
Sindrome di von Hippel-Lindau barbara pasini Università di Torino Dipartimento di Genetica Biologia...
37
Sindrome di von Hippel-Lindau barbara pasini Università di Torino Dipartimento di Genetica Biologia e Biochimica
-
Upload
floriano-russo -
Category
Documents
-
view
244 -
download
6
Transcript of Sindrome di von Hippel-Lindau barbara pasini Università di Torino Dipartimento di Genetica Biologia...

Sindrome di von Hippel-Lindau
barbara pasini
Università di TorinoDipartimento di Genetica
Biologia e Biochimica

iperplasia /adenomaparatiroidi
feocromocitoma
carcinomamidollare della tiroide
MEN2
tumori endocrinipancreatici
adenoma ipofisi
gastrinomi duodenalicarcinoidi ecc.
MEN1
angiomatosi retinicaemangioblastoma SNCcarcinoma renale (CC)
VHL
HPT-JT
AIPPRKAR1A
CDK-Inhibitors
SDHB-SDHD SDHC, SDHAF2SDH5, SDHATMEM127 e NF1
paraganglioma

caratteristiche generali delleneoplasie endocrine multiple
• autosomiche dominanti • combinazione di “lesioni” specifiche
- istologia / clinica caratteristica- PT: iperplasia in MEN2A / adenomi in MEN1 / adenoma cistico HPT-JT- PiA: frequente PRL in MEN1 – frequente GH in AIP- PNET: spesso secernenti in MEN1 / non secernenti VHL- MTC con CCH in MEN2
- età caratteristiche di insorgenza- più manifestazioni presenti contemporaneamente- sviluppo di manifestazioni in successione
- manifestazioni cutanee, cistiche ecc.
- malformazioni, dismorfismi (MEN2B)
• penetranza alta ma espressività variabile• correlazione genotipo-fenotipo
- MEN2, VHL, SDH (no MEN1)
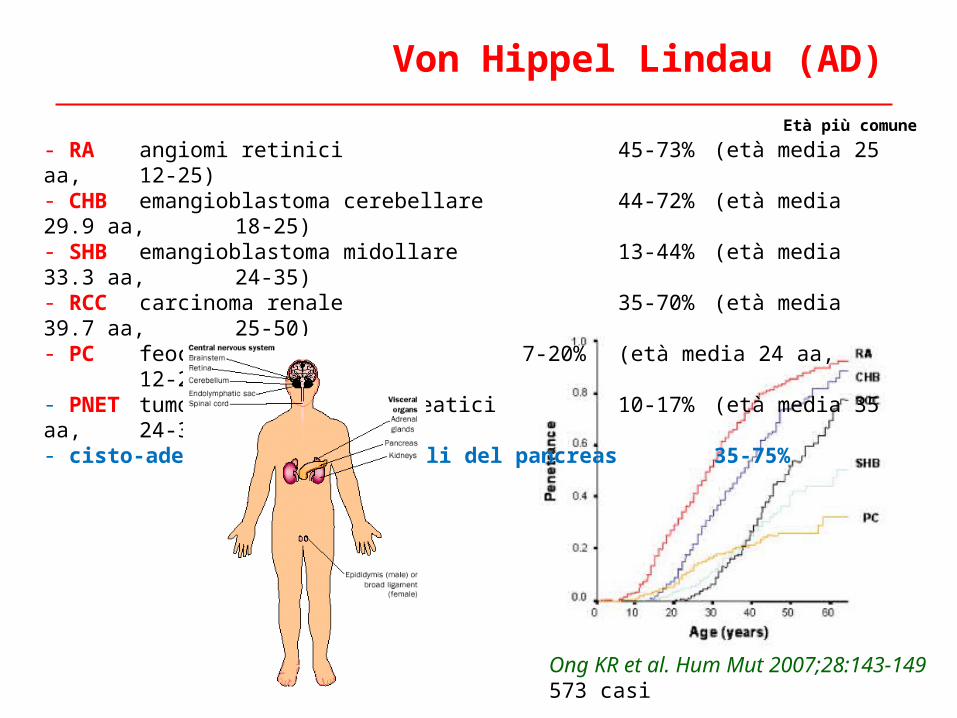
- RA angiomi retinici 45-73% (età media 25 aa, 12-25)- CHB emangioblastoma cerebellare 44-72% (età media 29.9 aa, 18-25)- SHB emangioblastoma midollare 13-44% (età media 33.3 aa, 24-35) - RCC carcinoma renale 35-70% (età media 39.7 aa, 25-50)- PC feocromocitoma 7-20% (età media 24 aa, 12-25)- PNET tumori endocrini pancreatici 10-17% (età media 35 aa, 24-35)- cisto-adenomi sierosi multipli del pancreas 35-75%
Ong KR et al. Hum Mut 2007;28:143-149573 casi
Von Hippel Lindau (AD)
Età più comune

gene VHL
Localizzato nel 1988 in 3p (3p25-26), clonato nel 1993
proteina VHL di 213 aa - 28-30 kDa
(isoforma 2 di 19 kDa)
espressa in molti tessuticitoplasmatica- ubiquitinazione HIF- riduzione dell’espressione delle integrine (5)- riduzione dell’espressione della ciclina D1 (aumento di p27)- legame con p53 ……
5’ 3’-UTR1 32
ATG stop at nt. 852
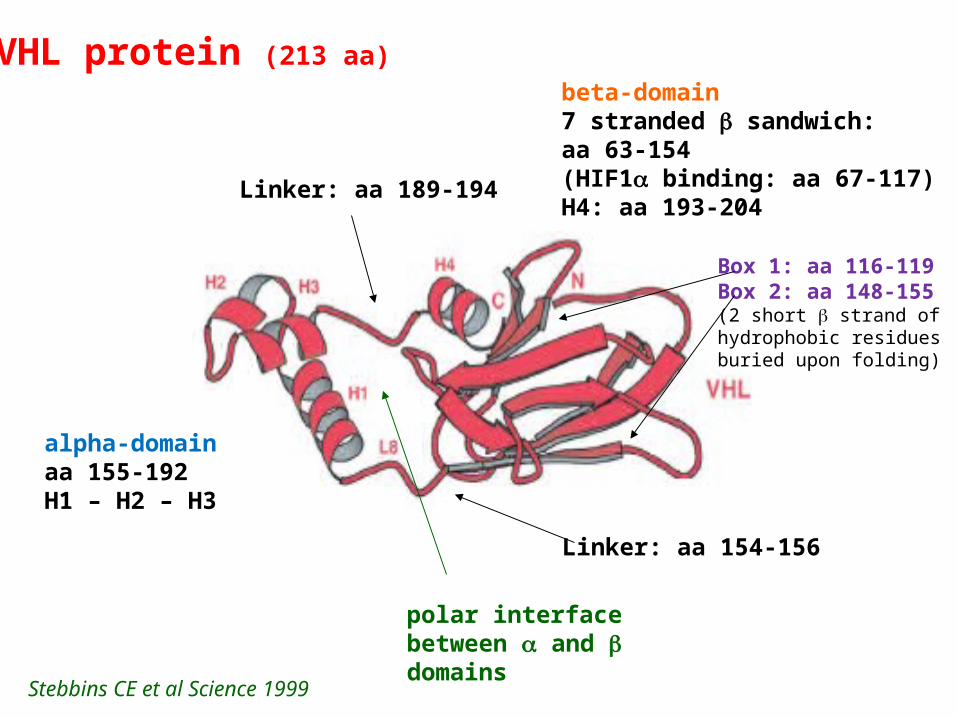
VHL protein (213 aa)beta-domain7 stranded sandwich:aa 63-154(HIF1 binding: aa 67-117)H4: aa 193-204
polar interfacebetween and domains
Linker: aa 154-156
alpha-domainaa 155-192H1 – H2 – H3
Linker: aa 189-194
Stebbins CE et al Science 1999
Box 1: aa 116-119Box 2: aa 148-155(2 short strand of hydrophobic residuesburied upon folding)
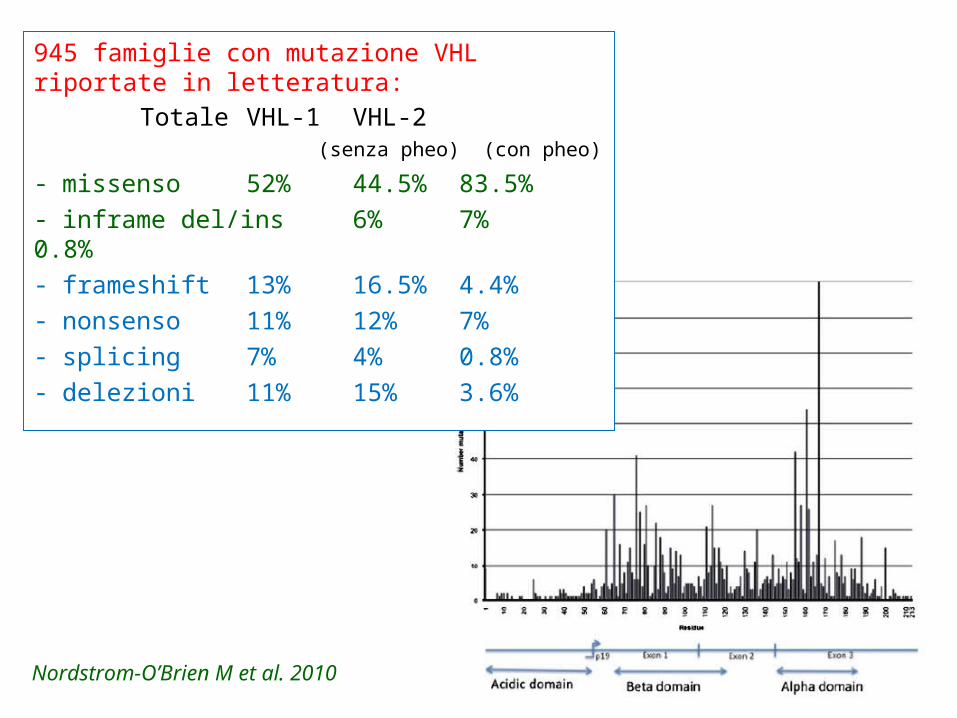
945 famiglie con mutazione VHLriportate in letteratura:
Totale VHL-1 VHL-2 (senza pheo) (con pheo)
- missenso 52% 44.5% 83.5%- inframe del/ins 6% 7% 0.8%- frameshift 13% 16.5% 4.4%- nonsenso 11% 12% 7%- splicing 7% 4% 0.8%- delezioni 11% 15% 3.6%
Nordstrom-O’Brien M et al. 2010

Von Hippel Lindau (AD)
Tipo 2 con pheo
2A HB, raro RCC
2B HB + RCC
2C “solo” pheo
Tyr98His Germany - Black ForestTyr112His-domain (HIF) bassa destabilizzazione-domain (El C) bassa destabilizzazione
mutazioni troncanti(HB, RA, RCC più precoci)
missense -domain (HIF)delezioni parziali o completedelezioni C3orf10 (1B raro RCC)
missense “surface”-domain (El C) alta destabilizzazionecodons 74-90 > 130-136
Arg64Pro, Ser68Trp, Val84Leu, Gly93Ser, Val155Leu, Leu188Val, Arg238Try
omozigosi o eterozigosi compostaArg200Trp, His191Asp, Pro192Ser ecc.
Policitemia congenita(AR: eritrocitosi famigliare tipo 2)
Tipo 1 senza pheo

Tyr98Asn 292 T>AHIF binding Hyp564(liner of the pochet)
slightly increased levels of HIF2a, increased levels of integrins and slightly disorganized tight junctionshigher destabilization ofHIF binding domain
VHL-2B
Tyr98His 292 T>CHIF binding Hyp564(liner of the pochet)
low levels of HIF2a, only slightly increased levels of integrins and adequate tight junctionslower destabilization ofHIF binding domain
VHL-2A (Black Forest)insPheoVHL with RCC (6)VHL without RCC (14)VHL with PheoPGL-HNPGL (HB-PHEO in fam)
VHL – HIF1 interface
Hyp564 pocket: H115 – S111 (HB, floor)lined by W88 – Y98 – W117
with right chian orientation of H115 by S65, Y112, R113, G114
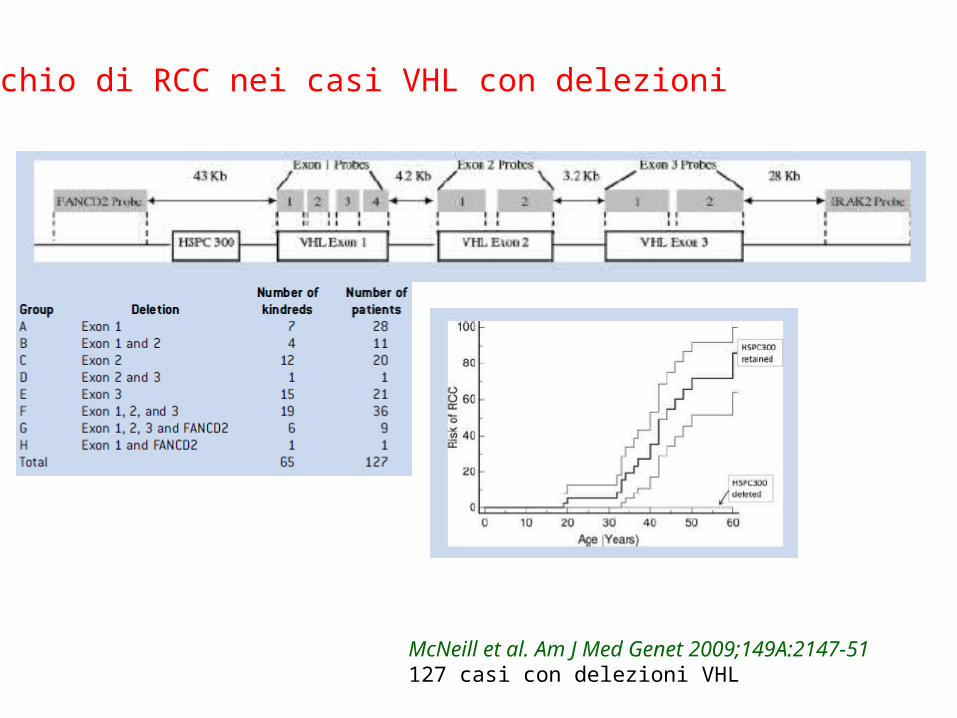
Rischio di RCC nei casi VHL con delezioni
McNeill et al. Am J Med Genet 2009;149A:2147-51127 casi con delezioni VHL
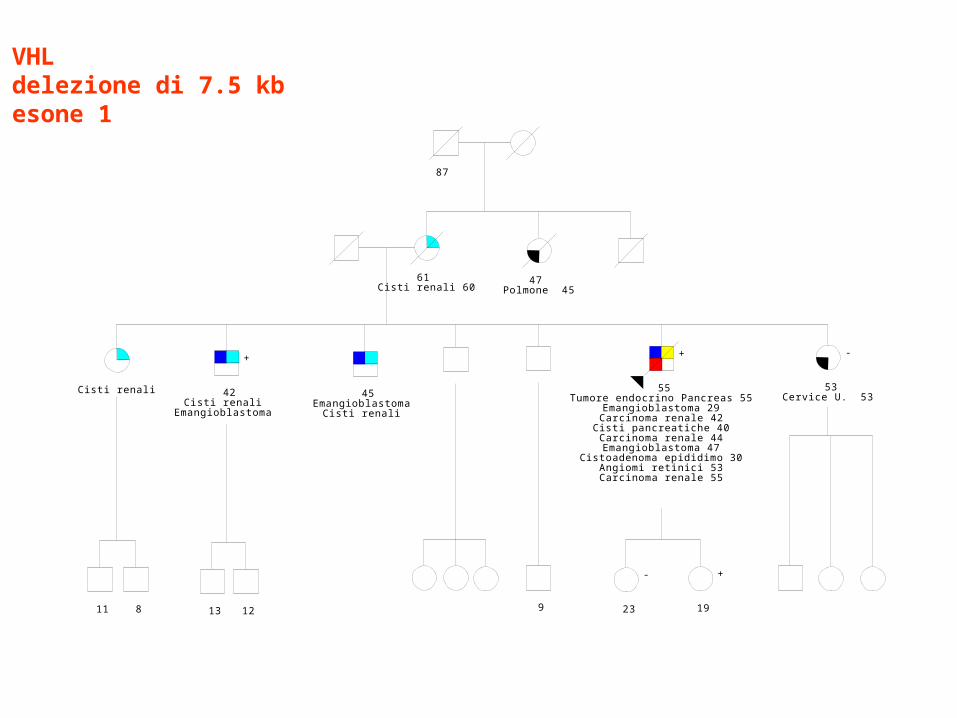
VHLdelezione di 7.5 kbesone 1
18/03/2008
55Tumore endocrino Pancreas 55
Emangioblastoma 29Carcinoma renale 42Cisti pancreatiche 40Carcinoma renale 44Emangioblastoma 47
Cistoadenoma epididimo 30Angiomi retinici 53
Carcinoma renale 55
+
23
-
19
+
9
45Emangioblastoma
Cisti renali
42Cisti renali
Emangioblastoma
+
13 12
Cisti renali
11 8
61 Cisti renali 60
47 Polmone 45
87
53Cervice U. 53
-
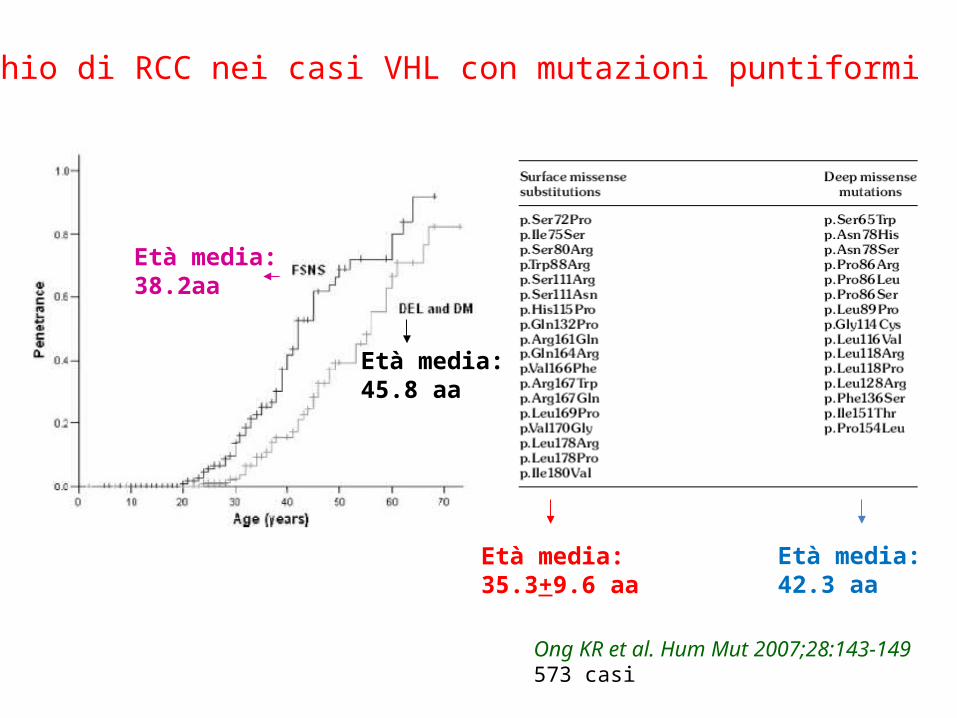
Età media:35.3+9.6 aa
Età media:42.3 aa
Età media:45.8 aa
Età media:38.2aa
Rischio di RCC nei casi VHL con mutazioni puntiformi
Ong KR et al. Hum Mut 2007;28:143-149573 casi
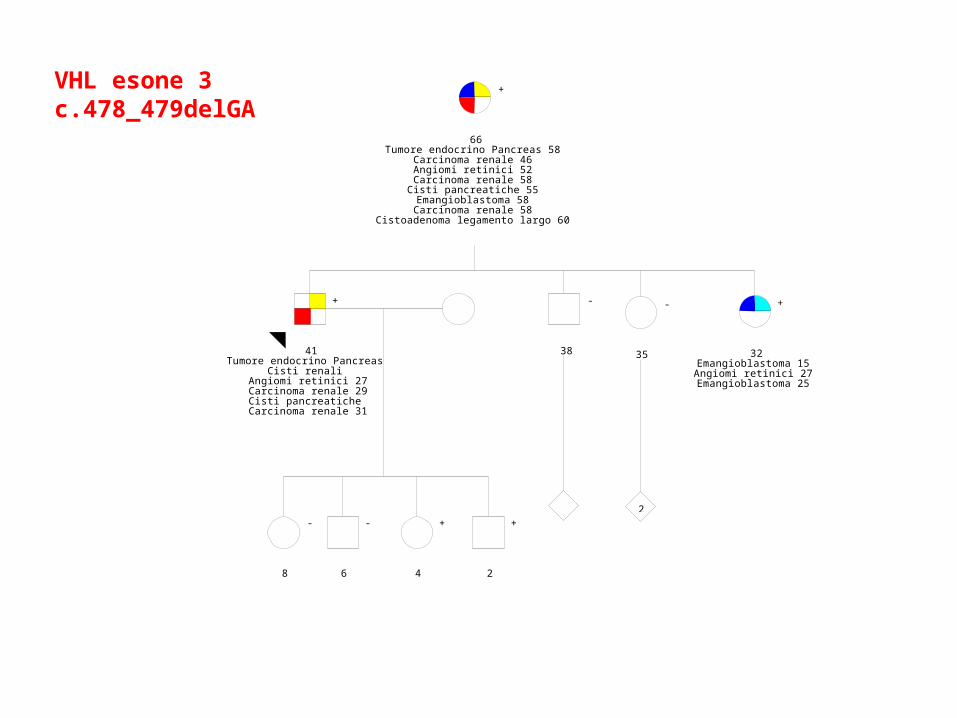
09/10/2009
66Tumore endocrino Pancreas 58
Carcinoma renale 46Angiomi retinici 52
Carcinoma renale 58Cisti pancreatiche 55Emangioblastoma 58Carcinoma renale 58
Cistoadenoma legamento largo 60
+
41Tumore endocrino Pancreas
Cisti renali Angiomi retinici 27
Carcinoma renale 29Cisti pancreatiche
Carcinoma renale 31
+
38
-
35
-
32Emangioblastoma 15Angiomi retinici 27
Emangioblastoma 25
+
8
-
6
-
4
+
2
+
2
VHL esone 3c.478_479delGA
Forman JR et al. Proteins 2009;77:84-96
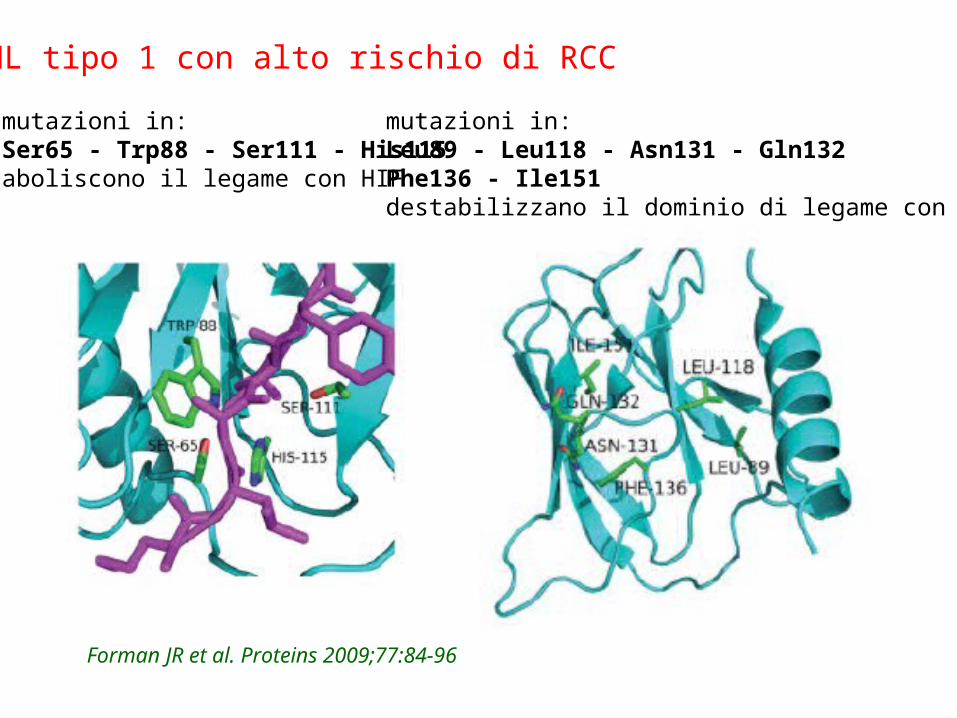
mutazioni in:Ser65 - Trp88 - Ser111 - His115aboliscono il legame con HIF
mutazioni in:Leu89 - Leu118 - Asn131 - Gln132Phe136 - Ile151destabilizzano il dominio di legame con HIF
VHL tipo 1 con alto rischio di RCC

Rischio di PHEO nei casi VHL con mutazioni puntiformi
Ong KR et al. Hum Mut 2007;28:143-149573 casi

Forman JR et al. Proteins 2009;77:84-96
VHL con alto rischio di PHEO
mutazioni in:Val155 - Arg161 - Val166che aboliscono il legame con elonghina CGln164che destabilizza il dominio di legamecon elonghina C

17/05/2005
26 Feocromocitoma 17Feocromocitoma 19Angiomi retinici 20
Emangioblastoma 20Emangioblastoma 20Emangioblastoma 20
Cisti renali 26
+
57
-
55
-
30
-
VHL esone 3p.Arg161Gln de novo

21/03/2005
48
18Feocromocitoma 13Feocromocitoma 13
13Feocromocitoma 13
46 1 2 1
74 35
51 53
77 73
VHL esone 3p.Tyr156Serlegame con elong C
18/03/2008
53 Feocromocitoma Midollare surrene 49Feocromocitoma Midollare surrene 50
Tumore endocrino Pancreas 53Angiomi retinici 50
Carcinoma renale 50Carcinoma renale 53
+
23 Feocromocitoma Midollare surrene 13Feocromocitoma Midollare surrene 13
+
18 Feocromocitoma Midollare surrene 13
+
51 1 2 1
74 35
56 58
82 73

Forman JR et al. Proteins 2009;77:84-96
VHL con alto rischio per tutte le manifestazioni
mutazioni:Arg167Gln , Arg167Trpdestabilizzano la struttura secondaria della proteina(Arg167 forma ponti idrogeno con Asp187, Leu188 e Phe190)
mutazioni in Arg167sono responsabili del45% circa dei casi VHL-2nella popolazione caucasica

31/03/2011
23 Feocromocitoma Midollare surrene 22Feocromocitoma Midollare surrene 22
Angiomi retinici (multipli dex) 22Angiomi retinici (multipli sin) 22Cisti renali (multiple bilat.) 22
+
28
56 62 65 63
2
3
2
35SNC 33
86 69
55
4
VHL esone 3p.Arg167Trp
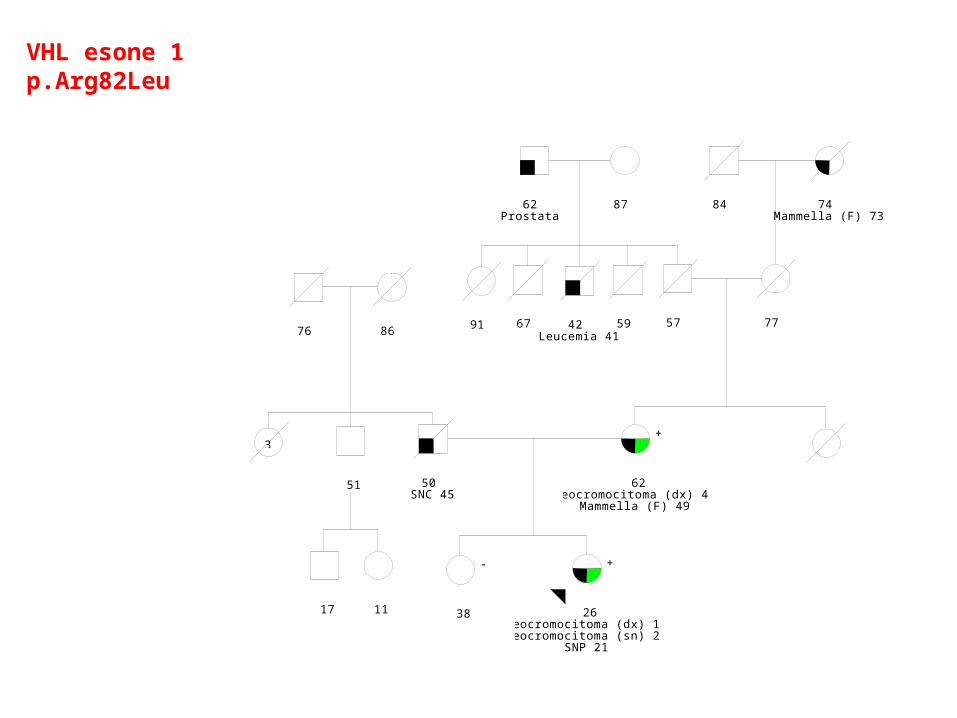
15/02/2006
26Feocromocitoma (dx) 19Feocromocitoma (sn) 23
SNP 21
+
38
-
50 SNC 45
62Feocromocitoma (dx) 46
Mammella (F) 49
+
57 77
84 74 Mammella (F) 73
59 42 Leucemia 41
67 91
51
17 11
3
76 86
62 Prostata
87
VHL esone 1p.Arg82Leu
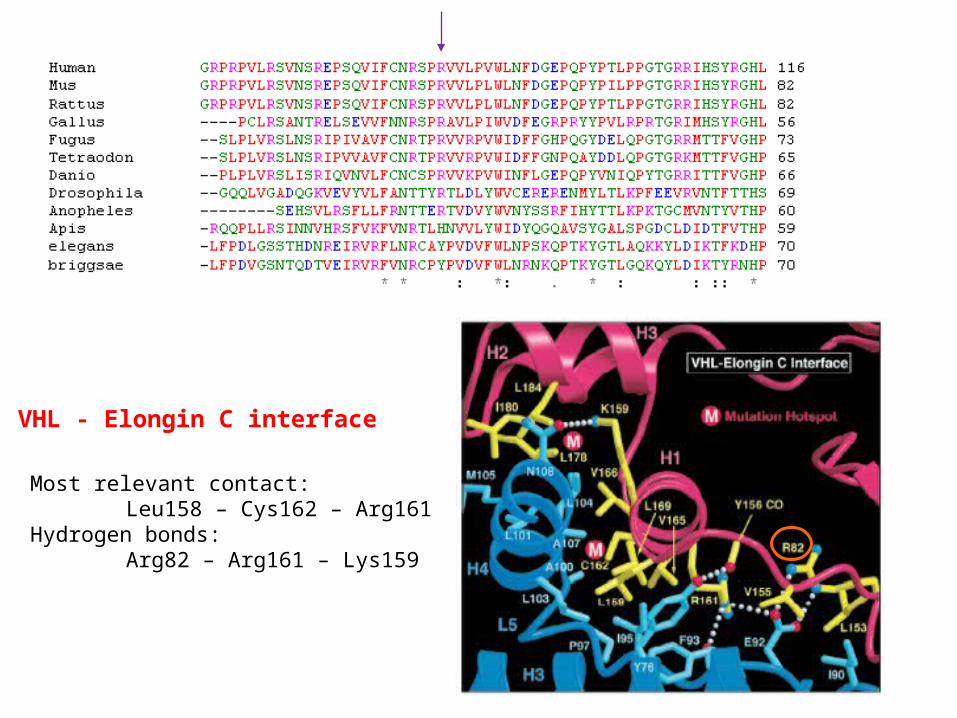
VHL - Elongin C interface
Most relevant contact:Leu158 – Cys162 – Arg161
Hydrogen bonds:Arg82 – Arg161 – Lys159
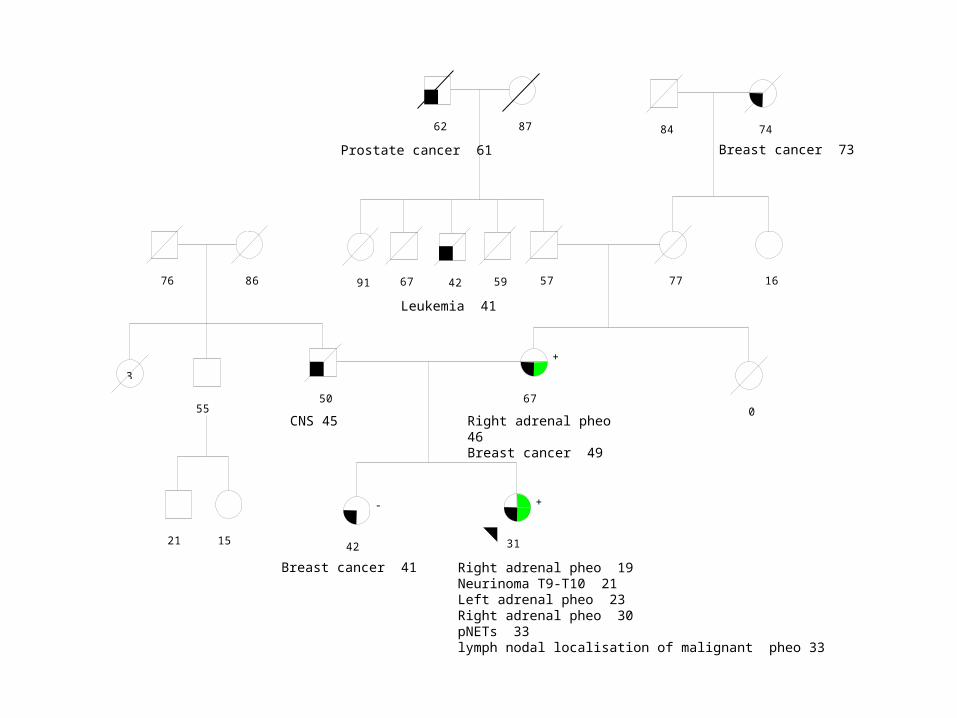
14/09/2010
31
+
42
-
50 67
+
0
57 77
84 74
59 42 67 91
55
21 15
3
76 86
62 87
16
Right adrenal pheo 19Neurinoma T9-T10 21Left adrenal pheo 23Right adrenal pheo 30pNETs 33lymph nodal localisation of malignant pheo 33
Breast cancer 41
Right adrenal pheo 46Breast cancer 49
CNS 45
Leukemia 41
Breast cancer 73 Prostate cancer 61
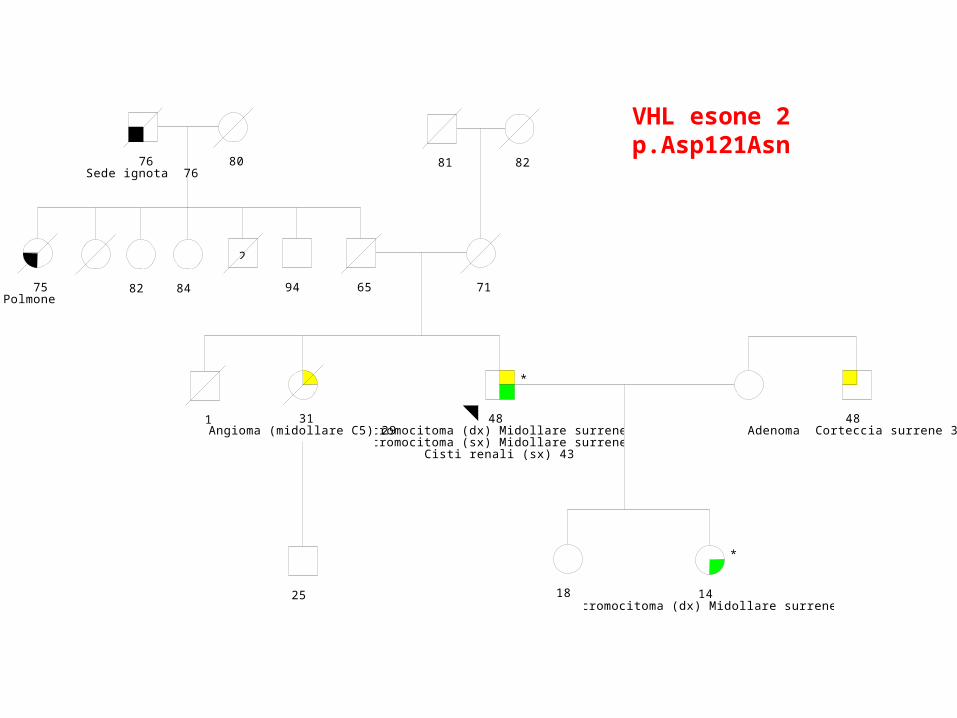
24/03/2009
48 Feocromocitoma (dx) Midollare surrene 43Feocromocitoma (sx) Midollare surrene 43
Cisti renali (sx) 43
*
18 14 Feocromocitoma (dx) Midollare surrene 14
*
31Angioma (midollare C5) 29
1
25
65 71
81 82
84 94
2
76Sede ignota 76
80
82 75Polmone
48 Adenoma Corteccia surrene 37
VHL esone 2p.Asp121Asn
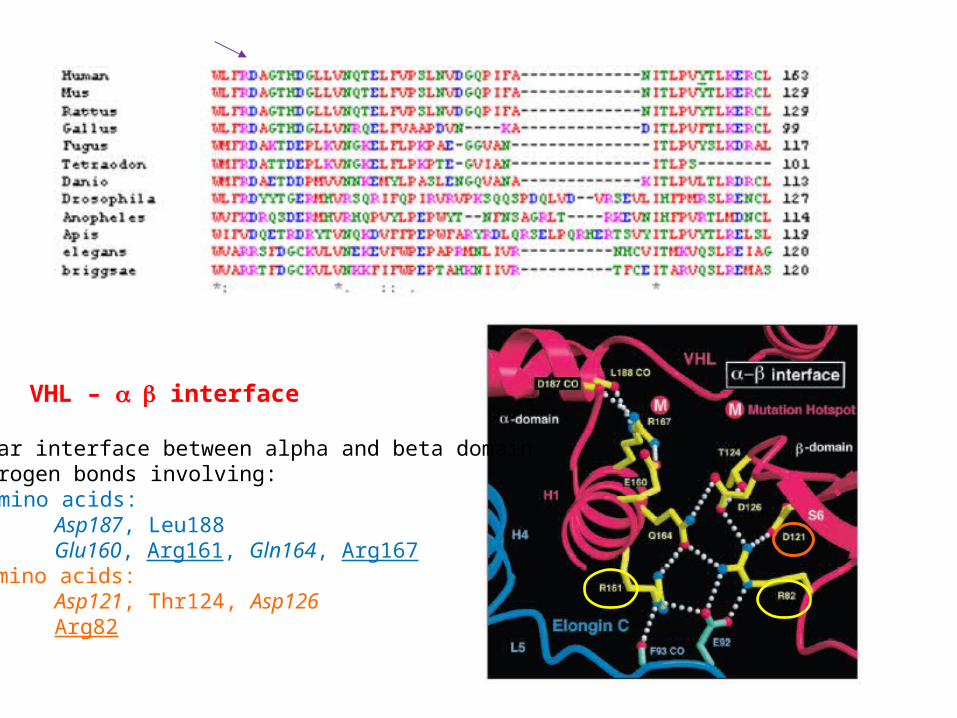
VHL – interface
Polar interface between alpha and beta domainHydrogen bonds involving: amino acids: H3 Asp187, Leu188H1 Glu160, Arg161, Gln164, Arg167 amino acids:L6 Asp121, Thr124, Asp126L2 Arg82
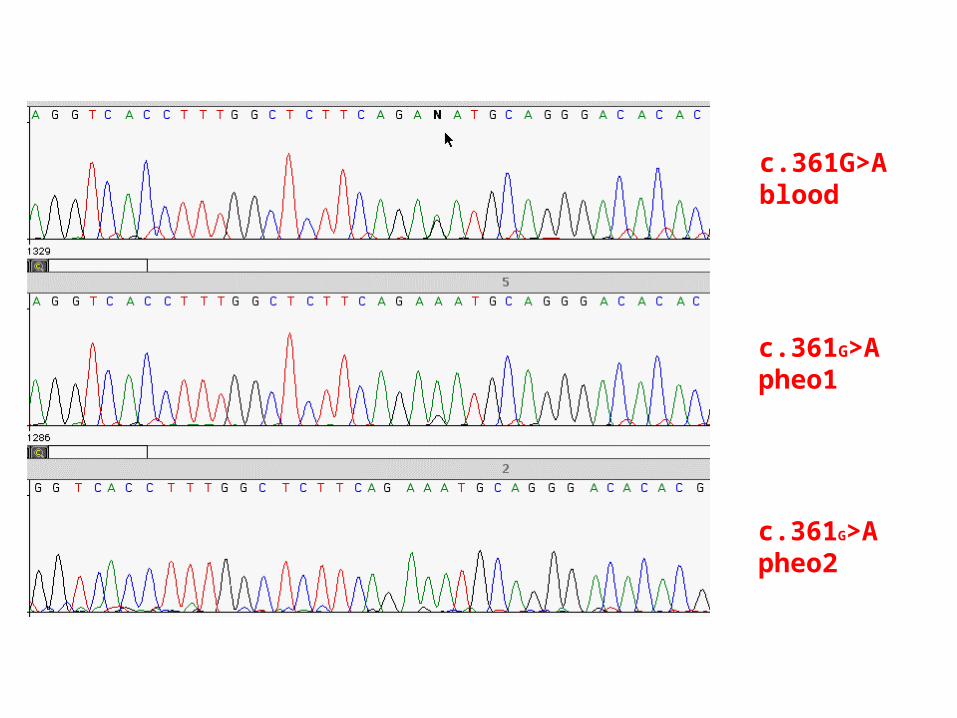
c.361G>Ablood
c.361G>Apheo1
c.361G>Apheo2
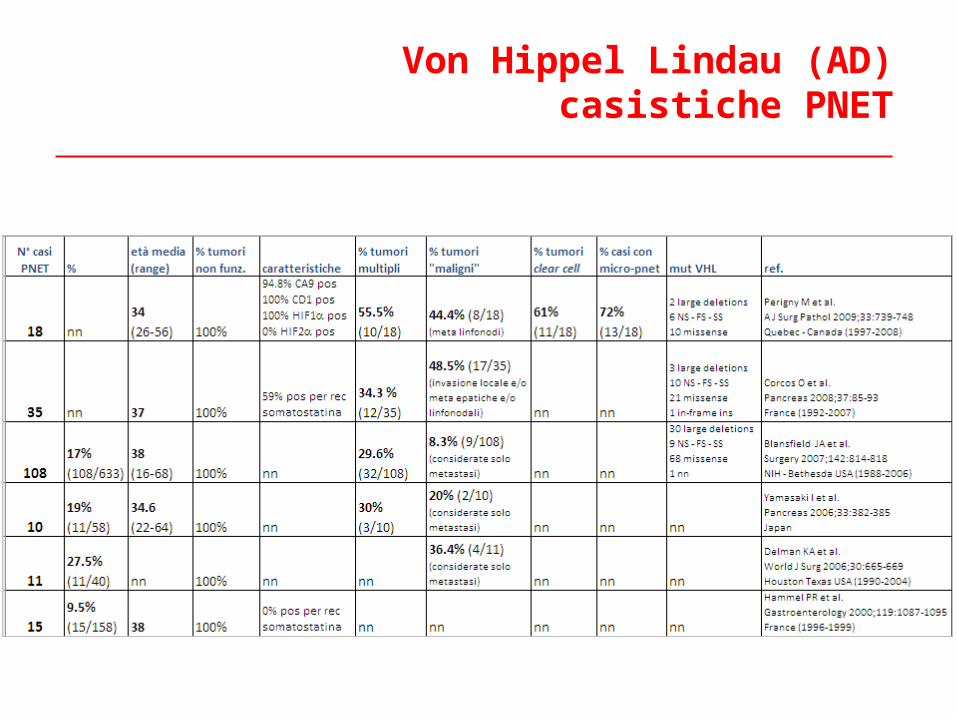
Von Hippel Lindau (AD)casistiche PNET
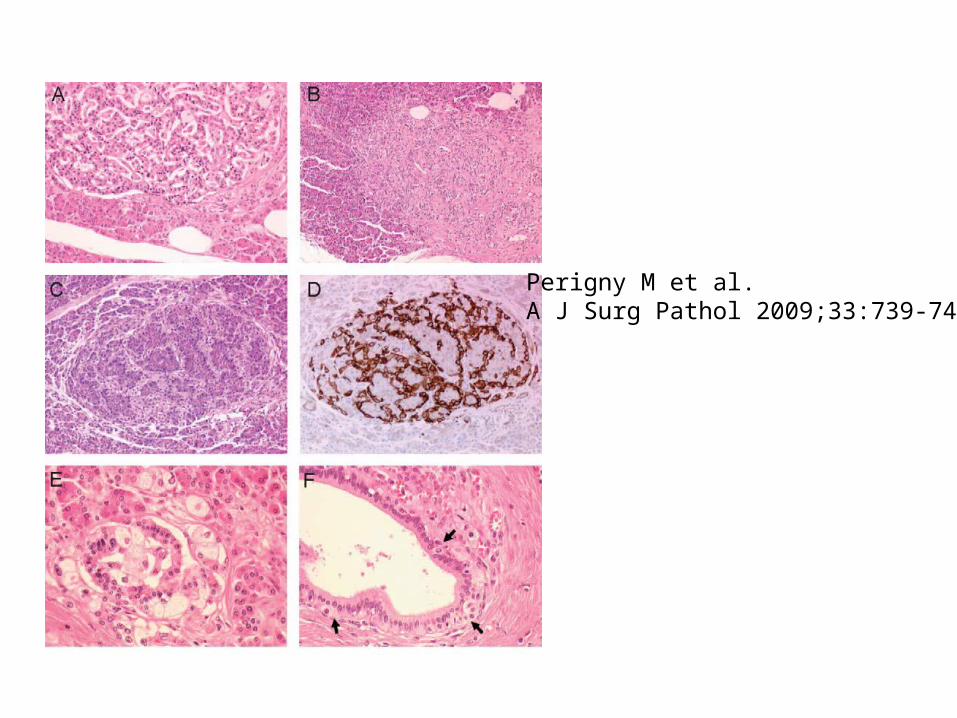
Perigny M et al. A J Surg Pathol 2009;33:739-748

Dati inerenti 108 PNET in VHL – casistica NIHBlansfield JA et al. Surgery, 2007;142:814-818

Von Hippel Lindau (AD)

21/05/2009
33
*
34
*
12 7
54
*
57
-
60
*
57
-
51
-
32
-
2
80Polmone
94
3 2
Neuroblastoma (tum. neuroblastico G3) 1
27 anni: emangioblastoma cerebellare29 anni: policitemia vera
VHL IVS1+5G>C
analisi dell’mRNA: difetto parzialedi splicing dell’allele mutatoattivazione di sito accettore criptico304 bp a monteframe-shift e stop prematuro(c.37_340del304bp, p.Gly14ThrfsX44)
mutazione somatica (second hit ?)nell’emangioblastoma(esone 3: c. 486C>G, p.Cys162Trp)
mutazioni con effetto “moderato” fenotipi particolari ?

test genetico nelleneoplasie endocrine multiple - VHL
• indicazione al test• sospetto clinico: almeno due segni di malattia o famigliarità• vantaggio del test genetico rispetto alla diagnosi clinica
- paziente giovane alla prima manifestazione
- le altre manifestazioni attese sono più tardive
- lo screening clinico è costoso / impegnativo
- il test genetico è informativo
• utilità del test• confermare la diagnosi• correlazioni genotipo-fenotipo• stimare il rischio per ulteriori manifestazioni di malattia• curare in modo opportuno le manifestazioni di malattia• screening dei famigliari a rischio (età precoce)• diagnosi prenatale

Criteri di accesso al test genetico
• manifestazioni isolate “giovanili” o multiple– angioma retinico < 30 anni o multiplo-bilaterale
– ca. renale cellule chiare < 30 anni o multiplo-bilaterale
– emangioblastoma cerebellare-spinale < 50 anni o multiplo-bilaterale
– feocromocitoma < 50 anni o bilaterale
• due manifestazioni di malattia o famigliarità positiva– tra le precedenti
– e/o cisti multiple pancreatiche, renali
• diagnosi clinica della sindrome
Von Hippel Lindau (AD)

• cisti renali– mutiple/bilat. 0/3
• tumore renale– sporadico 0/2– multipli/bilat. 0/4– famigliare 0/1– con PNET 0/2– con cisti pancreatiche 0/1– con emangioblastoma 0/3– con angiomi retinici 1/1– multipli e sindromici 6/6
(100%)(HB, AR, CP, CE, PNET)
analisi VHL in 79 casi indice
• feocromocitoma– sporadico 0/25– bilaterale/con PGL 4/8
(50%)– famigliare 2/2
(100%)– sindromico 3/3 (100%)
(AR o PNET o HB+RCC)
• PNET/cistoadenoma/cisti– PNET/cistoA 0/3– cisti pancreatiche 2/3 (66%)
• angiomi retinici/emangiobl.– AR mutipli/fam. 0/4– HB sporadico 0/7– HB multipli 0/1– HB sindromico 2/2 (100%)
(AR o policitemia)
screening dei famigliari permutazione VHL nota in famiglia (50):- 20 mutati- 30 non portatori
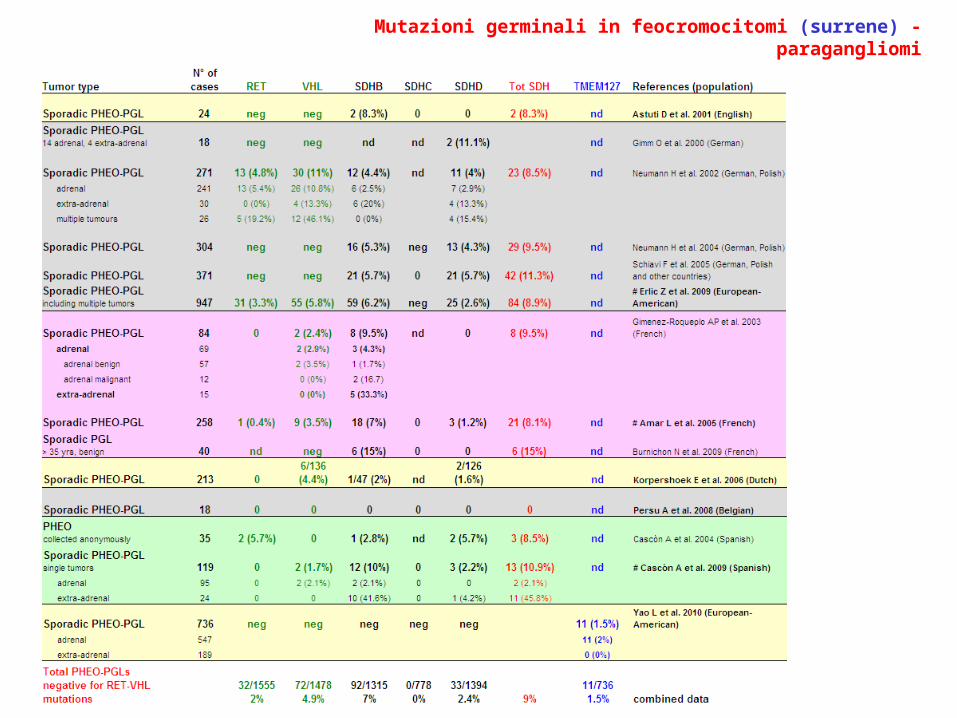
Mutazioni germinali in feocromocitomi (surrene) - paragangliomi

Mutazioni germinali in feocromocitomi (surrene) – paragangliomi maligni
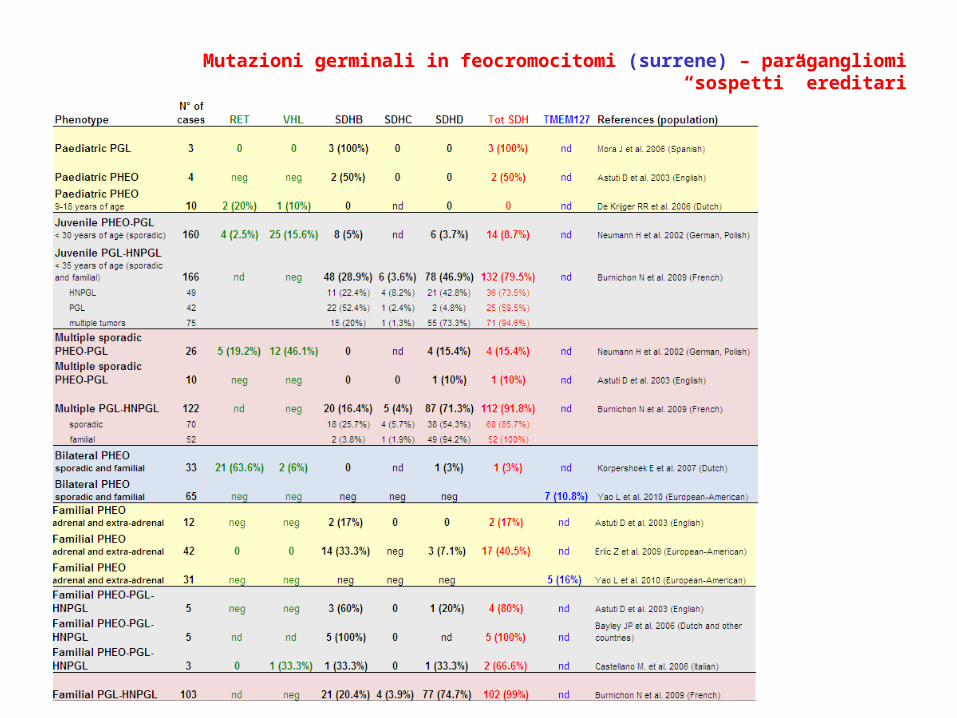
Mutazioni germinali in feocromocitomi (surrene) – paragangliomi “sospetti” ereditari



![[Arthur Robert Von Hippel] Dielectrics and Waves](https://static.fdocuments.net/doc/165x107/55cf99df550346d0339f986b/arthur-robert-von-hippel-dielectrics-and-waves.jpg)














