
The role of specific maternal nutritional factors on ...€¦ · on epigenetic programming of fetal...
155
The role of specific maternal nutritional factors on epigenetic programming of fetal immune development N. D. D. Manori Amarasekera MBBS, MPhil This thesis is presented for the degree of Doctor of Philosophy of University of Western Australia School of Paediatrics and Child Health 2015
Transcript of The role of specific maternal nutritional factors on ...€¦ · on epigenetic programming of fetal...

The role of specific maternal nutritional factors
on epigenetic programming of fetal immune
development
N. D. D. Manori Amarasekera MBBS, MPhil
This thesis is presented for the degree of Doctor of Philosophy of
University of Western Australia
School of Paediatrics and Child Health
2015

ii
DEDICATION
This thesis is dedicated to my mother, father and my sister who motivated me to go for
my highest potential and to Buddhika and Dehemi who continue to be my greatest
support and Nethmi for bearing up long hours of lab work with me in my womb for nine
months.

iii
STATEMENT OF CANDIDATE CONTRIBUTION
The author performed all the work presented in this thesis except where contributions
from others have been acknowledged below.
The work presented in this thesis was performed between February 2011 and December
2014 at the School of Paediatrics and Child Health, University of Western Australia
under the supervision of Professor Susan Prescott, Dr. Deborah Strickland, Dr. David
Martino and Professor Meri Tulic.
The Childhood Allergy and Immunology Research team (CAIR) assisted with
participant recruitment, data and blood collection, blood processing and data entry.
Professor Susan Prescott performed all clinical diagnoses.
Assistance with FACS was provided by Dr. Michelle Tourigny at Telethon Kids
Institute, Perth.
Dr. David Martino, Murdoch Children’s Research Institute, Melbourne (under the
supervision of Dr. Richard Saffery) was responsible for Illumina 450k methylation
array including designing and data analysis.
Hani Harb and Dr. Dorthe Kesper, Institute of Laboratory Medicine and
Pathobiochemistry, Molecular Diagnostics, Philipps University of Marburg, Germany
(under the supervision of Professor Harald Renz) performed and analysed the histone
acetylation experiments.
This work is original and has not been submitted previously for any other degree.
____________________ ____________________
Professor Susan Prescott N.D.D.Manori Amarasekera
Coordinating supervisor Candidate

iv
THESIS SUMMARY
The burden of non-communicable diseases (NCD) including cardiovascular, metabolic,
allergic and chronic lung diseases has reached pandemic proportions, as the major
global health challenge in the 21st century. Understanding the causes and mechanisms of
these diverse but pathogenically related inflammatory conditions remains a key
challenge in overcoming these trends. The rise in these NCD is strongly related to
modern lifestyle changes, and shared environmental risk factors, many of which disrupt
early metabolic immune programming and predisposed to subsequent disease.
The ‘Developmental Origin of Health and Disease’ (DOHaD) paradigm recognises the
crucial importance of the early environment in shaping both structural, developmental
and physiological response patterns in ways that may determine future health, biological
reserve and disease susceptibility. Among many environmental exposures, the quality of
early life nutrition is one of the most important factors influencing all aspects of
development during critical formative periods, particularly during gestation. While the
role of early metabolic adaptations has been long recognised as a predisposing factor,
the impact of the early environment on the developing immune system in the
subsequent predisposition to inflammation, has only been recognised more recently.
Identifying and optimising early factors that influence the patterns of immune
development is central in understanding disease pathogenesis and in implementing
better prevention strategies. In particular, variations in maternal nutrition can
significantly modify both immune and metabolic programming, to determine the long-
term health outcomes of the offspring. Only recently has it been recognised that many
of these effects are mediated through epigenetic mechanisms such as DNA methylation
and histone acetylation. The strong link between changes in the modern diet and
escalating rate of allergic and other NCD underscores the importance of identifying
specific in utero nutritional exposures affecting the fetal epigenome leading to an altered
immune profile.
The work of this thesis is amongst the first to explore the role of maternal nutrition on
neonatal epigenetic profile and subsequent risk for allergic outcomes using state-of-the-
art epigenomic profiling technologies.

v
The studies herein aimed to investigate epigenetic variation in neonatal immune cells in
association with variation in utero exposure to folate (chapters 3 and 4) and determined
whether maternal supplementation with fish oil containing omega-3 polyunsaturated
fatty acids (n-3 PUFA) modify the neonatal DNA methylome (chapter 5).
In study 1 (chapters 3 and 4), an ‘extremes of phenotype’ approach was employed to
retrospectively select a cohort of 23 cord blood samples from a prospective birth cohort
(n=420), categorised by high in utero folate (High Folate; HF, n=11) and low folate
(Low Folate; LF, n=12) exposure based on maternal serum folate levels measured in the
last trimester of pregnancy. DNA from both neonatal CD4+ T-cells and antigen
presenting cells (APC) isolated from cord blood were then bisulphite converted and
profiled for genome-wide methylation using Illumina Human Methylation 450 arrays.
In parallel, using a novel chromatin immunoprecipitation (ChIP) assay, histone 3 (H3)
and histone 4 (H4) acetylation levels at selected loci were measured in CD4+ T-cells as
an additional level of epigenetic regulation. A comparison of genome-wide DNA
methylation profiles between HF and LF groups revealed differential methylation at
seven regions across the genome. By far, the biggest effect observed was
hypomethylation of a 923bp region 3kb upstream of the ZFP57 transcript, regulator of
DNA methylation during development. Consistent differences in folate-sensitive
differentially methylated regions (fDMR) were reported in both CD4+ T-cells and APC,
which are derived from distinct hematopoietic cell lineages, suggesting these
differences have originated at the common hematopoietic progenitor stage. Additional
functional studies of mRNA expression and histone acetylation revealed that HF
exposures was associated with relatively higher mRNA expression and concomitant
higher H3 and H4 acetylation levels at the downstream ZFP57 gene. These findings
were replicated in an additional independent set of samples derived from the same
prospective birth cohort using Sequenom Mass Spectrometry and the findings from
these experiments were reported in FASEB medical journal.
Parallel studies of H3 and H4 acetylation levels at loci associated with allergy
development were undertaken to address a different hypothesis, was high folate
exposure is associated with variations in the activity of allergy-associated genes.. Using
targeted assays, these studies revealed significantly higher histone acetylation levels at
GATA3 locus, a master regulator of allergic immune responses, in neonates of the HF
group, suggesting increased propensity for activation of these pathways. In the same

vi
group a significant increase in H4 acetylation at the IL9 locus, another allergy-
promoting cytokine was observed. Together these findings suggest that exposure to high
gestational folate is associated with more transcriptionally permissive chromatin at
allergy-associated genes in CD4+ T-cells.
In study 2 (chapter 5) we studied the role of n-3 PUFA supplementation during
pregnancy, which previous studies in the host lab have shown to reduce the severity of
clinical symptoms in infants at high risk of allergic diseases. Experiments were carried
out to investigate whether these health promoting effects are mediated through DNA
methylation by analysing the CD4+ T-cells purified from cryo-preserved cord blood
samples selected from a randomised controlled trial of maternal fish oil
supplementation. In this double-blind trial, mothers were given either fish oil or placebo
from 20 weeks of pregnancy until delivery. Of the 80 mother-infant pairs who
completed the initial trial, cord blood samples of 70 neonates were available for
genome-wide DNA methylation profiling. Analysis of cord red cell fatty acid
parameters provided robust evidence that the maternal supplementation modified fetal
lipid profiles. There was very little evidence to support a role for DNA methylation in
mediating these effects. Comparison of DNA methylation profiles between treatment
and control groups did not reveal any statistically significant differences in CpG
methylation, at the single-CpG or regional level. Effect sizes for top-ranked candidates
were small and of unknown biological significance. Further studies would be required
to address the role of other epigenetic or post-transcriptional mechanisms that could
mediate these effects, however the experiments in chapter 5 argue against a substantial
role for DNA methylation.
Using cutting-edge epigenomic profiling technologies, this is the first study to
undertake extensive profiling of the neonatal epigenome in purified immune cells of
different lineages to unravel the combinatorial epigenetic effects of specific maternal
nutritional factors. This study provides evidence that epigenetic mechanisms play an
important role in mediating environmentally driven effects on neonatal genome. The
epigenomic regions identified in this thesis will provide the framework for future
studies in the evolving area of nutri-epigenomics to understand the origin of NCD and
help identify targets for treatment and prevention strategies.

vii
TABLE OF CONTENTS
DEDICATION............................................................................................................................ II
STATEMENT OF CANDIDATE CONTRIBUTION ........................................................... III
THESIS SUMMARY ................................................................................................................ IV
TABLE OF CONTENTS ....................................................................................................... VII
LIST OF FIGURES .................................................................................................................... X
LIST OF TABLES .................................................................................................................. XII
ABBREVIATIONS ................................................................................................................ XIII
DECLARATION OF PUBLISHED WORK ANDWORK PREPARED FOR
PUBLICATION ....................................................................................................................... XV
ACKNOWLEDGEMENTS ............................................................................................... XVIII
CHAPTER 1: LITERATURE REVIEW .................................................................................. 1
1.1 FETAL LIFE: THE CRITICAL PERIOD OF IMMUNE DEVELOPMENT ...................................... 1
1.1.1 Immunology of feto-maternal interface ...................................................................... 1
1.1.2 Ontogeny of immune development ............................................................................. 2
1.1.3 Transition of the immune profile from prenatal to postnatal life ................................ 3
1.1.4 Environmental exposures modulate immune responses: the effects of dietary factors
on immune development ...................................................................................................... 5
1.2 DEVELOPMENTAL PROGRAMING AND THE ‘DEVELOPMENTAL ORIGIN OF HEALTH AND
DISEASE (DOHAD)’ HYPOTHESIS ............................................................................................. 8
1.3 GENE-ENVIRONMENT INTERACTIONS IN DETERMINING DISEASE RISK ............................. 10
1.4 EPIGENETICS: WHAT IS IT? ................................................................................................ 11
1.4.1 Molecular mechanisms of epigenetics ...................................................................... 12
1.4.2 DNA methylation ...................................................................................................... 12
1.4.3 Histone modifications ............................................................................................... 14
1.4.4 Regulation by non-coding RNA (ncRNA) ................................................................ 15
1.4.5 Chromatin remodelling complexes ........................................................................... 16
1.5 FUNCTIONAL IMPLICATIONS OF EPIGENETIC DYNAMICS .................................................. 16
1.5.1 Epigenetic regulation during embryo development and haematopoietic development
............................................................................................................................................ 17
1.5.2 Epigenetic profile of the neonatal immune system and epigenetic changes during
postnatal development ........................................................................................................ 19
1.5.3 Evidence of disruption of epigenetic pathways in immune diseases......................... 20

viii
1.6 EPIGENETICS AS AN UNDERLYING MECHANISM OF DEVELOPMENTAL ORIGIN OF CHRONIC
DISEASES ................................................................................................................................ 22
1.7 MATERNAL ENVIRONMENTAL EXPOSURES THAT MODULATE THE NEONATAL EPIGENOME
................................................................................................................................................ 23
1.7.1 Nutritional exposures in early life and the fetal epigenome ...................................... 23
1.7.1.1 Folate and other nutrients involved in one-carbon metabolism ......................... 24
1.7.1.2 Polyunsaturated fatty acids (PUFA) .................................................................. 29
1.7.2 Other maternal exposures that affect the fetal epigenome ........................................ 30
1.8 CHALLENGES OF EPIGENOME-WIDE ASSOCIATION STUDIES ............................................. 33
1.9 CONCLUDING REMARKS .................................................................................................... 34
CHAPTER 2: STUDY RATIONALE, STUDY DESIGN AND METHODS ...................... 36
2.1 STUDY RATIONALE ............................................................................................................ 36
2.2 STUDY COHORTS ............................................................................................................... 37
2.3 DATA AND SAMPLE COLLECTION ...................................................................................... 38
2.3.1 Questionnaire data ..................................................................................................... 38
2.3.2 Maternal and infant skin prick test (SPT) ................................................................. 38
2.3.3 Clinical assessment for allergic diseases ................................................................... 39
2.3.4 Collection of cord and maternal peripheral blood ..................................................... 39
2.4 LABORATORY METHODS ................................................................................................... 39
2.4.1 Harvesting and cryopreservation of mononuclear cells ............................................ 39
2.4.2 Thawing of cryopreserved mononuclear cells ........................................................... 40
2.4.3 Cord blood immune responses .................................................................................. 41
2.4.4 Purification of CD4+ T-cells ...................................................................................... 41
2.4.5 Purification of antigen presenting cells ..................................................................... 42
2.4.6 Purification of genomic DNA and total RNA ........................................................... 43
2.4.7 Quantitative polymerase chain reaction (qPCR) ....................................................... 43
2.4.8 DNA methylation analysis ........................................................................................ 45
2.4.9 Validation of methylation array targets ..................................................................... 45
2.4.10 Histone 3 and Histone 4 acetylation analysis .......................................................... 46
2.5 STATISTICAL ANALYSIS .................................................................................................... 46
2.6 ETHICS APPROVAL ............................................................................................................ 46
CHAPTER 3: PAPER 1 ........................................................................................................... 47
3.1 ABSTRACT ......................................................................................................................... 48
3.2 INTRODUCTION ................................................................................................................. 49
3.3 MATERIALS AND METHODS .............................................................................................. 50
3.4 RESULTS ............................................................................................................................ 54
3.5 DISCUSSION ....................................................................................................................... 62

ix
3.6 REFERENCES ..................................................................................................................... 64
CHAPTER 4: PAPER 2 ........................................................................................................... 67
4.1 ABSTRACT ......................................................................................................................... 68
4.2 INTRODUCTION ................................................................................................................. 69
4.3 METHODS .......................................................................................................................... 70
4.4 RESULTS ............................................................................................................................ 73
4.5 DISCUSSION ....................................................................................................................... 77
4.6 REFERENCES ..................................................................................................................... 79
CHAPTER 5: PAPER 3 ........................................................................................................... 81
5.1 ABSTRACT ......................................................................................................................... 82
5.2 INTRODUCTION ................................................................................................................. 83
5.3 MATERIALS AND METHODS .............................................................................................. 84
5.4 RESULTS ............................................................................................................................ 86
5.5 DISCUSSION ....................................................................................................................... 90
5.6 REFERENCES ..................................................................................................................... 93
CHAPTER 6: GENERAL DISCUSSION ............................................................................... 96
6.1 DISCUSSION OF KEY FINDINGS IN THE CONTEXT OF EXISTING LITERATURE ..................... 96
6.2 LIMITATIONS OF THE STUDY ........................................................................................... 100
6.3 DIRECTIONS FOR FUTURE STUDIES IN THIS AREA ........................................................... 102
6.4 CONCLUDING REMARKS .................................................................................................. 103
BIBLIOGRAPHY ................................................................................................................... 105
APPENDIX 1 ........................................................................................................................... 133
APPENDIX 2 ........................................................................................................................... 134
APPENDIX 3 ........................................................................................................................... 137

x
LIST OF FIGURES
Figure 1.1 Schematic representation of the organisation and packaging of nucleosome Figure 1.2 Schematic illustration of one carbon metabolism Figure 2.1 Outline of purification steps used to isolate CD4+ T-cells and antigen presenting cells (APC) from cord blood mononuclear cells (CBMC) for the studies in this thesis Figure 2.2 Outline of screening strategy for isolating antigen presenting cells (APC) using FACS analysis Figure 3.1 Relationship between maternal serum folate and cord serum folate levels Figure 3.2 Differential methylation analysis between CD4+ and APC cells Figure 3.3 Analysis of global patterns of DNA methylation between folate groups Figure 3.4 Folate sensitive differentially methylated region (fDMR) in chromosome 6 Figure 3.5 This figure shows validation data of chromosome 6 fDMR and functional assessment of ZFP57 in CD4+ cells Figure 3.6 Sequenom Epityper analysis of DNA methylation at the chromosome 6 fDMR in an independent set of samples (n=19) Figure 4.1 Outline of the ChIP assay and shearing efficiency Figure 4.2 Lower limit of quantification and freeze-thawing effect of chromatin Figure 4.3 Levels of H3 (A) and H4 (B) histone acetylation at key genes associated with allergic inflammation in neonatal CD4+ T-cells Figure 5.1 Cord red blood cell fatty acid measurements in fish oil and control groups Figure 5.2 Regression analysis of CpG sites that co-associate with cord red cell total n-3 fatty acid levels
12 25 41 43 54 56 57 58 59 60 73 74 75 87 89

xi
Figure 5.3 Comparative analysis of DNA methylation profiles between treatment and intervention groups
89

xii
LIST OF TABLES
Table 1.1 Gene loci that show epigenetic modulation in association
with immune diseases
Table 3.1 Population characteristics of the mother and infants in
the discovery cohort
Table 3.2 Differentially methylated regions (DMR) according to
the folate status
Table 4.1 The effect of temperature and long-term storage on per
cent enrichment of H3 and H4 histone acetylation ± SEM
Table 5.1 Population characteristics of the mothers and infants in
the cohort (n=70)
21
55
58
74
86

xiii
ABBREVIATIONS
2ME 2-mercaptoethanol
5hmC 5-hydroxymethylcytosine
5mC 5-methylcytosine
AHRR aryl hydrocarbon receptor repressor
APC antigen presenting cells
bp base pairs
CBMC cord blood mononuclear cells
cDNA complementary DNA
ChIP chromatin immunoprecipitation
CpG cytosine followed by a guanine, linked together by a phosphate
DC dendritic cells
DEP diesel exhaust particles
DHA docosahexanoic acid
DMP differentially methylated positions
DMR differentially methylated region
DMSO dimethyl sulphoxide
DNMT DNA methyltransferases
DOHaD developmental origin of health and disease
EPA eicosapentaenoic acid
EWAS epigenome-wide association studies
FCS fetal calf serum
fDMR folate sensitive-differentially methylated regions
FOXP3 forkhead box 3
GATA3 GATA binding protein 3
gDNA genomic DNA
GWAS genome-wide association studies
H3 histone 3
H3K4me/me2/me3 mono/di/tri-methylation of lysine 4 of Histone 3
H3Kn lysine (n) of Histone 3
H4 histone 4
HAT histone acetyltransferases
HF high folate
HIFCS heat-inactivated fetal calf serum

xiv
HKG house keeping gene
IFNG interferon gamma
IGF2 insulin-like growth factor-2
IL- interleukin
IP mmunoprecipitation
LCPUFA long chain polyunsaturated fatty acids
LF low folate
MBD methyl binding domain
MC mononuclear cells
MDS multidimensional scaling
miRISC miRNA-associated RNA-induced silencing complex
miRNA microRNA
n-3 PUFA omega 3 polyunsaturated fatty acids
NCD non-communicable diseases
ncRNA non coding RNA
NIMA non-inherited maternal alloantigens
OVA ovalbumin
PCR polymerase chain reaction
PGC primordial germ cells
PM particulate matter
qPCR quantitative polymerase chain reaction
RCT randomised controlled trial
RPMI Roswell Park Memorial Institute
RT room temperature
SAM S-adenosylmethionine
SCFA short chain fatty acids
SDS sodium dodecyl sulfate
SNPs single nucleotide polymorphisms
SPT skin prick test
TET ten-eleven translocation proteins
Th1/2 T helper1/2
Tregs regulatory T-cells
TSLP thymic stromal lymphopoietin
UTR untranslated region

xv
DECLARATION OF PUBLISHED WORK AND
WORK PREPARED FOR PUBLICATION
This thesis contains published work and work prepared for publication, all of which has
been co-authored. The bibliographical details of the work, where it appears in the thesis
and author contributions are outlined below.
1. Genome-wide DNA methylation profiling identifies a folate-sensitive region of
differential methylation upstream of ZFP57 imprinting regulator in humans
Manori Amarasekera, David Martino, Sarah Ashley, Hani Harb, Dörthe Kesper,
Deborah Strickland, Richard Saffery, and Susan L. Prescott
Published in: FASEB J. 2014 Jun 2. pii: fj.13-249029. [Epub ahead of print] and
constitutes Chapter 3 of the thesis.
M. Amarasekera was involved in study conception and design, isolation of CD4+ T-
cells, sorting APC, DNA extraction, qPCR assay, target gene validation for methylation,
data analysis and manuscript preparation. All authors assisted in editing the manuscript
content and approved the final manuscript.
2. Folate status as a modifier of H3/H4 histone acetylation marks at allergy-
associated genes in human neonatal CD4+ T-cells
Hani Harb* Manori Amarasekera*, Sarah Ashley, Meri K. Tulic, Petra Ina Pfefferle,
Daniel P. Potaczek , Harald Renz , David Martino, Dörthe A. Kesper & Susan L.
Prescott
*HH and MA contributed equally to this work and share first authorship.
In preparation to be submitted to Epigenetics and constitutes Chapter 4 of the thesis.
M. Amarasekera was involved in study conception and design, isolation of CD4+ T-
cells, qPCR assay, cell culture experiments, cytokine analysis, data analysis and
manuscript preparation. All authors assisted in editing the manuscript content and
approved the final manuscript.

xvi
3. Epigenome-wide analysis of neonatal CD4+ T-cell DNA methylation sites
potentially affected by maternal fish oil supplementation
Manori Amarasekera, Paul Noakes, Deborah Strickland, Richard Saffery, David J
Martino, Susan L Prescott
Published in: Epigenetics. 2014 Dec 7:0. [Epub ahead of print] and constitutes Chapter
5 of the thesis.
M. Amarasekera assisted in study conception and design, isolation of CD4+ T cells,
flow cytometry analysis, DNA extraction, data analysis and manuscript preparation. All
authors assisted in editing the manuscript content and approved the final manuscript.
4. Nutrition in early life, immune-programming and allergies: the role of
epigenetics
Manori Amarasekera, Susan L. Prescott and Debra J. Palmer
Published in: Asian Pac J Allergy Immunol. 2013 Sep;31(3):175-82.
M. Amarasekera drafted and assisted in editing the manuscript.
5. Epigenetic aberrations in human allergic diseases
Manori Amarasekera, David Martino, Meri K Tulic, Richard Saffery and Susan
Prescott
Published in: Tryve O. Tollesfsbol, editor. Epigenetics in human disease. Philadelphia:
Elsevier Inc. 2012. p371-387.
M. Amarasekera drafted and assisted in editing the manuscript.
6. Epigenetics in immune development and in allergic and autoimmune diseases
David Martino, Dorthe A. Kesper, Manori Amarasekera, Hani Harb, Harald Renz and
Susan Prescott.

xvii
Published in: J Reprod Immunol. 2014 Oct;104-105:43-8.
M. Amarasekera assisted in drafting and editing the manuscript.
____________________ ____________________
Professor Susan Prescott N.D.D.Manori Amarasekera
Coordinating supervisor Candidate

xviii
ACKNOWLEDGEMENTS
I would like to thank the study participants and the obstetricians and midwives who
dedicated their time and effort to make this study possible.
My sincere gratitude and eternal thanks go to my primary supervisor Professor Susan
Prescott for offering me the opportunity to work on this project. You motivated me to
believe in myself and in my potentials. Thank you for the guidance and generous
opportunities along the way. I am very grateful also to Dr. Deborah Strickland and Prof.
Meri Tulic, my co- supervisors, for the time, advice and support. Many thanks to Dr.
David Martino for being so patient with me. I could always ring you to get advice in
difficult situations. Without your support and advice I would not have been able to
perform complex molecular work involved in this thesis.
Many thanks go to Dr. Anthony Kicic, Dr. Erika Sutanto and Thomas Iosifidis for their
excellent advice and assistance with qPCR. Thanks to Nina D’Vaz for her advice and
assistance with Luminex protein assays. I would like to thank Dr. Michelle Tourigny for
her assistance in FACS. Thanks to the Cancer and Disease Epigenetic Group, Murdoch
Children’s Research Institute, Melbourne for their assistance in Sequenom work and Dr.
Richard Saffery for his advice on methylation data analysis and interpretation. I would
also like to thank the Childhood Allergy and Immunology Research Group – Debra
Palmer, Paul Noakes, Suzanne Meldrum, Rachel West, Sarah Miller, Catherine Haigh,
Emma Prescott and Emma Snelgar. Thanks go to my fellow PhD students – Alexandra
Heaton, Valene See, Jessica Metcalfe and Anderson Jones for your support.
I would also like to acknowledge the financial support I received from the Australian
Government (Endeavour Postgraduate scholarship).
Finally I would like to thank my husband and my two gorgeous daughters for being
there for me as a constant source of strength.

1
Chapter 1
Literature Review
This literature review examines evidence for in utero epigenetic variations as mediators
of environment-induced changes that may modify later health outcomes, particularly
immune diseases. As a prelude to this, the initial sections of this review briefly
summarise the ontogeny of the immune system, how epigenetics are involved in
immune development, and how disruptions in epigenetic regulation are associated with
disease states.
1.1 Fetal life: the critical period of immune development
There has been a significant increase in immune-mediated diseases over the past 4-6
decades (1). This has been clearly associated with the marked environmental changes
associated with transition to more modern life-styles (2). The dramatic rise in infant
immune diseases, most notably allergy, indicates the specific vulnerability of the
immune system to early environmental changes. The associated parallel rise in
metabolic diseases that are characterised by low-grade inflammation, including obesity,
childhood type 2-diabetes and non-alcoholic fatty liver disease (collectively known as
non-communicable diseases, NCD) further highlights the importance of the finely
balanced development of the immune system (2, 3). Allergic diseases frequently
manifest within the first months of life (4) and this strongly suggests that the developing
immune system is exquisitely sensitive to environmental changes and that events
occurring during pregnancy play a central role in determining immune development.
1.1.1 Immunology of feto-maternal interface
The mammalian fetus can be considered as a highly successful allograft. The placenta is
the immunologically active interface coordinating fetal and maternal immune systems to
coexist during pregnancy (5). The placental cytokine milieu (higher levels of IL-4, and
IL-5) and hormonal environment (progesterone and prostaglandin E2) directs adaptation
of cellular and humoral immune response at the feto-maternal interface to favour
maternal tolerance to allogenic fetal cells (6-8). Furthermore, there is also expansion of
maternal regulatory T-cells (Tregs) during pregnancy, and these are also recruited to the
feto-maternal interface where they orchestrate immune tolerance to the fetus (9). There

2
is accumulating evidence that epigenetic mechanisms (section 1.5.2) provide the
biological mechanisms underpinning the establishment and maintenance of the distinct
immunological environment that is essential for optimal fetal development. As the
primary interface between maternal and fetal circulations, placenta is potentially
exposed to a wide range of environmental factors through maternal exposures. There is
evidence that maternal factors influence the placental function and affect the fetal
immune and metabolic development, indicating the importance of intrauterine
environment in shaping the immune development. (10-12).
1.1.2 Ontogeny of immune development
The fetal immune system was long considered as an immature version of the adult
immune system (13), however, Mold and McCune recently challenged this notion (14).
While many aspects of fetal immune responses are immature, there is now evidence of
more active immune regulation and subtle complexities that are adaptive for the unique
environment of the fetus. Several lines of evidence suggest that development of the
mammalian immune system occurs in distinct layers, with unique functions at different
stages, rather than a simple progression from an ‘immature state’ to a ‘mature state’ in a
linear fashion (14). This “layered immune hypothesis” has been supported by
observations from animal models that revealed the existence of unique waves of
lymphocyte production during fetal and neonatal development. In a chick chimeric
model, it was initially observed that populations of thymocytes with different
characteristics appear in the thymus at different stages of fetal development, completely
replacing earlier populations (15). This suggests that functionally different cell types are
acquired during different stages of development. Subsequently, these observations were
extended to murine models providing further evidence that the immune system develops
in stratified layers (16-18).
In humans, Mold and colleagues recently revealed similar developmental processes in
ontogeny of human haematopoiesis. Notably, they found that fetal T-cells show a
stronger developmental bias towards tolerance compared to adult T-cells. This is likely
to reflect the need to develop an initial broad repertoire of regulatory T-cells that are
tolerant to self-antigens and other newly encountered harmless antigens (19). Thus, a
more tolerogenic fetal immune system provides survival benefit in preventing a
deleterious response to non-inherited maternal alloantigens (NIMA) that can be passed
to the fetus (19, 20). Nevertheless, there is much to be explored as to how and when

3
these developmental variants occur and what triggers the switch from one phase to the
other. It is intriguing to explore whether the tolerogenic properties of fetal immunity
extend beyond self-antigens and NIMA to foreign antigens, since it may have both
beneficial and detrimental consequences. Detailed examination of human
haematopoiesis is beyond the scope of this thesis, however, studies further exploring
fetal immune development will have the capacity to fill the gaps in our understanding of
pathogenesis of NCD including allergic diseases.
1.1.3 Transition of the immune profile from prenatal to postnatal life
The active ‘tolerogenic’ state of the fetal immune system is maintained through a
complex and coordinated network of immune cells (14, 19, 21). While maintaining
immunological ‘quiescent’ state, this active surveillance network also appears to shape
the fetal antigen-specific effector responses to promote fetal survival (22, 23) by
providing an appropriate level of immune defence while limiting excessive effector
responses to self-antigens and NIMA. Fetal dendritic cells (DC) and Tregs provide
signals directing T-helper (Th) cell differentiation along the Type-2 helper (Th2)
pathway, away from potentially destructive Type-1 helper (Th1) responses (21-23).
This Th2 / regulatory-biased immune environment is observed both in the peripheral
immune system (24, 25) as well as in the thymic microenvironment (26) highlighting
the tight and coordinated regulation of activation-induced immune responses at multiple
levels to confer survival benefits.
A large body of data has demonstrated differences in immune function at birth in
neonates who subsequently develop allergic disease, indicating in utero events are
playing a critical role in shaping the fetal immune development (4). Allergy-prone
neonates demonstrate altered functions in adaptive (24), innate (27-29) and Tregs (26,
30) compartments suggesting disease result from disruption to an extensive immune
network rather than from a defect in an isolated cell subset. Reduced capacity for Th1
function and Th2-skewed allergen-specific responses are prominent in high risk
neonates (24). As further discussed below a number of maternal factors such as
maternal dietary nutrients, microbial exposure and exposure to pollutants have shown to
modify immune profiles at birth (31). While the origin and the functional relevance of
specific immune responses to exogenous antigens including allergens during fetal life
are not clear, it highlights, together with recognised associations of maternal exposures

4
with risk of developing allergic disease (2), that in utero exposures can have a greater
impact on immune development.
With the transition to the postnatal environment the developing immune system faces
another series of challenges, as the infant encounters a microbe-rich extra-uterine
environment. The immune system must establish balanced mutualism with colonising
commensals, while maintaining defence against potential pathogens. As such, the
immediate postnatal period is another crucial time for the induction of an expanded
repertoire of specific immunological tolerance to a wider range of nearly encountered
harmless antigens, and a broad repertoire of memory responses to potential pathogens
(32). To tailor these functional requirements of postnatal life, there is a gradual “shift”
of the default responses over the first years of life. One of the most obvious transitions
is from a relatively Th2 dominant default response in the perinatal period, to a more
Th1 dominated responses over the preschool years (24, 27) to eventually achieve the
‘mature’ patterns of response observed in adult life. As part of this process, adaptive
changes can be observed to occur in innate (22, 27, 33, 34) and Treg function (26, 35)
and the patterns of effector T-cell responses (24, 36), to achieve functionally distinct
immune cells that converge to meet the developmental stage-appropriate requirements.
Early interaction with the microbial environment is arguably the most critical factor
driving normal maturation of the immune system in favour of increasing Th1 propensity
as well as mature regulatory function (1). The likely importance of microbial exposure
is demonstrated in animal models which show grossly abnormal development of the
intestinal immune systems of mice raised in germ-free environments (37). There is also
evidence in humans that altered patterns of gut microbiota in the early postnatal period
are associated with dysregulated immune development in childhood (38, 39) and that
reduced biodiversity of gut microflora in the first months of life is associated with
subsequent allergic outcomes (32, 38, 39).
In summary, evidence suggests that immune development evolves in distinct functional
waves that serve different purposes at different developmental stages. During this
process, fetal and early postnatal period represent critical periods that are susceptible to
modulation. Nutritional patterns and a plethora of other early environmental factors
influence this immune development and maturation both by modulating the microbiota
and through direct immunomodulating effects. As discussed further below, some of

5
these mediate their effects by modulating the epigenome (described in detail in section
1.7).
1.1.4 Environmental exposures modulate immune responses: the effects of dietary
factors on immune development
Complex environmental and lifestyle changes are implicated in the dramatic increase in
NCD (2). Of these, modern dietary changes are among the most likely environmental
culprits implicated in the rising risk of so many diverse NCD (3, 40). Although there are
wide regional variations, many modern diets typically contain more processed and
synthetic foods rich in fats and refined carbohydrates with lower amounts of fibre, fresh
fish, fruits and vegetables compared to more traditional diets. These changes have been
associated with changes in the gut microbiome, metabolic responses and immune
function - all of which may contribute to chronic low-grade inflammation and altered
homeostatic mechanisms which are common risk factors for virtually all NCD.
Epidemiological studies have revealed how favourable dietary patterns, such as the
Mediterranean diet in pregnancy and in early childhood can have a protective effect on
persistent wheeze and atopy in children (41, 42). This dietary pattern is also associated
with reduced risk of diabetes (43), cardiovascular disease, some cancers and other NCD
(44). Notably, these beneficial effects reflect composite dietary patterns, and are
difficult to attribute to a single dietary element. On the other hand, there are also a
number of studies that have taken a ‘component’ approach to demonstrate the specific
immunomodulatory properties of individual dietary components such as vitamins,
minerals, short chain fatty acids (SCFA), and long-chain polyunsaturated fatty acids
(LCPUFA) (40). A key aim of this thesis was to explore the role of key dietary
components identified as risk factors in these studies, and to examine their potential
effects on the developing epigenome.
The local tissue microenvironment is an important determinant of immune
development. Changes in the local tissue milieu can modify the pattern of effector
response (45). Environmental factors which modify the local microenvironment,
therefore, have significant potential to alter immune development and the propensity for
inflammation. Of these, a range of dietary factors (such as LCPUFA and antioxidants)
have recognised effects on tissue milieu as a result of their influence as metabolic
components, substrates or structural components of cells and tissues, with downstream
effects on gene expression through a number of different pathways (46, 47). LCPUFA

6
are good examples of dietary factors that have multiple metabolic, immune and
structural functions that influence the propensity for inflammation. As structural
components of cell membranes, they influence membrane fluidity and cell signalling
(48). As substrates for prostanoid production, they influence the level of inflammatory
prostaglandins (49) and as substrates for resolvins they influence the local control of
inflammation (50). Animal studies demonstrate that dietary modulation with LCPUFA
(a combination of omega-3; n-3, and omega-6; n-6, PUFA) can change the local
production of immunomodulatory factors by the skin keratinocytes including IL-10 and
thymic stromal lymphopoietin (TSLP) thereby influencing skin inflammation (51). In
humans, it has been demonstrated that high dose n-3 PUFA supplementation in
pregnancy can modulate fetal oxidative stress (52), leukotriene metabolism (53) with
associated effects on immune function in cord blood (54). Clinical trials in pregnancy
and during the early postnatal period using fish oil supplementation have shown that
these immunomodulatory properties of n-3 PUFA have been associated with a reduction
in some allergic outcomes suggesting that the biological effects have clinical relevance
(54-57). This adds credence to the epidemiological observations that reduced intake of
n-3 PUFA and increased intake of nutrients rich in n-6 PUFA such as margarine has
shown to be associated with the rise in allergic disease (58-60). Section 1.7.1.2 of this
thesis explores the role of n-3 PUFA supplementation during pregnancy and its effects
on the epigenome during fetal immune development.
Antioxidants (such as selenium, zinc, vitamin C and vitamin E) are other examples of
dietary nutrients that have immunomodulatory effects potentially through changes in the
local tissue milieu. Studies using isolated human cells have shown that by favourably
altering the ‘redox’ status of cells, antioxidants can enhance IL-12 production by
antigen presenting cells (APC) to promote Th1 differentiation (61), although it is not
clear if this can be extrapolated to the in vivo setting. Observational studies suggest that
higher dietary intakes of antioxidant rich foods (such as fresh fruits and vegetables) or
higher antioxidant levels measured in pregnancy (62, 63) and early childhood (64, 65)
may reduce the risk of wheezing, asthma and/or eczema. As yet, there are no
intervention studies in early life to directly examine potential preventive effects. In part
this has been because there are two contrary hypotheses around the role of dietary
antioxidant intake and immune outcomes (66). While antioxidants have been suggested
to protect against allergic disease, there has also been conjecture that oxidative stress,
which increases the production of reactive oxygen species by macrophages could favour

7
Th1 immune differentiation. This alternative hypothesis proposes a theoretical concern
that antioxidant supplementation could increase the probability of Th2 differentiation
(by inhibiting oxidative stress) and favour the development of asthma and allergic
disease (67).
Changing dietary patterns are also implicated in altered microbiota. While this was
initially attributed to more ‘hygienic environments’, antibiotic use (68, 69) and reduced
exposure to infectious agents (70), it is now recognised that dietary profiles are also a
major determinant of the gut microbiome and biodiversity (71). There is now good
evidence that high-fat, low-fibre diets can alter the gut microbiome promoting low-
grade chronic inflammation (72, 73). Experimental mouse models have been used to
demonstrate the importance of neonatal colonisation with a diversified intestinal
microbiota for successful induction of oral tolerance to ovalbumin (OVA) (74, 75).
They also demonstrate the beneficial effects of dietary fibre and SCFA which are by-
products of fermentation of dietary fibre, in modulating the microbiome and anti-
inflammatory effects (76-78). In humans, use of prebiotic fibre to promote more
favourable colonisation in infancy has been associated with demonstrable reduction in
allergic disease (77, 79), underscoring the role of dietary nutrients in modulating
immune development through effects on the intestinal microbiota.
There are also interactions between metabolic and immune pathways and the gut
microbiota. The changes in gut immune regulation implicated in the rise in allergy and
autoimmunity (38, 39) have also been linked to the rise in obesity and metabolic disease
(80, 81). The changes in the gut microbiome induced by a high-fat, low-fibre diets have
been associated with changes in gut permeability, higher systemic antigenic load (low
grade endotoxaemia) and higher serum levels of inflammatory cytokines (72, 73).
Animal studies demonstrate how gut microbiota can regulate expression of genes that
affect fatty acid oxidation and fat deposition in host adipocytes (82). In humans, obese
individuals have documented differences in gut microbiota compared to lean
individuals, suggesting a role of gut microbiota in maintaining host metabolic
homeostasis (83). These observations collectively highlight the complex interplay
between metabolic and immune programming in the gut and provide new perspectives
on how diet-induced alterations to gut microbiome can have multisystem effects.

8
A range of other specific nutrients have also been implicated in the rise of allergic
conditions and many other inflammatory diseases (40). Some of these, such as vitamin
D, have recognised effects on immune function, as well as epidemiological associations
with allergic disease and other NCD (84). Others, such as folate, have been of particular
interest because of epigenetic effects on immune programming in animal models
(below). Collectively these studies underscore the importance of early events during
immune programming and how dietary factors can influence these processes. This is a
key theme of the current thesis, and is explored in detail in chapters 3, 4 and 5.
1.2 Developmental programing and the ‘Developmental Origin of Health and
Disease (DOHaD)’ hypothesis
Extensive data from both human and animal studies indicate that mammalian
development involves a number of periods of critical “programing” that shapes
physiologic and metabolic functions in ways that determines adaptive responses and
resilience to subsequent challenges, and susceptibility to disease in later life (85). While
these adaptations may be to promote optimal development, or ensure survival under
adverse uterine conditions, some of these effects may have deleterious effects on long-
term health increasing vulnerability to disease in later life (86).
Barker and colleagues were the first to highlight this phenomenon by demonstrating
nutritional patterns in early life are linked to the risk of cardio-vascular and metabolic
diseases many decades later (86, 87). Barker referred this to the ‘fetal origins of chronic
diseases’ hypothesis (87) but it is now recognised that these influences extend before
conception and also developmental plasticity continues into the postnatal period, as
reflected in the now broader nomenclature of the “Developmental Origin of Health and
Disease’ (DOHaD) (88). The notions of DOHaD established through this new field of
research, have led to more cross-disciplinary approaches to investigate mechanisms
underpinning developmental programming of many inter-related organ systems, which
are influenced by many common risk factors in early life (87, 89).
The original observations that formed the basis of the DOHaD hypothesis were centred
around the effects of maternal under-nutrition at critical stages in early life, leading to
persisting physiological adaptation as well as growth patterns. Barker’s observation led
to the realisation that some consequences may only become apparent later in adult life.
Specifically, a series of observational studies conducted in United Kingdom found that

9
low birth weight, as a crude marker of fetal under-nutrition was associated with greater
risk of heart disease, stroke, hypertension and diabetes in later life (87). These findings
have been since supported by animal experiments using dietary manipulation to
replicate the effects of a wide variety of early risk factors associated with metabolic and
cardiovascular disease (90-93). While DOHaD hypothesis has provided logical
explanation for the growing epidemiological literature on associations between early-
life exposures and later health status, “predictive adaptive response’ hypothesis that was
proposed by Bateson and Gluckman (94) has attracted attention of the epidemiologists
working in the DOHaD field. It posits that the developing organism have the capacity
to receive the information about the quality of the external environment, and in
response, formulate predictions as to future ecological conditions. When the predicted
and actual environments differ, the mismatch between the individual's phenotype and
the conditions in which it finds itself can have adverse consequences for later health.
Observations in animals have supported this model (95), however, it is not clear how
relevant is this in humans.
More recently, chronic-low grade inflammation has been recognised as a key element in
both the initiation and perpetuation of cardiovascular, metabolic and immune diseases.
This has driven interest in early environmental factors that may determine the dynamics
of inflammation in adulthood. In this context, McDade and colleagues found that low
birth weight, in addition to effects on metabolic dysregulation, is also an independent
risk factor for elevated baseline C-reactive protein (as a measure of low-grade systemic
inflammation) in adulthood (96). This suggested early effects on immune development
as well as metabolic pathways, providing additional mechanisms for the observed
associations between adverse early life conditions and increased incidence of
cardiovascular and other NCD in later life. Chronic low-grade inflammation as a
characteristic feature of virtually all NCD, also highlights the central role of the immune
system in the risk and pathogenesis of NCD, and further underscores the need to
investigate early life origin of immune disease and immune dysregulation.
In this context, the recent epidemic of allergic diseases takes on new significance.
Allergic conditions are typically the earliest onset NCD often presenting within the first
months of life (4), as a clear early indicator of the rising propensity for inflammation
and immune dysregulation and the very early vulnerability to modern environmental
changes. Many studies now show significant differences in the neonatal immune

10
function of children who subsequently develop allergy, adding to evidence that allergic
disease also has origins in utero. This is reflected in well-documented differences in
adaptive (24, 97-99) and innate immune compartments (27) as well as regulatory
networks (10, 26) in allergic children at birth. Many of these studies suggest relative
immaturities of T-cell function compared to non-allergic children. Consistent with this,
in a recent study, T-cell signalling pathways showed to be differently expressed at birth
between allergic and non-allergic children with marked down regulation of NF-кB
genes in allergic individuals (100). Furthermore, many risk factors for other NCD
(microbial exposures, diet and pollutants), also have been associated with differences in
immune function at birth, also suggesting in utero effects (40, 70, 101-103).
Thus, the initial observations of Barker and colleagues, together with many subsequent
studies clearly indicate that a broad range of inflammatory diseases have their origins in
fetal life. It is also evident that events in utero and the early postnatal period are
important in determining the propensity for inflammation that contributes to the
subsequent disease risk. This relatively new field of medicine holds much promise for
explaining the rise of NCD. However, many key questions remain. To what extent do
transient exposures really result in long-term adverse outcomes in the offspring? Which
exposures are important and what is the mechanism through which their effects are
exerted? This thesis aims to contribute to this area by examining effects of specific
nutritional exposures on early gene expression in humans using cutting edge epigenetic
profiling technologies.
1.3 Gene-environment interactions in determining disease risk
Despite the extensive body of data linking modern lifestyles to chronic inflammatory
diseases, not all individuals living in modern environments acquire these conditions.
One proposed explanation for this suggests genetic factors are important for
determining an individual’s susceptibility profile. The ‘gene-environment’ interaction
paradigm is a longstanding model through which to understand complex diseases. This
paradigm supposes “a different effect of an environmental exposure on disease risk in
persons of different genotypes” (104). Following the completion of the human genome
project in 2003, a decade of genome-wide association studies (GWAS) studies sought to
identify disease-causing variants that might serve as predictive markers or preventive
targets. However, despite a large investment in human population genetics, for most
complex diseases, these endeavours have failed to attribute any substantive proportion

11
of risk to genetics, or consistent gene-environment interactions (105-107). Moreover, a
genetic-based model cannot sufficiently account for the rapid cumulative rise in many
immune-related diseases since, in evolutionary terms, emergence of genetic traits
conferring disease risk may take many generations, much more than the documented
time frame for escalating rise in NCD. Therefore, the focus has been in identifying
mechanisms that have the potential to regulate expression of genes, independently of the
primary DNA sequence, that interact with the changing environment.
The emerging field of epigenetics provides a new frontier for understanding how
environment influences the expression of genes. Unlike DNA marks, epigenetic marks
encode information from both the inherited genotype and environmental exposures and
thus represent a promising approach to explain pathogenesis of complex diseases.
1.4 Epigenetics: what is it?
Epigenetic processes are key determinants of gene expression, and as such they now
appear to be the key mediators of gene-environment interactions, providing new
dimensions of interest in epigenetic mechanisms of disease risk.
Epigenetics can be broadly defined as a series of complex, dynamic and interconnected
biological processes that regulate the expression of genes, to produce changes in cellular
function from a constant and unchanging underlying DNA sequence (108). This allows
the extensive cellular diversity within organisms and allows the functional flexibility in
response to changes in internal and external conditions. New insights into the
mechanisms of how gene expression can be altered by a range of environmental factors
has become the cornerstone of the DOHaD paradigm (109). These epigenetic processes
include DNA methylation, post-translational modification to histone tails and chromatin
remodelling factors and non-coding RNAs (ncRNA). Modification of gene expression
through these epigenetic processes controls a vast array of biological functions such as
cell differentiation, genomic imprinting and aging. Furthermore, information stored in
the epigenetic “code” can be propagated through mitosis and meiosis (110). Thus,
epigenetic information is not only maintained in daughter cells throughout life, but also
has the potential to be passed on to subsequent generations. The latter, also known as
epigenetic inheritance has been demonstrated in plants, in which a defined methylation
pattern has shown to propagate for many generations (111) and also in animals (112)
however, the extent and mechanisms of such inheritance in humans is yet not clear.

12
1.4.1 Molecular mechanisms of epigenetics
Eukaryotic genomes are packaged into chromatin, whose basic repeating unit is a
nucleosome that consists of a histone octamer wrapped around 147 base pairs (bp) of
DNA (Figure 1.1). The functional outcome of the genetic code largely depends on the
accessibility of the DNA to transcriptional machinery. Epigenetics provide a mechanism
by which the access to DNA is regulated resulting in either transcriptional activation or
repression of the gene. Through these mechanisms, over 200 cell types that comprise
the human body can be specified from a single genetic code. The epigenetic processes
that modulate the access to DNA in response to upstream signals include DNA
methylation, covalent modification of histones and chromatin interaction with
regulatory ncRNA and are described in detail in the following sections.
Figure 1.1 Schematic representation of the organisation and packaging of
nucleosome.
1.4.2 DNA methylation
DNA methylation in mammals, the most widely studied epigenetic process, consists of
transfer of a methyl moiety from S-adenosylmethionine (SAM) to the 5-position of the
cytosine residues in certain CpG (cytosine followed by a guanine, linked together by a
phosphate) dinucleotides. This transfer reaction is catalysed by DNA
methyltransferases (DNMT) that include family members DNMT1, DNMT2 and
DNMT3. The addition of methyl groups to DNA generally induces transcriptional
silencing by blocking the binding of transcription factors and/ or through promoting the
binding of methyl CpG-binding domain (MBD) proteins, which recognise 5-methyl
cytosine (5-mC) on DNA. Such proteins bind to 5-mC, which in turn recruits histone-
modifying complexes to the site resulting in a closed chromatin structure and
transcriptional silencing (113). Recently, Severin et al reported that DNA methylation
may also regulate gene expression through altering mechanical properties of DNA

13
including flexibility of DNA which is important for DNA to wrap around histones,
hence accessibility of DNA (114).
CpG dinucleotides are unevenly distributed throughout the genome. In general, CpG are
under-represented in mammalian genome due to evolutionary spontaneous deamination
of 5-mC to thymine (115). However, certain regions of DNA contain a high density of
CpG and are referred to CpG islands. They often span sites of transcriptional initiation.
Typically 60-70% of annotated gene promoters are associated with a CpG island,
including most house-keeping genes. These CpG islands are mostly unmethylated
favouring a constant expression of these important genes. In contrast, the majority of
non-CpG island sites are methylated, such as repetitive sequences and mobile elements
ensuring genome stability through silencing these transposable elements (116).
However, new data suggest that methylation is not exclusive to CpG sites as previously
thought, but also occurs at non CpG sites such as CpA (cytosine followed by an
adenosine) and CpT (cytosine followed by thymine) (117, 118).
DNMT1 family is important for establishment and regulation of tissue-specific patterns
of methylated cytosine residues. This enzyme copies the pre-existing methyl marks on
to the daughter strands during DNA replication to maintain the established methylation
patterns across successive cell divisions and given the name “maintenance
methyltransferases” since they add a methyl group to DNA of the unmethylated strand
when the other strand is already methylated (hemimethylated) (119). DNMT3 family is
different to DNMT1 such that it is mainly responsible for de novo methylation and is
abundantly expressed in early embryo. This family of DNMT include, DNMT3A and
DNMT3B and one regulatory factor DNMT3-like protein (DNMT3L), which has some
structural similarities and differences to the rest of DNMT3 proteins. DNMT3 is
important during embryogenesis to establish de novo methylation marks in the zygote
that were lost globally shortly after fertilisation (120).
DNA methylation is a relatively stable epigenetic mark established primarily during cell
differentiation. Each cell type in the mammals contains a unique DNA methylation
signature that confers cell-specific functions. Despite these being very long-lived
modifications, DNA methylation is also dynamic and responsive to environmental
factors, conferring phenotypic plasticity among related cell types (121). Removal of
methyl groups (DNA demethylation) is an active process mediated by ten-eleven

14
translocation proteins (TET) proteins that remove or hydrolyse the methyl group from
5-mC (122). Also, passive DNA demethylation has been observed during successive
rounds of replication in the absence of functional DNA methylation maintenance
machinery. Animal experiments have provided evidence that 5-hemimethyl cytosine (5-
hmC) may not merely be an intermediate in the demethylation pathway but may have a
distinct epigenetic role (123). However, the exact mechanism and functional
consequences of TET-mediated DNA demethylation remain to be clarified.
The work of this thesis involves genome-wide profiling of DNA methylation patterns in
neonatal immune cells to identify areas of methylome that are sensitive to early
environmental cues, and that may modulate immune development.
1.4.3 Histone modifications
Chromatin is a dynamic structure that responds to upstream signals to regulate access to
DNA. DNA is organised into nucleosome subunits of packaged DNA via histone
proteins. The nucleosome core consists of DNA wound around an octamer of 2 copies
each of the core histone proteins H2A, H2B, H3 and H4 (Figure 1.1). Unstructured tail
domains of histones are the sites where modifications take place. These modifications
represent major pathways in gene transcription, replication and repair. The structure of
chromatin can be altered in several ways including, covalent modifications of histone
tails, replacement of core histones with specialized variants (124) and repositioning or
eviction of histones from DNA by ATP-dependent chromatin remodelling enzymes
(125). Of these, modifications to histone tails has been extensively studied given the
fact that such modifications correlate well with the transcriptional activity of genes and
a large number of modifications have been described to date (126).
Acetylation of histone lysine tail residues carried out by histone acetyltransferases
(HAT) facilitates gene transcription. To describe how acetylation could favour gene
expression, it has been proposed that acetylation neutralises the positive charge of lysine
residues, weakening charge-dependent interactions between a histone and nucleosomal
DNA, linker DNA and/or adjacent histones, thereby increasing the accessibility of DNA
to the transcription machinery (127). Recent data show, in addition to direct influence
on increased accessibility of DNA, histone acetylation could favour binding of protein
modules that regulate transcription and chromatin dynamics (128). Advent of such
histone binding proteins may provide opportunities to discover molecules/particles that

15
have capacity to interact with histones to modify the protein-histone interactions to
influence the gene expression.
Unlike acetylation of histone lysine residues that are associated with transcriptional
activation, lysine methylation can be associated with either transcriptional activation
(H3K4me3 and H3K36me3) or repression (H3K9me3 and H3K27me3). Histone
methylation inhibits recruitment of polycomb repressive complexes, a family of
polycomb group proteins that through their enzymatic activity initiate and maintain
transcriptional silencing. Interaction of polycomb repressive complexes with methylated
lysine residues renders chromatin refractory to transcriptional repression (129). In
contrast, histones that are associated with repressive methylation marks show increased
affinity to certain DNA binding proteins including polycomb repressive complexes that
promote gene silencing (130). Other covalent histone modifications including
phosphorylation, ribosylation, glycosylation and ubiquitylation have been described,
however, we still know relatively little about their specific role in relation to
transcription and their relevance to biological processes.
Analysis of acetylation status of histones gives invaluable information on epigenetic
regulation of gene expression. Comparison of acetylation marks between healthy and
disease status further enables discovery of causal pathways and targets for therapeutics.
This project also analyses the acetylation status of histones that are associated with
known-immune genes.
1.4.4 Regulation by non-coding RNA (ncRNA)
There is accumulating evidence that ncRNA such as microRNA (miRNA), small
interfering RNA, and long ncRNA represent an important class of newly recognised
regulatory molecules (131, 132). Of these, miRNA has been the interest for their
association, when dysregulated, with progression of human diseases. MiRNA are small
non-coding single stranded RNA molecules of 19-25 nucleotides in length and have
shown to control major cellular processes, including metabolism, apoptosis,
differentiation and development (133). Regulation by miRNA, often negative on target
genes, is achieved in one of two ways; miRNA can cleave target mRNA transcripts by a
miRNA-associated RNA-induced silencing complex (miRISC) or miRNA binding to
sites within the 3′ untranslated regions (UTR) of target mRNA, thus inhibiting the
initiation of translation via the miRISC (134, 135). Subsets of miRNAs are thought to

16
act as tumour suppressor genes or oncogenes, and their dysregulation is a common
feature of human cancers (136). There are some fascinating features of biogenesis and
regulation of miRNA. A cluster of miRNA may be controlled by one promoter, but one
miRNA may be encoded by multiple pre-miRNA. Also, one miRNA may target
multiple genes, and one gene may be targeted by multiple miRNAs (137, 138). Because
miRNA directly regulate protein expression without transcription, miRNA may respond
more rapidly than other epigenetic regulators that require gene transcription in addition
to protein translation. Epigenetic modulation by ncRNA is becoming increasingly
recognised as a significant processes in disease development, however, it does not fall
within the main scope of this thesis.
1.4.5 Chromatin remodelling complexes
A group of chromatin remodelling complexes have been described that physically alter
chromatin structure, thereby influencing the function of the chromatin (139). These
complexes contain a central ATPase component that harnesses ATP hydrolysis to
physically remove or slide histones from DNA. The chromatin-remodelling complexes
are categorized into four distinct groups: ISWI (Imitation SWItch), INO80/SWR1
(INOsitol requiring/ Sick With Rat8 ts), CHD (chromodomain helicase DNA binding
protein) and SWI/SNF (SWItch/Sucrose Non- Fermentable). ISWI complex, the best
characterized chromatin remodelling complex, has shown to affect the DNA repair,
DNA replication and transcriptional regulation (repression/activation) through
interactions with transcription factors, co-regulators or components of the transcription
machinery (139).
The list of epigenetic mechanisms being discovered continues to grow. However, DNA
methylation and covalent modifications of histones have so far been a central focus in
epigenetic research. While different epigenetic mechanisms follow different pathways
to regulate gene expression, they tend to be interrelated as part of a coordinated
regulation of chromatin dynamics.
1.5 Functional implications of epigenetic dynamics
The phenotypic outcome of a particular epigenetic process depends on the biological
pathway/pathways they are linked to. There is increasing evidence that epigenetic
regulation continues to regulate across the whole life span, from the first moments of

17
life until death. The following sections focus on epigenetic control involved in
development and immune maturation.
1.5.1 Epigenetic regulation during embryo development and haematopoietic
development
The erasure and re-establishment of DNA methylation patterns during mammalian
embryonic development demonstrates the dynamic nature of epigenetic regulation.
Remarkable changes to the epigenetic signature occur naturally during this period,
particularly when gametes fuse to form the zygote, and when gamete precursors, the
primordial germ cells (PGC) develop and migrate in the embryo (140).
Initially, in the zygote, the paternal and maternal genomes remain physically separated
and undergo different epigenetic reprogramming. The paternal genome is subjected to
both histone modifications and global DNA demethylation in the first cell of the zygote.
These changes are evident at a later stage in the maternal genome (in preimplantation
embryo) causing early asymmetry of the two epigenomes (141, 142). This epigenetic
asymmetry appears to be essential for normal embryonic development (143). Although
demethylation appears to be widespread across the genome, some genomic sequences
such as centromeric heterochromatin, imprinted loci and some retrotransposons escape
DNA demethylation during this preimplantation development (144, 145). These loci
being resistant to demethylation prevents activation of the silenced genes otherwise be
deleterious to the developing embryo. Sequence specific-DNA binding factors
cooperate with histone modifying machinery to protect these sites from loss of 5-mC.
Zfp57 and Trim 28, which are essential for maintaining methylation at imprinted loci
recruit DNMT and histone modifiers to maintain the methylation signature at imprinted
loci (146-148).
As the cleavage divisions progress, totipotency is lost and differentiation process
commences coinciding with the specification of the first cell lineages (149). This
process is largely accomplished by up-regulation of the de novo methylases, DNMT3a
and DNMT3b (150). The resulting new pattern of DNA methylation is then maintained
throughout subsequent cell divisions. The gene-specific epigenetic reprogramming is
essential to lock in gene expression programmes that specify cell fate. After
implantation, de novo methylation also ensures X-chromosome inactivation in female

18
embryos. Random X-chromosome inactivation involves chromosome-wide changes
silencing genes in the chosen allele (151, 152).
A second wave of epigenetic reprograming occurs at the stage of germ cell
development. The cells that are already specified to become PGC show first sign of
reprogramming during their migration to the genital ridges of the developing embryo
(153). Then they undergo extensive and rapid loss of 5-mC including demethylation of
the imprinted genes when they enter the gonads (154). In addition to DNA
demethylation, changes to histone proteins have also been documented. During their
migration PGC undergo a reciprocal loss of H3K9me2 and increase in H3K27me3
(155). Nevertheless, whether the histone modifications initiate the DNA demethylation
or these are two parallel epigenetic processes is not clearly known.
Through this process PGC genomes are ‘wiped clean’ of most epigenetic marks
associated with somatic gene regulation to acquire the pleuripotential capacity of the
germ cell-specific signature. During the reprogramming period PGC lose parent-of-
origin-specific differential methylation marks at imprinted loci and assume imprinting
marks appropriate to their own sex. While this erasure of epigenetic marks occur in a
similar time frame in males and females, reestablishment program diverge according to
sex. For example, in males, de novo methylation of germ cells in fetal testis is largely
completed at birth (156, 157). In contrast, in females, imprints in primary oocytes are
quiescent until they enter the growth phase in the postnatal period (158, 159).
Collectively, data points to the importance of epigenetic mechanisms in controlling
critical developmental processes during early phases of embryogenesis and disruption
of this regulation may have detrimental consequences for development.
Haematopoietic cell development is another key process governed by the epigenetic
machinery during fetal life. Epigenetic mechanisms regulate the development of
haematopoietic stem cells (HSC), myeloid and lymphoid progenitors (160, 161). Both
DNA methylation and histone modifications governed by polycomb repressor
complexes play a significant role in these processes (162, 163). There are other
examples of how epigenetic processes govern the fate of progenitors as they
differentiate into various cell types (164-166). Given the important role of epigenetic
mechanisms in governing the ordered process of hematopoietic development, there is
speculation that disruption to these processes may alter the function of key immune cell

19
types, and their resulting response capacity. In the present study we have focused on
leukocyte methylation profiles as they may harbour epigenetic variants relevant to
immune function.
1.5.2 Epigenetic profile of the neonatal immune system and epigenetic changes
during postnatal development
Epigenetic mechanisms play an important role in maintaining the unique immune
profile at the feto-maternal interface, supporting and facilitating successful pregnancy
through the distinct immune profile characterised by a Th2 dominant cytokine milieu. In
particular, hypomethylation of Fork head box protein 3 (FOXP3) in the placental tissue
(10) and specific DNA methylation profile of macrophages (167) are important for
maintaining an anti-inflammatory environment at the feto-maternal interface.
It is now also evident that epigenetic mechanisms play a crucial role in directing
neonatal responses, controlling the expression of key cytokine genes in Th cells towards
Th2-biased neonatal responses, relative to the more Th1-dominant responses of adults
to the same stimuli (24). CD4+ T-cells are central to these effector immune responses
and many epigenetic mechanisms converge to build a unique epigenetic profile in
neonatal CD4+ T-cells to meet the functional requirements of this critical developmental
stage (168-170). In addition to epigenetic differences in CD4+ T-cell compartment,
epigenetic changes also underlie the functional differences in innate immune cells such
as DC (171, 172) and monocytes (173). A previous study in my host laboratory
characterised the longitudinal changes in DNA methylation of mononuclear cells (MC),
revealing significant methylation changes in genes associated with immune pathways
from birth to 5 years of life (174). Collectively, these observations suggest that
epigenetic mechanisms make an important contribution in the regulation of immune
maturation.
Collectively, these studies provide an epigenetic basis for the significant reprogramming
of immune function required as the fetus transits from the uterine environment, where
immune tolerance is key to survival, to the extra uterine environmental where defence
against potential infectious threats becomes increasingly important.

20
1.5.3 Evidence of disruption of epigenetic pathways in immune diseases
An increasing number of studies show that epigenetic variations are associated with
many chronic diseases such as asthma and allergic diseases. Such association studies are
helpful to identify potential biomarkers or targets for therapy. Analysis of DNA
methylation profiles of patients with seasonal allergic rhinitis was able to clearly
distinguish patients from healthy controls indicating the potential of epigenetic analysis
to identify methylation marks as biomarkers in a clinical setting (175). Epigenetic marks
on disease-associated genes also vary with severity of the disease or the response to
treatment indicating the diversified benefits of epigenetic analysis in a broad spectrum
of clinical scenarios (176-178).
Genome-wide epigenetic profiling, when combined with pathway analysis, provides
information regarding potential biological pathways linked to disease pathogenesis. For
an example monocytes (179) and keratinocytes (180) from patients with atopic
dermatitis show aberrant DNA methylation patterns associated with IgE pathway and
TSLP pathway respectively. Further studies on atopic dermatitis revealed that miR-155
was up-regulated in skin lesions indicating disease pathogenesis involves disruption of
many complex and coordinated epigenetic processes (181). A recent study mapping
histone modifications in peripheral blood CD4+ T-cell subsets from asthmatic
individuals identified disease-specific enhancers and enhancers that are associated with
known single nucleotide polymorphisms (SNPs) related to asthma showed significant
changes in histone marks along the Th2 differentiation pathway (182). This is an
example of how epigenetic studies bring new insight into mechanisms of complex
diseases.
Genome-wide or candidate gene-specific case control studies have identified additional
loci that undergo differential epigenetic modifications and are summarised in Table 1.1.
These findings, together support that immune pathways are under epigenetic control and
development of disease parallels disruption of these epigenetic dynamics.
Human observational studies continue to provide a link between aberrant epigenetic
pathways and immune dysregulation, however, it is difficult to determine whether
epigenetic variation is the true cause or simply an effect of disease development in
human studies. Animal experimental work provides direct evidence that key pathways
in immune response are epigenetically regulated and that these are causally related to
21
Table 1.1 Gene loci that show epigenetic modulation in association with immune
diseases
Disease/ Phenotype
Tissue/ cell type
Gene loci associated with immune
disease/pathway
Associated epigenetic changes
Asthma Buccal cells ARG promoter DNA methylation inversely correlated with exhaled nitric oxide (183)
Asthma Treg cells and effector T-cells
FOXP3 and IFNG Increased DNA methylation in asthmatics (184)
Asthma Airway epithelial cells
STAT5A and CRIP1
Differential methylation in asthmatics (185)
Asthma Whole blood IL4R Methylation of cg09791102 site showed an association with asthma (186)
Asthma CBMC ASCL3 Methylation at 5’-CpG island of ASCL3 was associated with asthma (187)
Asthma Whole blood ADRB2 Higher methylation at 5’ UTR of ADRB2 was associated with asthma (188)
Asthma CD19+ B lymphocytes
CYP26A1 Differential methylation in house dust mite allergic asthma (189)
Systemic Lupus Erythrocytosis
CD4+ T-cells NRLP2, CD300LB and S1PR3
Differential DNA methylation profiles correlated with clinical phenotype (190)
disease pathogenesis. Mice deficient in Th2 locus controlling region in CD4+ T-cells
show a loss of general histone H3 acetylation and histone H3-K4 methylation, and
demethylation of DNA in the Th2 cytokine locus (191). In a mouse model of allergic
asthma this is associated with a marked reduction in eosinophil and lymphocyte
recruitment in the airways, decreased serum IgE levels and lung inflammation when
challenged with ovalbumin (191). Another murine study demonstrated that allergen
challenge is associated with an increase in DNA methylation at interferon gamma
(IFNG) promoter in CD4+ T-cells, with concomitant decrease in IFN-γ production
(192). This study also provided evidence that pharmacological treatment can reverse
these epigenetic changes. Others have shown that silencing of the IFNG gene may also
involve histone modifications at this locus (193). Specifically, in a mouse model of
Th2-driven allergic asthma, the chemical inhibition or loss of SUV39H1, a histone
methylase that trimethylates H3K9, skews T-cell response towards Th1 response and

22
decreases lung pathology (193). Several other animal experiments also show that
pathogenesis of allergic airway inflammation involves pathways regulated at the level
of miRNA (194-196).
Thus, epigenetics plays a significant role in regulating many important biological
processes. In particular, animal models provide convincing evidence that disruption of
epigenetically regulated pathways are causally related to immune diseases. In humans
there is also growing evidence that disease states are associated with underlying
dysregulation of functionally relevant epigenetic pathways. There is a recognised need
to understand how environmental risk factors may be driving these underlying
epigenetic changes to promote disease, as targets for intervention.
1.6 Epigenetics as an underlying mechanism of Developmental Origin of Chronic
Diseases
As described in section 1.2 a significant component of the risk of chronic disease is
conditioned during initial programming of physiological and metabolic responses to
environmental cues perceived during critical stages of early development (86, 87, 197).
Evidence both from human observational studies and animal experiments now suggest
epigenetics is the likely mediator of early gene-environment interactions that may
modify the susceptibility to NCD (89, 198). Furthermore, aberrant epigenetic regulation
during fetal period may be implicated in disease pathogenesis, as suggested by detection
of altered disease-associated epigenetic marks at birth (199, 200). Thus, factors that
influence the epigenetic processes during fetal development may alter gene expression
profiles resulting in phenotypic and functional changes that may predispose to disease
risk. Diet, smoking, exposure to microbes and exposure to endocrine disruptors and
other pollutants during pregnancy are some of the maternal factors that are associated
with modulation of the neonatal epigenome and are further discussed below. These
environmental culprits have also been associated with developing immune diseases
indicating the role of epigenetic regulation mediating the environment induced-disease
risk. Understanding the epigenetic effects of maternal exposures help to identify
epigenetic processes amenable to environmental modulation that might serve as
preventive or treatment targets.

23
1.7 Maternal environmental exposures that modulate the neonatal epigenome
The fetus is completely dependent on the mother, for all nutrients. Maternal health and
the maternal environment are of critical importance for all aspects of development, and
a broad range of associated factors have now been identified to affect the fetal
epigenome. As the major focus of the thesis, maternal nutritional factors that modulate
offspring epigenome are discussed in detail in section 1.7.1 and an outline for other
factors known to affect the neonatal epigenome is given in section 1.7.2.
1.7.1 Nutritional exposures in early life and the fetal epigenome
Nutrition is arguably the most influential environmental factor during fetal
development. Prenatal nutrition influences fetal growth and the development of
physiological functions of all organ systems (92, 201-205). Similarly, postnatal
nutritional exposures are critical for the ongoing developmental maturation of many
organ systems and for optimal physiological functions (203, 206).
While it has been recognised for some time that nutritional changes could modify
patterns of gene expression with lasting influences on phenotype and function, it has
been only relatively recently identified that epigenetic mechanisms play a key role in
this process. External environmental pressures such as dietary exposures can lead to
subtle variations in epigenetic regulation of immune gene expression, which can
potentially lead to more profound effects on subsequent immune function, clinical
phenotype and disease risk. There are now good examples in animal models (93, 207)
of how dietary changes can directly influence the epigenetic machinery by inhibiting the
enzymes that catalyse DNA methylation or histone modifications or by altering the
availability of substrates necessary for enzymatic reactions. These alterations in
epigenetic marks lead to either enhanced or supressed gene expression with an altered
phenotype depending on the nature of the affected biological pathways (108). Mathers
has described the sequence of events that link nutritional exposures to later health
outcomes through epigenomic modifications, by the concept of four ‘Rs’: nutritional
factors are Received and Recorded by the epigenome, evidence of these exposures are
Remembered across successive cell generations and the consequences of these
exposures are Revealed as altered gene expression, cell function and ultimately, health
(208). This concept reiterates that fetal epigenome is sensitive to nutritional exposures,
thus can be modulated leading to an altered phenotype.

24
Using a mouse model, Li and colleagues showed that early postnatal over-nutrition
leads to a reduction in spontaneous physical activity in female mice that sustained into
adulthood (209). These changes were associated with extensive changes in
hypothalamic DNA that was also persisted into adulthood, providing potential
mechanistic basis for persisted physiological effects. Similarly, prenatal exposure to
famine (under-nutrition) has also been associated with persistent epigenetic differences
in humans. Individuals who were prenatally exposed to Dutch Hunger Winter in 1944-
45 had, 6 decades later, less DNA methylation of the imprinted insulin-like growth
factor-2 (IGF2) gene compared to their unexposed, same-sex siblings (210). A growing
number of studies have reported that epigenetic marks serve as a memory of exposure in
early life to inadequate or inappropriate nutritional factors as reviewed in (211) and
(212). These exposures may modify a range of epigenetic marks including, DNA
methylation, post-translational histone modifications, chromatin remodelling complexes
and miRNA.
As noted in section 1.1.4, changing patterns of maternal diet also has a major impact on
changing microbial diversity, specifically gut microbiome with effects on both maternal
immune and metabolic homeostasis with consequences for fetal immune and metabolic
development. It is intriguing to speculate that developmental influences associated with
altered gut microbiome may be induced by effects on host epigenetic machinery. This is
based on the observations in animal models that gut microbiota can directly modulate
host immune gene expression through epigenetic changes. (213-215). The murine
experiments show that microbial short chain fatty acid fermentation products regulate
host Treg development through modulation of histones (216, 217). Collectively these
studies suggest that changes in the gut microbiome modulate immune development
potentially through epigenetic effects.
Identifying individual components of the diet which exert epigenetic effects,
importantly affecting fetal developmental programing, is a huge challenge. This thesis
examines two important maternal dietary factors namely folate and n-3 PUFA which
potentially affect developmental programing through epigenetic mechanisms.
1.7.1.1 Folate and other nutrients involved in one-carbon metabolism
As a major focus of this thesis, dietary components such as folate, methionine and
choline are of much interest because of their role in DNA methylation through 1-carbon

25
metabolism (218). Folate is a B vitamin (vitamin B9), and it is needed both to
synthesize DNA – 3 out of the 4 nucleotides needed to synthesize DNA require a one-
carbon that is carried by folate – and the methyl groups that are used to regulate gene
expression that converts cytosine to methylcytosine (also come out of one-carbon
metabolism, or folate metabolism) (Figure 1.2).
Consequently, folate metabolism is intimately tied to the genome by affecting both
genome stability as well as epigenetic marks on DNA. Dietary folate, through a series
of reactions, is processed into metabolic co-factors that carry chemically activated
single carbons, referred to as ‘one-carbon’. In this metabolism, a carbon unit from serine
or glycine is transferred to folate-derived co-factors.. Other nutrients and metabolites
that can provide one-carbon metabolites include choline and its degradation products
betaine, dimethylglycine and sarcosine. Other co-factors such as niacin (vitamin B3),
riboflavin (vitamin B2), vitamin B6 and cobolamin (vitamin B12) are also required to
complete the oxidation cycle of one-carbon to either synthesize nucleotides or to
generate methyl groups. In eukaryotes, folate metabolism is compartmentalized in the
cytoplasm, mitochondria and nucleus (219). While one-carbon metabolism in the
cytoplasm is required for the synthesis of purines and thymidylate and the remethylation
of homocysteine to methionine, mitochondrial events are much complex, with varying
metabolic outcomes depending on the tissue type and the development stage.
Folate is readily available in fresh fruits and vegetables, but destroyed by cooking.
Deficiency of folate is associated with intrauterine growth retardation, preterm or fetal
death, Down syndrome, and neural cleft-related birth defects including neural tube
defects and oro-facial clefting (220-222). Based on evidence that improving folate
status from preconception lowers the risk of neural tube defects, daily folic acid
supplementation 400µg has been recommended for all women planning for pregnancy
(223, 224). Identification of the protective effects of periconceptional folic acid
supplementation against neural tube defects in neonates and the high rates of unintended
pregnancy led to the mandated fortification of flour and other grain products with folic
acid first in the United States and then in several other countries (225). This initiation
has been a success in preventing occurrence of neural tube defects, however, total folate
intake exceeding the recommended limit has been reported among women of child-
bearing age (226, 227).

26
Figure 1.2 Schematic illustration of one carbon metabolism. The diet contains folate monoglutamates and polyglutamates, which need to be hydrolysed to monoglutamtes by glutamate carboxypeptidase II (GCPII). Monoglutamates are subsequently absorbed through the apical membrane of the intestinal epithelium. Folic acid, a synthetic form of folate is more readily absorbed than folate. The monoglutamate form of 5-Me THF is the predominant folate in the circulation and stored in liver until redistributed to other tissues. Uptake of folate by peripheral tissues occur via Reduced Folate Carrier (RFC) and once enter the cell, 5-Me THF acts as a carrier of methyl groups unit in the folate-methionine cycle. Subsequently the methyl groups are utilised to synthesize S-adenosylmethionine (SAM), the universal methyl donor in all tissues.
Previous studies by our group have shown that many women continue to consume
higher than necessary doses of folic acid later in pregnancy, well beyond the window of
risk for neural tube defects (228). There is little evidence of any significant beneficial
effects of folic acid supplementation after the first trimester (229), and some studies
suggest that this may be associated with an increased risk of allergic outcomes
(discussed further below).
There has also been a specific interest in effects of maternal folic acid supplementation
on the immune development of the offspring. This gathered momentum following the
seminal study, by Hollingsworth et al. who demonstrated that maternal folate

27
supplementation induced an allergic phenotype, with associated epigenetic changes, in
the offspring (93). This folate-rich maternal diet induced methylation changes in 82-
gene loci in the offspring, resulting in increased airway hyperesponsiveness, airway
eosinophilia, and production of inflammatory cytokines. This trait was then inherited to
the subsequent F2 generation. Among these genes, RUNX3, a gene known to regulate T-
lymphocyte development and to suppress airways inflammation (230) was
hypomethylated with concordant transcriptional silencing of this gene in the progeny
(93). The effects of nutritional impact on F1 and F2 generation is largely considered as
“parental effects” as the developing embryo and its germline are simultaneously
exposed to environmental signal that may also influence the F1 and F2 epigenomes. The
“epigenetic memory” retained in F1 and F2 generations may translate to phenotypic
variation in later life (231). This is different from true transgenerational epigenetic
inheritance that is seen from F3 generation and beyond who were not exposed to initial
environmental signals that triggered the epigenetic changes.
It is not clear how acquired epigenetic signature successfully pass through epigenetic
reprograming (erasure and re-establishment of epigenetic marks) in the developing
embryo and is difficult to address in human studies, however, elegant animal
experiments provide evidence for transgenerational epigenetic inheritance with
phenotypic consequences. In this regard agouti mouse model in which mice carrying the
viable yellow agouti metastable epiallele (Avy) causing variable expression of agouti
protein in isogenic individuals is an elegant animal model to study the epigenetic
inheritance (231). The variable coat colour of isogenic Avy mice correlates to
methylation status at Avy locus and using this murine model Morgan et al found that the
distribution of phenotypes among offspring is related to the phenotype of the dam that
was inherited through the female germline (231). Studies investigating this model
further observed that offspring born to dams fed a methyl-supplemented diet had brown
(pseudoagouti) rather than yellow coats in parallel with hypermethylation at Avy locus
(232, 233). Of most importance maternal nutritional change not only influence their
immediate offspring’s coat colour, but the effect was augmented throughout subsequent
five generations without a quantitative change in the exposure i.e. with the exact same
diet (231). Transgenerational amplification of environmental effects through changes in
the epigenome may have important health implications and provide a new hypothesis
that a stable environmental exposure could lead to a persistently increase in related
disorders such as allergic diseases.

28
Subsequent observational studies in humans have also linked folate intake with a higher
risk of allergic diseases (228, 234, 235) although these findings have not been entirely
consistent (236, 237). Several human studies reported an association of altered DNA
methylation in offspring born to mothers with folic acid supplementation supporting the
findings of the animal studies. Steegers-Theunissen et al (238) found a positive
correlation between periconceptional maternal folic acid supplementation and infant
DNA methylation of IGF2 differentially methylated region (DMR). While a lower
DNA methylation of H19 DMR has also been reported in association with higher
maternal folic acid intake during pregnancy (239), other studies have found no
association between either maternal folate intake or cord folate levels and DNA
methylation of IGF2 DMR (239, 240) or specific promoter regions of IGF2 (241) or
LINE1 (242, 243). Interestingly, Fryer et al (244) analysed the CpG dinucleotide
methylation in 12 cord blood samples using a genome-wide methylation profiling and
found an association between cord plasma homocysteine levels (inversely correlated
with serum folate) and methylation patterns, with 298 CpG sites showing significant
associations with plasma homocysteine level. Chang and colleagues investigated the
influence of maternal serum folate on DNA methylation in 18-28 weeks old fetuses
following termination of pregnancy for fetal anomalies (245). They found a positive
correlation between the levels of maternal serum folate and DNA methylation in brain
tissue but not in other tissues indicating that exposure to folic acid during pregnancy
may have tissue-specific effects in the developing offspring.
Studies investigating underlying epigenetic mechanisms of in utero folate exposure on
offspring effects have mostly focused on genomic regions surrounding imprinted genes
and only a few human studies have used a genome-wide approach in mapping DNA
methylation changes at birth in relation to variation in folate exposure. The folic acid
pathway plays a critical role in fetal development yet its effects on the epigenome and
gene expression is incompletely understood. This study investigates the genome-wide
effects of in utero folate exposure on DNA methylation in neonatal immune cells. It is a
timely need to understand the epigenetic effects of folate intake in pregnancy on
regulation of immune maturation which plays a central role in pathogenesis of NCD.
In addition to DNA methylation, histone modifications are another important epigenetic
modification that is associated with changes in nutritional exposures. Of histone
modifications, changes to histone acetylation is closely associated with variation in

29
dietary components (212).Therefore, examination of combinatorial effects of DNA
methylation and histone acetylation of nutritional manipulation will give a better
understanding of the epigenetic programming and subsequent health outcomes of such
interventions.
1.7.1.2 Polyunsaturated fatty acids (PUFA)
Dietary content of more “western” diet is changing and of immediate relevance to this
thesis, decreasing intake of n-3 PUFA has been implicated in rising propensity of
inflammatory diseases (2). Along with a reduction in food rich in n-3 PUFA such as
oily fish, increased intake of food high in n-6 PUFA has shifted the n-3/n-6 PUFA from
around 1:1 to 1:5-1:2 (246). This change in fatty acid balance has a profound effect on
immune response as a result of the relatively inflammatory properties of the n-6 PUFA
relative to the anti-inflammatory products of n-3 PUFA. To address the relative
deficiency of n-3 PUFA as a potential culprit of dysregulated immune development
efforts are being made to restore the PUFA balance through supplementing with fish oil
in pregnancy and early infancy. Previous studies from our laboratory have consistently
shown that n-3 PUFA supplementation in pregnancy (in the form of fish oil) reduce
oxidative stress and inflammation (52) , with effects on T cell activation (247) which
include reducing Th2 propensity in neonatal immune cells (52-54, 247) and have
supported the finding other groups (248, 249). While this dietary intervention has
shown to affect many biochemical and physiological properties of cells and organs
(250) the exact molecular mechanism by which maternal intake of n-3 PUFAs exert
their multi-system benefits in the offspring has not been clearly elucidated.
Emerging data from animal studies suggest maternal PUFA intake has epigenetic effects
in the offspring (251, 252) indicating offspring immunomodulatory effects of such
dietary interventions may have an epigenetic basis. This is a poorly investigated area
albeit the enormous potential of epigenetic studies to explore the immunomodulatory
pathways and molecules of PUFA supplementation.
In a recent study involving pregnant women supplemented with n-3 docosahexanoic
acid (DHA) or a placebo from the second trimester until delivery, Lee et al measured
the DNA methylation changes in genes associated with Th1, Th2, Th17, and Treg
development, as well as LINE1 repetitive elements of cord blood mononuclear cells
(253). Although there were no significant differences in DNA methylation pattern in

30
genes when comparing the treatment and the placebo groups, overall n-3 PUFA
supplementation did have effects on methylation in the subgroup of neonates whose
mothers smoked during pregnancy. Specifically, in this risk group, n-3 PUFA
supplementation was associated with differences in methylation levels of LINE1
repetitive element. Collectively, these data suggests maternal n-3 PUFA intake during
pregnancy may modify the fetal epigenome, and that this may depend on other
environmental exposures.
1.7.2 Other maternal exposures that affect the fetal epigenome
In addition to maternal nutritional exposures there are number of other maternal
environmental factors affecting fetal epigenome that have been described. Studies
employing genome-wide epigenetic analysis to investigate such effects have made a
significant contribution to discover areas in the fetal epigenome that are sensitive to
maternal exposures.
Evidence is accumulating for a clear association between maternal smoking to specific
changes in off spring DNA methylation, some of which were in line with the findings in
non-pregnant adult smokers. This has important health implications as exposure to
cigarette smoke in pregnancy has many adverse effects on the fetus, including effects on
lung function and asthma risk (254, 255). Smoking in the last trimester has been
associated with early onset of airway hyper-reactivity (likely asthma) by the age of 1
year (256). Moreover, both maternal and grand-maternal smoking during pregnancy are
associated with increased risk of childhood asthma, suggesting a persistent heritable
effect (257).
Human studies linking methylome profiles and maternal smoking have employed
available fetal derived tissues such as placenta, buccal cells or cord blood. Recent
several studies have linked exposure to smoking in pregnancy with DNA methylation
change in the Aryl hydrocarbon receptor repressor (AHRR) gene, a gene that is involved
in the detoxification of chemicals found in tobacco smoke. A study that involved
screening of 1062 newborn cord blood samples using the Infinium
HumanMethylation450 platform (HM450) found hypomethylation of the AHRR and
Growth factor independent 1 transcription repressor, and hypermethylation of
cytochrome P450–1A1 and myosin IG (258). These findings have been replicated in
other birth cohorts (258, 259). Interestingly, other recent studies in adults have also

31
identified hypomethylation in the same region of the AHRR gene in association with
smoking in many tissues or cell types (260-263). Furthermore, there is now evidence
that prenatal tobacco exposure not only have effects on epigenetic profile at birth, these
effects can also persist into infancy (259) or adolescence (264). In addition to the gene-
specific effects of prenatal tobacco exposure, there is emerging evidence of maternal
smoking could alter the fetal epigenome in a global scale (265). However, genetic
constitution appears to influence over the epigenetic-environmental interactions in this
context (265, 266).
Polycyclic aromatic hydrocarbons (PAH) are one of the most wide spread organic
pollutants and also a major component of particulate matter (PM) of urban aerosol.
They are found in oil, coal, and tar deposits and they are also formed by the incomplete
combustion of carbon-containing fuels such as wood, coal, diesel, fat, tobacco, and
incense. Grilled, smoked or barbecued meat also appears to contain high levels of PAH
(267, 268). In addition to its carcinogenic properties (269), it has been found not only to
impair functions of airway cells and smooth muscle cells but also diminish
responsiveness to standard therapy given to asthmatics (270) and lead to cognitive
defects and fetal growth impairment with in utero exposure (271, 272). A novel
epigenetic marker for PAH-associated asthma has been identified and cord blood
mononuclear cells (CBMC) of children born to mothers who had been exposed to
considerable level of PAH showed hypermethylation of the acyl-CoA synthetase long-
chian family member 3 promoter (187). Furthermore, the exposure level was highly
correlated with increased risk of asthma symptoms in the offspring before age 5 years.
A recent study reported hypermethylation of the IFNG promoter to be associated with
PAH levels in cord blood of offspring of the exposed mother (273).
The largest contributor of traffic-related PM is diesel exhaust particles (DEP). Exposure
of mice to inhaled DEP for 3 weeks can augment the production of IgE upon intranasal
administration of Aspergillus fumigates (274). Hypersensitisation occurred through
hypermethylation of IFNG promoter with concomitant hypomethylation of IL4
promoter in DNA from splenic CD4+ T-cells. The effects of PM could be mediated in
the airways through induction of oxidative stress. Treatment of A549 cells
(adenocarcinomic human alveolar basal epithelial cells) with either PM-10 or H2O2
increased the expression and release of IL-8 which increased the HAT activity, hence
remodelled the IL8 promoter region (275) suggesting PM exert their effects through

32
chromatin remodelling of the susceptible genes. Given the widespread exposure of
human population to environmental pollutants, both indoors and outdoors, it is
important to research the epigenetic effects of these exposures as a potential mechanism
underpinning the maternal transmission of adverse health outcomes to the next
generations.
As noted earlier (in sections 1.1.3 and 1.1.4) prenatal and postnatal microbial exposure
provides a primary signal for the maturation of a balanced immune system, and now
there is evidence that these effects are mediated through microbe-induced modulation of
the host epigenome. In support of this, offspring of mothers living in rural farming
environments have shown to carry a different epigenetic profile compared to those who
were born to mothers not living on a farm (276-279). In mice, maternal exposure to
different microbes during pregnancy led to attenuation of asthmatic symptoms in the
offspring due to increased IFN-γ production accompanied by hyperacetylation at the
IFNG promoter (280). These findings together, are highly suggestive of the fact that
microbial exposure during pregnancy can modify fetal immune responses through
epigenetic mechanisms.
* * *
The epigenetic profile at birth reflects the accumulated changes over fetal life in
response to in utero exposures. Over the course of pregnancy many exposures can affect
the epigenetic profile to varying degrees to influence phenotype, resilience or
vulnerability to disease. This identifies the need to more fully investigate the effects and
associations of individual environmental exposures and epigenetic marks at birth, and
beyond. In this context, well-controlled randomised trials provide an ideal opportunity
to examine specific factors while minimising the effects of other confounding factors.
Ultimately, it is also recognised that none of these exposures varies in isolation and that
an understanding of the composite effects of early environmental interactions will be
needed. However a component approach is an important part of these initial
investigations. To that end, the studies of this thesis focus on the effects of key dietary
exposures, namely folate and n-3 PUFA, including analysis of samples collected in the
course of randomised controlled trials.

33
1.8 Challenges of epigenome-wide association studies
Epigenome-wide association studies (EWAS) have recently been enabled following
developments in new technology including microarray technology and next-generation
sequencing. These platforms offer unprecedented opportunities to study the epigenetic
effects on a genome scale. EWAS designs have become popular approaches to identify
potential epigenetic mechanisms in an unbiased fashion, with a view to hypothesis
generation. Despite their recent popularity, EWAS study designs are unique and contain
a number of unique challenges.
Given the fact that epigenetic marks are tissue specific it is logical to examine
appropriate tissues relevant to the disease of interest, however, this is not always
possible due to limited access to tissue samples in humans. It is even more difficult to
explore tissue-specific epigenetic aberrations in fetal programming and epigenetic
analysis may have to be carried out on readily accessible fetal tissues such as cord blood
and placental tissues collected at birth. On the other hand, cellular heterogeneity of
blood and other tissues is a potential confounder for epigenetic analyses. A number of
algorithms have now been developed to account for the inter-individual variation in the
composition of whole blood during data analysis (281). Use of purified cell populations
is a better alternative to overcome this problem in epigenetic studies.
Conclusively identifying causal from consequential epigenetic variants is a considerable
issue in EWAS as epigenetic variation can be causal for disease or can arise as a
consequence of disease (reverse causation). While some disease-associated epigenetic
variations have been detected prior to disease onset (282, 283) this is generally not
achievable for most phenotype-based retrospective studies and large longitudinal
cohorts provide an approach to rule out reverse causation of epigenetic variation. The
presence of highly correlated DNA methylation patterns across multiple tissue or cell
types, which otherwise have a unique epigenetic profile, in association with a disease
also suggest a causative link (284). Therefore, it is worth considering screening two or
more cell or tissue types for epigenomic analysis to gather supportive evidence for
causation.
Another aspect to consider in EWAS design is measuring heritable contribution to the
epigenetic variation (285). Underlying DNA sequence, at least for some loci is
important in determining the epigenetic status and therefore may influence how the fetal

34
epigenome respond to environmental exposures. Integration of genomic, epigenomic
and environmental data is a useful strategy to uncover the effects of genotype on the
epitype in an altered environment. Technology is now available to determine the precise
nucleotide positioning, genome-wide spread of methylated cytosines and histone
modifications across the genome enabling integration of multitude of information
during the analysis, but these strategies are likely to require large sample sizes to
achieve adequate power (286, 287). On the other hand, randomised controlled trials
offer unique opportunity to control the genetic confounding of epigenetic effects
induced by specific exposures through analysing repeated epigenetic measurements
before and after the intervention.
Despite the issues surrounding designing the studies, EWAS holds promise to unravel
gene-environment interactions in disease pathogenesis. While the field of epigenetic
epidemiology is still in its infancy, EWAS is likely to evolve as information and
experience accumulate and human epigenetic studies are essential to advance our
understanding of DOHaD.
1.9 Concluding remarks
It is clear that developing immune system is most vulnerable for modulation by
environmental factors during fetal and early postnatal periods as it is a period with the
potential to program or ‘steer’ the developing immune system towards an altered
phenotype.
Although some inherent plasticity remains across the life course, the appearance of
disease phenotypes in the first years of life, namely infant allergic disease, indicates
early consequences of different trajectories of immune development (4). While genetic
predisposition is important, surging rates of disease indicate that changes in the early
environment are driving these modifiable effects on early immune function. Because
inflammation and immune dysregulation are predisposing elements of many other later-
onset NCD, understanding how early events modulate the developing immune system is
also relevant to understanding the dynamics of inflammation later in life (96), and the
multisystem consequences. In particular, understanding how common risk factors for
many NCD modulate gene expression in early life is a research priority, as this may
provide better strategies for more targeted and more effective disease prevention.

35
Maternal dietary changes, whether as a general part of lifestyle change or as a specific
intervention, has consistently shown to affect the immune outcomes in the neonate (41,
53, 54, 288, 289). Nevertheless, the underlying mechanism of environment-induced
immune modulation is far from clear and only recently epigenetics emerged as likely
mechanism that underpin such effects. The work of this thesis aims to measure
epigenetic variation associated with in utero dietary exposures, specifically exposure to
folate and n-3 PUFA, which are known to influence immune development. This will
firstly, identify epigenomic regions that are sensitive to these specific environmental
factors and thus susceptible to modulation, and secondly, it will add to the scientific
evidence needed to inform evidence-based recommendations on dietary practice and
other strategies to promote favourable early immune development, ultimately aimed at
reducing the burden of both early and later onset inflammatory NCD.

36
Chapter 2
Study Rationale, Study Design and Methods
2.1 Study rationale
Based on clear evidence that periconceptional folic acid intake significantly reduces the
occurrence of neural tube defects in neonates, folic acid in doses of 400 µg daily has
been recommended to women of child bearing age in many countries (223, 224).
Mandatory folic acid fortification of flour and grain products in many countries has
been a public health success story to date, resulting in a significant reduction in
incidence of neural tube defects in newborns (290). Whilst these beneficial effects are
clearly warranted, post fortification studies have also raised some concerns over the
combined effects of public health recommendations and fortification of the food supply.
In particular there are legitimate concerns that pregnant women may potentially be
exposed to higher than recommended doses of daily folic acid (227, 291). Evidence
supporting these concerns was published in studies reporting a link between higher
folate intake during pregnancy, and an increased risk of asthma and allergic diseases in
the offspring. These data from observational studies have been inconsistent (228, 234-
237, 292), however mouse models have presented convincing mechanistic evidence for
such associations (93). Clearly more research is needed in this area in order to better
understand the role of folate in pregnancy and in the long-term health of the offspring.
The clear role of folate in one-carbon metabolism suggests a plausible mechanistic link
between excessive folate levels, aberrant DNA methylation at important immunity
genes, and potentially altered immune responses. In line with the DOHaD hypothesis,
these effects could potentially be programed through disrupting DNA methylation
levels during the establishment of the fetal epigenome. However, many studies in this
area have taken a candidate approach, limiting analysis for few target genes (238, 239),
and the full spectrum of genomic regions modulated by folate exposure are unclear.
This thesis investigates the effects of in utero folate exposure on DNA methylation
particularly in neonatal immune cells using an unbiased genome-wide approach. It also
investigates the effects of folate exposure on histone acetylation in the same population,
to understand the combinatorial effects of nutritional manipulation on different but
intimately related epigenetic processes.

37
In addition to the effects of folate there has been increasing interest in fish oil
supplementation during pregnancy and early infancy as a potential intervention to
restore the imbalance between relatively anti-inflammatory n-3 PUFA and more
inflammatory n-6 PUFA typically associated with modern diets. While n-3 PUFA have
been shown to exert multisystem effects promoting a healthy immune maturation, the
underlying mechanism is not clearly elucidated. Studies by Sadli et al (293) and
Ceccarelli et al (294) provided key evidence that fish oil may exert its effects through
DNA methylation. This thesis contributes to this emerging body of work, by
investigating the potential effects of maternal fish oil supplementation on the neonatal
epigenome, with a view to identifying previously unknown pathways in the
inflammatory cascade.
2.2 Study cohorts
The cohorts of this study were derived from participants from two previous research
projects carried out in our laboratory. The participants were recruited through Princess
Margaret Hospital for Children’s clinical research facility. An outline of the two studies
is given below.
Allergy Prevention Program (APP)
This double-blind placebo control trial was performed by Dr. Janet Dunstan (under the
supervision of Prof Susan Prescott) during the period between 1999 and 2002 at the
School of Paediatrics and Child Health, University of Western Australia to investigate
the effects of fish oil supplementation in pregnancy on infant outcomes. Ninety eight
atopic pregnant women were recruited to the study and received either fish oil or
placebo from 20 weeks of gestation until delivery. A total of 70 cord blood samples
collected from neonates of this cohort were available as cryopreserved samples and
were used to investigate the effects of maternal fish oil supplementation during
pregnancy on neonatal epigenetic profile as described in chapter 5.
Infant Fish Oil Study (IFOS)
This postnatal double-blind randomized clinical trial was performed by Dr. Nina D’vaz
and Dr. Suzanne Meldrum (also under the supervision of Prof Susan Prescott) during
the period between February 2008 and January 2011 at the School of Paediatrics and
Child Health, University of Western Australia. This study included 420 infants at high

38
risk of allergic disease (born to atopic mothers) who were supplemented with either fish
oil or placebo oil daily from birth to six months of age to determine whether infant fish
oil supplementation in the early postnatal period can favourably modify infant immune
development away from the allergic phenotype.
As this population did not have any interventions in pregnancy, it was the suitable
population to examine the effects of variations in maternal folate status in pregnancy, in
relation to the cord blood epigenetic marks. To investigate this, subgroups of neonates
with either high or low folate levels were selected from this cohort (based on the criteria
described in chapter 3). Since these neonates were supplemented with fish oil or placebo
in the postnatal period, the intervention did not influence on epigenetic marks at birth.
2.3 Data and sample collection
An overview of the available clinical and questionnaire data of aforementioned two
cohorts as well as available relevant samples is provided below.
2.3.1 Questionnaire data
Questionnaires were given to the mothers at their antenatal visits to collect data on
factors that may affect offspring immune development including familial allergy,
ethnicity, parental ages, number and age of siblings, socioeconomic and educational
status, use of medications, illnesses, home environment and lifestyle. A detailed account
of supplement intake and diet during pregnancy was collected at the recruitment through
semi-quantitative food frequency questionnaires.
Data on neonatal birth parameters, delivery mode, drugs used during the delivery,
complications during or after the delivery, and congenital abnormalities were available
for both cohorts. Follow-up questionnaires had been administered at 12 months to
collect data on potential symptoms of infant allergy (including eczema, food allergy,
and recurrent wheezing), infections, medication use, vaccination, daycare attendance
and exposure to other children, pets and tobacco smoke, feeding practices, solids
introduction and any changes that may have occurred in the home environment.
2.3.2 Maternal and infant skin prick test (SPT)
Maternal and infant (at 12 months) allergy was assessed by skin prick test to a panel of
inhalant and food allergens using a standardized technique (295). Allergens tested in the

39
mothers included cockroach, house dust mite, mould, southern grasses, rye grasses, cat,
dog and feathers. The infants were tested to egg, peanut, milk, house dust mite, cat, rye
grass and southern grasses. Saline was included as a negative control and histamine as a
positive control. A wheal diameter of ≥2 mm was considered positive.
2.3.3 Clinical assessment for allergic diseases
Infants were assessed clinically at 12 months of age, and the findings were reviewed by
a paediatric allergist. A child was classified as having “allergic disease” if they had a
physician diagnosis of IgE-mediated food allergy, eczema or asthma (although the
limitations in diagnosing asthma at this age are recognised). IgE-mediated food allergy
was defined by a history of immediate symptoms within 1-2 hours following contact
and/or ingestion and a positive SPT to the implicated food. . The diagnosis of atopic
dermatitis in infants was based on a history of typical skin lesions. The severity of
eczema was assessed using the modified SCORAD index (296). A diagnosis of asthma
was based on a history of recurrent wheeze (>2 episodes of wheezing) that was
demonstrated to be responsive to bronchodilator medications, but the limitations are
recognised at this age.
2.3.4 Collection of cord and maternal peripheral blood
The umbilical cord was cleaned and swabbed with 70% ethanol to prevent
contamination and cord blood was then collected by venepuncture (19G needle) of
placental vessels, and placed into heparinised (500 µl of preservative-free heparin)
Roswell Park Memorial Institute (RPMI) (Invitrogen, California, USA) tissue culture
medium. Twenty millilitres of maternal blood was collected via venepuncture of the
cubital fossa vein, into an equivolume of heparinised (500 µl of preservative-free
heparin) RPMI tissue culture medium. Cord and maternal serum was also collected and
stored separately for subsequent analyses.
2.4 Laboratory methods
Detailed methods are included in relevant chapters. Information relating to the methods
which are not described in detail elsewhere is described below:
2.4.1 Harvesting and cryopreservation of mononuclear cells
For logistic reasons and to allow batched analysis, cryopreserved cells were used for in
vitro analyses. These methods have been established and validated in the host laboratory

40
(297). Mononuclear cells were isolated from cord and maternal peripheral blood and
cryopreserved within 4 hrs and 12 hrs of collection respectively, as outlined below.
Cord blood samples were separated into plasma, mononuclear cell and erythrocyte
fractions by gradient centrifugation. Blood/RPMI was layered over Lymphoprep
(NYcomed Pharmacia, Norway) and centrifuged at 500g for 30 minutes. CBMC were
isolated from the layer above the concentration gradient and washed once with RPMI
and then layered over Lymphoprep and centrifuged at 500g for 20 minutes. This
“double-layer” technique ensures almost complete removal of contaminating
erythrocytes and nucleated erythroid precursors. CBMC were isolated again and
washed twice with RPMI + 2% heat-inactivated fetal calf serum (HIFCS) (Australian
Biosearch, Australia). A sample of cells were stained with white cell counting fluid and
were enumerated using an Improved Neubauer haematocytometer (weber Scientific,
West Sussex, England) and diluted to 30-40 x 106 cells/ml RPMI+2% HIFCS and
placed on ice. For cryopreservation an equal volume of cooled 15% dimethyl
sulphoxide (DMSO) (BDH, Australia) in HIFCS was added drop wise. Heat
inactivation of FCS (56ºC for 60 minutes) was performed prior to use.
Samples were frozen in 1ml aliquots each containing 15-20 x 106 cells. The samples
were cooled to – 80 ºC at a rate of 1ºC/minute in a specialized freezing container
(Nalgene cryocontainer) containing iso-propyl alcohol (BDH, Australia). Samples were
then transferred to a liquid nitrogen tank for long-term storage.
2.4.2 Thawing of cryopreserved mononuclear cells
Vials of cryopreserved CBMC were thawed rapidly in a 37ºC water bath. Cells were
transferred to a 10ml polypropylene tube and cold RPMI supplemented with DNAseI
(5µg/ml) (Sigma, Australia) was added drop wise (at 1ml/min) until the tube was filled.
The tubes were incubated for 10 min at room temperature to digest gDNA from dead
cells. The cells were centrifuged at 500g for 5 min and then resuspended to 1 ml in
AIM-V serum-free tissue culture medium (Invitrogen, California, USA) supplemented
with 2-mercaptoethanol ) (2ME) (4 x 10-5M final concentration) (Sigma, Australia) or
RPMI supplemented with 10% non-heat-inactivated FCS for determination of in vitro
tissue culture immune responses as detailed below. A sample of cells was stained with
(trypan Blue) and cell number and viability were determined using an Improved
Neubauer haematocytometer.

41
2.4.3 Cord blood immune responses
Cell suspensions were diluted in AIM-V supplemented with 2ME (for adaptive immune
responses) or RPMI + 10% non-HIFCS (for innate immune responses) to a
concentration of 1 x 106 cells/ml and seeded into 96-well round-bottom tissue grade
polystyrene plates at a volume of 250 µl/well alone or with optimal stimulating doses of
allergens or innate stimulants. The samples were cultured in duplicate where adequate
numbers of cells were available. Optimal allergen concentrations have been previously
determined by the host laboratory. Cells were incubated for 24 hrs and 48 hrs at
physiological 37ºC, 5% CO2 for innate and adaptive studies respectively. At the end of
the incubation period, 200 µl of culture supernatant was collected into microfuge tubes
and frozen at -20ºC for multiplex analysis of cytokine production.
2.4.4 Purification of CD4+ T-cells
CD4+ T-cells were chosen for this study to investigate epigenetic perturbations as they
play a key role in immune responses and have been shown to be susceptible to in utero
perturbation from environmental exposures (298).
The flow chart (Figure 2.1) below outlines the cell purification steps that were used to
isolate cell subtypes and indicates where the collected data on these cell subtypes are
represented in this thesis. CD4+ T-cells were isolated from CBMC using a two-stage
isolation strategy with magnetic DYNAL beads (Invitrogen, Victoria, Australia). 5 ml
of thawed primary mononuclear cells in AIM-V + 2ME tissue culture medium at a
concentration of 1 x 106 cells/ml was transferred each to two 10ml polypropylene tubes.
The volumes of buffers for this procedure were determined based on the starting CBMC
as per instructions from the manufacturer. The tubes were filled with PCF buffer
(Appendix 1) and centrifuged at 500g for 5 min at 8ºC to pellet cells. Supernatants were
aspirated and pellets were resuspended in 160uL of PCFD buffer (Appendix 1) and
combined with 33.6 µl of pre-washed magnetic beads coated with a monoclonal
antibody specific for human CD8. Cells were incubated at 4ºC for 20 min on a circular
rotor. Cells were gently resuspended and placed on a DYNAL magnet for 2 min before
removing the CD8- supernatant fraction. This fraction was added directly to 50.4 µl of
pre-washed magnetic beads coated with a monoclonal antibody specific for human
CD4. An extra 200µl of PCFD buffer was added to cells and incubated for 20 min at 4
ºC on a circular rotor. Meanwhile 350uL of RLT lysis buffer (Qiagen) supplemented
with 2ME (10 µl/ml) was added directly to CD8+ T-cell fraction, vortexed and frozen at

42
-80 ºC. Bead-bound CD4+ cells were washed twice in 1ml of PCF buffer and 350 µl of
RLT lysis buffer supplemented with 2ME was added directly onto beads, vortexed and
frozen at -80 ºC.
Figure 2.1 Outline of purification steps used to isolate CD4+ T-cells and antigen presenting cells (APC) from cord blood mononuclear cells (CBMC) for the studies in this thesis.
2.4.5 Purification of antigen presenting cells
Antigen presenting cells (APC) are critical regulators of host first line of defence and
dendritic cells (DC) in particular play a key role in the generation of adaptive immunity.
DC epigenome has shown to be sensitive to environmental cues (299) and variation in
epigenetic marks in DC and monocytes has been associated with disease risk (179, 199,
300). For epigenetic studies in this thesis, DC and monocytes were obtained from cell
sorting as described below.
The CD4-CD8- supernatant obtained following magnetic bead cell depletion (section
2.4.4) was transferred to a fresh 2 ml screw-cap tube and placed on a DYNAL magnet
for 1 min to remove any residual beads in the cell suspension that may interfere with the
process of cell sorting. Cells were transferred to a 5 ml polystyrene round-bottom tube
and were washed once with FACS buffer (Appendix 1). Cells were resuspended in
CD4+T-cells
CD8+T-cells
CD4-CD8-T-cells
APC cells
Fluorescence activated cell sorting
Methylation
Acetylation
CBMC
Methylation
Epigenetic
study
Representation
in the thesis
Chapter 3
Chapter 5
Chapter 3
Chapter 4
Chapte 3
Magnetic bead separation

43
100µl of antibody mix (20µl of anti-CD19-FITC, 10µl of anti-CD3-PE, 10µl of anti-
HLADR-PerCP, 2.5µl of anti-CD11c-PECy7, 2.5µl of anti-CD14-APCCy7 and 55µl of
FACS buffer). Whole CBMC were stained for single- colour controls (either with the
monoclonal antibody or with the appropriated concentration of matched-isotype control)
and used for determining the appropriate gating strategy. All antibodies were obtained
from BD Biosciences, CA, USA. After 30 min incubation in the dark at 4C, the cells
were washed twice with FACS buffer, and resuspended in 300µl PBS+20%FCS and
immediately placed on ice until run on the cell sorter (FACSAria) on the same day.
Shown in Figure 2.2 are representative FACS plots illustrating the sorting strategy that
was adopted to collect the monocyte/DC cell subsets from the CD4-CD8- fraction of the
CBMC samples. Figure 2.2A illustrates the FSC/SSC characteristics of the CD4-CD8-
fraction of CBMC. Figure 2.2B shows that the supernatant obtained following magnetic
bead cell isolation is depleted for CD3+ cells. From the CD3/CD8/CD4 depleted cell
population CD14+ (monocytes) and CD11c+HLADR+ dendritic cells were collected
(Figures 2.2 C, D and E) and were immediately resuspended with 350 µl of RLT lysis
buffer supplemented with 2ME, vortexed and frozen at -80ºC. Purities of sorted samples
containing DC and monocytes were 99-100% CD3-, 97-100% CD19-, 97-100% CD11c+
and 95-100% HLADR+.
2.4.6 Purification of genomic DNA and total RNA
Genomic DNA (gDNA) and total RNA were extracted from the frozen CD4+ T-cells
and APC using DNA/RNA mini columns (Qiagen, Australia) according to the protocol
recommended by the manufacturer. gDNA and RNA yield and purity were assessed by
spectrophotometry.
2.4.7 Quantitative polymerase chain reaction (qPCR)
First strand synthesis of complementary DNA (cDNA)
First-strand cDNA was generated by reverse transcription of 500 ng of total RNA per
sample with random hexamers using SuperScript VILO (Invitrogen, CA, USA)
according to the manufacturer’s protocol. RNA was treated with Deoxyribonuclease I
(Invitrogen, CA, USA) to remove any contaminating DNA prior to reverse
transcription. Reverse transcription was performed on an Applied Biosystems
thermocycler (GeneAmp 7000). cDNA were diluted 1/5 in RNAse free water and stored
at -20ºC.

44
Figure 2.2 Outline of screening strategy for isolating antigen presenting cells (APC) using FACS cell sorting. (A) FSC/SSC gating on the CD4 and CD8 depleted live cell population (B) CD3-CD19- cell population was selected and further gated for (C) CD14+ and HLA-DR. We collected the CD14+ monocytes (shown in C and D) and pooled these with the CD11c+HLA-DR+ cells as illustrated in (E) from the CD14-cell gate as per (C).

45
Real time polymerase chain reaction (PCR)
Guaranteed primer assays were purchased from Invitrogen and the sequence
information is proprietary of that company. Relative quantitation of gene expression
was performed using pre-designed TaqMan assays (Applied Biosystems, Foster City,
CA, USA) on the ABI 7300 real-time PCR system in triplicate. All samples were
normalized to the house-keeping gene PPIA.
2.4.8 DNA methylation analysis
A total of 500ng of purified genomic DNA (gDNA) from each cell type was bisulphite
converted using the Methyl Xceed kit (Human Genomic Signatures, NSW, Australia).
This procedure converts unmethylated cytosine residues into uracil while not affecting
5-methylcytosine. Successful bisulphite conversion was evaluated for all samples using
an in-house bisulphite-specific PCR. Bisulphite DNA was sent to Australian Genome
Research Facility (Parkville, Melbourne, Australia) for labelling, staining and
hybridization to Illumina Human Methylation 450 arrays (Illumina). Raw iDAT files
were processed using the Minfi package from the bioconductor project
(http://www.bioconductor.org) in the R statistical environment (http://cran.r-
project.org/). The minfi package was used for array pre-processing using the stratified
quantile normalization method. Technical bias attributable to different probe chemistries
between Type 1 and Type II probes were adjusted in this procedure. Probes with a
detection P-value call >0.01 in 1 or more samples were removed. Probes on the X and
Y-chromosomes were removed to eliminate gender bias. Probes previously
demonstrated to potentially cross-hybridize non-specifically in the genome were
removed (301). Probes containing a polymorphic single nucleotide polymorphism
(SNP) at the single-base extension site with a minor allele frequency of <0.05 were
removed. The log2 ratio for methylated probe intensity to unmethylated probe intensity,
the M value, was subsequently derived and used for statistical inference. Beta values
were derived from intensities as defined by the ratio of methylated to unmethylated
probes given by B=M/(U/M*100) and were used to compliment M-value as a measure
of effect size. Cluster analysis was used in conjunction with the chip-wide medians of
the methylated and unmethylated channels to identify any outlying samples.
2.4.9 Validation of methylation array targets
Probes that were shown to be differentially methylated in the 450k array were then
tested by mass spectrometry to determine the validity of the array findings. Amplicons

46
for targets were designed using the Sequenom EpiDesigner software
(http://www.epidesigner.com/) and performed using the Sequenom EpiTYPER platform
in triplicate. EpiTYPER (Sequenom, San Diego, USA) is a tool for quantitative analysis
of methylated DNA based on bisulfite conversion. Through bisulfite treatment
unmethylated cytosine residues are de-aminated to uracil and transformed into thymine
during PCR amplification, whereas methylated cytosine residues still appear as
cytosines. Consequently, bisulfite treatment results in methylation dependent sequence
variations of C to T after PCR amplification. Amplification products were treated with
shrimp alkaline phosphatase to dephosphorylate unincorporated deoxyribonucleotide
triphosphates from PCR. Subsequently, in vitro transcription was performed followed
by RNase A specific cleavage to produce smaller fragments. Cleavage products were
prepared for analysis in the mass spectrometer according to manufacturer’s protocol.
MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass
spectrometry) detects the 16 Da mass difference between guanine and adenine residues
(resulting from C/T variations at the opposite strand) due to methylated and
unmethylated DNA templates). MALDI-TOF MS data are then processed using the
EpiTYPER software generating quantitative results for each cleavage product. Probes
showing a % mean methylation of less than 5 and all probes overlapping with SNP were
removed from the analysis.
2.4.10 Histone 3 and Histone 4 acetylation analysis
Chromatin immunoprecipitation (ChIP) assay for histone acetylation studies requires a
higher starting numbers of freshly isolated cells. This has limited the use of this assay
primarily to cell lines rather than primary cell analysis. Therefore an in-house assay of
histone acetylation was developed and validated by our collaborating research group at
Philips University Marburg, Germany (described in detail in chapter 4). Using this
assay, levels of H3 and H4 acetylation of isolated neonatal CD4+ T–cells were measured
and analysed against the variation in folate exposure as described in chapter 4.
2.5 Statistical analysis
Statistical analysis is outlined in detail in the relevant chapters.
2.6 Ethics approval
Ethical approval for this study was obtained from the ethics committee of the Princess
Margaret Hospital for Children, Perth, Western Australia.

47
Chapter 3
Paper 1
Genome-wide DNA methylation profiling identifies a folate-sensitive
region of differential methylation upstream of ZFP57 imprinting
regulator in humans
Manori Amarasekera,1 David Martino,2, 3, 4 Sarah Ashley,2, 5 Hani Harb,6 Dörthe
Kesper,6 Deborah Strickland,7 Richard Saffery,2, 3 and Susan L. Prescott,1
1School of Paediatrics and Child Health, University of Western Australia, Perth,
Australia 2Cancer and Disease Epigenetics Group, Murdoch Children’s Research Institute,
Melbourne, Australia 3 Department of Paediatrics, University of Melbourne, Melbourne, Australia 4Centre for Food and Allergy Research, Melbourne, Australia 5Institute for Medical Research, Monash University, Melbourne, Australia. 6Institute of Laboratory Medicine, Pathobiochemistry and Molecular Diagnostics,
Philipps University Marburg, Germany 7Telethon Institute for Child Health Research, Perth, Australia
All authors are members of inFLAME (International Inflammation Network) of the
Worldwide University Network.

48
3.1 Abstract
Folate intake during pregnancy may affect the regulation of DNA methylation during
fetal development. The genomic regions in the offspring that may be sensitive to folate
exposure during in utero development have not been characterized. Using genome-scale
profiling we investigated DNA methylation in two immune cell types (CD4+ & antigen
presenting cells, APCs) isolated from neonatal cord blood, selected on the basis of in
utero folate exposure. High (HF, n=11) and low folate (LF, n=12) groups were selected
from opposite extremes of maternal serum folate levels measured in the last trimester of
pregnancy. A comparison of these groups revealed differential methylation at seven
regions across the genome. By far, the biggest effect observed was hypomethylation of
a 923bp region 3kb upstream of the ZFP57 transcript, a regulator of DNA methylation
during development, observed in both cell types. Levels of H3/H4 acetylation at ZFP57
promoter and ZFP57 mRNA expression were higher in CD4+ cells in HF group relative
to the LF group. Hypomethylation at this region was replicated in an independent
sample set. This data suggests that exposure to folate has effects on the regulation of
DNA methylation during fetal development, and this may be important for health and
disease.

49
3.2 Introduction
DNA methylation is a key epigenetic mechanism controlling fetal growth and
development (1). Animal studies suggest that dietary intake of nutrients involved in
one-carbon metabolism such as folic acid during pregnancy can influence DNA
methylation in the developing offspring that may lead to an altered phenotype (2-4).
In humans, periconceptional supplementation with folic acid is routinely recommended
to prevent the occurrence of neural tube defects (5). However, emerging data has linked
folic acid intake during pregnancy with disease outcomes in the offspring, including
atopic dermatitis (6, 7) childhood wheeze (8) and asthma (9). Parallel investigations into
potential underlying epigenetic mechanisms have mostly focused on genomic regions
surrounding imprinted genes that regulate growth and development. Several studies
have now reported variations in DNA methylation at these regions in offspring exposed
to periconceptional folic acid. Steegers-Theunissen et al reported the average
methylation of the imprinted insulin-like growth factor 2 (IGF2) differentially
methylated region (DMR) was 4.5% higher in infants whose mothers reported
periconceptional intake of folic acid at a dose of 400µg/day compared with infants
whose mothers did not take folic acid (10). Another study reported that average
methylation at the imprinted H19 DMR in cord blood leukocytes was significantly
lower in neonates of mothers who took folic acid either before or during pregnancy
compared to neonates of mothers who reported no folic acid intake as ascertained from
self-administered questionnaires (11). Together, these findings suggest a complex
relationship between maternal folic acid intake and DNA methylation levels at
imprinted genes and variation in folate exposure during development may have effects
on neonatal growth and susceptibility to diseases in later life (12). Adding another layer
of complexity to the association between folate and methylation profile is that the
effects of folate exposure appear to be tissue specific. Chang and colleagues
investigated the influence of maternal serum folate on DNA methylation in 18-28 weeks
old fetuses following termination of pregnancy for fetal anomalies. They found a
positive correlation between the levels of DNA methylation and maternal serum folate
levels in brain tissue but not in other tissues (13) indicating that exposure to folic acid
during pregnancy may have tissue-specific effects in the developing offspring.
Very few human studies have used a genome-wide approach in mapping the DNA
methylation changes at birth that are associated with variation in folate exposure during

50
pregnancy (14, 15). The concern has therefore been such that changes in epigenetic
marks in fetal epigenome induced by variations in maternal folic acid intake may have
long-term health consequences in accordance with the Developmental Origin of Health
and Disease hypothesis (DOHaD) (16). To advance this area, we conducted a genome-
scale profiling of 450,000 CpG sites in two key neonatal immune cell types (CD4+ T-
cells and antigen presenting cells, APC) purified from cord blood. We chose to study
two cell types representing distinct hematopoietic lineages with well-defined roles in
regulating the host immune response to the environment (17). Importantly by using
purified cell populations in this study, we were able to avoid potential confounding
effects of cell heterogeneity that may affect the measured epigenetic changes (18).
Neonates for this study were selected from the extremes of the maternal serum folate
distribution curve from a Western Australian birth cohort. Using an explorative
hypothesis-free approach coupled with an extreme of exposure design we have
identified several folate-sensitive differentially methylated regions (fDMRs) in the
genome.
3.3 Materials and Methods
Selection of the study population
The study population included 23 neonates selected from a larger prospective birth
cohort of mother-infant pairs recruited through the allergy research clinic in the Princess
Margaret Hospital for Children (7). Mothers were recruited during the last trimester
(>=28 weeks) of pregnancy at which time maternal blood samples were collected, and
cord blood samples were subsequently collected at the time of birth. Peripheral blood
mononuclear cells were harvested from blood samples within 12 hours of collection
according to standard protocols (19). Matched serum folate measurements were
available for both maternal and cord blood for n=222 mother-infant pairs. Extensive
clinical and dietary data collected at recruitment through semi-quantitative food
frequency questionnaires and maternal antenatal and socio-demographic factors were
also available.
The sample population was selected based on the following criteria: Infants were ‘non-
atopic’ and otherwise healthy based on clinical assessments and skin prick test to a
range of inhalant and dietary allergens conducted at 12 months. The high (HF) and low
folate (LF) groups were defined according to the first and third quartiles from the
distribution of maternal serum folate levels in conventional extremes of exposure

51
design. Infants were excluded from the study if exposed to maternal smoking in
pregnancy, or had evidence of congenital birth defects. All study procedures were
carried out in accordance with full institutional ethics.
Serum folate measurement
Folate levels were measured in maternal serum from blood samples collected during the
last trimester of pregnancy, and neonatal serum folate was measured from cord blood
samples using the Immulite 2000 competitive immunoassay platform (Siemans Medical
Solutions Diagnostics, Flanders, NJ, USA).
Isolation of CD4+ T cells
CD4+ T cells were isolated from cord blood mononuclear cells (CBMC) using a two-
stage positive isolation strategy with magnetic DYNAL beads (Invitrogen, Victoria,
Australia). CBMC were incubated with CD8+ magnetic beads as per the recommended
protocol (Invitrogen, CA, USA), and the CD8- fraction was then incubated with CD4+
magnetic beads as per the recommended protocol. Routine purity tests were conducted
by flow cytometry using antibodies CD19-FITC, CD3-PE, CD8-PerCP, CD11c-PE
Cy7, CD4-APC and CD14-APC Cy7 (BD Biosciences, CA, USA) and appropriate
concentration matched isotype controls. CD4+ cell purities ranged from 89-96% pure. A
fraction of isolated CD4+ cells were frozen with 15% dimethyl sulphoxide in heat-
inactivated fetal calf serum and stored in liquid nitrogen until transported to Marburg,
Germany for chromatin immunoprecipitation (ChIP) analysis. The rest of the CD4+ T
cells were lysed with RLT buffer containing 2-mercaptoethanol (Qiagen Allprep kit,
Qiagen, Victoria, Australia)) and stored at -80 ºC until genomic DNA was extracted for
methylation analysis.
Isolation of APC cells
CD3-CD19-CD14+/- CD11c+ HLADR+ myeloid APCs were sorted from CD4+ and CD8+
depleted cell fraction using FACSAria instrument and FACSDiva software (BD
Biosciences, San Jose, CA, USA).
Nucleic acid extraction
Genomic DNA and mRNA were co-purified from CD4+ T and APC cells using Qiagen
Allprep kits according to manufacturer’s instructions.

52
DNA methylation analysis
A total of 500ng of purified genomic DNA from each cell type was bisulphite converted
using the Methyl Xceed kit from Human Genomic Signatures. Successful bisulphite
conversion was evaluated for all samples using an in-house bisulphite-specific PCR.
Bisulphite DNA was sent to Australian Genome Research Facility (Parkville,
Melbourne, Australia) for labelling, staining and hybridization to Illumina Human
Methylation 450 arrays (Illumina). Raw iDAT files were processed using the Minfi
package from the bioconductor project (http://www.bioconductor.org) in the R
statistical environment (http://cran.r-project.org/). The minfi package was used for array
pre-processing using the stratified quantile normalization method. Technical bias
attributable to different probe chemistries between Type 1 and Type II probes were
adjusted in this procedure. Probes with a detection P-value call >0.01 in 1 or more
samples were removed. Probes on the X and Y-chromosomes were removed to
eliminate gender bias. Probes previously demonstrated to potentially cross-hybridize
non-specifically in the genome were removed (20). Probes containing a polymorphic
SNP at the single-base extension site with a minor allele frequency of <0.05 were
removed. The log2 ratio for methylated probe intensity to unmethylated probe intensity,
the M value, was subsequently derived and used for statistical inference. Beta values
were derived from intensities as defined by the ratio of methylated to unmethylated
probes given by B=M/(U/M*100) and were used to compliment M-value as a measure
of effect size. Cluster analysis was used in conjunction with the chip-wide medians of
the methylated and unmethylated channels to identify any outlying samples, and 1
sample was removed from the data set.
Quantitative PCR
First-strand cDNA was generated by reverse transcription of 500 ng of total RNA per
sample with random hexamers using Superscript VILO (Invitrogen, CA, USA)
according to the manufacturer’s instructions. cDNA was diluted 1:5 in RNAse free
water for gene expression analysis. Relative quantitation of gene expression was
performed using pre-designed TaqMan assays (Applied Biosystems, Foster City, CA,
USA) for ZFP57 on the ABI 7300 real-time PCR system in triplicate. All samples were
normalized to the house-keeping gene PPIA. Absolute levels of ZFP57 mRNA
expression were low and not measurable for 8/21 individuals and these were assigned a
maximum CT value of 45 for statistical analysis and interpretation at the group level.

53
Data analysis
Characteristics of genome-wide DNA methylation were assessed by principal
components analysis in the presence of potential confounding factors. The first fifteen
principal components capturing over 65% of the total variance were derived and tested
for associations with clinical variables. Clinical variables associated with variation in
DNA methylation are provided in the supplement and were included as covariates in
regression modelling. To identify differentially methylated regions (DMRs) we used the
dmrFind algorithm in the charm package under the default settings with the cluster
marker function. This algorithm combines surrogate variable analysis (21) for
modelling unexplained variability due to batch effects or covariates, with regression
modelling fitted to the entire dataset in the presence of covariates and surrogate
variables (22). Comparisons are made at the regional level as opposed to single CpG
analysis whereby methylation estimates are smoothed over spatial blocks accounting for
correlation between nearby CpG. Ontology enrichment analysis was performed using
the GREAT ontology tool under the default basal plus extension settings. Significant
ontologies are reported as raw -log10 binomial P-value <0.05.
Validation of array targets
Target validation was performed using the Sequenom EpiTYPER platform in triplicate.
Amplicons were designed using the Sequenom EpiDesigner software
(http://www.epidesigner.com/). Amplification conditions were as follows: 95° C for 10
min, 95° C for 15 s, 56° C for 30 s, 72° C for 2 min for 5 cycles, 95° C for 10 s, 60° C
for 30 s, 72° C for 1 min 30 s for 30 cycles, 72° C for 7 min. The target sequences of the
chromosome 6 fDMR were; aggaagagagGAGGATTTTAGAGGTTGGAAGTTTT (left)
and cagtaatacgactcactatagggagaaggctCCCTCTCATCTAAATCAAAAAACAC (right ).
Histone 3 (H3) and Histone 4 (H4) acetylation analysis
Using a validated ChIP analysis for H3/H4 acetylation in human immune cells we
analysed the levels of H3/H4 acetylation at selected loci. In brief, DNA-histone
interactions in isolated CD4+ T cells were first cross-linked with 1% formaldehyde.
Subsequently, chromatin was sonicated to achieve the optimal DNA length for the
immunoprecipitation which was done using antibodies against acetylated H3 and H4
(Millipore, Darmstadt, Germany). IgG mock control was bought from Abcam, San
Francisco, CA, USA. Quantitative PCR was performed to measure the precipitated
DNA of investigated loci. Primers were designed for known transcripts of ZFP57 and

54
are as follows; ZFP57-001 distal (ZFP57_1D) cac cac cgg cta act ttt gt (forward) and cct
ggg caa aaa gag tga aa (reverse), ZFP57-002 proximal (ZFP57_2P) ccc agg ctg gtg ttg
tta ct (forward) and ggt ttg atg tgg ctt cct gt (reverse), ZFP57-002 distal (ZFP57_2D) cat
gga aga gat ctt aga gag tg (forward) and acc taa tgc aga gat gct aaa taa (reverse).
3.4 Results
Clinical characteristics of the cohort
Maternal serum folate levels ranged from 4.1 nmol/l to 172.0 nmol/l in the cohort from
which the study samples were derived, with first and third quartiles 25.6 nmol/l and
50.5 nmol/l respectively. The high (HF) and low folate (LF) groups were identified
from this cohort and defined according to the first and third quartiles from the
distribution of maternal serum folate levels in conventional extremes of exposure
design. The mean maternal serum folate measurement at recruitment was 74.6 nmol/l
and 16.8 nmol/l in the HF and LF group (P < .001), with corresponding cord blood
serum levels of 78.2 and 36.4 nmol/l (P < .001) respectively (Table 3.1). There were no
significant differences in the key neonatal parameters between HF and LF groups, with
the exception of folate intake during pregnancy (either as food, P = .02 or as
supplement, p = .006), and cord blood serum vitamin D levels (P = .04), which were
found to be significantly higher in the HF group (Table 3.1). Based on this, all
subsequent analysis was adjusted for any potential effects of vitamin D. There was a
significant linear relationship between maternal serum folate and cord blood folate
levels in the sample population (r = 0.75, P < .001) (Figure 3.1A).
Characteristics of genome-scale DNA methylation profiles
Genome-scale DNA methylation data were generated from two blood cell types
representing both the innate (APC) and the adaptive (CD4+) immune cell compartments.
These cell types play a well-defined role in antigen presentation and processing and
mediation of effector immune cell responses. Principal components analysis on Beta
methylation values was used to explore variation in neonatal epigenetic profiles. The
most substantial source of variation in the data set was attributable to cell type (APC or
CD4+). Multidimensional scaling (MDS) and hierarchical clustering of samples showed
widespread differences between cell types reflecting the alternative lineages of APC
(myeloid origin) and CD4+ cells (lymphoid origin) (Figure 3.2A). Differential analysis
of CD4+ and APC cells revealed a signature of 13, 424 CpG sites (approximately 3.5%
of the CpG sites measured) that differed by a minimum delta beta value of at least 20%

55
and reached genome-wide significance (5048 hypermethylated; 8376 hypomethylated
sites). Localization of these sites to functional regions in the genome revealed the
majority of these differentially methylated positions (DMPs) were localized to gene
bodies as opposed to promoters, and overlapped with flanking regions of CpG islands
(CpG island shore and shelves), or were found in open sea regions (not island-
associated) (Figure 3.2B). The DMPs were significantly enriched for genes involved in
leukocyte function and antigen mediated pathways (binomial P value <0.05) (data not
shown). Birth mode, maternal serum folate levels, folate status (high or low) and serum
vitamin D levels were included as covariates in data modelling.
Figure 3.1 Relationship between maternal serum folate and cord serum folate levels. (A) Samples in the discovery cohort and (B) samples in the replication cohort. The graph shows the correlation plot between maternal serum folate levels measured at the recruitment visit during the 3rd trimester and cord serum folate levels collected at birth.

56
Table 3.1 Population characteristics of the mother and infants in the discovery
cohort
Characteristic
High folate group (n=11)
Low folate group (n=12)
P value
Mothers at recruitment Age, (yrs [SE]) 31.9 (1.25) 32.1 (0.82) 0.95
History of allergic disease 8 (73%) 8 (67%) 0.75 Asthma 4 (36%) 3 (25%) 0.55 Hay fever 5 (45%) 7 (58%) 0.54 Eczema 5 (45%) 4 (33%) 0.55 IgE mediated food allergy 1 (9%) 1 (8%) 0.95 Sensitization 9 (82%) 10 (83%) 0.92 Had tertiary education 8 (73%) 11 (92%) 0.23 Not exposed to passive smoke 36% 33% 0.44 Infants
Male (%) 4 (36%) 5 (42%) 0.80 Gestation (wk [SE]) 39.7 (0.3) 39.2 (0.3) 0.28 Delivery method
Vaginal 6 (54%) 4 (40%) Caesarean section 5 (45%) 6 (60%) 0.51
Neonatal growth parameters Birth weight (g [SE]) 3420.3 (79) 3570.9 (128) 0.43
Length (cm [SE] 50.2 (0.5) 49.9 (0.9) 0.79 Head circumference (cm [SE]) 35.0 (0.3) 35.6 (0.5) 0.34 Serum folate
Maternal serum folate level at recruitment (nmol/l [SE]) 74.59 (6.1) 16.8 (1.6) <0.001* Cord serum folate (nmol/l [SE]) 78.15 (5.1) 36.4 (2.3) <0.001* Cord serum Vitamin D (nmol/ [SE]) 59.9 (7.8) 39.0 (5.7) 0.04* Maternal daily intake from supplements (from questionnaire data)
Folic acid (µg DFE/day [SE]) 730.9 (127) 236.6 (120.7) 0.006*
Maternal daily intake from diet (from semi-quantitative food frequency questionnaire)
Folate (µg DFE/day [SE]) 310.7 (36.4) 199.2 (33.7) 0.02* Vitamin D (µg [SE]) 3.8 (0.8) 4.4 (1.7) 0.56 Total Energy (kcal [SE]) 2271.9 (126) 2019.2 (294) 0.71 Protein (g [SE]) 88.9 (5.2) 83.7 (12.8) 0.71 Total fat (g [SE]) 88.8 (8.4) 79.4 (12.9) 0.37 Copper (mg [SE]) 2.2 (0.1) 1.8 (0.3) 0.22 Zinc (mg [SE]) 12.0 (0.8) 10.5 (1.3) 0.22 Retinol (µg [SE]) 512.3 (84) 538.4 (157) 0.56 Vitamin B6 (mg [SE]) 2.0 (0.2) 1.5 (0.3) 0.22 Vitamin B12 (mg [SE]) 3.4 (0.4) 3.7 (1.0) 0.96 Alcohol (g [SE]) 1.8 (0.8) 1.8 (1.1) 0.78 Coffee (g [SE]) 84.2 (58.8) 144.9 (69.2) 0.25 Vitamin A (µg [SE]) 1390.2 (106) 1215.4 (268) 0.5
Statistical comparisons by Chi square and Mann-Whitney U-tests. DFE, dietary folate
equivalent units.
* indicates significant P-values (P < 0.05)

57
Figure 3.2 Differential methylation analysis between CD4+ and APC cells. (A) MDS analysis of sample relationships based on whole genome DNA methylation profile (left panel). Hierarchical clustering of samples based on Euclidean distance (right panel). Samples are coloured according to cell type (green, APC; orange, CD4+). (B) Differential hypomethylation (left panel) and hypermethylation (right panel) according to genomic region. APC; Antigen presenting cells, MDS; Multidimensional scaling.
Differential methylation according to folate status
We first examined general features of DNA methylation between folate groups at
several regions in the genome including repeat elements, non-repeat CpGs and
imprinted genes. Folate status was not associated with changes in DNA methylation
levels at long interspersed nucleotide element-1 (LINE-1) repeat elements in the
genome of either cell type (Figure 3.3A). The average level of DNA methylation in the
genome as measured by the mean of all CpGs on the array did not differ according to
serum folate status in either cell type (Figure 3.3B). Similarly, average and total levels
of DNA methylation levels across CpG sites targeting known imprinted genes did not
vary according to folate status (Figure 3.3C). Collectively this data suggests the general
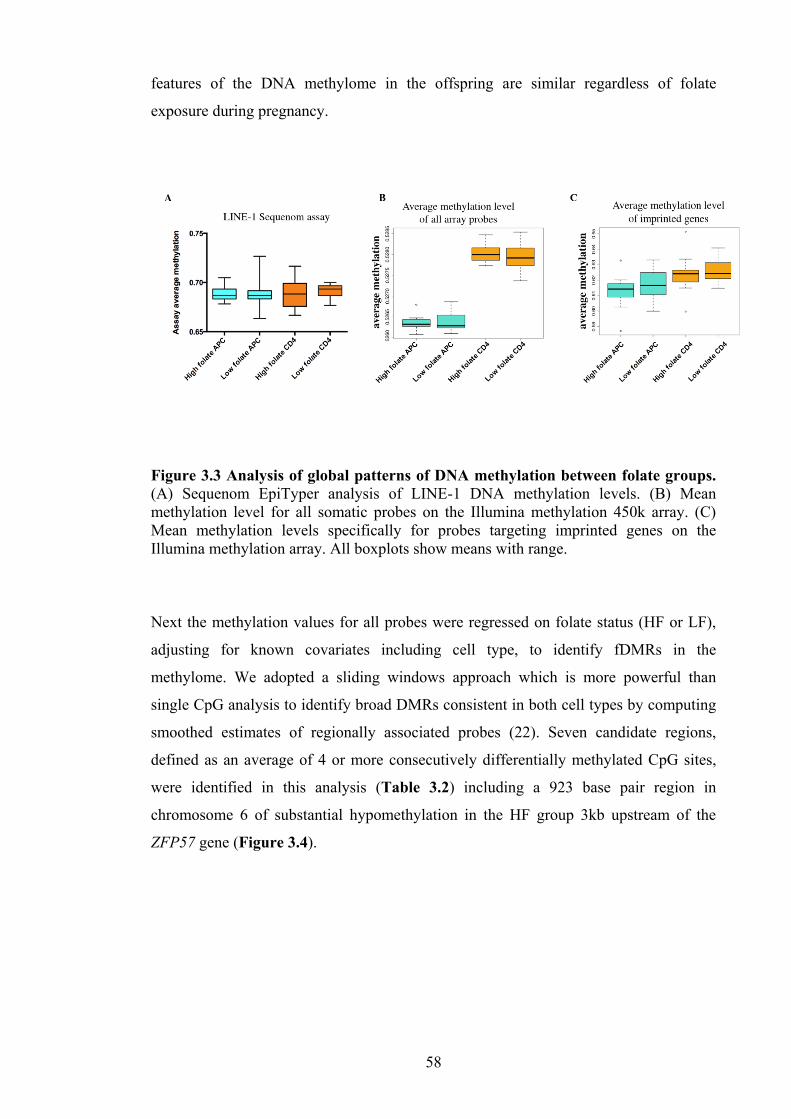
58
features of the DNA methylome in the offspring are similar regardless of folate
exposure during pregnancy.
Figure 3.3 Analysis of global patterns of DNA methylation between folate groups. (A) Sequenom EpiTyper analysis of LINE-1 DNA methylation levels. (B) Mean methylation level for all somatic probes on the Illumina methylation 450k array. (C) Mean methylation levels specifically for probes targeting imprinted genes on the Illumina methylation array. All boxplots show means with range.
Next the methylation values for all probes were regressed on folate status (HF or LF),
adjusting for known covariates including cell type, to identify fDMRs in the
methylome. We adopted a sliding windows approach which is more powerful than
single CpG analysis to identify broad DMRs consistent in both cell types by computing
smoothed estimates of regionally associated probes (22). Seven candidate regions,
defined as an average of 4 or more consecutively differentially methylated CpG sites,
were identified in this analysis (Table 3.2) including a 923 base pair region in
chromosome 6 of substantial hypomethylation in the HF group 3kb upstream of the
ZFP57 gene (Figure 3.4).

59
Table 3.2 Differentially methylated regions (DMR) according to the folate status
aHypomethylation, bHypermethylation
Figure 3.4 Folate sensitive differentially methylated region (fDMR) in chromosome 6. The figure shows smoothed Beta methylation values for LF (red) and HF (blue) groups, averaged for both cell types. The location and size of this DMR is shown in the genome browser track above the sliding window. The ideogram track shows the location of this region on chromosome 6. LF, Low; Low folate, HF; High folate, DMR; Differentially methylated region.
Validation of candidate loci and functional assessment of ZFP57
We measured DNA methylation levels in CD4+ from the initial discovery cohort of
samples interrogated by the DNA microarray using the Sequenom EpiTyper platform as
Table 3.2 Differentially methylated regions (DMRs) according to the folate status
DMR according to the folate status, irrespective of cell type
DMR Chr start end size (bp)
no. of probes
average DM
maximum DM
HF vs LF gene assocations function
1 6 29648161 29649084 923 19 0.19 0.28
hypo a
ZFP57 (-3692)
Maintenance of DNA methylation and genomic imprinting
2 17 37123638 37123949 311 8 -0.08 -0.12
hyper b
LASP1 (+97682)
Regulation of dynamic actin-based, cytoskeletal activities
3 1 76189707 76190008 301 5 -0.13 -0.23
hyper ACADM
(-185)
mitochondrial fatty acid beta-oxidation pathway
4 8 144120106 144120706 600 7 0.08 0.10 hypo
LY6E (+20504) Lymphocyte antigen complex
5 1 228075423 228075749 326 5 -0.10 -0.13 hyper WNT9A
(+60090) development and cell fate
6 21 47604052 47604654 602 5 0.09 0.11 hypo
C21orf56 (+20) unknown
7 2 202901045 202901470 425 5 -0.08 -0.11 hyper FZD7
(+1948) Wnt protein binding

60
a gold standard technology. We designed an amplicon spanning a 321 base pair region
capturing 8 CpG sites in the chromosome 6 DMR upstream of ZFP57 identified by
HM450 analysis as broadly hypomethylated in HF samples. Data from the EpiTyper
was in agreement with observations from the array with hypomethylation observed in
HF relative to LF groups (Figure 3.5A). To determine whether hypomethylation at this
fDMR was indeed associated with the functional activity of the distal ZFP57 gene, we
measured both mRNA gene expression and histone acetylation in the same CD4+
samples that were analysed by microarray. A TaqMan PCR amplicon and an in-house
histone H3/H4 acetylation assay targeted to the transcriptional start site of ZFP57 were
designed. A comparison of HF and LF groups revealed increased levels of histone H3
and H4 acetylation at ZFP57, a marker of increased transcriptional activity (Figure
3.5B). In general absolute mRNA expression levels were low but relative differences
were in the order of 4-fold between groups which was statistically significant. (Figure
3.5C). Collectively these lines of evidence suggest a functional role for observed
differences in DNA methylation associated with ZFP57.
Figure 3.5 This figure shows validation data of chromosome 6 fDMR and functional assessment of ZFP57 in CD4+ cells. (A) Consistent with findings from the Illumina array, data from Sequenom EpiTyper platform showing the trend of lower methylation in HF group and higher methylation in LF group. (B) mRNA expression of ZFP57 in CD4+ cells shows an upregulation of the gene in HF group. (C) Levels of H3 and H4 acetylation at ZFP57 promoter region are higher in the HF group indicating a more open chromatin state in this group. Details of the primer sequences are given in the Appendix 2. fDMR; Folate sensitive differentially methylated region, HF; High folate, LF; Low folate.

61
Assessment of reproducibility in an independent set of samples
An independent sample population was next drawn from the source cohort in order to
determine the potential reproducibility of the findings from our initial discovery cohort.
We sampled an additional set of 19 samples from the first and third quartiles of the
maternal serum folate distribution curve from the source population. The characteristics
of these additional samples were similar to the discovery cohort including a linear
relationship between maternal serum folate levels and cord serum folate levels (r = 0.76,
P < 0.001) (Figure 3.1B) but with the notable exception that vitamin D levels were not
different in this group (P = 0.2). Both CD4+ and APC populations were purified from
cord blood as described previously. In agreement with data obtained from the discovery
cohort, levels of DNA methylation were consistently lower at the ZFP57 fDMR in
samples from the HF group (Figure 3.6).
Figure 3.6 Sequenom Epityper analysis of DNA methylation at the chromosome 6 fDMR in an independent set of samples (n=19). Data shows mean DNA methylation level at each CpG site with standard errors. Statistics represent paired two-tailed t-test for average DNA methylation level across all 8 CpG sites (assay average). fDMR; folate sensitive differentially methylated region.

62
3.5 Discussion
The folic acid metabolic pathway plays a critical role in fetal development (23) yet its
effects on regulation of fetal DNA methylation and gene expression are incompletely
understood. In this study we investigated the effects of third trimester maternal folate
status on the DNA methylation profile of neonatal immune cells, with a view to
identifying fDMRs in the genome. We began with a hypothesis free discovery approach
with subsequent replication of candidate loci in an independent set of samples. With this
strategy we have identified regions of the methylome that appear sensitive/responsive to
folic acid status in pregnancy.
Using a genome-scale platform we report several interesting features of the methylation
landscape in neonates stratified by folate exposure in gestation. It is noteworthy that
average DNA methylation levels, methylation levels at imprinted and repeat elements of
the genome appear unperturbed. Despite this, our analysis identified seven folate-
sensitive regions of methylation change in either direction. Folate plays a well-known
role as a carrier of one-carbon units that ultimately resulting in providing methyl groups
to methylate DNA, hence higher folate levels are expected to increase the levels of
DNA methylation. However, many studies, both in humans and animals, have reported
that high folate is associated with hypo- and hyper methylation either globally or in a
gene-specific manner (3, 11, 24). Ontology enrichment analysis of fDMRs suggests
these differences affect important genes involved in the regulation of DNA methylation
during fetal development. Principal among these is ZFP57, a gene that plays a central
role in regulation and maintenance of imprinting-associated DNA methylation (25, 26).
Both data from the microarray and the EpiTyper platform suggest HF exposure was
associated with hypomethylation of a 923bp region localized within a CpG dense
cluster 3kb upstream of the currently annotated ZFP57 transcription start site. Analysis
of histone acetylation profiles in the promoter of ZFP57 and gene expression levels
suggest hypomethylation at this fDMR is associated with markers of increased
transcriptional activity. It is therefore tempting to speculate that a compensatory
mechanism or a negative feed-back loop may exist to regulate the activity of the ZFP57
gene in response to folate exposure to stabilize DNA methylation levels at imprinting
control regions. This assertion is based on the observation that imprinted regions
targeted by the Illumina array did not differ according to folate status. However, within
what range of folate exposure that this mechanism is biologically plausible and what
other factors that may interfere with this compensatory mechanism is yet to be explored.

63
Accordingly, a recent study revealed that ZFP57 is expressed in low but detectable
levels in adult tissues including peripheral blood mononuclear cells and suggests that
ZFP57 may have other functions beyond imprinting establishment and maintenance
(27). More recently, a study focusing on genes down-regulated in association with
embryonic stem cell differentiation in mice, identified ZFP57-mediated IGF2 up
regulation in anchorage-independent growth of cancer cells (28). The authors conclude
that ZFP57 acts as an oncogene in many cancer types. Further studies are needed to
reconcile these findings.
Our two-cell type screen suggests many of the fDMRs identified in this study are
present more broadly throughout the hematopoietic compartment, and given that APCs
and CD4+ represent distinct lineages, suggest that folate driven effects on the epigenome
have originated at the common hematopoietic progenitor stage. In addition we identified
a small number of cell-specific CpG sites associated with folic acid exposure, however
it is likely we are underpowered to characterize these smaller effects in sufficient detail.
Analysis of a second sample set drawn from the source population suggests the findings
reported here are likely to be reproducible to future investigators. Despite this, we have
used an extremes of exposure design in which candidates were selected on the basis of
serum folate levels, and therefore we cannot extend the effect sizes reported here to the
general population. Nevertheless this study reports novel targets that represent viable
research avenues for more large-scale population based studies in the future. These will
be important to determine the extent to which periconceptional folate exposure modify
the disease risk in offspring.

64
3.6 References
1. Foley DL, Craig JM, Morley R, Olsson CA, Dwyer T, Smith K, et al. Prospects
for epigenetic epidemiology. Am J Epidemiol. 2009 Feb 15;169(4):389-400.
2. Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, et al. In
utero supplementation with methyl donors enhances allergic airway disease in
mice. J Clin Invest. 2008 Oct;118(10):3462-9.
3. Cho CE, Sanchez-Hernandez D, Reza-Lopez SA, Huot PS, Kim YI, Anderson
GH. High folate gestational and post-weaning diets alter hypothalamic feeding
pathways by DNA methylation in Wistar rat offspring. Epigenetics. 2013
Jul;8(7):710-9.
4. Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and
imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition.
2004 Jan;20(1):63-8.
5. Lumley J, Watson L, Watson M, Bower C. Periconceptional supplementation with
folate and/or multivitamins for preventing neural tube defects. Cochrane Database
Syst Rev. 2001(3):CD001056.
6. Kiefte-de Jong JC, Timmermans S, Jaddoe VW, Hofman A, Tiemeier H, Steegers
EA, et al. High circulating folate and vitamin B-12 concentrations in women
during pregnancy are associated with increased prevalence of atopic dermatitis in
their offspring. J Nutr. 2012 Apr;142(4):731-8.
7. Dunstan JA, West C, McCarthy S, Metcalfe J, Meldrum S, Oddy WH, et al. The
relationship between maternal folate status in pregnancy, cord blood folate levels,
and allergic outcomes in early childhood. Allergy. 2012 Jan;67(1):50-7.
8. Haberg SE, London SJ, Stigum H, Nafstad P, Nystad W. Folic acid supplements
in pregnancy and early childhood respiratory health. Arch Dis Child. 2009
Mar;94(3):180-4.
9. Whitrow MJ, Moore VM, Rumbold AR, Davies MJ. Effect of supplemental folic
acid in pregnancy on childhood asthma: a prospective birth cohort study. Am J
Epidemiol. 2009 Dec 15;170(12):1486-93.
10. Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C,
Steegers EA, et al. Periconceptional maternal folic acid use of 400 microg per day
is related to increased methylation of the IGF2 gene in the very young child. PLoS
One. 2009;4(11):e7845.
11. Hoyo C, Murtha AP, Schildkraut JM, Jirtle RL, Demark-Wahnefried W, Forman
MR, et al. Methylation variation at IGF2 differentially methylated regions and

65
maternal folic acid use before and during pregnancy. Epigenetics. 2011
Jul;6(7):928-36.
12. Girardot M, Feil R, Lleres D. Epigenetic deregulation of genomic imprinting in
humans: causal mechanisms and clinical implications. Epigenomics. 2013
Dec;5(6):715-28.
13. Chang H, Zhang T, Zhang Z, Bao R, Fu C, Wang Z, et al. Tissue-specific
distribution of aberrant DNA methylation associated with maternal low-folate
status in human neural tube defects. J Nutr Biochem. 2011 Dec;22(12):1172-7.
14. Fryer AA, Emes RD, Ismail KM, Haworth KE, Mein C, Carroll WD, et al.
Quantitative, high-resolution epigenetic profiling of CpG loci identifies
associations with cord blood plasma homocysteine and birth weight in humans.
Epigenetics. 2011 Jan;6(1):86-94.
15. Binder AM, Michels KB. The causal effect of red blood cell folate on genome-
wide methylation in cord blood: a Mendelian randomization approach. BMC
Bioinformatics. 2013;14:353.
16. Barker DJ. In utero programming of chronic disease. Clin Sci (Lond). 1998
Aug;95(2):115-28.
17. Zaghouani H, Hoeman CM, Adkins B. Neonatal immunity: faulty T-helpers and
the shortcomings of dendritic cells. Trends Immunol. 2009 Dec;30(12):585-91.
18. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al.
Differential DNA methylation in purified human blood cells: implications for cell
lineage and studies on disease susceptibility. PLoS One. 2012;7(7):e41361.
19. Martino DJ, Tulic MK, Gordon L, Hodder M, Richman T, Metcalfe J, et al.
Evidence for age-related and individual-specific changes in DNA methylation
profile of mononuclear cells during early immune development in humans.
Epigenetics. 2011 Sep 1;6(9).
20. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et
al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina
Infinium HumanMethylation450 microarray. Epigenetics. 2013 Feb;8(2):203-9.
21. Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by
surrogate variable analysis. PLoS Genet. 2007 Sep;3(9):1724-35.
22. Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump
hunting to identify differentially methylated regions in epigenetic epidemiology
studies. Int J Epidemiol. 2012 Feb;41(1):200-9.

66
23. Gueant JL, Namour F, Gueant-Rodriguez RM, Daval JL. Folate and fetal
programming: a play in epigenomics? Trends Endocrinol Metab. 2013
Jun;24(6):279-89.
24. Sie KK, Li J, Ly A, Sohn KJ, Croxford R, Kim YI. Effect of maternal and
postweaning folic acid supplementation on global and gene-specific DNA
methylation in the liver of the rat offspring. Mol Nutr Food Res. 2013
Apr;57(4):677-85.
25. Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, et al. Zinc
finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and
maintains DNA methylation imprint in embryonic stem cells via its transcriptional
repression domain. J Biol Chem. 2012 Jan 13;287(3):2107-18.
26. Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et
al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide
to affect chromatin and DNA methylation of imprinting control regions. Mol Cell.
2011 Nov 4;44(3):361-72.
27. Plant K, Fairfax BP, Makino S, Vandiedonck C, Radhakrishnan J, Knight JC. Fine
mapping genetic determinants of the highly variably expressed MHC gene ZFP57.
Eur J Hum Genet. 2013 Nov 6.
28. Tada Y, Yamaguchi Y, Kinjo T, Song X, Akagi T, Takamura H, et al. The stem
cell transcription factor ZFP57 induces IGF2 expression to promote anchorage-
independent growth in cancer cells. Oncogene. 2014 Jan 27.

67
Chapter 4
Paper 2
Folate status as a modifier of H3/H4 histone acetylation marks at
allergy-associated genes in human neonatal CD4+ T-cells
Hani Harb1*, Manori Amarasekera2*, Sarah Ashley3, Meri K. Tulic2,4, Petra Ina
Pfefferle1, Daniel P. Potaczek1 , Harald Renz1 , David Martino3,5, Dörthe A. Kesper1* &
Susan L. Prescott2*
1 Institute of Laboratory Medicine, Pathobiochemistry and Molecular Diagnostics,
Philipps University Marburg, Germany 2 School of Paediatrics and Child Health, University of Western Australia, Perth,
Australia 3 Murdoch Children’s Research Institute, Melbourne, Australia. 4 Tolerance Immunitaire, Université de Nice Sophia-Antipolis, Hôpital de l’Archet,
Nice, France 5 University of Melbourne, Melbourne, Australia
*HH and MA contributed equally to this work and share first authorship. DK and SP
contributed equally to this work and share senior authorship.
All authors are members of inFLAME (International Inflammation Network) of the
Worldwide University Network (WUN).

68
4.1 Abstract
In animal models, exposure to high maternal folate can modify developmental
programing of the fetal immune system and the risk of allergic diseases, through
epigenetic changes. While effects on the neonatal methylome have been investigated
(Chapter 3), little is known about effects on histone marks which are also sensitive to in
utero nutritional exposures. The aim of this study was to determine if variations in folate
exposure during pregnancy modulate histone acetylation of genes associated with
allergic inflammation. An in-house method was established and validated which allows
quantitative assessment of histone acetylation in candidate genes in CD4+ T-cells.
Using this protocol, histone 3 (H3) and histone 4 (H4) acetylation levels were analysed
in genes associated with allergy development in neonates exposed to high folate (HF,
n=11) and low folate (LF, n=12) in pregnancy. Baseline mRNA expression levels and
allergen and mitogen stimulated cytokine responses were measured in CD4+ T-cells and
cord blood mononuclear cells (CBMC) respectively. H3 and H4 acetylation at GATA3
and H4 acetylation at IL9 were significantly higher in neonates born to mothers with
high serum folate levels. However, there were no significant differences in baseline
mRNA expression of the corresponding genes between the folate groups indicating that
they might represent loci poised for increased transcriptional activity in response to
activation-induced signals. CBMC cytokine profiles did not reveal significant
differences between the groups, however, generally large sample sizes are required to
detect functional differences between groups. In conclusion, maternal serum folate
levels are likely influencing the developing fetal immune system by modifying histone
acetylation marks. High folate levels are associated with transcriptional permissive
chromatin state at allergy-associated loci and might thus affect disease development in
later life. The established protocol can now be employed in other cohort studies to
measure potential differences on the level of acetylation in genes of interest.

69
4.2 Introduction
In utero epigenetic variation has emerged as a key mechanism underpinning different
patterns of fetal ‘programming’ in response to variable environmental cues (1). While
this appears intended to facilitate adaption to the environmental context for short-term
survival, environment-induced epigenetic variation may also adversely influence the
susceptibility to disease later in life (2,3). Understanding environmental modulation of
the epigenetic programing during early development may lead to a better understanding
of both the developmental origins and the prevention of disease. Early dietary patterns,
and specific nutrients, have been a logical area of focus. In particular, there has been
growing interest in the role of nutrients such as folate, which have plausible epigenetic
effects. Folate is a key mediator in one-carbon metabolism that ultimately provides
methyl groups for DNA methylation (4, 5), however, its role in other aspects of
chromatin modification is less well-known.
Maternal folic acid supplementation is recommended pre-conception and in early
pregnancy to reduce the risk of neural tube defects (6). However, many women continue
to consume higher than necessary doses of folic acid later in pregnancy, well beyond the
window of risk for birth anomalies (7). Studies in mice provided compelling evidence
that supplementation with methyl donors in pregnancy (including folic acid) has
epigenetic effects on asthma-associated genes in the offspring, and increases the allergic
phenotype in the offspring (4). Subsequent observational studies in humans have also
linked folate intake with a higher risk of allergic diseases (7-9) although these findings
have not been entirely consistent (10, 11).
To explore this further, we recently conducted a genome-wide DNA methylation
analysis in immune cells isolated from neonatal cord blood, selected on the basis of in
utero folate exposure (12). Using a conventional ‘extremes of exposure’ design we
identified several regions of the neonatal methylome that appear to be sensitive/
responsive to folate status. Specifically we observed hypomethylation of a 923bp region
3kb upstream of the ZFP57 transcript, a regulator of DNA methylation during
development in neonatal immune cells. However DNA methylation machinery appeared
to be refractory to changes in folate status at allergy-associated genes in this study
population. Dietary availability of methyl donors – in addition to its effects on DNA
methylation – also changes the pattern of histone modifications (13). Therefore, we
hypothesised that folate exposure during pregnancy may have effects on H3/H4

70
acetylation marks and was examined in the same cohort of neonates. Here we developed
and validated a new chromatin immunoprecipitation method, in order to compare
acetylation marks in neonates exposed to high versus low folate levels in pregnancy.
4.3 Methods
Selection of the study population
Details of selection of the study population have been published elsewhere (12). In
brief, 23 neonates were selected from a large birth cohort according to their levels of
folate exposure. The HF (n=11) and LF (n=12) groups were defined according to the
first and third quartiles from the distribution of maternal serum folate levels in
conventional ‘extremes of exposure’ design. All study procedures were carried out in
accordance with full institutional ethics approval (by the Princess Margaret Hospital
Ethics Committee).
Serum folate measurement
Folate levels were measured in maternal and cord serum using a competitive
immunoassay (Immulite 2000; Siemans Medical Solutions Diagnostics, Flanders, NJ,
USA).
Establishment and validation of chromatin immunoprecipitation method
We developed this protocol using CD4+ T-cells from adult volunteers that was approved
by the ethics committee of the Medical Faculty of the Philipps University Marburg,
Germany.
Preparation of CD4+ T-cell chromatin for ChIP
CD4+ T-cells were isolated from CBMC using a two-stage isolation strategy with
magnetic DYNAL beads (Invitrogen). To crosslink histones with DNA, cells were fixed
at RT using 1% formaldehyde for 8 min. The reaction was stopped using 55 µl of 2.5 M
glycine. The cross-linked chromatin was treated with 5M sodium butyrate for 3 min and
then centrifuged at 8000 rpm at room temperature (RT) for 5 min to enhance the
binding of the acetyl group to histones. The chromatin was then incubated with lysis
buffer I at RT for 20 min. After centrifugation at 8 000 rpm at RT for 5 min, the
chromatin was incubated with lysis buffer II containing 1% sodium dodecyl sulfate
(SDS) at RT for 5 min followed by 3 min on ice. The chromatin was then placed
directly in the power-up bioruptor (Diagenode, Belgium) for 30 cycles, where each

71
cycle was 30” on and 30 “off (high power). Afterwards, the chromatin was centrifuged
for 15 min at 28000 rpm to remove any debris that may interfere with the process. The
supernatant was transferred to a new reaction tube and diluted to an SDS concentration
of 0.1%.
Chromatin Immunoprecipitation assay
Sepharose beads (GE Healthcare Bio-sciences AB) were washed once with lysis buffer
II and centrifuged at 1800 rpm for 2 min. After removing the supernatant, the beads
were blocked by incubating overnight at 4oC with 1 mg/ml BSA and 400 µg of salmon
sperm DNA. After the incubation, the beads were centrifuged at 1800 rpm for 5 min and
washed once with 5 ml of lysis buffer II. 30 µl of the beads per immunoprecipitation
(IP) per sample were transferred to new reaction tubes and stored until used. 20 µl beads
per antibody per sample were then added to the chromatin and incubated at 4oC for 2
hours to remove non-specific chromatin bound to Sepharose beads (pre-clearing 1).
After the incubation, supernatant with pre-cleared chromatin was transferred to new 1.5
ml reaction tubes.
The remaining beads were mixed with a combination of 500 µl of lysis buffer II and 1
µg of non-specific IgG antibody (Abcam) per sample. The beads bound to non-specific
antibody were incubated at 4oC for 1 hour. Subsequently, the beads coupled with IgG
antibody were centrifuged at 1800 rpm for 5 min and washed 3 times each with 5 ml of
lysis buffer II and centrifuged at 1800 rpm for 5 min. Next, 20 µl of IgG-coupled beads
were added to the pre-cleared chromatin and incubated at 4oC for another 2 hours to
remove chromatin bound to non-specific IgG antibodies (pre-clearing 2). At the end of
the incubation time, the pre-cleared chromatin was centrifuged at 8000 rpm at 4oC for 5
min. The supernatant containing chromatin was transferred to a new reaction tube and
10% of it was set aside for the input control.
H3 or H4 pan acetylation antibodies (4 µg) (Millipore) were added to the chromatin
obtained at the end of the cleaning process and incubated at 4oC overnight along with a
negative control prepared using 0.5 µg of IgG (Abcam). After the overnight incubation,
30 µl of the IgG-coupled beads were added to each IP and incubated at 4oC for 2 hours.
At the end of the incubation period, the beads were centrifuged at 8000 rpm for 2 min
and washed two times with wash buffer I, two times with wash buffer II, three times
with wash buffer III, and finally two times with 1X TE buffer. After discarding the

72
supernatant, 500 µl of the elution buffer were added to the mixture, vortexed and
incubated at RT for 30 min. The mixture was centrifuged at 8000 rpm for 2 min. The
supernatant containing the precipitated chromatin was transferred to a new reaction
tube.
To reverse the crosslinking, the following mixture was added to each IP product as well
as to the input controls: 20 µl of 5 M NaCl, 10 µl of 0.5M EDTA at pH 8, 20 µl of 1M
Tris at pH7.2, 1 µl of Proteinase K (20 mg/mL), and1 µl of RNase A (10 mg/mL). This
mixture ensures detachment of histones from the DNA and denaturation of the rest of
proteins and any remaining RNA. The samples were incubated at 55oC for 3 hours and
then at 65oC overnight. On the following day, DNA was purified using QIAquick kit
according to the manufacturer’s protocol. All incubation steps were performed on an
overhead rotator. Purified DNA was used in real time polymerase reaction to measure
the enrichment of different promoter regions of genes of interest. All primers are listed
in Appendix 2. The enrichment was calculated using the following formula: %
Enrichment = 100*2[(CTinput – 3.3) - CTsample] .The per cent enrichment of the negative
control (IgG) was subtracted from this value and was normalized to the house keeping
gene (HKG), RPL32 as the ratio of % Enrichment of the gene of interest to %
Enrichment to the HKG. We used RPL32 as the house keeping gene which is always
transcriptionally active and should always be associated with acetylated histone H3 and
H4. MS4A2 encodes a gene that is transcriptionally silent in CD4+ T cells and was used
as a negative control.
Quantitative PCR
First-strand cDNA was generated by reverse transcription of 500 ng of total RNA per
sample with random hexamers using SuperScript VILO (Invitrogen) according to the
manufacturer’s instructions. cDNA was diluted 1:5 in RNAse free waster for gene
expression analysis. Relative quantitation of gene expression was performed using pre-
designed TaqMan assays (Applied Biosystems) for target genes on the ABI 7300 real-
time PCR system in triplicate. All samples were normalized to the house-keeping gene.
Assessment of immune responses
Cryopreserved stocks of CBMC were thawed in RPMI media (Invitrogen, CA, USA)
and treated with DNAseI (5µg/ml) for 10 minutes to digest extracellular DNA. Cells
were washed and resuspended to a concentration of 1x106 cells/ml in appropriate culture

73
media and seeded in 96-well polystyrene plates (Thermo Scientific, MA, USA). For
assessment of immune responses, CBMC were cultured alone or with optimised doses
of house dust mite (HDM; 40µg/ml; Greer, NC, USA), ovalbumin (OVA; 100µg/ml;
Sigma Aldrich, Dartford, UK) or mitogen phytohaemaglutinin (PHA; 1µg/ml; H16,
Murex Biotech Ltd, Dartford, UK) for 48 hours in AIM-V media plus β-
mercaptoethanol (4 x 10-5mol/l). Lymphocyte cultures were maintained in 5% CO2 at 37
°C before supernatants were collected and stored at -20 °C for subsequent quantification
of cytokines (IL-5, IL-13, IFN-γ and TNF) with BioPlex200 system (Bio-Rad
Laboratories, NSW, Australia). The lower limit of detection was 3pg/ml for all the
assays.
Statistical analysis
Qualitative variables were compared by chi-square test. Relationship between two
continuous parameters was analysed using Spearman rank correlation coefficient. Initial
comparisons of quantitative variables between the two folate groups were made by
Mann-Whitney U-test. Adjustment analysis was conducted by multiple linear
regression, in which continuous variables having distribution other than normal
according to Shapiro-Wilk test (positively skewed in all cases) were approximated to
normality by square-root transformation before entering the respective model. All
statistical analyses were done using SPSS 21 software (IBM, Armonk, NY, USA).
4.4 Results
Typically Chromatin immunoprecipitation (ChIP) assays have a high requirement for
large number of freshly isolated cells. This has limited the use of this assay primarily to
cell lines rather than primary cell analysis. To address this, we developed and validated
a novel histone acetylation assay that is amenable to analysis of patient samples in a
clinical setting. We targeted this approach to specific genes previously associated with
the development of allergy. We validated this approach in a study involving 10 non
atopic healthy adult volunteers (male: female=1:1). Here we report the key features of
the protocol that influence the optimum output of the ChIP assay for human CD4+ T-
cells.
As non-activated CD4+ T- cells have a compact chromatin structure, and are resistant to
sonication (14), relatively high SDS concentrations and prolonged sonication has been a
requirement for successful fragmentation of the chromatin. Nevertheless, we were able

74
to demonstrate that pre-incubation of the cells with 1% SDS and sonication of the
chromatin in 1% SDS for 30 min (30 sec on 30 sec off at high power) can produce
chromatin with a fragment length of 100-1000bp (Figure 4.1) which is a prerequisite
for the initial steps of the assay.
In a titration experiment we quantified the lower limits of starting material necessary for
this assay. The lowest number of cells that can be used for the method is 1x105 cells per
assay (Figure 4.2A), below this number the ChIP loses its sensitivity. With this starting
number of cells, we identified the lowest enrichment measureable by the used method
for analysed genes in comparison to the positive and negative controls (Appendix 2).
Additionally, samples were frozen and thawed to estimate the freeze-thawing effect on
the enrichment percentage in comparison to freshly isolated and processed samples.
There was a trend towards lower enrichment after freezing the chromatin at some loci,
but this was not significant (Figure 4.2B) indicating that frozen samples can be used for
this assay without affecting the yield.
Figure 4.1 Outline of the ChIP assay and shearing efficiency. (A) Flow Chart outlining the main steps in ChIP method. (B) Shearing efficiency of chromatin at different sodium dodecyl sulfate (SDS) concentrations.

75
There was a significant decrease in the enrichment percentage at RPL32 (positive
control) and IL4 promoter in the 24 h RT chromatin in comparison with fresh chromatin
indicating that the samples should be analysed or frozen immediately (Table 4.1). There
was no difference in the enrichment percentage between freshly isolated and 7 or 30
days frozen chromatin or cells, thus, stored samples can be readily utilised for batch
analysis.
Figure 4.2 Lower limit of quantification and freeze-thawing effect of chromatin. (A) The lowest number of cells that can be used for this method is 1x105 cell per assay. (B) There was no significant effect of freezing on the ChIP assay.

76
Table 4.1 The effect of temperature and long-term storage on per cent enrichment of H3 and H4 histone acetylation ± SEM
Having established the ChIP assay and validating this methodology, we next applied the
technique to our clinical folate cohort to address our primary research question. We
compared H3 and H4 acetylation levels between the HF and LF groups for a panel of
key allergy-associated genes.
The characteristics of the neonates selected for this study have been published
elsewhere (12). In summary, the maternal serum folate measurement at the recruitment
visit were HF= 74.6 nmol/l, LF=16.8 nmol/l (P < .001) and cord serum levels were
HF=78.2nmol/l, LF= 36.4 nmol/l (P<.001) with clear linear relationship between
maternal serum folate and cord blood folate levels in the sample population (R=.75,
P<.001).
The level of H3 and H4 acetylation at the GATA3 locus and H4 at the IL9 locus were
higher in neonate in HF group (Figure 4.3). Additionally, there was a consistent
tendency for increased acetylation of H3 histone at the IL9 locus although this did not
reach full significance. To determine whether increased acetylation level, a marker of
increased transcriptional activity is associated with functional activity of the gene, we
measured mRNA gene expression of the loci that were significantly differently
acetylated between groups. However, there were no significant differences in baseline
mRNA expression of the analysed genes between the folate groups (data not shown). In
order to determine the functional consequences of differences in the acetylation profile
on cytokine production, bulk cord blood mononuclear cells were stimulated with house
dust mite (HDM), ovalbumin (OVA) and phytohaemaglutinin (PHA). We did not
observe any difference in cytokine profile between folate groups (data not shown).

77
Figure 4.3 Levels of H3 (A) and H4 (B) histone acetylation at key genes associated with allergic inflammation in neonatal CD4+ T-cells. All data are presented as mean relative enrichment to the house keeping gene, RPL32 ± SEM. (*P < .05, **P< 0.01)
4.5 Discussion
Recent concerns over the biological effects of in utero exposure to high levels of folate
as a potential risk factor for allergic diseases have been so far limited to animal models
(4) and a small number of human observational studies (7-11). In a recent genome-wide
methylation study, we have provided evidence for exposure to folate has effects on the
regulation of DNA methylation during fetal development (12), however, the exposure
did not appear to modulate the offspring DNA methylation at loci associated with
development of allergic inflammation. Using a candidate approach, we extended the
analysis to examine histone acetylation as an underlying mechanism for possible effects
of gestational folate exposure on the patterns of immune response in the neonate. In
particular, we examined isolated CD4+T-cells, which are central determinants of the
relative polarisation of adaptive effector responses and focused on loci which are known
to be involved in the development of allergic disease. Importantly, by using purified cell
populations in this study, we were able to avoid potential confounding effects of cell
heterogeneity that may affect the measured epigenetic changes (15, 16).
Higher acylation levels at GATA3 and IL9 loci were observed in neonates who had been
exposed to relatively higher levels of folate. GATA-3 directs CD4+ T-cell polarisation
towards Th2 pathway (17) and IL-9, initially described as a Th2 cytokine but now
shown to be produced by an array of Th subsets, is associated with allergic
inflammation and autoimmune diseases (18, 19). The data suggest that exposure to high
gestational folate is associated with more transcriptionally permissive chromatin at

78
allergy-associated genes in neonatal CD4+ T-cells, thus potentially program the
immune profile towards an allergic phenotype. The level of gene expression in resting
cells did not reflect the differences in histone acetylation; however this does not rule out
the possibility that activation-induced expression of these targets may be altered
between groups and should be addressed in future studies. The cytokine responses of
CBMC also did not reveal any difference between the folate groups. However, the
sample size in this study is smaller than generally required to detect significant
functional differences. Collectively these findings suggest that maternal folate status
may influence the fetal immune trajectory by modulating the histone acetylation marks
that may affect the disease development in later life.
This is the first study that investigated variation in histone acetylation in neonatal
immune cells according to in utero folate exposure. The folate-sensitive targets we have
identified in this study provides the future direction upon which more detailed
investigation can be undertaken particularly with large cohorts, for which, the
established protocol can now be employed.

79
4.6 References
1. Gluckman PD, Hanson MA. Developmental origins of disease paradigm: a
mechanistic and evolutionary perspective. Pediatr Res. 2004;56(3):311-7.
2. Barker DJ. In utero programming of chronic disease. Clin Sci (Lond).
1998;95(2):115-28.
3. Barker DJ. The intrauterine environment and adult cardiovascular disease. Ciba
Found Symp. 1991;156:3-10; discussion -6.
4. Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, et al. In
utero supplementation with methyl donors enhances allergic airway disease in
mice. J Clin Invest. 2008;118(10):3462-9.
5. Cho CE, Sanchez-Hernandez D, Reza-Lopez SA, Huot PS, Kim YI, Anderson
GH. High folate gestational and post-weaning diets alter hypothalamic feeding
pathways by DNA methylation in Wistar rat offspring. Epigenetics.
2013;8(7):710-9.
6. Jagerstad M. Folic acid fortification prevents neural tube defects and may also
reduce cancer risks. Acta Paediatr. 2012;101(10):1007-12.
7. Dunstan JA, West C, McCarthy S, Metcalfe J, Meldrum S, Oddy WH, et al. The
relationship between maternal folate status in pregnancy, cord blood folate levels,
and allergic outcomes in early childhood. Allergy. 2012;67(1):50-7.
8. Kiefte-de Jong JC, Timmermans S, Jaddoe VW, Hofman A, Tiemeier H, Steegers
EA, et al. High circulating folate and vitamin B-12 concentrations in women
during pregnancy are associated with increased prevalence of atopic dermatitis in
their offspring. J Nutr. 2012;142(4):731-8.
9. Whitrow MJ, Moore VM, Rumbold AR, Davies MJ. Effect of supplemental folic
acid in pregnancy on childhood asthma: a prospective birth cohort study. Am J
Epidemiol. 2009;170(12):1486-93.
10. Bekkers MB, Elstgeest LE, Scholtens S, Haveman-Nies A, de Jongste JC,
Kerkhof M, et al. Maternal use of folic acid supplements during pregnancy, and
childhood respiratory health and atopy. Eur Respir J. 2012;39(6):1468-74.
11. Magdelijns FJ, Mommers M, Penders J, Smits L, Thijs C. Folic acid use in
pregnancy and the development of atopy, asthma, and lung function in childhood.
Pediatrics. 2011;128(1):e135-44.
12. Amarasekera M, Martino D, Ashley S, Harb H, Kesper D, Strickland D, et al.
Genome-wide DNA methylation profiling identifies a folate-sensitive region of

80
differential methylation upstream of ZFP57-imprinting regulator in humans.
FASEB J. 2014.
13. Pogribny IP, Tryndyak VP, Muskhelishvili L, Rusyn I, Ross SA. Methyl
deficiency, alterations in global histone modifications, and carcinogenesis. J Nutr.
2007;137(1 Suppl):216S-22S.
14. Rawlings JS, Gatzka M, Thomas PG, Ihle JN. Chromatin condensation via the
condensin II complex is required for peripheral T-cell quiescence. EMBO J.
2011;30(2):263-76.
15. Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir
G, et al. Heterogeneity in white blood cells has potential to confound DNA
methylation measurements. PLoS One. 2012;7(10):e46705.
16. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al.
Differential DNA methylation in purified human blood cells: implications for cell
lineage and studies on disease susceptibility. PLoS One. 2012;7(7):e41361.
17. Wilson CB, Westall J, Johnston L, Lewis DB, Dower SK, Alpert AR. Decreased
production of interferon-gamma by human neonatal cells. Intrinsic and regulatory
deficiencies. J Clin Invest. 1986;77(3):860-7.
18. Pan HF, Leng RX, Li XP, Zheng SG, Ye DQ. Targeting T-helper 9 cells and
interleukin-9 in autoimmune diseases. Cytokine & growth factor reviews.
2013;24(6):515-22.
19. Brough HA, Cousins DJ, Munteanu A, Wong YF, Sudra A, Makinson K, et al. IL-
9 is a key component of memory TH cell peanut-specific responses from children
with peanut allergy. J Allergy Clin Immunol. 2014;134(6):1329-38 e10.

81
Chapter 5
Paper 3
Epigenome-wide analysis of neonatal CD4+ T-cell DNA methylation
sites potentially affected by maternal fish oil supplementation
Manori Amarasekera1, Paul Noakes1, Deborah Strickland2, Richard Saffery3,4, David J
Martino3,4,5*, Susan L Prescott1,2*
1School of Paediatrics and Child Health, University of Western Australia, Perth,
Australia 2Telethon Kids Institute, Perth, Australia 3Murdoch Children’s Research Institute, Melbourne, Australia 4Department of Paediatrics, University of Melbourne, Melbourne, Australia
5Centre for Food and Allergy Research, Melbourne, Australia
*DM and SLP contributed equally to this work and share senior authorship.
All authors are members of inFLAME (International Inflammation Network) of the
Worldwide University Network.

82
5.1 Abstract
Supplementation of fish oil rich in omega-3 polyunsaturated fatty acids (n-3 PUFA)
during pregnancy has been shown to confer favourable health outcomes in the offspring.
In a randomised controlled trial, we have previously shown that n-3 PUFA
supplementation in pregnancy was associated with modified immune responses and
some markers of immune maturation. However, the molecular mechanisms underlying
these heritable effects are unclear. To determine whether the biological effects of
maternal n-3 PUFA supplementation are mediated through DNA methylation we
analysed the CD4+ T-cells purified from cryo-banked cord blood samples from
previously conducted clinical trial. Of the 80 mother-infant pairs completed the initial
trial, cord blood samples of 70 neonates were available for genome-wide DNA
methylation profiling. Comparison of purified total CD4+ T-cell DNA methylation
profiles between the supplement and control groups did not reveal any statistically
significant differences in CpG methylation, at the single-CpG or regional level. Effect
sizes among top-ranked probes were less than 5% and did not warrant further
validation. Tests for association between methylation levels and key n-3 PUFA
parameters docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA) or total n-3
PUFAs were suggestive of dose-dependent effects, but these did not reach genome-wide
significance. Our analysis of the microarray data did not suggest strong modifying
effects of in utero n-3 PUFA exposure on CD4+ T-cell methylation profiles, and no
probes on the array met our criteria for further validation. Other epigenetic mechanisms
may be more relevant mediators of functional effects induced by n-3 PUFA in early life.

83
5.2 Introduction
Supplementation of polyunsaturated fatty acids (PUFAs) during pregnancy and during
early development has been shown to be potentially beneficial in preventing allergic (1-
3) cardiovascular (4, 5) and metabolic disorders (6) in the offspring. We have
previously shown that supplementation with n-3 PUFA DHA (22:6 n-3) and EPA (20:5
n-3) from 20 weeks of gestation until delivery modified inflammatory immune
responses in neonatal immune cells (1, 7-9). While this dietary intervention affects
many biochemical and physiological properties of cells and organs, the exact molecular
mechanism by which n-3 PUFAs exert their multi-system benefits is yet to be fully
elucidated. Specifically, in the context of heritable effects of maternal exposure, the
proposed pathways do not provide a clear molecular link.
There is mounting evidence that dietary factors can change cellular epigenetic marks in
association with more favourable clinical outcomes (10). Dietary PUFAs have also been
shown to regulate gene expression through epigenetic mechanisms (11-13). Treatment
of U937 leukemic macrophage cells with EPA has been shown to increase mRNA
expression of CCAAT/enhancer-binding protein delta with concomitant demethylation
of specific CpG loci in the gene promoter region (11) whereas treatment of M17
neuroblastoma cells with DHA tended to induce histone modifications that are
consistent with active transcription (12). Extending these findings to human cohorts, a
recent study reported 27 differentially methylated CpG sites at biologically relevant
regions that were associated with n-3 PUFA intake using an “extreme phenotypes”
model in a cohort of 185 adults (14). Another study that investigated the association of
n-6 PUFA intake and central obesity in 40 young women found that the level of TNF
promoter methylation of peripheral white cells was associated with n-6 PUFA intake
(15). Of greatest relevance here, emerging data from animal studies support a role for
maternal intake of PUFA in pregnancy in the modulation of offspring epigenetic profile
(16, 17). In a recent study involving pregnant women who had been supplemented with
DHA or placebo from the second trimester until delivery, Lee et al measured the DNA
methylation changes in genes associated with Th1, Th2, Th17, and regulatory T cell
development, as well as LINE1 repetitive elements of cord blood mononuclear cells
(18). Although there were no significant differences in DNA methylation pattern in
genes when comparing the treatment and the placebo groups, overall n-3
supplementation modified effects on methylation in the subgroup of neonates whose
mothers smoked during pregnancy. Specifically in this risk group, n-3 supplementation

84
was associated with differences in methylation levels of LINE1 repetitive element.
Collectively, these data suggests maternal n-3 PUFA intake during pregnancy may
modify the fetal epigenome, and that this may depend on other environmental
exposures.
To determine whether exposure to n-3 PUFA intake during pregnancy alters the fetal
epigenome, we compared whole genome DNA methylation profiles from neonatal
CD4+ T-cells derived from previously carried out randomized controlled trial of
maternal supplementation of fish oil during pregnancy. CD4+ T-cells were chosen as
they play a key role in immune responses and have been shown to be susceptible to in
utero perturbation from environmental exposures (19, 20). We hypothesised that
maternal n-3 PUFA supplementation may modulate epigenetic programming of CD4+
T-cells, and that this may modulate later health outcomes.
5.3 Materials and Methods
Study population
We used samples of the well-established study cohort which was a double-blind
randomised trial of pregnant mothers who were atopic and had clinical allergy (either
allergic rhinitis or asthma) but were otherwise healthy (1). They were randomised to
receive either 3.7g of fish oil (with 56.0% as DHA and 27.7% as EPA) or placebo in
capsules daily from 20 weeks of gestation until delivery. 83 women and their healthy
full-term babies completed the trial. All study procedures were carried out in
accordance with full institutional ethics. Full details of this cohort have been published
elsewhere (1).
Data and blood sample collections
Maternal fasting blood samples were collected at 20 weeks of gestation to determine the
maternal baseline fatty acids levels and at 37 weeks to calculate the increment in fatty
acids levels during the study period. Women also completed a validated semi-
quantitative food frequency questionnaire.
Cord blood samples were collected at the time of birth. Cord blood mononuclear cells
(CBMC) were harvested from blood samples within 12 hours of collection according to
standard protocols (21). Of the completed participants, a total of 70 cord bloods were
available for the present study. Maternal antenatal and socio-demographic factors and
infant clinical outcomes at 1year were also available.

85
Erythrocyte fatty acid composition analysis
Both maternal and cord blood samples were collected and processed according to
standard protocols to determine the fatty acid composition in red cells (22). The fatty
acids were expressed as a percentage of the weight of the total fatty acids measured. The
total sum of n-3 PUFAs (20:5n-3, 22:5n-3, and 22:6n-3) and n-6 PUFAs (18:2n-6,
20:3n-6, 20:4n-6, 22:3n-6, and 22:4n-6) as well as the ratio of n3 to n6 fatty acids were
also expressed.
Isolation of CD4+ T-cells
CD4+ T cells were isolated from CBMC using a two-stage positive isolation strategy
with magnetic DYNAL beads (Invitrogen). CBMC were incubated with CD8+ magnetic
beads as per the recommended protocol (Invitrogen), and the CD8- fraction was then
incubated with CD4+ magnetic beads as per the recommended protocol. Routine purity
tests were conducted by flow cytometry using antibodies CD19-FITC, CD3-PE, CD8-
PerCP, CD11c-PE Cy7, CD4-APC and CD14-APC Cy7 (BD Biosciences) and
appropriate concentration matched istoype controls. CD4+ cell purities ranged from 91-
96% pure. CD4+ T cells were lysed with RLT buffer containing 2-mercaptoethanol
(Qiagen Allprep kit) and stored at -80 ºC until genomic DNA was extracted for
methylation analysis.
Nucleic acid extraction
Genomic DNA was purified from CD4+ T- cells using Qiagen Allprep kits according to
manufacturer’s instructions.
DNA methylation analysis
DNA samples were submitted to the NXT-Dx service facility (Belgium) for bisulphite
conversion and DNA methylation profiling using the Illumina Human Methylation 450k
platform. Samples were randomized across arrays according to treatment group, age and
maternal allergy status. Raw data (iDAT files) were processed using the Minfi package
(23) from the bioconductor project (http://www.bioconductor.org) in the R statistical
environment (http://cran.r-project.org/). The minfi package was used evaluate control
probes on the array and both sample-dependent and assay-dependent control probes
indicated high quality data. Arrays were pre-processed using the stratified quantile
normalization method. Technical bias attributable to different probe chemistries
between Type 1 and Type II probes were adjusted in this procedure. Probes with a

86
detection P-value call >0.01 in 1 or more samples were removed. Probes on the X and
Y-chromosomes were removed to eliminate gender bias. Probes previously
demonstrated to potentially cross-hybridize non-specifically in the genome were
removed (24) Probes containing a polymorphic SNP at the single-base extension site
with a minor allele frequency of <0.05 were removed. The log2 ratio for methylated
probe intensity to unmethylated probe intensity, the M value, was subsequently derived
and used for statistical inference. Beta values were derived from intensities as defined
by the ratio of methylated to unmethylated probes given by B=M/(U/M*100) and were
used to compliment M-value as a measure of effect size. Cluster analysis was used in
conjunction with the chip-wide medians of the methylated and unmethylated channels
to identify any outlying samples, all samples passed QC. The final data set included 424
803 probes.
Data analysis
Following pre-processing a surrogate variable analysis was conducted on normalized
M-values to estimate any residual variation not associated with the primary outcomes of
interest using the method of Storey and Leek (25). This method estimates unwanted
variation empirically from the observed data and latent variables constructed from this
analysis were included in the regression model as covariates to negate their effects. To
identify CpG sites associated with treatment group or total n-3 fatty acid levels, all
probes were regressed on these independent variables and the empirical Bayes method
of Smyth et al (26) was used to compare treatment groups. For continuous variables, we
used the CpGassoc statistical package (27) and included surrogate variables as the
covariate matrix. QQ-plots of P-value distributions and scatterplots were used to
visualise data. Adjustment for multiple testing was performed using the Benjamini
Hochberg method (28) with adjusted P-Values < 0.1 considered significant. Our criteria
for identifying significant hits for further validation studies included a P-value < 0.05
after correction for multiple testing, and an effect size greater than 5%.
5.4 Results
Characteristics of the cohort
Of the total 83 mother-infant pairs who completed the initial randomised controlled
trial, 70 had cord blood available for this epigenome-wide DNA methylation analysis.
Table 5.1 shows the characteristics of the 70 mother-infant pairs; 36 in the fish oil

87
group and 34 in the control group. There were no significant differences in the key
maternal and neonatal parameters between treatment and control groups.
Table 5.1 Population characteristics of the mothers and infants in the cohort
(n=70)
Characteristic
Fish oil
(n=36)
Control
(n=34) P value
Mothers at recruitment Age, (yrs [SE]) 31.1 (0.7) 32.6 (0.6) 0.09
BMI at entry (kg/m2 [SE]) 25.5 (0.8) 25.9 (0.7) 0.726 Asthma (n [%]) 15 (42%) 12 (35%) Allergic rhinitis (n [%]) 21 (58%) 22 (65%) 0.584 Exposed to passive smoke (n [%]) 20 (56%) 19 (56%) 0.811 Infants
Male (n [(%]) 16 (44%) 21 (62%) 0.147 Gestation (days [SE]) 274.5 (1.4) 273.7 (1.4) 0.687 Delivery method
Vaginal (n [(%]) 26 (72%) 27 (79%) Caesarean section (n [(%]) 10 (28%) 7 (21%) 0.483
Neonatal growth parameters Birth weight (g [SE]) 3491 (57.1) 3481 (63.6) 0.906
Length (cm [SE] 50.4 (0.3) 49.7 (0.3) 0.117 Head circumference (cm [SE]) 35.2 (0.2) 34.6 (0.2) 0.065 Fatty acid composition in maternal
erythrocytes at 37 weeks of gestation
eicosapentaenoic acid, EPA (SE) 2.51 (0.11) 0.68 (0.02) < 0.001*
docosahexaenoic acid, DHA (SE) 10.75 (0.33) 6.39 (0.15) < 0.001* Arachidonic acid, AA (SE) 10.37 (0.28) 13.83 (0.16) < 0.001* Total n-3 PUFA (SE)b 22.31 (0.58) 15.98 (0.29) < 0.001* Total n-6 PUFA (SE)c 21.38 (0.53) 28.44 (0.27) < 0.001* Total n-3 to n-6 (SE)d 1.06 (0.03) 0.57 (0.01) < 0.001*
Statistical comparisons by Chi square and Man-Whitney U-test . a Values for fatty acids are expressed as a percentage of total fatty acids. b Sum 20:5, 22:5, 22:3. c Sum 18:2, 20:3, 20:4, 22:3, 22:4. d Ratio [Sum 20:5, 22:5, 22:3] to [18:2, 20:3, 20:4, 22:3, 22:4].
* indicates significant P-values (P < 0.05)
Maternal supplementation with n-3 PUFA modifies fetal red cell parameters
To determine effectiveness of the maternal intervention fatty acid parameters in cord red
blood cells were compared between treatment and intervention group. There was clear
evidence of significantly higher total n-3 (fish oil=17.8; control 13.6; P < 0.001) and
lower total n-6 (fish oil=25.2; control 29.6; P < 0.001) fatty acid levels in treatment

88
group. Key n-3 PUFAs EPA and DHA were also higher in the treatment group while
arachidonic acid a key n-6 PUFA were lower compared to that of the control group
(Figure 5.1). These readouts provided robust evidence that the maternal
supplementation modified fetal lipid profiles.
Figure 5.1 Cord red blood cell fatty acid measurements in fish oil and control groups. Boxplots represent median and range. Statistical analysis by Man-Whitney U-test. EPA-eicosapentaenoic acid, DHA-docosahexaenoic acid, AA- arachidonic acid, total n-3 PUFAs (Sum 20:5, 22:5, 22:3), total n-6 PUFAs (Sum 18:2, 20:3, 20:4, 22:3, 22:4)
Identification of CpG sites that co-associate with n-3 red cell fatty acid levels
To identify CpG sites associated with n-3 supplementation in the neonatal CD4+ T-cell
epigenome, we regressed all probes on neonatal total n-3 red cell fatty acid levels as a
continuous variable in the presence of latent covariates (methods). We did not identify
any CpG sites that were significantly associated with total n-3 levels that survived
multiple testing (Figure 5.2A). We therefore ranked all CpG sites on the array by un-
adjusted P-values and examined the top-ranked CpG sites for evidence of an effect
using scatterplots. The top 10 ranked CpG were examined by plotting methylation
versus total n-3 fatty acid level. A dose-response relationship was apparent among the
CpG sites ranked at the top of the list and these relationships were bi-directional at the
individual CpG level, with some CpG positively associated with n-3 fatty acid levels

89
and others negatively associated with increasing n-3 fatty acid levels (Figure 5.2B, 4
CpG shown for convenience). Individually these CpG sites exhibited a significant
association with total n-3 fatty acid levels although the effect sizes were small (less than
5% change in methylation) (Figure 5.2C) and not significant at the genome scale, and
therefore of unknown biological relevance. We found this to be the case regardless of
whether the data was modelled on cord red cell DHA levels or EPA levels, or whether
covariates were included in the model or not. We filtered the data set to include only the
top 25% most variable probes on the array and repeated the analysis with the same
result (data not shown).
Figure 5.2 Regression analysis of CpG sites that co-associate with cord red cell total n-3 fatty acid levels. (A) qq plot of p-values from the association test between CpGs and total n-3 fatty acids did not deviate from the null hypothesis. (B) Scatterplots of the top four CpGs ranked by p-value for the association with total n-3 fatty acids suggested a dose-response relationship. (C) Histogram of the coefficients from the regression model indicated a small effect size.
Comparison of methylation profiles between treatment and control groups
We next compared genome-wide methylation profiles between treatment and control
groups using multidimensional scaling analysis. Neither genome-wide methylation
profiles, nor the top 5000 most variable probes discriminated the two groups (Figure
5.3A). We compared treatment and control groups for differential methylation in a case
control comparison by regressing all probes on treatment group in the presence of

90
covariates (described in methods). Genome-wide profiles of methylation were not
significantly different (Figure 5.3B), nor did we detect significant differences when we
restricted the analysis to promoter regions or CpG island-associated regions only. We
ranked CpG sites by un-adjusted P-value and examined the top ranked candidates.
There was no clear evidence for a substantial effect of fish oil on the neonatal CD4+ T-
cell methylome (Figure 5.3C). This analysis was repeated for the top 25% most
variable probes, which did not show a significant difference between groups (not
shown). We also report no evidence against the null hypothesis using a sliding windows
analysis to test for regional differences in adjacent CpG sites (not shown).
Figure 5.3 Comparative analysis of DNA methylation profiles between treatment and intervention groups. (A) MDS scaling analysis did not discriminate treatment groups. Samples are indicated by open circles and projected along axes of the first two principal components of the top 5000 most variable CpG sites. (B) qq plot of p-values from the moderated-t test between intervention and control groups did not deviate from the null hypothesis. (C) Boxplots of data from six top-ranked CpG sites for the between-group comparison did not indicate substantive differences in methylation levels. Boxplots show median and range.
5.5 Discussion
This study builds upon an emerging literature suggesting dietary components can
potentially modify epigenetic regulation in humans (10). Foundational work by others

91
(11-14) suggests dietary n-3 PUFAs may alter promoter methylation in blood cells (14).
Here we examined this in the context of supplementation of n-3 PUFA during
pregnancy. In line with the emerging evidence of maternal dietary factors can influence
the neonatal epigenetic profile in modulating immune development (29, 30), we
investigated a potential role of DNA methylation as an underlying mechanism
mediating biological effects of maternal n-3 fish oil supplementation. Our primary
hypothesis was that high maternal supplementation of n-3 PUFA modifies neonatal
CD4+ T-cell DNA methylation profiles. Despite the clear evidence of a modifying effect
of maternal supplementation on red cell lipid profiles, we did not observe substantial
changes in the methylome, at least not at baseline in CD4+ T-cells. Analysis of CpG
sites that co-vary with total n-3 red cell fatty acid levels suggests there may be small
effects at specific CpG sites, but these did not appear to differ in a biologically
meaningful way between the treatment and control groups in this study. Our data do not
support the idea that n-3 PUFA supplementation in pregnancy alters the developing
fetal methylome. Whilst our analysis was restricted to a single purified cell type, we
expect than any effects due to n-3 PUFA exposure during in utero development would
be likely to propagate throughout the hematopoietic system and be present in other
blood cell types as we have reported before (19). Despite this it remains possible that
other blood cell components such as erythrocyte populations may exhibit cell-type
specific effect not observed in CD4+ T-cells. Moreover, investigating the immune
response capacity of CD4+ T-cells was beyond the scope of the present study, and
therefore it remains possible that neonatal fish oil exposure may modify activation-
induced effects on the neonatal DNA methylome. Follow-up studies are required to
address this given our data suggests any effects on the establishment and maintenance
of DNA methylation marks in the neonatal epigenome are likely to be subtle. Other
epigenetic marks such as histone modifications, which in general terms are more
dynamically responsive to environment than DNA methylation, may be key modulators
of n-3 PUFA induced effects on gene transcription. These represent further avenues of
investigation.
To date only a few animal studies have reported the potential effects of maternal PUFA
intake on the fetal epigenome (16, 17). One such study reported differing levels of
FADS2 promoter methylation in the liver tissue from offspring exposed to linoleic acid
during gestation (17). This was replicated in a rat model in which dietary fatty acid
exposure during gestation was associated with differential methylation of 4 CpG

92
dinucleotides within the FADS2 promoter (16). In humans the evidence for these effects
on the neonatal epigenome are lacking, and certainly our own study did not reproduce
these findings in animal models. Similarly, Lee and coworkers analysed a
comparatively larger birth cohort (n=261) and reported that maternal intervention
significantly modulated the LINE1 methylation which is a surrogate marker of global
methylation (18). However, the difference in the methylation level of LINE1 sequences
between supplemented and control group was about 1%, in line with our own findings
in non-repeat regions of the genome and therefore of unknown biological significance.
This is a critical issue in DNA methylation studies as the finite distribution of
methylation measurements is readily amenable to achieving statistical significance, in
situations where the variance distribution across cases and controls is small. However it
is unclear whether these small effects bear any biological relevance at all. DNA
methylation levels mitigate functional effects in specific genomic contexts, and
therefore it is imperative that functionality is established through examination of effect
sizes (with > 10% widely considered to be biologically meaningful), and genomic
context (location of methylation site relative to gene promoter regions, splice sites etc.).
Further research is therefore warranted to uncover the molecular effects of dietary fatty
acid on neonatal immune development. Based on our observations, we anticipate large
cohorts or animal models will be required to address this using genome-wide
association tests and in this respect our study informs future EWAS designs with similar
goals.
Strength of this study is the design in which a priori evidence for a modification effect
on neonatal red blood cell lipid profiles was a driver of our primary investigation.
Moreover the analysis of purified CD4+ T-cells is more homogenous than whole blood
or mononuclear cells, is therefore less subjected to unwanted variation due to cell
composition and is thus expected to provide good sensitivity (31). In summary, we
conducted a hypothesis-free epigenome-wide search to identify genomic regions in
neonatal immune cells that is modulated by maternal n-3 PUFA supplementation. Our
data argues against substantial modifying effects on the neonatal CD4+ T-cell
methylome. This suggests that the well described biological effects of n-3 PUFA are
more likely to be mediated by other epigenetic or post-transcriptional effects that
modulate cellular function.

93
5.6 References
1. Dunstan JA, Mori TA, Barden A, Beilin LJ, Taylor AL, Holt PG, et al. Fish oil
supplementation in pregnancy modifies neonatal allergen-specific immune
responses and clinical outcomes in infants at high risk of atopy: a randomized,
controlled trial. J Allergy Clin Immunol. 2003;112(6):1178-84.
2. Palmer DJ, Sullivan T, Gold MS, Prescott SL, Heddle R, Gibson RA, et al. Effect
of n-3 long chain polyunsaturated fatty acid supplementation in pregnancy on
infants' allergies in first year of life: randomised controlled trial. BMJ.
2012;344:e184.
3. Klemens CM, Berman DR, Mozurkewich EL. The effect of perinatal omega-3
fatty acid supplementation on inflammatory markers and allergic diseases: a
systematic review. BJOG. 2011;118(8):916-25.
4. Skilton MR, Ayer JG, Harmer JA, Webb K, Leeder SR, Marks GB, et al.
Impaired fetal growth and arterial wall thickening: a randomized trial of omega-3
supplementation. Pediatrics. 2012;129(3):e698-703.
5. Forsyth JS, Willatts P, Agostoni C, Bissenden J, Casaer P, Boehm G. Long chain
polyunsaturated fatty acid supplementation in infant formula and blood pressure
in later childhood: follow up of a randomised controlled trial. BMJ.
2003;326(7396):953.
6. Innis SM. Metabolic programming of long-term outcomes due to fatty acid
nutrition in early life. Matern Child Nutr. 2011;7 Suppl 2:112-23.
7. Prescott SL, Barden AE, Mori TA, Dunstan JA. Maternal fish oil supplementation
in pregnancy modifies neonatal leukotriene production by cord-blood-derived
neutrophils. Clin Sci (Lond). 2007;113(10):409-16.
8. Barden AE, Mori TA, Dunstan JA, Taylor AL, Thornton CA, Croft KD, et al.
Fish oil supplementation in pregnancy lowers F2-isoprostanes in neonates at high
risk of atopy. Free Radic Res. 2004;38(3):233-9.
9. Prescott SL, Irvine J, Dunstan JA, Hii C, Ferrante A. Protein kinase Czeta: a novel
protective neonatal T-cell marker that can be upregulated by allergy prevention
strategies. J Allergy Clin Immunol. 2007;120(1):200-6.
10. McKay JA, Mathers JC. Diet induced epigenetic changes and their implications
for health. Acta Physiol (Oxf). 2011;202(2):103-18.
11. Ceccarelli V, Racanicchi S, Martelli MP, Nocentini G, Fettucciari K, Riccardi C,
et al. Eicosapentaenoic acid demethylates a single CpG that mediates expression

94
of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia
cells. J Biol Chem. 2011;286(31):27092-102.
12. Sadli N, Ackland ML, De Mel D, Sinclair AJ, Suphioglu C. Effects of zinc and
DHA on the epigenetic regulation of human neuronal cells. Cell Physiol Biochem.
2012;29(1-2):87-98.
13. Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. Colon
cancer cell apoptosis is induced by combined exposure to the n-3 fatty acid
docosahexaenoic acid and butyrate through promoter methylation. Exp Biol Med
(Maywood). 2014;239(3):302-10.
14. Aslibekyan S, Wiener HW, Havel PJ, Stanhope KL, O'Brien DM, Hopkins SE, et
al. DNA methylation patterns are associated with n-3 fatty acid intake in Yup'ik
people. J Nutr. 2014;144(4):425-30.
15. Hermsdorff HH, Mansego ML, Campion J, Milagro FI, Zulet MA, Martinez JA.
TNF-alpha promoter methylation in peripheral white blood cells: relationship with
circulating TNFalpha, truncal fat and n-6 PUFA intake in young women.
Cytokine. 2013;64(1):265-71.
16. Hoile SP, Irvine NA, Kelsall CJ, Sibbons C, Feunteun A, Collister A, et al.
Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic
regulation of Fads2 in offspring liver. J Nutr Biochem. 2013;24(7):1213-20.
17. Niculescu MD, Lupu DS, Craciunescu CN. Perinatal manipulation of alpha-
linolenic acid intake induces epigenetic changes in maternal and offspring livers.
FASEB J. 2013;27(1):350-8.
18. Lee HS, Barraza-Villarreal A, Hernandez-Vargas H, Sly PD, Biessy C,
Ramakrishnan U, et al. Modulation of DNA methylation states and infant immune
system by dietary supplementation with omega-3 PUFA during pregnancy in an
intervention study. Am J Clin Nutr. 2013;98(2):480-7.
19. Amarasekera M, Martino D, Ashley S, Harb H, Kesper D, Strickland D, et al.
Genome-wide DNA methylation profiling identifies a folate-sensitive region of
differential methylation upstream of ZFP57-imprinting regulator in humans.
FASEB J. 2014.
20. Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, et al.
Epigenome-wide association study reveals longitudinally stable DNA methylation
differences in CD4+ T cells from children with IgE-mediated food allergy.
Epigenetics. 2014;9(7).

95
21. Martino DJ, Tulic MK, Gordon L, Hodder M, Richman T, Metcalfe J, et al.
Evidence for age-related and individual-specific changes in DNA methylation
profile of mononuclear cells during early immune development in humans.
Epigenetics. 2011;6(9).
22. Dunstan JA, Roper J, Mitoulas L, Hartmann PE, Simmer K, Prescott SL. The
effect of supplementation with fish oil during pregnancy on breast milk
immunoglobulin A, soluble CD14, cytokine levels and fatty acid composition.
Clin Exp Allergy. 2004;34(8):1237-42.
23. Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen
KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the
analysis of Infinium DNA methylation microarrays. Bioinformatics.
2014;30(10):1363-9.
24. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et
al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina
Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203-9.
25. Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by
surrogate variable analysis. PLoS Genet. 2007;3(9):1724-35.
26. Smyth GK. Linear models and empirical bayes methods for assessing differential
expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3.
27. Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: an R function for
analysis of DNA methylation microarray data. Bioinformatics. 2012;28(9):1280-
1.
28. Benjamini Y HY. Controlling the False Discovery Rate: A Practical and Powerful
Approach to Multiple Testing. JSTOR. 1995;57(1):289-300.
29. Martino DJ, Prescott SL. Silent mysteries: epigenetic paradigms could hold the
key to conquering the epidemic of allergy and immune disease. Allergy.
2010;65(1):7-15.
30. Amarasekera M, Prescott SL, Palmer DJ. Nutrition in early life, immune-
programming and allergies: the role of epigenetics. Asian Pac J Allergy Immunol.
2013;31(3):175-82.
31. Adalsteinsson BT, Gudnason H, Aspelund T, Harris TB, Launer LJ, Eiriksdottir
G, et al. Heterogeneity in white blood cells has potential to confound DNA
methylation measurements. PLoS One. 2012;7(10):e46705.

96
Chapter 6
General Discussion
This thesis describes a unique series of studies that investigate the role of maternal
dietary factors and their capacity to alter the neonatal epigenome. In the broader
context, this thesis aims to establish a mechanistic link between early life exposures, the
developing fetal epigenome and neonatal immune responses, and their role in the risk
for immune diseases. The key findings are summarised and discussed in the context of
existing literature in Section 6.1. Study limitations and suggested directions for future
research are detailed in Section 6.2 and 6.3 respectively and conclusions in Section 6.4.
6.1 Discussion of key findings in the context of existing literature
The general hypothesis underpinning this thesis was that maternal nutritional factors
have the capacity to modify the neonatal epigenome and potentially modify the risk of
immune diseases. We focused on allergic disease as one of the earliest manifestations of
NCD, with a high prevalence in paediatric populations, and thus directed our studies
towards genes and pathways important in the development of allergy. We chose to
study two main dietary factors with well-documented developmental effects that were
readily measurable in pregnant women recruited to this study. The primary aims of this
thesis were to, firstly, examine the variation in the neonatal epigenome according to
levels of in utero folate exposure and, secondly, to investigate epigenetic mechanisms as
the likely mediator of favourable immune outcomes of maternal n-3 PUFA
supplementation during pregnancy.
We took different approaches to test our hypotheses: for the study of DNA methylation
we adopted a hypothesis free approach and utilised cutting edge DNA microarray
technology to discover novel regions of the methylome that may be modified by in
utero exposures to folate. For the study of histone modifications, we adopted a
hypothesis driven approach and targeted assays to key cytokine and transcription factor
loci known to be involved in the development of allergic disease. This was in part due
to the technologies available, logistics and expenses.

97
In chapter 3, we performed a comparison of DNA methylation profiles from two
neonatally derived leukocyte populations according to the level of in utero folate
exposure. Our research efforts were directed by increasing concerns over inadvertent
adverse effects of potentially excessive maternal folate intake on offspring long-term
health outcomes and evidence from animal studies suggesting DNA methylation is the
likely mechanism of such effects. It was necessary to test this in human cohorts as
animal studies, although more tractable, may not precisely reflect the events occurring
in human epigenome.
Analysis of the microarray data revealed that DNA methylation levels at long
interspersed nucleotide element-1 (LINE-1) repeat elements and average level of DNA
methylation in the genome were not different according folate status in both CD4+ T-
cells and APC. This broad view of the neonatal methylome suggests that the neonatal
methylome is in general, resistant to changes in utero folate exposure.
Despite the global methylation status being largely unperturbed, we detected several
DMR associated with folate status, the most striking of which was a 923 bp region
broadly hypomethylated in neonates exposed to higher levels of folate in utero.
Interestingly this de-methylated region was located in a CpG island upstream of the
promoter region of ZFP57, a gene with a known role in the regulation of DNA
methylation. Analysis of H3 and H4 histone acetylation in the promoter region of
ZFP57 revealed relatively higher acetylation levels in neonates from HF group
indicating that hypomethylation of the upstream region is associated with a more
transcriptionally permissive histone mark in this group. ZFP57 mRNA expression was
higher in samples from the HF group and this is consistent with a reduction in DNA
methylation in the upstream region. We validated this key finding in an independent set
of samples using an alternative technology, Sequenom EpiTyper. Both the microarray
screen and the EpiTyper data were concordant for de-methylation in this region in both
CD4+ T-cell and APC cells.
ZFP57 plays a central role in regulating imprinting marks during development through
preferentially binding to methylated DNA in imprinted domains. This binding leads to
recruitment of DNMT to target sites to initiate or maintain the methylation of the
genomic region rendering the region transcriptionally inactive (146, 147). Aberrant
expression of imprinted genes are associated with a number of developmental disorders

98
(302), thus maintaining stable expression pattern of this region is crucial for the
development. Our genome-wide screening did not observe difference in DNA
methylation levels at imprinted control regions in high vs low folate groups indicating a
compensatory mechanism or a negative feed-back loop may exist to regulate the activity
of the ZFP57 gene in response to folate exposure to stabilise DNA methylation levels at
imprinting control regions. It is also plausible that the magnitude of difference in the
ZFP57 transcript was not sufficient to induce changes in imprinted genes or the level of
ZFP57 protein was not changed sufficiently to alter the methylation of other genes.
Analysis of dose-response relationship of folic acid on the activity of ZFP57 in
maintaining DNA methylation at imprinting control regions in mouse models that are
knocked out for the upstream regulatory region would be useful to study the precise role
of this region and is an area for future research.
Emerging evidence suggests that ZFP57 may have other functions beyond imprinting
establishment and maintenance; ZFP57 is expressed in low but detectable levels in adult
tissues including peripheral blood mononuclear cells (303), ZFP57 mediates up-
regulation in anchorage-independent growth of cancer cells in mice pointing to
oncogenic properties of ZFP57 (304). Interestingly, a recent study by Andrew
Feinberg’s group found up-stream region of ZFP57, a similar region to that we have
reported here, is differentially methylated in post mortem brain tissues from individuals
with autism spectrum disorders compared to brain tissue of healthy individuals (305).
This together with our finding indicates that aberrant methylation in this nutritionally
sensitive genomic region may be involved in disease pathogenesis and is a logical
candidate for studies investigating DOHaD.
Data from histone acetylation assays performed on the same sample population (chapter
4) revealed higher histone acetylation levels at GATA3 and IL9 loci in neonates exposed
to higher folate levels. GATA-3, the master regulator of Th2 cell lineage, activates
transcription of Th2 cytokine loci and stabilizes the differentiated state by repressing
activation of the alternative Th1 cell fate (306). Initially described as a Th2 cytokine,
IL-9 has now shown to be produced by other Th subsets and is associated with allergic
inflammation and autoimmune diseases (307, 308). These histone modifications did not
appear to alter the gene activity as determined by analysis of mRNA expression levels
in both groups. Furthermore we did not observer any concurrent changes in DNA
methylation using the microarray screen. Therefore, the biological significance of these

99
differentially acetylated histones is unclear. It is possible that they represent loci poised
for increased transcriptional activity in response to activation-induced signals. Future
studies should aim to study the activation response in addition to baseline histone
acetylation and DNA methylation levels.
Our data provide direct evidence that maternal folic acid consumption has the capacity
to modify developmental DNA methylation profiles at site-specific loci. This is the first
genome-wide study to describe these effects in humans and contribute to a growing
body of data regarding epigenetic programing of in utero nutritional exposures. Despite
these effects described here, contribution of higher levels of maternal folate to increased
risk of allergic diseases is not clear. Based on the lack of strong evidence for functional
epigenetic changes at allergy-associated loci in the experiments described here, it is
unlikely that high folic acid levels modify the risk for disease via this mechanism which
is in contrast to the findings of the highly cited mouse study by Hollingsworth et al.
Rather, it is plausible that ZFP57 may play a role in compensating the effects of folate
exposure by maintaining a stable DNA methylation level at these loci. Would this
mechanism be biologically plausible and within what range of folate exposure it would
occur are questions that need to be addressed in future studies.
In chapter 5, we performed a between-group comparison of genome-wide DNA
methylation profiles from a randomised controlled trial of maternal fish oil
supplementation during pregnancy. We studied global methylation profiles from CD4+
T-cells in neonates exposed or not exposed to n-3 PUFA in utero. Our main hypothesis
was that the beneficial effects of maternal fish oil intake during pregnancy are mediated
through epigenetic modifications. However, analysis of DNA methylation microarray
data did not provide clear evidence of substantial epigenetic effects of maternal fish oil
intake on the neonate. There were no CpG sites that were substantially modified
between treatment groups, detected effects were small and unlikely to be of biological
significance. These observations were made in a well-designed study cohort, in which
priori evidence of favourable immune outcomes of maternal fish oil supplementation
has been reported. Based on the available data, there was no convincing evidence that
biological pathways involved in mediating anti-inflammatory effects of n-3 PUFA are
likely to be regulated by DNA methylation, at least in CD4+ T-cells. It is possible that
neonatal erythrocytes, in which direct effects of the supplementation on lipid profile
were observed, may harbour CpG sites or regions amenable for modulation by maternal

100
fish oil, affecting downstream biological processes. Nevertheless, such modulations in
red cells, if present, are unlikely to have direct effects on immune development.
Folate is a crucial component of the one-carbon mediated pathway, therefore, is a
potential candidate for epigenetic regulation whereas the links with fish oil and DNA
methylation are less clear. It remains possible that the well described biological effects
of n-3 PUFA are more likely to be mediated by other epigenetic or post-transcriptional
effects that modulate cellular function. In animals, feeding dams with high fat diets was
associated with alterations in histone marks (309, 310) and reduced expression of
ncRNA (310) in the offspring supporting this notion. Studies are currently underway by
our group to extend the findings of animal experiments to human cohorts.
Collectively, the data suggest that some maternal nutritional factors have the capacity to
modulate the fetal epigenome, of which with direct effects on one-carbon mediated
pathway have the potential to exert biologically significant effects through direct effects
on DNA methylation.
6.2 Limitations of the study
The field of epigenetic epidemiology is a relatively new area of research and its
application to DOHaD research has not been extensively elaborated. While there has
been rapid advancement in technology to characterise and analyse epigenomic data over
the past 4-5 years, further evidence is required to conceptualise the study design to
achieve the maximum research output in a cost-effective manner.
In human populations the most powerful study design is the randomised controlled trial
(RCT), which can provide level one evidence regarding the effect of specific exposures.
In chapter 5 we were able to utilise such a study design, however, in chapter 4 this was
not possible given the ethical limitations of a RCT of folic acid during pregnancy.
Therefore, we were restricted to sampling from a normally distributed population. For
the discovery phase of this study we sampled individuals from the extremes of the
distribution curve in a classical ‘extremes of exposure’ design. In comparison to random
sampling, selecting individuals from the phenotypic extremes of a binary trait has
shown to increase the statistical power to discover genetic variation contributing to the
trait in GWAS (311-313). Given the high cost of genomics experiments, this type of
design maximises power to detect biological effects in a small population, however,

101
replication of findings becomes necessary due to the potential to sample individuals
who may differ in some way by chance alone. The disadvantages of such an approach
are that the results may not represent the true effect size in the total population and the
analysis is limited to the selected phenotype (313, 314). Regardless, this approach is
useful to identify relevant candidates in small cohort which then can be assessed in a
large population using a more cost-effective targeted approach. The data generated here
by this approach can now form the basis of large scale follow-up studies.
The methylation studies of this thesis were of exploratory in nature enabling
identification of DMR in response to in utero nutritional exposures. We were restricted
to employ a selected candidate approach to study histone modifications due to
requirement of higher starting material for genome-wide histone assay and to limited
available funds and it is possible that loci other than reported here may harbour histones
potentially modulated by maternal folate. We did not observe any difference in
stimulated-CBMC cytokine profile between folate groups. However, it is likely that
significant changes in the cytokine profile may not be apparent with the small number
of individuals examined for each group (11 vs 12).
Analysis of peripheral blood is considered appropriate for investigating epigenetic
modulation in immune-related diseases/phenotypes, however, only relevant tissue
samples would inform the site-specific epigenetic changes occurred following an
environmental exposure. Cord blood is one of the relatively easily accessible neonatal
biological samples, therefore, in this thesis cord blood derived leukocytes were analysed
as the reference cellular material to study the epigenetic immune programing of the
neonate. Analysis of multiple neonatal tissues would provide the opportunity to study
the degree to which different tissues show varying sensitivities to the same maternal
exposures and the extent to which this interaction influence the epigenetic programing
and disease risk, however, may not be practical in human studies. Also, it remains
unclear whether postnatal accumulation of environmentally mediated epigenetic
changes have the potential to ‘overwrite’ the epigenetic programming established at
birth. The epigenetic ‘blue print’ in cord blood derived immune cells may undergo
developmental changes in the first years of life which also pose a window of
opportunity for immune development and maturation that influence the later life health
and disease risk. Longitudinal birth cohorts with detailed collection of environmental
and other data and epigenetic analysis of multiple biospecimens will greatly facilitate

102
our understanding of nutritionally mediated epigenetic programing in influencing the
disease risk.
Despite these limitations this thesis has identified a number of neonatal epigenetic
marks that are amenable for modifications by maternal nutritional exposures upon
which detailed investigations can be carried out in future studies.
6.3 Directions for future studies in this area
The results obtained through the present studies make a substantial contribution to
advance our knowledge of nutritional programming of the fetal epigenome. Our data
provides direct evidence in humans that these effects occur but also highlight the
complexity of epigenetic dynamics. They provide direction for further studies in this
evolving area of nutri-epigenetics and recommendations for future studies are given
below.
Epigenetic regulation of gene expression occurs at multiple levels, therefore,
investigations should not be limited to DNA methylation and histone modifications.
Future studies in this area should consider investigating the contribution from other
epigenetic mechanisms such as miRNA and chromatin remodelling complexes possibly
on a genome-wide scale. It is necessary to develop cross-platform workflows for this
purposes, however, integration of multiple genome-wide molecular data sets pose huge
challenges during data processing, statistical analyses and interpreting the data.
Limited access to relevant tissue samples is an issue in human epigenetic studies, more
in studies in fetal programming. Non-invasive techniques (i.e. imaging techniques) of
epigenetic profiling will revolutionise the field of epigenetic studies enabling profiling
epigenetic marks across multiple fetal (and maternal) tissues simultaneously. Such
technology will also enable tracking dynamic epigenetic changes at different time points
during pregnancy that facilitates quantification of individual epigenetic variation to a
specific exposure.
Recent data from animal experiments points towards a role of maternal diet in shaping
the offspring gut microbiome (315-318). In mice, feeding dams a diet rich in n-3 fatty
acids have resulted in altered offspring gut microbiome, anti-inflammatory immune
response and reduced allergic reactivity to allergens suggesting that immune modulating

103
effects of maternal diet may mediate through gut microbiome. While, evidence from
animal studies are suggestive, it is necessary to replicate these findings in humans as
such randomised clinical trials of maternal supplementations with relevant
biospecimens would provide the opportunity to test this hypothesis. A recent landmark
study by Aagaard and colleagues revealed that the placenta is not devoid of microflora
as we previously thought and the pattern of placental microbiome resembles tissue-
specific maternal microbiome (319). While, it requires more studies in support of this
observation before any conclusions are made, it is intriguing to speculate that a
microbiome at the feto-maternal interface could play an important role in shaping the
fetal immune development. Furthermore, maternal dietary factors such as fish oil
supplementation may have effects on the placental microbiome with consequences on
fetal immune programing and should be investigated in future studies.
6.4 Concluding remarks
The dramatic rise in a whole range of NCD including cardiovascular, metabolic and
allergic diseases is a major health crisis of the modern world. Developmental Origin of
Health and Disease hypothesis proposed that these conditions have their origin in fetal
life and the events occurring during pregnancy play a crucial role in determining the
disease risk. The programming effects of maternal nutrition has been clearly linked with
infant immune diseases highlighting the importance of maternal nutrition in influencing
immune development and maturation. There is hope that intense research interest in
elaborating mechanistic links of dysregulated immune development following early
exposure to a number of environmental factors will shed light on better understanding
disease development and on future intervention and/or prevention strategies. In this
context, studies investigating epigenetic mechanisms as the likely mediator of
environment induced-aberrant immune development are likely to provide valuable
information on understanding disease pathogenesis.
Using cutting-edge epigenomic profiling techniques, this thesis has provided evidence
for maternal nutritional factors can modulate fetal developmental epigenome that may
have clear implications for developmental health. It further highlights the complex
interplay between different layers of epigenetic processes in regulating downstream
effects of environmental exposures and should be an important consideration in all
future studies of this nature.

104
This thesis has identified several epigenomic regions that are susceptible to modulation
by maternal nutritional factors, providing a frame work for more detailed investigations
towards identifying strategies to promote optimum immune development that will
ultimately aid reducing the burden of inflammatory NCD.

105
BIBLIOGRAPHY 1. Bach JF. The effect of infections on susceptibility to autoimmune and allergic
diseases. N Engl J Med. 2002;347(12):911-20.
2. Prescott SL. Early-life environmental determinants of allergic diseases and the
wider pandemic of inflammatory noncommunicable diseases. J Allergy Clin
Immunol. 2013;131(1):23-30.
3. Kolb H, Mandrup-Poulsen T. The global diabetes epidemic as a consequence of
lifestyle-induced low-grade inflammation. Diabetologia. 2010;53(1):10-20.
4. Prescott SL. Early origins of allergic disease: a review of processes and influences
during early immune development. Curr Opin Allergy Clin Immunol.
2003;3(2):125-32.
5. Saito S. Cytokine network at the feto-maternal interface. J Reprod Immunol.
2000;47(2):87-103.
6. Breckler LA, Hale J, Jung W, Westcott L, Dunstan JA, Thornton CA, et al.
Modulation of in vivo and in vitro cytokine production over the course of
pregnancy in allergic and non-allergic mothers. Pediatr Allergy Immunol.
2010;21(1 Pt 1):14-21.
7. Wegmann TG, Lin H, Guilbert L, Mosmann TR. Bidirectional cytokine
interactions in the maternal-fetal relationship: is successful pregnancy a TH2
phenomenon? Immunol Today. 1993;14(7):353-6.
8. Piccinni MP, Scaletti C, Maggi E, Romagnani S. Role of hormone-controlled
Th1- and Th2-type cytokines in successful pregnancy. J Neuroimmunol.
2000;109(1):30-3.
9. Schumacher A, Brachwitz N, Sohr S, Engeland K, Langwisch S, Dolaptchieva M,
et al. Human chorionic gonadotropin attracts regulatory T cells into the fetal-
maternal interface during early human pregnancy. J Immunol. 2009;182(9):5488-
97.
10. Prescott SL, Tulic M, Kumah AO, Richman T, Crook M, Martino D, et al.
Reduced placental FOXP3 associated with subsequent infant allergic disease. J
Allergy Clin Immunol. 2011;128(4):886-7 e5.
11. Lewis RM, Demmelmair H, Gaillard R, Godfrey KM, Hauguel-de Mouzon S,
Huppertz B, et al. The placental exposome: placental determinants of fetal
adiposity and postnatal body composition. Annals of nutrition & metabolism.
2013;63(3):208-15.

106
12. Novakovic B, Saffery R. The ever growing complexity of placental epigenetics -
role in adverse pregnancy outcomes and fetal programming. Placenta.
2012;33(12):959-70.
13. Koning H, Baert MR, Oranje AP, Savelkoul HF, Neijens HJ. Development of
immune functions related to allergic mechanisms in young children. Pediatr Res.
1996;40(3):363-75.
14. Mold JE, McCune JM. At the crossroads between tolerance and aggression:
Revisiting the "layered immune system" hypothesis. Chimerism. 2011;2(2):35-41.
15. Le Douarin NM, Jotereau FV. Tracing of cells of the avian thymus through
embryonic life in interspecific chimeras. J Exp Med. 1975;142(1):17-40.
16. Havran WL, Allison JP. Developmentally ordered appearance of thymocytes
expressing different T-cell antigen receptors. Nature. 1988;335(6189):443-5.
17. Havran WL, Allison JP. Origin of Thy-1+ dendritic epidermal cells of adult mice
from fetal thymic precursors. Nature. 1990;344(6261):68-70.
18. Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, Chien YH, et al. A
developmental switch in thymic lymphocyte maturation potential occurs at the
level of hematopoietic stem cells. Cell. 1990;62(5):863-74.
19. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina
SA, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell
lineages in humans. Science. 2010;330(6011):1695-9.
20. Claas FH, Gijbels Y, van der Velden-de Munck J, van Rood JJ. Induction of B
cell unresponsiveness to noninherited maternal HLA antigens during fetal life.
Science. 1988;241(4874):1815-7.
21. Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation
of regulatory T cells in the human fetus. Eur J Immunol. 2005;35(2):383-90.
22. Upham JW, Lee PT, Holt BJ, Heaton T, Prescott SL, Sharp MJ, et al.
Development of interleukin-12-producing capacity throughout childhood. Infect
Immun. 2002;70(12):6583-8.
23. Langrish CL, Buddle JC, Thrasher AJ, Goldblatt D. Neonatal dendritic cells are
intrinsically biased against Th-1 immune responses. Clin Exp Immunol.
2002;128(1):118-23.
24. Prescott SL, Macaubas C, Smallacombe T, Holt BJ, Sly PD, Holt PG.
Development of allergen-specific T-cell memory in atopic and normal children.
Lancet. 1999;353(9148):196-200.

107
25. Chen L, Cohen AC, Lewis DB. Impaired allogeneic activation and T-helper 1
differentiation of human cord blood naive CD4 T cells. Biology of blood and
marrow transplantation : journal of the American Society for Blood and Marrow
Transplantation. 2006;12(2):160-71.
26. Tulic MK, Andrews D, Crook ML, Charles A, Tourigny MR, Moqbel R, et al.
Changes in thymic regulatory T-cell maturation from birth to puberty: differences
in atopic children. J Allergy Clin Immunol. 2012;129(1):199-206 e1-4.
27. Tulic MK, Hodder M, Forsberg A, McCarthy S, Richman T, D'Vaz N, et al.
Differences in innate immune function between allergic and nonallergic children:
new insights into immune ontogeny. J Allergy Clin Immunol. 2011;127(2):470-8
e1.
28. Prescott SL, Noakes P, Chow BW, Breckler L, Thornton CA, Hollams EM, et al.
Presymptomatic differences in Toll-like receptor function in infants who have
allergy. J Allergy Clin Immunol. 2008;122(2):391-9, 9 e1-5.
29. Prescott SL, Taylor A, King B, Dunstan J, Upham JW, Thornton CA, et al.
Neonatal interleukin-12 capacity is associated with variations in allergen-specific
immune responses in the neonatal and postnatal periods. Clin Exp Allergy.
2003;33(5):566-72.
30. Smith M, Tourigny MR, Noakes P, Thornton CA, Tulic MK, Prescott SL.
Children with egg allergy have evidence of reduced neonatal
CD4(+)CD25(+)CD127(lo/-) regulatory T cell function. J Allergy Clin Immunol.
2008;121(6):1460-6, 6 e1-7.
31. Devereux G, Barker RN, Seaton A. Antenatal determinants of neonatal immune
responses to allergens. Clin Exp Allergy. 2002;32(1):43-50.
32. Pfefferle PI, Prescott SL, Kopp M. Microbial influence on tolerance and
opportunities for intervention with prebiotics/probiotics and bacterial lysates. J
Allergy Clin Immunol. 2013;131(6):1453-63; quiz 64.
33. Martino D, Holt P, Prescott S. A novel role for interleukin-1 receptor signaling in
the developmental regulation of immune responses to endotoxin. Pediatr Allergy
Immunol. 2012;23(6):567-72.
34. Yerkovich ST, Wikstrom ME, Suriyaarachchi D, Prescott SL, Upham JW, Holt
PG. Postnatal development of monocyte cytokine responses to bacterial
lipopolysaccharide. Pediatr Res. 2007;62(5):547-52.
35. Fujimaki W, Takahashi N, Ohnuma K, Nagatsu M, Kurosawa H, Yoshida S, et al.
Comparative study of regulatory T cell function of human CD25CD4 T cells from

108
thymocytes, cord blood, and adult peripheral blood. Clinical & developmental
immunology. 2008;2008:305859.
36. Delespesse G, Yang LP, Ohshima Y, Demeure C, Shu U, Byun DG, et al.
Maturation of human neonatal CD4+ and CD8+ T lymphocytes into Th1/Th2
effectors. Vaccine. 1998;16(14-15):1415-9.
37. Maeda Y, Noda S, Tanaka K, Sawamura S, Aiba Y, Ishikawa H, et al. The failure
of oral tolerance induction is functionally coupled to the absence of T cells in
Peyer's patches under germfree conditions. Immunobiology. 2001;204(4):442-57.
38. Nylund L, Satokari R, Nikkila J, Rajilic-Stojanovic M, Kalliomaki M, Isolauri E,
et al. Microarray analysis reveals marked intestinal microbiota aberrancy in
infants having eczema compared to healthy children in at-risk for atopic disease.
BMC Microbiol. 2013;13:12.
39. Bjorksten B, Naaber P, Sepp E, Mikelsaar M. The intestinal microflora in allergic
Estonian and Swedish 2-year-old children. Clin Exp Allergy. 1999;29(3):342-6.
40. West CE, Videky DJ, Prescott SL. Role of diet in the development of immune
tolerance in the context of allergic disease. Curr Opin Pediatr. 2010;22(5):635-41.
41. Castro-Rodriguez JA, Garcia-Marcos L, Alfonseda Rojas JD, Valverde-Molina J,
Sanchez-Solis M. Mediterranean diet as a protective factor for wheezing in
preschool children. J Pediatr. 2008;152(6):823-8, 8 e1-2.
42. Chatzi L, Torrent M, Romieu I, Garcia-Esteban R, Ferrer C, Vioque J, et al.
Mediterranean diet in pregnancy is protective for wheeze and atopy in childhood.
Thorax. 2008;63(6):507-13.
43. Viscogliosi G, Cipriani E, Liguori ML, Marigliano B, Saliola M, Ettorre E, et al.
Mediterranean Dietary Pattern Adherence: Associations with Prediabetes,
Metabolic Syndrome, and Related Microinflammation. Metab Syndr Relat Disord.
2013.
44. Sofi F, Abbate R, Gensini GF, Casini A. Accruing evidence on benefits of
adherence to the Mediterranean diet on health: an updated systematic review and
meta-analysis. Am J Clin Nutr. 2010;92(5):1189-96.
45. Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance.
Nat Rev Immunol. 2011;11(3):221-30.
46. Prescott SL, Dunstan JA. Prenatal fatty acid status and immune development: the
pathways and the evidence. Lipids. 2007;42(9):801-10.

109
47. Seifried HE, Anderson DE, Fisher EI, Milner JA. A review of the interaction
among dietary antioxidants and reactive oxygen species. J Nutr Biochem.
2007;18(9):567-79.
48. Stulnig TM, Zeyda M. Immunomodulation by polyunsaturated fatty acids: impact
on T-cell signaling. Lipids. 2004;39(12):1171-5.
49. Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of
prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on
COX-2 expression and IL-6 secretion. Proc Natl Acad Sci U S A.
2003;100(4):1751-6.
50. Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute
inflammation. Trends Immunol. 2007;28(4):176-83.
51. Weise C, Heunemann C, Loddenkemper C, Herz U, van Tol EA, Worm M.
Dietary docosahexaenoic acid in combination with arachidonic acid ameliorates
allergen-induced dermatitis in mice. Pediatr Allergy Immunol. 2011;22(5):497-
504.
52. Barden AE, Mori TA, Dunstan JA, Taylor AL, Thornton CA, Croft KD, et al.
Fish oil supplementation in pregnancy lowers F2-isoprostanes in neonates at high
risk of atopy. Free Radic Res. 2004;38(3):233-9.
53. Prescott SL, Barden AE, Mori TA, Dunstan JA. Maternal fish oil supplementation
in pregnancy modifies neonatal leukotriene production by cord-blood-derived
neutrophils. Clin Sci (Lond). 2007;113(10):409-16.
54. Dunstan JA, Mori TA, Barden A, Beilin LJ, Taylor AL, Holt PG, et al. Fish oil
supplementation in pregnancy modifies neonatal allergen-specific immune
responses and clinical outcomes in infants at high risk of atopy: a randomized,
controlled trial. J Allergy Clin Immunol. 2003;112(6):1178-84.
55. Palmer DJ, Sullivan T, Gold MS, Prescott SL, Heddle R, Gibson RA, et al. Effect
of n-3 long chain polyunsaturated fatty acid supplementation in pregnancy on
infants' allergies in first year of life: randomised controlled trial. BMJ.
2012;344:e184.
56. Furuhjelm C, Warstedt K, Larsson J, Fredriksson M, Bottcher MF, Falth-
Magnusson K, et al. Fish oil supplementation in pregnancy and lactation may
decrease the risk of infant allergy. Acta Paediatr. 2009;98(9):1461-7.
57. D'Vaz N, Meldrum SJ, Dunstan JA, Martino D, McCarthy S, Metcalfe J, et al.
Postnatal fish oil supplementation in high-risk infants to prevent allergy:
randomized controlled trial. Pediatrics. 2012;130(4):674-82.

110
58. Sausenthaler S, Koletzko S, Schaaf B, Lehmann I, Borte M, Herbarth O, et al.
Maternal diet during pregnancy in relation to eczema and allergic sensitization in
the offspring at 2 y of age. Am J Clin Nutr. 2007;85(2):530-7.
59. Oien T, Storro O, Johnsen R. Do early intake of fish and fish oil protect against
eczema and doctor-diagnosed asthma at 2 years of age? A cohort study. J
Epidemiol Community Health. 2010;64(2):124-9.
60. Hodge L, Salome CM, Peat JK, Haby MM, Xuan W, Woolcock AJ. Consumption
of oily fish and childhood asthma risk. Med J Aust. 1996;164(3):137-40.
61. Utsugi M, Dobashi K, Ishizuka T, Endou K, Hamuro J, Murata Y, et al. c-Jun N-
terminal kinase negatively regulates lipopolysaccharide-induced IL-12 production
in human macrophages: role of mitogen-activated protein kinase in glutathione
redox regulation of IL-12 production. J Immunol. 2003;171(2):628-35.
62. Devereux G, Turner SW, Craig LC, McNeill G, Martindale S, Harbour PJ, et al.
Low maternal vitamin E intake during pregnancy is associated with asthma in 5-
year-old children. Am J Respir Crit Care Med. 2006;174(5):499-507.
63. Martindale S, McNeill G, Devereux G, Campbell D, Russell G, Seaton A.
Antioxidant intake in pregnancy in relation to wheeze and eczema in the first two
years of life. Am J Respir Crit Care Med. 2005;171(2):121-8.
64. Forastiere F, Pistelli R, Sestini P, Fortes C, Renzoni E, Rusconi F, et al.
Consumption of fresh fruit rich in vitamin C and wheezing symptoms in children.
SIDRIA Collaborative Group, Italy (Italian Studies on Respiratory Disorders in
Children and the Environment). Thorax. 2000;55(4):283-8.
65. Okoko B, Burney P, Newson R, Potts J, Shaheen S. Childhood asthma and fruit
consumption in South London. The European Respiratory Journal. 2007:1-24.
66. Nurmatov U, Devereux G, Sheikh A. Nutrients and foods for the primary
prevention of asthma and allergy: systematic review and meta-analysis. J Allergy
Clin Immunol. 2011;127(3):724-33 e1-30.
67. Murr C, Schroecksnadel K, Winkler C, Ledochowski M, Fuchs D. Antioxidants
may increase the probability of developing allergic diseases and asthma. Med
Hypotheses. 2005;64(5):973-7.
68. Alm JS, Swartz J, Lilja G, Scheynius A, Pershagen G. Atopy in children of
families with an anthroposophic lifestyle. Lancet. 1999;353(9163):1485-8.
69. Wickens K, Pearce N, Crane J, Beasley R. Antibiotic use in early childhood and
the development of asthma. Clin Exp Allergy. 1999;29(6):766-71.

111
70. Strachan DP. Hay fever, hygiene, and household size. BMJ.
1989;299(6710):1259-60.
71. Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the
gut microbiome and the immune system. Nature. 2011;474(7351):327-36.
72. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic
endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761-
72.
73. Shi L, Li M, Miyazawa K, Li Y, Hiramatsu M, Xu J, et al. Effects of heat-
inactivated Lactobacillus gasseri TMC0356 on metabolic characteristics and
immunity of rats with the metabolic syndrome. Br J Nutr. 2013;109(2):263-72.
74. Gaboriau-Routhiau V, Raibaud P, Dubuquoy C, Moreau MC. Colonization of
gnotobiotic mice with human gut microflora at birth protects against Escherichia
coli heat-labile enterotoxin-mediated abrogation of oral tolerance. Pediatr Res.
2003;54(5):739-46.
75. Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of
intestinal bacterial flora for the development of an IgE production system fully
susceptible to oral tolerance induction. J Immunol. 1997;159(4):1739-45.
76. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of
inflammatory responses by gut microbiota and chemoattractant receptor GPR43.
Nature. 2009;461(7268):1282-6.
77. Arslanoglu S, Moro GE, Schmitt J, Tandoi L, Rizzardi S, Boehm G. Early dietary
intervention with a mixture of prebiotic oligosaccharides reduces the incidence of
allergic manifestations and infections during the first two years of life. J Nutr.
2008;138(6):1091-5.
78. Gruber C, van Stuijvenberg M, Mosca F, Moro G, Chirico G, Braegger CP, et al.
Reduced occurrence of early atopic dermatitis because of immunoactive prebiotics
among low-atopy-risk infants. J Allergy Clin Immunol. 2010;126(4):791-7.
79. Moro G, Arslanoglu S, Stahl B, Jelinek J, Wahn U, Boehm G. A mixture of
prebiotic oligosaccharides reduces the incidence of atopic dermatitis during the
first six months of age. Arch Dis Child. 2006;91(10):814-9.
80. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An
obesity-associated gut microbiome with increased capacity for energy harvest.
Nature. 2006;444(7122):1027-31.

112
81. Holmes E, Li JV, Marchesi JR, Nicholson JK. Gut microbiota composition and
activity in relation to host metabolic phenotype and disease risk. Cell Metab.
2012;16(5):559-64.
82. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut
microbiota as an environmental factor that regulates fat storage. Proc Natl Acad
Sci U S A. 2004;101(44):15718-23.
83. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al.
A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480-4.
84. Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266-81.
85. Lucas A. Programming by early nutrition: an experimental approach. J Nutr.
1998;128(2 Suppl):401S-6S.
86. Barker DJ, Fall CH. Fetal and infant origins of cardiovascular disease. Arch Dis
Child. 1993;68(6):797-9.
87. Barker DJ. In utero programming of chronic disease. Clin Sci (Lond).
1998;95(2):115-28.
88. Gluckman PD, Hanson MA. Developmental origins of disease paradigm: a
mechanistic and evolutionary perspective. Pediatr Res. 2004;56(3):311-7.
89. Martino DJ, Prescott SL. Silent mysteries: epigenetic paradigms could hold the
key to conquering the epidemic of allergy and immune disease. Allergy.
2010;65(1):7-15.
90. Desai M, Crowther NJ, Lucas A, Hales CN. Organ-selective growth in the
offspring of protein-restricted mothers. Br J Nutr. 1996;76(4):591-603.
91. Lucas A, Baker BA, Desai M, Hales CN. Nutrition in pregnant or lactating rats
programs lipid metabolism in the offspring. Br J Nutr. 1996;76(4):605-12.
92. Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al.
DNA methylation, insulin resistance, and blood pressure in offspring determined
by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci
U S A. 2007;104(49):19351-6.
93. Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, et al. In
utero supplementation with methyl donors enhances allergic airway disease in
mice. J Clin Invest. 2008;118(10):3462-9.
94. Gluckman PD, Hanson MA, Spencer HG. Predictive adaptive responses and
human evolution. Trends Ecol Evol. 2005;20(10):527-33.
95. Lee TM, Zucker I. Vole infant development is influenced perinatally by maternal
photoperiodic history. Am J Physiol. 1988;255(5 Pt 2):R831-8.

113
96. McDade TW. Early environments and the ecology of inflammation. Proc Natl
Acad Sci U S A. 2012;109 Suppl 2:17281-8.
97. Rinas U, Horneff G, Wahn V. Interferon-gamma production by cord-blood
mononuclear cells is reduced in newborns with a family history of atopic disease
and is independent from cord blood IgE-levels. Pediatr Allergy Immunol.
1993;4(2):60-4.
98. Tang ML, Kemp AS, Thorburn J, Hill DJ. Reduced interferon-gamma secretion in
neonates and subsequent atopy. Lancet. 1994;344(8928):983-5.
99. D'Vaz N, Ma Y, Dunstan JA, Lee-Pullen TF, Hii C, Meldrum S, et al. Neonatal
protein kinase C zeta expression determines the neonatal T-Cell cytokine
phenotype and predicts the development and severity of infant allergic disease.
Allergy. 2012.
100. Martino DJ, Bosco A, McKenna KL, Hollams E, Mok D, Holt PG, et al. T-cell
activation genes differentially expressed at birth in CD4+ T-cells from children
who develop IgE food allergy. Allergy. 2012;67(2):191-200.
101. Braun-Fahrlander C, Riedler J, Herz U, Eder W, Waser M, Grize L, et al.
Environmental exposure to endotoxin and its relation to asthma in school-age
children. N Engl J Med. 2002;347(12):869-77.
102. Nowak D, Heinrich J, Jorres R, Wassmer G, Berger J, Beck E, et al. Prevalence of
respiratory symptoms, bronchial hyperresponsiveness and atopy among adults:
west and east Germany. Eur Respir J. 1996;9(12):2541-52.
103. Ponsonby AL, Dwyer T, Kemp A, Couper D, Cochrane J, Carmichael A. A
prospective study of the association between home gas appliance use during
infancy and subsequent dust mite sensitization and lung function in childhood.
Clin Exp Allergy. 2001;31(10):1544-52.
104. Ottman R. Gene-environment interaction: definitions and study designs.
Preventive medicine. 1996;25(6):764-70.
105. Maher B. Personal genomes: The case of the missing heritability. Nature.
2008;456(7218):18-21.
106. McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et
al. Genome-wide association studies for complex traits: consensus, uncertainty
and challenges. Nat Rev Genet. 2008;9(5):356-69.
107. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al.
Finding the missing heritability of complex diseases. Nature.
2009;461(7265):747-53.

114
108. Waterland RA, Michels KB. Epigenetic epidemiology of the developmental
origins hypothesis. Annu Rev Nutr. 2007;27:363-88.
109. Gluckman PD, Hanson MA, Mitchell MD. Developmental origins of health and
disease: reducing the burden of chronic disease in the next generation. Genome
Med. 2010;2(2):14.
110. Kota SK, Feil R. Epigenetic transitions in germ cell development and meiosis.
Developmental cell. 2010;19(5):675-86.
111. Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V, Simon M, Agier N, et
al. Assessing the impact of transgenerational epigenetic variation on complex
traits. PLoS Genet. 2009;5(6):e1000530.
112. Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance
via the gametes in mammals. Nat Rev Genet. 2012;13(3):153-62.
113. Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene
repression. Eur J Biochem. 2001;268(1):1-6.
114. Severin PM, Zou X, Gaub HE, Schulten K. Cytosine methylation alters DNA
mechanical properties. Nucleic Acids Res. 2011;39(20):8740-51.
115. Holliday R, Grigg GW. DNA methylation and mutation. Mutat Res.
1993;285(1):61-7.
116. Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, et al. The DNA methylome of human
peripheral blood mononuclear cells. PLoS Biol. 2010;8(11):e1000533.
117. Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, et al. Hotspots of
aberrant epigenomic reprogramming in human induced pluripotent stem cells.
Nature. 2011;471(7336):68-73.
118. Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-
CpG methylation is prevalent in embryonic stem cells and may be mediated by
DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97(10):5237-42.
119. Chen T, Li E. Establishment and maintenance of DNA methylation patterns in
mammals. Curr Top Microbiol Immunol. 2006;301:179-201.
120. Cantone I, Fisher AG. Epigenetic programming and reprogramming during
development. Nat Struct Mol Biol. 2013;20(3):282-9.
121. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell.
2007;128(4):635-8.
122. Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of
CpG islands? EMBO Rep. 2012;13(1):28-35.

115
123. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet
proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass
specification. Nature. 2010;466(7310):1129-33.
124. Talbert PB, Henikoff S. Histone variants--ancient wrap artists of the epigenome.
Nat Rev Mol Cell Biol. 2010;11(4):264-75.
125. Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics,
genomics and mechanisms. Cell Res. 2011;21(3):396-420.
126. Berger SL. The complex language of chromatin regulation during transcription.
Nature. 2007;447(7143):407-12.
127. Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a
simple histone H4 acetylation code. Proc Natl Acad Sci U S A.
2005;102(15):5501-6.
128. Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology
and gene transcription. Curr Opin Drug Discov Devel. 2009;12(5):659-65.
129. Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, et al.
Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell.
2011;42(3):330-41.
130. Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular
basis for the discrimination of repressive methyl-lysine marks in histone H3 by
Polycomb and HP1 chromodomains. Genes Dev. 2003;17(15):1870-81.
131. Lu TX, Hartner J, Lim EJ, Fabry V, Mingler MK, Cole ET, et al. MicroRNA-21
limits in vivo immune response-mediated activation of the IL-12/IFN-gamma
pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J
Immunol. 2011;187(6):3362-73.
132. Fitzgerald KA, Caffrey DR. Long noncoding RNAs in innate and adaptive
immunity. Curr Opin Immunol. 2014;26C:140-6.
133. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation.
Nat Rev Genet. 2004;5(7):522-31.
134. Lucas K, Raikhel AS. Insect microRNAs: biogenesis, expression profiling and
biological functions. Insect biochemistry and molecular biology. 2013;43(1):24-
38.
135. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by
adenosines, indicates that thousands of human genes are microRNA targets. Cell.
2005;120(1):15-20.

116
136. Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat
Rev Cancer. 2006;6(4):259-69.
137. Wang JW, Li K, Hellermann G, Lockey RF, Mohapatra S. Regulating the
Regulators: microRNA and Asthma. World Allergy Organ J. 2011;4(6):94-103.
138. Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell
differentiation and plasticity. Nat Rev Immunol. 2013;13(9):666-78.
139. Erdel F, Rippe K. Chromatin remodelling in mammalian cells by ISWI-type
complexes--where, when and why? FEBS J. 2011;278(19):3608-18.
140. Bergman Y, Cedar H. DNA methylation dynamics in health and disease. Nat
Struct Mol Biol. 2013;20(3):274-81.
141. Arney KL, Bao S, Bannister AJ, Kouzarides T, Surani MA. Histone methylation
defines epigenetic asymmetry in the mouse zygote. Int J Dev Biol.
2002;46(3):317-20.
142. Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic
paternal genome. Nature. 2000;403(6769):501-2.
143. Gill ME, Erkek S, Peters AH. Parental epigenetic control of embryogenesis: a
balance between inheritance and reprogramming? Curr Opin Cell Biol.
2012;24(3):387-96.
144. Tremblay KD, Duran KL, Bartolomei MS. A 5' 2-kilobase-pair region of the
imprinted mouse H19 gene exhibits exclusive paternal methylation throughout
development. Mol Cell Biol. 1997;17(8):4322-9.
145. Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, et al. Resistance of
IAPs to methylation reprogramming may provide a mechanism for epigenetic
inheritance in the mouse. Genesis. 2003;35(2):88-93.
146. Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et
al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide
to affect chromatin and DNA methylation of imprinting control regions. Mol Cell.
2011;44(3):361-72.
147. Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, et al. Zinc
finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and
maintains DNA methylation imprint in embryonic stem cells via its transcriptional
repression domain. J Biol Chem. 2012;287(3):2107-18.
148. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ, 3rd. SETDB1: a
novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that

117
contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-
finger proteins. Genes Dev. 2002;16(8):919-32.
149. Santos F, Hendrich B, Reik W, Dean W. Dynamic reprogramming of DNA
methylation in the early mouse embryo. Dev Biol. 2002;241(1):172-82.
150. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and
Dnmt3b are essential for de novo methylation and mammalian development. Cell.
1999;99(3):247-57.
151. Keohane AM, Lavender JS, O'Neill LP, Turner BM. Histone acetylation and X
inactivation. Dev Genet. 1998;22(1):65-73.
152. Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, et al.
Role of histone H3 lysine 27 methylation in X inactivation. Science.
2003;300(5616):131-5.
153. Seki Y, Hayashi K, Itoh K, Mizugaki M, Saitou M, Matsui Y. Extensive and
orderly reprogramming of genome-wide chromatin modifications associated with
specification and early development of germ cells in mice. Dev Biol.
2005;278(2):440-58.
154. Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, et al. Epigenetic
reprogramming in mouse primordial germ cells. Mech Dev. 2002;117(1-2):15-23.
155. Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, et al.
Chromatin dynamics during epigenetic reprogramming in the mouse germ line.
Nature. 2008;452(7189):877-81.
156. Davis TL, Yang GJ, McCarrey JR, Bartolomei MS. The H19 methylation imprint
is erased and re-established differentially on the parental alleles during male germ
cell development. Hum Mol Genet. 2000;9(19):2885-94.
157. Henckel A, Chebli K, Kota SK, Arnaud P, Feil R. Transcription and histone
methylation changes correlate with imprint acquisition in male germ cells. EMBO
J. 2012;31(3):606-15.
158. Lucifero D, Mann MR, Bartolomei MS, Trasler JM. Gene-specific timing and
epigenetic memory in oocyte imprinting. Hum Mol Genet. 2004;13(8):839-49.
159. Hiura H, Obata Y, Komiyama J, Shirai M, Kono T. Oocyte growth-dependent
progression of maternal imprinting in mice. Genes Cells. 2006;11(4):353-61.
160. Liu B, Yee KM, Tahk S, Mackie R, Hsu C, Shuai K. PIAS1 SUMO ligase
regulates the self-renewal and differentiation of hematopoietic stem cells. EMBO
J. 2014;33(2):101-13.

118
161. Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, et al.
DNA methylation protects hematopoietic stem cell multipotency from
myeloerythroid restriction. Nat Genet. 2009;41(11):1207-15.
162. Konuma T, Oguro H, Iwama A. Role of the polycomb group proteins in
hematopoietic stem cells. Dev Growth Differ. 2010;52(6):505-16.
163. Santourlidis S, Graffmann N, Christ J, Uhrberg M. Lineage-specific transition of
histone signatures in the killer cell Ig-like receptor locus from hematopoietic
progenitor to NK cells. J Immunol. 2008;180(1):418-25.
164. Maier H, Ostraat R, Gao H, Fields S, Shinton SA, Medina KL, et al. Early B cell
factor cooperates with Runx1 and mediates epigenetic changes associated with
mb-1 transcription. Nat Immunol. 2004;5(10):1069-77.
165. Cao Y, Li H, Sun Y, Chen X, Liu H, Gao X, et al. Interferon regulatory factor 4
regulates thymocyte differentiation by repressing Runx3 expression. Eur J
Immunol. 2010;40(11):3198-209.
166. Rui J, Liu H, Zhu X, Cui Y, Liu X. Epigenetic silencing of CD8 genes by
ThPOK-mediated deacetylation during CD4 T cell differentiation. J Immunol.
2012;189(3):1380-90.
167. Kim SY, Romero R, Tarca AL, Bhatti G, Kim CJ, Lee J, et al. Methylome of fetal
and maternal monocytes and macrophages at the feto-maternal interface. Am J
Reprod Immunol. 2012;68(1):8-27.
168. White GP, Hollams EM, Yerkovich ST, Bosco A, Holt BJ, Bassami MR, et al.
CpG methylation patterns in the IFNgamma promoter in naive T cells: variations
during Th1 and Th2 differentiation and between atopics and non-atopics. Pediatr
Allergy Immunol. 2006;17(8):557-64.
169. Kaminuma O, Kitamura F, Miyatake S, Yamaoka K, Miyoshi H, Inokuma S, et al.
T-box 21 transcription factor is responsible for distorted T(H)2 differentiation in
human peripheral CD4+ T cells. J Allergy Clin Immunol. 2009;123(4):813-23 e3.
170. Weitzel RP, Lesniewski ML, Haviernik P, Kadereit S, Leahy P, Greco NJ, et al.
microRNA 184 regulates expression of NFAT1 in umbilical cord blood CD4+ T
cells. Blood. 2009;113(26):6648-57.
171. Goriely S, Van Lint C, Dadkhah R, Libin M, De Wit D, Demonte D, et al. A
defect in nucleosome remodeling prevents IL-12(p35) gene transcription in
neonatal dendritic cells. J Exp Med. 2004;199(7):1011-6.

119
172. Porras A, Kozar S, Russanova V, Salpea P, Hirai T, Sammons N, et al.
Developmental and epigenetic regulation of the human TLR3 gene. Mol
Immunol. 2008;46(1):27-36.
173. Chassin C, Kocur M, Pott J, Duerr CU, Gutle D, Lotz M, et al. miR-146a
mediates protective innate immune tolerance in the neonate intestine. Cell Host
Microbe. 2010;8(4):358-68.
174. Martino DJ, Tulic MK, Gordon L, Hodder M, Richman T, Metcalfe J, et al.
Evidence for age-related and individual-specific changes in DNA methylation
profile of mononuclear cells during early immune development in humans.
Epigenetics. 2011;6(9).
175. Nestor CE, Barrenas F, Wang H, Lentini A, Zhang H, Bruhn S, et al. DNA
methylation changes separate allergic patients from healthy controls and may
reflect altered CD4+ T-cell population structure. PLoS Genet.
2014;10(1):e1004059.
176. Morales E, Bustamante M, Vilahur N, Escaramis G, Montfort M, de Cid R, et al.
DNA hypomethylation at ALOX12 is associated with persistent wheezing in
childhood. Am J Respir Crit Care Med. 2012;185(9):937-43.
177. Ito K, Caramori G, Lim S, Oates T, Chung KF, Barnes PJ, et al. Expression and
activity of histone deacetylases in human asthmatic airways. Am J Respir Crit
Care Med. 2002;166(3):392-6.
178. Gaffin JM, Raby BA, Petty CR, Hoffman EB, Baccarelli AA, Gold DR, et al.
beta-2 adrenergic receptor gene methylation is associated with decreased asthma
severity in inner-city schoolchildren: asthma and rhinitis. Clin Exp Allergy.
2014;44(5):681-9.
179. Liang Y, Wang P, Zhao M, Liang G, Yin H, Zhang G, et al. Demethylation of the
FCER1G promoter leads to FcepsilonRI overexpression on monocytes of patients
with atopic dermatitis. Allergy. 2012;67(3):424-30.
180. Luo Y, Zhou B, Zhao M, Tang J, Lu Q. Promoter demethylation contributes to
TSLP overexpression in skin lesions of patients with atopic dermatitis. Clin Exp
Dermatol. 2014;39(1):48-53.
181. Sonkoly E, Janson P, Majuri ML, Savinko T, Fyhrquist N, Eidsmo L, et al. MiR-
155 is overexpressed in patients with atopic dermatitis and modulates T-cell
proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4.
J Allergy Clin Immunol. 2010;126(3):581-9 e1-20.

120
182. Seumois G, Chavez L, Gerasimova A, Lienhard M, Omran N, Kalinke L, et al.
Epigenomic analysis of primary human T cells reveals enhancers associated with
TH2 memory cell differentiation and asthma susceptibility. Nat Immunol.
2014;15(8):777-88.
183. Breton CV, Byun HM, Wang X, Salam MT, Siegmund K, Gilliland FD. DNA
methylation in the arginase-nitric oxide synthase pathway is associated with
exhaled nitric oxide in children with asthma. Am J Respir Crit Care Med.
2011;184(2):191-7.
184. Runyon RS, Cachola LM, Rajeshuni N, Hunter T, Garcia M, Ahn R, et al. Asthma
discordance in twins is linked to epigenetic modifications of T cells. PLoS One.
2012;7(11):e48796.
185. Stefanowicz D, Hackett TL, Garmaroudi FS, Gunther OP, Neumann S, Sutanto
EN, et al. DNA methylation profiles of airway epithelial cells and PBMCs from
healthy, atopic and asthmatic children. PLoS One. 2012;7(9):e44213.
186. Soto-Ramirez N, Arshad SH, Holloway JW, Zhang H, Schauberger E, Ewart S, et
al. The interaction of genetic variants and DNA methylation of the interleukin-4
receptor gene increase the risk of asthma at age 18 years. Clin Epigenetics.
2013;5(1):1.
187. Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, et al. Relation of
DNA methylation of 5'-CpG island of ACSL3 to transplacental exposure to
airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One.
2009;4(2):e4488.
188. Fu A, Leaderer BP, Gent JF, Leaderer D, Zhu Y. An environmental epigenetic
study of ADRB2 5'-UTR methylation and childhood asthma severity. Clin Exp
Allergy. 2012;42(11):1575-81.
189. Pascual M, Suzuki M, Isidoro-Garcia M, Padron J, Turner T, Lorente F, et al.
Epigenetic changes in B lymphocytes associated with house dust mite allergic
asthma. Epigenetics. 2011;6(9):1131-7.
190. Zhao M, Liu S, Luo S, Wu H, Tang M, Cheng W, et al. DNA methylation and
mRNA and microRNA expression of SLE CD4+ T cells correlate with disease
phenotype. J Autoimmun. 2014.
191. Koh BH, Hwang SS, Kim JY, Lee W, Kang MJ, Lee CG, et al. Th2 LCR is
essential for regulation of Th2 cytokine genes and for pathogenesis of allergic
asthma. Proc Natl Acad Sci U S A. 2010;107(23):10614-9.

121
192. Brand S, Kesper DA, Teich R, Kilic-Niebergall E, Pinkenburg O, Bothur E, et al.
DNA methylation of TH1/TH2 cytokine genes affects sensitization and progress
of experimental asthma. J Allergy Clin Immunol. 2012;129(6):1602-10 e6.
193. Allan RS, Zueva E, Cammas F, Schreiber HA, Masson V, Belz GT, et al. An
epigenetic silencing pathway controlling T helper 2 cell lineage commitment.
Nature. 2012;487(7406):249-53.
194. Kumar M, Ahmad T, Sharma A, Mabalirajan U, Kulshreshtha A, Agrawal A, et
al. Let-7 microRNA-mediated regulation of IL-13 and allergic airway
inflammation. J Allergy Clin Immunol. 2011;128(5):1077-85 e1-10.
195. Polikepahad S, Knight JM, Naghavi AO, Oplt T, Creighton CJ, Shaw C, et al.
Proinflammatory role for let-7 microRNAS in experimental asthma. J Biol Chem.
2010;285(39):30139-49.
196. Collison A, Mattes J, Plank M, Foster PS. Inhibition of house dust mite-induced
allergic airways disease by antagonism of microRNA-145 is comparable to
glucocorticoid treatment. J Allergy Clin Immunol. 2011;128(1):160-7 e4.
197. Barker DJ. The intrauterine environment and adult cardiovascular disease. Ciba
Found Symp. 1991;156:3-10; discussion -6.
198. Martino D, Prescott S. Epigenetics and prenatal influences on asthma and allergic
airways disease. Chest. 2011;139(3):640-7.
199. Fedulov AV, Kobzik L. Allergy risk is mediated by dendritic cells with congenital
epigenetic changes. Am J Respir Cell Mol Biol. 2011;44(3):285-92.
200. Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, et al.
Epigenome-wide association study reveals longitudinally stable DNA methylation
differences in CD4+ T cells from children with IgE-mediated food allergy.
Epigenetics. 2014;9(7):998-1006.
201. Harding JE, Johnston BM. Nutrition and fetal growth. Reprod Fertil Dev.
1995;7(3):539-47.
202. Stein AD, Zybert PA, van de Bor M, Lumey LH. Intrauterine famine exposure
and body proportions at birth: the Dutch Hunger Winter. Int J Epidemiol.
2004;33(4):831-6.
203. Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure
in utero and early infancy. N Engl J Med. 1976;295(7):349-53.
204. Smith CA. Effects of maternal under nutrition upon the newborn infant in Holland
(1944-1945). J Pediatr. 1947;30(3):229-43.

122
205. Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, et al.
Schizophrenia after prenatal famine. Further evidence. Archives of general
psychiatry. 1996;53(1):25-31.
206. Caron E, Ciofi P, Prevot V, Bouret SG. Alteration in neonatal nutrition causes
perturbations in hypothalamic neural circuits controlling reproductive function. J
Neurosci. 2012;32(33):11486-94.
207. Nian H, Delage B, Ho E, Dashwood RH. Modulation of histone deacetylase
activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane
and garlic organosulfur compounds. Environ Mol Mutagen. 2009;50(3):213-21.
208. Mathers JC. Session 2: Personalised nutrition. Epigenomics: a basis for
understanding individual differences? Proc Nutr Soc. 2008;67(4):390-4.
209. Li G, Kohorst JJ, Zhang W, Laritsky E, Kunde-Ramamoorthy G, Baker MS, et al.
Early postnatal nutrition determines adult physical activity and energy
expenditure in female mice. Diabetes. 2013;62(8):2773-83.
210. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al.
Persistent epigenetic differences associated with prenatal exposure to famine in
humans. Proc Natl Acad Sci U S A. 2008;105(44):17046-9.
211. McKay JA, Mathers JC. Diet induced epigenetic changes and their implications
for health. Acta Physiol (Oxf). 2011;202(2):103-18.
212. Choi SW, Friso S. Epigenetics: A New Bridge between Nutrition and Health. Adv
Nutr. 2010;1(1):8-16.
213. Shenderov BA. Gut indigenous microbiota and epigenetics. Microbial ecology in
health and disease. 2012;23.
214. Xue X, Feng T, Yao S, Wolf KJ, Liu CG, Liu X, et al. Microbiota downregulates
dendritic cell expression of miR-10a, which targets IL-12/IL-23p40. J Immunol.
2011;187(11):5879-86.
215. Kim GW, Gocevski G, Wu CJ, Yang XJ. Dietary, metabolic, and potentially
environmental modulation of the lysine acetylation machinery. Int J Cell Biol.
2010;2010:632739.
216. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al.
Metabolites produced by commensal bacteria promote peripheral regulatory T-cell
generation. Nature. 2013;504(7480):451-5.
217. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al.
Commensal microbe-derived butyrate induces the differentiation of colonic
regulatory T cells. Nature. 2013;504(7480):446-50.

123
218. Gueant JL, Namour F, Gueant-Rodriguez RM, Daval JL. Folate and fetal
programming: a play in epigenomics? Trends Endocrinol Metab. 2013;24(6):279-
89.
219. Christensen KE, MacKenzie RE. Mitochondrial one-carbon metabolism is
adapted to the specific needs of yeast, plants and mammals. Bioessays.
2006;28(6):595-605.
220. Weingartner J, Lotz K, Fanghanel J, Gedrange T, Bienengraber V, Proff P.
Induction and prevention of cleft lip, alveolus and palate and neural tube defects
with special consideration of B vitamins and the methylation cycle. J Orofac
Orthop. 2007;68(4):266-77.
221. Gueant JL, Gueant-Rodriguez RM, Anello G, Bosco P, Brunaud L, Romano C, et
al. Genetic determinants of folate and vitamin B12 metabolism: a common
pathway in neural tube defect and Down syndrome? Clin Chem Lab Med.
2003;41(11):1473-7.
222. Blom HJ, Smulders Y. Overview of homocysteine and folate metabolism. With
special references to cardiovascular disease and neural tube defects. J Inherit
Metab Dis. 2011;34(1):75-81.
223. Berry RJ, Li Z, Erickson JD, Li S, Moore CA, Wang H, et al. Prevention of
neural-tube defects with folic acid in China. China-U.S. Collaborative Project for
Neural Tube Defect Prevention. N Engl J Med. 1999;341(20):1485-90.
224. Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by
periconceptional vitamin supplementation. N Engl J Med. 1992;327(26):1832-5.
225. Crane NT, Wilson DB, Cook DA, Lewis CJ, Yetley EA, Rader JI. Evaluating
food fortification options: general principles revisited with folic acid. Am J Public
Health. 1995;85(5):660-6.
226. Lewis CJ, Crane NT, Wilson DB, Yetley EA. Estimated folate intakes: data
updated to reflect food fortification, increased bioavailability, and dietary
supplement use. Am J Clin Nutr. 1999;70(2):198-207.
227. Quinlivan EP, Gregory JF, 3rd. Effect of food fortification on folic acid intake in
the United States. Am J Clin Nutr. 2003;77(1):221-5.
228. Dunstan JA, West C, McCarthy S, Metcalfe J, Meldrum S, Oddy WH, et al. The
relationship between maternal folate status in pregnancy, cord blood folate levels,
and allergic outcomes in early childhood. Allergy. 2012;67(1):50-7.
229. Fekete K, Berti C, Trovato M, Lohner S, Dullemeijer C, Souverein OW, et al.
Effect of folate intake on health outcomes in pregnancy: a systematic review and

124
meta-analysis on birth weight, placental weight and length of gestation. Nutr J.
2012;11:75.
230. Fainaru O, Shseyov D, Hantisteanu S, Groner Y. Accelerated chemokine receptor
7-mediated dendritic cell migration in Runx3 knockout mice and the spontaneous
development of asthma-like disease. Proc Natl Acad Sci U S A.
2005;102(30):10598-603.
231. Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at
the agouti locus in the mouse. Nat Genet. 1999;23(3):314-8.
232. Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl
supplements affect agouti gene expression in Avy/a mice. FASEB J.
1998;12(11):949-57.
233. Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional
effects on epigenetic gene regulation. Mol Cell Biol. 2003;23(15):5293-300.
234. Kiefte-de Jong JC, Timmermans S, Jaddoe VW, Hofman A, Tiemeier H, Steegers
EA, et al. High circulating folate and vitamin B-12 concentrations in women
during pregnancy are associated with increased prevalence of atopic dermatitis in
their offspring. J Nutr. 2012;142(4):731-8.
235. Whitrow MJ, Moore VM, Rumbold AR, Davies MJ. Effect of supplemental folic
acid in pregnancy on childhood asthma: a prospective birth cohort study. Am J
Epidemiol. 2009;170(12):1486-93.
236. Bekkers MB, Elstgeest LE, Scholtens S, Haveman-Nies A, de Jongste JC,
Kerkhof M, et al. Maternal use of folic acid supplements during pregnancy, and
childhood respiratory health and atopy. Eur Respir J. 2012;39(6):1468-74.
237. Magdelijns FJ, Mommers M, Penders J, Smits L, Thijs C. Folic acid use in
pregnancy and the development of atopy, asthma, and lung function in childhood.
Pediatrics. 2011;128(1):e135-44.
238. Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C,
Steegers EA, et al. Periconceptional maternal folic acid use of 400 microg per day
is related to increased methylation of the IGF2 gene in the very young child. PLoS
One. 2009;4(11):e7845.
239. Hoyo C, Murtha AP, Schildkraut JM, Jirtle RL, Demark-Wahnefried W, Forman
MR, et al. Methylation variation at IGF2 differentially methylated regions and
maternal folic acid use before and during pregnancy. Epigenetics. 2011;6(7):928-
36.

125
240. McKay JA, Groom A, Potter C, Coneyworth LJ, Ford D, Mathers JC, et al.
Genetic and non-genetic influences during pregnancy on infant global and site
specific DNA methylation: role for folate gene variants and vitamin B12. PLoS
One. 2012;7(3):e33290.
241. Ba Y, Yu H, Liu F, Geng X, Zhu C, Zhu Q, et al. Relationship of folate, vitamin
B12 and methylation of insulin-like growth factor-II in maternal and cord blood.
Eur J Clin Nutr. 2011;65(4):480-5.
242. Fryer AA, Nafee TM, Ismail KM, Carroll WD, Emes RD, Farrell WE. LINE-1
DNA methylation is inversely correlated with cord plasma homocysteine in man:
a preliminary study. Epigenetics. 2009;4(6):394-8.
243. Boeke CE, Baccarelli A, Kleinman KP, Burris HH, Litonjua AA, Rifas-Shiman
SL, et al. Gestational intake of methyl donors and global LINE-1 DNA
methylation in maternal and cord blood: prospective results from a folate-replete
population. Epigenetics. 2012;7(3):253-60.
244. Fryer AA, Emes RD, Ismail KM, Haworth KE, Mein C, Carroll WD, et al.
Quantitative, high-resolution epigenetic profiling of CpG loci identifies
associations with cord blood plasma homocysteine and birth weight in humans.
Epigenetics. 2011;6(1):86-94.
245. Chang H, Zhang T, Zhang Z, Bao R, Fu C, Wang Z, et al. Tissue-specific
distribution of aberrant DNA methylation associated with maternal low-folate
status in human neural tube defects. J Nutr Biochem. 2011;22(12):1172-7.
246. Simopoulos AP. The importance of the ratio of omega-6/omega-3 essential fatty
acids. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie.
2002;56(8):365-79.
247. Prescott SL, Irvine J, Dunstan JA, Hii C, Ferrante A. Protein kinase Czeta: a novel
protective neonatal T-cell marker that can be upregulated by allergy prevention
strategies. J Allergy Clin Immunol. 2007;120(1):200-6.
248. Noakes PS, Vlachava M, Kremmyda LS, Diaper ND, Miles EA, Erlewyn-
Lajeunesse M, et al. Increased intake of oily fish in pregnancy: effects on neonatal
immune responses and on clinical outcomes in infants at 6 mo. Am J Clin Nutr.
2012;95(2):395-404.
249. Childs CE, Romijn T, Enke U, Hoile S, Calder PC. Maternal diet during
pregnancy has tissue-specific effects upon fetal fatty acid composition and alters
fetal immune parameters. Prostaglandins Leukot Essent Fatty Acids. 2010;83(4-
6):179-84.

126
250. Calder PC. Mechanisms of action of (n-3) fatty acids. J Nutr. 2012;142(3):592S-
9S.
251. Hoile SP, Irvine NA, Kelsall CJ, Sibbons C, Feunteun A, Collister A, et al.
Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic
regulation of Fads2 in offspring liver. J Nutr Biochem. 2013;24(7):1213-20.
252. Niculescu MD, Lupu DS, Craciunescu CN. Perinatal manipulation of alpha-
linolenic acid intake induces epigenetic changes in maternal and offspring livers.
FASEB J. 2013;27(1):350-8.
253. Lee HS, Barraza-Villarreal A, Hernandez-Vargas H, Sly PD, Biessy C,
Ramakrishnan U, et al. Modulation of DNA methylation states and infant immune
system by dietary supplementation with omega-3 PUFA during pregnancy in an
intervention study. Am J Clin Nutr. 2013;98(2):480-7.
254. Magnusson LL, Olesen AB, Wennborg H, Olsen J. Wheezing, asthma, hayfever,
and atopic eczema in childhood following exposure to tobacco smoke in fetal life.
Clin Exp Allergy. 2005;35(12):1550-6.
255. Bisgaard H, Loland L, Holst KK, Pipper CB. Prenatal determinants of neonatal
lung function in high-risk newborns. J Allergy Clin Immunol. 2009;123(3):651-7,
7 e1-4.
256. Sears MR, Holdaway MD, Flannery EM, Herbison GP, Silva PA. Parental and
neonatal risk factors for atopy, airway hyper-responsiveness, and asthma. Arch
Dis Child. 1996;75(5):392-8.
257. Li YF, Langholz B, Salam MT, Gilliland FD. Maternal and grandmaternal
smoking patterns are associated with early childhood asthma. Chest.
2005;127(4):1232-41.
258. Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al.
450K epigenome-wide scan identifies differential DNA methylation in newborns
related to maternal smoking during pregnancy. Environ Health Perspect.
2012;120(10):1425-31.
259. Novakovic B, Ryan J, Pereira N, Boughton B, Craig JM, Saffery R. Postnatal
stability, tissue, and time specific effects of AHRR methylation change in
response to maternal smoking in pregnancy. Epigenetics. 2014;9(3):377-86.
260. Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH, et al.
Coordinated changes in AHRR methylation in lymphoblasts and pulmonary
macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet.
2012;159B(2):141-51.

127
261. Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, et
al. Epigenome-wide association study in the European Prospective Investigation
into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated
with smoking. Hum Mol Genet. 2013;22(5):843-51.
262. Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon
receptor repressor as a biomarker for nascent smokers. Epigenetics.
2012;7(11):1331-8.
263. Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al.
Tobacco smoking leads to extensive genome-wide changes in DNA methylation.
PLoS One. 2013;8(5):e63812.
264. Toledo-Rodriguez M, Lotfipour S, Leonard G, Perron M, Richer L, Veillette S, et
al. Maternal smoking during pregnancy is associated with epigenetic
modifications of the brain-derived neurotrophic factor-6 exon in adolescent
offspring. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(7):1350-4.
265. Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco
smoke exposure affects global and gene-specific DNA methylation. Am J Respir
Crit Care Med. 2009;180(5):462-7.
266. Patil VK, Holloway JW, Zhang H, Soto-Ramirez N, Ewart S, Arshad SH, et al.
Interaction of prenatal maternal smoking, interleukin 13 genetic variants and
DNA methylation influencing airflow and airway reactivity. Clin Epigenetics.
2013;5(1):22.
267. Liu G, Niu Z, Van Niekerk D, Xue J, Zheng L. Polycyclic aromatic hydrocarbons
(PAHs) from coal combustion: emissions, analysis, and toxicology. Rev Environ
Contam Toxicol. 2008;192:1-28.
268. Simko P. Factors affecting elimination of polycyclic aromatic hydrocarbons from
smoked meat foods and liquid smoke flavorings. Mol Nutr Food Res.
2005;49(7):637-47.
269. Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA.
Environmental pollutants and breast cancer: epidemiologic studies. Cancer.
2007;109(12 Suppl):2667-711.
270. Factor P, Akhmedov AT, McDonald JD, Qu A, Wu J, Jiang H, et al. Polycyclic
Aromatic Hydrocarbons Impair 2AR Function in Airway Epithelial and Smooth
Muscle Cells. Am J Respir Cell Mol Biol. 2011.

128
271. Choi H, Jedrychowski W, Spengler J, Camann DE, Whyatt RM, Rauh V, et al.
International studies of prenatal exposure to polycyclic aromatic hydrocarbons
and fetal growth. Environ Health Perspect. 2006;114(11):1744-50.
272. Perera FP, Rauh V, Whyatt RM, Tang D, Tsai WY, Bernert JT, et al. A summary
of recent findings on birth outcomes and developmental effects of prenatal ETS,
PAH, and pesticide exposures. Neurotoxicology. 2005;26(4):573-87.
273. Tang WY, Levin L, Talaska G, Cheung YY, Herbstman J, Tang D, et al. Maternal
exposure to polycyclic aromatic hydrocarbons and 5'-CpG methylation of
interferon-gamma in cord white blood cells. Environ Health Perspect.
2012;120(8):1195-200.
274. Liu J, Ballaney M, Al-alem U, Quan C, Jin X, Perera F, et al. Combined inhaled
diesel exhaust particles and allergen exposure alter methylation of T helper genes
and IgE production in vivo. Toxicol Sci. 2008;102(1):76-81.
275. Gilmour PS, Rahman I, Donaldson K, MacNee W. Histone acetylation regulates
epithelial IL-8 release mediated by oxidative stress from environmental particles.
Am J Physiol Lung Cell Mol Physiol. 2003;284(3):L533-40.
276. Michel S, Busato F, Genuneit J, Pekkanen J, Dalphin JC, Riedler J, et al. Farm
exposure and time trends in early childhood may influence DNA methylation in
genes related to asthma and allergy. Allergy. 2013;68(3):355-64.
277. Slaats GG, Reinius LE, Alm J, Kere J, Scheynius A, Joerink M. DNA methylation
levels within the CD14 promoter region are lower in placentas of mothers living
on a farm. Allergy. 2012;67(7):895-903.
278. Schaub B, Liu J, Hoppler S, Schleich I, Huehn J, Olek S, et al. Maternal farm
exposure modulates neonatal immune mechanisms through regulatory T cells. J
Allergy Clin Immunol. 2009;123(4):774-82 e5.
279. Douwes J, Cheng S, Travier N, Cohet C, Niesink A, McKenzie J, et al. Farm
exposure in utero may protect against asthma, hay fever and eczema. Eur Respir J.
2008;32(3):603-11.
280. Brand S, Teich R, Dicke T, Harb H, Yildirim AO, Tost J, et al. Epigenetic
regulation in murine offspring as a novel mechanism for transmaternal asthma
protection induced by microbes. J Allergy Clin Immunol. 2011;128(3):618-25 e1-
7.
281. Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in
analysis of DNA methylation data. Bioinformatics. 2014;30(10):1431-9.

129
282. Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, et al.
Genome-wide survey reveals predisposing diabetes type 2-related DNA
methylation variations in human peripheral blood. Hum Mol Genet.
2012;21(2):371-83.
283. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al.
Identification of type 1 diabetes-associated DNA methylation variable positions
that precede disease diagnosis. PLoS Genet. 2011;7(9):e1002300.
284. Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano
M, et al. Season of conception in rural gambia affects DNA methylation at
putative human metastable epialleles. PLoS Genet. 2010;6(12):e1001252.
285. Meagher RB, Mussar KJ. The influence of DNA sequence on epigenome-induced
pathologies. Epigenetics & chromatin. 2012;5(1):11.
286. Laird PW. Principles and challenges of genomewide DNA methylation analysis.
Nat Rev Genet. 2010;11(3):191-203.
287. Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies
for common human diseases. Nat Rev Genet. 2011;12(8):529-41.
288. Dunstan JA, Prescott SL. Does fish oil supplementation in pregnancy reduce the
risk of allergic disease in infants? Curr Opin Allergy Clin Immunol.
2005;5(3):215-21.
289. D'Vaz N, Meldrum SJ, Dunstan JA, Lee-Pullen TF, Metcalfe J, Holt BJ, et al.
Fish oil supplementation in early infancy modulates developing infant immune
responses. Clin Exp Allergy. 2012;42(8):1206-16.
290. Pfeiffer CM, Caudill SP, Gunter EW, Osterloh J, Sampson EJ. Biochemical
indicators of B vitamin status in the US population after folic acid fortification:
results from the National Health and Nutrition Examination Survey 1999-2000.
Am J Clin Nutr. 2005;82(2):442-50.
291. Shane B. Folate fortification: enough already? Am J Clin Nutr. 2003;77(1):8-9.
292. Haberg SE, London SJ, Stigum H, Nafstad P, Nystad W. Folic acid supplements
in pregnancy and early childhood respiratory health. Arch Dis Child.
2009;94(3):180-4.
293. Sadli N, Ackland ML, De Mel D, Sinclair AJ, Suphioglu C. Effects of zinc and
DHA on the epigenetic regulation of human neuronal cells. Cell Physiol Biochem.
2012;29(1-2):87-98.
294. Ceccarelli V, Racanicchi S, Martelli MP, Nocentini G, Fettucciari K, Riccardi C,
et al. Eicosapentaenoic acid demethylates a single CpG that mediates expression

130
of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia
cells. J Biol Chem. 2011;286(31):27092-102.
295. Dreborg S. The skin prick test in the diagnosis of atopic allergy. Journal of the
American Academy of Dermatology. 1989;21(4 Pt 2):820-1.
296. Severity scoring of atopic dermatitis: the SCORAD index. Consensus Report of
the European Task Force on Atopic Dermatitis. Dermatology. 1993;186(1):23-31.
297. Prescott SL, Macaubas C, Holt BJ, Smallacombe TB, Loh R, Sly PD, et al.
Transplacental priming of the human immune system to environmental allergens:
universal skewing of initial T cell responses toward the Th2 cytokine profile. J
Immunol. 1998;160(10):4730-7.
298. Martino D, Joo JE, Sexton-Oates A, Dang T, Allen K, Saffery R, et al.
Epigenome-wide association study reveals longitudinally stable DNA methylation
differences in CD4+ T cells from children with IgE-mediated food allergy.
Epigenetics. 2014;9(7).
299. Hung CH, Yang SN, Kuo PL, Chu YT, Chang HW, Wei WJ, et al. Modulation of
cytokine expression in human myeloid dendritic cells by environmental
endocrine-disrupting chemicals involves epigenetic regulation. Environ Health
Perspect. 2010;118(1):67-72.
300. Gunawardhana LP, Gibson PG, Simpson JL, Benton MC, Lea RA, Baines KJ.
Characteristic DNA methylation profiles in peripheral blood monocytes are
associated with inflammatory phenotypes of asthma. Epigenetics. 2014;9(9):1302-
16.
301. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et
al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina
Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203-9.
302. Butler MG. Genomic imprinting disorders in humans: a mini-review. Journal of
assisted reproduction and genetics. 2009;26(9-10):477-86.
303. Plant K, Fairfax BP, Makino S, Vandiedonck C, Radhakrishnan J, Knight JC. Fine
mapping genetic determinants of the highly variably expressed MHC gene ZFP57.
Eur J Hum Genet. 2013.
304. Tada Y, Yamaguchi Y, Kinjo T, Song X, Akagi T, Takamura H, et al. The stem
cell transcription factor ZFP57 induces IGF2 expression to promote anchorage-
independent growth in cancer cells. Oncogene. 2014.

131
305. Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP.
Common DNA methylation alterations in multiple brain regions in autism.
Molecular psychiatry. 2014;19(8):862-71.
306. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell
differentiation. Nat Rev Immunol. 2009;9(2):91-105.
307. Pan HF, Leng RX, Li XP, Zheng SG, Ye DQ. Targeting T-helper 9 cells and
interleukin-9 in autoimmune diseases. Cytokine & growth factor reviews.
2013;24(6):515-22.
308. Brough HA, Cousins DJ, Munteanu A, Wong YF, Sudra A, Makinson K, et al. IL-
9 is a key component of memory T cell peanut-specific responses from children
with peanut allergy. J Allergy Clin Immunol. 2014.
309. Gabory A, Ferry L, Fajardy I, Jouneau L, Gothie JD, Vige A, et al. Maternal diets
trigger sex-specific divergent trajectories of gene expression and epigenetic
systems in mouse placenta. PLoS One. 2012;7(11):e47986.
310. Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the
metabolic syndrome-like phenomenon in mouse offspring through epigenetic
changes in adipocytokine gene expression. Endocrinology. 2012;153(6):2823-30.
311. Kryukov GV, Shpunt A, Stamatoyannopoulos JA, Sunyaev SR. Power of deep,
all-exon resequencing for discovery of human trait genes. Proc Natl Acad Sci U S
A. 2009;106(10):3871-6.
312. Lander ES, Botstein D. Mapping mendelian factors underlying quantitative traits
using RFLP linkage maps. Genetics. 1989;121(1):185-99.
313. Van Gestel S, Houwing-Duistermaat JJ, Adolfsson R, van Duijn CM, Van
Broeckhoven C. Power of selective genotyping in genetic association analyses of
quantitative traits. Behavior genetics. 2000;30(2):141-6.
314. Guey LT, Kravic J, Melander O, Burtt NP, Laramie JM, Lyssenko V, et al. Power
in the phenotypic extremes: a simulation study of power in discovery and
replication of rare variants. Genet Epidemiol. 2011.
315. Myles IA, Pincus NB, Fontecilla NM, Datta SK. Effects of parental omega-3 fatty
acid intake on offspring microbiome and immunity. PLoS One. 2014;9(1):e87181.
316. Myles IA, Fontecilla NM, Janelsins BM, Vithayathil PJ, Segre JA, Datta SK.
Parental dietary fat intake alters offspring microbiome and immunity. J Immunol.
2013;191(6):3200-9.
317. Hallam MC, Barile D, Meyrand M, German JB, Reimer RA. Maternal high-
protein or high-prebiotic-fiber diets affect maternal milk composition and gut

132
microbiota in rat dams and their offspring. Obesity (Silver Spring).
2014;22(11):2344-51.
318. Ma J, Prince AL, Bader D, Hu M, Ganu R, Baquero K, et al. High-fat maternal
diet during pregnancy persistently alters the offspring microbiome in a primate
model. Nat Commun. 2014;5:3889.
319. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta
harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra65.

133
APPENDIX 1
BUFFER RECIPIES
PBS – Citrate (0.6%)
500ml 1x PBS
3g tri-sodium citrate dihydrate
PCF
50ml PBS-Citrate
1ml FCS (heat inactivated)
PCFD
5ml PCF
50ul DNAse (1mg/ml)
PBS-BSA (1%)
100ml PBS
1g BSA
FACS buffer
500ml PBS
2.5g BSA

134
APPENDIX 2
Primer sequences used in validation studies in Chapter 3
Validation of candidate loci by Sequenome EpiTYPER platform Primer name Tag + Sequence Chromosome 6 DMR – Left primer
aggaagagagGAGGATTTTAGAGGTTGGAAGTTTT
Chromosome 6 DMR – Right primer
cagtaatacgactcactatagggagaaggctCCCTCTCATCTAAATCAAAAAACAC
Primers for H3/H4 acetylation by Chromatin Immunoprecipitation (ChIP) Primers for ZFP57 transcripts (Ensemble)
Sequence Sequence start and end sites relative to the transcription start site
ZFP57-001 distal (ZFP57_1D) - forward
cac cac cgg cta act ttt gt starts at bp -801 and ends at bp -545
- reverse cct ggg caa aaa gag tga aa
ZFP57-002 proximal (ZFP57_2P) - forward
ccc agg ctg gtg ttg tta ct starts at bp -173 and ends at bp +20
-reverse
ggt ttg atg tgg ctt cct gt
ZFP57-002 distal (ZFP57_2D) - forward
cat gga aga gat ctt aga gag tg starts at bp -603 and ends at -366
reverse acc taa tgc aga gat gct aaa taa

135
Human promoter primers used for the acetylation experiments in Chapter 5
Locus Forward Reverse
RPL32 5‘-GGAAGTGCTTGCCTTTTTCC-3‘ 5‘-GGATTGCCACGGATTAACAC-3‘
IL1B 5‘-CGTGGGAAAATCCAGTATTTTAATG-3‘
5‘-CAAATGTATCACCATGCAAATATGC-3‘
IL4 5‘-TGGGTAAGGACCTTATGGACC-3‘ 5‘-GGTGGCATCTTGGAAACTGTC-3‘
IL5 5‘-AGGAGATCTTTTTAGTCACTGGCAACA-3‘
5‘-CGTCTCGAGGGCAAAGAAAGTGCATAG-3‘
IL9 5‘-CGTTAGAACACCCATGAC-3‘ 5‘-TTCTGGTTGTGAGAGTTAG-3‘
IL10 5‘-GACAACACTACTAAGGCTCCTTTGGGA-3‘
5‘-GTGAGCAAACTGAGGCACAGAAAT-3‘
IL13 5‘-TGTGGGAGATGCCGTGGG-3‘ 5‘-TCTGACTCCCAGAAGTCTGC-3‘
IL17A 5‘-AATTTCTGCCCTTCCCATTT-3‘ 5‘-CCCAGGAGTCATCGTTGTTT-3‘
GATA3 5‘-CACATTTAAAGGGCCAGAGC-3‘ 5‘-AAGGAAACTGCAACCCAAAC-3‘
TBX21 5‘-TGGCCAAGAGCGTAGAATTT-3‘ 5‘-GCTTTGCTGTGGCTTTATGA-3‘
RORC 5‘-TCTCCCCTATGCCTGTCACCTG-3‘ 5‘-TGATTTTGCCCAAGGACTCACAC-3‘
FOP3 5‘-ATCGTGAGGATGGATGCATTAATA-3‘ 5‘-CCACTGGGAAGGTCCCTAGC-3‘
IRF1 5‘-GTACTTCCCCTTCGCCG-3‘ 5‘-GCGTACTCACCTCTGCTGC-3‘
TNF 5‘-AGAAGACCCCCCTCGGAACC-3‘ 5‘-ATCTGGAGGAAGCGGTAGTG-3‘
IFNG 5‘-AATCCCACCAGAATGGCACAGGTG-3‘
5‘-GAACAATGTGCTGCACCTCCTCTGG-3‘
MS4A2 5‘-CTTAGGGGTTAGATTTTATGTGTTTGAACCCCAA-3‘
5‘-CCATCTTCTTCATGGACTCCTGGTGCTTAC-3‘

136
The reference ranges for lower limit of detection (LOD), sensitivity and specificity
for analysed genes for ChIP in Chapter 5

137
APPENDIX 3
Purity of isolated cells by magnetic bead separation
1st
experiment 2nd
experiment 3rd
experiment 4th
experiment PBMC before subjected to magnetic bead separation CD4 44.3 26.6 50.7 48.9 CD8 20.7 24.8 11.8 11.4 CD14 11.5 18.4 18.9 14.5 CD19 4.54 6.11 6.7 6.4 Extracted CD4 CD4 95.9 92.3 90.5 89.6 CD8 0.054 0 0.03 0.0 CD14 0.481 4.51 6.41 4.9 CD19 3.08 1.11 2.43 0.6 Extracted CD8 CD4 1.47 4.31 10.4 17.9 CD8 92.8 79.5 78.8 62.7 CD14 0.029 2.6 2.38 3.4 CD19 3.63 3.95 2.43 2.5
Remaining cell mix after magnetic bead separation CD3 7.62 17.1 3.6 CD14 25.7 35.7 51.1 CD19 4.76 8.42 11.3


















