
Septic Shock Is Associated with Receptor for Advanced Glycation
11
of January 21, 2019. This information is current as of LPS Advanced Glycation End Products Ligation Septic Shock Is Associated with Receptor for Yonekura and Hiroshi Yamamoto Okamoto, Satoshi Shimura, Tadahiro Karasawa, Hideto Takuo Watanabe, Masahide Asano, Shin Takasawa, Hiroshi Tsuneyama, Seiichi Munesue, So Motoyoshi, Dong Han, Yasuhiko Yamamoto, Ai Harashima, Hidehito Saito, Koichi ol.1002253 http://www.jimmunol.org/content/early/2011/01/26/jimmun published online 26 January 2011 J Immunol Material Supplementary 3.DC1 http://www.jimmunol.org/content/suppl/2011/01/26/jimmunol.100225 average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2011 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on January 21, 2019 http://www.jimmunol.org/ Downloaded from by guest on January 21, 2019 http://www.jimmunol.org/ Downloaded from
Transcript of Septic Shock Is Associated with Receptor for Advanced Glycation

of January 21, 2019.This information is current as
of LPSAdvanced Glycation End Products Ligation Septic Shock Is Associated with Receptor for
Yonekura and Hiroshi YamamotoOkamoto, Satoshi Shimura, Tadahiro Karasawa, Hideto Takuo Watanabe, Masahide Asano, Shin Takasawa, HiroshiTsuneyama, Seiichi Munesue, So Motoyoshi, Dong Han, Yasuhiko Yamamoto, Ai Harashima, Hidehito Saito, Koichi
ol.1002253http://www.jimmunol.org/content/early/2011/01/26/jimmun
published online 26 January 2011J Immunol
MaterialSupplementary
3.DC1http://www.jimmunol.org/content/suppl/2011/01/26/jimmunol.100225
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on January 21, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Septic Shock Is Associated with Receptor for AdvancedGlycation End Products Ligation of LPS
Yasuhiko Yamamoto,*,1 Ai Harashima,*,1 Hidehito Saito,* Koichi Tsuneyama,†
Seiichi Munesue,* So Motoyoshi,* Dong Han,* Takuo Watanabe,* Masahide Asano,‡
Shin Takasawa,x Hiroshi Okamoto,{ Satoshi Shimura,‖ Tadahiro Karasawa,‖,2
Hideto Yonekura,# and Hiroshi Yamamoto*
Septic shock is a severe systemic response to bacterial infection. Receptor for advanced glycation end products (RAGE) plays a role
in immune reactions to recognize specific molecular patterns as pathogen recognition receptors. However, the interaction between
LPS, the bioactive component of bacterial cell walls, and RAGE is unclear. In this study, we found direct LPS binding to RAGE by
a surface plasmon resonance assay, a plate competition assay, and flow cytometry. LPS increased TNF-a secretion from peritoneal
macrophages and an NF-kB promoter-driven luciferase activity through RAGE. Blood neutrophils and monocytes expressed
RAGE, and TLR2 was counterregulated in RAGE2/2 mice. After LPS injection, RAGE+/+ mice showed a higher mortality, higher
serum levels of IL-6, TNF-a, high mobility group box 1, and endothelin-1, and severe lung and liver pathologies compared with
RAGE2/2 mice without significant differences in plasma LPS level. Administration of soluble RAGE significantly reduced the
LPS-induced cytokine release and tissue damage and improved the LPS-induced lethality even in RAGE2/2 as well as RAGE+/+
mice. The results thus suggest that RAGE can associate with LPS and that RAGE system can regulate inflammatory responses.
Soluble RAGE would be a therapeutic tool for LPS-induced septic shock. The Journal of Immunology, 2011, 186: 000–000.
Septic shock is a systemic response to severe bacterial in-fection. The shock caused by LPS, the major component ofthe cell surface of Gram-negative bacteria, is often asso-
ciated with a high mortality (1). LPS consists of three covalentlylinked regions: lipid A, core oligosaccharide, and O Ag poly-
saccharide (2). Lipid A resides at the inner most layer and is re-sponsible for toxicity of LPS, thus having been known asendotoxin (2). LPS can induce massive production of cytokines,such as TNF-a and IL-6, by immune and nonimmune cells,thereby leading to inflammatory tissue injuries and finally multi-organ failure, the clinical hallmarks of septic shock (3–5). It iswell known that TLR4, a member of pathogen recognitionreceptors (PRRs), mediates LPS-induced activation of the innateimmune system and subsequent immunostimulation (6). Whencomplexed with CD14 and MD-2, TLR4 has been shown to par-ticipate in the cellular recognition of LPS (7). It is also reportedthat TLR2 could sensitize LPS (8). Other LPS receptors and re-ceptor clusters are recently reported; these include heat shockproteins 70 and 90, CXCR4 and growth differentiation factor 5cluster (9), RP105-MD-1 complex (10), CD11c (11), CD55 (12),L-selectin (CD62L) (13), P-selectin (14), and scavenger receptorclass A (15). On the basis of function, PRRs may be put into twocategories of signaling type and endocytic type. TLRs and scav-enger receptor class A are regarded as representative of the formerand the latter, respectively.The receptor for advanced glycation end products (RAGE) is
a cell-surface receptor belonging to the Ig superfamily. It iscomposed of an N-terminal extracellular domain with a V region-like domain and two C region-like domains, a single trans-membrane domain, and a C-terminal highly charged short cyto-plasmic domain, which is essential for signal transduction (16).RAGE is known to be a multiligand receptor participating ina wide variety of physiological and pathological processes, such asdiabetic complications, atherosclerosis, cancer, neurodegenerativedisorders, and inflammation (16–18). RAGE is engaged not onlyby advanced glycation end product (AGE) but also by amphoterin/high mobility group box 1 (HMGB1) (19), S100 proteins (20),transthyretin (21), b2 integrin Mac-1 (22), and amyloid b-proteins(23). Binding of these ligands to RAGE initiates intracellularsignaling to activate transcription factors NF-kB (16). Among
*Department of Biochemistry and Molecular Vascular Biology, Kanazawa UniversityGraduate School of Medical Science, Kanazawa 920-8640, Japan; †Department ofDiagnostic Pathology, Graduate School of Medicine and Pharmaceutical Science,University of Toyama, Toyama 930-0194, Japan; ‡Division of Transgenic AnimalScience, Kanazawa University Advanced Science Research Center, Kanazawa 920-8640, Japan; xDepartment of Biochemistry, Nara Medical University, Kashihara 634-8521, Japan; {Tohoku University, Sendai 980-8577, Japan; ‖Division of Health Sci-ences, Department of Clinical Laboratory Science, Kanazawa University GraduateSchool of Medical Science, Kanazawa 920-0942, Japan; and #Department of Bio-chemistry, Kanazawa Medical University, Uchinada 920-0293, Japan
1Y.Y. and A.H. contributed equally to this work.
2Current address: Fujimi Kogen Hospital, Fujimi Town, Nagano, Japan.
Received for publication July 6, 2010. Accepted for publication December 25, 2010.
This work was supported in part by grants-in-aid from the Ministry of Education,Science, Sports, Culture and Technology of Japan (13670113) and for scientific re-search from the Japan Society for the Promotion of Sciences (16790183, 17590241,and 16570113).
Y.Y., A.H., and H.Y. designed research; Y.Y., A.H., H.S., S.M., S.M., D.H., and S.S.performed research; K.T. performed histology; M.A., S.T., and H.O. contributedreagents and animal supply; T.W., T.K. and H.Y. analyzed data; and Y.Y., A.H.,and H.Y. wrote the paper.
Address correspondence and reprint requests to Dr. Yasuhiko Yamamoto, Departmentof Biochemistry and Molecular Vascular Biology, Kanazawa University GraduateSchool of Medical Science, 13-1 Takara-machi, Kanazawa 920-8640, Japan. E-mailaddress: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: AGE, advanced glycation end product; esRAGE,endogenous secretory receptor for advanced glycation end products; ET-1, endothe-lin-1; HMGB1, high mobility group box 1; KDO, 3-deoxy-D-manno-octulosonicacid; MPO, myeloperoxidase; PRR, pathogen recognition receptor; RAGE, receptorfor advanced glycation end products; siRNA, small interfering RNA; SPR, surfaceplasmon resonance; sRAGE, soluble receptor for advanced glycation end products;WT, wild-type.
Copyright� 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1002253
Published January 26, 2011, doi:10.4049/jimmunol.1002253 by guest on January 21, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

those ligands, HMGB1, S100 proteins, and Mac-1 are reported toyield proinflammatory reactions through RAGE (16, 19, 20, 22).RAGE is now recognized as one of PRRs capable of recognizingpathogen-associated molecular patterns. HMGB1 is a nuclear ar-chitectural chromatin binding and a cytosolic protein releasedfrom necrotic cells and activated macrophages and has been re-lated to LPS-induced lethality (16, 19). In an LPS-induced septicshock model, administration of anti-HMGB1 neutralizing Ab wasshown to prevent the death and RAGE-null mice exhibited aprominent decrease in the lethality compared with wild-type (WT)mice (24). In another septic shock model induced by polymicro-bial peritonitis following cecal ligation and puncture, RAGE-nullmice were less susceptible to the septic shock than WT mice (25).These observations have prompted us to investigate whether
RAGE could directly bind LPS, mediate LPS-induced signalingand proinflammatory reactions, and eventually lead to the septicshock. The results obtained have clearly defined RAGE as a newreceptor of LPS and a decoy form of RAGE (soluble RAGE[sRAGE]) as an effective therapeutic means against LPS-inducedseptic shock.
Materials and MethodsLPS
LPS (Escherichia coli 055:B5, E. coli 0127:B8, E. coli 0111:B4, Klebsiellapneumoniae, and Salmonella enterica serotype typhimurium) and 3-deoxy-D-manno-octulosonic acid (KDO)2-lipid A were purchased from Sigma-Aldrich and Avanti Polar Lipids, respectively. Surface plasmon resonance(SPR) assay showed that anti-AGE Abs such as anti-pentosidine Ab (clonePEN-12; TransGenic), anti-pyrraline Ab (clone H-12; TransGenic), andanti–Nε-carboxymethyllysine Ab (clone 6D12; TransGenic) did not react toLPS (Supplemental Fig. 1). Pretreatment of these AGE Abs also did notshow any inhibitory effects on LPS-mediated NF-kB activation, suggestingno AGEmodification on LPS employed in the assays (Supplemental Fig. 1).
Mice and a septic shock model
Male RAGE2/2 mice crossbred with the CD-1 strain over seven gen-erations and their WT littermates were used at 8–10 wk of age (26). Forinduction of septic shock, LPS serotype 055:B5 from E. coli (Sigma-Aldrich) was administered i.p. at 50 mg/kg body weight as described(27). Recombinant mouse sRAGE was prepared for animal experiments toavoid any immunological reactions and kindly provided by MitsubishiPharma. Mice received a single i.p. injection of sRAGE protein (35.0 mg/mouse) at 30 min after the LPS challenging. Survival was monitored for 72h. Survival as the end point in these experiments was calculated from thetime of LPS treatment using the product limit Kaplan-Meier method (28).Animals were treated in accordance with the Fundamental Guidelines forProper Conduct of Animal Experiment and Related Activities in AcademicResearch Institutions under the jurisdiction of the Ministry of Education,Culture, Sports, Science and Technology of Japan, and animal experimentswere approved by the Committee on Animal Experimentation of Kana-zawa University.
SPR assay
Purified human endogenous secretory RAGE (esRAGE) proteins having theligand-binding domain (29) were immobilized to a BIAcore CM5 research-grade sensor chip with the amine coupling kit (GE Healthcare) to a densityof ∼5000 response units. LPS (E. coli 0111:B4) was also coupled to thesensor chip by amine coupling. LPS binding to the immobilized RAGEprotein or esRAGE binding to the immobilized LPS was examined with theBIAcore 2000 system (GE Healthcare) as described previously (26, 29); theflow buffer used contained 10 mM HEPES (pH 7.4), 0.15 M NaCl, 3 mMNa-EDTA, and 0.005% (v/v) surfactant P-20. Association and dissociationwere measured at 25˚C at a flow rate of 20 ml/min. The sensor chips wereregenerated by washing with 10 mM NaOH and 0.5% (w/v) SDS.
Plate competition assay
LPS competition of AGE-RAGE association was assayed with AGE-BSA–coated 96-well plates, human esRAGE proteins, and Eu-conjugated anti-human RAGE Ab as previously described (26). AGE–BSA competition for LPS–RAGE interaction was also assayed withLPS-coated 96-well plates.
Peritoneal macrophages
Thioglycolate-elicited murine peritoneal macrophages were isolated fromRAGE+/+ and RAGE2/2 mice as previously described (30). The macro-phages were washed and cultured overnight in DMEM supplemented with10% FBS, 100 IU/ml penicillin, and 100 mg/ml streptomycin and stimu-lated with LPS 100 ng/ml for 24 h.
Flow cytometry
For flow cytometric analysis, peritoneal macrophages were washed andresuspended in staining buffer (PBS containing 2% FCS) containingFcBlock (BD Biosciences), and cells were stained with the following Abs(15 min at 4˚C in the dark): CD45-biotin (eBioscience), CD11b–APC-Cy7(BD Biosciences), TLR4-PE (eBioscience), TLR2–PE-Cy7 (eBioscience),and polyclonal rabbit anti-RAGE Ab (18). Biotinylated Ab and the RAGEAb were visualized following a second incubation with streptavidin-PE–Texas Red (BD Biosciences) and anti-rabbit IgG-FITC (eBioscience), re-spectively. Cells were resuspended in 200 ml staining buffer containing 0.2mg/ml propidium iodide (Sigma-Aldrich), filtered through a 100-mm mesh,and analyzed by FACSAria II (BD Biosciences). Blood were collectedwith 5 mM EDTA, treated with FcBlock (BD Biosciences), and stainedwith same Abs above and additional CD3-eFluor450 (eBioscience), CD8-APC (eBioscience), and CD19-PerCyP–Cy5.5 (eBioscience). For detectionof LPS binding, RAGE Ab and anti-rabbit IgG-FITC were replaced withLPS-FITC (Sigma-Aldrich). RBCs were then lysed with FACS lysing so-lution (BD Biosciences). Cells were resuspended in 200 ml staining buffer,filtered through a 100-mm mesh, and analyzed by FACSAria II (BD Bio-sciences). For detection of LPS binding to sRAGE in vivo, we used anti-mouse Ig, k beads of polystyrene particles (BD Biosciences), and thenegative control beads (BD Biosciences) with a mouse monoclonal RAGEAb (clone 278-13G4) (31) to detect the complex of sRAGE with FITC-labeled LPS (Sigma-Aldrich) using flow cytometry. Plasma was obtainedafter 2 h of i.p. injection of sRAGE (35.0 mg/mouse) and FITC-LPS (50mg/kg) into mice and then incubated with the RAGE Ab-coupled or thecontrol beads. The beads were washed three times and analyzed withFACSAria II (BD Biosciences). Data were transferred and reanalyzed withFlowJo software (Tree Star).
Plasma LPS concentration
Transition of i.p.-injected FITC-labeled LPS (Sigma-Aldrich) into plasmawas determined using a Labsystems Fluoroscan Ascent FL fluorescencemeter (Labsystems).
ELISAs for TNF-a, IL-6, endothelin-1, and HMGB1
Serum concentrations of TNF-a and IL-6 were measured by ELISAaccording to the manufacturer’s protocol (R&D Systems, Wiesbaden-Nordenstadt, Germany). Serum endothelin-1 (ET-1) and serum and culturemedium HMGB1 levels were measured with Peninsula Laboratories (SanCarlos, CA) and Shino test (Tokyo, Japan) ELISA systems, respectively.
NF-kB luciferase assay
C6 rat glioma cells were used for this assay according to the procedures asdescribed previously (26, 32). In brief, RAGE-expressing C6 cells stablytransformed with NF-kB promoter-driven luciferase construct were pre-incubated for 24 h in DMEM supplemented with 0.1% FBS and thenstimulated by LPS serotype 0111:B4 (50 ng/ml) (Sigma-Aldrich) for 4 h.RAGE expression was silenced by the small interfering RNA (siRNA)expression system using the pSilencer 3.0-H1 vector (Ambion, Austin,TX) and a dominant-negative RAGE lacking cytoplasmic domain was over-expressed (26). TLR2 and TLR4 expressions were silenced by FlexitubesiRNAs (Qiagen Rn_Tlr2-1, SI02045967, and Rn_Tlr4-2, SI02046002, re-spectively) according to the manufacturer’s protocol. The luciferase activitywas assayed with Luciferase Assay System (Promega). The resultant fluo-rescent products were measured with a Fluoroscan Ascent FL luminometer(Labsystems).
Histopathological examination
Animals were sacrificed for evaluation of tissue damage at 25 h after LPSchallenge with or without sRAGE treatment. Excised organs were fixedwith 4% paraformaldehyde, embedded in paraffin, and cut into 4-mm-thicksections followed by H&E staining for microscopic analysis. The neutro-phil infiltration was assessed by immunostaining for the myeloperoxidase(MPO). Primary Ab of rabbit-polyclonal anti-MPO (1:100; Santa CruzBiotechnology, Santa Cruz, CA) and peroxidase-conjugated Envision(Envision-PO, Envision System; DakoCytomation) were used. The sec-
2 RAGE MEDIATES LPS SHOCK
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
tions were immersed in DAB solution (Sigma-Aldrich) with H2O2, coun-terstained with hematoxylin (DakoCytomation), and mounted under cov-erslips.
Statistical analysis
Data obtained by the Kaplan-Meier method were analyzed by the log-ranktest. Differences between groups were assessed for statistical significanceusing the ANOVA test or the unpaired, two-tailed Student t test withStatview software (Abacus Concepts). p , 0.05 was considered to indicatestatistical significance.
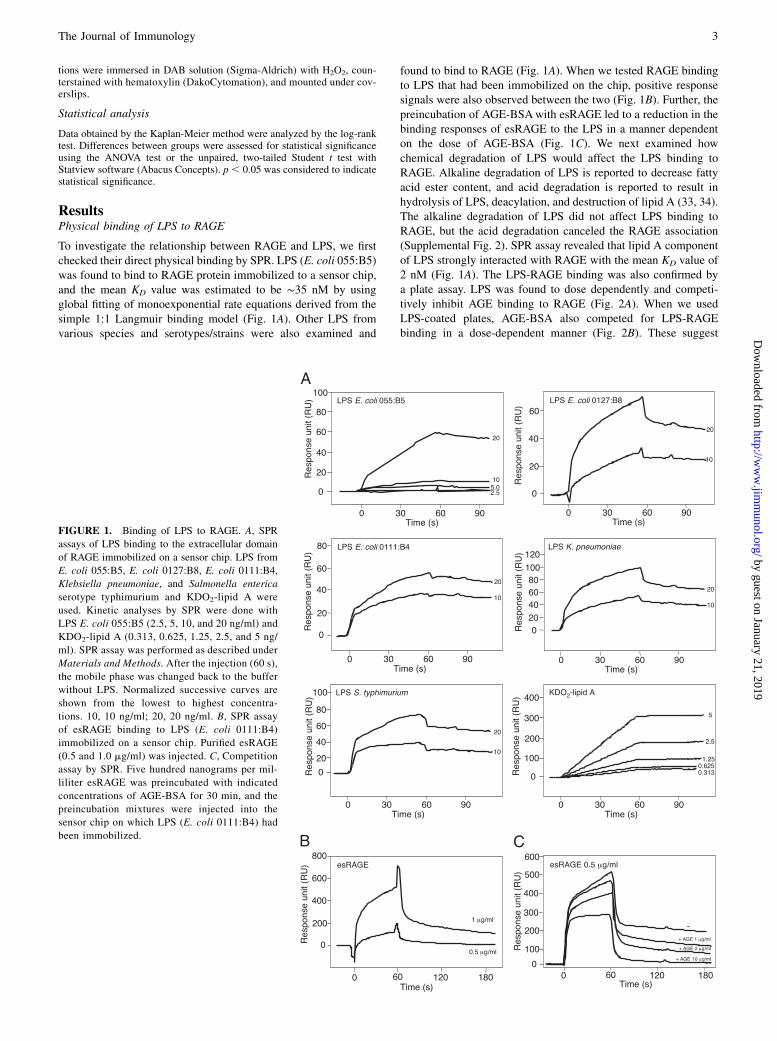
ResultsPhysical binding of LPS to RAGE
To investigate the relationship between RAGE and LPS, we firstchecked their direct physical binding by SPR. LPS (E. coli 055:B5)was found to bind to RAGE protein immobilized to a sensor chip,and the mean KD value was estimated to be ∼35 nM by usingglobal fitting of monoexponential rate equations derived from thesimple 1:1 Langmuir binding model (Fig. 1A). Other LPS fromvarious species and serotypes/strains were also examined and
found to bind to RAGE (Fig. 1A). When we tested RAGE bindingto LPS that had been immobilized on the chip, positive responsesignals were also observed between the two (Fig. 1B). Further, thepreincubation of AGE-BSAwith esRAGE led to a reduction in thebinding responses of esRAGE to the LPS in a manner dependenton the dose of AGE-BSA (Fig. 1C). We next examined howchemical degradation of LPS would affect the LPS binding toRAGE. Alkaline degradation of LPS is reported to decrease fattyacid ester content, and acid degradation is reported to result inhydrolysis of LPS, deacylation, and destruction of lipid A (33, 34).The alkaline degradation of LPS did not affect LPS binding toRAGE, but the acid degradation canceled the RAGE association(Supplemental Fig. 2). SPR assay revealed that lipid A componentof LPS strongly interacted with RAGE with the mean KD value of2 nM (Fig. 1A). The LPS-RAGE binding was also confirmed bya plate assay. LPS was found to dose dependently and competi-tively inhibit AGE binding to RAGE (Fig. 2A). When we usedLPS-coated plates, AGE-BSA also competed for LPS-RAGEbinding in a dose-dependent manner (Fig. 2B). These suggest
FIGURE 1. Binding of LPS to RAGE. A, SPR
assays of LPS binding to the extracellular domain
of RAGE immobilized on a sensor chip. LPS from
E. coli 055:B5, E. coli 0127:B8, E. coli 0111:B4,
Klebsiella pneumoniae, and Salmonella enterica
serotype typhimurium and KDO2-lipid A were
used. Kinetic analyses by SPR were done with
LPS E. coli 055:B5 (2.5, 5, 10, and 20 ng/ml) and
KDO2-lipid A (0.313, 0.625, 1.25, 2.5, and 5 ng/
ml). SPR assay was performed as described under
Materials and Methods. After the injection (60 s),
the mobile phase was changed back to the buffer
without LPS. Normalized successive curves are
shown from the lowest to highest concentra-
tions. 10, 10 ng/ml; 20, 20 ng/ml. B, SPR assay
of esRAGE binding to LPS (E. coli 0111:B4)
immobilized on a sensor chip. Purified esRAGE
(0.5 and 1.0 mg/ml) was injected. C, Competition
assay by SPR. Five hundred nanograms per mil-
liliter esRAGE was preincubated with indicated
concentrations of AGE-BSA for 30 min, and the
preincubation mixtures were injected into the
sensor chip on which LPS (E. coli 0111:B4) had
been immobilized.
The Journal of Immunology 3
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
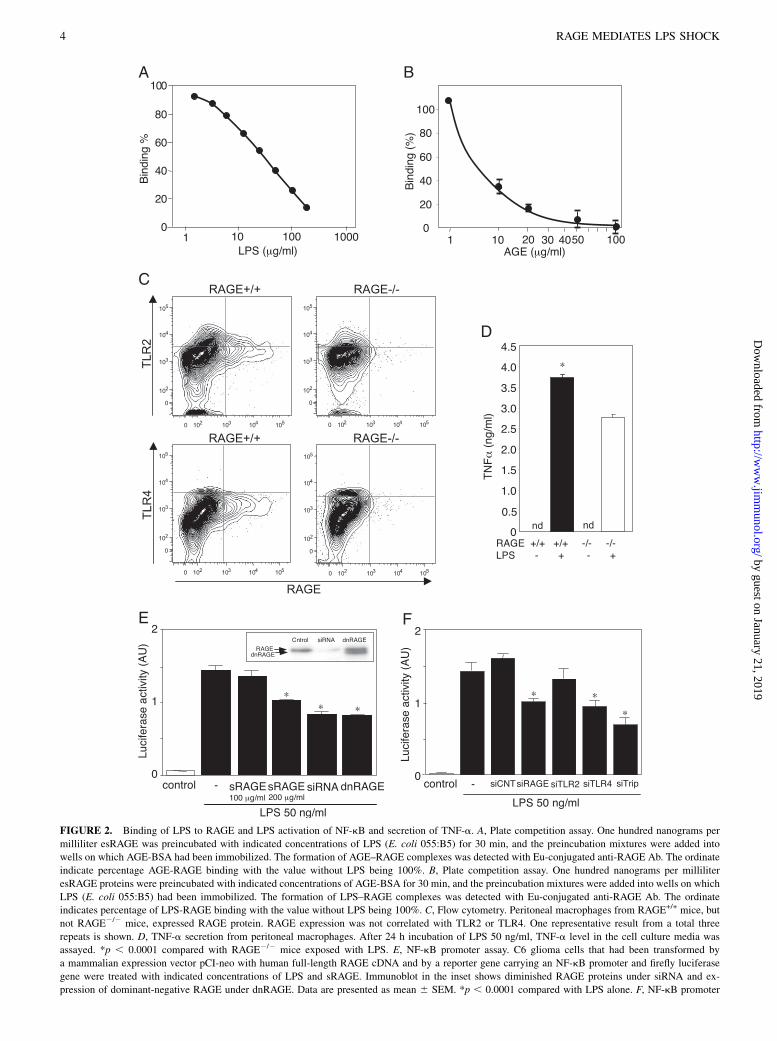
FIGURE 2. Binding of LPS to RAGE and LPS activation of NF-kB and secretion of TNF-a. A, Plate competition assay. One hundred nanograms per
milliliter esRAGE was preincubated with indicated concentrations of LPS (E. coli 055:B5) for 30 min, and the preincubation mixtures were added into
wells on which AGE-BSA had been immobilized. The formation of AGE–RAGE complexes was detected with Eu-conjugated anti-RAGE Ab. The ordinate
indicate percentage AGE-RAGE binding with the value without LPS being 100%. B, Plate competition assay. One hundred nanograms per milliliter
esRAGE proteins were preincubated with indicated concentrations of AGE-BSA for 30 min, and the preincubation mixtures were added into wells on which
LPS (E. coli 055:B5) had been immobilized. The formation of LPS–RAGE complexes was detected with Eu-conjugated anti-RAGE Ab. The ordinate
indicates percentage of LPS-RAGE binding with the value without LPS being 100%. C, Flow cytometry. Peritoneal macrophages from RAGE+/+ mice, but
not RAGE2/2 mice, expressed RAGE protein. RAGE expression was not correlated with TLR2 or TLR4. One representative result from a total three
repeats is shown. D, TNF-a secretion from peritoneal macrophages. After 24 h incubation of LPS 50 ng/ml, TNF-a level in the cell culture media was
assayed. *p , 0.0001 compared with RAGE2/2 mice exposed with LPS. E, NF-kB promoter assay. C6 glioma cells that had been transformed by
a mammalian expression vector pCI-neo with human full-length RAGE cDNA and by a reporter gene carrying an NF-kB promoter and firefly luciferase
gene were treated with indicated concentrations of LPS and sRAGE. Immunoblot in the inset shows diminished RAGE proteins under siRNA and ex-
pression of dominant-negative RAGE under dnRAGE. Data are presented as mean 6 SEM. *p , 0.0001 compared with LPS alone. F, NF-kB promoter
4 RAGE MEDIATES LPS SHOCK
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

that the LPS-binding site on RAGE might overlap with the site towhich AGE ligands bind. To identify potential LPS-binding sitesof RAGE, we employed synthetic peptides of human RAGEprotein. VN1 peptide (KGAPKKPPQRLEWKLN), but not VN2peptide (WKLNTGRTEAWKVLSPQG), was revealed by SPR tobind lipid A as well as various types of LPS (Supplemental Fig. 3).
Induction of TNF-a secretion and NF-kB activation
Next, we isolated peritoneal macrophages from RAGE+/+ andRAGE2/2 mice and examined LPS-induced TNF-a secretion intothe culture media. Flow cytometry showed the RAGE proteinexpressed on the cell surface of the macrophage from RAGE+/+
mice (Fig. 2C). TLR2 and -4 expressions were also observed inmacrophages from both and were independent of RAGE expres-sion (Fig. 2C). LPS stimulation significantly increased TNF-asecretion in RAGE+/+ mouse-derived macrophages when com-pared with that in RAGE-null macrophages (Fig. 2D). We furtherchecked whether LPS could induce a post-RAGE signaling byusing an RAGE-expressing C6 glioma cells carrying a luciferasereporter gene under a control of NF-kB promoter. Addition of LPSin the cell-culture media markedly increased NF-kB–driven lu-ciferase activity (Fig. 2E). This activation was significantly inhib-ited in the coexistence of sRAGE, although the inhibition waspartial in the conditions employed. RAGE dependency of the LPS-induced NF-kB activation was also demonstrated with RAGEsiRNA and dominant-negative RAGE lacking the intracellular do-main. The C6 glioma cells expressing RAGE siRNA or dominant-negative RAGE showed significant reduction in the LPS-inducedluciferase activity. We performed gene knockdown experimentswith RAGE siRNA, TLR2 siRNA, TLR4 siRNA, or the allsiRNAs in the C6 rat glioma cells. The LPS-induced NF-kBluciferase activity in the C6 glioma cells were downregulatedby the treatment with RAGE siRNA, TLR2 siRNA, TLR4 siRNA,or the combination of three siRNAs by 37.2, 17.6, 41.1, and57.1%, respectively (Fig. 2F). These observations suggest thatRAGE and TLR4 contribute in a comparable manner to the LPS-induced NF-kB activation in this cell line.
RAGE expression on blood neutrophils and monocytes
We also checked RAGE expression levels in mouse circulatingneutrophils and monocytes by flow cytometry. The neutrophils andmonocytes were defined as CD45+CD11b+Gr1++CD32CD192 andCD45+CD11b+CD32CD192, respectively. Both cell types fromRAGE+/+ mice expressed RAGE protein (Fig. 3A). Neutrophilswere found to have more abundant cell-surface RAGE proteinthan monocytes (Fig. 3B). FITC-labeled LPS efficiently bound toRAGE+/+ mouse-derived neutrophils and monocytes (Fig. 3C–E).In addition, this ligation was significantly lowered in RAGE2/2
mouse-derived cell and by the pretreatment of unlabeled LPS (Fig.3D, 3F), indicating sufficient evidence of RAGE-specific LPSbinding on the inflammatory cell surface. We next examined re-lationship between RAGE and TLR2 and -4 on blood neutrophilsand monocytes. TLR2-positive population was significantly andcompensatedly increased in neutrophils from RAGE2/2 micewhen compared with RAGE+/+ mice (Fig. 3G, 3H). This tendencywas also seen in monocytes (Fig. 3G, 3H). However, we could notobserve such an increase in TLR4 in RAGE2/2 mice (Fig. 3G,3H).
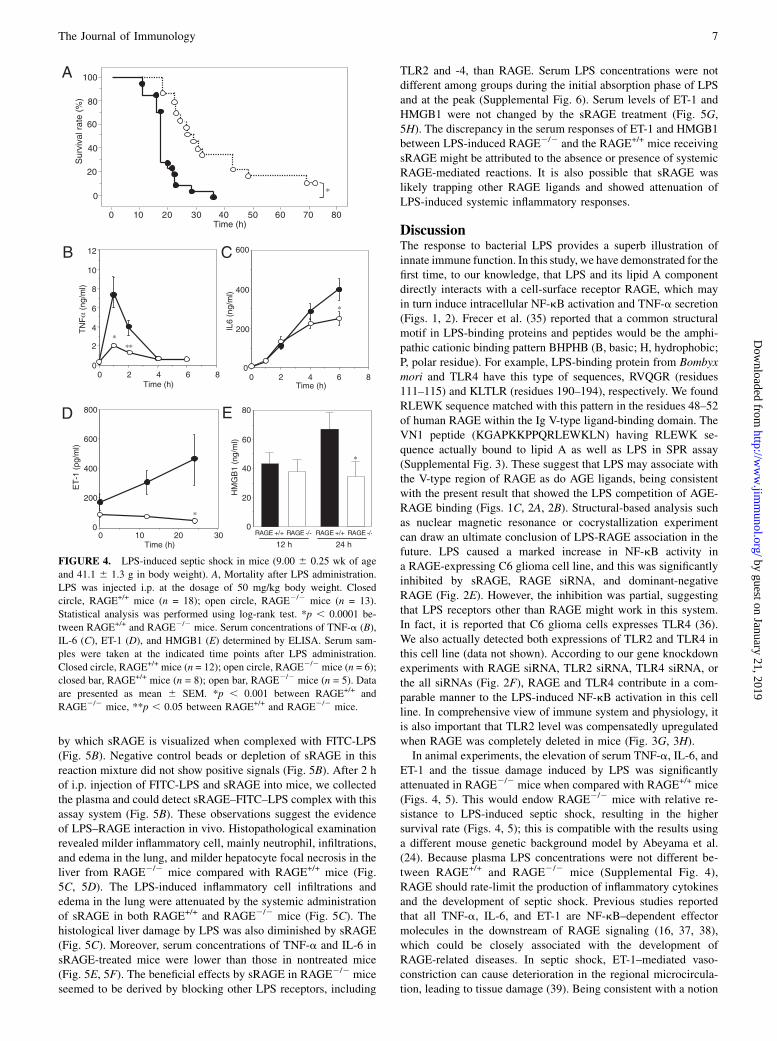
LPS-induced septic shock in RAGE+/+ and RAGE2/2 mice
To examine in vivo role of LPS–RAGE interactions in an LPS-loading septic shock model, RAGE2/2 and RAGE+/+ mice werei.p. injected with LPS and observed. All of the mice that receivedLPS showed apparent signs of distress such as apathy, fur ruffling,and diarrhea. These symptoms were more prominent in RAGE+/+
mice. All RAGE+/+ mice succumbed to shock within the periodfrom 24–38 h after LPS injection (Fig. 4A). However, RAGE2/2
mice began to die after 18 h of LPS injection, and 10% of theanimals still survived at 72 h (p, 0.003) (Fig. 4A), indicating thatRAGE2/2 mice are relatively resistant to septic shock. CirculatingLPS concentrations did not differ between RAGE+/+ and RAGE2/2
mice during the initial absorption phase of LPS and at the peak(Supplemental Fig. 4).Next, we investigated serum levels of acute inflammation
markers and a vascular tone regulator closely related to LPS-induced septic shock. Serum levels of TNF-a and IL-6 weresignificantly lower in RAGE2/2 mice than in RAGE+/+ mice at 1and 2 h and at 6 h, respectively, after injection of LPS (Fig. 4B,4C), the time points at which the serum concentrations of therespective cytokines are known to peak (5, 27). Moreover, serumET-1 level was also significantly decreased in RAGE2/2 micecompared with RAGE+/+ mice (Fig. 4D). The difference in therelease of acute-phase cytokines, TNF-a and IL-6, and vascularconstrictor ET-1 could influence the difference in the survival rate.Additionally, we checked serum level of HMGB1, a late-phasecytokine associated with the survival in septic shock and knownas a bioactive molecule binding to RAGE. Serum HMGB1 in-creased at 24 h after LPS injection in RAGE+/+ mice but remainedunchanged in RAGE2/2 mice (Fig. 4E). We also measured serumlevels of S100A8/A9 and S100B, other RAGE ligands, by ELI-SAs. S100B level before LPS injection (background level) wasfound to be significantly lower in RAGE2/2 mice than in RAGE+/+
mice (Supplemental Fig. 5). After LPS injection, both serumlevels of S100A8/A9 and S100B were dramatically increased(Supplemental Fig. 5). However, serum S100A8/A9 level at 24 hafter LPS injection was 2-fold higher in RAGE2/2 mice than inRAGE+/+ mice (Supplemental Fig. 5); this suggests that increasedserum S100A8/A9 and S100B might not settle the severe in-flammation by LPS in RAGE+/+ mice, and RAGE deficiency eli-cited a significant increase in serum S100A8/A9 level after LPSloading.
sRAGE treatment attenuated LPS-induced septic shock
We next examined whether the treatment with sRAGE, which actsas a decoy receptor for RAGE ligands, could improve the survivalof the LPS-injected RAGE+/+ mice as well as RAGE2/2 mice. Inthis experiment, we reproduced the same survival difference seenin Fig. 4A between RAGE+/+ and RAGE2/2 mice. However, themean survival time of RAGE+/+ or RAGE2/2 mice was longer inFig. 5A than in Fig. 4A; this might be caused by the difference inbody weight (41.1 6 1.3 and 36.0 6 0.5 g in Figs. 4A, 5A, re-spectively; p , 0.0001). As a result, the treatment of sRAGE wasfound to attribute significantly longer survival rate not only inRAGE+/+ mice but also in RAGE2/2 mice (Fig. 5A). For detectionof LPS–sRAGE complex formed in vivo, we established a newassay system with the anti-mouse Ig, k beads, and the RAGE Ab,
assay with the RAGE expressing C6 glioma cells. Data are presented as mean 6 SEM. *p , 0.03 compared with LPS alone and siCNT. AU, arbitrary unit;
dnRAGE, the C6 glioma cells further transformed by pCI-neo expressing dominant-negative RAGE that lacked the cytoplasmic domain; nd, not detected;
siCNT, control siRNA; siRAGE, RAGE siRNA; siRNA, the C6 glioma cells further transformed by RAGE siRNA expression vector; siTLR2, TLR2 siRNA;
siTLR4, TLR4 siRNA; siTrip, a combination of RAGE siRNA, TLR2 siTNA, and TLR4 siRNA.
The Journal of Immunology 5
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
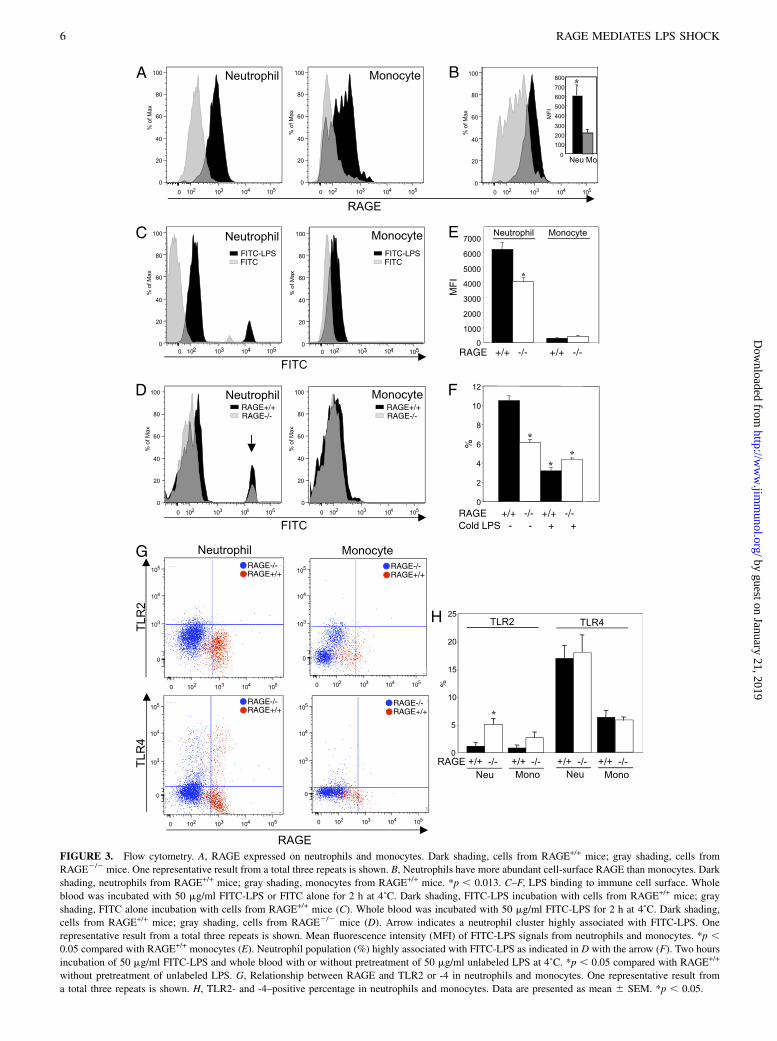
FIGURE 3. Flow cytometry. A, RAGE expressed on neutrophils and monocytes. Dark shading, cells from RAGE+/+ mice; gray shading, cells from
RAGE2/2 mice. One representative result from a total three repeats is shown. B, Neutrophils have more abundant cell-surface RAGE than monocytes. Dark
shading, neutrophils from RAGE+/+ mice; gray shading, monocytes from RAGE+/+ mice. *p , 0.013. C–F, LPS binding to immune cell surface. Whole
blood was incubated with 50 mg/ml FITC-LPS or FITC alone for 2 h at 4˚C. Dark shading, FITC-LPS incubation with cells from RAGE+/+ mice; gray
shading, FITC alone incubation with cells from RAGE+/+ mice (C). Whole blood was incubated with 50 mg/ml FITC-LPS for 2 h at 4˚C. Dark shading,
cells from RAGE+/+ mice; gray shading, cells from RAGE2/2 mice (D). Arrow indicates a neutrophil cluster highly associated with FITC-LPS. One
representative result from a total three repeats is shown. Mean fluorescence intensity (MFI) of FITC-LPS signals from neutrophils and monocytes. *p ,0.05 compared with RAGE+/+ monocytes (E). Neutrophil population (%) highly associated with FITC-LPS as indicated in D with the arrow (F). Two hours
incubation of 50 mg/ml FITC-LPS and whole blood with or without pretreatment of 50 mg/ml unlabeled LPS at 4˚C. *p , 0.05 compared with RAGE+/+
without pretreatment of unlabeled LPS. G, Relationship between RAGE and TLR2 or -4 in neutrophils and monocytes. One representative result from
a total three repeats is shown. H, TLR2- and -4–positive percentage in neutrophils and monocytes. Data are presented as mean 6 SEM. *p , 0.05.
6 RAGE MEDIATES LPS SHOCK
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by which sRAGE is visualized when complexed with FITC-LPS(Fig. 5B). Negative control beads or depletion of sRAGE in thisreaction mixture did not show positive signals (Fig. 5B). After 2 hof i.p. injection of FITC-LPS and sRAGE into mice, we collectedthe plasma and could detect sRAGE–FITC–LPS complex with thisassay system (Fig. 5B). These observations suggest the evidenceof LPS–RAGE interaction in vivo. Histopathological examinationrevealed milder inflammatory cell, mainly neutrophil, infiltrations,and edema in the lung, and milder hepatocyte focal necrosis in theliver from RAGE2/2 mice compared with RAGE+/+ mice (Fig.5C, 5D). The LPS-induced inflammatory cell infiltrations andedema in the lung were attenuated by the systemic administrationof sRAGE in both RAGE+/+ and RAGE2/2 mice (Fig. 5C). Thehistological liver damage by LPS was also diminished by sRAGE(Fig. 5C). Moreover, serum concentrations of TNF-a and IL-6 insRAGE-treated mice were lower than those in nontreated mice(Fig. 5E, 5F). The beneficial effects by sRAGE in RAGE2/2 miceseemed to be derived by blocking other LPS receptors, including
TLR2 and -4, than RAGE. Serum LPS concentrations were notdifferent among groups during the initial absorption phase of LPSand at the peak (Supplemental Fig. 6). Serum levels of ET-1 andHMGB1 were not changed by the sRAGE treatment (Fig. 5G,5H). The discrepancy in the serum responses of ET-1 and HMGB1between LPS-induced RAGE2/2 and the RAGE+/+ mice receivingsRAGE might be attributed to the absence or presence of systemicRAGE-mediated reactions. It is also possible that sRAGE waslikely trapping other RAGE ligands and showed attenuation ofLPS-induced systemic inflammatory responses.
DiscussionThe response to bacterial LPS provides a superb illustration ofinnate immune function. In this study, we have demonstrated for thefirst time, to our knowledge, that LPS and its lipid A componentdirectly interacts with a cell-surface receptor RAGE, which mayin turn induce intracellular NF-kB activation and TNF-a secretion(Figs. 1, 2). Frecer et al. (35) reported that a common structuralmotif in LPS-binding proteins and peptides would be the amphi-pathic cationic binding pattern BHPHB (B, basic; H, hydrophobic;P, polar residue). For example, LPS-binding protein from Bombyxmori and TLR4 have this type of sequences, RVQGR (residues111–115) and KLTLR (residues 190–194), respectively. We foundRLEWK sequence matched with this pattern in the residues 48–52of human RAGE within the Ig V-type ligand-binding domain. TheVN1 peptide (KGAPKKPPQRLEWKLN) having RLEWK se-quence actually bound to lipid A as well as LPS in SPR assay(Supplemental Fig. 3). These suggest that LPS may associate withthe V-type region of RAGE as do AGE ligands, being consistentwith the present result that showed the LPS competition of AGE-RAGE binding (Figs. 1C, 2A, 2B). Structural-based analysis suchas nuclear magnetic resonance or cocrystallization experimentcan draw an ultimate conclusion of LPS-RAGE association in thefuture. LPS caused a marked increase in NF-kB activity ina RAGE-expressing C6 glioma cell line, and this was significantlyinhibited by sRAGE, RAGE siRNA, and dominant-negativeRAGE (Fig. 2E). However, the inhibition was partial, suggestingthat LPS receptors other than RAGE might work in this system.In fact, it is reported that C6 glioma cells expresses TLR4 (36).We also actually detected both expressions of TLR2 and TLR4 inthis cell line (data not shown). According to our gene knockdownexperiments with RAGE siRNA, TLR2 siRNA, TLR4 siRNA, orthe all siRNAs (Fig. 2F), RAGE and TLR4 contribute in a com-parable manner to the LPS-induced NF-kB activation in this cellline. In comprehensive view of immune system and physiology, itis also important that TLR2 level was compensatedly upregulatedwhen RAGE was completely deleted in mice (Fig. 3G, 3H).In animal experiments, the elevation of serum TNF-a, IL-6, and
ET-1 and the tissue damage induced by LPS was significantlyattenuated in RAGE2/2 mice when compared with RAGE+/+ mice(Figs. 4, 5). This would endow RAGE2/2 mice with relative re-sistance to LPS-induced septic shock, resulting in the highersurvival rate (Figs. 4, 5); this is compatible with the results usinga different mouse genetic background model by Abeyama et al.(24). Because plasma LPS concentrations were not different be-tween RAGE+/+ and RAGE2/2 mice (Supplemental Fig. 4),RAGE should rate-limit the production of inflammatory cytokinesand the development of septic shock. Previous studies reportedthat all TNF-a, IL-6, and ET-1 are NF-kB–dependent effectormolecules in the downstream of RAGE signaling (16, 37, 38),which could be closely associated with the development ofRAGE-related diseases. In septic shock, ET-1–mediated vaso-constriction can cause deterioration in the regional microcircula-tion, leading to tissue damage (39). Being consistent with a notion
FIGURE 4. LPS-induced septic shock in mice (9.00 6 0.25 wk of age
and 41.1 6 1.3 g in body weight). A, Mortality after LPS administration.
LPS was injected i.p. at the dosage of 50 mg/kg body weight. Closed
circle, RAGE+/+ mice (n = 18); open circle, RAGE2/2 mice (n = 13).
Statistical analysis was performed using log-rank test. *p , 0.0001 be-
tween RAGE+/+ and RAGE2/2 mice. Serum concentrations of TNF-a (B),
IL-6 (C), ET-1 (D), and HMGB1 (E) determined by ELISA. Serum sam-
ples were taken at the indicated time points after LPS administration.
Closed circle, RAGE+/+ mice (n = 12); open circle, RAGE2/2 mice (n = 6);
closed bar, RAGE+/+ mice (n = 8); open bar, RAGE2/2 mice (n = 5). Data
are presented as mean 6 SEM. *p , 0.001 between RAGE+/+ and
RAGE2/2 mice, **p , 0.05 between RAGE+/+ and RAGE2/2 mice.
The Journal of Immunology 7
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
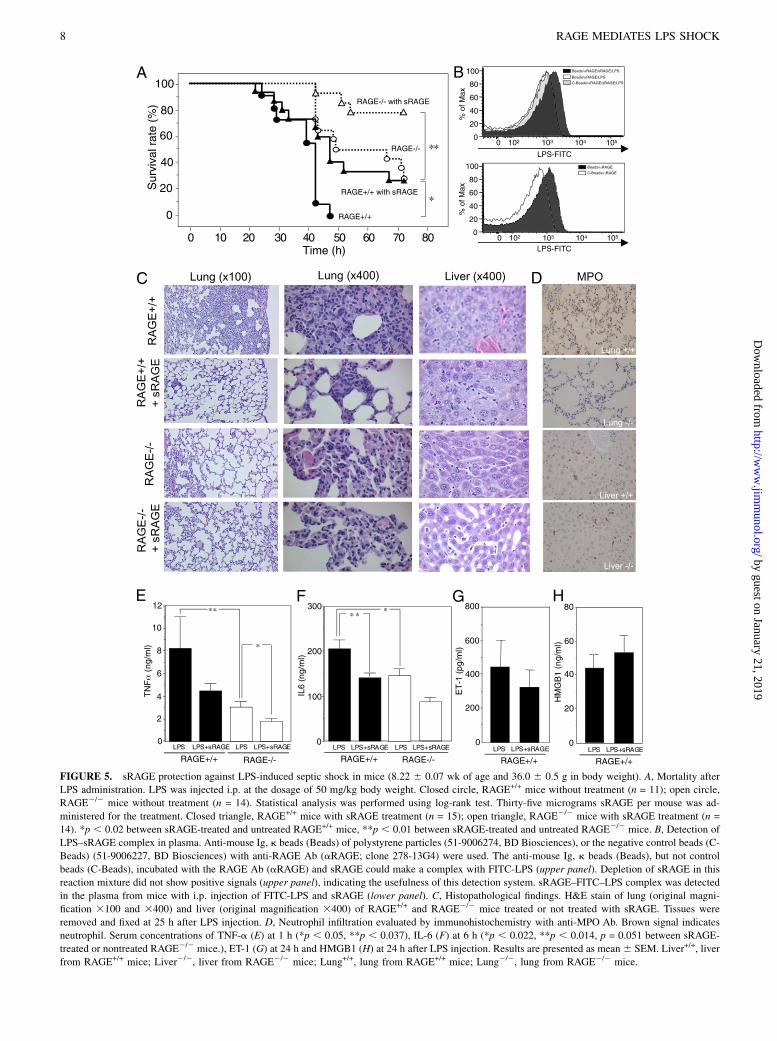
FIGURE 5. sRAGE protection against LPS-induced septic shock in mice (8.22 6 0.07 wk of age and 36.0 6 0.5 g in body weight). A, Mortality after
LPS administration. LPS was injected i.p. at the dosage of 50 mg/kg body weight. Closed circle, RAGE+/+ mice without treatment (n = 11); open circle,
RAGE2/2 mice without treatment (n = 14). Statistical analysis was performed using log-rank test. Thirty-five micrograms sRAGE per mouse was ad-
ministered for the treatment. Closed triangle, RAGE+/+ mice with sRAGE treatment (n = 15); open triangle, RAGE2/2 mice with sRAGE treatment (n =
14). *p , 0.02 between sRAGE-treated and untreated RAGE+/+ mice, **p , 0.01 between sRAGE-treated and untreated RAGE2/2 mice. B, Detection of
LPS–sRAGE complex in plasma. Anti-mouse Ig, k beads (Beads) of polystyrene particles (51-9006274, BD Biosciences), or the negative control beads (C-
Beads) (51-9006227, BD Biosciences) with anti-RAGE Ab (aRAGE; clone 278-13G4) were used. The anti-mouse Ig, k beads (Beads), but not control
beads (C-Beads), incubated with the RAGE Ab (aRAGE) and sRAGE could make a complex with FITC-LPS (upper panel). Depletion of sRAGE in this
reaction mixture did not show positive signals (upper panel), indicating the usefulness of this detection system. sRAGE–FITC–LPS complex was detected
in the plasma from mice with i.p. injection of FITC-LPS and sRAGE (lower panel). C, Histopathological findings. H&E stain of lung (original magni-
fication 3100 and 3400) and liver (original magnification 3400) of RAGE+/+ and RAGE2/2 mice treated or not treated with sRAGE. Tissues were
removed and fixed at 25 h after LPS injection. D, Neutrophil infiltration evaluated by immunohistochemistry with anti-MPO Ab. Brown signal indicates
neutrophil. Serum concentrations of TNF-a (E) at 1 h (*p , 0.05, **p , 0.037), IL-6 (F) at 6 h (*p , 0.022, **p , 0.014, p = 0.051 between sRAGE-
treated or nontreated RAGE2/2 mice.), ET-1 (G) at 24 h and HMGB1 (H) at 24 h after LPS injection. Results are presented as mean6 SEM. Liver+/+, liver
from RAGE+/+ mice; Liver2/2, liver from RAGE2/2 mice; Lung+/+, lung from RAGE+/+ mice; Lung2/2, lung from RAGE2/2 mice.
8 RAGE MEDIATES LPS SHOCK
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

that TNF-a is an initiator of the cascade of proinflammatoryresponses to LPS loading (5), circulating TNF-a level was ob-served to peak as rapidly as 1 h after LPS challenge (Fig. 4B). Anelevation of the late mediator HMGB1, which is also a RAGEligand, was marked at 24 h after LPS loading (Fig. 4E). HMGB1is known to be released readily from necrotic or damaged cellsand serves as a local signal for inflammation. Such paracrine-likeactions of HMGB1 should also be taken into account even in thissystemic inflammatory model. These findings suggest that LPS-mediated RAGE signaling accelerates acute inflammatory reac-tions and vascular dysregulation leading to tissue damages, whichthen mediate HMGB1 release, resulting in a pernicious cycle ofRAGE-dependent lethality in septic shock.This study has also shown that sRAGE, a truncated soluble form
carrying extracellular ligand-binding domain of RAGE, has pro-tective effects against LPS-induced septic shock in mice. Previousstudies using sRAGE demonstrated its beneficial effects on RAGE-associated diseases such as atherosclerosis and inflammation (20,40, 41). The treatment with sRAGE reduced the LPS induction ofTNF-a and IL-6 and of liver and lung damages and significantlyimproved survival rate (Fig. 5). It may be reasonable to posit thatsRAGE trapping of LPS may also inhibit non-RAGE LPS receptorsignaling as well. In addition, sRAGE may also neutralize late-phase HMGB1 function, leading to prevention of LPS-inducedseptic shock. Liliensiek et al. (25) have also shown that RAGE2/2
mice from delayed-type hypersensitivity-triggered inflammation byusing sRAGE. Further studies are needed to reveal whether sRAGEdirectly can block other receptors than RAGE or sRAGE can an-tagonize ligand/LPS binding to the other receptors.A recent report using the alive E. coli infection model in contrast
to this LPS shock model demonstrated that intact RAGE signalingcontributed to an effective antibacterial defense such as inhibitionof bacterial outgrowth and dissemination, and RAGE deficiencyresulted in enhanced organ injuries such as liver necrosis (42). Ingeneral, the innate immune response to severe bacterial infectioncan act as a double-edged sword, on the one hand protectingthe host against invading pathogens, and on the other hand po-tentially destroying cells and tissues. Even though the LPS shockmodel is an extreme exaggerated inflammation model, RAGE canparticipate in sensing pathogens and controlling a delicate balancebetween clearance of invading pathogens and exaggerated in-flammation.In conclusion, the results obtained indicate that RAGE partic-
ipates in inflammatory responses. Upon LPS stimulation, sRAGEseems to antagonize ligand binding not only to RAGE, but also toTLR2 and TLR4, resulting in inhibition of LPS-induced inflam-mation. sRAGEmay become an effective remedy for treating septicshock.
AcknowledgmentsWe thank S. Matsudaira, R. Kitamura, and Y. Niimura for assistance.
DisclosuresThe authors have no financial conflicts of interest.
References1. Morrison, D. C., and J. L. Ryan. 1987. Endotoxins and disease mechanisms.
Annu. Rev. Med. 38: 417–432.2. Raetz, C. R. H. 1990. Biochemistry of endotoxins. Annu. Rev. Biochem. 59: 129–170.3. Glauser, M. P., G. Zanetti, J. D. Baumgartner, and J. Cohen. 1991. Septic shock:
pathogenesis. Lancet 338: 732–736.4. Parrillo, J. E. 1993. Pathogenetic mechanisms of septic shock. N. Engl. J. Med.
328: 1471–1477.5. Engelberts, I., E. J. von Asmuth, C. J. van der Linden, and W. A. Buurman. 1991.
The interrelation between TNF, IL-6, and PAF secretion induced by LPS in anin vivo and in vitro murine model. Lymphokine Cytokine Res. 10: 127–131.
6. Poltorak, A., X. He, I. Smirnova, M. Y. Liu, C. Van Huffel, X. Du, D. Birdwell,E. Alejos, M. Silva, C. Galanos, et al. 1998. Defective LPS signaling in C3H/HeJand C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088.
7. da Silva Correia, J., K. Soldau, U. Christen, P. S. Tobias, and R. J. Ulevitch.2001. Lipopolysaccharide is in close proximity to each of the proteins in itsmembrane receptor complex. transfer from CD14 to TLR4 and MD-2. J. Biol.Chem. 276: 21129–21135.
8. Chassin, C., M. Picardeau, J. M. Goujon, P. Bourhy, N. Quellard, S. Darche,E. Badell, M. F. d’Andon, N. Winter, S. Lacroix-Lamande, et al. 2009. TLR4-and TLR2-mediated B cell responses control the clearance of the bacterialpathogen, Leptospira interrogans. J. Immunol. 183: 2669–2677.
9. Triantafilou, K., M. Triantafilou, and R. L. Dedrick. 2001. A CD14-independentLPS receptor cluster. Nat. Immunol. 2: 338–345.
10. Miyake, K., H. Ogata, Y. Nagai, S. Akashi, and M. Kimoto. 2000. Innate rec-ognition of lipopolysaccharide by Toll-like receptor 4/MD-2 and RP105/MD-1.J. Endotoxin Res. 6: 389–391.
11. Wright, S. D., S. M. Levin, M. T. Jong, Z. Chad, and L. G. Kabbash. 1989. CR3(CD11b/CD18) expresses one binding site for Arg-Gly-Asp-containing peptidesand a second site for bacterial lipopolysaccharide. J. Exp. Med. 169: 175–183.
12. El-Samalouti, V. T., J. Schletter, H. Brade, L. Brade, S. Kusumoto,E. T. Rietschel, H. D. Flad, and A. J. Ulmer. 1997. Detection of lipopolysac-charide (LPS)-binding membrane proteins by immuno-coprecipitation with LPSand anti-LPS antibodies. Eur. J. Biochem. 250: 418–424.
13. Malhotra, R., and M. I. Bird. 1997. L-selectin: a novel receptor for lipopoly-saccharide and its potential role in bacterial sepsis. Bioessays 19: 919–923.
14. Malhotra, R., R. Priest, M. R. Foster, and M. I. Bird. 1998. P-selectin binds tobacterial lipopolysaccharide. Eur. J. Immunol. 28: 983–988.
15. Hampton, R. Y., D. T. Golenbock, M. Penman, M. Krieger, and C. R. Raetz.1991. Recognition and plasma clearance of endotoxin by scavenger receptors.Nature 352: 342–344.
16. Bierhaus, A., P. M. Humpert, M. Morcos, T. Wendt, T. Chavakis, B. Arnold,D. M. Stern, and P. P. Nawroth. 2005. Understanding RAGE, the receptor foradvanced glycation end products. J. Mol. Med. 83: 876–886.
17. Inagi, R., Y. Yamamoto, M. Nangaku, N. Usuda, H. Okamato, K. Kurokawa,C. van Ypersele de Strihou, H. Yamamoto, and T. Miyata. 2006. A severe di-abetic nephropathy model with early development of nodule-like lesions inducedby megsin overexpression in RAGE/iNOS transgenic mice. Diabetes 55: 356–366.
18. Yamamoto, Y., I. Kato, T. Doi, H. Yonekura, S. Ohashi, M. Takeuchi,T. Watanabe, S. Yamagishi, S. Sakurai, S. Takasawa, et al. 2001. Developmentand prevention of advanced diabetic nephropathy in RAGE-overexpressing mice.J. Clin. Invest. 108: 261–268.
19. Scaffidi, P., T. Misteli, and M. E. Bianchi. 2002. Release of chromatin proteinHMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195.
20. Hofmann, M. A., S. Drury, C. Fu, W. Qu, A. Taguchi, Y. Lu, C. Avila,N. Kambham, A. Bierhaus, P. Nawroth, et al. 1999. RAGE mediates a novelproinflammatory axis: a central cell surface receptor for S100/calgranulin pol-ypeptides. Cell 97: 889–901.
21. Sousa, M. M., S. D. Yan, D. Stern, and M. J. Saraiva. 2000. Interaction of thereceptor for advanced glycation end products (RAGE) with transthyretin triggersnuclear transcription factor kB (NF-kB) activation. Lab. Invest. 80: 1101–1110.
22. Chavakis, T., A. Bierhaus, N. Al-Fakhri, D. Schneider, S. Witte, T. Linn,M. Nagashima, J. Morser, B. Arnold, K. T. Preissner, and P. P. Nawroth. 2003. Thepattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins:a novel pathway for inflammatory cell recruitment. J. Exp. Med. 198: 1507–1515.
23. Yan, S. D., X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao,M. Nagashima, J. Morser, et al. 1996. RAGE and amyloid-beta peptide neuro-toxicity in Alzheimer’s disease. Nature 382: 685–691.
24. Abeyama, K., D. M. Stern, Y. Ito, K. Kawahara, Y. Yoshimoto, M. Tanaka,T. Uchimura, N. Ida, Y. Yamazaki, S. Yamada, et al. 2005. The N-terminaldomain of thrombomodulin sequesters high-mobility group-B1 protein,a novel antiinflammatory mechanism. J. Clin. Invest. 115: 1267–1274.
25. Liliensiek, B., M. A. Weigand, A. Bierhaus, W. Nicklas, M. Kasper, S. Hofer,J. Plachky, H. J. Grone, F. C. Kurschus, A. M. Schmidt, et al. 2004. Receptor foradvanced glycation end products (RAGE) regulates sepsis but not the adaptiveimmune response. J. Clin. Invest. 113: 1641–1650.
26. Myint, K. M., Y. Yamamoto, T. Doi, I. Kato, A. Harashima, H. Yonekura,T. Watanabe, H. Shinohara, M. Takeuchi, K. Tsuneyama, et al. 2006. RAGE controlof diabetic nephropathy in a mouse model: effects of RAGE gene disruption andadministration of low-molecular weight heparin. Diabetes 55: 2510–2522.
27. Hill, M. R., S. Clarke, K. Rodgers, B. Thornhill, J. M. Peters, F. J. Gonzalez, andJ. M. Gimble. 1999. Effect of peroxisome proliferator-activated receptor alphaactivators on tumor necrosis factor expression in mice during endotoxemia. In-fect. Immun. 67: 3488–3493.
28. Kaplan, E., and P. Meier. 1958. Nonparametric estimation from incompleteobservations. J. Am. Stat. Assoc. 53: 457–816.
29. Yonekura, H., Y. Yamamoto, S. Sakurai, R. G. Petrova, M. J. Abedin, H. Li,K. Yasui, M. Takeuchi, Z. Makita, S. Takasawa, et al. 2003. Novel splice variantsof the receptor for advanced glycation end-products expressed in human vascularendothelial cells and pericytes, and their putative roles in diabetes-inducedvascular injury. Biochem. J. 370: 1097–1109.
30. Randriamampita, C., and A. Trautmann. 1987. Ionic channels in murine mac-rophages. J. Cell Biol. 105: 761–769.
31. Sakurai, S., Y. Yamamoto, H. Tamei, H. Matsuki, K. Obata, L. Hui, J. Miura,M. Osawa, Y. Uchigata, Y. Iwamoto, et al. 2006. Development of an ELISA foresRAGE and its application to type 1 diabetic patients. Diabetes Res. Clin.Pract. 73: 158–165.
The Journal of Immunology 9
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

32. Harashima, A., Y. Yamamoto, C. Cheng, K. Tsuneyama, K. M. Myint,A. Takeuchi, K. Yoshimura, H. Li, T. Watanabe, S. Takasawa, et al. 2006.Identification of mouse orthologue of endogenous secretory receptor for ad-vanced glycation end-products: structure, function and expression. Biochem. J.396: 109–115.
33. Niwa, M., K. C. Milner, E. Ribi, and J. A. Rudbach. 1969. Alteration of physical,chemical, and biological properties of endotoxin by treatment with mild alkali. J.Bacteriol. 97: 1069–1077.
34. Zahringer, U., B. Lindner, Y. A. Knirel, W. M. van den Akker, R. Hiestand,H. Heine, and C. Dehio. 2004. Structure and biological activity of the short-chainlipopolysaccharide from Bartonella henselae ATCC 49882T. J. Biol. Chem. 279:21046–21054.
35. Frecer, V., B. Ho, and J. L. Ding. 2000. Interpretation of biological activity dataof bacterial endotoxins by simple molecular models of mechanism of action. Eur.J. Biochem. 267: 837–852.
36. Ogura, M., N. Nakamichi, K. Takano, H. Oikawa, Y. Kambe, Y. Ohno, H. Taniura,and Y. Yoneda. 2006. Functional expression of A glutamine transporter responsiveto down-regulation by lipopolysaccharide through reduced promoter activity incultured rat neocortical astrocytes. J. Neurosci. Res. 83: 1447–1460.
37. Deane, R., S. Du Yan, R. K. Submamaryan, B. LaRue, S. Jovanovic, E. Hogg,D. Welch, L. Manness, C. Lin, J. Yu, et al. 2003. RAGE mediates amyloid-beta
peptide transport across the blood-brain barrier and accumulation in brain. Nat.Med. 9: 907–913.
38. Cataldegirmen, G., S. Zeng, N. Feirt, N. Ippagunta, H. Dun, W. Qu, Y. Lu,L. L. Rong, M. A. Hofmann, T. Kislinger, et al. 2005. RAGE limits regenerationafter massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J. Exp. Med. 201: 473–484.
39. Shindo, T., H. Kurihara, Y. Kurihara, H. Morita, and Y. Yazaki. 1998. Upregu-lation of endothelin-1 and adrenomedullin gene expression in the mouse endo-toxin shock model. J. Cardiovasc. Pharmacol. 31(Suppl 1): S541–S544.
40. Bucciarelli, L. G., T. Wendt, W. Qu, Y. Lu, E. Lalla, L. L. Rong, M. T. Goova,B. Moser, T. Kislinger, D. C. Lee, et al. 2002. RAGE blockade stabilizesestablished atherosclerosis in diabetic apolipoprotein E-null mice. Circulation106: 2827–2835.
41. Zhang, H., S. Tasaka, Y. Shiraishi, K. Fukunaga, W. Yamada, H. Seki, Y. Ogawa,K. Miyamoto, Y. Nakano, N. Hasegawa, et al. 2008. Role of soluble receptor foradvanced glycation end products on endotoxin-induced lung injury. Am. J.Respir. Crit. Care Med. 178: 356–362.
42. van Zoelen, M. A., A. M. Schmidt, S. Florquin, J. C. Meijers, R. de Beer,A. F. de Vos, P. P. Nawroth, A. Bierhaus, and T. van der Poll. 2009. Receptor foradvanced glycation end products facilitates host defense during Escherichia coli-induced abdominal sepsis in mice. J. Infect. Dis. 200: 765–773.
10 RAGE MEDIATES LPS SHOCK
by guest on January 21, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
![Modulation of soluble receptor for advanced glycation end ...€¦ · healthy population [39]. Healthy centenarians have a significant higher plasma sRAGE amount than healthy young](https://static.fdocuments.net/doc/165x107/601bd06f5c35006e1c658ee4/modulation-of-soluble-receptor-for-advanced-glycation-end-healthy-population.jpg)

















