
ln compliance with the - McGill Universitydigitool.library.mcgill.ca/thesisfile83088.pdf ·...
169
ln compliance with the Canadian Privacy Legislation sorne supporting forms may have been removed from this dissertation. While these forms may be included in the document page count, their removal does not represent any loss of content from the dissertation.
Transcript of ln compliance with the - McGill Universitydigitool.library.mcgill.ca/thesisfile83088.pdf ·...

ln compliance with the Canadian Privacy Legislation
sorne supporting forms may have been removed from
this dissertation.
While these forms may be included in the document page count,
their removal does not represent any loss of content from the dissertation.


EIlhancing Hydrolase Activity and Selectivity
by Medium, Substrate, and Protein
Engineering
SeQngsoon Park
A thesis submitted ta the Faculty afGraduate Studies and Research of McGill University
in partialfulfillment of the requirements of the degree of Dactar of Philasaphy
Department of Chemistry Mc Gill University Montréal, Québec Canada
January 2003
© Seongsoon Park, 2003

1+1 National Library of Canada
Bibliothèque nationale du Canada
Acquisitions and Bibliographie Services
Acquisisitons et services bibliographiques
395 Wellington Street Ottawa ON K1A ON4 Canada
395, rue Wellington Ottawa ON K1A ON4 Canada
The author has granted a nonexclusive licence allowing the National Library of Canada to reproduce, loan, distribute or sell copies of this thesis in microform, paper or electronic formats.
The author retains ownership of the copyright in this thesis. Neither the thesis nor substantial extracts from it may be printed or otherwise reproduced without the author's permission.
Canada
Your file Votre référence ISBN: 0-612-88548-8 Our file Notre référence ISBN: 0-612-88548-8
L'auteur a accordé une licence non exclusive permettant à la Bibliothèque nationale du Canada de reproduire, prêter, distribuer ou vendre des copies de cette thèse sous la forme de microfiche/film, de reproduction sur papier ou sur format électronique.
L'auteur conserve la propriété du droit d'auteur qui protège cette thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou aturement reproduits sans son autorisation.

Abstract
Researchers use enzymes for enantio- and regioselective reactions because of
their high selectivity and activity toward natural substrates. However, researchers
sometimes need to modify the reaction system or the enzyme itself to get reliable
selectivity and activity when they deal with unnatural substrates. To obtain researcher's
need, one can change the solvent, modify the substrates, or alter the enzyme itself. These
processes are called medium, substrate, and protein engineering, respectively.
This thesis deals with hydrolases, which are classified by EC 3. We applied the
proper approach to improve their activity and selectivity depending on the reactions. For
the first approach, highly polar ionic liquids were applied to lipase-catalyzed acylation.
Ionie liquids worked reliably in enantio- and regioselective lipase-catalyzed reactions. In
particular, ionic liquids dissolved polar substrates such as glucose and L-ascorbic acid,
thereby facilitating their acylations. In the second approach to improving
enal1tioselectivity of CAL-B (Candida antarctica lipase B) in p-Iactam ring opemng
. reactions, we changed the nucleophile from water to a range of alcohols. Longer,
·secondary alcohols increased the reaction rate as weil as the enantioselectivity. Molecular
modeling revealed that the high enantioselectivity of CAL-B and the critical role of
. alcohols. For the last approach, structure-guided random mutagenesis was applied to
increase the enantioselectivity of PFE (Pseudomonas fluorescens esterase) toward
MBMP (methyl 3-bromo-2-methylpropionate). The homology model was used to select
amÎl10 acid residues for mutagenesis near the stereocenter of the docked tetrahedral
intermediate of the substrate. RandomÎzation of these residues yielded a Va1122Ser
mutant with E increased to 61 (from 12 of wild type enzyme), as weIl as a Va1122Met
mutant to 36.
11

Résumé
Les chercheurs utilisent les enzymes pour les réactions d'enantio- et regioselective
en raison de leur sélectivité et activité élevées vers les substrats naturels. Cependant,
quand ils traitent les substrats artificiels les chercheurs doivent parfois modifier le
système de réaction ou l'enzyme lui-même pour obtenir la sélectivité et l'activité fiables.
Pour obtenir le besoin du chercheur, on peut changer le solvant, modifier les substrats, ou
changer l'enzyme lui-même. Ces processus s'appellent et la technologie du milieu, du
substrat, de la protéine.
Cette thèse traite les hydrolases, qui sont classifiées par EC 3. Nous avons
appliqué l'approche appropriée pour améliorer leur activité et sélectivité selon les
réactions. Pour la première approche, des liquides ioniques fortement polaires ont été
appliiqués à l'acylation catalysée par un lipase. Les liquides ioniques ont fonctionné
sûrement dans les réactions regioselective et enantioselective - et par un catalysée lipase-.
En particulier, les liquides ioniques ont dissous les substrats polaires tels que le glucose et
l'acide L-~scorbique, facilitant de ce fait leurs acylations. Pour la deuxième approche
visant à améliorer l'enantioselectivity de CAL-B (la lipase B de Candida antarctica) dans
des réactions d'ouverture d'anneau de p-Iactame, nous avons changé le nucleophile de
l'eau en gamme des alcools. Plus longs, les alcools secondaires ont augmenté les taux de
réaction aussi bien que l'enantioselectivité. Modeler moléculaire montra que la haute
enantioselectivity de CAL-B et le rôle critique des alcools. Pour la dernière approche, la
mutagénèse aléatoire structure-guidée a été appliquée pour augmenter l'enantioselectivity
de PFE (estérase de Pseudomonas fluorescens) vers MBMP (3-bromo-2-methyl
propionate méthylique). Le modèle d'homologie a été employé pour choisir des résidus
d'acide aminé pour la mutagénèse près du stereocenter de l'intermédiaire tétraédrique
accouplée du substrat. Randomisation de ces résidus a rapporté un mutant de Va1l22Ser
avec E augmenté jusqu'à 61, aussi bien qu'un mutant de Va1122Met à 36.
III

Acknowledgements
"And Gad said, Let there be /ight: and there was light. And Gad saw the /ight, that
it was good: and Gad divided the light trom the darkness"
GENESIS 1:3-4
First, 1 would like to thank my supervisor, Dr. Romas J. Kazlauskas, for aIl his
help, support, and guidance during my studies. He always encouraged me, ev en wh en 1
made an absUfd idea. 1 do not believe that 1 could finish this long work without his
encouragement.
1 would also like to thank my parents for all of their support. My father always
says, "Never give up yOuf endeavor and do yOuf best although you may not succeed." His
words will be in my mind forever. 1 truly appreciate my mom's full of love and support.
And also 1 should thank my brother and sister for their love and support.
1 thank the people that 1 have collaborated with during my time here, namely Dr.
Eniko Forr6, Geoffrey P. Horsman, Krista Morley, and Harjap Grewal. Their innovative
ideas and suggestion keep me awake to work. And 1 should thank Vladimir and Geoff for
OUf small research to find the best place for making innovative ideas in Montreal. 1
appreciate the collaboration and help of Eniko and Krista.
1 also thank Prof. Karl HuIt and Fredrik Viklund who collaborated during my
staying in Stockholm. Prof. Karl HuIt directed me to finish up my work in Stockholm.
And Fredrik and Joke's kind help made me get successful stay in Stockholm.
Finally 1 would like to thank all of people in the Kazlauskas research group
(Alessandra, Shu, Ebru, Paul, Jeremy, and Chris) also the people whom 1 met during my
staying in Montreal. 1 will not forget initial help of Alessandra, Shu, and Ebru when 1
joined OUf group. Jeremy, Chris, Cedric, and Oleh, they are my true coworkers in the lab
of Thomson house. And also 1 greatly thank my Korean friends, Dr. Kyunil Rah, Dr.
Taeho Lee, and Dr. Yongkyun Cho for sharing their knowledges.
IV

l'm Ieaving Montreal now, but believe that 1 will see themall in the world of
Chemistry. And my joumey to find the truth will be continued un der the direction of
God. Thank God and please keep watching me.
"Ami he said unto them, Go ye into ail the world, and preach the gospel to evety
creature. "
MARK 16:15
v

Thesis Formatting
The following text, concernmg the inclusion of manuscripts m a thesis, is
reproduced from the "Guidelines for Thesis Preparation".
"As an alternative to the traditional thesis fonnat, the dissertation can consist of a
collection of papers that have a cohesive, unitary character making them a report of a
single program of research. The structure for the manuscript-based thesis must confonn
to the following:
Candidates have the option of including, as part of the thesis, the text of one or
more papers submitted, or to be submitted, for publication, or the clearly-duplicated text
(not the reprints) of one or more published papers. These texts must confonn to the
Thesis Preparation Guidelines with respect to font size, line spacing, and margin sizes
and must be bound together as an integral part of the thesis. (Reprints of published papers
can be included in the appendices at the e:Qd of the thesis.)
The thesis must be more than a collection of manuscripts. AlI components must be
integrated into a cohesive unit with a logical progression from one chapter to the next. In
order to ensure that the thesis has continuity, connecting texts that provide logical
bridges between the different papers are mandatory.
The thesis must conform to aIl other requirements of the "Guidelines for Thesis
Preparation" in addition to the manuscripts. The thesis must include the following: a table
of contents; an abstract in English and French; an introduction which clearly states the
rational and objectives of the research, a comprehensive review of the literature (in
addition to that covered in the introduction to each paper); a final conclusion and
summary. Students are not required to organize their references into one
comprehensive Iist in a manuscript-based thesis. They are free to choose wh ether to
make a comprehensive Iist or to put references after each paper.
VI

As manuscripts for publication are frequently very concise documents, where
appropriate, addition al material must be provided (e.g., in appendices) in sufficient detail
to allow a clear and precise judgement to be made of the importance and originality of the
research reported in the thesis.
In general, when co-authored papers are included in a thesis the candidate must
have made a substantial contribution to all papers included in the thesis. In addition, the
candidate is required to make an explicit statement in the thesis as to who
contributed to such work and to what extent. This statement should appear in a single
section entitled "Contribution of Authors" as a preface to the thesis. The supervisor
must attest to the accuracy of the statement at the doctoral oral defence. Since the task of
the examiners is made more difficult in these cases, it is in the candidate's interest to
clearly specify the responsibilities of aIl the authors of the co-authored material.
When previously published copyright material is presented in the thesis, the
candidate must obtain, if necessary, signed waivers from the co-authors and publishers
and submit these to the Thesis Office with the final deposition.
Irrespective of the internaI and external examiners reports, if the oral defence
committee feels that the thesis has major omissions \vith regard to the above guidelines,
the candidate may be required to resubmit an amended version of the thesis.
In no case can a co-author of any component ef such a thesis serve as an external
examiner for that thesis."
vii

Contribution of Authors
This thesis represents a collection of one manuscript, three drafts, and one
appendix. Chapter 1 and Chapter 3 appendix have been published or accepted,
respectively, and three drafts (Chapter 3, 4, and 5) will be submitted for publication
shortly. AlI of the work contained in these drafts has been completed as part of my
research for the degree of Doctor of Philosophy.
The work in Chapter 2 was done under supervision of my research director, Dr.
Romas J. Kazlauskas. 1 synthesized ionic liquids, measured polarities of ionic liquids, and
performed aIl lipase-catalyzed reactions. In addition, 1 interpreted all the experimental
and structural determination data such as GC analysis, IH, and 2-D NMR.
1 conducted the work in Chapter 3 under supervision of Dr. Karl HuIt as weIl as
Dr. Romas J. Kazlauskas and with the collaboration of Fredrik Viklund. Fredrik Viklund
optimized the separation condition of reaction mixtures for HPLC analysis and performed
a large-scale synthetic reaction of L-ascorbyl oleate in tert-amyl alcohol. 1 synthesized
ionic liquids and performed aIl lipase-catalyzed reactions in ionie liquids.
The work in Chapter 4 was done with collaboration of Dr. Eniko Forro and
Harjap Grewal un der the guidance of Dr. Romas J. Kazlaus~as. One collaborator, Dr.
Eniko Forro, performed sm aIl scale and 0.5 g-scale reactions. Harjap Grewal did initial
computer modeling. 1 performed initial screening with commercial hydrolases and sorne
smaH scale reactions. 1 also did the detailed computer modeling.
In Chapter 5, 1 collaborated with Geoffrey P. Horsman and Krista Morley un der
supervision of Dr. Romas J. Kazlauskas. Geoffrey P. Horsman chose the amino acid
residues for mutagenesis and Krista Morley measured the end-point E value of compound
2, methyl 2-methylbutyrate. 1 perfonned mutagenesis work, screened mutant Iibraries,
optimized a docking model, designed the substrate analogues, determined most end-point
E values, and measured the kinetic parameters.
viii

Table of Contents
Abstract ............................................................................................................................... ii
Résumé ............................................................................................................................... iii
Acknowledgements ............................................................................................................ iv
Thesis Formatting .............................................................................................................. vi
Contribution of Authors ................................................................................................... viii
Table of Contents ............................................................................................................... ix
Glossary of Frequently Used Symbols and Abbreviations .............................................. xiii
Chapter 1 ............................................................................................................................. 1
Chapter 1. Introduction ....................................................................................................... 2
1.1. Hydrolases ................................................................................................................ 2
1.1.1. Lipase (EC 3.1.1.3) ........................................................................................... 4
l.l.2. Esterase (Carboxyester hydrolase, EC 3. l. l. 1) ................................................. 6
1.1.3. The mechanism for lipase- and esterase-catalyzed hydrolysis ......................... 7
LIA. Lipase from Pseudomonas cepacia (PCL) ....................................................... 9
1.1.5. Lipase B from Candida antarctica (CAL-B) .................................................... 9
1.1.6. Esterase from Pseudomonas fluorescens (PFE) ............................................. Il
l.2. Enzymatic Reactions in Non-aqueous Media ........................................................ 13
1.2.1. Enzyme activity in non-aqueous media .......................................................... 14
1.2.2. Enzyme-catalyzed reactions in ionic liquids ................................................... 16
1.3. Substrate Engineering for Enhancing Enantioselectivity of Hydrolases ............... 18
lA. Protein Engineering to Improve Enantioselectivity ............................................... 21
1.4.1. Rational design for altering enantioselectivity of enzyme .............................. 21
ix

1.4.2. Direeted evolution for improving enantioseleetivity ...................................... 23
1.4.3 Combining rational protein design and direeted evolution .............................. 26
1.5. High-throughput Sereening: Quick E ..................................................... ................ 27
1.6. Moleeular Modeling ............................................................................................... 31
1.6.1. Moleeular mechanies vs Quantum mechanies ................................................ 31
1.6.2. Energy minimization ....................................................................................... 33
1.6.3. Conformational searching ............................................................................... 36
1.7. Enhancing Hydrolase Aetivity and Seleetivity - outline of this thesis .................. 38
Chapter 2 ........................................................................................................................... 40
Chapter 2. lmproved Preparation and Use of Room-Temperature Ionie Liquids in Lipase-
Catalyzed Enantio- and Regioseleetive Aeylations .......................................................... 41
Introduction ................................................................................................................... 41
Results ........................................................................................................................... 43
Discussion .................................................................................................................. : .. 55
Experimental Section .................................................................................................... 59
Chapter 3 ............................................................................................................................ 63
Chapter 3. Lipase-Catalyzed Direct Condensation of L-Aseorbie Aeid and Fatty Acids in
Ionie Liquids with Assistance of Hydrophobie Additives ................................................ 64
Introduction ................................................................................................................... 64
ResuIts ........................................................................................................................... 66
Discussion ..................................................................................................................... 72
Experimental Section .................................................................................................... 73
Chapter 3. Appendix 1 ....................................................................................................... 76
Ionie Liquids Create New Opportunities for Nonaqueous Bioeatalysis with Polar
Substrates .......................................................................................................................... 77
x

Introduction ................................................................................................................... 78
Results ........................................................................................................................... 80
Purification of Ionic Liquids ..................................................................................... 81
Polarity of Ionic Liquids is Similar to that for Polar Organic Sol vents .................... 82
High Activity of Lipases in Ionic Liquids in Spite of their High Polarity ................ 84
More Regioselective Acylation of Glucose in Ionic Liquids .................................... 85
Regioselective Acylation of Ascorbic Acid in Ionic Liquids ................................... 87
Discussion ..................................................................................................................... 89
Experimental Section .................................................................................................... 92
Chapter 4 ........................................................................................................................... 95
Chapter 4. Enantioselective Ring Opening of p-Lactams Catalyzed by Candida
antarctica Lipase B: Molecular Basis and Optimization .................................................. 96
Introduction ................................................................................................................... 96
Results ......................................................................................................................... 100
Discussion ................................................................................................................... 110
Experimental Section .................................................................................................. 114
Chapter 5 ......................................................................................................................... 119
Chapter 5. Discovery and Molecular Basis of Enantioselectivity of Va1122Ser Mutant of
PFE toward Methyl 3-Bromo-2-methylpropionate ......................................................... 120
Introduction ................................................................................................................. 120
Results ......................................................................................................................... 124
Discussion ................................................................................................................... 134
Experimental Section .................................................................................................. 138
Conclusions and Summary ............................................................................................. 145
Contributions to Knowledge ........................................................................................... 148
Xl

Appendix ......................................................................................................................... 150
xii
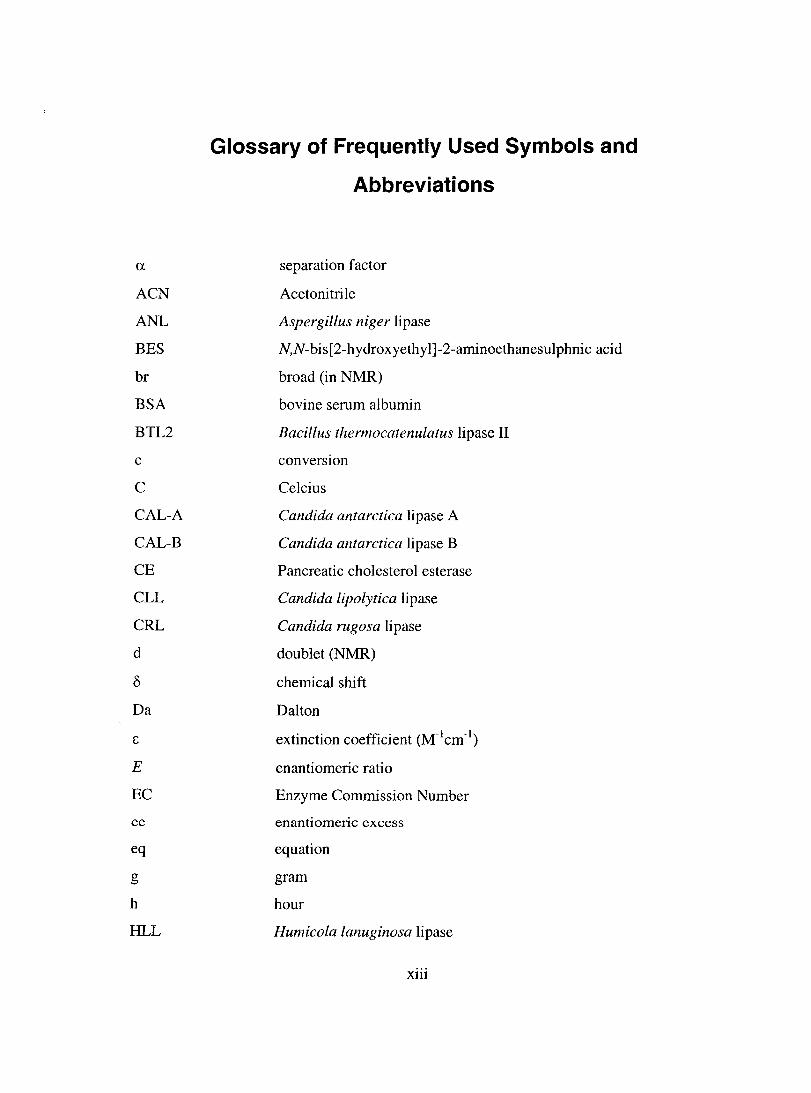
a
ACN
ANL
BES
br
BSA
BTL2
c
C
CAL-A
CAL-B
CE
CLL
CRL
d
Ô
Da
E
EC
ee
eq
g
h
HLL
Glossary of Frequently Used Symbols and
Abbreviations
separation factor
Acetonitrile
Aspergillus niger lipase
N,N-bis[2-hydroxyethyl]-2-aminoethanesulphnic acid
broad (in NMR)
bovine serum albumin
Bacillus thermocatenulatus lipase II
converSlOn
Celcius
Candida antarctica lipase A
Candida antarctica lipase B
Pancreatic cholesterol esterase
Candida lipolytica lipase
Candida rugosa lipase
doublet (NMR)
chemical shift
Dalton
extinction coefficient (M-1cm-1)
enantiomeric ratio
Enzyme Commission Number
enantiomeric excess
equation
gram
hour
Humicola lanuginosa lipase
xiii

HPLC
Hz
In
J
k
k'
L
m
f.!
M
mm
mol
na
nd
nr
%
p
P
Pcaml
PCL
PCR
PFE
PFL
pH
pKa
pNP
pNPAc
high performance liquid chromatography
hertz
indicator
coupling constant (in NMR)
kilo (103)
capacity factor
Michaelis-Menten constant
dissociation constant
enzyme turnover number
pathlength
liter
meter
mIcro
molar
minute(s)
mole(s)
not available
not determined
no reaction
percent (parts per hundred)
para
product
Penicillium camembertii lipase
Pseudomonas cepacia lipase
Polymerase Chain Reaction
Pseudomonas jluorescens esterase
Pseudomonas jluorescens lipase
negative logarithm of hydrogen ion concentration
negative logarithm of equilibrium constant for association
para-ni trophen 01
para-ni trophen y 1 acetate
XIV

pp
PPL
q
R
Rf
RML
ROL
Rs
s
sec
t
Td
TLC
TRIS
V
V/mg
v
V rnax
w/v
pages
porcine pancreatic lipases
quartet (in NMR)
Alkyl or aryl group
retenti on factor
Rhizomucor miehei lipase
Rhizopus oryzae lipase
Resolution factor
singlet (in NMR)
secondes)
triplet (in NMR)
tetrahedral intermediate
thin-layer chromatography
tris(hydroxymethyl)-aminomethane
unit
)..lmol of ester hydrolyzed per minute per mg protein.
initial velocity
maximum velocity
weight-to-volume
xv

Chapter 1
'Th~m said Jesus ta those Jews which believed on him, If ye continue in my
ward, then are ye my disciples indeed;
And ye shall know the truth, and the truth shall make you free."
JOHN 8:31-32
1

Chapter 1. Introduction
Biocatalysts are biological molecules that activate or speed up a chemical
reaction. They can be an enzyme, a cell organelle, or a whole cell, which can be from
microbial, plant or animal origin. However, chemical reactions within living cells are
catalyzed by enzymes 1, which are called biological catalysts.
Enzymes are highly selective. They can recognize a specific molecule among the
thousands of different compounds in a cell and transform it into a new product. In
addition, they are very effective catalysts. For example, the half-life of a typical
phosphate diester bond in DNA in neutral water at 25 oC is expected to be on the order of
tens to hundreds of billions of years. However, an enzyme (e.g., DNAse) can hydrolyze
DNA within 1 sec.
The work in this thesis deals with hydrolases (in particular, lipases and esterases),
which are enzymes that catalyze hydrolysis. The goal of this thesis is enhancing
hydrolase activity and selectivity. The purpose can be achieved by three approaches:
medium, substrate, and protein engineering. This thesis describes those approaches
depending on the reactions.
1.1. Hydrolases
Hydrolases are enzymes that catalyze digestion of nutrients through hydrolysis in
nature and are classified into the EC32 category. Three classes of hydrolases, namely
lipases, esterases, and proteases, are most common biocatalysts in industrial and
acadlemic research because of their low cost, commercial availability, good stability, lack
of co-factors, broad substrate tolerance, and wide range of catalytic activities. About 75%
1 Before the beginning of the 1980's, aIl enzymes were thought to be proteins. However,
it has been found that RNA molecules can also function as enzymes.
2 Ee: Enzyme Commission Number
2

of aIl enzyme-catalyzed syntheses have used hydrolases. 3 Less common hydrolases are
phospholipases, epoxide hydrolases, amidases, and nitrilases. The work in this thesis
explores increasing the activity and selectivity of two hydrolases: lipase and esterase.
Although both esterases and lipases catalyze hydrolysis of esters,4 lipases
preferentially hydrolyze water-insoluble esters or triglycerides composed of long chain
fatty acids while esterases usually accept water-soluble esters or short-chain fatty acid
triglycerides like tributyrin. Apart from their basic role in biological systems, hydrolases
can catalyze the reverse reaction of hydrolysis (i.e., condensation) and other reactions
such as transesterification, aminolysis, and lactone ring-opening reaction (Scheme 1).
Hydrolysis
0
RAO,H
0
Go
o RAO,R'
11 HO-Ser-Enz
o RAo/ser-Enz acyl enzyme
Transesterification
R'OH 0
Esterification RAO,R'
H20 0
Lactone ring-opening HO~O,H reaction
Aminolysis
Scheme 1. Reactions catalyzed by lipases or esterases
3 Jaeger, K-E.; Scheneidinger, B.; Rosenan, F.; Werner, M.; Lang, D.; Dijkstra, B. W.;
Schimmosek, K; Zonta, A.; Reetz, M. T. J. Mol. Catal. B: Enzym. 1997,3,3-12.
4 Faber, K Biotransformations in Organic Chemistry, 4th ed.; Springer-Verlag: Berlin,
Germany, 2000. Roberts, S. M. Preparative Biotransformations; Wiley: Chichester,
1992-1998. Wong, C.-H.; Whitesides, G. M. Enzymes in Synthetic Organic Chemistry;
Elsevier Science: Tarrytown, NY, 1994.
3

1.1.1. Lipase (EC 3.1.1.3)
Lipases, found in a wide range of organisms inc1uding animaIs, plants, fungi, and
bacteria, catalyze the hydrolysis of esters. Although lipases preferentially catalyze
hydrolysis of water-insoluble esters like triglycerides, lipases also catalyze the hydrolysis
of a broad range of unnatural esters, which are synthetically important building blocks,
with high enantio- or regioselectivity. The broad substrate range and high selectivity of
lipases make them useful catalysts for the hydrolysis of esters, transesterification,
synthesis of esters and peptides and the resolution of racemic mixtures.5 The most useful
lipases for organic synthesis are porcine pancreatic lipase (PPL) , lipase from
Pseudomonas cepacia, lipase from Candida rugosa (CRL), and lipase B from Candida
antarctica (CAL-B).
Apart from other hydrolases, a unique feature of lipases is that most of lipases
possess a lid covering the active site and require a water-oil interface in reaction media
(exceptions inc1ude: lipase B from Candia antarctica (CAL-B) and cutinase from
Fusarium solanis, which have only a small or no lid). Since most lipases have the active
site that is buried beneath a helical segment (i.e., the lid), lipases need to change their
conformations (from c10sed to open)6 for the productive reaction. Without the
5 a) Vulfson, E. N. In Lipases: Their Structure, Biochemistry and Applications; Woolley,
P.; Petersen, S. B., Eds; Cambridge University Press: Cambridge, 1994; Chapter 13. b)
Gilbert, E. J. Enzyme Microb. Technol. 1993, 15, 634-645. c) Soberon-Chavez, G.;
Palmeros, B. Crit. Rev. Microbiol. 1994, 20, 95-105. d) Jaeger, K.-E.; Ransac, S.;
Dijkstra, B. W.; Colson, c.; van Heuvel, M.; Misset, O. FEMS Microbiol. Rev. 1994,
15,29-63.
6 The crystal structures for both conformations of sorne lipases have been solved. For
example, the open and c10sed conformations of Candida rugosa lipase are available.
Grochulski, P; Li, Y; Schrag, J. D.; Bouthillier, F.; Smith, P.; Harrison, D.; Rubin, B.;
Cygler, M. J. Biol. Chem. 1993,268, 12843-12947. Grochulski, P.; Li, Y. Schrag, J. D.;
Cygler, M. Prote in, Sei. 1994,3,82-91.
4

conformational change, the lid remains closed and the substrate can not reach the active
site.? Upon exposure of the lipase to a lipid interface, lipases change a conformation to
open the lid and expose the catalytic machinery to the substrate. This process is called as
the interfacial activation of lipases and explains that lipases typically show low activity
towards soluble substrates in aqueous media where there is no water-lipid interface
(Figure 1).
soluble
insoluble
substrate concentration substrate concentration
a) Esterase b) Lipase
Figure 1. a) Esterase kinetics following normal Michaelis-Menten kinetics8 and b) Lipase
kinetics where interfacial activation9 is observed.
Lipases are classified according to their microbial source or protein sequence
alignments. Classification using protein sequence alignments is consistent with the 3-D
structures of lipases and is more reliable (Table 1).
? Faber, K. Biotransformations in Organic Chemistry, 4th ed.; Springer-Verlag: Berlin,
Germany, 2000; pp 94-98.
8 Fersht, A. Enzyme Structure and Mechanism, 2nd ed.; W. H. Freeman and Co.: New
York, 1985, pp 98-103.
9 Verger, R. Trends Biotechnol. 1997,15,32-38.
5

Table 1. Classification of Commercial Lipases According to Similarities in Protein
Sequence10
Classification Characteristics Mammalian (pancreatic) lipases 50 kDa
Fungallipases Candida rugosa family Rhizomucor family Unclassified
Baderiallipases Pseudomonas family Staphylococcus family
60-65 kDa 30-35 kDa
30-35 kDa 40-45 kDa
Examples PPLa
CRL,GCL,CE CAL-B, RML, ROL, 1ll..L, PcamL ANL, CAL-A, CLL
PCL, PFL, CYL BTL2
a abbreviation: PPL: porcine pancreas lipase; CRL: Candida rugosa lipase; GCL:
Geotrichum candidum lipase; CE: pancreatic cholesterol esterase; CAL-B: Candida
antarctica lipase B; RML: Rhizomucor miehei lipase: ROL: Rhizopus oryzae lipase;
HLL: Humicola lanuginosa lipase; PcamL: Penicillium camembertii lipase; ANL:
Aspergillus niger lipase; CAL-A: Candida antarctica lipase A; CLL: Candida lipolytica
lipase; PCL: Pseudomonas cepacia lipase; PFL: Pseudomonas fluorescens lipase; CYL:
Chromobacterium viscosum lipase; BTL2: Bacillus thermocatenulatus lipase II
1.1.2. Esterase (Carboxyester hydrolase, EC 3.1.1.1)
With exception of acetyl- and butyryl choline esterases, which hydrolyze the
neurotransmitters acetyl- and butyryl choline in vivo, the physiological role of most
esterases is still unclear.
Esterases and lipases show many biochemical and structural similarities. Esterases
catallyze the hydrolysis of water-soluble carboxylic acid esters. AlI esterases have a
characteristic a/p-hydrolase fold11 and a similar catalytic triad that typicalIy consists of a
10 Bornscheuer, U. T.; Kazlauskas, R. J. Hydrolases in Organic Synthesis: Regio- and
Stereoselective Biotransformations; Wiley-VCH: Weinheim, Germany, 1999; pp 20-21.
11 OlIis, D. L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.; Hare!, M.;
Remington, S. J.; Silman, 1. Protein Eng. 1992, 5, 197-211. Holmquist, M. Curr. Prot.
6

nuc1eophilic serine (in the consensus sequence G-X-S-X-G), histidine, and
aspartatefglutamate. AIl known structures of lipases also show the afp-hydrolase fold
(Figure 2).12
The afp-hydrolase fold consists of a core of eight mostly-pararellei p-sheets,
which are surrounded on both sides by a-helices. The connectivity of the sheets and
helices is the same in aIl afp-hydrolases.
oxyanion
n 3 4 5 6 7 8
a./f}-hydrolase fold
Figure 2. a/p Hydrolase fold found in esterases and lipases. The arrows and squares
represent P sheets and a helices, respectively. Oxyanion: residues that stabilize the
oxyanion; Nu: nuc1eophilic residue; His: catalytic histidine; acid: a residue that activates
the catalytic histidine.
1.1.3. The mechanismfor lipase- and esterase-catalyzed hydrolysis
The catalytic machinery consists of a catalytic triad, Ser, His, and Asp/Glu, and
an oxyanion hole composed of back-bone amide protons and/or amino acid side chains.
These residues occur in the same order in aIl lipase (serine esterase) amino acid
Pept. Sei. 2000, 1, 209-235.
12 Cygler, M.; Schrag, J. D.; Ergan, F. Biotechnol. Genet. Eng. Rev. 1992,10, 143-184.
7
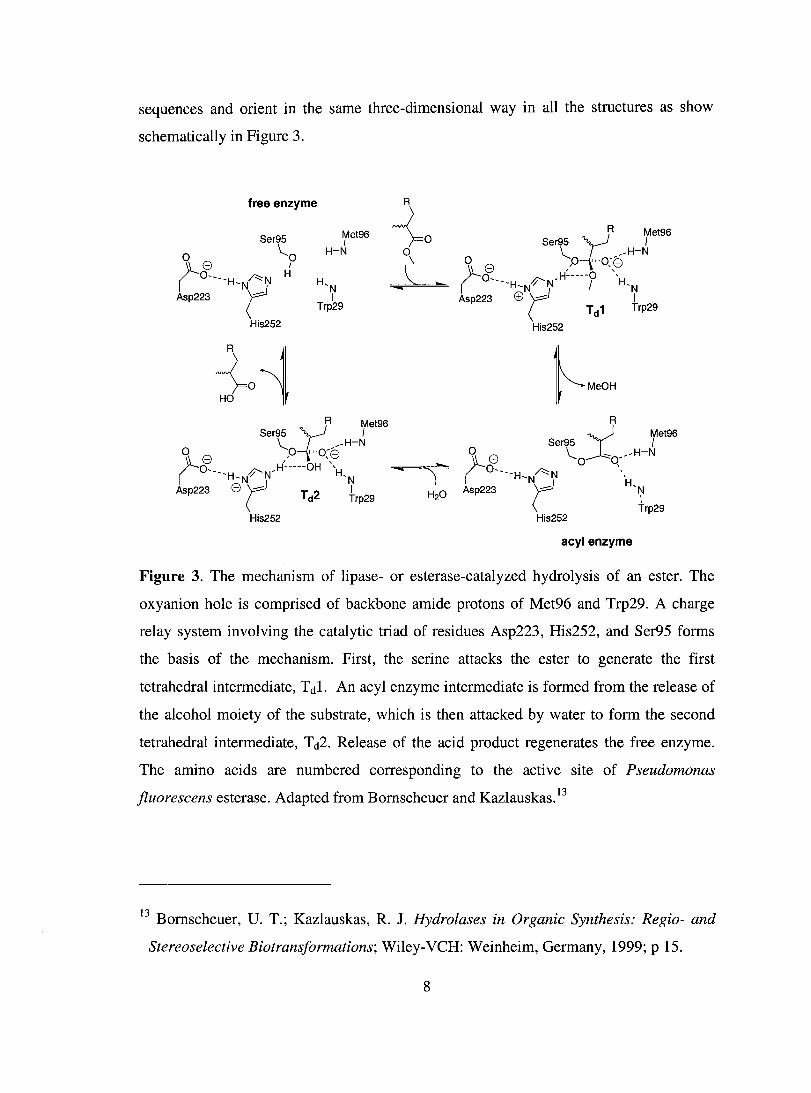
sequences and orient in the same three-dimensional way in aH the structures as show
schematicaHy in Figure 3.
free enzyme
Met96 1
H-N
H'N 1
Trp29
R Met96 Ser95 ~ 1 _-H-N
o "0 " '0-8-=--!-8 " , , 0 - H-----OH '
--- ij'N' H H-N 'N Asp223 <±l 'yi T 2 1
( d Trp29
His252
-
R Met96
Ser95 ~ 1 H-N o "0 " '0-8-~-!-8 " , ,
0-_ 'H-----O, --H-Nij'N / H-N
Asp223 <±l 'yi 1
( Td1 Trp29
His252
R
X Met96 Ser95 1
~ 8 "0 _q __ -H-N
( O----H_NAN '
Asp223 'yi k~ ( Trp29 His252
acyl enzyme
Figure 3. The mechanism of lipase- or esterase-catalyzed hydrolysis of an ester. The
oxyanion hole is comprised of backbone amide protons of Met96 and Trp29. A charge
relay system involving the catalytic triad of residues Asp223, His252, and Ser95 forms
the basis of the mechanism. First, the serine attacks the ester to generate the first
tetrahedral intermediate, T dl. An acyl enzyme intermediate is formed from the release of
the a1cohol moi et y of the substrate, which is then attacked by water to form the second
tetrahedral intermediate, T d2. Release of the acid product regenerates the free enzyme.
The amino acids are numbered corresponding to the active site of Pseudomonas
fluorescens esterase. Adapted from Bomscheuer and Kazlauskas. 13
13 Bomscheuer, U. T.; Kazlauskas, R. J. Hydrolases in Organic Synthesis: Regio- and
Stereoselective Biotransformations; Wiley-VCH: Weinheim, Germany, 1999; p 15.
8

The hydrolytic mechanism involves two tetrahedral intermediates and an acyl
enzyme. Attack of the ester by the nuc1eophilic serine produces the first tetrahedral
intermediate (T dl), which is stabilized by hydrogen bonds between catalytic histidine and
the alcohol, and between the oxyanion and the backbone of the oxyanion hole residues.
The alcohol is subsequently lost to pro duce the acyl enzyme, which is attacked by water
to form the second tetrahedral intermediate (Td2). The release of the acid product
regenerates the free enzyme.
1.1.4. Lipase from Pseudomonas cepacia (PCL)
PCL (also known as Burkholderia cepacia lipase) has 320 amino acids with a
moiecular weight of 33 kDa. It has been c10ned and expressed from 4 different strains.
Several X-ray crystal structures are available of the lipase and they show that it has a
typical afp-hydrolase fold. The catalytic triad of the enzyme is Ser87-His286-Asp264
and the backbone amides of OIn88 and Leu17 contribute to the stabilization of the
oxyanion. PCL, like common lipases, has a large lid that covers the large hydrophobic
active site and shows interfacial activity; -25 foid activity increased when bound to an
oil-water interface.
While purified crystalline PCL does not show activity in orgamc solvents,
immobilized or crude PCLs are fairly active in nonpolar organic solvents. 14 Generally, it
shows good selectivity toward secondary alcohols and carboxylic acids with E> 50.
1.1.5. Lipase B from Candida antarctica (CAL-B)
CAL-Bis a fungal lipase with 317 amino acid length and 33 kDa weight. It also
belongs to afp-hydrolase fold hydrolase family. The lipase originates from the yeast
Candida antarctica. Two fairly different lipases were characterized from this yeast:
14 Bomscheuer, u.; Reif, O.-W.; Lausch, R.; Freitag, R.; Scheper, T.; Kolisis, F. N.;
Menge, U. Biochim. Biophys. Acta 1994, 1201, 55-60.
9

component A (Candida antarctica lipase A) and component B (Candida antarctica lipase
B). The three-dimensional structures of the free enzyme of CAL-B as weIl as of inhibitor
binding have been reported (Protein Data Base entries: lLBS, lLBT, 1 TCA, 1 TCB,
1 TCC). No major conformational differences were observed between the free enzyme
and the enzyme covalently bonded with a phosphonate transition-state analog: both are
considered as the open-conformation. Although CAL-B does not display interfacial
activation, which is a common feature of lipases, it is not only active toward water
soluble substrates but also toward water-insoluble substrates. In addition, since CAL-B
has :a very smalllid, it may be also regarded as an esterase.
The active site is buried at the bottom of a tunnel-shaped binding site. The
catalytic machinery is placed at the bottom of the active site and consists of the catalytic
triad plus the oxyanion hole. The catalytic triad of CAL-B is Serl05-His224-AspI87
structure. The oxyanion hole is composed of the backbone amide proton of Glnl06 and
the backbone amide proton and side chain hydroxyl proton of Thr40 for hydrogen
bonding with the oxyanion.
CAL-B immobilized preparation (trade name Novozym 435) is stable at 60-80 oC
for extended periods of time. In addition to its high thermal stability, CAL-B retains most
of its activity in a wide range of nonaqueous solvents. It shows very high activity and
high enantioselectivity toward secondary alcohols15 while its enantioselectivity toward
carboxylic acids is usually low. CAL-B shows regioselectivity toward the primary
alcohol in glucose or ascorbic acid. 16
15 Rotticci, D.; Ottosson, J.; Norin, T.; HuIt, K. In Methods in Biotechnology: Enzymes in
Nonaqueous Solvents, Vulfson, E. N., Halling, P. J., Holland, H. L., Eds.; Humana;
Totowa, 2001, Vol. 15, pp 261-276.
16 a) Cao, L.; Bomscheuer, U. T.; Schmid, R. D. J. Mol. Catal. B: Enzym. 1999, 6, 279-
285. b) Pulido, R.; Goto, V. J. Chem. Soc. Perkin Trans. 1 1993, 589-592. c) Yan, Y.;
Bomscheuer. U. T.; Schmid, R. D. Biotechnol. LeU. 1999,21,1051-1054.
10

1.1.6. Esterase from Pseudomonasfluorescens (PFE)
At least four kinds of esterase have been isolated from Pseudomonas fluorescens
according to their properties such as location within the cell and substrate specificity.17
The esterase used in this thesis is an arylesterase (EC 3.1.1.2), which was first isolated,
cloned, and expressed in E. coli by Choi et al. and later resequenced ta correct errors.18
While this enzyme (pure form) is not stable in organic solvent, it is stable in a wide range
oftemperature (up to 70 OC) and pH (5-10) with an optimum pH 7.5-8 and 45 °C. 19
PFE is 272 amino acids long and its weight is 29.5 kDa. It has sequence similarity
to bacterial non-heme (or cofactor-free) haloperoxidases enzymes, which catalyze in vitro
halogenation of organic substrates in the presence of peroxides such as H20 2 and halide
ions. Both PFE and non-heme haloperoxidases have catalytic similarity such that PFE has
low brominating activity and a bromoperoxidase shows a low rate of hydrolysis of p
nitrophenyl acetate (PNPAc). X-ray crystal structures of non-heme haloperoxidases have
revealed an a/p hydrolase fold and catalytic triad characteristic of hydrolases. 20, 21
17 Choi, K. D.; Jeohn, G. H.; Rhee, J .S; Yoo, O. J. Agrie. Biol. Chem. 1990, 54, 2039-
2045.
18 Microorganism: SIK-Wl; 816 nucleotide sequence; GenBank accession: U12537;
http://www.ncbi.nlm.nih.govlEntrez/: Pelletier, I.; Altenbuchner, J. Mierobiol. 1995,
141,459-468.
19 Krebsfanger, N.; Zocher, F.; Altenbuchner, J.; Bomscheuer, U. T. Enzyme Mierob.
Technol. 1998,22,641-646.
20 a) Hecht, H. J.; Sobek, H.; Haag, T.; Pfeifer, O.; van Pée, K. H. Nat. Struet. Biol. 1994,
1, 532-537. b) Hofmann, B.; TOlzer, S.; Pelletier, I.; Altenbuchner, J.; van Pée, K. H.;
Hecht, H. J. J. Mol. Biol. 1998,279,889-900.
21 A mechanism for the halogenation reaction of the non-heme haloperoxidases has been
proposed by Hofmann et al. (ref 16 b) The reaction proceeds by attack of the catalytic
serine on the carbonyl carbon of the carboxylic acid. Elimination of water pro duces the
acyl-enzyme complex. Subsequently, the nucleophilic attack of hydrogen peroxide
generates a peroxoacid. Subsequent formation of hypohalous acid followed by
11

The substrate binding site of a bromoperoxidase22 and several chloroperoxidases
is composed of four D' helices which form two sides of a triangle, while the third side is
provided by loops carrying the active site residues aspartate and histidine. The
nucleophilic serine is at the bottom of a pocket in the middle of the triangle. Sequence
alignment among haloperoxidases and PFE (46-51% amino acid sequence identity)
indicates that conserved residues are limited to the central p-sheet and covering helices A
to F. The catalytic triad of PFE is Ser95-His252-Asp223 and the oxyanion hole contains
two stabilizing backbone amides of Met96 and Trp29. The active site of the enzyme is
very restricted and has little exposure to the solvent.
Substrate mapping of PFE19, 23 reveals that it prefers short-chain fatty acid esters,
acetyl esters of aromatic alcohols, and activated esters (vinyl or halogenated) over
aromatic acid esters. Compared to the activity toward the hydrolysis of ethyl acetate, PFE
showed only 2% activity towards esters of aromatic acids, acetates of 2-phenylethanol,
and esters of benzyl alcohol.
While PFE shows good activity but no selectivity toward chiral primary alcohols
such as solketal (- 0.5 V/mg, E24 = -1), it shows highly enantioselective hydrolysis for
halogenation of an organic substrate are considered as not catalyzed by the enzyme
because a specifie halide binding site does not seem to be present in the enzyme.
However, the size and hydrophobie environment of the active site pocket suggest an
important role, as the environment may protect the peroxoacid against hydrolysis while
the halide is transported to the active site.
22 http://www.rcsb.org/pdb/a)pdbcode:1BRO;b)pdbcode:1A8S.lA88.lA8Q
23 Krebsfanger, N.; Schierholz, K.; Bomscheuer, V. T. J. Biotechnol. 1998, 60, 105-112.
Zocher, F.; Krebsfanger, N.; Yoo, O. J.; Bomscheuer, U. T. J. Mol. Catal. B: Enzym.
1998,5, 199-202. Liu, A. M. F.; Somers, N. A.; Kazlauskas, R. J.; Brush, T. S.; Zocher,
F.; Enzelberger, M. M.; Bomscheuer, U. T.; Horsman, G. P.; Mezzetti, A.; Schmidt
Dannert, c.; Schmid, R. D. Tetrahedron: Asymmetry 2001,12,545-556.
24 E = (kea/KM) fast
(kcat / KM) slow
12

secondary a1cohol such as 1-phenylethyl acetate (E = 58). PFE has moderate
enantioselectivity towards chiral carboxylic acids having a stereocenter at a carbon,
however, E is only 12 in favor of the (S)-enantiomer for methyl 3-bromo-2-
methylpropionate (MBMP).
1.2 .. Enzymatic Reactions in Non-aqueous Media
Before the 1980s, researchers generally believed that enzymes were inactive in
organic sol vents. One common technique for denaturing enzymes in water was, and is,
adding a water-miscible organic solvent such as acetonitrile or propanol into the enzyme
solution?5 Since the early 1980s, researchers discovered that enzymes especially tolerate
water-immiscible solvents?6 Indeed, most enzymes, such as lipase, terpene cyclase,
cytochrome oxidase, A TPase, and chymotrypsin, are more thermostable in dry organic
solvents than in water. 27
ln addition to their thermostability in water-immiscible organic solvents, using
these organic sol vents rather than water for enzymatic reactions has many potential
advantages. Many substrates, which are not soluble in water or are water-sensitive, can be
25 Generally around 50% acetonitrile denatures enzymes. Griebenow, K; Klibanov, A M.
J. Am. Chem. Soc. 1996, 118,11695- 11700.; Partridge, J.; Moore, B. D.; Haling, P. J.
J. Mol. Catal. B: Enzym. 1999,6, 11-20.
26 a) Antonini, E.; Carrea, G.; Cremonesi, P. Enzyme Microb. Technol. 1981,3,291-296.
b) Martinek, K.; Semenov, AN.; Berezin, 1. V. Biochim. Biophys. Acta 1981,658, 76-
89 .. c) Martinek, K.; Levashov, A V.; Khmelnitsky, Y. L.; Klyachko, N. L.; Berezin, 1.
V. Science 1982, 218, 889-891. e) Zaks, A; Klibanov, A M. Proc. Natl. Acad. Sci.
U.S.A. 1985,82,3192-3196.
27 a) Zaks, A; Klibanov, A M. Science 1984, 224, 1249-1251. b) Wheeler, C. J.;
Croteau, R. Arch. Biochem. Biophys. 1986, 248, 429-434. c) Ayala, G.; de G6mez
Puyou, M. T.; Darszon, A FEBS Lett. 1986,203,41-43. d) Zaks, A; Klibanov, A M.
J. Biol. Chem. 1988,263,3194-320.
13

dissolved or are stable in organic sol vents. The insolubility of enzymes in most organic
sol vents with the exception of a few polar organic solvents28, such as dimethyl sulfoxide,
formamide, and sorne hydrophilic organic solvents, makes it very easy to simply recover
and reuse them.
Organic sol vents can reverse the thermodynamic equilibrium of hydrolysis
reactions and make new synthetic transformations available. For example, hydrolases can
catalyze an esterification in organic solvents, which is the reverse reaction of hydrolysis
catalyzed by hydrolytic enzymes. Additionally, using organic sol vents often simplifies
work-up procedures and avoids microbial contamination of the reaction.29
1.2.1. Enzyme aetivity in non-aqueous media
Enzymatic activity tends to be lower in organic sol vents than in water?O Several
theories rationalize this reduced enzyme activity in organic media.
One theory proposes that the suspended enzymes in organic solvents encounter
diffusional limitation. This diffusional limitation limits the mass transfer and decreases
the substrate-accessibility to the active site, thereby lowering the activity.31 Another
theory proposes that the enzyme preparation, such as lyophilization, could denature
enzymes by changing the conformation and thereby decrease enzyme activity in organic
solvents.32 Altematively, the nonpolar organic solvent could change the conformation of
28 Enzymes dissolve in these polar solvents but are completely denatured. a) Singer, S. J.
Adv. Protein Chem. 1961, 17, 1-68. b) Chin, J. T.; Wheeler, S. L.; Klibanov, A. M.
Bioteehnol. Bioeng. 1994,44, 140-145.
29 Klibanov, A. M. Nature 2001, 409,241-246.
30 a) Schmitke, J. L.; Wescott, C. R.; Klibanov, A. M. J. Am. Chem. Soc. 1996, 118,
3360-3365. b) Klibanov, A. M. Trens Bioteehnol. 1997, 15, 97-101.
31 Karmat, S.; Beckman, E. J.; Russell, A. J. Enzyme Mierob. Teehnol. 1992,14,265-271.
32 a) Dong, A.; Prestrelski, S. J.; Allison, S. D.; Carpenter, J. F. J. Phann. Sei. 1995, 84,
415-424. b) Griebenow, K.; Klibanov, A. M. Proe. Nat!. Aead. Sei. U.S.A. 1995, 92,
14

enzymes33 , reduce the flexibility of enzymes3\ or alter the interaction process between
enzyme and substrate. Yet a further theory focuses on the stabilization of ground state of
substrate and destabilization of transition state by hydrophobic organic solvents.35 The
hydrophobic substrates may be stabilized by a solvation effect of hydrophobic sol vents at
ground state. The stabilized substrates may be more partitioned in a hydrophobic solvent,
such as hexane, rather than in the active site of enzyme. In addition, hydrophobic solvents
may destabilize the charge-developed transition state.
To increase the enzymatic activity in nonpolar organic sol vents , an effective
strategy has been developed using additives such as sugars, polyethylene glycol,
inorganic salts, substrate-resembling ligands, and crown ethers during enzyme
preparation (or lyophilization).36 These additives prevent enzyme denaturation or
conformational change during dehydration and may keep enzymes in native-like
conformation. Another effective approach is based on a hypothesis of introducing
flexibility of enzymes in anhydrous nonpolar solvents. Enzymatic activity has been
increased by up to two to three orders of magnitude by adding small quantities of water37
or denaturing organic solvents such as formamide and dimethyl sulfoxide.38
10969-10976.
33 a) Fitzpatrick, P. A; Klibanov, A M. J. Am. Chem. Soc. 1991,113,3166-3171. b) Wu,
H. S.; Chu, F. Y.; Wang, K Bioorg. Med. Chem. Lett. 1991,1,399.
34 a) Affleck, R.; Haynes, C. A; Clark, D. S. Proc. Natl. Acad. Sci. U.S.A. 1992, 89,
5167-5170. b) Hartsough, D. S.; Merz, Jr., K M. J. Am. Chem. Soc. 1992,114, 10113-
10116.
35 Wangikar, P. P.; Rich, J. O.; Clark, D. S.; Dordick, J. S. Biochemistry 1995, 34, 12302-
12310.
36 Review: Theil, F. Tetrahedron 2000,56,2905-2919.
37 a) Zaks, A; Klibanov, A M. J. Biol. Chem. 1988,263,3194-3201. b) Affleck, R.; Xu,
Z.-F.; Suzawa, V.; Focht, K; Clark, D. S.; Dordick, J. S. Proc. Natl. Acad. Sei. U.S A.
1992,89,1100-1104.
38 Almarsson, O.; Klibanov, A M. Biotechnol. Bioeng. 1996,49,87-92.
15

However, enzymes in polar organic solvents, such as dimethyl sulfoxide and
formamide, remain inactive or have far lower activity due to the induction of significant
changes in active-site structure. 39 The fact that polar organic solvents denature enzymes
has limited the use of polar substrates, which are insoluble in nonpolar organic solvents,
for the enzymatic transformation.
1.2.2. Enzyme-eatalyzed reaetions in ionie liquids
Ionie liquids are organic salts remaining liquid at room temperature or slightly
higher.40 The first ionic liquid, EtNH3'N03, was reported in 1914. A large number of
ionic liquids have been reported in the literature recently. A new type of ionic liquids
such as EMIM·BF4 (1-ethyl-3-methylimidazolium tetrafluoroborate) was prepared by
metathesis in 1992.41 Ionie liquids are completely nonvolatile and can be recyc1ed and
reused. The combination of non volatile and recyc1able properties makes ionic liquids a
"green" solvent.42 Organic solvents are generally volatile and can release toxic vapors.
The non volatile and recyc1able properties of room temperature ionic liquids enable their
use as alternative solvent.
In addition to their nonvolatile properties, ionic liquids are highly polar and strong
solvating agents. According to their paired cation and anion structure, ionic liquids can
dissolve a wide range of both inorganic and organic substrates of interest. These physical
properties of ionic liquids can be used as potential sol vents for synthesis such as
oligomerization of butene, alkylation of olefins, Diels-Alder reactions, hydrogenation,
39 Review: Dordick, J. S. Bioteehnol. Prog. 1992,8,259-267.
40 Review: Seddon, K. R. J. Chem. Teehnol. Bioteehnol. 1997,68,351-356. Welton, T.
Chem. Rev. 1999,99, 2071-2083. Wasserscheid, P.; Keim, W. Angew. Chem., Inti. Ed.
Engl. 2000,39, 3772-3789.
41 Wilkes, J. S.; Zaworotko, M. J. J. Chem. Soc. Chem. Commun. 1992,965-967.
42 Adam. D. Nature 2000, 407, 938-940.
16

hydroformylation, and Friedel-Crafts reactions.43 For these reactions, ionic liquids often
show faster reaction and higher selectivity.
Recently, enzyme-catalyzed reactions in ionic liquids have been reported.44 The
first use of ionic liquids for enzyme-catalyzed reactions was reported by Erbeldinger et
al.45 They reported that, for peptide synthesis, thermolysin was more stable in ionic
liquids than in ethyl acetate, but the reaction rates were lower. On the other hand, the
SheIdon group demonstrated a variety of lipase-catalyzed reactions, such as alcoholysis,
ammoniolysis and perhydrolysis, in ionic liquids.46 Similarly, SchOfer et al. reported
lipase-catalyzed reactions in sorne ionic liquids, but no reaction at an in others, even
when the structures were very similar. In addition, they as weIl as Kim et al. found that
the enantioselectivity of lipase-catalyzed acetylation of secondary alcohols was higher in
sorne ionic liquids.47 Itoh et al. reported reuse of ionic liquids for lipase-catalyzed
reactions.48
Although enzyme-catalyzed reactions work in ionic liquids, the advantages,
besides environmental ones, of ionic liquids over non-polar organic sol vents for enzyme
catalyzed reactions are not yet clear. In addition, to identify the benefit properties of ionic
liquids for enzyme-catalyzed reactions, their proper preparation (or purification) is
required. If enzyme-catalyzed reactions in ionic liquids do not work because of unknown
impurities, which may be produced during their synthesis, it is difficult to realize the
advantage of ionic liquids.
43 Holbrey, J. D.; Seddon, K. R. Clean Prod. ProC. 1999,1,223-236.
44 Review: Sheldon, R. A.; Lau, R. M.; Sorgedrager, M. J.; van Rantwijk, F.; Seddon, K.
R. Green Chem. 2002,4, 147-151.
45 Erbeldinger, M.; Mesiano, M.; Russell, A. J. Biotechnol. Prog. 2000, 16, 1129-1131.
46 Lau, R. M.; van Rantwijk, F.; Seddon, K. R.; Sheldon, R. A. Org. Lett. 2000,2,4189-
4191.
47 a) Schofer, S. H.; Kaftzik, N.; Wasserscheid, P.; Kragl, U. Chem. Commun. 2001, 425-
426. b) Kim, K.-W.; Song, B.; Choi, M.-Y.; Kim, M.-J. Org. Lett. 2001,3,1507-1509.
48 Itoh, T.; Akasaki, E.; Kudo, K.; Shirakami, S. Chem. Lett. 2001, 262-263.
17

The work in this thesis (Chapter 2) explores an improved preparation of ionic
liquids that yields ionic liquids that work reliably in enzyme-catalyzed reactions. In
addition, Chapter 2 and 3 deals with the application for lipase-catalyzed acylation of
polar substrates such as glucose and ascorbic acid, which are insoluble in non polar
organic solvents.
1.3. Substrate Engineering for Enhancing Enantioselectivity of
Hydrolases
Enantiomers of a chiral substrate may orient and bind differently in an enzyme
active site, hence the substrate engineering is one of the most important tools and
sometimes easier than other approaches for increasing enzyme activity or selectivity.
In the enzyme-catalyzed kinetic resolution of chiral secondary a1cohols, the
substrate engineering of the a1cohol itself, or of the acyl donor has been studied. In an
example of the substituent effect on chiral secondary a1cohols, a series of secondary
a1cohols were used to determine the effect of enantioselectivity of CAL_B.49 Reactions
with a series of 1-X-heptanol show that the size of substituent (X) affects the
enantioselectivity as well as the reactivity. While smaller groups on the substituent, such
as methyl and ethyl, gave high enantioselectivity (E = 340), secondary a1cohols with
larger substituents, such as propyl, but yi, isopropyl or tert-butyl, showed lower
enantioselectivity (E < 10) with a hundred-fold decrease in reaction rate, or did not react
at all. Reactions with a series of 1-X-ethanol, larger substituents such as isopropyl, tert
but yi or propyl groups, gave higher enantioselectivity (E> 300).
OH
X~ 1-X-heptanol
X = methyl, ethyl, propyl, but yi, isopropyl, tert-butyl
OH
X~ 1-X-ethanol
X = ethyl, isopropyl, tert-butyl, propyl
49 Orrenius, C.; Haeffner, F.; Rotticci, D.; Ohmer, N.; Norin, T.; HuIt, K. Biocatal.
Biotrans. 1998, 16, 1-15.
18

On the other hand, the acyl chain length of vinyl esters employed as acyl donors
was also shown to have a strong effect on the enantioselectivity of CAL-B-catalyzed
kinetic resolution toward 3-methyl-2-butanol and on its thermodynamic components.50
The highest enantioselectivity (E = 810) was achieved with the longest acyl chain, vinyl
octanoate. AIthough the relationship between enantioselectivity and acyl chain length is
not linear, shorter chain acyl donors decreased the enantioselectivity: vinyl hexanoate (E
= 720), butanoate (E = 390), and vinyl propanoate (E = 470). This discrepancy between
vinyl propanoate and butanoate is due to the change of different thermodynamic
components (i.e., enthalpic and entropic components).51 While the longer acyl chains
(butanoyl, hexanoyl and octanoyl) mainly aItered their entropie components, vinyl
propanoate changed both the enthalpic and the entropic components.
'- 10H + #' jL ,/ - '- l.oji, ,/ + '- J'OH + 0\ 1 . -;:7 0' \In CAL·S 1 ~ , \In 1 n = 1,2,4,6
To get a productive reaction, the substrates must be folded as a hairpin structure
to enter into the hydrolase.52 Since two moieties become c10sed in the narrow active site
of a hydrolase (especially lipases and esterases), thereby interacting each other, the
substrate engineering of both moieties influences the enantioselectivity of hydrolases.
For the hydrolase-catalyzed kinetic resolution of chiral carboxylic acids, the basis
of enantioselectivity is more complicated than for that of chiral alcohols because two
diastereomeric acyl enzymes form during hydrolase-catalyzed reaction. Since both
tetrahedral intermediates involved in the reaction mechanism inc1ude the chiral segment
50 Ottosson, J.; HuIt, K. J. Mol. Catal. B: Enzym. 2001,11,1025-1028.
51 The enantioselectivity (E) can be described by thermodynamic expression: i1i10:j: =
- RTlnE= i1i1H:j: - Ti1i1S:j:; Phillips, R. S. Trends Biotechnol. 1996,14,13-16.
52 For example, an inhibitor binding X-ray structure of CAL-B shows the hairpin
structure. Uppenberg, J.; Ohmer, N.; Norin, M.; Huit, K.; Kleywegt, G. J.; Patkar, S.;
Waagen, V.; Anthonsen, T.; Jones, T. A. Biochemistry 1995, 34, 16838-16851.
19

of the substrate, both transition stages may influence the enantioselectivity.53 Therefore,
substrate engineering of the acid moiety for the enzyme-catalyzed kinetic resolution of a
chiral carboxylic acid may influence the enantioselectivity more than substrate
engineering for that of a chiral alcohol does. In an example of substrate engineering of
carboxylic acids, it has been reported that the change of the substrate can invert the
enantiomeric preference.54 Molecular modeling suggests that the phenoxy group binding
pocket in Humieolar lanuginosa lipase (JilL) would be different according to each
enantiomer of 2-phenoxyalkanoic acid. Structural changes of substrates can alter the
binding interaction between the substrate and HHL. For example, the change of substrate
from ethyl 2-phenoxyacetate to ethyl 2-phenoxypropionate shows inverted
enantioselectivity. On the other hand, the engineering of the alcohol moiety in the
enzyme-catalyzed kinetic resolution of chiral carboxylic acids esters also shows increase
of enantioselectivity.55 In the resolution of 2-methyloctanoic acid by Candida rugosa
lipase (CRL), the enantioselectivity was linearly related to the chain length of alcohol.
The enantioselectivity increased from 20 (with butanol) to 126 (with hexadecanol).
Presumably, increasing the size of alcohol increases steric hinderance in the active site of
CRL, thereby changing the interaction between substrates and the enzyme.
Substrate engineering was studied for ring opening of p-Iactams in this thesis
(Chapter 4). The influence of a series of alcohols and water as a nucleophile was explored
on the reaction rate as well as selectivity.
53 Sih, C. J.; Wu, S.-H. In Topies in Stereoehemistry; Eliel, E. L., Wilen, S. H. Eds.;
Wiley: New York, 1989; Vol. 19, pp 63-125.
54 Berglund, P.; Vallikivi, I.; Fransson, L.; Dannacher, H.; Holmquist, M.; Martinelle, M.;
Bjorkling, F.; Parve, O.; Huit, K. Tetrahedron: Asymmetry 1999, 10,4191-4202.
55 Berglund, P.; Holmquist, M.; Hedenstrom, E.; Huit, K.; Hogberg, H.-E. Tetrahedron:
Asymmetry 1993,4, 1869-1878.
20

1.4" Protein Engineering to Improve Enantioselectivity
Although biocatalysts are useful to produce enantiopure compounds, natural
enzymes do not always fit the demands of this process and often need to be modified.
Improved enzymes may be obtained by mutagenesis as weIl as chemical modification of
amino acid residues. Although mutagenesis is only possible when a gene is available in a
suitable expression system, it is used more often because of its predictable results and
because greater diversity can be created. Protein engineering through mutagenesis is
generally performed using two approaches, rational design and directed evolution.
1.4.1. Rational design for altering enantioselectivity of enzyme
In rational design, precise changes in amino acid sequence require a detailed
knowledge of protein structure (an X-ray crystal structure), function, and mechanism. On
the basis of that information, researchers identify a target site using molecular modeling56
for a desirable change in enantioselectivity in the enzyme and then change the amino
acids using site-directed mutagenesis.57
Although information on the mechanisms of enantioselectivity is available58, only
a few rational protein designs have succeeded in altering the enantioselectivity of
enzymes. One rational approach to invert the stereospecificity of a vanillyl-alcohol
oxidase was reported by van den Heuvel et al. 59 Double mutants, Aspl70Ala/Thr457Glu
56 a) Kazlauskas, R. J. Curr. Opin. Chem. Biol. 2000,4,81-88. b) Kazlauskas, R. Seience
2001,293,2277-2279.
57 Chen, R. Trends Biotechnol. 1999,17,344-345.
58 a) Kazlauskas, R. J.; Weissfloch, A. N. E.; Rappaport, A. T.; Cuccia, L. A. J. Org.
Chem. 1991,56,2656-2665 b) Cygler, M.; Grochulski, P.; Kazlauskas, R. J.; Schrag, J.
D.:; Bouthillier, F.; Rubin, B.; Serreqi, A. N.; Gupta, A. K. J. Am. Chem. Soc. 1994,116,
3180-3186.
59 van den Heuvel, R. H. H.; Fraaije, M. W.; Ferrer, M.; Mattevi, A.; van Berkel, W. J. H.
Proc. Natl. Acad. Sei. U.S.A. 2000, 97, 9455-9460.
21

and Asp170SerlThr457Glu, created by site-directed mutagenesis showed an inverted
stereopreference of the hydroxylation of 4-ethylphenol. The mutation relocated the
putative active site to the opposite face of the active site cavity. In another ex ample of
inversion of enantioselectivity, amino acid sequence comparison between two enzymes
having different enantiopreference was used to identify the target sites.60 A triple mutant,
Val266LeulLeu287Ile/Phe221Leu, shows completely opposite enantio-selectivity in
hydrolysis of 1,4-dihydropyridine -3,5-dicarboxylic acid ester. Additional examples of
reversaI of the stereoselectivity by rational approach were obtained by altering the size of
enzyme active site pockets in hydrolysis of organophosphates.61 .
In an example of improving enantioselectivity of enzymes, molecular modeling
indicates two amine acid residues, which have steric interaction with triradylglycerol
substrate. The double mutant, Leu258PhelLeu254Phe, of Rhizopus oryzae lipase
increases the steric repulsion with acyl groups of the substrate and show moderate
improvement in enantioselectivity (E = from 8 to 25).62 Similarly, a rational approach
was used to increase the enantioselectivity of Candida antarctica lipase B (CAL-B)
towards bromo- or chlorohydrin.63 On the basis of a crystal structure and molecular
modeling, the alcohol-binding region was identified as the possible binding pocket for the
bromo or chloro group of the fast-reacting enantiomer. In this binding pocket, there are
four hydrophilic amino acid residues such as Thr40, Ser47 , Thr42 , and Trp104. Those
hydrophilic amino acid residues would make a repulsive interaction with the fast
enantiomer due to partial negative charge on bromo or chloro group. One of mutants,
Ser47 Ala, can remove this repulsive interaction, thereby stabilizing the fast-reacting
60 Hirose, Y.; Kariya, K; Nakanishi, Y.; Kurono, Y.; Achiwa, K Tetrahedron Lett. 1995,
36,. 1063-1066.
61 Chen-Goodspeed, M.; Sogorb, M. A.; Wu, F.; Raushel, F. M. Biochemistry 2001, 40,
1332-1339.
62 Scheib, H.; Pleiss, J.; Stadler, P.; Kovac, A.; Potthoff, A. P.; Haalck, L.; Spener, F.;
Paltauf, F.; Schmid, R. D. Prote in Eng. 1998,11,675-682.
63 Rotticci, D.; Rotticci-Mulder, J. c.; Denman, S.; Norin, T.; HuIt, K ChemBioChem
2001, 2, 766-770.
22

enantiomer in the productive conformation and shows doubly increased enantioselectivity
towards I-bromo-2-octanol or l-chloro-2-octanol. However, this approach was not
successful for Trp104His mutant. The Trp104His mutant showed -70% lower enantio
selectivity than wild type enzyme.
The other example by Magnusson et al. shows the enhancement of
enantioselectivity of CAL-B through substrate-assisted catalysis.64 The hydroxyl group of
Thr40 in CAL-B makes a key hydrogen bond that stabilizes the oxyanion of substrates
(esters) in transition state. The Thr to Val mutation eliminates this hydrogen bond.
However, the hydroxyl group of one enantiomer of the substrate ethyl 2-hydroxy
propanoate restores this missing hydrogen bond, thereby reacting faster than the other
enantiomer. AIthough the enantioselectivity was greatly improved from E = 1.6 to E = 22, the activity of the mutant enzyme was hundred times lower than that of wild type
enzyme.
The rational protein design approach generates a small number of mutant
enzymes and avoids screening large numbers of mutants. However, if a detailed
theoretical understanding of enzymatic catalysis is not available, rational design
approaches are often misguided and fail. Additionally, the approach is not always
successful because of unpredictable structural changes from site-directed mutagenesis.
Since molecular modeling cannot model protein folding or stability, these substitutions
based on molecular modeling might ignore the structural properties of enzymes, thereby
simultaneously changing the activity of enzymes.
1.4.2. Directed evolution for improving enantioselectivity
Detailed structural and mechanistic information of enzymes is not required for
directed evolution (recursive generation and screening of mutants, Figure 4).65 Directed
evolution employs a random process of mutagenesis by error-prone PCR or DNA
64 Magnusson, A.; HuIt, K.; Holmquist, M. J. Am. Chem. Soc. 2001, 125,4354-4355.
65 Petrounia, 1. P.; Arnold, F. H. Curr. Opin. Biotechnol. 2000, 11, 325-330.
23

shuffling to create a library of mutagenized genes. Subsequent high-throughput screening
identifies or selects improved mutant enzymes. The selected mutants may be subjected to
further rounds of mutation and screening to enhance the original beneficial mutation.
Random Screen Mutagenesis for function
~ En In: En
ild Type zyme;
, Mutant Library 1 Better enzyme ldequate zyme
" Idealenzyme
t
Figure 4. Directed evolution. Random mutations are generated followed by screening to
identify improvements. The improved mutant may be used as a parent for the next round
of evolution.
Although directed evolution successfully modified catalytic activity and a wide
range of properties of enzymes, only a few examples improved the enantioselectivity.
The Reetz group reported the first example for increasing enantioselectivity by
directed evolution.66 The wild type of a lipase from Pseudomonas aeruginosa (PAL) has
E = 1.1 towards hydrolysis of p-nitrophenyl 2-methyldecanoate. Four generations of
random mutagenesis using error prone peR and screening 1000-2400 colonies per each
generation (total 5600 colonies) increased the enantioselectivity from 1.1 to 11.3. Later,
two additional generations of random mutagenesis identified a mutant having E = 13.5.
The combination of saturation mutagenesis with random mutation further increased E to
25.8. They proposed that a region of loops, which is involved the conformational change
66 a) Reetz, M. T.; Zonta, A.; Schimossek, K.; Liebeton, K.; Jaeger, K.-E. Angew. Chem.,
Int. Ed. Engl. 1997,36, 2830-2832. b) Liebeton, K.; Zonta, A.; Schimossek, K.; Nardini,
M.; Lang, D.; Dijkstra, B. W. ; Reetz, M. T. ; Jaeger, K.-E. Chem. Biol. 2000, 7,709-718.
24

from closed to open structure, may be important for enantioselectivity. In another
approach (combination of cassette mutagenesis and DNA shuffling), they reported
increased enantioselectivity of up to 51 after screening 40,000 colonies.67
Similarly, the Bornscheuer group improved the enantioselectivity of an esterase
from Pseudomonas fluorescens from 3.5 to 6.6 by error-prone PCR and a mutator
strain.68 This esterase was used for the work in this thesis (Chapter 5).
The Arnold group inverted the enantioselectivity of hydantoinase by directed
evolution. 69 After the first generation of random mutagenesis, they screened 10,000
colonies and found two less D-selective mutants. A second generation of random
mutagenesis showed two active mutants, which have the less L-selectivity with same
stereoprefernece but more activity, from 10,000 colonies. Additional second generation
mutation (another screening of 10,000 colonies) using a higher error rate did not affect
the L-selectivity. On the basis of the second generation of random mutagenesis,
saturation mutagenesis was performed to introduce aIl amino acids (sorne of which may
not be present in the library because of nonconservative substitutions by PCR
mutagenesis at low error rates). According to this site saturation mutagenesis, they
identified one L-selective mutant (I95F/Q251R1VI80A) with low enantioselectivity (20%
eeL at -30% conversion).
A bene fit of directed evolution is the lack of a requirement for structural and
mechanistic information. In addition, it may create a new unexpected selectivity
mechanism. However, the disadvantages are imparted by the challenging task of library
construction and screening of large numbers of mutants. In addition, random mutagenesis
favOirs mutations away from the active site because the number of amino acids increases
as one moves away from the active site and more amino acids lie far from the center than
close to the center (Table 2). In PFE enzyme, most of the amino acids (64%) lie at least
67 Reetz, M. T.; Wilensek, S.; Zha, D.; Jaeger, K.-E. Angew. Chem., Int. Ed. Engl. 2001,
40, 3589-3591.
68 Henke, E.; Bornscheuer, U. T. Biol. Chem. 1999,380,1029-1033.
69 May, O.; Nguyen, P. T.; Arnold, F. H. Nat. Biotechnol. 2000,18,317-320.
25

15 A from the active site and <10% lie within 10 Â of the active site. The mutation away
from the active site may not give effective change of enzyme properties. For example, the
Arnold group found that the mutation 26 Â away from active site of hydantoinase did not
change any enzyme properties. More effective changes were found in the mutation within
1O-1l5 Â. Although a molecular basis for the enantioselectivity change is likely different
in each case, mutations close to the active site are likely to affect the enantioselectivity
more strongly than mutations far from the active site.
Table 2. The number of amino acids with respect to distance from the active site of PFE.
distancea number of amino acidé fraction of aU amino acids within 7 Â within 10 Â within 15 Â within 17.5 Â an (within 31 Â)
8 24 98 137 272
2.6% 8.5% 36% 50% 100%
a The distances are between Ca of the amino acid residues and the stereocenter of
a bound substrate (methyl 3-bromo-2-methylpropionate).
b The numbers are accumulated according to distance.
1.4.3 Combining rational protein design and directed evolution
When the X-ray structure of an enzyme is available, the hybrid approach of a
rational design and directed evolution can be employed. Molecular modeling of a
substrate bound enzyme structure helps researchers to select sites for introducing a
functional amino acid. A directed evolution may th en not only fix the unexpected
structural change from rational design but also improve an additional property of
enzymes. For example, Copinus cinereus heme peroxidase used as a dye-transfer
inhibitor in laundry detergent was made more stable to washing machine conditions using
a combination of computer modeling to identify important sites to be changed for
improving the stability and error-prone PCR to increase the additional stability.70
70 Cherry, J. R.; Lamsa, M. H.; Schneider, P.; Vind, J.; Svendsen, A.; Jones, A.; Pedersen,
26

Although an approach of rational design and refinement by directed evolution would be
successful for enhancing enantioselectivity of enzymes, both intensive structural analysis
and extensive screening are still required.
However, using approximate structural information, which can be easily accessed
by a homology model generated from amino acid sequence7!, to select a small number of
amino acid residues around substrate binding region for random mutagenesis would be
the best combination of both approaches since the detailed molecular modeling and
extensive screening can be avoided. An approach like that just mentioned was applied in
this thesis to increase enantioselectivity of a hydrolase. The work (chapter 5) in this thesis
deals with the hydrolysis of MBMP (methyl 3-bromo-2-methylpropionate) by PFE
(Pseudomonas fluorescens esterase) as a model system for developing protein
engineering methods to efficiently improve enantioselectivity.
1.5" High-throughput Screening: Quiek E
The stereospecificity of enzymes was first formulated as the ratio of the reactivity
index (l<cat/KM) for each substrate enantiomer.72 Later, Sih and coworkers expressed this
ratio as the enantiomeric ratio, E, and described the equation to determine the ratio by the
degree of conversion and the enantiomeric excesses of starting material or produCt.73
However, this conventional determination of enantioselectivity requires chromatographic
analysis to determine enantiomeric excesses, thereby taking a few hours for analysis of
one reaction. Therefore, it is not suitable for rapid screening of large enzyme or substrate
libraries.
A. H. Nat. Biotechnol. 1999,17,379-384.
71 Guex, N.; Diemand, A.; Peitsch, M. C. Trends Biochem. Sei. 1999,24,364-367.
72 Hein, G. E.; Niemann, C. J. Am. Chem. Soc. 1962,84,4487-4494.
73 Chen, C.-S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1982, 104,
7294-7299.
27

Over the past few years high-throughput screening based on a wide range of
detection systems have been developed for rapid analysis of the large numbers of
enzyme-catalyzed reactions.74 A convenient colorimetrie screening method was
developed by the Reetz group. This UV/Vis-based screening system is based on separate
measurement of the initial rate of hydrolysis of each enantiomer containing a
chromogenic moiety, p-nitrophenol. Although this screening method is simple and
effective in identifying selective enzyme, it neglects substrate competition with respect to
the enzyme and requires chromogenic substrates such as p-nitrophenyl esters.
Recently in our group, a new high-throughput screening method, called Quick E, has
been developed to consider competitive conditions of an enzymatic process.
The Quick E screening method for enantioselective ester hydrolysis has been
developed using a pH indicator to detect the reaction rate and a chromogenic resorufin
ester, which competes with the substrate for enzyme binding sites (Figure 5).75 The
resorufin ester is present with enantiopure substrate during the reaction, providing a
reference by which to compare the rates of each pure enantiomer and therefore
accounting for competitive binding.
74 Review: Reetz, M. T. Angew. Chem., [nt. Ed. Engl. 2001, 40, 284-310.
75 a) Janes, L. E.; Kazlauskas, R. J. J. Org. Chem. 1997, 62, 4560-4561. b) Janes, L. E.;
Lë~wendahl, AC.; Kazlauskas, R. J. Chem. Eur. J. 1998,4, 2317-2324. c) Janes, L. E.;
Cimpoia, A; Kazlauskas, R. J. J. Org. Chem. 1999, 64, 9019-9029. d) Janes, L. E.;
Lowendahl, AC.; Kazlauskas, R. J. PCT/US99/14448.
28
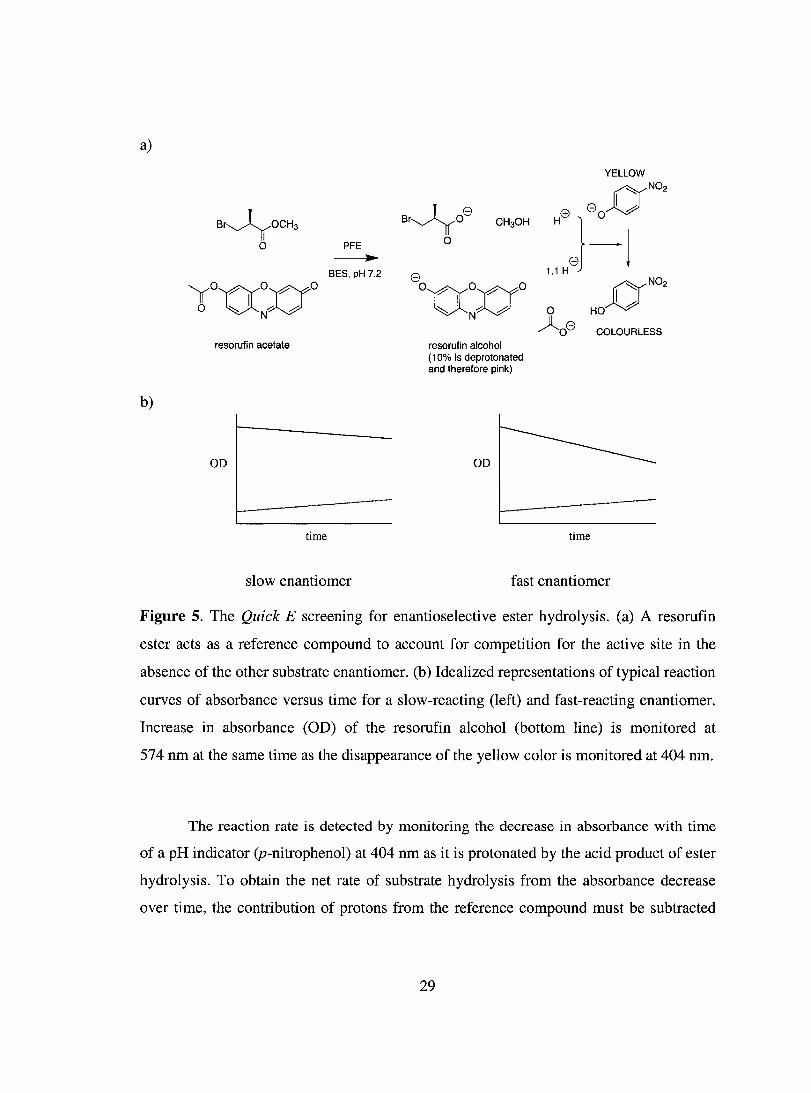
a)
BES, pH 7.2
resorufin acetate
b)
OD
time
slow enantiomer
YELLOW ON02
H$$}~î 8 1.1 H
0yyOyyO ~N02
V~ ° HO~ N Il 8
resorufin alcohol (10% is deprotonated and therefore pink)
OD
/"'--0 COLOURLESS
time
fast enantiomer
Figure 5. The Quick E screening for enantioselective ester hydrolysis. (a) A resorufin
ester acts as a reference compound to account for competition for the active site in the
absence of the other substrate enantiomer. (b) Idealized representations of typical reaction
curves of absorbance versus time for a slow-reacting (left) and fast-reacting enantiomer.
Increase in absorbance (OD) of the resorufin a1cohol (bottom line) is monitored at
574 nm at the same time as the disappearance of the yellow color is monitored at 404 nm.
The reaction rate is detected by monitoring the decrease in absorbance with time
of a pH indicator (p-nitrophenol) at 404 nm as it is protonated by the acid product of ester
hydrolysis. To obtain the net rate of substrate hydrolysis from the absorbance decrease
over time, the contribution of protons from the reference compound must be subtracted
29

(eq 1.1).76 The rate of hydrolysis of the resorufin ester is monitored as its absorbance at
574 nm increases (eq 1.2). The rates relative to the reference compound may be
compared to calculate the enantioselectivity (eq 1.3).
ratesubstrate[ ~Ol] = (dA404 X 1 x V x 106
Jlmol x QJ -l.l(ratereference) (1.1) mm dt 11& 404 x f mol
where
dA40Jdt = change in absorbance with time at 404 nm,
~&404 = difference in extinction coefficient between protonated and
deprotonated forms of p-nitrophenol (PNP) at 404 nm (M-1 cm-1),
f = pathlength (cm),
v = reaction volume (L),
Q = [BES] + [pNP] . [pNP]
ratereference -- = --x x V x --'---[ JlmOI] dA574 1 10
6 Jlmol
min dt &574 x f mol
where
&574 = extinction coefficient of resorufin at 574 nm.
( ratesubstratel [reJerence Jor 1] J
QuickE = ratereferenceforl x [substrate1]
( ratesubstrate2 [reJerence Jor2] J
ratereferencefor2 x [substrate2]
(1.2)
(L3)
76 At the pH of reaction condition (pH 7.2), while the acid products (pKa = - 5) are fully
30

This high-throughput screening method (Quick E) was applied to identify
improved enantioselective mutants of PFE generated by structure guided random
mutagenesis (Chapter 5).
1.6" Molecular Modeling
Molecular modeling is a set of computational chemistry techniques that deal with
displaying molecules, minimizing their structure, estimating their properties, visualizing
molecular motion, and predicting binding modes of molecules. Since these techniques are
useful in prediction and interpretation of the transition state of chemical reactions, they
are a popular tool for experimentalists. In addition, molecular modeling can be used to
explain molecular basis of enzyme selectivity and suggest changes for improvement of
enzyme-catalyzed reactions.
1.6.1. Molecular mechanics vs Quantum mechanics
The molecular models for ca1culating molecular energy are divided into quantum
mechanics and molecular mechanics models (i.e., empirical force field model). 77
Quantum mechanics (e.g., ab initio) offers a model for chemical reactions involved in
bond making or breaking, which depend on the electronic distribution. However,
quantum mechanics is limited to small systems since it deals with subatomic particles
(i.e., electrons), thereby requiring strong computational power. In a minimization
ca1culation (recursive energy ca1culation and slight conformation al change of a
molecule), ab initio ca1culations may become time-consuming because the free motion of
the nucIei incIuding electrons causes enormous computation al calculations. Approximate
deprotonated, 10% of resorufin (pKa = 8.15) can be deprotonated.
77 Leach, A. R. Molecular Modeling: Principles and Applications; Longman:Harlow,
u.K., 1996, Chapter 2.
31

quantum mechanical methods can significantly reduce computational needs.
Incorporation of parameters derived from experirnental data enables the prograrn to
calculate sorne properties more accurately than ab initia methods. Currently, MINDO/3,
MNDO, and AMI are widely used as semi-empirical quantum mechanics methods.
Although sorne of the electrons can be ignored by the semi-empirical methods to
decrease enormous cornputational efforts, a large number of partic1es should still be
considered in quantum mechanics. As empirical force field methods (i.e., molecular
mechanics) only deal with the nuc1ear positions without considering the electronic
motions based on c1assical mechanics and experimental data, molecular mechanics is able
to calculate a system, such as protein and DNA, containing significant numbers of atorns
with reasonable accuracy. For this reason, structure minimization usually uses molecular
mechanics.
An empirical force field can be defined by its functional form and its parameters.
These two parts are different depending on the force field type. The functional form is
most simply characterized by four component terms of the intra- and intermolecular
forces in the system. More sophisticated force field may have more terms. The four key
contributions to a molecular mechanics force field are bond stretching, angle bending,
torsional terms and non-bonded interactions.
The other part (i.e., pararneters) in a force field is expressed by the atorn type. The
atom type contains information about its hybridization state and sometimes about the
local environment. For example, the AMBER force field78 uses different atom types for
carbon atorns of histidine depending on its protonation state while other force fields
78 a) Weiner, S. J.; Kollrnan, P. A; Case, D. A; Singh, U. c.; Ghio, c.; Alagona, G.;
Profeta, Jr., S.; Weiner, P. J. Am. Chem. Soc. 1984, 106, 765-784. b) Cornell, W. D.;
Cieplak, D. P.; Bayly, C. I.; Gould, I. R.; Merz, Jr., K. M.; Ferguson, D. M.;
Spellmeyer, D.C.; Fox, T.; Caldwell, J. W.; Kollman, P. A J. Am. Chem. Soc. 1995,
117,5179-5197.
32

would define these atoms as the same carbon atom type.79 Specific force fields should be
used for modeling of specific classes of molecules.
The AMBER force field has been specifically designed for biopolymers such as
proteins and DNA. This force field is composed of six energy terms. The first three terms
are related to the internaI coordinates of bonds, angles, and dihedrals (eq. 1.4). The next
two terms describe van der Waals and electrostatic interactions. Hydrogen bond energy is
mostly calculated by dipole-dipole interaction in AMBER while the sixth term de scribes
addition al hydrogen bonding interactions.
EIOlal = L K2(b-bo)2 + LHe(()-()O)2 + L v" [1 + cos(nqj-qjo)] bonds angles dihedral 2
(1.4)
1.6.2. Energy minimization
The most important feature in molecular modeling is finding the most stable
structure, (i.e., the global minima) of a molecule or a system. In addition to the most
stable structure, the system may adopt many other stable structures (i.e., local minima).
The energy minimization algorithm is an approach used to find lower potential
energy by slightly changing the coordinates of atoms in a system. Once the potential
energy of the initial structure of the system is evaluated, the energy is re-evaluated after
each atom is slightly moved. The position of minimum can be estimated by the direction
of the first derivative of the energy (i.e., the gradient). Two first-order minimization
algorithms (using the first derivatives) are frequently used in molecular modeling:
steepest descent and the conjugate gradient methods.8o. These two algorithms were
formulated depending on the way in searching the direction of new coordinates of atoms.
79 Leach, A. R. Molecular Modeling: Principles and Applications; Longman: Harlow, U.
K., 1996, Chapter 3.
80 a) Insight II user guide, BiosymlMSI; San Diego, CA, 1995. b) Leach, A. R. Molecular
33

1.6.2.1. Steepest descents method
The steepest descents method changes the coordinates of the atoms in the
direction parallel to the net force. In addition to the direction to move, it is necessary to
decide how far to move along the gradient. For example, in the two-dimensional energy
surface (Figure 6), the gradient direction from the starting point (a) is along the line
indicated. To locate the minimum point, two minimization paths are available: a line
search and a step of arbitrary size.
((x, y)
Figure 6. Aline search to locate the minimum in the direction of the gradient.
Aline search is to find the minimum along a specified direction (i.e., along a line
through the multidimensional space). After finding three points along the line such that
the energy of the middle point is lower than the energy of the two outer points, an
iterative procedure can be used to decrease the distance between the three points. The
gradient at the minimum point obtained from the line search will be perpendicular to the
previous direction. Thus, when the line search method is used to locate the minimum
along the gradient then the next direction in the steepest descents algorithm will be
orthogonal to the previous direction (Figure 7 a). This orthogonal property the directions
oscillate along the way to the minimum.
Modeling: Principles and Applications; Longman: Harlow, u.K., 1996; Chapter 4.
34

On the other hand, the position for the new gradient may be updated at any point
if the trial point along the gradient had a lower energy. Constant change of the direction
to match the current gradient, oscillations along the minimization path might be
decreased. The result of such a minimization path is shown in Figure 7b. Each line search
needs to use two points for the energy evaluations. When the trial point has a higher
energy, the step is adjusted downward and a new trial point generated. The arbitrary step
method needs fewer the total function evaluations than the rigorous line search method.
a b
Figure 7. Minimization path following a steepest-descents path. a) with line
searches b) without line searches
This steepest descents method is generally robust even when the starting point is
far from a minimum. However, it requires many small steps when proceeding down a
long narrow valley. Convergence is slow and the path oscillates near the minimum.
1.6.2.2. Conjugate gradient method (conjugate directions method)
The conjugate gradient method is an approach to find the right direction to the
minimum with avoiding the reverse progress in an earlier iteration, which is a character
of the steepest descents method. In the conjugate gradients, although the gradients at each
point are orthogonal, the directions are conjugate. The direction (Vk) can be calculated
from the gradient (gk) at the point k and the previous direction (Vk-l). The Yk at point k is a
scalar constant from the gradient vectors (gk and gk-l).
Yk =
35

The conjugate gradients method produces a set of directions without the
oscillatory behavior of the steepest descents method (Figure 8).
Figure 8. A minimization path following conjugate gradient method
While the conjugate gradient method is more accurate and faster in finding close
minima, the steepest descent method is stable for the system far away from minima.
Generally, researchers use the steepest descent in the initial steps and then switch to
conjugate gradient method.
1.6.3. Conformational searching
Since most minimization algorithms find the nearest stable geometries from an
initial structure of the system, additional methods, such as a random search (e.g., Monte
Carlo method), molecular dynamics, and manual search, have been used for producing
larger changes in larger systems (e.g., proteins or DNAs).81
Monte Carlo method randomly moves individual atoms with random-number
generating algorithm and evaluates the energy of the resulting configurations. After the
evaluation of energy, it decides whether the new configuration is accepted or not. When
new configuration is lower than the energy of the previous one, the new configuration is
accepted. However, if the energy of the new configuration is higher than the energy of
the previous one, another treatment is required. First, the Boltzman factor of the energy
difference is ca1culated. Then, a random number is then generated between 0 and 1 and
compared with this Boltzman factor. While the move with higher random number than
the Boltzman factor is rejected and the previous configuration is retained for the next
81 Young, D. Computational Chemistry: A Practical Guide for Applying Techniques to
Real World Problems; Wiley-Interscience: New York, 2001.
36

iteration, the move with lower random number is accepted and the new configuration
becomes the next state. This procedure allows moves to states of higher energy.
However, the smaller uphill move has higher probability for acceptance. Monte Carlo
method is particularly useful for an intermediate size system since the random motions
are irrespective of potentially large barrier to rotation. However, this is not suitable for
large systems such as enzymes because the cooperative motions about internaI
coordinates inhibit random motions of atoms.
On the other hand, molecular dynamics is a simulation of the time-dependent
behavior of a molecular system. Molecular dynamics uses Newton's law of motion and
generates successive configurations of a system. 82 The result is a trajectory that includes
the change of the positions and velocities of the atom in the system with time. The time
interval should be longer than the computation time. But if the time interval is too long,
the atom moves too far along a given trajectory, thereby poorly simulating the motion.
The time interval is generally limited to between 101_102 ps for large molecular systems
such as enzymes because the computational ca1culation becomes enormous for large
systems. For the modeling of molecular basis of enantioselectivity, researchers generally
use molecular dynamics in the narrow area of substrate binding active site of enzyme,
thereby saving the computational ca1culation.
Alternately, when the substrate has fairly rigid structure (i.e., the number of
possible conformations is small), the manual conformational search can be applied to
save enormous computational efforts.
In this thesis (Chapter 4), the molecular modeling technique has been used to
elucidate the role of the a1cohol and molecular basis of enantioselectivity in ring opening
r2 F 82 a Xi _ x -- _i where Xl.: a coordinate of a particle, Fx ,.: the force on the particle in that dt 2
- mi
direction, mi: the mass of the particle. The new coordinates can be generated by this
equation in given time. Leach, A. R. Molecular Modeling: Principles and Applications;
Longman: Harlow, UK., 1996; Chapter 6.
37

of ~-lactam. Because the structure of ~-lactam is fairly rigid, manual search has been
usecl for the conformational search.
1. 7. Enhancing Hydrolase Activity and Selectivity - outline of this thesis
Biocatalysts are useful for synthesis because of their high selectivity and the mild
reaction condition. However, natural enzymes are often not suitable for aIl purposes of
synthesis in terms of their activity and selectivity. Enhancing their activity or selectivity
can be approached in different ways depending on the reactions. For example, enzymes
are inactive in polar solvents (e.g., dimethyl sulfoxide) that can dissolve polar substrates
such as sugars and peptides, which are becoming important starting materials for
pharmaceutical chemistry. For the reliable enzyme-catalyzed reactions of polar
substrates, alternative sol vents are required. On the other hand, when enzymes show low
selectivity to a substrate, the selectivity might be increased through substrate engineering.
In addition, if the gene of an enzyme is available, the enzyme itself can be altered to
improve its selectivity.
This thesis deals with three approaches (i.e., medium, sub strate , and protein
engineering) to enhance hydrolase activity and enantioselectivity. Chapter 2 and 3
describe alternative solvents that reliably work for hydrolase-catalyzed reactions of polar
substrates. Ionie liquids as an alternative solvent dissolve polar substrates such as glucose
and ascorbic acid, thereby giving reliable reaction rates and regioselectivity (in case of
acylation of glucose). In chapter 4, substrate engineering was applied to increase reaction
rate and enantioselectivity for ~-lactam ring opening reaction. The change of
nuc1eophiles (i.e., alcohols or water) showed dramaticaIly altering reaction rate and
enantioselectivity. In addition, molecular modeling was used to elucidate the important
role of alcohol and high enantioselectivity of CAL-B. Chapter 5 deals with an efficient
approach to improve the enantioselectivity of PFE through protein engineering. Structure
guided random mutagenesis is more efficient to discover more enantioselective mutants
than rational approach and directed evolution. In order to rationalize the high
38

enantioselectivity of Va1122Ser mutant, the effect of changing the shape or the electronic
character of substrate on the enantioselectivity was investigated.
39

Chapter 2
Currently, research on green technology, which avoids using volatile orgamc
solvents, has been increased. One promising class of solvents is ionic liquid, which is
completely nonvolatile. In addition, ionic liquids are highly polar and therefore dissolve a
broad range of substrates, thereby facilitating many reactions.
In this chapter, we apply medium engineering to obtain reliable activity and
selectivity in enzyme-catalyzed reactions of polar substrates. This chapter describes
improved preparation method of ionic liquids for enzyme-catalyzed reactions and their
applications. Newly purified ionic liquids work reliably in lipase-catalyzed reactions.
Lipase-catalyzed reaction of glucose in ionic liquids shows higher regioselectivity than in
organic solvents.
Reproduced with permIssIOn from The Journal of Organic Chemistry, Vol. 66, Seongsoon Park and Romas J. Kazlauskas, "Improved Preparation and Use of RoomTemperature Ionic Liquids in Lipase-Catalyzed Enantio- and Regioselective Acylations", 8395-8401. Copyright 2001 American Chemical Society.
40

Chapter 2. Improved Preparation and Use of Room
Temperature Ionie Liquids in Lipase-Catalyzed Enantio
and Regioseleetive Acylations
Abstract: Polar organic sol vents such as methanol or N-methylfonnamide inactivate
lipases. Although ionic liquids such as 3-alkyl-l-methylimidazolium tetrafluoroborates
have polarities similar to these polar organic solvents, they do not inactivate lipases. To
get reliable lipase-catalyzed reactions in ionic liquids, we modified their preparation by
adding a wash with aqueous sodium carbonate. Lipase-catalyzed reactions that previously
did not occur in untreated ionic liquids now occur at rates comparable to those in
nonpolar organic sol vents such as toluene. Acetylation of I-phenylethanol catalyzed by
lipase from Pseudomonas cepacia (PCL) was as fast and as enantioselective in ionic
liquids as in toluene. Ionic liquids pennit reactions in a more polar solvent than
previously possible. Acetylation of glucose catalyzed by lipase B from Candida
antarctica (CAL-B) was more regioselective in ionic liquids because glucose is up to one
hundred times more soluble in ionic liquids. Acetylation of insoluble glucose in organic
sol vents yielded the more soluble 6-0-acetyl glucose, which underwent further
acetylation to give 3,6-0-diacetyl glucose (2-3:1 mixture). However, acetylation of
glucose in ionic liquids yielded only 6-0-acetyl glucose (>13:1 and up to >50:1).
Introduction
Although enzymes are environmentally friendly reagents, sorne enzyme-catalyzed
reactions require environmentally harmful organic solvents. One potential solution is to
replace organic sol vents with room-temperature ionic liquids. Room-temperature ionic
liquids are organic salts whose ions do not pack weIl and remain liquid at room
41

temperature. Ionic liquids are completely nonvolatile and can usually be recycled and
reused. 1
Several groups recently reported enzyme-catalyzed reactions in ionic liquids and
identified sorne potential advantages besides environmental ones. Thermolysin for
peptide synthesis was more stable in ionic liquids as compared to ethyl acetate, but the
reaction rates were lower. 2 On the other hand, reaction rates of lipase-catalyzed
a1coholysis, ammoniolysis and perhydrolysis were comparable or slightly better in ionic
liquids as compared to organic solvents? Similarly, Schofer et al. reported faster
reactions in sorne ionic liquids, but no reaction at aIl in others, even when the structures
were very similar. In addition, they as weIl as Kim et al. found that the enantioselectivity
of lipase-catalyzed acetylation of secondary a1cohols was higher in sorne ionic liquids.4
Although one can finely tune the properties of an ionic liquid by varying its structure, one
must first identify which solvent properties are important. Wh en enzyme-catalyzed
reactions work in one ionic liquid, but not in a similar one, it is impossible to identify the
most important solvent properties for enzyme-catalyzed reactions.
In this paper, we report an improved preparation of ionic liquids that yields ionic
liquids that work reliably in enzyme-catalyzed reactions. Minor changes in structure of
1 Review: Seddon, K. R J. Chem. Technol. Biotechnol. 1997, 68, 351-356. Welton, T.
Chem. Rev. 1999,99,2071-2083. Wasserscheid, P.; Keim, W. Angew. Chem., IntI. Ed.
Engl. 2000, 39, 3772-3789.
2 Erbeldinger, M.; Mesiano, A. J.; Russell, A. J. Biotechnol. Prog. 2000, 16, 1131-1133.
Aiston, W. C., II; Ng, K. Book of Abstracts; 217th ACS National Meeting, Anaheim,
CA, March 21-25 1999; American Chemical Society: Washington, DC, 1999; BIOT-
131.
3 Lau, R M.; Rantwijk, F. van; Seddon, K. R; Sheldon, R A. Org. Lett. 2000, 2, 4189-
4191.
4 Itoh, T.; Akasaki, E.; Kudo, K.; Shirakami, S. Chem. Lett. 2001, 262-263. SchOfer, S.
H.; Kaftzik, N.; Wasserscheid, P.; Kragl, U. Chem. Commun. 2001,425-426. Kim, K.
W.; Song, B.; Choi, M.-Y.; Kim, M.-J. Org. Lett. 2001,3, 1507-1509.
42

the ionic liquids no longer cause dramatic changes in reaction rate. We also measured the
polarity of common ionic liquids and show that they are comparable to methanol and N
methylformamide. As an example of a lipase-catalyzed transformation of a polar
substrate, we report the lipase-catalyzed acetylation of glucose in ionic liquids.
Results
Synthesis of Ionie Liquids. Ten ionic liquids were prepared either by literature
procedures or by straightforward modification of literature procedures, Scheme 1.5 For
example, alkylation of N-methylimidazole with an alkyl halide yielded 3-alkyl-l-methyl
imidazolium halides as white solids. Metathesis with sodium tetrafluoroborate yielded the
desired tetrafluoroborate salts as viscous oils. Unfortunately, lipase-catalyzed reactions in
these unpurified ionic liquids were either slow or did not occur; see below. We suspected
that an impurity in the se ionic liquids might inhibit the lipase-catalyzed reactions, so we
tested several purification methods.
5 BMIM'BF4 or BMIM'PF6 via metathesis of the halide with NaBF4, NaPF6, or HPF6: (a)
Huddleston, J. G.; Willauer, H. D.; Swatloski, R. P.; Visser, A. E.; Rogers, R. D. Chem.
Commun. 1998, 1765-1766. (b) Suarez, P. A. Z.; DuIlius, J. E. L.; Einloft, S.; De Souza,
R. F.; Dupont, J. Polyhedron 1996, 15, 1217-1219. EMIM·BF4 or PMIM·BF4 via
AgBF4: (c) Holbrey, J. D.; Seddon, K. R. J. Chem. Soc., Dalton Trans. 1999, 2133-
2139.
43

® ® (';,N R-X ~N_R NaBF4 ~~_Re NJ~N3 e-N....:I
1 1 X 1 BF4 H3C H3C white solid H3C ionic liquid
(X = CI or Br) 1 purify 'f
Et n-Pr n-Bu s-Bu MeOCH2CH2
Abbreviation EMIM-BF4 PMIM-BF4 BMIM-BF4 sBMIM-BF4 MOEMIM-BF4
B1..= B2..= Abbreviation H n-Pr PPYR-BF 4 H n-Bu BPYR-BF 4
Me n-Pr PNPYRoBF 4
Me n-Bu BMPYR-BF4
MethodA 1) add AgBF4, filter 2) column chromatography on silical gel
Method B 1) filter through silica gel 2) wash with sat'd. Na2C03
Scheme 1. Ten Ionic Organic Liquids Investigated in This Paper. The cations are either
3-alkyl-l-methylimidazolium or N-alkylpyridinium. The anions were tetrafluoroborate in
all nine salts shown, but the tenth was a hexafluorophosphate salt: 3-butyl-l-methyl
imidazolium hexafluorophosphate, BMIM·PF6•
One known impurity in these crude ionic liquids is 3-alkyl-l-methylimidazolium
halide, which remains from an incomplete metathesis reaction.5 We confirmed that crude
ionic liquids contain halide because a precipitate formed upon addition of aqueous silver
nitrate. Previous researchers removed the contaminating halide either by precipitation
with silver tetrafluoroborate or by carrying out the metathesis in acetone from which the
tetrafluoroborate salts separated as viscous oils. The acetone method does not completely
remove the halide, so we used the silver tetrafluoroborate method. Precipitation of the
halide using silver tetrafluoroborate followed by chromatography on silica gel, which we
call purification method A, removed the halide as shown by no precipitate upon addition
of sil ver nitrate solution. Although this purification method improved sorne ionic liquids,
others still gave slow reactions or, in sorne cases, no reaction; see below.
We developed an alternative purification that avoids the use of silver ion, which
we call purification method B. The ionic liquid was diluted with methylene chloride,
filtered through silica gel to remove the 3-alkyl-l-methylimidazolium halide, and washed
44

with saturated aqueous sodium carbonate.6 Finally, we dried the ionic liquid with
anhydrous magnesium sulfate followed evaporation of the methylene chloride under
vacuum. This purification method B yielded ionic liquids that worked reliably in aIl
lipase-catalyzed reactions tested in this paper.
These procedures yielded five 3-alkyl-1-methylimidazolium tetrafluoroborate
ionic liquids. One additional ionic liquid, 3-butyl-1-methylimidazolium hexafluoro
phosphate, was prepared from the halide by metathesis with hexafluorophosphoric acid
followed by purification using method B. Similar reactions and purification methods
yielded four more ionic liquids based pyridinium and 4-methylpyridinium cations for a
total of 10 different ionic liquids, Scheme 1.
Lip:ase-Catalyzed Enantioselective Acetylation Reactions in Ionie Liquids. As a
model reaction, we used the acetylation of 1-phenylethanol with vinyl acetate catalyzed
by lipase from Pseudomonas cepacia, peL, eq 1. This is a highly enantioselective
reaction so the maximum conversion was 50%. We compared the rates of reaction and
enantioselectivities in ionic solvents to those in normal organic solvents such as toluene
and acetone, Table 1. Lipases did not dissolve in ionic liquids, but remained suspended as
powders as they do in organic solvents. The enantioselectivity of the acetylation
remained high, E > 200, in all ionic liquids. However, the reaction rates, as measured by
the degree of conversion after 24 h, varied dramaticaIly. Reaction rates in unpurified
ionic liquids (Table 1, entries 9-12) were at least two to five times slower than in toluene,
THF, or acetone (Table 1, entries 1-4). In many ionic liquids, of which one example is
shown, PMIM·BF4 (Table 1, entry 12), no reaction occurred. Upon purification of the
ionic liquids using method A (Table 1, entries 13-16), the reaction rate in BMIM·BF4
(compare entries 9 and 14 of Table 1) doubled, while that in PMIM·BF4 increased from
no reaction to a rate similar to that in toluene or acetone (compare entries 12 and 15 of
6 A1though BMIM·PF6 is not miscible with water, the tetrafluoroborate ionic liquids
dissolve in water. The ionic liquids were diluted with methylene chloride to permit
washing with an aqueous solution.
45

Table 1). Nevertheless, a number of structurally similar ionic liquids still gave no
reaction after purification by method A (Table 1, entries 13 and 16).
CAl·B = YD PClor Xo ()H '-'::+ --.. '-'::+ '-'::+0
1 ,.,;; bAc solvent 1 A V \ (1)
On the other hand, ionic liquids purified by method B showed consistent behavior
(Table 1, entries 21-30). Reaction rates varied moderately with moderate changes in
structure of the ionic liquid, and the fastest rates were the same as in toluene or acetone.
To identify the impurities that cause of the lack of reaction in sorne ionic solvents,
we measured the effect of additives on the reaction, Table 1, entries 31-43. For ionic
sol vents purified by method B, addition of silica gel, bromide salt of the ionic liquid, or
sodium carbonate had no detectable effect on either the rate or enantioselectivity of the
reaction. However, addition of sil ver tetrafluoroborate completely stopped the reaction,
while addition of acetic acid slowed the reaction by approximately a factor of 2. Thus,
sil ver ion stops the PCL-catalyzed reaction, while acetic acid slows it down. Another
lipase, lipase B from Candida antarctica, CAL-B, showed a similar inactivation upon
addition of silver tetrafluoroborate.7 (Data not shown.) Thus, the most likely causes of
slow reaction or no reaction in ionic sol vents purified by method A are traces of
remaining silver ion or acidic impurities.
Consistent with this explanation, the addition of solid sodium carbonate to ionic
liquid purified by method A dramatically increased the rate of reaction to the same level
7 The inactivation of these two lipases by traces of silver ion is not surprising. PCL
contains two cysteine residues that form a disulfide link on the surface of the protein
(C190 and C270). Similarly, CAL-B contains six cysteine residues that form three
disulfide links on the lipase surface (C22 and C64, C216 and C258, C293 and C311).
Silver presumably disrupts these links and inactivates the lipase. Unlike the other ionic
liquids, lipases PCL and CAL-B remained active in sBMIM·BF4 even after the addition
of silver ion, Table 1, entry 38. (Data for CAL-B not shown.) We do not understand the
reason for this phenomenon.
46
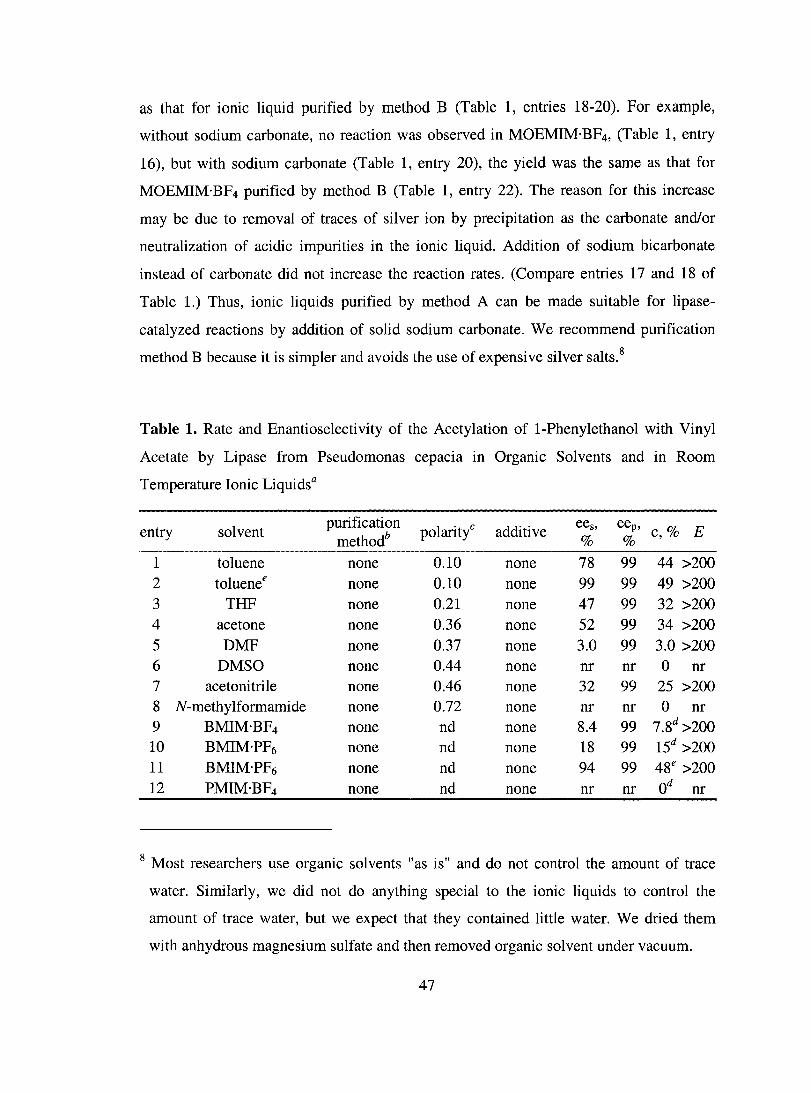
as that for ionic liquid purified by method B (Table 1, entries 18-20). For example,
without sodium carbonate, no reaction was observed in MOEMIM'BF4, (Table 1, entry
16), but with sodium carbonate (Table 1, entry 20), the yield was the same as that for
MOEMIM'BF4 purified by method B (Table 1, entry 22). The reason for this increase
may be due to removal of traces of silver ion by precipitation as the carbonate and/or
neutralization of acidic impurities in the ionic liquid. Addition of sodium bicarbonate
instead of carbonate did not increase the reaction rates. (Compare entries 17 and 18 of
Table 1.) Thus, ionic liquids purified by method A can be made suitable for lipase
catalyzed reactions by addition of solid sodium carbonate. We recommend purification
method B because it is simpler and avoids the use of expensive silver salts.8
Table 1. Rate and Enantioselectivity of the Acetylation of I-Phenylethanol with Vinyl
Acetate by Lipase from Pseudomonas cepacia in Organic Solvents and in Room
Temperature Ionic Liquidsa
entry solvent purification
polaritl additive ees, eep,
c,% E methodb % %
1 toluene none 0.10 none 78 99 44 >200 2 toluenee none 0.10 none 99 99 49 >200 3 THF none 0.21 none 47 99 32 >200 4 acetone none 0.36 none 52 99 34 >200 5 DMF none 0.37 none 3.0 99 3.0 >200 6 DMSO none 0.44 none nr nr 0 nr 7 acetonitrile none 0.46 none 32 99 25 >200 8 N-methylformamide none 0.72 none nr nr 0 nr 9 BMIM'BF4 none nd none 8.4 99 7.8d >200 10 BMIM'PF6 none nd none 18 99 15d >200 11 BMIM'PF6 none nd none 94 99 48e >200 12 PMIM'BF4 none nd none nr nr Od nr
8 Most researchers use organic solvents "as is" and do not control the amount of trace
water. Similarly, we did not do anything special to the ionic liquids to control the
amount of trace water, but we expect that they contained little water. We dried them
with anhydrous magne sium sulfate and then removed organic solvent under vacuum.
47

13 EMIM·BF4 A nd none nr nr 0 nr 14 BMIM·BF4 A nd none 15 99 13 >200 15 PMIM·BF4 A nd none 63 99 39 >200 16 MOEMIM·BF4 A nd none nr nr 0 nr 17 BPyr-BF4 A nd NaHC03 nr nr 0 nr
18 BPyr-BF4 A nd Na2C03 47 99 32 >200 19 EMIM·BF4 A nd Na2C03 87 99 46 >200 20 MOEMIM·BF4 A nd Na2C03 71 99 42 >200 21 EMIM·BF4 B 0.71 none 85 99 46 >200 22 MOEMIM·BF4 B 0.70 none 73 99 42 >200 23 PMIM·BF4 B 0.69 none 62 99 38 >200 24 BMIM·BF4 B 0.68 none 55 99 36 >200 25 sBMIM·BF4 B 0.68 none 54 99 35 >200 26 BMIM·PF6 B 0.68 none 41 99 29 >200 27 BMPyr·BF4 B 0.63 none 34 99 25 >200 28 PMPyr-BF4 B 0.67 none 47 99 33 >200 29 BPyr-BF4 B 0.64 none 62 99 38 >200 30 PPyr-BF4 B 0.66 none 59 99 37 >200 31 EMIM·BF4 B nd silica gel 81 99 45 >200 32 MOEMIM·BF4 B nd silica gel 77 99 43 >200 33 EMIM·BF4 B nd EMIM·Br 75 99 43 >200 34 MOEMIM·BF4 B nd MOEMIM·Cl 63 99 39 >200 35 EMIM·BF4 B nd AgBF4 nr nr 0 nr 36 PMIM·BF4 B nd AgBF4 nr nr 0 nr 37 BMIM·BF4 B nd AgBF4 nr nr 0 nr 38 sBMIM·BF4 B nd AgBF4 37 99 27 >200 39 MOEMIM·BF4 B nd AgBF4 nr nr 0 nr 40 EMIM·BF4 B nd Na2C03 77 99 44 >200 41 MOEMIM·BF4 B nd Na2C03 71 99 42 >200 42 EMIM·BF4 B nd acetic acicY 28 99 22 >200 43 MOEMIM·BF4 B nd acetic acicY 25 99 20 >200
a Conditions: 1 mmol of vinyl acetate, 1 mmol of sec-phenethyl a1cohol, 1 mL of solvent, 20 mg of PCL or 5 mg of CAL-B, 10 mg of additive, 24 h, room temperature, stirred with magnetic stirring bar. PCL was the lipase unless otherwise noted. Sorne reactions were run at twice this scale. nr = no reaction, nd = not determined, E = enantiomeric ratio as defined by Chen, C. S.; Fujimoto, Y.; Girdaukas, G.: Sih, C. J. J. Am. Chem. Soc. 1982, 104,7294-7299. These reactions are highly enantioselective, so the maximum conversion is 50%. The values of conversion, c, were ca1culated using the measured enantiomeric excess of the starting material (ees) and product (eep). The values in the box are our recommended reaction conditions for lipase-catalyzed reactions in ionic solvents. These data in the box as weIl as the data for normal organic sol vents are also plotted in Figure 1.b Purification method A: add silver tetrafluoroborate, remove silver halide precipitate by filtration, followed by chromatography on silica gel. Purification method B: filtration
48

through silica gel plug, wash with saturated aqueous sodium carbonate.c Solvent polarity according to Reichardt's normalized polarity scale, ET
N• On this scale, tetramethylsilane
has a polarity of 0 and water has a polarity of 1. The values for the organic sol vents were taken from a recent review (Reichardt, C. Chem. Rev. 1994,94,2319-2358.). The values for the ionic liquids were calculated using the measured absorbance maximum of the long-wavelength transition of 2,6-diphenyl-4-(2,4,6-triphenylpyridinio)phenolate as described in the Experimental Section.d 108 h reaction time, but using only 5 mg of PCL. e CAL-B used in place of PCL.! 20 )lL added.
Polarity of Ionie Liquids As Compared to Organie Solvents. The color of Reichardt's
dye (a substituted N-(4-oxidophenyl)pyridinium, Chart 1) varies strongly with the
polarity of the solvent-from Âmax = 453 nm in water to Âmax = 810 nm in diphenyl ether.9
The ground state of this dye is highly polarized, while the first excited state is less
polarized due to charge transfer. Polar solvents, especially those that form a hydrogen
bond to the phenoxide oxygen, stabilize the ground state, thereby increasing the
difference between the ground and excited states and increasing the energy of the
absorption. In nonpolar solvents, the energy difference between ground and excited state
is much smaller and the absorption is at lower energy.
~~ Ph Ph
Chart 1. Reichardt's Dye
We measured the polarity of the different ionic solvents by measuring the color of
Reic:hardt's dye dissolved in the different ionic solvents. We used Reichardt's normalized
scale where the tetramethylsilane has a value of zero and water has a value of one. The
polarity values for the 10 ionic liquids in Scheme 1 ranged from 0.63 to 0.71 with the
most polar being EMIM'BF4 and the least polar being BMPYf'BF4, Table 1. Muldoon
et al. IO recently measured the polarity of seve raI ionic liquids using this dye. Carmichael
9 Reichardt, C. Chem. Soc. Rev. 1992, 147-153. Reichardt, C. Chem. Rev. 1994,94,2319-
2358.
10 Muldoon, M. J.; Gordon, C. M.; Dunkin, 1. R. J. Chem. Soc., Perkin Trans. 2 2001,
433-434.
49

and Seddon recently measured the polarity of several ionic liquids using another
solvatochromic dye, Nile Red, Il and Aki et al. used fluorescent probes to measure the
polarity of ionic solvents. 12 Although only a few ionic liquids are the same as the ones we
measured, our values are similar and the relative ranking of the polarities is the same.
Ionic liquids permit researchers to run lipase-catalyzed reactions in a solvent
polarity range that was previously inaccessible. Organic sol vents with polarities similar to
the ionic liquids inc1ude the following: methanol, 2-chloroethanol, N-methylformamide,
diethylene glycol, or 1,2-propanediol. Most of these are hydroxylic solvents, which are
not suitable for acylation reactions since the solvent would compete with the substrate
a1cohol for the acyl donor. The one potentially suitable organic solvent, N
methylformamide, showed no reaction presumably because it denatured the lipase,
Table 1.
With normal organic solvents, the trend is toward higher reaction rates in less
polar solvents. However, for the PCL-catalyzed acetylation in ionic liquids the trend was
in the opposite direction-toward higher reaction rates in the more polar ionic liquids,
Figure 1. However, for the acetylation of glucose below, the reaction rate showed no
correlation with the polarity of the ionic solvent. In this case, the solubility of the
substrate glucose and acetylated products likely influences reaction rates.
11 Carmichael, A. J.; Seddon, K. R. J. Phys. Org. Chem. 2000, 13, 591-595.
12 Aki, S. N. V. K.; Brennecke, J. F.; Samanta, A. Chem. Commun. 2001,413-414.
50

50
45
40 .cl ..,.
35 ("l
'"' Q,j
;t:
= 30 ---~ 25 '-'
== Q ... 20 '" '"' Q,j
~
== 15 Q U
10
5
0 0 0.1
organic solvents
Tlrnc(h)
• acetone
• ACN
DMF· DMSO
0.2 0.3 0.4
ionic liquids
0.5 0.6
polarity (Reichardt's scale)
NMF
0.7 t 0.8
methanol, 2-chloroethanol
Figure 1. Rates of lipase-catalyzed reactions as a function of solvent polarity for normal
organic solvents and for ionic liquids. The model reaction is a PCL-catalyzed acetylation
of racemic I-phenylethanol with vinyl acetate, which is highly enantioselective, so the
maximum conversion is 50%. Inset: A comparison of the time course of the acetylation
reaction in several solvents. The reaction rate in both toluene and the polar ionic liquid,
EMIM'BF4, are similar, but the initial reaction rate in a less polar ionic liquid,
BMIM'PF6, was approximately three times slower. (Reaction conditions: 4 mL of
solvent, 30 mg of peL, 4 mmol of vinyl acetate, 4 mmol of I-phenylethanol, 96 h, room
temperature.) Main graph: Correlation between degree of conversion in a lipase-catalyzed
acylation and solvent polarity (Reichardt's normalized polarity scale) for organic solvents
and ionic liquids. For normal organic solvents, the acetylation reaction proceeds weIl in
nonpolar solvent, but not in polar solvents. The reaction is nearly completed in toluene
51

and partially eompleted in tetrahydrofuran (THF) , aeetone, or acetonitrile (ACN), but
proceeds very slowly or not at an in the more polar N,N-dimethylformamide (DMF) ,
dimethyl sulfoxide (DMSQ), or N-methylformamide. Although the ionie liquids are
highly polar sol vents (similar to N-methylformamide), the acetylation reaction proeeeds
welll in an ionic liquids tested. Alcoholic solvents sueh as methanol or 2-ehloroethanol
have polarities similar to the ionic liquids, but are not suitable for this acylation reaction
because they would react with the aeyl donor. The trend for ionic liquids is for higher
degrees of conversion as the polarity of the ionic liquid increases, while the trend for
organic sol vents is the opposite-Iower degrees of conversion as the polarity increases.
The trend lines are not a fit to any theory, but are included only to guide the eye. Using
ionic liquids as sol vents permits lipase-catalyzed reaetions to be run in a previously
inaccessible polarity region. (Reactions are similar to those in Table 1: 1 mL of solvent,
20 mg of PCL, 1 mmol of vin yi acetate, 1 mmol of I-phenylethanol, 24 h, room
temperature) .
Regioselective 6-0-Acetylation of ~-D-Glucose in Ionie Liquids. Since ionie liquids
are polar sol vents that do not denature lipases, they may be ideal for lipase-catalyzed
transformations of polar substrates. To test this idea, we examined the lipase-catalyzed
acylation of glucose, eq 2, Table 2.
OH ~ ~ ~O OAc
H~o40H ionic liquid
CALoS
OAc
H~~q .O~OH
6-Q-acetyl D-glucose (mixture of anomers)
+
OAc
HO~q ACO~OH
3,6-Q-diacetyl D-glucose (mixture of anomers)
52
(2)
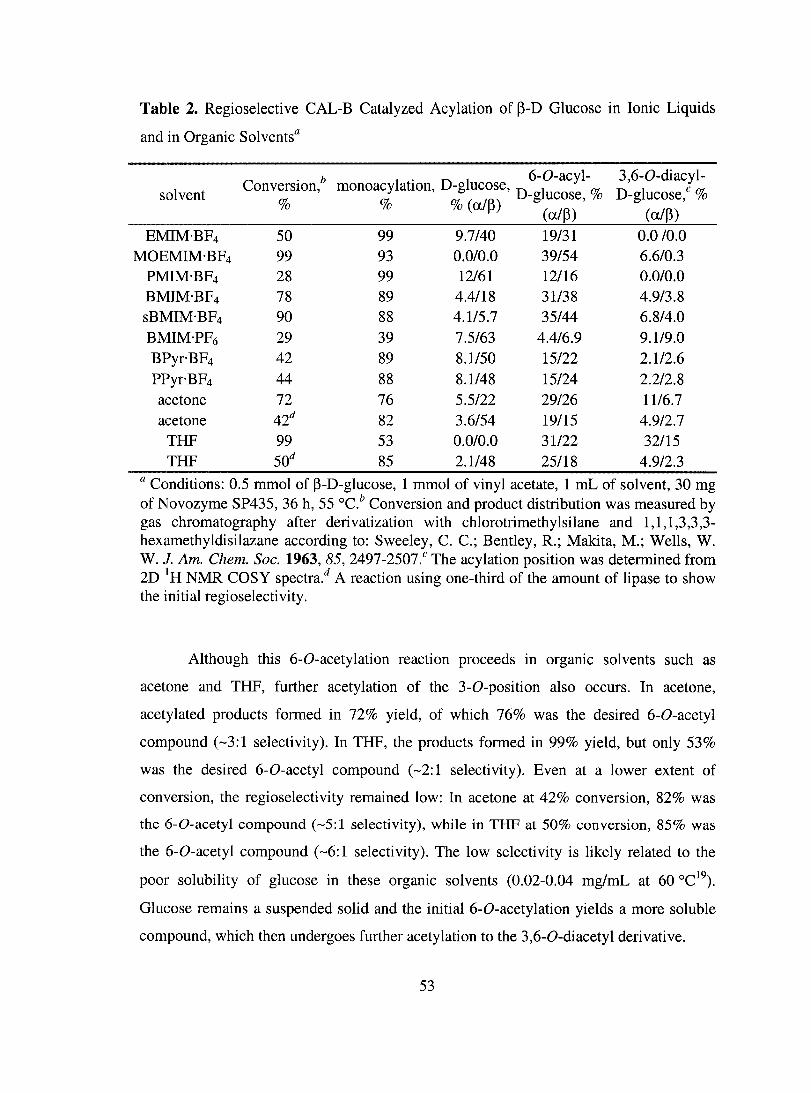
Table 2. Regioselective CAL-B Catalyzed Acylation of p-D Glucose in Ionie Liquids
and in Organic Solventsa
Conversion,b . 6-0-acyl- 3,6-0-diacyl-
solvent monoacylatlOn, D-glucose, D 1 ~ D-glucose, C %
% ~ ~ (/P) -g ucose, 0 o 0 a (a/p) (a/p)
EMIM'BF4 50 99 9.7/40 19/31 0.0/0.0 MOEMIM'BF4 99 93 0.0/0.0 39/54 6.6/0.3
PMIM'BF4 28 99 12/61 12/16 0.0/0.0 BMIM·BF4 78 89 4.4/18 31/38 4.9/3.8 sBMIM'BF4 90 88 4.1/5.7 35/44 6.8/4.0 BMIM·PF6 29 39 7.5/63 4.4/6.9 9.1/9.0 BPyr'BF4 42 89 8.1/50 15/22 2.1/2.6 PPyr'BF4 44 88 8.1/48 15/24 2.2/2.8 acetone 72 76 5.5/22 29/26 11/6.7 acetone 42d 82 3.6/54 19/15 4.9/2.7
THF 99 53 0.0/0.0 31/22 32/15 THF 50d 85 2.1/48 25/18 4.9/2.3
a Conditions: 0.5 mmol of P-D-glucose, 1 mmol of vinyl acetate, 1 mL of solvent, 30 mg of Novozyme SP435, 36 h, 55 °C.b Conversion and product distribution was measured by gas chromatography after derivatization with chlorotrimethylsilane and 1,1,1,3,3,3-hexamethyldisilazane according t~: Sweeley, C. c.; Bendey, R.; Makita, M.; Wells, W. W. J. Am. Chem. Soc. 1963,85, 2497-2507.c The acylation position was determined from 2D IH NMR COSY spectra.d A reaction using one-third of the amount of lipase to show the initial regioselectivity.
Although this 6-0-acetylation reaction proceeds in organic solvents such as
acetone and THF, further acetylation of the 3-0-position also occurs. In acetone,
acetylated products formed in 72% yield, of which 76% was the desired 6-0-acetyl
compound (-3:1 selectivity). In THF, the products formed in 99% yield, but only 53%
was the desired 6-0-acetyl compound (-2:1 selectivity). Even at a lower extent of
conversion, the regioselectivity remained low: In acetone at 42% conversion, 82% was
the 6-0-acetyl compound (-5:1 selectivity), while in THF at 50% conversion, 85% was
the 6-0-acetyl compound (-6: 1 selectivity). The low selectivity is likely related to the
poor solubility of glucose in these organic sol vents (0.02-0.04 mg/mL at 60 °C19).
Glucose remains a suspended solid and the initial 6-0-acetylation yields a more soluble
compound, which then undergoes further acetylation to the 3,6-0-diacetyl derivative.
53

No reaction occurred in unpurified ionic liquids. However, acylation of glucose
proceeded smoothly in aIl ionic liquids after purification by method B. In addition, the
selectivity for monoacetylation is much higher in ionic liquids than in organic solvents.
The 6-0-acetylation proceeds with 88-99% selectivity in the seven ionic liquids
containing a tetrafluoroborate anion. The degree of conversion varied from 42 to 99%.
The best ionic liquid for this reaction was MOEMIM·BF4, where the acetylated products
formed in 99% yield, of which 93% was the desired 6-0-acetyl compound. The one ionic
liquid with a hexafluorophosphate anion, BMIM'PF6, showed both slow reaction (29%
conversion) and low selectivity (39% monoacetyl).
Approximately 100 times more glucose dissolves in the best ionic liquid,
MOEMIM'BF4 -5 mg/mL at 55 oC, than in acetone or THF. On the other hand, glucose
is not very soluble in the worst ionic liquid, BMIM'PF6, <1 mg/mL at 55 oC. For the
acetylation of glucose in ionic liquids, the ability of the ionic liquid to dissolve glucose is
an important factor.
Although we started with the ~-anomer of glucose, we observed anomerization of
both the starting materials and the products in both ionic liquids and in organic solvents.
The higher temperature of the reaction (55 oC, 36 h) and traces of acetic acid formed by
lipase-catalyzed hydrolysis of vinyl acetate most likely caused this anomerization.
Hydrolysis of vinyl acetate is a significant side reaction even in "dry" organic solvents. 13
Initial experiments show that CAL-B also catalyzes the regioselective acylation of
maltose monohydrate, a disaccharide that is even more polar than glucose. Using the
reaction conditions as in Table 2, but only half the amount of maltose (0.25 mmol instead
of 0 .. 5 mmol) and MOEMIM'BF4 as the solvent, yielded 50% of acetylated products.
13 b We er, H. K, Weber, H., Kazlauskas, R. J. Tetrahedron: Asymmetry 1999, 10, 2635-
2638. Chaudhary, A. K.; Beckman, E. J.; Russell, A. J. AIChE J. 1998,44,753-764.
54

Discussion
Besides potential environmental benefits, ionic liquids permit enzyme-catalyzed
reactions in a solvent polarity range that was previously inaccessible. Although there was
no reaction in a polar organic solvent like N-methylformamide, ionic liquids with similar
polarities on Reichardt's polarity scale gave excellent reactions. These more polar
solvents offer major advantages with polar substrates such as glucose and maltose.
Reactions with these polar substrates either do not proceed at aIl in organic solvent or
proceed with low regioselectivity due to further acylation of the more soluble products.
The higher solubility of glucose and maltose in ionic liquids facilitates their reaction.
Previous enzyme catalyzed acylations of maltose required refIuxing tert-butyl a1cohol
(82 OC) as the solvent. 14
Although the catalyst usually controls the regioselectivity of a reaction, with
poorly soluble substrates and products such as glucose and its derivatives, the relative
solubility also contributes to the regioselectivity. For example, most researchers chose to
acylate not glucose, but the more organic-solvent soluble alkyl glucosides (e.g., 1-0-ethyl
glucoside).15 These acylations show high regioselectivity for the primary a1cohol because
the CAL-B favors the primary a1cohol position, and both the starting alkyl glucosides and
14 Woudenberg-van Oosterrom, M.; van Rantwijk, F.; Sheldon, R. A. Biotechnoi. Bioeng.
1996,49, 328-333. H. Ianuginosa lipase in tert-amyl a1cohol/dimethyl sulfoxide: Ferrer,
M.; Cruces, M. A.; Plou, F. J.; Bernabé, M.; Ballesteros, A. Tetrahedron 2000, 56,
4053-4061. Subtilisin in dimethylformamide at 45 oC: Riva, S.; Chopineau, J.;
Kieboom, A. P. G.; Klibanov, A. M. J. Am. Chem. Soc. 1988,110,584-589.
15 Adelhorst, K.; Bjorkling, F.; Godtfredsen, S.; Kirk, O. Synthesis 1990, 2, 112-115.
Theil, F. Schick, H. Synthesis 1991, 533-535. Pelenc, V. P.; Paul, F. M. B.; Monsan, P.
F. World Patent WO 93/04185, 1993. Degoede, A. T. J. W.; Vanoosterom, M.;
Vandeurzen, M. P. J.; Sheldon, R. A.; Vanbekkum, H.; Vanrantwijk, F. Biocatalysis
1994, 9, 145-155. Danieli B.; Luisetti M.; Sampognaro G.; Carrea G.; Riva S. 1. Mol.
Catai. B: Enzym. 1997,3, 193-201.
55

the products 6-0-acyl-I-O-alkyl glucosides have similar solubilities in the reaction
media. Similarly, acylation of other organic solvent soluble derivatives of glucose such as
borate complexes16 or isopropylidene ether derivatives17 also shows high regioselectivity
for the primary alcohol. In these cases, the relative solubilities of the starting glucose
derivative and the product are similar and do not significantly affect the regioselectivity.
On the other hand, acylation of unmodified glucose varies with length of the acyl
group because the solubility of the product 6-0-acyl derivative varies with the length of
the acyl group. Acylation with shorter chain acyl groups (e.g., C2 to C6) gives a mixture
of regioisomers because the initially formed 6-0-acyl derivative is soluble and undergoes
further acylation. Pig pancreatic lipase catalyzed acylation of glucose in pyridine with
activated acetyl esters gave a 5.6: 1 mixture of regioisomers. 18 A CAL-B catalyzed
acylation of glucose with shorter chain acids such as caproic acid gave a "small" amount
ofthe diester. 19
However, acylation with longer chain acyl groups (~ CI2) showed high
regioselectivity for monacylation at the 6-position because the product 6-0-acyl glucose
is also poody soluble. Pig pancreatic lipase catalyzed acylation in pyridine with activated
lauryl esters (CI2) gave a 20: 1 mixture of regioisomers instead of the 5.6: 1 mixture with
the acetyl ester. 18 A CAL-B-catalyzed acylation of glucose with longer chain acids such
as palmitic (C16) gave only monacylation at the 6-position.19 Similarly, Tsitsimpokou et
16 Ikeda, 1.; Klibanov, A. M. Biotechnol. Bioeng. 1993,42,788-791; Oguntimein, G. B.;
Erdmann, H.; Schmid, R. D. Biotechnol. Lett. 1993,15,175-80.
17 Fregapane, G.; Sarney, D. B.; Vulfson, E. N. Biocatalysis 1994, 11, 9-18. Sarney, D. B.
Kapeller, H. Fregapane, G. Vulfson, E. N. J. Am. Oil Chem. Soc. 1994,71,711-714.
18 Therisod, M.; Klibanov, A M. J. Am. Chem. Soc. 1986,108,5638-5640.
19 Cao, L.; Fischer, A; Bornscheuer, U. T.; Schmid, R. D. Biocatal. Biotransform. 1997,
14, 269-283. Arcos, J. A; Bernabé, M.; Otero, C. Biotechnol. Bioeng. 1998, 57, 505-
509.
56

al. acylated glucose absorbed on silica gel with lauric acid using CAL-B and observed
high regioselectivity for the 6-position.20
To increase the regioselectivity for acylation of unmodified glucose with short
acyl chains, one needs a solvent where either the glucose is more soluble or the 6-0-acyl
derivative is less soluble. Degn et al. found that tert-butyl alcohol dissolves glucose to
2.4 mg/mL at 45°C and CAL-B was highly regioselective for the primary alcohol
position in this solvent.21 However, these reaction conditions required dilute solutions -
acylation of 100 mg of glucose would require 40 mL of solvent. Ionic liquids also
increase the solubility of glucose in the reaction medium and thereby increase in
regioselectivity. Our reaction conditions were about 40 times more concentrated than
those of Degn et al. Acylation of 100 mg of glucose would require only 1 mL of ionic
liquid. There is no need to propose a special interaction of the ionic liquid and lipase to
explain the increased regioselectivity.
When the acyl chains are long, 6-0-acyl glucose and similar derivatives are
surfactants,22 but the surface-active properties make it an inconvenient synthetic
intennediate. Derivatives with a short acyl chain are more useful as synthetic
intennediates. Although researchers have also developed chemical methods for selective
6-0-acylation of unprotected glucose, these methods are less selective and lower yi el ding
than enzymatic methods.23 The ionic solvent method here may be the best way to protect
the 6-position of glucose and quite likely other sugars also.
As with organic solvents, one ionic liquid is not best for aIl reactions. By varying
the structure of the ionic liquid, one can optimize both the rates and the selectivities for
20 Tsitsimpikou, C.; Daflos, H.; Kolisis, F. N. J. Mol. Catal. B: Enzym. 1997,3, 189-192.
21 Degn, P.; Pedersen, L. H.; Duus, J. 0.; Zimmermann, W. Biotechnol. Lett. 1999,21,
275-280.
22 Andresen, O.; Kirk, O. Prog. Biotechnol. 1995,10,343-349.
23 Reinefeld, E.; Kom, H. F. Die Stiirke 1968, 20, 181-189. Yoshimoto, K; Tahara, K;
Suzuki, S.; Sasaki, K; Nishikawa, Y.; Tsuda, Y. Chem. Pharm. Bull. 1979, 27, 2661-
2674. Plusquellec, D.; Baczko, K Tetrahedron Lett. 1987,28,3809-3812.
57

each reaction. For I-phenylethanol, acetylation was fastest in EMIM·BF4 and slowest, by
about a factor of 2, in BMIM·PF6. The enantioselectivity remained high in aIl ionic
liquids. For glucose, acetylation was fastest in MOEMIM'BF4 and slowest, by about a
factor of 3, in either PMIM'BF4 or BMIM·PF6. The regioselectivity was high in aIl ionic
liquids except for one, BMIM·PF6•
The key structural features that control enzyme-catalyzed reactions ln IOnIC
liquids remain unc1ear at this time. For the enantioselective acetylation of 1-
phenylethanol, we found a correlation between the polarity of the ionic solvent and the
reaction rate. A possible explanation relates to the relative solvation of the substrate in
the solvent vs enzyme. A more polar ionic liquid does not solvate a nonpolar substrate
welll, so the substrate binds to the enzyme and reacts. However, for the acetylation of
glucose, we did not see a correlation between reaction rate and solvent polarity. For this
poorly soluble substrate, reaction was fastest in the solvent most able to dissolve the
substrate glucose. The nature of the anion had no effect on the enantioselectivity of the
acetylation of I-phenylethanol, but the hexafluorophosphate anion caused lower
regioselectivity in the acylation of glucose.
The wash with aqueous sodium carbonate yields ionic solvents that are suitable
for :reactions in ionic liquids. For example, in the acetylation of I-phenylethanol SchOfer
et al. reported no reaction for CAL-B-catalyzed reaction in BMIM'BF4 or BMIM'PF6, no
reac:tion for the PCL-catalyzed reaction in BMIM'PF6 and slow reaction in BMIM·BF4.
Upon preparation of ionic liquids using the purification method B involving the wash
with aqueous sodium carbonate, an these reactions proceeded at rates comparable to
those in nonpolar organic solvents. We do not know why the sodium carbonate wash
improves the reaction rates, but speculate that it may add smaIl amounts of a buffer or
water to the ionic liquid.
58

Experimental Section
General Methods. IH NMR spectra were recorded in acetone-d6 at 400 MHz. Lipase
from Pseudomonas cepacia (commercial name PS30) was purchased from Amano USA
(Lombard, IL). An immobilized form of lipase B from Candida antarctica (SP435) was
used for acylation of glucose and maltose, while a soluble form was used to test the
acetylation of I-phenylethanol. Other chemicals were purchased from Sigma-Aldrich.
Synthesis of Ionie Liquids. BMIM'PF6 was prepared according to a literature procedure
using hexafluorophosphoric acid. The tetrafluoroborate salts of the ionic liquids were
made according to literature procedures.5 A mixture of alkyl halide (0.40 mol) and 1-
methylimidazole (0.40 mol, 31.9 mL) was stirred at 70-80 oC (50 oC for EMIM'BF4) for
1 or 2 days under nitrogen. The mixture was cooled to room temperature, and ethyl
acetate (70 mL) was added causing precipitation of l-alkyl-3-methyl imidazolium halide
as a white solid. This solid was recovered by filtration and washed with ethyl acetate
followed by ethyl ether: the crude yield was 90-100%.
To prepare the tetrafluoroborate salts, the l-alkyl-3-methylimidazolium halide salt
(0.40 mol) was added to a suspension of NaBF4 (1.2 equiv, 52.7 g, 0.48 mol) in acetone
(150 mL). After the mixture was stirred for 48 h at room temperature, the sodium halide
precipitate was removed by filtration and the filtrate concentrated to an oil (-100 mL) by
rotary evaporation. This oil still contained some l-alkyl-3-methyl imidazolium halide
because it gave a precipitate when mixed with aqueous silver nitrate.
Purification of Tetrafluoroborate Salts: Method A.5b•c The oil (-100 mL) was
dissolved in methyl alcohol (100 mL), and an aqueous solution of AgBF4 (generated from
Ag20 and HBF4) was added dropwise until no more precipitate was formed. The mixture
was filtered through Celite (no. 545), concentrated by rotary evaporation, dissolved in
dichloromethane (100 mL), and filtered again to remove insoluble material. The product
was purified by column chromatography in three portions on silica gel (-400 g) eluted
59

with methylene chloride/methanol (9:1). The solvent removed under vacuum yielded pale
yellow oil, 60-80% yield.
Puriification of Tetrafluoroborate Salts: Method B. The crude ionic liquid was diluted
with methylene chloride (200 mL) and filtered through silica gel (-100 g). This step
removed the l-alkyl-3-methylimidazolium halide since the filtrate no longer gave a
precipitate mixed with aqueous silver nitrate. The solution was washed twice with
saturated sodium carbonate aqueous solution (40 mL) and dried over anhydrous
magnesium sulfate. Removal of solvent under vacuum yielded a pale yellow oil, 50-70%
yield. Washing a solution of EMIM'BF4 in methylene chloride with aqueous sodium
carbonate yielded three layers: water on the top, EMIM'BF4 in the middle, and methylene
chloride at the bottom. The two bottom layers were separated, evaporated to remove
water dissolved in the EMIM'BF4 layer, diluted with methylene chloride (200 mL), dried
over anhydrous magne sium sulfate, and concentrated to an oil.
EMIM·BF4. IH NMR: 8 8.98 (IH, s); 7.75 (IH, dd); 7.68 (IH, dd); 4.37 (2H, q); 4.03
(3H, s); 1.54 (3H, t).
MOEMIM·BF4. IH NMR: 88.95 (IH, s); 7.71 (IH, dd); 7.68 (IH, dd); 4.51 (2H, t); 4.05
(3H, s); 3.80 (2H, t); 3.34 (3H, s).
PM][M·BF4. IH NMR: 8 8.99 (IH, s); 7.75 (IH, dd); 7.71 (IH, d); 4.31 (2H, t); 4.04 (3H,
s); 1.95 (2H, m); 0.95 (3H, t).
BMllM·BF4. IH NMR: 8 8.99 (IH, s); 7.75 (IH, d); 7.70 (IH, d); 4.35 (2H, t); 4.04 (3H,
s); 1.91 (2H, m); 1.37 (2H, m); 0.94 (3H, t).
sBl\HM·BF4. IH NMR: 8 9.05 (IH, s); 7.82 (IH, dd); 7.73 (IH, dd); 4.57 (IH, m); 4.04
(3H, s); 1.94 (2H, m); 1.60 (3H, d); 0.87 (3H, t).
BMIM·PF6• IH NMR: 8 8.99 (IH, s); 7.76 (IH, dd); 7.71 (IH, dd); 4.36 (2H, t); 4.06
(3H, s); 1.93 (2H, m); 1.38 (2H, m); 0.94 (3H, t).
PMPyr·BF4• IH NMR: 8 8.95 (2H, d); 8.06 (2H, d); 4.70 (2H, t); 2.73 (3H, s); 2.09 (2H,
q); 0.99 (3H, t).
BMPyr·BF4. IH NMR: 8 8.96 (2H, d); 8.05 (2H, d); 4.73 (2H, t); 2.72 (3H, s); 2.06 (2H,
m); 1.42 (2H, m); 0.96 (3H, t).
60

PPyr·BF4. IR NMR: Ô 9.13 (2R, d); 8.72 (IR, t); 8.26 (2R, t); 4.78 (2R, t); 2.13 (2R, m);
1.00 (3R, t).
BPyr·BF4. IR NMR: Ô 9.16 (2R, d); 8.73 (IR, t); 8.27 (2R, t); 4.83 (2R, t); 2.10 (2R, m);
1.45 (2R, m); 0.97 (3R, t).
Transesterification of 1-Phenylethanol. Vinyl acetate (92 ilL, 1.0 mmol) and 1-
phenylethanol (120 ilL, 1.0 mmol) were added to a suspension of PCL (20 mg) in solvent
(1.0 mL of either organic solvents or ionic liquids) and stirred at 25 oc. The reactions
were monitored by TLC (ethyl acetate/hexane, 1:3). After 24 h, the reaction mixture was
extracted with hexane (3 mL), and the hexane extract was analyzed by GC with a chiral
capillary column (Chromopak Chiralsil-Dex CB column (25 m x 0.25 mm, Raritan, NJ):
initial column temperature 125 oc for 2 min, then ramp to 150 oC over 10 min: 1-
phenylethanol (a = 1.06, kR = 3.32, ks = 3.35); 1-methylbenzyl acetate (a = 1.12, kR = 2.53, ks = 2.26) The conversion, c, was ca1culated from the enantiomeric excess of the
product, eep, and of the starting material, ees, using the equation below. 24
c=
Transesterification of p-D-Glucose. Vinyl acetate (92 ilL, 1.0 mmol), P -D-glucose (90
mg, 0.5 mmol), and Novozym SP435 (30 mg) were mixed with solvent (1.0 mL of either
organic sol vents or ionic liquids) and stirred at 55 oc. After 36 h, pyridine (2 mL),
1,1,1,3,3,3-hexamethyldisilazane (1 mL), and chlorotrimethylsilane (1 mL) were added to
the reaction mixture, and the mixture was extracted with hexane (5 mL). The hexane
extract was analyzed by gas chromatography on the column noted above. The derivatives
of glucose and acetyl glucose were separated using the following temperature program:
initially 2 min at 180 oC, then gradient to 190 oC at 1°C/min, then held at 190 oC for 28
24 Chen, C. S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1982, 104,
7294-7299.
61

min. In a separate experiment, we confirmed that this derivatization method does not
cause anomerization of glucose.
Polarity Determination Using Reichardt's Polarity Scale. Reichardt's dye (2,6-
diphenyl-4-(2,4,6-triphenylpyridinio )phenolate, 0.4 mg) was dissolved in ionic liquid (0.5
mL), and an aliquot was transferred to a 96-well microplate. The wavelength of the
absorption maximum of the long-wavelength transition was measured at 25 oc using a
microplate reader (Spectra Max 340, Molecular Deviees Co., Sunnyvale, CA).
Normalized polarity values (ENT) , which range from 0.0 for tetramethylsilane to 1.0 for
water, were ca1culated using the equation
=
ET(solvent) - ET(TMS)
ET(water) - ET(TMS)
ET(solvent) - 30.7
32.4
where ET(solvent) is the energy, in kilocalories per mole, of the maximum of the long
wavelength transition and is given by
Er(solvent)(kcal/mol) =
62
28591
Âmax (nm)

Chapter 3
Enzyme-catalyzed reactions of polar substrates under nonaqueous conditions have
a long-standing problem due to their poor solubility in nonpolar organic solvents, which
do not denature enzymes. In Chapter 2, we presented that ionic liquids, in which a lipase
shows comparable activity to that in nonpolar solvents, worked in lipase-catalyzed
enantio- and regioselective acylations. These high polar ionic liquids can dissolve polar
substrates, thereby facilitating lipase-catalyzed reactions of polar substrates.
Fatty acid esters of L-ascorbic acid are potentially important because of their high
reducing and antitumor-promoting activities. However, it is difficult to make them by
chemical methods because the reaction must be regioselective and also avoid the fast air
oxidlation of L-ascorbic acid and its ester. It is also difficuIt to make them by enzymatic
method because L-ascorbic acid does not dissolve in nonpolar organic sol vents where
enzymes are normally stable. In this chapter, we used ionic liquids to dissolve L-ascorbic
acid for the lipase-catalyzed direct esterification of L-ascorbic acid. This lipase-catalyzed
direct esterification in ionic liquids yielded fatty acid esters of L-ascorbic acid with high
conversion.
Contributions
This work was done under supervision of Dr. Karl HuIt and Dr. Romas J.
Kazlauskas. One collaborator, Fredrik Viklund, optimized the separation condition of
reaction mixtures for HPLC analysis and performed a large-scale synthetic reaction of L
ascorbyl oleate in tert-amyl alcohol. 1 synthesized ionic liquids and performed most
lipase-catalyzed reactions in ionic liquids.
63

Chapter 3. Lipase-Catalyzed Direct Condensation of
L-Ascorbic Acid and Fatty Acids in Ionie Liquids with
Assistance of Hydrophobie Additives
Abstract: Although L-ascorbic acid (Vitamin C) is a potentially useful compound for
food chemistry or cosmetics because of its high reducing activity, it dissolves only in
water and thus cannot be used in applications that require solubility in fats. Modification
to its fatty acid esters would enable its use as a fat-soluble antioxidant. Researchers have
tried to synthesize L-ascorbyl fatty acid esters un der a mild reaction conditions such as
lipase-catalyzed esterification, but the poor solubility of L-ascorbic acid in nonpolar
organic sol vents prevented an efficient synthesis. Highly polar ionic liquids permit a
lipase-catalyzed esterifcation with high conversion because they dissolve L-ascorbic acid
(e.g .. , -130 mg/mL in sBMIM·BF4 at 60 OC) but do not inactivate the lipase. The product,
L-ascorbyl fatty acid esters, inhibited the reaction by precipitating on the lipase partic1es.
To avoid a product inhibition, we used small amount of hydrophobic additives such as
hexane or polypropylene. With assistance of these additives, lipase B from Candida
antarctica (immobilized on macroporous acrylic resin) catalyzed a direct esterification of
ascorbic acid with 83% conversion and 65% yield to pro duce L-ascorbyI6-oleate.
HO HO R'v"0H ~,. 0 CALoS ~,. 0
Il + HO ~ R + H20 o - Ionie Liquid \( -
HO OH 0 HO OH
Introduction
Although L-ascorbic acid is widely used natural antioxidant with strong reducing
activity, its hydrophilicity limits its application in cosmetics or in the presence of fats and
oils. Using the fatty acid esters of L-ascorbic acid having amphiliphilic property can
64

avoid this limitation. In addition to their fat solubility, fatty acid esters of L-ascorbic acid
possess antioxidant, antimutagenic, and antitumor-promoting activities. 1 Synthesis of
fatty acid esters of L-ascorbic acid requires both mild reaction condition to pre vent fast
air oxidation of L-ascorbic acid and its esters, and regioselective control.
Several groups reported an enzyme-catalyzed synthesis of ascorbic acid ester
through direct condensation or transesterification using excess fatty acids or their
activated forms in polar organic solvents. Humeau et al. reported syntheses of ascorbyl
palmitate and eicosapentaenoate in 2-methyl-2-butanol using CAL-B (Candida
antarctica lipase B).2 Using 9-fold excess of palmitate methyl ester gave the maximum
yield (around 40% yield after 5 h). Similarly, Yan et al. used a 3-fold excess of activated
acyl donor such as fatty acid vinyl esters in acetone or t-butyl alcohol and achieved 63 -
91 % yield after 48 h. 3 Watanabe et al. synthesized L-ascorbyl 6-0-eicosapentaenoate
through condensation of L-ascorbic acid with a 5-fold excess of eicosapentaenoic acid in
acetone and achieved 47% yield after 32 h. 4 Both fatty acids and fatty acid esters bind
similarly to the lipase, 5 so it would be best to use the less expensive fatty acids. 6
Although lipases can catalyze the esterification of ascorbic acid through trans
esterification or direct condensation in moderately polar organic solvents such as acetone
1 Rao, C. V.; Rivenson, A; Kelloff, G. J.; Reddy, B. S. Anticancer Res. 1995, 15, 1199-
1204.
2 (a) Humeau, C.; Girardin, M.; Rovel, B.; Mic1o, A J. Biotechnol. 1998, 63, 1-8. (b)
Humeau, c.; Girardin, M.; Rovel, B.; Mic1o, A J. Mol. Catal. B: Enzym. 1998,5, 19-
23.
3 Yan, y.; Bomscheuer. U. T.; Schmid, R. D. Biotechnol. Lett. 1999,21,1051-1054.
4 Watanabe, Y.; Minemoto, Y.; Adachi, S.; Nakanishi, K.; Shimada, Y.; Matsuno, R.
Biotechnol. Lett. 2000, 22, 637-640.
5 Luhong, T.; Hao, Z.; Shehate, M. M.; Yunfei, S. Biotechnol. Appl. Biochem. 2000,32,
35--39.
6 For example, methyl oleate costs twice as much as oleic acid. TCI Catalog 2002-2003;
TCI America: Portland, OR, 2002, P 1292.
65

or tertiary alcohols, the reactions are slow. Lipases are generally more active in nonpolar
organic sol vents than these organic solvents. 7 However, enzyme-catalyzed reactions in
ionic liquids are as fast as those in nonpolar organic sol vents such as toluene. 8 On the
other hand, since highly hydrophilic L-ascorbic acid does not dissolve in nonpolar
organic sol vents such as hexane and toluene, these sol vents are unsuitable for reactions
involving ascorbic acid. Ionic liquids are polar enough to dissolve ascorbic acid and
permit lipase-catalyzed direct esterification with high conversion.
In this paper, we de scribe a CAL-B-catalyzed direct esterification of L-ascorbic
acid using fatty acids in ionic liquids. We focused on the synthesis of ascorbyl oleate,
which is an interesting compound because of its antibiotic and anticancer activity.
Product inhibition due to precipitation of the products in ionic liquids was avoided by
adding 10-20 vol% hexane, which releases the products from the surface of the
immobilized lipase. We did not observe any other regioisomers of the product.
Results
Synthesis of Ionie Liquids. This paper focuses on two ionic liquids (Chart 1) which we
prepared by modified literature procedures. In previous our study, we washed the ionic
liquid solution in methylene chloride with saturated aqueous sodium carbonate solution
7 For example, the initial rate of an esterification by porcine pancreatic lipase in acetone
is ten times lower than that in hexane or toluene. Zaks, A.; Klibanov, A. M. Proe. Natl.
Aead. Sei. U.S.A. 1985,82,3192-3196.
8 a) Erbeldinger, M.; Mesiano, A. J.; Russell, A. J. Bioteehnol. Prog. 2000, 16, 1131-
1133. b) Lau, R M.; Rantwijk, F. van; Seddon, K. R; Sheldon, R A. Org. Lett. 2000,
2,4189-4191. c) Itoh, T.; Akasaki, E.; Kudo, K.; Shirakami, S. Chem. Lett. 2001, 262-
263. d) SchOfer, S. H.; Kaftzik, N.; Wasserscheid, P.; Kragl, U. Chem. Commun. 2001,
425-426. e) Kim, K.-W.; Song, B.; Choi, M.-Y.; Kim, M.-J. Org. Lett. 2001,3, 1507-
1509. f) Park, S; Kazlauskas, R J. J. Org. Chem. 2001,66,8395 -8401.
66

to remove trace amount of acid impurities which inactivate the enzyme. This purification
step may leave a small amount of sodium carbonate in the purified ionic liquids. To avoid
using sodium carbonate9, we instead filtered the solution through neutral alumina. These
newly purified ionie liquids give similar enzyme activity to the Ionie liquids purified by
washing with saturated aqueous sodium carbonate solution (see below).
F\® -~~
BF~ 1-seo-Butyl-3-methyl-imidazolium tetrafluoroborate
sBMIM'BF4
F\J?J-J -~A\
BF~ 3-Methyl-1-( 1-methyl-butyl)-3-imidazolium tetrafluoroborate
2PentMIM'8F4
Chart 1. Ionie Liquids Used in This Paper
Lipase-Catalyzed Regioselective Esterifications of Ascorbic Acid in Ionie Liquids.
As a model reaction for synthesis of ascorbyl oleate, we used palmitic acid as an
acylating reagent. Initially, we screened several ionic liquids towards acylation of
ascorbie acid with palmitic acid (Table 1, entry 1-4). We used molecular sieves to remove
water to shift the equilibrium toward product formation.
9 Ascorbic acid and its esters are well-known antioxidants. The oxidation of ascorbie acid
or its esters is catalyzed by heat, light, high pH, and transition metals: Jung. M. Y.;
Kim, S. K.; Kim, S. y. Food Chem. 1995, 53, 397-403. Bisby, R. H.; Morgan, C. G.;
Hamblett, 1.; Gorman, A. A. J. Phys. Chem. A 1999, 103, 7454-7459. Refer also
references therein. Sodium carbonate may catalyze their oxidation. Indeed, the reaction
mixture tumed brown when the reaction was not protected from air.
67

Table 1. Initial screening acylation reaction of ascorbic acid in ionic liquidsa
Entry SolventlO C . b onverSlOn Yieldb
1 MOEMIM·BF4c 24% 18% 2 PMIM·BF/ 20% 12% 3 BMIM·PF6
c 11% 10% 4 sBMIM·BF4
c 42% 16% 5 sBMIM·BF/ 43% 40% 6 2PentMIM·BF/ 74% 53%
a Conditions: 200 mM of ascorbic acid, 240 mM of palmitic acid, 0.5 mL of solvent, 20 mg of CAL-B (Novozyme 435), 50 mg of molecular sieve 4 Â. 2 mg of internaI standard (9-fluorenone), 24 h, 60 oC, under nitrogen, stirred with magnetic stirring bar. b Conversion and yield were determined by comparison with internaI standard by HPLC. Conversion was calculated by the decease in the amount of ascorbic acid while yield was determined by the increase in the amount of product ester. C Purified by washing with saturated aqueous sodium carbonate solution. d Purified by filtration through neutral alumina.
Presumably, the activity of CAL-B in these ionic liquids would be similar because
the structure of ionic liquids is similar except for BMIM·PF6. However, the conversion
after 24 h in sBMIM·BF4 was higher by a factor of 1.8 ~ 4 than that in other ionic liquids
such as MOEMIM·BF4, PMIM·BF4, and BMIM·PF6. Since ascorbic acid dissolved
readily in an ionic liquids 11, the solubility of the other reactant, palmitic acid, may
influence the reaction conversion. In fact, we observed that palmitic acid remained
10 Abbreviations: MOEMIM·BF4, 1-(2-methoxyethyl)-3-methylimidazolium tetrafluoro
borate; PMIM·BF4, I-propyl-3-methylimidazolium tetrafluoroborate; BMIM·PF6, 1-
butyl-3-methylimidazolium hexafluorophosphate; sBMIM·BF4, l-sec-butyl-3-methyl
imidazolium tetrafluoroborate; 2PentMIM·BF4, 3-methyl-l-(1-methyl-butyl)-3-
imidazolium tetrafluoroborate.
11 Solubility of L-ascorbic acid: >130 mg/mL In sBMIM·BF4 and -60 mg/mL in
2PentMIM·BF4 at 60 oC
68

suspended in the other ionic liquids but not in sBMIM·BF4. To increase the solubility of
palmitic acid in ionic liquid, we designed a new ionic liquid, 2PentMIM·BF4, which has
similar structure to sBMIM·BF4 but one more carbon. The extra carbon in
2PentMIM·BF4 increases the solubility of palmitic acid, thereby giving higher conversion
(Table 1, compare entries 5 and 6).
The discrepancy between the conversion (amount of ascorbic acid consumed) and
the yield (amount of product formed) is presumably due to the fast air oxidation of the
product L-ascorbyl fatty acid ester. For this reason, the conversion was always higher
than the yield.
The time course of the esterification showed a decreases in the reaction rate by a
factor of 3.5 after 3 h and a further dramatic decrease after 10 h (see Figure 1). Even after
24 h, the conversion was only 43%, which is still far from the maximum of 100 %. Since
ionic liquids do not denature lipases, another factor may cause the reaction to slow down.
To identify the cause, we tested the solubility of the product, ascorbyl palmitate, in ionic
liquids. Less than 2.5 mg of ascorbyl palmitate dissolves in 0.5 mL of sBMIM.BF4. 12
The amount of the product produced after 3 h (i.e., 16.4% yield) is around 6.8 mg, which
is over the solubility limit of the product in sBMIM·BF4. Presumably, as the reaction
progresses, the product precipitates, accumulates on the surface of the immobilized
enzyme, and prevents the substrates from reaching the lipase, thereby inhibiting or
stopping the reaction. Releasing the accumulated product from the surface of
immobilized enzyme would prevent the reaction slow down.
12 Solubility of ascorbyl palmitate: < 5 mg/mL in sBMIM·BF4; < 10 mg/mL in
2PentMIM·BF4 at 60 oc.
69

100% c .2
75% UI ... CI)
-e-Yield
~ Conversion > c 0 50% u ... 0
:2 25% CI)
>= 0%
0 5 10 15 20 25
Time (h)
Figure 1. The time course of the esterification of ascorbic acid with palmitie aeid in
sBMIM·BF4. Conditions are same as those in Table 1. Reaction rates: 0-3 h: 0.27
Ilmol/h/mg; 3-6 h: 0.078 Ilmol/h/mg; 6-10 h: 0.075 Ilmol/h/mg; 10-24 h: 0.045
Ilmol/h/mg.
With this assumption, we introduced a hydrophobie additive, sueh as hexane or
porous polypropylene, to the reaction mixture. These additives are more hydrophobie and
have stronger interaction with the hydrophobie products than macroporous acrylie resin,
which is the material for immobilization of CAL-B. The degree of conversion was
determined according to different amount of additives (Figure 2). In the both cases there
was an optimum amount of additive - 10-20 vol% and 50 wt% for hexane and poly
propylene, respectively. The conversion of the reaction with 10-20 vol% hexane was
increased by a factor of 2. Similarly 50 wt% polypropylene increases the conversion by a
factor of 1.5.
We applied the hexane additive to synthesize ascorbyl oleate as well as aseorbyl
palmitate (Table 2). We used sBMIM·BF4 or 2PentMIM·BF4 inc1uding 10-20 vol%
hexane as a sol vent and molecular sieves ta remove water re1eased from the reaction. The
reaction was performed under nitrogen to prevent air oxidation of the produet. The
optimized reaction condition in 2PentMIM·BF4 gave 83% conversion and 65% yield for
ascorbyl oleate (Table 2, entry 8). CAL-B showed highly regioseleetivity and produeed
only L-aseorbyl 6-0leate without any other regioisomers.
70

100%
80% c 0 60% .j!! (1)
> 40% c 0 0 20% ~Hexane
--- Polypropylene 0%
0% 20% 40% 60% 80% 100%
Relative amount of additives
Figure 2. The effect of hydrophobic additives on the conversion of esterification of
ascorbic acid with palmitic acid in sBMIM·BF4. Conditions are the same as those in
Table 1. The amount of hexane and polypropylene is relative to that of sol vents and the
enzyme used, respectively.
Table 2. Optimized acylation of ascorbic acida
Entry Solvent Hexane content Acylating reagent Time
Conversionb Yieldb
(h) 1 sBMIM·BF4 10% Palmitic acid 10 66% 54% 2 sBMIM·BF4 10% Oleic acid 10 72% 44% 3 2PentMIM·BF4 10% Palmitic acid 10 73% 62% 4 2PentMIM·BF4 10% Oleic acid 10 78% 65% 5 sBMIM·BF4 20% Palmitic acid 14 73% 43% 6 sBMIM·BF4 20% Oleic acid 14 76% 42% 7 2PentMIM·BF4 20% Palmitic acid 14 81% 44% 8 2PentMIM·BF4 20% Oleic acid 14 83% 65%
a Reaction conditions are similar to those in Table 1: 200 mM of ascorbic acid, 240 mM of palmitic acid or oleic acid, 0.5 mL of solvents, 10-20 vol% hexane, 45 mg of CAL-B (Novozyme 435), 50 mg of molecular sieve 4 Â. 2 mg of internaI standard (9-fluorenone), 60 oC, under nitrogen, stirred with magnetic stirring bar b Conversion and yield were determined as in Table 1.
71

Discussion
It is difficult to make the potentially useful L-ascorbyl oleate by chemical
methods because of requirement of regioselective control as weIl as the fast air oxidation
of L-ascorbic acid and oleic acid. On the other hand, the enzyme-catalyzed reaction is not
easy due to the low solubility of ascorbic acid in non-polar organic sol vents and the low
activity of enzymes in polar solvents. As a compromise, most researchers chose
moderately polar solvents, such as acetone, THF and tertiary alcohols, for lipase
catalyzed synthesis of the ascorbyl ester. However, the conversion or the yield in lipase
catalyzed direct esterifications was less than 50% in most cases. 13 The high solubility of
L-ascorbic acid in ionic liquids, in which a lipase shows comparable activity to that in
'nonpolar solvents, facilitates the reaction.
Although ionic liquids reliably permit lipase-catalyzed reaction, their preparation
methods or impurities in ionic liquids strongly affect the activity of lipase or the reaction
process. To get the reliable lipase activity or the proper reaction product, their purification
is critical to use ionic liquids in a lipase-catalyzed reaction. For example, halide or acid
impurities in ionic liquids stop or slow a lipase-catalyzed reaction. On the other hand, the
presence of sodium carbonate in ionic liquids causes undesired reactions (i.e., its
oxidation and following decompositions)14 in esterification of ascorbic acid. Ascorbic
acid is easily oxidized in aerobic condition. The oxidation reaction is catalyzed by heat
and base. In addition, the rate of oxidation of ascorbyl palmitate is three orders of
magnitude faster than that of ascorbic acid. 15 This faster oxidation of the product causes
discrepancy between the degree of conversion and the yield. Consistent with this
assumption, the yield increased after modifying the purification by filtering through
13 Researchers have not reported the causes of low yield or conversion.
14 The decomposition of ascorbic acid was studied by Kimoto, E.; Tanaka, H.; Ohmoto,
T.:; Choami, M. Anal. Biochem. 1993,214, 38-44. Chen, F.; Yuan, J.-P. J. Agric. Food
Chem. 1998,46,5078-5082.
15 Liu, X. Y.; Guo, F. L.; Liu, Y. c.; Liu, Z. L. Chem. Phys. Lipids 1996, 83, 39-43.
72

alumina instead of washing with saturated sodium carbonate. Additionally, to get high
yield, the reaction should be strictly protected from air. The reactions in this chapter were
done on a small scale where it is difficult to exc1ude air completely. If this reaction was
scaled up to an indus trial scale, protection from oxygen would be more efficient and the
yield would match the conversion.
As for the substrates, the solubility of the product also influences the conversion
or the yield of reactions. In homogeneous catalysis, if the product precipitates out from
the reaction media, the equilibrium could be shifted to product formation, thereby
increasing the reaction rate or conversion. However, in heterogeneous catalysis,
precipitation of the product blocks the contact between substrates and the catalyst, and
inhibits the reaction. Proper selection of additives can avoid this product inhibition. The
product of esterification of ascorbic acid with fatty acids is hydrophobic fatty esters.
Addling hydrophobic additives can prevent the product oiling-out on the immobilized
enzyme. Small amount of hexane (10-20%) increases the conversion of reaction by a
factor of 2.
Besides highly solvating ability of ionic liquids, ionic liquids are completely
nonvolatile. In this research, we used molecular sieves to remove water during the
reaction progress, thereby shifting its equilibrium toward product formation. Alternately,
using vacuum can be used on the same purpose. Currently we are investigating to use
vacuum to remove water and use polypropylene beads instead of hexane.
Experimental Section
General Methods. IH NMR spectra were recorded in acetone-d6 or CDCh at 400 MHz
(M400, Varian) and 500 MHz (Bruker). An immobilized form of lipase B from Candida
antarctica (Novozym 435) was donated from Novozymes AIS (Denmark). Other
chemicals were purchased from Sigma-Aldrich. In HPLC analysis, the substrates,
prodlucts, and the internaI standard used were identified by comparison of the retenti on
time of authentic samples.
73

Synthesis of sBMIM·BF4 and 2PentMIM·BF4• The bromide salts, 1-alkyl-3-
methylimidazolium bromide, of corresponding ionic liquids were prepared according to
the literature procedure. I6 The tetrafluoroborate salts were prepared and purified
according to a literature procedure with slight modification. The 1-alkyl-3-
methylimidazolium bromide salt (0.40 mol) was added to a suspension of NaBF4 (1.2
equiv, 52.7 g, 0.48 mol) in acetone (150 mL). After the mixture was stirred for 48 h at
room temperature, the sodium bromide precipitate was removed by filtration and the
filtrate concentrated to an oil (-100 mL) by rotary evaporation. The cru de product was
diluted with methylene chloride (200 mL) and filtered through silica gel (40-50 g). The
solution was filtered again through neutral aluminum oxide (30-40 g) to remove trace
amounts of silica gel and other acidic impurities from the ionic liquid. Removal of
solvent under vacuum yielded a pale yellow oil: yield 60%.
2PentMIM·BF4. IH NMR (400 MHz, acetone-d6, 8): 9.04 (s, 1H); 7.82 (dd, 1H); 7.72
(dd, 1H); 4.66 (m, 1H); 4.04 (s, 3H); 1.92 (m, 2H); 1.60 (d, 3H); 1.30 (m, 2H); 0.92 (t,
3H). 13C NMR (100 MHz, acetone-d6, 8): 136.89, 124.22, 120.64, 57.50, 38.70, 36.01,
20.87, 19.15, 13.28.
Esterification of Ascorbic Acid. Oleic acid (38 ~L, 0.12 mmol) or palmitic acid (31 mg,
0.12 mmol), ascorbic acid (18 mg, 0.1 mmol), 9-fluorenone (2 mg), molecular sieve (50
mg), and Novozym 435 (20 mg) were mixed with solvent (0.5 mL of ionic liquids) and
stirred at 60 oC un der nitrogen. After 24 h, methanol (10 mL) was added to stop the
reaction. Analysis of the mixture was performed by high performance liquid
chromatography on a C-18 column (SUPELCO, Bellefonte, PA) with isocratic elution
16 (a) Suarez, P. A. Z.; DuIlius, J. E. L.; Einloft, S.; De Souza, R. F.; Dupont, J.
Polyhedron 1996, 15, 1217-1219. (b) Holbrey, J. D.; Seddon, K. R. J. Chem. Soc.,
Dalton Trans. 1999, 2133-2139.
74

using 95% methanoI/lO% water containing 0.5% acetic acid at 1 mL/min. Detection was
achieved by a UV detector at 254 nm.
Retention time: ascorbic acid, 1.92 min; internaI standard (9-fluorenone), 2.41 min;
ascorbyl oleate, 3.66 min; ascorbyl palmitate, 3.60 min.
L-Ascorbyl 6-0-o1eate. IH NMR (500 MHz, CDCh, Ô)17: 5.40 (m, 2H); 4.86 (d, IH);
4.45 (m, IH); 4.26 (d, 2H); 2.81 (m, 3H); 2.40 (t, 2H); 2.15 (m, 4H); 1.65 (m, 2H); 1.30
(br s, 20H); 0.90 (t, 3H).
17 The NMR data was obtained from a separate large-scale reaction in tert-amyl alcohol:
isolated yield 23%.
75

Chapter 3. Appendix 1
This article is a verbatim copy of an accepted article and is reproduced with automatic permission from the symposium proceedings of "Ionie Liquids as Green Solvents: Progress & Prospects", Seongsoon Park, Viklund, Karl HuIt, Romas J. Kazlauskas, "Ionie Liquids Create New Opportunities for Nonaqueous Biocatalysis with Polar Substrates". Copyright 2003 American Chemical Society.
76

Ionie Liquids Create New Opportunities for Nonaqueous
Bioeatalysis with Polar Substrates
Acylation of Glucose and Ascorbic Acid
Seongsoon Park1, Fredrik Viklund2
, Karl Rult2, Romas J. Kazlauskas 1
,2*
1 McGill University, Department of Chemistry, 801 Sherbrooke St. W., Montréal, QC H3A 2K6 Canada
2Royallnstitute of Technology (KTH), Department of Biotechnology, AlbaNova University Centre, SE-106 91 Stockholm, Sweden
Abstract: Lipase-catalyzed reactions of polar substrates are inefficient in organic
solvents. Nonpolar organic solvents do not dissolve polar substrates, while polar organic
sol vents inactivate lipases. Ionic liquids such as l-alkyl-3-methyl imidazolium
tetrafluoroborate are as polar as N-methyl formamide or methanol, but, unlike these
solvents, ionic liquids do not inactivate lipases. This unusual feature creates opportunities
for nonaqueous biocatalysis with polar substrates. First, we describe a simple purification
involving filtration through silica gel, which yields ionic liquids that work reliably as
solvents in lipase-catalyzed reactions. Next, we report two examples that exploit these
unique advantages of ionic liquids. First, lipase-catalyzed acetylation of glucose was up
to twelve times more regioselective in ionic liquids than in acetone. Second, lipase
catalyzed the acylation of ascorbic acid to make fat-soluble antioxidants. In sorne cases,
reactions in ionic liquids were comparable or slower than in tert-amyl alcohol, but in
typical cases, the reactions in ionic liquids were twice as fast and proceeded to higher
conversion. Ionic liquids also offer the possibility to use vacuum to remove water formed
by the esterification and drive the equilibrium even further toward product.
77

Introduction
A long-standing problem in biocatalysis is reactions of polar substrates under
nonaqueous conditions. Reactions such as acylation of an alcohol require nonaqueous
conditions to avoid competing hydrolysis. However, polar substrates such as sugars
dissolve only in the most polar organic sol vents such as dimethylsulfoxide. Unfortunately
enzymes such as lipases inactivate in such polar organic sol vents. Current solutions
involve compromises. In sorne cases, researchers use moderately polar organic solvents,
where the substrate dissolves slightly and the enzymes retain sorne activity. Such
reactions are usually too slow for preparative use. Another alternative is to modify the
substrates (e.g., use an alkyl glycoside instead of a glycoside) and use a less polar organic
solvent where the enzyme remains active. However, this approach yields an analog of the
desired product.
In spite of these difficulties, the ability to catalyze reactions on polar substrates in
nonaqueous media is becoming increasingly important. Natural building blocks -
peptides, sugars, nucleotides, biochemical intermediates - are important starting materials
for pharmaceuticals, fine chemicals and materials. These building blocks are becoming
increasingly important in a bio-based economy, where chemicals and materials come
from plants and microorganisms.
This paper focuses on room temperature ionic liquids l as a solution to biocatalysis
reactions with polar substrates un der nonaqueous conditions. Ionic liquids are polar
solvents (comparable to methanol) and readily dissolve polar substrates. However, for
reasons that are still not clear, ionic liquids do not denature lipases, as would an organic
solvent of comparable polarity. For this reason lipase-catalyzed reactions of polar
substrates proceed more efficiently or more selectively in ionic liquids. Several groups
1 Review: Welton, T. Chem. Rev. 1999,99,2071-2083.
78

have reported enzyme-catalyzed reactions in ionic liquids2-5
• The advantages of using
ionic liquids over an organic solvent varied for each case and inc1uded increased
enantioselectivity3, increased stability of the enzyme4 or increased molecular weight of
the product polymer5. Here we focus on advantages related to reactions of polar
substrates.
The first example is the acetylation of glucose with vinyl acetate catalyzed by lipase
B from Candida antarctica (CAL-B)6. This acetylation is more regioselective in ionic
liquids than in moderately polar organic sol vents such as tetrahydrofuran. This increased
regioselectivity yields only 6-0-acetyl D-glucose instead of a mixture of 6-0-acetyl- and
3,6-0-diacetyl D-glucose. The increased solubility of glucose relative to 6-0-acetyl D
glucose in ionic liquids accounts for the increased regioselectivity.
2 Lau, R. M.; van Rantwijk, F.; Seddon, K. R.; Sheldon, R. A. Organic Lett. 2000, 2,
4189-4191; Itoh, T.; Akasaki, E.; Kudo, K.; Shirakami, S. Chem. Lett. 2001, 262-263;
Husum, T. L.; Jorgensen, C. T.; Christensen, M. W.; Kirk, O. Biocatal. Biotransform.
2001,19,331-338.
3 Kim, K.-W.; Song, B.; Choi, M.-Y.; Kim, M.-J. Org. Lett. 2001,3,1507-1509; Schofer,
S. H.; Kaftzik, N.; Wasserscheid, P.: Kragl, U. Chem. Commun. 2001, 425-426;
Kielbasinski, P.; Albrycht, M.; Luczak, J.; Mikolajczyk, M. Tetrahedron Asymm. 2002,
13,735-738.
4 Erbeldinger, M.; Mesiano, A. J.; Russell, A. J. Biotechnol. Prog. 2000, 16, 1131-1133;
Lozano, P.; de Diego, T.; Guegan, J.-P.; Vaultier, M.; Iborra, J. L. Biotechnol. Bioeng.
2001, 75, 563-569; Kaftzik, Nicole; Wasserscheid, P.; Kragl, U. Org. Process Res. Dev.
2002,6,553-557.
5 Uyama, H.; Takamoto, T.; Kobayashi, S. Polymer J. (Tokyo) 2002, 34, 94-96.
6 Park, S.; Kazlauskas, R. J. J. Org. Chem. 2001,66,8395-8401.
79

hc
HO 6-o-acetyl D-glucose HO
OH OH
The second example is the CAL-B-catalyzed acylation of L-ascorbic acid (vitamin C)
with unactivated fatty acids to make fat-soluble antioxidants, such as 6-0-ascorbyl
palmitate or 6-0-ascorbyl oleate. Reaction of the polar L-ascorbic acid with nonpolar
fatty acids proceeds to higher conversion in ionic liquids than in an organic solvent (tert
amyl a1cohol). In addition, since ionic liquids are nonvolatile, they offer the possibility of
using vacuum to remove water and shift the equilibrium of the reaction more toward
product formation.
ResuUs
OH 0 0 0
~
Ionic liquids, prepared either by literature procedures 7 or straightforward
modifications, did not work reliably as sol vents for lipase-catalyzed reactions. In sorne
cases, reactions proceeded weIl; in other cases reactions proceeded slowly or not at aIl.
7 Huddleston, J. G.; Willauer, H. D.; Swatloski, R. P.; Visser, A. E.; Rogers, R. D. J.
Chem. Soc., Chem. Commun. 1998, 1765-1766; Suarez, P. A. Z.; DuIlius, J. E. L.;
Einloft, S.; De Souza, R. F.; Dupont, J. Polyhedron, 1996, 15, 1217-1219. Aiso see:
Dupont, 1.; Consorti, C. S.; Suarez, P. A. Z.; de Sousa, R. F. Org. Synth. 2002, 79, 236-
243:: Law, M. C.; Wong, K. Y.; Chan, T. H. Green Chem. 2002,4,328-330.
80

Sinœ the structures of the ionic liquids were similar, we suspected that impurities might
cause the unpredictable behavior.
The synthesis of ionic liquids involved initial preparation of the halide salt followed
by exchange of the halide with tetrafluoroborate, Scheme 1. A likely impurity in ionic
liquids is the halide salt due to incomplete exchange. Indeed, ionic liquids gave a
precipitate with sil ver nitrate solution, thereby confirming the presence of halide. For this
reason, we purified aIl ionic liquids to remove halide salts.
Purification of Ionie Liquids
Purification involved filtration of the diluted ionic liquid through silica gel to remove
traces of 3-alkyl-l-methylimidazolium halide and then washing with saturated aqueous
sodium carbonate to remove cloudiness due to fine particles of silica gel. FinaIly, drying
the solution over magnesium sulfate and removing the diluent (methylene chloride) by
vacuum yielded the purified ionic liquid. An alternative purification replaced the sodium
carbonate wash and drying with magne sium sulfate with a filtration through neutral
alumina. This second method avoided traces of the basic carbonate anion in the ionic
liquid. Both methods yielded ionic liquids that work reliably in aIl lipase-catalyzed
reactions that we tested. These procedures yielded six 3-alkyl-l-methylimidazolium
tetrafluoroborate ionic liquids, Scheme 1.
81

Œ> Œ> ~N R-X ~WR NaBF4 ~WR N--.!J -N--.!J 8- N--.!J 8 , ,x ,BF4
H3C H3C while solid H3C ionic liquid
El
(X = CI or Br) Ipurity
Abbreviation • EMIMoBF 1) dilule w/ CH2CI2
4 2) tiller Ihrough silica gel PMIMoBF4 or tiller Ihrough {3) wash wilh sal'd. Na2C03 BMIMoBF 4 neulral alumina 4) dry over MgS04
8.
n-Pr n-Bu s-Bu MeOCH2CH2 2-penlyl
sBMIMoBF4 5) evaporale CH2CI2 MOEMIMoBF4 2PeniMIMoBF4
8.' 8.2 Abbreviation H n-Pr PPYRoBF4 -oŒ> H n-Bu BPYRoBF4 R' _ N-R2
Me n-Pr PNPYRoBF4 e Me n-Bu BMPYRoBF4
BF4
Scheme 1. Preparation and purification of 3-alkyl-l-methylimidazolium tetrafluoroborate
ionic liquids for biocatalysis. Similar methods yielded the related N-alkylpyridinium
tetrafluoroborate ionic liquids.
Similar reactions and purifications gave five other ionic liquids: one hexafluoro
phosphate salt, 3-n-butyl-l-methylimidazolium hexafluorophosphate, and the four based
on pyridinium and 4-methylpyridinium cations shown below.
Polarity of Ionie Liquids is Similar to that for Polar Organie Solvents
To compare the polarity of ionic liquids and organic solvents, we measured their
polarities with Reichardt's dye8. Polar solvents stabilize the polar ground state of
Reichardt's dye thereby shifting its color to shorter wavelengths. We compared the
polarities of the different sol vents using Reichardt's normalized scale where
tetramethylsilane has a value of zero and water has a value of one. The polarity values for
the ionic liquids we used ranged from 0.63 to 0.71 with the most polar being EMIM-BF4
and the least polar being BMPYR-BF4, see x-axis of Figure 1. BMIM-BF4 is more polar
that BMIM-PF6.
8 Reichardt, C. Chem. Rev. 1994,94,2319-2358.
82
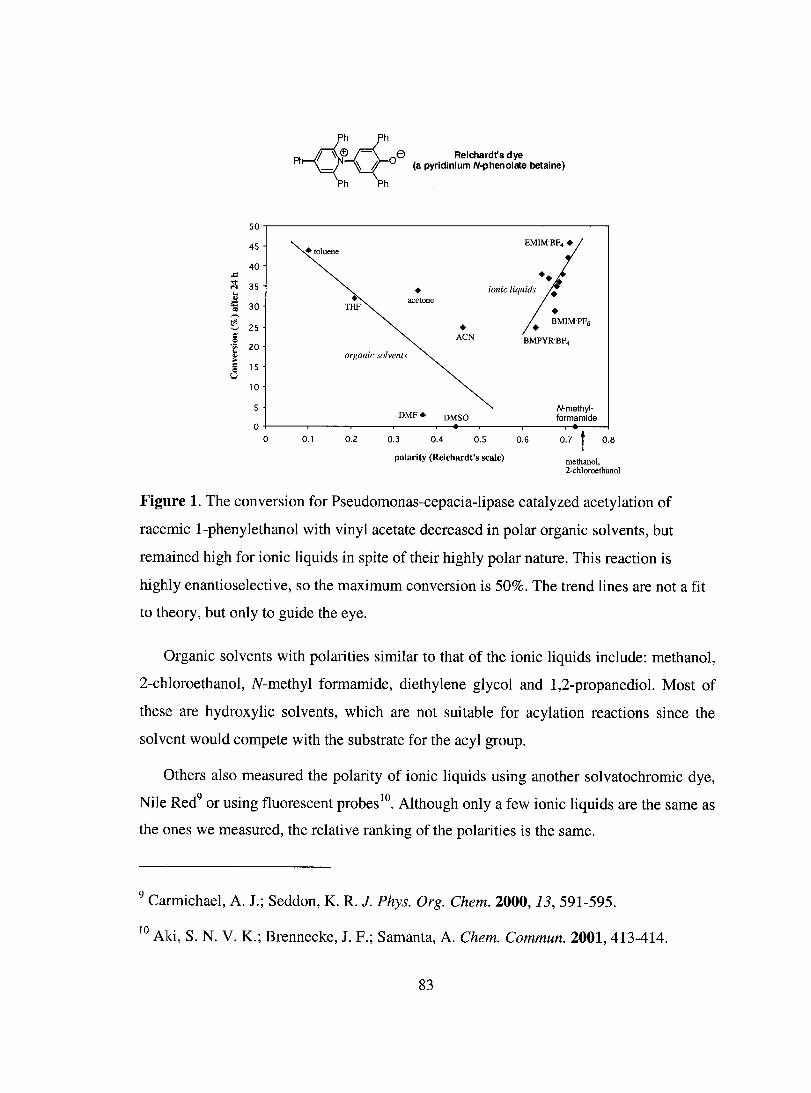
50
45 .toluene
40 ..c: ~ 35 .. .. 0::
'" 30
~ 25
= . ~ 20 .. § 15 U
10
5
0 0 0.1
Reichardt's dye (a pyridinium N-phenolate betaine)
• acetone
• ACN
EMIMBF ••
• • BMIMPF6
BMPYRBF •
organic solvellts
DMF. DMSO
0.2 0.3 0.4 0.5
polarity (Reichardt's scale)
0.6
N-methyl· formamide
0.7 t 0.8
methanol, 2·chloroethanol
Figure 1. The conversion for Pseudomonas-cepacia-lipase catalyzed acetylation of
racemic 1-phenylethanol with vinyl acetate decreased in polar organic solvents, but
remained high for ionic liquids in spite of their highly polar nature. This reaction is
highly enantioselective, so the maximum conversion is 50%. The trend lines are not a fit
to theory, but only to guide the eye.
Organic sol vents with polarities similar to that of the ionic liquids include: methanol,
2-chloroethanol, N-methyl formamide, diethylene glycol and 1,2-propanediol. Most of
these are hydroxylic solvents, which are not suitable for acylation reactions since the
solvent would compete with the substrate for the acyl group.
Others also measured the polarity of ionic liquids using another solvatochromic dye,
Nile Red9 or using fluorescent probes lO• Although only a few ionic liquids are the same as
the ones we measured, the relative ranking of the polarities is the same.
9 Carmichael, A. J.; Seddon, K. R. J. Phys. Org. Chem. 2000, 13, 591-595.
10 Aki, S. N. V. K.; Brennecke, J. F.; Samanta, A. Chem. Commun. 2001,413-414.
83

High Aetivity of Lipases in Ionie Liquids in Spite oftheir High Polarity
Lipases showed good activity in ionic liquids even though they showed no, or little,
activity in normal organic solvents with similar polarities. As a model reaction, we used
the acetylation of racemic I-phenylethanol with vinyl acetate catalyzed by lipase from
Pseudomonas cepacia, PCL, equation 1. This reaction is highly enantioselective so the
maximum conversion was 50%. We compared the rates of reaction and
enantioselectivities in ionic sol vents to those in normal organic solvents. In aU cases, the
enantioselectivity of the reaction remained high: E >200.
+ ~ (1)
Surprisingly, the conversion after 24 h also remained high in ionic liquids despite their
high polarity, Figure 1 above.
For normal organic solvents, the acetylation reaction proceeds weU in nonpolar
solvent, but not in polar solvents. The reaction is nearly complete in toluene, partiaUy
complete in tetrahydrofuran (THF) , acetone or acetonitrile (ACN), but proceeds very
slowly or not at aU in the more polar N,N-dimethylformamide (DMF), dimethylsulfoxide
(DMSO), or N-methylformamide. Although the ionic liquids are highly polar (similar to
N-rnethyl-formamide), the acetylation reaction proceeds weU in aU ionic liquids tested.
The reaction is nearly complete in EMIM-BF4 and MOEMIM-BF4 and partiaUy complete
in aIl other ionic liquids. In addition, the trend for ionic liquids is for higher degrees of
conversion as the polarity of the ionic liquid increases, while the trend for organic
sol vents is the opposite -lower degrees of conversion as the polarity increases.
Since the substrates for this model reaction dissolve in both nonpolar organic solvent
and in ionic liquids, there is no obvious advantage to using ionic liquids in this case.
84

More Regioselective Acylation of Glucose in Ionie Liquids
Since ionic liquids are polar sol vents that do not denature lipases, they may be ideal
for Iipase-catalyzed transformations of polar substrates. As a first example, we examined
the lipase-catalyzed 6-0-acetylation of glucose catalyzed by lipase B from Candida
antarctica (CAL-B), equation 2, Table 1.
~OAc J3-D-glucose ..
CAL-B
HOhC
+ HO ~ ~AC _n HO A~~
OH OH OH OH (2) organic solvenls: 2-3: 1 ionic liquids: >13: 1
In organic sol vents such as acetone and tetrahydrofuran (THF) , the 6-0-acetylation
reaction proceeded along with further acetylation of the 3-position. In acetone, acetylated
products formed in 72% yield, of which 76% was the desired 6-0-acetyl compound (-3: 1
selectivity). In THF, glucose reacted completely, but only 53% was the desired 6-0-
acetyl compound ( - 2: 1 selecti vit Y ). Even at a lower extent of conversion, the
regioselectivity remained low. In acetone at 42% conversion, 82% was the 6-0-acetyl
compound (-5:1 selectivity), while in THF at 50% conversion, 85% was the 6-0-acetyl
compound (-6: 1 selectivity). The low selectivity is likely related to the poor solubility of
glucose in the se organic solvents (0.02-0.04 mg/mL at 60 OC ll). Glucose remains a
suspended solid and the initial 6-0-acetylation yields a more soluble compound, which
then undergoes further acetylation to the 3,6-0-diacetyl derivative.
11 Cao, L., Fischer, A., Bomscheuer, U. T., Schmid, R. D. Biocatal. Biotransform. 1997,
14,269-283.
85

Table 1. Regioselective CAL-B-Catalyzed Acetylation of Glucosea
Solvent
EMIM'BF4
[MOEMIM'BF4
PMIM'BF4
BMIM·BF4
sBMIM'BF4
BMIM'PF6
BPYR·BF4
PPYRBF4
Acetone THF
Final composition of reaction mixture 36-0-
6-0-Acetyl- . ' D-Glucose, % D GI nt. Dzacetyl-- ucose -/0
D-Glucose, % 49.6
0.0 72.2 22.4 9.8 70.5 58.0 56.4 27.7
0.0
50.3 93.0 27.8 68.9 79.2 11.3 37.3 38.6 55.0 52.6
0.0 6.9 0.0 8.7
10.8 18.1 4.7 5.0
17.4 47.4
MonoConversion
% acylation, %
50.4 99.9 99.9 93.1 27.8 99.8 77.6 88.8 90.1 87.9 29.5 38.5 42.0 88.7 43.6 88.4 72.3 76.1 99.9 52.6
a Conditions: 0.5 mmol P-D-glucose, 1 mmol vinyl acetate, 1 mL solvent, 30 mg Novozyme SP435, 36 h, 55 oC, Data from reference 6. After the reaction, both remaining glucose and the acetylated products were a mixture of anomers. The conversion was measured by gas chromatography after derivatization with chlorotrimethylsilane and 1,1,1,3,3,3-hexamethyldisilazaneI2
. The acylation positions were determined by COSY experiments.
On the other hand, acetylation of glucose proceeded with much higher selectivity for
monoacetylation in ionic liquids than in organic solvents. In the seven ionic liquids
containing a tetrafluoroborate anion, the 6-0-acetylation proceeded with 42-99%
conversion, of which 88-99% was the desired 6-0-acetyl glucose (-7: 1 to -100: 1
selectivity). The best ionic liquid was MOEMIM-BF4, where aIl the glucose was
acetylated and 93% was the desired 6-0-acetyl compound (-13: 1 selectivity). The one
ionic liquid with a hexafluorophosphate anion, BMIM-PF6, showed both slow reaction
(29% conversion) and low selectivity (39% monoacetyl, -0.6: 1 selectivity).
The higher solubility of glucose in ionic liquids correlates with the higher
regioselectivity in these solvents. Approximately 100 times more glucose dissolves in the
best ionic liquid, MOEMIM-BF4 -5 mg/mL at 55 oC, than in acetone or THF. On the
12 Sweeley, C. c.; Bentley, R.; Makita, M.; Wells, W. W. J. Am. Chem. Soc. 1963, 85,
2497-2507.
86

other hand, glucose is not very soluble in the worst ionic liquid, BMIM-PF6, <1 mg/mL
at 55 oc. The increased solubility of glucose increases the relative concentration of the
desired reactant, glucose, relative to the undesired reactant, 6-0-acetyl glucose.
Initial experiments also showed that CAL-B catalyzes the regioselective acylation of
maltose monohydrate, a disaccharide that is ev en more polar than glucose. Using the
reaction conditions in Table l, but only half the amount of maltose (0.25 mmol instead of
0.5 mmol) and MOEMIM-BF4 as the solvent, yielded 50% of acetylated products.
Regioseleetive Aeylation of Aseorbie Acid in Ionie Liquids
Another example of a lipase-catalyzed acylation of a polar substrate is the acylation
of ascorbic acid (vitamin C) with a fatty acid to make a fat-soluble antioxidant, equation
3. The choice of solvent for this reaction is more difficult because one reactant is polar
(ascorbic acid), the other is nonpolar (fatty acid) and the product is amphiphilic.
palmitie aeid or
oleie aeid
OH
HO~~ " 0 H' _
HO OH
CAL·S moleeular sieves solvent, 60 oc
OH
Rr~ HO OH (3)
6-0-ascorbyl palmitate or
6-o-ascorbyl oleate
Although ascorbic acid dissolved readily in aIl ionic liquids (e.g., >130 mg/mL of
sBMIM·BF4 at 60 OC), the other reactant, palmitic or oleic acid, dissolved only in the
most hydrophobic ionic liquids, sBMIM-BF4 or 2PentMIM-BF4. Not surprisingly
therefore, the initial reaction rate was 1.8-8 times faster in sBMIM-BF4 or
2PentMIM-BF4 than in other ionic liquids. (Data not shown.) Reactions in the more
hydrophobic 2PentMIM-BF4, which we prepared specifically for this reaction, showed
higher conversions than reactions in sBMIM-BF4. (Compare entries 1 and 3 of Table II).
In the best cases, the rates of reaction and conversion were slightly better in an
organic solvent, tert-amyl alcohol, than in ionic liquids (entry 10, Table II). However,
these best cases were difficult to reproduce and more typical reactions in tert-amyl
87

a1cohol were up to two times slower than in ionic liquids and reached only 25-40%
conversion (entry 9, Table II). We suspect that incomplete drying of the tert-amyl a1cohol
causes the lower conversions. Acylation occurred only at the primary alcohol position of
ascorbic acid in aIl cases.
Consistent with their role as antioxidants, the product ascorbyl fatty acid esters were
very sensitive to oxidation. For this reason, the conversion (amount of ascorbic acid
consumed) was always higher than the yield (amount of product formed). For example,
entry 2 in Table II shows 42% conversion, but only 16% yield. To minimize this
oxidation, we used an alternate purification method for the ionic liquid, filtration through
neutral alumina, which avoids the wash with the strongly basic sodium carbonate
solution. This modified purification gave a higher yield: 43% conversion, 40% yield
(entry 1, Table II).
Table II. CAL-B-Catalyzed Acylation of Ascorbic Acida
AcylatingConv., Approx. Yield, Entry Solvent Additive
Rati acid % %
1 sBMIM-BF4 c Palmitic 43 0.090 40 2 sBMIM-BF4 C c, d Palmitic 42 0.088 16 3 2PentMIM-BF4 c Palmitic 74 0.15 53 4 sBMIM-BF4 polypropyleneC Palmitic 63 0.13 43
5 sBMIM-BF4 10 vol%
Palmitic 66 0.15 54 hexane
6 sBMIM-BF4 10 vol%
Oleic 72 0.16 44 hexane
7 2PentMIM-BF4 10 vol%
Palmitic 73 0.16 62 hexane
8 2PentMIM-BF4 10 vol%
Oleic 78 0.17 65 hexane
9 t-Amyl alcohol Palmitic 25-40 0.079 -
nd e 0.13
10 t-Amyl alcohol f Palmitic 71-86 0.22 -
nd 0.27
a Conditions: 200 mM (100 f.lmol) of ascorbic acid, 240 mM of palmitic acid, 0.5 mL of solvent, 45 mg of CAL-B, 50 mg of molecular sieve 4Â, 2 mg of internaI standard (9-fluorenone), 60 oC, 10 h, under nitrogen, stirred with magnetic stirring bar. The conversion (amount of starting material consumed) and yield (amount of product formed) were determined using HPLC by comparison with internaI standard. Unless otherwise
88

note:d, ionic liquids were purified by the filtration-through-neutral-alumina method. b !.IlIlol/h/mg CAL-B for the consumption of starting material. C Only 20 mg of CAL-B were used, but the time was extended to 24 h. d Ionie liquid was purified by filtration through silica gel and washing with saturated sodium carbonate solution. e Typical yields using the following conditions: 57 mM (570 !J1Ilol) of ascorbic acid, 57 mM of palmitic acid, 10 ml solvent, 50 mg of CAL-B, 36 h, 100 mg of molecular sieve 3A. f Best yields under the conditions in note e.
Another factor that limited conversion was the poor solubility of ascorbyl palmitate in
ionic liquids «10 mg/mL of 2PentMIM·BF4). In sorne cases, this precipitate occluded the
catalyst and stopped the reaction. To minimize the interaction between the product ester
and the hydrophobic support of the lipase, we added either hexane or polypropylene
beads. The hexane did not dissolve the ascorbyl palmitate, but prevented the oiling out on
the immobilized lipase. For example, under similar conditions condensation of ascorbic
acid with palmitic acid in sBMIM-BF4 with no additive gave 43% conversion (entry 1),
while addition of 10 vol% hexane increased the conversion to 73% (entry 5). As an
alternative, addition of 50 wt% polypropylene beads increased the conversion to 63%
(entry 4).
Discussion
Solvent purification is a key first step in most organic synthesis laboratories. Not
surprisingly, solvent purification is also critical for reproducible results when working
with ionic liquids. The purification methods outlined here involve filtration through silica
gel followed by either a wash with aqueous sodium carbonate or a filtration through
neutral alumina. These methods remove traces of chloride salts from the ionic liquids and
possibly other unidentified impurities. Ionic liquids prepared in this manner worked
reliably and consistently as solvents for lipase-catalyzed reactions.
Besides potential environmental benefits, ionic liquids also expand the accessible
solvent polarity range for lipase-catalyzed reactions. Lipase-catalyzed acylations did not
proc:eed in a polar organic solvent like N-methylformamide, but did proceed in ionic
89

liquids with similar polarities. As researchers have previously noted13, ionic liquids can
dissolve polar molecules such as carbohydrates. Lipase-catalyzed acylations of these
polar substrates work better in ionic liquids than in organic solvents, but the precise
advantage differs for each case.
Acetylation of I-phenylethanol Ca non polar substrate) was as fast in ionic liquids as
in organic sol vents like toluene. There was no obvious advantage to carrying out this
reaction in an ionic liquid.
Acetylation of glucose was more regioselective for the 6-hydroxyl group in ionic
liquids because of the higher solubility of glucose. Although the catalyst usually controls
the regioselectivity of a reaction, with poorly soluble substrates and products such as
glucose and its derivatives, the relative solubility also contributes. The product 6-0-
acetyl glucose is much more soluble in organic sol vents than glucose and therefore
underwent further acetylation. In ionic liquids, glucose is more soluble so acetylation of
glucose is fast enough to compete with the further acetylation of the product. A less
attractive alternative is a dilute reaction mixture where the glucose dissolves. For
example, tert-butyl a1cohol dissolves glucose to 2.4 mg/mL at 45 oC and CAL-B was
highly regioselective for the primary a1cohol position in this solvent14, but the conditions
are about fort Y times more dilute than our conditions in ionic liquid.
13 Kimizuka, N.; Nakashima, T. Langmuir 2001,17,6759-6761; Swatloski, R P.; Spear,
S. K.; Holbrey, J. D.; Rogers, R. D. J. Am. Chem. Soc 2002, 124,4974-4975; Khan, N.;
Moens, L. In lonic Liquids; Rogers, R. D.; Seddon, K. R, Eds. ACS Symposium Series
818:: American Chemical Society: Washington, DC, 2002, pp 360-372.
14 Degn, P.; Pedersen, L. H.; Duus, J. 0.; Zimmermann, W. Biotechnol. Lett. 1999,21,
275··280.
90

The higher solubility of maltose, a disaccharide, in ionic liquids also facilitates
acetylation. Previous lipase-catalyzed acylations of maltose required refluxing tert-butyl
a1cohol as the solventl5.
Acylation of ascorbic acid with fatty acids proceeded to higher conversion in ionic
liquids that dissolved both substrates than a typical reaction in tert-amyl a1cohol. The
advantage of this reaction in ionic liquids as compared to previous reports in organic
sol vents is that fatty acids can be used directly as acyl donors l6. Further, since ionic
liquids are not volatile, one could shift equilibrium toward synthesis by vacuum removal
of water. Researchers previously used the vacuum removal of water to increase the
molecular weight of condensation polymers17•
The best ionic liquid differed for each reaction, but was usually the one that best
dissolved the substrates. For glucose, acetylation was fastest in MOEMIM,BF4 and
slowest, by about a factor of 3, in either PMIM-BF4 or BMIM-PF6. The regioselectivity
was high in an ionic liquids except for one, BMIM-PF6. For acylation of ascorbic acid
with fatty acids, the best ionic liquid was 2PentMIM-BF4. This liquid dissolved both the
ascorbic acid and the fatty acid and reactions were up to eight times faster than in other
ionic liquids, which dissolved the ascorbic acid, but not the fatty acid. In both cases, the
ability to dissolve the substrates was a key parameter, but perhaps not the only one.
15 Woudenberg-van Oosterrom, M.; van Rantwijk, F.; Sheldon, R. A. Bioteehnol. Bioeng.
1996, 49, 328-333; Revew: Plou, F. J.; Crucesa, M. A.; Ferrera, M.; Fuentesa, G.;
Pastora, E.; Bernabé, M.; Christensen, M.; Comelles, F.; Parrad, J. L.; Ballesteros, A. J.
Bioteehnol. 2002, 96, 55-66.
16 Humeau, c.; Girardin, M.; Rovel, B.; Miclo, A. J. Bioteehnol. 1998,63, 1-8; idem, J.
Mol. Catal. B: Enzymatic 1998, 5, 19-23; Yan, y.; Bomscheuer. U. T.; Schmid, R. D.
Bioteehnol. Lett. 1999, 21, 1051-1054; Watanabe, Y.; Minemoto, Y.; Adachi, S.;
Nakanishi, K.; Shimada, Y.; Matsuno, R. Bioteehnol. Lett. 2000, 22, 637-640; Luhong,
T.; Hao, Z.; Shehate, M. M.; Yunfei, S. Bioteeh. Appl. Bioehem. 2000,32,35-39.
17 Brazwell, E. M.; Filos, D.; Morrow, C. J. J. Polym. Sei., A, 1995,33,89-95.
91

This research focused on efficient reactions and did not address how best to isolate
proclucts from ionic liquids. Possibilities inc1ude crystallization, extraction with a polar
organic solvent or even a supercritical fluid I8.
Experimental Section
General. IH NMR spectra were recorded in acetone-d6 or CDCh at 400 MHz (M400,
Varian) and 500 MHz (Bruker). An immobilized form of lipase B from Candida
antarctica (Novozym SP435) was donated from Novo Nordisk (Denmark). Other
chemicals were purchased from Sigma-Aldrich.
Synthesis of Ionie Liquids. l-Alkyl-3-methyl-imidazolium bromide was prepared from
N-methylimidazole and alkyl bromide by literature methods7. The tetrafluoroborate salts
were prepared by a slight modification of literature procedures. l-Alkyl-3-
methylimidazolium-bromide (0.40 mol) was added to a suspension of NaBF4 (1.2 equiv,
52.7 g, 0.48 mol) in acetone (150 mL). After the mixture was stirred for 48 h at room
temperature, the sodium bromide precipitate was removed by filtration and the filtrate
concentrated by rotary evaporation to an oil (-100 mL). This oil gave a precipitate when
mixed with aqueous silver nitrate indicating that it still contained sorne sorne l-alkyl-3-
methyl imidazolium halide.
Purification of MOEMIM·BF4• The crude ionic liquid was diluted with methylene
chloride (200 mL) and filtered through silica gel (-50 g). This step removed the l-alkyl-
3-methyl imidazolium halide since the filtrate no longer gave a precipitate mixed with
aqueous sil ver nitrate. The solution was washed twice with sat' d sodium carbonate
aqueous solution (40 mL) and dried over anhydrous magnesium sulfate. Removal of
18 Blanchard, L. A.; Hancu, D.; Beckman, E. J.; Brennecke, J. F. Nature 1999, 399, 28-
29.
92

solvent under vacuum yielded a pale yellow oil, 50-70% yield. 1 H-NMR: Ù 8.95 (1H, s);
7.71 (IH, dd); 7.68 (1H, dd); 4.51 (2H, t); 4.05 (3H, s); 3.80 (2H, t); 3.34 (3H, s).
Purification of (±)-2PentMIM-BF4• The crude ionic liquid was diluted with methylene
chloride (200 mL), filtered through silica gel (-50 g) and then filtered through neutral
aluminum oxide (-50 g) to remove traces of silica gel. Removal of solvent under vacuum
yielded a pale yellow oil, 60% yield. IH NMR (400 MHz, acetone-d6): Ù 9.04 (s, IH);
7.82 (dd, 1H); 7.72 (dd, 1H); 4.66 (m, 1H); 4.04 (s, 3H); 1.92 (m, 2H); 1.60 (d, 3H); 1.30
(m, 2H); 0.92 (t, 3H). 13C NMR: 136.89, 124.22, 120.64, 57.50, 38.70, 36.01, 20.87,
19.15, 13.28.
Transesterification of sec-Phenethyl alcohol. Vinyl acetate (92 !JL, 1.0 mmol) and sec
phenethyl aicohoi (13 !JL, 1.0 mmol) were added to a suspension of lipase from
Pseudomonas cepacia (PCL, Amano Pharmaceuticai Co. Nagoya, Japan, 20.0 mg) in
solvent (1.0 mL of either organic soivents or ionic liquids) and stirred at 25 oc. The
reactions were monitored by TLC (ethyl acetate:hexane, 1:3). After 24 h, the reaction
mixture was extracted with hexane (3 mL) and the hexane extract was analyzed by gas
chromatography on a Chiralsil-Dex CB column (Chromopak). The conversion, c, was
ca1culated from the enantiomeric excess of the product, eep' and of the starting materiaI,
ees, using the equation below19.
c = ee y
ees + eep
Acetylation of Glucose. Vinyi acetate (92 !JL, 1.0 mmol), P-D-glucose (90 mg, 0.5
mmoI), and Novozym SP435 (30 mg) were mixed with soivent (1.0 mL of either organic
solvents or ionic liquids) and stirred at 55 oC. After 36 h, pyridine (2 mL), 1,1,1,3,3,3-
hexamethyIdisilazane (1 mL) and chioromethyIsiIane (1 mL) were added to the reaction
19 Chen, C. S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1982, 104,
7294-7299.
93

mixture. The mixture was extraeted with hexane (5 mL) and analyzed by gas
chromatography on the column above. Temperature pro gram: initial temperature 180 oC
for 2 min, increase to 190 oC over 10 min, and hold for 28 min.
Acylation of Ascorbic Acid. Oleic acid (38 j.!L, 0.12 mmol) or palmitic acid (31 mg,
0.12 mmol), ascorbic acid (18 mg, 0.1 mmol), 9-fluorenone (2 mg, internaI standard),
molecular sieve (50 mg), and Novozym SP435 (20 mg) were mixed with ionie liquid (0.5
mL) and stirred at 60 oC under nitrogen. After 24 h, methanol (10 mL) was added to the
reaction mixture. Analysis of the mixture was performed by high performance liquid
chromatography on a C-18 column (4.6 mm id x 25 cm) eluted with 95%
methanol/5%water containing 0.5% acetie acid at 1 mL/min. Peaks were detected by UV
absorbance at 254 nm. Retention time: ascorbic acid, 1.92 min; internaI standard (9-
fluorenone), 2.41 min; ascorbyl oleic acid ester, 3.66 min; ascorbyl palmitic acid ester,
3.60 min.
6-0-L-Ascorbyloleate. IH NMR (500 MHz, CDCI3): 85.40 (m, 2H); 4.86 (d, 1H); 4.45
(m, 1H); 4.26 (d, 2H); 2.81 (m, 3H); 2.40 (t, 2H); 2.15 (m, 4H); 1.65 (m, 2H); 1.30 (br s,
20H); 0.90 (t, 3H).
94

Chapter 4
To enhance enzyme selectivity and activity, one can slightly alter substrates
because substrate engineering is more diverse than medium engineering but does not
need the gene of enzyme, which is required for protein engineering. However, substrates
of interest should be altered slightly to be regenerated easily.
ln this chapter, we apply substrate engineering to improve the activity and
enantioselectivity of CAL-catalyzed p-Iactam ring opening reaction. CAL-B showed low
enantioselectivity (E = -5) of p-Iactam ring opening reaction in water. However, when
the reaction was performed with a long secondary alcohol instead of water, the reaction
was faster by a factor of 2 and more enantioselective (E > 200). In addition, we
rationalized the high enantioselectivity and a critical role of alcohol by molecular
modeling.
Contributions
This work was done under supervision of Dr. Romas J. Kazlauskas. One
collaborator, Dr. Eniko Forro, performed small scale and 0.5 g-scale reactions. Harjap
Grewal did initial computer modeling. 1 performed initial screening with commercial
hydrolases and a part of small scale reactions. 1 aiso did the detailed computer modeling.
95

Chapter 4. Enantioselective Ring Opening of ~·Lactams
Catalyzed by Candida antarctica Lipase B: Molecular
Basis and Optimization
Abstract: Lipase B from Candida antarctica (CAL-B) catalyzed the slow, but highly
enantioselective (E > 200), ring-opening a1coholysis of two bicyclic and two 4-
arylsubstituted p-lactams. A 0.5-g scale reaction under optimized conditions (2-octanol as
the nucleophile in diisopropyl ether at 60 OC) yielded the unreacted p-lactam in 39-46%
yield (max yield is 50%) with ~ 96% ee. The product p-amino acid esters reacted further
by polymerization (not isolated or characterized) or by hydrolysis due to small amounts
of water in the reaction mixture yielding p-amino acid (7-11 % yield, ~ 96% ee ). The
favored enantiomer of aIl four p-lactams had similar 3-D orientation of substituents, as
did most previously reported p-lactams and p-lactones in similar ring-opening reactions.
Computer modeling of the ring opening of 4-phenyl-azetidin-2-one, 3, suggests that the
reaction proceeds via an unusual substrate-assisted transition state, where the substrate
a1cohol bridges between the catalytic histidine and the nitrogen of the p-lactam.
Computer modeling also suggested that the molecular basis for the high
enantioselectivity is a severe steric clash between Ile189 in CAL-B and the phenyl
substituent on the slow-reacting enantiomer of the p-lactam.
Introduction
f3-Lactams, key structures in f3-1actam antibiotics, are also important synthetic
intennediates for f3-amino acids,t short peptide segments,2 natural products,3 and
1 a) Fülop, F. Chem. Rev. 2001, 101, 2181-2204; Fülop, F. In Studies in Natural Product
Chemistry Vol. 22, Atta-ur-Rahman, (Ed.) Elsevier Science Publishers, 2000, pp 273-
96

heterocyc1es.4 For this reason, many researchers are searching for good enantioselective
routes to p-Iactams.5 Although lipase-catalyzed reactions are often a good
enantioselective route to many chiral intermediates,6 lipases and esterases do not usually
catalyze the ring opening of the p-Iactams. In contrast, p-Iactams and p-Iactones inhibit
serine hydrolases by forming a stable acyl-enzyme intermediate. The natural protein
target of p-Iactam antibiotics is the serine transpeptidase and possibly other penicillin
binding proteins, which catalyzes the last step in bacterial cell wall biosynthesis.
Similarly, medicinal chemists used p-Iactams as mechanism-based inhibitors of serine
proteases7 and p-Iactones as mechanism-based inhibitors of lipases, which also have a
nuclleophilic serine. 8
306. b) Sonnet, P.; Dallemagne, P.; Guillon, J.; Enguehard, C.; Stiebing, S.; Tanguy, J.;
Bureau, R.; Rault, S.; Auvray, P.; Moslemi, S.; Sourdaine, P.; Séralini, G-E. Bioorg.
Med. Chem. 2000, 8, 945-955. c) Palomo, C.; Aizpurua, J.M.; Ganboa, 1.; Oiarbide, M.
Synlett 2001,12, 1813-1826.
2 a) Palomo, c.; Oiarbide, M.; Landa, A.; Esnal, A.; Linden, A. J. Org. Chem. 2001,66,
4180-4186. b) Palomo, c.; Ganboa, 1.; Oiarbide, M.; Sciano, G. T.; Miranda, J. L.
Arkivoc 2002, 5, 8-16; http://www.arkat-usa.org/ark/joumaI12002/Mmanas?MM-334C/
MM-334C.pdf.
3 Lee, H. K.; Chun, J. S.; Park, C. S. Tetrahedron Lett. 2001,42,3483-3486.
4 a) Cabell, L. A.; McMurray, J. S. Tetrahedron Lett. 2002,43,2491-2493. b) Alcaide, B.;
Almendros, P.; Alonso, J. M.; Aly, M. F. J. Org. Chem. 2001, 66, 1351-1358.
5 Review: Magriotis, P. A. Angew. Chem. Intl. Ed. 2001, 40, 4377-4379; Palomo, C.;
Aizpurua, J. M.; Ganboa, 1.; Oiarbide, M. Eur. J. Org. Chem. 1999,3223-3235.
6 Bomscheuer, U. T.; Kazlauskas, R. J. Hydrolases in Organic Synthesis Regio and
Enantioselective Reactions, Wiley-VCH, 1999.
7 Example: W. B. Knight, B. G. Green, R. M. Chabin, P. Gale, A. L. Maycock, H.
Weston, D. W. Kuo, W. M. Westler, C. P. Dom, Finke, P. E.; Hagmann, W. K.; Hale, J.
J.; Liesch, J.; MacCoss, M.; Navia, M. A.; Shah, S. K.; Underwood, D.; Doherty, J. B.
97

However, researchers can use lipase-catalyzed enantioselective reactions with
pendant groups to resolve ~-lactams. For example, lipase from Pseudomonas cepacia
catalyzes the highly enantioselective acylation of the N-hydroxymethylated ~-lactams
even though the reaction site (the hydroxyl group) is far from the stereocenter, eq. 1.9,10
In a related approach, Achilles et al. resolved ~-lactams by chymotrypsin-catalyzed
hydrolysis of the ester in a pendant N-CH2COOEt group. 1 1
Equation 1
Biochemistry 1992,31,8160-8170; Wilmouth, R. c.; Kassamally, S.; Westwood, N. J.;
Sheppard, R. J.; Claridge, T. D.; Aplin, R. T.; Wright, P. A; Pritchard, G. J.; Shofield,
C. J. Biochemistry 1999, 38, 7989-7998.
8 Tetrahydrolipstatin: Lüthi-Peng, Q.; Marki, H. P.; Hadvâry, P. FEBS Lett. 1992, 299,
111-115; LDL phopholipase A2: Tew, D. G.; Boyd, H. F.; Ashmman, S.; Theobald, C.;
Leach, C. A Biochemistry 1998, 37, 10087-10093.
9 Nagai, H.; Shiozawa, T.; Achiwa, K.; Terao, Y. Chem. Pharm. Bull. 1993,41, 1933-
1938.
10 a) Csomos, P.; Kanerva, L. T.; Bemâth, G.; FülOp, F. Tetrahedron: Asymmetry 1996,7,
1789-1796. b) Kâmân, J.; Forro, E.; Fülop, F. Tetrahedron: Asymmetry 2000, 11, 1593-
1600. c) FülOp, F.; Palk6, M.; Kaman, J.; Lazar, L.; Sil1anpaa, R. Tetrahedron: Asymmetry
2000,11,4179-4187. d) Forro, E.; Ârva, J.; Fülop, F. Tetrahedron: Asymmetry 2001,12,
643 .. 649. e) Forro, E.; FülOp, F. Tetrahedron: Asymmetry 2001,12,2351-2358.
11 Achilles, K.; Schirmeister, T.; Otto, H-H. Arch. Pharm. Pharm. Med. Chem. 2000,333,
243--253.
98

Surprisingly, two groups reported lipase- or esterase-catalyzed ring opening of p
lactams. Jones and Page reported the ring opening of the p-Iactam in benzylpenicillin
catalyzed by pig liver esterase. 12 Adam et al. recently reported the direct ring opening of
a-methylene p-Iactams by lipase B from Candida antarctica. The reactions were slow,
but highly enantioselective (E usually > 100).13 This p-Iactam ring is unusual due to the
a-methylene group, which increases ring strain, flattens the conformation of the ring and
withdraws electrons from the carbonyl group.
In this paper, we report similar enantioselective ring opening of p-Iactams, but for
the unstrained normal p-Iactams: the bicyclic (±)-1 and (±)-2 and 4-arylsubstituted p
lactams (±)-3 and (±)-4, Scheme 1. Ring opening yields the ring-opened p-amino acids
la - 4a and unreacted p-lactam enantiomers 1 - 4. Change of nucleophiles, alcohols,
causes dramatic changes in reaction rate. We used modeling to propose a possible
transition state for this unusual reaction and propose a molecular basis for the
enantioselectivity.
R"" )(NH o
(±)-1 - (±)-4
CAL-B R /'-.. R'OH/H20 ~ NH2 +
COOH
(1 R,25)-1a, 2a (R)-3a,4a
R"" )(NH o
(1S,6R)-1,2 (5)-3,4
4-MeP~
yNH o
(R)-4
Scheme 1. Enantioselective ring opening of p-Iactams yields a p-amino acid and
unreacted p-Iactam. The initial product may be the p-amino acid ester, but this ester
12 Jones, M.; Page, M. J. J. Chem. Soc., Chem. Commun. 1991,316-317.
13 Adam, W.; Oroer, P.; Rumpf, R-V.; Saha-M6ller, C. R. J. Org. Chem. 2000,65,4919-
4922; Another ring opening reaction, but for five-membered y-Iactams, is the subtilisin
catalyzed hydrolysis of two N-substituted bicyclic y-Iactams: Mahmoudian, M.; Lowdon,
A.; Jones, M.; Dawson, M.; Wallis, C. Tetrahedron: Asymmetry 1999, 10,1201-1206.
99

hydrolyzes to the acid under the reaction conditions. The structures show the fast reacting
enantiomers.
Results
CAL-B-catalyzed ring opening of fJ-lactams
1,2-Dipolar cyc1oaddition of chlorosulfonyl isocyanate to 1,3- and 1,4-
cyc1ohexadiene, styrene, and 4-methylstyrene yielded the bicyc1ic p-Iactams (±)-1 and
(±)-2 and the 4-arylsubstituted p-lactams (±)-3 and (±)-4, Scheme 2. 14 Initial screening of
commercial lipases, esterases and proteases for their ability to catalyze hydrolysis of (±)-
1 re:vealed several that catalyzed the hydrolysis, but with low enantioselectivity.15 The
most promising hydrolase was lipase B from Candida antarctica (Novozym 435, CAL-B)
since it was slightly enantioselective (E = 5, Table 1, row 1). Changing the solvent from
water to toluene and using ethanol as the nuc1eophile increased the enantioselectivity (E
> 40, Table 1, row 2), but the conversion was approximately ten-fold lower.
14 a) Bestian, H.; Biener, H.; Clauss, K.; Heyn, H. Justus Liebigs Ann. Chem. 1968, 718,
94-100. b) Singh, R; Cooper, R D. G. Tetrahedron 1994,50, 12049-12064. c) Furet, P.;
Garcia-Echeverria, c.; Gay, B.; Scoepfer, J.; Zeller, M.; Rahuel, J. J. Med. Chem. 1999,
42, 2358-2363. d) Palomo, c.; Oiarbide, M.; Bindi, S. J. Org. Chem. 1998, 63, 2469-
2474.
15 Other hydrolases that catalyzed hydrolysis of (±)-1 inc1ude: lipase from wheat germ
(type l, Sigma), rennin from Mucor meihei (mucorpeptin, Fluka), protease from
Aspergillus oryzae (Type XIII, Sigma), bacterial proteinase (Fluka), protease subtilisin
Carlsberg (bacterial type VIII from Bacillus licheniformis, Sigma), cholesterol esterase
from bovine pancreas (Genzyme), a-chymotrypsin (Sigma), and subtilisin from Bacillus
licheniformis (Fluka).
100

5,6
7,8 Ar = Ph, 4-Me-Ph
~NH o
{±)-3, {±)-4
Scheme 2. Synthesis of racemic p-lactams.
Table 1. Conversion and enantioselectivity of CAL-B-catalyzed ring opening of (±)_la
CAL-B Solvent: R'OH (1:15, v/v)
Time Conv.b eesc ee d
Row p (mg mL-1
) (h) (%) (%) (%) 1 20 H20 72 36e 32 57 2 20 Toluene:CH3CH2OH 44 2 2 >95 3 20 Toluene:CH3(CH2)60H 44 14 16 >95 4 20 Toluene:CH3(CH2)110H 44 31 43 >95 5 20 Toluene:Ci)CCH2OH 44 16 18 >95 6 20 Toluene:(CH3)3COH 46 14 16 >95 7 20 Toluene:(CH3hCHOH 48 3 3 >95 8 20 Toluene:CH3(CH2)5CH(CH3)OH 44 34 48 >95 9 20 Toluene:C6H5CH(CH3)OH 46 20 24 >95 10 20 Toluene:(CICH2)2CHOH 44 5 1 19 11 20 Toluene:(BrCH2)(CH3CH2)CHOH 44 36 Rac Rac 12 20 Toluene:C6H5OH 44 No reaction 13 20 iPr20:CH3(CH2)5CH(CH3)OH 44 36 53 >95
14 30 iPr20:CH3(CH2)5CH(CH3)OH + 43 39 71 >95 Et3N 68 46 81 >95
15 10 iPr20:CH3(CH2)5CH(CH3)OH 43 15 17 >95 16 10 iPr20:CH3(CH2)5CH(CH3)OH 40t 28 36 >95 17 10 iPr20:(+)-CH3(CH2)5CH(CH3)OH 40t 28 37 >95 18 10 iPr20:( -)-CH3(CH2)5CH(CH3)OH 40t 28 37 >95
E
5 >40 >46 >60 >47 >45 >40 >63 >49 -1 1
>66 >73 >98 >46 >56 >56 >56
a 0.05 M substrate (approx 15 mg in 2 mL), 60 oc b Conversion was determined using intemal standard by comsuming the starting compound. C According to GC on a Chiralsil Dex CB column. d Ca1culated from the conversion and the enantiomeric purity of the remaining starting material, which were measured by GC. e Determined by IH-NMR. f At 70°C
101

We optimized this reaction first by varying the alcohol. Replacing ethanol with
longer chain alcohols (heptanol and dodecanol (rows 3 and 4)), 2,2,2-trichloroethanol
(row 5) or tert-butanol (row 6) increased the conversion while maintaining high
enantioselectivity (E> 45). Sorne secondary alcohols (2-octanol and I-phenyl-l-ethanol
(rows 7-9)) showed even higher conversions while maintaining high enantioselectivity (E
> 40). Two halogenated secondary alcohols ((rows 10 and 11) showed low
enantioselectivity, while no reaction occurred with phenol (row 12). We concluded that
2-octanol showed the best combination of enantioselectivity and reaction rate (row 8).
Later experiments in diisopropyl ether as the solvent showed that either enantiomer of
enantiopure 2-octanol gave the same rate and enantioselectivity as racemic 2-octanol (rows
16-1l8).
Next we optimized the solvent. The CAL-B-catalyzed alcoholysis of (±)-1 with 2-
octanol was very slow when toluene was replaced by acetonitrile, tetrahydrofuran or
dichloromethane (1-2% conversion after 24 h; data not shown), but the conversion was
marginally higher in diisopropyl ether (row 8 vs. 13) and we chose to continue our
studlies in diisopropyl ether. Although Parker et al. reported faster CAL-B-catalyzed ring
opening of 4-substitued oxazol-5( 4H)-ones upon addition of triethylamine,16 in our
reaction adding triethylamine had little effect on conversion (row 13-14).
Finally, we optimized the temperature of the reaction. CAL-B catalyzed
alcoholysis of (±)-1 with racemic 2-octanol at 35-50 oc was slower and much less
enantioselective (Table 2, rows 1-4). At 55-75 oC, the ring opening showed excellent
enantioselectivity, but dropped again at 80 oc. We chose 60 oC as the optimum
temperature. Ring opening of the other p-Iactams - (±)-2 - (±)-4 also showed excellent
enantioselectivity under these conditions.
16 Paker, M. C.; Brown, S. A.; Robertson, L.; Turner, N. J. Chem. Commun. 1998,2247-
2248.
102
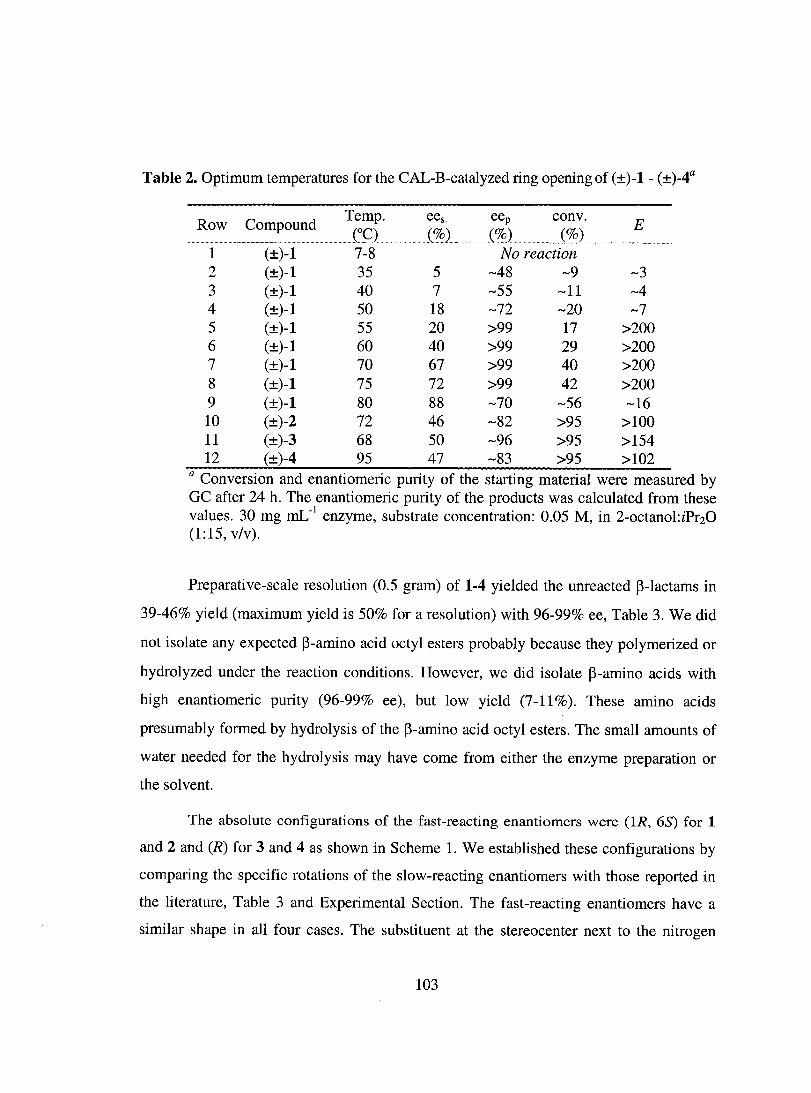
Table 2. Optimum temperatures for the CAL-B-catalyzed ring opening of (±)-1 - (±)_4a
Row Compound Temp. ees. eep conv.
E (OC) (%) (%) (%) 1 (±)-1 7-8 No reaction 2 (±)-1 35 5 -48 -9 -3 3 (±)-1 40 7 -55 -11 -4 4 (±)-1 50 18 -72 -20 -7 5 (±)-1 55 20 >99 17 >200 6 (±)-1 60 40 >99 29 >200 7 (±)-1 70 67 >99 40 >200 8 (±)-1 75 72 >99 42 >200 9 (±)-1 80 88 -70 -56 -16 10 (±)-2 72 46 -82 >95 >100 11 (±)-3 68 50 -96 >95 >154 12 (±)-4 95 47 -83 >95 >102
a Conversion and enantiomeric purity of the starting material were measured by GC after 24 h. The enantiomeric purity of the products was ca1culated from the se values. 30 mg mL- l enzyme, substrate concentration: 0.05 M, in 2-octanol:iPr20 (1:15, v/v).
Preparative-scale resolution (0.5 gram) of 1-4 yielded the unreacted p-Iactams in
39-46% yield (maximum yield is 50% for a resolution) with 96-99% ee, Table 3. We did
not isolate any expected p-amino acid oct yI esters probably because they polymerized or
hydrolyzed under the reaction conditions. However, we did isolate p-amino acids with
high enantiomeric purity (96-99% ee), but low yield (7-11%). These amino acids
presumably formed by hydrolysis of the p-amino acid oct yI esters. The smaIl amounts of
water needed for the hydrolysis may have come from either the enzyme preparation or
the solvent.
The absolute configurations of the fast-reacting enantiomers were (IR, 6S) for 1
and 2 and (R) for 3 and 4 as shown in Scheme 1. We established these configurations by
comparing the specific rotations of the slow-reacting enantiomers with those reported in
the literature, Table 3 and Experimental Section. The fast-reacting enantiomers have a
similar shape in aIl four cases. The substituent at the stereocenter next to the nitrogen
103

(position 6 for 1 and 2, position 4 for 3 and 4) points down in Scheme 1 in aIl four cases,
even though the sequence priority mIes yield a 6S configuration for 1 and 2 and a 4R
configuration for 3 and 4.
Table 3. Preparative-scale CAL-B-catalyzed ring openinga of (±)-1- (±)-4
B-Lactam recovered B-Amino acid produced Time Conv.
E (1- 4) (la - 4a)
(h) (%) Yield Isomer
eé [a]6'
Yield Isomer
eec
[a]6' (%) (%) (%) (%) (±)-l 44 50 >200 39 lS,6R 99 +161d 11 1R,2S 97 +120e
(±)-2 47 50 >200 42 lS,6R 99 -2gf 9 1R,2S 99 -398
(±)-3 20 50 >200 46 S 99 _139h 11 R 96 +6.8 i
(+)-41 48 50 >200 40 S 96 -12i 7 R 98 _8k
a 0.5 g substrate, 4.0 g immobilized CAL-B, 80 mL 2-octanol:iPr20 (1:15, v/v) at 60 oc. b Ac:cording to GC. C According to HPLC. d c = 0.29; CHCh. e c = 0.27; H20. f c = 0.26;
h . . k CHCh. 8 c = 0.5; H20. c = 0.19; EtOH. 1 c = 0.45; H20. ] c = 0.5; EtOH. c = 0.1; H2O.
Molecular modeling
To rationalize the reactivity and high enantioselectivity of CAL-B toward these B
lactams and the critical role of the a1cohol, we used computer modeling of the ring
opening step for B-Iactam 3. The lipase-catalyzed reaction of B-Iactams presumably
proc:eeds in two stages, Scheme 3. First, ring opening of the B-Iactam forms an acyl
enzyme intermediate and second, deacylation of the enzyme yields the product ester.
(This ester may undergo subsequent polymerization or hydrolysis by small amounts of
water in the reaction medium. Indeed, we isolated the hydrolysis product in low yield as
discussed above.) The first step invoives the B-Iactams, while the second step invoives
the more flexible B-amino acyl group. Since the less flexible B-Iactams would be more
difficult to accommodate in the active site, we hypothesized that enantioselectivity
originates in the ring-opening step and focused our computer modeling on this step.
104

~ 13-I~~~am ~NH2 dea~~~tion~NH2 opemng _
NH • ° 0, 's 105 R er HO-Ser105 °
° ° HO, 105 acyl enzyme Ser intermediate
Scheme 3. The CAL-B catalyzed transesterification of ~-lactams with alcohols proceeds
in two steps: ring opening of the ~-lactam followed by deacylation of the acyl enzyme
intermediate.
First, we identified a catalytically productive conformation of (R)-3, the fast
reac:ting enantiomer. We modeled a phosphonate as an analog of the tetrahedral
intermediate that results upon attack of the active site serine at the carbonyl of ~-lactam.
We define a catalytically productive conformation as one that maintains all the key
hydrogen bonds required for catalysis (see below) and avoids severe steric clashes with
the enzyme. It was difficult to find a catalytically productive conformation for the fast
reacting enantiomer of 3.
The tetrahedral intermediate for hydrolysis of (R)-( + )-3 (fast enantiomer) is
relatively rigid and can adopt only two conformations, which differ in the pucker of the
~-lactam ring, Scheme 4. Neither conformation contained all of the key hydrogen bonds,
Table 4. For conformation 1, the hydrogen bond between the N-H of Gln106 and the
phosphonyl oxygen was weak or missing. The N-Q distance is 3.36 Â, beyond the limit
for a hydrogen bond of -3.2 Â. More importantly, the phenyl substituent encountered
severe steric strain with Ile285. Because of the missing hydrogen bond and the severe
steric clash, conformation 1 cannot be catalytically productive.
105

""\ Glnl06
Serl05 C/H
~O-- p\--"O:::, d
b
~Ph ,N __ ~
+ .}.... ~ '\:"H_
H-N~H---b----NH,)~
~ (R)-3 '1.,,rJ"
His224 conformation 2
Scheme 4. Line diagrams of possible catalytically productive confonnations for the ring
opening of p-Iactam (R)-(+)-3 (fast enantiomer) catalyzed by CAL-B showing the
phosphonate analogs of the tetrahedral intennediates. a) Confonnation 1 orients the
lactam amide towards His224. Hydrogen bond 'c' (red) between the main chain amide N
H of Gln106 and the phosphonyl oxygen, which mimics the oxyanion of the tetrahedral
intermediate is weak or missing in this structure. In addition, the phenyl group encounters
severe steric strain with Ile285 (not shown) b) Confonnation 2 orients the amide away
from His224. Hydrogen bond 'b' (red) between His 224 NE-H and the p-Iactam nitrogen
is weak or missing. c) The substrate alcohol fonns a hydrogen-bonded bridge (green)
between His 224 NE-H and the lactam nitrogen of confonnation 2. (A sixth key hydrogen
bond present in all structures is between His 224 Nô-H and the carboxylate of Asp264.
For clarity, neither this hydrogen bond nor Asp264 are shown in the line diagrams.)
106

Table 4. Key hydrogen bonds in possible catalytically productive models of the CAL-B
catalyzed ring opening of the fast reacting enantiomer p-Iactam (R)-(+)-3.
Conformation 1
Conformation 2
Alcohol bridged
Alcohol bridged tautomer
Hydrogen H-bond distance, bondsa  (angle)b
a 3.02 (130°) b 3.19 (146°) c 3.36 (148°) d 2.73 (157°) e 2.74 (163°) a 2.81 (127°) b 3.37 (140°) c 2.79 (144°) d 2.80 (168°) e 2.74 (175°) a 2.82 (125°) b 3.70 (125°) c 2.82 (147°) d 2.81 (168°) e 2.75 (171°) f 2.96 (125°) g 3.25 (136°)
Comments
H-bond oc' is weak or missing. Shows severe steric strain between Ile285 and phenyl substituent of the p-Iactam. Unlikely to be a catalytically productive structure.
H-bond ob' is weak or missing.
H-bond ob' is missing, but may not be needed because 2-octanol forms a bridge of altemate hydrogen bonds, but H-bond 'g' is weak.
a 2.81 (126°) H-bond Ob' is missing, but may not b 3.76 (130°) be needed because 2-octanol forms c 2.77 (139°) a bridge of altemate hydrogen d 2.81 (168°) bonds. Bridging H-bonds 'f' and 'g' e 2.74 (173°) are stronger because 2-octanol is f 2.86 (125°) deprotonated and the p-Iactam g 3.00 (145°) nitrogen is protonated.
a Scheme 3 defines the hydrogen bonds. b Distance between non-hydrogen atoms (N-N, N-O or 0-0). Distances of 2.7 - 3.2 Â are consistent with a hydrogen bond. Angle refer to the N-H-O or similar angle. For an ideal hydrogen bond, this angle is 180°, but angle of >120° are consistent with a hydrogen bond.
Conformation 2 is also unlikely to be catalytically productive because the
hydrogen bond between His 224 NE-H and the p-lactam nitrogen is weak or missing. The
calculated N-N distance is 3.37 A, also beyond the limit of -3.2 A. Without this
hydrogen bond, the leaving group would be the highly basic RNIr, which is chemically
very unlikely. However, conformation 2 fit weIl in the active site of CAL-B and did not
encounter any steric clashes.
107

However, upon adding the co-substrate 2-octanol to the conformation-2 model,
we did find a catalytically productive conformation. The 2-octanol formed a hydrogen
bond bridge between the His 224 NE-H and the p-Iactam nitrogen and restored the
missing hydrogen bond (not shown). One hydrogen bond of this bridge was weak
(3.25Â), Table 4. This hydrogen bond strengthened (to 3.00 Â), when transferred the
proton from the 2-octanol (leaving the alkoxide) to the p-Iactam nitrogen creating a
protonated amine, Figure lc. This transfer of a proton strengthened the hydrogen bond
because it created complementary charges at each end of the hydrogen bond. Either the
alcohol-bridged structure or the alkoxide-bridged structure could be the catalytically
productive conformation of the fast-reacting enantiomer of 3. This complex is an
example of substrate-assisted catalysis.
108

Figure 1. Proposed tetrahedral intermediate analogs of the enantiomers of p-Iactam 3.
CAL-B is shown in space-filling representation, except for Ile189, which is shown as
stick representations. The tetrahedral intermediate analog for the p-Iactam is shown in
stick representation as is the 2-octanol in part a. a) The proposed productive conformation
for the fast-reacting enantiomer, (R)-3, corresponds to structure c in Scheme 3. The
phenyl ring points upward and toward the reader and makes good hydrophobic
interactions with Ile189, Ala141, Thr138. b) We did not identify any catalytically
productive structures for the slow-reacting enantiomer, (S)-3. The picture shows a non
energy minimized structure of the slow enantiomer in an orientation similar to that in part
a. Upon energy minimization, the phenyl ring distorts to an unrealistic non-pl anar
geometry due to severe steric clash with the side chains of Va1190 and Ile189.
109

Rationalizing the observed enantioselectivity was straightforward since the slow
reacting enantiomer, (S)-3, encountered severe steric clashes in both conformations. In
conformation 1, the phenyl substituent of the p-Iactam clashed with Ile189 and Leu278
(on the lower left side of the binding pocket in Figure 1), while in conformation 2, it
clashes with Ile189 and Va1190 (structure shown in Figure lb). Energy minimization of
either structure distorted the phenyl group to a non-planar geometry. On the other hand,
the fast-reacting enantiomer fit well in conformation 2 as discussed above and in Figure
la. The phenyl ring makes favorable hydrophobie contacts to Ile189 (3.3 Â between
carbon atoms), Ala14l (4.0 Â), and Thr138 (3.4 Â).
Discussion
CAL-B catalyzes the highly enantioselective (E > 200) nng openmg of
unactivated bicyclic and 4-arylsubstituted p-Iactams in diisopropyl ether at 60 oc. Adding
2-octanol to this reaction increased both the enantioselectivity and reaction rate. We
previously resolved the same p-Iactams using Pseudomonas-cepacia-lipase-catalyzed
butyrylation of the N-hydroxymethyl derivatives. Both preparative methods show high
enantioselectivity and favor the same enantiomer. The CAL-B catalyzed direct ring
opening does not require the addition and removal of the N-hydroxymethyl group, but
does require more enzyme because it is slower. Aiso a disadvantage was the low yield (7-
Il %) of the ring opening products, the p-amino acids la - 4a.
Our experimental results and computer modeling suggest that this ring opening of
p-Iactams 1 - 4 proceeds through an unusual transition state, which involves added 2-
octanol to form a hydrogen-bond bridge for catalysis. Other researchers also reported
substrate-assisted catalysis in subtilisin and even in the same lipase CAL-B. Carter
110

et al. 17 removed the catalytic histidine (His64) in subtilisin BPN' by site-directed
mutagenesis. The mutant had 105 -fold lower activity toward normal substrates, but only
ten-fold lower activity with a histidine-containing substrate. Apparently the histidine in
the substrate restores the missing hydrogen bond. In another example, Magnusson et al. 18
increased the enantioselectivity of CAL-B through substrate-assisted catalysis. The side
chain hydroxyl of Thr40 makes a key hydrogen bond in the transition state. Mutation
from Thr40 to Val decreased the reaction rate dramatically. However, for ethyl 2-
hydroxypropanoate, which contained a pendant hydroxyl group, the mutant showed
increased enantioselectivity (E = 1.6 to E = 22). Presumably, only one enantiomer can
restore the key hydrogen bond.
Since CAL-B also catalyzed the hydrolysis of p-Iactams, a water molecule can
also serve as a hydrogen-bond bridge. There are several well-characterized examples of
proton transfer via water bridges in proteins. 19 In our case the long chain a1cohols such as
octanol and dodecanol were more effective that short-chain a1cohols like ethanol
presumably because the longer chain a1cohols bind to the active site more strongly due to
their higher hydrophobicity. Kobayashi and coworkers also noted that addition of long
chain a1cohols (l-octanol in his case) increased the rate of ring opening polymerization of
P-butyrolactone (4-methyl-oxetan-2-one).20 Their increased rate may also be due to a
simüar a1cohol-assisted catalysis.
Although the proposed transition state is unusual, this ring-opening reaction is
slow and uncommon. The inability of most esterases and lipases to cleave p-Iactams is
17 Carter, P.; Abrahmsen, L.; Wells, J. A. Biochemistry, 1991,30,6142-6148.
18 Magnusson, A.; Huit, K.; Holmquist, M. J. Am. Chem. Soc. 2001,123,4354-4355.
19 Examples: carbonic anhydrase: Jude, K. M.; wright, S. K.; Tu, c.; Silverman, D. N.;
Violar, R. E.; Christianson, D. W. Biochemistry 2002, 41, 2458-2491; gramicidin:
Pomès, R. [sr. J. Chem. 1999, 39, 387-395; uracil DNA glycosylase: Luo, N.; Mehler,
E.:; Osman, R. Biochemisty 1999,38,9209-9220.
20 Uyama, H.; Suda, S.; Kikuchi, H.; Kobayashi, S. Chem. Lett. 1997, 1109-1110;
Kikuchi, H.; Uyama, H.; Kobayashi, S. Macromolecules 2000,33,8971-8975.
111

likely due to the difficulties in forming key hydrogen bonds as identified by our
modeling. Normally, p-Iactamases cleave p-Iactams using either a zinc hydrolase
mec:hanism (class B p-Iactamases) or modified serine hydrolase mechanisms (class A, C,
and D p-Iactamases). Even though the both serine esterases/lipases and serine
p-Iactamases have a serine as the nucleophile, their protein folds and catalytic machinery
differ. 21 Although bacteria have not developed resistance to p-Iactam antibiotics using
esterases, proteases, or lipases, it is possible that they may in the future since sorne of
these enzymes can catalyze cleavage of the p-Iactam ring.
The favored enantiomer in the CAL-B-catalyzed ring opening of other p-Iactams
and the related p-Iactones usually has a similar three-dimensional shape to the four p
lactams in this paper, Scheme 5. As discussed above, steric strain of the substituent with
the walls of the active site pocket account for the enantioselectivity. The
enantioselectivity is lowest for the methyl-substituted compound presumably because the
smaller steric stain caused by small methyl group. The isopropyl-substituted p-Iactam is
either an exception or a misassignment of the absolute configuration.22
21 Knox, J. R. ; Moews, P. C.; Frère, J. M. Chem. Biol. 1996,3,937-947; Maveyraud, L.;
Golemi, D.; Kotra, L. P.; Tranier, S.; Vakulenko, S.; Mobashery, S.; Samama, J.-P.
Structure 2000, 8, 1289-1298.
22 Adam, W.; Salgado, V. O. N.; Wegener, B.; Winterfeldt, E. Chem. Ber. 1993, 126,
1509-1510. The authors assigned the absolute configuration of this lactone on the basis
of the IH-NMR spectra of an adduct with an enantiomerically pure cyclopentadiene
derivative.
112

a 8
~NH o
R= Me, Et
==10 ~ R
yo o
b
yx{0 o ®
secondary alcohols and related amines
o o R = Me (E = 40) R = CF2CI (E >50)
R
çx o
3-substitued J3-lactones and J3-lactams
Scheme 5. Enantiopreference of CAL-B in the ring opening of ~-lactams and ~-lactones.
a) Examples of enantioselective CAL-B catalyzed ring opening of ~-lactams and ~
lactones. The structures show the fast reacting enantiomer. AlI but one ex ample have the
substituent pointing back. An asterisk marks the exception. b) Generalized structure for
the fast reacting enantiomer. The secondary a1cohol rule cannot be used for lactones
because the stereocenter lies in a different position. Acyclic esters adopt a syn
conformation along the carbonyl C-a1cohol-O-bond. The crystal structure of transition
state analogs bound to lipases suggest that this conformation persists in the active site. On
the other hand, the lactone ring forces an anti conformation along the carbonyl
C-alcohol-O-bond, which places the stereocenter in a different part of the enzyme. In
particular, the lactone stereocenter appears to lie entirely within the L-pocket of the
a1cohol-binding crevice. Indeed, many of the lactone examples in this section do not
folIow the secondary a1cohol rule.
Although the a1cohol portion of y- and b-Iactones is a secondary a1cohol, the
secondary a1cohol rule cannot be used here because the stereocenter lies in a different
position, Scheme 5. Indeed the molecular basis for enantioselectivity is likely different in
the two cases: steric strain in the acyl binding pocket for ~-lactones and ~-lactams, and
113

fit of the medium substituent in the stereospecificity pocket of the alcohols binding site
for secondary alcohols.23
Experimental Section
General. Esterase, lipase and protease were from Fluka, Sigma, while chemicals were
from Aldrich. Solvents were of the highest analytical grade. CAL-B immobilized on a
mac:roporous poly(acrylic) beads (Novozym 435) was from Novozymes AIS (Denmark).
1H-NMR spectra were run at 400 MHz in CDCh unless otherwise noted. Chemical shift
values, 8, are in ppm. The p-Iactams (±)-1 - (±)-4 were prepared by 1,2-dipolar
cycloaddition of chlorosulfonyl isocyanate to the corresponding cyclohexadiene, styrene and
4-rnethylstyrene.10e,14
Initial Screening. A mixture of enzyme (10 mg) and racemic p-Iactam (10 mg) in
potassium phosphate buffer (100 mM, pH 7, 1 mL) was stirred for 7 days at 25 oC. The
conversion was estimated by TLC analysis (CHCh:MeOH:AcOH:H20 = 70:20:8:2). Before
50% conversion, unreacted p-Iactam was extracted with ethyl acetate (3 mL). The ee of the
unreacted p-Iactam was determined by gas chromatography on a Chromopak Chiralsil-Dex
CB column (25 m x 0.25 mm, Raritan, NJ).
Typical Small-scale Experiment. A mixture of racemic p-Iactam (0.05 M solution) in an
organic solvent (2 mL), Novozyme 435 (10-75 mg mL-1), a1cohol (65 ~L mL-1
) and n
decane as an internaI standard (1 ~L) was stirred magnetically at the selected temperature.
The progress of the reaction was followed by gas chromatography on a Chromopak
23 Cygler, M.; Grochulski, P.; Kazlauskas, R. J.; Schrag, J. D.; Bouthillier, F.; Rubin, B.;
Serreqi, A. N.; Gupta, A. K. J. Am. Chem. Soc. 1994,116,3180-3186.
114

Chiralsil-Dex CB column (25 m x 0.25 mm, Raritan, NJ). The enantiomeric purity of the
remaining starting material was measured directly and the conversion was measured by
comparison to the internaI standard n-decane. The enantiomeric purity of the products (ring
opened ~-arnino acid derivatives) was calculated from the enantiomeric purity of the starting
materiai and the conversion?4
The enantiomeric purity of the isolated p-arnino acids was deterrnined by HPLC on
a reversed-phased (C18) column after derivatization of the sample with (lS,2S)-1,3-
diacetoxy-1-( 4-nitrophenyl)-2-propylisothiocyanate (DANI).25
Preparative-scale Resolution of 7-Azabicyclo[4.2.0]oct-4-en-8-one, (±)-1. Racemic 1
(0.500 g, 4.06 mmol) was dissolved in diisopropyl ether (80 mL). CAL-B (4 g, 50
mg mL-I) and 2-octanol (5.3 mL) were added and the mixture was shaken in an incubator
shaker at 60 oc for 44 h. The reaction was stopped by filtering off the enzyme at 50%
conversion (ee-1 = 99%). The soivent was evaporated and the residue was
chromatographed on silica eluted with ethyl acetate:hexane (7:3) yieiding unreacted
(lS,6R)-1 [0.19 g, 39%; [a]l] = +161 (c = 0.29; CHCh); mp 111-113 oC (recrystallized
from diisopropyl ether); ee 99%]. IH NMR 8: 1.63-2.11 (4H, m, 2xCHz), 3.51 (lH, m,
H-1), 4.01-4.04 (IH, m, H-2), 5.93-6.14 (2H, m, CRCIl) , 5.94 (lH, bs, NH). Analysis:
calculated for C7H9NO: C, 68.27; H, 7.37; N, 11.37; found: C, 67.99; H, 7.34; N, 11.28.
The fiitered enzyme was washed with distilled water (5xlO mL), and the water was
evaporated, yieiding the crystalline p-amino acid (lR,2S)-1a [65 mg, 11%; [a]l] = +120
(c =: 0.27; H20); mp 236-238 oC (recrystallized from water); lit lOb mp 220-221 oC; ee =
97%].
24 Footnote 17 in Chen, C. S., Fujimoto, Y., Girdaukas, G., Sih, C. J. J. Am. Chem. Soc.
1982,104,7294-7299.
25 Péter, M.; Péter, A.; Fülop, F. J. Chromatogr. A 2000,871,115-126.
115

IH NMR (D20 ) ô: 1.83-2.17 (4H, m, 2xCH2) 2.72-2.76 (IH, m, H-l) 3.99-4.01 (IH, m,
H-2) 5.72-6.13 (2H, m, CHCH). Analysis: calculated for C7HllN02: C, 59.56; H, 7.85;
N, 9.92; found: C, 59.44; H, 7.80; N, 9.79.
Preparative-scale Resolution of 7-Azabicyclo[4.2.0]oct-3-en-8-one, (±)-2. With the
procedure described above, the ring opening of racemic 2 (0.500 g, 4.06 mmol) with 2-
octanol (5.3 mL) in diisopropyl ether (80 mL) in the presence of CAL-B (4 g, 50
mgmL-I) at 60 oC for 47 h afforded unreacted (lS,6R)-2 [0.2 g, 42%; [a]6' = -29 (c =
0.26; CHCb); mp 152-153 oC (recrystallized from diisopropyl ether); ee = 99%]; IH
NMR 8 1.58-2.11 (4H, m, 2xCH2), 3.51 (lH, m, H-1), 4.01-4.04 (lH, m, H-2), 5.93-6.15
(2H, m, CHCH), 6.01 (lH, bs, NH). Analysis: calculated for C7H9NO: C, 68.27; H, 7.37;
N, 11.37; found: C, 68.12; H, 7.33; N, 11.37.and the amino acid (IR, 2S)-2a [55 mg, 9%;
[a]6' = -39 (c = 0.5, H20); lit lOb [a]6' = -36.2 (c = 0.5, H20); mp 233-235 oC
(recrystallized from water/acetone); lit lOb mp 224-225 oC; ee = 99%]; IH NMR (D20) 8
2.23-2.51 (4H, m, 2xCH2) 2.74-2.78 (lH, m, H-1) 3.76-3.79 (lH, m, H-2) 5.63-5.83 (2H,
m, CHCH). Analysis: calculated for C7HllN02: C, 59.56; H, 7.85; N, 9.92; found: C,
59.51; H, 7.69; N, 9.89.
Preparative-scale Resolution of 4-Phenyl-2-azetidinone, (±)-3. With the procedure
desc:ribed above, the ring opening of racemic 3 (0.500 g, 3.39 mmol) with 2-octanol
(4.6 mL) in diisopropyl ether (70 mL) in the presence of CAL-B (3.5 g, 50 mg mL-I) at
60 oC for 20 h afforded unreacted (S)-3 [0.23 g, 46%; [a]6' = -139 (c = 0.19; EtOH); mp
114 oC (recrystallized from diisopropyl ether); ee = 99%]; IH NMR 8: 2.85-2.89 (lH, dd,
J = 2, 14.8, CHAH) 3.41-3.47 (lH, ddd, J = 2.4; 5.2; 7.6, CHBH), 4.71-4.73 (lH, dd, J = 2.5; 5.3, CH), 6.27 (lH, bs, NH) 7.26-7.40 (5H, m, Ph). Analysis: calculated for
C9H9NO: C, 73.45; H, 6.16; N, 9.52; found: C, 73.23; H, 6.41; N, 9.62.and the amine
acid (R)-3a [59 mg, Il %; [a]6' = +6.8 (c = 0.45, H20); lit26 for (R)-~-phenyl-~-alanine
26 Soloshonok, V. A.; Fokina, N. A.; Rybakova, A. V.; Shiskina, 1. P.; Galushko, S. V.;
116

[an? = +6.5 (c = 0.9; H20); mp 233-235 oc (recrystallized from water/acetone); IH NMR
(D20) ô 2.84-2.91 (2H, m, CH2) 4.65-4.66 (IH, m, CHNH2) 7.46-7.50 (5H, m, Ph).
Analysis: ca1culated for C9HllN02: C, 65.44; H, 6.71; N, 8.48; found: C, 65.23; H, 6.72;
N,8.33.
Preparative-scale Resolution of 4-(p-tolyl)-2-Azetidinone, (±)-4. With the procedure
described above, the ring opening of racemic 4 (0.500 g, 3.1 mmol) with 2-octanol (4 mL)
in diisopropyl ether (60 mL) in the presence of CAL-B (3 g, 50 mg mL-1) at 60 oc for 48
h afforded unreacted (S)-4 [0.2 g, 40%; [a]6' = -121.9 (c = 0.5; EtOH); mp 56 oc
(recrystallized from diisopropyl ether); ee 96%]; IH NMR ô: 2.35 (3H, s, CH3) 2.83-2.88
(IH, dd, J = 1.7; 14.8, CHAH) 3.39-3.44 (lH, ddd, J = 2.4; 5.2; 7.6, CHBH) , 4.68-4.69
(lH, dd, J = 2.3; 5.2, CH), 6.12 (lH, bs, NH) 7.17-7.27 (4H, m, Ph). Analysis: ca1culated
for ClQHllNO: C, 74.51; H, 6.88; N, 8.69; found: C, 74.62; H, 6.89; N, 8.66 and the
amino acid (R)-4a [39 mg, 7%; [a]6' = -8 (c = 0.1, H20); mp 241-243 oC (recrystallized
from water/acetone); ee = 98%]; IH NMR ô: 2.35 (3H, s, CH3) 2.82-2.89 (2H, m, CH2)
4.60-4.64 (IH, m, CHNH2) 7.31-7.37 (4H, m, Ph). Analysis: calculated for ClQH13N02:
C, 67.02; H, 7.31; N, 7.82; found: C, 66.89; H, 7.22; N, 7.77.
Computer Modeling of Transition State Analogues in CAL-B. All modeling was done
with Discover, version 2.9.7 (Accelrys, San Diego, CA) using the AMBER27 force field.
Results were displayed using Insight II version 95.0 (Accelrys). The starting structure
was the x-ray crystal structure of CAL-B containing a covalently linked phosphonate
inhibitor (Protein Data Bank28 file llbs). Using the biopolymer module of Insight II,
Sorochinsky, A E.; Kukhar, V. P. Tetrahedron: Asymmetry 1995,5, 1601-1610.
27 Weiner, S. J.; Kollman P.A; Case, D. A; Singh, U. c.; Ghio, c.; Alagona, G.; Profeta,
S. Jr.; Weiner, P. A J. Am. Chem. Soc. 1984,106,765-784.
28 Berman, H. M.; Westbrook, J.; Feng, Z.; G. Gilliland, G.; Bhat, T. N.; Weissig, H.;
Shindyalov, 1. N.; Boume, P. E. Nucleic Acids Res. 2000, 28, 235-242;
117

hydrogen atoms were added to correspond to pH 7.0. Histidines were uncharged,
aspartateds and glutamates were negatively charged and arginines and lysines were
positively charged. The catalytic histidine (His) was protonated. The phosphonate group,
which covalently linked to Ser105 in the x-ray structure, was replaced by a phosphonate
analog of ~-lactam 3.
Energy minimization proceeded in four stages. First, 100 iterations of steepest
descent algorithm, aIl protein atoms constrained with a force constant of 10 kcal morl
k 2; second, 500 iterations of conjugate gradients algorithm with the same constraints;
and third, 500 iterations of conjugate gradients algorithm with only the backbone
constrained by a 10 kcal morl k 2 force constant. For the fourth stage, minimization was
continued using conjugate gradients algorithm without any constraints until the rms
derivatives reached less than 0.005 kcal morl k l. CrystaIlographic water molecules were
included in aIl minimizations. Water molecules and the substrate were not constrained
through any of the minimization cycles.
http://www .rcsb.org/pdb/
118

Chapter 5
Although enzymes are highly reglO- and enantioselective toward natural
substrates, they sometimes are not selective enough toward unnatural substrates. Medium
and substrate engineering can sometimes increase the selectivity of such reactions, but in
other cases, one needs to use protein engineering.
ln this chapter, we apply protein engineering to increase enantioselectivity of PFE
(Pseudomonas fluorescens esterase) toward an important building block (i.e., methyl 3-
bromo-2-methylpropionate). We developed an efficient protein tailoring strategy to
improve enantioselectivity of a hydrolase by a combination of rational design and
directed evolution. In addition, to rationalize high enantioselectivity of mutants
discovered, we designed and tested a series of substrate analogues.
Contributions
This work was done under supervision of Dr. Romas J. Kazlauskas and
collaboration with Geoffrey P. Horsman and Krista Morley. Geoffrey P. Horsman chose
the amino acid residues for mutagenesis and Krista Morley measured the end-point E
value of compound 2. 1 performed mutagenesis work, optimizing a docking model,
designed the substrate analogues, determined end-point E values, and measured kinetic
parameters.
119

Chapter 5. Discovery and Molecular Basis of
Enantioselectivity of Val122Ser Mutant of PFE toward
MethyI3-Bromo-2-methylpropionate
Abstract: A mutant of an esterase from Pseudomonas fluorescens (PFE) , in which the
Va1122 is replaced by Ser (Va1122Ser), greatly enhances the enantioselectivity (E = 61
from 12 of wild type enzyme) towards methyl (S)-3-bromo-2-methylpropionate. The
mutant libraries were generated by structure-guided saturation mutagenesis in substrate
binding region (Va1122 and Va1226) in active site of PFE. The substrate-binding region
was identified by docking the substrate into a homology model of PFE. Quick E high
throughput screening identified the more enantioselective mutants. In order to rationalize
the high enantioselectivity of Va1122Ser mutant, we investigated the effect of changing
the shape or electronic character of substrate on the enantioselectivity of wild type and
Va1l22Ser mutant enzyme. The origin of the improved enantioselecti vit Y is the
combination of substituent size effect of substrates and electronic effect of their
functional groups. The overall energy change (~~~G:j: = 0.96 kcal/mol) between wild
type: and V122Ser mutant enzyme is the sum of electronic effect (0.65 kcal/mol) and
substituent size effects (0.26-0.36 kcal/mol).
Introduction
Since biocatalysts can perform organic reactions with high stereo- and
regioselectivity under mild condition, their application in both industrial, especially
pharmaceutical and agrochemical, and academic research has increased dramatically.l
1 a) Liese, A.; Filho, M. V. Curr. Opin. Biotechnol. 1999, 10, 595-603. b) Schmid, A.;
Dordick, J. S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Nature 2001, 409,258-
120

Although biocatalysts are useful to produce enantiopure compounds under the mild
conditions, the natural enzymes are not always suited to this task. This limitation has
driven protein engineering for the last few decades.z The most popular methods of protein
engineering to improve the enzyme enantioselectivity are 1) rational modification by site
directed mutagenesis based on crystal structure and 2) directed evolution by error prone
PCR, mutator strain, or DNA shuffling.
Rational protein design requires detailed structural and mechanistic understanding
to choose the mutations.3 Although the rational approach can avoid the need for huge
screening, it is not always successful due to unpredictable structural changes from site
directed mutagenesis. For example, the HuIt group reported a rational approach to
increase enantioselectivity of lipase B from Candida antarctica towards bromo- or
chlorohydrin.4 On the basis of a crystal structure and molecular modeling, the a1cohol
binding region was identified as was the possible binding pocket for the bromo or chloro
group of the fast-reacting enantiomer. In this binding pocket, there are four hydrophilic
amino acids such as Thr40, Ser47, Thr42, and Trp104. They proposed those hydrophilic
residues would make repulsive interaction because of partial negative charge on bromo or
chloro group. One of mutants, Ser47 Ala, showed doubly increased enantioselectivity
towards 1-bromo-2-octanol or 1-chloro-2-octanol. But another mutant for Trp104His
showed only ~30% enantioselectivity of wild type enzyme. This suggests that an
unexpected structural change may have occurred.
268. c) McCoy, M. Chem. Eng. News 1999, 77,10-14.
2 Taylor, S.; Kast, P.; Hilvert, D. Angew. Chem., [nt. Ed. Engl. 2001,40,3310-3335.
3 a) Holmquist, M.; Martinelle, P.; Berglund, P.; Clausen, 1. G.; Patkar, S.; Svensden, A.;
HuIt, K. J. Protein Chem. 1993, 11, 749-757. b) Scheib, H.; Pleiss, J.; Stadler, P.;
Kovac, A.; Potthoff, P.; Haa1ck, L.; Spener, F.; Paltauf, F.; Schmid, R. D. Protein Eng.
1998, Il,675-682.
4 Rotticci, D.; Rotticci-Mulder, J. C.; Denman, S.; Norin, T.; HuIt, K. ChemBioChem
2001,2, 766-770.
121

Although directed evolution (recursive generation and screening of mutants)
requires no structural and mechanistic information, and may reveal new discrimination
mechanisms, it requires extensive screening of mutants. For example, if the enzyme
contains 300 amino acids, one amino acid substitution creates 5000 mutants.5 The Reetz
group reported the first example of increased enantioselectivity by directed evolution.6
The wild type of a lipase from Pseudomonas aeruginosa (PAL) has E = 1.1 towards
hydrolysis of p-nitrophenyl 2-methyldecanoate. They improved enantioselectivity to II.3
after four rounds of random mutagenesis using the error prone PCR and screening 1000-
2400 colonies per each round (total 5600 colonies). Later they did additional two more
generations of random mutagenesis and identified a mutant having E = 13.5. The
combination of saturation mutagenesis and random mutation gave better result such as E
= 25.8. On the basis of this result they proposed the region of loops, which is involved
the conformational change from c10sed to open structure, may be important for the
enantioselectivity. In another approach (combination of cassette mutagenesis and DNA
shuffling), they reported the increased enantioselectivity of up to 51 after screening
40,000 colonies.7
On the other hand, when a crystal structure of a enzyme is available, a hybrid
approach can be applied to improve enzymatic properties. First, a small number of
residues can be selected for rational design using computer modeling. Second, random
5 The number of possible mutants can be calculated by the following equation:
P = (l9M·300!)/((300-M)!·M! where P: the number of possible mutant, M: the number of
substitution of amino acid. Moore, J. C.; Jin, H.-M.; Kuchner, O.; Arnold, F. H. J. Mol.
Biol. 1997,272,336-347.
6 a) Reetz, M. T.; Zonta, A.; Schimossek, K; Liebeton, K; Jaeger, K-E. Angew. Chem.,
Int. Ed. Engl. 1997, 36, 2830-2832. b) Liebeton, K; Zonta, A.; Schimossek, K;
Nardini, M.; Lang, D.; Dijkstra, B. W. ; Reetz, M. T. ; Jaeger, K-E. Chem. Biol. 2000,
7,709-718.
7 Reetz, M. T.; Wilensek, S.; Zha, D.; Jaeger, K-E. Angew. Chem., Int. Ed. Engl. 2001,
40,3589-3591.
122

mutagenesis can be applied to fix unexpected structural change of the enzyme or to
improve additional enzymatic properties. However, only a few examples have been
reported,8 and no examples of enhancing enantioselectivity. In an example, Copinus
einereus heme peroxidase used as a dye-transfer inhibitor in laundry detergent was made
more stable to washing machine conditions using a combination of rational design and
directed evolution. Using computer modeling of crystal structure, Cherry et al. selected
several sites for site-directed mutagenesis in order to remove oxidizable residues around
the active site and to reduce a potentially destabilizing interaction between two
structurally adjacent glutamic acid side chains. Subsequently, applying random
mutagenesis (e.g., error-prone PCR and gene shuffling in vivo) improved additional
thermal and oxidative stability of the peroxidase.
Although a combinational approach of rational design and directed evolution
would be successful for enhancing enantioselectivity of enzymes, detailed computer
modeling and enormous screening effort are still required. However, to select a small
number of amino acid residues around substrate binding region for random mutagenesis,
using approximate structural information, which can be easily accessed by a homology
model generated from amino acid sequence9, would be the most efficient combination of
both approaches since the detailed molecular modeling and extensive screening can be
avoided.
In this paper, we describe an efficient strategy to generate diverse mutant enzyme
with assistance of a homology model from one-dimensional structural information (i.e.,
amino acid sequence). We selected mutation sites that are close to the substrate binding
region in the active site of PFE. From this information, we generated mutant libraries by
site··saturation mutagenesis and identified enantioselective mutants through relatively
small scale of screening. To rationalize dramatic enantioselectivity change of mutants, we
8 a) Cherry, J. R.; Lamsa, M. H.; Schneider, P.; Vind, J.; Svendson, A.; Jones, A.;
Pedersen, A. H. Nat. Bioteehnol. 1999, 17, 379-384. b) Akanuma, S.; Yamagishi, A.;
Tanaka, N.; Oshima, T. Eur. J. Bioehem. 1999,260,499-504.
9 Guex, N.; Diemand, A.; Peitsch, M. C. Trends Bioehem. Sei. 1999,24,364-367.
123

tested a series of substrate analogues and determined kinetic parameters for the fast
reacting enantiomer of MBMP (methyl 3-bromo-2-methylpropionate).
Results
Target Sites of PFE for Random Mutagenesis. A doc king model of the first tetrahedral
intermediate of PFE homology model with the slow-reacting enantiomer of MBMP
suggested that Va1122 and Val226 are closely located to the acyl group of substrate
binding region and may affect the enantioselectivity of PFE. Val226 is also close to
Asp223, which is part of the catalytic triad and essential amino for catalysis. The amide
proton of Val226 hydrogen-bonds to one of 08 atoms of Asp223. This hydrogen bond
may be essential to orient Asp223 correctly for catalysis. Va1122 has no special
interaction with any catalytic amino acids. However, these suggestions are speculative
since the docking model is based on a homology model.
Saturation Mutagenesis and Hydrolase Activity of Mutant Library. Saturation
mutagenesis was performed by QuickChange® Site-Directed Mutagenesis10 with
complementary primers containing a degenerate codon (NNK)l1. The theoretical fraction
of possible codons was determined by Warren and Bankovic's equation. 12 We screened
192 colonies, which represents 99.8% of the possible 32 codons (corresponding to an
amino acid possibilities) assuming that mutation rate is 100%. One hundred and ninety
10 Papworth, c.; Braman, J.; Wright, D. A. Strategies 1996, 9, 3-4.
Il The degenerate primer is a mixture of 32 primers, each being a permutation of the
degenerate codon NNK. N: a mixture of an for nUcleotides; K: a mixture of guanosine
(G) and thymidine (T); M: a mixture of cytidine (C) and adenosine (A).
12 Mn(1-lIn) = In(1-P), where N = the theoretical number of colonies, n = each possible
new codon and P = probability: Warren, M. S.; Benkovic, S. J. Protein Eng. 1997, 10,
63-68.
124

two transformant colonies from saturation mutagenesis procedure were picked for protein
expression in a 96-well assay block, subsequently screened towards pNPAc (p
nitrophenyl acetate) to identify mutants having hydrolase activity, and then measured
hydrolysis of MBMP.
The Va1122 library contained a larger fraction of mutants active toward pNPAc
than the Val226 library. The active mutants were 55% in Va1122 library and 35% in
Val226 library (Figure 1). Similarly, the fraction of the active mutants toward hydrolysis
of MBMP in Val226library was half of that in Va1122library: 15% in Val226library and
32% in Va1122 library. The enantioselectivity of the active mutants toward MBMP was
determined by Quick el3.
Va/122 Library
Activity toward pNPAc Active
Activity toward MBMP Active 1
QuickE QE>50 1 Ser, Met, other
Va/226 Library
Activity toward pNPAc Active
Activity toward MBMP Active l QuickE 20> QE > 12 1
lIe,other
0% 25%
1
1 Inactive
Inactive
50> QE > 12 1
Leu,other
Inactive
Inactive
12> QE
Ser, Ala, other
50%
Fraction of Mutant
75%
12> QE
100%
Figure 1. Fraction of active mutants in libraries of PFE. The hydrolase activity was
measured by the hydrolysis rate towards pNPAc. Then the mutants active toward pNPAc
13 a) Janes, L. E.; Kazlauskas, R. J. J. Org. Chem. 1997, 62, 4560-4561. b) Janes, L. E.;
Lowendahl, A. c.; Kazlauskas, R. J. Chem. Eur. 1. 1998,4, 2317-2324. c) Janes, L. E.;
Cimpoia, A.; Kazlauskas, R. J. J. Org. Chem. 1999,64,9019-9029.
125

were selected to determine Quick Evalues toward hydrolysis of MBMP. Active: more
than 9.90xlO's ~mol/min, inactive: less than 9.90xlO's ~mol/min under screening
condition. Data are for cell free extracts of PFE colonies. Control experiments showed
that DH5a without PFE gene did not pro duce hydrolases and had no activity toward
pNPAc.
Emmtioselectivity of Mutants. Quick E screening of each mutant library revealed
enantioselective mutants. The highest and lowest Quick E values in the Val226 library
were 18 and 8, respectively. The enantioselectivity of mutants in the Val226 library is not
varied, but similar to the value (i.e., 12) of wild type PFE. In the library of Va1122, we
found much more enantioselective mutants (Quick E > 50) than wild type. The DNA
sequencing revealed Va1122Ser and Va1122Met.
To confirm the enantioselectivities found by Quick E screening, endpoint E
determination14 was carried out for Va1122Ser and Va1122Met (see Table 1). The most
enantioselective mutant, Va1122Ser, has E = 61, which is increased by a factor of 5
without loosing activity towards the fast-reacting enantiomer (see Table 3 below). The
enantioselectivity of the other mutant (Va1122Met) was 36.
Table 1. Summary of mutants discovered from saturation mutagenesis at Va1122 and
Va1226.
PFEEnzyme Numbera
Wild-type Va1122Ser V122-4
V122-36 V122-69
V122-128 Val 122Met V122-80
a Refers to the microwell position of the mutant. b n.d. = not determined
Codon Endpoint E 12
TCT 61 TCT n.d.b
TCG n.d. TCG n.d. ATG 36
14 Chen, C. S.; Fujimoto, Y.; Girdaukas, G.; Sih, C. J. J. Am. Chem. Soc. 1982, 104,
126

Kinetic Parameters of Va1122Ser toward the Fast-reacting Enantiomer of MBMP.
The efficiency of mutant enzymes as a synthetic catalyst can be estimated by the specifie
activity and kinetic parameters of the fast-reacting enantiomer15 of MBMP (Table 2). The
mutant Va1122Ser is more active (122%) than wild type while the mutant Va1122Met is
less active (74% activity of wild type). While both mutants have lower KM values (22
mM for both) than wild type (27 mM), Va1122Ser has higher kcat value (1.44 x 10-2 min-1)
than wild type (1.34 x 10-2 min-1) but Va1122Met has lower kcat value (0.81x 10-2 min-1
).
With using Va1122Ser mutant to produce the same amount of the fast-reacting
enantiomer of MBMP, one needs 22% less reaction time or 20% less amount of enzyme.
In addition, this suggests that the mutation hinders the slow-reacting enantiomer. For
example, becauseof the mutation of Va1122Ser increased the enantioselectivity by a
factor of 5 and the specificity constant for the fast-reacting enantiomer by a factor of 1.3,
it decreased the specificity constant of the slow-reacting enantiomer by a factor of 3.8. 16
7294-7299.
15 The kinetic parameters for slow-reacting enantiomer could not be measured since a
range of concentration for the reliable reaction rates were way below KM.
16 E can be defined by the ratio of the specificity constants Ckcat/KM) for both enantiomers.
Faber, K. Biotransformations in Organic Chemistry, 4th ed.; Springer-Verlag: Berlin,
Germany, 2000, p 40.
127

Table 2. The specific activity of purified enzyme and kinetic parameters for the fast
reacting enantiomer from noncompetitive measurementa
Specific Relative kcat x 102c kcaJKM
Enzyme Activitl specific KM (mM)C (min-1
) (min-1 M-1) (~mol/min/mg) activitl
Wild type 1.59 ± 0.08 1 27 1.34 0.49 Va1122Ser 1.94 ± 0.01 1.22 22 1.44 0.65 Val 122Met 1.18 ± 0.04 0.74 22 0.81 0.37
a AllI measurements were performed four times under the following conditions: in a reaction well, 7% MeCN; 4.65 mM BES buffer (pH 7.2); 0.29 mM Triton-X100; 3.13 ~g/rnL enzyme; total volume, 100 ~L; enzyme volume, 10 ~L; 0.42 mM pNP; 25 oc b Enrors are reported as standard deviations. Substrate concentration: 1.15 mM C Errors are within 10%. Substrate concentration: 0.23-5.75 mM
Origin of enantioselectivity of Va1122Ser mutant. The mutation Va1122Ser changes
the size and the electronic characteristics of the residue because serine is smaller17 than
valine and has different electronic character (HO- vs. CH3). To rationalize the mutational
effect on the high enantioselectivity of Va1122Ser mutant toward MBMP, we designed a
series of substrate analogues, Chart 1. Enantioselectivity may originate from two main
features: electronic effects (e.g., hydrogen bonding or electronic repulsion) and
substituent size recognition by attraction or repulsion. MBMP (i.e., Compound 1) has two
different substituents: bromomethyl and methyl. Bromomethyl and methyl group have
different sizes as well as different electronic characteristics. To distinguish the effects
from these features (i.e., size and electronic characteristic), the substrate analogues were
designed. The substitution of bromo by methyl group of MBMP gives compound 2,
which has same electronic characteristic on the substituents but different sizes. The
enantioselectivity toward compound 2 reveals the substituent size effect on altering the
enantioselectivity. On the other hand, compound 3 and 5 were designed to identify the
(kcaJKM) fast E = ---'=------"';........0.:;_::;:..
(kcat / KM) slow
17 van der Waals volume, Val: 138 A3; Ser: 94 A3
. Tsai, J.; Taylor, R.; Chothia, C.;
Gerstein, M. J. Mol. Biol. 1999,290, 253-266.
128

electronic effect since bromo and methyl group have similar sizes,18 but different
electronic characteristics. Compound 4 has similar situation to MBMP. It has different
sizes and different electronic characteristics between two substituents (i.e., ethyl and
bromo).
.+ Ëlr
2 3 4 5
Chart 1. Substrates used to rationalize the increased enantioselectivity of Va1122Ser
mutant. The configuration is shown for the fast-reacting enantiomer.
The Echange between wild type and Va1122Ser was different depending on
substrate analogues (Table 3). The E values of the wild type and Va1122Ser mutant for
compound 2 were 32 and 50, respectively. In the case of compound 3, the
enantioselectivity could not be measured because PFE did not react with it. The E
changes for compound 4 and 5 were from 32 of wild type to 59 of Va1122Ser and from 4
of wild type to 12 of Va1122Ser, respectively. The Echange can be converted to the
_~~~GU9. The -~~~G:j: values were 0.96 kcal/mol for MBMP, 0.26 kcal/mol for
compound 2, 0.36 kcal/mol for compound 4, and 0.65 kcal/mol for compound 5 (Table
3).
18 The van der Waals volume of C14 and HBr are 17.1 and 17.9 cm3/mol, respectively.
AccessPerry's Home Page. http://www.accessperrys.com (accessed Dec 2002).
19 The difference in activation free energy for the two enantiomers (~~G:j:) can be
caIculated from enantiomeric ratio (E) by following equation: ~~G:j: = -RTlnE; Phillips,
R. S. Trends Biotechnol. 1996, 14, 13-16. On the other hand, the difference (i.e.,
~i\~G:j:) between ~~G:j: values represents the energetic relationship of the enantiomeric
discrimination between different enzymes.
129

Table 3. Evalues towards MBMP substrate and its analogues.
Compound Compound Compound Compound Compound 1 2 3 4 5
Wild Type 12 (st 32 (R) n.r. b 32 (R) 4 (R) V122S Mutant 61 (S) 50 (R) n.r. 59 (R) 12 (R}
-ôôôG+ 0.96 0.26 0.36 0.65
(kcal/mol) n.r.
a The letter in the parenthesis is the absolute configuration of the fast-reacting enantiomer. b n.r. = no reaction.
The -ôôôG+ value for MBMP is the biggest value and close to the numerical
summation of 0.26-0.36 (for compound 2 and 4, respectively) and 0.65 kcal/mol (for
compound 5). Since substituents of MBMP have different size and electronic
characteristics, presumably two recognition processes contribute to the -ôôôG+ value
(Figure 2a). PFE wild type and Va1122Ser mutant enzymes did not react with compound
3. Presumably, PFE does not have enough space to accept two ethyl (or ethyl and
bromomethyl) groups in acyl-binding region. Compound 2 and 4 showed similar energy
differences between wild type and Va1122Ser mutant enzyme. The main contribution to
the increase of enantioselectivity by Va1122Ser mutant towards compound 2 and 4 is
substituent size effect. Both substrates would bind to PFE in same way to MBMP such
that larger substituent binds in a same pocket. This same binding mode of larger
substituent makes reverse enantioselectivity compared to MBMP. For the compound 5,
Ver122Ser mutant is still more selective than wild type by a factor of 3 (-ôôôG+ = 0.65
kcal/mol). Although the difference of size between methyl and bromo is small, the
change of Evalue between wild type and Va1122Ser mutant toward compound 5 is bigger
than those towards compound 2 and 4.
130

a)
l +=
-~~G of Val122Ser mutant
0.65 kcal/mol (electronic contribution)
0.26-0.36 kcal/mol (size contribution)
b) SIOWM -- - -- - - - - -.---.------------- - - - - - - - - - -------- - - - ---;-- SIOWM
0.96 kcal/mol 0.65 k~al/mol 0.65 kcal/mol
--r- SIOWM' 1 0.14 kcal/mol
fastwt--r- slowwt 0.31 kdal/mol fastwt~ -- slowwt fastM-+- 0.17 kcal/mo~SIOWM' ___________ : _________ fastM~ 17 kcaVmol
fast enantiomer slow enantiomer fast enantiomer slow enantiomer
Hypothesis 1 Hypothesis 2
Figlllre 2. a) Schematic representation for the contribution of enhancing recognition
process by Va1122Ser mutant. b) Proposed mutational effects on each enantiomer.
Hypothesis 1: Mutation stabilizes both enantiomers by 0.17 kcal/mol and then
destabilizes only the slow enantiomer by 0.96 kcal/mol. Hypothesis 2: Mutation stabilizes
only the fast enantiomer by 0.17 kcal/mol and then destabilizes the slow enantiomer by
0,79 kcal/mol.
On the other hand, the specificity constants (kcatfKM)20 of the fast-reacting
enantiomer revealed that Va1122Ser mutant stabilizes more the fast-reacting enantiomer
by 0.17 kcal/mol and destabilizes more the slow-reacting enantiomer by 0.79 kcal/mol at
transition state than wild type enzyme (Table 4 and Figure 2b).
20 The difference in activation free energy for the fast-reacting enantiomer between wild
type and mutant enzymes can be calculated from the ratio of the specificity constants:
~~G:j:fast = - RTln((kcatIKM)mutant)/(kcat/KM)wt) For example, the ratio between 0,65
131

Table 4. The converted energy values from the specificity constants.
Wild type Va1122Ser
(l<catlKM)fast (min-1 M-1)
0.49
0.65
R TIn C (kcatIKM)mutantl C kcatIKM)wt) for the fast enantiomer
0.17 kcal/mol
-RTln( Ckcat/KM)mutantl (kcatlKM)wtt for the slow enantiomer
0.79 kcal/mol
a The value for the slow enantiomer was calculated from the value for the fast enantiomer and -AAAG*: (0.96 - 0.17) kcal/mol.
Proposed Substrate Binding Mode. The binding mode of MBMP to PFE is still
speculative since the x-ray crystal structure is not known. However, the docking model of
PFE homology model can give a general idea. The transition state structure (Tdl) of
MBMP may be one of three rotamers for each enantiomer in PFE according to the
rotaltion of the axis between Cl and C2 (a-carbon). Each rotamer makes different
orientations of bromomethyl group in active site of PFE: towards Va1122 residue (S-A
and R-A in Figure 3), Trp 29 (S-B and R-B in Figure 3), and Val226 residue (S-C and R
C in Figure 3).
A docking model of substrate binding to PFE shows that Val226 residue is close
to the a carbon (within 3.7 Â) (Figure 3). Both substituents (i.e., bromomethyl and
methyl groups) of MBMP may not fit in this region because of not enough space (S-C
and R-C in Figure 3). In addition, if any substituent of MBMP locates in this region, the
substituent will bump the catalytic histidine (His252), thereby interrupting the catalytic
role of His252.
With excluding substituents of MBMP orienting toward Va1226, substituents
(bromomethyl or methyl) should orient toward Va1122 or Trp29. A docking model shows
that the region of Va1122 residue has less space than that of Trp29 residue. Consequently,
the smaller substituent (i.e., methyl group) and larger one (i.e., bromomethyl group) of
MBMP may orient towards Va1122 residue and Trp29, respectively. This substituent
orientation provides the configuration of the fast-reacting enantiomer of MBMP (S-B in
(l<c:atlKM of Va1122S) and 0.49 Ckcat/KM of wild type) accounts for 0.17 kcal/mol.
132
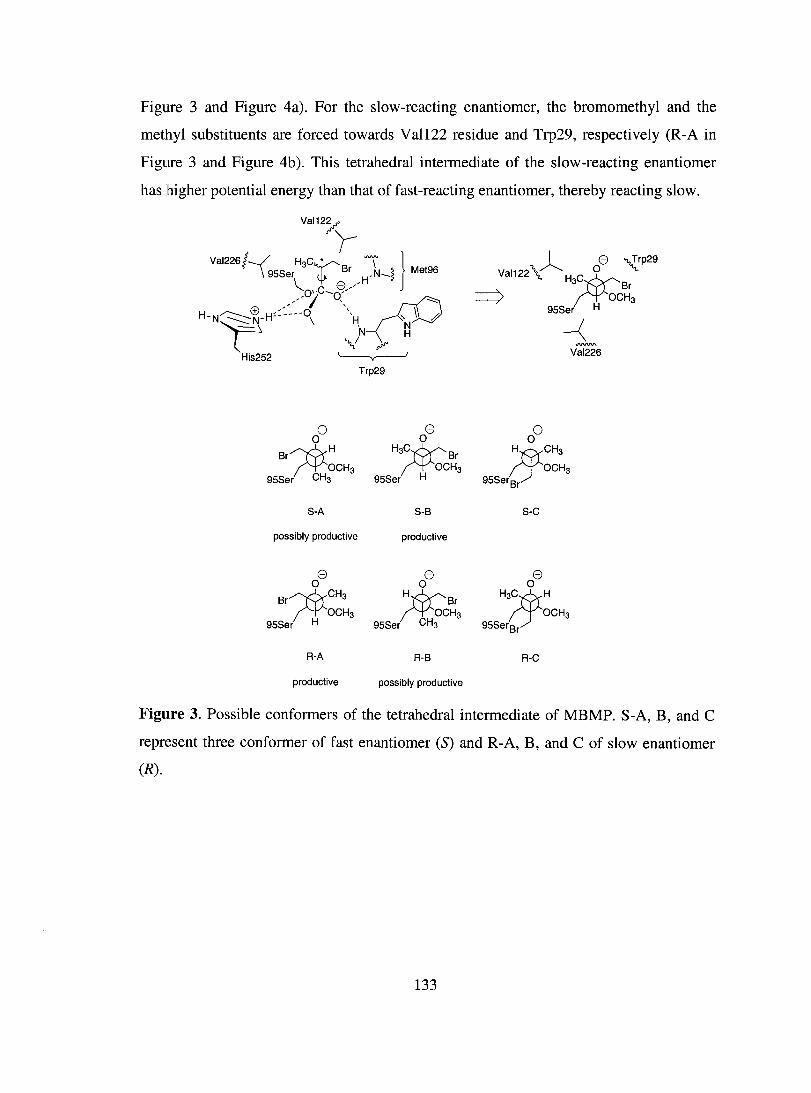
Figure 3 and Figure 4a). For the slow-reacting enantiomer, the bromomethyl and the
methyl substituents are forced towards Va1122 residue and Trp29, respectively (R-A in
Figure 3 and Figure 4b). This tetrahedral intermediate of the slow-reacting enantiomer
has higher potential energy than that of fast-reacting enantiomer, thereby reacting slow.
~ 08 "{rp29 Va1122\: H~
3 Br
===> OCH3 95Ser H
~ Val226
8 8 8 0 0 0
B~H H~Br ~CH3 r OCH3 OCH3 OCH3
95Ser CH3 95Ser H 95SerBr
S-A S-B SoC
possibly productive productive
8 8 8 0 0 0
Br~CH3 %B
H~H OCH3 OC~3 OCH3
95Ser H 95Ser CH3 95SerBr
R-A R-B R-C
productive possibly productive
Figure 3. Possible conformers of the tetrahedral intermediate of MBMP. S-A, B, and C
represent three conformer of fast enantiomer (S) and R-A, B, and C of slow enantiomer
(R).
133

(a) fast-reacting enantiomer (S) (b) slow-reacting enantiomer (R)
Figure 4. Proposed binding mode of MBMP to PFE wild type enzyme. A homology
model of PFE into which has been built the first tetrahedral intermediate (Td l) of MBMP
hydrolysis. Five residues, Va1226, Va1122, catalytic His252, Ser95, and Trp29 , were
represented as sticks. The Va1122 and Va1226 are located around the acyl region of
MBMP (space-filling representation). The oxyanion hole is composed of backbone amide
protons of Trp29 and Met96. The figure was made using RasMac v2.6 software. 21
Discussion
A structure-guided random mutagenesis approach increased enantioselectivity
much more efficiently than rational protein design or directed evolution. Use of a docking
model of a homology model of PFE avoided the requirement of crystal structure and the
detailed computer modeling in rational computer design. In addition, random
mutagenesis avoided loss of enzyme activity or stability due to unexpected structural
change in rational approach. On the other hand, the selection of small number of target
21 Sayle, R.; Milner-White, E. J. Trends Biochem. Sei. 1995,20,374-376.
134

sites for mutagenesis avoided the enormous screening effort in directed evolution. We
identified a higher enantioselective as weIl as more active mutant through relatively small
screening (i.e., 192 colonies per each library).
Val226 mutant library showed 85% inactive mutants and unvaried
enantioselectivity. This region (DQIV) is conserved in chloroperoxidase (DQVV),22
which has 51% homology identity with PFE. The Asp223 has a catalytic role by
stabilizing the transient positive charge on the histidine. The crystal structure of
chloroperoxidase shows that this Asp223 has two hydrogen bonds: one to the catalytic
histidine and one to the backbone amide proton of Va1226. The conserved valine can be
found in other hydrolases such as CAL-B (Va1190) and PCL (VaI266). The substitution
of valine to others in Val226 residue may affect the catalytic role of Asp223 (or the
hydrolytic activity of PFE) by interruption of this hydrogen bond. The change of valine to
othe:r amino acids with similar or smaller side chain presumably maintains the hydrogen
bond, and thus the hydrolytic activity. In fact, we found that the mutants containing
alanine, isoleucine, and serine showed the hydrolytic activity. On the other hand,
unvaried enantioselectivity in Val226 library may be because it is far from the methyl as
weIl as bromomethyl substituents of MBMP in a docking model of PFE homology
model. Changes at position 226 may not change the interaction between the protein and
the substituents of the substrate. In addition, very small number of active mutants may
cause unvaried enantioselectivity of the library because the number of active mutants in
Val226 library is perhaps only 3 (i.e., 15% of 20 amino acids).
Va1122 mutant library shows more varied and higher enantioselectivity than
Val226 library. In this library, we identified high enantioselective mutants: Va1122Ser (E
= 61) and Va1122Met (E = 36). Since the crystal structure of PFE is not known to date,
the substrate binding mode of MBMP in PFE is not clear. However, more variant and
higher enantioselectivity in Va1122 library can be explained by study of a docking model
of PFE as weIl as changes of enantioselectivity according to a series of substrate
22 Hofmann, B.; Tolzer, S.; Pelletier, 1.; Altenbuchner, J.; van Pée, K. H.; Hecht, H. J. J.
Mol. Biol. 1998,279,889-900.
135

analogues. A docking model of PFE suggests that the substituents of tetrahedral
intennediate analog of MBMP are more closely located toward Va1122 than Va1226. The
change of Va1122 residue causes stronger influence on the interaction between PFE and
substrates than that of Va1226.
Rationalization of enhancing enantioselectivity of Va1122Ser mutant compared to
wild type can be approached through substrate engineering in consideration of substituent
shape and electronic character. A study of enantioselectivity altered by a series of
substrate analogues revealed that introducing serine instead of valine changed the
substrate recognition process by substituent size and electronic effects.
The Va1122Ser mutant more sensitively recognizes the size difference between
two substituents (e.g., methyllethyl or bromo/ethyl) than wild type enzyme, thereby
increasing the -LlLlG+ value by 0.26 - 0.36 kcal/mol. Presumably, changing volume of the
side chain by mutation increases the efficiency of size-recognition process of PFE.
However, understanding mutational effect of the size contribution on each enantiomer is
more complicated because the mutation creates more space in acyl binding region of
PFE. The van der Waals volume of serine is smaller than that of valine, but yet
enantioselectivity is higher. One possible explanation for the substituent size contribution
is that serine residue could accept a water molecule through a strong hydrogen bond. It is
conceivable that the serine residue with a water molecule can repulse the acyl moiety of
the slow-reacting enantiomer. In the same way, the introduction of methionine may
reduce space in the region and push out the bromomethyl group of the slow-reacting
enantiomer.
In addtion, Va1122Ser better distinguishes the electronic difference between
bromomethyl and methyl groups than wild type. This improved recognition process
causes to increase the -LlLlG+ value by 0.65 kcal/mol. The hydroxyl side chain in serine
residue in Va1122Ser mutant can have direct electronic interaction with bromo group.
This interaction (repulsion or attraction) causes to improve the electronic recognition
process of PFE. The kinetic study showed that the mutation increase enantioselectivity by
decreasing the rate of reaction of the slow-reacting enantiomer. The electronic effect on
the slow-reacting enantiomer can be explained by repulsive electronic interaction. The
136

proposed binding mode of the slow-reacting enantiomer suggests that the bromomethyl
group of MBMP orients toward Ser122 in Va1122Ser mutant. The side chain (i.e., -
CH20H) of the serine residue can have the strong repulsive interaction with the
bromomethyl substituent of the slow-reacting enantiomer because of the partial charges
on them. This repulsive interaction in Va1122Ser mutant can make the slow-reacting
enantiomer slower, thereby enhancing enantioselectivity. Similarly, Rotticci et al.4, 23
proposed that a repulsive electronic interaction may contribute the enantioselectivity of
CAL-B toward 1-bromo-2-butanol. 1-Bromo-2-butanol has two of isosteric substituents
(-CH2CH3 and -CH2Br) at the stereocenter but CAL-B shows high enantioselectivity (E
= 81). The a1cohol binding region in CAL-B is composed of Ser47, Thr42, and Trp104
residues. The hydroxyl group in Ser47 and Thr42 residues may have strong repulsive
interaction with the bromo group of the slow-reacting enantiomer because of their partial
negative charges. Consequently, electronic interactions in the active site contribute to the
enantiodiscrimination of CAL-B.
Although quantitative analysis of the mutational effects (substituent size and
electronic effects) in Va1122 residue on each enantiomer is complicated, it may be
explained by two possible hypotheses. One is that the mutation may stabilize both
transition states of each enantiomer by 0.17 kcal/mol and then destabilizes the slow
reacting enantiomer by 0.96 kcal/mol (both size and electronic effects. The mutation may
cause the conformational change of PFE and introduce better substrate binding by 0.17
kcal/mol. The mutation destabilizes only the transition state of the slow-reacting
enantiomer by two recognition processes (i.e., 0.96 kcal/mol). The other possibility is that
the size contribution (-0.31 kcal/mol) affects both enantiomers such that the mutation
stabilizes the transition state of the fast-reacting enantiomer by 0.17 kcal/mol and
destabilizes that of the slow-reacting enantiomer by 0.14 kcal/mol. Then the electronic
contribution affects only the slow-reacting enantiomer at transition state by 0.65
kcal/mol. However, the both hypotheses are still speculated because the binding mode of
PFE is not c1ear.
23 Rotticci, D.; Orrenius c.; Huit, K.; Norin, T. Tetrahedron: Asymmetry 1997, 8, 359-
137

More detailed or quantitative analysis of the mutational effects on each
enantiomer may be possible by a study of the accurate substrate binding mode based on
crystal structure and extended kinetics for other substrates. Currently, we are trying to
solve the X-ray crystal structure of wild type of PFE to understand the detailed substrate
binding mode.
Experimental Section
General Methods. 1 H NMR spectra were recorded in CDCh at 400 MHz, 300 MHz, or
200 MHz (M400, M300, or M200, respectively, Varian). All chemicals, buffers and
lysozyme were purchased from Sigma-Aldrich. LB media was obtained from Difco.
RNase A was purchased from USB and DNase 1 was purchased from Gibco BRL. The
Sheldon Biotechnology Centre (McGill University, Montreal, Canada) provided primers
and performed dideoxy termination sequencing. The absolute configuration of the fast
reacting enantiomer of substrates was determined by comparing the retenti on time with
pure samples. Methyl (S)-2-bromo-2-methyl propionate was prepared from (S)-2-bromo-
2-methyl propanoic acid and methyl (S)-2-bromobutyrate from (S)-2-aminobutanoic
acid. 24
Synthesis of Methyl (±)-3-Bromo-2-methylpropionate (MBMP, 1). Racemic methyl 3-
bromo-2-methylpropionate was made according to literature procedure25 with slight
modification as follows. To a solution of HBr (30% in AcOH, 9.3 mL, 47 mmol) in 70
mL of ethyl ether was added methyl methacrylate (47 mmol, 4.7 g, 5.0 mL) slowly. The
reaction mixture was stirred ovemight at r.t. After washed with a saturated NaHC03
aqueous solution, the ether layer was dried over MgS04. The product was purified by
362.
24 Compagnone, R. S.; Rapoport, H. J. Org. Chem. 1986,51,1713-1719.
25 Coutts, R. T.; Midha, K. K. J. Pharm. Sei. 1969, 58,949-951.
138

column chromatography using hexane/ethyl acetate/dichloromethane (18:1:1) eluent:
yield 73%.
IH NMR (400 MHz): ù 3.72 (3H, s); 3.70-3.43 (2H, m); 2.92-2.89 (lH, m); 1.29 (2H, d).
Synthesis of Methyl (±)-2-Bromomethylbutyrate, 3. 2-Ethyl acrylic acid was made
according to literature procedure as follows. 26 To a solution of 2-ethyl malonic acid (5.77
g, 44 mmol) in ethyl acetate (50 mL) was added dimethyl amine (88 mmol, 2 M in THF)
and paraformaldehyde (2 g, 66 mmol) on ice-bath. The reaction mixture was removed
from the ice bath and stirred for 10 min at room tempo and then refluxed for 3 h. The
reaction mixture was concentrated by rotavap, diluted with water, acidified by conc. HCI,
and extracted with ethyl acetate (3x 50 mL). The organic layer was dried over anhydrous
MgS04 and concentrated to give a white powder, 2-ethyl acrylic acid: yield 72%.
IH NMR (200 MHz, ù): 6.29 (lH, d); 5.66 (lH, d); 2.35 (2H, q); 1.11 (3H, t).
To a solution of 2-ethyl acrylic acid (3.15 g, 0.0315 mol) in methanol (40 mL)
was added a few drop of sulfuric acid (-0.1 mL). The reaction mixture was refluxed for 3
h and concentrated by rotavap. The residue oil was neutralized by adding 5% NaHC03
aqueous solution (40 mL), and extracted with ethyl ether (3x 50 mL). The ether layer was
combined and dried over anhydrous MgS04. Without purification of the ester product,
RBr (7.5 mL, 30% in acetic acid) was added to the ether solution. The mixture was
stirred ovemight at room tempo and washed with 5% NaHC03 aqueous solution until
neUitralized. The ether was evaporated by rotavap to give an oil. The product was purified
by column chromatography using hexane/ethyl acetate/dichloromethane (18:1:1) eluent:
yield 13%. IH NMR (400 MHz, ù): 3.72 (3H, s); 3.70-3.44 (2H, m); 2.77-2.70 (lH, m);
1.77-1.65 (2H, m); 0.93 (3H, t). l3C NMR (100 MHz, ù): 173.62,52.28,49.79,32.45,
24.84, 11.74.
26 Huntington, K. M.; Yi, T.; Wei, Y.; Pei, D. Biochemistry 2000,39,4543-4551.
139

Scalle-up Reaction for End-point E. Substrate solution (100 ~L of 1 Min acetonitrile)
was added to a solution of 1 mM BES buffer (9200 ~L, pH 7.2) and acetonitrile (600
~L)" After adding enzyme solution (100 ~L, refer to details below, pH 7.2), the reaction
conversion was monitored by the amount of base (0.1 N NaOH) added. The reaction was
quenched by extracting with ethyl acetate (3 x 10 mL) at -40% conversion. The organic
layers containing starting esters were combined, dried over MgS04 and concentrated by
rotavap. The aqueous layer (product acid) was acidified to pH 2 by adding conc. HCl
solution (-1 mL) and extracted with ethyl acetate (3 x 10 mL), dried over MgS04 and
concentrated. In order to convert the product acid to methyl ester, the acid product was
dissolved in methyl a1cohol (40 mL) containing few drops of concentrated H2S04 (-0.1
mL). The mixture was refluxed for 2 h and concentrated by rotavap. The concentrated
solution was diluted with ethyl acetate (20 mL) and washed with 10% aqueous NaHC03
solution. The ethyl acetate layer was dried over MgS04 and concentrated by rotavap.
Both of starting and product parts were analyzed by gas chromatography with a chiral
capillary column (Chrompak Chirlasil-Dex CB column, 25 m x 0.25 mm, Raritan, NJ):
initial column temperature 85 oC (for 1 and 4) or 95 oC (for 2 and 3) for 2 min, then ramp
to 120 oC over 14 min (or 12 min): methyl 3-bromo-2-methylpropionate (a = 1.03, ks =
4.80, kR = 4.92). Enantiomeric ratio, E, and conversion, c, were calculated from the
enantiomeric excess of the product, eep, and of the starting material, ees, using the
equations below.14
E = ln[(l-c )(l-ees)]
In[(l-c )(1 +ees)]
c = ----
Saturation Mutagenesis. pJOE279227 was used as a template and The QuickChange
Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used according to the
27 (a) Krebsfanger, N.; Zocher, F.; Altenbuchner, J.; Bomscheuer, U. T. Enzyme Microb.
Technol. 1998,22, 641-646. (b) Krebsfanger, N.; Schierholz, K.; Bomscheuer, U. T. J.
Biotechnol. 1998,60, 105-112.
140

manufacturer's instructions, with complementary primers V122 (5' -CCTGGTGCTGCT
GGGCGCCNNKACCCCGCTGTTCGGCCAGAAGC-3'), cV122 (5' -GCTTCTGGCC
GAACAGCGGGGTMNNGGCGCCCAGCAGCACCAGG-3 ,)28, V226 (5' -GGCGATG
GCGACCAGATCNNKCCGTTCGAGACCACCGGC-3') and cV226 (5'-GCCGGTGG
TCTCGAACG GMNNGATCTGGTCGCCATCGCC-3').
PFE Expression in 96-well Assay Blocks. Ovemight culture (10 ilL) was added to each
weIl in 96-weU assay blocks (2 mL, Costar #3960), foUowed by the addition of 1 mL of
LB broth (ampicillin, 100 Ilg/mL). rhe assay block was sealed with a ThermoweU sealer
(Costar #6570) and incubated at 37 Oc and 325 rpm for 2.5-3 h (OD600 was visuaUy
estimated to be 0.5). Protein expression was induced by the addition of filtered (0.22 Ilm)
sterile L-rhamnose solution (50 ilL; 4 w/v %) to each weU, foUowed by incubation for
additional 6 h. The assay blocks were centrifuged (10 min, 4000 rpm, 4 OC), and the
supematant was removed with an 8-channel pipette (1.2 mL, Effendorf). Lysozyme
solution (250 ilL, 1 mg/mL) in BES buffer (5 mM, pH 7.2) was added to each weU and
the block sealed with a ThermoweU sealer prior to vortexing to resuspend the cell pellets.
The assay blocks were incubated for 45 min at 37 Oc with shaking at 325 rpm and then
frozen at - 20 oC for a freeze-thaw cycle. The lysed ceUs were then treated with nuclease
solution (RNase A, 0.2 mg/mL; DNase l, 3.3 ilL/mL) in BES buffer (5mM, pH 7.2) for
15 min at 37 Oc with 325 rpm, and then centrifuged (45 min, 4000 rpm, 4 OC). The
supematant was transferred with an 8-channel pipette to a new 96-well assay block (1
mL) for storage.
Screening toward p-Nitrophenyl Acetate. The assay solutions were prepared by mixing
4-nitrophenylacetate (20 ilL of 200 mM solution in acetonitrile), BES buffer (1110 ilL of
5.0 mM, pH 7.2), and acetonitrile (870 ilL) and then vortexing the solution. Mutant
enzyme solutions (5 llL/well) were transferred from the culture plate to a 96 well
28 M: a mixture of cytidine (C) and adenosine (A); N: a mixture of aU for nucleotides.
141

microtiter plate using an eight-channel pipette. Assay solution (100 J.lLlwell) was added
to each weIl using a 1,200 J.lL eight-channel pipette. Each microplate weIl contained 105
J.lL total volume (100 J.lL assay solution, 5 J.lL enzyme solution; BES, 4.65 mM, pH 7.2;
pNPAc, 0.32 mM; acetonitrile, 7 vol%). The plate was placed in the microplate reader
and shaken for 5 s to ensure complete rnixing, and the simultaneous decrease in
absorbance at 404 nm was monitored at 25 oc. Data were collected for 15 min. The first
10 s of data were excluded because the initial data were sometimes erratic, possibly due
to dissipation of bubbles created during shaking.
Screening toward Methyl 3-Bromo-2-methylpropionate by Quick E. Enzymatic
hydrolysis of pure enantiomers of MBMP was monitored colorimetrically in the presence
of a reference compound (resorufin acetate) as previously described. 13 Rates of
hydrolysis at 25 oC of substrate (11 mM (R)-enantiomer or 1.1 mM (S)-enantiomer) and
resorufin acetate (0.11 mM) in buffer solution (100 J.lL; BES, 5 mM, pH 7.2; Triton X-
100,,0.33 mM; acetonitrile, 8 vol%; enzyme solution, 10 vol%) were determined from the
change in absorbance at 404 nm and 574 nm as a function of time using a microplate
reader.
Purification of PFE Enzyme. The ovemight culture (lmL) was added to 100 mL LB
and grown (37 oC, 200 rpm) to an OD600 of 0.5. PFE expression was induced by adding
sterile rhamnose (1 mL; 20 w/v %) and incubated for 6 h at 37 oC at 200 rpm. The cells
were harvested by centrifugation (15 min; 4300 rpm; 4 OC) and the supematant was
discarded. The cell pellet was resuspended in Buffer A (4 mL; NaH2P04, 50 mM; NaCI,
300 mM; imidazole, 10 mM; adjusted to pH 8.0 with NaOH), to which lysozyme was
added (1 mg/mL; 47000 U/mg). Incubation at 37 oC, 200 rpm, 45 min was followed by a
freeze-thaw cycle at - 20 oC and room temperature.
The viscosity of the lysate was reduced by repeatedly passing the solution through
a needle (Becton-Dickinson; 20 gauge) attached to a syringe in order to shear DNAIRNA
released from the lysed cells. After centrifugation (45 min; 4300 rpm; 4 OC), the
142

supematant (4 mL) was collected. To the remaining cleared lysate was added Ni-NTA
agarose slurry (Qiagen Inc, Califomia, USA; 2 mL; provided as a 50% slurring in
ethanol), and the mixture was stirred at 4 oC for 1 h. The mixture was loaded on a Poly
Prep column (BioRad), allowed to settle, and then drained. The flow-through was
retained for SDS-PAGE analysis. The Ni-NTA column containing bound His6-PFE was
then washed two times with Buffer B (4 mL; NaH2P04, 50 mM; NaCl, 300 mM;
imidazole, 20 mM; adjusted to pH 8.0 with NaOH) to remove any non-specifically bound
contaminants. The His6-PFE enzyme was eluted from the column with Buffer C (15 mL;
NaI-hP04, 50 mM; NaCl, 300 mM; imidazole, 250 mM; adjusted to pH 8.0 with NaOH).
Eluate from the Ni-NTA column containing purified PFE was buffer-exchanged
from Buffer C to BES (5 mM, pH 7.2) using a centrifugaI concentrator (MACROSEP,
Pall Filtration Co., Northborough, MA) with lOK molecular weight cutoffs. After
concentrated to -3 mL by centrifuge (4300 rpm, 90 min), the enzyme solution was
washed 3 times with 5 mM BES buffer (10 mL, pH 7.2).
Homology Model Containing Tetrahedral Intermediate in Active Site
The primary amino acid sequence (excluding the His6-tag) of PFE was submitted
to the SWISS-MODEL automated homology modeling server of the Swiss Institute of
Bioilnformatics (http://www.expasy.chfswissmod/SM_FIRST.htmli9 and the results
retumed in pdb file format. The homology model was based on 46-51 % amino acid
identity with non-heme haloperoxidases3o which have the afp-hydrolase fold and exhibit
low esterase activity. The quality of the model was judged to be acceptable based on the
low energy of the backbone and side chain residues for the entire protein except for a few
loops distant from the active site. The tetrahedral intermediate formed after nucleophilic
attack by the catalytic serine (Ser95) on the carbonyl carbon of methyl 3-bromo-2-
methylpropionate was built into the homology model using Insight II, version 97
29 Guex, N.; Diemand, A.; Peitsch, M. C. Trends Bioehem. Sei. 1999,24,364-367.
30 pdb code: lA8S, lA88, lA8Q. Hofmann, B.; TOlzer, S.; Pelletier, 1.; Altenbuchner, J.;
van Pée, K. H.; Hecht, H. J. J. Mol. Biol. 1998,279,889-900.
143

(Accelrys, San Diego, CA). The substrate was positioned such that the oxyanion was
stabilized by two hydrogen bonds: one to the backbone amide proton of Met96, and one
to the backbone amide proton of Trp29. The protonated catalytic His252 formed
hydrogen bonds with both the Ser95 oxygen atom and the alcohol leaving group of the
substrate. The docking model was simply minimized by optimize mode using
Builder/Biopolymer with CVFF force field31.
31 Dauber-Osguthorpe, P.; Roberts, V. A.; Osguthorpe, D. J.; Wolff, J.; Genest, M.;
Hagler, A. T. Proteins 1988,4,31-47.
144

Conclusions and Summary
Enzymes are an excellent biocatalyst for the regio- and enantioselective reactions
because of their inherent activity and selectivity toward natural substrates under mild
condition. However, natural enzymes sometimes do not have enough activity or
selectivity toward unnatural substrates. To enhance their activity or selectivity, mainly
three approaches, such as medium, substrate, and protein engineering, are available
depending on the reactions. In this thesis, those three approaches are described in
hydrolase-catalyzed reactions.
The first approach was applied to lipase-catalyzed reactions of polar substrates.
The application of lipase-catalyzed reactions toward polar substrates has a long-stand
problem due to their solubility in nonpolar solvents, which do not denature lipases. Polar
substrates only dissolve in polar sol vents such as methanol and N-methylformamide.
However, the se polar solvents denature enzymes and cannot be used for lipase-catalyzed
reactions. Although ionic liquids such as 3-alkyl-l-methylimidazolium tetrafluoroborates
have polarities similar to these polar organic solvents, they do not denature lipases. We
focused on use of ionic liquids in lipase-catalyzed reactions of polar substrates because
ionic liquids dissolve polar substrates but do not denature lipases. To get reliable lipase
catalyzed reactions in ionic liquids, we developed new purification method by adding a
wash with aqueous sodium carbonate or by filtering through neutral alumina. While
lipase-catalyzed reactions did not occur in untreated ionic liquids, they occur in newly
purified ionic liquids at rates comparable to those in nonpolar organic sol vents such as
toluene. Acetylation of I-phenylethanol catalyzed by lipase from Pseudomonas cepacia
(PCL) was as fast and as enantioselective in ionic liquids as in toluene. Acetylation of
glucose catalyzed by lipase B from Candida antarctica (CAL-B) was more regioselective
(>13:1 and up to >50:1) in ionic liquids than in THF or acetone (2-3:1 mixture) because
glucose is more soluble in ionic liquids. A direct esterification of L-ascorbic acid
catalyzed by CAL-B gave higher yield (e.g., 83% conversion and 65% yield to produce
L-ascorbyl 6-0-0Ieate) in ionic liquids than that (generally less than 50% conversion) in
145

moderately polar sol vents such as acetone and tert-amyl a1cohol because of its high
solubility in ionic liquids (e.g., -130 mg/mL in sBMIM·BF4 at 60 OC).
To improve enzyme selectivity and activity, one can slightly alter substrates
because this is sometimes easier and faster than other approaches. We applied this
approach to lipase-catalyzed ring opening p-Iactams. CAL-B catalyzed the slow and low
enantioselective (E = -5) ring opening reaction of inactivated p-Iactams in water. When
the nuc1eophile was changed to the long secondary a1cohol (i.e., 2-octanol), the reaction
was accelerated by a factor of 2 and highly selective (E > 200). A 0.5-g scale reaction
under optimized conditions (2-octanol as the nuc1eophile in diisopropyl ether at 60 OC)
yielded the unreacted p-Iactam in 39-46% yield (max yield is 50%) with ~ 96% ee.
Computer modeling of transition state analogs for ring opening of 4-phenyl-azetidin-2-
one was used to rationalize the high enantioselectivity and a critical role of a1cohol.
Unusual transition state of inactivated p-Iactam in computer modeling does not have all
key hydrogen bonds for the productive reaction. We propose that the role of a1cohol is
restoring the lack of the key hydrogen bond as a bridge as well as attacking as a
nuclleophile.
Although medium and substrate engineering can be applied to improve hydrolase
activity and selectivity, one may need to modify hydrolase itself if the gene of the
hydrolase is available. We developed an efficient protein engineering strategy to improve
enantioselectivity of PFE (an esterase from Pseudomonas fluorescens) by a combination
of rational design and directed evolution. The mutant libraries were generated by
structure-guided saturation mutagenesis in substrate-binding region (Va1122 and Va1226)
in active site of PFE based on a docking model of a homology model of PFE. Use of a
docking model of a homology model of PFE avoided the requirement of crystal structure
and the detailed computer modeling in rational computer design. On the other hand,
selecting small number of target sites for mutagenesis based on the doc king model saved
the enorrnous screening effort in directed evolution. We identified a higher
enantioselective mutant through relatively small screening (i.e., 192 colonies per each
library). A mutant, in which the Va1122 is replaced by Ser (Va1122Ser), greatly enhances
the enantioselectivity (E = 61 from 12 of wild type enzyme) towards methyl 3-bromo-2-
146

methylpropionate. In order to rationalize the high enantioselectivity of Va1122Ser mutant,
we investigated the effect of changing the shape or electronic character of substrate on
the enantioselectivity of wild type and Va1122Ser mutant enzyme. The origin of
improving enantioselectivity is due to altering the enantiodiscrimination process of PFE
such as substituent size effect and electronic effect of substrates.
147

Contributions to Knowledge
1. We developed preparation methods of ionic liquids to use in lipase-catalyzed reactions.
Newly purified ionic liquids reliably work in regio- and enantioselective lipase-catalyzed
reactions. Lipase-catalyzed reactions in ionic liquids occurred at rates comparable to
those in nonpolar organic sol vents such as toluene.
2. Lipase-catalyzed reactions in ionic liquids of polar substrates such as glucose and
L-ascorbic acid are more regioselective (>13: 1 and up to >50: 1 for monoacylation of
glucose) and higher conversion (83% conversion and 65% yield to produce L-ascorbyl
6-0-0Ieate) than in moderately polar organic solvents such as acetone and tertiary
a1cohols.
3. We developed an efficient synthesis of unactivated enantiopure p-lactams by CAL-B
catalyzed ring opening reactions. A 0.5-g scale reaction under optimized conditions (2-
octanol as the nucleophile in diisopropyl ether at 60 OC) yielded the unreacted p-Iactam in
39-46% yield (max yield is 50%) with ~ 96% ee.
4. We proposed molecular basis of high enantioselectivity and the role of a1cohols in p
lactam ring opening reactions. Molecular modeling of transition state analogs for ring
opening of 4-phenyl-azetidin-2-one suggested that a severe steric clash between Ile189 in
CAL-B and the phenyl substituent on the slow-reacting enantiomer of the p-Iactam
accounts for the high enantioselectivity. In addition, molecular modeling of transition
state analogs suggested a key hydrogen bond is missing and perhaps restored by the
nuclleophilic a1cohol.
5. To improve the enantioselectivity of PFE (esterase from Pseudomonas jluorescens)
toward MBMP (methyl 3-bromo-2-methylpropionate), we developed an efficient protein
148

engineering technique by a combination of rational design and directed evolution. We
discovered a mutant (Va1122Ser) having high enantioselecitivty (E = 61 from 12 of wild
type: enzyme)
6. We rationalized the high enantioselectivity of Va1122Ser mutant of PFE toward
MBMP by investigating the effect of changing the shape or electronic character of
substrate on the enantioselectivity of wild type and Val 122Ser mutant enzyme. The origin
of improving enantioselectivity is the combination of substituent size and electronic
effects.
149

Appendix
Copyright waivers for published articles
150

ACS PUBLICATIONS DIVISION GUIDELINES
FOR THESES AND DISSERTATIONS
ATIN: STUDENTS. STUDENT ADVISORS, AND TEACHERS
Permission is now automatically granted to inclllde your paper(s) or portions ofyollr paper(s) in your thesis; please pay special attention to the implications paragraph below. The Joint Board/Council Committees on Copyrights and Publications recently approved the following:
Copyright permission for published and submitted material from theses and dissertations ACS extends blanket permission to students to include in their theses and dissertations their own articles, or portions thereof, that have been published in ACS journals or submitted to ACS journals for publication, provided that the ACS copyright credit li ne is noted on the appropriate page(s).
Publishing implications of electronic publication of theses and dissertation material Students and their mentors should be aware that posting of theses and dissertation material on the Web prior to submission of malerial from that thesis/dissertation to an ACS journal may affect publication in that journal. Whether Web posting is considered prior publication may be evaluated on a case-by-case basis by the journal's editor. If an ACS journal editor considers Web posting to be "prior publication", the paper will not be accepted for publication in that journal.
Ifyour paper has not vet been published bv ACS, we have no objection to your including part or all of it in your thesis in print and microfilm formats: please note, however, that electronic distribution or Web posting orthe unpublished paper as part oryour thesis in electronic formats mightjeopardize publication ofyour paper by ACS. Please prin! the rollowing credit line on the first page ofyour article: "Reproduced (or 'Reproduced in part') \Vith permission from [JOURNAL NAME], in press (or 'submitted for publication'). Unpublished work copyright [CURRENT YEAR] American Chemical Society." Indude appropriate information.
-, If your paper has alreadv been published by ACS and you want to include part or aU of it in your thesis or dissertation~ please print the ACS copyright credit line on the first page ofyour article: "Reproduced (or 'Reproduced in part') with permission from [FULL REFERENCE CITATION.] Copyright [YEAR] American Chemical Society." Include appropriate information.
Note: Ifyou plan to submit your thesis to UMI or to another dissertation distributor, you should not inc1ude the unpublished ACS paper in your thesis if the thesis will be disseminated electronically, until ACS has published your paper. After publication of the paper by ACS, you may release the entire thesis for electronic dissemination: ACS's copyright credit tine should be printed on the first page of the ACS paper.
Permission is not granted to post any published or unpublished ACS articles on any Web site.
Questions? Cali ACS Publications Division Copyright Office staff at
Ç.Q'p',xrjgbKlJ)J~Ç.~:.Q.r.g· e-mail us at
August 1998


















