
Reversibly Photoswitchable Nucleosides: Synthesis and ... · Reversibly Photoswitchable...
17
Electronic Supporting Information Reversibly Photoswitchable Nucleosides: Synthesis and Photochromic Properties of Diarylethene-functionalized 7-Deazaadenosine Derivatives Marco Singer, Andres Jäschke Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Im Neuenheimer Feld 364, Heidelberg 69120, Germany. Content Synthetic procedures: ......................................................................................................... 2 Base pairing: ..................................................................................................................... 12 Photochromism:................................................................................................................ 13 Acidichromism: ................................................................................................................ 16 Supporting References: .................................................................................................... 17 List of abbreviations: BuLi = n-butyl lithium, DCM = dichloromethane, DMSO = dimethylsulfoxide, Et 2 O = diethyl ether, MeCN = acetonitrile, MeOH = methanol, TLC = thin layer chromatography, TDA-1 = tris[2-(2-methoxyethoxy)ethyl]amine, TEA = triethylamine, TEAA = triethyl- ammonium acetate, TFA = trifluoroacetic acid, THF = tetrahydrofurane, TMS = trimethyl- silane.
Transcript of Reversibly Photoswitchable Nucleosides: Synthesis and ... · Reversibly Photoswitchable...

Electronic Supporting Information
Reversibly Photoswitchable Nucleosides: Synthesis and Photochromic Properties of
Diarylethene-functionalized 7-Deazaadenosine Derivatives
Marco Singer, Andres Jäschke
Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Im Neuenheimer
Feld 364, Heidelberg 69120, Germany.
Content
Synthetic procedures: ......................................................................................................... 2 Base pairing:..................................................................................................................... 12 Photochromism:................................................................................................................ 13 Acidichromism: ................................................................................................................ 16 Supporting References: .................................................................................................... 17
List of abbreviations:
BuLi = n-butyl lithium, DCM = dichloromethane, DMSO = dimethylsulfoxide, Et2O = diethyl
ether, MeCN = acetonitrile, MeOH = methanol, TLC = thin layer chromatography,
TDA-1 = tris[2-(2-methoxyethoxy)ethyl]amine, TEA = triethylamine, TEAA = triethyl-
ammonium acetate, TFA = trifluoroacetic acid, THF = tetrahydrofurane, TMS = trimethyl-
silane.

S2
Synthetic procedures:
Commercially available reagents and laboratory grade solvents were used without further
purification unless otherwise noted. Dry solvents were purchased in sealed bottles over
molecular sieves. Solvents for oxygen-sensitive reactions were degassed prior use by repeated
freeze-pump-thaw cycling. All reactions were performed under inert atmosphere (argon) and
monitored by TLC using silica plates coated with fluorescent indicator. Visualisation was
performed with UV-light (254 nm) or blue-shift solution (Ce(SO4)2; molybdatophosphoric
acid; sulfuric acid). Reactions under high pressure were conducted in an autoclave (Büchi)
equipped with a manometer. Flash chromatography was performed according to Still et al. 1
with silica gel 60 (0.04-0.063 mm) using laboratory grade solvents. HPLC analysis was
performed on an Agilent system (HP1100) with reverse phase columns (Phenomenex Luna
5 µm, C18, 4.6 x 250 mm) using a gradient of buffer A (0.1 M TEAA in water) and buffer B
(0.1 M TEAA in methanol or acetonitrile) as eluent. IR-spectra were aquired on a Jasco FT-IR
4100 using neat samples. NMR-spectra were recorded on a Varian Systems 500 instrument.
The chemical shifts (δ) are indicated in parts per million (ppm) downfield of TMS and
referenced to the respective residual undeuterated solvent peak as follows: CDCl3 = 7.26 ppm;
DMSO-d6 = 2.50 ppm; MeCN-d3 = 1.94 ppm for 1H-NMR and CDCl3 = 77.0 ppm; DMSO-d6
= 39.4 ppm; MeCN-d3 = 1.2 ppm for 13C-NMR. Apparent coupling constants (J) are reported
in Hz. The assignment of proton and carbon resonances is based on two dimensional
correlation NMR experiments (COSY, HSQC, HMBC). Low-resolution mass spectra were
recorded on a JEOL JMS 700 sectorfield machine (EI and FAB), whereas high-resolution
spectra were obtained on a Finnigan TSQ 700 instrument (ESI).

S3
4-Chloro-6-methyl-7H-pyrrolo[2,3-d]pyrimidine (3)
A dried flask was charged with 1.39 g (8.29 mmol) of 6-methyl-7H-
pyrrolo[2,3-d]pyrimidine-4-one (2),2 which was suspended in 15 mL POCl3.
The reaction mixture was refluxed for 4 h yielding a clear brown solution.
The excess of POCl3 was evaporated and the residue was slowly poured on
ice. After neutralizing with solid NaHCO3, the aqueous solution was repeatedly extracted with
Et2O and the combined organic phase dried over Na2SO4. Evaporation of the solvent yielded
1.28 g (83%) of a yellowish solid.
Rf = 0.33 (DCM/MeOH: 95/5). IR: 3104 (w); 2962 (w); 2848 (w); 1897 (vw); 1607 (w); 1568
(m); 1540 (m); 1340 (m); 1212 (m) cm-1. 1H-NMR (500 MHz, (CD3)2SO): δ 2.43 (d, J=1.1
Hz, 3H, CH3); 6.29 (t, J=1.0 Hz, 1H, NHCCH); 8.48 (s, 1H, NCH); 12.38 (bs, 1H, NH) ppm. 13C-NMR (125 MHz, (CD3)2SO): δ 13.36; 96.11; 117.32; 139.31; 148.27; 149.27; 152.00
ppm. FAB-MS: m/z calculated for [M+H]+: 168; found : 168.
4-Chloro-5-iodo-6-methyl-7H-pyrrolo[2,3-d]pyrimidine (4)
A suspension of 150 mg (0.895 mmol ) 4-chloro-6-methyl-7H-pyrrolo[2,3-
d]pyrimidine (3) in 10 mL dry DCM was treated with 242 mg (1.07 mmol) N-
iodosuccinimide, then stirred for 1h at room temperature. After filtration the
residual white solid was washed with cold DCM and dried under vacuum to yield 234 mg
(89 %) of a white powder.
Rf = 0.34 (DCM/MeOH: 95/5). IR: 3087 (w); 2950 (w); 2813 (w); 1681 (m); 1602 (w); 1556
(m); 1337 (w); 1317 (w); 1214 (m); 1185 (m) cm-1. 1H-NMR (500 MHz, (CD3)2SO): δ 2.42
(s, 3H, CH3); 8.51 (s, 1H, CH); 12.92 (bs, 1H, NH) ppm. 13C-NMR (125 MHz, (CD3)2SO): δ
14.53; 53.00; 116.53; 141.43; 148.77; 149.39; 151.80 ppm. FAB-MS: m/z calculated for
[M]+: 293; found: 293. HR-MS (ESI+) [C7H6ClIN3]+: 293.9284; found: 293.9291.
NH N
N
ClI
NH N
N
Cl

S4
4-Chloro-7-[2-deoxy-3,5-di-O-(p-toluoyl)-β-d-erythro-pentofuranosyl]-5-iodo-6-methyl-7H-pyrrolo[2,3-d]pyrimidine (5)
A solution of 293 mg (1.00 mmol) 4-chloro-5-iodo-6-
methyl-7H-pyrrolo[2,3-d]pyrimidine (4) and 40 µL TDA-1
in 12 mL MeCN was treated with freshly powdered 225 mg
(3.50 mmol) KOH (87%). After stirring for 1 h at room
temperature 505 mg (1.30 mmol) of 2-deoxy-3,5-di-O-(p-
toluoyl)-α-D-erythro-pentofuranosyl chloride3 was added
portionwise. The reaction mixture was filtered, concentrated, and purified by flash
chromatography (DCM). The product was obtained in 63% yield (408 mg) as a yellowish
foam.
Rf = 0.17 (hexanes/ethyl acetate: 4/1). 1H-NMR (500 MHz, CDCl3): δ 2.41, 2.44 (2s, 6H, 2
Ar-CH3); 2.54-2.60 (m,1H, C(2’)-H); 2.62(s, 3H, CH3); 3.75 (td, J=7.4 14.6 Hz, 1H, C(2’)-H);
4.53 (q, J=4.2 Hz, 1H, C(4’)-H); 4.60 (dd, J=4.9 11.9 Hz, 1H, C(5’)-H); 4.80 (dd, J=4.1 11.9
Hz, 1H, C(5’)-H); 5.95 (td, J=3.7 7.4 Hz, 1H, C(3’)-H); 6.56 (t, J=7.2 Hz, 1H, C(1’)-H); 7.19-
7.20 (m, 2H, 2 Ar-H); 7.27-7.29 (m, 2H, 2 Ar-H); 7.85-7.87 (m, 2H, 2 Ar-H); 7.96-7.98 (m,
2H, 2 Ar-H); 8.47 (s, 1H, NCHN) ppm. 13C-NMR (125 MHz, CDCl3): δ 15.07; 21.68; 21.72;
35.94; 56.79; 63.49; 74.50; 81.92; 84.87; 117.55; 126.56; 126.78; 129.08; 129.24; 129.27;
129.64; 129.77; 141.26; 143.91; 144.37; 149.65; 151.28; 151.78; 165.99; 166.14 ppm. FAB-
MS: m/z calculated for [M+H]+: 646; found: 646. HR-MS (ESI+) [C28H26ClIN3O5]+:
646.0600; found: 646.0594.
3-(2-Bromocyclopent-1-enyl)-2-methylbenzo[b]thiophene (7a)
A solution of 500 mg (2.20 mmol) 3-bromo-2-methylbenzo[b]thiophene
(6a4) in 10 mL dry THF was cooled to -78 °C. After dropwise addition of
990 µL n-BuLi (2.5 M in hexane; 2.48 mmol), the stirring was continued
for 1 h at the same temperature. The yellow solution was then treated with
1.03 mL (3.85 mmol) tributylborate and stirred for 1.5 h, while slowly heating to room
temperature. Then 2 mL aqueous Na2CO3 (20%), 400 mg (1.77 mmol) 1,2-
dibromocyclopentene and 245 mg (0.22 mmol) Pd(PPh3)4 were added and the heterogeneous
mixture was heated under reflux to 70 °C and stirred for another 12 h. The reaction mixture
was finally diluted with water and extracted with ethyl acetate thrice. The combined organic
S
Br
O
O
Op-Tolyl
O
O
p-Tolyl
N N
N
ClI

S5
fractions were dried over Na2SO4 and evaporated onto silica, followed by flash
chromatography (hexanes), affording 44% (229 mg) of the desired product as a colorless oil.
Rf = 0.48 (hexanes/DCM, 1:1). 1H-NMR (500 MHz, CDCl3): δ 2.15-2.23 (m, 2H,
CH2CH2CH2); 2.47 (s, 3H, CH3); 2.52-2.60 (m, 1H, CH2CH2CHH); 2.77-2.79 (m, 1H,
CH2CH2CHH); 2.89-2.93 (m, 2H, CH2CH2CH2); 7.25-7.34 (m, 2H, 2 Ar-H); 7.46-7.48 (m,
1H, Ar-H); 7.75-7.77 (m, 1H, Ar-H) ppm. 13C-NMR (125 MHz, CDCl3): δ14.77; 22.81;
36.21; 40.57; 121.60; 122.06; 122.10; 123.57; 123.92; 128.77; 135.91; 136.67; 138.84 ppm.
FAB-MS: m/z calculated for [M]+: 292; found: 292. HR-MS (ESI+) [C14H13BrNaS]+:
314.9814; found: 314.9815.
3-(2-Bromocyclopent-1-enyl)-2-methyl-5-phenylthiophene (7b)
Compound 7b was prepared from 2.20 g (8.69 mmol) 3-bromo-2-
methyl-5-phenylthiophene (6b)5 as described for 7a in 55 mL dry
THF, using 3.82 mL (2.5M in Hexan; 9.56 mmol) n-BuLi, 3.28 mL (
12.7 mmol) tributylborate, 10.0 mL aqueous Na2CO3 (20%), 2.94 g
(13.0 mmol) 1,2-dibromocyclopentene, and 355 mg (0.435 mmol)
[1,1’-bis(diphenylphosphino)ferrocene]dichloro palladium(II). After flash chromatography
(hexanes/ethyl acetate, 1:0 to 9:1) the desired compound was obtained in 39% (1.09 g) yield
as a colorless oil.
Rf = 0.38 (hexanes). 1H-NMR (500 MHz, CDCl3): δ 2.06-2.12 (m, 2H, CH2CH2CH2); 2.43 (s,
3H, CH3); 2.64-2.68 (m, 2H, CH2CH2CH2); 2.80-2.84 (m, 2H, CH2CH2CH2); 7.13 (s, 1H,
SCCH); 7.23-7.26 (m, 1H, Ar-H); 7.34-7.37 (m, 2H, 2 Ar-H); 7.54-7.56 (m, 2H, 2 Ar-H)
ppm. 13C-NMR (125 MHz, CDCl3): δ 14.95; 22.47; 36.83; 40.94; 118.79; 123.56; 125.42;
127.07; 128.75; 134.34 134.40; 135.42. 136.50; 139.99 ppm. FAB-MS: m/z calculated for
[M+H]+: 319; found: 319. HR-MS (ESI+) [C16H15BrKS]+: 356.9709; found: 356.9711.
3-(2-Bromocyclopent-1-enyl)-2-methyl-5-(4-pyridyl)thiophene (7c)
Compound 7c was prepared from 2.00 g (7.87 mmol) 3-bromo-2-
methyl-5-(4’-pyridyl)thiophene (6c)6 as described for 7a, in 50 mL
dry THF using 3.78 mL (2.5M in hexane; 9,44 mmol) n-BuLi,
2.98 mL (11.0 mmol) tributylborate, 10.0 mL aqueous Na2CO3
(20%), 2.67 g (11.8 mmol) 1,2-dibromocyclopentene, and 320 mg (0.39 mmol)
S N
Br
S
Br

S6
[1,1’-bis(diphenylphosphino)ferrocene]dichloro palladium(II). After flash chromatography
(hexanes/acetone, 9:1) the desired compound was obtained in 41% yield (1.04 g) as a yellow
oil.
Rf = 0.47 (hexanes/acetone, 8:2)1H-NMR (500 MHz, CDCl3): δ 2.06-2.12 (m, 2H,
CH2CH2CH2); 2.45 (s, 3H, CH3); 2.63-2.67 (m, 2H, CH2CH2CH2); 2.80-2.84 (m, 2H,
CH2CH2CH2); 7.33 (s, 1H, SCCH); 7.39-7.40 (m, 2H, 2 Ar-H); 8.54-8.55 (m, 2H, 2 Ar-H)
ppm. 13C-NMR (125 MHz, CDCl3): δ 15.09; 22.46; 36.81; 40.92; 119.34; 119.62; 126.04;
135.09; 135.96; 136.72; 138.24; 141.32; 150.23 ppm. FAB-MS: m/z calculated for [M+H]+:
320; found: 320.
2-(2-Methylbenzo[b]thien-3-yl)cyclopent-1ene-boronic acid pinacol ester (8a)
A solution of compound 7a (25.0 mg; 0.085 mmol) in 1.0 mL dry
THF was cooled to -78 °C and treated with 37.5 µL (2.5M in Hexan;
0.094 mmol) n-BuLi under constant stirring. After 30 minutes,
21.0 µL ( 0.102 mmol) 2-isopropoxyboronic acid pinacol ester were
added and stirring was continued for 60 minutes, while heating the solution slowly to room
temperature. The reaction was quenched by addition of 100 µL MeOH and diluted in ethyl
acetate. The reaction mixture was washed twice with water, once with brine and was dried
over Na2SO4. The white solid (31.1 mg, quant.) obtained after evaporating the solvent was
sufficiently pure for analysis and further transformation.
Rf = 0.27 (hexanes/DCM, 1:1). 1H-NMR (500 MHz, CDCl3): δ 1.01 (s, 12H, 4 OCCH3); 2.00-
2.06 (m, 2H, CH2CH2CH2); 2.43 (s, 3H, SCCH3); 2.71-2.74 (m, 4H, CH2CH2CH2); 7.20-7.27
(m, 2H, 2 Ar-H); 7.44-7.46 (m, 1H, Ar-H); 7.70-7.72 (m, 1H, Ar-H) ppm. 13C-NMR (125
MHz, CDCl3): δ 14.57; 24.51; 24.64; 36.64; 82.60; 121.62; 122.33; 123.01; 123.53; 132.06;
134.65; 138.13; 140.51; 151.66 ppm. FAB-MS: m/z calculated for [M]+: 340; found: 340. HR-
MS (ESI+) [C20H25BKO2S]+: 379.1304; found: 379.1300.
2-[2-Methyl-5-phenylthiophen-3-yl]cyclopent-1-ene boronic acid pinacol ester (8b)
The synthesis of compound 8b was performed as described for 8a, starting from 900 mg
(2.82 mmol) compound 7b in 20 mL dry THF, with 1.24 mL (2.5M in hexane; 3.10 mmol)
n-BuLi, and 690 µL (3.38 mmol) 2-isopropoxyboronic acid pinacol ester. The crude product
S
BO
O

S7
was chromatographed (hexanes/ethyl acetate, 98:2) and yielded
914 mg (89 %) of the desired product as colorless crystals.
Rf = 0.58 (hexanes/acetone, 7:3). 1H-NMR (500 MHz, CDCl3):
δ 1.19 (s, 12H, 4 OCCH3); 1.90-1.96 (m, 2H, CH2CH2CH2);
2.39 (s, 3H, SCCH3); 2.62-2.71 (m, 4H, CH2CH2CH2); 7.14 (s, 1H, SCCH); 7.20-7.23 (m, 1H,
Ar-H); 7.31-7.35 (m, 2H, 2 Ar-H); 7.53-7.55 (m, 2H, 2 Ar-H) ppm. 13C-NMR (125 MHz,
CDCl3): δ 14.59; 24.30; 24.80; 37.11; 40.24; 82.87; 125.15; 126.68; 128.69; 134.39; 134.72;
138.42; 152.05 ppm. FAB-MS: m/z calculated for [M]+: 366; found: 366. HR-MS (ESI+)
[C22H27BKO2S]+: 405.1460; found: 405.1460.
2-[2-Methyl-5-(4-pyridyl)thiophen-3-yl]cyclopent-1-ene boronic acid pinacol ester (8c)
The synthesis of compound 8c was performed as described for
8a, starting from 1.10 g (3.45 mmol) compound 7c in 25 mL
dry THF, with 1.86 mL (2.5M in hexane; 4.66 mmol) n-BuLi,
and 1.06 mL (5.18 mmol) 2-isopropoxyboronic acid pinacol
ester. The crude product was chromatographed
(hexanes/acetone, 9:1 to 8:2) and yielded 773 mg (61 %) of the desired product as an orange
oil, that cristallized slowly at -20°C.
Rf = 0.33 (hexanes/acetone, 7:3). 1H-NMR (500 MHz, CDCl3): δ 1.17 (s, 12H, 4 OCCH3);
1.91-1.97 (m, 2H, CH2CH2CH2); 2.41 (s, 3H, SCCH3); 2.62-2.69 (m, 4H, CH2CH2CH2); 7.34
(s, 1H, SCCH); 7.40-7.41 (m, 2H, 2 Ar-H); 8.51-8.52 (m, 2H, 2 Ar-H) ppm. 13C-NMR (125
MHz, CDCl3): δ 14.76; 24.25; 24.77; 37.06; 40.18; 82.93; 119.19; 128.13; 134.84; 137.87;
138.42; 142.36; 149.35; 151.40 ppm. HR-MS (ESI+) [C21H27BNO2S]+: 368.1854; found:
368.1851.
4-Chloro-7-[2-deoxy-3,5-di-O-(p-toluoyl)-β-D-erythro-pentofuranosyl]-6-methyl-5-[2-(2-methylbenzo[b]thien-3-yl)cyclopent-1-enyl]-7H-pyrrolo[2,3-d]pyrimidine (9a)
Compound 5 (300 mg, 0.464 mmol) and compound 8a
(190 mg, 0.558 mmol) were dissolved in 60 mL THF in the
presence of 118 mg (0.558 mmol) powdered K3PO4 and
38 mg (0.046 mmol) [1,1’-bis(diphenylphosphino)-
ferrocene]dichloropalladium(II) followed by the addition of
S
BO
O
S N
BO
O
O
O
Op-Tolyl
O
O
p-Tolyl
N N
N
Cl
S

S8
6 mL H2O and 322 µL (2.30 mmol) TEA. The reaction mixture was stirred at reflux for 24 h
at 75 °C. After the dilution in ethyl acetate the organic phase was washed twice with water,
once with brine and dried over Na2SO4. The crude product was evaporated onto silica and
purified by flash chromatography (hexanes/ethyl acetate, 9:1 to 8:2). The desired compound
was obtained in 52 % yield (174 mg) as a yellowish oil.
Rf = 0.22 (hexanes/DCM, 1:1)1H-NMR (500 MHz, CDCl3): δ 1.88 (s, 2H); 2.06-2.12 (m, 3H);
2.22-2.25 (m, 2H); 2.41-2.44 (m, 7H); 2.63-2.70 (m, 2H); 3.20-3.26 (m, 2H); 4.40-4.73 (m,
3H); 5.71, 5.91 (2bs, 1H); 6.06 (t, J=7.1 Hz, 0.5H); 6.50-6.63 (m, 0.5H); 7.17-7.33 (m, 6H);
7.56-7.71 (m, 2H); 7.86-7.92 (m, 4H); 8.35-8.49 (m, 1H) ppm. 13C-NMR (125 MHz, CDCl3):
δ 11.52; 12.04; 14.46; 15.06; 21.66; 21.67; 21.68; 24.00; 24.05; 26.89; 34.87; 37.83; 40.40;
63.64; 63.77; 74.35; 74.50; 75.07; 77.20; 81.33; 81.78; 84.63; 105.03; 109.74; 117.10;
122.04; 122.20; 123.23; 126.52; 126.69; 126.87; 126.96; 129.00; 129.09; 129.14; 129.19;
129.67; 129.70; 129.72; 135.04; 135.88; 136.48; 138.29; 138.39; 138.46; 139.30; 139.40;
143.72; 144.16; 144.29; 149.46; 149.69; 150.05; 150.16; 151.90; 152.12; 165.91; 166.18 ppm.
HR-MS (ESI+) [C42H39ClN3O5S]+: 732.2294; found: 732.2313.
4-Chloro-7-[2-deoxy-3,5-di-O-(p-toluoyl)-β-D-erythro-pentofuranosyl]-5-[2-(2-methyl-5-phenylthien-3-yl)cyclopent-1-enyl]-6-methyl-7H-pyrrolo[2,3-d]pyrimidine (9b)
Compound 9b was prepared from 5 (250 mg, 0.387 mmol)
and 8b (170 mg, 0.464 mmol) in 30 mL THF as described
for 9a, using 99 mg (0.464 mmol) of K3PO4 and 32 mg (
0.039 mmol) [1,1’-bis(diphenylphosphino)ferrocene]-
dichloropalladium(II) in the presence of 268 µL (1.94 mmol)
TEA and 3 mL H2O. Flash chromatography
(hexanes/acetone, 7:3) afforded 252 mg (86 %) of 9b as a
yellowish oil.
Rf = 0.47 (hexanes/acetone, 7:3). 1H-NMR (500 MHz, CDCl3):1.99 (s, 3H); 2.03 (s, 3H); 2.11
(2s, 6H); 2.12-2.16 (m, 4H); 2.37-2.44 (m, 15H); 2.45-2.60 (m, 5H); 2,71-2,79 (m, 2H); 2.96-
3.08 (m, 4H); 3.42-3.48 (m, 1H); 4.04 (td, J=7.4, 14.4 Hz, 1H); 4.46-4.49 (m, 2H); 4.56-4.81
(m, 5H); 5.80-5.82 (m, 1H); 5.93-5.96 (m, 1H); 6.42 (t, J=7.2 Hz, 1H); 6.55-6.59 (m, 1H);
6.90 (s, 1H); 6.91 (s, 1H); 7.15-7.31 (m, 15H); 7.39-7.40 (m, 4H); 7.86-7.98 (m, 10H); 8.41
(s, 1H); 8.49 (s, 1H) ppm. 13C-NMR (125 MHz, CDCl3): 11.23; 11.69; 14.43; 14.51; 15.08;
21.66; 21.67; 21.70; 21.73; 23.61; 23.63; 29.27; 35.17; 35.84; 38.10; 38.17; 40.47; 40.51;
N N
N
Cl
S
O
O
Op-Tolyl
O
O
p-Tolyl

S9
53.40; 63.66; 63.86; 74.61; 75.01; 81.50; 81.82; 81.92; 83.73; 84.55; 84.87; 110.50; 111.10;
116.82; 117.20; 123.52; 123.59; 125.15; 125.27; 126.60; 126.70; 126.89; 126.96; 126.98;
128.74; 128.76; 129.02; 129.05; 129.09; 129.18; 129.21; 129.25; 129.65; 129.70; 129.73;
129.74; 129.78; 131.39; 131.83; 134.16; 134.30; 134.33; 135.60; 135.89; 135.95; 136.16;
138.71; 138.91; 139.54; 139.68; 143.75; 143.83; 144.21; 144.29; 149.60; 149.66; 149.77;
150.37; 150.45; 152.00; 152.15; 165.98; 166.22 ppm. FAB-MS: m/z calculated for [M+H]+:
758; found 758. HR-MS (ESI+) [C44H41ClN3O5S]+: 758.2450; found: 758.2469.
4-Chloro-7-[2-deoxy-3,5-di-O-(p-toluoyl)-β-d-erythro-pentofuranosyl]-5-{2-[2-methyl-5-(4-pyridyl)thien-3-yl]cyclopent-1-enyl}-6-methyl-7H-pyrrolo[2,3-d]pyrimidine (9c)
Compound 9c was prepared from 5 (101 mg, 0.156 mmol)
and 8c (69 mg 0.188 mmol) in 20 mL THF as described for
9a, using 40 mg (0.188 mmol) of K3PO4 and 13 mg
(0.016 mmol) [1,1’-bis(diphenylphosphino)ferrocene]-
dichloropalladium(II) in the presence of 108 µL
(0.779 mmol) TEA and 2 mL H2O. Flash chromatography
(hexanes/acetone, 9:1 to 7:3) afforded 81 mg (68 %) of 9c as
an orange oil.
Rf = 0.47 (hexanes/acetone, 7:3). 1H-NMR (500 MHz, CDCl3): δ 2.02 (s, 3H); 2.05 (s, 3H);
2.11 (s, 3H); 2.12 (s, 3H); 2.13-2.15 (m, 2H); 2.38 (s, 3H); 2.40 (s, 3H); 2.42 (2s, 6H); 2.46-
2.53 (m, 2H); 2.55-2.62 (m, 2H); 2.71-2.79 (m, 2H); 2.95-3.09 (m, 4H); 3.51 (td, J=7.7, 14.3
Hz, 1H); 3.96 (td, J= 7.5, 14.6 Hz, 1H); 4.46-4.49 (m, 2H); 4.55-4.59 (m, 2H); 4.70 (dd,
J=4.5, 11.7 Hz, 1H); 4,74 (dd, J= 4.4, 11.7 Hz, 1H); 5.82 (td, J= 3.3, 7.0 Hz, 1H); 5.93 (td,
J= 3.4, 7.0 Hz, 1H); 6.28 (t, J= 7.1 Hz, 1H) 6.53 (dd, J= 6.7, 8.1 Hz, 1H); 7.13 (s, 1H); 7.15-
7.19 (m, 4H); 7.24-7.28 (m, 8H); 7.85-7.88 (m, 4H); 7.92-7.94 (m, 4H); 8.42 (s, 1H); 8.48 (s,
1H); 8.49 (m, 4H) ppm. 13C-NMR (125 MHz, CDCl3): δ 11.32; 11.65; 14.65; 14.71; 21.67;
21.70; 23.55; 35.34; 38.15; 38.19; 40.53; 40.55; 63.61; 63.77; 74.59; 74.87; 81.50; 81.76;
83.82; 84.43; 110.29; 110.73; 116.70; 116.99; 119.21; 119.31; 126.23; 126.28; 126.55;
126.63; 126.88; 126.90; 129.03; 129.04; 129.20; 129.21; 129.69; 129.73; 132.36; 132.71;
135.49; 135.78; 136.19; 136.25; 136.77; 136.94; 137.35; 137.50; 138.07; 138.24; 141.70;
141.78; 143.81; 143.86; 144.29; 144.34; 149.56; 149.62; 149.74; 149.89; 150.36; 150.44;
152.02; 152.13; 165.94; 165.97; 166.16; 166.17 ppm. FAB-MS: m/z calculated for [M+H]+:
759; found: 759. HR-MS (ESI+) [C43H40ClN4O5S]+: 759.2403; found: 759.2408.
N N
N
Cl
SN
O
O
Op-Tolyl
O
O
p-Tolyl

S10
4-Amino-7-(2-deoxy-β-D-erythro-pentofuranosyl)-6-methyl-5-[2-(2-methylbenzo[b]thien-3-yl)cyclopent-1-enyl]-7H-pyrrolo[2,3-d]pyrimidine (1a)
An autoclave was charged with 30 mg (41.0 µmol) of 9a and 1 mL
of dioxane were added, followed by 3 mL 25% aqueous ammonia
solution. The suspension cleared upon heating to 120 °C and was
maintained stirring under pressure (5-6 bar) at this temperature for
12 h. The reaction mixture was then cooled to room temperature,
evaporated onto silica and purified by flash chromatography
(DCM/MeOH, 9:1) to give 10 mg (51 %) of a yellowish solid.
Rf = 0.35 (DCM/MeOH, 9:1). 1H-NMR (500 MHz, CD3CN): δ 2.00-2.04 (m, 1H); 2.15-2.29
(m, 8H); 2.74-3.24 (m, 6H); 3.56-3.71 (m, 2H); 3.92, 3.94 (2s, 1H); 4.48 (d, J=5.2 Hz, 1H);
5.56, 5.60 (2bs, 2H); 6,03 (1bs, 1H); 6.17, 6.33 (2d, J=10.8 Hz, 1H); 7.15-7.34 (m, 2H); 7.62-
7.71 (m, 2H); 7.93 (bs, 1H) ppm. 13C-NMR (125 MHz, CD3CN): δ 11.50; 24.08, 24.13, 38.52;
28.56; 39.94; 40.22; 40.65; 40.74; 63.97; 64.06; 73.62; 73.67; 86.74; 86.86; 89.66; 89.72;
109.27; 109.42; 122.80; 122.87; 123.36; 123.39; 124.39; 124.44; 124.64; 130.83; 130.97;
138.92; 138.98; 140.25; 150.99; 151.03; 157.52 ppm. HR-MS (ESI+) [C26H29N4O3S]+:
477.1955; found: 477.1950.
4-Amino-7-(2-deoxy-β-D-erythro-pentofuranosyl)-6-methyl-5-[2-(2-methyl-5-phenylthien-3-yl)cyclopent-1-enyl]-7H-pyrrolo[2,3-d]pyrimidine (1b)
Compound 1b was prepared as described for 1a starting from
150 mg (198 µmol) 9b in 2 mL dioxane and 4.5 mL 25%
aqueous ammonia solution. The final product was purified by
flash chromatography (DCM/MeOH, 99:1 to 98:2) to give
64 mg (65 %) of yellowish solid.
Rf = 0.38 (DCM/MeOH: 9/1).1H-NMR (500 MHz, CD3CN): 2.04-2.14 (m, 3H); 2.09 (s, 3H);
2.15, 2.18 (2s, 3H); 2.71-2.79 (m, 2H); 2.88-2.93 (m, 2H); 2.95-3.05 (m, 1H); 3.62-3.77 (m,
2H); 3.99 (m, 1H); 4.53 (t, J=5.3 Hz, 1H); 5.70 (bs, 2H); 6.17, 6.19 (2t, J=5.4 Hz, 1H); 7.01,
7.03 (2s, 1H); 7.20-7.23 (m, 1H); 7.28-7.32 (m, 2H); 7.36-7.41 (m, 2H); 7.97, 7.98 (2s, 1H)
ppm. 13C-NMR (125 MHz, CD3CN): 11.23; 11.26; 14.66; 14.71; 23.78; 38.64; 40.25; 40.53;
40.64; 64.06; 73.68; 73.73; 86.86; 86.89; 89.74; 103.44; 103.48; 109.90; 110.07; 125.42;
O
OH
HO N N
N
NH2
S
N N
N
NH2
S
O
OH
HO

S11
125.82; 125.93; 128.09; 129.83; 131.10; 131.26; 133.73; 134.97; 134.99; 135.42; 135.63;
136.82; 136.97; 139.25; 139.69; 139.89; 150.16; 150.22; 150.84; 150.87; 157.76 ppm. HR-
MS (ESI+) [C28H31N4O3S]+: 503.2111; found:. 503.2106.
4-Amino-7-(2-deoxy-β-D-erythro-pentofuranosyl)-5-{2-[2-methyl-5-(4-pyridyl)thien-3-yl]cyclopent-1-enyl}-6-methyl-7H-pyrrolo[2,3-d]pyrimidine (1c)
Compound 1c was prepared as described for 1a starting from
65 mg (85.6 µmol) 9c in 1 mL dioxane and 3 mL 25%
aqueous ammonia solution. The crude product was purified
by flash chromatography (DCM/MeOH, 9:1) to give 28 mg
(65 %) of a redish solid.
Rf = 0.30 (DCM/MeOH, 9:1). 1H-NMR (500 MHz, CD3CN): 2.13-2.21 (m, 1H); 2.21, 2.22
(2s, 3H); 2.23-2.29 (m, 2H); 2.26, 2.28 (2s, 3H); 2.81-2.93 (m, 2H); 3.00-3.04 (m, 2H); 3.07-
3.15 (m, 1H); 3.74 (d, J=12.4 Hz, 1H); 3.85 (td, J= 1.6, 12.4 Hz, 1H); 4.10 (dd, J= 1.7, 3.0
Hz, 1H); 4.64-4.65 (m, 1H); 5.63, 5.65 (2bs, 2H); 6.25-6.29 (m, 1H); 7.37, 7.39 (2s, 1H);
7.39-7.43 (m, 2H); 8.08 (2s, 1H); 8.56-8.57 (m, 2H) ppm. 13C-NMR (125 MHz, CD3CN):
11.22; 11.25; 14.82; 14.87; 23.75; 23.76; 38.61; 40.22; 40.55; 40.67; 64.07; 73.68; 86.86;
86.91; 89.76; 103.37; 103.40; 109.64; 109.77; 119.86; 119.95; 128.02; 128.04; 131.09;
131.23; 134.41; 134.51; 136.67; 136.80; 137.45; 137.58; 138.17; 138.35; 138.80; 139.22;
141.76; 141.78; 150.27; 150.32; 151.07; 151.10; 151.16; 157.75 ppm. FAB-MS: m/z
calculated for [M+H]+: 504; found: 504. HR-MS (ESI+) [C27H30N5O3S]+: 504.2064; found:
504.2059.
Figure S1: HPLC traces of compounds 1a-c after final purification. Detection wavelength 260 nm.
N N
N
NH2
SN
O
OH
HO

S12
Base pairing:
Figure S2: Partial NMR spectra showing downfield shifts of NH in thymidine upon addition of 1b (left) and 1c (middle). No shift is observed in the presence of 5 (right) which lacks the amino group required for base pairing with T.
Table S1: Interaction shifts of thymidine (T) with adenosine (A) and adenosine-analogs 1a-c and 5.
Thymidine Adenosine-analogs
NH / ppm Δ ppm Δ Hz NH2 /
ppm Δ ppm Δ Hz
T 8.85 – – – – –
A+T 9.05 0.20 100 6.08 0.04 20a
1a+T 9.03 0.18 90 5.58 0.02 10a
1b+T 8.99 0.14 70 5.53 0.03 15a
1c+T 8.99 0.14 70 5.53 0.02 10a
5+T 8.86 0.01 5 – – – Downfield shifts of imine and amine protons were measured in Hz at 500 Mhz in 5 mM (each component) d3-MeCN solutions at 25 °C. a Reference chemical shifts of NH2 were measured at equal concentrations with the respective single compounds.

S13
Photochromism:
Figure S3: HPLC traces of compounds 1a-c before irradiation and in the photostationary state after irradiation at 300 nm. The detection wavelength was set to the respective isosbestic points of each compound to enable direct quantification of closed and opened isomers.
S14
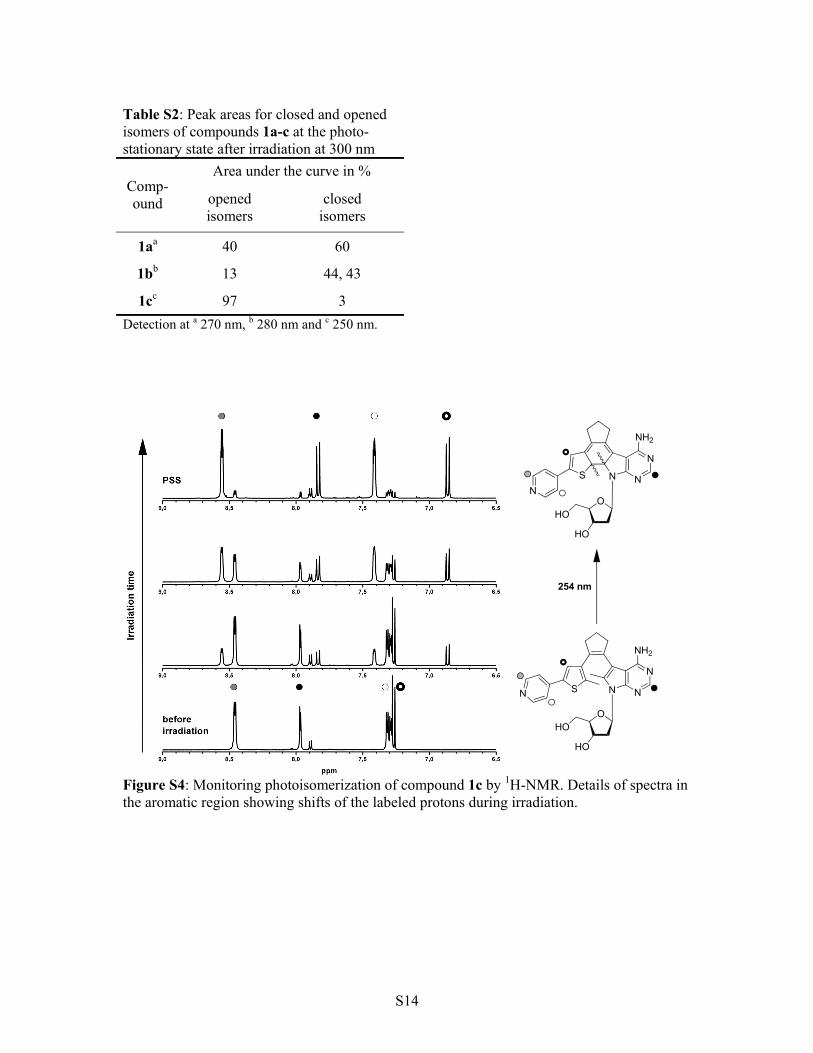
Table S2: Peak areas for closed and opened isomers of compounds 1a-c at the photo-stationary state after irradiation at 300 nm
Area under the curve in % Comp-ound opened
isomers closed
isomers
1aa 40 60
1bb 13 44, 43
1cc 97 3
Detection at a 270 nm, b 280 nm and c 250 nm.
N
N
N
NH2
OHO
HO
SN
NS N
N
NH2
NO
HO
HO
254 nm
Figure S4: Monitoring photoisomerization of compound 1c by 1H-NMR. Details of spectra in the aromatic region showing shifts of the labeled protons during irradiation.
S15
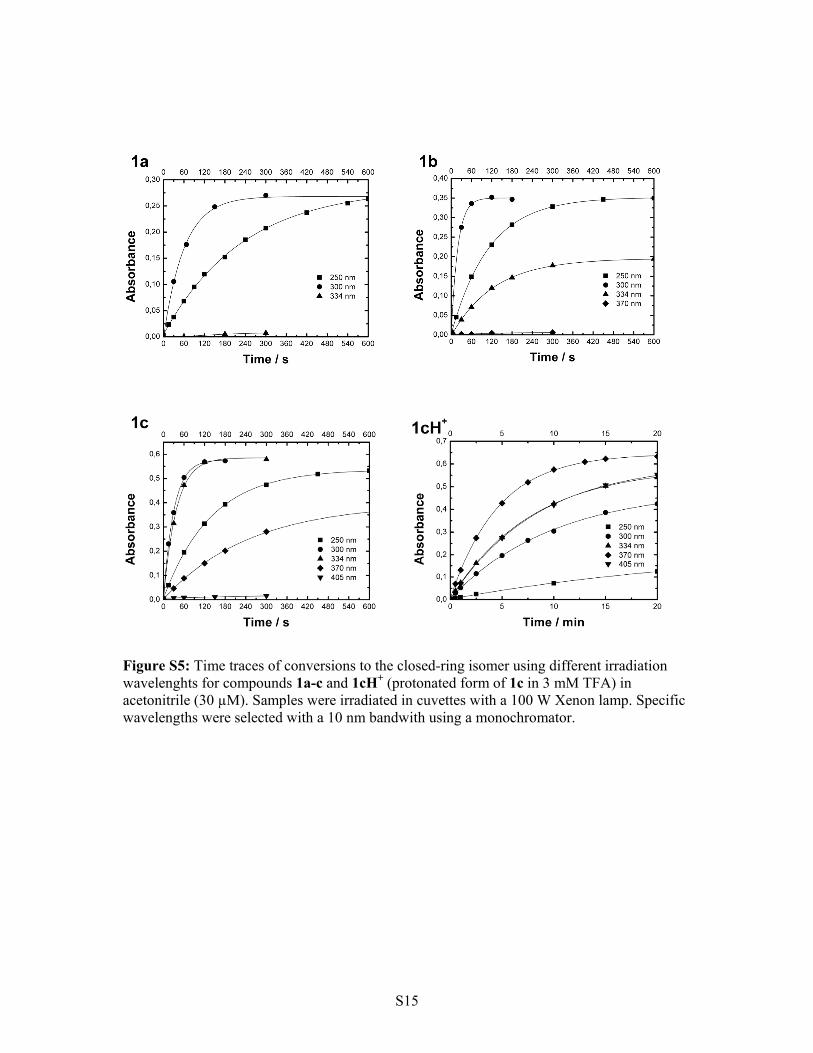
Figure S5: Time traces of conversions to the closed-ring isomer using different irradiation wavelenghts for compounds 1a-c and 1cH+ (protonated form of 1c in 3 mM TFA) in acetonitrile (30 µM). Samples were irradiated in cuvettes with a 100 W Xenon lamp. Specific wavelengths were selected with a 10 nm bandwith using a monochromator.
S16
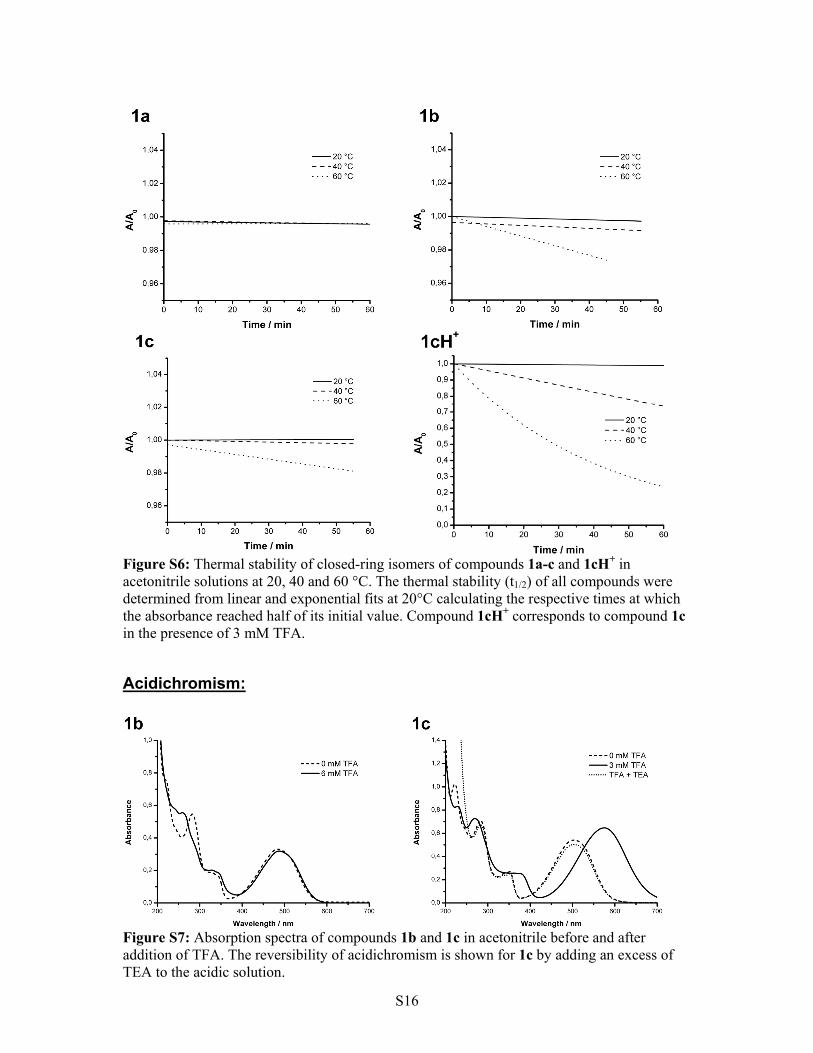
Figure S6: Thermal stability of closed-ring isomers of compounds 1a-c and 1cH+ in acetonitrile solutions at 20, 40 and 60 °C. The thermal stability (t1/2) of all compounds were determined from linear and exponential fits at 20°C calculating the respective times at which the absorbance reached half of its initial value. Compound 1cH+ corresponds to compound 1c in the presence of 3 mM TFA.
Acidichromism:
Figure S7: Absorption spectra of compounds 1b and 1c in acetonitrile before and after addition of TFA. The reversibility of acidichromism is shown for 1c by adding an excess of TEA to the acidic solution.

S17
Supporting References: (1) Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923-2925. (2) Davoll, J. J. Chem. Soc. 1960, 131-138. (3) Hoffer, M. Chem. Ber. 1960, 93, 2777-2781. (4) Katritzky, A. R.; He, H.-Y.; Long, Q.; Cui, X.; Level, J.; Wilcox, A. L. ARKIVOC
2000, 1, 240-251. (5) Irie, M.; Lifka, T.; Kobatake, S.; Kato, N. J. Am. Chem. Soc. 2000, 122, 4871-4876. (6) Gilat, S. L.; Kawai, S. H.; Lehn, J. M. Chem.-Eur. J. 1995, 1, 275-284.


















