
Modulatoren epigenetischer Regulationsmechanismen · Modulatoren epigenetischer...
239
Modulatoren epigenetischer Regulationsmechanismen: Medizinische Chemie neuer KDM4-Inhibitoren und Methodenentwicklung zur SFC-MS-Analytik von Ketaminmetaboliten I n a u g u r a l d i s s e r t a t i o n zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. Nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Universität Greifswald vorgelegt von Georg Michael Fassauer Greifswald, Juli 2018
Transcript of Modulatoren epigenetischer Regulationsmechanismen · Modulatoren epigenetischer...

Modulatoren epigenetischer Regulationsmechanismen:
Medizinische Chemie neuer KDM4-Inhibitoren und Methodenentwicklung zur
SFC-MS-Analytik von Ketaminmetaboliten
I n a u g u r a l d i s s e r t a t i o n
zur
Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
(Dr. rer. Nat.)
der
Mathematisch-Naturwissenschaftlichen Fakultät
der
Universität Greifswald
vorgelegt von
Georg Michael Fassauer
Greifswald, Juli 2018

Dekan: Prof. Dr. Werner Weitschies
1. Gutachter: Prof. Dr. Andreas Link
2. Gutachter: Prof. Dr. Gerhard Scriba
Tag der Promotion: 19. Oktober 2018

„Der Wille öffnet die Türen zum Erfolg.“
Louis Pasteur (1822-1895)

Inhaltsverzeichnis
II
Inhaltsverzeichnis Abkürzungsverzeichnis V
1 Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen 1
1.1 Einleitung 1
1.1.1 Aufbau des Chromatins und epigenetische Modifikationen 1
1.1.1.1 Lysin-spezifische Histon-Demethylasen 7
1.1.1.2 JumonjiC-Domäne enthaltende Histon-Demethylasen 8
1.1.2 Literaturübersicht über bisher publizierte Histon-Demethylase-Inhibitoren 12
1.1.2.1 Inhibitoren der Lysin-spezifischen Histon-Demethylase 12
1.1.2.2 Inhibitoren der JumonjiC-Domäne enthaltenden Histon-Demethylasen 14
1.1.3 Konzept der Bioisosterie 19
1.2 Untersuchungen, Ergebnisse und Diskussion 22
1.2.1 Synthese von Tetrazol- und Carbonsäurehydrazid-beinhaltenden
KDM4-Inhibitoren 22
1.2.1.1 Anthranilsäurederivate 22
1.2.1.2 Essigsäurederivate 32
1.2.2 Synthese von Tetrazol- und Pyridin-beinhaltenden KDM4-Inhibitoren 38
1.2.2.1 4-(1H-Tetrazol-5-yl)pyridin-2-amine/thiole/thioether/ether 38
1.2.2.2 4-(1H-Tetrazol-5-yl)picolinamide 48
1.2.2.3 4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure 51
1.2.2.4 [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamine 52
1.2.2.5 4-(1H-Tetrazol-5-yl)picolinsäure/2,4-Di(1H-tetrazol-5-yl)pyridin 65
1.2.3 Synthese von Carbonsäure- und Pyridin-beinhaltenden KDM4-Inhibitoren 67
1.2.4 Biologische Testung/Analyse der Kristallstrukturen 69
1.3 Zusammenfassung und Ausblick Kapitel 1 81
2 Auswirkungen von epigenetischer (Fehl-)Regulation auf psychische Erkrankungen 83
3 Analytische Untersuchungen von Ketamin und dessen Metaboliten 86
3.1 Vorbetrachtungen 86
3.1.1 Depressive Erkrankungen und leitliniengerechte Therapie 86

Inhaltsverzeichnis
III
3.1.2 Ketamin als neue Therapiestrategie zur Behandlung (therapieresistenter)
depressiver Erkrankungen 89
3.1.3 Allgemeines zur Superkritischen Fluidchromatographie und ihre Vorteile 94
3.1.4 Besonderheiten der Analyten bei der Detektion mit einem
Single-Quadrupol-Massenspektrometer 97
3.1.5 Parameter der Validierung von chromatographischen Methoden 98
3.1.5.1 Selektivität 99
3.1.5.2 Kalibrierfunktion 99
3.1.5.3 Unterstes Quantifizierungslevel 100
3.1.5.4 Richtigkeit 100
3.1.5.5 Präzision 100
3.1.5.6 Wiederfindung, Matrix-Effekte und Stabilität 101
3.2 Methodenentwicklung und Validierung 101
3.2.1 Entwicklung und Optimierung einer SFC-MS-Methode 101
3.2.2 Validierung der SFC-MS-Methode 108
3.2.2.1 Selektivität 108
3.2.2.2 Kalibrierfunktion 110
3.2.2.3 Unterstes Quantifizierungslevel 114
3.2.2.4 Präzision und Richtigkeit 114
3.2.2.5 Wiederfindung, Matrix-Effekte und Stabilität 116
3.3 Anwendung der SFC-MS-Methode auf Proben einer klinischen Studie als
Proof-of-Concept 117
3.4 Gegenüberstellung der SFC-MS-Methode mit literaturbekannten
LC-Methoden 120
3.5 Zusammenfassung und Ausblick Kapitel 3 122
4 Experimenteller Teil 123
4.1 Geräteparameter 123
4.1.1 NMR-Spektroskopie 123
4.1.2 Schmelzverhalten 123
4.1.3 Dünnschichtchromatographie/Schwerkraftsäulenchromatographie 124
4.1.4 MIR-Spektroskopie 124

Inhaltsverzeichnis
IV
4.1.5 Mikrowellenreaktor 124
4.1.6 MPLC 125
4.1.7 Präparative HPLC 125
4.1.8 Hochauflösende Massenspektrometrie (HRMS) 125
4.1.9 SFC-MS 126
4.1.10 Visualisierung der Kristallstrukturen 126
4.1.11 Kristallographie (Institut für Biochemie, Greifswald) 126
4.1.12 Kristallographie (BESSY II, Berlin) und isothermer Titrationskalorimetrie 127
4.1.13 LANCE-Assay (Institut für Pharmazeutische Wissenschaften, Freiburg) 128
4.2 Chemikalien 128
4.3 Beschreibung der Synthesen 130
4.3.1 Synthese von Tetrazol- und Carbonsäurehydrazid-beinhaltenden
Substanzen 130
4.3.2 Synthese von Tetrazolylpyridinen und Isonicotinsäure-beinhaltenden
Substanzen 143
4.4 Beschreibung der validierten SFC-MS-Methode 195
4.4.1 Probenvorbereitung 195
4.4.2 Säulenauswahl 195
4.4.3 Chromatographische Parameter 196
4.4.4 MS-Parameter 196
4.4.5 Mathematische Prüfung auf Linearität 196
4.4.6 Sonstige experimentelle Daten 198
5 Literaturverzeichnis 202
Eigenständigkeitserklärung 225
Tabellarischer Lebenslauf 226
Eigene Publikationen, Poster und Vorträge 227
Danksagung 229

Abkürzungsverzeichnis
V
Abkürzungsverzeichnis
±I-Effekt Induktiver Substituenteneffekt
±M-Effekt Mesomerer Substituenteneffekt
2OG 2-Oxoglutarat
AcOH Konzentrierte Essigsäure
ACN Acetonitril
Äq. Molare Äquivalente
AMPA α-Amino-3-hydroxy-5-methyl-4-isoxazolpropionsäure
API Active Pharmaceutical Ingredient
BfArM Bundesinstitut für Arzneimittel und Medizinprodukte
Bn Benzyl-Schutzgruppe
BnCl Benzylchlorid
BPR Back Pressure Regulator (Rückdruckregulator)
CRS Chemische Referenzsubstanz
δ Chemische Verschiebung in der NMR-Spektroskopie
d Dublett
DC, dc Dünnschichtchromatographie, dünnschichtchromatographisch
DCM Dichlormethan
DEPT135 Distortionless Enhancement of Polarization Transfer
DIBAL Diisobutylaluminiumhydrid
DIPEA N,N-Diisopropylethylamin
DMF N,N-Dimethylformamid
DMSO Dimethylsulfoxid
DNA Deoxyribonucleic acid
DNK Dehydronorketamin
DNMT DNA-Methyltransferase
ELISA Enzyme-linked Immunosorbent Assay
EMA European Medicines Agency
ESI Elektronenspray-Ionisation
EtOAc Ethylacetat
EtOH Ethanol
FAD Flavin-Adenin-Dinukleotid (oxidiert)
FADH2 Flavin-Adenin-Dinukleotid (reduziert)
FDA Food and Drug Administration
FDH Formaldehyd-Dehydrogenase
FTIR Fourier-Transformations-Infrarotspektroskopie
GC Gas Chromatographie

Abkürzungsverzeichnis
VI
HMBC Heteronuclear Multiple Bond Correlation
HNK Hydroxynorketamin
HPLC High Performance Liquid Chromatography
HSQC Heteronuclear Single Quantum Correlation
IPA Propan-2-ol
IS Interner Standard
IT Ion Trap
ITC isotherme Titrationskalorimetrie
IUPAC International Union of Pure and Applied Chemistry
i. v. intravenös
J Kopplungskonstante
Jmj Jumonji-(Domäne)
KDM Histon-Lysin-Demethylase
KET Ketamin
LANCE-Assay Lanthanide-Chelate-Excite-Assay
LLOD Lower Limit of Detection
LLOQ Lower Limit of Quantification
LM Laufmittel
LSD Lysin-spezifische Histon-Demethylase
m Multiplett (NMR-Spektroskopie), medium (MIR-Spektroskopie)
MAO Monoaminoxidase
MeOH Methanol
MPLC Medium Pressure Liquid Chromatography
MS Massenspektrometrie
MTBE Methyl-tert-butylether (IUPAC: 2-Methoxy-2-methylpropan)
MW Mittelwert
NIH National Institutes of Health
NK Norketamin
NMDA N-Methyl-D-aspartat
NMP N-Methyl-2-pyrrolidon
NMR Nuclear Magnetic Resonance �� Wellenzahl
PDA Photo Diode Array Detector
PDCA Pyridin-2,4-dicarbonsäure
PHD Plant-Homöo-Domäne
p. o. peroral
ppm parts per million
q Quartett

Abkürzungsverzeichnis
VII
QC Quality Control
SQMS Single-Quadrupol-Massenspektrometer
RSD Relative Standardabweichung (Relative Standard Deviation)
s Singulett (NMR-Spektroskopie), strong (MIR-Spektroskopie)
SC Säulenchromatographie
sCO2 Überkritisches CO2
SD Standardabweichung (Standard Deviation)
SFC Supercritical Fluid Chromatography
SIM Selected Ion Monitoring
SPE Solid Phase Extraction
SSRI Selective Serotonine Reuptake Inhibitor
TEA Triethylamin
TEA∙HCl Triethylammoniumchlorid
THF Tetrahydrofuran
TOF Time of Flight
TZA Tri- und tetrazyklische Antidepressiva
ULOQ Upper Limit of Quantification
UV/Vis Ultraviolet/Visible
w weak (MIR-Spektroskopie)
WHO World Health Organization


Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 1
1 Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
1.1 Einleitung
Bereits im Jahr 1940 wurde der Begriff Epigenetik geprägt, um die Bedeutung der
Entwicklung von verschiedenartigen Zelltypen aus ein und demselben Genom zu
adressieren und die Entstehung des Phänotyps in Korrelation von Genen und Umwelt zu
begründen1. Im modernen Kontext wird Epigenetik als potentiell vererbbare, aber durch
Umwelteinflüsse veränderbare Regulation der genetischen Funktion und Expression,
welche auf nicht DNA-codierten Mechanismen beruht, verstanden2. Heute lassen sich viele
Beispiele von epigenetischen Modifikationen aufführen, so werden unter anderem kausale
Zusammenhänge mit zahlreichen Krankheitsgeschehen angenommen. Besonders
eingängig war die Entdeckung, dass Bienenlarven sich auf Grund von differenzierter
Nutrition entweder zur Arbeiterbiene oder zur Bienenkönigin entwickeln konnten. Das
Genom war jeweils identisch, die unterschiedliche Ernährung führte jedoch zu
andersartigen Methylierungsmustern der DNA und damit zu heterogenen Phänotypen3.
Obwohl schon einige Arzneistoffe, welche in epigenetische Regulationsvorgänge
eingreifen, zur Verfügung stehen, ist das Forschungsfeld nach wie vor attraktiv. Im
Folgenden sollen die verschieden Arten von bekannten Modifikationen aufgezählt und
teilweise erörtert werden. Da oftmals chromatinische Strukturen Angriffspunkt von
epigenetischen Veränderungen sind, sollen auch diese kurz erläutert werden.
1.1.1 Aufbau des Chromatins und epigenetische Modifikationen
Auf Grund der Fülle an Informationen, welche im Erbgut gespeichert ist, müssen die
entsprechend langen, linearen molekularen Informationsträger, die DNA-Helices, „gepackt“
(kondensiert) werden. Dieses geschieht in Form von Chromatin, einer Assoziation von
DNA, Histonen, Nicht-Histon-Proteinen und RNA. Strukturell besteht eine Chromatin-
Einheit (Nucleosom) aus einem Histon-Oktamer, zusammengesetzt aus einem Tetramer
von jeweils zwei Histonen H3/H4, zwei Dimeren bestehend aus Histon H2A/H2B und etwa
147 Basenpaaren DNA in helikaler Form, die um das Oktamer gewunden sind4 (Abb. 1
oben). Mit dem fünften Histon H1 wird ein Linker zu Verfügung gestellt, welcher die weitere
Komprimierung zu Chromatinstrukturen höherer Ordnung ermöglicht. Die Packungsdichte
des Chromatins wird unter anderem durch epigenetische Modulatoren beeinflusst und
damit auch eine Möglichkeit zur Regulation der Transkription eröffnet. Das für die Mitose
essentielle, dichter gepackte Heterochromatin ist für Ableseprozesse weniger gut geeignet,
während das aufgelockerte Euchromatin für Transkriptionsabläufe besser zugänglich ist.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
2 | Einleitung
Ein ausgeglichenes Verhältnis beider Chromatinarten ist für die störungsfreie
Genexpression von großer Bedeutung5.
Abb. 1: Oben: Aufbau eines Chromosomen-Kern-Komplexes mit umwindender DNA (PDB: 1KX5) (Farben der Histone entsprechen der unteren Abbildung). Unten: Organisation des Chromatins und N-terminale Histonstränge (modifiziert nach Sun et al.6): Grundlage von eukaryotischen Genomen ist die DNA, welche gewunden um Histone vorliegt. Diese Anordnungen können weiter komprimiert werden und formen so höhere Ordnungen wie Solenoide oder Chromosomen aus. Die vom Histon-Oktamer (bestehend aus jeweils zwei Kopien von H2A, H2B, H3, H4) hinausstehenden N-terminalen Enden können Ziel von posttranslationalen Modifikationen wie Acylierungen und Methylierungen sein (Alterationsmöglichkeiten von Lysinen graphisch markiert, die gezeigten Längen der Histone entsprechen nur einem Ausschnitt).
Posttranslationale Modifikationen sind reversible Vorgänge, so steht in der Regel jedem
„Writer“ (Einfügen einer Modifikation) ein „Eraser“ (Entfernen einer Modifikation)
gegenüber. Mit Hilfe von „Readern“ werden die Modifikationen erkannt und
nachgeschaltete Kaskaden gestartet7. Da der Fokus dieser Arbeit auf den Histon-
Demethylasen liegt, sollen andere epigenetische Modifikationen an dieser Stelle nur
umrissen werden.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 3
Ein nicht direkt Histon-modifizierender epigenetischer Eingriff ist die Methylierung von DNA.
Diese findet hauptsächlich an Position C5 von Cytosin statt und wird von dem Enzym DNA-
Methyltransferase (DNMT), unter Verwendung von S-Adenosylmethionin (SAM) als
Methylgruppendonor, katalysiert. Es sind verschiedene Varianten des Enzyms bekannt
(DNMT1, DNMT3a und DNMT3b8), welche jeweils eigene Erkennungsmuster besitzen9. Die
Entwicklung von Modulatoren der DNA-Methyltransferasen ist fortgeschritten, so dass
bereits Arzneistoffe eine Zulassung erhielten. Mit Decitabin (E1, Abb. 2), einem Cytidin-
Desoxynucleosidanalogon, werden DNMTs gehemmt und so eine Gen-Promoter-
Hypomethylierung provoziert, die in einer Reaktivierung der Tumorsuppressor-Gene und
einer Induktion der Zelldifferenzierung oder der Zellseneszenz, gefolgt von programmiertem
Zelltod, resultieren kann10. Azacitidin (E2) wirkt über einen aktuell weniger gut geklärten
Mechanismus, greift aber ebenfalls mit hemmender Wirkung in die Funktion von DNMT ein,
was eine Hypomethylierung der inadäquat methylierten Gene zu Folge hat und somit in
Krebszellen zur Re-Expression und zur Aktivierung Krebs-unterdrückender Funktionen
führen könnte11. Beide Inhibitoren besitzen eine Zulassung für die Indikation akute
myeloische Leukämie (AML), Azacitidin wird zusätzlich noch angewendet bei
myelodysplastischen Syndromen (MDS) und chronischer myelomonozytärer Leukämie
(CMML). Als „Eraser“ der DNMT können Ten-Eleven-Translocation (TET) Methylcytosin-
Dioxygenasen angesehen werden, welche die neu eingefügte Methylgruppe des
5-Methylcytosins zwar nicht entfernen, 5-Methylcytosin durch Hydroxylierung aber in
5-Hydroxymethylcytosin überführen können12. Interessanterweise ist dieses Enzym
abhängig von 2-Oxoglutarat (2OG) und Fe(II)13. Damit weist es eine gewisse Ähnlichkeit zu
den JmjC-Domänen enthaltenden Histon-Demethylasen auf. Da auch Mutationen der TETs
in Tumoren nachgewiesen werden konnten14, erscheint der Nutzen einer Hemmung oder
Aktivierung von DNMTs beziehungsweise der TETs in hohem Maße individuell zu sein.
Abb. 2: DNA-Methylase-Inhibitoren Decitabin und Azacitidin, Histon-Desacylase-Inhibitoren Vorinostat, Belinostat und Panobinostat.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
4 | Einleitung
Ein weiteres „Writer-Eraser-Paar“ modifiziert freie Aminogruppen der Aminosäure Lysin von
N-terminalen Enden der Histonproteine mit Acylresten (einige Substrate in Abb. 1 unten).
Histon-Acetyltransferasen (HATs) fügen unter Zuhilfenahme von Acetyl-Coenzym A eine
Acetyl-Gruppe ein. Die basisch reagierende, leicht protonierbare Aminofunktion wird
dadurch in ein nicht mehr basisches Carbonsäureamid umgewandelt, was theoretisch zu
einer Verminderung der elektrostatischen Wechselwirkung zwischen positiv geladenem
Protein und negativ geladener DNA führt. Daraus resultieren eine Auflockerung des
Chromatins und eine erhöhte Transkriptionsbereitschaft. Die Rückreaktion, also das
Entfernen von Acetyl- und auch anderen kleinen Acylgruppen, wird durch Histon-
Desacylasen (HDACs) katalysiert. Diese arbeiten entweder Zink-abhängig (Klasse I, II und
IV) oder mittels NAD+ (Klasse III oder Sirtuine)15,16. Für die Zink-abhängigen HDACs
konnten ebenfalls Inhibitoren für die Arzneimitteltherapie zugelassen werden. So zielen die
drei Hydroxamsäurederivate Vorinostat (E3), Belinostat (E4) und Panobinostat (E5, Abb. 2)
auf eine Komplexierung des zentralen Zn2+-Kations ab17. Bei dem cyclischen Peptid
Romidepsin (nicht abgebildet) können nach reduktiver Spaltung einer Disulfidbrücke in vivo
entstandene Thiolgruppen an das metallische Zentrum koordiniert werden18. Auch hier sind
die Indikationen in der Therapie von Tumorerkrankungen angesiedelt (u. a. T-Zell-
Lymphome und Multiples Myelom). Interessanterweise sind darüber hinaus Nicht-Histon-
Proteine wie das Tumorsuppressor-Gen p53 oder der Transkriptionsfaktor NF-κB mögliche
Substrate der HATs/HDACs19. Weitere Modulationen von den vor allem aus dem
Chromosomeninneren herausragenden Histon-Enden können ferner Phosphorylierungen
(„Writer“: Histon-Kinasen, „Eraser“: Histon-Phosphatasen)20 und ADP-Ribosylierungen
(„Writer“: Poly(ADP-Ribose)-Polymerase, „Eraser“: Poly(ADP-Ribose)-Glycohydrolase)21
sein. Modifikationen mit anderen Proteinen wie Ubiquitin („Writer“: E1 (Aktivierendes
Enzym), E2 (Konjugierendes Enzym) und E3 (Ligase), „Eraser“: Ubiquitin-Hydrolasen)22
oder „Small Ubiquitin-related Modifier“ (SUMO) („Writer“: E1, E2 und E3, „Eraser“: Sentrin-
spezifische Proteasen)23 wurden ebenfalls beschrieben.
Die namensgebende posttranslationale Modifikation im Rahmen dieser Arbeit ist jedoch die
Demethylierung von Lysinen an N-terminalen Enden der Histone. Die dazu
entgegengesetzten „Writer“, die Lysin- und Arginin-Methyltransferasen (HMTs), fügen
Methylgruppen an die entsprechenden Aminosäuren an24. Als Cofaktor dient in der Regel
S-Adenosylmethionin, die Unterteilung der Methyltransferase erfolgt nach Aufbau (u. a.
Suv39, G9a oder EZH1) und nach Substrat, wobei hauptsächlich die Histone H3K9, H3K27
und H3K79 das Methylierungsziel darstellen25. Die Auswirkungen auf die Transkription
fallen jedoch weniger einheitlich als bei der Acylierung von Histonen oder Methylierung von
DNA aus. Das liegt auch daran, dass die basischen Eigenschaften der Aminofunktionen
durch das Anfügen einer oder mehrerer Methylgruppen nur geringfügig verändert werden.
So kommt es durch Einfach-Methylierung von H3K9 zu einer Aktivierung, bei einer Zwei-

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 5
oder Dreifach-Methylierung aber zur Drosselung der transkriptionellen Regulation26,27.
Ähnlich verhält es sich mit H3K79, wobei prinzipiell alle drei Methylierungsmuster
aktivierend wirken können, für H3K79me3 aber auch eine Unterdrückung der
Genexpression festgestellt wurde28.
Die Entfernung der Methylgruppen wird durch Histon-Demethylasen katalysiert. Diese
können in Lysin-spezifische und JumonjiC-Domäne (JmjC) enthaltende Demethylasen
unterteilt werden. Letztere erhielt ihre Bezeichnung aus dem Japanischen und bedeutet
wörtlich übersetzt „Kruzifix“, weil bei der Erstentdeckung anormale, Kreuz-ähnliche Gebilde
der Neuralrinnen in Jumonji-mutierten Mäusen gefunden wurden29,30. Erst durch die
Entdeckung von KDM1 (LSD1, Lysin-spezifische Histon-Demethylase) konnte bewiesen
werden, dass die Methylierung von Histonproteinen keinen irreversiblen Prozess darstellt31.
Weitere Forschungsarbeiten förderten Informationen über Vertreter der deutlich größeren
Gruppe der JmjC-Domäne enthaltenden Histon-Lysin-Demethylasen zu Tage. Beide
Klassen unterschieden sich primär in ihrem Mechanismus, mit welchem sie Methylgruppen
von Lysinen entfernen können. Die Substrate hingegen überschneiden sich zum Teil und
sind hauptsächlich am Histon 3 (H3K4, K9, K27, K36 und K79) und geringfügig am Histon
4 (H4K20) lokalisiert32. Der allgemeine Aufbau der verschiedenen Enzyme wurde bereits in
Übersichtsarbeiten zusammengefasst33,34 (Tab. 1). Neben der für die katalytische Funktion
essentiellen Aminoxidase- und JmjC-Domäne sind zahlreiche weitere Motive zu finden,
welche Substrat- oder Protein-bindende und zum Teil namensgebende Eigenschaften
besitzen (z. B. JARID durch Jmj-At-rich-interactive-Domäne).
Über die Auswirkungen von (fehlgeleiteten) repressiven oder aktivierenden Effekten auf die
Transkription von verschiedenen Vertretern unter- oder überregulierter Histon-
Demethylasen wurden ebenfalls mehrere Übersichtsarbeiten publiziert32,35,36. Indirekt
lassen sich daraus auch mögliche Indikationen für Inhibitoren oder Aktivatoren
interpretieren. Diese gehen in eine ähnliche Richtung wie die bereits zugelassenen DNMT-
und HDAC-Inhibitoren, so konnten beispielsweise Überexpressionen von KDM1 in Brust-,
Prostata-, und Blasenkarzinomen festgestellt werden37,38. Auch für die KDM4-Enzymreihe
wurden Hinweise gefunden, dass es unter anderem zu einer Überregulierung in
Prostatatumoren gekommen war39-41.
Im Folgenden soll genauer auf die katalytischen Mechanismen der KDM1- und KDM4-
Enzymreihe eingegangen werden. Erstere ist ebenfalls für den zweiten Teil dieser Arbeit
von Interesse. Im Falle der KDM4 soll der Fokus für diesen Abschnitt besonders auf die
Varianten A und D gelegt werden.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
6 | Einleitung
Tab. 1: Übersicht, struktureller Aufbau und Substrate der KDM-Familie (modifiziert nach33,34).
Bezeichnung Domänen Substrat(e) KDM1A (LSD1) H3K4me1/me2, H3K9me1/me2 KDM1B (LSD2) H3K4me1/me2 KDM2A (JHDM1A) H3K36me1/me2 KDM2B (JHDM1B) H3K4me3, H3K36me1/me2 KDM3A (JMJD1A) H3K9me1/me2 KDM3B (JMJD1B) H3K9me1/me2 KDM3C (JMJD1C) H3K9me1/me2 KDM4A (JMJD2A) H3K9me2/me3, H3K36me2/me3,
H1.4K26me2/me3 KDM4B (JMJD2B) H3K9me2/me3, H3K36me2/me3,
H1.4K26me2/me3 KDM4C (JMJD2C) H3K9me2/me3, H3K36me2/me3,
H1.4K26me2/me3 KDM4D (JMJD2D) H3K9me2/me3,
H1.4K26me2/me3 KDM4E (JMJD2E) H3K9me2/me3,
H3K56me3 KDM5A (JARID1A) H3K4me2/me3 KDM5B (JARID1B) H3K4me2/me3 KDM5C (JARID1C) H3K4me2/me3 KDM5D (JARID1D) H3K4me2/me3 KDM6A (UTX) H3K27me2/me3 KDM6B (JMJD3) H3K27me2/me3 KDM7A (JHDM1D) H3K9me1/me2, H3K27me1/me2 KDM7B (PHF8) H3K9me1/me2, H4K20me1 KDM7C (PHF2) H3K9me2 KDM8 (JMJD5) H3K36me2
Aminoxidase-Domäne
JmjC-Domäne Plant-Homöo-DomäneJmjN-Domäne
Tudor-Domäne
CXXC Zinkfinger-Domäne Leucin-rich-repeat-Domäne
At-rich-interactive-Domäne C5HC2 Zinkfinger-Domäne
Tetratricopeptid-Domäne
SWIRM-Domäne CW-Typ Zinkfinger-Domäne
F-Box-Domäne

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 7
1.1.1.1 Lysin-spezifische Histon-Demethylasen
Der Mechanismus, mit welchem LSD1 Methylgruppen von Lysinresten an N-terminalen
Enden vom Histon H3 entfernt, ist abhängig von Flavin-Adenin-Dinukleotid (FAD) und läuft
wie folgt ab (Abb. 3): Die Oxidation eines mono- oder dimethylierten Lysins wird dadurch
katalysiert, dass FAD ein Hydridion aufnehmen kann. Das unter der Hydridabgabe
gebildete Iminiumion wird im weiteren Verlauf unter nichtkatalytischen Bedingungen mit
Wasser zum niedersubstituierten Amin, einem Proton und einem Molekül Formaldehyd
gespalten. Entstandenes FADH2 kann dann unter Einwirkung von molekularem Sauerstoff
über eine radikalische Spezies unter Abgabe von Wasserstoffperoxid wieder regeneriert
werden. Zur Oxidation des methylierten Lysinrestes wird ein freies Elektronenpaar benötigt,
weshalb nur mono- oder dimethylierte, aber keine trimethylierten Lysinreste als Substrat in
Frage kommen42. Es konnte jedoch nachgewiesen werden, dass auch Nicht-Histone, wie
Protein p5343 oder DNMT144, als Ziel adressiert werden.
Abb. 3: FAD-abhängiger Mechanismus der LSD1 (Erklärungen siehe Text).
LSD1 und 2 sind Monoaminoxidasen und stehen daher in enger Verwandtschaft zu anderen
Enzymen, die dieses katalytische Zentrum aufweisen45. Die Monoaminoxidasen (MAO) A
und B stellen beispielsweise schon seit längerer Zeit ein Target für Arzneistoffdesign dar.
Mit der Inhibition dieser Enzyme können sowohl depressive Erkrankungen als auch Morbus
Parkinson behandelt werden. Dass die Entwicklung von Inhibitoren der LSD1 davon stark
beeinflusst wurde, wird in Abschnitt 1.1.2.1 herausgearbeitet.
Strukturell enthält LSD1 neben der für die katalytische Aktivität notwendigen FAD- und
Substrat-bindenden Subdomäne noch eine SWIRM- (in den Proteinen Swi3p, Rsc8p und
Moira als erstes entdeckt) und eine Tower-Domäne (Abb. 4). Letztere ermöglicht die
Bindung anderer Proteine, wie zum Beispiel von CoREST (Repressor element-1 silencing

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
8 | Einleitung
transcription factor corepressor)46. LSD2 weist diese Tower-Domäne nicht auf, verfügt dafür
aber über eine Zinkfinger-Domäne47.
Abb. 4: Bändermodell der humanen LSD1 mit Cofaktor FAD (rot), ohne N-flexible Region und Abschnitte des C-Terminus (PDB: 2HKO).
1.1.1.2 JumonjiC-Domäne enthaltende Histon-Demethylasen
Die Klasse der JmjC-Domänen enthaltenden Enzyme ist in ihrer Funktion abhängig von
den Cofaktoren Fe(II) und 2OG. Wie der Name bereits suggeriert, besitzen alle Enzyme
dieser Reihe mindestens die katalytisch aktive JmjC-Domäne48. Sie sind im Gegensatz zur
LSD1/2 in der Lage auch trimethylierte Lysinreste zu demethylieren. Der Mechanismus läuft
dabei wie folgt ab (Abb. 5): Die durch zwei Histidinreste, einen Glutamat- oder Aspartatrest
und drei Wassermoleküle komplexierte Eisen(II)-Spezies wird im ersten Schritt durch ein
2OG-Molekül chelatisiert. Mit der Annäherung eines Substrates kann molekularer
Sauerstoff über Fe2+ gebunden werden. Es erfolgt eine Decarboxylierung des 2OG unter
Oxidation des Eisens zu einer reaktiven Fe(IV)-oxo-Spezies (vgl. auch49,50). Mit Abgabe des
Sauerstoffatoms auf die Methylgruppe des Substrates und der Bildung eines Halbaminals,
wird das Eisen wieder auf Stufe II reduziert. Im letzten Schritt wird durch Wassermoleküle
der Ausgangszustand der Komplexierung wiederhergestellt und das um eine Methylgruppe
verringerte Substrat unter Abgabe von Formaldehyd und dem Decarboxylierungsprodukt
des 2OG, Bernsteinsäure, freigegeben.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 9
Abb. 5: Katalytischer Mechanismus der JmjC-Domäne enthaltenden Histon-Demethylasen (Erklärungen siehe Text).
Die JmjC-Domäne enthaltenden Enzyme werden der Cupin-Superfamilie der
Dioxygenasen zugeordnet51. Eine Übersicht über den verschiedenen Aufbau von Vertretern
dieser Klasse wurde bereits weiter oben gegeben (Tab. 1). Neben der JmjC-Domäne
besitzen alle Enzyme der KDM4-Reihe, wie auch die Vertreter der KDM5-Reihe, die für die
strukturelle Integrität wichtige N-terminale Jumonji-Domäne (JmjN). Zusätzlich können
Plant-Homöo- und Tudor-Domänen vorhanden sein, welche für die Bindung von
methylierten Lysinresten und damit für die Substratselektivität zuständig sind33. KDM4D
und E fehlen diese beiden Domänen, sie sind daher bedeutend kürzer als KDM4A, B und C.
Mittlerweile konnten einige röntgenkristallographische Untersuchungen von verschiedenen
KDM4-Enzymen mit und ohne 2OG oder mit Inhibitoren vorgenommen und Strukturdaten
generiert werden (u. a.52-54). Häufig wird wegen der Praktikabilität nicht unter Luftausschluss
gearbeitet und deshalb aus Stabilitätsgründen bei der Kristallisation Ni2+- anstelle von Fe2+-
Kationen eingesetzt, da diese nicht von Luftsauerstoff oxidiert werden. Im eigentlichen
Zentrum der KDM4-Enzymreihe befinden sich die Motive der JmjN- und JmjC-Domäne,

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
10 | Einleitung
eine β-Haarnadel und ein kurzes C-terminales Ende. Letzteres formt mit zwei Cysteinresten
der JmjC-Domäne ein Zinkfinger-Motiv, es ist also neben dem Fe2+- auch ein Zn2+-Ion
vorhanden53 (Abb. 6, PDB: 2GP5).
Abb. 6: Katalytisches Zentrum der humanen KDM4A (PDB: 2GP5): Cofaktor 2OG (rot) komplexiert das zentrale Fe2+-Ion.
Für den weiteren Verlauf dieser Arbeit sind die Enzyme KDM4A und KDM4D von speziellem
Interesse, weshalb der genaue Aufbau der katalytischen Zentren an dieser Stelle verglichen
werden soll. Wie bereits erwähnt, fehlen KDM4D (UniProt: Q6B0I6) die beiden PHD- und
Tudor-Domänen, weshalb dieses Protein mit insgesamt 523 Aminosäuren deutlich kürzer
ausfällt als KDM4A (UniProt: O75164) mit 1064 Aminosäuren. Da KDM4A in dem
verwendeten biologischen Assay nicht in nativer Form zum Einsatz kam, sondern ein
katalytisch aktives, auf die Aminosäuren 1-359 gekürztes Fragment eingesetzt wurde, kann
dieses gut mit KDM4D verglichen werden (Abb. 7). Es wird deutlich, dass die drei zur
Komplexierung des Fe2+-Ions notwendigen Aminosäuren in beiden Strukturen identisch
sind. Ferner decken sich die Bestandteile, welche für relevante Interaktionen wichtig und
im weiteren Verlauf dieser Arbeit von Bedeutung sind. Lediglich das Aminosäurepaar
Histidin/Asparagin 90 unterscheidet sich dabei. Das heißt, dass Ergebnisse, welche mit
einem der beiden Enzyme erhalten werden, prinzipiell auch auf das andere übertragbar
sein sollten.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 11
Physiologisch werden KDM4A, B und C weitreichend in humanen Geweben, wie
beispielsweise in der Milz, in Ovarien oder im Darm, exprimiert. Hingegen findet sich eine
ausgeprägte Expression von KDM4D und E vor allen im Hoden wieder, wobei die Funktion
dort nach wie vor noch nicht genauer geklärt ist33. In Experimenten mit Knockout-Mäusen
konnte durch Eingriff in die Regulation von KDM4A entdeckt werden, dass die Bildung von
hypertrophem Herzmuskelgewebe beeinflussbar war55. Durch einen Knockout von KDM4A
in Brusttumorzelllinien konnte deren Proliferationsbereitschaft gestoppt werden56. Ähnliche
Beobachtungen wurden auch mit Colontumorzelllinien gemacht57. Des Weiteren spielen die
Vertreter der KDM4-Enzymreihe bei der Entwicklung von embryonalen Stammzellen eine
wichtige Rolle58.
Auf Grundlage der bisherigen Befunde scheint die Klasse der KDM4-Enzyme ein attraktives
Ziel für aktuelle Forschungen darzustellen. Trotz der mittlerweile mehr als eine Dekade
zurückliegenden Erstbeschreibung sind bisher noch keine regulierend eingreifenden
Moleküle über den experimentellen Status hinaus gelangt. Bei der LSD1 hingegen konnten
1 10 20 30 40 50 60 70
---MASES-E TLNPSARIMT FYPTMEEFRN FSRYIAYIES QGAHRAGLAK VVPPKEWKPR ASYDDIDDLV
METMKSKANC AQNPNCNIMI FHPTKEEFND FDKYIAYMES QGAHRAGLAK IIPPKEWKAR ETYDNISEIL
# # ## ## # ## ### # #### ## ########## ###### # ## #
71 80 90 100 110 120 130 140
IPAPIQQLVT GQSGLFTQYN IQKKAMTVRE FRKIANSDKY CTPRYSEFEE LERKYWKNLT FNPPIYGADV
IATPLQQVAS GRAGVFTQYH KKKKAMTVGE YRHLANSKKY QTPPHQNFED LERKYWKNRI YNSPIYGADI
# # ## # # #### ###### # # ### ## ## ## ######## # ######
141 150 160 170 180 190 200 210
NGTLYEKHVD EWNIGRLRTI LDLVEKESGI TIEGVNTPYL YFGMWKTSFA WHTEDMDLYS INYLHFGEPK
SGSLFDENTK QWNLGHLGTI QDLLEKECGV VIEGVNTPYL YFGMWKTTFA WHTEDMDLYS INYLHLGEPK
# # ## # # ## ## ### # ######### ####### ## ########## ##### ####
211 220 230 240 250 260 270 280
SWYSVPPEHG KRLERLAKGF FPGSAQSCEA FLRHKMTLIS PLMLKKYGIP FDKVTQEAGE FMITFPYGYH
TWYVVPPEHG QRLERLAREL FPGSSRGCGA FLRHKVALIS PTVLKENGIP FNRITQEAGE FMVTFPYGYH
## ###### ###### #### # # ##### ### # ## ### # ###### ## #######
281 290 300 310 320 330 340 350
AGFNHGFNCA ESTNFATRRW IEYGKQAVLC SCRKDMVKIS MDVFVRKFQP ERYKLWKAGK DNTVIDHTLP
AGFNHGFNCA EAINFATPRW IDYGKMASQC SCGEARVTFS MDAFVRILQP ERYDLWKRGQ DRAVVDHMEP
########## # #### ## # ### # # ## # # ## ### ## ### ### # # # ## #
Abb. 7: Vergleich der Aminosäuresequenzen von KDM4A und KDM4D: erste Reihe: Position; zweite Reihe: KDM4A; dritte Reihe: KDM4D; vierte Reihe: bei Übereinstimmung Markierung mit #, usw. Die grün hervorgehobenen Aminosäuren werden zum Komplexieren des Fe2+-Ions benötigt, die rot markierten Stellen repräsentieren relevante Wechselwirkungspartner für die in dieser Arbeit synthetisierten Inhibitoren. Die Zählweise der jeweiligen Positionen der Aminosäuren ist an KDM4D angepasst.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
12 | Einleitung
bereits erste klinische Studien mit Inhibitoren durchgeführt werden. Das nächste Kapitel soll
eine Übersicht über die aktuelle Entwicklung von KDM- und LSD-Inhibitoren geben.
1.1.2 Literaturübersicht über bisher publizierte Histon-Demethylase-Inhibitoren
Die Aktualität des hier behandelten Themas wird auch dadurch unterstrichen, dass in den
letzten Jahren zahlreiche Übersichtsarbeiten publiziert worden sind. So lassen sich
allgemeine59,60, spezifische61 und drei hochaktuelle Arbeiten, welche im Jahr 2018
erschienen sind62-64, finden.
1.1.2.1 Inhibitoren der Lysin-spezifischen Histon-Demethylase
Da nach der Entdeckung und Aufklärung von LSD1 relativ schnell klar wurde, dass eine
Homologie zur MAO-A und -B bestand, war es nur folgerichtig, bekannte MAO-Inhibitoren
auch auf ihre biologische Aktivität gegen LSD1/2 zu testen. Der irreversible MAO-Hemmer
Tranylcypromin (E5) zeigte in einem orientierenden Versuch einen IC50-Wert von 2 µM und
diente damit als Leitstruktur für weitere Derivatisierungen65 (Abb. 8). Ein durch nucleophilen
Angriff an den Cyclopropylrest erhaltenes FAD-Addukt sorgte bei LSD1/2 ebenfalls für
einen irreversiblen Bindungsmodus. Da E5 bereits ein zugelassener und somit gut
untersuchter Arzneistoff war, konnten mehrere klinische Studien zur Behandlung von akuter
myeloischer Leukämie (AML) vergleichsweise schnell initiiert werden66,67. Auch die Derivate
GSK2879552 (E8) und ORY-1001 (E9) sind beziehungsweise waren ebenfalls Gegenstand
von Phase-I-Studien68,69. Damit stellen die Substanzen auf Tranylcypromin-Basis bisher die
am weitesten fortgeschrittenen Inhibitoren aller Histon-Lysin-Demethylasen in der Klinik
dar. Ferner wurden auch die MAO-Hemmer Phenelzin und Pargylin weiter modifiziert, um
beispielsweise die Potenz oder die Selektivität gegen LSD1 zu steigern. Mit Bizine (E10)
und dem Propargyl-Lysin-derivatisierten Peptid E11 (basierend auf einer modifizierten
H3-Substratpeptidsequenz) wurden zwei Analoga veröffentlicht70,71. Beide Inhibitoren
weisen ebenfalls irreversibles Bindungsverhalten auf, da der „Warhead“ (ein Hydrazin- oder
Propargyl-Rest) eine kovalente Bindung mit dem Enzym ausbildet.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 13
Abb. 8: Auswahl einiger auf Tranylcypromin, Phenelzin oder Pargylin basierender LSD-Inhibitoren.
Gleichermaßen wurden reversible Inhibitoren der LSD1/2 synthetisiert und publiziert, wobei
die Heterogenität der Substanzen höher ist als bei den irreversiblen Inhibitoren (Abb. 9). So
wies das relativ große und unselektive (Bis)thioharnstoff-Derivat E14 einen IC50-Wert von
4,8 µM gegen LSD1 auf. Das mit einem Benzoesäurehydrazid ausgestatte Molekül E12 war
mit einem IC50-Wert von 0,013 µM deutlich potenter72 und konnte in einer Folgearbeit durch
Modifikationen noch verbessert werden73. Substanz E13 mit 3-(Piperidin-4-yl-
methoxy)pyridin-Struktur besaß wiederum völlig andere Bindungsmotive, konnte die LSD1
aber mit einem IC50-Wert von 0,029 µM potent und reversibel hemmen74.
Abb. 9: Auswahl von reversiblen LSD1-Inhibitoren.
Cl
OH
NNH
O
SN
O
O
O
E12
IC50 = 0,013 µM
N
N
O
E13
IC50 = 0,029 µM
HN
HN
S
HN
HN
HN
S
HN
E14
IC50 = 4,8 µM
NH

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
14 | Einleitung
1.1.2.2 Inhibitoren der JumonjiC-Domäne enthaltenden Histon-Demethylasen
Mittlerweile ist die Anzahl an Inhibitoren der Fe(II)- und 2OG-abhängigen KDMs nicht nur
durch Originalarbeiten, sondern vor allem durch Patentliteratur merklich angestiegen.
Herausfordernd bei der Entwicklung ist zum einen die Selektivität, da die Enzyme mit der
katalytisch wirksamen JmjC-Domäne einen sehr ähnlichen Aufbau besitzen. Zum anderen
kam oftmals hinzu, dass sehr polare oder amphotere Grundkörper als Leitstrukturen genutzt
wurden und deren schlecht zellmembrangängigen Eigenschaften nur mit Mühe beseitigt
werden konnten.
Der theoretische Ansatz für viele Inhibitoren war die physiologische Komplexierung von
Eisen durch 2OG. Durch Verdrängung dieses Cofaktors durch ein ebenfalls Metall-
chelatisierendes Molekül wird die Bindung von 2OG unterbunden, weshalb für die
Demethylierung dann weniger 2OG zur Verfügung steht. Da dieser Vorgang prinzipiell bei
allen Vertretern der Enzymklasse möglich ist, wird diese Gruppe (zumindest die nicht weiter
derivatisierten Vertreter) zu den pan-Inhibitoren gezählt (Abb. 10).
Abb. 10: Auswahl an pan-Inhibitoren der JmjC-Domäne enthaltenden KDMs.
Einen der ersten Inhibitoren stellte N-Oxalylglycin (NOG), das Aza-Analogon von 2OG, dar.
Neben der mangelnden Selektivität innerhalb der KDM-Enzymreihe, wurden auch andere
2OG-abhängige Proteine wie die humane HIF Prolyl-Hydroxylase PHD2 oder die
Asparaginyl-Hydroxylase FIH gehemmt54. In derselben Publikation wurden ebenfalls
Pyridin-2,4-dicarbonsäure (2,4-PDCA) und das Bipyridin E15 vorgestellt. Für 2,4-PDCA
konnte sogar eine Röntgenkristallstruktur gelöst werden, welche zeigte, dass das zentrale
Fe2+-Ion von dem Pyridinstickstoffatom und der Carboxygruppe in Position 2 chelatisiert
wurde. E15 komplexierte das katalytische Metall über seine beiden Pyridinstickstoffatome.
In einer Folgearbeit wurden dann verschiedene Monoamide von E15 hergestellt, wodurch
HO
O
O
OH
O
2-Oxoglutarat (2OG)
HO
O
NH
O
OH
O
N-Oxalylglycin (NOG)
IC50(KDM4E) = 24 µM
N
O
O
OH
OH
Pyridin-2,4-dicarbonsäure (2,4-PDCA)
IC50(KDM4A) = 0,7 µM
IC50(KDM4E) = 1,4 µM
N
O O
N
OH
O
E15
IC50(KDM4E) = 6,6 µM
N
OH
O OH
N
N
HN
N
Cl
JIB-04, E17
IC50(KDM4A) = 0,445 µM
IC50(KDM4E) = 0,34 µM
IC50(KDM5A) = 0,23 µM
IOX1, E16
IC50(KDM4A) = 1,7 µM
IC50(KDM4E) = 2,4 µM

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 15
eine Inhibition im nanomolaren Bereich erreicht werden konnte75. Ein weiterer unselektiver
Inhibitor und Chelator von Eisen, basierend auf dem Zusammenwirken einer
Hydroxyfunktion und einem Chinolinstickstoffatom, wurde als IOX1 (5-Carboxy-8-
hydroxychinolin, E16) veröffentlicht. Die IC50-Werte lagen hierbei im einstelligen
mikromolaren Bereich76. Auch dieses Molekül sollte später, wie fast alle der hier genannten,
als Ausgangsstruktur für weitere Modifikationen genutzt werden. Das Pyridin-Hydrazon-
enthaltende Molekül JIB-04 (E17) kann ebenfalls zu den pan-Inhibitoren gezählt werden.
Die IC50-Werte lagen im submikromolaren Bereich77, erreicht wahrscheinlich durch die
Komplexierung des Eisens durch jeweils ein Stickstoffatom vom Pyridinring und
Hydrazonelement.
Als Weiterentwicklungen mit 8-Hydroxychinolin-Grundkörper (8HQ) wurden unter anderem
die nano/mikromolaren Inhibitoren E18 und E19 publiziert. Durch Derivatisierungen mit
lipophilen Resten an Position 6 oder 7 konnten Selektivität und Potenz gesteigert
werden78,79.
Abb. 11: Leitstrukturanaloga mit 4-Hydroxychinolin und 2,4-PDCA-Grundstuktur (Auswahl).
Die wohl am häufigsten aufgegriffene Struktur stellte die Isonicotinsäure dar. Zwei Beispiele
für direkte Arylverknüpfungen waren die Substanzen E20 und E21 (Abb. 11). Die
Derivatisierung fand jeweils an Position 2 des Pyridinrings statt und resultierte in
N
OH
HN
O
N
OH
Cl
SO
E18
IC50(KDM4B) = 0,01 µM
E19
IC50(KDM4C/E) = 5 µM
N
O OH
HN
O
N
O OH
N
NHN
O
N
O OH
HN
HOO
OH
E22
IC50(KDM4A-E) < 0,1 µM
IC50(KDM5C) = 0,1 µM
E20
IC50(KDM4A) = 0,37 µM
E21
IC50(KDM4C) = 12 µM
N
N
HNO O
N
N
E23
IC50(KDM4C) = 0,5 µM
IC50(KDM5C) = 0,06 µM

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
16 | Einleitung
unterschiedlichen Komplexierungsmustern des Fe2+-Ions. E20 konnte ein zusätzliches
Stickstoffatom des Pyrimidinrings dafür nutzen80, während bei E21 eine phenolische
Gruppe verwendet wurde81. Auch hier lagen die IC50-Werte im nanomolaren Bereich. Eine
Kopplung an Position 3 der Isonicotinsäure mit einem Amin führte gleichermaßen zu einem
potenten Inhibitor (E22) mit nanomolaren IC50-Werten82. Die zwei Stickstoffatome waren
hierbei räumlich nicht mehr in der Lage, eine Komplexierung des Eisens vorzunehmen.
Kristallstrukturanalysen zeigten, dass offenbar eine alleinige Koordinierung des
Pyridinstickstoffatoms ausreichte, um eine potente Inhibition zu garantieren. Der gleichen
Arbeitsgruppe um Westaway gelang es im Anschluss, die für die Zellpermeation hinderliche
Säure-Base-Amphoterie zu unterdrücken, indem sie die Carbonsäure in einen Heterozyklus
(E23) überführten83. Damit wurde gleichzeitig auch klar, dass eine saure Gruppe in
Position 4 des Pyridins nicht unverzichtbar ist.
Daminozid (E23), ein Pflanzenwachstumsregulator, zeigte eine verhältnismäßig hohe
Selektivität und hemmte insbesondere KDM2A und 7A84. Die Struktur ähnelt dem 2OG,
anstatt der Carbonsäurefunktion ist ein dimethyliertes Hydrazid vorhanden. Darauf
aufbauend, konnten Rüger et al. durch einen bioisosteren Austausch der zweiten
Carboxyfunktion mit einem Tetrazol fragmentartige Leitstrukturen entwickeln. E25 zeigte
die stärkste inhibitorische Wirkung gegen KDM4A auf, war gleichzeitig aber deutlich
schwächer wirksam gegen KDM5A und 6B und konnte daher im gewissem Maße als
tendenziell selektiv bezeichnet werden85.
Abb. 12: 2-Oxoglutaratmimetika mit bioisosterem Austausch als Grundlage.
Dem Prinzip der Chelatisierung ließ sich nicht nur mit Pyridinen, Hydraziden und Phenolen
nachgehen, sondern auch mit den von HDAC-Inhibitoren häufig verwendeten
Hydroxamsäuren. E26 war ein Beispiel dafür, dass mit so einem Strukturelement JmjC-
Domäne enthaltende Enzyme ebenfalls gehemmt werden konnten. Als Grundlage diente
wieder 2OG, welches über Seitenketten verschiedener Länge derivatisiert wurde86 (Abb.
13).
Weniger homogen ging es bei der Entdeckung von Motiven natürlichen Ursprungs zu.
Nachdem diskutiert wurde, ob Curcuminioide als Inhibitoren von Histon-Demethylasen
verwendet werden könnten87, ergab ein virtuelles Screening mit anschließender Testung
eines Nitro-Curcumin-Derivats (E27), dass auch diese Substanz inhibitorische
Eigenschaften gegen die KDM4-Enzymreihe aufweist88. Das in zahlreichen Pflanzen als

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 17
Sekundärmetabolit vorkommende Epigallocatechingallat (EGCG, nicht gezeigt) konnte in
biologischen Testungen ebenfalls inhibitorische Eigenschaften (IC50(KDM4E) = 3 µM)
aufzeigen89. Kritisch muss dazu jedoch angemerkt werden, dass beide Stoffklassen
Merkmale von Pan-Assay Interference Compounds (PAINS) in sich tragen (u. a.
Polyphenole und Michael-Systeme).
E28 führte ein neues Strukturmotiv ein: Ein mit einem Styrenrest modifiziertes Pyridinium
konnte trotz fehlender Komplexierung potente Inhibitionen gegen KDM2A und 4A ergeben
und könnte daher noch interessant für zukünftige Untersuchungen werden90. Der Habitus
eines Iridium(III)-Komplexes (E29) unterschied sich von den bisher vorgestellten Inhibitoren
grundsätzlich, daher war es überraschend, dass auch E29 inhibitorische Aktivität
gegenüber verschiedenen Histon-Demethylasen zeigte. Der Wirkungsmechanismus
konnte jedoch noch nicht geklärt werden, ein Austausch des Fe(II) gegen Ir(III) sollte aber
nicht der Grund für dessen biologische Aktivität sein91. Einen nicht Eisen-involvierenden
Mechanismus besaß das in der Alkoholabusus-Therapie eingesetzte Disulfiram (E30). Hier
wurde das Entfernen des Zinkkations aus dem Zinkfinger-Motiv der KDM4 als
Mechanismus postuliert92. Die damit erreichte Inhibition im mikromolaren Bereich ist
beachtlich, wie jedoch die Selektivität gegen andere Zinkfinger-enthaltenden Proteine
aussieht, ist fraglich. Auch zyklische Peptide (nicht gezeigt) konnten bereits mit einer
Hemmung von KDM4 (IC50 = 0,6 µM) in Verbindung gebracht werden93.
Abb. 13: Auswahl an strukturell heterogenen KDM-Inhibitoren.
Die bisher vorgestellten Inhibitoren entstammen der wissenschaftlichen Fachliteratur. Da
aber vor allem in der Patentliteratur in verhältnismäßig kurzer Zeit zahlreiche Modulatoren
NN OH
O
OH
O
E26, n = 8
IC50(KDM4A) = 3,0 µM
IC50(KDM4C) = 1,0 µM Ir
N
N
N
N
O
O
O2N NO2
Cl
Cl
Cl
Cl
N
S
SS
S
N
F
N+
N
I-
E27
IC50(KDM4A) = 6,4 µM
IC50(KDM4B) = 9,3 µM
E28
IC50(KDM4A) = 2,58 µM
E29
IC50(KDM4D) = 15 µM
Disulfiram, E30
IC50(KDM4A) = 3,3 µM
n
+ PF6-

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
18 | Einleitung
von JmjC-Domäne enthaltenden Enzymen vorgestellt wurden, sollen auch diese kurz
präsentiert werden (Abb. 14). Es wurden zwei Übersichtsarbeiten dazu ausgewertet, eine
aus dem Jahr 201394 und eine etwas aktuellere von 201595.
Schofield et al. publizierten ihre zahlreichen Inhibitoren nicht nur in der Fachliteratur,
sondern patentierten auch viele Derivate. So wurde unter anderem der Grundkörper der
2,4-PDCA wieder aufgenommen und mit verschiedenen Aminen in Position 3 modifiziert.
In der Regel erreichten diese Inhibitoren IC50-Werte im dreistelligen nanomolaren Bereich
(E31)96. Labellé et al. von der Firma Epitherapeutics nahmen sich der Struktur der
Isonicotinsäure an und alternierten sie an Position 2 mit einer Methylenbrücke und
verschiedenen Aminen97,98. E32 stellte ein hochpotentes Molekül dar, welches gegen
nahezu alle JmjC-Domäne enthaltenden Enzyme inhibitorisch aktiv war (u. a. KDM2B,
KDM4A/B/C, KDM5A). Die Veresterung der Carboxyfunktion führte in der Regel zu einem
Abfall der Potenz, durch die Seitenketten konnten Selektivitäten gegenüber verschiedenen
Enzym-Klassen erreicht werden.
Abb. 14: Auswahl an Inhibitoren von JmjC-Domäne enthaltenden Enzymen aus der Patentliteratur.
Quanticel Pharmaceuticals war das zweite größere Pharmaunternehmen, welches
reihenweise Derivate von Isonicotinsäure patentierte. Diese trugen in Position 2 einen
Pyrazol- bzw. Imidazolrest, welcher mannigfaltig substituiert wurde99. Auch diese Reihe von
Inhibitoren war hochpotent, wobei sich unter ihnen wenige Tetrazol-Derivate mit geringerer,
aber immer noch beachtlicher Aktivität befanden (E33). Die im Pharmabereich gängigen
Übernahmen betrafen dann wenig später die beiden genannten Unternehmen, so wurde
Quanticel Pharmaceuticals von Celgene und Epitherapeutics von Gilead Sciences für
Millionenbeträge übernommen100,101. Celgene und Gilead beabsichtigten dadurch ihr
Portfolio und Knowhow im Bereich epigenetische Regulationsmechanismen-

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 19
beeinflussender Wirkstoffe zu erweitern und unterstrichen damit zusätzlich, welches
Potential den Inhibitoren der Histon-Demethylasen zugeschrieben wurde.
Eine Variation der bisher genannten Moleküle stellten die Pyrido[3,4-d]pyrimidine dar (E34).
Die IC50-Werte befanden sich hier ebenfalls im nanomolaren Bereich. Durch die nicht mehr
vorhandene Amphoterie konnte Zellgängigkeit erwirkt werden, was durch Angabe von
zellulären IC50-Werten ausgedrückt wurde102. Ferner fand eine massenhafte Patentierung
von an Position 3 derivatisierten Isonicotinsäuren von Chen et al. im Namen der nun
vereinigten Firma Celgene Quanticel Research statt103 (E35). Viele dieser Inhibitoren
zeigten wiederum nanomolare IC50-Werte. Zwar war auch eine Synthesevorschrift für
Tetrazolderivate vorhanden, Ergebnisse aus biologischen Testungen fehlten jedoch. Hier
bieten sich Anhaltspunkte für mögliche Ziele dieser Arbeit, inwiefern Tetrazolderivate von
Isonicotinsäuren auf der Suche nach neuen, innovativen Inhibitoren der KDM4-Enzymreihe
nutzbar sein können.
1.1.3 Konzept der Bioisosterie
Ein häufig genutztes Tool in der Entwicklung und im Design von neuartigen Arzneistoffen
ist die Verwendung von bioisosteren funktionellen Gruppen. Darunter wird häufig
verstanden, dass zwei ähnliche Moleküle eine vergleichbare biologische Wirkung entfalten,
wenn sie jeweils mit einer unterschiedlichen Gruppe analoger räumliche Ausdehnung und
zumeist auch ähnlicher chemischer und physikalischer Eigenschaften verknüpft sind.
Dieses Konzept wird unter vielen Gesichtspunkten eingesetzt, so können Moleküle mit
einem bioisosteren Austausch in Hinsicht auf ihr pharmakologisches Profil verändert und
Einfluss auf physikochemische Eigenschaften wie Acidität/Basizität, Lipophilie,
metabolische Stabilität und nicht zuletzt auch auf den Bindungsmodus am Zielprotein
genommen werden.
Abb. 15: Beispiele für bioiosteren Ersatz von Carbonsäuren (modifiziert nach104,105).

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
20 | Einleitung
Bei der Leitstruktur 2,4-PDCA bot es sich auf Grund der ausgeprägten Hydrophilie an, die
Carboxygruppe an Position 4 des Pyridinrings zu ersetzen. Für Carboxygruppen sind
etliche funktionelle Gruppen publiziert worden, mit denen vergleichbare Eigenschaften
erzeugt werden konnten104,106,107 (Abb. 15). Neben den bekannteren Vertretern wie
beispielsweise Sulfonamide (u. a. in Sulfamethoxazol, Sulfasalazin, Dorzolamid,
Hydrochlorothiazid, etc.), Acylsulfonamide (u. a. in Paritaprevir, Denoprevir),
Sulfonylharnstoffe (u. a. in Glimepirid) und Oxazolidindione (u. a. in Ethadion,
Paramethadion) wurden auch nicht mehr acide Oxetane oder Thietane beziehungsweise
deren Derivate als Substitutionsmöglichkeit vorgestellt105.
Im Arbeitskreis Link konnten bereits zahlreiche Erfahrungen mit der Synthese und den
biologischen Aktivitäten von Tetrazolen gesammelt werden. Die cyclischen Systeme mit
vier Stickstoffatomen besitzen einige Vorteile gegenüber Carbonsäuren. So verfügen sie
bei gesteigerter Lipophilie über eine etwas geringere Acidität, was zumindest theoretisch
zu einer besseren Membranpermeabilität führen könnte107. Daneben spielen metabolische
Parameter eine Rolle, denn Carbonsäuren können über zahlreiche Wege im Organismus
im Rahmen von Phase-II-Reaktionen erst mit Coenzym-A aktiviert und dann zum Beispiel
mit Glycin konjugiert werden. Weiterhin bestehen die Möglichkeiten der Decarboxylierung
und der β-Oxidation108. Tetrazole gehen hauptsächlich N1- oder N2-Glucuronidierungen
(letzteres bevorzugt) ein109, welche durch das Enzym UDP-Glucuronosyltransferase
katalysiert werden, und sind daher als metabolisch stabiler anzusehen.
Ein weiterer Unterschied zwischen den beiden sauren Gruppen liegt in der effektiven
Länge. Bei röntgenkristallographischen Untersuchungen von Propellan-haltigen
Glutaminsäureanaloga (E37/E38) konnte gezeigt werden, dass Tetrazole etwas
raumeinnehmender waren110 (Abb. 16).
Abb. 16: Pharmazeutisch chemische Entwicklungsstufen von Losartan und Glutaminanaloga als Vermessungsstruktur für Tetrazole (1 am Angiotensin-II-Rezeptor-Subtyp-1).
Ferner existieren rechnerische Modelle für elektrostatische Umgebungspotentiale, welche
vor allem offenbarten, dass die im deprotonierten Zustand generierte Aromatizität für eine
gleichmäßigere Ladungsverteilung sorgte111. Dieser Punkt war letztendlich auch der Grund
für die Erfolgsgeschichte der Entwicklung des Biaryltetrazols und AT1-

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Einleitung | 21
Rezeptorantagonisten Losartan: Im deprotonierten Zustand besaß das Tetrazol gegenüber
der Carbonsäure eine größere Fähigkeit der Ladungsverteilung, die dann zu einer besseren
Wechselwirkung mit positiven Ladungen in der Bindetasche führten112. Daher zeigte das
Tetrazolderivat Losartan eine etwa zehnfach höhere inhibitorische Potenz als das
ursprüngliche Carbonsäure-haltige Molekül E35.
Auf Grund der zahlreichen Heteroatome besitzen Tetrazole mehr
Wasserstoffbrückenakzeptoren und zahlreiche Koordinierungsmöglichkeiten vor allem als
Tetrazolatanion. Im aromatischen Zustand sind zusätzlich noch π-π-Interaktionen mit
arylischen Aminosäuren möglich und können zu „π-π-Sandwich“-Komplexen führen113. Da
Tetrazole nach wie vor zu den unterrepräsentierten Motiven im Arzneistoffdesign gezählt
werden114, ist die Erstellung von Bibliotheken mit diesen Heterozyklen besonders reizvoll.
Eine gesonderte Formulierung der Zielstellung findet an dieser Stelle nicht statt, es wird
daher auf die Eingangsbemerkung bei den jeweiligen Synthesekapiteln verwiesen.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
22 | Untersuchungen, Ergebnisse und Diskussion
1.2 Untersuchungen, Ergebnisse und Diskussion
1.2.1 Synthese von Tetrazol- und Carbonsäurehydrazid-beinhaltenden KDM4-Inhibitoren
Die in Abschnitt 1.1.2.2 beschriebenen, fragmentartigen KDM4-Inhibitoren mit
carbonsäuremimetischem Tetrazol- und chelatisierendem Carbonsäurehydrazid-Anteil
stellten wegen der zwar nur moderaten, im Verhältnis zur Molekülmasse aber beachtlichen
biologischen Aktivität, geeignete Leitstrukturen für weitere Untersuchungen dar85,113.
Aufgrund der flachen Struktur-Aktivitätsbeziehungen bei bisher von Rüger et al.
dargestellten Derivaten, die es nicht ermöglicht hatten, eng verwandte Analoga mit
gesteigerter Aktivität zu erhalten, wurde eine Erhöhung der Diversität im verbindenden
Molekülteil angestrebt. Erhaltene Verbindungen mit Variation der Kettenlänge des
aliphatischen Molekülrückgrats führten zunächst zu Hinweisen, die eine kürzere
Kettenlänge als im Substrat 2OG als optimal suggerierten. Spätere Re-Evaluation erwies
diesen Befund als vorläufig und eine um eine Methylengruppe längere Rückgratstruktur
lieferte bessere Hemmstoffe der KDM4A. Da eine weitere Verlängerung oder Kürzung
dieser 2-Oxoglutarsäureanaloga sich als ungünstig erwiesen hatte, wurde Anstelle des
flexiblen aliphatischen Linkers ein aromatischer Ring als Rückgrat ausgewählt, um weitere
Testsubstanzen bereitzustellen. Hierfür sollte ein Syntheseweg basierend auf
Anthranilsäure im Rahmen der vorliegenden Arbeit neu etabliert werden.
1.2.1.1 Anthranilsäurederivate
Die Zielstruktur der neuen Syntheseroute stellte 5-(1H-Tetrazol-5-yl)anthranilsäurehydrazid
(IX) dar (Abb. 17). Die Aminogruppe der Anthranilsäure verfügt theoretisch über zwei
Neuerungen gegenüber unsubstituierten (1H-Tetrazol-5-yl)benzoesäurehydraziden: Das
freie Elektronenpaar am Stickstoffatom könnte ebenfalls dafür genutzt werden, um an das
katalytisch wirkende Metallion zu koordinieren. Zusammen mit dem Carbonsäurehydrazid
wäre somit ein dreizähniger Chelator möglich. Außerdem sind Aminogruppen wertvolle
Bestandteile einer divergenten Synthesestrategie, d. h. dass erst im letzten oder vorletzten
Schritt der Syntheseroute eine Verzweigung stattfindet und somit der Weg zur zügigen
Erstellung einer Substanzbibliothek geebnet ist. Weiterhin war der Ausgangsstoff
Anthranilsäure preisgünstig kommerziell erhältlich. Die einzelnen Syntheseschritte werden
nachfolgend ausgeführt und erörtert (Abb. 17).
Der wohl wichtigste Schritt der geplanten Synthese war die Einführung einer Cyanogruppe
an Position 5 des aromatischen Systems. Aliphatische Nitrile sind häufig relativ einfach über
eine Kolbe-Nitril-Synthese zugänglich, aromatische Nitrile können unter anderem durch
eine Sandmeyer-Reaktion oder nucleophile aromatische Substitution (SNAr) dargestellt

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 23
werden. Erstere würde sich schwierig gestalten, da dann zwei Aminofunktionen vorhanden
sein müssten und eine selektive Diazotierung nur einer Aminogruppe hohen
experimentellen Aufwand erfordern würde. Für Reaktionen vom Typ der SNAr sollten
Aromaten deaktiviert, also mit stark elektronenziehenden Gruppen substituiert, sein, was
bei der Anthranilsäure nicht zutrifft. Durch die Rosenmund-von-Braun-Reaktion sollte es
allerdings möglich werden, ein Arylbromid in ein Arylnitril zu überführen. Basierend auf
Annahme, dass ein Bromatom relativ einfach an Position 5 der Anthranilsäure einzuführen
war, wurde daher dieser Weg eingeschlagen.
Abb. 17: Geplante Syntheseroute zur Darstellung von 5-(1H-Tetrazol-5-yl)anthranilsäurehydrazid (IX): i) Acetanhydrid, Rückfluss, 60 min; ii) Br2, H2O, Raumtemperatur, 18 h; iii) NaOH, H2O, Rückfluss, 4 h; iv) CuCN, NMP, Rückfluss, 2 h; v) MeOH, SOCl2, Rückfluss, 15 h; vi) NaN3, TEA∙HCl, Toluen, 105 °C, 24 h; vii) EtOH, N2H4∙H2O, Rückfluss, 8 h.
Die direkte Bromierung von Anthranilsäure konnte aufgrund der Substituenteneinflüsse zu
zwei an den Positionen 3 beziehungsweise 5 monobromierten Produkten oder auch zu
einer dibromierten Anthranilsäure führen115. Um dies zu vermeiden, wäre eine Kühlung der
Reaktionslösung sinnvoll116,117. Sollten bereits mono- und disubstituierte Produkte
nebeneinander vorliegen, scheint eine Trennung mit Hilfe eines Auszugs mit kochendem
Wasser möglich118. Durch vorherige Acetylierung der Aminofunktion zum entsprechenden
Amid, konnte jedoch eine Regioselektivität für Position 5 erreicht werden119-121. Da dieser
Schritt zügig und quantitativ durch Umsetzung mit Acetanhydrid im Überschuss verlief,
wurde dieser Weg gewählt. Das acetylierte Intermediat II wurde anschließend mit
Bromwasser behandelt. Dabei zeigte eine bleibende Rotbraunfärbung das Ende der
NH2
OH
O NH
OH
O
O
NH
OH
O
O
Br
NH2
OH
O NH2
O
O
Br
NH2
O
O
N
NH2
O
O
N
N N
NHNH2
NH
NH2
NN
NNH
O
i ii
iii
iv v
iv
vivii
N
I II III
IVV VI
VIIVIIIIX
NH2
OH
O
Br

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
24 | Untersuchungen, Ergebnisse und Diskussion
Notwendigkeit der Bromzugabe an. Nach weiterem Rühren und Abdampfen des
überschüssigen Broms im Laborabzug, konnte ein leicht rosafarbener Feststoff abfiltriert
werden. Dieser enthielt wie angenommen ausschließlich das an Position 5 monobromierte
Produkt III. Da die Aminofunktion als erstes ohne Derivatisierung auf Interaktionen mit
KDM4-Enzymen untersucht werden sollte, musste die Acetylgruppe jedoch wieder entfernt
werden. Dies geschah im Sinne einer klassischen Amidspaltung mit einem Überschuss an
Natriumhydroxid in wässriger Lösung unter Rückfluss122.
Es lag nahe, die erhaltene 5-Bromanthranilsäure (IV) direkt zum entsprechenden Nitril
umzusetzen. In der Patentliteratur wurde das Zielmolekül aus eben diesem Edukt mit Hilfe
von Kupfer(I)-cyanid und dem hochsiedenden polaren Lösungsmittel N-Methyl-2-pyrrolidon
(NMP) erhalten123. Diese Bedingungen entsprachen der klassischen Rosenmund-von-
Braun-Reaktion. Der allgemeine Mechanismus ist dabei noch nicht abschließend geklärt.
Es werden eine Bildung von Cu(III)-Spezies und ein Additions-Eliminations-Mechanismus
diskutiert. Die Reaktionskinetik wurde bereits genauer untersucht124. Hohe Temperaturen
sind notwendig für das Gelingen der Synthese, obgleich dadurch ein erhöhtes Maß an
Nebenreaktionen in Kauf genommen werden muss. Lösungsmittelfreie Varianten der
Synthese, welche mit einem Zusammenschmelzen der Edukte arbeiteten, wurden ebenso
beschrieben wie Abwandlungen mit ionischen Flüssigkeiten als Reaktionsmedium125,126.
Die Umsetzung von temperaturempfindlichen Stoffen ist mit dieser Methode limitiert,
weshalb verschiedene Cyanidquellen, Katalysatoren und Liganden untersucht worden sind.
Kaliumhexacyanidoferrat(II) als CN–-Donor hatte den großen Vorteil, dass die Toxizität im
Gegensatz zu Natrium-, Kalium- oder Kupfer(I)-cyanid minimal war. Zur erfolgreichen
Darstellung wurden aber in der Regel Katalysatoren auf Palladium-Basis benötigt127,128.
Spuren von CN– können jedoch durch die freie Carbonsäure protoniert werden, was zur
Bildung von HCN führt. Neben sicherheitsrelevanten Aspekten kann Cyanwasserstoff den
Palladium-Katalysator deaktivieren, da HCN eine besonders hohe Affinität zu Pd(0)-
Spezies aufweist129,130. Als weitere Cyanidquellen kamen Cyanhydrine zum Einsatz,
gegebenenfalls in Kombination mit Kaliumiodid, welches zu einem katalytischen
Halidaustausch führen sollte131. Zinkcyanid und Liganden auf Ethylendiamin-Basis konnten
ebenfalls dazu beitragen, dass die benötigten Reaktionstemperaturen abgesenkt werden
können132,133. Eine im Jahr 2014 erschienene Übersichtsarbeit fasst die zahlreichen
Möglichkeiten der vor allem Kupfer-katalysierten Cyanierungsreaktionen zusammen134.
Von den vorgestellten Möglichkeiten wurden unter anderem die klassische Rosenmund-
von-Braun-Reaktion, im Arbeitskreis auch auf Kaliumhexacyanidoferrat(II)-basierende
Methoden und die Ethylendiamin-gestützte Synthese ausprobiert. In keinem Falle konnte
die gewünschte 5-Cyananthranilsäure (V) erhalten werden. Da sehr wahrscheinlich die freie
Carboxygruppe den Grund für diese Fehlschläge darstellte, musste diese als Ester
geschützt werden, welcher zugleich für die Hydrazinolyse im finalen Schritt eine Aktivierung

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 25
der Carboxygruppe bewirkte. Für die Veresterung wurden zwei Wege untersucht: Die
klassische Fischer-Veresterung mit Methanol und Schwefelsäure als Katalysator und eine
Umsetzung mit Methanol und Thionylchlorid135,136. Die moderate Umsetzung von bis zu
65 % konnte mit sterischer Hinderung der Carbonsäure durch die Aminofunktion (bzw. das
entsprechende Ammoniumsalz mit Gegenion bei saurem Katalysator) begründet werden,
weshalb es in den Experimenten mit Hinblick auf die Ausbeuten auch keine Rolle spielte,
welche der beiden Veresterungsmöglichkeiten genutzt wurde. Der Thionylchlorid-Weg
besaß aber zusätzlich den Vorteil, dass etwas weniger harzige Rückstände entstanden.
Carbonsäure und Ester ließen sich zweckdienlich durch Flüssig-Flüssig-Extraktion mit
Ethylacetat und Natriumhydrogencarbonat-Lösung trennen.
Der so erhaltene 5-Bromanthranilsäuremethylester (VI) konnte dann einer Substitution mit
Cyanid unterzogen werden, wofür ebenfalls die Rosenmund-von-Braun-Reaktion genutzt
wurde137-139. Durch die hohen Temperaturen von über 200 °C kam es zu einer
Schwarzfärbung des Reaktionsansatzes, welche sich auch im weiteren
Aufarbeitungsverlauf als störend erwies. Neben dem eigentlichen Produkt entstanden
relativ große Mengen an Nebenprodukten, die weder in Wasser noch in Ethylacetat löslich
waren. Bei der DC-Verlaufskontrolle zeigten diese Stoffe keinerlei Elution mit
verschiedenen Laufmitteln und verblieben an der Startlinie. Das könnte darauf hinweisen,
dass es zu einer Polymerisation gekommen sein könnte, zumindest wäre es denkbar, dass
aus Ester und Amin eines zweiten Moleküls Polyamid-Strukturen entstanden sind. Diese
konnten zwar einfach durch Filtration aus dem Gemisch entfernt werden, sind aber
wahrscheinlich der Grund, warum die Ausbeuten weder sehr hoch, noch reproduzierbar
waren (19-54 %). Zusätzlich war die Massendifferenz von Bromid und Cyanid
verhältnismäßig hoch, sodass große Einwaagen an Edukt eingesetzt werden mussten, um
anschließend über genügend 5-Cyananthranilsäuremethylester (VII) für weitere Schritte zu
verfügen. Der Anfall von erheblichen Mengen an cyanidhaltigen Schwermetallabfällen war
ein weiterer Nachteil dieser Reaktion.
Auf die Diskussion des Tetrazolringschlusses soll in diesem Kapitel verzichtet werden, da
diese noch ausführlich im Abschnitt 1.2.2.1 durchgeführt wird. Lediglich auf die
Besonderheit, dass die im Arbeitskreis etablierte Methode mit Natriumazid,
Ammoniumchlorid und Dimethylformamid (vgl.85) für die hier gegeben Syntheseroute nicht
übertragbar war, soll an dieser Stelle hingewiesen werden. Diese führte zu einem
Hauptprodukt, dessen 1H-NMR-Spektrum nicht mehr das für einen Methylester typische
3H-Integral bei einer chemischen Verschiebung δ von etwa 3,8 ppm zeigte. Weil dazu noch
ein zusätzliches 1H-Integral bei etwa δ = 13,0 ppm vorhanden war und alle anderen Signale
denen des Edukts ähnelten, ist davon auszugehen, dass eine Esterhydrolyse eintrat. Bevor
die Gründe dafür gefunden wurden, konnte erfolgreich ein Tetrazolringschluss mit
Natriumazid, Triethylammoniumchlorid (TEA∙HCl) und Toluen etabliert werden140. Durch

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
26 | Untersuchungen, Ergebnisse und Diskussion
das Einführen der sauren Tetrazolgruppe änderten sich jedoch auch die
physikochemischen Eigenschaften des Stoffes. So besaß 5-(1H-Tetrazol-5-
yl)anthranilsäuremethylester (VIII) einen zwitterionischen Charakter und ließ sich nicht
mehr ohne weiteres in Ethylacetat lösen. Dafür konnte die Protonierung zum Tetrazol
genutzt werden, um Tetrazolat-beinhaltende Stoffe aus einer basischen Lösung zu fällen,
ein Prozess, der im Laufe dieser Arbeit häufiger genutzt wurde.
Die Hydrazinolyse barg wie die Cyanierung Gefahren, welche hier durch ein mögliches
karzinogenes Potential des Hydrazins begründet war. Es galt daher, alle
arbeitssicherheitstechnischen Gegebenheiten zu beachten und die Belastung für das
Laborpersonal auf dem niedrigsten Level zu halten. In einer alkoholischen Hydrazinlösung
(Methanol oder Ethanol sind unter anderem möglich141,142) konnte der Methylester durch
längeres Erhitzen unter Rückfluss zu dem gewünschten 5-(1H-Tetrazol-5-
yl)anthranilsäurehydrazid (IX) umgesetzt werden. Die Ausbeute des Syntheseschrittes
(15 %) spiegelte jedoch nicht die vollständige Umsetzung wider, sondern war vielmehr ein
Ausdruck der anspruchsvollen Aufarbeitung des Rohproduktes. Durch eine weitere
basische Gruppe im Molekül wurde der amphotere Charakter gestärkt, wodurch auch die
Polarität anstieg. Es war damit nicht mehr ohne weiteres möglich, eine
säulenchromatographische Aufreinigung an unmodifiziertem Kieselgel durchzuführen, da
das Laufmittel hohe Anteile an Methanol benötigte. Es hat sich daher als zweckmäßig
erwiesen, das Endprodukt aus wässriger Methanol-Lösung umzukristallisieren und Verluste
in der Ausbeute dafür in Kauf zu nehmen. Es ist aber dennoch gelungen, den
siebenstufigen Syntheseentwurf umzusetzen und das gewünschte Molekül zu erhalten.
Mit Blick auf die mäßigen Ausbeuten und mangelnde Reproduzierbarkeit der Cyanierung
wurde versucht, diese mit Modifikationen der Syntheseroute zu verbessern. Ein Kellerfund
brachte 5-Iodanthranilsäure (X) hervor. Diese besaß zwar nur eine mäßige Reinheit
(schwarzer Feststoff statt farblose Prismen143), da das 1H-NMR-Spektrum aber eine
saubere Grundlinie zeigte, war wahrscheinlich lediglich überschüssiges Iod der Grund für
die Verfärbung. Es sollte untersucht werden, ob der Iodsubstituent besser gegen Cyanid
austauschbar war als ein Bromsubstituent. Der Ablauf ähnelte dem bisherigen Weg und
verlief über eine Veresterung und die Rosenmund-von-Braun-Reaktion (Abb. 18). Für die
Veresterung wurde diesmal die schwefelsäurekatalysierte Variante verwendet (modifiziert
nach Takalo et al.144), welche vergleichbare Ausbeuten wie bei 5-Bromanthranilsäure (IV)
aufwies. Die anschließende Cyanierung führte dann zu einer Ausbeute von 58 %, was
einen minimal besseren Wert darstellte als beim Bromderivat. Da die Bromierung eines
Aromaten aufgrund der höheren Reaktivität von Br2 in der Regel besser gelingt als die
Iodierung, war es aber fraglich, ob sich die minimal besseren Ausbeuten der Cyanierung
auch im Endergebnis niederschlagen würden.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 27
Abb. 18: Cyanierung ausgehend von 5-Iodanthranilsäure (X): i) MeOH, H2SO4, Rückfluss, 24 h; ii) CuCN, NMP, Rückfluss, 2 h.
Aufgrund einer Anmerkung von Škoch et al.145, dass freie Amine bei einer
palladiumkatalysierten Cyanierung störende Einflüsse ausüben können, lag es nahe, dass
auch kupferkatalysierte Reaktionen davon negativ beeinflusst werden. Folgerichtig sollte
untersucht werden, ob die Rosenmund-von-Braun-Reaktion mit einer Acetamido-Gruppe
effizienter durchgeführt werden kann (Abb. 19). Hierfür wurde der bereits vorhandene
5-Bromanthranilsäuremethylester (VI) quantitativ mit Acetanhydrid acetyliert. Wider
Erwarten führte die anschließende Umsetzung mit Kupfer(I)-cyanid zu einer
unterdurchschnittlichen Ausbeute von nur 15 %. Auch hierbei sind große Mengen an
Wasser- und Ethylacetat-unlöslichen Feststoffen angefallen. Es gilt also zu resümieren,
dass die Einführung von Cyano-Gruppen in die gegebenen Moleküle prinzipiell möglich war,
die Erwartungen an Effizienz und Umweltverträglichkeit aber nicht vollständig erfüllt werden
konnten.
Abb. 19: Die Syntheseroute für N-Acetyl-5-(1H-tetrazol-5-yl)anthranilsäurehydrazid (XV) führte zu 3-Amino-2-methyl-6-(1H-tetrazol-5-yl)chinazolin-4(3H)-on (XVI): i) Acetanhydrid, Rückfluss, 30 min; ii) CuCN, NMP, Rückfluss, 1 h; iii) NaN3, TEA∙HCl, Toluen/DMF, 105 °C, 24 h; iv) N2H4∙H2O, MeOH, Rückfluss, 8 h.
Als weiterer negativer Punkt stellte sich die Tetrazolbildung aus N-Acetyl-5-
cyananthranilsäuremethylester (XIII) heraus. Das gewünschte Tetrazol XIV konnte nur in
Spuren erhalten werden. Die dafür genutzte Variante mit Toluen als Lösungsmittel hatte
wahrscheinlich dazu geführt, dass die Löslichkeit des Eduktes nicht ausreichte. Es erschien
NH2
O
O
I
i
X XI
NH2
OH
O
I
NH2
O
O
N
ii
VII
NH
O
O
BrVI XII
NH2
O
O
Br
O NH
O
O
XIII
O
N
NH
O
O
XIV
O
N
N N
NH
NH
NH
NH2
NN
NNH
O
O
N
NNH2
NN
NNH
O
XVI
i ii iii
iv iv
XV

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
28 | Untersuchungen, Ergebnisse und Diskussion
daher sinnvoller, die Derivatisierung der Aminofunktion erst nach dem Tetrazolringschluss
vorzunehmen (Abb. 20). Würde die Acylierung im Anschluss an die Hydrazinolyse
stattfinden, bestünde die Gefahr, dass das Carbonsäurehydrazidstrukturelement ebenfalls
modifiziert würde.
Die Hydrazinolyse von N-Acetyl-5-(1H-tetrazol-5-yl)anthranilsäuremethylester (XIV)
brachte ein unvermutetes Molekül hervor: Statt des korrespondierenden
Carbonsäurehydrazids bildete sich der Heterozyklus 3-Amino-2-methyl-6-(1H-tetrazol-5-
yl)chinazolin-4(3H)-on (XVI) aus. Dieser Sachverhalt konnte mit Hilfe der hochauflösenden
Massenspektrometrie (HRMS), 2D-NMR-Spektren und Literaturhinweisen aufgeklärt
werden146. Auffällig war vor allem, dass ein 2H-Integral bei δ = 5,9 ppm im 1H-NMR-
Spektrum sichtbar wurde. Die terminale Aminogruppe von Carbonsäurehydraziden wurde
aber aufgrund ihres protonenaustauschenden Charakters bisher im 1H-NMR-Spektrum
nicht beobachtet. Dennoch stellte das derivatisierte Chinazolin ebenfalls ein interessantes
Molekül dar, das theoretisch im Besitz der notwendigen Eigenschaften zur Bindung an das
katalytische Zentrum der KDM4A war und daher unbedingt einer biologischen Testung
unterzogen werden sollte.
Abb. 20: Acylierungen von Tetrazolen können zu 1,3,4-Oxadiazolen (Umlagerungen nach Huisgen) (XVII) führen: ia) Acetanhydrid, Rückfluss, 1 h; ib) Acetylchlorid, Pyridin, 0 °C bis 40 °C, 2 h.
Da der Tetrazolringschluss des N-Acetyl-5-cyananthranilsäuremethylesters (XIII) fast
ausschließlich zu einem anderen als dem gewünschten Produkt führte, lag es nahe, die
Acetylierung erst nach der Tetrazolbildung durchzuführen. Aber auch bei dieser Route
(Abb. 20) galt es, Fallstricke zu beachten. Huisgen et al. untersuchten im Jahr 1960 unter
anderem die Stabilität von Tetrazolen147. Dabei wurde die Erkenntnis gewonnen, dass sich
Tetrazole mit Carbonsäurechloriden oder -anhydriden an Position 2 acylieren lassen. Diese
Gebilde sind thermolabil und lagern sich verhältnismäßig einfach bei Temperaturen von 60-
130 °C unter Abgabe von molekularem Stickstoff zu 1,3,4-Oxadiazolen um. So ließen sich
mit diesem Syntheseweg z. B. Methyl- oder Phenyl-substituierte 1,3,4-Oxadiazole in guten
Ausbeuten herstellen148,149. Selbiges ließ sich bei der Umsetzung von 5-(1H-Tetrazol-5-

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 29
yl)anthranilsäuremethylester (VIII) mit Acetanhydrid unter Rückfluss beobachten: Das
resultierende Molekül war ein 5-Methyl-1,3,4-oxadiazol (XVII), welches aufgrund des
Überschusses an Acetanhydrid aber wie gewünscht auch an dem primären aromatischen
Amin acetyliert wurde. Diese sauerstoffhaltigen Heterozyklen besitzen zwar nicht die
Acidität eines Tetrazols, könnten aber aufgrund der freien Elektronenpaare von Sauerstoff-
und Stickstoffatomen interessante Wechselwirkungen eingehen und sollten daher auch für
biologische Testungen gegen KDM4A in Betracht gezogen werden. Um synthetisch das
eigentlich gewollte Acetamido-Derivat des Tetrazoles (XIV) zugänglich zu machen, wurde
eine Methode herangezogen, die gewissermaßen für die Herstellung von
Carbonsäureestern gedacht ist. Bei der Einhorn-Variante (auf Grundlage der Schotten-
Baumann-Methode150) zur Herstellung von Estern aus Säurechloriden wurde Pyridin als
Lösungsmittel und Katalysator gleichzeitig genutzt. Weiterhin war es wichtig, dass der
Reaktionsansatz bei Zugabe des Säurechlorides gekühlt und danach nur gelinde erwärmt
wurde. Durch anschließende Zugabe von verdünnter HCl-Lösung wurde das Tetrazolat
wieder protoniert und ausgefällt, während das Pyridin als wasserlösliches Hydrochlorid
ausgewaschen werden konnte. Aufgrund der milden Reaktionsbedingungen blieb das
Tetrazol unverändert, jedoch mussten dafür auch mäßige Ausbeuten von 30-60 % in Kauf
genommen werden. Dennoch zeigte dieses Beispiel, wie wichtig die richtige Auswahl der
Bedingungen für die Selektivität einer Synthese von Tetrazolderivaten sein konnte. Ein
anderes Kriterium stellte die Auswahl des Lösungsmittels dar und soll im Folgenden kurz
erklärt werden.
Die Idee der Benutzung von Anthranilsäure als Grundkörper ging auch darauf zurück, dass
die freie primäre aromatische Aminogruppe genutzt werden sollte, um verschiedenste
Derivate darzustellen. Die Substituenten sollten möglichst universell gewählt werden
können, neben aliphatischen kamen daher ebenfalls aromatische Reste zum Einsatz.
Fluorsubstitutionen sind sinnvoll, um metabolische Stabilität zu gewähren und
physikochemische Eigenschaften aufgrund der starken Elektronegativität des Fluors zu
modifizieren151. Die Umsetzung von 5-(1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII) mit
4-Fluorbenzoylchlorid wird nachfolgend gezeigt (Abb. 21). Neben der bereits
beschriebenen Einhorn-Variante, welche hier zum gewünschten Produkt N-4-Fluorbenzoyl-
5-(1H-tetrazol-5-yl)anthranilsäuremethylester (XIX) führte (modifiziert nach152,153), sollten
zudem die Auswirkungen eines aprotischen Lösungsmittels untersucht werden.
Literaturhinweise gaben Aufschluss über die Durchführbarkeit der Acylierung in
Dichlormethan154, allerdings konnte schon gezeigt werden, dass Benzoyltetrazole durchaus
isolierbar waren, wenn sie im aprotischen Lösungsmittel und unter Eiskühlung hergestellt
wurden155.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
30 | Untersuchungen, Ergebnisse und Diskussion
Abb. 21: Die Wahl des Lösungsmittels beeinflusst die Selektivität der Acylierungen von Tetrazolen: ia) 4-Fluorbenzoylchlorid, CH2Cl2, TEA, 0 °C bis Raumtemperatur, 18 h; ib) 4-Fluorbenzoylchlorid, Pyridin, 0 °C bis 50 °C, 3 h; ii) N2H4∙H2O, MeOH, Rückfluss, 5 h.
Ein Grundproblem bei der Nutzung von aprotischen Lösungsmitteln war die schlechte
Löslichkeit von (1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII) in diesen Solvenzien.
Durch Zusatz einer Hilfsbase (welche ohnehin zugesetzt werden musste, um beim
Reaktionsverlauf mit einem Carbonsäurechlorid entstehendes HCl abzufangen) wurde die
Löslichkeit nur minimal verbessert. Trotz des Vorliegens einer Suspension aus Edukt in
Dichlormethan war nach Zugabe des 4-Fluorbenzoylchlorids eine Umsetzung detektierbar.
Nach Aufarbeitung des Reaktionsansatzes zeigte das 1H-NMR-Spektrum des Produktes
eine Überraschung: Das Fehlen eines Amid-NH-Signals und das Vorhandensein eines
2H-Integrals der NH2-Gruppe indizierten eine Acylierung des Tetrazols (Abb. 23). Der
zusätzliche Test mit Ehrlich-Sprühreagenz (Dimethylaminobenzaldehyd) war positiv, das
heißt, es hatte sich ein orangebraunes Iminoderivat gebildet. Die primäre aromatische
Aminofunktion wurde also nachweislich zunächst nicht umgesetzt und dann mit dem
Farbreagenz derivatisiert. Dies ist deshalb nicht unbedingt zu erwarten gewesen, weil ein
Tetrazolat über eine Mesomeriestabilisierung verfügt (Abb. 22). In der Regel sind solche
Moleküle dadurch reaktionsträger, weshalb angenommen wurde, dass die NH2-Gruppe als
erstes eine Acylierung unterlaufen würde. Auch dieses Beispiel zeigte, wie wichtig und
zeitaufwendig die Optimierungen der Reaktionsbedingungen in diesem Arbeitsfeld sind,
und dass mit nur wenigen Änderungen bereits völlig andere Moleküle erhalten werden
können.
Abb. 22: Mesomeriestabilisierung des Tetrazolats.
Mit dem Lösungsmittel Pyridin konnte Intermediat XIX zwar erhalten werden, der
anschließende Schritt der Hydrazinolyse gestaltete sich aber erneut schwierig (Abb. 21).

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 31
Die Untersuchungen der Produkte wurden auch von den vorherigen Befunden geleitet, dass
eventuell ein Chinazolin-Grundgerüst entstanden sein könnte. Die Umsetzung war jedoch
marginal und ein sauberer Stoff konnte auch nach intensiver Aufarbeitung nicht erhalten
werden. Gegebenenfalls war der aromatische Rest sterisch zu anspruchsvoll, weshalb ein
Ringschluss nicht gut gelang.
Abb. 23: In Bezug auf NH-Protonen deutlich unterschiedliche 1H-NMR-Spektren aus der Acylierung von (1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII).
Weiterhin wurde versucht, die freie Aminofunktion für eine Alkylierung zu nutzen. Dafür
wurde nach einer Literaturangabe vorgegangen, welche Chloressigsäure im Wässrigen und
Natriumcarbonat als Hilfsbase (Abb. 24) vorschlug156. Die zusätzlich eingeführte

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
32 | Untersuchungen, Ergebnisse und Diskussion
Carbonsäurefunktion sollte internen computergestützten Berechnungen zufolge ein
erhöhtes Interaktionspotential mit KDM4A ermöglichen.
Abb. 24: Fehlgeschlagene Alkylierung des primären aromatischen Amins (XXI): i) Chloressigsäure, Na2CO3, H2O, 90 °C, 3 h.
Die zitierte Literatur setzte aber nicht wie hier vorgestellt den Anthranilsäureester um,
sondern die freie Säure. Aufgrund der hohen Temperaturen und dem basischen pH-Wert
durch das eingesetzte Carbonat-Salz, kam es jedoch fast ausschließlich zur
Esterhydrolyse. Eine Alkylierung konnte nicht nachgewiesen werden. Zukünftige
Alkylierungsversuche sollten also wässrige Systeme meiden. Wegen Bedenken, dass eine
zweite saure Gruppe am Molekül zu einer erheblichen Steigerung der ohnehin schon hohen
Polarität des Moleküls geführt hätte, wurden Alternativen für die misslungene Umsetzung
nicht untersucht.
Für den vorgestellten Anthranilsäureweg ist festzuhalten, dass dieser erfolgreich verfolgt
werden konnte. Es wurden unterschiedliche Aspekte der kritischen Syntheseschritte (unter
anderem Cyanierung und Acylierung) untersucht und von verschiedenen Seiten beleuchtet.
Insgesamt standen mit 5-(1H-Tetrazol-5-yl)anthranilsäurehydrazid (IX) und 3-Amino-2-
methyl-6-(1H-tetrazol-5-yl)chinazolin-4(3H)-on (XVI) zwei vollständig charakterisierte und
aufgereinigte Verbindungen zur Verfügung, welche auf inhibitorische Eigenschaften gegen
KDM4A getestet werden sollen.
1.2.1.2 Essigsäurederivate
In Zusammenarbeit mit der Arbeitsgruppe Makromolekulare Kristallographie um Dr.
Manfred Weiss von der Photonenquelle BESSY II des Helmholtz-Zentrum Berlin für
Materialien und Energie konnten im Rahmen von Diplomarbeiten und Dissertationen im
Arbeitskreis bereits mehrere Kristallstrukturen aufgelöst werden113,157. Die Kristallisation
erfolgte im katalytischen Zentrum der KDM4D, wobei aus Stabilitätsgründen das zentrale
Fe(II)-Kation gegen ein Ni(II)-Kation ausgetauscht wurde. KDM4D besitzt zwar einen sehr
ähnlichen Aufbau wie KDM4A, es konnte allerdings mit der Struktur AA040 gezeigt werden,
dass ein Phenylrest, welcher mit fünf Kohlenstoffkettengliedern aus dem Zentrum
herausragt, eine weitere Interaktion mit dem Aminosäurerest Histidin 90 eingeht (Abb. 25).
Dieses π-π-Stacking könnte einen zusätzlichen Energiegewinn bei der Bindung einbringen
und, noch wichtiger, eine gewisse Selektivität für die sonst sehr homogen aufgebaute
NH2
O
NN
NNH
O
VIII
i
NH
O
NN
NNH
O
XXI
O
OH

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 33
KDM4-Enzymreihe bedeuten, da KDM4A an dieser Position eine andere
Aminosäure(sequenz) aufweist. Daher ließ sich schlussfolgern, dass unbedingt noch ein
Derivat vom 2-Amino-2-(1H-tetrazol-5-yl)essigsäurehydrazid-Typ (vgl.113) mit
π-Elektronenüberschussaromaten bei gleicher Kettenlänge synthetisiert werden sollte.
Einen zweiten Punkt stellte die Untersuchung der Notwendigkeit der Carbonsäurehydrazid-
Gruppe von AA040 dar. Die chelatisierende Wirkung auf das zentrale Eisen-Kation wird
ohne Zweifel eine der gewinnbringendsten Molekül-Liganden-Interaktion repräsentieren.
Da aber bei AA040 offensichtlich noch weitere Interaktionspartner außerhalb des
katalytischen Zentrums der KDM4D vorhanden sind, wird die Frage aufgeworfen, ob ein
Verzicht auf die promiskuitiv bindende Hydrazid-Partialstruktur möglich wäre. Die
Synthesewege für beide Moleküle sollen im Folgenden besprochen werden.
Abb. 25: Kristallstruktur von AA040 (2-(4-Phenylbutylamido)-2-(1H-tetrazol-5-yl)essigsäurehydrazid) in KDM4D: Der Phenylrest zeigt ein zusätzliches π-π-Stacking mit His90.
Die zentrale Frage bei der Synthese von AA040-Analoga ohne Hydrazidfunktion war, ob
der Elektronenzug auf das Nitril ausreichen würde, um eine zufriedenstellende
Tetrazolbildung zu gewährleisten. Auf den genauen Mechanismus dieser Reaktion wird in
Abschnitt 1.2.2.1 eingegangen, es soll aber vorweggenommen werden, dass der
Tetrazolringschluss ohne ausreichende –I- oder –M-Effekte der benachbarten Gruppen
erschwert ist. Im Falle der Synthesestrategie von AA040 wurde der Elektronenzug von
einem angrenzenden Oxim generiert, welcher eine gute Ausbeute ermöglichte. Entfällt
dieser, müssen die Einflüsse des Amids den notwendigen Elektronenzug zu Verfügung
stellen (Abb. 26).

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
34 | Untersuchungen, Ergebnisse und Diskussion
Im ersten Schritt wurde 5-Phenylvaleriansäure (XXII) mit Hilfe von Thionylchlorid in sein
korrespondierendes Carbonsäurechlorid (XXIII) übergeführt157. Ohne Aufarbeitung dieses
Intermediats erfolgte die Umsetzung mit 2-Aminoacetonitril-Hydrochlorid (XXIV) zum
entsprechenden Amid (XXV) (modifiziert nach Palumbo et al.158) mit guten Ausbeuten. Bei
der anschließenden Tetrazolbildung wurde zuerst eine drastischere Variante mit der Lewis-
Säure Aluminiumchlorid untersucht159. Dadurch sollte die Bildung von Aluminiumazid
realisiert werden, welches höhere Ausbeuten bei niedrigerer Reaktionstemperatur in
Aussicht stellte. Angewendet auf die gegebene Struktur fand jedoch keine Umsetzung statt
und das Edukt konnte nahezu verlustfrei wiedergewonnen werden. In der Literatur werden
strukturell verwandte Produkte erzielt, wenn die Umsetzung des Nitrils mit
metallorganischen Aziden wie Trimethylsilylazid oder Tributylzinnazid erfolgte160,161. Diese
Reagenzien besitzen in der Regel eine hohe Toxizität und sollten deshalb nur als Ultima
Ratio zum Einsatz kommen. Eine ältere Publikation zeigte jedoch, dass es eventuell
möglich sein könnte, das Amidonitril mit den „klassischen“ Reaktionsbedingungen
(Natriumazid, Ammoniumchlorid und Dimethylformamid) umzusetzen162.
Überraschenderweise führte dies zu verhältnismäßig guten Ausbeuten von über 50 %,
wobei das nicht abreagierte Edukt ebenfalls wiedergewonnen werden konnte. Der
ausgeübte Elektronenzug des Amids war offenbar ausreichend, um eine erfolgreiche
Tetrazolsynthese zu initiieren.
Abb. 26: Synthesestrategie für N-(1H-Tetrazol-5-yl)methyl-5-phenylvalerianamid (XXVI): i) SOCl2, Raumtemperatur, 24 h; ii) Pyridin, CH2Cl2, Raumtemperatur, 20 h; iii) NaN3, NH4Cl, DMF, 105 °C, 24 h.
Bezüglich der Wahl des π-Elektronenüberschussaromaten gab es Hinweise darauf, dass
die π-π-Wechselwirkung eines Imidazolrings (Histidin) mit einem Indolrest (Tryptophan)
etwa vier Mal größer sein konnte als die Interaktion eines Imidazolrings mit einem
Benzenrest (Phenylalanin)163. Es sollte daher mit hoher Priorität versucht werden, einen
Indolring an Stelle des Phenylrestes zu integrieren. Die Kettenlänge sollte dabei
unverändert bleiben, dies entsprach einer an Position 4 modifizierten Buttersäure, welche
ein Indol-3-yl-Rest trägt (Abb. 27).

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 35
Die Synthese des Grundkörpers 2-Amino-2-(1H-tetrazol-5-yl)essigsäureethylester-
Hydrochlorid (XXIX) erfolgte nach einem zweistufigen Schema und wurde bereits
ausführlich in der Dissertation von N. Rüger besprochen, weshalb hierauf verzichtet werden
soll113. Die Umsetzung mit (1H-Indol-3-yl)buttersäure (XXVII), beziehungsweise seinem
korrespondierenden Säurechlorid (XXVIII), sollte zum gewünschten Amid (XXX) führen. Die
lösungsmittelfreie Reaktion mit SOCl2 führte dabei ebenso wenig zum Erfolg wie die
Variante, in welcher Tetrahydrofuran als Lösungsmittel verwendet wurde164. In beiden
Fällen kam es zur Schwarzfärbung des Edukts, ohne dass das gewollte Säurechlorid
entstand.
Abb. 27: Syntheseschema für 2-[4-(1H-Indol-3-yl)butylamido]-2-(1H-tetrazol-5-yl)essigsäure-hydrazid (XXXI): i) SOCl2, DMF, Raumtemperatur, 24 h; ii) XXVII, DIC, DIPEA, CH2Cl2, 0 °C bis Raumtemperatur, 20 h; iii) N2H4∙H2O, MeOH, Rückfluss, 6 h.
Die Überführung von Carbonsäuren in Säurechloride zur Amidsynthese stellte einen
verhältnismäßig simplen Ansatz dar. Mittlerweile ist die Auswahl an leistungsfähigen
Kupplungsreagenzien zur Knüpfung von Peptidbindungen derart groß, dass hier alternative
Syntheseoptionen zur Verfügung standen. So wurden ähnliche Amide bereits unter der
Verwendung von 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-Hydrochlorid (EDCI∙HCl)
(Abb. 28), welches eine Variante des bewährten Dicyclohexylcarbodiimids (DCC) darstellt,
synthetisiert165,166. Unter Zusatz von 1-Hydroxybenzotriazol (HOBt) können sich Aktivester
mit Carbodiimiden formen, welche für bessere Ausbeuten und weniger Nebenreaktionen
sorgen sollen167. Die Kupplung mit EDCI konnte auf das genannte Molekül erfolgreich
übertragen werden, jedoch nur in geringen Ausbeuten (15 %). Wichtig bei diesem
Reaktionstyp ist der konsequente Ausschluss von Wasser. Für die Experimente wurde
EDCI∙HCl verwendet, welches als farbloser Feststoff vorliegen sollte. Das verwendete
Reagenz war aber von wachsartige Konsistenz. Eine partielle Hydrolyse des EDCI∙HCl
konnte deshalb nicht ausgeschlossen werden und war sehr wahrscheinlich der Grund für
die niedrigen Ausbeuten. Unter Zuhilfenahme von Diisopropylcarbodiimid (DIC) und
4-(Dimethylamino)pyridin (4-DMAP) (Abb. 28) gelang es schließlich, 2-[4-(1H-Indol-3-
yl)butylamido]-2-(1H-tetrazol-5-yl)essigsäureethylester (XXX) in guten Ausbeuten (etwa

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
36 | Untersuchungen, Ergebnisse und Diskussion
80 %) zu erhalten. Diese Kombination ist eigentlich für die Veresterung von Alkoholen und
Carbonsäuren entwickelt worden und auch unter der Bezeichnung Steglich-Veresterung
bekannt168. Allen Carbodiimid-gestützten Synthesen sind sehr milde
Reaktionsbedingungen eigen. Größter Nachteil ist in der Regel die zum Teil schwierige
Abtrennung der entstehenden Harnstoffderivate. Der bei der Reaktion mit DIC entstandene
Diisopropylharnstoff konnte aber über eine Flüssig-Flüssig-Extraktion und Fällung des
Produktes mit Salzsäure entfernt werden. Ein weiterer interessanter Ansatz war die
Verwendung von Cyanurchlorid, welches Carbonsäuren ebenfalls in Carbonsäurechloride
überführen kann. Für Indolessigsäure wurde bereits eine Kupplung mit einem Amin unter
Verwendung dieses Reagenzes beschrieben169. Diese Reaktionsbedingungen konnten
ebenfalls auf die hier vorgestellten Edukte übertragen werden, führten aber nur zu mäßigen
Ausbeuten von etwa 20 %. Damit gilt es zusammenzufassen, dass die Kupplung von 2-
Amino-2-(1H-tetrazol-5-yl)essigsäureethylester-Hydrochlorid (XXIX) mit (1H-Indol-
3-yl)buttersäure (XXVII) am besten mit DIC und 4-DMAP gelang und der Syntheseroute
über das in situ erzeugte Carbonsäurechlorid hinsichtlich Ausbeuten und
Reaktionsbedingungen klar überlegen war.
Abb. 28: Hilfsreagenzien für die Amidsynthese (v. l. n. r.): Thionylchlorid und Cyanurchlorid (Trichlortriazin, TCT) dienen der Herstellung von Carbonsäurechloriden; 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-Hydrochlorid (EDCI∙HCl) und Diisopropylcarbodiimid (DIC) vermögen Carbonsäuren direkt an Amine zu kuppeln; 4-(Dimethylamino)pyridin (4-DMAP) dient als Katalysator dieser Reaktion.
Die nachfolgende Hydrazinolyse von Estern wie XXX wurde mit N2H4∙H2O in
methanolischer Lösung unter Rückfluss für mehrere Stunden durchgeführt. Die Umsetzung
konnte mit dünnschichtchromatographischer Unterstützung verfolgt werden und gelang in
der Regel quantitativ. Die Aufreinigung des Tetrazolylhydrazids stellte die größte
Herausforderung dar. Aufgrund des amphoteren Charakters war die Polarität von 2-[4-(1H-
Indol-3-yl)butylamido]-2-(1H-tetrazol-5-yl)essigsäurehydrazid (XXXI) trotz lipophiler
Seitenkette relativ hoch, weshalb sich eine säulenchromatographische Aufarbeitung
schwierig gestaltete. Unter Normalphasen-Bedingungen waren die Wechselwirkungen mit
dem Kieselgel stark ausgeprägt, wodurch große Anteile an Methanol im Laufmittel benötigt
wurden. Auf der anderen Seite wurde mit einer RP18-Phase kaum Retention erzielt. In
vorherigen Arbeiten konnte das Aufarbeitungsproblem mit einem zusätzlichen
Derivatisierungsschritt mit Methylethylketon zum Hydrazon und anschließender Spaltung
(mit zum Teil nur mäßigen Ausbeuten) gelöst werden113,157. Im Rahmen dieser Synthese
wurde erst eine Schwerkraftsäulenchromatographie mit 30 % Methanol im Laufmittel
S
O
Cl Cl
N
N
N
Cl
ClClN
N N HClN
N
N
N

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 37
durchgeführt. Die erzielte Reinheit reichte hierbei jedoch noch nicht aus. Erst der Vergleich
mit bisher beschriebenen HPLC-Methoden für das verhältnismäßig ähnlich aufgebaute
Tryptophan (amphoterer Charakter, Indolrest) brachte eine Möglichkeit zu Tage, mit
welcher das synthetisierte Molekül auch auf einer RP18-Phase reterniert werden konnte170.
Die Einstellung des pH-Wertes der wässrigen mobilen Phase auf 2,7 mit Phosphorsäure
führte offensichtlich zu einem günstigen Protonierungsgrad. Dennoch zeigte dieses
Beispiel, wie wichtig ein universelles Portfolio an verschiedenartigen Packmaterialien für
HPLC-Säulen sein kann. Ein HILIC-Trennmaterial wäre bei dieser Fragestellung mit
Sicherheit geeigneter gewesen, die hohen Anschaffungspreise deckeln aber den
weitreichenden Einsatz in der präparativen Anwendung.
In zukünftigen Synthesen sollte darüber nachgedacht werden, die für die Aufreinigung
problematische Amphoterie zumindest partiell einzuschränken, zum Beispiel durch
Umsetzung des Hydrazids mit Phosgen171-173 oder dem weniger toxischen
Carbonyldiimidazol174 (CDI) (Abb. 29). Dadurch verliert das Molekül seinen basischen
Charakter, denn das entstehende 1,3,4-Oxadiazol-2-ol ist eher saurer Natur. Es ist dann
zwar fraglich, in wie weit die komplexierenden Eigenschaften noch gegeben sind, sollte es
aber zu erheblichen Vorteilen in der Produktaufarbeitung kommen, wäre diese Variante in
Erwägung zu ziehen. In Modellreaktionen wurde versucht, 2-(1H-Tetrazol-5-
yl)essigsäurehydrazid (XXXII) sowohl mit Phosgen als auch mit CDI in verschiedenen
Lösungsmitteln umzusetzen. Eine erfolgreiche Isolierung des Oxadiazols gelang jedoch
nicht. Aufgrund der Fokussierung auf eine andere Grundstruktur ohne Hydrazidfunktion
bleibt die Zyklisierung zu Oxadiazolen zukünftigen Arbeiten vorbehalten.
Abb. 29: Nicht gelungene Überführung des Carbonsäurehydrazids in das entsprechende 1,3,4-Oxadiazol-2-ol bzw. das tautomere 1,3,4-Oxadiazol-2(3H)-on: i) CDI, THF/DMF, Rückfluss, 20 h oder Phosgen, Chlorbenzen, Rückfluss, 2 h.
Es konnten somit die beiden vielversprechenden Abwandlungen von AA040 ohne
Carbonsäurehydrazid (N-(1H-Tetrazol-5-yl)methyl-5-phenylvalerianamid (XXVI)) und mit
π-Elektronenüberschussaromaten (2-[4-(1H-Indol-3-yl)butylamido]-2-(1H-tetrazol-5-
yl)essigsäurehydrazid (XXXI)) synthetisiert und für die Cokristallisation zur Verfügung
gestellt werden. Diese Moleküle könnten als Tools für die Investigation neuer Bindungsmodi
ohne Chelator und für erweiterte π-π-Interaktionen in KDM4D dienen.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
38 | Untersuchungen, Ergebnisse und Diskussion
1.2.2 Synthese von Tetrazol- und Pyridin-beinhaltenden KDM4-Inhibitoren
Wie bereits in Abschnitt 1.1.2.2 aufgezeigt, stand mit Pyridin-2,4-dicarbonsäure ein
hochpotenter experimenteller pan-Inhibitor für die KDM4-Enzymreihe als Ausgangspunkt
zur Verfügung. Auch verschiedenste Derivate davon, die vor allem aus der Patentliteratur
stammten, konnten bereits synthetisiert und auf ihre Aktivität getestet werden. In der Regel
trugen diese Inhibitoren eine Carboxygruppe in Position 4 des Pyridinrings. Der Austausch
dieser Carboxygruppe durch einen aromatischen Tetrazolring war bisher nur vereinzelt
durchgeführt worden, und wenn, dann meistens als hintenangestellte Einzelmonographie
einer reihenweisen Patentierung von Pyridin-4-carbonsäuren99,103. Systematische
Untersuchungen der 4-(1H-Tetrazol-5-yl)pyridine auf inhibitorische Eigenschaften gegen
KDM4-Enzyme fehlten und stellten somit ein Ziel dieses Abschnitts dar. Des Weiteren sollte
auch die Untersuchung eines zweckmäßigen Aufbaus der Seitenkette in Position 2 des
Pyridins adressiert werden. Neben der direkten Verknüpfung über ein Heteroatom
(Stickstoff-, Schwefel- oder Sauerstoffatom), waren Stoffe mit Carbonylgruppe als
verknüpfendem Element oder deren reduzierte Varianten mit Methylenbrücke eine Option
(Tab. 2). Ein direkter Austausch der Carboxygruppe durch einen Tetrazolring in Position 2
oder 4 sollte auch erfolgen, um direkte Vergleiche der Aktivitäten erhalten zu können. Die
zum Teil nur geringfügigen Änderungen im Molekül machten zahlreiche neue
Synthesewege notwendig, welche im Folgenden besprochen werden sollen.
Tab. 2: Geplante Substitutionsmuster der (1H-Tetrazol-5-yl)pyridine.
Grundkörper Reste Strukturen
R1
R2
1.2.2.1 4-(1H-Tetrazol-5-yl)pyridin-2-amine/thiole/thioether/ether
Als Ausgangstoff für die 4-(1H-Tetrazol-5-yl)pyridin-2-amine diente 2-Chlorisonicotinnitril
(XXXIV), welches im ersten Schritt zum korrespondierenden Tetrazol XXXV (Abb. 30) und
anschließend mit verschiedenen Nucleophilen im Sinne einer SNAr umgesetzt werden
sollte. Eine vorangestellte Reaktion des Nitrils war wichtig, da dieses ebenfalls mit
Nucleophilen reagieren kann, wie beispielsweise die Synthese des Biguanids Metformin

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 39
und die Pinner-Reaktion zeigten175,176. Allerdings wurde dadurch ein amphoterer Charakter
generiert, welcher im Folgenden vor allem die Auswahl des Lösungsmittels stark
einschränkte.
Abb. 30: Tetrazolringschluss zum 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV): i) NaN3, TEA∙HCl, Toluen, 110 °C, 18 h.
Die Formation des Tetrazols XXXV erfolgte bei der Umsetzung des aromatischen Nitrils
XXXIV mit Natriumazid in nahezu quantitativen Ausbeuten. Ein genauerer Blick auf den
Mechanismus gab Erklärungsansätze, warum dieser Syntheseschritt mit aromatischen
Systemen deutlich besser realisierbar war als mit aliphatischen Grundkörpern. Himo et al.
berechneten dafür Energiebarrieren für konzertierte 1,3-dipolare Cycloadditionen (Huisgen-
Cycloaddition177) und Tetrazolbildungen, welche über eine Imido-Azid-Zwischenstufe
abliefen178 (Abb. 31). Letztere wurde dann bevorzugt durchlaufen, wenn die Reaktion
protonenkatalysiert ablief. Das erforderliche Proton wurde unter den hier genannten
Reaktionsbedingungen vom Triethylammoniumchlorid zur Verfügung gestellt. Dabei greift
ein Azid-Stickstoffatom das Kohlenstoffatom des Nitrils nucleophil an. Je positiver das
Kohlenstoffatom polarisiert war, desto besser konnte die Tetrazolformung also gelingen.
Abb. 31: Wahrscheinlicher Mechanismus des Tetrazolringschlusses unter der Verwendung von Brønsted-Säuren (modifiziert nach Himo et al.178): Es wird ein achtgliedriger Übergangszustand und eine Imido-Azid-Zwischenstufe durchlaufen.
Bei aliphatischen Nitrilen erbrachten die benachbarten Alkylreste schwache +I- und
Hyperkonjugations-Effekte, weshalb die Reaktion hier in der Regel träge ablief.
Aromatische Reste können jedoch Elektronen über Mesomerieeffekte delokalisieren, das
heißt, dass das Kohlenstoffatom von aromatischen Nitrilen deutlich stärker positiv polarisiert
sein sollte. Pyridin als Substituent hatte zusätzlich eine elektronenziehende Wirkung, da ein

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
40 | Untersuchungen, Ergebnisse und Diskussion
ausgeprägter –I-Effekt vorhanden war. Dies führte schlussendlich dazu, dass die
Umsetzung zum Tetrazol nahezu quantitativ gelang.
Die in dieser Arbeit hauptsächlich verwendete Kombination aus Toluen, Natriumazid und
Triethylammoniumchlorid (vgl. auch179) hatte aber laut der Originalarbeit von Koguro et al.
einen leicht abweichenden Ablauf140. So bildet sich aus dem Triethylammoniumchlorid ein
Triethylammoniumazid, welches in Toluen eine relevante Löslichkeit zeigte. Dieses wurde
in der Reaktion mit dem Nitril verbraucht, es entsteht das entsprechende
Triethylammoniumtetrazolat. Damit würde die Brønsted-Säure einen äquimolaren
Reaktionspartner darstellen und nicht nur katalytischer Natur sein.
Neben der sehr häufigen Behandlung von Nitrilen mit Natriumazid, Ammoniumchlorid und
Dimethylformamid für den Tetrazolringschluss180, kamen auch Zusätze von Lewis-Säuren
und organometallische Azide zum Einsatz (siehe auch Abschnitt 1.2.1.2). Auf der anderen
Seite muss das Edukt nicht immer ein Nitril sein, beispielsweise sind Tetrazole auch
ausgehend von Carbonsäureamiden zugänglich181, was wegen der großen Vielfalt an
verfügbaren Vertretern ein Vorteil wäre. Andererseits stünde anders als bei den Nitrilen bei
Carbonsäureamiden das Repertoire der Click-Chemie als besonders effizientes Verfahren
nicht zur Verfügung182. Aufgrund der Vielseitigkeit und Möglichkeiten des
Themenkomplexes wird an dieser Stelle auf drei Übersichtsarbeiten verwiesen108,183,184.
Das synthetisierte 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) sollte dann mit zahlreichen
Aminen unterschiedlichster Diversität umgesetzt werden (Abb. 32 und Tab. 3). Neben
aliphatischen Resten, kamen auch aromatische Substituenten zum Einsatz. Eine zweite
funktionelle Gruppe konnte durch die Verwendung von Diaminen oder Aminoalkoholen
eingeführt werden. Diese sollten im weiteren Verlauf der Kettenverlängerung dienen.
Erfreulicherweise kam es bei der SNAr mit Diaminen nicht in nennenswertem Umfang zu
befürchteten Dimerisierungen. Wahrscheinlich wurde diese mögliche Folgereaktion durch
einen ausreichend großen Überschuss an Diamin während der Reaktion verhindert. Wäre
eine Dimerisierung so nicht vermeidbar gewesen, hätten Schutzgruppenstrategien für die
Darstellung von Substanzen dieses Typs entwickelt werden müssen.
Abb. 32: Darstellung der 4-(1H-Tetrazol-5-yl)pyridin-2-amine: i) Amin, bis zu 220 °C, bis zu 48 h, Mikrowellenreaktor oder Rundkolben.
Die Synthesen der verschiedenen 4-(1H-Tetrazol-5-yl)pyridin-2-amine wurden in der Regel
umweltfreundlich ohne Verwendung von Lösungsmitteln im Mikrowellenreaktor und im Falle

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 41
der längeren Diamine/Aminoalkohole im Rundkolben durchgeführt. Durch Fällung aus
zumeist wässriger Lösung mit Salzsäure konnten zahlreiche Produkte isoliert werden. Die
Löslichkeit dieser Produkte war jedoch zum Teil noch schlechter als die des betreffenden
Eduktes. So mussten viele NMR-Spektren von den entsprechenden Natrium-Salzen der
4-(1H-Tetrazol-5-yl)pyridin-2-amine in D2O aufgenommen werden, da eine ausreichende
Löslichkeit in DMSO-d6 nicht gegeben war.
Tab. 3: Übersicht der dargestellten 4-(1H-Tetrazol-5-yl)pyridin-2-amine mit jeweiligem Rest R.
Alkylamine
XXXVI XXXVII XXXVIII
Benzylamine
XXXIX XL XLI XLII
Diamine185-187 XLIII XLIV XLV
Aminoalkohole188 XLVI XLVII
Piperazine189/ Morpholine
XLVIII XLIX L
Einen Hinweis auf die molekularen und supramolekularen Ursachen für die schlechte
Löslichkeit der 4-(1H-Tetrazol-5-yl)pyridin-2-amine lieferte die röntgenkristallographische
Untersuchung von N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII)
(Abb. 33). Neben der kompletten Planarität und den zwitterionischen Eigenschaften des
Tetrazolylpyridins wurden zahlreiche intra- und intermolekulare
Wasserstoffbrückenbindungen beobachtet, welche zu starken Wechselwirkungen im
Kristallgitter führten und damit wohl ursächlich für Löslichkeitsprobleme waren. Dennoch
konnte mit der Kristallstruktur bewiesen werden, dass der entworfene Syntheseweg mit den
zum Teil harschen Bedingungen (u. a. Mikrowellenreaktor bei Temperaturen bis 220 °C)
bis hierhin umsetzbar war. Zusätzlich zeigten die Derivate XLVIII, XLIX und L mit Piperazin-
oder Morpholin-Partialstrukturen auf Grund des räumlichen und sp³-reichen Charakters der
Heterozyklen an der Seitenkette, dass auch mit dem coplanaren Tetrazolylpyridin-
Grundgerüst eine gute Wasserlöslichkeit erreicht werden konnte.
N
O

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
42 | Untersuchungen, Ergebnisse und Diskussion
Abb. 33: Röntgenkristallographische Untersuchungen von N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII).
Bei der Einführung eines Schwefelatoms an Position 2 des Pyridinrings wurde ein anderer
Weg eingeschlagen als bei den oben genannten Aminsubstituenten. Da sich Thiole
(beziehungsweise Thiolate) als relativ weiche und gute Nucleophile bequem alkylieren
lassen und im Gegensatz zu Aminen allgemein keine Mehrfachalkylierung befürchtet
werden muss, gestaltete es sich zweckdienlicher, das entsprechende Mercaptoderivat des
2-Chlor-4-(1H-tetrazol-5-yl)pyridins (LI) herzustellen (Abb. 34).
Abb. 34: Die Substitution durch ein Schwefelatom an Position 2 des 2-Chlor-4-(1H-tetrazol-5-yl)pyridins (XXXV) schlug fehl, war aber mit 2-Chlorisonicotinnitril (XXXIV) möglich: i) Thioharnstoff, EtOH, 135 °C, 60 min, Mikrowellenreaktor; ii) Benzylbromid, TEA, ACN, Raumtemperatur, 4 h; iii) NaN3, TEA∙HCl, Toluen, 100 °C, 17 h.
Es gelang jedoch nicht, die Substitution mit bereits vorhandenem Tetrazolring
durchzuführen, obwohl die prinzipielle Machbarkeit ähnlicher Umsetzungen mit freier
Carbonsäurefunktion bereits gezeigt wurde190. Daher sollte das Nitrilderivat XXXIV zuerst
mit einem Schwefelatom modifiziert und dann erst nach Alkylierung in das Tetrazolderivat
übergeführt werden. Dies funktionierte mit Hilfe des Mikrowellenreaktors passabel

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 43
(modifiziert nach191,192), die anschließende Alkylierung war ebenfalls gut umsetzbar193.
Damit konnte der Einsatz von oftmals sehr unangenehm riechenden Thiolen umgangen
werden194.
Mit diesem Syntheseweg war es möglich, das schwefelhaltige Benzylderivat LIV mit
Tetrazolylpyridin-Grundkörper darzustellen. Allerdings nahm die hohe Divergenz der
Synthesestrategie, wie es bei den Aminen noch der Fall war, mit einer späten Ausbildung
des Tetrazolrings ab. Es wurde nur eine Thioether-enthaltende Substanz synthetisiert, da
zuerst die biologischen Testungen abgewartet werden sollten, um zu entscheiden, ob
weitere Untersuchungen gerechtfertigt erscheinen.
Als Fehlschlag erwies sich die versuchte Substitution von 2-Chlor-4-(1H-tetrazol-5-
yl)pyridin (XXXV) mit Alkoholen bzw. Alkoholaten (Abb. 35). Es sollte versucht werden,
sowohl eine Umsetzung mit Methanolat als auch mit Phenolat zu erzielen. Für letzteres
wurde eine Vorschrift mit Phenol und tert-Butanolat in DMSO verwendet195. Hierbei konnten
außer unangenehm riechenden Abbauprodukten des DMSO keine weiteren
Umsetzungsprodukte detektiert werden. Bei der Reaktion mit Methanolat in Methanol
konnte ebenfalls keine Bildung des Zielprodukts beobachtet werden. Literaturhinweisen zu
Folge wäre es effektiver gewesen, bereits das Nitril XXXIV mit Methanolat reagieren zu
lassen196.
Abb. 35: Fehlgeschlagene Substitution von 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) mit Alkoholaten/Phenolaten: i) Natriummethanolat in Methanol/Phenol mit Kalium-tert-Butanolat, DMSO, bis zu 150 °C, bis zu 2 h, Mikrowellenreaktor.
Die zum Teil sehr niedrigen Ausbeuten bei der Substitution mit einigen Aminen (3-75 %)
deuteten bereits an, dass das 2-Chlorpyridin durch den Tetrazolring für die SNAr offenbar
deaktiviert wurde. Ein Grund dafür könnte sein, dass unter den stets basischen
Reaktionsbedingungen das acide Tetrazol-Proton entfernt wurde. Das resultierende
Tetrazolat erhöhte mit einem +I-Effekt die Elektronendichte im Pyridinring, was für die SNAr
als negativ einzuschätzen war. Dieser Gedankengang ließ folgende Schlüsse zu: Entweder
wird als Edukt das Nitril eingesetzt (siehe 2-(Benzylthio)-4-(1H-tetrazol-5-yl)pyridin (LIV)),

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
44 | Untersuchungen, Ergebnisse und Diskussion
mit Katalysatoren wie Palladium gearbeitet197-199 (Experimente, bei denen BrettPhos-
Katalysatoren verwendet wurden, schlugen aber ebenfalls fehl) oder das acide Tetrazol-
Proton durch eine Schutzgruppe ausgetauscht. Letzteres spielte im weiteren Verlauf dieser
Arbeit eine erhebliche Rolle und soll an späterer Stelle diskutiert werden.
Um die chemische Diversität der Testsubstanzsammlung zu erweitern, sollten die durch
Substitution mit Diaminen und Aminoalkoholen erfolgreich dargestellten Derivate über
deren neu eingeführte Amino- oder Hydroxyfunktionen weiter funktionalisiert werden. Dafür
wurden Carbon-, Sulfon- oder verschiedene Kohlensäurechloride eingesetzt, welche zu
den entsprechenden Amiden beziehungsweise Carbamaten führten (Abb. 36 und Tab. 4).
Abb. 36: Umsetzung von N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII) zu den entsprechenden Derivaten: i) Carbon-, Sulfon- oder Kohlensäurechlorid, NaOH, Wasser/ACN, 0 °C bis Raumtemperatur, bis zu 24 h.
Tab. 4: Übersicht der auf N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII) basierenden dargestellten Derivate mit jeweiligem Rest R.
Carbonsäureamide
LVII LVIII
Sulfonsäureamide
LIX
Carbamate
LX
Auf Grund der sehr geringen Löslichkeit des Edukts XLIII gestaltete sich eine Acylierung
schwierig. Der Ausgangstoff war grundsätzlich nur dann in Wasser oder Methanol
solvatisierbar, wenn zusätzlich ein basisches Agens wie Natriumhydroxid oder Ammoniak
dazugegeben wurde. Die protischen Lösungsmittel stellen ebenfalls Reaktionspartner für
Säurechloride dar, selbiges gilt für die basischen Zusätze, die entweder die
Hydrolyse/Alkoholyse unterstützten oder zur Bildung des entsprechenden primären Amids
O

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 45
führten. Trotz Eiskühlung und mehrfacher Nachdosierung der Säurechloridäquivalente
waren deshalb die Ausbeuten sehr gering (ab 6 %).
Bei Substanz LX wurde deshalb eine Weiterentwicklung des präparativen Vorgehens
vorgenommen: Das Edukt XLIII wurde dafür erst in Acetonitril suspendiert und
anschließend mit verdünnter Natronlauge versetzt. Diese löste den Feststoff unter
Ausbildung eines Zweiphasen-Systems. Acetonitril und Wasser sind eigentlich vollständig
miteinander mischbar, da aber durch das hochkonzentrierte Natriumtetrazolat eine hohe
Ionenstärke erreicht wurde, bildete sich eine Grenzschicht aus, welche die Acetonitril- und
die wässrige Phase von einander abtrennte. Die deutlich lipophileren Säurechloride
reicherten sich hingegen in der organischen Phase an. Da Acetonitril zu den nicht
protischen Lösungsmitteln gezählt wird, waren die Säurechloride weitestgehend vor
Hydrolyse/Alkoholyse geschützt. An der Grenzfläche kam es dann zur Reaktion mit dem in
der wässrigen Phase gelösten Edukt XLIII. Wahrscheinlich fand auch eine partielle
Hydrolyse statt, die höhere Ausbeute (56 %) und die Verwendung weniger Äquivalente
Säurechlorid sprachen aber für die Überlegenheit dieser präparativen Variante. Es galt
jedoch zu beachten, dass aus den abreagierten Säurechloriden bei Umsetzung äquimolare
Mengen an HCl freigesetzt wurden. Daher war es unter Umständen nötig, Natronlauge zu
ergänzen, um erneutes Ausfällen der Edukte zu verhindern.
Diese Technik sollte auch für Reaktionen mit dem an der Seitenkette um ein
Kohlenstoffatom verlängerten Edukt XLIV zum Einsatz kommen (Abb. 37 und Tab. 5). Bei
den verhältnismäßig lipophilen Derivaten LXIII und LXIV wiesen die Edukte 4-Methoxy- und
4-Brombenzoylchlorid keine ausreichende Löslichkeit mehr in Acetonitril auf. Durch einen
Austausch mit Tetrahydrofuran konnte auf diesen Befund reagiert und die negativen
Auswirkungen abgewendet werden. Tetrahydrofuran bildete ebenso wie Acetonitril trotz
eigentlicher Mischbarkeit mit Wasser auf Grund der hohen Salzkonzentration ein
Zweiphasen-System aus. Zumindest für das Bromderivat konnte mit dem
Lösungsmittelaustausch eine beachtliche Ausbeutensteigerung auf 82 % erreicht werden.
Abb. 37: Umsetzung von N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV) zu den entsprechenden Derivaten: i) Carbonsäure-, Sulfonsäure oder Kohlensäurechlorid, NaOH, Wasser/ACN/THF, Raumtemperatur, bis zu 24 h.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
46 | Untersuchungen, Ergebnisse und Diskussion
Tab. 5: Übersicht der dargestellten Derivate basierend auf N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV) mit jeweiligem Rest R.
Carbonsäureamide
LXI LXII LXIII LXIV
Sulfonsäureamide
LXV
Carbamate
LXVI LXVII
Neben der erfolgreichen Umsetzung der Diamin-Seitenketten zu Amiden oder Carbamaten,
sollte ebenfalls die freie Hydroxygruppe der Aminoalkohol-Seitenkette für weitere
Reaktionen adressiert werden. Die Bildung eines Esters fungierte dafür als eine Möglichkeit
(Abb. 38). Der Vorteil lag darin, dass das sehr hydrophile Molekül XLVI durch den unpolaren
Ester eine ausgeglichenere Polarität erhielt. Als nachteilig war die Hydrolyseempfindlichkeit
einzuschätzen. Da zum Zeitpunkt dieser Synthese die oben beschriebene Arbeitstechnik
unter Verwendung eines Zweiphasen-Systems noch nicht etabliert war und deshalb nur auf
eine Kaliumhydroxid-Lösung als Lösungsmittel zurückgegriffen wurde, war die Ausbeute
der Synthese entsprechend niedrig (3 %). Erst nach Vorliegen biologischer Testdaten sollte
entschieden werden, ob die Synthese einer ganzen Reihe von Estern des Typs LXVIII
sinnvoll wäre. Bei positiven Resultaten hätte dann mit der Zweiphasen-Arbeitstechnik ein
Weg zur Verfügung gestanden, mit welchem in kürzester Zeit eine Substanzbibliothek hätte
erstellt werden können.
Abb. 38: Derivatisierung von 2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethan-1-ol-Hydrochlorid (XLVI) zum entsprechenden Benzylester: i) Benzoylchlorid, KOH-Lösung, Raumtemperatur, 18 h.
Neben der Synthese der hydrolyselabilen Ester war auch die Darstellung verschiedener
Ether geplant. Die klassische Ethersynthese mit Natriumhydrid und Alkylbromid (modifiziert

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 47
nach Bream et al.200) im apolaren Lösungsmittel wurde dabei ebenso untersucht wie die
Umsetzung mit Iodmethan katalysiert durch Silber(I)-oxid201,202 (Abb. 39). Beide
Etherbildungen schlugen aber fehl, weshalb weiterhin versucht wurde, zwei Alkohole unter
Sulfonsäurekatalyse in einer Dean-Stark-Apparatur zu einem Ether zu kondensieren203.
Auch diese Synthese gestaltete sich nicht erfolgreich. Ein letzter Versuch wurde deshalb
mit dem Ziel unternommen, den Alkohol mittels Thionylchlorid zunächst in sein Alkylchlorid
umzuwandeln204. Dies gelang jedoch ebenfalls nicht. Allen diesen nicht wie geplant
verlaufenden Synthesen ist die Verwendung eines apolaren, nicht protischen
Lösungsmittels gemein. Das Edukt XLVI ist aber wie die Edukte XLIII und XLIV nahezu
unlöslich in aprotischen Lösungsmitteln und kann wie bereits beschrieben nur in basischem
Methanol oder Wasser gelöst werden. Diese Eigenschaft wurde als Hauptgrund für die
fehlgeschlagenen Etherbildungen in Betracht gezogen. Ein Alkylierungsversuch in
protischen Lösungsmitteln erschien jedoch als keine sinnvolle Alternative, da dort das
Alkylanz vor allem mit dem Lösungsmittel abreagieren würde. Weiterhin bestand die
interessante Fragestellung, ob es bei einer Alkylierung überhaupt zu einer Derivatisierung
des Alkohols oder eventuell doch des Tetrazols gekommen wäre. Dass sich Tetrazole leicht
alkylieren lassen, wird im weiteren Verlauf dieser Arbeit aufgezeigt.
Abb. 39: Fehlgeschlagene Synthese von Ethern und Azomethinen: i) Iodmethan, NaH/Ag2O, aprotisches Lösungsmittel, bis 100 °C, mehrere h; ii) Thionylchlorid, Toluen, Rückfluss, mehrere h; iii) entsprechender Aldehyd bzw. Keton (hier Propionaldehyd), ohne Lösungsmittel/MeOH/ACN/Dioxan, Raumtemperatur bis 120 °C, mehrere Tage, Mikrowellenreaktor.
Ähnliche Beobachtungen konnten bei der Umsetzung von XLIII mit Aldehyden oder
Ketonen gemacht werden (Abb. 39). Der Azomethinbildung, eine sehr pH-empfindliche
Reaktion wie Untersuchungen zeigten205, sollte eine Reduktion mit Natriumboranat
nachgestellt werden, um ein sekundäres Amin zu erhalten. Trotz großer Überschüsse an
Aldehyd/Keton und intensiver Energiezufuhr über den Mikrowellenreaktor gelang es nicht,
die Bildung eines Azomethins nachzuweisen. Es wurde daher auch probiert, methanolische
Suspensionen mit Natronlauge oder Salzsäure gerade so einzustellen, dass sich die Edukte

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
48 | Untersuchungen, Ergebnisse und Diskussion
lösten, das pH-Optimum der Reaktion aber noch nicht verlassen wurde. Hierbei blieb der
Erfolg jedoch aus.
Daher kann zusammengefasst werden, dass die 4-(1H-Tetrazol-5-yl)pyridin-2-amine
und -thiole bzw. -thioether in großer Mannigfaltigkeit synthetisierbar waren. Die über die
Substitution mit Diaminen oder Aminoalkoholen neu eingeführte funktionelle Gruppe konnte
nach Optimierung der Reaktionsbedingungen für zahlreiche Acylierungen genutzt werden.
1.2.2.2 4-(1H-Tetrazol-5-yl)picolinamide
Der amphotere Charakter der Tetrazolylpyridine besaß einen eher negativen Einfluss vor
allem auf die Löslichkeit der Moleküle. Es war daher angebracht, neben den bislang
verwendeten +M-Substituenten auch solche einzubringen, die durch einen Elektronenzug
die basischen Eigenschaften des Pyridinstickstoffatoms abschwächen. Eine in Position 2
befindliche Carbonylkomponente übt einen –M- und –I-Effekt auf das Restmolekül aus und
kann damit einen Beitrag zur Herabsetzung der Basizität leisten.
Als zweckdienlich hatte sich dafür eine radikalische Alkoxycarbonylierung im Zweiphasen-
System erwiesen, welche als Abwandlung der Minisci-Reaktion aufgefasst werden kann206
(Abb. 40). Anders als in den vorherigen Synthesen kam als Edukt hierbei das
unsubstituierte Isonicotinnitril (LXXII) zum Einsatz. Mit Hilfe von Ethylpyruvat und der
Radikalstarterkombination aus Wasserstoffperoxid und Eisen(II)-sulfat wurde ein
Ethoxycarbonylradikal erzeugt, welches hauptsächlich das in Position 2 vorhandene Proton
substituierte. Heinisch und Lötsch gaben zu beachten, dass sowohl die Verhältnisse von
Peroxid zu Substrat, als auch die Volumina von Wasser und Dichlormethan eingehalten
werden müssen207-209. Zwar hätte die Empfehlung ein Verhältnis von 1:10 (Substrat zu
Peroxid) einzuhalten die besten Ausbeuten generieren sollen, auf Grund der
unökonomischen Verhältnisse wurde jedoch eine Substrat-Peroxid-Konstellation von 1:3
gewählt. Die damit erhaltene Ausbeute lag bei etwas mehr als 50 %. Hierbei liefen jedoch
kaum Nebenreaktionen ab, so dass weder Dimere noch doppelt substituierte Produkte
gefunden wurden. Die Aufreinigung beschränkte sich daher auf Basisanwendungen wie
Flüssig-Flüssig-Extraktion und Waschen mit Diethylether. Das so erhaltene in Position 2
modifizierte Derivat LXXIII konnte dann mit der etablierten Synthesemethode in sehr guten
Ausbeuten zum entsprechenden Tetrazol LXXIV umgesetzt werden.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 49
Abb. 40: Syntheseschema für die 4-(1H-Tetrazol-5-yl)picolinamide: i) Ethylpyruvat, FeSO4∙7H2O, H2O2, H2SO4, Dichlormethan, Wasser, –5 °C, 30 min; ii) NaN3, TEA∙HCl, Toluen, 110 °C, 16 h; iii) Amin, Ethanol, 165-185 °C, bis zu 2 h, Mikrowellenreaktor; iv) NaOH, MeOH/Wasser, Rückfluss, 3 h; v) Amin, HATU, DIPEA, DMF, bis zu 50 °C, bis zu 24 h.
Tab. 6: Übersicht der dargestellten 4-(1H-Tetrazol-5-yl)picolinamide mit jeweiligem Rest R.
Carbonsäureamide aus Aminolyse Carbonsäureamide aus HATU-Kupplung
LXXVI
LXXX
LXXVII
LXXXI
LXXVIII
LXXIX
Die eingeführte Esterfunktion bot eine Grundlage für Umsetzungen mit Aminen zu
entsprechenden 4-(1H-Tetrazol-5-yl)picolinamiden (Tab. 6). Die direkte Aminolyse war
jedoch nur mit flüssigen und benzylischen oder aliphatischen Aminen im Überschuss unter
Einsatz des Mikrowellenreaktors möglich. Bereits bei dem Derivat LXXIX, dessen Edukt
4-(Thiophen-2-yl)benzylamin als Feststoff vorlag und nur etwas mehr als äquimolar

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
50 | Untersuchungen, Ergebnisse und Diskussion
zugesetzt wurde, war eine Umsetzung kaum mehr durchführbar. Es konnte zwar das
gewünschte Produkt isoliert werden, allerdings reichte die erhaltene Menge gerade nur für
die Aufnahme eines 1H-NMR-Spektrums zur Überprüfung der Identität des Stoffes aus. Bei
einem eher elektronenarmen primären aromatischen Amin wie 2-Aminobenzimidazol
gelang die Aminolyse dann wie erwartet nicht mehr. Das Derivat LXXX sollte aber
dargestellt werden, da es nach theoretischen Betrachtungen interessante
Wasserstoffbrücken-Donoreigenschaften und -Akzeptoreigenschaften erwarten ließ210. Um
dieses Ziel zu erreichen, musste deshalb eine alternative Syntheseroute gesucht und
beschritten werden.
Diese Herausforderung wurde erneut unter Zuhilfenahme eines Kupplungsreagenzes
angegangen (Abb. 40). Dafür musste als erstes die freie Säure LXXV durch Esterhydrolyse
hergestellt werden. Diese wurde mit methanolischer Natronlauge unter Rückfluss mit
nahezu quantitativen Ausbeuten durchgeführt. Die anschließende Kupplung fand dann mit
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium-hexafluorphosphat (HATU, Abb.
41) statt (modifiziert nach Dakin et al.211). Bei dieser Synthese greift das Carboxylat von
LXXV (deprotoniert durch die Hilfsbase DIPEA) den Uronium-Teil von HATU nucleophil an
und bildet dabei ein acyliertes und geladenes Isoharnstoffderivat aus. Dieses wird rasch
von dem entstandenen 7-Azabenzotriazol-1-olat zu einem Aktivester umgesetzt, wobei
Tetramethylharnstoff als Nebenprodukt generiert wird. Der Aktivester steht dann zur
weiteren Umsetzung mit einem Amin oder Alkohol zur Verfügung212,213.
Durch die Verwendung von HATU konnte das Benzimidazolderivat LXXX wie angestrebt
synthetisiert werden. Die Ausbeute von nur 8 % war der aufwendigen Aufreinigung
geschuldet und spiegelte nicht die gute Umsetzung wider. Mit dem Butylderivat LXXXI
konnte noch ein zweites Produkt unter Zuhilfenahme von HATU hergestellt werden.
Abb. 41: O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium-hexafluorphosphat (HATU) (gezeigt ist die Uronium-Form, eine Guanidinium-Form ist ebenfalls beschrieben)
Die dargestellten 4-(1H-Tetrazol-5-yl)picolinamide zeigten wie vermutet eine bessere
Löslichkeit in diversen Lösungsmitteln. So war es möglich, die NMR-Spektren komplett in
DMSO-d6 aufzunehmen, so dass diese nicht wieder umständlich als Na+-Salz in D2O
vermessen werden mussten. Diese Tatsache ist sehr wahrscheinlich der Absenkung der
Basizität des Pyridinstickstoffatoms zu verdanken. Die coplanare Anordnung des
Tetrazolylpyridins ist nach wie vor gegeben und wurde sogar um das sp2-hybridisierte
Kohlenstoffatom des Carbonsäureamids erweitert. Eventuell kann das Sauerstoffatom

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 51
dieser Gruppe sogar dafür genutzt werden, um zusammen mit dem Pyridinstickstoffatom
einen Chelatkomplex mit dem katalytischen Eisen-Kation der KDM4-Enzymreihe zu bilden.
Sollte sich dieser Befund in den biologischen Testungen erhärten, stünde mit der Aminolyse
oder der HATU-Kupplung ein etablierter Weg zur Verfügung, um zügig neue Derivate mit
diversen Seitenketten zu synthetisieren.
1.2.2.3 4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure
Trotz ähnlicher Synthesewege von 4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure und der
4-(1H-Tetrazol-5-yl)picolinamide, muss die Darstellung in einem gesonderten Unterkapitel
behandelt werden, da hinsichtlich Polarität und Komplexierungseigenschaften zum Teil
ausgeprägte Unterschiede existieren.
Als Edukte standen wieder der Ester LXXIV und die Carbonsäure LXXV zur Verfügung
(Abb. 42). In der Patentliteratur war die Synthese einer Pyridin-2-hydroxamsäure
ausgehend von dem entsprechenden Ester beschrieben214. Die Umsetzung mit wässriger
Hydroxylamin-Lösung sollte von katalytischen Mengen Kaliumcyanid profitieren. Bei dem
Versuch, diese Vorschrift auf das hier vorgestellte Molekül zu übertragen, konnten jedoch
lediglich die Ausgangsstoffe zurückgewonnen werden. Aus diesem Grund wurde sich einer
Methode bedient, bei welcher die Carbonsäure mit Hilfe von Oxalylchlorid in das
entsprechende Carbonsäurechlorid übergeführt wurde215. Hierbei kam jedoch
Dichlormethan als Lösungsmittel zum Einsatz, in dem die Pyridincarbonsäure LXXV
erwartungsgemäß als kleines und sehr polares Molekül nahezu keine Löslichkeit zeigte.
Deshalb wurde ein Austausch des Lösungsmittels notwendig. In Dimethylformamid war
LXXV ausreichend gut löslich, da Dimethylformamid aber selber mit Oxalylchlorid reagieren
konnte, stellte diese Wahl nur einen Kompromiss dar. Anhand bläulich-grüner
Verfärbungen während der Synthese ließen sich die Nebenreaktionen optisch verfolgen.
Abb. 42: Die Synthese von 4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure gelang nur durch Umsetzung des korrespondierenden Säurechlorids: i) NH2OH-Lsg., KCN, THF/MeOH, bis 40 °C, 28 h; ii) Oxalylchlorid, NH2OH∙HCl, DIPEA, DMF, Raumtemperatur, 18 h.
Die nach Fällung mit Salzsäure notwendige Säulenchromatographie zeigte die wohl
markanteste Eigenschaft der Hydroxamsäuren auf: Es kam zu einer Violettfärbung des
Zielprodukts noch während der chromatographischen Aufarbeitung, womöglich durch
Schwermetallspuren im Kieselgel. Diese für Hydroxamsäuren typische Färbung von deren

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
52 | Untersuchungen, Ergebnisse und Diskussion
Komplexen mit vielen Metallkationen kann zur Analytik und das Konzept der Chelatisierung
mittlerweile auch zur Therapie genutzt werden. Durch Umkristallisation aus Wasser wurde
dann aus dem gefärbten Rohprodukt ein ausreichend reines Endprodukt erhalten.
Dass es sich bei Hydroxamsäuren oftmals um promiskuitive Liganden vieler
Metalloenzyme, sogenannte „Dirty Drugs“, handelt, ist durchaus verständlich, da prinzipiell
viele unterschiedliche Kationen adressiert werden können. Im Falle der schon
besprochenen Histon-Desacylase-Hemmer scheint es aber durch einen spezifischen
Aufbau möglich zu sein, eine gewisse Selektivität zu erreichen, obgleich mit ähnlichen
Strukturen auch DNA-Methyltransferasen inhibiert werden konnten216,217. In der
Fachinformation zu Panobinostat, einem in Deutschland zugelassenen HDAC-Inhibitor für
Patienten mit rezidiviertem und/oder refraktärem Multiplen Myelom, heißt es218: „Die
Behandlung von Tumorzellen mit Panobinostat führte zu einem dosisabhängigen Anstieg
der Acetylierung der Histone H3 und H4 sowohl in vitro als auch in präklinischen Xenograft-
Tiermodellen, was auf eine zielgerichtete Hemmung hinweist.“ Die allgemeine Aussage,
dass es sich bei Hydroxamsäuren immer um unselektive Inhibitoren handeln muss, scheint
mittlerweile nicht mehr haltbar zu sein.
Ob die hier vorgestellte 4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure einen inhibierenden
Einfluss auf die KDM4-Enzymreihe zeigen konnte, sollte durch biologische Tests geklärt
werden. Bei positiven Befunden bleibt die Frage nach der Selektivität aber unausweichlich.
1.2.2.4 [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamine
Neben den bisher vorgestellten Tetrazolylpyridinen, welche an Position 2 direkt mit einem
Hetero- oder einem Carbonylkohlenstoffatom verknüpft waren, sollten im Folgenden
Strategien entwickelt werden, um eine Methylengruppe als Spacer zwischen Pyridinring
und einem neu eingeführten Stickstoffatom zu integrieren. Als Grundlage dieser
Betrachtung dienten zahlreiche, bereits eingangs erwähnte Inhibitoren mit Pyridin-4-
carbonsäure-enthaltenden Strukturen vor allem aus der Patentliteratur. Diese wiesen in der
Regel IC50-Werte im einstelligen mikromolaren Bereich gegen verschiedene Vertreter der
KDM4-Enzymreihe auf. Die Methylenbrücke verhindert eine weitere Beteiligung des freien
Elektronenpaares des eingeführten Heteroatoms am mesomeren System des Pyridins. Die
Auswirkungen auf physikochemische Eigenschaften wie Wasserlöslichkeit und
Acidität/Basizität sollten auch deshalb untersucht werden, weil die bisher dargestellten
4-(1H-Tetrazol-5-yl)pyridin-2-amine eine generell schlechte Löslichkeit zeigten.
Die Einführung der Methylengruppe wurde über verschiedene Wege realisiert. Für die
Synthese des nicht weiter derivatisierten [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamins
(LXXXVIII) gelang dies nach zahlreichen Optimierungsversuchen mit einem Austausch

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 53
eines Chlorsubstituenten gegen eine Cyanogruppe mit anschließender Reduktion (Abb.
43).
Abb. 43: Syntheseschema für [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin (LXXXVIII): i) u. a. CuCN, NMP, 240 °C, 30 min, Mikrowellenreaktor; ii) Tritylchlorid, TEA, THF, Raumtemperatur, 17 h; iii) Zn(CN)2, Tetrakis(triphenylphosphin)palladium(0), DMF, 160-190 °C, 20-30 min, Mikrowellenreaktor; iv) Benzylbromid, K2CO3, Aceton, 50 °C, 1,5 h; v) H2, Pd/C, MeOH, Raumtemperatur, 24h.
Die Cyanierung eines Bromaromaten wurde bereits in Abschnitt 1.2.1.1 diskutiert und stellte
eine gewisse Herausforderung dar. Bei dem Edukt XXXV handelte es sich aber um einen
elektronenarmen Aromaten, welcher zusätzlich in Position 2 mit einem Chloratom eine
verhältnismäßig gute Abgangsgruppe besaß. Ein Austausch im Sinne einer klassischen
SNAr lag deshalb nahe. Zusätzlich wurden weiterhin die Bedingungen der ebenfalls schon
erwähnten Rosenmund-von-Braun-Reaktion angewendet. Überraschenderweise konnte
damit kein Zielprodukt erhalten werden. Da jedoch CN– ein gutes Nucleophil darstellte,
wurde die Vermutung gestützt, dass 2-Chlor-4-(1H-tetrazol-5-yl)pyridin für die nucleophile
aromatische Substitution nicht mehr zugänglich war. Um die Aktivierungsenergie zu
senken, wurden deshalb unter anderem zwei Katalysatoren eingesetzt:
Tetrakis(triphenylphosphin)palladium(0) mit Kaliumcyanid als Cyanidquelle219 und
BrettPhos Pd G3, welches Palladium in der Oxidationsstufe +2 enthielt, in Kombination mit
Kaliumhexacyanidoferrat(II)220. Weiterhin wurde auch beachtet, dass sich Katalysatoren-
deaktivierender Cyanwasserstoff aus der Reaktion von saurem Tetrazol und der
Cyanidquelle bilden konnte. Deshalb wurden die Experimente auch mit dem
N Cl
N
NN
HN
i
N
N
NN
HN
N
N Cl
N
NN
N
ii
N Cl
N
NN
N
iv
iii
N
N
NN
N
N
iii
N
N
NN
N
NN
N
NN
HN
NH2
v
XXXV
LXXXIV LXXXV
LXXXIII
LXXXVI LXXXVII LXXXVIII

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
54 | Untersuchungen, Ergebnisse und Diskussion
entsprechenden Natriumtetrazolat durchgeführt. Es war jedoch nicht möglich, das cyanierte
Derivat LXXXIII zu synthetisieren. Neben Spuren von Wasser, welche zur Protonierung von
Cyanidionen geführt haben könnten, durfte weiterhin die Cyanidquelle nicht im Überschuss
eingesetzt werden, da sich dies negativ auf die Umsetzung ausübte129. Eine andere
mögliche Ursache für das Scheitern der Reaktion lag in der Koordinierungsbereitschaft des
Tetrazols. Palladium-haltige Tetrazolkomplexe sind bekannt und verhältnismäßig leicht
herzustellen221. Es war daher nicht auszuschließen, dass die eingesetzten Palladium-
Liganden-Kombinationen zu nicht katalytisch wirkenden Palladium-Tetrazol-Komplexen
umgesetzt wurden. Mit einer Alkylierung des Tetrazolrings würde die Fähigkeit zu
Koordinierung an Metalle stark eingeschränkt werden, weshalb die Suche nach einer
passenden Schutzgruppe an dieser Stelle unausweichlich wurde.
Getestet wurden hierzu zum einen die Triphenylmethyl- (Trityl-) und zum anderen die
Benzyl-Gruppe (Bn). Beide Reste konnten durch klassische Alkylierungsreaktionen mit den
jeweiligen Chloriden oder Bromiden und einer Hilfsbase zum Abfangen des substituierten
Protons eingefügt werden. Der Austausch mit der sperrigen Trityl-Gruppe erfolgte mit nur
mäßigen Ausbeuten von etwa 25 % (modifiziert nach Nisnevich et al.222). Ein Grund hierfür
könnte die Durchführung der Umsetzung bei Raumtemperatur gewesen sein, wobei durch
Erhitzen eventuell die Ausbeuten hätten gesteigert werden können. Dieser Punkt erschien
aber unerheblich, da beim Einfügen der Cyanidgruppe eine unerfreuliche Beobachtung
gemacht wurde: Statt des derivatisierten Pyridins wurde Tritylcyanid als Hauptprodukt
erhalten. Dementsprechend ließ sich schlussfolgern, dass die Trityl-Schutzgruppe an
Tetrazolen sehr empfindlich gegenüber nucleophilen Angriff reagierte und somit ungeeignet
für die gegebene Fragestellung war. Dieser Befund wurde auch von bisherigen
Untersuchungen im Arbeitskreis gestützt, da es bereits bei der Umkristallisation aus
Methanol zur Umsetzung des Trityl-geschützten Tetrazols zum Methylether des
Triphenylmethans kam113.
Die Benzyl-Schutzgruppe entwickelte sich klassischerweise aus dem Kontext der
Alkoholfunktionen und schützt diese als benzylischen Ether223. Die Entschützung kann
unter Palladium-katalysierten, reduktiven Bedingungen mit Wasserstoff erfolgen und hat
damit gleichzeitig den Vorteil, dass das Nitril zum entsprechenden Amin umgesetzt werden
kann224. Somit können Abspaltung der Schutzgruppe und der letzte geplante
Reaktionsschritt unter denselben Bedingungen und damit auch in einem Ansatz
abgehandelt werden (Abb. 43).
Da Tetrazole in einem Tautomerengemisch von 1H- und 2H-Isomer vorliegen, sind
folgerichtig auch Alkylierungen an Position 1 und 2 möglich. Moutevelis-Minakakis et al.
publizierten eine Benzylierung eines C-substituierten Tetrazols mit Hilfe von Benzylbromid
und Kaliumcarbonat in Aceton225. Die Autoren gingen in ihrer Arbeit ausschließlich von einer
Alkylierung in Position 1 des Tetrazols aus. Im Gegensatz dazu zeigten Nelson et al., dass

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 55
Alkylierungen in Acetonitril mit Triethylamin zu einem Regioisomerengemisch führte226. Es
sind ferner systematische Untersuchungen vorhanden, die verdeutlichten, dass die
entsprechenden Reste an Position 1 und 5 der Tetrazole ausschlaggebend für die
Verhältnisse der Produkte waren227. Eine Umsetzung mit Benzylalkohol im stark Sauren
sollte ausschließlich zu einer Alkylierung an Position 2 führen228. Um ein isomerenreines,
selektiv an Position 1 alkyliertes Produkt zu erhalten, könnte beispielsweise von einem
N-Benzylamid ausgegangen werden229.
Anmerkung: Aus Gründen der Übersicht werden in dieser Arbeit nur die
Alkylierungsprodukte der 1H-Tetrazoltautomere aufgezeigt. Der Sinn einer
Schutzgruppe besteht darin, Reaktionen zu ermöglichen, die ohne diese nicht oder
nur schlecht durchführbar sind, oder um Nebenreaktionen zu vermeiden. Da es in
der Natur einer Schutzgruppe liegt, in späteren Reaktionsschritten wieder aus dem
Molekül entfernt zu werden, erschien eine röntgenkristallographische Untersuchung
als nicht zwingend notwendig. Aus den NMR-Spektren wird das Alkylierungsmuster
nicht deutlich, es wurde aber stets nur mit einem Regioisomer gearbeitet.
Wie bei der benannten Alkylierung nach Moutevelis-Minakakis et al. konnte nach der
Flüssig-Flüssig-Extraktion mit Ethylacetat und verdünnter Natronlauge nur ein alkyliertes
Produkt durch dünnschichtchromatographische Kontrolle aufgefunden werden. Dieses
wurde zwar noch weiter säulenchromatographisch gereinigt, aber nur um noch
überschüssiges Benzylbromid und farbige Nebenprodukte zu entfernen. Dementsprechend
wurde offenbar kein Regioisomerengemisch erhalten, was für die weiteren
Reaktionsschritte einen positiven Aspekt darstellte.
Die weitere Umsetzung gestaltete sich ebenfalls erfolgreich, wenn gleich nur mit mäßigen
Ausbeuten. Die Substitution des Chloratoms von LXXXVI gegen eine Cyanogruppe
funktionierte mit Kupfer(I)-cyanid bei hohen Temperaturen nur in Spuren. Besser gelang
die Variante mit Zinkcyanid und Tetrakis(triphenylphosphin)palladium(0) im
Mikrowellenreaktor230-232. Limitierend hierbei waren aber zum einen die hohen Kosten des
Katalysators, der zwar nur zu 5 mol%, aber aufgrund der großen molaren Masse bis zu
200 mg je Ansatz zugesetzt wurde. Zum anderen konnte die Ausbeute von knapp 25 %
nicht weiter gesteigert werden, obwohl unter anderem längere Reaktionszeiten bei höheren
Temperaturen und eine Erhöhung der Katalysatormenge ausprobiert wurden.
Die darauffolgende Reduktion des Nitrils und die gleichzeitige Abtrennung der
Schutzgruppe wurden dann mit Wasserstoff und Pd/C in Methanol mit einer Ausbeute von
knapp 50 % realisiert. Bei der Aufreinigung zeigte sich erneut, dass der amphotere
Charakter, welcher bei der benzylierten Vorstufe aufgrund der fehlenden Acidität des
Tetrazols nicht gegeben war, zu negativen Einflüssen auf die chromatographischen

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
56 | Untersuchungen, Ergebnisse und Diskussion
Eigenschaften in Form von starkem Tailing führte. Trotzdem konnte das eigentliche
Zielmolekül [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin (LXXXVIII) synthetisiert werden
und stünde theoretisch für weitere Kettenverlängerungen über das mit einer
Methylenbrücke verknüpfte primäre Amin zur Verfügung. Die Gesamtausbeute von etwa
8 % für alle drei Schritte reichte praktisch aber nicht annähernd, um eine genügende Menge
an Ausgangsstoff bereitzustellen, um die geplanten Derivate zu synthetisieren. Aus diesem
Grund wurden drei neue Synthesewege entwickelt, welche im Folgenden vorgestellt
werden sollen (Abb. 44).
Abb. 44: Geplante Syntheserouten für [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Derivate: i) Reduktion von 4-(1H-Tetrazol-5-yl)picolinamiden; ii) reduktive Aminierung von 4-(1H-Tetrazol-5-yl)picolinaldehyd; iii) nucleophile Substitution von Sulfonaten ausgehend von [4-(1H-Tetrazol-5-yl-pyridin-2-yl)methanol.
Eine alternative Möglichkeit stellte die Reduktion der Carbonsäureamide der zum Teil
bereits vorhandenen Picolinamide dar. Grundlegendes Problem hierbei war die überaus
schlechte Löslichkeit der Stoffe in Lösungsmitteln wie Diethylether oder Tetrahydrofuran.
Carbonsäureamide sind verhältnismäßig stabil gegenüber reduktiven Angriffen und
benötigen daher starke Reduktionsmittel wie beispielsweise Lithiumalanat. Da protische
Lösungsmittel gänzlich ungeeignet für die Reduktion mit Lithiumalanat sind, wird
üblicherweise ein aliphatischer Ether als Lösungsmittel benutzt233-235.
Diese Reaktionsbedingungen kamen bereits bei den Verbindungen LVII und LXI zum
Einsatz, wobei die Amide an den Seitenketten zu den entsprechenden sekundären Aminen
reduziert werden sollten. Wahrscheinlich auf Grund der Löslichkeit schlug dieser Versuch
jedoch fehl. Auch eine fortlaufende Benutzung einer Soxhlet-Apparatur über 96 Stunden,
welche das Lösungsmittel kontinuierlich recyclierte, ermöglichte es nicht, das gewünschte
reduzierte Amid zu erhalten. Bei der Reduktion der Verbindungen LVII und LXI kamen
sowohl Aluminiumchlorid als katalytisch wirkende Lewis-Säure236, als auch der polarere
Bis(2-methoxyethyl)ether (Diglyme) mit komplexierenden Eigenschaften237 erfolglos zum

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 57
Einsatz. Eine Teilumsetzung konnte bei der Verwendung von (Di-)Boran beobachtet
werden, welches zweckdienlich aus Natriumboranat und elementarem Iod in
Tetrahydrofuran in situ hergestellt wurde238. Der resultierende Boran-Tetrahydrofuran-
Komplex ist ein effektives Reduktionsagenz und vermag auch Amide zu reduzieren239.
Jedoch gelang es nicht, die intermediären Boran-Amin-Komplexe so zu spalten, dass das
gewünschte Amin isoliert werden konnte. Stattdessen wurde das ursprüngliche Amid
erhalten. Die Reduktion mit dem BH3-THF-Komplex war also geprägt durch die
Reversibilität der einzelnen Reaktionsschritte240, was die Umsetzung generell schwierig
gestaltete.
Da es immer wieder zu Schwierigkeiten vor allem auf Grund der Löslichkeit (Amphoterie)
kam und Erfahrungen zur Tetrazolalkylierung bereits vorlagen, wurde auch bei dem Edukt
LXXIV das acide Tetrazolproton gegen eine Benzylgruppe substituiert (Abb. 45). Nach
Aufbereitung durch Flüssig-Flüssig-Extraktion mit Ethylacetat und Kaliumcarbonat-Lösung
konnte im Übrigen wieder nur ein Regioisomer mittels Dünnschichtchromatographie
nachgewiesen werden.
Abb. 45: Benzylierung von 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV): i) Benzylbromid, K2CO3, Aceton, Rückfluss, 2 h.
Das resultierende Benzyltetrazolderivat LXXXIX besaß nun eine ausreichend hohe
Lipophilie, um in vielen aprotischen Lösungsmitteln solvatisierbar zu sein. LXXXIX konnte
anschließend in sehr guten Ausbeuten einer weiteren Aminolyse unterzogen werden, um
die Morpholin-enthaltende Verbindung XC zu generieren (Abb. 46).
Die direkte Überführung in das korrespondierende Amin XCII mit Hilfe von Lithiumalanat
schlug allerdings auch hier fehl. Löslichkeitsprobleme konnten nun nicht mehr die Ursache
für den Misserfolg darstellen, da eine klare Lösung des Edukts XC in Tetrahydrofuran
vorlag. Die Vermutungen gingen eher dahin, dass das verwendende Lithiumalanat trotz
Lagerung im Exsikkator schon partiell hydrolytisch zersetzt worden war. Bei der Reaktion
mit Wasser kam es zwar zur Gasentwicklung, jedoch nicht annähernd in der heftigen Weise,
wie es in den Sicherheitsdatenblättern erwähnt wird. Da die Zielmoleküle jedoch später
über andere Wege erzielt werden konnten, wurden keine weiteren Untersuchungen mit
Lithiumalanat mehr vorgenommen.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
58 | Untersuchungen, Ergebnisse und Diskussion
Abb. 46: Reduktion eines Amids mit Hilfe von PCl5 und NaBH4: i) 3-Morpholinopropylamin, EtOH, 55 °C, 72 h; ii) LiAlH4, THF, Rückfluss, 24 h; iii) PCl5, DCM, Rückfluss, 5 h; iv) NaBH4, EtOH, 0 °C-Rückfluss, 45 min; v) H2, Pd/C, MeOH, 35 °C, 6 h.
Da sich mittlerweile einige Reduktionsmöglichkeiten verschiedener Amide in dieser Arbeit
als sehr zeitintensiv und erfolglos herausgestellt hatten, sollte eine letzte und eher weniger
bekannte Variante auf ihre Eignung untersucht werden. Carbonsäureamide können auch
mit dem in protischen Lösungsmitteln verhältnismäßig stabilen Reduktionsmittel
Natriumboranat in guten Ausbeuten reduziert werden, wenn sie vorher durch ein
Chlorierungsagenz wie Phosphoroxychlorid241 oder Phosphorpentachlorid242 „aktiviert“
wurden. Diese Chlorierungen fanden überwiegend in Dichlormethan durch Erhitzen unter
Rückfluss statt. Das resultierende Chloriminium-Ion (auch als Vilsmeier-Komplex bekannt)
war ausreichend reaktiv, um in Ethanol mit Natriumboranat oder auch elementarem Zink
reduziert zu werden243. Eine gesonderte Isolierung der Spezies war nicht vorgesehen, es
wurde lediglich das Lösungsmittel im Teilvakuum entfernt. Der Rückstand konnte dann in
Ethanol aufgenommen und anschließend erfolgreich zum sekundären Amin reduziert
werden. Die Rohausbeute von etwa 35 % wäre dann als ausreichend angesehen worden,
wenn die Entschützung des Tetrazols quantitativ verlaufen wäre. Da es hier allerdings zu
einer noch geringeren Ausbeute kam, konnte gerade nur so viel Zielprodukt XCIII isoliert

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 59
werden, dass die Analytik vervollständigt und eine ausreichende Menge für die biologische
Testung zur Verfügung gestellt werden konnte. Damit stellte dieser Weg zwar die erste
erfolgreiche Reduktion einen Amids mit Tetrazolylpyridin-Grundstruktur in dieser Arbeit dar,
gleichermaßen musste aber auch festgestellt werden, dass die Ausbeuten nicht optimal
waren. Ferner ging die Diversität der Syntheseroute dahingehend verloren, dass erst jedes
einzelne Picolinamid hergestellt werden musste. Eine Aufzweigung des Synthesewegs erst
auf letzter oder vorletzter Stufe wäre für die Effizienz in Hinblick auf die Darstellung einer
Substanzbibliothek wünschenswert gewesen. Es wurde daher beschlossen, vergleichbare
Reduktionen anderer Amide nicht weiter zu verfolgen.
Stattdessen erfolgten Experimente, die eine nucleophile Substitution am Aliphaten
vorsahen, um die entsprechenden [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Derivate
zu erhalten (Abb. 44, Weg iii). Durch das Überführen einer Alkoholfunktion in einen
Sulfonsäureester, sollte letzteres als gutes Nucleofug genutzt werden, um eine Substitution
mit einem Amin zu ermöglichen. Hierbei würde erst im letzten Schritt eine Verzweigung des
Synthesewegs erfolgen, eine hohe Diversität der so darstellbaren Produkte wäre also
gegeben.
Eine Besonderheit von 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV), welche auch die
Grundlage des hier beschriebenen Wegs der nucleophilen Sulfonatsubstitution
mitbegründete, ist die spezielle Reduktionsanfälligkeit der Esterfunktion. Diese ist an
Position 2 des Pyridinrings lokalisiert und damit einem starken Elektronenzug ausgesetzt.
Dadurch kommt es zu einer Intensivierung der ohnehin vorhandenen positiven
Polarisierung des Carbonylkohlenstoffatoms, welches auf Grund dessen von dem
verhältnismäßig milden Reduktionsmittel Natriumboranat angegriffen werden kann. Der
Vorteil von Natriumboranat wiederum ist seine Stabilität gegenüber protischen
Lösungsmitteln. Es war hierbei also nicht notwendig, den Ester LXXIV mit einer
Schutzgruppe zu derivatisieren. Stattdessen konnte dieser direkt in einem Tetrahydrofuran-
Methanol-System mit Natriumboranat in guten Ausbeuten zum korrespondieren Alkohol
XCIV umgesetzt werden (Abb. 47).
Die Reduktion von an Elektronenmangelaromaten lokalisierten Carbonsäureestern wäre
auch in anderen Medien als in einem Tetrahydofuran-Methanol-Gemisch möglich
gewesen244,245. Es wurde ebenfalls eine Kombination aus Dioxan und Wasser
ausprobiert246. Hierbei kam es allerdings durch den basischen pH-Wert, verursacht durch
die partielle Hydrolyse von Natriumboranat zu Natriumhydroxid im Wässrigen, auch zu einer
Esterhydrolyse. Die freie Säure ließ sich von dem ebenfalls sehr polaren Alkohol nur schwer
abtrennen, weshalb das wasserfreie Medium zu bevorzugen war. Mit Polyethylenglycol 400
stünde dafür ein geeignetes Lösungsmittel zur Verfügung247.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
60 | Untersuchungen, Ergebnisse und Diskussion
Abb. 47: Syntheseschema für [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Derivate über eine nucleophile Substitution von Sulfonaten: i) NaBH4, THF/Methanol, Rückfluss, 20 h; ii) Methansulfonylchlorid oder Tosylchlorid, TEA, 4-DMAP, THF/DMF, 0 °C-Raumtemperatur, bis zu 36 h (Intermediate nicht isoliert); iii) entsprechendes Amin, DMF, 60-80 °C, bis zu 72 h.
Die Hydrophilie des Alkohols XCIV erwies sich hingegen bei dem Schritt der Darstellung
des Sulfonsäureesters als hinderlich. In Tetrahydrofuran kam es lediglich zu einer
Suspendierung, erst durch Zugabe von Dimethylformamid wurde eine nahezu feststofffreie
Lösung erhalten. Bedingt durch die Ergänzung von Dimethylformamid kam es aber
wahrscheinlich zu Nebenreaktionen, welche auch durch Zugabe des Katalysators 4-DMAP
nicht verhindert werden konnte.
Es war nicht vorgesehen, die empfindlichen Sulfonate zu isolieren, dennoch wurde ein
Versuch unternommen, diese auf Grund der entstandenen Nebenprodukte mittels
Säulenchromatographie aufzureinigen. Der Beweis, dass es sich um reaktive Intermediate
handelte, wurde auch direkt erbracht, da nach Eluieren über Kieselgel kein
Zwischenprodukt mehr gefunden werden konnte. Es kam wohl zu einer Reaktion bereits
während der Chromatographie, was jedoch als Beweis dafür gesehen wurde, dass das
gewünschte Sulfonat vorlag.
Das ungereinigte Sulfonat konnte alternativ direkt durch Zugabe eines entsprechenden
Amins als Eintopfsynthese zum sekundären Amin umgesetzt werden (modifiziert nach
Chelucci et al.248). Vier Derivate waren in Planung (Tab. 7), wobei die Umsetzung mit
Vertretern von aliphatischen, benzylischen und primären aromatischen Aminen erprobt
wurde. Nur mit den 3-Aminochinolin- und 4-Fluorbenzylamin-Substituenten konnte das
gewünschte Produkt erhalten werden. Allerdings waren die Ausbeuten der Reaktion so
gering, dass die erhaltene Menge nur für eine vollumfängliche Analytik der Stoffe
N
N
NN
HN
O
O
LXXIV
N
N
NN
HN
OH
iii
N
N
NN
HN
OS
O
O
i
N
N
NN
HN
OS
O
O
N
N
NN
HN
HN
R
ii
XCIV
XCV
XCVI

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 61
ausreichte. Über diesen Weg war es jedoch nicht möglich, genügend Substanz für eine
biologische Testung zur Verfügung zu stellen. Bei der Reaktion mit 3-Aminopropan-1-ol und
3-Picolylamin konnte überhaupt kein Zielprodukt isoliert werden.
Tab. 7: Übersicht der Derivate mit [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Grundstruktur hergestellt über den Sulfonsäureester-Weg mit jeweiligem Rest R.
Erfolgreiche Substitution Fehlgeschlagene Substitution
XCVII XCIX
XCVIII C
Einen möglichen Grund für die niedrigen Ausbeuten stellte die aufwendige Aufreinigung
dar. Dadurch, dass sich bereits bei der Veresterung Nebenprodukte bildeten, konnten diese
dann ebenfalls mit dem Amin weiterreagieren. Eine Abtrennung der Zielprodukte war daher
aufwendig und nur mit zwei aufeinanderfolgenden chromatographischen Verfahren
möglich. Der eingangs erwähnte Vorteil der Reduktionsmöglichkeit des Edukts in Methanol
und damit auch die Abwesenheit einer Schutzgruppe am Tetrazolring führte dazu, dass die
Löslichkeit im zweiten Schritt des Sulfonsäureester-Wegs nicht mehr ausreichend war, um
auf Dimethylformamid als Lösungsmittel verzichten zu können. Somit wäre folglich eine
Benzylierung auch hier eventuell sinnvoll gewesen, um nicht nur die Nebenreaktion zu
begrenzen und damit die Ausbeuten zu steigern, sondern auch um eine größere Vielfalt an
Derivaten zu ermöglichen.
An dieser Stelle kann zusammengefasst werden, dass es mit den bisher vorgestellten
Wegen der Amidreduktion und der nucleophilen Substitution von Sulfonsäureestern nicht
möglich war, eine ausreichende Anzahl und Menge an gewünschten [4-(1H-Tetrazol-5-
yl)pyridin-2-yl]methanamin-Derivate zu gewinnen. Diese Struktur ist aber in Hinblick auf die
Inhibition der KDM4-Enzymreihe äußerst vielversprechend und sollte daher nicht verworfen
werden. In der Patentliteratur wird häufig mit einer reduktiven Aminierung gearbeitet (u. a.
in98), um entsprechende methylenverbrückte Amine zu erhalten. Ob diese Methode auch
auf die hier präsentierten Tetrazolylpyridine übertragbar war, soll im Folgenden erläutert
werden (Abb. 44, Weg ii).
Dafür musste im ersten Schritt der entsprechende Ester selektiv zum korrespondieren
Aldehyd reduziert werden. Erfahrungen aus dem Sulfonsäureester-Weg zeigten, dass eine
Reaktion mit Natriumboranat in methanolischer Lösung bereits zur Reduktion bis zum
Alkohol führte. Ein selektiveres Reduktionsmittel stellt hingegen Diisobutylaluminiumhydrid

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
62 | Untersuchungen, Ergebnisse und Diskussion
(DIBAL, Abb. 48) dar. Durch die sperrigen Substituenten soll ein Mehrfach-Angriff auf das
Ester-Carbonylkohlenstoffatom verhindert und damit eine Weiterreduktion zum Alkohol
vermieden werden. Üblicherweise erfolgt die Reaktion bei tiefen Temperaturen (bis –75 °C)
in Toluen249 oder Tetrahydofuran250,251. Die Verwendung dieser Lösungsmittel machte es
erforderlich, dass hier wieder mit der benzylgeschützten Verbindung LXXXIX gestartet
werden musste, um eine ausreichende Löslichkeit zu garantieren und um
Neutralisationsreaktionen mit dem basisch reagierenden DIBAL zu unterbinden. Nach
einigen Versuchen konnten dann die optimalen Bedingungen für die Reduktion zum
Aldehyd CI gefunden werden (Abb. 48). Die Reaktionstemperatur wurde mit einem
Ethylacetatbad kontrolliert, welches mit flüssigem Stickstoff bis kurz über den Schmelzpunkt
(–83 °C) heruntergekühlt wurde. An Stelle der vorgeschlagenen vier Äquivalente DIBAL
(nach Swahn et al.251) wurden nur drei Äquivalente benutzt. Theoretisch konnte dieser
Überschuss bereits ausreichen, um eine Weiterreduktion zu initiieren. Wahrscheinlich auf
Grund der starken Kühlung wurde diese jedoch nicht beobachtet.
Abb. 48: Optimierte reduktive Aminierung als dritter Weg der Herstellung von [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Derivaten: i) DIBAL, THF, –80 °C, 5 h; ii) entsprechendes Amin, NaBH4, MeOH, 0 °C-Rückfluss, 2-4 h; iii) H2, Pd/C, MeOH, Raumtemperatur, 1,5-4,5 h. Rechts: Strukturformel von DIBAL (Diisobutylaluminiumhydrid).
Durch Zugabe von Wasser und Essigsäure wurde überschüssiges DIBAL nach erfolgter
Reaktion zersetzt. Bei der anschließenden Flüssig-Flüssig-Extraktion gab es zu beachten,
dass die wässrige Phase nicht zu stark alkalisch eingestellt wurde, da es ansonsten zu
einer Gelbildung kam, vermutlich auf Grund von Aluminium- und Aluminiumhydroxid-
haltigen Verbindungen. Die hohe Viskosität machte eine Extraktion mit Ethylacetat
unmöglich, war jedoch durch Zugabe von wenigen Tropfen Essigsäure reversibel. Die
orangene Färbung nach Ansprühen des isolierten Produktes mit 2,4-Dinitrophenylhydrazin-
Reagenz auf der Kieselgelplatte zeigte an, dass der Aldehyd wie erhofft nicht weiter zum

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 63
Alkohol reduziert wurde, da die entsprechenden Alkohole nicht zu orangefarbenen
Hydrazonen reagieren können. Auf Grund von farbigen Verunreinigungen, die vor allem bei
der Hydrolyse des überschüssigen DIBAL entstanden, musste noch eine
Säulenchromatographie zur Aufreinigung durchgeführt werden. Die Gesamtausbeute der
Reduktion ist mit 80 % als gut zu bewerten.
Tab. 8: Übersicht der dargestellten [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Derivate über den Weg der reduktiven Aminierung mit jeweiligem Rest R.
Dargestellte Verbindungen Nichterhaltene Zielstruktur
XCVIII
C
CII
CIII
CIV
CV
Der Aldehyd CI konnte dann mit einem primären Amin zum entsprechenden Azomethin
umgesetzt werden. Diese Reaktion wurde in Methanol unter Rückfluss durchgeführt und
war je nach Gegebenheiten des Amins mehr oder weniger vollständig. Auch hier war es
nicht vorgesehen, das Azomethin zu isolieren, sondern den Glasaufbau im Sinne einer
Eintopfreaktion direkt mit Wasserstoff zu fluten, damit sowohl das Azomethin zum
sekundären Amin reduziert als auch die benzylische Schutzgruppe abgespalten werden
konnte. Somit beinhaltete diese Syntheseoperation wiederum gleich zwei sequentielle
Reaktionsschritte, wodurch diese Durchführung gewisse ökonomische Vorteile aufwies. Auf
diese Art konnte das Amin CII mit zwei Methoxysubstituenten in mittelmäßigen Ausbeuten
hergestellt werden (Tab. 8). Das bereits über den Sulfonsäureester-Weg dargestellte
O
O

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
64 | Untersuchungen, Ergebnisse und Diskussion
Derivat XCVIII musste auf Grund der geringen Ausbeuten erneut synthetisiert werden. Mit
dem Weg der reduktiven Aminierung waren die Erträge allerdings nicht wesentlich höher.
Gänzlich unmöglich war es, das Amin C mit einer zweiten Pyridinstruktur herzustellen.
Bei der Darstellung einiger [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin-Derivate kam es
während des Syntheseverlaufs zu einer unerwünschten Abspaltung benzylischer
Strukturelemente. Dabei konnte ein Trend beobachtet werden: Je stärker
elektronenziehend der Substituent war (3,4-Dimethoxy < 4-Fluor < Pyridin), desto eher
wurde auch die Amin-verbindende Methylengruppe durch die reduktiven Bedingungen
abgetrennt. Exemplarisch konnte dafür ein Bruchstück aus der Synthese von C isoliert
werden, welches im 1H- und 13C-NMR-Spektrum die gleichen Signale aufzeigte, wie das
bereits zuvor synthetisierte [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin (LXXXVIII) (Abb.
49). Als Hauptgrund konnten die zu lang gewählten Reduktionszeiten ausgemacht werden.
Bei den Aminen XCVIII, C und CIII betrug der Kontakt mit Wasserstoffatmosphäre bis zu
24 h. Diese sollten daher im Folgenden gekürzt werden. Da es sich bei der Schutzgruppe
aber ebenfalls um eine benzylische Struktur handelte, waren solche Arten von
Nebenreaktionen nicht gänzlich auszuschließen.
Abb. 49: Bei der Synthese von C und CIII kam es unter den Reaktionsbedingungen von H2 und Pd/C nicht nur zur Abspaltung der Schutzgruppe, sondern auch zur Abtrennung anderer Strukturen (rot).
Auf Grund dieser Befunde wurde dazu übergegangen, einen weiteren Isolierungsschritt zu
integrieren. Hintergrund waren auch die nicht immer vollständig gelungenen
Azomethinbildungen, weshalb jetzt zwei Äquivalente Amin anstelle eines äquimolaren
Verhältnisses eingesetzt wurden. Es wurde ferner davon abgesehen, Reduktion und
Entschützung in einem Schritt durchzuführen. Stattdessen sollte die Doppelbindung des
Azomethins erst selektiv durch Natriumboranat reduziert werden (Abb. 48). Damit das
überschüssige Amin und die anorganischen Salze keine störendenden Einflüsse auf die
Entschützung ausübten, wurde eine Flüssig-Flüssig-Extraktion mit Ethylacetat und
Natriumhydrogencarbonat-Lösung zwischengeschaltet. Das noch geschützte und damit
lipophile Amin reicherte sich in der organischen Phase an, während die polaren Amine und
Salze in der Wasserphase verblieben. Nach Entfernen des Ethylacetats wurde dann die
Schutzgruppe nach bekannter Methode mit Wasserstoff an Pd/C entfernt (eine gesonderte
Analytik der noch benzylgeschützten Amine war nicht vorgesehen). Mit den bereits
gesammelten Erfahrungen der Entschützung wurde in der Folge die Reaktion mit Hilfe der

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 65
Dünnschichtchromatographie detailliert verfolgt. Statt der bisherigen 24 h musste die
Hydrogenolysezeit auf maximal 4,5 h heruntergesetzt werden, wobei auch hier große
individuelle Schwankungen auftraten. So konnte bei dem Cyclopropylderivat CIII bereits
nach 1,5 h ein methylsubstituiertes Tetrazolylpyridin isoliert und analytisch ausgewertet
werden (Abb. 49). Die Ausbeute des Zielprodukts CIII betrug aber trotzdem etwa 34 %,
weshalb sich die aufwendigen Verlaufskontrollen als unabdingbar erwiesen. Die Amine CIV
und CV konnten dann in noch besseren Ausbeuten dargestellt werden (Hydrogenolysezeit
hier bis 4,5 h) (Tab. 8). Mit der Einführung des Tyramin-Restes bei Derivat CV konnte ferner
bewiesen werden, dass dieser Weg auch mit reaktiven Gruppen, wie einem Phenol,
erfolgreich durzuführen war.
Dass die Abspaltung von Benzylgruppen ein kritischer Parameter sein kann, konnte bereits
gezeigt werden252. Die Entschützung ist unter anderem abhängig von der Zeit, Temperatur,
Palladiumbeladung und Größe der Kohlepartikel. Bei sehr ähnlichen Strukturen wie einem
benzylgeschützten Tetrazol und einem benzylischen Amin war es daher schwierig, eine
ausreichende Selektivität zu erreichen. Für zukünftige Versuche wäre es daher eventuell
ertragreicher, wenn nicht mit einem unsubstituierten Schutzgruppenaromaten gearbeitet
würde, sondern dieser mit elektronenziehenden Gruppen verknüpft wäre. Dadurch könnte
das Tetrazol in geringerer Zeit entschützt werden, während das restliche Molekül (noch
nicht) angegriffen würde. In der hiesigen Arbeit konnten aber mit dem Weg der reduktiven
Aminierung fünf Derivate mit einem methylenverbrückten Amin hergestellt werden, welche
auf ihre inhibitorischen Eigenschaften gegenüber der KDM4-Enzymreihe getestet werden
sollten. Damit stellte dieser Weg zugleich den erfolgreichsten dar, da bei den anderen zwei
präsentierten Syntheserouten für die Darstellung von Derivaten mit [4-(1H-Tetrazol-5-
yl)pyridin-2-yl]methanamin-Struktur entweder die Ausbeuten oder die Substanzanzahl stark
eingeschränkt war. Bei erfolgreicher Testung stünde dann mit dem hier präsentierten Weg
eine Möglichkeit zur Verfügung, mit welcher eine große Anzahl an Derivaten in kurzer Zeit
dargestellt werden könnte. Mit einer Optimierung der Schutzgruppe wäre die Erstellung
einer solchen Substanzbibliothek eventuell noch einfacher möglich.
1.2.2.5 4-(1H-Tetrazol-5-yl)picolinsäure/2,4-Di(1H-tetrazol-5-yl)pyridin
Bei den bisher synthetisierten Substanzen ging es vor allem um die Art der Seitenkette in
Position 2 des Pyridinrings und damit auch um den Erkenntnisgewinn hinsichtlich der Wahl
der Länge, des Verbrückungstyps oder des Heteroatoms, um an dieser Stelle günstige
Einflüsse auf die inhibitorischen Eigenschaften gegen die KDM4-Enzymreihe erzielen zu
können. Um aber ebenfalls einen direkten Vergleich mit der Leitsubstanz Pyridin-2,4-
dicarbonsäure (Abb. 50) ziehen zu können, mussten entweder beide
Carbonsäurefunktionen oder nur eine durch einen Tetrazolring substituiert werden.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
66 | Untersuchungen, Ergebnisse und Diskussion
4-(1H-Tetrazol-5-yl)picolinsäure (LXXV) wurde bereits im Rahmen der Untersuchungen zu
Picolinamiden (Abschnitt 1.2.2.2) synthetisiert und stand deshalb in ausreichender Menge
zur Verfügung.
Abb. 50: Bioisosterer Austausch der Carboxygruppen von Pyridin-2,4-dicarbonsäure: i) CuCN, NMP, 240 °C, 30 min; ii) NaN3, TEA∙HCl, Toluen, 105 °C, 24 h.
Für die Darstellung von 2,4-Di(1H-tetrazol-5-yl)pyridin (CVII) musste hingegen erst Pyridin-
2,4-dicarbonitril (CVI) hergestellt werden, um im Anschluss beide Cyanogruppen
gleichzeitig in ein Tetrazol zu überführen (Abb. 50). Zum Zeitpunkt der Synthese lagen
durch die vorherigen Cyanierungsexperimente Erfahrungen über die Gegebenheiten dieser
anspruchsvollen Reaktion vor. In einer weiteren Umsetzung von 2-Chlorisonicotinnitril
(XXXIV) im Sinne einer SNAr mit Kupfer(I)-cyanid in Dimethylformamid (modifiziert nach
Alemparte et al.253) konnte zwar ein Produkt erhalten werden. Dieses zeigte aber im 1H-NMR-Spektrum ein 6H-Integral bei einer chemischen Verschiebung von δ = 3,05 ppm,
was mit den zwei Methylgruppen des Dimethylformamids in Verbindung gebracht werden
könnte. Durch einen Wechsel des Lösungsmittels zu N-Methyl-2-pyrrolidon konnte das
Dicyano-Produkt CVI letztlich erhalten werden. Auf Grund der geringen Ausbeute wurde
noch eine zweite Variante ausprobiert. In der Originalarbeit kamen bei dieser Kaliumcyanid,
Dimethylformamid und [18]Krone-6 als Kalium-Chelatbildner zum Einsatz254. Letzteres
wurde aber durch das ebenfalls chelatisierende Eigenschaften gegenüber kleineren
Kationen besitzende Lösungsmittel Diglyme ersetzt. Die Erträge lagen hierbei mit 10 %
jedoch noch niedriger als bei der Reaktion mit Kupfer(I)-cyanid (25 % Ausbeute). Da
allerdings die gleichzeitige Tetrazolierung beider Cyanogruppen gut von statten ging und
das Molekül ohnehin nicht weiter derivatisiert werden sollte, waren die erhaltenen Mengen
an Dicyano-Produkt CVI aus beiden Reaktionen ausreichend. Eventuell wäre auch ein Weg
über das entsprechende Pyridin-N-oxid des Isonicotinnitrils (LXXII) möglich gewesen255.
Durch die Oxidation wird der elektronenarme Charakter des Pyridinrings an den Positionen
i
N Cl
N
N
ii
N
N
NN
HNN
NN N
N
HN
N N
N
NN
HN
OH
O
O
OH
O
OH
LXXV
XXXIV CVI CVII
2,4-PDCA

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 67
2 und 4 aufgehoben und der Heteroaromat wäre damit an diesen Stellen für eine
elektrophile aromatische Substitution zugänglich.
1.2.3 Synthese von Carbonsäure- und Pyridin-beinhaltenden KDM4-Inhibitoren
Neben dem direkten Vergleich der Pyridin-2,4-dicarbonsäure mit 2,4-Di(1H-tetrazol-5-
yl)pyridin, welcher zur Abschätzung der Auswirkungen der bioisosteren Substitution dienen
sollte, wurde ferner das literaturbekannte Motiv der Pyridin-4-carbonsäure auf zwei der
schon synthetisierten Tetrazolylpyridine übertragen. Dabei sollten die Substanzen 3-{[4-
(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propan-1-ol-Hydrochlorid (XLVII) und N-⟨2-{[4-(1H-
Tetrazol-5-yl)pyridin-2-yl]amino}ethyl⟩methansulfonamid-Hydrochlorid (LIX) so verändert
werden, dass sich statt der Tetrazol- eine Carbonsäurefunktion an Position 4 des
Pyridinrings befindet.
Abb. 51: Darstellung der 4-Pyridincarbonsäurederivate: i) NaOH, EtOH, Wasser, Rückfluss, 3 h; ii) Ethylendiamin, 180 °C, 1 h, Mikrowellenreaktor; iii) Methansulfonylchlorid, ACN, Wasser, 0 °C-Raumtemperatur, 20 h; iv) 3-Aminopropan-1-ol, 120 °C, 22 h.
Für den ersten Schritt des Synthesewegs konnte auf dasselbe Edukt zurückgegriffen
werden, welches auch schon bei den 4-(1H-Tetrazol-5-yl)pyridin-2-aminen genutzt wurde
(Abb. 51). Hierbei wurde allerdings nicht mehr mit Natriumazid gearbeitet, statt dessen kam
eine Mischung wässriger Natronlauge mit Ethanol zum Einsatz194,256. Die basenkatalysierte
Hydrolyse führte unter Abgabe von Ammoniak zur Ausbildung einer Carboxygruppe.
Letztgenannte konnte über die Öffnung der Rückflussapparatur gut wahrgenommen
werden, weshalb die Vollständigkeit der Reaktion nicht mittels

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
68 | Untersuchungen, Ergebnisse und Diskussion
Dünnschichtchromatographie detektiert, sondern auf Grund des abklingenden
Ammoniakgeruchs erkannt wurde.
Die anschließende nucleophile Substitution mit dem entsprechenden Amin im
Mikrowellenreaktor oder im Rundkolben brachte interessante Beobachtungen hervor. Zum
einen besaßen alle synthetisierten 2-Aminoisonicotinsäurederivate eine hervorragende
Wasserlöslichkeit. Die dazu äquivalenten Tetrazolylpyridine hingegen waren nahezu
unlöslich in Wasser. Das galt sowohl für Hydrochlorid-Salze als auch für Nicht-
Hydrochloride. Lediglich durch die Zugabe von Ammoniak oder NaOH/KOH konnte das
Tetrazol in das Tetrazolat-Salz überführt und damit wasserlöslich gemacht werden. Da die
4-Pyridincarbonsäuren CIX-CXI aber folglich nicht mehr als Hydrochloride gefällt werden
konnten, erschwerte sich damit zum einen auch die Isolierung dieser Stoffe. Zum anderen
konnten durch massenspektrometrische Untersuchungen Hinweise darauf gefunden
werden, dass Amidbildungen mit der freien Carbonsäurefunktion bei der Synthese von CIX
auftraten (Abb. 51, CXII). Prinzipiell sollte die Formung von Amiden aus der verhältnismäßig
unreaktiven Carbonsäure ohne weitere Zugabe von Katalysatoren erschwert sein. Aufgrund
der harschen Reaktionsbedingungen erschien der massenspektrometrische Nachweis von
Amiden aber plausibel. Dieses Nebenprodukt musste in einem weiteren Schritt hydrolysiert
werden und wurden daher mit Salzsäure im Überschuss unter Rückfluss für mehrere
Stunden erhitzt. Durch anschließendes Einengen des Reaktionsansatzes auf einen
Bruchteil des Ausgangsvolumens und durch Lagerung im Kühlschrank konnte letztendlich
eine Ausfällung des Produkts aus der noch stark sauren Lösung provoziert werden. Die
weitere Derivatisierung des Diamin-Derivates CIX wurde dann mit dem schon
beschriebenen Zweiphasen-System (Abschnitt 1.2.2.1) erfolgreich durchgeführt.
Tab. 9: Gegenüberstellung von (berechneten) physikochemischen Eigenschaften der Substanzen XLVII und CXI. 1 Osiris Property Explorer, 2 Molinspiration bioactivity score v2016.03, 3 ChemBioDraw v. 13.
Substanz XLVII CXI
Summenformel C9H12N6O C9H12N2O3 Molare Masse 220,23 196,20 Wasserlöslichkeit Sehr schwer löslich Leicht löslich Anzahl H-Donatoren 3 3 Anzahl H-Akzeptoren 7 5 clogP1 –0,14 –0,09 clogP2 –0,09 0,40 clogP3 0,01 0,06
Eine direkte Gegenüberstellung der 3-Aminopropan-1-ol-Derivate zeigte einige
Unterschiede in den physikochemischen Eigenschaften auf (Tab. 9). Neben der
Wasserlöslichkeit wurde nochmals deutlich, dass Tetrazole mehr Wasserstoffbrücken-
Akzeptoren besitzen. Die allgemeine Annahme, dass Tetrazolsysteme gegenüber ihren

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 69
Carbonsäureanaloga lipophiler sind, konnte zumindest mit den drei verwendeten
Kalkulationsprogrammen nicht gestützt werden. Dabei gilt es aber auch zu berücksichtigen,
dass die Algorithmen auf Inkrementrechnungen und Datenbanken beruhen und für
Tetrazole schlichtweg weniger Daten-Input vorhanden ist. Unbestritten blieb aber, dass die
synthetisierten Moleküle zu den polareren Vertretern ihrer Art gehörten.
Mit den zwei hier präsentierten 4-Pyridincarbonsäuren lagen weitere Moleküle vor, welche
in einem Direktvergleich zeigen können, ob die entsprechenden Tetrazolanaloga ebenso
für die Hemmung der KDM4-Enzymreihe geeignet sind. Dadurch sollten Struktur-
Aktivitätsbeziehungen generiert werden, welche in zukünftigen Arbeiten bei der Darstellung
von weiteren KDM4-Inhibitoren benutzt und weiterentwickelt werden können.
1.2.4 Biologische Testung/Analyse der Kristallstrukturen
Für die Evaluation der inhibitorischen Aktivitäten wurden die in dieser Arbeit vorgestellten
Substanzen an die Albert-Ludwigs-Universität Freiburg versendet. Im Arbeitskreis von Prof.
Dr. Manfred Jung konnte auf zwei verschiedene Testsysteme zurückgegriffen werden. Der
Formaldehyd-Dehydrogenase-Assay (FDH-Assay), welcher auf einer Reduktion von NAD+
zu NADH unter Oxidation des freiwerdenden Formaldehyds zu Ameisensäure beruht,
konnte für die Substanzen zumeist auf Grund von Eigenfluoreszenz nicht verwendet
werden. Stattdessen kam ein Antikörper-basierter Lanthanide-Chelate-Excite-Assay
(LANCE-Assay) zum Einsatz. Die Funktionsweise soll im Folgenden kurz erläutert werden
(Abb. 52).
Das Prinzip basiert auf dem Förster-Resonanzenergietransfer (FRET), bei dem ein Donor
nach entsprechender Anregung seine Energie strahlungslos über Dipol-Dipol-
Wechselwirkungen auf einen Akzeptorfarbstoff überträgt (vgl.257). Dieser wiederum kann
die ihm übertragene Energie in Form von Licht einer bestimmten Wellenlänge abgeben,
welche zur Messung genutzt werden kann. Der Donor ist in diesem Falle Europium. Das
Lanthanoid ist an einen Antikörper gebunden, welcher dimethyliertes Lysin in Position 9 am
humanen Histon H3 (H3K9me2) erkennt. Dieses liegt nur dann vor, wenn das ursprünglich
hinzugegebene Histonfragment (H3K9me3) vorher durch KDM4A im Assay demethyliert
werden konnte. Das Histonfragment besitzt ferner ein Biotin-Label, welches dazu dient, im
zweiten Schritt des Assays von Streptavidin gebunden zu werden. Dieses ist wiederum
Träger des Akzeptorfarbstoffs LANCE Ultra ULight. Sind sowohl Eu-Antikörper als auch
Streptavidin-ULight gebunden und in räumlicher Nähe zueinander (~10 nm), kann nach
entsprechender Anregung der FRET eintreten. Als Positivkontrolle wurde im Assay
2,4-PDCA verwendet, als Negativkontrolle diente DMSO. Zum Teil wurden lediglich

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
70 | Untersuchungen, Ergebnisse und Diskussion
Vortests durchgeführt, das heißt, dass hier nur prozentuale Inhibitionen bei gegebener
Konzentration an Substanz vorlagen.
Abb. 52: Funktionsweise des LANCE-Assays: i) Durch die Zugabe von KDM4A, 2OG, FeSO4 und Ascorbinsäure wird die enzymatische Reaktion gestartet, welche ein H3K9me3-Fragment in ein H3K9me2-Fragment unter Abgabe von Formaldehyd demethyliert; ii) der Europium-markierte Antikörper bindet spezifisch an das H3K9me2-Fragment, während Streptavidin an Biotin bindet; iii) durch Anregung von Europium mit Licht und der räumlichen Nähe zum Ultra-ULight-Farbstoff kann es zu einem FRET kommen, welcher eine Emission von Strahlung auslöst.
Das aromatische Tetrazolhydrazid IX mit einer zusätzlichen primären aromatischen
Aminofunktion zeigte keine wesentlichen inhibitorischen Eigenschaften gegenüber KDM4A
auf (Tab. 10). Dies ist auch deshalb verwunderlich, weil die bereits im Arbeitskreis
synthetisierten Aryltetrazolhydrazide nachweislich im katalytischen Zentrum von KDM4D
binden können113. Diese zeigten im LANCE-Assay jedoch ebenfalls kaum Inhibition. Die
Unterschiede der KDM4A mit der KDM4D sind jedoch nur außerhalb des katalytischen
Zentrums zu finden (Abschnitt 1.1.1.2) und sollten daher nicht Grund für die
gegensätzlichen Beobachtungen sein. Die zusätzliche Aminofunktion ist damit offenbar
nicht in der Lage, das Chelatisierungspotential des Carbonsäurehydrazids mit einem
weiteren freien Elektronenpaar zu unterstützen.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 71
Tab. 10: Biologische Testung der Aryltetrazolhydrazide, 1 Enzyminhibition bei 250 µmol/L.
Substanz Strukturformel Inhibition gegen KDM4A IX
IC50 = 206,0 ± 42,3 μM
XVI
86 %1
Dazu nicht kongruent war, dass das zweite Aryltetrazolhydrazid XVI basierend auf einem
Chinazolingerüst eine gute, wenn auch nicht komplette Hemmung von KDM4A bei einer
Konzentration von 250 µmol/L zeigte. Damit war diese Substanz deutlich stärker
inhibitorisch wirksam als die Variante mit freier Aminofunktion. Eventuell war die
Erweiterung des planaren Systems ursächlich für den Befund. Es wurde jedoch kein IC50-
Wert bestimmt, was zu Klärung der genauen Aktivität aber unbedingt nachgeholt werden
sollte.
Tab. 11: Ergebnisse der biologischen Testung der 4-(1H-Tetrazol-5-yl)pyridin-2-amine.
Substanz Seitenkette Enzyminhibition gegen KDM4A bei 250 µmol/L
XXXVI
22 %
XLV
51 %
XLVII
42 %
LVIII
37 %
LIX
54 %
LXI
25 %
Als nicht inhibitorisch wirksam zeigten sich viele Vertreter der 4-(1H-Tetrazol-5-yl)pyridin-
2-amine und -2-thioether. Die wenigen schwach aktiven Substanzen tragen aliphatische
Reste unterschiedlicher Kettenlänge (Tab. 11). Diese endeten sowohl mit freien Amino- als
auch Hydroxygruppen, konnten aber auch von amidischer Struktur sein. Am stärksten
inhibitorisch gegen KDM4A zeigte sich die Verbindung LIX mit einer terminalen
Methylsulfonamid-Gruppe. Diese verfügte über einen Wasserstoffbrückendonor und drei
H-Akzeptoren. Stoffe mit aromatischen Seitenketten konnten keine Inhibitionen aufzeigen,

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
72 | Untersuchungen, Ergebnisse und Diskussion
obwohl literaturbekannte Substanzen mit Arylseitenkette durchaus in der Lage waren, eine
starke Hemmung auf die enzymatische Aktivität von KDM4A auszuüben. Auch deshalb liegt
es nahe, dass der Einfluss einer direkten Verknüpfung von Heteroatom und Pyridinring nicht
günstig war. Ob der bioisostere Austausch ebenfalls im kausalen Zusammenhang mit den
mäßigen Ergebnissen stand, wird aus den folgenden Testreihen ersichtlich.
Die 4-(1H-Tetrazol-5-yl)picolinamide LXXVI-LXXXII zeichneten sich durch eine im
Vergleich zu den bisher beschriebenen Tetrazolderivaten bessere Löslichkeit in vielen
Medien aus, wahrscheinlich durch Absenkung der Basizität des Pyridinstickstoffatoms.
Auf die biologischen Daten hatte diese Änderung allerdings wenig Einfluss. Die getesteten
Picolinamide waren ebenfalls nur mäßige Inhibitoren der KDM4A (Tab. 12). Die Substanz
LXXVII mit Morpholinrest zeigte mit einem IC50-Wert von etwa 100 µM noch die beste
Inhibition. Im Vergleich zu den bereits bekannten, auf Pyridin-4-carbonsäure basierenden
Hemmstoffen, ist die inhibitorische Potenz als gering einzustufen.
Tab. 12: Biologische Testung der 4-(1H-Tetrazol-5-yl)picolinamide und -hydroxamsäure. 1 Es konnte keine vollständige Sigmoidfunktion erhalten werden, weshalb die Werte nur als Näherung zu betrachten sind (eventuell kam es bei höheren Konzentration zu Aggregation der Substanzen). 2 Enzyminhibition bei 250 µmol/L.
Substanz Seitenkette Inhibition gegen KDM4A LXXVI
IC50 = 161,3 ± 8,6 µM1
LXXVII
IC50 = 100,0 ± 28,8 µM1
LXXVIII
26 %2
LXXX
43 %2
LXXXI
keine Inhibition
LXXXII
Assay-Interferenz
Gänzlich überraschend neben den bisher eher mäßigen Ergebnissen war, dass bei der
Testung der Hydroxamsäure LXXXII kein auswertbares Ergebnis erhalten werden konnte.
Für diesen Stoff wurde angenommen, dass es durch die zusätzlichen chelatisierenden
Eigenschaften der Hydroxamsäurefunktion in jedem Falle zu einer Inhibition kommen

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 73
würde. Weshalb es zu einer Überlagerung im Assay kam, konnte nicht geklärt werden.
Stünden nur Daten aus dem LANCE-Assay bezüglich der 4-(1H-Tetrazol-5-yl)picolinamide
und 4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure zur Verfügung, müsste davon
ausgegangen werden, dass auch diese für weitere Forschungsarbeiten wenig Relevanz
besäßen. Dass dem aber nicht so war, lag vor allem an Ergebnissen, welche durch
isotherme Titrationskalorimetrie und Röntgenkristallstrukturanalyse gewonnen wurden. Die
Resultate werden weiter unten in diesem Kapitel aufgezeigt.
Eine große Erwartungshaltung bestand gegenüber den Strukturen der [4-(1H-Tetrazol-5-
yl)pyridin-2-yl]methanamine, weil die Ergebnisse der Substanz XXXVIII mit freier terminaler
Aminofunktion recht früh vorlagen (Tab. 13). Mit einem IC50-Wert von etwa 21 µM bot das
verhältnismäßig kleine Molekül eine gute Grundlage für weitere Derivatisierungs-
experimente. Aus diesem Grund wurden auch die drei Wege zur Darstellung der
Methanamine eingehend untersucht, da unbedingt ein effizienter Syntheseweg für diese
Testkandidaten gefunden werden sollte. Die Route der reduktiven Aminierung des
Aldehyds wurde allerdings erst als letztes eingeschlagen.
Tab. 13: Biologische Testung der [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamine. 1 Enzyminhibition bei 125 µmol/L, 2 Enzyminhibition bei 250 µmol/L.
Substanz Seitenkette Inhibition gegen KDM4A XXXVIII IC50 = 20,9 ± 3,6 µM XCII
(Tetrazol mit Bn-Gruppe)
57 %1
XCIII
Assay-Interferenz
XCVIII
51 %2
CII
19 %2
CIII
keine Inhibition
CIV
49 %2
CV
41 %2 HN
OH

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
74 | Untersuchungen, Ergebnisse und Diskussion
Methanamine mit aromatischen Seitenketten (XCVIII, CIV und CV) zeigten eine moderate
Inhibition, was verdeutlichte, dass bei den 4-(1H-Tetrazol-5-yl)pyridin-2-aminen nicht
unbedingt die Seitenkette ursächlich für die schwache Hemmung von KDM4A war. Die
Cyclopropylgruppe von CIII hob offenbar die hemmenden Einflüsse auf, eine
underivatisierte Aminogruppe war damit Bestandteil des potentesten Inhibitors. Eine
Überraschung zeigte sich bei der noch benzylgeschützten Substanz XCII: Trotz der nicht
vorhandenen Acidität des Tetrazolrings konnte eine Inhibition im Vortest beobachtet
werden. Dies widersprach den Erwartungen, denn in vorherigen Befunden im Arbeitskreis
hatte sich gezeigt, dass eine Methylierung des Tetrazols zur Inaktivität des Moleküls in
Hinblick auf hemmende Einflüsse gegenüber KDM4A führte85. Eine Ermittlung des
IC50-Wertes mit Mehrfachbestimmung wäre deshalb wünschenswert gewesen, um etwaige
zufällige Phänomene auszuschließen. Die entschützte Verbindung XCIII führte dann
wiederum zu einer Interferenz im Assay und damit zu einem nicht auswertbaren Ergebnis.
Es gelang also nicht, die Aktivität von [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin
(XXXVIII) durch Derivatisierung der Seitenkette zu steigern. Die bisher erhaltenen
Resultate ließen an der Eignung des bioisosteren Austausches der Carbonsäure durch ein
Tetrazol zweifeln. Aus diesem Grund wurden mehrere Paare von ansonsten gleichen
Molekülen synthetisiert (Tab. 14).
Bei dem Molekülpaar LIX und CX zeigte sich eindeutig, dass das Tetrazolderivat der
Carbonsäure unterlegen war. Hier betrug der Faktor etwa zwei. Noch drastischer fiel der
Vergleich von XLVII und CXI aus, wobei sich das Molekül mit Tetrazolsubstituent etwa
sechsfach weniger inhibitorisch wirksam zeigte. Erschwerend kam hinzu, dass die
Löslichkeiten der Tetrazolylpyridine einer vollständigen Hemmung der KDM4A im Wege
standen.
Der Vergleich der direkten Mimetika von 2,4-PDCA stützte die Theorie, dass 4-(1H-
Tetrazol-5-yl)pyridine deutlich weniger gut als Inhibitoren der KDM4A agierten als
vergleichbare Isonicotinsäuren. Das Monotetrazolderivat LXXV war etwa um den Faktor
200 weniger inhibitorisch wirksam als 2,4-PDCA, das Bistetrazol CVII wiederum zwei Mal
weniger aktiv als LXXV. Da die Inhibition mit 2,4-PDCA im gewählten Assay sehr
ausgeprägt ausfällt (aufgrund der niedrigen Enzymkonzentration im LANCE-Assay werden
im Vergleich zu vielen anderen Assays merklich niedrigere Werte erhalten), weichen die
Werte von literaturüblichen IC50-Werten von 2,4-PDCA (etwa 0,7 µM) deutlich ab. Ein
Vergleich mit Literaturwerten wäre daher weniger ungünstig ausgefallen, es zeigt sich aber
einmal mehr, dass der Vergleich von IC50-Werten schwierig ist und am besten Daten aus
ein und demselben Testsystem in Relation gesetzt werden sollten, um Fehlinterpretationen
zu vermeiden.

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 75
Tab. 14: Direkte Gegenüberstellung der Tetrazolylpyridine mit Isonicotinsäuren, 1 Auf Grund von Sättigung wegen geringer Löslichkeit konnte eine vollständige Hemmung des Enzyms nicht erreicht werden. Die Werte sind daher nur als Näherung anzusehen.
Substanz Seitenkette Inhibition gegen KDM4A LIX
IC50 = 206 ± 5 µM1
CX
IC50 = 90,7 ± 27,1 µM
XLVII
IC50 = 314 ± 124 µM1
CXI
IC50 = 57,5 ± 20,3 µM
2,4-PDCA
IC50 = 0,027 ± 0,012 µM
LXXV
IC50 = 6,44 ± 2,90 µM
CVII
IC50 = 13,6 ± 0,6 µM
Zusammenfassend zeigten die Ergebnisse des LANCE-Assays, dass die Kombination von
Tetrazol und Pyridin offenbar keine gleichwertige Alternative gegenüber der Verknüpfung
aus Carbonsäure und Pyridin war. Der direkte Austausch an Position 4 führte zu einer

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
76 | Untersuchungen, Ergebnisse und Diskussion
deutlichen Aktivitätsminderung gegenüber der Inhibition von KDM4A. Eindeutige Aussagen
zur Beschaffenheit der Seitenkette waren jedoch deutlich schwieriger zu treffen. Die beste
Inhibition konnte mit einer unsubstituierten Aminofunktion mit Methylenbrücke erzielt
werden. Dennoch zeigten auch einige Picolinamide und Pyridine, welche mit einem
Stickstoffatom direkt verknüpft waren und einen aliphatischen Rest trugen, moderate
Aktivitäten. Die biologischen Testungen lieferten also wertvolle Hinweise darauf, dass
einige der synthetisierten Substanzen das Potential besaßen, an das katalytische Zentrum
von KDM4A zu binden. Die aber zum Teil flachen Struktur-Aktivitätsbeziehungen sollten mit
Hilfe der Röntgenstrukturanalyse verifiziert und damit auch ein beträchtlicher Zugewinn an
Informationen über die Bindungsmodi generiert werden.
In der Folge konnten am Helmholtz-Zentrum Berlin für Materialien und Energie mit Hilfe der
Photonenquelle BESSY II in der Arbeitsgruppe Makromolekulare Kristallographie um
Dr. Manfred Weiss vier Kristallstrukturen von KDM4D (gekürzt auf Aminosäure 1-342) mit
den Isonicotinsäuren CX und CXI sowie mit den Tetrazolen LXXVII und LXXXII
(Picolinamid/Hydroxamsäure) gelöst werden. Zusätzlich wurden in Experimenten mit
isothermer Titrationskalorimetrie (ITC) für drei der vier Stoffe Bindungsaffinitäten bestimmt.
Als Diskussionsgrundlage für die Bindungsmechanismen soll im Folgenden die
Kristallstruktur von Krojer et al. (PDB: 4D6Q) dienen. Diese zeigt den pan-Inhibitor
2,4-PDCA im katalytischen Zentrum von KDM4D. Allgemein werden zur Komplexierung des
zentralen Nickel(II)-Kations (aus Stabilitätsgründen für das physiologische Eisen(II)-Kation
eingesetzt) die Aminosäuren Histidin 192 und 280 (bei KDM4A His 188/276) sowie
Glutaminsäure 194 (bei KDM4A Glu 190) benötigt. Das Pyridin-Stickstoffatom und ein
Sauerstoffatom der Carbonsäure an Position 2 der 2,4-PDCA chelatisieren das Kation, so
dass mit einem zusätzlichen Wassermolekül das Ni2+-Atom von insgesamt sechs Liganden
umschlossen wird. Eine zusätzliche Wechselwirkung der 2,4-PDCA erfolgt über die
Carbonsäure in Position 4 mit Tyrosin 136 und Lysin 210 (bei KDM4A Tyr 132 und Lys 206).
Eine weitere Interaktion kann ferner vom zweiten Sauerstoffatom der ortho-ständigen
Carbonsäure mit Lysin 245 (bei KDM4A Lys 241) erfolgen. Damit besetzt der Ligand
2,4-PDCA die Bindetasche mit zahlreichen Wechselwirkungen und formt dabei starke
Interaktionen wie Chelate und Wasserstoffbrücken aus. Tendenziell ist auch eine
Salzbrücke aus negativ geladenem Carboxylat und positiv geladener Aminogruppe des
Lysins 210 eine Möglichkeit (Gesamtübersicht in Abb. 53 und Tab. 15).

Untersuchungen, Ergebnisse und Diskussion | 77
Abb. 53: Kristallstrukturen der Liganden CX (o. l.), CXI (o. r.), LXXVII (u. l.) und LXXXII (u. r.) im katalytischen Zentrum von KDM4D (Erklärungen siehe Text und Tab. 15).

78 | Untersuchungen, Ergebnisse und Diskussion
Tab. 15: Übersicht der Bindungsmodi der Liganden 2,4-PDCA, CX, CXI, LXXVII und LXXXII in KDM4D. 1 über Wassermoleküle mit Ligand in Wechselwirkung.
Interaktion mit in Position 2,4-PDCA CX CXI LXXVII LXXXII
Ni2+ - Chelat über Pyridin-N-Atom und 2-Carboxygruppe
Koordination über Pyridin-N-Atom
Koordination über Pyridin-N-Atom
Chelat über Pyridin-N- und 2-Carbonyl-O-Atom
Chelat über Pyridin-N- und 2-Hydroxamsäure-N-Atom
Tyrosin 136 4-Carboxygruppe 4-Carboxygruppe 4-Carboxygruppe - 4-Tetrazol Lysin 210 4-Carboxygruppe 4-Carboxygruppe 4-Carboxygruppe 4-Tetrazol 4-Tetrazol Histidin 90 - Seitenkette-
Sulfonamid-O-Atom
- - -
Tyrosin 179 - - - Seitenkette-Morpholin-O-Atom
-
Phenylalanin 189 gegebenenfalls π-π-Stacking Alanin 190 - Seitenkette-
Sulfonamid-O-Atom
Seitenkette-OH - -
Glutaminsäure 194 - - - - 2-Hydroxamsäure-OH Lysin 245 2-Carboxygruppe - - - 2-Hydroxamsäure-
Carbonyl-O-Atom Sonstiges1 - Asparagin 202 Glutamin 88,
Asparaginsäure 139 und Lysin 245
Lysin 245 - Asparagin 202 und Asparagin 284

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Untersuchungen, Ergebnisse und Diskussion | 79
Bei den Isonicotinsäurederivaten CX und CXI ist das Stickstoffatom der Seitenkette direkt
an den Pyridinring gebunden. Dieser Abstand führte dazu, dass eine Komplexierung über
zwei Heteroatome nicht mehr stattfinden konnte, lediglich das Pyridin-Stickstoffatom
koordinierte an das zentrale Metallkation. Die Carbonsäure in Position 4 interagierte in
gleicher Weise wie es bei 2,4-PDCA vorzufinden ist (Wasserstoff- oder Salzbrücke mit
Tyrosin 136 und Lysin 210). Bei den Seitenketten kam es ebenfalls zur Ausbildung von
Wasserstoffbrücken, wobei beide Stoffe Alanin 190 adressierten. Bei Substanz CX diente
dafür ein Sauerstoffatom des Sulfonamids, bei CXI konnte die Brücke durch die terminale
Hydroxyfunktion ausgebildet werden. Ferner war Verbindung CX in der Lage, mit Hilfe des
anderen Sulfonamid-Sauerstoffatoms mit Histidin 90 in Wechselwirkung zu treten. Die
Fähigkeit zum π-π-Stacking mit Phenylalanin 189 blieb bei beiden Verbindungen, wie auch
bei 2,4-PDCA, wahrscheinlich gegeben. Insgesamt betrachtet entsprachen damit die
Bindungsmodi größtenteils den Erwartungen. Durch den Verlust der Chelatbildung wurde
allerdings ein großer Teil des Bindungspotentials unterbunden, weshalb es spätestens hier
eindeutig wurde, dass eine direkte Verknüpfung der Seitenkette in Position 2 über ein
Stickstoffatom mit dem Pyridinsystem keine optimale Kombination darstellte.
Das Picolinamid LXXVII hingegen war in der Lage, das Ni2+-Kation mit Hilfe des Pyridin-
Stickstoffatoms und des Carbonyl-Sauerstoffatoms zu komplexieren. Allerdings rückte mit
Tyrosin 136 ein wichtiger Interaktionspartner für das Tetrazol aus der Bindetasche heraus.
Somit stand nur Lysin 210 für eine Salz- oder Wasserstoffbrücke zur Verfügung. Es wurde
ferner ersichtlich, dass sich das Tetrazol nicht in die gleiche Ebene wie das Pyridin
orientierte. Die längere Seitenkette fand in der Bindetasche genügend Platz und wurde
terminal durch das Morpholin-Sauerstoffatom am Tyrosin 179 über eine Wasserstoffbrücke
fixiert (nicht gezeigt). Interessant war weiterhin, dass sich die Seitenkette in eine andere
Richtung ausrichtete als es bei jener von Substanz CXI der Fall war. Dies spricht für die
Größe der Bindetasche und damit auch für die mögliche Diversität der dort
akkomodierbaren Seitenketten. Durch die fehlende Wechselwirkung mit Tyrosin 136 wurde
aber klar, dass die Bindetasche durch LXXVII nicht optimal besetzt wurde. Worin die
Ursache des „Herausdrückens“ der Aminosäure aus der Bindetasche zu begründen war,
konnte nur gemutmaßt werden. Eventuell war es bei der Komplexierung durch Pyridin-
Stickstoffatom und Carbonyl-Sauerstoffatom zu einer Verschiebung weg von der optimalen
Position gekommen. Ein Indiz dafür könnte sein, dass die Carbonylfunktion ebenfalls nicht
mit dem Pyridinring in einer Ebene stand. Auch hier lag eine gewisse Torsion vor.
Die wohl interessantesten Befunde kamen durch die Hydroxamsäure LXXXII zu Tage (auch
in Abb. 54 mit Überlagerung von CXI). Nach der ungeklärten Assay-Interferenz war nicht
klar, ob diese Substanz das Potential besitzen würde, um überhaupt im katalytischen
Zentrum von KDM4A zu binden. Die röntgenkristallographischen Untersuchungen konnten
aber zeigen, dass sich LXXXII trotz des Tetrazols in vergleichbarer Art und Weise wie

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
80 | Untersuchungen, Ergebnisse und Diskussion
2,4-PDCA in die Bindetasche der KDM4D einfügen konnte. Die wichtige Chelatisierung des
Ni2+-Kations erfolgte durch die Pyridin- und Hydroxamsäure-Stickstoffatome. Besonders
relevant war dabei, dass die Hydroxamsäure nicht über ihren üblichen Mechanismus (beide
Sauerstoffatome) komplexierte, sondern untypischerweise dazu das Stickstoffatom
benutzte und wahrscheinlich als tautomere Imidsäure vorlag. Dies könnte auch in Hinblick
auf die Selektivität einen wichtigen Aspekt darstellen, da Hydroxamsäuren mitunter zu den
Pan-Assay Interference Compounds (PAINS), welche zu unspezifischen falsch-positiven
Ergebnissen in Screening-Tests führen können, gezählt werden. Erfreulicherweise war nun
sowohl das Lysin 210 als auch das Tyrosin 136 an einer Wasserstoff- oder Salzbrücke mit
dem Tetrazol beteiligt. Damit wurde auch zum ersten Mal der Beweis erbracht, dass die
Tetrazolylpyridinstruktur in Kontrast zu Isonicotinsäureverbindungen einen vergleichbaren
Bindungscharakter aufwies. Ferner konnten über beide Sauerstoffatome der
Hydroxamsäure noch Glutaminsäure 194 und Lysin 245 mit einer Wasserstoff- oder
Salzbrücke adressiert werden. Die erhoffte Bildung eines „π-π-Sandwich“-Komplexes mit
Phenylalanin 189 und Tyrosin 181, wie es bei der ursprünglichen Leitstruktur 2-(1H-
Tetrazol-5-yl)essigsäurehydrazid noch der Fall war, blieb jedoch aus.
Abb. 54: Superimposition der Liganden CXI und LXXXII: Der Tetrazolring nahm mehr Platz als die Carbonsäuregruppe in Anspruch und war minimal verschoben. Die Seitenkette der Hydroxamsäure orientierte sich nach vorn, während der Hydroxypropyl-Rest nach hinten in die Bindetasche gelenkt wurde.
Es war damit unstrittig, dass die vier hier vorgestellten Inhibitoren im katalytischen Zentrum
der KDM4D binden würden. Zusätzlich konnten KD-Werte erhalten werden, welche am Max-
Delbrück-Centrum für Molekulare Medizin in Berlin durch Dr. Piotr Małecki in ITC-
Experimenten erhoben wurden. Die Liganden CX und CXI führten jeweils zu mittelmäßigen

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
Zusammenfassung und Ausblick Kapitel 1 | 81
Werten von KD = 35 µM und KD = 37 µM. Auf Grund der nicht vorhandenen Chelatisierung
überraschte dieses Ergebnis allerdings nicht. Ein Wert für LXXVII wurde nicht ermittelt. Die
Hydroxamsäure LXXXII zeigte jedoch mit KD = 0,5 µM eine Bindungsaffinität im
submikromolaren Bereich. Damit bietet LXXXII zugleich eine Grundlage für mögliche
weitere Inhibitoren, welche zum Beispiel von einer längeren Seitenkette profitieren könnten.
Das Tetrazolessigsäurederivat XXVI ohne Hydrazidfunktion verschob bei den Soaking-
Experimenten mit KDM4D das Metallkation im katalytischen Zentrum. Das heißt, dass der
Ligand dort kurzeitig lokalisiert war, jedoch nicht mitkristallisiert werden konnte. Das spricht
dafür, dass das Hydrazid ein obligatorisches Element der Tetrazolhydrazide darstellte und
dass die übrigen Bindungsmechanismen nicht ausreichten, um ein „Festhalten“ im
katalytischen Zentrum zu garantieren. Das Indolderivat XXXI zerstörte bei den Soaking-
Experimenten die Kristalle von KDM4D, weshalb hier keine Röntgenuntersuchungen
stattfinden konnten.
1.3 Zusammenfassung und Ausblick Kapitel 1
Im Rahmen dieser Promotionsarbeit sollten Inhibitoren der KDM4-Enzymreihe mit
unterschiedlichen Kombinationen von funktionellen Gruppen synthetisiert werden.
Basierend auf einer im Arbeitskreis gefundenen fragmentartigen Leitstruktur aus der Klasse
der Tetrazolhydrazide wurden aromatische und aliphatische Motive ausgewählt und
erfolgreich synthetisiert. Neben den aliphatischen Essigsäurederivaten konnte ein komplett
neuer Weg basierend auf Anthranilsäure etabliert werden. Als zweiter Grundkörper,
welcher wiederum auf dem literaturbekannten Inhibitor Pyridin-2,4-dicarbonsäure basierte,
wurde Tetrazolylpyridin gewählt. Es sollte evaluiert werden, welchen Einfluss und welche
synthetischen Herausforderungen der bioisostere Austausch der Carboxygruppe durch
einen Tetrazolring besaß beziehungsweise mit sich brachte. Ferner ging es darum,
systematisch den Einfluss der Seitenkette auf mögliche Bindungsmechanismen zu
ergründen. Neben der direkten Anknüpfung von Heteroatomen an das Tetrazolylpyridin-
Grundgerüst konnten außerdem Syntheserouten etabliert werden, welche Bindungen über
Carbonylgruppen ermöglichten und auch zur Darstellung von methylenverbrückten Aminen
als Seitenkette genutzt werden konnten. Die zahlreichen Verbindungen sollten dann zum
einen an der Albert-Ludwigs-Universität Freiburg im Arbeitskreis von Prof. Dr. Manfred Jung
in einem biologischen Assay auf ihre inhibitorischen Eigenschaften gegenüber KDM4A
getestet und zum anderen am Helmholtz-Zentrum Berlin für Materialien und Energie in der
Arbeitsgruppe Makromolekulare Kristallographie um Dr. Manfred Weiss im Rahmen von
Kristallisationsexperimenten begutachtet werden.
Die Ergebnisse der biologischen Testungen fielen insgesamt durchschnittlich aus. Bei den
Tetrazolhydraziden konnte mit 3-Amino-2-methyl-6-(1H-tetrazol-5-yl)chinazolin-4(3H)-on

Synthese und Evaluation neuer möglicher Inhibitoren von Histon-Demethylasen
82 | Zusammenfassung und Ausblick Kapitel 1
(XVI) eine verhältnismäßig aktive Substanz gefunden werden, welche in zukünftigen
Arbeiten noch näher untersucht und gegebenenfalls weiter derivatisiert werden sollte. Für
die Tetrazolylpyridine war festzustellen, dass der bioisostere Austausch zu Änderungen von
physikochemischen Eigenschaften führte, gleichzeitig aber auch die inhibitorische Potenz
gegenüber KDM4A erniedrigte. Bei dem Design der Seitenkette waren Verknüpfungen des
Pyridinrings mit Carbonylgruppen und methylenverbrückten Aminen zu bevorzugen.
Durch die Ergebnisse der Röntgenkristallographie konnte eindeutig gezeigt werden, dass
die direkte Verknüpfung von Pyridin und Stickstoffatom mit einem Verlust der Fähigkeit zur
Chelatisierung einherging. Der Bindungsmodus der Hydroxamsäure LXXXII konnte jedoch
beweisen, dass Tetrazolylpyridine grundsätzlich auch in der Lage waren, wichtige
Interaktionen mit KDM4D auszubilden. Mit einer Dissoziationskonstanten im
submikromolaren Bereich lag sie ferner in vergleichbarer Größenordnung wie die
Leitstruktur Pyridin-2,4-dicarbonsäure.
Daher konnten im Rahmen dieser Arbeit nicht nur weitere Bindungsmodi aufgeklärt,
sondern ein aktives Molekül mit bioisosterem Tetrazolring synthetisiert werden. Auf dessen
Grundlage wäre es unter anderem möglich, die Seitenkette über einen Ester der
Hydroxamsäure zu verlängern. Weiterhin könnte die ausgeprägte Hydrophilie des
Tetrazolylpyridins mit dem Konzept des Prodrug-Designs gesenkt werden, unter anderem
durch die Verwendung eines Doppelester-Konzeptes258. Damit wäre nicht nur die
Wahrscheinlichkeit der Penetration von Zellmembranen erhöht, sondern auch einige
Syntheseschritte, die von der Amphoterie der Tetrazolylpyridine negativ beeinflusst waren,
einfacher durchführbar.
Die Kristallstrukturen konnten inzwischen in der Protein Data Bank hinterlegt werden (6F5T,
6F5R, 6F5S, 6F5Q).

Auswirkungen von epigenetischer (Fehl-)Regulation auf psychische Erkrankungen
Überleitung | 83
2 Auswirkungen von epigenetischer (Fehl-)Regulation auf psychische Erkrankungen
Die bisherigen Betrachtungen von epigenetischer Regulation waren vor allem auf
neoplastische Ereignisse fokussiert. Deshalb überrascht es auch nicht, dass die derzeit
zugelassenen Arzneistoffe (DNMT- und HDAC-Inhibitoren, LSD1-Inhibitoren in klinischer
Prüfung) tumorspezifische Indikationen besitzen. Da das Wissen um das Epigenom in den
letzten Jahren aber rasant zugenommen hat, wurden in jüngster Zeit zahlreiche
experimentelle Befunde erhalten, welche vermuten lassen, dass mit niedermolekularen
Modulatoren von epigenetische Regulationsmechanismen-beeinflussenden Enzymen eine
Vielzahl von Krankheiten behandelt werden könnte.
Ein Schwerpunkt zielt dabei auf die (indirekte) Verstrickung von Histon-Demethylasen und
depressiven Erkrankungen ab. In einer Veröffentlichungen von Ambrosio et al. wurde der
Zusammenhang zwischen LSD1 und Autophagozytose hergestellt259. Die Autoren
beabsichtigten damit zwar auf mögliche Konsequenzen bei der Regulation von
Neuroblastomen hinzuweisen, stellten jedoch einen Signalweg vor, welcher
Überschneidungen mit möglichen Therapieoptionen von depressiven Patienten nahe legt.
Ambrosio et al. fanden heraus, dass LSD1 an die Promoterregion von Sestrin2 (SESN2)
bindet und so dessen Expression unterdrückt. Wurde nun die Funktion von LSD1 mit
Tranylcypromin oder E12 (Abb. 9) unterdrückt, stieg die Expressionsrate von SESN2.
Dieser üblicherweise Stress-induzierte Vertreter der Sestrin-Familie wiederum hemmte
dann als wichtiger Regulator des „mammalian target of rapamycin complex 1“ (mTORC1)
Signalwegs dessen Aktivität. Es wird vermutet, dass die Wirkmechanismen neuartiger
Antidepressiva wie NV-5138 (Struktur noch nicht veröffentlicht) oder Ketamin (Arzneistoff
bisher als Analgetikum/Anästhetikum zugelassen) ebenfalls einen Sestrin/mTORC1-
Mechanismus beinhalten und somit, zumindest theoretisch, Parallelen mit der Regulation
von LSD1 auftreten könnten260,261.
Für die JmjC-enthaltende KDM7B konnte ebenfalls eine Verbindung mit dem mTOR-
Signalweg nachgewiesen werden. Mäuse mit ausgeschalteter KDM7B zeigten ein
gemindertes Lernverhalten und kognitive Defizite, welche sich unter Gabe des mTOR-
Inhibitors Sirolimus merklich besserten262. Walsh et al. führten gleichermaßen Experimente
mit KDM7B-defizienten Mäusen durch, fanden dabei jedoch keine Anhaltspunkte für eine
nachteilige Entwicklung. Dafür entdeckten sie, dass durch den Knockout eine erhöhte
Resilienz gegenüber Stress-induzierten Angst- und Depressions-ähnlichen
Verhaltensmustern erreicht werden konnte. Dadurch, dass sie ebenfalls einen
Zusammenhang zwischen Serotoninrezeptoren und KDM7B sahen, stellten sie für KDM7B
eine Rolle als mögliches neues Target für die weitere Erforschung von depressiven
Erkrankungen in Aussicht263.

Auswirkungen von epigenetischer (Fehl-)Regulation auf psychische Erkrankungen
84 | Überleitung
Auch Auswirkungen der Regulation der KDM4-Enzymreihe auf die Pathologie der
Depression wurden im Mausmodell bereits untersucht und sind im Hinblick auf die in dieser
Arbeit synthetisierten Inhibitoren von besonderem Interesse264. In ersten Versuchsreihen
führten Pathak et al. mit Nagern gängige fünf- oder zehntägige Angst- und Stresstests
durch, wie bespielweise einen Aufenthalt im Labyrinth oder eine erzwungene
Schwimmprüfung. Neben resilienten Mäusen konnten durch die Tests ebenfalls
depressionsähnliche Phänotypen (u. a. Anhedonie und sozialer Rückzug) beobachtet
werden. Diese zeigten im Nucleus accumbens, einer wichtigen Region des
Belohnungszentrums, unterschiedliche Expressionsprofile der mRNA von KDM4A-D auf.
Anschließend sollte die Herunterregulation mancher KDMs durch die Stresstests nur mit
Hilfe der Applikation von den KDM-Inhibitoren Dimethyloxalylglycin (DMOG, Diester von
N-Oxalylglycin um Zellgängigkeit zu erreichen), ML-324 (ein Derivat von E18 mit
8-Hydroxychinolingrundkörper) oder JIB-04 (E17) (Abb. 10) untersucht werden. Unter der
Gabe von DMOG und ML-324 kam es wie in den Stresstests zur Ausbildung von
Depressions-ähnlichen Phänotypen. Die Anwendung von JIB-04 führte jedoch nicht zu
Merkmalen depressiver Erkrankungen, was die Autoren damit begründeten, dass JIB-04
als unselektiver pan-Inhibitor (u. a. gegen KDM5A/6B) galt. Auch wenn die letzte
Begründung durchaus fraglich erscheint (NOG wird ebenfalls als pan-Inhibitor eingeordnet),
so konnte die Arbeitsgruppe um Pathak zeigen, dass die Ausbildung von Depressions-
ähnlichen Phänotypen mit der (Herunter-)Regulation der Aktivitäten von KDM4-Enzymen
zusammenhing und sich durch die Gabe von deren Inhibitoren ebenfalls initiieren ließ.
Trotzdem diese Befunde nur experimenteller Natur waren und im Nagermodell durchgeführt
wurden, so hätten KDM4-Inhibitoren jedoch, wenn sie zur Behandlung von
Tumorerkrankungen eingesetzt würden, ein erhebliches Potential für Nebenwirkungen,
welches beachtet werden müsste.
Dass psychosozialer Stress sogar Auswirkungen auf Folgegenerationen haben könnte,
zeigten Serpeloni et al. mit ihren Forschungsergebnissen265. Sie belegten, dass
Misshandlungen während der Schwangerschaft zu Änderungen im DNA-
Methylierungsmuster führten. Diese Strukturen konnten dann nicht nur bei den Kindern,
sondern auch bei den Kindeskindern nachgewiesen werden. Dabei wurden Genregionen
methyliert, welche mit Symptomen von Depressionen und Posttraumatischen
Belastungsstörungen assoziiert waren. Die Autoren wiesen zwar klar aus, dass damit nicht
automatisch ein kausaler Zusammenhang zwischen der Entwicklung des Phänotyps der
Enkelkinder und dem großmütterlichen Stress bestand, konnten aber dennoch ihre
Vermutungen untermauern, dass die mit psychischen Erkrankungen assoziierte
Grundlagen (vererbbar) im Epigenom gespeichert wurden.

Auswirkungen von epigenetischer (Fehl-)Regulation auf psychische Erkrankungen
Überleitung | 85
Die genannten Literaturbeispiele stellen sehr wahrscheinlich nur den Anfang des
epigenomischen Verständnisses in Hinblick auf psychische Erkrankungen, speziell der
Depression, dar. Es konnte experimentell gezeigt werden, dass verschiedene Epigenetik-
assoziierte Enzyme an Krankheitsgeschehen und Ausbildung Depressions-ähnlicher
Phänotypen beteiligt waren. Die weitere Erforschung der Depression und deren Therapie
ist angesichts der Anzahl der Betroffenen und des hohen Leidensdrucks von eminenter
Bedeutung. Der zweite Teil dieser Arbeit soll einen Beitrag zur Analyse einer neuen
Therapieoption bei der Behandlung von depressiven Patienten leisten, in dem eine
leistungsfähige Nachweismethode für möglicherweise antidepressiv wirkende Metaboliten
eines bekannten Arzneistoffs zur Verfügung gestellt wird. Trennung, qualitative und
quantitative Erfassung von chiralen Metaboliten des Arzneistoffs Ketamin können zukünftig
zur Aufklärung der molekularen Targets beitragen, die an der ungewöhnlich schnell
eintretenden antidepressiven Wirkung im menschlichen Organismus beteiligt sind.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
86 | Vorbetrachtungen
3 Analytische Untersuchungen von Ketamin und dessen Metaboliten
3.1 Vorbetrachtungen
3.1.1 Depressive Erkrankungen und leitliniengerechte Therapie
Bei der Depression handelt es sich um eine psychische Störung, welche durch einen
Zustand deutlich gedrückter Stimmung, Interessenlosigkeit und Antriebsminderung über
einen längeren Zeitpunkt gekennzeichnet ist266. Die World Health Organization beziffert die
Prävalenz der Depression weltweit auf 322 Millionen Menschen. Davon werden in
Deutschland 4,1 Millionen mit der Diagnose Depression konfrontiert, was etwa 5,2 % der
deutschen Gesamtpopulation ausmacht267. Erkrankte leiden häufig an verringerter
Leistungsfähigkeit268, einem Mangel an gesundheitsbezogener Lebensqualität269 und
gesteigerter Mortalität270. Der von der Erkrankung ausgehende Leidensdruck für die
Patienten ist hoch, da in zentraler Art und Weise das Wohlbefinden und Selbstwertgefühl
beeinträchtigt wird271. Neben den starken persönlichen Einschränkungen entstehen den
Volkwirtschaften durch depressionsassoziierte direkte und indirekte Kosten Schäden in
Milliardenhöhe272.
Die aktuelle S3-Leitline „Unipolare Depression“ der AWMF273 bietet umfassende
Informationen zur Einteilung verschiedener Depressionsarten, Diagnosekriterien und
Therapieoptionen und soll im Folgenden nur partiell besprochen werden. Neben
zahlreichen nicht-medikamentösen Behandlungsoptionen wie Psychotherapie,
somatischer Therapieverfahren und Aufklärungsangebote für Patienten und Angehörige,
stellt die Pharmakotherapie eine existenzielle Säule der Interventionen dar.
Wie häufig in der Entwicklung der Arzneimittel erfolgte auch die Entdeckung der ersten
antidepressiv wirkenden Substanzen nach dem Serendipitätsprinzip. So wurde bei der
Behandlung von Tuberkulosepatienten mit dem aus Isoniazid abgeleiteten Iproniazid die
Beobachtung gemacht, dass sich ein großer Teil des Patientenkollektives einer
gesteigerten Fröhlichkeit erfreute274. Die Bezeichnung des entdeckten APIs als „Psychic
Energizer“ deutet schon an, dass das Wissen um die Depression als komplexes
Krankheitsgeschehen im Jahr 1957 als eher begrenzt einzuschätzen ist. Trotzdem
resultierte daraus der erste zugelassene MAO-Hemmer. Diese Klasse von Arzneistoffen ist
jedoch heute nicht mehr Mittel der ersten Wahl, da sie zu viele Interaktionen mit anderen
Medikamenten verursacht (unter anderem Serotonin-Syndrom) und penibel auf eine
tyraminarme Zusammensetzung der Nahrung geachtet werden muss. An dieser Stelle soll
noch einmal Tranylcypromin hervorgehoben werden, da es eben auch einen potenten
Inhibitor der LSD1 darstellt. Dieser Bestandteil der epigenetischen Regulation wurde schon
im ersten Teil dieser Arbeit ausführlich besprochen. Es soll nur noch darauf hingewiesen

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 87
werden, dass sich bei erfolgreichem Abschluss der klinischen Pilotstudien zur Therapie der
akuten myeloischen Leukämie der Indikationsbereich des MAO-Hemmers schlagartig
ändern könnte66,67.
Relativ zeitgleich zur Entdeckung von MAO-Inhibitoren wurde an der Weiterentwicklung von
Antipsychotika mit Phenothiazin-Grundgerüst geforscht. Der erste Vertreter der Tri- und
tetrazyklischen Antidepressiva (TZA) war Imipramin und wurde ursprünglich nicht für die
Indikation Depression sondern Schizophrenie entwickelt. Während erster Studien an
Patienten konnte jedoch beobachtet werden, dass diese sich gegenseitig unterhielten, an
Spielen teilnahmen oder sogar teilweise wieder lachen konnten275. Nach offizieller
Zulassung waren Iproniazid und Imipramin damit die ersten beiden Vertreter von APIs,
welche erfolgreich für die Therapie von depressiven Erkrankungen eingesetzt wurden. Mit
der Aufklärung ihrer Mechanismen trugen sie ferner zur Erkenntnis bei, dass das
Krankheitsgeschehen an einen Mangel von Serotonin, Noradrenalin und Dopamin im
zentralen Nervensystem geknüpft sein muss (Monoamin-Hypothese)276. Auf dieser
Hypothese aufbauend, konnten mittlerweile mehrere Klassen von Antidepressiva entwickelt
werden.
Heute kommen neben den MAO-Hemmern und TZA weitere Gruppen von Antidepressiva
zum Einsatz: Selektive Serotonin-Rückaufnahme-Inhibitoren (SSRI), Selektive Serotonin-
/Noradrenalin-Rückaufnahme-Inhibitoren (SSNRI), α2-Rezeptor-Antagonisten, Selektive
Noradrenalin- und Dopamin-Rückaufnahme-Hemmer, Melatonin-Rezeptor-Agonisten
sowie Lithiumsalze, Trazodon und Johanniskraut(-extrakte) (Tab. 16).
Tab. 16: Übersicht über pharmakotherapeutische Möglichkeiten bei Depression (modifiziert nach Hiemke et al.277).
Wirkstoffgruppe Beispiele Tri- und tetrazyklische Antidepressiva Amitriptylin, Amitriptylinoxid, Clomipramin
Desipramin, Doxepin, Imipramin, Maprotilin, Nortriptylin, Trimipramin
Selektive Serotonin-Rückaufnahme-Inhibitoren
Citalopram, Escitalopram, Fluoxetin, Fluvoxamin, Paroxetin, Sertralin, Vortioxetin
Monoaminoxidase-Inhibitoren Moclobemid, Tranylcypromin Selektive Serotonin-/Noradrenalin-Rückaufnahme-Inhibitoren
Venlafaxin, Duloxetin
α2-Rezeptor-Antagonisten Mianserin, Mirtazapin Selektiver Noradrenalin- und Dopamin-Rückaufnahme-Hemmer
Bupropion
Melatonin-Rezeptor-Agonisten Agomelatin Sonstige Trazodon, Lithiumsalze, Phytopharmaka
(Hypericum perforatum)

Analytische Untersuchungen von Ketamin und dessen Metaboliten
88 | Vorbetrachtungen
Cipriani et al. ermittelten und verglichen in einer aktuelle Metaanalyse über 500 Studien mit
mehr als 100000 Patienten zur Therapieeffektivität und -verträglichkeit von Antidepressiva
zum Teil untereinander oder gegen Placebo, und kamen dabei zu einer eindeutigen
Aussage: Jedes Antidepressivum ist wirksamer als Placebo278! Gleichzeitig wurde auch
erwähnt, dass in Sachen Verträglichkeit nahezu jedes Verum dem Placebo unterlegen sei.
Gelistet wurden teils stark therapieeinschränkende unerwünschte Arzneimittelwirkungen
(UAW) wie anticholinerge Effekte, Gewichtszunahme, Sedierung, erhöhte
Blutungsneigung, Verlängerung der QTc-Zeit im EKG mit Risiko potentiell
lebensbedrohlicher Herzrhythmusstörungen, Bluthochdruckkrisen,
Leberfunktionsstörungen oder mögliche Induktion von Agranulozytose273.
Neben den zahlreichen UAWs spielt die „Lag-Time“, also die Zeit, welche benötigt wird
damit das Antidepressivum seine gewünschte Wirkung entfaltet, eine große Rolle.
Allgemein wird mindestens eine Woche, eher jedoch zwei bis vier Wochen, therapiert, bevor
mit den beabsichtigten Therapieeffekten zu rechnen ist279,280. Diese Zeitspanne kann vor
allem für Patienten mit ausgeprägten Suizidgedanken bereits zu lang sein.
Einen letzten herausgenommenen Punkt stellen die therapieresistenten Depressionen dar.
Nach vier- bis sechswöchiger Therapiedauer nach Neueinstellung eines Patienten, soll die
Wirksamkeit systematisch überprüft werden281. Bei Nichteintreten der gewünschten Effekte
stehen zum Teil evidente Behandlungsoptionen zur Verfügung: Durch ein therapeutisches
Drug Monitoring (TDM) können Non-Adhärenz oder Resorptionsstörungen bei Patienten
aufgedeckt, der Wechsel zu einem anderen Antidepressivum bzw. eine Dosiseskalation
kann erwogen werden. Die Augmentation mit Lithium bzw. Antipsychotika der 2. Generation
nimmt neben der Kombination zweier Antidepressiva einen hohen Stellenwert ein281.
Dennoch gelingt es mit den vorhandenen Medikationsschemata nicht immer eine
Chronifizierung der Erkrankung zu verhindern.
Damit gilt zwar zusammenfassend, dass die bisher in der Therapie eingesetzten
Antidepressiva wirksam sind, wenngleich auch nicht immer optimal. Daher ist es sinnvoll
weitere Substanzen in klinischen Studien zu testen, die auf den ersten Blick eventuell nicht
direkt in das nach bisherigem Kenntnisstand gelehrte Krankheitsgeschehen involviert sind,
wodurch sich neue molekulare Mechanismen eröffnen können. Das Beispiel des NMDA-
Rezeptorantagonisten Ketamin zeigt auf, dass es sich dabei nicht immer um neue APIs
handeln muss, sondern dass das Prinzip des „Repurposing“ ein weiterer Weg sein kann,
Arzneistoffe für eine neue Indikation zu entdecken.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 89
3.1.2 Ketamin als neue Therapiestrategie zur Behandlung (therapieresistenter) depressiver Erkrankungen
Ketamin (rac-2-(2-Chlorphenyl)-2-(methylamino)cyclohexan-1-on, KET) (Abb. 58) wurde in
den 1960er Jahren das erste Mal beschrieben282 und ging chemisch aus dem Anästhetikum
Phenylcyclohexylpiperidin (Phenylcyclidin, PCP) (Abb. 57) hervor. Dieses zeigte ein
ungünstiges Risiko-Nutzen-Verhältnis, weshalb eine nebenwirkungsärmere und weniger
missbrauchsanfälligere Variante gesucht wurde283. Die WHO führt KET als Anästhetikum in
der Liste der unentbehrlichen Arzneimittel auf284. Des Weiteren ist es für verschiedene Arten
der Analgesie zugelassen285.
Abb. 55: Aufbau des heterotetrameren NMDA-Rezeptor-Komplexes286: GluN1- (blau) und GluN2-Untereinheit (orange) unterteilt in aminoterminale Domäne (ATD), Ligandenbindungsdomäne (LBD) und transmembranäre Domäne (TDM).
Seine therapeutische Wirkung entfaltet KET hauptsächlich über einen Antagonismus an
Glutamat-Rezeptoren. Diese können in G-Protein-gekoppelte und ionotrope Rezeptoren
unterteilt werden. Letztere stellen die wichtigere Untergruppe für den molekularen
Mechanismus von KET dar. Hier kann weiter in N-Methyl-D-aspartat- (NMDA, Abb. 55),
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionsäure- (AMPA) und Kainat-Rezeptoren
unterteilt werden. Die Namen rühren daher, dass die entsprechende Substanz unter
experimentellen Bedingungen als Agonist am jeweiligen Rezeptor wirkt. Glutamat als
wichtigster exzitatorischer Neurotransmitter ist unter anderem essentiell für Lern- und
Gedächtnisvorgänge287.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
90 | Vorbetrachtungen
Abb. 56: Schematische Darstellung der Funktionsweise des NMDA-Rezeptors: Im offenem Zustand (u. a. durch Bindung von L-Glutamat und Glycin in den jeweiligen Bindetaschen der LBD) wird die Rezeptorpore von einem Mg(II)-Kation blockiert, durch Depolarisation der Zelle wird das blockierende Mg(II)-Kation kurzzeitig ausgetauscht und ermöglicht den Wechsel von Na+ und Ca2+ in und K+ aus der Zelle, „Porenblocker“ (nicht-kompetitive Antagonisten) wie Ketamin und MK-801 binden an der PCP-Bindestelle und beeinflussen somit das Öffnungsverhalten und die Signalweiterleitung (Polyamin- und Zn2+-Bindetasche nicht gezeigt).
KET blockiert den geöffneten NMDA-Rezeptor nicht-kompetitiv nahe der Position, an der
unter physiologischen Bedingungen ein Magnesium(II)-Kation die Ein- bzw. Auslasspore
besetzt (Abb. 56). Es verhindert damit den Austausch von Ca2+, Na+ und K+-Ionen und
beeinflusst auch die nachgeschaltete Bindung von Ca2+ an Calmodulin, welche in noch
weiteren Schritten unter anderem zu einer Aktivierung verschiedener Kinasen und zur
Freisetzung von Stickstoffmonoxid führt283. Es konnte gezeigt werden, dass das S-Isomer
von KET gegenüber dem R-Enantiomer eine drei bis vier Mal höhere Affinität zum NMDA-
Rezeptor besitzt288. Andere Wirkungen von KET wie z. B. an Opioid- und Sigmarezeptoren
oder dopaminerge Effekte werden ebenfalls beschrieben289.
Erste Untersuchungen aus dem Jahr 1990 zeigen eventuelle Zusammenhänge zwischen
antidepressiven Verhaltensmustern und NMDAR-Antagonismus290. Mäuse, mit denen ein
erzwungener Schwimmtest durchgeführt wurde, zeigten bei Gabe eines kompetitiven
(AP-7) und eines nicht-kompetitiven (MK-801, Abb. 57) NMDAR-Antagonisten
dosisabhängig eine Linderung der Symptome, die durch den Stresstest provoziert wurden.
Abb. 57: Strukturformeln von ausgewählten NMDAR-Antagonisten.
Die Hinweise, dass NMDA-Rezeptoren im Krankheitsgeschehen der Depression mitwirken,
verdichteten sich weiter in Untersuchungen, bei denen Imipramin über zwei Wochen
appliziert wurde291. Dabei kam es zu Modulationen des NMDAR-Komplexes, woraus die
Autoren schlossen, dass auch glutamaterge Signalwege von Antidepressiva betroffen sein

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 91
müssen. Im Jahr 2000 konnten die Folgerungen aus Grundlagenforschung und
Tierversuchen dann in einer kleinen klinischen Studie bewiesen werden292: Einem
Patientenkollektiv von neun depressiven Freiwilligen wurde eine subanästhetische Dosis
Ketamin i. v. verabreicht. Innerhalb von drei Tagen zeigten sich robuste Verbesserungen
der durch die Depression verursachten Symptome. Damit wurde der Weg für einen
regelrechten „Ketaminboom“ geebnet, welcher bis heute anhält und mittlerweile auch in der
Populärpresse angekommen ist293.
KET wird im ersten Schritt des Metabolismus (Abb. 58) überwiegend durch Cytochrom
P450 3A4 zu Norketamin (NK) demethyliert. NK besitzt noch etwa ein Fünftel der Potenz
von KET. Ausgehend von NK ist ferner eine Dehydrierung sowohl zu Dehydronorketamin
(DNK), als auch eine Hydroxylierung an den Positionen 4, 5 und 6, möglich. Jede
eingeführte OH-Funktion resultiert in einem zweiten Chiralitätszentrum, welches in der
Summe eine Bildung von zwölf verschiedenartigen Hydroxynorketaminen (HNK)
ermöglicht. Theoretisch ist ebenfalls die direkte Hydroxylierung von KET denkbar und
würde zu zwölf Hydroxyketaminen (HK) führen. Über die Alkoholfunktionen lässt sich
außerdem eine glykosidische Bindung mit Glucuronsäure ausbilden (vgl.294-296).
Mittlerweile existieren Übersichtsarbeiten und Reviews, welche bisherige
Probandenstudien zusammenfassen und analysieren297-299. Danach ist mit einem Eintritt
der antidepressiven Wirkung nach einmaliger intravenöser Gabe nach zwei bis vier Stunden
zu rechnen und hält bis zu sieben Tage an. Bei therapieresistenter Depression scheint sich
KET zu einer substanziellen Alternative zu entwickeln, was sich auch in der Anzahl und
Ausrichtung momentan laufender klinischen Studien widerspiegelt300-302. Völlig
nebenwirkungsfrei ist KET dabei jedoch nicht: Trotz guter Verträglichkeit wurden
psychotomimetische Effekte (u. a. Agitation, Paranoia und Psychosen, jedoch viel seltener
als unter anästhetischer Dosierung), Schläfrigkeit/Sedierung, erhöhter Blutdruck und
dissoziative Zustände während und bis zu einer Stunde nach der Infusion registriert.
Letzteres erklärt auch das erhöhte Missbrauchspotential des Wirkstoffes (u. a.
Halluzinationen und Out-of-Body-Phänomene). Eventuell steht die Inzidenz der
Nebenwirkungen im Zusammenhang mit der Art und Häufigkeit der Applikation. So musste
eine Studie mit mehrfacher intranasaler Gabe von KET auf Grund von schwerwiegenden
Nebenwirkungen abgebrochen werden303, bei den bisher häufiger durchgeführten Studien
mit einmaliger i. v. Dosierung war das Nebenwirkungsprofil deutlich günstiger.

92 | Vorbetrachtungen
Abb. 58: In vivo Metabolismus von Ketamin (mod. nach294,296,304,305): i) Demethylierung von Ketamin führt zu Norketamin, ii) durch anschließende Dehydrierung wird Dehydronorketamin erhalten, iii) die Hydroxylierung von Norketamin kann an insgesamt drei Positionen erfolgen und führt somit zu 12 Hydroxynorketamin-Metaboliten, iv) auch eine vorangestellte Hydroxylierung ist möglich, welche 12 Hydroxyketamin-Derivate zur Folge hat, v) eine anschließende Demethylierung führt wiederum zu den 12 Hydroxynorketaminen. Die neu eingeführte Alkoholfunktion kann für eine Kupplung mit Glucuronsäure genutzt werden. Racemisches Norketamin-d4 wurde als interner Standard benutzt. Nur die hervorgehobenen Metaboliten waren für die SFC-MS-Methode relevant, weshalb auch auf die explizite Darstellung aller möglichen Enantiomere bzw. Diastereomere verzichtet wurde.
O
NH
Cl
O
NH2
Cl
O
NH2
Cl
HO
OCl
D
D
D
DNH2
O
NH
Cl
O
NH2
Cl
HO
O
NH2
Cl
HO
O
NH2
Cl
HO
O
NH2
Cl
O
NH2
Cl
O
NH2
Cl
O
NH
Cl
O
NH
ClO
NH
Cl
HO
OHHO
O
NH2
Cl O
NH2
Cl
HOOH
R-Ketamin S-Ketamin
2R,4R-Hydroxyketamin2S,4S-Hydroxyketamin2R,4S-Hydroxyketamin2S,4R-Hydroxyketamin
2R,5R-Hydroxyketamin2S,5S-Hydroxyketamin2R,5S-Hydroxyketamin2S,5R-Hydroxyketamin
2R,6R-Hydroxyketamin2S,6S-Hydroxyketamin2R,6S-Hydroxyketamin2S,6R-Hydroxyketamin
R-Norketamin S-Norketamin
R-Dehydronorketamin
S-Dehydronorketamin
2R,6R-Hydroxynorketamin 2S,6S-Hydroxynorketamin
2R,6S-Hydroxynorketamin 2S,6R-Hydroxynorketamin
weitere Konjugation mit Glucuronsäure mgl.
rac-Norketamin-d4
2R,4R-Hydroxynorketamin2S,4S-Hydroxynorketamin2R,4S-Hydroxynorketamin2S,4R-Hydroxynorketamin
2R,5R-Hydroxynorketamin2S,5S-Hydroxynorketamin2R,5S-Hydroxynorketamin2S,5R-Hydroxynorketamin
weitere Konjugation mit Glucuronsäure mgl. weitere Konjugation mit Glucuronsäure mgl.
i
iiiii
iii
iv
v

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 93
In einer Konsenserklärung, welche aber explizit keine Leitlinie darstellen soll, werden
Hinweise für die potentielle Off-Label-Therapie und die damit zusammenhängende
Patientensicherheit genannt306. Die Dosierung ist hierbei bedeutend geringer, in der Regel
0,5 mg/kg, als bei der Allgemeinanästhesie (1,0-2,0 mg/kg285), jedoch vergleichbar mit den
in der Schmerztherapie üblichen Mengen. S-KET wird üblicherweise in der Anästhesie mit
0,5-1,0 mg/kg dosiert307, in der Therapie von depressiven Erkrankungen wurde bisher
größtenteils racemisches KET untersucht.
Ob die Wirkung von KET ausschließlich über einen Antagonismus von NMDA-Rezeptoren
ausgeübt wird, ist mittlerweile umstritten. Der in der Alzheimer-Therapie eingesetzte
moderate NMDAR-Antagonist Memantin (Abb. 57) konnte in kleineren Studien nicht die
gleichen positiven Effekte bei Depression aufzeigen wie KET308. Eine im Jahr 2016
veröffentlichte Studie wies im Nagermodell nach, dass 2R,6R-HNK (Abb. 58) ebenfalls
durch Stresstests verursachte depressionsähnliche Symptome lindern kann305. Interessant
ist dabei der nicht NMDA-abhängige Mechanismus, stattdessen konnte eine Beeinflussung
der glutamatergen Signalgebung über den AMPA-Rezeptor nachgewiesen werden. In der
Folge kam es zu wissenschaftlichen Diskussionen, unter anderem darüber, dass
Nagermodelle nicht ohne weiteres auf den Menschen übertragbar seien und dass R-KET
in Experimenten eine mitunter länger anhaltende antidepressive Wirkung gezeigt hätte309-
313. In einer weiteren klinischen Studie konnte außerdem dargelegt werden, dass intranasal
appliziertes S-KET ebenfalls gute Wirkungen bei therapieresistenter Depression aufzeigt314.
Da es in vivo nicht zur Racemisierung kommt, können dementsprechend bei der Applikation
von S-KET an Position 2 keine R-konfigurierten Metabolite entstehen315. Die Idee jedoch,
dass 2R,6R-HNK bei ähnlicher Effektivität nebenwirkungsärmer als die Muttersubstanz sein
könnte, veranlasste die erstbeschreibenden Autoren zur Patentierung der Substanz316.
Andere molekulare Ansätze untersuchen eine Beeinflussung von glutamatergen Neuronen
in der lateralen Habenula im Epithalamus317 oder auch antiinflammatorische und
immunmodulierende Eigenschaften von KET318. Es bleiben also noch vielfältige Fragen auf
molekularer Ebene offen. Die für die Patienten wichtigen Punkte der Dosierung,
Langzeitanwendung, -effektivität und -sicherheit, sowie die Frage, ob es sinnvoller ist KET
oder einen Metaboliten zu applizieren, scheinen einer offiziellen Zulassung für die Indikation
(therapieresistente) Depression bislang noch im Weg zu stehen. Aber gerade für die
Analytik der biologischen Matrices von Probandenproben unter anderem zur Dosisfindung
und Metabolitenaufklärung ist es von Wichtigkeit, dass Tools und Methoden zur Verfügung
gestellt werden, die den hohen Ansprüchen von Richtigkeit und Präzision genügen.
Aufgrund der mittlerweile über 50-jährigen Geschichte der Anwendung von KET als
Arzneistoff, sind demgemäß diverse analytische Methoden beschrieben. Neben
zahlreichen gaschromatographischen Methoden (u. a. 294,295,319,320), soll vor allem auf
flüssigchromatographische Verfahren eingegangen werden. Hierbei wird das

Analytische Untersuchungen von Ketamin und dessen Metaboliten
94 | Vorbetrachtungen
Hauptaugenmerk häufig auf die Trennung von KET und NK, welches als Hauptmetabolit
angesehen wurde, gelegt321,322, wobei auch enantioselektive Methoden für beide Analyten
zahlreich beschrieben wurden323-325. Dass es sich dabei nicht immer um Analysetechniken
für den bestimmungsgemäßen Gebrauch von KET handeln muss, wird unter anderem in
Methoden gezeigt, welche neben der Erfassung von KET auch andere basische
Substanzen mit Missbrauchspotential wie Amphetamine oder Opioide einschließen326.
Weitere Methoden beschreiben die Mitauftrennung des Metaboliten DHK327. Ein aktueller
Review gibt eine Übersicht über chromatographische Methoden seit dem Jahr 2000328. HK-
und HNK-Metabolite wurden mittlerweile ebenfalls untersucht304,329. Im Abschnitt 3.4
werden einzelne Publikationen im Kontext der hier vorgestellten SFC-MS-Methode genauer
verglichen und diskutiert.
Jeder Entwicklung eines chromatographischen Verfahrens liegt eine möglich exakte
Problemstellung zu Grunde. Wichtige Parameter sind neben der Anzahl und Art der
Analyten auch die Probenmatrix, zu erwartende Messbereiche und Anforderungen an
Analysengeschwindigkeit und Probenvorbereitung. Auch das Umweltbewusstsein rückt
immer mehr in den Vordergrund. Mit konkreten Bezug auf diese Arbeit lassen sich folgende
Prämissen zentralisieren: Eine parallele und enantioselektive Quantifizierung von rac-KET,
rac-DHK, rac-NK, rac-6-HNK und rac-NK-d4 innerhalb einer Methode soll ermöglicht
werden. Für die Probenmatrix wird Urin gewählt, der Messbereich muss so bemessen sein,
dass bei einer Infusion von 5 mg rac-KET alle Metaboliten quantifizierbar sind. Ferner soll
die Probenvorbereitung einen sinnvollen Beitrag zur Reduktion der Matrixbelastung leisten.
Die Methode sollte dabei schnell sein und einen unnötig hohen Verbrauch an Lösungsmittel
vermeiden. Mit Hilfe bisher wenig genutzter Tools, wie der Superkritischen
Fluidchromatographie, können neue Akzente in der Analytik gesetzt werden. Im Folgenden
wird daher eine kurze Einführung in die Möglichkeiten gegeben.
3.1.3 Allgemeines zur Superkritischen Fluidchromatographie und ihre Vorteile
Die Superkritische Fluidchromatographie (SFC) nutzt neben der GC und der HPLC eine
dritte Art der Interaktionsmöglichkeit von stationärer und mobiler Phase. Die Bezeichnung
leitet sich aus der Verwendung von überkritischen (im englischen Sprachgebrauch auch
superkritischen) Stoffen als mobile Phase ab. Zur Überführung von chemischen
Verbindungen aus ihrer Gas- oder Flüssigphase in den überkritischen Zustand, muss der
kritische Punkt überschritten werden. Gleichen sich die Dichten von Gas- und Flüssigphase
bis zu diesem Punkt noch an, kann der Aggregatzustand nach Überschreiten dieses Punkts
nicht mehr unterschieden werden. Der kritische Punkt ist stoffspezifisch und abhängig von
der Temperatur und dem Druck. In der SFC wird vor allem Kohlenstoffdioxid verwendet.
Bereits bei 31,0 °C und 73,8 bar kann hier der überkritische Zustand erreicht werden. Im

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 95
Gegensatz zu den in dieser Hinsicht ähnlichen Gasen Distickstoffmonoxid oder Ammoniak
sind weitere Vorteile vorhanden: Kohlenstoffdioxid ist nicht brennbar beziehungsweise
brandfördernd, nicht toxisch, relativ inert und kostengünstig. Des Weiteren steht auch die
Senkung des Verbrauchs von Methanol und Acetonitril als klassische organische
Bestandteile von mobilen Phasen in der Chromatographie im Fokus. Aus diesem Grund
wird die SFC der Green Chemistry zugeordnet.
Die Idee der Benutzung von überkritischen Stoffen als mobile Phase ist nicht neu. Bereits
im Jahr 1958 wurde die Nutzung überkritischer Fluide für die chromatographische
Separierung von Analyten vorgeschlagen330. Es dauerte anschließend vier Jahre bis zur
ersten konkreten Umsetzung dieser Idee331. Die erste kommerziell erhältliche SFC-Anlage
wurde in den frühen 1980er Jahren von der Firma Hewlett-Packard eingeführt332, während
die ersten Kopplungen von SFC-Geräten mit Massenspektrometern in den späten
1980er/frühen 1990er Jahren beschrieben wurde333,334. Doch trotz des großen Potentials
und zahlreicher Publikationen konnte sich die SFC gegenüber der HPLC und GC bisher
nicht durchsetzen. Seit einigen Jahren bieten nun die drei großen Hersteller von
Chromatographiesystemen Agilent, Waters und Shimadzu technische Lösungen für die
SFC an. Durch die verbesserte und weiterentwickelte Technik sollen Unterschiede in
Zuverlässigkeit und Robustheit von HPLC- und GC-Systemen im Gegensatz zu SFC-
Anlagen minimiert werden.
Abb. 59: Schematischer Aufbau eines SFC-MS-Systems (BPR: Back Pressure Regulator, PDA: Photo Diode Array Detektor).
Der Aufbau von SFC-Maschinen ähnelt im Wesentlichen dem der HPLC (Abb. 59). Ein
neues Bauteil ist die CO2-fördernde Pumpe, welche im Falle der in dieser Arbeit
verwendeten Shimadzu-Anlage das Kohlendioxid in flüssiger Form über ein Steigrohr aus
der CO2-Vorratsflasche entnimmt. Um sicherzustellen, dass das Kohlendioxid erst nach der
Analyse wieder in den gasförmigen Zustand übergeht, ist dem System ein Restriktor (Back
Pressure Regulator, BPR) nachgeschaltet, welcher standardmäßig einen Rückdruck von
150 bar (individuell anpassbar) erzeugt. Da sich das CO2 nach der Passage des BPR
entspannt und so gegebenenfalls nicht genügend Flüssigkeit für ein konstantes Spray für
die Elektrospray-Ionisation (ESI) zur Verfügung steht, ist noch eine weitere Pumpe

Analytische Untersuchungen von Ketamin und dessen Metaboliten
96 | Vorbetrachtungen
notwendig. Diese wird Makeup-Pumpe genannt, greift nicht in die eigentliche Analyse ein
und ist ausschließlich für die Aufrechterhaltung eines konstanten Sprühnebels in der ESI
zuständig.
Überkritische Fluide besitzen ähnliche Dichten wie Flüssigkeiten, jedoch auch das
Diffusionsvermögen, die Oberflächenspannung und Viskosität von Gasen. Somit vereinen
sie das Lösungsvermögen und die Elutionskraft einer Flüssigkeit mit dem hohen
Massentransfer und der guten Penetration in poröse stationäre Phasen von Gasen335.
Diese Eigenschaften ermöglichen hohe Flussraten, geringe Rückdrücke über die Säule und
kurze Analysezeiten bei nicht benachteiligter Auflösung und Peakform.
Dass überkritisches CO2 (sCO2) die gleichen Eigenschaften wie kurzkettige
Kohlenwasserstoffe innehat, stimmt nur bezogen auf die Polarität. Beide besitzen einen
streng hydrophob ausgeprägten Charakter. Hingegen können beispielsweise die freien
Elektronenpaare an den Sauerstoffatomen des Kohlendioxids genutzt werden, um
Wasserstoffbrückenbindungen auszubilden, was wiederum Auswirkung auf Selektivität und
Retention haben kann336. Um auch stärker polare Analyten eluieren zu können, kann dem
sCO2 ein Modifier zugesetzt werden, welcher über die Modifier-Pumpe der mobilen Phase
beigemengt wird. Üblicherweise werden dafür Meth-, Eth- oder Isopropanol verwendet.
Auch der Zusatz von Acetonitril oder sogar kleinen Anteilen von Wasser können eine Option
darstellen. Hier ergibt sich jedoch ein Punkt, welcher nicht nur zu Terminologiefragen führt:
Bei Zunahme des Modifiers ändert sich auch der kritische Punkt der mobilen Phase. So
kann es eintreten, dass beispielsweise in einem steilen Gradienten der überkritische
Zustand unterschritten wird. Hier wird dann, vor allem in der englischen Literatur, von einem
subkritischen Zustand gesprochen, welcher so jedoch nicht definiert ist, da unterhalb des
kritisches Punkts lediglich der gasförmige oder flüssige Zustand existiert337. Es gibt jedoch
Untersuchungen, bei denen die CO2-Phase bewusst nur in einem flüssigen Zustand
gehalten wurde338, mit dem Ergebnis, dass die Chromatogramme mit über- oder
subkritischen mobilen Phasen kaum differieren. In diesem Zusammenhang fiel auch der
Begriff der Convergence Chromatography. Es muss aber in jedem Falle gewährleistet sein,
dass, sofern mehrere Komponenten für das Fließmittel verwendet werden, keine
Phasentrennung eintritt. Diese würde nicht nur zu einem erhöhten Rauschen im
Detektorsignal führen, sondern kann auch die eigentliche Trennung nachhaltig negativ
beeinflussen339,340.
Ein Review und ein Editorial aus dem Jahr 2014 heben die besondere Stellung der SFC in
der Analyse von chiralen Molekülen hervor341,342. Es konnte gezeigt werden, dass vor allem
die Kombination von hohen Flussraten und Polysaccharidphasen einen gewissen
Synergismus besitzt. Allerdings sind durchaus auch Nachteile vorhanden: das allgemeine
Knowhow in der Methodenentwicklung ist verglichen mit dem in der HPLC marginal, die
Anschaffungskosten von SFC-Geräten sind nach wie vor ungleich höher als für etabliertere

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 97
Flüssigchromatographen und durch die komplexen Zusammenhänge von Druck,
Temperatur, Dichte und damit auch Polarität von mobilen Phasen gibt es viele Parameter,
welche die SFC sehr vielschichtig machen. So ist es wenig verwunderlich, dass selbst in
einem aktuellen Review aus dem Jahr 2018 die Hauptanwendung der SFC bei der Analyse
von klinischen Proben im chiralen Bereich gesehen wird343.
Zusammenfassend sollte gesagt werden, dass die SFC keinesfalls als Ersatz von HPLC
oder GC zu behandeln ist, sondern vielmehr ein ergänzendes Verfahren darstellt. Die Vor-
und Nachteile sind seit Jahrzehnten bekannt, die Umsetzung nach wie vor noch
zurückhaltend. Es soll somit auch ein Ziel dieser Arbeit sein, weitere Erfahrungen im
Umgang mit der SFC zu sammeln und diese dem Fachpublikum zugänglich zu machen.
3.1.4 Besonderheiten der Analyten bei der Detektion mit einem Single-Quadrupol-Massenspektrometer
Eine Besonderheit der Analyten findet sich in ihrer unterschiedlichen
Isotopenzusammensetzung, begründet vor allem in den zwei stabilen Chlor-Isotopen 35Cl
und 37Cl. Die relative Molekülmasse eines 37Cl-enthaltenden KET-Moleküls ähnelt dem
eines 35Cl-enthaltenden 6-HNK-Moleküls, wenn beide ansonsten aus den häufigen
Stabilisotopen 1H, 12C, 14N und 16O zusammengesetzt sind. Auch Überschneidungen
hinsichtlich eines NK- und eines DNK-Analyten mit entsprechender
Isotopenzusammensetzung sind vorhanden (Tab. 17). Ein solches Auftreten von isobaren
Überlappungen kann die Selektivität einer Methode erheblich beeinträchtigen. Das Single-
Quadrupol-Massenspektrometer (SQMS), welches für diese Arbeit genutzt wurde, kann
aufgrund unzureichender Massengenauigkeit nicht zwischen den genannten
Isotopenzusammensetzungen unterscheiden.
Dieses Phänomen kann jedoch instrumentell oder chromatographisch gelöst werden: durch
die Benutzung eines hochauflösenden Massenspektrometers, z. B. durch die Kombination
einer Quadrupol-Ionenfalle und eines Flugzeitmassenanalysators (IT-TOF), ist die
Auflösung der Molekülmassen so exakt, dass auch im ppm-Bereich differenziert werden
kann. Ein Triple-Quadrupol-Massenspektrometer (QqQ, Tandem-MS oder MS/MS) erreicht
diese Massengenauigkeit zwar nicht, durch das Vorhandensein einer Kollisionszelle, in
welcher Fragmentierungen der Moleküle möglich sind, weist das Tandem-MS aber eine
erhöhte Selektivität auf. Das Mutter-Ion wird dabei von dem ersten Quadrupol durch sein
Masse-Ladungsverhältnis (m/z) selektiert und anschließend in der Kollisionszelle
fragmentiert. Die entstanden Tochter-Ionen werden zur Quantifizierung (Quantifier-Ion) und
zur Bestätigung der Messung (Qualifier-Ion) genutzt. Diese Massenübergänge (Transition)
sind in der Regel spezifisch für jedes Molekül, weshalb das Tandem-MS eine sehr hohe
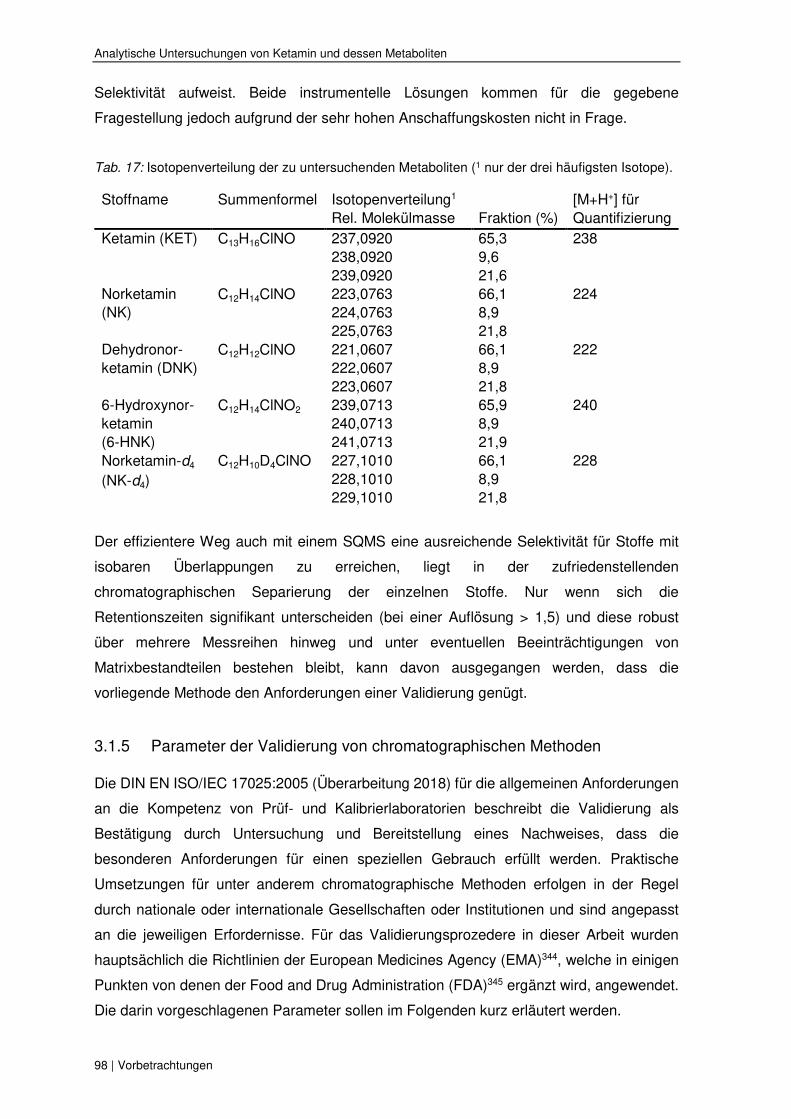
Analytische Untersuchungen von Ketamin und dessen Metaboliten
98 | Vorbetrachtungen
Selektivität aufweist. Beide instrumentelle Lösungen kommen für die gegebene
Fragestellung jedoch aufgrund der sehr hohen Anschaffungskosten nicht in Frage.
Tab. 17: Isotopenverteilung der zu untersuchenden Metaboliten (1 nur der drei häufigsten Isotope).
Stoffname
Summenformel Isotopenverteilung1 [M+H+] für Quantifizierung Rel. Molekülmasse Fraktion (%)
Ketamin (KET) C13H16ClNO 237,0920 238,0920 239,0920
65,3 9,6 21,6
238
Norketamin (NK)
C12H14ClNO 223,0763 224,0763 225,0763
66,1 8,9 21,8
224
Dehydronor-ketamin (DNK)
C12H12ClNO 221,0607 222,0607 223,0607
66,1 8,9 21,8
222
6-Hydroxynor-ketamin (6-HNK)
C12H14ClNO2 239,0713 240,0713 241,0713
65,9 8,9 21,9
240
Norketamin-d4
(NK-d4) C12H10D4ClNO 227,1010
228,1010 229,1010
66,1 8,9 21,8
228
Der effizientere Weg auch mit einem SQMS eine ausreichende Selektivität für Stoffe mit
isobaren Überlappungen zu erreichen, liegt in der zufriedenstellenden
chromatographischen Separierung der einzelnen Stoffe. Nur wenn sich die
Retentionszeiten signifikant unterscheiden (bei einer Auflösung > 1,5) und diese robust
über mehrere Messreihen hinweg und unter eventuellen Beeinträchtigungen von
Matrixbestandteilen bestehen bleibt, kann davon ausgegangen werden, dass die
vorliegende Methode den Anforderungen einer Validierung genügt.
3.1.5 Parameter der Validierung von chromatographischen Methoden
Die DIN EN ISO/IEC 17025:2005 (Überarbeitung 2018) für die allgemeinen Anforderungen
an die Kompetenz von Prüf- und Kalibrierlaboratorien beschreibt die Validierung als
Bestätigung durch Untersuchung und Bereitstellung eines Nachweises, dass die
besonderen Anforderungen für einen speziellen Gebrauch erfüllt werden. Praktische
Umsetzungen für unter anderem chromatographische Methoden erfolgen in der Regel
durch nationale oder internationale Gesellschaften oder Institutionen und sind angepasst
an die jeweiligen Erfordernisse. Für das Validierungsprozedere in dieser Arbeit wurden
hauptsächlich die Richtlinien der European Medicines Agency (EMA)344, welche in einigen
Punkten von denen der Food and Drug Administration (FDA)345 ergänzt wird, angewendet.
Die darin vorgeschlagenen Parameter sollen im Folgenden kurz erläutert werden.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Vorbetrachtungen | 99
3.1.5.1 Selektivität
Selektivität ist die Fähigkeit, alle Analyten von Interesse nebeneinander und ohne
gegenseitige Störungen zu bestimmen. Nicht nur die bereits herausgearbeitete Thematik
der isobaren Überlappungen bei der Detektion mit dem SQMS (Abschnitt 3.1.4), sondern
auch die Komplexität der Matrix erschweren den Nachweis dieses Parameters. Neben dem
Vermessen von Leerwerten zur Ermittlung von Systempeaks wird weiterhin die Erfassung
von Matrixsignalen aus sechs verschiedenen Leermatrices gefordert. Es geht dabei nicht
um die völlige Abwesenheit von Fremdsignalen, was bei einer komplexen Matrix wie Urin
ohnehin nicht möglich wäre, sondern vielmehr um das Verhältnis der Signale von Analyt zu
Matrix, welches eine bestimmte Schwelle, bezogen auf die Bestimmungsgrenze und den
internen Standard (IS), nicht überschreiten darf. Dem Goldstandard, nämlich die
Identifizierung der unbekannten Peaks anhand von zertifiziertem Referenzmaterial
vorzunehmen, konnte größtenteils entsprochen werden, da zum einen die Wege des
Metabolismus bekannt und zum anderen alle Standards (bis auf die 6-HNK-Enantiomere)
in zertifizierter Qualität käuflich zu erwerben waren.
3.1.5.2 Kalibrierfunktion
Die Kalibrierfunktion gibt den Zusammenhang von Messsignal zur Analytenkonzentration
über den Messbereich an. Für den Unterpunkt wurde bewusst auf die Bezeichnung
„Linearität“ verzichtet, da diese einen Zusammenhang auf Grundlage der mathematischen
Gleichung � = � + ∗ � impliziert. Es ist aber keineswegs vorgeschrieben, ein lineares
Modell für die Kalibierfunktion zu wählen. So spricht die EMA-Richtlinie von einem
„einfachen und adäquaten Zusammenhang des Messsignals und der Konzentration der
Analyten“. Es ist also nicht vorgeschrieben, welche mathematische Regression zu wählen
ist, solange die notwendige Reproduzierbarkeit erreicht wird. Es soll lediglich der
einfachste, aber auch der akkurateste Zusammenhang gewählt werden. Als
Beurteilungsparameter können beispielsweise die optische Betrachtung der
Geraden/Kurve, die Analyse der Korrelationskoeffizienten bzw. des Bestimmtheitsmaßes
oder auch mathematische Tests auf Linearität (z. B. F-Test nach Mandel) herangezogen
werden.
Die Proben der Kalibrierung sollen auf dem gleichen Weg hergestellt werden, wie auch die
Probandenproben. Neben sechs unterschiedlichen Kalibrierkonzentrationen, werden
ebenfalls eine Blank-Probe (double blank, verarbeitete Matrix ohne Analyten und ohne IS)
und eine Null-Probe (single blank, verarbeitete Matrix ohne Analyten aber mit IS)
vorgeschlagen. Der Arbeitsbereich soll den zu erwartenden Bereich abdecken und wird
nach unten von der Bestimmungsgrenze (LLOQ, Lower Limit Of Quantification) und nach
oben von dem Upper Limit Of Quantification (ULOQ) begrenzt.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
100 | Vorbetrachtungen
Die durch die mathematische Funktion berechneten Messpunkte sollen von den
Nominalmesswerten (die Werte, die als richtig angesehen werden) nicht mehr als ±15 %
abweichen, im Bereich des LLOQ sind Abweichungen auf ±20 % zu begrenzen. Die
Herstellung der Proben für die Kalibrierfunktion erfolgt vorzugsweise frisch an jedem
Messtag.
3.1.5.3 Unterstes Quantifizierungslevel
Die Bestimmungsgrenze oder auch LLOQ ist die Konzentration, bei welcher der Analyt mit
hinreichender Richtigkeit und Präzision noch zuverlässig quantifiziert werden kann. Sie
sollte den zu erwartenden Konzentrationen angepasst sein und bildet den niedrigsten
Kalibrierlevel ab. Praktisch sollte das LLOQ mindestens dem fünffachen Signal der Blank-
Probe entsprechen. Ältere Richtlinien sehen aber zum Teil auch das Signal/Rausch-
Verhältnis als maßgebend an346. Dieses sollte mindestens 10 betragen.
3.1.5.4 Richtigkeit
Die Richtigkeit ist einer der wichtigsten Parameter einer Validierung und ist ein Maß für die
Übereinstimmung von gemessenem Wert gegenüber dem Nominalwert. Sie ist somit
Ausdruck für den systematischen Fehler und wird üblicherweise in Prozent (als Abweichung
vom wahren Wert) angegeben. Ermittelt wird die Richtigkeit mit Hilfe von Qualitätskontrollen
(QC), welche in Analogie zu den Probanden- und Kalibrierproben, aber separat, hergestellt
werden. Es wird ferner unterschieden, ob die QCs innerhalb eines Analysenlaufs (within-
run) oder verschiedener Analysenläufe (between-run) gemessen werden. Für beide sollen
mindestens jeweils 5 Proben vermessen werden, welche den Bereich vom LLOQ, über ein
mittleres (medium QC, 30-50 %) bis in ein hohes Niveau (high QC, ab 75 %) der Kalibration
abdecken. Die erhaltenen Werte sollen auch hier nicht mehr als ±15 % von den
Nominalwerten, am LLOQ ±20 %, abweichen.
3.1.5.5 Präzision
Die Präzision ist als Maß für die Übereinstimmung unabhängiger Analysenergebnisse
untereinander anzusehen (Streuung der Analysenergebnisse). Mit ihr kann der zufällige
Fehler, üblicherweise als Standardabweichung oder relativer Standardabweichung
(Variationskoeffizient), ausgedrückt werden.
Praktisch kann die Präzision mit denselben Proben erfasst werden, mit denen auch die
Richtigkeit bestimmt wird. Wichtig ist hierbei, dass within-run und between-run QCs
verwendet werden, welche sich über den gesamten Kalibrierbereich erstrecken. Die
Grenzen von ±15 % bzw. ±20 % (LLOQ) vom Nominalwert sind einzuhalten.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 101
3.1.5.6 Wiederfindung, Matrix-Effekte und Stabilität
Die Wiederfindung eines Analyten einer Methode entspricht dem Verhältnis der
Messsignale einer Probe, welcher im Rahmen der Probenvorbereitung mit Matrix
verarbeitet wurde und der gleichen Probe, welche den Nominalwert repräsentiert (im
Normalfall eine Probe aus der Verdünnungsreihe ohne Matrix). Die Richtlinie sagt explizit,
dass die Wiederfindung nicht 100 % betragen muss, es geht vielmehr darum, dass Analyt
und IS reproduzierbar, einheitlich und präzise isoliert werden. Auch hier sollen drei
Konzentrationsbereiche (niedrig, mittel und hoch) abgedeckt werden. Der Vergleich soll mit
nicht extrahierten Proben vorgenommen werden, für welche eine 100%ige Wiederfindung
vermutet wird.
Vor allem bei massenspektrometrischen Methoden kann es durch Anwesenheit von Matrix
zur Erhöhung oder Erniedrigung der Ionenausbeute kommen347-350. Deshalb lässt die EMA-
Richtlinie auch auf Effekte der Matrix auf die Ionisierung von Analyten prüfen. Praktisch ist
dafür ein Abgleich der Signale von einem Analyten, welcher erst nach Aufarbeitung einer
Blank-Matrix zugesetzt wurde, und eines Analyten, welcher matrixfrei verarbeitet wurde,
möglich. Insgesamt sollen die Matrices von sechs individuellen Spendern stammen. Die
Konzentrationen orientieren sich wieder an niedrigen, mittleren und hohen Bereichen der
Kalibriergeraden. Die relative Standardabweichung zwischen den sechs interindividuellen
Matrices soll nicht mehr als 15 % betragen.
Detaillierte Untersuchungen zur Stabilität sind für die Qualität der Methode unabdingbar.
Es werden folgende Stabilitäten unterschieden: Einfrier-Auftau-Stabilität (Freeze-and-
Thaw-Stability), Bench-Top-Stabilität, Langzeitstabilität, Stammlösungsstabilität und
Stabilität der verarbeiteten Proben. Die zu lagernden Proben sollen mit einer frisch
zubereiteten Kalibrierfunktion vor und nach der Lagerung verglichen werden und dabei nicht
mehr als ±15 % des Nominalwertes abweichen.
3.2 Methodenentwicklung und Validierung
3.2.1 Entwicklung und Optimierung einer SFC-MS-Methode
Bei der Wahl der stationären Phase spielen mehrere Faktoren eine Rolle. So muss die
Säule SFC-kompatibel sein. Viele HPLC-Säulen sind auch SFC-beständig, zum Teil
unterscheiden sie sich aber in ihrem Dichtungsmaterial. Proteinphasen wie beispielsweise
immobilisiertes humanes Serum Albumin (HSA) oder α1-acid Glycoprotein (AGP) auf
porösem Silicagel sind laut Herstellerangaben nicht mit der SFC kombinierbar. Jedoch
bedienen sich bereits Methoden, die erfolgreich KET und einige KET-Metabolite getrennt
haben, letzterem Säulenmaterial304,329. Hingegen sind Polysaccharid-Phasen sehr gut
geeignet für überkritische Fließmittel. Besonders vorteilhaft scheinen Kieselgele zu sein,

Analytische Untersuchungen von Ketamin und dessen Metaboliten
102 | Methodenentwicklung und Validierung
welche mit einer Cellulose- oder Amyloseschicht überzogen wurden. So konnte bisher in
einer Säulenevaluation Ketamin zumindest im LC-Modus zufriedenstellend chiral getrennt
werden351. Gleichzeitig schlug jedoch die Trennung mit überkritischen mobilen Phasen fehl.
2R,6R-HNK und sein Enantiomer wurden erstmals auf einer Amylose-Phase separiert329,
allerdings nicht im SFC-Modus. Dennoch könnte die Lux Amylose-2 der Firma Phenomenex
mit Amylose-tris(5-chlor-2-methylphenylcarbamat) als Packungsmaterial eine geeignete
stationäre Phase darstellen (Abb. 60). Neben der helikalen Struktur der Amylose, werden
Möglichkeiten der Interaktion zwischen Analyt und stationärer Phase über
H-Brückenbindungen, Dipol-Dipol- sowie π-π-Wechselwirkungen ermöglicht. Auch
Halogen-Brücken wären über das chlorierte Phenylcarbamat denkbar.
Abb. 60: Amylose-tris(5-chlor-2-methylphenylcarbamat).
Um einen ersten Eindruck davon zu bekommen, ob die gewählte stationäre Phase geeignet
war, wurde die Trennung mit Standardparametern begonnen. So wurde zuerst auf das
Massenspektrometer als Detektor verzichtet, da die Optimierung von Ionisation und
Desolvatation für einen Gesamtüberblick an dieser frühen Stelle der Entwicklung zu
komplex wäre. Die Bedingungen sind in Tab. 18 angegeben und führten zu dem
Chromatogramm in Abb. 61. Die Analyten waren mit 50 µg/mL bewusst hoch konzentriert,
um in jedem Falle Signale zu erhalten.
Tab. 18: SFC-Parameter für den Scoutlauf mit der Lux Amylose-2.
Parameter Wert Flussrate (mL/min) 3 Mob. Phase (%) A (CO2) 90
B (Methanol + 0,1 % NH3) 10 Modus isokratisch Detektion (DAD) (nm) 220

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 103
Abb. 61: Erster Trennungsversuch von Ketamin-, Norketamin- und 6-Hydroxynorketamin-Enantiomeren mit 10 % MeOH (+0,1 % NH3) als Modifier.
Das Hauptaugenmerk bei der Bewertung des erhaltenen Chromatogramms lag auf der
Grundlinientrennung der beiden 6-HNK-Enantiomere. Unter den gegeben Bedingungen
wurde dieser Anforderung vollkommen entsprochen. Beide Stoffe wiesen eine einwandfreie
Peaksymmetrie ohne Tailing mit Basislinientrennung auf. Erfreulicherweise waren auch die
NK-Enantiomere unter diesen Parametern trennbar. Es zeigte sich aber eine
Überschneidung mit S-KET, was jedoch für die massenspektrometrische Analyse später
nicht relevant war, da zwischen KET und NK keine isobaren Überlappungen auftreten.
Weiterhin wurde klar, dass die Separierung der KET-Enantiomere die größte
Herausforderung darstellte, da hier keine Basislinientrennung erzielt werden konnte. Die
Analyten waren in diesen Scoutläufen nur teilweise gemischt und nicht mit Matrix belastet,
des Weiteren fehlte zu diesem Zeitpunkt noch DNK als weiterer Metabolit. Dennoch zeigten
diese ersten Versuche, dass die stationäre Phase in Kombination mit der SFC für die
Bearbeitung der Problemstellung geeignet zu sein schien.
Um die bisherige ungenügende Trennung der KET-Enantiomere zu optimieren, sollte der
Effekt verschiedener Modifier untersucht werden. Neben den Alkoholen Methanol und
Isopropanol (IPA), sollte auch der Einfluss von Acetonitril (ACN) evaluiert werden. Letzteres
verfügt über zwei π-Bindungen, welche mit den delokalisierten π-Elektronen des
aromatischen Systems des KETs in Wechselwirkung treten können. Abb. 62 zeigt die
Ergebnisse der isokratischen Trennung mit einer Flussrate von 3 mL/min und einem
Modifier-Anteil von 10 % unter Zusatz von 0,1 % NH3.
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0 5,5 6,0 6,5 7,0 7,5 8,0 8,5 9,0 9,5 min
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
uV
R-KET S-KET
R/S-NK 2S,6S-HNK 2R,6R-HNK

Analytische Untersuchungen von Ketamin und dessen Metaboliten
104 | Methodenentwicklung und Validierung
Abb. 62: Untersuchungen des Einflusses von verschiedenen Modifiern auf die Trennleistung des Systems in Bezug auf die Trennung von racemischem Ketamin.
Die Auflösung war zweifelsohne bei der Verwendung von Isopropanol als organischen
Modifier am besten. Sie überschritt auch den kritischen Wert von 1,5, welcher eine
Basislinientrennung indiziert. Acetonitril als Modifier führte zu einer geringfügig besseren
Trennung als bei Verwendung von Methanol, allerdings zu Lasten der Peakform.
Interessant war ebenfalls der Vergleich der Retentionszeiten: Mit Methanol als Modifier
eluierten die Peaks im Mittel schon nach 2,4 min, mit Isopropanol nach 3,4 min und mit
Acetonitril nach 3,5 min. Das würde auch der Reihenfolge der Elutionskraft an einer
Normalphase entsprechen. Wird sCO2 simplifiziert als Hexan angenommen, erscheint
diese Betrachtungsweise nicht allzu abwegig. Weiterhin wird diese Theorie von dem Fakt
unterstützt, dass die erhaltene Reihenfolge der Retentionszeiten (erst KET, dann NK und
deutlich später HNK) einem Abfall der clogP-Werte (kalkuliert mit ChemBioDraw,
Version 13) entsprach (Abb. 61). Ketamin (clogP=2,93) eluierte als erstes, während das
weniger lipophile demethylierte NK (clogP=2,41) eine in diesem Dreierfeld mittlere
Retentionszeit aufwies. Das deutlich polarere HNK (clogP=1,58) wurde folglich mit
größerem Abstand eluiert. Es scheint also durchaus Parallelen zwischen den Mechanismen
von Normalphasen-Chromatographie und SFC zu geben. Dies sollte aber keinesfalls
verallgemeinert werden, da sich die Befunde ausschließlich auf die Lux Amylose-2 als
stationäre Phase beziehen.
Neben dem Modifier selber spielt auch das Additiv, welches dem Modifier zugegeben wird,
eine beachtliche Rolle für die Selektivität und Auflösung. So wurde z. B. bei einer chiralen
Trennung von rac-Phenylalaninmethylester auf einer Chiralpak AD Säule mit 20 % Ethanol
0,0 1,0 2,0 3,0 4,0 5,0 min-0,25
0,00
0,25
0,50
0,75
1,00
1,25
1,50
1,75
2,00
2,25
2,50
2,75
3,00
3,25
3,50
3,75
4,00
4,25
4,50
4,75
5,00
5,25
5,50
5,75
(x1,000,000)
ACN: RS=0,99
MeOH: RS=0,87
IPA: RS=1,54
cps

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 105
als Modifier nur ein Peak erhalten. Unter Zusatz von 0,1 % Cyclohexylamin zum Modifier
erfolgte schon eine teilweise Trennung der Enantiomere und bei einer Additivkonzentration
von 1,0 % Cyclohexylamin wurde das Racemat basisliniengetrennt352. Somit könnte der
Effekt eines Additivs eventuell den des Modifiers noch übersteigen, ist aber in jedem Falle
eine Untersuchung wert. Aufgrund der direkten Kopplung mit dem Massenspektrometer
sollten allzu schwer verdampfbare Amine vermieden werden, weshalb sich die Auswahl des
Additivs auf wässrige Ammoniaklösung beschränkte.
Die Auswirkungen auf die Auflösung von den KET-Enantiomeren bei unterschiedlichen
Konzentration von NH3 im Modifier waren deutlich (Abb. 63). Als organische Komponente
wurde hier aufgrund der erfolgreichen Vortests Isopropanol gewählt. Die NH3-Beigaben
betrugen 0,02, 0,05 und 0,075 %, die Flussrate 3 mL/min bei 8 % organischem Modifier.
Bei der niedrigsten Konzentration an NH3-Zusatz erfolgte keine Trennung zwischen den
KET-Enantiomeren. Bei 0,05 % kam es immerhin zu einer teilweisen Trennung, welche bei
0,075 % noch einmal zunahm. Die erhaltene Basislinientrennung wurde offensichtlich nur
ab einer Additivkonzentration von über 0,1 % NH3 erreicht (Abb. 62). Damit können die
Befunde aus der eingangs erwähnten Literatur, dass das Additiv einen ausgeprägten
Einfluss auf das Trennergebnis hat, bestätigt werden.
Abb. 63: Verbesserung der Auflösung bei der Trennung von racemischen Ketamin durch unterschiedliche NH3-Konzentrationen.
Folglich wäre es sinnvoll gewesen, mit einer NH3-Konzentration von 0,1 % weiter zu
arbeiten. Allerdings wurde dies erschwert durch drastisch ansteigende Rückdrücke, die
nach etwa fünf bis zehn Injektionen auftraten. Ein anschließender Spülgang mit über 50 %
Methanol vermochte das Problem nur kurzzeitig zu lösen. Laut Hersteller soll die Säule mit
0,0 1,0 2,0 3,0 4,0 5,0 6,0 7,0 min
0,00
0,25
0,50
0,75
1,00
1,25
1,50
1,75
2,00
2,25
2,50
2,75
3,00
3,25
3,50
3,75
4,00
4,25
4,50
4,75
5,00
5,25
5,50
5,75
6,00
6,25
6,50
6,75
(x100,000)cps
RS=0 bei 0,02 % NH3
RS=0,47 bei 0,05 % NH3
RS=0,87 bei 0,075 % NH3

Analytische Untersuchungen von Ketamin und dessen Metaboliten
106 | Methodenentwicklung und Validierung
basischen Zusätzen bis zu einer Konzentration von 0,5 % im organischen Laufmittel
kompatibel sein353. Da es auch nach vielen Injektionen nicht zu einer Änderung des
Trennungsbildes kam, war die chemische Degradation des chiralen Rückgrats der
stationären Phase oder der Kieselgelbasis auszuschließen. Um dennoch den steigenden
Rückdrücken entgegenzuwirken, wurde ein Gradient implementiert und ein Spülmethode
alle 10-15 Injektionen mit 25 % Methanol ohne Zusätze mit anschließender 20-minütiger
Einspülphase zwischengeschaltet (Tab. 19). Ferner wurde die Startkonzentration des
Modifiers auf 8 % festgelegt, da damit die besten Peakformen aller Analyten erzielt wurden.
Tab. 19: Integration des finalen Gradienten für die SFC-Methode.
Zeit (min) A (CO2) (%) B (IPA+0,075 % NH3) (%)
0–7 92 8 7,01–7,25 80 20 7,26–11 80 20 11,01–11,25 92 8 11,26–15 92 8
Bisher lag das Augenmerk vor allem auf der Optimierung der chromatographischen
Eigenschaften der Methode. Da die zu erwartenden Zielkonzentrationen von KET
beziehungsweise seiner Metaboliten im Urin bekannt waren (niedrige zweistellige ng/mL),
mussten die Nachweis- bzw. Bestimmungsgrenzen der Methode auch entsprechend
angepasst werden. Massenspektrometer besitzen in der Regel höhere Sensitivitäten als
PDAs. Außerdem sind sie weniger anfällig gegenüber Drifts oder Rauschen, wenn CO2 ein
Bestandteil der mobilen Phase ist und die SFC im Gradientenmodus betrieben wird.
Ein Parameter bei der Optimierung der Empfindlichkeit des SQMS ist das Drying-Gas.
Dieses dient der Entfernung von Lösungsmittelmolekülen während des
Ionisierungsprozesses. Sind die Analyten nicht vollständig desolvatisiert, kann die
Ionisierung und somit auch die Empfindlichkeit der Methode ungünstig beeinflusst
werden354.
Im Folgenden wurden mehrere Injektionen mit verschiedenen Drying-Gas-Flüssen (0, 5 und
10 L/min) und (2S,6S)-HNK bei einer Konzentration von 100 ng/mL durchgeführt (Abb. 64).
Während bei Abwesenheit des Trocknungsgases nicht einmal die Nachweisgrenze erreicht
wurde, lag bei einen Drying-Gas-Fluss von 10 L/min das Signal/Rausch-Verhältnis (S/N)
bei über 60. Allein mit dieser Einstellung konnte die Empfindlichkeit der Methode um mehr
als den Faktor 20 gesteigert werden.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 107
Abb. 64: Auswirkungen des Drying-Gas-Flusses auf das Signal/Rausch-Verhältnis.
Des Weiteren wurde auch der Effekt der Heat-Block-Temperatur untersucht. Dieses Bauteil
umschließt die Ansaugöffnung des SQMS und dient ebenfalls der Desolvatation. Die
Variation der Temperatur von 200 auf 300 und 400 °C zeigte jedoch keine Unterschiede in
den S/N-Verhältnissen. Ein ähnlicher Befund zeigte sich nach Änderung der Interface
Voltage von 4,5 kV auf 4,8 kV, bei dem das S/N-Verhältnis ebenfalls nicht verbessert
werden konnte.
Da Ketamin bzw. dessen Metaboliten durch die Aminofunktion eine mäßig stark basische
Gruppe besitzen, ist es empfehlenswert durch Säurezugabe den Protonierungsprozess der
ESI zu unterstützen. Es wurde bereits gezeigt, dass die Zugabe von NH3 in den Modifier
unabdingbar für ein gutes Trennungsbild war, weshalb es nicht sinnvoll gewesen wäre, dem
Elutionsmittel auch noch Säure hinzuzufügen. Dafür bot sich jedoch die Makeup-Pumpe
an, da diese die Flüssigkeit erst nach Passage der Säule zusetzt. Aus diesem Grund wurde
Methanol mit 0,1 % Ameisensäure als Makeup-Flüssigkeit verwendet.
Ein letzter Schritt zum Erreichen der notwendigen Nachweisgrenze lag in der
Probenvorbereitung. Durch Aufkonzentrieren der Proben war es möglich die Methode
sensitiver zu gestalten. In der Praxis wurde eine Flüssig-Flüssig-Extraktion mit Methyl-tert-
butylether (MTBE) unter vorheriger Zugabe einer Natriumcarbonat-Lösung, welche die
Protonierung der Aminofunktion der verschiedenen Analyten zurückdrängen sollte,
gewählt. Bei einem Ausgangsvolumen von 1 mL Probe und einem Rekonstitutionsvolumen
von 100 µL Methanol für den eingeengten MTBE, konnte die Nachweisgrenze erneut um
den Faktor 10 gesenkt werden.
7,0 7,5 8,0 8,5 9,0 9,5 10,0 10,5 11,0 11,5 12,0 12,5 13,0 min
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
(x100,000)
S/N=2,6 bei 0 L/min
S/N=18,8 bei 5 L/min
S/N=61,1 bei 10 L/min
cps

Analytische Untersuchungen von Ketamin und dessen Metaboliten
108 | Methodenentwicklung und Validierung
3.2.2 Validierung der SFC-MS-Methode
3.2.2.1 Selektivität
Neben den im Optimierungsteil präsentierten Wechselwirkungen der Analyten
untereinander, kann auch die Matrix einen erheblichen Grund für Interferenzen und
fluktuierende Retentionszeiten darstellen355. Urin als Matrix beinhaltet hauptsächlich polare
Bestandteile wie anorganische Salze, Abbauprodukte aus dem Purinstoffwechsel,
Harnstoff und Kreatinin. Des Weiteren können unphysiologische Exkretionen bei
Krankheitsgeschehen stattfinden (z. B. Glucose, Proteine oder Bakterien). Bei Einnahme
von Xenobiotika können auch diese oder davon abstammende Metaboliten die Analyse
stören.
Der Schritt der Probenvorbereitung war bereits essentiell: durch die Flüssig-Flüssig-
Extraktion mit dem sehr lipophilen MTBE verteilten sich die Analyten in der organischen
Phase, während die polaren Matrixbestandteile in der wässrigen Phase verblieben. Damit
konnten Interaktionsmöglichkeiten von Analyt und Matrix bereits vor dem eigentlichen SFC-
Lauf minimiert werden. Der simplifizierte Ansatz der ausschließlichen Verdünnung von
Matrix als Probenvorbereitung (Dilute-and-Shoot) hat große Vorteile hinsichtlich
Zeitersparnis und Lösungsmittelverbrauch. Allerdings wird damit auch ein erhöhtes
Wechselwirkungspotential in Kauf genommen. So konnte für basische Analyten gezeigt
werden, dass es mitunter zu Verlängerung von Retentionszeiten kam356, wenn Urinproben
im Rahmen der Probenvorbereitung ausschließlich verdünnt werden.
Die Analyse der Null-Probe (verarbeitete Matrix versetzt mit IS) zeigte, dass sich die
Grundlinien der Massenspuren der Analyten weitestgehend im Bereich des üblichen
Rauschens befanden (Abb. 65). Lediglich für die KET-Spur (schwarz) konnte ein
Matrixsignal im frühen Bereich des Chromatogramms festgestellt werden. Die NK-d4-
Enantiomere wurden einwandfrei aufgetrennt und zeigten ebenfalls keine
Signalüberschneidungen mit der Matrix.
Eine Auftrennung aller Analyten war möglich (Abb. 66). NK-, DNK- und HNK-Enantiomere
wurden basisliniengetrennt. Sehr wahrscheinlich führten die Beeinflussung der Substanzen
untereinander und die Anwesenheit von Matrix dazu, dass, trotz vorheriger Optimierungen
zur Trennung der KET-Enantiomere, diese nicht mehr basisliniengetrennt wurden. Solange
dieser Befund aber robust ist, sollte eine Validierung trotzdem möglich sein, da durch die
Integrationstools softwareseitig eine Reihe an Möglichkeiten zur Verfügung gestellt werden,
um auch nicht grundliniengetrennte Peaks reproduzierbar zu integrieren.
Die bereits in Absatz 3.1.4 vorgestellte Thematik der isobaren Überlappung war ebenfalls
gut ersichtlich (Abb. 66). So erschienen in der HNK-Spur (blau) KET-Signale, sowie DNK-
Peaks in der NK-Massenspur (Magenta). Da die entsprechenden Stoffe aber bestens

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 109
voneinander abgetrennt wurden, war davon auszugehen, dass die Methode die nötige
Selektivität für die gegebene Aufgabenstellung besaß.
Abb. 65: Repräsentative Null-Probe mit racemischem Norketamin-d4 als internen Standard.
Abb. 66: Repräsentative Probe (Matrix mit Analyt versetzt) aus einem Validierungslauf bei einer Konzentration von 5 ng/mL je Analyt.
Unterstützt wurden diese Daten von den Standardabweichungen und
Variationskoeffizienten der Retentionszeiten aus den Validierungsläufen (Tab. 20). Die
relativen Standardabweichungen lagen zwischen 0,9 und 1,8 %. Obwohl die in dieser Arbeit
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 min0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
65000
70000
75000
80000
85000
90000
95000
100000
105000
110000
115000
120000 R/S-NK-d4
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 min
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
65000
70000
75000
80000
85000
90000
95000
100000
105000
110000
115000
R-KET S-KET
R/S-NK
2S,6S-HNK 2R,6R-HNK
R/S-DNK

Analytische Untersuchungen von Ketamin und dessen Metaboliten
110 | Methodenentwicklung und Validierung
eingangs zitierten Richtlinien dahingehend keine spezifizierten Grenzen vorschreiben, sind
Werte unter 2 % als akzeptabel anzusehen.
Da zum Zeitpunkt der Methodenerstellung bzw. -validierung nur KET und HNK als
enantiomerenreine Verbindungen zur Verfügung standen, konnte auch nur für diese eine
einwandfreie Zuordnung der Stereodeskriptoren stattfinden. DHK und NK waren
ausschließlich als Racemat käuflich zu erwerben, weshalb im Folgenden zum Teil die
stereochemischen Deskriptoren gegen die anonymisierende Bezeichnungen En1 und En2
ausgetauscht werden. In Absatz 3.3 wird ergänzend erläutert, wie zumindest für DHK
anhand von stereoselektiver Verstoffwechslung eine Zuordnung der Enantiomere erfolgen
könnte.
Tab. 20: Mittelwerte und relative Standardabweichungen der Retentionszeiten aus den Kalibrierläufen (5 Läufe mit jeweils 6 Werten, � = 30).
Substanz MW Retentionszeit ± SD (min) RSD (%)
R-Ketamin 4,514±0,061 1,36 S-Ketamin 4,691±0,053 1,14 Norketamin En1 4,844±0,056 1,16 Norketamin En2 6,088±0,092 1,51 Dehydronorketamin En1 6,964±0,131 1,87 Dehydronorketamin En2 7,389±0,137 1,85 2S,6S-Hydroxynorketamin 8,355±0,092 1,09 2R,6R-Hydroxynorketamin 9,047±0,087 0,96 Norketamin-d4 En1 4,798±0,057 1,19 Norketamin-d4 En2 6,025±0,088 1,46
3.2.2.2 Kalibrierfunktion
Aufgrund der besonderen Anfälligkeit von Massendetektoren gegenüber komplexen
Matrices, gilt die Verwendung von internen Standards bei der Quantifizierung von Analyten
als notwendig. In dieser Arbeit wurde vierfach deuteriertes Norketamin (Austausch aller
vorhandenen Aromatenprotonen) als IS verwendet. Dieser Massenunterschied von 4 u ist
positiv zu bewerten, da bereits gezeigt werden konnte, dass bei einem Massenunterschied
von nur 2 u Interferenzen auftreten können357. Ferner wurde im Absatz 3.1.4
herausgearbeitet, dass keine weiteren isobaren Überschneidungen mit anderen Analyten
auftreten. Strenggenommen handelte es sich beim IS um zwei Substanzen, da auch dieser
nur als Racemat erhältlich war. Die Trennung beider Enantiomere gelang aber
reproduzierbar, weshalb entschieden wurde, das früher eluierende Enantiomer des IS auf
das zuerst eluierende Enantiomer eines chiralen Analytenpaares zu beziehen. Die zweite
eluierende Substanz des IS wurden dann auch auf den zweiten Stoff des jeweiligen
Enantiomerenpaares bezogen (vgl. Tab. 20, z. B. R-KET/NK-d4 En1 und S-KET/NK-d4
En2).

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 111
Abb. 67: Vergleich der quadratischen (links) gegenüber der linearen (rechts) Kalibrierfunktion für 2S,6S-Hydroxynorketamin, 2R,6R-Hydroxynorketamin und S-Ketamin.
Zur Ermittlung des mathematischen Zusammenhangs zwischen Analytenkonzentration und
Messsignal wurde das Verhältnis aus den Peakflächen des Analyten zum
korrespondierenden IS gegen den Quotienten aus Konzentration des Analyten und
Konzentration des IS aufgetragen und im ersten Schritt subjektiv beurteilt. Anhand der drei
repräsentativen Beispiele 2S,6S-Hydroxynorketamin, 2R,6R-Hydroxynorketamin und
S-Ketamin eines Validierungstages wurde deutlich, dass die Zuhilfenahme einer
quadratischen Regression eventuell favorisiert werden sollte. Es konnte gezeigt werden,
dass bei einer Kurve die Messpunkte häufiger Überschneidungen mit dieser zeigten, als es
bei einer Geraden der Fall war (Abb. 67). Die Analyse der Regressionsvariablen und des
Bestimmtheitsmaßes (fett hervorgehoben in Tab. 21) ließ ferner vermuten, dass die lineare
Regression der quadratischen im gegebenen Zusammenhang unterlegen war. Es sollte ein
Wert für R² von über 0,995 angestrebt werden. Die erhaltenen Bestimmheitsmaße für die
2S,6S-HNK
2R,6R-HNK 2R,6R-HNK
2S,6S-HNK
S-KET S-KET
0.0 25.0 50.0 75.0 100.0 125.0 150.0 175.0 Conc. Ratio0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
Area Ratio
1 2 3
4
5
6
7
0.0 25.0 50.0 75.0 100.0 125.0 150.0 175.0 Conc. Ratio0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
Area Ratio
1 2 3
4
5
6
7
0.0 25.0 50.0 75.0 100.0 125.0 150.0 175.0 Conc. Ratio0
1
2
3
4
5
6
7
8
9
10
11
Area Ratio
1 2
4
5
6
7
0.0 25.0 50.0 75.0 100.0 125.0 150.0 175.0 Conc. Ratio0
1
2
3
4
5
6
7
8
9
10
11
Area Ratio
1 2
4
5
6
7
0.0 25.0 50.0 75.0 100.0 125.0 150.0 175.0 Conc. Ratio0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
Area Ratio
1 2 3
4
5
6
7
0.0 25.0 50.0 75.0 100.0 125.0 150.0 175.0 Conc. Ratio0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
Area Ratio
1 2 3
4
5
6
7

Analytische Untersuchungen von Ketamin und dessen Metaboliten
112 | Methodenentwicklung und Validierung
linearen Regressionen der Beispiele erreichten diesen Wert nicht und waren somit auch
denen der quadratischen unterlegen.
Tab. 21: Variablen und Bestimmtheitsmaß bei quadratischer und linearer Kalibrierfunktion für 2S,6S-Hydroxynorketamin, 2R,6R-Hydroxynorketamin und S-Ketamin.
Substanz Variable Quadratische Funktion � = � ∗ �� + ∗ � + � Lineare Funktion � = � ∗ � + � 2S,6S-Hydroxynorketamin a −2,1034e−5 0,0130
b 0,0172 - c −0,0265 0,0463 R² 0,9995 0,9922
2R,6R-Hydroxynorketamin a −2,6715e−5 0,0130 b 0,0183 - c −0,0517 0,0408 R² 0,9996 0,9879
S-Ketamin a −8,1469e−5 0,0546 b 0,0701 - c −0,0870 0,2322 R² 0,9997 0,9932
Eine weitere graphische Aufarbeitung der Daten war mit Hilfe eines Residuenplots möglich.
Dieser zeigte anschaulich, wie ausgeprägt die einzelnen Messpunkte von den berechneten
Werten der Kalibrationskurve/-gerade abwichen (Abb. 68). Je dichter die Residuen um die
Abszissenachse streuten, desto eher konnte davon ausgegangen werden, dass das
mathematische Modell den Sachverhalt zufriedenstellend widerspiegelte. Bei allen drei
Graphen wurde deutlich, dass die quadratische Regression (rote Vierecke) vor allem im
oberen Kalibrierbereich besser beschreibend war als die lineare (blaue Rauten).
Die mathematische Signifikanz der Regression kann nach Mandel berechnet werden
(Abschnitt 4.4.5). Sie ist ein letzter Indikator dafür, welches Modell benutzt werden sollte.
Der Vergleich der Reststandardabweichungen (Tab. 22) der drei Beispiele zeigte bereits
an, dass eine Regression weniger Streuung besaß, wenn sie nach 2. Ordnung erfolgte.
Somit war es dann auch nicht mehr verwunderlich, dass die Prüfwerte in allen Fällen größer
waren als die Vergleichsgröße der F-Tabelle. Daher war die quadratische Regression als
signifikant besser anzusehen als die lineare.

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Methodenentwicklung und Validierung | 113
Abb. 68: Residuenplots für die entsprechenden linearen und quadratischen Kalibrationen aus Abb. 67 für 2S,6S-Hydroxynorketamin, 2R,6R-Hydroxynorketamin und S-Ketamin.
Es wurde also visuell und mathematisch deutlich, dass es zu einer Art Sättigung kam.
Häufig wird an dieser Stelle der Kalibrierbereich verkleinert, um den Zusammenhang zu
linearisieren. Da die Richtlinien eine andersartige Regression als die lineare aber nicht
kategorisch ausschließen, sondern verständlicherweise nur eine robuste Methode und
konstante experimentelle Bedingungen fordern, wurde die Regression 2. Ordnung für die
Validierung gewählt.
Tab. 22: Reststandardabweichung sy² und Mandel-Linearitätstest für die Kalibration aus Abb. 67 (6-Hydroxynorketamin 7-Punkt-Kalibration, S-Ketamin 6-Punkt-Kalibration) (1 � = 99%, �� = 1, �� =� − 3).
Substanz Reststandardabweichung (sy²) Prüfwert PW (nach Mandel)358
Vergleichsgröße1 linear quadratisch
2S,6S-HNK 0,00956 0,00082 53,85 21,19 21,19 2R,6R-HNK 0,01400 0,00143 44,86
S-KET 0,20084 0,01100 70,01 34,11
Eine Übersicht über den Kalibrierbereich und die Mittelwerte der Bestimmtheitsmaße der
Kalibrierkurven konnte aufgezeigt werden (Tab. 23). Die zurückgerechneten
-0,100
-0,050
0,000
0,050
0,100
0,150
0,00 20,00 40,00 60,00 80,00 100,00 120,00 140,00 160,00 180,00 200,00
Res
idue
n d i
Conc. Ratio
Residuenplot 2S,6S-Hydroxynorketamin
Residuen linear
Residuen quadratisch
-0,150
-0,100
-0,050
0,000
0,050
0,100
0,150
0,200
0,00 20,00 40,00 60,00 80,00 100,00 120,00 140,00 160,00 180,00 200,00
Res
idue
n d i
Conc. Ratio
Residuenplot 2R,6R-Hydroxynorketamin
Residuen linear
Residuen quadratisch
-0,450
-0,300
-0,150
0,000
0,150
0,300
0,450
0,600
0,00 20,00 40,00 60,00 80,00 100,00 120,00 140,00 160,00 180,00 200,00
Res
idue
n d i
Conc. Ratio
Residuenplot S-Ketamin
Residuen linear
Residuen quadratisch

Analytische Untersuchungen von Ketamin und dessen Metaboliten
114 | Methodenentwicklung und Validierung
Konzentrationen für jeden Messpunkt auf der jeweiligen Kalibrierkurve am entsprechenden
Validierungstag sind in Absatz 4.4.6 angegeben. Hierbei konnten die Zielvorgaben der
Richtlinien (±15 % bzw. ±20 % am LLOQ für die Richtigkeit) eingehalten werden, weshalb
die Methode für den Punkt Kalibrierfunktion als valide anzusehen war.
Tab. 23: Übersicht der Arbeitsbereiche und Bestimmtheitsmaße für die Validierungsläufe (� = 5).
Substanz MW R²±SD Arbeitsbereich (ng/mL)
R-Ketamin 0,9997±0,0001
5-200
S-Ketamin 0,9990±0,0009 Norketamin En1 0,9995±0,0004 Norketamin En2 0,9995±0,0006 Dehydronorketamin En1 0,9990±0,0010 Dehydronorketamin En2 0,9991±0,0007 2S,6S-Hydroxynorketamin 0,9994±0,0005 2R,6R-Hydroxynorketamin 0,9992±0,0005
3.2.2.3 Unterstes Quantifizierungslevel
Aus dem Arbeitsbereich der Kalibriergeraden ergab sich das LLOQ von 5 ng/mL. Bei dieser
Konzentration konnte den Anforderungen an Präzision und Richtigkeit noch zuverlässig
entsprochen werden. Es wurde ersichtlich, dass die Signale von den HNK-Enantiomeren
bei gleicher Konzentration kleiner waren als die der anderen Analyten (Abb. 66).
Folgerichtig ließen die Validierungsdaten auch vermuten, dass zumindest für KET, NK und
DHK theoretisch ein LLOQ von 3 ng/mL möglich gewesen wäre. Dieses hätte aber zu einem
unverhältnismäßigen Mehraufwand bei der Erstellung der Kalibrierkurven geführt, weshalb
darauf verzichtet wurde. Die Nachweisgrenze (Lower Limit of Detection, LLOD) betrug für
KET, NK und DHK 1 ng/mL und für HNK 3 ng/mL. Diese wurde optisch aus dem Vergleich
von Messsignal und Rauschen bestimmt.
3.2.2.4 Präzision und Richtigkeit
Der Nachweis der Präzision und Richtigkeit wurde an dieser Stelle zusammengelegt, da
die Erfassung der beiden Parameter mit den gleichen Proben geschah. Zur Abdeckung des
gesamten Kalibrierbereiches wurden drei Konzentrationen gewählt, welche ein niedriges
(dreifacher LLOQ, 9 oder 15 ng/mL), ein mittleres (100 ng/mL) und ein hohes
Konzentrationslevel (ULOQ, 200 ng/mL) abbildeten. Für die within-run Evaluierung wurden
sechs Proben an einem Tag vermessen, während die between-run Bestimmungen mit fünf
Proben an fünf unterschiedlichen Tagen erfolgte (Tab. 24). Die vorgeschlagenen Grenzen
von ±15 %, bzw. 20 % am LLOQ, wurden in allen Fällen eingehalten. Die Methode konnte
daher in Hinsicht auf Präzision und Richtigkeit als valide eingestuft werden.

Methodenentwicklung und Validierung | 115
Tab. 24: Übersicht der Validierungsparameter Präzision und Richtigkeit (between-run � = 6, within-run � = 5).
Substanz Nominalkonz. (ng/mL)
Gemessene Konz. ± SD (ng/mL) Präzision (%) Richtigkeit (%) between-run within-run between-run within-run between-run within-run
KET
R 9 8,79±0,45 9,06±0,29 5,16 3,17 −2,28 0,71 15 15,17±2,15 15,61±1,17 14,20 7,51 1,15 4,11 200 186,75±13,09 193,28±5,37 7,01 2,78 −6,62 −3,36
S 9 8,36±0,45 8,87±0,65 5,38 7,31 −7,10 −1,40 15 15,56 ±1,58 15,46±1,19 10,13 7,70 3,74 3,06 200 213,83±15,70 197,26±5,15 7,34 2,61 6,92 −1,37
NK
En1 15 14,54±0,95 14,94±0,99 6,53 6,61 −3,07 −0,43 100 103,54±4,06 98,77±4,06 3,92 4,11 3,54 −1,23 200 196,19±5,97 192,45±4,17 3,04 2,17 −1,90 −3,77
En2 15 15,03±0,69 15,96±0,49 4,59 3,04 0,19 6,43 100 102,83±3,76 108,66±6,79 3,65 6,25 2,83 8,66 200 199,13±6,41 195,10±7,31 3,22 3,74 −0,43 −2,45
DNK
En1 15 14,95±0,79 15,33±1,55 5,27 10,10 −0,35 8,13 100 104,58±7,44 95,98±7,27 7,12 7,58 4,58 −4,11 200 203,73±14,03 201,99±10,40 6,89 5,15 1,87 0,99
En2 15 15,00±0,29 15,97±0,71 1,96 4,42 0,01 6,45 100 102,84±3,61 103,25±5,57 3,51 5,39 2,84 3,25 200 202,72 ±19,27 191,48±4,86 9,51 2,54 1,36 −4,26
HNK
2S,6S 15 14,97±0,42 15,87±0,49 2,82 3,07 −0,23 5,77 100 95,49 ±7,21 100,96±8,57 7,55 8,49 −4,51 0,96 200 181,94±15,42 196,73±10,45 8,48 5,31 −9,03 −1,63
2R,6R 15 15,29±1,21 14,83±0,88 7,89 5,95 1,93 −1,09 100 96,88±7,61 92,02±2,11 7,86 2,29 −3,12 −7,98 200 183,56±14,45 178,56±5,11 7,87 2,87 −8,22 −10,72

Analytische Untersuchungen von Ketamin und dessen Metaboliten
116 | Methodenentwicklung und Validierung
3.2.2.5 Wiederfindung, Matrix-Effekte und Stabilität
Die Wiederfindung der Analyten wurde bei drei Konzentrationsstufen (9, 15, 200 ng/mL) durch
Vergleich von Proben ohne Matrixbelastung (100 % Wert) mit Matrix-belasteten Proben
durchgeführt (� = 6). Folgende Resultate ergaben sich bei dem Vergleich: KET (91-108 %), NK
(78-94 %), DNK (76-96 %) und HNK (62-81 %). Damit waren die Wiederfindungen für KET, NK
und DNK als gut zu bewerten, es werden in den Richtlinien jedoch keine spezifischen Grenzen
vorgegeben. Für HNK lag die Wiederfindung niedriger, was eventuell dem Umstand geschuldet
sein könnte, dass dieser Metabolit durch die zusätzliche Hydroxy-Gruppe am Molekül eine
erhöhte Polarität aufweist und sich deshalb weniger stark in der lipophilen Phase bei der Flüssig-
Flüssig-Extraktion anreicherte. Entscheidend ist aber vor allem das konstante Verhältnis bei der
Extraktion von Analyt zu IS. Wäre dieses nicht reproduzierbar gewesen, hätten auch Präzision
und Richtigkeit der Methode nicht gezeigt werden können. Aus diesem Grund war davon
auszugehen, dass die Wiederfindung während allen Analysenläufen robust war.
Die Evaluation der Matrix-Effekte erfolgte mit einem Abgleich einer Matrix-unbelasteten Probe
(100 % Wert) und einer Matrix-Probe, welche erst nach der Extraktion mit Analyt versetzt wurde
(post-extraction sample) (Tab. 25). Es kam bis auf zwei Werte (bei 15 ng/mL für HNK-
Enantiomere) zu keiner Verstärkung oder Abschwächung des Messsignals, weshalb davon
ausgegangen wurde, dass die Probenaufarbeitung in Kombination mit der Auswertung unter
Nutzung eines IS und Massendetektion geeignet für die Quantifizierung der vorliegenden
Analyten war.
Tab. 25: Ergebnisse der Matrixeffekt-Experimente (� = 6).
Substanz Matrixeffekt (%) bei entsprechender Konzentration 9 ng/mL 15 ng/mL 200 ng/mL MW
R-Ketamin 89,7 94,2 90,2 91,4 S-Ketamin 89,7 113,9 95,7 99,8 Norketamin En1 98,9 105,3 95,8 99,9 Norketamin En2 98,7 105,7 97,5 100,7 Dehydronorketamin En1 95,8 101,4 89,3 98,6 Dehydronorketamin En2 99,5 105,7 95,1 100,1 2S,6S-Hydroxynorketamin 116,9 126,2 98,2 121,5 2R,6R-Hydroxynorketamin 113,0 122,4 96,3 110,6
Auf die Stabilitätsdaten soll hier nur kurz eingegangen werden, da diese bereits ausführlich
besprochen wurden329. Es konnte gezeigt werden, dass die Analyten mindestens drei Einfrier-
Auftau-Vorgänge überstehen und auch bei Raumtemperatur bis zu 3 h stabil sind. Dazu
ergänzend wurde die Autosampler-Stabilität bei 6 °C für 96 h untersucht, mit dem Ergebnis,
dass es hierbei zur keiner nennenswerten Degradation kam (Gehalt zwischen 85 und 106 %).

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Anwendung der SFC-MS-Methode auf Proben einer klinischen Studie als Proof-of-Concept | 117
Anhand der Resultate des Validierungsprozesses war davon auszugehen, dass die präsentierte
SFC-MS-Methode den Anforderungen der o. g. Richtlinien entsprach. Die Überprüfungen zu
Selektivität, Kalibrierfunktion, Nachweis- und Bestimmungsgrenzen, Präzision, Richtigkeit,
Wiederfindung, Matrix-Effekte und Stabilität konnten in den geforderten Maßen erbracht
werden. Daher besaß die Methode die nötige Zuverlässigkeit und Robustheit und sollte geeignet
sein, auch Urinproben von Probanden, welche an einer pharmakokinetischen Studie zur
Untersuchung von Ketaminmetabolismus teilgenommen haben, zu analysieren.
3.3 Anwendung der SFC-MS-Methode auf Proben einer klinischen Studie als Proof-of-Concept
Im Rahmen einer pharmakokinetischen Pilotstudie, sollten die Besonderheiten des
Metabolismus von Ketamin evaluiert werden359. Hierbei lag ein Fokus auch darauf, ob es zu
einer favorisierten Verstoffwechslung eines Enantiomeres kommt und sich somit bestimmte
chirale Metabolite eher bilden als andere. Die Studie wurde in der Klinischen Pharmakologie
Greifswald unter Aufsicht von Prof. Dr. Werner Siegmund und Prof. Dr. Stefan Oswald
durchgeführt.
Das zu dieser Arbeit zugehörige Probandenkollektiv bestand aus drei männlichen Freiwilligen
(25-29 Jahre alt, mit einem BMI von 20,7±1,0 kg/m²), welche im Rahmen nationaler und
internationaler Regulatorien ihre Zustimmung zur Teilnahme abgegeben hatten. Die Studie
wurde von der Ethik-Kommission der Universitätsmedizin Greifswald (MV01/14) bewilligt. Den
Probanden wurden 5 mg rac-Ketamin (Ketamin-ratiopharm, Ratiopharm GmbH) als 30-minütige
Infusion i. v. verabreicht. Die Urinproben wurden vor und 24, 48 und 72 h nach der
Arzneimittelapplikation gesammelt.
An dieser Stelle soll noch betont werden, dass das Ziel der Analyse der Studienproben vor allem
war, die Anwendbarkeit der validierten SFC-MS-Methode aufzuzeigen. Repräsentative klinische
Aussagen waren aufgrund der geringen Probandenanzahl nicht möglich und auch nicht Fokus
dieser Arbeit. Deshalb werden nur die Konzentrationen der Urinproben zum entsprechenden
Zeitpunkt angegeben, nicht aber die Gesamtexkretionsmengen.
Die Chromatogramme der Proben eines Probanden werden beispielhaft vorgestellt (Abb. 69 bis
Abb. 72). Die Zuordnungen gestalten sich wie folgt: braune Spur zum Zeitpunkt vor, blaue Spur
24 h nach, magenta Spur 48 h nach und die schwarze Spur 72 h nach Applikation von Ketamin.
Alle Metaboliten beziehungsweise die Ursubstanzen konnten einwandfrei identifiziert und der
Verlauf der Bildung bzw. Elimination verfolgt werden. Die Wahl der Matrix erfolgte nicht
grundlos, da die Konzentrationen von Metaboliten gegenüber Ketamin im Urin deutlich erhöht
sind. Bei Plasmauntersuchungen wurde jedoch hauptsächlich die Ursubstanz gefunden304. So
verwunderte es auch nicht, dass die KET-Enantiomere die kleinsten Konzentrationen aufzeigten

Analytische Untersuchungen von Ketamin und dessen Metaboliten
118 | Anwendung der SFC-MS-Methode auf Proben einer klinischen Studie als Proof-of-Concept
und bereits nach 48 h unter das LLOQ fielen. NK und vor allem DNK wurden in deutlich höheren
Konzentrationen ausgeschieden. Mit Fokus auf den wichtigsten Metaboliten 2R,6R-HNK fielen
noch zwei weitere HNK-Metaboliten auf. Eine genauere Zuordnung dieser Metaboliten gleicher
Masse war aber ohne authentisches Referenzmaterial nicht ohne weiteres möglich, es handelte
sich aber sehr wahrscheinlich um Diastereomere (2S,6R-HNK und 2R,6S-HNK) oder
Regioisomere (4- oder 5-HNK) von 2S,6S/2R,6R-HNK.
Abb. 69: Repräsentative Probe eines Studienteilnehmers, Massenspur von Ketamin.
Abb. 70: Repräsentative Probe eines Studienteilnehmers, Massenspur von Norketamin.
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 min
-25000
0
25000
50000
75000
100000
125000
150000
175000
200000
225000
250000
275000
300000
325000
350000
375000
400000
R-KET S-KET
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 min
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
110000
120000
130000
140000
150000
160000
170000 R/S-NK

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Anwendung der SFC-MS-Methode auf Proben einer klinischen Studie als Proof-of-Concept | 119
Abb. 71: Repräsentative Probe eines Studienteilnehmers, Massenspur von 6-Hydroxynorketamin.
Abb. 72: Repräsentative Probe eines Studienteilnehmers, Massespur von Dehydronorketamin.
Die Mittelwerte der Analyten aus den Urinproben der drei Probanden wurden ermittelt (Tab. 26).
In Übereinstimmung mit Literaturangaben war DNK als Hauptmetabolit anzusehen304,329. Ferner
findet sich in diesen Quellen ein Verhältnis von etwa 1 zu 1,15-1,30 der DNK-Enantiomere.
Diese Werte konnten nicht nur bestätigt, sondern auch dazu genutzt werden, um den chiralen
Deskriptor zuzuordnen. Somit würde es sich bei DHK En 1 um das S-konfigurierte und bei DHK
En2 um das R-konfigurierte Enantiomer handeln. Die erhobenen Daten ließen weiterhin
vermuten, dass es sich bei 2R,6R-HNK um das bevorzugte Isomer handelte. Der Abgleich mit
der Literatur war hierbei jedoch deutlich schwieriger, da bisher nur eine HPLC-Methode
beschrieben wurde, die in der Lage ist, die 6-HNK-Enantiomere chiral zu trennen.
Es kann also zusammengefasst werden, dass die validierte SFC-MS-Methode für die Analyse
von Urinproben aus klinischen Studien einsetzbar war. Neben qualitativen Aussagen zum
Auftreten von Metaboliten, ließen sich außerdem die Enantiomere getrennt quantifizieren und
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 min
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
60000
65000
70000
75000
80000
85000
90000
2S,6S-HNK 2R,6R-HNK
weiterer HNK-Metabolit weiterer HNK-Metabolit
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 min
0
25000
50000
75000
100000
125000
150000
175000
200000
225000
250000
275000
300000
325000
350000 R/S-DNK

Analytische Untersuchungen von Ketamin und dessen Metaboliten
120 | Gegenüberstellung der SFC-MS-Methode mit literaturbekannten LC-Methoden
aus diesen Daten Verhältnisse von Enantiomerenpaaren beziffern. Damit kann die präsentierte
Methode einen großen Benefit für zukünftige klinische Studien leisten.
Tab. 26: Übersicht der Konzentrationen aus den Probandenproben (Werte unterhalb des LLOQ sind als Schätzung anzusehen).
Substanz Gemessene Konzentration ± SD (ng/mL) 0-24h 24-48h 48-72h
R-Ketamin 10,32±1,94 1,05±0,12 0,79±0,00 S-Ketamin 12,51±3,41 1,82±0,24 1,66±0,32 Norketamin En1 16,65±4,18 2,64±0,15 1,95±0,33 Norketamin En2 17,60±4,44 1,76±4,44 2,40±2,38 Dehydronorketamin En1 36,54±7,66 5,69±1,23 2,85±0,76 Dehydronorketamin En2 42,19±2,42 6,90±2,02 3,50±0,36 2S,6S-Hydroxynorketamin 24,16±6,68 3,70±0,57 3,11±1,51 2R,6R-Hydroxynorketamin 29,86±9,75 7,37±4,62 5,04±0,95
3.4 Gegenüberstellung der SFC-MS-Methode mit literaturbekannten LC-Methoden
Da Ketamin kein neues API darstellt, sondern im Rahmen des „Repurposing“ für eine neue
mögliche Indikation entdeckt wurde, sind bereits zahlreiche HPLC-Methoden publiziert. Ein
Review aus dem Jahr 2017 (Publikation erst 2018) trägt dem Rechnung328 und wurde schon
genauer im Absatz 3.1.2 besprochen. Daher sollen an dieser Stelle nur Vergleiche mit den
wichtigsten Publikationen vor (Moaddel et al.304 und Hasan et al.329) und nach (Ramiole et al.360
und Toki et al.361) Veröffentlichung der präsentierten Methode im Journal of Pharmaceutical and
Biomedical Analysis erörtert werden.
Moaddel et al.304 publizierten die wohl komplexeste Methode gemessen an der Anzahl der
getrennten Analyten. An einer Eclipse XDB-C18 Säule konnten neben KET, NK, DNK und
2S,6S/2R,6R-HNK auch die sich dazu diastereomer verhaltenden 2S,6R/2R,6S-HNK sowie die
Regioisomere 4- und 5-HNK getrennt werden. Die entsprechenden Glucuronsäure-Konjugate
wurden ebenfalls mit betrachtet. Neben der verhältnismäßig langen Laufzeit von 30 min ist der
größte Nachteil, dass hierbei keine chirale Separierung ermöglicht wurde. Dafür stellten die
Autoren eine zweite Methode zur Verfügung, welche in der Lage war, KET, NK und DNK
innerhalb von 20 min chiral (Chiral-AGP Säule) zu trennen. Zugegebenermaßen lag der
physiologische Hintergrund der Methoden nicht bei depressiven Erkrankungen, sondern bei der
Therapie des komplexen regionalen Schmerzsyndroms. Das könnte ein Grund sein, warum sich
nicht weiter der enantioselektiven Analyse von hydroxylierten Metaboliten gewidmet wurde. Da
aber mittlerweile genug Befunde vorhanden sind, die 2R,6R-HNK ein großes Potential in der

Analytische Untersuchungen von Ketamin und dessen Metaboliten
Gegenüberstellung der SFC-MS-Methode mit literaturbekannten LC-Methoden | 121
Therapie von Depressionen prognostizieren, sind diese Methoden weniger für die aktuelle
Problemstellung geeignet.
Hasan et al.329 übernahmen beide Methoden von Moaddel et al. größtenteils, konnten dabei die
Laufzeit der achiralen Methode (XTerra MS C18 Säule) auf 6 min deutlich verkürzen,
verzichteten dafür aber auf HNK und Hydroxyketamin als Analyten. Zusätzlich wurden auch das
LLOQ und der Verbrauch an Probenmaterial abgesenkt. Die größte Neuerung lag allerdings in
der Implementierung einer dritten Methode, welche die 6-HNK Enantiomere trennen konnte.
Hierfür wurde von uns die Lux Amylose-2 als Säule vorgeschlagen und beschafft. Der größte
Nachteil ist zweifelsohne die ausgedehnte Laufzeit von 60 min. Damit war es die erste
Publikation, welche die Trennung dieses Enantiomerenpaares aufzeigte, gleichwohl die
Verwendung von drei Methoden nebeneinander mit einem effizienten Zeitmanagement nur
schwerlich zu verbinden ist.
Ramiole et al.360 legten ihren Schwerpunkt weniger auf die chromatographische Entwicklung als
vielmehr auf die vergleichende Probenvorbereitung. Dafür evaluierten sie die Effekte von
Flüssig-Flüssig-Extraktion mit Chloroform, Acetonitril-induzierter Proteinfällung und
Festphasenextraktion. Die HPLC-Methode benutzte die Atlantis T3C18 als Säule und schafft die
Trennung von DNK, NK und KET in 7 min. Hierbei fand keine chirale Separation der einzelnen
Enantiomere statt.
Eine der neuesten publizierten Methoden im Rahmen der Ketaminanalyse stammt von Toki et
al.361. Mit Hilfe einer Chiralpak AS-3R konnten KET und NK in einer sehr kurzen Zeit von nur
5 min enantioselektiv voneinander getrennt werden. Der Fokus lag primär auf der Reduktion
von Probenmaterial auf wenige µL pro Analyse. Dies gelang, in dem die bereits angesprochene
Dilute-and-Shoot-Variante zur Probenvorbereitung berücksichtigt wurde. Wohlwissend, dass
dabei die Belastung mit Matrixbestandteilen für den Massendetektor außerordentlich hoch war,
wurde der Fluss der mobilen Phase nur über eine bestimmte Zeit in den Detektor zugelassen.
Eine Analyse weiterer Metabolite erfolgte deshalb nicht, um die bei diesem Vorgehen
unvermeidlichen Verunreinigungen des Detektors zu begrenzen.
Aus den vier Publikationen wird ersichtlich, dass jede Arbeit einer dezidierten Fragestellung
nachging und deshalb dort auch jeweils andere Schwerpunkte gesetzt wurden. Im Kontext der
chiralen Metabolitentrennung mit Fokus auf antidepressive Therapien, konnte keine der
vorgestellten Verfahren gänzlich überzeugen, da entweder mehrere Methoden pro Analyt
benötigt oder aber nicht alle relevanten Metaboliten in die Analyse eingeschlossen wurden.
Daher lässt sich der Schluss ziehen, dass die hier beschriebene SFC-MS-Methode zwar nicht
in allen Belangen überlegen war (z. B. LLOQ oder benötigte Zeit bzw. verwendetes
Matrixvolumen für Probenaufbereitung), in der gemeinsamen chiralen Analyse von 6-HNK, KET,
NK und DNK innerhalb einer Methode aber ein Alleinstellungsmerkmal besaß. Ferner waren
auch die Bezeichnungen effizient und umweltfreundlich zutreffend, da die Analyse mit 15 min

Analytische Untersuchungen von Ketamin und dessen Metaboliten
122 | Zusammenfassung und Ausblick Kapitel 3
bei vermindertem Verbrauch von organischem Lösungsmittel im direkten Vergleich sehr gut
abschneidete.
Einen gänzlich anderen Punkt betraf die leitliniengerechte Validierung der SFC-MS-Methode. In
den letzten Jahren wurden immer wieder SFC-Verfahren vorgestellt, die nur teilweise validiert
wurden, so in drei aktuellen Arbeiten aus dem Jahr 2018: die Publikationen über die Analyse
von aromatischen Kohlenwasserstoffen in kaffeehaltigen Getränken bzw. Dunkelbier362, die
enantioselektive Bestimmung von Fenbuconazol einschließlich Metaboliten in Früchten und
Gemüse363 und das Monitoring des Insektizids Thiacloprid in Treibhausgemüse364 brachten
allesamt Validierungsparameter zu Sprache, wenngleich sie in der Regel nicht ausführlich nach
EMA- oder FDA-Kriterien ausgeführt wurden. Dazu passend wurden in der Zusammenfassung
einer aktuellen Übersichtsarbeit über die SFC in der Bioanalytik noch einmal die inhärenten
Möglichkeiten hervorgehoben, aber auch festgestellt, dass die bisherigen Methoden oftmals
unzureichend validiert seien365. Bevor sich die SFC nicht mit den entsprechenden
Empfehlungen von ICH, FDA und EMA vollumfänglich decke, folgerten die Autoren, würde eine
Implementierung in die Routineanalytik nicht möglich seien.
3.5 Zusammenfassung und Ausblick Kapitel 3
Es scheint nur noch eine Frage der Zeit zu sein, bis Ketamin den Status „off-label“ für die
Therapie von depressiven Erkrankungen verlässt und Einzug in die Leitlinien findet. Der
Mechanismus und das tatsächlich wirksame Agens sind nach wie vor aber nicht genau bekannt
und benötigen noch weitere pharmakodynamische und analytische Untersuchungen. Die in
dieser Arbeit vorgestellte Methode kann einen wichtigen Beitrag dazu leisten. Anhand der
gängigen Validierungsparameter der EMA und FDA wurde die SFC-MS-Methode systematisch
optimiert und die Nachweise für Brauchbarkeit bzw. Zuverlässigkeit erbracht. Dabei wurden die
Vorteile, welche in der Natur der SFC liegen, ausgenutzt und führten zu einer Anwendung, die
vergleichbare LC-Methoden hinsichtlich Analysengeschwindigkeit, Umweltfreundlichkeit und
Selektivität zum Teil erheblich übertrifft. Des Weiteren wurde mit der erfolgreichen Publikation
der Ergebnisse dazu beigetragen, die Anwendungsmöglichkeiten zu präsentieren und damit
auch aufzuzeigen, dass die benötigte Robustheit für die Routineapplikation der SFC in der
Bioanalytik erreicht werden kann.
Zukünftig können solche Methoden ferner davon profitieren, dass sich die Probenvorbereitung
von den „klassischen“ Vorgehensweisen abwendet und mit neuen kombiniert wird. Dies konnte
bereits in einer Weiterentwicklung der SFC-MS-Methode im eigenen Arbeitskreis gezeigt
werden, bei der die Flüssig-Flüssig-Extraktion gegen eine Extraktion mit sCO2 ausgetauscht
wurde366. Durch die on-line Kopplung von Probenvorbereitung und Analysenlauf eröffnen sich
neue Möglichkeiten bezüglich Zeit- und Lösungsmittelersparnis.

Experimenteller Teil
Geräteparameter | 123
4 Experimenteller Teil
Die Wiedergabe von Gebrauchs-, Marken- und Handelsnamen oder Warenbezeichnungen in
dieser Arbeit berechtigt auch ohne besondere Kennzeichnung nicht zu der Annahme, dass
solche Namen im Sinne der Warenzeichen- und Markenschutz-Gesetzgebung als frei zu
betrachten wären und daher von jedermann benutzt werden dürften.
4.1 Geräteparameter
4.1.1 NMR-Spektroskopie
Gerät Bruker Biospin FT-NMR-Spektrometer Avance III 400 MHz
Messfrequenz 1H: 400,1 MHz; 13C, DEPT-135, HSQC, HMBC: 100,6 MHz
Messtemperatur 25 °C
Interner Standard Tetramethylsilan oder Lösungsmittelbasissignal
Software Bruker TopSpin (Version 2.1)
Angaben zur Kopplungskonstante J gelten für H,H-Kopplungen (i. d. R. werden nur die
Hauptkopplungen betrachtet), sofern nicht anders angegeben (allgemein: XJY,Z). Chemische
Verschiebungen sind in ppm (δ-Skala) angegeben und gelten in Relation zu Tetramethylsilan
bzw. dem Lösungsmittelbasissignal. Bei 13C-Spektren, die in D2O aufgenommen wurden, liegt
kein Basissignal vor, sodass hier die Spektren unkalibriert ausgewertet werden. Die
verwendeten Lösungsmittel sind bei den jeweiligen Spektrendaten angegeben. Die Zuordnung
der Signale in allen NMR-Spektren wurde wenn nötig mittels DEPT-135-, HSQC- und/oder
HMBC-Experimenten verifiziert. Die angegebenen Nummerierungen der Verbindungen
beziehen sich nur auf die Zuordnung der NMR-Signale und sind nicht IUPAC-konform.
Ungenauigkeiten der Kopplungskonstanten sind der Unschärfe der Spektren geschuldet
(gerundet auf Vielfache von 0,4 Hz).
4.1.2 Schmelzverhalten
Gerät Büchi Schmelzpunkt M-565
Die manuelle Bestimmung erfolgt mit Hilfe der Präzisionslupe mit zweieinhalbfacher
Vergrößerung, die automatische Bestimmung über eine Videokamera mit Videoaufzeichnung
mit sechsfacher Vergrößerung. Die Temperaturauflösung beträgt 0,1 °C. Der Schmelzpunkt
wird mit einer Messgenauigkeit von ± 0,3 °C bis 250 °C und von ± 0,3 °C bis ± 0,5 °C bei 250 °C
bis 400 °C, bei einer Aufheizrate von 0,5 °C/min bestimmt. Sämtliche Analysen wurden mit Hilfe

Experimenteller Teil
124 | Geräteparameter
des Probenverdichters M-569 vorbereitet. Bei allen Substanzen wurde vorher der ungefähre
Schmelzbereich durch eine Schnellmessung (5 °C/min) bestimmt und danach ab 10 °C
unterhalb der niedrigsten Schmelztemperatur mit der Rate von 1,0 °C/min gemessen.
4.1.3 Dünnschichtchromatographie/Schwerkraftsäulenchromatographie
Alle Analysen erfolgten auf TLC Silicagel 60 F254-Platten (Kieselgel auf Aluminiumfolie,
Schichtdicke 0,20 mm, Porengröße 60 Å, Partikelgröße 10–12 μm) der Firma Merck KGaA. Die
Detektion erfolgte mittels UV- (λ= 254 und 366 nm) oder Vis-Licht. Bei Bedarf wurden Ninhydrin-
Reagenz (0,15 g Ninhydrin in 47,5 mL Isopropanol und 2,5 mL Essigsäure), Ehrlich-Reagenz
(1 g 4-Dimethylaminobenzaldehyd in 25 mL Salzsäure und 75 mL Methanol), Eisen(III)-chlorid-
Reagenz (1 g FeCl3 in 50 mL Wasser und 50 mL Methanol) oder DNPH-Reagenz (0,2 g 2,4-
Dinitrophenylhydrazin in 40 mL Eisessig, 40 mL 25%iger Salzsäure und 20 mL Methanol) mit
eventuell anschließender Erwärmung (>150 °C) eingesetzt.
Bei der präparativen Säulenchromatographie kam Kieselgel 60 (Korngröße 20–45 µm,
Porengröße 60 Å) der Firma Carl Roth zum Einsatz. Dieses wurde auch zum Selberpacken der
Glassäulen (230×15 mm, Büchi) mit dem Büchi Sepacore Dry-Filling Set für die MPLC benutzt.
Die Bezeichnung der Durchführung erfolgt nach: Auftragsart (flüssig oder adsorbiert an
Kieselgur (aus Lösungsmittel)), qualitative Laufmittelzusammensetzung und quantitative
Laufmittelzusammensetzung und ist bei den jeweiligen Synthesen mit angegeben.
4.1.4 MIR-Spektroskopie
Gerät Bruker Optics Alpha FT-IR-Spektrometer
Probenkopf Platinum-ATR-Modul (Diamant)
Software Bruker Optics Opus (Version 7.5)
4.1.5 Mikrowellenreaktor
Gerät Anton Paar Monowave 300
Alle Synthesen wurden im Closed-Vessel-Verfahren in G30- (max. 20 mL) oder G10-Vials (max.
6 mL) durchgeführt. Die Temperaturkontrolle erfolgte mittels IR-Sensor am Boden des
Reaktionsraumes, die Druckkontrolle mit Hilfe eines Detektors oberhalb des Septums des
Reaktionsgefäßes. Alle Synthesen wurden unter Rühren durchgeführt. Über die Reaktor-interne
Software (Version 3.20) wurden die Maximalwerte für Temperatur, Druck und Leistung
eingestellt. Die Kameraüberwachung erfolgte über das Programm VLC-Mediaplayer.

Experimenteller Teil
Geräteparameter | 125
4.1.6 MPLC
Gerät Büchi Sepacore Flash Chromatograph
Bestandteile Pump Manager: C-615, Pump Module: C-601, UV-Monitor: C-630,
Fraction Collector: C-660
Software Büchi SepacoreRecord (Version 1.3.1000.500)
4.1.7 Präparative HPLC
Gerät Shimadzu Prep LC
Bestandteile System Controller: CBM-20A, Solvent Delivery Module: LC-20AP,
Autosampler: SIL-20A, UV-Detector: SPD-20A, Fraction Collector:
FRC-10A, manuelles 6-Wege-Injektionsventil mit 5 mL Probenschleife
Software Shimadzu LabSolutions (Version 5.75 SP2)
Säule LiChrospher100 RP-18 endcapped, 5 μm, 250×25 mm
Die entsprechenden Laufmittelzusammensetzungen sind bei den einzelnen Substanzen
angegeben. Vor Injektion der Produktlösung wurde eine Filtration mittels PTFE-Filter (0,45 µm)
der Firma VWR vorgenommen.
4.1.8 Hochauflösende Massenspektrometrie (HRMS)
Gerät Shimadzu LCMS-IT-TOF
Bestandteile System Controller: CBM-20A, Solvent Delivery Module: LC-20AD,
Autosampler: SIL-20AC HT, Column Oven: CTO-20A, PDA: SPD-
M20A
Software Shimadzu LCMS Solution (Version 3.41)
Säule Chromolith SpeedRod RP-18 endcapped, 50×4,6 mm
Eine hohe Massenauflösung ist Bedingung für eine hohe Massengenauigkeit in der
Massenspektrometrie. Hochauflösende Massenspektren wurden nur von nicht
literaturbekannten Substanzen aufgenommen, um deren Masse mit hoher Genauigkeit zu
bestimmen und zu bestätigen. Alle Verbindungen wurden mittels ESI ionisiert. Die
monoisotopischen Massen gelten als bestätigt, wenn die experimentellen Daten ≤ 5 ppm vom
berechneten Wert abweichen. Die angegebenen Massen beziehen sich bereits auf die um ein
Proton korrigierten Massen (entsprechend der Ionisierung im positiven oder negativen Modus).
Als mobile Phase kam ein Methanol-Wasser-Gemisch (+0,1 % Ameisensäure) in angepasster
Zusammensetzung an die Polarität der Analyten zum Einsatz.

Experimenteller Teil
126 | Geräteparameter
4.1.9 SFC-MS
Gerät Shimadzu Nexera SFC/UHPLC switching system
Bestandteile System Controller: CBM-20A, Solvent Delivery Module: LC-20ADXR,
CO2 Delivery Module: LC-30ADSF, Back Pressure Regulator: SFC-30A,
Autosampler: SIL-30AC, Column Oven: CTO-20AC, Degasser:
DGU-20A5R
Software Shimadzu LabSolution (Version 5.82)
4.1.10 Visualisierung der Kristallstrukturen
Software Molecular Operating Environment 2018.1 (Chemical Computing Group)
Software PyMOL Molecular Graphics System Version 1.8.4.2 (Schrödinger LLC.)
4.1.11 Kristallographie (Institut für Biochemie, Greifswald)
Die röntgenkristallographische Untersuchung der Substanz N-[4-(1H-Tetrazol-5-yl)pyridin-2-
yl]ethan-1,2-diamin-Hydrochlorid (XLIII) erfolgte im Arbeitskreis von Prof. Dr. Carola Schulzke
am Institut für Biochemie der Universität Greifswald nach folgendem Ablauf:
Die Beugungsdaten wurden mit dem Einkristalldiffraktometer STOE-IPDS 2T bei
Raumtemperatur mit graphit-monochromatischer MoKα-Strahlung (λ = 0.71073 Å)
aufgenommen. Die Struktur wurde mittels direkter Methoden gelöst (SIR-92; Journal of Applied
Crystallography, 1994, 27, 435-436) und durch die Voll-Matrix-Methode der kleinsten Quadrate
(KQ-Methode) verfeinert (SHELXL-13; Acta Crystallographica Section A: Foundations of
Crystallography, 2008, 64, 112-122). Alle Nicht-Wasserstoffatome wurden durch anisotrope
Auslenkungsparameter verbessert. Alle Stickstoff-gebundenen Wasserstoffatome wurden
gefunden und frei verfeinert. Die Kohlenstoff-gebundenen Wasserstoffatome wurden ebenfalls
gefunden, dann jedoch als „riding“ behandelt und in ideale Positionen gesetzt mit einer
Restriktion der Auslenkungsparameter auf Uiso = 1,2 Ueq der entsprechenden gebundenen
Kohlenstoffatome.
Hergestellt wurde der Kristall mit Hilfe der Vapor-Diffusion-Methode. Dafür wurden etwa 20 mg
Substanz in leicht ammoniakalischem Methanol gelöst und in ein offenes Schnappdeckelgefäß
gegeben. Dieses wurde in ein größeres Becherglas, welches etwa bis zur Hälfte mit Diethylether
gefüllt war, gestellt und dann luftdicht verschlossen. Der Wachstumsprozess dauerte etwa 2
Wochen.

Experimenteller Teil
Geräteparameter | 127
4.1.12 Kristallographie (BESSY II, Berlin) und isotherme Titrationskalorimetrie
Im Arbeitskreis Makromolekulare Kristallographie um Dr. Manfred Weiss (Helmholtz-Zentrum
Berlin für Materialien und Energie, Photonenquelle BESSY II) wurden die
Röntgenkristallstrukturen der Substanzen LXXVII, LXXXII, CX und CXI vermessen und
nachberechnet. Dafür wurde folgenden Methoden genutzt:
KDM4D-Proteinkristalle (KDM4D gekürzt auf Aminosäuresequenz 1-342) wurden unter
Verwendung der Sitting-Drop-Methode bei einer Temperatur von 291 K erhalten. Es wurden
dafür gleiche Volumina (0,4 µL) Protein- und Nicht-Proteinlösung in eine 96-Well-Mikrotiterplatte
(Intelli-Plate, Art Robbin Instruments) pipettiert (Kristallisierroboter Gryphon, Art Robbins
Instruments). Die Zusammensetzung der Nicht-Proteinlösung betrug 24 % PEG 3350,
180 mmol/L Ammoniumsulfat und 0,1 mol/L HEPES-Puffer (pH 7), während die Proteinlösung
eine Konzentration von 19 mg/mL KDM4D, 0,5 mol/L NaCl, 5 % Glycerol, 1 mmol/L Tris(2-
carboxyethyl)phosphin (TCEP) und 10 mmol/L HEPES-Puffer (pH 7,5) aufwies. Für die Soaking-
Experimente wurden die erhaltenen KDM4D-Kristalle in Nicht-Proteinlösung mit 10 mmol/L
NiCl2 eingeweicht und anschließend in Liganden-Lösung (hergestellt aus Nicht-Proteinlösung
mit 100-150 mmol/L Ligand) transferiert. Die Inkubationszeit betrug zwischen 30 Minuten und
24 Stunden. Anschließend wurden die Kristalle kurzzeitig in eine Frostschutzlösung bestehend
aus Nicht-Proteinlösung und 20 % Ethylenglycol getaucht und dann schockgefroren.
Zur Erhebung der Röntgenbeugungsdaten wurden die Abschnitte 14.1 und 14.3 am BESSY II
Synchrotron Berlin mit den Pilatus 6M bzw. Rayonix MX225 CCD Detektoren benutzt (The
European Physical Journal Plus, 2015, 130, 141). Integration und Anpassung der Daten wurden
mit der XDSAPP Software durchgeführt (Journal of Applied Crystallography, 2016, 49,
1085-1092). Die Strukturen wurden mit Hilfe von einfachen molekularen Ersatzmethoden mit
den gängigen Modellen aufgelöst und mit den Programmen refmac bzw. phenix.refine verfeinert
(Acta Crystallography Section D Biological Crystallography, 2011, 67, 355-367 und 2012, 68,
352-367). Die finalen Strukturen wurden anschließend manuell mit der Software Coot adjustiert
und in der Protein Data Bank hochgeladen.
Die Messungen der Bindungsaffinitäten von KDM4D mit den Liganden LXXXII, CX und CXI
erfolgte mit dem MicroCal PEAQ-ITC Mikrokalorimeter (Malvern Panalytical GmbH). Die
Experimente wurden in einer Lösung von 10 mmol/L HEPES-Puffer (pH 7,5), 5 % Glycerol und
1 mmol/L TCEP bei 20 °C durchgeführt. Die Proteinkonzentration wurde spektroskopisch
(λ = 280 nm) bestimmt. Die DMSO-Stammlösungen der Liganden wurden entsprechend mit
Puffer-Lösung auf Arbeitskonzentration verdünnt (Gesamtgehalt DMSO 0,4 bis 1 %). Um
thermische Mischungseffekte auszuschließen, wurden die KDM4D-Lösungen vorher den
entsprechenden DMSO-Konzentrationen der Liganden-Lösungen angepasst. Die eigentliche
Titration verlief in 13 bis 19 Schritten zu je etwa 2-3 µL Liganden-Lösung (bei 180 Sekunden

Experimenteller Teil
128 | Chemikalien
Spacing Time und 5 Sekunden Filtering Period). Die Datenauswertung aller Experimente
erfolgte mit der Software MicroCal PEAQ-ITC Analysis.
4.1.13 LANCE-Assay (Institut für Pharmazeutische Wissenschaften, Freiburg)
Die biologischen Daten wurden im Arbeitskreis von Prof. Dr. Manfred Jung an der Albert-
Ludwigs-Universität Freiburg nach folgender Vorgehensweise erhoben:
Der kommerziell erhältliche Antikörper-basierte LANCE-Assay (PerkinElmer) wurde in einem
Volumen von 10 μL in weißen OptiPlate-384-Mikrotiterplatten (PerkinElmer) durchgeführt. Der
verwendete 50 mmol/L HEPES-Puffer (pH = 7,5) enthielt 0,01 % Tween-20 und 0,01 % bovines
Serumalbumin (BSA). Die zu untersuchenden Substanzen (0-1000 μmol/L in DMSO) wurden
für 10 min bei Raumtemperatur mit einer Lösung aus 60 nmol/L KDM4A AS1–359 inkubiert.
Anschließend erfolgte der Zusatz von 100 μmol/L Ascorbinsäure, 5 μmol/L FeSO4, 1 μmol/L
2OG sowie 400 nmol/L biotinyliertem H3K9me3-Substrat (ARTKQTARK(me3)-
STGGKAPRKQLA-K(Biotin)) (BPS Bioscience). Der finale DMSO-Gehalt der Lösungen betrug
in allen Fällen 5 %. Die Proben wurden für 45 min bei Raumtemperatur inkubiert und die
Reaktion daraufhin durch Zugabe von 10 μL einer Lösung aus 2 nmol/L Europium-markiertem
anti-H3K9me2-LANCE-Antikörper (PerkinElmer), 50 nmol/L ULight-Streptavidin-Farbstoff
(PerkinElmer) und 1 mmol/L EDTA in 1X-LANCE-Detektionspuffer (PerkinElmer) gestoppt (alle
angegebenen Konzentrationen entsprechen den finalen Konzentrationen). Anschließend
erfolgte eine weitere Inkubation bei Raumtemperatur für 60 min. Für die Messung der FRET-
Intensität diente ein EnVision 2102-Multilabel-Plattenlesegerät (PerkinElmer) bei λex = 340 nm
und λem = 665 nm mit einer Verzögerung von 100 μs. Die Auswertung erfolgte mithilfe der
Software GraphPad Prism 4.00, wobei die IC50-Werte als Mittelwert von zwei unabhängigen
Experimenten errechnet wurden.
Der FDH-Assay kam auf Grund von Eigenfluoreszenz der getesteten Substanzen nicht zum
Einsatz. Um sicherzustellen, dass keine Interferenzen mit dem LANCE-Assay auftraten, wurde
als Negativkontrolle der Assay mit den Substanzen ohne Enzym durchgeführt, wobei keine
Änderung des Messsignals beobachtet werden konnte.
4.2 Chemikalien
Für den synthetischen Teil dieser Arbeit wurden Chemikalien so eingesetzt, wie vom Händler
geliefert wurde, sofern keine Trocknungsverfahren angegeben sind, und besaßen mindestens
die Reinheit „zur Synthese“. Bei dem verwendeten Wasser handelt es sich ausschließlich um
voll entsalztes Wasser.

Experimenteller Teil
Chemikalien | 129
Eine Auflistung der Chemikalien für den analytischen Teil findet sich in der nachfolgenden
Tabelle. Einzelne Standards aus dieser Tabelle wurden unentgeltlich vom National Center for
Advancing Translational Sciences des National Instiute of Health der USA zur Verfügung
gestellt.
Chemikalie Reinheit (%) oder Bezeichnung Hersteller Acetonitril 99,9 Fluka Analytical Ameisensäure 99,5 Fisher Chemical Ammoniak-Lösung, wässrig (25 %)
LC-MS grade Sigma-Aldrich
rac-Dehydronorketamin∙HCl Certified reference material Sigma-Aldrich (2S,6S)-Hydroxynorketamin (2R,6R)-Hydroxynorketamin
99,5 National Center for Advancing Translational Sciences
Isopropanol 99,95 Carl Roth rac-Ketamin∙HCl Certified reference material Sigma-Aldrich S-Ketamin∙HCl Arzneimittel Pfizer Kohlenstoffdioxid 99,995 Air Liquide Methanol 99,98 Carl Roth Methyl-tert-butylether 99,5 Merck Natriumcarbonat 99,9 VWR rac-Norketamin∙HCl Certified reference material Sigma-Aldrich rac-Norketamin-d4∙HCl Certified reference material Sigma-Aldrich Wasser ≤0,055 µS/cm, pH 5.0–6.0 In-Haus

Experimenteller Teil
130 | Beschreibung der Synthesen
4.3 Beschreibung der Synthesen
Hinweis: Bei nichtäquimolaren Verhältnissen oder nicht kompletten Umsätzen von Aziden
während der Tetrazolsynthese, müssen Azidreste unschädlich gemacht werden, da von ihnen
akute Gefahr für Mensch und Umwelt ausgeht. In dieser Arbeit wurden die zu neutralisierenden
Azidreste mit Salpetriger Säure (in situ Herstellung aus Natriumnitrit und verdünnte
Schwefelsäure) zu molekularem Stickstoff und Distickstoffmonoxid umgesetzt367. Bei
Kombination mit dem Lösungsmittel Dimethylformamid muss eine mögliche Entstehung von
potentiell kanzerogenen Nitrosaminen aus Aminverunreinigungen des Dimethylformamids
beachtet werden, welche wahrscheinlich auch ursächlich für einen aktuellen Rückruf von mit
Nitrosaminen verunreinigten Valsartan-Chargen war368.
4.3.1 Synthese von Tetrazol- und Carbonsäurehydrazid-beinhaltenden Substanzen
N-Acetylanthranilsäure (II)
Anthranilsäure (I) (20,0 g, 145 mmol) wird in einem Überschuss Acetanhydrid (etwa 200 mL)
suspendiert und unter Rückfluss für etwa 60 min erhitzt, wobei das Edukt komplett in Lösung
geht. Nach Erkalten des Ansatzes wird die Lösung in etwa 800 mL Eiswasser gegossen,
wodurch das Reaktionsprodukt in feinen Nadeln auskristallisiert. Diese werden abfiltriert und,
wenn nötig, aus 20%igem Ethanol (Wasser/Ethanol, 80:20, v/v) umkristallisiert.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 184-187 °C (184-185 °C116).
Ausbeute: 99 %. 1H-NMR (DMSO-d6): δ = 2,13 (s, 3H, C(9)H3), 7,13 (t, 1H, C(5)H, 3J = 8,4 Hz), 7,57 (t, 1H, C(4)H,
3J = 8,0 Hz), 7,96 (d, 1H, C(3)H, 3J = 8,0 Hz), 8,45 (d, 1H, C(6)H, 3J = 8,4 Hz), 11,05 (s, 1H,
N(7)H), 13,56 (s, 1H, C(10)OOH). 13C-NMR (DMSO-d6): δ = 24,9 (C9), 116,5 (C1), 119,9 (C3), 122,5 (C5), 131,0 (C6), 133,9 (C4),
140,8 (C2), 168,4 (C8), 169,4 (C10).
MIR (ṽ): 3330 (w, νN-H), 3100-2300 (s, νO-H, assoziiert) cm−1.

Experimenteller Teil
Beschreibung der Synthesen | 131
N-Acetyl-5-bromanthranilsäure (III)
N-Acetylanthranilsäure (II) (15,8 g, 88 mmol) wird in so viel Wasser aufgeschlämmt, dass eine
komplette Durchmischung durch einen Magnetrührer möglich ist. Anschließend erfolgt die
langsame Zugabe von 1,3 Äq. Br2-Lösung (18,2 g, 114 mmol; Lösung aus 30,0 g KBr und
200 mL Wasser) mit Hilfe eines Tropftrichters über 1,5 h unter starkem Rühren. Nach erfolgter
Bromzugabe wird der Reaktionsansatz über Nacht unter einem gut arbeitenden Laborabzug
weitergerührt. Das erhaltene Produkt wird abfiltriert und, wenn nötig, aus 20%igem Ethanol
(Wasser/Ethanol, 80:20, v/v) umkristallisiert.
Beschaffenheit: Leicht rosafarbener amorpher Feststoff.
Schmelzbereich: 214-215 °C (215-216 °C121).
Ausbeute: 86 %. 1H-NMR (DMSO-d6): δ = 2,13 (s, 3H, C(9)H3), 7,75 (dd, 1H, C(4)H, 3J = 8,8 Hz), 8,03 (s, 1H,
C(6)H), 8,40 (d, 1H, C(3)H, 3J = 8,8 Hz), 10,95 (s, 1H, N(7)H), 13,90 (s, 1H, C(10)OOH). 13C-NMR (DMSO-d6): δ = 24,9 (C9), 113,9 (C1), 118,8 (C5), 122,1 (C3), 133,0 (C6), 136,4 (C4),
139,8 (C2), 168,0 (C8), 169,5 (C10).
MIR (ṽ): 3318 (w, νN-H), 3100-2300 (s, νO-H, assoziiert) cm−1.
5-Bromanthranilsäure (IV)
N-Acetyl-5-bromanthranilsäure (III) (18,5 g, 72 mmol) wird in 400 mL einer 1 mol/L Natronlauge
gelöst und unter Rückfluss für 4 h erhitzt. Nach Erkalten der Lösung wird mit HCl bis auf einen
pH-Wert von etwa 1-2 eingestellt. Dabei kommt es zur Fällung des Reaktionsproduktes. Dieses
wird abfiltriert und aus 25%igem Ethanol (Wasser/Ethanol, 75:25, v/v) umkristallisiert.
Beschaffenheit: Feine farblose Nadeln.

Experimenteller Teil
132 | Beschreibung der Synthesen
Schmelzbereich: 216-217 °C (219-220 °C122).
Ausbeute: 88 %. 1H-NMR (DMSO-d6): δ = 6,73 (d, 1H, C(3)H, 3J = 8,8 Hz), 7,34 (dd, 1H, C(4)H, 3J = 8,8 Hz), 7,75
(s, 1H, C(6)H), 8,72 (s, 2H, N(7)H2).
13C-NMR (DMSO-d6): δ = 104,5 (C1), 111,1 (C5), 118,6 (C3), 132,8 (C6), 136,1 (C4), 150,5
(C2), 168,3 (C8).
MIR (ṽ): 3474 (s, νN-H2), 3100-2300 (s, νO-H, assoziiert) cm−1.
5-Bromanthranilsäuremethylester (VI)
Es werden 10,0 g 5-Bromanthranilsäure (IV) (46 mmol) in 150 mL MeOH gelöst und im Eisbad
gerührt. Dazu werden langsam 22 mL Thionylchlorid (303 mmol) mit Hilfe eines Tropftrichters
zugegeben. Der anfangs gebildete Feststoff löst sich beim Erhitzen unter Rückfluss vollständig.
Nach 15 h Siedehitze wird das Methanol im Teilvakuum entfernt (es ist eine Waschflasche mit
Natronlauge zu benutzen, um die Vakuumpumpe zu schonen). Der Überstand wird in
Ethylacetat aufgenommen und mit gesättigter NaHCO3-Lösung extrahiert. Durch
anschließendes Trocknen über Na2SO4 und Einengen im Teilvakuum wird aus der organischen
Phase das Zielprodukt erhalten.
Beschaffenheit: Gelblich glänzender Feststoff.
Schmelzbereich: 68-70 °C (68-69 °C135).
Ausbeute: 64 %. 1H-NMR (DMSO-d6): δ = 3,79 (s, 3H, C(9)H3), 6,76 (d, 1H, C(3)H, 3J = 8,8 Hz und 2H, N(7)H2),
7,37 (dd, 1H, C(4)H, 3J = 8,8 Hz), 7,76 (s, 1H, C(6)H). 13C-NMR (DMSO-d6): δ = 51,7 (C9), 104,7 (C1), 110,1 (C5), 118,9 (C3), 132,2 (C6), 136,4 (C4),
150,4 (C2), 166,6 (C8).
MIR (ṽ): 3452 (s, νN-H2), 1684 (s, νC=O) cm−1.

Experimenteller Teil
Beschreibung der Synthesen | 133
5-Cyananthranilsäuremethylester (VII)
5-Bromanthranilsäuremethylester (VI) (2,9 g, 13 mmol) und 1,5 g CuCN (17 mmol, 1,3 Äq.)
werden trocken in einem Zweihalskolben vermengt. Die gesamte Apparatur mitsamt
Rückflusskondensator wird evakuiert und anschließend mit Stickstoff geflutet (insgesamt 3 Mal).
Die Zugabe von 20 mL NMP erfolgt über den mit einem Septum verschlossenen Seitenhals.
Unter Rückfluss (etwa 210 °C) wird für 2 h erhitzt. Das entstandene dunkle Rohprodukt wird in
300 mL gesättigte NH4Cl-Lösung gegossen. Anschließend wird die wässrige Phase mit
Ethylacetat extrahiert (gegebenenfalls können störende, weder in Wasser noch Ethylacetat
lösliche Feststoffe, abfiltriert werden). Nach dem Trocknen über Na2SO4 und Einengen der org.
Phase im Teilvakuum, wird eine zähe dunkle Masse erhalten, aus welcher mit siedendem
10%igem ACN (Wasser/ACN, 90:10, v/v) das gewünschte Produkt extrahiert werden kann. Bei
mitunter auftretender Harzbildung sollte die ACN-Wasser-Mischung abdekantiert werden. Aus
dieser kristallisiert beim Abkühlen der 5-Cyananthranilsäuremethylester.
Beschaffenheit: Farbloser watteartiger Feststoff.
Schmelzbereich: 124-126 °C.
Ausbeute: bis zu 54 %. 1H-NMR (DMSO-d6): δ = 3,82 (s, 3H, C(9)H3), 6,87 (d, 1H, C(3)H, 3J = 8,8 Hz), 7,45 (s, 2H,
N(7)H2), 7,57 (dd, 1H, C(4)H, 3J = 8,8 Hz), 8,05 (s, 1H, C(6)H).
13C-NMR (DMSO-d6): δ = 51,9 (C9), 95,8 (C5), 108,8 (C1), 117,4 (C3), 119,4 (C10), 136,1 (C6
und C4), 153,9 (C2), 166,3 (C8).
MIR (ṽ): 3464 (s, νN-H2), 2211 (s, νC≡N), 1707 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C9H8N2O2 + H]+: 176,0586; gefunden: 176,0579.

Experimenteller Teil
134 | Beschreibung der Synthesen
5-(1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII)
Es werden 1,9 g 5-Cyananthranilsäuremethylester (VII) (11 mmol) in 20 mL Toluen gelöst. Dazu
erfolgt die Zugabe von 880 mg NaN3 (14 mmol) und 1,9 g TEA∙HCl (14 mmol) (beide 1,3 Äq.).
Nach etwa 24 h Erhitzen bei 100 °C wird das Toluen im Teilvakuum eingeengt. Der
resultierende Überstand wird in Wasser aufgenommen und mit so viel Na2CO3 versetzt, bis ein
pH-Wert von 7-8 resultiert. Die wässrige Phase wird anschließend mit Ethylacetat extrahiert und
dann mit Salzsäure auf einen pH-Wert von 2 eingestellt. Der entstandene Niederschlag
entspricht dem gewünschten Produkt und kann durch Filtration gewonnen werden.
Beschaffenheit: Khakifarbener amorpher Feststoff.
Schmelzbereich: 231-233 °C unter Zersetzung.
Ausbeute: 78 %. 1H-NMR (DMSO-d6): δ = 3,87 (s, 3H, C(9)H3), 6,96 (d, 1H, C(3)H, 3J = 8,8 Hz), 7,2 (s, 2H,
N(7)H2), 7,88 (dd, 1H, C(4)H, 3J = 8,8 Hz), 8,45 (s, 1H, C(6)H).
13C-NMR (DMSO-d6): δ = 51,7 (C9), 108,6 (C1), 110,1 (C5), 117,3 (C3), 130,2 (C6), 132,22 (C4),
153,1 (C2), 156,0 (C10), 167,2 (C8).
MIR (ṽ): 3452 (s, νN-H2), 1678 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C9H9N5O2 + H]+: 219,0756; gefunden: 219,0756.
5-(1H-Tetrazol-5-yl)anthranilsäurehydrazid (IX)
Es werden 550 mg 5-(1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII) (2,5 mmol) in 10 mL
EtOH suspendiert und 2,2 mL N2H4∙H2O (80%ige Lösung in Wasser) hinzugegeben. Es
resultiert eine Lösung, welche für mindestens 8 h unter Rückfluss erhitzt wird. Nach Abkühlen
wird das Lösungsmittel azeotrop an einer Drehschieberpumpe, gegebenenfalls unter erneuter
Zugabe von 10 mL EtOH, entfernt. Der Überstand wird mit Hilfe von 50%igem Methanol

Experimenteller Teil
Beschreibung der Synthesen | 135
(Wasser/Methanol, 50:50, v/v) kristallisiert und aus dem gleichen Lösungsmittelgemisch
umkristallisiert. Nach Abfiltration wird das Produkt mit wenigen mL kaltem Aceton gewaschen.
Beschaffenheit: Khakifarbender amorpher Feststoff.
Schmelzbereich: 253-254 °C unter Zersetzung.
Ausbeute: 15 %. 1H-NMR (DMSO-d6): δ = 6,77 (s, 2H, N(7)H2), 6,85 (d, 1H, C(3)H, 3J = 8,4 Hz), 7,75 (dd, 1H,
C(4)H, 3J = 8,4 Hz), 8,08 (s, 1H, C(6)H), 9,66 (s, 1H, N(9)H). 13C-NMR (DMSO-d6): δ = 109,9 (C1), 114,3 (C5), 116,3 (C3), 127,6 (C6), 130,0 (C4), 151,2
(C2), 154,9 (C10), 167,8 (C8).
MIR (ṽ): 3475 (s, νN-H2), 1622 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C8H9N7O + H]+: 219,0869; gefunden: 219,0878.
5-Iodanthranilsäuremethylester (XI)
8,0 g 5-Iodanthranilsäure (X) (30 mmol) werden in 120 mL MeOH gelöst und mit 9 mL H2SO4
versetzt. Der Reaktionsansatz wird anschließend für mindestens 24 h unter Rückfluss erhitzt.
Nach Einengen des Lösungsmittels im Teilvakuum, wird der Überstand in Wasser
aufgenommen, mit NaOH basisch eingestellt und mit Ethylacetat extrahiert. Durch
anschließendes Trocknen über Na2SO4 und Einengen im Teilvakuum wird aus der organischen
Phase das Zielprodukt erhalten.
Beschaffenheit: Dunkler amorpher Feststoff.
Schmelzbereich: ab 50 °C (83-85 °C144).
Ausbeute: 73 %. 1H-NMR (DMSO-d6): δ = 3,78 (s, 3H, C(9)H3), 6,64 (d, 1H, C(3)H, 3J = 8,8 Hz), 6,77 (s, 2H,
N(7)H2), 7,48 (dd, 1H, C(4)H, 3J = 8,8 Hz), 7,93 (s, 1H, C(6)H).
13C-NMR (DMSO-d6): δ = 51,6 (C9), 74,3 (C5), 111,0 (C1), 119,2 (C3), 138,3 (C6), 141,7 (C4),
150,6 (C2), 166,5 (C8).
MIR (ṽ): 3460 (s, νN-H2), 1695 (s, νC=O) cm−1.

Experimenteller Teil
136 | Beschreibung der Synthesen
N-Acetyl-5-bromanthranilsäuremethylester (XII)
5-Bromanthranilsäuremethylester (VI) (2,3 g, 10 mmol) wird mit Acetanhydrid im Überschuss
(20 mL) 30 min unter Rückfluss erhitzt. Die abgekühlte Lösung wird in 500 mL Eiswasser
gegossen, wobei das gewünschte Produkt in feinen Nadeln auskristallisiert. Diese werden
abfiltriert und, wenn nötig, unter Verwendung von 50%igem Methanol (Wasser/Methanol, 50:50,
v/v) umkristallisiert.
Beschaffenheit: Feine farblose Nadeln.
Schmelzbereich: 128-129 °C (134-135 °C369)
Ausbeute: 88 %. 1H-NMR (DMSO-d6): δ = 2,12 (s, 3H, C(9)H3), 3,85 (s, 3H, C(11)H3), 7,77 (dd, 1H, C(4)H,
3J = 8,8 Hz), 7,96 (s, 1H, C(6)H), 8,13 (d, 1H, C(3)H, 3J = 8,8 Hz), 10,46 (s, 1H, N(7)H). 13C-NMR (DMSO-d6): δ = 24,5 (C9), 52,6 (C11), 114,6 (C1), 120,2 (C5), 123,3 (C3), 132,5 (C6),
136,3 (C4), 138,6 (C2), 166,2 (C10), 168,6 (C8).
MIR (ṽ): 3270 (m, νN-H), 1695 und 1681 (s, νC=O) cm−1.
N-Acetyl-5-cyananthranilsäuremethylester (XIII)
Die Herstellung erfolgt nach der Vorschrift für 5-Cyananthranilsäuremethylester (VII) mit zwei
Änderungen: N-Acetyl-5-bromanthranilsäuremethylester (XII) (2,4 g, 8,8 mmol) wird mit 1,0 g
CuCN (11 mmol) vermengt und in 15 mL NMP gelöst. Das Lösungsmittel zur Extraktion ist
50%iger IPA (Wasser/Isopropanol, 50:50, v/v).
Beschaffenheit: Blassgelber amorpher Feststoff.

Experimenteller Teil
Beschreibung der Synthesen | 137
Schmelzbereich: 135-136 °C.
Ausbeute: 15 %. 1H-NMR (DMSO-d6): δ = 2,17 (s, 3H, C(9)H3), 3,88 (s, 3H, C(11)H3), 8,02 (dd, 1H, C(4)H,
3J = 8,4 Hz), 8,26 (s, 1H, C(6)H), 8,39 (d, 1H, C(3)H, 3J = 8,4 Hz), 10,77 (s, 1H, N(7)H). 13C-NMR (DMSO-d6): δ = 24,7 (C9), 52,8 (C11), 105,2 (C5), 118,0 (C1 und C12), 121,2 (C3),
134,8 (C6), 137,1 (C4), 143,1 (C2), 166,1 (C10), 169,1 (C8).
MIR (ṽ): 3253 (m, νN-H), 2225 (s, νC≡N), 1700 und 1682 (s, νC=O) cm−1.
N-Acetyl-5-(1H-tetrazol-5-yl)anthranilsäuremethylester (XIV)
Variante A: siehe Vorschrift für 5-(1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII) mit
N-Acetyl-5-cyananthranilsäuremethylester (XIII) (275 mg, 1,3 mmol), 110 mg NaN3 (1,7 mmol)
und 240 mg (1,7 mmol) TEA∙HCl als Edukte. Aufgrund der minimalen Ausbeute ist Variante B
zu bevorzugen.
Variante B (nach Einhorn150): 600 mg 5-(1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII)
(2,7 mmol) werden in 5 mL Pyridin gelöst und unter Eiskühlung langsam mit 277 µL (3,9 mmol,
1,4 Äq.) Acetylchlorid versetzt. Nach etwa 30-minütigem Rühren wird auf 40 °C für 2 h mit
aufgesetztem Trockenrohr erhitzt. Anschließend wird die Lösung in Eiswasser gegossen und
mit Salzsäure auf einen pH-Wert von 2 eingestellt. Der entstandene Niederschlag wird abfiltriert
und aus 50%igem MeOH (Wasser/MeOH, 50:50, v/v) umkristallisiert.
Beschaffenheit: Blassgelber amorpher Feststoff.
Schmelzbereich: 241-243 °C unter Zersetzung.
Ausbeute: 30 %. 1H-NMR (DMSO-d6): δ = 2,18 (s, 3H, C(9)H3), 3,92 (s, 3H, C(11)H3), 8,23 (dd, 1H, C(4)H,
3J = 8,8 Hz), 8,45 (d, 1H, C(3)H, 3J = 8,8 Hz), 8,59 (s, 1H, C(6)H), 10,73 (s, 1H, N(7)H). 13C-NMR (DMSO-d6): δ = 24,7 (C9), 52,7 (C11), 118,2 (C1), 118,8 (C5), 121,6 (C3), 129,1 (C6),
132,0 (C4), 141,6 (C2), 153,6 (C12), 166,9 (C10), 168,8 (C8).
MIR (ṽ): 3261 (m, νN-H), 1690 (s, νC=O) cm−1.

Experimenteller Teil
138 | Beschreibung der Synthesen
HRMS (ESI, m/z), berechnet [C11H11N5O3 + H]+: 261,0862; gefunden: 261,0870.
3-Amino-2-methyl-6-(1H-tetrazol-5-yl)chinazolin-4(3H)-on (XVI)
500 mg N-Acetyl-5-(1H-tetrazol-5-yl)anthranilsäuremethylester (XIV) (1,9 mmol) werden in
10 mL MeOH gelöst und mit 2,5 mL N2H4∙H2O (80%ige Lösung in Wasser) versetzt.
Anschließend erfolgt Erhitzen unter Rückfluss für mindestens 8 h. Mit Hilfe einer
Drehschieberpumpe wird das Lösungsmittel azeotrop abdestilliert, gegebenenfalls unter
erneuter Zugabe von 10 mL MeOH. Der Überstand wird erst mit reichlich Ethylacetat, dann mit
Wasser, welches mit Salzsäure auf einen pH-Wert von 2 eingestellt wurde, gründlichst
gewaschen. Durch anschließendes Lösen des Stoffes in verdünnter Natronlauge und erneutem
Fällen aus der Lösung mit Salzsäure, wird der gewünschte Stoff als Präzipitat erhalten. Wenn
nötig, kann der Stoff mit der präparativen HPLC weiter aufgereinigt werden (gelöst in ACN/H2O,
50:50, v/v; A: H2O mit 0,1 % HCOOH und B: ACN, isokratisch, 30 % B).
Beschaffenheit: Bräunlicher amorpher Feststoff.
Schmelzbereich: ab 280 °C unter Zersetzung.
Ausbeute: 8 %. 1H-NMR (DMSO-d6): δ = 2,62 (s, 3H, C(8)H3), 5,88 (s, 2H, N(10)H2), 7,77 (d, 1H, C(3)H,
3J = 8,8 Hz), 8,39 (dd, 1H, C(4)H, 3J = 8,8 Hz), 8,78 (s, 1H, C(6)H). 13C-NMR (DMSO-d6): δ = 22,0 (C8), 120,2 (C1), 122,9 (C5), 124,4 (C6), 127,8 (C3), 131,8 (C4),
147,7 (C2), 155,8 (C11), 157,0 (C7), 159,9 (C9).
MIR (ṽ): 3324 (m, νN-H2), 1624 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C10H9N7O + H]+: 243,0869; gefunden: 243,0869.
N-Acetyl-5-(5-methyl-1,3,4-oxadiazol-2-yl)anthranilsäuremethylester (XVII)

Experimenteller Teil
Beschreibung der Synthesen | 139
Es werden 400 mg 5-(1H-Tetrazol-5-yl)anthranilsäuremethylester (VIII) (1,8 mmol) in 7 mL
Acetanhydrid suspendiert und unter Rückfluss für mindestens 1 h erhitzt. Der Überschuss an
Acetanhydrid wird anschließend im Teilvakuum entfernt. Die Umkristallisation des Überstandes
erfolgt aus 30%igem MeOH (Wasser/Methanol, 70:30, v/v) mit Heißfiltration. Der beim Abkühlen
aus der filtrierten Lösung auskristallisierende Stoff entspricht dem Zielprodukt.
Beschaffenheit: Blassgelbe feine Nadeln.
Schmelzbereich: 186-188 °C.
Ausbeute: 52 %. 1H-NMR (DMSO-d6): δ = 2,18 (s, 3H, C(12)H3), 2,58 (s, 3H, C(14)H3), 3,92 (s, 3H, C(9)H3), 8,16
(dd, 1H, C(4)H, 3J = 8,8 Hz), 8,42 (s, 1H, C(6)H), 8,47 (d, 1H, C(3)H, 3J = 8,8 Hz), 10,75 (s, 1H,
N(7)H). 13C-NMR (DMSO-d6): δ = 10,6 (C14), 24,7 (C12), 52,7 (C9), 117,9 (C1), 121,5 (C3), 128,4 (C6),
131,4 (C4), 142,1 (C2), 162,9/163,9 (C10 und C13), 166,7 (C8), 168,9 (C11).
MIR (ṽ): 3262 (m, νN-H), 1709 und 1683 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C13H13N3O4 + H]+: 275,0906; gefunden: 275,0896.
N-4-Fluorbenzoyl-5-(1H-tetrazol-5-yl)anthranilsäuremethylester (XIX)
Die Herstellung erfolgt nach der Vorschrift für N-Acetyl-5-(1H-tetrazol-5-
yl)anthranilsäuremethylester (XIV) Variante B mit folgenden Änderungen: 500 mg 5-(1H-
Tetrazol-5-yl)anthranilsäuremethylester (VIII) (2,3 mmol) werden in 5 mL Pyridin gelöst und mit
340 µL 4-Fluorbenzoylchlorid (2,9 mmol, 1,3 Äq.) unter Eiskühlung versetzt. Das Lösungsmittel
für die Umkristallisation ist Methanol.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 257-259 °C unter Zersetzung.
Ausbeute: 57 %. 1H-NMR (DMSO-d6): δ = 3,96 (s, 3H, C(9)H3), 7,45 (t, 2H, 2×C(14)H, 3J = 8,8 Hz), 8,04 (t, 2H,
2×C(13)H, 3J = 8,8 Hz), 8,29 (dd, 1H, C(4)H, 3J = 8,4 Hz), 8,68 (s, 1H, C(6)H), 8,73 (d, 1H,
C(3)H, 3J = 8,4 Hz), 11,71 (s, 1H, N(7)H).

Experimenteller Teil
140 | Beschreibung der Synthesen
13C-NMR (DMSO-d6): δ = 53,0 (C9), 116,1 (2JC,F = 22 Hz, 2×C14), 117,8 (C1), 121,5 (C3), 129,2
(C6), 130,0 (3JC,F = 9 Hz, 2×C13), 130,5 (4JC,F = 3 Hz, C12), 132,4 (C4), 142,0 (C2), 155,5 (C10),
163,3 und 165,8 (1JC,F = 249 Hz, C15), 163,9 (C11), 167,4 (C8).
MIR (ṽ): 3255 (w, νN-H), 1688 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C16H12FN5O3 + H]+: 341,0924; gefunden: 341,0923.
N-Cyanmethyl-5-phenylvalerianamid (XXV)
700 mg Aminoacetonitril-Hydrochlorid (XXIV) (7,5 mmol) werden in einer Mischung aus 10 mL
Pyridin und 40 mL CH2Cl2 gelöst. Anschließend werden 2,1 g 5-Phenylvaleriansäurechlorid
(XXIII) (11 mmol, 1,5 Äq.) tropfenweise hinzugegeben und mindestens 20 h bei
Raumtemperatur gerührt. Durch Einengen des Reaktionsansatzes im Teilvakuum wird eine
sirupöse Masse erhalten, welche durch Flüssig-Flüssig-Extraktion mit Wasser (pH 2 mit
Salzsäure eingestellt) und Ethylacetat gewaschen wird. Die organische Phase wird mit Na2SO4
getrocknet und anschließend das Lösungsmittel im Teilvakuum entfernt. Der Überstand wird
anschließend säulenchromatographisch aufgearbeitet (gelöst in wenigen mL EtOAc, LM:
PE/EtOAc, 50:50, v/v).
Beschaffenheit: Blassgelber wachsartiger Feststoff.
Schmelzbereich: 70-71 °C.
Ausbeute: 74 %. 1H-NMR (CDCl3): δ = 1,67 (m, 4H, C(3)H2 und C(4)H2, 3J = 7,2 Hz), 2,24 (t, 2H, C(2)H2,
3J = 7,2 Hz), 2,63 (t, 2H, C(5)H2, 3J = 7,2 Hz), 4,13 (s, 2H, C(11)H2), 6,00 (s, 1H, N(10)H), 7,17-
7,28 (m, 5H, 2×C(7)H und 2×C(8)H und C(9)H). 13C-NMR (CDCl3): δ = 25,0 (C3), 27,5 (C11), 31,0 (C4), 35,7 (C5), 36,0 (C2), 116,3 (C12), 126,1-
128,6 (2×C7 und 2×C8 und C9), 142,2 (C6), 173,1 (C1).
MIR (ṽ): 3285 (s, νN-H), 2852 (m, νC-H2), 1655 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C13H16N2O + H]+: 216,1263; gefunden: 216,1254.

Experimenteller Teil
Beschreibung der Synthesen | 141
N-(1H-Tetrazol-5-yl)methyl-5-phenylvalerianamid (XXVI)
Es werden 575 mg N-Cyanmethyl-5-phenylvalerianamid (XXV) (2,6 mmol) mit 190 mg NaN3
(2,9 mmol, 1,1 Äq.) und 160 mg NH4Cl (2,9 mmol, 1,1 Äq.) eingewogen. Nach Zugabe von
10 mL DMF wird für mindestens 24 h bei 105 °C erhitzt. Nach Einengen des Reaktionsansatzes
im Teilvakuum wird dieser mit K2CO3-Lösung aufgenommen und mehrfach mit Ethylacetat im
Scheidetrichter gewaschen. Ansäuern der wässrigen Phase (langsam wegen CO2- und
möglicher HN3-Entwicklung, Abzug!) führt zur Fällung des gewünschten Produktes. Nach
Abfiltration und Trocknung kann das Produkt, wenn nötig, aus 50%igem MeOH
(Wasser/Methanol, 50:50, v/v) umkristallisiert werden.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 157-158 °C.
Ausbeute: 53 %. 1H-NMR (DMSO-d6): δ = 1,53 (t, 4H, C(3)H2 und C(4)H2, 3J = 6,8 Hz), 2,17 (t, 2H, C(2)H2,
3J = 6,8 Hz), 2,55 (t, 2H, C(5)H2, 3J = 6,8 Hz), 4,53 (s, 2H, C(11)H2), 7,16-7,27 (m, 5H, 2×C(7)H
und 2×C(8)H und C(9)H), 8,55 (s, 1H, N(10)H). 13C-NMR (DMSO-d6): δ = 24,5 (C3), 30,5 (C4), 32,3 (C11), 34,8 (C2 und C5), 125,6-128,3 (2×C7
und 2×C8 und C9), 142,0 (C6), 154,5 (C12), 172,6 (C1).
MIR (ṽ): 3283 (s, νN-H), 2853 (m, νC-H2), 1656 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C13H17N5O − H] −: 259,1433; gefunden: 259,1432.
2-[4-(1H-Indol-3-yl)butylamido]-2-(1H-tetrazol-5-yl)essigsäureethylester (XXX)
510 mg 2-Amino-2-(1H-tetrazol-5-yl)essigsäureethylester-Hydrochlorid (XXIX) (2,5 mmol) und
500 mg 4-(1H-Indol-3-yl)buttersäure (XXVII) (2,5 mmol, 1,0 Äq.) werden in einem Eisbad in
30 mL wasserfreiem CH2Cl2 gelöst. Dazu werden erst 770 µL Diisopropylcarbodiimid (5,0 mmol,
2,0 Äq.) hinzugegeben und dann tropfenweise 840 µL DIPEA (5,0 mmol, 2,0 Äq.) addiert. Nach

Experimenteller Teil
142 | Beschreibung der Synthesen
30-minütigem Rühren bei 0 °C wird der Reaktionsansatz bei Raumtemperatur für weitere 20 h
durchmengt. Nach Einengen des Gemisches wird der Überstand mit K2CO3-Lösung
aufgenommen und im Scheidetrichter mit Ethylacetat gewaschen. Die wässrige Phase scheidet
nach Ansäuern mit Salzsäure ein bräunliches Öl ab, welches unter Eiskühlung zügig erstarrt.
Dieser Feststoff (gewünschtes Produkt) wird abfiltriert und mit heißem Diethylether mehrfach
gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 179-181 °C.
Ausbeute: 77 %. 1H-NMR (DMSO-d6): δ = 1,16 (t, 3H, C(18)H3, 3J = 7,2 Hz), 1,88 (m, 2H, C(6)H2, 3J = 7,2 Hz),
2,27 (t, 2H, C(5)H2, 3J = 7,2 Hz), 2,67 (t, 2H, C(7)H2, 3J = 7,2 Hz), 4,17 (q, 2H, C(17)H2, 3J = 7,2 Hz), 5,92 (d, 1H, C(2)H, 3J = 7,6 Hz), 6,95-7,07 (2×t, C(14)H und C(15)H, 3J = 7,6 Hz),
7,10 (s, 1H, C(9)H), 7,32 (d, 1H, C(16)H, 3J = 8,0 Hz), 7,48 (d, 1H, C(13)H, 3J = 8,0 Hz), 9,07
(d, 1H, N(3)H, 3J = 7,6 Hz), 10,75 (s, 1H, N(10)H). 13C-NMR (DMSO-d6): δ = 13,8 (C18), 24,1 (C6), 25,9 (C7), 38,9 (C5), 47,4 (C2), 61,8 (C17),
111,3 (C16), 114,0 (C8), 118,1 (C13 und C14), 120,8 (C15), 122,3 (C9), 127,1 (C12), 136,3
(C11), 154,0 (C19), 167,3 (C1), 172,7 (C4).
MIR (ṽ): 3357 und 3265 (m, νN-H), 1740 und 1640 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C17H20N6O3 + H]+: 356,1597; gefunden: 356,1582.
2-[4-(1H-Indol-3-yl)butylamido]-2-(1H-tetrazol-5-yl)essigsäurehydrazid (XXXI)
Es werden 2-[4-(1H-Indol-3-yl)butylamido]-2-(1H-tetrazol-5-yl)essigsäureethylester (XXX)
(560 mg, 1,6 mmol) in 40 mL MeOH gelöst und mit 2,3 mL N2H4∙H2O (80%ige Lösung in
Wasser) versetzt. Die Umsetzung erfolgt unter Rückfluss für mindestens 6 h. Der
Reaktionsansatz wird im Hochvakuum an einer Drehschieberpumpe eingeengt und
anschließend über Nacht in einem Exsikkator (H2SO4!) von weiteren Hydrazinresten befreit. Es
erfolgt erst eine säulenchromatographische Aufarbeitung (adsorbiert an Kieselgur (aus MeOH),
LM: EtOAc/MeOH/NH3, 70:30:1, v/v/v), ehe die vorgereinigten Produktfraktionen vereint und mit
HN
NH
O
NH2
NN
NNH
ONH
12
3 45
67
19
89
10
11
16
15
12
13
14
1718
XXXI

Experimenteller Teil
Beschreibung der Synthesen | 143
Hilfe der präparativen HPLC weiter aufgearbeitet werden (gelöst in ACN/H2O/DMSO, 10:80:10,
v/v/v; A: H2O mit H3PO4 auf pH 2,7 eingestellt und B: ACN, isokratisch, 20 % B).
Beschaffenheit: Gräulicher amorpher Feststoff.
Schmelzbereich: 129-131 °C unter Zersetzung.
Ausbeute: 22 %. 1H-NMR (DMSO-d6): δ = 1,87 (m, 2H, C(6)H2, 3J = 7,6 Hz), 2,30 (t, 2H, C(5)H2, 3J = 7,6 Hz),
2,67 (t, 2H, C(7)H2, 3J = 7,6 Hz), 5,83 (d, 1H, C(2)H, 3J = 7,6 Hz), 6,95-7,05 (2×t, C(14)H und
C(15)H, 3J = 7,2 Hz), 7,10 (s, 1H, C(9)H), 7,32 (d, 1H, C(16)H, 3J = 8,0 Hz), 7,49 (d, 1H, C(13)H, 3J = 7,6 Hz), 8,73 (d, 1H, N(3)H, 3J = 7,6 Hz), 10,75 (s, 1H, N(10)H). 13C-NMR (DMSO-d6): δ = 24,3 (C6), 24,8 (C7), 34,6 (C5), 46,9 (C2), 111,3 (C16), 114,1 (C8),
118,2 (C13 und C14), 120,8 (C15), 122,3 (C9), 127,2 (C12), 136,3 (C11), 154,9 (C19), 165,5
(C1), 172,6 (C4).
MIR (ṽ): 3403 und 3273 (s, νN-H), 1704 und 1651 (s, νC=O) cm−1.
HRMS (ESI, m/z), berechnet [C15H18N8O2 + H]+: 342,1553; gefunden: 342,1554.
4.3.2 Synthese von Tetrazolylpyridinen und Isonicotinsäure-beinhaltenden Substanzen
2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV)
Es wird 2-Chlorisonicotinnitril (XXXIV) (20,0 g, 144 mmol) in 200 mL Toluen gelöst.
Anschließend werden 12,2 g Natriumazid (1,3 Äq., 187 mmol) und 25,8 g
Triethylammoniumchlorid (1,3 Äq., 187 mmol) hinzugegeben und der Reaktionsansatz für 18 h
bei 110 °C gerührt. Nach Abkühlen des Reaktionsgemisches auf Raumtemperatur wird so viel
wässrige Natronlauge zugesetzt, bis ein pH-Wert von etwa 12 eingestellt ist. Die wässrige
Phase wird dreimal mit 50 mL Ethylacetat gewaschen und anschließend mit konzentrierter
Salzsäure angesäuert, wodurch 2-Chlor-4-(1H-tetrazol-5-yl)pyridin ausgefällt wird. Nach
Filtration kann der resultierende Rückstand aus wässrigem Methanol (Wasser/MeOH, 85:15,
v/v) umkristallisiert werden. Die Lagerung erfolgt unter Lichtausschluss.
Beschaffenheit: Farbloser amorpher Feststoff.

Experimenteller Teil
144 | Beschreibung der Synthesen
Schmelzbereich: 166-168 °C unter Zersetzung.
Ausbeute: 92 %. 1H-NMR (DMSO-d6): δ = 8,02 (d, 1H, C(3)H, 3J = 4,0 Hz), 8,07 (s, 1H, C(5)H), 8,66 (d, 1H,
C(2)H, 3J = 4,4 Hz). 13C-NMR (DMSO-d6): δ = 120,3 (C5), 121,2 (C3), 135,7 (C4), 151,3 (C6) 151,4 (C2), 154,8 (C7).
MIR (ṽ): 1611 (s, νC=N), 1400 (s, νC-Cl) cm−1.
HRMS (ESI, m/z), berechnet [C6H4N5Cl + H]+: 181,0155; gefunden: 181,0161.
N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
Es werden 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol) mit 2,2 mL 70%iger
wässriger Ethylamin-Lösung (10 Äq., 27 mmol) versetzt und 25 Minuten bei 190 °C im
Mikrowellenreaktor erhitzt. Nach Abkühlen auf Raumtemperatur wird das Reaktionsgemisch in
20 mL Eiswasser gegossen und anschließend mit konzentrierter Salzsäure auf einen pH-Wert
von etwa 2 eingestellt, wobei das N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin als
Hydrochloridsalz auskristallisiert. Das Zielprodukt wird abfiltriert und mit wenig heißem Methanol
gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 280-282 °C unter Zersetzung.
Ausbeute: 5 %. 1H-NMR (DMSO-d6): δ = 1,14 (t, 3H, C(9)H3, 3J = 7,2 Hz), 3,27 (m, 2H, C(8)H2), 6,39 (t, 1H,
N(10)H, 3J = 5,6), 7,04 (m, 2H, C(3)H und C(5)H, 3J = 5,2 Hz), 7,94 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 14,8 (C9), 35,5 (C8), 104,0 (C5), 109,3 (C3), 140,0 (C4), 147,6 (C2),
159,3 (C6), 159,6 (C7).
MIR (ṽ): 3199 (m, νN-H), 1638 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C8H10N6 + H]+: 190,0967; gefunden: 190,0967.
Nachfolgende Synthesebeschreibungen, welche auf N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin
(XXXVI) Bezug nehmen, meinen ebenfalls eine mikrowellengestützte Umsetzung im Closed-

Experimenteller Teil
Beschreibung der Synthesen | 145
Vessel-Verfahren und, wenn nicht anders angegeben, eine anschließende Fällung des
Hydrochloridsalzes aus einer entsprechenden Lösung.
N-Allyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVII)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 2,1 mL Allylamin (10 Äq., 28 mmol) bei 200 °C für 30 min umgesetzt. Die Fällung
des Hydrochlorids kann durch wenige mL Ethylacetat unterstützt werden. Als Waschflüssigkeit
wird Isopropanol verwendet. Das Zielprodukt kann, wenn nötig, aus Wasser umkristallisiert
werden.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 238-239 °C unter Zersetzung.
Ausbeute: 9 %. 1H-NMR (DMSO-d6): δ = 3,98 (s, 2H, C(8)H2), 5,12 (dd, 1H, C(10)H2, 3J = 1,6 und 10,4 Hz), 5,24
(dd, 1H, C(10)H2, 3J = 2,0 und 18,4 Hz), 5,94 (m, 1H, C(9)H, 3J = 1,6 und 10,4 und 18,4 Hz),
7,14 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,25 (s, 1H, C(5)H), 7,51 (s, 1H, N(11)H), 8,13 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (DMSO-d6): δ = 43,1 (C8), 105,6 (C5), 108,9 (C3), 115,4 (C10), 134,9 (C4), 135,3
(C9), 146,3 (C2), 155,8 (C6), 157,8 (C7).
MIR (ṽ): 3080 (m, νC=C-H), 1635 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C9H10N6 + H]+: 202,0967; gefunden: 202,0962.
N-Cyclohexyl-4-(1H-tetrazol-5-yl)pyridin-2-amin (XXXVIII)
N NH
N
NN
HN
1
2
34
5
6
7
8
910
11
XXXVIII

Experimenteller Teil
146 | Beschreibung der Synthesen
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 2,8 mL Cyclohexylamin (9 Äq., 24 mmol) und 0,9 mL DIPEA (2,0 Äq., 5,4 mmol) bei
190 °C für 1 h gerührt. Nach Abkühlen auf Raumtemperatur wird im Teilvakuum eingeengt und
der Überstand chromatographisch aufgereinigt (adsorbiert an Kieselgur (aus MeOH), LM:
EtOAc/Aceton/MeOH/Eisessig, 75:15:10:1, v/v/v/v). Das Zielprodukt kann, wenn nötig, aus
Methanol umkristallisiert werden.
Beschaffenheit: Leicht bräunlicher amorpher Feststoff.
Schmelzbereich: 255-257 °C unter Zersetzung.
Ausbeute: 6 %. 1H-NMR (D2O): δ = 0,92 (m, 3×1H, 2×C(9)H2 und C(11)H2), 1,10 (m, 2×1H, 2×C(10)H2), 1,34
(m, 1H, C(11)H2), 1,47 (m, 2×1H, 2×C(10)H2), 1,66 (m, 2×1H, 2×C(9)H2), 3,17 (m, 1H, C(8)H),
6,83 (s, 1H, C(5)H), 6,91 (d, 1H, C(3)H, 3J = 6,0 Hz), 7,73 (d, 1H, C(2)H, 3J = 6,0 Hz). 13C-NMR (D2O): δ = 24,4 (2×C10), 25,2 (C11), 32,3 (2×C9), 50,1 (C8), 106,0 (C5), 110,0 (C3),
137,8 (C4), 147,6 (C2), 158,5 (C6), 160,5 (C7).
MIR (ṽ): 3245 (m, νN-H), 1638 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C12H16N6 + H]+: 244,1436; gefunden: 244,1435.
N-Benzyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXIX)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 750 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (4,1 mmol)
werden mit 3,6 mL Benzylamin (8 Äq., 33 mmol) bei 190 °C für 30 min erhitzt. Das nach
Ansäuern und Abfiltrieren anfallende Zielprodukt (mitunter längere Lagerung im Kühlschrank
notwendig) kann aus Methanol umkristallisiert werden.
Beschaffenheit: Leicht gelblicher amorpher Feststoff.
Schmelzbereich: 205-206 °C.
Ausbeute: 9 %.

Experimenteller Teil
Beschreibung der Synthesen | 147
1H-NMR (D2O, Na+-Salz): δ = 4,27 (s, 2H, C(8)H2), 6,95 (s, 1H, C(5)H), 7,01 (d, 1H, C(3)H,
3J = 5,6 Hz), 7,13 (m, 1H, C(12)H), 7,19 (m, 4H, 2×C(10)H und 2×C(11)H), 7,83 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 45,1 (C8), 105,6 (C5), 110,5 (C3), 127,1-128,6 (2×C10 und
2×C11 und 12), 138,0 (C4), 139,1 (C9), 147,7 (C2), 158,9 (C6), 160,5 (C7).
MIR (ṽ): 3322 (m, νN-H), 1615 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C13H12N6 + H]+: 252,1123; gefunden: 252,1119.
N-(Pyridin-3-yl-methyl)-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XL)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 2,5 mL β-Picolylamin (2-(Aminomethyl)pyridin, 9 Äq., 24 mmol) bei 190 °C für 30
min durchmengt. Nach Abkühlen auf Raumtemperatur wird im Teilvakuum eingeengt und der
Überstand chromatographisch aufgereinigt (adsorbiert an Kieselgur (aus ammoniakalischem
MeOH), LM: EtOAc/Aceton/MeOH/Ammoniak, 50:30:20:1, v/v/v/v). Das erhaltene Produkt wird
mit Salzsäure in sein Hydrochloridsalz überführt und mit reichlich heißem Isopropanol
nachgewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 240-242 °C.
Ausbeute: 10 %. 1H-NMR (D2O, Na+-Salz): δ = 4,22 (s, 2H, C(8)H2), 6,89 (s, 1H, C(5)H), 6,96 (d, 1H, C(3)H,
3J = 5,6 Hz), 7,10 (q, 1H, C(11)H, 3J = 4,8 und 8,0 Hz), 7,51 (d, 1H C(10)H, 3J = 8,0 Hz), 7,78
(d, 1H, C(2)H, 3J = 5,6 Hz), 8,15 (d, 1H, C(12)H, 3J = 4,4 Hz), 8,25 (s, 1H, C(13)H). 13C-NMR (D2O, Na+-Salz): δ = 42,4 (C8), 105,6 (C5), 110,6 (C3), 124,0 (C11), 135,2 (C9), 136,1
(C10), 137,9 (C4), 147,1 (C12), 147,4 (C13), 147,7 (C2), 158,5 (C6), 160,3 (C7).
MIR (ṽ): 3247 (m, νN-H), 1621 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C12H11N7 + H]+: 253,1076; gefunden: 253,1073.

Experimenteller Teil
148 | Beschreibung der Synthesen
N-(3-Methoxybenzyl)-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XLI)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 2,9 mL 3-Methoxybenzylamin (8 Äq., 23 mmol) bei 190 °C für 1 h erhitzt. Die
Präzipitation des Zielproduktes im Kühlschrank aus der salzsauren Lösung kann bis zu 14 Tage
betragen. Dieses Produkt wird aus einer Ethylacetat-Methanol-Mischung (EtOAc/MeOH, 70:30,
v/v) umkristallisiert.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 219-220 °C unter Zersetzung.
Ausbeute: 3 %. 1H-NMR (D2O, Na+-Salz): δ = 3,35 (s, 3H, C(15)H3), 4,03 (s, 2H, C(8)H2), 6,36 (d, 1H, C(14)H,
3J = 8,0 Hz), 6,52 (s, 1H, C(10)H), 6,56 (d, 1H, C(12)H, 3J = 7,6 Hz), 6,78 (s, 1H, C(5)H), 6,82
(m, 1H, C(13)H, 3J = 8,0 Hz), 6,86 (d, 1H, C(3)H, 3J = 4,8 Hz), 7,70 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (D2O, Na+-Salz): δ = 44,9 (C8), 54,9 (C15), 105,4 (C5), 110,4 (C3), 112,2 (C10), 112,4
(C12), 119,6 (C14), 129,6 (C13), 137,9 (C4), 140,8 (C9), 147,6 (C2), 158,7 (C6 und C11), 160,3
(C7).
MIR (ṽ): 3254 (m, νN-H), 1657 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H14N6O + H]+: 282,1229; gefunden: 282,1236.
N-(4-Chlorbenzyl)-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XLII)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
N NH
N
NN
HN
Cl
HCl
1
2
34
5
6
7
89
1011
12
XLII

Experimenteller Teil
Beschreibung der Synthesen | 149
werden mit 2,6 mL 4-Chlorbenzylamin (8 Äq., 21 mmol) für 30 min bei 190 °C durchmengt. Nach
Abfiltration des Hydrochloridsalzes wird mit Aceton und Isopropanol nachgewaschen.
Beschaffenheit: Leicht gelblicher amorpher Feststoff.
Schmelzbereich: 258-260 °C unter Zersetzung.
Ausbeute: 32 %. 1H-NMR (D2O, Na+-Salz): δ = 4,04 (s, 2H, C(8)H2), 6,80 (s, 1H, C(5)H), 6,86 (d, 2×2H, 2×C(10)H
und 2×C(11)H), 6,91 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,74 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 44,3 (C8), 105,4 (C5), 110,5 (C3), 128,2 (2×C10 und 2×C11),
131,8 (C12), 137,5 (C9), 137,9 (C4), 147,6 (C2), 158,6 (C6), 160,3 (C7).
MIR (ṽ): 1641 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C13H11N6Cl + H]+: 286,0734; gefunden: 286,0731.
N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 1,8 mL Ethylen-1,2-diamin (10 Äq., 27 mmol) für 25 min bei 190 °C erhitzt. Das
Zielprodukt wird nach Abfiltration mit reichlich Methanol gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 272-273 °C unter Zersetzung.
Ausbeute: 62 %. 1H-NMR (D2O, Na+-Salz): δ = 2,79 (t, 2H, C(9)H2, 3J = 6,0 Hz), 3,21 (t, 2H, C(8)H2, 3J = 6,0 Hz),
6,89 (s, 1H, C(5)H), 6,99 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,82 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 38,8 (C9), 39,0 (C8), 105,9 (C5), 110,6 (C3), 137,8 (C4), 146,8
(C2), 158,0 (C6), 160,0 (C7).
MIR (ṽ): 3428 (m, νN-H), 1643 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C8H11N7 + H]+: 205,1076; gefunden: 205,1069.
N NH
N
NN
HN
NH2
1
2
34
5
6
7
8
9
XLIII
HCl

Experimenteller Teil
150 | Beschreibung der Synthesen
N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV)
Es werden 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol) mit 2,2 mL Propan-
1,3-diamin (6 Äq., 16 mmol) im Rundkolben eingewogen und mit 0,5 mL Pyridin bei 135 °C für
10 h erhitzt. Anschließend wird das Pyridin im Teilvakuum entfernt, der Überstand mit Wasser
aufgenommen und so viel Salzsäure hinzugegeben bis ein pH-Wert von etwa 2 eingestellt ist.
Es erfolgt die Filtration des Präzipitats. Dieses wird in Ammoniak-Lösung gelöst und
anschließend erneut mit Salzsäure ausgefällt. Das Zielprodukt wird abfiltriert und mit Wasser
nachgewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 292-294 °C unter Zersetzung.
Ausbeute: 52 %. 1H-NMR (D2O, Na+-Salz): δ = 1,70 (m, 2H, C(9)H2, 3J = 6,8 und 7,2 Hz), 2,87 (t, 2H, C(10)H2,
3J = 7,2 Hz), 3,00 (t, 2H, C(8)H2, 3J = 6,8 Hz), 6,72 (s, 1H, C(5)H), 6,89 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,69 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (D2O, Na+-Salz): δ = 27,3 (C9), 37,4 (C10), 38,3 (C8), 105,5 (C5), 110,1 (C3), 137,5
(C4), 147,2 (C2), 158,7 (C6), 160,3 (C7).
MIR (ṽ): 3468 (m, νN-H), 1639 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C9H13N7 + H]+: 219,1232; gefunden: 219,1226.
N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]butan-1,4-diamin-Hydrochlorid (XLV)
Butan-1,4-diamin (4,7 g, 20 Äq., 54 mmol) wird bei etwa 50 °C geschmolzen und mit 500 mg
2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol) versetzt. Die Lösung wird für 48 h bei
100 °C gerührt. Anschließend erfolgt nach Abkühlen eine Verdünnung mit etwa 10 mL Wasser.

Experimenteller Teil
Beschreibung der Synthesen | 151
Durch Zugabe von Salzsäure wird das Zielprodukt ausgefällt und kann nach Abfilitration aus
einer Methanol-Lösung (MeOH/Wasser, 85:15, v/v) umkristallisiert werden.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 265-266 °C unter Zersetzung.
Ausbeute: 23 %. 1H-NMR (D2O, Na+-Salz): δ = 1,27 (m, 4H, C(9)H2 und C(10)H2), 2,41 (t, 2H, C(11)H2,
3J = 6,8 Hz), 3,55 (t, 2H, C(8)H2, 3J = 6,8 Hz), 6,85 (s, 1H, C(5)H), 6,98 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,77 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 25,8 (C10), 29,5 (C9), 40,4 (C11), 41,6 (C8), 105,4 (C5), 110,0
(C3), 137,7 (C4), 147,6 (C2), 159,1 (C6), 160,6 (C7).
MIR (ṽ): 3207 (m, νN-H), 1626 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C10H15N7 + H]+: 233,1389; gefunden: 233,1384.
2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethan-1-ol-Hydrochlorid (XLVI)
Für die Synthese werden 1,0 g 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (5,4 mmol) in 8,0 mL
2-Aminoethan-1-ol (25 Äq., 135 mmol) gelöst und für 16 h bei 120 °C im Rundkolben erhitzt.
Nach Abkühlen wird mit 75 mL Wasser verdünnt und Salzsäure bis zur deutlich sauren Reaktion
hinzugegeben. Das gewünschte Produkt wird anschließend abfiltriert und aus Methanol
umkristallisiert.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 244-245 °C unter Zersetzung.
Ausbeute: 75 %. 1H-NMR (D2O, Na+-Salz): δ = 3,21 (t, 2H, C(9)H2, 3J = 5,6 Hz), 3,62 (t, 2H, C(8)H2, 3J = 5,6 Hz),
6,86 (s, 1H, C(5)H), 6,94 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,77 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (D2O, Na+-Salz): δ = 43,5 (C9), 60,2 (C8), 105,6 (C5), 110,3 (C3), 137,8 (C4), 147,4
(C2), 159,0 (C6), 160,4 (C7).
MIR (ṽ): 3177 (m, νO-H), 1620 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C8H10N6O + H]+: 206,0916; gefunden: 206,0907.

Experimenteller Teil
152 | Beschreibung der Synthesen
3-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propan-1-ol-Hydrochlorid (XLVII)
Es werden 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol) in 7,5 mL
3-Aminopropan-1-ol (37 Äq., 99 mmol) gelöst und zusammen für 48 h bei 120 °C erhitzt.
Anschließend erfolgt eine Verdünnung des Reaktionsansatzes mit 75 mL Wasser. Durch
Ansäuern mit Salzsäure wird das Hydrochloridsalz des Zielprodukts erhalten (Kristallisation
kann einige Tage in Anspruch nehmen). Das Endprodukt wird abfiltriert und aus methanolischer
Lösung (MeOH/Wasser, 85:15, v/v) umkristallisiert.
Beschaffenheit: Bräunlicher amorpher Feststoff.
Schmelzbereich: 210-211 °C unter Zersetzung.
Ausbeute: 26 %. 1H-NMR (D2O, Na+-Salz): δ = 1,63 (m, 2H, C(9)H2, 3J = 6,8 und 6,4 Hz), 3,03 (t, 2H, C(8)H2,
3J = 6,8 Hz), 3,55 (t, 2H, C(10)H2, 3J = 6,4 Hz), 6,81 (s, 1H, C(5)H), 6,93 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,75 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 30,9 (C9), 38,4 (C8), 59,4 (C10), 105,4 (C5), 110,0 (C3), 137,7
(C4), 147,4 (C2), 159,0 (C6), 160,4 (C7).
MIR (ṽ): 3194 (m, νO-H), 1624 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C9H12N6O + H]+: 220,1073; gefunden: 220,1069.
1-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]-4-methylpiperazin-Hydrochlorid (XLVIII)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 7,2 mL Methylpiperazin (24 Äq., 65 mmol) und 1,8 mL Pyridin (22 mmol) bei 220 °C
N N
N
NN
HN
N
HCl
1
2
34
5
6
7
8
9
10
XLVIII

Experimenteller Teil
Beschreibung der Synthesen | 153
für 30 min erhitzt. Die resultierende Lösung wird im Teilvakuum eingeengt. Der Überstand wird
anschließend in salzsaurem Methanol aufgenommen, wobei eine Suspension erhalten wird.
Das Zielprodukt kann aus dieser durch Filtration isoliert werden. Es erfolgt eine
säulenchromatographische Aufarbeitung des vom Lösungsmittel befreiten Filtrats (adsorbiert an
Kieselgur (aus ammoniakalischem MeOH), LM: EtOAc/MeOH/Ammoniak, 75:25:1, v/v/v). Das
Produkt wird mit Hilfe von Salzsäure in das Hydrochloridsalz überführt und abschließend mit
Methanol gewaschen.
Beschaffenheit: Leicht gelblicher amorpher Feststoff.
Schmelzbereich: 260-262 °C unter Zersetzung.
Ausbeute: 23 %. 1H-NMR (D2O): δ = 2,97 (s, 3H, C(10)H3), 3,33 (t, 2×1H, 2×C(9)H2, 3J = 12,0 Hz), 3,66 (t, 2×1H,
2×C(8)H2, 3J = 12,0 Hz), 3,73 (d, 2×1H, 2×C(9)H2, 3J = 14,8 Hz), 4,33 (d, 2×1H, 2×C(8)H2, 3J = 14,8 Hz), 7,47 (d, 1H, C(3)H, 3J = 6,8 Hz), 7,77 (s, 1H, C(5)H), 8,04 (d, 1H, C(2)H, 3J = 6,8 Hz). 13C-NMR (D2O): δ = 43,0 (C10), 43,5 (2×C8), 52,0 (2×C9), 110,3 (C5), 111,8 (C3), 138,2 (C2),
141,0 (C4), 152,5 (C6), 156,6 (C7).
MIR (ṽ): 1611 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C11H15N7 + H]+: 245,1389; gefunden: 245,1377.
2-{4-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]piperazin-1-yl}ethan-1-ol-Hydrochlorid (XLIX)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 2 mL 1-(2-Hydroxyethyl)piperazin (6 Äq., 17 mmol) bei 220 °C für 45 min
durchmengt. Nach Ansäuern des Reaktionsansatzes mit Salzsäure wird dieser im Teilvakuum
eingeengt. Durch Zugabe von Methanol und kräftiges Durchmischen wird eine Suspension
erhalten, aus welcher durch Abfiltrieren das Rohprodukt gewonnen wird. Der resultierende
Feststoff wird chromatographisch gereinigt (adsorbiert an Kieselgur (aus Wasser),
LM: EtOAc/MeOH/Ammoniak, 70:30:1, v/v/v) und abschließend, nach Überführung in das
Hydrochloridsalz, mit Aceton und Ethanol gewaschen.

Experimenteller Teil
154 | Beschreibung der Synthesen
Beschaffenheit: Leicht gelblicher amorpher Feststoff.
Schmelzbereich: 233-235 °C unter Zersetzung.
Ausbeute: 63 %. 1H-NMR (DMSO-d6): δ = 3,24 (s, 2×1H und 2H, 2×C(9)H2 und C(10)H2), 3,57 (m, 2×1H,
2×C(8)H2), 3,63 (d, 2×1H, 2×C(9)H2, 3J = 12,0 Hz), 3,83 (t, 2H, C(11)H2, 3J = 5,2 Hz), 4,56 (d,
2×1H, 2×C(8)H2, 3J = 13,6 Hz), 7,45 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,98 (s, 1H, C(5)H), 8,34 (d,
1H, C(2)H, 3J = 5,6 Hz), 10,92 (s, 1H, N(12)H+). 13C-NMR (DMSO-d6): δ = 42,3 (2×C8), 50,6 (2×C9), 55,0 (C11), 57,8 (C10), 106,8 (C5), 110,8
(C3), 134,7 (C7), 146,3 (C2), 156,8 (C6).
MIR (ṽ): 3308 (m, νO-H), 1646 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C12H17N7O + H]+: 275,1495; gefunden: 275,1493.
N-(3-Morpholinopropyl)-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (L)
Die Synthese erfolgt analog zu N-Ethyl-4-(1H-tetrazol-5-yl)pyridin-2-amin-Hydrochlorid (XXXVI)
mit folgenden Änderungen: 500 mg 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (2,7 mmol)
werden mit 3,6 mL 3-Morpholinopropylamin (9 Äq., 24 mmol) für 1 h bei 190 °C erhitzt.
Anschließend wird mit 40 mL Ethanol verdünnt und mit Salzsäure angesäuert. Das Präzipitat
entspricht dem Zielprodukt und kann nach Abfiltrieren aus methanolischer Lösung
(MeOH/Wasser, 85:15, v/v) umkristallisiert werden.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 250-251°C unter Zersetzung.
Ausbeute: 48 %. 1H-NMR (DMSO-d6): δ = 2,14 (t, 2H, C(9)H2, 3J = 7,6 Hz), 3,10 (s, 2×1H, 2×C(11)H2), 3,27 (t,
2H, C(10)H2, 3J = 7,6 Hz), 3,42 (s, 2×1H, 2×C(11)H2), 3,61 (s, 2H, C(8)H2), 3,92 (s, 2×2H,
2×C(12)H2), 7,46 (d, 1H, C(3)H, 3J = 6,4 Hz), 7,86 (s, 1H, C(5)H), 8,13 (d, 1H, C(2)H, 3J = 6,8 Hz), 9,49 (s, 1H, N(13)H), 11,31 (s, 1H, N(14)H+). 13C-NMR (DMSO-d6): δ = 22,2 (C9), 39,4 (C8), 51,1 (2×C11), 53,4 (C10), 63,1 (2×C12), 109,1
(C5), 110,6 (C3), 137,5 (C4), 138,2 (C2), 153,3 (C6), 155,3 (C7).

Experimenteller Teil
Beschreibung der Synthesen | 155
MIR (ṽ): 3523 (m, νN-H), 1626 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C13H19N7O + H]+: 289,1651; gefunden: 289,1646.
2-Mercaptoisonicotinnitril (LII)
Es werden 500 mg (3,6 mmol) 2-Chlorisonicotinnitril (XXXIV) mit 410 mg (1,5 Äq., 5,4 mmol)
Thioharnstoff eingewogen und in 7,5 mL Ethanol gelöst. Der Ansatz wird bei 135 °C für 60 min
im Mikrowellenreaktor erhitzt und anschließend abgekühlt. Das Zielprodukt kristallisiert aus der
erkaltenden Reaktionslösung aus, wird anschließend filtriert und mit 50 mL Toluen in drei
Portionen gewaschen. Der Feststoff kann aus methanolischer Lösung (MeOH/Wasser, 90:10,
v/v) umkristallisiert werden.
Beschaffenheit: Intensiv gelbe wollige Nadeln.
Schmelzbereich: 221-223 °C unter Zersetzung.
Ausbeute: 65 %. 1H-NMR (DMSO-d6): δ = 6,99 (d, 1H, C(3)H, 3J = 6,0 Hz), 7,71 (s, 1H, C(5)H), 7,80 (d, 1H,
C(2)H, 3J = 6,0 Hz), 14,04 (s, 1H, N(1)H). 13C-NMR (DMSO-d6): δ = 112,1 (C3), 116,2 (C4), 120,0 (C7), 137,6 (C5), 139,7 (C2), 178,4
(C6).
MIR (ṽ): 3147 (m, νN-H), 2236 (s, νC≡N) cm−1.
HRMS (ESI, m/z), berechnet [C6H4N2S − H] −: 136,0095; gefunden: 136,0096.
Anmerkung: Die Daten deuten darauf hin, dass es sich hierbei um das entsprechende Thioamid-
Tautomer handelt. Dafür sprechen der hohe Schmelzpunkt und vor allem die starke
Entschirmung des Signals des Schwefelatom-tragenden Kohlenstoffatoms im 13C-NMR-
Spektrum, welche für Carbonyl-Kohlenstoffatome charakteristisch ist.

Experimenteller Teil
156 | Beschreibung der Synthesen
2-(Benzylthio)isonicotinnitril (LIII)
2-Mercaptoisonicotinnitril (LII) (500 mg, 3,6 mmol) wird in 10 mL ACN gelöst. Dazu werden
620 µL Triethylamin (1,2 Äq., 4,5 mmol) pipettiert. Anschließend werden unter kräftigem Rühren
510 µL Benzylbromid (1,2 Äq., 4,3 mmol), welches in 5 mL ACN vorgelöst ist, hinzugegeben.
Nach mindestens 4 h Rühren bei Raumtemperatur wird der Ansatz im Teilvakuum eingeengt,
der Rückstand in Ethylacetat aufgenommen, filtriert und wieder eingeengt. Es folgt Waschen
mit Wasser und nach vollständiger Trocknung Umkristallisation aus n-Hexan.
Beschaffenheit: Farblose Nadeln.
Schmelzbereich: 70-71 °C (69-71 °C192).
Ausbeute: 46 %. 1H-NMR (DMSO-d6): δ = 4,46 (s, 2H, C(8)H2), 7,23-7,43 (m, 5H, 2×C(10)H und 2×C(11)H und
C(12)H), 7,55 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,86 (s, 1H, C(5)H), 8,67 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 33,3 (C8), 116,4 (C4), 119,8 (C7), 121,0 (C5), 123,4 (C3), 127,2 (C12),
128,4-128,9 (2×C10 und 2×C11), 137,2 (C9), 150,4 (C2), 160,2 (C6).
MIR (ṽ): 2238 (s, νC≡N) cm−1.
2-(Benzylthio)-4-(1H-tetrazol-5-yl)pyridin (LIV)
Es werden 400 mg (1,7 mmol) 2-(Benzylthio)isonicotinnitril (LIII) mit 150 mg NaN3 (1,3 Äq.,
2,2 mmol) und 315 mg TEA∙HCl (1,3 Äq., 2,2 mmol) eingewogen und in 10 mL Toluen
suspendiert. Der Reaktionsansatz wird bei 100 °C für etwa 17 h mit aufgesetztem N2-Ballon
gerührt. Anschließend wird das Toluen im Teilvakuum entfernt. Der Überstand wird in
verdünnter Natronlauge aufgenommen und zwei Mal mit je 30 mL EtOAc gewaschen. Die
wässrige Phase wird dann mit Salzsäure auf einen pH-Wert von etwa 2 eingestellt, wobei das

Experimenteller Teil
Beschreibung der Synthesen | 157
Zielprodukt auskristallisiert. Anschließend erfolgt eine Aufreinigung mittels präparativer HPLC
(gelöst in MeOH/DMSO, 80:20, v/v; A: H2O und B: MeOH, Gradient, 0–7 min (B) 75 %, –9 min
(B) 100%, –15 min (B) 100 %, –16 min (B) 75%).
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 160-161 °C.
Ausbeute: 75 %. 1H-NMR (DMSO-d6): δ = 4,50 (s, 2H, C(8)H2), 7,23-7,45 (m, 5H, 2×C(10)H und 2×C(11)H und
C(14)H), 7,72 (d, 1H, C(3)H, 3J = 4,8 Hz), 7,88 (s, 1H, C(5)H), 8,70 (d, 1H, C(2)H, 3J = 4,8 Hz). 13C-NMR (DMSO-d6): δ = 33,4 (C8), 117,0 (C3), 118,3 (C5), 127,1 (C12), 128,4-128,9 (2×C10
und 2×C11), 132,6 (C4), 137,6 (C9), 150,6 (C2), 154,8 (C7), 159,7 (C6).
MIR (ṽ): 1607 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C13H11N5S + H]+: 269,0735; gefunden: 269,0729.
N-⟨2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethyl⟩benzamid-Hydrochlorid (LVII)
Es werden 350 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII)
(1,5 mmol) und 220 μL Benzoylchlorid (1,2 Äq., 1,9 mmol) unter Eiskühlung in 2 mol/L
Natronlauge gelöst und in der Kälte gerührt. Nach 3 h werden weitere 40 μL Benzoylchlorid
(0,2 Äq.) hinzugegeben. Die Reaktionslösung wird für insgesamt 24 h bei Raumtemperatur
gerührt. Anschließend erfolgt die Zugabe von Salzsäure bis zur sauren Reaktion und wenigen
mL Ethylacetat. Das ausgefallene Hydrochloridsalz wird abfiltriert und abschließend mit
Ethylacetat, Methanol und Aceton gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 248-250 °C unter Zersetzung.
Ausbeute: 53 %. 1H-NMR (D2O, Na+-Salz): δ = 3,37 (m, 4H, C(8)H2 und C(9)H2), 6,89-6,90 (s, 2H, C(5)H und
C(3)H), 7,13 (m, 2H, 2×C(11)H), 7,27 (t, 1H, C(13)H, 3J = 7,6 Hz), 7,33 (d, 2H, 2×C(12)H, 3J = 7,6 Hz), 7,77 (d, 1H, C(2)H, 3J = 6,0 Hz).

Experimenteller Teil
158 | Beschreibung der Synthesen
13C-NMR (D2O, Na+-Salz): δ = 40,2 (C8), 40,7 (C9), 105,33 (C5), 110,3 (C3), 126,7 (2×C12),
128,3 (2×C11), 131,8 (C13), 133,1 (C10), 138,0 (C4), 147,6 (C2), 159,2 (C6), 160,3 (C7), 170,8
(C14).
MIR (ṽ): 3317 (m, vN-H), 1659 (s, vC=O), 1625 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C15H15N7O + H]+: 309,1338; gefunden: 309,1330.
N-⟨2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethyl⟩buttersäureamid-Hydrochlorid (LVIII)
N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII) (500 mg, 2,1 mmol)
wird unter Eiskühlung in 2 mol/L Natronlauge gelöst. Anschließend werden 250 μL Butyrylchlorid
(1,1 Äq., 2,3 mmol) in die weiterhin gut gekühlte Lösung hinzugetropft. Die Reaktion findet bei
Raumtemperatur für weitere 7 h statt. Danach werden erneut 120 μL Butyrylchlorid (0,5 Äq.)
hinzugegeben. Nachdem der Ansatz insgesamt 21 h reagieren konnte, wird konzentrierte
Salzsäure bis zur sauren Reaktion ergänzt. Die Kristallisationsdauer des Hydrochloridsalzes
kann bis zu 3 Wochen im Kühlschrank betragen. Das Präzipitat wird abfiltriert und mit
Ethylacetat gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 244-246 °C unter Zersetzung.
Ausbeute: 6 %. 1H-NMR (D2O, Na+-Salz): δ = 0,53 (t, 3H, C(12)H3, 3J = 7,2 Hz), 1,22 (q, 2H, C(11)H2,
3J = 7,2 und 7,6 Hz), 1,87 (t, 2H, C(10)H2, 3J = 7,6 Hz), 3,15 (s, 4H, C(8)H2 und C(9)H2), 6,81
(s, 1H, C(5)H), 6,89 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,73 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (D2O, Na+-Salz): δ = 12,6 (C12), 18,7 (C11), 37,6 (C10), 38,9 (C9), 40,6 (C8), 105,3
(C5), 110,3 (C3), 137,9 (C4), 147,6 (C2), 159,0 (C6), 160,5 (C7), 177,2 (C13).
MIR (ṽ): 1672 (s, vC=O), 1625 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C12H17N7O + H]+: 275,1495; gefunden: 275,1497.

Experimenteller Teil
Beschreibung der Synthesen | 159
N-⟨2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethyl⟩methansulfonamid-Hydrochlorid (LIX)
Es werden 500 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII)
(2,1 mmol) unter Eiskühlung in 2 mol/L Natronlauge gelöst und anschließend mit 220 μL
Methansulfonylchlorid (1,4 Äq., 2,9 mmol) in 5 mL 2 mol/L Natronlauge versetzt. Der
Reaktionsansatz wird für 2 h in der Kälte und 4 h bei Raumtemperatur gerührt. Danach werden
erneut 315 µL Methansulfonylchlorid (2,0 Äq.) zugetropft und für weitere 24 h bei
Raumtemperatur gerührt. Durch Ansäuern mit Salzsäure erfolgt die Kristallisation des
Hydrochloridsalzes, welches erst abfiltriert, dann mit einem Wasser-Methanol-Gemisch
(Wasser/MeOH, 80:20, v/v) gewaschen und abschließend aus Wasser umkristallisiert wird.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 262-263 °C unter Zersetzung.
Ausbeute: 15 %. 1H-NMR (D2O, Na+-Salz): δ = 2,66 (s, 3H, C(10)H3), 3,01 (t, 2H, C(8)H2, 3J = 6,4 Hz), 3,24 (t,
2H, C(9)H2, 3J = 6,4 Hz), 7,01 (s, 1H, C(5)H), 7,06 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,91 (d, 1H,
C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 38,0 (C10), 43,5 (C8), 44,0 (C9), 105,6 (C5), 110,3 (C3), 138,0
(C4), 147,8 (C2), 159,2 (C6), 160,6 (C7).
MIR (ṽ): 3238 (m, vN-H), 1637 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C9H13N7O2S + H]+: 283,0851; gefunden: 283,0846.
Ethyl-⟨2-{[4-(1H-tetrazol-5-yl)pyridin-2-yl]amino}ethyl⟩carbamat-Hydrochlorid (LX)
Es werden 200 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]ethan-1,2-diamin-Hydrochlorid (XLIII)
(0,9 mmol) in 20 mL ACN suspendiert und mit 2 mL Natronlauge (2 mol/L) versetzt. Bei

Experimenteller Teil
160 | Beschreibung der Synthesen
Raumtemperatur werden 120 μL und nach 1 h erneut 50 μL Chlorameisensäureethylester
(zusammen 2,0 Äq., 1,8 mmol) hinzugegeben. Nach insgesamt 2 h wird der Reaktionsansatz
mit konzentrierter Salzsäure angesäuert, der ausfallende Feststoff abfiltriert und verworfen. Das
Filtrat wird im Teilvakuum eingeengt und der Überstand aus wässrigem Methanol
(MeOH/Wasser, 90:10, v/v) umkristallisiert.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 222-223 °C unter Zersetzung.
Ausbeute: 56 %. 1H-NMR (DMSO-d6): δ = 1,12 (t, 3H, C(11)H3, 3J = 7,2 Hz), 3,29 (q, 2H, C(8)H2, 3J = 6,0 Hz),
3,53 (d, 2H, C(9)H2, 3J = 5,2 Hz), 3,98 (q, 2H, C(10)H2, 3J = 7,2 Hz), 7,26 (t, 1H, N(13)H, 3J = 5,2 Hz), 7,40 (d, 1H, C(3)H, 3J = 6,4 Hz), 7,73 (s, 1H, C(5)H), 8,12 (d, 1H, C(2)H, 3J = 6,4 Hz), 9,00 (s, 1H, N(12)H). 13C-NMR (DMSO-d6): δ = 14,6 (C11), 39,0 (C9), 41,8 (C8), 59,8 (C10), 109,0 (C5), 110,1 (C3),
137,4 (C4), 138,9 (C2), 153,9 (C6), 155,5 (C7), 156,4 (C14).
MIR (ṽ): 3293 (m, vN-H), 1699 (s, vC=O), 1627 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C11H15N7O2 + H]+: 277,1287; gefunden: 277,1290.
N-⟨3-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩acetamid-Hydrochlorid (LXI)
Es werden 200 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV)
(0,8 mmol) in 20 mL ACN suspendiert. Zur Suspension werden 2 mL verdünnte Natronlauge
(2 mol/L) hinzugegeben, anschließend 110 μL Acetylchlorid (2,0 Äq., 1,6 mmol) ergänzt. Nach
3 h werden erneut 65 µL Acetylchlorid (1,2 Äq.) hinzugegeben, nach weiteren 3 h werden
nochmals 2 mL verdünnte Natronlauge und 65 µL Acetylchlorid addiert. Nach insgesamt 24 h
wird das Reaktionsgemisch im Teilvakuum eingeengt und der Überstand chromatographisch
aufgearbeitet (adsorbiert an Kieselgur (aus ammoniakalischem Methanol), LM:
EtOAc/Aceton/MeOH/Ammoniak, 60:20:20:1, v/v/v/v). Die Produktfraktionen werden mit
Salzsäure in ihr Hydrochloridsalz überführt und aus Methanol umkristallisiert.
Beschaffenheit: Farbloser amorpher Feststoff.
N NH
N
NN
HN
NH
O
HCl
1
2
34
5
6
7
8
9
10
1112
LXI

Experimenteller Teil
Beschreibung der Synthesen | 161
Schmelzbereich: 238-240 °C unter Zersetzung.
Ausbeute: 31 %. 1H-NMR (D2O, Na+-Salz): δ = 1,56 (t, 2H, C(9)H2, 3J = 6,8 Hz), 1,82 (s, 3H, C(11)H3), 3,01 (t,
2H, C(10)H2, 3J = 7,2 Hz), 3,06 (t, 2H, C(8)H2, 3J = 6,8 Hz), 6,84 (s, 1H, C(5)H), 6,95 (d, 1H,
C(3)H, 3J = 5,6 Hz), 7,77 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 21,8 (C11), 27,8 (C9), 37,2 (C10), 39,0 (C8), 105,4 (C5), 110,1
(C3), 137,8 (C4), 147,6 (C2), 159,0 (C6), 160,5 (C7), 173,9 (C12).
MIR (ṽ): 1666 (m, vC=O), 1620 (s, νC=N) cm−1.
N-⟨3-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩pivalinamid-Hydrochlorid (LXII)
N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV) (250 mg, 1,0 mmol)
wird in 20 mL ACN suspendiert, bevor 3 mL verdünnte Natronlauge (2 mol/L) hinzugegeben
werden. Im Anschluss werden 145 μL Pivalinsäurechlorid (1,2 Äq., 1,2 mmol) zugetropft und die
Reaktion für 4 h bei Raumtemperatur gerührt. Das Reaktionsgemisch wird mit Salzsäure
versetzt und für 72 h im Kühlschrank zur Kristallisation belassen. Die Umkristallisation des
ausgefallenen Hydrochloridsalzes erfolgt aus wässrigem Methanol (MeOH/Wasser, 90:10, v/v).
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 204-205 °C unter Zersetzung.
Ausbeute: 27 %. 1H-NMR (D2O, Na+-Salz): δ = 0,71 (s, 9H, C12(H)3 und C(13)H3 und C(14)H3), 1,33 (m, 2H,
C(9)H2, 3J = 6,4 und 6,8 Hz), 2,78 (t, 2H, C(8)H2, 3J = 6,8 Hz), 2,84 (t, 2H, C(10)H2, 3J = 6,8 Hz),
6,64 (s, 1H, C(5)H), 6,74 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,55 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 26,4 (C12 und C13 und C14), 27,9 (C9), 37,0 (C10), 38,1 (C11),
38,9 (C8), 105,2 (C5), 109,9 (C3), 137,7 (C4), 147,5 (C2), 158,8 (C6), 160,3 (C7), 181,9 (C15).
MIR (ṽ): 1657 (m, vC=O), 1625 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H21N7O + H]+: 303,1808; gefunden: 303,1806.

Experimenteller Teil
162 | Beschreibung der Synthesen
N-⟨3-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩-4-methoxybenzamid-Hydrochlorid
(LXIII)
Es werden 500 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV)
(2,0 mmol) in 15 mL ACN suspendiert und mit 4 mL verdünnter Natronlauge (2 mol/L) versetzt.
Dann werden 470 mg 4-Methoxybenzoylchlorid (1,5 Äq., 3,0 mmol) in 15 mL Tetrahydrofuran
gelöst und zur ersten Lösung hinzugetropft. Der Ansatz rührt für etwa 3,5 h bei Raumtemperatur.
Die ACN/THF-Phase wird abgenommen, mit verdünnter Salzsäure angesäuert und am
Rotationsverdampfer eingeengt. Abschließend erfolgt eine säulenchromatographische
Aufarbeitung (adsorbiert an Kieselgur (aus wässriger Ammoniaklösung), LM:
EtOAc/MeOH/Ammoniak, 75:25:1, v/v/v), die Überführung in das Hydrochloridsalz und
Waschen mit Ethylacetat und Ethanol.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 236-237 °C unter Zersetzung.
Ausbeute: 17 %. 1H-NMR (D2O, Na+-Salz): δ = 1,56 (t, 2H, C(9)H2, 3J = 6,4 Hz), 2,98 (t, 2H, C(8)H2, 3J = 6,4 Hz),
3,10 (t, 2H, C(10)H2, 3J = 6,4 Hz), 3,34 (s, 3H, C(15)H3), 6,33 (d, 2H, 2×C(13)H, 3J = 8,4 Hz),
6,58 (s, 1H, C(5)H), 6,74 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,13 (d, 2H, 2×C(12)H, 3J = 8,8 Hz), 7,61
(d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 27,5 (C9), 37,9 (C10), 39,3 (C8), 55,0 (C15), 105,6 (C5), 109,8
(C3), 113,3 (2×C13), 125,3 (C11), 128,4 (2×C12), 137,5 (C4), 147,2 (C2), 158,7 (C6), 160,1
(C7), 161,3 (C14), 169,1 (C16).
MIR (ṽ): 1661 (m, vC=O), 1620 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C17H19N7O2 + H]+: 353,1600; gefunden: 353,1598.

Experimenteller Teil
Beschreibung der Synthesen | 163
N-⟨3-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩-4-brombenzamid-Hydrochlorid
(LXIV)
N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV) (400 mg, 1,6 mmol)
wird in 15 mL Tetrahydrofuran suspendiert und mit 3 mL verdünnter Natronlauge (2 mol/L)
versetzt. Dann werden 525 mg 4-Brombenzoylchlorid (1,5 Äq., 2,4 mmol), gelöst in 10 mL
Tetrahydrofuran, und weitere 2 mL verdünnte Natronlauge hinzugegeben. Nach 2,5 h Rühren
bei Raumtemperatur wird die organische Phase abgenommen, mit konzentrierter Salzsäure
versetzt und am Rotationsverdampfer eingeengt. Der Überstand wird chromatographisch
aufgereinigt (adsorbiert an Kieselgur (aus ammoniakalischem Methanol), LM:
EtOAc/MeOH/Ammoniak, 85:15:1, v/v/v). Nach Überführen in das Hydrochloridsalz erfolgt
abschließend ein Waschgang mit Ethanol und Diethylether.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 240-241 °C unter Zersetzung.
Ausbeute: 82 %. 1H-NMR (DMSO-d6): δ = 1,90 (t, 2H, C(9)H2, 3J = 6,8 Hz), 3,38 (d, 2H, C(10)H2, 3J = 6,4 Hz),
3,43 (d, 2H, C(8)H2, 3J = 6,4 Hz), 7,35 (d, 1H, C(3)H, 3J = 6,4 Hz), 7,65 (s, 1H, C(5)H), 7,66 (d,
2H, 2×C(13)H, 3J = 8,8 Hz), 7,84 (d, 2H, 2×C(12)H, 3J = 8,4 Hz), 8,11 (d, 1H, C(2)H, 3J = 6,4 Hz),
8,71 (t, 1H, N(17)H, 3J = 5,6 Hz), 8,84 (s, 1H, N(16)H). 13C-NMR (DMSO-d6): δ = 27,9 (C9), 36,8 (C10), 38,9 (C8), 108,8 (C3), 109,0 (C5), 124,8 (C14),
129,3 (2×C12), 131,2 (2×C13), 133,5 (C11), 137,2 (C4), 139,7 (C2), 154,2 (C6), 155,7 (C7),
165,4 (C15).
MIR (ṽ): 1660 (s, vC=O), 1621 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C16H16N7OBr + H]+: 401,0600; gefunden: 401,0580.

Experimenteller Teil
164 | Beschreibung der Synthesen
N-⟨3-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩-4-methylbenzensulfonamid-
Hydrochlorid (LXV)
Für die Synthese werden 500 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-
Hydrochlorid (XLIV) (2,0 mmol) in 20 mL ACN suspendiert und 4 mL verdünnte Natronlauge
(2 mol/L) hinzugegeben. 570 mg para-Toluolsulfonsäurechlorid (1,5 Äq., 3,0 mmol) werden
ebenfalls in 20 mL ACN gelöst und in kleinen Portionen der ersten Lösung hinzugetropft. Nach
30 min Reaktionszeit bei Raumtemperatur wird der Reaktionsansatz mit Salzsäure angesäuert
und anschließend am Rotationsverdampfer eingeengt. Der Überstand wird in sehr wenig
Wasser aufgeschlämmt, abfiltriert und mit Ethylacetat gewaschen. Die abschließende
Aufreinigung erfolgt mit Hilfe der präparativen HPLC (gelöst in ammoniakalischem MeOH/H2O,
50:50, v/v; A: H2O und B: ACN, isokratisch, 45 % B).
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 221-223 °C unter Zersetzung.
Ausbeute: 17 %. 1H-NMR (D2O, Na+-Salz): δ = 1,49 (t, 2H, C(9)H2, 3J = 6,4 Hz), 1,78 (s, 3H, C(15)H3), 2,69 (t,
2H, C(10)H2, 3J = 6,8 Hz), 2,95 (t, 2H, C(8)H2, 3J = 6,0 Hz), 6,68 (s, 1H, C(5)H), 6,78 (d, 2H,
2×C(13)H, 3J = 8,0 Hz), 7,00 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,32 (d, 2H, 2×C(12)H, 3J = 8,0 Hz),
7,79 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 20,1 (C15), 29,9 (C9), 38,9 (C8), 42,1 (C10), 105,7 (C5), 109,9
(C3), 126,1 (2×C12), 128,9 (2×C13), 137,6 (C4), 139,3 (C11), 141,6 (C14), 147,4 (C2), 159,0
(C7), 160,6 (C6).
MIR (ṽ): 1639 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C16H19N7O2S + H]+: 373,1321; gefunden: 373,1312.
N NH
N
NN
HN
NH
S
1
2
34
5
6
7
8
9
10 12
1113
14
15LXV
O O
HCl

Experimenteller Teil
Beschreibung der Synthesen | 165
Isobutyl-⟨3-{[4-(1H-tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩carbamat-Hydrochlorid (LXVI)
Es werden 350 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV)
(1,4 mmol) in 20 mL ACN suspendiert und 3 mL verdünnte Natronlauge (2 mol/L) hinzugegeben.
Anschließend werden 290 μL Isobutylchlorformiat (1,6 Äq., 2,2 mmol) addiert und bei
Raumtemperatur gerührt. Nach 1 h werden weitere 200 μL (0,7 Äq., 1,0 mmol) ergänzt.
Insgesamt rührt die Reaktionslösung 2 h, wird mit Salzsäure versetzt und am
Rotationsverdampfer eingeengt. Der zurückbleibende Feststoff wird aus einer Isopropanol-
Methanol-Mischung (IPA/MeOH, 50:50, v/v) umkristallisiert, wobei der in der Siedehitze nicht
lösliche Überstand abfiltriert und verworfen wird. Aus dem Filtrat kristallisiert beim Abkühlen das
Zielprodukt aus.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 248-249 °C unter Zersetzung.
Ausbeute: 33 %. 1H-NMR (DMSO-d6): δ = 0,87 (d, 6H, C(13)H3 und C(14)H3, 3J = 6,8 Hz), 1,72 (m, 2H, C(9)H2,
3J = 6,8 Hz), 1,82 (m, 1H, C(12)H, 3J = 6,8 Hz), 3,09 (q, 2H, C(10)H2, 3J = 6,4 Hz), 3,31 (d, 2H,
C(8)H2, 3J = 6,8 Hz), 3,72 (d, 2H, C(11)H2, 3J = 6,8 Hz), 7,14 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,15
(s, 1H, N(16)H), 7,26 (s, 1H, C(5)H), 7,48 (s, 1H, N(15)H), 8,09 (d, 1H, C(2)H, 3J = 6,0 Hz). 13C-NMR (DMSO-d6): δ = 18,9 (C13 und C14), 27,6 (C12), 28,9 (C9), 38,0 (C10), 38,7 (C8),
69,7 (C11), 105,8 (C5), 108,7 (C3), 135,9 (C4), 145,2 (C2), 156,0 (C7), 156,5 (C17), 157,4 (C6).
MIR (ṽ): 1685 (s, vC=O), 1665 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H21N7O2 + H]+: 319,1757; gefunden: 319,1756.
Benzyl-⟨3-{[4-(1H-tetrazol-5-yl)pyridin-2-yl]amino}propyl⟩carbamat-Hydrochlorid (LXVII)

Experimenteller Teil
166 | Beschreibung der Synthesen
Es werden 400 mg N-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]propan-1,3-diamin-Hydrochlorid (XLIV)
(1,6 mmol) in 15 mL ACN suspendiert und mit 4 mL verdünnter Natronlauge (2 mol/L) versetzt.
Anschließend werden 315 μL Chlorameisensäurebenzylester (1,4 Äq., 2,2 mmol)
hinzugegeben. Nach 22 h bei Raumtemperatur wird Salzsäure addiert und am
Rotationsverdampfer eingeengt. Der Überstand wird aus Ethanol umkristallisiert (Heißfiltration).
Abschließend erfolgt die Aufreinigung mit Hilfe der präparativen HPLC (gelöst in MeOH/DMSO,
90:10, v/v; A: H2O und B: MeOH, Gradient, 0–2 min (B) 35 %, –5 min (B) 60 %, –6 min (B) 100
%, –14 min (B) 100 %, –14,5 min (B) 35 %, –20min (B) 35 %).
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 280-281 °C unter Zersetzung.
Ausbeute: 39 %. 1H-NMR (DMSO-d6): δ = 1,73 (m, 2H, C(9)H2, 3J = 6,8 Hz), 3,12 (d, 2H, C(10)H2, 3J = 6,8 Hz),
3,32 (d, 2H, C(8)H2, 3J = 6,8 Hz), 5,02 (s, 2H, C(11)H2), 7,14 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,26
(s, 1H, C(5)H), 7,35 (m, 5H, 2×C(13)H und 2×C14H und C15H), 7,46 (s, 2×1H, N(17)H und
N(16)H), 8,09 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (DMSO-d6): δ = 28,9 (C9), 38,2 (C10), 38,7 (C8), 65,2 (C11), 105,8 (C5), 108,7 (C3),
127,7-128,3 (2×C13 und 2×C14 und C15), 135,8 (C4), 137,2 (C12), 145,3 (C2), 156,0 (C7),
156,2 (C18), 157,5 (C6).
MIR (ṽ): 1684 (s, vC=O), 1661 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C17H19N7O2 + H]+: 353,1600; gefunden: 353,1591.
2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethylbenzoat-Hydrochlorid (LXVIII)
Für die Synthese werden 500 mg 2-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]amino}ethan-1-ol-
Hydrochlorid (XLVI) (2,1 mmol) in 2 mol/L Kalilauge gelöst und anschließend mit 1 mL in
Kalilauge verdünntem Benzoylchlorid (4,0 Äq., 8,4 mmol) versetzt. Der Ansatz wird bei
Raumtemperatur über Nacht gerührt. Durch Ansäuern mit Salzsäure wird das Hydrochloridsalz
ausgefällt, anschließend abfiltriert und mit Toluen gewaschen. Die abschließende
Umkristallisation erfolgt aus Methanol.
N NH
N
NN
HN
O
O1
2
34
5
6
7
8
9
10
1112
13
14
LXVIII
HCl

Experimenteller Teil
Beschreibung der Synthesen | 167
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: ab 198 °C unter Zersetzung.
Ausbeute: 3 %. 1H-NMR (D2O, Na+-Salz): δ = 3,27 (t, 2H, C(8)H2, 3J = 5,2 Hz), 3,65 (t, 2H, C(9)H2, 3J = 5,2 Hz),
6,94 (s, 1H, C(5)H), 7,01 (d, 1H, C(3)H, 3J = 5,2 Hz), 7,36 (t, 2H, 2×C(12)H, 3J = 5,6 Hz), 7,41
(t, 1H, C(13)H, 3J = 6,0 Hz), 7,76 (d, 2H, 2×C(11)H, 3J = 6,0 Hz) 7,84 (d, 1H, C(2)H, 3J = 5,6 Hz). 13C-NMR (D2O, Na+-Salz): δ = 43,5 (C8), 60,3 (C9), 105,7 (C5), 110,3 (C3), 128,3 (2×C12),
128,8 (2×C11), 131,2 (C13), 136,2 (C10), 137,9 (C4), 147,5 (C2), 159,1 (C6), 160,5 (C7), 175,7
(C14).
MIR (ṽ): 1715 (s, vC=O), 1624 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C15H15N7O + H]+: 309,1338; gefunden: 309,1330.
4-Cyanpicolinsäureethylester (LXXIII)
Für die Synthese werden 3,1 g Isonicotinnitril (30 mmol) (LXXII) zusammen mit 25 g
FeSO4∙7H2O (90 mmol) in ein hohes Becherglas eingewogen. Dazu werden 150 mL
Dichlormethan gegeben und unter Kühlung (–5 °C, NaCl/Eis) gerührt. Der Suspension werden
weiterhin 9 g H2SO4 (90 mmol) und 24 g Wasser hinzugefügt. In einem zweiten Gefäß erfolgt
die Vermengung von 16 g Ethylpyruvat (135 mmol) mit 10 g H2O2 (90 mmol, 30%ig in Wasser),
welches langsam und ebenfalls unter Eiskühlung (–5 °C, NaCl/Eis) hinzugegeben wird. Die
letztere Lösung wird dann der ersten unter kräftigem Rühren hinzugetropft und nach kompletter
Zugabe für etwa 15 min unter weiterer Eiskühlung gerührt. Anschließend wird der
Reaktionsansatz in Eiswasser gegossen, die Phasen getrennt und noch zwei Mal mit je 150 mL
Dichlormethan extrahiert. Nach Trocknen über Na2SO4 und Einengen der vereinigten
organischen Phasen im Teilvakuum wird eine zähe gelbe Masse erhalten, welche über Nacht
im Laborabzug kristallisiert. Es erfolgen abschließend 2-3 Waschgänge mit –21 °C kaltem
Diethylether. Der Feststoff wird dadurch entfärbt, die Waschlösung ist zu verwerfen.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 89-91 °C (89-93 °C209).
Ausbeute: 53 %.

Experimenteller Teil
168 | Beschreibung der Synthesen
1H-NMR (CDCl3): δ = 1,47 (t, 3H, C(9)H3, 3J = 7,2 Hz), 4,52 (q, 2H, C(8)H2, 3J = 7,2 Hz), 7,72
(d, 1H, C(3)H, 3J = 4,8 Hz), 8,35 (s, 1H, C(5)H), 8,96 (d, 1H, C(2)H, 3J = 4,8 Hz). 13C-NMR (CDCl3): δ = 14,4 (C9), 62,9 (C8), 115,8 (C7), 122,0 (C4), 126,9 (C5), 128,2 (C3),
149,8 (C6), 151,0 (C2), 163,7 (C10).
MIR (ṽ): 2241 (w, νC≡N), 1715 (s, vC=O), cm−1.
4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV)
Es werden 3,0 g 4-Cyanpicolinsäureethylester (LXXIII) (17 mmol) in 50 mL Toluen gelöst.
Anschließend werden 1,4 g NaN3 (1,3 Äq., 22 mmol) und 3,0 g TEA∙HCl (1,3 Äq., 22 mmol)
hinzugegeben und der Reaktionsansatz für etwa 16 h bei 110 °C gerührt. Der Reaktionsansatz
wird im Teilvakuum eingeengt und mit so viel wässriger Natronlauge versetzt, bis ein pH-Wert
von etwa 12 eingestellt ist. Die wässrige Phase wird dreimal mit 50 mL Ethylacetat gewaschen
und anschließend mit konzentrierter Salzsäure angesäuert, wodurch 4-(1H-Tetrazol-5-
yl)picolinsäureethylester ausgefällt wird. Nach Abfiltration erfolgt Waschen mit Isopropanol.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 251-252 °C unter Zersetzung.
Ausbeute: 88 %. 1H-NMR (DMSO-d6): δ = 1,38 (t, 3H, C(9)H3, 3J = 7,2 Hz), 4,42 (q, 2H, C(8)H2, 3J = 7,2 Hz), 8,24
(d, 1H, C(3)H, 3J = 4,8 Hz), 8,64 (s, 1H, C(5)H), 8,96 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 14,8 (C9), 61,6 (C8), 121,6 (C5), 124,0 (C3), 133,5 (C4), 148,9 (C6),
151,1 (C2), 155,0 (C7), 164,1 (C10).
IR (ṽ): 1724 (s, vC=O), 1610 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C9H9N5O2 + H]+: 219,0756; gefunden: 219,0745.
N
N
NN
HN
O
O
1
2
34
5
6
7
8
910
LXXIV

Experimenteller Teil
Beschreibung der Synthesen | 169
4-(1H-Tetrazol-5-yl)picolinsäure (LXXV)
4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (500 mg, 2,3 mmol) wird in 10 mL Methanol
suspendiert. Dazu werden 365 mg Natriumhydroxid (4,0 Äq., 9,2 mmol), gelöst in 10 mL
Wasser, addiert und anschließend unter Rückfluss für 3 h erhitzt. Durch Ansäuern mit Salzsäure
fällt das Zielprodukt aus der Lösung aus und wird nach Abfiltrieren mit wässrigem Methanol
(MeOH/Wasser, 50:50, v/v) gewaschen.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 255-257 °C unter Zersetzung.
Ausbeute: 94 %. 1H-NMR (DMSO-d6): δ = 8,23 (d, 1H, C(3)H, 3J = 4,8 Hz), 8,65 (s, 1H, C(5)H), 8,96 (d, 1H,
C(2)H, 3J = 4,8 Hz). 13C-NMR (DMSO-d6): δ = 121,6 (C5), 123,7 (C3), 133,5 (C4), 149,7 (C6), 150,9 (C2), 155,0
(C7), 165,6 (C8).
MIR (ṽ): 3352 (m, vO-H), 1722 (m, vC=O), 1630 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C7H5N5O2 + H]+: 191,0443; gefunden: 191,0445.
N-(4-Chlorbenzyl)-4-(1H-tetrazol-5-yl)picolinamid (LXXVI)
Es werden 470 mg 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (2,1 mmol) mit 10 mL
Ethanol und 2,6 mL 4-Chlorbenzylamin (10 Äq., 21 mmol) versetzt. Der Reaktionsansatz wird
im Mikrowellenreaktor bei 175 °C für 30 min erhitzt. Nach Abkühlen der Lösung wird mit
Salzsäure angesäuert und anschließend abfiltriert. Durch Waschen mit 3×10 mL Ethanol wird
überschüssiges 4-Chlorbenzylamin∙HCl aus dem Überstand entfernt. Die Umkristallisation (zwei

Experimenteller Teil
170 | Beschreibung der Synthesen
Mal) des Filterkuchens erfolgt abschließend aus wässrigem Ethanol (EtOH/Wasser, 75:25, v/v)
(Heißfiltration in Siedehitze wegen nicht lösbaren Produkten notwendig).
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 271-273 °C.
Ausbeute: 36 %. 1H-NMR (DMSO-d6): δ = 4,53 (d, 2H, C(8)H2, 3J = 6,4 Hz), 7,38 (s, 4H, 2×C(10)H und 2×C(11)H),
8,23 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,67 (s, 1H, C(5)H), 8,91 (d, 1H, C(2)H, 3J = 4,8 Hz), 9,57 (t,
1H, N(14)H, 3J = 6,4 Hz). 13C-NMR (DMSO-d6): δ = 41,9 (C8), 119,0 (C5), 123,4 (C3), 128,2 (2×C11), 129,2 (2×C10),
131,3 (C12), 133,7 (C4), 138,4 (C9), 149,9 (C2), 151,2 (C6), 155,0 (C7), 163,4 (C13).
MIR (ṽ): 3357(s, vN-H), 1649 (s, vC=O), 1614 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H11N6OCl − H]−: 314,0683; gefunden: 314,0672.
N-(3-Morpholinopropyl)-4-(1H-tetrazol-5-yl)picolinamid-Hydrochlorid (LXXVII)
Die Synthese erfolgt analog zu N-(4-Chlorbenzyl)-4-(1H-tetrazol-5-yl)picolinamid (LXXVI) mit
folgenden Änderungen: 915 mg 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (4,1 mmol)
werden mit 3,0 mL 3-Morpholinopropylamin (5,0 Äq., 20,5 mmol) und 15 mL Ethanol im
Mikrowellenreaktor für 45 min bei 165 °C erhitzt. Der Ansatz wird nach Abkühlen auf insgesamt
80 mL Ethanol Gesamtvolumen verdünnt und mit Salzsäure sauer eingestellt. Die
anschließende Fällung aus Ethanol findet bei –21 °C für etwa eine Stunde statt. Nach
Abfiltrieren erfolgt zweimaliges Umkristallisieren aus Ethanol. Das so erhaltene Produkt wird
abschließend mit reichlich Toluen, Tetrahydrofuran und Aceton gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 129-130 °C.
Ausbeute: 18 %. 1H-NMR (DMSO-d6): δ = 2,30 (m, 2H, C(9)H2, 3J = 8,0 Hz), 3,06 (s, 2×1H, 2×C(11)H2), 3,14 (m,
2H, C(10)H2, 3J = 8,0 Hz), 3,43 (m, 2H und 2×1H, C(8)H2 und 2×C(11)H2), 3,85 (2×s, 2×2H,

Experimenteller Teil
Beschreibung der Synthesen | 171
2×C(12)H2), 8,27 (d, 1H, C(3)H, 3J = 4,8 Hz), 8,67 (s, 1H, C(5)H), 8,88 (d, 1H, C(2)H, 3J = 5,2 Hz), 9,16 (t, 1H, N(14)H, 3J = 6,4 Hz), 10,98 (s, 1H, N(15)H+). 13C-NMR (DMSO-d6): δ = 23,4 (C9), 36,3 (C8), 50,9 (2×C11), 53,9 (C10), 63,2 (2×C12), 118,9
(C5), 123,3 (C3), 133,9 (C4), 149,8 (C2), 151,1 (C6), 155,1 (C7), 163,5 (C13).
MIR (ṽ): 3344 (s, vN-H), 1644 (s, vC=O), 1613 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H19N7O2 + H]+: 317,1600; gefunden: 317,1591.
N-(4-Fluorphenylethyl)-4-(1H-tetrazol-5-yl)picolinamid (LXXVIII)
Die Synthese erfolgt analog zu N-(4-Chlorbenzyl)-4-(1H-tetrazol-5-yl)picolinamid (LXXVI) mit
folgenden Änderungen: 1,0 g 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (4,6 mmol)
werden mit 2,8 g 4-Fluorphenylethylamin (4,3 Äq., 20 mmol) und 6 mL Ethanol im
Mikrowellenreaktor für 45 min bei 180 °C erhitzt. Anschließend werden der Lösung 15 mL
Ethanol hinzugefügt und mit Salzsäure angesäuert, worauf ein Präzipitat gebildet wird. Dieses
wird abfiltriert und mit etwa 20 mL Ethanol nachgewaschen. Abschließend erfolgt
Umkristallisation aus Ethanol.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 229-231 °C.
Ausbeute: 70 %. 1H-NMR (DMSO-d6): δ = 2,89 (t, 2H, C(9)H2, 3J = 7,6 Hz), 3,57 (q, 2H, C(8)H2, 3J = 7,6 Hz), 7,11
(t, 2H, 2×C(12)H, 3J = 8,8 Hz), 7,29 (t, 2H, 2×C(11)H, 3J = 8,4 Hz), 8,20 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,64 (s, 1H, C(5)H), 8,87 (d, 1H, C(2)H, 3J = 5,2 Hz), 9,01 (t, 1H, N(15)H, 3J = 6,0 Hz). 13C-NMR (DMSO-d6): δ = 34,1 (C9), 40,4 (C8), 115,0 (2JC,F = 21 Hz, 2×C12), 118,8 (C5), 123,2
(C3), 130,4 (3JC,F = 8 Hz, 2×C11), 133,6 (C4), 135,4 (4JC,F = 3 Hz, C10), 149,8 (C2), 151,1 (C6),
155,0 (C7), 159,6 und 162,0 (1JC,F = 240 Hz, C13), 163,0 (C14).
MIR (ṽ): 3378 (s, vN-H), 1640 (s, vC=O), 1612 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C15H13N6OF + H]+: 312,1135; gefunden: 312,1120.

Experimenteller Teil
172 | Beschreibung der Synthesen
4-(1H-Tetrazol-5-yl)-N-[4-(thiophen-2-yl)benzyl]picolinamid (LXXIX)
Die Synthese erfolgt analog zu N-(4-Chlorbenzyl)-4-(1H-tetrazol-5-yl)picolinamid (LXXVI) mit
folgenden Änderungen: 385 mg 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (1,8 mmol)
werden mit 480 mg 4-(Thiophen-2-yl)benzylamin (1,4 Äq., 2,5 mmol), 120 µL Triethylamin
(0,4 Äq., 0,8 mmol) und 3 mL Ethanol im Mikrowellenreaktor für 2 h bei 175 °C erhitzt.
Anschließend wird der Ethanol im Teilvakuum entfernt, der Überstand mit K2CO3-Lösung
aufgenommen und mit EtOAc extrahiert. Durch Ansäuern der wässrigen Phase wird das
Zielprodukt aus dieser Lösung ausgefällt. Abschließend erfolgt eine säulenchromatographische
Aufreinigung (adsorbiert an Kieselgur (aus Methanol), LM: EtOAc/DCM/MeOH/Essigsäure,
60:30:10:2, v/v/v/v), wobei das gewünschte Produkt aus den Zielfraktionen spontan
auskristallisiert.
Beschaffenheit: Farbloser kristalliner Feststoff.
Ausbeute: Spuren. 1H-NMR (DMSO-d6): δ = 4,54 (d, 2H, C(8)H2, 3J = 6,4 Hz), 7,12 (m, 1H, C(15)H, 3J = 3,6 Hz und
4,8 Hz), 7,39 (d, 2H, 2×C(10)H, 3J = 8,0 Hz), 7,47 (d, 1H, C(14)H, 3J = 3,2 Hz), 7,52 (d, 1H,
C(16)H, 3J = 4,8 Hz), 7,61 (d, 2H, 2×C(11)H, 3J = 8,0 Hz), 8,22 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,67
(s, 1H, C(5)H), 8,90 (d, 1H, C(2)H, 3J = 5,2 Hz), 9,54 (s, 1H, N(18)H, 3J = 6,4 Hz).
Auf Grund der minimalen Ausbeute ist es nur gelungen, ein 1H-NMR-Spektrum zu vermessen
und auszuwerten. Die Konzentration war für das 13C-Spektrum bereits zu niedrig, weshalb
dieses nicht interpretiert werden konnte.
N-(1H-Benzimidazol-2-yl)-4-(1H-tetrazol-5-yl)picolinamid (LXXX)

Experimenteller Teil
Beschreibung der Synthesen | 173
Für die Synthese werden 250 mg 4-(1H-Tetrazol-5-yl)picolinsäure (LXXV) (1,3 mmol) und
175 mg 2-Aminobenzimidazol (1,0 Äq., 1,3 mmol) in 7 mL DMF gelöst. Dazu werden erst 1,1 mL
DIPEA (5,0 Äq., 6,5 mmol) und dann 990 mg HATU (2,0 Äq., 2,6 mmol) addiert. Der Ansatz
reagiert bei 50 °C für 15 h. Anschließend wird die dunkle Lösung mit Hilfe einer
Säulenchromatographie vorgereinigt (adsorbiert an Kieselgur (aus DMF, direkt aus dem
Ansatz), LM: EtOAc/DCM/MeOH/Ammoniak, 50:30:20:1, v/v/v/v). Abschließend erfolgt mit den
eingeengten Zielfraktionen eine Aufreinigung mittels präparativer HPLC (gelöst in
ammoniakalischem H2O; A: H2O+0,05 % NH3 und B: ACN, isokratisch, 15 % B) mit
nachfolgender Lyophilisation.
Beschaffenheit: Weißlich-gelber amorpher Feststoff.
Schmelzbereich: 270-272 °C unter Zersetzung.
Ausbeute: 8 %. 1H-NMR (DMSO-d6): δ = 7,19 (m, 2H, C(11)H und C(12)H, 3J = 6,8 Hz), 7,56 (m, 2H, C(10)H
und C(13)H, 3J = 6,8 Hz), 8,29 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,80 (s, 1H, C(5)H), 8,93 (d, 1H,
C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 114,1 (C10 und C13), 119,5 (C5), 121,9 (C11 und C12), 123,9 (C3),
135,2 (C9 und C14), 136,9 (C4), 145,7 (C8), 149,5 (C6), 149,9 (C2), 156,5 (C7), 163,4 (C15).
MIR (ṽ): 3570 (w, vN-H), 1715 (m, vC=O), 1626 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H10N8O + H]+: 306,0978; gefunden: 306,0985.
N-Butyl-4-(1H-tetrazol-5-yl-)picolinamid (LXXXI)
Die Synthese erfolgt analog zu N-(1H-Benzimidazol-2-yl)-4-(1H-tetrazol-5-yl)picolinamid
(LXXX) mit folgenden Änderungen: 165 mg 4-(1H-Tetrazol-5-yl)picolinsäure (LXXV) (0,9 mmol)
werden mit 90 µL Butylamin (1,0 Äq., 0,9 mmol) in 6 mL DMF gelöst. Dazu werden erst 770 µL
DIPEA (5,0 Äq., 4,5 mmol) und dann 650 mg HATU (2,0 Äq., 1,8 mmol) addiert. Der Ansatz
reagiert bei Raumtemperatur für 24 h. Anschließend wird das DMF bestmöglich im Teilvakuum
entfernt, der Überstand mit 30 mL Wasser aufgenommen und mit Salzsäure sauer eingestellt.
Nach Abfiltrieren des Präzipitats wird dieser Feststoff aus Ethanol umkristallisiert. Abschließend
erfolgt eine Aufreinigung mittels präparativer HPLC (gelöst in ammoniakalischem H2O;

Experimenteller Teil
174 | Beschreibung der Synthesen
A: H2O+0,05 % NH3 und B: ACN, isokratisch, 20 % B) mit nachfolgender Lyophilisation der
Zielfraktionen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 234-236 °C unter Zersetzung.
Ausbeute: 37 %. 1H-NMR (DMSO-d6): δ = 0,91 (t, 3H, C(11)H3, 3J = 7,2 Hz), 1,32 (m, 2H, C(10)H2, 3J = 7,2 Hz),
1,54 (m, 2H, C(9)H2, 3J = 7,2 Hz), 3,33 (dd, 2H, C(8)H2, 3J = 6,4 und 7,2 Hz), 8,19 (d, 1H, C(3)H, 3J = 4,8 Hz), 8,64 (s, 1H, C(5)H), 8,86 (d, 1H, C(2)H, 3J = 4,8 Hz), 8,91 (t, 1H, N(13)H, 3J = 6,4 Hz). 13C-NMR (DMSO-d6): δ = 13,7 (C11), 19,6 (C10), 31,2 (C9), 38,6 (C8), 118,8 (C5), 123,0 (C3),
134,2 (C4), 149,7 (C2), 151,4 (C6), 155,3 (C7), 163,1 (C12).
MIR (ṽ): 3362 (m, vN-H), 1651 (s, vC=O), 1618 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C11H14N6O + H]+: 246,1229; gefunden: 246,1219.
4-(1H-Tetrazol-5-yl)pyridin-2-hydroxamsäure (LXXXII)
4-(1H-Tetrazol-5-yl)picolinsäure (LXXV) (200 mg, 1,0 mmol) wird in 10 mL DMF gelöst und
unter Eiskühlung gerührt. Dieser Mischung werden 120 µL Oxalylchlorid (1,4 Äq., 1,4 mmol)
hinzugefügt. Nach etwa 30 min Rühren im Eisbad wird im Teilvakuum der Überschuss an
Oxalylchlorid entfernt und anschließend werden erst 525 µL DIPEA (3,0 Äq., 3 mmol) und dann
100 mg Hydroxylaminhydrochlorid (1,4 Äq., 1,4 mmol) addiert. Die Reaktion findet bei
Raumtemperatur für etwa 18 h statt. Die resultierende Lösung wird mit jeweils 10 mL Ethanol
und Wasser verdünnt und das Rohprodukt mit Hilfe von Salzsäure ausgefällt. Dieses wird
säulenchromatographisch aufgearbeitet (adsorbiert an Kieselgur (aus ammoniakalischem
Methanol), LM: EtOAc/MeOH/Essigsäure, 50:50:1, v/v/v) und abschließend aus Wasser
umkristallisiert.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 241-242 °C unter Zersetzung.
Ausbeute: 21 %.

Experimenteller Teil
Beschreibung der Synthesen | 175
1H-NMR (DMSO-d6): δ = 8,18 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,60 (s, 1H, C(5)H), 8,85 (d, 1H,
C(2)H, 3J = 5,2 Hz), 9,23 (s, 1H, N(8)H), 11,64 (s, 1H, O(9)H). 13C-NMR (DMSO-d6): δ = 118,8 (C5), 123,0 (C3), 133,8 (C4), 149,9 (C2), 151,3 (C6), 154,8
(C7), 160,6 (C10).
MIR (ṽ): 3335 (m, vN-H), 1643 (s, vC=O), 1610 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C7H6N6O2 + H]+: 206,0552; gefunden: 206,0544.
2-Chlor-4-(1-trityl-1H-tetrazol-5-yl)pyridin (LXXXIV)
Für die Synthese werden 3 g 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (17 mmol) und 4,7 mL
Triethylamin (2,0 Äq., 34 mmol) abgewogen und in 20 mL THF vorgelöst. Dazu wird eine Lösung
aus 5,2 g Tritylchlorid (1,1 Äq., 18,7 mmol) in 15 mL THF hinzugefügt. Der Ansatz wird 17 h bei
Raumtemperatur gerührt. Entstandene Feststoffe werden abfiltriert und mit Tetrahydrofuran
nachgewaschen. Anschließend wird das Filtrat mit Natriumcarbonat-Lösung verdünnt und mit
Ethylacetat extrahiert. Nach Trocknen über Na2SO4 und Einengen der vereinigten organischen
Phasen im Teilvakuum wird der Überstand mittels Säulenchromatographie aufgearbeitet
(adsorbiert an Kieselgur (aus EtOAc), LM: Petrolether/EtOAc/Ammoniak, 90:10:1, v/v/v). Das
Zielprodukt kann, wenn nötig, aus Acetonitril umkristallisiert werden.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 162-164 °C.
Ausbeute: 24 %. 1H-NMR (CDCl3): δ = 7,14 (m, 6H, 6×C(10)H), 7,37 (m, 9H, 6×C(11)H und 3×C(12)H), 7,94 (d,
1H, C(3)H, 3J = 5,2 Hz), 8,04 (s, 1H, C(5)H), 8,51 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (CDCl3): δ = 84,2 (C8), 119,9 (C5), 121,8 (C3), 128,1-130,5 (6×C10 und 6×C11 und
3×C12), 138,0 (C6), 141,1 (3×C9), 150,7 (C2), 152,6 (C4), 161,3 (C7).
MIR (ṽ): 1605 (s, νC=N) cm−1.
N Cl
N
NN
N
1
2
34
5
6
7
89
1011
12
LXXXIV

Experimenteller Teil
176 | Beschreibung der Synthesen
4-(1-Trityl-1H-tetrazol-5-yl)picolinnitril (LXXXV)
Anmerkung: Statt dem gewünschten Produkt wurde Tritylcyanid (2,2,2-Triphenylacetonitril)
erhalten!
2-Chlor-4-(1-trityl-1H-tetrazol-5-yl)pyridin (LXXXIV) (300 mg, 0,7 mmol), 85 mg Zn(CN)2
(1,0 Äq., 0,7 mmol) und 20 mg Tetrakis(triphenylphosphin)palladium(0) (2,5 mol%) werden in
ein Mikrowellengefäß eingewogen und mit 2 mL DMF versetzt. Anschließend wird im
Mikrowellenreaktor für 30 min bei 160 °C erhitzt. Die Feststoffe werden über Kieselgur abfiltriert
und der Filterkuchen mit Wasser und Ethylacetat nachgewaschen. Die zwei resultierenden
Filtrat-Phasen werden getrennt, die wässrige Phase anschließend erneut mit Ethylacetat
extrahiert. Nach Trocknen über Na2SO4 und Einengen der vereinigten organischen Phasen im
Teilvakuum, wird der Überstand mittels Säulenchromatographie aufgearbeitet (gelöst in
wenigen mL EtOAc, LM: Petrolether/EtOAc, 90:10, v/v).
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 126-128 °C (127-129 °C370).
Ausbeute: nicht bestimmt. 1H-NMR (CDCl3): δ = 7,22 (m, 6H, 6×C(3)H), 7,35 (m, 9H, 6×C(4)H und 3×C(5)H).
13C-NMR (CDCl3): δ = 57,6 (C1), 123,7 (C6), 128,3-129,0 (6×C3 und 6×C4 und 3×C5),
140,4 (3×C2).
MIR (ṽ): 2232 (w, νC≡N) cm−1.
4-(1-Benzyl-1H-tetrazol-5-yl)-2-chlorpyridin (LXXXVI)

Experimenteller Teil
Beschreibung der Synthesen | 177
Es werden 3 g 2-Chlor-4-(1H-tetrazol-5-yl)pyridin (XXXV) (17 mmol), 2,8 g Kaliumcarbonat
(1,2 Äq., 20 mmol) und 2,4 mL Benzylbromid (1,2 Äq., 20 mmol) in einen Rundkolben gegeben
und in etwa 50 mL Aceton suspendiert. Der Ansatz wird bei 50 °C für 90 min unter
Stickstoffatmosphäre erhitzt. Nach Abkühlen wird der ausgefallene Feststoff abfiltriert und mit
Aceton nachgewaschen. Das organische Lösungsmittel wird anschließend im Teilvakuum
entfernt, der Überstand in verdünnter Natronlauge aufgenommen und mehrfach mit Ethylacetat
extrahiert. Nach Trocknen über Na2SO4 und Einengen der vereinigten organischen Phasen im
Teilvakuum wird der Überstand mittels Säulenchromatographie aufgearbeitet (gelöst in wenigen
mL EtOAc, LM: Petrolether/EtOAc, 80:20, v/v). Das Zielprodukt kann, wenn nötig, aus Methanol
umkristallisiert werden.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 88-89 °C.
Ausbeute: 70 %. 1H-NMR (CDCl3): δ = 5,83 (s, 2H, C(8)H2), 7,41 (m, 5H, 2×C(10)H und 2×C(11)H und C(12)H),
7,94 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,06 (s, 1H, C(5)H), 8,52 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (CDCl3): δ = 57,5 (C8), 119,7 (C5), 121,7 (C3), 128,7-129,5 (2×C10 und 2×C11 und
C12), 132,9 (C9), 137,8 (C6), 150,8 (C2), 152,7 (C4), 162, 6 (C7).
MIR (ṽ): 1604 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C13H10N5Cl + H]+: 271,0625; gefunden: 271,0611.
4-(1-Benzyl-1H-tetrazol-5-yl)picolinnitril (LXXXVII)
In ein Mikrowellengefäß werden 800 mg 4-(1-Benzyl-1H-tetrazol-5-yl)-2-chlorpyridin (LXXXVI)
(2,9 mmol) mit 170 mg Tetrakis(triphenylphosphin)palladium(0) (5 mol%) und 410 mg Zn(CN)2
(1,2 Äq., 3,5 mmol) eingewogen. Unter Stickstoffatmosphäre werden 10 mL DMF hinzugegeben
und anschließend das Gefäß noch einmal mit Stickstoff gespült. Die Reaktion findet im
Mikrowellenreaktor bei 190 °C für 20 min statt. Anschließend wird der Ansatz mit der fünffachen
Menge an Wasser verdünnt, über Kieselgur filtriert und der Filterkuchen mit reichlich Wasser
und Ethylacetat nachgewaschen. Nach Trennen der beiden flüssigen Phasen, wird die wässrige

Experimenteller Teil
178 | Beschreibung der Synthesen
Phase nochmals mit Ethylacetat extrahiert. Durch anschließendes Trocknen der vereinigten
organischen Phasen über Na2SO4 und Einengen im Teilvakuum wird das Rohprodukt erhalten,
welches nachfolgend chromatographisch aufgearbeitet wird (adsorbiert an Kieselgur (aus
EtOAc), LM: n-Hexan/EtOAc, 75:25, v/v). Die vereinigten, vom Lösungsmittel befreiten
Fraktionen werden in wenigen mL Ethanol gelöst und mit Hilfe von etwa 150 mL Petrolether bei
–21°C ausgefällt.
Beschaffenheit: Farbloser watteartiger Feststoff.
Schmelzbereich: 79-80°C.
Ausbeute: 24 %. 1H-NMR (CDCl3): δ = 5,85 (s, 2H, C(8)H2), 7,42 (m, 5H, 2×C(10)H und 2×C(11)H und C(12)H),
8,23 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,39 (s, 1H, C(5)H), 8,84 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (CDCl3): δ = 57,6 (C8), 116,9 (C13), 123,9 (C3), 125,7 (C5), 128,8-129,6 (2×C10 und
2×C11 und C12), 132,7 (C9), 135,1 (C6), 136,5 (C4), 152,1 (C2), 162,0 (C7).
MIR (ṽ): 2237 (w, νC≡N), 1610 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H10N6 + H]+: 262,0967; gefunden: 262,0954.
[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanamin (LXXXVIII)
Für die Synthese werden 210 mg 4-(1-Benzyl-1H-tetrazol-5-yl)picolinnitril (LXXXVII) (0,8 mmol)
mit 300 mg Palladium auf Aktivkohle-Pulver (Pd/C, 10 %, wassernass) in einen Rundkolben mit
Seitenhals, welcher durch ein Septum verschlossen wurde, eingewogen. Anschließend wird der
Aufbau drei Mal evakuiert und mit Wasserstoff geflutet. Durch das Septum werden 50 mL
Methanol hinzugegeben und der Ansatz bei Raumtemperatur für etwa 24 h gerührt
(gegebenenfalls muss der H2-Ballon mehrmals nachgefüllt werden). Nachfolgend wird über
Kieselgur abfiltriert, der Überstand mit reichlich Methanol nachgewaschen und anschließend
das Filtrat vom Lösungsmittel im Teilvakuum befreit. Die abschließende Aufreinigung erfolgt mit
Hilfe der MPLC (adsorbiert an Kieselgur (aus ammoniakalischem MeOH), LM:
EtOAc/MeOH/Ammoniak, 65:35:1, v/v/v; stat. Phase: Biotage Snap Cartridge KP-Sil 25 g
(50 µm)).

Experimenteller Teil
Beschreibung der Synthesen | 179
Beschaffenheit: Leicht beigefarbener amorpher Feststoff.
Schmelzbereich: ab 250°C Zersetzung.
Ausbeute: 49 %. 1H-NMR (DMSO-d6): δ = 4,48 (s, 2H, C(8)H2), 7,25 (s, 2H, N(9)H2), 7,94 (d, 1H, C(3)H,
3J = 5,2 Hz), 8,09 (s, 1H, C(5)H), 8,64 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 50,6 (C8), 119,4 (C5), 119,8 (C3), 140,3 (C4), 149,4 (C2), 152,6 (C6),
158,6 (C7).
MIR (ṽ): 3335 (m, vN-H), 1618 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C7H8N6 − H]−: 176,0810; gefunden: 176,0810.
4-(1-Benzyl-1H-tetrazol-5-yl)picolinsäureethylester (LXXXIX)
Es werden 2,3 g 4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (10 mmol), 1,7 g
Kaliumcarbonat (1,2 Äq., 12 mmol) und 1,4 mL Benzylbromid (1,2 Äq., 12 mmol) in einen
Rundkolben gegeben und in 45 mL Aceton suspendiert. Der Ansatz wird unter Rückfluss für
etwa 2 h unter Stickstoffatmosphäre erhitzt. Das organische Lösungsmittel wird anschließend
im Teilvakuum entfernt, der Überstand in Ethylacetat suspendiert und mehrfach mit verdünnter
K2CO3-Lösung gewaschen. Nach Trocknen über Na2SO4 und Einengen der organischen Phase
im Teilvakuum wird der Überstand mittels Säulenchromatographie aufgearbeitet (gelöst in
wenigen mL EtOAc, LM: DCM/EtOAc, 80:20, v/v). Die Kristallisation des Zielprodukts kann bis
zu vier Wochen dauern.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 58-59°C.
Ausbeute: 91 %. 1H-NMR (CDCl3): δ = 1,47 (t, 3H, C(9)H3, 3J = 7,2 Hz), 4,51 (q, 2H, C(8)H2, 3J = 7,2 Hz), 5,85 (s,
2H, C(11)H2), 7,41 (m, 5H, 2×C(13)H und 2×C(14)H und C(15)H), 8,19 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,81 (s, 1H, C(5)H), 8,89 (d, 1H, C(2)H, 3J = 4,8 Hz).
N
N
NN
N
O
O
LXXXIX
1
2
34
5
6
711
1213
14
15
8
910

Experimenteller Teil
180 | Beschreibung der Synthesen
13C-NMR (CDCl3): δ = 14,6 (C9), 57,5 (C11), 62,4 (C8), 122,5 (C5), 123,9 (C3), 128,8-129,5
(2×C13 und 2×C14 und C15), 133,0 (C12), 136,3 (C4), 149,5 (C6), 150,9 (C2), 163,0 (C7),
165,0 (C10).
MIR (ṽ): 1740 (s, vC=O), 1607 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C16H15N5O2 + H]+: 309,1226; gefunden: 309,1211.
4-(1-Benzyl-1H-tetrazol-5-yl)-N-(3-morpholinopropyl)picolinamid (XC)
Für die Synthese werden 2,7 g 4-(1-Benzyl-1H-tetrazol-5-yl)picolinsäureethylester (LXXXIX)
(8,7 mmol) in 60 mL Ethanol gelöst und mit 5,1 mL 3-Morpholinopropylamin (4,0 Äq., 35 mmol)
versetzt. Der Ansatz reagiert bei 55 °C für etwa 72 h. Nach Entfernen des Lösungsmittels erfolgt
eine säulenchromatographische Aufarbeitung des Überstandes (gelöst in wenigen mL EtOAc,
LM: EtOAc/DCM/MeOH, 65:25:10, v/v/v). Wenn erwünscht, kann die freie Base in Ethylacetat
gelöst und unter starkem Rühren HCl-Gas in den Rundkolben zum Ausfällen des
Hydrochloridsalzes eingeleitet werden.
Folgende Daten beziehen sich auf die freie Base:
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 77-78°C.
Ausbeute: 93 %. 1H-NMR (CDCl3): δ = 1,83 (m, 2H, C(9)H2, 3J = 6,0 Hz), 2,55 (m, 2H und 4H, C(10)H2 und
2×C(11)H2, 3J = 6,4 Hz), 3,61 (q, 2H, C(8)H2, 3J = 6,0 Hz), 3,82 (t, 4H, 2×C(12)H2), 5,84 (s, 2H,
C(14)H2), 7,41 (m, 5H, 2×C(16)H und 2×C(17)H und C(18)H), 8,16 (d, 1H, C(3)H, 3J = 5,2 Hz),
8,69 (d, 1H, C(2)H, 3J = 5,2 Hz), 8,86 (s, 1H, C(5)H), 9,02 (s, 1H, N(19)H). 13C-NMR (CDCl3): δ = 25,5 (C9), 39,5 (C8), 54,0 (2×C11), 57,4 (C14), 58,1 (C10), 67,0 (2×C12),
119,8 (C5), 123,1 (C3), 128,9-129,4 (2×C16 und 2×C17 und C18), 133,0 (C15), 136,5 (C4),
149,0 (C2), 151,2 (C6), 163,4 (C13), 164,0 (C7).
MIR (ṽ): 3270 (m, vN-H), 1655 (s, vC=O), 1609 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C21H25N7O2 + H]+: 407,2070; gefunden: 407,2054.
N
O
N
NN
N
HN N
O
XC
1
2
34
5
6
714
15
16
17
18
8
913
1012
11
19

Experimenteller Teil
Beschreibung der Synthesen | 181
N-{[4-(1-Benzyl-1H-tetrazol-5-yl)pyridin-2-yl]methyl}-3-morpholinopropan-1-amin-
Dihydrochlorid (XCII) (ohne vorherige Isolation von XCI)
Schritt I (XCI): 4-(1-Benzyl-1H-tetrazol-5-yl)-N-(3-morpholinopropyl)picolinamid (XC) (400 mg,
1,0 mmol) und 415 mg PCl5 (2,0 Äq., 2,0 mmol) werden in 25 mL Dichlormethan gelöst und für
etwa 5 h unter Rückfluss erhitzt. Die farblose Lösung nimmt dabei eine rötliche Färbung an. Das
Lösungsmittel wird anschließend im Teilvakuum entfernt. Es resultiert ein bräunlicher,
schaumähnlicher Feststoff, welcher direkt weiter verarbeitet wird.
Schritt II (XCII): Der Überstand aus der Synthese von XCI wird in 50 mL Ethanol aufgenommen
und im Eisbad gekühlt. Zu der Lösung werden langsam und in gleichmäßigen Portionen 225 mg
NaBH4 (6 Äq., 6 mmol) gegeben. Nach etwa 15 min Rühren im Eisbad und Erliegen der
Gasentwicklung wird für 30 min unter Rückfluss erhitzt. Anschließend erfolgen das Entfernen
des Lösungsmittels und eine Flüssig-Flüssig-Extraktion mit Ethylacetat und K2CO3-Lösung.
Nach Trocknen über Na2SO4 und Einengen der organischen Phasen im Teilvakuum wird der
Überstand mittels Säulenchromatographie aufgearbeitet (adsorbiert an Kieselgur (aus EtOAc),
LM: DCM/EtOAc/MeOH/Ammoniak, 45:45:10:1, v/v/v/v). Zum Überführen der freien Base in das
Dihydrochlorid wird diese in Ethylacetat gelöst und anschließend HCl-Gas unter kräftigem
Rühren eingeleitet. Das Präzipitat wird anschließend abfiltriert und mit kaltem Aceton und wenig
Isopropanol gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 211-213 °C unter Zersetzung.
Ausbeute: 35 %. 1H-NMR (DMSO-d6): δ = 2,28 (m, 2H, C(9)H2, 3J = 7,2 Hz), 3,05 (s, 2H, C(11)H2), 3,14 (s, 2H,
C(10)H2), 3,20 (d, 2H, C(8)H2, 3J = 6,8 Hz), 3,37 (s, 2H, C(11)H2), 3,92 (m, 4H, 2×C(12)H2), 4,44
(s, 2H, C(13)H2), 6,08 (s, 2H, C(14)H2), 7,41 (m, 5H, 2×C(16)H und 2×C(17)H und C(18)H), 8,04
(d, 1H, C(3)H, 3J = 4,8 Hz), 8,19 (s, 1H, C(5)H), 8,83 (d, 1H, C(2)H, 3J = 5,2 Hz), 9,72 (s, 2H,
N(19)H2+), 11,64 (s, 1H, N(20)H+).

Experimenteller Teil
182 | Beschreibung der Synthesen
13C-NMR (DMSO-d6): δ = 19,6 (C9), 44,1 (C8), 50,1 (C13), 50,8 (2×C11), 52,9 (C10), 56,5 (C14),
63,0 (2×C12), 120,2 (C3 und C5), 128,4-128,9 (2×C16 und 2×C17 und C18), 133,8 (C15), 134,8
(C4), 150,4 (C2), 153,5 (C6), 162,3 (C7).
MIR (ṽ): 3382 (m, vN-H), 1607 (m, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C21H27N7O + H]+: 393,2277; gefunden: 393,2262.
N-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methyl}-3-morpholinopropan-1-amin-Dihydrochlorid
(XCIII)
N-{[4-(1-Benzyl-1H-tetrazol-5-yl)pyridin-2-yl]methyl}-3-morpholinopropan-1-amin (XCII)
(350 mg, 0,9 mmol) wird mit 100 mg Pd/C (10 %, wassernass) in einen Rundkolben mit
Seitenhals eingewogen. Der Ansatz wird evakuiert und über ein Septum am Seitenhals etwa
50 mL Methanol zugesetzt. Anschließend wird der Aufbau mit Wasserstoff geflutet. Die
Reaktionszeit beträgt etwa 6 h bei 35 °C. Nach Abfiltrieren über Kieselgur wird der Filterkuchen
großzügig mit Methanol gewaschen, das Lösungsmittel anschließend im Teilvakuum entfernt
und der ölige Rückstand mit Hilfe von zugesetztem Ethylacetat kristallisiert. Die abschließende
Aufreinigung erfolgt mittels präparativer HPLC (gelöst in H2O; A: H2O mit 0,05 % NH3 und B:
MeOH, isokratisch, 15 % B) mit nachfolgender Lyophilisation der Zielfraktionen und Überführen
in das Dihydrochloridsalz.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 219-221 °C unter Zersetzung.
Ausbeute: 11 %. 1H-NMR (DMSO-d6): δ = 1,82 (m, 2H, C(9)H2, 3J = 6,4 Hz), 2,42 (m, 2H und 4H, C(10)H2 und
2×C(11)H2, 3J = 6,4 Hz), 3,10 (t, 2H, C(8)H2, 3J = 6,4 Hz), 3,59 (s, 4H, 2×C(12)H2), 4,40 (s, 2H,
C(13)H2), 7,93 (d, 1H, C(3)H, 3J = 4,8 Hz), 8,04 (s, 1H, C(5)H), 8,61 (d, 1H, C(2)H, 3J = 5,2 Hz),
9,04 (s, 1H, N(14)H+). 13C-NMR (DMSO-d6): δ = 21,6 (C9), 46,6 (C8), 50,5 (C13), 53,0 (2×C11), 55,9 (C10), 65,9
(2×C12), 119,1 (C5), 119,8 (C3), 140,1 (C4), 149,3 (C2), 152,6 (C6), 159,0 (C7).
MIR (ṽ): 2952 (m, νC-H, aliphatisch), 1614 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H21N7O + H]+: 303,1808; gefunden: 303,1795.

Experimenteller Teil
Beschreibung der Synthesen | 183
[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanol (XCIV)
4-(1H-Tetrazol-5-yl)picolinsäureethylester (LXXIV) (1,1 g, 5 mmol) wird mit 1,5 g NaBH4 (8 Äq.,
40 mmol) in einen Rundkolben eingewogen und in 100 mL Tetrahydrofuran suspendiert. Nach
fünfminütigem Erhitzen auf 70 °C werden dem Reaktionsansatz 25 mL Methanol zugesetzt. Die
Suspension klart unter heftiger Gasentwicklung auf. Es wird für weitere 20 h bei 70 °C erhitzt.
Nach Abkühlen werden 5 mL Wasser und so viel Salzsäure zugesetzt, bis ein neutraler pH-Wert
resultiert. Durch Entfernen der Lösungsmittel im Teilvakuum wird ein Überstand erhalten,
welcher durch eine kurze Säulenchromatographie von überschüssigen anorganischen Salzen
befreit wird (adsorbiert an Kieselgel (aus ammoniakalischem Methanol), LM:
EtOAc/MeOH/DCM/Essigsäure, 50:30:20:1, v/v/v/v). Nach Einengen der Zielfraktionen im
Teilvakuum wird das Zielprodukt mit reichlich Ethylacetat gewaschen.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 258-259 °C unter Zersetzung.
Ausbeute: 83 %. 1H-NMR (DMSO-d6): δ = 4,69 (s, 2H, C(8)H2), 5,79 (s, 1H, O(9)H), 8,03 (d, 1H, C(3)H,
3J = 5,2 Hz), 8,21 (s, 1H, C(5)H), 8,72 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 63,6 (C8), 117,3 (C5), 119,2 (C3), 133,6 (C4), 149,1 (C2), 155,2 (C7),
163,1 (C6).
MIR (ṽ): 3350-3100 (s, vO-H, assoziiert), 1628 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C7H7N5O + H]+: 177,0651; gefunden: 177,0657.
N-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methyl}-chinolin-3-amin (XCVII)

Experimenteller Teil
184 | Beschreibung der Synthesen
Für die Synthese werden 150 mg [4-(1H-Tetrazol-5-yl)pyridin-2-yl]methanol (XCIV) (0,8 mmol)
zusammen mit 225 µL Triethylamin (2,0 Äq., 1,6 mmol) und 10 mg 4-(Dimethylamino)pyridin
(0,08 mmol) in einer Mischung aus 10 mL THF und 10 mL DMF gelöst. Dazu werden unter
Eiskühlung 95 µL Methansulfonylchlorid (1,5 Äq., 1,2 mmol) gegeben und unter langsamer
Erwärmung auf Raumtemperatur für mindestens 36 h gerührt. Zeigt die DC-Kontrolle eine
vollständige Umsetzung an, erfolgt die Zugabe von 460 mg 3-Aminochinolin (4,0 Äq.,
3,2 mmol). Anschließend wird für mindestens 48 h bei 80 °C erhitzt. Die resultierende dunkle
Lösung wird mit Hilfe einer Säulenchromatographie vorgereinigt (adsorbiert an Kieselgur (aus
DMF, direkt aus dem Ansatz), LM: EtOAc/MeOH/DCM/Essigsäure, 50:30:20:1, v/v/v/v).
Abschließend erfolgt mit den eingeengten Zielfraktionen eine Aufreinigung mittels präparativer
HPLC (gelöst in MeOH/DMSO, 70:30, v/v; A: H2O mit 0,05 % NH3 und B: ACN, Gradient, 0–4
min (B) 15 %, –10 min (B) 85%, –25 min (B) 85 %, –26 min (B) 15%) mit nachfolgender
Lyophilisation.
Beschaffenheit: Gelber amorpher Feststoff.
Schmelzbereich: 223-226 °C unter Zersetzung.
Ausbeute: 9 %. 1H-NMR (DMSO-d6): δ = 4,55 (s, 2H, C(8)H2), 7,02 (s, 1H, C(17)H), 7,09 (s, 1H, N(18)H), 7,33
(m, 2H, C(13)H und C(14)H, 3J = 8,4 Hz), 7,57 (d, 1H, C(12)H, 3J = 8,0 Hz), 7,77 (d, 1H, C(15)H, 3J = 7,6 Hz), 7,81 (s, 1H, C(3)H), 8,01 (s, 1H, C(5)H), 8,63 (s, 2H, C(2)H und C(10)H). 13C-NMR (DMSO-d6): δ = 48,1 (C8), 108,2 (C17), 117,6 (C5), 118,8 (C3), 124,0 (C13),
125,7 (C15), 126,5 (C14), 128,5 (C12), 129,3 (C16), 137,8 (C9), 140,1 (C11), 142,2 (C4), 143,7
(C10), 149,6 (C2), 157,5 (C7), 159,5 (C6).
MIR (ṽ): 3356 (w, vN-H), 1611 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C16H13N7 + H]+: 303,1232; gefunden: 303,1233.
[4-(1H-Tetrazol-5-yl)pyridin-2-yl]-N-(4-fluorbenzyl)methanamin (XCVIII)
Variante A (Sulfonsäureester-Weg): Es werden 150 mg [4-(1H-Tetrazol-5-yl)pyridin-2-
yl]methanol (XCIV) (0,8 mmol) zusammen mit 225 µL Triethylamin (2,0 Äq., 1,6 mmol) und
10 mg 4-(Dimethylamino)pyridin (0,08 mmol) in 15 mL Tetrahydrofuran suspendiert. Dazu
werden unter Eiskühlung 70 µL Methansulfonylchlorid (1,1 Äq., 0,9 mmol) gegeben und unter

Experimenteller Teil
Beschreibung der Synthesen | 185
langsamen Erwärmen auf Raumtemperatur für mindestens 24 h gerührt. Zeigt die DC-Kontrolle
eine vollständige Umsetzung an, erfolgt die Zugabe von 365 µL 4-Fluorbenzylamin (4,0 Äq.,
3,2 mmol) und 20 mL DMF. Anschließend wird für mindestens 72 h bei 60 °C erhitzt. Die
resultierende orangefarbene Lösung wird mit Hilfe einer Säulenchromatographie vorgereinigt
(adsorbiert an Kieselgur (aus DMF, direkt aus dem Ansatz), LM: EtOAc/MeOH/DCM/Ammoniak,
50:30:20:1, v/v/v/v). Die abschließende Aufreinigung erfolgt mit Hilfe der präparativen HPLC
(gelöst in MeOH/H2O, 50:50, v/v; A: H2O mit 0,05 % NH3 und B: ACN, Gradient, 0–6 min (B)
10 %, –10 min (B) 85%, –25 min (B) 85 %, –26 min (B) 15%) und nachfolgender Lyophilisation.
Variante B (reduktive Aminierung): 4-(1-Benzyl-1H-tetrazol-5-yl)picolinaldehyd (CI) (200 mg,
0,8 mmol) wird mit 90 µL 4-Fluorbenzylamin (1,0 Äq., 0,8 mmol) in 20 mL Methanol gelöst und
für 24 h unter Rückfluss erhitzt. Nach Abkühlen der Lösung werden dem Ansatz 300 mg
Palladium auf Aktivkohle-Pulver (10 %, wassernass) und weitere 20 mL Methanol zugesetzt,
das System evakuiert und mit Wasserstoff geflutet. Der Ansatz reagiert bei Raumtemperatur für
etwa 22 h, gegebenenfalls muss während der Reaktion der Wasserstoff-Ballon wieder befüllt
werden. Der Katalysator wird anschließend über einem Bett aus Kieselgur abfiltriert und das
erhaltene Filtrat im Teilvakuum eingeengt. Es erfolgen zwei Säulenchromatographien (erste:
adsorbiert an Kieselgur (aus Methanol), LM: EtOAc/MeOH/DCM/Ammoniak, 50:30:20:1, v/v/v/v;
zweite: adsorbiert an Kieselgur (aus Methanol), LM: EtOAc/Aceton/MeOH/Essigsäure,
40:30:30:1, v/v/v/v) zur Aufreinigung des Rohprodukts. Die nach Einengen der Zielfraktionen
erhaltene ölige Substanz wird mit Hilfe von wenig Ethanol und reichlich Diethylether kristallisiert.
Beschaffenheit: Gräulicher amorpher Feststoff.
Schmelzbereich: 230-233°C unter Zersetzung.
Ausbeute: 5 % (A). 9 % (B). 1H-NMR (DMSO-d6): δ = 4,00 (s, 2H, C(9)H2), 4,09 (s, 2H, C(8)H2), 7,20 (t, 2H, 2×C(12)H,
3J = 8,0 Hz), 7,49 (t, 2H, 2×C(11)H, 3J = 7,6 Hz), 7,82 (d, 1H, C(3)H, 3J = 4,4 Hz), 8,01 (s, 1H,
C(5)H), 8,52 (d, 1H, C(2)H, 3J = 4,8 Hz), 8,73 (s, 1H, N(14)H). 13C-NMR (DMSO-d6): δ = 49,7 (C9), 50,5 (C8), 115,4 (2JC,F = 21 Hz, 2×C12), 119,3 (C5), 119,7
(C3), 129,1 (C10), 132,2 (3JC,F = 9 Hz, 2×C11), 140,5 (C4), 149,4 (C2), 153,1 (C6), 158,7 (C7),
161,0 und 163,5 (1JC,F = 244 Hz, C13).
MIR (ṽ): 1615 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H13N6F + H]+: 284,1186; gefunden: 284,1185.

Experimenteller Teil
186 | Beschreibung der Synthesen
4-(1-Benzyl-1H-tetrazol-5-yl)picolinaldehyd (CI)
4-(1-Benzyl-1H-tetrazol-5-yl)picolinsäureethylester (LXXXIX) (1,0 g, 3,2 mmol) wird in einen
Rundkolben mit Seitenhals und aufgesetztem Trockenrohr eingewogen und in 20 mL trockenem
Tetrahydrofuran gelöst. Der Reaktionsansatz wird mit einer Mischung aus Ethylacetat und
flüssigem Stickstoff in einem Dewar-Gefäß auf etwa –80 °C gekühlt. Anschließend werden dazu
über den Seitenhals langsam 8,0 mL DIBAL (3,0 Äq., 9,6 mmol) (1,2 mol/L Lösung in Toluen)
hinzugegeben und für 5 h unter fortlaufender Kühlung gerührt. Die noch kalte Lösung wird dann
in eine Mischung aus 20 mL Wasser und 3 mL Essigsäure gegossen und für weitere 15 min
gerührt, bis die Gasentwicklung zum Erliegen kommt. Anschließend erfolgt eine Flüssig-Flüssig-
Extraktion mit Ethylacetat, die wässrige Phase wird vorher auf einen pH-Wert von etwa 8 mit
K2CO3 eingestellt. Nach Trocknen über Na2SO4 und Einengen der vereinigten organischen
Phasen im Teilvakuum wird der Überstand mittels Säulenchromatographie aufgearbeitet (gelöst
in wenigen mL EtOAc, LM: Petrolether/EtOAc, 50:50, v/v).
Beschaffenheit: Ockerfarbener amorpher Feststoff.
Schmelzbereich: 96-97 °C.
Ausbeute: 79 %. 1H-NMR (CDCl3): δ = 5,85 (s, 2H, C(9)H2), 7,38 (m, 5H, 2×C(11)H und 2×C(12)H und C(13)H),
8,25 (d, 1H, C(3)H, 3J = 4,8 Hz), 8,64 (s, 1H, C(5)H), 8,92 (d, 1H, C(2)H, 3J = 4,8 Hz), 10,14 (s,
1H, C(8)H). 13C-NMR (CDCl3): δ = 57,5 (C9), 119,2 (C5), 124,9 (C3), 128,8-129,5 (2×C11 und 2×C12 und
C13), 132,9 (C10), 136,4 (C4), 151,3 (C2), 153,9 (C6), 163,0 (C7), 193,0 (C8).
MIR (ṽ): 1700 (s, vC=O), 1612 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C14H11N5O + H]+: 265,0964; gefunden: 265,0967.
N
N
NN
N
O
H
1
2
34
5
6
79
1011
12
13
8
CI

Experimenteller Teil
Beschreibung der Synthesen | 187
[4-(1H-Tetrazol-5-yl)pyridin-2-yl]-N-(3,4-dimethoxybenzyl)methanamin (CII)
Für die Synthese werden 250 mg 4-(1-Benzyl-1H-tetrazol-5-yl)picolinaldehyd (CI) (0,9 mmol) in
20 mL Methanol gelöst und mit 140 µL 3,4-Dimethoxybenzylamin (1,0 Äq., 0,9 mmol) versetzt.
Es wird für 15 h unter Rückfluss erhitzt. Nach erfolgter Azomethinbildung wird der Ansatz nach
Abkühlen mit 300 mg Pd/C (10 %, wassernass) versetzt, die Glasapparatur evakuiert und mit
Wasserstoff geflutet. Nach 8 h Reaktionszeit bei Raumtemperatur wird der Katalysator über
Kieselgur abfiltriert und das Filtrat im Teilvakuum eingeengt. Die abschließende Aufreinigung
erfolgt mittels präparativer HPLC (gelöst in H2O/DMSO, 50:50, v/v; A: H2O und B: MeOH,
isokratisch, 50 % B) und anschließender Lyophilisation.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 153-156 °C unter Zersetzung.
Ausbeute: 26 %. 1H-NMR (DMSO-d6): δ = 3,77 (s, 6H, 2×C(16)H3), 4,18 (s, 2H, C(9)H2), 4,32 (s, 2H, C(8)H2),
7,03 (m, 2H, C(14)H und C(15)H), 7,17 (s, 1H, C(11)H), 7,92 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,05
(s, 1H, C(5)H), 8,62 (d, 1H, C(2)H, 3J = 5,2 Hz), 9,33 (s, 1H, N(17)H). 13C-NMR (DMSO-d6): δ = 49,9 und 50,2 (C8 und C9), 55,5 (2×C16), 111,6 (C14), 113,6 (C11),
119,5 (C5), 119,9 (C3), 122,8 (C15), 123,9 (C10), 140,5 (C4), 148,6 und 149,3 (C12 und C13),
149,5 (C2), 152,5 (C6), 158,6 (C7).
MIR (ṽ): 2835 (w, νC-H, aliphatisch), 1612 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C16H18N6O2 + H]+: 326,1491; gefunden: 326,1488.
N-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methyl}cyclopropanamin (CIII)
4-(1-Benzyl-1H-tetrazol-5-yl)picolinaldehyd (CI) (250 mg, 0,9 mmol) wird in 20 mL Methanol
gelöst und mit 130 µL Cyclopropylamin (2,0 Äq., 1,8 mmol) versetzt. Der Ansatz wird für etwa

Experimenteller Teil
188 | Beschreibung der Synthesen
1,5 h unter Rückfluss erhitzt. Anschließend wird im Eisbad heruntergekühlt, 140 mg NaBH4
(4,0 Äq.), welches in 5 mL Wasser suspendiert wurde, hinzugefügt und für 30 min weitergerührt.
Durch Einengen im Teilvakuum wird das Lösungsmittel entfernt. Der Überstand ist in verdünnter
NaHCO3-Lösung aufzunehmen und mit Ethylacetat zu extrahieren. Anschließend werden die
organischen Phasen wiederum vom Lösungsmittel befreit und der resultierende Überstand mit
300 mg Pd/C (10 %, wassernass) und 35 mL Methanol versetzt. Die Glasapparatur wird
evakuiert und mit Wasserstoff geflutet. Die Reaktionszeit beträgt 1,5 h bei Raumtemperatur. Der
Katalysator wird über Kieselgur abfiltriert, mit reichlich Methanol nachgewaschen und nach
Ansäuern mit Salzsäure wird das Filtrat im Teilvakuum eingeengt. Der resultierende Überstand
wird mit Ethanol digeriert, die Nebenprodukte abfiltriert und das Zielprodukt-enthaltende Filtrat
vom Lösungsmittel befreit. Die finale Aufreinigung erfolgt mittels präparativer HPLC (gelöst in
H2O/MeOH, 50:50, v/v; A: H2O und B: MeOH, isokratisch, 15 % B) und anschließender
Lyophilisation.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: ab 220 °C Zersetzung.
Ausbeute: 34 %. 1H-NMR (DMSO-d6): δ = 0,74 (m, 2H, C(11)H2,
3J = 6,8 Hz), 0,83 (m, 2H, C(10)H2, 3J = 7,2 Hz),
2,75 (m, 1H, C(9)H, 3J = 7,2 Hz), 4,44 (s, 2H, C(8)H2), 7,92 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,07 (s,
1H, C(5)H), 8,60 (d, 1H, C(2)H, 3J = 5,2 Hz). 13C-NMR (DMSO-d6): δ = 3,5 (C10 und C11), 29,9 (C9), 51,6 (C8), 119,4 (C5), 119,8 (C3), 140,4
(C4), 149,4 (C2), 153,0 (C6), 158,5 (C7).
MIR (ṽ): 3249 (s, vN-H), 1617 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C10H12N6 + H]+: 216,1123; gefunden: 216,1118.
1-[4-(1H-Tetrazol-5-yl)pyridin-2-yl]-N-[3-(trifluormethyl)benzyl]methanamin (CIV)
Die Synthese erfolgt analog zu N-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methyl}cyclopropanamin
(CIII) mit folgenden Änderungen: 4-(1-Benzyl-1H-tetrazol-5-yl)picolinaldehyd (CI) (250 mg,
0,9 mmol) wird mit 260 µL 3-(Trifluormethyl)benzylamin (2,0 Äq., 1,8 mmol) in 20 mL Methanol
für 3 h unter Rückfluss erhitzt. Im Eisbad wird das Azomethin anschließend mit 140 mg NaBH4,

Experimenteller Teil
Beschreibung der Synthesen | 189
welches suspendiert in 5 mL Wasser vorliegt, reduziert. Es folgt eine Flüssig-Flüssig-Extraktion
mit Ethylacetat und NaHCO3-Lösung, wobei die erhaltenen organischen Phasen nach Trocknen
über Na2SO4 im Teilvakuum eingeengt werden. Der Überstand wird in etwa 50 mL Methanol
aufgenommen und mit 300 mg Pd/C (10 %, wassernass) versetzt. Nach Evakuieren der
Glasapparatur wird mit Wasserstoff geflutet und bei Raumtemperatur für 4 h unter Wasserstoff-
Atmosphäre gerührt. Anschließend wird über Kieselgur abfiltriert, das Lösungsmittel entfernt
und der ölige Rückstand mit Diethylether kristallisiert. Die abschließende Aufreinigung erfolgt
mittels präparativer HPLC (gelöst in Methanol; A: H2O und B: MeOH, isokratisch, 50 % B) und
anschließender Lyophilisation.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 221-223 °C unter Zersetzung.
Ausbeute: 46 %. 1H-NMR (DMSO-d6): δ = 4,36 (s, 2H, C(9)H2), 4,39 (s, 2H, C(8)H2), 7,68 (t, 1H, C(14)H,
3J = 7,6 Hz), 7,78 (d, 1H, C(13)H, 3J = 8,0 Hz), 7,83 (d, 1H, C(15)H, 3J = 8,0 Hz), 7,93 (d, 1H,
C(3)H, 3J = 5,2 Hz), 7,96 (s, 1H, C(11)H), 8,07 (s, 1H, C(5)H), 8,62 (d, 1H, C(2)H, 3J = 5,2 Hz),
9,39 (s, 1H, N(17)H). 13C-NMR (DMSO-d6): δ = 49,8 (C9), 50,7 (C8), 119,5 (C5), 119,9 (C3), 122,7 und 125,4
(1JC,F = 270 Hz, C16), 125,5 (3JC,F = 4 Hz, C13), 126,9 (3JC,F = 4 Hz, C11), 129,2 (2JC,F = 32 Hz,
C12), 129,6 (C14), 133,8 (C10), 134,3 (C15), 140,4 (C4), 149,4 (C2), 152,8 (C6), 158,6 (C7).
MIR (ṽ): 1615 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C15H13N6F3 + H]+: 334,1154; gefunden: 334,1156.
4-⟨2-{[(4-(1H-Tetrazol-5-yl)pyridin-2-yl)methyl]amino}ethyl⟩phenol (CV)
Die Synthese erfolgt analog zu N-{[4-(1H-Tetrazol-5-yl)pyridin-2-yl]methyl}cyclopropanamin
(CIII) mit folgenden Änderungen: 4-(1-Benzyl-1H-tetrazol-5-yl)picolinaldehyd (CI) (350 mg,
1,3 mmol) wird mit 360 mg Tyramin (2,0 Äq., 2,6 mmol) in 20 mL Methanol für 2,5 h unter
Rückfluss erhitzt. Im Eisbad wird das Azomethin anschließend mit 210 mg NaBH4, vorliegend
als Suspension in 5 mL Wasser, reduziert. Es folgt eine Flüssig-Flüssig-Extraktion mit
Ethylacetat und NaHCO3, wobei die erhaltenen organischen Phasen nach Trocknen über

Experimenteller Teil
190 | Beschreibung der Synthesen
Na2SO4 im Teilvakuum eingeengt werden. Der Überstand wird in etwa 50 mL Methanol
aufgenommen und mit 300 mg Pd/C (10 %, wassernass) versetzt. Nach Evakuieren der
Glasapparatur wird mit Wasserstoff geflutet und bei Raumtemperatur für 4,5 h unter
Wasserstoff-Atmosphäre gerührt. Anschließend wird über Kieselgur abfiltriert, das
Lösungsmittel entfernt und der Rückstand in wenig Ethanol aufgenommen, um aus dieser
Lösung das Zielprodukt mit Hilfe von Aceton/Diethylether (30:70, v/v) zu fällen. Das gewünschte
Produkt wird abfiltriert und abschließend mit wenig Isopropanol und reichlich Aceton
gewaschen.
Beschaffenheit: Leicht bräunlicher amorpher Feststoff.
Schmelzbereich: 263-265 °C unter Zersetzung.
Ausbeute: 54 %. 1H-NMR (DMSO-d6): δ = 2,86 (t, 2H, C(10)H2, 3J = 7,6 Hz), 3,16 (t, 2H, C(9)H2, 3J = 7,6 Hz),
4,37 (s, 2H, C(8)H2), 6,72 (d, 2H, 2×C(13)H, 3J = 8,4 Hz), 7,04 (d, 2H, 2×C(12)H, 3J = 8,4 Hz),
7,92 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,05 (s, 1H, C(5)H), 8,60 (d, 1H, C(2)H, 3J = 5,2 Hz), 8,83 (s,
1H, N(16)H), 9,38 (s, 1H, O(15)H). 13C-NMR (DMSO-d6): δ = 31,1 (C10), 48,6 (C9), 50,9 (C8), 115,4 (2×C13), 119,3 (C5), 119,8
(C3), 127,3 (C11), 129,6 (2×C12), 140,5 (C4), 149,4 (C2), 153,1 (C6), 156,1 (C14), 158,7 (C7).
MIR (ṽ): 2957 (w, νC-H, aliphatisch), 1615 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C15H16N6O + H]+: 296,1386; gefunden: 296,1389.
Pyridin-2,4-dicarbonitril (CVI)
Es werden 360 mg 2-Chlorisonicotinnitril (XXXIV) (2,6 mmol) in 13 mL NMP gelöst und mit
510 mg CuCN (2,2 Äq., 5,7 mmol) versetzt. Der Ansatz reagiert im Mikrowellenreaktor bei
240 °C für 30 min, wobei es zu einer ausgeprägten Schwarzfärbung kommt. Die
Reaktionslösung wird in ein Eisbad mit etwa 100 mL Wasser (+ wenige Tropfen NH3) gegossen
und mit Ethylacetat extrahiert. Nach Trocknen über Na2SO4 und Einengen der vereinigten
organischen Phasen im Teilvakuum wird der Überstand mittels Säulenchromatographie
aufgearbeitet (adsorbiert an Kieselgur (aus MeOH), LM: Petrolether/EtOAc, 80:20, v/v). Eine
abschließende Umkristallisation erfolgt aus n-Hexan mit wenig Diethylether.

Experimenteller Teil
Beschreibung der Synthesen | 191
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 89-90 °C (88-91 °C255).
Ausbeute: 25 %. 1H-NMR (DMSO-d6): δ = 8,25 (d, 1H, C(3)H, 3J = 5,2 Hz), 8,64 (s, 1H, C(5)H), 9,01 (d, 1H,
C(2)H, 3J = 4,8 Hz). 13C-NMR (DMSO-d6): δ = 115,4 und 116,3 (C7 und C8), 121,3 (C4), 129,9 (C3), 130,8 (C5),
133,6 (C6), 152,3 (C2).
MIR (ṽ): 2246 (m, νC≡N) cm−1.
2,4-Di(1H-tetrazol-5-yl)pyridin (CVII)
Pyridin-2,4-dicarbonitril (CVI) (200 mg, 1,5 mmol) wird in 30 mL Toluen gelöst. Anschließend
werden 230 mg Natriumazid (2,3 Äq., 3,5 mmol) und 485 mg Triethylammoniumchlorid (2,3 Äq.,
3,5 mmol) hinzugegeben und der Reaktionsansatz für etwa 24 h bei 105 °C gerührt. Nach
Abkühlen des Reaktionsgemisches auf Raumtemperatur wird das Lösungsmittel im Teilvakuum
entfernt, der Überstand mit wässriger Natronlauge aufgenommen und einmal mit Ethylacetat
gewaschen. Anschließend wird die wässrige Phase im Eisbad mit konzentrierter Salzsäure
angesäuert und die entstandene Suspension für etwa 15 min weitergerührt. Nach Abfiltrieren
des Zielprodukts wird dieses mit Hilfe der präparativen HPLC aufgereinigt (gelöst in
MeOH/DMSO, 50:50, v/v; A: H2O mit 0,1 % Essigsäure und B: ACN, isokratisch, 15 % B).
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 259-262 °C unter Zersetzung.
Ausbeute: 68 %. 1H-NMR (DMSO-d6): δ = 8,23 (d, 1H, C(3)H, 3J = 4,4 Hz), 8,83 (s, 1H, C(5)H), 9,03 (d, 1H,
C(2)H, 3J = 4,8 Hz). 13C-NMR (DMSO-d6): δ = 119,4 (C3), 122,8 (C5), 134,1 (C4), 145,0 (C6), 151,5 (C2), 154,6 und
155,1 (C7 und C8).
MIR (ṽ): 1619 (s, νC=N) cm−1.
HRMS (ESI, m/z), berechnet [C7H5N9 + H]+: 215,0668; gefunden: 215,0662.

Experimenteller Teil
192 | Beschreibung der Synthesen
2-Chlorisonicotinsäure (CVIII)
Für die Synthese werden 2,8 g 2-Chlorisonicotinnitril (XXXIV) (20 mmol) in 20 mL Ethanol gelöst
und mit 2,4 g NaOH (3 Äq., 60 mmol) in 10 mL Wasser versetzt. Die Lösung reagiert 3 h unter
Rückfluss, wobei die Reaktion dann beendet ist, wenn kein Ammoniakgeruch mehr
wahrgenommen werden kann. Anschließend wird das Lösungsmittel im Teilvakuum entfernt
und der resultierende Überstand ausgiebig mit Ethylacetat gewaschen. Das Zielprodukt wird in
Wasser aufgenommen, mit Salzsäure gefällt und anschließend abfiltriert. Wenn nötig, kann die
Substanz aus Wasser umkristallisiert werden.
Beschaffenheit: Farbloser kristalliner Feststoff.
Schmelzbereich: 235-236 °C (234-235 °C371).
Ausbeute: 64 %. 1H-NMR (DMSO-d6): δ = 7,83 (d, 2H, C(3)H und C(5)H, 3J = 4,8 Hz), 8,61 (s, 1H, C(2)H,
3J = 4,8 Hz), 14,01 (s, 1H, C(7)OOH). 13C-NMR (DMSO-d6): δ = 122,2 (C5), 123,5 (C3), 141,8 (C4), 151,1 (C2 und C6), 164,8 (C7).
MIR (ṽ): 1707 (s, vC=O), 1367 (s, νC-Cl) cm−1.
2-[(2-Aminoethyl)amino]isonicotinsäure-Hydrochlorid (CIX)
2-Chlorisonicotinsäure (CVIII) (1,4 g, 8,9 mmol) wird mit 5,9 mL Ethylendiamin (10 Äq.,
89 mmol) im Mikrowellenreaktor für 60 min bei 180 °C erhitzt. Anschließend wird im
Feinvakuum bei 70 °C überschüssiges Amin entfernt, der Überstand mit verdünnter Salzsäure
bis zur sauren Reaktion aufgenommen und am Rotationsverdampfer wieder eingeengt. Der so
erhaltene Feststoff wird mit reichlich Methanol digeriert, die Suspension filtriert und das
Zielprodukt-enthaltende Filtrat erneut im Teilvakuum eingeengt. Dem Rohprodukt werden etwa
15 mL Salzsäure (25 %ig) zugesetzt und auf Grund eventuell eingetretener Amidbildung wird
der Ansatz für 3 h unter Rückfluss erhitzt. Die Menge des Lösungsmittels wird in der Folge auf
etwa die Hälfte reduziert und der Ansatz über Nacht im Kühlschrank gelagert, wobei das

Experimenteller Teil
Beschreibung der Synthesen | 193
Zielprodukt aus der noch stark sauren Lösung auskristallisiert. Der Feststoff wird abfiltriert und
mit einer Mischung aus Tetrahydrofuran und Diethylether (50:50, v/v) mehrmals gewaschen.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 271-273 °C unter Zersetzung.
Ausbeute: 72 %. 1H-NMR (D2O): δ = 3,28 (t, 2H, C(9)H2, 3J = 6,4 Hz), 3,75 (t, 2H, C(8)H2, 3J = 6,0 Hz), 7,27 (d,
1H, C(3)H, 3J = 6,4 Hz), 7,53 (s, 1H, C(5)H), 7,90 (d, 1H, C(2)H, 3J = 6,8 Hz). 13C-NMR (D2O): δ = 37,6 (C9), 39,5 (C8), 112,1 (C5 und C3), 136,4 (C2), 145,8 (C4), 153,3
(C6), 166,6 (C7).
MIR (ṽ): 3348 (m, νN-H), 1727 (s, vC=O) cm−1.
HRMS (ESI, m/z), berechnet [C8H11N3O2 + H]+: 181,0851; gefunden: 181,0852.
2-{[2-(Methylsulfonamido)ethyl]amino}isonicotinsäure (CX)
2-[(2-Aminoethyl)amino]isonicotinsäure-Hydrochlorid (CIX) (400 mg, 1,8 mmol) wird in 10 mL
Acetonitril suspendiert. Dazu wird so viel verdünnte Natronlauge gegeben, bis eine klare Lösung
entsteht. Dem Ansatz werden unter Eiskühlung 420 µL Methansulfonylchlorid (3 Äq., 5,4 mmol)
hinzugefügt und anschließend bei Raumtemperatur für 20 h gerührt. Das Lösungsmittel wird
dann im Teilvakuum entfernt und der Überstand chromatographisch aufgearbeitet (adsorbiert
an Kieselgur (aus ammoniakalischem Methanol), LM: EtOAc/MeOH/Ammoniak, 60:40:1, v/v/v).
Die Zielfraktionen werden vom Laufmittel befreit und anschließend aus wässrigem Ethanol
(EtOH/Wasser, 95:5, v/v) umkristallisiert. Abschließend wird das Zielprodukt mittels präparativer
HPLC gereinigt (gelöst in Wasser; A: H2O mit 0,1 % Essigsäure und B: MeOH, isokratisch, 10 %
B) und lyophilisiert.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 273-275 °C unter Zersetzung.
Ausbeute: 29 %. 1H-NMR (DMSO-d6): δ = 2,89 (s, 3H, C(10)H3), 3,10 (t, 2H, C(9)H2, 3J = 6,4 Hz), 3,39 (t, 2H,
C(8)H2, 3J = 6,4 Hz), 6,88 (d, 1H, C(3)H, 3J = 5,6 Hz), 6,97 (m, 2H, C(5)H und N(12)H), 7,09 (d,
1H, C(2)H, 3J = 6,0 Hz), 8,10 (s, 1H, N(11)H), 13,25 (s, 1H, C(7)OOH).

Experimenteller Teil
194 | Beschreibung der Synthesen
13C-NMR (DMSO-d6): δ = 39,5 (C10), 40,9 (C9), 41,8 (C8), 108,5 (C5), 110,3 (C3), 138,8 (C2),
148,4 (C4), 159,1 (C6), 166,7 (C7).
MIR (ṽ): 3156 (m, vN-H), 1677 (s, vC=O) cm−1.
HRMS (ESI, m/z), berechnet [C9H13N3O4S + H]+: 259,0627; gefunden: 259,0626.
2-[(3-Hydroxypropyl)amino]isonicotinsäure (CXI)
Es werden 1,0 g 2-Chlorisonicotinsäure (CVIII) (7,2 mmol) mit 10 mL 3-Aminopropan-1-ol
(18 Äq., 130 mmol) im Rundkolben für 22 h bei 120 °C erhitzt. Anschließend wird der Ansatz
mit 50 mL Wasser und 25 mL konzentrierter Salzsäure verdünnt (bis etwa pH 1-2) und während
Erhitzen unter Rückfluss für weitere 16 h gerührt. Das Gesamtvolumen wird dann im Teilvakuum
auf etwa ein Viertel des Ausgangswertes reduziert und der Ansatz bei 5 °C zur Kristallisation
belassen. Nach 12 h wird das Rohprodukt abfiltriert und mit Tetrahydrofuran nachgewaschen.
Die abschließende Aufreinigung erfolgt mittels präparativer HPLC (gelöst in Wasser; A: H2O und
B: MeOH, isokratisch, 30 % B) und Lyophilisation.
Beschaffenheit: Farbloser amorpher Feststoff.
Schmelzbereich: 194-196 °C.
Ausbeute: 26 %. 1H-NMR (DMSO-d6): δ = 1,73 (m, 2H, C(9)H2, 3J = 6,4 Hz), 3,39 (s, 2H, C(8)H2), 3,50 (t, 2H,
C(10)H2, 3J = 6,4 Hz), 6,96 (d, 1H, C(3)H, 3J = 5,6 Hz), 7,28 (s, 1H, C(5)H), 8,02 (d, 1H, C(2)H, 3J = 5,6 Hz), 8,14 (s, 1H, N(11)H). 13C-NMR (DMSO-d6): δ = 31,5 (C9), 38,6 (C8), 58,1 (C10), 109,8 (C5), 111,7 (C3), 140,8 (C4),
142,4 (C2), 156,3 (C6), 165,5 (C7).
MIR (ṽ): 3240 (m, νO-H), 3182 (m, νN-H), 1713 (s, vC=O) cm−1.
HRMS (ESI, m/z), berechnet [C9H12N2O3 + H]+: 196,0848; gefunden: 196,0841.

Experimenteller Teil
Beschreibung der validierten SFC-MS-Methode | 195
4.4 Beschreibung der validierten SFC-MS-Methode
Die nachfolgenden Arbeitsanweisungen beziehen sich auf die optimierte und validierte SFC-
MS-Methode für die Trennung von rac-Ketamin, rac-Norketamin, rac-Norketamin-d4, rac-
Dehydronorketamin und rac-6-Hydroxynorketamin aus humanem Urin.
4.4.1 Probenvorbereitung
Eingefrorene Proben (−80 °C) oder tiefgekühlte Stammlösungen in Methanol (−21 °C, 1 g/mL
oder 100 µg/mL) werden langsam bei Raumtemperatur aufgetaut. Zu 1 mL Probe werden 10 µL
NK-d4 (Gesamtkonzentration 1 µg/mL), 750 µL Natriumcarbonat-Lösung (gesättigte Lösung
1 zu 1 mit Wasser verdünnt) und 4 mL MTBE gegeben. Nach dem automatisierten
Ausschüttelvorgang von 15 min und anschließendem fünfminütigen Abzentrifugieren bei
3200×g, wird die organische Phase abgenommen und im Stickstoff-Strom bei Raumtemperatur
abgedampft. Die Rekonstitution des Überstandes erfolgt in 100 µL Methanol.
Für die Kalibiergerade werden pro Messtag 6 Punkte gewählt (5, 10, 25, 50, 100 und 200 ng/mL,
Stammlösungen in Methanol). Die Proben werden wie oben beschrieben hergestellt, mit dem
Unterschied, dass statt 1 mL Probandenurin 1 mL eines Ketamin-unbelasteten Urins (zur
Verfügung gestellt von Labormitarbeitern) mitsamt zugesetzter Stammlösung verwendet wird.
Zur Überprüfung der Messungen eines Tages sollen weiterhin Qualitätskontrollen hergestellt
werden, welche mindestens drei verschiedene Konzentrationen (gemessen als Duplikat) oder
mindestens 5 % der Gesamtanzahl der Proben repräsentieren und den gesamten Messbereich
abdecken (z. B. 15, 100 und 200 ng/mL). Die Herstellung erfolgt auch hier analog den
Probanden- bzw. Kalibrierproben.
4.4.2 Säulenauswahl
Für die Methode wird die Säule Phenomenex Lux Amylose-2 (150×4,6 mm, 5 µm) verwendet.
Diese wird von einer Vorsäule (gleiches Material, 4×3,0 mm, 5 µm), eingespannt im
SecurityGuard System von Phenomenex, geschützt.

Experimenteller Teil
196 | Beschreibung der validierten SFC-MS-Methode
4.4.3 Chromatographische Parameter
Gerät Parameter Wert Pumpen Mob. Phase (A):
CO2 0–7 min → 8 %B 7,01–7,25 min → auf 20 %B 7,26–11 min → 20 %B 11,01–11,25 min → auf 8 %B 11,26–15 min → 8 %B
Mob. Phase (B): Isopropanol + 0,075 % NH3
Flussrate (A+B) 3,0 mL/min Mob. Phase (C): Methanol + 0,1 % HCOOH
0–15 min → 0,3 mL/min
Autosampler Temperatur 6 °C Injektionsvolumen 5 µL Säulenofen Temperatur 30 °C Rückdruckregulator A Temperatur 50 °C Druck 150 bar Rückdruckregulator B Temperatur 50 °C Druck 400 bar
4.4.4 MS-Parameter
Parameter Wert ESI Positiver Modus, SIM [M + H]+ 222, 224, 228, 238, 240 Event Time 0,05 sec Interface Voltage 4,5 kV Nebulizing-Gas 1,5 L/min Drying-Gas 12 L/min Interface-Temperatur 350 °C Heat-Block-Temperatur 300 °C Desolvation-Line-Temperatur 250 °C
4.4.5 Mathematische Prüfung auf Linearität358
Nach dem optischen Abgleich der linearen und quadratischen Regression gegeneinander und
dem Vergleich des Bestimmheitsmaßes R² für beide Arten der Kalibration, dient der
Linearitätstest nach Mandel der Abschätzung des richtigeren mathematischen Modells. Es wird
geprüft, ob die Regression 2. Ordnung (quadratisch) signifikant besser zutrifft als die Regression
1. Ordnung (linear). Zur Prüfung werden die jeweiligen Quadrate der Reststandardabweichung
aus linearer und quadratischer Regression hinzugezogen.
Im ersten Schritt werden dafür alle erforderlichen Variablen für den Regressionstyp 1. Ordnung � = � ∗ � + � und 2. Ordnung � = � ∗ �� + ∗ � + � bestimmt. Mit Hilfe dieser Regressionen
können dann die Residuen (entspricht dem vertikalen Abstand eines gemessenen Punktes von
der errechneten Geraden bzw. Kurve) wie folgt berechnet werden:

Experimenteller Teil
Beschreibung der validierten SFC-MS-Methode | 197
�� = �� − ��� �� = Residuen �� = Messsignal an i-ter Stelle ��� = der zu �� passende berechnete Wert aus der Regressionsfunktion
Die Standardabweichung der Residuen für eine Regression wird als Reststandardabweichung
sy bezeichnet. Diese Reststandardabweichungen können an dieser Stelle (für Funktion 1. und
2. Ordnung) bereits miteinander verglichen werden. Kleinere Werte geben an, dass die
Streuung der Messwerte um die Regressionsgerade bzw. um die Kurve kleiner und damit auch
geeigneter zur Beschreibung des Verhältnisses von Messsignal zur Analytenkonzentration ist.
Die Reststandardabweichungen werden wie folgt berechnet:
��� = 1� − 2"(��$���)²'�(�
für lineare Regressionen oder
���) = 1� − 3"(��$���)²'�(�
für quadratische Regressionen.
Wird hierbei herausgefunden, dass die Reststandardabweichung der linearen Regression
größer ist als die der quadratischen Regression, so sollte eine Funktion 2. Ordnung gewählt
werden.
Um eine mathematische Signifikanz nachzuweisen, kann der Prüfwert PW ermittelt werden.
Vorher ist jedoch die Differenz der Abweichungsvarianzen DS² zu ermitteln:
*+� = (� − 2) ∗ ���� − (� − 3) ∗ ���)² �, = *+²���)²
Der erhaltene Prüfwert wird mit dem zutreffenden Wert aus einer F-verteilten Tabelle verglichen
(� = 99%, �� = 1, �� = � − 3). Für den Fall PW < F-Wert gilt, dass der Unterschied der linearen
Regression gegenüber der quadratischen nicht signifikant ist. Hierbei sollte das lineare Modell
verwendet werden. Im umgekehrten Fall PW > F-Wert gilt, dass die quadratische Regression
der linearen signifikant überlegen ist. Hierbei besteht keine direkte Proportionalität zwischen
Messsignal und Analytenkonzentration.

198 | Beschreibung der validierten SFC-MS-Methode
4.4.6 Sonstige experimentelle Daten
Zurückgerechnete Werte aus den Kalibrierläufen der Validierung der SFC-MS-Methode
Norketamine En1
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5
Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 5,180 5,647 4,393 5,022 5,600 5,168 0,509 9,9 3,4 10 9,345 10,861 9,038 9,929 10,683 9,971 0,800 8,0 –0,3 25 24,349 24,984 27,505 24,958 24,574 25,274 1,275 5,0 1,1 50 52,129 45,437 51,049 50,472 47,709 49,359 2,733 5,5 –1,3 100 98,808 103,408 98,467 99,682 101,874 100,448 2,121 2,1 0,4 200 200,173 199,366 200,266 200,051 199,664 199,904 0,378 0,2 –0,1
Norketamine En2
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 4,764 5,498 4,657 5,248 5,329 5,099 0,368 7,2 2,0 10 10,301 10,252 8,884 10,128 10,866 10,086 0,730 7,2 0,9 25 23,721 24,814 27,041 24,546 24,518 24,928 1,250 5,0 –0,3 50 56,789 48,731 51,773 49,377 49,675 51,269 3,290 6,4 2,5 100 96,091 100,934 98,134 100,605 100,433 99,239 2,079 2,1 –0,8 200 200,471 199,85 200,312 199,895 199,914 200,088 0,283 0,1 0,1

Beschreibung der validierten SFC-MS-Methode | 199
2S,6S-Hydroxynorketamine
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 5,185 5,698 5,784 4,799 5,612 5,416 0,415 7,7 8,3 10 8,909 10,375 8,345 8,390 11,177 9,439 1,272 13,5 –5,6 25 24,203 28,717 23,923 27,152 24,703 25,740 2,097 8,1 3,0 50 50,649 54,896 51,875 53,201 47,540 51,632 2,779 5,4 3,3 100 99,998 96,019 99,207 97,511 101,788 98,905 2,229 2,3 –1,1 200 199,980 200,505 200,131 200,327 199,728 200,134 0,301 0,2 0,1
2R,6R-Hydroxynorketamine
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 6,503 5,740 5,429 6,121 6,052 5,969 0,406 6,8 19,4 10 9,963 7,588 8,577 9,398 11,131 9,331 1,346 14,4 –6,7 25 21,673 28,043 24,201 29,194 23,778 25,378 3,136 12,4 1,5 50 48,600 63,663 51,780 54,355 47,675 53,215 6,413 12,1 6,4 100 102,383 90,157 99,106 96,116 102,416 98,036 5,123 5,2 –2,0 200 199,532 200,948 200,189 200,522 199,455 200,129 0,640 0,3 0,1

200 | Beschreibung der validierten SFC-MS-Methode
R-Ketamine
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 4,688 5,373 4,947 4,429 5,316 4,951 0,404 8,2 –1,0 10 9,635 10,330 8,940 10,007 10,168 9,816 0,553 5,6 –1,8 25 26,042 24,474 27,610 24,466 25,579 25,634 1,302 5,1 2,5 50 47,654 47,637 47,672 53,078 47,492 48,707 2,445 5,0 –2,6 100 101,290 101,865 100,729 98,022 101,426 100,666 1,533 1,5 0,7 200 199,810 199,689 199,932 200,305 199,813 199,910 0,237 0,1 0,0
S-Ketamine
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 4,980 4,630 4,521 4,412 5,549 4,818 0,461 9,6 –3,6 10 9,923 10,323 9,523 9,764 9,643 9,835 0,310 3,2 –1,6 25 26,456 24,204 28,709 24,272 23,208 25,370 2,212 8,7 1,5 50 54,580 52,954 56,207 53,873 49,698 53,462 2,418 4,5 6,9 100 96,465 97,975 94,956 97,507 101,098 97,600 2,273 2,3 –2,4 200 200,542 200,400 200,685 200,450 199,689 200,353 0,387 0,2 0,2
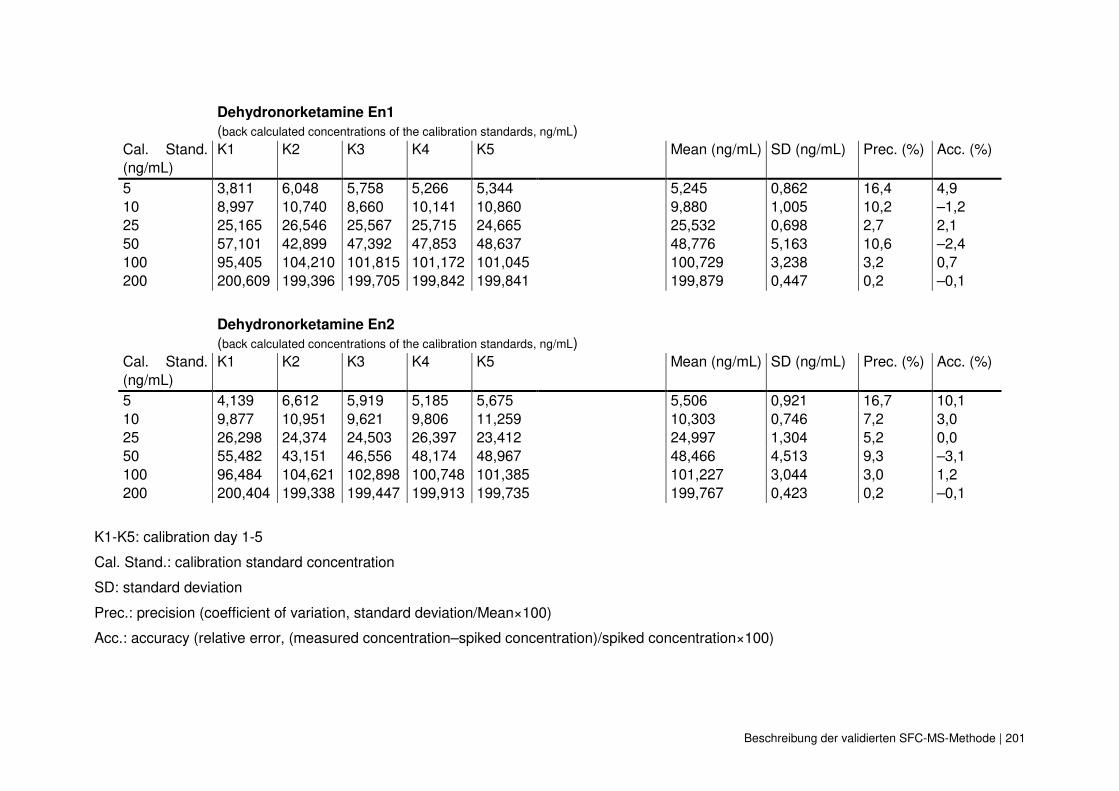
Beschreibung der validierten SFC-MS-Methode | 201
Dehydronorketamine En1
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 3,811 6,048 5,758 5,266 5,344 5,245 0,862 16,4 4,9 10 8,997 10,740 8,660 10,141 10,860 9,880 1,005 10,2 –1,2 25 25,165 26,546 25,567 25,715 24,665 25,532 0,698 2,7 2,1 50 57,101 42,899 47,392 47,853 48,637 48,776 5,163 10,6 –2,4 100 95,405 104,210 101,815 101,172 101,045 100,729 3,238 3,2 0,7 200 200,609 199,396 199,705 199,842 199,841 199,879 0,447 0,2 –0,1
Dehydronorketamine En2
(back calculated concentrations of the calibration standards, ng/mL)
Cal. Stand. (ng/mL)
K1 K2 K3 K4 K5 Mean (ng/mL) SD (ng/mL) Prec. (%) Acc. (%)
5 4,139 6,612 5,919 5,185 5,675 5,506 0,921 16,7 10,1 10 9,877 10,951 9,621 9,806 11,259 10,303 0,746 7,2 3,0 25 26,298 24,374 24,503 26,397 23,412 24,997 1,304 5,2 0,0 50 55,482 43,151 46,556 48,174 48,967 48,466 4,513 9,3 –3,1 100 96,484 104,621 102,898 100,748 101,385 101,227 3,044 3,0 1,2 200 200,404 199,338 199,447 199,913 199,735 199,767 0,423 0,2 –0,1
K1-K5: calibration day 1-5
Cal. Stand.: calibration standard concentration
SD: standard deviation
Prec.: precision (coefficient of variation, standard deviation/Mean×100)
Acc.: accuracy (relative error, (measured concentration–spiked concentration)/spiked concentration×100)

Literaturverzeichnis
202 | Literatur
5 Literaturverzeichnis
1. Waddington, C. H., Organisers and Genes. Cambridge, 1940.
2. Russo, V. E. A.; Martienssen, R. A.; Riggs, A. D., Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbour, NY, 1997.
3. Kucharski, R.; Maleszka, J.; Foret, S.; Maleszka, R., Nutritional Control of Reproductive Status in Honeybees via DNA Methylation. Science 2008, 319, 1827-1830.
4. Mariño-Ramírez, L.; Kann, M. G.; Shoemaker, B. A.; Landsman, D., Histone structure and nucleosome stability. Expert Review of Proteomics 2005, 2, 719-729.
5. Moss, T. J.; Wallrath, L. L., Connections between epigenetic gene silencing and human disease. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis 2007, 618, 163-174.
6. Sun, H.; Kennedy, P. J.; Nestler, E. J., Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology 2013, 38, 124-137.
7. Arrowsmith, C. H.; Bountra, C.; Fish, P. V.; Lee, K.; Schapira, M., Epigenetic protein families: a new frontier for drug discovery. Nature Reviews Drug Discovery 2012, 11, 384-400.
8. Okano, M.; Xie, S.; Li, E., Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nature Genetics 1998, 19, 219-220.
9. Jin, B.; Li, Y.; Robertson, K. D., DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes & Cancer 2011, 2, 607-617.
10. Janssen-Cilag International NV, Fachinformation Dacogen 50 mg Pulver für ein Konzentrat zur Herstellung einer Infusionslösung (Decitabin). Stand August 2017.
11. Celgene Europe Ltd, Fachinformation VIDAZA 25 mg/ml Pulver zur Herstellung einer Injektionssuspension (Azacytidin). Stand Mai 2017.
12. Jones, P. A.; Issa, J.-P. J.; Baylin, S., Targeting the cancer epigenome for therapy. Nature Reviews Genetics 2016, 17, 630-635.
13. Tahiliani, M.; Koh, K. P.; Shen, Y.; Pastor, W. A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L. M.; Liu, D. R.; Aravind, L.; Rao, A., Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930-935.
14. Delhommeau, F.; Dupont, S.; Valle, V. D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; Lécluse, Y.; Plo, I.; Dreyfus, F. J.; Marzac, C.; Casadevall, N.; Lacombe, C.; Romana, S. P.; Dessen, P.; Soulier, J.; Viguié, F.; Fontenay, M.; Vainchenker, W.; Bernard, O. A., Mutation in TET2 in Myeloid Cancers. New England Journal of Medicine 2009, 360, 2289-2301.
15. Lombardi, P. M.; Cole, K. E.; Dowling, D. P.; Christianson, D. W., Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Current Opinion in Structural Biology 2011, 21, 735-743.
16. Shi, Y.; Zhou, Y.; Wang, S.; Zhang, Y., Sirtuin Deacetylation Mechanism and Catalytic Role of the Dynamic Cofactor Binding Loop. The Journal of Physical Chemistry Letters 2013, 4, 491-495.
17. Richon, V. M., Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. British Journal Of Cancer 2006, 95, S2-S6.
18. VanderMolen, K. M.; McCulloch, W.; Pearce, C. J.; Oberlies, N. H., Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. The Journal Of Antibiotics 2011, 64, 525-531.

Literaturverzeichnis
Literatur | 203
19. Glozak, M. A.; Sengupta, N.; Zhang, X.; Seto, E., Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15-23.
20. Rossetto, D.; Avvakumov, N.; Côté, J., Histone phosphorylation. Epigenetics 2012, 7, 1098-1108.
21. Hassa, P. O.; Haenni, S. S.; Elser, M.; Hottiger, M. O., Nuclear ADP-Ribosylation Reactions in Mammalian Cells: Where Are We Today and Where Are We Going? Microbiology and Molecular Biology Reviews 2006, 70, 789-829.
22. Cao, J.; Yan, Q., Histone Ubiquitination and Deubiquitination in Transcription, DNA Damage Response, and Cancer. Frontiers in Oncology 2012, 2, 1-9.
23. Cubeñas-Potts, C.; Matunis, M. J., SUMO: A Multifaceted Modifier of Chromatin Structure and Function. Developmental Cell 2013, 24, 1-12.
24. Spannhoff, A.; Sippl, W.; Jung, M., Cancer treatment of the future: Inhibitors of histone methyltransferases. The International Journal of Biochemistry & Cell Biology 2009, 41, 4-11.
25. Liu, Y.; Liu, K.; Qin, S.; Xu, C.; Min, J., Epigenetic targets and drug discovery: Part 1: Histone methylation. Pharmacology & Therapeutics 2014, 143, 275-294.
26. Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D. E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K., High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823-837.
27. Rosenfeld, J. A.; Wang, Z.; Schones, D. E.; Zhao, K.; DeSalle, R.; Zhang, M. Q., Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics 2009, 10, 1-11.
28. Steger, D. J.; Lefterova, M. I.; Ying, L.; Stonestrom, A. J.; Schupp, M.; Zhuo, D.; Vakoc, A. L.; Kim, J.-E.; Chen, J.; Lazar, M. A.; Blobel, G. A.; Vakoc, C. R., DOT1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled with Gene Transcription in Mammalian Cells. Molecular and Cellular Biology 2008, 28, 2825-2839.
29. Takeuchi, T.; Yamazaki, Y.; Katoh-Fukui, Y.; Tsuchiya, R.; Kondo, S.; Motoyama, J.; Higashinakagawa, T., Gene trap capture of a novel mouse gene, jumonji, required for neural tube formation. Genes & Development 1995, 9, 1211-1222.
30. Takeuchi, T.; Watanabe, Y.; Takano‐Shimizu, T.; Kondo, S., Roles of jumonji and jumonji family genes in chromatin regulation and development. Developmental Dynamics 2006, 235, 2449-2459.
31. Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J. R.; Cole, P. A.; Casero, R. A.; Shi, Y., Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941-953.
32. Rotili, D.; Mai, A., Targeting Histone Demethylases: A New Avenue for the Fight against Cancer. Genes & Cancer 2011, 2, 663-679.
33. Labbé, R. M.; Holowatyj, A.; Yang, Z.-Q., Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. American Journal of Translational Research 2014, 6, 1-15.
34. Kooistra, S. M.; Helin, K., Molecular mechanisms and potential functions of histone demethylases. Nature Reviews Molecular Cell Biology 2012, 13, 297-311.
35. Pedersen, M. T.; Helin, K., Histone demethylases in development and disease. Trends in Cell Biology 2010, 20, 662-671.
36. Johansson, C.; Tumber, A.; Che, K.; Cain, P.; Nowak, R.; Gileadi, C.; Oppermann, U., The roles of Jumonji-type oxygenases in human disease. Epigenomics 2014, 6, 89-120.

Literaturverzeichnis
204 | Literatur
37. Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J., Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512-520.
38. Hayami, S.; Kelly John, D.; Cho, H. S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field Helen, I.; Neal David, E.; Yamaue, H.; Ponder Bruce, A. J.; Nakamura, Y.; Hamamoto, R., Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. International Journal of Cancer 2010, 128, 574-586.
39. Cloos, P. A. C.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K. H.; Helin, K., The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307-311.
40. Fodor, B. D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R. J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K.; Schotta, G.; Jenuwein, T., Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes & Development 2006, 20, 1557-1562.
41. Guerra-Calderas, L.; González-Barrios, R.; Herrera, L. A.; Cantú de León, D.; Soto-Reyes, E., The role of the histone demethylase KDM4A in cancer. Cancer Genetics 2015, 208, 215-224.
42. Stavropoulos, P.; Blobel, G.; Hoelz, A., Crystal structure and mechanism of human lysine-specific demethylase-1. Nature Structural & Molecular Biology 2006, 13, 626-632.
43. Huang, J.; Sengupta, R.; Espejo, A. B.; Lee, M. G.; Dorsey, J. A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M. T.; Jenuwein, T.; Berger, S. L., p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105-108.
44. Wang, J.; Hevi, S.; Kurash, J. K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; Gaudet, F.; Li, E.; Chen, T., The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nature Genetics 2008, 41, 125-129.
45. Gaweska, H.; Fitzpatrick, P. F., Structures and Mechanism of the Monoamine Oxidase Family. Biomolecular Concepts 2011, 2, 365-377.
46. Chen, Y.; Yang, Y.; Wang, F.; Wan, K.; Yamane, K.; Zhang, Y.; Lei, M., Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proceedings of the National Academy of Sciences 2006, 103, 13956-13961.
47. Zhang, Q.; Qi, S.; Xu, M.; Yu, L.; Tao, Y.; Deng, Z.; Wu, W.; Li, J.; Chen, Z.; Wong, J., Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Research 2012, 23, 225-241.
48. Tsukada, Y.-i.; Fang, J.; Erdjument-Bromage, H.; Warren, M. E.; Borchers, C. H.; Tempst, P.; Zhang, Y., Histone demethylation by a family of JmjC domain-containing proteins. Nature 2005, 439, 811-816.
49. Sinnecker, S.; Svensen, N.; Barr, E. W.; Ye, S.; Bollinger, J. M.; Neese, F.; Krebs, C., Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates in Taurine: α-Ketoglutarate Dioxygenase from Escherichia coli and Its His99Ala Ligand Variant. Journal of the American Chemical Society 2007, 129, 6168-6179.
50. Tian, G.; Su, H.; Liu, Y., Mechanism of Sulfoxidation and C–S Bond Formation Involved in the Biosynthesis of Ergothioneine Catalyzed by Ergothioneine Synthase (EgtB). ACS Catalysis 2018, 8, 5875-5889.
51. Clissold, P. M.; Ponting, C. P., JmjC: cupin metalloenzyme-like domains in jumonji, hairless and phospholipase A2β. Trends in Biochemical Sciences 2001, 26, 7-9.
52. Couture, J.-F.; Collazo, E.; Ortiz-Tello, P. A.; Brunzelle, J. S.; Trievel, R. C., Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nature Structural & Molecular Biology 2007, 14, 689-695.

Literaturverzeichnis
Literatur | 205
53. Chen, Z.; Zang, J.; Whetstine, J.; Hong, X.; Davrazou, F.; Kutateladze, T. G.; Simpson, M.; Mao, Q.; Pan, C.-H.; Dai, S.; Hagman, J.; Hansen, K.; Shi, Y.; Zhang, G., Structural Insights into Histone Demethylation by JMJD2 Family Members. Cell 2006, 125, 691-702.
54. Rose, N. R.; Ng, S. S.; Mecinović, J.; Liénard, B. M. R.; Bello, S. H.; Sun, Z.; McDonough, M. A.; Oppermann, U.; Schofield, C. J., Inhibitor Scaffolds for 2-Oxoglutarate-Dependent Histone Lysine Demethylases. Journal of Medicinal Chemistry 2008, 51, 7053-7056.
55. Skarnes, W. C.; Rosen, B.; West, A. P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A. O.; Thomas, M.; Harrow, J.; Cox, T.; Jackson, D.; Severin, J.; Biggs, P.; Fu, J.; Nefedov, M.; de Jong, P. J.; Stewart, A. F.; Bradley, A., A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337-342.
56. Li, B.-X.; Zhang, M.-C.; Luo, C.-L.; Yang, P.; Li, H.; Xu, H.-M.; Xu, H.-F.; Shen, Y.-W.; Xue, A.-M.; Zhao, Z.-Q., Effects of RNA interference-mediated gene silencing of JMJD2A on human breast cancer cell line MDA-MB-231 in vitro. Journal of Experimental & Clinical Cancer Research 2011, 30, 1-9.
57. Kim, T. D.; Shin, S.; Berry William, L.; Oh, S.; Janknecht, R., The JMJD2A demethylase regulates apoptosis and proliferation in colon cancer cells. Journal of Cellular Biochemistry 2011, 113, 1368-1376.
58. Wan, M.; Liang, J.; Xiong, Y.; Shi, F.; Zhang, Y.; Lu, W.; He, Q.; Yang, D.; Chen, R.; Liu, D.; Barton, M.; Songyang, Z., The Trithorax Group Protein Ash2l Is Essential for Pluripotency and Maintaining Open Chromatin in Embryonic Stem Cells. Journal of Biological Chemistry 2013, 288, 5039-5048.
59. Maes, T.; Carceller, E.; Salas, J.; Ortega, A.; Buesa, C., Advances in the development of histone lysine demethylase inhibitors. Current Opinion in Pharmacology 2015, 23, 52-60.
60. McAllister, T. E.; England, K. S.; Hopkinson, R. J.; Brennan, P. E.; Kawamura, A.; Schofield, C. J., Recent Progress in Histone Demethylase Inhibitors. Journal of Medicinal Chemistry 2016, 59, 1308-1329.
61. Zheng, Y. C.; Ma, J.; Wang, Z.; Li, J.; Jiang, B.; Zhou, W.; Shi, X.; Wang, X.; Zhao, W.; Liu, H. M., A Systematic Review of Histone Lysine‐Specific Demethylase 1 and Its Inhibitors. Medicinal Research Reviews 2015, 35, 1032-1071.
62. Lin, H.; Li, Q.; Li, Q.; Zhu, J.; Gu, K.; Jiang, X.; Hu, Q.; Feng, F.; Qu, W.; Chen, Y.; Sun, H., Small molecule KDM4s inhibitors as anti-cancer agents. Journal of Enzyme Inhibition and Medicinal Chemistry 2018, 33, 777-793.
63. Kaniskan, H. Ü.; Martini, M. L.; Jin, J., Inhibitors of Protein Methyltransferases and Demethylases. Chemical Reviews 2018, 118, 989-1068.
64. Hauser, A.-T.; Robaa, D.; Jung, M., Epigenetic small molecule modulators of histone and DNA methylation. Current Opinion in Chemical Biology 2018, 45, 73-85.
65. Lee, M. G.; Wynder, C.; Schmidt, D. M.; McCafferty, D. G.; Shiekhattar, R., Histone H3 Lysine 4 Demethylation Is a Target of Nonselective Antidepressive Medications. Chemistry & Biology 2006, 13, 563-567.
66. Kohlweyer, U. Study of Sensitization of Non-M3 AML Blasts to ATRA by Epigenetic Treatment With Tranylcypromine (TCP) (TRANSATRA) (NCT02717884). (accessed März 2018, clinicaltrials.gov/ct2/show/NCT02717884?term=tranylcypromin&draw=3&rank=5).
67. Watts, J. Phase 1 Study of TCP-ATRA for Adult Patients With AML and MDS (TCP-ATRA) (NCT02273102). (accessed März 2018, clinicaltrials.gov/ct2/show/NCT02273102?term=tranylcypromin&draw=3&rank=7).

Literaturverzeichnis
206 | Literatur
68. GlaxoSmithKline. Investigation of GSK2879552 in Subjects With Relapsed/Refractory Small Cell Lung Carcinoma (NCT02034123). (accessed Juni 2018, clinicaltrials.gov/ct2/show/NCT02034123?term=GSK2879552&rank=3).
69. Oryzon Genomics. A phase I study of Human Pharmacokinetics and Safety of ORY-1001, and LSD1 inhibitor, in relapsed or refractory acute leukaemia (AL) (EudraCT Number: 2013-002447-29). (accessed Juni 2018, clinicaltrialsregister.eu/ctr-search/trial/2013-002447-29/GB).
70. Culhane, J. C.; Szewczuk, L. M.; Liu, X.; Da, G.; Marmorstein, R.; Cole, P. A., A Mechanism-Based Inactivator for Histone Demethylase LSD1. Journal of the American Chemical Society 2006, 128, 4536-4537.
71. Prusevich, P.; Kalin, J. H.; Ming, S. A.; Basso, M.; Givens, J.; Li, X.; Hu, J.; Taylor, M. S.; Cieniewicz, A. M.; Hsiao, P.-Y.; Huang, R.; Roberson, H.; Adejola, N.; Avery, L. B.; Casero, R. A.; Taverna, S. D.; Qian, J.; Tackett, A. J.; Ratan, R. R.; McDonald, O. G.; Feinberg, A. P.; Cole, P. A., A Selective Phenelzine Analogue Inhibitor of Histone Demethylase LSD1. ACS Chemical Biology 2014, 9, 1284-1293.
72. Sorna, V.; Theisen, E. R.; Stephens, B.; Warner, S. L.; Bearss, D. J.; Vankayalapati, H.; Sharma, S., High-Throughput Virtual Screening Identifies Novel N′-(1-Phenylethylidene)-benzohydrazides as Potent, Specific, and Reversible LSD1 Inhibitors. Journal of Medicinal Chemistry 2013, 56, 9496-9508.
73. Zhou, Y.; Li, Y.; Wang, W.-J.; Xiang, P.; Luo, X.-M.; Yang, L.; Yang, S.-Y.; Zhao, Y.-L., Synthesis and biological evaluation of novel (E)-N′-(2,3-dihydro-1H-inden-1-ylidene) benzohydrazides as potent LSD1 inhibitors. Bioorganic & Medicinal Chemistry Letters 2016, 26, 4552-4557.
74. Wu, F.; Zhou, C.; Yao, Y.; Wei, L.; Feng, Z.; Deng, L.; Song, Y., 3-(Piperidin-4-ylmethoxy)pyridine Containing Compounds Are Potent Inhibitors of Lysine Specific Demethylase 1. Journal of Medicinal Chemistry 2016, 59, 253-263.
75. Chang, K. H.; King Oliver, N. F.; Tumber, A.; Woon Esther, C. Y.; Heightman Tom, D.; McDonough Michael, A.; Schofield Christopher, J.; Rose Nathan, R., Inhibition of Histone Demethylases by 4‐Carboxy‐2,2′‐Bipyridyl Compounds. ChemMedChem 2011, 6, 759-764.
76. King, O. N. F.; Li, X. S.; Sakurai, M.; Kawamura, A.; Rose, N. R.; Ng, S. S.; Quinn, A. M.; Rai, G.; Mott, B. T.; Beswick, P.; Klose, R. J.; Oppermann, U.; Jadhav, A.; Heightman, T. D.; Maloney, D. J.; Schofield, C. J.; Simeonov, A., Quantitative High-Throughput Screening Identifies 8-Hydroxyquinolines as Cell-Active Histone Demethylase Inhibitors. PLOS ONE 2010, 5, e15535.
77. Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A. M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; Babinski, D. J.; Frantz, D. E.; Brekken, R. A.; Quinn, A. M.; Simeonov, A.; Easmon, J.; Martinez, E. D., A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nature Communications 2013, 4, 2035.
78. Duan, L.; Rai, G.; Roggero, C.; Zhang, Q.-J.; Wei, Q.; Ma, Shi H.; Zhou, Y.; Santoyo, J.; Martinez, Elisabeth D.; Xiao, G.; Raj, Ganesh V.; Jadhav, A.; Simeonov, A.; Maloney, David J.; Rizo, J.; Hsieh, J.-T.; Liu, Z.-P., KDM4/JMJD2 Histone Demethylase Inhibitors Block Prostate Tumor Growth by Suppressing the Expression of AR and BMYB-Regulated Genes. Chemistry & Biology 2015, 22, 1185-1196.
79. Thinnes, C. C.; Tumber, A.; Yapp, C.; Scozzafava, G.; Yeh, T.; Chan, M. C.; Tran, T. A.; Hsu, K.; Tarhonskaya, H.; Walport, L. J.; Wilkins, S. E.; Martinez, E. D.; Muller, S.; Pugh, C. W.; Ratcliffe, P. J.; Brennan, P. E.; Kawamura, A.; Schofield, C. J., Betti reaction enables efficient synthesis of 8-hydroxyquinoline inhibitors of 2-oxoglutarate oxygenases. Chemical Communications 2015, 51, 15458-15461.

Literaturverzeichnis
Literatur | 207
80. Roatsch, M.; Robaa, D.; Pippel, M.; Nettleship, J. E.; Reddivari, Y.; Bird, L. E.; Hoffmann, I.; Franz, H.; Owens, R. J.; Schüle, R.; Flaig, R.; Sippl, W.; Jung, M., Substituted 2-(2-aminopyrimidin-4-yl)pyridine-4-carboxylates as potent inhibitors of JumonjiC domain-containing histone demethylases. Future Medicinal Chemistry 2016, 8, 1553-1571.
81. Korczynska, M.; Le, D. D.; Younger, N.; Gregori-Puigjané, E.; Tumber, A.; Krojer, T.; Velupillai, S.; Gileadi, C.; Nowak, R. P.; Iwasa, E.; Pollock, S. B.; Ortiz Torres, I.; Oppermann, U.; Shoichet, B. K.; Fujimori, D. G., Docking and Linking of Fragments To Discover Jumonji Histone Demethylase Inhibitors. Journal of Medicinal Chemistry 2016, 59, 1580-1598.
82. Westaway, S. M.; Preston, A. G. S.; Barker, M. D.; Brown, F.; Brown, J. A.; Campbell, M.; Chung, C.-w.; Diallo, H.; Douault, C.; Drewes, G.; Eagle, R.; Gordon, L.; Haslam, C.; Hayhow, T. G.; Humphreys, P. G.; Joberty, G.; Katso, R.; Kruidenier, L.; Leveridge, M.; Liddle, J.; Mosley, J.; Muelbaier, M.; Randle, R.; Rioja, I.; Rueger, A.; Seal, G. A.; Sheppard, R. J.; Singh, O.; Taylor, J.; Thomas, P.; Thomson, D.; Wilson, D. M.; Lee, K.; Prinjha, R. K., Cell Penetrant Inhibitors of the KDM4 and KDM5 Families of Histone Lysine Demethylases. 1. 3-Amino-4-pyridine Carboxylate Derivatives. Journal of Medicinal Chemistry 2016, 59, 1357-1369.
83. Westaway, S. M.; Preston, A. G. S.; Barker, M. D.; Brown, F.; Brown, J. A.; Campbell, M.; Chung, C.-w.; Drewes, G.; Eagle, R.; Garton, N.; Gordon, L.; Haslam, C.; Hayhow, T. G.; Humphreys, P. G.; Joberty, G.; Katso, R.; Kruidenier, L.; Leveridge, M.; Pemberton, M.; Rioja, I.; Seal, G. A.; Shipley, T.; Singh, O.; Suckling, C. J.; Taylor, J.; Thomas, P.; Wilson, D. M.; Lee, K.; Prinjha, R. K., Cell Penetrant Inhibitors of the KDM4 and KDM5 Families of Histone Lysine Demethylases. 2. Pyrido[3,4-d]pyrimidin-4(3H)-one Derivatives. Journal of Medicinal Chemistry 2016, 59, 1370-1387.
84. Rose, N. R.; Woon, E. C. Y.; Tumber, A.; Walport, L. J.; Chowdhury, R.; Li, X. S.; King, O. N. F.; Lejeune, C.; Ng, S. S.; Krojer, T.; Chan, M. C.; Rydzik, A. M.; Hopkinson, R. J.; Che, K. H.; Daniel, M.; Strain-Damerell, C.; Gileadi, C.; Kochan, G.; Leung, I. K. H.; Dunford, J.; Yeoh, K. K.; Ratcliffe, P. J.; Burgess-Brown, N.; von Delft, F.; Muller, S.; Marsden, B.; Brennan, P. E.; McDonough, M. A.; Oppermann, U.; Klose, R. J.; Schofield, C. J.; Kawamura, A., Plant Growth Regulator Daminozide Is a Selective Inhibitor of Human KDM2/7 Histone Demethylases. Journal of Medicinal Chemistry 2012, 55, 6639-6643.
85. Rüger, N.; Roatsch, M.; Emmrich, T.; Franz, H.; Schüle, R.; Jung, M.; Link, A., Tetrazolylhydrazides as Selective Fragment‐Like Inhibitors of the JumonjiC‐Domain‐Containing Histone Demethylase KDM4A. ChemMedChem 2015, 10, 1875-1883.
86. Hamada, S.; Suzuki, T.; Mino, K.; Koseki, K.; Oehme, F.; Flamme, I.; Ozasa, H.; Itoh, Y.; Ogasawara, D.; Komaarashi, H.; Kato, A.; Tsumoto, H.; Nakagawa, H.; Hasegawa, M.; Sasaki, R.; Mizukami, T.; Miyata, N., Design, Synthesis, Enzyme-Inhibitory Activity, and Effect on Human Cancer Cells of a Novel Series of Jumonji Domain-Containing Protein 2 Histone Demethylase Inhibitors. Journal of Medicinal Chemistry 2010, 53, 5629-5638.
87. Kim, T.-D.; Fuchs, J. R.; Schwartz, E.; Abdelhamid, D.; Etter, J.; Berry, W. L.; Li, C.; Ihnat, M. A.; Li, P.-K.; Janknecht, R., Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. American Journal of Translational Research 2014, 6, 236-247.
88. Chu, C.-H.; Wang, L.-Y.; Hsu, K.-C.; Chen, C.-C.; Cheng, H.-H.; Wang, S.-M.; Wu, C.-M.; Chen, T.-J.; Li, L.-T.; Liu, R.; Hung, C.-L.; Yang, J.-M.; Kung, H.-J.; Wang, W.-C., KDM4B as a Target for Prostate Cancer: Structural Analysis and Selective Inhibition by a Novel Inhibitor. Journal of Medicinal Chemistry 2014, 57, 5975-5985.
89. Sakurai, M.; Rose, N. R.; Schultz, L.; Quinn, A. M.; Jadhav, A.; Ng, S. S.; Oppermann, U.; Schofield, C. J.; Simeonov, A., A miniaturized screen for inhibitors of Jumonji histone demethylases. Molecular BioSystems 2010, 6, 357-364.

Literaturverzeichnis
208 | Literatur
90. Wang, W.; Marholz, L. J.; Wang, X., Novel Scaffolds of Cell-Active Histone Demethylase Inhibitors Identified from High-Throughput Screening. Journal of Biomolecular Screening 2015, 20, 821-827.
91. Liu, L.-J.; Lu, L.; Zhong, H.-J.; He, B.; Kwong, D. W. J.; Ma, D.-L.; Leung, C.-H., An Iridium(III) Complex Inhibits JMJD2 Activities and Acts as a Potential Epigenetic Modulator. Journal of Medicinal Chemistry 2015, 58, 6697-6703.
92. Sekirnik, R.; Rose, N. R.; Thalhammer, A.; Seden, P. T.; Mecinovic, J.; Schofield, C. J., Inhibition of the histone lysine demethylase JMJD2A by ejection of structural Zn(II). Chemical Communications 2009, 6376-6378.
93. Leurs, U.; Lohse, B.; Rand, K. D.; Ming, S.; Riise, E. S.; Cole, P. A.; Kristensen, J. L.; Clausen, R. P., Substrate- and Cofactor-independent Inhibition of Histone Demethylase KDM4C. ACS Chemical Biology 2014, 9, 2131-2138.
94. Harrison, C., Recent patents related to histone demethylases. Nature Reviews Drug Discovery 2013, 12, 899.
95. Chin, Y.-W.; Han, S.-Y., KDM4 histone demethylase inhibitors for anti-cancer agents: a patent review. Expert Opinion on Therapeutic Patents 2015, 25, 135-144.
96. Schofield, C.; McDonough, M.; Rose, N.; Thalhammer, A. WO2010/043866A2, Histone Lysine Demethylase Inhibitors. 2010.
97. Labelle, M.; Boesen, T.; Mehrotra, M.; Khan, Q.; Ullah, F. WO2014/053491A1, Inhibitors of Histone Demethylases. 2014.
98. Zhang, R.; Martyr, C. D.; Saraswat, N.; Boesen, T. WO2015/153498A1, Inhibitors of Histone Demethylases. 2015.
99. Nie, Z.; Stafford, J.; Veal, J.; Wallace, M. WO2014/089364A1, Histone Demethylase Inhibitors. 2014.
100. Celgene Corporation. Celgene to Acquire Quanticel Pharmaceuticals. (accessed Februar 2018, ir.celgene.com/releasedetail.cfm?releaseid=908643).
101. Gilead Sciences Inc. Gilead Sciences Acquires EpiTherapeutics. (accessed Februar 2018, gilead.com/news/press-releases/2015/5/gilead-sciences-acquires-epitherapeutics).
102. Boloor, A.; Kanouni, T.; Stafford, J.; Veal, J.; Wallace, M. WO2017/161012A1, Histone Demethylase Inhibitors. 2017.
103. Young, C. K.; Kanouni, T.; Nie, Z.; Stafford, J.; Veal, J.; Wallace, M. US9611221B2, Histone Demethylase Inhibitors. 2017.
104. Ballatore, C.; Huryn, D. M.; Smith, A. B., Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385-95.
105. Lassalas, P.; Oukoloff, K.; Makani, V.; James, M.; Tran, V.; Yao, Y.; Huang, L.; Vijayendran, K.; Monti, L.; Trojanowski, J. Q.; Lee, V. M. Y.; Kozlowski, M. C.; Smith, A. B.; Brunden, K. R.; Ballatore, C., Evaluation of Oxetan-3-ol, Thietan-3-ol, and Derivatives Thereof as Bioisosteres of the Carboxylic Acid Functional Group. ACS Medicinal Chemistry Letters 2017, 8, 864-868.
106. Meanwell, N. A., Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. Journal of Medicinal Chemistry 2011, 54, 2529-2591.
107. Lassalas, P.; Gay, B.; Lasfargeas, C.; James, M. J.; Tran, V.; Vijayendran, K. G.; Brunden, K. R.; Kozlowski, M. C.; Thomas, C. J.; Smith, A. B.; Huryn, D. M.; Ballatore, C., Structure Property Relationships of Carboxylic Acid Isosteres. Journal of Medicinal Chemistry 2016, 59, 3183-3203.
108. Herr, R. J., 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorganic & Medicinal Chemistry 2002, 10, 3379-3393.

Literaturverzeichnis
Literatur | 209
109. Alonen, A.; Finel, M.; Kostiainen, R., The human UDP-glucuronosyltransferase UGT1A3 is highly selective towards N2 in the tetrazole ring of losartan, candesartan, and zolarsartan. Biochemical Pharmacology 2008, 76, 763-772.
110. Costantino, G.; Maltoni, K.; Marinozzi, M.; Camaioni, E.; Prezeau, L.; Pin, J.-P.; Pellicciari, R., Synthesis and biological evaluation of 2-(3′-(1H-tetrazol-5-yl)bicyclo[1.1.1]pent-1-yl)glycine (S-TBPG), a novel mGlu1 receptor antagonist. Bioorganic & Medicinal Chemistry 2001, 9, 221-227.
111. Allen, F. H.; Groom, C. R.; Liebeschuetz, J. W.; Bardwell, D. A.; Olsson, T. S. G.; Wood, P. A., The Hydrogen Bond Environments of 1H-Tetrazole and Tetrazolate Rings: The Structural Basis for Tetrazole–Carboxylic Acid Bioisosterism. Journal of Chemical Information and Modeling 2012, 52, 857-866.
112. Carini, D. J.; Duncia, J. V.; Aldrich, P. E.; Chiu, A. T.; Johnson, A. L.; Pierce, M. E.; Price, W. A.; Santella, J. B.; Wells, G. J., Nonpeptide angiotensin II receptor antagonists: the discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives. Journal of Medicinal Chemistry 1991, 34, 2525-2547.
113. Rüger, N. Dissertation, Synthese und Charakterisierung potentieller Histon-Demethylase-Inhibitoren. Universität Greifswald, 2018.
114. Rüger, N.; Fassauer, G. M.; Bock, C.; Emmrich, T.; Bodtke, A.; Link, A., Substituted tetrazoles as multipurpose screening compounds. Molecular Diversity 2017, 21, 9-27.
115. Wheeler, A. S.; Oates, W. M., The Bromination of Anthranilic Acid. Journal of the American Chemical Society 1910, 32, 770-773.
116. Dunn, G. E.; Prysiazniuk, R., Rates of Decarboxylation of Substituted Anthranilic Acids in Nitrobenzene Solution. Canadian Journal of Chemistry 1961, 39, 285-296.
117. Rajveer, C. H.; Kumaraswamy, D.; Sudharshini, S.; Benjamin, S. R., Synthesis of 6-bromo-oxo quinazolinone derivatives and their pharamcological activities. International Journal of Chemistry Research 2010, 1, 21-24.
118. Tailor, H. S., Synthesis of some Novel Heterocyclic Compound and their Antimicrobial Activity. Journal of Scientific Research 2014, 3, 35-36.
119. Bogert, M. T.; Hand, W. F., 5-Brom-2-Aminobenzoic Acid and some of its Derivatives. Journal of the American Chemical Society 1905, 27, 1476-1484.
120. Jackson, O. R., Ueber das Methylketol. Berichte der deutschen chemischen Gesellschaft 1881, 14, 879-888.
121. Alt, H., Ueber Bromirung der o‐Acetylamidobenzoësäure. Berichte der deutschen chemischen Gesellschaft 1889, 22, 1643-1647.
122. Böttcher, S.; Thiem, J., Indoxylic Acid Esters as Convenient Intermediates Towards Indoxyl Glycosides. European Journal of Organic Chemistry 2014, 2014, 564-574.
123. Landry, D. W.; Deng, S.; Arancio, O.; Wasmuth, A. WO2013/109738A1, Novel Phosphodiesterase Inhibitors and Uses Thereof. 2013.
124. Koelsch, C. F.; Whitney, A. G., The Rosenmund-von Braun Nitrile Synthesis. The Journal of Organic Chemistry 1941, 6, 795-803.
125. v. Braun, J.; Manz, G., Fluoranthen und seine Derivate. III. Mitteilung. Justus Liebigs Annalen der Chemie 1931, 488, 111-126.
126. Wu, J. X.; Beck, B.; Ren, R. X., Catalytic Rosenmund–von Braun reaction in halide-based ionic liquids. Tetrahedron Letters 2002, 43, 387-389.
127. Gerber, R.; Oberholzer, M.; Frech Christian, M., Cyanation of Aryl Bromides with K4[Fe(CN)6] Catalyzed by Dichloro[bis{1‐(dicyclohexylphosphanyl)piperidine}]palladium, a Molecular Source of Nanoparticles, and the Reactions Involved in the Catalyst‐Deactivation Processes. Chemistry – A European Journal 2012, 18, 2978-2986.

Literaturverzeichnis
210 | Literatur
128. Ren, Y.; Liu, Z.; He, S.; Zhao, S.; Wang, J.; Niu, R.; Yin, W., Development of an Open-Air and Robust Method for Large-Scale Palladium-Catalyzed Cyanation of Aryl Halides: The Use of i-PrOH to Prevent Catalyst Poisoning by Oxygen. Organic Process Research & Development 2009, 13, 764-768.
129. Erhardt, S.; Grushin, V. V.; Kilpatrick, A. H.; Macgregor, S. A.; Marshall, W. J.; Roe, D. C., Mechanisms of Catalyst Poisoning in Palladium-Catalyzed Cyanation of Haloarenes. Remarkably Facile C−N Bond Activation in the [(Ph3P)4Pd]/[Bu4N]+ CN- System. Journal of the American Chemical Society 2008, 130, 4828-4845.
130. Ushkov, A. V.; Grushin, V. V., Rational Catalysis Design on the Basis of Mechanistic Understanding: Highly Efficient Pd-Catalyzed Cyanation of Aryl Bromides with NaCN in Recyclable Solvents. Journal of the American Chemical Society 2011, 133, 10999-11005.
131. Cristau, H. J.; Ouali, A.; Spindler, J. F.; Taillefer, M., Mild and Efficient Copper‐Catalyzed Cyanation of Aryl Iodides and Bromides. Chemistry – A European Journal 2005, 11, 2483-2492.
132. Jin, F.; Confalone, P. N., Palladium-catalyzed cyanation reactions of aryl chlorides. Tetrahedron Letters 2000, 41, 3271-3273.
133. Zanon, J.; Klapars, A.; Buchwald, S. L., Copper-Catalyzed Domino Halide Exchange-Cyanation of Aryl Bromides. Journal of the American Chemical Society 2003, 125, 2890-2891.
134. Wen, Q.; Jin, J.; Zhang, L.; Luo, Y.; Lu, P.; Wang, Y., Copper-mediated cyanation reactions. Tetrahedron Letters 2014, 55, 1271-1280.
135. Leiby, R. W., Synthesis of 3-amino-4(3H)-quinazolinones from N-(2-carbomethoxyphenyl)imidate esters. The Journal of Organic Chemistry 1985, 50, 2926-2929.
136. Ramakrishna, N.; Abdul, M. R.; Ishtiyaque, A.; Pradeep, J. US2012/0277216A1, 1,2-Dihydro-2-oxoquinoline Compounds as 5-HT4 Receptor Ligands. 2012.
137. Yang Katherine, S.; Budin, G.; Tassa, C.; Kister, O.; Weissleder, R., Bioorthogonal Approach to Identify Unsuspected Drug Targets in Live Cells. Angewandte Chemie International Edition 2013, 52, 10593-10597.
138. Tiang, M.; Khan, S. H.; Menchen, S. M.; Rosenblum, B. B. US7429651B2, Fluorescent Nucleobase Conjugates Having Anionic Linkers. 2008.
139. Wall, M. J.; Player, M. R.; Patch, R. J. WO2005/009967A2, Quinolinone Derivatives as Inhibitors of C-FMS Kinase. 2005.
140. Koguro, K.; Oga, T.; Mitsui, S.; Orita, R., Novel Synthesis of 5-Substituted Tetrazoles from Nitriles. Synthesis 1998, 1998, 910-914.
141. Zhou, Y.; Wang, B.; Di, F.; Xiong, L.; Yang, N.; Li, Y.; Li, Y.; Li, Z., Synthesis and biological activities of 2,3-dihydro-1,3,4-oxadiazole compounds and its derivatives as potential activator of ryanodine receptors. Bioorganic & Medicinal Chemistry Letters 2014, 24, 2295-2299.
142. Yadagiri, B.; Holagunda, U. D.; Bantu, R.; Nagarapu, L.; Guguloth, V.; Polepally, S.; Jain, N., Rational design, synthesis and anti-proliferative evaluation of novel benzosuberone tethered with hydrazide–hydrazones. Bioorganic & Medicinal Chemistry Letters 2014, 24, 5041-5044.
143. Borsche, W.; Weußmann, H.; Fritzsche, A., Untersuchungen über Isatin und verwandte Verbindungen, VI: Über 5‐Jod‐isatin und 5.5′‐Dijod‐indigo. Berichte der deutschen chemischen Gesellschaft (A and B Series) 1924, 57, 1770-1775.
144. Takalo, H.; Kankare, J.; Hänninen, E., Synthesis of Some Substituted Dimethyl and Diethyl 4-(Phenylethynyl)-2,6-pyridinedicarboxylates. Acta Chemica Scandinavica 1988, 42b, 448-454.

Literaturverzeichnis
Literatur | 211
145. Škoch, K.; Císařová, I.; Štěpnička, P., Phosphinoferrocene Ureas: Synthesis, Structural Characterization, and Catalytic Use in Palladium-Catalyzed Cyanation of Aryl Bromides. Organometallics 2015, 34, 1942-1956.
146. Khan, M. T. H.; Khan, R.; Wuxiuer, Y.; Arfan, M.; Ahmed, M.; Sylte, I., Identification of novel quinazolin-4(3H)-ones as inhibitors of thermolysin, the prototype of the M4 family of proteinases. Bioorganic & Medicinal Chemistry 2010, 18, 4317-4327.
147. Huisgen, R.; Sauer, J.; Sturm Hans, J.; Markgraf John, H., Ringöffnungen der Azole, II. Die Bildung von 1.3.4‐Oxdiazolen bei der Acylierung 5‐substituierter Tetrazole. Chemische Berichte 1960, 93, 2106-2124.
148. Jursic, B. S.; Zdravkovskif, Z., Preparation of 5-Substituted 2-Methyl-1,3,4-oxadiazoles from 5-Substituted Tetrazoles and Acetic Anhydride. Synthetic Communications 1994, 24, 1575-1582.
149. Shi, L.-F.; Li, B.; Zhang, L.-M.; Zuo, Q.-H.; Yue, S., Synthesis, characterization and striking photoluminescence variation of a series of copper(I) complexes containing oxadiazole ligand. Inorganica Chimica Acta 2013, 400, 91-98.
150. Becker, H. G. O.; Berger, W.; Günter Domschke et al., Organikum. Weinheim, 24. Auflage, 2015; p 491-492.
151. Shah, P.; Westwell, A. D., The role of fluorine in medicinal chemistry. Journal of Enzyme Inhibition and Medicinal Chemistry 2007, 22, 527-540.
152. Thor, M.; Beierlein, K.; Dykes, G.; Gustavsson, A.-L.; Heidrich, J.; Jendeberg, L.; Lindqvist, B.; Pegurier, C.; Roussel, P.; Slater, M.; Svensson, S.; Sydow-Bäckman, M.; Thornström, U.; Uppenberg, J., Synthesis and pharmacological evaluation of a new class of peroxisome proliferator-activated receptor modulators. Bioorganic & Medicinal Chemistry Letters 2002, 12, 3565-3567.
153. Yee, Y. K.; Tebbe, A. L.; Linebarger, J. H.; Beight, D. W.; Craft, T. J.; Gifford-Moore, D.; Goodson, T.; Herron, D. K.; Klimkowski, V. J.; Kyle, J. A.; Sawyer, J. S.; Smith, G. F.; Tinsley, J. M.; Towner, R. D.; Weir, L.; Wiley, M. R., N2-Aroylanthranilamide Inhibitors of Human Factor Xa. Journal of Medicinal Chemistry 2000, 43, 873-882.
154. Hsieh, P.-W.; Hwang, T.-L.; Wu, C.-C.; Chiang, S.-Z.; Wu, C.-I.; Wu, Y.-C., The evaluation and structure–activity relationships of 2-benzoylaminobenzoic esters and their analogues as anti-inflammatory and anti-platelet aggregation agents. Bioorganic & Medicinal Chemistry Letters 2007, 17, 1812-1817.
155. Bhat, B. US005767270A, Acylation of Nucleosides with N-Acyl tetrazoles. 1998.
156. Devendra, L. P.; Gaikar, V. G., Purification of Forskolin by Adsorptive Separation Using Functionalized Polymer Bearing Specific Ligands Designed by Molecular Simulation. Industrial & Engineering Chemistry Research 2011, 50, 11667-11676.
157. Arndt, A. Diplomarbeit, Darstellung und Charakterisierung potentieller Histon-Demethylase-Inhibitoren mit einer Tetrazolteilstruktur. Universität Greifswald, 2016.
158. Palumbo Piccionello, A.; Musumeci, R.; Cocuzza, C.; Fortuna, C. G.; Guarcello, A.; Pierro, P.; Pace, A., Synthesis and preliminary antibacterial evaluation of Linezolid-like 1,2,4-oxadiazole derivatives. European Journal of Medicinal Chemistry 2012, 50, 441-448.
159. Behringer, H.; Kohl, K., Über Tetrazole, I. Mitteil.: Die Synthese des 5‐[β‐Amino‐äthyl]‐tetrazols und einiger Tetrazoly‐(5)‐methylderivate. Chemische Berichte 1956, 89, 2648-2653.
160. Ortar, G.; Cascio, M. G.; Moriello, A. S.; Camalli, M.; Morera, E.; Nalli, M.; Di Marzo, V., Carbamoyl tetrazoles as inhibitors of endocannabinoid inactivation: A critical revisitation. European Journal of Medicinal Chemistry 2008, 43, 62-72.
161. Wang, Y.; Patil, P.; Dömling, A., Easy Synthesis of Two Positional Isomeric Tetrazole Libraries. Synthesis 2016, 48, 3701-3712.

Literaturverzeichnis
212 | Literatur
162. Grzonka, Z.; Rekowska, E.; Liberek, B., Tetrazole analogues of amino acids and peptides— V: Syntheses of peptide derivatives containing tetrazole analogues of amino acids in C-terminal position. Tetrahedron 1971, 27, 2317-2322.
163. Liao, S.-M.; Du, Q.-S.; Meng, J.-Z.; Pang, Z.-W.; Huang, R.-B., The multiple roles of histidine in protein interactions. Chemistry Central Journal 2013, 7, 44.
164. Naik, N.; Kumar, H. V.; Swamya, J. R., Novel indole-3-butyric acid analogues: Synthesis and a new insights into their antioxidant activity. Journal of Pharmacy Research 2012, 5, 4504-4509.
165. Cho, J.-H.; Jung, K.-Y.; Jung, Y.; Kim, M. H.; Ko, H.; Park, C.-S.; Kim, Y.-C., Design and synthesis of potent and selective P2X3 receptor antagonists derived from PPADS as potential pain modulators. European Journal of Medicinal Chemistry 2013, 70, 811-830.
166. Elliott, G. I.; Fuchs, J. R.; Blagg, B. S. J.; Ishikawa, H.; Tao, H.; Yuan, Z. Q.; Boger, D. L., Intramolecular Diels−Alder/1,3-Dipolar Cycloaddition Cascade of 1,3,4-Oxadiazoles. Journal of the American Chemical Society 2006, 128, 10589-10595.
167. Audia, J. E.; Britton, T. C.; Folmer, B. K.; Wu, J. US006207710B1, Compounds for Inhibiting beta-Amyloid Peptide Release and/or its Synthesis. 2001.
168. Neises, B.; Steglich, W., Einfaches Verfahren zur Veresterung von Carbonsäuren. Angewandte Chemie 1978, 90, 556-557.
169. Siddalingamurthy, E.; Mahadevan, K. M.; Jagadeesh, N. M.; M. N. Kumara, Synthesis of Indolecarboxamides and Their Docking Studies with H1,5HT and CCR2 Antagonist Receptors. American Journal of Pharmacy & Health Research 2014, 2, 245-258.
170. Sultana, N.; Arayne, M. S.; Khan, M. M.; Saleem, D. M.; Mirza, A. Z., Determination of Tryptophan in Raw Materials, Rat Brain and Human Plasma by RP-HPLC Technique. Journal of Chromatographic Science 2012, 50, 531-537.
171. Lieser, T.; Nischk, G., Die Umsetzung von Hydrazin‐Derivaten mit Phosgen. Chemische Berichte 1949, 82, 527-530.
172. Diehr, H.-J.; Lantzsch, R.; Jelich, K. EP0872450A1, Verfahren zur Herstellung von substituierten Oxadiazolonen. 1998.
173. Caldwell, H. C.; Seiwald, R. J.; Burckhalter, J. H., The Synthesis and Proof of Structure of N-Alkylated Oxadiazolones. Journal of the American Pharmaceutical Association 1958, 47, 799-802.
174. Jansen, M.; Rabe, H.; Strehle, A.; Dieler, S.; Debus, F.; Dannhardt, G.; Akabas, M. H.; Lüddens, H., Synthesis of GABAA Receptor Agonists and Evaluation of their α-Subunit Selectivity and Orientation in the GABA Binding Site. Journal of Medicinal Chemistry 2008, 51, 4430-4448.
175. Werner, E. A.; Bell, J., CCXIV.-The preparation of methylguanidine, and of ββ-dimethylguanidine by the interaction of dicyanodiamide, and methylammonium and dimethylammonium chlorides respectively. Journal of the Chemical Society, Transactions 1922, 121, 1790-1794.
176. Pinner, A.; Klein, F., Umwandlung der Nitrile in Imide. Berichte der deutschen chemischen Gesellschaft 1877, 10, 1889-1897.
177. Huisgen, R., Proceedings of the Chemical Society. Proceedings of the Chemical Society 1961, 1961, 357-396.
178. Himo, F.; Demko, Z. P.; Noodleman, L.; Sharpless, K. B., Mechanisms of Tetrazole Formation by Addition of Azide to Nitriles. Journal of the American Chemical Society 2002, 124, 12210-12216.

Literaturverzeichnis
Literatur | 213
179. Schnute, M. E.; O’Brien, P. M.; Nahra, J., Discovery of (pyridin-4-yl)-2H-tetrazole as a novel scaffold to identify highly selective matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. Bioorganic & Medicinal Chemistry Letters 2010, 20, 576-580.
180. Finnegan, W. G.; Henry, R. A.; Lofquist, R., An Improved Synthesis of 5-Substituted Tetrazoles. Journal of the American Chemical Society 1958, 80, 3908-3911.
181. Duncia, J. V.; Pierce, M. E.; Santella, J. B., Three synthetic routes to a sterically hindered tetrazole. A new one-step mild conversion of an amide into a tetrazole. The Journal of Organic Chemistry 1991, 56, 2395-2400.
182. Demko Zachary, P.; Sharpless, K. B., A Click Chemistry Approach to Tetrazoles by Huisgen 1,3‐Dipolar Cycloaddition: Synthesis of 5‐Sulfonyl Tetrazoles from Azides and Sulfonyl Cyanides. Angewandte Chemie International Edition 2002, 41, 2110-2113.
183. Wittenberger, S. J., Recent Developments in Tetrazole Chemistry. A Review. Organic Preparations and Procedures International 1994, 26, 499-531.
184. Sarvary, A.; Maleki, A., A review of syntheses of 1,5-disubstituted tetrazole derivatives. Molecular Diversity 2015, 19, 189-212.
185. Geyer, R.; Kaske, M.; Baumeister, P.; Buschauer, A., Synthesis and Functional Characterization of Imbutamine Analogs as Histamine H3 and H4 Receptor Ligands. Archiv der Pharmazie 2013, 347, 77-88.
186. Arambula, J. F.; Ramisetty, S. R.; Baranger, A. M.; Zimmerman, S. C., A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proceedings of the National Academy of Sciences 2009, 106, 16068-16073.
187. Ife, R. J.; Catchpole, K. W.; Durant, G. J.; Robin Ganellin, C.; Harvey, C. A.; Meeson, M. L.; Owen, D. A. A.; Parsons, M. E.; Slingsby, B. P.; Theobald, C. J., Non-basic histamine H1-antagonists. I. Synthesis and biological evaluation of some substituted 2-(2-pyridylaminoalkylamino) pyrimidones and related compounds. European Journal of Medicinal Chemistry 1989, 24, 249-257.
188. Cantello, B. C. C.; Cawthorne, M. A.; Cottam, G. P.; Duff, P. T.; Haigh, D.; Hindley, R. M.; Lister, C. A.; Smith, S. A.; Thurlby, P. L., [[ω-(Heterocyclylamino)alkoxy]benzyl]-2,4-thiazolidinediones as potent antihyperglycemic agents. Journal of Medicinal Chemistry 1994, 37, 3977-3985.
189. Berry, A.; Chen, Z.; Lombaert, S. D.; Takahashi, H. WO2012/082817A1, Biarylamide Inhibitors of Leukotriene Production. 2012.
190. Yoshida, T.; Moriyama, Y.; Nakano, S., Fluorescence Properties of 2-Substituted 6-, 7- or 8-Methoxyquinoline-4-carboxylic Acid Derivatives. Chemical and Pharmaceutical Bulletin 1992, 40, 1322-1324.
191. Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Lozach, O.; Loaëc, N.; Meijer, L.; Besson, T., Synthesis and biological evaluation of N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. European Journal of Medicinal Chemistry 2012, 58, 171-183.
192. Klimešová, V.; Otčenášek, M.; Waisser, K., Potential antifungal agents. Synthesis and activity of 2-alkylthiopyridine-4-carbothioamides. European Journal of Medicinal Chemistry 1996, 31, 389-395.
193. Furukawa, N.; Takahashi, F.; Kawai, T.; Kishimoto, K.; Ogawa, S.; Oae, S., Sytheses and some Properties of Sulfoxides, Sulfilimines, Aminosulfonium Salts and Sulfoximines containing Pyridine Nuclei. Phosphorus and Sulfur and the Related Elements 1983, 16, 167-180.
194. Miletín, M.; Doležal, M.; Opletalová, V.; Hartl, J.; Král’ová, K.; Macháček, M., Some Anilides of 2-Alkylthio- and 2-Chloro-6-Alkylthio-4-Pyridinecarboxylic Acids: Synthesis and Photosynthesis-Inhibiting Activity. Molecules 2001, 6, 603-613.

Literaturverzeichnis
214 | Literatur
195. Yang, S.; Wu, C.; Ruan, M.; Yang, Y.; Zhao, Y.; Niu, J.; Yang, W.; Xu, J., Metal- and ligand-free Ullmann-type C–O and C–N coupling reactions promoted by potassium tert-butoxide. Tetrahedron Letters 2012, 53, 4288-4292.
196. Tan, L.; Tao, Y.; Wang, T.; Zou, F.; Zhang, S.; Kou, Q.; Niu, A.; Chen, Q.; Chu, W.; Chen, X.; Wang, H.; Yang, Y., Discovery of Novel Pyridone-Conjugated Monosulfactams as Potent and Broad-Spectrum Antibiotics for Multidrug-Resistant Gram-Negative Infections. Journal of Medicinal Chemistry 2017, 60, 2669-2684.
197. Zhao, M.; Yin, J.; Huffman, M. A.; McNamara, J. M., A very concise synthesis of a potent N-(1,3-thiazol-2-yl)pyridin-2-amine KDR kinase inhibitor. Tetrahedron 2006, 62, 1110-1115.
198. Maiti, D.; Fors, B. P.; Henderson, J. L.; Nakamura, Y.; Buchwald, S. L., Palladium-catalyzed coupling of functionalized primary and secondary amines with aryl and heteroaryl halides: two ligands suffice in most cases. Chemical Science 2011, 2, 57-68.
199. Shen, Q.; Shekhar, S.; Stambuli James, P.; Hartwig John, F., Highly Reactive, General, and Long‐Lived Catalysts for Coupling Heteroaryl and Aryl Chlorides with Primary Nitrogen Nucleophiles. Angewandte Chemie International Edition 2005, 44, 1371-1375.
200. Bream, R. N.; Hayes, D.; Hulcoop, D. G.; Whiteman, A. J., Development of Synthetic Routes, via a Tropinone Intermediate, to a Long-Acting Muscarinic Antagonist for the Treatment of Respiratory Disease. 2013, 17, 641-650.
201. Walker, H. G.; Gee, M.; McCready, R. M., Complete Methylation of Reducing Carbohydrates. The Journal of Organic Chemistry 1962, 27, 2100-2102.
202. Broka, C. A.; Hawley, R. C. US2010/0324056A1, Biphenyl and Phenyl-Pyridine Amides as P2X3 and P2X2/3 Antagonists. 2010.
203. Ohkawa, S.; Hashimoto, T.; Fukatsu, K. WO98/08842, Cyclic Ether Compounds as Sodium Channel Modulators. 1998.
204. Vennerstrom, J. L.; Dong, Y.; Charman, S. A.; Wang, X.; Charman, W. N. WO2009/058859A2, Dispiro 1,2,4-Trioxolane Antimalarials. 2009.
205. Crugeiras, J.; Rios, A.; Riveiros, E.; Richard, J. P., Substituent Effects on Electrophilic Catalysis by the Carbonyl Group: Anatomy of the Rate Acceleration for PLP-Catalyzed Deprotonation of Glycine. Journal of the American Chemical Society 2011, 133, 3173-3183.
206. Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M., Nucleophilic character of alkyl radicals—VI: A new convenient selective alkylation of heteroaromatic bases. Tetrahedron 1971, 27, 3575-3579.
207. Heinisch, G.; Lötsch, G., Homolytic Alkoxycarbonylation Reactions in Two-phase Systems. Introduction of a Single Carboxylic Acid Ester Function into Cyano- or Alkoxycarbonyl Substituted N-Heteroaromatics. Heterocycles 1987, 26, 731-744.
208. Heinisch, G.; Lötsch, G., Homolytic alkoxycarbonylation reactions in two-phase systems.: Part II Studies on the ethoxycarbonylation of some selected π-deficient n-heteroaromatic systems. Tetrahedron 1986, 42, 5973-5977.
209. Heinisch, G.; Lötsch, G., Entscheidende Steigerung der Regioselektivität bei radikalischen Substitutionen: Minisci‐Reaktion im Zweiphasensystem. Angewandte Chemie 1985, 97, 694-695.
210. Kuhn, B.; Mohr, P.; Stahl, M., Intramolecular Hydrogen Bonding in Medicinal Chemistry. Journal of Medicinal Chemistry 2010, 53, 2601-2611.
211. Dakin, L.; Dowling, J. E.; Lamb, M.; Read, J.; Zheng, X. WO2010/001169A2, Chemical Compounds 251. 2010.

Literaturverzeichnis
Literatur | 215
212. Carpino, L. A.; Imazumi, H.; El‐Faham, A.; Ferrer, F. J.; Zhang, C.; Lee, Y.; Foxman, B. M.; Henklein, P.; Hanay, C.; Mügge, C.; Wenschuh, H.; Klose, J.; Beyermann, M.; Bienert, M., The Uronium/Guanidinium Peptide Coupling Reagents: Finally the True Uronium Salts. Angewandte Chemie International Edition 2002, 41, 441-445.
213. Carpino, L. A., 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. Journal of the American Chemical Society 1993, 115, 4397-4398.
214. Benenato, K. E.; Choy, A. L.; Hale, M. R.; Miller, M. WO2010/100475A1, Hydroxamic Acid Derivatives as Gram-negative Antibacterial Agents. 2010.
215. Flipo, M.; Charton, J.; Hocine, A.; Dassonneville, S.; Deprez, B.; Deprez-Poulain, R., Hydroxamates: Relationships between Structure and Plasma Stability. Journal of Medicinal Chemistry 2009, 52, 6790-6802.
216. Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P. K.; Veerasamy, R.; Sharma, P. C.; Dixit, A.; Mishra, P., A Structural Insight into Hydroxamic Acid Based Histone Deacetylase Inhibitors for the Presence of Anticancer Activity. Current Medicinal Chemistry 2014, 21, 2642-2664.
217. Yuan, Z.; Sun, Q.; Li, D.; Miao, S.; Chen, S.; Song, L.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y., Design, synthesis and anticancer potential of NSC-319745 hydroxamic acid derivatives as DNMT and HDAC inhibitors. European Journal of Medicinal Chemistry 2017, 134, 281-292.
218. Novartis Pharma GmbH, Fachinformation Farydak Hartkapseln (Panobinostat). Stand Juni 2017.
219. Akita, Y.; Shimazaki, M.; Ohta, A., Einführung der Cyano-Gruppe in Pyrazine. Synthesis 1981, 12, 974-975.
220. Senecal Todd, D.; Shu, W.; Buchwald Stephen, L., A General, Practical Palladium‐Catalyzed Cyanation of (Hetero)Aryl Chlorides and Bromides. Angewandte Chemie International Edition 2013, 52, 10035-10039.
221. Jonassen, H. B.; Nelson, J. H.; Schmitt, D. L.; Henry, R. A.; Moore, D. W., Platinum- and palladium-tetrazole complexes. Inorganic Chemistry 1970, 9, 2678-2681.
222. Nisnevich, G.; Rukhman, I.; Pertsikov, B.; Kaf-Tanov, J.; Dolitzky, B.-Z. WO2004/065383A2, Novel Synthesis of Irbesartan. 2004.
223. Heathcock, C. H.; Ratcliffe, R., Stereoselective total synthesis of the guaiazulenic sesquiterpenoids α-bulnesene and bulnesol. Journal of the American Chemical Society 1971, 93, 1746-1757.
224. Koval, I. A.; van der Schilden, K.; Schuitema, A. M.; Gamez, P.; Belle, C.; Pierre, J.-L.; Lüken, M.; Krebs, B.; Roubeau, O.; Reedijk, J., Proton NMR Spectroscopy and Magnetic Properties of a Solution-Stable Dicopper(II) Complex Bearing a Single μ-Hydroxo Bridge. Inorganic Chemistry 2005, 44, 4372-4382.
225. Moutevelis-Minakakis, P.; Papavassilopoulou, E.; Michas, G.; Georgikopoulou, K.; Ragoussi, M.-E.; Neophytou, N.; Zoumpoulakis, P.; Mavromoustakos, T.; Hadjipavlou-Litina, D., Synthesis, in silico docking experiments of new 2-pyrrolidinone derivatives and study of their anti-inflammatory activity. Bioorganic & Medicinal Chemistry 2011, 19, 2888-2902.
226. Nelson, D. W.; Gregg, R. J.; Kort, M. E.; Perez-Medrano, A.; Voight, E. A.; Wang, Y.; Grayson, G.; Namovic, M. T.; Donnelly-Roberts, D. L.; Niforatos, W.; Honore, P.; Jarvis, M. F.; Faltynek, C. R.; Carroll, W. A., Structure−Activity Relationship Studies on a Series of Novel, Substituted 1-Benzyl-5-phenyltetrazole P2X7 Antagonists. Journal of Medicinal Chemistry 2006, 49, 3659-3666.
227. Huff, L.; Henry, R. A., Alkylation of 5-substituted tetrazoles. Journal of Medicinal Chemistry 1970, 13, 777-779.

Literaturverzeichnis
216 | Literatur
228. Koren, A. O.; Gaponik, P. N., Selective N(2) alkylation of tetrazole and 5-substituted tetrazoles by alcohols. Chemistry of Heterocyclic Compounds 1990, 26, 1366-1370.
229. Rao, S. N.; Babu, K. S., An improved and efficient synthesis of Irbesartan, an antihypertensive drug. Organic Communications 2011, 4, 105-111.
230. Gundersen, L.-L., Synthesis of Purinecarbonitriles by Pd(0)-Catalysed Coupling of Halopurines with Zinc Cyanide. Acta Chemica Scandinavica 1996, 50, 58-63.
231. Galley, G.; Norcross, R.; Pflieger, P. WO2012/168265A1, Substituted Benzamide Derivatives. 2012.
232. Price, S.; Heald, R.; Savy, P. P. A. WO2009/085980A1, 8-Anilinoimidazopyridines and Their Use as Anti-cancer and/or Anti-inflammatory Agents. 2009.
233. Huang, W.; Tang, L.; Shi, Y.; Huang, S.; Xu, L.; Sheng, R.; Wu, P.; Li, J.; Zhou, N.; Hu, Y., Searching for the Multi-Target-Directed Ligands against Alzheimer’s disease: Discovery of quinoxaline-based hybrid compounds with AChE, H3R and BACE 1 inhibitory activities. Bioorganic & Medicinal Chemistry 2011, 19, 7158-7167.
234. Nemoto, T.; Matsumoto, T.; Masuda, T.; Hitomi, T.; Hatano, K.; Hamada, Y., P-Chirogenic Diaminophosphine Oxide: A New Class of Chiral Phosphorus Ligands for Asymmetric Catalysis. Journal of the American Chemical Society 2004, 126, 3690-3691.
235. Aquino, C. J.; Armour, D. R.; Berman, J. M.; Birkemo, L. S.; Carr, R. A. E.; Croom, D. K.; Dezube, M.; Dougherty, R. W.; Ervin, G. N.; Grizzle, M. K.; Head, J. E.; Hirst, G. C.; James, M. K.; Johnson, M. F.; Miller, L. J.; Queen, K. L.; Rimele, T. J.; Smith, D. N.; Sugg, E. E., Discovery of 1,5-Benzodiazepines with Peripheral Cholecystokinin (CCK-A) Receptor Agonist Activity. 1. Optimization of the Agonist “Trigger”. Journal of Medicinal Chemistry 1996, 39, 562-569.
236. Larionov, E.; Achrainer, F.; Humin, J.; Zipse, H., The Catalytic Potential of Substituted Pyridines in Acylation Reactions: Theoretical Prediction and Experimental Validation. ChemCatChem 2012, 4, 559-566.
237. Brown, H. C.; Rao, B. C. S., A New Powerful Reducing Agent—Sodium Borohydride in the Presence of Aluminum Chloride and Other Polyvalent Metal Halides. Journal of the American Chemical Society 1956, 78, 2582-2588.
238. Prasad, A. S. B.; Kanth, J. V. B.; Periasamy, M., Convenient methods for the reduction of amides, nitriles, carboxylic esters, acids and hydroboration of alkenes using NaBH4/I2System. Tetrahedron 1992, 48, 4623-4628.
239. Lane, C. F., Reduction of organic compounds with diborane. Chemical Reviews 1976, 76, 773-799.
240. Manku, S.; Laplante, C.; Kopac, D.; Chan, T.; Hall, D. G., A Mild and General Solid-Phase Method for the Synthesis of Chiral Polyamines. Solution Studies on the Cleavage of Borane−Amine Intermediates from the Reduction of Secondary Amides. The Journal of Organic Chemistry 2001, 66, 874-885.
241. Kuehne, M. E.; Shannon, P. J., Reduction of amides and lactams to amines by reactions with phosphorus oxychloride and sodium borohydride. The Journal of Organic Chemistry 1977, 42, 2082-2087.
242. Atta ur, R.; Basha, A.; Waheed, N.; Ahmed, S., A new method for the reduction of amides to amines with sodium borohydride. Tetrahedron Letters 1976, 17, 219-222.
243. Basha, A.; Atta Ur, R., A new method for the reduction of amides with zinc-ethanol. Experientia 1977, 33, 101-102.
244. da Costa, J. C. S.; Pais, K. C.; Fernandes, E. L.; de Oliveira, P. S. M.; Mendonça, J. S.; de Souza, M. V. N.; Peralta, M. A.; Vasconcelos, T. R. A., Simple reduction of ethyl, isopropyl and benzyl aromatic esters to alcohols using sodium borohydride-methanol system. ARKIVOC 2006, 2006, 128-133.

Literaturverzeichnis
Literatur | 217
245. Boechat, N.; da Costa, J. C. S.; de Souza Mendonça, J.; de Oliveira, P. S. M.; Vinı́cius Nora De Souza, M., A simple reduction of methyl aromatic esters to alcohols using sodium borohydride–methanol system. Tetrahedron Letters 2004, 45, 6021-6022.
246. Bianco, A.; Passacantilli, P.; Righi, G., Improved Procedure for the Reduction of Esters to Alcohols by Sodium Borohydride. Synthetic Communications 1988, 18, 1765-1771.
247. Santaniello, E.; Ferraboschi, P.; Sozzani, P., Reduction of esters to alcohols by means of sodium borohydride in polyethylene glycols. The Journal of Organic Chemistry 1981, 46, 4584-4585.
248. Chelucci, G.; Cabras, M. A.; Saba, A., Synthesis of optically active hydroxyalkylpyridines and related pyridyl amines. Tetrahedron: Asymmetry 1994, 5, 1973-1978.
249. Szántay, C.; Töke, L.; Kolonits, P., Synthesis of Protoemetine. A New Total Synthesis of Emetine. The Journal of Organic Chemistry 1966, 31, 1447-1451.
250. Burgey, C. S.; Crowley, B. N.; Denk, Z. J.; Paone, D. V.; Potteiger, C. M., WO2010/111060A1, P2X3 Receptor Antagonists for Treatment of Pain. 2010.
251. Swahn, B.-M.; Kolmodin, K.; Karlström, S.; von Berg, S.; Söderman, P.; Holenz, J.; Berg, S.; Lindström, J.; Sundström, M.; Turek, D.; Kihlström, J.; Slivo, C.; Andersson, L.; Pyring, D.; Rotticci, D.; Öhberg, L.; Kers, A.; Bogar, K.; von Kieseritzky, F.; Bergh, M.; Olsson, L.-L.; Janson, J.; Eketjäll, S.; Georgievska, B.; Jeppsson, F.; Fälting, J., Design and Synthesis of β-Site Amyloid Precursor Protein Cleaving Enzyme (BACE1) Inhibitors with in Vivo Brain Reduction of β-Amyloid Peptides. Journal of Medicinal Chemistry 2012, 55, 9346-9361.
252. Albano, G.; Evangelisti, C.; Aronica Laura, A., Hydrogenolysis of Benzyl Protected Phenols and Aniline Promoted by Supported Palladium Nanoparticles. ChemistrySelect 2017, 2, 384-388.
253. Alemparte-Gallardo, C.; Barfoot, C.; Barros-Aguirre, D.; Remuinan-Blanco, J. M. WO2009/141398A1, Tricyclic Nitrogen Containing Compounds and Their Use as Antibacterials. 2009.
254. Stadlbauer, W.; Prokopcova, H.; Sefcovikova, J.; Täubl, A. E., Nucleophilic Introduction of Functional Carbon Substituents into Quinolines, Quinolones and Related Heterocycles In The 7th International Electronic Conference on Synthetic Organic Chemistry (ECSOC-7), Basel, 2003.
255. Feely, W. E.; Beavers, E. M., Cyanation of Amine Oxide Salts. A New Synthesis of Cyanopyridines. Journal of the American Chemical Society 1959, 81, 4004-4007.
256. Bannen, L. C.; Chan, D. S.-M.; Mac, M. B.; Xu, W. US2010/0249071A1, Modulators of S1P and Methods of Making and Using. 2010.
257. Arcand, M.; Blouin, J.; Caron, M.; Labonté, A.; Normand, C.; Beaudet, L.; Padrós, J., LANCE Ultra JMJD2A Histone H3-Lysine 36 Demethylase Assay, Technical Note, PerkinElmer. 2011.
258. Arns, S.; Gibe, R.; Moreau, A.; Monzur Morshed, M.; Young, R. N., Design and synthesis of novel bone-targeting dual-action pro-drugs for the treatment and reversal of osteoporosis. Bioorganic & Medicinal Chemistry 2012, 20, 2131-2140.
259. Ambrosio, S.; Sacca, C. D.; Amente, S.; Paladino, S.; Lania, L.; Majello, B., Lysine-specific demethylase LSD1 regulates autophagy in neuroblastoma through SESN2-dependent pathway. Oncogene 2017, 36, 6701-6711.
260. Hiolski, E., New possible target for rapid antidepressants found. Chemical & Engineering News 2017, 95, 6-6.

Literaturverzeichnis
218 | Literatur
261. Duman, R. S., Ketamine and rapid-acting antidepressants: a new era in the battle against depression and suicide. F1000Research 2018, 7, 659.
262. Chen, X.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L.; Song, L.; Chen, A. P.; Song, J.; Takahashi, E.; He, G.; He, L.; Li, W.; Chen, C. D., Phf8 histone demethylase deficiency causes cognitive impairments through the mTOR pathway. Nature Communications 2018, 9, 114.
263. Walsh, R. M.; Shen, E. Y.; Bagot, R. C.; Anselmo, A.; Jiang, Y.; Javidfar, B.; Wojtkiewicz, G. J.; Cloutier, J.; Chen, J. W.; Sadreyev, R.; Nestler, E. J.; Akbarian, S.; Hochedlinger, K., Phf8 loss confers resistance to depression-like and anxiety-like behaviors in mice. Nature Communications 2017, 8, 15142.
264. Pathak, S. S.; Maitra, S.; Chakravarty, S.; Kumar, A., Histone Lysine Demethylases of JMJD2 or KDM4 Family are Important Epigenetic Regulators in Reward Circuitry in the Etiopathology of Depression. Neuropsychopharmacology 2017, 42, 854-863.
265. Serpeloni, F.; Radtke, K.; de Assis, S. G.; Henning, F.; Nätt, D.; Elbert, T., Grandmaternal stress during pregnancy and DNA methylation of the third generation: an epigenome-wide association study. Translational Psychiatry 2017, 7, e1202.
266. Cassano, P.; Fava, M., Depression and public health: An overview. Journal of Psychosomatic Research 2002, 53, 849-857.
267. World Health Organization, Depression and Other Common Mental Disorders - Global Health Estimates, WHO/MSD/MER/2017.2. 2017.
268. Alexopoulos, G. S.; Vrontou, C.; Kakuma, T.; Meyers, B. S.; Young, R. C.; Klausner, E.; Clarkin, J., Disability in geriatric depression. American Journal of Psychiatry 1996, 153, 877-885.
269. Unützer, J.; Patrick, D. L.; Diehr, P.; Simon, G.; Grembowski, D.; Katon, W., Quality Adjusted Life Years in Older Adults With Depressive Symptoms and Chronic Medical Disorders. International Psychogeriatrics 2000, 12, 15-33.
270. Schulz, R.; Drayer, R. A.; Rollman, B. L., Depression as a risk factor for non-suicide mortality in the elderly. Biological Psychiatry 2002, 52, 205-225.
271. Wittchen, H. U.; Müller, N.; Schmidkunz, B.; Winter, S.; Pfister, H., Erscheinungsformen, Häufigkeit und Versorgung von Depressionen. Ergebnisse des bundesweiten Gesundheitssurveys „Psychische Störungen”. Fortschritte der Medizin Originalien 2000, 118, S4-10.
272. König, H.-H.; Luppa, M.; Riedel-Heller, S., Die Kosten der Depression und die Wirtschaftlichkeit ihrer Behandlung. Psychiatrische Praxis 2010, 37, 213-215.
273. DGPPN, BÄK, KBV, AWMF (Hrsg.) für die Leitliniengruppe Unipolare Depression, S3-Leitlinie/Nationale VersorgungsLeitlinie Unipolare Depression – Langfassung, 2. Auflage. 2015.
274. Loomer, H. P.; Saunders, J. C.; Kline, N. S., A clinical and pharmacodynamic evaluation of iproniazid as a psychic energizer. Psychiatric Research Reports 1957, 8, 129-141.
275. Hillhouse, T. M.; Porter, J. H., A brief history of the development of antidepressant drugs: from monoamines to glutamate. Experimental and Clinical Psychopharmacology 2015, 23, 1-21.
276. Delgado, P. L., Depression: the case for a monoamine deficiency. Journal of Clinical Psychiatry 2000, 61, 7-11.
277. Hiemke, C.; Baumann, P.; Bergemann, N.; Conca, A.; O. Dietmaier et al., AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry: Update 2011. Pharmacopsychiatry 2011, 44, 195-235.

Literaturverzeichnis
Literatur | 219
278. Cipriani, A.; Furukawa, T. A.; Salanti, G.; Chaimani, A.; Atkinson, L. Z.; Ogawa, Y.; Leucht, S.; Ruhe, H. G.; Turner, E. H.; Higgins, J. P. T.; Egger, M.; Takeshima, N.; Hayasaka, Y.; Imai, H.; Shinohara, K.; Tajika, A.; Ioannidis, J. P. A.; Geddes, J. R., Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. The Lancet 2018, 391, 1357-1366.
279. Machado-Vieira, R.; Baumann, J.; Wheeler-Castillo, C.; Latov, D.; Henter, I. D.; Salvadore, G.; Zarate, C. A., The Timing of Antidepressant Effects: A Comparison of Diverse Pharmacological and Somatic Treatments. Pharmaceuticals (Basel) 2010, 3, 19-41.
280. Nierenberg, A. A.; Farabaugh, A. H.; Alpert, J. E.; Gordon, J.; Worthington, J. J.; Rosenbaum, J. F.; Fava, M., Timing of Onset of Antidepressant Response With Fluoxetine Treatment. American Journal of Psychiatry 2000, 157, 1423-1428.
281. Bschor, T.; Bauer, M.; Adli, M., Chronische und therapieresistente Depression. Deutsches Ärzteblatt 2014, 45, 766-776.
282. Stevens, C. L. US3254124, Aminoketones and Methods for Their Production. 1966.
283. Sinner, B.; Graf, B. M., Handbook of Experimental Pharmacology 182: Modern Anesthetics. Berlin, Heidelberg, 2008; p 313-333.
284. World Health Organization, WHO Model List of Essential Medicines (20th List). 2017.
285. Hameln Pharma Plus GmbH, Fachinformation Ketamin-hameln 50 mg/ml Injektionslösung. Stand Januar 2015.
286. Lee, C.-H.; Lü, W.; Michel, J. C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E., NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 2014, 511, 191-197.
287. Danysz, W.; Zajaczkowski, W.; Parsons, C. G., Modulation of learning processes by ionotropic glutamate receptor ligands. Behavioural Pharmacology 1995, 6, 455-474.
288. Ulrich Zeilhofer, H.; Swandulla, D.; Geisslinger, G.; Brune, K., Differential effects of ketamine enantiomers on NMDA receptor currents in cultured neurons. European Journal of Pharmacology 1992, 213, 155-158.
289. Persson, J., Ketamine in Pain Management. CNS Neuroscience & Therapeutics 2013, 19, 396-402.
290. Trullas, R.; Skolnick, P., Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. European Journal of Pharmacology 1990, 185, 1-10.
291. Nowak, G.; Trullas, R.; Layer, R. T.; Skolnick, P.; Paul, I. A., Adaptive changes in the N-methyl-D-aspartate receptor complex after chronic treatment with imipramine and 1-aminocyclopropanecarboxylic acid. Journal of Pharmacology and Experimental Therapeutics 1993, 265, 1380-1386.
292. Berman, R. M.; Cappiello, A.; Anand, A.; Oren, D. A.; Heninger, G. R.; Charney, D. S.; Krystal, J. H., Antidepressant effects of ketamine in depressed patients. Biological Psychiatry 2000, 47, 351-354.
293. Oaklander, M. New Hope for Depression, Time Magazine 2017, 190. (accessed Februar 2018, time.com/4876098/new-hope-for-depression/).
294. Adams, J. D.; Baillie, T. A.; Trevor, A. J.; Castagnoli, N., Studies on the biotransformation of ketamine, 1—Identification of metabolites produced in vitro from rat liver microsomal preparations. Biological Mass Spectrometry 1981, 8, 527-538.

Literaturverzeichnis
220 | Literatur
295. Woolf, T. F.; Adams, J. D., Biotransformation of ketamine, (Z)-6-hydroxyketamine, and (E)-6-hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica 1987, 17, 839-847.
296. Turfus, S. C.; Parkin, M. C.; Cowan, D. A.; Halket, J. M.; Smith, N. W.; Braithwaite, R. A.; Elliott, S. P.; Steventon, G. B.; Kicman, A. T., Use of Human Microsomes and Deuterated Substrates; An Alternative Approach for the Identification of Novel Metabolites of Ketamine by Mass Spectrometry. Drug Metabolism and Disposition 2009, 37, 1769-1778.
297. Zarate, C. A.; Niciu, M. J., Ketamine for depression: evidence, challenges and promise. World Psychiatry 2015, 14, 348-350.
298. McGirr, A.; Berlim, M. T.; Bond, D. J.; Fleck, M. P.; Yatham, L. N.; Lam, R. W., A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychological Medicine 2015, 45, 693-704.
299. Grady, S. E.; Marsh, T. A.; Tenhouse, A.; Klein, K., Ketamine for the treatment of major depressive disorder and bipolar depression: A review of the literature. Mental Health Clinician 2017, 7, 16-23.
300. Gamble, J. ECT With Ketamine Anesthesia vs High Intensity Ketamine With ECT Rescue for Treatment-Resistant Depression (NCT03272698). (accessed März 2018, clinicaltrials.gov/ct2/show/NCT03272698?term=mood+disorder&cond=ketamine&draw=5&rank=4).
301. Blier, P. Action of Ketamine in Treatment-Resistant Depression (NCT01945047). (accessed März 2018, clinicaltrials.gov/ct2/show/NCT01945047?term=mood+disorder&cond=ketamine&draw=5&rank=2).
302. Williams, N. Double-Blind, Cross-Over Trial of Ketamine Therapy Plus or Minus Naltrexone in Treatment Resistant Depression (TRD) (Ket_Nal) (NCT02911597). (accessed März 2018, clinicaltrials.gov/ct2/show/NCT02911597?term=mood+disorder&cond=ketamine&draw=3&rank=25).
303. Gálvez, V.; Li, A.; Huggins, C.; Glue, P.; Martin, D.; Somogyi, A. A.; Alonzo, A.; Rodgers, A.; Mitchell, P. B.; Loo, C. K., Repeated intranasal ketamine for treatment-resistant depression – the way to go? Results from a pilot randomised controlled trial. Journal of Psychopharmacology 2018, 32, 397-407.
304. Moaddel, R.; Venkata, S. L. V.; Tanga, M. J.; Bupp, J. E.; Green, C. E.; Iyer, L.; Furimsky, A.; Goldberg, M. E.; Torjman, M. C.; Wainer, I. W., A parallel chiral–achiral liquid chromatographic method for the determination of the stereoisomers of ketamine and ketamine metabolites in the plasma and urine of patients with complex regional pain syndrome. Talanta 2010, 82, 1892-1904.
305. Zanos, P.; Moaddel, R.; Morris, P. J.; Georgiou, P.; Fischell, J.; Elmer, G. I.; Alkondon, M.; Yuan, P.; Pribut, H. J.; Singh, N. S.; Dossou, K. S. S.; Fang, Y.; Huang, X.-P.; Mayo, C. L.; Wainer, I. W.; Albuquerque, E. X.; Thompson, S. M.; Thomas, C. J.; Zarate Jr, C. A.; Gould, T. D., NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481-486.
306. Sanacora, G.; Frye, M. A.; McDonald, W.; Mathew, S. J.; Turner, M. S.; Schatzberg, A. F.; Summergrad, P.; Nemeroff, C. B., A consensus statement on the use of ketamine in the treatment of mood disorders. JAMA Psychiatry 2017, 74, 399-405.
307. Pfizer Pharma PFE GmbH, Fachinformation Ketanest® S. Stand September 2017.
308. Newport, D. J.; Carpenter, L. L.; McDonald, W. M.; Potash, J. B.; Tohen, M.; Nemeroff, C. B., Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. American Journal of Psychiatry 2015, 172, 950-966.

Literaturverzeichnis
Literatur | 221
309. Yang, C.; Qu, Y.; Abe, M.; Nozawa, D.; Chaki, S.; Hashimoto, K., (R)-Ketamine Shows Greater Potency and Longer Lasting Antidepressant Effects Than Its Metabolite (2R,6R)-Hydroxynorketamine. Biological Psychiatry 2017, 82, e43-e44.
310. Abdallah, C. G., What’s the Buzz About Hydroxynorketamine? Is It the History, the Story, the Debate, or the Promise? Biological Psychiatry 2017, 81, e61-e63.
311. Collingridge, G. L.; Lee, Y.; Bortolotto, Z. A.; Kang, H.; Lodge, D., Antidepressant Actions of Ketamine Versus Hydroxynorketamine. Biological Psychiatry 2017, 81, e65-e67.
312. Zanos, P.; Moaddel, R.; Morris, P. J.; Wainer, I. W.; Albuquerque, E. X.; Thompson, S. M.; Thomas, C. J.; Zarate, C. A.; Gould, T. D., Reply to: Antidepressant Actions of Ketamine Versus Hydroxynorketamine. Biological Psychiatry 2017, 81, e69-e71.
313. Singh, N. S.; Zarate, C. A.; Moaddel, R.; Bernier, M.; Wainer, I. W., What is hydroxynorketamine and what can it bring to neurotherapeutics? Expert Review of Neurotherapeutics 2014, 14, 1239-1242.
314. Daly, E. J.; Singh, J. B.; Fedgchin, M.; Cooper, K.; Lim, P.; Shelton, R. C.; Thase, M. E.; Winokur, A.; Nueten, L. V.; Manji, H.; Drevets, W. C., Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: A randomized clinical trial. JAMA Psychiatry 2018, 75, 139-148.
315. Geisslinger, G.; Hering, W.; Thomann, P.; Knoll, R.; Kamp, H. D.; Brune, K., Pharmacokinetics and pharmacodynamics of ketamine enantiomers in surgical patients using a stereoselective analytical method. British Journal of Anaesthesia 1993, 70, 666-671.
316. Wainer, I. W.; Moaddel, R.; Bernier, M.; Zarate, C. A.; Torjman, M. C. US9867830B2, The use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine and other stereoisomeric dehydro and hydroxylated metabolites of (R,S)-ketamine in the treatment of depression and neuropathic pain. 2018.
317. Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H., Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317-322.
318. Kock, M.; Loix, S.; Lavand'homme, P., Ketamine and Peripheral Inflammation. CNS Neuroscience & Therapeutics 2013, 19, 403-410.
319. Clements, J. A.; Nimmo, W. S., Pharmacokinetics and analgesic effect of ketamine in man. British Journal of Anaesthesia 1981, 53, 27-30.
320. Moreno, I.; Barroso, M.; Martinho, A.; Cruz, A.; Gallardo, E., Determination of ketamine and its major metabolite, norketamine, in urine and plasma samples using microextraction by packed sorbent and gas chromatography-tandem mass spectrometry. Journal of Chromatography B 2015, 1004, 67-78.
321. Wang, K.-C.; Shih, T.-S.; Cheng, S.-G., Use of SPE and LC/TIS/MS/MS for rapid detection and quantitation of ketamine and its metabolite, norketamine, in urine. Forensic Science International 2005, 147, 81-88.
322. Legrand, T.; Roy, S.; Monchaud, C.; Grondin, C.; Duval, M.; Jacqz-Aigrain, E., Determination of ketamine and norketamine in plasma by micro-liquid chromatography–mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis 2008, 48, 171-176.
323. Geisslinger, G.; Menzel-Soglowek, S.; Kamp, H. D.; Brune, K., Stereoselective high-performance liquid chromatographic determination of the enantiomers of ketamine and norketamine in plasma. Journal of Chromatography B: Biomedical Sciences and Applications 1991, 568, 165-176.
324. Svensson, J.-O.; Gustafsson, L. L., Determination of ketamine and norketamine enantiomers in plasma by solid-phase extraction and high-performance liquid chromatography. Journal of Chromatography B: Biomedical Sciences and Applications 1996, 678, 373-376.

Literaturverzeichnis
222 | Literatur
325. Yanagihara, Y.; Ohtani, M.; Kariya, S.; Uchino, K.; Aoyama, T.; Yamamura, Y.; Iga, T., Stereoselective high-performance liquid chromatographic determination of ketamine and its active metabolite, norketamine, in human plasma. Journal of Chromatography B: Biomedical Sciences and Applications 2000, 746, 227-231.
326. Lin, H.-R.; Liao, C.-C.; Lin, T.-C., Improved identification of multiple drugs of abuse and relative metabolites in urine samples using liquid chromatography/triple quadrupole mass spectrometry coupled with a library search. Rapid Communications in Mass Spectrometry 2014, 28, 2043-2053.
327. Bolze, S.; Boulieu, R., HPLC determination of ketamine, norketamine, and dehydronorketamine in plasma with a high-purity reversed-phase sorbent. Clinical Chemistry 1998, 44, 560-564.
328. Göktaş, E. F.; Arıöz, F., A review of chromatographic methods for ketamine and its metabolites norketamine and dehydronorketamine. Biomedical Chromatography 2018, 32, e4014.
329. Hasan, M.; Hofstetter, R.; Fassauer, G. M.; Link, A.; Siegmund, W.; Oswald, S., Quantitative chiral and achiral determination of ketamine and its metabolites by LC-MS/MS in human serum, urine and fecal samples. Journal of Pharmaceutical and Biomedical Analysis 2017, 139, 87-97.
330. Smith, R. M., Supercritical fluids in separation science – the dreams, the reality and the future. Journal of Chromatography A 1999, 856, 83-115.
331. Klesper, E.; Corwin, A. H.; Turner, D. A., High Pressure Gas Chromatography above Critical Temperatures. The Journal of Organic Chemistry 1962, 27, 700-701.
332. Berger, T. A., Supercritical Fluid Chromatography. Agilent Technologies, Ed. 2015.
333. Sadoun, F.; Virelizier, H.; Arpino, P. J., Packed-column supercritical fluid chromatography coupled with electrospray ionization mass spectrometry. Journal of Chromatography A 1993, 647, 351-359.
334. Arpino, P., Coupling techniques in LC/MS and SFC/MS. Fresenius' Journal of Analytical Chemistry 1990, 337, 667-685.
335. Lozowoski, D. Supercritical CO2: A Green Solvent. (accessed März 2018, chemengonline.com/supercritical-co2-a-green-solvent/).
336. Phillips, J. H.; J. Robey, R., Solvent strength and selectivity properties of supercritical carbon dioxide relative to liquid hexane. Journal of Chromatography A 1989, 465, 177-188.
337. Bieber, S.; Letzel, T. SFC – Superkritische Fluid Chromatographie oder Science Fiction Chromatographie? (accessed März 2018, analytik-news.de/Fachartikel/2015/2.html).
338. Tarafder, A.; Hill, J. F.; Baynham, M., Convergence Chromatography Versus SFC - What’s in a Name? Chromatography Today 2014, 34-36.
339. Berger, T. A., Density of methanol-carbon dioxide mixtures at three temperatures: Comparison with vapor-liquid equilibria measurements and results obtained from chromatography. Journal of High Resolution Chromatography 1991, 14, 312-316.
340. Page, S. H.; Goates, S. R.; Lee, M. L., Methanol/CO2 phase behavior in supercritical fluid chromatography and extraction. The Journal of Supercritical Fluids 1991, 4, 109-117.
341. Płotka, J. M.; Biziuk, M.; Morrison, C.; Namieśnik, J., Pharmaceutical and forensic drug applications of chiral supercritical fluid chromatography. Trends in Analytical Chemistry 2014, 56, 74-89.
342. Parr, M. K.; Schmidt, A. H., What is the potential of measuring the enantiomeric ratio of drugs using supercritical fluid chromatography–MS? Bioanalysis 2014, 6, 3267-3270.

Literaturverzeichnis
Literatur | 223
343. Kocova Vlckova, H.; Pilarova, V.; Svobodova, P.; Plisek, J.; Svec, F.; Novakova, L., Current state of bioanalytical chromatography in clinical analysis. Analyst 2018, 143, 1305-1325.
344. European Medicines Agency (EMA), Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009 Rev.1 Corr.2**, 2011.
345. Food and Drug Administration (FDA), Guidance for Industry - Bioanalytical Method Validation. Draft Guidance Revision 1, 2013.
346. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Validation of Analytical Procedures: Text and Methodology Q2(R1). Current Step 4 version, 2005.
347. Annesley, T. M., Ion Suppression in Mass Spectrometry. Clinical Chemistry 2003, 49, 1041.
348. Matuszewski, B. K.; Constanzer, M. L.; Chavez-Eng, C. M., Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Analytical Chemistry 2003, 75, 3019-3030.
349. Taylor, P. J., Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography–electrospray–tandem mass spectrometry. Clinical Biochemistry 2005, 38, 328-334.
350. Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A., An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrometry Reviews 2011, 30, 491-509.
351. Nelander, H.; Andersson, S.; Öhlén, K., Evaluation of the chiral recognition properties as well as the column performance of four chiral stationary phases based on cellulose (3,5-dimethylphenylcarbamate) by parallel HPLC and SFC. Journal of Chromatography A 2011, 1218, 9397-9405.
352. Ye, Y. K.; Lynam, K. G.; Stringham, R. W., Effect of amine mobile phase additives on chiral subcritical fluid chromatography using polysaccharide stationary phases. Journal of Chromatography A 2004, 1041, 211-217.
353. Phenomenex. HPLC Column Protection Guide. (accessed März 2018, phx.phenomenex.com/lib/gu54810610.pdf).
354. Page, J. S.; Kelly, R. T.; Tang, K.; Smith, R. D., Ionization and Transmission Efficiency in an Electrospray Ionization–Mass Spectrometry Interface. Journal of the American Society for Mass Spectrometry 2007, 18, 1582-1590.
355. Schlittenbauer, L.; Seiwert, B.; Reemtsma, T., Matrix effects in human urine analysis using multi-targeted liquid chromatography–tandem mass spectrometry. Journal of Chromatography A 2015, 1415, 91-99.
356. Deventer, K.; Pozo, O. J.; Verstraete, A. G.; Van Eenoo, P., Dilute-and-shoot-liquid chromatography-mass spectrometry for urine analysis in doping control and analytical toxicology. Trends in Analytical Chemistry 2014, 55, 1-13.
357. Duxbury, K.; Owen, L.; Gillingwater, S.; Keevil, B., Naturally occurring isotopes of an analyte can interfere with doubly deuterated internal standard measurement. Annals of Clinical Biochemistry 2008, 45, 210-212.
358. Kromidas, S., Validierung in der Analytik. Weinheim, 2. überarbeitete Auflage, 2011; p 142-147.
359. Hasan, M. Dissertation, Pharmacokinetics and Disposition of Prolonged-Release Ketamine Tablets for Treatment of Neuro-Psychiatric Diseases. Universität Greifswald, 2017.

Literaturverzeichnis
224 | Literatur
360. Ramiole, C.; D'Hayer, B.; Boudy, V.; Legagneux, J.; Fonsart, J.; Houzé, P., Determination of ketamine and its main metabolites by liquid chromatography coupled to tandem mass spectrometry in pig plasma: Comparison of extraction methods. Journal of Pharmaceutical and Biomedical Analysis 2017, 146, 369-377.
361. Toki, H.; Ichikawa, T.; Mizuno-Yasuhira, A.; Yamaguchi, J.-i., A rapid and sensitive chiral LC–MS/MS method for the determination of ketamine and norketamine in mouse plasma, brain and cerebrospinal fluid applicable to the stereoselective pharmacokineticstudy of ketamine. Journal of Pharmaceutical and Biomedical Analysis 2018, 148, 288-297.
362. Yoshioka, T.; Nagatomi, Y.; Harayama, K.; Bamba, T., Development of an analytical method for polycyclic aromatic hydrocarbons in coffee beverages and dark beer using novel high-sensitivity technique of supercritical fluid chromatography/mass spectrometry. Journal of Bioscience and Bioengineering 2018, 126, 126-130.
363. Tao, Y.; Zheng, Z.; Yu, Y.; Xu, J.; Liu, X.; Wu, X.; Dong, F.; Zheng, Y., Supercritical fluid chromatography–tandem mass spectrometry-assisted methodology for rapid enantiomeric analysis of fenbuconazole and its chiral metabolites in fruits, vegetables, cereals, and soil. Food Chemistry 2018, 241, 32-39.
364. Li, R.; Chen, Z.; Dong, F.; Xu, J.; Liu, X.; Wu, X.; Pan, X.; Tao, Y.; Zheng, Y., Supercritical fluid chromatographic-tandem mass spectrometry method for monitoring dissipation of thiacloprid in greenhouse vegetables and soil under different application modes. Journal of Chromatography B 2018, 1081-1082, 25-32.
365. Dispas, A.; Jambo, H.; Andre, S.; Tyteca, E.; Hubert, P., Supercritical fluid chromatography: a promising alternative to current bioanalytical techniques. Bioanalysis 2018, 10, 107-124.
366. Hofstetter, R.; Fassauer, G. M.; Link, A., Supercritical fluid extraction (SFE) of ketamine metabolites from dried urine and on-line quantification by supercritical fluid chromatography and single mass detection (on-line SFE–SFC–MS). Journal of Chromatography B 2018, 1076, 77-83.
367. Clusius, K.; Knopf, H., Reaktionen mit 15N. XXII. Die Zersetzung von Azid mit Salpetriger Säure und die Azid-Bildung aus Distickstoffoxyd und Natriumamid. Chemische Berichte 1956, 89, 681-685.
368. Bundesinstitut für Arzneimittel und Medizinprodukte. Valsartan: chargenbezogener Rückruf valsartanhaltiger Arzneimittel, deren Wirkstoff von dem chinesischen Hersteller Zhejiang Huahai Pharmaceutical produziert wurde. (accessed Juli 2018, bfarm.de/SharedDocs/Pressemitteilungen/DE/2018/pm5-2018.html).
369. Osadchii, S. A.; Shul’ts, E. E.; Polukhina, E. V.; Shakirov, M. M.; Tolstikov, G. A., Study of alkaloids of the Siberian and Altai flora. Russian Chemical Bulletin 2006, 55, 1077-1084.
370. Wieland, H.; Rosenfeld, B., Triphenyl‐chlormethan und Knallsilber. Justus Liebigs Annalen der Chemie 1930, 484, 236-245.
371. Büchi, J.; Labhart, P.; Ragaz, L., Zur Kenntnis lokalanästhetisch wirksamer Pyridin‐4‐carbonsäure‐Derivate. Helvetica Chimica Acta 1947, 30, 507-519.

Eigenständigkeitserklärung
Eigenständigkeitserklärung | 225
Eigenständigkeitserklärung
Hiermit erkläre ich, dass diese Arbeit bisher von mir weder an der Mathematisch-
Naturwissenschaftlichen Fakultät der Universität Greifswald noch einer anderen
wissenschaftlichen Einrichtung zum Zwecke der Promotion eingereicht wurde.
Ferner erkläre ich, dass ich diese Arbeit selbstständig verfasst und keine anderen als die
darin angegebenen Hilfsmittel und Hilfen benutzt und keine Textabschnitte eines Dritten
ohne Kennzeichnung übernommen habe.
Unterschrift des Promovenden

Tabellarischer Lebenslauf
226 | Lebenslauf
Tabellarischer Lebenslauf
Hinweis: Der Lebenslauf wurde aus der elektronischen Version der Arbeit entfernt.

Eigene Publikationen, Poster und Vorträge
Publikationsliste | 227
Eigene Publikationen, Poster und Vorträge
Peer-Review-Artikel
Robert Hofstetter, Georg M. Fassauer and Andreas Link, Supercritical fluid extraction (SFE)
of ketamine metabolites from dried urine and on-line quantification by supercritical fluid
chromatography and single mass detection (on-line SFE–SFC–MS), Journal of
Chromatography B 2018, 1076, 77-83.
Georg M. Fassauer, Robert Hofstetter, Mahmoud Hasan, Stefan Oswald, Christina Modeß,
Werner Siegmund and Andreas Link, Ketamine metabolites with antidepressant effects:
Fast, economical, and eco-friendly enantioselective separation based on supercritical-fluid
chromatography (SFC) and single quadrupole MS detection, Journal of Pharmaceutical and
Biomedical Analysis 2017, 146, 410-419.
Mahmoud Hasan, Robert Hofstetter, Georg M. Fassauer, Andreas Link, Werner Siegmund
and Stefan Oswald, Quantitative chiral and achiral determination of ketamine and its
metabolites by LC–MS/MS in human serum, urine and fecal samples, Journal of
Pharmaceutical and Biomedical Analysis 2017, 139, 87-97.
Nicole Rüger, Georg Michael Fassauer, Christian Bock, Thomas Emmrich, Anja Bodtke and
Andreas Link, Substituted tetrazoles as multipurpose screening compounds, Molecular
Diversity 2017, 21, 9-27.
Corina Pfeifer, Sylvia Noll, Hagen Gerecke, Georg Fassauer, Thomas Jira, Yvonne
Remane, Jan Vogel, Roberto Frontini and Robert Reinhardt, A stability study of
amphotericin B, colistin and tobramycin in a hydrophilic suspension commonly used for
selective decontamination of the digestive tract by HPLC and in vitro potency
measurements, European Journal of Hospital Pharmacy 2017, 24, 235-241.
Corina Pfeifer, Georg Fassauer, Hagen Gerecke, Thomas Jira, Yvonne Remane, Roberto
Frontini, Jonathan Byrne and Robert Reinhardt, Purity determination of amphotericin B,
colistin sulfate and tobramycin sulfate in a hydrophilic suspension by HPLC, Journal of
Chromatography B 2015, 990, 7-14.

Eigene Publikationen, Poster und Vorträge
228 | Publikationsliste
Nicht-Peer-Review-Artikel
G. M. Fassauer and R. K. Hofstetter, Determination and quantification of ketamine and its
metabolites by means of supercritical-fluid chromatography and single quadrupole MS
detection, Shimadzu Application News 2018, No. SCA_190_038.
Vorträge
Georg M. Fassauer, A Comparative View of the Separation and Quantification of Ketamine
Metabolites Highlighting the Advantages of a Novel SFC-MS Method in Contrast to
Established LC-MS/MS Methods, 32nd International Symposium on Chromatography 2018,
Cannes, Frankreich (Accepted oral presentation).
Georg M. Fassauer, Substituted Tetrazoles as Multipurpose Screening Compounds,
Departmental Seminar Max Delbrück House 2017, Max-Delbrück-Centrum für Molekulare
Medizin, Berlin-Buch.
Poster
G. M. Fassauer, R. K. Hofstetter and A. Link, Ketamine metabolites with antidepressant
effects: fast, economical, and eco-friendly enantioselective separation based on SFC and
single quadrupole MS detection, DPhG Annual Meeting 2017, Saarbrücken.

Danksagung
Danksagung | 229
Danksagung
Als erstes möchte ich mich bei Prof. Dr. Andreas Link für die Aufnahme in seinen
Arbeitskreis, die Betreuung und jede Art von Unterstützung bedanken.
Dem NCATS des NIH, Division of Preclinical Innovation, aus Rockville, MD, USA,
insbesondere Patrick Morris, Ph.D., gilt mein Dank für die großzügige Überlassung
synthetisch hergestellter Ketaminmetaboliten, die nicht kommerziell verfügbar aber für
diese Arbeit von großer Bedeutung waren.
Mein Dank gilt ferner Sandra Wenzler, Dr. Martin Roatsch sowie allen beteiligten
Mitarbeitern aus dem Arbeitskreis von Prof. Dr. Manfred Jung der Albert-Ludwigs-
Universität Freiburg für die Durchführung der biologischen Testungen. Bei Drs. Piotr
Malecki und Manfred Weiss vom Helmholtz-Zentrum Berlin möchte ich mich für die
Bereitstellung von Röntgenkristallstrukturen und ITC-Daten herzlichst bedanken.
Auch Master-Student Patrik Zeyen hat mit seiner tatkräftigen Unterstützung und seinen
intensiven Internetrecherchen zum Erfolg dieser Arbeit beigetragen, wofür ich mich gerne
bei ihm bedanken möchte.
Für die Bearbeitung und Fertigstellung jeglicher NMR-, IR- oder HRMS-Anfragen möchte
ich mich bei Dr. Anja Bodtke, Maria Hühr und Michael Eccius bedanken. Sylvia Ewert und
Ruth Ball danke ich für die Bereitstellung und Organisation von Laborverbrauchsmaterial,
Glasgeräten und Chemikalien.
Meinen Kollegen im Praktikum der organischen Chemie PD Dr. Gregor Radau, Dr. Anja
Bodtke, Christoph Grathwol, Christian Bock und Dr. Christian Lemmerhirt danke ich für die
Kooperationsbereitschaft und angenehme Atmosphäre. Vielen Dank an alle Weggefährten
meiner Doktorandenzeit aus den Arbeitskreisen Link und Bednarski: Steffen Vojacek,
Christian Bock, Rupert Hofstetter, Christoph Grathwol, Abdrrahman Surur, Nicole Rüger,
Lukas Schulig, Stefan Lohmann, Dr. Thomas Emmrich, Drs. Heidi und Christian
Lemmerhirt, Dr. Steven Behnisch, Carsten Lange, Patrick Werth, Kristin Beirow und Lisa
Wolff, sowie alle Diplomanden und Wahlpflichtstudenten aus den Arbeitskreisen. Danke für
die Unterstützung und Gesellschaft innerhalb und außerhalb der Arbeitszeiten. Vielen Dank
dabei auch an alle Mitglieder der unterhaltsamen und konstruktiven Kaffeepausen.
Drs. Simon Merdivan und Mahmoud Hasan möchte ich ferner für die wertvollen Hinweise
rund um die HPLC danken. Erik Wollmer und Steffen Vojacek gebührt als immerwährende
Motivationsstütze an dieser Stelle noch ein besonderes Dankeschön. Des Weiteren möchte
ich mich bei den Korrekturlesern des Manuskripts PD Dr. Gregor Radau, Prof. Dr. Thomas
Jira und meinen Eltern bedanken.
Meinen Eltern, Großeltern und meiner Freundin Berit gilt ebenso ein herzlichstes
Dankeschön. Nicht nur für die Unterstützung während der Promotion, sondern auch auf den
vorherigen und zukünftigen Lebensabschnitten.


















![NEUARTIGE SILICIUM-HALTIGE ALLOSTERISCHE MODULATOREN …hss.ulb.uni-bonn.de/2002/0106/0106.pdf · losterische Wirkung über diese Bindungsstelle zu entfalten scheinen [27]. In den](https://static.fdocuments.net/doc/165x107/5e1f64a9d100e0281c3db3f4/neuartige-silicium-haltige-allosterische-modulatoren-hssulbuni-bonnde200201060106pdf.jpg)