
Molecular tools for zoology and botany -...
74
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA --- The use of molecular tools for taxonomic research in zoology & botany
Transcript of Molecular tools for zoology and botany -...

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
The use of molecular tools for taxonomic research in
zoology & botany
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
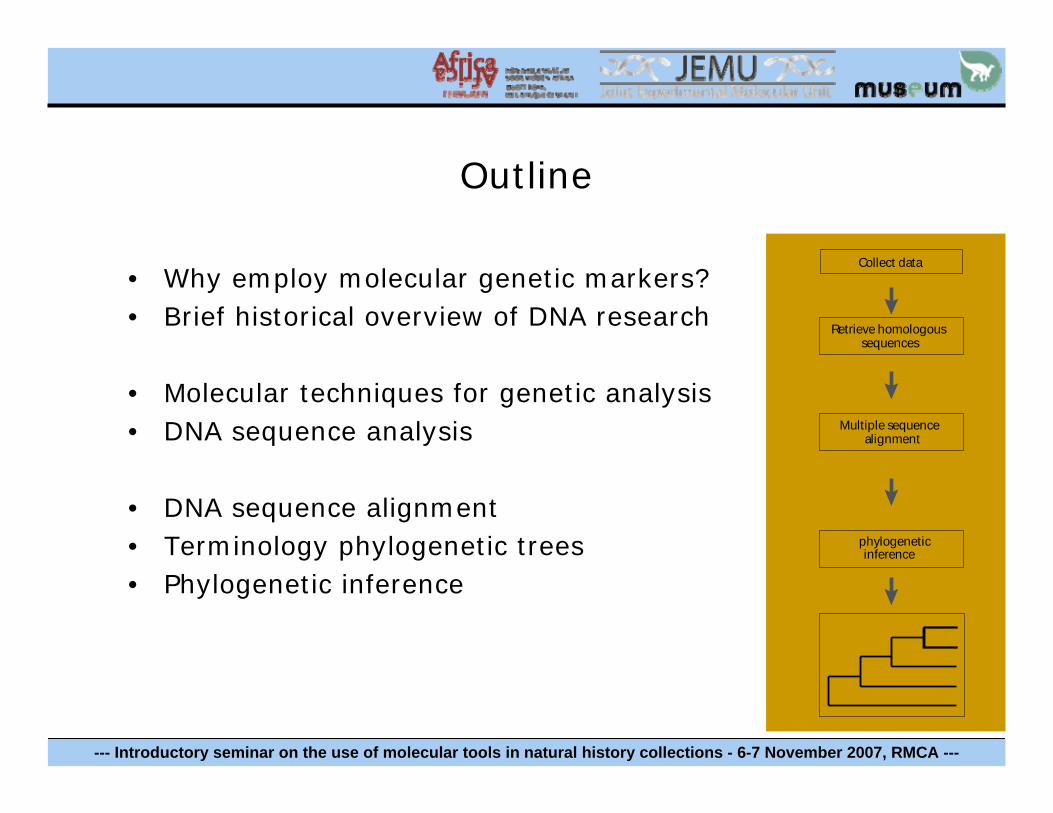
Outline
• Why employ molecular genetic markers? • Brief historical overview of DNA research
• Molecular techniques for genetic analysis• DNA sequence analysis
• DNA sequence alignment• Terminology phylogenetic trees• Phylogenetic inference
Multiple sequencealignment
Retrieve homologoussequences
Collect data
inferencephylogenetic

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Why employ Molecular Genetic Markers
• Systematics: the biological discipline that is devoted to characterizing the diversity of life and organizing our knowledge about this diversity
• Tools– Morphology– Physiology– Behaviour– Embryology– Other organismal characteristics– Genomic information
Carolus von Linnaeus(1707-1778)
Swedish scientist who laid the foundation for modern
taxonomy

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Why employ Molecular Genetic Markers
• Genomic information- Human genome
• 3.000.000.000 bp (3 billion bp)• 20.000 – 25.000 genes• 1.5 % coding for proteins
– Fungi, plants, animals• 10 million bp – 200 billion bp
– Bacterial genomes• 0.5 million bp – 10 million bp
– Protists• 20 million bp – 500 billion bp

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Why employ Molecular Genetic Markers
• Levels of genetic variation– Randomly drawn pairs of homologues DNA sequences from the
human gene pool differ typically at about 0.1% of nucleotide positions
– Two random human genomes differ approximately at 3 million nucleotide positions
– Most other species display higher levels of nucleotide diversity

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Why NOT employ Molecular Genetic Markers
• Molecular Laboratory• Trained staff
– Genetic analysis– Data analysis
• Cost

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Historical overview
• 1944: experimental evidence that DNA is genetic material• 1953: Watson and Crick propose a molecular model for
DNA structure• 1966: Margoliash determines amino acid sequence of
cytochrome c in several taxa and generates the first phylogenetic tree
• 1968: Kimura proposes the neutral theory of molecular evolution
• 1977: Maxam & Gilbert and Sanger et al describe laboratory methods for DNA sequencing
• 1979: Avise et al and Brown et al introduce mtDNAapproaches to study natural populations
• 1981: Palmer et al initiate the use cpDNA for molecular phylogenetic reconstruction in plants

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Historical overview
• 1985: Saiki and Mullis et al report the enzymatic in vitro amplification of DNA via the polymerase chain reaction (PCR)
• 1989: Kocher et al discover conserved PCR-primers to amplify mtDNA fragments from many species (insert picture mtDNA)
• 2001: Publication of draft sequence of the human genome by Lander et al and Venter et al
• 2005: Margulies et al developed a high-throughput parallel sequence technology for sequencing full genomes (454 sequencing)
May 31, 2007– 454 Life Sciences Corporation, in collaboration with scientists at the Human Genome
Sequencing Center, Baylor College of Medicine, announced today in Houston, Texas, the completion of a project to sequence the genome of James D. Watson, Ph.D., co-discoverer of the double-helix structure of DNA. The mapping of Dr. Watson’s genome was completed using the Genome Sequencer FLX™ system and marks the first individual genome to be sequenced for less than $1 million. “When we began the Human Genome Project, we anticipated it would take 15 years to sequence the 3 billion base pairs and identify all the genes,” said Richard Gibbs, Ph.D. , director, Human Genome Sequencing Center, Baylor College of Medicine. “We completed it in 13 years in 2003 – coinciding with the 50th anniversary of the publication of the work of Watson and Dr. Francis Crick that described the double helix. Today, we give James Watson a DVD containing his personal genome – a project completed in only two months. It demonstrates how far sequencing technology has come in a short time.”

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Historical overview
• Interactive Timelines:
– HUGO (http://www.genome.gov/25019887)
– http://www.dnai.org/timeline/index.html

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Molecular techniques
• Protein immunology (since 1904)– Immunological distance between taxa– First method used for phylogenetics
• Protein electrophoresis (mid-1960s)– Starch-gel electrophoresis (SGE)– Allozyme polymorphisms

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Molecular techniques
• DNA technology:– DNA-DNA hybridization
• Yields mean genetic differences across a large fraction of any two genomes
• Source of phylogenetic information (30 000 DNA-DNA hybridizations on 1700 avian species)
– Restriction analysis• Discovery of restriction endonucleases (1968)• Cleave duplex DNA at particular oligonucleotide sequences
(EcoRI 5’-GAATTC-3’)• RFLP: Restriction Fragment Length Polymorphism

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Molecular techniques
• DNA technology:– RAPD Randomly Amplified Polymorphic DNA– AFLP Amplified Fragment-Length Polymorphism– SSCP Single-Strand Conformational Polymorphism– SINE Short Interspersed Elements– STR Short Tandem Repeat (microsatellites)– SNP Single Nucleotide Polymorphism– DNA sequencing

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
DNA sequencing
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Outline
• Why employ molecular genetic markers? • Brief historical overview of DNA research
• Molecular techniques for genetic analysis• DNA sequence analysis
• DNA sequence alignment• Terminology phylogenetic trees• Phylogenetic inference
Multiple sequencealignment
Retrieve homologoussequences
Collect data
inferencephylogenetic

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
DNA sequence alignment
GCGGCCCA TCAGGTAGTT GGTGGGCGGCCCA TCAGGTAGTT GGTGGGCGTTCCA TCAGCTGGTT GGTGGGCGTCCCA TCAGCTAGTT GGTGGGCGGCGCA TTAGCTAGTT GGTGA******** ********** *****
TTGACATG CCGGGG---A AACCGTTGACATG CCGGTG--GT AAGCCTTGACATG -CTAGG---A ACGCGTTGACATG -CTAGGGAAC ACGCGTTGACATC -CTCTG---A ACGCG******** ?????????? *****

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
What is a Multiple Alignment?
• An Alignment is an hypothesis of positional homology between nucleotide bases / Amino Acids.
GCGGCCCA TCAGGTAGTT GGTGGGCGGCCCA TCAGGTAGTT GGTGGGCGTTCCA TCAGCTGGTT GGTGGGCGTCCCA TCAGCTAGTT GGTGGGCGGCGCA TTAGCTAGTT GGTGA******** ********** *****
TTGACATG CCGGGG---A AACCGTTGACATG CCGGTG--GT AAGCCTTGACATG -CTAGG---A ACGCGTTGACATG -CTAGGGAAC ACGCGTTGACATC -CTCTG---A ACGCG******** ?????????? *****

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Multiple Sequence Alignment- Methods
• Manual• Automatic• Combined

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Overview of ClustalW procedure
Quick pairwise alignment: calculate distance matrix
Neighbor-joining tree(guide tree)
Progressive alignment following guide tree
Hbb_Human 1 -Hbb_Horse 2 .17 -Hba_Human 3 .59 .60 -Hba_Horse 4 .59 .59 .13 -Myg_Whale 5 .77 .77 .75 .75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
1
2 3 4
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
1
2 3 4

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW- First pair
• Align the two most closely-related sequences first.• This alignment is then ‘fixed’ and will never
change. If a gap is to be introduced subsequently, then it will be introduced in the same place in both sequences, but their relative alignment remains unchanged.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW- Decision time
• Next consult the guide tree to see what alignment is performed next.
• It can either be two different sequences that are aligned together or a third sequence can be aligned to the first two.
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
1
2 3 4

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW- Alternative 1
• If the situation arises where a third sequence is aligned to the first two, then when a gap has to be introduced to improve the alignment, each of these two entities are treated as two single sequences.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW- Alternative 2
• If, on the other hand, two separate sequences have to be aligned together, then the first pairwise alignment is placed to one side and the pairwise alignment of the other two is carried out.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW- Progression
• The alignment is progressively built up in this way, with each step being treated as a pairwise alignment, sometimes with each member of a ‘pair’ having more than one sequence.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW-Good points/Bad points
• Advantages:– Speed.
• Disadvantages:– No objective function.– No way of quantifying whether or not the
alignment is good– No way of knowing if the alignment is
‘correct’.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
ClustalW- User-supplied values
• Two penalties are set by the user (there are default values, but you should know that it is possible to change these).
• GOP- Gap Opening Penalty is the cost of opening a gap in an alignment.
• GEP- Gap Extension Penalty is the cost of extending this gap.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Advice on alignments
• Treat cautiously• Can be improved by eye (usually)• Often helps to have colour-coding.• Depending on the use, the user should be able to make a
judgement on those regions that are reliable or not.• For phylogeny reconstruction, only use those positions whose
hypothesis of positional homology is unimpeachable

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Outline
• Why employ molecular genetic markers? • Brief historical overview of DNA research
• Molecular techniques for genetic analysis• DNA sequence analysis
• DNA sequence alignment• Terminology phylogenetic trees• Phylogenetic inference
Multiple sequencealignment
Retrieve homologoussequences
Collect data
inferencephylogenetic

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Terminology
• A tree is a mathematical structure that is used to model the actual evolutionary history of a group of sequences or organisms.
• Represents phylogenetic relationship between organisms or genes, consists of nodes connected by braches:
• Terminal nodes, leaves, OTUs (Operational Taxonomic Units) or terminal taxa.
• Internal nodes represent hypothetical ancestors

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Terminology
• Types of phylogenetic trees– Cladogram: shows relative recency of common ancestry– Additive trees (or metric or phylograms): contains additional
information, namely branch lengths, which correspond to the amount of evolutionary change.
– Ultra metric trees (or dendrograms): special kind of additive tree in which the tips are all equidistant from the root

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Terminology• Rooted versus Unrooted trees
– Rooted tree: • root node• direction = evolutionary time.
– Unrooted tree: • specifies relationship between
OTUs• does not define the evolutionary
path.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Terminology

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Terminology
• Homoplasy– Parallel evolution– Convergent evolution– Secondary loss
• Similarity:
– Any 2 sequences can be compared and the similarity computed (% nucleotide identity).
– Allowing gaps, 2 non-homologous nt sequences can have a similarity of up to 50%; for aa sequence this can be up to 20%.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Outline
• Why employ molecular genetic markers? • Brief historical overview of DNA research
• Molecular techniques for genetic analysis• DNA sequence analysis
• DNA sequence alignment• Terminology phylogenetic trees• Phylogenetic inference
Multiple sequencealignment
Retrieve homologoussequences
Collect data
inferencephylogenetic

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Phylogenetic Inference
• Commonly used methods are usually classified into four major groups: – parsimony methods – distance methods– likelihood methods– Bayesian methods

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Phylogenetic Inference

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Cluster methods vs. search methods
• Cluster methods use an algorithm (set of steps) to generate a tree.– easy to implement – computationally efficient– produce a single tree – tree depends upon the order in which we add sequences to the tree
• Search methods use some sort of optimality criteria to choose among the set of all possible trees. – The optimality criteria gives each tree a score that is based on the
comparison of the tree to data– Advantage: search methods use an explicit function relating the trees
to the data – Disadvantage: computationally very expensive (NP complete
problem).

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum Parsimony
• Aims to find the tree topology that can be explained with the smallest number of character changes
• The most parsimonous or most simple explanation is evolutionary also the most likely one
• Given a set of characters, such as aligned sequences, parsimony analysis works by determining the fit (number of steps) of each character on a given tree
• The sum over all characters is called Tree Length
• Most parsimonious trees (MPTs) have the minimum tree length needed to explain the observed characters
• Evaluation of the tree length for all possible topologies

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum Parsimony
Site
1 2 3 4 5
seq 1 A T A T Tseq 2 A T C G Tseq 3 G C A G Tseq 4 G C C G T
1
2
3
4
G A
1
3
2
4
A A
1
4
2
3
A A
Site
Tree 1 2 3 4 5 Total
((1,2),(3,4)) 1 1 2 1 0 5((1,3),(2,4)) 2 2 1 1 0 6((1,4),(2,3)) 2 2 2 1 0 7

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum Parsimony
• Results:
– One or more most parsimonious trees
– Hypotheses of character evolution associated with each tree (where and how changes have occurred)
– Branch lengths (amounts of change associated with branches)
– Various tree and character statistics describing the fit betweentree and data

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum Parsimony
• Advantages:
– is a simple method - easily understood operation
– does not seem to depend on an explicit model of evolution
– gives trees and associated hypotheses of character evolution
– reliable results if the data is well structured and homoplasy is either rare or widely (randomly) distributed on the tree
• Disadvantages
– May give misleading results if homoplasy is common
– Underestimates branch lengths
– Model of evolution is implicit - behaviour of method not well understood

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Distance methods

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Distance methods
• Distance estimates attempt to estimate the mean number of changes per site since 2 taxa last shared a common ancestor
• During evolution, multiple hits can have happened at a single position: the evolutionary distance is almost always larger thanthe dissimilarity (% nucleotide divergence)
Correction
Expected difference based on number of
mutations that happened
Observed difference
Time/Evolutionary distance
Seq
uen
ce d
iffe
rence

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Distance methods
• Computation of evolutionary distances
Convert dissimilarity to evolutionary distanceby correcting for multiple events per site according to a certain model of evolution
T C A A G T C A G G T T C G A
T C C A G T T A G A C T C G A
T T C A A T C A G G C C C G A
1
2
3
1 2 3
2
3
0.266
0.333 0.333
dissimilarity
1 2 3
2
3
0.328
0.44 0.44
evolutionarydistance
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
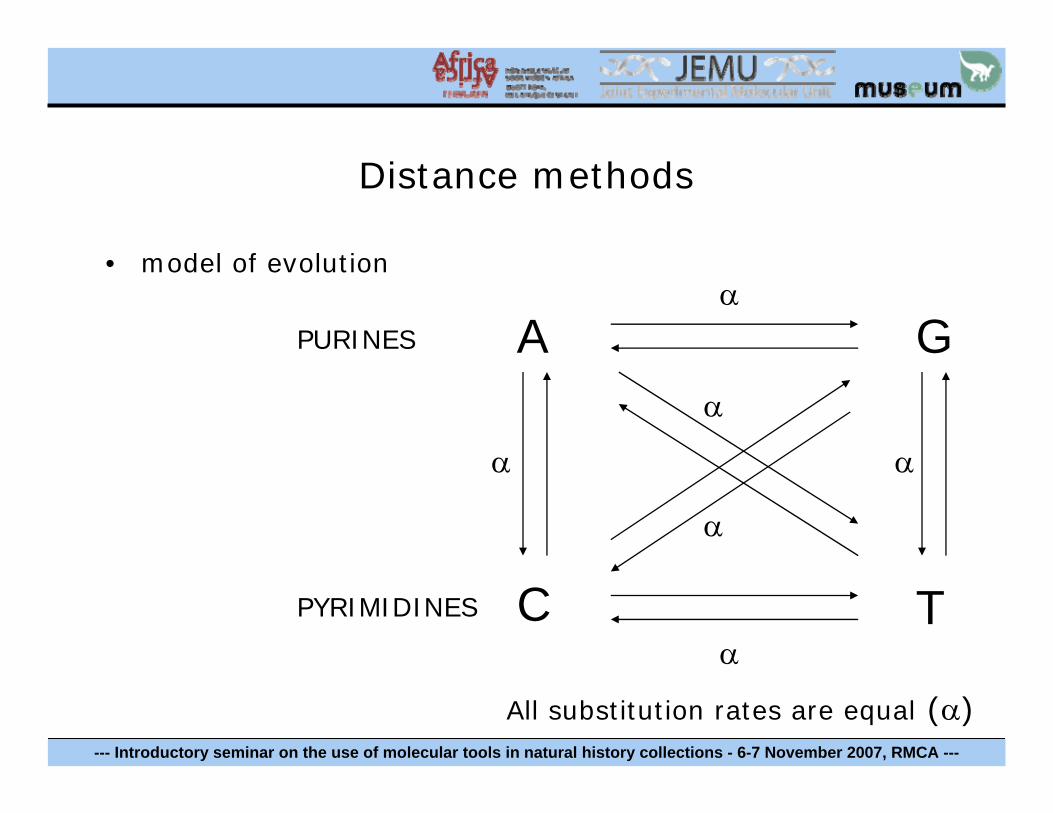
Distance methods
• model of evolution
PURINES
PYRIMIDINES
A
C T
Gα
α
α
α
αα
All substitution rates are equal (α)

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Distance methods
• 4 possible transitions:
– A→G
– C→T
• 8 possible transversions:
– A→C
– A→T
– G→C
– G→T
• Thus if mutations were random, transversions are 2 times more likely than transitions. Due to steric hindrance and chemical properties, the opposite is true, transitions occur in general 2times more often.
• Transversions result in more disruptive amino acid changes

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Distance methods
• model of evolution
PURINES
PYRIMIDINES
A
C T
Gα
α
β
β
ββ
Rate for transitions (α) is different from transversions (β)
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
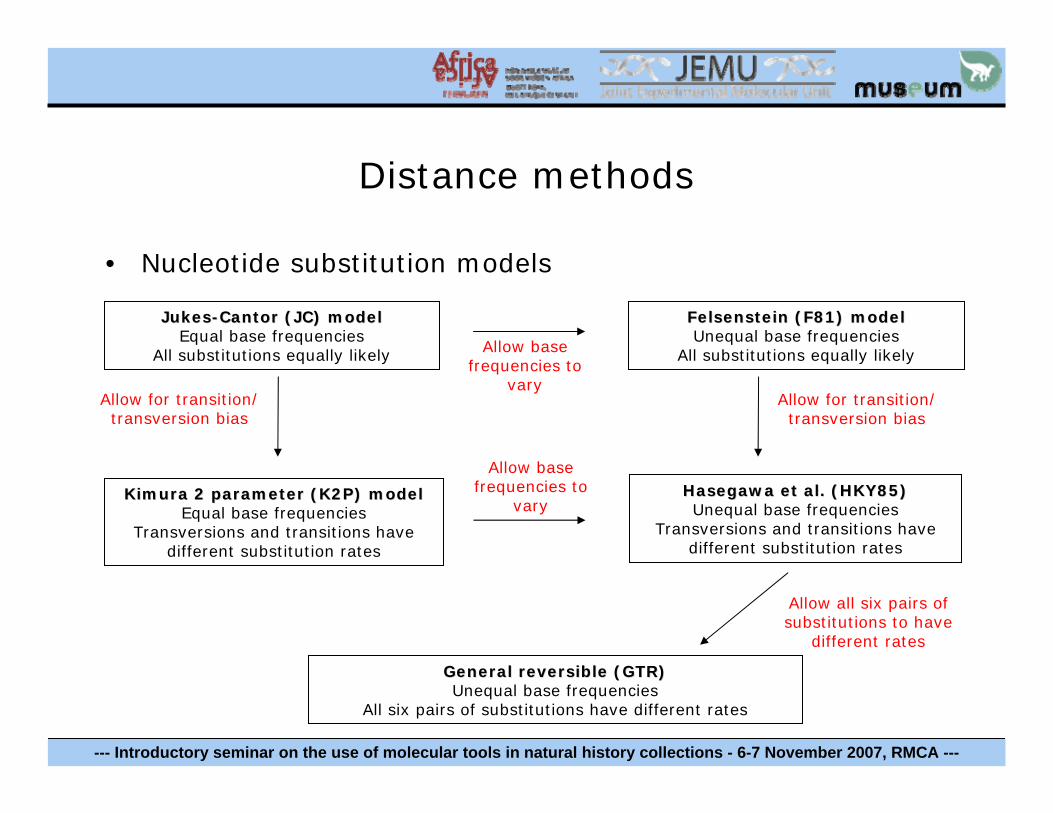
Distance methods
• Nucleotide substitution models
JukesJukes--Cantor (JC) modelCantor (JC) modelEqual base frequencies
All substitutions equally likely
Kimura 2 parameter (K2P) modelKimura 2 parameter (K2P) modelEqual base frequencies
Transversions and transitions have different substitution rates
Allow for transition/transversion bias
FelsensteinFelsenstein (F81) model(F81) modelUnequal base frequencies
All substitutions equally likelyAllow basefrequencies to
vary
Hasegawa et al. (HKY85)Hasegawa et al. (HKY85)Unequal base frequencies
Transversions and transitions have different substitution rates
General reversible (GTR)General reversible (GTR)Unequal base frequencies
All six pairs of substitutions have different rates
Allow basefrequencies to
vary
Allow for transition/transversion bias
Allow all six pairs of substitutions to have
different rates

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Distance methods
• Advantages:– Fast - suitable for analysing data sets which are too large for
ML– A large number of models are available with many parameters
- improves estimation of distances
• Disadvantages:– Information is lost - given only the distances it is impossible to
derive the original sequences– Only through character based analyses (ML, parsimony) can
the most informative positions be inferred– Generally outperformed by Maximum likelihood methods in
choosing the correct tree in computer simulations

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum likelihood methods
• Maximum likelihood methods of phylogenetic inference evaluate a hypothesis about evolutionary history (the branching order and branch lengths of a tree) in terms of a probability that a proposed model of the evolutionary process and the hypothesised history (tree) would give rise to the data we observe
• The likelihood of observing a given set of sequence data for a specific substitution model is maximized for each topology and the topology that gives the highest maximum likelihood is chosen as the final tree.
• The method requires a probabilistic model for the process of nucleotide substitutions.
• Maximum likelihood methods of tree building must solve two problems:– For a given topology, what set of branch lengths makes the observed
data most likely (what is the maximum likelihood value for that tree)?– Which tree of all the possible trees has the greatest likelihood?

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum likelihood methods
• A set of aligned nucleotide sequences for four OTU’s
• What is the probability that this tree could have generated the data under our chosen model of evolution.
• Under the assumption that nucleotide sites evolve independently, we can calculate we can calculate the likelihood for each site separately, and combine the likelihoods into a total value.
• To calculate the likelihood for some site j– consider all possible scenarios – there are 16 possibilities to consider.
• Having calculated the likelihoods at each site, the joint probability that the tree and model confer upon all sites is computed as the product of the individual site likelihoods
• Because the probability of any single observation is an extremely small number, we almost always evaluate the log of the likelihood instead, so the probabilities are accumulated as the sum of the logs of the single site likelihoods.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum likelihood methods
• Advantages:– Mathematically rigorous & performs well in computer simulations– Allows investigation of the fit between model and data – Provides a simple way of comparing trees according to their likelihoods
(difference tests - Kishino Hasegawa Test)
• Disadvantages:– Maximum likelihood will only be consistent (converge on the true tree)
if evolution proceeds according to the assumed model: How well does the model fit the data ?
– Becomes impossible computationally if many taxa or many model parameters

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Choosing Models
• Models can be made more parameter rich to increase their realism:– But the more parameters you estimate from the data the more time
needed for an analysis and the more sampling error accumulates– One might have a realistic model but large sampling errors– Realism comes at a cost in time and precision!– Fewer parameters may give an inaccurate estimate, but more
parameters decrease the precision of the estimate
• In general use the simplest model which fits the data• Compare nested models incorporating additional parameters for
their likelihoods

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Cluster methods vs. search methods
• Cluster methods use an algorithm (set of steps) to generate a tree.– easy to implement – computationally efficient– produce a single tree – tree depends upon the order in which we add sequences to the tree
• Search methods use some sort of optimality criteria to choose among the set of all possible trees. – The optimality criteria gives each tree a score that is based on the
comparison of the tree to data– Advantage: search methods use an explicit function relating the trees
to the data – Disadvantage: computationally very expensive (NP complete
problem).

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Cluster methods
• UPGMA– Unweighted pair group method with arithmetic means– Clustering is done by searching for the smallest distance in pairwise
distance matrix– Only one tree is obtained
• Neighbour-joining
– The NJ algorithm uses as branch length criterion a corrected average of an OTU with all other OTUs:
– unequal branch length are allowed
– Only one tree is obtained
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
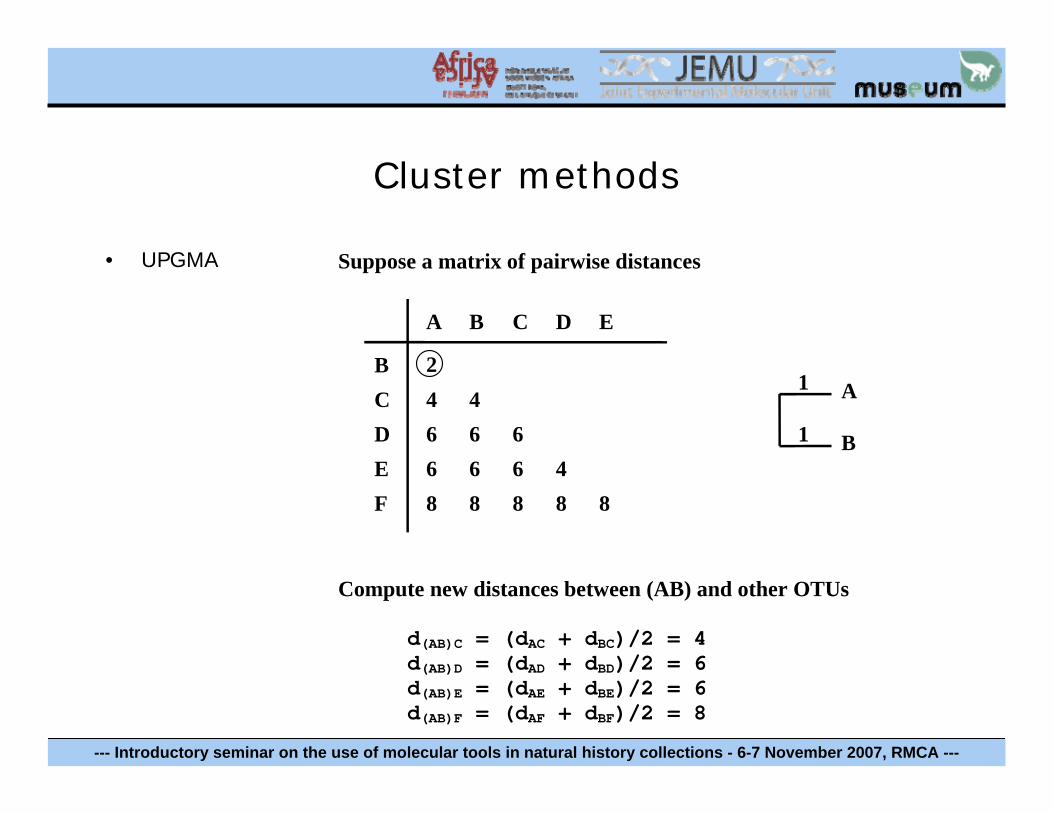
Cluster methods
• UPGMA Suppose a matrix of pairwise distances
Compute new distances between (AB) and other OTUs
d(AB)C = (dAC + dBC)/2 = 4d(AB)D = (dAD + dBD)/2 = 6d(AB)E = (dAE + dBE)/2 = 6d(AB)F = (dAF + dBF)/2 = 8
A B C D E
BCDEF 8 8 8 8 8
6 6 6 46 6 64 42
1
1
A
B

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Clustering methods
• UPGMA(AB) C D E
CDEF 8 8 8 8
6 6 46 64 2
2
D
E
Compute new distances between (DE) and other OTUs
d(DE)(AB) = (dD(AB) + dE(AB))/2 = 6d(DE)C = (dDC + dEC)/2 = 6d(DE)F = (dDF + dEF)/2 = 8

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Clustering methods
• UPGMA
Compute new distances between (ABC) and otherOTUs
d(ABC)(DE) = (d(AB)(DE) + dC(DE))/2 = 6d(ABC)F = (d(AB)F + dCF)/2 = 8
(AB) C (DE)
C
(DE)F 8 8 8
6 64
1
1
A
B1
2 C

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Clustering methods
• UPGMA
(ABC) (DE)
(DE)F 8 8
6
1
1
A
B1
2 C
2
2
D
E
1
1
Compute new distances between (ABCDE) and OTU F
d(ABCDE)F = (d(ABC)F + d(DE)F)/2 = 8

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Clustering methods
• UPGMA
(ABC),(DE)
F 8
1
1
A
B1
2 C
2
2
D
E
1
1
1
4 F

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
search methods
• Exhaustive search: guaranteed to find the minimum tree because all tree topologies are evaluated. Not possible for morethan ±10 sequences
• Branch and bound: guaranteed to find the minimum tree without evaluating all tree topologies: a larger number of taxa can be evaluated but still limited (depends on the dataset)
• Heuristic searches: not guaranteed to find the minimal tree– Uses stepwise addition of taxa– and rearrangement process (branch swapping)

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
search methods
A
B
C
D
A
C
B
D
A
BC
A
D
B
C
A
B
C
D
A
B
C
D
A
B
C
D
A
B
C
D
A
B
C
D
A
C
B
D
A
C
B
D
A
C
B
D
A
C
B
D
A
C
B
D
A
D
B
C
A
D
B
C
A
D
B
C
A
D
B
C
A
D
B
C
E
E
E E
E EE E E
E E E
E E E
1
3
15 105
....
( )Nn
nU n=−−−
( ) !!
2 52 33

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
search methods
• Branch and bound
A
C
B
D16 subst
A
D
B
C17 subst
A
B
C
D15 subst
E
A
B
C
D13 subst
A
B
C
DE
E
EE
E
15 substitutions 15 substitutions with 5 with 5 taxataxa
16 substitutions 16 substitutions with 4 with 4 taxataxa
17 substitutions 17 substitutions with 4 with 4 taxataxa
Do not retain topologies with more Do not retain topologies with more substitutions than encountered in a next substitutions than encountered in a next
step: Only 5 topologies have to be step: Only 5 topologies have to be investigated instead of 15 !investigated instead of 15 !
--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
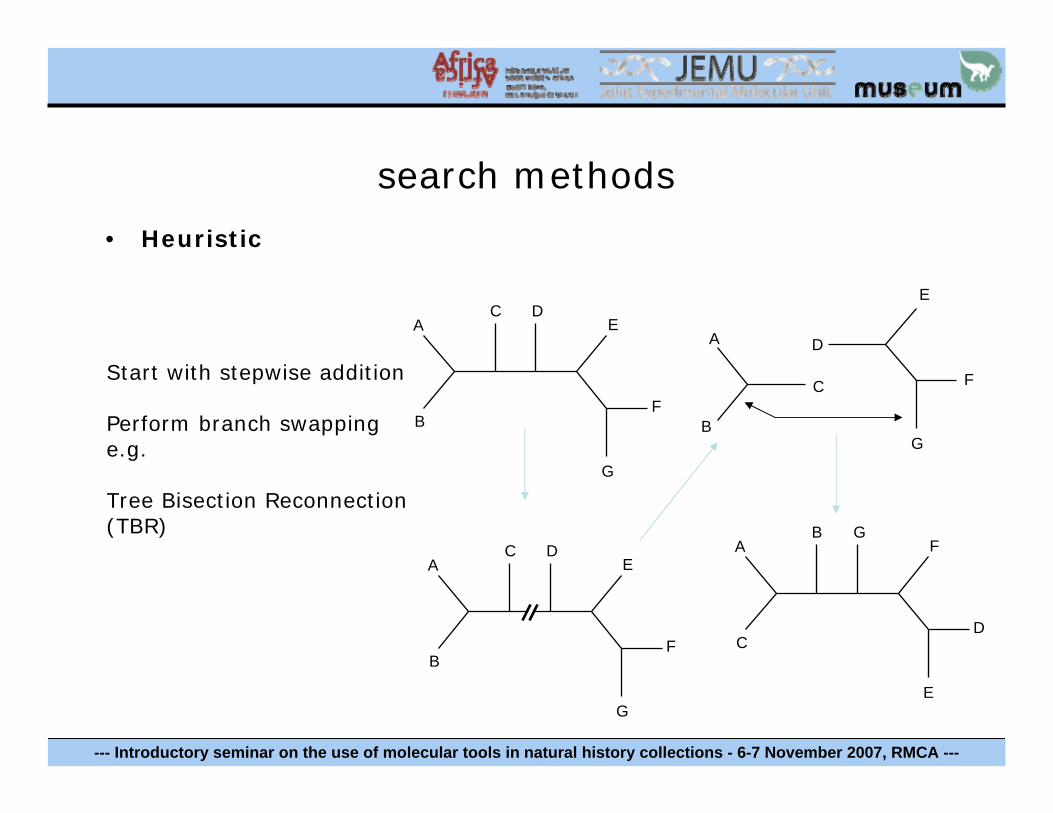
search methods
• Heuristic
A
B
C DE
F
G
A
B
C DE
F
G
A
B
C
D
E
F
G
A
C
B GF
D
E
Start with stepwise addition
Perform branch swapping e.g.
Tree Bisection Reconnection (TBR)

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
search methods
• Heuristic

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Bootstrapping

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Bayesian phylogenetics
– Prior probability Pr[Tree i] :• Probability of tree before observations have
been made– Likelihood Pr[Data|Tree i]
• Proportional to the probability of the observations (=alignment)
• Requires specific assumptions about the process generating the observations (=parameters evolutionary model)
– Posterior probability Pr[Tree i|Data] :• The probability of the tree conditional on the
observations (=alignment)• Obtained by combining prior & likelihood for
each tree using Bayes formula:

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Bayesian phylogenetics
– The optimal tree is the one that maximizes the posterior probability
– Bayesian methods allow complex methods of evolution to be implemented (ML methods have problems when the ratio of data points to parameters is low)
– Baysian methods rely on an algorithm (MCMC, Markov Chain Monte Carlo) that does not attempt to find the highest point in the space of all parameters
– Treats parameters in a different way compared to ML methods. (marginal vs joint estimation)
– Provides support measures (no bootstrapping)

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Bayesian phylogenetics

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Summary
Holder & Lewis 2003 Nature Reviews Genetics (4) 275-283

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Terminology• Gene trees and species trees
– The divergences of genes is longer than the time of species divergence.
– Topology of gene tree can be different from the species tree due to lineage sorting
– depends on • long-term effective population
size• generation time • interval between successive
speciations– When the speciation event occurs
every 1 or 2 million years it is unlikely that the species tree differs from the gene tree.

--- Introductory seminar on the use of molecular tools in natural history collections - 6-7 November 2007, RMCA ---
Maximum likelihood methods
A non-biological example: coin tossing• If the probability of an event X
dependent on model parameters p is written:
P ( X | p )• then we would talk about the likelihood
L ( p | X )• that is, the likelihood of the parameters
given the data.
• Likelihood is the hypothetical probability that an event that has already occurred would yield a specific outcome. The concept differs from that of a probability in that a probability refers to the occurrence of future events, while a likelihood refers to past events with known outcomes.


















