







Languages
Pages
Legal


DATOS PERSONALESIDENTIF.PACIENTE
5010100000001005
FECHA DENACIMIENTO
01/01/1980
SEXO FETNIA Caucásica
PROFESIONAL DE SALUDSOLICITANTEJoseph Voland M.D.
39981 Sorrento Valley Blvd
Washington, DC 20266 US
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
Array:Director del Laboratorio,Linda Wasserman, M.D., Ph.D.
METODOLOGÍA DEL EXAMEN
Genotipificación a gran escala utilizandosondas moleculares múltiples
INFORMACIÓN PARA EL LABORATORIO
NÚMERO DEREGISTRO
D3515003
CÓDIGO DEACTIVACIÓN
CONVR-ABABA
CÓDIGO DEMUESTRA
SALIVA
FECHA DERECOLECCIÓN
20/04/2013
FECHA DELINFORME
10/05/2013
FECHA DERECEPCIÓN
07/05/2013
Estado De Portador FIBROSIS QUÍSTICA
Gen Analizado - CFTR
DescripciónEl paciente es portador de esta enfermedad, pero no es propenso a presentar síntomas.Hay una probabilidad de 50% de que el paciente transmita esta mutación a uno de sushijos. En este caso, su hijo también sería portador de la enfermedad. Si la pareja delpaciente también es portador/a de una mutación para esta enfermedad, hay unaprobabilidad de 25% de que cada uno de sus hijos herede la mutación de ambos padresy desarrolle la enfermedad. Debido a que existen muchas mutaciones poco frecuenteses posible que el paciente sea portador de una mutación no analizada de este examen.
HOMOZIGÓTICO
HETEROZIGÓTICOCOMPUESTO
PORTADOR
NO PORTADOR
► NO PORTADOR:
El paciente no es portador de la(s) mutación(es) analizada(s) para esta enfermedad.Debido a que existen muchas mutaciones poco frecuentes, es posible que, aparte de lasmutaciones detectadas, el paciente sea portador de una mutación no analizada en esteexamen.
Estado De Portador NO PORTADOR DE LAS SIGUIENTES ENFERMEDADES
NOPORTADOR
Acidemia Glutárica, Tipo 1
Acidemia Metilmalónica
Acidemia Propiónica
Acrodermatitis Enteropática
Anemia De Fanconi
Beta-talasemia
Citrulinemia Tipo I
Deficiencia De 3-metilcrotonil-CoACarboxilasa
Deficiencia De Acil-CoA DeshidrogenasaDe Cadena Corta
Deficiencia De Acil-CoA DeshidrogenasaDe Cadena Media
Deficiencia De Acil-CoA DeshidrogenasaDe Cadena Muy Larga
Deficiencia De Alpha-1 Antitripsina
Deficiencia De Argininosuccinato Liasa
Deficiencia De Beta-cetotiolasa
Deficiencia De Biotinidasa
Deficiencia De DihidropirimidinaDeshidrogenasa
Deficiencia De Galactocinasa
Deficiencia De La HMG-CoA Liasa
Deficiencia De Lipoproteína Lipasa,Familiar
Deficiencia De Precalicreína
Deficiencia De Protrombina
Deficiencia Del Factor XI
Deficiencia Múltiple De Carboxilasa
Deficiencia Primaria De CarnitinaSistémica
Diabetes, Neonatal Permanente
Disautonomía Familiar
Enfermedad De Canavan
HOMOZIGÓTICO
HETEROZIGÓTICOCOMPUESTO
PORTADOR
NO PORTADOR
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 1 de 15
INFORME DE MUESTRA

Estado De Portador NO PORTADOR DE LAS SIGUIENTES ENFERMEDADESEnfermedad De Células Falciformes
Enfermedad De Gaucher
Enfermedad De La Hemoglobina C
Enfermedad De La Hemoglobina E
Enfermedad De Niemann-Pick
Enfermedad De Pompe
Enfermedad De Sandhoff
Enfermedad De Tay-Sachs
Enfermedad De Von Willebrand Tipo 2Normandía
Enfermedad De Von Willebrand Tipo 3
Enfermedad Del Almacenamiento DeGlucógeno Tipo 1A
Enfermedad Renal Poliquística
Enfermedad Urinaria De Jarabe De Arce
Esclerosis Lateral Amiotrófica
Esferocitosis, Hereditaria
Fenilcetonuria
Fiebre Mediterránea Familiar
Galactosemia
Gangliosidosis-GM1
Hemocromatosis
Homocistinuria, Clásico
Homocistinuria, Tipo CblE
Mucolipidosis II
Mucolipidosis III
Mucolipidosis IV
Pseudodeficiencia De Tay-Sachs
Pérdida Auditiva DFNB1 Y DFNB9 No-sindrómica
Pérdida Auditiva DFNB59 No-sindrómica
Raquitismo, Deficiencia DePseudovitamina D
Síndrome De Bartter Tipo 4A
Síndrome De Bloom
Síndrome De Crigler-Najjar
Síndrome De Dubin-Johnson
Síndrome De Ehlers-Danlos TipoCifoescoliosis
Síndrome De Ehlers-Danlos TipoDermatosparaxis
Síndrome De Ehlers-Danlos TipoHiperlaxitud
Síndrome De Hurler
Síndrome De Rh Nulo
Síndrome Del Nódulo Sinusal Enfermo
Síndrome Nefrótico Resistente AEsteroides
Síndrome Poliglandular Autoinmune, TipoI
Tirosinemia
Trombocitopenia, CongenitalAmegacariocítica
Xantomatosis Cerebrotendinosa
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 2 de 15
INFORME DE MUESTRA

DETALLE GENOTIPO/HAPLOTIPO
ESTADO DE PORTADOREsta lista contiene las mutaciones analizadas para el estado de portador del paciente. Las mutaciones analizadas están organizadas según laenfermedad y aparecen entre paréntesis al lado del gen respectivo.
• Si el paciente es portador de la mutación analizada, el resultado aparecerá en color rojo en la sección "Portador de".
• Si el paciente no es portador de la mutación analizada, el resultado aparecerá en color negro en la sección "No portador de".
• Si no se pudo obtener un resultado para la mutación, el resultado aparecerá en la sección "No hay datos".
• Un resultado "Pendiente" indica que no se ha terminado el análisis para esta enfermedad en particular.
• "No se puede reportar" indica que el laboratorio no puede proporcionar un resultado.
Riesgo residuo: Debido a que existen muchas mutaciones poco frecuentes, es posible que el paciente sea portador de alguna mutación noincluida en el examen.
FIBROSIS QUÍSTICA
Portador de: CFTR [deltaF508]No portador de: CFTR [1078delT,1677delTA, 1717-1G>A, 1812-1G>A,1898+1G>A, 1949del84, 2043delG,2055del9>A, 2105del13ins5, 2108delA,2184delA, 2307insA, 2789+5G>A,2869insG, 3120+1G>A, 3120G>A,3171delC, 3272-26A>G, 3659delC,3667ins4, 3791delC, 3849+10kbC>T,3876delA, 3905insT, 394delTT,405+1G>A, 405+3A>C, 444delA,574delA, 621+1G>T, 663delT,711+1G>T, 712-1G>T, 846delT,935delA, 936delTA, A455E, A561E,C524X, D1152H, deltaF311, deltaI507,G1349D, G178R, G330X, G542X, G551D,G622D, G85E, K710X, L206W, L558S,M1101K, N1303K, P205S, P574H, P750L,Q1100P, Q1238X, Q359K/T360K, Q493X,R1158X, R1162X, R117H, R334W, R347P,R352Q, R553X, R560T, R709X, S1196X,S1251N, S364P, S549N, S549R (A>C),S549R (T>G), V232D, V520F/I, W1089X,W1204X, W1282X]
ACIDEMIA GLUTÁRICA, TIPO 1
No portador de: GCDH [A293T, A421V,R227P, R402W, V400M]
ACIDEMIA METILMALÓNICA
No portador de: MMAA [503delC,R145X]; MUT [E117X, G717V, N219Y,R108C, R369C]
ACIDEMIA PROPIÓNICA
No portador de: PCCA [R399Q]; PCCB[1172_1173insT, 1218del14ins12,R410W, T428I]
ACRODERMATITISENTEROPÁTICA
No portador de: SLC39A4[1223_1227delCCGGG, L48X]
ANEMIA DE FANCONI
No portador de: FANCC [322delG,IVS4+4A>T, L554P, Q13X, R185X,R548X]
BETA-TALASEMIA
No portador de: HBB [-28A>G, -29A>G,-87C>G, -88C>T, 17A>T, 41/42-TTCT,cd24T>A, cd39C>T, cd44-C, cd8-AA,cd8/9+G, Hb Malay, IVS1+110G>A,IVS1+1G>A, IVS1+5G>T, IVS1+6T>C,IVS2+1G>A, IVS2+654C>T, IVS2+745C>G,IVS2+849A>C, IVS2+849A>G]
CITRULINEMIA TIPO I
No portador de: ASS1 [952delG,E191K, G390R, IVS6-2A>G, K310Q,Q380X, R272C, R304W, R307C, R86H,R95S, S180N, V269M, V345G, Y190D]
DEFICIENCIA DE3-METILCROTONIL-COACARBOXILASA
No portador de: MCCC1 [A289V, D532H,L437P, R385S]; MCCC2 [E99Q, I437V,R193C, S173L, V339M]
DEFICIENCIA DE ACIL-COADESHIDROGENASA DE CADENACORTA
No portador de: ACADS [A199V, I390M,M370V, Q365H, R107C, R139C, R380W,R46W, S353L, T169P, W177R]
DEFICIENCIA DE ACIL-COADESHIDROGENASA DE CADENAMEDIA
No portador de: ACADM [K304E, Y42H]
DEFICIENCIA DE ACIL-COADESHIDROGENASA DE CADENAMUY LARGA
No portador de: ACADVL [V283A]
DEFICIENCIA DE ALPHA-1ANTITRIPSINA
No portador de: SERPINA1 [S allele,Z allele]
DEFICIENCIA DEARGININOSUCCINATO LIASA
No portador de: ASL [D87G,IVS5+1G>A, Q116X, Q354X, R193Q,R385C, V178M]
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 3 de 15
INFORME DE MUESTRA

DEFICIENCIA DE BETA-CETOTIOLASA
No portador de: ACAT1 [149delC,G152A, G183R, IVS11+2T>C, IVS8+1G>T,Q272X, R208X, T297M]
DEFICIENCIA DE BIOTINIDASA
No portador de: BTD [A171T,C33FfsX36, D444H, Q456H, R538C]
DEFICIENCIA DEDIHIDROPIRIMIDINADESHIDROGENASA
No portador de: DPYD [A777S, E386X,H978R, I560S, IVS11+1G>T,IVS14+1G>A, M182K, P86L, R235W,R886H, V335L]
DEFICIENCIA DEGALACTOCINASA
No portador de: GALK1 [G349S, Q382X,R256W, T344M]
DEFICIENCIA DE LA HMG-COALIASA
No portador de: HMGCL [504_505delCT,E37X, R41Q]
DEFICIENCIA DE LIPOPROTEÍNALIPASA, FAMILIAR
No portador de: LPL [G188E]
DEFICIENCIA DEPRECALICREÍNA
No portador de: KLKB1 [C529Y, W383X]
DEFICIENCIA DE PROTROMBINA
No portador de: F2 [C181Y, D161Y,E352K, R263C, R2W, R314C, R457Q,R538C]
DEFICIENCIA DEL FACTOR XI
No portador de: F11 [C128X, E117X,F283L, IVS14+1G>A]
DEFICIENCIA MÚLTIPLE DECARBOXILASA
No portador de: HLCS [780delG,D571N, G581S, L237P, R508W, R665X,V550M]
DEFICIENCIA PRIMARIA DECARNITINA SISTÉMICA
No portador de: SLC22A5 [N32S, P46S,R169W, R254X, T219fsX284, T440M,T468R, W283C, Y211C, Y4X]
DIABETES, NEONATALPERMANENTE
No portador de: ABCC8 [E382K, N72S,P45L]; GCK [IVS8+2T>G, R397L]
DISAUTONOMÍA FAMILIAR
No portador de: IKBKAP [IVS20+6T>C,R696P]
ENFERMEDAD DE CANAVAN
No portador de: ASPA [245insA,433-2A>G, 827delGT, A305E, C218X,E285A, F295S, G274R, M195R, P280S,Y109X, Y231X]
ENFERMEDAD DE CÉLULASFALCIFORMES
No portador de: HBB [Hemoglobin S]
ENFERMEDAD DE GAUCHER
No portador de: GBA [84GG, D409H,IVS2+1G>A, N370S, R463C, V394L]
ENFERMEDAD DE LAHEMOGLOBINA C
No portador de: HBB [Hemoglobin C]
ENFERMEDAD DE LAHEMOGLOBINA E
No portador de: HBB [Hemoglobin E]
ENFERMEDAD DE NIEMANN-PICK
No portador de: NPC1 [G992W,I1061T]; NPC2 [E20X]; SMPD1[deltaR608, H421Y, L302P,P330SfsX382, R496L]
ENFERMEDAD DE POMPE
No portador de: GAA [2741AG>CAGG,D645E, G309R]
ENFERMEDAD DE SANDHOFF
No portador de: HEXB [76delA,IVS2+1G>A, S62L]
ENFERMEDAD DE TAY-SACHS
No portador de: HEXA [1278insTATC,613delC, C458Y, deltaTTC910-912,G269S, I335F, IVS12+1G>C, IVS2+1G>C,IVS5-1G>T, IVS9+1G>A, IVS9-1G>T,R170Q, R170W, R178H/L, R499H, R504C,S210F, V192L, W329X]
ENFERMEDAD DE VONWILLEBRAND TIPO 2NORMANDÍA
No portador de: VWF [C1060R, C1225G,C788R, C788Y, C804F, D879N, E1078K,E787K, G785E, H817Q, M771V, P812L,Q1053H, R763G, R782W, R816Q, R816W,R854Q, T791M, Y795C]
ENFERMEDAD DE VONWILLEBRAND TIPO 3
No portador de: VWF [1384delG,1657dupT, 191delG, 2016_2019delCTCT,2157delA, 2269_2270delCT, 276delT,3258_3259insT, 3736_3737dupCC,374del14, 4092_4093delAC,4324dupAGTGTGGA, 6182delT, 7139dupT,7172_7173insT, 7674dupC, 7683delT,892dupG, C1071F, C2174G, C2362F,C2671Y, C2739Y, C2754W, C2804Y,D47H, E620X, IVS28+1G>A,IVS29+10C>T, IVS40-1G>C, IVS45+7C>T,IVS50+3G>T, IVS7+1G>A, IVS9-1G>A,L1267X, Q1346X, Q218X, Q2544X,Q565X, R1315C, R1853X, R2434X,R2535X, R273W, R324X, R365X, R373X,S71X, V1314F/3940delG, W222X, W377C,Y1456X, Y1542X, Y357X, Y610X]
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 4 de 15
INFORME DE MUESTRA

ENFERMEDAD DELALMACENAMIENTO DEGLUCÓGENO TIPO 1A
No portador de: G6PC [378_379dupTA,79delC, deltaF327, G188R, G270V,Q242X, Q347X, R83C, R83H]
ENFERMEDAD RENALPOLIQUÍSTICA
No portador de: PKHD1 [D3230fs,I222V, I2944fs, I2957T, I3177T,P805L, Q3392X, R3482C, R496X, T36M,V3471G]
ENFERMEDAD URINARIA DEJARABE DE ARCE
No portador de: BCKDHB [E372X,G278S, R183P]
ESCLEROSIS LATERALAMIOTRÓFICA
No portador de: ALS2 [1867delCT]
ESFEROCITOSIS, HEREDITARIA
No portador de: ANK1 [5703+16C>T,V463I]; EPB42 [A142T, D175Y,IVS6+1G>A, R310Q, R317C, W119X]
FENILCETONURIA
No portador de: PAH [A403V, E280K,F39L, I65T, IVS10-11G>A, L48S,P281L, R158Q, R243X, R261Q, R408Q,R408W, V245A, Y414C]
FIEBRE MEDITERRÁNEAFAMILIAR
No portador de: MEFV [A744S, K695R,M680I, M694I, M694V, R653H, R761H,V726A]
GALACTOSEMIA
No portador de: GALT [E203K, F171S,IVS2-2A>G, K285N, L195P, L218L(c.652C>T), N314D, Q188R, S135L,Y209C]
GANGLIOSIDOSIS-GM1
No portador de: GLB1 [R59H]
HEMOCROMATOSIS
No portador de: HFE [C282Y, H63D,S65C]; HFE2 [G320V]; TFR2 [M172K,Y250X]
HOMOCISTINURIA, CLÁSICO
No portador de: CBS [1566delG,1591delTTCG, 1622ins4, 298fsX329,A114V, A155V, A226T, A355P, C109R,C165Y, D376N, D444N, D47E, E128D,E144K, E176K, E239K, E302K, G116R,G139R, G148R, G151R, G307S, G347S,G85R, H232D, I278T, I435T,IVS11-2A>C, IVS12+1G>A, IVS8+1G>A,IVS9+1G>T, K102N, K384E, L101P,L539S, M126V, P145L, P290L, P49L,P78R, P88S, R121C, R266G, R266K,R336C (C>T), R336C (delCC/insTT),R336H, R369C, R369H, R379W, R491C,R58W, S217F, S349N, S466L, T191M,T257M, T353M, V168A, V320A, V354M,V371M, W43X]
HOMOCISTINURIA, TIPO CBLE
No portador de: MTRR[1622_1623dupTA, 1726delTTG,1953-6_1953-2del5, R3W, R525X]
MUCOLIPIDOSIS II
No portador de: GNPTAB [1581delC,3503_3504delTC, 616_619delACAG,Q104X, Q845X, R1189X, R1205X]
MUCOLIPIDOSIS III
No portador de: GNPTAB [IVS17+6T>G,K4Q]; GNPTG [347_349_delACA,499dupC]
MUCOLIPIDOSIS IV
No portador de: MCOLN1 [IVS3-2A>G]
PSEUDODEFICIENCIA DE TAY-SACHS
No portador de: HEXA [R247W, R249W]
PÉRDIDA AUDITIVA DFNB1 YDFNB9 NO-SINDRÓMICA
No portador de: GJB2 [167delT,235delC, 35delG, L90P, V37I]; OTOF[Q829X]
PÉRDIDA AUDITIVA DFNB59 NO-SINDRÓMICA
No portador de: DFNB59 [113dupT,509_512delCACT, 726delT, 988delG,L244R, R167X]
RAQUITISMO, DEFICIENCIA DEPSEUDOVITAMINA D
No portador de: CYP27B1[3398dupCCCACCC, 958delG, IVS3+1G>A,R389H]
SÍNDROME DE BARTTER TIPO4A
No portador de: BSND [G47R]
SÍNDROME DE BLOOM
No portador de: BLM [blmAsh, Q645X,Q975fsX, R836fsX, R899X, S186X,W428X, W567X, W803fsX]
SÍNDROME DE CRIGLER-NAJJAR
No portador de: UGT1A1 [1043delA,1186delG, 1220delA, 1223insG,397_402delCAACAA, 517delC, 652insT,801delC, 878_890del, 973delG, A292V,A368T, A401P, A478D, C177R, C280X,E463A, F170del, G377V, G493R, H376R,H39D, I294T, I370V, IVS1+1G>C,IVS3-2A>G, IVS4+1G>T, IVS4-1G>A,K428E, K437X, L131P, L175Q, L233R,L443P, M204V, N279Y, N400D, P34Q,P387R, Q185P, Q239fsX256, Q283X,Q331R, Q331X, Q357R, Q357X, R209W,R336W, R341X, R403C, S375F, S381R,S488F, V160E, V225G, W335X, W40R,W483X (TAG), W483X (TGA), Y192X,Y486D, Y74X]
SÍNDROME DE DUBIN-JOHNSON
No portador de: ABCC2 [I1173F,R1150H]
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 5 de 15
INFORME DE MUESTRA

SÍNDROME DE EHLERS-DANLOS TIPO CIFOESCOLIOSIS
No portador de: PLOD1 [1362delC,153dupC, 1702insC, 467-2delA,975+2_975+3insTT, A667T, G678R,H706R, Q327X, Q49X, R319X, R670X,W446G, W612C, Y142X, Y511X]
SÍNDROME DE EHLERS-DANLOS TIPODERMATOSPARAXIS
No portador de: ADAMTS2 [Q225X,W795X]
SÍNDROME DE EHLERS-DANLOS TIPO HIPERLAXITUD
No portador de: TNXB[2116_2117dupGT, 3551_3552delAA]
SÍNDROME DE HURLER
No portador de: IDUA [1044delCGACAA,1695del11, 1814_1815delTT, Q70X,W402X]
SÍNDROME DE RH NULO
No portador de: RHAG [V270I]
SÍNDROME DEL NÓDULOSINUSAL ENFERMO
No portador de: SCN5A [G1408R,P1298L, R1632H, T220I]
SÍNDROME NEFRÓTICORESISTENTE A ESTEROIDES
No portador de: NPHS2 [1036delC,436delA, R138Q]
SÍNDROME POLIGLANDULARAUTOINMUNE, TIPO I
No portador de: AIRE [64-69del6,967-979del13, A502fsX519,C311fsX376, C311Y, C446G,C449fsX502, E298K, F77S, G218fsX284,G228W, H415fsX422, IVS11+1G>A,IVS3+2T>C, IVS3-2A>T, IVS7+1G>A,IVS8+5G>T, K83E, L28P, L29P,L323fsX372, L397fsX478, L417fsX478,L93R, L97P, M1L, M388fsX422, P252L,
SÍNDROME POLIGLANDULARAUTOINMUNE, TIPO I
P370fsX370, P539L, Q358X, R139X,R15C, R203X, R257X, R303P, R433/C434fsX, S135fsX147, T16M, V80L,W78R, X546C, Y85C, Y90C]
TIROSINEMIA
No portador de: FAH [G337S, P261L,Q64H, W262X]
TROMBOCITOPENIA,CONGENITALAMEGACARIOCÍTICA
No portador de: MPL [R102P, R43X]
XANTOMATOSISCEREBROTENDINOSA
No portador de: CYP27A1 [A216P,D354G, E195X, E408X, G145G, G472A,IVS2+1G>A, IVS4+1G>A, IVS6+1G>A,IVS6-1G>T, IVS7+1G>A, IVS7+5G>T,K284X, P384L, P401R, P441S, Q159X,Q461X, R127Q, R127W, R137W, R231X,R270X, R395C, R405Q, R405W, R474W,T339M, W260X]
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 6 de 15
INFORME DE MUESTRA
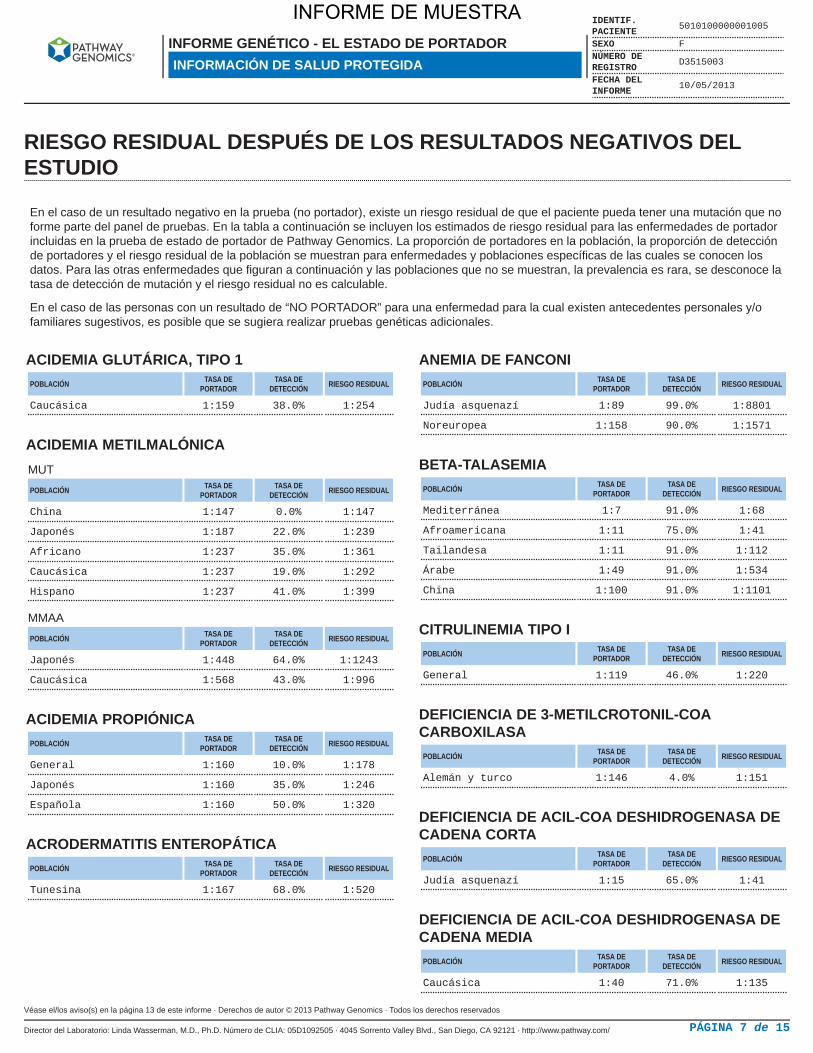
RIESGO RESIDUAL DESPUÉS DE LOS RESULTADOS NEGATIVOS DELESTUDIO
En el caso de un resultado negativo en la prueba (no portador), existe un riesgo residual de que el paciente pueda tener una mutación que noforme parte del panel de pruebas. En la tabla a continuación se incluyen los estimados de riesgo residual para las enfermedades de portadorincluidas en la prueba de estado de portador de Pathway Genomics. La proporción de portadores en la población, la proporción de detecciónde portadores y el riesgo residual de la población se muestran para enfermedades y poblaciones específicas de las cuales se conocen losdatos. Para las otras enfermedades que figuran a continuación y las poblaciones que no se muestran, la prevalencia es rara, se desconoce latasa de detección de mutación y el riesgo residual no es calculable.
En el caso de las personas con un resultado de “NO PORTADOR” para una enfermedad para la cual existen antecedentes personales y/ofamiliares sugestivos, es posible que se sugiera realizar pruebas genéticas adicionales.
ACIDEMIA GLUTÁRICA, TIPO 1POBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:159 38.0% 1:254
ACIDEMIA METILMALÓNICAMUT
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
China 1:147 0.0% 1:147
Japonés 1:187 22.0% 1:239
Africano 1:237 35.0% 1:361
Caucásica 1:237 19.0% 1:292
Hispano 1:237 41.0% 1:399
MMAA
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Japonés 1:448 64.0% 1:1243
Caucásica 1:568 43.0% 1:996
ACIDEMIA PROPIÓNICAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
General 1:160 10.0% 1:178
Japonés 1:160 35.0% 1:246
Española 1:160 50.0% 1:320
ACRODERMATITIS ENTEROPÁTICAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Tunesina 1:167 68.0% 1:520
ANEMIA DE FANCONIPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:89 99.0% 1:8801
Noreuropea 1:158 90.0% 1:1571
BETA-TALASEMIAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Mediterránea 1:7 91.0% 1:68
Afroamericana 1:11 75.0% 1:41
Tailandesa 1:11 91.0% 1:112
Árabe 1:49 91.0% 1:534
China 1:100 91.0% 1:1101
CITRULINEMIA TIPO IPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
General 1:119 46.0% 1:220
DEFICIENCIA DE 3-METILCROTONIL-COACARBOXILASA
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Alemán y turco 1:146 4.0% 1:151
DEFICIENCIA DE ACIL-COA DESHIDROGENASA DECADENA CORTA
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:15 65.0% 1:41
DEFICIENCIA DE ACIL-COA DESHIDROGENASA DECADENA MEDIA
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Caucásica 1:40 71.0% 1:135
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 7 de 15
INFORME DE MUESTRA
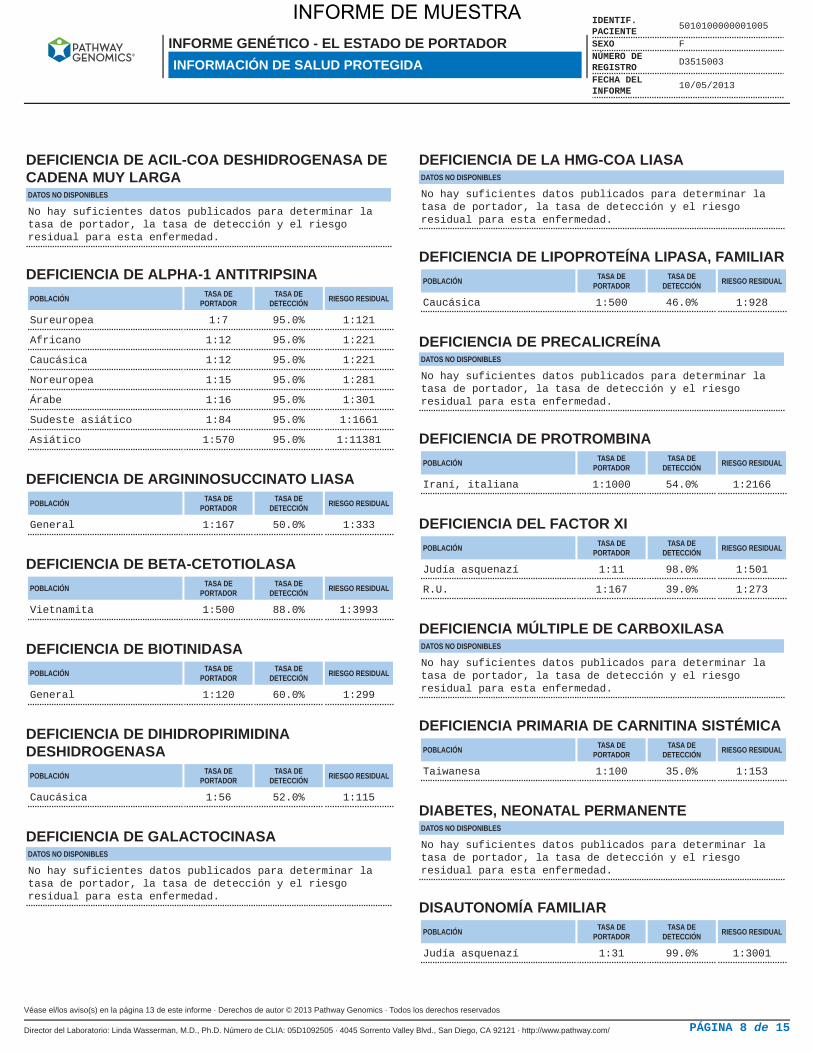
DEFICIENCIA DE ACIL-COA DESHIDROGENASA DECADENA MUY LARGADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
DEFICIENCIA DE ALPHA-1 ANTITRIPSINAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Sureuropea 1:7 95.0% 1:121
Africano 1:12 95.0% 1:221
Caucásica 1:12 95.0% 1:221
Noreuropea 1:15 95.0% 1:281
Árabe 1:16 95.0% 1:301
Sudeste asiático 1:84 95.0% 1:1661
Asiático 1:570 95.0% 1:11381
DEFICIENCIA DE ARGININOSUCCINATO LIASAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
General 1:167 50.0% 1:333
DEFICIENCIA DE BETA-CETOTIOLASAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Vietnamita 1:500 88.0% 1:3993
DEFICIENCIA DE BIOTINIDASAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
General 1:120 60.0% 1:299
DEFICIENCIA DE DIHIDROPIRIMIDINADESHIDROGENASA
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Caucásica 1:56 52.0% 1:115
DEFICIENCIA DE GALACTOCINASADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
DEFICIENCIA DE LA HMG-COA LIASADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
DEFICIENCIA DE LIPOPROTEÍNA LIPASA, FAMILIARPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:500 46.0% 1:928
DEFICIENCIA DE PRECALICREÍNADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
DEFICIENCIA DE PROTROMBINAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Iraní, italiana 1:1000 54.0% 1:2166
DEFICIENCIA DEL FACTOR XIPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:11 98.0% 1:501
R.U. 1:167 39.0% 1:273
DEFICIENCIA MÚLTIPLE DE CARBOXILASADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
DEFICIENCIA PRIMARIA DE CARNITINA SISTÉMICAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Taiwanesa 1:100 35.0% 1:153
DIABETES, NEONATAL PERMANENTEDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
DISAUTONOMÍA FAMILIARPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:31 99.0% 1:3001
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 8 de 15
INFORME DE MUESTRA
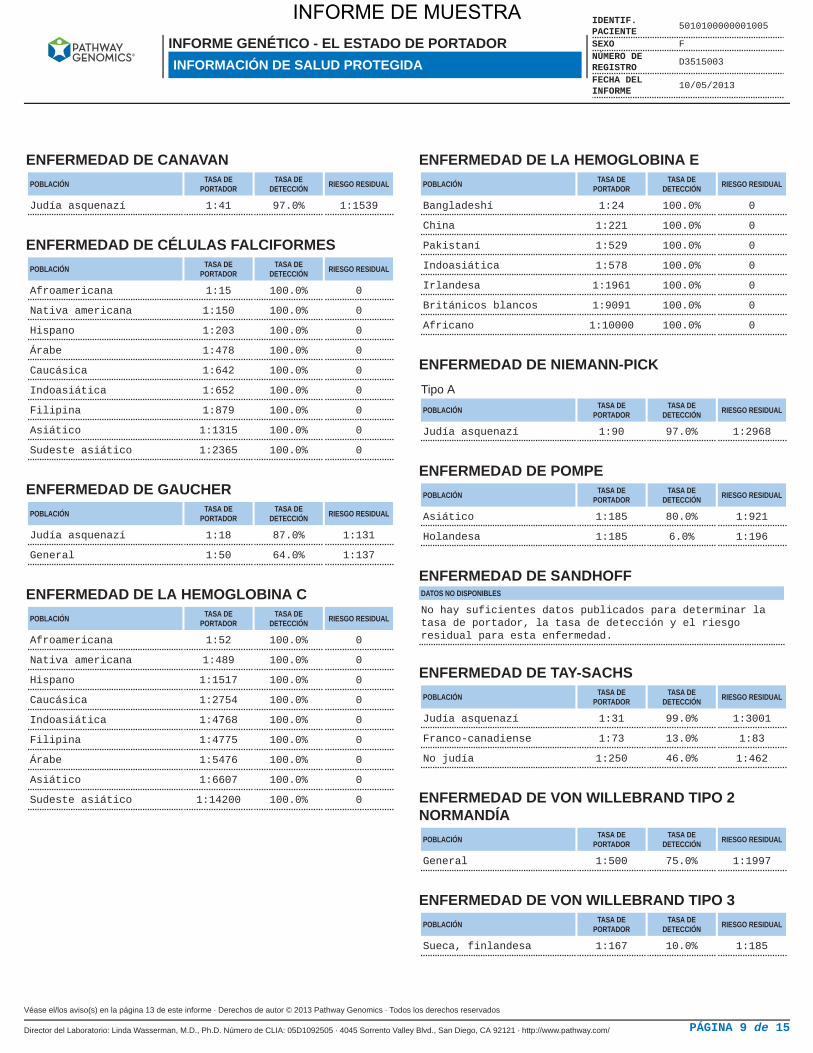
ENFERMEDAD DE CANAVANPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:41 97.0% 1:1539
ENFERMEDAD DE CÉLULAS FALCIFORMESPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Afroamericana 1:15 100.0% 0
Nativa americana 1:150 100.0% 0
Hispano 1:203 100.0% 0
Árabe 1:478 100.0% 0
Caucásica 1:642 100.0% 0
Indoasiática 1:652 100.0% 0
Filipina 1:879 100.0% 0
Asiático 1:1315 100.0% 0
Sudeste asiático 1:2365 100.0% 0
ENFERMEDAD DE GAUCHERPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:18 87.0% 1:131
General 1:50 64.0% 1:137
ENFERMEDAD DE LA HEMOGLOBINA CPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Afroamericana 1:52 100.0% 0
Nativa americana 1:489 100.0% 0
Hispano 1:1517 100.0% 0
Caucásica 1:2754 100.0% 0
Indoasiática 1:4768 100.0% 0
Filipina 1:4775 100.0% 0
Árabe 1:5476 100.0% 0
Asiático 1:6607 100.0% 0
Sudeste asiático 1:14200 100.0% 0
ENFERMEDAD DE LA HEMOGLOBINA EPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Bangladeshí 1:24 100.0% 0
China 1:221 100.0% 0
Pakistaní 1:529 100.0% 0
Indoasiática 1:578 100.0% 0
Irlandesa 1:1961 100.0% 0
Británicos blancos 1:9091 100.0% 0
Africano 1:10000 100.0% 0
ENFERMEDAD DE NIEMANN-PICKTipo A
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:90 97.0% 1:2968
ENFERMEDAD DE POMPEPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Asiático 1:185 80.0% 1:921
Holandesa 1:185 6.0% 1:196
ENFERMEDAD DE SANDHOFFDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
ENFERMEDAD DE TAY-SACHSPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:31 99.0% 1:3001
Franco-canadiense 1:73 13.0% 1:83
No judía 1:250 46.0% 1:462
ENFERMEDAD DE VON WILLEBRAND TIPO 2NORMANDÍA
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
General 1:500 75.0% 1:1997
ENFERMEDAD DE VON WILLEBRAND TIPO 3POBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Sueca, finlandesa 1:167 10.0% 1:185
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 9 de 15
INFORME DE MUESTRA
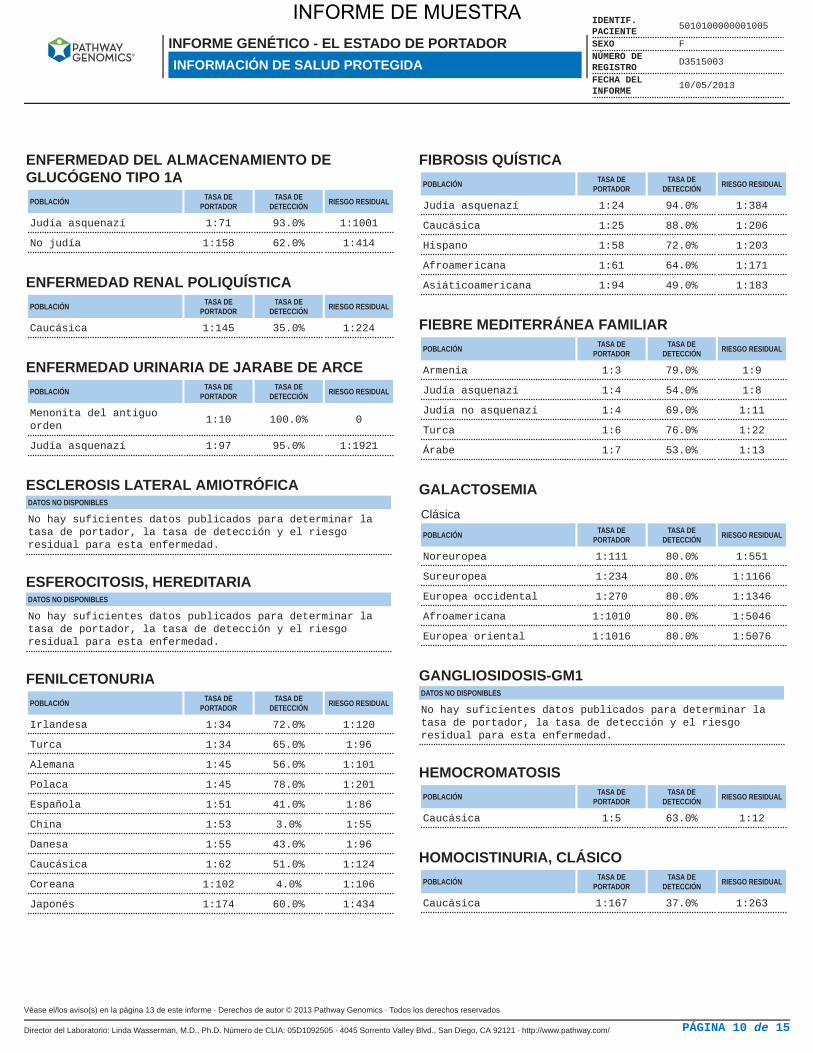
ENFERMEDAD DEL ALMACENAMIENTO DEGLUCÓGENO TIPO 1A
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:71 93.0% 1:1001
No judía 1:158 62.0% 1:414
ENFERMEDAD RENAL POLIQUÍSTICAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:145 35.0% 1:224
ENFERMEDAD URINARIA DE JARABE DE ARCEPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Menonita del antiguoorden
1:10 100.0% 0
Judía asquenazí 1:97 95.0% 1:1921
ESCLEROSIS LATERAL AMIOTRÓFICADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
ESFEROCITOSIS, HEREDITARIADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
FENILCETONURIAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Irlandesa 1:34 72.0% 1:120
Turca 1:34 65.0% 1:96
Alemana 1:45 56.0% 1:101
Polaca 1:45 78.0% 1:201
Española 1:51 41.0% 1:86
China 1:53 3.0% 1:55
Danesa 1:55 43.0% 1:96
Caucásica 1:62 51.0% 1:124
Coreana 1:102 4.0% 1:106
Japonés 1:174 60.0% 1:434
FIBROSIS QUÍSTICAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:24 94.0% 1:384
Caucásica 1:25 88.0% 1:206
Hispano 1:58 72.0% 1:203
Afroamericana 1:61 64.0% 1:171
Asiáticoamericana 1:94 49.0% 1:183
FIEBRE MEDITERRÁNEA FAMILIARPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Armenia 1:3 79.0% 1:9
Judía asquenazí 1:4 54.0% 1:8
Judía no asquenazí 1:4 69.0% 1:11
Turca 1:6 76.0% 1:22
Árabe 1:7 53.0% 1:13
GALACTOSEMIAClásica
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
Noreuropea 1:111 80.0% 1:551
Sureuropea 1:234 80.0% 1:1166
Europea occidental 1:270 80.0% 1:1346
Afroamericana 1:1010 80.0% 1:5046
Europea oriental 1:1016 80.0% 1:5076
GANGLIOSIDOSIS-GM1DATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
HEMOCROMATOSISPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:5 63.0% 1:12
HOMOCISTINURIA, CLÁSICOPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:167 37.0% 1:263
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 10 de 15
INFORME DE MUESTRA

HOMOCISTINURIA, TIPO CBLEDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
MUCOLIPIDOSIS IIPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
General 1:500 56.0% 1:1138
Japonés 1:500 60.0% 1:1249
MUCOLIPIDOSIS IIIDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
MUCOLIPIDOSIS IVPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:127 77.0% 1:549
PSEUDODEFICIENCIA DE TAY-SACHSPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía no asquenazí 1:370 100.0% 0
PÉRDIDA AUDITIVA DFNB1 Y DFNB9 NO-SINDRÓMICADFNB1 (GJB2)
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
General 1:33 80.0% 1:165
DFNB9 (OTOF)DATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
PÉRDIDA AUDITIVA DFNB59 NO-SINDRÓMICADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
RAQUITISMO, DEFICIENCIA DE PSEUDOVITAMINAD
POBLACIÓN TASA DEPORTADOR
TASA DEDETECCIÓN RIESGO RESIDUAL
General 1:2000 61.0% 1:5127
SÍNDROME DE BARTTER TIPO 4ADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
SÍNDROME DE BLOOMPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:107 99.0% 1:10601
SÍNDROME DE CRIGLER-NAJJARPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:500 39.0% 1:819
SÍNDROME DE DUBIN-JOHNSONPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía iraní 1:18 100.0% 0
Judía marroquí 1:18 100.0% 0
SÍNDROME DE EHLERS-DANLOS TIPOCIFOESCOLIOSISDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
SÍNDROME DE EHLERS-DANLOS TIPODERMATOSPARAXISDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
SÍNDROME DE EHLERS-DANLOS TIPOHIPERLAXITUDDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 11 de 15
INFORME DE MUESTRA
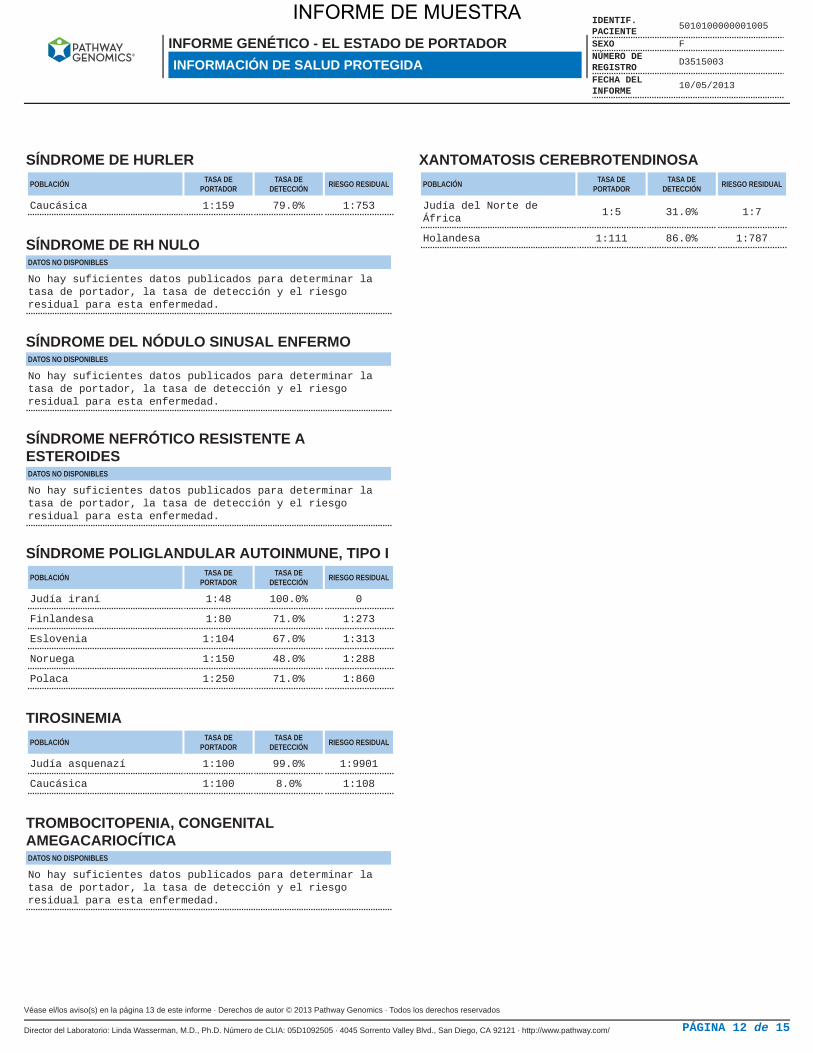
SÍNDROME DE HURLERPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Caucásica 1:159 79.0% 1:753
SÍNDROME DE RH NULODATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
SÍNDROME DEL NÓDULO SINUSAL ENFERMODATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
SÍNDROME NEFRÓTICO RESISTENTE AESTEROIDESDATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
SÍNDROME POLIGLANDULAR AUTOINMUNE, TIPO IPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía iraní 1:48 100.0% 0
Finlandesa 1:80 71.0% 1:273
Eslovenia 1:104 67.0% 1:313
Noruega 1:150 48.0% 1:288
Polaca 1:250 71.0% 1:860
TIROSINEMIAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía asquenazí 1:100 99.0% 1:9901
Caucásica 1:100 8.0% 1:108
TROMBOCITOPENIA, CONGENITALAMEGACARIOCÍTICADATOS NO DISPONIBLES
No hay suficientes datos publicados para determinar latasa de portador, la tasa de detección y el riesgoresidual para esta enfermedad.
XANTOMATOSIS CEREBROTENDINOSAPOBLACIÓN TASA DE
PORTADORTASA DE
DETECCIÓN RIESGO RESIDUAL
Judía del Norte deÁfrica
1:5 31.0% 1:7
Holandesa 1:111 86.0% 1:787
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 12 de 15
INFORME DE MUESTRA

ACLARACIÓN
Este informe no pretende ser utilizado únicamente por el paciente sin consultarlo con un profesional autorizado. El laboratorio desarrolló esteexamen y estableció sus características de rendimiento. Este examen no ha sido supervisado o aprobado por el Departamento de Control deAlimentos y Medicamentos de los Estados Unidos (FDA por sus siglas en inglés).
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 13 de 15
INFORME DE MUESTRA

RIESGOS Y LIMITACIONES
Riesgos para errores en el laboratorio o problemas de índole técnica
El laboratorio certificado opera bajo procedimientos estándares para evitar problemas de índole técnica y operativa. Sin embargo, siempreexiste la posibilidad de que estos problemas ocurran. El laboratorio que realiza los exámenes genéticos recibe muestras que fueronrecolectadas por pacientes y médicos. Por lo tanto, hay una posibilidad de que ocurran problemas durante el envío o durante la preparación delas muestras. Estos problemas incluyen, pero no se limitan al daño al espécimen o a los documentos necesarios, problemas con las etiquetasrequeridas, demoras, o la pérdida de la muestra. También existe la posibilidad de que ocurran problemas en el laboratorio que pueden impedirla obtención de resultados. Ejemplos para esto son, pero no se limitan a, problemas con las etiquetas requeridas, contaminación del ADN,resultados no interpretables, tal como errores humanos o del sistema informático. En estos casos el laboratorio puede pedir una nueva muestradel paciente. Sin embargo, es posible que el laboratorio no puede obtener un resultado a pesar de este esfuerzo.
Como es el caso con otros exámenes clínicos de laboratorio, hay una pequeña posibilidad de que el laboratorio reporte información incorrecta.Por ejemplo, es posible que el laboratorio reporte la presencia de un cierto genotipo cuando no está presente. Cualquier error de laboratoriopuede llevar a decisiones incorrectas en cuanto al tratamiento médico del paciente, o a la dieta o el ejercicio que se recomiendan. Si sesospecha o se detecta un error de laboratorio, un profesional médico puede considerar análisis adicionales u otro tipo de examen. Siempre sepueden considerar análisis adicionales para confirmar cualquier resultado.
Limitaciones
Dependiendo de los exámenes que el médico esté solicitando, el objetivo de este examen es informar al paciente sobre cómo sus genesafectan su estado de portador para algunas enfermedades heredadas, sus respuestas a medicamentos, sus riesgos para enfermedadescomunes, sus respuestas al ejercicio y a la nutrición, y le permite aprender más sobre sus antiguos ancestros. El paciente no debe cambiar sucuidado médico basado en estos resultados sin antes consultar con un profesional médico (esto incluye pero no se limita a cambios en la dosisde los medicamentos o en la frecuencia en la que estos estén tomados, el régimen de dieta o ejercicio, o la planificación de un embarazo).
La ciencia que apoya el significado o la interpretación de ciertos resultados sigue evolucionando. A pesar de que los advances científicos hayansido significantes en cuanto a los exámenes genéticos, todavía hay mucha información que debe ser descubierta. Este examen genético sebasa en la información, los descubrimientos y las técnicas de análisis disponibles en este momento. Es posible que el futuro traerá cambios a lainterpretación de los resultados que fueron analizados anteriormente. Cualquier examen genético se limita a las variantes analizadas. Esposible que la interpretación de algunas variantes cambie a base de nuevos descubrimientos. Debido a la falta de estudios genéticos, esposible que algunas variantes asociadas con las enfermedades, la respuesta a medicamentos, dieta, nutrición o ejercicio no hayan sidoidentificadas por estudios genéticos.
Muchas de las enfermedades y respuestas a medicamentos analizados dependen tanto de factores genéticos como no genéticos. Estosincluyen la edad, el historial personal y familiar, la dieta, y la etnia del paciente. Por lo tanto, es posible que una persona no presente larespuesta (sea a un medicamento, dieta, ejercicio) o enfermedad indicada por el resultado genético de este examen.
Otra limitación para algunas condiciones, sobre todo en las áreas de dieta y ejercicio, es que las asociaciones genéticas han sido estudiadas yobservadas sólo en poblaciones caucásicas. En este caso, las interpretaciones y recomendaciones aparecen en el contexto de estudios concaucásicos. Esto significa que estos resultados pueden o no ser relevantes para personas etnias no caucásicas o mixtas.
Basado en los resultados y el conocimiento médico de la persona, los profesionales médicos pueden considerar análisis independientesadicionales o consultar con otro médico o con un asesor en genética.
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 14 de 15
INFORME DE MUESTRA

DEFINICIONES DE RESULTADOS
Modificado Los resultados y/o los datos del paciente han sido actualizados sin afectar elsignificado clínico de los resultados y/o el diagnóstico, tratamiento o cuidadomédico del paciente.
Corregido Los resultados y/o los datos del paciente han sido actualizados, lo cual puedeimpactar el significado clínico de los resultados y/o el diagnóstico, tratamiento ocuidado del paciente.
Final Los resultados que fueron disponibles a la hora de la publicación del informe oque fueron actualizados de un estado pendiente a un estado final.
Pendiente Resultados que no fueron disponibles a la hora de publicación del informe.Todos los resultados pendientes estarán indicados en el informe.
INFORME GENÉTICO - EL ESTADO DE PORTADORINFORMACIÓN DE SALUD PROTEGIDA
IDENTIF.PACIENTE
5010100000001005
SEXO FNÚMERO DEREGISTRO
D3515003
FECHA DELINFORME
10/05/2013
Véase el/los aviso(s) en la página 13 de este informe · Derechos de autor © 2013 Pathway Genomics · Todos los derechos reservados
Director del Laboratorio: Linda Wasserman, M.D., Ph.D. Número de CLIA: 05D1092505 · 4045 Sorrento Valley Blvd., San Diego, CA 92121 · http://www.pathway.com/ PÁGINA 15 de 15
INFORME DE MUESTRA
Top Related