Signal transduction signal transduction system composes: receptor, enzyme,pathway

THE JOURNAL OF BIOL~CICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc
Val. 269, No. 49, Issue of December 9, PP . 31183-31189, 1994 Printed in U.S.A.
Potassium Channel Induction By the Ras/Raf Signal Transduction Cascade*
(Received for publication, July 12, 1994, and in revised form, September 2, 1994)
Yi Huang and Stanley G. RaneS From the Department of Biological Sciences, Purdue University, West Lafayette, Indiana 47907
p21- plays a critical role in cell growth, differentia- tion, and oncogenic transformation. However, the final physiological effectors of p21rm-mediated signal trans- duction remain to be determined. We have used patch clamp electrophysiology, pharmacological agents, and transfection with specific Ras or Raf plasmids, to dem- onstrate that induction of a unique Ca2+-activated K+ channel in murine fibroblast cell lines depends on p21- and its immediate downstream target, the Raf kinase. The importance of this channel in mitogenic signaling is further indicated by its induction in nontransformed cells by epidermal growth factor and platelet-derived growth factor and the ability of K+ channel blockers to inhibit cell proliferation. We suggest that this Ca2+-acti- vated K+ channel is one ultimate physiological target of p2l"mediated signal transduction and that it may play a role in cell proliferation and ras transformation.
p21" plays a pivotal role in controlling cell proliferation in both normal and transformed states (1,2). Membrane localiza- tion of p21"", via C-terminal isoprenylation (3,4), allows for its interaction with activated receptor tyrosine kinases, Grb2 and Ras guanine nucleotide releasing factor, and subsequent con- version from a GDP- to GTP-bound form (5-7). GTP-bound activated ~ 2 1 " ~ stimulates Raf serinehhreonine kinase (8-10) leading to activation of mitogen-activated protein kinase cas- cades and ultimately certain transcription factors (8, 11, 12- 16). In this way, proliferative signals at the cell surface are transmitted to the nucleus via p21"" and a series of kinases. Because oncogenic ~ 2 1 " ~ exists in a GTP-bound state independ- ent of tyrosine kinase activation, one model for rus transforma- tion is that oncogenic p21" provides constitutive stimulation of downstream response elements such as Raf and mitogen-acti- vated protein kinases, and presumably rus-responsive genes. Thus ras-transformed cells should constitutively express the final effectors of the signal cascade. Although the early events in p21m"-mediated signaling are now relatively clear, these fi- nal effector molecules and their contribution to cell transfor- mation remain to be determined
Because Ras proteins are in many ways similar to the het- erotrimeric G-proteins, which have been extensively shown to modulate ion channels (17, 181, we investigated whether ion channels could be potential targets for regulation by ~21"". This was based on the idea that p21" may have acute signaling
* This work was supported by a National Institutes of Health Grant ROlGM43462 (to S. R.) and by graduate fellowships (to Y. H.) from Purdue University Research Foundation and the Purdue University Cancer Center supported by the American Cancer Society (IN-1731). The costs of publication of this article were defrayed in part by the
"aduertisement" in accordance with 18 U.S.C. Section 1734 solely to payment of page charges. This article must therefore be hereby marked
indicate this fact. $ To whom correspondence should be addressed. Tel.: 317-494-8191;
Fax: 317-494-0876 E-mail: [email protected].
functions at the membrane in addition to its long term genetic action. Our finding that, in murine fibroblast cell lines, a unique Ca2+-activated K+ channel was constantly upregulated in conjunction with ras transformation was consistent with the model described above for constitutive signaling by oncogenic p21m9 (19,20). Work from other labs has also implicated p21"" in the regulation of other ion channels types in proliferating (21-23) as well as terminally differentiated cells (24, 25), and there are indications that mitogen signaling or cell cycle regu- lation may also involve ion channel regulation (26, 27). How- ever, the mechanisms and significance of ion channel regula- tion by the mitogenic pathways subserved by p21"", and particularly the role of Raf kinase, remain to be defined.
In this study we show that unlike the rather direct actions of the heterotrimeric G-proteins on ion channel activities, K+ channel upregulation by oncogenic p21"" appears to be due to Raf kinase-dependent induction of channel expression. Fur- thermore, K+ channel induction by EGF' and PDGF in non- transformed cells and the inhibition of proliferation in these cells by K+ channel blockers suggest that endogenous cellular Ras proteins also regulate the channel with consequences for cell growth. These results suggest that this Caz+-activated K+ channel is one final physiological target activated by the Rad Raf signal transduction pathway, and they hint at a role for the K+ channel in regulating cell growth in response to oncogenic and possibly normal p21"
MATERIALS AND METHODS Cell Culture and Thnsfections-The following cell lines were
used for this study: NIH 3T3 and C3HlOT1/2, as well as their H-ras- and v-src-transformed counterparts (Dr. E. J . Taparowsky, Purdue University), NIH 3T3 K-ras12V186S and NIH 3T3 K-rasl2V (Dr. J . H. Jackson, Scripps Institute), NIH 3T3 ZCR (Dr. L. Feig, Tufts University School of Medicine), NIH 3T3 22W (Dr. G. M. Cooper, Harvard Univer- sity), Balb 3T3 (A31 clone) and its K-ras-transformed counterpart (ATCC CCL 163.3). Cultures were maintained in a humidified, 5% CO, atmosphere in Dulbecco's modified Eagle's medium (Life Technologies, Inc.) containing either 10% calf serum (NIH 3T3 and Balb 3T3 cells) or 10% fetal bovine serum (C3HlOT1/2). The following plasmids were used for transfection: pKOpT24 containing activated human H-ras and neo- mycin resistance gene (Dr. E J. Taparowsky, Purdue University); RSV- raf-301 and RSV-raf-C4 (Dr. U. R. Rapp, NIH). Cells were transfected with calcium phosphate precipitation as described previously (28), and stably transfected cells were selected for resistance to 400 pg/ml G418 (geneticin; Life Technologies, Inc.). Foci of surviving cells were isolated and cloned. In some experiments, cells were cotransfected with pKOpT24 (50 ng1100-mm plate) and RSV-raf-301 or RSV-raf-C4 (500 ng/lOO-mrn plate) at a 1 : l O ratio.
For serum starvation, cells were cultured in Dulbecco's modified Eagle's medium containing 0.1% bovine serum albumin for 18-24 h to achieve quiescence. Cell proliferation was determined by hemacytome- ter counts of cells.
Immunofluorescence-Cells cultured overnight on glass coverslips were permeabilized for 20 min in cold methanol and 5 mM EGTA at
The abbreviations used are: EGF, epidermal growth factor; PDGF, platelet-derived growth factor; ChTX, charybdotoxin; PA, perillic acid; TEA, tetraethylammonium; TMA, tetramethylammonium.
31183

31184
FIG. 1. Ca2+-activated K+ current is present at higher levels in ras-trans- formed versus nontransformed fibro- blasts. A, whole cell assay for Ca2+-acti- vated K+ currents. Overlaid current records were obtained before and after ex- tracellular application of 1 p~ A23187. B, mean K+ current amplitudes and densi- ties (&E.) determined by whole cell re- cordings from four fibroblast cell lines and their rus-transformed counterparts: neo- natal rat kidney, C3HlOT1/2, Balb 3T3, and NIH 3T3. Current density for each cell was determined by normalizing cur- rent amplitude to cell capacitance, which is proportional to cell surface area. K+ channel activity is significantly elevated in each of the rus-transformed lines. Each bur represents recordings from tens to hundreds of cells.
Ras lRaf Induction of K+ Channel
A Ras transformed NIH 3T3 1.51 + ~23187
B
0 normal cells I rascells
non-transformed NIH 3T3
a + A23187
I
-20 "C and then washed in phosphate-buffered saline and incubated for 4 h at 37 "C with a rat anti-Ras antibody Y13-259 (Oncogene Science) and a rabbit anti-tubulin antiserum (30). After a 15-min wash in phos- phate-buffered saline (5 min for 3 times), cells were incubated for 1 h at 37 "C with fluorescein-labeled goat anti-rat IgG and rhodamine-labeled anti-rabbit IgG secondary antibody (Kirkegaard & Perry Laboratories). Cells were then washed in phosphate-buffered saline for 3 times, 5 min each time. Coverslips were mounted on glass slides. Mounted specimens were examined using a Zeiss microscope equipped with epiillumination and nonoverlapping filters for fluorescein, rhodamine, and 4,g-diamin- idino-2-phenylindole. Images were recorded on TMAX 400 film (East- man Kodak Corp.).
Western Blot of Rus Proteins-Cell homogenates (200 pg of proteid lane) were electrophoresed (SDS-l2% polyacrylamide gel) and trans- ferred electrophoretically to nitrocellulose blotting membrane (Schleicher & Schuell). Immunodetection of Ras proteins was done us- ing specific anti-H-Ras monoclonal antibody F235-1.7.1 (Oncogene Sci- ence), anti-mouse IgG horseradish peroxidase conjugate, and ECLTM Western blot detection reagents (Amersham Corp.). Visualization was achieved by chemiluminescence (Amersham Corp.).
Electrophysiology-Preparation of cells for electrophysiological re- cordings and details for pipette manufacture and data acquisition have been described previously (19, 20). Whole cell currents were recorded every 3 s by a step to 0 mV from a holding potential of -70 mV, and currents were filtered a t 3 kHz (-3 db) with an 8-pole Bessel filter. For all experiments, analog compensation was used both to attenuate ca- pacitive transients and to determine whole cell capacitance. All statis- tical results are given as the mean f S.E., with significant differences between means (t test at t ~ ) indicated by *.
The patch pipette solution consisted of the following: 150 mM KCl, 1 mM MgCl,, 10 mM HEPES, and 0.1 m~ EGTA (and 1 mM MgATP and 0.5 mM GTP for protein kinase C or protein kinase A experiments), while the bath solution was 138 mM NaCl, 9 mM KC1, 10 mM HEPES, 1 mM MgCl,, and 1 m~ CaCl, for recording whole cell Ca2+-activated K+ cur- rents. For inside-out patch experiments, the bath solution was 150 mM KCl, 1 mM MgCl,, 10 mM HEPES, and 10 p~ free Ca2+ as CaCl, buffered with 0.1 mM EGTA. All solutions were at pH 7.3, and experiments were carried out a t room temperature, 20-23 "C. A23187 (Sigma) was pre- pared fresh from a frozen stock by dilution to a final concentration of 1 VM, and it was applied from blunt-tipped micropipettes (19, 20). The following reagents were used in the study: tetraethylammonium (TEA), tetramethylammonium (TMA), 8-(4-chlorophenylthio)adenosine-cAMP, arachidonic acid, staurosporine, and cycloheximide (all from Sigma); charybdotoxin (Research Biochemicals Inc.); perillic acid (Biomol); EGF, PDGF, and lavendustin A (all from Life Technologies, Inc.); diC8 and phor- bo1 12-myristate 13-acetate (Molecular Probes, Inc.); okadaic acid and ge- nistein (LC Laboratories). Argl2Ras protein was kindly provided by Dr. L. A. Feig ( M s University School of Medicine). Lovastatin (gift from Merck) was prepared as described previously (29) and stored at -20 "C as 10 mM stock.
RESULTS
Ras-dependent Induction of K+ Currents-We have previ- ously shown that stable transformation of Balb 3T3, C3HlOT1/2 and neonatal rat kidney cells with oncogenic ras genes results in the selective appearance of a unique Ca2+- activated K+ channel (19, 20). We have continued our experi- ments with these cell lines and have also extended our original findings to NIH 3T3 cells (Fig. 1). Thus in all four cell lines tested so far, ras transformation was consistently correlated with appearance of the K+ channel. To exclude the possibility that K+ channel induction was a nonspecific effect of long-term cell transformation rather than a consequence of p21"" signal- ing, we performed standard patch clamp experiments on newly transformed NIH 3T3 cells. Before transfection, K+ current density (given as K+ current amplitude normalized to cell ca- pacitance) was 7.6 2 2.0 pA/pF ( n = 24), while the density 2 weeks after transfection with oncogenic H-ras was 66.0 2 7.4 pA/pF (n = 3) and 75.6 2 11.9 pA/pF (n = 4) in two stably transformed colonies. These densities were comparable with the density in long term ras-transformed NIH 3T3 cells (78.3 2 3.8 pA/pF, n = 52). Although the elevation in K+ current density varied with each transfection, the increase was always at least 3-fold and could be as much as 10-fold.
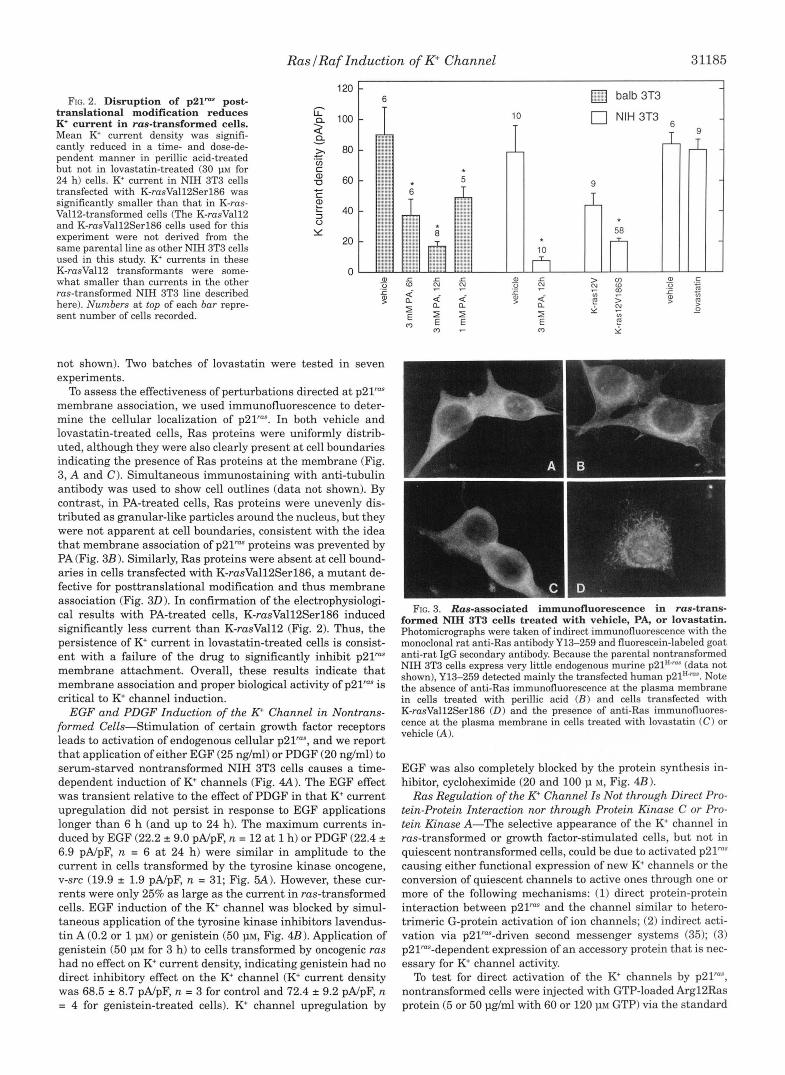
To further demonstrate that the appearance of the K+ current is due to signaling by membrane-associated p21", ras-trans- formed cells were treated with two inhibitors of ~ 2 1 ~ " post- translational modification. Perillic acid (PA), an inhibitor of farnesyltransferase for small G-proteins (31), reduced whole cell K+ current by as much as 85% in a time- and dose-depend- ent manner (Fig. 2). To rule out effects on signaling elements downstream of p21" or on the channel itself, we showed that PA did not reduce K+ current induced by constitutively acti- vated raf (22W Ref. 32). This N-terminal deleted form of Raf, the activity of which is p2Yas-independent, also greatly in- creased K+ current density (see Fig. 5 A ) . K+ current density in untreated 22W cells was 137.5 2 15.7 pA/pF (n = 71, and after treatment with perillic acid (3 m ~ ) for 12 h, K+ current density was 152.7 2 16.7 pA/pF ( n = 7). Lovastatin, which inhibits posttranslational processing by preventing isoprenyl group for- mation (33, 34), had surprisingly little effect on K+ current density in ras-transformed NIH (Fig. 2) or Balb 3T3 cells (data
Ras lRaf Induction of K+ Channel 31185
what smaller than currents in the other ras-transformed NIH 3T3 line described here). Numbers at top of each bar repre- sent number of cells recorded.
a, 0 8
>
- ._
not shown). Two batches of lovastatin were tested in seven experiments.
To assess the effectiveness of perturbations directed at p21"" membrane association, we used immunofluorescence to deter- mine the cellular localization of ~ 2 1 " ~ . In both vehicle and lovastatin-treated cells, Ras proteins were uniformly distrib- uted, although they were also clearly present a t cell boundaries indicating the presence of Ras proteins at the membrane (Fig. 3, A and C). Simultaneous immunostaining with anti-tubulin antibody was used to show cell outlines (data not shown). By contrast, in PA-treated cells, Ras proteins were unevenly dis- tributed as granular-like particles around the nucleus, but they were not apparent at cell boundaries, consistent with the idea that membrane association of p21" proteins was prevented by PA (Fig. 3B 1. Similarly, Ras proteins were absent at cell bound- aries in cells transfected with K-rasVall2Serl86, a mutant de- fective for posttranslational modification and thus membrane association (Fig. 30). In confirmation of the electrophysiologi- cal results with PA-treated cells, K-rasVall2Serl86 induced significantly less current than K-rusVall2 (Fig. 2). Thus, the persistence of K+ current in lovastatin-treated cells is consist- ent with a failure of the drug to significantly inhibit p21" membrane attachment. Overall, these results indicate that membrane association and proper biological activity of p21" is critical to K+ channel induction.
EGF and PDGF Induction of the R Channel in Nontrans- formed Cells-Stimulation of certain growth factor receptors leads to activation of endogenous cellular p21", and we report that application of either EGF (25 ng/ml) or PDGF (20 ng/ml) to serum-starved nontransformed NIH 3T3 cells causes a time- dependent induction of K+ channels (Fig. 4A). The EGF effect was transient relative to the effect of PDGF in that K+ current upregulation did not persist in response to EGF applications longer than 6 h (and up to 24 h). The maximum currents in- duced by EGF (22.2 * 9.0 pA/pF, n = 12 at 1 h) or PDGF (22.4 * 6.9 pA/pF, n = 6 at 24 h) were similar in amplitude to the current in cells transformed by the tyrosine kinase oncogene, v-src (19.9 1.9 pA/pF, n = 31; Fig. 5A). However, these cur- rents were only 25% as large as the current in ras-transformed cells. EGF induction of the K+ channel was blocked by simul- taneous application of the tyrosine kinase inhibitors lavendus- tin A (0.2 or 1 J~M) or genistein (50 p ~ , Fig. 4B). Application of genistein (50 PM for 3 h) to cells transformed by oncogenic ras had no effect on K+ current density, indicating genistein had no direct inhibitory effect on the K+ channel (K+ current density was 68.5 * 8.7 pA/pF, n = 3 for control and 72.4 2 9.2 pA/pF, n = 4 for genistein-treated cells). K+ channel upregulation by
10
T
O b alb 3T3
0 NIH 3T3
m
58 n v)
Y m
formed NIH 3T3 cells treated with vehicle, PA, or lovastatin. FIG. 3. Ras-associated immunofluorescence in ms-trans-
Photomicrographs were taken of indirect immunofluorescence with the monoclonal rat anti-Ras antibody Y13-259 and fluorescein-labeled goat anti-rat IgG secondary antibody. Because the parental nontransformed NIH 3T3 cells express very little endogenous murine p21".m" (data not shown), Y13-259 detected mainly the transfected human ~21"'"". Note the absence of anti-Ras immunofluorescence at the plasma membrane in cells treated with perillic acid ( B ) and cells transfected with K-rasVall2Serl86 (D) and the presence of anti-Ras immunofluores- cence at the plasma membrane in cells treated with lovastatin ( C ) or vehicle (A ).
EGF was also completely blocked by the protein synthesis in- hibitor, cycloheximide (20 and 100 p M, Fig. 4B).
Ras Regulation of the K+ Channel Is Not through Direct Pro- tein-Protein Interaction nor through Protein Kinase C or Pro- tein Kinase A-The selective appearance of the K+ channel in ras-transformed or growth factor-stimulated cells, but not in quiescent nontransformed cells, could be due to activated p21"" causing either functional expression of new K+ channels or the conversion of quiescent channels to active ones through one or more of the following mechanisms: (1) direct protein-protein interaction between p21" and the channel similar to hetero- trimeric G-protein activation of ion channels; (2) indirect acti- vation via p21"-driven second messenger systems (35); (3) p21""-dependent expression of an accessory protein that is nec- essary for K+ channel activity.
To test for direct activation of the K+ channels by p21"", nontransformed cells were injected with GTP-loaded Argl2Ras protein (5 or 50 pg/ml with 60 or 120 p~ GTP) via the standard

31186
A
Ras lRaf Induction of R Channel
- 1s
B I
c . i
30 l h 2h 3h 6h 24h
EGF(25nglml) + + + + + + - LEV A (1IM) - 0 . 2 1 - - - - Genistein (50 pM) . - . + - - - Cycloheximide(~M) . - - . 20 100 -
FIG. 4. EGF and PDGF induce K' current in serum-stanred nontransformed NIH 3T3 cells. A, EGF (25 ng/ml) or PDGF (20 ng/ml) were applied to serum-starved nontransformed NIH 3T3 cells for the times indicated. Growth factors were removed prior to K+ current
with resolvable K+ currents (>30 PA, Ref. 19) over total number of cells recordings. The numbers at top of each bar represent number of cells
tested (*, significantly different from nontreated cells). B , cells were stimulated for 1 h with either EGF alone or with tyrosine kinase or protein synthesis inhibitors added 10-15 min prior to EGF addition. Numbers at top of each bar represent number of cells recorded (*, significantly different from EGF-treated cells).
whole cell patch clamp configuration. Protein injection was assured by application of positive pressure to the suction tube attached to the patch pipette holder until cells swelled by about 40-50% of their initial diameter. At 5 min to 1 h after injection, whole cell K+ currents were measured in response to 1 p~ A23187. No significant increase in mean current was observed in injected (4.3 2 1.3 pA/pF, n = 34 cells for Balb 3T3; 0.6 2 0.6 pA/pF, n = 4 for C3HlOT1/2) versus control (6.1 2 1.5 pA/pF, n = 28 for Balb 3T3; 2.0 2 2.0 pA/pF, n = 2 for C3HlOT1/2) cells. Furthermore, K+ currents in rus-transformed cells (55.4 2 9.2 pA/pF, n = 6 for NIH 3T3; 33.4 2 3.2 pA/pF, n = 9 for Balb 3T3) were not reduced by similarly timed injection experiments with a neutralizing anti-p21"" antibody (Y13-259; 0.5 or 1 w; 60.2 2 13.8 pA/pF, n = 4 for NIH 3T3; 33.3 2 7.1 pA/pF, n = 8 for Balb 3T3). Finally, inside-out patches from cells transformed by overexpression of cellular rus (ZCR) were excised into a solu- tion containing 1 mM M e , and the activity of single Ca2+- activated K+ channels was monitored. Under this condition, the half-life for GTP hydrolysis by membrane-bound cellular p21" is 10 min (36); however, no reduction of channel activity was detected for up to 25 min (4 channels, 2/2 patches). In addition, inside-out patches from cells transformed by oncogenic rus were excised into a solution with 0.5 mM GDP-p-S and low Mg2+ (0.1 mM EDTA, no added M e ) . Under this condition, the half- time for guanine nucleotide exchange for oncogenic p21"" is approximately 1 rnin (36); however, channel activity persisted for up to 20 min (16 channels, 4/4 patches). Therefore, K+ chan- nel activity was not suppressed either by GTP hydrolysis by normal ~21"" or by guanine nucleotide exchange on oncogenic p21"", arguing against a direct interaction of p21"" and the channel.
To determine whether or not protein kinase C is involved in regulating the K+ current, cells were treated with diC8, an activator of protein kinase C. diC8 (25 p~ in the bath for 1-3 h) had little effect on K+ currents in either rus-transformed (39.5 2 6.9 pA/pF, n = 9 in diC8 treated cells versus 39.6 2 5.3 pA/pF, n = 6 in control cells) or nontransformed cells (5.9 2 2.0 pA/pF, n = 22 in diC8 treated cells versus 7.0 2 3.0 pA/pF, n = 11 in control cells). Inclusion of the protein kinase C inhibitor stau- rosporine (0.1 p) in the patch pipette solution or the bath also failed to modulate the K+ current in rus-transformed cells (40.6 2 7.2 pA/pF, n = 8 in staurosporine treated cells versus 42.5 2 6.6 pA/pF, n = 7 in control cells). Finally, down-regulation of protein kinase C with phorbol 12-myristate 13-acetate (100 ng/ml for 26 h) did not affect K+ current in either rus-trans- formed or nontransformed cells (data not shown). In addition, we found that 8-(4-chlorophenylthio)adenosine-cAMP (100 p~ in pipette or in bath), an activator of protein kinase Nokadiac acid (1 p in pipette), an inhibitor of serindthreonine phospha- tase, and arachidonic acid (10 or 100 p ~ ) , were all ineffective in modulating the K+ channel (data not shown). Therefore, it is unlikely that the regulation of the K+ channel by p21"" is through those second messenger systems.
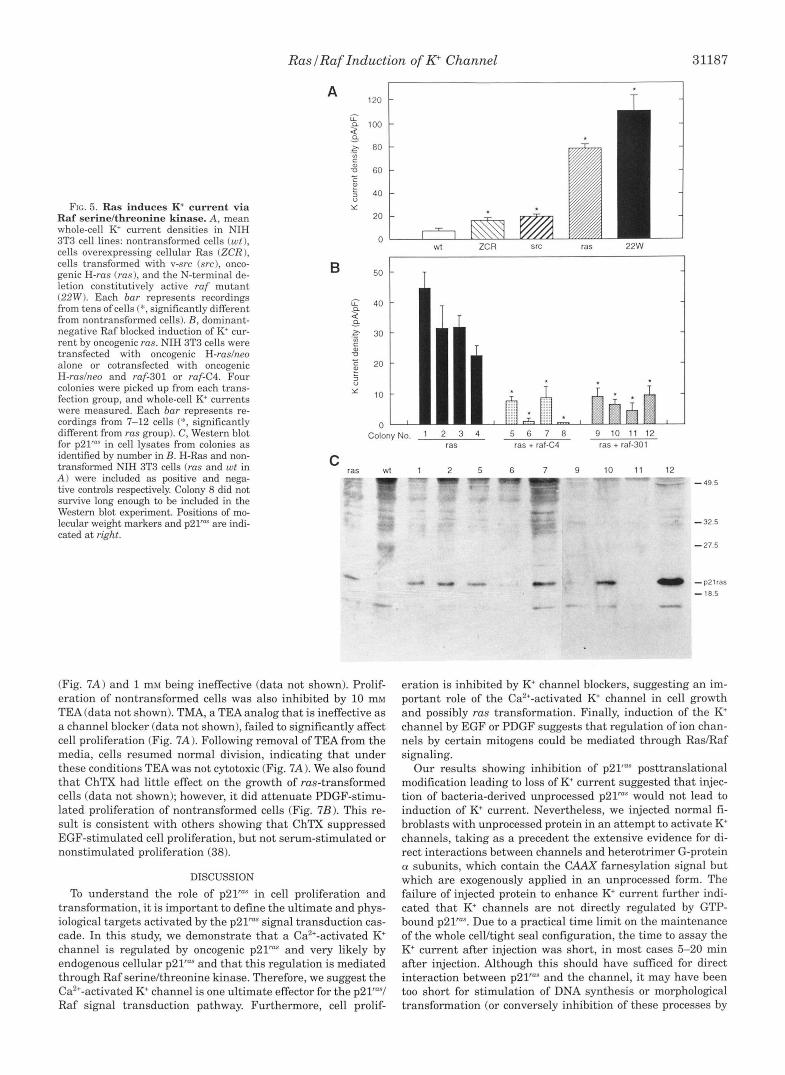
Rus Regulates the K+ Channel through Ruf SerinelThreonine Kinuse-As a first step in testing whether Raf kinase mediates p21" induction of K+ channel activity, we examined whether the ruf N-terminal deletion mutant, 22W, which has constitu- tive kinase activity, could induce K+ channel activity. Indeed, K+ current density in 22W cells was even greater than that in rus-transformed cells (Fig. 5 A ) . Besides being calcium acti- vated, the channel induced in 22W cells was blocked by ChTX (data not shown) and had the same open channel current-volt- age relationship as the channel in rus-transformed cells (Fig. 6; Ref. 20, Fig. 11, strongly suggesting that the K+ channels in 22W and rus-transformed cells are identical. Furthermore, in cells transformed by overexpression of cellular rus or by v-src, K+ channel densities were 100% greater than those in non- transformed cells, but they were still only 25% of that observed in rus- or ruf-transformed cells. Thus, signaling through the p21raS/Raf cascade consistently activates the K+ channel, but the magnitude of this activation may be determined by the position of the activated component in the cascade.
To determine if Raf is necessary for p21"" activation of the channel, nontransformed NIH 3T3 cells were transfected with oncogenic H-ruslneo alone or cotransfected with oncogenic H-ruslneo and one of two dominant negative ruf mutants, ruf- 301 or ruf-C4, which block signals transmitted by p21"" (8,371. Fewer colonies were formed, and cells grew more slowly in the dominant negative Raf cotransfected groups relative to the cells in the Ras group. Whole cell K+ currents were measured from four colonies from each transfection group within 21-24 days after transfection (Fig. 5B). All four colonies in the rus- transfected group had elevated K' channel activity relative to vector-transfected controls (11.2 f 4.8 pA/pF, n = 5 cells); how- ever, K+ channel activity was not elevated in the cotransfected groups. Survival of the cotransfected colonies in selective me- dium, as well as Western blot for p21"", indicate incorporation and expression of the transfected rus gene. Therefore, the ab- sence of elevated K+ channel activity in these groups resulted from disruption of ~ 2 1 " ~ to Raf kinase signaling caused by the dominant inhibitory Raf proteins.
Cell Proliferation Is Attenuated by the Ki Channel Blockers TEA and ChTX-rus-transformed cells were treated with TEA, and cell proliferation was determined by counting cell numbers each day after the treatment. TEA inhibited cell proliferation with the same dose dependence as for channel blockade (19, 20), with 10 mM TEA completely inhibiting cell proliferation
RaslRaf Induction of R Channel 31187
FIG. 5. Ras induces K' current v ia Raf serinelthreonine kinase. A, mean whole-cell K current densities in NIH 3T3 cell lines: nontransformed cells (wt ), cells overexpressing cellular Ras (ZCR), cells transformed with v-src (src), onco- genic H-rus (rus), and the N-terminal de- letion constitutively active ruf mutant (22W). Each bur represents recordings from tens of cells (*, significantly different from nontransformed cells). B, dominant- negative Raf blocked induction of K' cur- rent by oncogenic rus. NIH 3T3 cells were transfected with oncogenic H-ruslneo alone or cotransfected with oncogenic H-ruslneo and ruf-301 or ruf-C4. Four colonies were picked up from each trans- fection group, and whole-cell K+ currents were measured. Each bur represents re- cordings from 7-12 cells (*, significantly different from rus group). C, Western blot for p21'"" in cell lysates from colonies as identified by number in B. H-Ras and non- transformed NIH 3T3 cells (rus and wt in A ) were included as positive and nega- tive controls respectively. Colony 8 did not survive long enough to be included in the Western blot experiment. Positions of mo- lecular weight markers and p21"" are indi- cated at right.
A 120 1
" 1 T ii 40
2 1 ._ $ 30 C a, -0
E 20
3 0
10
n " Colony No. 1 2 3 4 5 6 7 8 9 1 0 1 1 1 2
ras ras + ral-C4 ras + ral-301
C ras wt 1 2 5 6 7 9 10 11 12 - """"."" - 49.5
- 32.5 -27.5
"p2lras - 18.5
(Fig. 7A) and 1 mM being ineffective (data not shown). Prolif- eration of nontransformed cells was also inhibited by 10 mM TEA (data not shown). TMA, a TEA analog that is ineffective as a channel blocker (data not shown), failed to significantly affect cell proliferation (Fig. 7A). Following removal of TEA from the media, cells resumed normal division, indicating that under these conditions TEA was not cytotoxic (Fig. 7A). We also found that ChTX had little effect on the growth of rus-transformed cells (data not shown); however, it did attenuate PDGF-stimu- lated proliferation of nontransformed cells (Fig. 7B). This re- sult is consistent with others showing that ChTX suppressed EGF-stimulated cell proliferation, but not serum-stimulated or nonstimulated proliferation (38).
DISCUSSION
To understand the role of p21"" in cell proliferation and transformation, it is important to define the ultimate and phys- iological targets activated by the p21"" signal transduction cas- cade. In this study, we demonstrate that a Ca2+-activated K+ channel is regulated by oncogenic p21"" and very likely by endogenous cellular p21"" and that this regulation is mediated through Raf serinelthreonine kinase. Therefore, we suggest the Ca2+-activated K+ channel is one ultimate effector for the p21""I Raf signal transduction pathway. Furthermore, cell prolif-
eration is inhibited by K' channel blockers, suggesting an im- portant role of the Ca2+-activated K' channel in cell growth and possibly rus transformation. Finally, induction of the K+ channel by EGF or PDGF suggests that regulation of ion chan- nels by certain mitogens could be mediated through RasIRaf signaling.
Our results showing inhibition of ~21'"" posttranslational modification leading to loss of K+ current suggested that injec- tion of bacteria-derived unprocessed p21"" would not lead to induction of K+ current. Nevertheless, we injected normal fi- broblasts with unprocessed protein in an attempt to activate K+ channels, taking as a precedent the extensive evidence for di- rect interactions between channels and heterotrimer G-protein (Y subunits, which contain the CAAX farnesylation signal but which are exogenously applied in an unprocessed form. The failure of injected protein to enhance K+ current further indi- cated that K' channels are not directly regulated by GTP- bound ~21"". Due to a practical time limit on the maintenance of the whole cellltight seal configuration, the time to assay the K+ current after injection was short, in most cases 5-20 min after injection. Although this should have suficed for direct interaction between p21"" and the channel, it may have been too short for stimulation of DNA synthesis or morphological transformation (or conversely inhibition of these processes by

31188 Ras f Raf Induction of K' Channel
and single channel records for the FIG. 6. Current-voltage relationship
Cas+-activated K+ channel from raf- transformed cells (22W). Inside-out patch records in symmetric 150 m~ KC1 with 10 p free Ca2+ in the bath solution. Numbers beside records show the holding potential, and bars indicate the closed channel level. See Ref. 20, Fig. 1 for comparison.
- -90mv
- 6 0 m V
- 30mV
- mmv
A
, , , , , , , ,
0 1 2 3 4 5 6 7
Days R%nomI of TEA
B 18
16 - NIH3T3
p ' 4 - I 1 : : 12
- E IO
-
i P
5 - 8 - /gj 0 mnIlOl f ,;/ v PDOF + ChTX-
4 0 1 2 3
Days
FIG. 7. Cell proliferation is inhibited by TEA and ChTx A, ras- transformed Balb 3T3 cells were seeded at 2.5 x lo5 per 35-mm plate in Dulbecco's modified Eagle's medium + 10% CS and treated with vehicle, TEA(10 mM), or TMA(10 mM) on day 0. Trypan blue exclusion was used to verify that cell viability was not affected by TEA or TMA. Medium was changed, and TEA and TMA were replenished each day. Data points represent mean S.E. of counts from duplicate plates, with lack of error bars indicating errors within the size of the symbol. Results are
NIH 3T3 cells were seeded at 5 x lo4 celldwell in Dulbecco's modified representative of three other identical experiments. B, nontransfonned
were stimulated with PDGF (20 ng/ml) in the absence or presence of 200 Eagle's medium + 0.1% bovine serum albumin on day 0. On day 1, cells
n~ ChTX. Controls received medium only. Medium was changed and PDGF, ChTX were replenished every day. Data presentation is the same as for A. Results were repeated in another identical experiment.
injection of anti-p21"" antibody into rus-transformed cells). Therefore, these results remain consistent with our conclusion that p21"" induces K+ channel expression, and they are not in conflict with other studies showing altered DNA synthesis or morphology many hours following p21" or Y13-259 microin- jections (39, 40).
It remains to be determined whether p2lYRaf signaling activates otherwise quiescent K+ channels or induces new K+ channel expression. Attempts to get at the first possibility by
-4 1.
labeling the channel with '261-Ch'IX have been unsuccessful due to the impairment of ChTX binding affinity associated with iodination (data not shown). However, other data favor the idea that Ras/Raf induces K+ channel expression. First, protein in- jection experiments indicate that regulation of the K+ channel by p21"* is not an acute process. Second, AMPPNP, a nonhy- drolyzable ATP analog did not affect channel activity in ruf- transformed cells (data not shown), suggesting that regulation of the K+ channel by Raf is also not an acute, phosphorylation- dependent process. Lastly, transient induction of K+ channels by EGF was blocked by the protein synthesis inhibitor cyclo- heximide. Although this last result could be interpreted as evi- dence for p2lTRaf-mediated expression of accessory proteins critical to K+ channel function, we feel that the most probable conclusion is that K+ channel expression is a direct consequence of Ras/Raf signaling.
The importance of ion channels in mitogenesis has long been recognized (26) in particular, stimulation of K+ channel activity has been observed with mitogenic stimulation, and in some instances cell proliferation is inhibited by K+ channel blockers (38,4244). The Ca2+-activated K+ channel type we have char- acterized is unique from other large (BK) or small conductance (SKI Ca2+-activated K+ channel classes, both in its association with rus-transformation and its physiology and pharmacology (19,20). The constant presence of this channel at high density, regardless of the growth state of transformed cells, suggests that it may play a role in the transformation process. Indeed, a very similar if not identical channel has been found as a pre- dominant channel type in tumor-derived cells (45, 46). Induc- tion of the K+ channel by EGF and PDGF suggests that it may also be important in the proliferation process of normal cells. Interestingly, PDGF, a full mitogen (471, stimulated prolonged upregulation of the Ca2+-activated K+ current, while only a very transient upregulation was observed in response to the partial mitogen EGF.
Our results showing ChTX inhibition of proliferation in nor- mal but not ms-transformed fibroblasts may be explained by work showing ChTX inhibition of mitogenesis in human lym- phocytes, but only if it is added within the first few hours after mitogen stimulation. In addition, serum as low as 0.1% abol- ishes this action of ChTX (44). Since oncogenic p21" is consti- tutively activated, rus-transformed cells are in a sense similar to nontransformed cells under constitutive mitogen stimula-

RaslRaf Induction of K+ Channel 31189
tion. We speculate that failure of ChTX to block proliferation in ras-transformed fibroblasts may be due to this persistent mi- togen-stimulated-like state andor the presence of serum. Two models have been proposed to suggest a role for K+ channels in cell growth: the channels may maintain a negative resting membrane potential, which in turn enhances mitogenic Ca2+ influx (48), or they may contribute to volume regulatory re- sponse linked to initiation of DNA synthesis (26). In PC12 cells, calcium influx leads to mitogen-activated protein kinase acti- vation via ras, suggesting a direct link between ion flux mecha- nisms and this proliferative signal transduction pathway (49). Our results with K+ channel blockers suggest that the p21"V Raf regulated Ca2+-activated K+ channel contributes to growth regulation under certain conditions, but a mechanistic inter- pretation of this phenomenon is still not possible.
The signal transduction systems responsible for mitogen regulation of ion channel expression and activity remain to be elucidated. Nerve growth factor-induced expression of voltage- dependent sodium channels in PC12 cells has been shown to be p2l"'"independent (50,511, and acute regulation of channels by tyrosine kinases has been demonstrated (52-54). We have shown that a unique Ca2+-activated K+ channel is upregulated by growth factors known to stimulate the p2l"VRaf signaling pathway as well as by oncogenic forms of ~ 2 1 " ~ and Raf, and overexpression of cellular p21". Therefore, a role for p2YaS/Raf signaling in mitogen induction of the Ca2+-activated K+ channel seems likely. Since the p21""lRaf signal transduction pathway appears to be highly conserved among eukaryotic organisms, we postulate that the K+ channel regulation described here could also extend beyond murine fibroblasts and account for other reports of ion channel modulation by mitogens or oncogenes.
Acknowledgments-We thank Dr. E. Taparowsky for help with cell transfection, Dr. D. Asai for help with immunofluorescence, and Dr. N. Mascarenhas for help with Western blotting. We also thank DeAnn Black for technical assistance.
REFERENCES 1. Smith, M. R., DeGudicibus, S. J., and Stacey, D. W. (1986) Nature 320,540-543 2. Stacey, D. W., Toudebush, M., Day, R., Mosser, S. D., Gibhs, J. B., and Feig,
3. Hancock, J. F., Magee, A. I., Childs, J. E., and Marshall, C. J. (1989) Cell 57,
4. Jackson, J. H., Cochrane, C. G., Bourne, J. R., Solski, P. A,, Buss, J. E., and Der,
5. Li, N., Batzer, A,, Daly, R., Yajnik, V., Skolnik, E., Chardin, P., Bar-Sagi, D.,
6. Gale, N. W., Kaplan, S., Lowenstein, E. J., Schlessinger, J., and Bar-Sagi, D.
7. Rozakis-Adcock, M., Fernley, R., Wade, J., Pawson, T., and Bowtell, D. (1993)
8. Bruder, J. T., Heidecker, G., and Rapp, U. R. (1992) Genes & Deu. 6, 54S556 9. Aelst, L. V., Barr, M., Marcus, S., Polverino, A., and Wigler, M. (1993) Proc.
Natl. Acad. Sei. U. S. A. 90, 62134217 10. Zhang, X., Settleman, J., Kyriakis, J. M., Takeuchi-Suzuki, E., Elledge, S. J.,
Marshall, M. S., Bruder, J. T., Rapp, U. R., and Avruch, J. (1993) Nature
L. A. (1991) Oncogene 6,2297-2304
1167-1177
C. J . (1990) Proc. Natl. Acad. Sei. U. S. A. 87,3042-3046
Margolis, B., and Schlessinger, J. (1993) Nature 363, 8 5 4 8
(1993) Nature 363, 88-92
Nature 363, 83-85
364,308-313 11. Kyriakis, J. M., App, H., Zhang, X,, Banejee, P., Brautigan, D. L., Rapp, U. R.,
12. Dent, P., Haser, W., Haystead, T. A. J., Vincent, L. A,, Roberts, T. M., and and Avruch, J. (1992) Nature 558, 417-421
13.
15. 14.
16. 17. 18.
20. 19.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30. 31.
32.
33.
34.
35. 36.
37.
38.
39.
41. 40.
42.
43.
44.
45.
46.
47. 48. 49.
50.
51.
52.
53. 54.
Samuels, M. L., Weber, M. J., Bishop, J. M., and McMahon, M. (1993) Mol. Cell.
Hill, C. S., Marais, R., John, S., Wynne, J., Dalton, S., andTreisman, R. (1993) Li, S., and Sedivy, J. M. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 9247-9251
Marais, R., Wynne, J., and Treisman, R. (1993) Cell 73, 381-394 Bourne, H. R., Sanders, D. A,, and McCormick, F. (1991) Nature 349, 117-127 Birnbaumer, L., Abramowitz, J., and Brown, A. M. (1990) Biochim. Biophys.
Rane, S. G. (1991)Am. J. Physiol. 260, ClOpC112 Huang, Y., and Rane, S. G. (1993) J. Physiol. 461,601418 Chen, C., Corbley, M. J. , Roberts, T. M., and Hess, P. (1988) Science 239,
Flamm, R. E., Birnberg, N. C., and Kaczmarek, L. K. (1990)PflugersArch. 416,
Hemmick, L. M., Perneym, T. M., Flamm, R. E., Kaczmarek, L. K., and
Collin, C., Papageorge, A. G., Lowy, D. R., and Alkon, D. L. (1990) Science 250,
Yatani, A., Okabe, K., Polakis, P., Halenbech, R., McCormick, F., and Brown, A.
Dubois, J.-M., and Rouzaire-Dubois, B. (1993) Prog. Biophys. Mol. B i d . 59,
Day, M. L., Pickering, S. J., Johnson, M. H., and Cook, D. I. (1993) Science 365,
Vaidya, T. B., Weyman, C. M., Teegarden, D., Ashendel, C. L., and Taparowsky,
Keyomarsi, K., Sandoval, L., Band, V., and Pardee,A. B. (1991) Cancer Res. 51,
Asai, D. J., and Brokaw, C. J. (1980) J. Cell Biol. 87, 114-123 Crowell, P. L., Chang, R. R., Ren, Z., Elson, C. E., and Gould, M. N. (1991)
Stanton, V. P., Jr., Nichols, D. W., Laudano,A. P., and Cooper, G. M. (1989) Mol.
Sinensky, M., Beck, L.A., Leonard, S., and Evans, R. (1990) J. Biol. Chem. 265,
Cox, A. D., Hisaka, M. M., Buss, J. E., and Der, C. J. (1992) Mol. Cell. Biol. 12,
Bollag, G., and McCormick, F. (1991)Annu. Reu. Cell B i d . 7, 601-632 Hall, A., Cales, C., Hancock, J. F., Lloyd, A., Self, A,, Gardener, S., Houslay,
M. D., Wakelam, M. J. O., and Marshall, C. J. (1988) Cold Spring Harbor
Kolch, W., Heidecker, G., Lloyd, P., and Rapp, U. R. (1991) Science 349 ,426 Symp. Quant. Biol. 53,855462
428 Magni, M., Meldolesi, J., and Pandiella, A. (1991) J. Biol. Chem. 266, 6329-
6335 Feramisco, J. R., Gross, M., Kamata, T., Rosenberg, M., and Sweet, R. W.
(1984) Cell 38, 109-117 Mulcahy, L. S., Smith, M. R., and Stacey, D. W. (1985) Nature 313, 241-243 Gallego, C., Gupta, S. K., Heasley, L. E., Qian, N.-X., and Johnson, G. L. (1992)
Lee, S. C., Sabath, D. E., Deutsch, C., and Prystowsky, M. B. (1986) J. Cell Biol.
Sturgill, T. W. (1992) Science 257, 1404-1407
Biol. 13, 6241-6252
Cell 73,395406
Acta 1031, 163-224
1024-1026
120-125
Birherg, N. C. (1992) J. Neumsci. 12,2007-2014
1743-1745
M. (1990) Cell 61, 769-776
1-21
560-562
E. J. (1991) J. Cell B i d . 114, 809420
3602-3609
J. B i d . Chem. 266,17679-17685
Cell. B i d . 9, 639-647
19937-19941
2606-2615
Proc. Natl. Acad. Sci. U. S. A. 89, 7355-7359
102,1200-1208 DeCoursey, T. E., Chandy, K. G., Gupta, S., and Cahalan, M. D. (1987) J. Gen.
Phvsiol. 89. 405420 Price, M., Lee, S. C., and Deutsch, C. (1989) Proc. Natl. Acad. Sci. U. S. A. 86,
Enomoto, K., Furuya, K., Maeno, T., Edwards, C., and Oka, T. (1991)J. Membr
Sauv6, R., Simoneau, C., Monette, R., and Roy, G. (1986) J. Membr Biol. 92,
Rozengurt, E. (1986) Science 234, 161-166 Grissmer, S., Lewis, R. S., and Cahalan, M. D. (1992) J. Gen. Physiol. 99,6344 Rosen, L. B., Ginty, D. D., Weber, M. J., and Greenberg, M. E. (1994) Neuron
Fanger, G. R., Erhardt, P., Cooper, G . M., and Maue, R. A. (1993) J. Neurochem. 12, 1207-1221
DArcangelo, G., Paradiso, K., Shepherd, D., Brehm, P., Halegoua, S., and 61,1977-1980
Repp, H., Draheim, H., Ruland, J., Seidel, G., Beise, J., Presek, P., and Dreyer, Mandel, G. (1993) J. Cell Biol. 122, 915-921
Catarsi, S., and Drapeau, P. (1993) Nature 363, 353-355 F. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 34034407
Huang, X., Morielli, A. D., and Peralta, E. G. (1993) Cell 75, 1145-1156
~~
10171-10175
B i d . 119, 133-139
269-282