
Microarray-based characterization of differential gene expression ... · differential gene...
11
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 311-321 doi:10.1242/dmm.018366 311 ABSTRACT The vocal fold (VF) mucosa confers elegant biomechanical function for voice production but is susceptible to scar formation following injury. Current understanding of VF wound healing is hindered by a paucity of data and is therefore often generalized from research conducted in skin and other mucosal systems. Here, using a previously validated rat injury model, expression microarray technology and an empirical Bayes analysis approach, we generated a VF-specific transcriptome dataset to better capture the system-level complexity of wound healing in this specialized tissue. We measured differential gene expression at 3, 14 and 60 days post-injury compared to experimentally naïve controls, pursued functional enrichment analyses to refine and add greater biological definition to the previously proposed temporal phases of VF wound healing, and validated the expression and localization of a subset of previously unidentified repair- and regeneration-related genes at the protein level. Our microarray dataset is a resource for the wider research community and has the potential to stimulate new hypotheses and avenues of investigation, improve biological and mechanistic insight, and accelerate the identification of novel therapeutic targets. KEY WORDS: Fibrosis, Larynx, Scar formation, Tissue repair, Transcriptome INTRODUCTION Wound healing is a complex biological process that is characterized by a dynamic series of molecular, cellular and extracellular events (Martin, 1997). These events primarily occur at the wound site but are also orchestrated, in part, at the organ and organism levels (Kisseleva and Brenner, 2008; Mori et al., 2005; Song et al., 2010). The wound healing process is generally conceptualized as consisting of at least three overlapping phases (Gurtner et al., 2008): inflammation (granulocyte and leukocyte infiltration, inflammatory factor secretion), fibroblast and epithelial cell proliferation [provisional extracellular matrix (ECM) deposition, reepithelialization, tissue contraction] and remodeling (ECM reorganization, crosslinking and maturation). In RESOURCE ARTICLE 1 Department of Surgery, Division of Otolaryngology, University of Wisconsin School of Medicine and Public Health, Madison, WI 53792, USA. 2 Department of Biostatistics and Medical Informatics, University of Wisconsin School of Medicine and Public Health, Madison, WI 53706, USA. *Present address: Department of Biostatistics, University of Alabama- Birmingham, Birmingham, AL 35294, USA. ‡ Present address: Department of Otolaryngology, Kitano Hospital, Tazuke Kofukai Medical Research Institute, Osaka, 530-8480 Japan. § Author for correspondence ([email protected]) This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed. Received 17 September 2014; Accepted 5 January 2015 different biological contexts, wound healing can result in a number of outcomes ranging from complete regeneration with restoration of uninjured morphology and function (e.g. fetal wounds) to impaired healing resulting in chronic pathology (e.g. diabetic wounds) (Colwell et al., 2008; Occleston et al., 2010; Schultz et al., 2011; Sullivan et al., 1995). Furthermore, evidence suggests that both the local injury response and eventual repair outcome differ as a function of tissue type. For example, injured adult skin heals more slowly, and with more scar formation, than adult oral mucosa (Schrementi et al., 2008; Szpaderska et al., 2003) and, under permissive conditions, the regeneration-privileged adult liver (Periwal et al., 2014). The vocal fold (VF) mucosa is a specialized tissue with distinctive repair and regeneration needs. Unlike other upper airway mucosae, it is routinely subject to high-frequency oscillation driven by aerodynamic forces, resulting in compressive, tensile and shear stresses (Gunter, 2004; Titze, 1988). Its ability to generate voice is predominantly a function of favorable tissue viscoelasticity, which in turn is conferred by the organization and structure of its ECM (Gray et al., 2000; Gray et al., 1999). Following injury, the matrix typically undergoes disordered repair, resulting in impaired phonation (Benninger et al., 1996; Choi et al., 2012; Welham et al., 2011a; Welham et al., 2007) and, in humans, significant morbidity (Cohen et al., 2006; Cohen et al., 2012). Most existing research in VF wound healing is based on a small set of candidate genes, proteins, signaling molecules and cell types, adopted from work in skin and other mucosal systems. System-level datasets are emerging in other areas of VF biology (Thibeault et al., 2003; Welham et al., 2013; Welham et al., 2011b) and show potential in generating new hypotheses and model refinements, as well as identifying new disease biomarkers and therapeutic candidates. Here, using a previously validated rat injury model (Ling et al., 2010a; Tateya et al., 2005; Tateya et al., 2006b), expression microarray technology and an empirical Bayes analysis approach (Kendziorski et al., 2003), we generated a VF-specific transcriptome dataset to better capture the system-level complexity of VF wound healing and serve as a resource for the wider research community. We characterized transcript-level differential expression in injured and experimentally naïve VF mucosae over time, pursued functional enrichment analyses to refine and add greater biological definition to the previously proposed temporal phases of VF wound healing, and validated the expression and localization of a subset of previously unidentified repair- and regeneration-related genes at the protein level. RESULTS Within-time-point analysis We generated a microarray dataset using rat VF mucosae harvested at three post-injury (PI) time points, and VF mucosae from experimentally naïve age-matched controls (Fig. 1). Injured VF Microarray-based characterization of differential gene expression during vocal fold wound healing in rats Nathan V. Welham 1,§ , Changying Ling 1 , John A. Dawson 2, *, Christina Kendziorski 2 , Susan L. Thibeault 1 and Masaru Yamashita 1,‡ Disease Models & Mechanisms
Transcript of Microarray-based characterization of differential gene expression ... · differential gene...

© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 311-321 doi:10.1242/dmm.018366
311
ABSTRACTThe vocal fold (VF) mucosa confers elegant biomechanical functionfor voice production but is susceptible to scar formation followinginjury. Current understanding of VF wound healing is hindered by apaucity of data and is therefore often generalized from researchconducted in skin and other mucosal systems. Here, using apreviously validated rat injury model, expression microarraytechnology and an empirical Bayes analysis approach, we generateda VF-specific transcriptome dataset to better capture the system-levelcomplexity of wound healing in this specialized tissue. We measureddifferential gene expression at 3, 14 and 60 days post-injurycompared to experimentally naïve controls, pursued functionalenrichment analyses to refine and add greater biological definition tothe previously proposed temporal phases of VF wound healing, andvalidated the expression and localization of a subset of previouslyunidentified repair- and regeneration-related genes at the proteinlevel. Our microarray dataset is a resource for the wider researchcommunity and has the potential to stimulate new hypotheses andavenues of investigation, improve biological and mechanistic insight,and accelerate the identification of novel therapeutic targets.
KEY WORDS: Fibrosis, Larynx, Scar formation, Tissue repair,Transcriptome
INTRODUCTIONWound healing is a complex biological process that is characterizedby a dynamic series of molecular, cellular and extracellular events(Martin, 1997). These events primarily occur at the wound site but arealso orchestrated, in part, at the organ and organism levels (Kisselevaand Brenner, 2008; Mori et al., 2005; Song et al., 2010). The woundhealing process is generally conceptualized as consisting of at leastthree overlapping phases (Gurtner et al., 2008): inflammation(granulocyte and leukocyte infiltration, inflammatory factor secretion),fibroblast and epithelial cell proliferation [provisional extracellularmatrix (ECM) deposition, reepithelialization, tissue contraction] andremodeling (ECM reorganization, crosslinking and maturation). In
RESOURCE ARTICLE
1Department of Surgery, Division of Otolaryngology, University of WisconsinSchool of Medicine and Public Health, Madison, WI 53792, USA. 2Department ofBiostatistics and Medical Informatics, University of Wisconsin School of Medicineand Public Health, Madison, WI 53706, USA. *Present address: Department of Biostatistics, University of Alabama-Birmingham, Birmingham, AL 35294, USA. ‡Present address: Department ofOtolaryngology, Kitano Hospital, Tazuke Kofukai Medical Research Institute,Osaka, 530-8480 Japan.
§Author for correspondence ([email protected])
This is an Open Access article distributed under the terms of the Creative CommonsAttribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricteduse, distribution and reproduction in any medium provided that the original work is properlyattributed.
Received 17 September 2014; Accepted 5 January 2015
different biological contexts, wound healing can result in a numberof outcomes ranging from complete regeneration with restorationof uninjured morphology and function (e.g. fetal wounds) toimpaired healing resulting in chronic pathology (e.g. diabeticwounds) (Colwell et al., 2008; Occleston et al., 2010; Schultz et al.,2011; Sullivan et al., 1995). Furthermore, evidence suggests thatboth the local injury response and eventual repair outcome differ asa function of tissue type. For example, injured adult skin heals moreslowly, and with more scar formation, than adult oral mucosa(Schrementi et al., 2008; Szpaderska et al., 2003) and, underpermissive conditions, the regeneration-privileged adult liver(Periwal et al., 2014).
The vocal fold (VF) mucosa is a specialized tissue with distinctiverepair and regeneration needs. Unlike other upper airway mucosae,it is routinely subject to high-frequency oscillation driven byaerodynamic forces, resulting in compressive, tensile and shearstresses (Gunter, 2004; Titze, 1988). Its ability to generate voice ispredominantly a function of favorable tissue viscoelasticity, whichin turn is conferred by the organization and structure of its ECM(Gray et al., 2000; Gray et al., 1999). Following injury, the matrixtypically undergoes disordered repair, resulting in impairedphonation (Benninger et al., 1996; Choi et al., 2012; Welham et al.,2011a; Welham et al., 2007) and, in humans, significant morbidity(Cohen et al., 2006; Cohen et al., 2012).
Most existing research in VF wound healing is based on a smallset of candidate genes, proteins, signaling molecules and cell types,adopted from work in skin and other mucosal systems. System-leveldatasets are emerging in other areas of VF biology (Thibeault et al.,2003; Welham et al., 2013; Welham et al., 2011b) and showpotential in generating new hypotheses and model refinements, aswell as identifying new disease biomarkers and therapeuticcandidates. Here, using a previously validated rat injury model (Linget al., 2010a; Tateya et al., 2005; Tateya et al., 2006b), expressionmicroarray technology and an empirical Bayes analysis approach(Kendziorski et al., 2003), we generated a VF-specific transcriptomedataset to better capture the system-level complexity of VF woundhealing and serve as a resource for the wider research community.We characterized transcript-level differential expression in injuredand experimentally naïve VF mucosae over time, pursued functionalenrichment analyses to refine and add greater biological definitionto the previously proposed temporal phases of VF wound healing,and validated the expression and localization of a subset ofpreviously unidentified repair- and regeneration-related genes at theprotein level.
RESULTSWithin-time-point analysisWe generated a microarray dataset using rat VF mucosae harvestedat three post-injury (PI) time points, and VF mucosae fromexperimentally naïve age-matched controls (Fig. 1). Injured VF
Microarray-based characterization of differential gene expressionduring vocal fold wound healing in ratsNathan V. Welham1,§, Changying Ling1, John A. Dawson2,*, Christina Kendziorski2, Susan L. Thibeault1 andMasaru Yamashita1,‡
Dis
ease
Mod
els
& M
echa
nism
s

312
mucosa samples were collected and processed at 3, 14 and 60 daysPI, based on previously reported rat data suggesting that these timepoints represent inflammatory, proliferative and remodeling woundhealing phases, respectively (Lim et al., 2006; Ling et al., 2010a;Tateya et al., 2006a; Tateya et al., 2005; Tateya et al., 2006b).Using a ‘within-time-point’ analysis strategy, we first evaluatedprobes and genes that were differentially expressed across the PIand control conditions at individual time points. We observed anoverall decrease in differentially expressed probes and genes withwound healing progression: the numbers of differentiallyexpressed probes and genes were greatest at 3 days PI (4773probes; 2311 genes) and progressively decreased at 14 days PI(1113 probes; 714 genes) and 60 days PI (78 probes; 49 genes)(Fig. 2). The majority of differentially expressed probes and genesat 3 days PI were exclusive to the 3-day-PI time point (4183probes; 1931 genes); less than half of the differentially expressedprobes and genes at 14 days PI were exclusive to the 14-day-PItime point (501 probes; 315 genes); the minority of differentiallyexpressed probes and genes at 60 days PI were exclusive to the 60-day-PI time point (23 probes; 10 genes). A total of 18 genes,represented by 25 probes, were differentially expressed at all timepoints.
Across-time-point analysisWe also performed a parallel analysis of the gene-level data using an‘across-time-point’ analysis strategy, focusing on genes that weredifferentially expressed over time within the PI and control conditions,respectively. Genes identified as differentially expressed within eithercondition were further categorized into one of four patterns: 3d-outgroup (3d-out; differentially expressed at the 3-day time pointcompared with at 14 days and 60 days); 14d-out (differentiallyexpressed at the 14-day time point compared with at 3 days and 60days); 60d-out (differentially expressed at the 60-day time pointcompared with at 3 days and 14 days); and distinct (differentiallyexpressed across all three time points). A total of 3605 differentiallyexpressed genes were identified in the PI condition (Fig. 3A): themajority (3272 genes; 90.8%) exhibited a 3d-out pattern (Fig. 3B),consistent with predominant transcriptional activity during theinflammatory wound healing phase. In contrast, 139 differentiallyexpressed genes were identified in the control condition (Fig. 3A): themajority (126 genes; 90.6%) exhibited a 60d-out pattern (Fig. 3C),consistent with slow-changing transcriptional activity that might beattributable to maturation or aging. Further analysis of the 76 genesthat were differentially expressed in both PI and control conditionsrevealed that 12 genes (15.8%) showed no change in differentialexpression pattern across conditions, whereas the majority (31 genes;41.8%) switched from a 60d-out pattern (the predominant pattern inthe control condition) to a 3d-out pattern (the predominant pattern inthe PI condition) following injury (Fig. 3D).
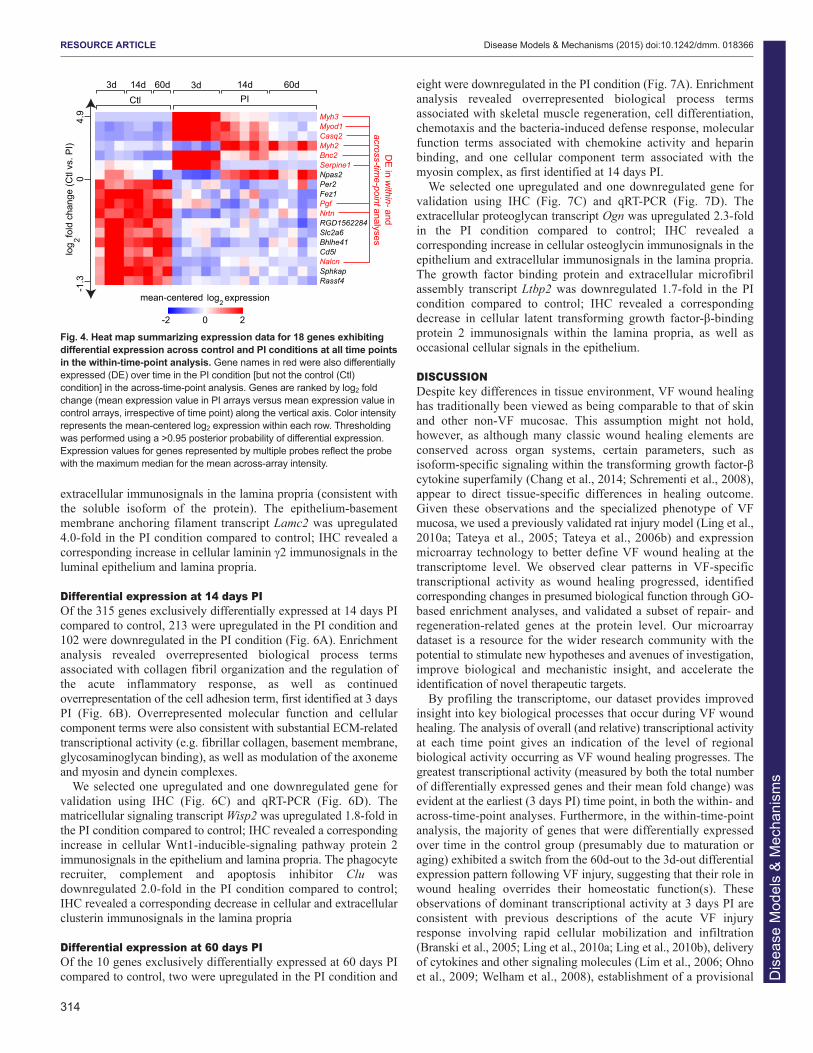
A subset of genes exhibit sustained differential expressionthroughout the wound healing process Next, we examined the subset of 18 genes that were consistentlydifferentially expressed at 3, 14 and 60 days PI in the within-time-
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
RESOURCE IMPACTBackgroundInjury to the vocal fold (VF) mucosa initiates a series of wound healingevents that can lead to scar formation. Scarred VF mucosa ischaracterized by disordered tissue biomechanics and vibratory function,which can result in loss of voice (dysphonia) and reduced quality of life.There are currently no effective therapies for the scarred VF mucosa. Asignificant hindrance to progress in this area is the lack of VF-specific‘omic’ data, particularly at the level of transcriptomics. The availability of‘omic’ datasets would help to advance our understanding of the system-level complexity of wound healing as it occurs in the dynamicbiomechanical environment of the VF mucosa.
ResultsIn this study, the authors used a previously validated in vivo rat model ofVF injury. They combined expression microarray technology with anempirical Bayes analysis approach to examine differential geneexpression at 3, 14 and 60 days post-injury (PI) compared toexperimentally naïve controls. Overall, transcriptional activity was highestat 3 days PI and then tapered over time. Early transcription events at 3days PI were primarily associated with cell division and proliferation, aswell as cell adhesion to the provisional extracellular matrix (ECM). Acuteinflammatory activity, first detected at this time point, continued furtherand was accompanied by the acceleration of ECM-related transcriptionat 14 days PI. The authors observed clear resolution of inflammation-and proliferation-driven transcriptional activity by 60 days PI, by whichtime they could detect only ten differentially expressed genes. A geneset that is predominantly associated with muscle differentiation,contractile function and repair exhibited sustained differential expressionthroughout the entire experimental time course.
Implications and future directionsThe microarray dataset produced in this study is publically available atthe Gene Expression Omnibus (GEO) repository and thus represents avaluable resource for the wider research community with the potential tostimulate new hypotheses and avenues of investigation, improvebiological and mechanistic insight, and accelerate the identification ofnovel therapeutic targets for VF injury and scarring. The analysis offersa more accurate biological definition of the temporal phases of VF woundhealing compared to that previously proposed. Future work with this‘omic’ dataset might include the refinement and validation of existing invitro models, the examination of tissue-specific differences in woundhealing outcome and, ultimately, the identification of a therapeutically-relevant transcriptomic signature for scarless tissue regeneration.
bila
tera
l V
F in
jury
14d-PI 60d-PI3d-PI
age-
mat
ched
cont
rol
bilateral VF injury
within-tim
e-point analysis
across-time-point analysis
Fig. 1. Experimental design and analysis strategy. A set of 4-month-old ratsreceived bilateral vocal fold (VF) mucosal stripping injuries, followed by tissueharvest at 3, 14 and 60 days post-injury (d-PI). A parallel set of experimentallynaïve age-matched rats were used as controls. Blue arrows indicate theprimary comparisons of interest in the within-time-point analysis; green arrowsindicate the primary comparisons of interest in the across-time-point analysis.
Dis
ease
Mod
els
& M
echa
nism
s

point analysis, compared with genes that were differentiallyexpressed over time in the PI condition but not the control conditionin the across-time-point analysis. A total of 9 genes weredifferentially expressed under both scenarios (Fig. 4), including themuscle-related genes Myh2, Myh3, Myod1 and Casq2. In additionto exhibiting consistent differential expression across conditions andtime points, several genes within this subset showed temporaltranscription variation. For example, Myh3, Myod1 and Casq2, aswell as the fibrinolysis inhibitor Serpine1 and the basal epithelialcell transcription factor Bnc2, showed the highest expression levelsat 3 days PI followed by tapering as wound healing progressed,whereas Myh2 was highly expressed beginning at 14 days PI. Othergenes in this subset, such as the angiogenesis promotor Pgf, theneurotrophic factor Nrtn and the ion channel Nalcn, showed moreuniform differential expression over the experimental time course.
Differential expression at 3 days PIOf the 1931 genes exclusively differentially expressed at 3 days PIcompared to control, 1130 were upregulated in the PI condition and801 were downregulated in the PI condition (Fig. 5A). Enrichmentanalysis using the Gene Ontology (GO) annotations (Gene OntologyConsortium, 2013) revealed a cluster of overrepresented biologicalprocess terms associated with cell adhesion, cell cycle and mitoticprocesses, as well as cellular component terms associated with thecytoskeleton, chromosomes and ECM (Fig. 5B). We identified nooverrepresented molecular function terms at 3 days PI.
We selected four highly upregulated genes for immunovalidationand localization at the protein level using immunohistochemistry(IHC) (Fig. 5C), alongside validation at the transcript level usingquantitative real-time PCR (qRT-PCR) (Fig. 5D). The extracellularglycoprotein transcript Tnc was upregulated 41.6-fold in the PIcondition compared to control; IHC revealed a correspondingincrease in cellular and extracellular tenascin C immunosignals inthe lamina propria. The matrix metalloproteinase (specifically,collagenase) transcript Mmp13 was upregulated 11.9-fold in the PIcondition compared to control; IHC revealed a correspondingincrease in extracellular matrix metalloproteinase 13 immunosignalsin the subepithelium and superficial lamina propria. The Toll-interleukin receptor transcript Il1rl1 (also known as St2) wasupregulated 6.7-fold in the PI condition compared to control; IHCrevealed a corresponding increase in cellular interleukin 1 receptor-like 1 immunosignals in the epithelium (consistent with themembrane-bound receptor isoform of the protein) as well as
313
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
ProbesTotal DE = 5323
(100.0%)
GenesTotal DE = 2656
(100.0%)
3d-PI4773
(89.7%)
60d-PI78
(1.5%)
14d-PI1113
(20.9%)
4183(78.6%)
26(0.5%)
4(0.1%)
501(9.4%)
561(10.5%)
23(0.4%)
25(0.5%)
3d-PI2311
(94.5%)
60d-PI49
(1.8%)
14d-PI714
(26.9%)
1931(72.7%)
1(< 0.1%)
315(11.9%)
361(13.6%)
10(0.4%)
18(0.7%)
20(0.8%)
A
B
Fig. 2. Within-time-point analysis summary. Venn diagrams illustrating theoverlap in differentially expressed (DE) probes (A) and genes (B) at 3, 14 and60 days post-injury (d-PI) compared to age-matched controls. Thresholdingwas performed using a >0.95 posterior probability of differential expression.Owing to rounding errors, not all percentages add up to 100.0%.
3529(96.2%)
63(1.7%)
76(2.1%)
Total DE3668 genes (100.0%)
PI3605 genes
(98.3%)
Ctl139 genes
(3.8%)
A
0
10
20
30
40
DE
gen
es (%
)
3d-ou
t
3d-ou
t60
d-out
60d-o
ut
3d-ou
t
60d-o
utdis
tinct
60d-o
ut
3d-ou
t
14d-o
ut14
d-out
60d-o
ut
3d-ou
t
distin
ct
no DE-pattern changeDE-pattern change
DE in PI and Ctl76 genes
D
11(14.5%)
1(1.3%)
31(40.8%)
24(31.6%)
4(5.2%) 3
(3.9%)2
(2.6%)
DE in Ctl139 genes
0
20
40
60
80
100
DE
gen
es (%
)3d-out DE1 gene(0.7%)
14d-out DE7 genes(5.0%)
60d-out DE126 genes(90.6%)
distinct DE5 genes(3.6%)
C
0
20
40
60
80
100
3d-out DE3272 genes(90.8%)
14d-out DE18 genes(0.5%)
60d-out DE94 genes(2.6%)
distinct DE221genes(6.1%)
DE
gen
es (%
)
DE in PI3605 genes
B
Fig. 3. Across-time-point analysis summary. (A) Venn diagram illustratingoverlap in differentially expressed (DE) genes in post-injury (PI) and control(Ctl) conditions, irrespective of time point. (B) Distribution of differentialexpression patterns for 3605 differentially expressed genes in the PIcondition. The majority of genes (90.8%) exhibited a 3d-out pattern.(C) Distribution of differential expression patterns for 139 differentiallyexpressed genes in the control condition. The majority of genes (90.6%)exhibited a 60d-out pattern. (D) Change in differential expression pattern for76 differentially expressed genes in both control and PI conditions. Themajority of genes (40.8%) changed from a 60d-out pattern in the controlcondition to a 3d-out pattern in the PI condition. Differential expressionpatterns are defined in the main text. Thresholding was performed using a>0.95 posterior probability of differential expression. Owing to roundingerrors, not all percentages add up to 100.0%.
Dis
ease
Mod
els
& M
echa
nism
s
314
extracellular immunosignals in the lamina propria (consistent withthe soluble isoform of the protein). The epithelium-basementmembrane anchoring filament transcript Lamc2 was upregulated4.0-fold in the PI condition compared to control; IHC revealed acorresponding increase in cellular laminin γ2 immunosignals in theluminal epithelium and lamina propria.
Differential expression at 14 days PIOf the 315 genes exclusively differentially expressed at 14 days PIcompared to control, 213 were upregulated in the PI condition and102 were downregulated in the PI condition (Fig. 6A). Enrichmentanalysis revealed overrepresented biological process termsassociated with collagen fibril organization and the regulation of the acute inflammatory response, as well as continuedoverrepresentation of the cell adhesion term, first identified at 3 daysPI (Fig. 6B). Overrepresented molecular function and cellularcomponent terms were also consistent with substantial ECM-relatedtranscriptional activity (e.g. fibrillar collagen, basement membrane,glycosaminoglycan binding), as well as modulation of the axonemeand myosin and dynein complexes.
We selected one upregulated and one downregulated gene forvalidation using IHC (Fig. 6C) and qRT-PCR (Fig. 6D). Thematricellular signaling transcript Wisp2 was upregulated 1.8-fold inthe PI condition compared to control; IHC revealed a correspondingincrease in cellular Wnt1-inducible-signaling pathway protein 2immunosignals in the epithelium and lamina propria. The phagocyterecruiter, complement and apoptosis inhibitor Clu wasdownregulated 2.0-fold in the PI condition compared to control;IHC revealed a corresponding decrease in cellular and extracellularclusterin immunosignals in the lamina propria
Differential expression at 60 days PIOf the 10 genes exclusively differentially expressed at 60 days PIcompared to control, two were upregulated in the PI condition and
eight were downregulated in the PI condition (Fig. 7A). Enrichmentanalysis revealed overrepresented biological process termsassociated with skeletal muscle regeneration, cell differentiation,chemotaxis and the bacteria-induced defense response, molecularfunction terms associated with chemokine activity and heparinbinding, and one cellular component term associated with themyosin complex, as first identified at 14 days PI.
We selected one upregulated and one downregulated gene forvalidation using IHC (Fig. 7C) and qRT-PCR (Fig. 7D). Theextracellular proteoglycan transcript Ogn was upregulated 2.3-foldin the PI condition compared to control; IHC revealed acorresponding increase in cellular osteoglycin immunosignals in theepithelium and extracellular immunosignals in the lamina propria.The growth factor binding protein and extracellular microfibrilassembly transcript Ltbp2 was downregulated 1.7-fold in the PIcondition compared to control; IHC revealed a correspondingdecrease in cellular latent transforming growth factor-β-bindingprotein 2 immunosignals within the lamina propria, as well asoccasional cellular signals in the epithelium.
DISCUSSIONDespite key differences in tissue environment, VF wound healinghas traditionally been viewed as being comparable to that of skinand other non-VF mucosae. This assumption might not hold,however, as although many classic wound healing elements areconserved across organ systems, certain parameters, such asisoform-specific signaling within the transforming growth factor-βcytokine superfamily (Chang et al., 2014; Schrementi et al., 2008),appear to direct tissue-specific differences in healing outcome.Given these observations and the specialized phenotype of VFmucosa, we used a previously validated rat injury model (Ling et al.,2010a; Tateya et al., 2005; Tateya et al., 2006b) and expressionmicroarray technology to better define VF wound healing at thetranscriptome level. We observed clear patterns in VF-specifictranscriptional activity as wound healing progressed, identifiedcorresponding changes in presumed biological function through GO-based enrichment analyses, and validated a subset of repair- andregeneration-related genes at the protein level. Our microarraydataset is a resource for the wider research community with thepotential to stimulate new hypotheses and avenues of investigation,improve biological and mechanistic insight, and accelerate theidentification of novel therapeutic targets.
By profiling the transcriptome, our dataset provides improvedinsight into key biological processes that occur during VF woundhealing. The analysis of overall (and relative) transcriptional activityat each time point gives an indication of the level of regionalbiological activity occurring as VF wound healing progresses. Thegreatest transcriptional activity (measured by both the total numberof differentially expressed genes and their mean fold change) wasevident at the earliest (3 days PI) time point, in both the within- andacross-time-point analyses. Furthermore, in the within-time-pointanalysis, the majority of genes that were differentially expressedover time in the control group (presumably due to maturation oraging) exhibited a switch from the 60d-out to the 3d-out differentialexpression pattern following VF injury, suggesting that their role inwound healing overrides their homeostatic function(s). Theseobservations of dominant transcriptional activity at 3 days PI areconsistent with previous descriptions of the acute VF injuryresponse involving rapid cellular mobilization and infiltration(Branski et al., 2005; Ling et al., 2010a; Ling et al., 2010b), deliveryof cytokines and other signaling molecules (Lim et al., 2006; Ohnoet al., 2009; Welham et al., 2008), establishment of a provisional
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
Ctl PI3d 60d
DE
in with
in- and
acro
ss-tim
e-p
oin
t analyses
3d 14d 60d14d
Myh3
Myod1
Casq2
Myh2
Bnc2
Serpine1
Npas2
Per2
Fez1
Pgf
Nrtn
RGD1562284
Slc2a6
Bhlhe41
Cd5l
Nalcn
Sphkap
Rassf4
log
fold
cha
nge
(Ctl
vs. P
I)2
4.9
-1.3
0
mean-centered
-2 20
log expression2
Fig. 4. Heat map summarizing expression data for 18 genes exhibitingdifferential expression across control and PI conditions at all time pointsin the within-time-point analysis. Gene names in red were also differentiallyexpressed (DE) over time in the PI condition [but not the control (Ctl)condition] in the across-time-point analysis. Genes are ranked by log2 foldchange (mean expression value in PI arrays versus mean expression value incontrol arrays, irrespective of time point) along the vertical axis. Color intensityrepresents the mean-centered log2 expression within each row. Thresholdingwas performed using a >0.95 posterior probability of differential expression.Expression values for genes represented by multiple probes reflect the probewith the maximum median for the mean across-array intensity.
Dis
ease
Mod
els
& M
echa
nism
s

315
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
log
fold
cha
nge
(3d-
PI v
s. 3
d-C
tl)2
-3.0
0B
Il1rl1
epith
eliu
mla
min
a pr
opria
3d-Ctl 3d-PI 3d-Ctl 3d-PILamc2H&E Tnc
3d-Ctl 3d-PI 3d-Ctl 3d-PIMmp13
3d-PI
microarray qRT-PCR
Mmp13 (3d-PI) -8-6-4-202468
microarray qRT-PCR
Tnc (3d-PI)log
fold
cha
nge
2
microarray qRT-PCR
Lamc2 (3d-PI)
microarray qRT-PCR
Il1rl1 (3d-PI)
Ctl PI
3d 60d14d
Ctl PI Ctl PITnc
Mmp13
Il1rl1
Lamc2
5.4
A
C
D
mean-centered
-2 20
log expression2
Fig. 5. See next page for legend. Dis
ease
Mod
els
& M
echa
nism
s

316
ECM (Branski et al., 2005; Tateya et al., 2006b) and initiation ofreepithelialization (Chang et al., 2014; Ling et al., 2011). The sharpreduction in differential expression seen at 60 days PI (i.e. 10differentially expressed genes, exhibiting log2 fold changes of −1.6to 1.3) is consistent with previous work showing that the majorityof wound healing events are completed by this time point in the ratVF (Tateya et al., 2005). Reports in other injury systems (e.g. skin)have suggested that ECM remodeling can continue beyond 1 yearafter injury (Gurtner et al., 2008); some of these remodeling events,such as fibrous protein crosslinking, prolination and glycosylation,occur at the protein and post-translational levels and are thereforebest examined with complementary assays.
As noted above, the conceptualization of VF wound healing intoinflammatory, proliferative and remodeling phases (the timing andduration of which were used to select our experimental time points)is based on the extrapolation of classic descriptions in other modelsystems, as well as limited transcript, protein and histological datafrom injured VF tissue (Lim et al., 2006; Ling et al., 2010a; Tateyaet al., 2006a; Tateya et al., 2005; Tateya et al., 2006b). It is apparentfrom our rat VF microarray data, however, that these phases are notdiscrete. Early transcription events (i.e. at 3 days PI) were primarilyassociated with cell division and proliferation, as well as celladhesion to the provisional ECM; acute inflammatory activitycontinued at 14 days PI and was accompanied by the acceleration ofECM-related transcription. This overlapping of biological eventsacross wound healing phases is consistent with currentunderstanding in the general literature (Gurtner et al., 2008; Martin,1997) and reinforces the dynamic complexity of tissue repair in theVF microenvironment. We observed clear resolution of thisinflammation- and proliferation-driven transcriptional activity by 60days PI, by which time there were just 10 differentially expressedgenes. Sampling at additional post-injury time points would provideimproved temporal characterization of the VF injury response,
particularly during the abovementioned transitional periods thatconnect overlapping wound healing phases.
A subset of transcripts exhibited sustained differential expressionthroughout the entire experimental time course. Notably, severalmembers of this gene set, such as Myh2, Myh3, Myod1 and Casq2,are associated with muscle differentiation, contractile function andrepair. This finding suggests that thyroarytenoid muscle repair is arelatively slow process compared to that of the VF mucosa, anobservation that is further supported by the enriched skeletal muscleregeneration ontology term seen at 60 days PI. The detection ofmuscle-specific gene transcription in our dataset is not surprisinggiven that: (1) our surgical injury procedure involved progressivelystripping the VF mucosa to achieve muscle exposure, and (2) priorproteomic studies have consistently reported contaminating muscleproteins in microdissected VF mucosa (Welham et al., 2011b;Welham et al., 2013).
In future work, it would be valuable to compare thesetranscriptomic data with those generated from VF mucosal woundsof varying severity (Imaizumi et al., 2014; Mau et al., 2014),including in humans (Hirano et al., 2009), as many clinical patientsundergo mucosal resections that are more conservative than thestripping procedure used in this study. Future studies might alsoconsider the correspondence between select aspects of VF woundhealing in vivo and the behavior of cultured VF fibroblasts andepithelial cells harvested from naïve, injured or scarred tissue.Classic expression microarray-based work with naïve human dermalfibroblasts, for example, has shown that the in vitro response ofthese cells to serum stimulation is consistent with certain aspects ofthe initial (i.e. hemostatic) injury response in vivo (Iyer et al., 1999);more recent research using this experimental approach suggests thatthere are distinct gene sets expressed by fibroblasts isolated from theregeneration-privileged oral mucosa, naïve skin and non-healingchronically wounded skin (Peake et al., 2014). The identification ofsuch transcriptomic signatures for biologically relevant conditionsin VF mucosa might lead to improved evaluation of emerging andfuture therapies: specifically, their capacity to drive the injured VFmucosa towards regeneration, rather than fibrosis.
MATERIALS AND METHODSMucosal injury procedure and tissue harvestAll animal experiments were performed in accordance with the PublicHealth Service Policy on Humane Care and Use of Laboratory Animals andthe Animal Welfare Act (7 U.S.C. et seq.); the animal use protocol wasapproved by the Institutional Animal Care and Use Committee of theUniversity of Wisconsin-Madison.
Four-month-old Fischer 344 male rats (Charles River, Wilmington, MA)were used for all experiments. Bilateral VF mucosal stripping injuries werecreated under endoscopic guidance as previously reported (Ling et al., 2010a;Tateya et al., 2005); experimentally naïve age-matched rats were used ascontrols. Animals were killed and tissue harvested at three time points tocapture global expression profiles characteristic of the inflammatory phase (3days PI), proliferative phase (14 days PI), and maturation and remodelingphase (60 days PI) of wound healing. A total of 20 PI rats (five arrays, n=4pooled animals per array) and 12 control rats (three arrays, n=4 pooled animalsper array) were reserved for microarray analysis and qRT-PCR validation ateach time point (total n=96). An additional three PI rats and one control ratwere reserved for histological and IHC analyses at each time point (totaln=12). Our pooling strategy and sample size were based on typical RNAyields and VF mucosal injury-induced transcription changes reported inprevious studies (Chang et al., 2014; Chang et al., 2010).
VF mucosa samples intended for RNA isolation were microdissected inan RNase-free environment, immersed in 10 μl RNAlater (Qiagen, Valencia,CA) at 4°C overnight, and then transferred to −80°C. Whole-mount laryngesintended for histology and IHC were embedded in optimal cutting
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
Fig. 5. Expression, enrichment and validation analyses of genesexhibiting differential expression at 3 days PI compared to age-matchedcontrols. (A) Heat map summarizing expression data for 1931 differentiallyexpressed genes exclusive to the 3-day PI (3d-PI) time point. Genes areranked by log2 fold change [mean expression value in 3d-PI arrays versusmean expression value in age-matched control (3d-Ctl) arrays] along thevertical axis. Color intensity represents mean-centered log2 expression withineach row. Thresholding was performed using a >0.95 posterior probability ofdifferential expression. Expression values for genes represented by multipleprobes reflect the probe with the maximum median for the mean across-arrayintensity. (B) Hierarchical map summarizing relationships betweenoverrepresented gene ontology terms at 3d-PI compared to 3d-Ctl.Overrepresented biological process terms are red; overrepresented cellularcomponent terms are green; interconnecting terms are gray. Arrows betweenrelated terms signify ‘is a’ (I), ‘part of’ (P) or ‘regulates’ (R). Overrepresentedterms were enriched for at least 10 differentially expressed genes andsurpassed a z-score threshold of 5. No overrepresented molecular functionterms were identified at this time point. (C) Images showing the morphologyof the VF mucosa and immunovalidation of proteins corresponding to fourrepresentative gene transcripts of interest at 3d-PI. Frozen VF mucosacoronal sections were stained with H&E, anti-tenascin C (Tnc), anti-matrixmetalloproteinase 13 (Mmp13), anti-interleukin 1 receptor-like 1 (Il1rl1) oranti-laminin γ2 (Lamc2) antibodies (red), and the nuclear dye DAPI (blue).Black dashed boxes in the H&E-stained image indicate the approximateanatomic orientation of all immunostained images. White dashed lines in theupper set of immunostained images indicate the boundary between theepithelium (right) and superficial lamina propria (left). Arrows indicatepositively labeled cells; arrowheads indicate positive extracellular signals.Scale bar: 300 μm (H&E-stained image); 30 μm (immunostained images).(D) qRT-PCR-based validation of the four representative gene transcriptshighlighted in A and analyzed at the protein level in C (mean±s.e.m.).
Dis
ease
Mod
els
& M
echa
nism
s

temperature (OCT) compound (Tissue Tek, Sakura, Tokyo, Japan), frozenwith acetone and dry ice, and stored at −80°C until sectioning. Frozensections (8 μm thick) were prepared in the coronal plane using a Leica CM-3050S cryostat (Leica, Wetzlar, Germany).
RNA isolationTotal RNA was isolated using the RNeasy Micro kit (Qiagen) according tothe manufacturer’s instructions. RNA yield and integrity were evaluatedusing a NanoDrop ND-1000 spectrophotometer (NanoDrop, Wilmington,
317
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
BCtl PI
3d 60d14d
Ctl PI Ctl PI
Clu
Wisp2
log
fold
cha
nge
(14d
-PI v
s. 1
4d-C
tl)2
3.5
-3.0
0A
CWisp2
epith
eliu
mla
min
a pr
opria
14d-Ctl 14d-PI 14d-Ctl 14d-PICluH&E
14d-PI
-4-3-2-101234
microarray qRT-PCR
Wisp2 (14d-PI)log
fold
cha
nge
2
microarray qRT-PCR
Clu (14d-PI)
D
mean-centered
-2 20
log expression2
Fig. 6. See next page for legend.
Dis
ease
Mod
els
& M
echa
nism
s

318
DE), and samples meeting the following three criteria were retained: aconcentration above 40 ng/ml, an OD260:280 of 1.8-2.0 and an OD260:230
above 1.8. Samples were further evaluated using the Agilent 2100Bioanalyzer and RNA 6000 Pico kit (Agilent, Santa Clara, CA) accordingto the manufacturer’s instructions. Samples with electropherogramsexhibiting sharp 18S and 28S rRNA peaks and no evidence of degradationwere retained.
MicroarraysTotal RNA yield in the 60-day PI group was sufficient to run just four of thefive arrays intended for this condition. All other arrays and conditions wererun as planned. Biotinylated antisense cRNA was prepared by single round invitro amplification of 1.2 μg input RNA using the MessageAmp II-BiotinEnhanced aRNA kit (Ambion, Austin, TX) according to the manufacturer’sinstructions (the in vitro transcription reaction was performed at 37°C for 14hours). Polyadenylated RNA controls (Affymetrix, Santa Clara, CA) werespiked into each reaction. Fragmented cRNA sample quality was confirmedby using 2% agarose gel electrophoresis, an Agilent 2100 Bioanalyzer analysis(Pico kit) and hybridization to Affymetrix GeneChip Test3 arrays. Sampleswere hybridized to Affymetrix GeneChip Rat Genome 230 2.0 arrays at 45°Cfor 16 hours. Post-processing was performing using the GeneChip FluidicsStation 450, arrays were scanned using the GC3000 G7 scanner andfluorescence intensity data were background-corrected and extracted usingExpression Console software (Affymetrix). All hybridization, post-processingand scanning procedures were performed according to Affymetrix protocols;all control parameters for Test3 and rat genome arrays were withinmanufacturer guidelines. Microarray data have been deposited with the GeneExpression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accessionnumber GSE62204.
qRT-PCRReverse transcription was performed using the QuantiTect RT kit (Qiagen)with 300 ng input total RNA per 20 μl reaction, according to themanufacturer’s instructions. Negative controls were prepared without RNAtemplate and without reverse transcriptase. qRT-PCRs were run using thefollowing rat-specific commercial primers (QuantiTect, Qiagen):QT01081297 (Clu), QT00178955 (Il1rl1), QT00379260 (Lamc2),
QT00192220 (Ltbp2), QT00385686 (Mmp13), QT00435015 (Ogn),QT00195958 (Sdha), QT02340814 (Tnc), QT00189840 (Wisp2). Reactionswere performed on a 7500 Fast Real-Time PCR system (AppliedBiosystems, Foster, CA) using the QuantiTect SYBR Green PCR kit(Qiagen). Each 25 μl total volume reaction contained 12.5 μl 2× QuantiTectMaster Mix, 2.5 μl 10× QuantiTect Primer Assay and 10 μl cDNA template(diluted 1:10 with nuclease-free H2O). Amplifications were performed inMicroAmp Fast Optical 96-well reaction plates with optical adhesive filmcovers (Applied Biosystems) according to cycling conditions suggested forthe Applied Biosystems 7500 instrument in the QuantiTect SYBR Greenhandbook (initial activation at 95°C for 15 minutes; 40 cycles of 94°C for15 seconds, 55°C for 30 seconds, 72°C for 30 seconds).
PCR runs were performed in duplicate using cDNA synthesized from thesame batch and starting amount of total RNA. Negative controls containingno cDNA template were included for each gene within each PCR run. Toavoid the influence of variation in amplification conditions across runs, allreactions for a given gene of interest were performed in the same 96-wellplate. Amplification specificity for each gene was confirmed by a singledistinct melting curve.
qRT-PCR data were analyzed using the 2–ΔΔCT method (Livak andSchmittgen, 2001). Mean cycle threshold (CT) values from duplicate runswere used as input data (duplicate CT values consistently varied by <0.1).Sdha, previously validated as stably expressed in our rat VF injury model(Chang et al., 2010), was used as the reference gene. Data were presentedas log2-transformed mean±s.e.m. fold change
Histology and IHCRoutine hematoxylin and eosin (H&E) staining was performed to evaluateoverall tissue morphology. Sections intended for IHC were fixed in 4%paraformaldehyde, washed with phosphate-buffered saline (PBS), andincubated with Block-Ace (AbD Serotech, Raleigh, NC) and 5% donkeyserum (Jackson ImmunoResearch, West Grove, PA) for 30 minutes to blocknon-specific binding. Next, sections were sequentially incubated with aprimary antibody for 90 minutes followed by a relevant secondary antibodyfor 60 minutes, with thorough washing between each incubation step.Finally, slides were covered with anti-fade mounting medium containingDAPI (Vectashield, Vector Labs, Burlingame, CA) and coverslips wereadded. Control sections stained with an isotype control or without theprimary or secondary antibody showed no immunoreactivity.
The primary antibodies used were: rabbit anti-osteoglycin (Ogn), cloneM-70 (1:50; sc-67170, Santa Cruz Biotechnology, Santa Cruz, CA); goatanti-latent transforming growth factor-β-binding protein 2 (Ltgp2), clone E-18 (1:100; sc-18343, Santa Cruz Biotechnology); rabbit anti-Wnt1-inducible-signaling pathway protein 2 (Wisp2), clone H-74 (1:50; sc-25442,Santa Cruz Biotechnology); rabbit anti-clusterin (Clu), clone H-330 (1:150;sc-8354, Santa Cruz Biotechnology); goat anti-tenascin C (Tnc), clone F-17(1:50, sc-9872; Santa Cruz Biotechnology); goat anti-laminin γ2 (Lamc2),clone G-16 (1:50; sc-31092, Santa Cruz Biotechnology); goat anti-interleukin 1 receptor-like 1 (Il1rl1), clone C-20 (1:50; sc-18687, Santa CruzBiotechnology); and goat anti-matrix metalloproteinase 13 (Mmp13), (1:50;AB8120, Millipore, Billerica, MA). The secondary antibodies used were:Texas-Red-conjugated donkey anti-goat- and donkey anti-rabbit-IgG (1:200;Jackson ImmunoResearch).
Brightfield and fluorescent microscopy images were captured using amicroscope (E-600, Nikon, Melville, NY) connected to a digital microscopycamera (Olympus DP-71, Center Valley, PA). All images were captured withconsistent exposure settings. Representative images from each experimentalgroup were selected for presentation.
Statistical analysisMicroarray data were analyzed within the R statistical computingenvironment (R Development Core Team, 2007). Affymetrix probe-leveldata were preprocessed using Robust Multi-Array Analysis (RMA) (Irizarryet al., 2003), based on evidence of improved precision over defaultAffymetrix algorithms (Wu and Irizarry, 2004). Probes without acorresponding gene symbol were purged from all gene-level analyses. Incases where multiple probes corresponded to a single gene symbol, weselected the probe with the median mean across-array intensity; in the case
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
Fig. 6. Expression, enrichment and validation analyses of genesexhibiting differential expression at 14 days PI compared to age-matched controls. (A) Heat map summarizing expression data for 315differentially expressed genes exclusive to the 14-day PI (14d-PI) time point.Genes are ranked by log2 fold change [mean expression value in 14d-PIarrays versus mean expression value in age-matched control (14d-Ctl)arrays] along the vertical axis. Color intensity represents mean-centered log2
expression within each row. Thresholding was performed using a >0.95posterior probability of differential expression. Expression values for genesrepresented by multiple probes reflect the probe with the maximum medianfor the mean across-array intensity. (B) Hierarchical map summarizingrelationships between overrepresented gene ontology terms at 14d-PIcompared to 14d-Ctl. Overrepresented biological process terms are red;overrepresented cellular component terms are green; overrepresentedmolecular function terms are blue; interconnecting terms are gray. Arrowsbetween related terms signify ‘is a’ (I), ‘part of’ (P) or ‘regulates’ (R).Overrepresented terms were enriched for at least 10 differentially expressedgenes and surpassed a z-score threshold of 5. (C) Images showingmorphology of the VF mucosa and immunovalidation of proteinscorresponding to two representative gene transcripts of interest at 14d-PI.Frozen VF mucosa coronal sections were stained with H&E, anti-Wnt1-inducible-signaling pathway protein 2 (Wisp2) or anti-clusterin (Clu)antibodies (red), and the nuclear dye DAPI (blue). Black dashed boxes in theH&E-stained image indicate the approximate anatomic orientation of allimmunostained images. White dashed lines in the upper set ofimmunostained images indicate the boundary between the epithelium (right)and superficial lamina propria (left). Arrows indicate positively labeled cells;arrowheads indicate positive extracellular signals. Scale bar: 300 μm (H&E-stained image); 30 μm (immunostained images). (D) qRT-PCR-basedvalidation of the two representative gene transcripts highlighted in A andanalyzed at the protein level in C (mean±s.e.m.).
Dis
ease
Mod
els
& M
echa
nism
s

of an even number of matched probes, we selected the larger of the twomedian probe intensities. The resulting normalized data were clustered tocheck for consistency prior to formal analysis.
Expression analysis was performed using an empirical Bayes approach asimplemented in the R package EBarrays (Kendziorski et al., 2003). Alognormal-normal moderated variance (LNNMV) model was fitted to the
data; parameter estimates were obtained through 20 iterations of anexpectation-maximization (EM) algorithm where convergence was achievedafter 10 iterations. We pursued two analysis strategies in parallel: a within-time-point analysis was used to compare PI and control data at individualtime points, and an across-time-point analysis was used to evaluate changein expression over time in the PI and control groups, respectively. For all
319
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
Ctl PI
3d 60d14d
Ctl PI Ctl PI
log
fold
cha
nge
2 (6
0d-P
I vs.
60d
-Ctl)
Ogn
Ltbp2
1.3
0-1
.6A
Ogn
epith
eliu
mla
min
a pr
opria
60d-Ctl 60d-PI 60d-Ctl 60d-PILtbp2H&E
60d-PI
C
-4-3-2-101234
microarray qRT-PCR
Ogn (60d-PI)
microarray qRT-PCR
Ltbp2 (60d-PI)
log
fold
cha
nge
2
D
B
mean-centered
-2 20
log expression2
Fig. 7. Expression, enrichment and validation analyses of genes exhibiting differential expression at 60 days post-injury compared to age-matchedcontrols. (A) Heat map summarizing expression data for 10 differentially expressed genes exclusive to the 60-day PI (60d-PI) time point. Genes are ranked bylog2 fold change [mean expression value in 60d-PI arrays versus mean expression value in age-matched control (60d-Ctl) arrays] along the vertical axis. Colorintensity represents mean-centered log2 expression within each row. Thresholding was performed using a >0.95 posterior probability of differential expression.Expression values for genes represented by multiple probes reflect the probe with the maximum median for the mean across-array intensity. (B) Hierarchicalmap summarizing relationships between overrepresented gene ontology terms at 60d-PI compared to 60d-Ctl. Overrepresented biological process terms arered; overrepresented cellular component terms are green; overrepresented molecular function terms are blue; interconnecting terms are gray. Arrows betweenrelated terms signify ‘is a’ (I), ‘part of’ (P) or ‘regulates’ (R). Overrepresented terms were enriched for at least 10 differentially expressed genes and surpassed az-score threshold of 5. (C) Images showing morphology of the VF mucosa and immunovalidation of proteins corresponding to two representative genetranscripts of interest at 60d-PI. Frozen VF mucosa coronal sections were stained with H&E, anti-osteoglycin (Ogn) or anti-latent transforming growth factor-βbinding protein 2 (Ltbp2) antibodies (red), and the nuclear dye DAPI (blue). Black dashed boxes in the H&E-stained image indicate the approximate anatomicorientation of all immunostained images. White dashed lines in the upper set of immunostained images indicate the boundary between the epithelium (right)and superficial lamina propria (left). Arrows indicate positively labeled cells; arrowheads indicate positive extracellular signals. Scale bar: 300 μm (H&E-stainedimage); 30 μm (immunostained images). (D) qRT-PCR-based validation of the two representative gene transcripts highlighted in A and analyzed at the proteinlevel in C (mean±s.e.m.).
Dis
ease
Mod
els
& M
echa
nism
s

320
comparisons, thresholding was performed using a >0.95 posteriorprobability of differential expression, providing false discovery rate controlat the 5% level. Heat maps summarizing expression data for differentiallyexpressed gene lists of interest were generated using Matrix2png (Pavlidisand Noble, 2003).
Tests of enrichment through overrepresentation were conducted using theR package allez (Newton et al., 2007), the GO dataset, and genes identifiedas exclusively differentially expressed at 3, 14 and 60 days PI in the within-time-point analysis. Overrepresented ontology terms required at least 10distinct differentially expressed genes and a z-score >5. Ontology maps weregenerated using OBO-Edit (Day-Richter et al., 2007).
Initial diagnostic testing was performed using quantile-quantile (QQ) plotsof log intensity data versus a standard normal distribution. We further usedQQ plots and comparisons of theoretical and empirical densities to evaluatethe assumption of a scaled inverse chi-square prior on the gene-specificvariances used in the LNNMV model. The diagnostics did not showevidence of an unacceptable model fit.
AcknowledgementsWe gratefully acknowledge Toshi Kinoshita (Department of Pathology, University ofWisconsin School of Medicine and Public Heath, Madison, WI) for assistance withhistology.
Competing interestsThe authors declare no competing or financial interests.
Author contributionsN.V.W. conceived the study and obtained funding. N.V.W., S.L.T. and C.K.designed the experiments. N.V.W. and M.Y. conducted the in vivo experiments.C.L. processed all RNA and tissue samples, and performedimmunohistochemistry. J.A.D. performed all statistical analyses under the directionof C.K. N.V.W. wrote the manuscript. All authors reviewed and approved the finalversion.
FundingThis work was funded by grants from the National Institute on Deafness and otherCommunication Disorders [grant numbers R01 DC004428, R01 DC010777]; andthe Clinical and Translational Science Award (CTSA) program of the NationalCenter for Research Resources [grant number UL1 RR025011].
ReferencesBenninger, M. S., Alessi, D., Archer, S., Bastian, R., Ford, C., Koufman, J.,
Sataloff, R. T., Spiegel, J. R. and Woo, P. (1996). Vocal fold scarring: currentconcepts and management. Otolaryngol. Head Neck Surg. 115, 474-482.
Branski, R. C., Rosen, C. A., Verdolini, K. and Hebda, P. A. (2005). Acute vocal foldwound healing in a rabbit model. Ann. Otol. Rhinol. Laryngol. 114, 19-24.
Chang, Z., Ling, C., Yamashita, M. and Welham, N. V. (2010). Microarray-drivenvalidation of reference genes for quantitative real-time polymerase chain reaction ina rat vocal fold model of mucosal injury. Anal. Biochem. 406, 214-221.
Chang, Z., Kishimoto, Y., Hasan, A. and Welham, N. V. (2014). TGF-β3 modulatesthe inflammatory environment and reduces scar formation following vocal foldmucosal injury in rats. Dis. Model. Mech. 7, 83-91.
Choi, S. H., Zhang, Y., Jiang, J. J., Bless, D. M. and Welham, N. V. (2012).Nonlinear dynamic-based analysis of severe dysphonia in patients with vocal foldscar and sulcus vocalis. J. Voice 26, 566-576.
Cohen, S. M., Dupont, W. D. and Courey, M. S. (2006). Quality-of-life impact of non-neoplastic voice disorders: a meta-analysis. Ann. Otol. Rhinol. Laryngol. 115, 128-134.
Cohen, S. M., Kim, J., Roy, N., Asche, C. and Courey, M. (2012). Direct health carecosts of laryngeal diseases and disorders. Laryngoscope 122, 1582-1588.
Colwell, A. S., Longaker, M. T. and Peter Lorenz, H. (2008). Identification ofdifferentially regulated genes in fetal wounds during regenerative repair. WoundRepair Regen. 16, 450-459.
Day-Richter, J., Harris, M. A., Haendel, M., Lewis, S.; Gene Ontology OBO-EditWorking Group (2007). OBO-Edit – an ontology editor for biologists. Bioinformatics23, 2198-2200.
Gene Ontology Consortium (2013). Gene Ontology annotations and resources.Nucleic Acids Res. 41, D530-D535.
Gray, S. D., Titze, I. R., Chan, R. and Hammond, T. H. (1999). Vocal foldproteoglycans and their influence on biomechanics. Laryngoscope 109, 845-854.
Gray, S. D., Titze, I. R., Alipour, F. and Hammond, T. H. (2000). Biomechanical andhistologic observations of vocal fold fibrous proteins. Ann. Otol. Rhinol. Laryngol.109, 77-85.
Gunter, H. E. (2004). Modeling mechanical stresses as a factor in the etiology ofbenign vocal fold lesions. J. Biomech. 37, 1119-1124.
Gurtner, G. C., Werner, S., Barrandon, Y. and Longaker, M. T. (2008). Wound repairand regeneration. Nature 453, 314-321.
Hirano, S., Minamiguchi, S., Yamashita, M., Ohno, T., Kanemaru, S. and Kitamura,M. (2009). Histologic characterization of human scarred vocal folds. J. Voice 23,399-407.
Imaizumi, M., Thibeault, S. L. and Leydon, C. (2014). Classification for animal vocalfold surgery: resection margins impact histological outcomes of vocal fold injury.Laryngoscope 124, E437-E444.
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf,U. and Speed, T. P. (2003). Exploration, normalization, and summaries of highdensity oligonucleotide array probe level data. Biostatistics 4, 249-264.
Iyer, V. R., Eisen, M. B., Ross, D. T., Schuler, G., Moore, T., Lee, J. C., Trent, J. M.,Staudt, L. M., Hudson, J., Jr, Boguski, M. S. et al. (1999). The transcriptionalprogram in the response of human fibroblasts to serum. Science 283, 83-87.
Kendziorski, C. M., Newton, M. A., Lan, H. and Gould, M. N. (2003). On parametricempirical Bayes methods for comparing multiple groups using replicated geneexpression profiles. Stat. Med. 22, 3899-3914.
Kisseleva, T. and Brenner, D. A. (2008). Mechanisms of fibrogenesis. Exp. Biol. Med.(Maywood) 233, 109-122.
Lim, X., Tateya, I., Tateya, T., Muñoz-Del-Río, A. and Bless, D. M. (2006). Immediateinflammatory response and scar formation in wounded vocal folds. Ann. Otol. Rhinol.Laryngol. 115, 921-929.
Ling, C., Yamashita, M., Waselchuk, E. A., Raasch, J. L., Bless, D. M. andWelham, N. V. (2010a). Alteration in cellular morphology, density and distribution inrat vocal fold mucosa following injury. Wound Repair Regen. 18, 89-97.
Ling, C., Yamashita, M., Zhang, J., Bless, D. M. and Welham, N. V. (2010b).Reactive response of fibrocytes to vocal fold mucosal injury in rat. Wound RepairRegen. 18, 514-523.
Ling, C., Raasch, J. L. and Welham, N. V. (2011). E-cadherin and transglutaminase-1epithelial barrier restoration precedes type IV collagen basement membranereconstruction following vocal fold mucosal injury. Cells Tissues Organs 193, 158-169.
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression datausing real-time quantitative PCR and the 2–ΔΔCT Method. Methods 25, 402-408.
Martin, P. (1997). Wound healing – aiming for perfect skin regeneration. Science 276,75-81.
Mau, T., Du, M. and Xu, C. C. (2014). A rabbit vocal fold laser scarring model fortesting lamina propria tissue-engineering therapies. Laryngoscope 124, 2321-2326.
Mori, L., Bellini, A., Stacey, M. A., Schmidt, M. and Mattoli, S. (2005). Fibrocytescontribute to the myofibroblast population in wounded skin and originate from thebone marrow. Exp. Cell Res. 304, 81-90.
Newton, M. A., Quintana, F. A., den Boon, J. A., Sengupta, S. and Ahlquist, P.(2007). Random-set methods identify distinct aspects of the enrichment signal ingene-set analysis. Ann. Appl. Stat. 1, 85-106.
Occleston, N. L., Metcalfe, A. D., Boanas, A., Burgoyne, N. J., Nield, K., O’Kane,S. and Ferguson, M. W. J. (2010). Therapeutic improvement of scarring:mechanisms of scarless and scar-forming healing and approaches to the discoveryof new treatments. Derm. Res. Pract. 2010, 405262.
Ohno, T., Hirano, S. and Rousseau, B. (2009). Gene expression of transforminggrowth factor-β1 and hepatocyte growth factor during wound healing of injured ratvocal fold. Laryngoscope 119, 806-810.
Pavlidis, P. and Noble, W. S. (2003). Matrix2png: a utility for visualizing matrix data.Bioinformatics 19, 295-296.
Peake, M. A., Caley, M., Giles, P. J., Wall, I., Enoch, S., Davies, L. C., Kipling, D.,Thomas, D. W. and Stephens, P. (2014). Identification of a transcriptional signaturefor the wound healing continuum. Wound Repair Regen. 22, 399-405.
Periwal, V., Gaillard, J. R., Needleman, L. and Doria, C. (2014). Mathematical modelof liver regeneration in human live donors. J. Cell. Physiol. 229, 599-606.
R Development Core Team (2007). R: A Language and Environment for StatisticalComputing. Vienna: R Foundation for Statistical Computing.
Schrementi, M. E., Ferreira, A. M., Zender, C. and DiPietro, L. A. (2008). Site-specific production of TGF-β in oral mucosal and cutaneous wounds. Wound RepairRegen. 16, 80-86.
Schultz, G. S., Davidson, J. M., Kirsner, R. S., Bornstein, P. and Herman, I. M.(2011). Dynamic reciprocity in the wound microenvironment. Wound Repair Regen.19, 134-148.
Song, G., Nguyen, D. T., Pietramaggiori, G., Scherer, S., Chen, B., Zhan, Q.,Ogawa, R., Yannas, I. V., Wagers, A. J., Orgill, D. P. et al. (2010). Use of theparabiotic model in studies of cutaneous wound healing to define the participation ofcirculating cells. Wound Repair Regen. 18, 426-432.
Sullivan, K. M., Lorenz, H. P., Meuli, M., Lin, R. Y. and Adzick, N. S. (1995). A modelof scarless human fetal wound repair is deficient in transforming growth factor beta.J. Pediatr. Surg. 30, 198-202, discussion 202-203.
Szpaderska, A. M., Zuckerman, J. D. and DiPietro, L. A. (2003). Differential injuryresponses in oral mucosal and cutaneous wounds. J. Dent. Res. 82, 621-626.
Tateya, T., Tateya, I., Sohn, J. H. and Bless, D. M. (2005). Histologic characterizationof rat vocal fold scarring. Ann. Otol. Rhinol. Laryngol. 114, 183-191.
Tateya, I., Tateya, T., Lim, X., Sohn, J. H. and Bless, D. M. (2006a). Cell productionin injured vocal folds: a rat study. Ann. Otol. Rhinol. Laryngol. 115, 135-143.
Tateya, T., Tateya, I., Sohn, J. H. and Bless, D. M. (2006b). Histological study ofacute vocal fold injury in a rat model. Ann. Otol. Rhinol. Laryngol. 115, 285-292.
Thibeault, S. L., Hirschi, S. D. and Gray, S. D. (2003). DNA microarray geneexpression analysis of a vocal fold polyp and granuloma. J. Speech Lang. Hear.Res. 46, 491-502.
Titze, I. R. (1988). The physics of small-amplitude oscillation of the vocal folds. J.Acoust. Soc. Am. 83, 1536-1552.
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
Dis
ease
Mod
els
& M
echa
nism
s

Welham, N. V., Dailey, S. H., Ford, C. N. and Bless, D. M. (2007). Voice handicapevaluation of patients with pathologic sulcus vocalis. Ann. Otol. Rhinol. Laryngol.116, 411-417.
Welham, N. V., Lim, X., Tateya, I. and Bless, D. M. (2008). Inflammatory factor profilesone hour following vocal fold injury. Ann. Otol. Rhinol. Laryngol. 117, 145-152.
Welham, N. V., Choi, S. H., Dailey, S. H., Ford, C. N., Jiang, J. J. and Bless, D. M.(2011a). Prospective multi-arm evaluation of surgical treatments for vocal fold scarand pathologic sulcus vocalis. Laryngoscope 121, 1252-1260.
Welham, N. V., Yamashita, M., Choi, S. H. and Ling, C. (2011b). Cross-samplevalidation provides enhanced proteome coverage in rat vocal fold mucosa. PLoSONE 6, e17754.
Welham, N. V., Chang, Z., Smith, L. M. and Frey, B. L. (2013). Proteomic analysis ofa decellularized human vocal fold mucosa scaffold using 2D electrophoresis andhigh-resolution mass spectrometry. Biomaterials 34, 669-676.
Wu, Z. and Irizarry, R. A. (2004). Preprocessing of oligonucleotide array data. Nat.Biotechnol. 22, 656-658, author reply 658.
321
RESOURCE ARTICLE Disease Models & Mechanisms (2015) doi:10.1242/dmm. 018366
Dis
ease
Mod
els
& M
echa
nism
s


















