
Stem cell based therapies for diabetes
28
StemCell Based Therapies For Diabetes Aman Kumar Naik Integrated MS. SBS, NISER
-
Upload
aman-kumar-naik -
Category
Science
-
view
207 -
download
15
Transcript of Stem cell based therapies for diabetes
StemCell Based Therapies For Diabetes
Aman Kumar NaikIntegrated MS.
SBS, NISER
notable that total pancreatectomy with intrahepatic autoislettransplantation for non-T1D patients results in only 30% insulinindependence at 3 years [17]. Although this is in part becauseof low islet yield in somepatients [17], failure toachievehigh ratesof insulin independence in autologous, autoimmunity-free set-tings highlights the current limitations of islet transplantation.With further improvement, islet transplantation could providea long-term therapy for patients with insulin deficiency.
An additional key issue is the persistent shortage of postmor-tempancreatic tissue for islet isolation. Thiswill be amajor hurdlewhen islet transplantation becomes the standard therapy forT1D. One solution would be the successful combination of anopt-out system and the priority rule to increase organ donation.Additionally, in an effort to obtain a sustainable source of insulin-producing cells, various alternative sources have been explored,including islet transplantation from living donors [18] and xeno-geneic islets [19]. Insulin-producing surrogate cells from diversestemcell sources havealsoemergedas alternativebiotherapeuticcandidates for diabetes care [8, 20]. Among adult stem cells, ex-tensive experience has been obtained with multipotent mesen-chymal stem cells (MSCs) [21]. In addition to generation ofb-like cells from MSCs [21, 22], their potent anti-inflammatoryor immune-suppressive effects have been increasingly evaluated.This includes their potential for immunomodulation and tissuedamage protection to suppress autoimmunity in T1D or to en-hance islet engraftment and survival [23–25], underscoring versa-tility in their mechanism of action and benefit potential. Becausethe current efforts using adult mesenchymal stem cells, and alsohematopoietic or pancreatic stem cells, have been extensively
reviewed [8, 20, 21, 26, 27], this synopsis highlights natural andbioengineered pluripotent stem cells, underscoring their transla-tional potential for diabetes therapies.
EMBRYONIC STEM CELL-BASED THERAPIES FOR DIABETES
The capacity for unlimited self-renewal and pluripotent lineagespecification renders embryonic stem cells (ESCs) a unique plat-form for regenerative medicine. Various protocols have shownsuccessful and efficient differentiation into ectoderm, endoderm,and mesoderm. Currently, there are two main approaches forgenerating insulin-producing cells from ESCs. The first is to lever-age the spontaneous differentiation propensity of pluripotentcells through embryoid body (EB) formation, followed by selec-tion for b-cell/b-cell-progenitor marker-expressing cells. Use ofinsulin-producing cells from ESC-derived progeny, selected forb-cell-specific gene expression, has been shown to normalize hy-perglycemia in diabetic mice [28, 29]. The second strategy in-volves stepwise, lineage-specific differentiation protocols, largelyadapted from in utero b-cell developmental blueprints. In thisway, guided differentiation of ESCs has achieved generation ofinsulin-producing cells [30], although subsequent studies havequestioned the authenticity of derived progeny [31]. Througha marriage of both strategies, spontaneous differentiation of EBsandcoaxeddifferentiationof earlypancreatic progenitors, success-ful generationof insulin-producing cells has alsobeendocumented[32].
As spontaneous differentiation is limited by inefficiency,guided differentiation has been the primary strategy used for dif-ferentiation of human ESC into insulin-producing cells. Withrenewed focus on decoding embryonic development, the pro-gressive evolution of endoderm into primitive gut tube and ulti-mately discrete pancreatic b cells has become increasinglydefined [33]. The critical first step of guided differentiation pro-tocols is the induction of endoderm from human ESCs [34, 35],which is typically achieved through stimulation with activin A (aNodal surrogate), Nodal, and/or Wnts, under low serum condi-tions [36–38]. Derived definitive endoderm expresses markerssuchas FOXA2, SOX17, andCXCR4 [33]. Further guidanceachievesgeneration of pancreatic multihormonal endocrine cells throughforegut, pancreatic endoderm, and endocrine progenitor stages[39]. However, resulting cells demonstrate immature b-cell-like phenotypes [39]; that is, these human ESC-derived b-likecells produce high levels of intracellular C peptide comparableto human islets and respond to insulin secretagogs but fail to re-spond to high glucose stimulation. Modified or improved proto-cols have been established using combinations of cytokines andsmall molecules, such as fibroblast growth factors, noggin,KAAD-cyclopamine Sonic hedgehog pathway inhibitors (KAAD-cyclopamine or SANT-1), retinoic acid, nicotinamide, and GLP-1(Table 1) [40–51]. Notable improvements in pancreatic differen-tiation have been reported with use of a small-molecule Indolac-tam V, which accelerates induction of pancreatic progenitor cellsfrom definitive endoderm through protein kinase C (PKC) activa-tion [52], or suppression of the transforming growth factor(TGF)b/activin/bone morphogenetic protein signaling pathwaysat specific stages by noggin or SB431542/ALK5 inhibitors [43,44]. Accordingly, use of noggin, PKC activator (2S,5S)-(E,E)-8-(5-(4-(trifluoromethyl)phenyl)-2,4-pentadienoylamino)benzolac-tam (TPB) and TGFb inhibitor in combination has proven effective
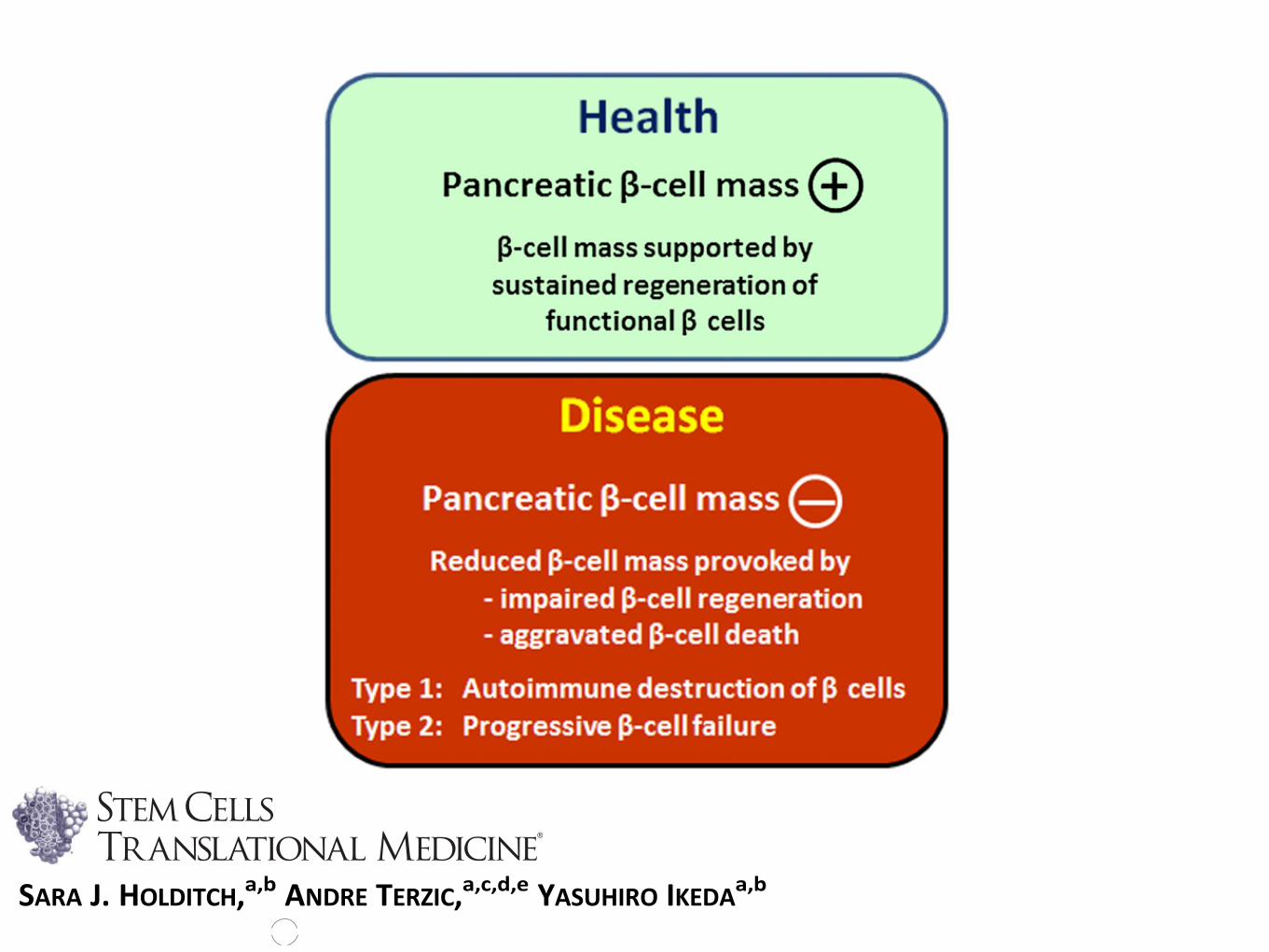
Figure 1. Inadequate b-cell regeneration because of b-cell destruc-tion and/or insufficient b-cell replenishment causes diabetes. Thehealthy state is characterized by sustained and adequate regenera-tion of the pancreatic b-cell mass. Disease is precipitated by loss ofb-cell mass, through impaired innate regeneration or excessive apo-ptosis, underlying the pathological substrate of type 1 and type 2diabetes.
2 Pluripotent Stem Cell Applications for Diabetes
©AlphaMed Press 2014 STEM CELLS TRANSLATIONAL MEDICINE
at Tufts Hirsh H
ealth Sciences Library on September 25, 2014
http://stemcellstm
.alphamedpress.org/
Dow
nloaded from
Enabling Technologies for Cell-Based Clinical Translation
Concise Review: Pluripotent Stem Cell-BasedRegenerative Applications for Failing b-Cell Function
SARA J. HOLDITCH,a,b ANDRE TERZIC,a,c,d,e YASUHIRO IKEDAa,b
Key Words. Diabetes mellitus x Regenerative medicine x Stem cells x Translation x Transplantation
ABSTRACT
Diabetes engenders the loss of pancreatic b-cell mass and/or function, resulting in insulin deficiencyrelative to the metabolic needs of the body. Diabetic care has traditionally relied on pharmacother-apy, exemplified by insulin replacement to target peripheral actions of the hormone. With growingunderstanding of the pathogenesis of diabetic disease, alternative approaches aiming at repair andrestoration of failing b-cell function are increasingly considered as complements to current diabetestherapy regimens. To this end, emphasis is placed on transplantation of exogenous pancreas/islets orartificial islets, enhanced proliferation and maturation of endogenous b cells, prevention of b-cellloss, or fortified renewal ofb-like-cell populations fromstemcell pools andnon-b-cell sources. In lightof emerging clinical experienceswith human embryonic stem cells and approval of the first in-humantrial with induced pluripotent stem cells, in this study we highlight advances in b-cell regenerationstrategies with a focus on pluripotent stem cell platforms in the context of translational applications.STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9
INTRODUCTION
Diabetes is a staggering health problem affectingmore than 300 million people worldwide. By2030, an estimated 440 million adults will beafflicted with diabetes [1, 2]. Premature morbidityand mortality create a substantial and escalatingburden on the global health system and society.Type 1 diabetes (T1D) is defined by insulin defi-ciency brought about by autoimmune destructionof islet b cells. Type 2 diabetes (T2D) is defined bythe progressive inability of the insulin secretory ca-pacity tomatch peripheral insulin needs. Defectiveinnateb-cell regeneration, because of eitherb-celldestruction or insufficient b-cell replenishment, isincreasingly recognized as central to thepathobiol-ogy of both T1D and T2D (Fig. 1) [3–6].
Pharmacological means that promote insulinproduction or replace insulin function have com-prised a primary line of therapy for insulin insuf-ficiency in T1D and a large proportion of patientswith T2D, in particular those with advanced dis-ease. Case in point, injection of exogenous insulinis required for people with T1D and advancedT2D. Glucagon-like peptide-1 (GLP-1) analogshave been designed more recently to augmentinsulin secretion and preserve b-cell function. Al-ternatively, DPP-4 inhibitors, which prevent inac-tivation of GLP-1, have been used to promoteendogenous insulinproduction inT2D[7].Whereascollectively the spectrum of preventive and pallia-tive approaches has led to improved diabeticcare, a fail-safe physiological regulation of sys-temic blood glucose levels remains challenging.
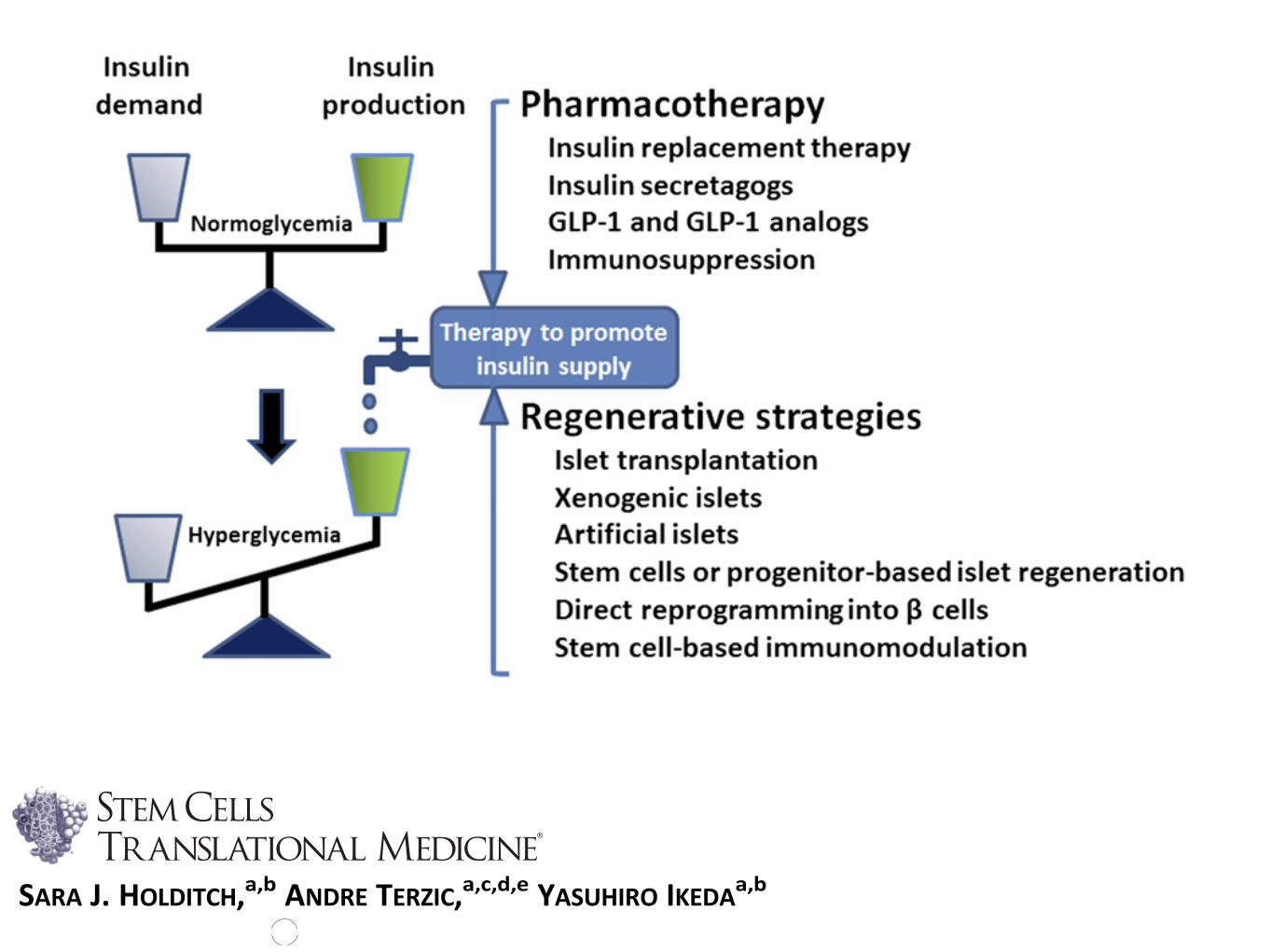
Frequent fluctuations in blood glucose levelshave been implicated as a culprit of heteroge-neous (co)morbidities, such as retinopathy,nephropathy, neuropathy, or cardiovascularcomplications. In this context, various nonpharma-cological approaches have been recently exploredto restore functionality of the failing b-cell mass.Such regenerative approaches have evolved rap-idly, from prototypic cell replacement therapiesthrough pancreas/islet transplantation to thepotential use of artificial insulin-producing cellsderived from stemcells or pancreatic progenitorcells (Fig. 2).
ADULT CELL-BASED THERAPIES FOR DIABETES
Islet transplantation has provided a foundationfor next-generation cell-based therapeutics fordiabetes [8]. The Edmonton protocol, a flagshipislet transplantation protocol, has achieved long-term islet survival. In early studies, more than50%ofsubjectsgained insulin independence1yearpost-transplantation and experienced improvedglycated hemoglobin levels and protection fromhypoglycemia [9]. Twenty percent of islet recipi-ents are insulin therapy-free 5 years after trans-plantation [10]. Recent studies have shownimprovements in primary efficacy, safety out-comes, and insulin independence 3 years post-transplant [11, 12]. Despite notable progress inthe field, several issues still need to be addressed.These include the islet isolation methods, sitesfor transplantation, immunosuppression, or im-munoisolation strategies [13–16]. It is also
aCenter for RegenerativeMedicine, bDepartment ofMolecular Medicine,cDivision of CardiovascularDiseases, Department ofMedicine, dDepartment ofMolecular Pharmacology andExperimental Therapeutics,and eDepartment of MedicalGenetics, Mayo Clinic,Rochester, Minnesota, USA
Correspondence: Yasuhiro Ikeda,D.V.M., Ph.D., Center forRegenerative Medicine, MayoClinic, 200 First Street SW,Rochester, Minnesota 55905,USA. Telephone: 507-538-0153;E-Mail: [email protected]
Received October 8, 2013;accepted for publicationDecember 30, 2013.
©AlphaMed Press1066-5099/2014/$20.00/0
http://dx.doi.org/10.5966/sctm.2013-0184
STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9 www.StemCellsTM.com ©AlphaMed Press 2014
ENABLING TECHNOLOGIES FOR CELL-BASED CLINICALTRANSLATION
http://stemcellstm.alphamedpress.org/cgi/doi/10.5966/sctm.2013-0184The latest version is at Published Ahead of Print on March 19, 2014 as 10.5966/sctm.2013-0184
at Tufts Hirsh H
ealth Sciences Library on September 25, 2014
http://stemcellstm
.alphamedpress.org/
Dow
nloaded from
Enabling Technologies for Cell-Based Clinical Translation
Concise Review: Pluripotent Stem Cell-BasedRegenerative Applications for Failing b-Cell Function
SARA J. HOLDITCH,a,b ANDRE TERZIC,a,c,d,e YASUHIRO IKEDAa,b
Key Words. Diabetes mellitus x Regenerative medicine x Stem cells x Translation x Transplantation
ABSTRACT
Diabetes engenders the loss of pancreatic b-cell mass and/or function, resulting in insulin deficiencyrelative to the metabolic needs of the body. Diabetic care has traditionally relied on pharmacother-apy, exemplified by insulin replacement to target peripheral actions of the hormone. With growingunderstanding of the pathogenesis of diabetic disease, alternative approaches aiming at repair andrestoration of failing b-cell function are increasingly considered as complements to current diabetestherapy regimens. To this end, emphasis is placed on transplantation of exogenous pancreas/islets orartificial islets, enhanced proliferation and maturation of endogenous b cells, prevention of b-cellloss, or fortified renewal ofb-like-cell populations fromstemcell pools andnon-b-cell sources. In lightof emerging clinical experienceswith human embryonic stem cells and approval of the first in-humantrial with induced pluripotent stem cells, in this study we highlight advances in b-cell regenerationstrategies with a focus on pluripotent stem cell platforms in the context of translational applications.STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9
INTRODUCTION
Diabetes is a staggering health problem affectingmore than 300 million people worldwide. By2030, an estimated 440 million adults will beafflicted with diabetes [1, 2]. Premature morbidityand mortality create a substantial and escalatingburden on the global health system and society.Type 1 diabetes (T1D) is defined by insulin defi-ciency brought about by autoimmune destructionof islet b cells. Type 2 diabetes (T2D) is defined bythe progressive inability of the insulin secretory ca-pacity tomatch peripheral insulin needs. Defectiveinnateb-cell regeneration, because of eitherb-celldestruction or insufficient b-cell replenishment, isincreasingly recognized as central to thepathobiol-ogy of both T1D and T2D (Fig. 1) [3–6].
Pharmacological means that promote insulinproduction or replace insulin function have com-prised a primary line of therapy for insulin insuf-ficiency in T1D and a large proportion of patientswith T2D, in particular those with advanced dis-ease. Case in point, injection of exogenous insulinis required for people with T1D and advancedT2D. Glucagon-like peptide-1 (GLP-1) analogshave been designed more recently to augmentinsulin secretion and preserve b-cell function. Al-ternatively, DPP-4 inhibitors, which prevent inac-tivation of GLP-1, have been used to promoteendogenous insulinproduction inT2D[7].Whereascollectively the spectrum of preventive and pallia-tive approaches has led to improved diabeticcare, a fail-safe physiological regulation of sys-temic blood glucose levels remains challenging.
Frequent fluctuations in blood glucose levelshave been implicated as a culprit of heteroge-neous (co)morbidities, such as retinopathy,nephropathy, neuropathy, or cardiovascularcomplications. In this context, various nonpharma-cological approaches have been recently exploredto restore functionality of the failing b-cell mass.Such regenerative approaches have evolved rap-idly, from prototypic cell replacement therapiesthrough pancreas/islet transplantation to thepotential use of artificial insulin-producing cellsderived from stemcells or pancreatic progenitorcells (Fig. 2).
ADULT CELL-BASED THERAPIES FOR DIABETES
Islet transplantation has provided a foundationfor next-generation cell-based therapeutics fordiabetes [8]. The Edmonton protocol, a flagshipislet transplantation protocol, has achieved long-term islet survival. In early studies, more than50%ofsubjectsgained insulin independence1yearpost-transplantation and experienced improvedglycated hemoglobin levels and protection fromhypoglycemia [9]. Twenty percent of islet recipi-ents are insulin therapy-free 5 years after trans-plantation [10]. Recent studies have shownimprovements in primary efficacy, safety out-comes, and insulin independence 3 years post-transplant [11, 12]. Despite notable progress inthe field, several issues still need to be addressed.These include the islet isolation methods, sitesfor transplantation, immunosuppression, or im-munoisolation strategies [13–16]. It is also
aCenter for RegenerativeMedicine, bDepartment ofMolecular Medicine,cDivision of CardiovascularDiseases, Department ofMedicine, dDepartment ofMolecular Pharmacology andExperimental Therapeutics,and eDepartment of MedicalGenetics, Mayo Clinic,Rochester, Minnesota, USA
Correspondence: Yasuhiro Ikeda,D.V.M., Ph.D., Center forRegenerative Medicine, MayoClinic, 200 First Street SW,Rochester, Minnesota 55905,USA. Telephone: 507-538-0153;E-Mail: [email protected]
Received October 8, 2013;accepted for publicationDecember 30, 2013.
©AlphaMed Press1066-5099/2014/$20.00/0
http://dx.doi.org/10.5966/sctm.2013-0184
STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9 www.StemCellsTM.com ©AlphaMed Press 2014
ENABLING TECHNOLOGIES FOR CELL-BASED CLINICALTRANSLATION
http://stemcellstm.alphamedpress.org/cgi/doi/10.5966/sctm.2013-0184The latest version is at Published Ahead of Print on March 19, 2014 as 10.5966/sctm.2013-0184
at Tufts Hirsh H
ealth Sciences Library on September 25, 2014
http://stemcellstm
.alphamedpress.org/
Dow
nloaded from

Figure 1 illustrates what many believe to be the course ofislet inflammation. Many lines of evidence indicate thatantigen-presenting cells (APC), especially dendritic cells(DC) are pathologically activated to orchestrate theinsulitic process.24 It is thought that islet-resident APCrespond to a microenvironmental anomaly (perhaps b-cell death and/or impaired b-cell turnover or apopto-sis25–27) and initiate the insulitis process by migrating outof the islets and into the peripheral pancreatic lymphnodes. By presenting the b-cell antigens they haveacquired, the APC interact with b-cell-reactive T cells,which escaped thymic deletion and trigger their activa-tion and proliferation (Figure 1). Once activated, these Tcells act as one pole of a chronic tug-of-war between b-cell-specific autoimmunity and peripheral mechanismsof tissue-specific tolerance. Regulatory T cells of variouscell surface phenotypes and cytokine secretion profilesmay also be involved in modulating this unstableequilibrium. Ultimately, this chronic process ends infavor of the b-cell-reactive T cells, which eventually endup destroying enough b-cell mass to render the patientinsulin-dependent. This process has been extensivelystudied in the NOD mouse and is believed to occur inhumans as well.
Lessons to be learned: gene vectors or cellsor both?
An appreciable amount of work has focused on usingviral vectors to infect intact islets in culture prior totransplantation into recipients to impede the allogeneic
rejection (reviewed in Giannoukakis et al28,29). Theexcitement generated by these studies, however, wastempered by the appreciation that permanent allograftsurvival was generally not achieved. Often, to explainthis limited success, investigators invoke the immuno-genicity of the particular vector used, although recentevidence suggests that the quality of the islets may bemore crucial than the vector choice in determining thepresence and grade of inflammation in and around thegraft.30 Table 1aa lists the vectors that have been used todate to transduce intact human islets as well as their prosand cons (Table 1c). (Table 1b lists their properties). Thelist indirectly demonstrates that no ‘ideal’ vector yetexists. New technology, including small interferenceRNA (siRNA),31 adeno-associated virus inverted term-inal repeat (AAV ITR)-based plasmids,32,33 novel classesof lentivirus (equine infectious anemia virus-EIAV; felineimmunodeficiency virus- FIV),34–40 lentivirus–herpes-virus hybrids and other viral vectors is in development,but their efficiency has yet to be reported in the context of
Steady-state flux of APC through islets
Microenvironmental anomaly:Activation and mobilisationof residentand extra-islet APC into islet
β-cell apoptosis:Uptake of autoantigensby APC
Migration of activated APCto peripheral lymph nodes
Activation of autoreactive CD4+ and CD8+ T-cellsin pancreatic lymph nodes
Cytokine-induced β-cell impairment/further apoptosis promotion
T-cell and cytokine-dependentβ cell impairment and apoptosis
Alpha cell
Beta cell
Delta cell
APC
1
2
3
4
56
cytokines
CD8+ TcCD4+ Tc
Figure 1 Multistep process of insulitis. During ontogeny, a population of thymocytes whose TCR recognize b-cell-specific antigens are either not deleted inthe thymus, or fail to be tolerized subsequently, in the periphery. These T cells may circulate dormant, or may be active, but suppressed by regulatory T-cellnetworks. Islet-resident APC (DC or macrophages) are normally in a steady-state flux sampling the microenvironment. An as-yet unidentifiedmicroenvironmental anomaly shifts their phenotype into activators of an inflammatory response, as they migrate out of the islet environment and into theperipheral lymphoid organs. There, they eventually encounter the autoreactive T cells. In the meantime, antigen nonspecific inflammation progresses withinislets because of macrophage and DC secretion of soluble mediators of b-cell dysfunction and apoptosis activation.
Table 1a Gene vectors that transduce islets (with references)
Plasmid DNA202!205
Adenovirus206!219
Adeno-associated virus114,115,130,132,220!228
MoLV retrovirus229
Lentivirus230!232
Herpes simplex virus233,234
Cationic liposomes204,205,214
Peptide fusion domains118,235
Therapeutics for type I diabetes mellitusR Bottino et al
876
Gene Therapy
REVIEW
Gene- and cell-based therapeutics for type I diabetesmellitus
R Bottino1,2, P Lemarchand3, M Trucco1,2 and N Giannoukakis2,41Department of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA; 2Diabetes Institute, University ofPittsburgh School of Medicine, Pittsburgh, PA, USA; 3INSERM, U533, Nantes, France; and 4Department of Pathology, University ofPittsburgh School of Medicine, Pittsburgh, PA, USA
Type 1 diabetes mellitus, an autoimmune disorder is anattractive candidate for gene and cell-based therapy. Fromthe use of gene-engineered immune cells to inducehyporesponsiveness to autoantigens to islet and beta cellsurrogate transplants expressing immunoregulatory genes toprovide a local pocket of immune privilege, these strategieshave demonstrated proof of concept to the point wheretranslational studies can be initiated. Nonetheless, along with
the proof of concept, a number of important issues havebeen raised by the choice of vector and expression systemas well as the point of intervention; prophylactic ortherapeutic. An assessment of the current state of thescience and potential leads to the conclusion that somestrategies are ready for safety trials while others requirevarying degrees of technical and conceptual refinement.Gene Therapy (2003) 10, 875–889. doi:10.1038/sj.gt.3302015
Introduction
Of all the autoimmune diseases, none has been aspopular a target of cell and gene-based prophylacticand therapeutic interventions as type I diabetes mellitus.The delineation of the cellular effectors and the mole-cular pathways involved in the breakdown of central andperipheral tolerance has promoted interventions asdiverse as bone marrow transplantation with or withoutantibody-based immunosuppression for tolerance induc-tion, transplantation of genetically engineered islets ofLangerhans to restore insulin production, embryonic andpancreatic stem cells and non pancreatic progenitors assurrogate b cells. Almost all of these strategies have beenrealized in the non obese diabetic (NOD) mouse model,the workhorse of this area of investigation and, inparallel, yet to a smaller degree pursued in the diabetes-prone BioBreeding (DP-BB) rat. It is worth noting thatmany of these strategies have successfully preventeddiabetes with varying effects on the degree of insulitis(the cellular inflammation in and around the islets) asoutlined by Atkinson and Leiter.1 in a very notable list. Inhumans, to date, the only clinically acceptable treatmentfor type I diabetes, other than insulin replacement,remains islet transplantation under the cover of pharma-cologic immunosuppression. A very recent safety trialhas begun using an anti-CD3 antibody, but the resultsrequire confirmation and further safety analysis. Whetherthe cell- and gene-based strategies published in the lastdecade will ever be clinically translatable is not yet clear.We undertook to review these strategies and their potentialclinical utility with the ultimate objective of giving aperspective on the field from the view of gene therapy.
Type I diabetes mellitus: the autoimmuneprocess
There is no doubt that type I diabetes mellitus (TIDM) isgenetically determined and is triggered by an as-yetunidentified postnatal determinant, very likely environ-mental in nature. The genetics of the disease is multi-factorial and involve two loci (IDDM1 and IDDM2)confirmed to be in linkage with the disease. IDDM1encompasses the HLA gene complex and it alone definesthe most important risk factor. In humans, the disease isassociated with the inheritance of DR3/DR4 haplotypes(DR3: DQA1*0501, DQB1*0201 and DR4: DQA1*0301,DQB1*0302).2,3 IDDM2 has been mapped to a variablenumber of tandem repeats (VNTR) polymorphism up-stream of the insulin gene promoter, which can deter-mine thymic levels of insulin.4,5 In fact, a recent studydemonstrated that the number of active copies of insulinin a transgenic mouse can influence the degree ofimmune cell reactivity towards insulin, a putativeautoantigen.6 A number of other loci have demonstratedsuggestive associations, but to date, none of these resultshave been replicated to establish significant linkage withthe disease.7–9
A number of earlier hypotheses with some supportingevidence have been put forward to explain the possiblemechanism of action of the environmental triggerincluding b-cell death secondary to virally triggeredinflammation, molecular mimicry, superantigens anddiet.10–19 What is certain is that at some point postnatally,the immune system of a genetically predisposed indivi-dual is activated to infiltrate chronically the islets ofLangerhans. While the initial phase of infiltration maynot involve b-cell destruction, a number of studies in vivoand in vitro suggest that immune cells become able torender b-cells dysfunctional through the actions ofcytokines they produce such as interleukin-1b.20–23
Correspondence: Dr. N Giannoukakis, Department of Pathology, Uni-versity of Pittsburgh School of Medicine, Diabetes Institute, RangosResearch Center 5102, 3460 Fifth Avenue, Pittsburgh, PA, 15213, USA
Gene Therapy (2003) 10, 875–889& 2003 Nature Publishing Group All rights reserved 0969-7128/03 $25.00
www.nature.com/gt
REVIEW
Gene- and cell-based therapeutics for type I diabetesmellitus
R Bottino1,2, P Lemarchand3, M Trucco1,2 and N Giannoukakis2,41Department of Pediatrics, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA; 2Diabetes Institute, University ofPittsburgh School of Medicine, Pittsburgh, PA, USA; 3INSERM, U533, Nantes, France; and 4Department of Pathology, University ofPittsburgh School of Medicine, Pittsburgh, PA, USA
Type 1 diabetes mellitus, an autoimmune disorder is anattractive candidate for gene and cell-based therapy. Fromthe use of gene-engineered immune cells to inducehyporesponsiveness to autoantigens to islet and beta cellsurrogate transplants expressing immunoregulatory genes toprovide a local pocket of immune privilege, these strategieshave demonstrated proof of concept to the point wheretranslational studies can be initiated. Nonetheless, along with
the proof of concept, a number of important issues havebeen raised by the choice of vector and expression systemas well as the point of intervention; prophylactic ortherapeutic. An assessment of the current state of thescience and potential leads to the conclusion that somestrategies are ready for safety trials while others requirevarying degrees of technical and conceptual refinement.Gene Therapy (2003) 10, 875–889. doi:10.1038/sj.gt.3302015
Introduction
Of all the autoimmune diseases, none has been aspopular a target of cell and gene-based prophylacticand therapeutic interventions as type I diabetes mellitus.The delineation of the cellular effectors and the mole-cular pathways involved in the breakdown of central andperipheral tolerance has promoted interventions asdiverse as bone marrow transplantation with or withoutantibody-based immunosuppression for tolerance induc-tion, transplantation of genetically engineered islets ofLangerhans to restore insulin production, embryonic andpancreatic stem cells and non pancreatic progenitors assurrogate b cells. Almost all of these strategies have beenrealized in the non obese diabetic (NOD) mouse model,the workhorse of this area of investigation and, inparallel, yet to a smaller degree pursued in the diabetes-prone BioBreeding (DP-BB) rat. It is worth noting thatmany of these strategies have successfully preventeddiabetes with varying effects on the degree of insulitis(the cellular inflammation in and around the islets) asoutlined by Atkinson and Leiter.1 in a very notable list. Inhumans, to date, the only clinically acceptable treatmentfor type I diabetes, other than insulin replacement,remains islet transplantation under the cover of pharma-cologic immunosuppression. A very recent safety trialhas begun using an anti-CD3 antibody, but the resultsrequire confirmation and further safety analysis. Whetherthe cell- and gene-based strategies published in the lastdecade will ever be clinically translatable is not yet clear.We undertook to review these strategies and their potentialclinical utility with the ultimate objective of giving aperspective on the field from the view of gene therapy.
Type I diabetes mellitus: the autoimmuneprocess
There is no doubt that type I diabetes mellitus (TIDM) isgenetically determined and is triggered by an as-yetunidentified postnatal determinant, very likely environ-mental in nature. The genetics of the disease is multi-factorial and involve two loci (IDDM1 and IDDM2)confirmed to be in linkage with the disease. IDDM1encompasses the HLA gene complex and it alone definesthe most important risk factor. In humans, the disease isassociated with the inheritance of DR3/DR4 haplotypes(DR3: DQA1*0501, DQB1*0201 and DR4: DQA1*0301,DQB1*0302).2,3 IDDM2 has been mapped to a variablenumber of tandem repeats (VNTR) polymorphism up-stream of the insulin gene promoter, which can deter-mine thymic levels of insulin.4,5 In fact, a recent studydemonstrated that the number of active copies of insulinin a transgenic mouse can influence the degree ofimmune cell reactivity towards insulin, a putativeautoantigen.6 A number of other loci have demonstratedsuggestive associations, but to date, none of these resultshave been replicated to establish significant linkage withthe disease.7–9
A number of earlier hypotheses with some supportingevidence have been put forward to explain the possiblemechanism of action of the environmental triggerincluding b-cell death secondary to virally triggeredinflammation, molecular mimicry, superantigens anddiet.10–19 What is certain is that at some point postnatally,the immune system of a genetically predisposed indivi-dual is activated to infiltrate chronically the islets ofLangerhans. While the initial phase of infiltration maynot involve b-cell destruction, a number of studies in vivoand in vitro suggest that immune cells become able torender b-cells dysfunctional through the actions ofcytokines they produce such as interleukin-1b.20–23
Correspondence: Dr. N Giannoukakis, Department of Pathology, Uni-versity of Pittsburgh School of Medicine, Diabetes Institute, RangosResearch Center 5102, 3460 Fifth Avenue, Pittsburgh, PA, 15213, USA
Gene Therapy (2003) 10, 875–889& 2003 Nature Publishing Group All rights reserved 0969-7128/03 $25.00
www.nature.com/gt
for induction of pancreatic endoderm and endocrine precursors[46].
INDUCED PLURIPOTENT STEM CELL-BASED THERAPIESFOR DIABETES
Despitenotableprogress, the clinical developmentof cell replace-ment therapies using human ESCs has been surroundedby ethicalconcerns. The use of allogeneic ESC-derived cells is also associ-ated with immunological mismatch. Although a recent studyhas demonstrated successful generation of personalized humanESCs through somatic cell nuclear transfer (NT-ESCs) [53], deriva-tionofhumanNT-ESCs remains challenging. In this regard, nuclearreprogramming technology, which allows generation of pluripo-tent stem cells from adult somatic cells, has opened a new pathfor generating patient-specific pluripotent stem cells [54, 55].The induced pluripotent stem cell (iPSC) technology relies on ge-netic introduction of selected pluripotency-associated factors inadult somatic cell sources, which reprogram cell fate enabling de-differentiation into a pluripotent stem cell state [56, 57]. Derivedhuman iPSC lines show characteristics similar to human ESCs, in-cluding morphology, global gene expression profiles, elongatedtelomeres, and the propensity to differentiate into all three germlayers [58, 59], offering a self-renewable source of new tissues de-rived from the patient’s own cell pool [60].
PATIENT-DERIVED IPSCS AND THEIR DIFFERENTIATION INTOINSULIN-PRODUCING CELLS
Unlike their natural counterparts, iPSCs carry patient-specific ge-netic traits, providing a unique autologous pluripotent platform.Accordingly, differentiation of patient-derived iPSCs into disease-relevant cell types would allow for patient-specific modeling ofdisease progression and patient-specific drug screening [59,61]. Several studies have demonstrated the efficacy of recapitu-lating disease phenotypes using patient-specific iPSCs [62–64],verifying the utility of iPSC technology for in vitro disease model-ing. Patient-specific iPSCs would also create the opportunity for
immunosuppression-free, autologous stem cell-based regenera-tive approaches for degenerative disorders.
Derivation of diabetes-specific iPSCs and their differentiationinto functional b cells provide the foundation for new diagnosticand therapeutic applications. Diabetes-specific iPSCs have beenderived from both T1D and T2D patients [61, 65–68], which dem-onstrate similar genome-wide gene expression profiles to thoseof human ESCs [69]. Importantly, iPSC clones derived frompatients of different age groups and sex are capable of generatinginsulin-producing cells [65, 68, 69], a prerequisite in establishinga broader translational platform for diabetes-specific iPSCs. Pa-tient iPSC-derived b-like cells would enable detailed analysis ofpatient-specific immunity against b cells at the cellular level,whereas autologous properties would facilitate use as a cell-based therapy for diabetes. A recent study demonstrates thatiPSC-derived b cells from subjects with maturity-onset diabetesof the young type 2 (MODY2), characterized by impaired glucoki-nase activity, recapitulate the b-cell-autonomous phenotypes ofMODY2 [70].
CHALLENGES FOR CLINICAL APPLICATIONS OF
DIABETES-SPECIFIC IPSCS
Immunogenicity and Epigenetic Abnormalities
Recent iPSC studies have raised several possible concerns forbroader application. In particular, the reprogramming processand subsequent expansionof iPSCshavebeenassociatedwithpo-tential genetic and epigenetic abnormalities [71–74]. Moreover,iPSC-derived cells have been reported to showabnormal gene ex-pression patterns, capable of inducing T-cell-dependent immu-nity in syngeneic recipients [75]. Yet, these initial studies needfurther confirmation as, for example, only limited immunogenic-ity of transplanted iPSC-derived cells has been reported [76].
Teratoma Formation
Another biosafety concern surrounding the therapeutic use ofiPSCs or derivatives is the risk of teratoma formation upon trans-plantation. The primary source of teratoma is the residual
Figure 2. Balancing insulin demand and production is a central objective of diabetes therapy. Complementing pharmacological approaches,a series of regenerative strategies have been developed ranging from islet transplantation to stem cell-based platforms aimed at restoring in-sulin homeostasis and normoglycemia. Abbreviation: GLP-1, glucagon-like peptide-1.
Holditch, Terzic, Ikeda 3
www.StemCellsTM.com ©AlphaMed Press 2014
at Tufts Hirsh H
ealth Sciences Library on September 25, 2014
http://stemcellstm
.alphamedpress.org/
Dow
nloaded from
Enabling Technologies for Cell-Based Clinical Translation
Concise Review: Pluripotent Stem Cell-BasedRegenerative Applications for Failing b-Cell Function
SARA J. HOLDITCH,a,b ANDRE TERZIC,a,c,d,e YASUHIRO IKEDAa,b
Key Words. Diabetes mellitus x Regenerative medicine x Stem cells x Translation x Transplantation
ABSTRACT
Diabetes engenders the loss of pancreatic b-cell mass and/or function, resulting in insulin deficiencyrelative to the metabolic needs of the body. Diabetic care has traditionally relied on pharmacother-apy, exemplified by insulin replacement to target peripheral actions of the hormone. With growingunderstanding of the pathogenesis of diabetic disease, alternative approaches aiming at repair andrestoration of failing b-cell function are increasingly considered as complements to current diabetestherapy regimens. To this end, emphasis is placed on transplantation of exogenous pancreas/islets orartificial islets, enhanced proliferation and maturation of endogenous b cells, prevention of b-cellloss, or fortified renewal ofb-like-cell populations fromstemcell pools andnon-b-cell sources. In lightof emerging clinical experienceswith human embryonic stem cells and approval of the first in-humantrial with induced pluripotent stem cells, in this study we highlight advances in b-cell regenerationstrategies with a focus on pluripotent stem cell platforms in the context of translational applications.STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9
INTRODUCTION
Diabetes is a staggering health problem affectingmore than 300 million people worldwide. By2030, an estimated 440 million adults will beafflicted with diabetes [1, 2]. Premature morbidityand mortality create a substantial and escalatingburden on the global health system and society.Type 1 diabetes (T1D) is defined by insulin defi-ciency brought about by autoimmune destructionof islet b cells. Type 2 diabetes (T2D) is defined bythe progressive inability of the insulin secretory ca-pacity tomatch peripheral insulin needs. Defectiveinnateb-cell regeneration, because of eitherb-celldestruction or insufficient b-cell replenishment, isincreasingly recognized as central to thepathobiol-ogy of both T1D and T2D (Fig. 1) [3–6].
Pharmacological means that promote insulinproduction or replace insulin function have com-prised a primary line of therapy for insulin insuf-ficiency in T1D and a large proportion of patientswith T2D, in particular those with advanced dis-ease. Case in point, injection of exogenous insulinis required for people with T1D and advancedT2D. Glucagon-like peptide-1 (GLP-1) analogshave been designed more recently to augmentinsulin secretion and preserve b-cell function. Al-ternatively, DPP-4 inhibitors, which prevent inac-tivation of GLP-1, have been used to promoteendogenous insulinproduction inT2D[7].Whereascollectively the spectrum of preventive and pallia-tive approaches has led to improved diabeticcare, a fail-safe physiological regulation of sys-temic blood glucose levels remains challenging.
Frequent fluctuations in blood glucose levelshave been implicated as a culprit of heteroge-neous (co)morbidities, such as retinopathy,nephropathy, neuropathy, or cardiovascularcomplications. In this context, various nonpharma-cological approaches have been recently exploredto restore functionality of the failing b-cell mass.Such regenerative approaches have evolved rap-idly, from prototypic cell replacement therapiesthrough pancreas/islet transplantation to thepotential use of artificial insulin-producing cellsderived from stemcells or pancreatic progenitorcells (Fig. 2).
ADULT CELL-BASED THERAPIES FOR DIABETES
Islet transplantation has provided a foundationfor next-generation cell-based therapeutics fordiabetes [8]. The Edmonton protocol, a flagshipislet transplantation protocol, has achieved long-term islet survival. In early studies, more than50%ofsubjectsgained insulin independence1yearpost-transplantation and experienced improvedglycated hemoglobin levels and protection fromhypoglycemia [9]. Twenty percent of islet recipi-ents are insulin therapy-free 5 years after trans-plantation [10]. Recent studies have shownimprovements in primary efficacy, safety out-comes, and insulin independence 3 years post-transplant [11, 12]. Despite notable progress inthe field, several issues still need to be addressed.These include the islet isolation methods, sitesfor transplantation, immunosuppression, or im-munoisolation strategies [13–16]. It is also
aCenter for RegenerativeMedicine, bDepartment ofMolecular Medicine,cDivision of CardiovascularDiseases, Department ofMedicine, dDepartment ofMolecular Pharmacology andExperimental Therapeutics,and eDepartment of MedicalGenetics, Mayo Clinic,Rochester, Minnesota, USA
Correspondence: Yasuhiro Ikeda,D.V.M., Ph.D., Center forRegenerative Medicine, MayoClinic, 200 First Street SW,Rochester, Minnesota 55905,USA. Telephone: 507-538-0153;E-Mail: [email protected]
Received October 8, 2013;accepted for publicationDecember 30, 2013.
©AlphaMed Press1066-5099/2014/$20.00/0
http://dx.doi.org/10.5966/sctm.2013-0184
STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9 www.StemCellsTM.com ©AlphaMed Press 2014
ENABLING TECHNOLOGIES FOR CELL-BASED CLINICALTRANSLATION
http://stemcellstm.alphamedpress.org/cgi/doi/10.5966/sctm.2013-0184The latest version is at Published Ahead of Print on March 19, 2014 as 10.5966/sctm.2013-0184
at Tufts Hirsh H
ealth Sciences Library on September 25, 2014
http://stemcellstm
.alphamedpress.org/
Dow
nloaded from
Enabling Technologies for Cell-Based Clinical Translation
Concise Review: Pluripotent Stem Cell-BasedRegenerative Applications for Failing b-Cell Function
SARA J. HOLDITCH,a,b ANDRE TERZIC,a,c,d,e YASUHIRO IKEDAa,b
Key Words. Diabetes mellitus x Regenerative medicine x Stem cells x Translation x Transplantation
ABSTRACT
Diabetes engenders the loss of pancreatic b-cell mass and/or function, resulting in insulin deficiencyrelative to the metabolic needs of the body. Diabetic care has traditionally relied on pharmacother-apy, exemplified by insulin replacement to target peripheral actions of the hormone. With growingunderstanding of the pathogenesis of diabetic disease, alternative approaches aiming at repair andrestoration of failing b-cell function are increasingly considered as complements to current diabetestherapy regimens. To this end, emphasis is placed on transplantation of exogenous pancreas/islets orartificial islets, enhanced proliferation and maturation of endogenous b cells, prevention of b-cellloss, or fortified renewal ofb-like-cell populations fromstemcell pools andnon-b-cell sources. In lightof emerging clinical experienceswith human embryonic stem cells and approval of the first in-humantrial with induced pluripotent stem cells, in this study we highlight advances in b-cell regenerationstrategies with a focus on pluripotent stem cell platforms in the context of translational applications.STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9
INTRODUCTION
Diabetes is a staggering health problem affectingmore than 300 million people worldwide. By2030, an estimated 440 million adults will beafflicted with diabetes [1, 2]. Premature morbidityand mortality create a substantial and escalatingburden on the global health system and society.Type 1 diabetes (T1D) is defined by insulin defi-ciency brought about by autoimmune destructionof islet b cells. Type 2 diabetes (T2D) is defined bythe progressive inability of the insulin secretory ca-pacity tomatch peripheral insulin needs. Defectiveinnateb-cell regeneration, because of eitherb-celldestruction or insufficient b-cell replenishment, isincreasingly recognized as central to thepathobiol-ogy of both T1D and T2D (Fig. 1) [3–6].
Pharmacological means that promote insulinproduction or replace insulin function have com-prised a primary line of therapy for insulin insuf-ficiency in T1D and a large proportion of patientswith T2D, in particular those with advanced dis-ease. Case in point, injection of exogenous insulinis required for people with T1D and advancedT2D. Glucagon-like peptide-1 (GLP-1) analogshave been designed more recently to augmentinsulin secretion and preserve b-cell function. Al-ternatively, DPP-4 inhibitors, which prevent inac-tivation of GLP-1, have been used to promoteendogenous insulinproduction inT2D[7].Whereascollectively the spectrum of preventive and pallia-tive approaches has led to improved diabeticcare, a fail-safe physiological regulation of sys-temic blood glucose levels remains challenging.
Frequent fluctuations in blood glucose levelshave been implicated as a culprit of heteroge-neous (co)morbidities, such as retinopathy,nephropathy, neuropathy, or cardiovascularcomplications. In this context, various nonpharma-cological approaches have been recently exploredto restore functionality of the failing b-cell mass.Such regenerative approaches have evolved rap-idly, from prototypic cell replacement therapiesthrough pancreas/islet transplantation to thepotential use of artificial insulin-producing cellsderived from stemcells or pancreatic progenitorcells (Fig. 2).
ADULT CELL-BASED THERAPIES FOR DIABETES
Islet transplantation has provided a foundationfor next-generation cell-based therapeutics fordiabetes [8]. The Edmonton protocol, a flagshipislet transplantation protocol, has achieved long-term islet survival. In early studies, more than50%ofsubjectsgained insulin independence1yearpost-transplantation and experienced improvedglycated hemoglobin levels and protection fromhypoglycemia [9]. Twenty percent of islet recipi-ents are insulin therapy-free 5 years after trans-plantation [10]. Recent studies have shownimprovements in primary efficacy, safety out-comes, and insulin independence 3 years post-transplant [11, 12]. Despite notable progress inthe field, several issues still need to be addressed.These include the islet isolation methods, sitesfor transplantation, immunosuppression, or im-munoisolation strategies [13–16]. It is also
aCenter for RegenerativeMedicine, bDepartment ofMolecular Medicine,cDivision of CardiovascularDiseases, Department ofMedicine, dDepartment ofMolecular Pharmacology andExperimental Therapeutics,and eDepartment of MedicalGenetics, Mayo Clinic,Rochester, Minnesota, USA
Correspondence: Yasuhiro Ikeda,D.V.M., Ph.D., Center forRegenerative Medicine, MayoClinic, 200 First Street SW,Rochester, Minnesota 55905,USA. Telephone: 507-538-0153;E-Mail: [email protected]
Received October 8, 2013;accepted for publicationDecember 30, 2013.
©AlphaMed Press1066-5099/2014/$20.00/0
http://dx.doi.org/10.5966/sctm.2013-0184
STEM CELLS TRANSLATIONAL MEDICINE 2014;3:1–9 www.StemCellsTM.com ©AlphaMed Press 2014
ENABLING TECHNOLOGIES FOR CELL-BASED CLINICALTRANSLATION
http://stemcellstm.alphamedpress.org/cgi/doi/10.5966/sctm.2013-0184The latest version is at Published Ahead of Print on March 19, 2014 as 10.5966/sctm.2013-0184
at Tufts Hirsh H
ealth Sciences Library on September 25, 2014
http://stemcellstm
.alphamedpress.org/
Dow
nloaded from

Citation: Xi Y, Bu S (2014) Stem Cells Therapy in Diabetes Mellitus. J Stem Cell Res Ther 4: 199. doi:10.4172/2157-7633.1000199
Page 4 of 6
Volume 4 • Issue 5 • 1000199J Stem Cell Res TherISSN: 2157-7633 JSCRT, an open access journal
markers, and their efficiency of differentiation greatly relies on the purity of cell source sorted by cell surface markers. Reports suggest that approximately 100,000 cells are needed for each recipient, but a low differentiation rate necessitate longer time in in vitro culture to develop adequate numbers of cells for transplantation. However, the longer culture time can increase the possibility of malignancy. Indeed, new technologies to improve differentiation efficiency are essential.
Monitoring clinical trials closely will be key. Development of a transplant registry in combination with assessment and optimization of clinical protocols will help identify optimal cell types and cell surface markers for characterization, and may ultimately lead to safe, effective treatments. Acknowledgments
This work was supported in part by National Natural Science Foundation of China (81370165 and 31301068), Natural Science Foundation of Ningbo (2013A610207 and 2013A610209), and K.C. Wong Magna Fund in Ningbo University.
References
1. Abu-Rmeileh NM, Husseini A, Capewell S, O’Flaherty M (2013) Preventing type 2 diabetes among Palestinians: comparing five future policy scenarios. BMJ Open 3(12): e003558.[PubMed]
2. Vetere A, Choudhary A, Burns SM, Wagner BK (2014) Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov 13(4): 278-289.[PubMed]
3. Power C, Thomas C (2011) Changes in BMI, duration of overweight and obesity, and glucose metabolism: 45 years of follow-up of a birth cohort. Diabetes Care 34(9):1986-1991.[PubMed]
4. El-Kaissi S, Sherbeeni S (2011) Pharmacological management of type 2 diabetes mellitus: an update. Curr Diabetes Rev 7(6): 392-405.[PubMed]
5. Odawara M, Hamada I, Suzuki M (2014) Efficacy and safety of vildagliptin as add-on to metformin in Japanese patients with type 2 diabetes mellitus. Diabetes Ther.[PubMed]
6. Kobayashi K, Yokoh H, Sato Y, Takemoto M, Uchida D, et al. (2014) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin compared with α-glucosidase inhibitor in Japanese patients with type 2 diabetes inadequately controlled on sulfonylurea alone (SUCCESS-2): a multicenter, randomized, open-label, non-inferiority trial. Diabetes Obes Metab.[PubMed]
7. Ghosh A, Sengupta N, Sahana P, Giri D, Sengupta P, et al. (2014) Efficacy and safety of add on therapy of bromocriptine with metformin in Indian patients with type 2 diabetes mellitus: A randomized open labeled phase IV clinical trial. Indian J Pharmacol 46(1): 24-28.[PubMed]
8. Rizos EC, Ntzani EE, Papanas N, Tsimihodimos V, Mitrogianni Z, et al. (2014) Combination Therapies of DPP4 Inhibitors and GLP1 Analogues with Insulin in Type 2 Diabetic Patients: A Systematic Review. Curr Vasc Pharmacol 11(6): 992-1000.[PubMed]
9. Dabhi AS, Bhatt NR, Shah MJ (2013) Voglibose: an alpha glucosidase inhibitor.J Clin Diagn Res 7(12): 3023-3027.[PubMed]
10. Gitt AK, Bramlage P, Binz C, Krekler M, Deeg E, et al. (2013) Prognostic implications of DPP-4 inhibitor vs. sulfonylurea use on top of metformin in a real world setting - results of the 1 year follow-up of the prospective DiaRegis registry.Int J Clin Pract 67(10): 1005-1014.[PubMed]
11. Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, et al. (2014) Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124(2): 499-508.[PubMed]
12. Kim JJ, Kido Y, Scherer PE, White MF, Accili D (2007) Analysis of compensatory beta-cell response in mice with combined mutations of Insr and Irs2. Am J Physiol Endocrinol Metab 292(6): E1694-1701.[PubMed]
13. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, et al. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes.Diabetes 52(1): 102-110.[PubMed]
14. Ferrannini E (2010) The stunned beta cell: a brief history. Cell Metab 11(5): 349–352.[PubMed]
15. Butler AE, Galasso R, Meier JJ, Basu R, Rizza RA, et al (2007) Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia 50(11): 2323-2331.[PubMed]
16. Gepts W (1995) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14(10): 619-633.[PubMed]
17. American Diabetes Association (2013) Diagnosis and classification of diabetes mellitus. Diabetes Care 36 (Suppl 1): S67-74.[PubMed]
18. Malmgren S, Spégel P, Danielsson AP, Nagorny CL, Andersson L, et al. (2013) Coordinate changes in histone modifications, mRNA levels, and metabolite profiles in clonal INS-1 832/13 β-cells accompany functional adaptations to lipotoxicity. J Biol Chem 288(17): 11973-11987.[PubMed]
19. Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42(5): 105-116.[PubMed]
20. Imaizumi M, Sato Y, Yang DT, Thibeault SL (2013) In vitro epithelial differentiation of human induced pluripotent stem cells for vocal fold tissue engineering. Ann Otol Rhinol Laryngol 122(12): 737-747.[PubMed]
21. Budniatzky I, Gepstein L (2014) Concise review: reprogramming strategies for cardiovascular regenerative medicine: from induced pluripotent stem cells to direct reprogramming. Stem Cells Transl Med 3(4): 448-457.[PubMed]
22. Powers JM, Trobridge GD (2013) Identification of hematopoietic stem cell engraftment genes in gene therapy studies. J Stem Cell Res Ther 2013(Suppl 3).[PubMed]
23. Parsons XH (2014) Direct conversion of pluripotent human embryonic stem cells under defined culture conditions into human neuronal or cardiomyocyte cell therapy derivatives.Methods Mol Biol.[PubMed]
24. Bruin JE, Erener S, Vela J, Hu X, Johnson JD, et al. (2014) Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res 12(1): 194-208.[PubMed]
25. Jagtap S, Meganathan K, Wagh V, Natarajan K, Hescheler J, et al. (2013) All-trans retinoic acid and basic fibroblast growth factor synergistically direct pluripotent human embryonic stem cells to extraembryonic lineages. Stem Cell Res 10(2): 228-240.[PubMed]
26. Payne C, King J, Hay D (2011) The role of activin/nodal and Wnt signaling in endoderm formation. Vitam Horm 85: 207-216.[PubMed]
27. Zhou J, Su P, Li D, Tsang S, Duan E, et al. (2010) High-efficiency induction of neural conversion in human ESCs and human induced pluripotent stem cells with a single chemical inhibitor of transforming growth factor beta superfamily receptors. Stem Cells 28(10):1741-1750.[PubMed]
Abbreviations: ESCs, embryonic stem cells; iPS, induced pluripotent stem; MSCs, mesenchymal stem cells.
Figure 1: Treatment to diabetes.
Volume 4 • Issue 5 • 1000199J Stem Cell Res TherISSN: 2157-7633 JSCRT, an open access journal
Open AccessReview Article
Stem CellResearch & Therapy
Xi and Bu, J Stem Cell Res Ther 2014, 4:5http://dx.doi.org/10.4172/2157-7633.1000199
Stem Cells Therapy in Diabetes MellitusYang Xi and Shizhong Bu*
Diabetes Center, and Zhejiang Provincial Key Laboratory of Pathophysiology, Institute of Biochemistry and Molecular Biology, School of Medicine, Ningbo University, Ningbo 315211, China
*Corresponding author: Shizhong Bu, 818 Fenghuai Rd., Jiangbei District, Ningbo 315211, China, Tel: 0086-574-87609607; Fax: 0086-574-87608638; E-mail: [email protected]
Received March 17, 2014; Accepted April 24, 2014; Published April 26, 2014
Citation: Xi Y, Bu S (2014) Stem Cells Therapy in Diabetes Mellitus. J Stem Cell Res Ther 4: 199. doi:10.4172/2157-7633.1000199
Copyright: © 2014 Xi Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords: Stem cells; Diabetes mellitus; Cell therapy; Insulin
Abbreviations: BMSCs: Bone Marrow-derived Stem Cells; c-MYC: Myc Proto-oncogene Protein; CXCr: CXC-Chemokine Receptor; DE: Definitive Endoderm; DPP4: Dipeptidyl Peptidase 4; EB: Embryoid Body; EGF: Epidermal Growth Factor; ES: Embryonic Stem; ESCs: Embryonic Stem Cells; FACS: Fluorescence-activated Cell Sorting; FGF: Fibroblast Growth Factor; FoXa2: Forkhead Box Protein A2; GLP1: Glucagon-like Peptide 1; hESCs: Human Embryonic Stem Cells; De1: Definitive Endoderm 1; IPCs: International Programme on Chemical Safety; iPS: Induced Pluripotent Stem; KlF4: Kruppel-like Factor 4; LIF: Leukemia Inhibitory Factor; LIN28: Lin-28 Homolog; MAFA: Transcription Factor MAFA; MSCs: Mesenchymal Stem Cell; Mtpn: Myotrophin; NeuroD: Neurogenic Differentiation Factor; Ngn3: Neurogenin-3; OCT: Octamer-binding Transcription Factor; Pax4: Paired Box Protein 4; Pdx1: Pancreatic and Duodenal Homeobox1; PKU: Phenylketonuria; SGLT2: Sodium-dependent Glucose Cotransporter 2; SOX2: Sry related HMG box-2; T1DM: Type 1 Diabetes Mellitus; TGFβ: Transforming Growth Factorβ; TZDs: Thiazolidinediones
IntroductionDiabetes mellitus is a devastating and complex metabolic disease,
expected to affect over 500 million people worldwide by the year 2030; up from 350 million in 2010 [1]. Approximately 95% of patients suffer from type 2 diabetes, and its prevalence is expected to increase in the future [2]. Furthermore, the age of onset for type 2 diabetic patients is trending toward earlier onset in adulthood [3]. Diabetes is associated with severe long-term micro- and macrovascular complications, and carries a high rate of morbidity and mortality. Indeed, both type 1 and 2 diabetes are a significant public health concern with numerous debilitating complications, leading to a constant increase in treatment costs.
Currently, both type 1 and type 2 diabetes can be treated with insulin analogues and Pramlintide. Pramlintide or Amylin is a 37-residue peptide hormone that delays gastric emptying, and endorses satiety and inhibits glucagon secretion; averting post-prandial prickles in blood glucose levels. Recombinant modifications of insulin can act faster and longer, similar to endogenous insulin [4]. The following drugs are used to treat type 2 diabetes: 1) Metformin, augments insulin release, 2) Sulphonylureas (Thiazolidinediones (TZDs) and Meglitinides), increase insulin sensitivity, 3) Bromocriptine, antagonizes dopamine
D2 and serotonin receptors, 4) Glucagon-like peptide 1 (GLP1) analogues, 5) Alpha-glucosidase inhibitors, 6) Dipeptidyl peptidase 4 (DPP4) inhibitors, and 7) Sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors [5-11].
The physiological control of blood glucose levels can only be restored effectively by replacing the β-cell mass [12]. β-cells in the pancreatic islets of Langerhans are responsible for the production of insulin and much of the pathology of diabetes losses can be attributed to the loss of β-cell number and function [13,14]. In patients with type 1 diabetes, the onset of overt disease is assumed to occur when the β-cell mass falls below 20% of the normal range [15,16]; whereas in patients with type 2 diabetes, the β-cell mass is unable to meet the increased insulin demands of the body [17]. Eventually, the β-cell mass in type 2 diabetes also declines to 40–60% of the normal range. Indeed, in both type 1 and type 2 diabetes, restoration of a functional β-cell mass constitutes the central goal of diabetes therapy [18,19]. Exploring ways to protect or expand pancreatic β-cell mass and function could be an effective therapeutic approach, and β-cell replacement represents an attractive therapeutic prospect. Stem cells, particulary the pluripotent stem cells, demonstrate strong self-renewal abilities and potential to differentiate into all cell types of the body, making them a supreme cell source for regenerative medicine and tissue engineering [20-22]. In this review, we address some of the major advancements that could lead to regenerative therapy for diabetes mellitus.
Human Embryonic Stem CellsHuman embryonic stem cells (hESCs) have the ability to form
cells derived from all three germ layers [23]. hESCs can be induced to differentiate into fetal-like pancreatic cells in vitro using a 33-day, 7-stage protocol [24]. The differentiation protocols for inducing hESCs
AbstractDiabetes is one of the top 10 leading causes of morbidity and mortality, affecting nearly 350 million people
worldwide. β-cell replacement represents an attractive prospect for diabetes therapy but treatment options remain quite limited. There is increasing hope placed on insulin producing cells derived from human pluripotent stem cells, even as the approach faces continued challenges. The most effective protocols thus far have produced cells that express insulin, and have molecular characteristics that closely resemble genuine insulin-secreting cells. However, these cells demonstrate little sensitivity to glucose – an issue that will hopefully be resolved in coming years. This review summarizes recent progress in obtaining cells that express insulin from different progenitor sources, and highlights the major pathways and genes involved in diabetic patients.
Volume 4 • Issue 5 • 1000199J Stem Cell Res TherISSN: 2157-7633 JSCRT, an open access journal
Open AccessReview Article
Stem CellResearch & Therapy
Xi and Bu, J Stem Cell Res Ther 2014, 4:5http://dx.doi.org/10.4172/2157-7633.1000199
Stem Cells Therapy in Diabetes MellitusYang Xi and Shizhong Bu*
Diabetes Center, and Zhejiang Provincial Key Laboratory of Pathophysiology, Institute of Biochemistry and Molecular Biology, School of Medicine, Ningbo University, Ningbo 315211, China
*Corresponding author: Shizhong Bu, 818 Fenghuai Rd., Jiangbei District, Ningbo 315211, China, Tel: 0086-574-87609607; Fax: 0086-574-87608638; E-mail: [email protected]
Received March 17, 2014; Accepted April 24, 2014; Published April 26, 2014
Citation: Xi Y, Bu S (2014) Stem Cells Therapy in Diabetes Mellitus. J Stem Cell Res Ther 4: 199. doi:10.4172/2157-7633.1000199
Copyright: © 2014 Xi Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords: Stem cells; Diabetes mellitus; Cell therapy; Insulin
Abbreviations: BMSCs: Bone Marrow-derived Stem Cells; c-MYC: Myc Proto-oncogene Protein; CXCr: CXC-Chemokine Receptor; DE: Definitive Endoderm; DPP4: Dipeptidyl Peptidase 4; EB: Embryoid Body; EGF: Epidermal Growth Factor; ES: Embryonic Stem; ESCs: Embryonic Stem Cells; FACS: Fluorescence-activated Cell Sorting; FGF: Fibroblast Growth Factor; FoXa2: Forkhead Box Protein A2; GLP1: Glucagon-like Peptide 1; hESCs: Human Embryonic Stem Cells; De1: Definitive Endoderm 1; IPCs: International Programme on Chemical Safety; iPS: Induced Pluripotent Stem; KlF4: Kruppel-like Factor 4; LIF: Leukemia Inhibitory Factor; LIN28: Lin-28 Homolog; MAFA: Transcription Factor MAFA; MSCs: Mesenchymal Stem Cell; Mtpn: Myotrophin; NeuroD: Neurogenic Differentiation Factor; Ngn3: Neurogenin-3; OCT: Octamer-binding Transcription Factor; Pax4: Paired Box Protein 4; Pdx1: Pancreatic and Duodenal Homeobox1; PKU: Phenylketonuria; SGLT2: Sodium-dependent Glucose Cotransporter 2; SOX2: Sry related HMG box-2; T1DM: Type 1 Diabetes Mellitus; TGFβ: Transforming Growth Factorβ; TZDs: Thiazolidinediones
IntroductionDiabetes mellitus is a devastating and complex metabolic disease,
expected to affect over 500 million people worldwide by the year 2030; up from 350 million in 2010 [1]. Approximately 95% of patients suffer from type 2 diabetes, and its prevalence is expected to increase in the future [2]. Furthermore, the age of onset for type 2 diabetic patients is trending toward earlier onset in adulthood [3]. Diabetes is associated with severe long-term micro- and macrovascular complications, and carries a high rate of morbidity and mortality. Indeed, both type 1 and 2 diabetes are a significant public health concern with numerous debilitating complications, leading to a constant increase in treatment costs.
Currently, both type 1 and type 2 diabetes can be treated with insulin analogues and Pramlintide. Pramlintide or Amylin is a 37-residue peptide hormone that delays gastric emptying, and endorses satiety and inhibits glucagon secretion; averting post-prandial prickles in blood glucose levels. Recombinant modifications of insulin can act faster and longer, similar to endogenous insulin [4]. The following drugs are used to treat type 2 diabetes: 1) Metformin, augments insulin release, 2) Sulphonylureas (Thiazolidinediones (TZDs) and Meglitinides), increase insulin sensitivity, 3) Bromocriptine, antagonizes dopamine
D2 and serotonin receptors, 4) Glucagon-like peptide 1 (GLP1) analogues, 5) Alpha-glucosidase inhibitors, 6) Dipeptidyl peptidase 4 (DPP4) inhibitors, and 7) Sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors [5-11].
The physiological control of blood glucose levels can only be restored effectively by replacing the β-cell mass [12]. β-cells in the pancreatic islets of Langerhans are responsible for the production of insulin and much of the pathology of diabetes losses can be attributed to the loss of β-cell number and function [13,14]. In patients with type 1 diabetes, the onset of overt disease is assumed to occur when the β-cell mass falls below 20% of the normal range [15,16]; whereas in patients with type 2 diabetes, the β-cell mass is unable to meet the increased insulin demands of the body [17]. Eventually, the β-cell mass in type 2 diabetes also declines to 40–60% of the normal range. Indeed, in both type 1 and type 2 diabetes, restoration of a functional β-cell mass constitutes the central goal of diabetes therapy [18,19]. Exploring ways to protect or expand pancreatic β-cell mass and function could be an effective therapeutic approach, and β-cell replacement represents an attractive therapeutic prospect. Stem cells, particulary the pluripotent stem cells, demonstrate strong self-renewal abilities and potential to differentiate into all cell types of the body, making them a supreme cell source for regenerative medicine and tissue engineering [20-22]. In this review, we address some of the major advancements that could lead to regenerative therapy for diabetes mellitus.
Human Embryonic Stem CellsHuman embryonic stem cells (hESCs) have the ability to form
cells derived from all three germ layers [23]. hESCs can be induced to differentiate into fetal-like pancreatic cells in vitro using a 33-day, 7-stage protocol [24]. The differentiation protocols for inducing hESCs
AbstractDiabetes is one of the top 10 leading causes of morbidity and mortality, affecting nearly 350 million people
worldwide. β-cell replacement represents an attractive prospect for diabetes therapy but treatment options remain quite limited. There is increasing hope placed on insulin producing cells derived from human pluripotent stem cells, even as the approach faces continued challenges. The most effective protocols thus far have produced cells that express insulin, and have molecular characteristics that closely resemble genuine insulin-secreting cells. However, these cells demonstrate little sensitivity to glucose – an issue that will hopefully be resolved in coming years. This review summarizes recent progress in obtaining cells that express insulin from different progenitor sources, and highlights the major pathways and genes involved in diabetic patients.
Volume 4 • Issue 5 • 1000199J Stem Cell Res TherISSN: 2157-7633 JSCRT, an open access journal
Open AccessReview Article
Stem CellResearch & Therapy
Xi and Bu, J Stem Cell Res Ther 2014, 4:5http://dx.doi.org/10.4172/2157-7633.1000199
Stem Cells Therapy in Diabetes MellitusYang Xi and Shizhong Bu*
Diabetes Center, and Zhejiang Provincial Key Laboratory of Pathophysiology, Institute of Biochemistry and Molecular Biology, School of Medicine, Ningbo University, Ningbo 315211, China
*Corresponding author: Shizhong Bu, 818 Fenghuai Rd., Jiangbei District, Ningbo 315211, China, Tel: 0086-574-87609607; Fax: 0086-574-87608638; E-mail: [email protected]
Received March 17, 2014; Accepted April 24, 2014; Published April 26, 2014
Citation: Xi Y, Bu S (2014) Stem Cells Therapy in Diabetes Mellitus. J Stem Cell Res Ther 4: 199. doi:10.4172/2157-7633.1000199
Copyright: © 2014 Xi Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords: Stem cells; Diabetes mellitus; Cell therapy; Insulin
Abbreviations: BMSCs: Bone Marrow-derived Stem Cells; c-MYC: Myc Proto-oncogene Protein; CXCr: CXC-Chemokine Receptor; DE: Definitive Endoderm; DPP4: Dipeptidyl Peptidase 4; EB: Embryoid Body; EGF: Epidermal Growth Factor; ES: Embryonic Stem; ESCs: Embryonic Stem Cells; FACS: Fluorescence-activated Cell Sorting; FGF: Fibroblast Growth Factor; FoXa2: Forkhead Box Protein A2; GLP1: Glucagon-like Peptide 1; hESCs: Human Embryonic Stem Cells; De1: Definitive Endoderm 1; IPCs: International Programme on Chemical Safety; iPS: Induced Pluripotent Stem; KlF4: Kruppel-like Factor 4; LIF: Leukemia Inhibitory Factor; LIN28: Lin-28 Homolog; MAFA: Transcription Factor MAFA; MSCs: Mesenchymal Stem Cell; Mtpn: Myotrophin; NeuroD: Neurogenic Differentiation Factor; Ngn3: Neurogenin-3; OCT: Octamer-binding Transcription Factor; Pax4: Paired Box Protein 4; Pdx1: Pancreatic and Duodenal Homeobox1; PKU: Phenylketonuria; SGLT2: Sodium-dependent Glucose Cotransporter 2; SOX2: Sry related HMG box-2; T1DM: Type 1 Diabetes Mellitus; TGFβ: Transforming Growth Factorβ; TZDs: Thiazolidinediones
IntroductionDiabetes mellitus is a devastating and complex metabolic disease,
expected to affect over 500 million people worldwide by the year 2030; up from 350 million in 2010 [1]. Approximately 95% of patients suffer from type 2 diabetes, and its prevalence is expected to increase in the future [2]. Furthermore, the age of onset for type 2 diabetic patients is trending toward earlier onset in adulthood [3]. Diabetes is associated with severe long-term micro- and macrovascular complications, and carries a high rate of morbidity and mortality. Indeed, both type 1 and 2 diabetes are a significant public health concern with numerous debilitating complications, leading to a constant increase in treatment costs.
Currently, both type 1 and type 2 diabetes can be treated with insulin analogues and Pramlintide. Pramlintide or Amylin is a 37-residue peptide hormone that delays gastric emptying, and endorses satiety and inhibits glucagon secretion; averting post-prandial prickles in blood glucose levels. Recombinant modifications of insulin can act faster and longer, similar to endogenous insulin [4]. The following drugs are used to treat type 2 diabetes: 1) Metformin, augments insulin release, 2) Sulphonylureas (Thiazolidinediones (TZDs) and Meglitinides), increase insulin sensitivity, 3) Bromocriptine, antagonizes dopamine
D2 and serotonin receptors, 4) Glucagon-like peptide 1 (GLP1) analogues, 5) Alpha-glucosidase inhibitors, 6) Dipeptidyl peptidase 4 (DPP4) inhibitors, and 7) Sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors [5-11].
The physiological control of blood glucose levels can only be restored effectively by replacing the β-cell mass [12]. β-cells in the pancreatic islets of Langerhans are responsible for the production of insulin and much of the pathology of diabetes losses can be attributed to the loss of β-cell number and function [13,14]. In patients with type 1 diabetes, the onset of overt disease is assumed to occur when the β-cell mass falls below 20% of the normal range [15,16]; whereas in patients with type 2 diabetes, the β-cell mass is unable to meet the increased insulin demands of the body [17]. Eventually, the β-cell mass in type 2 diabetes also declines to 40–60% of the normal range. Indeed, in both type 1 and type 2 diabetes, restoration of a functional β-cell mass constitutes the central goal of diabetes therapy [18,19]. Exploring ways to protect or expand pancreatic β-cell mass and function could be an effective therapeutic approach, and β-cell replacement represents an attractive therapeutic prospect. Stem cells, particulary the pluripotent stem cells, demonstrate strong self-renewal abilities and potential to differentiate into all cell types of the body, making them a supreme cell source for regenerative medicine and tissue engineering [20-22]. In this review, we address some of the major advancements that could lead to regenerative therapy for diabetes mellitus.
Human Embryonic Stem CellsHuman embryonic stem cells (hESCs) have the ability to form
cells derived from all three germ layers [23]. hESCs can be induced to differentiate into fetal-like pancreatic cells in vitro using a 33-day, 7-stage protocol [24]. The differentiation protocols for inducing hESCs
AbstractDiabetes is one of the top 10 leading causes of morbidity and mortality, affecting nearly 350 million people
worldwide. β-cell replacement represents an attractive prospect for diabetes therapy but treatment options remain quite limited. There is increasing hope placed on insulin producing cells derived from human pluripotent stem cells, even as the approach faces continued challenges. The most effective protocols thus far have produced cells that express insulin, and have molecular characteristics that closely resemble genuine insulin-secreting cells. However, these cells demonstrate little sensitivity to glucose – an issue that will hopefully be resolved in coming years. This review summarizes recent progress in obtaining cells that express insulin from different progenitor sources, and highlights the major pathways and genes involved in diabetic patients.

1. Human embryonic stem cells
2. Induced pluripotent stem cells
3. Umbilical cord blood
4. Mesenchymal stromal cells
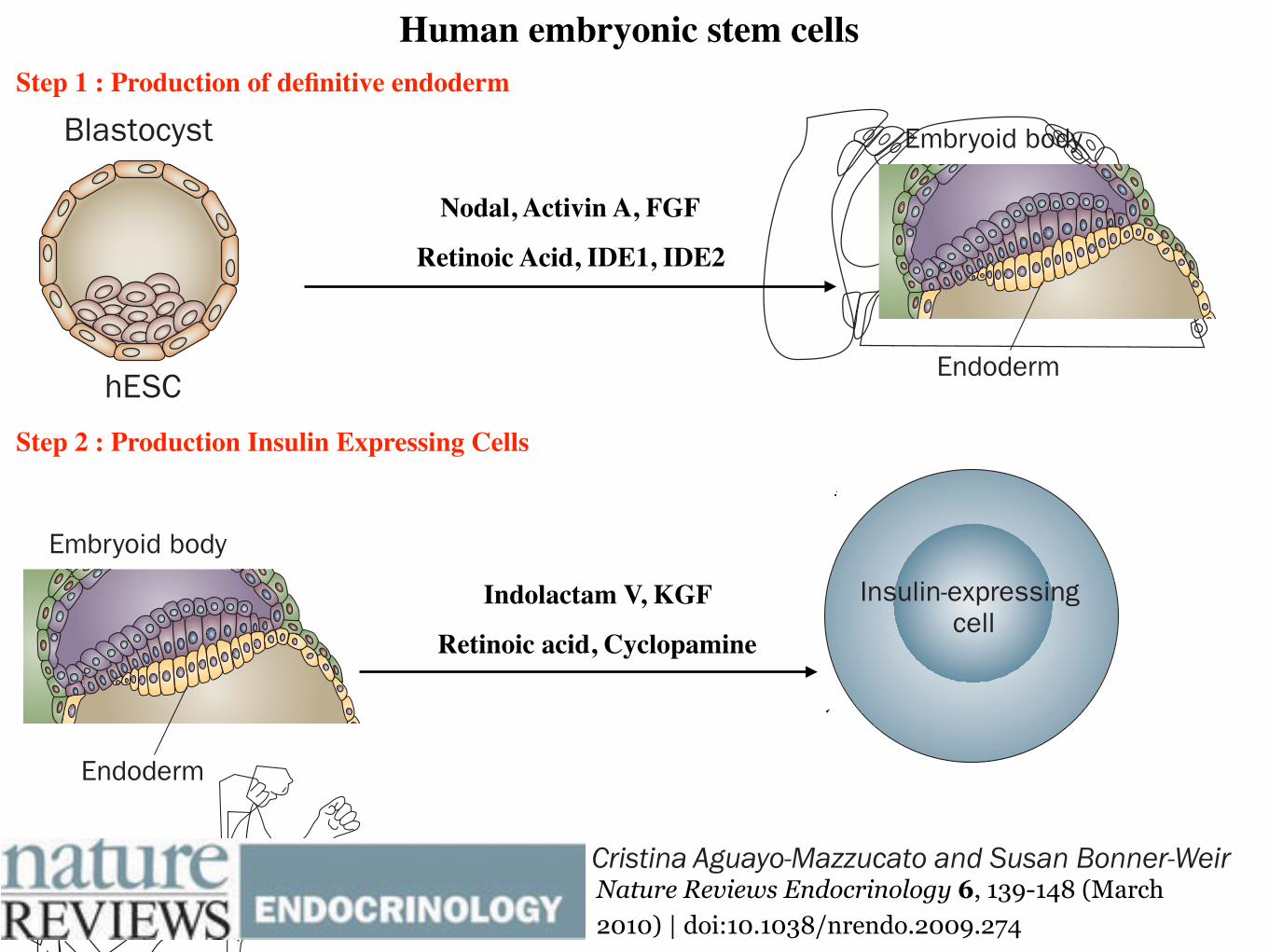
Human embryonic stem cells Step 1 : Production of definitive endoderm
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
Nodal, Activin A, FGF
Retinoic Acid, IDE1, IDE2
Step 2 : Production Insulin Expressing Cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
Indolactam V, KGF
Retinoic acid, Cyclopamine
3/25/16, 8:19 AMAccess : Stem cell therapy for type 1 diabetes mellitus : Nature Reviews Endocrinology
Page 1 of 3http://www.nature.com/nrendo/journal/v6/n3/full/nrendo.2009.274.html
ARTICLETOOLS
Send to afriend
Exportcitation
Exportreferences
Rights andpermissions
Ordercommercialreprints
SEARCHPUBMEDFOR
CristinaAguayo-Mazzucato
Susan
AccessTo read this article in full you may need to login, make a payment or gain access through asite license (see right).
ReviewNature Reviews Endocrinology 6, 139-148 (March2010) | doi:10.1038/nrendo.2009.274
Stem cell therapy fortype 1 diabetesmellitusCristina Aguayo-Mazzucato &Susan Bonner-Weir
The use of stem cells inregenerative medicine holdsgreat promise for the cure ofmany diseases, including type 1diabetes mellitus (T1DM). Anypotential stem-cell-based curefor T1DM should address theneed for β-cell replacement, aswell as control of theautoimmune response to cellswhich express insulin. The exvivo generation of β cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 139
Section of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, 1 Joslin Place, Boston, MA 02215, USA. (C. Aguayo-Mazzucato, S. Bonner-Weir).
Correspondence to: S. Bonner-Weir [email protected]
Stem cell therapy for type 1 diabetes mellitusCristina Aguayo-Mazzucato and Susan Bonner-Weir
Abstract | The use of stem cells in regenerative medicine holds great promise for the cure of many diseases, including type 1 diabetes mellitus (T1DM). Any potential stem-cell-based cure for T1DM should address the need for β-cell replacement, as well as control of the autoimmune response to cells which express insulin. The ex vivo generation of β cells suitable for transplantation to reconstitute a functional β-cell mass has used pluripotent cells from diverse sources, as well as organ-specific facultative progenitor cells from the liver and the pancreas. The most effective protocols to date have produced cells that express insulin and have molecular characteristics that closely resemble bona fide insulin-secreting cells; however, these cells are often unresponsive to glucose, a characteristic that should be addressed in future protocols. The use of mesenchymal stromal cells or umbilical cord blood to modulate the immune response is already in clinical trials; however, definitive results are still pending. This Review focuses on current strategies to obtain cells which express insulin from different progenitor sources and highlights the main pathways and genes involved, as well as the different approaches for the modulation of the immune response in patients with T1DM.
Aguayo-Mazzucato, C. & Bonner-Weir, S. Nat. Rev. Endocrinol. 6, 139–148 (2010); doi:10.1038/nrendo.2009.274
IntroductionType 1 diabetes mellitus (T1DM) is characterized by the autoimmune destruction of the insulin- expressing, pan-creatic β cells; as a result, patients with T1DM are depen-dent on exogenous insulin for their blood glucose control. Although the discovery of insulin has changed the outlook and survival of this group of individuals for almost a century,1 patients with T1DM are still not exempt from the development of diabetic complications.2,3 These com-plications are thought to result, in part, from the lack of physiological oscillations in insulin secretion and higher than normal glucose levels; thus, the search for effective ways to re establish a functional β-cell mass in patients with T1DM is important. Transplantation of whole human pancreas or isolated, cadaveric human islets4,5 has allowed patients with T1DM to become insulin-independent and has thereby shown proof of concept for β-cell replacement therapies. The shortage of donor pancreata and islets,6 however, results in the search for alternative cell sources, and differen tiation of stem cells (Box 1) into functional β cells is an attractive alternative.
A definitive cure for T1DM needs to address both the β-cell deficit and the autoimmune response to cells that express insulin. As clearly shown by the transplantation of segmental pancreatic grafts from identical twins, the autoimmune response remains many years after disease onset.7 Thus, any β-cell replacement therapy will need some modulation of the immune system, whether by drugs or cell-based approaches.
This Review focuses mainly on the different approaches to obtain functional, insulin-producing cells from various types of progenitor cells. Although the goal of the field is to
direct differentiation of human stem cells into functional β cells, some animal studies are included here, as they could lay the framework for success ful future interventions in humans. Although stem cell- derived, insulin-positive cells have not yet reached clinical trial, clinical data from stem cell-derived therapies that target the autoimmune response have been reported and are discussed.
Many roads lead from stem cells to β cellsEfforts have been directed at the development of efficient protocols for the differentiation of stem cells of either embryonic (Figure 1) or adult origin (Figure 2) into func-tional cells that secrete insulin. A caveat to many of these studies is that some developmental pathways generate insulin-expressing cells that will never become true β cells, for example, cells of the fetal liver, yolk sac and brain.8–10 A β cell should be defined by its ability to store large amounts of insulin and to secrete it in a regulated manner in response to a demand, such as glucose stimulation.11
Human embryonic stem cellsEfforts to generate β cells from human embryonic stem cells (hESCs) have focused on the fundamentals of normal embryonic development as a basis to understand the early stages of endoderm formation. The first step in the differen tiation of hESCs into β cells is the production of definitive endoderm12–17 rather than visceral endoderm, which expresses the same markers (for example, sex determining region Y box [SOX] 17 and forkhead box protein A2 [FOXA2]) but forms different tissues.18
Current protocols for hESC differentiation into β cells aim to reproduce the developmentally active signaling pathways Wnt and transforming growth factor β (TGFβ) (Box 2).17,19–22 Growth factors, such as Activin A, fibroblast
Competing interestsThe authors declare no competing interests.
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 139 4/2/10 17:00:58
© 20 Macmillan Publishers Limited. All rights reserved10

Human embryonic stem cells
Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
the same or a very similar developmental potential to its in vivocounterpart. The induction of endoderm from ESCs by IDEs isconserved between mouse and human species. Though wehave yet to achieve complete stepwise differentiation fromESCs to pancreatic b cells by chemical means, this studyfocuses on an important first step: formation of endoderm.
RESULTS
Screening with ESCs for Endoderm FormationTo obtain a reporter for endoderm formation, we generateda mouse ESC line with the red fluorescent protein dTomato,a variant of DsRed (Shaner et al., 2004) coding sequence undercontrol of the Sox17 promoter (Figure S1 available online).Several lines of evidence show that Sox17 is an endodermalmarker, both definitive and extraembryonic. A null mutation inmice is devoid of foregut endoderm, and mid- and hindgut endo-derm fails to expand (Kanai-Azuma et al., 2002). Gene expres-sion analysis of isolated endoderm confirms that Sox17 is
Figure 1. High-Throughput Screening(A) Scheme of differentiation into endoderm and
evaluation of an endoderm reporter line. Treat-
ment with either Activin A or Nodal induces endo-
derm in mouse ESC cultures, and at day 6 of treat-
ment, 45% of total cells are Sox17/dsRed double
positive. Every dsRed+ cell stains positively for
Sox17 antibody.
(B) Overview of the identification of endoderm
inducers from small molecule collection. Out of
>4000 screened compounds, 27 primary hits
were selected and further evaluated for specificity
and toxicity. Markers for definitive endoderm (DE)
and extraembryonic endoderm (EE) were tested
by Q-RT-PCR and immunohistochemistry, and
two compounds that induced high levels of DE
were indentified. R 3 SD = more than three stan-
dard deviations.
a marker of endoderm, both definitiveand extraembryonic (Sherwood et al.,2007). However, it is important to notethat Sox17 expression is not restrictedexclusively to endoderm; genetic lineagetracing shows that Sox17 is expressed inthe endodermal lineage as early as E7.5but later on marks the gut tube as wellas other organs (Park et al., 2006; Kimet al., 2007; Liu et al., 2007; R.M., unpub-lished data).
In vitro, application of TGF-b family ofgrowth factors, including Activin A orNodal, to both mouse and human ESCcultures leads to the preferential differen-tiation into endodermal lineages (Kuboet al., 2004; Yasunaga et al., 2005;D’Amour et al., 2005). Consistent withthose previous studies, we observeendoderm induction when Sox17-dsRedmouse ESCs are treated with Activin A
for 6 days in low-serum conditions (see Experimental Proce-dures). At 6 days, 45% of cells stain positively for Sox17 andexpress dsRed (Figure 1A). All dsRed+ cells also expressSox17 protein as judged by Sox 17 antibody staining, whichshows that the reporter line accurately reflects endogenousSox17 expression. The coexpression of the fluorescent markerand endogenous Sox17 was also confirmed at all time pointsin vitro as well as in E6.5 and E7.5 embryos (data not shown).There is a minor population of cells (10% ± 3.6%) that stainedfor Sox17 but did not express dsRed; however, no false positiveexpression of the transgene (DsRed positive but Sox17 antibodynegative) was observed.
The endoderm differentiation protocol used in this screen isbased on previously published protocols (Kubo et al., 2004;Yasunaga et al., 2005; D’Amour et al., 2005) with several modifi-cations to allow endoderm lineage induction in a high-throughput format. Specifically, we cultured the mouse ESCsas a monolayer (in contrast to embryoid bodies) in gelatin-coated384-well plates without any feeder cell layer. We also adjusted
Cell Stem Cell
Small Molecules Induced Definitive Endoderm
Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc. 349
Figure 2. Two Small Molecule Inducers ofEndoderm in Mouse and Human ESCCultures(A) Chemical structure of IDE1 and IDE2 that
induce endoderm from mouse ESCs. The lower
panel shows dose response curves of Sox17
expression (based on immunofluorescence)
following treatment with compound for 6 days.
The EC50 values and curve fitting were performed
with Graph Prism software. Data are presented as
mean ± SD. n = 4.
(B) Representative images of aSox17 immunofluo-
rescence show the highest Sox17 induction in
mouse ESCs by compound at day 6 of treatment,
quantified as the percent of cells expressing
Sox17 (upper panel) out of the total cells. The
majority of Sox17+ cells (R95%) induced by
chemical treatment coexpress another definitive
endoderm marker, FoxA2.
(C) HUES cell cultures were treated for 6 days with
IDE1 or IDE2. The endoderm markers Sox17 and
FoxA2 were analyzed by immunofluorescence.
Treatment with either compound leads to Sox17
expression in 55%–65% of total cells; R95% of
Sox17+ cells coexpress FoxA2.
Cell Stem Cell
Small Molecules Induced Definitive Endoderm
Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc. 351
Figure 2. Two Small Molecule Inducers ofEndoderm in Mouse and Human ESCCultures(A) Chemical structure of IDE1 and IDE2 that
induce endoderm from mouse ESCs. The lower
panel shows dose response curves of Sox17
expression (based on immunofluorescence)
following treatment with compound for 6 days.
The EC50 values and curve fitting were performed
with Graph Prism software. Data are presented as
mean ± SD. n = 4.
(B) Representative images of aSox17 immunofluo-
rescence show the highest Sox17 induction in
mouse ESCs by compound at day 6 of treatment,
quantified as the percent of cells expressing
Sox17 (upper panel) out of the total cells. The
majority of Sox17+ cells (R95%) induced by
chemical treatment coexpress another definitive
endoderm marker, FoxA2.
(C) HUES cell cultures were treated for 6 days with
IDE1 or IDE2. The endoderm markers Sox17 and
FoxA2 were analyzed by immunofluorescence.
Treatment with either compound leads to Sox17
expression in 55%–65% of total cells; R95% of
Sox17+ cells coexpress FoxA2.
Cell Stem Cell
Small Molecules Induced Definitive Endoderm
Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc. 351
the same or a very similar developmental potential to its in vivocounterpart. The induction of endoderm from ESCs by IDEs isconserved between mouse and human species. Though wehave yet to achieve complete stepwise differentiation fromESCs to pancreatic b cells by chemical means, this studyfocuses on an important first step: formation of endoderm.
RESULTS
Screening with ESCs for Endoderm FormationTo obtain a reporter for endoderm formation, we generateda mouse ESC line with the red fluorescent protein dTomato,a variant of DsRed (Shaner et al., 2004) coding sequence undercontrol of the Sox17 promoter (Figure S1 available online).Several lines of evidence show that Sox17 is an endodermalmarker, both definitive and extraembryonic. A null mutation inmice is devoid of foregut endoderm, and mid- and hindgut endo-derm fails to expand (Kanai-Azuma et al., 2002). Gene expres-sion analysis of isolated endoderm confirms that Sox17 is
Figure 1. High-Throughput Screening(A) Scheme of differentiation into endoderm and
evaluation of an endoderm reporter line. Treat-
ment with either Activin A or Nodal induces endo-
derm in mouse ESC cultures, and at day 6 of treat-
ment, 45% of total cells are Sox17/dsRed double
positive. Every dsRed+ cell stains positively for
Sox17 antibody.
(B) Overview of the identification of endoderm
inducers from small molecule collection. Out of
>4000 screened compounds, 27 primary hits
were selected and further evaluated for specificity
and toxicity. Markers for definitive endoderm (DE)
and extraembryonic endoderm (EE) were tested
by Q-RT-PCR and immunohistochemistry, and
two compounds that induced high levels of DE
were indentified. R 3 SD = more than three stan-
dard deviations.
a marker of endoderm, both definitiveand extraembryonic (Sherwood et al.,2007). However, it is important to notethat Sox17 expression is not restrictedexclusively to endoderm; genetic lineagetracing shows that Sox17 is expressed inthe endodermal lineage as early as E7.5but later on marks the gut tube as wellas other organs (Park et al., 2006; Kimet al., 2007; Liu et al., 2007; R.M., unpub-lished data).
In vitro, application of TGF-b family ofgrowth factors, including Activin A orNodal, to both mouse and human ESCcultures leads to the preferential differen-tiation into endodermal lineages (Kuboet al., 2004; Yasunaga et al., 2005;D’Amour et al., 2005). Consistent withthose previous studies, we observeendoderm induction when Sox17-dsRedmouse ESCs are treated with Activin A
for 6 days in low-serum conditions (see Experimental Proce-dures). At 6 days, 45% of cells stain positively for Sox17 andexpress dsRed (Figure 1A). All dsRed+ cells also expressSox17 protein as judged by Sox 17 antibody staining, whichshows that the reporter line accurately reflects endogenousSox17 expression. The coexpression of the fluorescent markerand endogenous Sox17 was also confirmed at all time pointsin vitro as well as in E6.5 and E7.5 embryos (data not shown).There is a minor population of cells (10% ± 3.6%) that stainedfor Sox17 but did not express dsRed; however, no false positiveexpression of the transgene (DsRed positive but Sox17 antibodynegative) was observed.
The endoderm differentiation protocol used in this screen isbased on previously published protocols (Kubo et al., 2004;Yasunaga et al., 2005; D’Amour et al., 2005) with several modifi-cations to allow endoderm lineage induction in a high-throughput format. Specifically, we cultured the mouse ESCsas a monolayer (in contrast to embryoid bodies) in gelatin-coated384-well plates without any feeder cell layer. We also adjusted
Cell Stem Cell
Small Molecules Induced Definitive Endoderm
Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc. 349

Human embryonic stem cells
which time the lateral walls of the embryonic gut fold ventrallyand the gut tube closes—around E9.5. ES-derived endodermalcells induced by small molecules integrated into the developinggut tube, whereas control-treated ESCs did not. Moreover, ESCderived expressed markers of gut tube FoxA2 and Claudin6 andshowed the characteristic morphology of gut cells (Andersonet al., 2008) (Figure 6B). Cells induced by IDE1 treatment inte-grated into the gut in 8 out of 35 cases and in 7 out of 29 embryosfor IDE2. Conversely, DMSO (control)-treated cells never inte-grate into the developing gut tube (0 out of 10 and 0 out of 11,respectively) and instead remain in the lumen (Figure 6B).
In Vitro Potential of Compound-Induced EndodermTo further evaluate the developmental potential of compound-induced endoderm, we tested whether it can differentiatein vitro into pancreatic progenitors. Pancreatic progenitorsappear in the pancreatic bud at E9.5 and are marked by theexpression of the transcription factor Pdx1. Pdx1 is requiredfor pancreas development and b cell formation because a nullmutation of Pdx1 in mice results in a failure to form a pancreas(Jonsson et al., 1994; Offield et al., 1996), and lineage tracingstudies show that Pdx1 marks progenitors that give rise to allpancreatic cell types (Gu et al., 2003). Using Pdx1-GFP knockin(Micallef et al., 2005) mouse ESCs, we induced definitive endo-
Figure 7. Developmental Potential ofChemically Derived Endoderm(A) Scheme of mouse ESC differentiation into
Pdx1+ pancreatic progenitors. Endoderm was
first derived through treatment with either IDE1
or IDE2, and then formation of pancreatic progen-
itors was monitored using a Pdx1-GFP ES reporter
line.
(B) Endoderm-enriched cultures were grown for
another 6 days in chemically defined media con-
taining: DMSO without any additional growth
factors or compounds (control), in the presence
of growth factors (FGF10, CYC, RA), or in the pres-
ence of Indolactam V to induce the expression of
Pdx1. At day 12, in cultures treated initially with
IDE1 or IDE2 and followed by ILV, 50% of the total
cells were Pdx1+, a 10-fold increase above control
treatment.
derm using either of the hit compoundsand cultured cells in various conditionsfor an additional 6 days.
Published protocols for differentiationof ESCs into pancreatic progenitors arebased on studies of the factors thatmodulate signaling during pancreaticorganogenesis (Stafford and Prince,2002; Lau et al., 2006; Bhushan et al.,2001). For example, in one protocol,endoderm is induced with Activin A,followed by treatment with FGF10 andthe Hedgehog signaling inhibitor KAAD-cyclopamine for 3 days, and is thenexposed to a posteriorizing factor, reti-noic acid, for an additional 3 days to
induce Pdx1 expression (D’Amour et al., 2006). Using thisregimen, 12.1% ± 4% of the mouse ESCs differentiate furtherinto pancreatic progenitors, defined by Pdx1+ expression. Bycomparison, when compound-induced endoderm was used asa starting population, rather than Activin A-induced endoderm,25% ± 6% of cells expressed Pdx1. Spontaneous differentiationin the absence of hit compounds, additional growth factors, orsignaling modulators occurs at a lower level, producing GFPexpression in 4.6% ± 1.1% of cells (Figure 7).
Finally, a separate chemical screen performed using humancells has identified a compound, Indolactam V (Chen et al.,2009), that can induce pancreatic progenitors from endodermthat has been produced from HUES. Indolactam V is also ableto efficiently induce pancreatic progenitors (Pdx1+ cells) inendoderm derived by chemical treatment of mouse ESC. Treat-ment of IDE1- or IDE2-induced mouse endoderm yields 51% ±7.4% Pdx1-GFP-expressing pancreatic progenitors at day 6(Figure 7). When cultures were stained with Pdx1 antibodies,we observed a similar number (53% ± 6.4%) of positive cellsafter two-step treatment with small molecules. Notably, this isa 10-fold increase in cell number compared to the controlDMSO treated and a 4-fold increase compared to publishedprotocols. The majority of Pdx1+ (92% ± 5%) cells also expressother pancreatic progenitor markers, including HNF6 (Figure S4).
Cell Stem Cell
Small Molecules Induced Definitive Endoderm
Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc. 355
and upregulating Nodal expression through a Smad2/FAST-1-dependent autoregulatory loop that feeds on Nodal transcription(Agius et al., 2000; Pogoda et al., 2000).
In Vivo Competency of Chemically Derived EndodermFor chemically derived endoderm to be useful, it is important todetermine its developmental potential beyond the expression ofendodermal markers. The ultimate goal is to direct the cells toform functional b cells. To begin to address this possibility andthe functional potential of derived populations, we inducedmouse ESCs ubiquitously expressing enhanced yellow fluores-cent protein (EYFP) (Hadjantonakis et al., 2002) to form endo-derm with IDE1, IDE2, or DMSO (control). At day 6 of treatment,we dissociated and injected the cells into the gut tube (a deriva-tive of endoderm) of live E8.75 embryos. The lumen of the guttube can be accessed prior to the completion of embryonic
turning and gut tube closure and, therefore, provides a develop-mental window for functional assessment of ES-derived endo-derm cells. Chemically derived endoderm was injected into theprimitive gut tube at the anterior and posterior intestinal portals.At this time, the gut tube is still open, and anterior intestinal portalis accessible (A.E.C. and D.A.M., unpublished data, andFigure 6A). We culture the embryos ex vivo for 24–30 hr, during
Figure 5. Small Molecule Inducers of Endoderm Activate TGF-b
Signaling(A) Phosphorylation of Smad2 was analyzed in lysates of ESCs treated with
IDE1, IDE2, DMSO, Activin A, or Nodal or in the presence of the ALK4/5/7
inhibitor SB431542. Treatment with IDE1 or IDE2 leads to activation of the
TGF-b pathway after 24 hr, similar to either Nodal or Activin A treatment. Phos-
phorylation of Smad2 by either of the two compounds is significantly attenu-
ated in the presence of SB431542.
(B) Increase in Nodal expression after treatment with small molecules IDE2,
IDE1, and Nodal. Relative expression over DMSO treatment is shown as
a mean of triplicate experiments ± SD.
Figure 6. Functional Evaluation of Chemically Derived Endoderm(A) Scheme of in vivo assay to assess the functional potential of compound-
induced endoderm. Mouse ESCs treated with chemical inducers incorporate
into the developing host gut tube. Cultures of mouse ESC reporter lines ex-
pressing constitutive YFP were differentiated into endoderm with IDE1 or
IDE2, producing 60%–70% Sox17+ cells, and then trypsinized and injected
into the nascent gut lumen of E8.75 mouse embryos. Dashed line shows an
approximate plane of section.
(B) After 24–30 hr ex vivo culture, mouse embryos were fixed, transversally
sectioned, and stained with antibodies against FoxA2 and Cldn6 to detect
gut epithelial cells and anti-YFP antibodies to visualize injected cells. IDE1
and IDE2 induced endodermal cells incorporate into gut tube and show
expression of gut tube markers. In contrast, DMSO-treated cells remain clus-
tered in the gut tube lumen 30 hr after injection and do not incorporate into the
gut epithelia or express gut tube markers.
Cell Stem Cell
Small Molecules Induced Definitive Endoderm
354 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Invito assay Invivo assay
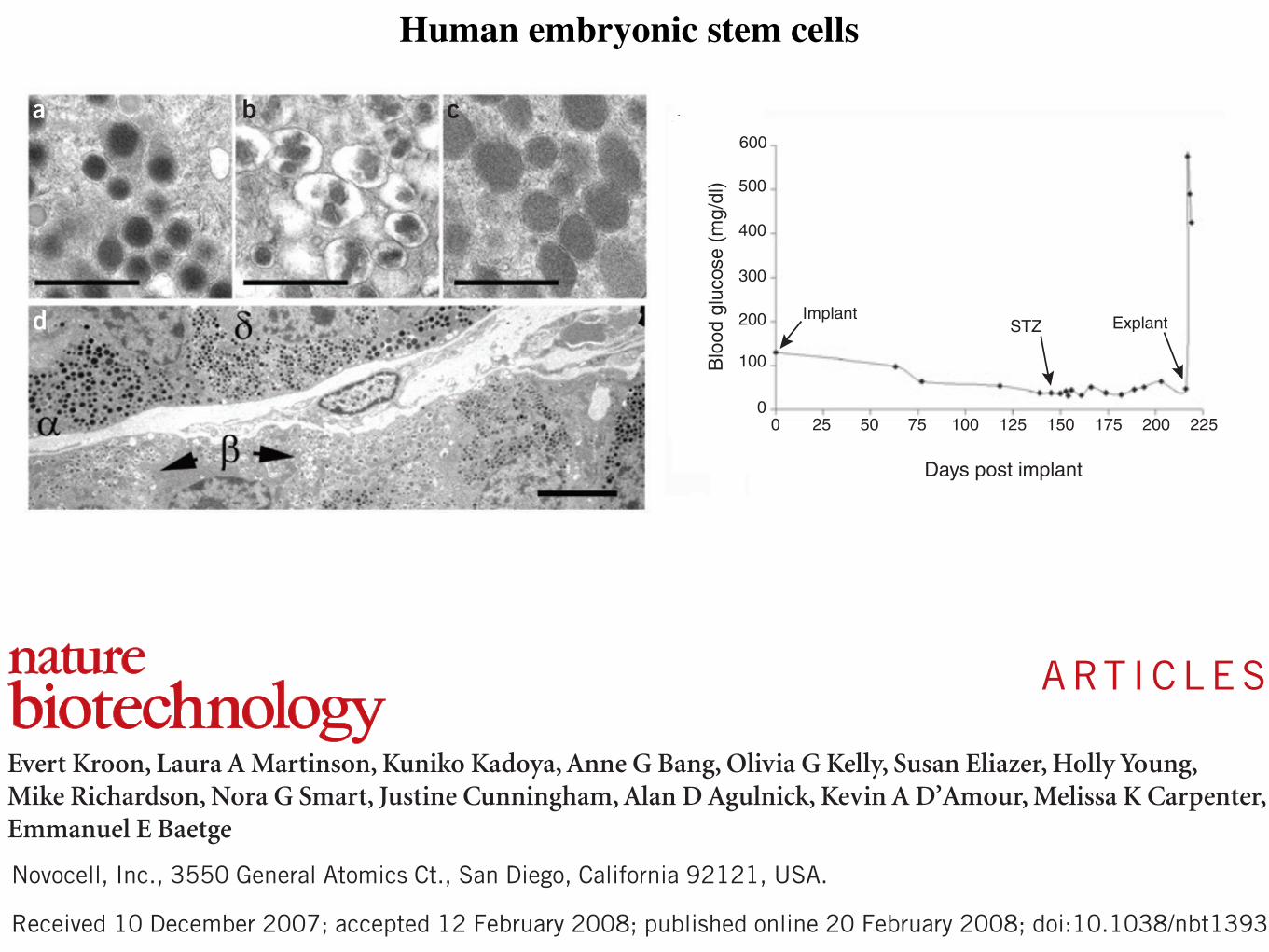
Human embryonic stem cells
©20
08 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
©20
08 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
©20
08 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
©20
08 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
©20
08 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy©
2008
Nat
ure
Publ
ishi
ng G
roup
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy
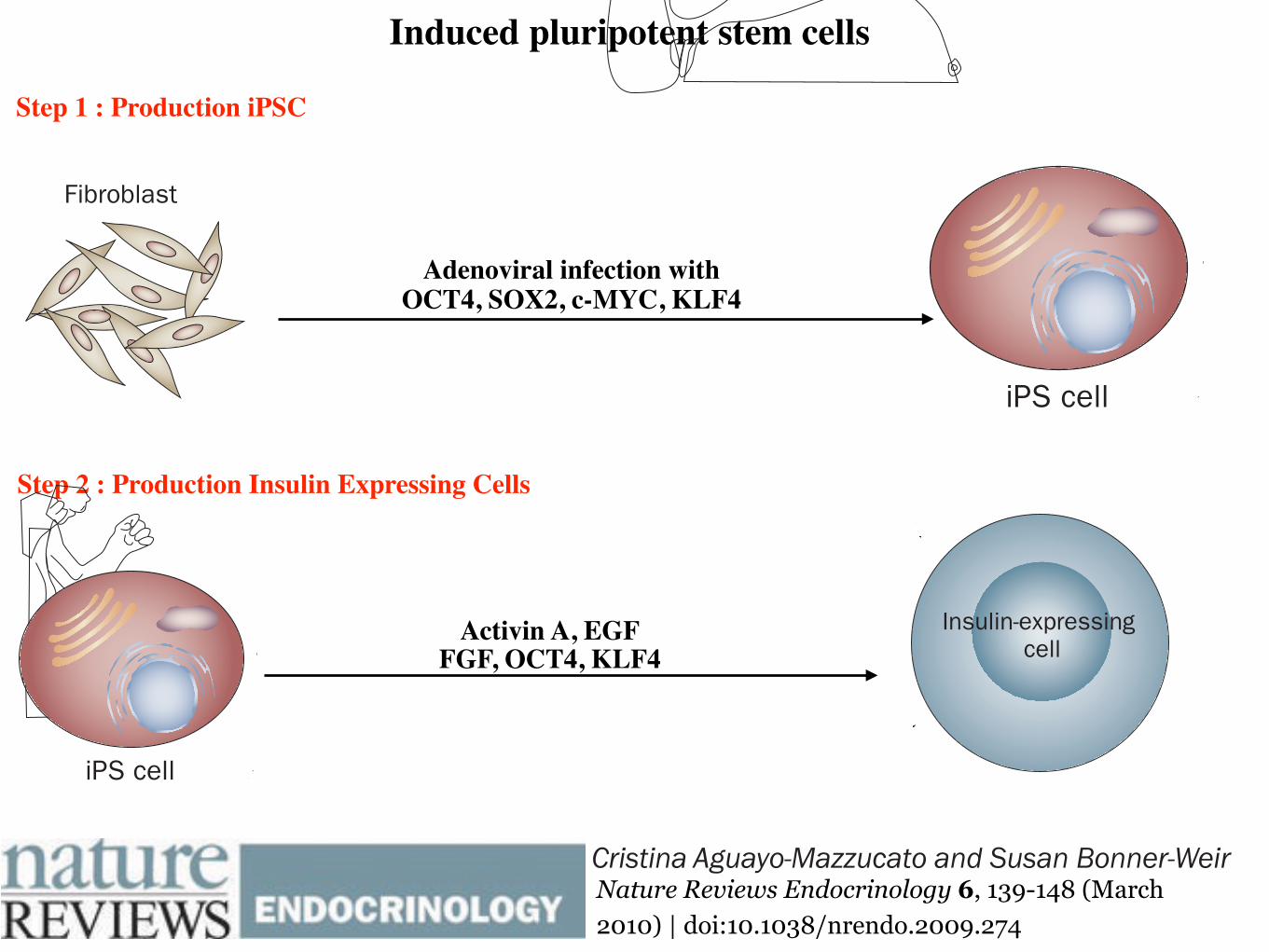
Induced pluripotent stem cells
Step 1 : Production iPSC
Step 2 : Production Insulin Expressing Cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
Adenoviral infection with OCT4, SOX2, c-MYC, KLF4
Activin A, EGF FGF, OCT4, KLF4
3/25/16, 8:19 AMAccess : Stem cell therapy for type 1 diabetes mellitus : Nature Reviews Endocrinology
Page 1 of 3http://www.nature.com/nrendo/journal/v6/n3/full/nrendo.2009.274.html
ARTICLETOOLS
Send to afriend
Exportcitation
Exportreferences
Rights andpermissions
Ordercommercialreprints
SEARCHPUBMEDFOR
CristinaAguayo-Mazzucato
Susan
AccessTo read this article in full you may need to login, make a payment or gain access through asite license (see right).
ReviewNature Reviews Endocrinology 6, 139-148 (March2010) | doi:10.1038/nrendo.2009.274
Stem cell therapy fortype 1 diabetesmellitusCristina Aguayo-Mazzucato &Susan Bonner-Weir
The use of stem cells inregenerative medicine holdsgreat promise for the cure ofmany diseases, including type 1diabetes mellitus (T1DM). Anypotential stem-cell-based curefor T1DM should address theneed for β-cell replacement, aswell as control of theautoimmune response to cellswhich express insulin. The exvivo generation of β cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 139
Section of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, 1 Joslin Place, Boston, MA 02215, USA. (C. Aguayo-Mazzucato, S. Bonner-Weir).
Correspondence to: S. Bonner-Weir [email protected]
Stem cell therapy for type 1 diabetes mellitusCristina Aguayo-Mazzucato and Susan Bonner-Weir
Abstract | The use of stem cells in regenerative medicine holds great promise for the cure of many diseases, including type 1 diabetes mellitus (T1DM). Any potential stem-cell-based cure for T1DM should address the need for β-cell replacement, as well as control of the autoimmune response to cells which express insulin. The ex vivo generation of β cells suitable for transplantation to reconstitute a functional β-cell mass has used pluripotent cells from diverse sources, as well as organ-specific facultative progenitor cells from the liver and the pancreas. The most effective protocols to date have produced cells that express insulin and have molecular characteristics that closely resemble bona fide insulin-secreting cells; however, these cells are often unresponsive to glucose, a characteristic that should be addressed in future protocols. The use of mesenchymal stromal cells or umbilical cord blood to modulate the immune response is already in clinical trials; however, definitive results are still pending. This Review focuses on current strategies to obtain cells which express insulin from different progenitor sources and highlights the main pathways and genes involved, as well as the different approaches for the modulation of the immune response in patients with T1DM.
Aguayo-Mazzucato, C. & Bonner-Weir, S. Nat. Rev. Endocrinol. 6, 139–148 (2010); doi:10.1038/nrendo.2009.274
IntroductionType 1 diabetes mellitus (T1DM) is characterized by the autoimmune destruction of the insulin- expressing, pan-creatic β cells; as a result, patients with T1DM are depen-dent on exogenous insulin for their blood glucose control. Although the discovery of insulin has changed the outlook and survival of this group of individuals for almost a century,1 patients with T1DM are still not exempt from the development of diabetic complications.2,3 These com-plications are thought to result, in part, from the lack of physiological oscillations in insulin secretion and higher than normal glucose levels; thus, the search for effective ways to re establish a functional β-cell mass in patients with T1DM is important. Transplantation of whole human pancreas or isolated, cadaveric human islets4,5 has allowed patients with T1DM to become insulin-independent and has thereby shown proof of concept for β-cell replacement therapies. The shortage of donor pancreata and islets,6 however, results in the search for alternative cell sources, and differen tiation of stem cells (Box 1) into functional β cells is an attractive alternative.
A definitive cure for T1DM needs to address both the β-cell deficit and the autoimmune response to cells that express insulin. As clearly shown by the transplantation of segmental pancreatic grafts from identical twins, the autoimmune response remains many years after disease onset.7 Thus, any β-cell replacement therapy will need some modulation of the immune system, whether by drugs or cell-based approaches.
This Review focuses mainly on the different approaches to obtain functional, insulin-producing cells from various types of progenitor cells. Although the goal of the field is to
direct differentiation of human stem cells into functional β cells, some animal studies are included here, as they could lay the framework for success ful future interventions in humans. Although stem cell- derived, insulin-positive cells have not yet reached clinical trial, clinical data from stem cell-derived therapies that target the autoimmune response have been reported and are discussed.
Many roads lead from stem cells to β cellsEfforts have been directed at the development of efficient protocols for the differentiation of stem cells of either embryonic (Figure 1) or adult origin (Figure 2) into func-tional cells that secrete insulin. A caveat to many of these studies is that some developmental pathways generate insulin-expressing cells that will never become true β cells, for example, cells of the fetal liver, yolk sac and brain.8–10 A β cell should be defined by its ability to store large amounts of insulin and to secrete it in a regulated manner in response to a demand, such as glucose stimulation.11
Human embryonic stem cellsEfforts to generate β cells from human embryonic stem cells (hESCs) have focused on the fundamentals of normal embryonic development as a basis to understand the early stages of endoderm formation. The first step in the differen tiation of hESCs into β cells is the production of definitive endoderm12–17 rather than visceral endoderm, which expresses the same markers (for example, sex determining region Y box [SOX] 17 and forkhead box protein A2 [FOXA2]) but forms different tissues.18
Current protocols for hESC differentiation into β cells aim to reproduce the developmentally active signaling pathways Wnt and transforming growth factor β (TGFβ) (Box 2).17,19–22 Growth factors, such as Activin A, fibroblast
Competing interestsThe authors declare no competing interests.
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 139 4/2/10 17:00:58
© 20 Macmillan Publishers Limited. All rights reserved10
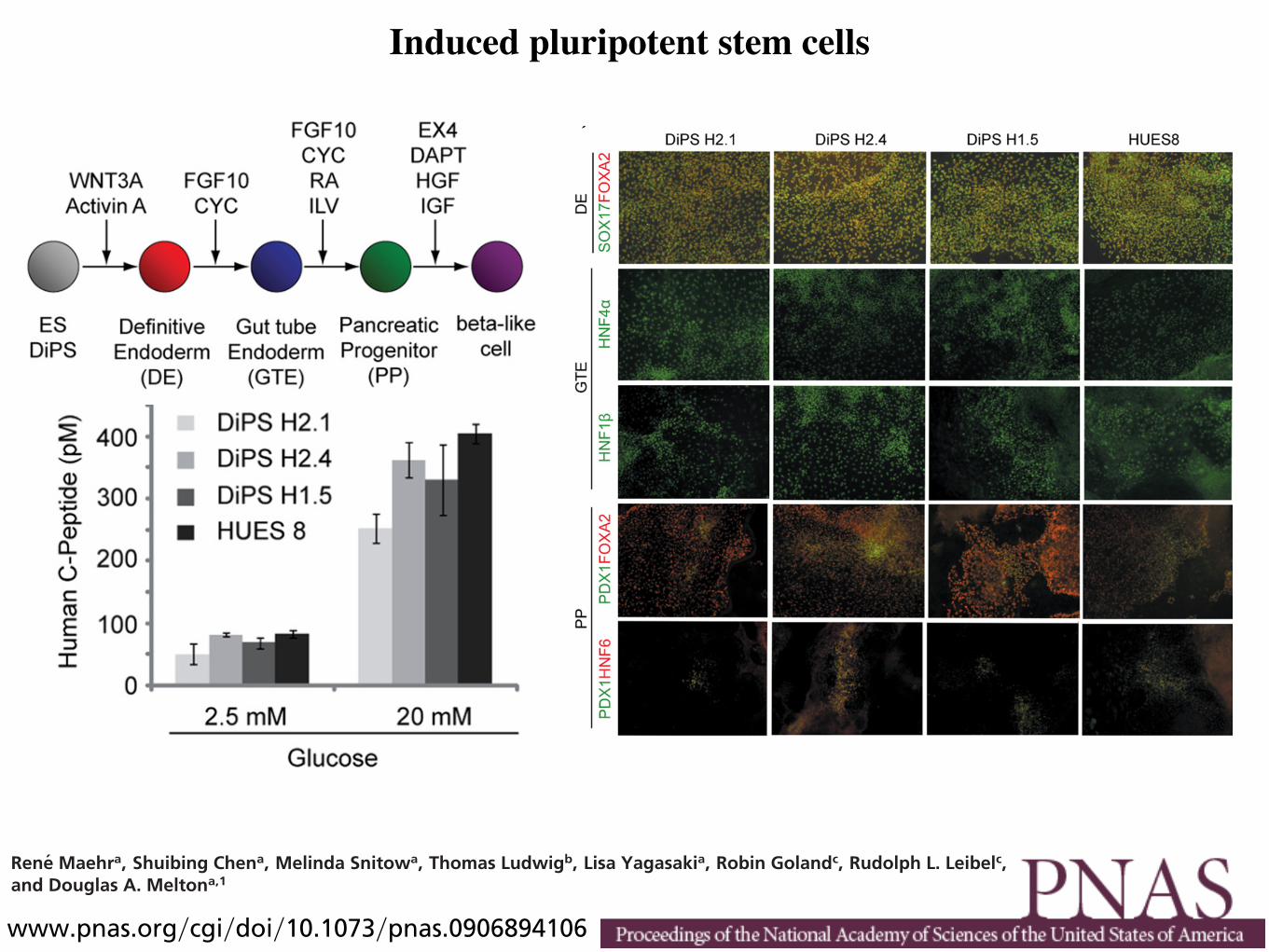
Induced pluripotent stem cells
variation in differentiation propensities, potential deviations inT1D disease onset and progression will require the generation ofmultiple DiPS lines to reflect the human population afflictedwith T1D. The two cell lines from different patients describedhere represent a starting point for this larger task.
Differentiation of DiPS cells to !-like cells is relevant not onlyfor the long term possibility of autologous cell replacementtherapy, but also for disease modeling. We propose to extend thisinvestigation to generate cell types of the immune system thatmay allow the generation of a cellular interaction model of T1D.This approach should provide a way to investigate T1D diseaseonset and progression in vitro and/or in reconstituted animalmodels. These in vitro and in vivo systems will also be useful fortesting of preventative and therapeutic strategies.
Materials and MethodsCell Culture. Skin fibroblasts from T1D patients and controls were derived fromexplants of 3-mm dermal biopsies after informed consent under protocols
approved both by Harvard University and Columbia University College ofPhysicians and Surgeons. Briefly, 3-mm skin biopsies were minced with scalpelsinto smaller pieces, and tissue fragments were placed into a 60-mm tissueculture dish under a sterile coverslip held down by sterilized silicon greaseunder one corner. Media was added to completely immerse the coverslip, anddishes were incubated at 37 °C in a humidified incubator (5% CO2). Media waschanged every 5 days without disturbing the coverslip. Fibroblasts grew out ofthe tissue fragments, and when sufficiently numerous, cells were trypsinizedand expanded. Subsequently, fibroblasts were maintained in fibroblast me-dium (DMEM supplemented with 10% FBS, glutamine, sodium pyruvate,nonessential amino acids, and penicillin/streptomycin). The resulting fibro-blasts lines are referred to as fibroblast Harvard (H) lines 1 and 2.
Human ES cell and DiPS lines were cultured in human ES media (knockoutDMEM supplemented with 10% knockout serum replacement, 10% humanplasma fraction, 10 ng/mL bFGF, nonessential amino acids, !-mercaptoetha-nol, L-glutamine, and penicillin/streptomycin). Cultures were maintained onmouse embryonic fibroblast feeders and passaged enzymatically using either0.05% Trypsin (GIBCO) or Collagenase type IV.
Fig. 3. Spontaneous differentiation of DiPS cells into cells of different germ layer origin. (A) In vitro differentiation of DiPS lines H1.5, H2.1, and H2.4 in EB assayswas followed by monolayer culture and immunostaining for markers of ectoderm (TUJ1), mesoderm (SMA), and endoderm (FOXA2 and SOX17). An overlay witha nuclear stain (DAPI) is displayed. (B) Teratoma formation occurred after injection of DiPS into immunocompromised mice. Hematoxylin and Eosin staining ofteratoma sections shows nerve fibers (N), melanocytes (M), pigmented epithelium (P), cartilage (C), and glandular structures (G).
Fig. 4. Stepwise differentiation of ES/DiPS cells toward !-like cells. (A) Schematic representation of stepwise differentiation of human PS cells to !-like cells.DiPS cell lines H1.5, H2.1, and H2.4 differentiation to definitive endoderm (DE), gut tube endoderm (GTE) and pancreatic progenitors (PPs) indicated by (B)immunostaining and (C) RT-PCR. SOX, SRY (sex determining region Y)-box; FOXA2, forkhead box protein A2; HNF, hepatocyte nuclear factor; PDX1, pancreaticand duodenal homeobox 1; HB9, homeobox gene HLXB9; NKX6.1, NK6 transcription factor related, locus 1.
Maehr et al. PNAS ! September 15, 2009 ! vol. 106 ! no. 37 ! 15771
DEV
ELO
PMEN
TAL
BIO
LOG
YSE
ECO
MM
ENTA
RY
variation in differentiation propensities, potential deviations inT1D disease onset and progression will require the generation ofmultiple DiPS lines to reflect the human population afflictedwith T1D. The two cell lines from different patients describedhere represent a starting point for this larger task.
Differentiation of DiPS cells to !-like cells is relevant not onlyfor the long term possibility of autologous cell replacementtherapy, but also for disease modeling. We propose to extend thisinvestigation to generate cell types of the immune system thatmay allow the generation of a cellular interaction model of T1D.This approach should provide a way to investigate T1D diseaseonset and progression in vitro and/or in reconstituted animalmodels. These in vitro and in vivo systems will also be useful fortesting of preventative and therapeutic strategies.
Materials and MethodsCell Culture. Skin fibroblasts from T1D patients and controls were derived fromexplants of 3-mm dermal biopsies after informed consent under protocols
approved both by Harvard University and Columbia University College ofPhysicians and Surgeons. Briefly, 3-mm skin biopsies were minced with scalpelsinto smaller pieces, and tissue fragments were placed into a 60-mm tissueculture dish under a sterile coverslip held down by sterilized silicon greaseunder one corner. Media was added to completely immerse the coverslip, anddishes were incubated at 37 °C in a humidified incubator (5% CO2). Media waschanged every 5 days without disturbing the coverslip. Fibroblasts grew out ofthe tissue fragments, and when sufficiently numerous, cells were trypsinizedand expanded. Subsequently, fibroblasts were maintained in fibroblast me-dium (DMEM supplemented with 10% FBS, glutamine, sodium pyruvate,nonessential amino acids, and penicillin/streptomycin). The resulting fibro-blasts lines are referred to as fibroblast Harvard (H) lines 1 and 2.
Human ES cell and DiPS lines were cultured in human ES media (knockoutDMEM supplemented with 10% knockout serum replacement, 10% humanplasma fraction, 10 ng/mL bFGF, nonessential amino acids, !-mercaptoetha-nol, L-glutamine, and penicillin/streptomycin). Cultures were maintained onmouse embryonic fibroblast feeders and passaged enzymatically using either0.05% Trypsin (GIBCO) or Collagenase type IV.
Fig. 3. Spontaneous differentiation of DiPS cells into cells of different germ layer origin. (A) In vitro differentiation of DiPS lines H1.5, H2.1, and H2.4 in EB assayswas followed by monolayer culture and immunostaining for markers of ectoderm (TUJ1), mesoderm (SMA), and endoderm (FOXA2 and SOX17). An overlay witha nuclear stain (DAPI) is displayed. (B) Teratoma formation occurred after injection of DiPS into immunocompromised mice. Hematoxylin and Eosin staining ofteratoma sections shows nerve fibers (N), melanocytes (M), pigmented epithelium (P), cartilage (C), and glandular structures (G).
Fig. 4. Stepwise differentiation of ES/DiPS cells toward !-like cells. (A) Schematic representation of stepwise differentiation of human PS cells to !-like cells.DiPS cell lines H1.5, H2.1, and H2.4 differentiation to definitive endoderm (DE), gut tube endoderm (GTE) and pancreatic progenitors (PPs) indicated by (B)immunostaining and (C) RT-PCR. SOX, SRY (sex determining region Y)-box; FOXA2, forkhead box protein A2; HNF, hepatocyte nuclear factor; PDX1, pancreaticand duodenal homeobox 1; HB9, homeobox gene HLXB9; NKX6.1, NK6 transcription factor related, locus 1.
Maehr et al. PNAS ! September 15, 2009 ! vol. 106 ! no. 37 ! 15771
DEV
ELO
PMEN
TAL
BIO
LOG
YSE
ECO
MM
ENTA
RY
Reprogramming. VSVG-coated retroviruses were generated according to stan-dard procedures. One day before infection, 10E5 fibroblasts were seeded perwell of a six well plate. Fibroblasts were infected on days 1 and 2 with acombination of OCT4, SOX2, and KLF4 containing Moloney viruses (constructswere obtained from Addgene) (15). The media was changed on day 3 toDMEM supplemented with 10% FBS, L-glutamine, penicillin/streptomycin,nonessential amino acids, and sodium pyruvate. A day later the cells were splitonto gelatinized 10-cm cell culture dishes. Subsequently, the cells were fedevery other day with human ES cell media. Generation of DiPS lines H2.4, H2.3,and H2.1b occurred with supplementation of the media with 1 mM valproicacid during the reprogramming process as described before (18), whereas DiPSlines H2.1, H1.1, H1.5 were generated without addition of the chemical.Colonies were picked starting !4 weeks after infection.
Spontaneous Differentiation. Spontaneous differentiation through EB forma-tion was initiated by dissociation of human DiPS cells using collagenase IVtreatment, and subsequent transfer to low attachment 6-well plates in knock-out DMEM supplemented with 20% knockout serum replacement, nonessen-tial amino acid, !-mercaptoethanol, L-glutamine, and penicillin/streptomycin.After 8–10 days suspension culture, EBs were transferred to gelatin-coatedplates and cultured for an additional 8–10 days as attachment culture.
For teratoma formation assays DiPS cells were collected by collagenase IVtreatment, and injected s.c. into immunocompromised mice (NOD-SCID orSCID-Beige mice). Teratomas were collected 7–10 weeks after injection, andprocessed according to standard procedures for paraffin embedding andhematoxylin and eosin staining.
Directed Differentiation. Directed differentiation was conducted as described(13, 29) with the following modifications: human DiPS cells were cultured onMEF feeder cells to 70–80% confluency, then treated with 25 ng/mL WNT3A(R&D systems) " 100 ng/mL Activin A (R&D systems) in advanced RPMI (A-RPMI;Invitrogen) supplemented with 1#L-Glu and 1#PS for 1 day, followed bytreatment with 100 ng/mL Activin A in A-RPMI supplemented with 1#L-Glu,1#PS and 0.2% FBS (Invitrogen). Two days later, the media was changed to 50ng/mL FGF10 (R&D systems) " 0.25 "M KAAD-CYC (Calbiochem) in A-RPMI
supplemented with 1#L-Glu, 1#PS and 2% FBS and maintained for additional2 days. Cells were then transferred to 50 ng/mL FGF10 " 0.25 "M KAAD-CYC " 2"M RA (Sigma) in DMEM supplemented with 1#L-Glu, 1#PS, 1# B27 (Invitro-gen) and cultured for an additional 4 days. The media was then changed to 50ng/mL FGF10 " 300 nM ILV (Axxora) in DMEM supplemented with 1#L-Glu,1#PS, 1# B27 and cultured for an additional 4 days. Then, cells were trans-ferred to 50 ng/mL EX-4 (Sigma) " 10 "M DAPT (Sigma) in DMEM supple-mented with 1#L-Glu, 1#PS, 1# B27 and cultured for an additional 6 days.Cells were then cultured in 50 ng/mL HGF (R&D systems) " 50 ng/mL IGF1 (R&Dsystems) in CMRL-1066 (Invitrogen) supplemented with 1#L-Glu, 1#PS, 1#B27 for 6 days.
Immunofluorescence. Immunofluorescence staining was performed usingprimary antibodies against C-peptide (4020-01; Linco), FOXA2 (07-633;Upstate), glucacon (4031; Linco), HNF6 (sc-13050; Santa Cruz Biotechnol-ogy), insulin (A0564; Dako), NANOG (ab21624; Abcam), NKX2.5 (sc-14033;Santa Cruz Biotechnology), OCT4 (sc-5279; Santa Cruz Biotechnology),PDX1 (AF2419; R&D systems), SMA (A5228; Sigma), somatostatin (A0566;Dako), SOX2 (sc-17320; Santa Cruz Biotechnology), SOX17 (AF1924; R&Dsystems), SSEA4 (MAB4304; Chemicon), TRA-1– 60 (MAB4360; Chemicon),TRA-1– 81 (MAB4381; Chemicon), and TUJ-1 (MMS-435P; Covance ResearchProducts). Appropriate secondary antibodies were obtained from Molec-ular Probes.
Gene Expression Analysis. RNA was isolated from cells using RNAeasy kit(Qiagen). For quantitative and semiquantitative PCR analysis, cDNA synthesiswas performed using SuperScript III Reverse Transcriptase and Oligo (dT)primers (Invitrogen). Primers used for amplification are listed in Table S3.
For whole-genome expression analysis, Illumina Total Prep RNA amplificationKit (Ambion) was used according to manufacturer’s guideline. Hybridization toWhole-GenomeExpressionBeadChips (HumanRef-8)was followedbyanalysisonan Illumina Beadstation 500. All samples were prepared in duplicates. Dataanalysis was conducted using manufacturer’s Beadstudio software.
C-Peptide Release Assay. C-peptide release was measured by incubating thecells in Krebs–Ringer solution containing bicarbonate and Hepes (KRBH; 129
Fig. 5. DiPS cell lines H1.5, H2.1, and H2.4 differentiate to hormone-expressing endocrine cells indicated by (A) immunostaining and (B) semiquantitative PCR.(C) The DiPS-derived C-peptide-expressing cells secreted C-peptide on glucose stimulation. The DiPS-derived populations were stimulated with 2.5 and 20 mMD-glucose, and the amount of human C-peptide released to culture supernatant was analyzed by ELISA. C-PEP, C-peptide; INS, insulin; GLU, glucagon; SS,somatostatin.
15772 ! www.pnas.org"cgi"doi"10.1073"pnas.0906894106 Maehr et al.
Generation of pluripotent stem cells from patientswith type 1 diabetesRene Maehra, Shuibing Chena, Melinda Snitowa, Thomas Ludwigb, Lisa Yagasakia, Robin Golandc, Rudolph L. Leibelc,and Douglas A. Meltona,1
aDepartment of Stem Cell and Regenerative Biology, Howard Hughes Medical Institute, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue,Cambridge, MA 02138; and bDepartment of Pathology and Cell Biology, and cDivision of Molecular Genetics and Naomi Barrie Diabetes Center, Collegeof Physicians and Surgeons, Columbia University, New York, NY 10032
Contributed by Douglas A. Melton, July 8, 2009 (sent for review May 18, 2009)
Type 1 diabetes (T1D) is the result of an autoimmune destructionof pancreatic ! cells. The cellular and molecular defects that causethe disease remain unknown. Pluripotent cells generated frompatients with T1D would be useful for disease modeling. We showhere that induced pluripotent stem (iPS) cells can be generatedfrom patients with T1D by reprogramming their adult fibroblastswith three transcription factors (OCT4, SOX2, KLF4). T1D-specificiPS cells, termed DiPS cells, have the hallmarks of pluripotency andcan be differentiated into insulin-producing cells. These results area step toward using DiPS cells in T1D disease modeling, as well asfor cell replacement therapy.
! cell ! disease model ! autoimmune ! directed differentiation ! endoderm
The study of human disease is often hindered by the lack of agood model system. The initiation of the primary disease
process often occurs long before the patient shows any sign ofdisease. Also, relevant patient tissue can be limited and difficultto obtain. Although rodent models can give valuable insights,these rarely fully recapitulate the human disease. Although thenonobese diabetic (NOD) mouse has been enormously useful,there are justifiable concerns regarding its validity as a model forhuman type 1 diabetes (T1D) (1–3). Recently, human inducedpluripotent stem (iPS) cells with disease genotypes have beengenerated as a tool for human disease modeling (4–7). Disease-relevant cell types can be generated via in vitro differentiationprotocols, and in the best cases, these protocols enable an in vitroanalysis of the disease pathology. Although ES cells are the goldstandard for pluripotent stem (PS) cells, ES cells only modeldiseases that can be diagnosed or predicted by simple Mendeliangenetics [e.g., cystic fibrosis (8) and Fanconi Anemia (9)]. Ourfocus lies in understanding T1D, a disease with complex under-lying genetics and unidentified environmental triggers. For T1D,as well as other multigenic diseases, iPS cells are the best startingpoint, because they are derived from patient cells and, thereby,capture the disease genotype in a stem cell. To this date, it is notclear whether different forms of type 1 diabetes exist, and howgenetics and environment factor influence each other in diseaseonset and progression.
T1D results from the destruction of insulin-producing ! cellsby the body’s own immune system. A cure could be achieved bycombining ! cell replacement therapy with induction of toler-ance to such cells. Cell replacement therapy for T1D requires asource of glucose-responsive, insulin-secreting cells. Promisingresults have been obtained by transplantation of pancreatic isletsof Langerhans or pancreatic tissue, but this approach is circum-scribed by the limited and irregular supply of cadaveric donortissue, as well as the risks of treatment with immunosuppressantdrugs (10, 11). An alternative source of insulin-producing cellsare PS cells that can be differentiated into pancreatic ! cells. Forexample, ES cells have been differentiated in monolayer culturealong the endodermal lineage toward insulin-producing cells(12, 13). However, ! cells derived from immunologically un-matched ES cells will likely be the targets of both allograft
reactions and the autoimmune response that caused the initial !cell destruction.
Mouse and human fibroblasts can be used to generate iPS cells(14–16). Recently, iPS cells have been generated from fibro-blasts obtained from patients with various diseases (4–7), but notfor T1D. T1D-specific iPS (DiPS) cells derived from patientsoffer several significant advantages. First, DiPS cells wouldunquestionably contain the genotype responsible for the humandisease. Second, DiPS cells would provide an immunologicallymatched autologous cell population, although dependent onimprovements in differentiation protocols. Third, and thepresent focus of our work, patient-specific cells make possiblepatient-specific disease modeling wherein the initiation andprogression of this poorly understood disease can be studied.Because DiPS cells can be manipulated and studied in vitro, oneshould be able to assess how the different cell types, includingdifferentiated ! cells, and immunocytes interact to produce apathological phenotype. The purpose of the present study was toderive DiPS cells from patients with T1D, and to test whetherthese cells can be differentiated into the major target cell type,the pancreatic ! cell. Extending this approach to all cell typesinvolved in T1D could lead to an understanding of the rootcauses of the disease and to the development of effectiveprophylactic and therapeutic strategies.
ResultsGeneration of iPS Cells from Patients with T1D. Skin biopsies wereobtained from two Caucasian males with T1D of 11- and27-years duration, respectively. Patient 1 was presented at age 21with polyuria, polydypsia, and a blood glucose concentration of680 mg/dL. Patient 2 was presented at age 3 in diabetic ketoac-idosis requiring hospitalization. For both individuals, the bodymass index did not exceed 22 or 23, respectively. HLA haplo-types, and other clinical data, are given in Table 1. Fibroblastsobtained from skin biopsies were cultured and infected with acombination of retroviruses encoding the transcription factorsOCT4, SOX2, and KLF4 (15). Starting 4 weeks after infections,colonies were picked based on their morphological resemblanceto human ES cell colonies and expanded. To test whether the celllines expressed pluripotency markers, we verified the presence ofalkaline phosphatase (AP) activity, as well as staining forantibodies to OCT4, NANOG, SOX2, TRA1-60, TRA1-81, andSSEA4. The reprogrammed cells were positive for AP activity,and were reactive to antibodies against all pluripotency markers
Author contributions: R.M., R.G., R.L.L., and D.A.M. designed research; R.M., S.C., M.S., andL.Y. performed research; T.L., R.G., and R.L.L. contributed new reagents/analytic tools; R.M.,S.C., L.Y., R.L.L., and D.A.M. analyzed data; and R.M. and D.A.M. wrote the paper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.
See Commentary on page 15523.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0906894106/DCSupplemental.
15768–15773 ! PNAS ! September 15, 2009 ! vol. 106 ! no. 37 www.pnas.org"cgi"doi"10.1073"pnas.0906894106
Generation of pluripotent stem cells from patientswith type 1 diabetesRene Maehra, Shuibing Chena, Melinda Snitowa, Thomas Ludwigb, Lisa Yagasakia, Robin Golandc, Rudolph L. Leibelc,and Douglas A. Meltona,1
aDepartment of Stem Cell and Regenerative Biology, Howard Hughes Medical Institute, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue,Cambridge, MA 02138; and bDepartment of Pathology and Cell Biology, and cDivision of Molecular Genetics and Naomi Barrie Diabetes Center, Collegeof Physicians and Surgeons, Columbia University, New York, NY 10032
Contributed by Douglas A. Melton, July 8, 2009 (sent for review May 18, 2009)
Type 1 diabetes (T1D) is the result of an autoimmune destructionof pancreatic ! cells. The cellular and molecular defects that causethe disease remain unknown. Pluripotent cells generated frompatients with T1D would be useful for disease modeling. We showhere that induced pluripotent stem (iPS) cells can be generatedfrom patients with T1D by reprogramming their adult fibroblastswith three transcription factors (OCT4, SOX2, KLF4). T1D-specificiPS cells, termed DiPS cells, have the hallmarks of pluripotency andcan be differentiated into insulin-producing cells. These results area step toward using DiPS cells in T1D disease modeling, as well asfor cell replacement therapy.
! cell ! disease model ! autoimmune ! directed differentiation ! endoderm
The study of human disease is often hindered by the lack of agood model system. The initiation of the primary disease
process often occurs long before the patient shows any sign ofdisease. Also, relevant patient tissue can be limited and difficultto obtain. Although rodent models can give valuable insights,these rarely fully recapitulate the human disease. Although thenonobese diabetic (NOD) mouse has been enormously useful,there are justifiable concerns regarding its validity as a model forhuman type 1 diabetes (T1D) (1–3). Recently, human inducedpluripotent stem (iPS) cells with disease genotypes have beengenerated as a tool for human disease modeling (4–7). Disease-relevant cell types can be generated via in vitro differentiationprotocols, and in the best cases, these protocols enable an in vitroanalysis of the disease pathology. Although ES cells are the goldstandard for pluripotent stem (PS) cells, ES cells only modeldiseases that can be diagnosed or predicted by simple Mendeliangenetics [e.g., cystic fibrosis (8) and Fanconi Anemia (9)]. Ourfocus lies in understanding T1D, a disease with complex under-lying genetics and unidentified environmental triggers. For T1D,as well as other multigenic diseases, iPS cells are the best startingpoint, because they are derived from patient cells and, thereby,capture the disease genotype in a stem cell. To this date, it is notclear whether different forms of type 1 diabetes exist, and howgenetics and environment factor influence each other in diseaseonset and progression.
T1D results from the destruction of insulin-producing ! cellsby the body’s own immune system. A cure could be achieved bycombining ! cell replacement therapy with induction of toler-ance to such cells. Cell replacement therapy for T1D requires asource of glucose-responsive, insulin-secreting cells. Promisingresults have been obtained by transplantation of pancreatic isletsof Langerhans or pancreatic tissue, but this approach is circum-scribed by the limited and irregular supply of cadaveric donortissue, as well as the risks of treatment with immunosuppressantdrugs (10, 11). An alternative source of insulin-producing cellsare PS cells that can be differentiated into pancreatic ! cells. Forexample, ES cells have been differentiated in monolayer culturealong the endodermal lineage toward insulin-producing cells(12, 13). However, ! cells derived from immunologically un-matched ES cells will likely be the targets of both allograft
reactions and the autoimmune response that caused the initial !cell destruction.
Mouse and human fibroblasts can be used to generate iPS cells(14–16). Recently, iPS cells have been generated from fibro-blasts obtained from patients with various diseases (4–7), but notfor T1D. T1D-specific iPS (DiPS) cells derived from patientsoffer several significant advantages. First, DiPS cells wouldunquestionably contain the genotype responsible for the humandisease. Second, DiPS cells would provide an immunologicallymatched autologous cell population, although dependent onimprovements in differentiation protocols. Third, and thepresent focus of our work, patient-specific cells make possiblepatient-specific disease modeling wherein the initiation andprogression of this poorly understood disease can be studied.Because DiPS cells can be manipulated and studied in vitro, oneshould be able to assess how the different cell types, includingdifferentiated ! cells, and immunocytes interact to produce apathological phenotype. The purpose of the present study was toderive DiPS cells from patients with T1D, and to test whetherthese cells can be differentiated into the major target cell type,the pancreatic ! cell. Extending this approach to all cell typesinvolved in T1D could lead to an understanding of the rootcauses of the disease and to the development of effectiveprophylactic and therapeutic strategies.
ResultsGeneration of iPS Cells from Patients with T1D. Skin biopsies wereobtained from two Caucasian males with T1D of 11- and27-years duration, respectively. Patient 1 was presented at age 21with polyuria, polydypsia, and a blood glucose concentration of680 mg/dL. Patient 2 was presented at age 3 in diabetic ketoac-idosis requiring hospitalization. For both individuals, the bodymass index did not exceed 22 or 23, respectively. HLA haplo-types, and other clinical data, are given in Table 1. Fibroblastsobtained from skin biopsies were cultured and infected with acombination of retroviruses encoding the transcription factorsOCT4, SOX2, and KLF4 (15). Starting 4 weeks after infections,colonies were picked based on their morphological resemblanceto human ES cell colonies and expanded. To test whether the celllines expressed pluripotency markers, we verified the presence ofalkaline phosphatase (AP) activity, as well as staining forantibodies to OCT4, NANOG, SOX2, TRA1-60, TRA1-81, andSSEA4. The reprogrammed cells were positive for AP activity,and were reactive to antibodies against all pluripotency markers
Author contributions: R.M., R.G., R.L.L., and D.A.M. designed research; R.M., S.C., M.S., andL.Y. performed research; T.L., R.G., and R.L.L. contributed new reagents/analytic tools; R.M.,S.C., L.Y., R.L.L., and D.A.M. analyzed data; and R.M. and D.A.M. wrote the paper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.
See Commentary on page 15523.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0906894106/DCSupplemental.
15768–15773 ! PNAS ! September 15, 2009 ! vol. 106 ! no. 37 www.pnas.org"cgi"doi"10.1073"pnas.0906894106

Umbilical cord blood
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
Lentiviral infection with OCT4, SOX2, NANOG, LIN28
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
Insulin, High GlucoseActivin A, Retinoic Acid, FGF
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
Activin A, EGF FGF, OCT4, KLF4
3/25/16, 8:19 AMAccess : Stem cell therapy for type 1 diabetes mellitus : Nature Reviews Endocrinology
Page 1 of 3http://www.nature.com/nrendo/journal/v6/n3/full/nrendo.2009.274.html
ARTICLETOOLS
Send to afriend
Exportcitation
Exportreferences
Rights andpermissions
Ordercommercialreprints
SEARCHPUBMEDFOR
CristinaAguayo-Mazzucato
Susan
AccessTo read this article in full you may need to login, make a payment or gain access through asite license (see right).
ReviewNature Reviews Endocrinology 6, 139-148 (March2010) | doi:10.1038/nrendo.2009.274
Stem cell therapy fortype 1 diabetesmellitusCristina Aguayo-Mazzucato &Susan Bonner-Weir
The use of stem cells inregenerative medicine holdsgreat promise for the cure ofmany diseases, including type 1diabetes mellitus (T1DM). Anypotential stem-cell-based curefor T1DM should address theneed for β-cell replacement, aswell as control of theautoimmune response to cellswhich express insulin. The exvivo generation of β cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 139
Section of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, 1 Joslin Place, Boston, MA 02215, USA. (C. Aguayo-Mazzucato, S. Bonner-Weir).
Correspondence to: S. Bonner-Weir [email protected]
Stem cell therapy for type 1 diabetes mellitusCristina Aguayo-Mazzucato and Susan Bonner-Weir
Abstract | The use of stem cells in regenerative medicine holds great promise for the cure of many diseases, including type 1 diabetes mellitus (T1DM). Any potential stem-cell-based cure for T1DM should address the need for β-cell replacement, as well as control of the autoimmune response to cells which express insulin. The ex vivo generation of β cells suitable for transplantation to reconstitute a functional β-cell mass has used pluripotent cells from diverse sources, as well as organ-specific facultative progenitor cells from the liver and the pancreas. The most effective protocols to date have produced cells that express insulin and have molecular characteristics that closely resemble bona fide insulin-secreting cells; however, these cells are often unresponsive to glucose, a characteristic that should be addressed in future protocols. The use of mesenchymal stromal cells or umbilical cord blood to modulate the immune response is already in clinical trials; however, definitive results are still pending. This Review focuses on current strategies to obtain cells which express insulin from different progenitor sources and highlights the main pathways and genes involved, as well as the different approaches for the modulation of the immune response in patients with T1DM.
Aguayo-Mazzucato, C. & Bonner-Weir, S. Nat. Rev. Endocrinol. 6, 139–148 (2010); doi:10.1038/nrendo.2009.274
IntroductionType 1 diabetes mellitus (T1DM) is characterized by the autoimmune destruction of the insulin- expressing, pan-creatic β cells; as a result, patients with T1DM are depen-dent on exogenous insulin for their blood glucose control. Although the discovery of insulin has changed the outlook and survival of this group of individuals for almost a century,1 patients with T1DM are still not exempt from the development of diabetic complications.2,3 These com-plications are thought to result, in part, from the lack of physiological oscillations in insulin secretion and higher than normal glucose levels; thus, the search for effective ways to re establish a functional β-cell mass in patients with T1DM is important. Transplantation of whole human pancreas or isolated, cadaveric human islets4,5 has allowed patients with T1DM to become insulin-independent and has thereby shown proof of concept for β-cell replacement therapies. The shortage of donor pancreata and islets,6 however, results in the search for alternative cell sources, and differen tiation of stem cells (Box 1) into functional β cells is an attractive alternative.
A definitive cure for T1DM needs to address both the β-cell deficit and the autoimmune response to cells that express insulin. As clearly shown by the transplantation of segmental pancreatic grafts from identical twins, the autoimmune response remains many years after disease onset.7 Thus, any β-cell replacement therapy will need some modulation of the immune system, whether by drugs or cell-based approaches.
This Review focuses mainly on the different approaches to obtain functional, insulin-producing cells from various types of progenitor cells. Although the goal of the field is to
direct differentiation of human stem cells into functional β cells, some animal studies are included here, as they could lay the framework for success ful future interventions in humans. Although stem cell- derived, insulin-positive cells have not yet reached clinical trial, clinical data from stem cell-derived therapies that target the autoimmune response have been reported and are discussed.
Many roads lead from stem cells to β cellsEfforts have been directed at the development of efficient protocols for the differentiation of stem cells of either embryonic (Figure 1) or adult origin (Figure 2) into func-tional cells that secrete insulin. A caveat to many of these studies is that some developmental pathways generate insulin-expressing cells that will never become true β cells, for example, cells of the fetal liver, yolk sac and brain.8–10 A β cell should be defined by its ability to store large amounts of insulin and to secrete it in a regulated manner in response to a demand, such as glucose stimulation.11
Human embryonic stem cellsEfforts to generate β cells from human embryonic stem cells (hESCs) have focused on the fundamentals of normal embryonic development as a basis to understand the early stages of endoderm formation. The first step in the differen tiation of hESCs into β cells is the production of definitive endoderm12–17 rather than visceral endoderm, which expresses the same markers (for example, sex determining region Y box [SOX] 17 and forkhead box protein A2 [FOXA2]) but forms different tissues.18
Current protocols for hESC differentiation into β cells aim to reproduce the developmentally active signaling pathways Wnt and transforming growth factor β (TGFβ) (Box 2).17,19–22 Growth factors, such as Activin A, fibroblast
Competing interestsThe authors declare no competing interests.
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 139 4/2/10 17:00:58
© 20 Macmillan Publishers Limited. All rights reserved10
Flow Cytometry
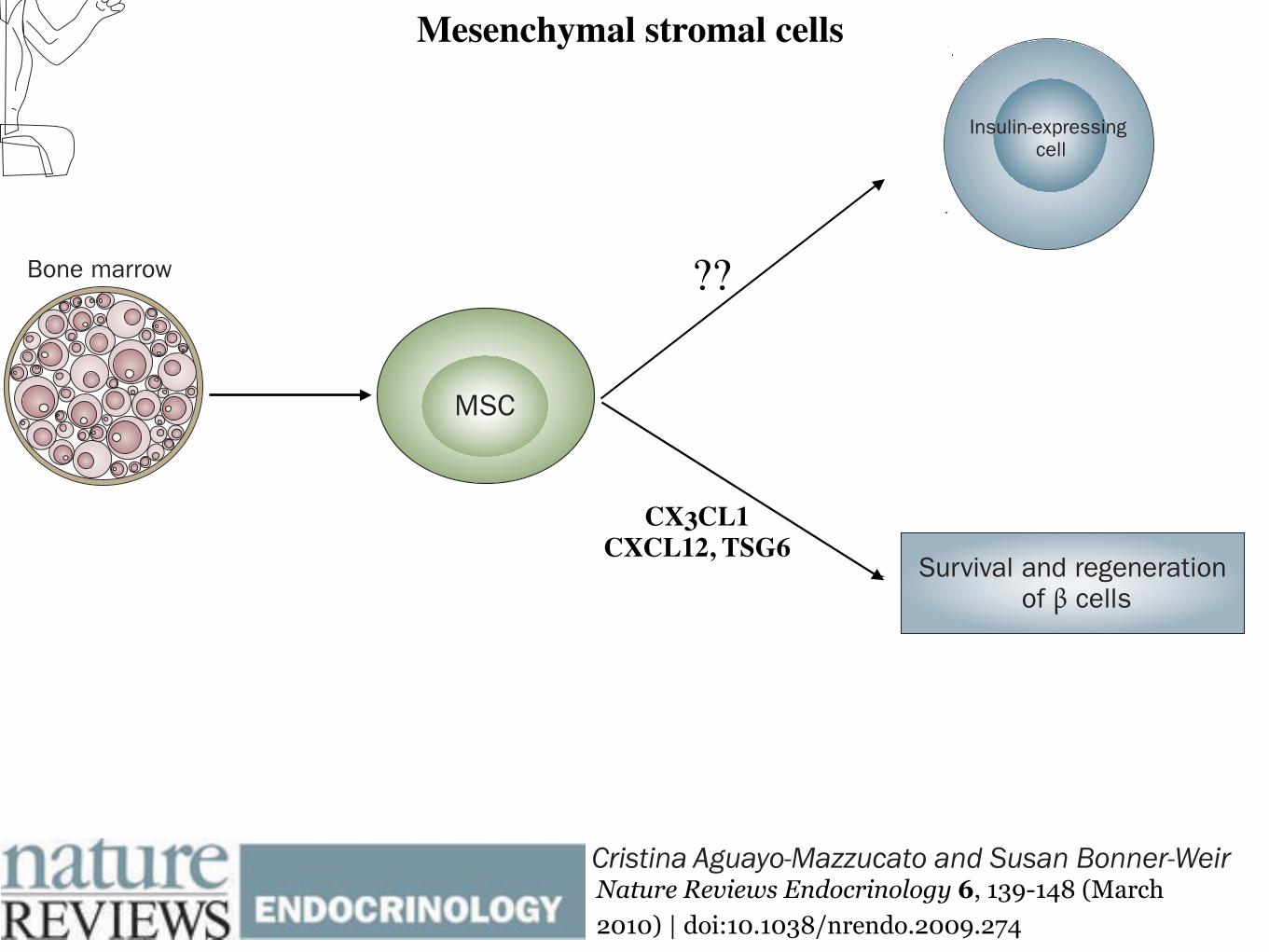
Mesenchymal stromal cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 141
two factors, OCT4 and SOX2, thus making therapeutic use of reprogrammed cells potentially safer and more practi-cal.35 The initial use of retroviruses or lenti viruses to deliver the transcription factor genes raised concerns about viral integration into the host genome increasing tumorigenicity risk. Novel protocols use repeated transfection of expres-sion plasmids that resulted in iPS cells without evidence of plasmid integration.36 Nevertheless, although the protocols for this reprogramming are evolving rapidly and no longer require the use of oncogenes and viral vectors, whether iPS cells are truly equivalent to hESCs with respect to pluripoten cy is not yet fully elucidated.
iPS cells have also been generated from umbilical cord blood by lentiviral overexpression of the reprogramming factors OCT4, SOX2, NANOG and LIN28.37,38 The effi-ciency of reprogramming was similar to that of keratino-cytes and fibroblasts. Since umbilical cord blood is a juvenescent cell source, its use addresses the concerns that arise with the use of adult somatic cells, which can accumulate mutations over the lifetime of an organism.
The first studies to use iPS cells for the generation of insulin-positive cells are only just emerging. In one study hESC-like iPS cells, derived from skin cells by retroviral expression of OCT4, SOX2, c-MYC and KLF4, under-went a serum-free in vitro culture protocol that resulted in islet-like cell clusters that expressed insulin.39 Two of the four cell lines derived in this study could differentiate, one of which released low amounts of C-peptide (0.15 ng/μg DNA) in response to 40 mM glucose. Although the researchers verified that the rate of apoptosis was not elevated at this extremely high glucose level, no osmotic controls were presented and the C-peptide levels were extremely low (equivalent of 2–3 pg C-peptide per 100 islets). The investigators’ conclusion that these cells were glucose-responsive, therefore, is still questionable. In a different study,40 iPS cells derived from skin biopsies of patients with T1DM were differentiated into cells which express insulin, C-peptide, glucagon and somatostatin. Three transcription factors (OCT4, SOX2 and KLF4) were used to reprogram these adult fibroblasts into iPS
Figure 1 | Generation of insulin-expressing cells from pluripotent cells. Different differentiation protocols recapitulate the embryological development of β cells. Although robust insulin secretion in response to glucose has only been shown in hESC-derived insulin-positive cells, reprogramming of fibroblasts and umbilical cord cells into iPS cells could potentially represent an approach to obtain a renewable and available source of undifferentiated cells that can provide reliable models of disease in vitro and might eventually lead to a cell-derived therapy for T1DM. The generation of insulin-expressing cells from bone marrow-derived MSCs remains to be confirmed; however, these cells promote survival and regeneration of β cells. Abbreviations: c-MYC, Myc proto-oncogene protein; CX3CL1, CX3C-chemokine ligand 1; CXCL12, CXC-chemokine ligand 12; EGF, epidermal growth factor; FGF, fibroblast growth factor; hESC, human embryonic stem cell; IDE, induce definitive endoderm; iPS, induced pluripotent stem cells; KGF, keratinocyte growth factor; KLF4, Kruppel-like factor 4; LIN28, lin-28 homolog A; MSC, mesenchymal stromal cell; OCT4, Octamer-binding transcription factor 4; SOX2, transcription factor sex determining region Y box; T1DM, type 1 diabetes mellitus; TSG6, tumor necrosis factor-inducible gene 6 protein.
Blastocyst
Fibroblast
Umbilical cord
Bone marrow
hESC Endoderm
Embryoid body
Factors: Activin A, FGF,retinoic acid, IDE1, IDE2
Factors: indolactam V,KGF, retinoic acid,
cyclopamine
Factors: Activin A, EGF,FGF, OCT4, KLF4
Adenoviral infectionwith OCT4, SOX2,
c-MYC, KLF4
Lentiviral infectionwith OCT4, SOX2,
NANOG, LIN28Factors: insulin,
high glucose, Activin A,retinoic acid, FGF
?
iPS cell
Insulin-expressing cell
CD133+
CD34+
MSCSurvival and regeneration
of cells
CX3CL1, CXCL12, TSG6
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 141 4/2/10 17:01:13
© 20 Macmillan Publishers Limited. All rights reserved10
CX3CL1CXCL12, TSG6
??
3/25/16, 8:19 AMAccess : Stem cell therapy for type 1 diabetes mellitus : Nature Reviews Endocrinology
Page 1 of 3http://www.nature.com/nrendo/journal/v6/n3/full/nrendo.2009.274.html
ARTICLETOOLS
Send to afriend
Exportcitation
Exportreferences
Rights andpermissions
Ordercommercialreprints
SEARCHPUBMEDFOR
CristinaAguayo-Mazzucato
Susan
AccessTo read this article in full you may need to login, make a payment or gain access through asite license (see right).
ReviewNature Reviews Endocrinology 6, 139-148 (March2010) | doi:10.1038/nrendo.2009.274
Stem cell therapy fortype 1 diabetesmellitusCristina Aguayo-Mazzucato &Susan Bonner-Weir
The use of stem cells inregenerative medicine holdsgreat promise for the cure ofmany diseases, including type 1diabetes mellitus (T1DM). Anypotential stem-cell-based curefor T1DM should address theneed for β-cell replacement, aswell as control of theautoimmune response to cellswhich express insulin. The exvivo generation of β cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 139
Section of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, 1 Joslin Place, Boston, MA 02215, USA. (C. Aguayo-Mazzucato, S. Bonner-Weir).
Correspondence to: S. Bonner-Weir [email protected]
Stem cell therapy for type 1 diabetes mellitusCristina Aguayo-Mazzucato and Susan Bonner-Weir
Abstract | The use of stem cells in regenerative medicine holds great promise for the cure of many diseases, including type 1 diabetes mellitus (T1DM). Any potential stem-cell-based cure for T1DM should address the need for β-cell replacement, as well as control of the autoimmune response to cells which express insulin. The ex vivo generation of β cells suitable for transplantation to reconstitute a functional β-cell mass has used pluripotent cells from diverse sources, as well as organ-specific facultative progenitor cells from the liver and the pancreas. The most effective protocols to date have produced cells that express insulin and have molecular characteristics that closely resemble bona fide insulin-secreting cells; however, these cells are often unresponsive to glucose, a characteristic that should be addressed in future protocols. The use of mesenchymal stromal cells or umbilical cord blood to modulate the immune response is already in clinical trials; however, definitive results are still pending. This Review focuses on current strategies to obtain cells which express insulin from different progenitor sources and highlights the main pathways and genes involved, as well as the different approaches for the modulation of the immune response in patients with T1DM.
Aguayo-Mazzucato, C. & Bonner-Weir, S. Nat. Rev. Endocrinol. 6, 139–148 (2010); doi:10.1038/nrendo.2009.274
IntroductionType 1 diabetes mellitus (T1DM) is characterized by the autoimmune destruction of the insulin- expressing, pan-creatic β cells; as a result, patients with T1DM are depen-dent on exogenous insulin for their blood glucose control. Although the discovery of insulin has changed the outlook and survival of this group of individuals for almost a century,1 patients with T1DM are still not exempt from the development of diabetic complications.2,3 These com-plications are thought to result, in part, from the lack of physiological oscillations in insulin secretion and higher than normal glucose levels; thus, the search for effective ways to re establish a functional β-cell mass in patients with T1DM is important. Transplantation of whole human pancreas or isolated, cadaveric human islets4,5 has allowed patients with T1DM to become insulin-independent and has thereby shown proof of concept for β-cell replacement therapies. The shortage of donor pancreata and islets,6 however, results in the search for alternative cell sources, and differen tiation of stem cells (Box 1) into functional β cells is an attractive alternative.
A definitive cure for T1DM needs to address both the β-cell deficit and the autoimmune response to cells that express insulin. As clearly shown by the transplantation of segmental pancreatic grafts from identical twins, the autoimmune response remains many years after disease onset.7 Thus, any β-cell replacement therapy will need some modulation of the immune system, whether by drugs or cell-based approaches.
This Review focuses mainly on the different approaches to obtain functional, insulin-producing cells from various types of progenitor cells. Although the goal of the field is to
direct differentiation of human stem cells into functional β cells, some animal studies are included here, as they could lay the framework for success ful future interventions in humans. Although stem cell- derived, insulin-positive cells have not yet reached clinical trial, clinical data from stem cell-derived therapies that target the autoimmune response have been reported and are discussed.
Many roads lead from stem cells to β cellsEfforts have been directed at the development of efficient protocols for the differentiation of stem cells of either embryonic (Figure 1) or adult origin (Figure 2) into func-tional cells that secrete insulin. A caveat to many of these studies is that some developmental pathways generate insulin-expressing cells that will never become true β cells, for example, cells of the fetal liver, yolk sac and brain.8–10 A β cell should be defined by its ability to store large amounts of insulin and to secrete it in a regulated manner in response to a demand, such as glucose stimulation.11
Human embryonic stem cellsEfforts to generate β cells from human embryonic stem cells (hESCs) have focused on the fundamentals of normal embryonic development as a basis to understand the early stages of endoderm formation. The first step in the differen tiation of hESCs into β cells is the production of definitive endoderm12–17 rather than visceral endoderm, which expresses the same markers (for example, sex determining region Y box [SOX] 17 and forkhead box protein A2 [FOXA2]) but forms different tissues.18
Current protocols for hESC differentiation into β cells aim to reproduce the developmentally active signaling pathways Wnt and transforming growth factor β (TGFβ) (Box 2).17,19–22 Growth factors, such as Activin A, fibroblast
Competing interestsThe authors declare no competing interests.
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 139 4/2/10 17:00:58
© 20 Macmillan Publishers Limited. All rights reserved10
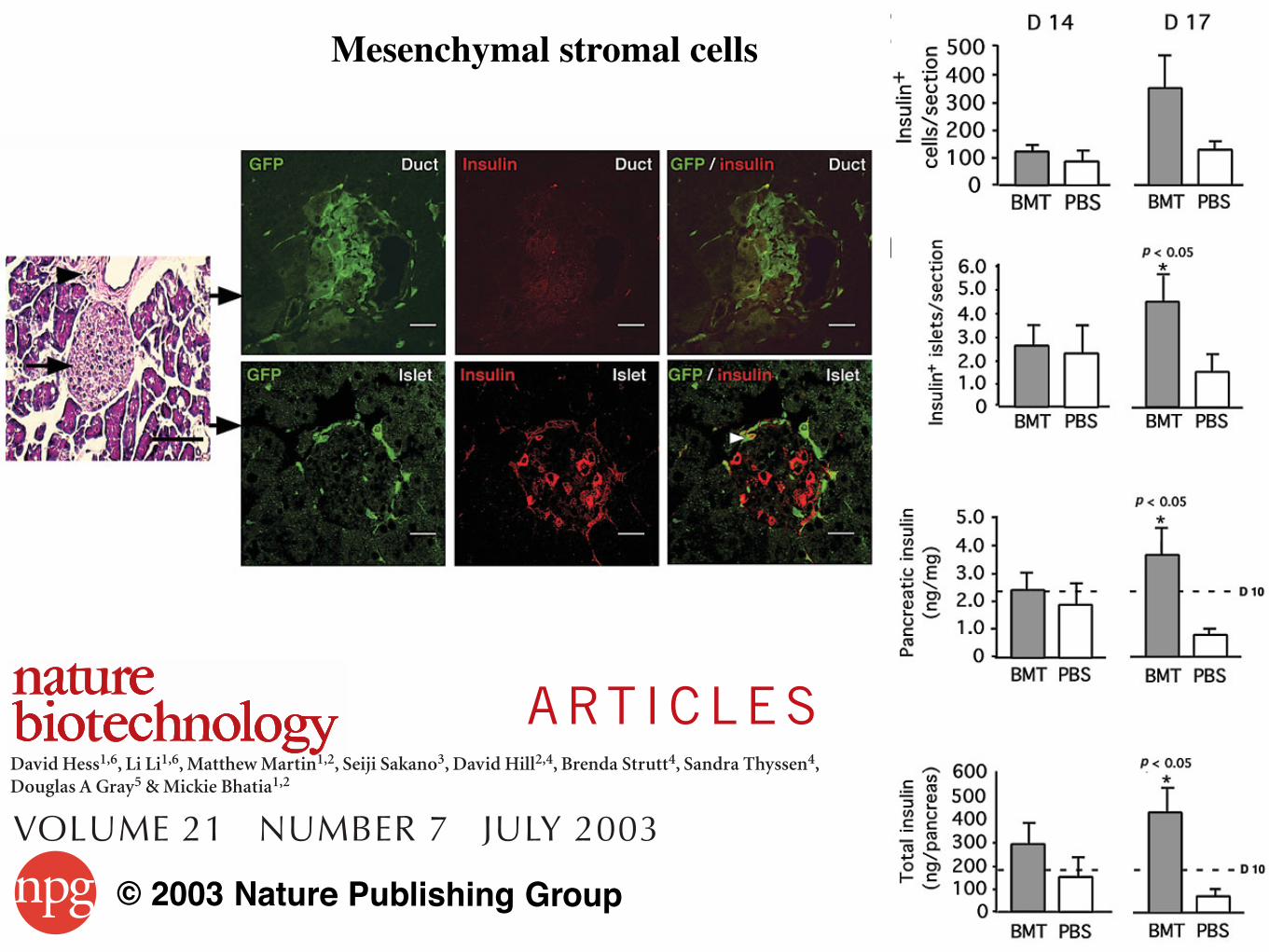
Mesenchymal stromal cells A RT I C L E S
was observed in both nuclear and cytoplasmic compartments,whereas insulin was found exclusively in the cytoplasm and not in4,6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue, Fig. 3b).Incremental z-series images indicated that single GFP+, insulin-pos-itive cells could be dissected to reveal cellular localization of insulinthat followed a punctate cytoplasmic pattern (Fig. 3c).
We compared the frequency of donor GFP+ bonemarrow–derived cells coexpressing insulin in STZ-treated and vehi-cle-treated recipients (Fig. 3d). Among mice transplanted withGFP+ bone marrow–derived cells, GFP+ insulin-positive cells (cellsof donor origin) were nearly absent in the pancreas of non-STZ-treated mice but made up 2.5% of insulin-positive cells in the pan-creas of STZ-treated animals. The presence of insulin-positivedonor cells only in STZ-treated mice suggests that pancreatic dam-age was required for this phenotype to be observed.
Because embryonic stem cells have demonstrated immunoreac-tivity to insulin resulting from the uptake of exogenous insulin
available in vitro25, we sought to determine whether observableinsulin-positive, donor bone marrow–derived cells expressed PDX-1,a transcription factor that binds to the insulin promoter and isessential for glucose-stimulated insulin production26,27. DonorGFP+ and endogenous GFP– cells were isolated from pancreatic tis-sues and analyzed for PDX-1 expression by RT-PCR. RecipientGFP– pancreatic cells expressed PDX-1, whereas donor GFP+ cellsderived from the bone marrow did not (Fig. 3e). Thus, althoughbone marrow–derived cells were capable of adopting a β-cell pheno-type (insulin positive), they did not express a molecular factor asso-ciated with β-cell development28.
Based on the inability to demonstrate significant insulin produc-tion from transplanted bone marrow–derived cells, we postulatethat endogenous pancreatic cells of the recipient may be primarilyresponsible for amelioration of hyperglycemia in our model.
766 VOLUME 21 NUMBER 7 JULY 2003 NATURE BIOTECHNOLOGY
a
b
c
d
Figure 3 Transplanted donor bone marrow (BM) cells engraft ductal andislet regions in recipient pancreas and produce a low frequency of donorinsulin-positive (insulin+) cells devoid of PDX-1 expression. (a) GFP andinsulin expression of donor cells engrafting the ductal epithelial region andsurrounding recipient-derived insulin producing cells. White arrowheadsindicate insulin-producing donor cells in the ductal (black arrowhead, top left) and islet (black arrow, middle left) regions. Scale bars, 50 µm. (b) Visualization of insulin-producing donor (insulin+, GFP+) cell. Scale bars, 10 µm. (c) z-series of a single insulin-producing donor cell. Cytoplasmicstaining of insulin granules with GFP expression was observed throughoutthe nuclear and cytoplasmic regions. Each panel represents serial images (5 µm) through the thickness of the tissue. (d) Frequency of insulin-producing donor cells in the pancreas of NOD/SCID recipients. (e) RT-PCRshowing PDX-1 mRNA levels in donor and recipient cells in the pancreas ofNOD/SCID recipients; β-actin mRNA was assessed as input templatecontrol. Data are shown as mean ± s.e.m. Tissue sectioning analysis wasperformed as detailed in methods.
a b
c g
d
e
f
h
i
j
Figure 4 Donor bone marrow–derived stem cells rapidly engraft the pancreasof recipient mice and induce endogenous pancreatic insulin production.(a,b) Analysis of the number of insulin-positive cells per section and thenumber of islets per mm2 in the pancreas of STZ-treated NOD/SCIDrecipients at day 42. BMT, bone marrow transplant. (c) Experimental designfor the detection of insulin-producing cells, PECAM-1-expressing donor cellsand BrdU-labeled proliferating cells in the pancreases of hyperglycemicmice 4 and 7 d after transplant with GFP+ bone marrow (BM) cells or mocktransplanted with PBS. (d–f) Representative analysis of GFP and insulinexpressing cells detected in pancreatic ductal and islets regions at 4 (day 14(D 14)) and 7 days (day 17 (D 17)) after intravenous (IV) transplantation ofeither BM or PBS. (g,h) Quantitative analysis of the number of insulin-producing cells (g) and the number of islet structures per section (h) in thepancreas of STZ-treated mice, 4 and 7 d after transplantation with eitherBM or PBS. (i,j) Quantitative analysis of the pancreatic mass (i) and totalpancreatic insulin content (j) in STZ-treated mice 4 and 7 d after BMtransplant or PBS injection. Horizontal dotted line indicates levels at day 10before transplantation. All data are mean ± s.e.m. (n = 5 mice per group). Asignificant (P < 0.05) increase in pancreas insulin content was observed inBM transplanted mice within 7 d. Horizontal dotted line indicates levels at d 10 before transplantation.
©20
03 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
A RT I C L E S
was observed in both nuclear and cytoplasmic compartments,whereas insulin was found exclusively in the cytoplasm and not in4,6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue, Fig. 3b).Incremental z-series images indicated that single GFP+, insulin-pos-itive cells could be dissected to reveal cellular localization of insulinthat followed a punctate cytoplasmic pattern (Fig. 3c).
We compared the frequency of donor GFP+ bonemarrow–derived cells coexpressing insulin in STZ-treated and vehi-cle-treated recipients (Fig. 3d). Among mice transplanted withGFP+ bone marrow–derived cells, GFP+ insulin-positive cells (cellsof donor origin) were nearly absent in the pancreas of non-STZ-treated mice but made up 2.5% of insulin-positive cells in the pan-creas of STZ-treated animals. The presence of insulin-positivedonor cells only in STZ-treated mice suggests that pancreatic dam-age was required for this phenotype to be observed.
Because embryonic stem cells have demonstrated immunoreac-tivity to insulin resulting from the uptake of exogenous insulin
available in vitro25, we sought to determine whether observableinsulin-positive, donor bone marrow–derived cells expressed PDX-1,a transcription factor that binds to the insulin promoter and isessential for glucose-stimulated insulin production26,27. DonorGFP+ and endogenous GFP– cells were isolated from pancreatic tis-sues and analyzed for PDX-1 expression by RT-PCR. RecipientGFP– pancreatic cells expressed PDX-1, whereas donor GFP+ cellsderived from the bone marrow did not (Fig. 3e). Thus, althoughbone marrow–derived cells were capable of adopting a β-cell pheno-type (insulin positive), they did not express a molecular factor asso-ciated with β-cell development28.
Based on the inability to demonstrate significant insulin produc-tion from transplanted bone marrow–derived cells, we postulatethat endogenous pancreatic cells of the recipient may be primarilyresponsible for amelioration of hyperglycemia in our model.
766 VOLUME 21 NUMBER 7 JULY 2003 NATURE BIOTECHNOLOGY
a
b
c
d
Figure 3 Transplanted donor bone marrow (BM) cells engraft ductal andislet regions in recipient pancreas and produce a low frequency of donorinsulin-positive (insulin+) cells devoid of PDX-1 expression. (a) GFP andinsulin expression of donor cells engrafting the ductal epithelial region andsurrounding recipient-derived insulin producing cells. White arrowheadsindicate insulin-producing donor cells in the ductal (black arrowhead, top left) and islet (black arrow, middle left) regions. Scale bars, 50 µm. (b) Visualization of insulin-producing donor (insulin+, GFP+) cell. Scale bars, 10 µm. (c) z-series of a single insulin-producing donor cell. Cytoplasmicstaining of insulin granules with GFP expression was observed throughoutthe nuclear and cytoplasmic regions. Each panel represents serial images (5 µm) through the thickness of the tissue. (d) Frequency of insulin-producing donor cells in the pancreas of NOD/SCID recipients. (e) RT-PCRshowing PDX-1 mRNA levels in donor and recipient cells in the pancreas ofNOD/SCID recipients; β-actin mRNA was assessed as input templatecontrol. Data are shown as mean ± s.e.m. Tissue sectioning analysis wasperformed as detailed in methods.
a b
c g
d
e
f
h
i
j
Figure 4 Donor bone marrow–derived stem cells rapidly engraft the pancreasof recipient mice and induce endogenous pancreatic insulin production.(a,b) Analysis of the number of insulin-positive cells per section and thenumber of islets per mm2 in the pancreas of STZ-treated NOD/SCIDrecipients at day 42. BMT, bone marrow transplant. (c) Experimental designfor the detection of insulin-producing cells, PECAM-1-expressing donor cellsand BrdU-labeled proliferating cells in the pancreases of hyperglycemicmice 4 and 7 d after transplant with GFP+ bone marrow (BM) cells or mocktransplanted with PBS. (d–f) Representative analysis of GFP and insulinexpressing cells detected in pancreatic ductal and islets regions at 4 (day 14(D 14)) and 7 days (day 17 (D 17)) after intravenous (IV) transplantation ofeither BM or PBS. (g,h) Quantitative analysis of the number of insulin-producing cells (g) and the number of islet structures per section (h) in thepancreas of STZ-treated mice, 4 and 7 d after transplantation with eitherBM or PBS. (i,j) Quantitative analysis of the pancreatic mass (i) and totalpancreatic insulin content (j) in STZ-treated mice 4 and 7 d after BMtransplant or PBS injection. Horizontal dotted line indicates levels at day 10before transplantation. All data are mean ± s.e.m. (n = 5 mice per group). Asignificant (P < 0.05) increase in pancreas insulin content was observed inBM transplanted mice within 7 d. Horizontal dotted line indicates levels at d 10 before transplantation.
©20
03 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
A RT I C L E S
Bone marrow is an important source of easily procurable adult stemcells1. In addition to the ability of bone marrow–derived stem cells toreconstitute the hematopoietic system1, cells derived from the bonemarrow compartment have been shown to possess endothelial2,3,mesenchymal4 and pluripotent capabilities5. Other recent studieshave indicated that transplanted bone marrow–derived stem cellscan generate cells with unexpected phenotypes, including muscle6,liver7, brain8 and epithelial lineages9, suggesting greater develop-mental plasticity than previously believed10. However such eventsoccur at low frequency11, and the apparent plasticity has been shownin one case to be explained by cellular fusion12,13. Despite the obscu-rity of the cellular mechanism, the ability of transplanted bone mar-row to correct physiological function in the whole animal has beendocumented7,13,14.
In the context of regenerative therapies, the role of adult stem cellsmay not be limited to direct replacement of damaged cells. Recent ani-mal and clinical studies have suggested that tissue regeneration canoccur upon the introduction of bone marrow–derived stem cells thathave multiple effects, including the vascularization of damaged tis-sue15. Vascularization may be facilitated by angiogenic factors releasedfrom endothelial cells originating from transplanted bone marrowstem cells15,16. In addition, embryonic organogenesis is known to bedependent on tissue-specific endothelium, and endothelial interac-tions and signaling may play a role in post-natal stem cell engraftmentand differentiation16. Accordingly, regenerative ability of adult tissuemay be directed by essential factors provided by transplanted endothe-lial cells of bone marrow origin15,16.
Given the potential therapeutic benefit of bone marrow transplan-tation, we sought to determine the cellular mechanism and physiolog-ical relevance of bone marrow–derived stem cells for the restoration of
tissue function after pancreatic injury. Using a mouse model of chem-ically induced pancreatic damage that causes hyperglycemia, we showthat transplantation of bone marrow–derived stem cells initiatesendogenous pancreatic regeneration. Engraftment of bone marrow-derived cells to ductal and islet structures was accompanied by rapidproliferation of recipient pancreatic cells and neogenesis of insulin-positive cells of recipient origin.
RESULTSModel of pancreatic damage and repairTo evaluate the potential capacity of bone marrow–derived stem cellsto restore tissue function after injury, we adopted a mouse model ofstreptozotocin (STZ)-induced pancreatic damage causing hyper-glycemia17 and designed an experimental model to assess pancreaticrepair upon transplantation of allogeneic green fluorescent protein(GFP)-expressing bone marrow–derived stem cells (Fig. 1a).Recipients were treated daily for 5 d with 35 mg STZ per kg bodyweight, which induced pancreatic damage (Fig. 1b) and secondaryhyperglycemia (blood glucose >13.9 mmol/l; Fig. 1c). Morphologicalassessment of pancreatic sections stained with hematoxylin and eosin(H+E) and for insulin showed that pancreatic damage by STZ wasassociated with the destruction of pancreatic islets at day 10 and apaucity of insulin-expressing cells at day 42 (Fig. 1b).
Hyperglycemic mice were intravenously transplanted with trans-genic GFP+ bone marrow–derived cells, and blood glucose and seruminsulin were monitored (Fig. 1a). By day 10 (day of i.v. transplant),blood glucose levels in STZ-treated animals were significantly elevated(P < 0.05), and at day 42 blood glucose levels were above 30 mmol/l(Fig. 1c). Increased blood glucose correlated with marked reductionsin serum insulin by day 10, and these remained attenuated at day 42 as
1Robarts Research Institute, Stem Cell Biology and Regenerative Medicine, 100 Perth Drive, London, Ontario N6A 5K8, Canada. 2The University of Western Ontario,1151 Richmond Street, London, Ontario N6A 4B8, Canada. 3Asahi Kasei Corporation, Central R&D and Central Research Laboratory, 2-1 Samajima, Fuji, Shizuoka416-8501, Japan. 4Lawson Health Research Institute, 268 Grosvenor Street, London, Ontario N6A 4V2, Canada, and 5Centre For Cancer Therapeutics, 503 SmythRoad, Ottawa, Ontario K1H 1C4, Canada. 6These authors contributed equally to this work. Correspondence should be addressed to M.B. ([email protected]).
Bone marrow–derived stem cells initiate pancreaticregenerationDavid Hess1,6, Li Li1,6, Matthew Martin1,2, Seiji Sakano3, David Hill2,4, Brenda Strutt4, Sandra Thyssen4,Douglas A Gray5 & Mickie Bhatia1,2
We show that transplantation of adult bone marrow–derived cells expressing c-kit reduces hyperglycemia in mice withstreptozotocin-induced pancreatic damage. Although quantitative analysis of the pancreas revealed a low frequency of donorinsulin-positive cells, these cells were not present at the onset of blood glucose reduction. Instead, the majority of transplantedcells were localized to ductal and islet structures, and their presence was accompanied by a proliferation of recipient pancreaticcells that resulted in insulin production. The capacity of transplanted bone marrow–derived stem cells to initiate endogenouspancreatic tissue regeneration represents a previously unrecognized means by which these cells can contribute to the restorationof organ function.
NATURE BIOTECHNOLOGY VOLUME 21 NUMBER 7 JULY 2003 763
©20
03 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
A RT I C L E S
Bone marrow is an important source of easily procurable adult stemcells1. In addition to the ability of bone marrow–derived stem cells toreconstitute the hematopoietic system1, cells derived from the bonemarrow compartment have been shown to possess endothelial2,3,mesenchymal4 and pluripotent capabilities5. Other recent studieshave indicated that transplanted bone marrow–derived stem cellscan generate cells with unexpected phenotypes, including muscle6,liver7, brain8 and epithelial lineages9, suggesting greater develop-mental plasticity than previously believed10. However such eventsoccur at low frequency11, and the apparent plasticity has been shownin one case to be explained by cellular fusion12,13. Despite the obscu-rity of the cellular mechanism, the ability of transplanted bone mar-row to correct physiological function in the whole animal has beendocumented7,13,14.
In the context of regenerative therapies, the role of adult stem cellsmay not be limited to direct replacement of damaged cells. Recent ani-mal and clinical studies have suggested that tissue regeneration canoccur upon the introduction of bone marrow–derived stem cells thathave multiple effects, including the vascularization of damaged tis-sue15. Vascularization may be facilitated by angiogenic factors releasedfrom endothelial cells originating from transplanted bone marrowstem cells15,16. In addition, embryonic organogenesis is known to bedependent on tissue-specific endothelium, and endothelial interac-tions and signaling may play a role in post-natal stem cell engraftmentand differentiation16. Accordingly, regenerative ability of adult tissuemay be directed by essential factors provided by transplanted endothe-lial cells of bone marrow origin15,16.
Given the potential therapeutic benefit of bone marrow transplan-tation, we sought to determine the cellular mechanism and physiolog-ical relevance of bone marrow–derived stem cells for the restoration of
tissue function after pancreatic injury. Using a mouse model of chem-ically induced pancreatic damage that causes hyperglycemia, we showthat transplantation of bone marrow–derived stem cells initiatesendogenous pancreatic regeneration. Engraftment of bone marrow-derived cells to ductal and islet structures was accompanied by rapidproliferation of recipient pancreatic cells and neogenesis of insulin-positive cells of recipient origin.
RESULTSModel of pancreatic damage and repairTo evaluate the potential capacity of bone marrow–derived stem cellsto restore tissue function after injury, we adopted a mouse model ofstreptozotocin (STZ)-induced pancreatic damage causing hyper-glycemia17 and designed an experimental model to assess pancreaticrepair upon transplantation of allogeneic green fluorescent protein(GFP)-expressing bone marrow–derived stem cells (Fig. 1a).Recipients were treated daily for 5 d with 35 mg STZ per kg bodyweight, which induced pancreatic damage (Fig. 1b) and secondaryhyperglycemia (blood glucose >13.9 mmol/l; Fig. 1c). Morphologicalassessment of pancreatic sections stained with hematoxylin and eosin(H+E) and for insulin showed that pancreatic damage by STZ wasassociated with the destruction of pancreatic islets at day 10 and apaucity of insulin-expressing cells at day 42 (Fig. 1b).
Hyperglycemic mice were intravenously transplanted with trans-genic GFP+ bone marrow–derived cells, and blood glucose and seruminsulin were monitored (Fig. 1a). By day 10 (day of i.v. transplant),blood glucose levels in STZ-treated animals were significantly elevated(P < 0.05), and at day 42 blood glucose levels were above 30 mmol/l(Fig. 1c). Increased blood glucose correlated with marked reductionsin serum insulin by day 10, and these remained attenuated at day 42 as
1Robarts Research Institute, Stem Cell Biology and Regenerative Medicine, 100 Perth Drive, London, Ontario N6A 5K8, Canada. 2The University of Western Ontario,1151 Richmond Street, London, Ontario N6A 4B8, Canada. 3Asahi Kasei Corporation, Central R&D and Central Research Laboratory, 2-1 Samajima, Fuji, Shizuoka416-8501, Japan. 4Lawson Health Research Institute, 268 Grosvenor Street, London, Ontario N6A 4V2, Canada, and 5Centre For Cancer Therapeutics, 503 SmythRoad, Ottawa, Ontario K1H 1C4, Canada. 6These authors contributed equally to this work. Correspondence should be addressed to M.B. ([email protected]).
Bone marrow–derived stem cells initiate pancreaticregenerationDavid Hess1,6, Li Li1,6, Matthew Martin1,2, Seiji Sakano3, David Hill2,4, Brenda Strutt4, Sandra Thyssen4,Douglas A Gray5 & Mickie Bhatia1,2
We show that transplantation of adult bone marrow–derived cells expressing c-kit reduces hyperglycemia in mice withstreptozotocin-induced pancreatic damage. Although quantitative analysis of the pancreas revealed a low frequency of donorinsulin-positive cells, these cells were not present at the onset of blood glucose reduction. Instead, the majority of transplantedcells were localized to ductal and islet structures, and their presence was accompanied by a proliferation of recipient pancreaticcells that resulted in insulin production. The capacity of transplanted bone marrow–derived stem cells to initiate endogenouspancreatic tissue regeneration represents a previously unrecognized means by which these cells can contribute to the restorationof organ function.
NATURE BIOTECHNOLOGY VOLUME 21 NUMBER 7 JULY 2003 763
©20
03 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
©20
08 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
A RT I C L E S
Bone marrow is an important source of easily procurable adult stemcells1. In addition to the ability of bone marrow–derived stem cells toreconstitute the hematopoietic system1, cells derived from the bonemarrow compartment have been shown to possess endothelial2,3,mesenchymal4 and pluripotent capabilities5. Other recent studieshave indicated that transplanted bone marrow–derived stem cellscan generate cells with unexpected phenotypes, including muscle6,liver7, brain8 and epithelial lineages9, suggesting greater develop-mental plasticity than previously believed10. However such eventsoccur at low frequency11, and the apparent plasticity has been shownin one case to be explained by cellular fusion12,13. Despite the obscu-rity of the cellular mechanism, the ability of transplanted bone mar-row to correct physiological function in the whole animal has beendocumented7,13,14.
In the context of regenerative therapies, the role of adult stem cellsmay not be limited to direct replacement of damaged cells. Recent ani-mal and clinical studies have suggested that tissue regeneration canoccur upon the introduction of bone marrow–derived stem cells thathave multiple effects, including the vascularization of damaged tis-sue15. Vascularization may be facilitated by angiogenic factors releasedfrom endothelial cells originating from transplanted bone marrowstem cells15,16. In addition, embryonic organogenesis is known to bedependent on tissue-specific endothelium, and endothelial interac-tions and signaling may play a role in post-natal stem cell engraftmentand differentiation16. Accordingly, regenerative ability of adult tissuemay be directed by essential factors provided by transplanted endothe-lial cells of bone marrow origin15,16.
Given the potential therapeutic benefit of bone marrow transplan-tation, we sought to determine the cellular mechanism and physiolog-ical relevance of bone marrow–derived stem cells for the restoration of
tissue function after pancreatic injury. Using a mouse model of chem-ically induced pancreatic damage that causes hyperglycemia, we showthat transplantation of bone marrow–derived stem cells initiatesendogenous pancreatic regeneration. Engraftment of bone marrow-derived cells to ductal and islet structures was accompanied by rapidproliferation of recipient pancreatic cells and neogenesis of insulin-positive cells of recipient origin.
RESULTSModel of pancreatic damage and repairTo evaluate the potential capacity of bone marrow–derived stem cellsto restore tissue function after injury, we adopted a mouse model ofstreptozotocin (STZ)-induced pancreatic damage causing hyper-glycemia17 and designed an experimental model to assess pancreaticrepair upon transplantation of allogeneic green fluorescent protein(GFP)-expressing bone marrow–derived stem cells (Fig. 1a).Recipients were treated daily for 5 d with 35 mg STZ per kg bodyweight, which induced pancreatic damage (Fig. 1b) and secondaryhyperglycemia (blood glucose >13.9 mmol/l; Fig. 1c). Morphologicalassessment of pancreatic sections stained with hematoxylin and eosin(H+E) and for insulin showed that pancreatic damage by STZ wasassociated with the destruction of pancreatic islets at day 10 and apaucity of insulin-expressing cells at day 42 (Fig. 1b).
Hyperglycemic mice were intravenously transplanted with trans-genic GFP+ bone marrow–derived cells, and blood glucose and seruminsulin were monitored (Fig. 1a). By day 10 (day of i.v. transplant),blood glucose levels in STZ-treated animals were significantly elevated(P < 0.05), and at day 42 blood glucose levels were above 30 mmol/l(Fig. 1c). Increased blood glucose correlated with marked reductionsin serum insulin by day 10, and these remained attenuated at day 42 as
1Robarts Research Institute, Stem Cell Biology and Regenerative Medicine, 100 Perth Drive, London, Ontario N6A 5K8, Canada. 2The University of Western Ontario,1151 Richmond Street, London, Ontario N6A 4B8, Canada. 3Asahi Kasei Corporation, Central R&D and Central Research Laboratory, 2-1 Samajima, Fuji, Shizuoka416-8501, Japan. 4Lawson Health Research Institute, 268 Grosvenor Street, London, Ontario N6A 4V2, Canada, and 5Centre For Cancer Therapeutics, 503 SmythRoad, Ottawa, Ontario K1H 1C4, Canada. 6These authors contributed equally to this work. Correspondence should be addressed to M.B. ([email protected]).
Bone marrow–derived stem cells initiate pancreaticregenerationDavid Hess1,6, Li Li1,6, Matthew Martin1,2, Seiji Sakano3, David Hill2,4, Brenda Strutt4, Sandra Thyssen4,Douglas A Gray5 & Mickie Bhatia1,2
We show that transplantation of adult bone marrow–derived cells expressing c-kit reduces hyperglycemia in mice withstreptozotocin-induced pancreatic damage. Although quantitative analysis of the pancreas revealed a low frequency of donorinsulin-positive cells, these cells were not present at the onset of blood glucose reduction. Instead, the majority of transplantedcells were localized to ductal and islet structures, and their presence was accompanied by a proliferation of recipient pancreaticcells that resulted in insulin production. The capacity of transplanted bone marrow–derived stem cells to initiate endogenouspancreatic tissue regeneration represents a previously unrecognized means by which these cells can contribute to the restorationof organ function.
NATURE BIOTECHNOLOGY VOLUME 21 NUMBER 7 JULY 2003 763
©20
03 N
atur
e Pu
blis
hing
Gro
up h
ttp://
ww
w.n
atur
e.co
m/n
atur
ebio
tech
nolo
gy
AR
TIC
LE
S
Bon
e marrow
is an im
portant source of
easily procurable adult stemcells 1.In
addition to the ability ofbon
e marrow
–derived stem cells to
reconstitu
te the hematopoietic system
1,cells derived from the bon
em
arrow com
partmen
t have been show
n to possess en
dothelial 2,3,m
esenchym
al 4an
d pluripoten
t capabilities 5.O
ther recent stu
dieshave in
dicated that transplan
ted bone m
arrow–derived stem
cellscan
generate cells w
ith un
expected phenotypes,in
cludin
g mu
scle6,
liver 7,brain
8an
d epithelial lineages 9,
suggestin
g greater develop-m
ental plasticity than
previously believed
10.H
owever such even
tsoccur at low
frequency
11,and the apparen
t plasticity has been show
nin
one case to be explain
ed by cellular fusion12,13.D
espite the obscu-rity of
the cellular mechan
ism,the ability of
transplan
ted bone m
ar-row
to correct physiological fun
ction in
the whole an
imal has been
documen
ted7,13,14.
In the context ofregenerative therapies,the role of
adult stem cells
may not be lim
ited to direct replacement ofdam
aged cells.Recent ani-
mal and clinical studies have suggested that tissue regeneration can
occur upon the introduction ofbone m
arrow–derived stem
cells thathave m
ultiple effects,including the vascularization of
damaged tis-
sue15.V
ascularization may be facilitated by angiogenic factors released
from endothelial cells originating from
transplanted bone marrow
stem cells 15,16.In addition,em
bryonic organogenesis is known to be
dependent on tissue-specific endothelium,
and endothelial interac-tions and signaling m
ay play a role in post-natal stem cell engraftm
entand differentiation
16.Accordingly,regenerative ability of
adult tissuem
ay be directed by essential factors provided by transplanted endothe-lial cells ofbone m
arrow origin
15,16.G
iven the potential therapeutic benefit ofbone m
arrow transplan-
tation,we sought to determ
ine the cellular mechanism
and physiolog-ical relevance ofbone m
arrow–derived stem
cells for the restoration of
tissue function
after pancreatic in
jury.Usin
g a mouse m
odel ofchem-
ically induced pan
creatic damage that causes hyperglycem
ia,we show
that transplan
tation of
bone m
arrow–derived stem
cells initiates
endogen
ous pancreatic regen
eration.E
ngraftm
ent of
bone m
arrow-
derived cells to ductal and islet structures was accom
panied by rapidproliferation of
recipient pancreatic cells and neogenesis ofinsulin-
positive cells ofrecipient origin.
RESU
LTSM
odel of pancreatic damage and repair
To evaluate the potential capacity ofbone m
arrow–derived stem
cellsto restore tissue function after injury,w
e adopted a mouse m
odel ofstreptozotocin
(STZ
)-induced pancreatic
damage
causing hyper-
glycemia
17and designed an experim
ental model to assess pancreatic
repair upon transplantation ofallogeneic green fluorescent protein
(GFP
)-expressing bone
marrow
–derived stem
cells
(Fig.1a).
Recipients w
ere treated daily for 5 d with 35 m
g STZ
per kg bodyw
eight,w
hich induced pancreatic damage (Fig.
1b) and secondaryhyperglycem
ia (blood glucose >13.9 m
mol/l;Fig.1c).M
orphologicalassessm
ent ofpancreatic sections stained w
ith hematoxylin and eosin
(H+
E) and for insulin show
ed that pancreatic damage by ST
Z w
asassociated w
ith the destruction ofpancreatic islets at day 10 and a
paucity ofinsulin-expressing cells at day 42 (Fig.1b).H
yperglycemic m
ice were intravenously transplanted w
ith trans-genic G
FP+
bone marrow
–derived cells,and blood glucose and seruminsulin w
ere monitored (Fig.1a).B
y day 10 (day ofi.v.transplant),
blood glucose levels in STZ
-treated animals w
ere significantly elevated(P <
0.05),and at day 42 blood glucose levels were above 30 m
mol/l
(Fig.1c).Increased blood glucose correlated with m
arked reductionsin serum
insulin by day 10,and these remained attenuated at day 42 as
1Robarts R
esearch Institute, Stem
Cell B
iology and Regenerative M
edicine, 10
0 P
erth Drive, London, O
ntario N6
A 5
K8
, Canada. 2The U
niversity of Western O
ntario,1
15
1 R
ichmond S
treet,London, Ontario N
6A
4B
8, C
anada. 3Asahi K
asei Corporation, C
entral R&
D and C
entral Research Laboratory, 2
-1 S
amajim
a, Fuji, Shizuoka
41
6-8
50
1, Japan. 4Law
son Health R
esearch Institute, 26
8 G
rosvenor Street, London, O
ntario N6
A 4
V2, C
anada, and 5Centre For C
ancer Therapeutics, 50
3 S
myth
Road, O
ttawa, O
ntario K1
H 1
C4
, Canada. 6These authors contributed equally to this w
ork. Correspondence should be addressed to M
.B. (m
Bone m
arrow–derived stem
cells initiate pancreaticregenerationD
avid Hess 1,6,Li Li 1,6,M
atthew M
artin1,2,Seiji Sakano
3,David H
ill 2,4,Brenda Strutt 4,Sandra T
hyssen4,
Douglas A
Gray
5&
Mickie B
hatia1,2
We show
that transplantation of adult bone marrow
–derived cells expressing c-kit reduces hyperglycemia in m
ice with
streptozotocin-induced pancreatic damage. Although quantitative analysis of the pancreas revealed a low
frequency of donorinsulin-positive cells, these cells w
ere not present at the onset of blood glucose reduction. Instead, the majority of transplanted
cells were localized to ductal and islet structures, and their presence w
as accompanied by a proliferation of recipient pancreatic
cells that resulted in insulin production. The capacity of transplanted bone marrow
–derived stem cells to initiate endogenous
pancreatic tissue regeneration represents a previously unrecognized means by w
hich these cells can contribute to the restorationof organ function.
NA
TUR
E BIO
TECH
NO
LOG
YV
OLU
ME 2
1
NU
MB
ER 7
JU
LY 2
00
37
63
© 2003 Nature Publishing Group http://www.nature.com/naturebiotechnology

CHEMOKINES
Bone marrow mesenchymal stem cells express a restricted set of functionallyactive chemokine receptors capable of promoting migration to pancreatic isletsValeria Sordi, Maria Luisa Malosio, Federica Marchesi, Alessia Mercalli, Raffaella Melzi, Tiziana Giordano, Nathalie Belmonte,Giuliana Ferrari, Biagio Eugenio Leone, Federico Bertuzzi, Gianpaolo Zerbini, Paola Allavena, Ezio Bonifacio, and Lorenzo Piemonti
Bone marrow–derived mesenchymal stemcells (BM-MSCs) are stromal cells withthe ability to proliferate and differentiateinto many tissues. Although they repre-sent powerful tools for several therapeu-tic settings, mechanisms regulating theirmigration to peripheral tissues are stillunknown. Here, we report chemokine re-ceptor expression on human BM-MSCsand their role in mediating migration totissues. A minority of BM-MSCs (2% to25%) expressed a restricted set of chemo-kine receptors (CXC receptor 4 [CXCR4],CX3C receptor 1 [CX3CR1], CXCR6, CC
chemokine receptor 1 [CCR1], CCR7) and,accordingly, showed appreciable chemo-tactic migration in response to the chemo-kines CXC ligand 12 (CXCL12), CX3CL1,CXCL16, CC chemokine ligand 3 (CCL3),and CCL19. Using human pancreatic is-lets as an in vitro model of peripheraltissue, we showed that islet supernatantsreleased factors able to attract BM-MSCsin vitro, and this attraction was princi-pally mediated by CX3CL1 and CXCL12.Moreover, cells with features of BM-MSCswere detected within the pancreatic isletsof mice injected with green fluorescent
protein (GFP)–positive BM. A populationof bona fide MSCs that also expressedCXCR4, CXCR6, CCR1, and CCR7 couldbe isolated from normal adult human pan-creas. This study defines the chemokinereceptor repertoire of human BM-MSCsthat determines their migratory activity.Modulation of homing capacity may beinstrumental for harnessing the therapeu-tic potential of BM-MSCs. (Blood. 2005;106:419-427)
© 2005 by The American Society of Hematology
Introduction
Bone marrow (BM) is a complex tissue containing hematopoieticprogenitor cells and a connective-tissue network of stromal cells.Marrow stroma includes a subpopulation of undifferentiated cellsthat are capable of becoming one of a number of phenotypes,including bone and cartilage, tendon, muscle, fat, and marrowstromal connective tissue that supports hematopoietic cell differen-tiation.1,2 These cells are referred to as mesenchymal stem cells(MSCs), since they are known to have capacity of proliferation anddifferentiation into the mesenchymal lineage. Due to their potentialfor differentiation into different tissues, MSCs have emerged as apromising tool for clinical applications such as tissue engineeringand cell and gene therapy.3-5
Several reports underline the ability of MSCs to migrate.6-13
MSCs are thought to migrate in the bloodstream to seed new sitesof hematopoiesis and to various tissues during embryonic and fetaldevelopment.14,15 MSCs are present in large numbers in humanblood from at least 7 weeks’ gestation and they persist untilapproximately 12 weeks’ gestation.14 Although circulating MSCsdecrease after 12 weeks, there is evidence that a very low-frequency population of circulating multipotent nonhematopoieticcells resembling the classical MSCs persist through to adultlife.16-18 MSCs migrate efficiently to hematopoietic tissues (BMand spleen) after transplantation in some experimental animal
models,19,20 whereas reports of BM homing in humans are inconsis-tent.21-26 Of particular interest for tissue remodeling, intravenousdelivery of MSCs results in their specific migration to a site ofinjury.6-8,10,27 This ability of implanted MSCs to seek out the site oftissue damage has been demonstrated in bone or cartilage frac-ture,28 myocardial infarction,8,29 and ischemic cerebral injury.6,10,11
Because MSCs have been shown to give rise to many tissues (suchas bone, cartilage, fat, endothelia, muscle, brain, and pancreaticislet cells30,31), migrating MSCs may represent a source of pluripo-tent cells that are constantly available for the repair of damagedorgans. The mechanisms that guide homing of implanted cells areunclear. In this study, we examined the role of chemokines andtheir receptors in the migration of human MSCs. Moreover theinteraction between human pancreatic islets and MSCs was investi-gated as a model of tissue cross talk.
Material and methods
Human bone marrow mesenchymal stem cell culture
Human bone marrow mesenchymal stem cells (BM-MSCs) were obtainedfrom Cambrex (Baltimore, MD). There were 3 different batches used for thestudy. Before use, the cells were analyzed for morphology, marker
From the Telethon-Juvenile Diabetes Research Foundation Center for ! CellReplacement, the H. S. Raffaele-Telethon Institute for Gene Therapy(HSR-TIGET), and the Renal Pathophysiology Laboratory, Division ofMedicine, San Raffaele Scientific Institute, Via Olgettina, Milan Italy; theDepartment of Immunology and Cell Biology, “Mario Negri” Institute, Milan,Italy; and the Department of Surgery, University of Milano-Bicocca, Milan, Italy.
Submitted September 15, 2004; accepted March 11, 2005. Prepublished onlineas Blood First Edition Paper, March 22, 2005; DOI 10.1182/blood-2004-09-3507.
Supported by grants from Cassa di RIsparmio delle Province Lombarde
(CARIPLO), Istituto Superiore di Sanita (CS93 and CS81), and the JuvenileDiabetes Research Foundation/Telethon Italy (JT-01).
Reprints: Lorenzo Piemonti, Laboratory of Experimental Surgery, S. RaffaeleScientific Institute, Via Olgettina 60, 20132 Milan, Italy; e-mail: [email protected].
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 U.S.C. section 1734.
© 2005 by The American Society of Hematology
419BLOOD, 15 JULY 2005 ! VOLUME 106, NUMBER 2
For personal use only.on March 24, 2016. by guest www.bloodjournal.orgFrom March 22, 2005
originally published onlinedoi:10.1182/blood-2004-09-35072005 106: 419-427
Zerbini, Paola Allavena, Ezio Bonifacio and Lorenzo PiemontiGiordano, Nathalie Belmonte, Giuliana Ferrari, Biagio Eugenio Leone, Federico Bertuzzi, Gianpaolo Valeria Sordi, Maria Luisa Malosio, Federica Marchesi, Alessia Mercalli, Raffaella Melzi, Tiziana to pancreatic islets
migrationfunctionally active chemokine receptors capable of promoting Bone marrow mesenchymal stem cells express a restricted set of
http://www.bloodjournal.org/content/106/2/419.full.htmlUpdated information and services can be found at:
(3360 articles)Hematopoiesis and Stem Cells (564 articles)Chemokines, Cytokines, and Interleukins
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society
For personal use only.on March 24, 2016. by guest www.bloodjournal.orgFrom

Organ-specific stem or progenitor cells
1. Duct epithelial cells
2. Acinar cells
3. Liver cells

Generation of mTert-GFP mice as a model to identifyand study tissue progenitor cellsDavid T. Breault*†, Irene M. Min‡, Diana L. Carlone*, Loredana G. Farilla*, Dana M. Ambruzs*, Daniel E. Henderson*,Selma Algra§, Robert K. Montgomery§, Amy J. Wagers‡, and Nicholas Hole¶
Divisions of *Endocrinology and §Gastroenterology, Children’s Hospital Boston, Harvard Medical School, Boston, MA 02115; ‡Section on Developmentaland Stem Cell Biology, Joslin Diabetes Center and Harvard Stem Cell Institute, Boston, MA 02215; and ¶School of Biological and Biomedical Sciences,Durham University, Durham DH1 3LE, United Kingdom
Communicated by Patricia K. Donahoe, Massachusetts General Hospital, Boston, MA, May 23, 2008 (received for review August 9, 2007)
Stem cells hold great promise for regenerative medicine, butremain elusive in many tissues in part because universal markersof ‘‘stemness’’ have not been identified. The ribonucleoproteincomplex telomerase catalyzes the extension of chromosomeends, and its expression is associated with failure of cells toundergo cellular senescence. Because such resistance to senes-cence is a common characteristic of many stem cells, we hypoth-esized that telomerase expression may provide a selectivebiomarker for stem cells in multiple tissues. In fact, telomeraseexpression has been demonstrated within hematopoietic stemcells. We therefore generated mouse telomerase reverse tran-scriptase (mTert)-GFP-transgenic mice and assayed the ability ofmTert-driven GFP to mark tissue stem cells in testis, bonemarrow (BM), and intestine. mTert-GFP mice were generated byusing a two-step embryonic stem cell-based strategy, whichenabled primary and secondary screening of stably transfectedclones before blastocyst injection, greatly increasing the prob-ability of obtaining mTert reporter mice with physiologicallyappropriate regulation of GFP expression. Analysis of adult miceshowed that GFP is expressed in differentiating male germ cells,is enriched among BM-derived hematopoietic stem cells, andspecifically marks long-term BrdU-retaining intestinal crypt cells.In addition, telomerase-expressing GFP! BM cells showed long-term, serial, multilineage BM reconstitution, fulfilling the func-tional definition of hematopoietic stem cells. Together, thesedata provide direct evidence that mTert-GFP expression marksprogenitor cells in blood and small intestine, validating thesemice as a useful tool for the prospective identification, isolation,and functional characterization of progenitor/stem cells frommultiple tissues.
intestinal stem cell ! telomerase ! tissue stem cells
S tem cells hold great promise for regenerative medicine andtissue repair, but have been difficult to identify in many
tissues. Traditional methods used to isolate stem cells havelargely relied on FACS by using complex combinations ofantibodies to cell surface antigens. This approach has enabledthe characterization of hematopoietic stem cells (HSCs), alongwith a detailed understanding of hematopoietic lineage devel-opment (1). More recently, the development of methods basedon particular biochemical properties of stem cells (e.g., theside-population phenotype) (2) has provided additional tools toenrich for stem cell populations. A model system that enables thedirect isolation and/or enrichment of stem cells from multipletissues would greatly facilitate the identification, purification,and functional characterization of novel stem cell populations.
Relative resistance to cellular senescence despite multiple roundsof cell division is a common characteristic of stem cells (3).Telomerase is a ribonucleoprotein complex that helps maintain thetelomeric ends of chromosomes, normally shortened with each celldivision. Because loss of telomeric DNA beyond a critical thresholdinduces senescence in most somatic cells, maintenance or inductionof telomerase activity provides a means of preventing cellular
senescence (4) that may be relevant for the self-renewal of tissuestem cells. Consistent with this hypothesis, telomerase activity hasbeen demonstrated in self-renewing ES cells (5), HSCs (6), hema-topoietic progenitor and lymphoid cells (6, 7), germ cells (8),regenerating tissues such as intestine and skin (9–11), and self-renewing tissues from urochordates, suggesting an evolutionarilyconserved mechanism (12).
Mouse telomerase reverse transcriptase (mTert) is tightlyregulated and correlates with telomerase activity (11). mTertexpression is down-regulated upon differentiation in most so-matic cells (11), and telomerase-deficient mice exhibit a defectin stem cell maintenance, such that tissues highly dependent onstem cell function throughout life [e.g., bone marrow (BM),testis, and intestine] undergo organ failure (10). Several reportshave suggested a direct role for mTert in the regulation of adultstem cell proliferation and mobilization (13, 14). Because mTertgene expression is the regulatory step in the assembly of afunctional telomerase complex (11), we hypothesized that areporter gene system using the mTert promoter would allow forthe identification of telomerase-expressing cells and thusprovide a useful biomarker for stem cells in adult tissues.
In this article, we describe the generation of mTert-GFP-transgenic mice using a two-step ES cell-based screening strat-egy and validate GFP expression as a marker of two classicalstem cells: male germ cells and HSCs. In addition, these micehave been used to mark the intestinal stem cell and may allow forthe subsequent isolation and characterization of this highlyelusive cell. Thus, mTert-GFP-transgenic mice represent a modelsystem to facilitate the identification, purification, and functionalanalysis of novel stem cell populations.
ResultsGeneration of mTert-GFP-Transgenic Mice. Given that previousattempts to generate mTert-GFP mice (using the same 4.4-kbpromoter fragment used in this article) failed to show reportergene expression in adult tissues (15), we used an alternativemethod to generate transgenic mice with the goal to screen foroptimal levels of GFP expression before the production of liveanimals. Because ES cells normally express telomerase at highlevels (5), the use of ES cell transgenesis (rather than pronuclearoocyte injection) allowed for the selection of maximal transgeneexpression (1° screen) and exclusion of clones that underwenttransgene silencing upon genomic integration. G418-resistent EScell clones expressing high levels of GFP fluorescence were
Author contributions: D.T.B., I.M.M., D.L.C., R.K.M., A.J.W., and N.H. designed research;D.T.B., I.M.M., D.L.C., L.G.F., D.M.A., D.E.H., S.A., and R.K.M. performed research; N.H.contributed new reagents/analytic tools; I.M.M., D.L.C., L.G.F., R.K.M., and A.J.W. analyzeddata; and D.T.B., D.L.C., and A.J.W. wrote the paper.
The authors declare no conflict of interest.†Towhomcorrespondenceshouldbeaddressed.E-mail:[email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0804800105/DCSupplemental.
© 2008 by The National Academy of Sciences of the USA
10420–10425 ! PNAS ! July 29, 2008 ! vol. 105 ! no. 30 www.pnas.org"cgi"doi"10.1073"pnas.0804800105
results demonstrate that GFP expression in LT HSCs is not afunction of cell cycle regulation.
Functional Analysis of GFP Expression in HSCs. To validate thatmTert-GFP expression marks a functionally significant subset ofHSCs, we assessed the capacity of GFP! BM cells to exhibitlong-term, serial, multilineage reconstitution, the functionaldefinition of the HSC. BM was harvested from adult mTert-GFPmale mice and sorted by FACS into GFP! and GFP" fractions(Fig. 4a). The cell-sorting gate for GFP! cells corresponds to theGFPhi population defined to be enriched for HSCs (Fig. S1a). Anequal number of GFP! or GFP" cells were injected into the tailvein of prewarmed, sublethally irradiated isogenic (129/SvImJ)female mice (GFP! recipients, n # 4; GFP" recipients, n # 5).Long-term engraftment, defined as the persistence of donor cells$5 months after BM transplantation (BMT), was demonstratedin 50% of recipient mice receiving GFP! cells by FACS analysisof BM and PBCs (data not shown). In contrast, recipient micereceiving GFP" BM cells showed no evidence for engraftmentas late as 3.5 months after BMT (data not shown). Together,these data support the conclusion that (i) mTert-GFP! HSCsmaintain their capacity to compete for the HSC niche, (ii) giverise to long-term engraftment, and (iii) are enriched in the GFPhi
population. These data do not, however, rule out the presenceof HSCs within the GFP" population, as demonstrated by thephenotypic analysis (see Fig. 3).
To further demonstrate that GFP marks HSCs in mTert-GFPmice, we next performed BMT into secondary female recipientsusing GFP! BM cells obtained 5 months after the initial BMT(Fig. 4b). FACS-purified GFP! cells (10,000 or 40,000) wereinjected into lethally irradiated isogenic female recipients (toestablish the capacity of GFP! cells for radioprotection). Thesecondary recipient receiving 10,000 GFP! cells survived for 28days, compared with control mice, which died at 13–15 days(data not shown). In contrast, the female recipient receiving40,000 GFP! cells demonstrated long-term HSC engraftment,surviving 5 months after 2° BMT, when she was killed for analysisof blood chimerism. Multilineage reconstitution was assessed at2 and 5 months after 2° BMT (Fig. 4c). At 2 months, PBCs weresorted into myeloid (Mac-1! and Gr-1!) or lymphoid (CD4! andB220!) populations, followed by FISH analysis for the Y chro-mosome, to confirm that cells were derived from the originalmale donor. The results demonstrated chimerism for bothmyeloid (9.8% % 0.8) as well as lymphoid cells (11.4% % 2.4)(Fig. 4e). The presence of Y chromosome-containing myeloidcells 2 months after serial BMT (7 months after the initial BMT)demonstrates GFP is expressed within the HSC because anytransplanted myeloid progenitors would be exhausted by thistime (21). Chimerism also was confirmed at 5 months (10 monthsafter the initial BMT) by demonstrating GFP! myeloid (Mac-1!) and lymphoid (CD4! and B220!) cells in the peripheralblood (Fig. 4f ) at levels comparable to the frequency of GFPexpression in these subsets in unmanipulated donor mice (datanot shown). These results demonstrate the capacity of GFP! BMcells to support long-term, multilineage reconstitution in 2°transplants, and thus functionally establish mTert-GFPexpression as a marker for the HSC.
Analysis of GFP Expression in Intestinal Stem Cells. For $30 years, ithas been accepted that the continuously renewing intestinalepithelium is maintained by pluripotent stem cells located in theintestinal crypts (23). Despite this well established model, intes-tinal stem cells (ISCs) have remained highly elusive (24). Todetermine whether GFP expression could serve as a marker forISCs, we used the only established method to label these cells,long-term (LT) BrdU retention (25). mTert-GFP mice wereadministered a BrdU pulse during intestinal regeneration, fol-lowed by a period of chase. Immunohistochemical analysis of
sequential sections from small intestine revealed singleGFP!BrdU! cells in the lower portion of the intestinal crypts(Fig. 5, arrowhead), consistent with previous reports demon-strating single, LT BrdU-retaining cells in the lower crypt (25).Analysis of &15,700 crypts demonstrated 1 in 157 to containsingle GFP! cells and 1 in 25 to contain single LT BrdU! cells,similar to prior estimates of intestinal stem cell frequency (25).Of the identified GFP-expressing crypt cells, 17% coexpressedLT BrdU, suggesting that GFP is marking a subpopulation ofISCs. Expression of GFP within single cells provides us with auseful model system in which to isolate viable cells and func-tionally characterize this elusive stem cell population.
Telomerase Activity Colocalizes with GFP-Expressing Cells. Finally, toensure that GFP expression in vivo correlates with telomeraseactivity, isolated cells from BM, testis, and intestine were fraction-ated into GFP! and GFP" populations by FACS and assayed fortelomerase activity by using the Telomeric Repeat AmplificationProtocol (TRAP) (Fig. 4d and Fig. S3). Telomerase activity wasdetected within each GFP! cell fraction, whereas low or undetect-able levels of activity were observed in each GFP" cell fraction,indicating that mTert-GFP is a valid biomarker for mTert activity.
DiscussionWe have generated mTert-GFP-transgenic mice as a modelsystem to facilitate the identification, isolation, and functionalanalysis of adult stem cells. To ensure adequate levels oftransgene expression in adult mice, we used a two-step EScell-based visual screening method, which resulted in the effi-cient generation of two informative transgenic founder lines.GFP expression was then shown to mark three well acceptedstem cell populations (male germ cells, HSCs, and ISCs). Theseresults illustrate the power of this model system to identify andenrich for putative stem cell populations and to subsequentlyvalidate their functional significance.
Using multiple techniques, mTert-GFP-transgene expressionwas demonstrated in male germ cells at various stages ofspermatogenesis, with the greatest percentage of GFP! cellsdemonstrated within meiotically active primary spermatocytes,followed by secondary spermatocytes. Haploid spermatidsshowed the lowest percentage of GFP! cells. These results areconsistent with previous reports describing telomerase expres-sion in meitotically active male germ cells (16). The expressionof GFP within spermatids, previously suggested to be mTert-negative (26), may be explained by the long half-life of GFP(&24 h) or by aberrant transgene expression within these cells.
ba
Fig. 5. GFP expression in intestinal stem cells. Immunohistochemical analysisfor GFP and BrdU using serial sections from mTert-GFP mice having undergoneBrdU pulse–chase. (a) Staining with anti-GFP antibody, VIP (dark purple)chromagen substrate. (b) Staining with anti-BrdU antibody, DAB (dark brown)chromagen substrate. (Magnification: '40.)
Breault et al. PNAS ! July 29, 2008 ! vol. 105 ! no. 30 ! 10423
DEV
ELO
PMEN
TAL
BIO
LOG
Y

Pancreatic Duct epithelial cells
progenitors that give rise to both new islets and acini after birthand after injury.
ResultsWe generated Tg mice in which the CAII promoter drivesexpression of CAII-Cre or inducible CAII-CreERTM (Fig. 1B).Upon mating these transgenic mice with a reporter strain,ROSA26 loxP-Stop-loxP LacZ (R26R) (24), Cre-mediated re-combination should result in the permanent marking of CAII-expressing cells and their progeny. Therefore, if the CAIItransgene is specifically expressed only in ductal cells and theyserve as pancreatic progenitors after birth, we should find!-galactosidase-positive cells not only in ducts but also in isletsand acini in double-Tg mice; however, if they do not act asprogenitors, only ducts should be labeled (Fig. 1B).
To determine whether the transgene expression was faithful tothat of the human promoter, we undertook several assessments.In double-heterozygous CAII-R26R mice, !-galactosidase im-munostaining was seen throughout the ducts at birth (Fig. 2 A–Cand H) and only rarely at embryonic day 18.5 [supportinginformation (SI) Fig. S1]; these structures were confirmed asducts by morphology and by immunostaining (Fig. S2). At birth!-galactosidase expression in pancreatic cell types was restrictedto the ducts: its expression was not found in insulin-positive betacells (Fig. 2 A and B and Figs. S3 and S4), glucagon-positive alphacells (Fig. 2C and Fig. S5), or acini (Fig. 2 A–C and Figs. S3–S5).CAII is expressed in both the peripheral and central nervoussystems (25) but is irrelevant to the current analysis, and CAIIneural expression serves as positive control (Fig. 2 A and B).However, because ganglia are intimately associated with islets inthe mouse (26), we could not use RNA from isolated islets toexclude the possibility of Cre expression in beta cells of our mice.Therefore, we crossed our Tg mice with MIP-GFP mice (27),dispersed the excised pancreas, and FACS purified GFP! betacells from 1-day-old, 2-week-old, 4-week-old, and 8-week-oldCAII-Cre:MIP-GFP mice (Fig. S6). In FACS-purified beta cells,no CAII or Cre mRNA was found, even after 40 cycles of PCR
amplification, but this mRNA was found in both Tg kidneys (22)and GFP" pancreatic cells (Fig. 2 D and F). The RNA integrityof these samples was verified by probing for pdx1 at 28 cycles(Fig. 2E). To further confirm the specificity of transgene ex-pression, we immunostained for Cre recombinase in pancreasfrom CAII-Cre and CAII-CreERTM mice: the only Cre-positivecells were in ducts (Fig. 2 G–J) and ganglia (Fig. 2 K and L). Thus,transgene expression appears appropriately restricted.
Pancreatic weight increases about 15-fold between birth and 4weeks of age (28). Using data from our longitudinal study of betacell mass and its determinants (28), we estimated that more than30% of the new beta cells seen at day 31 did not arise fromreplication of preexisting beta cells (3). Therefore, pancreases ofdouble-heterozygous CAII-Cre:R26R mice were analyzed at day0 and at 4 weeks to determine the contribution of new islets orlobes during normal postnatal development (Fig. 3A). At 4 weeks!-galactosidase expression was found in many ducts, patches ofacinar cells, and some islets (Fig. 3 B–D). Within marked islets,both beta and alpha cells were !-galactosidase-positive (Fig. 3 Band C). Islets are formed by coalescence of endocrine cells andare polyclonal in origin (29), so it was expected that islets wouldhave various proportions of !-galactosidase-positive cells. Pos-itive acinar cells were scattered as single cells or small clusters,
Fig. 1. Experimental approach to test the hypothesis of ductal cells aspancreatic progenitors. (A) Our hypothesis is that with replication, matureduct cells regress to a less differentiated phenotype and then act as pancreaticprogenitors to form new acini, islets, and ducts. (B) Tg mice in which humanCAII promoter drives Cre or CreERTM expression were generated (CAII-Cre andCAII-CreERTM) and crossed with a Cre-mediated recombination reporter strain,R26R. In the double-Tg mice (CAII-Cre; R26R), if CAII-expressing ductal cellsserve as pancreatic progenitors after birth, we should find !-galactosidase-positive cells not only in ducts but also in islets and acini; however, if duct cellsdo not act as progenitors, only ducts should be labeled.
A
G H I J
K L
B C
D EF
Fig. 2. Characterization of transgene expression. (A–C) At birth !-galacto-sidase immunostaining (green) was seen in ducts (d) and nerve ganglion (g)but not in insulin-positive beta cells (red; A and B), glucagon-positive cells (red;C) or acini of CAII-Cre mice. (D) GFP-expressing beta cells from CAII-Cre:MIP-GFP mice FACS purified for RNA analysis. At day 0 and at 2 and 4 weeks of age,the GFP! sorted beta cells showed no band for Cre or CAII, even with 40 cyclesof amplification. Kidneys from the transgenic (Tg) and non-Tg (WT) animalswere positive and negative controls; RT " indicates controls for genomiccontamination. (E) The 2- and 4-week RNA samples shown in D were probedfor the transcription factor Pdx-1 at 28 cycles to show the integrity of RNA. (F)RNA from 10,000 pancreatic cells, either GFP! beta cells or GFP" cells from an8-week-old CAII-Cre:MIP-GFP mouse, was similarly probed for Cre at 40 cycles.!C indicates positive control. (G) Cre immunostaining found in the cytoplasmand nuclei of small ducts (here 4 weeks) in CAII-Cre mice. (H) Expression of!-galactosidase in day 0 small ducts in CAII-Cre mice. (I and J) In CAII-CreERTM
mice killed 3 weeks after the end of tamoxifen treatment, Cre and !-galac-tosidase immunostaining was found in larger ducts (I and J, adjacent sections).Cre staining here was cytoplasmic but not nuclear, and was only in some ductalcells. (K) The only other cells expressing Cre protein were ganglia (insulin,green; Cre, red). (L) The same section as shown in K with overlay of anti-BMPR1A also in green; ganglionic cells expressing BMPR1A are now yellow ifexpressing Cre or green (*) if not immunostained in previous image. (Scalebars, 50 "m in A–C, G, and I–L and 100 "m in H.)
19916 ! www.pnas.org"cgi"doi"10.1073"pnas.0805803105 Inada et al.
Carbonic anhydrase II-positive pancreatic cells areprogenitors for both endocrine and exocrinepancreas after birthAkari Inadaa,1, Cameron Nienabera, Hitoshi Katsutaa, Yoshio Fujitanib,2, Jared Levinea, Rina Moritaa, Arun Sharmaa,and Susan Bonner-Weira,3
aSection of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, One Joslin Place, Boston, MA 02215; andbDepartment of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 37232
Edited by Donald F. Steiner, University of Chicago, Chicago, IL, and approved October 20, 2008 (received for review June 17, 2008)
The regenerative process in the pancreas is of particular interestbecause diabetes results from an inadequate number of insulin-producing beta cells and pancreatic cancer may arise from theuncontrolled growth of progenitor/stem cells. Continued and sub-stantial growth of islet tissue occurs after birth in rodents andhumans, with additional compensatory growth in response toincreased demand. In rodents there is clear evidence of pancreaticregeneration after some types of injury, with proliferation ofpreexisting differentiated cell types accounting for some replace-ment. Additionally, neogenesis or the budding of new islet cellsfrom pancreatic ducts has been reported, but the existence andidentity of a progenitor cell have been debated. We hypothesizedthat the progenitor cells are duct epithelial cells that after repli-cation undergo a regression to a less differentiated state and thencan form new endocrine and exocrine pancreas. To directly testwhether ductal cells serve as pancreatic progenitors after birth andgive rise to new islets, we generated transgenic mice expressinghuman carbonic anhydrase II (CAII) promoter: Cre recombinase(Cre) or inducible CreERTM to cross with ROSA26 loxP-Stop-loxPLacZ reporter mice. We show that CAII-expressing cells within thepancreas act as progenitors that give rise to both new islets andacini normally after birth and after injury (ductal ligation). Thisidentification of a differentiated pancreatic cell type as an in vivoprogenitor of all differentiated pancreatic cell types has implica-tions for a potential expandable source for new islets for replen-ishment therapy for diabetes.
diabetes ! islets of Langerhans ! lineage tracing
Regeneration studies in mammals have focused on tissue-specific stem and progenitor cells. The regenerative process
in the pancreas is of particular interest because diabetes resultsfrom an inadequate number of insulin-producing beta cells (1)and pancreatic cancer may arise from the uncontrolled growthof progenitor/stem cells (2). Continued and substantial growth ofislet tissue occurs after birth in rodents and humans, withadditional compensatory growth in response to increased de-mand (3). In rodents there is clear evidence of pancreaticregeneration after some types of injury, with proliferation ofpreexisting differentiated cell types accounting for some replace-ment (4–7). The mechanisms thought to be responsible for betacell growth are replication of preexisting beta cells and differ-entiation from precursor cells or neogenesis, defined as islethormone-positive cells budding from ducts. For the latter,differentiated ductal cells have been hypothesized to act aspancreatic progenitor cells (3, 8). Whether adult stem cellscontribute to this replacement is unclear.
In adult mice replication is the major mechanism for expand-ing the beta cell mass in pregnancy (9), obesity/insulin resistance(10), or normal growth and aging. Using inducible RIP-CreERTM mice to label the beta cells in adult mice, Dor et al. (11)confirmed the predominance of replication in the adult mouseand concluded that neogenesis does not occur after embryonic
or early postnatal life and that solely self-duplication replenishedbeta cells. However, they neither examined the neonatal periodnor clearly defined new lobes after partial pancreatectomy (12),both of which are reported in rats to have highly active neogen-esis. Additionally, it is difficult to conclude that new isletformation does not occur by marking a small fraction of cells andattempting to show a reduction in that fraction. Recently, Xu etal. (13) reported multipotent islet progenitor cells of unknownorigin within the adult pancreas by showing the induction ofneurogenin 3 after ductal ligation, isolating these neurogenin3-positive cells and showing that they could differentiate to isletcells. Additionally, increased neogenesis is reported in adultrodents given exendin-4 (14) or betacellulin (15), overexpressingIFN-! (16) or TGF-" (17), or after partial pancreatectomy (12).The neogeneic pathway may be more important in adult humansfor compensatory expansion of beta cell mass (1, 18, 19) becauseadult human beta cells have a very low replication (20).
We hypothesize that the progenitors of these new islets weredifferentiated pancreatic ductal cells that regressed to a lessdifferentiated phenotype after replication and then functionedas progenitors (Fig. 1A) (3, 8). To test this hypothesis we took adirect approach of genetically marking ductal cells by generatingtransgenic (Tg) mice in which the human carbonic anhydrase II(CAII) promoter drives expression of Cre recombinase (CAII-Cre) or tamoxifen-inducible CAII-CreERTM (Fig. 1B). CAII hasbeen used previously as a marker of differentiated ductal cells(21), which are distinct from the embryonic pancreatic progen-itors. Starting late in gestation (embryonic day 18.5), CAIIprotein is expressed throughout the ductal tree in adult mice butnot in the beta cells (22). The human promoter was chosen toprovide specificity for tracing the progeny of ductal cells becausein human pancreas CAII is expressed only throughout the ductaltree (23), whereas in rodents glucagon-expressing alpha cells alsoexpress CAII (22). Thus, in these transgenic mice, if the trans-gene expression is faithful to that of the promoter, Cre recom-binase should be expressed only in the ducts and not the islets.Here, we show that the CAII-expressing cells act as pancreatic
Author contributions: A.I., Y.F., A.S., and S.B.-W. designed research; A.I., C.N., H.K., J.L.,R.M., and S.B.-W. performed research; A.I. and S.B.-W. analyzed data; and A.I., A.S., andS.B.-W. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1Present address: SSP Stem Cell Unit, Graduate School of Medicine, Kyushu University,Fukuoka, Japan.
2Present address: Center for Therapeutic Innovations in Diabetes, Department of Medicine,Metabolism and Endocrinology, Juntendo University Graduate School of Medicine, Tokyo,Japan.
3To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0805803105/DCSupplemental.
© 2008 by The National Academy of Sciences of the USA
www.pnas.org"cgi"doi"10.1073"pnas.0805803105 PNAS ! December 16, 2008 ! vol. 105 ! no. 50 ! 19915–19919
MED
ICA
LSC
IEN
CES
Carbonic anhydrase II-positive pancreatic cells areprogenitors for both endocrine and exocrinepancreas after birthAkari Inadaa,1, Cameron Nienabera, Hitoshi Katsutaa, Yoshio Fujitanib,2, Jared Levinea, Rina Moritaa, Arun Sharmaa,and Susan Bonner-Weira,3
aSection of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, One Joslin Place, Boston, MA 02215; andbDepartment of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 37232
Edited by Donald F. Steiner, University of Chicago, Chicago, IL, and approved October 20, 2008 (received for review June 17, 2008)
The regenerative process in the pancreas is of particular interestbecause diabetes results from an inadequate number of insulin-producing beta cells and pancreatic cancer may arise from theuncontrolled growth of progenitor/stem cells. Continued and sub-stantial growth of islet tissue occurs after birth in rodents andhumans, with additional compensatory growth in response toincreased demand. In rodents there is clear evidence of pancreaticregeneration after some types of injury, with proliferation ofpreexisting differentiated cell types accounting for some replace-ment. Additionally, neogenesis or the budding of new islet cellsfrom pancreatic ducts has been reported, but the existence andidentity of a progenitor cell have been debated. We hypothesizedthat the progenitor cells are duct epithelial cells that after repli-cation undergo a regression to a less differentiated state and thencan form new endocrine and exocrine pancreas. To directly testwhether ductal cells serve as pancreatic progenitors after birth andgive rise to new islets, we generated transgenic mice expressinghuman carbonic anhydrase II (CAII) promoter: Cre recombinase(Cre) or inducible CreERTM to cross with ROSA26 loxP-Stop-loxPLacZ reporter mice. We show that CAII-expressing cells within thepancreas act as progenitors that give rise to both new islets andacini normally after birth and after injury (ductal ligation). Thisidentification of a differentiated pancreatic cell type as an in vivoprogenitor of all differentiated pancreatic cell types has implica-tions for a potential expandable source for new islets for replen-ishment therapy for diabetes.
diabetes ! islets of Langerhans ! lineage tracing
Regeneration studies in mammals have focused on tissue-specific stem and progenitor cells. The regenerative process
in the pancreas is of particular interest because diabetes resultsfrom an inadequate number of insulin-producing beta cells (1)and pancreatic cancer may arise from the uncontrolled growthof progenitor/stem cells (2). Continued and substantial growth ofislet tissue occurs after birth in rodents and humans, withadditional compensatory growth in response to increased de-mand (3). In rodents there is clear evidence of pancreaticregeneration after some types of injury, with proliferation ofpreexisting differentiated cell types accounting for some replace-ment (4–7). The mechanisms thought to be responsible for betacell growth are replication of preexisting beta cells and differ-entiation from precursor cells or neogenesis, defined as islethormone-positive cells budding from ducts. For the latter,differentiated ductal cells have been hypothesized to act aspancreatic progenitor cells (3, 8). Whether adult stem cellscontribute to this replacement is unclear.
In adult mice replication is the major mechanism for expand-ing the beta cell mass in pregnancy (9), obesity/insulin resistance(10), or normal growth and aging. Using inducible RIP-CreERTM mice to label the beta cells in adult mice, Dor et al. (11)confirmed the predominance of replication in the adult mouseand concluded that neogenesis does not occur after embryonic
or early postnatal life and that solely self-duplication replenishedbeta cells. However, they neither examined the neonatal periodnor clearly defined new lobes after partial pancreatectomy (12),both of which are reported in rats to have highly active neogen-esis. Additionally, it is difficult to conclude that new isletformation does not occur by marking a small fraction of cells andattempting to show a reduction in that fraction. Recently, Xu etal. (13) reported multipotent islet progenitor cells of unknownorigin within the adult pancreas by showing the induction ofneurogenin 3 after ductal ligation, isolating these neurogenin3-positive cells and showing that they could differentiate to isletcells. Additionally, increased neogenesis is reported in adultrodents given exendin-4 (14) or betacellulin (15), overexpressingIFN-! (16) or TGF-" (17), or after partial pancreatectomy (12).The neogeneic pathway may be more important in adult humansfor compensatory expansion of beta cell mass (1, 18, 19) becauseadult human beta cells have a very low replication (20).
We hypothesize that the progenitors of these new islets weredifferentiated pancreatic ductal cells that regressed to a lessdifferentiated phenotype after replication and then functionedas progenitors (Fig. 1A) (3, 8). To test this hypothesis we took adirect approach of genetically marking ductal cells by generatingtransgenic (Tg) mice in which the human carbonic anhydrase II(CAII) promoter drives expression of Cre recombinase (CAII-Cre) or tamoxifen-inducible CAII-CreERTM (Fig. 1B). CAII hasbeen used previously as a marker of differentiated ductal cells(21), which are distinct from the embryonic pancreatic progen-itors. Starting late in gestation (embryonic day 18.5), CAIIprotein is expressed throughout the ductal tree in adult mice butnot in the beta cells (22). The human promoter was chosen toprovide specificity for tracing the progeny of ductal cells becausein human pancreas CAII is expressed only throughout the ductaltree (23), whereas in rodents glucagon-expressing alpha cells alsoexpress CAII (22). Thus, in these transgenic mice, if the trans-gene expression is faithful to that of the promoter, Cre recom-binase should be expressed only in the ducts and not the islets.Here, we show that the CAII-expressing cells act as pancreatic
Author contributions: A.I., Y.F., A.S., and S.B.-W. designed research; A.I., C.N., H.K., J.L.,R.M., and S.B.-W. performed research; A.I. and S.B.-W. analyzed data; and A.I., A.S., andS.B.-W. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1Present address: SSP Stem Cell Unit, Graduate School of Medicine, Kyushu University,Fukuoka, Japan.
2Present address: Center for Therapeutic Innovations in Diabetes, Department of Medicine,Metabolism and Endocrinology, Juntendo University Graduate School of Medicine, Tokyo,Japan.
3To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0805803105/DCSupplemental.
© 2008 by The National Academy of Sciences of the USA
www.pnas.org"cgi"doi"10.1073"pnas.0805803105 PNAS ! December 16, 2008 ! vol. 105 ! no. 50 ! 19915–19919
MED
ICA
LSC
IEN
CES

DTZ-staining
Pellet
Ficoll-gradient
Pancreas
(Digestion)
Preculture in 10% FCS(Removal of fibroblasts)
Floating cells
Culture in 0.5% FCS with EGF
(Removal of β-cells)
DTZ (-) cells
A
400
600
800
Cel
lula
r ins
ulin
con
tent
(ng/
mg
prot
ein)
Monolayer Suspension0
200
D
Insulin
B
C
Acinar cells
activation of these promoters has been established (31–33) andalso confirmed by us (see Fig. 6, which is published as supportinginformation on the PNAS web site). Immediately after enzy-matic dissociation, pancreatic exocrine cells were infected withthe adenoviruses at a multiplicity of infection of 100 for 6 h.Thereafter, the cells were cultured for 4 days in suspension asdescribed.
Cell Analyses. After culture, the cells were collected and subjectedto analyses for immunocytochemistry, immunoblotting, RT-PCR (see Table 1, which is published as supporting informationon the PNAS web site), and insulin secretion (see SupportingText).
ResultsGeneration of Insulin-Producing Cells from Exocrine Pancreas. Theprotocol for induction of insulin-producing cells from exocrinepancreas is outlined in Fig. 7, which is published as supportinginformation on the PNAS web site. By Ficoll gradient centrif-ugation, the pancreatic acinar cell-enriched exocrine fractionwas recovered as a pellet. Although the pellet contained only asmall amount of native pancreatic ! cells (!0.5% of total cellsrecovered), we further removed native ! cells by staining thepancreatic islets and their fragments with dithizone (DTZ) (34,35), followed by handpicking under a dissecting microscope.After this procedure, the frequency of insulin-positive cells was!0.01%, as assessed by immunostaining and quantitative real-time RT-PCR for insulin genes. Almost no insulin response tohigh glucose, high KCl, or the sulfonylurea glibenclamide wasdetectable in the DTZ (") fraction (data not shown).
We then cultured the acinar cell-enriched DTZ (") fractionat a low concentration (0.5%) of FCS and 20 ng!ml EGF. Underthese conditions, the cells readily formed aggregates and becamesmooth spheroids. The cells began to adhere within 3 days andformed small monolayer colonies between days 5 and 7. Wefound that a subset of the cells in these colonies expressed insulinon day 7 (Fig. 7). However, insulin secretion was undetectableunder these culture conditions.
In the course of the study, we found that suspension cultureusing dishes treated with 2-methacryloyloxyethyl phosphoryl-choline (MPC), which interferes with cell attachment (36), incombination with 10 mM nicotinamide increases the cellularinsulin content (Fig. 7). Nicotinamide is known to prevent ! celldamage due to activation of poly(ADP-ribose) synthethase!polymerase (37) and induce endocrine differentiation in cul-tured human fetal pancreatic islet cells and an increase in insulingene expression (38). When cultured on MPC dishes, the cells
did not adhere to the surface of the dish, and smooth spheroidswere maintained throughout the experiments. Quantitative anal-ysis by real-time RT-PCR showed that the expression of theinsulin genes was gradually increased by the culture (Fig. 1A).The frequency of insulin-positive cells was also increased (Fig.1B), to !5% of total cells on day 4. The insulin-positive cells alsoexpressed C-peptide (Fig. 1C), indicating insulin biosynthesis inthese cells. Cells with proliferating ability, assessed by cellproliferating marker Ki67-staining, were rare (#1% of totalcells) in the culture. Insulin!Ki67 double-positive cells were notfound (Fig. 8, which is published as supporting information onthe PNAS web site).
Because insulin content in native mouse islets is 252 $ 8"g!mg protein, that in the exocrine pancreas-derived cells(0.617 $ 0.083 "g!mg protein, including both insulin-positiveand -negative cells) is 1!400th that of native pancreatic islets.Because the frequency of insulin-positive cells is !5% of totalcells in the culture (Fig. 1B), the insulin content of the newlymade exocrine pancreas-derived insulin-positive cells is !1!20ththat of a native ! cell.
Fig. 1. Generation of insulin-producing cells from adult mouse exocrinepancreas. (A) Quantitative real-time RT-PCR analysis for insulin genes. Insulinexpression in the culture was gradually increased. (B) Immunostaining ofexocrine pancreas-derived cells. Insulin-positive cells are increased by theculture. (Scale bars, 50 "m.) (C) Double immunostaining of insulin and C-peptide. The insulin-positive cells are also positive for C-peptide. (Scale bar,20 "m.)
Fig. 2. Cell lineage tracing by the Cre!loxP-based system. (A) The scheme ofpancreatic acinar cell specific cell marking. In cells from the R26R-ECFP mouse,expression of the fluorescent protein (ECFP) is activated through the action ofCre recombinase to remove a transcriptional ‘‘stop’’ sequence. When amylase!elastase-expressing acinar cells are infected with adenovirus expressing Crerecombinase under control of either amylase or elastase promoter, the cellsare labeled permanently with ECFP. (B–D) Lineage tracing of labeled acinarcells. Pancreatic acinar cells from R26R-ECFP were labeled by infection ofAd-pAmy-Cre at !50% efficiency (B). Because fluorescence of ECFP is dimin-ished after fixation, ECFP expression was detected by using anti-GFP antibody.Cells positive for insulin (arrowheads), ECFP (arrows), and both insulin andECFP (asterisks) are observed (B). Photographs of higher magnification ofinsulin!ECFP double-positive cells are shown (C). CK!ECFP double-positivecells are also observed (D). (Scale bars, 20 "m.)
Minami et al. PNAS " October 18, 2005 " vol. 102 " no. 42 " 15117
CELL
BIO
LOG
Y
Kohtaro Minami*, Masaaki Okuno*, Kazumasa Miyawaki*, Akinori Okumachi*, Katsuhiko Ishizaki†, Kazunobu Oyama†, Miho Kawaguchi†, Nobuko Ishizuka‡, Toshihiko Iwanaga§, and Susumu Seino*†¶
Lineage tracing and characterization ofinsulin-secreting cells generated from adultpancreatic acinar cellsKohtaro Minami*, Masaaki Okuno*, Kazumasa Miyawaki*, Akinori Okumachi*, Katsuhiko Ishizaki†, Kazunobu Oyama†,Miho Kawaguchi†, Nobuko Ishizuka‡, Toshihiko Iwanaga§, and Susumu Seino*†¶
*Department of Experimental Therapeutics, Translational Research Center, Kyoto University Hospital, Kyoto 606-8507, Japan; †Division of Cellularand Molecular Medicine, Kobe University Graduate School of Medicine, Kobe 650-0017, Japan; ‡Department of Diabetes and Clinical Nutrition,Kyoto University Graduate School of Medicine, Kyoto 606-8507, Japan; and §Laboratory of Anatomy, Graduate School of Medicine, HokkaidoUniversity, Sapporo 060-8638, Japan
Communicated by Donald F. Steiner, University of Chicago, Chicago, IL, August 31, 2005 (received for review June 27, 2005)
Although several studies have suggested that insulin-secretingcells can be generated in vitro from cells residing in adult exocrinepancreas, neither the origin of these cells nor their precise insulinsecretory properties was obtained. We show here that insulin-secreting cells can be derived from adult mouse pancreatic exocrinecells by suspension culture in the presence of EGF and nicotin-amide. The frequency of insulin-positive cells was only 0.01% in theinitial preparation and increased to !5% in the culture conditions.Analysis by the Cre!loxP-based direct cell lineage tracing systemindicates that these newly made cells originate from amylase!elastase-expressing pancreatic acinar cells. Insulin secretion is stim-ulated by glucose, sulfonylurea, and carbachol, and potentiationby glucagon-like peptide-1 also occurs. Insulin-containing secre-tory granules are present in these cells. In addition, we found thatthe enzymatic dissociation of pancreatic acini itself leads to acti-vation of EGF signaling, and that inhibition of EGF receptor kinaseblocks the transdifferentiation. These data demonstrate that pan-creatic acinar cells can transdifferentiate into insulin-secreting cellswith secretory properties similar to those of native pancreatic !cells, and that activation of EGF signaling is required in suchtransdifferentiation.
Cre!loxP " diabetes " EGF " transdifferentiation
Pancreatic ! cells play an indispensable role in the mainte-nance of systemic glucose homeostasis, and ! cell turnover
slowly occurs throughout the lifetime of a mammal (1). Althoughit has been reported that replication is the major mechanism ofthe generation of new ! cells in adult mice (2, 3), various studieshave shown that ! cells also are generated by neogenesis (4–10),suggesting stem!progenitor cells in the adult pancreas yet to beidentified. On the other hand, many attempts have been maderecently to generate new insulin-producing ! cells in vitro fromnon-! cells (11–15). Pancreatic ! cells are characterized by wellregulated insulin secretion in response to various stimuli re-quired in the maintenance of blood glucose levels within anarrow physiological range. Although glucose responsiveness(16), metabolism-electrical activity coupling (17), and regulatedexocytosis of insulin granules (18) are the primary features of the! cell, they have been overlooked or underemphasized in studiesof in vitro-generated insulin-producing cells (15, 19).
Recent studies have revealed unexpected high plasticity ofadult cells in their differentiation capacity (20, 21). For example,differentiated cells can convert into cells of a different pheno-type by transdifferentiation (22–24). Several lines of evidencesuggest that pancreatic acinar and duct cells can transdifferen-tiate into endocrine cells in vivo (5–10). In certain experimentalmodels and pathological conditions, pancreatic lesion leads totransformation of acinar cells into duct-like structures (acino-ductal metaplasia), followed by islet neogenesis (5–10). Previousstudies (10, 25) have shown that pancreatic acinar cells can
transdifferentiate in vitro into cells with a ductal phenotype thatexpress Pdx1, a transcription factor critical in pancreatic devel-opment and insulin gene expression, but those transdifferenti-ated cells failed to produce insulin. Very recently, studies (26, 27)have suggested that rat pancreatic acinar cells can transdiffer-entiate into insulin-producing cells in vitro, but direct evidenceof the origin of the cells was not reported. Lineage tracing by theCre-loxP recombination system has been used successfully todetermine cell fate by following the progeny of labeled cellsthrough the differentiation process (2, 28, 29).
In the present study, we provide direct evidence that insulin-secreting cells can be generated in vitro from pancreatic acinarcells of adult mouse, using the Cre!loxP-based cell lineagetracing system. The newly made cells are equipped with theapparatus of glucose-induced insulin secretion and its potenti-ation, the principal mechanisms of insulin secretion. Further-more, EGF signaling is essential for this transdifferentiation.These data clearly show that pancreatic acinar cells possesssufficient plasticity to transdifferentiate into pancreatic endo-crine cells in vitro.
Materials and MethodsCulture of Isolated Pancreatic Exocrine Cells. Isolation and purifi-cation of pancreatic exocrine cells from male C57BL!6 mice aredescribed in Supporting Text, which is published as supportinginformation on the PNAS web site. Purified acinar-enrichedexocrine cells were cultured in RPMI medium 1640 (Sigma) with10% FCS. After 6–8 h of culture, f loating cells were collectedand replated on dishes treated with 2-methacryloyloxyethylphosphorylcholine (Nalge Nunc) in RPMI medium 1640 sup-plemented with 0.5% FCS!20 ng/ml EGF (R & D Systems)!10mM nicotinamide. The culture medium was not renewedthroughout the experiment. All animal experiments were ap-proved by the animal research committees of the Kyoto Uni-versity Graduate School of Medicine and the Kobe UniversityGraduate School of Medicine.
Adenoviral Infection. ROSA26 reporter mice, in which enhancedcyan fluorescent protein (ECFP) expression is activated uponremoval of the loxP site-f lanked stop cassette by Cre-recombinase (R26R-ECFP) (30), were provided by F. Costantini(Columbia University, New York). Cre-recombinase expressingadenoviruses under control of mouse amylase-2 promoter (31)and rat elastase-1 promoter (32) were generated by using anadenovirus expression vector kit (Takara Bio, Tokyo), accordingto the manufacturer’s instructions. Pancreatic acinar cell-specific
Abbreviations: ECFP, enhanced cyan fluorescent protein; EGFR, EGF receptor.¶To whom correspondence should be addressed. E-mail: [email protected].
© 2005 by The National Academy of Sciences of the USA
15116–15121 " PNAS " October 18, 2005 " vol. 102 " no. 42 www.pnas.org!cgi!doi!10.1073!pnas.0507567102

Liver cells
Animal models used for repopulation studies are sum-marized in Table 2. Each model shares a common theme:transplanted cells have a strong selective advantage overendogenous host hepatocytes. In many models, this selec-tion occurs because of genetic differences between the trans-planted and endogenous cells (transgene/knockout). Inhi-bition of endogenous hepatocyte proliferation with DNA-damaging agents, including chemicals and !-irradiation,has also been used. Targeted liver irradiation (ie, not totalbody irradiation) has been shown to be particularly effectivein animal models and might eventually be used in a clinicalsetting.99 Recently, it has been shown that mouse liver canbe repopulated with human hepatocytes.100–102 Immune-deficient mice have been crossed with strains that providean environment that selects for transplanted cells, such astransgenic mice that express albumin–urokinase plasmino-gen activator 101 or are deficient for a liver-specific enzyme,fumarylacetoacetate hydrolase (Fah), involved in tyrosine catab-olism.102 Some chemical exposure regimens can also beused to select for transplanted hepatocytes.103 These modelswill serve as in vivo assays for the development of humanhepatocyte progenitors.
In vivo models have been used to determine the iden-tity of transplantable liver-repopulating cells; it wasshown that these cells could be serially transplanted for!100 cell doublings without loss of functionality.104
Interestingly, hepatocytes were the only donor-derivedcells that were found to have liver repopulation capacity.No biliary epithelium or other cell types of donor originwere found to have this ability, indicating that the hepa-tocyte itself might be a “unipotential” stem cell.
Hepatocytes as Liver-Repopulating CellsLiver repopulation is unique among organ sys-
tems. In contrast to the hematopoietic system, wheremature adult blood cells have minimal proliferative ca-
pacity, differentiated hepatocytes themselves have highcapacity for liver repopulation. First, mature hepatocytes(identified by size fractionation, retroviral marking, orserial transplantation) were transplanted into livers ofFah knockout mice.105 Large, binucleated hepatocytesthat represented "70% of the hepatocyte populationwere found to mediate most of the liver repopulation.Secondly, fusion-derived hepatocytes, which by definitionare at least tetraploid, that were serially transplanted werecapable of at least 30 cell divisions without loss of func-tion.106 Finally, fluorescence-activated cell sorting–iso-lated polyploid hepatocytes proliferated extensively aftertransplantation into livers of urokinase plasminogen ac-tivator transgenic mice.107 Together, these experimentsshow that mature, polyploid adult hepatocytes have ahigh degree of replicative potential. It is important tonote that the nature of the human cells that are capableof liver repopulation102 has not been definitively deter-mined. However, in human patients with hereditary ty-rosinemia, the large size of clonal revertant nodules (ie,healthy liver tissue derived from spontaneous hepatocytemutations that correct the underlying genetic defect)indicates that mature human hepatocytes also have ex-tensive regenerative potential.108
Some studies have indicated that hepatocytes are bipo-tential. In vitro and in vivo transplantation experimentshave shown hepatocyte differentiation into ductular epi-thelium.109 These results are provocative, suggesting thatthere are no stem cells in the liver at all, but that allregenerative responses, including the oval cell response,rely on hepatocytes. Taken together, the data stronglysuggest that fully differentiated hepatocytes, which con-stitute the majority of liver cells, are efficient in liverrepopulation and have stem cell–like capacity for celldivision.
Figure 4. Cells with adult liver-repopulating potential. (A) Unipo-tential liver-repopulating cells.Hepatocytes and bile duct epithe-lial cells regenerate during normaltissue turnover. Under defined ex-perimental conditions, pancreaticprogenitor cells and MSCs differen-tiate into hepatocytes, and HSC-derived myelomonocytic cells fusewith hepatocytes. (B) Bipotentialliver-repopulating cells. Intrahe-patic liver stem cells and/or ovalcells differentiate into hepatocytesand bile duct epithelial cells (ie,oval cell response). Fetal hepato-blasts, derived from pluripotentESCs,alsodifferentiate intohepato-cytes and bile duct epithelial cells intransplantation experiments.
REVIEW
SIN
BASIC
AN
DCLIN
ICAL
GA
STROEN
TEROLO
GY
474 DUNCAN ET AL GASTROENTEROLOGY Vol. 137, No. 2
REVIEWS IN BASIC AND CLINICALGASTROENTEROLOGY
John P. Lynch and David C. Metz, Section Editors
Stem Cells and Liver Regeneration
ANDREW W. DUNCAN,* CRAIG DORRELL,* and MARKUS GROMPE*,‡
*Oregon Stem Cell Center, Oregon Health & Science University, Portland; and ‡Papé Family Research Institute, Department of Pediatrics, Oregon Health & ScienceUniversity, Portland, Oregon
One of the defining features of the liver is the capacityto maintain a constant size despite injury. Althoughthe precise molecular signals involved in the mainte-nance of liver size are not completely known, it isclear that the liver delicately balances regenerationwith overgrowth. Mammals, for example, can survivesurgical removal of up to 75% of the total liver mass.Within 1 week after liver resection, the total numberof liver cells is restored. Moreover, liver overgrowthcan be induced by a variety of signals, including he-patocyte growth factor or peroxisome proliferators;the liver quickly returns to its normal size when theproliferative signal is removed. The extent to whichliver stem cells mediate liver regeneration has beenhotly debated. One of the primary reasons for thiscontroversy is the use of multiple definitions for thehepatic stem cell. Definitions for the liver stem cellinclude the following: (1) cells responsible for normaltissue turnover, (2) cells that give rise to regenerationafter partial hepatectomy, (3) cells responsible forprogenitor-dependent regeneration, (4) cells that pro-duce hepatocyte and bile duct epithelial phenotypesin vitro, and (5) transplantable liver-repopulatingcells. This review will consider liver stem cells in thecontext of each definition.
The adult mammalian liver is composed of diverse celltypes that arise from various embryologic origins. In
this review, the discussion of the “liver stem cell” or“hepatic stem cell” focuses on precursors of 2 liver epi-thelial cell types: hepatocytes and bile duct epithelialcells.
Organization and Functions of AdultMammalian LiverAn appreciation of liver architecture is essential to
the understanding of hepatic stem cell biology. An exten-sive description of liver organization/function is foundelsewhere.1 Briefly, the primary functional unit of theliver is the hepatic lobule (Figure 1A and B).2 Locatedalong the lobule perimeter, the portal triad consists of a
small portal vein, hepatic artery, and bile duct. Bloodenters the liver from the portal vein and hepatic artery,and it flows through liver sinusoids toward the centralvein. Rows of hepatocytes form a hepatic plate. Thebasolateral hepatocyte surface is lined with fenestratedendothelium, unique among capillary beds, forming si-nusoidal vessels. These vessels facilitate interactions be-tween blood and the hepatocyte cell surface.3 The apicalface of adjacent hepatocytes forms a bile canaliculus. Bileis secreted by hepatocytes into the bile canaliculus andthen drains toward bile ducts, which are lined by ductepithelial cells. Bile canaliculi connect with bile ducts viathe Canal of Hering, which is also believed to be a nichefor liver progenitor cells (see following text).
The liver is responsible for the synthesis of serumproteins; intermediary metabolism of amino acids, lipids,and carbohydrates; and detoxification of xenobiotic com-pounds. These functions are performed primarily byhepatocytes. Hepatocyte function (eg, gene expressionprofile and biochemical activities), however, is not iden-tical among all hepatocytes. Rather, hepatocytes performdifferent roles depending on their physical locationwithin the hepatic lobule. “Metabolic zonation” refers tothe differential properties of periportal (adjacent to theportal triad) and pericentral (adjacent to the central vein)hepatocytes.4 Periportal hepatocytes, for example, expressurea cycle enzymes and convert ammonia to urea.5,6 Incontrast, pericentral hepatocytes express glutamine syn-thase and utilize ammonia to generate glutamine.5
Abbreviations used in this paper: AFP, !-fetoprotein; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; ECFC, epithelial colony-formingcell; ESC, embryonic stem cell; HSC, hematopoietic stem cell; iPS,induced pluripotent stem cell; MSC, mesenchymal stem cell; TWEAK,tumor necrosis factor–like weak inducer of apoptosis.
© 2009 by the AGA Institute0016-5085/09/$36.00
doi:10.1053/j.gastro.2009.05.044
REV
IEWS
INBA
SICA
ND
CLINICA
LG
ASTR
OEN
TERO
LOG
Y
GASTROENTEROLOGY 2009;137:466–481
REVIEWS IN BASIC AND CLINICALGASTROENTEROLOGY
John P. Lynch and David C. Metz, Section Editors
Stem Cells and Liver Regeneration
ANDREW W. DUNCAN,* CRAIG DORRELL,* and MARKUS GROMPE*,‡
*Oregon Stem Cell Center, Oregon Health & Science University, Portland; and ‡Papé Family Research Institute, Department of Pediatrics, Oregon Health & ScienceUniversity, Portland, Oregon
One of the defining features of the liver is the capacityto maintain a constant size despite injury. Althoughthe precise molecular signals involved in the mainte-nance of liver size are not completely known, it isclear that the liver delicately balances regenerationwith overgrowth. Mammals, for example, can survivesurgical removal of up to 75% of the total liver mass.Within 1 week after liver resection, the total numberof liver cells is restored. Moreover, liver overgrowthcan be induced by a variety of signals, including he-patocyte growth factor or peroxisome proliferators;the liver quickly returns to its normal size when theproliferative signal is removed. The extent to whichliver stem cells mediate liver regeneration has beenhotly debated. One of the primary reasons for thiscontroversy is the use of multiple definitions for thehepatic stem cell. Definitions for the liver stem cellinclude the following: (1) cells responsible for normaltissue turnover, (2) cells that give rise to regenerationafter partial hepatectomy, (3) cells responsible forprogenitor-dependent regeneration, (4) cells that pro-duce hepatocyte and bile duct epithelial phenotypesin vitro, and (5) transplantable liver-repopulatingcells. This review will consider liver stem cells in thecontext of each definition.
The adult mammalian liver is composed of diverse celltypes that arise from various embryologic origins. In
this review, the discussion of the “liver stem cell” or“hepatic stem cell” focuses on precursors of 2 liver epi-thelial cell types: hepatocytes and bile duct epithelialcells.
Organization and Functions of AdultMammalian LiverAn appreciation of liver architecture is essential to
the understanding of hepatic stem cell biology. An exten-sive description of liver organization/function is foundelsewhere.1 Briefly, the primary functional unit of theliver is the hepatic lobule (Figure 1A and B).2 Locatedalong the lobule perimeter, the portal triad consists of a
small portal vein, hepatic artery, and bile duct. Bloodenters the liver from the portal vein and hepatic artery,and it flows through liver sinusoids toward the centralvein. Rows of hepatocytes form a hepatic plate. Thebasolateral hepatocyte surface is lined with fenestratedendothelium, unique among capillary beds, forming si-nusoidal vessels. These vessels facilitate interactions be-tween blood and the hepatocyte cell surface.3 The apicalface of adjacent hepatocytes forms a bile canaliculus. Bileis secreted by hepatocytes into the bile canaliculus andthen drains toward bile ducts, which are lined by ductepithelial cells. Bile canaliculi connect with bile ducts viathe Canal of Hering, which is also believed to be a nichefor liver progenitor cells (see following text).
The liver is responsible for the synthesis of serumproteins; intermediary metabolism of amino acids, lipids,and carbohydrates; and detoxification of xenobiotic com-pounds. These functions are performed primarily byhepatocytes. Hepatocyte function (eg, gene expressionprofile and biochemical activities), however, is not iden-tical among all hepatocytes. Rather, hepatocytes performdifferent roles depending on their physical locationwithin the hepatic lobule. “Metabolic zonation” refers tothe differential properties of periportal (adjacent to theportal triad) and pericentral (adjacent to the central vein)hepatocytes.4 Periportal hepatocytes, for example, expressurea cycle enzymes and convert ammonia to urea.5,6 Incontrast, pericentral hepatocytes express glutamine syn-thase and utilize ammonia to generate glutamine.5
Abbreviations used in this paper: AFP, !-fetoprotein; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; ECFC, epithelial colony-formingcell; ESC, embryonic stem cell; HSC, hematopoietic stem cell; iPS,induced pluripotent stem cell; MSC, mesenchymal stem cell; TWEAK,tumor necrosis factor–like weak inducer of apoptosis.
© 2009 by the AGA Institute0016-5085/09/$36.00
doi:10.1053/j.gastro.2009.05.044
REVIEW
SIN
BASIC
AN
DCLIN
ICAL
GA
STROEN
TEROLO
GY
GASTROENTEROLOGY 2009;137:466–481
RE
VIE
WS
INB
AS
ICA
ND
CLI
NIC
AL
GA
STR
OE
NTE
RO
LOG
YJo
hnP
. Lyn
chan
dD
avid
C. M
etz,
Sec
tion
Edito
rs
Ste
mC
ells
and
Live
rR
egen
erat
ion
AN
DR
EWW
.D
UN
CA
N,*
CR
AIG
DO
RR
ELL,
*an
dM
AR
KU
SG
RO
MP
E*,‡
*Ore
gon
Ste
mC
ell C
ente
r,O
rego
nH
ealth
&S
cien
ceU
nive
rsity
, Por
tland
; and
‡P
apé
Fam
ilyR
esea
rch
Inst
itute
, Dep
artm
ent o
f Ped
iatr
ics,
Ore
gon
Hea
lth&
Sci
ence
Uni
vers
ity, P
ortla
nd, O
rego
n
On
eof
the
defi
nin
gfe
atur
esof
the
live
ris
the
capa
city
tom
ain
tain
aco
nst
ant
size
desp
ite
inju
ry.
Alt
hou
ghth
epr
ecis
em
olec
ular
sign
als
invo
lved
inth
em
ain
te-
nan
ceof
live
rsi
zear
en
otco
mpl
etel
ykn
own
,it
iscl
ear
that
the
live
rde
lica
tely
bala
nce
sre
gen
erat
ion
wit
hov
ergr
owth
. Mam
mal
s,fo
rex
ampl
e,ca
nsu
rviv
esu
rgic
alre
mov
alof
upto
75%
ofth
eto
tal
live
rm
ass.
Wit
hin
1w
eek
afte
rli
ver
rese
ctio
n, t
he
tota
ln
umbe
rof
live
rce
lls
isre
stor
ed.
Mor
eove
r,li
ver
over
grow
thca
nbe
indu
ced
bya
vari
ety
ofsi
gnal
s,in
clud
ing
he-
pato
cyte
grow
thfa
ctor
orpe
roxi
som
epr
olif
erat
ors;
the
live
rqu
ickl
yre
turn
sto
its
nor
mal
size
wh
enth
epr
olif
erat
ive
sign
alis
rem
oved
.T
he
exte
nt
tow
hic
hli
ver
stem
cell
sm
edia
teli
ver
rege
ner
atio
nh
asbe
enh
otly
deba
ted.
On
eof
the
prim
ary
reas
ons
for
this
con
trov
ersy
isth
eus
eof
mul
tipl
ede
fin
itio
ns
for
the
hep
atic
stem
cell
.D
efin
itio
ns
for
the
live
rst
emce
llin
clud
eth
efo
llow
ing:
(1) c
ells
resp
onsi
ble
for
nor
mal
tiss
uetu
rnov
er, (
2)ce
lls
that
give
rise
tore
gen
erat
ion
afte
rpa
rtia
lh
epat
ecto
my,
(3)
cell
sre
spon
sibl
efo
rpr
ogen
itor
-dep
ende
nt r
egen
erat
ion
, (4)
cell
sth
atpr
o-du
ceh
epat
ocyt
ean
dbi
ledu
ctep
ith
elia
lph
enot
ypes
invi
tro,
and
(5)
tran
spla
nta
ble
live
r-re
popu
lati
ng
cell
s.T
his
revi
eww
ill
con
side
rli
ver
stem
cell
sin
the
con
text
ofea
chde
fin
itio
n.
The
adul
t mam
mal
ian
liver
isco
mpo
sed
ofdi
vers
ece
llty
pes
that
aris
efr
omva
riou
sem
bryo
logi
cor
igin
s.In
this
revi
ew,
the
disc
ussi
onof
the
“liv
erst
emce
ll”or
“hep
atic
stem
cell”
focu
ses
onpr
ecur
sors
of2
liver
epi-
thel
ial
cell
type
s:he
pato
cyte
san
dbi
ledu
ctep
ithe
lial
cells
.
Org
aniz
atio
nan
dFu
nctio
nsof
Adu
ltM
amm
alia
nLi
ver
An
appr
ecia
tion
ofliv
erar
chit
ectu
reis
esse
ntia
l to
the
unde
rsta
ndin
gof
hepa
tic
stem
cell
biol
ogy.
An
exte
n-si
vede
scri
ptio
nof
liver
orga
niza
tion
/fun
ctio
nis
foun
del
sew
here
.1B
riefl
y,th
epr
imar
yfu
ncti
onal
unit
ofth
eliv
eris
the
hepa
tic
lobu
le(F
igur
e1A
and
B).2
Loca
ted
alon
gth
elo
bule
peri
met
er, t
hepo
rtal
tria
dco
nsis
tsof
a
smal
lpo
rtal
vein
,he
pati
car
tery
,an
dbi
ledu
ct.
Blo
oden
ters
the
liver
from
the
port
alve
inan
dhe
pati
car
tery
,an
dit
flow
sth
roug
hliv
ersi
nuso
ids
tow
ard
the
cent
ral
vein
.R
ows
ofhe
pato
cyte
sfo
rma
hepa
tic
plat
e.T
heba
sola
tera
lhe
pato
cyte
surf
ace
islin
edw
ith
fene
stra
ted
endo
thel
ium
,un
ique
amon
gca
pilla
rybe
ds,
form
ing
si-
nuso
idal
vess
els.
The
seve
ssel
sfa
cilit
ate
inte
ract
ions
be-
twee
nbl
ood
and
the
hepa
tocy
tece
llsu
rfac
e.3
The
apic
alfa
ceof
adja
cent
hepa
tocy
tes
form
sa
bile
cana
licul
us. B
ileis
secr
eted
byhe
pato
cyte
sin
toth
ebi
leca
nalic
ulus
and
then
drai
nsto
war
dbi
ledu
cts,
whi
char
elin
edby
duct
epit
helia
l cel
ls. B
ileca
nalic
uli c
onne
ctw
ith
bile
duct
svi
ath
eC
anal
ofH
erin
g,w
hich
isal
sobe
lieve
dto
bea
nich
efo
rliv
erpr
ogen
itor
cells
(see
follo
win
gte
xt).
The
liver
isre
spon
sibl
efo
rth
esy
nthe
sis
ofse
rum
prot
eins
; int
erm
edia
rym
etab
olis
mof
amin
oac
ids,
lipid
s,an
dca
rboh
ydra
tes;
and
deto
xific
atio
nof
xeno
biot
icco
m-
poun
ds.
The
sefu
ncti
ons
are
perf
orm
edpr
imar
ilyby
hepa
tocy
tes.
Hep
atoc
yte
func
tion
(eg,
gene
expr
essi
onpr
ofile
and
bioc
hem
ical
acti
viti
es),
how
ever
, is
not
iden
-ti
cal a
mon
gal
l hep
atoc
ytes
. Rat
her,
hepa
tocy
tes
perf
orm
diff
eren
tro
les
depe
ndin
gon
thei
rph
ysic
allo
cati
onw
ithi
nth
ehe
pati
clo
bule
. “M
etab
olic
zona
tion
”re
fers
toth
edi
ffer
enti
alpr
oper
ties
ofpe
ripo
rtal
(adj
acen
tto
the
port
altr
iad)
and
peri
cent
ral (
adja
cent
toth
ece
ntra
l vei
n)he
pato
cyte
s.4
Peri
port
alhe
pato
cyte
s,fo
rex
ampl
e,ex
pres
sur
eacy
cle
enzy
mes
and
conv
ert
amm
onia
tour
ea.5
,6In
cont
rast
, per
icen
tral
hepa
tocy
tes
expr
ess
glut
amin
esy
n-th
ase
and
utili
zeam
mon
iato
gene
rate
glut
amin
e.5
Abb
revi
atio
nsus
edin
this
pape
r:A
FP,
!-fe
topr
otei
n;D
DC,
3,5-
diet
hoxy
carb
onyl
-1,4
-dih
ydro
colli
dine
;EC
FC,
epith
elia
lco
lony
-form
ing
cell;
ESC,
embr
yoni
cst
emce
ll;H
SC,
hem
atop
oiet
icst
emce
ll;iP
S,in
duce
dpl
urip
oten
tst
emce
ll;M
SC, m
esen
chym
alst
emce
ll;TW
EAK
,tu
mor
necr
osis
fact
or–l
ike
wea
kin
duce
rof
apop
tosi
s.©
2009
byth
eAG
AIn
stitu
te00
16-5
085/
09/$
36.0
0do
i:10.
1053
/j.g
astr
o.20
09.0
5.04
4
REVIEWS INBASIC AND CLINICALGASTROENTEROLOGY
GA
STR
OEN
TER
OLO
GY
2009
;137
:466
–48
1

142 | MARCH 2010 | VOLUME 6 www.nature.com/nrendo
cells. These were then subjected to a directed differen-tiation protocol to produce cells that released human C-peptide and showed a fivefold increase in secretion of C-peptide in response to 20 mM glucose, which sug-gests that functional β cells can eventually be derived from iPS cells.
Nevertheless, iPS cells, if generated from patients with T1DM and transplanted back into the donor, would still be targeted by the immune system. Particular in terest, therefore, lies in the generation of both immune cells and β cells from the same iPS cell to broaden our un derstanding of the autoimmune destruction of β cells.
Umbilical cord bloodIn addition to its use for generating iPS cells, umbilical cords represent a potential and readily available source of mesenchymal stromal cells (MSCs) and blood stem cells that have been explored both as modulators of the immune response in T1DM and as potential sources of insulin-positive cells. Islet-like clusters derived from MSCs in the human umbilical cord released very low amounts of insulin in vitro.41 The more selective approach of flow cytometry showed that cells from umbilical cord blood that express the human hematopoietic precursor markers CD133 and CD34 rapidly differentiated into cells that were positive for C-peptide and insulin by
immunohistochemistry; however, insulin content and insulin secretion were not evaluated.42 Our previous caveat about what defines a β cell must be remembered when interpreting these studies.
Bone marrow-derived mesenchymal stromal cellsBone marrow-derived cells, including MSCs, have been shown to differentiate into various lineages and are readily available, which makes them an attractive source for regenerative cell-replacement strategies for T1DM. Transplantation of bone marrow-derived cells increased levels of serum insulin and reduced blood glucose levels in hyperglycemic mice with streptozotocin-induced pan-creatic tissue damage, which improved the metabolic state and survival of recipients.43 A careful histological analy-sis of the pancreas of these animals suggested that cells transplanted from the bone marrow homed to the site of pancreatic injury and promoted the formation of insulin-positive cells of recipient origin, probably by secreting products of the endothelial precursor cells. By contrast, several reports suggest that bone marrow- derived cells themselves differentiate into insulin- positive cells,44,45 but this cell lineage switch remains to be confirmed.
Other studies, which used rat MSCs46 or multipotent stromal cells from human bone marrow,47 have shown similar results of enhanced insulin secretion and repair of
Replication of preexisting cells
Acinartransdifferentiation
to cells
Differentiationof stem and progenitorsin the ductal epithelium
Growth factors:GLP1, exendin-4, Activin A,
HGF, betacellulinTranscription factors:
PDX1, NEUROD1, NGN3, PAX4
Growth factors:EGF, LIF
Transcription factors:NGN3, PDX1, MAFA
Growth factors:Activin A
Transcription factors:PAX1, NEUROD1
GLP1, exendin-4
Liver
Differentiation ofstem and progenitors
(not islet, duct or acinar cells)
Pancreas
Insulin-expressing cell
Figure 2 | Strategies to obtain β cells from organ-specific stem or progenitor cells. The pancreas itself can be a source of new insulin-expressing β cells either via replication of pre-existing β cells, transdifferentiation of acinar tissue into β cells, differentiation of progenitors within the ductal epithelium or differentiation of pancreatic stem or progenitor cells. The liver represents another source of embryologically related tissue that has been transdifferentiated into insulin-expressing cells. Abbreviations: EGF, epidermal growth factor; GLP1, glucagon-like peptide 1; HGF, hepatocyte growth factor; hESC, human embryonic stem cell; iPS, induced pluripotent stem cells; LIF, leukemia inhibitory factor; NEUROD1, neurogenic differentiation factor 1; NGN3, Neurogenin-3; MAFA, transcription factor MafA; PAX4, paired box protein; PDX1, pancreas and duodenum homeobox protein 1. Permission obtained from Macmillan Publishers Ltd © Bonner-Weir, S. & Weir, G. C. Nature Biotechnol. 23, 857–861 (2005).
REVIEWS
nrendo_274_MAR10.indd 142 4/2/10 17:01:36
© 20 Macmillan Publishers Limited. All rights reserved10
Generation of β cells from organ-specific stem or progenitor cells
3/25/16, 8:19 AMAccess : Stem cell therapy for type 1 diabetes mellitus : Nature Reviews Endocrinology
Page 1 of 3http://www.nature.com/nrendo/journal/v6/n3/full/nrendo.2009.274.html
ARTICLETOOLS
Send to afriend
Exportcitation
Exportreferences
Rights andpermissions
Ordercommercialreprints
SEARCHPUBMEDFOR
CristinaAguayo-Mazzucato
Susan
AccessTo read this article in full you may need to login, make a payment or gain access through asite license (see right).
ReviewNature Reviews Endocrinology 6, 139-148 (March2010) | doi:10.1038/nrendo.2009.274
Stem cell therapy fortype 1 diabetesmellitusCristina Aguayo-Mazzucato &Susan Bonner-Weir
The use of stem cells inregenerative medicine holdsgreat promise for the cure ofmany diseases, including type 1diabetes mellitus (T1DM). Anypotential stem-cell-based curefor T1DM should address theneed for β-cell replacement, aswell as control of theautoimmune response to cellswhich express insulin. The exvivo generation of β cells
NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | MARCH 2010 | 139
Section of Islet Transplantation and Cell Biology, Joslin Diabetes Center, Harvard Medical School, 1 Joslin Place, Boston, MA 02215, USA. (C. Aguayo-Mazzucato, S. Bonner-Weir).
Correspondence to: S. Bonner-Weir [email protected]
Stem cell therapy for type 1 diabetes mellitusCristina Aguayo-Mazzucato and Susan Bonner-Weir
Abstract | The use of stem cells in regenerative medicine holds great promise for the cure of many diseases, including type 1 diabetes mellitus (T1DM). Any potential stem-cell-based cure for T1DM should address the need for β-cell replacement, as well as control of the autoimmune response to cells which express insulin. The ex vivo generation of β cells suitable for transplantation to reconstitute a functional β-cell mass has used pluripotent cells from diverse sources, as well as organ-specific facultative progenitor cells from the liver and the pancreas. The most effective protocols to date have produced cells that express insulin and have molecular characteristics that closely resemble bona fide insulin-secreting cells; however, these cells are often unresponsive to glucose, a characteristic that should be addressed in future protocols. The use of mesenchymal stromal cells or umbilical cord blood to modulate the immune response is already in clinical trials; however, definitive results are still pending. This Review focuses on current strategies to obtain cells which express insulin from different progenitor sources and highlights the main pathways and genes involved, as well as the different approaches for the modulation of the immune response in patients with T1DM.
Aguayo-Mazzucato, C. & Bonner-Weir, S. Nat. Rev. Endocrinol. 6, 139–148 (2010); doi:10.1038/nrendo.2009.274
IntroductionType 1 diabetes mellitus (T1DM) is characterized by the autoimmune destruction of the insulin- expressing, pan-creatic β cells; as a result, patients with T1DM are depen-dent on exogenous insulin for their blood glucose control. Although the discovery of insulin has changed the outlook and survival of this group of individuals for almost a century,1 patients with T1DM are still not exempt from the development of diabetic complications.2,3 These com-plications are thought to result, in part, from the lack of physiological oscillations in insulin secretion and higher than normal glucose levels; thus, the search for effective ways to re establish a functional β-cell mass in patients with T1DM is important. Transplantation of whole human pancreas or isolated, cadaveric human islets4,5 has allowed patients with T1DM to become insulin-independent and has thereby shown proof of concept for β-cell replacement therapies. The shortage of donor pancreata and islets,6 however, results in the search for alternative cell sources, and differen tiation of stem cells (Box 1) into functional β cells is an attractive alternative.
A definitive cure for T1DM needs to address both the β-cell deficit and the autoimmune response to cells that express insulin. As clearly shown by the transplantation of segmental pancreatic grafts from identical twins, the autoimmune response remains many years after disease onset.7 Thus, any β-cell replacement therapy will need some modulation of the immune system, whether by drugs or cell-based approaches.
This Review focuses mainly on the different approaches to obtain functional, insulin-producing cells from various types of progenitor cells. Although the goal of the field is to
direct differentiation of human stem cells into functional β cells, some animal studies are included here, as they could lay the framework for success ful future interventions in humans. Although stem cell- derived, insulin-positive cells have not yet reached clinical trial, clinical data from stem cell-derived therapies that target the autoimmune response have been reported and are discussed.
Many roads lead from stem cells to β cellsEfforts have been directed at the development of efficient protocols for the differentiation of stem cells of either embryonic (Figure 1) or adult origin (Figure 2) into func-tional cells that secrete insulin. A caveat to many of these studies is that some developmental pathways generate insulin-expressing cells that will never become true β cells, for example, cells of the fetal liver, yolk sac and brain.8–10 A β cell should be defined by its ability to store large amounts of insulin and to secrete it in a regulated manner in response to a demand, such as glucose stimulation.11
Human embryonic stem cellsEfforts to generate β cells from human embryonic stem cells (hESCs) have focused on the fundamentals of normal embryonic development as a basis to understand the early stages of endoderm formation. The first step in the differen tiation of hESCs into β cells is the production of definitive endoderm12–17 rather than visceral endoderm, which expresses the same markers (for example, sex determining region Y box [SOX] 17 and forkhead box protein A2 [FOXA2]) but forms different tissues.18
Current protocols for hESC differentiation into β cells aim to reproduce the developmentally active signaling pathways Wnt and transforming growth factor β (TGFβ) (Box 2).17,19–22 Growth factors, such as Activin A, fibroblast
Competing interestsThe authors declare no competing interests.
FOCUS ON NONINSULIN THERAPIES FOR T1DM
nrendo_274_MAR10.indd 139 4/2/10 17:00:58
© 20 Macmillan Publishers Limited. All rights reserved10

Stem cells and autoimmunity
T cells may facilitate direct or bystander suppression ofeffector T cells or allow for restoration of tolerance by theirinhibitory effects on multiple cell types [45].
The study has been designed as a 2-year, unblinded ob-servational study with peak c-peptide following a standardmixed-meal tolerance test, hemoglobin A1c (HbA1c), anddaily insulin requirement set as the primary outcomevariables. Subjects undergo mixed-meal tolerance testingimmediately before cord blood infusion and then every3 months during the first postinfusion year and every6 months in the second postinfusion year (Fig. 2). Inaddition to measures of insulin production and metaboliccontrol, we study the immunological effects of autologouscord blood infusion by obtaining blood for peripheral bloodmononuclear cell (PBMC) analysis at each visit. PBMCs
are analyzed by flow cytometry with staining for the cellsurface markers CD3, CD4, CD8, CD25, and the intracellu-lar marker FOXP3. In addition, suppression assays are per-formed to measure the function of peripherally collectedTregs.
Our study has been designed with broad inclusion crite-ria, as little preclinical data are available to guide selectionof specific subjects. As such, we allow any child older than1 year of age with T1D, stored autologous cord blood,normal screening labs (complete blood count and basicmetabolic profile), and no other significant past medicalhistory to participate in the study. Similarly, no specific cri-teria as to length of time since diagnosis or baseline insulinproduction are included. To be usable, the cord blood cellviability must be at least 50%, and both the unit and the
CORD BLOOD IN T1D: STUDY TIMELINE
CORD BLOODINFUSION
Year 1 – q 3 month MMTT andblood draw
Year 2 – q 6 month MMTT andblood draw
3 mo 6 mo 9 mo 12 mo 18 mo 24mo
T1DDIAGNOSIS
Figure 2. Cord blood in type 1 diabetes (T1D) study timeline. Our study was designed to be a 2-year observational study of the effects of autologous cordblood infusion in children with T1D. We follow each child every 3 months during the first year postinfusion and every 6 months during the second yearpostinfusion. Blood is obtained for metabolic and immunologic studies at each visit. MMTT 5 mixed-meal tolerance testing.
Treg Teff
Treg
Teff Treg
Teff
Autoimmunity
Homeostasis
Induced
Tolerance
CORD BLOOD MEDIATED RESTORATION OF
IMMUNE TOLERANCE IN T1D
CD4+CD25
+ Treg
CD4+CD25
- Teff
CORD
BLOOD
Figure 1. Cord blood–mediated restoration of immune tolerance in type 1 diabetes (T1D). This theoretical model of the basic imbalance between regulatoryT cell (Treg) number or function seen in autoimmune disease demonstrates the simple concept that a rich source of Treg, such as cord blood, may have thepotential to tip the scales back in favor of immune tolerance. If true, this concept could be applied in the treatment of many autoimmune diseases.(Figure produced, with permission, using software from www.servier.com).
713M.J. Haller et al./ Experimental Hematology 2008;36:710–715
Autologous umbilical cord blood infusion for type 1 diabetes
Michael J. Hallera, Hilla-Lee Vienerb, Clive Wasserfallb,Todd Bruskob, Mark A. Atkinsonb, and Desmond A. Schatza
aDepartment of Pediatrics; bDepartment of Pathology, University of Florida, Gainesville, Fla., USA
(Received 31 October 2007; revised 31 October 2007; accepted 23 January 2008)
Objective. The physical, emotional, and economic costs of type 1 diabetes (T1D) mandate con-tinued efforts to develop effective strategies to prevent or reverse the disease. Herein, we de-scribe the scientific and therapeutic rationale underlying efforts utilizing umbilical cord blood(UCB) as a therapy for ameliorating the progression of this autoimmune disease.
Materials and Methods. We recently embarked on a pilot study to document the safety andpotential efficacy of autologous UCB infusion in subjects with T1D. Under this protocol,patients recently diagnosed with the disease and for whom autologous cord blood is stored,undergo infusion. Studies are performed before infusion and every 3 to 6 months postinfusionfor immunologic and metabolic assessment. To date, 15 autologous infusions have beenperformed.
Results. Preliminary observations suggest that autologous cord blood transfusion is safe andprovides some slowing of the loss of endogenous insulin production in children with T1D.Mechanistic studies demonstrate that umbilical cord blood contains highly functional popula-tions of regulatory T cells (Treg) and that increased Treg populations may be found in theperipheral blood of subjects more than 6 months after cord blood infusion. We provide therationale for cord blood–based therapies, a summary of our initial protocol, and plans forfuture studies designed to explore the potential of cord blood–derived regulatory T cells totreat T1D.
Conclusions. Prolonged follow-up and additional mechanistic efforts are urgently needed todetermine if umbilical cord blood–derived stem cells can be used as part of safe and effectivetherapies for T1D. ! 2008 ISEH - Society for Hematology and Stem Cells. Published byElsevier Inc.
Type 1 diabetes (T1D) is an autoimmune disease character-ized by T-cell–mediated destruction of insulin producingb cells and lifelong dependence on exogenous insulin ad-ministration. T1D affects nearly 1 in 300 within the UnitedStates and the incidence of the disease continues to rise at ap-proximately 3% per year [1,2]. On an international level, in-cidence of T1D varies dramatically; as much as 500-fold [3].
Since the discovery of insulin by Banting, Best, Collip,and McCleod in 1921, T1D has evolved from a uniformlyfatal disease to a chronic disease; one with a continuouslyevolving armamentarium of devices and insulin analogsthat have greatly improved care. Nevertheless, the over-whelming majority of patients with T1D are still unableor unwilling to utilize currently available resources that
would minimize their risk of diabetes-related complications[4]. With this, the tremendous physical, emotional, and eco-nomic costs of T1D demand that we explore novel strate-gies for the prevention and reversal of T1D.
During the last 25 years, a majority of efforts seeking toameliorate the autoimmune process and reverse disease inthose recently diagnosed have focused on the use of immu-nosuppressive or immunomodulatory drugs [5,6]. Whileseveral of these agents have shown, and continue to show,promise (e.g., anti-CD3), no single agent has succeeded indemonstrating long-term success in preventing or reversingT1D. More recently, additional efforts have focused on theuse of both autologous and allogeneic stem cells as sourcesof new islets, and perhaps more intriguingly, as potentialsources of safe and effective immunomodulation [7–9].Among the broad array of potential cell-based therapies,use of autologous umbilical cord blood (UCB) as a sourceof immunomodulatory cells for treatment of autoimmune
Offprint requests to: Michael J. Haller, M.D., Department of Pediatrics,University of Florida, P.O. Box 100296, Gainesville, FL 32610; E-mail:[email protected]
0301-472X/08 $–see front matter. Copyright ! 2008 ISEH - Society for Hematology and Stem Cells. Published by Elsevier Inc.doi: 10.1016/j.exphem.2008.01.009
Experimental Hematology 2008;36:710–715
Autologous umbilical cord blood infusion for type 1 diabetes
Michael J. Hallera, Hilla-Lee Vienerb, Clive Wasserfallb,Todd Bruskob, Mark A. Atkinsonb, and Desmond A. Schatza
aDepartment of Pediatrics; bDepartment of Pathology, University of Florida, Gainesville, Fla., USA
(Received 31 October 2007; revised 31 October 2007; accepted 23 January 2008)
Objective. The physical, emotional, and economic costs of type 1 diabetes (T1D) mandate con-tinued efforts to develop effective strategies to prevent or reverse the disease. Herein, we de-scribe the scientific and therapeutic rationale underlying efforts utilizing umbilical cord blood(UCB) as a therapy for ameliorating the progression of this autoimmune disease.
Materials and Methods. We recently embarked on a pilot study to document the safety andpotential efficacy of autologous UCB infusion in subjects with T1D. Under this protocol,patients recently diagnosed with the disease and for whom autologous cord blood is stored,undergo infusion. Studies are performed before infusion and every 3 to 6 months postinfusionfor immunologic and metabolic assessment. To date, 15 autologous infusions have beenperformed.
Results. Preliminary observations suggest that autologous cord blood transfusion is safe andprovides some slowing of the loss of endogenous insulin production in children with T1D.Mechanistic studies demonstrate that umbilical cord blood contains highly functional popula-tions of regulatory T cells (Treg) and that increased Treg populations may be found in theperipheral blood of subjects more than 6 months after cord blood infusion. We provide therationale for cord blood–based therapies, a summary of our initial protocol, and plans forfuture studies designed to explore the potential of cord blood–derived regulatory T cells totreat T1D.
Conclusions. Prolonged follow-up and additional mechanistic efforts are urgently needed todetermine if umbilical cord blood–derived stem cells can be used as part of safe and effectivetherapies for T1D. ! 2008 ISEH - Society for Hematology and Stem Cells. Published byElsevier Inc.
Type 1 diabetes (T1D) is an autoimmune disease character-ized by T-cell–mediated destruction of insulin producingb cells and lifelong dependence on exogenous insulin ad-ministration. T1D affects nearly 1 in 300 within the UnitedStates and the incidence of the disease continues to rise at ap-proximately 3% per year [1,2]. On an international level, in-cidence of T1D varies dramatically; as much as 500-fold [3].
Since the discovery of insulin by Banting, Best, Collip,and McCleod in 1921, T1D has evolved from a uniformlyfatal disease to a chronic disease; one with a continuouslyevolving armamentarium of devices and insulin analogsthat have greatly improved care. Nevertheless, the over-whelming majority of patients with T1D are still unableor unwilling to utilize currently available resources that
would minimize their risk of diabetes-related complications[4]. With this, the tremendous physical, emotional, and eco-nomic costs of T1D demand that we explore novel strate-gies for the prevention and reversal of T1D.
During the last 25 years, a majority of efforts seeking toameliorate the autoimmune process and reverse disease inthose recently diagnosed have focused on the use of immu-nosuppressive or immunomodulatory drugs [5,6]. Whileseveral of these agents have shown, and continue to show,promise (e.g., anti-CD3), no single agent has succeeded indemonstrating long-term success in preventing or reversingT1D. More recently, additional efforts have focused on theuse of both autologous and allogeneic stem cells as sourcesof new islets, and perhaps more intriguingly, as potentialsources of safe and effective immunomodulation [7–9].Among the broad array of potential cell-based therapies,use of autologous umbilical cord blood (UCB) as a sourceof immunomodulatory cells for treatment of autoimmune
Offprint requests to: Michael J. Haller, M.D., Department of Pediatrics,University of Florida, P.O. Box 100296, Gainesville, FL 32610; E-mail:[email protected]
0301-472X/08 $–see front matter. Copyright ! 2008 ISEH - Society for Hematology and Stem Cells. Published by Elsevier Inc.doi: 10.1016/j.exphem.2008.01.009
Experimental Hematology 2008;36:710–715
Autologous umbilical cord blood infusion for type 1 diabetes
Michael J. Hallera, Hilla-Lee Vienerb, Clive Wasserfallb,Todd Bruskob, Mark A. Atkinsonb, and Desmond A. Schatza
aDepartment of Pediatrics; bDepartment of Pathology, University of Florida, Gainesville, Fla., USA
(Received 31 October 2007; revised 31 October 2007; accepted 23 January 2008)
Objective. The physical, emotional, and economic costs of type 1 diabetes (T1D) mandate con-tinued efforts to develop effective strategies to prevent or reverse the disease. Herein, we de-scribe the scientific and therapeutic rationale underlying efforts utilizing umbilical cord blood(UCB) as a therapy for ameliorating the progression of this autoimmune disease.
Materials and Methods. We recently embarked on a pilot study to document the safety andpotential efficacy of autologous UCB infusion in subjects with T1D. Under this protocol,patients recently diagnosed with the disease and for whom autologous cord blood is stored,undergo infusion. Studies are performed before infusion and every 3 to 6 months postinfusionfor immunologic and metabolic assessment. To date, 15 autologous infusions have beenperformed.
Results. Preliminary observations suggest that autologous cord blood transfusion is safe andprovides some slowing of the loss of endogenous insulin production in children with T1D.Mechanistic studies demonstrate that umbilical cord blood contains highly functional popula-tions of regulatory T cells (Treg) and that increased Treg populations may be found in theperipheral blood of subjects more than 6 months after cord blood infusion. We provide therationale for cord blood–based therapies, a summary of our initial protocol, and plans forfuture studies designed to explore the potential of cord blood–derived regulatory T cells totreat T1D.
Conclusions. Prolonged follow-up and additional mechanistic efforts are urgently needed todetermine if umbilical cord blood–derived stem cells can be used as part of safe and effectivetherapies for T1D. ! 2008 ISEH - Society for Hematology and Stem Cells. Published byElsevier Inc.
Type 1 diabetes (T1D) is an autoimmune disease character-ized by T-cell–mediated destruction of insulin producingb cells and lifelong dependence on exogenous insulin ad-ministration. T1D affects nearly 1 in 300 within the UnitedStates and the incidence of the disease continues to rise at ap-proximately 3% per year [1,2]. On an international level, in-cidence of T1D varies dramatically; as much as 500-fold [3].
Since the discovery of insulin by Banting, Best, Collip,and McCleod in 1921, T1D has evolved from a uniformlyfatal disease to a chronic disease; one with a continuouslyevolving armamentarium of devices and insulin analogsthat have greatly improved care. Nevertheless, the over-whelming majority of patients with T1D are still unableor unwilling to utilize currently available resources that
would minimize their risk of diabetes-related complications[4]. With this, the tremendous physical, emotional, and eco-nomic costs of T1D demand that we explore novel strate-gies for the prevention and reversal of T1D.
During the last 25 years, a majority of efforts seeking toameliorate the autoimmune process and reverse disease inthose recently diagnosed have focused on the use of immu-nosuppressive or immunomodulatory drugs [5,6]. Whileseveral of these agents have shown, and continue to show,promise (e.g., anti-CD3), no single agent has succeeded indemonstrating long-term success in preventing or reversingT1D. More recently, additional efforts have focused on theuse of both autologous and allogeneic stem cells as sourcesof new islets, and perhaps more intriguingly, as potentialsources of safe and effective immunomodulation [7–9].Among the broad array of potential cell-based therapies,use of autologous umbilical cord blood (UCB) as a sourceof immunomodulatory cells for treatment of autoimmune
Offprint requests to: Michael J. Haller, M.D., Department of Pediatrics,University of Florida, P.O. Box 100296, Gainesville, FL 32610; E-mail:[email protected]
0301-472X/08 $–see front matter. Copyright ! 2008 ISEH - Society for Hematology and Stem Cells. Published by Elsevier Inc.doi: 10.1016/j.exphem.2008.01.009
Experimental Hematology 2008;36:710–715
Autologous umbilical cord blood infusion for type 1 diabetes
Michael J. Hallera, Hilla-Lee Vienerb, Clive Wasserfallb,Todd Bruskob, Mark A. Atkinsonb, and Desmond A. Schatza
aDepartment of Pediatrics; bDepartment of Pathology, University of Florida, Gainesville, Fla., USA
(Received 31 October 2007; revised 31 October 2007; accepted 23 January 2008)
Objective. The physical, emotional, and economic costs of type 1 diabetes (T1D) mandate con-tinued efforts to develop effective strategies to prevent or reverse the disease. Herein, we de-scribe the scientific and therapeutic rationale underlying efforts utilizing umbilical cord blood(UCB) as a therapy for ameliorating the progression of this autoimmune disease.
Materials and Methods. We recently embarked on a pilot study to document the safety andpotential efficacy of autologous UCB infusion in subjects with T1D. Under this protocol,patients recently diagnosed with the disease and for whom autologous cord blood is stored,undergo infusion. Studies are performed before infusion and every 3 to 6 months postinfusionfor immunologic and metabolic assessment. To date, 15 autologous infusions have beenperformed.
Results. Preliminary observations suggest that autologous cord blood transfusion is safe andprovides some slowing of the loss of endogenous insulin production in children with T1D.Mechanistic studies demonstrate that umbilical cord blood contains highly functional popula-tions of regulatory T cells (Treg) and that increased Treg populations may be found in theperipheral blood of subjects more than 6 months after cord blood infusion. We provide therationale for cord blood–based therapies, a summary of our initial protocol, and plans forfuture studies designed to explore the potential of cord blood–derived regulatory T cells totreat T1D.
Conclusions. Prolonged follow-up and additional mechanistic efforts are urgently needed todetermine if umbilical cord blood–derived stem cells can be used as part of safe and effectivetherapies for T1D. ! 2008 ISEH - Society for Hematology and Stem Cells. Published byElsevier Inc.
Type 1 diabetes (T1D) is an autoimmune disease character-ized by T-cell–mediated destruction of insulin producingb cells and lifelong dependence on exogenous insulin ad-ministration. T1D affects nearly 1 in 300 within the UnitedStates and the incidence of the disease continues to rise at ap-proximately 3% per year [1,2]. On an international level, in-cidence of T1D varies dramatically; as much as 500-fold [3].
Since the discovery of insulin by Banting, Best, Collip,and McCleod in 1921, T1D has evolved from a uniformlyfatal disease to a chronic disease; one with a continuouslyevolving armamentarium of devices and insulin analogsthat have greatly improved care. Nevertheless, the over-whelming majority of patients with T1D are still unableor unwilling to utilize currently available resources that
would minimize their risk of diabetes-related complications[4]. With this, the tremendous physical, emotional, and eco-nomic costs of T1D demand that we explore novel strate-gies for the prevention and reversal of T1D.
During the last 25 years, a majority of efforts seeking toameliorate the autoimmune process and reverse disease inthose recently diagnosed have focused on the use of immu-nosuppressive or immunomodulatory drugs [5,6]. Whileseveral of these agents have shown, and continue to show,promise (e.g., anti-CD3), no single agent has succeeded indemonstrating long-term success in preventing or reversingT1D. More recently, additional efforts have focused on theuse of both autologous and allogeneic stem cells as sourcesof new islets, and perhaps more intriguingly, as potentialsources of safe and effective immunomodulation [7–9].Among the broad array of potential cell-based therapies,use of autologous umbilical cord blood (UCB) as a sourceof immunomodulatory cells for treatment of autoimmune
Offprint requests to: Michael J. Haller, M.D., Department of Pediatrics,University of Florida, P.O. Box 100296, Gainesville, FL 32610; E-mail:[email protected]
0301-472X/08 $–see front matter. Copyright ! 2008 ISEH - Society for Hematology and Stem Cells. Published by Elsevier Inc.doi: 10.1016/j.exphem.2008.01.009
Experimental Hematology 2008;36:710–715
Autologous umbilical cord blood infusion for type 1 diabetes
Michael J. Hallera, Hilla-Lee Vienerb, Clive Wasserfallb,Todd Bruskob, Mark A. Atkinsonb, and Desmond A. Schatza
aDepartment of Pediatrics; bDepartment of Pathology, University of Florida, Gainesville, Fla., USA
(Received 31 October 2007; revised 31 October 2007; accepted 23 January 2008)
Objective. The physical, emotional, and economic costs of type 1 diabetes (T1D) mandate con-tinued efforts to develop effective strategies to prevent or reverse the disease. Herein, we de-scribe the scientific and therapeutic rationale underlying efforts utilizing umbilical cord blood(UCB) as a therapy for ameliorating the progression of this autoimmune disease.
Materials and Methods. We recently embarked on a pilot study to document the safety andpotential efficacy of autologous UCB infusion in subjects with T1D. Under this protocol,patients recently diagnosed with the disease and for whom autologous cord blood is stored,undergo infusion. Studies are performed before infusion and every 3 to 6 months postinfusionfor immunologic and metabolic assessment. To date, 15 autologous infusions have beenperformed.
Results. Preliminary observations suggest that autologous cord blood transfusion is safe andprovides some slowing of the loss of endogenous insulin production in children with T1D.Mechanistic studies demonstrate that umbilical cord blood contains highly functional popula-tions of regulatory T cells (Treg) and that increased Treg populations may be found in theperipheral blood of subjects more than 6 months after cord blood infusion. We provide therationale for cord blood–based therapies, a summary of our initial protocol, and plans forfuture studies designed to explore the potential of cord blood–derived regulatory T cells totreat T1D.
Conclusions. Prolonged follow-up and additional mechanistic efforts are urgently needed todetermine if umbilical cord blood–derived stem cells can be used as part of safe and effectivetherapies for T1D. ! 2008 ISEH - Society for Hematology and Stem Cells. Published byElsevier Inc.
Type 1 diabetes (T1D) is an autoimmune disease character-ized by T-cell–mediated destruction of insulin producingb cells and lifelong dependence on exogenous insulin ad-ministration. T1D affects nearly 1 in 300 within the UnitedStates and the incidence of the disease continues to rise at ap-proximately 3% per year [1,2]. On an international level, in-cidence of T1D varies dramatically; as much as 500-fold [3].
Since the discovery of insulin by Banting, Best, Collip,and McCleod in 1921, T1D has evolved from a uniformlyfatal disease to a chronic disease; one with a continuouslyevolving armamentarium of devices and insulin analogsthat have greatly improved care. Nevertheless, the over-whelming majority of patients with T1D are still unableor unwilling to utilize currently available resources that
would minimize their risk of diabetes-related complications[4]. With this, the tremendous physical, emotional, and eco-nomic costs of T1D demand that we explore novel strate-gies for the prevention and reversal of T1D.
During the last 25 years, a majority of efforts seeking toameliorate the autoimmune process and reverse disease inthose recently diagnosed have focused on the use of immu-nosuppressive or immunomodulatory drugs [5,6]. Whileseveral of these agents have shown, and continue to show,promise (e.g., anti-CD3), no single agent has succeeded indemonstrating long-term success in preventing or reversingT1D. More recently, additional efforts have focused on theuse of both autologous and allogeneic stem cells as sourcesof new islets, and perhaps more intriguingly, as potentialsources of safe and effective immunomodulation [7–9].Among the broad array of potential cell-based therapies,use of autologous umbilical cord blood (UCB) as a sourceof immunomodulatory cells for treatment of autoimmune
Offprint requests to: Michael J. Haller, M.D., Department of Pediatrics,University of Florida, P.O. Box 100296, Gainesville, FL 32610; E-mail:[email protected]
0301-472X/08 $–see front matter. Copyright ! 2008 ISEH - Society for Hematology and Stem Cells. Published by Elsevier Inc.doi: 10.1016/j.exphem.2008.01.009
Experimental Hematology 2008;36:710–715Autologous umbilical cord blood infusion for type 1 diabetes
Michael J. Hallera, Hilla-Lee Vienerb, Clive Wasserfallb,Todd Bruskob, Mark A. Atkinsonb, and Desmond A. Schatza
aDepartment of Pediatrics; bDepartment of Pathology, University of Florida, Gainesville, Fla., USA
(Received 31 October 2007; revised 31 October 2007; accepted 23 January 2008)
Objective. The physical, emotional, and economic costs of type 1 diabetes (T1D) mandate con-tinued efforts to develop effective strategies to prevent or reverse the disease. Herein, we de-scribe the scientific and therapeutic rationale underlying efforts utilizing umbilical cord blood(UCB) as a therapy for ameliorating the progression of this autoimmune disease.
Materials and Methods. We recently embarked on a pilot study to document the safety andpotential efficacy of autologous UCB infusion in subjects with T1D. Under this protocol,patients recently diagnosed with the disease and for whom autologous cord blood is stored,undergo infusion. Studies are performed before infusion and every 3 to 6 months postinfusionfor immunologic and metabolic assessment. To date, 15 autologous infusions have beenperformed.
Results. Preliminary observations suggest that autologous cord blood transfusion is safe andprovides some slowing of the loss of endogenous insulin production in children with T1D.Mechanistic studies demonstrate that umbilical cord blood contains highly functional popula-tions of regulatory T cells (Treg) and that increased Treg populations may be found in theperipheral blood of subjects more than 6 months after cord blood infusion. We provide therationale for cord blood–based therapies, a summary of our initial protocol, and plans forfuture studies designed to explore the potential of cord blood–derived regulatory T cells totreat T1D.
Conclusions. Prolonged follow-up and additional mechanistic efforts are urgently needed todetermine if umbilical cord blood–derived stem cells can be used as part of safe and effectivetherapies for T1D. ! 2008 ISEH - Society for Hematology and Stem Cells. Published byElsevier Inc.
Type 1 diabetes (T1D) is an autoimmune disease character-ized by T-cell–mediated destruction of insulin producingb cells and lifelong dependence on exogenous insulin ad-ministration. T1D affects nearly 1 in 300 within the UnitedStates and the incidence of the disease continues to rise at ap-proximately 3% per year [1,2]. On an international level, in-cidence of T1D varies dramatically; as much as 500-fold [3].
Since the discovery of insulin by Banting, Best, Collip,and McCleod in 1921, T1D has evolved from a uniformlyfatal disease to a chronic disease; one with a continuouslyevolving armamentarium of devices and insulin analogsthat have greatly improved care. Nevertheless, the over-whelming majority of patients with T1D are still unableor unwilling to utilize currently available resources that
would minimize their risk of diabetes-related complications[4]. With this, the tremendous physical, emotional, and eco-nomic costs of T1D demand that we explore novel strate-gies for the prevention and reversal of T1D.
During the last 25 years, a majority of efforts seeking toameliorate the autoimmune process and reverse disease inthose recently diagnosed have focused on the use of immu-nosuppressive or immunomodulatory drugs [5,6]. Whileseveral of these agents have shown, and continue to show,promise (e.g., anti-CD3), no single agent has succeeded indemonstrating long-term success in preventing or reversingT1D. More recently, additional efforts have focused on theuse of both autologous and allogeneic stem cells as sourcesof new islets, and perhaps more intriguingly, as potentialsources of safe and effective immunomodulation [7–9].Among the broad array of potential cell-based therapies,use of autologous umbilical cord blood (UCB) as a sourceof immunomodulatory cells for treatment of autoimmune
Offprint requests to: Michael J. Haller, M.D., Department of Pediatrics,University of Florida, P.O. Box 100296, Gainesville, FL 32610; E-mail:[email protected]
0301-472X/08 $–see front matter. Copyright ! 2008 ISEH - Society for Hematology and Stem Cells. Published by Elsevier Inc.doi: 10.1016/j.exphem.2008.01.009
Experimental Hematology 2008;36:710–715

Cell Stem Cell
Article
Small Molecules Efficiently DirectEndodermal Differentiation of Mouseand Human Embryonic Stem CellsMalgorzata Borowiak,1,2 Rene Maehr,1,2 Shuibing Chen,1,2 Alice E. Chen,1,2 Weiping Tang,4 Julia L. Fox,1
Stuart L. Schreiber,2,3,4 and Douglas A. Melton1,2,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute2Howard Hughes Medical Institute, Harvard University3Department of Chemistry and Chemical Biology, Harvard University7 Divinity Avenue, Cambridge, MA 02138, USA4Broad Institute of Harvard and MIT, 7 Cambridge Center, Cambridge, MA 02142, USA*Correspondence: [email protected] 10.1016/j.stem.2009.01.014
SUMMARY
An essential step for therapeutic and research appli-cations of stem cells is the ability to differentiate theminto specific cell types. Endodermal cell derivatives,including lung, liver, and pancreas, are of interestfor regenerative medicine, but efforts to producethese cells have been met with only modest success.In a screen of 4000 compounds, two cell-permeablesmall molecules were indentified that direct differen-tiation of ESCs into the endodermal lineage. Thesecompounds induce nearly 80% of ESCs to form defin-itive endoderm, a higher efficiency than that achievedby Activin A or Nodal, commonly used proteininducers of endoderm. The chemically induced endo-derm expresses multiple endodermal markers, canparticipate in normal development when injectedinto developing embryos, and can form pancreaticprogenitors. The application of small molecules todifferentiate mouse and human ESCs into endodermrepresents a step toward achieving a reproducibleand efficient production of desired ESC derivatives.
INTRODUCTION
Type I diabetes results from the destruction of insulin-producingpancreatic b cells, and, therefore, there are several approachesaimed at cell-based strategies to replace these cells and rejuve-nate the pancreas. The spontaneous or undirected differentia-tion of embryonic stem cells (ESCs) produces very smallnumbers of insulin-producing cells, barely enough for researchstudy and far short of the numbers needed for therapeutic appli-cation. One strategy to increase the efficiency of b cell formationis to mimic embryonic development by exposing ESCs and theirderivatives to factors that they would normally encounter in vivo.The starting point for this strategy is differentiating ESCs intodefinitive endoderm.
Our understanding of the developmental pathways thatcontrol endoderm formation has so far been guided by studies
in Xenopous laevis, zebrafish, and mice (reviewed by Wells andMelton, 1999; Lewis and Tam, 2006). Collectively, these studiessuggest a conserved mechanism for the commitment to endo-derm/mesoderm, utilizing signaling proteins from the Wnt, fibro-blast growth factor (FGF), and transforming growth factorb (TGF-b) families. Similarly, in vitro application of Activin A orNodal to mouse or human ESC cultures leads to endoderminduction (Kubo et al., 2004; Yasunaga et al., 2005; D’Amouret al., 2005). Other molecules that influence endoderm formationin vitro include Wnts (D’Amour et al., 2005), bone morphogenicproteins (BMPs), and members of the AKT/P13K pathway(McLean et al., 2007).
Permeable small molecules can control cellular processes bymodulating signal transduction pathways, gene expression, ormetabolism and have been effectively used in ESC differentia-tion protocols. Small molecules can be synthesized in high quan-tity and purity, as well as conveniently supplied or removed,giving them great potential to be useful for therapeutic applica-tions. High-throughput screens have been performed to identifynovel small molecules that can support self-renewal of ESCs(Chen et al., 2006; Desbordes et al., 2008) and the specificationof cardiomyocytes (Wu et al., 2004) and neural progenitors(Diamandis et al., 2007; Ding and Schultz, 2004).
We describe here two small molecules, IDE1 and IDE2, thatcan efficiently induce definitive endoderm (IDE) from mouseand human ESCs. Treatment of mouse ESC monolayer cultureswith either compound yields high quantities of endoderm-expressing multiple endodermal marker genes. We show thatchemically derived endoderm develops into pancreatic progen-itors in vitro in response to the growth factor FGF10, retinoicacid, and hedgehog inhibitors, a commonly used combinationto induce pancreatic progenitors in vitro. Moreover, we applya recently identified small molecule, Indolactam V (Chen et al.,2009), which induces pancreatic progenitors in human ESCculture, to either IDE1- or IDE2-derived endoderm and inducehigher yields of pancreatic progenitors compared to a growthfactor-based approach. Finally, we show that compound-induced endoderm can contribute to gut tube formation in vivowhen the cells are injected into the developing gut tube of mouseembryos. All together, we introduce two small molecules thatinduce a robust differentiation of ESCs into endoderm that has
348 Cell Stem Cell 4, 348–358, April 3, 2009 ª2009 Elsevier Inc.
Resource
Generation of Functional HumanPancreatic b Cells In VitroFelicia W. Pagliuca,1,3 Jeffrey R. Millman,1,3 Mads Gurtler,1,3 Michael Segel,1 Alana Van Dervort,1 Jennifer Hyoje Ryu,1
Quinn P. Peterson,1 Dale Greiner,2 and Douglas A. Melton1,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge,MA 02138, USA2Diabetes Center of Excellence, University of Massachusetts Medical School, 368 Plantation Street, AS7-2051, Worcester, MA 01605, USA3Co-first author*Correspondence: [email protected]://dx.doi.org/10.1016/j.cell.2014.09.040
SUMMARY
The generation of insulin-producing pancreatic bcells from stem cells in vitro would provide an un-precedented cell source for drug discovery and celltransplantation therapy in diabetes. However, insu-lin-producing cells previously generated from humanpluripotent stem cells (hPSC) lack many functionalcharacteristics of bona fide b cells. Here, we reporta scalable differentiation protocol that can generatehundreds of millions of glucose-responsive b cellsfrom hPSC in vitro. These stem-cell-derived b cells(SC-b) express markers found in mature b cells, fluxCa2+ in response to glucose, package insulin intosecretory granules, and secrete quantities of insulincomparable to adult b cells in response to multiplesequential glucose challenges in vitro. Furthermore,these cells secrete human insulin into the serum ofmice shortly after transplantation in a glucose-regu-latedmanner, and transplantation of these cells ame-liorates hyperglycemia in diabetic mice.
INTRODUCTION
The discovery of human pluripotent stem cells (hPSC) openedthe possibility of generating replacement cells and tissuesin the laboratory that could be used for disease treatment anddrug screening. Recent research has moved the stem cell fieldcloser to that goal through development of strategies to generatecells that would otherwise be difficult to obtain, like neurons orcardiomyocytes (Kriks et al., 2011; Shiba et al., 2012; Sonet al., 2011). These cells have also been transplanted into animalmodels, in some caseswith a beneficial effect like suppression ofarrhythmias with stem-cell-derived cardiomyocytes (Shiba et al.,2012), restoration of locomotion after spinal injury with oligoden-drocyte progenitors (Keirstead et al., 2005), or improved visionafter transplantation of retinal epithelial cells into rodent modelsof blindness (Lu et al., 2009).
One of the rapidly growing diseases that may be treatable bystem-cell-derived tissues is diabetes, affecting >300 million peo-ple worldwide, according to the International Diabetes Federa-
tion. Type 1 diabetes results from autoimmune destruction of bcells in the pancreatic islet, whereas themore common type 2 dia-betes results from peripheral tissue insulin resistance and b celldysfunction. Diabetic patients, particularly those suffering fromtype 1 diabetes, could potentially be cured through transplanta-tion of new b cells. Patients transplantedwith cadaveric human is-lets can be made insulin independent for 5 years or longer via thisstrategy, but this approach is limited because of the scarcity andquality of donor islets (Bellin et al., 2012). The generation of anunlimited supply of human b cells from stem cells could extendthis therapy to millions of new patients and could be an importanttest case for translating stem cell biology into the clinic. This isbecause only a single cell type, the b cell, likely needs to be gener-ated, and the mode of delivery is understood: transplantation to avascularized location within the body with immunoprotection.Pharmaceutical screening to identify new drugs that improve b
cell function, survival, or proliferation is also hindered by limitedsupplies of islets and high variability due to differential causesof death, donor genetic background, and other factors in theirisolation. A consistent, uniform supply of stem-cell-derived b cellswould provide a unique and valuable drug discovery platform fordiabetes. Additionally, genetically diverse stem-cell-derived bcells could be used for disease modeling in vitro or in vivo.Studies on pancreatic development in model organisms
(Gamer and Wright, 1995; Henry and Melton, 1998; Ninomiyaet al., 1999; Apelqvist et al., 1999; Kim et al., 2000; Hebroket al., 2000; Murtaugh et al., 2003) identified genes and signalsimportant for the pancreatic lineage, and these have been effec-tively used to form cells in the b cell lineage in vitro from hPSC.Definitive endoderm and subsequent pancreatic progenitorscan now be differentiated with high efficiencies (Kroon et al.,2008; D’Amour et al., 2005, 2006; Rezania et al., 2012). Thesecells can differentiate into functional b cells within 3–4 monthsfollowing transplantation into rodents (Kroon et al., 2008; Rezaniaet al., 2012), indicating that some cells in the preparation containthe developmental potential to develop into b cells if providedenough time and appropriate cues. Unfortunately, the months-long process the cells undergo in vivo is not understood, and itis unclear whether this process of in vivo differentiation wouldalso occur in human patients. Attempts to date at generatinginsulin-producing (INS+) cells from human pancreatic progeni-tors in vitro have generated cells with immature or abnormalphenotypes. These cells either fail to perform glucose-stimulated
428 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.
Resource
Generation of Functional HumanPancreatic b Cells In VitroFelicia W. Pagliuca,1,3 Jeffrey R. Millman,1,3 Mads Gurtler,1,3 Michael Segel,1 Alana Van Dervort,1 Jennifer Hyoje Ryu,1
Quinn P. Peterson,1 Dale Greiner,2 and Douglas A. Melton1,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge,MA 02138, USA2Diabetes Center of Excellence, University of Massachusetts Medical School, 368 Plantation Street, AS7-2051, Worcester, MA 01605, USA3Co-first author*Correspondence: [email protected]://dx.doi.org/10.1016/j.cell.2014.09.040
SUMMARY
The generation of insulin-producing pancreatic bcells from stem cells in vitro would provide an un-precedented cell source for drug discovery and celltransplantation therapy in diabetes. However, insu-lin-producing cells previously generated from humanpluripotent stem cells (hPSC) lack many functionalcharacteristics of bona fide b cells. Here, we reporta scalable differentiation protocol that can generatehundreds of millions of glucose-responsive b cellsfrom hPSC in vitro. These stem-cell-derived b cells(SC-b) express markers found in mature b cells, fluxCa2+ in response to glucose, package insulin intosecretory granules, and secrete quantities of insulincomparable to adult b cells in response to multiplesequential glucose challenges in vitro. Furthermore,these cells secrete human insulin into the serum ofmice shortly after transplantation in a glucose-regu-latedmanner, and transplantation of these cells ame-liorates hyperglycemia in diabetic mice.
INTRODUCTION
The discovery of human pluripotent stem cells (hPSC) openedthe possibility of generating replacement cells and tissuesin the laboratory that could be used for disease treatment anddrug screening. Recent research has moved the stem cell fieldcloser to that goal through development of strategies to generatecells that would otherwise be difficult to obtain, like neurons orcardiomyocytes (Kriks et al., 2011; Shiba et al., 2012; Sonet al., 2011). These cells have also been transplanted into animalmodels, in some caseswith a beneficial effect like suppression ofarrhythmias with stem-cell-derived cardiomyocytes (Shiba et al.,2012), restoration of locomotion after spinal injury with oligoden-drocyte progenitors (Keirstead et al., 2005), or improved visionafter transplantation of retinal epithelial cells into rodent modelsof blindness (Lu et al., 2009).
One of the rapidly growing diseases that may be treatable bystem-cell-derived tissues is diabetes, affecting >300 million peo-ple worldwide, according to the International Diabetes Federa-
tion. Type 1 diabetes results from autoimmune destruction of bcells in the pancreatic islet, whereas themore common type 2 dia-betes results from peripheral tissue insulin resistance and b celldysfunction. Diabetic patients, particularly those suffering fromtype 1 diabetes, could potentially be cured through transplanta-tion of new b cells. Patients transplantedwith cadaveric human is-lets can be made insulin independent for 5 years or longer via thisstrategy, but this approach is limited because of the scarcity andquality of donor islets (Bellin et al., 2012). The generation of anunlimited supply of human b cells from stem cells could extendthis therapy to millions of new patients and could be an importanttest case for translating stem cell biology into the clinic. This isbecause only a single cell type, the b cell, likely needs to be gener-ated, and the mode of delivery is understood: transplantation to avascularized location within the body with immunoprotection.Pharmaceutical screening to identify new drugs that improve b
cell function, survival, or proliferation is also hindered by limitedsupplies of islets and high variability due to differential causesof death, donor genetic background, and other factors in theirisolation. A consistent, uniform supply of stem-cell-derived b cellswould provide a unique and valuable drug discovery platform fordiabetes. Additionally, genetically diverse stem-cell-derived bcells could be used for disease modeling in vitro or in vivo.Studies on pancreatic development in model organisms
(Gamer and Wright, 1995; Henry and Melton, 1998; Ninomiyaet al., 1999; Apelqvist et al., 1999; Kim et al., 2000; Hebroket al., 2000; Murtaugh et al., 2003) identified genes and signalsimportant for the pancreatic lineage, and these have been effec-tively used to form cells in the b cell lineage in vitro from hPSC.Definitive endoderm and subsequent pancreatic progenitorscan now be differentiated with high efficiencies (Kroon et al.,2008; D’Amour et al., 2005, 2006; Rezania et al., 2012). Thesecells can differentiate into functional b cells within 3–4 monthsfollowing transplantation into rodents (Kroon et al., 2008; Rezaniaet al., 2012), indicating that some cells in the preparation containthe developmental potential to develop into b cells if providedenough time and appropriate cues. Unfortunately, the months-long process the cells undergo in vivo is not understood, and itis unclear whether this process of in vivo differentiation wouldalso occur in human patients. Attempts to date at generatinginsulin-producing (INS+) cells from human pancreatic progeni-tors in vitro have generated cells with immature or abnormalphenotypes. These cells either fail to perform glucose-stimulated
428 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.
Resource
Generation of Functional HumanPancreatic b Cells In VitroFelicia W. Pagliuca,1,3 Jeffrey R. Millman,1,3 Mads Gurtler,1,3 Michael Segel,1 Alana Van Dervort,1 Jennifer Hyoje Ryu,1
Quinn P. Peterson,1 Dale Greiner,2 and Douglas A. Melton1,*1Department of Stem Cell and Regenerative Biology, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge,MA 02138, USA2Diabetes Center of Excellence, University of Massachusetts Medical School, 368 Plantation Street, AS7-2051, Worcester, MA 01605, USA3Co-first author*Correspondence: [email protected]://dx.doi.org/10.1016/j.cell.2014.09.040
SUMMARY
The generation of insulin-producing pancreatic bcells from stem cells in vitro would provide an un-precedented cell source for drug discovery and celltransplantation therapy in diabetes. However, insu-lin-producing cells previously generated from humanpluripotent stem cells (hPSC) lack many functionalcharacteristics of bona fide b cells. Here, we reporta scalable differentiation protocol that can generatehundreds of millions of glucose-responsive b cellsfrom hPSC in vitro. These stem-cell-derived b cells(SC-b) express markers found in mature b cells, fluxCa2+ in response to glucose, package insulin intosecretory granules, and secrete quantities of insulincomparable to adult b cells in response to multiplesequential glucose challenges in vitro. Furthermore,these cells secrete human insulin into the serum ofmice shortly after transplantation in a glucose-regu-latedmanner, and transplantation of these cells ame-liorates hyperglycemia in diabetic mice.
INTRODUCTION
The discovery of human pluripotent stem cells (hPSC) openedthe possibility of generating replacement cells and tissuesin the laboratory that could be used for disease treatment anddrug screening. Recent research has moved the stem cell fieldcloser to that goal through development of strategies to generatecells that would otherwise be difficult to obtain, like neurons orcardiomyocytes (Kriks et al., 2011; Shiba et al., 2012; Sonet al., 2011). These cells have also been transplanted into animalmodels, in some caseswith a beneficial effect like suppression ofarrhythmias with stem-cell-derived cardiomyocytes (Shiba et al.,2012), restoration of locomotion after spinal injury with oligoden-drocyte progenitors (Keirstead et al., 2005), or improved visionafter transplantation of retinal epithelial cells into rodent modelsof blindness (Lu et al., 2009).
One of the rapidly growing diseases that may be treatable bystem-cell-derived tissues is diabetes, affecting >300 million peo-ple worldwide, according to the International Diabetes Federa-
tion. Type 1 diabetes results from autoimmune destruction of bcells in the pancreatic islet, whereas themore common type 2 dia-betes results from peripheral tissue insulin resistance and b celldysfunction. Diabetic patients, particularly those suffering fromtype 1 diabetes, could potentially be cured through transplanta-tion of new b cells. Patients transplantedwith cadaveric human is-lets can be made insulin independent for 5 years or longer via thisstrategy, but this approach is limited because of the scarcity andquality of donor islets (Bellin et al., 2012). The generation of anunlimited supply of human b cells from stem cells could extendthis therapy to millions of new patients and could be an importanttest case for translating stem cell biology into the clinic. This isbecause only a single cell type, the b cell, likely needs to be gener-ated, and the mode of delivery is understood: transplantation to avascularized location within the body with immunoprotection.Pharmaceutical screening to identify new drugs that improve b
cell function, survival, or proliferation is also hindered by limitedsupplies of islets and high variability due to differential causesof death, donor genetic background, and other factors in theirisolation. A consistent, uniform supply of stem-cell-derived b cellswould provide a unique and valuable drug discovery platform fordiabetes. Additionally, genetically diverse stem-cell-derived bcells could be used for disease modeling in vitro or in vivo.Studies on pancreatic development in model organisms
(Gamer and Wright, 1995; Henry and Melton, 1998; Ninomiyaet al., 1999; Apelqvist et al., 1999; Kim et al., 2000; Hebroket al., 2000; Murtaugh et al., 2003) identified genes and signalsimportant for the pancreatic lineage, and these have been effec-tively used to form cells in the b cell lineage in vitro from hPSC.Definitive endoderm and subsequent pancreatic progenitorscan now be differentiated with high efficiencies (Kroon et al.,2008; D’Amour et al., 2005, 2006; Rezania et al., 2012). Thesecells can differentiate into functional b cells within 3–4 monthsfollowing transplantation into rodents (Kroon et al., 2008; Rezaniaet al., 2012), indicating that some cells in the preparation containthe developmental potential to develop into b cells if providedenough time and appropriate cues. Unfortunately, the months-long process the cells undergo in vivo is not understood, and itis unclear whether this process of in vivo differentiation wouldalso occur in human patients. Attempts to date at generatinginsulin-producing (INS+) cells from human pancreatic progeni-tors in vitro have generated cells with immature or abnormalphenotypes. These cells either fail to perform glucose-stimulated
428 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.
insulin secretion in vitro, fail to express appropriate b cell markerssuch as NKX6-1 or PDX1, abnormally coexpress other hormoneslike glucagon (GCG), fail to function after transplantation in vivo,or display a combination of these abnormal features (D’Amouret al., 2006; Cheng et al., 2012; Hrvatin et al., 2014; Narayananet al., 2014; Xie et al., 2013; Nostro et al., 2011).Herein, we report the discovery of a strategy for large-scale
production of functional human b cells from hPSC in vitro. Byusing sequential modulation of multiple signaling pathways in athree-dimensional cell culture system, without any transgenesor genetic modification, we generate glucose-responsive,monohormonal insulin-producing cells that show key featuresof a bona fide b cell, including coexpression of key b cell markersand b cell ultrastructure. Furthermore, these cells mimic thefunction of human islets both in vitro and in vivo. Finally, wedemonstrate the potential utility of these cells for in vivo trans-plantation therapy for diabetes.
RESULTS
Generation of Glucose-Sensing Insulin-Secretingb Cells In VitroOur strategy to generate functional b cells from hPSC in vitro isoutlined in Figure 1A. To produce large numbers, we used a scal-
able suspension-based culture system that can generate >108
hPSCs and later differentiated cell types (modified from Schulzet al., 2012). Clusters of cells (!100–200 mm in diameter, eachcluster containing several hundred cells) from a human embry-onic stem cell (hESC) line (HUES8) or two human-induced plurip-otent stem cell (hiPSC) lines (hiPSC-1 and hiPSC-2) wereinduced into definitive endoderm (>95% SOX17+ cells, DE cellsin Figure 1A) and subsequently early pancreatic progenitors(>85% PDX1+ cells, PP1 cells in Figure 1A).Transplantation of pancreatic progenitors expressing PDX1+/
NKX6-1+ (PP2 in Figure 1A) into mice gives rise to functional bcells in vivo after 3–4 months (Kroon et al., 2008; Rezania et al.,2012). And previous studies had shown that these PDX1+/NKX6-1+ pancreatic progenitors (PP2) could be further differen-tiated in vitro into some INS+ cells along with INS+/GCG+ orINS+/SST+ polyhormonal (PH) cells (Nostro et al., 2011; Rezaniaet al., 2012; Thowfeequ et al., 2007; Aguayo-Mazzucato et al.,2013; D’Amour et al., 2006; Hrvatin et al., 2014). We use thenomenclature PH (polyhormonal, Figure 1A) to refer to this cellpopulation of in-vitro-differentiated hPSCs. Transcriptional anal-ysis of in-vitro-differentiated PH cells showed that these cellsresemble human fetal and not adult b cells (Hrvatin et al.,2014). Because these PH cells show neither glucose-stimulatedinsulin secretion (GSIS) nor other key properties of bona fide b
Figure 1. SC-b Cells Generated In VitroSecrete Insulin in Response to MultipleSequential High-Glucose Challenges likePrimary Human b Cells(A) Schematic of directed differentiation from
hPSC into INS+ cells via new or previously pub-
lished control differentiations.
(B–D) Representative ELISA measurements of
secreted human insulin from HUES8 SC-b cells
(B), PH cells (C), and primary b (1"b) cells (D)
challenged sequentially with 2, 20, 2, 20, 2, and
20 mM glucose, with a 30 min incubation for each
concentration (see Experimental Procedures). Af-
ter sequential low/high-glucose challenges, cells
were depolarized with 30 mM KCl.
(E–G) Box and whisker plots of secreted human
insulin from different biological batches of HUES8
(open circles) and hiPSC SC-b (black circles) cells
(E; n = 12), biological batches of PH cells (F; n = 5),
and primary b cells (G; n = 4). Each circle is the
average value for all sequential challenges with
2 mM or 20 mM glucose in a batch. Insulin
secretion at 20 mM ranged 0.23–2.7 mIU/103 cells
for SC-b cells and 1.5–4.5 mIU/103 cells for human
islets, and the stimulation index ranged 0.4–4.1 for
SC-b cells and 0.6–4.8 for primary adult. The thick
horizontal line indicates the median.
SeealsoFiguresS1andS2AandTableS1. *p<0.05
when comparing insulin secretion at 20 mM versus
2 mM with paired t test. Act A, activin A; CHIR,
CHIR99021, aGSK3a/b inhibitor; KGF, keratinocyte
growth factor or FGF family member 7; RA, retinoic
acid; SANT1, sonic hedgehog pathway antagonist;
LDN, LDN193189, a BMP type 1 receptor inhibitor;
PdbU, Phorbol 12,13-dibutyrate, a protein kinase C
activator; Alk5i, Alk5 receptor inhibitor II; T3, triio-
dothyronine, a thyroid hormone; XXI, g-secretase
inhibitor; Betacellulin, EGF family member.
Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc. 429
in vivo and in vitro. The characterization of SC-b cell physiologycan be extended in future studies by performing a time course ofinsulin synthesis and secretion and determining how long thesecells maintain this phenotype in vitro and in vivo.
We note that the insulin detected in serum of transplantedSC-b cells was generally lower than that of animals transplantedwith cadaveric donor islets on a per cell basis (Tables S2 and S3).One possibility is that the other cell types in islets, mesenchyme,endothelial cells, or other endocrine cell types may help engraft-ment or the vascularization necessary for glucose and insulinexchange from the mouse vasculature. Another possibility isthat the slightly larger size of the SC-b clusters compared tothe human islets that we received may influence relative insulin
amounts secreted, as it has been observed that smaller isletssecrete more insulin per cell than larger islets (Fujita et al.,2011). Nonetheless, the fact that transplantation of SC-b cellsconsistently results in rapid and glucose-responsive insulin pro-duction and that these cells function to treat diabetes in a mousemodel is encouraging.The results described here suggest that SC-b cells present an
opportunity for cell therapy. Limited supplies of donated cadav-eric islets and the very small amount of human b cell replicationachieved in vitro have severely limited human b cell supplies todate. This limitation has restricted transplantation options for pa-tients as well as high-throughput drug screening and diseasemodeling. A single 68 kg (150 lb) patient requires roughly 340–750 million transplanted islet cells to effectively resolve type 1diabetes via islet transplantation (McCall and Shapiro, 2012;Shapiro et al., 2006). The strategy that we describe here mayaddress this limitation, as it utilizes a renewable cell sourceand a scalable method, with cells grown 300 million at a timein suspension in a single 500 ml flask. Thus, one or two flasksmay be sufficient for treatment of a patient. An additional clinicaladvantage of SC-b cells compared to previously proposed ther-apies using pancreatic progenitors is that these cells do notrequire an extended posttransplantation period before obser-ving insulin production. Immediate insulin production makes ittheoretically possible to reduce patient exogenous insulin re-quirements from the first few days posttransplantation.These cells also present new opportunities for b cell studies. A
single flask could provide enough cells for screening 30,000compounds in a 384-well format seeded with 10,000 cells perwell. Unlike primary human b cells, SC-b cells can also be gener-ated from pluripotent cells of any desired genetic background.hiPSC, which function like hESC, can be generated from any fi-broblasts or other somatic cell types through introduction of asmall set of pluripotency genes (Takahashi and Yamanaka,2006). hiPSC cells from patients with diabetes or other metabolicsyndromes have been derived. Work in progress in our labora-tory has demonstrated that this protocol can be applied to differ-entiate SC-b cells from diabetic hiPSC lines, which could beused for diseasemodeling and the study of b cell function or sus-ceptibility to stress or immune attack. Furthermore, technologieslike TALEN and CRISPR enable genome editing of hESC orhiPSC cells to test variants identified by genome-wide asso-ciation studies (GWAS). SC-b cell drug responses could becompared between genetically matched pairs of mutant andnonmutant b cells (Ding et al., 2013). Identification and testingof novel biomarkers for b cell function or pharmacogenetics isalso enabled by the combination of these technologies. ThusSC-b cells provide a new platform for in vitro drug discoveryand characterization for human metabolism and diabetes.The generation of SC-b cells provides a potentially useful step
toward the generation of islets and pancreatic organs. Incorpo-rating pancreatic niche cells, such as mesenchymal or endothe-lial cells, into cultures of stem-cell-derived pancreatic cells maybe beneficial (Sneddon et al., 2012; Lammert et al., 2001). Otherevidence suggests that the presence of a and g cells may beimportant for precise tuning of normal b cell function (Rodri-guez-Diaz et al., 2011). Tissue engineering of a pancreatic organwill require incorporation of exocrine and ductal tissue, possibly
Figure 6. Transplanted SC-b Cells Rapidly Ameliorate Hyperglyce-mia in Diabetic Mice(A) Fasting blood glucose measurements of progressively diabetic NRG-Akita
mice transplanted with HUES8 SC-b cells (53 106 cells; open circles; n = 6) or
PH cells (53 106 cells; closed square; n = 6 for 0, 18, and 28 days and n = 4 for
53, 84, and 112 days). Glucose measurements were saturated at 600 mg/dl.
*p < 0.05 comparing the two cell groups on the same day with unpaired t test.
Data presented as mean ± SEM.
(B) Blood glucose measurements of mice that have been transplanted
137 days prior with HUES8 SC-b cells (n = 5) or PH cells (n = 1) and a separate
independent cohort of mice transplanted 34 days prior with human islets
(4,000 IEQ; closed triangle; n = 6). Note: It was not possible to coordinate
receipt of human cadaveric islets and the production of SC-b cells. So the data
in Figure 6B come from a different animal cohort. One mouse that received
HUES8 SC-b cells died after a blood draw taken in (C), and five mice that
received PH cells died over the duration of the observation period. Data pre-
sented as mean ± SEM.
(C) ELISA measurements of human insulin from the serum of individual mice
transplanted with HUES8 SC-b cells (n = 6) or PH cells (n = 2). Mice were fasted
for 16 hr, and then blood glucose wasmeasured before (white bars) and 30min
after (black bars) a glucose injection of mice, 126 days posttransplantation. p <
0.05 for transplanted HUES8 SC-b cells insulin concentration 30 min versus
0 min after glucose injection with paired t test.
436 Cell 159, 428–439, October 9, 2014 ª2014 Elsevier Inc.

cPB
cPFFib
cPE
cDd0
cPB
d7 cPF
cPE
Transfection 4 days
Initiation 7 days
Conversion 2–3 weeks
Colonies Intermediates Intermediates Fibroblasts
EGF, bFGF, CHIR, NECA,NaB, Par, RG
Activin A, CHIR, NECA,NaB, Par, RG
cPF specification 2–4 passages
CHIR, A83, EGF, and bFGF
cPF expansion up to 3 months
CHIR, A83, EGF, and bFGF
cPE differentiation
5 days
FGF7, FGF10, A83, C-E, LDN, RA, and GDC for 2 days EGF, EX4, A83, C-E, LDN, PBDu, and NIC for 3 days
cPE expansion 2 months
EGF, bFGF, and A83
cPB differentiation 14+7 days
Alk5i, T3, LDN, C-E, Vc, BayK for 7 days
Alk5i, T3, AXLi, N-Cys, Vc, BayK for 7 days
cPB differentiation 10+8–12 days
Ex-4, A83, NIC, FSK, DEX,C-E, Vc,
and BayK
Transplantation of cPB cells
Evaluate beta cell function in vivo
and stem cell-derived beta-like cells6–8. Therefore, our resultsdemonstrate that human adult fibroblasts can be used toefficiently and rapidly generate functional cPB cells, a findingthat opens up opportunities for patient-specific analysis of beta-cell properties and cell therapy approaches.
DiscussionVarious strategies towards the generation of functional beta cellsare currently being pursued, including directed differentiationof pluripotent stem cells2–4,6–8 and direct reprogramming of
different endodermal cell types48–51. In this study, we (I)developed a simple, robust and non-integrating approach forthe inter-lineage conversion of mesodermal human fibroblastsinto definitive endodermal progenitor cells (cDE cells) followingthe CASD transdifferentiation approach; (II) showed that cDEcells could be further specified into posterior foregut-likeprogenitor (cPF) cells that can be greatly expanded;(III) demonstrated that cPF cells can be further differentiatedinto expandable pancreatic endodermal progenitor (cPE) cells;(IV) reported that these cPE cells can give rise to functional
MergePDX1C-pep DAPI
MergeNKX6.1C-pep DAPI
MergeGCGC-pep DAPI
MergeSSTC-pep DAPI
Transplantationof cPB cells
Evaluate beta cell function in vivo
cPB
a
0
100
200
300
400
500
600
* * * *
Weeks post STZ
Blo
od g
luco
se (
mg
dl–1
)c
1 2 3 54 6
cPB FibIn vivo GSIS
Fib
Fast
2.2-Fold, P=0.01
Hum
an C
-pep
tide
(pM
)
0
100
200
300
400
500
Gluc Fast Gluc
Nephrectomy
db
cPB
Figure 6 | Transplanted cPB cells remain functional and protect mice from chemically induced diabetes. (a) Schematic representation of thetransplantation of cPB into immunodeficient mice. (b) ELISA analysis of serum from fasted and glucose-challenged mice 2 months post transplantation witheither fibroblasts (Fib) or cPB are shown. cPB graft-bearing mice exhibit significant higher levels of circulating human C-peptide in serum after a glucosebolus, indicating that transplanted cPB cells remain functional in vivo. Mice transplanted with Fib controls do not exhibit circulating human C-peptide. n¼ 12mice for cPB and n¼ 5 mice for Fib. P value was calculated using a two-tailed Student’s t-test. (c) Immunofluorescence analysis of 2-month-old cPB cellgrafts shows co-expression of C-peptide (C-pep) and the beta-cell transcription factors PDX1 and NKX6.1 but not the hormones glucagon (GCG) andsomatostatin (SST). Scale bar, 50mm. Data shown are representative of two mice. (d) Fed blood glucose levels of mice-bearing cPB grafts with circulatinghuman C-peptide levels above 200 pM after glucose stimulation and control fibroblasts are shown. Mice were treated with the mouse-specific beta-celltoxin streptozotocine (STZ) to ablate endogenous beta cells. Uni-lateral nephrectomy of cPB graft-bearing mice 5 weeks after STZ treatment resulted in arapid rise in blood glucose levels, directly demonstrating euglycemic control due to cPB grafts after STZ treatment in these mice. n¼ 6 mice for cPB andn¼ 6 mice for Fib. P value was calculated using a two-tailed Student’s t-test. *Po0.05.
NATURE COMMUNICATIONS | DOI: 10.1038/ncomms10080 ARTICLE
NATURE COMMUNICATIONS | 7:10080 | DOI: 10.1038/ncomms10080 | www.nature.com/naturecommunications 9
and stem cell-derived beta-like cells6–8. Therefore, our resultsdemonstrate that human adult fibroblasts can be used toefficiently and rapidly generate functional cPB cells, a findingthat opens up opportunities for patient-specific analysis of beta-cell properties and cell therapy approaches.
DiscussionVarious strategies towards the generation of functional beta cellsare currently being pursued, including directed differentiationof pluripotent stem cells2–4,6–8 and direct reprogramming of
different endodermal cell types48–51. In this study, we (I)developed a simple, robust and non-integrating approach forthe inter-lineage conversion of mesodermal human fibroblastsinto definitive endodermal progenitor cells (cDE cells) followingthe CASD transdifferentiation approach; (II) showed that cDEcells could be further specified into posterior foregut-likeprogenitor (cPF) cells that can be greatly expanded;(III) demonstrated that cPF cells can be further differentiatedinto expandable pancreatic endodermal progenitor (cPE) cells;(IV) reported that these cPE cells can give rise to functional
MergePDX1C-pep DAPI
MergeNKX6.1C-pep DAPI
MergeGCGC-pep DAPI
MergeSSTC-pep DAPI
Transplantationof cPB cells
Evaluate beta cell function in vivo
cPB
a
0
100
200
300
400
500
600
* * * *
Weeks post STZ
Blo
od g
luco
se (
mg
dl–1
)
c
1 2 3 54 6
cPB FibIn vivo GSIS
Fib
Fast
2.2-Fold, P=0.01
Hum
an C
-pep
tide
(pM
)
0
100
200
300
400
500
Gluc Fast Gluc
Nephrectomy
db
cPB
Figure 6 | Transplanted cPB cells remain functional and protect mice from chemically induced diabetes. (a) Schematic representation of thetransplantation of cPB into immunodeficient mice. (b) ELISA analysis of serum from fasted and glucose-challenged mice 2 months post transplantation witheither fibroblasts (Fib) or cPB are shown. cPB graft-bearing mice exhibit significant higher levels of circulating human C-peptide in serum after a glucosebolus, indicating that transplanted cPB cells remain functional in vivo. Mice transplanted with Fib controls do not exhibit circulating human C-peptide. n¼ 12mice for cPB and n¼ 5 mice for Fib. P value was calculated using a two-tailed Student’s t-test. (c) Immunofluorescence analysis of 2-month-old cPB cellgrafts shows co-expression of C-peptide (C-pep) and the beta-cell transcription factors PDX1 and NKX6.1 but not the hormones glucagon (GCG) andsomatostatin (SST). Scale bar, 50mm. Data shown are representative of two mice. (d) Fed blood glucose levels of mice-bearing cPB grafts with circulatinghuman C-peptide levels above 200 pM after glucose stimulation and control fibroblasts are shown. Mice were treated with the mouse-specific beta-celltoxin streptozotocine (STZ) to ablate endogenous beta cells. Uni-lateral nephrectomy of cPB graft-bearing mice 5 weeks after STZ treatment resulted in arapid rise in blood glucose levels, directly demonstrating euglycemic control due to cPB grafts after STZ treatment in these mice. n¼ 6 mice for cPB andn¼ 6 mice for Fib. P value was calculated using a two-tailed Student’s t-test. *Po0.05.
NATURE COMMUNICATIONS | DOI: 10.1038/ncomms10080 ARTICLE
NATURE COMMUNICATIONS | 7:10080 | DOI: 10.1038/ncomms10080 | www.nature.com/naturecommunications 9
ARTICLEReceived 11 Jul 2015 | Accepted 2 Nov 2015 | Published 6 Jan 2016
Human pancreatic beta-like cells converted fromfibroblastsSaiyong Zhu1,*, Holger A. Russ2,*, Xiaojing Wang1, Mingliang Zhang1, Tianhua Ma1, Tao Xu1, Shibing Tang1,
Matthias Hebrok2 & Sheng Ding1,3
Pancreatic beta cells are of great interest for biomedical research and regenerative medicine.
Here we show the conversion of human fibroblasts towards an endodermal cell fate by
employing non-integrative episomal reprogramming factors in combination with specific
growth factors and chemical compounds. On initial culture, converted definitive endodermal
progenitor cells (cDE cells) are specified into posterior foregut-like progenitor cells
(cPF cells). The cPF cells and their derivatives, pancreatic endodermal progenitor cells
(cPE cells), can be greatly expanded. A screening approach identified chemical compounds
that promote the differentiation and maturation of cPE cells into functional pancreatic
beta-like cells (cPB cells) in vitro. Transplanted cPB cells exhibit glucose-stimulated insulin
secretion in vivo and protect mice from chemically induced diabetes. In summary, our study
has important implications for future strategies aimed at generating high numbers of
functional beta cells, which may help restoring normoglycemia in patients suffering from
diabetes.
DOI: 10.1038/ncomms10080 OPEN
1 Gladstone Institute of Cardiovascular Disease, University of California, San Francisco, California 94158, USA. 2 Diabetes Center, Department of Medicine,University of California, San Francisco, San Francisco, California 94143, USA. 3 Department of Pharmaceutical Chemistry, University of California SanFrancisco, 600 16th Street, San Francisco, California 94158, USA. * These authors contributed equally to this work. Correspondence and requests for materialsshould be addressed to M.H. (email: [email protected]) or to S.D. (email: [email protected]).
NATURE COMMUNICATIONS | 7:10080 | DOI: 10.1038/ncomms10080 | www.nature.com/naturecommunications 1
ARTICLEReceived 11 Jul 2015 | Accepted 2 Nov 2015 | Published 6 Jan 2016
Human pancreatic beta-like cells converted fromfibroblastsSaiyong Zhu1,*, Holger A. Russ2,*, Xiaojing Wang1, Mingliang Zhang1, Tianhua Ma1, Tao Xu1, Shibing Tang1,
Matthias Hebrok2 & Sheng Ding1,3
Pancreatic beta cells are of great interest for biomedical research and regenerative medicine.
Here we show the conversion of human fibroblasts towards an endodermal cell fate by
employing non-integrative episomal reprogramming factors in combination with specific
growth factors and chemical compounds. On initial culture, converted definitive endodermal
progenitor cells (cDE cells) are specified into posterior foregut-like progenitor cells
(cPF cells). The cPF cells and their derivatives, pancreatic endodermal progenitor cells
(cPE cells), can be greatly expanded. A screening approach identified chemical compounds
that promote the differentiation and maturation of cPE cells into functional pancreatic
beta-like cells (cPB cells) in vitro. Transplanted cPB cells exhibit glucose-stimulated insulin
secretion in vivo and protect mice from chemically induced diabetes. In summary, our study
has important implications for future strategies aimed at generating high numbers of
functional beta cells, which may help restoring normoglycemia in patients suffering from
diabetes.
DOI: 10.1038/ncomms10080 OPEN
1 Gladstone Institute of Cardiovascular Disease, University of California, San Francisco, California 94158, USA. 2 Diabetes Center, Department of Medicine,University of California, San Francisco, San Francisco, California 94143, USA. 3 Department of Pharmaceutical Chemistry, University of California SanFrancisco, 600 16th Street, San Francisco, California 94158, USA. * These authors contributed equally to this work. Correspondence and requests for materialsshould be addressed to M.H. (email: [email protected]) or to S.D. (email: [email protected]).
NATURE COMMUNICATIONS | 7:10080 | DOI: 10.1038/ncomms10080 | www.nature.com/naturecommunications 1
ARTICLEReceived 11 Jul 2015 | Accepted 2 Nov 2015 | Published 6 Jan 2016
Human pancreatic beta-like cells converted fromfibroblastsSaiyong Zhu1,*, Holger A. Russ2,*, Xiaojing Wang1, Mingliang Zhang1, Tianhua Ma1, Tao Xu1, Shibing Tang1,
Matthias Hebrok2 & Sheng Ding1,3
Pancreatic beta cells are of great interest for biomedical research and regenerative medicine.
Here we show the conversion of human fibroblasts towards an endodermal cell fate by
employing non-integrative episomal reprogramming factors in combination with specific
growth factors and chemical compounds. On initial culture, converted definitive endodermal
progenitor cells (cDE cells) are specified into posterior foregut-like progenitor cells
(cPF cells). The cPF cells and their derivatives, pancreatic endodermal progenitor cells
(cPE cells), can be greatly expanded. A screening approach identified chemical compounds
that promote the differentiation and maturation of cPE cells into functional pancreatic
beta-like cells (cPB cells) in vitro. Transplanted cPB cells exhibit glucose-stimulated insulin
secretion in vivo and protect mice from chemically induced diabetes. In summary, our study
has important implications for future strategies aimed at generating high numbers of
functional beta cells, which may help restoring normoglycemia in patients suffering from
diabetes.
DOI: 10.1038/ncomms10080 OPEN
1 Gladstone Institute of Cardiovascular Disease, University of California, San Francisco, California 94158, USA. 2 Diabetes Center, Department of Medicine,University of California, San Francisco, San Francisco, California 94143, USA. 3 Department of Pharmaceutical Chemistry, University of California SanFrancisco, 600 16th Street, San Francisco, California 94158, USA. * These authors contributed equally to this work. Correspondence and requests for materialsshould be addressed to M.H. (email: [email protected]) or to S.D. (email: [email protected]).
NATURE COMMUNICATIONS | 7:10080 | DOI: 10.1038/ncomms10080 | www.nature.com/naturecommunications 1
Saiyong Zhu1,*, Holger A. Russ2,*, Xiaojing Wang1, Mingliang Zhang1, Tianhua Ma1, Tao Xu1, Shibing Tang1, Matthias Hebrok2 & Sheng Ding1,3
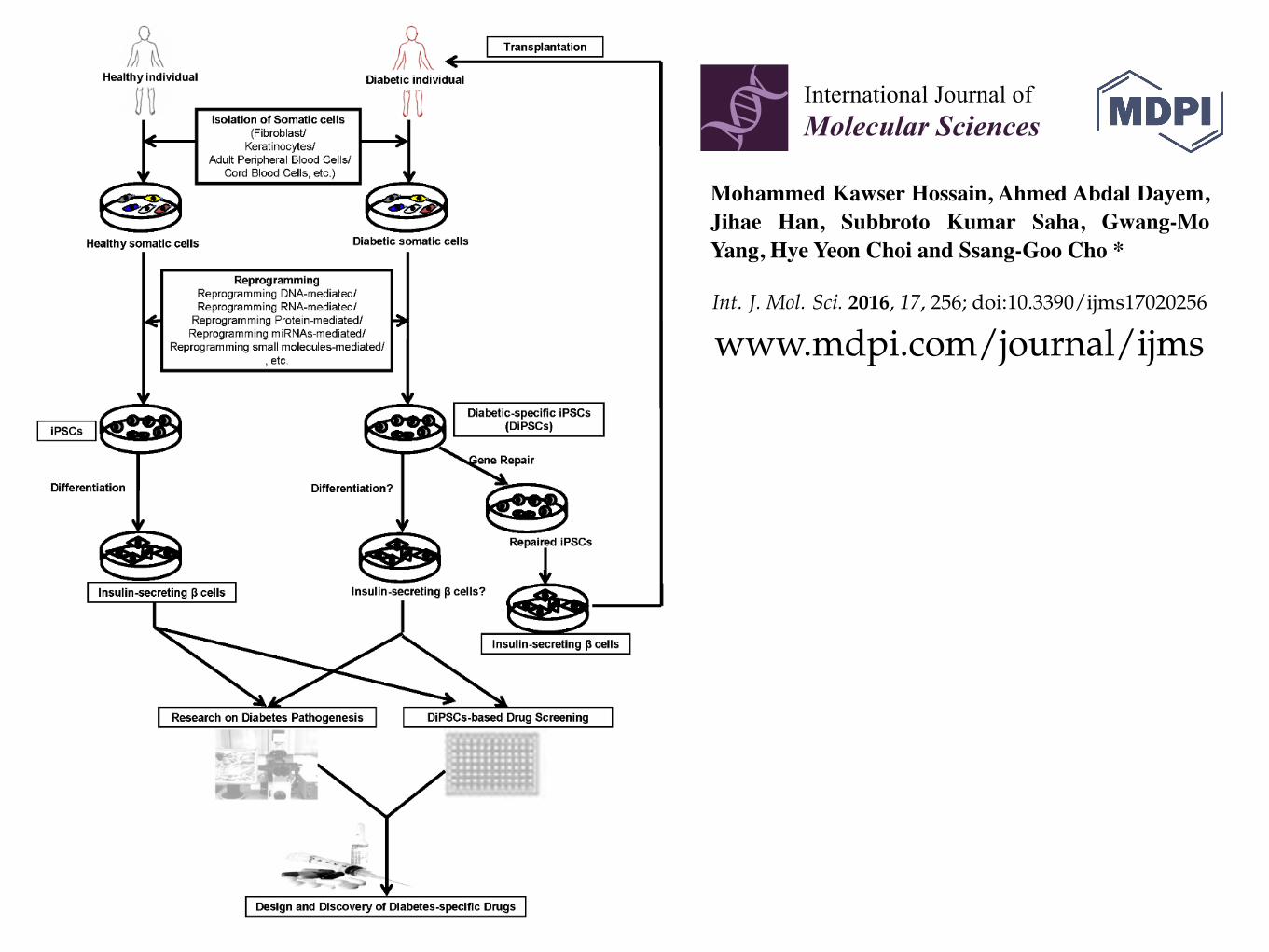
Int. J. Mol. Sci. 2016, 17, 256 4 of 17
order to solve the problem of complication of the organogenesis process that hampers the in vitroderivation of organs from patient’s pluripotent stem cells, Kobayashi et al. succeeded to generatepluripotent stem cell-derived pancreas via compensation of the empty space of the pancreaticdevelopmental niche by the injection of mouse wild type pluripotent stem cells into the blastocystof the pancreatogenesis-disabled mouse (Pdx1´{´) [44]. Interestingly, they confirmed the possibilityof interspecific chimera production between mouse and rat with injection of mouse or rat PSCs intoembryos from the other species. The injected pluripotent stem cell-derived cells were distributedthroughout the body and appeared to have normal function. In 2012, Ohmine et al. were able togenerate another type of DiPSCs from the keratinocytes of an elderly T2DM patient, opening up a newvenue in regenerative medicine for elder diabetic patients [45].
Int. J. Mol. Sci. 2016, 17, 256 4 of 17
possibility of interspecific chimera production between mouse and rat with injection of mouse or rat PSCs into embryos from the other species. The injected pluripotent stem cell-derived cells were distributed throughout the body and appeared to have normal function. In 2012, Ohmine et al. were able to generate another type of DiPSCs from the keratinocytes of an elderly T2DM patient, opening up a new venue in regenerative medicine for elder diabetic patients [45].
Figure 1. Schematic presentation of generation of iPSCs (induced pluripotent stem cells) from healthy and diabetic patients and their application in the patient-specific iPSC-based diabetic therapy. Footprint-free iPSCs can be generated from healthy individual- or diabetic patient-derived somatic cells using reprogramming DNA-, RNA-, protein-, miRNA-, or small molecule-mediated reprogramming system. DiPSCs (iPSCs derived from diabetic patients) can be further differentiated into insulin-secreting pancreatic β cells for cell-based diabetic drug screening or for transplantation into diabetic patients for cell therapy. DiPSCs can also be repaired by gene correction and then be differentiated into functional insulin-secreting pancreatic β cells, which can be transplanted into a specific diabetic patient. For disease modeling, DiPSCs can be differentiated into insulin-secreting pancreatic β cells for drug screening or pathogenesis studies to develop the compounds or therapies to treat specific type of diabetes.
Figure 1. Schematic presentation of generation of iPSCs (induced pluripotent stem cells) fromhealthy and diabetic patients and their application in the patient-specific iPSC-based diabetictherapy. Footprint-free iPSCs can be generated from healthy individual- or diabetic patient-derivedsomatic cells using reprogramming DNA-, RNA-, protein-, miRNA-, or small molecule-mediatedreprogramming system. DiPSCs (iPSCs derived from diabetic patients) can be further differentiatedinto insulin-secreting pancreatic � cells for cell-based diabetic drug screening or for transplantationinto diabetic patients for cell therapy. DiPSCs can also be repaired by gene correction and then bedifferentiated into functional insulin-secreting pancreatic � cells, which can be transplanted into aspecific diabetic patient. For disease modeling, DiPSCs can be differentiated into insulin-secretingpancreatic � cells for drug screening or pathogenesis studies to develop the compounds or therapies totreat specific type of diabetes.
International Journal of
Molecular Sciences
Review
Recent Advances in Disease Modeling and DrugDiscovery for Diabetes Mellitus Using InducedPluripotent Stem Cells
Mohammed Kawser Hossain, Ahmed Abdal Dayem, Jihae Han, Subbroto Kumar Saha,Gwang-Mo Yang, Hye Yeon Choi and Ssang-Goo Cho *
Department of Animal Biotechnology, Animal Resources Research Center,and Incurable Disease Animal Model and Stem Cell Institute (IDASI), Konkuk University, Gwangjin-gu,Seoul 05029, Korea; [email protected] (M.K.H.); [email protected] (A.A.D.);[email protected] (J.H.); [email protected] (S.K.S.); [email protected] (G.-M.Y.);[email protected] (H.Y.C.)* Correspondence: [email protected]; Tel.: +82-2-450-4207
Academic Editor: Wenbin DengReceived: 3 January 2016; Accepted: 15 February 2016; Published: 19 February 2016
Abstract: Diabetes mellitus (DM) is a widespread metabolic disease with a progressive incidenceof morbidity and mortality worldwide. Despite extensive research, treatment options for diabeticpatients remains limited. Although significant challenges remain, induced pluripotent stem cells(iPSCs) have the capacity to differentiate into any cell type, including insulin-secreting pancreatic �
cells, highlighting its potential as a treatment option for DM. Several iPSC lines have recently beenderived from both diabetic and healthy donors. Using different reprogramming techniques, iPSCswere differentiated into insulin-secreting pancreatic �cells. Furthermore, diabetes patient-derivediPSCs (DiPSCs) are increasingly being used as a platform to perform cell-based drug screening inorder to develop DiPSC-based cell therapies against DM. Toxicity and teratogenicity assays basedon iPSC-derived cells can also provide additional information on safety before advancing drugs toclinical trials. In this review, we summarize recent advances in the development of techniques fordifferentiation of iPSCs or DiPSCs into insulin-secreting pancreatic � cells, their applications in drugscreening, and their role in complementing and replacing animal testing in clinical use. Advancesin iPSC technologies will provide new knowledge needed to develop patient-specific iPSC-baseddiabetic therapies.
Keywords: diabetes mellitus; induced pluripotent stem cells; insulin-secreting � cells; cell-baseddrug screening; iPSC-based diabetic therapy
1. Introduction
Diabetes mellitus (DM), commonly referred to as diabetes, is the most widespread metabolicdisease worldwide [1]. DM develops from glucose regulation defects, which cause high blood sugarlevels over a prolonged period. DM leads to severe hyperglycemia and several complications, includingdiabetic ketoacidosis, cardiovascular disease, kidney failure, foot ulcers, and tissue or organ damage.More than three hundred million people are suffering from DM and the number of DM patients issteadily increasing.
DM is classified into three types: type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus(T2DM), and gestational diabetes [2–6]. T1DM, previously known as insulin-dependent DM (IDDM) orjuvenile diabetes, results from the destruction of insulin-secreting pancreatic � cells as a consequence
Int. J. Mol. Sci. 2016, 17, 256; doi:10.3390/ijms17020256 www.mdpi.com/journal/ijms
International Journal of
Molecular Sciences
Review
Recent Advances in Disease Modeling and DrugDiscovery for Diabetes Mellitus Using InducedPluripotent Stem Cells
Mohammed Kawser Hossain, Ahmed Abdal Dayem, Jihae Han, Subbroto Kumar Saha,Gwang-Mo Yang, Hye Yeon Choi and Ssang-Goo Cho *
Department of Animal Biotechnology, Animal Resources Research Center,and Incurable Disease Animal Model and Stem Cell Institute (IDASI), Konkuk University, Gwangjin-gu,Seoul 05029, Korea; [email protected] (M.K.H.); [email protected] (A.A.D.);[email protected] (J.H.); [email protected] (S.K.S.); [email protected] (G.-M.Y.);[email protected] (H.Y.C.)* Correspondence: [email protected]; Tel.: +82-2-450-4207
Academic Editor: Wenbin DengReceived: 3 January 2016; Accepted: 15 February 2016; Published: 19 February 2016
Abstract: Diabetes mellitus (DM) is a widespread metabolic disease with a progressive incidenceof morbidity and mortality worldwide. Despite extensive research, treatment options for diabeticpatients remains limited. Although significant challenges remain, induced pluripotent stem cells(iPSCs) have the capacity to differentiate into any cell type, including insulin-secreting pancreatic �
cells, highlighting its potential as a treatment option for DM. Several iPSC lines have recently beenderived from both diabetic and healthy donors. Using different reprogramming techniques, iPSCswere differentiated into insulin-secreting pancreatic �cells. Furthermore, diabetes patient-derivediPSCs (DiPSCs) are increasingly being used as a platform to perform cell-based drug screening inorder to develop DiPSC-based cell therapies against DM. Toxicity and teratogenicity assays basedon iPSC-derived cells can also provide additional information on safety before advancing drugs toclinical trials. In this review, we summarize recent advances in the development of techniques fordifferentiation of iPSCs or DiPSCs into insulin-secreting pancreatic � cells, their applications in drugscreening, and their role in complementing and replacing animal testing in clinical use. Advancesin iPSC technologies will provide new knowledge needed to develop patient-specific iPSC-baseddiabetic therapies.
Keywords: diabetes mellitus; induced pluripotent stem cells; insulin-secreting � cells; cell-baseddrug screening; iPSC-based diabetic therapy
1. Introduction
Diabetes mellitus (DM), commonly referred to as diabetes, is the most widespread metabolicdisease worldwide [1]. DM develops from glucose regulation defects, which cause high blood sugarlevels over a prolonged period. DM leads to severe hyperglycemia and several complications, includingdiabetic ketoacidosis, cardiovascular disease, kidney failure, foot ulcers, and tissue or organ damage.More than three hundred million people are suffering from DM and the number of DM patients issteadily increasing.
DM is classified into three types: type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus(T2DM), and gestational diabetes [2–6]. T1DM, previously known as insulin-dependent DM (IDDM) orjuvenile diabetes, results from the destruction of insulin-secreting pancreatic � cells as a consequence
Int. J. Mol. Sci. 2016, 17, 256; doi:10.3390/ijms17020256 www.mdpi.com/journal/ijms
International Journal of
Molecular Sciences
Review
Recent Advances in Disease Modeling and DrugDiscovery for Diabetes Mellitus Using InducedPluripotent Stem Cells
Mohammed Kawser Hossain, Ahmed Abdal Dayem, Jihae Han, Subbroto Kumar Saha,Gwang-Mo Yang, Hye Yeon Choi and Ssang-Goo Cho *
Department of Animal Biotechnology, Animal Resources Research Center,and Incurable Disease Animal Model and Stem Cell Institute (IDASI), Konkuk University, Gwangjin-gu,Seoul 05029, Korea; [email protected] (M.K.H.); [email protected] (A.A.D.);[email protected] (J.H.); [email protected] (S.K.S.); [email protected] (G.-M.Y.);[email protected] (H.Y.C.)* Correspondence: [email protected]; Tel.: +82-2-450-4207
Academic Editor: Wenbin DengReceived: 3 January 2016; Accepted: 15 February 2016; Published: 19 February 2016
Abstract: Diabetes mellitus (DM) is a widespread metabolic disease with a progressive incidenceof morbidity and mortality worldwide. Despite extensive research, treatment options for diabeticpatients remains limited. Although significant challenges remain, induced pluripotent stem cells(iPSCs) have the capacity to differentiate into any cell type, including insulin-secreting pancreatic �
cells, highlighting its potential as a treatment option for DM. Several iPSC lines have recently beenderived from both diabetic and healthy donors. Using different reprogramming techniques, iPSCswere differentiated into insulin-secreting pancreatic �cells. Furthermore, diabetes patient-derivediPSCs (DiPSCs) are increasingly being used as a platform to perform cell-based drug screening inorder to develop DiPSC-based cell therapies against DM. Toxicity and teratogenicity assays basedon iPSC-derived cells can also provide additional information on safety before advancing drugs toclinical trials. In this review, we summarize recent advances in the development of techniques fordifferentiation of iPSCs or DiPSCs into insulin-secreting pancreatic � cells, their applications in drugscreening, and their role in complementing and replacing animal testing in clinical use. Advancesin iPSC technologies will provide new knowledge needed to develop patient-specific iPSC-baseddiabetic therapies.
Keywords: diabetes mellitus; induced pluripotent stem cells; insulin-secreting � cells; cell-baseddrug screening; iPSC-based diabetic therapy
1. Introduction
Diabetes mellitus (DM), commonly referred to as diabetes, is the most widespread metabolicdisease worldwide [1]. DM develops from glucose regulation defects, which cause high blood sugarlevels over a prolonged period. DM leads to severe hyperglycemia and several complications, includingdiabetic ketoacidosis, cardiovascular disease, kidney failure, foot ulcers, and tissue or organ damage.More than three hundred million people are suffering from DM and the number of DM patients issteadily increasing.
DM is classified into three types: type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus(T2DM), and gestational diabetes [2–6]. T1DM, previously known as insulin-dependent DM (IDDM) orjuvenile diabetes, results from the destruction of insulin-secreting pancreatic � cells as a consequence
Int. J. Mol. Sci. 2016, 17, 256; doi:10.3390/ijms17020256 www.mdpi.com/journal/ijms
International Journal of
Molecular Sciences
Review
Recent Advances in Disease Modeling and DrugDiscovery for Diabetes Mellitus Using InducedPluripotent Stem Cells
Mohammed Kawser Hossain, Ahmed Abdal Dayem, Jihae Han, Subbroto Kumar Saha,Gwang-Mo Yang, Hye Yeon Choi and Ssang-Goo Cho *
Department of Animal Biotechnology, Animal Resources Research Center,and Incurable Disease Animal Model and Stem Cell Institute (IDASI), Konkuk University, Gwangjin-gu,Seoul 05029, Korea; [email protected] (M.K.H.); [email protected] (A.A.D.);[email protected] (J.H.); [email protected] (S.K.S.); [email protected] (G.-M.Y.);[email protected] (H.Y.C.)* Correspondence: [email protected]; Tel.: +82-2-450-4207
Academic Editor: Wenbin DengReceived: 3 January 2016; Accepted: 15 February 2016; Published: 19 February 2016
Abstract: Diabetes mellitus (DM) is a widespread metabolic disease with a progressive incidenceof morbidity and mortality worldwide. Despite extensive research, treatment options for diabeticpatients remains limited. Although significant challenges remain, induced pluripotent stem cells(iPSCs) have the capacity to differentiate into any cell type, including insulin-secreting pancreatic �
cells, highlighting its potential as a treatment option for DM. Several iPSC lines have recently beenderived from both diabetic and healthy donors. Using different reprogramming techniques, iPSCswere differentiated into insulin-secreting pancreatic �cells. Furthermore, diabetes patient-derivediPSCs (DiPSCs) are increasingly being used as a platform to perform cell-based drug screening inorder to develop DiPSC-based cell therapies against DM. Toxicity and teratogenicity assays basedon iPSC-derived cells can also provide additional information on safety before advancing drugs toclinical trials. In this review, we summarize recent advances in the development of techniques fordifferentiation of iPSCs or DiPSCs into insulin-secreting pancreatic � cells, their applications in drugscreening, and their role in complementing and replacing animal testing in clinical use. Advancesin iPSC technologies will provide new knowledge needed to develop patient-specific iPSC-baseddiabetic therapies.
Keywords: diabetes mellitus; induced pluripotent stem cells; insulin-secreting � cells; cell-baseddrug screening; iPSC-based diabetic therapy
1. Introduction
Diabetes mellitus (DM), commonly referred to as diabetes, is the most widespread metabolicdisease worldwide [1]. DM develops from glucose regulation defects, which cause high blood sugarlevels over a prolonged period. DM leads to severe hyperglycemia and several complications, includingdiabetic ketoacidosis, cardiovascular disease, kidney failure, foot ulcers, and tissue or organ damage.More than three hundred million people are suffering from DM and the number of DM patients issteadily increasing.
DM is classified into three types: type 1 diabetes mellitus (T1DM), type 2 diabetes mellitus(T2DM), and gestational diabetes [2–6]. T1DM, previously known as insulin-dependent DM (IDDM) orjuvenile diabetes, results from the destruction of insulin-secreting pancreatic � cells as a consequence
Int. J. Mol. Sci. 2016, 17, 256; doi:10.3390/ijms17020256 www.mdpi.com/journal/ijms
Mohammed Kawser Hossain, Ahmed Abdal Dayem, Jihae Han, Subbroto Kumar Saha, Gwang-Mo Yang, Hye Yeon Choi and Ssang-Goo Cho *
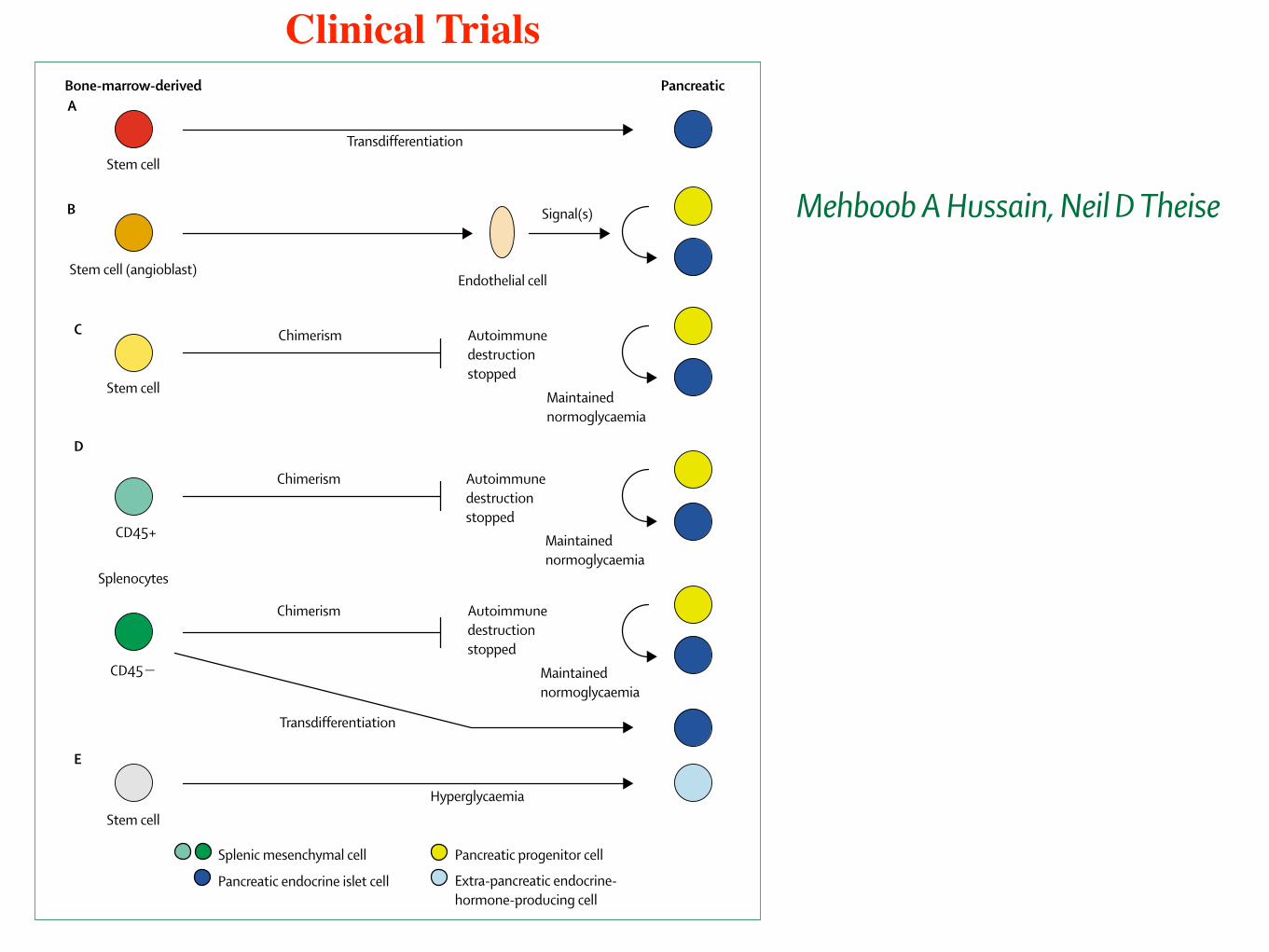
Rapid Review
204 www.thelancet.com Vol 364 July 10, 2004
Although these studies show the possibility of bone-marrow transplantation as a therapeutic approach for!-cell replacement, the immunological destruction ofnewly regenerated ! cells in type 1 diabetes remains aproblem. Bone-marrow transplantation induces micro-chimerism. In non-obese diabetic (NOD) mice, an auto-immune model of type 1 diabetes, transplanted withmarrow before development of autoimmune diabetes,chimerism prevents diabetes mellitus, presumably bymechanisms involving donor immunoregulatory cellsthat prevent the host cells from becoming autoreactiveagainst ! cells. By contrast, in NOD mice that are alreadydiabetic, induction of chimerism by sublethal or lethalirradiation followed by marrow transplantation does notresult in recovery from diabetes.21 However, when thesediabetic transplant-recipients are kept normoglycaemic byinsulin therapy, they ultimately recover from diabetes.
Pancreatic tissue showed increased proliferative activityand regeneration of ! cells. Thus marrow transplantationto induce immunological control plus maintenance ofnormoglycaemia allowed local ! or progenitor cells toproliferate as an adaptive response (figure, C).21
Transplantation of mesenchymal cells from the spleencombined with complete Freund’s adjuvant led to reversalof diabetes accompanied by regeneration of insulin-producing islets.22 The transplanted splenic mesenchymalcells differentiate into ! cells.23 Thus splenic mesen-chymal cells transplanted under certain conditions seemnot only to keep immune destruction of islets in check,but also can transdifferentiate into pancreatic ! cells. Therelative functions seemed to arise from different sub-populations (figure, D).23
Bone-marrow cells can differentiate in vitro under con-trolled conditions into insulin-expressing cells.25,26 Suchcells, transplanted under the kidney capsule of diabeticrodents, correct glucose. Removal of the grafted kidneyreturned the animals to a diabetic state.27
Cell fusion has been suggested as a mechanism ofapparent adaption of bone-marrow-derived cells into anextramedullary phenotype.24,28 Studies involving pancreaticendocrine-cell differentiation from haemopoietic-organderivatives largely rule out cell-fusion events as a mech-anism of transdifferentiation.19–23 Bone-marrow-derivedextramedullary parenchymal tissue is not always found.28
Some groups find little if any transdifferentiation29,30 orbone-marrow derivation of intra-islet endothelial cells indiabetic mice.31 The last finding was made recently byAndreas Lechner and colleagues. One group reports thegeneration of insulin-producing cells in liver, adiposetissue, spleen, and bone marrow in rodent models ofdiabetes mellitus.32 Bone-marrow transplantation showsthat most if not all extrapancreatic insulin-producing cellsderive from donor bone-marrow (figure, E).32
Stem cells in liver and pancreasIn isolated pancreatic tissue, pancreas-resident progenitorcells might give rise to endocrine islet cells. Human androdent pancreatic-duct cells,33,34 islet-derived cells,35,36 andexocrine tissue37 contain cells that can differentiatetowards a pancreatic endocrine phenotype. These tissues,cultured and differentiated in vitro, have been trans-planted and can reverse diabetes mellitus in rodents.
Rodent-liver stem cells and human fetal-liver cells havebeen differentiated in vitro into insulin-secreting cells byculture methods and/or introduction of !-cell-specificgenes. When transplanted, these cells reverse diabetesmellitus in rodents.39 Cells within liver that can differ-entiate into insulin-secreting cells after introduction ofß-cell-specific genes have also been seen in vivo afteradenoviral gene-delivery into rodents that have beenrescued from diabetes for long periods.40,41
A bone-fide pancreatic stem cell for !-cell regenerationremains elusive. A genetic-marking study in mice castsdoubt on the existence of any !-cell progenitor cells and
Pancreatic progenitor cell
Pancreatic endocrine islet cell Extra-pancreatic endocrine-hormone-producing cell
Splenic mesenchymal cell
Stem cell
Bone-marrow-derived Pancreatic
Transdifferentiation
A
E
Hyperglycaemia
Stem cell
Stem cell
Autoimmunedestructionstopped
Maintainednormoglycaemia
ChimerismC
CD45+
Chimerism Autoimmunedestructionstopped
Maintainednormoglycaemia
D
CD45"
Splenocytes
Chimerism Autoimmunedestructionstopped
Maintainednormoglycaemia
Transdifferentiation
Stem cell (angioblast)Endothelial cell
Signal(s)B
Figure: Pancreatic islet-cell regeneration after transplantation of cells derived from haemopoietic organsA=direct differentiation (transdifferentiation) of bone-marrow-derived stem cells into pancreatic endocrine cells.19
B=bone marrow contributes to generation of endothelial cells in damaged islets; newly formed endothelial cellsstimulate differentiation of locally present host-pancreatic progenitor cells.20 C=chimerism achieved by bone-marrow transplantation in NOD mice induces immunological tolerance towards ! cells.21 D=spleen mesenchymal-cell transplantation induces immunological tolerance towards ! cells.22,23 E=bone-marrow-derived cells producepancreatic endocrine hormones at various extrapancreatic sites in hyperglycaemic animals. Adapted from reference24 with permission.
Clinical Trials
Rapid Review
www.thelancet.com Vol 364 July 10, 2004 203
Embryonic stem cells (ESC) can be differentiated intoinsulin-producing cells by manipulating culture con-ditions. In-vitro differentiation of mouse ESC can gen-erate embryoid bodies, which, after selection for nestin-expressing ESC, were stimulated to differentiate towardsa !-cell-like phenotype.1 The addition of phosphoinositidekinase inhibitors promoted differentiation of largernumbers of ESC towards functional ! cells.2 Variations inESC-culture conditions generate cells with properties of! cells.3–5 With manipulation of culture conditions anduse of pax4 or pdx-1, transcription factors associated with!-cell lineage6,7 yield promising results.
Some doubt has been cast on whether ESC different-iation protocols truly yield cells that produce insulin, orcells that merely absorb insulin from the medium.8 Thesedifferentiated cells must actively synthesise and secreteinsulin rather than insulin being detected. Functioningmolecular components of regulated secretion of insulinand insulin-containing vesicles are additional featuresthat indicate a !-cell phenotype. Transplantation of ESC-derived insulin-producing cells reverses diabetes in rod-ents,2,6 indicating that these cells do synthesise andrelease insulin. The early and uncontrolled introductionof transcription factors into ESC during in-vitro differ-entiation might not yield the desired results.5 Regulatedexpression of introduced transcription factors that can beturned on during in-vitro differentiation of ESC might bemore successful.7 ESC, genetically selected for insulinexpression and injected into diabetic rats, improveglucose control.10
Human ESC produce insulin under different cultureconditions.11,12 Techniques that do not require murinefeeder cells have been developed, allowing for single-species propagation of ESC and avoiding possible zoo-notic infection of cells intended for clinical use.13 Prob-lems in control of differentiation and teratoma formation
from ESC-derived insulin-producing cells remain to beovercome.14 Existing ESC lines are not assumed to beidentical or ideal for generating islets or ! cells. Henceadditional ESC lines continue to be generated.15 Ethicalconcerns about the use of ESC need to be addressed andresolved in the face of this powerful technology.
Stem cells derived from haemopoietic organsBone marrow harbours cells that can become parench-ymal cells after entering the liver, intestine, skin, lung,skeletal muscle, heart muscle, and central nervoussystem,16 in rodent models and in human recipients ofmarrow or organ transplantation.17,18 In rodents, haemo-poietic organs harbour cells that can also differentiate intofunctional pancreatic endocrine cells.19–24
1–2 months after bone-marrow transplantation, donor-derived cells are found in pancreatic islets of recipientmice.19 These cells express insulin and genetic markers of! cells. In culture, the cells secrete insulin in response toglucose, and show intracellular calcium fluctuationssimilar to normal ! cells. However, only about 1–3% ofthe islet cells originate from the transplanted marrow(figure, A).19 A marrow-derived cell-type with pluri-potential capacity to transdifferentiate into various pheno-types has been described.25 This or a similar cell typemight be able to differentiate into pancreatic ! cells.
Similar experiments have been done in overtly diabeticmice whose ! cells have been destroyed by streptozotocin.After bone-marrow transplantation, blood glucose andinsulin concentrations were normal, and survival wasbetter.20 In islets, marrow-derived cells had differentiatedinto endothelial cells and occasionally into insulin-expressing cells.20 Endothelial engraftment was spec-ulated to stimulate the proliferation of local pancreaticprogenitors, leading in turn to the increased insulin-producing cell-mass (figure, B).
Context Curative therapy for diabetes mellitus mainly implies replacement of functional insulin-producing pancreatic! cells, with pancreas or islet-cell transplants. However, shortage of donor organs spurs research into alternative meansof generating ! cells from islet expansion, encapsulated islet xenografts, human islet cell-lines, and stem cells. Stem-cell therapy here implies the replacement of diseased or lost cells from progeny of pluripotent or multipotent cells. Bothembryonic stem cells (derived from the inner cell mass of a blastocyst) and adult stem cells (found in the postnatalorganism) have been used to generate surrogate ! cells or otherwise restore !-cell functioning.
Starting point Recently, Andreas Lechner and colleagues failed to see transdifferentiation into pancreatic ! cells aftertransplantation of bone-marrow cells into mice (Diabetes 2004; 53: 616–23). Last year, Jayaraj Rajagopal and colleaguesfailed to derive ! cells from embryonic stem cells (Science 2003; 299: 363). However, others have seen such effects.
Where next? As in every emerging field in biology, early reports seem confusing and conflicting. Embryonic and adultstem cells are potential sources for !-cell replacement and merit further scientific investigation. Discrepancies betweendifferent results need to be reconciled. Fundamental processes in determining the differentiation pathways of stemcells remain to be elucidated, so that rigorous and reliable differentiation protocols can be established. Encouragingstudies in rodent models may ultimately set the stage for large-animal studies and translational investigation.
Stem-cell therapy for diabetes mellitusMehboob A Hussain, Neil D Theise Lancet 2004; 364: 203–05
Liver and Stem Cell ResearchLaboratory, Division ofDigestive Diseases, Departmentof Internal Medicine, Beth IsraelMedical Center, Albert EinsteinCollege of Medicine, New York,NY 10003, USA (M A HussainMD, N D Theise MD)
Correspondence to: Dr MehboobHussain [email protected]

Thanks for your Patient ear


















