
US Regulation of In Vitro Diagnostic Devices (IVDs) Martinez... · US Regulation of In Vitro...
20
US Regulation of In Vitro Diagnostic Devices (IVDs) SoGAT- Clinical Diagnostic NIBSC, June 25, 2008 Francisco Martínez Murillo, PhD Office of In Vitro Diagnostic Device Evaluation (OIVD) Center for Devices and Radiological Health (CDRH) US Food and Drug Administration (FDA)
Transcript of US Regulation of In Vitro Diagnostic Devices (IVDs) Martinez... · US Regulation of In Vitro...

US Regulation of In Vitro
Diagnostic Devices (IVDs)
SoGAT- Clinical Diagnostic
NIBSC, June 25, 2008
Francisco Martínez Murillo, PhD
Office of In Vitro Diagnostic Device Evaluation (OIVD)
Center for Devices and Radiological Health (CDRH)
US Food and Drug Administration (FDA)

2
Highlights
• Regulatory process
• Device requirements
• Review challenges
3
FDA’s Mission
Get safe and effective
devices to market as
quickly as possible
Ensure that devices
on the market are
safe and effective

4
• Class I: common, low risk devicese.g., vitamin A deficiency test
– General Controls
– Most exempt from premarket submission
Risk-Based Regulation of IVDs
Substantial Equivalence
Safe & Effective vs Pred.
510(k) sect. of the Act
Evidence of Safety and Effectiveness
Impact on patient
Controlled clinical trials
Device stands on its own
• Class II: moderate riske.g. prognosis, monitoring in already diagnosed
cancer patients
– Special Controls
– Premarket Notification [510(k)]
• Class III: most complex, highest riske.g. cancer diagnosis or screening
– Safety, effectiveness, supported by valid scientific evidence
– Premarket Approval [PMA]
5
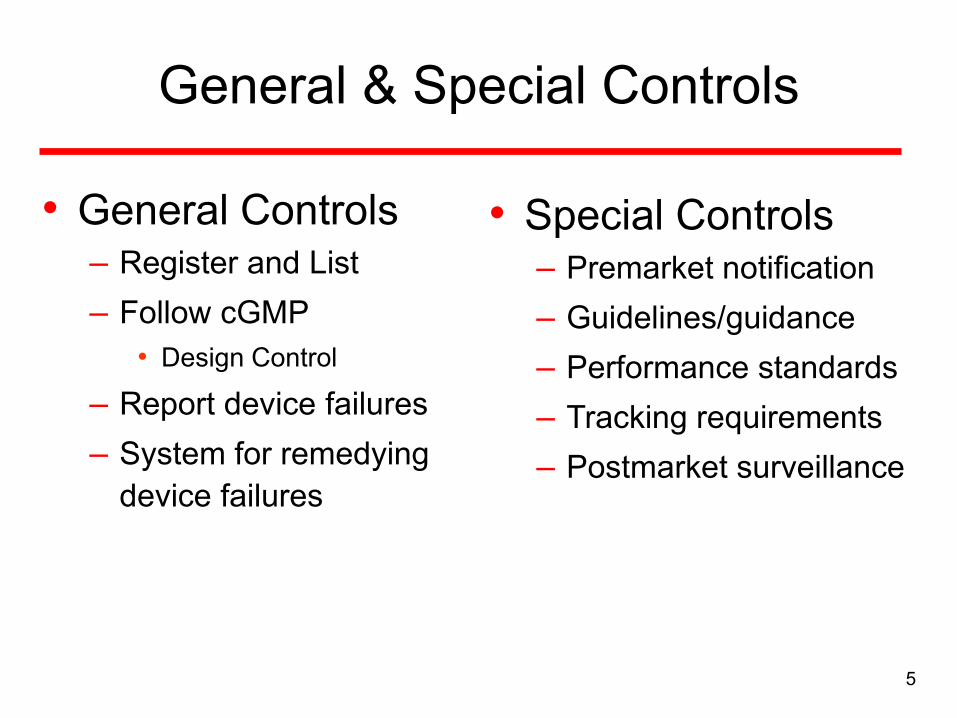
General & Special Controls
• General Controls– Register and List
– Follow cGMP
• Design Control
– Report device failures
– System for remedying
device failures
• Special Controls– Premarket notification
– Guidelines/guidance
– Performance standards
– Tracking requirements
– Postmarket surveillance

6
Key Elements of a Submission
• Intended use/indications for use
• Device description
• Analytical validation
• Clinical validity/clinical utility
• Instrument and software validation, if applicable
• Labeling (package insert)
• Manufacturing, design controls, quality system
requirements (QSRs/cGMP) (For PMA)

7
Major Review Issues
• Analytical performance– How reliably and correctly test measures analyte
• Clinical performance– How reliably test measures clinical condition
• Labeling– Intended use, directions for use, warnings, limitations,
interpretation of results, performance summary

8
Challenges for the Review
• Lack of “gold standards”/performance standards
• Cutting edge new technology - multiplex, bioinformatics, nanotechnology
– Paucity of standard methods and materials
• New biological/clinical knowledge
– Changes in practice of medicine
• Overt and latent bias
• Issues of traceability

9
IVDs and Standards
• Determination of analytical sensitivity
• Viral load/NAT detection vs. infectivity
• Determination of IU vs. SI

10
Lack of Standards/Ref. Materials
• Microbiology
– TB (NAT and infectivity)
– Malaria, WNV. Parasites (Chagas, Trypanosomiasis)
– CMV, EBV, VZV, HSV
• Chemistry
– Glucose
– Troponin
• Immunology
– PSA/AFP (for low level)
– Other specific cancer, allergen, autoimmune marker
– Copy number

11
Current Tentative Solutions
• Analytical calibration related to infective units
• Clinical performance referred to a cleared device
• Clinical performance related to clinical truth
(clinical trial outcome)
• Use of consensus of literature
• Composite reference + other lab tests/clinical history
• Use of recognized reference panels (CDC)
• Generation of Standards by accredited organization

12
Quest for Standards
• Standards already in documentation
– Founding member of NCCLS (CLSI)
– QSReg (21 CFR 820) (~ISO 13485/9100)
• Traceability-Commutability-Robustness
• Working with NIST

13
Handling the Challenges
• Regulatory trail is well lit - Literature, Guidance
• Broad menu of flexible regulatory tools
- Pre-IDE - Exempt
- IDE - de novo
- 510(k) - Real time
- PMA - Expedited
• Review not outcome oriented
• Mandate to be least burdensome
• New scientific resources/programs
• Evidence, risk, and knowledge base
Eye on Public Health
Risk-Based Regulation
14
Transparency

www.fda.gov/cdrh/oivd
www.fda.gov/cdrh/devadvice
• Guidance
– Documents/Recommendations
• Regulation
– Device Requirements
• Databases
– 510(k)/PMA/GGP/CLIA
e-mail: [email protected]

16
Supplementary Information

17
FREQUENTLY USED WEBSITES – June 2007
21 CFR Parts 1 - 1499 www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm
510(k) Paradigm www.fda.gov/cdrh/ode/parad510.html
CDRH Databases www.fda.gov/cdrh/databases.html
CDRH Home Page www.fda.gov/cdrh/
Code of Federal Regulations (CFR) www.fda.gov/cdrh/devadvice/365.html
Consumer Information/CDRH www.fda.gov/cdrh/consumer
Device Advice www.fda.gov/cdrh/devadvice/
Device Specific Contacts www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfReferral/referral.cfm
Device Standards Program www.fda.gov/cdrh/stdsprog.html
DSMICA Staff Directory www.fda.gov/cdrh/dsma/dsmastaf.html#contents
Electronic Products Radiation Control www.fda.gov/cdrh/radhealth/
Establishment Registration and U.S. Agent www.fda.gov/cdrh/devadvice/341.html
Export of Medical Devices from U.S. www.fda.gov\cdrh\devadvice\39.html
FDA Home Page www.fda.gov/
FDA’s Centers Small Business Contacts www.fda.gov/ora/fed_state/Small_Business/sb_guide/centerco.htm
Federal Food, Drug, and Cosmetic Act www.fda.gov/opacom/laws/fdcact/fdctoc.htm
Global Harmonization www.ghtf.org
Good Guidance Practices (GGP) www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfggp/search.cfm
Index of CDRH Web Documents www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/topicindex/topindx.cfm
International Program www.fda.gov/cdrh/international/
Medical Device Exemptions www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/315.cfm
Medical Device Foreign Regulators www.ita.doc.gov/td/health/regulations.html
Medical Device Listing www.fda.gov/cdrh/devadvice/342.html
Medical Device Reporting Home Page www.fda.gov/cdrh/mdr/index.html
Medical Device User Fee and Modernization www.fda.gov/cdrh/mdufma
Act of 2002 (MDUFMA)
MEDWATCH www.fda.gov/medwatch/index.html
Premarket Approval http://www.fda.gov/cdrh/devadvice/pma/
Premarket Notification (510(k)) www.fda.gov/cdrh/devadvice/314.html
Quality Systems www.fda.gov/cdrh/devadvice/32.html
Reuse of Single Use Devices www.fda.gov/cdrh/reprocessing/
Third Party Inspection Programs www.fda.gov/cdrh/ap-inspection/index.html
Third Party Review Program www.fda.gov/cdrh/thirdparty

18
FDA Guidance Documents
• Factor V Leiden DNA Mutation Detection Systems – March 2004http://www.fda.gov/cdrh/oivd/guidance/1236.html
• Instrumentation for Clinical Multiplex Test Systems – Class II Special Controls Guidance Document – March 2005http://www.fda.gov/cdrh/oivd/guidance/1546.html
• Drug-Diagnostic Co-Development Concept Paper – April 2005http://www.fda.gov/cder/genomics/pharmacoconceptfn.pdf
• RNA Preanalytical Systems (RNA Collection, Stabilization and Purification Systems for RT-PCR used in Molecular Diagnostic Testing) -- September 2005. http://www.fda.gov/cdrh/oivd/guidance/1563.html
• CFTR Gene Mutation Detection Systems – October 2005. http://www.fda.gov/cdrh/oivd/guidance/1564.html

19
FDA Guidance Documents
• RNA Preanalytical Systems (RNA Collection, Stabilization and Purification Systems for RT-PCR used in Molecular Diagnostic Testing) -- September 2005. http://www.fda.gov/cdrh/oivd/guidance/1563.html
• CFTR Gene Mutation Detection Systems – October 2005. http://www.fda.gov/cdrh/oivd/guidance/1564.html
• Pharmacogenetic Tests and Genetic Tests for Heritable Markers – February 2006. http://www.fda.gov/cdrh/oivd/guidance/1549.html
• Guidance on Informed Consent for In Vitro Diagnostic Device Studies Using Leftover Human Specimens that are Not Individually Identifiable – April 2006. http://www.fda.gov/cdrh/oivd/guidance/1588.html
• In Vitro Diagnostic Multivariate Index Assays – September 2006. http://www.fda.gov/cdrh/oivd/guidance/1610.html

20
FDA Guidance Documents
• Quality Control Material for Cystic Fibrosis Nucleic Acid Assays – January 2007. http://www.fda.gov/cdrh/oivd/guidance/1614.html
• Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests – March 2007. http://www.fda.gov/cdrh/osb/guidance/1620.html
• Gene Expression Profiling Test system for Breast Cancer Prognosis – May 2007.
http://www.fda.gov/cdrh/oivd/guidance/1627.html
• Drug Metabolizing Enzyme Genotyping System - Class II Special Controls Guidance Document
http://www.fda.gov/cdrh/oivd/guidance/1551.html
• In Vitro Diagnostic (IVD) Device Studies – Frequently Asked Questions
http://www.fda.gov/cdrh/oivd/guidance/1587.html
• Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices
http://www.fda.gov/cdrh/ode/guidance/337.html


















