
Supercritical fluids in analytical chemistry : chromatography and … · SUPERCRITICAL FLUIDS IN...
190
Supercritical fluids in analytical chemistry : chromatography and extraction Citation for published version (APA): Lou, X. W. (1997). Supercritical fluids in analytical chemistry : chromatography and extraction. Eindhoven: Technische Universiteit Eindhoven. https://doi.org/10.6100/IR497368 DOI: 10.6100/IR497368 Document status and date: Published: 01/01/1997 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, please follow below link for the End User Agreement: www.tue.nl/taverne Take down policy If you believe that this document breaches copyright please contact us at: [email protected] providing details and we will investigate your claim. Download date: 13. Apr. 2020
Transcript of Supercritical fluids in analytical chemistry : chromatography and … · SUPERCRITICAL FLUIDS IN...

Supercritical fluids in analytical chemistry : chromatographyand extractionCitation for published version (APA):Lou, X. W. (1997). Supercritical fluids in analytical chemistry : chromatography and extraction. Eindhoven:Technische Universiteit Eindhoven. https://doi.org/10.6100/IR497368
DOI:10.6100/IR497368
Document status and date:Published: 01/01/1997
Document Version:Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers)
Please check the document version of this publication:
• A submitted manuscript is the version of the article upon submission and before peer-review. There can beimportant differences between the submitted version and the official published version of record. Peopleinterested in the research are advised to contact the author for the final version of the publication, or visit theDOI to the publisher's website.• The final author version and the galley proof are versions of the publication after peer review.• The final published version features the final layout of the paper including the volume, issue and pagenumbers.Link to publication
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
• Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal.
If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, pleasefollow below link for the End User Agreement:www.tue.nl/taverne
Take down policyIf you believe that this document breaches copyright please contact us at:[email protected] details and we will investigate your claim.
Download date: 13. Apr. 2020

Supercritical Fluids · • ID
Analytical Chemistry
Chromatography and Extraction
..
Xianwen Lou

SUPERCRITICAL FLUIDS IN
ANALYTICAL CHEMISTRY
Chromatography and Extraction
PROEFSCHRIFT
ter verkrijging van de graad van doctor aan de Technische Universiteit Eindhoven, op gezag van de Rector Magnificus, Prof.dr. M. Rem, voor een commissie aangewezen door het College van Dekanen in het openbaar te verdedigen op dinsdag 9 september 1997 om 16.00 uur
door
XianwenLou
geboren te Yiwu, Zhejiang, China

Dit proefschrift is goedgekeurd door de promotoren:
prof.dr.ir. C.A.M.G. Cramers en prof.dr. P.J.F. Sandra
Copromotor: dr.ir. J.G.M. Janssen
Druk: Universiteitsdrukkerij Technische Universiteit Eindhoven.
Lou, Xianwen
Supercritical fluids in analytical chernistry: chromatography and extraction I Xianwen Lou. -Eindhoven: Technische Universiteit Eindhoven, 1997. ISBN 90-386-0878-0 NUGI 813 Trefw: superkritische vloeistoeffen I chromatografie I extractie Subject heading: supercritical fluid chromatography /supercritical fluid extraction.

To Yi and our parents

Contents
1. General Introduetion
2. Supercritical Fluid Chromatography and Extraction 2.1 Analytica! SFE and its comparison with SFC and some other extraction 7
techniques 2.1. 1 Introduetion 7 2.1.2 Properties of supercritical fluids 8 2.1.3 Analytical SFE and SFC 9 2.1.4 Analytical SFE and preparative SFE 19 2.1. 5 Optimization of analytical SFE 21 2.1.6 Analytica! SFE and other Extraction techniques 30
2.2 Correlation of SFE extraction reeoverles with SFC retention data: 39 A fundamental study 2.2.1 Introduetion 39 2.2.2 Theory 42 2.2.3 Experimental 46 2.2.3 Results and discussion 47 2.2.4 Conclusions 53
3. Tempersture and Pressure Effects on Solubility in Supercritical 54 Carbon Dioxide and Retention in SFC 3.1 Introduetion 54 3.2 Experimental 56 3.3 Results and discussion 57
3.3.1 Solubility determination with on-line FID metbod 57 3.3 .2 Fundamental study of temperature and pressure effects on solubility 60 3.3.3 Effects oftemperature on solute affinity for the stationary phase in 62
SFC 3.4 Conclusions 65
4. Pressure Drop Effects on Selectivity and Resolution in Packed 67 ColumnSFC 4.1 Introduetion 67 4.2 Experimental 69 4.3 Results and discussion 70
4.3.1 in-situ derivatization ofthe column 70 4.3.2 Effects ofinlet pressure on separation 71 4.3 .3 Effects of supercritical tluid flow rate on separation 77 4.3.4 Varlation ofresolution along an imaginary long column 78
4.4 Conclusions 82

ii Contents
5. SFE Extraction of Polymer Additives and Oligomers from 84
Polymerie Samples 5.1 Investigation of parameters affecting the SFE extraction of polymer 84
additives from polyethylene 5 .1.1 Introduetion 84 5 .1.2 Theory 86 5 .1.3 Experimental 88 5 .1. 4 Results and discussion 89
5 .I. 4.1 Effects of pressure 89 5.1.4.2 Effects oftemperature 91 5 .1. 4. 3 Effects of solute molecular properties 93 5 .1. 4. 4 Effects of supercritical fluid flow rates 95 5 .1. 4. 5 Effects of benzene as a modifier 96
5.2 Effects of modifier addition and temperature varlation in SFE of polymerie 103 matenals 5.2.1 Introduetion 103 5.2.2 Theory 105 5.2.3 Experimental 106 5.2.4 Results and discussion 108
5 .2. 4.1 Effects of temperature using pure carbon dioxide I 08 5.2.4.2 Effects of modifier 110
5.2.5 Conclusions ll9 5.3 Accelerated Solvent Extraction ofPolymeric Samples 121
5.3.1 Introduetion 121 5.3.2 Experimental 123 5.3.3 Results and discussion 124
5.3 .3 .I Kinetics of mass transfer in ASE of polymerie samples 125 5.3.3.2 Effects oftemperature 127 5.3.3.3 Effects ofpressure 128 5.3 .3. 4 Effects of other operating parameters 129 5.3.3.5 Optimization in ASE of polymerie samples 131 5.3.3.6 Advantages of ASE over other sample preparation methods 133
5.3 .4 Conclusions 134
6. On-line Combination of SFE with Capillary Gas 136 Chromatography
6.1 Quantitative aspects of SFE-capillary GC with a conventional split/splitless 136 injector as the interface 6.1.1 Introduetion 136 6.1.2 Experimental 137 6.1.3 Results and discussion 138
6.2 Investigation of parameters affecting the on-line combination ofSFE- 145 capillary GC

6.2.1 Introduetion 6.2.2 Experimental 6.2.3 Results and discussion
Contents
6.2.3 .1 On-line SFE-cGC of environmental samples 6.2.3.2 On-line SFE-cGC of polymerie samples
6.2.4 Conclusions
7. General Conclusions and Future Developments
Abstract
Samenvatting
Acknowledgments
Curriculum Vitae
Author's Publications on Chromatography
iii
145 147 149 149 155 159
161
168
172
176
177
178

Cha ter 1
1 General Introduetion
Important tasks in analytical chemistry involve the separation, identification, and quantitation of targetcomponentsin complex samples. Within analytical chemistry, chromatography is by far the most widely used technique. Modem chromatographic methods have an excellent separation power, are versatile and allow the use of various deleetion techniques. For complex samples, however, a sample preparation step is frequently required before the samples can be introduced into the chromatographic system. Extraelions using liquids, such as Soxhlet extraction and liquid-liquid extraction, are routinely used in numerous laboratones all over the world. Unfortunately, these methods are generally time consuming and sometimes require large amounts of toxic and expensive organic solvents. For a successful . chromatographic analysis, both the sample preparation step and the chromatographic process should be optimized carefully. The continuons search for rapid, efficient, cost-effective and environment-friendly means of analytica! extractions and separations has introduced supercritical fluids into the field of analytical chemistry. From a practical point of view, supercritical fluids can be defined as gases that are at temperatures above their critical values and that are compressed to an extent that liquid-like interactions become significant. Intheideal case, supercritical fluids can offer liquid-like solvating powers in combination with gas-like diffusion coefficients and viscosities. This combination is very attractive for faster sample preparation and improved chromatographic separation [1-4]. During the past 15 years the application of supercritical fluids in analytical chemistry, e.g. supercritical fluid chromatography (SFC) and supercritical fluid extraction (SFE), has experienced a rapid advance. Environmental chemistry, food and polymer chemistry, and phàrmaceutical and agricultural research are just a few areas to which SFC and SFE have been applied.
The ability of a supercritical fluid to dissolve solid materials was reported as early as more than a century ago [5]. Considerable time passed, however, before this basic knowledge was utilized for chromatography or extraction. The fust experiments using supercritical fluids as the mobile phase for chromatography were

2 General introduetion
performed by Klesper, Corwin and Turner in 1962 [6]. It were Sie and Rijnders [7] who first coined the name SFC to this form of chromatography. SFC can generally be considered as an intermediate technique between gas chromatography (GC) and high performance liquid chromatography (HPLC) because supercritical fluids combine many characteristics of gases and liquids. Potential advantages of (carbon dioxide based) SFC in comparison with HPLC include the compatibility with various GC detectors and the increased speed of analysis. In comparison with GC, SFC is advantageous for the analysis of less volatile or thermally labile compounds. However, despite all these potential advantages, SFC is mainly limited to the analysis of components with relatively low polarities. For the analysis of highly polar solutes, the addition of polar modifiers is often found to be necessary. This largely cancels the advantages of SFC. In addition to this, the influence of the column pressure drop on separation efficiency in SFC is stilllargely unknown. This lack of fundamental knowledge hinders the proper selection of SFC conditions for a given analytica! problem. It is one aim of this thesis to investigate systematically the effects of pressure drop on SFC retention, selectivity, plate height and resolution using the numerical methods developed by lanssen et al. [8].
In addition to their use as a mobile phase in a chromatographic system, another application area of supercritical fluids in analytica! chemistry is the use as an extractant for sample pretreatment. The use of a supercritical fluid as an extraction solvent in large-scale processing techniques was first reported by Zosel [9] in 1963. Since then, there has been a steady growth in the number of applications of SFE in the field of chemica! engineering, leading to the construction of several plants designed, e.g., to decaffemate coffee and to process hops and spices. Analytica! SFE was first introduced by Stahl and Schilz [10] in 1976. Unfortunately, until the mid-1980s, the analytica! potential of supercritical fluids for extractions was not fully recognized. The number of reports on analytica! SFE was limited and only two publications were registered in the Chemical Abstracts prior to 1986 [11]. Since then SFE has developed rapidly. Compared to the traditional extraction methods, as for example, Soxhlet extraction, SFE has many advantages such as a reduced usage of toxic organic solvents, shorter extraction times, adjustable solvent strength, as

Ch ter I 3
wellas the ability for on-line combination with analytical instruments. SFE is now considered as an important alternative to traditional sample preparatien methods. Unfortunately, however, SFE also has a number of disadvantages. Among these are the price of the instrumentation and, more importantly, the time consuming nature of metbod development. Current optimization strategies for SFE are almost exclusively based on trial and error experiments. Many parameters can affect the SFE process while their actual influence is still largely unk:nown. In this thesis the kinetics of mass transfer in SFE are carefully studied with the aim to improve the understanding ofthe effects ofvarious experimental parameters on the SFE process. Faster metbod development procedures for SFE are proposed based on a better comprehension of the kinetics of mass transfer.
Although very often considered as different techniques, SFE and (packed column) SFC are very much similar in many respects. In both techniques a supercritical fluid is used to dissolve the components and transport the analytes through a packed bed of solid particles. Extraction of components in SFE is controlled by many of the same factors that control relention in SFC. For example, if a compound requires extreme elution conditions in SFC, extraction in SFE will certainly be di:fficult. SFC retentien data can, thus, provide a means of gaining knowledge of the extraction characteristics in SFE. In sectien 2.1 of this thesis, the properties of supercritical fluids are briefly reviewed. After this, the similarities and differences between analytical SFE and SFC with respect to the instrumentation used are identified. As analytical SFE is a relatively new sample preparatien technique, the general process of optimization in analytica! SFE is discussed in detail. In this sectien also a comparison of the advantages and disadvantages of analytical SFE and traditional extraction methods is presented. Based on the close relationship between SFE extraction recoveries and SFC retentien data, a model is developed in sectien 2.2 which allows the predietien of SFE extraction efficiencies from readily obtainable SFC retentien data. With this model, predictions for the extraction yields in SFE can be made for homogeneons samples by performing SFC experiments at the same pressure and temperature as the SFE experiments.

4 General introduetion
A parameter of crucial importallee in both SFE and SFC is the solubility of components in the supercritical fluid. The solubility of a component in a supercritical fluid is controlled by the vapor pressure of the component and by its interaction with the supercritical fluid, and is a complex function of temperature, pre ss ure ( or density) as well as the properties of the supercritical fluid and the component. In chapter 3, solubilities of a series of polycyclic aromatic hydrocarbons (P AHs) in supercritical carbon dioxide are measured using an on-line FID method. The contribution of vapor pressure to the overall solubility is measured by using helium as the carrier instead of supercritical carbon dioxide. In this way, detailed information can be obtained on the relative importance of either contribution to the overall solubility. This greatly impraves the understanding of the effects of pressure and temperature variations in SFE and SFC
As mentioned above, supercritical fluids can offer a liquid-like solvating strength in combination with gas-like ditfusion coefficients and viscosities. SFC can thus generally be considered as an intermediale between GC and HPLC. The pressure dependenee of retention is, however, a notabie exception to this rule. Retention factors are almost independent of pressure in GC and HPLC, while pressure is a very important parameter cantrolling retention in SFC. In genera!, the pressure drop in SFC is low if open-tubular columns are used, whereas significantly higher pressure drops are encountered with packed columns. The effects of pressure drop on the SFC separation process is still not fully understood. In chapter 4, the influence of pressure drop on retention, plate height and resolution in packed column SFC is investigated systematically. Numerical methods which enable the prediction of pressure gradients, diffusitivities, retention factors, plate heights along the length of the column are used for model calculations. The possibilities and limitations of using long packed columns in SFC for obtaining high plate number separations are discussed.
Chapter 5 deals with the extraction of polymer additives and oligomers from polymerie samples using SFE and accelerated solvent extraction (ASE). It is emphasized in this chapter that a thorough comprehension of the kinetics of mass

Cha ter 1 5
transfer in these methods is essential to lUlderstand the effects of vanous experimental parameters on the extraction efficiencies. This forms the basis for rapid method development. The two-fihn theory, which considers mass transfer across a phase boundary, is applied to describe qualitatively the kinetics of mass transfer from the core of polymer particles into the extractant. The effects of experimental parameters, such as temperature, pressure, extraction time ( static and dynamic) and extractant flow rate, on the extraction efficiencies in SFE and ASE are investigated systematically. Moreover, the influence of modifier addition, i.e. modifier identity and its concentration, on the SFE extraction efficiency is studied in detail in sections 5.1 and 5.2. Some guidelines for the optimization of SFE and ASE of polymerie samples are given in sections 5.2 and 5.3, respectively. The performance for SFE and ASE of polymerie samples is compared and discussed in section 5 .3.
An important advantage of SFE is its easy combination with other analytica! techniques, especially with GC. Chapter 6 focuses on the on-line combination of SFE with capillary GC. In order to provide guidelines for the selection of operating conditions for on-line SFE-GC, two different injectors, a split/splitless injector and a programmed temperature vaporizer (PTV) injector are investigated as the interfaces. The parameters affecting the chromatographic peak: shapes and the quantitative performance of the interfaces are identified and studied. It bas been found that online SFE-GC with a split interface is suitable for samples that have high concentrations of extraetabie compounds, while on-line SFE-PTV -GC is particularly attractive in trace analysis.
Finally, some general conclusions and future developments of SFC, SFE and ASE are presented and discussed in chapter 7.
References
1. M.L. Lee and K.E. Markides (eds), Analytica/ Superer/tical F/uid Chromatography and Extraction, Chromatography Conference, Provo, UT, 1990.
2. J.R. Dean (Ed), Application of Supercritical Fluids in lndustrial Analysis, CRC Press, Inc., Boca Raton, Florida, 1993.

6 General introduetion
3. F.V. Bright and M.E.P. McNally, (Eds), Supercritical Fluid Technology: Theoretica/ and Applied Approaches to Analytica/ Chemistry; ACS Symposium Series 488, American Chernical Society, Washington DC, 1992.
4. B. Wenclawlak (Ed), Analysis with Supercritical Fluids: Extraction and Chromatography, Springer-
Verlag, Berlin, 1992. 5. J.B. Hannay and J. Hogarth, Proc. R. Soc. London, 29 (1879) 324. 6. E. Klesper, A.H. Corwin and D.A. Turner, J Org. Chem., 27 (1962) 700.
7. S.T. Sie, W. Beersmun and G.W.A. Rijnders, Sep. Sci. Techno/., 1 (1966) 459. 8. H.-G. Janssen and C.A. Cramers, J Chromatogr., 505 (1990) 19. 9. K. Zosel, Austrian Patent Application, 16.4.1963. 10. E. Stahl and W. Schiltz, Z. Anal. Chem., 280 (1976) 99.
11. S.B. Hawthorne, Anal Chem., 62 (1990) 633A.

2.1 Analytical Supercröical Fluid Extraction: Basic principles and Comparison with Supercröical Fluid Chromatography and Some Other Extraction Techniques
2.1.1. Introduetion
Most of the samples that have to be analyzed by chromatography are too complex, too dilute, or in their original state incompatible with the chromatographic system. For these samples direct injection can not be used and sample pretteatment prior to injection of the sample into the chromatography system is needed. Sample preparation methods generally used by analytical chemists nowadays are both time and solvent consuming. According to a recent survey, two third of the analysis time in chromatographic analyses is devoted to sample preparation. Moreover, this step accounts for at least one third of the error generated by the analytica! method [1]. The improverneut of sample preparation methods or the development of new methods will reduce the analysis time and allow the analyst to produce more precise results.
One of the most important steps in sample pretteatment is generally the extraction process. Supercritical fluid extraction {SPE) is recognized as an important alternative to Soxhlet extraction and bas attracted significant attention [2]. Several hooks dealing with analytical SPE have been publisbed in recent years [3-6]. Chester et al. reviewed the developments of SPE in great detail [2, 7]. Applications of SPE in the analysis of environmental samples, fossil fuel samples and surface coating samples were summarized by several research groups [8-10].
As a new and developing technique, analytical SFE bas borrowed most of its theory from supercritical fluid chromatography (SPC) and preparative-scale SFE in chemica! engineering. In this chapter the theoretica! backgrounds of analytica! SFE are summarized. Moreover, an attempt is made to identizy similarities and differences between analytica! SPE and SPC, as wellas between analytical SPE and preparative-scale SFE. The process of optimization of SFE extraction conditions is discussed in more detail. Attempts are also made to compare analytical SFE with some more traditional sample preparation methods, such as Soxhlet extraction, liquid-liquid extraction and solid phase extraction {SPE). Advantages and disadvantages of analytica! SFE over traditional methods are discussed.

8 SFEand SFC
2.1.2. Properties of supercritical fluids
A fluid is said to be in its supercritical state when both its temperature and pressure are above their respective critical values. This definition can be visualized by reference to the pressure/temperature phase diagram of co2 shown in Figure 2.1. Supercritical fluids can be seen as intermediates between gases and liquids. Supercritical fluids offer, at least in principle, liquid-like solvating powers in combination with gas-likeditfusion coefficients and viscosities. Depending on the operating conditions, the supercritical fluids can exhibit either more gas-like or more liquid-like behavior. The properties of supercritical fluids make them attractive both in SFC and SPE. Physical properties of supercritical fluids which prove to be important in chromatography as well as in extraction are the density, the viscosity and the diffusitivity of the fluid. Table 2.1 gives a comparison of these properties for gases, supercritical fluids and liquids.
Pressure
Critica!
Pressure
Solid
. . . . . Liquid ~ Supercri ti cal
Fluid
Critica! Point
Critica!
Temperature Temperature
Figure 2.1. Pressure/temperature phase diagram of C02•
Table 2.1. Approximate values of densities, viscosities and diffusion coefficients of gases, supercritical fluids and liquids [ 11].
Fluid Density Viscosity Diffusion coefficient (g/cm3
) (g/cm s) (cm2/s)
Gas (0.6-2)10"3 (1-3)104 0.1-1.0 Supercritical fluid 0.2-0.9 (1-3)10"3 (0.1-5)104
Liquid 0.6-1.6 (0.2-3) 10·2 (0.2-3)10"5
Since supercritical fluids combine many characteristics of liquids on the one hand and gases on the other, SFC can be described as both a combination of, as well as

Chapter 2 9
being complementary to GC and HPLC [12]. The solvating nature of supercritical fluids enables the elution of relatively high molecular weight components or thermally labile components in SFC at mild conditions. This is an important advantage of SFC over GC. When compared with HPLC, the diffusion coefficients in supercritical media are approximately one order of magnitude larger than in a liquid. Hence, a separation in SFC is inherently much faster than in HPLC. Moreover, C02 based SFC is compatible with most sensitive and selective GC detectors.
The combination of liquid-like solvent strength and gas-like transport properties of supercritical fluids make SFE an important alternative to traditional Soxhlet extraction. The density of a supercritical fluid is determined by its temperature and pressure. As the solvent strength of a supercritical fluid is a function of density, the solvating capability of a supercritical fluid can easily be changed by adjusting the extraction pressure and/or temperature. This allows the analyst to obtain selective extraction of compounds by varying the extraction conditions. The gas-like transport parameters, high diffusitivity and low viscosity, on the other hand, result in improved rates of mass transfer for solutes in supercritical media, resulting in faster extractions.
Many supercritical fluids have been used in SFC and SFE. Among these carbon dioxide is by far the most widely used. Its preferenrial use is due tothefact that it is chemically inert, inexpensive, non-toxic, non-flammable and because it bas easily accessible critica! parameters. Carbon dioxide is also available in high purities at a reasonable price.
2.1.3. Analytical SFE and SFC
The objective of supercritical fluid chromatography is, similar to the situation for the other chromatographic techniques, to separate and detect components of interest in the sample. In contrast, analytica! SFE is a sample preparation method which aims at quantitative and selective extraction of the components of interest from various complex matrices. Due to their different objectives, many aspects of SFE and SFC are different. A brief comparison of some key issues in SFC and analytica! SFE is presented in Table 2.2. In the following sections, comparisons between SFC and analytica! SFE with respect to instrumentation and mobile phases, as well as relationships between SFC and analytical SFE are discussed.

10 SFEand SFC
Table-2.2. Comparison of some aspects of analytical SFE and packed column SFC.
SFE
Purpose Quantitative (and selective) extraction.
Packing material/matrix Environmental samples (soil, sediments, etc.); Food and biologica! samples (animal tissue, vegetable etc.); Sarbents (Tenax, ODS etc.); Polymers.
Diroension of the extraction Short and wide cells. cell or SFC column
Oven Chromatographic oven; Tube heater.
Analyte Adsorbed or naturally contaminated; Dispersed among the sample matrix.
Mobile phase Supercritical fluid; Supercritical fluid with modifiers.
Modifier Added to the supercritical fluid; Added directly to the matrix.
Flow rate Any rate not causing problems for collection.
2.1.3.1. lnstrumentation
Packed column SFC
Separation and detection.
Small size, well-defined packings (stationary phase).
Narrow and long columns.
Chromatographic oven.
Introduced through an injection valve; Present as a narrow band at the head of the column.
Supercritical fluid; Supercritical fluid with modifiers.
Added to the supercritical fluid.
Above optima! linear velocity.
The available SPC and analytica! SPE systems span a wide range of prices and possibilities. Typical schematic diagrams of instrumentation for SPC and analytica! SPE are shown in Pigure 2.2A and B, respectively. In both SPC and analytica! SPE systems, a pwnp (or a system of pwnps) is used to deliver the supercritical fluid. The SPC column or SPE extraction cell is placed in a thermostatted oven and a restcictor is used to maintain the critica! conditions inside the column or the extraction cell. Despite the great deal of similarity, the differences between SPC and analytica! SPE are evident. In SPC the samples are injected through an injection valve, while in SPE, no sample injection step is involved as the solutes are in the sample matrix. Moreover, in SPE the components that have been extracted have to be collected in a collection device,

Chapter 2 11
whlle in SFC the components eluted out of the chromatographlè column are directly fed to the detector. Apart from these, the requirements posed on the mobile phase, the methods used for introducing modifiers, the dimensions of the extraction cell in SPE and the column in SFC, the supercritical fluid flow rate etc. can all be different in SPE and SFC. In the following sections, some instromental aspects in SFC and analytical SPE are compared. In this discussion, special emphasis will be paid to the analytical SPE part.
co,
Microcomputer Oven
(A)
(B)
Figure 2.2. Schematic diagram of SFC and SFE systems. {A) SFC; (B) SFE.

Pump and oven
Syringe pumps and reciprocating pistol pumps are now the most widely used fluid delivery systems in both SFE and SFC. Less expensive pumps are available for SFE because it is normally performed at constant pressures without the sophisticated pressure/density ramp controller required for SFC. Gas compressors may also be useful in SFE, particularly for large scale extractions for which a syringe pump may have insufficient capacity. The temperature of the extraction cell in SFE is normally controlled by placing the cell in a chromatographic oven, similar to SFC, or in a simple tube heater [13].
Dimensions and paclàngs of SFE extraction cells and SFC columns
Both fused-silica open tubular capillary columns and packed columns are routinely used in SFC. The columns should be carefully coated or well packed to obtain high column efficiencies. For more information about SFC columns, the reader is referred to recent lirerature [14].
The extraction cells used in SFE initially were empty HPLC columns. Later, vessels specially designed for analytica! SFE have been introduced. The extraction cells typically range in size from 150 Jll to 50 mL and are constructed from stainless steel or materials of similar inertness [15]. Sample cell designs currently available basically fall into two categoties as shown in Fig. 2.3A and B. The design shown in Fig. 2.3A is the most widely used one. In this design, the cell is always completely tilled with the sample. The extraction cell in Fig. 2.3B is only partly occupied by the sample matrix and the supercritical Duid from the headspace is collected. The latter design is normally used for the extraction of large volume samples or for the extraction of liquid samples.
(A) (B)
Figure 2.3. Extraction cells used in analytical SFE.

Chapter 2 13
Generally, cylindrical extraction vessels are used for SFB. The effects of the extraction cell dimensions on the SFB extraction of polycyclic aromatic hydrocarbons (PARs) from octadecyl-bonded sorbents were studied by Purton and co-workers [16, 17]. These authors found that the observed SFB efficiencies for the largest P AHs studied were increased by more than a factor or two by increasing the vessel diameter-to-length ratio from 1:20 to 1:1. The results indicate that in SFB short and wide bore extraction cells may be of benefit This is contrary to the observations in analytica! SFC. Here long and narrow-bore columns are used almost exclusively. Besides the extraction vessel dimensions, the orientation of the extraction vessel (vertical or horizontal) may also slightly affect the observed SFB recoveries [18].
Contrary to SFC packing materials, most of the sample matrices that are extracted by SFB are much more irregular in size and are normally randomly packed into the extraction cell. The packing of the sample matrix into the extraction cell can influence the efficiency and the reproducibility of the SFB process [19]. Prudent practice is to completely fill the extraction cell since the influence of packing structure is reduced when the extraction cell is fully occupied. However, for polymerie samples, problerns can occur if the extraction cell is densely packed with a polymer that can swell in a supercritical fluid. Firstly, the polymer may extrude from the cell during extraction, and secondly, the extraction cell may be blocked due to polymer expansion. Most likely, the best means is to fill the extraction cell to a level that is low enough to accommodate any swelling that may occur, and then to fill the rest of the extraction cell with a compressible support material such as clean glass wool [20]. Similar methods have also been applied to other types of matrices. Por example, placing glass beads (50-100 J..llTI) at either end of an extraction cell can be used to minimize the dead volume of the cell as well as to prevent blockage of the outlet frit of the extraction cell [19]. Wet samples may be mixed with a drying agent to prevent blockage of the resttictor [21]. Mixing copper with the sample matrix was recommended for the extraction of sediments containing high elemental sulphur contents [22]. Berg et al. reported an on-line reaction and extraction metbod for fatty acids and triglycerides by packing the sample matrix together with different layers of reacting, supporting and dehydrolyzing materials [23].
Restrictors and solute colleefion
Restrictor

14 SFEand SFC
One of the most important parts of instrumentation for both SFC and analytica! SFE is the restrictor, through which the mass flow of supercritical fluid is controlled and the supercritical fluid returns to atmospheric conditions. The restrietars used in analytica! SFE and SFC are almost identical and can be divided into two general categories: fixed restrietars and variabie restrictors. Fixed restrietars are easier to operate and much cheaper. Variation of the flow rate is, however, not possible.
In SFC, the analytes eluting at higher pressures have lower ditfusion coefficients, both because of their larger size and because of the increased density of the mobile phase. To keep the efficiency constant during a pressure programmed SFC analysis, the linear velocity should be appreciably reduced as the program progresses. However, in chromatographic systems equipped with a fixed restrictor, the rnass-flow rate can not be controlled. Due to the increase in pressure drop over the restrictor during pressure programming, the rnass-flow rate increases. Systems with variabie resttictors can control the pressure and rnass-flow rate independently and are, therefore, recommended for the SFC separation of complex samples with pressure programming [24]. In contrast, the rnass-flow rate in analytica! SFE is not so crucial. For components which can be easily extracted by SFE, even a restrictor-free system with solventless colteetion and rapid depressurization has been described [25].
Another important aspect associated with restrictor performance in both SFE and SFC is plugging. Plugging of restrietars is rarely a problem in SFC unless highly nonvolatile components have to be analyzed. In general, the samples that are iiüected in SFC are relatively clean and the amount of component passing through the restrictor is extremely small (S20 ng per peak). Moreover, restrietars are usually mounted in the base of a heated detector, as for example a flame ionization detector (FID). A heated FID usually heats several inches of the restcictor and would therefore heat the fluid for a substantial distance in front of the location where the pressure drop occurs [3]. However, in analytica! SFE of samples with high contents of extraetabie components or of wet samples, restrictor blockage is frequently observed. During depressurization at the restrictor tip and inside the restrictor, the reduction of the density of the extraction fluid leads to a decrease in the solubility of the analytes which in turn may lead to analyte precipitation and plugging of the restrictor. With wet samples the restrictor can easily be blocked by the formation of ice at the restcictor outlet due to the Joule-Thomson cooling effect ofthe expanding extraction fluid.

Chopter 2 15
The following solurions have been suggested for prevenring restrictor blockage [26]: a. Hearing of the restrictor; b. Inserting the restrictor into a suitable solvent in which the components have a
good solubility; c. Extracting smaller amounts of the specimen; d. Using restrictors with larger orifices or with different length-to-diameter
ratios; e. Using special matcrials to trap water andlor other extraetabie matrix
components prior to the restcictor.
Solute col/eetion
In SPC, the components eluted from the column are directly sent to a detector and no solute coneetion step is needed. The coneetion of the extracted components from the depressurized supercritical fluid is, however, an important step in the overall extraction procedure in SPE. The components can be collected in two distinctly different ways: off-line SPE and on-line SFE.
a. Off-line SPE
Off-line SPE is generally simpler to operate than on-line SPE. A sample extracted off-line can be analyzed by any appropriate subsequent technique and is available for multiple analysis. Off-line SPE is therefore very often preferred for method development in SPE. In off-line SPE, the coneetion of the extracted solutes is a very important step. Significant losses of analyte can occur during this step, teading the analyst to believe that the actual extraction efficiency was poor. In recent years the understanding of the factors influencing off-line solute colleerion has been greatly improved. There are three options for off-line collection: (a) expanding into an empty container with or without cryogenic cooling; (b) colleerion in a solventand (c) trapping onto a solid surface or an actsorbent [27-29].
The first method was used in the early development of analyrical SPE [27]. With this method, sample loss during coneetion can occur due to aerosol formation. An improvement of this method was introduced by Milier et al. who rapidly depressurized the supercritical fluid into an empty vial without a restrictor after static extraction [25]. They claimed that recoveries exceeding those obtained using dynamic SFE with coneetion in a liquid solvent could be obtained.

16 SFEand SFC
Among these three methods, the most commonly used one is the metbod in which the supercritical fluid is depressurized into an organic solvent (method b). This metbod is relatively simple to perform, and the resulting extract is immediately available for analysis [30-32]. With this method, efficient coneetion can sametimes be a problem when high flow rates of supercritical fluids are used. This is especially the case when the coneetion solvent is viscous, for example if a viscous collection solvent or a viscous modifier is used. Viscosity increase can also be a problem if the sample has a high content of extraetabie compounds. Viscous solutions can easily be bubbled out of the coneetion vial by the large flow of supercritical fluid. Moreover, increasing of viscosity may result in slow mass transfer from the gas phase into the coneetion solvent. The following solutions can be used to improve the coneetion efficiency: (1) reduce the flow rate of the supercritical fluid; (2) extract smaller amounts of the sample specimen; (3) use longer or wider collection vials, and (4) trapontoa solid actsorbent such as glass beads or Tenax etc ..
Although trapping on an actsorbent (method c) is somewhat more complicated than trapping in a solvent, it is highly efficient and well suited for automation and, consequently, for routine analysis. With trapping onto a solid sorbent, it is possible to introduce additional selectivity into the sample preparation metbod by using special sorbents. Moreover, higher supercritical fluid flow rates can be used while still maintaining a good collection efficiency. It has been demonstrateel that SFE with solid phase trapping has the potential of sirnultaneous extraction, clean up and concentration of the components from different matrices [33, 34]. However, when modifiers are used, the trapping conditions have to be carefully optimized. After condensation, the modifier can remove trapped analytes from the sorbent trap when the trapping temperature is lower than the boiling point of the modifier. The use of higher trapping temperatures, on the other hand, may lead to losses of volatile components [34].
For some applications, a single collection metbod may be insufficient to yield quantitative trapping efficiency. In these cases tandem combination of different coneetion methods could be employed [35-38]. Tandem solid/liquid trapping has been proved to be advantageous at high modifier percentages [37].
b. On-line SFE
Generally, on-line SFE refers to the direct coupling of SFE to a chromatographic system. With on-line SFE, intermediate sample handling steps are eliminateel and the entire extract rnay be analyzed without sample splitting. On-line SFE methods

Chapter 2 17
are particularly attractive when only limited quantities of samples are available and maximum sensitivity is desired. On-line SFE techniques. especially SFE/GC and SFE/SFC have gained widespread interest. In on-line SFE/GC, attention mainly focused on combination of SFE with open-tubular capillary GC. The successful coupling of SFE with capillary GC requires introduetion of the extracted solutes into the chromatographic column as a narrow band. On-column GC injectors, splitlsplitless injectors as well as programmed temperature vaporizer (PTV) injectors have been successfully used to interface SFE to capillary GC [39-41]. Burford et al. studied the experimental parameters affecting split SFE/GC analysis, and developed a simple and reliable SFE/GC metbod for the analysis of petroleum hydrocarbons from environmental samples [42, 43]. In on-line SFE/SFC, the extracted components should be focused prior to commencing the SFC analysis [44]. Apart from combiflations of SFE with GC and SFC, combiflations of SFE withother analytical methods, such as HPLC, MS and FI'-IR have been reported in lirerature [45-48].
2.1.3.2. Mobile phases and modifiers
Basically, SFC and SFE can be performed using the same supercrideal fluids. In this respect, ho wever, two important differences can be observed when comparing SFC and SFE, i.e. the supercritical fluid flow rate and the methods used to introduce modifiers.
Supercritical jluid flow rates
The flow rate of the supercritical fluid can significantly influence the column performance in SFC. The highest efficiency is obtained at the optimal linear velocity. In practice, however, mobile phase veloeities above the optimum value are frequently used to minimize the separation time [49].
The effects of the supercritical fluid flow rate on the performance of SFE are not so pronounced as ·in SFC. SFE can be performed in two ways, viz. static and dynamic extraction. Static SFE is performed by pressurizing the extraction cell and extracting the sample with no out-flow of supercritical fluid. After a preset period of time, a valve is opened to allow the analytes to be swept into the collection device. In dynamic SFE, the supercritical fluid is continuously flowing through the extraction cell, and a resttictor is used to maintain pressure in the extraction cell and allow the supercritical fluid to depressurize into the coneetion device. In dynamic SFE, the influence of the fluid flow rate on the extraction

18 SFEand SFC
killetics can be distinctly different in the cases of absence or presence of a modifier. In SFE, the modifier is often spiked directly onto the sample matrix. In this case, higher supercritical fluid flow rates can decrease the extraction efficiency as a result of the reduced contact time between the modifier and the sample. Low supercritical fluid flow rates, on the other hand, entail longer extraction times. In contrast, when a pure supercritical fluid is used, the extraction rate will increase with increasing supercritical fluid flow rate if the rate-1imiting step in the extraction is the solubility of the components in the supercritical fluid. If the rate-limiting step is ditlusion of the components in the sample matrix, the supercritical fluid flow rate has little or no effect on the extraction rate [50]. In addition to the effects on extraction rates, higher flow rates will make trapping of the components from the expanding supercrideal fluid more difficult.
Introduetion of modijiers
In the present practice of both SFC and analytica! SFE, carbon dioxide is by far the most widely used medium. Unfortunately, the polarity of supercritical carbon dioxide is very low which limitsits ability to dissolve polar components. In order to be able to elute polar components in SFC or extract these solutes in SFE, modifiers are added to carbon dioxide to improve the characteristics of the supercritical fluid.
The effects of modifiers in packed and open-tubular SFC have been investigated by Janssen et al. [51]. In SFC, the following two conditions should be fulfilled when modifiers are used: (1) the modifier should be compatible with the SFC detector; (iz) the mobile phase should be a homogeneous supercritical phase. If the conditions employed during SFC produce phase separation, mobile phase flow and hence solute relention beoomes erratic, the baseline becomes noisy, and irregular and broadened solute peaks will be observed [52]. These demands greatly restriet the possible selections ofmodifiers in SFC. Till now only a Iimited number of modifiers has been successfully used in SFC. Among these, the lower alcohols and alkanes are most frequently used.
In analytica! SFE, the objective is to quantitatively extract the components of interest from a sample matrix as selectively and rapidly as possible. Here it is not necessary to keep the extracting fluid as a single supercritical phase. In fact, the amount of components that can be extracted by SFE is comparatively large, and therefore, phase separation can occur during extraction even if no modifier is used. This situation is most likely to occur at the beginning of an extraction.

Chapter 2 19
Therefore, it is obvious that in SFE there are no such limitations as in SPC as far as the use of modifiers is conceme<i. This makes the introduetion of modifiers in SFE easier and the range of possible modifiers wider. Basically, there are two ways to introduce modifiers in SFE: (1) actdition of the modifier to the sample matrix prior to extraction; (2) bleuding the modifier with the supercritical fluid. At present, modifiers are mostly spiked directly onto the sample matrix. Many different compounds including organic solvents and even derivatization, ion pairing and complexation agents have been used as modifiers in analytica! SFE [53-61]. Extraction of polar, and even ionic matenals with nonpolar fluids as carbon dioxide, is possible through the use of various types of (reactive) modifiers. Nevertheless, phase separation rnay also cause problems in SFE because it can. make the SFE extraction less selective and optimization more difficult. The roles of modifiers in SPE can be summarized as follows: (1) increase the density and/or polarity of the supercritical extractant; (2) deactivate active sites in/on the sample matrix; (3) swell the sample matrix; (4) react with the componentsof interest to form more extraetabie compounds.
A given modifier may have more than one of the above mentioned functions. The selection of modifiers in analytical SPE will be discussed in section 2.1.4 and 2.1.5.
Although very much different as discussed above, SFE and packed colwnn SPC are quite similar in many aspects. The relationships between SFE and SFC will be studied in detail in section 2.2.
2.1.4. Analytical SFE and preparative SFE
Analytica) SPE and preparative SPE have in comrnon the use of supercritical fluids for extracting the target compounds. The compounds should first be dissolved in the supercritical fluid, then eluted out of the extraction cell, separated from the supercritical fluid, and finally collected. However, the aims of analytica! and preparative SFE are clearly different. The purpose of analytical SFE is to extract the components of interest selectively and quantitatively for analysis, while the objective of preparative SFE is to selectively extract most (if not all) of the target components for preparation. Due to their distinctly different objectives, analytica! SPE and preparative SPE are different in many aspects. Among these,

20 SFEand SFC
the concentrations of the target compounds, the apparatus and the requirement for the introduetion of modifiers are the most significant ones.
Concentration level and selection of extraction conditions
In preparative SFE, the concentrations of the target compounds should be high enough for practical applications. The extraction conditions should be chosen such that the selective extraction of (most of) the target compounds is obtained. The trace amounts of product that are strongly adsorbed on the matrix surface are normally of no concern. In this case, the effect of the sample matrix is limited and the solubility of the compounds of interest in the supercritical fluid determine the rate of the overall extraction. Therefore, the selection of conditions in preparative SFE is based on selecting pressure and temperature conditions where the target compounds have the highest solubilities in the supercritical fluid. These conditions can be predicted if the solubility parameters of the compounds are known and if correlations such as that proposed by Giddings et al. are used [66]:
(2.1)
Where ö is the Rilderbrand solubility parameter, Pc is the critical pressure of the fluid, p is the density of supercritical fluid, and p, is the liquid density of the fluid. A good solubility can be obtained if the solubility parameter of the extractant is approximately equal to that of the solute.
Correlations as the one given above are useful in preparative SFE because they predict conditions at which maximum solubilities can be obtained. This is important because the target compounds represent a large percentage of the bulk sample. However, these correlations become less useful in analytica! SFE because the concentrations .of the target analytes are normally very low. For such samples, dissolving the maximum amount of target analytes in the supercritical fluid is not a souree of concern, and the analytes need only to be sufficiently soluble in the supercritical fluid to be transported out of the extraction cell. The objective of analytica! SFE is to extract the target analytes selectively, rapidly and quantitatively. The extraction should be complete, which implies that the trace amounts of strongly adsorbed components should also be extracted. Solubility considerations address only part of the extraction problem, because the extraction of an analyte depends on its distribution between the supercritical fluid and the matrix. The ability of the supercritical fluid to compete with the analytes for active sites on the matrix may be more important than solubility considerations for determining optimal extraction conditions in analytica! SFE [67].

Chapter 2 21
Apparatus
The apparatus used in analytica] SPE bas been discussed in detail in section 2.1.3.1. The extraction vessels are nonnally cylindrical cells with sizes ranging from 150 J,tl to 50 ml. The extracted components can be collected using either of the three different methods discussed in section 2.1.3 .1. After extraction the supercritical fluid is vented to waste. However, the apparatus used in preparative SPE is more complicated and extracrions are performed at much larger scales than in analytical SPE. Large volume reactors are mostly used as the extraction vessels in preparative SPE. After extraction the supercritical fluids should be recycled because of the larger amounts of supercritical fluid employed, and the extracted compounds should be separated from the supercritical fluids. Two approaches have been developed for separating the extracted components and the supercritical fluid: an isobaric metbod in which the loaded supercritical fluid is heated and an isothermal approach in which precipitation of the extracted material is induced by expanding the supercritical fluid phase [68].
Modifiers
In analytical SPE, the compoundsof interest are frequently present at trace levels and adsorbed on active sites of the sample matrix. Carbon dioxide is a non-polar fluid and, therefore, a poor fluid to overcome the interactions between the compounds and the sample matrix. lmproved extraction reeoverles can be obtained by using suitable modifiers. In principle, any kind of compound can be used as a modifier in analytical SPE as long as it can improve the extraction recovery and does not interfere with the subsequent analysis.
In preparative SPE modifiers are not as frequently used as in analytical SPE because of the following two reasons. Pirstly, in preparative SPE the target compounds normally represent large percentages of the bulk sample. The trace quantities of the compounds that are adsorbed on active sites of the sample matrix are of no concern. Moreover, in most applications the target compounds have a good solubility in pure carbon dioxide. In this case it is not necessary to complicate the SPE operation with the actdition of modifiers. The other reason is that separation of the modifiers from the products can be difficult, and modifiers may thus contaminate the extraction products.
2.1.5. Optimization

22 SFEand SFC
Optimization is, perhaps, the area of greatest concern in SPE since it impacts on the accuracy and precision that can be obtained with this technique. Unfortunately, however, optimization in SPE is rather difficult and time consuming. Many experimental variables must be considered, including sample size, the choice of supercritical fluid, pressure and temperature, supercritical fluid flow rate and extraction time, and the metbod used to colleer the extracted analytes. The process of optimizing these parameters is discussed in more detail below.
Sample size
The sample size is one of the first parameters that should be considered before performing an SPE extraction. Pirstly, the sample size should be large enough to ensure sample homogencity and to obtain sufficient sensitivity for trace analysis. However, larger samples require larger amounts of supercritical fluid for quantitative extraction, may easily block the restrictor and can make trapping of the extracted analytes more difficult (especially for volatile components). Analytica! SPE is most often conducted using fluids that are gases at ambient conditions. In general, low supercritical fluid flow rates facilitate quantitative trapping of the extracted compounds, especially of volatile compounds. This limitation of the supercritical fluid flow rate will extend the extraction time needed to obtain quantitative results for larger samples. Normally, a small sample size is preferred on the condition that the requirements for sample homogeneity and sensitivity are satisfied [69].
Selection of supercritical jluid
The characteristics of several fluids that have been used for SFE are listed in Table 2.3. Among these fluids, supercritical carbon dioxide should be the first choice for most SPE extractions. Many of the staled advantages of SPE, such as reduced usage of organic solvents are directly related to the use of carbon dioxide as the extracting fluid. In general terms, carbon dioxide is an excellent extraction medium for non-polar to moderately polar species. lt has been successfully used to extract a variety of compounds, such as alkanes, polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), fats, esters, and organochlorine pesticides from various matrices [2, 7, 71). However, carbon dioxide does not have sufficient solvent strength to dissolve more polar compounds. Moreover, in the SFE extraction of some real-world samples, less than quantitative extracrions were frequently reported even for moderately polar compounds with carbon dioxide as the extraction fluid. In such samples, the

Chapter 2 23
analytes are adsorbed on adsorptive sites of the sample matrix and carbon dioxide is a poor tluid to overcome the matrix/solute interactions. To achleve quantitative recoverles, the ability of the supercrltical tluid to overcome matrix/analyte interactions is often more important than a high solubility.
Table 2.3. Characteristics of selected supercritical fluids [70].
Fluid T"eq Pc(atm) Dipole moment (Debyes)
C02 31.3 72.9 0.00
N20 36.5 72.5 0.17
NH3 132.5 112.5 1.47
MeOH 240.0 78.5 1.70
Xe 16.6 58.4 0.00
CCI2F2 111.8 40.7 0.17
CCIF3 28.8 38.2 0.50
Ethane 32.2 48.2 0.00
9.9 50.5 0.00
Increased reeoverles may be achieved by using other, more po lar supercritical fluids such as Freons etc. [72, 73]. Unfortunately, no tluids have the same attractive characterlstics as carbon dioxide. Despite of their excellent properties as extraction fluids, there are some practical and environmental limitations in using these tluids. Although Freons show excellent reeoverles for the extraction of PARs and PCBs from environmental samples [72, 73], these solvents are not environmentally friendly [74]. Nitrous oxide has critical parameters and a molecular weight similar to those of carbon dioxide, yet it has a permanent dipole moment and, therefore, is a better solvent for many solutes than carbon dioxide. Forsome samples more rapid extracrions can be achieved with nitrous oxide [75]. However, nitrous oxide is a strongly oxidizing agent and can present a serious safety hazard [76]. Supercrltical methanol is an excellent solvent but it is liquid at ambient conditions which complicates sample coneetion and concentration after extraction. Supercrltical ammonia would be very attractive from the point of view of solvent strength, but it is chemically reactive and is likely to be too dangerous for routine use [ 69].

The actdition of modifiers to carbon dioxide is another alternative to increase the SPE recovery. Many different modifiers have been used in SPE. Unfortunately, little information is available to aid in the selection of modifiers and their concentrations. The solubilities of the analytes in the modified supercritical fluids, and the interactions between the modified supercritical fluid with the matrix and the target analytes are poorly understood. In selecting modifiers, both properties of the target solutes and of the matrix should be considered. Until the action of modifiers is better understood, the choice of the best modifier for the extraction of complex samples is based on empirical experience. Por a tentative survey of modifiers, some preliminary guidelines can be utilized. (1) In the extraction of components which are highly soluble in supercritical
carbon dioxide trom homogeneous matrices (where the components are only adsorbed on the matrix surface and the matrices do not contain active sites), modifiers are virtually not neerled (such as for example, in most spiking/recovery studies).
(2) The modifier should not interfere with the subsequent analysis. (3) In the extraction of inhomogeneous samples (containing active sites, such
as in most environmental samples), polar or reactive modifiers can be necessary to deactivate active sites on the surface of the sample matrix, even for the extraction of compounds which are highly soluble in supercritical carbon dioxide. In this case, a small amount of a suitable modifier may result in a significantly improved extraction yield.
(4) Por polymerie samples where modifiers are mainly used to increase ditfusion of analytes from the core of polymer to its surface, the modifier should be a good swelling agent for the polymer. Pairly frequently large amounts of modifier are necessary and continuous modifier actdition or repeated spiking of modifier may be of benefit.
(5) Por components which are not soluble in carbon dioxide, the modifier should be a good solvent for the target analytes, or a reactive modifier (derivatizing reagent) should be used to transfer the compounds into extractable, non-polar derivatives.
(6) Por ionic compounds, ion pairing or complexation modifiers should be used;
Till now, optimization of SPE methods using modified fluids frequently requires time consuming experiments in which modifiers with different polarities and concentrations are evaluated at various pressures and temperatures [69].
Pressure

Chapter 2 25
The fust and most obvious requirement of SFB conditions is the ability to dissolve the target analytes. The solvent strength of a supercritical fluid can easily be controlled by changing the pressure, and/or to a lesser extent, the temperarure [77]. Pressure is one of the main parameters that influences the · extraction recovery and selectivity in SFB. An increase in pressure at a constant temperature results in an increase in solvent strength, which means a better solubility of the solutes in the supercritical fluid. In addition, the higher the extraction pressure, the smaller is the volume of supercritical fluid needed for extraction [62]. Fairly frequently, the extraction pressure is selected at the upper pressure limit of the SFE system. This selection generally yields a high recovery in a short extraction time. High pressures, however, are not always recommendable because of the limited selectivity of SFE extraction at high pressures. Selective extraction can be achieved only by a proper selection of the extraction conditions. For example, alkanes can be extracted from urban air particulates with carbon dioxide at 75 bar ( 45°C) whereas the P AHs remaio unextracted until the pressure is raised to 300 bar. By sequentially extracting the air particulates at these two pressures, 85-90% selectivities can be achieved [78]. Successful selective extracrions have also been reported for extracting analytes from bulk matrix materials that are also soluble in the supercritical fluid under stronger conditions [79].
Temperature
At a constant pressure, the density of a supercritical fluid deercases when the temperature is increased. On the contrary, temperature can also affect the volatilities and diffusitivities of the solutes, the flexibility of the matrix, and the affmity of the solutes to active sites on the matrix. Hence, the effect of a temperature elevation on SFB recovery and selectivity is difficult to predict [80]. On the other hand, for a volatile solute there is a competition between its solubility and volatility and higher reeoverles can be obtained at higher · temperatures [63], lf the rate-limiting parameter in SFE is ditfusion of the components or desorption of the analytes from active sites of the matrix, increasing temperature will result in an improved extraction recovery because increased temperature will facilitate solute diffusitivity and/or reduce the interaction between the solute and the matrix. For example, in the extraction of polymer additives from polymerie materials, an increase in temperature normally gives a faster extraction [50, 81]. Langenfeld et al. reported that increased extraction efficiencies of PCBs from sediments and P AHs from air particulate materials were obtained by increasing the extraction temperature from 50°C to

26 SFEand SFC
200°C at 350 bar. According to Langemeld et al. the results indicate that the kinetics of the partitioning process are improved at higher temperatures [82]. The observed improved extraction efficiency could, however, also be due to the reduced interaction of the solutes with active sites at higher temperatures [51].
Flow rate and extraction time
The effects of the flow rate of the supercritical fluid on the SFE extraction efficiency has been discussed in more detail in section 2.1.3.2. Experiments at different flow rates can yield valuable information about whether the major limitation to achieve rapid extraction is of a thermodynamic nature (i.e. the distribution of the analytes between the supercritical fluid and the sample matrix at equilibrium), or is related to kinetics (i.e. the time required to approach that equilibrium). For samples that show a dramatic increase in extraction rate when the fluid flow rate is increased, the kinetics of extraction are apparently fast. Solubility appears to be the limiting factor, and the extraction can be improved by increasing the extraction pressure or by exposing the sample to a larger volume of fluid. In contrast, if there is no large effect of the fluid flow rate on the extraction rate, it appears that mass transfer is slow, and this slow mass transfer limits the overall extraction rate [83]. In SFE with carbon dioxide as the extraction fluid, typical supercritical fluid flow rates range from 0.1 mi/min to 1 mi/min.
The optimal extraction time depends on the experimental temperature and pressure, the modifier type and concentration as well as on the flow rate of the fluid through the extraction cell. For extractions where modifiers are added directly to the sample matrix, a period of static extraction is often found to be necessary to ensure good contact of the modifier with the solutes and the matrix. The static times used generally range from 5 minutes to 30 minutes depending on the properties of the solutes, the modifier and the matrix. For unknown samples, the extraction time can best be found by experimentally conducting successive extractions to determine the completeness of extraction. The use of a nondestruclive detector in tandem with SFE can also aid the analyst in determining the extent of extraction. Knowledge of the sample matrix and the solubility of the components in the supercritical fluid can be of assistance in choosing the proper extraction time, since the extraction recovery is a tunetion of the ratio of a solute distributed between the supercritical fluid and the matrix. For example, extraction times of more than one hour are always needed in the extraction of polymerie materials, while complete extraction could be obtained within one hour for most environmental samples under properly selected conditions.

Chapter 2 27
Collection
The solute collection methods in SPE have already been discussed in detail in section 2.1.3.1. The coneetion of the extracted components is a very important step in the SPE procedure. Accurate evaluations of extraction efficiency can only be obtained if coneetion is quantitative. Hawthome et al. suggested that the coneetion efficiency should be the first parameter to be studied in developing an SPE metbod [83]. Because of the large number of experimental variables that can affect collection efficiencies, the determination of the quantitative abilities of the coneetion device must be determined using appropriate spiking/recovery studies. In these studies, the extraction conditions to be used for the real-world samples should be used to extract the analytes of interest spiked at known concentrations onto a relatively inert matrix, such as clean sand. The matrix should retain the spiked analytes until the SFE extraction is begun, but should easily release the spiked analytes during the SFE extraction since the goal is to evaluate only the co Heetion method. Fortunately, even with relatively simp Ie co Heetion methods, such as bubbling the depressurized extraction fluid through a few mL of solvent, quantitative coneetion of analytes as volatile as n-octane and phenol, are relative easy to achleve [18, 26, 84]. When more volatile analytes are of interest or if very high supercritical fluid flow-rates are used, the use of sorbent trapping should be considered. Sorbent trapping is also easier to automate.
Developing and validating SFE methods
The development of an SPE metbod is a time consuming process, because many parameters affect the SFE process and the SFE mechanisms are only poorly understood. The chemical and physicochemical aspects of SPE that have been discussed above can only provide some general guidelines in developing quantitative SFE methods. Hawthome et al. proposed a sequentia! metbod for the development of SFE methods for environmental samples [83].
In the selection of initial SFE extraction conditions, the properties of the target analytes and matrix and any literature reports on successful SFE methods for similar samples should be considered. The initial selections can rarely be satisfactory because SPE extraction efficiencies are highly matrix dependent The following are some suggestions for selecting the initial conditions in SFE experiments:

(1) Pure carbon dioxide can extract non-polar to moderately polar components that can be analyzed with conventional capillary gas chromatography (GC). (An exception to this is fat components which can be easily extracted with supercritical carbon dioxide, but are difficult to analyze with convendonal GC.);
(2) Por polar or really non-volatile components, the addition of modifiers may be necessary to obtain quantitadve extraction. Por example, ion pairing reagents are needed for extracting ionic compounds;
(3) The starting supercritical fluid flow rate and extraction pressure should be between 0.5 to 2 ml/min of the supercrideal fluid at the maximum pressure of the SFB system;
(4) Por thermally labile compounds, low extraction temperatures are favorable; (5) In general, supercrideal fluids are more effecdve extraction agents when
the extraction is performed at a temperature above the meldng points of the target analytes;
(6) In the extraction of polymerie materials, the extraction temperature should be selected well above the glass-transidon temperature of the fluid-swollen polymer in order to increase ditfusion of solutes in the polymerie materiaL (Unfortunately, the glass-transition temperature of a polymer under supercritical conditions is rarely known).
(7) Por samples with high concentradons of water and/or other extraetabie matrix components, the restcictor should be heated or some types of drying agents should be added to the sample to avoid plugging of the restcictor during extraction.
(8) Por environmental samples, extraction times of 30-60 minutes are useful since longer extraction times generally do not yield substantially higher recoveries. Extraction times of at least 1 hour are normally needed for polymerie samples.
After the initial selection of extraction condidons, the colleedon efficiency should first be tested and optimized. Only when the trapping efficiency is quantitative can the extraction efficiency and selectivity be evaluated. The collection of the extracted components in SFB has been discussed in more detail in sections 2.1.3.1. Since samples with known concentrations are only scarcely available, the validadon of an extraction metbod is generally based on one of the following three approaches, each depending on assumptions that may or may not be valid [83]: (1) Determining the recovery of known concentrations of spiked compounds
from the sample (or similar) matrix;

Chapter 2 29
(2) Comparison of the recoveries of native analytes with those achieved using conventionally-accepted extraction methods;
(3) Perform multiple sequentia! extraction of the same sample under different extraction conditions.
Perhaps the least reliable technique for validaring the efficiency of SFE methods is the use of spik:ed samples [85]. It should also be noted that conventional extraction methods may not yield quantitative recoveries of the native analytes, and therefore a highly efficient SFE extraction metbod may yit:Id higher recoveries than conventional methods. The use of multiple extracrions with different conditions could be a very useful way to validate the SFE extraction efficiency. This could include extracting the residues from an SFE extraction by Soxhlet in an appropriate liquid solvent, by extracting the residue under more stringent SFB conditions, or by a combination of these two approaches [83, 85].
Under the conditions initially selected, SFE extraction may not be quantitative or sufficiently selective, and the optimization step may have to be repeated several times. Some experimental design approaches have been used to reduce the time and the number of experiments needed for the optimization of SFE [86, 87]. An optimization strategy for the extraction of polymerie samples was recently proposed (see Fig. 2.4) [88]. It is very well possible that the demands for quantitative and selective extraction can be very difficult to be met simultaneously. In this case, additional selectivity can be introduced by packing a suitable sorbent into the extraction cell or by depositing the analytes onto a sorbent, and then eluting them with properly selected conditions. This additional selectivity was demonstrared by Sandra et al. in the analysis of the organochloropesticides from tobacco leaves [89]. The chromatogram could be greatly simplified and the components of interest could be easily quantified by adding silica to the extraction cell to retain medium polar and polar solutes.

30
SFE of polymers-
SFEand SFC
Increase extraction temperature
Ditfusion Umited -E Decrease polymer partiele size
Add modifier (to swell polymer)
Increase extraction pressure
Adjust extraction temperature Solubility limited
Add modifier (improve solubility)
Increase SF flow rate
Figure 2.4. Optimization strategies for SFE of polymerie samples.
In spite of the difficulty in optimization, SPE has been successfully applied to a wide range of samples and a lot of interesting results have been obtained. SPE has the potendal to become an important sample preparation metbod in analytical chemistry. The future achievements of SPE will depend on the increased understanding of the mechanisms that control the SPE processes.
2.1.6. Analytical SFE and other extraction techniques
Extraction is a process of eluting, by means of an extraction solvent, one or more of the components from a mixture of substances. It is widely used as a sample preparation metbod in analytical chemistry. In this section, SPE is compared with other extraction techniques which are conventionally used in analytica! chemistry, such as Soxhlet extraction, liquid-liquid extraction and solid phase extraction (SPE). In order to allow a comparison between these techniques, the conventional methods are first introduced briefly.
Soxhlet extraction
In the extraction of solutes from a solid matrix, the easiest metbod is to extract the components of interest directly with a solvent. However, if the solubility of the components in the solvent is low, large volumes of solvent and long extraction

Chapter 2 31
times will be required. Soxhlet extraction is a widely used method for the extraction of solutes trom a solid matrix. It is also a typical example of a timeconsuming technique that requires large volumes of liquid solvents. In Soxhlet extraction, the material to be extracted is placed in an extraction thimble which has a liquid-permeable wall. In operation, the solvent refluxing trom the condenser soaks the material, the liquid level rises until it has reached the rim of the thimble, and is then siphoned off through the delivery tube into the boiling flask. From there, solvent again evaporates, condenses, and drops into the extraction thimble, etc .. In this way a relatively small amount of solvent (typically 50-250 ml) can extract substantial quantities of materiaL Soxhlet extraction is a simple and rugged technique. It is, however, ditticuit to automate. Moreover, the application of this technique to the extraction of thermally unstable compounds is limited because the extraction is being carried out at the boiling point of the solvent used.
Soxhlet extraction is not only ditticuit to automate, it is also ditticuit to combine this technique on-line with other analytica! methods. Prior to chromatographic analysis, the samples should trequently be preconcentrated, especially for trace analysis. This step may incur losses through evaporation or decomposition of unstable or volatile compounds, particularly when heating is required to speCel up evaporation.
Although a wide variety of solvents is available for Soxhlet extraction, the possibility of selective extraction of components trom a sample is limited because the solvent strength of a liquid is essentially constant regardless of the extraction conditions. Once the solvent bas been chosen, the selectivity can no longer be varied. Another disadvantage of Soxhlet extraction is the time-consuming nature of the technique. One extraction may require hours or even days to perform.
liquid-liquid extraction
Liquid-liquid extraction is a technique in which a solution is brought into contact with a second solvent, essentially immiscible with the first, in order to bring about transfer of one or more solutes into the second solvent. The separations that can be achieved by this method are simple, they are applicable equally to trace level analysis as well as to the extraction of a large amount of materials. The efficiency of extraction depends primarily on the distribution of the solute between the two phases, the phase ratio, and the number of extractions. It is well-known that

repeated extractions with sman portions of solvent can reeover much more analyte than a single batch extraction with a large volume of the extraction fluid. Por practical reasons, however, a sample would be extracted no more than triple in most applications.
Liquid-liquid extraction is an extremely versatile method of sample preparation for liquid matrices. The theory about liquid-liquid extraction has been wen studied. The matured theory, convenience and ease to use as well as the availability of highly purified organic solvents have contribute to its continued widespread use. However, liquid-liquid extraction is not a technique which lends itself wen to automation. This technique is relatively time-consuming. It ofien requires the removal of solvents by evaporation, and this lengthy step may lead to losses through vaporization or decomposition of unstable compounds, in particular when heating is required. Emulsion formation is another drawback of the technique, especially if a solvent of intermediate polarity is included in the extracting solvent, or if the volume ratio of organic solvent to aqueous phase is not sufficiently large. The formation of emulsion may be avoided by changing the phase ratio, or may be broken by tiltration over glass wool, centrifugation, or refrigeration. Nevertheless, these approachestend to be unsatisfactory in that they add substantially to the analysis time, and full recovery of the two separate phases is rarely achieved [90].
Solid-phase extraction (SPE)
The general approach to solid-phase extraction (SPE) is the adsorption of components from a liquid onto asolid adsorbent or stationary phase immobilized on a solid support. Nowadays, many materials are commerciany available for SPE. Silica gels or silica gels modified with a variety of functional groups, e.g. alkyl-, phenyl, cyano and diol-moieties, are commonly used to provide specific interaction with the analytes.
SPE methodology is based on chromatographic principles, differing only in that compounds in an analytically useful situation would have capacity factors between 1 and 10, whereas in an efficient extraction scheme, they should be either much greater than 10, where the analytes are totally retained, or much less than 1, where the analytes are eluted easily. After the selection of an appropriate sorbent on which to carry out the SPE, conditions should be optimized to maximize the retention of the compounds of interest, while rninimizing retention of intertering substances. The simultaneous achlevement of these two objectives is rarely possible, but selectivity can be maximized by carefut selection of the sorption and

elution conditions. After purification, the components of interest should be desorbed from the sorbent by solvent elution, SFE or thermal-desorption.
SPE is a technique which can circumvent many of the problems associated with liquid-liquid extraction. SPE can be combined either on-line or off-line with chromatographic techniques, such as GC, HPLC, SFC etc .. The commercial availability of disposable SPE cartridges, generally contigured as luer-tipped syringes in various sizes, makes SPE very convenient. Unfortunately, SPE is only suitable for the extraction of liquid samples. After extraction, one more step is needed to elute the components of interest from the sorbent after SPE. Less than quantitative recovery in this step has been frequently observed for polar or labile compounds due to the presence of active sizes on/in the packed sorbent. Another problem associated with SPE is that the extraction cartridge can be readily blocked if the sample matrix is very viscous, highly contaminated or contains high concentrations of proteins or suspended matenals [90].
Advantage and disadvantages of SFE
Analytica! SFE has been successfully used for a wide variety of matrix/solute combinations, especially in the extraction of various pollutants, e.g. PCBs, PAHs · and pesticides from environmental materials such as soil, sediments and from biologica! materials. In comparison with the analytica! extraction methods described above, SFE offers many attractive advantages, such as higher selectivity, shorter extraction time, reduced usage of toxic organic solvents, ease to be combined on-line to other analytica! instruments etc ..
(a) SFE is a selective extraction technique. The solvent strength of a supercritical fluid depends on the pressure and temperature used for the extraction. It can easily be controlled by changing pressure and/or temperature. At a constant temperature, extraction at lower pressures will favour smaller and non-polar molecules, whereas extraction at higher pressures will favour larger molecular weight solutes. This allows an extraction to be optimized for a particular class of compounds simply by changing the pressure and/or temperature of extraction [69]. By selecting suitable SFE conditions, the extraction can be made selective, avoiding the need for further sample clean-up. In contrast, the solventstrengthof a given liquid is essentially constant regardless of extraction conditions.
(b) SFE is fast. Mass transfer limitations ultimately determine the rate at which an extraction is carried out. Supercritical fluids have solute diffusitivities about an

34 SFEand SFC
order of magnitude higher and viscosities about an order of magnitude lower than liquid solvents. Therefore, supercritical fluids have much better mass transfer characteristics. Por samples in which the components are adsorbed on the smooth surface of the matrix, mass transfer properties of the extraction solvent are not too important because mass transfer can be greatly enhanced by simply shaking the extraction cell or by other mechanica! means. In this case, traditional liquid extraction is fast. For most real-world samples, however, the matrix has deep pores and/or deep ditches, in which mass transfer is almost exclusively controlled by molecular diffusion. Good mass transfer characteristics are, therefore, particularly important in these cases. Supercritical fluids can penetrate into the pores and/or ditches and extract the components much faster than liquid solvents. Por comparison, SPE extracrions are generally quantitative in 10-60 min, whereas extraction times in liquid extraction can range from several hours to several days. In addition, because supercritical fluids are gases at ambient conditions, preconcentration steps after SPE are greatly simplified and direct coupling of SPE to chromatographic techniques is convenient. In contrast, extracts from liquid solvent extractions frequently need to be preconcentrated prior to the determination of trace organic analytes, a step that requires additional time and can result in the loss of volatile or thermally labile analytes. In recent years large volume injection techniques have been reported that allow the injection of samples of up to at least 100 J.ll, which implies that in some cases dilute samples can also be introduced directly into the chromatographic system without preconcentration [91]. However, large volume injection techniques are still fairly complicated and require special skilis and instrumentation.
(c) The extraction temperature in SPE is adjustable. In SPE the extraction temperature can be changed continuously from the critica! point of the supercritical fluid to much higher temperatures. The wide range of extraction temperatures available in SPE is of particular importance. Por example, the low critica! temperature of supercritical carbon dioxide makes SPE an excellent candidate for extracting thermally labile compounds under conditions slightly above room temperature. The high extraction temperatures available, on the other hand, favorably affect the diffusion coefficients of compounds and thus increase the SPE extraction rate for samples in which the rate-limiting parameter is related to diffusion in the matrix, such as in the case of extracting polymer additives from polymerie materials. This is in contrast to Soxhlet extractions where the extraction temperature is limited by the boiling point of the solvent used [50].

Chapter 2 35
(d) SFB has additional practical advantages. Most supercritical fluids are inert, nontoxic, pure, inexpensive, and SFB is easy to be connected on-line to other analytical instruments. Recent concerns about the hazardous nature of many commonly used solvents, the costs and envirorunental dangers of waste solvent disposal, and the emission of hazardous solvents into the atmosphere during sample concentration greatly support the development of SPE as an alternative sample extraction method.
As a new developing technique, SFE also has several disadvantages. The following three may be the most serious ones. Firstly, the costs for instrumentation and personnet training in SFB are much higher than other methods. Lopez-Avila et al. compared the costs associated with SFB and those for Soxhlet extraction by considering the labor, extraction time, instrumentation and solvent disposal. They found that in the extraction of envirorunental samples routine SFB extracrions are two to three times more expensive than Soxhlet extraction, even though SFB extractions are much faster [92]. Por samples for which simple extraction methods exist that are convenient, rapid, quantitative, and do not require preconcentration of the extract for the determination of the target analytes, SFB has few advantages [69]. Therefore, SFB should not be used in those cases where other methods are definitely working better, e.g. as in SFB of water samples [89]. Secondly, the addition of modifiers may largely eliminate some of the advantages of SPE over other sample preparation methods. Por example, the introduetion of a modifier will increase the critical temperature of the fluid, and thus limit its usage for the extraction of thermally labile compounds. In addition, the presence of modifiers will make the on-line conneetion of SPE with other analytica! methods more difficult. Thirdly, optimization in SFB is rather complicated. Many parameters affect the SPE process, e.g. temperature, pressure, modifier type and concentration as well as solute parameters such as molecular weight, polarity and volatility, and matrix parameters as partiele size, pore structure, adsorptive strength etc.. The numerous parameters that influence the extraction process and the lack of fundamental knowledge about how these parameters affect the extraction are to blame for the fact that method development in SFB up till now is mainly empirical. Current optimization strategies are almost exclusively basedon trial and error experiments.
With the development of SFB systems and the accumulation of fundamental knowledge of SFB, · the disadvantages associated with SFB will greatly be alleviated. Por example, the cost per extraction will undoubtedly be significantly

36 SFEand SFC
reduced when the technique has been optimized and reptacement parts are available on a large scale.
REFERENCES
1. R.E. Majors, LC. GC Int., 4, No.2 (1991) 10. 2. T.L. Chester, J.D. Pinkston and D.E. Raynie, Anal. Otem., 64 (1992) 153R. 3. M.L. Lee and K.E. Markides (eds), Analytica/ Supercrideal Fluid Otromatography and
Extraction, Chromatography Conference, Provo, UT, 1990. 4. F.B. Bright, M.E.P. McNally, (eds), Supercritical Fhtid Technology: Theoretica/ and applied
approaches to analytica/ Otemistry, ACS Symposium Series 488, American Chemical Society, Washiton, DC, 1992.
5. J.R. Dean (ed), Application of supercrideal Fluids in Industrial Analysis, CRC Press, lnc., Boca Raton, Florida, 1993.
6. B.A. Charpentier and M.R. Sevevants (eds), Supercritical F/uid Extraction and Chromatography, American Chemical Society, Washington DC, 1988.
7. T.L. Chester, J.D. Pinkston and D.E. Raynie, Anal. Otem., 66 (1994) 106R. 8. V. Camel, A. Tambute and M. Caude, J. Otromatogr., 642 (1993) 263. 9. J.M. Levy, J. High Resohtt. Otromatogr., 17 (1994) 212. 10. M.W. Raynor and K.D. Bartle, J. Supercrideal F/uids, 6 (1993) 39. 11. L.G. Randall, Sep. Sd. Technol., 17 (1982) 1. 12. L.G. Randall, Ultra High Resohttion Otromatography, ACS Symposium Series 250, S. Abuja
(Ed), American Chemica! Society, Washington, DC, 1984, p.l35. 13. V. Janda, K.D. Bartie and A.A. Clifford, J. Otromatogr., 642 (1993) 283. 14. H.-G. Janssen and C.A. Cramers, J. Otromatogr., 505 (1990) 19. 15. J.R. Dean and M. Kane, in J.R. Dean (ed), Application of Supercritical Fluids in lndustrial
Analysis, CRC Press, Inc., Boca Raton, Florida, 1993, p.52. 16. J. Rein, C.M. Cork and K.J. Furton, J. Otromatogr., 545 (1991) 149. 17. K.G. Furton and Q. Lin, Chromatographia, 34 (1992) 185. 18. J.J. Langenfeld, M.D. Burford, S.B. Hawthome and D.J. Miller, J. Otromatogr., 594 (1992)
297. 19. M. Richard and R.M. Campbell, liq. Otromatogr. Gas Otromatogr., 4 (1991) 36. 20. D.E. Knowies and T.K. Hoge, in J.R. Dean (ed), Application of Supercrideal Fhtids in
Industrial analysis, CRC Press, Inc., Boca Raton, Florida, 1993, p.104. 21. J.W. King, J. Chromatogr. Sci., 28 (1990) 9. 22. S. Bowardt and B. Johansson, Anal. Otem., 66 (1994) 667. 23. B.E. Berg, E.M. Hansen, S. Gjorven and T. Greibrokk, J. High Resohtt. Otromatogr., 16
(1993) 358. 24. H.-G. Janssen, J.A. Rijks and C.A. Cramers, J. Microcohtmn Sep., 2 (1990) 26. 25. D.J. Miller, S.B. Hawthome and M.E.P. McNally, Anal. Chem., 65 (1993) 1038. 26. M.D. Burford, S.B. Hawthome, D.J. Milier and T. Braggins, J. Otromatogr., 609 (1992) 321. 27. B.W. Wright, C.W. Wright, R.W. Gale and R.D. Srnith, Anal. Otem., 59 (1987) 38. 28. P. Sandra, F. David and E. Stottmeister, J. High Resohtt. Otromatogr., 13 (1990) 284. 29. L.J. Mulcahey, J.L. Hedrick and L.T. Taylor, Anal. Otem., 63 (1991) 2225. 30. J.L. Langenfeld, M.D. Burford, S.B. Hawthome and D.J. Miller, J. Otromatogr., 594 (1992)
297. 31. P.G. Thompson, L.T. Taylor, B.E. Richter, N.L. Porter and J.L. Ezzell, J. High Resol.
Otromatogr., 16 (1993) 713.

Chapter 2 37
32. J. Vejrosta, J. Planeta, M. Mikesova, A. Ansorgova, P. Karasek, J. Fanta and V. Janda, J. Chromatogr., 685 (1994) 113.
33. M.E.P. McNally, Anal. Chem., 61 (1995) 308A. 34. L.I. Mulcahey and L.T. Taylor, AnaL Chem., 64 (1992) 2352. 35. J.H. Raymer and E.D. Pellizzari, Anal. Chem., 59 (1987) 1043. 36. J. Vejrosta, a. Ansorgova, M. Mikesova and K.D. Bartle, J. Chromatogr., 659 (1994) 209. 37. W.N. Moore and L.T. Taylor, Anal. Chem., 61 (1995) 2030. 38. A. Meyer, W. Kleibobmer and K. Cammann, J. High Resol. Ozromatogr., 16 (1993) 491. 39. S.B. Hawthome, D.J. Milier and M.S. Krieger, J. Otromatogr. Sci., 27 (1989) 347. 40. X. Lou, H.-G. Janssen and C.A. Cramers, J. Chromatogr., 750 (1996) 215. 41. R.J. Houben, H.-G. Janssen, P.A. Leclercq, J.A. Rijks and C.A. Cramers, J. High Resol.
Chromatogr., 13 (1990) 669. 42. M.D. Burford, S.B. Hawthome and D.J. Miller, J. Chromatogr., 685 (1994) 79. 43. M.D. Burford, S.B. Hawthome and D.J. Miller, J. Ozromatogr., 685 (1994)95. 44. I.M. Levy, M. Ashraf-Khorassani, J. Chromatogr.libr., 53 (1992) 197. 45. M.H. Liu, S. Kaplia, K.S. Nam and A.A. Elseewi, J. Chromatogr., 639 (1993) 151. 46. T.A. Barber, P.R. Bienk:owski and H.D. Cochran, Sep. Sci. Technol. 25 (1990) 2033. 47. C.H. Kirschner and L.T. Taylor, Anal. Chem., 65 (1993) 78. 48. D.L. Heglund, D.C. Tilotta, S.B. Hawthome and D.I. Miller, AnaL Own., 66 (1994) 3543. 49. C.F. Poole and S.K. Poole, Chromatography Today, Elsevier, Amsterdam, 1991, p. 19. 50. X. Lou, H.-G. Janssen and C.A. Cramers, J. Microcol. Sep., 7 (1995) 303. 51. H.-G. Janssen, P.J. Schoenmakers and C.A. Cramers, J. High Rosol. Chromatogr., 12 (1989)
645. 52. H.-G. Janssen, P.J. Schoenmakers, Mikrochim. Acta, 2 (1991) 337. 53. A.L. Howard and L.T. Taylor, J. high Resol. Otromatogr., 16 (1993) 39. 54. T.M. Fahmy, M.E. Paulaiti, D.M. Johson and M.E.P. McNally, Anal. Chem., 65 (1993) 1462. 55. J. Dank:ers, M. Groeneboom, L.H.A. Scholtis and C. van de Heiden, J. Chromatogr., 641
(1993) 357. 56. T.S. Oostdyk, R.L. Grob, J.L. Snyder and M.E. McNally, J. Chromatogr. Sci., 31 (1993) 177. 57. J.J. Langenfeld, S.B. Hawthorne, D.I. Milier and J. Pawliszyn, Anal. Own., 66 (1994) 909. 58. Y. Lin and C.M. Wai, Anal. Chem., 66 (1994) 1971. 59. K.E. Laintz and E. Tachikawa, Anal. Chem., 66 (1994) 2190. 60. J.W. Hills, H.H. Hili, D.R. Hansen and S.G. Metcalf, J. Ozromatogr., 619 (1994) 319. 61. B.E. Berg, E.M. Hansen, S. Gjorven and T. Greibrokk, J. High Resol. Chromatogr., 16 (1993)
358. 62. M.E.P. McNally and J.R. Wheeler, J. Otromatogr., 441 (1988) 53. 63. J.R. Wbeeler and M.E. McNally, J. Otromatogr. Sci., 27 (1989) 534. 64. K.G. Purton and Q. Lin, J. Otromatogr. Sci., 31 (1993) 201. 65. X. Lou, H.-G. lanssen and C.A. Cramers, J. High Resolut. Otromatogr., 18 (1995) 483. 66. J.C. Giddings, M.N. Myers, L. McLaren, and R.A. Keiler, Science, 162 (1968) 67. 67. J. Pawliszyn, J. Chromatogr. Sci., 31 (1993) 31. 68. W. Eisenbach, P.J. Gottsch, K. Niemann and K. Zosel, Fluid Phase Equilibria, 10 (1983) 315. 69. S.B. Hawthorne, Anal. Own., 62 (1990) 633A. 70. R.C. Weast (ed), CRC Handbook of Chemistry and Physics, 62th Ed., Chemical rubber
Company, Boca Raton, Florida, 1984. 71. R.E. Clement, G.A. Eiceman and CJ. Koester, Anal. Chem., 61 (1995) 221R. 72. S.B. Hawthorne, J.J. Langenfeld, D.J. Milier and M.D. Burford, Anal. Chem., 64 (1992) 1614.

38 SFE and SFC
73. A.L. Howard, WJ. Yoo, L.T. Taylor, F.K. Schweigbardt, A.P. Emery, S.N. Chester and W.A. MacCrehan, J. O!romatogr. Sci., 31 (1993) 401.
74. R.D. Chambrs and S.R. James, in J.F. staddart (ed), Comprehensive Organic O!emistry, A. Wheaton & Co. Ltd., Exeter, 1979, Vol. 1, p. 525.
75. S.B. Hawthorne, DJ. MiUer and J.J. Langenfeld, J. O!romatogr. Sci., 28 (1990) 2. 76. D.E. Raynie, Anal. O!em., 65 (1993) 3127. 77. J.W. King, J. O!romatogr. Sci., 27 (1989) 355. 78. S.B. hawthorne and D.J. Miller, J. O!romatogr. Sci., 24 (1986) 258. 79. M.T.G. Hierro and G. Santa-Maria, Food O!em. 45 (1992) 189. 80. R.T. Krunik, S.J. Holla and C.R. reid, J. O!em. Eng. Data, 26 (1981) 47. 81. N.J. Cotton, K.D. BartJe, A.A. Clifford and CJ. Dowle, J. O!romatogr., Sci., 31 (1993) 157. 82. J.J. Langenfeld, S.B. Hawthome, D.J. Milier and J. Pawliszyn, Anal. O!em., 65 (1993) 338. 83. S.B. Hawthorne, D.J. Miller, M.D. Burford, J.J. langenfeld, S. Eckert-Tilotta and P.K. Louie,
J. Ozr01natogr., 642 (1993) 301. 84. N.L. Porter, A.F. Rynaski, E.R. Campbell, M. Saunders, B.E. Richter, J.T. Swanson, R.B.
Nietsen and B.J. Murphy, J. O!romatogr. Sci., 30 (1992) 367. 85. M.D. Burford, S.B. Hawthome and D.j. Miller, Anal. O!em., 35 (1993) 1497. 86. N. Adasoglu, S. Dicer and E. Bolat, J. Supercr. Fluids, 7 (1994) 93. 87. I.J. Barnabas, J.R. Dean, W.R. Tomlinson and S.P. Owen, Anal. O!em., 61 (1995) 2064. 88. X. Lou, H.-G. lanssen and C.A. Cramers, J. O!romatogr. Sci., 34 (1996) 282. 89. P. Sandra, A. Kot, F. David, in P. Sandra (ed), Proceedings of Sixteenth International
Symposium on Capillary O!romatography, Rivadel garda, Italy, 1994, p. 1515. 90. M.T. Kelly, in M.R. Smyth (ed), O!emical analysis in Complex Matrices, ellis Horwood Ltd.,
Chichester, West Sussex, 1992, p. 17. 91. H.G.J. Mol, H.-G.M. Janssen, C.A. Cramers. J.J. Vreuls and U.A.Th Brinkman, J.
O!romatogr., 703 (1995) 277. 92. V. Lopez-Avila, N.S. dodhiwala and W.F. Beckert, J. O!romatogr. Sd., 28 (1990) 468.

2.2 Correlation of Supercritical-Fluid Extraction Recoveries witb Supercritical-Fluid Cbromatograpbic Retention Data1
>
ABSTRACT
The possibility of using supercritical-fluid chromatographic retention data for examining the effects of operational parameters, such as pressure and flow rate, on the extraction characteristics in supercritical-fluid extraction (SFE) was investigated. A model was derived for calculating the extraction efficiency in SFE from retention data and peak shapes measured in supercritical-fluid chromatography (SFC). By performing the SFC experiments at the same pressure and temperature as the SFE extractions using the SFE extraction cell as the SFC column, an accurate prediction of extraction efficiencies could be made. Finally, the effects of matrix composition and analyte concentration on extraction efficiency were studied.
2.2.11NTRODUCTION
Supercritical fluid extraction (SFE) is a relatively new and rapidly growing technique for sample preparation in analytical chernistry [1]. SFE bas been recognized as an important alternative to conventional liquid or Soxhlet extraction and provides many advantages, such as a reduced use of organic solvents, shorter extraction time, adjustable solventstrengthand the ability for on-line transfer of the extracted components to other analytical instruments [2-5].
In trace analysis, the ability to rapidly release the extraction fluid simply by reducing the pressure and venting the extraction fluid is a significant advantage of SFE over liquid extraction. In conventional extraction techniques, the extracts often have to be reconcentrated prior to introduetion into the analytica! separation system. SFE extracts can be collected in a very small volume of liquid or even in dry coneetion tubes. For this reason, SFE is particularly prornising for trace analysis. Unfortunately, however, SFE also has a number of disadvantages. Among these is the price of the instrumentation and, more importantly, the time consurning nature of metbod development. Many parameters affect the SFE process, e.g. temperature, pressure, modifier type and concentration as well as solute parameters such as molecular weight, polarity and volatility, and matrix properties such as partiele size, pore structure and adsorptive strength. The numerous parameters that influence the extraction process and the lack of
'1 X. Lou, H.-G. Janssen and C.A. Cramers, J. High ResoL O!romawgr., 18 (1995) 483.

Correlation between SFE and SFC 40
fundamental knowledge about how these parameters affect the extraction are to blame for the fact that method development in SPE up till now is mainly empirica!. Current optimization strategies for SPE are almost exclusively based on trial and error experiments.
The SPE process basically involves three subsequent steps. Pirst, the solutes have to diffuse from the core of the matrix-particles to the surface. lt is obvious that this step is absent if the components are adsorbed on the surface of a particle, which is for example the case for components that are extracted from an SPE cartridge or from sandy soil. Next, the components are transferred from the partiele surface into the extraction fluid. The key parameter that controls this process is the distribution coefficient of the solute between the matrix and the supercritical fluid phase. Pinally, the components are eluted from the extraction cell by the flow of supercritical extractant. The last two steps of the extraction process are somewhat similar to the process occurring in supercritical fluid chromatography (SPC). Hence, the extraction behaviour for samples in which these two steps determine the rate of extraction should be at least qualitatively related to the retention behaviour of the solutes in SPC.
Optimization of SPE has been the subject of a large number of papers in recent chromatographic literature [6-16]. Most of these articles rely on empirica! optimization. A limited number of more fundamental studies was published in which attempts were made to increase the basic knowledge of the thermodynamic and kinetic parameters that impact on SPE. Bartie et al. [6] derived a model that allowed the calculation of the extraction kinetics in an SPE system where the rategoverning step is slow mass transfer inside the matrix particles. King [7] published a mathematica! model that enables the calculation of the solubility of organic compounds in supercritical fluids from molecular parameters. McNally and Wheeler [8, 9] attempted to correlate SPC retention data of diuron and linuron measured on an octadecyl material with the extraction behaviour of these components from soil in SPE. Similar work was done by Purton et al. [10].
Bartie' s model yielded an excellent agreement between theory and practical results in the case where the first step in the extraction process, i.e. diffusion in the solid particle, is the rate-limiting parameter. A typical SPE application where this situation occurs is the extraction of polymer additives from polymerie particles. Insome other important application areas of SPE, the components are adsorbed on the surface of a partiele such as a sandy soil or an SPE sorbent. In these cases, no diffusion of the components from the core of the partiele to the surface has to occur. Therefore, the key parameter controlling the extraction behaviour for these samples is the distribution coefficient of the solute between

Chapter 2 41
matrix and supercritical fluid. The distribution coefficient is the result of competition between the supercritical fluid extractant and the matrix for the solute. In analytica! SPE, the analyte concentratien in the matrix is generally low. Hence, the solubility of the components in the extraction fluid is generally not a limiting parameter. The observation that small variations in sample matrix composition can require substantial adjustment of the extraction parameters appears to indicate that effects associated with the matrix are far more important [16].
In chromatography, the capacity factor, being the quotient of the distribution coefficient and the phase ratio, represents the ratio of the amount of the component retained in the stationary phase to the amount present in the mobile phase. Therefore, the extraction killetics in SPE should at least be qualitatively related to the capacity factors of the components measured in SFC using the matrix that is to be extracted as the stationary phase. In more general terms, the knowledge of extraction killetics in SPE can be improved by studying the retentien behaviour in packed column SFC.
In the packed-column SFC separation of polar components, very often asymmetrical peaks are observed. The cause for the poor peak shape is the presence of residual active sites on the packing matrix [17,18]. These active sites tend to interact strongly with the analytes thereby resulting in non-linear isotherms. Moreover, for such systems, the distribution coefficient beoomes concentration dependent If now a similar situation occurs in SPE, this would mean that the extraction yields obtained in a given extraction time under given experimental conditions could also be affected by the concentratien of the analytes in the matrix. Schoenmakers et al. [17, 18] studied the distribution isothermsof polar cornponents on inhomogeneous stationary phases in SFC and found that the retention decreased with increasing sample size. Up till now, the effects of concentration on tbe extraction efficiency in SFE have not been investigated yet.
The aim of the present work is to investigate whether SFC retention data of the components of interest measured on the sample matrix can provide a means of gaining knowledge of the extraction killetics in SFE. After extraction, the components are injected by means of an SFC injection valve incorporated in the SFE set-up and the retention data and peak shapes are determined. SFE elution profiles are then correlated with retention data and peak shapes of the components as obtained in the SFC mode. A model is derived that allows the calculation of extraction yields from retentien data and peak shapes measured under SFC conditions using the extraction cell as the chromatographic column. The predicted recoveries are compared with experimentally observed extraction yields.

Correlation between SFE and SFC 42
Furthermore, the effects of analyte concentration on the kinetics of extraction were investigated.
2.2.2 THEORY
Supercritical fluid chromatography and supercritical fluid extraction have in common the use of a supercritical fluid for eluting the analytes. In both techniques, there is a stagnant phase that tends to retain the probe molecules. The matrix in SFE can be looked upon as a kind of stationary phase that retains the analytes analogous toa true stationary phase in SFC. In this section a model will be derived that allows the calculation of extraction yields in SFE from retention data and peak shapes measured in the SFC mode using the extraction cell as the chromatographic column.
For components adsorbed on the surface of the matrix, equilibration can be reached rapidly. The distribution of the analytes between the matrix and the supercritical fluid after equilibration can be expressed by the distribution coefficient. The distribution coefficient, K, is defined as the ratio of concentrations in the two phases:
K=Cma=mmaVsr ~mma Csf msr V ma msr
(2.2)
where c and m are the concentration and weight of the components in the two phases, respectively. V is the volume of the matrix and the supercritical fluid, and B is the phase ratio. The subscripts "ma" and "sf" refer to "matrix" and "supercritical fluid" , respectively.
For a given compound under given experimental conditions, K is the thermodynamic distribution coefficient which can be measured by SFC. If now the SFE and SFC experiments are carried out on the same packing under identical experimental conditions, the distribution coefficient obtained in SFC is also valid for SFE. Hence,
(2.3)
Combining eqs. 2.2 and 2.3 yields
ffisf = ~sfe ffima ~sfcks'fc
(2.4)

Where Ill,;r and 11\na are the amounts of the test compound dissolved in the supercritical fluid and adsorbed on the matrix, respectively.
The fraction of solute present in the supercritical fluid, R, is given by:
R ffisf
msr+mma (2.5)
Here R actually represents the extraction yield obtained in a static extraction. Combining eqs. 2.4 and 2.5, yields
R = ~sfe ~sfe + ~sfcks'fc
(2.6)
We can now define a parameter a which represents the ratio of the phase ratios under SPE and SPC conditions:
(2.7)
In the present work all SPE and SFC experiments were carried out using the cartridge packed with the sample both as the extraction cell and as the SPC column. Hence, the a value was one throughout this work. This means that eqn. 2.6 can be simplified to:
R= 1 1 +ks'fc
(2.8)
where k'src is the capacity factor ofthe analyte as determined by SFC.
In order to calculate the extraction yields, the SPE elution profile was studied. Por systems in which the extraction rate is determined by transfer of the solute from the partiele surface into the fluid, it can be assumed that in a short period of static extraction, equilibrium between the supercrideal fluid and the matrix is established. Moreover, for these systems there is no concentration gradient along the cell. In the hypothetical case that we have an extraction cell with an infinite plate number, the time required to elute the components is identical to the time required to elute the sample molecules that were originally present at the very top of the extraction cell. This time equals the retention time in SFC. Under the conditions specified above, the analytes elute at a constant rate and extraction is complete at the analyte retention time. A plot of the extraction rate vs. time curve

Correlation between SFE and SFC 44
and of the corresponding SFC chromatagram for this hypothetical system are given in Fig. 2.5A -C.
peak peak
A D
elution profile elutlon profile
B E
I --100%~
extraction yield __.------iOO%
F
I
Figure 2.5. Relationship between peak shape, calculated SFE elution profile and extraction yield vs time curve. A - C: hypothetical extraction cell with an infinite plate number; D - F: extraction cell with a finite plate number.
In SFE systems with a finite plate number, the time required to quantitatively extract the component is increased due to band-spreading. As the degree of bandspreading in SFE of the components originally present at the very top of the extraction cell is identical to that in SFC, the time required to obtain quantitative extraction in SFE equals the time required to obtain complete elution in SFC. Due to the band-spreading, a gradual decrease in extraction rate occurs towards the end of the extraction resulting in a smooth transition in the extraction yield vs. time curve.
In mathematica! terms, the extraction yield vs. time curves as given in Fig. 2.5C, can be obtained by integration of the extraction rate vs. time curves given in Fig. 2.5B. The shape of the extraction rate vs. time curve for an SFE cell with an infinite plate number is easy to predict (Fig. 2.5B). The prediction of extraction rate vs. time curves for cells with a finite plate number requires the use of a mathematica! technique called convolution. Basically, convolution of two functions equals multiplication of these functions. Convolution of the extraction rate curve obtained for a system with an infinite plate number with a transfer function that represents the response of a more realistic system with a fmite plate number to a very narrow input signal gives the extraction rate vs. time curve for an SFE cell with a fmite plate number. The response of the system to a narrow input signal is simply the chromatographic peak.

Chapter 2 45
In static SFE after equilibrium between the supercritical fluid and the matrix has been established, the analytes elute at a constant rate from the beginning of the dynamic extraction. Using convolution techniques, the elution profile in SFE can be expressed as:
M 1
J6c(t)dt) r(t) = to(l + k') ( - J~ c(t)dt)
(2.9)
where r(t) is the extraction rate at timet, Mis the total amount of the analyte, 1:o is the time needed to elute the void volume of the cell, and c(t) is the SFC elution profile.
The extraction yield, Y, can be calculated from:
y = J6r(t)dt M
(2.10)
If the elution profile in SFC, c(t), can be represented by a Gaussian peak, eqn. 2.9 can be rewritten as:
r(t) M 1 ft 1 -<t-tr)2 dt to(l + k') ( - o &cr e 2cr2 )
(2.11)
where t. is the retention time of the component, and cr the standard deviation. Combining eqns. 2.10 and 2.11 yields:
t t 1 (t-t,)2 J0(1- J0 ~ e· 2cr2 dt)dt y = -v21tcr
toCI + k') (2.12)
The extraction rate and extraction yield vs. time curves calculated from an SFC peak for an SFE cell with a :finite plate number are shown in Fig 2.50-F.
Up till now, it bas been assumed that the capacity factors of the components are independent of the solute concentration and that the sample matrix to be extracted is well defined and homogenrous (e.g. the extraction of non-polar components from an SPE adsorbent). For inhomogeneous samples where the sample matrix contains active sites for the solutes, however, various interaction mechanisms between the analyte and the matrix might contribute independently to the overall retention of the analytes, e.g. interaction with organic material and with active

Correlation between SFE and SFC 46
sites present in the matrix. In this case, a mixed retention mechanism [17] must be used, where
kt•ot(c) = kp•art + ka•ds(c) (2.13)
Here k'.J:c) is the overall capacity factor, k'part and k'ads(c) are the capacity factors due to partitioning and adsorption, respectively. The addition of (c) indicates the dependenee of the parameter on the concentration of the analytes. Por SPC systems in which such a situation occurs, lower capacity factors are often observed at higher concentration levels. In the case that the capacity factor is concentration dependent, eqns. 2.9-2.12 are no longer valid. It is one of the aims of the present work to investigate whether the analyte concentration affects the extraction characteristics of SPE.
2.2.3 EXPERIMENTAL
The experimental work was carried out on a modified Carlo Erba SPC 3000 capillary SPC instrument (Carlo Erba, Milan, Italy). A stainless steel HPLC column (0.5 cm i.d. x 10 cm length) obtained from Knauer (Berlin, Germany) was used as the extraction cell. Pused-silica capillaries (20 J.1Ill i.d., with different lengths) were used as restrictors. To enable static extractions an on-off valve (Valco, Switzerland) was installed directly bebind the extraction cell. The supercritical fluid flow from the extraction cell was split into two streams in a low dead-volume T-piece (Gerstel GmbH, Mülheim a/d Ruhr, Germany). One stream (approximately 2%) was fed to the flame ionization detector (PID) for detection, while the other was directed towards the sample coneetion device. A schematic representation of the instrument is given in Pigure 2.6.
Figure 2.6. Schematic diagram of the experimental set-up.
I pump
2 injection val ve
3 extraction cell
4 SFCoven
5 on/offvalve
6 T-piece
7 restrictor
8FID
9 glass via!

Chapter 2 47
All experiments were performed at 50°C unless noted elsewhere. The analytes were extracted at 200 bar (from sand) or 300 bar (from ODS) aftera 5 minute static period at the same pressure. The carbon dioxide used in the experiments had a purity of 99.996% (lntermar B.V. Breda, The Netherlands). An octadecylsilane solid phase material (ODS, 40 J..1II1 partiele size, J.T. Baker Inc., Philipsburg, NJ, USA) and sand were selected as model matrices. The test solutes were octadecane, eicosane, cyclohexanone and hexadecanol (All obtained from Fluka AG., Buchs SG, Switzerland). The extracted material was collected by inserting the resttictor outlet into a glass vial (10 cm x 1 cm i.d.) containing an internat standard and 5 m1 organic solvent. Hexane was selected as the coneetion solvent for octadecane and eicosane while dichloromethane was used for the coneetion of hexadecanol.
After collection, the solution was concentrated under a gentle flow of nitrogen and analysed using a gas chromatograph equipped with an on-column injector and an FID (GC 8000 series, Carlo Erba Instruments). The gas chromatographic separation was achieved on a crosslinked PEG-20 M capillary column (25 m x 0.32 mm i.d., film thickness 1.2 J..Ull) purchased from Chrompack (Middelburg, The Netherlands ).
2.2.4 RESUL TS AND DISCUSSION
Although very different at first sight, SPE and packed column SFC are very much similar in many respects. In both techniques, a supercritical fluid is used for dissolving the components and transporting the analytes through a packed bed of solid particles, i.e. the stationary phase in SFC or the matrix in SFE. If an analyte exhibits a strong interaction with the matrix, this will result in a high capacity factor in SFC and a poor extractability in SFE. Similarly, a packed cell exhibiting a large band spreading in SFC is unlikely to yield sharp extraction curves in SFE. From this it is plausible to assume that retention and peak shapes observed in SFC on the one hand and the extraction profile in SFE on the other hand are correlated.
In the theory section, equations have been derived that allow the calculation of SFE extraction rates and yields from chromatographic retention data and peak shapes (eqs. 2.8, 2.9 and 2.11). From these equations, the following conclusions can be drawn: (a) the fraction of solute present in the supercritical fluid will decease with increasing k' src;

Correlation between SFE and SFC 48
(b) If static extraction is foliowed by solventless co Heetion of the analytes in a rapid depressurization step without subsequent dynamic extraction [19], quantitative recovery can only be obtained if k' is zero; (c) any parameter affecting k' in SFC will affect the SFE extraction kinetics. (d) the extraction rate will decrease with increasing k' and/or tQ. These conclusions are in agreement with observations that better recoveries for SFE can be achieved by working under conditions that lead to reduced capacity factors in SFC, such as increased supercritical fluid density or the addition of modifiers [10].
The extraction rate vs. time curve calculated from eqn. 2.9 shows that after an initial period in which elution occurs at a constant rate, the extraction rate starts to decrease and finally reaches zero at the time when extraction is complete. The width of the constant elution rate range is determined by both the solute retention time and the column efficiency of the extraction celL That is, the time needed for complete extraction is controlled by both the solute retention time and the extraction cell efficiency. For a Gaussian peak, the SFE extraction yield at the point of the retention time of the solute in SFC can be calculated by eqn. 2.12. The results can be approximated by:
1 y ",; 1- 2.fii
where N is the plate number of the extraction cell.
(2.14)
From this equation, it can be seen that the column efficiency of the extraction cell has a considerable effect on the SFE extraction behaviour. lf the plate number of the extraction cell would be infinite, the extraction would be complete at the solute relention time. For a more realistic extraction cell with a finite plate number, the extraction yield at the solute retention time is less than 100%.
In order to correlate the SFE extraction kinetics with SFC retention data and peak shapes, SFC experiments were carried out immediately after the SFE experiments using the SFE cell as the SFC column. In tigure 2. 7 overlays are shown of the SPE elution profiles and the SFC chromatograms of octadecane at 50°C and different pressures. The elution profiles calculated directly from the chromatographic peak shapes using the convolution technique are also shown in this figure. From these figures, a number of interesting conclusions can be drawn. First, it can be seen that a very good agreement between the experimental extraction profile and the calculated profile is obtained. As predicted, the analytes elute at a constant rate from the start of the dynarnic extraction until close to the
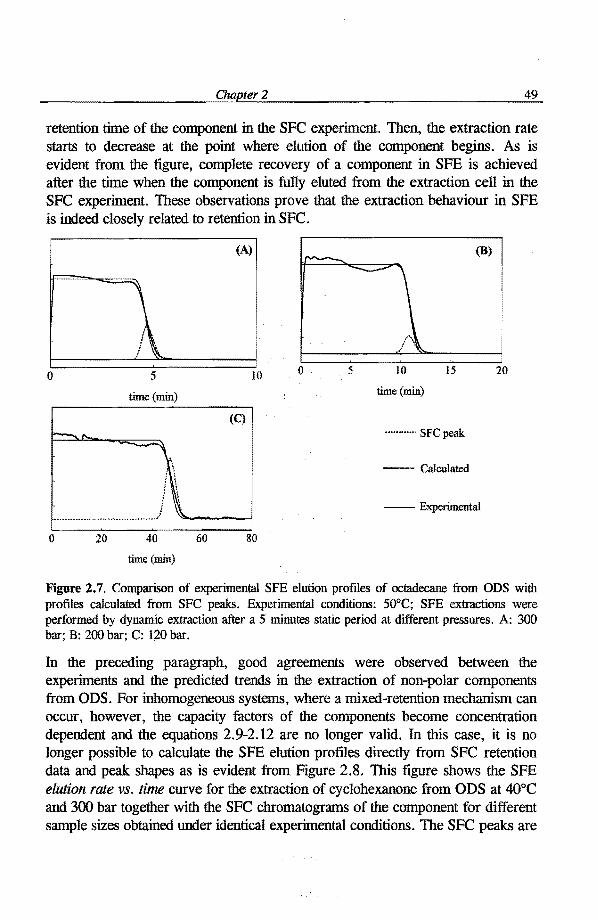
Chapter 2 49
retention time of the component in the SFC experiment. Then, the extraction rate starts to decrease at the point where elution of the component begins. As is evident from the figure, complete recovery of a component in SFE is achieved after the time when the component is fully eluted from the extraction cell in the SFC experiment. These observations prove that the extraction behaviour in SFE is indeed closely related to retention in SFC.
(A) (B)
0 5 10 0 5 10 15 20
time(min) time(min)
(C) ·······•···· SFCpeak
-- Calculated
Experimental
0 20 40 60 80
time (min)
ligure 2. 7. Comparison of experimental SFE elution profiles of octadecane from ODS with profiles calculated from SFC peaks. Experimental conditions: 50"C; SFE extracrions were performed by dynamic extraction after a 5 minutes static period at different pressures. A: 300 bar; B: 200 bar; C: lZO bar.
In the preceding paragraph, good agreements were observed between the experiments and the predicted trends in the extraction of non-polar components from ODS. For inhomogeneous systems, where a mixed-retention mechanism can occur, however, the capacity factors of the components become concentration dependentand the equations 2.9-2.12 are no longer valid. In this case, it is no longer possible to calculate the SFE elution profiles directly from SFC retention data and peak shapes as is evident from Figure 2.8. This tigure shows the SFE elution rate vs. time curve for the extraction of cyclohexanone from ODS at 40°C and 300 bar together with the SFC chromatograms of the component for different sample sizes obtained under identical experimental conditions. The SFC peaks are

Co"elation between SFE and SFC 50
broad and tailing due to the interaction of the solute with active sites present on the ODS material [20]. The SPE elution profile shows that the solute elutes at a constant rate from the start of the dynamic extraction until a time around to ( the time needed to elute the void volume of the extraction cell). Prom a comparison of the SPE elution profile with the SPC chromatogram, it can be seen that extraction is already complete before the time required to fully elute cyclohexanone in the SPC experiment.
SFE elution profile
0 5 10 15 20 25
time (min)
Figure 2.8. SFE elution profile of cyclohexanone from sand and SFC chromatograms at different sample sizes. Experimental conditions: 300 bar and 40°C; SFE was performed by dynamic extraction after a 5 minutes static period.
In order to investigate whether the SPE extraction yields are affected by the concentrations of the solutes in the sample, two model systems were studied in detail. The first system involved the extraction of non-polar solutes from ODS. The other system combined a polar solute, hexadecanol with sand as the matrix. The components were extracted at a gaseous carbon dioxide flow rate of approximately 60 ml/min for 3 minutes aftera 5 minute static period. The results are listed in Table 2.4 and Table 2.5, respectively. As can be seen from Table 2.4, no effect of the solute concentration on the SPE behaviour is observed for the extraction of dodecane or eicosane from ODS. Analogously, the capacity factors of dodecane and eicosane on ODS in SPC are independent of the concentration of the components. Another interesting condusion that can be drawn from the results presented in Table 2.4 is that the experimental extraction yields are in good agreement with the calculated data calculated according to eqn. 2.12.
Por the extraction of hexadecanol from sand, a different situation occurs. Here, the solute retention in the SPC mode is a function of solute concentration. Hence, the equations derived in the theory section can not be applied for this

Chapter 2 51
solute/matrix combination. The extraction yields at different concentration levels are listed in Table 2.5. From this table, it can beseen that higher extraction yields were obtained at higher hexadecanol concentrations. This observation is in agreement with expectations because the capacity factors of hexadecanol on sand were found to decrease at increasing concentration levels due to the non-linearity of the adsorption isotherm. The dependenee of extraction yield on analyte concentration in SFE further complicates SFE method development. Experimental conditions that give good recoveries for samples containing high solute concentrations might yield poor extraction recoveries for low-concentration samples.
Table 2.4.Comparison of experimental extraction yields and calculated data for octadecane and eicosane on ODS at different concentrations. (Extraction conditions: 5 minutes static extraction foliowed by 3 minutes dynamic extraction at 50°C and 300 bar.)
c1s•> C20bl
spiked experimental calculated experimental calculated yield amount {}.tg) yield(%) yield (%) yield (%) (%)
200 20.2 19.8 16.6 16.5
100 19.7 21.0 16.4 17.5
50 20.8 21.0 16.9 17.0
20 19.9 20.9 15.7 16.7
10 19.1 21.0 15.9 15.8
al with eicosane as the internal standard;bl with octadecane as the internal standard.
Table 2.5. Comparison of extraction yields of hexadecanol from sand at different concentrations. (Extraction conditions: 5 minutes static extraction foliowed by a 3 minutes dynamic extraction at 50°C and 200 bar.)
spiked amount (J.lg)
100
50
20
10
extraction yield (% )">
49.2
45.8
40.2
37.0
a> with eicosane as the internal standard.

Correlation between SFE and SFC 52
It is interesting to compare the SFE results obtained in the extraction of hexadecanol from sand with those from ODS. Hexadecanol can easily be eluted from sand even at fairly mild conditions (Figure 2.9A). On the contrary, it is strongly retained on ODS. On this material no peak was observed under the same experimental conditions (Figure 2.9B). In SFE after a 3 minutes dynamic extraction at 50"C and 200 bar, nearly 50% of hexadecanol was extracted from sand, while only a negligible percentage (1.5%) was extracted from ODS under identical experimental conditions (Table 2.6). This difference in extraction yields proves that variations in the composition of the sample matrix can have a large impact on the extraction rate in SFE. A practical consequence of this is that the extraction conditions required are strongly matrix dependent This again supports the presumption that the distribution coefficient is one of the most important parameters in SFE. Moreover, it emphasizes that SFC retention data can be useful in predicting SFE reeoverles as well as in optimizing SFE extraction conditions.
(A) (B)
0 10 20 30 time (min) 0 20 40 60 80 time (min) Figure 2.9. SFC chromatograrns of hexadecanol on sand and on ODS. Chromatographic conditions: 200 bar and 50"C. 5A: on sand; 5B: on ODS.
Table 2.6. Comparison of extraction yields of hexadecanol from sand and ODS. •> (Extraction conditions: 5 minutes static extraction foliowed by 3 minutes dynamic extraction at 50"C and 200 bar.) ·
sand ODS
49.2% 1.5%
with eicosane as the intemal standard.
For homogeneaus systems SFB conditions can be predicted directly from SFC experiments. For inhomogeneous systems that exhibit strong matrix effects or for systems where the extraction rate is detennined by diffusion in the sample, a direct transfer of the SFC retendon data to SFE extraction yields is not possible.

Chapter 2 53
However, SPC retention data can still be useful. If a solute requires extreme elution conditions in SPC, extraction in SPE will certainly be difficult.
2.2.5 CONCLUSIONS
Supercritical-tluid chromatography offers an attractive method to examine the effects of experimental parameters on the extraction behaviour in supercritical tluid extraction. A model is derived that enables the prediction of SPE extraction yields in both static and dynamic extraction from SPC retention data measured using the matrix that is to be extracted as the stationary phase. Por samples in which the extraction kinetics are controlled by transfer of the cornponents from the surface of the matrix into the extraction tluid and by subsequent elution out of the extraction cell, experimental extraction yields and predicted data are in good agreement. Extraction of components from solid sorbents is controlled by many of the same factors that control retention in SPC. Variations in sample matrix composition were found to require a substantial adjustment of the extraction parameters. Por inhomogeneous samples, solute concentration can have a considerable effect on SPE kinetics.
REFERENCES
1 T.L. Chester, J.D. Pinkston and D.E. Raynie, Anal. Otem., 64 (1992) 153. 2 M.L. Lee and K.E. Marleides (eds), Analytical Supercritical Otromatography and Extraction,
Chrornatography Conference, Provo, UT, 1990. 3 D.R. Gere, C.R. Knipe, P. Castelli, J. Hedrich, L.G. Randall, H. Schulenberg-Schell, R.
Schuster, L. Doherty, J. Orolin and H.B. Lee, J. Otromtogr. Sci., 31 (1993) 246. 4 X. Lou, H.-G. lanssen and C.A. Crarners, J. High Resol. Otromatogr., 16 (1993) 425. 5 R.J. Houben, H.-G.M. Janssen, P.A. Leclercq, J.A. Rijks and C.A. Crarners, J. High Resol.
Otromatogr., 13 (1990) 669. 6 K.D. Bartle, A.A. Clifford and G.F. Shilstone, J. Supercrit. Fluids (1989) 30. 7 J.W. King, J. Otromatogr. Sci., 27 (1989) 355. 8 M.E.P. McNally and J.R. Wheeler, J. Otromatogr., 447 (1988) 53. 9 J.R. Wheeler and M.E. McNally, J. Otromatogr. Sci., 27 (1989) 534. 10 K.G. Purton and Q. Lin, J. Otromatogr. Sci., 31 (1993) 201. 11 G.F. Shilstone, M.W. Raynor, K.D. Bartle, A.A. Clifford, I.L. David and S.A. Jafar,
Polycycl. Aromat. Compd., 1 (1990) 99. 12 J.C. Giddings, M.N. Myers, L. McLaren and R. A. Keiler, Science, 162 (1968) 67. 13 J.W. King and J.P. Friedrich, J. Otromatogr., 517 (1990) 449. 14 E. Stahl, W. Schilz, E. Schutzand E. Willing, Angew. Otem. Int. Ed. Engl., 17 (1978) 731. 15 J. Rein, C.M. Cork and K.G. Furton, J. Otromatogr., 545 (1991) 149. 16 J. Pawliszyn, J. Otromatogr. Sci., 31 (1993) 31. 17 P.J. Schoenmakers, L.G.M. Uunk and P.K. de Bokx, J. Otromatogr., 459 (1988) 201. 18 J.G.M. Janssen, P.J. Schoenrnakers and C.A. Crarners, J. High Resol. Otromatogr., 12 (1989)
645. 19 D.J. Miller, S.B. Hawthome and M.E.P. McNally, Anal. Otem., 38 (1993) 1038. 20 H.-G. Janssen, P.J. Schoenmakers and C.A. Crarners, J. Otromatogr., 552 (1991) 527.

Chapter 3 54
3 Temperature and Pressure Effects on Solubility in Supercritical Carbon Dioxide and Ketention in
Supercritical Fluid Chromatographf>
ABSTRACT
Solubilities of some polycyclic aromatic hydrocarbons (PAHs) in supercritical carbon dioxide were measured with a procedure based on a direct on-line combination of a saturation cell to a flame ionization detector (PID). Acenaphthene, anthrance and chrysene were selected as the test solutes. A method was developed and evaluated which enables the measurement of the contribution of solute vapor pressure to the overall solubility. The effects of temperature and pressure on solubility in supercritical carbon dioxide were investigated and discussed in detail. The trends of solubility changes in supercritical carbon dioxide and the variations in observed retention in SPC were correlated. Equations were derived to estirnate the effects of temperature on the solute's affinity for the stationary phase in SPC.
3.1 INTRODUCTION
In method development for supercritical fluid chromatography (SPC) and supercritical fluid extraction (SPE), one of the most important parameters to be considered is the solubility of the target compounds in the supercritical fluid. The solubility of a component in a supercritical fluid is generally assurned to be controlled by two parameters: the vapor pressure of the component and its interaction with the supercritical fluid. Many research groups have investigated the effects of various parameters on overall
solubilities in supercritical fluids [1-8].
The solubility data reported in literature are the sum of the contributions of the vapor pressure of the component and its interaction with the supercritical fluid. It is evident that these data are very important for method development in both SPC and SPE. Ho wever, to obtain a detailed insight into the effects of
z) X. Lou, H.-G. Janssen and C.A. Cramers, J Chromatogr., in press.

Temperafure and pressure effects on solubility andretention 55
temperature and pressure on solubility, it is also very important to distinguish these two different contributions to the overall solubility. Unfortunately, however, no reports on the differentiation between these two contributions have been publisbed so far. The explanations of the effects of experimental parameters on solubilities in supercritical fluids and retention in SFC are very often found to be incomplete at best. Por example, in SFC at a constant pressure, curves representing the effect of temperature on retention can generally be divided into two different regions. The ascending-descending shape of the curves are normally explained by two competing effects. Under conditions where the solute vapor pressure is not a dominant consideration, increasing temperature will lead to an increase in retention due to a reduced supercritical fluid density. If the solute has significant vapor pressure then increasing temperature will result in lower retention factors [9]. However, as will be shown later in this report, this widely accepted explanation of the influence of temperature on retention is over simplified and in some cases even incorrect. Por a more appropriate explanation, the contributions of i) the vapor pressure and ii) molecular interactions with the supercritical fluid to the overall solubility at different experimental conditions should be distinguished and assessed. In actdition to this, for a thorough understanding of retention in SPC, the effects of temperature on the solute's affinity for the stationary phase
should also be considered. Again, no reports on the influence of temperature on the affinity of a solute for a stationary phase have been publisbed so far.
In this article, overall solubilities of some polycyclic aromatic hydrocarbons (PARs) in supercritical carbon dioxide were determined over a wide range of temperatures and pressures. A method to measure the contribution of vapor pressure to the overall solubility was proposed and evaluated. The effects of temperature and pressure on solubilities in supercritical carbon dioxide were investigated and discussed in detail. Purthermore, the effects of experimental conditions on retention in SFC were studied and discussed. Finally, equations were derived to estimate the effects of temperature on affinity of the solute for the stationary phase in SFC.

Chapter 3 56
3.2 EXPERIMENTAL AU experiments were carried out on a Carlo Erba SFC 3000 instrument (Carlo Erba, Milan, Italy) equipped with a flame ionization detector (FID). A 3-mL stainless steel SFE extraction cell (Suprex Pittsburg, PA, USA) with handtight connectors (Suprex) was used as the saturation cell. Prior to be packed into the saturation cell, the solute to be investigated was actmixed with clean
sand at approximately 5 wt%. Stainless steel frits (3 J.tm) were located at either end of the saturation cell. Pressurized carbon dioxide was introduced into the saturation cell through a I-meter preheating coil made of stainless steel tubing (1/16 in. x 0.03 in.). A piece of fused silica capillary (50 cm x 200 J.lm i.d.) with one end carefully tapered was used as the restrictor to maintain supercritical conditions inside the saturation cell. The carbon dioxide flow rate was measured as gaseous flow exiting from the FID (with the FID gases off and FID temperature at 420°C). In order to prevent entrainment of the solutes by the supercritical fluid flow and to avoid incomplete vaporization in the FID, as well as to provide enough time for saturation, the carbon dioxide flow rate was adjusted to relatively low values (approximately 12 mL/min gaseous flow at 350 bar). For the determination of the contribution of vapor pressure to the overall solubility, similar experiments were repeated by using helium as the carrier fluid instead of supercritical carbon dioxide.
Calibration of the FID was performed by weighing approximately 5 mg of a test compound into the saturation cell foliowed by elution of the solute at a desired temperature and pressure. The FID response was recorded until the signal returned to baseline. Solubilities were calculated by:
(3.1)
where Ax is the area of the test compound, W0 the amount of test solute weighed into the saturation cell (g), Vco
2 the molar gas volume of carbon
dioxide (22.4 Llmol}, Ao the area of the calibration compound measured at an amount of W 0 , F the supercritical fluid flow rate (Limin), tx the width of the plateau-peak of the FID response (min) and Mx the molecular weight of the test solute (g/mol).

Tempera/ure and pressure effects on solubility and relention 57
The SFC experiments were perfonned on the same Carlo Erba SFC 3000 system. The column used for the SFC experiments was a Zorbax ODS reversed phase HPLC column (25 cm x 4.6 mm, 5 !Jlll particles) purchased from Rockland Technologies, Inc. (Chadds Ford, PA, USA). Prior to use, the column was deactivated with N ,0-bis(trimethylsilyl)-trifluoroacetamide) (BSTF A) as described previously [1 0]. The column effluent was split into two streams. One stream was fed to the FID via a linear fused silica restrietion capillary (45 cm x 10 !Jlll i.d.). The other was used to control the flow rate through the column. For the detennination of retention factors, methane was used as the dead time (tJ marker.
The PAHs (acenaphthene, anthracene and chrysene) were all purchased from Aldrich (Milwaukee, WI, USA) with the highest purity available. Carbon dioxide used in the experiments had a purity of 99.996% (lntennar B.V., Breda, The Netherlands).
3.3 RESULTS AND DISCUSSION
3.3.1 Solubility determination with on-line FID method Supercritical carbon dioxide has the capability to dissolve numerous compounds ranging in polarity from non-polar to moderately po lar. The aims of this artiele are threefold: i) measure the contribution of vapor pressure to the overall solubility; ii) investigate temperature and pressure effects on SFC retention and iii) correlate solubilities in supercritical fluids with relention in SFC. In the present investigation, three PAHs (acenaphthene, anthracene and chrysene) were selected as the test solutes. The method used for measuring solubility was an on-line FID method simHar to that described by Milier and Hawthorne [5]. Examples of FID solubility measurements are shown in Figures 3.1 and 3.2. Figure 3.1 shows the FID response versus pressure for anthracene at 40°C. Similar curves were also recorded at other temperatures and for the other PAHs. As expected, the solubilities of P AHs in carbon dioxide at constant temperature increase dramatically with increasing pressure.
Compared to the simple effects of pressure (at constant temperature), the effects of temperature on solubility in supercritical C02 at constant pressure are far more complicated. Somewhat controversial results on the influence of

Chapter 3 58
temperature on solubility were published in literature. Zhao et al [11] fmmd
that in the near-critical region the solubilities of some P AHs in supercritical C02 decrease significantly when increasing temperature at a constant pressure. In contrast to this, Milier and Hawthome [5] reported a continuous increase in
.AïT=4x: 350 bar
210
time (min)
Figure 3.1. FID response versus pressure for anthracene at 40°C.
0.70r------------, ATI=lx (A)
0.70 tso•c ATI JO x (B) 0.56. 0.56
M 150"C i 0.42
,wç~-~ 0.28
0.14 o•c so•c 6ooc
i 0.42
8 0.28 lz..
6QOC 700C
60 120 180 0.000 80 160
time(min) time(min)
0.70r---------------, ATT=40x (C)
0.56
i 0.42
150"C
El 0.28 lz..
0.14 4a•c so•c 600C 70"C
0'00o~---:::so:-----:-,60":-:---__j240
time(min)
Figure 3.2. FID response versus temperature for anthracene at different pressures. (A) 100 bar; (B) 200 bar; (C) 300 bar.
240

Terneerafure and eressure effects on solubilif:J!_ and relention 59
Table 3.1 Solubility of acenaphthene, anthracene and chrysene (mol/mol x 105) in carbon
dioxide at different temperatures and pressures measured by the on-line FID method.
Acenaphthene
Teq
P(bar) 40 50 60 70 80 100
75 2.94 2.79 3.81 5.66 9.18 17.0
80 5.61 4.12 5.07 7.75 11.0 19.0
85 12.7 7.37 6.91 11.4 14.6 23.3
90 Nt• 11.1 9.42 13.4 16.4 24.8
100 NT 33.4 23.4 NT NT 41.3 150 NT NT NT NT NT 237.0 Heb 0.0038 0.013 0.031 0.07 0.14 0.52
Chrysene
Teq P(bar) 40°C 50 60 70 100 150
80 0.242 0.168 NT NT NT NT 100 1.54 0.45 0.253 NT 0.733 7.16
150 4.98 4.62 NT NT 3.97 NT 200 5.92 7.43 9.19 10.6 13.4 40.3
300 8.23 11.6 15.9 21.2 43.2 126.0
350 8.53 13.3 NT NT 57.6 NT He NDC ND ND ND 0.032 0.977
Chrysene
Teq
P(bar) 40 50 70 100 120 150
80 0.0397 0.0204 NT 0.0422 NT NT 100 0.0452 0.0265 NT 0.077 NT NT 150 0.396 0.347 NT 0.367 NT NT
200 0.483 0.619 0.826 1.08 1.54 2.85 300 0.755 1.09 2.14 4.72 7.57 14.8
350 0.800 1.19 NT 6.13 NT NT He ND ND ND ND ND 0.0032 •: Not tested. b: Contribution of vapor pressure tested by using Helium as the carrier fluid. c:
Not detectable.
the solubility of some organic compounds in C02 when increasing temperature at a constant pressure, despite of the decrease in co2 density. In our
experiments, at pressures above 200 bar, the solubility of anthracene increased
continuously with increasing temperature (Figure 3.2B and C), while at the

Chapter 3 60
pressure of 100 bar the solubility of anthracene fiTst decreased and then increased with temperature (Pigure 3.2A). Prom these results, it can be concluded that the effects of temperature on solubility are quite complicated. Moreover, they can depend on the pressure and temperature range in which the experiments are performed. A somewhat qualitative explanation for this
observation will be discussed later in this contribution.
Table 3.1 lists solubility data of the PAHs measured using the on-line FID
metbod over a wide range of temperature and pressure conditions (40- 150°C and 80- 350 bar). Higher temperatures and pressures were not tested because of the limitations of the polymerie seals located at the ends of the saturation cell and the maximum allowable pressure of the pump. Por acenaphthene at temperatures below 1 oooc, an unstable PID signal was obtained at pressures above 100 bar. The reason for this is still unclear. The data listed in Table 3.1 are based on triplicate determinations. The relative standard deviations of all determinations were within 10%, and mostly within 5%.
3.3.2 Fundamental study oftemperature and pressure effects on solubility The solubility data obtained in the previous section can be modeled as the sum
of the contributions of the vapor pressure of the component and of its interaction with the supercritical fluid. Although this might be an oversimplification of the actual physicochemical back-grounds of the dissalution process, this way of looking at solubility in supercritical fluids is now widely accepted. Por a more thorough investigation of temperature and pressure effects on solubility, the two different contributions to the overall solubility should be differentiated. The contribution of vapor pressure at different experimental conditions was measured by using helium as the carrier fluid instead of supercritical carbon dioxide. Helium, even at high pressures, is an ideal gas that exhibits no interaction with the solute molecules. Hence, transport of the solute out of the saturation cell is solely due to its vapor pressure. The data of these experiments are presented in Table 3 .1. Evidently, the contribution of vapor pressure strongly depends on temperature and the properties of the solute. Por volatile compounds at high temperatures the contribution of vapor pressure to the overall solubility can be significant. Por the semi-volatile P AHs investigated, ho wever, the contribution of vapor

Temperature and pressure ~tfects on solubility andretention 61
pressure is very much limited (see Table 3.1). For example, for anthracene the vapor pressure contribution at the highest temperature tested (150°C) at 100 bar and 300 bar was only 13% and 0.8%, respectively. For the less volatile chrysene, only about 0.02% of the solubility was caused by vo1atility at 300 bar and 150°C. At lower temperatures, the contribution of vapor pressure to the overall solubility will be even less. No measurable contri bution of volatility was found for anthracene and chrysene at temperatures below 70°C and 1 oooc, respectively. From this it can be concluded that the contribution of vapor pressure to the solubilities of the P AHs is very much limited, even at relatively high temperatures. In literature the contribution of vapor pressure to the overall solubility is often overestimated, especially for semi-volatile or non-volatile compounds. Increased solubilities at elevated temperatures are mainly due to the variations of physicochemical properties of the solutes and the supercritical fluid. This phenomenon will be addressed in more detail below.
Table 3.2. Solubility parameters {MPa112) of acenaphthene, anthracene, chrysene and carbon dioxide.
Acenaphthenea anthracenea chrysenea carbon dioxideb
22.4 22.7 23.6 16.3
a: estimated according to Fedors [13]. b: at 40°C and 350 bar, calculated according to Giddings et al.[14].
By using the solubility parameter theory [12], the effects of temperature and density (or pressure) on solubility in supercritical fluids can be explained qualitatively. The basis of this theory is that a higher solubility is achieved when the solubility parameters of the solute and solvent are closer to each other. The solubility parameter of a given solute is mainly controlled by its
properties and temperature. It generally decreases monotonically with temperature [13]. For a supercritical fluid the solubility parameter can be calculated from [14]
(3.2)

Ch ter 3 62
where ÖsF is the solubility parameter of the supercritical fluid, Pc the tluid
critical pressure, Pr,sF the reduced density of the fluid and Pr,L the reduced density of the extraction fluid in the quasi-liquid state. Table 3.2 lists the
solubility parameters of the PAHs and carbon dioxide at 80°C and 340 bar.
When increasing pressure at a constant temperature, the solubility parameters of the P AHs remain virtually constant while that of the fluid increases and approaches those of the solutes. Therefore, at a constant temperature, the
solubilities of the P AHs increase with pressure. Increasing temperature at a constant density results in a decrease in the solubility parameters of the solutes while that of the tluid remains virtually constant. As a result the solubility will increase. In contrast to the continuous increase of solubility at a constant density, a more complicated situation arises when increasing temperature at a constant pressure. In this case, the solubility parameters of both the solutes and the supercritical fluid will decrease. The actual effects of temperature now depend on the temperature and pressure conditions as well as on the properties of the solutes and the tluid. The solubility parameter theory can, unfortunately, only provide a qualitative explanation for the observed effects of temperature and pressure on solubilities in supercritical fluids. For more quantitative explanations or predictions, more detailed physicochemical parameters that are generally not available, would be required.
3.3.3 Effects of temperature on solute affinity for the stationary phase in SFC As described in the Introduetion part, the explanations of the influence of experimental parameters on SFC retention is very often found to be · oversimplified. In this section the effects of temperature and pressure on SFC retention are investigated in more detail. Table 3.3 lists retention factors of the PAHs at different temperature and pressure conditions. From table 3.3 it is clear that the effects of temperature on retention can be quite different at different pressures. At pressures below 200 bar, the relention factors of all the PAHs tested increase rapidly and continuously when raising temperature from
40°C to 1 oooc. However, at pressures above 300 bar the retention factors first decrease and then increase with temperature. It is interesting to correlate the trends of solubility changes in supercritical carbon dioxide with the variations in observed SFC retention. As bas been demonstraled in the previous sections,
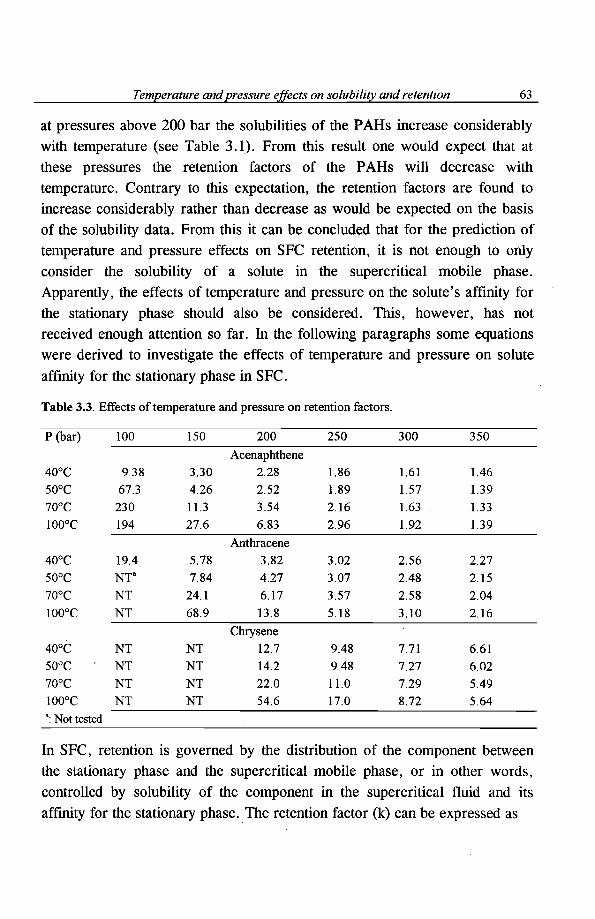
Temperafure and pressure effects on solubility and relention 63
at pressures above 200 bar the solubilities of the P AHs increase considerably with temperature (see Table 3.1). From this result one would expect that at these pressures the retentien factors of the P AHs will decrease with temperature. Contrary to this expectation, the retentien factors are found to increase considerably rather than decrease as would be expected on the basis of the solubility data. From this it can be concluded that for the predietien of temperature and pressure effects on SFC retention, it is not enough to only consicter the solubility of a solute in the supercritical mobile phase. Apparently, the effects of temperature and pressure on the salute's affinity for the stationary phase should also be considered. This, however, has nat received enough attention so far. In the following paragraphs some equations were derived to investigate the effects of temperature and pressure on solute affinity for the stationary phase in SFC.
Table 3.3. Effects oftemperature and pressure on retention factors.
P (bar) 100 150 200 250 300 350
Acenaphthene 40°C 9.38 3.30 2.28 1.86 1.61 1.46 50°C 67.3 4.26 2.52 1.89 1.57 1.39 70°C 230 11.3 3.54 2.16 1.63 1.33 100°C 194 27.6 6.83 2.96 1.92 1.39
Anthracene 40°C 19.4 5.78 3.82 3.02 2.56 2.27 50°C NT' 7.84 4.27 3.07 2.48 2.15 70°C NT 24.1 6.17 3.57 2.58 2.04 100°C NT 68.9 13.8 5.18 3.10 2.16
Chrysene 40°C NT NT 12.7 9.48 7.71 6.61 50°C NT NT 14.2 9.48 7.27 6.02 70°C NT NT 22.0 11.0 7.29 5.49 100°C NT NT 54.6 17.0 8.72 5.64 •: Not tested
In SFC, retention is governed by the distribution of the component between the stationary phase and the supercritical mobile phase, or in other words, controlled by solubility of the component in the supercritical fluid and its affinity for the stationary phase. The retentien factor (k) can be expressed as

K k=p (3.3)
where K is the distribution coefficient between the stationary phase and the
mobile phase and ~ is the phase ratio. It is evident that Kis determined by the
solubility of the component in the mobile phase and its affinity for the
stationary phase, which can be written as
s K=A_!i!_
Smp (3.4)
where A is a constant, Ssp and Smp are the affinity of the component for the
stationary phase and its solubility in the mobile phase, respectively.
Combining eqns. 3.3 and 3.4 yields
(3.5)
In eqn. 3.5, A and ~ are constants, while k and Smp can be measured
experimentally. Thus, the effect of temperature on Ssp can be estimated by
(3.6)
By using eqn. 3.6, the relative solute affinity values for the stationary phase at
different temperatures can be calculated. Table 3.4 lists the affinity values of
the P AHs for the stationary phase at various temperatures relative to those at
40°C. As can be seen from table 3.4, the affinities of the PAHs for the
stationary phase increase considerably with temperature at all pressures tested.
It is interesting to note that no effect of pressure on the relative affinity values
for the stationary phase was observed. This indicates that the interaction
between the solute and the ODS stationary phase is not affected by the
pressure of the mobile phase.
From the discussion above, it is clear that the effect of pressure (at constant
temperature) on SFC retention is relatively straightforward. A higher pressure
will lead to lower retention factors. The effects of temperature (at constant
pressure) are, on the contrary, very complicated. Temperature not only affects

Temperature and pressure effects on solubility and relention 65
the vapor pressure of the solute and the density of the supercritical fluid, but also influences the solubility parameters of both the solute and the supercritical fluid. Moreover, temperature changes can affect the affinity of the compound for the stationary phase. The actual effect of temperature on retention is a result of the various mechanisms identified above and will depend on the experimental conditions, the properties of the solutes and those of the supercritical fluid and the stationary phase.
Table 3.4. Relative affinities of anthracene and chrysene for an ODS stationary phase at
different temperatures•.
150 bar
250 bar 350 bar
200 bar 300 bar 350 bar
50°C
1.31
1.30 1.31
50°C 1.30 1.30 1.31
Anthracene 70°C 2.25
2.25 2.25
Chrysene 70°C 2.32 2.33 2.32
": Values relative to the affinities at 40°C.
3.4 CONCLUSIONS
wo oe 4.62
4.63 4.62
100°C 5.12 5.15 5.11
The interpretations of temperature effects on solubility in carbon dioxide and retention in SFC presented in literature are sometimes incomplete and oversimplified. The influence of temperature on solubility in supercritical fluids is determined by the properties of the solute and the supercritical fluid as well as by the experimental temperature and pressure conditions. Temperature variations will induce changes in the vapor pressure of the solute, the density of the supercritical fluid and the physicochemical properties of both the solute and the supercritical fluid. In literature, the contribution of vapor pressure to the overall solubility is frequently overestimated, especially for semi-volatile or non-volatile compounds. In actdition to the effects on solubility, temperature changes can also affect the affinity of solutes for the stationary phase in SFC.

Chapter 3 66
REFERENCES
1. S. Mitra, J.W. Chen and D.S. Viswanath, J. Chem. Eng. Data, 33 (198&) 35. 2. T.W. Zerda, B. Wiegang and J. Jonas, J. Chem. Eng. Data, 31 (1986) 274. 3. K.D. Bartle, A.A. Cliford and S.A .. :Iafar, J. Chem. Soc. Faraday Trans., 86 (1990) 855. 4. J. Yang and P.R. Griffiths, Anal. Chem., 68 (1996) 2353. 5. M. J. Milier and S.B. Hawthorne, Anal. Chem., 67 (1995) 273. 6. J.C. Giddings, M.N. Myers and J.W. King, J. Chromatogr. Sci., 7 {1969) 276. 7. R.D. Smith, H.R. Udseth and B.W. Wright, Supercritical Fluid Technology, J.M.L.
Penninger, M. Radosz, M.A. Mchugb and V.J. Krokonis (eds), Elsevier Science Publishers, Amsterdam, 1985, p.191
8. M.A. McHugh, Supercrideal Fluid Extraction, Butterworth Publisbers, 1986, p.69. 9. C.F. Poole and S.K. Poole, Chromatography Today, Elsevier Science Publishers, 1994,
p.624. 10. X. Lou, H.-G. Janssen, H. Snijders and C.A. Cramers, J. High Resol. Chromatogr., 19
(1996) 449. 11. S. Zhao, R. Wang and G. Yang, J. Supercritical Fluids, 8 (1995) 15. 12. A.F.M. Barton, CRC handhook of Solubility parameters and other cokesion parameters,
CRC press, lnc. Boca Raton, Fl., 1985. 13. R.F. Fedors, Polymer Eng. and Sci., 14 (1974) 147. 14. J.C. Giddings, M.N. Myers, L. McLaren and R.A. Keller, Science, 162 (1968) 67.

4
ABSTRACT
Chapter 4
Pressure Drop Effects on Selectivity and Resolution in Packed Column Supercritical Fluid Chromatographyl>
67
The influence of pressure drop on retention, selectivity, plate height and resolution was investigated systematically in packed supercritical fluid chromatography (SFC) using pure carbon dioxide as the mobile phase. Numerical methods developed previously which enabled the prediction of pressure gradients, diffusitivities, capacity factors, plate heights and resolutions along the length of the column were used for the model calculations. The effects of inlet pressure and supercritical fluid flow rate on selectivity and resolution are studied. In packed column SFC with pure carbon dioxide as the mobile phase, the pressure drop can have a significant effect on resolution. The flow rate is shown to have larger effect than generally realized. The calculated data are shown to be in good agreement with the experimental results. Finally, the variation of the chrornatographic parameters along a 5.5 meter long model SFC column is illustrated. The possibilities and limitations of using long packed columns in SFC are discussed. It is demonstraled that long columns with large plate numbers do not necessarily yield better separations.
4.1. INTRODUCTION
Chromatography is a metbod of separation which occurs as a result of repeated uptake/release during the movement of the components along the stationary phase bed. Separation is obtained due to differences in the distribution constants of the individual sample components. The actual quality of the separation or the resolution of two peaks is a function of the separation factor (a.-value) and the capacity factors (k-values) of the corresponding components as well as of the plate number of the column. In gas chromatography (GC) and high performance liquid chromatography (HPLC), the separation factor and capacity factor are constant for a given set of analytica! conditions (stationary phase, temperature, mobile
'l X. Lou, H.-G. Janssen, H. Snijders and C.A. Cramers, J. High Resol. Chromatogr., 19 (1996) 449.

Pressure drop effects in padeed column SFC 68
phase, etc.) and are independent of the column type and dimensions. The plate number, on the other hand, is increased approximately in proportion to the column length. Therefore better resolutions can normally be obtained with longer columns.
Supercritical :fluid chromatography (SFC) can generally be considered as an intermediate technique between GC and HPLC. The pressure dependenee of retention is, however, a notabie exception to this rule. Capacity factors and separation factors are almost independent of pressure in GC and HPLC, while pressure is a very important parameter controlling retention in SFC. In general, the pressure drop in SFC is low if open-tubular columns are used, whereas significantly higher pressure drops are encountered with paclred columns. The in:fluence of the pressure/density gradient along packed columns on retention and efficiency in SFC bas been studied by a number of research groups [1-12].. Moutier et al. [4] studied the effect of the pressure drop on the observed plate height. The considerable peak broadening observed at higher density drops was explained by an increase of the capacity factor along the column. Schoerunakers and Uunk [2] studied t:he effects of column pressure drop in packed column SFC in order to compare the potential applicability of packed and open-tubular columns and showed that the performance of packed columns is Iimited by the maximum allowable pressure drop. In contrasttothese observations other authors concluded that pressure drop has no actverse effect on the column performance in SFC as long as the column outlet pressure is high enough or in cases that modifiers are used. Koehier et al. [11, 12], for example, studied the influence of linear velocity, column length and pressure drop on SFC separations using modified co2 as the mobile phase and found that the plate number increased approximately in proportion to the increase in column length, irrespective of the pressure drop across the column. Berger [8] was able to achieve 220 000 theoretical plates in packed-column SFC by connecting ll conventional HPLC columns in series! In our previous work attempts were made to shed light on the mechanisrns of pressure drop in SFC. Numerical methods were presented which enabled the prediction of pressure gradients along packed and open tubular columns [9]. These methods were also used to calculate the pressure drop, the hold-up time in the column as well as the variation of density, diffusitivity, the local capacity factor and plate height along the length of the column. The predicted valnes for these parameters were found to be in excellent agreement with experimental data [10].

Chapter4 69
The munber of theoretica! plates is, evidently, very important for the quality of a chromatographic separation. However, the theoretica! plate number addresses only part of the separation quality. In many cases the selectivity of the separation system is of much more importance. As mentioned above, retention in SFC is a strong function of pressure/density. If now a significant pressure or density drop occurs along the column, not only the plate height but also the capacity factors and selectivity will vary along the column. This indicates that resolution improverneut in SFC obtained by using longer columns might be different from the improverneut obtained in GC or HPLC. In the latter two forms of chromatography resolution increases simply proportionally to the square root of the column length. The varlation of resolution in SFC with increasing column lengths is much more complicated. Here not only the plate number varies along the column, also the variation in the generally much more important factor, selectivity, has to be taken into account.
In this work the effects of pressure or density drops on SFC separations using pure carbon dioxide as the mobile phase are systematically investigated. Using model equations derived previously, the capacity factors, separation factors, plate heights and resolutions of test solutes in different samples were calculated. The varlation of these parameters along the column is illustrated and studied. The calculated values were compared with the experimental data. Finally, the effects of plate height and selectivity variations along the column on the separation quality are discussed.
4.2. EXPERIMENTAL
The column used in the experiments was a ZORBAX ODS reversed phase HPLC column (25 cm x 4.6 mm, 5 ~ particles) obtained from Rockland Technologies, Inc. (Chadds Ford, PA, USA). The column was deactivated with N,Obis(trimethylsilyl)-trifluoroacetamide (BSTFA) before being used in the experiments. Prior to deactivation the column wasdriedat l00°C under a flow of carbon dioxide at 140 bar for 1 hour. After this, 20 successive injections of 200 nL BSTFA were performed at the same temperature and pressure conditions.
All experiments were carried out on a Carlo Erba SFC 3000 instrument (Carlo Erba, Milan, Italy) equipped with a flame ionization detector (FID). The column

Pressure drop effects in packed column SFC 70
effluent is split into two streams. One stream is fed to the FID via a linear fusedsilica restrietion capillary (45 cm x 10 ).J.IU i.d.). The other is used to control the flow rate through and pressure drop across the column with a stainless steel capillary (210 ).J.IU i.d.) crimped at its end using a pair of pliers. The column flow rate is determined by the sum of the flow rate through the FID and the flow through the stainless steel capillary. A digital pressure sensor was installed between the column and the stainless steel capillary. The accuracy of the absolute pressure measurements was better than 0.15%. Carbon dioxide with a purity of 99.996% (Intermar B. V., Breda, The Netherlands) was used as the mobile phase. Two test samples were used in the experiments. One is a mixture of ethylbenzene, naphthalene, and bipheny1 (all from Aldrich, Milwaukee, Wisconsin, USA) dissolved in dichloromethane. The other is a mixture of liquid crystals used in LCD displays.
Accurate calculations of pressure gradients in packed column SFC require accurate values for both the column porosity and the column permeability. These values were determined as described previously [9]. The calculated values of column porosity and permeability for the column used in this study were 0.58 and 1240, respectively.
4.3. RESUL TS AND DISCUSSION
4.3.1. In-situ deactivation ofthe column
(B)!
___..____,u~LJ i
0 10 15
tune (min) time (mm)
Figure 4.1. The effectiveness of in-situ deactivation. Sample: liquid crystal mixture; Column
temperature: sooc; inlet pressure: 140 bar; outlet pressure: 133 bar. (A) before deactivation; (B)
after deactivation.l. NC-Ph-cH-Et 2. NC-Ph-cH-Pr 3. MeO-Ph-cH-Pr 4. NC-Ph-cH-Bu 5. NC
Ph-cH-Pe
Under SFC conditions, strong tailing is often observed when polar compounds are separated on packed columns using pure co2 as the mobile phase. This problem

Cha ter 4 71
is due to residual active sites on the paclring material [13]. To preclude erroneous results in the experiments, the packed column used was deactivated prior to use. The effectiveness of the in-situ deactivation with BSTF A is demonstrated in tigure 4.1. This tigure shows the chromatograms of the liquid crystal sample before and after deactivation. From this tigure it can be seen that, after deactivation, the peak shapes of the components in the liquid crystal mixture are greatly improved. Before deactivation only one peak and several humps were observed. On the contrary, for the aromatic test sample containing only apolar components (ethylbenzene, naphthalene and biphenyl), deactivation has no effect on the peak shapes. Symmetrical peaks are observed both before and after deactivation.
4.3.2. Capacity factors, separation factors, theoretica/ plate number and
resolulions at different inlet pressures The methods described previously enable the calculation of pressure, density and velocity gradients along an SFC column [9]. Once the density profile along the column is known, the local capacity factors of the solutes can be calculated at every position in the column from an appropriate relation between the density and the capacity factor. The following empirica! equation was used to relate the capacity factor to the density:
ln(k) =a+ bp+ cp2 (4.1)
where kis the capacity factor, p is the density and a, b, and c are constants at a constanttemperature.
In Table 4.1 the values for the coefficients, a, b and c are given for the solutes
studied at a column temperature of 50°C. These coefficients were determined by measuring the capacity factors of the solutes at various densities and then plotring In(k) versus density. These experiments were performed at very low linear veloeities where the effect of pressure drop on k is negligible. The hold-up times needed for determining the capacity factors were measured using methane as the
to marker.
Por the calculation of the plate height along the packed column a suitable plate height equation is required. Here we adapted the Horvathand Lin equation [14].
The procedure foliowed to calculate the local plate height at every position in the
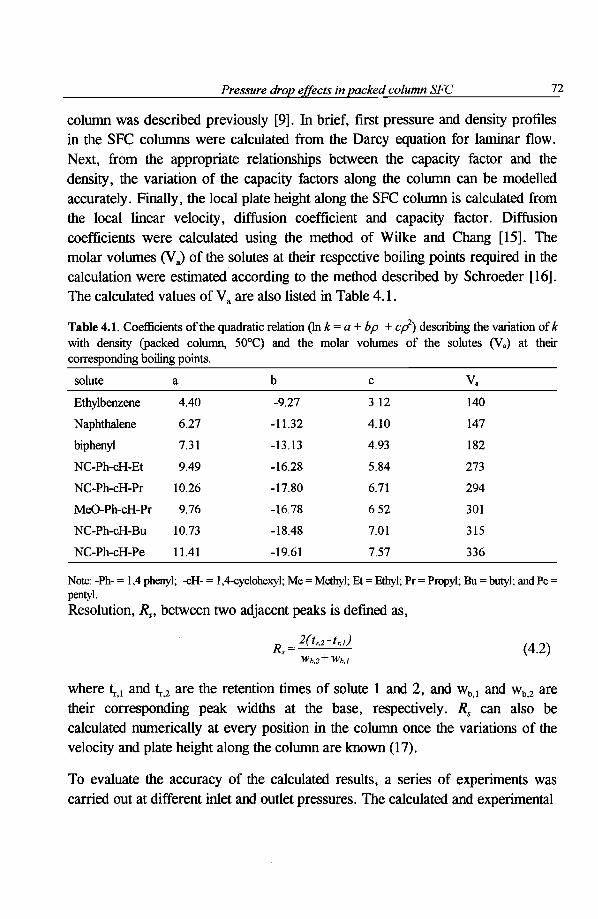
Pressure drop effects in packed column SFC 72
column was described previously [9]. In brief, first pressure and density profiles in the SFC columns were calculated from the Darcy equation for laminar flow. N ext, from the appropriate relationships between the capacity factor and the density, the variation of the capacity factors along the column can be modelled accurately. Finally, the local plate height along the SFC column is calculated from the local linear velocity, diffusion coefficient and capacity factor. Diffusion coefficients were calculated using the method of Wilke and Chang [15]. The molar volumes (V..) of the solutes at their respective boiling points required in the calculation were estimated according to the method described by Schroeder [16]. The calculated values of V a are also listed in Table 4.1.
Table 4.1. Coefficients ofthe quadratic relation (In k =a+ bp + crl) descrihing the variation of k with density (packed column, 50°C) and the molar volumes of the solutes (V.) at their corresponding boiling points.
solute a b c v. Ethylbenzene 4.40 -9.27 3.12 140
Naphthalene 6.27 -11.32 4.10 147
biphenyl 7.31 -13.13 4.93 182
NC-Ph-cH-Et 9.49 -16.28 5.84 273
NC-Ph-cH-Pr 10.26 -17.80 6.71 294
MeO-Ph-cH-Pr 9.76 -16.78 6.52 301
NC-Ph-cH-Bu 10.73 -18.48 7.01 315
NC-Ph-cH-Pe 11.41 -19.61 7.57 336
Note: -Ph-= 1,4 phenyl; -eH-= 1,4-cyclohexyl; Me= Methyl; Et= Ethyl; Pr = Propyl; Bu = butyl; and Pe = pentyl.
Resolution, R5 , between two adjacent peaks is defined as,
R. = 2(tr.rfr,J) wb,2+wb,J
(4.2)
where Îr, 1 and Îr,2 are the retention times of solute 1 and 2, and wb,I and wb,2 are their corresponding peak widths at the base, respectively. Rs can also be calculated numerically at every position in the column once the variations of the velocity and plate height along the column are known (17).
To evaluate the accuracy of the calculated results, a series of experiments was carried out at different inlet and outlet pressures. The calculated and experimental

Cha ter4 73
Table 4.2. Comparison between the calculated and experimental values of k, a, N and R. for
naphthalene (N) and biphenyl {B) at a constant fluid flow rate of600 miJmin (gaseous flow}.
Pï(bar)"> Po(bar)"J k(N) N(N) a(N,B) R.(N,B)
380 364.8 EXP 0.516 18710 1.08 0.91 CAL 0.527 19280 1.08 1.01
320 305.2 EXP 0.589 19520 1.09 1.17 CAL 0.599 18894 1.10 1.23
260 245.6 EXP 0.704 19360 1.11 1.59 CAL 0.716 18376 1.11 1.58
200 188.1 EXP 0.934 16230 1.15 2.23 CAL 0.948 17243 1.14 2.29
140 128.7 EXP 1.80 16260 1.24 4.75 CAL 1.86 16658 1.24 4.72
125 113.8 EXP 2.72 16220 1.30 6.08 CAL 2.94 16507 1.31 6.69
•J inlet pressure;b) outlet pressure; EXP = Experimenta1; CAL= Calculated.
Table 4.3. Comparison between calculated and experimental values of k and N for the components
in the liquid crystal at a constant fluid flow rate of600 miJmin (gaseous flow).
Pr'(bar) Pobj(bar) klc) k3d) k5• NI N3 N5
380 364.8 EXP 0.599 0.881 0.829 21630 19350 22230 CAL 0.609 0.890 0.841 21699 21843 22209
230 216.6 EXP 1.092 1.487 1.565 17360 16250 19533 CAL 1.139 1.556 1.629 20541 20739 21071
220 207.3 EXP 1.167 1.593 1.685 18790 19040 19440 CAL 1.220 1.656 1.753 20137 20363 20708
210 197.4 EXP 1.264 1.707 1.835 17520 18470 18052 CAL 1.319 1.779 1.908 20047 20227 20616
150 138.0 EXP 2.812 3.622 4.439 16890 18450 19110 CAL 2.945 3.768 4.626 19400 19663 19925
140 128.7 EXP 3.628 4.603 5.898 13080 19060 14550 CAL 3.834 4.843 6.218 18986 19272 19520
130 118.9 EXP 5.093 6.396 8.688 9195 17828 7400 CAL 5.651 7.027 9.644 18645 18947 19123
a) inlet pressure; bJ outlet pressure; cJ 1 represents NC-Ph-cH-Et; dJ 3 represents MeO-Ph-cH-Pr; eJ 5 represents NC-Ph-cH-Pe. Other notes see Tables 4 .I and 4 .2.

Pressure drop effects in paclced column SFC 74
values for the capacity factor, the theoretica! plate number, separation factor and resolution of the solutes at different inlet pressures are listed in tables 4.2 to 4.4, respectively. In these experiments the column temperature was kept constant at 50°C and flow rate at approximately 600 mL/min (gaseous flow under ambient conditions). As can beseen from these tables a very good agreement is generally observed between the calculated values and experimental data. Only for the liquid crystal mixture deviations in the theoretical plate number and resolution somelimes occurred when the inlet pressure is lower than 140 bar. Most likely, this is due to the tailing of the peaks caused by the remaining active sites. Nevertheless, even under these conditions, the calculated values of the capacity factors are still in good agreement with the experimental data.
Table 4.4. Comparison between experimental and calculated values of separation fàctor and
resolution for the components in the liquid crystal at a constant tluid flow rate of 600 mL/min (gaseous flow).
Pi"> (bar) Poh)(bar) a a a R. R. R. {2,3)") (3,4)d) (3,5)" (2,3) (3,4) (3,5)
380 364.8 EXP 1.33 0.83 0.94 4.45 2.90 1.06 CAL UI 0.84 0.94 4.39 2.88 0.98
230 216.6 EXP 1.22 0.92 1.05 3.69 1.55 1.01 CAL 1.22 0.92 1.05 4.25 1.87 1.02
220 207.3 EXP 1.22 0.92 1.06 3.92 1.66 1.20 CAL 1.22 0.93 1.06 4.18 1.41 1.29
210 197.4 EXP 1.21 0.94 1.08 3.72 1.32 LSI CAL 1.21 0.94 1.08 4.10 1.51 1.63
150 138.0 EXP 1.12 1.03 1.23 3.08 0.85 5.71 CAL 1.12 1.04 1.23 2.98 1.00 5.83
140 128.7 EXP 1.10 1.07 1.28 2.41 1.89 6.81 CAL 1.09 1.07 1.28 2.43 2.00 7.35
130 118.9 EXP 1.07 1.12 1.36 1.56 2.69 6.83 CAL 1.05 1.12 1.37 1.52 3.54 9.69
•l inlet pressure; bJ outlet pressure; cl 2 represents NC-Ph-cH-Pr; d} 4 represents NC-Ph-cH-Bu; Other notes see Tables 4.1 and 4.2.
Some representative chromatograms of the two samples at different inlet pressures are shown in figures 4.2 and 4.3, respectively. It can clearly be seen from these figures that the inlet pressure has a considerable effect on the separation obtained. The effects of inlet pressure on the resolution of ethylbenzene, naphthalene and biphenyl under the conditions tested are relatively

Cha ter 4 75
straightforward. Resolution is considerably irnproved when the inlet pressure is decreased. For the liquid crystal mixture, the situation is far more complicated as will be discussed later.
E
'-----
2 4
time (min)
N
(A)
B
\____) \..___
6 0 2
time (min)
4
Figure 4.2. SFC chromatograms of ethylbenzene (E), naphthalene (N) and biphenyl (B) at different inlet pressures. Column temperature: 50°C; supercritical fluid flow rate: 600 mL/min (gaseous flow at ambient conditions); Inlet pressures: (A) 125 bar, (B) 380 bar.
(A) (B)
time {min) time (min)
(C)
time(min) time (min)
Figure 4.3. SFC chromatograms ofthe liquid crystal mixture at different inlet pressures. Column temperature: 50°C; supercritical fluid flow rate: 600 mL/min (gaseous flow at ambient conditions). (A) 125 bar; (B) 140 bar; (C) 220 bar; (D) 380 bar. Peak identification as in tigure 4.1.

Pressure drop ejjects in packed column SFC 76
The final resolution obtained in a chromatographic separation is a function of column efficiency, selectivity and retention. For GC and HPLC the equation that describes resolution as a function of selectivity, retention and efficiency reads:
(4.3)
Unfortunately, however, this equation can not be applied to SFC because a, k and the plate height (H) are not constant along the column. To get a impression of the magnitude of the variation of these parameters with pressure, a, k and H values for the separation of naphthalene and biphenyl at inlet pressures of 380 bar and 125 bar are listed in table 4.5. When the inlet pressure was decreased from 380 bar to 125 bar, the value for (N)112 was only slightly lower. Opposedly, the values for k/(1 +k) and (a-1)/a were increased by more than 100% and 200%, respectively. From this it can be concluded that for these components, the better separation obtained at lower pressures is due to the larger capacity factors and, more importantly, to the increased selectivity at lower pressures. This implies that if a column is operated at a significant pressure drop, the first part of the column only has a small contribution to the resolution. In such a system, very high plate numbers can be obtained. The plate number, however, is no longer representative for the quality of the separation. In the first part of the column a lot of plates are generated. Unfortunately, as the components are only wealdy retained here, little separation is obtained. The last part of the column is operated at lower pressures and is therefore much more effective in separating the components. Of the overall separation observed, the largest part is obtained from the last part of the column. In this respect SFC is clearly different from both GC and HPLC where an increase in plate number always yields better separations and every part of the column contributes equally to the overall separation.
In contrast to the monotonous increasè in resolution at reduced pressures for the ethylbenzene, naphthalene and biphenyl mixture, the effects of inlet pressure on the separation of the liquid crystal mixture are much more complicated. By varying the inlet pressure, even the elution order of some of the components can be changed. At the supercritical fluid flow rate tested (around 600 mL/min, gaseous flow at ambient conditions), complete separation of the five peaks is only possible at inlet pressures around 220 bar or 140 bar.

Table 4.5. Effect ofinlet pressure on resolution ofnaphthalene and biphenyl•).
No. k a (a-1)/a k/(l+k) N R.
inlet pressure: 380 bar
N 0.516 0.340 18700 B 1.08 0.074 1.01
0.557 0.358 18800
inlet pressure: 125 bar
N 2.67 0.728 16600 1.30 0.231 6.08
B 3.47 0.776 14600 •l Supercritical fluid flow Iate, 600 mUmin (gaseous flow under ambient conditions).
4.3. 3. Effects of supercritical fluid flow rate on separation
In SFC, retention and resolution are strongly dependent on pressure, or as shown above, on the pressure gradient. For a given column at a constant temperature, the pressure gradient along the column is determined by the inlet pressure in combination with the supercritical fluid flow rate. In order to obtain a good separation both the supercritical fluid flow rate and the inlet pressure should be considered. In contrast to the situation in HPLC and GC where the flow rate affects resolution only via the plate nwnber, in SFC the flow rate influences both the plate height as well as the selectivity. In this section, the effects of flow rate on the resolution obtained were tested at two inlet pressures i.e. 220 bar and 140 bar at 50°C. As demonstrared above, only around these inlet pressures complete separation of the five peaks from the liquid crystal mixture could be obtained.
At the inlet pressure of 140 bar, the elution position of MeO-Ph-cH-Pr in the chromatagram could be varied between the positions of NC-Ph-cH-Pr and NCPh-cH-Bu by changing only the supercritical fluid flow rate. This is clearly in contrast to the situations in GC and HPLC where the relative elution positions in a non-programmed run can never be changed by changing the mobile phase flow rate. Complete separation of the five peak:s trom the liquid crystal mixture could only be obtained at a fluid flow rate around 600 rnL/rnin (see figure 4.3). At significantly higher or lower flow rates, either the separation between MeO-PhcH-Pr and NC-Ph-cH-Pr or between MeO-Ph-cH-Pr and NC-Ph-cH-Bu disappeared. Figure 4.4 shows the chromatagram of the liquid crystal mixture at a

Pressure drop effects in packed column SFC 78
flow rate of 150 mL/min. At this flow rate, MeO-Ph-cH-Pr and NC-Ph-cH-Bu almost coeluted. From these results it is clear that the resolution of these componentsis seriously affected by the fluid flow rate (or the pressure drop). As stated above, apart from affecting the plate number of the column the supercritical fluid flow rate can also influence the selectivity. In the present situation the effects of flow rate on selectivity are of much more importance to the separation. Similar effects of the flow rate were also observed at an inlet pressure of 220 bar where Meo-Ph-cH-Pr elute between the peaks of NC-Ph-cH-Bu and NC-Ph-cH-Pe.
10 20 50
time(min)
Figure 4.4. Chromatagram ofthe liquid crystal mixture at an inlet pressure of 140 bar. Column temperature: 50°C; supercritical fluid flow rate: 150 mL/min (gaseous flow at ambient conditions), Peak identification as in figure 4.1.
4.3.4. lllustration of varlation of the resolution along an imaginary long column In both GC and HPLC, resolution can generally be improved by using longer columns because of the larger plate numbers of such columns. As demonstraled above, resolution in SFC is considerably affected by the inlet pressure and the flow rate or pressure drop. In order to investigate the SFC separation quality obtained on long packed columns, the capacity factor, selectivity and resolution were calculated for a 5.5 meter long column using the models described previously. Through these calculations, the variations of k, a, N and Rs along the column can hence be investigated in detail.
Since the purpose of the calculations is to show the variation of k, a, N and ~ along the column, only one typical fluid flow rate of 2 x w-z gram/sec was used. In order to maintain supercritical conditions in the entire column, the outlet pressure was set to be higher than 80 bar. Taking the maximum allowable pressure of the system (i.e. 390 bar) into account, the resulting allowable

Chapter4 79
maximum column length was 5.5 meter. Therefore, this column length was used in all model calculations. From the calculations a number of interesting conclusions can be drawn. 1. Pressure drop is very large for long columns packed with small particles (5
Jliil). The pressure drop is almost linear along the column (figure 4.5).
~240 ] ~ 160
80
~.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5
(m)
Figure 4.5. Pressure profilefora 5.5 m long model column. Inlet pressure: 390 bar; flow rate: 2 x
10'2 gram/sec.
~
~ 8
i
500.----------------,
400 E
1 300
~ 200
100
~.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5
(m)
Figure 4.6. Varlation of plate numbers for ethylbenzene (E), naphthalene (N) and biphenyl (B)
along a 5.5 meter long model column. Inlet pressure: 390 bar; flow rate: 2 x 10-2 gram/sec.
2. Only slight changes in the calculated localplate height along the column were observed if the local pressure is above a certain value. The plate number increases approximately in proportion to the column length. However, at low
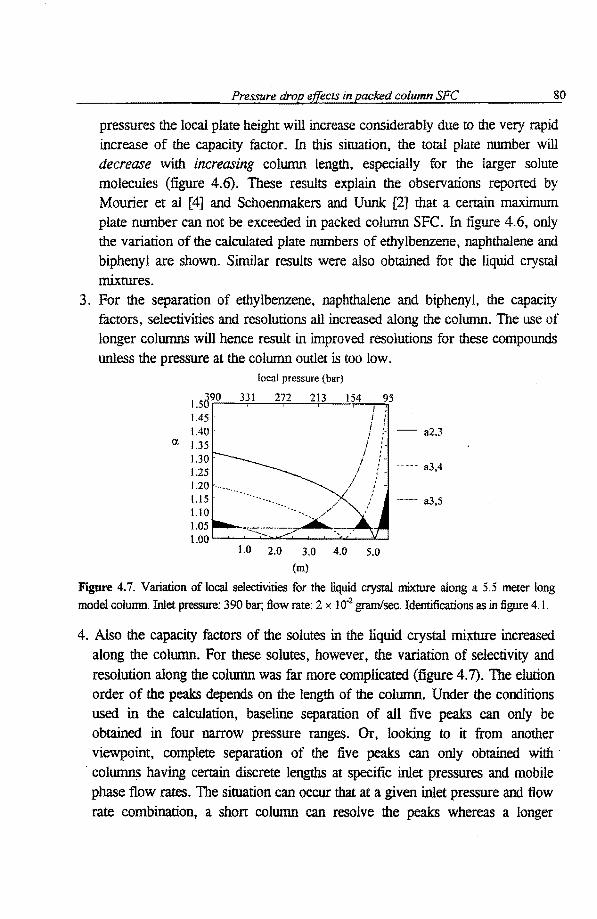
Pressure drop ef!ects in padred column SFC 80
pressures the local plate height will increase considerably due to the very rapid increase of the capacity factor. In this siruation, the total plare number will decrease with increasing column length, especially for the Iarger solute molecules (figure 4.6). These results explain the observations reponed by Mourier et al [4] and Schoenmakers and Uunk [2] that a certain maximum plate number can nat be exceeded in packed column SFC. In tigure 4.6, only the varlation of the calculated plate numbers of ethylbenzene, naphthalene and biphenyl are shown. Similar results were also obtained for the liquid crystal mixtures.
3. For the separation of ethylbenzene, naphthalene and biphenyl, the capacity factors, selectivities and resolutions all increased along the column. The use of langer columns will hence result in improved resolutions for these compounds unless the pressure at the column outlet is toa low.
local pressure (bar)
1.5d90 331 272 213 154 95
1.45 1.40·
u 1.35 . 1.30 1.25
1.20 ········· 1.15 •.•. / 1.10 ~--~" /
1.05 ··-··-----~---.....,.-'1--,111111 1.00 ....................... _ ........... ...;.;;;o· ....:;_.-..._....._...__.·· .... --~-"1.-..J
1.0 2.0 3.0 4.0 5.0
(m)
---- · a3,4
a3,5
Figure 4.7. Variation oflocaJ selectivities for the liquid crysta1 mixture along a 5.5 meter long model coluiml. Inlet pressure: 390 bar; flow rate: 2 x 10'2 gram/sec. Identifications as in figure 4.1.
4. Also the capacity factors of the solutes in the liquid crystal mixture increased along the column. For these solutes, however, the varlation of selectivity and resolution along the column was far more complicated (figure 4. 7). The elution order of the peaks depends on the length of the column. Under the conditions used in the calculation, baseline separation of all five peaks can only be obtained in four narrow pressure ranges. Or, looking to it from another viewpoint, complete separation of the five peaks can only obtained with ·.
· columns having certain discrete lengths at specific inlet pressures and mobile phase flow rates. The situation can occur that at a given inlet pressure and flow rate combination, a short column can resolve the peaks whereas a langer

Chapter4 81
column can not. This is because of the selectivity changes that occur in the column as a result of the pressure gradient along the column.
It is clear from the results of these calculations that for long columns the pressure drop is very large. The retention, selectivity and resolution can all vary considerably along the column as a result of the gradual decrease of pressure. Por the separations where the pressure drop has no negative effects on selectivity, (i.e. no changes in elution order), longer columns will yield better separations. This may be the case for the separation of structurally similar compounds, such as, homologues or enantiomers. In contrast, for the separation where the elution order of the components will depend on pressure, the increased pressure drop for a long column may show negative effects on selectivity. In this case, resolution may get worse when longer columns were used! In this respect SPC is clearly different from both GC and HPLC. Evidently, longer columns will also require longer analysis times. Por such situations, large pressure drops should be avoided and short columns or columns packed with large particles must be used.
In SFC, most practical separations are carried out with relatively short columns (i.e. 15 cm or 25 cm long). Prom the calculations presented here some very useful information with regard to method optimization can also be obtained. Por example, it can be seen from tigure 4. 7 that the optimum a-values of the five peaks in the liquid crystal sample are obtained at pressures around 390 bar, 220 bar, 140 bar and below 100 bar (shaded areas). However, at 390 bar the resolution of the five components is not satisfactory because the capacity factors of the components are very low. On the other hand, at pressures below 100 bar, very long analysis times are required because of the large capacity factors. That is, in practical separations using a standard column, the inlet pressure should be around 220 bar or around 140 bar. This prediction is in good agreement with the experimental data. Prom this it can be concluded that the models described here can also be used in method development in SPC. Prom a limited series of input runs, coefficients for the ln(p) vs. k equation can be obtained. This then allows the calculation of all relevant chromatographic parameters at any inlet pressure, flow rate and column length.
In this report only pure C02 was considered as the mobile phase. Por this mobile phase large effects of the pressure drop on selectivity and resolution were

Pressure drop effects in packed column SFC 82
observed. Por modified mobile phases, however, the effects of pressure gradients along the column can be significantly less because in these systems an equal pressure drop results in a lower reduction of the elution strength of the mobile phase.
4.4. CONCLUSIONS
In packed column SFC with pure carbon dioxide as the mobile phase, the pressure drop bas a significant effect on the chromatographic resolution. The data calculated with the numerical methods derived previously are in good agreements with the experimental results. The capacity factors increase along the column due to the pressure gradient. The variation of selectivity and resolution along the column is far more complicated. No simple equation is available for the calculation of resolution in packed column SFC because a, k and the plate height are not constant along the column. Increased plate number do not necessarily give a better separation.
For components for which the pressure drop has no adverse effect on selectivity, longer columns can yield better resolutions. On the contrary, for components for which the elution order is strongly dependent on pressure, resolution may get worse when longer columns are used. For such systems the effects of flow rate on resolution are much stronger than generally realized. In order to obtain a good separation, the inlet pressure, supercritical fluid flow rate and column length should all be optimized. Temperature is another important parameter that should be optimized. The model calculations presented can also be used for metbod development in SFC.
REFERENCES
1. S.M. Fields and M.L. Lee, J. Chromatogr., 349 (1985) 305. 2. P.J. Schoenmakers and F.C.C.J.G. Verhoeven, J. Chromatogr., 352 (1986) 315. 3. P.J. Schoenmakers and L.G.M. Uunk, Chromatographia, 24 (1987) 51.
4. P .A Mourier, M.H. Caude and R.H. Rosset, Chromatographia, 23 ( 1987) 21. 5. K.D. Bartle, T. Boddington, AA Cliffurd and G.F. Shilstone. J. Chromatogr., 411 (1989) 347. 6. D.P. Poeand D.E. Matire,J. Chromatogr., 511 (1990) 3. 7. T.A. Bergerand lF. Deye, Chromatographia, 30 (1990) 57.
8. T.A. Bergerand W.H. Wilson,Ana/. Chem., 65 (1993) 1451.
9. H.-G. Janssen, H.M.J. Snijders, JA Rijks, C.A. Cramers and P.J. Schoenmakers, J. High Resol. Chromatogr., 14 (1991) 438.
10. H.-G. Janssen, H.Snijders, C. Cramers and P. Schoenmakers, J. High Resol. Chromatogr., 15 (1992) 458.

Cha ter4 83
lL U. Koehler, P. Biermanns andE. Klesper,J. Chromatogr. Sci., 32 (1994) 461. 12. U. KoehlerandE. Klesper,J. Chromatogr. Sci., 32 (1994) 525. 13. H.-G. Janssen, P.J. Schoenmakers and C.A. Cramers,J. Chromatogr., 552 (1991) 527. 14. C. Horvth and H.-J. Lin, J. Chromatogr., 149 (1978) 43. 15. RC. Reil:i, J.M. Prausnitz and T.K Sherwood, The Properties of Gases and Liquids, 3rd ed.,
McGaw Hili, New York, 1977, p.544. 16. R.C. Reid, J.M. Prausnitz and T.K Sherwood, The Properties of Gases and Liquids, 3rd ed.,
McGaw Hili, New York, 1977, p.5L 17. J.G.M. Janssen, Ph.D. Thesis, Eindhoven University ofTecbnology (1991).

Chapter5 84
5.1 Investigation of parameters affecting the supercritical fluid extraction of polymer additives from polyethylene4)
Abstract
Polymer additives were extracted from polyethylene with supercritical carbon dioxide. The two-film theory, which considers mass transfer across a phase boundary, is applied to qualitatively describe the killetics of mass transfer from the core of the polymer particles into the supercritical fluid extractant. The effects of pressure, temperature, actdition of benzene as a modifier, properties and concentrations of the solutes, static time and supercritical fluid extraetafit flow rate on the extraction process are investigated systematically. At constant temperature the extraction rates first increase with increasing pressure. When pressure reaches a eertaio level, a further increase of the pressure does not further increase the extraction rates. At constant pressure, the extraction rates were found to increase first and then decrease with increasing temperature. In actdition to pressure and temperature, the SFE extraction killetics is also influenced by the solute concentration, and the rate-limiting parameter in the extraction can be changed from solubility to diffusion during the course of the extraction. The magnitude of the effects of the experimental parameters depends on the properties and molecular weights of the solutes. The role of benzene as a modifier in the extraction of polymer additives from polyethylene is swelling the polymer particles and improvement of the solvent strength of the supercritical fluid extractant. Modifier effects were found to be more pronounced at lower temperatures.
5.1.1 Introduetion
Polymers are widely used materials and are indispensible to mankind nowadays, being essential to clothing, shelter, transportation, and communication, as well as to the convenience of modem living [1]. Their properties can be improved by the presence of appropriately selected additives. Hitherto, Soxhlet extraction is normally used to determine the contents of polymer additives. However, this metbod is both time and solvent consuming. Additionally, after extraction the samples have to be concentrated and there is a requirement to dispose of the organic solvent in an appropriate manoer. Recent concern about the hazards
•l X. Lou, H.-G. Janssen and C.A. Cramers, J. Microcolumn separations, 7 (1991) 303.

5.1 SFE o(polymer additives [rom polyethylene 85
associated with most of the solvents used, the cost and the environmental dangers of waste solvent disposal, have led to the development of alternative sample extraction methods [2].
Supercritical fluid extraction (SFE) has many advantages over Soxhlet extraction. Besides advantages such as a reduced usage of organic solvent, shorter extraction time, adjustable solvent strength and the ability for on-line combination with analytica! instruments [3-5], the extraction temperature in SFE can be changed continuously from the critica! point of the supercritical fluid to temperatures well above the glass transition temperature of a polymerie materiaL This is in contrast to Soxhlet extraction where the extraction temperature is limited by the boiling point of the extraction solvent used. The wide range of extraction temperatures available in SFE is of particular importance. For example, the low critica! temperature of supercritical carbon dioxide makes SFE an excellent candidate for extracting thermally labile compounds under conditions slightly above room temperature [6]. The high extraction temperatures available, on the other hand, favourably affect the diffusion coefficients of the compounds and thus increase the SFE extraction rate for samples in which the rate-limiting parameter is related to diffusion in the matrix, such as in the case of extracting polymer additives from polymerie materials [7].
SFE has been applied successfully to a wide variety of matrixlanalyte combinations [8]. In the extraction of polymer additives from polymerie materials, the SFE process normally involves three steps. First, the solutes have to diffuse from the core of the polymerie material to the surface. Next, the compounds are transfered from the surface into the extraction fluid. Finally, the compounds are eluted out of the extraction cell by the flow of the supercritical extractant. Up tiH now, only a limited number of fundamental studies were carried out that aimed at increasing the knowledge of the various experimental parameters that affect the extraction behaviour in the SFE extraction of polymerie materials. Bartie et al. [9] derived a model for diffusion-limited extractions assuming that the matrix particles are spheres of a well-defined size and the initial distribution of the solutes within the spheres is uniform. Cotton et al. [10] and Kueppers [11] investigated temperature effects in SFE and found that higher extraction efficiencies could be obtained at elevated temperatures. Yenerna et al. [12] studied the effects of partiele size on the SFE extraction efficiency in the extraction of caprolactam and oligomers from nylon-6. Expectedly, higher

Chapter5 86
extraction rates were observed for smaller particles. In each of the four studies referred to above, pressure and temperature conditions were such that the ratelimiting factor for extraction was diffusion of the solutes in the polymerie materials. However, the actual mechanism that governs the extraction of compounds from polymerie matenals is far more complicated. Diffusion in the matrix particles is of course important, but other parameters such as solubility of the components in the supercritical extractant can also play an important role. Parameters affecting any one of the three subsequent steps in SPE identified above will influence the ultimate SPE efficiency.
In this article, the effects of various operational parameters such as temperature, pressure, supercritical fluid flow rate, static time, modifier concentration and solute characteristics on the SPE k:inetics in the extraction of polymer additives from polyethylene are investigated. Attempts are made to identify the ratedeterming step in the SPE process for the various sets of experimental conditions evaluated. The role of the modifier in the extraction of polymerie matenals is also investigated and discussed.
5.1.2 Theory
In the SPE extraction of polymerie materials, the solutes are extracted from the core of the polymer particles into the supercritical fluid. The k:inetics of mass transfer in this process can be represented by the two-film theory as is illustrated in tigure 5.1 [13]. The basis of this theory is the assumption that the zones in which the resistance to mass transfer lies can be replaced by two hypothetical layers, one on each side of the polymer surface. In these layers mass transfer is entirely by molecular diffusion. The concentration gradient is therefore linear in each of these layers and zero outside. The relative positions of the points C and D in tigure 5.1 are determined by the equilibrium distribution between the two phases. The two-film theory describes the general process occuring when a solute is transfered from one phase to another. In SPE, mass transfer from the core of the polymer particles to the surface is controlled by molecular diffusion. Here the diffusion coefficient is determined by parameters such as properties and stuctures of the polymerie material and the solutes, extraction temperature, presence of a modifier etc.. In general this step is slow because diffusion in the polymer particles is slow. Mass transfer from the polymer surface through the stagnant layer outside the polymer particles into the supercritical fluid stream, on the other

5.1 SFE ofpolymer additives .from polyethylene 87
hand, is extremely fast because supercritical fluids have high solute diffusivities. Moreover, the layer of stagnant extraction fluid around the particles is very thin. After being transfered into the supercritical fluid stream, the components are eluted out of the extraction cell by the flow of the extractant. The elution rate here is determined by tpe solubilities of the components and the flow rate of the supercritical fluid. Prom the discussions presenled above, it is clear that the SPE processcan be modelled as three subsequent steps. Pirst, the solutes must diffuse trom the core of the polymerie material to the surface. Next the solutes should be transfered from the surface into the supercritical fluid stream. Pinally the solutes are eluted out of the extraction cell. The SPE extraction rate is limited by the slowest of these three steps. As explained above, mass transfer from the surface of the polymer particles into the superctitical fluid extractant is very fust. Hence, the slowest, and therefore the rate-limiting step, is either ditfusion in the partiele or elution out of the extraction cell. The two possible extremes concerning the rate-limiting step are schematically shown in Pigure 5.2.
polymer surface
A B
supererhicalfhdd
polymer
F
Figure 5.1. Schematic representation of the two-film theory.
In Pigure 5.2A, the situation is depicted in which the rate-limiting step is diffusion of the solute in the polymer matrix. Increasing the diffusion coefficient of the solute by, for example, increasing the extraction temperature will increase the extraction rate. If the rate-limiting step is elution of the components out of the extraction cell (Figure 5.2B}, enhanced extraction rates can be obtained either by increasing the solvent strength of the extractant, (i.e. by increasing the extraction pressure or by the actdition of a modifier), or by increasing the supercritical fluid flow rate.

Chapter 5 88
surface surface polymer SF
1 polymer:;: ~ SF
ditfusion elution ditfusion I
elution
(A) (B)
Figure 5.2. Schematic diagram of two possible extremes conceming the rate-limiting step in SFE.
As discussed above, many experimental parameters affect the kinetics of SPE extraction. Among these, temperature, pressure and type and concentration of a modifier appear to be the most important ones. At low temperatures and high pressures (high densities), the solubility of the componentsin the extraction fluid is high but diffusion of the solutes in the polymerie material is slow. The extraction rate is now limited by diffusion inside the polymer particles. In contrast to this, at high temperatures and low pressures (low densities), ditfusion of the solutes in the polymerie material is fast while the solubility of the compounds in the supercritical fluid is low and solubility beoomes the rate-limiting parameter. When a modifier is used, the SPE mechanism becomes even more complicated as the modifier can affect both the matrix properties (swelling and deactivation) as wellas the fluid phase properties (polarity and density).
5.1.3 Experimental
SPE experiments were performed with a modified Carlo Erba SPC 3()(X) capillary SPC instrument (Carlo Erba, Milan, Italy). A 3 mi stainless steel extraction cell (Suprex, Pittsburgh, PA) was fitted with hand-tight connectors (Suprex, Pittsburgh, PA) for easy installation. Stainless steel frits (3 J..IIIl) were located at either end of the extraction cell. Pused-silica capillaries (20 J..1I11 i.d. or 50 J..IIIl i.d. with a length of 50 cm) were used as restrictors. To enable static extraction an on-off valve ('/alco, Switzerland) was installed directly bebind the extraction cell. The extracted material was collected by inserting the resttictor outlet into a glass

5.1 SFE o(polymer additives (rom polyethylene 89
vial (10 cm x 1 cm i.d.) containing 5 ml dichloromethane. This vial was changed every 30 minutes. Tetracontane was added to each vial as an internat standard. After collection, dichloromethane was evaporated under a gentle flow of nitrogen and the extracted material was redissolved in a suitable amount of hexane.
The polyethylene sample (powder) was obtained from DSM (Geleen, The Netherlands). The glass transition temperature of the polymer (under non-swollen conditions) is approximately -20°C. About 0.8 g polyethylene was weighed into the extraction cell before extraction. A static time of 30 minutes was used prior to dynamic extraction unless stateel otherwise. The highest extraction temperature tested in the experiments was 80°C. At higher temperatures the restcictor often blockeel due to parrial melting of the polymer. In the experiments with modifier, benzene was added directly to the extraction cell prior to extraction. The carbon dioxide used in the experiments had a purity of 99.996% (lntermar B.V. Breda, The Netherlands). Soxhlet extraction was performeel to produce a frame of reference. SFE extraction efficiencies were calculated relative to the Soxhlet data. For Soxhlet extraction, about 2 g sample was placed in the Soxhlet extractor and extracteet with hexane for 36 hours.
The extracted components were analysed using a gas chromatograph equipped with an on-column injector and an FID (GC 8000 series, Carlo Erba Instruments). The gas chromatographic separation was achieved with a HT
SIMDIST CB column (10 m x 0.53 mm i.d., film thickness 0.17J.UTI) purchased from Chrompack (Middelburg, The Netherlands). The initial temperature for GC separation was 40°C. The temperature was then programmeel to 425°C at
20°C/min.
5.1.4 Results and discussion
5.1. 4.1 Effects of pressure The extraction kinetics in SFE are determined by a number of experimental parameters. The fact that these parameters are generally interrelated is an additional complicating factor in metbod development in SFE. Figures 5.3 and 5.4 illustrate the intlucnee of the extraction pressure on the extraction yields of three polymer additives, Irgafos 168, Irganox 1076 and Irganox 1010, from polyethylene at temperatures of 50°C and 80°C, respectively. From these figures it is clear that the effect of pressure changes can be different at different temperatures. When pressure is increased from 150 bar to 300 bar at 50°C, no

Chapter5 90
significant varlation in the extraction yields of the three additives is observed (figure 5.3). This is because at 50°C the density of the supercritical fluid is relatively high, even at a mild pressure of only 150 bar (p=0.701 g/ml). As the
700.-------,
~600
~ 500 ~ 400 ><: ~300
[200 p.. 100
time (min)
-+- lrganox 168,300 bar
-!:.- lrganox 1076,300 bar
-0- lrganox 1010,300 bar
-+- lrgafos 168, ISO bar
-.6.- lrganox 1076,150 bar
-e- Irganox 1010,150 bar
Fïgure 5.3. Effects of pressure on SFE extraction rate at 50"C. Static time, 30 minutes; restrictor, 50 cm x 50 f..LID fused-silica capillary; collection solvent, 5 m1 dichloromethane.
30 60
time(min)
90 120
300r-------------.
0 30 60
time(min)
(C)
90 120
700r-----------,
600 .••••••••• ••••••••·• -········· •••.• ~~?. î 500 t_.....,."lil- =\tl-
~400 +----~300 +......--~200 +/
100 •
0 30
·-··· soxblet
-1- 300bar
-A- 250bar
-9-200bar
-1- ISObar
60
time(min)
90 120
Fïgure 5.4. Effects of pressure on SFE extraction rate at 80°C. A: lrgafos 168; B: lrganox 1076; C: Irganox 1010. Other details as in figure 5.3.

5.1 SFE o.fpolymer additives from polyethylene 91
diffusion coefficients of the components in the polymer are relatively smal!, the mass flow of components diffusing to the polymer surface is low. Molecules diffused to the surface are rapidly removed from the surface of the polymer particles and are rapidly carried out of the extraction cell by the flow of high density carbon dioxide. Under these conditions, the solubility of the components in the supercritical fluid is clearly not the rate-limiting parameter. The extraction rate is determined by diffusion of the additives in the polymer matrix. A different situation occurs at a temperature of 80°C (figure 5.4). Increasing the pressure from 150 bar to 200 bar at 80°C increases the extraction rates drastically for lrgafos 168 and lrganox 1076, especially in the first fraction of 30 minutes. A further increase of the pressure from 200 bar to 300 bar, however, gives no further improvement in extraction yields for these two components (figures 5 .4A and B). The effects of pressure on the extraction rate of Irganox 1010 are even more pronounced than those observed for lrgafos 168 and Irganox 1076 (figure 5.4C). Almost no extraction occurs for Irganox 1010 at 80°C and pressures below 150 bar. Increasing the pressure from 150 bar to 200 bar causes the extraction rate of this solute to increase significantly. An additional, but smaller increase in extraction rate is observed if the pressure is increased from 200 bar to 250 bar. At these conditions the extraction yield even exceeds that of Soxhlet extraction. No further increase in the extraction rateis observed upon further increasing the pressure from 250 bar to 300 bar. Summarizing, at a constant temperature, the extraction rate first increases with increasing pressure. After the pressure reaches a certain level, further increasing of the pressure does not result in a further increase of the extraction rate. Apparently above a certain pressure value, the solubility of the components in the supercritical fluid is no longer the rate-limiting parameter for extraction. The extraction rate is now limited by the rate of diffusion of the solutes in the polymer particles.
5.1.4.2 Effects oftemperature The effects of temperature at constant pressure are even more complicated than the effects of pressure at constant temperature described above. Increasing the temperature increases the diffusion coefficients of the solutes in the polymer whereas at the same time it also decreases the density and, related to this, the solvent strength of the supercritical fluid. lf the pressure is sufficiently high, the supercritical fluid is capable of rapidly dissolving the components diffused to the surface, which means that the solubility of the component in the supercritical fluid is not the rate-limiting parameter for extraction. Increasing the temperature will
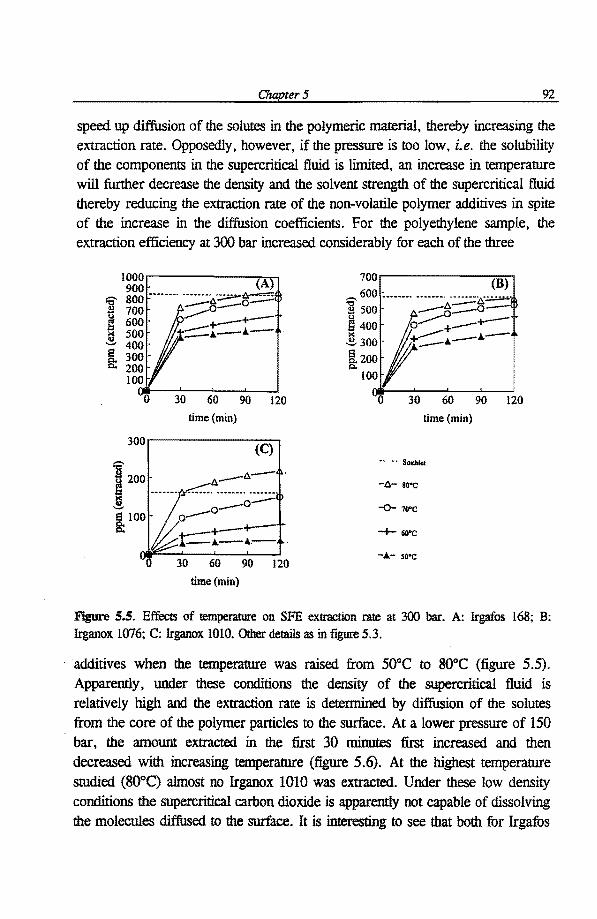
Chapter5 92
speed up ditfusion of the solutes in the polymerie material, thereby increasing the extraction rate. Opposedly, however, if the pressure is too low, i.e. the solubility of the components in the supercritical fluid is lirnited, an increase in temperature will further decrease the density and the solvent strengtil of the supercritical fluid thereby reducing the extraction rate of the non-volatile polymer additives in spite of the increase in the ditfusion coefficients. For the polyethylene sample, the extraction efficiency at 300 bar increased considerably for each of the three
1ggg (A_!. ........ ........ ·,;::.;:.-~ :a- 800 ..-a_o_
l i~~{j.~r-r-- 400• ~ 300. - 200
100 ~-:-':-----:'-----'--....J ""o 30 60 90 120
time (min)
300.--------(C~)-.
"'o 30 60 90 120
time (min)
700 (B) ..-. 600 ............................. ".. . ~ 500 . {j.--{j.:::::::g-s 400 ,~---;----1< --- ... -~300· +..-..t.-i2oo
100
~0 30 60 90 120
time(min)
•• •• Soxhlcl
-t:J.- 80"C
-Q- 70"C
Figure 5.5. Effects of temperarure on SFE extraction rate at 300 bar. A: Irgafos 168; B: lrganox 1076; C: lrganox 1010. Other details as in tigure 5.3.
additives when the temperatu.re was raised :from 50°C to 80°C (figure 5.5). Apparently. under these conditions the density of the supercritical fluid is relatively high and the extraction rate is detennined by diffusion of the solutes from the core of the polymer particles to the surface. At a lower pressure of 150 bar, the amount extracted in the first 30 rninutes first increased and then decreased with increasing temperature (figure 5.6). At the highest temperature studied (80°C) almast no Irganox 1010 was extracted. Under these low density conditions the supercritical carbon dioxide is apparently not capable of dissolving the molecules diffused to the surface. It is interesting to see that both for Irgafos

5.1 SFE o[polymer ad.di.tives from palyethylene 93
168 and Irganox 1076, the reeoverles in the first·fraction of 30 minutes at 150 bar and sooc are lower than those obtaineel at 150 bar and. 50°C, whereas, at proiongeel extraction times the yields approach and finally exceed the reeoverles at 50°C. Most lik:ely, during the first fraction of 30 minutes, the concentrations of the solutes on the surface of the polymer particles are relatively high, whereas their solubility in the supercritical tluid is limited. As the extraction proceeds, the components are continuousely transporteel out of the extraction cell by the flow of the supercritical tluid and their concentrations gradually decrease. At lower concentrations the solubilities of the components in the supercrltical tluid are no longer limiting factors and diffusion in the polymer now becomes important. From this it is clear that the rate-limiting parameter is also related to solute concentration. In practical extractions, it is very well possible that the ratelimiting parameter changes trom solubility to diffusion during the course of the extraction process.
9oo..----------, 800 (Á) _-()____.~-+- SO"C
i700 /0 *-~ ~ 600 · yA;;::::+- -A- 60"C
! !~ )/t . --o- 70"C
~ ~: + -+- SO"C
too fr/
600r----------r
500 · (B) a- -+- sooc 'Ó' 0--A-~ 400 · /.A-- _... -A- ro•c g ~ +?--~300,--:t:~ i200 /
100 -+- so•c
--Q- 70"C
""o 30 6o 90 120 lJIO 30 60 90 120
time (min) · time (min)
2oo.--------..., (C) -+- SO"C
-A- 60"C
-Q- 70"C
-+- 80"C
60 90 120
time(tnin)
Figure 5.6. Effects of temperature on SFE extraction rate at 150 bar. A. Irgafos 168; B: Irganox 1076; C: Irganox 1010. Other details as in tigure 5.3.
5.1.4.3 Effects qf solute molecular properties From the preceding paragraphs it can be seen that the actual intluence of

temperature and pressure on the extraction yield vs. time curve can depend on the properties of the additives to be extracted. To investigate the influence of the molecular structure on the extraction behaviour, the amounts extracted for the various additives in the first fraction of 30 minutes were studied. In order to allow a direct comparison, the amounts extracted were expressed relative to the amounts found in the Soxhlet extraction. The results are listed in Tables 5.1 and 5.2. Under conditions where the rate-limiting parameter is diffusion of the solutes in the polymer, e.g. 300 bar and temperatures between 50°C and 80°C, the amounts extracted in the first 30 minutes increase with temperature. The increases for Irgafos 168 and Irganox 1076 are of the same magnitude, whereas a more pronounced increase is found for Irganox 1010 (table 5.1). Most likely, this
difference is due to differences in the molecular structures or weights of the additives. It is remarkable to see that for lrganox 1010, 30 minutes SFE extraction at 80°C and 300 bar gives a slightly better extraction yield than 36 hours of Soxhlet extrcation. The molecular sizes of lrgafos 168 (relative molecular mass 647) and Irganox 1076 (relative molecular mass 537) are similar, whereas Irganox 1010 (relative molecular mass 1178) is clearly larger. Therefore, migration of Irganox 1010 in the polymer is restricted and the activation energy for diffussion will be higher. The activation energies for diffusion of lrgafos 168 and Irganox 1076, on the other hand, are of a similar magnitude. The variation of diffusion coefficient with temperature is generally described by an Arrhenius-type equation [14]:
(5.1)
where DT is the diffusion coefficient at temperature T, 0 0 is a constant related to the entropy of activation and E is the activation energy. From this equation, it can be seen that the diffusion coefficients increase with increasing temperature. This effect is more pronounced for components with a higher activation energy (larger molecules). Additionally, the apparent activation energy for diffusion decreases with increasing temperature as the activation energy is related to the amount of energy required to form transient voids in the matrix [15].
Changes in temperature affect the extraction process in two opposite ways. An
increase in temperature will, on the one hand, increase the diffusion of solutes inside the polymerie material and, on the other, decrease the density or solvent strength of the extractant. Because large molecules are generally less soluble in

5.1 SFE o(polymer additives.from polyethylene 95
supercritical carbon dioxide than smaller molecules, the density decrease at increased temperature is more likely to beoome a problem for the heavier solutes. This is confirmed by the experimental results. At 150 bar, the reduction in the recovery of lrganox 1010 at increased temperatures is more severe than that of Irgafos 168 or lrganox 1076. From this it is clear that in SFE the reeoverles of larger molecular solutes are more sensitive to temperature and pressure changes.
Table 5.1. Comparison of effects of temperature on the SFB extraction recoveries of different additives in the frrst 30 minutes fraction at 300 bar">.
Temp eq Irgafos 168 Irganox 1076 lrganox 1010
50 54.9% 46.4% 21.2%
60 60.6% 55.2% 29.6%
70 73.4% 69.7% 58.2%
80 86.1% 82.5% 100.4%
a) SFE extraction recoveries were calculated relative to Soxhlet data.
Table 5.2. Comparison of effects of temperature on the SFB extraction recoveries of different additives in the frrst 30 minute fraction at 150 bar.">
(OC) lrgafos 168 Irganox 1076 Irganox 1010
50 56.8% 46.6% 20.8%
60 60.6% 58.9% 31.7%
70 63.0% 57.7% 17.2%
80 27.9% 28.3% 1.0%
•> SFE extraction reeoverles were calculated relative to Soxhlet data.
5.1. 4. 4 Effects of supercritical fluid flow rate In the experiments described above, the restrictor used was a piece of 50 cm x 50 ).Ull fused-silica capillary. In order to investigate the effects of the supercritical fluid flowrateon the SFE kinetics, the resttictor was replaced by a 20 ).Ull fusedsilica capillary with the same length. The supercritical fluid flow rate with the 50 ).Ull capillary was approximately five times higher than that with the 20 J.U11 resttictor. As far as the extraction yields were concerned, no significant differences between these two resttictors were observed for the three additives at 300 bar and 50°C. When the same experiments were repeated at 300 bar and 80°C, the amounts of Irgafos 168 and Irganox 1076 extracted in the first 30 minutes at lower flow rates were reduced considerably in comparison with that

obtained at the higher flow rate. Opposed to this, under these conditions the extraction rate for Irganox 1010 did notchange noticeably when the restrictor was replaced (table 5.3). At prolonged extraction times, the difference in extraction yields of Irgafos 168 and Irganox 1076 between these two restrietars also disapeared. The explanation for these observations is that at 50°C ditfusion of the solutes in the polymer is relatively slow, whereas the density of the supercritical fluid is fairly high. Components diffused to the surface can easily be dissolved and eluted out of the extraction cell by thè high density carbon dioxide. Under these conditions ditfusion in the polymer partiele is the rate-lirniting parameter. Therefore, the recovery doesnotchange with the different supercritical fluid flow rates tested. A distinctly different situation occurs at 80°C where ditfusion of the solutes in the polymer is relatively fast and components can rapidly diffuse to the polymer surface. If now low flow rates are used, the ability of the extractant to dissolve the solutes and remove them from the extraction cell can become the rate-limiting parameter. This is most likely to occur for components that are present at high concentrations or have high ditfusion coefficients in the polymer. In our experiments, the concentrations of lrgafos 168 and Irganox 1076 in polyethylene are much higher than the concentration of lrganox 1010. Moreover, lrgafos 168 and Irganox 1076 have smaller molecular sizes and hence have higher ditfusion coefficients. These results indicates that if the rate-lirniting parameter is solubility, high supercritical fluid flow rates may be of benefit Unfonunately, high supercritical fluid flow rates will make the colleerion of the extracted components from the expanding gas stream more difficult. For this reason, in the following experiments a 50 J.UI1 fused-silica restrictor with a length of 50 cm was used as the resttictor.
Table 5.3. Effects of supercritical fluid flowrateon the SPE extraction recoveries of the three
additives in the frrst 30 minute fraction at 80"C and 300 bar"l.
Resttictor
50 11m x 50cm
20 11m x 50cm
Flow ratebl
350 mi/min
70 milmin
Irgafos 168
86.1%
71.5%
Irganox 1076 Irganox 1010
82.5% 100.4%
68.9% 97.4%
a> SFE extraction reeoverles were calculated relative to Soxhlet data; hl measured as gas flow after expansion.
5.1.4.5 Effects ofbenzene as a nwdifier In actdition to the parameters discussed above, the actdition of a modifier is another important factor affecting the SFE extraction process. In the present

5.1 SFE o[polymer additives from polyetlrYlene 97
smdy, the influence of modifiers on the SFE kinetics was investigated using benzene as the modifier. Benzene was selected because this solvent is knÓwn to
be a good swelling agent for polyethylene [16]. Figs. 5.7A and B illustrate the effects of the modifier on the extraction for Irgafos 168 at 300 bar and 50°C, and
300 bar and 80°C, respectively. Similar curves were also found for Irganox 1076 and Irganox 1010. The modifier was spiked onto the polymer sample prior to starting the SFE extraction. With increased modifier amounts added, the extraction yields for each of the three additives increased both at 50°C as well as at 80°C. The magnitude of the increase at 300 bar and 50°C was found to be larger than that at 300 bar and 80°C. The overall extraction yields at 50°C and
300 bar with 0.5 ml benzene as the modifier, however, are significant lower than that obtained at 80°C and 300 bar with pure carbon dioxide. Modifier amounts larger than 0.5 ml were not investigated because under these conditions the resttictor was found to block frequently presumably because a significant fraction of the low molecular weight polymer materials was co-extracted with the additives. As has been discussed in previous paragraphs, the rate-limiting parameter for extraction at an extraction pressure of 300 bar and extraction temperatures between 50°C and 80°C is diffusion of the solutes trom the core of the polymer particles to the surface. The impravement of the extraction yields observed by the addition of the modifier must hence be due to the fact that the ditfusion rates of the solutes increase when the polymer particles swell by the uptake of modifier. The smaller effects of the modifier addition at higher temperatures is most likely due to the fact that ditfusion of solutes in the polymer is already relatively fast at higher temperatures. Therefore, modifiers are more effective at lower extraction temperatures.
~~gg (A) IÖ' 800 ·······- ········ ····-··· ....... .
~ 700 + +-+-
1 ~gg ;==g==~= - 400 ~ 300 - 200
100 v~o~~30---6~0---9~0--~t20
time(min)
-+- SOOpl -e.- 200pl
-C.- lOOp! -Q- lOOp!
-Q- Opl -+- 20"1
... ... soxhlet -À- Otû
time(min)
Figure 5. 7. Effects of modifier amount on SFE extraction rate for Irgafos 168. A: 50"C and 300 bar; B: 80°C and 300 bar. Otherdetails as in tigure 5.3.

Chaoter5 98
Modifiers can not only swell rhe polymer matrix but also increase rhe supercritical fluid density and its polarity. If rhe solubility of rhe components in the supercritical fluid is not the rate-limiting parameter, rhe impravement of rhe
tooo.--------rr--n 900 ·······- ······-- ····---- ---~~!.
~ 800 n 1oo ~~~· .§ 600 __ +-:::;;;;;;;c-
~ ~g:;+~~ [ 300 0/ c. 200-
100 (JF--..1..----"------l..-.....J ..,.0 30 60 90 120
timc(tnin)
zoo.--------...,
~ ................ ········ .... ~9
~ /+-~ 100 ~
~ . .1.-A-A-
\)10 30 60 90 120
time (lnin)
7oo.------(-;;:B;:;:-,) ~ 600 ................ ········ ....... . g 500. tJ
.§400 ~~
~300r---+~ ~200
100
1}0 30 60 90 120
time (min)
-f-. SOOJil
-6- JOOpl
-o-o111
•• •• soxhlet
Figure 5:8. Effects of modifier amount on SFE extraction rate at 80°C and 150 bar. A~ Irgafos 168; B: Irganox 1076; C Irganox lOlO.Otberdetaïls as in tigure 5.3.
SFE recovery upon the addition of a modifier is mainly due to swelling of the polymer and enhanced. diffusion in the swollen polymer. The increase in rhe solvent strengtil of the supercritical fiuid is, in this case, only of minor importance. If the solubility of the components in the supercritical fluid is the rate-limiting parameter, i.e. at high temperamees and low pressures, the improved solvent strengtil of the supercritical fluid obtained by the addition of a modifier will increase the SFE extraction rate. Figure 5. 8 shows the effect of the modifier amount added to the extraction cell on the SFE extraction yields at 150 bar and
80°C. Under these conditions, the extraction rate-limiting parameter when pure carbon dioxide is used is the solubility of the components in the supercritical fluid. It can be seen from this figure that the amount extracted in the fust fraction of 30 minutes increased considerably upon the introduetion of the modifier. In particular this is true for Irganox 1010, which has a very low solubility in low

5.1 SFE o(polymer additives (rom polyethylene 99
density carbon dioxide. It can also be seen that for this solute relatively high modifier concentration are required. Here again the results for lrgafos 168 and lrganox 1076 are very much similar. Figure 5.8 also shows, as expected, that the degree of increase is larger at higher modifier concentrations. However, even with 0.5 mi benzene as modifier, the amount extracted at 80°C and 150 bar in the first 30 minutes is still less than that extracted at 80°C and 300 bar with pure carbon dioxide. Apparently, pure carbon dioxide at 80°C and 300 bar is a stronger solvent than carbon dioxide admixed with 0.5 m1 benzene at 80°C and 150bar.
Table 5.4. Effects of static time on SFE extraction recoveries in the frrst 30 minute fraction at 50"C and 300 bar">
without modifier with 0.5 mi benzene as modifier
static time 5 minutes 30 minutes 5 minutes 30 minutes
Irgafos 168 52.5% 54.9% 58.9% 75.1%
Irganox 1076 41.9% 46.4% 50.9% 70.6%
Irganox 1010 17.9% 21.2% 32.8% 47.0% •> SFE extraction recoveries were calculated relative to Soxhlet data.
Table 5.5. Effects of static time on SFE extraction reeoverles in the frrst 30 minute fraction at sooc and 300 bar. a)
without modifier with 0.5 mi bezene as modifier
static time 5 minutes 30 minutes 5 minutes 30 minutes
Irafos 168 82.2% 86.1% 92.9% 98.7%
Irganox 1076 79.7% 82.5% 89.0% 97.9%
Irganox 1010 90.5% 100.4% 128.6% 158.8% •l SFE extraction recoveries were calculated relative to Soxhlet data.
Another important factor affecting the SFB efficiency when a modifier is used is the static time as swelling of polymers is normally a slow process. The effects of the static time on the SFB extraction yields were studied at 300 bar and 50°C, as well as at 300 bar and 80°C using bath pure carbon dioxide and carbon dioxide modified with benzene. The results are listed in Tables 5.4 and 5.5, respectively. Variation of the static time from 5 minutes to 30 minutes was found to have only a margillal effect on the extraction yields when pure carbon dioxide was used. When a modifier is used, the polymer swells slowly thereby increasing ditfusion in the polymer. Now much larger amounts can diffuse into the supercritical tluid

Chapter 5 100
at langer static times. The magnirude of the static time effect was found to be larger at lower temperatures because at higher temperatures the polymer is already relatively flexible and ditfusion is hence relatively fast.
In the experiments described above, the modifier was directly spik:ed onto the solid sample in the extraction cell. During the extraction process, the modifier will continousely be removed from the cell by the flow of supercritical fluid. It is evident that the length of time that the modifier remains in the extraction cell is of importance for the extraction process. A series of experiments was performed to investigate how fast the modifier is removed from the extraction cell. The elution profiles of the modifier were tested by adding 0.5 mL benzene to the extraction cell packed with a polymer sample already extracted by SPE and monitoring the signal obtained on an FID connected to the outlet of the extraction cell. When helium at 150 bar was used as an inert transporting medium, benzene is retained in the extraction cell for about 60 min at 80°C, and about 90 min at 50°C. From this results, it is plausible to say that the modifier can penetrate into the polymer or can be adsorbed by the polymer. When supercritical carbon dioxide was pumped through the extraction cell, the modifier was eluted completely within 10 minutes under all experimental conditions tested. Apparently the modifier is rapidly extracted by the supercritical carbon dioxide. The rapid elution of modifier also explains why only the amount extracted in the first 30 minute fraction was increased when a modifier was added. The elution profiles of benzene at 80°C and 150 bar with heliwn and carbon dioxide are shown in tigure 5.9.
(A)
0 10 20 30 40 50 60 70 80 90
time(min)
0
~ 10
(B)
20 30 40 50 60
time(tnin)
Figure 5.9. Blution profile of the modifier with different transporting media at various conditions. A: Helium, SOOC and 150 bar; B: Carbon dioxide, 80°C and 150 bar; (A static time of 30 minutes was used prior to the elution.)

5.1 SFE o[polymer additives from polyethylene 101
From the results shown above, it can be seen that the main effects of modifier in the SFE extraction of polymerie matenals are related to increasing the diffusion coefficients of the solutes in the polymer, presumably due to swelling of the polymer particles, and to the improverneut of the solvent strength of the supercritical fluid. In the extraction of environmental samples, the modifier is used mainly for the deactivation of active sites on the surface and only small amounts of modifier can have a drastic effect on the extraction process [17]. As discussed above, a much larger amount of modifier is needed in the extraction of polymerie materials. This is especially the case at low temperatures where the modifier mechanism actually involves swelling of the polymer resulting in increased ditfusion coeffficients for the solutes.
In order to investigate the modifier role in the SFE extraction in more detail, the polymer additives were extracted with modifier using either pure helium (150 bar and 80°C) or pure carbon dioxide (70 bar and 80°C) as the carrier medium. No extraction accured under these conditions without modifier. It is interesting to see that the amount extracted at 80°C and 150 bar with carbon dioxide actmixed with 0.5 mi benzene is much larger than the sum of the amounts extracted under the same conditions without a modifier and the amount extracted with 0.5 ml benzene using either helium (150 bar and 80°C) or carbon dioxide (70 bar and 80°C) as the carrier medium (table 5.6). From this it can be concluded that the extraction rate is not simply the actdition of the extraction rates with only modifier and pure carbon dioxide.
Table 5.6. Comparison of extraction yields (ppm) in the frrst 30 min fraction at 80°C.
C02(150bar) + C02(150 bar) COz(70bar) + He(150bar) + 0.5 ml C6H6 0.5 mi 0.5 mi
lrgafos 168 499.3 233.3 45.1 66.6
lrganox 1076 266.5 162.0 9.0 12.2
Irganox 1010 136.1 1.7 3.2 4.2
Keferences
1 F.W. Billmeyer, Textbook of Polymer Science, John Wiley & Sons, Inc., 1984, p.3. 2 V. Camel, A. Tambute and M. Caude, J. Chromatogr., 642 (1993) 263. 3 D.R. Gere, C.R. Knipe, P. Castelli, J. Hendrich, L.G. Randall, H. Schulenberg-Schell, R.
Schuster, L. Doherty, J. Orolin and H.B. Lee, J. Chromatogr. Sci., 31 (1993) 246. 4 X. Lou, H.-G. lanssen and C.A. Cramers, J. High Resol. Chromatogr., 16 (1993) 425.

5 R.J. Houben, H.-G.M. Janssen, P.A. Leclercq, J.A. Rijks and C.A. Cramers, J. High Resol. Olromatogr, 13 (1990) 669.
6 M.L. Lee and K.E. Markides(eds), Analytica/ Supercritical Fluid Olromatography and Extraction, Chromatography Conference, Provo, Ut, 1990.
7 K.D. BartJe, T. Boddington, A.A. Clifford and N.J. Cotton, Anal. Olem. 63 (1991) 2371. 8 T.L. Chester, J.D. Pinkston and D.E. Raynle, Anal. Olem., 66 (1994) 106R. 9 K.D. BartJe, A.A. Clifford, S.B. Hawthorne, J.J. LangenfeJd, D.J. Milier and R.J. Robinson,
J. Supercrit. Fluids, 3 (1990) 143. 10 N.J. Cotton, K.D. BartJe, A.A. Clifford and C.J. Dowle, J. Olromatogr. Sci., 31 (1993) 157. 11 S. Kueppers, Olromatographia, 33 (1992) 434. 12 A. Venema, H.J.M.F. van de Ven, F. David and P. Sandra, J. High Resol. Olromatogr, 16
(1993) 552. 13 W.G. Whitman, Olem. andMet. Eng. 29 (1923) 147. 14 I. Goodman and B.F. Nesbit, J. Polym. Sci., 48 (1960) 423. 15 J.S. Verentas, J. Appl. Poly. Sci., 25 (1980) 1297. 16 R.A. Orwoll, in H.F. Mark, N.M. Bikales, C.G. Overberger, G. Menges and JJ. Kroschwitz
(eds), Encyclopaedia of Polymer Science and Engineering, John wiley & Sons, Inc., Vol. 15, p.231.
17 J.J. Langenfeld, S.B. Hawthorne, D.J. Milier and J. Pawliszyn, Anal. Olem., 66 (1994) 909.

Chapter5 103
5.2 Effects of Modifier Addition and Temperature Variation in the Supercritical Fluid Extraction of Polymerie MaterialsS>
ABSTRACT
The effects of modifier actdition and temperature variation on the supercritical fluid extraction (SFE) of nylon-6 and poly (1 ,4-butylene terephthalate) (PBT) samples are discussed. The modifiers studied here include hexane, chloroform, methanol and benzene. The influence of experimental parameters, such as extraction temperature and pressure, static time and supercritical fluid flow rate, are investigated systematically both with pure and with modified carbon dioxide. The actdition of a suitable modifier results in significantly enhanced extraction rates, in particular at low extraction temperatures. A simple experimental set-up for the introduetion of additional volumes of modifier during the extraction was developed and evaluated. Guidelines for the optimization of the SFE extraction of polymerie samples are given.
5.2.1. INTRODUCTION
Supercritical fluid extraction (SFE) has been recognized as an important alternative to extractions using liquid solvents [1-3]. The most widely used supercritical fluid in SFE is carbon dioxide because of its reasonable critica! properties, low toxicity, chemica! inertness, acceptable price and the ability to dissolve numerous compounds ranging in polarity from non-polar to moderately po lar. However, supercritical carbon dioxide does not have sufticient solvent strength for the extraction of polar analytes and it is a poor fluid to overcome the interaction between analytes and matrix.
The extracting ability of carbon dioxide can be greatly enhanced by using modifiers [4]. The effects of modifiers on SFE extraction efficiency have been the subject of a number of investigations. Numerous compounds, ranging from nonreactive modifiers, such as methanol, water, dichloromethane, organic amines and acids etc. to reactive modifiers, such as ion pairing agents and derivatizing agents, have been used as modifiers in analytica! SFE of mainly environmental samples [5-13]. Unfortunately, however, the role ofmodifiers in analytica! SFE is
Sl X. Lou, H.-G. Janssen and C.A. Cramers, J. Chromatogr. Sci., 34 (1996) 282.

104 5.2. Effects of modifier and temperature in SFE o{polymer
still not well understood. The selection of modifiers and their concentrations is still highly empirical. This is especially the case in the SFE extraction of polymerie samples where only very few applications of modifiers have been reported [14, 15].
SFE has been increasingly used for polymer applications, Bartie et al. derived a model for diffusion limited extractions assuming that the matrix particles are spheres of a well-defined size and the initia! distribution of the solutes within the spheres is uniform [16]. Cotton et al. [17] and Keuppers et al. [18] investigated temperature effects in SFE and found that higher extraction efficiency could be obtained at elevated temperatures. Via et al. [19] fractioned low molecular weight, high density polyethylene wax at different temperatures with three different supercritical fluids. They found that higher temperatures at constant density yielded extracts containing higher molecular weights. Similar work on fractioning of ethylene-methylacrylate copolymers using SFE was reported by Pratt et al. [20] .
Optimization of SFE methods using modified fluids frequently requires testing various modifiers at different concentrations as well as determining optima! temperature and pressure conditions. In the extraction of polymerie materials, the extraction pressure is relatively simple to select. High extraction pressures are generally advantageous. However, selection of optimal temperature conditions is quite complicated. Increasing the extraction temperature will, on the one hand, decrease the density of the supercritical fluid, while on the other hand it can also improve the kinetics of mass transfer of the analyte from the matrix to the supercritical extractant and increase vapour pressure of the analyte. Very often, the effects of a temperature variation will also depend on the extraction pressure and the properties of the analytes and matrix [14]. Temperature effects get even more complicated when modifiers are used. Yang et al. tested the combined effects of modifier actdition and temperature variation for the extraction of polycyclic aromatic hydrocarbons (PAHs) from environmental samples [21]. They found that for these samples the combination of a suitable modifier and elevated temperature was most effective. Unfortunately, for the extraction of polymerie materials, modifier effects and the combined influence of modifier and temperature till now have not been investigated in detail.
In this report, four modifiers with different chemical properties were tested at various temperatures in the extraction of two polymerie materials, nylon-6 and

Chapter5 105
poly (1 ,4-butylene terephthalate) (PBT). The effects of temperature when either pure or modified carbon dioxide was used are compared. The roles of modifier actdition and temperature variation in the extraction of polymerie samples are discussed. A simpte experimental set-up for the introduetion of additional volumes of modifier during the SPE extraction was proposed and evaluated.
5.2.2. THEORY
In a previous publication we applied the two-film theory for resistance to mass transfer to qualitatively describe the kinetics of extraction in SFB [14]. 1n that work, it was shown that the rate-limiting step for the SFB extraction of polymer additives from polymerie samples was either ditfusion in the polymer particles or transport of the extracted components out of the extraction cell. These two steps are related with two parameters, ditfusion and solubility, respectively. Which of these two parameters actually governs the extraction kinetics can be investigated by studying the influence of pressure or temperature on the extraction rate. If the rate-limiting parameter is solubility of the components in the supercritical fluid, an increase in pressure will increase the extraction rate. If, on the other hand, the rate-limiting parameter is ditfusion of the analytes in the polymer particles, extraction pressure will have little or no effect on the SPE extraction rate [14]. Apart from pressure, alsoother parameters affect the kinetics in SFB extractions. Among these, temperature, and type and concentration of the modifier appear to be the most important ones.
The strategy for optimizing the extraction conditions for the extraction of polymerie materials depends on which step in the extraction is the rate-limiting step. If the rate-limiting step is ditfusion of the analytes in the polymer particles, the extraction rate can be increased by increasing the extraction temperature as this results in increased ditfusion in the polymer particles. The actdition of a suitable modifier that swells the polymer is another method for enhancing ditfusion in the polymer particles. Evidently, the extraction rate can also be increased by decreasing the partiele size of the polymer. If the rate-limiting parameter for extraction is the solubility of the analytes in the supercritical fluid, extraction rates can be increased by increasing the solvent strength of the supercritical extractant. One way to achleve this is by introducing modifiers. Moreover, the extraction rate can also be increased by increasing the extraction pressure, or by increasing the supercritical fluid flow rate. A general strategy for

106 5.2. Effects of modifier anti temperature in SFE o{polymer
optimizing experimental parameters in the extraction of polymerie matcrials has already be shown in figure 2.4. As mentioned above, pressure effects are relatively easy to understand. Experimental pressure should be selected sufficiently high. However, the selection of temperature and modifier type and concentration is much more complicated because these parameters are highly matrix- as wellas analyte-dependent and their effects on extraction are very often interrelated.
5.2.3. EXPERIMENTAL
The polymer samples used in this study, nylon-6 {glass-transition temperature, Tg 62.5°C) and poly(l ,4-butylene terephthalate) (PBn (Tg = 66°C) were
purchased from Aldrich (Milwaukee, Wisconsin, USA). It should be emphasized here that Tg values under supercritical carbon dioxide conditions can be significantly different from the data specified above. The polymer samples were ground by milling under liquid nitrogen prior to extraction.
SPE experiments were performed with a modified Carlo Erba SFC 3000 capillary SFC instrument (Carlo Erba, Milan, Italy). A 3 ml stainless steel cell (Suprex, Pittsburgh, PA) was fitted with hand-tight connectors (Suprex) for easy installation. Stainless steel trits (3 ~m) were located at either end of the extraction cell. Fused-silica capillaries (20 IJ.m i.d. or 50 IJ.m i.d.) were used as restrictors. To enable static extraction an on-off valve (Valco, Schenkon, Switzerland) was installed directly behind the extraction cell. The extracted material was colleeled by inserting the restrictor outlet into a glass vial (10 cm x 1 cm i.d.) containing 5 mL dichloromethane. This vial was changed every 30 minutes and an internat standard was added. Eicosane and tetracontane were selected as the internat standards for the extracts from nylon-6 and PBT respectively. After collection, dichloromethane was evaporated under a gentie flow of nitrogen and the extracted material was redissolved in a suitable amount of chloroform. Quantitative trapping in the liquid coneetion trap was confirmed by spiking known amounts of the analytes onto clean sand and extract them for 30 minutes (5 min satatic + 30 min dynamic) with pure carbon dioxide at l50°C and 300 bar and at a supercritical fluid flow rate of 550 mL!min {gas flow measured at ambient conditions). The trapping efficiencies obtained this way were never lower than 92%.
For SPE extractions, about 1.9 g nylon-6 or 1.6 g PBT were weighed into the

Chapter5 107
extraction cell. A static time of 20 minutes was used prior to dynamic extraction. The highest extraction temperature tested for both polymers was 1700C. At higher temperatures the restcictor often blocked due to partial melting of the polymers. The carbon dioxide used in the experiments had a purity of 99.996% (Intermar B.V. Breda, The Netherlands). Soxhlet extractions were performed to produce frames of reference. SFE extraction efficiencies were calcu1ated relative to the Soxhlet data. For Soxhlet extracrions of both polymers, about 2 g samples were placed in the Soxhlet extractor and extracted for 36 hours. Methanol and chloroform were used as the Soxhlet extraction solvent for nylon-6 and PBT respectively [22].
ln order to introduce additional modifiers during SFE extraction, an LC injection valve (CH 6214, Valco) with a sample loop of 0.1 mL was installed before the extraction cell. The experimentalset-up is shown schematically in tigure 5.10 .
. . Microcomputer r u u j
Figure 5.10. Experimental set-up for SFE.
The extracted components were analyzed using a gas chromatograph equipped with an on-column injector and an FID (GC 8000 series, Carlo Erba Instruments). For the analysis of the extracts from nylon-6, a DB-1 column (15 m x 0.32 mm i.d., film thickness 1 j.Lm) purchased from J&W (Folsom, CA. USA) was used. The initial temperature for analysis was 40°C. Temperature wasthen programmed to 250°C at 20°C/min. A HT-SIMDIST CB column (10 m x 0.53 mm i.d., film thickness 0.17 j.Lm) purchased from Chrompack (Middelburg, The

108 5.2. Effects of modifier and temperature in SFE q{polymer
Netherlands) was used for the analysis of the extracts from PBT. The initial temperature for the GC separation was 40°C. Temperature wasthen programmed to 400°C at 20°C/min.
Many of the extracrions were repeated three times. Por the repeated experiments, the relative standard deviation was within 5% with pure carbon dioxide, and within 9% when modifiers are used.
5.2.4. RESUL TS AND DISCUSSION
5.2.4.1. Effects of temperaJure using pure carbon dioxide In the extraction of polymerie materials the extraction rate is controlled either by the rate of ditfusion of the analytes in the polymer or by the solubilities of the analytes in the supercritical fluid. When pressure was increased from 150 bar to 300 bar at 50°C, no considerable variatien in the extraction yields of the analytes from the polymers studied was observed. This is because at 50°C the density of the supercritical fluid is relatively high, even at a mild pressure of only 150 bar (p= 0.701 g/ml). As ditfusion of the components in the polymer is relatively slow at this low temperature, the mass flow of components ditfusing to the polymer surface is low. Molecules diffused to the surface are rapidly transferred to the supercritical fluid and carried out by the flow of high density carbon dioxide. Under these conditions the extraction rateis determined by ditfusion of the analytes in the polymer particles.
As discussed in the THEORY section, ditfusion coefficients of analytes in polymer particles can be increased either by increasing the extraction temperature or by the actdition of a suitable modifier. To investigate the influence of temperature, nylon-6 and PBT were extracted at different temperatures at a pressure of 300 bar. The results are illustrated in tigure 5 .11. The highest temperature tested for both polymers is 170°C. At higher temperatures frequent blocking of the restrictor occurred due to partial melting of the polymers. Por nylon-6, the extraction efficiency of caprolactam increased considerably when the temperature was raised from 50°C to 170°C at 300 bar (figure 5.11). No abrupt changes in extraction recoveries for both polymers were observed around their normal glass-transition temperatures, suggesting that the Tg values under supercritical carbon dioxide conditions are lower than the normal Tg values.

Chapter5 109
4000 recove -+-caprolactam, 30 minutes
3200 -+-caprolactam, 2 hours
-A- dimer, 30 minutes
-•- dimer, 2 hours
-o- trimer, 30 minutes
.... trimer, 2 hours
FigUre 5.11. Effects of temperature on the SFE extraction. Extractant: pure carbon dioxide; extraCtion pressure: 300 bar; static time: 20 minures; dynamic extraction time: 30 minutes or 2
hours; restrictor: 50 IJ.m x 70 cm fused-silica capillary; colleerion solvent: 5 mi dichloromethane.
It can be seen from tigure 5.11 that the effects of temperarure in the SFE . extraction of polymerie matenals are both matrix and analyte dependent. It appears that the effects of temperature on the SFE extraction of PBT are more complicated than on the extraction of nylon-6. In contrast to the monotonons increase in yield for the extraction of caprolactam from nylon-6, the total extraction recovery for the dimer obtained in two hours first increased considerably when the temperature was raised from 50°C to 150°C and then decreased at temperarures exceeding 150°C. Apparently, at low temperatures the rate-limiting parameter is diffusion of analytes in the polymer particles. At elevated temperatures diffusion will be enhanced. However, at the same time the density of the supercrideal fluid will decrease. Above a certain temperature, diffusion of the analytes in the polymer particles is relatively fast while at the same time the density of the supercrideal fluid is low. Now solubility of the components in the supercrideal fluid becomes the rate-limiting parameter. Solute molecules diffused to the surface can no langer be rapidly eluted by the flow of the low density extractant. Increasing temperature at constant pressure will further decrease the solubility of the relatively non-volatile oligomers in the supercrideal extractant, thus decreasing the SFE extraction rate. The temperarure where this occurs depends on the properties of both the analyte and the matrix, as well as on the extraction pressure. Similar effects of temperature were also observed for the extraction of the trimer from PBT. Here the temperature where the highest recovery was obtained in two hours was l10°C, which was significantly lower

110 5.2. Effects of modifier and temperature in SFE o{polymer
than that for the dimer (150°C). In the results discussed above extraction recoveries in two hours were compared at different temperatures. The highest yield was obtained at 150°C for the dimer and at ll0°C for the less soluble trim er. It is also interesting to compare the total extraction recoveries in two hours with the recoveries in the first half hour fraction. The highest extraction recovery in the first half hour was obtained at l10°C for the dimer and at 90°C for the trimer. Por both dimer and trimer, the temperatures where the highest recoveries could be obtained in the first half hour fractions were significantly lower than those in the total two hours. Apparently the influence of temperature on the extraction recovery can also be a function of analyte concentration. In the beginning of the extraction, the concentration of the analytes on the surface of the polymer particles is relatively high. Hence the solubility in the supercritical fluid is more likely to determine the extraction rate. During the extraction the concentration of the analytes gradually decreases. At lower concentrations the solubility of the analytes in the extractant becomes less critica!. In actdition to temperature, the extraction rates can also be greatly influenced by the actdition of modifiers as will be demonstrated below.
5.2.4.2. Effects ofmodifiers
5.2.4.2.1. /njluences of modifier identity and extraction temperature Apart from temperature, also the use of a modifier can result in a faster SFE extraction of polymer samples. The effects of different modifiers on the extraction efficiencies in the first 30 min fraction observed for the extraction of caprolactam from nylon-6 and the dimer and trimer from PBT at different temperatures are shown in figures 5.12 and 5.13, respectively. Figure 5.14 shows the effects of modifier actdition on the total extraction yields in 2 hours. In these experiments 0.5 mL of modifier was spiked to the extraction cell prior to extraction. The extraction recoveries with pure carbon dioxide are also shown in the figures for comparison. The highest temperature tested with modifiers for both polymers was 150°C. At higher temperatures restrictor blocking was observed frequently. From these figures a number of interesting conclusions can be drawn: (a) The influence of modifiers on the SFE extraction of polymerie materials is
significantly different with different modifiers. (b) The effects of modifiers in the extraction of polymerie materials are
strongly matrix dependent Among the four organic solvents tested, methanol is the most effective modifier for the extraction of nylon-6, while

Chapter5 111
chloroform is to be preterred for the extraction of PBT.
2000recove
-+- C02 + CHpH
-t:..- C02 + CHCl3
-o- co2 + C6H6
-'il- co a+ c6HI4
70 90 110 130 150 -<>-co
' 2
Figure 5.12. Modifier effects in the SFE extraction of caprolactam from nylon-6 as a function of temperature in the frrst half hour fraction. Extraction pressure: 300 bar; modifier amount: 0.5 roL; static time: 20 minutes; dynamic time: 30 minutes; restrictor: 50 j.Lm x 70 cm fused-silica capillary; collection solvent: 5 mL dichloromethane.
4000 recove
3200 (A) ----------2400 ·~ "
1000recov
-+- C02 + CHC!1
-t:J.- C02 + C
6H
6
-o- C02 + CHpH
-'il- col+ C6HI4
-<>- co2 70 90 110 130 150
("C)
Flgure 5.13. Modifier effects in the SFE extraction of the dimer and trimer from PBT as a function of temperature in the frrst half hour fraction. (A) dimer; (B) trimer. Other details as in figure 5.12.
(c) Modifiers are more effective at low temperatures. (d) No abrupt changes in extraction efficiencies were observed around the
normal glass-transition temperatures of the polymers irrespective of which modifier was used. Modifiers are still very effective at temperatures below the normal polymer glass-transition temperature.
(e) The influence of temperature on SFE extraction is different when different modifiers are used.
(f) At high temperatures modifiers can show negative effects on the extraction efficiency in the fractions after the first half hour. In the first half hour

112 5.2. Effects of modifier and temperature in SFE o[polymer
fraction at 150°C and 300 bar, slight incr:eases in the extraction recovery for caprolactam from nylon-6 as wellas for the dimer from PBT, and a larger increase for the less soluble triroer from PBT were observed when modifiers were added. However, the amounts extracted in later fracrions for all analytes were considerably lower than the amounts extracted in the corresponding fractions with pure carbon dioxide. This is especially the case in the extraction of the dimer from PBT, where the extraction recovery in two hours with a modifier was significantly lower than that with pure carbon dioxide.
The last phenomenon is very surprising as modifiers are generally expected to always increase the extraction efficiency in SFE, as reported for the extraction of environmental samples [1, 2, 23]. A possible reason for this is that when the modifier is "extracted" from the swollen polymer parricles, the particles will shrink and changes in the structure of the polymer particles may occur resulting in a more "closed" structure. These changes in structure can considerably slow down ditfusion of the analytes out of the polymer particles. This occurs especially at high temperatures and when good swelling agents are used as modifier.
From the results discussed above, it is clear that both optimization of temperature as well as of modifier identity are important in the extraction of polymerie materials. For the extraction of caprolactam from nylon-6 and the dimer from PBT, the highest extraction yields that can be obtained by the varlarion of extraction temperature or by the actdition of a modifier are of a similar magnitude. However, for the less soluble triroer, the actdition of a modifier is more effective than the varlation of temperature (figure 5.14).
4000 recove m
·-•-~t-<-3200 ~·/ /--- ~ 2400 /> 1600 --r+-:::::::*==-80 ~·::::::;;•....:::::::·-·= -o-o-0 _
0 70 90 110 130 150
coq
-+- caprolactam/C02
-+- caprolactarn/C02 + CH30H
-6.- dimer/C02
-•- dimer/C02 + CHCI3
-o- trimer/C02
-e- trimer/C02 + CHC13
Figure 5.14. Modifier effects on the SFE extraction yields. Dynamic extraction time: two hours. Other conditions as in figure 5.12.

Chapter5 113
5.2.4.2.2. Effects of modifier at dijfusion- and solubility-limiting conditions
5000 recove ( m)
(A 4000
3000
2000
1000
00 30 60 90 120
time( min)
(B) -+- 100 bar/CO,
-+- 200 bar/CO, + CHCJ,
-A- 25ft bar/C01
-•- 25ft bar/CO,+ CHCJ,
-o- 300 bar/CO,
0 30 60 90 120 ..... 300 bar/CO,+ CHCJ,
time( min)
Figure 5.15. Effects of pressure on SFE extraction of the dimer and trimer from PBT at 150°C using 0.5 mL chloroform as the modifier. (A) dimer; (B) trimer. Static time: 20 minutes, restrictor: 50 1-1m x 70 cm fused-silica capillary; collection solvent: 0.5 mi dichloromethane.
3000 recov
2000-
1000
30 60
time( min)
90
-+- 100 bar/C02
-+- 100 bar/C02 + CHpH
-e.- 200 bar/C02
_...__ 200 bar/C02 + CHpH
-o- 300 bar/C02
120 -e-- 300 bar/C02 + CHpH
Figure 5.16. Effects of pressure on SFE extraction of caprolactam from nylon-6 at 150°C using 0.5 mL methanol as the modifier. Other details as in figure 5.15.
The effects of modifiers on the extraction rate can be different depending on which parameter governs the extraction rate, i.e. ditfusion or solubility. In order to investigate the modifier effects in more detail, polymer samples were extracted with 0.5 mL modifier (methanol for nylon-6 and chloroform for PBT) at different pressures and at temperatures of 50°C and 150°C. The results are shown in the figures 5.15-5.18. At conditions where solubility is the rate limiting parameter, e.g. at 150°C with pure carbon dioxide, the extraction reeoverles for all analytes increased significantly at increased pressures (figures 5.15 and 5 .16). Besides pressure also modifiers can improve the density and polarity of the supercritical fluid and thus increase the solubility. In the extraction of PBT using 0.5 mL

114 5. 2. Effects of modifier and temperature in SFE ofpolymer
chloroform as modifier, the extraction recovery of the first half hour fraction for the diroer increased significantly at all pressures tested (figure 5.15A). Even greater increases in the extraction recovery were observed for the less soluble triroer (figure 5.15B). Opposedly, no significant increase in the extraction recovery in the first half hour fraction was observed for the extraction of caprolactam from nylon-6 when 0.5 mL methanol was added as the modifier (figure 5.16). Even more surprising is the observation that modifiers can also have negative effects at low pressures (100 and 200 bar) in the extraction of caprolactam from nylon-6. Similar negative effects of the modifier were also observed in the extraction of the diroer from PBT. From figures 5.15 and 5.16, it can also be seen that the extraction recovery found at 150°C using 0.5 mL methanol and 0.5 mL chloroform as modifiers increased significantly at increased pressures. This means that at this temperature the iroprovement of the solubility by the actdition of modifier directly into the extraction cell is not sufficient. The extraction rate is still limited by the solubility of the analytes in the supercritical fluid.
Also at conditions where diffusion in the polymer is the rate limiting parameter, the actdition of modifier can be very attractive. To investigate the effects of modifiers on diffusion in the polymer a series of extractions with modified carbon dioxide was performed at different pressures and a temperature of 50°C. As discussed before, no considerable variations in extraction recovery of the analytes from both polymers were observed at sooc when the extraction pressure was increased from 150 bar to 300 bar using pure carbon dioxide. This indicates that at this conditions the rate-limiting parameter is diffusion in the polymer. Suitable modifiers can swell the polymer thereby increasing diffusion of the analytes in the polymer. Much higher recoveries were observed for both polymers with modified carbon dioxide relative to those with pure carbon dioxide at all pressures tested. Figures 5.17 and 5.18 show the effects of pressure on SFE extraction of caprolactam from nylon-6 and the diroer from PBT. Similar effects were also observed for the triroer. With modified carbon dioxide, the extraction recoveries are already fhlrly high at 100 bar. Only slight increases of recoveries were observed when the extraction pressure was increased from 100 bar to 200 bar. A further increase in pressure gave no further improverneut in extraction recovery. This is probably because the density of the modified supercritical fluid is already high even at pressures as low as 100 bar. As the solubility of the components in the supercritical fluid is large, the rate-limiting parameter at 50°C with modified

Chapter5 115
carbon dioxide is ditfusion of the analytes in the polymer particles.
2000 Recove
-+- 300 bar/C02
-+- 100 bar/C02 + CHpH
-·- 200 bar/C02 + CH30H
0 30 60 90 120 -·- 300 bar/C02 + CH30H
time (min)
Figure 5.17. Effects of pressure on SFE extraction of caprolactam from nylon-6 at 50°C using 0.5 mL methanol as the modifier. Other details as in tigure 5.15.
~rec~o~v_e~ry~(~pp1m~1)~-------. 4000 .--
3000~-·-·-2000 +--+--+--
1000 +-+-+--
0 -0 30 60 90 120
time( min)
-+- 300 bar/C02
-+- 100 bar/C02 + CHC1
3
-·- 200 bar/C02 + CHC1
3
-·- 300 bar/C02
+ CHC13
Figure 5.18. Effects of pressure on SFE extraction of the dimer from PBT at 50°C using 0.5 mL chloroform as the modifier. Other details as in tigure 5.15.
Tö investigate the contribution of the modifier to the overall extraction, a series of experiments was performed in which the same modifier was used but the carbon dioxide was replaced by helium. When the experiments described above were repeated using helium (150 bar) as the carrier medium instead of supercritical carbon dioxide, only a small amount of caprolactam and no dimer or trimer could be extracted. It is clear from these results that the extracting ability of the modified supercritical fluid is not simply an actdition of that of the pure supercritical carbon dioxide and of the neat modifier.
5.2.4.2.3. lnjluence of modifier concentration, static time and supercritical jluid flow rate

ll6 5.2. Effects qfmod((ier and temperature in SFcc:'E~of'-'P::c:::O:.:;.lyc:.:m:.:.:e"-r ____ _
From the preceding paragraphs it can be seen that the effects of a modifier are: (a) swelling the polymer particles thereby enhancing ditfusion of analytes inside the particles; (b) increasing the solvent strength of the supercritical extractant. The relative extent of these two effects is not only determined by the modifier identity and its concentration but also by its residence time in the extraction cell (if modifiers are spiked onto the sample). The effects of different amounts of modifier on the extraction recoveries are listed in tables 5.7 and 5.8, respectively. These experiments were performed at 300 bar and temperatures of 50°C and 150°C. The largest amount of modifier added directly into the extraction cell was 0.8 mL. Larger amounts could not be accommodated by the polymer and leaked out of the extraction cell immediately. Expectedly, the extraction recoveries increased considerably at higher modifier concentrations.
Table 5.7. Effects of modifier amount on the SFE extraction yield in the fiTSt half hour
fractions at 50"C and 300 ba.r"l.
Modifer amount Nylon-6/Caprolactam PBT/Dimer PBT!frimer
(mL)
0 89.5 268.0 27.8
0.2 883.5 1238.9 264.5
0.5 1308.4 2629.3 683.1
0.8 1574.2 3577.2 1051.9
al Static time, 20 minutes; dynamic time: 30 minutes. •> extracted amount relative to the mass of polymer weighed into the extraction cell.
Table 5.8. Effects of modifier amount on the SFE extraction yield in the first half hour fraction at i50"C and 300 bar.">
Polymer/ Analyte
Modifier amount Nylon-6/Caprolactam PBT"l /Dimer PBT!frimer
(mL) (ppm)bl
0 1518.7 1110.2 47.4
0.2 1525.4 1319.0 84.8
0.5 1614.5 1977.2 484.9
0.8 NT"') 2793.8 1203.0
a) Static time, 20 minutes; dynamïc time, 30 minutes. bl extracted amount relative to the mass of polymer weighed into the extraction cell; c) not tested.

Chapter5 117
In our experiments modifiers were added directly into the extraction cell. The contact time of the modifier and the matrix depends on the static time, the modifier amount and identity, matrix properties, the extraction temperature and pressure as well as the supercritical fluid flow rate. At 50°C and 300 bar, the amounts extracted in the first half hour fraction increased continuously when the static time was increased from 10 minutes to 30 minutes (table 5.9). Opposed to this, no considerable changes in extraction recovery with different static times were observed at 150°C and 300 bar. At 50°C and 300 bar the effects of the modifier result mainly from swelling of the polymer. Swelling normally is a slow process which means that longer static times can be of benefit. On the other hand, the main effect of modifiers at 150°C and 300 bar is to increase the solvent strength of the supercritical fluid. Therefore, the extraction recovery at 150°C and 300 bar will not change considerably with increasing static time.
Table 5.9. Effects of static time on the SFE extraction yield in the frrst half hour fraction at 50°C and 300 bar.a)
Polymer/ Analyte
Static time Nylon-6 PBT/Dimer PBT/Trimer
(min} /Caprolactam (ppm) (ppm) (ppm)bJ
10 1264.2 2400.5 589.9
20 1308.4 2629.3 683.1
30 1433.6 2989.0 787.5
•> dynamic time, 30 minutes. bl: extracted amount relative to the mass of polymer weighed into the extraction een.
In the experiments described above a piece of 50 f..UU i.d. x 70 cm fused silica capillary was used as the flow resttictor. In order to investigate the effects of the supercritical fluid flow rate on the extraction recovery of the analytes, this restrictor was replaced by 20 IJ.m i.d. fused-silica capillaries with lengtbs of 70 cm and 20 cm, respectively. The effects of the fluid flow rates on the extraction reeoverles ofboth polymers at 50°C and 300 bar are listed in table 5.10. In these experiments 0.5 mL modifier was used. At 150oC the effects of fluid flow rate could not be tested because at these conditions the 20 IJ.m i.d. fused silica restrictor easily blocked. The extraction recoveries in the first half hour fraction

118 5.2. Effects of modifier and temperature in SFE o(polymer
increased considerably when the supercritical fluid flow rate was decreased from 550 mL/min (gas flow measured at ambient conditions) to 110 mL!min. However, no further increase in the extraction recovery was observed when the flow rate was further decreased from 110 mL/min to 35 mL/min. Nevertheless, the total extraction recovery obtained in two hours still increased slightly in this case. As the trapping efficiency even at the highest supercritical fluid flow rate tested is quantitative, the increased extraction recoveries must be due to the longer contact time of the modifier with the polymer matrix at lower supercritical fluid flow rates. It is clear from these data that the extraction recovery is a function of the extractant volume swept through the extraction cell, as well as of the contact time of the modifier with the matrix. Maximizing the contact time either by continuous addition of a modifier or by repeated spik:ing is advantageous as will be demonstraled below.
Table 5.10. Effects of supercritical fluid flowrateon the SFE extraction yield in the first half hour fraction at 50°C and 300 bar.•l
Polymer/Analyte
Flow rate Nylon-6 PBT/Dimer PBT!Trimer
(mL/min)bl /Caprolactam (ppm)c) (ppm) (ppm)
550 1308.4 2629.3 683.1
110 1420.2 2850.8 771.8
35 1429.6 2898.7 771.3
•l Static time, 20 minutes; dynamic time 30 minutes; modifier, methanol for nylon-6 and chloroform for PBT. b! gas flow; c) extracted amount relative to the mass of polymer weighed into the extraction cel!.
5.2.4.2.4. Introduetion of an additiofUll amount of modifier through an LC injection valve
From the results described above, it can be seen that the kinetics of SFE extraction of polymerie matenals are complex. Optimization of the extraction conditions should include, at least, the selection of the proper extraction temperature and modifier type and concentration. Modifiers are more effective at low temperatures and at higher concentrations. However, the modifier amount that can be added is limited if the modifiers are spiked directly into the extraction cell. In order to be able to introduce larger amounts of modifier, an LC if1iection valve was installed before the extraction cell. In this series of experiment, only the amounts extracted in the first half hour fractions were investigated. Flow

restrietion was provided by a piece of 20 IJ.m x 20 cm fused-silica. The results are listed in table 5.11. It can beseen from this table that if 0.8 mL of modifier is adcted ctirectly to the extraction cell prior to extraction foliowed by 2.0 mL introduced through the injection valve, the SPE extraction reeoverles of caprolactam from nylon-6 and the dimer from PBT in 30 minutes are already almost identical to those found for Soxhlet extraction with methanol or chloroform in 30 hours. However, the recovery for the less soluble trimer from PBT in 30 minutes is still considerably lower than that of Soxhlet extraction with chloroform in 30 hours. In the SPE extraction of less soluble and large solutes from polymer samples, continuous modifier actdition is likely to be more effective. This will be the subject of future investigations.
Table 5.11. Comparison of recoveries obtained in SFE and soxhlet extraction.
Extraction metbod
SFEa)
Soxhlet
Nylon-6/ Caprolactam (ppmt
1816.8
1827.8
Polymer/ Analyte PBT/Dimer
(ppm)
4810.8
4886.0
PBT!frimer
(ppm)
1484.7
2039.0
•> Extraction conditions, 50°C and 300 bar; static time, 20 minutes; dynamic time, 30 minutes: modifier, methanol for nylon-6 and chloroform for PBT. ~>> extracted amount relative to the mass of polymer weighed into the extraction cell.
5.2.4. CONCLUSIONS The extraction rate in SPE extraction of polymerie samples is govemed either by diffusion of the solutes in the polymer particles or by their solubility in the extraction fluid. If the rate-limiting parameter is diffusion of the analytes in the polymer particles increasing extraction temperature will increase the extraction rate. On the other hand, however, if the rate-limiting parameter is solubility of the analytes in the supercritical extractant, increasing temperature will have an actverse effect on extraction yielcts. In actdition to varlation of temperature, the actdition of modifiers can also strongly affect the SPE extraction rate. The results in this paper suggest that the role of modifier in the extraction of polymerie samples is twofold. A suitable modifier increases the solvent strength of the supercritical fluid anct swells the polymer particles resulting in enhanced ctiffusion in the polymer. Both modifier identity anct concentration were found to be important for increasing the extraction efficiency. Moctifiers are generally more

120 5.2. Effects of modifier and temperature in SFE o{potymer
effective at low temperatures. At high temperatures, modifiers may even sometimes have negative effects on the extraction efficiency. Also the contact time between the modifier and the matrix is of importance. Continuous modifier actdition or repeated spiking of a modifier may be advantageous if spiked modifiers are eluted out rapidly or if higher modifier concentrations are needed.
REFERENCES
1. S.B. Hawthome, Anal. Chem., 62 (1990) 633A. 2. T.L. Chester, J.D. Pinkston and D.E. Raynie, Anal. Olem., 66 (1994) 106R.
3. M.L. Lee and K.E. Markides (eds), Analytical Supercritical Fluid Chromatography and
Extraction, Cbromatography Conference, Provo, UT, 1990.
4. J.G.M. Janssen, P.J. Schoenmakers and C.A. Cramers, J. High Resolut. Chromatogr., 12 (1989) 645.
5. J.M. Levy, L. Dolata, R.M. Ravey, E. Storozynsky and H.A. Holowczak, J. High Resolut. Chromatogr., 16 (1993) 368.
6. T.M. Fahmy, M.E. Paulaitis, D.M. Johnson and M.E.P. McNally, Anal. Olem., 65 (1993)
1462.
7. J. Dankers, M. Groeneboom, L.H.A. Scholtis and C. van de Heiden, J. Chromatogr. 641
(1993) 375.
8. T.S. Oostdyk, R.L. Grob, J.L Snyder and M.E. McNally, J. Chromatogr. Sd., 31 (1993) 177.
9. J.J. Langenfeld, S.B. hawthorne, D.J. Milier and J. Pawliszyn, Anal. Chem., 66 (1994) 909.
10. Y. Lin and C.M. Wai, Anal. Chem., 66 (1994) 1971.
11. K.E. Laintz and E. Tachikawa, Anal. Chem., 66 (1994) 2190. 12. J.W. Hills, H.H. Hili, D.R. Hansen and S.G. Metcalf, J. Chromatogr., 679 (1994) 319.
13. B.E. Berg, E.M. Hansen, S. Gjorven and T. Greibrokk, J. High Resolut. Chromatogr., 16 (1993) 358.
14. X. Lou, H.-G. Janssen and C.A. Cramers, J. Microcol. Sep., 7 (1995) 303.
15. A. Venema, H.J.F.M. van de Ven, F. David and P. Sandra, J. High Resolut. Chromatogr., 16 (1993) 522.
16. K.D. Bartle, A.A. Clifford, S.B. Hawthorne, J.J. Langenfeld, DJ. Milier and R. Robinson, J. Supercriti. Fluids., 3 (1990) 143
17. N.J. Cotton, K.D. Bartle, A.A. Clifford and C.J. Dowle, J. Chromatogr. Sd., 31 (1993) 157. 18. St. Kueppers, Chromatographia, 33 (1992) 434.
19. J.C. Via, C.L. Braue and L.T. Taylor, Anal. Chem., 66 (1994) 603.
20. J.A. Pratt, S.H. Lee and M.A. Mchugh, J. Appl. Polym. Sci., 49 (1993) 953.
21. Y. Yang, A. Gharaibeh, S.B. Hawthome and D.J. MiJler, Anal. Chem., 67 (1995) 641.
22. R.A. Orwell, in H.F. Mark, N.M. Bikales, C.G. Overberger, G. Menges and J.I. Kroschwitz
(eds), Encyclopedia of Polymer Sdence and Engineering, John Wiley & Sons, Inc., 1989, Vol.
15, p.380. 23. M.E.P. McNally, Anal. Chem. 67 (1995) 308A.

Chapter5 121
5.3 A Fundamental Study of Parameters Affecting the Accelerated Solvent Extraction of Polymerie Samples6)
ABSTRACT
Accelerated solvent extraction (ASE) is applied for the extraction of monoroers and oligomers from polymerie samples. Two polymers, nylon-6 and poly(1,4-butylene terephthalate) (PBn areselectedas the model samples. The killetics of mass transfer in ASE of polymerie samples are discussed. The effects of various experimental parameters, such as temperature, pressure, static time, flow rate, etc, on the ASE extraction efficiency are investigated systematically. Furthermore, some general guidelines for the optimization in ASE extraction of polymerie samples are given. The extraction temperature and the type of solvent used are found to be the most important parameters affecting the ASE extraction efficiency of polymerie samples.
5.3.1. INTRODUCTION
Most of the samples that have to be analyzed by chromatography are too complex, too dilute, or in their original state incompatible with the chromatographic system. For these samples direct injection can not be used and sample pretreatment prior to injection of the sample into the chromatography system is required. Sample preparation methods generally used by analytica! chemists nowadays are both time and solvent consuming. According to a recent survey, two third of the analysis time in chromatographic analyses is devoted to sample preparation. Moreover, this step accounts for at least one third of the error generated by the analytica! method [1]. The importallee of sample preparation in analytica! chemistry, therefore, can not be overemphasized. The improverneut of sample preparation methods or the development of new methods will reduce the analysis time and allow the analyst to produce more precise results.
For solid samples, the most widely used extraction method is Soxhlet extraction. In the last few years, various new sample preparation techniques, such as microwave-assisted extraction (MAE) [2, 3], somcation extraction [4, 5], supercritical fluid extraction (SFB) [6-8] and accelerated solvent extraction (ASE) 6) X. Lou, H.-G. Janssen and C.A. Cramers, Anal. Chem., 69 (1997) 1598.

122 5.3. ASE of polymerie samples
[9] have been developed as alternatives for Soxhlet. Compared to Soxhlet extraction, each of the new techniques reduces the amount of solvent required and/or shortens the sample preparation time. The time and solvent consuming nature of Soxhlet extraction is generally imputed to the slow ditfusion of the analytes from the sample matrix into the extraction fluid and/or to the slow desorption of the components from the sample matrix. By the introduetion of microwave or sonication, by using supercritical fluids, or by extracting the components at elevated temperatures, the rates of ditfusion and desorption can be increased significantly. Among the new sample preparation methods, ASE is the metbod introduced most recently. Only very few reports on ASE have been publisbed so far [9, 10]. One of the general conclusions of these reports is that metbod development in ASE is relatively straightforward. The number of experimental parameters that have to be optimized is reduced and, at least for the extraction of the environmental samples studied, no matrix dependency of recoveries was observed. Additionally, the solvents used for conventional Soxhlet extraction can also be used for ASE.
In ASE, a solid sample is packed into the extraction cell and is than extracted with a suitable solvent at elevated temperature and pressure conditions. Richter et al [9] investigated the ASE extraction of polycyclic aromatic hydrocarbons, polychlorinated biphenyls and total petroleum hydrocarbons from environmental samples and found that the extraction temperature, pressure and volume of solvent used are important parameters affecting the extraction efficiency in ASE. In their experiments, the samples were fust extracted statically. After the static period, fresh solvent was introduced to flush the cell and the Iines. Liquid rernained in the celland the tubing was purged out with pressurized nitrogen gas. In the extraction of these environmental samples, ASE gave recoveries comparable to or better than those obtained with Soxhlet and other conventional techniques. So far no reports on ASE of polymerie samples have been published.
In this contribution, the ASE extraction of monomers and oligomers from polymerie samples is studied. A simpte experimental setup is proposed which allows ASE extraction to be performed in the static mode, the dynamic mode, or as a combination of these two modes. Two polymerie materials, nylon-6 and poly (1 ,4-butylene terephthalate) (PB1) are selected as the model samples. The mechanisrns of ASE extraction of polymerie samples are discussed. The effects of various experimental parameters, such as temperature, pressure, flow rate, static

Chapter5 123
time, and type and volume of solvent used are investigated systematically. Furthermore, guidelines for the optimization of ASE extracrions of polymerie samples are given.
5.3.2 EXPERIMENTAL
fu the experiments a home-built accelerated solvent extraction (ASE) system was used. Figure 5.19 shows a schematic diagram of this system. Apart from being suited for ASE, the system cou1d also be used for supercritical fluid extraction (SFE). A modified Varian 8500 syringe pump (Varian Associates, Sunnyvale, California, USA) was used to deliver the extraction solvent. The pump was equipped with a safety ropture disk and was operated in the pressure controlled mode. A 3 mL stainless steel cell (Suprex, Pittsburg, PA) was used as the extraction cell, unless stated otherwise. The extraction cell was fitted with handtight connectors (Suprex, Pittsburg, PA) for easy installation. Stainless steel frits (3 IJ.m) were located at either end of the extraction cell. Fused silica capillaries with different internal diameters and lengtbs were used as the restrictors to maintain the pressure inside the extraction cell during dynamic extraction. fu the extraction of PBT at extraction temperatures higher than llO"C, the resttictor was heated to prevent restrictor blockage. Both the purge/pump valve and the static/dynamic valve were six way valves (CH6214, VICI AG, Schenkon, Switzerland). The extracted material was collected by directly inserting the restrictor outlet into a glass vial (18 cm x 1.2 cm i.d.). After collection, the extraction solvent was evaporated under a gentie flow of nitrogen, and the extracted material was redissolved in a suitable amount of chloroform.
Pump
Oven w glass via!
Fïgure 5.19. Schematic illustration ofthe accelerated solvent extraction (ASE) system.

124 5.3. ASE o[polymeric samples
The polymerie samples used in this study, nylon-6 (glass transition temperature, Tg = 62.5.C) and poly(1 ,4-butylene terephthalate) (PBT) (Tg = 66'C) were purchased from Aldrich (Milwaukee, WI). The polymers were ground by rnilling under liquid nitrogen before extraction. The highest extraction temperature tested for both polymers was 170T. At higher temperatures the resttictor or the static/dynamic valve often blocked due to partial melting of the polymers. Soxhlet extraelions and SFE extraelions were performed to provide frames of reference. The details of the Soxhlet and SPE extractions of the polymers were reported previously [11]. Extraction efficiencies for ASE were calculated relative to the Soxhlet data.
The extracted components were analyzed using a gas chromatograph equipped with an on-column injector and a flame ionization detector (PID) (GC 8000 series, Carlo Erba, Milan, ltaly). A Carbowax column (25 m x 0.32 mm i.d., 1.2 Jlm film thickness) purchased from Chrompack (Middelburg, The Netherlands) was used for the analysis of the extracts from nylon-6. The initia! temperature for the analyses was 50T. Temperature was then programmed to 250T at 20.C/min. Por the analysis of the PBT extracts, an HT SimDist-CB column (10 m x 0.53 mm i.d., 0.17 Jlm film thickness) from Chrompack was used. The initial temperature for the gas chromatographic separation was 50T. Temperature was then programmed to 400T at 20"C/min.
5.3.3 RESUL TS AND DISCUSSION
The first challenge in ASE method development is the selection of a suitable extraction solvent. Por environmental samples the selection of the solvent is relatively straightforward. Por this type of samples, it was found that ASE can be performed with the same solvent as that employed in conventional extraction methods (such as Soxhlet) [9]. However, in the ASE extraction of polymerie samples a different situation might occur. The solvents used for Soxhlet extraction of polymers are normally good swelling agents for the polymer to be extracted. U se of these solvents can be troublesome in ASE, because the polymers may (partially) dissolve in the solvents at the high extraction temperatures used. Moreover, the connecting tubing in the ASE system can be blocked by polymer molecules dissolved in the hot solvent. In this article, therefore, hexane, which is a poor swelling solvent for both nylon-6 and PBT, is chosen as the extraction solvent. Despite the poor swelling capability of hexane, with this solvent at high temperatures excellent ASE extraction reeoverles can be obtained in a short time

Chapter5 125
as will bedemonstrared later in this article. In the following sections, the kinetics of mass transfer in the ASE extraction process are discussed. The various operational parameters, such as temperature, pressure, static time, flow rate, etc, on ASE extraction recovery are investigated systematically with the ultimate aim to improve the understanding of the processes occurring during ASE extraction of polymerie samples.
5.3. 3.1 Kinetics of mass transfer in ASE of polymerie samples
The extraction process in ASE can be modelled as three subsequent steps. First the solutes must diffuse from the core of the polymerie particles to the surface. Next they should be transferred from the surface into the extraction fluid. Finally the solutes are eluted out of the extraction cell. A schematic representation of these subsequent steps is depicted in tigure 5.20. The ASE extraction rate is limited by the slowest of these three steps. Extensive investigations on mass transfer in polymer particles have already been conducted by chemical engineers [12], and more recently by chromatographers [13-15]. In previous work [15], we used the two-film kinetic model to qualitatively describe the extraction mechanism in supercritical fluid extraction (SPE). In this section, the kinetic model is used to predict the effects of experimental parameters on the ASE extraction efficiency of polymerie samples.
.. .. ..........
~1 1~ ë . . l . ~ á3 ...................... en
e Polymer partiele
0 Stagnant solvent layer
Figure 5.20. Schematic presentation of the three subsequent steps in the ASE extraction process.
In ASE, mass transfer from the core of the polymer particles to the surface and from the surface through the layer of stagnant solvent outside the polymer particles into the extractant is controlled by molecular diffusion. The respective

126 5.3. ASE o[polymeric samples
diffusion coefficients are determined by parameters such as properties and structures of the polymer and the solutes, extraction temperature, type of extraction solvent, etc. Diffusion in both the polymer and through the layer of stagnant solvent around the polymer particles can be increased by raising the extraction temperature. Evidently, the time required for mass transfer from the core of the polymer particles to the surface will also be shorter if the size of the polymer particles is reduced. After being transferred into the extractant, the components are eluted out of the extraction cell by the flow of solvent. This elution process closely resembles a chromatographic separation. A solute is distributed between the extractant (the mobile phase) and the surface of the polymer particles (the stationary phase). Blution is faster if the solubility (the solute concentration at saturation) of the component in the extraction solvent is increased, if the interaction between the solute and the polymer surface is reduced, or if higher solvent flow rates are used. Temperature is a very effective parameter for increasing the extraction rate of the components. Elevated temperatures will increase the solubility of the solutes in the extractant [16] and reduce their interaction with the polymer surface. Moreover, as already discussed above, the use of higher temperatures also enhances diffusion inside the polymer and through the stagnant solvent layer around the polymer particles. From the discussion presented above, it is evident that the kinetics of mass transfer in all the three steps governing the ASE process can be improved by operaring at higher temperatures. To keep the extractant in the liquid state at temperatures above its normal boiling point, a certain pressure is required.
An alternative approach to enhance molecular diffusion and transport of the extracted solutes out of the extraction cell in ASE is to use organic solvents that are both good swelling agents for the polymer and good solvents for the analytes. In the swollen polymer particles diffusion is faster. Good solubility characteristics, on the other hand, result in rapid "elution" out of the extraction cell. Unfortunately, however, in ASE the positive effects of these solventscan not be fully exploited because at the elevated extraction temperatures used the polymer can dissolve in the solvents. In this situation, plugging of the system tubing, valves or the restrictor easily occurs.
Although SFE and ASE are different in many respects, the mechanisms of extraction in these two techniques are very much similar. lt was reported that in SFE the effects of solvent flow rate on the extraction efficiency can be used to

Chapter5 127
· identify which step controls the SPE extraction rate [15, 17]. A similar approach can also be adopted in ASE to determine whether the extraction is limited by ditfusion inside the polymer, by transfer from the polymer surface to the flowing extraction fluid, or by the solubility in the extractant. The main ditterenee between ASE and SFE lies in the ditfusion coefficients and viscosities of the extraction fluids used. In SFE, mass transfer from the polymer surface to the supercritical fluid extractant is very fast because of the high ditfusivity in supercritical fluids. Moreover, the layer of stagnant supercritical fluid around the partiele is very thin. Therefore, the rate-limiting step in SPE is either ditfusion in the polymer particles or solubility in the supercritical fluid. Which step actually controts the extraction ldnetics can be identified by monitoring the extraction yields at different temperatures or different flow rates [15]. Compared to SPE, a slightly more complicated situation prevails in ASE. In ASE, mass transfer from the surface of the polymer particles through the layer of stagnant solvent around the particles into the extraction solvent is much slower because of the lower solute ditfusivity in the extraction solvent and the relatively thick layer of stagnant solvent around the particles. In the ASE extraction of polymerie samples, the ratelimiting step hence can be either ditfusion inside the polymer particles, transfer from the surface into the extractant, or solubility in the extractant. If the extraction rates are controlled by solubility, larger solvent flow rates will yield higher extraction recoveries. In contrast, if the extraction rates are controlled by ditfusion inside the polymer particles and/or transfer through the layer of stagnant extractant around the polymer particles, the solvent flow rate will show little or no influence on the extraction recovery unless unpractically high flow rates are used to reduce the thickness of the stagnant layer.
5.3.3.2 Effects oftemperature in ASE
An important operational ditterenee between ASE and Soxhlet extraction is the extraction temperature. In ASE, extraction temperatures well above the normal boiling point of the solvent are used. Contrarily, in Soxhlet extraction the extraction temperature is limited by the boiling point of the solvent used. At elevated temperatures, ditfusion of the components from the inside of the polymer particles to their surface is enhanced. Moreover, transfer from the surface of the particles into the extraction solvent will be faster. In addition to these, also the solubility of the components in the extraction fluid will be improved when worldng at increased temperatures. Hence, the extraction rate at elevated

128 5.3. ASE o{polvmeric samples
extraction temperatures will be significantly higher. In order to investigate the effects of temperature on the extraction rates of polymers under ASE conditions, nylon-6 and PBT were extracted with hexane at different temperatures at a pressure of 200 bar. In these extractions, a static time of 20 minutes and a dynamic time of 30 minutes were used. The results are illustrated in figure 5.21. From this figure it can clearly be seen that the effect of temperature in ASE is relative straightforward. The extraction recoveries for both polymers increase significantly when the extraction temperature was raised from so·c to 17o·c. In this figure, the extraction recoveries obtained in SFE with pure carbon dioxide are also shown. Later in this article, a brief comparison of ASE and SFE will be given.
6000 .--------r--..,...-----;---;---.----,
Ê 5000
84000
s 3000
8 2000 Q)
~ 1000
Temperature ec)
Figure 5.21. Effects of temperature on extraction recoveries in ASE and in SFE. Extraction conditions: ASE with hexane at 200 bar, resttictor 1.5 m x 50 IJ.m i.d. fused silica capillary; SFE with pure carbon dioxide at 300 bar, resttictor 70 cm x 50 IJ.m i.d. fused silica capillary. Extraction time: static 20 minutes and dynamic 30 minutes. + SFE of caprolactam from nylon-6; + ASE of caprolactam from nylon-6, A SFE of the dimer from PBT; ... ASE of the dimer from PBT, 0 SFE of the trimer from PBT; e ASE of the trimer from PBT.
5.3.3.3 Effects ofpressure
In ASE a certain minimum pressure is required to maintain the extraction solvent in the liquid state at a temperature above the atmospheric boiling point. It was reported by Richter et al. [9] that pressure had no effect on the ASE recoveries in the extraction of P AHs from dry soil samples. Also in our experiments no difference in extraction yields was observed when pressure was varied in the range from 100 bar to 300 bar.

Chapter5 129
5.3.3.4 Effects of static time, solvent flow rate and air inside the extraction eelt
ASE can be performed in the static mode, the dynamic mode, or as a combination of these two modes. In static ASE, the sample is extracted with a solvent at elevated temperature and pressure conditions without any outflow of solvent. When the extraction has reached equilibrium the analytes are collected by rapidly flushing the extraction cell with solvent and an inert gas. In dynamic ASE, the extraction solvent is continuously flowing through the extraction cell. Some type of a flow resttictor is used to maintain the pressure inside the extraction cell at a desired value. An advantage of dynamic ASE is that the solvent is continuously refreshed during the extraction. Evidently this technique requires a larger volume of solvent than static ASE and is, therefore, less suited for trace analysis. Static ASE, on the other hand, may lead to incomplete extraction because of the limited volume of solvent used. In our experiments, a combination of static and dynamic extraction was used. In this regard the practice of ASE is very much similar to standard practice in SFE. Afier a certain period of static extraction, the samples were forther extracted in the dynamic mode. The effects of static time on ASE extraction reeoverles are listed in table 5.12. In these experiments a dynamic time
Table 5.12. Effects of static time on ASE extraction
Analyte/Polymer
Static time Caprolactam/nylon-6 Dimer/PBT Trimer/PBT (min) ppmb> ppm ppm
10 878.7 1978.0 387.7 (6.5)"' (3.0) (4.7)
20 1135.2 2425.2 496.1 (5.6) (4.7) (5.8)
40 1124.0 2333.8 534.3 (0.5) (1.2) (1.6)
•l Extraction solvent: Hexane; Temperature 150'C; Pressure 200 bar; Dynamic time lO minutes; Restrictor: 1.5 m x 50 j.Lm i.d. fused silica capillary. bJ Extracted amounts relative to the mass of polymer weighed into the extraction cell. "1 Relative standard deviation (RSD%) basedon three repeated extractions.
of 10 minutes was used. From table 5.12 it can be seen that the extraction recoveries for both polymers increase considerably when the static time was increased from 10 minutes to 20 minutes. Opposedly, increasing the static time from 20 minutes to 40 minutes, no forther improverneut of extraction recoveries was observed. Most likely, after 20 minutes of static extraction an equilibrium

130 5.3. ASE o[polymeric samples
state has been established. Therefore, in the following experiments, a static time of 20 minutes was used.
After static extraction, the polymerie samples were extracted dynamically. In order to investigate the effects of the extractant flow rate on ASE efficiency, fused silica capillary restrictors (50 j..l.m i.d.) with different lengths were used to adjust the flow rate while keeping pressure constant. Table 5.13 lists the effects of the extractant flow rate on the ASE extraction reeoverles at 150T and 200 bar. No changes in extraction yields were observed when the flow rate was changed from approximately 0.4 mL/min to about 2 mL/min. Flow rates lower than 0.4 mL/min were not tested. Similar results were also observed at other extraction conditions tested. Apparently, under the experimental conditions used, the ASE extraction rate was not controlled by solubility of the components. This condusion is further supported by a separate series of independent experiments where no differences in extraction reeoverles were observed whether the extraction cell was either fully or only partly packed with the polymerie samples. When lower (relative) extraction recoveries are obtained for a fully packed cell, this is an indication that solubility in the fluid is the limiting parameter for extraction [17].
Table 5.13. Effects of solvent flowrateon ASE extraction
Analyte/Polymer
Flow ratebi Caprolactam/nylon-6 Dimer/PBT Trimer/PBT (mL!min)
2.0 1195.1 2517.0 510.2 (1.8)d) (1.1) (3.1)
0.9 1134.6 2485.5 509.6 (4.1) (2.0) (2.9)
0.4 1135.2 2425.2 496.1 (5.6) (4.7) (5.8)
a) Extraction solvent: Hexane; Temperature 150T; Pressnre 200 bar; Dynarnic time 10 minutes. bJ
Approximate values obtained by adjusting the length of the fused silica restrietion capillaries of 50 j.Lm i.d .. Lengths, 43 cm, 74 cm and 150 cm, respectively. c) Extracted amounts relative to the mass of polymer weighed into the extraction cell. d) Relative standard deviation (RSD%) based on three repeated extractions.
In all experiments described above, the static/dynamic valve was opened prior to starting the extraction. This allows the air inside the extraction cell to be eliminated by the extraction solvent. After the removal of the air, the

Chapter 5 131
static/dynamic valve was closed and tbe oven was heated. Slightly lower extraction recoveries were observed for botb nylon-6 and PBT if tbe air inside tbe system was not eliminated prior to extraction. Most likely residual air can block direct contact between tbe solvent and the sample matrix. Therefore, it is recommended tbat air inside tbe system be eliminated before tbe oven temperature is raised. This is particularly important for samples tbat are susceptible to reactions witb air at elevated temperatures.
5.3.3.5 Optimization in ASE of polymerie samples
Optimization is, perhaps, tbe area of tbe greatest concern in ASE metbod development. In tbis section, some general guidelines for tbe optimization of ASE extractions of polymerie samples will be given. Due to tbe fact tbat ASE is still in its early stage of development, tbe discussion here can only be generaL
-Sample size. The sample size is one of tbe parameters tbat should be considered before performing an ASE extraction. Evidently, tbe sample size should be large enough to ensure sample homogeneity and to obtain sufficient sensitivity for trace analysis. However, larger samples require large amounts of solvent for quantitative extraction and may more easily block tbe system or its restrictor. Normally, a small sample size is preterred provided tbat tbe requirements for sample homogeneity and sensitivity are satisfied.
-Particle size. The in:fluence of partiele size on ASE extraction yields depends on which parameter controls tbe extraction efficiency. It is obvious tbat if tbe ASE extraction rate is limited by diffusion of the analytes in the polymer particles, the extraction rate can be greatly increased by decreasing the partiele size. Therefore, polymers should generally be ground or cut into small particles prior to extraction.
- Extractant. The selection of a suitable extraction solvent is probably tbe most difficult step in metbod development in ASE. The solvents used in conventional Soxhlet extraction may not be suited for ASE because tbe polymerie materials may dissolve or swell to too large an extent at tbe higher temperatures used in ASE. In the selection of the extraction solvent, tbe following two demands should be fulfilled: (a) The polymer should not be soluble in the solvent selected at the ASE temperatures used; (b) The analytes of interest must have a good solubility in tbe solvent under ASE conditions. As almost no data on the solubility of

132 5.3. ASE o[polymeric samples
polymers or solutes in high temperature solvents are available, solvent selection in ASE is stilllargely empirica!.
- Temperature. As discussed above, temperature is the most important parameter affecting the killetics of mass transfer in ASE. The ASE extraction rates can be increased significantly at higher temperatures. Evidently, however, the extraction temperature should not be above the melting point of the polymer that is to be extracted. Unfortunately, again no information of melting points of polymers under ASE conditions is available. Care should also be taken not to exceèd the temperature values where either the polymer or the analytes start to degrade. Information on the allowable temperature is only scarcely available. The optimal extraction temperature depends on the polymer, the extraction solvent and the target analytes, and thus, can only be determined experimentally.
- Pressure. In ASE, a certain minimum pressure is required to maintain the extraction solvent in the liquid state. Fortunately, these pressures need not to be ex.cessive. For example, 20 atm is sufficient to keep hexane (atmospheric bp 68.TC) in the liquid state at 209T. The minimum pressure required can be estimated from simple equations as described by Hass and Newton [18]. These authors also listed boiling points of many organic solvents at various pressures greater than one atm. In our experiments, pressure was found to have no influence on the ASE extraction efficiency for the polymerie samples studied. This implies that the selection of extraction pressure is fairly easy. Pressures wellabove the values required to keep the extraction solvent from boiling should be used.
- Static!dynamic. In the process of ASE extraction, the analytes are extracted from the inside of the polymer particles into the extraction solvent. Dynamic ASE might be expected to yield faster extractions by continuously providing fresh extraction solvent to the sample, but this technique requires more solvent than static ASE, particularly when extracting large samples. Therefore, a combination of static and dynamic extraction will often be the best choice in practice.
- Flow rate. The effect of flow rate on ASE extraction rates can be used to determine whether the extraction is limited by analyte solubility in the extractant or by diffusion in the polymer particles and/or transfer from the polymer surface to the extractant. If the extraction recoveries do not change when the flow rate is increased, the extraction is not solubility limited. The rate limiting step is

Chapter5 133
ditfusion inside the polymer particles and/or transfer from the polymer surface into the extractant. The extraction rate now can be enhanced by raising the extraction temperature. The use of very low flow rates is not recommended because it may easily lead to blockage of the resttictor. From a practical view of point, the initia! solvent flow rate can be selected at around 0.5 mL!min.
5.3.3.6Advantages of ASE over other sample preparation methods
In addition to the reduced solvent consumption, shorter extraction time required and the ease of method development, another important advantage of ASE over Soxhlet extraction is the wide range of possible extraction solvents. In ASE the samples are extracted at temperatures well above the boiling point of the extractant used. The killetics of mass transfer can be greatly improved at elevated temperatures. Therefore, it is very well possible that a poor extraction solvent in a Soxhlet extraction can be a good solvent in ASE at high temperatures. As demonstrated in table 5.14, hexane, which is a poor extraction solvent in the Soxhlet extraction of nylon-6 and PBT, yields excellent extraction recoveries in ASR The extraction recoveries of caprolactam from nylon-6 and the dimer from PBT in 50 minutes (20 minutes static and 30 minutes dynamic) are comparable with those found after extensive Soxhlet extraction with methanol and chloroform. Por the extraction of the large trimer molecule from PBT, the extraction recovery at an extraction duration of 50 minutes is stilllower than that of Soxhlet extraction with chloroform. To obtain higher extraction recoveries for the trimer, a longer extraction time is required.
Table 5.14. Comparison of extraction recoveries between ASE and Soxhlet extraction.
Analyte/Polymer
Caprolactarn/nylon-6 Dimer/PBT Trimer/PBT ppmd) ppmdl ppmd)
ASE•> 1956.1 4795.5 1358.5
Soxhlef"l 158.7 137.4 2.6
Soxhlet<> 1827.8 4886.0 2039.0
•> Extraction conditions: hexane, 170"C and 200 bar; static time 20 minutes; dynarnic time 30 minutes; Restrictor: 1.5 m x 50 ~-tm i.d. fused silica capillary. bl Soxhlet extraction, hexane, 40 hours. c) Soxhlet extraction, 30 hours, methanol for nylon-6 and chloroform for PBT. dl Extracted arnounts relative to the mass of polymer weighed into the extraction cell.
In Soxhlet extractions, considerable amounts of precipitate were observed in the

134 5.3. ASE ofpolymeric samples
extracts when methanol and chloroform were used as the extraction solvent for nylon-6 and PBT, respectively. Moreover, the quality of the GC columns used notably deterlorated after a few injections of these extracts, probably because large molecules that could not be eluted out the GC column were coextracted with the target analytes and injected into the GC system. In contrast to this, the extracts of ASE with hexane as the extractant are much cleaner. No precipitation of the extracts and deterioration of the GC columns were observed with ASE using hexane as the extractant. Prom this it is clear that due to the flexibility in the selection of the extraction solvent in ASE, some additional selectivity can be introduced by using different solvents.
It is interesting to compare the extraction reeoverles of SPE and ASE at identical extraction temperatures (figure 5.21). Hexane is the organic solvent having polarity properties most similar to supercritical carbon dioxide. It can be seen from figure 5.21 that at high extraction temperatures the extraction recoveries for both nylon-6 and PBT obtained in ASE are considerably higher than those obtained in SPE. This is especially the case for the relatively large molecules (dimer and trimer from PBT). The extraction reeoverles in ASE of caprolactam from nylon-6, and the dimer and trimer from PBT at 170"C, the highest temperature tested, are approximately 1.1, 6.5 and 37.6 times higher, than those obtained in SPE. Prom the discussion above it can be concluded that in the extraction of polymerie samples, ASE is more effective than SPE with pure carbon dioxide, particularly in the extraction of components having a poor solubility in supercritical carbon dioxide and/or when high extraction temperatures are necessary to increase ditfusion of the analytes in the polymer particles. Nevertheless, SPE has its unique advantages such as adjustable selectivity by tuning the extraction conditions, ease of on-line combination with other analytica! techniques, etc.
5.3.4 CONCLUSIONS
Accelerated solvent extraction (ASE) is a promising technique for sample preparation. In the process of ASE extraction of polymerie samples, three subsequent stepscan be identified, i.e. ditfusion of the components from the core of the polymer particles to their surface, transfer of the components from the surface into the extraction fluid and elution of the components out of the extraction cell. The kinetics of mass transfer in these three subsequent steps are improved significantly at higher temperatures. ASE gives reeoverles comparable

Chapter5 135
to those obtained with Soxhlet extraction. Metbod development in ASE is relatively straightforward because less parameters affect the extraction efficiency. Besides the selection of the extraction solvent, temperature is the most important parameter to be optimized. The range of solvents applicable in ASE is much wider than that in Soxhlet extraction. This is because a poor solvent for Soxhlet extractions can be a good solvent in ASE due to the higher extraction temperatures used. Some additional selectivity can be introduced in ASE by carefut selection of the extraction solvent and the temperature.
REFERENCES
1. R.E. Major, LC.GC Int., 4 (1991) 10. 2. K. Ganzler, J. Bati and K. Valko, J. Otromatogr., 371 (1986) 299. 3. V. Lopez-Avila, R. Young, R. Kim and W.F. Beckert, J. Otromatogr. Sci., 33 (1995) 481. 4. A.S.Y. Chau and L.J. Babjak, J. Assoc. OjJ. Anal. Chem., 62 (1979) 107. 5. F.M. Dunnivant and A.W. Elzerman, J. Assoc. OjJ. Anal. Chem., 71 (1988) 551. 6. S.B. Hawthorne, Anal. Chem., 62 (1990) 633A. 7. T.L. Chester, J.D. Pinkston and D.E. Raynie, Anal. Chem., 64 (1992) 153R. 8. M.L. Lee and K.E. Markides (Eds), Analytica/ Supercritical Fluid Otromatography and
Extraction, Chromatography Conference, Provo, UT, 1990, Chapter 5. 9. B.E. Richter, B.A. Jones, J.L. Ezzell, N.L. Porter, N. Avdalovic and C. Pohl, Anal. Chem.,
68 (1996) 1033. 10. F. Hoefler, J. Ezzell and B. Richter, Proceedings ofthe 3rd European Symposium on Analytical
SFC and SFE, Uppsala, Sweden, 1995, p.017. 11. X. Lou, H.-G. Janssen and C.A. Crarners, J. Otromatogr. Sci., 34 (1996) 282. 12. J. Crank and G.S. Park (Eds), Dijfusion in Polymers, John Wright and Sons Ltd., Bristol. Great
Britain, 1968. 13. N.J. Cotton, K.D. BartJe, A.A. Clifford and C.J. DowleJ. Chromatogr. Sci., 31 (1993) 157. 14. S. Kueppers, Chromatographia, 33 (1992) 434. 15. X. Lou, H.-G. Janssen and C.A. Crarners, J. Microcol. Sep., 7 (1995) 303. 16. D.J.W. Grant and T. Higuchi, Soluhility Behavior ofOrganic Compounds, John Wiley & Sons,
Inc., New York, 1990. 17. S.B. Hawthorne, A.B. Galy, V.O. Schrnitt and D.I. Miller, Anal. Chem., 67 (1995) 2723. 18. H.B. Hass and R.F. Newton. in R.C. Weast and M.I. Astle, (eds), Haruibook of Chemistry and
Physics, 62th ed., CRC Press Inc., Boca Raton, Florida, 1982, p.l89.

136
6.1
Chapter6
Quantitative Aspects of Directly Coupled Supercritical Fluid Extraction-Capillary Gas Chromatography with a Conventional Split/splitless Injector as the lnterface7l
ABSTRACT
The quantitative aspects of on-line supercritical fluid extraction-capillary gas chromatography (SFE-GC) based on the use of a split/splitless injector as the interface were investigated. Special attention was paid to the discrimination behaviour and the reproducibility of the split/splitless interface. A simpte experimentalset-up is proposed that allows accurate quantitation in on-line SPEsplit GC. The results obtained in on-line SFE-GC compare favourably with those from conventional GC with split injection. Discrimination was found to be absent when working at sufficiently high interface temperatures. Finally, the effects of the carbon dioxide flow rate, interface temperature and split ratio on both discrimination and reproducibility were studied.
6.1.1 INTRODUCTION
Supercritical fluid extraction (SFE) is a rapidly growing technique for sample preparadon [1]. SFE has a number of important advantages over conventional liquid extractions. Among these, the ease with which on-line combination of SFE sample preparation with chromatographic analysis can be obtained is one of the most important ones. Various interfaces for on-line SFE-GC have been described in literature [2-4]. Of these interfaces the split injector is the easiest to use and the most ruggest device.
In SFE-GC with a split injector as the interface, the SPE restrictor is inserted directly through the septurn into a conventional split/splitless injection port. The injector is heated to prevent plugging of the restrictor. The GC column is cooled to refocus the extracted components [5]. Split SFE-GC bas been shown to work well for samples that have high concentrations of extraetabie components, wet samples and for extractions using modifiers [ 6-8].
In a hyphenated method, the interface is the key to the technique. In conventional GC with split injection, the flow split ratio is often not a correct representation of
TJ X. Lou, H.-G. Janssen and C.A. Cramers. J. High Resolut. Chromatogr., 16(1993)425.

6.1 Quantitative aspectsof SFEIGC with spilt!splitless interface 137
the true sample split ratio. Moreover, discrimination, caused by large differences in volatility, can occur. In SFE-GC with a split/splitless injector as the interface, all the solutes are initially dissolved in the supercritical fluid. In transtering the sample to the GC, the supercritical fluid is expanded which results in a large volume of gaseous sample. In this regard, on-line SFE-split GC is clearly different from a conventional split injection in capillary GC.
Little attention bas been paid in lirerature to the quantitative aspects of the split injector when used as the interface in on-line SFE-GC. The discrimination behaviour and reproducibility of the split injector in on-line SFE-GC have not been investigated yet. Calibration of on-line SPE-split GC is frequently carried out by introducing a calibration standard directly into the split injector using a syringe [9]. In that case, however, the volume of sample that is introduced into the injector is not known accurately and there could be discrimination against volatility. Hence, accurate calibration can not be obtained. To circumvent these problems, we recently proposed to incorporate an injection valve in the SFE-GC set-up. With this valve accurately known volumes of a calibration standard can be introduced without discrimination [10].
In this paper, the ability to analyse components with different volatilities by online SPE-split GC is evaluated by model experiments. Furthermore, the reproducibility of SFE-GC using the split injector as the interface is evaluated. Also the influence of the carbon dioxide flow rate and the carrier gas flow on the performance of the system is investigated.
6.1.2 EXPERIMENTAL
Figure 6.1 shows a schematic diagram of the system used in the experiments. No extraction cell was included in the system. On-line SFE-GC experiments were modelled by injecting a standard solution of n-alkanes in hexane directly into the carbon dioxide flow stream at ambient temperature using an LC injection valve (CH 6214, VICI AG, Schenkon, Swizerland). In this way, solutions with known concentrations of the test components could be introduced directly into the split/splitless interface. By using this approach, the discrimination behaviour and reproducibility of the interface could be studied accurately as all errors introduced by the SFE part of the experimental set-up were excluded.

138 Chapter 6
Column
Figure 6.1. Schematic diagram of on-line SFE-GC model system used for evaluation of the split interface.
All GC analyses were carried out on a HRGC 5300 (Mega series, Carlo Erba Instrumentazione, Milane, Italy) equipped with a flame ionization detector. helium was used as the carrier gas. A crosslinked methyl-silicone fused-silica capillary column (25m x 0.32 mm i.d., film thickness 0.52 J.UTI) from HewlettPackard (A vondale, USA) was used for all GC analyses. The carbon dioxide used in the experiments had a purity of 99.996% (lntermar B. V., Breda, The Netherlands). A modified Varian 8500 syringe pump (Varian Associates, Sunnyvale, California, USA) was used to deliver the carbon dioxide. The carbon dioxide pressure was maintained at 300 atm. A stainless steel capillary (210 J.UTI i.d.) was used as the restrictor. The end of the resttictor was crimped to give the desired flow rate. The carbon dioxide gas flow rate was measured with a flow meter after the restrictor was inserted through the septurn into the heated liner of the split injector. A liner with a glass frit was used. Por the SFE-GC analyses, the column temperature was initially set at 30°C to trap the components on the head of the GC column. After 15 minutes, which was found to be sufficient for transferring the components to the GC column, the oven temperature was programmed to 325°C at 10°C/min. The SPE restrictor was left in the injector during the entire period of experimental work. Except in the experiments to study the effect of the split ratio, the split flow was kept at 240 mi/min (helium and carbon dioxide).
6.1.3 RESULTS AND DISCUSSION
The split/splitless injector has been widely used as a sample introduetion system

6.1 Quantitative aspectsof SFEIGC with spiltlsplitless interface 139
for capillary · GC. It has also been found to be a simple and useful interface for SFE-GC. In order to compare the SFE-GC interface with a conventional split/splitless injector with regard to discriminadon and reproducibility of peak areas, a solution of n-alkanes ranging from Cg to C36 at concentrations of approximately 0.80 mg/ml in hexane was injected directly with a syringe and through the SFE restrictor, respectively. The components were selected to cover a wide span of volatilities. The boiling points range from 126°C (Cg, atmosperic pressure) to 265°C (C36, 1 mm mercury). Except C8 and C10, all the solutes were effectively trapped on the head of the GC column and showed up as sharp, symetrical peaks. In the discrimination studies, c22 (0.80 mg/ml in hexane) was selected as the internat standard. The concentrations of the other components were calculated from the ratiosoftheir peak areas relative to C22• The results are shown in table 6.1 and tigure 6.2.
Table 6.1. Comparison of concentration values obtained with simulated on-line split SFE-GC and conventional split injection in GC.
SFE-GCd> split injection GC c•,bl
c•l RSD%cl c•> RSD%cl
C12 0.90 0.90 1.4 1.19 6.5
Ct4 0.92 0.91 1.3 1.16 5.4
Ct6 0.91 0.91 0.6 1.16 5.6
Cts 0.92 0.93 1.7 1.08 2.3
c20 0.76 0.76 0.8 0.81 2.6
Cn 0.80 internat standard
C24 0.85 0.85 0.5 0.80 4.2
C2s 0.77 0.77 0.8 0.65 9.7
c32 0.81 0.80 1.6 0.62 13.1
c36 0.81 0.78 1.3 0.58 15.1
•l concentration (rnglml in hexane); b) values obtained by on-column injection; <l relative standard deviation ofthe concentration values (n = 5); dl carbon dioxide gas flow= 93 mi/min.
In tigure 6.2, a chromatogram of a simulated on-line SFE-split GC experiment (tigure 6.2A) is compared with that of a conventional split injection in GC (tigure 6.2B). A summary of the quantitative data is given in Table 6.1. As can be seen from this table, significant discrimination occurs in the conventional split
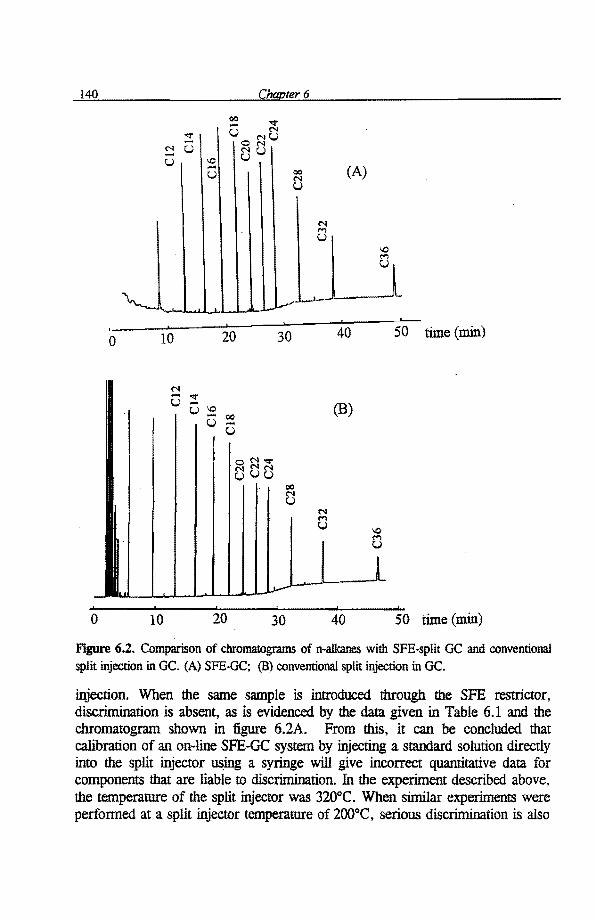
140 Chaeter6
co
u N "<T
o ~u -N u au -u ..0 -u co N
(A) u
v·
0 ___ 1 ...... 0 --7.20:----::30:---:4;n0--5-:ro\ time (min)
N - "<T u - (B) u..o - co u -u
oN"<T NNN uuu
co N u
N M u \0
M u
1 -0 10 20 30 40 50 time (min)
Figure 6.2. Comparison of chromatograms of n-alkanes with SFE-split GC and conventional split injection in GC. (A) SFE-GC; (B) conventional split injection in GC.
injection. When the same sample is introduced through the SFE resttictor, discriminanon is absent, as is evidenced by the data given in Table 6.1 and the chromatagram shown in tigure 6.2A. From this, it can be concluded that calibration of an on-line SFE-GC system by injecting a standard solution directly into the split injector ~ing a syringe will give incorrect quantitative data for components that are liable to discrimination. In the experiment described above, the temperature of the split injector was 320°C. When similar experiments were performed at a split injector temperature of 200°C, serious discrimination is also
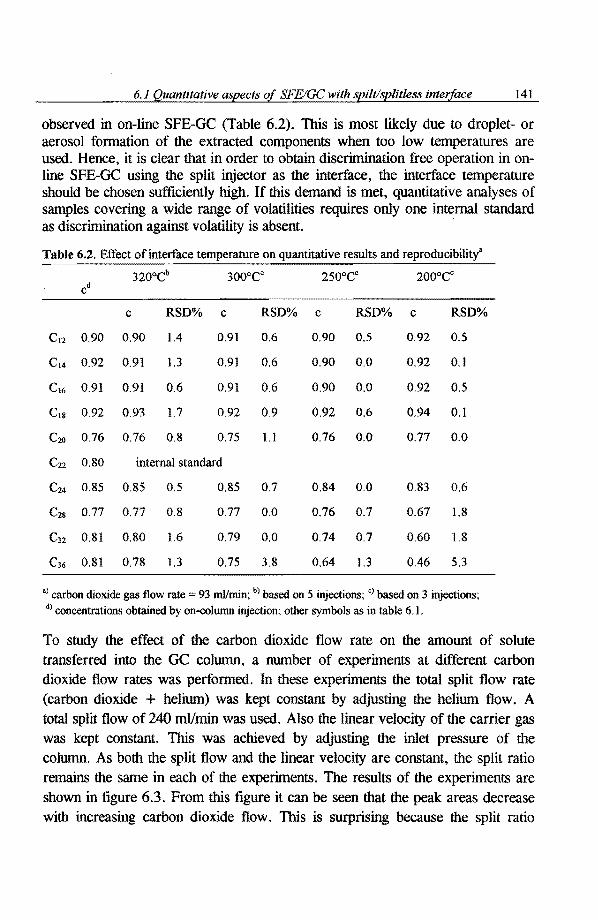
6.1 Quantitative aseects ot SFEIGC with Sf!.iltlsf!.litless inteiface 141
observed in on-line SFE-GC (Table 6.2). This is most likely due to droplet- or aerosol formation of the extracted components when too low temperatures are used. Hence, it is clear that in order to obtain discrimination free operation in on-line SFE-GC using the split injector as the interface, the interface temperature should be chosen sufficiently high. If this demand is met, quantitative analyses of samples covering a wide range of volatilities requires only one internat standard as discrimination against volatility is absent.
Table 6.2. Effect of interface temperature on quantitative results and reproducibility•
32oocb Jooocc 250°C0 200°CC cd
c RSD% c RSD% c RSD% c RSD%
Cn 0.90 0.90 1.4 0.91 0.6 0.90 0.5 0.92 0.5
C14 0.92 0.91 1.3 0.91 0.6 0.90 0.0 0.92 0.1
C16 0.91 0.91 0.6 0.91 0.6 0.90 0.0 0.92 0.5
C1s 0.92 0.93 1.7 0.92 0.9 0.92 0.6 0.94 0.1
C2o 0.76 0.76 0.8 0.75 1.1 0.76 0.0 0.77 0.0
c22 0.80 internal standard
C24 0.85 0.85 0.5 0.85 0.7 0.84 0.0 0.83 0.6
C2s 0.77 0.77 0.8 0.77 0.0 0.76 0.7 0.67 1.8
c32 0.81 0.80 1.6 0.79 0.0 0.74 0.7 0.60 1.8
c36 0.81 0.78 1.3 0.75 3.8 0.64 1.3 0.46 5.3
a) carbon dioxide gas flow rate = 93 mi/min; b) based on 5 injections; c) based on 3 injections; d) concentrations obtained by on-column injection; other symbols as in table 6.1.
To study the effect of the carbon dioxide flow rate on the amount of solute transferred into the GC column, a number of experiments at different carbon dioxide flow rates was performed. In these experiments the total split flow rate (carbon dioxide + helium) was kept constant by adjusting the helium flow. A total split flow of 240 mi/min was used. Also the linear velocity of the carrier gas was kept constant. This was achieved by adjusting the inlet pressure of the column. As both the split flow and the linear velocity are constant, the split ratio remains the same in each of the experiments. The results of the experiments are shown in figure 6.3. From this figure it can be seen that the peak areas deercase with increasing carbon dioxide flow. This is surprising because the split ratio

142 Cha ter 6
remains constant. Hence, one would expect the peak areas to be independent of the carbon dioxide flow.
Cll-t:l.- CI4-0- C16-+- CJ8-&- C20
·-•~ cu-v- cu-o- C18-o- 02-•- C36
50 t. 45'
based on 3 injections, RSD%<7o/o
~ ~- 40~ .. 35
t 30 25
20
carbon dioxide gas flow rate(ml/min)
Figure 6.3. Effect of carbon dioxide flow rare on peak areas.
Table 6.3.Effect of split flow and carrier gas velocity on peak area a)
uo 26.5 cm/minhl Uo 26.1 cm/minbl Uo 64.5 cm/minhl
Fsplit 450 Fsplit = 75 ml/minc) Fsplit 75 ml/mincl ml/minc)
area RSD% area RSD% area RSD%
cl2 36974 4.6 35865 2.1 93143 5.2
Ct4 36964 4.5 36022 1.8 92915 6.2
C16 36887 4.7 36105 1.8 93860 6.2
Cts 37406 4.7 36778 1.8 94952 6.2
C2o 30685 4.7 30185 1.5 78048 6.2
Cn 32465 4.8 31802 1.8 82998 6.8
c24 34542 4.7 33889 1.7 88219 6.5
C2s 31499 4.7 30639 2.7 80412 6.5
Cn 32257 4.6 31578 3.2 83091 6.3
c36 31089 5.0 29979 3.9 79395 6.2
a) carbon dioxide gas flow: 78 mi/min, interfuce temperature: 320°C, based on 3 injections; b) carrier gas velocity; c) split flow rate.
From the results presented in tigure 6.3, it appears that the behaviour of a split injector in on-line SFE-GC is markedly different from that normally observed in

6.1 Quantitative aspects q{ SFEIGC with spilt!splitless interface 143
conventional GC. To study the effect of the split flow on peak areas in more detail, a second series of experiments was performed. In these experiments, the split flow was changed from 450 mi/min to 75 rnl/min at a constant carbon dioxide flow and colwnn flow rate. Only the flow rates of helium were different in the two experiments. Hence, the split ratios are different in these two cases. The results are summarized in table 6.3. As can be seen from this table, almost no changes in absolute peak areas occur despite the large difference in the split ratios. Also this observation is in contrast to expectation. If, on the other hand, the colwnn flow rate was increased, the peak areas increased as is expected.
Figure 6.4. Schematic representation of the solute transfer process in the interface.
Most likely, the effects described above are caused by the flow pattem in the split injector. In figure 6.4, a flow pattem is proposed that could account for those observations. In the center of the liner, the linear velocity of the carbon dioxide expanding from the SFE resttictor is extremely high. Due to this high velocity the carbon dioxide flow is not homogeneously mixed with the helium. Hence, the gas in the central region of the liner almost exclusively consists of carbon dioxide. Thus, in our experiments, the carrier gas is mainly carbon dioxide, regardless of the magnitude of the helium flow through the liner. At increased carbon dioxide flow rates, the central region tends to expand, and the fraction of the solutes transfered to the capillary colwnn is reduced, again regardless of the magnitude of the helium flow rate. This explains the decrease of the peak areas at increased carbon dioxide flow rates as observed in figure 6.3. From the flow pattem in the liner it is also easily understood why the amount of solute entering the colwnn is almost independent of the split flow (see table 6.3). lf the split flow rate is increased while keeping the velocity of the carrier gas and the carbon dioxide

144 Chapter6
flow constant, only the flow rate in region 2 increases and there is almost no change in the central region (region 1}.
No significant in:fluence of the carbon dioxide flow rate, split flow and carrier gas velocity on the reproducibilities of absolute peak areas and on the discrimination behaviour was found in our experiments.
The complex flow pattem in the liner complicates quantitation in on-line SPE-split GC, because the actual sample split ratio is not known. Hence, accurate calibration can only be obtained by introducing the calibration standard in the same way as the real sample is introduced, i.e. through the SPE restrictor. Also method development in on-line SFE-GC is seriously complicated by the fact that the sample split ratio is a tunetion of the carbon dioxide flow. Por example, when the extraction pressure is varled in order to establish the in:fluence of this parameter on the extraction yield, the carbon dioxide flow and, hence, the fraction of the extracted components that are transferred into the column, changes.
REFERENCES
l. T. L. Chester, J. D. Pinkston and D. E. Raynie, Anal. 0/.em., 64 (1992) 153R. 2. S. B. Hawthome, D. J. Milier and M. S. Krieger, J. Otromarogr. Sci., 27 (1989) 347. 3. J. M. Levy, J. P. Guzowski and W. E. Huhak:, J. High Resofut. Otromatogr., 10 (1987) 337. 4. R. J. Houben, H. G. M. Janssen, P. A. Leclercq, J. A. Rijks and C. A. Cramers, J. High
Resofut. Otromatogr., 3 (1990) 669. 5. J. M. Levy and A. C. Rosselli, Otromatographia, 28 (1989) 613. 6. M. L. Lee and K. E. Markides (eds.), Analytical Supercritical Fluid Otromatography and
Extraction, Chromatography Conference, Provo, UT, 1990, p.339. 7. S. B. Hawthome, D. J. Milier and J. J. Langenfeld, J. Otromatogr. Sd., 28 (1990) 2. 8. J. M. Levy, R. A. Cavalier, T. N. Bosch, A. F. Rynaski and W. E. Huhak:, J. O!romatogr.
Sd., 27 (1989) 341. 9. J. M. Levy, A. C. Rosselli, D. S. Boyer and K. Cross, J. High Resofut. Otromatogr., 13
(1990) 418. 10. H.-G. Janssen, C. A. Cramers, L. M. van der Meulen and A. L. C. Smit, 15th International
Symposium on Capillary Otromatography, Rivadel Garda, ltaly, May 1993.

6.2
Abstracts
Chapter6 145
Investigation of parameters affecting the on-line combination of supercritical tluid extraction with capillary gas chromatographyS>
Two different injectors, a split/splitless injector and a programmed temperature vaporizer (PTV) injector were investigated as tbe interface in on-line SPE/CGC. The parameters affecting tbe chromatographic peak shapes as well as tbe quantitative performance of tbe interface in on-line SPE/CGC were identified and studied. Particular attention was paid to tbe case where modified extraction fluids were used. Bxperiments were performed on two different samples. The first sample consisted of PARs spiked on sand at different concentration levels. The otber sample was a polymerie materiaL
6.2.1. Introduetion
Supercritical fluid extraction (SFB) is an attractive sample preparation metbod for chromatography. It is fast and enables tbe introduetion of some selectivity in tbe extraction process. Moreover, tbe most commonly used fluid (COJ is inert, nontoxic and inexpensive. Furthermore, supercritical C02 is gaseous under ambient conditions. Therefore, tbe solute separation and concentration process is simplified and direct coupling of SFB to chromatographic techniques, especially to capillary gas chromatography (CGC), is greatly facilitated. The direct combination of SPE to CGC can, in many cases, be a straightforward procedure [1,2].
SFE can be combined witb CGC eitber in tbe off-line or tbe on-line fashion. Offline SFE/CGC is generally simpler to perform and allows tbe extracts to be analyzed at different CGC conditions or by any otber appropriate technique. Therefore, off-line SPE/CGC should be tbe first choice during SPE metbod development [3]. To date, tbe majority of SFB applications is performed off-line. On tbe otber hand, on-line SFB/CGC is more attractive in routine analysis since no sample handling steps are inc1uded between tbe SFE and CGC. On-line SFB/CGC operation basically involves three steps. First, tbe components are extracted by SFE. Next, tbe extracted components have to be transferred to tbe 8l X. Lou, H.-G. Janssen and C.A. Cramers, J. Olromatogr., 750 (1996) 215.

146 6.2. Parameters affecting SFE/CGC
CGC colunm via a suitable interface. Finally, the components must be separated and detected by the CGC instrument. Each of the conditions in these three steps, i.e. SFB parameters, analyte transfer conditions, and the chromatographic separation parameters must be optimized carefully before the analysis can be completed successfully.
Optimization in SFB is rather complicated because many parameters affect the extraction kinetics. A number of research groups has investigated the parameters affecting the extraction processin SFB. Langenfeld et al. [4] studied the influence of temperature and pressure on the SFB extraction of PCBs and P AHs from certified environmental reference materials using pure carbon dioxide and found that for acmeving high extraction efficiencies temperature is more important than
pressure. This is especially true if the interactions between the analytes and the matrices are strong. Similar results have been reported for the SFB extraction of polymerie samples. If pure C02 is used, high extraction recoveries can only be obtained at elevated temperatures [5, 6]. Alternatively, higher yields can be obtained by the actdition of a suitable modifier. Unfortunately, however, the effects of modifiers are highly matrix- and solute dependent In the extraction of some environmental samples, for example, it was found that the modifier idernity was more important than its concernration [7]. In contrast to this, both modifier idernity and concernration were found to be important in the extraction of polymerie samples [8]. Summarizing, high SFE extraction efficiencies can be obtained either at high extraction temperature and pressure conditions or with the actdition of a suitable modifier.
In a hyphenated method, the interface is the key to the technique. Several different interfaces have been described for on-line SFE/CGC. Among these are the split/splitless injector, the on-column injector and the programmed temperature vaporizer (PTV) injector [9-11]. Burford et al. [12, 13] developed a simpte and reliable on-line SFB/CGC system for the quantitative extraction and analysis of gasoline and diesel-range organics from environmental samples using a normal split/splitless injector as the interface. Hansen et al. [14] used coupled SFB/CGC with an on-column injector as the interface for the analysis of organic compounds in atmospheric aerosols. Houben et al. [11] showed applications of on-line SFB/CGC for atmospheric and cigarette smoke particles with a PTV injector as the interface. Levy et al. [15] reported the use of split SFB/GC in the analysis of a solid hydrocarbon waste with formic acid-modified C02• In all on-

Chapter 6 147
line SPE/CGC studies referred to above, only pure C02 or C02 admixed with small amounts of modifiers was used as the extractant. Little attention was paid to the quantitative performance of on-line SFE/CGC when modifiers were used. In this article, the quantitative aspects of the split/splitless and the PTV injector for on-line SFE/CGC with pure and modified C02 are studied and compared.
After selection of the interface, the CGC parameters, such as column temperature and split ratio (for the split/splitless injector), the stationary phase type and film thickness, columnlengthand inner diameter should all be optimized. Compared to the optimization of the SFE process, the optimization of the CGC separation is relatively simple. Prior to starting the CGC separation, the GC column should be set at a low temperature to refocus the extracted components at the head of the column. In the ideal case, the optimum extraction conditions determined by offline SPE can be used for on-line SPE/CGC without having to change or compromise.
The aim of this contribution is twofold. Firstly, the experimental parameters affecting the colteetion and chromatographic focusing of the extracted components in on-line SFE/CGC are investigated and optimized. Secondly, the quantitative aspects of on-line SPE/CGC with pure and modified C02 are studied and compared. Off-line SFE was used to optimize the SPE extraction conditions. Experiments were performed on two different samples. The first sample consists of P AHs spiked on clean sand at different concentration levels. This sample is representative for samples in which the components of interest are adsorbed onto the outer surface of the particles. The other sample is a polymerie material. Here the solutes are present in the particles to be extracted.
6.2.2. EXPERIMENTAL
SPE experiments were performed on a PrepMaster SPE instrument (Suprex Corporation, Pittsburgh, PA, USA). A 3 mL stainless steel extràction cell (Suprex) was fitted with hand-tight connectors (Suprex) for easy installation. Stainless steel frits (3 j.Lm) were located at either end of the extraction cell. A fused silica capillary (50 jlm i.d. with a length of 50 cm) was used as the restrictor. The carbon dioxide used in the experiments had a purity of 99.996% (Scott Specialty Gases, Breda, The Netherlands).
In the off-line SFE experiments, the extracted material was colleered by inserting

148 6.2. Parameters q[[ecting SFEICGC
the resttictor outlet into a glass vial (10 cm x 1 cm i.d.) containing 5 mL dichloromethane. After collection, dichloromethane was evaporated under a gentie flow of nitrogen and the extracted material was redissolved in a suitable amount of chloroform and analyzed using a gas chromatograph equipped with an on-column injector and a flame ionization detector (FID) (GC 8000 series, Fisons Instruments, Milan, Italy). On-line SFE/CGC was carried out by inserting the resttictor directly into the injector of a gas chromatograph (GC-17 A, Shimadzu Corporation, Kyoto, Japan) equipped with an FID detector and a PTV injector (Optie, Ai Cambridge, Cambridge, UK). The PTV injector liner was packed with Dexsil 300 (12% coated on Chrornsorb 750, 80-100 mesh) purchased from Chrompack (Middelburg, The Netherlands). The split SFE/CGC experiments were also performed on the PTV injector. Forthese experiments the packed liner was replaced with an empty one. Prior to extraction, the GC oven and the PTV injector were brought at the desired temperatures. The PTV could be cooled using liquid C02. The carrier gas (helium) was shut off during extraction and turned on afterwards using a controlled event from the GC.
Six PAHs, e.g. naphthalene, acenaphthene, anthracene, pyrene, chrysene and benzo[a]pyrene, all from Supelco (Bellefonte, PA, USA) were selectedas the test solutes for the analysis of P AHs in spiked sand. Two standard solutions (250 ppm and 25 ppm of each of the six PAHs) were prepared in hexane. For on-line SPE/split CGC and SFE/PTV CGC, different amounts of PAHs (10 !J.L of 250 ppm and 1 !J.L of 25 ppm standard solutions, respectively) were spiked on approximately 3 g of sand which was then tilled into the extraction cell. The extraction conditions for the spiked samples were 5o·c and 300 bar, both for pure and modified co2. Three organic solvents, dichloromethane, chloroform and methanol were investigated as modifiers. 0.5 mL of these modifiers was spiked onto the sample prior to extraction.
The polymer samples used in this study, nylon-6 and poly (1,4-butylene terephthalate) (PBT) were purchased from Aldrich (Milwaukee, Wisconsin, USA). Approximately 0.05 g of the polymerie sample was weighed into the extraction cell. The rest of the extraction cell was filled with glass wool. The SPE extraction conditions used were previously determined to be optimum settings [8]. These conditions were: (1) 15o·c and 300 bar for both nylon-6 and PBT with pure C02; (2) 5o·c and 300 bar with modified C02. 0.5 mL methanol and 0.5 mL chloroform were used as the modifiers for nylon-6 and PBT, respectively.

Chopter6 149
The GC separations were started after the SFE extractions. Two fused-silica capillary columns coated with methyl silicone (both 25 m x 0.32 mm i.d. with 0.52 j.Lm and 0.18 j.Lm film thickness, respectively) trom Hewlett Packard (Palo Alto, CA, USA) were used for the analysis of the PAHs. For SFE/CGC with the PTV injector, the PTV was cooled to the appropriate temperatures prior to commencing extraction. The column temperature was kept at 50"C during the extraction. After extraction, the PTV was heated to 320"C at 8T/sec. A splitless time of 4.5 minutes was used and the column temperature was programmed from 50"C (5 minute) to 320"C at lO"C/min. For split SFE/CGC, the injection port was operated isothermally at 350"C and the column was kept at the appropriate low temperature. The column temperature was programmed to 320"C after extraction.
In on-line SFE/CGC of the polymerie samples, only the split injector was investigated as the interface, since the concentrations of the analytes in the polymer matrix are relatively high. Two Carbowax columns (both 25 m x 0.32 mm i.d. with 1.2 j.Lm and 0.18 !J.m film thickness, respectively) from Chrompack were used for the analysis of caprolactam in nylon-6. The injection port was operated at 260"C. The column was cooled to low temperatures during extraction and then programmed to 250"C at lOT/min after extraction. For the analysis of the PBT sample, the column employed for the PAHs (crosslinked methyl silicone, 25 m x 0.32 mm i.d., 0.5 j.Lm film thickness) was used under identical temperature conditions as were used in the split SFE/CGC experiments of the PAHs.
6.2.3. RESULTS AND DISCUSSION
6. 2.3.1. On-line SFEICGC of environmental samples As the aim of this contribution is to investigate the coneetion and focusing of the extracted components and the quantitative aspects of the interfaces in on-line SFE/CGC, spiked samples were used as model samples. For samples spiked at relatively high levels of the target analytes, the split/splitless injector was used as the interface. On the other hand, for samples spiked with small amounts of analytes, the PTV injector was used.
On-line SFE/split CGC of spiked samples The split injector has been widely used as a sample introduetion system in CGC. It has also been found to be a simpte, rugged and useful interface for on-line SFE/CGC using pure C02 [9]. Figure 6.5 shows the comparison of the

150 6.2. Parameters atfecting SFE/CGC
chromatographic peak: shapes obtained by convenrional split CGC and on-line SFE/CGC using the split injector as the interface. From this tigure it can clearly beseen that excellent peak: shapes can also be obtained with modified C02• The modifiers used have no adverse effects on the peak shapes under the experimental condirloos tested. Except for naphthalene, all components are successfully trappeel at the inlet of the CGC column. In tigure 6.5, the initia! temperature of the CGC column was 35·c, which is below the boiling points of all modifiers tested. Also at higher trapping temperatures of 50"C and 7o·c, no adverse effect of the modifier on the peak: shapes was observed.. However, at a trapping temperature of 50"C the naphthalene peaks are seriously broadened., and at 70"C no naphthalene peaks could be detected.
2 (A) (B)
I s 7
.I 6
i 0 10 20 30 0 JO 20 30
time(min) time(min)
(C) (D) ., " "' .. .. = 8. ~ ..
~ ~
~ 9 "" I 11 I l
0 10 20 30 0 10 20 30 time (min) time (min)
FigUre 6.5. Comparison of peak shapes obtained using conventional split înjection with those obtained. using on-line SFE/CGC. A standard solution was spileed onto clean sand and exuacted. on-line with modified. C01• Exilaction time: static 20 min + dynamic 30 min. Column: 25 m x 0.32 mm i.d., 0.52 IJ.m ftlm. 1. Naphthalene, 2. Acenaphthene, 3. C16, 4. Anthracene, 5. Pyrene, 6. Chrysene, 7. Benzo[a]pyrene, 8. C32• (A) Split injection; (B) CO:z + CHzC~; (Q C01 + CH30H; (D) C02 + CHC13•
The effects of a modifier on the peak: shapes will depend on the amount of modifier transferred into the column. In split SFE/CGC only a small fracrion of the modifier is actually transferred to the column. The vast majority is vented via the split line. In our experiments, the split ratio during extraction was between

1 :2(~Y300 which means that only approximately 2 to 3 IJL of modifier was transferred to the column. Therefore, the effect of the modifier on the peak shapes are limited. When looking to the influence of a modifier on band focusing in the GC column, two counteracting mechanism can be distinguished. On the one hand modifierscan diston the trapping process by "washing" the trapped components along the CGC column. On the other hand, the uptake of a modifier by the stationary phase will facilitate focusing. The latter effect is called solvent trapping and is generally found to be more significant at trapping temperatures well below the boiling point of the solvent [16]. Moreover, at low temperatures the trapping efficiency of the stationary phase itself is higher. Prom the results and the discussion presented above, it can be concluded that in split SPE/CGC low trapping temperatures are generally beneficial for collecting and focusing the extracted components at the inlet of the CGC column. This is especially true for the volatile components, irrespective whether modifiers are used or not. This makes the selection of trapping conditions in split SPE/CGC relatively easy. It is interesting to see that the conclusions reached here are in contrast to the observations in off-line SPE when employing solid phase trapping. In the latter case, the optimum trapping temperatures should be selected above the boiling point of the modifier used, otherwise the liquified modifiers can rinse the target components from the trapping bed [17].
A secoud parameter that can affect the ability of the CGC column to focus the extracted analytes is the dynamic extraction time used in the extraction. Evidently, the shorter the dynamic extraction time, the easier is trapping. Pigure 6.6 shows the effects of the dynamic time on the peak shapes in split SFE/CGC at an initia! column temperature of 35·c. At a dynamic time of 15 minutes all peak shapes, except that of naphthalene, compare favourably with the peak shapes obtained by conventional split CGC. Por the SPE extraction of certain environmental samples, 15 minutes dynamic extraction can already be sufficient if the extraction conditions are suitably selected [2, 3]. If longer dynamic times of 30 minutes and 60 minutes are employed, the peak shape of naphthalene gets sigrii:ficantly worse and also the acenaphthene peak starts to broaden. At a dynamic time of 60 minutes the naphthalene peak can no Jonger be detected, most likely because it is far too wide or already eluted from the column during the SPE process. Similar, but stronger, effects of the dynamic extraction time on the peak shapes were also observed at higher trapping temperatures. Prom these results it is evident that the components can be more easily trapped at shorter dynamic extraction times.
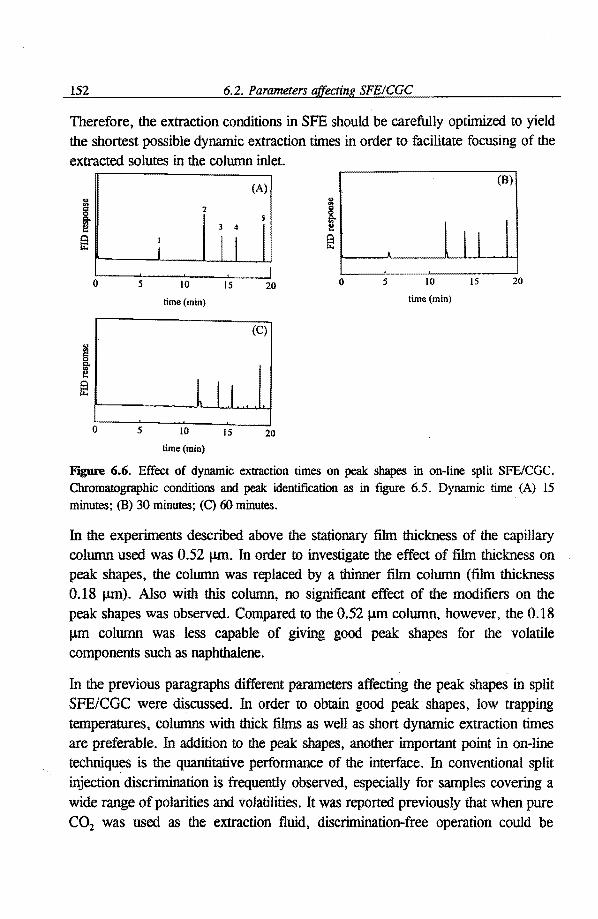
152 6.2. Parameters qf[ecting SFEICGC
Therefore, the extraction condirlans in SFE should be carefully optimized to yield the shortest possible dynamic extraction times in order to facilitate focusing of the extracted solutes in the column inlet.
(A) (B)
.. ä .. 2 § a. f s
3 4
I !
IÈ ! I I IÈ I
0 5 10 15 20 0 5 10 15 20
time (min) time(min)
(C) .,
J IÈ l I .1
0 5 10 15 20 time(min)
Figure 6.6. Effect of dynamic extraction times on peak shapes in on-line split SFE/CGC. Chromatographic conditions and peak identification as in tigure 6.5. Dynamic time (A) 15 minutes; (B) 30 minutes; (C) 60 minutes.
In the experiments described above the stationary fi1m thickness of the capillary column used was 0.52 J.i.m. In order to investigate the effect of fi1m thickness on peak: shapes, the column was replaced by a thinner film column (film thickness 0.18 J.l.m). Also with this column, no significant effect of the modifiers on the peak: shapes was observed. Compared to the 0.52 J.l.m column, however, the 0.18 IJ.ID column was less capable of giving good peak: shapes for the volatile components such as naphthalene.
In the previous paragraphs different parameters affecting the peak: shapes in split SFE/CGC were discussed. In order to obtain good peak: shapes, low trapping temperatures, columns with thick fi1ms as wellas short dynamic extraction times are preferable. In actdition to the peak shapes, another important point in on-line techniques is the quantitative performance of the interface. In conventional split injection discrimination is frequently observed, especially for samples covering a wide range of polarities and volatilities. It was reported previously that when pure co2 was used as the extraction fluid, discrimination-free operation cou1d be

Chapter6 153
obtained in on-line split SFE/CGC by operating the interface at sufficiently high temperatures [9]. Unfortunately, no evaluation of the quantitative aspects of the split/splitless interface is reported in literature for situations in which modifiers are used. A summary of the reproducibility and the quantitative performance of the split/splitless interface for on-line SFE/CGC using both pure and modified C02 is given in table 6.4. As can beseen from this table, the reproducibility of
Table 6.4. Quantitative aspectsof SFE/split CGC of PAHs from spiked sand using pure and modified co2 a).
Compo- na ph- ace na ph- c,. anthr- pyrene chr- benzo [a] nent thaiene thene acene
Pure C02 P.A.b 570142 912237 540121 545437 898845 658779 1442560 (RSD%)' (7.1) (5.5) (2.1) (5.4) (4.7) (2.6) (2.4)
R" 1.094 0.998 internat 1.059 1.079 1.070 1.072 (RSD%) (7.1) (3.4) standard (3.2) (2.6) (2.4) (1.9)
C02 + P.A. 600976 1043962 603702 583483 936719 632504 1556069 CH2CI. (RS[)%) (4.7) (5.6) (1.0) (0.2) (0.2) (1.0) (1.2)
R 1.040 1.022 internat 1.016 1.019 0.919 0.972 (RSD%) (5.2) (4.8) standard {0.7) (0.8) (1.7) (1.0)
co2 + P.A. 551701 937967 526600 519347 811894 579543 1317986 CHCI3 (RSD%) (1.9) (8.4) (2.1) (2.4) (2.7) (4.0) (3.2)
R 1.095 1.053 internat 1.036 1.012 0.966 1.005 (RSD%) (0.3) (7.9) standard (0.4) (0.8) (4.1) (2.7)
co2 + P.A. 1136573 2164132 1193804 978799 1367386 739941 1511771 CH30H (RSD%) (1.9) (16.2) (17.8) (19.3) (17.6) (7.0) (8.3)
R' 1.015 1.073 interna I 0.859 0.719 0.549 0.513 (RSD%) (17.2) (2.0) standard (3.2) (7.8) (9.3) (8.3)
''rapping at SO'C; bl Peak. area; "'Relative standard deviation, based on three experiments; <IJ Values relarive 10
standard data.
split SFE/CGC is excellent. Only in the case that methanol is used as the modifier, the relative standard deviations (RSD%) of some peak. areas are slightly above 10%. The RSD% of peak areas is better than 10% under all other conditions tested. In table 6.4 the trapping temperature was 50T. Similar results were also observed at 35"C and 70T. Another interesting point that becomes evident from this table is that the quantitative aspects of the interface depend on which modifier is used. For the SFE extraction of PAHs, discrimination is absent if dichloromethane or chloroform is used as the modifier. If methanol is used, however, significant discrimination occurs. The reason for this is not yet completely clear. The discrimination observed for methanol might be due to the poor solubility of the PAHs in methanol, as discrimination-free operation can be obtained in the extraction of caprolactam from nylon-6 with methanol as the modifier as will be demonstraled below. Hence, by carefut selection of the

154 6.2. Parameters a{fecting SFE/CGC
modifier, discrimination-free operation can be obtained in split SFE/CGC.
On-line SFEIPIV CGC of spiked samples The PTV injector has proven to be a useful and flexible injection system in GC. Apart from split and splitless injections, this device can also be used for large volume sample introduetion in GC. In that mode the solvent can selectively be
eliminated from the sample [16]. In on-line SFE/CGC using the PTV injector as the interface, the extracted components are first collected on the solid trapping material packed into the PTV liner. The extraction tluid, C02 and the modifier (if used), are vented through the split line. The trapped components are then transferred to the CGC column by heating the irtlector and sweeping the components in the splitless mode with carrier gas. With the PTV interface, all of the extracted components are transferred to the CGC column, resulting in a higher sensitivity ooropared to split SFE/CGC. Hence, on-line SFE/PTV CGC is particularly attractive fortrace analysis.
Figure 6.7 shows the chromalogram of the PAHs obtained using on-line SFE/PTV CGC. The quantitative performance of the PTV interface is shown in Table 6.5. In order to allow a direct comparison, identical amounts of the standard solution as in the spiking experiments were injected using a syringe. The peak areas obtained in on-line SFE/PTV CGC were expressed relative to the peak areas found in the conventional PTV splitless injection. From this table, it can be seen that when pure co2 is used as the extraction fluid, higher trapping efficiencies for the volatile components could be obtained by using lower trapping temperatures. For the less volatile components trapping is relatively easy and the trapping temperatures tested do not have a considerable effect on the trapping efficiency. When dichloromethane was used as the modifier, similar results were observed when the trapping temperature was above the boiling point of the modifier. When trapping was performed at a temperature below the boiling point of the modifier, however, a different situation occurred. No components could be trapped in the PTV at a trapping temperature of OT. This is easy to understand. At o·c dichloromethane is liquified and rinses the extracted components from the PTV. From the results shown above it can be concluded that when pure C02 is used as the extraction fluid, the selection of trapping conditions is relatively straightforward. When modifiers are used, however, the selection of trapping conditions is more difficult. On the one hand, the trapping temperature should be selected above the boiling point of the modifier used, while on the other hand, the

Chapter6 155
trapping temperarure should be selected as low as possible for trapping the volatile components. Therefore, when a modifier is required in on-line SFE/CGC using a PTV interface, organic solvents with low boiling points, such as dichloromethane, should first be considered. Alternatively, a liner packed with a selective and strong adsorbent could be used.
(A) (B) 2 s 1 3
4 8 1 6
''---.. I t 1 ... 0 5 10 15 20 25 30 35 0 5 10 15 20 25 30 35
time (min) time (min)
F:agure 6.7. Chromatogra.ms of the spiked PAHs obtained using on-line SFE/PTV CGC. A standard solution was spik:ed onto clean sand and extracted with on-line SPElPTV CGC. The PTV was kept at O'C when pure C01 was used and at 45'C when dichloromethane was used as the modifier. Other chromatographic conditions and peak identification as in tigure 6.5. (A) Pure C01; (B) C01 + CH2Cl2
Table 6.5. Recovery of spiked PAHs and alkanes in on-line SFE/PTV CGC.
Extraction fluid PureC02 C02 + CH2Cl2
Trapping o·c 45'C 70'C 45'C tem:perature
naphthalene 106"1 85 3 59
acenaphthene 105 100 85 88
cl6 106 103 90 91
anthracene 107 108 96 94
pyrene 120 120 107 98
chrysene 104 112 97 100
benzo[a ]pyrene 95 102 95 109
c32 104 120 106 109 •l Recovery(%) expressedas peak areasin on-line SFEJPTV COC relative to the peak areas obtained in conventional PTV splitless injection.
6.2.3.2. On-line SFEICGC of polymerie samples Polymers are widely used materials nowadays. Their properties can be

156 6.2. Parameters ajfecting SFEICGC
considerably affected by the presence of additives and/or oligomers. The increased diffusirivity of supercrirical fluids over liquid, the adjustable extraction temperature as well as the variabie solvent strength, have made SFE attractive for the polymer applicarions. Till now, most of the SFE applications to polymerie samples were carried out in off-line mode. To our knowledge, there are no publications on directly coupled SFE/CGC analysis of polymerie samples.
(A) (C) ~ intemal caprolactam 31
& standani !
~
~ ~·
'î 10 15 20 25 30 10 15 20 25 30
tîme(min) time(min)
(B) (D)
~ . .., "'
~ ! ~
~ ~
10 15 20 25 30 JO 15 20 25 30 time(min) time(min)
Figure 6.8. Effects of extraction time and stationary phase film thickness on the chromatographic peak shapes of caprolactam in SFE/CGC wîth pure C02• lnternal standard: Pentadecanoic acid methyl ester. (A) Split injection, 0.18 J.Lm column; (B) SFE/CGC, static 20 min + dynamic 30 min, 0.18 J.Lm column, trapping at 7o·c; (C) SFE/CGC, static 20 min + dynamic 60 min, 0.18 J.Lm column, trapping at 7o·c.(D) SFE/CGC, static 20 min + dynamic 60 min, 1.2 J.Lm column, trapping at 35·c.
In coupled SFE/CGC of polymerie samples, the splitlsplitless injector is the fust choice for the interface. This mainly because the concentrarions of the addirives or oligomers are relarively high (from hWldreds to thousands of ppm). The ability of on-line split SFE/CGC to give good peak: shapes at optimized SFE condirlans is invesrigated by comparing the chromatograms from conventional split injecrions of the extracts obtained by off-line SFE with those from on-line SFE/CGC analyses. In the extraction of polymerie samples relarively long extraction times

Chapter6 157
are frequently requireel. In order to investigate the effects of extraction time on the peak shapes, different extraction times were investigateel experimentally. Figure 6.8 shows the effects of the extraction time and stationary phase film thickness on the peak shapes generateel by SFE/CGC of nylon-6. If nylon-6 was extracteel for 50 minutes, very good peak shapes were observeel for both the 0.18 IJ.m and the 1.2 j.lm columns at all trapping temperatures testeel. However, if the polymer was extracted for 80 minutes, considerably broadeneel peaks were observeel with the 0.18 j.lm column at all trapping temperatures tested. In contrast to this, if the column with the thicker film (dr = 1.2 IJ.m) was useel at trapping temperatures below 60·c, good peak shapes were obtaineel. Nevertheless, the chromatographic peaks obtained with the thicker film column were also slightly broadened when trappeel at 7o·c. These observations support the conclusions reacheel insection 6.2.3.1 that thick film columns and low trapping temperatures are profitable for obtaining good chromatographic peak shapes in on-line SFE/CGC.
lO
10
caprolac:tam intemal
standatd
IS
15
20
time(min)
20
time(min)
25
2S
(A)
30
(C)
30
(8)
l ~
e ~ r--
10 IS 20 2S 30
time(min)
Figure 6.9. Effects of trapping temperature and stationary phase film thickness on chromatographic peak shapes of caprolactam in on-line split SFE/CGC using modified C02•
Internal standard: Pentadecanoic acid methyl ester. Extraction time: static 20 min + dynamic 30 min. (A) Trapping at 35•c, 0.18 J..Lm column; (B) Trapping at 60·c, 0.18 J..Lm column; (C) Tapping at 60·c, 1.2 J..Lm column.

158 6.2. Parameters af[ecting SFE/CGC
A similar influence of trapping temperature and film thickness on the peak shapes was found when a modifier was used (see fig. 6.9). If nylon-6 is extracted at 50'C and 300 bar with 0.5 mL methanol as the modifier, very good chromatographic peak shapes were observed for both the 0.18 J.Lm and the 1.2 J.Lm columns at the lowest trapping temperature tested (35'C). However, for the 0.18 J.Lm column the chromatographic peaks were seriously broadened and splitted at trapping temperatures higher than 60'C. Opposedly, for the 1.2 J.Lm column very good peak shapes were obtained when trapping at 60'C. Only slight splitting of the peaks was observed at the trapping temperature of 70'C. It was shown in tigure 6.9 that when nylon-6 was extracted with pure co2 for the same period of time, good peak shapes were obtained at all trapping temperatures tested on both columns. Therefore, the peak splitting must be due to the presence of the modifier. As discussed in section 6.2.3 .1, the effects of a modifier on the focusing of the extracted components at the head of the CGC column are twofold. The enhancement of retention power of the stationary phase by the up-take of modifier is more important for columns with thinner film thickness. For thick film columns this effect is less significant as the retention power of these columns is already relatively strong. From the results shown above, it is clear that when a modifier is necessary for the SFE extraction, it is better to select the trapping temperature well below the boiling point of the modifier. This is especially true when columns with thick films are not available or can not be used because of the presence of high molecular weight components in the sample.
Table 6.6. Quantitative aspects of on-line split SFE/CGC analysis of caprolactam in nylon-6.
Trapping 35'C temperature
SFE conditions ppmdl (RSD%)'>
50'C and 300 2014 bar"> (5.9)
150'C and 300 2244 bar") (5.9)
50'C
2051 (7.4)
2318 (7.9)
70'C
2118 (8.0)
2300 (9.1)
on-column•>
1852 (7.5)
2272 (3.6)
•> Obtained with off-line SFE/CGC; b>o.s mL methanol as the modifier, extraction time: static 20 minutes and dynamic 30 minutes; c) Pure C02, extraction time, static 20 minutes and dynamic 60 minutes; dl
Amount found relative to the mass of polymer weighed into the extraction cel!; •> Based on three experiments.
In order to investigate the quantitative performance of the coupled SFE/CGC

Chapter6 159
analysis of nylon-6, the results obtained by on-line SFE/CGC were compared with those from off-line SFE/on-column injection CGC (table 6.6). For both the on-line and the off-line mode, identical SFE conditions were used. It is obvious from table 6.6 that the results obtained by on-line SFE/CGC agree very well with those obtained by off-line SFE/on-column injection GC.
On-line SFE/CGC was also applied to the analysis of the dimer and trimer in PBT. Also here very good peak shapes were observed at all the extraction conditions and trapping temperatures tested (figure 6.10). The boiling points of the dimer and trimer from PBT are much higher than that of caprolactam. It is therefore evident that it is much easier to trap these components.
(A) (B)
~ intemal
I <timer Slandard t· ~ ~
I triroer I
10 20 30 40 50 60 10 20 30 40 50 60 time (min) time(min)
Figure 6.10. Comparison of peak shapes obtained by conventional split injection with those by on-line split SFE/CGC analysis of the dimer and trimer in PBT. InternaJ standard: Irgnox 1076. (A) split injection; (B) SFE/CGC, C02 + CHCI3 at so·c and 300 bar, static 20 min + dynamic 30 min.
6.2.4 CONCLUSIONS
On-line SFE/CGC with a split interface was found to be suitable for samples that have high concentrations of extraetabie components. Under optimized conditions, modifiers have no actverse effects on the chromatographic peak shapes in split SFE/CGC. Discrimination-free operation could also be obtained in on-line split SFE/CGC when modified C02 was used as the extractant. Short dynamic extraction times, low trapping temperatures and columns with thick films were found to be beneficia! for obtaining good peak shapes in split SFE/CGC. For online SFE/CGC with a PTV injector as the interface, the most important parameter to be optimized is the initial PTV trapping temperarure. Low trapping temperatures will yield high trapping efficiencies when pure co2 is used. In contrast, when modified co2 is used, the trapping temperature should be selected above the boiling point of the modifier. On-line SFE/PTV CGC is particularly

160 6.2. Parameters qf[ecting SFEICGC
attractive for trace analysis.
REFERENCES
1. T.L. Chester, J.D. Pinleston and D.E. Raynie, Anal. Chem., 66 (1994) 106R.
2. S.B. Hawthome, Anal. O!em., 62 (1990) 633A.
3. M.L. Lee and K.E. Mark:ides, Analytica/ Supercritical Fluid O!romatography and E.xtraction, Chromatography Conference, Provo, UT, 1990.
4. J.J. Langenfeld, S.B. Hawthome, D.J. Milier and J. Pawliszyn, Anal. O!em., 65 (1993) 338.
5. N.J. Cotton, K.D. Bartle, A.A. Clifford and C.J. Dowle, J. O!romatogr. Sci., 31 (1993) 157.
6. X. Lou, H.-G. Janssen and C.A. Cramers. J. Microcolumn Separations, 7 (1995) 303. 7. J.J. Langenfeld, S.B. Hawthome, D.J. Milier and J. Pawliszyn, Anal. O!ern., 66 (1994) 909. 8. X. Lou, H.-G. Janssen and C.A. Cramers, J. O!romatogr. Sci., 34 (1996) 282.
9. X. Lou, H.-G. Janssen and C.A. Cramers, J. High Reso/ut. Chromatogr., 16 (1993) 425.
10. S.B. Hawthome, D.J. Milier and M.S. Krieger, J. O!romatogr. Sci., 27 (1989) 347.
11. R.J. Houben, H.-G. M. Janssen, P.A. Leclercq, J.A. Rijks and C.A. Cramers, J. High Reso/ut. Chromatogr., 13 (1990) 669.
12. M.D. Burford, S.B. Hawthome and D.J. Miller, J. Chromatogr., 685 (1994) 79.
13. M.D. Burford, S.B. Hawthome and D.J. Miller, J. O!romatogr., 685 (1994) 95. 14. K.J. Hansen, B.N. Hansen, E. Cravens and R.E. Sievers, Anal. Chern., 67 (1995) 3541.
15. J.M. Levy, R.A. Cavalier, T.N. Bosch, A.F. Rynaski and W.E. Huhak:, J. O!romatogr. Sci., 27 (1989) 341.
16. H.G.J. Mol, H.-G. Janssen, C.A. Cramers and U.A.Th. Brinkman, J. High Reso/ut. O!romatogr., 18 (1995) 19.
17. M.N. Moore and L.T. Taylor, Anal. Chern., 67 (1995) 2030.

Cha ter 7 161
7 General Conclusions and Future Developments
7.1. General conclusions
The application of supercritical fluids, particularly carbon dioxide, in analytica! chemistry has experienced exciting developments during the last 15 years and the interest intheir application still continues to grow. The main application areas of supercritical fluids in analytica! chemistry include the employment of supercritical fluids as mobile phases for chromatography (SFC) and as solvents for extraction (SFE). SFC can generally be considered as an intermediale technique between GC and HPLC. Potential advantages of SFC over HPLC include the compatibility with various GC detectors and the increased speed of analysis. In comparison with GC, SFC is advantageous for the analysis of high molecular weight or thermally labile compounds. SFE is a powerful alternative to extractions using liquid solvents. Compared to conventional extraction methods SFE has many advantages, such as reduced usage of toxic organic solvents, · shorter extraction times, adjustable selectivities, and the ability for on-line combination with other analytica! techniques and instruments.
Although very different at fust sight, SFE and (packed column) SFC are very much similar in many aspects. In both techniques, a supercritical fluid is used to dissolve the components and transport the analytes through a packed bed of solid particles. It is well known that retention in SFC is not only controlled by the supercritical mobile phase, but also by the nature of the stationary phase. Similar statements are also correct in SFE, where variations in the composition of the sample matrix often require a substantial adjustment of the extraction conditions. Extraction of components from solid matrices is controlled by many of the same parameters controlling retention in SFC. SFC can hence be used to speed up method development in SFE. If a solute requires extreme elution conditions in SFC, extraction in SFE will certainly be difficult. For homogeneous samples, in which the
extraction kinetics are controlled by elution of the extracted solutes out of the extraction cell, SFE extraction conditions can be predicted directly from SFC

162 General conclusions and future deve/opments
retention data. For inhomogeneons samples in SFC, lower capacity factors are observed at higher concentration levels [ 1]. Analogously, solute concentration can have a considerable effect on the extraction kinetics for inhomogeneons samples in SFE.
Supercritical fluids are sometimes considered to he "super-solvents" because of their liquid-like solvating powers in combination with more gas-like ditfusion coefficients and viscosities. One of the most important parameters that must be considered in supercritical fluid technology is the solubility of target compounds in the supercritical phase. The solubility of a component in a supercritical fluid is controlled by (i) its vapor pressure and (ii) its interaction with the supercritical fluid. Solubility can he determined by using various instrumental techniques including gravimetrie, spectroscopie, chromatographic and on-line measurements using a variety of chromatographic detectors. Among the chromatographic detectors, it was found that a flame ionization detector (FID) coupled to a saturation cellis a simple and reliable method for the determination of solubilities of organic compounds of low to moderate solubilities in supercritical carbon dioxide [2]. With the on-line FID method, the contribution of vapor pressure to the overall solubility can be estimated by using helium as the carrier fluid in stead of supercritical carbon dioxide. In this way, the two different contributions to the overall solubility can he differentiated. The experiments performed clearly illustrate that in literature the contribution of vapor pressure to overall.solubility is often overestimated.
Comparatively speaking, the influence of pressure on solubility is relatively straightforward. Increasing pressure generally results in a higher solubility due to stronger interactions between the supercritical fluid and the solute molecules. In
contrast to this, the effects of temperature on solubility are much more complicated. Higher temperatures will, on the one hand, increase the vapor pressure of the components, while on the other hand decrease the supercritical fluid density. Moreover, temperature can also affect the physicochemical properties of both the supercritical fluid and the solutes. The actual effects of temperature depend on the properties of the solute and the supercritical fluid as well as the experimental conditions. In addition to the effects on solubility, temperature also influences the

Chapter 7 163
affinity of solutes for the stationary phase in SFC, and ditfusion and desorption rates
of analytes in SFE.
One parameter of great concern in SFC is the pressure gradient along the column. In SFC relention and resolution are strongly controlled by the pressure of the supercritical fluid. Large pressure drops are encountered in SFC if packed columns are used, which means that many chromatographic parameters can vary significantly along the column. The capacity factors increase along the column because of the
pressure drop. The variation of selectivity and resolution along the column can be
much more complicated. No simple equation is available for the calculation of
resolution in packed column SFC, since selectivity (a.), retention (k) and plate height
(H) are all changing along the column. Increased plate numbers obtained by using
longer columns do not necessarily yield a better separation due to the variations of a. and k. For components for which the pressure drop has no adverse effect on selectivity, longer columns can give better separations. On the contrary, for
components for which the elution order is strongly dependent on pressure, resolution
may even get worse when longer columns are used. In order to obtain a good separation in SFC, all the parameters that can affect the pressure gradient along the
column, e.g., the inlet pressure, supercritical fluid flow rate and column length should all he carefully optimized. Evidently, temperature is another parameter that
must be considered. By using the numerical methods developed previously [3], the
variation of various chromatographic parameters along the SFC column can be predicted accurately. The model calculations using these equations can be used for
method development in SFC.
Compared to SFC, the growth in SFE has been even faster. SFE has successfully
been applied to a wide variety of matri:xlanalyte combinations. In the extraction of polymer additives and oligomers from polymerie samples, the SFE process normally
involves three steps. First, the solutes have to diffuse from the core of the polymer particles to the surface. Next, the compounds are transferred from the surface into
the e~action fluid. Finally, the compounds are eluted out of the extraction cell by
the flow of extraction fluid. The SFE extraction rate is limited by the slowest of
these three steps. Because of the gas-like properties of supercritical fluids, mass

164 General conclusions and future developments
transfer from the surface of the polymer particles into the supercritical extractant is very fast. Hence, the slowest, and therefore the rate limiting step, is either diffusion inside the polymer particles or elution out of the extraction cell. A thorough understanding of the kinetics of mass transfer can greatly facilitate methad development in SFE and is essential to comprehend the effects of various experimental parameters on the extraction efficiency. Ifthe rate limiting parameter is diffusion in the polymer particles, the extraction rate can be increased by raising the extraction temperature or by the addition of a suitable modifier which can swell the polymer particles resulting in faster diffusion. Evidently, the extraction rate can also be increased by reducing the partiele size of the polymer sample. On the other hand, if the rate limiting step is solubility in the supercritical extractant, the extraction rate can be increased by increasing the extraction pressure, adjusting the extraction temperature, increasing the supercritical fluid flow, or adding a suitable modifier to enhance the solvent strength of the extractant. The role of modifiers in the SFE extraction of polymerie samples is twofold. A suitable modifier increases the solvent strength of the supercritical fluid and swells the polymer particles. Bath modifier identity and concentration were found to be important for improving the extraction efficiency. Modifiers are generally more effective at low temperatures. At high temperatures, modifiers may sametimes even have negative effects on the extraction efficiency. Continuous modifier addition or repeated spiking of a suitable modifier may be advantageous if a spiked modifier elutes rapidly or if high modifier concentrations are required.
In addition to SFE, some polymerie samples were also extracted with ASE. ASE is a sample preparation methad developed very recently. In ASE, a solid sample is packed into an extraction cell and then extracted with a suitable solvent at elevated temperature and pressure conditions. Higher extraction temperatures result in an improved solubility and faster diffusion and desorption rates, yielding more efficient extractions. Elevated pressures are required to maintain the extraction solvent in the liquid state at temperatures above its normal boiling point. The kinetics of mass transfer in ASE are quite similar to those in SFE. The main difference is the use of different extractants, i.e. supercri ti cal fluids in SFE and high temperature liquids in ASE. Methad development in ASE is relatively straightforward because less

parameters affect the extraction efficiency. Extraction solvent and temperature were found to he the most important parameters that should be optirnized in ASE. A poor solvent for Soxhlet extraction can be a good solvent in ASE due to the high extraction temperatures used. Some additional selectivity can be introduced in ASE by careful selection of the extraction solvent and temperature. Although ASE is still in its early stage of development, it is a very promising technique for sample preparation.
One of the most attractive advantages of SFE is its easy combination with other analytica! instruments, especially with GC. In the on-line combination of SFE to GC, three essential steps, i.e. SFE, transfer the extracts to the GC column, and GC separation can be identified. Each of the conditions in these steps should be optimized carefully before the analysis can be completed successfully. Optimization in SFE bas been discussed in the preceding paragraphs. The performance of transferring the extracts to the GC column is largely determined by the quantitative aspects of the interface and the chromatographic peak shapes. Several different interfaces have been described for on-line SFE-GC. The behavior of a split injector when used as an interface is remarkably different from its normal behavior in conventional GC. In on-line SFE-GC with a split/splitless interface, the split ratio bas no influence on the peak area if the column flow rate and the carbon dioxide flow rate are kept constant. The peak areas were found to decrease with increasing carbon dioxide flow rate. With a split/splitless injector as the interface, discrimination free operation can he obtained both for pure and modified carbon dioxide as the extractant. Under optimized conditions, modifiers have no adverse effects on peak shapes in split SFE-GC. Split on-line SFE-GC is suitable for samples having high concentrations of extraetabie components. For on-line SFE-GC with a prograrnmed temperature vaporizer (PTV) injector as the interface, low trapping temperatures will yield high trapping efficiencies when pure carbon dioxide is used. In contrast, when a modifier is used, the trapping temperature should he selected above the boiling point of the modifier. On-line SFE-PTV -GC is particularly attractive fortrace analysis.

166 General conclusions and future developments
7.2. Future developments
The recent concerns about decreased operation and analysis time, Jower waste generation and disposal costs will further support the application of supercritical fluids in analytica! chemistry. However, the biggest obstructions in the growth of supercritical fluid techniques have been and will continue to be the high price of the commercial instruments and the complex physicochemical properties of supercritical fluids. The lack of fundamental knowledge about supercritical fluids conceming phase behavier ( especially in multi-component systems) and the prediction of solubilities makes metbod development in supercritical fluid techniques very difficult and tîme-consuming. In the following paragraphs, the trends in future developments in SFC, SFE and ASE are briefly discussed.
7.2.1. Future developments in SFC
In SFC both open-tubular columns and packed columns are routinely used. A definite advantage of open-tubular columns over packed column is the high degree of surface deactivation which enables the elution of more solutes with pure carbon dioxide. However, the application of open-tubular SFC is complicated in some cases because of the lack of injection systems capable of delivering very narrow input bands and of highly sensitive detectors. Compared to open-tubular SFC, packed column SFC is better developed in view of instrumentation and ease of operation. Moreover, analyses with packed columns are generally faster. One important area in packed column SFC where a lot ofwork is still required is column technology. The development of homogeneons packing materials exhibiting reduced number of active sites on the surface will continue. Another area that requires further development in packed column SFC is optimization. Many parameters, such as the addition of modifiers and the pressure drop along the column, can influence the result of separation. Optimization and method development nowadays are stiH largely empirica!.
7.2.2. Future developments in SFE and ASE
Although SFE has successfully been applied to a wide variety of analyte/matrix combinations, a lot of work is still required before it can routinely be used as a

Cha ter 7 167
sample preparation method in analyticallaboratories. This is mainly because method
development in SFE is rather complicated. Smalt variations in sample matrix
composition frequently require substantial adjustment of the extraction conditions.
The addition of a suitable modifier is o:ften found to be necessary to yield
quantitative extraction as supercritical carbon dioxide does not have suftleient
solvent strength for the extraction of polar analytes, and it is a poor solvent for
overcoming the interaction between analytes and the matrix. The selection of
modifier type and concentration as well as other experimental parameters such as
extraction temperature and pressure are still largely based on trial and error
experiments. Moreover, the quantitative collection of the extracted components
from the large volume of gas a:fter depressurization from the supercritical fluids is by
no means easy, especially for volatile components and when modifiers are used. In
addition to these, the SFE restrictor can be blocked in the extraction of some
complex samples. Despite all these difficulties, the application of SFE will surely
continue to grow because of its attractive advantages over the traditional extraction
methods. In order to speed up method development in SFE, more attention should
be paid to improve the fundamental understanding of experimental parameters that
affect the extraction kinetics and the coneetion efficiency.
ASE is a new sample preparation technique developed only very recently. The use
of water as the extractant in ASE is very attractive since water is inexpensive,
widely available in a high purity, and friendly to the environment and the analyst. In
addition to these, the standard methods developed for water analysis can directly be
used for further pretTeatment and analysis of the extracts.
References
l. P.J. Schoenmaker, L.G.M. Uunk and P.K. de Bok:x, J. Chromatogr., 459 (1988) 201. 2. M.J. Milier and S.B. Hawthome, Anal. Chem., 67 (1995) 273. 3. H.-G. Janssen, H. Snijders, C.A. Cramers and P.J. Schoenmaker, J. High Resolut. Chromatogr., 15
(1992) 458.

168 Summary
Summary
A fluid is said to he in its supercritical state when both its temperature and pressure are above their respective critica! values. Supercritical fluids can offer liquid-like solvating powers in combination with gas-like ditfusion coefficients and viscosities. These properties make supercritical fluids very attractive to he used in both chromatography and extraction.
Chapter 1 opens with a general introduction. In this chapter the history of supercritical fluids in analytical chemistry is briefly reviewed and the scope of the thesis is presented. The basic principles of SFC and analytica! SFE are described in chapter 2.
In section 2.1, the properties of supercritical fluids are briefly reviewed. Attempts are made to identifY similarities and differences between analytica! SFE and SFC, as well as between analytica! SFE and preparative-scale SFE. Special attention is paid to the instrumental requirements and operation of analytical SFE. As analytica! SFE is a new and developing method, the process of its optirnization is discussed in detail. Attempts are also made in this section to compare analytical SFE with some traditional sample preparation methods. SFE offers many attractive advantages, such as short extraction times, high selectivity, reduced usage of toxic organic solvents, easy on-line combination withother analytica! instruments, etc .. However, as a new developing technique, SFE also suffers from some disadvantages. Among these, the high cost of the instrumentation and the difficulty in method development are the most important ones. In spite of the shortcomings of SFE, it has successfully heen used in a wide range of applications and a lot of interesting results have been obtained. SFE surely has the potenrial to become an important sample preparation method in analytica! chemistry.
Although very different at fust sight, SFE and (packed column) SFC are very much sirnilar in many aspects. In section 2.2, the possibility of using SFC retention data for method development in SFE is investigated. A model is derived that enables the predierion of SFE extraction yields from SFC

Summary 169
retention data measured using the matrix that is to be extracted as the stationary phase. For samples in which the extraction kinetics are controlled by elution out of the extraction cell, experimental extraction yields and predicted data are in excellent agreements. It is demonstrated in this section that the matrix composition and analyte concentration can significantly affect the SFE extraction efficiency.
The estimation of solubility in supercritical fluids is of crucial importance in metbod development for both SFC and SFE. In chapter 3 solubilities of a series of polycyclic aromatic hydrocarbons (P AHs) in supercritical carbon dioxide are measured using an on-line FID technique. A metbod is developed which enables the measurement of the contribution of solute vapor pressure to its overall solubility. The effects of temperature and pressure on solubility are fundamentally investigated. Furthermore, equations are derived which can be used for tbe estimation of tbe temperature influence on affinity of a solute for tbe stationary phase in SFC. In literature, the contribution of vapor pressure to the overall solubility is frequently overestimated, especially for semi-volatile or non-volatile compounds. In addition to its effects on solubility, temperature variations can also strongly affect tbe affinity of solutes for tbe stationary phase in SFC.
In chapter 4, tbe influence of pressure drop along a packed column on tbe quality of SFC separations obtained using pure carbon dioxide as tbe mobile phase is discussed. Numerical metbods developed previously are used to model the variation of pressure along the column and, to investigate its impact on efficiency and selectivity. The results of the tbeoretical studies are in good agreement with tbe experimental verifications. In packed column SFC, as tbe chromatographic parameters, retention factor, selectivity and plate height all vary along tbe column, longer colunms do not necessarily give better separations. For components for which tbe pressure drop has no adverse effect on selectivity, longer columns may yield better separations. On tbe contrary, for components for which the elution order is strongly pressure dependent, resolution may get worse when longer columns are used.

The extraction kinetics and strategies for rnethod developrnent in SFE and accelerated solvent extraction (ASE) of polymerie samples are discussed in detail in Chapter 5. The two-film theory, which considers mass transfer across a phase boundary, is applied to describe qualitatively the kinetics of mass transfer from the polymer particles into the extractant. In SFE, the ratelimiting step is either ditfusion inside the polymer particles or elution out of the extraction cell. If the rate-lirniting parameter is ditfusîon, the extraction rate can be increased by raising the extraction temperature. If, on the other hand, the rate-lirniting step is solubility, the extraction rate can be increased by increasing the extraction pressure. In addition to the variations of temperature and pressure, the addition of a modifier can also strongly influence the extraction rate in SFE. Both modifier identity and concentration are found to be important for improving the SFE extraction efficiency. Sorne general guidelines for method development in SFE of polymerie samples are given in section 5 .2. In the ASE extraction of polymerie samples, the selection of the extraction solvent and temperature are the most important parameters to be optirnized. A poor solvent in Soxhlet extraction may be a good solvent in ASE. The optirnization strategies in ASE extraction of polymerie samples are presented in section 5.3. Finally in this section, the performance of SFE and ASE for polymerie samples is compared and discussed.
Chapter 6 deals with the on-line combination of SFE with GC. A split/splitless injector and a temperature programmed vaporizer (PTV) injector are tested as interfaces for on-line SFE/GC. The parameters affecting the cbrornatographic peak shapes and the quantitative performance of the interfaces are identified and investigated. Special attention is paid to the situations where modified extraction fluids are used. In addition to these, the application of the on-line SFE/GC technique in the analysis of some real samples is dernonstrated. The results obtained are compared with those from conventional off-line extraction/Ge methods. With a split/splitless injector as the interface, discrirnination-free operation can be obtained both with pure and modified C02 as the extractant. When a PTV injector is used as the

interface, low trapping temperatures will yield high trapping efficiencies with pure C02 extractant. In contrast, when a modifier is used, the trapping temperature should he selected above the boiling point of the modifier. Under optimized conditions, modifiers have no adverse effect on the chromatographic peak shapes. Split SFE/OC is suitable for samples having high concentrations of extraetabie components, while SFE/PTV IOC is particularly attractive for trace analysis, for example in the environmental field.
Finally, some general conclusions and expected trends for future developments regarding the use of supercritical fluids in analytical chemistry are presented in Chapter 7.

SAMENVATTING
Een medium verkeert in de superkritische toestand wanneer zowel de druk als de temperatuur van de fase zich boven de voor deze stof geldende kritische waarden bevinden. Superkritische fasen combineren in principe de 'oplosmiddelsterkte' van een vloeistof, met de diffusiecoëfficiënten en viscositeiten van een gas. Deze combinatie van eigenschappen maakt superkritische media bijzonder aantrekkelijk voor gebruik als mobiele fase in een chromatografisch scheidingssysteem en als extractiemiddel in extractieprocessen.
In hoofdstuk 1 van dit proefschrift wordt een algemene inleiding gegeven die dient als basis voor hetgeen beschreven wordt in latere hoofdstukken. De historie van het gebruik van superkritische media in de analytische chemie wordt kort samengevat en de doelstelling van dit proefschrift wordt verduidelijkt. De basisprincipes van SFC en analytische SFE worden beschreven in hoofdstuk 2.
In paragraaf 2.1 worden de eigenschappen van superkritische media nader belicht. Er wordt gepoogd overeenkomsten en verschillen tussen SFE en SFC, en tussen analytische SFE en preparatieve SFE te identificeren om te komen tot een betere onderlinge positionering van deze technieken. Strategieën voor methode-ontwikkeling in de analytische SFE worden in detail besproken. Verder wordt een vergelijk gepresenteerd van analytische SFE met een aantal traditionele methoden voor monstervoorbewerking. Uit deze vergelijking komen de voordelen van analytische SFE overtuigend naar voren: SFE is snel, reduceert het verbruik aan toxische organische oplosmiddelen, biedt de mogelijke bepaalde stofklassen selectief te extraheren en maakt een directe koppeling met andere analytische instrumentatie mogelijk. SFE kent echter óok een aantal nadelen. Daartoe behoren met name de hoge kosten voor de vereiste instrumentatie en de moeilijke en tijdrovende methode-ontwikkeling. Ondanks deze problemen wordt SFE nu reeds succesvol toegepast voor een nog steeds groeiend aantal analytische toepassingen. Naar het zich laat aanzien zal SFE in de toekomst dan ook een belangrijke plaats gaan innemen in het brede scala van monstervoorbewerkingstechnieken voor de analytische chemie.
Hoewel SFE en (gepakte) SFC op het eerste gezicht twee sterk verschillende technieken lijken te zijn, vertonen deze technieken op een aantal gebieden een duidelijke overeenkomst. In paragraaf 2.2 wordt de

Samenvatting 173
mogelijkheid om SFC-retentiedata te gebruiken voor methodeontwikkeling in SFE nader bestudeerd. Er wordt een model afgeleid waannee SFE extractie-opbrengsten voorspeld kunnen worden uit SFC retentiedata gemeten met de te extraheren matrix als de stationaire fase. Voor monsters waarbij de extractiesnelheid wordt bepaald door de snelheid waarmee de geëxtraheerde verbindingen uit de extractiecel geëlueerd kunnen worden, komen de voorspelde extractie-rendementen zeer goed overeen met de voorspelde data. De experimenten beschreven in deze paragraaf tonen op overtuigende wijze aan dat de samenstelling van de matrix en de concentratie van de doel-stof in het monster, de extractiesnelheid en -opbrengst sterk kunnen beïnvloeden.
Informatie over de oplosbaarheden van de te extraheren of te scheiden stoffen in het superkritisch medium is van essentieel belang voor methodeontwikkeling in zowel SFC als SFE. In hoofdstuk 3 worden de oplosbaarheden van een reeks poly aromatische koolwaterstoffen (PAK' s) in superkritisch koolstofdioxide gemeten met een 'on-line' FID methode. Toepassing van een hier ontwikkelde methode maakt het mogelijk om de bijdrage van de dampspanning van een stof aan zijn totale 'oplosbaarheid' te bepalen. De effecten van temperatuur en druk op de oplosbaarheden in superkritische media worden nader bestudeerd. Een belangrijke conclusie uit deze studies is dat de bijdrage van de dampspanning van een component aan het transport van de component door de SFC kolom aanzienlijk kleiner is dan in de literatuur vaak wordt aangenomen. In dit hoofdstuk worden verder vergelijkingen afgeleid waannee de invloed van de temperatuur op de affiniteit van een stof voor de stationaire fase in SFC kan worden bepaald. Duidelijk is dat een verandering van temperatuur niet alleen de oplosbaarheid van een stof in de mobiele fase van een superkritisch-chromatografisch systeem beinvloedt, maar tevens een verandering in het retentiegedrag kan bewerkstelligen via een beïnvloeding van de affiniteit van de stoffen voor de stationaire fase.
Hoofdstuk 4 beschrijft de invloed van de drukgradiënt over een gepakte SFC kolom op de kwaliteit van de verkregen scheiding. In eerder werk ontwikkelde numerieke methoden worden gebruikt om de afname van de druk in de kolom-lengterichting te modelleren en de effecten daarvan op de efficiëntie en selectiviteit te beschrijven. De resultaten van de modelberekeningen zijn in zeer goede overeenstemming met de experimenteel gevonden data. Een opmerkelijke conclusie is dat in gepakte SFC, door de sterke invloed van druk(val) op efficiënties, selectiviteiten, retentiefactoren en schotelhoogten, langere kolommen niet

174 Samenvatting
noodzakelijkerwijze een betere scheiding leveren. Voor componenten waarbij de drukval geen nadelig effect heeft op de selectiviteit kunnen langere kolommen betere scheidingen geven. Voor componenten daarentegen waarbij de elutievolgorde sterk drukafhankelijk is kan een slechtere resolutie verkregen worden bij een toegenomen kolomlengte.
De extractiekinetiek in SFE en ASE en strategieën voor methodeontwikkeling in deze beide technieken zijn het onderwerp van hoofdstuk 5. De twee-film theorie, die massatransport door een fasen-grens beschrijft, wordt toegepast voor een kwalitatieve beschrijving van de snelheid van massatransport vanuit polymere deeltjes naar het extractiemiddeL In SFE is de snelheidsbepalende stap voor extractie de diffusie in het polymere deeltje danwel de elutie van de geëxtraheerde componenten uit de extractieceL Indien diffusie de snelheidsbepalende stap vormt, zal het werken bij verhoogde temperatuur een positief effect op de extractiesnelheid hebben. Indien echter de oplosbaarheid in de superkritische fase de beslissende factor is, is verhoging van de extractie druk de aangewezen weg om de extractie te versnellen. Naast variatie van druk en temperatuur kan ook de toevoeging van een organische modifier de extractiesnelheid positief beïnvloeden. Daarbij blijken zowel de identiteit van de modifier alsook de concentratie van belang te zijn. Enige algemene richtlijnen voor methode-ontwikkeling in de superkritische extractie van polymere monsters worden gegeven in paragraaf 5.2. Belangrijke parameters in de ASE extractie van polymeren zijn het te gebruiken extractiemiddel en de temperatuur. Opmerkelijk is dat een matig extractiemiddel in de Soxhlet extractie uitstekende resultaten kan geven in ASE. Optimalisatiestrategieën voor de ASE extractie van polymere monsters worden besproken in paragraaf 5.3. Tot slot worden de kenmerken van SFE en ASE voor de extractie van polymere monsters besproken.
Hoofdstuk 6 handelt over de on-line combinatie van SFE met capillaire GC. Van de split/splitless injector en de temperatuur programmeerbare PTV injector wordt de bruikbaarheid als interface in deze on-line koppeling bestudeerd. De parameters die de chromatografische piekvormen en de kwantitatieve prestaties van het interface bepalen worden geïdentificeerd en bestudeerd. Speciale aandacht wordt besteed aan het gebruik van binaire extractiemedia. Daarnaast wordt de toepasbaarheid van de on-line SFE-GC technique voor een aantal 'echte' monsters onderzocht. De resultaten verkregen voor deze monsters worden vergeleken met die van conventionele off-line extractie-Ge methoden. Met

een splitlsplitless injector als interface kan een discriminatie-vrije koppeling verkregen worden. Indien een PTV injector gebruikt wordt voor de interfacing, wordt kwantitatieve overdracht bij het gebruik van zuiver co2 als extractiemiddel slechts verkregen indien een lage initiële injectortemperatuur gebruikt wordt. Wordt daarentegen de extractie uitgevoerd met C02/modifier mengsels dan moet de interface-temperatuur boven het kookpunt van de modifier gekozen worden. Onder zorgvuldig geoptimaliseerde omstandigheden kan ook dan een kwantitatieve overdracht en goede chromatografische piekvormen verkregen worden. Split SFE-GC is geschikt voor monsters waarin zich hoge concentraties van de te extraheren stof bevinden. SFE-PTV-GC is meer geschikt voor sporenanalyse, bijvoorbeeld in de milieuanalyse.
Tot slot worden in hoofdstnk 7 enkele algemene conclusies en verwachte trends voor toekomstige ontwikkelingen met betrekking tot het gebruik van superkritische media in de analytische chemie weergegeven.

176 Acknowledgments
Acknowledgments
No thesis is the work only of its author. I am deeply indebted to Prof. Carel A Cramers for offering me the opportunity to carry out a doctorale project in his laboratory. It is his continuous support and supervision that led to the completion of this thesis. Special thanks go to my coach, Dr. Hans-Gerd Janssen. I appreciate his generous support, consistent guidance, constructive suggestions and supercritical comments far more than he could ever know.
I am grateful to Henri Snijders, Mark van Lieshout, Piet Leclercq, Jan de Haan, Denise Tjallema, Huub van Leuken, Anja Huygen, Hans van Rijsewijk, Harrie Maatbuis, Anton Bombeeck, and Dick Kroonenberg for their invaluable assistance. I also want to acknowledge my roommates and colleagues, Hans Mol, Al ex Schotten, Peter van Y sacker, Pham Tuan Hai, Erik Baltussen, Manuel Mertens, Gerard Rutten, Do Quang Huy, Angela Bickei and Cindy van den Boom for their kind co-operations, ideas and suggestions. I really enjoyed the working atmosphere in our group. Piet Leclercq, Henri Snijders, Dick Kroonenberg, Peter van Y sacker, Jan de Haan and Angela Bickei deserve my special appreciation for giving me lifts to symposia or for sight-seeing.
Special thanks are also due to my teachers in China, particularly Profs. Liangmo Zhou, Qinghai Wang, Daoqian Zhu and Yafeng Guan, for their support and encouragement during my stay in Dalian.
I owe a great deal to my parents who supported my education profusely even when they were struggling against poverty. I could not :find words to express how much I love them.
Last, but absolutely not least, I wish to thank the support from my wife, Yi Cui, who has become a major part of my life in the last five years. I can promise her that I will try to do more housework from now on.

Curriculum Vitae
The author was bom on 23 December 1966 in Yiwu county, Zhejiang province, P. R. China. After receiving bis B.S. in Chemistry from Zhejiang Institute of Engineering in 1985, he continued bis graduate study at Dalian Institute of Chemica! Physics (DICP), Chinese Academy of Sciences. He received bis M.S. in Analytica! Chemistry in 1988 from DICP and, a:ft:erwards worked there as a research assistant. For bis work on the chromatograpbic separation of ebiral compounds carried out at DICP, he received the second prize on Natura! Science Research from the Chinese Academy of Sciences in 1992. In December 1992, he came to the Labaratory of Instrumental Analysis, Eindhoven University of Technology, the Netherlands, to continue bis Ph. D. study. His work under the supervision of Prof. dr. ir. C.A. Cramers and Dr. ir. J.G.M. Janssen bas resulted in the present thesis.

178 Bibliography
Author's publications on Chromatography
1. Xianwen Lou, Zhengyun Zhan and Liangmo Zhou, Effect of the structures of chiral stationary phases on enantioselectivity, SEPU (Chinese J. Chromatogr.), 8 (1990) 240-243 (in Chinese).
2. Xianwen Lou, Yufeng Sheng and Liangmo Zhou, Investigation of parameters in the separation of amino acid enantiomers by supercritical fluid chromatography, J. Cbromatogr., 514 (1990) 253-257.
3. Xianwen Lou, Y ouqin Liu and Liangmo Zhou, Investigation of crosslinking chiral stationary phases within capillary columns, J. Cbromatogr., 552 (1991) 153-160.
4. Xianwen Lou, Xueliang Liu, Suizhi Zhang and Liangmo Zhou, Enantiomeric separation of a-phenylethylamine and its substituted isomers by gas chromatography, J. Chromatogr., 586 (1991) 139-144.
5. Liangmo Zhou, Xianwen Lou, Yufeng Sheng, Qinghai Wang and Daoqian Zhu, A crosslinked chiral stationary phase for the separation of enantiomers by gas and supercritical jluid chromatography, Chinese J. Cbem., 9 (1991) 322-326.
6. Yufeng Shen, Yuxin Zhou, Xianwen Lou, Qinghai Wang and Liangmo Zhou, The separation and determination of glycerides by supercritical fluid chromatography, SEPU (Chinese J. Chromatogr.), 9 (1991) 379-382 (in Chinese).
7. Xianwen Lou, Xueliang Liu, Yufeng Sheng and Liangmo Zhou, Direct enantiomeric separation of phenylalanine, DOPA and their intermediales by supercritical jluid chromatography, J. Chromatogr., 605 (1992) 103-107.
8. Xianwen Lou, Xueliang Liu, Qinghai Wang, Daoqian Zhu and Liangmo Zhou, Jnvestigation of the effect of acylation on the enantiomeric separation of amino acid isopropyl esters by gas chromatography, J. Cbromatogr., 626 (1992) 231-238.
9. Xianwen Lou and Liangmo Zhou, Study on crosslinked eh i ral capillary columns, SEPU (Chinese J. Chromatogr.), 10 (1992) 90-92 (in Chinese).

Bibliography 179
10. Xianwen Lou, Y ouqin Liu, Qinghai Wang, Daoqian Zhu and Liangmo Zhou, Determination of amino acids with enantiomer labelling by gas chromatography, SEPU (Chinese J. Chromatogr.), 10 (1992) 195-198 (in Chinese).
11. Liangmo Zhou, Xianwen Lou, Y ouqin Liu, Qinghai Wang and Daoqian Zhu, Investigation on chiral capillary columns and their application in separation of enantiomers, Chinese J. Chem., 10 (1992) 430-433.
12. Xianwen Lou, Xueliang Liu and Liangmo Zhou, Series-coupled capillary columns for the separation of N,(O)-trifluoroacetyl isopropyl derivatives of D,L-aspartic acid and L-hydroxyproline by gas chromatography, J. Chromatogr., 634 (1993) 281-288.
13. Xianwen Lou, Xueliang Liu and Liangmo Zhou, Chiral recognition of enantiomeric amides on a diamide chiral stationary phase by gas chromatography, J. Chromatogr., 634 (1993) 345-349.
14. Xianwen Lou, Tongfeng Zhang, Qinghai Wang, Daoqian Zhu · and Liangmo Zhou, Determination of fatty acids in glycerides by capillary gas chromatography, SEPU (Chinese J. Chromatogr.), 11 (1993) 346-347 (in Chinese).
15. Xueliang Liu, Xianwen Lou and Liangmo Zhou, Cyclodextrin derivatives as chiral stationary phases in high resolution capillary gas chromatography, HUAXUE SHIJI (Chinese J. of Chemical Reagents), 16 (1994) 90-95 (in Chinese).
16. Xueliang Liu, Fangqiu Guo, Xianwen Lou and Liangmo Zhou, A new chiral stationary phase - Heptakis(2,6-0-n-butyl-3-0-trifluoroacetyl)-f3-cyclodextrin for gas chromatography, HUAXUE XUEBAO (Chinese Chem. Bulletin), 52 (1994) 588-594 (in Chinese).
17. Xianwen Lou, Gas chromatographic separation of enantiomers, in New Techniques in Gas Chromatography, in Liangmo Zhou (editor), Academie Pressof China, Beijing, 1994, p.142-181 (in Chinese).
18. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Quantitative aspects of directly coupled supercritical jluid extraction-capillary gas chromatography with a conventional splitlsplitless injector as interface, J. High Resol. Chromatogr., 16 (1993) 425-428.

180 Bibliography
19. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Correlation of supercritical fluid extraction recoveries with supercritical fluid chromatographic retention data: A fundamental study, J. High Resol. Chromatogr., 18 (1995) 483-489.
20. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Investigation of parameters affecting the supercritical fluid extraction of polymer additives from polyethylene, J. Microcolumn Separations, 7 (1995) 303-317.
21. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Effects of modifier addition and temperafure variation in SFE of polymerie materials, J. Chromatogr. Sci., 34 (1996) 282-290.
22. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Investigation of parameters affecting supercritical .fluid extraction combined with capillary gas chromatography, J. Chromatogr., 750 (1996) 215-226.
23. Xianwen Lou, Hans-Gerd Janssen, Henri Snijders and Carel A. Cramers, Pressure drop effects on selectivity and resolution in packed column supercritical fluid chromatography, J. High Resol. Chromatogr., 8 (1996) 449-456.
24. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Parameters affecting the accelerated solvent extraction of polymerie samples, Anal. Chem., 69 (1997) 1598-1603.
25. Xianwen Lou, Hans-Gerd Janssen and Carel A. Cramers, Temperafure and pressure effects on solubility in supercritical carbon dioxide and relention in supercritical .fluid chromatography, J. Chromatogr., in press.
26. Hans-Gerd Janssen and Xianwen Lou, Packed columns in SFC: Mobile and stationary phases and further requirements, in D. Thiebaut and M. Caude (eds), Practical SFC-SFE, Harwood Academie Publishers, Accepted for publication.

Stellingen
Behorende bij het proefschrijft
Supercritical Fluids in Analytical Chemistry: Chromatography and Extraction
van Xianwen Lou
1. Extraction in SFE is controlled by many of the same parameters cantrolling retention in SFC. Chapter 2, this thesis.
2. In SFC, it is widely accepted that under conditions where the solute vapor pressure is not a dominant consideration, increasing temperature will lead to an increase in retention due to the reduced supercritical fluid density. This is, however, found to be over simplified and in some cases even incorrect. C.F. Poole and S.K. Poole, Chromatography Today, Elsevier Science Publishers, 1994, p.624. Chapter 3, this thesis.
3. In packed column SFC, long columns do not necessarily yield better separations. Chapter 4, this thesis.
4. In contrast to the SFE extraction of environmental samples where modifier identity was found to be more important than its concentration, both modifier identity and concentration are important for the SFE extraction of polymerie samples. J.J. Langenfeld, et al. Anal. Chem., 66(1994)909. Chapter 5, this thesis.
5. Supercritical fluids can sametimes become too critica/ to be used on routine bases.

6. No instrument alone can improve the analytica! performance without the efforts of analytica! chemists.
7. In the biologica! circle, hybridization can generally yield new species of superiority. Quite similarly, the coupling of different analytica! techniques or hyphenation, such as chromatography/mass speetrometry and electrophoresis/chromatography, have sparked extraordinary capability in Analytica! Chemistry.
8. In our complicated world, it is very important to keep balancing between Contradietory elements, for example, between cooperation and competition, environment and development, etc ..
9. It is a good cat, no matter white or black, as long as it can catch mice. DENG Xiaoping (1904-1997), former Chinese paramount leader.
lD.Referees always spoil sport games. However, without a referee no match can take place.
ll.People become quite illogical when they decide what can he eaten and what can not he eaten. Hardly any product can find worldwide acceptance.


















