
Progress in Polymer Science - web.ist.utl.pt
35
Progress in Polymer Science 36 (2011) 568–602 Contents lists available at ScienceDirect Progress in Polymer Science journal homepage: www.elsevier.com/locate/ppolysci Dye-labelled polymer chains at specific sites: Synthesis by living/controlled polymerization Mariana Beija d,∗ , Marie-Thérèse Charreyre a,b,∗∗ , José M.G. Martinho c a Laboratoire Joliot-Curie, ENS de Lyon, IFR128 BioSciences Gerland - Lyon Sud, 46 Allée d’Italie, Lyon Cedex 69364, France b Laboratoire Ingénierie des Matériaux Polymères, UMR CNRS 5223, INSA-Lyon, Université de Lyon, Domaine Scientifique de la Doua, Villeurbanne Cedex 69622, France c Centro de Química-Física Molecular and IN-Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal d Laboratoire des Intéractions Moléculaires et Réactivité Chimique et Photochimique, UMR 5623, Université Paul Sabatier, 31062 Toulouse Cedex 9, France article info Article history: Received 19 March 2010 Received in revised form 9 July 2010 Accepted 21 July 2010 Available online 27 July 2010 Keywords: CRP Fluorescence End-functionalized polymers Dye Labelled polymers abstract Dye-labelled polymer chains are extremely useful in many fields, such as optical imaging, signal amplification in biological diagnostics, light-harvesting and photochromic materials as well as in fluorescence studies about intra- and inter-molecular polymer chain asso- ciations, conformations and dynamics of polymer chains. However, in many cases, it is particularly useful that the dye is localized at a specific site, such as the chain-end or the junction between blocks. With the development of living/controlled polymerization techniques, end- and junction-functionalized polymers can be prepared with controlled molecular weights from a huge variety of monomers. This review highlights the state of the art in the strategies leading to one and only one precisely localized dye per polymer chain. Such dye can be introduced at three different steps of the polymerization: i) at the very beginning via the initiator or a chain transfer agent, ii) during polymerization via a functional monomer or a quencher, or iii) after polymerization via covalent binding of a dye-derivative. © 2010 Elsevier Ltd. All rights reserved. Contents 1. Introduction ........................................................................................................................ 569 2. Post-modification of polymer chains with a dye ................................................................................... 571 2.1. Polymers synthesized by living anionic polymerization ................................................................... 572 2.2. Polymers synthesized by NMP .............................................................................................. 572 2.3. Polymers synthesized by ATRP ............................................................................................. 573 2.4. Polymers synthesized by RAFT polymerization ............................................................................ 574 2.4.1. -end-labelling .................................................................................................... 574 2.4.2. -end-labelling .................................................................................................... 574 2.4.3. Junction-labelling ................................................................................................. 575 2.4.4. Conclusion on the post-modification of polymer chains synthesized by the RAFT process ..................... 576 ∗ Corresponding author. Tel.: +33 5 61 55 61 35; fax: +33 5 61 55 81 55. ∗∗ Corresponding author. Tel.: +33 4 72 72 89 38; fax: +33 4 72 72 87 87. E-mail addresses: [email protected] (M. Beija), [email protected] (M.-T. Charreyre), [email protected] (J.M.G. Martinho). 0079-6700/$ – see front matter © 2010 Elsevier Ltd. All rights reserved. doi:10.1016/j.progpolymsci.2010.06.004
Transcript of Progress in Polymer Science - web.ist.utl.pt

Dl
Ma
b
Vc
1d
3
a
ARRAA
KCFEDL
C
0d
Progress in Polymer Science 36 (2011) 568–602
Contents lists available at ScienceDirect
Progress in Polymer Science
journa l homepage: www.e lsev ier .com/ locate /ppolysc i
ye-labelled polymer chains at specific sites: Synthesis byiving/controlled polymerization
ariana Beijad,∗, Marie-Thérèse Charreyrea,b,∗∗, José M.G. Martinhoc
Laboratoire Joliot-Curie, ENS de Lyon, IFR128 BioSciences Gerland - Lyon Sud, 46 Allée d’Italie, Lyon Cedex 69364, FranceLaboratoire Ingénierie des Matériaux Polymères, UMR CNRS 5223, INSA-Lyon, Université de Lyon, Domaine Scientifique de la Doua,illeurbanne Cedex 69622, FranceCentro de Química-Física Molecular and IN-Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico, Av. Rovisco Pais 1,049-001 Lisboa, PortugalLaboratoire des Intéractions Moléculaires et Réactivité Chimique et Photochimique, UMR 5623, Université Paul Sabatier,1062 Toulouse Cedex 9, France
r t i c l e i n f o
rticle history:eceived 19 March 2010eceived in revised form 9 July 2010ccepted 21 July 2010vailable online 27 July 2010
eywords:RP
a b s t r a c t
Dye-labelled polymer chains are extremely useful in many fields, such as optical imaging,signal amplification in biological diagnostics, light-harvesting and photochromic materialsas well as in fluorescence studies about intra- and inter-molecular polymer chain asso-ciations, conformations and dynamics of polymer chains. However, in many cases, it isparticularly useful that the dye is localized at a specific site, such as the chain-end orthe junction between blocks. With the development of living/controlled polymerizationtechniques, end- and junction-functionalized polymers can be prepared with controlled
luorescencend-functionalized polymersyeabelled polymers
molecular weights from a huge variety of monomers. This review highlights the state ofthe art in the strategies leading to one and only one precisely localized dye per polymerchain. Such dye can be introduced at three different steps of the polymerization: i) at thevery beginning via the initiator or a chain transfer agent, ii) during polymerization via afunctional monomer or a quencher, or iii) after polymerization via covalent binding of a
dye-derivative.© 2010 Elsevier Ltd. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5692. Post-modification of polymer chains with a dye . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 571
2.1. Polymers synthesized by living anionic polymerization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5722.2. Polymers synthesized by NMP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5722.3. Polymers synthesized by ATRP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5732.4. Polymers synthesized by RAFT polymerization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 574
2.4.1. �-end-labelling. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .2.4.2. �-end-labelling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .2.4.3. Junction-labelling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .2.4.4. Conclusion on the post-modification of polymer c
∗ Corresponding author. Tel.: +33 5 61 55 61 35; fax: +33 5 61 55 81 55.∗∗ Corresponding author. Tel.: +33 4 72 72 89 38; fax: +33 4 72 72 87 87.
E-mail addresses: [email protected] (M. Beija), marie-therese.charreyr
079-6700/$ – see front matter © 2010 Elsevier Ltd. All rights reserved.oi:10.1016/j.progpolymsci.2010.06.004
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 574
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 574. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 575hains synthesized by the RAFT process . . . . . . . . . . . . . . . . . . . . . 576
[email protected] (M.-T. Charreyre), [email protected] (J.M.G. Martinho).

M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602 569
2.5. Polymers synthesized by TERP. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5762.6. Polymers synthesized by CMRP. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5762.7. Polymers synthesized by ROMP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5772.8. Conclusions on the post-polymerization labelling strategy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 577
3. Introduction of a dye (as a monomer or as a quencher) during polymerization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5824. Use of a dye-labelled initiator . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 582
4.1. Dye-labelled initiators for anionic polymerization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5834.2. Dye-labelled initiators for nitroxide-mediated polymerization (NMP) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5834.3. Dye-labelled initiators for atom transfer radical polymerization (ATRP) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5844.4. Conclusions on dye-labelled initiators. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 585
5. Use of a dye-labelled RAFT chain transfer agent . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5855.1. �-end-labelled polymers: modification on the R group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 585
5.1.1. Linear approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5855.1.2. Convergent approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5905.1.3. Radical approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 592
5.1.4. Conclusions on labelling at the R group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5925.2. �-end-labelled polymers: modification on the Z group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 592
5.2.1. Dithiocarbamates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5935.2.2. Dithioesters. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 593
5.2.3. Conclusions on labelling at the Z group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5985.3. Conclusions on dye-labelled RAFT chain transfer agents. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 598
6. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 598References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 599
1. Introduction
In the past years, a huge variety of polymers bear-ing several pendant dyes along the backbone have beenreported and used in optical imaging [1,2], signal amplifi-cation in diagnostics tests [3], light-harvesting or antennae[4]. Another very interesting kind of dye–polymer conju-gates are polymer chains carrying dyes at specific positions,such as the chain-end or the mid-chain. Indeed, for someapplications, the precise location of the dye is crucial toprovide a given property to the material or to under-stand some physicochemical phenomena. For instance, forlight-harvesting materials, a dye located at the polymerchain-end will act as the energy acceptor of an array ofchromophores present along the backbone [4,5]. In pho-tochromic systems, the attachment of a single dye to alow glass transition temperature polymer chain is one ofthe strategies to control the switching speed of a pho-tochromic dye in a polymer matrix of high glass transitiontemperature [6–9]. In addition, dye-end-labelled polymersare valuable to study chain dynamics, conformational tran-sitions, intra- and inter-molecular associations [10,11] andfilm formation from aqueous polymer dispersions [12–14],especially for quantitative studies of polymer systems [15].
Such end-functionalized (co)polymers can bear a dye attheir �- or �-end or at both ends. While both chain-endsmust be labelled for polymer cyclization studies [16–18],only one labelled chain-end is needed for radical polymer-ization model reaction studies [18–20]. The low percentageof labelling and the precise location of the dye have addi-tional advantages: i) the intrinsic properties (solubility,conformation) of the polymer chain will remain almostunaffected; ii) the energy migration and self-quenching
erties of the dye and, conversely, the influence of the dye onthe polymer chain conformation. For instance, the dye cansense a different environment due to the polymer chain andsome functional groups present along the chain may act asfluorescence quenchers. On the other hand, the presenceof a dye at the polymer chain-end may change the chainproperties, namely when the characteristics of the dye aresubstantially different from those of the polymer as well aswhen the affinity of the polymer and the dye to the solventare substantially different.
Dyes include fluorescent compounds (fluorophores)and non-fluorescent ones (chromophores) and must beadequately selected to provide the desired property tothe dye–polymer conjugate or the desired information ofa given system. For biomedical imaging purposes, dyesabsorbing and emitting in the red and near-infrared regionare preferred, such as rhodamine [21] or Texas Red [22].In the case of light-harvesting systems, after captureof sunlight by appropriate groups, the energy migratesthroughout them to finally be trapped by a suitable termi-nal dye, such as coumarin [23] and Ru complexes [24]. Forphotochromic systems, dyes of the families of azobenzenes,spiropyrans, spirooxazines, diarylethenes and fulgides areusually employed [25]. On the other hand, pyrene isthe most often chosen dye to study polymer dynamicsbecause it exhibits a very high intrinsic lifetime (>100 nsfor 1-pyrene derivative) and it is capable of forming anew electronic excited species, the excimer (electronicexcited dimer with a disssociative ground state), upon theencounter of an electronic excited pyrene with anotherpyrene molecule in the ground state. For local dynamicsof polymer chains, adequate polarization probes such asphenanthrene must be used in order to follow the fluo-
between closely located dyes will be avoided (or limited tointer-chain phenomena); iii) the binding of another kind ofentity along the chain will still be possible.
One important point to keep in mind is the possibleinfluence of the polymer chain on the spectroscopic prop-
rescence depolarization that reflects the local motion of
the chain [26,27]. For Förster Resonance Energy Trans-fer (FRET) studies, adequate energy donor/acceptor pairs(phenanthrene/anthracene; rhodamine/fluorescein) mustbe chosen. FRET is a very powerful tool that allows the
570 M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602
Nomenclature
AcN acenaphthylAnt anthraceneAPE aryl-modified phenylethyleneATRP atom transfer radical polymerizationAzB azobenzeneBTX benzothioxantheneCAN ceric ammonium nitrateCarb carbazoleCou coumarinCMRP cobalt-mediated radical polymerizationCRP controlled radical polymerizationCTA chain transfer agentDCC N,N′-dicyclohexylcarbodiimideDMA N,N-dimethylacrylamideDMAP 4-dimethylaminopyridineDMF N,N-dimethylformamideDP degree of polymerizationDPAnt diphenylanthraceneFCS fluorescence correlation spectroscopyFluo fluoresceinFRET Förster resonance energy transferInd indoleLY Lucifer YellowMA methyl acrylateMADIX macromolecular design via interchange of
xanthatesMALDI-ToF matrix-assisted laser
desorption/ionisation-time of flightMn number average molecular weightMS mass spectrometryMw weight average molecular weightMW molecular weightNaph naphthaleneNaphPyr naphtopyranNaphSulf naphthalene sulfonateNBD nitrobenzoxadiazoleNIPAM N-isopropylacrylamideNIR near infraredNMP nitroxide-mediated polymerizationNp naphtalimideOG488 Oregon green 488PAA poly(acrylic acid)PAcN polyacenaphthylenePAETMAC poly[(2-acryloxyethyl)trimethylammonium
chloride]PAPS poly[3-(acryloyloxy)propanesulfonate
potassium salt]PAzMA poly(2-azidoethyl methacrylate)PB polybutadienePBzMA poly(benzyl methacrylate)PCEM poly[2-(9-carbazol-9-yl)ethyl methacry-
late]PDcA poly(N-decylacrylamide)PDEA poly(N,N-diethylacrylamide)PDI polydispersity indexPDLLA poly(d,l-lactide)PDMAEMA poly[(2-dimethylaminoethyl)
methacrylate]
PDMAPMA poly[N-3-(dimethylamino)propylmethacrylamide]
PDS pyridyldisulfidePEA poly(ethyl acrylate)PEG poly(ethylene glycol)PEGMEA poly[(ethylene glycol) methyl ether acry-
late]PEHA poly(2-ethylhexyl acrylate)PEO poly(ethylene oxide)PerDI perylene diimidePFP pentafluorophenylPhAzoInd phenyl azo indazolePhe phenanthrenePHPMA poly[N-(2-hydroxypropyl) methacry-
lamide]PI polyisoprenePMA poly(methyl acrylate)PMaAN poly(maleic anhydride)PMM poly(methylidene malonate 2.1.2)PMMA poly(methyl methacrylate)PnBA poly(n-butyl acrylate)PNIPAM poly(N-isopropylacrylamide)POA polyoctylacrylatePol polymerPS polystyrenePSSS poly(sodium styrene sulfonate)PSt-Cou poly(coumarin-derived styrene)PtBA poly(tert-butyl acrylate)PVA poly(vinyl alcohol)PVAc poly(vinyl acetate)PVBTMA poly(vinyl benzyl trimethylammonium)PVPPS poly[3-(2-vinylpyridinio)propanesulfonate]PVTPA poly(4-vinyl triphenylamine)Py pyreneRAFT reversible addition-fragmentation chain
transferRho 110 Rhodamine 110Rho B Rhodamine BROMP ring opening metathesis polymerizationSEC size exclusion chromatographySFRP stable free radical polymerizationSpi spirooxazinetBA tert-butyl acrylatetBDMSPrLi 3-(tert-butyldimethylsilyloxy)-1-
propyllithiumTCEP tris(2-carboxyethyl)phosphineTERP telluride-mediated radical polymerizationTGA thermogravimetric analysisTHF tetrahydrofuranTR Texas Red
VTPA 4-vinyl triphenylamineVP N-vinyl pyrrolidonedetermination of distances (1–10 nm) between specificpoints in a polymer chain (spectroscopic ruler) and the sizeof nanodomains formed at the interface between differ-ent components of polymeric systems, either as a result oftheir processing (e.g., in polymer blends and particle dis-

olymer Science 36 (2011) 568–602 571
A
M. Beija et al. / Progress in P
persions) or by design (as in films and micelles obtained byself-assembly of block copolymer chains) [15,28–32].
Before the development of controlled radical polymer-ization (CRP) techniques some fifteen years ago, there weremainly two strategies to synthesize end-functionalizedpolymer chains: i) the use of a functional initiator (oftenan azo-one for free radical polymerization or an organo-lithium derivative for anionic polymerization) [33–35] orii) the use of a functional transfer agent (usually a thiolor a disulfide) [36]. However, the resulting polymers wereeither not controlled in term of molecular weight (MW)(in case of free radical polymerization) or limited to a fewkinds of monomers (in case of anionic polymerization).
CRP techniques [37,38] overcome these two limita-tions and mediate the synthesis of end-functionalizedpolymers with accurate control of both MW (with pre-dictable molecular weights and very narrow polymerchain distributions) and macromolecular structure (withthe possibility to easily synthesize block copolymers).Moreover, CRP techniques apply to a large variety ofmonomer families and use versatile and simple experi-mental conditions. Such improvements have yielded thesynthesis of well-defined polymer chains (especially blockcopolymers and end-functionalized (co)polymers) withunprecedented properties for multiple applications. There-fore, end-functionalized (co)polymers have been preparedwith a huge variety of entities of interest includingdyes but also biomolecules, such as peptides, (phos-pho)lipids, biotin, carbohydrates, nucleosides and nucleicacids, for applications in diagnostics, drug delivery or imag-ing [39–44]. Moreover, end-functionalized polymers havebeen used for preparation of inorganic–polymer hybridmaterials [43,45,46] and for grafting of carbon nanotubes[47–49].
Depending on the chosen CRP technique, NMP(nitroxide-mediated polymerization) [50–53], ATRP(atom transfer radical polymerization) [54,55] and RAFT(reversible addition-fragmentation chain transfer) poly-merization [56,57]/MADIX (macromolecular design viainterchange of xanthates) [58], the entity of interest can beintroduced at three different stages of the polymerization:i) at the very beginning via the initiator or a chain transferagent (CTA), ii) during polymerization via a functionalmonomer or a quencher, or iii) after polymerization viaa post-modification strategy. Thus, in order to preparepolymer chains bearing one (or two) dye(s) at specificsites, the following approaches can be envisaged:
. Post-modification of polymer chains with a dye: a well-defined (co)polymer presenting a reactive group at thechain-end (� or �) or at the junction between blocks canbe labelled by reaction with a functionalized dye. Themajor drawback of this strategy resides in the moder-ate reaction yields generally achieved, which are evenlower when the polymer chain length increases. Thisimplies the use of an excess of dye molecules and time-
consuming purification processes, both increasing costs.B. Introduction of a dye-labelled monomer or quencher dur-ing polymerization: this methodology is particularlyemployed in the preparation of block copolymers dye-labelled at the junction between blocks. Usually, when
Scheme 1. Summary of the different methodologies that have been usedfor the synthesis of �-, junction- and �-dye-labelled (co)polymers.
the first monomer has been consumed, one equivalent ofa dye-functionalized comonomer (relatively to the con-centration of chains) is added before introduction of thesecond monomer. Besides, �-dye-labelled chains can beobtained by addition of a dye bearing a functional groupthat will react with the propagating chains and inducetermination (quencher). This method distinguishes itselffrom the previous one since purification of the polymeris not carried out before the dye-labelling reaction.
C. Use of a dye-labelled initiator or chain transfer agent:when using living or controlled polymerization meth-ods, the functionalities that are present in the initiatoror in the chain transfer agent (CTA, of the general formulaR S C( S) Z) are usually preserved at one (or both)polymer chain-end(s). Thus, it is possible to obtain dye-labelled polymer chains at either �- or �-end by using aninitiator or a CTA (bearing the desired dye respectivelyin the R or Z group). Moreover, mid-chain dye-labelledpolymers may be synthesized by attaching two chaintransfer agents or two initiators to a single dye molecule(leading to a Y-geometry).
An exhaustive overview of dye-labelled polymers atspecific positions will be presented in the following sec-tions, together with the living/controlled polymerizationtechniques that were employed and their correspondingscopes and limitations (Scheme 1). Conversely, polymerscarrying several pendant dyes along the chain will notbe covered by this review. The three tables that summa-rize each part of the review can be viewed from differentperspectives: the chosen labelling strategy, the type ofpolymerization process, the nature of the dye, the corre-sponding maximum excitation and emission wavelengths,the nature of the polymer chain and the incorporation yieldof the dye.
2. Post-modification of polymer chains with a dye
Depending on the polymerization technique, the result-ing polymer chains bear various groups at their ends thatcan still be reactive. Then, it is possible to introduce a
dye through a chemical reaction between that reactivemoiety and a complementary function present in the dyemolecule. Similarly, a reactive group can be introduced dur-ing polymerization at the junction of a diblock copolymerand further modified with a dye. The most used chemi-
5 olymer
clcs
sishondsAsdR
trmt
2p
lpcm“gicatApiFcctwpaoap
RdcopcldFi
72 M. Beija et al. / Progress in P
al reactions involve the formation of ester, ether or amideinkages, but other possibilities, such as “click” reactionsan be envisaged depending on the reactivity of the con-idered functional groups.
Historically, dye-labelled polymer chains at a specificite have been prepared using living anionic polymer-zation and selected examples are presented in the firstection. However, in the last fifteen years, CRP techniquesave become the methods of choice for designing polymersf different architectures from numerous monomers andew post-modification methods adapted for CRP have beeneveloped. In the following sections, the post-modificationtrategies related to polymer chains synthesized by NMP,TRP, RAFT, TERP and CMRP are exhaustively reviewed andeveral detailed examples of dye-labelling procedures areescribed. In addition, one section has been devoted toOMP presenting a few works.
In all examples, not only the application and the syn-hesis procedure of the dye-labelled polymer chains areeported, but also the employed purification methods sinceost of the post-modification strategies require removal of
he excess of free dye.
.1. Polymers synthesized by living anionicolymerization
Generally, to perform a post-polymerization dye-abelling of polymer chains synthesized by living anionicolymerization, the “living” carbanionic moiety must beonverted into a reactive functional group. Most com-only, after total consumption of the monomer, the
living” moiety is quantitatively converted into a hydroxylroup through a ring-opening reaction with an added epox-de. After purification, the hydroxyl terminated polymerhains can react with an appropriate dye derivative (usu-lly, an acyl chloride). This strategy can be used not onlyo prepare �-labelled chains, but also �-�-labelled chains.s reported by Winnik and co-workers [17], when anionicolymerization is initiated by naphtylpotassium, a “liv-
ng” dianion is formed, leading to diol-modified polymers.ollowing this procedure, �,�-pyrene-labelled polystyrenehains were synthesized and, taking advantage of theapability of pyrene to form excimers, used to study end-o-end cyclization processes [16]. The labelling reactionas carried out with an excess of five to tenfold molar ofyrenebutyryl chloride. After purification by precipitationnd preparative size exclusion chromatography (SEC) inrder to eliminate all free dye molecules, UV absorbancend 1H NMR analyses indicated that ∼90% chains carried ayrene dye at both ends.
Similarly, an �-Rhodamine B-labelled polystyrene (PS-ho B) was synthesized in Krausch’s group to study theiffusion of polymer chains in organic solutions by fluores-ence correlation spectroscopy (FCS) [35]. After conversionf the carbanionic moieties into hydroxyl groups andurification of the polymer, a 2 M excess of Rho B acyl
hloride was added. Preparative SEC was used to separateabelled polymer from free dye molecules. However, someetails about the post-modification procedure are missing.irst, Rho B acyl chloride is not a commercial reagent andts synthesis is not described although the functionalizationScience 36 (2011) 568–602
of rhodamine dyes in the 2-position is not straightforward[59]. Second, the evidence of Rho B binding at the chain-end was tentatively given by MALDI-ToF MS analysis butthe results were ambiguous since the signal was very low(essentially at the background noise).
In some circumstances, the modification step of theliving moiety into a functional group is not necessarysince the living moiety is already an alcoxide. This isthe case for polymers, such as PEO and PDLLA, synthe-sized by ring-opening polymerization of ethylene oxideand d,l-lactide, respectively. Using this method, Kataokaand co-workers [60] labelled a PEO-block-poly(d,l-lactide)(PEO-block-PDLLA) by reaction of the �-hydroxyl groupwith pyrene-1-carbonyl cyanide (in the presence of quin-uclidine as catalyst). After purification by dialysis, thepolymer was analyzed by 1H NMR, indicating that almost90% of the chains were pyrene-�-end labelled.
Riess and co-workers recently developed a novel pro-cedure for the preparation of �-end-functionalized blockcopolymers with high yields (in the molecular weightrange of 2000–10,000 g mol−1) [61]. Using an amino alco-holate as initiator, PEO-block-poly(methylidene malonate2.1.2) (PEO-block-PMM) was synthesized, bearing a pri-mary amino group �-end, further used to bind fluoresceinisothiocyanate (Fluo-NCS). The labelling reaction (5 mol eq.of Fluo-NCS per polymer chain) was carried out eitherin a heterogeneous carbonate buffer solution at pH 9.5(micellar solution) or in THF. The samples were purifiedby dialysis and the functionalization yield was determinedby UV–Vis absorption; the percentage of fluorescein �-labelled chains was only slightly higher in THF (78%) thanin the micellar solution (68%).
The different dye-labelling strategies described abovefor post-modification of polymer chains synthesized by liv-ing anionic polymerization are summarized in Scheme 2.These post-modification procedures must be performedunder rigorous experimental conditions (similar to those ofliving anionic polymerization) otherwise the loss of activecentres will lead to a severe decrease of functionalizationyields.
2.2. Polymers synthesized by NMP
Concerning NMP polymerization technique, chain-endfunctionalization usually involves the synthesis of func-tionalized initiators prior to polymerization (Section 4-2).There are only a few methodologies for the introduc-tion of a specific molecule (e.g., a dye) at the �-terminusafter polymerization. A very clever strategy proposed byHarth et al. [62] is based on the reaction between apolymeric alkoxyamine and a pyrene-labelled maleimide.It relies on the possibility of sequential monomer poly-merization by NMP together with the impossibility ofmaleimide monomers to homopolymerize. Hence, whena series of alkoxyamine-ended PS chains (from 2000to 50,000 g mol−1) were mixed with 4 eq. of N-(4-
pyrenylbutyl)maleimide (at 120 ◦C), 98% of polymer chainswere terminated by a pyrene (determined by UV–Visabsorption and 1H NMR after purification by precipitationin methanol). Moreover, when this reaction was per-formed in polar solvents like DMF, an interesting result was
M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602 573
ion of po
Scheme 2. Dye-labelling strategies based on post-modificatobtained: the nitroxide unit was also eliminated simultane-ously to the introduction of a single maleimide at the poly-mer chain-end (Scheme 3A). This is a significant advantageof this procedure since the presence of the alcoxyamine atthe chain-end can lead to depolymerization.
Another strategy was recently reported, based on theconversion of the alcoxyamine end-group into an alco-hol moiety and subsequent coupling with a carboxylicacid-functionalized pyrene (Scheme 3B) [63]. In this pro-cedure, alcoxyamine-capped PS chains were treated withceric ammonium nitrate (CAN) in a THF/water mixtureunder reflux. After purification of the resulting hydroxyl-modified polymer (precipitation in methanol), reactionwith 10 eq. of 1-pyrenebutyric acid was carried outin the presence of N,N′-dicyclohexylcarbodiimide (DCC)and 4-dimethylaminopyridine (DMAP). UV–Vis absorptionindicated that 86% of polymer chains (purified by precipi-tation) were pyrene-end-labelled.
It should be noticed that in those two strategies, hightemperatures (around 100 ◦C) were required to remove thenitroxide unit. Furthermore, the polymers were purified byprecipitation and the absence of free dye molecules wasnot confirmed. Consequently, the coupling yield could beover-estimated since it is very difficult to eliminate all freedye molecules from a polymer sample only by precipitationprocedures.
2.3. Polymers synthesized by ATRP
The number of articles dealing with post-polymerization dye-labelling at a specific site of polymer
Scheme 3. Strategies of dye-labelling by post-modific
lymer chains synthesized by living anionic polymerization.
chains synthesized by ATRP is very scarce. In fact, thereported results describe the introduction of a C60 moiety(buckminsterfullerene) at a polymer chain-end, with theaim of increasing the fullerene solubility in commonorganic solvents or in water. In a first attempt, dormantpolymer chains of bromo-terminated PS or PMMA weremodified with C60 (at a C60:polymer ratio of 2:1) in thepresence of CuBr/2,2′-bipyridine at high temperatures(90–120 ◦C) [64]. The functionalization yield determinedby thermogravimetric analysis (TGA) was 80–100%. Sincefullerenes contain several reactive double bonds, an excessof C60 was used not only to increase the binding yield,but also to favour the synthesis of the monosubstitutedC60-labelled polymer.
Another strategy for introducing a C60 moiety ata specific position of the polymer chain involves the[3+2] cycloaddition reaction between an azide and thefullerene. It was used to prepare �- and junction-C60-labelled amphiphilic block copolymers. In the formercase, a bromine end-capped poly(tert-butyl acrylate)-block-PS was modified by reaction with NaN3 and thenwith 1.5 eq. of C60 at 130 ◦C, leading to a 40–80% C60incorporation yield (depending on the polymer molec-ular weight) as determined by TGA [65]. In the lattercase, a macromolecular ATRP initiator containing a PEOblock and a lateral azide moiety close to the bromide
end-group was used to polymerize styrene. The finaldiblock copolymer bearing the azide group at the junctionwas labelled using 3 eq. of C60 at 120 ◦C. The percent-age of chains carrying a C60 moiety was not given[66].ation of polymer chains synthesized by NMP.

5 olymer
Cbtdwsa5mr([
fps
2
ptccyamR
2
aebp[aapnp
MaewTo(c(trAod
tnt
74 M. Beija et al. / Progress in P
Recently, Li et al. used “click” chemistry to introduce60 moieties at the �-chain-end of polymers synthesizedy ATRP. Using ATRP (macro)initiators carrying one orwo azide groups, they prepared �-monoazide- and �,�-iazide-terminated PNIPAM chains or PEG-block-PNIPAMith an azide group at the junction between blocks. Sub-
equently, a reaction between these polymer precursorsnd 10 eq. of an alkynyl-modified C60 was carried out at0 ◦C, leading to �-, �,�- and junction-C60-labelled poly-ers. Although the labelling yields were high (>95%), the
equirement of large amounts of the C60 alkynyl derivativewhich has to be synthesized) limits the use of this strategy67].
It is worth mentioning that the use of large excess ofullerene is necessary to avoid the binding of more than oneolymer chain to each dye since there are many reactionites on the fullerene surface.
.4. Polymers synthesized by RAFT polymerization
An important feature of polymers synthesized by RAFTolymerization is the presence of a thiocarbonylthio func-ional group (at the end or in the middle of the polymerhain) that can be easily transformed into a thiol group. Thisharacteristic enables easy post-modification of polymers,ielding �-end-labelled chains or mid-labelled chains. It islso possible to get �-end-labelled polymer chains by post-odification if the used CTA bears a reactive moiety in thegroup.
.4.1. �-end-labellingNakayama and Okano were the first to take
dvantage of the presence of the thiocarbonylthiond-group to prepare pyrene-�-end-labelledlock copolymers [68]. In a one-pot procedure, aoly(NIPAM-co-DMA)-block-poly(benzylmethacrylate)P(NIPAM-co-DMA)-block-PBzMA] was modified byminolysis with ethanolamine (20 eq.), followed byddition of 20 eq. of N-(1-pyrenyl) maleimide. If theolymer-SH was first isolated, the authors observed sig-ificant coupling between chains (disulfide bridges) thatrompt them to use this one-pot procedure.
Such coupling between chains was also observed incCormick’s group. In order to overcome this problem,
nother procedure was proposed [69]. In a first step, the �-nd-group of PNIPAM homopolymer (Mn = 39,500 g mol−1)as reduced with NaBH4 and the polymer was isolated.
hen, before reaction with a maleimide derivative, 150 eq.f a mild reducing agent – tris(2-carboxyethyl)phosphineTCEP) – was added to convert all disulfide coupledhains into thiol-ended chains. After 24 h, 150 eq. of N-1-pyrenyl)maleimide was introduced. After purification,he binding yield was determined by SEC using bothefractive index and UV absorbance (at 340 nm) detectors.lthough a high yield was obtained (83%), the large excessf functionalized dye may result in a very costly proce-
ure.Later on, McCormick and co-workers [70] suggestedhat the use of a large excess of pyrene derivative wasecessary since the reduction of the thiocarbonylthio-erminated vinylic chain gave rise to a secondary thiol
Science 36 (2011) 568–602
less reactive than a primary one. Then, they proposedthat after reduction, the obtained secondary (or ter-tiary) thiol was converted into a primary amine througha disulfide exchange reaction between the polymer-SH and cystamine. For instance, after reduction of thechain-end of poly[N-(2-hydroxypropyl) methacrylamide]-block-poly[N-3-(dimethylamino) propylmethacrylamide](PHPMA-block-PDMAPMA; Mn = 39,700 g mol−1), an excessof cystamine was added (in Tris buffer, pH 9.5) andthe reaction proceeded for 92 h at 70 ◦C. After iso-lation of the amino-functionalized chains, the dyelabelling reaction was carried out with only 5 eq. of6-(fluorescein-5-carboxamido)hexanoic acid succinimidylester (Fluo-NHS) at 60 ◦C, resulting in a 80.1 ± 2.6% bindingyield (determined by UV–Vis absorption on the puri-fied fluorophore-�-end-labelled polymer). This syntheticpathway requires 30 times less dye derivative than theprevious one, but it has the inconvenience of linking thedye via a disulfide bond with limited stability in biologicalmedia.
Recently, an extremely efficient methodology wasreported by Segui et al. for the preparation of �,�-dipyrenyl-ended PNIPAM [71]. Instead of using a pyrene-maleimide derivative, 4-(1-pyrenyl)butyl iodide wasprepared and reacted with HS-PNIPAM-SH (reduced byreaction with a 10-fold excess of n-butylamine in thepresence of a catalytic amount of TCEP and recovered byprecipitation). Although only 1.5 eq. of 4-(1-pyrenyl)butyliodide (relatively to –SH groups) were used, the pyreneincorporation after purification was 95% for all sampleswith Mn < 45,200 g mol−1 (determined by UV–Vis absorp-tion). However, this strategy requires the prior synthesisof the dye iodide derivative.
A completely different approach for �-end-grouppost-modification reported by Chen et al. [72], involves“click” chemistry [73]. An azido-functionalized xan-thate was used to mediate the RAFT polymerization ofvinyl acetate, yielding an azido-�-end-capped poly(vinylacetate) (PVAc) (Mn = 4000 g mol−1). Then, the modifiedPVAc reacted with 1 eq. of a dye alkyne-derivative (7-propinyloxy coumarin) at 80 ◦C, in the presence of CuBrand N,N,N′,N′′,N′′-pentamethyl diethylenetriamine. UV–Visabsorption indicated that 69% of the polymer chainswere end-capped with the coumarin dye, correspond-ing to a coupling yield of 87%. However, since the dyeis linked to the �-chain-end by a spacer containing thethiocarbonylthio moiety (very sensitive to hydrolysis andaminolysis), the loss of some dye end-groups may occur.
The strategies that have been used for post-modificationof the thiocarbonylthio �-end-group of polymer chainssynthesized by RAFT polymerization are schematicallyillustrated in Scheme 4. Most of them lead to moderatebinding yields unless a large excess of dye-derivative isused and/or the resulting spacer arm between the dye andthe polymer chain presents limited stability in aqueousmedia.
2.4.2. ˛-end-labellingConcerning the post-modification strategies that lead
to �-dye-labelled polymer chains, a few articles were veryrecently published. For instance, Roth et al. used a CTA

M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602 575
post-m
Scheme 4. Dye-labelling strategies at the �-chain-end based oncarrying a pentafluorophenyl (PFP) activated ester as Rgroup (synthesized with a 51% yield) to control the poly-merization of MMA. The resulting polymer chains werefurther dye-labelled with 3.7 eq. of Texas Red (TR) aminoderivative. A robust amide bond was created betweenthe polymer chain and the dye, with a labelling yieldof 74% (determined by UV–Vis absorption). In a secondstep, aminolysis of the Z group was carried out by addi-tion of an excess of n-propyl amine in the presence ofmethyl methanethiosulfonate, which produced �-CH3-S-S-terminated PMMA chains (Scheme 5). Such polymerchains were used to decorate gold and semiconductor(quantum dots) nanoparticles [22].
A similar strategy was carried out in our group withanother kind of CTA bearing a N-succinimidyl activatedester as R group (synthesized with a 45% yield) [74],used to prepare �-end-reactive PDMA-block-PDEA ther-mosensitive block copolymers. Then, the chains were�-end-labelled with a rhodamine B piperazine amidederivative (3–6 eq.) at room temperature in the presenceof N,N-diisopropyl ethylamine to deprotonate the dye
derivative (Scheme 6). The binding yields were moderate(19–38%, respectively), considering this sterically hinderedsecondary amine [75]. In an improved strategy describedin Section 5.1.2, more than 90% labelling yield was reachedusing the corresponding dye-labelled CTA.Scheme 5. Dye-labelling strategy at the reactive �-pentafluorophenyl-
odification of polymer chains synthesized by the RAFT process.
A different approach was proposed by Liu et al., inwhich a symmetric trithiocarbonate bearing pyridyldisul-fide (PDS) moieties in the R group was first synthesized(56% yield) and further used to prepare a poly(PEG-A)-block-PS-block-poly(PEG-A) graft copolymer from anoligo(ethyleneglycol) acrylate macromonomer (PEG-A).This polymer was then dissolved in a dioxane:water(1:4) mixture, self-assembling in micelles with the PDSgroups accessible to react with a thiol-containing RhoB derivative (10 eq.). The dye was bound via a disul-fide bond (80% incorporation yield) [21]. It is worthnoting that this strategy preserved the dormant moi-ety.
Finally, “click” chemistry has also been employed tolabel polymers at the �-position. Tong et al. synthesized anazide-containing xanthate (20% yield) to mediate the RAFTpolymerization of vinyl acetate (VAc). The PVAc chainswere converted into poly(vinyl alcohol) by hydrolysis andreacted with 1.5 eq. of alkynyl-modified pyrene. The dyeincorporation was not determined, however the low syn-thesis yield of the azide-containing CTA is already a limiting
factor for this strategy [76].2.4.3. Junction-labellingRecently, Henry et al. proposed a post-modification
strategy to prepare both �- and junction-dye-labelled
chain-end of polymer chains synthesized by the RAFT process.

576 M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602
S n of thes
pmmfAmtrvj
atyacl
2c
setdoawtp
SR
cheme 6. Synthesis of dye-labelled polymer chains by post-modificatioized by RAFT polymerization.
olymers. Taking advantage from the fact that maleimidoonomers readily copolymerize but do not homopoly-erize, an amino-substituted maleimido monomer (in the
orm of a triflate salt) was introduced at the �-chain-end.lternatively, since the resulting chains were still dor-ant, a block copolymer bearing the maleimido unit at
he junction between blocks was also prepared. Then, aeaction between these polymer chains and a pyrenyl acti-ated ester derivative was carried out to obtain the �- andunction-pyrene-labelled polymers (Scheme 7) [77].
Such a synthetic route preserves the dormant moietynd leads to the formation of a stable amide bond betweenhe polymer chain and the dye. However, the couplingields were relatively low (44–86%) considering that highmounts of dye derivative were used (7–50 eq.), whichan be explained by the steric hindrance intrinsic to theabelling at the junction point.
.4.4. Conclusion on the post-modification of polymerhains synthesized by the RAFT process
The post-labelling strategies of polymer chains synthe-ized via RAFT polymerization are numerous and varied,ither at the �-end after an easy transformation of thehiocarbonylthio-function into a thiol one (using dyeerivatives bearing a disulfide, a maleimide or an iodide),
r at the �-end if the chosen CTA bears an activated ester,disulfide or an azide group that will respectively reactith an amino, thiol or alkyne dye derivative. Generally,he labelling procedure does not require elevated tem-eratures (room temperature may suffice); however, a
cheme 7. Dye-labelling strategy at the �-chain-end or at the junction betweenAFT process and a maleimido unit.
reactive �-succinimidyl-end PDMA-block-PDEA polymer chains synthe-
large excess of dye is usually necessary to get satisfyingyields.
2.5. Polymers synthesized by TERP
TERP is a recent method of CRP that involves organotel-lurium compounds. As soon as this technique was reported,a procedure for post-modification of polymer chains wassuggested. In fact, the �-end-group can be transformed intoa carboxylic acid group through tellurium–lithium trans-metalation (with butyllithium) followed by reaction withcarbon dioxide. Using this procedure, Yamago et al. mod-ified PS chains obtained by TERP, that subsequently werereacted with 4 eq. of pyrenebutanol to produce pyrene-�-end-labelled PS chains with 86% of pyrene incorporation asdetermined by UV–Vis absorption after purification. [78].
2.6. Polymers synthesized by CMRP
To produce well-controlled poly(vinyl acetate) (PVAc)leading to biocompatible poly(vinyl alcohol) after hydroly-sis, it is necessary to use xanthates/dithiocarbamates RAFTagents or cobalt-mediated radical polymerization (CMRP)that leads to high monomer conversion and relatively highmolecular weights [79].
The C–Co bond of PVAc chains end-capped by a cobaltcomplex can be easily cleaved (at 30 ◦C), providing reactivePVAc macroradicals for further end-functionalization. Forinstance, Detrembleur et al. have made such macroradi-cals react with fullerene (C60) to get PVAc/C60 nanohybrids
blocks based on post-modification of polymer chains synthesized by the

olymer
M. Beija et al. / Progress in P(since a molar excess of 8.6 was used, four arm fullerenewas prepared) [80]. After hydrolysis, an aqueous solutionof PVOH/C60 nanohybrid was irradiated at an appropri-ate wavelength that resulted in the production of singletoxygen, thus making this nanomaterial a candidate forphotodynamic cancer therapy. Later, a similar strategywas applied to poly(vinyl alcohol-co-N-vinyl pyrrolidone)copolymers [81].
The CMRP process may then be useful to prepare dye-end-labelled polymers from monomers that do not yieldwell-controlled polymer chains via the other CRP pro-cesses.
2.7. Polymers synthesized by ROMP
When using ruthenium based catalysts, ROMP canyield polymers of well-defined length [82] that canbe easily end-functionalized using appropriate cappingagents. Kiessling’s group used that property to intro-duce fluorescein at a glycopolymer �-end prepared byROMP and terminated by a bifunctional capping agentbearing a vinyl ether at one end and a masked car-boxylic acid at the other. After deprotection of thecarboxylic ester chain-end, an amino derivative of fluo-rescein was introduced (in the presence of a carbodiimideand sulfo-N-hydroxysuccinimide), with a binding yieldof 69% [83]. In a more recent strategy, a latent flu-orophore (alkyne derivative of bis(acetylated trimethyllock) Rho 110 [59]) was introduced by click chemistry ona glycopolymer �-chain-end synthesized by ROMP andterminated by an azide containing capping agent (yieldnot given). The resulting multivalent ligand was used tovisualize cellular internalization events (that occur on aminute time scale) since the dye becomes fluorescentby esterase catalysis [59] after cellular internalization[84].
Madkour et al. used an allyl ether instead of a vinyl etherto end-cap a polymer synthesized by ROMP, that yield anear quantitative end-group incorporation. Since that allylether was bearing a pentafluorophenyl activated ester atthe other end, an amino derivative of nitrobenzoxadiazolewas bound at the �-chain-end (room temperature, yieldnot given). As the polymer had antimicrobial activity, theresulting fluorescent polymer was used to label the cellmembrane of bacteria [85].
Another strategy of dye-labelling of ROMP polymers didnot require a functionalized capping agent since the post-modification was carried out at the �-chain-end [86]. Infact, a norbornyl derivative bearing a succinimidyl esterwas first polymerized with a 1:1 catalyst:monomer ratio,then a second norbornyl monomer bearing a protectedpeptide sequence was introduced in excess. After deprotec-tion of the peptide sequence, an amino derivative of Oregongreen 488 (OG488) dye was bound onto the first polymerunit at room temperature (74–80% yield). Such fluorescentnorbornyl oligopeptide polymers were used to visualize the
specific binding of the polymer to cell surface depending onthe nature of the oligopeptide sequence.The limitations of ROMP to prepare dye-end-functionalized polymers reside in the few number ofmonomers that can be polymerized by this technique
Science 36 (2011) 568–602 577
(even if the possibility to use a norbornyl derivative thatbears a succinimidyl ester extends the range of possiblelateral groups), the sensitivity to oxygen and the neces-sity to proceed to meticulous catalyst removal for someapplications.
2.8. Conclusions on the post-polymerization labellingstrategy
Post-modification strategies that lead to dye-labelledpolymers at specific positions (�- and �-end-groups, junc-tion between two blocks) have been used for many yearsand new methodologies are regularly proposed in the lit-erature. The main advantage of these approaches residesin the possibility of preparing families of monodispersepolymer samples that differ only in the choice of thedye end-group. However, most of the reported proce-dures require an excess of the dye derivative in orderto reach high binding yields. Since functionalized dyesare usually expensive or not commercially available andhave to be synthesized, the use of such large amountsis the major drawback of these approaches, togetherwith the need of tedious purification steps. Indeed, elim-ination of the dye excess can be extremely laboriousespecially if a water-soluble polymer is labelled with ahydrophobic dye and it is not straightforward to find theright purification conditions. A liquid–liquid extractionmay lead to an emulsion. Precipitation in apolar solventsmay lead to entrapment of dye molecules within poly-mer chains and dialysis may be non effective. Thus, insome cases, it is necessary to perform preparative sizeexclusion chromatography to get purified dye-labelledpolymers.
Table 1 exhibits all the dye-end-labelled polymerssynthesized by post-modification methods that weredescribed in the above sections. The binding yields are usu-ally high (between 80% and 100%). However, this methodwas performed with low molecular weight polymers, in therange of 2000–50,000 g mol−1. For longer chains, the reac-tive moiety becomes “protected” within the polymer coil,with a consequent decrease of the labelling yield.
The binding of a dye (very often hydrophobic) on ahydrophilic polymer may result in an amphiphilic poly-mer, especially difficult to characterize. In fact, two stepscan be distinguished: the modification of the polymer chainwith a dye (including the characterization of the resultinglabelled chain by various analytical techniques) and theuse of the labelled polymer chain for a specific applica-tion or fundamental study. Since most of the water-solublepolymers are also organo-soluble (in DMF or chlorinatedsolvents), the labelling reaction may often be carried outin an organic solvent (where both dye and polymer aresoluble) as well as the characterization of the dye-end-labelled chain. Then, after purification of the polymer(which may be time-consuming, as previously discussed)
and drying, dissolution in an aqueous phase may be per-formed if necessary. The possible amphiphilic characterof the dye-end-labelled chain should obviously be takeninto account during the photophysics studies in aqueousphase.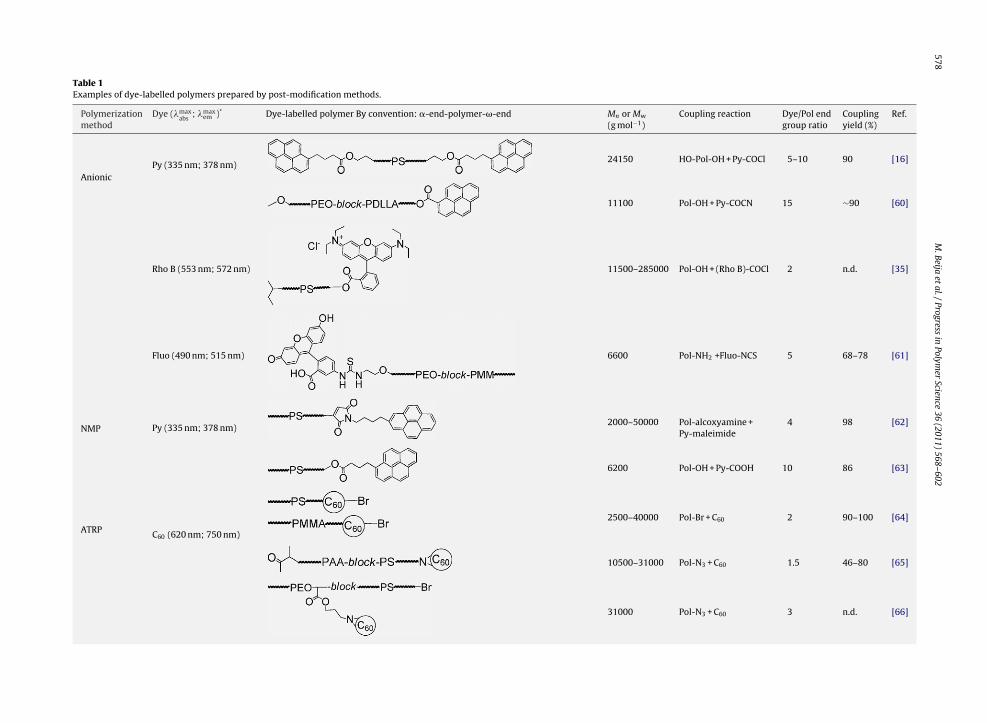
578M
.Beijaet
al./Progressin
Polymer
Science36 (2011) 568–602
Table 1Examples of dye-labelled polymers prepared by post-modification methods.
Polymerizationmethod
Dye (�maxabs
; �maxem )* Dye-labelled polymer By convention: �-end-polymer-�-end Mn or Mw
(g mol−1)Coupling reaction Dye/Pol end
group ratioCouplingyield (%)
Ref.
AnionicPy (335 nm; 378 nm)
24150 HO-Pol-OH + Py-COCl 5–10 90 [16]
11100 Pol-OH + Py-COCN 15 ∼90 [60]
Rho B (553 nm; 572 nm) 11500–285000 Pol-OH + (Rho B)-COCl 2 n.d. [35]
Fluo (490 nm; 515 nm) 6600 Pol-NH2 +Fluo-NCS 5 68–78 [61]
NMP Py (335 nm; 378 nm)2000–50000 Pol-alcoxyamine +
Py-maleimide4 98 [62]
6200 Pol-OH + Py-COOH 10 86 [63]
ATRP C60 (620 nm; 750 nm)
2500–40000 Pol-Br + C60 2 90–100 [64]
10500–31000 Pol-N3 + C60 1.5 46–80 [65]
31000 Pol-N3 + C60 3 n.d. [66]

M.Beija
etal./Progress
inPolym
erScience
36 (2011) 568–602579
Table 1 (Continued )
Polymerizationmethod
Dye (�maxabs
; �maxem )* Dye-labelled polymer By convention: �-end-polymer-�-end Mn or Mw
(g mol−1)Coupling reaction Dye/Pol end
group ratioCouplingyield (%)
Ref.
ATRP C60 (620 nm; 750 nm) 105001070012700
N3-Pol + C60-≡ 10 >95 [67]
RAFT
Py (335 nm; 378 nm)17300 Pol-SH + Py-maleimide 20 n.d. [68]
39500 Pol-SH + Py-maleimide 150 83 [69]
8150–45200 HS-Pol-SH + Py-I 1.5 95 [71]
Fluo (490 nm; 515 nm) 39700 Pol-S-S-CH2CH2NH2 + Fluo-NHS
5 80 [70]
Cou (320 nm; 370 nm) 4000 Pol-N3 + Cou-≡ 1 88 [72]
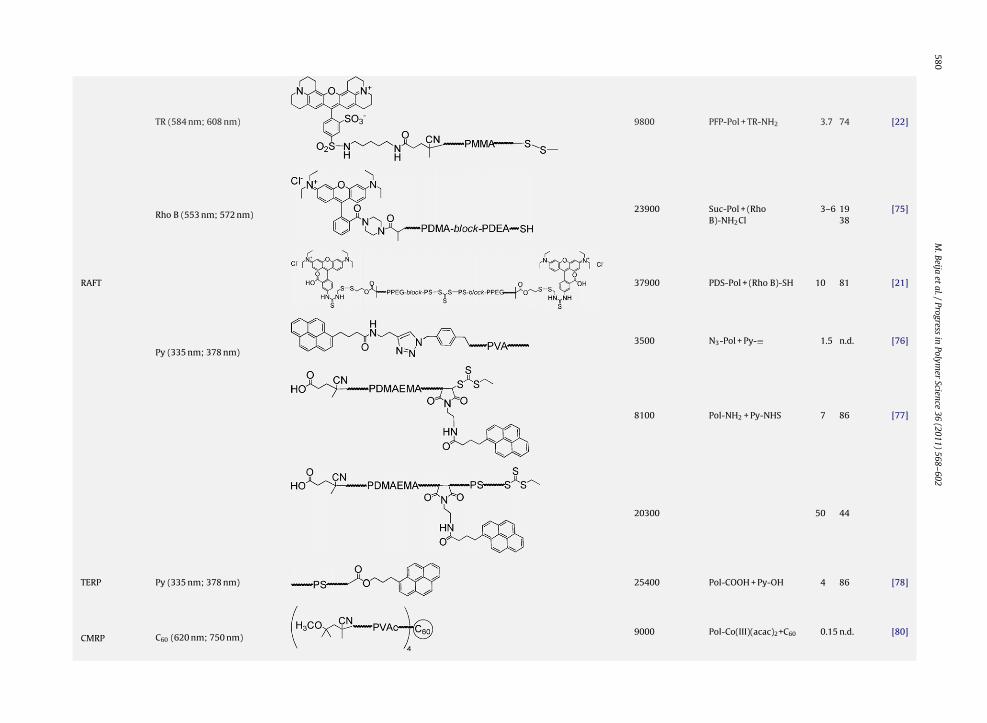
580M
.Beijaet
al./Progressin
Polymer
Science36 (2011) 568–602
TR (584 nm; 608 nm) 9800 PFP-Pol + TR-NH2 3.7 74 [22]
Rho B (553 nm; 572 nm)23900 Suc-Pol + (Rho
B)-NH2Cl3–6 19
38[75]
RAFT 37900 PDS-Pol + (Rho B)-SH 10 81 [21]
Py (335 nm; 378 nm)3500 N3-Pol + Py-≡ 1.5 n.d. [76]
8100 Pol-NH2 + Py-NHS 7 86 [77]
20300 50 44
TERP Py (335 nm; 378 nm) 25400 Pol-COOH + Py-OH 4 86 [78]
CMRP C60 (620 nm; 750 nm)9000 Pol-Co(III)(acac)2+C60 0.15 n.d. [80]

M.Beija
etal./Progress
inPolym
erScience
36 (2011) 568–602581
Table 1 (Continued )
Polymerizationmethod
Dye (�maxabs
; �maxem )* Dye-labelled polymer By convention: �-end-polymer-�-end Mn or Mw
(g mol−1)Coupling reaction Dye/Pol end
group ratioCouplingyield (%)
Ref.
9300 0.15–2 n.d. [81]
ROMP
Fluo (490 nm; 515 nm) 8200 Pol-COOH + Fluo-NH2 5 69 [83]
Pro-(Rho) 110 (497 nm; 520 nm) 3500 Pol-N3 + Pro-(Rho110)-≡
16 n.d. [84]
NBD (460 nm; 510 nm) 4200 Pol-PFP + NBD-NH2 1.2 n.d. [85]
OG488 (492 nm; 518 nm) 7200 Suc-Pol + OG488-NH2 4 74–80 [86]
acac: acetyl acetonate; Cou: coumarin; DMA: N,N-dimethylacrylamide; Fluo: fluorescein; NBD: nitrobenzoxadiazole; OG488: Oregon green 488; PAA: poly(acrylic acid); PBzMA: poly(benzyl methacrylate);PDLLA: poly(d,l-lactide); PDMAEMA: poly[(2-dimethylaminoethyl)methacrylate]; PDMAPMA: poly[N-3-(dimethylamino)propyl methacrylamide]; PDS: pyridyldisulfide group; PEG: poly(ethylene glycol); PEO:poly(ethylene oxide); PFP: pentafluorophenyl activated ester; PHPMA: poly[N-(2-hydroxypropyl)methacrylamide]; PMM: poly(methylidene malonate 2.1.2); PMMA: poly(methyl methacrylate); PNIPAM: poly(N-isopropyl acrylamide); Pol: polymer; Pro-(Rho 110): pro-rhodamine 110; PS: polystyrene; PVA: poly(vinyl alcohol); PVAc: poly(vinyl acetate); Py: pyrene; Rho B: rhodamine B; TR: Texas Red cadaverine; VP:N-vinyl pyrrolidone.
* Approximate values.

582 M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602
polymer
3q
isidrop[mcfimolt1i
pcwm
gt
Scheme 8. Synthesis of a dye-junction-labelled diblock co
. Introduction of a dye (as a monomer or as auencher) during polymerization
The strategy based on the introduction of a dye dur-ng polymerization has been mainly used for polymersynthesized by living anionic polymerization, especiallyn the case of block copolymers labelled with a FRETonor/acceptor pair at the junction between blocks. Aseported by Winnik and co-workers, if a 2-fold excessf a bulky dye-functionalized monomer is added afterolymerization completion of styrene [87] (or isoprene88]), one dye unit will be introduced at the poly-
er �-chain-end. The new living end is a less reactivearbanion (unable to homopolymerize), but remains suf-ciently reactive to initiate the polymerization of a secondonomer like methyl methacrylate (MMA). This method-
logy was applied to the synthesis of diblock copolymersabelled with phenantrene or anthracene at the junc-ion [using either 1-(9-phenanthryl)-1-phenylethylene or-(2-anthryl)-1-phenylethylene as monomer] with a dye
ncorporation efficiency of 92–100%.A similar approach was used by Horinaka et al. [89] for
reparation of anthracene-�-end-labelled PS. After totalonsumption of styrene, the living ends were terminated
ith a dye-functionalized quencher, 9-bromomethyl-10-ethylanthracene.The dye-labelling of a chain during polymerizationenerally gives a high dye incorporation yield. However,he choice of monomers is limited by the requirements
using a combination of anionic polymerization and ATRP.
of anionic (or cationic) polymerization. For instance, itwas not possible to obtain junction-labelled polyisoprene-block-PS (PI-block-PS) because the new carbanion formedafter addition of the dye derivative was not reactive enoughto initiate the polymerization of the second monomer.In order to circumvent this problem, Winnik and co-workers developed a strategy based on the combinationof anionic polymerization and ATRP [90]. After poly-merization of isoprene, the macrocarbanion was firstreacted with 1-aryl-1-phenylethylene phenanthrene (oranthracene) derivative and then treated with an excess of�,�′-dibromo-p-xylene (small and very reactive moleculeable to add onto the hindered carbanion), providing apolymer with a benzylic bromide end-group suitable to ini-tiate the polymerization of styrene by ATRP (Scheme 8).Using this strategy, they successfully synthesize PI-block-PS chains with 95% dye incorporation at the junctionbetween blocks.
4. Use of a dye-labelled initiator
Instead of introducing a dye after or during polymer-ization, a third strategy consists in using a dye-labelledinitiator or a dye-labelled CTA. The dye is chemically mod-
ified so that it becomes able either to initiate or to controla polymerization process. The design of the molecularstructure of the dye derivative depends on the chosen poly-merization technique. The main advantage of this strategyis the labelling of all chains (or a major part) at their �- or
olymer Science 36 (2011) 568–602 583
this latter monomer acts as an electron donor, the result-ing polymers exhibit photoluminescence quenching due toelectron transfer between the donor polymer chain and theperylene at the chain-end (electron acceptor) [96].
M. Beija et al. / Progress in P
�-chain-end. However, the derivatization of the dye andfurther polymerization should not induce the loss or sig-nificant modification of its spectroscopic properties, andconversely, the presence of the dye should not interferewith the polymerization mechanism, both conditions thatremain the major challenges of this strategy.
In the following sections, dye-labelled initiators thathave been used for living anionic polymerization, NMPand ATRP will be considered. The use of dye-labelledRAFT chain transfer agents will be discussed in a separatesection.
4.1. Dye-labelled initiators for anionic polymerization
Fluorescent initiators for anionic polymerization havealways been prepared using the reaction of a usualinitiator with a dye-modified vinyl monomer. Gener-ally, these fluorescent initiators were synthesized in situbefore addition of the monomer to be polymerized.This approach was first used in Webber’s group to syn-thesize a naphthalene-�-end-labelled PS-block-poly(tert-butylmethacrylate) diblock copolymer. Using cumylpotas-sium as initiator, a small amount of 2-vinylnaphthalenewas introduced and very short naphthalene-labelledoligomers were synthesized (carbanions with an averagepolymerization degree of 4) and further used as initiatorsfor the synthesis of PS blocks [91].
However, in order to achieve a reaction yield closeto 100%, an excess of dye-functionalized monomer mustbe used, with a high probability of addition of morethan one dye unit. This can result in polymer chainsbearing two fluorescent groups close to each other,enhancing dimer/excimer formation and consequentlyfluorescence quenching. To minimize this possibility, steri-cally hindered molecules are generally used. Indeed, Moonet al. synthesized an anthracene-labelled initiator foranionic polymerization by in situ reaction between sec-butyllithium and the bulky bisubstituted vinyl monomer,1-(2-anthryl)-1-phenylethylene, that was then used to ini-tiate polymerization of MMA [92].
More recently, Tong et al. prepared junction-labelleddiblock copolymers using heterobifunctional initiators forliving anionic polymerization and ATRP. Starting from analcohol protected initiator, 3-(tert-butyldimethylsilyloxy)-1-propyllithium (tBDMSPrLi), fluorescent anionic initiatorswere prepared by reaction with aryl-modified phenylethy-lene monomers (anthracene and phenantrene derivatives).After synthesis of a first PMMA block, elimination of thesilyl protecting group was performed, leading to hydroxyl-functionalized chains that were further modified intobromide �-dye-labelled polymer chains (ATRP macroini-tiator). Several dye-junction-labelled diblock copolymerswere then synthesized via ATRP of alkylacrylate monomers[93].
Finally, dye-labelled initiators for living anionic poly-
merization include a limited number of dyes, all veryhydrophobic (naphthalene, phenanthrene, anthracene)and without polar groups or labile protons in their structureto avoid the risk of interference with the polymerizationmechanism.Scheme 9. Simplified mechanism of NMP.
4.2. Dye-labelled initiators for nitroxide-mediatedpolymerization (NMP)
Among stable free radical polymerization (SFRP) tech-niques, nitroxide-mediated polymerization is the mostcommon. It is based on the property of nitroxides to scav-enge carbon-centered radicals by combination (formationof an alkoxyamine). This mechanism is reversible and thuscan be used to control the polymerization (Scheme 9)[50–53].
Typically, alkoxyamine derivatives are used as initiatorsfor NMP (Fig. 1). The polymerization is thermally initi-ated with dissociation of the alkoxyamine into an initiatingradical (R1) that will add to monomer and a nitroxide frag-ment (O-N-(R2)R3) that will control polymerization. Thus,when aiming to synthesize a dye-labelled NMP initiator, itis possible to introduce the chromophore at three differentpositions (R1, R2 or R3) that will originate either �-end (R1)or �-end (R2, R3) labelled polymer chains.
Concerning dye-labelled NMP initiators having a chro-mophore attached to the initiating fragment, Waymouthand co-workers synthesized perylene diimide deriva-tives with one or two alkoxyamine units [94,95]. Theseperylene-based initiators were used to polymerize styrene,butyl acrylate, butadiene and isoprene, giving labelledhomopolymers and block copolymers for single-moleculeimaging. Perylene diimide was chosen as a fluorescent labeldue to its high fluorescence quantum yield, resistance tophotobleaching and convenient absorption in the visible.Using the monosubstituted perylene initiator, polymerswith Mn ranging from 3000 to 42,000 g mol−1 were synthe-sized with low polydispersity index (PDI < 1.3). Conversely,with the bisubstituted perylene initiator, well-definedpolymers were only obtained when the targeted Mn waslower than 20,000 g mol−1. Another perylene diimide-derived monoalkoxyamine was synthesized (82% yield)by Lindner and Thelakkat and used to polymerize severalacrylates, styrene and 4-vinyltriphenylamine (VTPA). Since
Fig. 1. Alkoxyamine structure.

5 olymer Science 36 (2011) 568–602
tgpPowM9iddMlAns[ttcpwt
onpan
aOdsiiiodnmto
4p
acrhhmpf
wtf
84 M. Beija et al. / Progress in P
Pyrene and dansyl �-labelled alkoxyamines were syn-hesized (84% and 78% yields, respectively) by Hawker’sroup in order to investigate chain-end control in theolymerization of styrene and tert-butyl acrylate (tBA).olymer chains with molecular weights close to the the-retical ones and polydispersities between 1.05 and 1.15ere obtained. In addition, for PtBA and PS samples withn up to 70,000 g mol−1, dye incorporation was higher than
7%. Similar initiators containing the dye in the nitrox-de fragment (57% and 69% yields for pyrene and dansylerivatives, respectively) were prepared. In this case, theye incorporation was higher than 95% for polymers withn up to 50,000 g mol−1, while for those of higher molecu-
ar weights, the dye incorporation decreased to 81% [97].nother pyrene-labelled alkoxyamine (modified in theitroxide fragment) was quantitatively synthesized by Buc-iova et al. and used to mediate styrene polymerization98]. More recently, Chmela and Hrcová prepared an initia-or bearing a benzothioxanthene in the nitroxide fragmento evaluate the extent of livingness of PS chains (quantifi-ation of dye incorporation). They showed that the activeolymer chain-ends, the extent of “livingness”, decreasedith conversion even though the polydispersity was essen-
ially unchanged [99,100].Cunningham and co-workers reported the synthesis
f a naphthalene-labelled alkoxyamine (86% yield). Thisaphtoyloxy derivative was employed as a model com-ound to determine nitroxyl-exchange yields betweenlkoxyamine-terminated PS chains and a naphtoyloxyitroxide [101].
The synthesis of dye-labelled alkoxyamines at the initi-ting fragment is usually straightforward and very efficient.n the contrary, preparation of alkoxyamines bearing theye at the nitroxide fragment is a challenging task andeveral protection–deprotection steps are required, lead-ng to lower yields. Moreover, a higher dye incorporations reached when the initiator carries the dye in the initiat-ng fragment. In fact, a single reaction leads to the insertionf the dye at the polymer �-chain-end. By contrast, if theye is present in the nitroxide fragment (involved in a largeumber of cleavage/insertion/coupling cycles during poly-erization), there is a higher probability for chain-end loss
hrough occurrence of side reactions, such as terminationr transfer.
.3. Dye-labelled initiators for atom transfer radicalolymerization (ATRP)
Concerning ATRP, the control of polymerization ischieved by a reversible coupling mechanism. Polymerhains are deactivated through trapping of propagatingadicals by atom (or group) transfer (most commonly aalogen atom X, such as Cl, Br, I) from a metal complex in itsigher oxidation state (Scheme 10). On the contrary, poly-er chains become active by a redox reaction between the
olymer end group and the metal complex in its reduced
orm [102,103].Initiators used for ATRP are usually low moleculareight activated organic halides (R1X), where R1 is an alkyl
hat usually bears substituents capable of stabilizing theormed radical by resonance. Hence, in order to label an
Scheme 10. Simplified mechanism of ATRP.
ATRP initiator with a dye, R1 has to be appropriately modi-fied. Consequently, only �-dye-labelled polymer chains canbe prepared.
Dye-labelled ATRP initiators are usually synthesizedin a straightforward way by reaction of a hydroxyl-functionalized dye with an �-haloacyl halide (e.g.,�-bromopropionyl bromide). By this methodology,pyrene-labelled [104] and fluorescein-labelled [105]ATRP initiators were synthesized with 87% and 72%yields, respectively, and used to control the polymer-ization of NIPAM. A pyrene-modified ATRP initiatorwas also used to mediate the polymerization of (2-dimethylaminoethyl)methacrylate (DMAEMA) leading topolymer chains able to passivate colloidal CdSe nanocrys-tals (quantum dots). Using the fluorescence of pyrene,the average number of chains involved in the passiva-tion of these quantum dots was determined [106]. Morerecently, an ATRP initiator bearing a naphthalimide (Np)derivative was synthesized to control the polymerizationof DMAEMA. Both the Py- and Np-labelled polymers wereused to evaluate the separation of free polymer chains andpolymer-coated quantum dots by preparative HPLC [107].
Aiming to prepare photochromic materials, aspirooxazine-labelled bromo-initiator (85% yield)[108,109] and naphtopyran-labelled bromo- (78% yield)[110] or dibromo-initiators [9,111] were also synthesizedand used to control the polymerization of MMA [109,110],styrene [108], 2-ethylhexyl acrylate [110], n-butyl acry-late [9,111] and 2-(9H-carbazol-9-yl)ethyl methacrylate[110]. The photochromic spirooxazine-labelled polymerdemonstrated that the attached polymer could control theswitching speed of the dye without altering its electronicnature [108]. In the case of the naphtopyran dye, the designof a Y-branched geometry resulted in a decoloration speedenhancement in comparison with an end-group geometry[111]. Recently, Zhao et al. reported the synthesis of a newnaphthalene-labelled bifunctional initiator (43% yield) forpolymerization of MMA via the activator generated byelectron transfer for ATRP (AGET ATRP) [112].
Employing a completely different approach, Roof etal. used the photodimer of 9-bromoanthracene as anATRP initiator to polymerize styrene and n-butyl acry-late. After polymerization, �-anthracene-labelled polymerchains were obtained by photodimer cleavage. However,these chains presented a relatively broad distribution and anon-linear relationship between Mn and conversion [113].
More recently, these authors studied the influence of theligand nature during ATRP polymerization initiated bythose photodimers [114].
olymer
A
M. Beija et al. / Progress in P
Finally, according to the ATRP mechanism, the dye-modified fragment of the initiator will always appear atthe �-chain-end. The most important advantages of usingdye-labelled ATRP initiators are that they can be synthe-sized via simple procedures and that virtually all polymerchains will bear a dye.
4.4. Conclusions on dye-labelled initiators
Table 2 shows the molecular structures of the dye-labelled initiators that have been synthesized so far foranionic polymerization, NMP and ATRP. Depending onthe chosen polymerization technique, several syntheticpathways can be carried out to prepare the appropriate ini-tiators. In all cases, if the dye-labelled initiator exhibits highinitiation efficiency, the dye incorporation at the polymerchain-end will be greater than 90%.
5. Use of a dye-labelled RAFT chain transfer agent
Herein, dye-labelled polymers prepared by RAFT poly-merization using a dye-labelled chain transfer agent arereviewed. As well established, the control of RAFT polymer-ization is due to the presence of a chain transfer agent thatreacts with the propagating radicals, leading to an equi-librium between dormant and active chains [56,58,115].However, termination reactions compete with this equi-librium. As a consequence, in order to get a successfulcontrol, the concentration of those propagating radicalsshould be kept much lower than the concentration of dor-mant chains, that can be achieved by a careful choice ofthe polymerization conditions (initiator, temperature and[CTA]0/[initiator]0) [57,116].
In recent years, many thiocarbonylthio compoundshave been described in the literature and successfully usedas CTA for the RAFT polymerization of various monomers[57,116]. When designing dye-labelled RAFT agents, thisknow-how has to be considered in order to maintain thestructural features that will enable an efficient control ofthe polymerization [117,118].
In Scheme 11, the key steps of the RAFT mechanism arepresented, also depicting the general structure of a RAFTagent [ZC( S)SR]. During the conversion of the initial RAFTagent into a macroRAFT agent, both pre-equilibrium andmain equilibrium coexist. The pre-equilibrium is criticalfor a successful control of the RAFT polymerization and isstrongly influenced by the structure of the RAFT agent (Rand Z groups) [117]. Once all the RAFT agent molecules areconsumed, only the main equilibrium (ruled by the Z group)is present.
According to the RAFT mechanism, both groups, Z and R,will be present in the final dormant polymer chains. Hence,it is possible to synthesize �-, �- or �,�-dye-labelled poly-mer chains by the introduction of a dye respectively in theR, Z or R and Z groups of the initial RAFT agent. A similar dyeincorporation yield is expected for both types of modifica-
tion. However, in the case of a modification on the Z group,the resulting �-dye-labelled polymer chains have a lim-ited stability since the thiocarbonylthio end-group can beeasily degraded (by hydrolysis, nucleophilic attack or evena radical mechanism) inducing loss of the dye. Therefore,Science 36 (2011) 568–602 585
introduction of the dye in the R group of the RAFT agent ismore appropriate.
In order to have a high dye incorporation yield, the ini-tial RAFT agent has to be totally consumed and the majorityof the chains must be initiated by R•, i.e. only a few chainsmust be initiated by primary radicals arising from the ini-tiator. Hence, the ratio between initial concentration ofRAFT agent ([CTA]0) and the total concentration of radicalsarising from initiator (during all polymerization time) hasto be sufficiently high. These conditions are also requiredfor dye-labelled RAFT agents modified on the Z group, sinceit is expected that the number of dormant chains is equalto the number of chains initiated by R• (only consideringtermination by disproportionation).
The next section reviews the methodologies employedfor the synthesis of dye-labelled RAFT chain transfer agentstogether with the dye-labelled polymers that have beensynthesized so far by this approach.
5.1. ˛-end-labelled polymers: modification on the Rgroup
The R group corresponds to the initiating fragment aris-ing from the RAFT agent. Then, with the exception of afew chains that are initiated by primary radicals, all otherchains will bear the R group at their �-end. Consequently,dye-labelling at the R group can provide a high dye incor-poration yield.
Three synthesis strategies can be envisaged to pre-pare dye-labelled RAFT agents modified on the R group(Scheme 12):
. Linear approach: according to this strategy, the dyeundergoes a functionalization step followed by a reac-tion that leads to the formation of the thiocarbonylthiomoiety.
B. Convergent approach: a reactive functional group isintroduced in the dye molecule and, correspondingly, aRAFT agent is modified with a complementary functionto give a precursor RAFT agent. Then, a reaction betweenthe two functionalized molecules is carried out.
C. Radical approach: the dye is transformed into a monomerand undergoes a radical reaction with a RAFT agent (car-ried out with a 1:1 monomer/CTA ratio) in the presenceof an initiator that corresponds to the addition step ofthe RAFT polymerization mechanism.
Usually, the convergent method gives higher reactionyields than the linear procedure. Nevertheless, in thedesign of a dye-labelled RAFT agent, as the number of syn-thetic steps is generally small, the differences in yields arerelatively low. Some functionalized dyes are commerciallyavailable, making it possible to skip the first step for allthree methods. All of these methodologies have alreadybeen employed in the synthesis of dye-labelled RAFT agentsand will be discussed below.
5.1.1. Linear approachIn the linear approach, the introduction of the thiocar-
bonylthio group is carried out at the end of the syntheticpathway. As this group is very sensitive to both oxidation

586M
.Beijaet
al./Progressin
Polymer
Science36 (2011) 568–602
Table 2Dye-labelled initiators used in anionic, NMP and ATRP polymerizations.
Polymerizationmethod
Dye (�maxabs
; �maxem )* Dye-labelled initiator Initiator
synthesisyield (%)
Synthesized polymer Initiator efficiency or% dye incorporation
Ref.
Anionic
Naph (276 nm; 322 nm) – Naph-PS n.d. [91]
Ant (258 nm; 393 nm)n.i. Ant-PMMA n.d. [92]
85PMMA-Ant-PnBAPMMA-Ant-PEHA
91–100[93]
PtBA-Ant-PMMA 83
Phe (295 nm; 350 nm) 84 PMMA-Phe-PnBAPMMA-Phe-PEHAPMMA-Phe-PEA
95–100
NMPPerDI (490 nm; 537 nm) n.i. PerDI-(PS, PnBA, PB, PI) 99–100 [94–95]
PerDI (490 nm; 537 nm) n.i. PerDI-(PS, PnBA, PB, PI) 55–100 [94–95]
PerDI (490 nm; 537 nm) 82 PerDI-(PS, polyacrylates, PVTPA) n.d. [96]

M.Beija
etal./Progress
inPolym
erScience
36 (2011) 568–602587
Table 2 (Continued )
Polymerizationmethod
Dye (�maxabs
; �maxem )* Dye-labelled initiator Initiator
synthesisyield (%)
Synthesized polymer Initiator efficiency or% dye incorporation
Ref.
NMPPy (335 nm; 378 nm)
84 Py-PSPy-PtBAPS-PyPtBA-Py
95–100 [97]
57 81–100
Dansyl (337 nm; 492 nm)79 Dansyl-PS
Dansyl-PtBAPS-DansylPtBA-Dansyl
97–100 [97]
69 –
Py (335 nm; 378 nm) 100 PS-Py 33–81 [98]
Naph (276 nm; 322 nm) 86 PS-Naph – [101]
BTX (452 nm; 508 nm) n.i. PS-BTX 37–80 [99–100]

588M
.Beijaet
al./Progressin
Polymer
Science36 (2011) 568–602
Py (335 nm; 378 nm) 87 Py-PNIPAM n.d. [104]
ATRP Fluo (490 nm; 515 nm) 72 Fluo-PNIPAM n.d. [105]
Py (335 nm; 378 nm) 52 Py-PDMAEMA n.d. [106]
Np (410 nm; 505 nm) 72 Np-PDMAEMA n.d. [107]
Spi (350–400 nm; 650 nm) 85 Spi-PMMA n.d. [109]
NaphPyr (490 nm; n.a.) 78 NaphPyr-PMMANaphPyr-PEHANaphPyr-PCEM
n.d. [110]
Naph (276 nm; 322 nm) 43 PMMA-Naph-PMMA n.d. [112]
Ant (358 nm; 396 nm) Quantitative PS-Ant-Ant-PSPnBA-Ant-Ant-PnBA
20–85 [113–114]
a: depending on polymerization time; n.a.: not applicable; n.i.: not indicated; n.d.: not determined. Ant: anthracene; BTX: benzothioxanthene; Fluo: fluorescein; Naph: naphthalene; NaphPyr: naphtopyran; Np:naphthalimide; PB: poly(butadiene); PCEM: poly[2-(9-carbazol-9-yl)ethyl methacrylate]; PDMAEMA: poly[(2-dimethylaminoethyl)methacrylate]; PEA: poly(ethyl acrylate); PEHA: poly(2-ethylhexyl acrylate);PerDI: perylene diimide; Phe: phenanthrene; PI: polyisoprene; PMMA: poly(methyl methacrylate); PnBA: poly(n-butyl acrylate); PNIPAM: poly(N-isopropyl acrylamide); PS: polystyrene; PtBA: poly(tert-butylacrylate); PVTPA: poly(4-vinyltriphenylamine); Py: pyrene; Spi: spirooxazine.
* Approximate values.

M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602 589
orrespo
Scheme 11. Key steps of the RAFT mechanism. Pn• cand nucleophilic attack, degradation of the RAFT agent canbe avoided (or highly limited).
One of the investigated strategies is based on asingle-step procedure for the preparation of substituteddithioesters by reaction of carboxylic acids with alcoholsin the presence of phosphorus pentasulfide (P2S5 or P4S10in its dimeric form) [119]. In particular, dyes from thepolycyclic aromatic hydrocarbon class have been exten-sively employed to prepare anthracene [120] and pyrene[121] modified dithiobenzoates following the synthetic
Scheme 13:This method presents several disadvantages. First, it hasto be carried out in solvents such as toluene or CCl4 athigh temperatures (110 ◦C) for several hours (20–24 h). Inaddition, low yields are usually obtained (21% yield in the
Scheme 12. Synthesis strategies for int
nds to a propagating chain (oligo- or macroradical).
case of pyrenylmethyl dithiobenzoate). For the anthracenederivative, the reaction yield was not quoted.
Recently, the same authors reported the synthesis ofanthracene and naphthalene modified symmetric trithio-carbonates [122]. These dye-labelled RAFT agents wereprepared with yields higher than 92% by reaction of achloromethylderivative of the dye and CS2 in the presenceof a phase transfer agent in basic media (Scheme 14).
These four polycyclic aromatic hydrocarbon derivativeswere used in the RAFT polymerization of styrene. Polymer
chains with Mn ranging from 5000 to 20,000 g mol−1 andPDI < 1.3 were obtained. Since the dye-labelled trithiocar-bonates were symmetric, �,�-dye-labelled polymer chainswere formed. If the trithiocarbonate group was decom-posed by hydrolysis, aminolysis or other mechanism,roducing a dye on the R group.

590 M. Beija et al. / Progress in Polymer
Scheme 13. Synthesis of pyrene- and anthracene-labelled dithioben-zoates.
Sa
�Mavbcc
haabtpcats
ia2
containing an array of energy donors and a few (most com-
cheme 14. Synthesis of symmetric anthracene-labelled trithiocarbon-te.
-dye-labelled-�-thiol-derived chains with half of initialn were afforded. The authors determined the percent-
ge of dye incorporation by comparison between the Mn
alues obtained by 1H NMR, SEC or UV–Vis spectroscopy,eing usually higher than 76%. The living character of thehains was confirmed either by chain extension or by blockopolymerization with methyl acrylate (MA).
Another methodology involves the formation of aalide-derivative of the dye and further reaction with anlkyl (or aryl) dithiocarboxylic or trithiocarbonic acid (or,lternatively, the respective anion). Usually, these thiocar-onylthio acids are prepared in situ, immediately beforehe reaction with the halide-derived dye. Following thisrocedure, Such et al. prepared a photochromic trithio-arbonate from a spirooxazine bearing a hydroxyl moietyiming to develop new photochromic materials for oph-almic lenses [123]. The synthesis was carried out in twoteps (Scheme 15) with an overall yield of 81%.
This spirooxazine-labelled trithiocarbonate was usedn the preparation of diblock copolymers consisting of PSnd PnBA with molecular weights ranging from 3000 to8,000 g mol−1 and very low polydispersities (PDI < 1.18).
Scheme 15. Synthesis of a spirooxazi
Scheme 16. Synthesis of naphthalenesu
Science 36 (2011) 568–602
A dye incorporation of ca. 98% is expected considering thepolymerization conditions.
Using the same strategy, Mertoglu et al. synthesized adithiobenzoate bearing a naphthalenesulfonate dye [124].In a first step, a bromo-derivative was prepared in a het-erogeneous reaction mixture, followed by a reaction with adithiobenzoate salt (Scheme 16). After purification by frac-tionate precipitation, 55% of final product was obtained.This moderate yield is due to loss of product in the purifica-tion steps (and not during synthesis). Since this RAFT agentis an ionic aromatic compound (exhibiting an amphiphiliccharacter), the purification is indeed challenging.
This naphthalenesulfonate-labelled RAFT agent was fur-ther used to polymerize a series of water-soluble acrylate,acrylamide or styrenic monomers. The study was focusedon the evaluation of polymerization control as well as onkinetics. The dye incorporation was not determined, how-ever a yield higher than 94% is expected according to thepolymerization conditions.
5.1.2. Convergent approachThis strategy usually involves the synthesis of a car-
boxylic acid derived thiocarbonylthio compound thatreacts with an alcohol or amine dye-derivative. However,due to the low reactivity of the carboxylic acid functionalgroup, an activation step must be performed.
Ghigghino and co-workers prepared RAFT agentscarrying an anthracene [4] or Ru(II) polypyri-dine [5,24] chromophore by reaction of therespective hydroxyl-derivatives with 4-cyano-4-[(thiobenzoyl)sulfanyl]-pentanoic acid [125] in thepresence of DCC as coupling agent and DMAP as catalyst(Scheme 17). The reaction yield for the anthracene deriva-tive was not given while the Ru(II) polypyridine (RuPP)chromophore-RAFT agent was obtained with a 38% yield.
These dye-labelled RAFT agents were synthesized in thescope of a major work on “light-harvesting” polymers. A“light-harvesting” polymer comprises a polymer backbone
monly, a single one) energy acceptors usually present atthe polymer chain-end. The main purpose of such mate-rials is to mimic the natural photosynthetic process. Theycan be easily prepared by polymerization of a monomer
ne-labelled trithiocarbonate.
lfonate-labelled dithiobenzoate.

M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602 591
hiobenz
Scheme 17. Synthesis of dye-labelled ditcontaining an energy donor dye in the presence of a RAFTagent labelled with an energy acceptor dye. Hence, thesedithiobenzoates containing anthracene [4] and RuPP [5,24]were employed to control the polymerization of acenaph-thylene (AcN). Moreover, the RuPP-labelled RAFT agentwas used to control the polymerization of a coumarin-labelled styrene derivative, forming a star polymer that wasfurther used as macroRAFT agent to mediate the polymer-ization of AcN then NIPAM, yielding a star-shaped triblockcopolymer [5,24]. Although the experimental dye incorpo-ration yield was not determined, it is expected to be high(>80%) considering the chosen polymerization conditions.
Very recently, Liu et al. reported the synthesis ofa pyrene-labelled trithiocarbonate in two condensationsteps using DCC as coupling agent and DMAP as cat-alyst. First, a hydroxyl-terminated trithiocarbonate wasprepared (reaction of an acid-functionalized trithiocar-
oates by in situ carboxylic acid activation.
bonate with hydroxyl ethyl disulfide) and reacted withpyrene butyric acid (64% overall yield). Py-PNIPAM chainswere produced in the presence of the resulting RAFT agentand were subsequently used to prepare graphene–PNIPAMcomposites via �–� stacking interactions [126].
The main problem if one wish to use a similar strat-egy to prepare dye-labelled polymers for uses in aqueousmedia is the presence of an ester linkage that can be eas-ily hydrolyzed. An amide linkage is much more robustand should be preferred. However, when a primary aminereacts with a carboxylic acid derivative under these con-ditions, the dithioester moiety simultaneously undergoes
nucleophilic attack, leading to a (labelled) thioamide anda thiol [127]. In fact, dithioesters react much faster thanesters with primary and secondary aliphatic amines [128].To avoid this limitation, we proposed a different syn-thetic pathway based on a precursor RAFT agent bearing

592 M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602
nanthre
atcoaaldUyer
sprp
cwputPhsoaF
tLGtitdas
5
npa
Scheme 18. Synthesis of phe
succinimidyl activated ester group [74]. It was shownhat during the reaction of amino derivatives with this pre-ursor, no degradation of the thiocarbonylthio moiety wasbserved as long as the amino derivative/precursor RAFTgent molar ratio remained lower than unity. The maindvantages of this method are the formation of an amideinkage and the possibility of preparing a family of differentye-labelled RAFT agents from a single precursor molecule.sually, total conversion of the amine is observed and theields reach 65% after purification. In addition, this simplexperimental procedure is rapid and can be performed atoom temperature.
Such a method was first employed for the synthe-is of a phenanthrene-labelled dithiobenzoate [129]. Areviously prepared amino-derived phenanthrene [130]eacted with the precursor dithiobenzoate, yielding 65% ofhenanthrene-labelled RAFT agent (Scheme 18).
This phenanthrene-labelled dithiobenzoate was suc-essfully used to polymerize N-decylacrylamide (DcA)ith an expected dye incorporation of 91% from theolymerization conditions. The obtained polymer wassed as macroRAFT agent to control the polymeriza-ion of N,N-diethylacrylamide (DEA), affording a series ofDcA-block-PDEA block copolymers consisting of a highlyydrophobic block (11 units of DcA) and a thermorespon-ive block of PDEA with variable length (96, 180, 250, 390r 600 units) [129]. The formation of micelles in waternd their thermoresponsive behaviour has been studied byRET and light scattering [131].
Recently, the same procedure was extended to syn-hesize RAFT agents bearing water-soluble dyes, such asucifer Yellow (LY), Rhodamine B (Rho B) and Malachitereen (MG). Total conversion of the amino derivatives of
he dyes was observed. However, the purification of theseonic aromatic compounds appeared very challenging andime-consuming. In addition, the use of a secondary aminoerivative of Rho B led to a simultaneous nucleophilicttack at the thiocarbonyl centre, forming a thioamide aside product [75].
.1.3. Radical approachA third strategy takes advantage of the RAFT mecha-
ism. Indeed, a “RAFT polymerization-like” reaction can beerformed between a dye-labelled monomer and a RAFTgent, using a monomer to CTA molar ratio of 1:1 and a
ne-labelled dithiobenzoate.
small amount (1–2% molar equivalent) of a free radicalinitiator. Hence, RAFT agents carrying a coumarin [23], adiphenylanthracene [132], and an acenaphtene [133] dyeswere successfully synthesized in Ghiggino’s group with85%, 65% and 83% yields, respectively (Scheme 19).
This approach is very efficient and produces a very sta-ble C–C bond between the dye and the thiocarbonylthiomoiety. However, this procedure requires relatively hightemperatures and long reaction times (24 h) as well as thesynthesis of an alkenyl derivative of the dye which may bea very challenging step.
Such dye-labelled RAFT agents were also synthesized byGhiggino and co-workers for preparing “light-harvesting”polymers. In most cases, they were employed to controlthe polymerization of acenaphthylene. Moreover, statisti-cal and block copolymers were synthesized using acrylicacid (AA) [23] or maleic anhydride (MaAN) [132] ascomonomers. Alternatively, an oligomer of AcN was pre-pared by RAFT polymerization using a chain transfer agentbearing an acenaphthyl moiety in order to perform chaindynamics studies using the kinetics of excimer formation[133].
5.1.4. Conclusions on labelling at the R groupWhen synthesizing dye-labelled RAFT agents by modi-
fication on the R group, it is desirable that a strong link isestablished between the thiocarbonylthio moiety and thedye, such as a C–C bond or an amide bond. Thus, a carefulchoice of reactants and synthesis method must be made,and severe experimental conditions should be avoided.
5.2. �-end-labelled polymers: modification on the Zgroup
Although the Z group can be considered relatively labiledue to the sensitivity of the thiocarbonylthio moiety tonucleophilic addition, several examples of RAFT agentsbearing a dye on the Z group are described in the liter-ature. This is probably justified by the simplicity of their
synthesis. In all cases, the dye is directly linked to the thio-carbonylthio group, i.e. there is no spacer between thesetwo moieties. Thus, only carbazole and polycyclic aromatichydrocarbon (PAH) derived dyes have been used leadingrespectively to dithiocarbamates and dithioesters.
M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602 593
lled dith
Scheme 19. Synthesis of dye-labe5.2.1. DithiocarbamatesIn the dithiocarbamate family, mostly carbazole-
derived RAFT agents bearing different R groups, such asbenzyl [134], ethyl propionate [135], ethyl isobutyrate[136] have been synthesized motivated by the strong car-bazole fluorescence (exclusively in Zhu’s research group).They have been prepared through the same syntheticpathway. Using a basic catalysis, a nucleophilic attack ofcarbazole to CS2 occurs and the product reacts with an �-bromo-alkyl derivative at 0 ◦C to form the desired RAFTagent (Scheme 20). A double-labelled RAFT agent compris-ing a carbazole dye on the Z group and an azobenzenephotochromic dye on the R group was also synthesized bythis same procedure [136].
Alternatively, indole and phenylazo indazole-labelleddithiocarbamates were prepared with yields around20–40% and 51%, respectively [135,137]. These dye-labelled dithiocarbamates were used to control thepolymerization of styrene [134–136], MMA [134,136] or
acrylates (MA [136], nBA, n-octylacrylate [134]) with adye incorporation yield higher than 87%. In the case ofthe double-labelled dithiocarbamate, polymers with dif-ferent dyes at the �- and �-chain-ends were obtained[136].Scheme 20. Synthetic route for synthesis
iobenzoates via a radical reaction.
5.2.2. DithioestersThe methods used for the synthesis of dye-labelled
dithioesters modified on the Z group are more varied thanfor dithiocarbamates. Using the synthetic route depictedin Scheme 13, an anthracene-derived RAFT agent was pre-pared from anthracene-9-carboxylic acid and benzyl thiol[138].
Another current strategy involves a process previ-ously reported by Thang et al. [125], where a solu-tion of the appropriate bis(thiocarbonyl) disulfide isheated with 1.5 eq. of an azo compound (a radicalinitiator) in the absence of oxygen. Hence, 2-cyanoprop-2-yl 1-dithionaphthalate [139] and 2-cyanoprop-2-yl1-dithiophenanthrenate [140] were successfully synthe-sized with 40% and 33% yields, respectively (Scheme 21).
Another naphthalene-derived RAFT agent was also syn-thesized using a method similar to the one depicted inScheme 16. Accordingly, a reaction of 2-dithionaphtalateanion with 2-bromo-2-phenylacetonitrile gave the desired
product (yield: 63%), as shown in Scheme 22 [141,142].The anthracene-labelled dithioester was employed forthe RAFT polymerization of styrene (dye incorporation ofca. 75%) [138], the phenanthrene-labelled (dye incorpo-ration of 94%) [140] and the naphthalene-labelled [139]
of carbazole-derived RAFT agents.

594M
.Beijaet
al./Progressin
Polymer
Science36 (2011) 568–602
Table 3Dye-labelled RAFT agents modified either on R or Z group.
Modification on Dye (�maxabs
; �maxem )* Dye-labelled RAFT chain transfer agent R-S C( S) Z Synthesis
strategyCTA synthesisyield (%)
Synthesized polymer % dyeincorporation
Ref.
R
Py (335 nm; 378 nm)1 21 Py-PS
Py-PS-block-PMA80–90 [121]
c 64 Py-PNIPAM >93 [126]
Naph (276 nm; 322 nm) 1 90 Naph-PS-Naph 76 [122]
Ant (258 nm; 393 nm)1 92 Ant-PS-Ant 82–83
1 n.d. Ant-PS 96 [120]
Spi (605 nm; n.a.) 1 81 Spi-PnBA-block-PSSpi-PS-block- PnBA
98 [123]
NaphSulf (251 nm; 350nm)
l 55 NaphSulf-(PEGMEA;PAETMAC; PDMAPA;PVBTMA; PAPS; PVPPS;PSSS)
>94 [124]
Ant (258 nm; 393 nm) c n.d. Ant-PAcN 91 [4]

M.Beija
etal./Progress
inPolym
erScience
36 (2011) 568–602595
Diff34Table 3 (Continued )
Modification on Dye (�maxabs
; �maxem )* Dye-labelled RAFT chain transfer agent R-S C( S) Z Synthesis
strategyCTA synthesisyield (%)
Synthesized polymer % dyeincorporation
Ref.
RPhe (295 nm; 350 nm) c 65 Phe-PDcA
Phe-PDcA-block-PDEA91 [129]
LY (428 nm; 544 nm) c 47 – – [75]
RhoB (553 nm; 572 nm) c,l 31–58 Rho B-PDMARhoB-PDMA-block-PDEA
>95
MG (620 nm; n.a.) c n.d. MG-PDMAMG-PDMA-block-PDEA
37–88
RnPP (300 nm; n.a.) c 38 RuPP-PSt-CouRuPP-PSt-Cou-block-PAcNRuPP-PSt-Cou-block-PAcN-block-PNIPAM
70–95 [5,24]
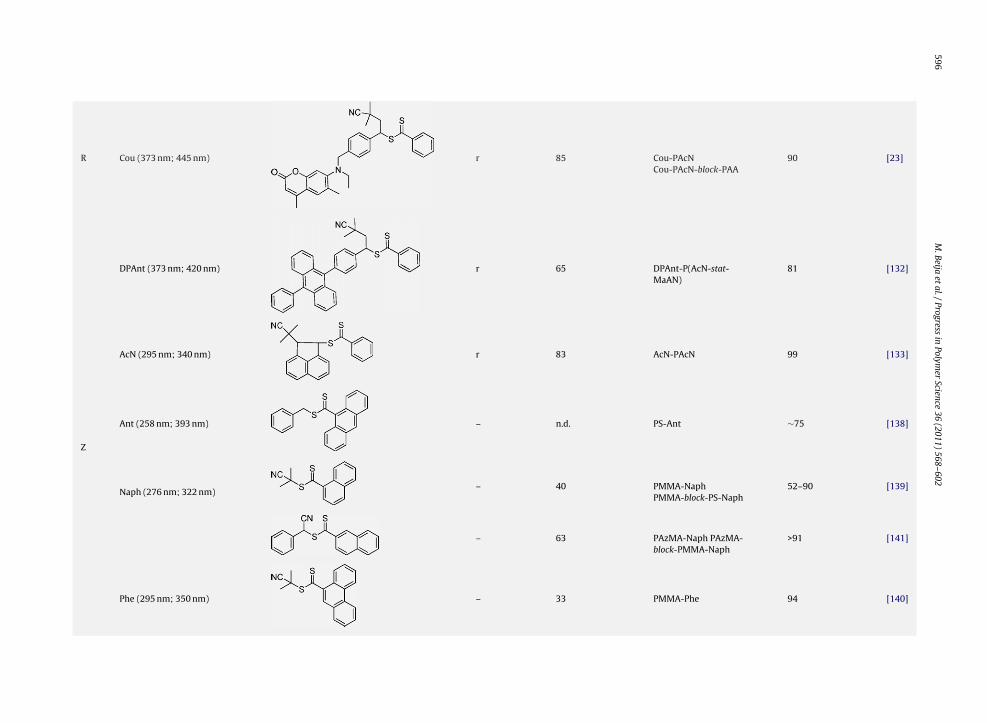
596M
.Beijaet
al./Progressin
Polymer
Science36 (2011) 568–602
R Cou (373 nm; 445 nm) r 85 Cou-PAcNCou-PAcN-block-PAA
90 [23]
DPAnt (373 nm; 420 nm) r 65 DPAnt-P(AcN-stat-MaAN)
81 [132]
AcN (295 nm; 340 nm) r 83 AcN-PAcN 99 [133]
Z
Ant (258 nm; 393 nm) – n.d. PS-Ant ∼75 [138]
Naph (276 nm; 322 nm)– 40 PMMA-Naph
PMMA-block-PS-Naph52–90 [139]
– 63 PAzMA-Naph PAzMA-block-PMMA-Naph
>91 [141]
Phe (295 nm; 350 nm) – 33 PMMA-Phe 94 [140]

M.Beija
etal./Progress
inPolym
erScience
36 (2011) 568–602597
Table 3 (Continued )
Modification on Dye (�maxabs
; �maxem )* Dye-labelled RAFT chain transfer agent R-S C( S) Z Synthesis
strategyCTA synthesisyield (%)
Synthesized polymer % dyeincorporation
Ref.
Carb (294 nm; 344 nm) – 20 PS-Carb; PMA-Carb;PnBA-Carb; POA-Carb
n.d. [134]
ZCarb (294 nm; 344 nm)
– 40 PMMA-Carb n.d. [136]
– n.d. PS-Carb 91–93 [135]
Ind (356 nm; 418 nm) – n.d. PS-Ind 88–90
PhAzInd (320 nm; 390 nm) – 51 PS-PhAzInd 79–89 [137]
R and Z AzB (319 nm; n.a.) andCarb (294 nm; 344 nm)
– 21 AzB-PS-Carb;AzB-PMA-Carb;AzB-PMMA-CarbAzB-PS-block-PMMA-Carb
∼97 [136]
n.a.: not applicable; n.d.: not determined; l: linear, c: convergent; r: radical; AcN: acenaphthyl; Ant: anthracene; AzB: azobenzene; Carb: Carbazole; Cou: coumarin 1; DPAnt: diphenylanthracene;Ind: indole; LY: Lucifer Yellow; Naph: naphthalene; NaphSulf: naphthalene sulfonate; PAA: poly(acrylic acid); PAcN: poly(acenaphthylene); PAETMAC: poly[(2-acryloxyethyl)trimethylammoniumchloride]; PAPS: poly[3-(acryloyloxy)propanesulfonate potassium salt]; PAzMA: poly(2-azidoethyl methacrylate); PDcA: poly(N-decylacrylamide); PDEA: poly(N,N-diethylacrylamide); PDMAPA: poly[N-(3-(dimethylaminopropyl)acrylamide]; PEGMEA: poly[(ethylene glycol) methyl ether acrylate]; PhAzoInd: phenyl azo indazole; Phe: phenanthrene; PMA: poly(methyl acrylate); PMaAN: poly(maleic anhydride);PMMA: poly(methyl methacrylate); PnBA: poly(n-butyl acrylate); POA: poly(octylacrylate); PS: polystyrene; PSSS: poly(sodium styrene sulfonate); PSt-Cou: poly(coumarin-derived styrene); PVBTMA:poly(vinylbenzyltrimethylammonium); PVPPS: poly[3-(2-vinylpyridinio)propanesulfonate]; Py: pyrene.
* Approximate values.

598 M. Beija et al. / Progress in Polymer Science 36 (2011) 568–602
Scheme 21. Synthesis of naphthalene- and
dnrohgAlp7
5
onttortfHytfmtmpc
5a
lRtip
Scheme 22. Synthesis of naphthalene-labelled dithioester.
ithioesters for the RAFT polymerization of MMA. The otheraphthalene-labelled dithioester was used for the prepa-ation of a “clickable” polymer by RAFT polymerizationf 2-(azidoethyl methacrylate) (dye incorporation yieldigher than 90%) [141]. The dye incorporation yield wasenerally estimated from the polymerization conditions.n extensive study was performed with the naphthalene-
abelled dithioester prepared by Zhu et al. [139] to produceolymers with different dye incorporation yields (between0% and 95%), depending on the polymerization conditions.
.2.3. Conclusions on labelling at the Z groupOnly a few methods have been used for the preparation
f RAFT agents labelled on the Z group. In most cases, aucleophilic addition of a dye derivative (bearing a nega-ive charge) on CS2 is carried out, then a further reactionakes place, such as a reaction with a bromo-derivativer the formation of disulfide followed by reaction with aadical initiator. The dye anion is obtained either by depro-onation of an amine (in the case of carbazoles) or byormation of a Grignard reagent (in the case of dithioesters).owever, these methods are not very efficient since lowields are generally achieved (<40%). Moreover, the syn-hesis of a RAFT agent labelled on the Z group is not aavorable strategy in order to prepare dye-labelled poly-
ers since heterolytic or homolytic degradation of thehiocarbonylthio moiety may occur, resulting in a detach-
ent of the dye from the polymer chain and the nearroximity of the thiocarbonylthio group to the dye mayause a severe fluorescence quenching.
.3. Conclusions on dye-labelled RAFT chain transfergents
Table 3 summarizes the molecular structures of dye-
abelled RAFT agents synthesized so far by modification onor Z group with the corresponding polymers. The syn-hesis yields vary dramatically, not only due to differencesn dye derivative reactivity but also to the used syntheticrocedures. If the polymerization conditions are carefully
phenanthrene-labelled dithioesters.
chosen, very high dye incorporation yields can be achieved(usually higher than 90%). However, it is not only importantto ensure a high dye incorporation yield but also to limitthe risks of dye loss during polymer synthesis and purifica-tion steps. Then, the link between the dye and the polymerchain has to be sufficiently robust for the aimed application.�-end-labelled polymers prepared from a Z-modified RAFTagent are very sensitive since the thiocarbonylthio functioncan be easily degraded by hydrolysis, nucleophilic attackor radical mechanisms. Similarly, in the case of �-end-labelled polymers, it may be preferable to have a stronglink, such as a C–C or amide bond between the R groupand the polymer (which is observed for the majority of theRAFT agents depicted in Table 3), especially for studies inaqueous media.
6. Conclusions
A dye-tag can be introduced at a precise site of a polymerchain before, during or after the polymerization process.Respectively, it requires the synthesis of a dye-labelled ini-tiator or CTA, the use of a dye-labelled (co)monomer orquencher and the use of a post-modification strategy.
The choice of one of these approaches relies on sev-eral parameters that include: the kind of monomer(s) to bepolymerized, the target molecular weight, the chosen dye,its reactivity and ability to be functionalized, and the poten-tial applications of the dye-labelled polymer. For instance,if the potential application does not require labelling of allthe chains, a simple synthesis method providing moderatelabelling yield may be chosen.
In many cases, the resulting linkages between the poly-mer chain and the dye are sensitive to hydrolysis (ester,dithioester) or easily exchangeable (disulfide exchange inthe presence of thiols). Consequently, the use of these poly-mers in aqueous solutions (especially at high pH values)or in biological media (where thiols are often present) islimited, since loss of dyes from polymer chain-ends couldeasily occur.
All the living/controlled polymerization techniques canbe employed to prepare dye-labelled polymers. The choiceof a specific technique mainly depends on the chosen
monomer(s). Nonetheless, the aimed application is also ofextreme importance for settling on a given strategy, sincesome functional groups or additive may promote severedetrimental effects. For the NMP process, alkoxyaminesbearing the dye at the initiating fragment rather than at
olymer
M. Beija et al. / Progress in Pthe nitroxide fragment should be preferred, though the useof high temperatures may be a limitation in some cases.For the ATRP process and some click chemistry reactions,the presence of residual metal may have an influence onthe dye fluorescence (quenching due to the heavy atomeffect). However, some recent procedures provide a rela-tively easy removal of the metal residues to a satisfyinglow level, at least for non in vivo biological applications.Concerning dye-labelled polymers prepared via the RAFTprocess, in the case of a dithiobenzoate �-end-group, areaction of elimination will be required before performingfluorescence studies.
Dyes from the polycyclic aromatic hydrocarbon family(such as pyrene, anthracene, phenantrene, naphthalene)are, so far, most often employed to end-label polymerchains. The main reason is probably the ready availabil-ity of functionalized PAH that can react with several otherfunctional groups. Moreover, their photophysical proper-ties are well known and, in the case of pyrene, the formationof excimers can provide important information about thepolymer conformation and dynamics. However, these dyesare highly hydrophobic, consequently, they can aggregateand induce changes in polymer properties, especially inaqueous solvents. There is a lack of polymers modified withwater-soluble dyes and only a few examples can be foundin the literature (with fluorescein and rhodamine deriva-tives). Since these molecules are simultaneously aromaticand hydrophilic (sometimes ionic), several problems areencountered during their synthesis and especially theirpurification. However, they could be very useful for manyapplications involving water-soluble polymers.
On the other hand, the majority of dye-labelled poly-mers that have been reported so far absorbs in theultraviolet region of electromagnetic spectrum. Neverthe-less, dye-labelled polymers absorbing in the visible ornear-infrared (NIR) are required, namely for biomedicalapplications and fluorescence imaging.
References
[1] Callahan J, Kopecek J. Semitelechelic HPMA copolymers function-alized with triphenylphosphonium as drug carriers for membranetransduction and mitochondrial localization. Biomacromolecules2006;7:2347–56.
[2] Kim K, Lee M, Park H, Kim JH, Kim S, Chung H, et al. Cell-permeableand biocompatible polymeric nanoparticles for apoptosis imaging.J Am Chem Soc 2006;128:3490–1.
[3] Charreyre MT, Mandrand B, Martinho JMG, Relógio P, Farinha JPS.Fluorescent polymers soluble in an aqueous solution and a methodfor the production thereof. WO2007003781, 2007.
[4] Chen M, Ghiggino KP, Mau AWH, Rizzardo E, Thang SH, WilsonGJ. Synthesis of light harvesting polymers by RAFT methods. ChemCommun 2002:2276–7.
[5] Chen M, Ghiggino KP, Thang SH, Wilson GJ. Star-shaped light-harvesting polymers incorporating an energy cascade. AngewChem Int Ed 2005;44:4368–72.
[6] Ercole F, Davis TP, Evans RA. Comprehensive modulation of naph-thopyran photochromism in a rigid host matrix by applyingpolymer conjugation. Macromolecules 2009;42:1500–11.
[7] Evans RA, Such GK. Research trends in photochromism: Control ofphotochromism in rigid polymer matrices and other advances. Aust
J Chem 2005;58:825–30.[8] Such GK, Evans RA, Davis TP. Rapid photochromic switching in arigid polymer matrix using living radical polymerization. Macro-molecules 2006;39:1391–6.
[9] Ercole F, Malic N, Harrisson S, Davis TP, Evans RA. Photochromicpolymer conjugates: the importance of macromolecular archi-
Science 36 (2011) 568–602 599
tecture in controlling switching speed within a polymer matrix.Macromolecules 2010;43:249–61.
[10] Relógio P, Martinho JMG, Farinha JPS. Effect of surfactanton the intra- and intermolecular association of hydrophobi-cally modified poly(N,N-dimethylacrylamide). Macromolecules2005;38:10799–811.
[11] Duhamel J. Polymer chain dynamics in solution probed with a flu-orescence blob model. Acc Chem Res 2006;39:953–60.
[12] Turshatov A, Adams J, Johannsmann D. Interparticle contact in dry-ing polymer dispersions probed by time resolved fluorescence.Macromolecules 2008;41:5365–72.
[13] Farinha JPS, Martinho JMG, Yekta A, Winnik MA. Direct nonradiativeenergy transfer in polymer interphases: fluorescence decay func-tions from concentration profiles generated by Fickian diffusion.Macromolecules 1995;28:6084–8.
[14] Winnik MA. Latex film formation. Curr Opin Colloid Interface Sci1997;2:192–9.
[15] Farinha JPS, Martinho JMG. Resonance energy transfer in polymernanodomains. J Phys Chem C 2008;112:10591–601.
[16] Winnik MA, Redpath T, Richards DH. The dynamics of end-to-endcyclization in polystyrene probed by pyrene excimer formation.Macromolecules 1980;13:328–35.
[17] Winnik MA. End-to-end cyclization of polymer chains. Acc ChemRes 1985;18:73–9.
[18] Sousa ATRE, Castanheira EMS, Fedorov A, Martinho JMG.Polystyrene cyclization using pyrene excimer formation. Effect ofgerminate pairsin good solvents. J Phys Chem A 1998;102:6406–11.
[19] Martinho JMG, Sienicki K, Blue D, Winnik MA. Transient effects indiffusion controlled reactions. 6. Intermolecular pyrene excimerformation in end-labelled polystyrene of MW 7800. The role of themutual diffusion coefficient. J Am Chem Soc 1988;110:7773–7.
[20] Stukelj M, Martinho JMG, Winnik MA, Quirk RP. Intermolecularexcimer formation for pyrene-end-capped polystyrene: a modelfor the termination process in free radical polymerization. Macro-molecules 1991;24:2488–92.
[21] Liu J, Liu H, Boyer C, Bulmus V, Davis TP. Approach to peptide dec-orated micelles via RAFT polymerization. J Polym Sci Part A PolymChem 2009;47:899–912.
[22] Roth PJ, Sim K-S, Bae SH, Sohn BH, Theato P, Zentel R.Hetero-telechelic dye-labeled polymer for nanoparticle decoration.Macromol Rapid Commun 2009;30:1274–8.
[23] Chen M, Ghiggino KP, Mau AWH, Rizzardo E, Sasse WHF, Thang SH,et al. Synthesis of functionalized RAFT agents for light harvestingmacromolecules. Macromolecules 2004;37:5479–81.
[24] Chen M, Ghiggino KP, Launikonis A, Mau AWH, Rizzardo E, SasseWHF, et al. RAFT synthesis of linear and star-shaped light harvest-ing polymers using di- and hexafunctional ruthenium polypyridinereagents. J Mater Chem 2003;13:2696–700.
[25] Ercole F, Davis TP, Evans RA. Photo-responsive systems and bio-materials: photochromic polymers, light-triggered self-assembly,surface modification, fluorescence modulation and beyond. PolymChem 2010;1:37–54.
[26] Viovy JL, Franck CW, Monnerie L. Fluorescence anisotropy decaystudies of local polymer dynamics in the melt. 2. Labeledmodel compounds of variable chain length. Macromolecules1985;18:2606–13.
[27] Viovy JL, Monnerie L, Merola F. Fluorescence anisotropy decay stud-ies of local polymer dynamics in the melt. 1. Labeled polybutadiene.Macromolecules 1985;18:1130–7.
[28] Valeur B. Molecular Fluorescence—Principles and Applications.Weinheim: WileyVCH; 2002.
[29] Farinha JPS, Spiro JG, Winnik MA. Energy transfer in the restrictedgeometry of lamellar block copolymer interfaces. J Phys Chem B2001;105:4879–88.
[30] Fredrickson GH. Intermolecular correlation function from Försterenergy-transfer experiments. Macromolecules 1986;19:441–7.
[31] Yekta A, Spiro JG, Winnik MA. A critical evaluation of direct energytransfer as a tool for analysis of nanoscale morphologies in poly-mers. Application to block copolymer interfaces. J Phys Chem B1998;102:7960–70.
[32] Yang J, Lu JP, Rharbi Y, Cao L, Winnik MA, Zhang YM,et al. Energy transfer study of the interface thickness insymmetrical isoprene–methyl methacrylate diblock copolymers.
Macromolecules 2003;36:4485–91.[33] Kitano H, Akatsuka Y, Ise N. pH-responsive liposomes which con-tain amphiphiles prepared by using lipophilic radical initiator.Macromolecules 1991;24:42–6.
[34] Ganachaud F, Theretz A, Erout MN, Llauro MF, Pichot C. Syn-thesis and characterization of bioreactive-end-group-containing

6 olymer
00 M. Beija et al. / Progress in Pazoinitiators and their use for preparing end-functionalizedpolyvinylpyrrolidone. J Appl Polym Sci 1995;58:1811–24.
[35] Zettl H, Hafner W, Boker A, Schmalz H, Lanzendorfer M, Muller AHE,et al. Fluorescence correlation spectroscopy of single dye-labeledpolymers in organic solvents. Macromolecules 2004;37:1917–20.
[36] Torchilin VP, Levchenko TS, Whiteman KR, Yaroslavov AA, TsatsakisAM, Rizos AK, et al. Amphiphilic poly-N-vinylpyrrolidones: syn-thesis, properties and liposome surface modification. Biomaterials2001;22:3035–44.
[37] Controlled/living radical polymerization: progress in ATRP, NMP,and RAFT.Matyjaszewski K, editor. ACS Symp Ser 768. Washington,DC: American Chemical Society; 2000.
[38] Advances in controlled/living radical polymeriza-tion.Matyjaszewski K, editor. ACS Symp Ser 854. Washington, DC:American Chemical Society; 2003.
[39] Stenzel MH, Davis TP, Fane AG. Honeycomb structured porous filmsprepared from carbohydrate based polymers synthesized via theRAFT process. J Mater Chem 2003;13:2090–7.
[40] Perrier S, Takolpuckdee P, Westwood J, Lewis DM. Versatile chaintransfer agents for reversible addition fragmentation chain transfer(RAFT) polymerization to synthesize functional polymeric architec-tures. Macromolecules 2004;37:2709–17.
[41] Nicolas J, Mantovani G, Haddleton DM. Living radical polymer-ization as a tool for the synthesis of polymer-protein/peptidebioconjugates. Macromol Rapid Commun 2007;28:1083–111.
[42] Barner L, Perrier S. Polymers with well-defined end groups viaRAFT—synthesis, applications and postmodification. In: Barner-Kowollik C, editor. Handbook of RAFT Polymerization. Weinheim:Wiley-VCH; 2008. p. 455–81.
[43] Favier A, De Lambert B, Charreyre MT. Toward new materials pre-pared via the RAFT process: from drug delivery to optoelectronics?In: Barner-Kowollik C, editor. Handbook of RAFT Polymerization.Weinheim: Wiley-VCH; 2008. p. 483–535.
[44] Le Droumaguet B, Nicolas J. Recent advances in the design of bio-conjugates from controlled/living radical polymerization. PolymChem 2010;1:563–98.
[45] Li Y, Schadler LS, Benicewicz BC. Surface and particle modificationvia the RAFT process: approach and properties. In: Barner-KowollikC, editor. Handbook of RAFT Polymerization. Weinheim: Wiley-VCH; 2008. p. 423–53.
[46] Huang Y, Liu Q, Zhou X, Perrier S, Zhao Y. Synthesis of silica particlesgrafted with well-defined living polymeric chains by combinationof RAFT polymerization and coupling reaction. Macromolecules2009;42:5509–17.
[47] Tasis D, Tagmatarchis N, Bianco A, Prato M. Chemistry of carbonnanotubes. Chem Rev 2006;106:1105–36.
[48] Singh P, Campidelli S, Giordani S, Bonifazi D, Biancoa A, PratoM. Organic functionalisation and characterisation of single-walledcarbon nanotubes. Chem Soc Rev 2009;38:2214–30.
[49] Sumerlin BS, Vogt AP. Macromolecular engineering through clickchemistry and other efficient transformations. Macromolecules2009;43:1–13.
[50] Solomon DH, Rizzardo E, Cacioli P. Polymerization process andpolymers produced thereby. US 4581429, 1986.
[51] Georges MK, Veregin RPN, Kazmaier PM, Hamer GK. Narrowmolecular-weight resins by a free-radical polymerization process.Macromolecules 1993;26:2987–8.
[52] Benoit D, Chaplinski V, Braslau R, Hawker CJ. Development of auniversal alkoxyamine for “living” free radical polymerizations. JAm Chem Soc 1999;121:3904–20.
[53] Hawker CJ, Bosman AW, Harth E. New polymer synthesis bynitroxide mediated living radical polymerizations. Chem Rev2001;101:3661–88.
[54] Wang JS, Matyjaszewski K. Contrlled living radicalpolymerization—Atom-Transfer Radical Polymerization inthe presence of transition-metal complexes. J Am Chem Soc1995;117:5614–5.
[55] Kato M, Kamigaito M, Sawamoto M, Higashimura T. Polymer-ization of methyl methacrylate with the carbon-tetrachloridedichlorotris(triphenylphosphine)ruthenium(II) methylaluminumbis(2,6-di-tert-butylphenoxide) initiating system—possibility of
living radical polymerization. Macromolecules 1995;28:1721–3.[56] Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, etal. Living free-radical polymerization by reversible addition-fragmentation chain transfer: the RAFT process. Macromolecules1998;31:5559–62.
Science 36 (2011) 568–602
[57] Moad G, Rizzardo E, Thang SH. Radical addition-fragmentationchemistry in polymer synthesis. Polymer 2008;49:1079–131.
[58] Corpart P, Charmot D, Biadatti T, Zard S, Michelet D. Method ofblock copolymer synthesis by radical controlled polymerisation.WO9858974, 1998.
[59] Beija M, Afonso CAM, Martinho JMG. Synthesis and applicationsof Rhodamine derivatives as fluorescent probes. Chem Soc Rev2009;38:2410–33.
[60] Yamamoto Y, Yasugi K, Harada A, Nagasaki Y, Kataoka K.Temperature-related change in the properties relevant to drugdelivery of poly(ethylene glycol)-poly(d,l-lactide) block copoly-mer micelles in aqueous milieu. J Controlled Release 2002;82:359–71.
[61] Studer P, Larras V, Riess G. Amino end-functionalized poly(ethyleneoxide)-block-poly(methylidene malonate 2.1.2) block copolymers:synthesis, characterization, and chemical modification for target-ing purposes. Eur Polym J 2008;44:1714–21.
[62] Harth E, Hawker CJ, Fan W, Waymouth RM. Chain end functional-ization in nitroxide-mediated “Living” free radical polymerizations.Macromolecules 2001;34:3856–62.
[63] O’Bryan G, Braslau R. Terminal functionalization of polymersvia single electron oxidation of N-alkoxyamines. Macromolecules2006;39:9010–7.
[64] Zhou P, Chen GQ, Hong H, Du FS, Li ZC, Li FM. Synthesis of C-60-end-bonded polymers with designed molecular weights andnarrow molecular weight distributions via atom transfer radicalpolymerization. Macromolecules 2000;33:1948–54.
[65] Yang D, Li L, Wang CC. Characterization and photoconductivitystudy of well-defined C-60 terminated poly(tert-butyl acrylate-b-styrene). Mater Chem Phys 2004;87:114–9.
[66] Wang XF, Zhang YF, Zhu ZY, Liu SY. Fabrication of fullerene-containing hybrid vesicles via supramolecular self-assembly of awell-defined amphiphilic block copolymer incorporated with a Sin-gle C-60 moiety at the diblock junction point. Macromol RapidCommun 2008;29:340–6.
[67] Li C, Hu J, Liu S. Click coupling fullerene onto thermorespon-sive water-soluble diblock copolymer and homopolymer chains atdefined positions. Macromolecules 2009;42:5007–16.
[68] Nakayama M, Okano T. Polymer terminal group effects on prop-erties of thermoresponsive polymeric micelles with controlledouter-shell chain lengths. Biomacromolecules 2005;6:2320–7.
[69] Scales CW, Convertine AJ, McCormick CL. Fluorescent label-ing of RAFT-generated poly(N-isopropylacrylamide) via afacile maleimide-thiol coupling reaction. Biomacromolecules2006;7:1389–92.
[70] York AW, Scales CW, Huang FQ, McCormick CL. Facile syntheticprocedure for omega, primary amine functionalization directly inwater for subsequent fluorescent labeling and potential biocon-jugation of RAFT-synthesized (Co)polymers. Biomacromolecules2007;8:2337–41.
[71] Segui F, Qiu XP, Winnik FM. An efficient synthesis of telechelicpoly(N-isopropylacrylamides) and its application to the prepa-ration of alpha,omega-dicholesteryl and alpha,omega-dipyrenylpolymers. J Polym Sci Part A Polym Chem 2008;46:314–26.
[72] Chen F, Cheng ZP, Zhu J, Zhang W, Zhu XL. Synthesis of poly(vinylacetate) with fluorescence via a combination of RAFT/MADIX and“click” chemistry. Eur Polym J 2008;44:1789–95.
[73] Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chem-ical function from a few good reactions. Angew Chem Int Ed2001;40:2004–21.
[74] Bathfield M, D’Agosto F, Spitz R, Charreyre MT, Delair T. Versatileprecursors of functional RAFT agents. Application to the synthe-sis of bio-related end-functionalized polymers. J Am Chem Soc2006;128:2546–7.
[75] Beija M. Synthesis of �-dye-labelled thermoresponsive blockcopolymers by RAFT polymerization. Behaviour at the air–waterinterface and in aqueous solutions. PhD thesis. Lisbon and Lyon Uni-versidade Técnica de Lisboa and Université Claude, Bernard Lyon 1,2009.
[76] Tong Y-Y, Wang R, Xu N, Du FS, Li ZC. Synthesis of well-definedazide-terminated poly(vinyl alcohol) and their subsequent mod-
ification via click chemistry. J Polym Sci Part A Polym Chem2009;47:4494–504.[77] Henry SM, Convertine AJ, Benoit DSW, Hoffman AS, Stayton PS.End-functionalized polymers and junction-functionalized diblockcopolymers via RAFT chain extension with maleimido monomers.Bioconjugate Chem 2009;20:1122–8.

olymer
M. Beija et al. / Progress in P[78] Yamago S, Iida K, Yoshida J. Organotellurium compounds as novelinitiators for controlled/living radical polymerizations. Synthesisof functionalized polystyrenes and end-group modifications. J AmChem Soc 2002;124:2874–5.
[79] Debuigne A, Caille J-R, Jérôme R. Highly efficient cobalt-mediatedradical polymerization of vinyl acetate. Angew Chem Int Ed2005;44:1101.
[80] Detrembleur C, Stoilova O, Bryaskova R, Debuigne A, Mouithys-Mickalad A, Jérôme R. Preparation of well-defined PVOH/C60
nanohybrids by cobalt-mediated radical polymerization of vinylacetate. Macromol Rapid Commun 2006;27:498–504.
[81] Hurtgen M, Debuigne A, Mouithys-Mickalad A, Jérôme R, JérômeC, Detrembleur C. Synthesis of poly(vinyl alcohol)/C60 and poly(N-vinylpyrrolidone)/C60 nanohybrids as potential photodynamiccancer therapy agents. Chem Asian J 2010;5:859–68.
[82] Grubbs RH, Trnka TM. The development of L2X2Ru=CHR olefinmetathesis catalysts: an organometallic success story. Acc ChemRes 2001;34:18–29.
[83] Gordon EJ, Gestwicki JE, Strong LE, Kiessling LL. Synthesisof end-labeled multivalent ligands for exploring cell-surface-receptor-ligand interactions. Chem Biol 2000;7:9–16.
[84] Mangold SL, Carpenter RT, Kiessling LL. Synthesis of fluoro-genic polymers for visualizing cellular internalization. Org Lett2008;10:2997–3000.
[85] Madkour AE, Koch AHR, Lienkamp K, Tew GN. End-functionalizedROMP polymers for biomedical applications. Macromolecules2010;43:4557–61.
[86] Roberts KS, Sampson NS. A facile synthetic method to prepare flu-orescently labeled ROMP polymers. Org Lett 2004;6:3253–5.
[87] Ni SR, Zhang P, Wang YC, Winnik MA. Energy transfer studies of theboudary layer interphase in polystyrene poly(methyl methacry-late) block copolymer films. Macromolecules 1994;27:5742–50.
[88] Tcherkasskaya O, Ni SR, Winnik MA. Direct energy transfer studiesof the domain-boundary interface in polyisoprene-poly(methylmethacrylate) block copolymer films. Macromolecules1996;29:610–6.
[89] Horinaka J, Maruta M, Ito S, Yamamoto M. Local motion of oligo- andpolystyrene chain end studied by the fluorescence depolarizationmethod. Macromolecules 1999;32:1134–9.
[90] Tong JD, Ni SR, Winnik MA. Synthesis of polyisoprene-b-polystyrene block copolymers bearing a fluorescent dye atthe junction by the combination of living anionic polymeriza-tion and atom transfer radical polymerization. Macromolecules2000;33:1482–6.
[91] Prochazka K, Kiserow D, Ramireddy C, Tuzar Z, Munk P, Web-ber SE. Time-resolved fluorescence studies of the chain dynamicsof naphthalene-labeled polystyrene-block-poly(methacrylic acid)micelles in aqueous media. Macromolecules 1992;25:454–60.
[92] Moon B, Hoye TR, Macosko CW. Anionic synthesis and detection offluorescence-labeled polymers with a terminal anhydride group. JPolym Sci Part A Polym Chem 2000;38:2177–85.
[93] Tong JD, Zhou CL, Ni SR, Winnik MA. Synthesis of meth(acrylate)diblock copolymers bearing a fluorescent dye at the junction usinga hydroxyl-protected initiator and the combination of anionic poly-merization and controlled radical polymerization. Macromolecules2001;34:696–705.
[94] Gavranovic GT, Csihony S, Bowden NB, Hawker CJ, WaymouthRM, Moerner WE, et al. Well-controlled living polymerization ofperylene-labeled polyisoprenes and their use in single-moleculeImaging. Macromolecules 2006;39:8121–7.
[95] Bowden NB, Willets KA, Moerner WE, Waymouth RM. Synthesisof fluorescently labeled polymers and their use in single-moleculeimaging. Macromolecules 2002;35:8122–5.
[96] Lindner SM, Thelakkat M. Fluorescent dye-labeled polymers carry-ing triphenylamine, styrene, or acrylate pendant groups. MacromolChem Phys 2006;207:2084–92.
[97] Rodlert M, Harth E, Rees I, Hawker CJ. End-group fidelity innitroxide-mediated living free-radical polymerizations. J Polym SciPart A Polym Chem 2000;38:4749–63.
[98] Bucsiova L, Yin MZ, Chmela S, Habicher WD. Nitroxide-mediatedliving radical polymerization of styrene with fluorescent initiator.J Macromol Sci Part A 2008;45:761–8.
[99] Hrdlovic P, Chmela S, Danko M, Sarakha M, Guyot G. Spectral
properties of probes containing benzothioxanthene chromophorelinked with hindered amine in solution and in polymer matrices. JFluor 2008;18:393–402.[100] Chmela S, Hrckova L. Nitroxide mediated styrene radical poly-merization using a fluorescence marked mediator. Eur Polym J2009;45:2580–6.
Science 36 (2011) 568–602 601
[101] Scott ME, Parent JS, Hennigar SL, Whitney RA, CunninghamMF. Determination of alkoxyamine concentrations in nitroxyl-mediated styrene polymerization products. Macromolecules2002;35:7628–33.
[102] Kamigaito M, Ando T, Sawamoto M. Metal-catalyzed living radicalpolymerization. Chem Rev 2001;101:3689–745.
[103] Matyjaszewski K, Xia JH. Atom transfer radical polymerization.Chem Rev 2001;101:2921–90.
[104] Duan Q, Miura Y, Narumi A, Shen X, Sato S, Satoh T, et al.Synthesis and thermoresponsive property of end-functionalizedpoly(N-isopropylacrylamide) with pyrenyl group. J Polym Sci PartA Polym Chem 2006;44:1117–24.
[105] Lu XJ, Zhang LF, Meng LZ, Liu YH. Synthesis of poly(N-isopropylacrylamide) by ATRP using a fluorescein-based initiator.Polym Bull 2007;59:195–206.
[106] Wang MF, Dykstra TE, Lou XD, Salvador MR, Scholes GD,Winnik MA. Colloidal CdSe nanocrystals passivated by a dye-labeled multidentate polymer: quantitative analysis by size-exclusion chromatography. Angew Chem Int Ed 2006;45:2221–4.
[107] Wang MF, Bardajee GR, Kumar S, Nitz M, Scholes GD, Winnik MA.Preparative size-exclusion chromatography for purification andcharacterization of colloidal quantum dots bound by chromophore-labeled polymers and low-molecular-weight chromophores. JChromatogr A 2009;1216:5011–9.
[108] Such GK, Evans RA, Davis TP. Control of photochromism throughlocal environment effects using living radical polymerization(ATRP). Macromolecules 2004;37:9664–6.
[109] Such GK, Evans RA, Davis TP. Tailoring photochromic performanceof polymer-dye conjugates using living radical polymerization(ATRP). Mol Cryst Liq Cryst 2005;430:273–9.
[110] Ercole F, Davis TP, Evans RA. Comprehensive modulation of naph-topyran photochromism in a rigid host matrix by applying polymerconjugation. Macromolecules 2009;42:1500–11.
[111] Malic N, Campbell JA, Evans RA. Superior photochromic per-formance of naphthopyrans in a rigid host matrix usingpolymer conjugation: fast, dark, and tunable. Macromolecules2008;41:1206–14.
[112] Zhao K, Cheng Z, Zhang Z, Zhu J, Zhu X. Synthesis of fluo-rescent poly(methyl methacrylate) via AGET ATRP. Polym Bull2009;63:355–64.
[113] Roof AC, Tillman ES, Malik RE, Roland AM, Miller DJ, SarryLR. Mechanistic investigation of 9-bromoanthracene photodimersas initiators in atom transfer radical polymerization. Polymer2006;47:3325–35.
[114] Cohen NA, Tillman ES, Thakur S, Smith JR, Eckenhoff WT, Pintauer T.Effect of the ligand in Atom Transfer Radical Polymerization reac-tions initiated by photodimers of 9-bromoanthracene. MacromolChem Phys 2009;210:263–8.
[115] Le TPT, Moad G, Rizzardo E, Thang SH. Polymerization with Livingcharacteristics. WO98/01478, 1998.
[116] Favier A, Charreyre MT. Experimental requirements for an effi-cient control of free-radical polymerizations via the reversibleaddition-fragmentation chain transfer (RAFT) process. MacromolRapid Commun 2006;27:653–92.
[117] Moad G, Rizzardo E, Thang SH. Living radical polymerization by theRAFT process—a first update. Aust J Chem 2006;59:669–92.
[118] Moad G, Solomon DH. The Chemistry of Radical Polymerization.2nd ed. Amsterdam Elsevier; 2006.
[119] Sudalai A, Kanagasabapathy S, Benicewicz BC. Phosphorus penta-sulfide: a mild and versatile catalyst-reagent for the preparation ofdithiocarboxylic esters. Org Lett 2000;2:3213–6.
[120] Zhou NC, Lu LD, Zhu XL, Yang XJ, Wang X, Zhu J, et al. Prepara-tion and characterization of anthracene end-capped polystyrenevia reversible addition-fragmentation chain transfer polymeriza-tion. Polym Bull 2006;57:491–8.
[121] Zhou N, Lu L, Zhu J, Yang X, Wang X, Zhu X, et al. Syn-thesis of polystyrene end-capped with pyrene via reversibleaddition-fragmentation chain transfer polymerization. Polymer2007;48:1255–60.
[122] Fu J, Cheng ZP, Zhou N, Zhu J, Zhang W, Zhu X. Reversibleaddition-fragmentation chain transfer polymerizations of styrenewith two novel trithiocarbonate as RAFT agents. Polymer 2008;49:
5431–8.[123] Such GK, Evans RA, Davis TP. The use of block copolymers tosystematically modify photochromic behavior. Macromolecules2006;39:9562–70.
[124] Mertoglu M, Laschewsky A, Skrabania K, Wieland C. New watersoluble agents for reversible addition-fragmentation chain transfer

6 olymer
02 M. Beija et al. / Progress in Ppolymerization and their application in aqueous solutions. Macro-molecules 2005;38:3601–14.
[125] Thang SH, Chong YK, Mayadunne RTA, Moad G, Rizzardo E. A novelsynthesis of functional dithioesters, dithiocarbamates, xanthatesand trithiocarbonates. Tetrahedron Lett 1999;40:2435–8.
[126] Li J, Yang W, Tao L, Li D, Boyer C, Davis TP. Thermosensitivegraphene nanocomposites formed using pyrene-terminal poly-mers made by RAFT polymerization. J Polym Sci Part A Polym Chem2010;48:425–33.
[127] ten Cate MGJ, Rettig H, Bernhardt K, Borner HG. Sequence-definedpolypeptide-polymer conjugates utilizing reversible additionfragmentation transfer radical polymerization. Macromolecules2005;38:10643–9.
[128] Deletre M, Levesque G. Kinetics and mechanism of poly-thioamidation in solution. 1. Reaction of mono(dithioester)sand bis(dithioester)s with excess amine. Macromolecules1990;23:4733–41.
[129] Prazeres TJV, Beija M, Farinha JPS, Charreyre MT, MartinhoJMG. RAFT polymerization and self-assembly of thermore-sponsive poly(N-decylacrylamide-b-N,N-diethylacrylamide) blockcopolymers bearing a phenanthrene alpha-end group. Polymer2010;51:355–67.
[130] Afonso CAM, Farinha JPS. Synthesis of 4-aryl-butylamine fluores-cent probes. J Chem Res (S) 2002:584–6.
[131] Marcelo G, Prazeres TJV, Charreyre MT, Martinho JMG, Far-inha JPS. Thermoresponsive micelles of phenanthrene-alpha-end-labeled poly(N-decylacrylamide-b-N,N-diethylacrylamide) in
water. Macromolecules 2010;43:501–10.[132] Chen M, Ghiggino KP, Mau AWH, Sasse WHF, Thang SH, WilsonGJ. Amphiphilic acenaphthylene-maleic acid light-harvestingalternating copolymers: reversible addition—fragmentationchain transfer synthesis and fluorescence. Macromolecules2005;38:3475–81.
Science 36 (2011) 568–602
[133] Chen M, Ghiggino KP, Thang SH, White J, Wilson GJ. Synthesis andfluorescence of a series of multichromophoric acenaphthenyl com-pounds. J Org Chem 2005;70:1844–52.
[134] Zhou D, Zhu XL, Zhu J, Hu LH, Cheng ZP. Synthesis of well-definedcarbazole group labelled polymer via RAFT polymerization andstudy on the optical properties. E-Polymers 2006:059/1-12.
[135] Zhou D, Zhu X, Zhu J, Cheng ZP. Synthesis and characterizationof fluorescence end-labeled polystyrene via reversible addition-fragmentation chain transfer (RAFT) polymerization. J Polym SciPart A Polym Chem 2008;46:6198–205.
[136] Wan XM, Zhu XL, Zhu J, Zhang ZB, Cheng ZP. Synthesis ofdithiocarbamate bearing azobenzene group and use for RAFT poly-merization of vinyl monomers. J Polym Sci Part A Polym Chem2007;45:2886–96.
[137] Ma F, Zhou N, Zhu J, Zhang W, Fan L, Zhu X. Light-drivenfluorescence enhancement of phenylazo indazole-terminatedpolystyrene. Eur Polym J 2009;45:2131–7.
[138] Zhou NC, Zhu J, Zhang ZB, Zhu XL. RAFT polymerization of styreneusing dithioester with anthracene Z group as chain transfer agent.E-Polymers 2007:137/1-10.
[139] Zhu J, Zhu XL, Cheng ZP, Liu F, Lu JM. Study on controlledfree-radical polymerization in the presence of 2-cyanoprop-2-yl1-dithionaphthalate (CPDN). Polymer 2002;43:7037–42.
[140] Zhu J, Zhu XL, Zhou D, Chen JY, Wang XY. Study on reversibleaddition-fragmentation chain transfer (RAFT) polymerization ofMMA in the presence of 2-cyanoprop-2-yl 1-dithlophenanthrenate(CPDPA). Eur Polym J 2004;40:743–9.
[141] Li Y, Yang JW, Benicewicz BC. Well-controlled polymerization of2-azidoethyl methacrylate at near room temperature and clickfunctionalization. J Polym Sci Part A Polym Chem 2007;45:4300–8.
[142] Li CZ, Benicewicz BC. alpha-Cyanobenzyl dithioester reversibleaddition-fragmentation chain-transfer agents for controlled radicalpolymerizations. J Polym Sci Part A Polym Chem 2005;43:1535–43.


















