
Pediatric Pathology QinshiPan Orange is stuff she said in class “know this, own this”
34
Pediatric Pathology QinshiPan Orange is stuff she said in class “know this, own this”
-
Upload
jody-bennett -
Category
Documents
-
view
218 -
download
2
Transcript of Pediatric Pathology QinshiPan Orange is stuff she said in class “know this, own this”

Pediatric Pathology
QinshiPanOrange is stuff she said in class
“know this, own this”

DefinitionsNeonatal: 0-4 weeks after birthPerinatal: -5 to 1 month (morbidity/ mortality)Infancy: 1st yearChildhood: Birth – legal adult ageCongenital Anomalies: morphological defects
present at birthMalformations: intrinsically abnormal
morphogenesis (anencephaly, heart defects)
Disruptions: destruction of previously normal structure (amniotic bands)
Deformations: extrinsic disturbance of development (leiomyomas/ oligohydramnios)
Sequence: a pattern of cascade anomalies set off by one initiating aberration (Potter sequence)
Malformation Syndrome: constellation of congential anomalies that are thought to be related but can’t be explained by single initiating event (viral infection)
Agenesis:complete absence of an organ and primordium(renal agenesis)
Aplasia: complete absence of an organ due to primordium development failure
Atresia: absence of an opening usually in hollow organ (trachea/ intestine)
Hyperplasia/Hypoplasia: increase/decrease in cell number (pulmonary hypoplasia)
Hypertrophy/hypotrophy: increase/decrease in cell size
Atrophy: decrease in size, wasting away, arrested development
Dysplasia: abnormal organization of cells (omphalocele [big defect], gastroschisis [sticking out of hole])

EpidemiologyInfant Mortality: lowest in Singapore, highest
in Angola, lowish in USUnder 1 year: Congenital abnormalities/ short
gestation/low birth weight1mo-1year: SIDS1-24 years: accidents and adverse effectsCauses of Congenital anomalies: unknown,
multifactorial, chromosomal abberations, mendelian inheritance, maternal disease state, maternal infections
Most common birth defects:Tri 21 (Down’s), Tri 13 (Patau), Tri 18
(Edwards), cleft palate/lip, tetrology of fallot, spinal bifida
Potter Sequence:Oligohydramnios (usually from renal aplasia/
amniotic leak)Potter Facies: • Ocular hypertelorism• Low-set ears• Receding chin• Flattening of the noseDeformed right foot (talipesequinovarus)Amnion Nodosum (nodules on fetal side of
amniontic sac)Pulmonary hypoplasiaBreech presentation

KaryotypicAbberations: usually during gametogenesis (not inherited)
Trisomy 21: Down’s• Simian Crease• Mental Retardation• Epitcanthic folds• Alzheimer’s @ 40• 4% from Robertsonian translocationDrugs and Chemicals:Retinoic Acid: Accutane, used in acne affects HOXThalomide: used to prevent nausea.
UpregulatesWNT(wingless)Antiepileptics:valproic acid disrupts HOX genesSmoking: low birth weight, prone to SIDSFetal Alcohol Syndrome: dose-dependent, short
palpebral fissures, maxillary hypoplasia, growth retardation, affects retinoic acid and SHH
High in Native American PopulationMCC of mental retardationPhalates: PVC ) can cross placenta and pass into
breast milk testicular problemsRadiation
Single Gene Mutations: 90% autosomal and arise during gametogenesis
SHH(Sonic Hedgehog Gene): Holoprosencephaly
GL13 (downstream of SHH):SynpolydactylyMaternal Diabetes: hyperglycemic mom
hyperinsulinemic baby = GIANT BABYMultifactorial:Cleft lip/palate (alcohol, rubella,
thalidomide)Neural tube defects (folic acid)Congenital dislocation of hip (shallow
acetabulum = genetic, breech =environment)
Congenital Anomalies: 3-9 weeks, peaks at 4-5 when all organ systems are developingPAX genes: DNA binding proteinsPAX 2: Renal-coloboma (kidney, ears, eyes, brain)PAX 3: Waardenburg (pigment and deafness), alveolar rhabdomyosarcoma (PAX3/7)PAX 5: Leukemia (Non-hodgkins B-cell)PAX6: Aniridia (no iris)PAX 7: Thyroid cancer (3 too)PAX8: thyroid cancer

APGAR: probability of survival (Done at 1 and 5 min following birth) Score 0-10, 0-2 for each category: appearance, pulse (100), grimace, activity, respiration
5min APGAR of 0-1 = 50% mortality in 1st 28 days
Birth Injuries: large babies (Maternal DM) in dangerCaput succedaneum (scalp edema)Cephalohematoma (hematoma on scalp)Intracranial hemorrhagesSkeletal fracturesNerve, liver, adrenal lacerationBirth WeightAGA: appropriate (10-90th %)SGA: small (<10th %) LGL: large (>90th %)Fetal Growth Restriction (FGR)1. Symmetrical: chromosomal disorder,
congenital anomaly, infection by a TORCH2. Assymmetric: uteroplacental insufficiency (esp
3rd tri) placenta, cord, insertion problems, Trisomy 7 mosaicism
1. Maternal Causes: preeclamsia, eclampsia, HTN, hypercoagulable states, alcohol, drugs, malnutrition
Gestational Age:Premature: < 37 weeksPostmature: > 42 weeksPremature: 2nd MCC of neonate deathRisks:1. Premature rupture of placental membranes
(PPROM/PROM- TNF and metalloproteinases 1,8,9)TLR-4 upregulated in placentas with chorioamnionitis. Also LPS binding TLR-4 may trigger preterm labor (said in class for us to look it up. R factor moment)
2. Intrauterine infection: can cause or be caused by (P)PROM, increased IL-6 or maternal G-CSF secretion of proteases and PGs (contractions)
3. Uterine, cervical, placental structural anomalies: fibroids, incomptent cervix that doesn’t stay close, placenta previa, placenta accreta
4. Multiple gestationsOrgans affected in preterm:Lungs (no surfactant)Kidneys (immature but OK)Liver (OK but too much bilirubinkernicterus :
treat with UV light)Brain: not developed, use temp/resp regulation to
fix

Respiratory Distress in the newborn. Causes: • Excessive maternal sedation• Fetal head injury• Blood/amniotic fluid aspiration• Intrauterine hypoxia from nuchal cord• MCC: Hyaline membrane disease
• Preterm/AGA, male, maternal DM, multiple gestation, C section before labor.
• Deficiency of pulmonary surfactant (normally lecithin, SP-B, SP-C) secreted by Type II pneumocytes to decrease surface tension in the lungs) induced by labor
• Increased diffusion gradient decreased O2 atelectesis, dilation of alvoili thickened hyaline membranes
Treatment:24-34 weeks: Antenatal treatment with steroids<26-28 weeks: Administer surfactantTherapy with O2= risk of retinopathy (increased
VEGF b/c decreased O2 after stopping O2 treatment) and bronchopulmonarydysplasia) (O2 decreases maturation so lungs stop at saccular stage
Necrotizing Enterocolitis:Occurs after ORAL feedingIntestinal ischemia bacterial colonization /formula aggravates mucosal injury inflammation PAF = mucosal breakdown invasion by bacteriaS&S: Babies present with poor feeding.Complications: May perforate and lead to peritonitis, sepsis, shock therefore SURGICAL EMERGENCYPostNEC strictures
Germinal Matrix Hemorrhage:Preterm infants: subependymal (periventricular hemorrhage) then into ventricles

Transplacental InfectionsT:oxoplasmaO:thers (T. Pallidum)R:ubella (Concep 16weeks)
Cataracts, cardiac, CNS, deafC:MV (MC 2nd trimester) owl eye inclusion,
mainly targets CNS, heptaosplenomegaly, myocarditis
H: erpes simplexParvo B19 (fifth disease) in mother occurs in 1-
5% of pregnancies***Transcervical InfectionsGroup B Strep (worst), some HSV IIPROM frequent with Preterm deliveryChorioamnionitis/funisitisPost delivery Early (0-7days): GBS: acquired at or shortly
before birth pneumonia, sepsis, meningitis
Late (7-90 days): Listeria, Candida, require latent growth period
***Pathognomonic for Parvo B19: infected cells are larger with intranuclear inclusions and rim of chromatin. Infects RBC precursors so RBCs never mature

Fetal Hydrops *** Accumulation of edema in fetus during intrauterine growth
Gross: generalized edema and cystic hygroma on neck
Immune Hydrops:Rh incompatibilityPathophysiology: Rh+ child sensitizes Rh- mother late in pregnancy IgM response (does not
cross placenta) 2ndRh+ child Mom makes IgG (does cross placenta and attacks fetal RBC) anemia, jaundice, hydrops, kernicterus (fetal cord blood is Coombs+)- Anemia extramedularyhematopoiesis and cardiac decompensationhydrops- Hb degradation excess bilirubin Jaundice Kernicterus
Treatment: Rhogam for mothers at 28 weeks and within 72 hours after birth. If hemolysis occurs in utero, treat with low dose of in utero transfusion
ABO IncompatibilityLow incidence b/c:- Most maternal anti-A/B are IgM- Fetal cells express A and B poorly- Most other cells also express A/B so Igs are sopped up- No effective prophylaxis (like Rhogam) therefore ABO incompatibility = MCC of immune
hemolysis in newbornsNon immune Hydrops: CV defects, chromosome anomalies, twintwin transfusion, fetal non-
immune anemia (alpha thalassemia, parvoB19 induced aplastic anemia)
Kernicterus: unconjucated (can cross BBB) bilirubin deposited in brain after birth >20mg/dL usually to basal ganglia, thalamus, cerebellum
Inborn Errors of MetabolismSelf-mutilation Lesch-Nyham Purine Degradation
Sweaty Feet Isovalericacidemia Isovaleric acid CoAdehydrogenase
Musty/mousy odor PKU Phenylalanine hydroxylase
Maple syrup urine Maple syrup disease Branched ketoaciddehydrogenase. Accum: ILV
Cherry Red macula Tay Sachs Hexominidase A
Cherry Red macula + hepatosplenomegaly
Nieman-Pick Sphingomyelinase
Dislocated lens, marfanoidhabitus
Homocysteinuria Cystathione beta synthase
CF: CFTR (MC lethal genetic disease in caucasians. Abnormal CFTR.S&S: pancreatic insufficiency, steatorrhea, malnutrition, heptatic cirrhosis, intestinal obstruction, male infertility, recurrent pulmonary infections, chronic lung disease
Sudden Infant Death Syndrome (SIDS):#1 cause of death 1mo-12mo. (esp 2-4mo) Diagnosis of exclusion (must have negative autopsy)Risks: paternal marijuana, maternal opiate, cocaine use; prone or side sleeping position, sleeping on soft surface, hyperthermia, co sleeping in 1st 3 months, male, premature, poorAutopsy findings:petechiae over thumus, viscera, epicardium, parietal pleura; congestion of lungs and pulmonary edema; astrogliosis of brainstem and cerebellum, hypoplasia of arcuate nucleus in BS
Sudden unexpected infant death: NOT SIDSFind something on autopsyEx: petechie in conjuctiva and lips of intentional suffocation
Pathogenesis (triple risk):1. Vulnerable infant2. Critical development in
homeostatic control3. Exogenous stressor (URT
infection, soft bedding, sleeping prone)
Hypothesis: delayed development of arousal and cardiorespiratory control

ALTE: apparent life threatening event (have been resuscitated)= increased risk of future respiratory death
S&S: prolonged apnea, diminished responses to hypercarbia or hypoxia
Protein-Energy Malnutrition (PEM):Marasmus: caloric deprivation (months)- Somatic compartment affected more- Normal/slightly reduced albumin- Loss of fat and muscle- Hypoplastic BM anemia
(hypochromicmicrocytic anemia)Kwashiorkor: protein deprivation (weeks)- Visceral compartment affected more- Low serum albumin, tranferrin, others- Generalized/dependent edema- Skin with zones of
hyper/hypopigmentation and desquamation
- Hair loss, large liver, lactase deficiency, anemia
Secondary PEM: advanced cancer, malsoprtion, diet restrictions, infections

Pediatric tumors: difficult to differentiate between true neoplasms and tumor-like collections of cells
present b/c of development (MC: benign)
Heterotopia (ectopic) or choristoma: normal cells from a tissue type in the wrong place
Hamartoma: overdevelopment of tissue normally present
Benign Tumors:Hemangiomas: most common tumor of infancy,
usually in skin, ass with von Hippel-Lindau and Sturgeweber, spontaneously regress
Lymphangiomas: skin or deeperFibrous tumor: Congenital infantile fibrosarcoma: t(12;15)
(p13;q25)= ETV6-NTKR3 fusion transcript TK that stimulates Ras (MapK: cell proliferation) and PI3K/AKT (PLC blc-2= inhibits apoptosis)
Teratomas: most common germ cell tumor of children (75% benign, 13% intermediate (immature), 12% frankly malignant (malignancy arising in teratoma))
Peaks at 2yo and late adolescence, younger= benign
Usually sacrococcygeal in females
Malignant Tumors: - Tendency to regress or differentiate (more cures in
children)- Small round blue cell tumors0-4yo: MC: neuroblastomaALL(#1 death) CNS5-9: ALL still most common add in hepatocellular
carcinoma, ewing sarcoma (t(11;22), lymphomaNeuroblastoma: N-myc amp, 17q gain, 1p del- Most common extracranial solid tumor of
childhood, usually in SNS, brain or adrenal medulla- 5 yo: 55%, median age 22 monthsHisto: Homer-Wright pseudorosettes, neurosecretory
granules, neuron-specific enolase (not helpful), Schwann cells (favorable prognosis)
S&S: abdominal mass, blueberry baby, catacholamine secretion (HTN less than pheo) Measure:VMA, HVA in urine
Prognosis: same except 4S: to skin, liver, BM = goodRetinoblastoma: MC malignant eye tumorGermline/somatic RB1 gene: white eye reflex and
Flexner-Wintersteiner rosettesNephroblastoma (Wilms Tumor): MC primary renal
tumor (feel bump in baby’s back), Histo is triphasic: stroma, tubules, blastemal elements)
Medulloblastoma (17p deletion)

WBC
Qinshi PanCytogentics, CD markers, pathognomic/uber
important, probably pretty impt.

CD MarkersCD1: Langerhangs CellsCD3/5: TcCD 4: helper TcCD 8: cytotoxicTcCD 10: immature Bc, follicular lymphomaCD11c: granulocytes, monos, hairy cell leukemiaCD 15: granulocytes and Reed-Sternberg (Hodgkins)CD 16: NK and granuloctyesCD 20/22: Mature Bc (Anti CD20= Rituxan) (Not in Hodgkin)CD 25: Increased in Adult T cell lymphoma (HTLV-1)CD 30: Reed-Sternberg CellCD 34: immature lympho/myelo cells (disappears on promyelocyte)CD 38: Multiple Myeloma, CLL/SLL (bad)CD 45 all leukocytes (Not in Hodgkin)yCD 56: NK and some TCD 79a: BcCD 103: Hairy Cell Leukemia sIg-: immature BcsIg+: mature Bc

Stains:Romanovsky stains: eosin +
methyleneWright Giemsa:
Lymphocytes, segmented neutrophil
Eosinophils
WBC Differential: count 100 WBC and relative # of each type expressed as %
Red marrow in vertebral bodies
Bone marrow aspirate: look at cytology and space occupying lesions
BM cellularity: 100-age until 20%Formed elements: cellular elements of bloodLeukemia: malignancy of BMLymphoma: Tumors from lymphoid tissue

Left Shift: absolute increase in # of immature npsHypersegmentation: PMNs have 5/6 lobes instead of ¾Toxic Granulation: more purple granules (usually bact or
G-CSF, reA change)Dohle bodies: blue patches of dilated ER (reA change)*Relative change in WBC doesn’t matter much, absolute
count matterNeutropenia: too few np (MCC: infection, MCC sig: drugs)
Agranulocytosis (severe neutropenia)Neutrophilia: 1. Increased release from marrow: acute infection/left
shift2. Increased circulation: epi, exercise3. Decreased to tissue: cortisone4. Increased # in marrow: chronic infection, CMLLeukemoid Reaction: elevated WBC and immature
precursor. LAP:(CML[low] vsleukemoid [high])Eosinophila: allergies/parasitesBasophilia: usually CML(9:22 philadelphia)Lymphopenia: usually HIV/SCIDLymphocytosis: chronic immunological stimulation, virus, pertussisMonocytosis: Chronic infections (Tb, SLE, UC)Atypical (Activated) Lymphocytes:mono
Life Span of WBCNp: 1-48 hrs (small% circulating)Bc: hours-daysTc: days to yearsEos: 1-48 hrs (small% circulating)Absolute counts:Np(seg/band): 1,700-7,000/uLLympho: 1,400-3,500/uLEos: 700/uL
Multipotent SC selective/commited cell (myeloid/lymphoid) Colony forming cell WBC/RBC/platelet**All of this depends on interaction with CK environment (GMCS: give to chemo ppl so they don’t get infections

Bone Marrow Transplant: eliminate own BM SC and replace with other SCsSource of SC supportBM: Rich, difficult to harvest, may be tumor contaminatedPeripheral blood: low counts, easy to harvest, mobilization with GFsBoth: not done unless donor is really far awayCord blood: lots of SC, low volume, need multiple donors
Autologous (own cells) Allogeneic (other’s cells)
Source of SC - Bone marrow,-peripheral blood SC harvest
- Identical twin (syngeneic)-HLA-matched sibling-Matched unrelated-Umbilical Cord (2/3 cords added but 1 will win)
- Give higher doses of chemo for cancer (solid tumors, lymphoma, acute leukemia post remission esp if no donors or old)- Allows manipulation of SC (gene therapy)
- Replaced damanged/depleted SC (aplastic anemia)- Replace defective SC (immunodeficiencies, Sickle,thalassemia, m0 storage disorder, myelodysplasia, acute/chronic leukemia)
Complications1. Host factors: Rejection (not enough CKs) 2. Donor: GVHD 3. Tumor factors: Relapse
4: Chemo factors: Organ damage5. Immunosuppresion: infection6: Other: sterility, 2ry malignancy, autoimmune disease

Bone Marrow Failure Syndromes: pancytopenia(all 3 lineages), bone marrow hypoproliferation (aplastic anemia), abnormal maturation (myelodysplasia)
Acquired Idiopathic Aplastic Anemia:Patho: depletion of SCs, suppression of SCs
by T cells (either Tc are whack or SCs are defective)
Causes: idiopathic, toxins, drugs (chloramphenicol), viral (hepB/C, EBV)
S&S: pancytopenia, normal physical, hypocellularmarrow replaced by fat and a few inflammatory cells (mimicked by post chemo)
Therapy: Immunosupression with ATG/cyclosporin, BMT, remove inciting agent (limit exp to drugs and blood transfusion [don’t want to hyperA])
Myelodysplastic Syndromes:>60 bone marrow failure, comes in tiredS&S: pancytopenia, pandysplasia(too many/few lobes, wrong shape), hypercellularity in BMLow grade: Low blast counts, ringed sideroblasts, refractory anemiaHigh grade: excess blasts AMLCytogenetics/Blast count to predict prognosisTreatment: supportive, targeted therapies, immunesuppression (low grade), chemo (high grade), allogenic SC transplantation
Associated with cytogenetic abnormalites (mutations in SC) and can progress to AML
Low platlets = bleedingLow RBC = fatigueLow WBC= infection

Acute Leukemias: malignant disorder of infiltration of marrow by blasts (these blasts secrete CKs that suppress normal SCs problems)
Clinical Feature: fatigue, fever, bleeding, bone and joint painPhysical Findings: B/Tc: splenomegaly, hepatomegaly, lymphadenopathyOther: gum swelling, skin nodules, sternal tenderness,
petechiaeLaboratory Findings:
Blood: pancytopenia& circulating blastsBone: hypercellular replaced by immature blastsLabs: elevated uric acid and LDH
Characteristics: monocloneclonalprogressionclonal dominance extinction of normal clones genetic instability
Pathogenesis: Environmental
RadiationChemicals (benzenes)Chemo drugs
Inherited disorders and DNA repair defects (Fanconi, Downs)Retroviruses (HTLV-1)Somatic Mutation (MCC): idiopathic
Oncogenes: cause tumors when they are activated (TKs and GF-R)Tumor Supressors: cause tumors when they are deleted (p53)Cytogenetics: grow cells in culture with mitogens then look at chromosomes
t (9:22):(Philadelphia chromosome) short= ALL; long=CMLBcr-abl tyrosine kinase: complete oncogene
T (15:17): Acute promyelocytic leukemiaPML-RARA (retinoic acid receptor): partial oncogene (need 2 hits)Mechanism: inhibits differentiation but promotes survival. High [Vit A] induces differentiation
11q23: AML, topo II inhibitorMLL: many different translocations with different cytologies
Secondary to alkylating chemotherapy5q: partial deletion

Classification of Acute Leukemia
ALL (acute lymphoblastic leukemia): committed SC of B/T
AML (acute myelogenous leukemia): multipotent/committed SC of myeloid lines (np, mono, RBC, megas)-Midadulthood, remission for 2 years, usually follows something else (ex. CML, toxins), AUER RODS- PML-RARA: good, Normal: intermediate, 5q/ 11q: poor
Acute leukemias of mixed lineage: multipotent SC that expresses lymphoid and myeloid
ALL/LBL (acute lymphoblastic leukemia/lymphoma): immature B/T cells (lymphoblasts)
- Leukemic: ALL: B-cell, childhood (MC malignancy, good prognosis), BM/LN/spleen, peak in 65-75 adults, poorer prognosis(Hyperdiploid: good (seen in kids), Normal: intermediate, translocation: poor)
- Lymphoma: LBL: T-cell, adolescent males, presents as mediastinal mass, thymic-Highly aggressive w/CNS
Diffuse chromatin, TdT, CD10, CD19, CD 22 markers
Treatment: chemotherapy to kill leukemia & normal but spare SCs (SCs are spared b/c they are supressed from division by CKs from leukemia)CNS: chemo infused into CSFNew: targeted therapies

Myeloproliferative Disorder (chronic leukemia): increase of 1 or more MATURE peripheral blood cell types
Commited germ cell markers: CFU-GM, CFU-E, BFU-E
SC Disorders: mutation in multipotent (mylodysplasia, aplastic anemia), mutation in commited (Acute leukemias, CML)
CML:30-35yoAsymptomatic: early Chronic:splenomegaly, hypermetabolic rate.PB: Increased granulocytes, np, baso (WBC>100,000 sludge decreased perfusion); more turnover= more LDH/uric acidBM: 100% hetergenous cellular (may go to fibrosis) Treat with oral chemo/ gleevec (imantinib) –I bcl-ablAcute:gains c-myc/ras goes to AML/ALLPB: immature np, anemiaBM: 100% homogenous cellular with blastsCytogenetics: Philadelphia 9: c-abloncogene; 22: unknown bcl. Short=ALL long= CML
P. Vera: 40-60 yoPB: increased RBC, np, plateletsBM: hypercellular and goes to fibrosis (looks like CML)Labs: less EPO, more LDH/uric acid)Cytogenetics: JAK2 mutation on many or both chromosomesDdx:1. P.vera: neoplastic2. Hypoxia: reactiveRelative. Stress in smokers: less
plasma V but same RBC so looks like P. Vera
Measure: 51Cr (RBC), CT/exam (spleen), leukocytosis, thrombocytosis, O2 sat.
Essential Thrombocytosis: 20 yo femalesplenomegalyPB: more platletsBM: normal/hypercellular with megakaryocytes in clusters (usually single)Cytogenetics: JAK2 mutation on 1 chrom. Or small # of cells
Idiopathic Myelofibrosis: Like P.Vera/ET w/o symptoms.PB: Nucleated RBCs and teardrop cells from squishing between fibrotic BMBM: thickenend, firbrosed. Increased reticulin and collagenCytogenetics: JAK2
Chronic: hetero Acute: homo

Reactive and NeoplasticLymphoid Conditions
Reactive Conditions: Normal: CD4>CD8; κ:λ 1:2 80% TcellsInfectious Mononucleosis: acute, MCC: EBV, rare: CMVEBV infects BcB/T response CD8 kill BcS&S: severe fatigue, lymphadenopathy, lymphocytosis
(days-weeks)PB: Downey cells (ballerina skirt): lymphocytes surrounded
by RBCs that push on PMDo Monospot test for heterophilIg testLymphopenia: Look at PB for morphology. If Hemogram
abnormality BMBx and aspirate, if immune deficiency flow and look for serum proteins
MCC: Chemo/Radi/Cortisone/EPO/AIDSAcute Nonspecific lymphadenitis:PAINFUL, sudden,
swollen nodes. Acute bacterial cervical/ mesentericLymphadeopathy:PAINLESS (viral unless >4cm then
malignant), sarcodoisis, mets (>60 yo)Chronic Nonspecific Lymphadenitis: PAINLESS
- Bc: follicular hyperplasia (RA, toxo, early HIV)- Tc: paracortical hyperplasia (mono, viral, vacc, dilatin) Histo: difuse- LN Sinusoids: sinus histocytosis (macrophages), look for breast cancer
LN: light zone is where B/T interact
Neoplastic Conditions:
SC
Lymphoid (ALL)
Myeloid (AML)
Bc(CLL)
B/T in LN (Lymphoma)
PC (MM)
Stuff(myeloproliferative disorders)
Within lymphoid:Hodkin: classic or NLPHLBcell: (im)matureTcell: (im)mature
Lymphoma: Dx needs Bx, confirm with flow
S&S:2/3 NHL + Hodkin enlarged nontender LN1/3 NHL extranodal symptoms (skin, stomach,
brain)1. Suppresion of normal hematopoeisi2. Due to circulationg factors (PC Ig, CK
B Symptoms : fever, night sweats, weight loss)
Other: BMF, CNS infl, Immune dysregulation (AIHA/thromb), compression (SC, ureters), obstruction (bowel), pleural/pericardial effusions, ascities
Epidemiology: Hodkin (stable) NHL (increasing)Indolent: CLL/SLL, MALT, follicular lymphoma Aggressive: Mantle Cell, diffuse large BcHighly Aggressive: Burkitt, lymphoblastic
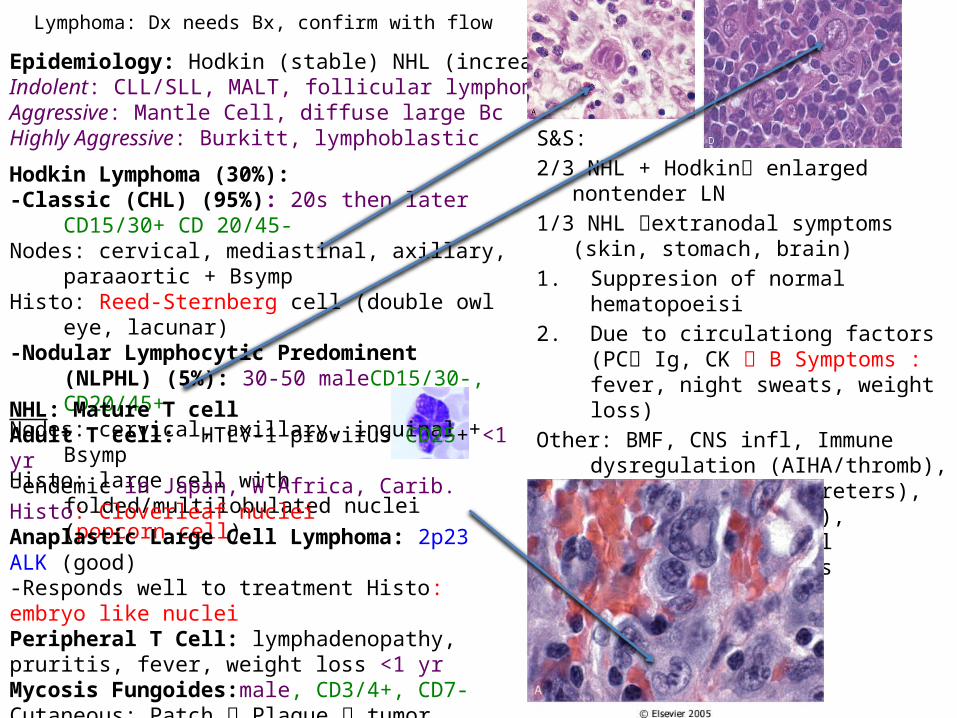
Hodkin Lymphoma (30%):-Classic (CHL) (95%): 20s then later CD15/30+ CD 20/45-Nodes: cervical, mediastinal, axillary, paraaortic + BsympHisto: Reed-Sternberg cell (double owl eye, lacunar)-Nodular Lymphocytic Predominent (NLPHL) (5%): 30-50
maleCD15/30-, CD20/45+Nodes: cervical, axillary, inguinal + BsympHisto: large cell with folded/multilobulated nuclei
(popcorn cell)NHL: Mature T cellAdult T cell: HTLV-1 provirus CD25+ <1 yr-endemic in Japan, W Africa, Carib. Histo: Cloverleaf nucleiAnaplastic Large Cell Lymphoma: 2p23 ALK (good)-Responds well to treatment Histo: embryo like nucleiPeripheral T Cell: lymphadenopathy, pruritis, fever, weight loss <1 yrMycosis Fungoides:male, CD3/4+, CD7-Cutaneous: Patch Plaque tumorSezary Syndrome: leukemic counterpart (general erythroderma)Pautriermicrabscess

NHL:Bc immature (ALL/LBL)Bc Mature:-Burkitts: 8:14 EBVc-mycIgHCD10/19+ Endemic (EBV+, jaw) Sporadic (EBV-, ileo-cecal, bilat breast/ovary), HIVHisto: Starry Sky (macrophage eating Bcells)-Follicular Lymphoma: white adults, 14:18 Bcl-2 Active, CD10+Generalized lymphadenopathy +BM, lots of nodules in spleen. 2 types: small/large Grade based on # of centroblasts-Mantle Cell: old man11:14 Bcl1(cyclin D) IgH CD5+, CD10/23 -, aggressive, hard to cure -Diffuse Large B cell: CD19/20+ No BM50% curable with chemo and Rituxan (Anti CD20), 1 tumor in spleen Subtypes (mediastinal, young female w/CNS, immune def w/EBV, body cavity w/KSHV/HHV8, 60-80% remission-MALT: extranodal inflammation (GI, skin, lung, cry and spit) 80% at 5yrs If gastic: ass w/H. Pylori therefore Abx sensitive-Lymphoplasmacytic: lymphadenopathy, hepatomegaly, splenomegaly, 50% with Waldenstrommacroglobulinemia(high IgM: cryoglobulinemia)Visual, dizzy, stupor, deaf, bleeding, NO LYTIC BONE LESIONS 4-5 yrs survivalHisto: differing degrees of PC maturation

CLL (chronic lymphoid leukemia)/SLL (small lymphocytic lymphoma)CD5+(Tc Ag)/CD23+(Activation marker) western 65yo female
SLL: CLL w/o leukemia Reactive lymphocytes @ fault.40% AsymptomaticLymphadenopathy (cervical/supraclav):
discrete, movable, painlessSplenohepatomegaly: tumor cell invadesInfections (pneumonia &osteomyelitis):
normal immune function disturbedB symptoms:esp when going to worseDdx:Tb: PPD & CultureB. Pertussis: young w/ coughEBV: HeterophileIg and teenagerLeukemic phase of NHL(FL): CD10+Hairy Cell Leukemia: CD 103+Lymphoplasmacytic Lymphoma: more
IgMLarge granular lymphocytic leukemia
Labs: lymphocytosis, anemia (autoimmune = +direct coombs), thrombocytopenia, increased b2m and LDHPB: nucleated RBC (b/c severe anemia), Smudge cell(fragile tumor cell), clump chromatin pattern (CLL) like soccer ball.BM: >30% lymphocytes. Patterns: interstitial, nodular, mixed, diffuse (bad)LN: not organized (effacement) of small lymphocytes with round nuclei, clumped chromatin, scant cytoplasmCytogenetics:-Del 13q (long survival 50%)-Del 11q (20%)bad-Tri 12bad-Del 17p (p53 locus) badTwo types of IgV genus-50-60% SHM of IgV (memory B) = better-40-50% no SHM of IgV (naive B)= bad
Staging: BEST PREDICTOR of survivalBinet: A15, B5(>2LN), C3 (<Hb/Platelet)Rai: Low (∧WBC), Inter (LN), High (∨RBC/Platlets)
Bad Prognosis:∧Prolymphocytes∧Doubling time∧Serum b2m11q/17q/tri12Diffuse BM involvementCD38+No SHM of IgVIndications for Treatment:-AIHA/thrombocytopenia that doesn’t respond to steroids-Repeated episodes of infection 2nd to hypoIgs-Increased risk (more bad)Treat: nucleoside analogs and Rituxin(anti CD20)Complications: LN, big liver, cytopenia, AIHA, Infections, Indolent aggressive (may go to prolymphocytic leukemia, diffuse B cell, Hodgkin (Richter Syndrome)

Other Chronic Lymphoid LeukemiasHairy Cell: CD103+, ∧surface hypermutationS&S: infiltrate BM, liver, spleen massive
splenomegaly, pancytopenia, infections, leukocytosis
Indolent, Sens. To chemo, long lasting remissionLarge Granular: Tγ lympoproliferative diseaseTc (Indolent) NK (aggressive)Histo: Lymphocytes with blue
cytoplasm/granules decreased np and RBC w/o BM
Ass w/ Felty disorder (RA, spleno, np)Extranodal NK/Tcell Lymphoma= lethal midline
granulomaEBV related, sinonasal lymphoma, MC in Asia,
S/Central America
Langerhangs Cell Histocytosis:S100+, CD1+, HLA-DR+, Birbeck Granules (tennis rackets)Multifocal Multisystem (Letterer-Siwe):<2yocutaneouslesion like seborrheicerruption, liver, spleen, pulm, bone lesionsChemo: 50% 5 year (w/o DIE)Unifocal:older children/adults, skeletal m, local excision/radiationMultifocal Unisystem: young children, multiple bony erosions, 50% have DI, spontaneous regression/chemoHand-Scheuller-Christian: calvarial bone defect, DI, exophtalmosPulmonary: 15-40yo, Bilateral interstitial, multiple fine nodules in upper/middle lung, regresses w/ stop smokingOther:Blasticplasmaytoid DC , highly agressive
Plasma Cell: normal PC are perivascularPC Dyscrasia: Bc clone that secretes single homogenous Ig/fragmentsBence Jones P: free light chains (in urine)M component: monoclonal Ig in blood
Plasmacytoma: single massWaldenstrom: more IgMHeavy(light): secretes free fragmentsAmyloid: free light amyloidMGUS: M comp in blood no S&S
Multiple Myeloma:14q32(IgH), CD38+, CD19-50-60 increases, then peak at 80-90, usually black maleS&S: hypercalcemia, infections (MCC death), renal insufficiency (2nd MCC), amyloids, BJP, path fractures, bone pain, Usually: IgG (60%) >IgAUntreated: 6-12mo, Chemo: <5yrMyeloma IL-6 RANKL/OPG/MIP-1 Osteoclastsresorption (BM: high cellular w/ sheets of PCs w/ER)Variants: Flame/Mott cell, Russell/Dutcher body

Spleen- Accessory common- Removes unwanted elements by
phagocytosis, harbors 30-40% platlets, can also undergo extramedullaryhematopoisis
- DCs in periarterial sheath trap Ag show to Tc T/B on edge of white pulp PC in sinus of red pulp
Splenomegaly- MCC: congestive- May rupture espw/mono, malaria, typhoid
fever, lymphoid neoplasm thrombocytopenia, anemia, leukopenia (treat with splenoctomy)
- Mild: Acute infections, congestion, etc- Moderate: everything else- Massive: lymphoma, hairy cell, malaria,
gaucher, primary spleen neoplasmNonspecific Acute Splenitis: blood borne infeCongestive: block vv, cirrhosisInfarcts: block aa (sickle or emboli)Neos:lymphangiomas&hemangiomasγδTc
Thymus- Medulla: 3rd pharyngeal pouch (endo)- Cortex: pharyngeal cleft (ecto)- Increase in size till puberty then get
smaller (replacd by fat)DiGeorge:Thymichypoplasia/aplasiaw/
parathyroid probs22q11(decreased cell mediated immunity)
Thymic Cysts: uncommon, incidental finding. If symptomatic (look for lymphoma/thymoma)
Hyperplasia: more Bc in thymus vs MyastheniaThymomas: >40yo. Neo thymic epithelial cells,
normal Tc. Minimal invasion = complete excision (90% 5yr)
Benign encapsulated: CD5+Malignant:
- Type I: Invasive thymomaCyto OK aggressive- Type II: thymic carcinoma Cyto BAD
Other Carciomas (CD5- so not Tc):MC(squamous), 2nd
(Lymphoepithelioma like w/50% EBV)

RBC
Qinshi Pan
Hematopoiesis: SC RBC/WBC/platlets-can increase 4-5 fold in 7-10 days-usually in BM, can also be in liver, spleen, LNRBCs in intravascular space (not stored)O2 in kidneyEPO(peritubular caps) SC (clusters of dark cells) RBC 40% of blood
RBC: transport O2 and CO2 for 100-120 daysAging: smaller, less elastic, spherical (removed in spleen)4 factos:1. Normal vs impaired Hb2. Acute vs Chronic loss3. Extent of RBC volume loss4. Indirect effects (Fe, spleen, etc)
Clinical Measurements:Hematocrit: RBC mass as % of blood volume (high as baby, dips at few months then men: 38.8-50/ female: 34.9-44.5)Hemoglobin: Total Hb/ volume bloodHct/3=HgbRed Cell Distribution (RDV): measure of anisocytosisLow Hb and Hct (amt of RBC)= anemiaLow MCV (size)= microcytosisLow MCH (color)= hypochromiaHyperchromic does not existHaptoglobin: protein that binds plasma circulating heme, in hemolysis taken out of circulation (acute phase reactant) therefore hemolysis= less haptoglobin
Hemolysis:-Intravascular (sudden, catastrophic): destruction of RBCs in BVsschistocytes, anemia, Hb goes up, Haptoglobin goes down, bilirubin increases, hemoglobinemia, hemoglobinuria Renal failure, DIC-- Immunehemolytic RBC, C’ mediated, severe osmotic stress-Extravascular(slow): chronic, enhancement, amp, of normal physiologic removal of RBCs anemia, elevated EPO, BM hyperplasia (rxn to decreased RBCs)
Anemia:Hb or Hct<2.5 percentile after being adjusted for age, sex, machine, altitude [therefore clinical definition]4 approaches: Etiology, RBC size/morphology, frequency of occurrence, practical (Fe/B12 shot)

Normal
Anisocytosis (size) Poikilocytosis (Shape)
Echinocytes: may be artifact from storing
Teardrop
Spherocytosis
Elliptocytosis
Stomacytosis
Rouleux
Basophilic Stippling: RNA
Howell-Jolly: DNA
Pappenheimer body: Iron
Heinz body: Denatured hemoglobin

Etiology:Blood Loss
- Acute: trauma (normal hgb/hct) 10-15% loss in <1hr: S&S from defect in
vascular volume not lack of O2 carrying capacity
20% loss in <1 hr: shock, postural hypoTN- Chronic: GI/Gyn: gradual b/c BM can increase production by 4-5 fold. S&S if Hb<7
Increased Destruction of RBCs- Intrinsic (Defective RBC):
-Hereditary:spherocytosis, G6PD thalassemia, sickle cell anemia
- Acquired: paroxysmal nocturnal hemoglobinuria- Extrinsic (Normal RBC): AIHA, mechanical trauma, Pb poisoning
Impaired Red Cell production
Intrinsic Defects
Hereditary Spherocytosis (memH): usu AD Ankryn, can also be spectrin. Unstable membrane shear stress in circulation loss of fragments
S&S: 6-9 Hb, spleno, cholelithiasisDx: osmotic fragility test Rx: SplenonectomySequelae: parvo infection aplastic crisisParoxysmal Nocturnal Hemoglobinuria (memA):
GPI anchor mutless CD55/59/C8BP C’ med chronic hemolysis WITHOUT severe hemoglobinuria acute at night
S&S:dark urine (morning), hemolysis and hemoglobinuria
Ass: venous thrombosis, AML
Peripheral Blood Congested Spleen
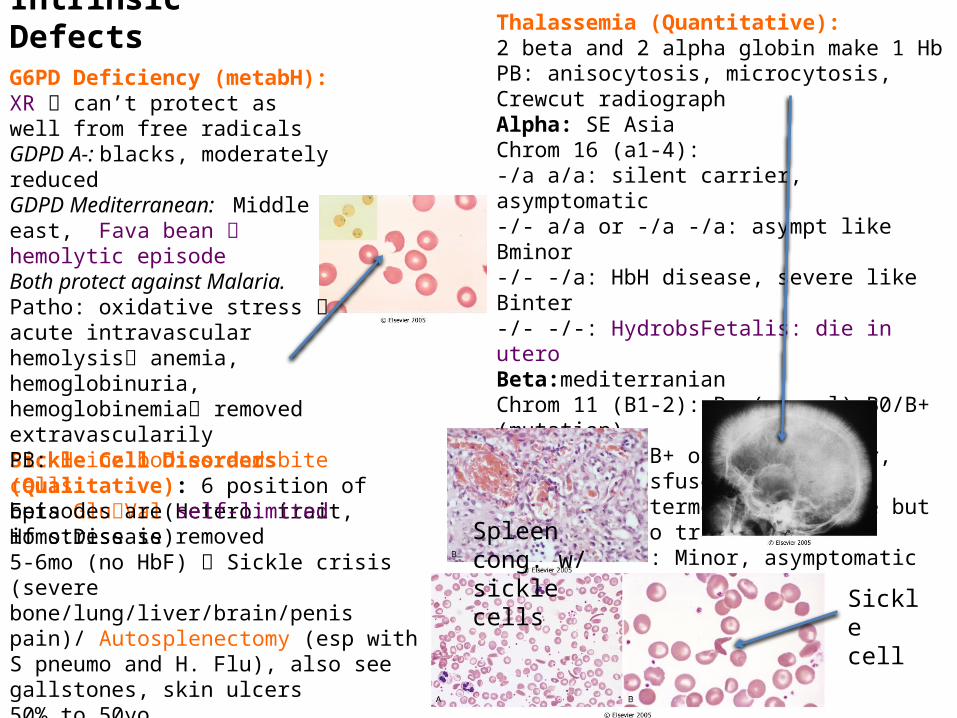
Sickle Cell Disorders (Qualitative): 6 position of beta GluVal(Hetero: trait, Homo:Disease)5-6mo (no HbF) Sickle crisis (severe bone/lung/liver/brain/penis pain)/ Autosplenectomy (esp with S pneumo and H. Flu), also see gallstones, skin ulcers50% to 50yoDx: Hb electrophoresis, DNA testing, HbS solubilityTreat with hydroxyurea
G6PD Deficiency (metabH): XR can’t protect as well from free radicalsGDPD A-: blacks, moderately reducedGDPD Mediterranean: Middle east, Fava bean hemolytic episodeBoth protect against Malaria.Patho: oxidative stress acute intravascular hemolysis anemia, hemoglobinuria, hemoglobinemia removed extravascularilyPB: Heinz bodies and bite cellsEpisodes are self-limited if stress is removed
Intrinsic Defects Thalassemia (Quantitative):2 beta and 2 alpha globin make 1 HbPB: anisocytosis, microcytosis, Crewcut radiographAlpha: SE AsiaChrom 16 (a1-4): -/a a/a: silent carrier, asymptomatic-/- a/a or -/a -/a: asympt like Bminor-/- -/a: HbH disease, severe like Binter-/- -/-: HydrobsFetalis: die in uteroBeta:mediterranianChrom 11 (B1-2): B- (normal) B0/B+ (mutation)B0/B0 or B+/B+ or B0/B+: Major, Severe, transfuseVariable: Intermediate, severe but don’t need to transfuseB0/B-, B+/B-: Minor, asymptomatic
Spleen cong. w/ sickle cells
Sickle cell

Extrinsic Defects
Chemical Toxicity (Pb):Patho: Pb binds sulfhydryl groups in ferrochelatase displaces iron amd makes zinc protoporphyrin/erytrocyteprotoporphyrinPB: hypochro, microcytic anemia, basophilic stipplingTraumatic Hemolytic Anemia: Path: trauma RBCs turn to fragments intravascular hemolysis-Mechanical: prostethic cardiac valve-Microangiopathic: TTP/HUS/DICImmunohemolytic:-Warm (IgG): idiopathic, SLE, drugs, CLL
-Severe, life threatening extravascularhemolysis (hard to treat b/c no compatible blood)
-Cold (IgM):-mycoplasma, mono (younger, abrupt, severe)- idiopathic, Waldenstrom, CLL, DLBCL, splenic lymphoma (older, mild, worse when cold)-Cold (IgG): Cold hemolysin Hemolytic Anemia paroxysmal cold hemoclobinuria. RARE
Anemia caused by impaired RBC productionDecreased(Diminished) or Ineffective Erythropoiesis
– Megaloblastic: B12 and folatedeficiency (nutritional)
– Iron Deficiency (nutritional)– Anemia of Chronic Disease (inhibits
hematopoeisis)– Aplastic anemia and pure red cell aplasia (destroy
SCs)Marrow failure: replacement/displacement marrow space
– Myelofibrosis, primary versus secondary– Space-occupying
• Hematologic malignancy• Metastatic non-hematologic malignancy

Megaloblastic Anemia (B12 and Folate):B12 Deficiency: elevated homocysteine and methylmalonic acid (better sensitivity than decreased serum cobalamin)Pernicious Anemia: Patients have antiparietal cell Ig No IF no Bwe
S&S: glossits, gastric atrophy, neurological (can occur before B12 goes too low) cortex/lateral columns
Usually 60Other causes: Nutritional, malsorption, competitive uptake by parasites, increased requirement (pregnancy/ hyperthyroidism)PB: hypersegmentednp with 6 lobes + leukopenia, severe macrocyticanemia, hyperbilirubinemia (extravascualrhemolysis)BM: ineffective erythropoiesisDx: serum B12, MMA, homocysteine (also up in B9 deficiency), Schillings test, pernicious needs Ig test
Folic Acid: common in alcoholics, pregnant, and those taking methotrexate.Does not present with neurological symptoms
Iron Deficiency:-insufficient intake (diet, malsorption, celiac, removal of ileium (Chrons), secondary to systemic- excessive loss (blood loss, hemodialysis)- excessive use (growing children, pregnancy, lactation)S&S: smooth tongue, spoon nailPB:hypochromatic, microcytic RBC, low serum ferritin/iron/transferrin saturation/stores, increased iron-binding capacityDx: Iron studies then look at reticulocytes to see if there’s BM problemBMBx:Prussian Blue stain for Iron
Anemia of Chronic Disease: Decreased EPO or can’t move Fe to erythroid precursor, MCC in hospitalized patients besides post-surgical and acute hemorrhage-Chronic infections: osteomyelitis/endocarditis-Immune (RA, Chrons)-Malig: Hodkin, Carcinoma of lung/breastHigh serum ferritin (Fe def has low)Definitive: BM has prussian blue macrophageAplastic Anemia: idiopathic, chemo (alkylatin), Chloramphenicol (ABX), Idiosyncratic, physical agents (Hep, CMV, VZ, fanconi, telomerase) FATTY BM- think back to WBCMyelophthsic Anemia: like aplastic but instead of messing with RBCs you have a mass in the bone

Polycythemia: RBC mass > 97.5 percentile (remember to adjust for altitude, age, sex)Erythrocytosis: Increased RBCs with no increase in WBC or plateletsMCC: smoking, high alt., blue bloatersPolycythemiarubravera: see WBC notes (comes with increase in WBC and platlets)-- increased viscosity, CV complications, thrombosis, hemmorrhageDx: phlebotomy
Blood TestingABO and RhDHep B/C, HIV, HTLV, Syphilis
Acute hemolytic Anemia:-Burning along vein, low back pain, chills, fever, shock, increase pulse rate, DIC-Lab: increased bilirubin and LDHFebrile, Nonhemolytic Reaction-Increase in temp by 1 degree, chillsAllergic Reaction:-Urticaria, pruritis, facial/glottal edemaAnaphylactic Reaction:-ANS dysregulation, dyspenea, pulmonary edema, bronchospasm, hypotensionTransfusion Related Acute Lung Injury (TRALI)-Acute respiratory distress in 6 hrs with hypoxemia and bilateral pulmonary infiltrates
ABO Compatibility:• O can only get O (universal donor)• AB can get every type (universal acceptor/ blood hogger)• A can’t have B, B can’t have A
Blood ProductsPlatelets: replaces platlets
10,000/ml if stable, >20,000/ml if unstable or > 50,000/ml in bleeding or surgical patients
Frozen Plasma: replace coagulation factorsactive bleeding or massive transfusions, emergency reversal warfarin effect,
Cryoprecipitatedantihemophilic factorReplace fibrinogen and Factor XIIIReplace factor VIII or vWF (if factor VIII and factor VIII/vWF
Cellular Therapy ProductsWhite cellsStem cells from bone marrow, cord blood, or apheresis


















