MECHANISTIC ANALYSIS OF THE TRYPTOPHAN BIOSYNTHETIC ENZYME ...
149
The Pennsylvania State University The Graduate School Eberly College of Science MECHANISTIC ANALYSIS OF THE TRYPTOPHAN BIOSYNTHETIC ENZYME INDOLE-3-GLYCEROL PHOSPHATE SYNTHASE A Dissertation in Chemistry by Margot J. Zaccardi ©2013 Margot J. Zaccardi Submitted in Partial Fullfillment of the Requirements for the Degree of Doctor of Philosophy December 2013
Transcript of MECHANISTIC ANALYSIS OF THE TRYPTOPHAN BIOSYNTHETIC ENZYME ...

The Pennsylvania State University
The Graduate School
Eberly College of Science
MECHANISTIC ANALYSIS OF THE TRYPTOPHAN BIOSYNTHETIC
ENZYME INDOLE-3-GLYCEROL PHOSPHATE SYNTHASE
A Dissertation in
Chemistry
by
Margot J. Zaccardi
©2013 Margot J. Zaccardi
Submitted in Partial Fullfillment
of the Requirements
for the Degree of
Doctor of Philosophy
December 2013

The dissertation of Margot J. Zaccardi was reviewed and approved* by the following:
David D. Boehr
Assistant Professor of Chemistry
Dissertation Advisor
Chair of Committee
Scott S. Showalter
Assistant Professor of Chemistry
Phillip C. Bevilacqua
Professor of Chemistry
Craig Cameron
Professor of Biochemistry and Molecular Biology
Eberly Chair in Biochemistry and Molecular Biology
Barbara J. Garrison
Shapiro Professor of Chemistry
Head of the Chemistry Department
*Signatures are on file in the Graduate School

iii
ABSTRACT
The design and production of enzymes that are capable of performing reactions in
an industrial setting is of profound importance for the advancement of technologies
including pharmaceuticals, biotechnology, agriculture, and materials. While some natural
enzymes have industrial relevance, the design of novel enzymes whose catalytic reactions
are not found in natural systems would greatly enhance the scope and efficiency of
industrial processes. The difficulty of engineering such enzymes is that it requires in
depth knowledge of all factors of catalysis and how they affect the reaction, including an
understanding of the natural enzyme scaffold to be used in new design. Indole-3-glycerol
phosphate synthase (IGPS), a tryptophan biosynthetic enzyme that catalyzes the ring
closure of 1-(o-carboxyphenylamino)-1-deoxyribulose 5-phosphate (CdRP) to form
indole-3-glycerol phosphate (IGP), is widely used as a scaffold in enzyme engineering
studies. However, the rate enhancements of these engineered enzymes are much lower
than those of natural enzymes. This work has analyzed the IGPS enzyme from the
thermophile Sulfolobus sulfataricus (ssIGPS) and gained a more complete understanding
of the kinetic and chemical mechanism for this enzyme that can be leveraged towards
enzyme engineering applications.
Steady-state kinetic assays were used to analyze wild type (WT) ssIGPS and
variants. The results showed a temperature dependent change in the rate-determining step
of the reaction; the ring closure is rate-determining at high temperatures and product
release is rate-determining at low temperatures. These studies also showed that
thermophilic ssIGPS and mesophilic IGPS from Escherichia coli (ecIGPS) have different
rate-determining steps at their biologically relevant temperatures.

iv
In addition to examining the temperature dependence of ssIGPS, amino acid
substituted variants of ssIGPS were used to probe the chemical mechanism of the
enzyme. While a proposed mechanism had been previously published, this analysis
provides additional and important details that were formerly unknown. The general acid
and base in the dehydration step of the reaction were reassigned to Lys53 and Glu51, as
opposed to the previously assigned Lys110 and Glu159 (or Glu210). This assignment
allowed for a more complete view of catalysis. First, Lys110 initiates the reaction by
donating a proton to the C2’ carbonyl of the substrate, which allows the ring closure to
occur and form the fleetingly stable I1 intermediate, which then undergoes
decarboxlation to form the I2 intermediate. The I2 undergoes a reorientation in the active
site to properly align it for dehydration assisted by the general acid and base, Lys53 and
Glu51. This step renders the product and leaves Lys53 neutral allowing for efficient
product release.
Finally, the role of the active site loop residues in ssIGPS catalysis was examined.
Lys53, the general acid in the dehydration step, is located on the dynamic β1α1 loop, and
Phe89, important for substrate binding, is on the β2α2 loop. Arg54 and Asn90, also on
these loops, were found to be coevolving by statistical coupling analysis (SCA), and
molecular dynamics (MD) simulations predicted their motions were also correlated. To
further assess the role of these residues, kinetic analysis of amino acid substituted
variants was performed. The results show that the interaction between Arg54 and Asn90
is important for the dehydration step of the reaction, mainly in the correct function of the
general acid and base, Lys53 and Glu51. The results also suggest that these residues play
a role in conformational exchange, as the effect on catalysis is temperature dependent,

v
suggesting that thermal energy at higher temperatures can help to overcome their
detrimental effect on catalysis. Together, these results provide a more complete
understanding of ssIGPS catalysis. The results can be leveraged towards the design of
novel enzymes as well as in the development of new antimicrobials.

vi
TABLE OF CONTENTS
LIST OF FIGURES……………………………………………………………………....ix
LIST OF TABLES……………………………………………………………………….xii
LIST OF ABBREVIATIONS…………………………………………………………...xiv
ACKNOWLEDGEMENTS……………………………………………………………..xvi
Chapter 1 Introduction to Indole-3-glycerol Phosphate Synthase………………..………1
1.1 Progress Towards Engineering Enzymes with New Functions………………1 1.2 The Conserved (β/α)8-Barrel Protein Fold as an Enzyme Engineering
Scaffold……….…………………………………………………………..5 1.3 (β/α)8-Barrel Enzymes in Tryptophan Biosynthesis………………………….6 1.4 The Tryptophan Biosynthetic Enzyme Indole-3-glycerol Phosphate
Synthase…………………………………………………………………..8 1.5 Previous Knowledge on the IGPS Mechanism....……………………………11 1.6 Conclusions...………………………………………………………...............20 1.7 References ……………………………………………………………………22 Chapter 2 The Temperature Dependent Kinetic Mechanism of Thermophilic and
Mesophilic IGPS Enzymes……………………………………………………....30 2.1 Abstract............................................................................................................30 2.2 Introduction…………………………………………………………………..31 2.3 Experimental Methods.....................................................................................34 2.3.1 Cloning of ssIGPS and ecIGPS…………………………………….34 2.3.2 Overexpression and Purification of ssIGPS, ecIGPS,
and tmIGPS………………………………………………………35 2.3.3 Steady-state Kinetic Assays for IGPS……………...……………....37
2.3.4 Solvent Viscosity Effects, Solvent Deuterium Kinetic Isotope Effects, and pH Effects………………………………………..…………39 2.3.5 Synthesis of CdRP…………………........………………………....41 2.3.6 Circular Dichroism...........................................................................42
2.4 Results……………………………………………………………………….43 2.4.1 Steady-state Kinetics of ssIGPS…………………………………...43 2.4.2 Solvent Viscosity Effects, Solvent Deuterium Kinetic Isotope
Effects, and pH Effects..…………………………………………45 2.5 Discussion……………………………………………………………………54 2.5.1 Temperature Dependent Kinetic Mechanism of ssIGPS…………..54 2.5.2 Differences in the Rate-Determining Step of Thermophilic ssIGPS
and Mesophilic ecIGPS at their Adaptive Temperatures………...55 2.6 Conclusions…………………………………………………………………..56

vii
2.7 References……………………………………………………………………56 Chapter 3 Functional Identification of the General Acid and Base in the Dehydration Step
of Indole-3-glycerol Phosphate Synthase Catalysis……………………………...59 3.1 Abstract............................................................................................................59 3.2 Introduction…………………………………………………………………..60 3.3 Experimental Methods….……………………………………………………63
3.3.1 Overexpression, Purification, and Kinetic Analysis of WT and Amino Acid Substituted IGPS.…………………………………..63
3.3.2 Overexpression and Purification of ε-13C-Lys Labeled ssIGPS…...64 3.3.3 Preparation of rCdRP………………………………………………65 3.3.4 13C-TROSY-HSQC Experiments on ssIGPS………………………66
3.4 Results………………………………………………………………………..67 3.4.1 Determination of the Rate-Determining Step of ssIGPS
Catalysis........................................................................................67 3.4.2 Analysis of Lys53 Indicates its Role as a General Acid…………...67 3.4.3 13C-Lys NMR to Determine pKas for Lys Residues in IGPS……...73 3.4.4 Analysis of Glu51 Identifies its Role as the General Base….……..81
3.5 Discussion……………………………………………………………………82 3.6 Conclusions…………………………………………………………………..89 3.7 References……………………………………………………………………89 Chapter 4 The Role of Active Site Loops in Catalysis by IGPS…..…………………….92
4.1 Abstract............................................................................................................92 4.2 Introduction…………………………………………………………………..92 4.3 Experimental Methods……………………………………………………….95 4.4 Results………………………………………………………………………..96 4.4.1 Investigation of Phe89 on the β2α2 Loop Identifies its Role in IGPS
Catalysis ………………………………...……………………….96 4.4.2 Interaction Between β1α1 and β2α2 Loops through Arg54 and
Asn90 is Important for Catalysis………………………………...98 4.4.3Analysis of Arg54Lys and Asn90Gln variants of ssIGPS………...103 4.4.4 Examination of the Interaction Between Coevolving Residues on the β2α2 Loop…………………………………………………..104 4.4.5 Structure and Stability of Loop Mutants………………………….105 4.5 Discussion…………………………………………………………………..107 4.6 Conclusions…………………………………………………………………110 4.7 References………………………………………………………………......111
Chapter 5 Conclusions………………………………………………………………….113
5.1 A New Understanding of Catalysis by IGPS……………………………….113 5.2 Implications for Understanding the Evolution of Thermophilic Versus Mesophilic Enzymes……………………………………………………115

viii
5.3 Engineering New Indole Derivatives and Improving Industrial Indole Synthesis with Biocatalysts…………………………………………….116 5.4 Improving Novel Enzyme Engineering Efforts…………………………….117 5.5 Future Studies..……………………………………………………………..120 5.6 Conclusions…………………………………………………………………123 5.7 References…………………………………………………………………..123
Appendix Solvent Deuterium Kinetic Isotope Effect Analysis ………………..............126

ix
LIST OF FIGURES
Figure 1.1: The three consecutive (β/α)8-barrel enzymes in tryptophan biosythnesis catalyze the fourth, fifth, and sixth steps of the pathway. PRAI (PDB 1Pll) catalyzes the amadori rearrangement of PRA to form CdRP. CdRP is then converted to IGP by IGPS (PDB 1Pll). Then, αTS (PDB 1V7Y) catalyzes the cleavage of IGP to form glyceraldehyde-3-phosphate and indole. ...........……...…………………………………..7 Figure 1.2: Indole-3-glyerol phosphate synthase (IGPS). (a) ssIGPS (PDB: 1IGP) is a (β/α)8-barrel enzyme that contains an additional 45 residue N-terminal extension. (b) IGPS catalyzes the conversion of CdRP to form IGP. The proposed mechanism contains three steps and two intermediates and utilizes a general acid and base (proposed as Lys110 and Glu159)……………………………………………………………………..12 Figure 1.3: Multiple sequence alignments between IGPS from S. sulfataricus (ssIGPS), Thermatoga maritima (tmIGPS), E. coli (ecIGPS), and Mycobacterium tuberculosis (mtIGPS). Secondary structure is denoted by boxes above the sequence with α-helices in green and β-sheets in blue. Conserved residues are bold and in blue. Catalytically relevant residues are denoted with a star. Sequence alignment was performed with Clustal Omega provided by The European Bioinformatics Institute at The European Molecular Biology Laboratory……………………………….................................………………..14 Figure 1.4: Active site of IGPS with reduced CdRP bound. Conserved and catalytically relevant residues are shown. Lys110 is the proposed general acid in the condensation and dehydration steps. Glu159 and Glu210 have both been proposed to act as general base. Phe89 and Arg182 are proposed to aid in substrate binding. Lys53 is also involved in substrate binding and may have additional roles in the chemistry. The role of Glu51 has not been extensively studied……………………………………………………………..15 Figure 1.5: IGPS in complex with rCdRP (PDB:1LBF) (yellow) and IGP (1A53) (blue) showing residues that interact with the anthranilate moiety. When CdRP binds in the active site, Trp8, Pro57, Phe89, Arg182, and Leu184 interact with the anthranilate. Conversely, when IGP binds, Phe89, Lys110, Phe112, Ile133, and Arg182 interact.…..18 Figure 1.6: Numbering for CdRP. Lys53 interacts with the C1 carboxyl and C3’ hydroxyl groups and is thought to aid in ring closure between C1 and C2’.....................19 Figure 2.1: Conserved structure and function of IGPS from E. coli (green) (PDB 1P11) and S. sulfataricus (blue) (PDB 1IGPS). Despite only 30% sequence identity and large differences in stability, ssIGPS and ecIGPS show strong structural similarity………….33 Figure 2.2: Standard curve of fluorescence units per nanomolar for converting cps/s to nM/s. The slope of the line (4036 cps/nM) was used to convert data for ssIGPS to the appropriate units. Curve was attained using IGPS from T. maritima, which does not display product inhibition..................................................................................................38

x
Figure 2.3: Representative data for ssIGPS assays. (a) Progress curves for ssIGPS at 75 °C at varying concentrations of CdRP (100, 400, 800, 1000, and 2000 nM). (b) Michaelis-Menton curve for ssIGPS at 75 °C...................................................................40 Figure 2.4: The rate-determining step of the IGPS catalyzed reaction can be determined using SVE and SDKIE experiments. Substrate binding and product release (green) are viscosity sensitive and isotope insensitive. Ring closure (blue) is viscosity insensitive and isotope sensitive. The decarboxylation and dehydration (red) are viscosity and isotope insensitive………………………………………………………………………………..46 Figure 2.5: Solvent viscosity effects for ssIGPS. At 25 °C (blue) there is an SVE of 1.0 ± 0.2, wherease at 75 °C (black) the SVE is no longer present (-0.2 ± 0.1). The SVE is defined by the slop of the line for vo/vi versus ni/no. The results indicate that at 25 °C product release is rate-determining but as temperature increases to 75 °C product release is no longer rate-determining, and a chemical step becomes rate-determining.................47 Figure 2.6: The pH dependence of WT ssIGPS at (a) 37 °C (pKa1 7.5 ± 0.2, pKa2 8.8 ± 0.3) and (b) 75 °C (pKa1 5.6 ± 0.2, pKa2 8.7 ± 0.2) and (c) ecIGPS at 37 °C (pKa1 6.7 ± 0.1, pKa2 8.8 ± 0.1) show an ascending and descending limb consistent with general base and general acid involvement, respectively....…………………………………………...49 Figure 2.7: The rate-determining step for ssIGPS at higher temperatures involves a single proton transfer event. (a) The maximum catalytic turnover of ki/ko versus mole fraction D2O:H2O at both 37 °C (blue) and 75 °C (green) show a linear fit. (b) The square root of ki/ko versus mole fraction D2O:H2O at 37 °C and 75 °C show a quadratic fit. These results indicate that one proton transfer event is involved in the rate-determining step of the reaction, namely the proton transfer from the general acid in the condensation step of the reaction.............................................................................................................53 Figure 3.1: Accepted mechanism for IGPS suggests that the reaction proceeds in three steps: condensation, decarboxylation, and dehydration, with two distinct intermediates……………………………………………………………………………..61 Figure 3.2: The pH profiles suggest that Lys53 and Glu51 act as the general acid and base, respectively, in the dehydration step of IGPS catalysis. Shown are the pH curves for IGPS (a) WT (pKa1 5.6 ± 0.2, pKa2 8.7 ± 0.1), (b) Lys53Arg (pKa1 6.9 ± 0.1, pKa2 > 9), and (c) Glu51Gln (pKa2 6.51 ± 0.3)……………………………………………………..71 Figure 3.3: 1H-13C HSQC on ε-13CH2-Lys labeled ssIGPS shows eighteen resonances, which is consistent with the number of lysine residues in the enzyme………………….74 Figure 3.4: Overlay of 1H-13C-HSQC Spectra for ssIGPS at pH 7.0 (black) and pH 10.5 (red). At high pH, the changes in the spectrum are likely caused by denaturation of ssIGPS................................................................................................................................75

xi
Figure 3.5: A representative plot of chemical shift versus pH for peak #2. The pKa value associated with this curve is 11.45 ± 0.14, although the change in chemical shift likely reflects denaturation of the enzyme rather than deprotonation of the lysine.....................76 Figure 3.6: pH dependence of the ssIGPS catalyzed reaction performed in H2O (pKa1 5.7 ± 0.1, pKa2 8.7 ± 0.1), shown in blue, and D2O (pKa1 5.3 ± 0.2, pKa2 8.9 ± 0.2), shown in green, display little difference in pKa values. Therefore, the use of D2O in NMR experiments does not explain the discrepancy in pKa values between the two methods…………………………………………………………………………………..78 Figure 3.7: Overlay of 1H-13C HSQCs of Lys53Arg (red) and WT ssIGPS (black) labeled with ε-CH2-Lys. Due to the low resolution of Lys53Arg ssIGPS spectrum, and its the poor alignment to WT, Lys53 and other resonances remain unassigned………………..79 Figure 3.8: The assigned role for the conserved and charged residues in the active site of IGPS. The ring closure is catalyzed by the general acid, Lys110, and assisted by Glu159 (blue). The dehydration is catalyzed be the general acid, Lys53 and general base, Glu51 (yellow). Arg182 and Glu210 are involved in substrate binding (orange)………………83 Figure 3.9: The modified mechanism of ssIGPS catalysis utilized Lys53 and Glu51 as the general acid and base pair in the dehydration step of the reaction. Additionally, the general base now attacks the amide hydrogen rather than the previously suggested alkyl hydrogen............................................................................................................................85 Figure 3.10: Rotation about the C3’-C4’ bond of ribose chain is required for the dehydration step in IGPS catalysis. Crystal structure of ssIGPS:IGP complex (Top) (PDB: 1LBF) shows the ligand bound in the active site such that Lys53 and Glu51 are not properly positioned for catalysis. The ribose chain must rotate (Bottom) to reposition the intermediate in the second binding pocket and allow for dehydration to form IGP…….86 Figure 3.11: Surface rendering of the IGPS binding pocket shows two distinct active sites for catalysis. In the first site (blue), Lys110 and Glu159 catalyze the ring closure step deep within the pocket. The intermediate then transitions to the second site (yellow), which is closer to where product exits the binding pocket, where Lys53 and Glu51 catalyze the dehydration step. (PDB: 1A53)…………………………………………….88

xii
Figure 4.1: The catalytically important residues in the dehydration step of IGPS are found on the β1α1 and β2α2 loops, which interact through a hydrogen bond between Arg54 and Asn90. (a) The ssIGPS catalyzed reaction has two distinct binding pockets for the two reaction steps. In step one, Lys110 (cyan) initiates the ring closure and decarboxylation. Following the formation of the intermediate, the anthranilate moiety is transferred to the second site where Lys53 and Glu51 (yellow) act as the active site acid and base. The role of the β1α1 and β2α2 loops (blue) including the interaction between Arg54 and Asn90 is examined herein. (b) This interaction is thought to have functional significance in IGPS since it is coevolving amongst IGPS species and exhibits correlated motion in MD simulations. This interaction is in close proximity to the conserved residues Lys53 and Phe89………………………………………………………………..94 Figure 4.2: pH profiles of WT and Asn90Ala ssIGPS show changes to the activity of the general acid and base. pH profile for (a) WT (pKa1 5.6, pKa2 8.7) shows two pKa values associated with general acid/base catalysis, whereas (b) Asn90Ala (pKa1 7.26 ) show a loss in the second ionization that was previously attributed to Lys53. This finding indicates that the Asn90 variant is affecting the dehydration step of the reaction, and interfering with proper function of the general acid Lys53…………..………………...100 Figure 4.3: Asn90Ala affects the thermal stability but not proper folding for ssIGPS. (a) Thermal inactivation curves show an increase in the thermal stability of the Asn90Ala variant compared to WT. (b) Circular dichroism curves indicate that the changes in activity for the Asn90Ala variant are not caused by gross structural changes to the enzyme and can be attributed to changes in the reaction……………………………….106

xiii
LIST OF TABLES
Table 1.1: Conserved active site residues in ssIGPS that are of interest to these studies
are shown along with their proposed roles in enzyme activity..........................................16
Table 2.1: Steady-state kinetic parameters for ssIGPS and ecIGPS at pH 7.5 indicate that
the rate-determining step changes as a function of temperature. ……......………………44
Table 2.2: pKa values for ssIGPS and ecIGPS..................................................................50
Table 3.1: Steady-state kinetics (at 75 °C) demonstrates that the dehydration step of
IGPS catalysis occurs through the general acid and base Lys53 and Glu51,
respectively. .........................................................………………...............……………..68
Table 3.2: pKa values for WT ssIGPS and Lys53Arg and Glu51Gln variants identify
Lys53 and Glu51 as the general acid and base in ssIGPS catalysis...................................72
Table 3.3: pKa values determined by 1H-13C HSQC on ε-13CH2-Lys labeled ssIGPS are
not in agreement with the pKa for the pH dependence of the enzymatic reaction
determined by steady-state kinetics……………………………………………………...77
Table 4.1: Steady-state kinetic parameters of WT ssIGPS and loop variants indicate an
important role for the β1α1 and β2α2 loop interaction in catalysis……………………...97
Table 4.2: pKa values for WT, Arg54Ala, and Asn90Ala indicate that the loop
interaction is important for general acid/base catalysis...................................................101

xiv
LIST OF ABBREVIATIONS
Amp Ampicillin
BICINE N,N-Bis(2-hydroxyethyl)glycine
CD Circular dichroism
CdRP 1-o-carboxylphenylamino deoxyribulose 5-phosphate
CHES N-cyclohexyl-2-aminoethanesulfonic acid
CPS Counts per second
DTT Dithiothreitol
ecIGPS Indole-3-glycerol phosphate synthase from Escherichia coli
EDTA Ethylenediaminetetraacetic acid
EI Enzyme-intermediate complex
EP Enzyme-product complex
ES Enzyme-substrate complex
Hepes 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
Hepps 3-[4-(2-hydroxyethyl)-1-piperazinyl] propanesulfonic acid
HPLC High pressure liquid chromatography
HSQC Heteronuclear single quantum coherence
I1 Intermediate 1 from the IGPS reaction
I2 Intermediate 2 from the IGPS reaction
IGP Indole-3-glycerol phosphate
IGPS Indole-3-glycerol phosphate synthase
IPTG Isopropyl β-D-1-thiogalactopyranoside
Kan Kanamycin
kcat Maximum catalytic turnover constant
KM Michaelis constant
LB Luria-Bertani
MD Molecular dynamics
MES 2-(N-morpholino)ethanesulfonic acid
mtIGPS Indole-3-glycerol phosphate synthase from Mycobacterium tuberculosis
NaBH4 Sodium borohydride

xv
NAC Near attack conformer
NMR Nuclear magnetic resonance
P Product
PCR Polymerase chain reaction
PDB Protein data bank
PMSF Phenylmethanesulfonyl fluoride
PRA Phosphoribosyl anthranilate
PRAI n-Phosphoribosyl anthranilate isomerase
rCdrP Reduced CdRP
S Substrate
SCA Statistical coupling analysis
SDKIE Solvent deuterium kinetic isotope effect
SDS-PAGE Sodium dodecylsulfate-polyacrylamide gel electrophoresis
ssIGPS Indole-3-glycerol phosphate synthase from Sulfolobus sulfataricus
SVE Solvent viscosity effect
TB Tuberculosis
TIM Triose Phosphate Isomerase
tmIGPS Indole-3-glycerol phosphate synthase from Thermotoga Maritima
tr-NOE Exchange transferred-nuclear Overhauser effect
TrpC IGPS encoding gene
WT Wild type
αTS Alpha subunit of tryptophan synthase

xvi
ACKNOWLEDGEMENTS
There are many people who deserve recognition for their support throughout my
graduate studies. I would first like to thank my advisor, David Boehr, whose mentorship
allowed me to develop into a thoughtful and independent scientist, as well as my
committee, Scott Showalter, Phil Bevilacqua, and Craig Cameron, for their thoughtful
comments on this work. I must also thank the other members of the Boehr laboratory,
especially Alyson Boehr, Jennifer Axe, Yan Mei Chan, Olga Manweiler, Laura Loggia,
and Alexander Chasin. Life in the Boehr lab as been an interesting and memorable
experience, and their support and entertainment has helped make the days more
interesting. I am honored to be the first person to complete my Ph.D. from the laboratory.
Without the use of the fluorometer in the Benkovic laboratory, I would not have been
able to complete this thesis, and so I would also like to thank Stephen Benkovic, Michelle
Spiering, and the other members of the Benkovic laboratory for allowing me into their
laboratory and for the generous loan of their instrumentation.
The mentorship I received at the Florida Institute of Technology from my
undergraduate research advisor, Dr. Joel Olson, is the reason I attended graduate school,
and also part of the reason I was able to succeed. He was the first person to spark my
curiosity for chemical research, and his famous words of wisdom, “research is like a
funnel,” have gotten me through the slow and tedious parts of this process and helped me
remember the bigger picture.
While the words presented in this dissertation reflect my scientific work, my
persistence in completing this thesis is largely due to those hours between experiments
spent with some amazing friends, especially Dr. Joy Gallagher, Dr. Jennifer Wilcox, Dr.

xvii
Jason Stephens, Kaitlin Haas, Erin Cullen, and Natalie Lamberton. My partner in crime,
Dr. Joy Gallagher, has always been there to listen and provide her perspective on my
research and my life. From our lunchtime study sessions in our first year to thesis review
sessions in our last few months, she has been an excellent confidant and provided endless
entertainment. My officemate, Dr. Jennifer Wilcox, has been my support in these last few
months. Together we have managed our stress, and I cannot think of a person I would
rather share my space or my snacks with.
I am fortunate the have the unwavering support of a wonderful family that has
been a source of incredible strength throughout my life. My father, Joseph Zaccardi, has
always given me his support and encouragement, and made me feel like I could tackle
seemingly insurmountable tasks. My mother, Joan Zaccardi, has been a sounding board
for my frustrations, and has provided great advice even when I did not think I needed it.
My siblings, Diane Baldwin and Joseph Zaccardi III, have been role models for me
throughout my life, and are excellent examples in attaining your goals. Lastly, I would
like to thank the greatest part of my life, my dog, Grace. She has learned more science
from holding my hand and listening as I practice all of my presentations that she probably
deserves her own dog doctorate. Taking care of her also forced me to take a break and get
some exercise on the busiest and most stressful days, allowing me to refocus and rest.
“This seems to be the law of progress in everything we do: it moves along a spiral
rather than a perpendicular; we seem to be actually going out of the way, and yet it
turns out that we were really moving upward all the time.”
Frances E. Willard

1
Chapter 1
Introduction to Indole-3-glycerol Phosphate Synthase
1.1 Progress Towards Engineering Enzymes with New Functions
Biocatalysts offer a unique solution to many common industrial synthesis
problems due to their high efficiencies and high degree of selectivity. They also allow for
the use of mild reaction conditions compared to typical industrial processes.1, 2 Because
of these properties, enzymes are being increasingly implemented in a variety of industries
including bioremediation, textiles, biofuel production, pharmaceuticals, and agriculture.3
For example, an optimized lipase is used to synthesize enantiomer specific precursors for
the production of diltiazem, a blood pressure medication.4 Much of the current industrial
application of enzymes has focused on improving stability, or changing specificity for
reactions already performed in natural enzyme systems. This focus is the result of a
desire to utilize biological systems amongst those industrial processes that have already
been optimized under conditions not typically suited for enzymes, such as high
temperatures, extreme pHs, high concentrations, and non-aqueous solvents.5 However,
there is also value in the production of “made to order” enzymes that can catalyze
reactions not catalyzed in nature; for many industrially important syntheses, a naturally
occurring enzyme capable of catalyzing the reaction does not exist.4, 6 Engineering
enzymes capable of catalyzing non-natural reactions at rates comparable to enzymes
found in nature challenges the fundamental understanding of enzyme function. In order to
recreate the mechanisms evolved in biological systems, we must completely understand

2
how sequence, structure, conformational dynamics, and function are intertwined, and
work together in catalyzing the reaction.
Over the last several years, a large number of studies have focused on the
synthesis of novel enzymes.7-17 The explosion in research on this subject largely began
with two renowned studies from the Baker laboratory.14, 15 In these studies, two non-
natural reactions were engineered onto natural enzyme scaffolds. First, plausible
chemical mechanisms along with appropriate transition states were established and then
computational methods were used to create an active site with the appropriate
architecture, using several different enzyme structures as a starting scaffold including the
(β/α)8-barrel and the jelly roll. In the first study, a retro-aldol enzyme capable of breaking
a carbon-carbon bond of a non-natural substrate was designed.14 The second study
engineered an enzyme for the Kemp elimination reaction, which requires the direct
removal of a hydrogen from a carbon, a process that is not possible through normal
synthetic routes due to its high activation energy barriers.15 While both studies were able
to engineer enzymes with an enhancement of the desired activity on the order of 106 for
the Kemp elimination and 104 for the retro-aldol reaction, these rate enhancements are
still very low compared to the rates of natural enzymes.14, 17 Reoptimization of both
systems has also been performed.17, 18 For the Kemp elimination, considerable increase in
catalytic rates was possible through directed evolution of the previously designed
catalysts. However, directed evolution studies are very time intensive, require screening
of a large number of inactive variants, and were still unable to match the rate
enhancement of biological systems.16

3
These studies are largely based on modeling the active site architecture, and
despite the high similarity of the crystal structures on the non-natural enzymes compared
to their models, reaction rate enhancements (105 to 106) are not able to match natural
enzymes (1023).19 This result may be because the algorithms used to design these systems
only integrate catalytically relevant residues, and disregard residues that may be
important for other processes. For example, molecular dynamics (MD) simulations show
that the enzymes designed for the Kemp elimination have dynamic fluctuations that
prevent proper active site configuration, and thus the enzymes show lower than expected
reaction rates.20 This finding highlights the need for enzyme engineering studies to also
take into account protein motions, and as such, many researchers have introduced
dynamics into their design algorithms. This advancement has allowed for some
improvements in rate enhancement, although a more complete understanding of how
sequence changes affect dynamic processes will be beneficial for the further development
of these techniques.20-22
Although enzymes are regularly depicted as rigid structures as they are found in
crystals, in solution enzymes are quite flexible, and undergo conformational fluctuations
that are thought to be important for regulating substrate binding, catalysis, folding, and
other processes. The role of flexibility in enzyme function has been widely studied, and
its importance in catalysis has been implicated in many different enzymes.23-29 However,
even with the growing knowledge of enzyme dynamics, reaction rates of engineered
enzymes were only enhanced by about five orders of magnitude over the rate of the
uncatalyzed reaction, whereas reaction rates of natural enzymes can be enhanced up to
twenty-three orders of magnitude.

4
Engineering new enzymes that are capable of performing non-natural reactions
will provide significant improvements to the technology in many different industries.
Industrial synthetic processes are typically harsher than those found in nature, and often
utilize high temperatures, extreme pH conditions, and non-aqueous solvents. The ability
to introduce enzymes that can withstand these extremes, or decrease the need for such
conditions, will drastically improve the efficiency of industrial processes.5 However, in
order to produce viable enzymes for these applications, a fundamental understanding of
the molecular determinants not just for catalysis, but also for structure, stability, and
dynamics is required. The process of changing the active site architecture from one
enzyme to another is intricate, and requires a very detailed understanding of all the
factors that can affect activity in the new enzyme including protein folding, flexibility,
stability, structure, and chemical interactions.
In order to engineer enzymes capable of catalyzing non-natural reactions at rates
closer to those found in nature, a more comprehensive approach is required that takes into
account not only the residues required for the new activity, but also the starting enzyme
architecture, and the role of both active site residues and residues more distant from the
active site in the scaffold enzyme. This undertaking requires an in depth knowledge for
the role of all residues in the protein, especially considering that semi-conserved or non-
conserved residues, in addition to those that are conserved, contribute to active site
architecture, affect the ability of an enzyme to catalyze the desired reaction, and are
involved in other processes like protein folding and stability.3

5
1.2 The Conserved (β/α)8-Barrel Protein Fold as an Enzyme Engineering Scaffold
Enzymes containing the (β/α)8-barrel (or TIM-barrel, named after triose
phosphate isomerase) fold have been widely used as enzyme engineering scaffolds due to
the fold’s high stability, diverse catalytic ability, and conserved structure. In fact, the
(β/α)8-barrel shows higher success in these studies (including the Kemp elimination and
retro aldolase reactions previously described) than other folds.14,15 The (β/α)8-barrel is the
most common enzyme fold in nature and is found in many different enzyme
superfamilies that are capable of catalyzing a diverse range of reactions.30-32 It generally
consists of eight units of alternating β-strands and α-helices that are connected by loops.
The β-strands form a barrel in the center and are surrounded by the α-helices. This
dualism of conserved structure with diverse function of (β/α)8-barrel enzymes provides an
excellent model for studying the relationship between enzyme sequence, structure,
function, and dynamics. Catalytically important residues are typically found on the C-
terminal ends of the β-sheets, and on the loops connecting the β-strands to the α-helices
(βα loops).33 This structure provides several different “take off” positions in enzyme
engineering studies that can be used for catalytic residues and transition state
stabilization, as all positions pointing towards the inside of the barrel can be used.15
The role of dynamics in enzymes containing the (β/α)8-barrel fold has been well
documented.34-36 In the iconic (β/α)8-barrel, triosephosphate isomerase (TIM), several
studies have examined loop dynamics that contribute to the catalysis by the enzyme.37-40
The β6α6 loop in TIM undergoes conformational exchange to allow for substrate binding
and product release. Similar behavior is seen for other (β/α)8-barrel enzymes including
alkanesulfaonate monooxygenase,41 imidazole glycerol phosphate synthase,42 and D-

6
ribulose 5-phosphate 3-epimerase.43 A better understanding of how specific amino acids
change the conformational dynamics of these enzymes would improve the ability to fine
tune the (β/α)8-barrel scaffold for new functions.
1.3 (β/α)8-Barrel Enzymes in Tryptophan Biosynthesis
In tryptophan biosynthesis, there are three consecutive enzymes that contain this
(β/α)8-barrel fold: N-(5’-phosphoribosyl)-anthranilate isomerase (PRAI), indole-3-
glycerol phosphate synthase (IGPS), and the alpha subunit of tryptophan synthase (αTS)
(Figure 1.1), which catalyze the fourth, fifth, and sixth committed steps of tryptophan
production.44, 45 PRAI catalyzes an Amadori rearrangement of phosphoribosyl
anthranilate (PRA) to form 1-(o-carboxyphenylamino)-1-deoxyribulose 5-phosphate
(CdRP). IGPS then performs a ring closure to form indole-3-glycerol phosphate (IGP).
Lastly, αTS removes the glyceraldehyde 3-phosphate from IGP to form the indole ring
that goes forward in the pathway and combines with L-serine to form tryptophan. It is
interesting from an evolutionary standpoint that these three enzymes catalyze different
types of reactions but all evolved with the same fold, leading to several possible
hypotheses for the evolution of the tryptophan biosynthetic pathway. First, all three
enzymes may have evolved divergently from a common ancestral enzyme that was
capable of catalyzing all three reactions with poor efficiency.30, 31, 46 In the divergent
mechanism, the enzymes may have evolved through gene duplication, where the
organism made multiple copies of a gene that could then evolve several different
functions.47, 48

7
Figure 1.1: The three consecutive (β/α)8-barrel enzymes in tryptophan biosythnesis catalyze the fourth, fifth, and sixth steps of the pathway. PRAI (PDB 1Pll) catalyzes the amadori rearrangement of PRA to form CdRP. CdRP is then converted to IGP by IGPS (PDB 1Pll). Then, αTS (PDB 1V7Y) catalyzes the cleavage of IGP to form glyceraldehyde-3-phosphate and indole.

8
Second, akin to proposals by Horowitz regarding the evolution of metabolism, the
pathway may have evolved backwards, with αTS evolving first, followed by IGPS, and
lastly PRAI.49 Several studies have explored the relationship between related (β/α)8-barrel
enzymes.47, 50-52 One such directed evolution study by Evran et al. was able to establish
PRAI activity using the α-TS scaffold.51 This study provided evidence of evolution of the
(β/α)8-barrel enzymes in tryptophan synthesis by divergence/gene duplication.52 Despite
the low sequence identity of these three enzymes (PRAI and IGPS are only about 22%
sequence identical, and αTS shows even lower sequence identity with both PRAI and
IGPS), they contain very conserved structural elements, including the phosphate binding
site located in the β7α7 and β8α8 loops. However, other loops have been shown to be
important for specific activity, as in one study in which loop swapping between PRAI
and α-TS changed the catalytic ability of the enzyme.34, 53
1.4 The Tryptophan Biosynthetic Enzyme Indole-3-glycerol Phosphate Synthase
In general the (β/α)8-barrel fold has shown greater success as an engineering
scaffold than other folds, and in several studies the largest number of active variants, as
well as those with the highest rate enhancement, were produced using IGPS from
Sulfolobus sulfataricus (ssIGPS) as a scaffold.14, 15 In fact, ssIGPS was used as a starting
scaffold in the studies from the Baker laboratory, and show the highest rate enhancement
compared to all other enzyme scaffolds examined (including TIM).14, 15 In addition to the
promise ssIGPS shows in novel enzyme design, its naturally catalyzed reaction is also
applicable for the industrial synthesis of indole and its derivatives. ssIGPS is a
thermophilic enzyme; therefore, its increased stability is desireable for industrial

9
processes, and the indole ring is a widely used structure in pharmaceuticals, agriculture,
and other industries.54
IGPS is also valuable in several other fields including the development of new
antimicrobial agents and the understanding of thermophilic enzymes.55 The tryptophan
biosynthetic pathway is not found in humans, but is found in pathogenic bacteria.
Therefore, the enzymes in the pathway are potential targets for new antimicrobial
compounds, particularly those pathogens that show a high occurance of multidrug
resistance such as Mycobacterium tuberculosis. Due to the widespread multidrug
resistance of the bacterium to currently available treatments, as well as the prevalence of
tuberculosis (TB) in impoverished areas of the world, the disease can be difficult to treat
and the currently available treatments are expensive, complex, and can have harsh side
effects.55 These characteristics lead to poor patient compliance and a push to identify new
targets for effective TB treatment.56
Studies by Smith et al. demonstrate that tryptophan auxotrophs of M. tuberculosis
were avirulent in mice, indicating that the bacterium may be unable to uptake these
amino acids in vivo.57 The gene that encodes for the IGPS enzyme (TrpC) was also
shown to be essential for growth of M. tuberculosis in vitro,58 which further demonstrates
the potential utility in targeting IGPS as a treatment for TB and other microbial diseases.
While there are several known inhibitors for IGPS,55, 59, 60 none are currently available for
treatment of tuberculosis or other bacterial infections. Studies towards the development
of new antimicrobials that target IGPS will benefit from a more in depth examination of
its mechanism and active site.

10
Thermophilic organisms have evolved robust mechanisms to overcome the
deleterious effects high temperature typically has on biomolecules, such as denaturation,
allowing life to exist under extreme conditions, including extreme temperatures. The
thermophilic archaeon, S. sulfolobus, is found at high temperatures (> 80 °C) and has
developed mechanisms in order to exist at extreme temperatures. For enzymes, this
includes an increase in stabilizing interactions such as hydrogen bonds and electrostatic
interactions, as well as higher packing efficiencies and increased burial of the
hydrophobic surfaces.61-64 Harnessing the stabilizing properties of thermophilic enzymes
without disrupting catalytic activity will provide major technological advancements for
the development of enzymes for biotechnology industries.
A fuller understanding of IGPS would also be useful for understanding
temperature adaptation, as IGPS has been widely studied in both thermophilic and
mesophilic organisms. Crystal structures and biochemical techniques have identified
structural differences between IGPS from S. sulfataricus and E. coli (ecIGPS),65, 66 as
well as Thermotoga maritima (tmIGPS),67 Thermatoga thermophilus,68 and
Thermococcus kadakarensis.69 Despite their high structural similarity, ssIGPS and
ecIGPS are only about 30% sequence identical, and ssIGPS contains additional
noncovalent interactions that prevent its denaturation at increased temperatures. ssIGPS
also shows decreased activity at lower temperatures compared to its mesophilic
counterpart in ecIGPS.64, 65, 70 Some studies suggest that the temperature dependent
activity differences between thermophilic ssIGPS and mesophilic ecIGPS is the result of
a decrease in flexibility of the protein caused by the stabilizers (e.g. salt bridges) that are
needed to adapt to the increase in temperature.71, 72 Indeed, Merz et al. semiquantitatively

11
examined the flexibility of ssIGPS at various temperatures through a limited proteolysis
study, and the results showed the less flexible the IGPS enzyme at lower temperatures,
the lower its rate of thermal inactivation, and the lower its activity at lower
temperatures.64
Despite the differences in activity for ssIGPS at lower versus higher temperatures,
little research has been performed on ssIGPS near its biologically relevant temperatures.
Studies on ssIGPS at lower temperatures suggest that the rate-determining step of the
overall reaction is product release.64, 73 However, there may be differences between the
kinetic or chemical reaction mechanism at lower versus higher temperatures as well as
between thermophilic and mesophilic homologs. A better understanding of catalysis for
both thermophiles and mesophiles over a range of temperatures will aid in the application
of the robust and stable enzymes in an industrial setting.
1.3 Previous Knowledge on the Mechanism of IGPS
The mechanism for the conversion of CdRP to form IGP by IGPS (Figure 1.2)
was originally proposed nearly forty years ago by Parry et al. The reaction is proposed to
occur in three steps (condensation, decarboxylation, and dehydration) with two distinct
intermediates (I1 and I2).74 This mechanism, particularly the formation of I1, was
motivated by the observation that the reaction does not occur for a substrate analog
lacking the carboxyl, which is evidence that the carboxyl is required for pyrrole ring
formation.75

12
Figure 1.2: Indole-3-glyerol phosphate synthase. (a) ssIGPS (PDB: 1IGP) is a (β/α)8-barrel enzyme that contains an additional 45 residue N-terminal extension compared to the standard (β/α)8-barrel fold. (b) IGPS catalyzes the conversion of CdRP to form IGP. The proposed mechanism contains three steps and two intermediates and utilizes a general acid and base (proposed as Lys110 and Glu159).74,77

13
Multiple sequence alignment of IGPS from various species shows several
conserved residues (Figure 1.3). Further insight into the possible role of these residues
and the mechanism of IGPS came from studies of amino acid substitutions in IGPS from
ecIGPS including Glu53, Lys55, Lys114, Glu163, Asn184, and Arg186 (corresponding
to Glu51, Lys53, Lys110, Glu159, Asn180, Arg182 in ssIGPS).76 The proposed general
acid, Lys114, is essential, and neither Arg nor His amino acid substitutions in this
position yielded active enzyme. Similar results were found for the Glu163Asp variant,
which lead to the assignment of Lys114 and Glu163 as the general acid-base pair in
ecIGPS. Amino acid substitutions at Lys55 and Glu53 also yielded interesting results,
with a 40-fold decrease in kcat for the Glu53Cys variant. This finding indicates that Glu53
is important for catalysis, and the authors suggest it may help with the proper positioning
of Lys114. The Lys55Ser variant shows a twenty-fold decrease in kcat and a 1800-fold
increase in KM, indicating a role for this residue in both chemistry and ligand binding.
While these experiments identified those residues that are catalytically required, they
were unable to discern the step of the reaction in which the residues were involved.
Additional insight about IGPS was gained from crystal structures of IGPS bound
with substrate, substrate analog, and product, which suggested roles for several amino
acids in IGPS catalysis (Figure 1.4, Table 1.1), including the proposed general acid and
base, Lys110 and Glu159, respectively (numbering according to ssIGPS).77 Arg182 and
Phe89 were predicted to be involved in substrate binding with Phe89 interacting with the
aromatic moiety and Arg182 interacting with the phosphate group. The roles for Glu51
and Lys53 were more ambiguous. The authors asserted that Lys53 helps bind the
substrate, but that Glu51 and Lys53 can also form a salt bridge triad with Lys110 that

14
Figure 1.3: Multiple sequence alignments between IGPS from S. sulfataricus (ssIGPS), Thermatoga maritima (tmIGPS), E. coli (ecIGPS), and Mycobacterium tuberculosis (mtIGPS). Secondary structure is denoted by boxes above the sequence with α-helices in green and β-sheets in blue. Conserved residues are bold and in blue. Catalytically relevant residues are denoted with a star. Sequence alignment was performed with Clustal Omega provided by The European Bioinformatics Institute at The European Molecular Biology Laboratory.

15
Figure 1.4: Active site of IGPS with reduced CdRP bound. Conserved and catalytically relevant residues are shown. Lys110 is the proposed general acid in the condensation and dehydration steps. Glu159 and Glu210 have both been proposed to act as general base. Phe89 and Arg182 are proposed to aid in substrate binding. Lys53 is also involved in substrate binding and may have additional roles in the chemical steps. The role of Glu51 has not been extensively studied.76,77

16
Table 1.1: Conserved active site residues in ssIGPS that are of interest to these studies are shown along with their proposed roles in enzyme activity.76,77,78
Conserved Residue Location in ssIGPS Proposed Role Glu51 β1 Strand Interacts with Lys110 and Lys53 Lys53 β1α1 Loop Substrate binding Phe89 β2α2 Loop Substrate binding Lys110 β3 Strand General acid Glu159 β5 Strand General base
(predicted by crystal structures) Arg182 β6α6 Loop Substrate binding Glu210 β7 Strand General base
(predicted by MD simulations)

17
may aid the activity of the general acid. Additionally, the crystal structure showed the
anthranilate group bound into a different hydrophobic pocket in IGPS complexed with
substrate than when complexed with product (Figure 1.5), introducing the idea that the
substrate may undergo conformational rearrangement in the active site during catalysis.
Lys53 is conserved and is located on the highly dynamic β1α1 loop. Crystal
structures predict that it is largely involved in substrate binding, as it can hydrogen bond
in multiple positions of CdRP including the C1 carboxyl and the C3’ hydroxyl
(numbering for CdRP, Figure 1.6). MD studies suggest Lys53 is important for structural
rearrangements that facilitate catalysis, and have suggested that the interaction of Lys53
with the substrate along with the flexibility of the β1α1 loop, may be involved in bridging
the gap between the C1 and C2’ allowing for the formation of the pyrrole ring.70, 78 The
authors asserted that enzyme flexibility is required for the formation of a near attack
conformer (NAC). NACs are groundstate substrate conformations that can convert most
efficiently into the transition state.78 For CdRP in the IGPS catalyzes reaction, the NAC is
defined by the reacting moeties, C1-C2’, within van der Waals contact distance (≤ 3.5 Å)
and at an approaching angle of 120° ± 20°.70,78 Similarly, work performed by Goodey and
Sterner indicates that the flexibility of this loop is coupled to enzyme activity.73
Other studies further highlight the potential role for dynamics and flexibility in
IGPS. Shen et al. examined correlated and coevolving residues in IGPS through a
combined study using statistical coupling analysis (SCA) and MD simulations.79 SCA
identifies residues that are not necessarily conserved, but covary between the enzymes
from different species. This method identified amino acid pairs whose interaction may be
important for protein folding, stability, or catalysis.80, 81 When combined with MD

18
Figure 1.5: IGPS in complex with rCdRP (PDB:1LBF) (yellow) and IGP (1A53) (blue) showing residues that interact with the aromatic, anthranilate moiety. When CdRP binds in the active site, Trp8, Pro57, Phe89, Arg182, and Leu184 interact with the anthranilate. Conversely, when IGP binds, Phe89, Lys110, Phe112, Ile133, and Arg182 interact.

19
Figure 1.6: Numbering for CdRP. Lys53 interacts with the C1 carboxyl and C3’ hydroxyl groups and is thought to aid in ring closure between C1 and C2’.

20
simulations, this study identified amino acid pairs in IGPS that showed the potential
importance of residues that are not necessarily conserved, but whose coordinated motion
is potentially important in regulating a variety of enzyme processes including
conformational exchange, enzyme folding, structural stability, or chemistry itself.82 The
authors assert that these amino acid pairs, which are all in van der Waals contact with one
another, form an amino acid network throughout the enzyme that aids in enzyme activity.
Most notable to this work are active site residues Arg54, Glu85, and Asn90. Their
location on dynamic active site loops, and their proximity to conserved and catalytic
residues may be indicative of a more direct role, potentially important for coordinating
functional motion in IGPS catalysis.
1.6 Conclusions
Considering the potential application for the IGPS enzyme across many different
industries, it is essential that the mechanism of IGPS be very well understood. For many
of these applications, a detailed understanding of the role of active site residues in the
catalytic mechanism is required, especially considering that a single mutation or small
structural change can cause large differences in enzyme function, folding, and/or
stability.36 In order to apply IGPS as an industrial enzyme, use it as a target for
antibacterial agents, or as a scaffold for enzyme engineering, we must understand all of
the active site residues, not just those that are conserved, but also those residues that may
contribute to enzyme architecture, dynamics, and other processes even if they are distal
from the active site. Even residues that are not conserved can play a large role in the
catalytic mechanism, or participate in other processes such as protein folding, stability, or

21
dynamics. Failure to consider how an enzyme undergoes catalysis will lead to less
efficient application of IGPS for enzyme engineering and as a antimicrobial target.
Despite the extensive research on IGPS, there are still many questions regarding
the kinetic and chemical mechanism as well as the specific role of several conserved
residues in catalysis. First, previous research only examined the kinetic mechanism of
ssIGPS at lower temperatures (25 °C). In Chapter 2, the kinetic differences between
thermophilic ssIGPS and mesophilic ecIGPS are examined. The results show that the
kinetic mechanism of ssIGPS is temperature dependent, with product release being rate-
determining at lower temperatures (25 °C) and the ring closure being rate-determining at
higher, biologically relevant temperatures (75 °C). Additionally, ssIGPS and ecIGPS
display different rate-determining steps at their adaptive temperatures.
Another issue with the mechanism of IGPS involves the general acid, Lys110,
which is suggested to donate a proton in both the condensation and dehydration steps,
although there has been no plausible mechanism suggested for its reprotonation. Several
studies are also at odds in the assignment of the general base, with crystallography
suggesting Glu159 performs this task and MD simulations suggesting Glu210. There are
also several other conserved residues in the active site whose roles are undefined. In
Chapter 3, the role of conserved, charged active site residues are examined. The results
show that Lys53 and Glu51 are the general acid and base in the dehydration step. This
finding is at odds with the previously published mechanism for IGPS that suggested the
general acid base pair was Lys110/Glu159. This new assignment also led to the proposal
that the substrate undergoes a reorientation in the active site after the first chemical step

22
in order to be properly aligned for catalysis. This study also suggests that the substrate
must undergo a reorientation in the active site.
Lastly, several studies have implicated a role for active site loops in catalysis
including computational research suggesting a functional role for coevolving residues in
catalysis was unspecific.78 A better understanding of the role for coevolving residues in
the IGPS is desirable for future applications. Therefore, in Chapter 4, the role of
coevolving, active site residues Arg54, Glu85, and Asn90 were examined. These residues
are not conserved, but our results suggest that they are still involved in proper function of
ssIGPS. Specifically, the interaction between Arg54 and Asn90 is involved in the proper
function of the general acid and base, Lys53 and Glu51, during dehydration. The research
presented in this dissertation has more completely examined the kinetic and chemical
mechanism for IGPS and has greatly improved the understanding of catalysis by this
enzyme.
1.6 References
1. Nestl, B. M.; Nebel, B. A.; Hauer, B., Recent progress in industrial biocatalysis. Current Opinion in Chemical Biology 15 (2), 187-‐193. 2. Wang, M.; Si, T.; Zhao, H., Biocatalyst development by directed evolution. Bioresource Technology 115, 117-‐125. 3. Kiss, G.; Roethlisberger, D.; Baker, D.; Houk, K. N., Evaluation and ranking of enzyme designs. Protein Science 2010, 19 (9), 1760-‐1773. 4. Bornscheuer, U. T.; Huisman, G. W.; Kazlauskas, R. J.; Lutz, S.; Moore, J. C.; Robins, K., Engineering the third wave of biocatalysis. Nature 2012, 485 (7397), 185-‐194. 5. Woodley, J. M., Protein engineering of enzymes for process applications. Current Opinion in Chemical Biology 2013, 17 (2), 310-‐316.

23
6. Campeotto, I.; Acevedo-‐Rocha, C. G., Hijacking nature-‐new approaches to unravel enzyme mechanisms and engineer improved biocatalysts. Embo Reports 2013, 14 (4), 299-‐301. 7. Linder, M.; Johansson, A. J.; Olsson, T. S. G.; Liebeschuetz, J.; Brinck, T., Designing a New Diels-‐Alderase: A Combinatorial, Semirational Approach Including Dynamic Optimization. Journal of Chemical Information and Modeling 2011, 51 (8), 1906-‐1917. 8. Alvizo, O.; Mittal, S.; Mayo, S. L.; Schiffer, C. A., Structural, kinetic, and thermodynamic studies of specificity designed HIV-‐1 protease. Protein Science 2012, 21 (7), 1029-‐1041. 9. Chen, M. M. Y.; Snow, C. D.; Vizcarra, C. L.; Mayo, S. L.; Arnold, F. H., Comparison of random mutagenesis and semi-‐rational designed libraries for improved cytochrome P450 BM3-‐catalyzed hydroxylation of small alkanes. Protein Engineering Design & Selection 2012, 25 (4), 171-‐178. 10. Privett, H. K.; Kiss, G.; Lee, T. M.; Blomberg, R.; Chica, R. A.; Thomas, L. M.; Hilvert, D.; Houk, K. N.; Mayo, S. L., Iterative approach to computational enzyme design. Proceedings of the National Academy of Sciences of the United States of America 2012, 109 (10), 3790-‐3795. 11. Smith, C. A.; Shi, C. A.; Chroust, M. K.; Bliska, T. E.; Kelly, M. J. S.; Jacobson, M. P.; Kortemme, T., Design of a Phosphorylatable PDZ Domain with Peptide-‐Specific Affinity Changes. Structure 2013, 21 (1), 54-‐64. 12. Karanicolas, J.; Com, J. E.; Chen, I.; Joachimiak, L. A.; Dym, O.; Peck, S. H.; Albeck, S.; Unger, T.; Hu, W.; Liu, G.; Delbecq, S.; Montelione, G. T.; Spiegel, C. P.; Liu, D. R.; Baker, D., A De Novo Protein Binding Pair By Computational Design and Directed Evolution. Molecular Cell 2011, 42 (2), 250-‐260. 13. Linder, M.; Johansson, A. J.; Olsson, T. S. G.; Liebeschuetz, J.; Brinck, T., Computational design of a Diels-‐Alderase from a thermophilic esterase: the importance of dynamics. Journal of Computer-Aided Molecular Design 2012, 26 (9), 1079-‐1095. 14. Jiang, L.; Althoff, E. A.; Clemente, F. R.; Doyle, L.; Rothlisberger, D.; Zanghellini, A.; Gallaher, J. L.; Betker, J. L.; Tanaka, F.; Barbas, C. F.; Hilvert, D.; Houk, K. N.; Stoddard, B. L.; Baker, D., De novo computational design of retro-‐aldol enzymes. Science 2008, 319 (5868), 1387-‐1391. 15. Rothlisberger, D.; Khersonsky, O.; Wollacott, A. M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J. L.; Althoff, E. A.; Zanghellini, A.; Dym, O.; Albeck, S.; Houk, K. N.; Tawfik,

24
D. S.; Baker, D., Kemp elimination catalysts by computational enzyme design. Nature 2008, 453 (7192), 190-‐U4. 16. Khersonsky, O.; Rothlisberger, D.; Dym, O.; Albeck, S.; Jackson, C. J.; Baker, D.; Tawfik, D. S., Evolutionary Optimization of Computationally Designed Enzymes: Kemp Eliminases of the KE07 Series. Journal of Molecular Biology 2010, 396 (4), 1025-‐1042. 17. Khersonsky, O.; Roethlisberger, D.; Wollacott, A. M.; Murphy, P.; Dym, O.; Albeck, S.; Kiss, G.; Houk, K. N.; Baker, D.; Tawfik, D. S., Optimization of the In-‐Silico-‐Designed Kemp Eliminase KE70 by Computational Design and Directed Evolution. Journal of Molecular Biology 2011, 407 (3), 391-‐412. 18. Althoff, E. A.; Wang, L.; Jiang, L.; Giger, L.; Lassila, J. K.; Wang, Z.; Smith, M.; Hari, S.; Kast, P.; Herschlag, D.; Hilvert, D.; Baker, D., Robust design and optimization of retroaldol enzymes. Protein Science 21 (5), 717-‐726. 19. Walsh, C., Enabling the chemistry of life. Nature 2001, 409, 226-‐231. 20. Ruscio, J. Z.; Kohn, J. E.; Ball, K. A.; Head-‐Gordon, T., The Influence of Protein Dynamics on the Success of Computational Enzyme Design. Journal of the American Chemical Society 2009, 131 (39), 14111-‐14115. 21. Correia, B. E.; Ban, Y.-‐E. A.; Friend, D. J.; Ellingson, K.; Xu, H.; Boni, E.; Bradley-‐Hewitt, T.; Bruhn-‐Johannsen, J. F.; Stamatatos, L.; Strong, R. K.; Schiefl, W. R., Computational Protein Design Using Flexible Backbone Remodeling and Resurfacing: Case Studies in Structure-‐Based Antigen Design. Journal of Molecular Biology 2011, 405 (1), 284-‐297. 22. Lassila, J. K., Conformational diversity and computational enzyme design. Current Opinion in Chemical Biology 2010, 14 (5), 676-‐682. 23. Ramanathan, A.; Agarwal, P. K., Evolutionarily Conserved Linkage between Enzyme Fold, Flexibility, and Catalysis. Plos Biology 2011, 9 (11). 24. Boehr, D. D.; Dyson, H. J.; Wright, P. E., An NMR perspective on enzyme dynamics. Chemical Reviews 2006, 106 (8), 3055-‐3079. 25. Boehr, D. D.; Nussinov, R.; Wright, P. E., The role of dynamic conformational ensembles in biomolecular recognition (vol 5, pg 789, 2009). Nature Chemical Biology 2009, 5 (12), 954-‐954. 26. Boehr, D. D.; McElheny, D.; Dyson, H. J.; Wright, P. E., The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313 (5793), 1638-‐1642.

25
27. Hammes, G. G., Multiple conformational changes in enzyme catalysis. Biochemistry 2002, 41 (26), 8221-‐8228. 28. Kempner, E. S., MOVABLE LOBES AND FLEXIBLE LOOPS IN PROTEINS -‐ STRUCTURAL DEFORMATIONS THAT CONTROL BIOCHEMICAL-‐ACTIVITY. Febs Letters 1993, 326 (1-‐3), 4-‐10. 29. Skliros, A.; Zimmermann, M. T.; Chakraborty, D.; Saraswathi, S.; Katebi, A. R.; Leelananda, S. P.; Kloczkowski, A.; Jernigan, R. L., The importance of slow motions for protein functional loops. Physical Biology 2012, 9 (1). 30. Gerlt, J. A.; Babbitt, P. C., Divergent evolution of enzymatic function: Mechanistically diverse superfamilies and functionally distinct suprafamilies. Annual Review of Biochemistry 2001, 70, 209-‐246. 31. Copley, R. R.; Bork, P., Homology among (beta alpha)(8) barrels: Implications for the evolution of metabolic pathways. Journal of Molecular Biology 2000, 303 (4), 627-‐640. 32. Vega, M. C.; Lorentzen, E.; Linden, A.; Wilmanns, M., Evolutionary markers in the (beta/alpha)8-‐barrel fold. Curr Opin Chem Biol 2003, 7 (6), 694-‐701. 33. Reisinger, B.; Bocola, M.; List, F.; Claren, J.; Rajendran, C.; Sterner, R., A sugar isomerization reaction established on various (beta alpha)(8)-‐barrel scaffolds is based on substrate-‐assisted catalysis. Protein Engineering Design & Selection 2012, 25 (11), 751-‐760. 34. Ochoa-‐Leyva, A.; Barona-‐Gomez, F.; Saab-‐Rincon, G.; Verdel-‐Aranda, K.; Sanchez, F.; Soberon, X., Exploring the Structure-‐Function Loop Adaptability of a (beta/alpha)(8)-‐Barrel Enzyme through Loop Swapping and Hinge Variability. Journal of Molecular Biology 2011, 411 (1), 143-‐157. 35. Malabanan, M. M.; Amyes, T. L.; Richard, J. P., A role for flexible loops in enzyme catalysis. Current Opinion in Structural Biology 2010, 20 (6), 702-‐710. 36. Glembo, T. J.; Farrell, D. W.; Gerek, Z. N.; Thorpe, M. F.; Ozkan, S. B., Collective Dynamics Differentiates Functional Divergence in Protein Evolution. Plos Computational Biology 2012, 8 (3). 37. Rozovsky, S.; McDermott, A. E., The time scale of the catalytic loop motion in triosephosphate isomerase. Journal of Molecular Biology 2001, 310 (1), 259-‐270. 38. Massi, F.; Wang, C.; Palmer, A. G., III, Solution NMR and computer simulation studies of active site loop motion in triosephosphate isomerase. Biochemistry 2006, 45 (36), 10787-‐10794.

26
39. Rozovsky, S.; Jogl, G.; Tong, L.; McDermott, A. E., Solution-‐state NMR investigations of triosephosphate isomerase active site loop motion: Ligand release in relation to active site loop dynamics. Journal of Molecular Biology 2001, 310 (1), 271-‐280. 40. Berlow, R. B.; Igumenova, T. I.; Loria, J. P., Value of a hydrogen bond in triosephosphate isomerase loop motion. Biochemistry 2007, 46 (20), 6001-‐6010. 41. Carpenter, R. A.; Xiong, J.; Robbins, J. M.; Ellis, H. R., Functional Role of a Conserved Arginine Residue Located on a Mobile Loop of Alkanesulfonate Monooxygenase. Biochemistry 2011, 50 (29), 6469-‐6477. 42. Lipchock, J.; Loria, J. P., Millisecond dynamics in the allosteric enzyme imidazole glycerol phosphate synthase (IGPS) from Thermotoga maritima. Journal of Biomolecular Nmr 2009, 45 (1-‐2), 73-‐84. 43. Liang, W.; Ouyang, S.; Shaw, N.; Joachimiak, A.; Zhang, R.; Liu, Z.-‐J., Conversion of D-‐ribulose 5-‐phosphate to D-‐xylulose 5-‐phosphate: new insights from structural and biochemical studies on human RPE. Faseb Journal 25 (2), 497-‐504. 44. List, F.; Sterner, R.; Wilmanns, M., Related (beta alpha)(8)-‐Barrel Proteins in Histidine and Tryptophan Biosynthesis: A Paradigm to Study Enzyme Evolution. Chembiochem 12 (10), 1487-‐1494. 45. Crawford, I. P., Evolution of a Biosynthetic-‐Pathway -‐ the Tryptophan Paradigm. Annual Review of Microbiology 1989, 43, 567-‐600. 46. Murzin, A. G.; Brenner, S. E.; Hubbard, T.; Chothia, C., SCOP -‐ A structural classification fo proteins database for the investigation of sequences and structures. Journal of Molecular Biology 1995, 247 (4), 536-‐540. 47. Leopoldseder, S.; Claren, J.; Jurgens, C.; Sterner, R., Interconverting the catalytic activities of (beta alpha)(8)-‐barrel enzymes from different metabolic pathways: Sequence requirements and molecular analysis. Journal of Molecular Biology 2004, 337 (4), 871-‐879. 48. Lang, D.; Thoma, R.; Henn-‐Sax, M.; Sterner, R.; Wilmanns, M., Structural evidence for evolution of the beta/alpha barrel scaffold by gene duplication and fusion. Science 2000, 289 (5484), 1546-‐1550. 49. Horowitz, N. H., ON THE EVOLUTION OF BIOCHEMICAL SYNTHESES. Proceedings of the National Academy of Sciences of the United States of America 1945, 31 (6), 153-‐157. 50. Claren, J.; Malisi, C.; Hocker, B.; Sterner, R., Establishing wild-‐type levels of catalytic activity on natural and artificial (beta alpha)(8)-‐barrel protein scaffolds.

27
Proceedings of the National Academy of Sciences of the United States of America 2009, 106 (10), 3704-‐3709. 51. Saab-‐Rincon, G.; Olvera, L.; Olvera, M.; Rudino-‐Pinera, E.; Benites, E.; Soberon, X.; Moretti, E., Evolutionary Walk between (beta/alpha)(8) Barrels: Catalytic Migration from Triosephosphate Isomerase to Thiamin Phosphate Synthase. Journal of Molecular Biology 2011, 416 (2), 255-‐270. 52. Evran, S.; Telefoncu, A.; Sterner, R., Directed evolution of (B/a)(8)-‐barrel enzymes: establishing phosphoribosylanthranilate isomerisation activity on the scaffold of the tryptophan synthase -‐subunit. Protein Engineering Design & Selection 2012, 25 (6), 285-‐293. 53. Ochoa-‐Leyva, A.; Soberon, X.; Sanchez, F.; Arguello, M.; Montero-‐Moran, G.; Saab-‐Rincon, G., Protein Design through Systematic Catalytic Loop Exchange in the (beta/alpha)(8) Fold. Journal of Molecular Biology 2009, 387 (4), 949-‐964. 54. Barden, T. C., Indoles: Industrial, agricultural and over-‐the-‐counter uses. Topics in Hetercyclic Chemistry 2011, 26, 31-‐46. 55. Shen, H.; Wang, F.; Zhang, Y.; Huang, Q.; Xu, S.; Hu, H.; Yue, J.; Wang, H., A novel inhibitor of indole-‐3-‐glycerol phosphate synthase with activity against multidrug-‐resistant Mycobacterium tuberculosis. Febs Journal 2009, 276 (1), 144-‐154. 56. Czekster, C. M.; Neto, B. A. D.; Lapis, A. A. M.; Dupont, J.; Santos, D. S.; Basso, L. A., Steady-‐state kinetics of indole-‐3-‐glycerol phosphate synthase from Mycobacterium tuberculosis. Archives of Biochemistry and Biophysics 2009, 486 (1), 19-‐26. 57. Smith, D. A.; Parish, T.; Stoker, N. G.; Bancroft, G. J., Characterization of auxotrophic mutants of Mycobacterium tuberculosis and their potential as vaccine candidates. Infection and Immunity 2001, 69 (2), 1142-‐1150. 58. Sassetti, C. M.; Boyd, D. H.; Rubin, E. J., Genes required for mycobacterial growth defined by high density mutagenesis. Molecular Microbiology 2003, 48 (1), 77-‐84. 59. Siddiqi, M. I.; Kumar, A., Review of knowledge for rational design and identification of anti-‐tubercular compounds. Expert Opinion on Drug Discovery 2009, 4 (10), 1005-‐1015. 60. Lu, J.; Yue, J.; Wu, J.; Luo, R.; Hu, Z.; Li, J.; Bai, Y.; Tang, Z.; Xian, Q.; Zhang, X.; Wang, H., In vitro and in vivo Activities of a New Lead Compound I2906 against Mycobacterium tuberculosis. Pharmacology 2010, 85 (6), 365-‐371. 61. Li, W. F.; Zhou, X. X.; Lu, P., Structural features of thermozymes. Biotechnology Advances 2005, 23 (4), 271-‐281.

28
62. Unsworth, L. D.; van der Oost, J.; Koutsopoulos, S., Hyperthermophilic enzymes -‐ stability, activity and implementation strategies for high temperature applications. Febs Journal 2007, 274 (16), 4044-‐4056. 63. Feller, G., Protein stability and enzyme activity at extreme biological temperatures. Journal of Physics-Condensed Matter 2010, 22 (32). 64. Merz, A.; Yee, M. C.; Szadkowski, H.; Pappenberger, G.; Crameri, A.; Stemmer, W. P. C.; Yanofsky, C.; Kirschner, K., Improving the catalytic activity of a thermophilic enzyme at low temperatures. Biochemistry 2000, 39 (5), 880-‐889. 65. Hennig, M.; Darimont, B.; Sterner, R.; Kirschner, K.; Jansonius, J. N., 2.0 A structure of indole-‐3-‐glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability. Structure 1995, 3 (12), 1295-‐306. 66. Knochel, T. R.; Hennig, M.; Merz, A.; Darimont, B.; Kirschner, K.; Jansonius, J. N., The crystal structure of indole-‐3-‐glycerol phosphate synthase from the hyperthermophilic archaeon Sulfolobus solfataricus in three different crystal forms: effects of ionic strength. J Mol Biol 1996, 262 (4), 502-‐15. 67. Knochel, T.; Pappenberger, A.; Jansonius, J. N.; Kirschner, K., The crystal structure of indoleglycerol-‐phosphate synthase from Thermotoga maritima -‐ Kinetic stabilization by salt bridges. Journal of Biological Chemistry 2002, 277 (10), 8626-‐8634. 68. Bagautdinov, B.; Yutani, K., Structure of indole-‐3-‐glycerol phosphate synthase from Thermus thermophilus HB8: implications for thermal stability. Acta Crystallographica Section D-Biological Crystallography 2011, 67, 1054-‐1064. 69. Gao, L.; Danno, A.; Fujii, S.; Fukuda, W.; Imanaka, T.; Fujiwara, S., Indole-‐3-‐Glycerol-‐Phosphate Synthase Is Recognized by a Cold-‐Inducible Group II Chaperonin in Thermococcus kodakarensis. Applied and Environmental Microbiology 78 (11), 3806-‐3815. 70. Mazumder-‐Shivakumar, D.; Bruice, T. C., Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole-‐3-‐glycerol phosphate synthase. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (40), 14379-‐14384. 71. Wolf-‐Watz, M.; Thai, V.; Henzler-‐Wildman, K.; Hadjipavlou, G.; Eisenmesser, E. Z.; Kern, D., Linkage between dynamics and catalysis in a thermophilic-‐mesophilic enzyme pair. Nature Structural & Molecular Biology 2004, 11 (10), 945-‐949. 72. Vemparala, S.; Mehrotra, S.; Balaram, H., Role of loop dynamics in thermal stability of mesophilic and thermophilic adenylosuccinate synthetase: A molecular

29
dynamics and normal mode analysis study. Biochimica Et Biophysica Acta-Proteins and Proteomics 1814 (5), 630-‐637. 73. Schlee, S.; Dietrich, S.; Kurcon, T.; Delaney, P.; Goodey, N. M.; Sterner, R., Kinetic Mechanism of Indole-‐3-‐glycerol Phosphate Synthase. Biochemistry 2012, 52 (1), 132-‐142. 74. Houlihan, W. J., Indoles. Wiley-‐Interscience: New York, 1972; Vol. 2. 75. Smith, O. H.; Yanofsky, C., 1-‐(ORTHO-‐CARBOXYPHENYLAMINO)-‐1-‐DEOXYRIBULOSE 5-‐PHOSPHATE, A NEW INTERMEDIATE IN THE BIOSYNTHESIS OF TRYPTOPHAN. Journal of Biological Chemistry 1960, 235 (7), 2051-‐2057. 76. Darimont, B.; Stehlin, C.; Szadkowski, H.; Kirschner, K., Mutational analysis of the active site of indoleglycerol phosphate synthase from Escherichia coli. Protein Science 1998, 7 (5), 1221-‐1232. 77. Hennig, M.; Darimont, B. D.; Jansonius, J. N.; Kirschner, K., The catalytic mechanism of indole-‐3-‐glycerol phosphate synthase: crystal structures of complexes of the enzyme from Sulfolobus solfataricus with substrate analogue, substrate, and product. J Mol Biol 2002, 319 (3), 757-‐66. 78. Mazumder-‐Shivakumar, D.; Kahn, K.; Bruice, T. C., Computational study of the ground state of thermophilic indole glycerol phosphate synthase: Structural alterations at the active site with temperature. Journal of the American Chemical Society 2004, 126 (19), 5936-‐5937. 79. Shen, H. B.; Xu, F.; Hu, H. R.; Wang, F. F.; Wu, Q.; Huang, Q.; Wang, H. H., Coevolving residues of (beta/alpha)(8)-‐barrel proteins play roles in stabilizing active site architecture and coordinating protein dynamics. Journal of Structural Biology 2008, 164 (3), 281-‐292. 80. Fodor, A. A.; Aldrich, R. W., Influence of conservation on calculations of amino acid covariance in multiple sequence alignments. Proteins-Structure Function and Bioinformatics 2004, 56 (2), 211-‐221. 81. Dekker, J. P.; Fodor, A.; Aldrich, R. W.; Yellen, G., A perturbation-‐based method for calculating explicit likelihood of evolutionary co-‐variance in multiple sequence alignments. Bioinformatics 2004, 20 (10), 1565-‐1572. 82. Estabrook, R. A.; Luo, J.; Purdy, M. M.; Sharma, V.; Weakliem, P.; Bruice, T. C.; Reich, N. O., Statistical colevolution analysis and molecular dynamics: Identification of amino acid pairs essential for catalysis. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (4), 994-‐999.

30
Chapter 2
The Temperature Dependent Kinetic Mechanism of Thermophilic and
Mesophilic IGPS Enzymes
[This Chapter was adapted from the paper entitled “Differences in the catalytic
mechanism between mesophilic and thermophilic indole-3-glycerol phosphate synthase
enzymes at their adaptive temperatures” by Margot J. Zaccardi, Olga Mannweiler, and
David D. Boehr in Biochemical and Biochemical Research Communications, 2012, 418,
324-329. Olga Mannweiler performed experiments on the IGPS enzyme from E. coli. All
other experiments were performed by Margot J. Zaccardi]1
2.1 Abstract
Thermophilic enzymes tend to be less catalytically-active at lower temperatures
relative to their mesophilic counterparts, despite having very similar crystal structures.
An often cited hypothesis for this general observation is that thermostable enzymes have
evolved a more rigid tertiary structure in order to cope with their more extreme, natural
environment, but they are also less flexible at lower temperatures, leading to their lower
catalytic activity under mesophilic conditions. An alternative hypothesis is that
complementary thermophilic–mesophilic enzyme pairs simply operate through different
evolutionary-optimized catalytic mechanisms. In this Chapter, we present evidence that
while the steps of the catalytic mechanisms for mesophilic and thermophilic indole-3-
glycerol phosphate synthase (IGPS) enzymes are fundamentally similar, the identity of
the rate-determining step changes as a function of temperature. Our findings indicate that

31
while product release is rate-determining at 25 °C for thermophilic IGPS, near its
adaptive temperature (75 °C), a proton transfer event, involving a general acid, becomes
rate-determining. The rate-determining steps for thermophilic and mesophilic IGPS
enzymes are also different at their respective, adaptive temperatures.
2.2 Introduction
Investigations into biological temperature adaptation and enzyme stability at
extreme temperatures can provide a deeper understanding of life under extreme
conditions, and can aid in the design of enzymes for industrial applications and new
biocatalysts with a wide range of temperature optima.2, 3 The use of thermophilic
enzymes for industrial processes provides the opportunity to improve a variety of
common synthesis issues including solubility, reaction time, and product yield.
Additionally, reactions performed at temperatures nearing 100 °C considerably decreases
the risk of bacterial contamination for food and drug related biosyntheses.4
High temperature increases the fluidity of membranes and destroys the normal
activity of biomolecules including proteins. Thermophilic organisms have evolved a
range of mechanisms to combat these problems in order to sustain life at higher
temeperatures.2 To maintain their three-dimensional structures at higher temperatures,
thermophilic enzymes tend to have an increased number of noncovalent interactions, such
as salt bridges and/or disulfide bonds, when compared to their mesophilic homologs.
Despite the similarities in amino acid sequences and three-dimensional structures, warm-
adapted enzymes have lower catalytic activity than their mesophilic counterparts when
assayed at lower temperatures.5, 6 The additional interactions required for stability are

32
thought to limit enzyme flexibility at lower temperatures, leading to a reduction in
enzyme activity.5-7 However, thermostable enzymes can still contain flexible regions that
are important or required for function as was seen by Wolf-Watz et al. in a comparison of
adenylate kinase from E. coli and the hyperthermophile Aquifex aeolicus.8
The purpose of this study is to investigate the differences in the kinetic
mechanism for IGPS enzymes from thermophilic versus mesophilic organisms, and to
determine whether other factors besides protein flexibility must be considered when
analyzing this temperature dependent activity. A more detailed explanation would
involve not only changes in enzyme flexibility, but also variations in the catalytic
mechanism due to adaptations required for structural stability and activity at higher
temperatures. Thermophilic enzymes from the tryptophan biosynthetic pathway,
including IGPS from Thermus thermophilis, T. maritima (tmIGPS), and S. sulfataricus
(ssIGPS) have been used as model systems to decipher the interactions responsible for
protein thermostability.9-12 S. sulfataricus, the thermophile of interest in these studies, is
found in hot sulfur beds,2 and ssIGPS is stable at temperatures above 85 °C.9
Despite a sequence identity of only 30%, ssIGPS and its mesophilic counterpart
from Escherichia coli (ecIGPS) show a strong structural similarity with a root mean
square deviaton (rmsd) of only 1.73 Å (Figure 2.1).9 The high thermostability of ssIGPS
is largely attributed to the increased occurrence of salt bridges (ssIGPS contains thirteen
additional salt bridges compared to ecIGPS) that connect loops and helices, creating a
tight network within the enzyme. The maximum catalytic turnover rate constant (kcat) for
ssIGPS is much lower at 37 °C compared to ecIGPS. The difference in activity was

33
Figure 2.1: Conserved structure and function of IGPS from E. coli (green) (PDB 1P11) and S. sulfobolus (blue) (PDB 1IGS). Despite only 30% sequence identity and large differences in stability, ssIGPS and ecIGPS show strong structural similarity.

34
previously attributed to the lower flexibility of ssIGPS at this temperature.9, 11 Previous
studies suggested that the rate-determining step of ssIGPS at 25 °C is product release.13
However, this temperature is not necessarily relevant for understanding the biological
activity of the thermophilic enzyme. We propose that while the chemical mechanisms for
thermophilic and mesophilic IGPS are similar, the kinetic parameters governing these
processes have different temperature dependencies. In this Chapter, we have examined
the kinetic mechanism for ssIGPS and ecIGPS over a range of temperatures. The results
show that the rate-determining step for ssIGPS is temperature dependent. While product
release is rate-determining at 25 °C, at higher, biologically relevant temperatures (i.e. 75
°C), the ring closure step of the chemical reaction is rate-determining. Additionally, the
rate-determining step for ecIGPS at its adaptive temperature (37 °C) is different from
ssIGPS.
2.3 Experimental Methods
2.3.1 Cloning of ssIGPS and ecIGPS
In E. coli, IGPS is found as the N-terminal domain in a bifunctional enzyme
covalently linked to the previous enzyme in the biosynthetic pathway, PRAI. IGPS in S.
sulfataricus occurs as a monofunctional enzyme. The ecIGPS monofunctional domain
has been shown to retain catalytic ability that is comparable to the bifunctional
complex;14 for the studies presented herein, only the ecIGPS domain was used (amino
acids 1–259). The E. coli codon usage-optimized transcript for both ssIGPS and ecIGPS
(GenScript) were PCR-amplified from the plasmid pSC101-trp (ATCC 31743), and
cloned into pET101 (Ampicillin, AmpR) using the Champion pET Directional TOPO

35
Expression Kit (Invitrogen). Unfortunately, preliminary protein expression trials
indicated a substantial overexpression of the β-lactamase protein. Consequently, the
genes were sub-cloned from pET101 into pET26 (Kanamycin, KanR) with the restriction
endonucleases XbaI and SacI using standard procedures. Due to low overexpression of
ssIGPS using the pET26 construct, the ssIGPS gene in pET21b (AmpR) was also obtained
as a kind gift from Dr. Reinhard Sterner at Universitaet Regensburg (Germany) along
with tmIGPS in the pET21b construct. The Lys110Arg ssIGPS variant was generated
using the QuikChange Lightning kit® (Stratagene) with appropriate primers for the E. coli
optimized pET26 ssIGPS construct. All sequences for wild type (WT) and amino acid
substituted IGPS were verified through DNA sequencing (Nucleic Acid Facility, The
Pennsylvania State University).
2.3.2 Overexpression and Purification of ssIGPS, ecIGPS, and tmIGPS
The overexpression of ssIGPS and ecIGPS was performed by transforming
plasmids (pET26) carrying the genes into E. coli BL21(DE3)star cells. A 10 mL Luria-
Bertani (LB) starter culture was inoculated with fresh transformations on LB-Agar
containing 50 µg/mL Kan and grown overnight at 37 °C. This culture was subsequently
used to inoculate 1L of LB media. The culture was grown at 37 °C to an optical density
(A600) between 0.500 and 0.600 at which time it was induced with isopropyl β-D-1-
thiogalactopyranoside (IPTG) and allowed to grow for approximately 20 hours at 25°C.
Cells were harvested by centrifugation (10,000 x g, 4°C, 20 minutes).
The purification of the enzymes followed protocols similar to those previously
described.9, 15 For ssIGPS, cell pellets were resuspended in 30 mL of 100 mM potassium

36
phosphate pH 7.8, 2 mM ethylenediamine tetraacetic acid (EDTA) and 1 mM
phenylmethanesulfonylfluoride (PMSF), and were lysed through sonication. Cell lysates
were then centrifuged at 30,000 xg and 4 °C for 30 min. The supernatant was heated at 75
°C for ten minutes to precipitate thermolabile host proteins, which were separated out via
centrifugation. The supernatant was then dialysed against 10 mM potassium phosphate,
pH 7.8, 2 mM EDTA (buffer A) and applied to a 20 mL HiPrep 16/10 Q-Sepharose anion
exchange column (GE Healthcare), washed with buffer A and eluted using a phosphate
gradient (0–100 mM phosphate over 20 column volumes, flow rate = 2 mL/min).
Fractions containing ssIGPS were selected using SDS–PAGE (sodium dodecylsulfate-
polyacrylamide electrophoresis), pooled and concentrated to approximately 1 mL using
Vivaspin-20 spin concentrators (Sartorius Stedium Biotech). Partially purified ssIGPS
was applied to a HiPrep 16/60 Sepharcryl S100 gel filtration column (GE Healthcare) and
eluted using buffer A containing 200 mM NaCl. Fractions containing ssIGPS were again
selected via SDS-PAGE, pooled, concentrated and then dialyzed against buffer A. The
protocol for ecIGPS was similar except that the cell pellet was reconstituted directly into
the Q-sepharose buffer A (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES) pH 7.5, 1 mM EDTA) and eluted on the anion exchange column with a 0 to
500M gradient of NaCl.
The pET21b ssIGPS construct is hexa-histidine tagged (His-tagged); the
overexpression and purification differed from that described for the pET26 construct.16
The growth was performed with BL21-CodonPlus(DE3)-RIPL (Agilent) E. coli cells in
LB media containing 100 µg/mL Amp following the same protocol as described for the
pET26 ssIGPS construct. These cells are advantageous because they are optimized for the

37
overexpression of heterologous proteins in E. coli. The cell pellet was reconstituted in 30
mL 100mM potassium phosphate pH 7.0, 300 mM potassium chloride (KCl) (lysis
buffer), and 1 mM PMSF, and were lysed through sonification. Cell lysates were then
centrifuged at 30,000 xg and 4 °C for 30 min, and the supertenant was heated at 75 °C for
ten minutes to precipitate thermolabile contaminants, which were removed via
centrifugation. The subsequent supernatant was applied to a column containing 2mL Ni-
NTA (HisPur-ThermoScientific). The column was then washed with lysis buffer plus 10,
20, 50, 100, and 250 mM Imidazole to elute bound protein. Fractions were selected by
SDS-PAGE, pooled, and concentrated as described.
The overexpression and purification protocols for tmIGPS, which is also in the
pET21b construct with a hexa-histidine tag, were identical to that for hexa-his tagged
ssIGPS except that no PMSF was added to the lysis buffer as described previously by
Schneider and coworkers.16
2.3.3 Steady-State Kinetic Assays for IGPS
ssIGPS activity was measured as previously described via fluorescence by
monitoring the formation of IGP.1, 14, 17 The sample was excited at 278 nm and emission
measured at 340 nm using a spectrofluorometer (Horiba Jobin Yvon). Assays were
performed in 50 mM 3-[4-(2-hydroxyethyl)-1-piperazinyl] propanesulfonic acid (HEPPS)
pH 7.5, 4 mM EDTA unless otherwise noted with an enzyme concentration of 10 nM and
a volume of 300 µL with substrate concentrations ranging from 30 nM to 4 µM. Raw
rates in cps/s were converted to nM/s using a standard curve for cps versus concentration
of CdRP (Figure 2.2). This standard curve was obtained using tmIGPS since that enzyme

38
Figure 2.2: Standard curve of fluorescence units per nanomolar for converting cps/s to nM/s. The slope of the line (4036 cps/nM) was used to convert data for ssIGPS to the appropriate units. Curve was attained using IGPS from T. maritima, which does not display product inhibition.

39
does not exhibit product inhibition.16 Assay times ranged from 30 seconds to 60 seconds.
ecIGPS activity was measured by monitoring the formation of IGP via absorbance
at 278 nm on a SpectraMax M2 plate reader (Molecular Devices). Assays were
performed in 50 mM HEPPS pH 7.5, 4 mM EDTA using a 96-well microtiter plate assay.
Assay volume was 250 µL, substrate concentrations ranged from 100 nM to 60 µM, and
assays were typically conducted for three to four minutes. Initial rates were fit to the
Michaelis–Menten equation (Equation 2.1) using nonlinear regression with
Kaleidograph,
€
v =kcat[E]T [S]KM + [S] (2.1)
where ν is the initial velocity, ET is the total enzyme concentration, and [S] is the
concentration of substrate, CdRP. All assays were performed in triplicate. Representative
progress curves and Michaelis-Menton curves for ssIGPS are shown in Figure 2.3.
2.3.4 Solvent Viscosity Effects, Solvent Deuterium Kinetic Isotope Effects, and pH
Effects
The solvent viscosity effects (SVE), solvent deuterium kinetic isotope effects
(SDKIE) and pH effects for IGPS enzymes were determined by varying the buffer
conditions of the standard assay. SVEs were determined using enzyme assays in 50 mM
HEPPS pH 7.5, 4 mM EDTA buffer with 0 to 30 % (w/v) glycerol. SVE experiments
were also performed in an alternate viscogen, sucrose, as well as in a microviscogen,
PEG 8000, as controls for nonspecific viscogen effects. Assays were performed at
saturating substrate concentrations (800 µM for ssIGPS; 12 µM for ecIGPS). The relative
viscosities of the buffer solutions were measured using an Ostwald viscometer. The SVE

40
Figure 2.3: Representative data for ssIGPS assays. (a) Progress curves for ssIGPS at 75 °C at varying concentrations of CdRP (100, 400, 800, 1000, and 2000 nM). (b) Michaelis-Menton curve for ssIGPS at 75 °C.

41
is defined as the slope of the line for the plot of (ratewithout viscogen/ratewith viscogen) versus
relative viscosity.
SDKIEs were obtained by comparing IGPS enzyme activities in H2O and D2O.
The pD was used instead of pH for solutions in D2O and was adjusted according to pD =
pH + 0.4. Proton inventory studies were performed by varying the mole fraction of D2O
in the buffer from 0 to 1.
pH studies for ssIGPS (37 and 75 °C) and ecIGPS (37 °C) were performed in
buffers with overlapping buffering ranges, including: 100 mM 2-(N-morpholino) ethane
sulfonic acid (MES) (pH 5.0-6.5), 100 mM HEPES (pH 6.5-8.0), N,N-Bis(2-
hydroxyethyl) glycine (BICINE) (pH 8.0-8.5), and 100 mM N-cyclohexyl-2-
aminoethanesulfonic acid (CHES) (pH 8.6-10.0). The pH rate profiles for both ecIGPS
and ssIGPS display two ionizations, and were fit to Eq. (2) using Kaleidograph:
(2.2)
where ν is the estimated kcat, C is the pH-independent rate value, and pKa1 and pKa2 are
the pKa values associated with the ascending and descending limbs of the pH profile,
respectively.
2.3.5 Synthesis of CdRP
Originally, the protocol described by Czekster et al.18 was used to synthesize the
solid barium salt of the substrate, CdRP. However, preliminary assays showed that this
form of the substrate was not active in assays measuring for IGPS activity and a new
protocol was obtained.15
!
" = C /(1+10pKa1# pH +10pH# pKa 2 )

42
In short, 47.7 mg of anthranilic acid was dissolved in 200 µL of ethanol and 58.8
mg of ribose-5-phosphate was dissolved in 200 µL of water. The two solutions were
combined and allowed to stand at room temperature in the dark for twenty hours. After
incubation, 4 mL of water was added to the reaction mixture and allowed to stand in the
dark for one hour. The product was washed with 5 mL ethyl acetate, allowing separation
of the aqueous and solvent layers in the separatory funnel. The aqueous phase containing
the CdRP was separated and the wash with ethyl acetate was repeated approximately five
to seven times until the solvent phase was colorless. The product was then purged with
nitrogen to remove any remaining ethyl acetate and stored in the dark at -80°C.
The concentration of CdRP was calculated using the tmGPS enzyme to convert
CdRP to IGP and measuring using absorbance both the decrease in CdRP at 327 nm
(ε=3.43 mM-1cm-1) and the increase in IGP at 278 nm (ε=4.48 mM-1cm-1).
2.3.6 Circular Dichroism
Circular dicroism (CD) experiments were performed on WT and Lys110Arg
ssIGPS on a Jasco J-810 Spectropolarimeter from 250 nm to 190 nm with 1 nm intervals
and a 1 nm bandwidth. The experiments were performed in 10 mM potassium phosphate
pH 7.0 with an enzyme concentration of 1.7 µM.

43
2.4 Results
2.4.1 Steady-state Kinetic of ssIGPS
The kinetic mechanism for IGPS can be described as follows:19
€
E + S⇔k−1
k1ES⇔
k−2
k2ES *⇔
k−3
k3EI1→
k4EI2⇔
k−5
k5EP *⇔
k−6
k6EP⇔
k−7
k7E + P
(2.3)
In this mechanism, the substrate (S = CdRP) binds to the enzyme to form the enzyme-
substrate complex (ES), which may then undergo a conformational change to form the
more active ES* complex, followed by irreversible chemistry involving the release of
CO2 gas to yield the enzyme-product (P = IGP) complex. This step likely reflects the
chemistry (condensation, decarboxylation, dehydration) proposed for IGPS, which occurs
through two intermediates, I1 and I2. The product complex then must undergo another
conformational change to allow product release in the final step.
To comprehensively compare the catalytic activities of ssIGPS and ecIGPS, the
steady-state kinetic parameters of the IGPS enzymes were determined across multiple
temperatures (25, 37, and 75 °C), ensuring that biologically relevant data was obtained
for both enzymes (Table 2.1). Consistent with previous observations, the maximum
turnover rate (kcat) for ecIGPS was 20-fold greater than that for ssIGPS at both 25°C and
37 °C, although the enzymes were similar in their catalytic efficiencies (kcat/KM).11 The
binding affinity of the substrate for ssIGPS is temperature independent, with similar KM
values at all temperatures assayed (~50 to 100 nM). Conversely, kcat decreases as
temperature decreases (0.67 s-1 at 75 °C versus 0.16 s-1 at 25 °C). The previously
proposed rate-determining step of ssIGPS at 25 °C is the release of product from the
enzyme,19 but the rate-determining step at more higher temperatures is not known.

44
Table 2.1: Steady-state kinetic parameters for ssIGPS and ecIGPS at pH 7.5 indicate that the rate-determining step changes as a function of temperature. Sample Temp
(°C) kcat (s-1) KM (nM) kcat/KM (x
106 M-1s -1) SVE SDKIE
ssIGPS 25 0.16 ± 0.02
74 ± 38 2.2 1.0 ± 0.2a 1.2 ± 0.2a
37 0.42 ± 0.04
88 ± 47 4.8 0.6 ± 0.3a 5.8 ± 0.1a
75 0.67 ± 0.03
44 ± 9 15 -0.2 ± 0.1a 3.6 ± 0.3a
ecIGPSc 25 4.1 ± 0.2a n.d.b n.d.b n.d.b n.d.b
37 9.3 ±0.6 1600 ± 300
5.7 0.2 ± 0.1a 1.0 ± 0.1a
aValues for kcat were determined using saturating substrate concentrations (800 nM for ssIGPS and 12 µM for ecIGPS) bValues not determined. cAssays on ecIGPS were performed by Olga Mannweiler

45
2.4.2 Solvent Viscosity Effects, Solvent Deuterium Kinetic Isotope Effects, and pH
Effects
There are various limiting scenarios for the kinetic mechanism of ssIGPS that will
determine the value of kcat : if product release is relatively fast compared to the chemical
step(s) (i.e. k3 << k5), then kcat will report on k3. Conversely, if product release is rate-
determining (i.e. k3 >> k5), then kcat will approach k5. These scenarios can be resolved
based on the sensitivity of the turnover rate to increasing solvent viscogen and the
introduction of deuterated solvent. Diffusion-limited processes including substrate
binding, product release, or large, global conformational changes will be dependent on
the viscosity of the solution, whereas processes independent of diffusion, like the
chemical steps, will be unaffected. Conversely, chemical processes whose rates are
dependent on the transfer of a solvent exchangeable proton will be affected by the
introduction of deuterated solvent as in the SDKIE experiments (Figure 2.2). In the IGPS
enzyme, the ring closure step is predicted to be isotope sensitive, due to the proton
donation from the general acid. Conversely, the dehydration the dehydration step is not
expected to produce a substantial SDKIE. Within the dehydration step, it is likely that the
removal of the non-exchangeable alkyl hydrogen is rate-determining over the loss of
water, causing the step to be isotope insensitive.
Solvent viscosity effects (SVEs) are defined by the slope of a plot of relative rate
(ratewithout viscogen/ratewith viscogen) versus relative viscosity, for which the theoretical
maximum effect is one. SVEs were determined for ecIGPS and ssIGPS enzymes across
multiple temperatures (Table 2.1, Figure 2.3). For ecIGPS, there is only a small SVE
(~0.20) at 37 °C. In contrast, there was a much larger SVE for ssIGPS at both 25 °C and

46
Figure 2.4: The rate-determining step of the IGPS reaction can be deciphered using SVE and SDKIE experiments. Substrate binding and product release (green) are viscosity sensitive and isotope insensitive. Ring closure (blue) is viscosity insensitive isotope sensitive. Decarboxylation and dehydration are both viscosity and isotope insensitive.

47
Figure 2.5: Solvent viscosity effects for ssIGPS. At 25 °C (blue) there is an SVE of 1.0 ± 0.2, wherease at 75 °C (black) the SVE is no longer present (-0.2 ± 0.1). The SVE is defined by the slop of the line for vo/vi versus ni/no. The results indicate that at 25 °C product release is rate-determining but as temperature increases to 75 °C product release is no longer rate-determining, and a chemical step becomes rate-determining.

48
37 °C, consistent with previous data suggesting that product release is rate-determining
for ssIGPS at these temperatures. However, there were not appreciable SVEs for ssIGPS
at higher temperatures (75 °C), reflecting a change in the identity of the rate-determining
step with increasing temperatures. The similar SVEs for ecIGPS and ssIGPS at their
respective, adaptive temperatures may indicate similar rate-determining step(s).
To test for any nonspecific effects of due to the introduction the microviscogen
used in these experiments (glycerol), the SVE experiments were also performed in
sucrose. The two different viscogens showed similar effects. At 37 °C, an SVE of 0.6 was
measured in glycerol for ssIGPS, and one of 0.4 was measured in sucrose. Similarly at 75
°C, an SVE of -0.2 was measured in both viscogens. This result indicates that the
viscosity effect is reporting on the diffusion controlled processes of IGPS catalysis.
Additionally, the experiments were performed in the presence of a macroviscogen, PEG
8000, in which the enzyme does not exhibit a viscosity effect (i.e. kcat(PEG 8000)/kcat(no
viscogen) was equal to 1.0 ± 0.2 for 1.9% (w/v) PEG 8000 at a relative viscosity of 1.7).
Differences in enzyme activities for ecIGPS and ssIGPS may be due to variations
in their catalytic mechanisms. The proposed mechanism for ssIGPS suggests the
involvement of both a general acid and a general base; if the chemical mechanism is the
same for ecIGPS and ssIGPS, a similar pH rate profile should be observed. The pH
dependence of the activity for ecIGPS and ssIGPS was determined (Figure 2.4, Table
2.2). The pH rate profiles for ecIGPS and ssIGPS at biologically relevant temperatures
(37 °C for ecIGPS and 75 °C for ssIGPS) exhibit a bell shaped curve with both ascending
and descending limbs, consistent with the involvement of general base and acid
chemistry, respectively. These results reflect the pH dependence of kcat. The pH rate
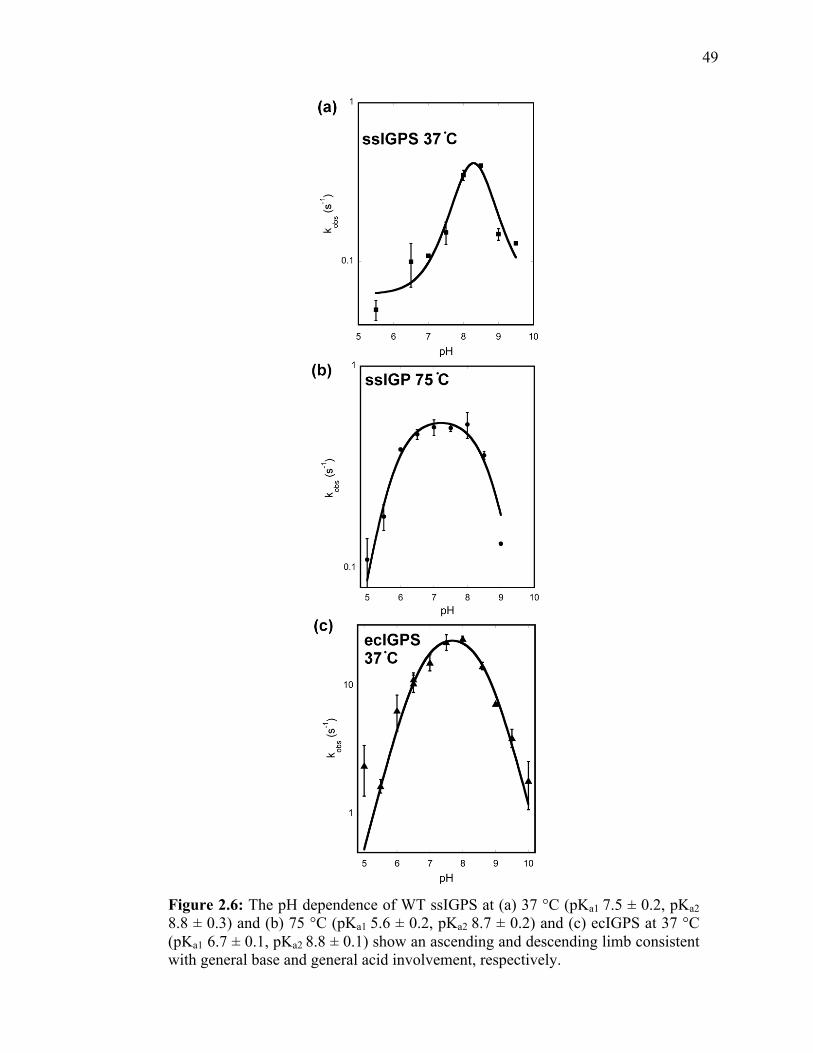
49
Figure 2.6: The pH dependence of WT ssIGPS at (a) 37 °C (pKa1 7.5 ± 0.2, pKa2 8.8 ± 0.3) and (b) 75 °C (pKa1 5.6 ± 0.2, pKa2 8.7 ± 0.2) and (c) ecIGPS at 37 °C (pKa1 6.7 ± 0.1, pKa2 8.8 ± 0.1) show an ascending and descending limb consistent with general base and general acid involvement, respectively.

50
Table 2.2: pKa values for ssIGPS and ecIGPS. Enzyme Temperature pKa1 pKa2
ecIGPS 37 °C 6.7 ± 0.1 8.8 ± 0.3
ssIGPS 37 °C 7.5 ± 0.2 8.8 ± 0.3
ssIGPS 75 °C 5.6 ± 0.2 8.7 ± 0.2

51
profile of kcat/KM for ssIGPS at 75 °C also yielded a bell-shaped curve with similar pKa
values (5.0 ± 0.4, 9.3 ± 0.5). It should be noted that for IGPS from M. tuberculosis
(mtIGPS), only the ascending limb in the pH rate profile is observed with a pKa of 6.8,18
which is similar to the pKa for ecIGPS (6.70 ± 0.08). At 75 °C, the pKa value (5.6 ± 0.2)
for the ascending limb of ssIGPS is 1 pH unit lower compared to the mtIGPS and ecIGPS
enzymes. The descending limb for the pH rate profiles yielded similar pKas for ecIGPS
(8.7 ± 0.1) and ssIGPS (37 °C, 8.8 ± 0.3; 75 °C, 8.7 ± 0.2).
The pKa for the descending limb of both ecIGPS and ssIGPS is in the range
expected for a Lys residue, such as the proposed general acid, Lys110. To further test the
importance of Lys110, the Lys110Arg substitution was assayed. An Arg at this position
is expected to make similar noncovalent interactions (e.g. hydrogen bond, electrostatic
interactions) with the CdRP, but would act as a much less effective general acid with a
higher pKa (~12). Consistent with this suggestion, the Lys110Arg variant of ssIGPS had
very low enzyme activity (kobs < 1.5 x 10-5 s-1 at 75 °C) compared to WT ssIGPS (kcat =
0.67 ± 0.03 s-1 at 75 °C). Circular dichroism experiments were performed on both WT
and Lys110Arg ssIGPS to ensure that the variant was properly folded and the loss in
activity was not due to gross changes in secondary structure.
The IGPS-catalyzed reaction is proposed to proceed through three chemical steps.
SDKIEs offer a way to determine which of the chemical steps may be rate-determining
for the IGPS enzymes. For ssIGPS at 25 °C, there was not a significant SDKIE for kcat
(Table 2.1). This finding is consistent with the large SVEs and previous studies
suggesting that product release is fully rate-determining at this temperature.13, 19
However, at higher temperatures there was a substantial SDKIE for kcat (kH2O/kD2O = 3.6

52
± 0.3 for ssIGPS at 75 °C). In contrast, ecIGPS does not have a substantial SDKIE for kcat
at 37 °C, suggesting that an isotope insensitive chemical step (and/or a viscosity-
independent conformational change) must be rate-determining. Consistent with the
ecIGPS results, mtIGPS also displayed a substantially lower SDKIE for kcat (~1.6)18
compared to ssIGPS. Parry’s proposed chemical mechanism (Figure 1.2) and the pH rate
profiles (Figure 2.4) for the IGPS enzymes suggest multiple proton transfer events that
may be responsible for the SDKIE observed for ssIGPS. To identify the responsible
proton transfer event(s) that are being reported on by the SDKIE experiment, proton
inventory studies and the pH dependence of the SDKIE were determined. At both 37 and
75 °C, the proton inventory study yielded a linear relationship between the relative rate
and mole fraction of D2O (Figure 2.5), indicating that a single proton transfer event is
responsible for the observed SDKIE for kcat. Additionally, for ssIGPS at pH 8.5, there
was not a substantial SDKIE for kcat (0.99 ± 0.07) at 37 °C, whereas at pH 6.5, an SDKIE
is still observed (2.18 ± 0.06), indicating that the SDKIE likely arises from the general
acid, Lys110.

53
Figure 2.7: The rate-determining step for ssIGPS at higher temperatures involves a single proton transfer event. (a) The maximum catalytic turnover of ki/ko versus mole fraction D2O:H2O at both 37 °C (blue) and 75 °C (green) show a linear fit. (b) The square root of ki/ko versus mole fraction D2O:H2O at 37 °C and 75 °C show a quadratic fit. These results indicate that one proton transfer event is involved in the rate-determining step of the reaction, namely the proton transfer from the general acid in the condensation step of the reaction.

54
2.5 Discussion
2.5.1 Temperature Dependent Kinetic Mechanism of ssIGPS
Thermophilic enzymes show lower activity at lower temperatures compared to
higher temperatures and as compared to their mesophilic counterparts. This finding is
often attributed to changes in the flexibility of the enzyme at lower temperatures. In the
case of ssIGPS, while flexibility may have a role in the activity of the enzyme, the results
also indicate that changes in the rate-determining step of the reaction are largely
responsible for the apparent changes in activity. At its biologically relevant temperature
(75 °C), ssIGPS does not exhibit an SVE. The substantial SDKIE indicates that chemistry
is rate-determining. These results, along with the proton inventory study and the studies
on Lys110Arg, suggest that a single proton transfer event involving the general acid,
Lys110, is rate-determining for ssIGPS at higher temperatures. This result suggests that
the identity of the rate-determining step of the ssIGPS reaction at biologically relevant
temperatures is the initial ring closure step. As temperature decreases, the rate-
determining step changes from chemistry to product release, as evidenced by the
presence of a SVE, and which is consistent with previously published work.11
At 37 °C, both an SVE and SDKIE are present, indicating that multiple steps are
contributing to the kinetic parameters at this temperature; product release and chemistry
are both partially rate-determining at this temperature. The pH rate profile at 37 °C still
shows both ascending and descending limbs that are indicative of acid base catalysis, but
the trend is more complex, particularly for the ascending limb. In mtIGPS, the ascending
limb was previously attributed to the general base (Glu159 according to ssIGPS
numbering). In ssIGPS, at 37 °C, the poor fit to the curve in this region (Figure 2.3) may

55
be explained by allowing more than one responsible ionizable group, which would be
consistent with multiple kinetic steps contributing to the overall rate. This finding
suggests that a different ionizable group may be responsible for this pKa at different
temperatures, and/or different microenvironments within the IGPS active site may have
different effects on similar ionizable group(s). The lower pKa observed for ssIGPS at 75
°C is closer to the range expected for a Glu or Asp residue. This is consistent with the
proposed mechanism in which Glu159 or Glu210 acts as the general base in the
dehydration step.
2.5.2 Differences in the Rate-Determining Step of Thermophilic ssIGPS and
Mesophilic ecIGPS
While IGPS from both thermophilic and mesophilic organisms is believed to
undergo the same chemical steps toward the product, the enzyme takes may have evolved
differently due to the environmental restrictions for each organism. These changes can
manifest as changes in the kinetic mechanism and the rate-determining step of the
reaction. In the ecIGPS enzyme, only a small SVE and no SDKIE is seen at its
biologically active temperature, 37 °C, suggesting that a different chemical step such as
dehydration which does not involve a proton transfer, or a viscosity-independent
conformational change, is largely rate-determining compared to ssIGPS. This result also
indicates differences in the rate-determining step for mesophilic and thermophilic IGPS
enzymes at their respective, adaptive temperatures.
The active site of IGPS enzymes are well shielded from solvent;9, 20 therefore,
conformational changes must accompany product release. At lower temperatures, these

56
conformational changes are slow and product release is rate-determining for ssIGPS, and
at higher temperatures, the protein motions are sufficiently fast to relieve this bottleneck
to ssIGPS catalysis. However, even at their respective, adaptive temperatures, the rate-
determining steps for ssIGPS and ecIGPS differ. It is unlikely that the fundamental
chemical mechanisms are substantially different considering the amino acid sequence and
structural similarity of the IGPS enzymes.
2.6 Conclusions
In this chapter, the kinetic mechanism of IGPS was examined in both
thermophilic and mesophilic enzymes. The results suggest that IGPS enzymes have
evolved to more efficiently catalyze different steps of the chemical reaction(s), likely due
to the different environmental stressors present. Engineering thermostable enzymes or
new enzyme activities on thermophilic proteins like ssIGPS21, 22 will require careful
consideration of not only protein stability-flexibility relationships, but also a thorough
understanding of how different physical and chemical barriers to catalysis respond to
temperature.
2.7 References
1. Zaccardi, M. J.; Mannweiler, O.; Boehr, D. D., Differences in the catalytic mechanisms of mesophilic and thermophilic indole-‐3-‐glycerol phosphate synthase enzymes at their adaptive temperatures. BBRC 2012, 418 (2), 324-‐329. 2. Rothschild, L. J.; Mancinelli, R. L., Life in extreme environments. Nature 2001, 409 (6823), 1092-‐1101. 3. Egorova, K.; Antranikian, G., Industrial relevance of thermophilic Archaea. Current Opinion in Microbiology 2005, 8 (6), 649-‐655.

57
4. Unsworth, L. D.; van der Oost, J.; Koutsopoulos, S., Hyperthermophilic enzymes -‐ stability, activity and implementation strategies for high temperature applications. Febs Journal 2007, 274 (16), 4044-‐4056. 5. Feller, G., Protein stability and enzyme activity at extreme biological temperatures. Journal of Physics-Condensed Matter 2010, 22 (32). 6. Sterpone, F.; Melchionna, S., Thermophilic proteins: insight and perspective from in silico experiments. Chemical Society Reviews 41 (5), 1665-‐1676. 7. Li, W. F.; Zhou, X. X.; Lu, P., Structural features of thermozymes. Biotechnology Advances 2005, 23 (4), 271-‐281. 8. Wolf-‐Watz, M.; Thai, V.; Henzler-‐Wildman, K.; Hadjipavlou, G.; Eisenmesser, E. Z.; Kern, D., Linkage between dynamics and catalysis in a thermophilic-‐mesophilic enzyme pair. Nature Structural & Molecular Biology 2004, 11 (10), 945-‐949. 9. Hennig, M.; Darimont, B.; Sterner, R.; Kirschner, K.; Jansonius, J. N., 2.0 A structure of indole-‐3-‐glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability. Structure 1995, 3 (12), 1295-‐306. 10. Knochel, T. R.; Hennig, M.; Merz, A.; Darimont, B.; Kirschner, K.; Jansonius, J. N., The crystal structure of indole-‐3-‐glycerol phosphate synthase from the hyperthermophilic archaeon Sulfolobus solfataricus in three different crystal forms: effects of ionic strength. J Mol Biol 1996, 262 (4), 502-‐15. 11. Merz, A.; Yee, M. C.; Szadkowski, H.; Pappenberger, G.; Crameri, A.; Stemmer, W. P. C.; Yanofsky, C.; Kirschner, K., Improving the catalytic activity of a thermophilic enzyme at low temperatures. Biochemistry 2000, 39 (5), 880-‐889. 12. Bagautdinov, B.; Yutani, K., Structure of indole-‐3-‐glycerol phosphate synthase from Thermus thermophilus HB8: implications for thermal stability. Acta Crystallographica Section D-Biological Crystallography 2011, 67, 1054-‐1064. 13. Merz, A.; Knochel, T.; Jansonius, J. N.; Kirschner, K., The hyperthermostable indoleglycerol phosphate synthase from Thermotoga maritima is destabilized by mutational disruption of two solvent-‐exposed salt bridges. Journal of Molecular Biology 1999, 288 (4), 753-‐763. 14. Eberhard, M.; Tsai-‐Pflugfelder, M.; Bolewska, K.; Hommel, U.; Kirschner, K., Indoleglycerol phosphate synthase-‐phosphoribosyl anthranilate isomerase: comparison of the bifunctional enzyme from Escherichia coli with engineered monofunctional domains. Biochemistry 1995, 34 (16), 5419-‐28.

58
15. Kirschner, K.; Szadkowski, H.; Jardetzky, T. S.; Hager, V., Phosphoribosylanthranilate Isomerase-‐Indoleglycerol-‐Phosphate Synthase from Escherichia-‐Coli. Methods in Enzymology 1987, 142, 386-‐397. 16. Schneider, B.; Knochel, T.; Darimont, B.; Hennig, M.; Dietrich, S.; Babinger, K.; Kirschner, K.; Sterner, R., Role of the N-‐terminal extension of the (betaalpha)8-‐barrel enzyme indole-‐3-‐glycerol phosphate synthase for its fold, stability, and catalytic activity. Biochemistry 2005, 44 (50), 16405-‐12. 17. Hommel, U.; Eberhard, M.; Kirschner, K., Phosphoribosyl anthranilate isomerase catalyzes a reversible amadori reaction. Biochemistry 1995, 34 (16), 5429-‐39. 18. Czekster, C. M.; Neto, B. A. D.; Lapis, A. A. M.; Dupont, J.; Santos, D. S.; Basso, L. A., Steady-‐state kinetics of indole-‐3-‐glycerol phosphate synthase from Mycobacterium tuberculosis. Archives of Biochemistry and Biophysics 2009, 486 (1), 19-‐26. 19. Schlee, S.; Dietrich, S.; Kurcon, T.; Delaney, P.; Goodey, N. M.; Sterner, R., Kinetic Mechanism of Indole-‐3-‐glycerol Phosphate Synthase. Biochemistry 2012, 52 (1), 132-‐142. 20. Wilmanns, M.; Priestle, J. P.; Niermann, T.; Jansonius, J. N., Three-‐dimensional structure of the bifunctional enzyme phosphoribosylanthranilate isomerase: indoleglycerolphosphate synthase from Escherichia coli refined at 2.0 A resolution. J Mol Biol 1992, 223 (2), 477-‐507. 21. Rothlisberger, D.; Khersonsky, O.; Wollacott, A. M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J. L.; Althoff, E. A.; Zanghellini, A.; Dym, O.; Albeck, S.; Houk, K. N.; Tawfik, D. S.; Baker, D., Kemp elimination catalysts by computational enzyme design. Nature 2008, 453 (7192), 190-‐U4. 22. Jiang, L.; Althoff, E. A.; Clemente, F. R.; Doyle, L.; Rothlisberger, D.; Zanghellini, A.; Gallaher, J. L.; Betker, J. L.; Tanaka, F.; Barbas, C. F.; Hilvert, D.; Houk, K. N.; Stoddard, B. L.; Baker, D., De novo computational design of retro-‐aldol enzymes. Science 2008, 319 (5868), 1387-‐1391.

59
Chapter 3
Functional Identification of the General Acid and Base in the
Dehydration Step of IGPS Catalysis
[This Chapter was adapted from the paper entitled “Functional identification
of the general acid and base in the dehydration step of indole-3-glycerol
phosphate synthase catalysis” by Margot J. Zaccardi, Eric M. Yezdimer, and
David D. Boehr in The Journal of Biological Chemistry 2013.]
3.1 Abstract
The chemical mechanism for IGPS was proposed by Parry to proceed through two
intermediates in a series of condensation, decarboxylation, and dehydration steps. X-ray
crystal structures have suggested that Lys110 behaves as a general acid both in the
condensation and dehydration steps, but did not reveal an efficient pathway for the
reprotonation of this critical residue. Amino acid substitutions and kinetic experiments
presented in this chapter suggest an alternative mechanism whereby Lys110 acts as a
general acid in the condensation step, but another invariant residue, Lys53, acts as the
general acid in the dehydration step. These studies also indicate that the conserved
residue Glu51 acts as the general base in the dehydration step. The revised mechanism
effectively divides the active site into discrete regions where the catalytic surfaces
containing Lys110 and Lys53/Glu51 catalyze the ring closure (i.e. condensation and
decarboxylation) and dehydration steps, respectively.

60
3.2 Introduction
The indole ring is a prevalent structure in biological systems and is found in
molecules that are relevant for many different industries including pharmaceuticals,
agriculture, and materials.1 Therefore, the synthesis of indoles remains a widely studied
area of research.2 New improvements for indole syntheses can be found through a better
understanding of the IGPS enzyme. The IGPS catalyzed reaction was previously
proposed to occur through two intermediates (I1, I2) in a series of condensation,
decarboxylation and dehydration steps (Figure 3.1).3, 4 Initiation of the enzyme-catalyzed
reaction is thought to begin with the protonation of the ketone in CdRP through general
acid catalysis by Lys110 (numbering according to ssIGPS), an absolutely conserved and
essential amino acid residue,4-6 which allows for electrophilic attack from the benzyl ring
that can drive the reaction forward to create the I1 intermediate; tandem mass
spectrometry has identified fragment patterms that were attributed to this intermediate.7
Following decarboxylation and the formation of the I2 intermediate, dehydration is
facilitated by a general acid and base (predicted to be Lys110 and Glu159, respectively),
to form the final product, IGP. Functional and structural studies are mostly consistent
with the currently accepted mechanism, but several unresolved issues remain, incuding
the proposition that Lys110 acts as the general acid in both the ring closure and
dehydration steps of the reaction. Not only is it atypical for an enzyme to use the same
residue for two separate proton donation steps, it also requires that Lys110 be
reprotonated in the middle of the catalytic reaction. An alternate hypothesis is that
another residue, such as Lys53, acts as the general acid in one of these steps.

61
Figure 3.1: Proposed mechanism for catalysis by IGPS suggests that the reaction proceeds in three steps: condensation, decarboxlation, and dehydration, with two intermediates. The proposed general acid and base are Lys110 and Glu159. Problems with this mechanism include the need to reprotonate Lys110 between the first and third steps and the direct removal of an alkyl hydrogen from the pyrrole ring.

62
Crystal structures of ssIGPS in complex with a reduced CdRP (rCdRP) analog
(i.e. a C2’ hydroxyl in place of the C2’ ketone) imply that the subtrate binds with the
enzyme in an extended, unproductive conformation, where the C1 and C2’ sites are
separated by about 4.5 Å, a distance too large for C-C bond formation.4 This finding
implies that CdRP must go through significant internal reorientation within the active site
after its activation by a general acid in order for condensation to occur. Interestingly, the
crystal structure of the ssIGPS:IGP binary complex shows that the anthranilate moiety is
docked into an adjacent hydrophobic pocket compared to crystal structures of the
IGPS:rCdRP complex, while the triosephosphate group remains secured in the same
position throughout the reaction. This result leads to the possibility that reorientation of
the substrate or intermediate may occur during catalysis. However, how or why this
reorientation occurs has not been resolved. A better understanding of this process would
be of utility in the industrial, biosynthetic production of new indole, the engineering of
new reactivity onto the IGPS active site, and the design of the most effective small
molecule inhibitors of IGPS for new antimicrobials.
One interaction that may be important in these conformational rearrangements of
the substrate is the salt bridge between Lys53 and the carboxylate moiety of the
anthranilate ring of CdRP; the enzyme is not able to catalyze the reaction with a substrate
analog lacking this carboxyl group.8 Lys53 is located on the highly dynamic β1α1 loop,
and Bruice and co-workers have proposed that the increased flexibility of this loop at
higher temperatures allows the substrate to form a more productive NAC for I1
formation.9, 10 The interaction between Lys53 and the carboxylate group may also be
involved in activating the C1 carbon for nucleophilic attack. The importance of Lys53

63
was further demonstrated in activity assays of ssIGPS variants with substitutions at
Lys53 in ecIGPS, which demonstrated that Lys53 (Lys55 in ecIGPS) is essential to IGPS
catalysis.11
In this study, a series of kinetic experiments were performed in order to elucidate
the role of the highly conserved, charged residue in the active site of IGPS and allow for
a more complete understanding of the chemical mechanism for this enzyme. The
experiments reveal that Lys53 and Glu51 act as the general acid-base pair in the reaction,
catalyzing the dehydration of I2 to IGP product. This assignment differs from the
previously proposed assignments of either Glu159 or Glu210 as determined from
crystallography4 and MD simulations.9, 10 In light of this new assignment, we also
concluded that there are separate catalytic surfaces in IGPS that catalyze the ring closure
and dehydration; the I2 intermediate must undergo a reorientation during the reaction in
order for dehydration to occur.
3.3 Experimental Methods
3.3.1 Overexpression, Purification, and Kinetic Analysis of WT and Amino Acid
Substituted IGPS
Site-directed mutagenesis was performed to obtain Glu51Gln, Lys53Arg,
Lys53Gln, Glu159Gln, Arg182Ala, and Glu210Gln variants of ssIPGPS as described in
Chapter 2. All sequences for WT and mutant ssIGPS were confirmed through DNA
sequencing (Nucleic Acid Facility, Pennsylvania State University). The overexpression,
purification, and steady-state assays followed previously published protocols and were
performed as described in Chapter 2.4, 6, 12-14 The original WT ssIGPS overexpressed

64
from the pET21b14 is N-terminal hexa-histidine tagged, but ssIPGS from pET26 is not.
Important to these studies, the His-tagged and non His-tagged versions of ssIGPS gave
essentially identical kinetic parameters (kcat(WT-hexahis)/kcat(WT-notag) = 1.2 ± 0.3).
All kinetic experiments were performed at 75 °C (unless otherwise noted). SVE,
SDKIE, and pH effects were determined by varying the buffer conditions of the original
assay as described in Chapter 2. Assays were performed at saturating substrate
concentrations of CdRP (800 nM for WT, 4000 nM for Glu210Gln, 8000 nM for
Glu51Gln, Lys53Arg, and Arg182Ala).
3.3.2 Overexpression and Purification of ε-13CH2-Lys Labeled ssIGPS
In order to selectively, isotopically label ssIGPS with ε-13CH2-Lysine, the enzyme
was overexpressed similarly to previously described.15 The following was dissolved into
950 mL water: 0.50 g alanine, 0.40 g aspartate, 0.40 g arginine, 0.05 g cystine, 0.40 g
glutamine, 0.65g glutamate, 0.55 g glycine, 0.10 g histidine, 0.23 g isoleucine, 0.23 g
leucine, 0.20 g 13C-ε-CH2-lysine (Cambridge Isotopes), 0.25 g methionine, 0.13 g
phenylalanine, 0.10 g proline, 0.50 g serine, 0.23 g threonine, 0.17 g tyrosine, 0.23 g
valine, 0.50 g adenine, 0.20 g thymine, 0.20 g cytosine, 0.50 g uracil, 1.50 g sodium
acetate, 1.50 g succinate, 0.50 ammonium chloride, 0.85 g sodium hydroxide (NaOH),
and 10.5 g dibasic potassium phosphate. This mixture was autoclaved and then allowed
to cool to room temperature. Then, 50 mL 40 % (w/v) glucose, 4 mL 1 M magnesium
sulfate, 1 mL 0.01 M iron chloride, and 10 mL of a vitamin mixture containing 2 mg
calcium chloride, 2 mg zinc sulfate, 2 mg manganese sulfate, 50 mg tryptophan, 50 mg
thiamine, 50 mg niacin, and 1 mg biotin was added. The pH was adjusted to

65
approximately 7.3 and the solution sterile filtered.
Plasmid (pET21b) containing the his-tagged ssIGPS gene was transformed into
BL21-CodonPlus(DE3)-RIPL E. coli cells as described in Chapter 2. Fresh
transformations were used to inoculate 5 mL LB starter cultures, which were grown
overnight at 37 °C. 1 mL of this starter culture was used to inoculate 50 mL of the
selectively labeled media which was grown for approximately 20 hours at 37 °C. This
culture was used to inoculate the remainder of the media, which was grown at 37 °C until
the optical density (A600) was between 0.500 and 0.600 at which time the culture was
induced with IPTG and grown for 16 to 20 hours at 25 °C. All cell cultures contained 100
µg/mL Ampicillin. Cells were harvested by centrifugation (10,000 xg, 4 °C, 20 minutes)
and IGPS was purified and concentrated as previously described for His-tagged IGPS.
3.3.3 Preparation of rCdRP
CdRP was reduced to form the unreactive substrate analog, rCdRP through
selective reduction of CdRP with sodium borohydride (NaBH4). A solution of 5 M
NaBH4 was prepared in 0.05 M NaOH and added to a solution of CdRP such that the
ratio of NaBH4:CdRP was 1:3. The pH of the solution was adjusted with 0.05 M NaOH
until basic (pH 8 to 9). The solution was allowed to react for approximately one hour at
room temperature in the dark. Then, the pH was adjusted to approximately 5.0 with 1 N
hydrochloric acid.
rCdRP was purified by high performance liquid chromatography (HPLC) using a
Hypersil Gold PFP preparatory column (Thermo Scientific). The rCdRP was diluted by
approximately 50% with water and 50 µL was injected onto the column. The molecule

66
was eluted using a gradient from 99% water, 1% tetrafluoroacetic acid (TFA) to 99%
acetonitrile, 1% TFA over 30 minutes with a flow rate of 2 mL/min. The fractions
containing purified rCdRP were identified using its absorbance at 327 nm and full
conversion to the reduced form was tested by adding a small portion to tmIGPS to
confirm that no turnover to IGP occured. The fractions were lyophilized to a powder and
reconstituted in D2O to a concentration of 5 mM.
3.3.4 13C-HSQC Experiments on ssIGPS
After purification and concentration of the sample, IGPS was diluted to
approximately 1 mM and exchanged into buffer containing 25 mM potassium phosphate
pH 7.0 (pD 6.6), 75 mM potassium chloride, 1 mM EDTA, 1 mM dithiothreitol (DTT),
0.02% sodium azide (prepared in 100% D2O) using ZEBA desalting spin columns
(Thermo Scientific). Standard 1H-13C-HSQC (heteronuclear single quantum coherence)
experiments were obtained at 306 K on a Bruker AV-III-500 MHz spectrometer. pH was
adjusted for the titration by adding small volumes (< 5µL) of 0.5 M NaOH prepared in
D2O. The pKas were determined by plotting the change in chemical shift of hydrogen for
each peak and fitting to the equation:
€
δ = C /(1+10pKa − pH ) (3.1)
where δ is 1H chemical shift and C is the pH-independent rate value.

67
3.4 Results
3.4.1 Determination of the Rate-Determining Step of ssIGPS Catalysis
There are five highly conserved, charged residues in the active site of IGPS:
Glu51, Lys53, Lys110, Glu159, Arg182, and Glu210. Previous kinetic experiments in
ecIGPS indicate that these residues are all important for catalysis, but that study was not
able to delineate the particular role for each residue. Using similar methodology as in
Chapter 2 (Figure 2.2), SVE and SDKIE experiments were used to define the specific
role for these residues. SVE report on steps that are diffusion-controlled (substrate
binding, product release, and large conformational changes), whereas SDKIEs report on
chemical steps involving proton transfer. At 75 °C, the rate-determining step of WT
ssIGPS is the ring closure, as evidenced by the presence of an SDKIE and lack of an
SVE, whereas at 25 °C, product release is rate-determining as evidenced by an SVE of
approximately one. The same methodology was used to examine the rate-determining
step for the ssIGPS variants.
3.4.2 Analysis of Lys53 Indicates its Role as a General Acid
The assignment of Lys110 as the general acid in both steps of the reaction was based on
crystal structures of the enzyme, but there is no obvious mode of reprotonation, leading to
our hypothesis that an alternate residue is responsible for donating a proton in one of
these steps. Excluding Lys110, the only remaining cationic residues in the active site
capable of performing the role of general acid are Lys53 and Arg182. Therefore, enzyme
variants with amino aids substitutions at these residues were kinetically examined (Table
3.1). The Arg182Ala substitution had only a minor effect on the catalytic turnover, kcat,

68
Table 3.1: Steady-state kinetics (at 75 °C) demonstrates that the dehydration step of IGPS catalysis occurs through the general acid and base Lys53 and Glu51, respectively.
aDue to the low activity of Lys53Gln, a full steady-state curve was not possible. The kcat reported is the kobserved at saturating substrate concentrations bActivity of Lys110Arg and Glu159Gln variants was below the limit of detection. The value reported is the lowest kobs able to be measured through the fluorescence assay
IGPS
Variant
akcat (s-1) aKM (nM) kcat/KM
(x106 M-1s-1)
SDKIE
(kH2O/kD2O)
SVE
WT 0.67 ± 0.03 44 ± 9 15 3.6 ± 0.3 -0.2 ± 0.1
Glu51Gln 0.007 ± 0.001 13.0 ±4.4 0.54 0.9 ± 0.1 0.16 ± 0.2
Lys53Arg 0.06 ± 0.01 2440 ± 3 0.03 1.0 ± 0.1 0.2 ± 0.1
Lys53Gln 0.001 ± 0.001 n.d.a n.d.a n.d.a n.d.a
Arg182Ala 0.41 ± 0.07 1062 ± 499 0.38 1.1 ± 0.6 0.7 ± 0.3
Glu210Gln 0.97 ± 0.07 386 ± 96 2.5 1.0 ± 0.3 0.7 ± 0.2
Lys110Arg < 1.5 x 10-5 b n.d.b n.d.b n.d.b n.d.b
Glu159Gln < 1.5 x 10-5 b n.d.b n.d.b n.d.b n.d.b

69
which suggests that this residue is not directly involved in the chemical steps of the
reaction. Conversely, this variant showed a substantial increase in KM (~20 fold). This
result, along with the considerable SVE and loss of SDKIE for this variant, implicates its
importance in substrate binding.
To test the importance of the positive charge on Lys53 and its ability to act as a
general acid, we assayed Lys53Arg and Lys53Gln ssIGPS variants; Arg retains the
positive charge and some ability to donate a proton, whereas Gln does not have either
function but would still be able to participate in CdRP binding through a hydrogen bond
interaction with the carboxylate moiety. Since Lys53 is known to participate in CdRP
binding, changes in substrate binding were expected for these variants since substitutions
at this position would create less optimal interactions with CdRP. The Lys53Arg
substitution led to a decrease in CdRP affinity, with a 55-fold increase in KM (Table 3.1).
In addition, there was a 10-fold decrease in catalytic turnover (kcat) for Lys53Arg ssIGPS,
which is consistent with a direct role for Lys53 in the chemical steps of IGPS catalysis.
The Lys53Gln substitution had an even more deleterious effect on IGPS catalysis,
resulting in an over 500 fold-decrease in activity, even at the very high CdRP
concentration of 12 µM (50 times KM for WT IGPS). This finding indicates that the
positive charge and likely the ability of Lys53 to act as a proton donor are critical for
IGPS catalysis.
SVE, SDKIE, and pH effects further established the role of Lys53 in the
chemistry of the IGPS catalyzed reaction. Lys53Arg ssIGPS had a slightly larger SVE
than WT ssIGPS, which is consistent with a decrease in the rate of substrate binding or
product release, causing these processes to make a slight contribution to the overall rate.

70
The pH and SDKIE results indicate the specific role for Lys53 in reaction chemistry. The
bell-shaped pH curve for WT IGPS is consistent with acid-base catalysis (Figure 3.2),
but Lys53Arg displays a substantially higher pKa for the descending limb compared to
the WT enzyme (Table 3.2), closer to what is expected for an Arg residue. This result,
together with the finding that the Gln substitution is much more detrimental than Arg
(Table 3.1), suggest that Lys53 is behaving as a general acid during the reaction. The loss
of an SDKIE in Lys53Arg IGPS compared to the WT enzyme indicates that the rate-
determining step of the Lys53Arg ssIGPS catalyzed reaction is the dehydration rather
than the ring closure; the ring closure is expected to be isotope sensitive due to the proton
donation from Lys110, but the dehydration step is proposed to be isotope insensitive (see
Appendix for kinetic analysis). This proposal is also consistent with the decreased
SDKIE in WT ssIGPS at higher pH values, and the finding that the SDKIE arises from a
single proton transfer event.6

71
Figure 3.2: The pH rate profiles suggest that Lys53 and Glu51 act as the general acid and base, respectively in the dehydration step of IGPS catalysis. Shown are the pH curves for IGPS (a) WT (b) Lys53Arg and (c) Glu51Gln at 25 °C (left) and 75 °C (right).

72
Table 3.2: pKa values for WT ssIGPS and Lys53Arg and Glu51Gln variants identify Lys53 and Glu51 as the general acid and base in ssIGPS catalysis. Enzyme Variant Temperature (°C) pKa1 pKa2
WT 25 5.4 ± 0.2 8.9 ± 0.2
75 5.6 ± 0.2 8.7 ± 0.1
Lys53Arg 25 6.9 ± 0.1 > 9.5
75 7.1 ± 0.1 > 9.5
Glu51Gln 25 5.7 ± 0.2 n/a
75 6.51 ± 0.3 n/a

73
3.4.3 13C-Lys NMR to Determine pKas for Lysine Residues in IGPS
In order to conclusively determine which residues in the active site were
responsible for the pKa2 observed in the steady-state pH experiments, 13C-1H HSQC
nuclear magnetic resonance (NMR) experiments were performed on an IGPS sample
labeled with 13C-Lys. Eighteen distinct resonances were identified in the spectrum
(Figure 3.3), which is consistent with the seventeen lysine residues in ssIGPS, though
some of these resonances may represent different protons from the same lysine, such as
peaks 2 and 3 which show the same 13C chemical shift but different 1H chemical shifts.
A pH titration was performed on the ssIGPS enzyme in order to assign pKa values
to each resonance in the spectrum and correlate the pKa in the steady-state assay to a
particular residue in the enzyme. The pKa values were measured by tracking the change
in chemical shift (Figure 3.4) and plotting chemical shift vs pH for each peak (Figure
3.5) determined using NMR were significantly higher than the pKa of the descending
limb for the pH profile (Table 3.3). The pH rate dependence experiments indicate that at
high pH values (>9.5) the enzyme begins to denature. It is likely that the pH titration and
pKa values are not valid due to this denaturation. In the pH range where ssIGPS is known
to be stable there was no change in chemical shift. Therefore, it is more likely that the
change in chemical shift at higher pH is due to denaturation, rather than the deprotonation
of the lysine residues.
To test whether the introduction of D2O in the NMR experiments was affecting
the titration, the pH dependence of the enzyme activity was performed in assay buffer
prepared in D2O. The results (Figure 3.5) showed that there was no difference in the pKa
values for the kinetic pH dependence in D2O versus H2O.

74
Figure 3.3: 1H-13C HSQC on ε-13CH2-Lys labeled ssIGPS shows seventeen resonances which is consistent with the number of lysine residues in the enzyme

75
Figure 3.4: Overlay of 1H-13C-HSQC Spectra for ssIGPS at pH 7.0 (black) and pH 10.5 (red). At high pH, the changes in the spectrum are likely caused by denaturation of ssIGPS.

76
Figure 3.5: A representative plot of chemical shift versus pH for peak #2. The pKa value associated with this curve is 11.45 ± 0.14, although the change in chemical shift likely reflects denaturation of the enzyme rather than deprotonation of the lysine.

77
Table 3.3: pKa values determined by 1H-13C HSQC on ε-13CH2-Lys labeled ssIGPS are not in agreement with the pKa for the pH dependence of the enzymatic reaction determined by steady-state kinetics.
Resonance pKa Resonance pKa
0 10.98 ± 0.34 9 10.94 ± 1.43
1 11.27 ± 0.16 10 9.63 ± 0.16
2 11.45 ± 0.14 11 10.86 ± 0.18
3 11.42 ± 0.13 12 11.43 ± 0.18
4 11.05 ± 0.10 13 11.88 ± 0.95
5 11.38 ± 0.12 14 11.34 ± 0.14
6 11.37 ± 0.13 15 11.06 ± 0.14
7 11.36 ± 0.12 16 11.53 ± 0.20
8 11.30 ± 1.05 17 10.72 ± 0.14

78
Figure 3.5: pH dependence of the ssIGPS catalyzed reaction performed in H2O (pKa1 5.7 ± 0.1, pKa2 8.7 ± 0.1), shown in blue, and D2O (pKa1 5.3 ± 0.2, pKa2 8.9 ± 0.2), shown in green, display little difference in pKa values. Therefore, the use of D2O in NMR experiments does not explain the discrepancy in pKa values between the two methods.

79
In addition to the NMR titration results not aligning with the steady-state assays,
assignment of the resonances in the spectrum also proved to be problematic. Assignments
were attempted by performing NMR experiments with amino acid substituted variants of
ssIGPS; it was expected that deletion of a Lys would lead to the loss of a resonance in the
spectrum. The Lys53Arg sample was not stable in solution at high concentrations (~1
mM) and much of the sample precipitated during these experiments. The spectrum for
Lys53Arg ssIGPS resembles that of WT ssIGPS, but the low resolution made it
impossible to assign the residue. While it is still unclear which resonance belongs to
Lys53Arg (Figure 3.6), it is clear that the Lys53Arg substitution changes the chemical
environment of several of the resonances compared with WT enzyme at the same pH.
In a final attempt to assign the residues, 1H-13C-HSQC experiments were
performed in the presence of rCdRP. The addition of this substrate analog may change
the chemical environment of some residues in the enzyme, leading to a more dispersed
spectrum. Unfortunately, even with high concentrations of rCdRP (5 times [IGPS]) the
spectrum did not show any significant differences from the ligand free WT ssIGPS
spectrum. Due to the poor alignment of the NMR data to the steady-state data, along with
the inability to assign resonances in the NMR spectrum, these experiments were
unsuccessful and did not provide any results that are useful in the interpretation of the
kinetic studies.

80
Figure 3.6: Overlay of 1H-13C HSQCs of Lys53Arg (red) and WT ssIGPS (black) labeled with ε-CH2-Lys. Due to the low resolution of Lys53Arg ssIGPS spectrum, and its the poor alignment to WT, Lys53 and other resonances remain unassigned.

81
3.4.4 Analysis of Glu51 Identifies its Role as the General Base
Both Glu159 and Glu210 were previously proposed to act as the general base in
the dehydration of I2 to IGP. Glu159 was proposed because it is essential to the reaction,
the analogous amino acid substitution in ecIGPS, Glu163Gln, showed a 540-fold
decrease in kcat,5 and because Glu159 would appear to be appropriately positioned if
Lys110 is the general acid in the dehydration. Glu210 was alternately predicted as the
general base through MD simulations, as both Gu159 and Glu210 are both a similar
distance from the C1’ of the substrate where attack by the general base is proposed to
occur.9, 10 Since our results demonstrate that Lys53, rather than Lys110, is acting as the
general acid in the dehydration, it is less likely that Glu159 or Glu210 could be
performing the role of general base due to their distal location from Lys53, which would
prevent appropriate contact of I2 with both Lys53 and Glu159/Glu210.4, 12, 16
To probe the role of these residues, Glu159Gln and Glu210Gln variants were
examined. Similar to the analogous change in ecIGPS, the Glu159Gln substitution in
ssIGPS led to a substantial decrease in catalytic activity (kobs< 1.5 x 10-5 s-1). This change
in activity is on the same order of magnitude that was previously seen for the Lys110Arg
variant,6 which likely indicates that Glu159 and Lys110 are operating on the same
chemical step of the reaction (ring closure), which is also consistent with previous
proposals by Bruice and coworkers.10 The Glu210Gln variant did not show any decrease
in kcat, indicating that Glu210 is not involved directly in the chemistry of the reaction.
However, the loss of binding affinity (four-fold increase in KM) for the Glu210Gln
variant suggests that Glu210 plays a role in substrate binding.
Besides Glu159 and Glu210, Glu51 is the only other conserved residue in the

82
active site that may be performing this function.4 Kinetic analysis of the Glu51Gln
variant shows a 100-fold decrease in activity compared to WT enzyme (Table 3.1). The
kinetic parameters are on the same order of magnitude as those seen for Lys53Arg, which
is expected for variants that affect the same step of the kinetic mechanism. The Glu51Gln
variant also shows a complete loss of the SDKIE for kcat, as was also observed for the
Lys53Arg variant, which indicates that this residue is also involved in the dehydration
step of the reaction. Most telling was the pH dependence of Glu51Gln activity; the
variant displays a complete loss in the ascending limb, which is indicative of a loss in
general base activity for this variant (Figure 3.2). Altogether our results implicate
Lys53/Glu51 as the general acid/base pair responsible for catalyzing the dehydration step
of the reaction.
3.5 Discussion
In this study, we have delineated the roles for the highly conserved, charged
residues present in the active site of ssIGPS (Figure 3.7). Consistent with previous
proposals, Lys110 acts as the general acid in the ring closure step, and is likely aided by
Glu159. Our studies indicate Lys53 and Glu51 are responsible for catalyzing the
dehydration step as the general acid and base, respectively. Additionally, Arg182 and
Glu210 participate in substrate binding.
The newly identified roles for Glu51 and Lys53 are at odds with the crystal
structure of the IGPS:IGP complex; in the crystallized conformation, neither Lys53 nor
Glu51 is appropriately positioned to facilitate catalysis. However, a rotation through the
C3’-C4’ bond of the ribose chain relocates the amine group of I2 and the C2’ hydroxyl

83
Figure 3.7: The assigned role for the conserved and charged residues in the active site of IGPS. The ring closure is catalyzed by the general acid, Lys110, and Glu159 (blue). The dehydration is catalyzed by the general acid, Lys53, and general base, Glu51 (yellow). Arg182 and Glu210 (orange) are involved in susbtrate binding.

84
within contact distance of Glu51 and Lys53, respectively. This rotation is reasonable if
the previously suggested internal bond rotations of CdRP required to close the initial 4.5
Å distance between the C1 and C2’ for the ring closure, the two previously identified
binding pockets for the anthranilate moiety of the substrate, and the weak
crystallographic electron density of the ribulose moiety in the bound protein are
considered. Additionally, there likely is sufficient thermal flexibility of the substrate
within the active site to reorient the I2 intermediate into a appropriate position with
respect to both the Glu51 and Lys53 residues.
Based on this study, we propose a revised mechanism for IGPS catalysis (Figure
3.8). Following the ring closure, internal rotations in the ribose ring reposition the I2
intermediate in a position where Lys53 is in close proximity to the C2’ hydroxyl, and
Glu51 is in close proximity to the amine hydrogen (Figure 3.9). During the final step, the
lone pair of electrons on Glu51 attacks the hydrogen bond of the amine group. The
original mechanism proposed the general base instead attacked the C1’ hydrogen;
however, the loss of a hydrogen from an amine (pKa ~21) is much more reasonable that
from a carbon (pKa ~44 to 51). This action starts an electron cascade that ends with the
proton donation by Lys53 to the hydroxyl of C2’, releasing H2O and forming the product,
IGP. The reaction leaves the side chain of Lys53 neutralized, weakening the electrostatic
attraction between the protein and the product, which likely acts as an important trigger
event for product release.

85
Figure 3.8: The modified mechanism of ssIGPS catalysis utilizes Lys53 and Glu51 as the general acid and base pair in the dehydration step of the reaction. Additionally, the general base now attacks the amine hydrogen rather than the previously suggested alkyl hydrogen.

86
Figure 3.9: Rotation about the C3’-C4’ bond of ribose chain is required for the dehydration step in IGPS catalysis. Crystal structure of ssIGPS:IGP (Top) (PDB: 1A53) complex shows the ligand bound in the active site such that Lys53 and Glu51 are not properly positioned for catalysis. The ribose chain must rotate (Bottom) to reposition the intermediate in the second binding pocket and allow for dehydration to form IGP.

87
Mapping this revised mechanism on to the active site reveals two distinct regions
of function (Figure 3.10). The first region contains the surfaces formed by the
combination of Lys110 and Glu159 and it is this region that is likely responsible for the
ring closure and decarboxylation step of the catalytic pathway. The second region is
located adjacent to the first and includes Glu51 and Lys53. This region is responsible for
completing the dehydration in the second step of the mechanism. After the dehydration
reaction, Lys53 is rendered neutral likely causing a decrease in binding affinity that leads
to the release of IGP from the enzyme. Additionally, the binding site for the dehydration
step is located closer to where the product will exit the active site following the reaction,
and it is likely that this aids in the release of IGP as well.

88
Figure 3.10: Surface rendering of the IGPS binding pocket shows two distinct active sites for catalysis. In the first site (blue), Lys110 and Glu159 catalyze the ring closure step deep within the pocket. The intermediate then transitions to the second site (yellow), which is closer to where product exits the binding pocket, where Lys53 and Glu51 catalyze the dehydration step. (PDB: 1A53).

89
3.6 Conclusions
The production of indole, tryptophan, and their derivatives is both a critical
biological process and one of extreme utility in the chemical and pharmaceutical
industries. The new experimental data on ssIGPS WT and variants presented in this
Chapter supports a modified catalytic mechanism for IGPS, whose previously proposed
mechanism had been standing for the past forty years. This mechanism divides the active
site into discrete regions where the first catalytic surface, containing Lys110 and Glu159,
catalyzes the ring closure step, and the second, containing and Lys53 and Glu51,
catalyzes the dehydration step. Since IGPS is a validated antimicrobial target, this new
mechanism can be used in the rational drug design of original IGPS inhibitors that target
one or both of the catalytic sites. IGPS has also been used as a superior starting scaffold
in the development of novel enzymes. Engineered IGPS enzymes should find utility in
the development of new synthetic schemes for novel indole derivatives that may find
medical and agricultural use.
3.7 References
1. Barden, T. C., Indoles: Industrial, agricultural and over-‐the-‐counter uses. Topics in Hetercyclic Chemistry 2011, 26, 31-‐46. 2. Humphrey, G. R.; Kuethe, J. T., Practical methodologies for the synthesis of indoles. Chemical Reviews 2006, 106 (7), 2875-‐2911. 3. Smith, M.; March, J., March's advanced organic chemistry : reactions, mechanisms, and structure. 5th ed.; John Wiley: New York, 2001; p xviii, 2083. 4. Hennig, M.; Darimont, B. D.; Jansonius, J. N.; Kirschner, K., The catalytic mechanism of indole-‐3-‐glycerol phosphate synthase: crystal structures of complexes of the enzyme from Sulfolobus solfataricus with substrate analogue, substrate, and product. J Mol Biol 2002, 319 (3), 757-‐66.

90
5. Darimont, B.; Stehlin, C.; Szadkowski, H.; Kirschner, K., Mutational analysis of the active site of indoleglycerol phosphate synthase from Escherichia coli. Protein Science 1998, 7 (5), 1221-‐1232. 6. Zaccardi, M. J.; Mannweiler, O.; Boehr, D. D., Differences in the catalytic mechanisms of mesophilic and thermophilic indole-‐3-‐glycerol phosphate synthase enzymes at their adaptive temperatures. Biochemical and Biophysical Research Communications 2012, 418 (2), 324-‐329. 7. Czekster, C. M.; Lapis, A. A. M.; Souza, G. H. M. F.; Eberlin, M. N.; Basso, L. A.; Santos, D. S.; Dupont, J.; Neto, B. A. D., The catalytic mechanism of indole-‐3-‐glycerol phosphate synthase (IGPS) investigated by electrospray ionization (tandem) mass spectrometry. Tetrahedron Letters 2008, 49 (41), 5914-‐5917. 8. Smith, O. H.; Yanofsky, C., 1-‐(ortho-‐carboxyphenylamino)-‐1-‐deoxyribulose 5-‐phosphate, a new intermediate in the biosynthesis of tryptophan. Journal of Biological Chemistry 1960, 235 (7), 2051-‐2057. 9. Mazumder-‐Shivakumar, D.; Kahn, K.; Bruice, T. C., Computational study of the ground state of thermophilic indole glycerol phosphate synthase: Structural alterations at the active site with temperature. Journal of the American Chemical Society 2004, 126 (19), 5936-‐5937. 10. Mazumder-‐Shivakumar, D.; Bruice, T. C., Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole-‐3-‐glycerol phosphate synthase. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (40), 14379-‐14384. 11. Eberhard, M.; Kirschner, K., Modification of a catalytically important residue of indole-‐3-‐glycerol phosphate synthase from Escherichia coli Febs Letters 1989, 245 (1-‐2), 219-‐222. 12. Hennig, M.; Darimont, B.; Sterner, R.; Kirschner, K.; Jansonius, J. N., 2.0 A structure of indole-‐3-‐glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability. Structure 1995, 3 (12), 1295-‐306. 13. Merz, A.; Yee, M. C.; Szadkowski, H.; Pappenberger, G.; Crameri, A.; Stemmer, W. P. C.; Yanofsky, C.; Kirschner, K., Improving the catalytic activity of a thermophilic enzyme at low temperatures. Biochemistry 2000, 39 (5), 880-‐889. 14. Schneider, B.; Knochel, T.; Darimont, B.; Hennig, M.; Dietrich, S.; Babinger, K.; Kirschner, K.; Sterner, R., Role of the N-‐terminal extension of the (betaalpha)8-‐barrel enzyme indole-‐3-‐glycerol phosphate synthase for its fold, stability, and catalytic activity. Biochemistry 2005, 44 (50), 16405-‐12.

91
15. Muchmore, D. C.; McIntosh, L. P.; Russell, C. B.; Anderson, D. E.; Dahlquist, F. W., Expression and N-‐15 labeling of proteins for proton and N-‐15 nuclear-‐magnetic resonance. Methods in Enzymology 1989, 177, 44-‐73. 16. Wilmanns, M.; Priestle, J. P.; Niermann, T.; Jansonius, J. N., Three-‐dimensional structure of the bifunctional enzyme phosphoribosylanthranilate isomerase: indoleglycerolphosphate synthase from Escherichia coli refined at 2.0 A resolution. J Mol Biol 1992, 223 (2), 477-‐507.

92
Chapter 4
The Role of Active Site Loops in Catalysis by IGPS
4.1 Abstract
Substrate binding, product release, and likely chemical catalysis in the tryptophan
biosynthetic enzyme indole-3-glycerol phosphate synthase (IGPS) are controlled through
the structural dynamics of the β1α1 loop. We have probed the role of the nearby β2α2
loop and the interaction between the β1α1 and β2α2 loops using amino acid substitutions
and kinetic analysis to gain insight into how loop-loop interactions regulate IGPS
catalysis. Our results indicate that interactions between co-evolving residues Arg54 and
An90 on these loops are important in coordinating the multistep reaction in IGPS, and
can modulate the general acid activity of the absolutely conserved Lys53 on the β1α1
loop.
4.2 Introduction
The IGPS structure consists of a (β/α)8-barrel, or TIM-barrel fold, the most
common fold found in structural biology. The fold typically consists of eight parallel β-
strands in the shape of a wheel surrounded by eight α-helices. TIM-barrel proteins are
catalytically diverse,1 but the structural elements are highly conserved, and most of the
chemically relevant amino acids residues are located on the inside of the β-barrel on the
C-terminal end of the β-strands or the loops connecting β-strands to α-helices (βα loops).
These βα loops are shown to be particularly important in the proper function of the
enzyme. In addition to containing many of the active site residues that participate in the

93
reaction, interactions between loops have been implicated in the function of many (β/α)8-
barrel enzymes2-4 including TIM where the structural dynamics of the β6α6 loop allows
for substrate binding and product release.5 Bioinformatics approaches and various
computational methods, including MD simulations, have also implicated an important
role for several loops in the (β/α)8-barrel enzyme IGPS,6-8 though the functional role of
these loops has not, until now, been investigated experimentally.
SCA combined with MD simulations identified a potentially important interaction
between residues on the β1α1 and β2α2 loops in IGPS.8 SCA identifies residue positions
that co-vary, and proposes that these residues are important in maintaining structure,
allowing proper protein folding, or more directly facilitating proper enzyme function and
catalysis.9 When used in conjunction with MD simulations, SCA can identify
functionally important amino acid pairs that are not absolutely conserved through all
species but whose interaction is conserved and whose coordinated motions may be
important in enzyme catalysis. A similar methodology also identified other co-varying
amino acids on IGPS, and mutational studies confirmed that these residues are
catalytically and/or structurally important.10 The co-varying residues in the β1α1 and
β2α2 loops may be especially important considering the functional role of the β1α1 loop
in IGPS catalysis.
Specifically, SCA-MD analysis suggests that interactions between Arg54 and
Asn90 on the β1α1 and β2α2 loops are important for enzyme function (Figure 4.1).
Structural comparison of the substrate-bound and product-bound forms of IGPS, together
with the structural modeling of the intermediate in the active site shows two distinct and
adjacent hydrophobic pockets, suggesting that the anthranilate group of the substrate

94
Figure 4.1: The catalytically important residues in the dehydration step of IGPS are found on the β1α1 and β2α2 loops, which interact through a hydrogen bond between Arg54 and Asn90. (a) The ssIGPS catalyzed reaction has two distinct binding pockets for the two reaction steps. In step one, Lys 110 (cyan) initiates the ring closure and decarboxylation. Following the formation of the intermediate, the anthranilate moiety is transferred to the second site where Lys53 and Glu51 (yellow) act as the active site acid and base. The role of the β1α1 and β2α2 loops (blue) including the interaction between Arg54 and Asn90 is examined herein. (b) This interaction is thought to have functional significance in IGPS since it is coevolving amongst IGPS species and exhibits correlated motion in MD simulations. This interaction is in close proximity to the conserved residues Lys53 and Phe89.

95
moves from one hydrophobic pocket to another during catalysis.11, 12 It is important to
note that the conserved Phe89 on the β2α2 loop comprises part of this anthranilate
binding pocket through π-π interations with the CdRP substrate. There is also a second
coevolving interaction between residues on the β2α2 loop through Glu85 and Asn90. We
hypothesize that the interaction between Arg54 and Asn90 (and perhaps Glu85) may help
to coordinate conformational changes in both the loops and the substrate or intermediate
itself.
In this Chapter, the importance of the β2α2 loop and its interaction with the β1α1
loop in ssIGPS catalysis was examined through the analysis of Arg54, Asn90, Glu85, and
Phe89 variants. The results indicate that the interaction between the two active site loops
in ssIGPS is important for proper function of the general acid/base pair, Lys53/Glu51, in
the dehydration step. It may also be involved in coordinating the transition of the
intermediate between the two binding sites during catalysis. These results provide insight
into the coordination of the multiple step reaction catalyzed by IGPS.
4.3 Experimental Methods
Site-directed mutagenesis of ssIGPS variants (Arg54Ala, Phe89Ala, Asn90Ala,
Asn90Gln. Arg54Lys, Arg54Ala/Asn90Ala, and Arg54Lys/Asn90Gln) was performed
using the QuikChange Lightning® Site-Directed Mutagenesis Kit (Agilent Technologies)
and appropriate primers as described in previous Chapters. All sequences (WT and
mutant) were confirmed through DNA sequencing (Nucleic Acid Facility, Pennsylvania
State University). Overexpression, purification, and steady-state kinetic analyses of WT
and variant enzymes were performed as previously described (see Chapter 2). 13,14

96
Thermal inactivation experiments for ssIGPS enzymes (non His-tagged, WT and
amino acid substituted) were performed by incubating the stock enzyme solution at 90 °C
for up to 30 minutes. Aliquots were removed every three minutes and the enzyme activity
was assayed in duplicate at 50 °C with a saturating CdRP concentration (800 nM). The
data was fit to a linear regression of relative rate (compared to the rate prior to
incubation) versus incubation time in seconds. The first-order rate constant of thermal
inactivation, kinact, is equal to the slope of this linear fit.
Circular dicroism (CD) experiments were performed out on a Jasco J-810
Spectropolarimeter from 250 nm to 190 nm with 1 nm intervals and a 1 nm bandwidth.
The experiments were performed in 10 mM potassium phosphate pH 7.0 with an enzyme
concentration of 1.7 µM.
4.4 Results
4.4.1 Investigation of Phe89 on the β2α2 Loop Identifies its Role in IGPS Chemistry
Since we are interested in the role of active site loops in IGPS catalysis, and
previous studies resolved the role of the conserved residues on the β1α1 loop and
adjecent β1-strand (Lys53 and Glu51), we examined the role of the essential and
invariant Phe89 residue located on the β2α2 loop with an alanine substitution. Phe89 is
on the β2α2 loop and makes a π-π interaction with the anthranilate moiety of CdRP.
Therefore, it is not surprising that the Phe89Ala amino acid substitution also led to an
approximately 24-fold increase in KM (Table 4.1). Interestingly, the substitution led to an
eleven-fold decrease in kcat associated with this variant, which suggests that this residue
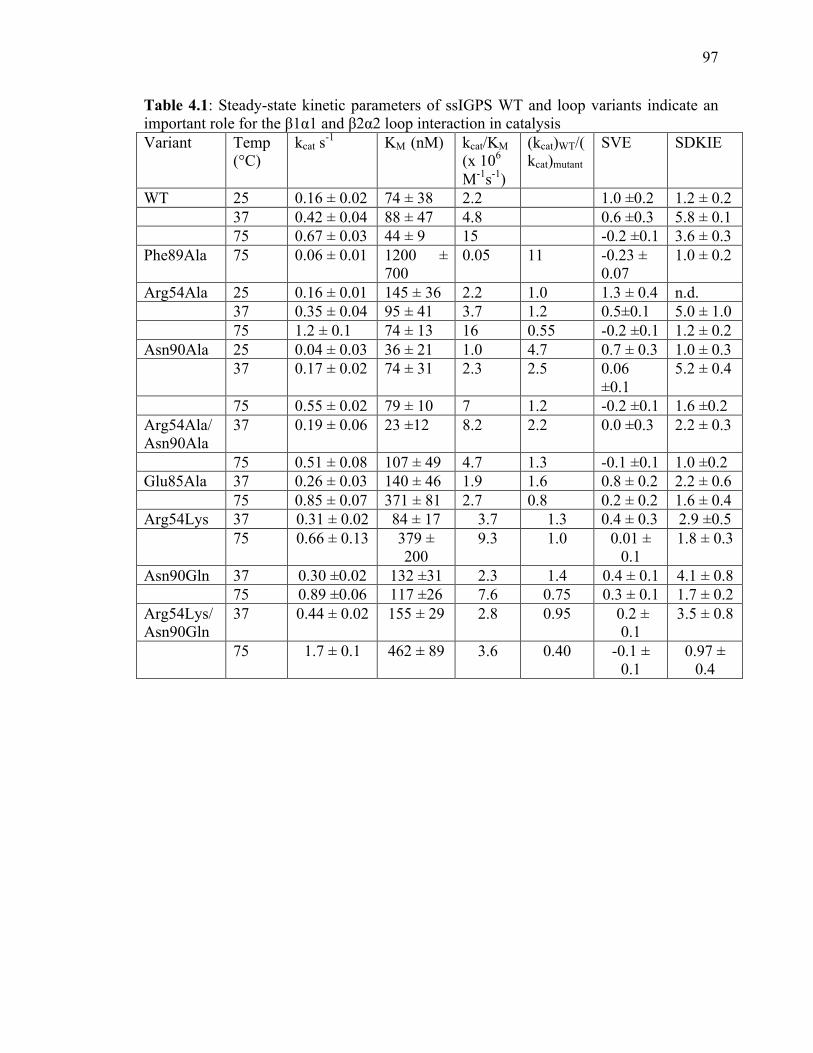
97
Table 4.1: Steady-state kinetic parameters of ssIGPS WT and loop variants indicate an important role for the β1α1 and β2α2 loop interaction in catalysis Variant Temp
(°C) kcat s-1 KM (nM) kcat/KM
(x 106 M-1s-1)
(kcat)WT/(kcat)mutant
SVE SDKIE
WT 25 0.16 ± 0.02 74 ± 38 2.2 1.0 ±0.2 1.2 ± 0.2
37 0.42 ± 0.04 88 ± 47 4.8 0.6 ±0.3 5.8 ± 0.1 75 0.67 ± 0.03 44 ± 9 15 -0.2 ±0.1 3.6 ± 0.3 Phe89Ala 75 0.06 ± 0.01 1200 ±
700 0.05 11 -0.23 ±
0.07 1.0 ± 0.2
Arg54Ala 25 0.16 ± 0.01 145 ± 36 2.2 1.0 1.3 ± 0.4 n.d. 37 0.35 ± 0.04 95 ± 41 3.7 1.2 0.5±0.1 5.0 ± 1.0
75 1.2 ± 0.1 74 ± 13 16 0.55 -0.2 ±0.1 1.2 ± 0.2 Asn90Ala 25 0.04 ± 0.03 36 ± 21 1.0 4.7 0.7 ± 0.3 1.0 ± 0.3 37 0.17 ± 0.02 74 ± 31 2.3 2.5 0.06
±0.1 5.2 ± 0.4
75 0.55 ± 0.02 79 ± 10 7 1.2 -0.2 ±0.1 1.6 ±0.2 Arg54Ala/Asn90Ala
37 0.19 ± 0.06 23 ±12 8.2 2.2 0.0 ±0.3 2.2 ± 0.3
75 0.51 ± 0.08 107 ± 49 4.7 1.3 -0.1 ±0.1 1.0 ±0.2 Glu85Ala 37 0.26 ± 0.03 140 ± 46 1.9 1.6 0.8 ± 0.2 2.2 ± 0.6 75 0.85 ± 0.07 371 ± 81 2.7 0.8 0.2 ± 0.2 1.6 ± 0.4 Arg54Lys 37 0.31 ± 0.02 84 ± 17 3.7 1.3 0.4 ± 0.3 2.9 ±0.5 75 0.66 ± 0.13 379 ±
200 9.3 1.0 0.01 ±
0.1 1.8 ± 0.3
Asn90Gln 37 0.30 ±0.02 132 ±31 2.3 1.4 0.4 ± 0.1 4.1 ± 0.8 75 0.89 ±0.06 117 ±26 7.6 0.75 0.3 ± 0.1 1.7 ± 0.2 Arg54Lys/Asn90Gln
37 0.44 ± 0.02 155 ± 29 2.8 0.95 0.2 ± 0.1
3.5 ± 0.8
75 1.7 ± 0.1 462 ± 89 3.6 0.40 -0.1 ± 0.1
0.97 ± 0.4

98
may also be involved in the chemistry of the reaction.
To gain additional insight into the binding and chemical steps reported on by the
kcat, SVEs and SDKIEs were examined for Phe89Ala using the same methodology as
presented in Chapter 2 and Chapter 3. The lack of SVE suggests that a chemistry step is
rate-determining, rather than substrate binding or product release as might be expected
considering the involvement of Phe89 in binding, and the observed kcat is reporting on a
chemical step. The lack of a SDKIE suggests that the dehydration step has now become
fully rate-determining, as opposed to WT ssIGPS, in which the rate-determining step at
75 °C is ring closure. Using the methodology defined in Chapter 2, the dehydration step
is isotope insensitive, and the ring closure is isotope sensitive. These findings indicate
that the Phe89Ala substitution affects both substrate binding and catalytic events
associated with the dehydration step.
4.4.2 Interaction Between β1α1 and β2α2 Loops through Arg54 and Asn90 is
Important for Catalysis
Our studies with Lys53 (Chapter 3) and Phe89 amino acid substitutions indicate
that these conserved β1α1 and β2α2 loop residues are important for the chemical steps of
ssIGPS catalysis. Considering the SCA-MD results demonstrating an important
interaction between these loops through Arg54 and Asn90, we were also interested in
how the amino acid interaction between these loops may impact ssIGPS function. This
interaction may be involved in modulating the role of the β1α1 and β2α2 loops during
catalysis.8 To probe these interactions further, we made amino acid substitutions at both
Arg54 and Asn90, and characterized the kinetics of the variant enzymes at 75 °C, the

99
biologically relevant temperature for the thermophile S. sulfataricus.
Arg54Ala and Asn90Ala display kinetic parameters very similar to WT enzyme at
75 °C, although Arg54Ala ssIGPS shows a small increase to kcat (~1.8 fold) compared to
WT ssIGPS. Although the overall rates were similar, it is still possible that the amino
acid substitutions could impact the individual steps of catalysis. To test this concept,
SVEs and SDKIEs were also measured in order to assign the rate-determining step for
each variant. In WT ssIGPS, the rate-determining step at 75 °C is the ring closure,
evidenced by the substantial SDKIE that was attributed the transfer of a single proton in
proton inventory studies (Figure 2.3).13 Neither Arg54Ala nor Asn90Ala exhibit an SVE
at 75 °C, suggesting kcat is reporting on a chemical event for both of these enzymes. Both
variants display a loss in the SDKIE at 75 °C that is similar to the behavior of previous
amino acid substitutions (Lys53Arg and Glu51Gln). Akin to these variants, the Arg54Ala
and Asn90Ala variants are likely affecting kinetics of the dehydration step, such that this
step becomes slower than the ring closure step and more rate-determining.
The Arg54Ala and Asn90Ala variants show changes to their pH profile (Figure
4.2), with an increase in the ascending pKa (attributed to general base activity by Gln51)
compared to WT enzyme occurring in both variants. The Asn90Ala variant also displays
a loss in the descending limb of the pH profile, whose pKa was previously attributed to
the general acid of the dehydration step, Lys53, although the same behavior is not seen
for Arg54Ala enzyme. The pH results provide evidence that the interaction between the
two residues is important for the dehydration step of the reaction. The
Arg54Ala/Asn90Ala double substitution was also examined and exhibited activity similar
to Asn90Ala. This result is evidence that the interaction between Arg54 and Asn90 is

100
Figure 4.2: pH profiles of WT, Arg54Ala, and Asn90Ala ssIGPS show changes to the activity of the general acid and base. pH profile for (a) WT (pKa1 5.6 ± 0.2, pKa2 8.7 ± 0.1) is different than (b) Arg54Ala (pKa1 6.5 ± 0.2, pKa2 8.5 ± 0.2) and (c) Asn90Ala (pKa1 7.3 ± 0.3, pKa2>>9 ) . The amino acid substitutions result in peturbations associated with the general acid and base in the dehydration step compared to WT enzyme.

101
Table 4.2: pKa values for WT, Arg54Ala, and Asn90Ala indicate that the loop interaction is important for general acid/base catalysis. Enzyme pKa1 pKa2
WT 5.6 ± 0.2 8.7 ± 0.1
Arg54Ala 6.5 ± 0.2 8.5 ± 0.2
Asn90Ala 7.3 ± 0.3 >9

102
important for function; but that Asn90 is the more vital residue, and the enzyme can more
easily overcome disruptions to Arg54 compared to Asn90.
The Arg54Ala and Asn90Ala variants change the rate-determining step of
catalysis at 75 °C. Our previous studies have also indicated that the rate-determining step
is temperature dependent. Therefore, to gain more insight into the effects of loop
substitutions, we also tested the temperature dependence of these variants. At lower
temperatures, Arg54Ala ssIGPS displays activity similar to WT ssIGPS. However, the
Asn90Ala variant shows a more substantial decrease in kcat compared to WT enzyme at
lower temperatures.
For WT ssIGPS, the rate-determining step at 75 oC is the ring closure; however, at
lower temperatures (25 oC) product release becomes rate-determining.13 At 37 °C both
the ring closure and product release contribute to the kinetic constants, so both are
partially rate-determining. The Asn90Ala variant shows no substantial SVE at 37 oC in
contrast to WT ssIGPS (Table 4.1). This finding suggests that kcat for the Asn90Ala
variant likely reflects a chemical step, and the effect of the Asn90Ala substitution on
chemistry is underestimated at the lower temperatures (i.e. for WT ssIGPS, kchemistry >>
kcat at 37 °C). Interestingly, there is still a significant SDKIE at 37 °C, as opposed to 75
°C at which both Asn90Ala and Arg54Ala enzymes display a loss in isotope effects. This
result indicates that the rate-determining step for these mutants is ring closure at 37 °C, as
opposed to 75 °C at which the dehydration step becomes rate-determining. For
Asn90Ala, the loss in SVE at 37 °C along with a significant SDKIE at this temperature
indicates that Asn90 is not only involved in dehydration, but also affects the rate of the
ring closure step, at least at lower temperatures.

103
4.4.3 Analysis of Arg54Lys and Asn90Gln Variants of ssIGPS
Since Arg54 and Asn90 are residues that coevolve between IGPS enzymes from
different organisms, we were interested in examining amino acid substitutions that
mimicked this process. In the mesophilic enzyme ecIGPS, the analogous residues to
Arg54 and Asn90 are Lys55 and Gln94, respectively. These residue changes keep
hydrogen bond interactions intact at these positions. Arg54Lys and Asn90Gln amino acid
substitutions of ssIGPS were examined in order to further evaluate the role of this
interaction. These substitutions are expected to make similar interactions with CdRP;
however, they may be less optimal than the residues in WT due to differences in size
(Lys is slightly smaller than Arg and Gln is slightly larger than Asn).
The Arg54Lys variant displays activity similar to WT IGPS (Table 4.2), which is
expected considering the more deleterious amino acid substitution at this position,
Arg54Ala, also showed similar kinetic parameters to the WT enzyme. Despite the
similarity in kcat, between WT ssIGPS and the Arg54Lys variant, there is a substantial
difference in the magnitude of the SDKIE compared to WT enzyme at 75 °C but not 37
°C. This finding siggests that there is a difference in the step of the chemical mechanism
that is affected at lower versus higher temperatures. At the mesophilic relevant
temperature, 37 °C, the rate-determining step of the reaction is the ring closure, similar to
WT ssIGPS. Conversely, at the thermophilic-relevant temperature, 75 °C, the Arg54Lys
substitution causes the dehydration step to become slowed, making it the rate-
determining step at this temperature.
The Asn90Gln ssIGPS variant showed catalytic activity more comparable to WT
enzyme than those of the Asn90Ala variant, although the kcat was slightly reduced at 37

104
°C compared to WT enzyme. Similar to the other substitutions evaluated, the SDKIE on
kcat was also substantially reduced compared to WT ssIGPS at 75 °C, indicating that the
Asn90Gln amino acid substitution also affects the dehydration step of the reaction,
despite the similar structure of Asn and Gln. At 37 °C, the SDKIE is present indicating
that the ring closure is rate-determining as shown previously. For completeness, the
double substitution, Arg54Lys/Asn90Gln ssIGPS, was also assayed. The kinetic
parameters are consistent with what was seen for the single substitutions, indicating that
the role of these residues are linked, and both are required for proper ssIGPS function.
4.4.4 Examination of the Interaction Between Coevolving Residues in the β2α2 Loop
In addition to the interaction between Arg54 and Asn90, there is a second active
site loop interaction identified using SCA-MD through Asn90 and Glu85, both of which
are located on the β2α2 loop. To further explore the role of this interaction, the Glu85Ala
variant was assayed. The steady-state kinetic parameters for Glu85Ala (Table 4.1) are
similar to WT ssIGPS. Interestingly, the KM at 75 °C is about six times higher than WT
enzyme, indicating that this residue may be involved in substrate binding. Similar to
Arg54Ala and Asn90Ala, Glu85Ala displayed a loss in the SDKIE at 75 °C, indicating
that this residue is also involved in the dehydration step of the reaction.
4.4.5 Structure and Stability of Loop Mutants
Amino acids residues like Arg54 and Asn90 may be important not only for
catalytic function, but may also for protein folding and stability, and changes to these
characteristics can also effect the activity of the enzyme.8 To assess the role of these

105
amino acids on the structure and stability of ssIGPS, thermal inactivation experiments
were performed for WT and amino acid substituted ssIGPS. The thermal inactivation
experiments showed a similar rate constant of inactivation for WT and Arg54Ala ssIGPS
(1.11 x 10-3 s-1 and 1.24 x 10-3 s-1, respectively) (Figure 4.3). However, a two-fold
decrease in the rate of inactivation was observed for Asn90Ala (6.91 x 10-4 s-1) and
Asn90Gln (3.59 x 10-4 s-1) implying that changes to Asn90 increases the thermal stability
of the enzyme. The reason for this increase in thermal stability is unclear. The CD spectra
for WT and Asn90Ala ssIGPS (Figure 4.3) were nearly identical, indicating that the
substitution did not result in any gross structural change. The kinetic changes reported for
these variants must be then to local changes in the catalytic environment, likely through
changes in hydrogen bonding, rather than changes in structure and folding.
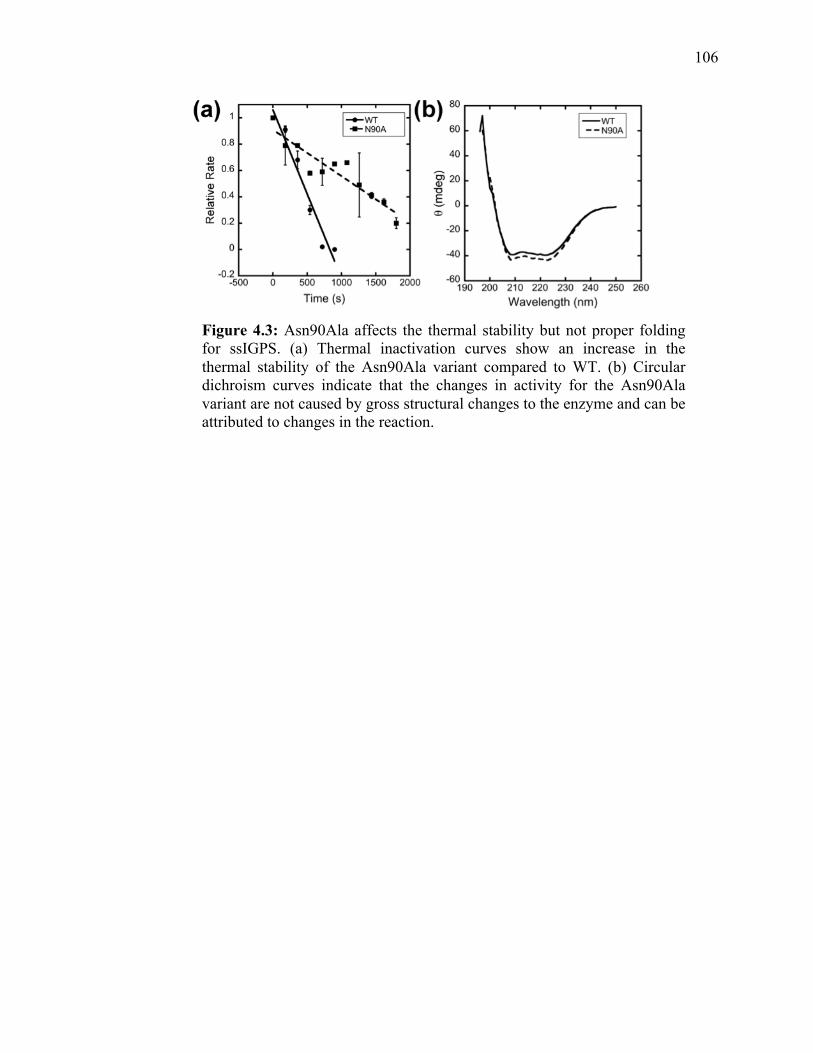
106
Figure 4.3: Asn90Ala affects the thermal stability but not proper folding for ssIGPS. (a) Thermal inactivation curves show an increase in the thermal stability of the Asn90Ala variant compared to WT. (b) Circular dichroism curves indicate that the changes in activity for the Asn90Ala variant are not caused by gross structural changes to the enzyme and can be attributed to changes in the reaction.

107
4.5 Discussion
Lys53 and Phe89 are conserved and important amino acid residues for the IGPS
catalyzed reaction, and are found on the active site β1α1 and β2α2 loops, respectively.
The loss of the SDKIE for amino acid substitutions to Lys53 (presented in Chapter 3)
and Phe89 indicate that both residues are involved in the dehydration step of the reaction.
MD simulations previously identified correlated motion in these loops, which occurs
through a hydrogen bond between Asn90 and Arg54. The results presented in this
Chapter help to further delineate the role for the active site loops and the interaction
between them through Arg54 and Asn90 through the examination of enzymes with amino
acid substitutions at these positions.
The Asn90Ala variant showed a decrease in kcat at 37 °C compared to WT
enzyme, whereas at 75 °C, kcat approached that of WT enzyme. Conversely, the
Arg54Ala variant showed kcat values similar to WT ssIGPS at low temperatures, with a
slight increase at 75 °C. The changes to kcat for the variants were modest, but this does
not necessarily indicate that these residues do not play an important role in catalysis. The
SDKIEs suggest that the identities of the rate-determining step for WT and variant
enzymes are different, indicating that steady-state kcat values are underrepresenting the
true effects on the individual kinetic steps of the reaction. The substantial decreases in
SDKIEs for Arg54Ala and Asn90Ala variants suggest that the dehydration step is
becoming more rate-limiting relative to ring closure. Steady-state kinetics and careful
consideration of the SDKIEs and SVEs is likely the most efficient way of teasing these
effects apart, especially considering that pre-steady-state kinetics by the Goodey and
Sterner groups were unable to deconvolute the rate constants for the chemical steps of the

108
reaction.
The temperature dependence of the Arg54Ala and Asn90Ala suggests that the
role of these residues may be affected by temperature. At 37 °C, there is still a substantial
SDKIE for both variants, indicating that ring closure is primarily rate-determining, as is
seen for the WT enzyme. This result further highlights the different temperature
dependencies of the chemical steps. We propose that at lower temperatures, the activation
energy for ring closure is much higher than the corresponding activation energy for
dehydration. At higher temperatures, the activation energies for the ring closure and the
dehydration steps are more similar. Upon the disruption of any amino acid involved in
dehydration, this step becomes rate-determining at 75oC, causing the loss of SDKIE seen
for the loop mutants. However, at lower temperatures, ring closure may still be primarily
rate-determining, leading to a less substantial effect on the SDKIE.
The decreased SDKIE for Arg54Ala and Asn90Ala variants along with the
changes to the pH profiles for these enzymes compared to WT ssIGPS indicates that
these coevolving residues effect the function of the general acid and base, Lys53 and
Glu51, in the dehydration step. The loss of the descending limb of the pH profile for
Asn90Ala is indicative of an increase in the pKa of the general acid, Lys53. Similarly, the
increase in pKa for the ascending limb of the pH profile for both Arg54Ala and Asn90Ala
variants suggests an effect on the general base, Glu51. Therefore, it is likely that Asn90 is
involved in proper function of Lys53, and affects the microenvironment and/or the
positioning of this general acid.
The results indicate that the interaction between the β1α1 and β2α2 loops helps to
properly bind the substrate and/or position Lys53 and Glu51 to act as the general acid and

109
base of the dehydration step, and to increase catalytic efficiency by both allowing
residues that bind the substrate to make more favorable interactions. The lack of a similar
pH effect for the descending limb of the pH profile for the Arg54Ala variant can be
explained either by noting that the Asn90 side chain may hydrogen bond to the backbone
of Arg54, or to another residue on the β1α1 loop that is free to make an interaction (i.e.
Lys55 or Ser56) in the Arg54Ala variant. In this way, the Arg54Ala variant still
maintains the interaction between the β1α1 and β2α2 loops.
The interaction between the β1α1 and β2α2 loops between Asn90 and Arg54
functions to correctly position conserved amino acid residues, Glu51, Lys53 and Phe89.
The deleterious effect of the substitutions at these positions can be overcome at higher
temperatures due to the ability of the enzyme to more easily sample the conformation that
is required for proper catalysis by Glu51 and Lys53. This idea is in agreement with the
findings from the Bruice lab that showed the presence of NACs increases for ssIGPS as
temperature increases, and so the appropriate conformation for catalysis is more available
at higher temperatures.70, 78
This study provides further evidence that proper catalysis by IGPS requires the
reorientation of the substrate in the active site. We propose that at lower temperatures, the
enzyme has a slower rate of conformational sampling and lower thermal energy makes it
more difficult to rearrange the substrate after it is captured; thus, the ring closure is rate-
determining. Conversely, at higher temperatures, this reorientation is easier, decreasing
the activation energy for this step relative to the dehydration step.
The amino acid substitutions that mimic the analogous residues in ecIGPS,
Arg54Lys and Asn90Gln, display similar kinetic parameters to WT enzyme but have

110
differences in the rate-determining step of the reaction at biologically relevant
temperatures. This finding indicates that the co-evolution of this loop and the particular
amino acids in these positions in ssIGPS are important to the kinetic mechanism of the
enzyme, and it is possible that these coevolving residues may play an important role in
the kinetic differences between the enzymes.
4.6 Conclusions
SCA-MD analysis provides additional insight into residue pairs that may not be
conserved but nonetheless play integral roles in enzyme catalysis. However, simply
identifying residues as important does not provide adequate information for application in
enzyme engineering, antibiotic development, or other industrial applications.
Experimental examination of the SCA-MD identified enzyme pairs provides a more in
depth understanding and additional details of the chemical mechanism that can be utilized
in other applications.
Our results provide an analysis of the interaction between the β1α1 and β2α2
loops through the SCA-MD identified Arg54 and Asn90. This interaction plays an
important role in the catalytic mechanism of ssIGPS and affects the role of Lys53 as a
general acid and Glu51 as a general base. Thus, it is likely that the interaction between
Asn90 and Arg54 is involved in guiding the anthranilate moiety of the substrate between
the two active sites and properly arranging the general acid for catalysis. These new
details can be used for the design of more efficient enzymes and perhaps new
antibacterial agents that target IGPS. Engineering new enzymes relies heavily in
understanding how a reaction occurs rather than just at what speed it occurs.

111
4.7 References
1. Hocker, B.; Jurgens, C.; Wilmanns, M.; Sterner, R., Stability, catalytic versatility and evolution of the (beta alpha)(8)-‐barrel fold. Current Opinion in Biotechnology 2001, 12 (4), 376-‐381. 2. Carpenter, R. A.; Xiong, J.; Robbins, J. M.; Ellis, H. R., Functional Role of a Conserved Arginine Residue Located on a Mobile Loop of Alkanesulfonate Monooxygenase. Biochemistry 2011, 50 (29), 6469-‐6477. 3. Malabanan, M. M.; Amyes, T. L.; Richard, J. P., A role for flexible loops in enzyme catalysis. Current Opinion in Structural Biology 2010, 20 (6), 702-‐710. 4. Lipchock, J.; Loria, J. P., Millisecond dynamics in the allosteric enzyme imidazole glycerol phosphate synthase (IGPS) from Thermotoga maritima. Journal of Biomolecular Nmr 2009, 45 (1-‐2), 73-‐84. 5. Rozovsky, S.; Jogl, G.; Tong, L.; McDermott, A. E., Solution-‐state NMR investigations of triosephosphate isomerase active site loop motion: Ligand release in relation to active site loop dynamics. Journal of Molecular Biology 2001, 310 (1), 271-‐280. 6. Mazumder-‐Shivakumar, D.; Bruice, T. C., Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole-‐3-‐glycerol phosphate synthase. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (40), 14379-‐14384. 7. Mazumder-‐Shivakumar, D.; Kahn, K.; Bruice, T. C., Computational study of the ground state of thermophilic indole glycerol phosphate synthase: Structural alterations at the active site with temperature. Journal of the American Chemical Society 2004, 126 (19), 5936-‐5937. 8. Shen, H. B.; Xu, F.; Hu, H. R.; Wang, F. F.; Wu, Q.; Huang, Q.; Wang, H. H., Coevolving residues of (beta/alpha)(8)-‐barrel proteins play roles in stabilizing active site architecture and coordinating protein dynamics. Journal of Structural Biology 2008, 164 (3), 281-‐292. 9. Estabrook, R. A.; Luo, J.; Purdy, M. M.; Sharma, V.; Weakliem, P.; Bruice, T. C.; Reich, N. O., Statistical colevolution analysis and molecular dynamics: Identification of amino acid pairs essential for catalysis. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (4), 994-‐999. 10. Dietrich, S.; Borst, N.; Schlee, S.; Schneider, D.; Janda, J. O.; Sterner, R.; Merkl, R., Experimental Assessment of the Importance of Amino Acid Positions Identified by an Entropy-‐Based Correlation Analysis of Multiple-‐Sequence Alignments. Biochemistry 2012, 51 (28), 5633-‐5641.

112
11. Hennig, M.; Darimont, B. D.; Jansonius, J. N.; Kirschner, K., The catalytic mechanism of indole-‐3-‐glycerol phosphate synthase: crystal structures of complexes of the enzyme from Sulfolobus solfataricus with substrate analogue, substrate, and product. J Mol Biol 2002, 319 (3), 757-‐66. 12. Schlee, S.; Dietrich, S.; Kurcon, T.; Delaney, P.; Goodey, N. M.; Sterner, R., Kinetic Mechanism of Indole-‐3-‐glycerol Phosphate Synthase. Biochemistry 2012, 52 (1), 132-‐142. 13. Zaccardi, M. J.; Mannweiler, O.; Boehr, D. D., Differences in the catalytic mechanisms of mesophilic and thermophilic indole-‐3-‐glycerol phosphate synthase enzymes at their adaptive temperatures. Biochemical and Biophysical Research Communications 2012, 418 (2), 324-‐329. 14. Hennig, M.; Darimont, B.; Sterner, R.; Kirschner, K.; Jansonius, J. N., 2.0 A structure of indole-‐3-‐glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: possible determinants of protein stability. Structure 1995, 3 (12), 1295-‐306.

113
Chapter 5 Conclusions
5.1 A New Understanding of Catalysis by IGPS The work presented in this dissertation focused on the kinetic and chemical
mechanism of ssIGPS, and has greatly improved the current understanding for this
enzyme. Previous to these studies, ssIGPS had only been extensively studied at lower
temperature (25 to 40 °C), which limits the biologically relevant information that can be
attained about catalysis since the enzyme is naturally found at higher temperatures.
Understanding the differences between thermophilic and mesophilic enzymes requires
rigorous analysis over a range of temperatures. The studies presented in Chapter 2
indicated that the identity of the rate-determining step of ssIGPS is temperature
dependent. At lower temperatures (25 °C), product release is rate-determining, but as
temperature increases to 75 °C, the ring closure step becomes rate-determining.
Interestingly, ecIGPS showed a different rate-determining step at its biologically relevant
temperature, indicating that differences in environment can affect the kinetic mechanism
of enzymes.1
Previous research also lacked a clear understanding of all conserved and
catalytically important active site residues. Some studies were at odds in the assignment
of the general base; crystal structures suggested Glu159 performed this role whereas MD
simulations implicated Glu210.2-4 There were also several conserved residues whose role
in catalysis was ambiguous including Glu51, Lys53, and Arg182. Lastly, the role of
Lys110 as the general acid in two different steps of the reaction is not typical in enzyme
catalysis and the mode of reprotonation was unknown. The comprehensive kinetic

114
analysis on WT ssIGPS and variants for each of these conserved residues as presented in
Chapter 3 allowed a more complete understanding of the role of these residues. The
experiments unambiguously assigned Glu51 and Lys53 as the general base and acid
responsible for catalyzing the dehydration of the I2 intermediate to form the product. This
finding is at odds with previous suggestions that Glu159 (or Glu210) and Lys110
catalyze this step. However, this mechanism resolves the need to reprotonate Lys110
following the ring closure (where it donates a proton to the C2’ carbonyl). This
identification also indicates that there are two distinct active site surfaces, requiring that
the substrate undergo a reorientation in the active site after the formation of the I2
intermediate in order to place Lys53 and Glu51 in an appropriate position in order to
catalyze the dehydration.
In addition to analyzing those residues that are conserved, Chapter 4 examined
residues that are not conserved but are coevolving and showed correlated motion, both of
which can be important to the catalytic mechanism.5 Considering the importance of
active site loop residues Lys53 and Phe89, coevolving residues Arg54 and Asn90 were
examined. At 75 °C, the rate-determining step of the amino acid substitution Asn90Ala
was dehydration, in contrast to ring closure in WT ssIGPS. Additionally, the activity of
Asn90Ala variant was decreased compared to WT enzyme at lower temperatures,
indicating that this amino acid substitution is more deleterious at lower compared to
higher temperatures.
Similar evaluation of amino acid substitutions at the Arg54 position in ssIGPS
showed an important role for this residue as well. The results suggest that the interaction
between Asn90 and Arg54 is important for proper function of the general acid-base pair,

115
Lys53-Glu51. Additionally, the deleterious effect of Asn90Ala variant is increased at
lower temperatures. This result along with MD simulations performed by Shen et al.
indicate that the interaction between the β1α1 and β2α2 loops through Arg54 and Asn90
may be involved in protein motions that are important for the chemical events of
catalysis.5 Together, these studies provide molecular level details on the role for active
site residues in ssIGPS catalysis, as well as a more complete view of catalysis by ssIGPS.
5.2 Implications for Understanding the Evolution of Thermophilic Versus Mesophilic Enzymes Efficient catalysis by enzymes requires a delicate balance between stability and
flexibility. Proteins from thermophilic organisms have developed an excess of stabilizing
interactions like salt bridges, in order to remain stable at high temperatures, which causes
a decrease in flexibility at lower temperatures. Conversely, their mesophilic homologs
lack these same structural elements, despite the similarity in structure and fold, allowing
increased flexibility at lower temperatures.6 The results from Chapter 2 shed some
insight into the evolutionary differences between thermophilic ssIGPS and mesophilic
ecIGPS. The two enzymes display differences in the rate-determining step of reaction. In
ssIGPS, the ring closure step is rate-determining, whereas this result is not seen for
ecIGPS.
This difference may be due to the added stabilizing interactions in ssIGPS that
create increased rigidity of the enzyme. In ecIGPS, there are fewer salt bridges, allowing
the enzyme to be more flexible. We propose that the ring closure step involves dynamic
motions in the enzyme that allow the bond between C1 and C2’ to form (as was also
proposed by Bruice and coworkers3, 4). ssIGPS cannot complete this step as quickly at

116
lower temperatures due to limited flexibility, while it can occur more quickly in ecIGPS.
Therefore, in ssIGPS this step has become rate-determining. In agreement with this idea
is the temperature dependence of the kinetic mechanism for ssIGPS. The decreased
motion at lower temperatures slows this step more severely than at higher temperatures
causing changes to the kinetic mechanism at different temperatures.
5.3 Engineering New Indole Derivatives and Improving Industrial Indole Synthesis with IGPS Many different industries including pharmaceuticals, agriculture, and materials
would benefit from the integration of enzyme technology into their processes.7 As a
thermophilic enzyme, ssIGPS is robust and can remain stable under high temperatures
and harsh conditions, which makes it quite advantageous for industrial processes
compared to its mesophilic homologs. Additionally, the IGPS catalyzed reaction is
industrially relevant, as indole is a widely used structure in many applications.8 In the
typical industrial catalysis, indole and its derivatives are made using the Fischer indole
synthesis or the Japp-Klingemann reaction.9 While both of these reactions have been
optimized for the desired product, they require the use of high heat, extreme pH,
nonaqueous solvent, and heavy metal catalysts. Additionally, they require the production
of intermediates that can be difficult to attain, and they can create byproducts, which
decrease the efficiency of the reaction and make purification more difficult.9 Introduction
of IGPS for indole derivative production would resolve many of these issues, and allow
for more environmentally friendly processes that do not require the use of nonaqueous
solvents or heavy metal catalysts. The issue that remains with this application is the
introduction of a substrate derivative into the catalyzed reaction. The widely relevant

117
indoles in industry are highly derivitized. Therefore, the active site of IGPS needs to be
adjusted to account for the new side chains.
The assignment of Lys53 and Glu51 as the general acid and base in the
dehydration step of the ssIGPS catalyzed reaction must be taken into consideration in the
design of enzymes for the industrial synthesis of indoles. Previous to this finding, the
unknown role for Glu51 may have resulted in its substitution in the design of an enzyme
for the production of an IGP derivative, which would prevent the proper catalysis by
IGPS. Similarly, side chains Arg182 and Glu210 are now known to be important for
substrate binding. The modification of the substrate may require that the position of these
residues be changed as well as the size of the side chain. For example, Arg182 may be
substituted with a Lys to accommodate a slightly larger substrate, or to Glu or Asp to
accommodate a positively charged group. Understanding how the WT residues impact
catalysis allows for more thoughtful design of a modified enzyme.
5.4 Improving Novel Enzyme Engineering Efforts
Despite the wide reaching applications of engineering novel enzymes capable of
catalyzing non-natural reactions, and the large amount of resources being used to
engineer such enzymes, scientists are still unable to match the rate enhancements
achievable by natural enzymes.10 Studies have shown that this may be due to a inability
to appropriately model the dynamics of the enzyme when designing the new enzyme
activity onto naturally occurring enzyme scaffolds; our understanding of enzyme
dynamics is still in its infancy.11 Bioinformatics approaches performed by Juritz et al.
examined the differences in dynamics of enzymes with similar tertiary structure and

118
correlated the results with protein sequence.12 The results suggest that sequence diversity
evolved among enzymes to allow for differences in dynamics. This finding suggests that
there is a specific relationship between sequence and dynamics that has yet to be
sufficiently understood, but will be essential in expanding the application of enzymes in
industry. Our ability to understand how sequence diversity contributes to dynamics and
catalysis is essential to further the knowledge of the role for enzyme dynamics in
catalysis, but requires extensive analysis of how individual residues contribute to
catalysis and dynamics. Enzymes containing the (β/α)8-barrel fold have very similary
structures, but catalyze a diverse range of reactions, which makes this fold an excellent
model for understanding the relationship between sequence, structure, and dynamics.
The idea that the sequence diversity is related to dynamics among enzymes with
the same three-dimensional structure may help explain the coevolution of residues in
enzymes. Many times changing residues does not change interactions; however, these
changes may alter dynamic fluctuations in the protein. Dynamic fluctuations can also be
thought of as the making and breaking of noncovalent interactions like hydrogen
bonds.13, 14 In ssIGPS, the hydrogen bond interaction between Arg54 and Asn90 helps to
aid in the dehydration step of the reaction. In ecIGPS this same interaction is between
Lys55 and Gln94. The difference in the interaction, caused by differences in the residues
at these positions, may create differences in how the enzymes fluctuate, and may
represent one reason for the coevolution, especially considering that comparison of
ssIGPS and ecIGPS has shown inherent differences in their kinetic mechanisms.
It has been long understood that amino acid residues that do not participate
directly in chemistry can still be important for folding and other processes. The result that

119
residues, like Arg54 and Asn90, can change the rate-determining step of the reaction
without showing significant changes in catalytic turnover also indicates that these
residues are important for the chemistry of the reaction. In enzyme engineering studies,
there is concern with understanding how a reaction proceeds. Based on the kinetic results
for the Asn90Ala mutant, it is clear that this variant affects proper function of the general
acid and base, Lys53 and Glu51. We propose that this amino acid is partially responsible
for the reorientation of the I2 intermediate in the active site. More specifically, Asn90Ala
and its interaction with the β1α1 loop, is involved in a conformational change that allows
for proper reorientation of the I2 intermediate prior to dehydration. The temperature
dependence of Asn90Ala ssIGPS activity indicates that its function is likely gated
through a dynamic process. Changing this residue (or other similar residues) in an
enzyme-engineering scaffold can create unexpected dynamic changes that can affect the
activity of the new model. Current algorithms for the design of enzymes are not able to
account for all of the dynamic changes in the enzyme that are caused by sequence since
we do not fully understand how sequence affects dynamics. This effect is perhaps part of
the reason current efforts toward engineering novel enzymes are falling short of natural
enzymatic rate enhancements. Studies like that presented here, along with experimental
studies of the enzymes dynamics in IGPS and other enzymes will help to develop this
understanding.

120
5.5 Future Studies
This research has evaluated the roles for both conserved and nonconserved
residues in ssIGPS catalysis. One of the most intriguing findings from this work is that
the substrate must undergo a reorientation during the reaction, which may be aided by the
enzyme. The next step is to understand how the enzyme undergoes dynamic fluctuation
in order to aid in this step of catalysis, especially considering the results by Goodey and
Sterner implicate dynamics of the β1α1 loop in catalysis even at low temperatures (25
°C).15 In these studies, the role of conformational motion in the active site β1α1 loop was
examined using the addition of fluorescent dyes. The authors suggest that the binding of
substrate and the chemical steps of catalysis are accompanied by conformational changes
in the enzyme.16
To further evaluate the role of dynamics, and gain additional site-specific
information about the conformational sampling for the IGPS enzyme, NMR can be
utilized. NMR is a robust technique and allows enzyme dynamics to be probed across
multiple timescales ranging from picosecond to second. Additionally, it allows the
measurement of parameters for almost every amino acid in an enzyme, providing site-
specific resolution of dynamics and conformational fluctuation.16 Preliminary NMR
experiments utilizing Carr-Purcell-Meiboom-Gill (CPMG) R2 relaxation dispersion
indicated that ssIGPS does not undergo very much conformational exchange on the
microsecond to millisecond timescale at the temperatures assayed (293 to 313K). The
lack of conformational exchange in the relaxation dispersion experiments can be
explained several ways. First, it is possible that the motions that contribute to IGPS
catalysis are occurring on a different timescale, and thus are not able to be measured

121
using this technique. Based on the kinetic results of WT and amino acid substituted
ssIGPS, we believe that loop motions help to reorient the substrate in the active site
during the reaction, but these motions may be too slow to be measured by relaxation
dispersion. Other NMR techniques that provide dynamic information on slower
timescales such as hydrogen-deuterium exchange spectroscopy may provide more
applicable information.
A second explanation for the lack of measurable conformational exchange by
relaxation dispersion is that these experiments were completed at low temperatures (20 to
40 °C) compared to the biologically relevant temperature of ssIGPS. Considering
previous results that indicate flexibility is decreased at lower temperatures and the
enzyme is more rigid, it is likely that even if the applicable conformational exchange of
IGPS is on the appropriate timescale, at lower temperatures the enzyme is too rigid and is
not undergoing the same exchange as at higher temperatures. This idea is consistent with
the results of Bruice et al. in which the number of NACs is lower at lower temperatures.3,
4 Unfortunately, the use of the cryoprobe in the NMR experiments prevents us from
performing the experiments at a temperature more biologically relevant for ssIGPS.
In order to overcome this issue, studying a mesophilic homolog like ecIGPS
would allow for NMR data to be collected at a temperature biologically appropriate for
the enzyme activity. However, NMR requires a large amount of protein compared to
traditional biochemical techniques, and the current ecIGPS construct in use in our
laboratory exhibits precipitation at high concentrations (~1 mM). The instability of the
enzyme may be caused by the truncation of the original gene, which contained both
ecIGPS and the preceding enzyme in the pathway, PRAI, which are found as one

122
bifunctional complex in E. coli naturally. Use of the entire bifunctional complex would
allow for the higher concentrations needed for NMR without instability problems, but
this enzyme is large by NMR standards (~50 kDa). To make these studies more feasible,
methodologies similar to those used in our laboratory to study RNA-dependent RNA
polymerase from poliovirus17 be used to examine the ecPRAI:IGPS complex. In these
experiments only certain residues in the enzyme are labeled (e.g. methionine), rather than
the entire backbone, and these resonances are used as probes for examining enzyme
dynamics and function. This will decrease spectral crowding and allow for the dynamics
of the entire complex be examined, as they may be different from the monofunctional
ecIGPS.
In addition to understanding the dynamics of ssIGPS, NMR can also allow the
orientation of the substrate in the active site to be probed. While crystal structures of
IGPS in complex with both rCdRP and IGP have been produced, it is clear from the
kinetic analysis that these structures likely do not represent the catalytically relevant
complex, as the C1 and C2’ carbons in rCdRP are located too far apart for a bond to
form, and IGP is bound such that Lys53 and Glu51 are not appropriately positioned for
dehydration of I2 to IGP. Exchange-transferred nuclear Overhauser effect (tr-NOE)
experiments can be explored as they allow for the measurement of inter-nuclear distances
in the ligand bound to substrate. Additionally, tr-NOE experiments do not require the use
of a cryoprobe, and thus experiments can be performed at temperatures more biologically
relevant to ssIGPS. These studies may provide valuable information about the orientation
of substrate at biologically relevant temperatures.
The relationship between sequence and dynamics will be important for the future

123
development of enzyme technologies. The kinetic analysis presented in this dissertation
provides the understanding for how amino acid substitutions in ssIGPS can affect the
activity, and the NMR studies on WT IGPS can provide insight into how motion
contribute to catalysis. In order to complete the understanding for how sequence in IGPS
or other (β/α)8-barrel enzymes is related to the dynamics, understanding how these
mutations change both the activity and the dynamics will complete the larger picture.
Therefore, utilization of NMR not only on WT enzyme but also on ssIGPS variants
should be explored.
5.6 Conclusions
This dissertation redefined the catalytic and kinetic mechanisms of the IGPS
enzyme. The results presented here will aid in a more complete picture of catalysis by
enzymes and will aid in the understanding of the relationship between sequence and
dynamics. It is also directly relevant to several different applications including enzyme
engineering and indole synthesis.
5.7 References
1. Zaccardi, M. J.; Mannweiler, O.; Boehr, D. D., Differences in the catalytic mechanisms of mesophilic and thermophilic indole-‐3-‐glycerol phosphate synthase enzymes at their adaptive temperatures. Biochemical and Biophysical Research Communications 2012, 418 (2), 324-‐329. 2. Hennig, M.; Darimont, B. D.; Jansonius, J. N.; Kirschner, K., The catalytic mechanism of indole-‐3-‐glycerol phosphate synthase: crystal structures of complexes of the enzyme from Sulfolobus solfataricus with substrate analogue, substrate, and product. J Mol Biol 2002, 319 (3), 757-‐66. 3. Mazumder-‐Shivakumar, D.; Bruice, T. C., Molecular dynamics studies of ground state and intermediate of the hyperthermophilic indole-‐3-‐glycerol phosphate

124
synthase. Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (40), 14379-‐14384. 4. Mazumder-‐Shivakumar, D.; Kahn, K.; Bruice, T. C., Computational study of the ground state of thermophilic indole glycerol phosphate synthase: Structural alterations at the active site with temperature. Journal of the American Chemical Society 2004, 126 (19), 5936-‐5937. 5. Shen, H. B.; Xu, F.; Hu, H. R.; Wang, F. F.; Wu, Q.; Huang, Q.; Wang, H. H., Coevolving residues of (beta/alpha)(8)-‐barrel proteins play roles in stabilizing active site architecture and coordinating protein dynamics. Journal of Structural Biology 2008, 164 (3), 281-‐292. 6. Feller, G., Protein stability and enzyme activity at extreme biological temperatures. Journal of Physics-Condensed Matter 2010, 22 (32). 7. Woodley, J. M., Protein engineering of enzymes for process applications. Current Opinion in Chemical Biology 2013, 17 (2), 310-‐316. 8. Barden, T. C., Indoles: Industrial, agricultural and over-‐the-‐counter uses. Topics in Hetercyclic Chemistry 2011, 26, 31-‐46. 9. Humphrey, G. R.; Kuethe, J. T., Practical methodologies for the synthesis of indoles. Chemical Reviews 2006, 106 (7), 2875-‐2911. 10. Ruscio, J. Z.; Kohn, J. E.; Ball, K. A.; Head-‐Gordon, T., The Influence of Protein Dynamics on the Success of Computational Enzyme Design. Journal of the American Chemical Society 2009, 131 (39), 14111-‐14115. 11. Lassila, J. K., Conformational diversity and computational enzyme design. Current Opinion in Chemical Biology 2010, 14 (5), 676-‐682. 12. Juritz, E.; Palopoli, N.; Silvina Fornasari, M.; Fernandez-‐Alberti, S.; Parisi, G., Protein Conformational Diversity Modulates Sequence Divergence. Molecular Biology and Evolution 2013, 30 (1), 79-‐87. 13. Boehr, D. D., During Transitions Proteins Make Fleeting Bonds. Cell 2009, 139 (6), 1049-‐1051. 14. Hammes, G. G., Multiple conformational changes in enzyme catalysis. Biochemistry 2002, 41 (26), 8221-‐8228. 15. Schlee, S.; Dietrich, S.; Kurcon, T.; Delaney, P.; Goodey, N. M.; Sterner, R., Kinetic Mechanism of Indole-‐3-‐glycerol Phosphate Synthase. Biochemistry 2012, 52 (1), 132-‐142.

125
16. Mittermaier, A. K.; Kay, L. E., Observing biological dynamics at atomic resolution using NMR. Trends in Biochemical Sciences 2009, 34 (12), 601-‐611. 17. Yang, X.; Welch, J. L.; Arnold, J. J.; Boehr, D. D., Long-‐Range Interaction Networks in the Function and Fidelity of Poliovirus RNA-‐Dependent RNA Polymerase Studied by Nuclear Magnetic Resonance. Biochemistry 2010, 49 (43), 9361-‐9371.

126
APPENDIX
Solvent Deuterium Kinetic Isotope Effect Analysis* *The following analysis was completed by Eric Yezdimer
To demonstrate the connection between the observed SDKIE and each discrete
mechanistic step, it is useful to consider the following generalized series of chemical
reactions as a feasible model for IGPS catalysis:
(A.1)
where S, ES, EI, EP, P, and E denote substrate, enzyme-substrate complex, enzyme-
intermediate complex, enzyme-product complex, product, and free enzyme, respectively.
Since the first intermediate is only thought to be fleetingly stable, and the release of
carbon dioxide is the first irreversible step of the reaction, the kinetic scheme was
simplified to only include one intermediate, the I2. The corresponding rate formulas for
each step are given by:
(A.2)
(A.3)
(A.4)
!
S + E"k#1
k1ES$
k2EI"
k#3
k3EP"
k#4
k4E + P
!
d[P]dt
= k4[EP] " k"4[E][P]
!
d[EP]dt
= k3[EI]+ k"4[E][P] " k"3[EP] " k4[EP]
!
d[EI]dt
= k2[EI]+ k"3[EP] " k3[EI]

127
(A.5)
(A.6)
Under conditions where the initial substrate concentration is much larger than the initial
free enzyme concentration, [S]o>>[E]o, the following steady-state approximations for the
enzyme bound species can be applied:
(A.7) Using the mass balance equations for the enzyme,
(A.8) and substrate/product concentrations,
(A.9)
Equation A.7 can be rewritten into the familiar Michaelis-Menton form.
(A.10)
where k’cat and K’M are parameters defined by,
(A.11)
!
d[ES]dt
= k1[E][S] " k"1[ES] " k2[ES]
!
d[S]dt
= k"1[ES] " k1[E][S]
!
d[EP]dt
=d[EI]dt
=d[ES]dt
= 0
!
[E] = [E]o " [ES] " [EI] " [EP]
!
[S] " [S]o # [P]
!
d[P]dt
= k 'cat[E]o([S]o " [P])K 'M +[S]o " [P]
!
k'cat =k2k3k4
k3k4 + k2(k3 + k"3k4 )

128
(A.12)
Within the limiting initial rate behavior of the Michaelis-Menton Equation (A.7), [P] is
assumed to be near zero, whichr reduces Equation A.12 to an extended Michaelis
constant,
(A.13)
K’M can be experimentally determined along with k’cat. Dividing equations A.11 and
A.13 provides an expression for catalytic efficiency, k’cat/K’M,
(A.14)
To understand the source of the experimental SDKIE for ssIGPS in the k’cat and K’M
constants, we assumed that ony rate constants for the chemical steps k2, k3, and k-3 are
potentially isotope sensitive. Both steps of IGPS catalysis involve proton transfers with
solvent exchangeable protons. Rate constants under H2O or D2O solvent conditions will
!
K 'M =(k2 + k"2)(k3k4 + k3k"4[P])k1(k2k3 + k2k4 + k3k4 + k2k"3)
!
K 'M =k3k4k2 + k"1k3k4
k1(k2k3 + k2k4 + k3k4 + k2k"3)
!
k 'catK 'M
=k1k2k3k4 (k2k3 + k2k4 + k3k4 + k2k"3)
(k2 + k"1)(k3k4 + k2(k3 + k4 + k"3))(k3k4 + k3k"4[P]+ k"3k"4[P])

129
be denoted by either “H” or “D” subscripts, respectively. The SDKIE for k’cat and K’M
are given by
(A.15)
(A.16)
This result illustrates the ability of the SDKIE for k’cat/K’M to report on steps after an
irreversible step. Furthermore, if k-1<<k2, as is predicted to occur for enzymes such as
ssIGPS with strong susbtrate binding affinity, then the SDKIE for k’cat/K’M is reduced to
one.
It is also valuable to consider k’cat,H/k’cat,D under various limiting conditions in
order to highlight other factors that could suppress the SDKIE. In cases where the
conversion of ES to EI represents a much slower catalytic step than the conversion of EI
to EP (k2<<k3), the SDKIE for k’cat simplifies to
(A.17)
!
k'cat,Hk'cat,D
=k2,Hk3,H (k2,Dk3,D + k2,Dk4 + k3,Dk4 + k2,Dk"3,D )
k2,Dk3,D (k2,Hk3,H + k2,Hk4 + k2,Hk"3,H )
!
k 'cat,HK 'M ,H
k 'cat,DK 'M ,D
=k2,Hk3,H (k2,D + k"1)(k3,Dk4 + k3,Dk"4[P]+ k"3,Dk"4[P])k2,Dk3,D (k2,H + k"1)(k3,Hk4 + k3,Hk"4[P]+ k"3,Hk"4[P])
!
k'cat,Hk'cat,D
=k2,H (k2,D + k4 )k2,D + k4

130
Under these conditions, the SDKIE should report on the conversion of ES to EI provided
that product release is similar to or faster than the irreversible formation of the
intermediate. If product release is very slow (k4<<k2<<k3), the SDKIE will reduce to one.
In the opposite situation where the conversion from EI to EP is much slower than
the conversion of ES to EI (k3<<k2), the SDKIE for k’cat is given by,
(A.18)
If product release is very slow under these conditions (k4<<k3<<k2)
(A.19)
This analysis indicates that under conditions of very slow product release (k4<<k2,k3), as
is predicted from SVE experiments for ssIGPS at 25 °C,1 the SDKIE for k’cat should
approach one if
(i) k2 << k3
Or
(ii) k3 << k2, and k-3 << k3
Under conditions of fast product release, as is predicted from SVE experiments for
ssIGPS at 75 °C, the SDKIE for k’cat should approach one if
!
k'cat,Hk'cat,D
=k3,H (k3,D + k4 + k"3,D )k3,D (k3,H + k4 + k"3,H )
!
k'cat,Hk'cat,D
=k3,H (k3,D + k"3,D )k3,D (k3,H + k"3,H )

131
(i) k2 << k3 and k2 is isotope insensitive
Or
(ii) k3 << k2 and k3, k-3 are isotope insensitive.
To differentiate these two scenaries, the effects of amino acid substitutions on
residues predicted to be important for these steps (i.e. Glu51 and Lys53) must be
examined. Amino acid substitutions are predicted to substantially slow the second step
chemical step such that k3<<k2. Considering that Glu51Gln and Lys53Arg substitutions
result in an SDKIE of approximately one, these results suggest that k3 and k-3 are solvent
isotope insensitive. It follows that the observation of an SDKIE for k’cat for WT at 75 °C,
k2<<k3 as equation A.18 will also reduce to one if k3, k-3 are not isotope sensitive. This
logic is also consistent with proton inventory studies on WT ssIGPS that indicate a single
proton transfer event if responsible for the SDKIE.

VITA
Margot J. Zaccardi
Education 2008-2013 Ph.D. in Chemistry The Pennsylvania State University, University Park, PA 2004-2008 B.S. in Chemistry, Research Option The Florida Institute of Technology, Melbourne, FL Publications Margot J. Zaccardi, Yezdimer O., Boehr, D.D. Functional identification of the general acid and base in the dehydration step of indole-3-glycerol phosphate synthase catalysis. JBC 2013, In Press. Margot J. Zaccardi, Mannweiler, O., Boehr, D.D. Differences in the catalytic mechanisms of mesophilic and thermophilic indole-3-glycerol phosphate synthase enzymes at their adaptive temperatures. BBRC. 2012, 418, 324-329. Margot J. Zaccardi, Olson, J.A., and Winkelmann, K. Preparation of Electrochemically Etched Tips for Ambient Instructional Scanning Tunneling Microscopy. J. Chem. Ed. 2010, 87(3), 303-310. Presentations Margot J. Zaccardi, Yezdimer, E.M., Boehr, D.D. “A new understanding of the catalytic mechanism of the tryptophan biosynthetic enzyme indole-3-glycerol phosphate synthase” Poster Presenter at the Enzyme Mechanisms Conference. San Diego, CA, January 3-7, 2013. Margot J. Zaccardi, Yezdimer, E.M., Boehr, D.D. “Understanding the catalytic function of protein dynamics in indole-3-glycerol phosphate synthase” Poster Presenter at American Chemical Society National Meeting. Philadelphia, PA, August 19-23, 2012. Margot J. Zaccardi, Axe, J.M., Boehr, D.D. “Mutational Analysis of Coevolving Residues in the Tryptophan Biosynthetic Enzyme Indole-3-glycerol Phosphate Synthase” Poster Presenter at Gordon Research Conference: Enzymes, Coenzymes, and Metabolic Pathways. Waterville Valley, NH, July 10-15, 2011.