyrate; binding was in- hibited by acridine orange and acridine yellow G (Table I).
Acridine Derivatives Inhibit Protein Kinase C in Human Platelets-To examine whether these compounds exert an inhibitory effect on protein kinase C activity in intact cell systems, the effect of acridine orange and acridine yellow G on protein kinase C-dependent phosphorylation in human platelets was investigated. Both compounds were found to inhibit the thrombin-induced phosphorylation of a 40-kDa protein (Fig. 8). Similar results were obtained when platelets

5128 Aminoacridines, Inhibitors of Protein Kinase C TABLE I
Inhibition of phorbol binding by acridine derivatives
Compound 13H]PDBu binding [3H]PDBu binding
kinase C ICma- domain IC,% to protein to lipid-binding
mM rnM
Acridine yellow 0.6* 0.4 Acridine orange 0.7 0.6 9-Acridine carboxylic acid NI’ NI Acridine NI NI Acriflavine HCI 1.5 NTd 9-Aminoacridine 1.25 NT 1-Aminoanthracene NI NT
a Concentrations inhibiting phorbol binding by 50%. bHigher concentrations of acridine compounds were needed to
displace phorbol binding than to inhibit protein kinase C activity because different assay conditions were used; 20 mol % PS was used for phorbol binding and only 6 mol % PS was used for kinase activity.
NI, not inhibitory. NT, not tested.
were stimulated with diCs. Acridine orange and acridine yel- low G also inhibited phorbol dibutyrate binding to human platelets (data not shown). Both effects on platelets were subject to surface dilution with inhibition decreasing as the number of platelets was increased (data not shown). Acridine orange had little effect on thrombin-induced 20-kDa polypep- tide phosphorylation (Fig. 8). This finding implies that acri- dine orange does not block thrombin-induced phosphoryli- nositol4,5-bisphosphate turnover, inositol 1,4,5-triphosphate generation, Ca2+ mobilization, and Ca2+-dependent phospho- rylation of the 20-kDa polypeptide. The specificity of ami- noacridine inhibition of protein kinase C in human platelets and other cell systems as it pertains to signal transduction mechanisms remains to be established. Acridines are potent DNA intercalators, and given their ability to inhibit protein kinase C in cells, their mechanism of action in vivo may be quite complex.
Structural Features of Actiue Acridine Derivatives-The structural features required for inhibition were investigated using a number of analogues (Fig. 9). Acriflavine HC1 and 9- aminoacridine also inhibited protein kinase C activity. All these compounds share an amphipathic nature as well as a charged amino group. o-Amsacrine and n-amsacrine were weak inhibitors, while 1-aminoanthracine, 9-acridine carbox- ylic acid, and the parent compound acridine did not inhibit protein kinase C activity (Fig. lo), nor did 1-amino-4-bromo- 2-methylanthraquinone (data not shown). The latter two compounds did not inhibit [‘Hlphorbol dibutyrate binding (Table I). These compounds lack the amino groups. Acridine, 9-acridine carboxylic acid, and 1-aminoanthracine had no effect on 40-kDa phosphorylation. These structure-function studies are consistent with the interactions between acridine and platelet protein kinase C having some specificity.
CONCLUDING REMARKS
The discovery and development of inhibitors of protein kinase C should permit critical analysis of specific functions of protein kinase C in cell systems and in animals. Inhibitors also extend and confirm knowledge of the molecular mecha- nisms of enzyme activation. A number of inhibitors of protein kinase C have been reported. These include dibucaine (31), chlorpromazine (31, 32), adriamycin (32), verapamil (31), amiloride (33), tamoxifen (34), alkyllysophospholipid (35), an anti-neoplastic lipoidal amine, CP-46,665-1 (36), bilirubin (37), palmitoylcarnitine (38), an isoquinolinesulfonamide de- rivative, H7 (39), and staurosporine (40). Except for the last two which appear to be competitive inhibitors with respect to
8
0 50 100 200 300 400 500 Acridine Yellow G (hM)
FIG. 8. Acridine inhibition of platelet protein phosphoryl- ation. A, effect of increasing concentrations of acridine orange on thrombin-induced 40- and 20-kDa polypeptide phosphorylation in human platelets. Lane 1, control platelets; lune 2, thrombin (1 unit/ ml); lune 3,25 pM acridine orange; lune 4,50 pM acridine orange; lune 5,100 pM. B, inhibition of phosphorylation was quantitated by cutting out the 40- and 20-kDa bands from sodium dodecyl sulfate-polyacryl- amide gel and counting 32P.
ATP, little data is available regarding the mechanisms by which these inhibitors affect protein kinase C activity. This has led to the assumption that most of these molecules “per- turb” the lipid bilayer and consequently inhibit enzyme activ- ity nonspecifically. We have recently reported on a new class of protein kinase C inhibitors, sphingosine and other sphin- goid bases (21,41,42) and the lysosphingolipids (22). In these studies, we applied Triton X-100 mixed micellar assays for protein kinase C activation and phorbol ester binding to analyze the mechanism of inhibition. These methods of analy- sis proved to be extremely useful tools in providing a homo- geneous and physically defined environment to study enzyme inhibition where artifacts due to “membrane perturbation” are eliminated. We have extended these tools to the study of other inhibitors of protein kinase C.
The results presented in this paper demonstrate the effects

Arninoacridines, Inhibitors of Protein Kinme C 5129
R I X R R ’ X
R
”-
Acrldlne - H - H -H
Acrldlne Orange -N(CH,), -H -H
Acrldlne Yellow G - NHz -CH, - H
Acrlflovine . HCI - NH
9 - amlnoacrldlne - H - H - H
9 . acrldinecarboxyllc acid - H - H - H
Y
- H
- H
- H
- H
-NHz.HCI
-C02H
& I -aminoan?hracene
0 NH,
w c H 3 I -amino-4-brarna-2merhy lanrhraquinone
0 Bf . .
FIG. 9. Structure of acridine, acridine derivatives, and re- lated compounds.
A A 0 - - - 0 1.0 2 .O 3.0 4.0
CONCENTRATION (mM)
FIG. 10. Inhibition of protein kinase C activity by acridine and related compounds. Mixed micelles were formed with 6 mol % PS and 2 mol % dic,,,. Compounds were added from aqueous solu- tions except for 1-aminoanthracene and acridine which were added from concentrated dimethyl sulfoxide solutions. o-AMSA, o-amsa- crine; m-AMSA, rn-amsacrine.
of a new class of protein kinase C inhibitors, acridine deriva- tives. These molecules proved to be potent inhibitors of both protein kinase C activation and of phorbol ester binding. A number of tools were used to examine the mechanism of this inhibition. Both the catalytic domain and the lipid-binding (or regulatory) domain of protein kinase C were affected, suggesting multiple sites of interaction of the acridine deriv- atives with protein kinase C. Acridine yellow G and acridine orange behaved as competitive inhibitors of the interaction of the catalytic fragment with MgATP but not with histone or MgZ’. This implies an interaction of acridine derivatives with the MgATP site of the catalytic fragment of protein kinase C. On the other hand, these compounds inhibited phorbol
dibutyrate binding to protein kinase C and to its purified regulatory domain implying, in this case, an interaction of the acridine derivatives with the lipid-binding domain of protein kinase C. This was also supported by the kinetic analysis of the inhibition which showed a competitive type of inhibition between the acridine derivatives and PS, DAG, and phorbol dibutyrate, the regulatory cofactors of protein kinase C. Al- though these results reveal two sites of interaction between the acridine derivatives and protein kinase C, evidence points to a predominant effect of the interaction at the Iipid-binding domain. First, kinetic analysis of the inhibition with respect to MgATP reveals that while the acridine derivatives are competitive with MgATP when the catalytic subunit is stud- ied, the inhibition is noncompetitive when the intact protein kinase C is studied. Second, the potency of inhibition is stronger against protein kinase C than against the catalytic subunit. Finally, detergent micelles or phospholipid vesicles overcome inhibition of the catalytic fragment as a conse- quence of the inhibitors partitioning into these surfaces. This last observation provides strong evidence that in the physio- logic state these inhibitors will preferentially partition into the bilayer and exert little effect on the active site.
Our previous studies on the stoichiometry and structure- function analysis of protein kinase C regulation by PS and DAG led us to postulate a model for protein kinase C regula- tion (43). In this model, protein kinase C is envisioned to interact with four PS molecules and Ca”. In this “primed” state, protein kinase C is surface-bound, but inactive (11). The addition of DAG (or phorbol ester) leads to activation by forming three bonds with the enzyme, PS, and Caz+ complex. Two bonds form between protein kinase C and DAG; the third critical bond is postulated to form between DAG and Ca2+ in the complex. According to this model, and from the kinetic analysis, aminoacridine derivatives likely interact with the protein kinase C .4PS. Ca2+ complex by two ionic bonds between the two amino groups present in acridine and two phosphates present in phosphatidylserine. The interatomic distances between the two amino groups present on acridine and the two phosphate moieties present within the postulated protein kinase C. 4PS. Ca2+ complex are fully consistent with the postulated ion pairs (Fig. 11). Aminoacridines are surface active compounds and partition into phospholipid bilayers (micelles). It is, therefore, likely, that the aminoacridines lie with the hydrophobic areas partitioned within the bilayer (micelle) and bridge the protein kinase C. 4PS. Caz+ complex. Such a bridge would physically prevent DAG from interacting with the protein kinase C. 4PS. Ca2+ complex and activating it. This mechanism would also explain why [’HH]phorbol- dibutyrate binding is inhibited.
Further, this mechanism of inhibition proposed is consist- ent with the structure activity relationships observed (Figs. 9 and 10). Thus, the most potent inhibitors contain amino functional groups in the 3 and 6 positions. Compounds which cannot bridge via two ion pairs were not effective inhibitors. Some of these possess primary amines or secondary amines (Figs. 9 and 10). 9-Amino-acridine is intermediate in its inhibitory ability and could bridge by formation of a complex of a different geometry than proposed for the 3,6-diaminoac- ridines.
The proposed model of protein kinase C activation by PS . Ca” and DAG does indeed make several predictions. One of these would be that compounds which bridge the protein kinase C .4PS. Ca2+ complex would be inhibitors. Thus, the aminoacridines employed can be viewed as chemical agents to test the proposed model. In fact, the observation that such agents are inhibitors is consistent with the model.

5130 Aminoacridines, Inhibitors of Protein Kinase C
I B "
C a I
i
FIG. 11. Model of protein kinase C interaction with acridine orange. A, space filling model of acridine orange. B, a view of four space-filling models of PS shown in the transverse plane of the membrane bilayer with Ca2+ liganding the four carboxyls of PS and DAG liganding PS and Ca2+. C, acridine orange is shown interacting with two PS molecules via two ionic bonds between the amino groups and phosphates (arrows) within the protein kinase C -4PS. Ca2+ complex. Note that the interatomic distances are in excellent agreement. D, acridine orange interacting with the 4PS. Caz+ complex. Such interactions would sterically prevent DAG binding and the formation of the protein kinase C.4PS.Ca2+ .DAG quaternary active complex when the hydrophobic protion of acridine partitions in the bilayer (micelle). Acridine interaction within the protein kinase C .4PS. Ca2+ complex would also block ['HJPDBu binding.
Because the acridine derivatives interact with protein ki- nase C a t two different and physically separable sites, and because they are fluorescent molecules, they may prove to be significant tools in further probing the mechanism of activa- tion and catalysis of protein kinase C when substrate amounts of enzyme become available.
The inhibition of protein kinase C by acridine derivatives may provide a mechanism by which these compounds exert some of their biologic effects. Notably, these compounds have a potent anti-tumor action thought to be related to their ability to intercalate into DNA (44). The ability of acridine compounds to interact with both DNA and the PS. Ca2+. DAG complex on a surface may not be totally fortuitous. The PS. Ca2+ surface with which protein kinase C interacts is not dissimilar to the surface of DNA. Certain anti-DNA antibod- ies that occur in autoimmune diseases also react with phos- pholipid vesicles, particularly PS and cardiolipin vesicles (45, 46). PS and cardiolipin are the two most active phospholipids in protein kinase C activation (12). The PS .Ca2+ surface contains phosphodiester bonds and an abrupt change in po- larity occurs as the hydrocarbon interior of the phospholipid bilayer is penetrated. DNA possesses phosphodiester bonds and a similar change in polarity is noted as a consequence of the stacked base pairs. These similarities may account for the
ability of acridine compounds to inhibit protein kinase C.5 As protein kinase C plays an important role in tumor promotion and cell regulation, its inhibition by the acridine derivatives may be the basis of their anti-tumor action. The inhibition of protein kinase C must now be added to the list of biological responses attributed to the aminoacridines.
Acknowledgments-We thank Linda Karolak and John Bloomen- thal for expert technical assistance.
REFERENCES
1. Nishizuka, Y. (1984) Nature 308,693-698 2. Berridge, M. J. (1984) Biockm. J. 220,345-360 3. Bell, R. M. (1986) Cell 45,631-632 4. Niedel, J. E., Kuhn, L., and Vandenbark, G. R. (1983) Proc. Natl.
Acad. Sei. U. S. A. 80,36-40 5. Castagna, M., Takai. Y., Kaibuchi, K., Sano, K., Kikkawa, U.,
and Nishizuka, Y. (1982) J. Bwl. Ckm. 257, 7847-7851 6. Sharkey, N. A., and Blumberg, P. M. (1985) Biochem. Biophys.
Res. Commun. 133,1051-1056 7. Ebeling, J. G., Vandenbark, G. R., Kuhn, L., Ganong, B., Bell, R.
M., and Niedel, J. (1985) Proc. Natl. Acad. Sci. U. S. A. 82, 815-819
We have found that a number of DNA intercalators also inhibit protein kinase C activation (Y. A. Hannun and R. M. Bell, unpub- lished observations).

Aminoacridines, Inhibitors of Protein Kinase C 5131
8. Nishizuka, Y. (1983) Philos. Trans. R. Soc. Lond. B. Biol. Sci. 27. Mavis, R. D., Bell, R. M., and Vagelos, P. R. (1972) J. Biol. Chem.
9. Preiss, J., Loomis, C. R., Bishop, W. R., Stein, R., Niedel, J. E., 28. wolf, M.7 Cuatrecasas, p., and SahYoun, N. (1985) J. Bid. Chem. 302,101-112 247,2835-2841
and Bell, R. M. (1986) J. Biol. Chem. 261,8597-8600 260,15718-15721
Science 231,407-410 ll. Hanun, y. A., Loomis, c. R., and Bell, R. M, (1985) J , Bioi, 30. Siess, W., Siegel, F. L., and Lapetina, E. G. (1983) J. Bid. Chem.
Chem. 260,10039-10043 12. Hannun, Y. A., Loomis, C. R., and Bell, R. M. (1986) J. Biol.
31. Mori, T., Takai, Y., Minakuchi, R., Yu, B., and Nishizuka, Y.
Chem. 261,7184-7190 (1980) J. Biol. Chem. 255,8378-8380
13. Lee, M.- H., and Bell, R. M. (1986) J. Bwl. Chem. 261, 14867- 32. Wise, B. C., Glass, D. B., Chou, C.- H. J., Raynor, R. L., Katoh,
14870 F. (1982) J. Bwl. Chem. 257,8489-8495 N., Schatzman, R. C., Turner, R. S., Kibler, R. F., and Kuo, J.
14. Hoshijima, M., Kikuchi, A., Yanimoto, T., Kaibuchi, K., and 33. Besterman, J. M., May, W. S., Jr., LeVine, H., 111, Cragoe, E. J., Takai, Y. (1986) Cancer Res. 46, 3000-3004 Jr., and Cuatrecasas, P. (1985) J. Bwl. Chem. 260, 1155-1159
15. Huang, K. P., and Huang, F. L. (1986) Biochem. Biophys. Res. 34. O'Brian, C. A., Liskamp, R. M., Solomon, D. H., and Weinstein, Commun. 139,320-326 I. B. (1985) Cancer Res. 45, 2462-2465
16. Inoue, M., Kishimoto, A., Takai, Y., and Nishizuka, Y. (1977) J. 35. Helfman, D. M., Barnes, K. C., Kinkade, J. M., Volger, W. R., Bwl. Chem. 252,7610-7616 Shoji, M., and Kuo, J. F. (1983) Cancer Res. 43,2955-2961
C. R., Hewick, R. M., and Bell, R. M. (1986) Cell 46,491-502 Res. Commun. 127,590-595
10. Fleishmann, L. E., Chahwala, S. B., and Cantley, L. (1986) 29. Hannun, y. A*, and Bell, R. M. (1986) J. Bioi. Chem. 261, 9341- 9347
258, 11236-11242
17. Knopf, J. L., b e , M. H., Sultzman, L. A., Kritz, R. W., Loomis, 36. Shoji, M., Volger, w. Rv and KUo, J. F. (1985) Bwchem. BioPhYs.
18. One, y., Kurokawa, T., Kawahara, K., Nishimura, O., Marumoto, 37. Sane, K., Nakamura, H., and Matsuo, T. (1985) Pediatr. Res. 19, R.7 b r a s h i , K-3 Su&no7 y.3 Kikkawa, u.9 o@% K., and Nish- 38. Katoh, N., wrenn, R, w., wise, B. c., shoji, M., and K ~ ~ , J. F.
19. Parker, p., Coussens, L., TottY, N., Rh% L., Young, s., Chen, 39. Hidaka, H., Inagaki, M., Kawamoto, S., and Sasaki, Y. (1984)
587-590
izuka, Y. (1986) FEBS Lett. 203,111-115
E., Stabel, S., Waterfield, M., and Ullrich, A. (1986) Science
20. Coussens, L., Parker, P., Rhee, L., Yang-Feng, T., Chen, E., 40. Tamaoki, T., Nomoto, H., Takahashi, I., Kato, Y., Morimoto, M.,
Waterfield, M., Francke, U., and Ullrich, A. (1986) Science and Tomita, P. (1986) Biochem. Biophys. Res. Commun. 135,
(1981) Proc. Natl. Acad. Sci. U. S. A. 78, 4813-4817
233,853-859 Biochembtv 23,5036-5041
397-402 233,859-866 41. Merril, A. H., Sereni, A., Stevens, W., Hannun, Y. A,, Bell, R.
21. Hannun, Y. A., Loomis, C. R., Merrill, A. H., Jr., and Bell, R. M. M., and Kinkade, J. (1986) J. Biol. Chem. 261,12610-12615 (1986) J. Biol. Chem. 261, 12604-12609 42. Wilson, E., Olcott, M. C., Bell, R. M., Merril, A. H., and Lambeth,
22. Hannun, Y. A., and Bell, R. M. (1987) Science 235,670-674 J. D. (1986) J. Biol. Chem. 261, 12616-12623 23. Mai, J., Schmidt-Kastner, R., and Tefett, H. (1984) J. Hktochem. 43. Ganong~ B. c. R . ~ Hannunl y- A-, and R. M.
24. Lippincott-Schwartz, J., and Fambrough, D. (1986) J. Cell Bwl. 44' Robertson' I.' Denny' w' A'' and B' c. (1980) J' Caneer16.1133-1139
25. Albert, A. (1966) The Acridines, 2nd ed., Edward Arnold, London 45. Lafer, E., Rauch, J., Andrzejewski, C., Mudd, D., Furie, B.,
26. Denny, W., Baguley, B., Cain, B., and Waring, M. (1983) in Schwartz, R. S., and Stollar, B. D. (1981) J. Exp. Med. 153,
~ o l e c u h r Aspects of Anti-Cancer Drug Action Weidle, s., and 46. Koike, T., Sueishi, M., Funaki, H., Tomioka, H., and Yoshida, S. Waring, M., eds) pp. 1-34, MacMillan Press, London (1984) Clin. Exp. Zmmuml. 56, 193-199
Cytochem. 32,97-104 (1986) Proc. Natl. Acad. Sci. U. S. A. 8 3 , 1184-1188
101,1593-1605
897-909


















