
La Revue Du Praticien-Endocrinologie Nutrition
121
Transcript of La Revue Du Praticien-Endocrinologie Nutrition



Endocrinologie - Métabolisme - Nutrition
1149L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
présentes même en cas de micro-adénomes, par mise entension du diaphragme sellaire.• Les réductions du champ visuel(campimétrie deGoldmann) sont observées seulement en cas de macro-adénome ayant une extension suprasellaire atteignantles voies optiques. Du fait de la répartition des fibresnerveuses au niveau du chiasma, le champ temporal dechaque côté est le premier touché. L’intensité de l’atteinte est croissante avec le degré de la compression :exclusion de la tache aveugle, aplatissement des iso-ptères, quadranopsie temporale supérieure, puis hémia-nopsie temporale, jusqu’à la cécité. L’expansion supra-sellaire étant souvent médiane, le retentissement est leplus souvent bilatéral, réalisant ainsi la classique hémia-nopsie bitemporale, caractéristique d’une compressionchiasmatique.
2. NeuroradiologieLes seules explorations morphologiques contributivessont un examen tomodensitométrique (scanner) ou derésonance magnétique (IRM) hypophysaire. L’imageriepar résonance magnétique a prouvé sa supériorité dansles micro-adénomes (limite de sensibilité : environ 2 mm)comme dans les macro-adénomes. Le scanner peut toutefois être utilisé pour des raisons d’accessibilité oupour mieux explorer le cadre osseux. Une anomalie designal arrondie intraparenchymateuse peut être observéechez près de 10 % de sujets témoins (« incidentalomeshypophysaires ») et ne doit donc être interprétée qu’enfonction des contextes clinique et biologique (fig. 1 à 3).La radiographie simple du crâne, même centrée sur laselle turcique, ne possède pas une sensibilité suffisanteet ne doit donc pas être demandée à titre diagnostique.
Syndromes d’hypersécrétion
1. Hyperprolactinémie• Le retentissement endocriniende l’hyperprolactinémiese manifeste assez précocement chez la femme nonménopausée sous la forme de troubles du cycle (oligo-spanio-ménorrhée, aménorrhée), d’une galactorrhée, detroubles sexuels (baisse de la libido, sécheresse vaginale,dyspareunie) et parfois seulement sous forme d’uneinfertilité par anovulation avec conservation des cycles.
Diagnostic
Selon leurs caractéristiques morphologiques (taille,extension tumorale) et fonctionnelles, les adénomeshypophysaires peuvent se manifester par un ou plusieursdes éléments de la triade symptomatique : syndrometumoral, avec ses manifestations cliniques et radio-logiques ; hypersécrétion d’une ou plusieurs hormonesanté-hypophysaires ; déficit hormonal touchant une ouplusieurs des lignées hormonales hypophysaires, avecleurs manifestations cliniques et biologiques.
Syndrome tumoral
1. Clinique• Les céphaléessont souvent frontales ou orbitaires.Peu spécifiques, non pulsatiles, elles sont généralementcalmées par les antalgiques habituels. Elles sont
Adénomes hypophysairesde l’adulteDiagnostic, complications
PR Thierry BRUE
Service d’endocrinologie, hôpital de la Timone, 13005 Marseille.
• Bénins, les adénomes hypophysaires sont des tumeurs bien différenciées, de croissancehabituellement lente sur plusieurs années,développées de manière monoclonale à partirdes cellules endocrines anté-hypophysaires.
• Fréquents, ils représentent 10 % des tumeursintracrâniennes.
• On distingue les micro-adénomes, dont le plus grand diamètre est inférieur à 10 mm,et les macro-adénomes qui peuvent représenterde volumineuses tumeurs envahissantes.
• Les adénomes peuvent être non sécrétants,révélés alors par le syndrome tumoral associééventuellement à des signes d’hypopituitarisme,ou sécrétants : les prolactinomes, les plus fréquents, entraînent le classique syndromeaménorrhée-galactorrhée ; les adénomes somatotropes sont responsables de l’acromégalie ;les adénomes corticotropes entraînent une maladie de Cushing et les adénomes thyréotropes, plus rares, une hyperthyroïdie.
Points Forts à comprendre

Le mécanisme de l’atteinte de la fonction gonadique estune inhibition de la libération de LH-RH (luteinizinghormone releasing hormone) hypothalamique induitepar l’excès de prolactine. Chez la femme ménopausée,la galactorrhée est rare et c’est le syndrome tumoral quiest révélateur. Chez l’homme, les manifestations,
conduisant plus tardivement au diagnostic que chez lafemme jeune, sont représentées par des troubles sexuels(baisse de libido, dysérection, impuissance), raréfactionde la pilosité faciale ou somatique et rarement gynéco-mastie voire galactorrhée.• Sur le plan biologique,la prolactinémie basale esttrouvée élevée, supérieure à 20 µg et généralement biencorrélée avec le volume tumoral, un taux supérieur à 200µg/L étant quasi spécifique d’un macroprolactinome. Aucontraire, un taux inférieur à 100 µg/L en présence d’unmacro-adénome volumineux est en faveur d’une hyper-prolactinémie accompagnant un adénome non sécrétantpar un mécanisme de compression de la tige pituitaire.En cas d’insuffisance gonadotrope lésion-nelle associée,les gonadotrophines sont abaissées en base ou après sti-mulation par LH-RH exogène (test à la LH-RH).
2. Acromégalie• Le tableau cliniquelié à l’hypersécrétion chroniquede GH (growth hormone) est caractérisé par l’installationprogressive de modifications morphologiques : progna-thisme, élargissement des mains et des pieds nécessitantdes changements de pointure de chaussures, épaississe-ment des traits, en particulier le nez et les lèvres. Ces
A D É N O M E S H Y P O P H Y S A I R E S D E L ’ A D U L T E
1150 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Micro-adénome corticotrope. En acquisition T1, aprèsinjection de produit de contraste, la lésion apparaît en coupeaxiale sous la forme d’une image arrondie avec hyposignalde 8 mm de diamètre au contact du sinus caverneux gauche.
1
Macro-adénome somatotrope. Sur cette coupe coronale,en acquisition T1, le parenchyme hypophysaire, refoulé versle haut et à droite, est plus fortement rehaussé par le produitde contraste que la masse adénomateuse qui occupe la quasi-totalité de la selle turcique et s’étend vers le bas dans le sinussphénoïdal. La tige pituitaire est déplacée vers la droite.
2
Macro-adénome non sécrétant. Coupe coronale, enacquisition T1, montrant un macro-adénome invasif.L’adénome envahit les 2 sinus caverneux, entourant lesiphon carotidien de chaque côté (flèches blanches) ; il existeune expansion suprasellaire dans la citerne optochiasma-tique refoulant le chiasma optique vers le haut (flèchesnoires). L’adénome apparaît hyperintense par rapport auparenchyme cérébral après injection de gadolinium.
3

4. Hyperthyroïdie hauteL’adénome thyréotrope entraîne les mêmes signes queles autres causes de thyrotoxicose : tachycardie, amai-grissement, hypersudation et thermophobie, diarrhéemotrice, nervosité, fatigabilité. Il s’y associe un goitre leplus souvent de volume modéré.Le profil biologique typique est celui d’une élévationdes fractions libres des hormones thyroïdiennes T3 etT4, associée à un taux de TSH dans les limites de la normale (mais inapproprié au taux de T3 et T4) oumodérément élevé. La sous-unité α libre de la TSH estélevée, avec un rapport molaire par rapport à la TSH supérieur à 1.
Syndromes d’hyposécrétion
1. Déficit corticotrope• Cliniquement, asthénie croissante au cours de la journée, hypotension orthostatique, pâleur, anorexie ounausées sont les principaux symptômes. À la différencedes insuffisances surrénales périphériques, il n’y a pasde mélanodermie ni d’anomalie ionique en dehors d’uneéventuelle décompensation. • L’exploration hormonale montre un cortisol libre urinaire bas, une cortisolémie abaissée le matin, ou lorsde mesures répétées, en regard de taux d’hormone corticotrope bas ou « normaux » mais inappropriés.L’atténuation de la réponse de l’hormone corticotrope etdu cortisol au cours d’une hypoglycémie insuliniquepeut confirmer le diagnostic en cas de doute.
2. Déficit gonadotrope• Au plan clinique,les troubles du cycle chez la femme,une dépilation chez l’homme, des troubles de la fonctionsexuelle et de la fertilité dans les 2 sexes sont les consé-quences de l’hypogonadisme par atteinte lésionnelle descellules gonadotropes.• Biologiquement,l’abaissement des stéroïdes sexuels(œstradiol chez la femme, testostérone chez l’homme)contraste avec des gonadotrophines basses, et ne s’élevantpas normalement au cours du test de stimulation à laLH-RH.
3. Déficit thyréotrope• Le tableau cliniqueest le même que celui de l’hypo-thyroïdie périphérique : asthénie, bradycardie, infiltra-tion tégumentaire avec prise de poids, constipation,ralentissement psychomoteur et état dépressif.• Sur le plan hormonal, les taux bas d’hormones thyroïdiennes contrastent avec des valeurs de TSH nonaugmentées, souvent dans l’intervalle de la normale. La TSH sécrétée est en effet caractérisée par une perted’activité biologique.
4. Déficit somatotropeLes éléments cliniques rapportés au déficit somatotropesont une fatigabilité accrue, une perte d’énergie, unebaisse des performances à l’exercice. Sur le plan objectif,on met en évidence une diminution de la masse maigre,
signes sont mis en évidence par la comparaison de clichés successifs. On note également une hypersuda-tion, une hyperséborrhée, parfois une hypertrichose ; destroubles de l’articulé dentaire et une macroglossie, avecfréquents ronflements nocturnes, une raucité de la voix ;des arthralgies, un syndrome du canal carpien; unehépato-splénomégalie. Une hypertension artérielle, uneintolérance au glucose ou un diabète sucré peuventapparaître.• La confirmation biologiqueest apportée par le test decharge orale en glucose au cours duquel le taux de GHest normalement freiné en dessous de 1 µg/L. Du fait dela pulsatilité de la sécrétion de GH, seuls des dosagesrépétés peuvent permettre d’apprécier le degré d’hyper-sécrétion de l’hormone. En revanche, un dosage uniquede l’effecteur périphérique de l’action de GH, l’IGF-1(insulin-like growth factor 1) permet de confirmer lediagnostic d’acromégalie lorsqu’il est trouvé supérieur àla normale pour l’âge et le sexe. On peut observer uneélévation paradoxale de la GH au cours du test à la TRH(thyrotrophin releasing hormone).
3. Maladie de Cushing• Cliniquement, l’hypercorticisme induit par l’hyper-sécrétion chronique d’hormone corticotrope (adreno-corticotropic hormoneou ACTH) entraîne une prise depoids de type androïde, c’est-à-dire prédominant à lapartie supérieure du corps (thorax, abdomen), contrastantavec des membres rendus grêles par l’amyotrophie. Lefaciès est rond, érythrosique. On note des vergeturespourpres, des ecchymoses apparaissant lors de trauma-tismes minimes, une hypertrichose. Les irrégularitésmenstruelles sont habituelles. Il peut survenir un étatdépressif ou des troubles du comportement. Une hyper-tension artérielle apparaît ou s’aggrave. Il peut existerune hypokaliémie.• Le diagnostic biologiqued’hypercortisolisme est souvent difficile et comporte 2 aspects :– le diagnostic positif de l’hypercorticisme repose sur
l’augmentation de la cortisolémie basale, de préférencemesurée le soir ou à plusieurs reprises au cours des 24 h, montrant une perte du rythme nycthéméral ; uneélévation du cortisol libre urinaire des 24 h ; l’absencede freinage de l’hypercorticisme au cours d’un test àla dexaméthasone « minute » (1 mg au coucher, etmesure du cortisol le lendemain à 8 h) ou « faible »(test de Liddle faible comportant la prise de 2 mg/j dedexaméthasone à raison de 0,5 mg toutes les 6 h pendant 48 h) ;
– le diagnostic étiologique repose sur un faisceau d’arguments cliniques, biologiques et radiologiquesqui permettent de distinguer l’hypercorticisme lié à unadénome corticotrope (dénommé maladie deCushing), qui représente environ les deux tiers descauses de syndromes de Cushing endogènes, d’uneautre cause, essentiellement adénome surrénal ousécrétion ectopique d’hormone corticotrope. Lesmoyens de ce diagnostic sont donc développés dans lapartie « Diagnostic différentiel ».
Endocrinologie - Métabolisme - Nutrition
1151L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

notamment osseuse et musculaire et une augmentationde la graisse viscérale. Le diagnostic requiert un effon-drement de la réponse de GH lors d’une hypoglycémieinsulinique.
5. PanhypopituitarismeL’association des différents déficits hormonaux réalisele panhypopituitarisme. Il combine les signes déjà évo-qués, avec en particulier une asthénie marquée, une peaupâle, sèche et fine, des troubles sexuels. Le diabète insi-pide ne complique un adénome hypophysaire, en règle,qu’à la suite d’une lésion post-hypophysaire ou de latige au cours d’une exérèse chirurgicale.
Diagnostic du type d’adénome
Le diagnostic repose sur l’étude histologique et immuno-histochimique de l’adénome lorsque celui-ci est retiréchirurgicalement. Dans le cas contraire, le diagnostic estfondé sur des arguments de présomption représentés parles données cliniques, biologiques et radiologiques.
1. Prolactinome (40 %)L’hyperprolactinémie, typiquement non stimulable (élévation du taux de prolactine [PRL] inférieure à100 % de la valeur basale) par le TRH ou le métoclopra-mide est associée à une lésion tumorale hypophysaire.Le volume de l’adénome est en règle proportionnel auxtaux de PRL. La forme la plus fréquente est le micro-prolactinome de la femme jeune. Chez l’homme ou lafemme ménopausée, il s’agit le plus souvent d’unmacroprolactinome.
2. Adénome somatotrope (15 % )Il s’agit dans la majorité des cas de macro-adénomesavec des extensions supra- ou para-sellaires. Du fait ducaractère insidieux des déformations progressives, leretard diagnostique est en moyenne de 5 à 10 ans.
3. Adénome corticotrope (10 %)La plupart sont des micro-adénomes. Il n’est pas rarequ’ils ne soient pas visualisés même par des examens enimagerie par résonance magnétique de qualité optimale.
4. Adénome thyréotrope (moins de 1 % )Il s’agit dans la majorité des cas d’un macro-adénomesouvent multisécrétant.
5. Adénome gonadotrope (environ 33 %)Révélés en général par un syndrome tumoral, ils repré-sentent le type le plus fréquent de macro-adénome.Autrefois qualifiés de «chromophobes», ils correspondenten fait le plus souvent à des adénomes gonadotropessécrétant des gonadotrophines intactes ou leurs sous-unités libres inactives (α, β-LH ou β-FSH). De tellessécrétions n’étant pas biologiquement actives, elles nedonnent lieu habituellement à aucun syndrome cliniqued’hypersécrétion. Le taux basal de FSH, LH ou sous-unité α est rarement très élevé.
Diagnostic différentiel
1. Devant un syndrome de masse hypophysaire
Les adénomes hypophysaires en sont la principale causechez l’adulte. Les principales autres causes sont rassem-blées dans le tableau ci-dessous. Parmi les plus impor-tantes, les craniopharyngiomes sont fréquents dans l’enfance et l’adolescence, mais près de 50 % sont néanmoins diagnostiqués chez l’adulte devant destroubles visuels associés parfois à un diabète insipide età des signes d’hypopituitarisme. Souvent révélés parune hypertension intracrânienne, un diabète insipide ouun hypopituitarisme chez un adulte jeune, les germi-nomes peuvent sécréter un marqueur biologique :β-hCG (human chorionic gonadotropin).
2. Devant une hyperprolactinémieL’hyperprolactinémie peut être due à un dysfonction-nement du tissu lactotrope normal, notamment par levéedu frein tonique inhibiteur dopaminergique. Environ25 % des cas d’aménorrhée secondaire sont liés à unehyperprolactinémie. • Lésions hypophysaires non lactotropesou lésionssuprahypophysaires : l’hyperprolactinémie résulte del’interruption de la voie tubéro-infundibulaire par unelésion tumorale ou mécanique (tableau).
A D É N O M E S H Y P O P H Y S A I R E S D E L ’ A D U L T E
1152 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Cause physiologique❑ Hyperplasie lactotrope gravidique
Autres tumeurs bénignes❑ craniopharyngiomes❑ méningiomes❑ hypothyroïdie périphérique
Tumeurs malignes❑ germinomes (pinéalomes ectopiques)❑ sarcomes❑ chordomes❑ adénocarcinomes hypophysaires❑ métastases hypophysaires
Kystes❑ kyste de la poche de Rathke❑ kyste dermoïde❑ kyste arachnoïdien
Lésions inflammatoires et infiltratives❑ hypophysite lymphocytaire❑ histiocytose X❑ abcès hypophysaire❑ tuberculome hypophysaire
Lésions responsables de syndrome de masse hypophysaire, en dehors
des adénomes hypophysaires
TABLEAU

Complications des syndromes d’hypersécrétion
1. Complications de l’hyperprolactinémieL’ostéoporose liée à l’hypogonadisme est la conséquenced’une hyperprolactinémie prolongée.
2. Complications de l’acromégalieElles sont surtout liées à l’ancienneté de l’hypersécrétionsomatotrope. L’insuffisance cardiaque peut être laconséquence de la cardiopathie acromégalique associéeà l’hypertension artérielle. Les déformations thoraciquespeuvent entraîner un syndrome restrictif et le syndromed’apnée du sommeil est fréquent, responsable notam-ment d’une somnolence diurne. Le diabète, le plus souvent non insulinodépendant, peut entraîner toutes sescomplications propres. La fréquence accrue des polypeset du cancer du côlon justifie un dépistage systématiqueinitial par colonoscopie. L’ensemble de ces complicationsrend compte d’une multiplication du taux de mortalitépar 2 ou 3 par rapport à une population de référence. Si le retentissement osseux et articulaire est irréversible,ses conséquences sur la mortalité peuvent être évitéespar un traitement approprié.
3. Complications de la maladie de Cushing Ce sont les mêmes complications que celles d’une corticothérapie au long cours : risque accru d’infection ;déminéralisation osseuse avec fractures vertébrales,cervico-fémorales ou des os longs ; décompensationpsychiatrique ; hypokaliémie sévère, hypertension arté-rielle ou diabète compliqués.
4. Complications des hyperthyroïdies hautesCe sont les mêmes que celles des autres causes de thyrotoxicose, essentiellement le risque de cardio-thyréose.
Complications des hypopituitarismes
• L’insuffisance surrénale aiguëest rarement révélatrice.Elle doit être prévenue par une bonne information dupatient et de son entourage sur les risques de décompen-sation de l’insuffisance corticotrope, même traitée, quepeuvent entraîner un stress important, par exemple chi-rurgical ou accidentel, un état de déshydratation, unepathologie grave intercurrente. Elle se manifeste par uneasthénie majeure, une hypotension artérielle entraînantun collapsus cardiovasculaire, des troubles digestifs àtype de nausées, douleurs abdominales, vomissements.Une hyponatrémie avec natriurèse conservée est alorsprésente.• Un accroissement de la morbi-mortalité globale etcardiovasculaire a été observé chez des patients hypo-pituitaires recevant un traitement substitutif des fonctions thyroïdienne, surrénale et gonadique. Le déficitsomatotrope associé, non traité, pourrait représenter unedes raisons de cette situation. ■
• Hyperprolactinémies iatrogéniques :très banales,elles sont le fait d’un grand nombre de médicaments antidopaminergiques (neuroleptiques, antidépresseurs,antiémétiques…) ou œstrogéniques (contraceptifsoraux…).• Hyperprolactinémies d’accompagnement :hypo-thyroïdie périphérique, dystrophie ovarienne poly-kystique, insuffisance rénale chronique, traumatismesthoraciques.• Macroprolactinémies :correspondant à un excès deformes lourdes de PRL, elles sont liées à des auto-anticorps anti-prolactine sans retentissement patho-logique.
3. Devant une thyrotoxicoseLes causes périphériques sont caractérisées par une TSH freinée en regard de valeurs élevées d’hormonesthyroïdiennes (T3 et [ou] T4).
4. Devant un hypercorticismeLa détermination du caractère de dépendance à l’hormone corticotrope ou non de l’hypercorticismerepose sur le dosage immunoradiométrique de l’hormone corticotrope. En regard d’une cortisolémiesupérieure à 15 µg/dL (415 nmol/L), un taux d’hormonecorticotrope inférieur à 5 pg/mL (1,1 pmol/L) signel’origine surrénale de l’hypersécrétion de cortisol,qui freine l’hormone corticotrope. Il faut alors rechercherune masse surrénale par un scanner ou une imagerie par résonance magnétique des surrénales. Si le dosagede l’hormone corticotrope est en faveur d’une tumeur,il faut déterminer si celle-ci est hypophysaire ou ectopique. Classiquement, la résistance à l’inhibitionpar les glucocorticoïdes étant partielle dans les adénomes corticotropes et totale dans les tumeurs ectopiques, on utilise pour les différencier le test à ladexaméthasone fort, ou test de Liddle fort (8 mg/j à raison de 2 mg toutes les 6 h pendant 48 h). En l’absence de visualisation d’une image hypophysairepar l’imagerie par résonance magnétique, on peut réaliserun cathétérisme des sinus pétreux pour s’assurer del’origine hypophysaire de l’hypersécrétion d’hormonecorticotrope.
Complications
Complications tumorales
• La diplopieest observée en cas de compression d’unnerf oculomoteur du fait d’une extension tumorale dansle sinus caverneux. Elle peut être explorée par un test deLancaster.• Une apoplexie hypophysaire,correspondant à unebrusque hémorragie intra-adénomateuse, peut entraînerun tableau évoquant une hémorragie méningée, aveccéphalées intenses d’apparition brutale, fébricule etdiplopie.
Endocrinologie - Métabolisme - Nutrition
1153L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

Étiopathogénie
L’étiopathogénie des adénomes hypophysaires a fait l’objet de nom-breux travaux récents. La démonstration du caractère monoclonal deces tumeurs a montré qu’elles prenaient leur origine dans une prolifé-ration des cellules hypophysaires, même si des facteurs extérieurs àces cellules (facteurs de croissance, hormones hypothalamiques…) peu-vent jouer un rôle promoteur. Ainsi, dans 40 % environ des adénomessomatotropes, on trouve une mutation activatrice de la sous-unité α de la protéine Gs, protéine de couplage du récepteur membranairede la somatolibérine hypothalamique (GHRH, ou growth hormonereleasing hormone) qui permet l’activation de l’adénylate cyclase et latransduction du signal hormonal. L’immense majorité des adénomeshypophysaires survient de manière sporadique, mais certaines patholo-gies familiales peuvent s’accompagner d’une fréquence accrue d’adé-nomes hypophysaires. C’est le cas de la néoplasie endocrinienne mul-tiple de type 1, dont le gène est désormais connu, qui associe unehyperparathyroïdie, quasi constante, à d’autres atteintes glandulaires,concernant essentiellement le pancréas endocrine (gastrinomes, insulinomes) et l’hypophyse.
Traitement
Il repose sur des moyens médicaux, chirurgicaux et radiothérapiques.En agissant sur les récepteurs de type D2, les agonistes dopaminer-giques (bromocriptine ou Parlodel, lisuride ou Dopergine, quinagolideou Norprolac, cabergoline ou Dostinex) permettent dans la majoritédes cas de prolactinomes de restaurer une fonction sexuelle et reproductive normale, de normaliser la prolactinémie et de réduire levolume tumoral sans toutefois entraîner la disparition des cellulesadénomateuses. Ils peuvent réduire les taux de GH chez moins de20 % des patients acromégales. Les dopaminergiques peuvent tousentraîner les mêmes types d’effets secondaires, partiellement prévenuspar l’augmentation progressive de la dose : nausées, hypotensionorthostatique, somnolence. Ils sont interrompus en cas de grossesse.Les agonistes somatostatinergiques sous forme injectable sous-cutanée(octréotide ou Sandostatine, en 3 injections/j), ou intramusculaire àlibération prolongée (lanréotide ou Somatuline LP, 1 injection tousles 10 à 14 j, octréotide ou Sandostatine LP, 1 injection par mois) per-mettent de réduire ou de bloquer l’hypersécrétion de GH chez uneforte proportion de patients acromégales, ou de TSH dans les adé-nomes thyréotropes, mais n’entraînent que rarement une réductionfranche du volume tumoral. Leur intérêt dans les adénomes nonsécrétants n’est pas démontré. Leur action s’exerce par l’intermédiai-re des récepteurs somatostatinergiques de types 2 et 5. Leurs effetssecondaires sont surtout digestifs : diarrhée, lithiase biliaire. Desexpérimentations en cours, encourageantes, permettront d’établir laplace des antagonistes de la GH (pegvisomant) dans le traitement del’acromégalie. Des traitements anticortisoliques peuvent être utilisésà titre essentiellement adjuvant dans la maladie de Cushing, en parti-culier OP’-DDD (mitotane) ou kétoconazole (Nizoral). À ces traite-ments doivent être ajoutées les thérapeutiques substitutives des défi-cits hormonaux éventuels. Le déficit corticotrope nécessite la priseorale de 15 à 40 mg/j de cortisol (Hydrocortisone, comprimés à 10mg) ; le déficit thyréotrope est traité par L-thyroxine (100 à 200 µg/jde Lévothyrox) ; le déficit gonadotrope relève sur le plan hormonaldes œstroprogestatifs chez la femme (sauf en cas de macroprolacti-nome) ou des androgènes chez l’homme, et d’une induction de la gamé-togenèse par gonadotrophines sur le plan de la fertilité ; le déficitsomatotrope peut être corrigé par injections sous-cutanées quoti-diennes d’hormone de croissance recombinante.
• Les abords chirurgicaux font essentiellement appel à la voietranssphénoïdale qui permet, dans une équipe neurochirurgicale
spécialisée, l’exérèse sélective totale des micro-adénomes, et l’exérèsepartielle ou parfois totale de nombreux macro-adénomes. D’autresvoies (sous-frontale, ptérionale) sont utilisées en cas de tumeursenvahissantes. La fréquence de l’envahissement microscopique desstructures adjacentes tel le sinus caverneux, même dans le cas desmicro-adénomes, explique la possibilité de récidive tumorale retar-dée et justifie une surveillance à long terme. La radiothérapie peutêtre administrée sur un résidu post-chirurgical soit sous forme d’uneirradiation externe conventionnelle (environ 50 Gy), qui entraîne leplus souvent un hypopituitarisme, soit sous des formes plus focaliséestelles que le Gamma-Unit.
• Les indications thérapeutiques dépendent du type et de la taillede l’adénome. Les microprolactinomes relèvent soit d’un traitementchirurgical qui peut seul être curateur, soit d’un traitement médicaldopaminergique au long cours. Les macroprolactinomes doivent êtretraités en premier par dopaminergiques, la chirurgie étant réservée,sauf urgence compressive, aux cas de résistance ou d’intolérance autraitement médical. Les adénomes somatotropes, thyréotropes oucorticotropes relèvent toujours d’un abord transsphénoïdal lorsqu’ilest possible, éventuellement précédé ou suivi d’un traitement médical.Les adénomes gonadotropes et non sécrétants relèvent d’une exérèsechirurgicale lorsqu’ils sont volumineux, et plus rarement d’une simplesurveillance.
A D É N O M E S H Y P O P H Y S A I R E S D E L ’ A D U L T E
1154 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• La classification repose sur le type de sécrétionsadénomateuses suggéré par les dosages plasmatiques et attesté le cas échéant par l’immunohistochimie. La plupart des adénomessont responsables d’une hypersécrétion hormonale symptomatique.
• La présentation clinique peut comporter unsyndrome tumoral (céphalées, troubles visuels),des signes d’hypersécrétion hormonale, oud’hypofonctionnement hypophysaire lié à deslésions de voisinage.
• Les adénomes cliniquement non fonctionnelscorrespondent le plus souvent à des adénomesgonadotropes, sécrétant des gonadotrophines(LH, FSH) ou leurs sous-unités (α ou β).
Points Forts à retenir
POUR APPROFONDIR
Bremont C. Mosnier-Pudar H. Luton JP. Maladie de Cushing. RevPrat 1996 ; 46 : 1490-7.
Brue T, Morange I, Jaquet P. L’adénome à prolactine. Rev Prat1996 ; 46 (12) : 1486-9.
Jaquet P. L’acromégalie : réflexions à propos du suivi de 104patients. Ann Endocrinol 1998 ; 59 (5) : 425-9.
Levy A, Lightman SL. Diagnosis and management of pituitarytumors. BMJ 1994 ; 308 : 1087-91.
Morange I, Jaquet P. Acromégalie. Rev Prat 1996 ; 46 : 1482-5.
Prevost G, Vantyghem MC, Hober C et al. Les adénomes thyréo-tropes : revue de la littérature. À propos de deux cas. AnEndocrinol 1996 ; 57 : 194-202.
POUR EN SAVOIR PLUS

Endocrinologie - Métabolisme - NutritionB 236
209L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
Valeur énergétique du vin et de la bièreL’apport énergétique des boissons alcoolisées dépendde leur teneur en alcool et de leur concentration en glucides :1 g d’alcool (éthanol) = 7 kcal ; 1 g de glucides = 4 kcal.Après oxydation, l’éthanol est utilisé à des fins énergé-tiques (thermogenèse en particulier) et de stockage. Seuleune faible partie est éliminée dans les urines et l’air expiré.La valeur calorique réelle in vivo n’est pas connue mais estsûrement inférieure aux 7 kcal/g mesurées in vitro.En France, l’apport énergétique des boissons alcooliséesreprésente en moyenne 9 % de la ration totale chezl’homme et 3 % chez la femme (tableau II).
Cinétique de l’alcool L’alcool ingéré est absorbé sans modification et entotalité dans le tube digestif : 30 % dans l’estomac et70 % dans la partie supérieure de l’intestin. Il en résul-te une augmentation rapide de la concentration d’al-cool dans le sang atteignant son maximum en 60 à90 min. Le pic maximal d’alcoolémie (Cmax) et ledélai de survenue (Tmax) dépendent de la teneur enalcool de la boisson et des modalités d’ingestion(vitesse, à jeun ou non, environnement alimentaire). Lacinétique de l’alcool est caractérisée par le pic maxi-mal d’alcoolémie qui est le rapport entre la quantité
Teneur moyenne en alcool
Le degré alcoolique (DA) d’une boisson correspond au volume en alcool (va) contenu dans l’ensemble duvolume (V). Il permet de calculer la quantité d’alcoolpur en prenant en compte sa densité qui est de 0,8.À titre d’exemple, 1 L de vin à 13˚ contient 130 mL et 104 g d’alcool pur :
DA = va x 100 soit 13° = va x 100 V 100 mL
soit va = 13 x 1000 mL = 130 mL x 0,8 = 104 g.100
La présentation traditionnelle des différentes boissonsapporte approximativement la même quantité d’alcoolpur : le type de verre est adapté à la boisson de telle sortequ’il contient environ 10 g d’alcool pur (tableau I).
AlcoolismeTeneur moyenne en alcool et valeur énergétique du vin et de la bière,cinétique de l’alcool, épidémiologie de la consommation et de la pathologieliée à la consommation d’alcool en France, dépistage de la consommation excessived’alcool, aspects médico-légaux, structures de prise en charge.
PR Jean-Louis SCHLIENGER,Thomas DERVAUX
Service de médecine interne et nutrition, CHU, hôpital de Hautepierre, 67098 Strasbourg Cedex.
• La consommation d’alcool – habitude,plaisir, moyen d’intégration sociale ou sourced’intoxication et d’imprégnation – pose unproblème médical qui est toujours d’actualité.
• En raison de la possibilitéd’une alcoolodépendance, l’alcool està présent considéré comme une drogue.L’excès aigu est sanctionné par des troublesdu comportement immédiats qui ont amenéle législateur à sévir.
• L’imprégnation alcoolique chroniquedes buveurs excessifs ou des buveurs dépendantsest à l’origine d’une surmorbi-mortalitédifférée qui pèse toujours lourd en termesde santé publique. Les conséquencesde la consommation de boissons alcooliséesne sont pas univoques et dépendentdu consommateur, des modalités et du niveaude consommation et des co-morbidités.
Points Forts à comprendre
❑ Vin 10° à 16°❑ Liqueur, apéritif 18° à 20°❑ Alcools « forts », digestifs 40° et plus
Bière❑ « ménage » < 4°❑ « luxe » 5-6°❑ « forte » 9°❑ « sans alcool » < 1,2°
Degré alcooliquedes principales boissons
TABLEAU I
d’alcool ingérée et l’espace de dilution qui, en l’occurrence,est presque identique au compartiment hydrique. La diffusion dépend du flux sanguin. Elle est rapide dans lepoumon, le cerveau, le rein et plus lente dans lesmuscles. La décroissance de l’alcoolémie est d’environ0,1 g/L/h. Ainsi, un homme de 70 kg peut éliminer environ 100 g d’alcool par 24 h.Le pic maximal d’alcoolémie est alors plus faible maisla présence d’alcool dans le sang plus prolongée. Lesboissons à faible teneur en alcool ont une absorptionralentie lorsqu’elles sont associées à un repas dans lamesure où la vitesse du passage de l’alcool vers le sangdépend de la vitesse d’évacuation gastrique. La consom-mation de certains glucides – notamment le fructose quirégénère plus rapidement la nicotinamide-adénine-dinucléotide (NAD) nécessaire à l’action de l’alcooldeshydrogénase – réduit le pic maximal d’alcoolémiepar une modification du métabolisme de l’alcool.
Métabolisme
La cinétique de l’alcool dépend, pour une bonne part, deson métabolisme. En effet, l’alcool ne peut être stocké.Le site principal du métabolisme de l’alcool est hépatique(90 à 95 %). La voie principale est l’oxydation de l’alcoolen acétaldéhyde par l’alcool deshydrogénase. Cetteenzyme dont le cofacteur est le nicotinamide-adénine-dinucléotide (NAD+) est surtout présente dans le foie.Cette oxydation libère de l’énergie qui peut être stockéesous forme d’adénosine triphosphate (ATP) et entraînela production de NADH+H+ qui limite la poursuite de ladégradation par inhibition enzymatique.La voie du système microsomal d’oxydation de l’alcool(MEOS) située dans le réticulum endoplasmique lisseintervient pour une alcoolémie supérieure à 0,3 g/L.L’énergie produite ne peut être stockée sous formed’ATP et est dissipée sous forme de chaleur.L’oxydation de l’alcool par la voie catalasique est une3e solution très accessoire. Cette enzyme ubiquitaireest une aldéhyde oxydase dont l’action nécessite laprésence d’H2O2 (peroxyde d’hydrogène).
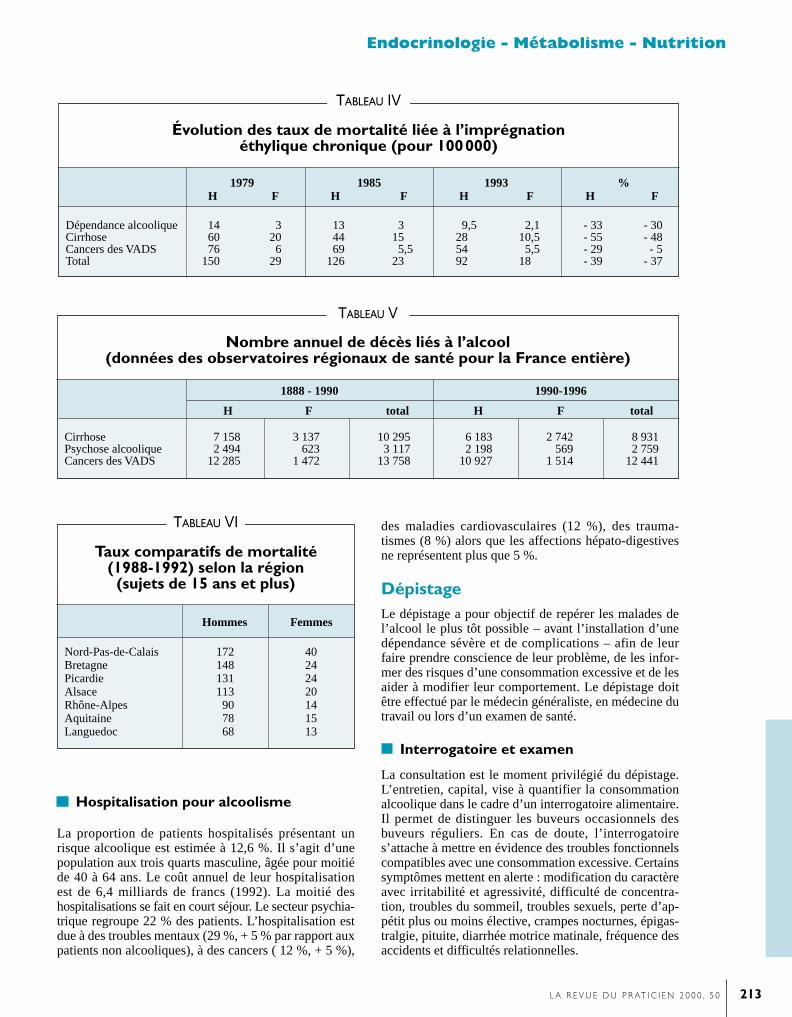
L’acétaldéhyde est le produit final de ces réactions, quelleque soit la voix d’oxydation de l’alcool. Ce composé trèstoxique pour l’organisme est rapidement métabolisé enacétate par une acétylaldéhyde deshydrogénase en présencede NAD. L’inhibition de cette enzyme par diversesmolécules, dont le disulfirame (Espéral) administré dansle cadre des cures de désintoxication alcoolique, est àl’origine d’un effet antabuse.L’acétate est catabolysé en acétyl-CoA (coenzyme A) sousl’influence d’une acétate thiokinase en présence d’ATP. Ilentre dans le cycle de Krebs pour former de l’acéto-acéta-te ou est utilisé pour la synthèse des lipides. La vitesse del’oxydation de l’alcool est peu accrue en cas de consom-mation chronique de boissons alcoolisées (figure).La cinétique de l’alcool dépend donc principalement de sonmétabolisme ; l’élimination par les urines, la respiration ou la sudation ne représentant que 5 à 10 % de l’alcoolingéré. Le pic maximal d’alcoolémie étant atteint, l’al-coolémie décroît en peu de temps. La première phase dedécroissance rapide est fonction du niveau de l’alcoolé-mie maximale qui correspond à l’action de l’alcooldeshydrogénase lorsqu’il est saturé. La concentration san-guine d’alcool décroît d’environ 0,15 g/L/h avec élimina-tion de 7 g d’alcool/h. Cette décroissance rapide est suivied’une décroissance exponentielle lorsque l’alcoolémie estinférieure à 0,30g/L ce qui correspond au fonctionnementde l’alcool deshydrogénase au-dessous de son seuil desaturation. Le système microsomial d’oxydation de l’alcoolet le système catalase n’interviennent plus à ce stade.
Épidémiologie
• En France, la consommation d’alcool pur par an et par habitant décroît rapidement depuis 1970.Estimée à 11,1 L/an, elle place la France au troisièmerang européen (la tendance est inverse dans la plupartdes autres pays européens). Il existe d’importantes disparités de consommation régionales et individuelles.Le vin est largement dominant (54 % de l’alcoolconsommé contre 27 % pour la bière et 19 % pour lesalcools forts). La bière est surtout consommée dans le
A L C O O L I S M E
210 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
Boisson Éthanol (g) Glucides (g) Énergie (kcal)
Vin rouge❑ 10° 80 0 560❑ 12° 96 0 672
Vin blanc 12° 96 7 (« sucre résiduel ») 700
Bière❑ sans alcool < 10 56 294❑ 5° 40 40 560
Apport énergétique du vin et de la bière (par litre)
TABLEAU II

Maladies liées à la consommationexcessive d’alcool :données épidémiologiquesL’alcoolisme est considéré comme une maladie dont laprise en charge thérapeutique, indispensable, a pourobjectif de prévenir les problèmes sociaux (familiaux etprofessionnels) et les complications pathologiques àl’origine d’une mortalité notable estimée à 20 % chezles hommes de 35 à 64 ans. Les conséquences éco-nomiques sont considérables (incapacité, traitement descomplications, réparation des conséquences, mortalité)et nettement supérieures au coût de la prise en charge(ratio coût-avantages très favorable).L’expression pathologique est contrastée (Pour appro-fondir 1). Il convient de distinguer les conséquencesd’une alcoolisation aiguë à l’origine de troubles du com-portement, avec dangerosité immédiate mais transitoire,et celles d’une imprégnation chronique responsable decomplications d’installation progressive et insidieusesouvent irréversibles évoluant, parfois, pour leur proprecompte. Le seuil de la consommation acceptable pré-servant de tout trouble est difficile à définir en dehors du cadre légal qui vise surtout à prévenir toutes lesconséquences aiguës. Il existe une grande variabilitéindividuelle de la tolérance vis-à-vis des répercussionspathologiques chroniques. On estime qu’une consom-mation quotidienne régulière supérieure à 35 g d’alcoolchez l’homme et 25 g chez la femme expose au risquede cirrhose.
Nord et l’Est de la France. La consommation n’est pasfigée. Ainsi, les vins de table sont peu à peu délaissés au profit de vins de qualité d’appellation d’originecontrôlée (AOC) ou de qualité supérieure.• Les modes de consommation sont variables. Les abstinents totaux constituent une petite minorité chez leshommes. La consommation coutumière est dominante.Les hommes consomment en moyenne 1,9 verre d’uneboisson alcoolisée par jour et les femmes 0,7 verre mais30 % des hommes et 6 % des femmes boivent 2 verresde vin ou plus par jour. Le pic de consommation se situevers la cinquantaine. Chez les adolescents, la consom-mation est de plus en plus précoce et s’effectue sur unmode discontinu par accès, avec excès : 20 % des garçons et 5 % des filles de 18 ans ont présenté desivresses multiples dans l’année.Les frontières entre la consommation culturelle et convi-viale, sans danger, et la consommation excessive ouinadaptée, posant d’importants problèmes de santépublique, sont difficiles à tracer.L’usage inadapté de l’alcool concerne 4 millions de per-sonnes en France : 2,5 millions sont à risque ou « menacées »bien qu’elles soient à même de contrôler leur consommationalors que 1,5 million ont perdu cette liberté parce qu’ellessont alcoolodépendantes. Une enquête récente situe à 20 %la prévalence de la consommation excessive d’alcoolparmi les consultants de médecine générale et à 15-25 %en milieu hospitalier. En 1994, les décès imputables à uneconsommation excessive étaient de plus de 30 000 dont23 400 pour « alcoolisme chronique ».
Endocrinologie - Métabolisme - Nutrition
211L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
Métabolisme de l’alcool chez l’alcoolique chronique.
Oxygène
NADPH
H2O2
Catalyse
ADH
Alcool
MEOS NADPH
Oxygène
Élimination directeRein
PoumonSueur
Acétaldéhyde
Acétate
Cycle de Krebs Synthèse lipidique Acétoacétate
ATP, H2 2O, CO

Paradoxalement, les résultats des grandes études de popu-lation ont mis en évidence un effet bénéfique de laconsommation régulière mais très modérée de boissonsalcoolisées (1 à 2 verres/j). Ces résultats, favorables, long-temps controversés mais confirmés par plusieurs étudesépidémiologiques, mettent en exergue la diminution durisque relatif de la morbi-mortalité cardiovasculaire chezles consommateurs par rapport aux abstinents, selon unecourbe en J. Toutefois, ce bénéfice est limité en-deça de laconsommation quotidienne de 3 verres d’une boissonalcoolisée ; au-delà la mortalité globale s’élève en raisond’une forte élévation de la mortalité par cancer, par accident, meurtre et suicide. La protection cardiovasculaire observée avec le vin et labière semble due à un effet de l’éthanol qui s’exerce parl’augmentation de la concentration du cholestérol HDL etpar un effet antithrombotique. L’effet antioxydant despolyphénols, contenus en grande quantité dans le vinrouge, accentue le potentiel de protection cardiovasculaire.Contrairement à ce qui a été avancé, la faible mortalitécoronaire observée en France – à consommation de graissesaturée et à cholestérolémie égales – n’est pas seulementle fait d’une consommation régulière de vin rouge. Celle-ci ne fait que contribuer au fameux French paradox.
Mortalité liée à la consommationexcessive de boissons alcoolisées(tableaux III à V)
Cirrhose
La part de la cirrhose dans la mortalité générale atteint unmaximum à 45-55 ans, quel que soit le sexe. Elle estalors responsable de 6 % des décès masculins et de 7 %des décès féminins. La fréquence maximale de décès est
atteinte à 70 ans chez l’homme (75/100 000) et à 60 anschez la femme (26/100 000). Les courbes de la cirrhoseen fonction de l’âge ressemblent à celles de l’alcoolisme.
Cancers des voies aérodigestives supérieures
Chez l’homme, ces cancers sont la 3e cause de mortalitéprématurée avant 65 ans, après le cancer du poumon et lesuicide. La surmortalité est maximale entre 35 et75 ans et l’incidence dépasse 120/100 000 après 55 ans.Chez les femmes, ces cancers sont beaucoup plus rares(10 à 20/100 000). Ils sont clairement liés à la consomma-tion conjointe de tabac et d’alcool avec un risque relatif de120 par rapport aux abstinents « tabac-alcool ».
Dépendance et psychose alcooliques
Les décès par alcoolisme sont maximaux vers 60 ans(20/100 000 chez les hommes et 4/100 000 chez lesfemmes).
Mortalité par alcool
La mortalité liée à la consommation excessive d’alcooldépend du sexe, du statut matrimonial et du statut socio-professionnel.Au cours des dernières années, on a pu observer une nettediminution du taux de mortalité liée à l’imprégnationéthylique chronique. Mais il persiste d’importantes dispa-rités régionales de la mortalité. Il existe un croissant géo-graphique de surmortalité allant de l’Ouest (Bretagne) àl’Est (Alsace) en passant par le Nord (Pas-de-Calais) quicontraste avec une zone de sous-mortalité dans la moitiéSud de la France (à l’exception de l’Auvergne). Chez leshommes, le rapport entre le Nord et le Sud de la Franceest de 3 et chez les femmes, il est de 4.
A L C O O L I S M E
212 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
Dépendance Cirrhose Cancer VADS Totalalcoolique
Sexe❑ hommes 11,5 34,6 66,6 112,7❑ femmes 2,6 13,0 6,8 22,4
Situationmatrimoniale hommes❑ mariés 5,1 24,7 56,4 86,2❑ célibataires 34,0 63,7 98,4 196,2❑ divorcés 39 82 124 245
Statut socioprofessionnel❑ cadres 1,0 2,8 5,2 9❑ agriculteurs 7 14 20 41❑ ouvriers et employés 9 25 51 85
Taux de mortalité (pour 100000) liée à l’imprégnation éthylique chronique chez les hommes
TABLEAU III
des maladies cardiovasculaires (12 %), des trauma-tismes (8 %) alors que les affections hépato-digestivesne représentent plus que 5 %.
DépistageLe dépistage a pour objectif de repérer les malades del’alcool le plus tôt possible – avant l’installation d’unedépendance sévère et de complications – afin de leurfaire prendre conscience de leur problème, de les infor-mer des risques d’une consommation excessive et de lesaider à modifier leur comportement. Le dépistage doitêtre effectué par le médecin généraliste, en médecine dutravail ou lors d’un examen de santé.
Interrogatoire et examen
La consultation est le moment privilégié du dépistage.L’entretien, capital, vise à quantifier la consommationalcoolique dans le cadre d’un interrogatoire alimentaire.Il permet de distinguer les buveurs occasionnels desbuveurs réguliers. En cas de doute, l’interrogatoires’attache à mettre en évidence des troubles fonctionnelscompatibles avec une consommation excessive. Certainssymptômes mettent en alerte : modification du caractèreavec irritabilité et agressivité, difficulté de concentra-tion, troubles du sommeil, troubles sexuels, perte d’ap-pétit plus ou moins élective, crampes nocturnes, épigas-tralgie, pituite, diarrhée motrice matinale, fréquence desaccidents et difficultés relationnelles.
Hospitalisation pour alcoolisme
La proportion de patients hospitalisés présentant unrisque alcoolique est estimée à 12,6 %. Il s’agit d’unepopulation aux trois quarts masculine, âgée pour moitiéde 40 à 64 ans. Le coût annuel de leur hospitalisation est de 6,4 milliards de francs (1992). La moitié des hospitalisations se fait en court séjour. Le secteur psychia-trique regroupe 22 % des patients. L’hospitalisation estdue à des troubles mentaux (29 %, + 5 % par rapport auxpatients non alcooliques), à des cancers ( 12 %, + 5 %),
Endocrinologie - Métabolisme - Nutrition
213L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
1979 1985 1993 %H F H F H F H F
Dépendance alcoolique 14 3 13 3 9,5 2,1 - 33 - 30Cirrhose 60 20 44 15 28 10,5 - 55 - 48Cancers des VADS 76 6 69 5,5 54 5,5 - 29 - 5Total 150 29 126 23 92 18 - 39 - 37
Évolution des taux de mortalité liée à l’imprégnation éthylique chronique (pour 100000)
TABLEAU IV
1888 - 1990 1990-1996
H F total H F total
Cirrhose 7 158 3 137 10 295 6 183 2 742 8 931Psychose alcoolique 2 494 623 3 117 2 198 569 2 759Cancers des VADS 12 285 1 472 13 758 10 927 1 514 12 441
Nord-Pas-de-Calais 172 40Bretagne 148 24Picardie 131 24Alsace 113 20Rhône-Alpes 90 14Aquitaine 78 15Languedoc 68 13
Nombre annuel de décès liés à l’alcool(données des observatoires régionaux de santé pour la France entière)
TABLEAU V
Hommes Femmes
Taux comparatifs de mortalité (1988-1992) selon la région
(sujets de 15 ans et plus)
TABLEAU VI

Examen physique
Il est souvent probant au stade de l’inspection : varicositéfaciale, injection conjonctivale, trémulation des extrémi-tés et de la langue, langue saburrale, hypersudation,haleine évocatrice, parotidomégalie, tachycardie, hyper-tension systolique et, parfois, hépatomégalie.L’intégration de ces signes, dont aucun n’est vraimentspécifique d’une consommation excessive chronique,conforte le bien-fondé d’une démarche ciblée sur la problématique de l’alcool avec recherche de marqueursbiologiques. Des questionnaires standardisés aident lerepérage (exemple CAGE/DETA) [Pour approfondir 2].Ils doivent être intégrés dans l’entretien clinique et peuvent contribuer à distinguer les consommateursexcessifs des patients alcoolodépendants.
Biologie
Les marqueurs biologiques de base sont : le volume globulaire moyen (VGM) et la gamma-glutamyl trans-peptidase (γGT). Ils permettent de confirmer une alcoo-lisation chronique excessive mais ne peuvent prétendreau diagnostic d’alcoolodépendance. L’augmentation de la gamma-glutamyl transpeptidaseest la conséquence d’une induction enzymatique parl’alcool qui survient après plus d’une semaine deconsommation excessive. Sa sensibilité n’est que de50 à 70 % et sa spécificité de 60 %. L’arrêt de la prised’alcool entraîne une diminution rapide et probante demoitié tous les 15 jours. L’augmentation du volume globulaire moyen au-delà de95 µm3 survient après 2 mois. Sa spécificité est élevée(90%) mais sa sensibilité est médiocre (50%). Sadécroissance après l’arrêt de l’alcool est lente. Au total, plus de 85 % des consommateurs excessifschroniques peuvent révéler l’un ou l’autre de ces2 marqueurs qui gagnent à être prescrits ensemble.L’élévation de l’acide urique, des triglycérides et destransaminases (en cas de stéatose) sont d’autres mar-queurs biologiques quelque peu discrédités par leurfaible valeur prédictive. La transferrine déficiente encarbo-hydrate a une sensibilité de l’ordre de 80 % etune spécificité de 70 %. Il s’agit d’un marqueur deréférence à réserver aux cas litigieux ; son élévationpermet de rattacher une élévation de la gamma-glutamyl transpeptidase inexpliquée à une consom-mation excessive. Ce dosage est coûteux et encore peudisponible. Ces marqueurs permettant de confirmer une suspicionde consommation excessive d’alcool ne doivent en aucun cas se substituer à l’entretien qu’ils complè-tent. Ils permettent d’amorcer un dialogue, d’assurerun suivi de sevrage et de repérer les consommateurs à risque. Il reste alors à établir les signes de dépen-dance et à rechercher les complications somatiquespsychologiques et sociales liées à cette consommationexcessive.
Aspects médico-légaux
Les mesures relatives à la lutte contre l’alcoolisme sonttrès nombreuses. Seules seront indiquées les principalesdispositions.
Seuils
Le rôle joué par l’imprégnation alcoolique des conduc-teurs dans la mortalité routière a inspiré des dispositionsréglementaires strictes visant à réprimer la conduitesous l’influence de l’alcool. Des contrôles préventifs enl’absence d’accident ou d’infraction ont été institués en1978 dans un but de dissuasion. Les contrôles se fontdans l’air expiré (éthylotest) et sont confirmés par unemesure de l’alcoolémie en cas de positivité. La loi du29 août 1995 stipule que « la conduite de tout véhiculemême en l’absence de tout signe d’ivresse manifeste »est un délit lorsque l’alcoolémie est supérieure ou égaleà 0,50 g/L ou lorsque la concentration d’alcool pur dansl’air expiré est supérieure ou égale à 0,25 g/L.
Circonstances
La lutte contre l’alcool au volant inspire le dispositifréglementaire national mais les circonstances de contrôlede l’imprégnation éthylique ne se limitent pas aux situa-tions d’accident de la circulation ou d’infraction au codede la route. Elle est obligatoire en cas de crime, délit ouaccident ayant entraîné un décès et peut être effectuée àl’initiative des agents de police administratifs ou judi-ciaires sur le responsable et la victime. Le refus ducontrôle est passible d’emprisonnement (1 an) et d’uneamende de 25 000 F.
Modalités du contrôle
• Alcootest : effectué à la demande d’un représentant del’autorité publique, il a pour but de dépister une impré-gnation éthylique. Négatif, il dispense de la mesurequantitative de l’alcool dans l’air expiré ou dans le sangmais ne constitue pas un préalable à ces mesures.• Éthylotest : il permet la mesure de la concentrationd’alcool dans l’air expiré à l’aide d’un appareil homologué.Le seuil légal est inférieur à 0,25 g/L. Une confirmationpar mesure de l’alcoolémie est souhaitable.• Alcoolémie : la prise de sang (et l’examen médicalpréalable) pour la mesure de l’alcoolémie est effectuéepar un médecin requis à cet effet au maximum dans les6 h suivant une infraction ou un accident. Les résultatssont consignés sur une fiche médicale ad hoc remise àl’autorité requérante. Le nécessaire de prise de sang estmis à la disposition du médecin par l’autorité – officierou agent de police administratif ou judiciaire – qui assisteau prélèvement sanguin. Le sang est réparti en 2 flaconsétiquetés et scellés. Le 1er échantillon est adressé pour dosage à un biolo-giste expert. Le 2e échantillon permet une éventuelleanalyse de contrôle pratiquée par un autre biologiste
A L C O O L I S M E
214 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0

Plus récemment, dans les mesures définissant un cadrelégal à l’évolution de la politique de lutte contre lesdrogues, des mesures concernant l’alcool ont étéincluses.• Alcoolisme et travail : le code du travail interdit l’intro-duction de boissons alcoolisées fortes sur le lieu de travailet l’employeur a l’obligation de mettre à disposition des boissons non alcoolisées et de fournir gratuitementde l’eau.En cas de suspicion d’imprégnation alcoolique sur lelieu de travail, le médecin du travail (ou éventuellementun médecin libéral) pratique un examen médical,propose la réalisation d’un alcootest dont le résultat nedoit pas être divulgué et préconise des mesures visant à supprimer le risque immédiat (repos à l’infirmerie,retour à domicile avec accompagnement, hospitali-sation) ou différé (cure de sevrage, suivi médical, chan-gement de poste de travail).
Structures de prise en charge
Le sevrage thérapeutique s’impose chaque fois qu’il y aalcoolodépendance. Il s’intègre dans une stratégie globale de soins visant à traiter les symptômes dedépendance physique et à prévenir les complicationsinduites par l’arrêt brutal de la consommation d’alcool.Un suivi thérapeutique prolongé est nécessaire en ambu-latoire ou en institution. Tout doit être mis en œuvrepour transformer la démarche de la prise de consciencede l’imprégnation alcoolique et du sevrage en une expé-rience positive et valorisante sur le plan personnel, fami-lial et professionnel. Diverses structures de prise encharge peuvent aider à la réalisation de ces objectifs.
Médecin généraliste
Son action est déterminante pour le repérage et le dia-gnostic de l’alcoolisme. Il lui est possible d’initier et desuivre le sevrage si sa disponibilité est suffisante. Il peutaussi orienter le patient vers d’autres structures.
Centre de cure ambulatoire en alcoologie (CCAA)
Anciennement dénommée centre d’hygiène alimentaire,cette structure a un rôle d’accueil, de dépistage, de pré-vention et de traitement et d’aide aux buveurs excessifsou alcoolodépendants. Son accès est gratuit. Elle com-porte une équipe médicale, paramédicale et sociale. Sonrecrutement est assuré par les médecins généralistes et dutravail, les centres hospitaliers et les services de la DDASen cas d’alcoolémie contrôlée supérieure à 0,5 g/L.
Hospitalisation
Elle permet de soustraire le patient à son environnementet garantit la réalité du sevrage tout en permettant lebilan et le traitement d’une affection associée. Elle
expert. Des formulaires comportant 3 fiches (A, B et C)encadrent ce contrôle. La fiche A, remplie par l’autoritérequérante, permet une description du comportement ;la fiche B, remplie par le médecin, concerne l’examenclinique médical. Le résultat du dosage d’alcoolémie estconsigné sur la fiche C. Il appartient au médecin expertd’établir un rapport final sur la base des fiches A, B et Cet d’adresser ses conclusions au procureur. Une analysede contrôle peut être effectuée dans les 5 j à la demandedes magistrats.
Conséquences de la mise en évidenced’une imprégnation alcoolique documentée
Si l’on constate une conduite en état d’imprégnation, levéhicule est immobilisé en l’absence d’un autre conduc-teur. Un retrait de 3 points du permis de conduire ainsiqu’une amende pouvant aller jusqu’à 5 000 F sont infligés.
Principales dispositions législatives
• Comportement dangereux : une mise sous contrôlede l’autorité sanitaire (DDAS) est prévue par la loi du15 avril 1954 qui considère l’alcoolique comme unmalade.La loi du 27 juin 1990 régit les hospitalisations sans leconsentement du malade. L’hospitalisation d’officeordonnée par les autorités est applicable aux personnes« dont les troubles mentaux compromettent l’ordrepublic ou la sûreté des personnes ». L’arrêté d’hospitali-sation d’office doit être motivé et circonstancié parl’existence de troubles mentaux attestés par un certificatmédical rédigé par un psychiatre. L’hospitalisation surdemande d’un tiers est faite à la demande de l’entouragemoyennant 2 certificats médicaux attestant que « l’étatde santé rend impossible le consentement et impose dessoins immédiats assortis d’une surveillance constante enmilieu hospitalier ».• Répression de l’ivresse : l’ivresse publique constitueune infraction passible de peines d’emprisonnement etd’amendes. Les mesures réglementaires imposent deprésenter l’impétrant à un médecin aux fins d’examenmédical. Un certificat de non-hospitalisation est délivrési l’hospitalisation n’est pas jugée nécessaire. La per-sonne est alors maintenue en chambre de sûreté jusqu’àcomplète récupération d’un état normal.• Protection des mineurs : plusieurs textes législatifsprotègent les mineurs contre l’alcool tels que : entrée desmineurs dans les débits de boisson, nature des consom-mations, zones protégées autour des établissements sco-laires, interdiction de publicité dirigée vers les jeunes.• Publicité : la loi du 10 janvier 1991 dite loi Évin fixeles conditions de la publicité pour les boissons alcooliséesselon le principe que le message publicitaire doit selimiter à quelques indications sur le degré alcoolique,l’origine et la dénomination, assortis d’une mentionavertissant du danger d’abuser de l’alcool.
Endocrinologie - Métabolisme - Nutrition
215L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0

s’impose en cas de syndrome de sevrage sévère. Limitéedans le temps, cette hospitalisation qui correspond à latraditionnelle « cure de désintoxication » n’a de sens ques’il existe un suivi post-hospitalisation par le médecintraitant, le centre de cure ambulatoire en alcoologie, unmouvement d’anciens buveurs ou un centre de post-cure.
Centres de cure
Les établissements spécialisés dans la prise en charge dela dépendance alcoolique réalisent un sevrage en unesemaine suivi d’une préparation à une vie sans alcoolpendant 3 semaines et sont destinés aux malades fragiles.
Centres de post-cure (long séjour)
Ils interviennent après la phase de sevrage pour laconsolider par un suivi de 1 à 3 mois. L’admission nedevrait s’y faire que sur la base du volontariat assortied’une prescription médicale pour permettre la prise encharge du séjour par l’assurance médicale. Un suivimédical et psychologique et des activités de réhabili-tation y sont proposés.
Associationsd’anciens malades alcooliques
Elles ont pour but de faciliter des relations amicales sansrisque de consommation d’alcool, de fournir un lieu oùl’on peut parler du problème d’alcool avec des per-sonnes qui le comprennent et présenter des exemples devie sans alcool. Elle se rapprochent d’une thérapie degroupe, permettent de mieux prendre conscience de laproblématique de l’alcool et de conforter l’abstinenceselon qu’elle s’adresse à des buveurs dépendants ou àdes buveurs sevrés. Les plus connues sont lesAlcooliques anonymes (AA), la Croix Bleue, Santé etFamille, la Croix d’Or. ■
A L C O O L I S M E
216 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
• La consommation excessive, aiguë ou chronique, et l’alcoolodépendance constituent un problème majeur de santépublique qu’il convient de repérer et traitercomme une maladie.
• Au-delà d’une législation répressive visant surtout l’alcoolisation aiguë et en dépitde la diminution de la fréquencedes complications, une vigilance extrêmereste de mise.
• Le médecin généraliste joue un rôle capitaldans le dépistage de l’alcoolisme. Il est aidépar diverses structures de prise en charge auxquelles il faut faire appel à bon escient.
Points Forts à retenir
1 / Répercussions de l’alcoolisme chronique
Atteinte hépatique :– stéatose (réversible) : 20 à 50 % des buveurs excessifs ;– cirrhose avec insuffisance hépatocellulaire et hypertension portale :10 à 30 % des alcooliques ;– expression plus retardée chez les hommes que chez les femmes
(20 à 25 ans d’alcoolisme).Cancers :– voies aérodigestives supérieures, potentialisés par le tabac ;– gastrique : non prouvé ;– foie : associé aux complications hépatiques de l’alcool ;– sein : même pour de faibles consommation ;– pancréas : pancréatite chronique ± calcifianteCœur :– cardiomyopathie hypokinétique dilatée ;– troubles du rythme (alcoolisation aiguë).Système nerveux central :– démence ;– atrophie cérébrale et cérébelleuse ;– comitialité (risque X 7) ;– psychopathologies diverses.Système nerveux périphérique :– polynévrite (10 %) ;– névrite optique (favorisée par le tabagisme).Muscle :– amyotrophie ;– rhabdomyolyse (alcoolisation aiguë).Os : ostéoporoseGlandes endocrines :– impuissance ;– hypogonadisme.
2 / Exemple d’un questionnaire standardisé
Questionnaire CAGE/DETA (version française)
Deux réponses positives ou plus incitent à suspecter un problèmed’alcool. Ces questions sont à intégrer dans l’entretien clinique chezdes patients qui reconnaissent être des consommateurs réguliersd’alcool.
POUR APPROFONDIR
Girre C, Hispard E. Législation se rapportant à l’alcoolisme et àsa prévention. Toxicologie pathologie professionnelle. EncyclMed Chir (Paris) 1995 ; 16-047-A-21 : 4 p.
Got C,Weill J. L’alcool à chiffres ouverts. Paris : Seli-Arslan, 1997.
Objectifs, indications et modalités du sevrage du patient alcoolo-dépendant. Conférence du Consensus. Concours Med 1999 ;121 : 2311-8.
Les malades de l’alcool.Monographie.Rev Prat 1999 ;49 :365-405.
POUR EN SAVOIR PLUS
Avez-vous déjà ressenti le besoin de diminuer votre consommation d’alcool ?Votre entourage vous a-t-il déjà fait remarquer que vousconsommez trop d’alcool ?Avez-vous déjà eu l’impression que vous buvez trop ?Avez-vous déjà eu besoin d’alcool le matin pour vous sentiren forme ?

Endocrinologie - MétabolismeB 335
881L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
Les apports moyens correspondent à la moyenne desbesoins individuels dans une population donnée.Les apports de sécurité (ou apports recommandés ou deréférence) permettent de couvrir les besoins de la quasi-totalité des sujets d’une classe d’âge donnée. Ils corres-pondent aux besoins moyens en leur ajoutant + 2 DS.Le nouveau-né (0-1 mois) étant exclu de cette question,seront abordés ici les besoins nutritionnels des nourris-sons (1 mois-1 an), des enfants (1-10 ans), et des adoles-cents (10-18 ans), ainsi que ceux de la femme enceinteet allaitante, et de la personne âgée (≥ 65 ans).La couverture des besoins nutritionnels a pour but d’as-surer un état de santé normal. Ceux-ci doivent corres-pondre à la dépense énergétique totale d’un individu,c’est-à-dire la dépense énergétique de repos, la thermo-régulation, la transformation des nutriments en sourced’énergie et l’activité physique, auxquelles il faut ajouter,chez l’enfant, la croissance. La définition des besoinsnutritionnels passe en pratique par la détermination desapports de sécurité (antérieurement dénommés apportsrecommandés). Ceux-ci sont très variables selon lespays, les périodes et les études, et il existe de plus unegrande variabilité individuelle justifiant donc de consi-dérer ces apports de sécurité comme des données statis-tiques utilisables à l’échelon d’une population mais avecprudence pour un individu donné.
Nourrisson, enfant, adolescent
Énergie
L’estimation des besoins énergétiques peut être faite parl’analyse des ingesta spontanés d’une population desujets en bonne santé ou par l’analyse des différentescomposantes de la dépense énergétique.
Définitions
L’apport minimal (moyenne - 2 DS) est la plus faiblequantité d’un nutriment assurant le maintien des fonc-tions et un état physiologique normal, en assurant unecroissance optimale chez l’enfant et un poids stable chez l’adulte.
Besoins nutritionnels du nourrisson,de l’enfant,de la femme enceinte et allaitant et de la personne âgée Énergie, fer, protéines, acides gras essentiels, vitamine D, calcium
PR Frédéric GOTTRAND1, DR David SÉGUY2
1. Clinique de pédiatrie, hôpital Jeanne-de-Flandre ; 2. service de nutrition, CHU Lille, 59037 Lille Cedex.
• Les besoins nutritionnels sont différents des apports de sécurité qui correspondent aux recommandations destinées à couvrir les besoins de 97,5 % d’une population donnée.
• Le coût énergétique de la croissance est maximal durant les 6 premiers mois de la vie où il représente 20 à 25 % des ingesta puis il diminue ensuite rapidement.
• Les besoins protéiques sont de 7,3 g/j en moyenne au cours de la 1re année.
• La grossesse comporte 2 phases sur un plannutritionnel : la première est une phase anabolique pour la mère lorsque la croissancedu fœtus est faible ; la seconde est une phasecatabolique pour la mère pendant laquelle le fœtus prélève sur les réserves maternelles une partie de l’énergie nécessaire à sa croissance.
• La dénutrition chez la personne âgée provientd’une réduction des apports alimentaires (causes physiques, métaboliques,environnementales et iatrogéniques),mais aussi d’une augmentation des besoins(maladies intercurrentes).
Points Forts à comprendre

Les besoins énergétiques en fonction de l’âge sont rap-portés dans le tableau. Sur un plan qualitatif, la réparti-tion des différents nutriments est un peu différente decelle de l’adulte avec 50 à 55 % de glucides, 30 à 35 %de lipides, et 9 à 10 % de protides.
Protéines
Les besoins protéiques correspondent à la somme desbesoins pour la maintenance et pour la croissance, c’est-à-dire les besoins en azote et en acides aminés essentielsnécessaires pour permettre une croissance normale de lataille et du poids sans compromettre l’équilibre dumilieu intérieur, ni dépasser les capacités hépatiques etrénales d’élimination des déchets. La détermination deces besoins peut passer par 2 méthodes : l’observationde la consommation spontanée d’enfants en bonne santéou la méthode factorielle qui consiste à faire la sommedes pertes obligatoires d’azote et de la quantité de protéines déposées au cours de la croissance. L’apportprotéique de maintenance, destiné à compenser lespertes obligatoires (sueurs, selles, urines, phanères, des-quamation cutanée), est estimé à 0,7 à 0,9 g/kg/j.L’apport protéique nécessaire au développement de lamasse musculaire et à l’accroissement squelettique estvariable en fonction de la vitesse de croissance. Il estestimé à 1,3 g/kg/j au cours du 1er mois de vie,0,56 g/kg/j de 2 à 3 mois, 0,29 g/kg/j de 5 à 6 mois,0,2 g/kg/j de 9 à 12 mois, et 0,08 g/kg/j de 2 à 3 ans.Ainsi au cours de la 1re année, la somme des besoins demaintenance (qui augmentent avec l’âge) et des besoinsde croissance (qui diminuent avec l’âge) reste constanteen moyenne à 7,3 g/j. Les apports conseillés actuelle-ment sont sensiblement inférieurs à ceux antérieurementétablis, en particulier chez le nourrisson (voir tableau).Les besoins protéiques de l’adolescent sont importants.L’apport protéique doit être associé à un apport éner-gétique suffisant pour favoriser la synthèse protéique,dans le cas contraire, une partie des protéines sert à laproduction de l’énergie. Le rapport optimal calories surazote n’est actuellement pas précisément déterminé, etl’on admet habituellement que les protéines doiventreprésenter environ 10 % de l’énergie totale. De 10 à 20 ans, le garçon fixe 1 350 g d’azote (7,5 kg de protéine),alors que la fille en fixe nettement moins : 750 g d’azotesoit 3,75 kg de protéine.À côté de ces aspects quantitatifs, il est nécessaire deprendre en compte des données qualitatives concernantl’apport protéique. En effet, un certain nombre de para-mètres doivent être envisagés : l’apport énergétique totalqui modifie les besoins azotés, la valeur nutritionnelledes protéines de l’alimentation, le coefficient d’utilisa-tion digestive, la teneur en acides aminés des protéines.L’apport spécifique en acides aminés, en particulier enacides aminés essentiels, n’est pas précisé. Les protéinesalimentaires constituant la base de l’alimentation del’enfant permettent, en effet, au niveau d’apportsconseillés, de couvrir tous les besoins en acides aminés.Ces acides aminés sont au nombre de 8 chez l’adulte :
leucine, thréonine, lysine, tryptophane, phénylalanine,valine, méthionine, isoleucine. Chez l’enfant en crois-sance, il faut y ajouter l’histidine. Le coefficient d’utili-sation protéique, défini par le rapport azote retenu surazote ingéré est de 100 et 90 % respectivement pourl’œuf et le lait de femme, qui sont ainsi considéréscomme les protéines de référence. Il est de 75 % pour lelait de vache et 52 % pour la farine de blé, les protéinesvégétales ayant en général un coefficient d’absorptionmoins bon que les protéines animales. L’indice pro-téique chimique est défini par le pourcentage du taux del’acide aminé limitant (acide aminé dont le taux est leplus bas par rapport à la protéine de référence) dans cetteprotéine par rapport à la protéine de référence. La plupartdes protéines végétales ont un indice protéique chi-mique médiocre.
Acides gras essentiels
Les acides gras essentiels (AGE) sont les constituantsindispensables des membranes cellulaires, en particulierdu tissu cérébral. Ce sont des acides gras polyinsaturés :l’acide linoléique (C18 : 2n-6) et l’acide α-linolénique(C18 : 3n-3). Leur carence, rare dans les pays dévelop-pés, se manifeste par un retard de croissance staturo-pondéral, des anomalies cutanéo-phanériennes, desinfections à répétition et des perturbations du dévelop-pement psychomoteur. Les apports recommandés sontde 3,5 à 5 % de l’apport énergétique total pour l’acidelinoléique, et de 0,5 à 1 % pour l’acide α-linolénique,avec un rapport entre ces 2 acides gras de 4 à 6. À l’in-verse, des apports excessifs de l’ordre de 10 % de l’ap-port énergétique total pour l’acide linoléique, et de plusde 3 % pour l’acide α-linolénique sont à déconseiller(inhibition des enzymes clés du métabolisme des acidesgras comme la D6-désaturase par l’acide α-linolénique,production de radicaux libres).
Fer
La carence en fer est la plus fréquente des carencesnutritionnelles dans les pays industrialisés. Quel quesoit l’âge, chez le sujet normal, l’absorption digestive dufer est basse, de l’ordre de 10 à 15 %, ce qui fait que desapports de 10 à 15 mg/j sont nécessaires pour couvrirdes besoins de 1 à 2 mg/j. Le fer héminique (viande,poisson) est mieux absorbé que le fer non héminique(lait, végétaux, œufs). La teneur en fer du lait de vacheet du lait maternel est faible, mais la biodisponibilité dece dernier est élevée (proche de 50 %), de sorte que chezle nourrisson au sein, aucune supplémentation n’estnécessaire jusqu’à l’âge de 6 mois.Bien que les besoins de l’adolescent en fer soient enthéorie identiques à ceux des adultes (12 à 13 mg/j), ilexiste cependant des risques de carence en fer, notam-ment chez le garçon au moment de la poussée de crois-sance et chez la fille à l’installation des premièresrègles. C’est pourquoi les recommandations sont plusimportantes à l’adolescence (voir tableau).
B E S O I N S N U T R I T I O N N E L S
882 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
Endocrinologie - Métabolisme
883L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
1-2 mois
3-5 mois
6-8 mois
9-11 mois
1-3 ans
4-6 ans
7-10 ans
11-14 ans
15-18 ans
Femmeenceinte
Femmeallaitante
Personne âgéestable
Personne âgéemalade
450
600
700
850
1 300
1 700
1 950
2 200 (fille)
2 500 (garçon)
2 300 (fille)
2 900 garçon)
2 150-2 250
2 500
Jamais < 1 500(25-30 kcal/kg/j)
(> 30 kcal/kg/j)
10
10
10
10
10-12
25
25
40 (fille)
42 (garçon)
52 (fille)
58 (garçon)
70
80
12-15 % aet(1 g/kg/j)
(> 1,5 g/kg/j)
4-6
4-6
4-6
4-6
?
?
?
??
??
5,5-5,7
5,5-5,7
2-3
2-3
6-10
6-10
6-10
6-10
10
10
10
1212
18 (fille)
15 (garçon)
20-100
13
10
10
1 000
1 000
1 000
1 000
400
400
400
400400
400400
800
600
480-800
480-800
400
400
600
600
800
800
800-1 000
1 000-1 2001 000-1 200
1 000-1 2001 000-1 200
1 200-1 500
1 200
1 000-1 200
1 000-1 200
kcal : 1 kilocalorie = 4,18 kilojoules.1 g de protéine = 6,25 g d’azote.AGE % aet : acides gras essentiels exprimés en % de l’apport énergétique total et représentant la somme de l’acide linoléique et de l’acide α-linoléniquedont le rapport est de 4 à 6 quel que soit l’âge.UI : unité internationale = 0,025 mg de vitamine D.
Âge Énergie(kcal/j)
Protéines(g/j)
AGE(% aet)
Fer(mg/j)
Vit D(UI/j)
Calcium(mg/j)
Apports de sécurité (recommandés) en France
TABLEAU

Vitamine D Les besoins en vitamine D, dont le rôle est fondamentalpour l’absorption intestinale du calcium, sont impor-tants à considérer au cours des 2 premières années de lavie, période où la croissance staturale est la plus rapide.Les réserves en vitamine D du nouveau-né dépendentétroitement de celles de leur mère et sont donc le plussouvent basses. Le lait maternel contient peu de vitamine D(25 à 70 UI/L). Les laits pour nourrisson et les laits desuite sont supplémentés depuis 1993 et contiennent 40 à120 UI/100 kcal. Une supplémentation de 400 à 1 000UI/j reste recommandée entre la naissance et 2 ans, etpendant les mois d’automne et d’hiver jusqu’à 5 ans.Pendant l’adolescence, une supplémentation orale envitamine D reste discutée et dépend de l’origine ethnique,de l’exposition au soleil et du type d’alimentation.
CalciumLes besoins en calcium varient selon la période de crois-sance considérée. Les besoins calciques sont très élevésà l’adolescence (tableau). Entre 9 et 16 ans, la filleconstitue 50 % de son capital osseux, dont on sait qu’ilest acquis pour toute sa vie et dont la qualité est proba-blement un facteur de protection des complicationsostéoporotiques de l’âge adulte.
Femme enceinte et allaitante
ÉnergieLes recommandations nutritionnelles chez la femmeenceinte sont destinées à couvrir les besoins propres de lagrossesse et à assurer au fœtus une croissance normale.La première moitié de la grossesse correspond à unephase anabolique pour la mère où la croissance du fœtusest faible et où la mère constitue des réserves énergétiques(lipide, glycogène). La seconde moitié est une phase cata-bolique pour la mère pendant laquelle le fœtus prélèveune partie de l’énergie nécessaire à sa croissance sur lesréserves maternelles. Le coût énergétique de la grossessea été évalué entre 70 000 et 80 000 kcal, ce qui représenteun apport théorique supplémentaire de 250 à 280 kcal/j. Du fait de mécanismes d’adaptation méta-bolique au cours de la grossesse, un supplément caloriquemodéré de 100 kcal/j au cours des 2 derniers trimestres dela grossesse est en fait suffisant pour mener à bien unegrossesse normale. Les recommandations habituelles res-tent cependant plus importantes avec une augmentationde la ration calorique de l’ordre de 100 à 300 kcal/j. Le coût de la production journalière de 800 mL de laitest de 500 à 600 kcal/j. L’apport énergétique supplémen-taire conseillé dépend en fait du gain de poids de la mèrependant la grossesse, et ne doit pas dépasser 500 kcal/j.Un apport énergétique insuffisant au cours de la lacta-tion entraîne principalement une réduction du volumede lait produit mais en modifie peu la composition.
ProtéinesLa femme enceinte est capable de mobiliser durant le der-nier trimestre de gestation, au moment de la période decroissance la plus rapide du fœtus, les réserves protéiquesaccumulées au début de la grossesse. Une augmentationdes apports protéiques de 10 g/j est cependant recomman-dée au cours de la grossesse (voir tableau).L’allaitement ne s’accompagne pas d’un mécanismed’épargne analogue à celui de l’anabolisme gravidique.La production de 850 mL de lait par jour correspond àune exportation protéique d’environ 10 g/j. Le rende-ment de synthèse protéique étant estimé à 50 %, unapport supplémentaire de 20 g/j est recommandé durantla période de lactation (voir tableau).
Acides gras essentielsCompte tenu des faibles réserves en acides gras polyinsa-turés en n-3 dans l’organisme humain adulte, il est prudentde recommander, au cours de la grossesse et de la lacta-tion, un apport en acide α-linolénique un peu supérieuraux recommandations de l’adulte, soit 1 à 1,2 % de l’éner-gie ingérée. De même, l’apport d’acide linoléique sera de4,5 % de l’apport énergétique total durant ces périodes,contre 3 % en dehors de la grossesse et de la lactation.
FerAu cours de la grossesse, les besoins en fer sont très éle-vés. Les pertes en fer au cours de la grossesse sont enmoyenne de 1 285 mg, réparties en : augmentation de lamasse des hématies (500 mg), fer fœtal (290 mg), fer pla-centaire (25 mg), hémorragie du post-partum (250 mg),pertes physiologiques (220 mg). Les besoins en ferabsorbé sont de 0,8 mg/j le 1er trimestre, 4,4 mg/j le 2e etde 6,3 mg/j le 3e. Cela explique que, malgré l’augmen-tation de l’absorption de fer en fin de grossesse, lesapports alimentaires ne sont habituellement pas suffisantspour compenser ces pertes, raison pour laquelle une sup-plémentation médicamenteuse précoce et systématiqueest recommandée (50 à 100 mg de fer élément par jour).Le lait de femme contient de 0,04 à 0,05 mg de fer/100 mL.Cela représente une perte de 2,8 à 3,2 mg/j de fer. Lesapports recommandés pendant la période d’allaitementsont de 21 mg/j.
Vitamine DLes situations de carence en vitamine D sont particuliè-rement fréquentes en fin de grossesse et à la fin de l’hi-ver chez les femmes d’Europe du Nord ou non exposéesau soleil. Une supplémentation à partir du 6e mois degrossesse est donc souhaitable dans ces conditions, sousforme d’un apport quotidien de 400 à 600 UI (10 à 15 µg/j)ou d’une dose de charge de 100 000 UI.
CalciumL’apport calcique recommandé est de 1,2 à 1,5 g/j chez lafemme enceinte, et de 1,2 g/j au cours de l’allaitement.
B E S O I N S N U T R I T I O N N E L S
884 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9

Protéines
Les protéines constituent 12 à 15 % de la ration énergé-tique totale chez le sujet stable. Cependant si la consom-mation énergétique diminue, il faut s’efforcer de main-tenir un apport protéique minimal de 1 g/kg/j. Cet apportpeut être augmenté au-delà de 1,5 g/kg/j en cas de mala-die (15 à 20 % de la ration énergétique totale). Commechez l’adulte, il est recommandé de diversifier lessources de protéines animales et végétales.
Acides gras essentiels
Les lipides ne doivent pas en théorie dépasser 30 % del’apport énergétique mais ils atteignent souvent 35 à40 % de la ration dans les pays occidentaux. Ces apportslipidiques ne doivent pas être inférieurs à 20 % au risquede diminuer l’apport énergétique et la palatabilité des ali-ments. Les besoins quotidiens en acides gras essentielssont de 10 g/j soit 2 à 3 % de l’apport énergétique total.
Fer
Les apports quotidiens recommandés ne semblent pasdifférer de ceux de l’adulte (10 mg) et sont générale-ment couverts par l’alimentation. La carence martialeest d’ailleurs rare (de l’ordre de 5 %) chez le sujet âgé,en dehors des cas où elle est secondaire à une pathologieentraînant un saignement chronique telle qu’une hémor-ragie digestive occulte.
Vitamine D
La diminution de l’absorption digestive et de la synthèsecutanée de vitamine D, la faible exposition au soleil etl’alitement favorisent la carence en vitamine D qui estfréquente chez le sujet âgé. L’apport alimentaire de vita-mine D est faible (huiles de poisson). Alors qu’un apportquotidien de 400 UI est suffisant chez l’adulte, il doitêtre au minimum de 480 chez la personne âgée. La dosesera augmentée à 800 UI/j en cas d’absence d’exposi-tion solaire surtout en hiver ou chez le sujet alité. Letraitement peut être administré quotidiennement ou defaçon cumulée par mois ou par trimestre.
Calcium
Les apports conseillés en calcium augmentent avecl’âge. La perte du calcium osseux lors du vieillissementfait intervenir 2 phénomènes :– l’ostéoporose sénile qui débute à 40 ans et se manifes-te après 70 ans dans les 2 sexes est secondaire à unediminution de l’activité ostéoblastique ; elle entraîne àlong terme la survenue de fractures du col du fémur ;– l’ostéoporose ménopausique par carence œstrogé-nique qui touche la femme après la ménopause et favori-se tassements vertébraux et fractures.Par ailleurs, le taux d’absorption intestinal du calciumdiminue avec l’âge tandis que la calcémie reste stable.
Personne âgée
Énergie
La dépense énergétique diminue en moyenne de 10 %par décennie à partir de 60 ans avec toutefois une grandevariabilité interindividuelle. Ce phénomène est expliquépar la diminution de la masse maigre et surtout de l’activité physique lors du vieillissement. Cependant, ilfaut souligner que, chez la personne âgée, le risquemajeur n’est pas l’obésité mais la dénutrition qui est très fréquente et peut atteindre 50 % des sujets hospita-lisés. Les facteurs qui provoquent cette dénutrition sontde 2 types : ceux qui vont diminuer les apports alimen-taires et ceux qui vont augmenter les besoins nutri-tionnels.La carence d’apports du sujet âgé a différentes origines :physique (difficultés d’approvisionnement, augmentationdu seuil de perception du goût et de l’olfaction, sécheressebuccale, altération de la denture, de la muqueuse et de lasécrétion d’enzymes digestives), métabolique (troublesde régulation de l’appétit à l’origine d’une baisse durabledes ingesta après un épisode d’anorexie temporaire, diffi-cultés d’adaptation métabolique au jeûne), environ-nementale (solitude, dépression, baisse des revenus) etiatrogénique (surconsommation médicamenteuse, régi-mes trop restrictifs ou désodés anorexigènes).Avec le vieillissement, la diminution du rendement mus-culaire et surtout les maladies intercurrentes vont aug-menter les besoins nutritionnels. La morbidité va aggra-ver l’état nutritionnel et faire entrer le sujet âgé dansl’anorexie chronique. La dénutrition protéino-energétiqueaugmente de 2,5 à 4 fois le risque de mortalité chez lesujet âgé lors de son hospitalisation. Sa prévention estessentielle dès qu’une anorexie est dépistée.Il faut distinguer les besoins du sujet sain et stable dontl’alimentation doit être suffisante pour maintenir un équi-libre satisfaisant et retarder le vieillissement, des besoinsdu sujet malade et affaibli qui sont augmentés. Lesapports doivent tenir compte de l’activité physique quimodifie beaucoup la dépense énergétique. Pour une acti-vité modérée, les besoins sont proches de 25 kcal/kg/j depoids corporel et atteignent 30 kcal/kg/j pour une activitéplus intense. Les apports recommandés pour un sujet de70 kg sont de 1 750 à 2 100 kcal/j en sachant qu’un apportinférieur à 1 500 kcal/j, quel que soit le poids, ne permetpas de couvrir les besoins en vitamines et en minéraux.En cas de pathologie les apports doivent être supérieurs à30 kcal/kg/j.Les glucides constituent 50 à 55 % de l’énergie néces-saire, au profit des sucres complexes. La proportion dessucres simples doit être limitée à moins de 20 % de l’ap-port énergétique total. En effet, l’âge aggrave la tendan-ce à l’hyperglycémie par retard de sécrétion du pic d’in-suline post-prandial et résistance périphérique auglucose. Le rapport glucides sur protides doit être com-pris entre 2,5 et 3 afin de permettre un métabolismesatisfaisant des protéines ingérées.
Endocrinologie - Métabolisme
885L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9

Chez le sujet âgé, l’absorption du calcium dépend essen-tiellement de la concentration intra-intestinale de cal-cium et par conséquent de l’apport calcique. Les apportsrecommandés sont de 1 000 à 1 200 mg/j chez les sujetsâgés. Cependant, l’obtention d’un apport dépassant 1 g/jn’est possible qu’en consommant quotidiennement 4 produits laitiers et des boissons riches en calcium ensachant qu’il existe souvent une intolérance au lactosepar insuffisance enzymatique chez la personne âgée. La prévention de l’ostéoporose doit commencer dèsl’enfance au moment de la constitution du capital cal-cique et se poursuivre à l’âge adulte grâce au maintiende l’activité physique, du statut hormonal en particulieren œstrogènes et grâce à un apport suffisant en calcium,en phosphate, en vitamine D, en fluor et en protéines.■
B E S O I N S N U T R I T I O N N E L S
886 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
• Les apports nutritionnels doivent couvrir la dépense énergétique de repos,la thermorégulation, la transformation des nutriments en source d’énergie et l’activitéphysique.
• Chez l’enfant, les apports nutritionnels doivent aussi couvrir la croissance.
• La prévention de la dénutrition protéino-énergétique est essentielle dès qu’une anorexieest dépistée chez le sujet âgé.
• Le sujet âgé, quel que soit son poids,ne doit jamais avoir un apport calorique inférieur à 1 500 kcal/j ni un apport protéiqueinférieur à 1 g/kg/j.
Points Forts à retenir
Comité de nutrition de la Société française de pédiatrie. Besoins en protéines des nourrissons et des enfants en bonne santé. Arch Pediatr 1997 ; 4 : 373-82.
Dupin H, Abraham J, Giachetti I. Apports nutritionnels conseilléspour la population française. CNRS-CNERMA (ed) 2e édition. Paris : Tec et Doc Lavoisier, 1992.
Ghisolfi J, Ricour C, Putet G, Goulet O. Apports nutritionnelsconseillés en France chez le nourrisson et l’enfant. In : Ricour C,Ghisolfi J, Putet G, Goulet O (eds). Traité de nutrition pédiatrique.Paris : Maloine, 1993 : 361-71.
Gottrand F, Loeuille GA, Douchain F, Turck D. Apports diététiquesinappropriés et erreurs nutritionnelles de l’enfance. In : Ricour C,Ghisolfi J, Putet G, Goulet O (eds). Traité de nutrition pédiatrique.Paris : Maloine, 1993 : 983-95.
Lesourd B, Ferry M. Le sujet âgé. In : SFNEP (eds). Traité de nutri-tion artificielle de l’adulte. Paris : Mariette Guéna,1998 : 647-63.
Putet G. Besoins énergétiques. In : Ricour C, Ghisolfi J, Putet G,Goulet O (eds). Traité de nutrition pédiatrique. Paris : Maloine, 1993 : 123-32.
Sachet P. Les besoins nutritionnels de l’adolescent. Med Enfance1994 ; 4 : 11-3.
POUR EN SAVOIR PLUS

875L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
B 339
Cancers de la thyroïdeAnatomie pathologique, diagnostic
Dr Jean-Michel ANDRIEU, Dr Line BALDET, Pr Claude JAFFIOLService d’endocrinologie, CHU, hôpital Lapeyronie, 371, av. du doyen G.-Giraud, 34295 Montpellier cedex 5
• Le diagnostic de cancer thyroïdien est évoquédevant tout nodule thyroïdien.• Le développement des techniqueséchographiques fait que l’incidence de ces nodulesest de plus en plus importante or le pourcentage decancer est faible.• Aucun examen ne permettant de déterminer avecune certitude absolue la malignité ou la bénignitéd’un nodule, lorsqu’existe une suspicion clinique,seule l’intervention chirurgicale assurera lediagnostic.
Points Forts à comprendre Dans 80 % des cas, il existe une inclusion correspondantà une invagination du cytoplasme. On observe souvent desconcrétions calcaires appelées psammomes. L’envahisse-ment ganglionnaire est fréquent et précoce. Les adénopa-thies récurrentielles sont les plus fréquentes. Les métastasesganglionnaires sont présentes chez 25 à 30 % des patientsau moment du diagnostic. L’invasion vasculaire est plusrare, les métastases viscérales s’observent chez 5 à 17 %des malades, principalement dans les poumons et le sque-lette.• Formes histologiques variantes :15 à 20 % des cancerspapillaires ont des caractéristiques histologiques moinstypiques mais sont classés comme cancers papillaires enraison de leurs caractéristiques nucléaires. On distingueainsi :– le cancer papillaire sclérosant diffus : rare, observé pré-férentiellement chez le sujet jeune, il s’étend à tout un lobevoire à l’ensemble de la thyroïde. L’aspect peut évoquerune thyroïdite. On observe une infiltration tumorale dontle caractère papillaire peut être difficile à affirmer en rai-son d’une métaplasie malpighienne ;– le carcinome papillaire de forme vésiculaire : on décritdeux sous-types: la forme macrovésiculaire encapsulée, faited’un mélange de grandes vésicules d’allure normale et depetites vésicules d’allure tumorale, de bon pronostic et laforme folliculaire diffuse atteignant toute la thyroïde et s’ac-compagnant d’un taux élevé de métastases viscérales ;– le carcinome papillaire à cellules hautes : les cellules sontdeux fois plus hautes que larges, le cytoplasme est granu-leux, éosinophile, abondant. Observé surtout chez le sujetâgé, il représente 5 à 10 % des cancers papillaires. Le pro-nostic serait péjoratif ;– le carcinome à cellules oncocytaires : il représente 3 % des carcinomes thyroïdiens. L’architecture est papil-laire le plus fréquemment. Les noyaux sont sombres,nucléolés, irréguliers, les facteurs pronostiques sont iden-tiques à ceux des papillaires courants. Il survient à un âgeplus avancé ;– le carcinome à cellules cylindriques : exceptionnel et depronostic sombre ;– le microcarcinome papillaire : tumeur de moins de 1 cm,soit symptomatique découvert lors de l’exploration d’unnodule, soit occulte de découverte fortuite. Histologique-ment d’architecture vésiculaire ou papillaire, plusieurssous-types sont définis en fonction du mode d’encapsula-tion et de croissance. Très souvent métastasé au momentdu diagnostic, l’atteinte ganglionnaire dépend de l’invasi-vité, de la multifocalité et de la taille de la tumeur.
Le cancer thyroïdien représente une pathologie tumoraleassez rare, de pronostic favorable dans les formes diffé-renciées lorsqu’elles sont précocement prises en charge.D’où l’intérêt d’un diagnostic précoce et d’une confirma-tion anatomo-pathologique précise pour engager rapide-ment la thérapeutique adéquate.
Anatomie pathologiqueLa classification utilisée est celle préconisée par l’Organi-sation mondiale de la santé ; elle repose essentiellement surdes données morphologiques (tableau I).
Cancers différenciés d’origine vésiculaireDeux formes histologiques de cancers différenciés sontindividualisées : les cancers papillaires et les cancers vési-culaires.1. Cancer papillaire de la thyroïde• Forme histologique habituelle :ce sont des tumeursmalignes épithéliales, de souche vésiculaire, typiquementconstituées de formations papillaires et (ou) vésiculaires etcomportant des modifications nucléaires caractéristiques.Macroscopiquement, ces cancers se présentent comme destumeurs dures, blanchâtres et invasives. Parfois kystiques(10 %), ils sont souvent multifocaux et bilatéraux avec unefréquence qui varie selon les séries.Microscopiquement, on observe des papilles et des vési-cules. Chaque papille est formée par un axe conjonctivo-vasculaire bordé de cellules dont les noyaux apparaissentchevauchant, fissurés, clairs au centre, dits en verre dépoli.

876 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
C A N C E R S D E L A T H Y R O Ï D E
Macroscopiquement il se présente comme une tumeur detaille variable, souvent localisée à la jonction des tiers supé-rieur et moyen des lobes thyroïdiens. Bien limitée, elle peutparfois présenter un aspect infiltrant et envahir les tissusadjacents.Microscopiquement la forme typique représente 80 % deslésions. Les cellules sont rondes ou polyédriques. Lesmitoses sont rares. Certains noyaux présentent une inclu-sion cytoplasmique intranucléaire. Les granules neurosé-crétoires sont mis en évidence en microscopie optique oupar l’existence d’un immunomarquage positif pour lachrommogranine. Le stroma tumoral, parfois émaillé decalcosphérites est fait d’un mélange de substance hyalineet amyloïde. Il existe une immunoréactivité pour la calci-tonine.Il existe des variantes intéressant soit l’aspect des cellulesqui peuvent être oxyphiles ou géantes, soit le matériel sécré-toire qui peut être du mucus ou de la mélanine. Dans lesformes très indifférenciées, c’est la positivité de l’immu-nomarquage pour la calcitonine ou l’antigène carcino-embryonnaire (ACE) qui permet le diagnostic.
Autres tumeurs1. Lymphomes de la thyroïdeIls représentent 2 % des lymphomes extra-ganglionnaireset moins de 5 % des tumeurs malignes de la thyroïde. Ils’agit de lymphomes diffus, à grandes cellules, de type Bet d’origine centro-folliculaire. Ils sont confirmés par lesmarqueurs des lymphocytes B. La majorité se développentà partir d’une thyroïdite auto-immune.
2. Carcinomes vésiculairesTumeurs malignes épithéliales, de souche vésiculaire quine présentent pas les caractéristiques cytologiques des can-cers papillaires.• Forme histologique habituelle : macroscopiquement, lescarcinomes vésiculaires se présentent sous la forme d’unnodule isolé, unique dans 90 % des cas, ferme et de colo-ration beige. Ils peuvent être kystiques, multifocaux.Microscopiquement, du fait d’un important degré de res-semblance avec la thyroïde normale, le diagnostic anatomo-pathologique est difficile, il n’existe en effet aucun critèrecellulaire formel permettant d’affirmer la malignité. Ondistingue :– le carcinome vésiculaire à invasion minime : il représenteplus de 50 % des cancers vésiculaires. Histologiquement,il s’agit d’un nodule hypercellulaire, trabéculaire ou micro-vésiculaire. Le diagnostic de malignité repose sur la décou-verte de signes d’invasion des vaisseaux et (ou) d’infiltra-tion de la capsule ;– le carcinome vésiculaire largement invasif : le diagnosticde malignité est plus facile en raison de l’important enva-hissement vasculaire, thyroïdien ou extrathyroïdien. Parfoisbien différenciées, les cellules sont le plus souvent atypiquesavec une anusocaryose et une activité mitotique élevée.Le cancer vésiculaire se dissémine par voie hématogène,plus rarement lymphatique. Les sites métastatiques préfé-rentiels sont le poumon, l’os et plus rarement le cerveau.• Variantes du cancer vésiculaires– Cancer vésiculaire à cellules oxyphiles (cellules deHurthle) : les cellules oxyphiles sont de grandes cellulespolyédriques à cytoplasme abondant très riche en mito-chondries, éosinophile et granuleux. Les noyaux sont aty-piques ;– Les carcinomes peu différenciés et carcinomes insulaires :leurs caractères morphologiques et évolutifs sont intermé-diaires entre ceux des carcinomes différenciés et anapla-siques. Très invasifs, la différence avec les tumeurs ana-plasiques est liée à la présence d’images tantôt vésiculairestantôt papillaires.
Cancer anaplasiqueTrès agressif, il correspond au stade terminal de la dédiffé-renciation d’une tumeur vésiculaire. Il représente de 5 à 15 % des cancers de la thyroïde, il est plus fréquent dans lesrégions où domine la carence iodée. La tumeur est volumi-neuse, hémorragique, infiltrant thyroïde et tissus adjacents.Les cellules sont fusiformes, polygonales, géantes. L’im-munohistochimie permet d’éliminer un lymphome, un can-cer médullaire ou un cancer vésiculaire indifférencié. L’ex-tension est locale, avec envahissement des structures du cou.Les métastases pulmonaires sont les plus fréquentes.
Cancer médullaire de la thyroïdeIl se développe à partir des cellules C parafolliculaires etreprésente 5 à 8 % des cancers de la thyroïde. On décrit laforme sporadique survenant à tout âge et la forme fami-liale qui représente 20 à 30 % des cancers médullaires dela thyroïde.
• Cancers différenciés de la thyroïde (70 %)Cancer papillaire (60 à 80 %) :– forme histologique habituelle– papillaire de forme vésiculaire– papillaire sclérosant diffus– à cellules hautes– à cellules oncocytaires– à cellules cylindriquesCancer vésiculaire (20 à 40 %) :– forme histologique habituelle :
. à invasion minime
. largement invasif– à cellules oxyphilles (cellules de Hurthle)– carcinome insulaire
• Cancer médullaire de la thyroïde (7 %)• Cancer anaplasique (15 %)• Autres tumeurs rares– Lymphome de la thyroïde (5 %)– Fibrosarcome primitif de la thyroïde– Tératrome malin– Métastases intrathyroïdiennes
Formes histologiquesdes cancers de la thyroïde
en fonction de leurs fréquences
TABLEAU I

877L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
2. Fibrosarcome primitif de la thyroïdeLa distinction avec le cancer anaplasique est difficile.
3. TératomesIls sont très souvent malins chez l’adulte.
4. Métastases intra-thyroïdiennesTrès fréquemment observées lors d’autopsies, elles peu-vent révéler un cancer en particulier rénal.
Diagnostic
Forme typique : le nodule thyroïdien
1. Circonstances de découverte et signes fonctionnelsDans sa forme typique, il s’agit d’une tuméfaction cervi-cale antérieure, d’apparition récente répondant au nodulethyroïdien. Le diagnostic est souvent fortuit lors d’un pal-per cervical ou lors de la réalisation d’une échographie cer-vicale. L’interrogatoire recherche les facteurs de risques decancer de la thyroïde (tableau II), l’âge du patient, l’exis-tence de signes de compressions (œsophagienne, trachéale,récurrentielle, veineuse), le mode d’apparition et l’évolu-tion du volume du nodule.
2. Examen cliniqueL’examen clinique débute par l’inspection puis la palpa-tion du cou en se plaçant derrière le patient, tête en exten-sion. Il faut apprécier le nombre, la taille et la consistancedes nodules. On recherche la présence d’adénopathiesjugulo-carotidiennes. Les arguments cliniques en faveur del’origine cancéreuse du nodule sont présentés dans letableau II. Toutefois, aucun de ces éléments n’est spéci-
fique. L’examen général recherche des signes d’hyper- oud’hypothyroïdie et d’éventuelles contre-indications à la chi-rurgie.
3. Examens complémentaires
Les nodules thyroïdiens sont très fréquents ; seuls 5 à 10 % d’entre eux sont cancéreux. La difficulté va donc rési-der dans la sélection des patients à opérer. Aucun examenne va apporter une certitude diagnostique mais parmi eux,la cytoponction apporte actuellement le plus d’informa-tions. Seul l’examen anatomo-pathologique de la pièce opé-ratoire permet un diagnostic de certitude.• Examen cytologique du produit de ponction à l’aiguillefine : la cytoponction, effectuée avec une aiguille fine, avecou sans aspiration à la seringue, à raison de 3 ponctions parnodule, réalisée par un médecin expérimenté, interprétéepar un cytologiste entraîné, est l’examen le plus fiable, avecune sensibilité et une spécificité proche de 95 %. Les pré-lèvements sont classés soit bénin, soit malin ou douteux.Les facteurs limitants sont un pourcentage incompressiblede prélèvements non contributifs (10 à 15 %) et la grandedifficulté à poser le diagnostic de malignité pour lestumeurs vésiculaires différenciées. La ponction peut êtreguidée sous échographie.• Échographie thyroïdienne :cet examen permet de déter-miner avec précision la position du nodule au sein du corpsthyroïde et ses dimensions. La nature liquidienne, solide oumixte du nodule sera précisée de même que son iso-, hypo-ou hyperéchogénicité par rapport au reste du parenchyme.Les limites, nettes (halo clair) ou non du nodule doivent êtreévaluées. Enfin il est indispensable que les aires ganglion-naires soient explorées, permettant parfois la mise en évi-dence d’adénopathies non palpables. Cette exploration n’ap-porte cependant pas d’élément de certitude en faveur de lamalignité. Toutefois les formations kystiques parfaitementdélimitées par une capsule et totalement anéchogènes peu-vent être considérées comme bénignes. Le nodule plein estplus suspect s’il est hypoéchogène et de contours irréguliers.La présence d’adénomégalies de plus de 1 cm de diamètreest un élément qui doit inciter à faire pratiquer l’exérèse. Lesmicrocalcifications ont été rapportées comme l’expressionéchographique des calcosphérites des cancers papillaires,mais cela n’a aucune valeur de certitude. L’écho-doppler per-met d’individualiser les nodules peu vascularisés avec encor-bellement vasculaire périphérique (apparemment bénin) desnodules richement vascularisés en leur sein considéréscomme suspect.L’échographie a une grande sensibilité pour détecter desnodules non palpables à côté de la formation tumorale cli-niquement individualisée. Leur découverte a un intérêt pourla stratégie chirurgicale et la conduite thérapeutique s’ils’agit de lésions bénignes, appelant la mise en route d’untraitement freinateur.• La scintigraphie, réalisée avec l’iode ou le Tc-99m, dif-férencie les nodules froids ne fixant pas le traceur desnodules chauds. Pour tout nodule à TSH normale, la scin-tigraphie avec ce type de traceur ne présente plus grandintérêt. Sa spécificité est effectivement trop basse. Lesnodules froids sont cancéreux dans 10 % des cas. Le carac-
• Antécédents– Irradiation cervicale dans l’enfance– Antécédents familiaux de cancer médullaire ou papillaire– Âge < 20 ou > 60 ans– Sexe masculin– Taille du nodule > 3 cm– Polypose colique
• Caractéristiques du nodule– Augmentation de taille, notamment sous traitement freinateur de la TSH– Consistance ferme ou dure, irrégulière– Fixation aux tissus avoisinants– Sensibilité
• Autres– Adénopathies cervicales– Dysphagie, voix rauque– Diarrhée, flush
Arguments en faveurde l’origine maligne d’un nodule
TABLEAU II

878 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
C A N C E R S D E L A T H Y R O Ï D E
tère chaud ne permet pas d’exclure ce diagnostic (1 à 4 %de ces nodules seraient cancéreux).D’autres traceurs sont en cours d’évaluation comme le thal-lium dont la spécificité pour la détection des nodules can-céreux serait proche de celle de la cytoponction. Cepen-dant le coût élevé de ce marqueur radioactif ne permet passon utilisation de première intention pour explorer unnodule thyroïdien.• La résonance magnétique nucléairea fait l’objet de tra-vaux confirmant son intérêt pour l’analyse du tissu thyroï-dien. Cependant sa spécificité qui n’est pas absolue et soncoût ne permettent pas de proposer systématiquement laréalisation de cet examen devant tout nodule thyroïdien.Lorsque la suspicion de malignité est forte cet examen per-met l’analyse des rapports de la tumeur avec les organesde voisinage sans nécessiter une injection iodée ce qui estle cas pour le scanner.• Biologiquement,l’augmentation de la thyrocalcitonineet de l’antigène carcino-embryonnaire (ACE) est évoca-trice de cancer médullaire. Le dosage préopératoire de lathyrobloguline n’apporte aucun élément diagnostique. Ledosage de la TSH renseigne uniquement sur le caractèrefonctionnel de la thyroïde.• L’examen extemporané constitue un temps essentiel dela démarche diagnostique en orientant le geste chirurgicalvers une attitude conservatrice ou radicale (thyroïdectomietotale) en cas de cancer confirmé. Il doit être toujours pra-tiqué même si la cytoponction affirme le caractère malin.
Formes cliniques
1. Cancer révélé par une adénopathie cervicaleisoléeLes principales voies de drainage sont les chaînes pré- etlatéro-trachéales, la partie basse de la chaîne jugulo-caro-tidienne, la chaîne cervico-transverse superficielle, auniveau sus-claviculaire ainsi qu’au niveau sous-digastriquepour la partie haute du lobe. La palpation thyroïdienne peutêtre normale, la ponction de l’adénopathie permet souventle diagnostic. Il s’agit très fréquemment de cancers papil-laires.
2. Cancer sur goitre multinodulaireLa présence de multiples nodules accroît le risque de can-cer. Tout nodule dont le volume augmente doit être consi-déré comme suspect. La cytoponction risque de mécon-naître le nodule cancéreux car il paraît impossible de tousles atteindre.
3. Formes révélées par une métastaseToute tumeur, apparemment secondaire peut révéler uncancer de la thyroïde. Cependant, les sites métastatiquesrévélateurs les plus souvent rencontrés sont par ordre defréquence, le poumon, l’os, le cerveau et très rarement lefoie pour les tumeurs épithéliales différenciées. La palpa-tion cervicale associée à une échographie doit faire partiedu bilan demandé pour rechercher la tumeur primitive. Siun accès biopsique ou chirurgical est possible, l’examenanatomo-pathologique et l’immunohistochimie à l’aide
d’anticorps anti-thyroglobuline ou anti-calcitonine assure-ront le diagnostic.
4. Formes évoluéesIl s’agit de formes histologiquement non différenciées, quipeuvent se présenter sous la forme d’une tuméfaction cer-vicale mal limitée, fixée aux structures de voisinage, défor-mant le cou. Révélées par des signes de compressions, laradiographie cervicale recherche une déviation trachéale,l’ingestion de baryte permet de mettre en évidence unecompression œsophagienne extrinsèque. Les métastasesosseuses et pulmonaires sont souvent présentes d’emblée.
5. Cancer de l’enfantExceptionnel, il s’agit d’un cancer papillaire après irra-diation (Tchernobyl) ou de cancer médullaire de la thy-roïde dans le cadre des tumeurs endocrines multiples detype 2 (NEM2).
6. Forme histologique• Le cancer médullaire de la thyroïde,cancer sporadiqueet cas index familial, est révélé par un nodule thyroïdienavec fréquemment des métastases ganglionnaires aumoment du diagnostic. Les métastases à distance sont pré-sentent dans 25 % des cas. On suspecte un cancer médul-laire de la thyroïde devant la localisation du nodule auniveau de la jonction tiers supérieur/tiers moyen du lobethyroïdien, son caractère sensible ou douloureux à la pal-pation douce, sur l’existence d’une diarrhée motrice ou deflush, sur des antécédents familiaux de tumeurs thyroï-dienne, de phéochromocytome. Le diagnostic est confirmépar le dosage de la thyrocalcitonine.• Les tumeurs endocrines multiples de type 2 (NEM2) cor-respondent à une maladie génétique à transmission auto-somique dominante, caractérisée en fonction du sous-typeclinique pour un cancer médullaire de la thyroïde souventrévélateur, un phéochromocytome et (ou) une hyperpara-thyroïdie.L’anomalie génétique responsable des formes familialesest connue : il existe une mutation somatique du gène RET(voir : pour approfondir / 1). Plusieurs types de mutationssont décrites permettant de réaliser un dépistage génétiquelorsque le cas index est porteur d’une mutation. En l’ab-sence de mutation, le dépistage repose sur la réalisationd’un test à la pentagastrine (0,5 µg/kg de poids corporel).Ce test, parfois responsable de manifestations généralesdésagréables, est contre-indiqué chez la femme enceinte,l’asthmatique, le coronarien, l’hypertendu sévère. Laréponse normale est un pic inférieur à 30 pg/mL. Uneréponse supérieure à 100 pg/mL est très suspecte. Uneréponse intermédiaire nécessite une surveillance régulière.
Diagnostic postopératoire des métastases
1. Cancers différenciésAprès thyroïdectomie totale, l’élévation persistante de lathyroglobuline fait suspecter la présence d’un tissu tumo-ral. Les profils isotopiques corps entiers, après sevrage enhormones thyroïdiennes, utilisant l’iode 131 sont considé-rés comme l’argument fondamental de repérage des réci-

879L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
dives et métastases. Mais certaines localisations risquentd’être méconnues soit en raison de la présence simultanéede métastases osseuses et pulmonaires qui vont fixer pré-férentiellement l’iode, soit en raison d’une faible différen-ciation de la métastase. L’échographie cervicale, les explo-rations par résonance magnétique et tomodensitométriqueont leur place pour la détection de ces lésions secondairesqui ne fixent pas mais aussi certains isotopes comme lethallium, le Sesta-MIBI ou la somatostatine marquée parl’indium (Octréoscan). Une étude récente souligne aussi lavaleur des scintigraphies couplées par le 201 T1 et l’hy-droxyméthylène diphosphonate marqué par le technétium.
2. Cancer médullaireLa présence de métastases après l’intervention est suspec-tée sur l’élévation persistante de la thyrocalcitonine et (ou)de l’endogène carcino-embryonnaire. En l’absence de mar-queurs isotopiques spécifiques, leur diagnostic repose surl’échographie cervicale, la tomodensitométrie ou l’image-rie par résonance magnétique.
Diagnostic différentielC’est celui d’un nodule thyroïdien.
1. Kyste thyroïdienAnéchogène en échographie, la ponction ramène un liquidecitrin et affaisse complètement le nodule.
2. Adénome colloïdeC’est la cause la plus fréquente de nodules. Il peut poserdes problèmes de diagnostic différentiel difficile avec uncancer vésiculaire différencié.
3. Nodules des thyroïdites auto-immunesLe contexte clinique et biologique (positivité des anticorpsantipéroxydase) est évocateur, toutefois certains de cesnodules peuvent être cancéreux. ■
Tumeurs de la thyroïde. Martin Schlumberger et Furio Pacini.Paris : Nucleon, 1997.Anatomo-pathologie et histopronostic des carcinomes thyroï-diens différenciés d’origine folliculaire. C de MICCO. Ann Endo-crinol 1997 ; 58 : 172.Wemeau JL, Bauters C. L’exploration isotopique des cancers thy-roïdiens : l’éventail s’élargit. Le journal faxé d’endocrinologie. Jan-vier 1998.
POUR EN SAVOIR PLUS
POUR APPROFONDIR
Le proto-oncogène RET, codé par un gène situé en position 10q11-12,est un récepteur transmembranaire à activité tyrosine kinase, exprimé parde nombreuses cellules dérivant de la crête neurale. Son ligand estinconnu. Il est constitué de 16 exons. Un spectre de mutations, à l’originedu développement des différentes associations pathologiques familialescaractérisant les tumeurs endocrines multiples de type II (NEM2), a étéétabli permettant de retrouver une corrélation génotype phénotype. Cegène est aussi impliqué dans les formes familiales de maladie de Hirschs-prung.
• Le terme cancer de la thyroïde regroupeplusieurs types histologiques au pronostic biendifférent. Les cancers différenciés sont les plusfréquents, notamment les formes papillaires, leurpronostic est habituellement excellent.
• Le traitement passe par la réalisation d’unethyroïdectomie totale avec curage ganglionnaire.Ce geste permet de réaliser une surveillance trèsrigoureuse de l’évolutivité tumorale grâce au dosage de la thyroglobuline.• Le caractère hormono-dépendant des formesdifférenciées impose la prescription d’untraitement par L-thyroxine à visée substitutive et freinatrice de la TSH.• La possibilité d’apparition de métastases à trèslong terme nécessite une surveillance prolongéedes patients.• Enfin, la recherche d’une mutation RET sur un prélèvement sanguin doit être systématiquedevant tout cancer médullaire de la thyroïde.
Points Forts à retenir

Endocrinologie - Métabolisme - NutritionB 332
443L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
La prise en charge adaptée des diabétiques, leur éducationet l’autocontrôle glycémique ont nettement réduit la fré-quence de cette complication (2 à 4 épisodes/100 patientsen 1 an), mais l’augmentation des nouveaux cas de diabèteinsulinodépendant pourrait avoir l’effet inverse.
Étiologie
La céto-acidose diabétique est la conséquence d’un déficitabsolu ou relatif en insuline.
1. Déficit absolu La céto-acidose est une forme révélatrice du diabète detype 1 dans 30 % des cas.Elle est due à un arrêt intempestif de l’insulinothérapie,soit volontaire (patient « manipulateur » créant de toutepièce la forme de diabète « instable » à céto-acidosesrépétées, par opposition au diabète instable avec hypo-glycémies sévères répétées), soit involontaire (mauvais fonctionnement d’un stylo à insuline, panne d’une pompeà insuline, avec dans ce dernier cas échappement rapideen céto-acidose du fait de l’absence d’insuline retard).On constate de rares cas de paralysie d’îlot par bêta-mimétique, diazoxide, hydantoïne. et pentamidine
2. Déficit relatif en insuline Les diabètes de type 2, très exceptionnellement (la cétoseest fréquente, la céto-acidose rarissime), mais surtout lesdiabètes de type 1 peuvent présenter une céto-acidose encas d’adjonction d’un facteur hyperglycémiant :– une infection (pieds diabétiques infectés) même
mineure, mais en général fébrile, souvent par compor-tement inadapté (baisse des doses d’insuline pour « compenser » la réduction des apports alimentairesliés à des nausées) ;
Céto-acidose diabétiqueLa céto-acidose (plutôt que acidocétose) se définit parl’association d’une hyperglycémie (> 2,5 g/L) ; d’unecétonémie positive ou cétonurie (� ++) ; d’un pH san-guin veineux supérieur à 7,25 ou artériel inférieur à 7,30ou de bicarbonates plasmatiques inférieurs à 15 mEq/L ;d’une nécessité d’un traitement médical non ambulatoire.
Complications métaboliquesaiguës du diabète sucré (acidocétose, hypoglycémie,hyperosmolarité,acidose lactique)Étiologie, physiopathologie, diagnosticTraitement d’urgence de l’acidocétose avec la posologie médicamenteuse
PR Jean-Louis SÉLAM
Service de diabétologie et des maladies endocriniennes, Hôtel-Dieu de Paris, 75181 Paris Cedex 04.
• La céto-acidose est le résultat d’une carenceabsolue ou relative en insuline chez le patientdiabétique de type 1 surtout. Les symptômessont progressifs, il n’y a pratiquement jamaisde coma complet. Le traitement est très stéréo-typé, suivant un protocole, il peut être effectuépar des non réanimateurs dans les formesmodérées sur bon terrain.
• Le coma hyperosmolaire est dû à une hyper-glycémie majeure et une déshydratation massivechez le sujet âgé, diabétique de type 2. Les traitements et les perfusions doivent êtreadministrés progressivement et avec prudence.
• L’acidose lactique est rarissime mais gravissime.Elle résulte d’une accumulation de biguanides.
• L’hypoglycémie est le prix à payer du boncontrôle glycémique dans le diabète de type 1.
• Les hypoglycémies sulfamidées du type 2 ne sont à redouter qu’en cas d’utilisation de sulfamides trop puissants, chez des sujetsfragiles (âge, insuffisance rénale).
Points Forts à comprendre

– un stress majeur comme l’infarctus du myocarde oules traumatismes, une gangrène artéritique ;
– une hyperthyroïdie, un hypercorticisme ou un phéo-chromocytome évolutif ;
– une corticothérapie sans augmentation compensatoiredes doses d’insuline (exemple : traitement anti-œdéma-teux post-chirurgie ophtalmologique du diabétique).
Dans 24 % des cas la cause déclenchante de la céto-acidose demeure indéterminée.
Physiopathologie
1. Métabolisme glucidiqueLa production hépatique est multipliée par 3 à 5 en raisonnotamment de l’augmentation du flux des substrats néo-glucogéniques (acides aminés, lactates, glycérol) vers lefoie.La réduction de la captation périphérique par mécanis-me de résistance est liée à l’excès d’acides gras et decorps cétoniques circulants.Les corps cétoniques plutôt que le glucose sont utilisésde préférence par le cerveau.
2. Métabolisme lipidiqueLa lipolyse est très fortement accrue avec une produc-tion en excès d’acétylcoenzyme A puis des 3 corps céto-niques (acéto-acétate, hydroxybutyrate et acétone), tousces corps étant incomplètement oxydés dans le cycle deKrebs du fait d’un manque de disponibilité de l’oxalo-acétate utilisé complètement pour la néoglucogenèse.L’excès d’ions H+ est partiellement éliminé dans lesurines avec perte de sel et de potassium. Au contraire, onconstate une mauvaise élimination de l’acide urique.Ce même excès d’ions H+ est responsable de l’accéléra-tion du rythme respiratoire, de la vasodilatation périphé-rique, de l’hypothermie éventuelle, d’un effet cardiaqueinotrope négatif, mais surtout de la sortie du potassiumintracellulaire vers les milieux extracellulaires. La bar-rière hémato-méningée est peu perméable aux ions H+.De ce fait, l’état de conscience est relativement conservépar rapport à des acidoses de même profondeur maisd’origine respiratoire, avec augmentation du CO2 quipasse bien. Inversement, il peut se produire au momentde la correction thérapeutique de l’acidose sanguine,une aggravation cérébrale paradoxale par arrêt de lapolypnée qui fait remonter les taux de CO2 qui, passantla barrière, vont créer une acidose cérébrale profonde.
3. Métabolisme hydro-ioniqueDes vomissements et une diurèse osmotique entraînentdes pertes hydriques de l’ordre de 50 à 150 mL/kg (jus-qu’à 10 % du poids corporel).Les pertes en sodium peuvent atteindre 7 à 10 mEq/kgavec hypovolémie et insuffisance rénale fonctionnelle,voire hyperlactacidémie.Les pertes potassiques par fuite rénale et vomissementspeuvent atteindre 3 à 12 mmol/kg, soit l’équivalent de30 g de chlorure de potassium à perfuser.
Diagnostic positif
1. CliniqueLa céto-acidose s’installe rarement en quelques heures(enfant, infarctus, pompe), le plus souvent en 2 à 3 jours.• Lors de la phase de cétose sans acidose, on observedes signes de manque d’insuline (fatigue, soif, polyurie,amaigrissement, quelquefois troubles visuels) associés àdes signes évocateurs de cétose (nausées, douleursabdominales, anorexie, crampes).Si le traitement n’est pas adapté (voir plus loin), onpasse au stade d’une céto-acidose constituée.• Lors de la phase de céto-acidose, on constate unedyspnée de Küssmaul plus ample, profonde et bruyanteque rapide (30 à 45/min). L’état de conscience est quel-quefois normal (20 %), le plus souvent c’est un état stu-poreux, avec parfois confusion, mais rarement comahypotonique calme sans signe de localisation (10 %). Ladéshydratation est le plus souvent extracellulaire (plicutané, hypotonie du globe oculaire, hypotension arté-rielle), souvent mixte avec une note intracellulaire(sécheresse des muqueuses).Des symptômes divers sont à prendre en compte : hypo-thermie relative d’acidose, vasodilatation faciale, odeuracétonique, « pomme reinette », de l’haleine, surtoutsignes abdominaux, quelquefois au premier plan avecpar exemple tableau d’iléus douloureux.
2. Biologique Le diagnostic de cétose est facile lors de la présenced’urines (bandelettes réactives semi-quantitatives,Kétodiabur ou Kétodiastix). Dans certains cas, on peutdoser les corps cétoniques directement sur plasma aprèscourte centrifugation par les mêmes bandelettes, soit aulaboratoire (corps cétoniques totaux ou mieux bêta-hydroxybutyrate, valeur comprise entre 5 et 30 mmol/L).On peut noter qu’en cas de cétose grave avec prédomi-nance de bêta-hydroxybutyrate les bandelettes, qui nedosent pas ce corps cétonique, peuvent donner des résultats artificiellement abaissés. Le taux de glycémie se situe entre 3 et 7 g/L.Le degré d’acidose est évalué par les bicarbonates vei-neux (< 15 mEq/L) et il suffit de faire le pH sur sang vei-neux (valeur seulement de 0,1 point plus basse que lesvaleurs artérielles) [voir encadré].Une pseudo-hyponatrémie est fréquente. Pour calculerla natrémie corrigée, il faut ajouter 1,6 mmol/g de glycémieau-dessus de la normale. Une valeur élevée de la natrémiecorrigée indique une déshydratation intracellulaire associéeà la céto-acidose.
C O M P L I C AT I O N S M É TA B O L I Q U E S A I G U Ë S D U D I A B È T E S U C R É
444 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
1 g de KCl = 13 mEq 1g de NaCl = 17 mEqNatrémie corrigée = Natrémie mesurée + 1,6 mM/g
de glycémie supérieure à la normaleOsmolarité = 2 (Na + K) + glycémie (mM) + urée (mM)Trou anionique = (Na + K) – (Cl + CO3H) � 17Déficit eau pure = 0,6 x poids (kg) x (1 – 140 )
natrémie corrigéeKaliémie corrigée = K mesurée – 1 mEq par 0,1 point de pH < 7,4
FORMULES UTILES

s’alimenter, une glycémie stabilisée entre 2 et 2,5 g/L,un ionogramme normalisé en particulier une réservealcaline atteignant 25 mEq/L. Il est habituel de voir per-sister quelques heures une petite cétonurie.L’évolution est défavorable voire mortelle dans 4 % descas surtout chez le sujet âgé, débilité. Infarctus du myo-carde et infection peuvent être méconnus.Parmi les complications iatrogéniques, il faut surtoutredouter les surcharges hydrosodées chez les sujets âgésinsuffisants cardiaques ou surtout insuffisants rénaux, etl’œdème cérébral avec aggravation paradoxale progres-sive du coma sous traitement par baisse trop rapide del’osmolarité plasmatique.
Traitement
1. PréventifEn phase de cétose, les règles éducatives qui doiventpermettre aux diabétiques d’éviter de passer en céto-acidose sont : connaître les signes et causes de l’hyper-glycémie ; savoir intensifier l’autosurveillance glycé-mique et acétonurique ; savoir faire la différence entrecétose de jeûne et cétose vraie ; ne jamais omettre sesinjections même en cas d’inappétence, voire augmenterles doses d’insuline retard ; maintenir boissons etapports glucidiques même en cas d’inappétence en frac-tionnant ces derniers (compote, purée, coca) ; savoir sesupplémenter en insuline rapide sous-cutanée toutes les3 h en cas de déséquilibre significatif (glycémie > 3g etacétone � ++) ; savoir faire appel au médecin si le problème n’est pas résolu avant la nuit.
2. Curatif Il peut être effectué en milieu diabétologique si le serviceest équipé, mais il est plus prudent de faire appel à unservice de réanimation s’il faut une catéthérisation cen-trale, si des troubles cardiovasculaires sont présents et sile pH artériel est inférieur ou égal à 7, et la kaliémie à 4. • L’insuline : après un bolus en intraveineuse de 10 uni-tés, elle est donnée sous perfusion à débit fixe, à laseringue électrique de 0,15 U/kg/h. Ce débit est maintenufixe pendant toute la réanimation.• La réanimation hydroélectrolytique : le volume àperfuser doit tenir compte de la perte de poids si elle estconnue, ou du calcul du déficit hydrique en cas d’hyper-natrémie (voir formule sur tableau), auquel il faut ajouterles besoins de base de 1,5 à 2 L/24 h. Le volume total (4 à 7 L) est passé pour moitié sur les 8 premières heureset pour moitié sur les 16 heures suivantes. Ce volume est réajusté en cours de réanimation en fonction de latolérance clinique. En cas d’hyperosmolarité associée,ou chez les sujets âgés il est prudent d’étaler cet apportvolumique non sur 24 mais 48, voire 72 h.Exceptionnellement les premiers solutés perfusés peuvent être des solutés d’expansion plasmatique en casde collapsus ou du bicarbonate isotonique (maximum500 cm3) seulement en cas de pH inférieur à 6,9.En particulier chez le sujet âgé, et si la diurèse n’est pas amorcée et n’atteint pas au moins 50 cm3/h après les
Une pseudo-normo- ou hyperkaliémie secondaire à l’aci-dose peut apparaître. Pour que la kaliémie reflète le poolpotassique, il faut enlever 1 mmol/L par 0,1 point de pH endessous de 7,4. Ainsi, le pool potassique d’un patient seprésentant avec une kaliémie à 6 et un pH à 7,1 corresponden fait à une kaliémie théorique de 3 mmol/L en situationde non-acidose, (c’est-à-dire en pratique au cours du traitement de la céto-acidose, s’il n’y a pas d’apport).Le trou anionique est habituellement inférieur à 3 etaugmente en cas d’acidose rénale et (ou) lactique associée.On constate des signes d’hypovolémie et de déshydratationextracellulaire : créatinine et urée modérément élevées,hémoconcentration avec hématocrite élevé. L’hyper-lipémie est souvent majeure avec sérum lactescent.D’autres éléments biologiques sont trompeurs : une hyper-leucocytose même en l’absence d’infection, une augmen-tation des enzymes pancréatiques, hépatiques et car-diaques, en l’absence d’atteinte spécifique de ces organes. Ces examens ne devraient donc être inclus dans le bilande départ qu’en cas de forte orientation.Un électrocardiogramme est indispensable (recherched’infarctus et de signe de dyskaliémie) avec monitoringcontinu si possible ainsi qu’une hémoculture, un examencytobactériologique des urines (ECBU), des prélèvementslocaux, radiographie du thorax, hCG (human chorionicgonadotrophin), une hémostase, un groupage uniquementen cas d’orientation clinique.
Diagnostic différentiel
La céto-acidose alcoolique est une complication peuconnue de l’alcoolisme aigu. La glycémie est générale-ment normale mais des formes associées (céto-acidosediabétique et céto-acidose alcoolique) sont possibles. Les autres comas diabétiques sont facilement éliminésmême s’il existe des formes intriquées.Une cétonurie « de jeûne » peut être prise à tort pour unecétonurie d’insulinopénie, chez un patient diabétiquemal éduqué en cure de restriction pondérale ou en périodede vomissement avec injection inopportune d’insulinesupplémentaire.Surtout, il faut savoir déceler derrière un tableau dedécompensation diabétique une autre urgence : unehypovolémie, des troubles digestifs, une hyponatrémie,une hyperkaliémie de l’insuffisance surrénale aiguë, unehyponatrémie, des vomissements de l’insuffisance réna-le aiguë, et surtout un infarctus du myocarde et de vraisproblèmes chirurgicaux abdominaux.
Évolution, complication
La surveillance clinique toutes les heures pendant 8 hpuis toutes les 2 h est essentielle : pouls, tension artérielle,auscultation pulmonaire, glycémie, acétone, diurèse. Le monitoring de l’électrocardiogramme, incluant fré-quence cardiaque et tension artérielle, est souhaitable.L’ionogramme doit être répété à 3, 6, 12 et 24 h. La fin de la réanimation doit survenir dans les premières24 h. Les critères de retour à l’insulinothérapie sous-cutanée sont les suivants : un sujet conscient pouvant
Endocrinologie - Métabolisme - Nutrition
445L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0

3 premières heures, il faut se méfier de la surchargehydrosodée et ralentir les débits, après avoir vérifié l’absence de globe vésical en posant une sonde urinaire(il s’agit de la seule indication licite de sondage). Dans les cas habituels, on passe au perfuseur électriquedu sérum physiologique qu’on remplace lorsque la gly-cémie atteint 2,5 g/L par du glucosé à 5 % additionné de4 à 6 g de chlorure de sodium par litre et accompagnéd’une perfusion en Y de glucosé 10 dont le débit, indé-pendant et variable par rapport au flacon de G5, permetde maintenir la glycémie dans la zone des 2 à 2,5 g/L,sans toucher au débit de base.Le potassium est passé idéalement indépendamment dessolutés à la seringue électrique. L’apport doit commencer dèsla réception de la kaliémie si elle est inférieure à 6, à la dosede 1 à 2 g/h. Si la kaliémie est inférieure à 4, il faut passerplus de 2 g/h, mais uniquement sur voie veineuse centrale.En cas d’hyperosmolarité majeure (forme mixte), oncommence par du sérum physiologique que l’on remplacepar du salé hypotonique à 4,5 pour 1 000 de façon à corrigerprogressivement sans brutalité le déficit en eau libre.Toujours pour être plus progressif, les apports en insulineet potassium pourront être moins importants.
3. SupplémentaireUn sonde gastrique peut être posée si l’acidose est pro-fonde, en cas de coma ou de troubles digestifs importants. Selon les cas et les écoles, on peut prescrire une antibio-thérapie, une héparinothérapie sous-cutanée ou une supplémentation vitaminique.Une supplémentation orale en potassium et une augmen-tation des doses d’insuline sous-cutanée sont souvent justifiées au cours des jours qui suivent la céto-acidose.
Coma hyperosmolaire
Dans sa forme pure, ce coma est 10 fois moins fréquentque la céto-acidose diabétique, mais de bien plus mauvaispronostic (mortalité 20 à 50 %). À l’inverse de la céto-acidose, il survient habituellement chez des patientsâgés porteurs d’un diabète non insulinodépendant (type 2). Il est défini par une hyperglycémie supérieure à 33 mmol/L (6 g/L), une osmolarité plasmatique supé-rieure à 350 mmol/L ou une natrémie corrigée supérieureà 155 mEq/L et un pH supérieur à 7,20 avec bicarbo-nates plasmatiques supérieurs à 15 mmol/L et une cétoseabsente ou modérée (acétonurie � +).
Étiologie
Le coma hyperosmolaire et la déshydratation résultentde la conjonction de 2 facteurs : une agression hypergly-cémiante : infection, diurétiques, corticoïdes, etc. ; unapport compensatoire en eau insuffisant : soif non per-çue (certains diabétiques seraient prédisposés au comahyperosmolaire par une dysrégulation préexistante desmécanismes centraux d’osmorégulation) ou impossibleà assouvir (isolement, détérioration de la conscience,réanimation).
Physiopathologie
L’hyperglycémie majeure sans compensation hydriqueentraîne une hyperosmolarité plasmatique avec déshy-dratation intracellulaire. La diurèse osmotique entraîne en outre une perte d’eauet de sel avec hypovolémie, insuffisance rénale fonction-nelle et quelquefois oligo-anurie.Les autres conséquences de cette déshydratation surtoutintracellulaire sont une diminution du volume cérébralavec troubles majeurs de la conscience, une réductiondu débit cardiaque, une augmentation de la viscositésanguine et une détérioration de l’insulino-sécrétion.Cette dernière n’est cependant pas suffisante pourdéclencher une céto-acidose, mais explique les acétonuriesfaibles usuelles d’accompagnement.
Diagnostic
1. CliniqueLes symptômes s’installent très progressivement surplusieurs jours, voire plusieurs semaines.Des troubles profonds de la conscience, de la léthargie aucoma parfois agité sont accompagnés de signes focaux.Une déshydratation massive intracellulaire prédominantes’installe avec perte de poids importante souvent supé-rieure à 10 kg. La déshydratation intracellulaire est aupremier plan avec peau sèche, voire cartonnée, hyper-thermie, langue rôtie. La déshydratation extracellulaireest responsable d’une hypotension et d’une oligurieparadoxaleLe signe négatif le plus important est qu’il n’existe pasde dyspnée de Küssmaul.
2. BiologiqueL’hyperglycémie majeure est supérieure à 6 g/L et peutdépasser 20 g/L.L’hypernatrémie corrigée majeure est supérieure à 155 mEq/L.On constate une insuffisance rénale fonctionnelle.La kaliémie est le plus souvent normale, même si ladéplétion potassique est constante.Le pH normal est bas (> 7,2) avec corps cétoniques pré-sents mais modérés (faits essentiellement d’hydroxybu-tyrate donc non détecté par les bandelettes urinaires).Il y a hémoconcentration avec élévation de l’hématocri-te des protides et des leucocytes et élévation inconstantedes enzymes par souffrance cellulaire.
Évolution, complicationsMême si la réanimation est précoce, appropriée et pro-gressive, la mortalité est fréquente du fait du terrain etdu grand âge.Le coma peut s’aggraver au cours de la réanimation parœdème cérébral lié à une correction plus lente de l’hyper-osmolarité intracellulaire que de l’hyperosmolaritéextracellulaire et donc une attraction de l’eau vers lescellules cérébrales.
C O M P L I C AT I O N S M É TA B O L I Q U E S A I G U Ë S D U D I A B È T E S U C R É
446 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0

– dans le type 2, le choc et l’hypoxie surviennent éven-tuellement au cours de l’acidose lactique. Le diabètefait partie des affections favorisant l’acidose lactiqueau même titre que l’insuffisance rénale ou hépatiquepar exemple et les biguanides, au même titre que l’alcool, le méthanol, les salicylés.
Physiopathologie
Les biguanides en bloquant la néoglucogenèse peuvententraîner une hyperproduction de lactates, mais il fauten outre une insuffisance d’élimination (insuffisancehépatique ou plus souvent rénale) pour générer une aci-dose lactique.L’acidose métabolique est ensuite entretenue parl’anoxie tissulaire résultant des troubles cardio-vasculaires.
Diagnostic
1. Clinique
Dans la phase prodromique, le patient souffre d’asthénieet de crampes pendant quelques heures à quelques jours.Puis, on constate une grande acidose métabolique avecpolypnée, une instabilité tensionnelle, une oligo-anurie,une hypothermie et des troubles de la consciencevariables.
2. BiologieLe pH est souvent inférieur à 7 avec une réserve alcalineinférieure à 10 mEq/L.Le trou anionique est important (> 15 mmol/L). La gly-cémie est variable et la cétose absente ou discrète ; lakaliémie est souvent élevée ; l’insuffisance rénalefranche et la calcémie supérieure à 6 mmol/L.
Traitement
Il consiste à corriger l’acidose et les troubles hémo-dynamiques.L’épuration extrarénale traite à la fois l’acidose, l’insuf-fisance rénale et l’excès de biguanides.Le traitement préventif consiste en un respect rigoureuxdes contre-indications des biguanides telles l’insuffisan-ce pulmonaire, cardiaque, hépatique et surtout rénale etle grand âge. Les biguanides doivent être interrompusavant l’examen radiologique avec opacification ou chi-rurgie, mais aussi en cas de problèmes circulatoires ouinfectieux importants. La prudence est recommandée encas d’association concomitante de médicaments poten-tiellement néphrotoxiques.
HypoglycémieSurtout pour les diabétiques insulinodépendants, l’hypo-glycémie constitue pratiquement le prix à payer pourl’obtention d’un bon contrôle glycémique moyen etl’obstacle majeur à l’obtention de ce bon contrôle.
L’hypotension aussi peut être aggravée pendant le traite-ment du fait du passage de l’eau extracellulaire vers lesecteur intracellulaire.D’autres complications peuvent apparaître telles descomplications de décubitus, une atélectasie pulmonaire,une thrombose, une infection ou une rhabdomyolyse.
Traitement
La surveillance est identique à la surveillance de la céto-acidose.L’insuline est injectée à la seringue électrique à desdoses moindres que lors de la céto-acidose : pas debolus initial, puis 5 unités/h, à diminuer en adaptant surles contrôles glycémiques dès que la glycémie atteint2,5 à 3 g/L.Le volume perfusé est généralement plus important quepour la céto-acidose (6 à 12 L) calculé sur le déficit en eaulibre (voir formule tableau) mais étalé sur au moins 36 à72 h (par exemple, première moitié dans les 12 premièresheures, seconde moitié dans les 24 heures suivantes).La perfusion est composée comme suit : au début saléisotonique 2 à 3 L (de toute façon hypotonique par rap-port à l’osmolarité plasmatique) puis hypotonique 4,5 ‰(moitié eau, moitié sérum physiologique) additionné depotassium 1 à 2 g/L. On remplace par du glucosé 5 %additionné de 3 à 4 g de NaCl/L et de potassium dès quela glycémie atteint 2,5 à 3 g/L. Contrairement au traitement de la céto-acidose, c’est ledébit d’insuline et non pas le débit de perfusion que l’ondoit adapter pour maintenir la glycémie dans la zone des2 à 3 g/L pendant la réanimation.Le traitement associé est une antibiothérapie, une hépa-rinothérapie à dose préventive, un nursage, une humidi-fication bronchique, des soins de bouche, etc.La réanimation est interrompue sans hâte uniquementaprès restauration complète de la conscience et de l’iono-gramme avec notamment une natrémie normalisée.L’insuline peut être poursuivie à petites doses sous-cutanéeset éventuellement relayée par des antidiabétiques orauxen l’absence de contre-indication.
Acidose lactiqueL’acidose lactique est une complication encore plus raremais encore plus grave que le coma hyperosmolaire. Lerespect scrupuleux des contre-indications des bigua-nides et la moindre toxicité de la metformine par com-paraison à la phenformine aujourd’hui retirée du marchéexplique cette rareté. L’acidose lactique est définie pardes taux plasmatiques de lactates supérieurs à 7 mmol/Let un pH artériel inférieur à 7,25.
Étiologie
On classe les acidoses lactiques en 2 types :– dans le type 1, l’anoxie et le choc surviennent avant
l’acidose lactique, qu’ils provoquent par hyperproduc-tion ;
Endocrinologie - Métabolisme - Nutrition
447L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0

L’hypoglycémie est fréquente, désagréable, impression-nante pour le patient et l’entourage, mais exceptionnel-lement dangereuse.
Étiologie
1. Diabète insulinodépendant• Diabète instable avec hypoglycémies sévères répétées :le cadre nosologique des diabètes instables est flou,mais selon les définitions les plus récentes, on définit desdiabètes instables avec céto-acidose récurrente (patientsprésentant au moins 3 céto-acidoses sur 2 ans), et desdiabètes instables avec hypoglycémie sévère récurrente(patients faisant plus de 3 hypoglycémies sévères dansl’année). Dans la majorité des cas et dans les 2 formes,les causes psychologiques, voire psychiatriques sont aupremier plan. • Diabète en insulinothérapie intensifiée avec hypo-glycémie sévère fréquente : le DCCT (Diabetes controland complications trial) a noté un triplement du risqued’hypoglycémie sévère chez leurs patients en insulino-thérapie intensifiée. Il semble cependant que le risquen’augmente réellement de façon inexorable et inévitableque pour des hémoglobines glycosylées inférieures à 7 %.Dans ce cadre, les autres facteurs de risque prédictifsd’hypoglycémie sévère sont l’âge jeune, l’importancedes doses d’insuline, la longue durée du diabète, maissurtout la mauvaise perception des hypoglycémies.Cette dernière serait la conséquence d’hypoglycémies,même modérées, répétées, notamment nocturnes (voirphysiopathologie).• Diabète insulinodépendant tout venant avec fréquenteshypoglycémies : chez un diabétique bien équilibré, avec unehémoglobine A1c dans la zone des 7 à 8 %, une fréquencehebdomadaire des hypoglycémies modérées supérieure à 3, amène à rechercher des facteurs favorisants :– repas ou collation insuffisants ou sautés ;– exercice physique non programmé ou avec une mau-
vaise adaptation des doses d’insuline et des apportsglucidiques supplémentaires ;
– repas (notamment du soir) insuffisamment glucidique,souvent par méconnaissance des règles d’équivalencediététique ;
– erreurs dans la réalisation de l’injection d’insuline ;– injection dans des zones de lipodystrophie ;– repas trop retardés par rapport à l’injection ;– schéma insulinique comportant trop d’insuline rapide
(pas plus de 50 % de la dose totale et, le soir pas plusde 30 % de la dose nocturne) ;
– objectifs glycémiques trop ambitieux par rapport à laprise en charge globale. Le fractionnement des injectionset la multiplication des autocontrôles glycémiquesdoivent notamment en être le corollaire ;
– adaptation des doses trop brutale, voire inappropriée,avec suppléments d’insuline rapide intempestifs,notamment au coucher ;
– autres erreurs éducatives comme la méconnaissancedes symptômes, la pratique d’un « resucrage » insuffisantou trop tardif ;
– prise de médicaments potentialisant les hypoglycémiestels que les inhibiteurs de l’enzyme de conversion (IEC),les anticalciques, les bêtabloquants non cardiosélectifs,le Di-Antalvic et certains antiarythmiques ; en fait celaa rarement été confirmé ;
– très rarement, cause organique telle qu’une gastroparésie,souvent accusée à tort, une insuffisance hormonale, uneinsuffisance hépatique, et surtout une insuffisance rénale.
2. Hypoglycémie du diabète non insulinodépendant (DNID)La fréquence est nettement moins importante que dansle diabète insulinodépendant, mais pour certains la gra-vité et le risque seraient plus grands en raison notam-ment de l’âge plus avancé et du terrain vasculaire.• Diabète non insulinodépendant traité par insuline :selon l’UKPDS (United Kingdom Diabetes ProspectiveStudy), 1 patient sur 3 a fait une hypoglycémie modéréedans l’année, mais seulement 2 % des patients unehypoglycémie sévère, soit une fréquence 20 foismoindre que chez le diabètique insulinodépendant.• Dans cette même étude, la fréquence des hypoglycé-mies sous sulfamides hypoglycémiants est de 20 % paran avec 0,5 % d’hypoglycémie sévère. Les causes habi-tuelles sont l’utilisation de sulfamides trop puissants ouà trop longue durée d’action (glibenclamide, glimépiride)chez un patient âgé, souvent insuffisant rénal ; un effortphysique ou un repas sauté ; une potentialisation parl’alcool, des anti-inflammatoires non stéroïdiens,Daktarin, Bactrim, fibrates, Di-Antalvic, des inhibiteursde l’enzyme de conversion (en pratique, exceptionnelle-ment confirmé) ; biguanides et acarbose peuvent êtreconsidérés comme non générateurs d’hypoglycémie.
Physiopathologie
En pratique, les cellules cérébrales n’utilisent comme sub-strat énergétique que le glucose circulant dont elles sontdonc très dépendantes. À environ 3 mmol/L chez un dia-bétique insulinodépendant traité conventionnellement et2,5 chez un diabétique insulinodépendant en traitementintensifié, apparaissent les signes neuroglucopéniques etles petits troubles cognitifs qui peuvent en cas d’hypo-glycémie profonde aboutir à la perte de connaissance.Le glucostat cérébral, déclencheur des réactions neuro-sympathiques et de la contre-régulation hormonale, estsitué dans l’hypothalamus ventromédian. Il se déclencheaux alentours de 3,5 mmol chez un diabétique insulino-dépendant en traitement conventionnel, mais à des tauxbeaucoup plus bas, de l’ordre de 2,5 mmol, chez le diabé-tique en traitement intensifié. Il retarde de ce fait les réac-tions de contre-régulation, la survenue des symptômesdysautonomiques, qui sont ceux qui en général permettentaux sujets d’identifier l’hypoglycémie, et donc la percep-tion de l’hypoglycémie, et la mise en train d’un comporte-ment correctif. Ce déficit de la contre-régulation et cetabaissement des seuils de déclenchement expliquent lafréquence des hypoglycémies non ou mal perçues des dia-bétiques en traitement intensifié et l’accroissement majeurdu risque d’hypoglycémie sévère dans de tels cas (figure).
C O M P L I C AT I O N S M É TA B O L I Q U E S A I G U Ë S D U D I A B È T E S U C R É
448 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
mation diagnostique par une glycémie capillaire prati-quée par le malade est conseillée, mais ne doit jamaisretarder le « resucrage ».
Diagnostic différentiel
Tout symptôme, toute manifestation rapide ou brutalechez un diabétique doivent être considérés a prioricomme une hypoglycémie.
Évolution et complications
Dans la très grande majorité des cas, les hypoglycémiesdu diabétique même non traitées sont réversibles sansséquelle.Les effets néfastes sur la qualité de vie sont probables,mais le seuil de tolérance est variable d’un sujet àl’autre, allant du simple désagrément à des réactionsd’angoisse, d’évitement social ou sexuel, de culpabilitéet de dépendance. Dans de trop nombreux cas, la phobiede l’hypoglycémie empêche toute possibilité de boncontrôle glycémique.La mortalité n’a jamais été retrouvée augmentée dansles études d’intensification de l’insulinothérapie, mêmesi quelques cas de mort subite du sujet jeune (dead inbed syndrome) ont pu lui être imputés. Des cas d’hypo-glycémie mortelle ont été constatés au cours d’uneintoxication alcoolique aiguë chez des diabétiques.Les risques d’accident et de traumatisme ne sont statisti-quement pas plus élevés, probablement en partie grâceaux restrictions légales à la conduite et aux activités àrisque (alpinisme par exemple).L’intensification de l’insulinothérapie a été retrouvéeassociée dans 20 % des cas à une aggravation transitoirede la rétinopathie non avancée. Par contre il existe unrisque réel d’aggravation avec passage au stade prolifé-rant de rétinopathie avancée préproliférante non laseriséeen cas d’équilibration glycémique trop rapide avec fré-quentes hypoglycémies.L’hypoglycémie a été accusée à tort de déclencher desépisodes ischémiques coronaires ou vasculocérébrauxnotamment chez les sujets âgés. Les plus grandes étudesrécentes (DCCT et UKPDS) ont infirmé cette assertion.En ce qui concerne la détérioration cognitive chronique,le risque de séquelle cérébrale patente après coma hypo-glycémique très profond (< 0,2 g/L), surtout prolongé
Ce syndrome de « maladaptation » cérébrale est provo-qué par la répétition des hypoglycémies mêmemineures. Il peut être réversible si elles sont prévenues.Il serait lié à une augmentation de l’apport cellulairecérébral de glucose, lui-même dû à une augmentation dudébit sanguin cérébral.
Diagnostic
Il faut distinguer les hypoglycémies asymptomatiques(ou biologiques) c’est-à-dire toute glycémie inférieure à0,6 ou 0,5 g/L selon les définitions, et les hypoglycé-mies symptomatiques modérées ou sévères. Ces der-nières sont définies par la nécessité de l’assistance d’unetierce personne et incluent donc les hypoglycémies pro-fondes au cours desquelles le sujet ne peut s’alimentertout seul et les formes dites très sévères, définies par lasurvenue d’un coma, la nécessité d’une hospitalisationou d’une injection de glucose ou de glucagon. On parled’hypoglycémie non ressentie ou mal perçue quand lessymptômes annonciateurs ont totalement ou partielle-ment disparu ou plus exactement surviennent tardive-ment pour des glycémies très basses.Les symptômes sont actuellement classés en :– signes dysautonomiques (dénomination plus appro-
priée qu’adrénergique) tels que des sueurs, des palpi-tations, des tremblements, une sensation de faim ;
– signes neuroglucopéniques tels les troubles de laconcentration, les difficultés à parler, une incoordina-tion motrice, une sensation d’ébriété ;
– signes non spécifiques comme une fatigue brutale, descéphalées, des nausées, des paresthésies notammentpéribuccales, des troubles de la vision, notammentune diplopie ;
– mais on peut aussi citer une nervosité, une irritabilité,une sensation de froid, une angoisse, une agressivité,des accès de rires ou de pleurs, une somnolence et, surtoutdétectable par l’entourage, pâleur, yeux fixes, regarddans le vide, ralentissement de la parole ou des actes.
Le coma hypoglycémique s’installe rapidement, précé-dé ou non des symptômes annonciateurs. Il est typique-ment agité avec signes d’irritation pyramidale, tachycar-die, sueurs, souvent crise convulsive et quelquefoismanifestations focalisées.Au total, une symptomatologie très polymorphe maissouvent stéréotypée chez un même malade. La confir-
449L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
Hiérarchie des seuils de contre-régulation en fonction du degré de contrôle glycémique.
5
4
3
2
1
Glycémie (mmol/L)
Utilisation cérébrale du glucoseAdrénaline+ système nerveux sympathique
Troubles cognitifs +Symptômes
+ perception
Signes EEG
Coma
Diabète insulinodépendanttraitement conventionnel
5
4
3
2
1
Glycémie (mmol/L)
Utilisation cérébrale du glucose
Adrénaline+ système nerveux sympathique
Troubles cognitifs
Symptômes + perceptionSignes EEG
Coma
Diabète insulinodépendanttraitement intensifié

(au moins 4 h) et aux âges extrêmes est indiscutablemais heureusement rarissime. En revanche, le risqued’installation, après hypoglycémies sévères répétées,d’un syndrome de détérioration intellectuelle progressiveavec ralentissement de l’idéation et détérioration duquotient intellectuel (encéphalopathie hypoglycémiquechronique), voire troubles auditifs, atrophie cérébraleavec plaque de leuco-araïose est très probablementinexistant chez l’adulte et reste controversé chez l’enfant.La prise de poids fréquemment constatée lors de l’inten-sification de l’insulinothérapie ne doit pas être attribuéeaux hypoglycémies, même fréquentes et « resucrées »,mais à une disparition de la perte calorique glucosée urinaire du fait du meilleur équilibre.
Traitement
1. Curatif• Sujet conscient et capable de s’alimenter : arrêt detoute activité et « resucrage » immédiat par 15 g desucre (3 sucres, 2 barres de pâte de fruit, un verre decoca ou soda ou jus de fruit en boîte). Ne pas utiliserfruits, chocolat, ou jus de fruits frais. Les symptômesdisparaissent en 10 à 15 minutes. Le sujet doit apprendreà patienter, à se recontrôler après 20 à 30 minutes, avantde se « resucrer » à nouveau si nécessaire. Si le repassuivant est éloigné, plutôt que la collation glucido-proti-dique classique, il faut conseiller au sujet de contrôler ànouveau sa glycémie.• Sujet inconscient : la voie per os est strictementcontre-indiquée du fait du risque de fausse route. On utilise le glucagon injectable, préférentiellement maispas obligatoirement intramusculaire, ou mieux le glucoséhypertonique à 30 % intraveineux (30 à 50 mL).• Dans le cas particulier d’une hypoglycémie sous sul-famides, le risque de prolongation de l’hypoglycémieimpose une perfusion prolongée de glucose.
2. Préventif• Diabète non insulinodépendant : chez le sujet âgé et(ou) en cas d’insuffisance rénale, il faut préférer les sul-famides à demi-vie courte et à métabolisation complètecomme le glipizide (Glibénèse) et prochainement lerepaglinide (Novonorm). L’instauration d’un traitementsulfamidé doit toujours être progressive en commençantpar de faibles doses. En cas d’insuffisance rénale avérée(< 30 mL/min), l’insulinothérapie est préférable. Danstous les cas, le patient doit être conscient du risque d’hypo-glycémie en cas de repas sauté, même s’il ne prend passon comprimé, et en cas d’activité physique prolongée.• Diabète insulinodépendant : les moyens principauxde réduction du risque hypoglycémique sont nombreux.La sélection des patients candidats à l’intensification del’insulinothérapie permet aux seuls patients disciplinés,éduqués et suivis régulièrement, d’être justiciables del’insulinothérapie intensifiée.En cas d’hypoglycémie mal ou non perçue, il faut cher-cher à éviter les petites hypoglycémies méconnues,notamment nocturnes (glycémie à 3 h du matin, objectif
glycémique à jeun légèrement plus élevé) et rééduquerle patient aux petits symptômes annonciateurs qui n’ontpeut-être pas tous disparu.L’éducation, et en premier lieu l’autocontrôle glycé-mique (voir chapitre étiologie), doit être renforcée.De la même façon, le suivi doit être intensifié : un suivirapproché mensuel, associé éventuellement à des contactstéléphoniques, permet de renforcer la motivation et d’aiderle patient à adapter ses doses d’insuline.Le schéma insulinique peut être optimisé. L’intensificationde l’insulinothérapie impose comme corollaire le fraction-nement des doses en au moins 3 injections journalières. Lepassage à l’analogue ultrarapide apporte un petit bénéfice(réduction des hypoglycémies de l’ordre de 10 %) quin’est pas négligeable. Enfin, la persistance d’hypoglycé-mies répétées chez un patient très discipliné, bien éduqué,et bénéficiant d’une prise en charge déjà globalementintensifiée peut conduire à l’indication d’une pose d’unepompe à insuline externe sous-cutanée, voire implantableintrapéritonéale, qui ont, surtout pour cette dernière,prouvé leur efficacité à réduire les fluctuations glycé-miques et les épisodes d’hypoglycémie. ■
C O M P L I C AT I O N S M É TA B O L I Q U E S A I G U Ë S D U D I A B È T E S U C R É
450 L A R E V U E D U P R AT I C I E N 2 0 0 0 , 5 0
• La céto-acidose est la seule complication métabolique aiguë non hypoglycémique encorefréquente.
• Les risques iatrogéniques principaux sont liés à la kaliémie, à la surcharge sodée ou une réhydratation trop brutale.
• Le calcul de la perte du déficit hydrique par la natrémie est important,surtout dans le coma hyperosmolaire.
• Les contre-indications des biguanides pour prévenir l’acidose lactique doivent être respectées.
• L’hypoglycémie n’est pas à proprement parlerune complication, mais un effet incontournableet indésirable du traitement intensif par insulineou sulfamides.
• Elle est l’obstacle majeur à la bonne équilibration des diabétiques.
• L’éducation est nécessaire, mais pas suffisantepour prévenir ces complications.
Points Forts à retenir
Sélam JL. La céto-acidose diabétique. Encycl Med Chir 1997 ; 10(366-H-10) : 6 p.Sélam JL. Risque cognitif des hypoglycémies répétées chez le diabétique. Diabetes Metab Rev 1998 ; 24 : 167-72.Sélam JL. Réévaluation du risque hypoglycémique au cours de l’in-sulinothérapie intensive. Journées de diabétologie de l’Hôtel-Dieu,1998. Paris : Flammarion Médecine Sciences : 257-80.Sélam JL. Traitement du diabète de type 1 (diabète insulino-dépendant). Encycl Med Chir (Elsevier, Paris). Endocrinologie-Nutrition 1999 ; 10 (366-R-30) : 10 p.
POUR EN SAVOIR PLUS

Endocrinologie - Métabolisme - NutritionB 334
1593L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Étiologie, physiopathologie
La dénutrition résulte d’un bilan négatif, entre apportsnutritionnels et besoins métaboliques, notamment protéino-énergétiques. Elle est le plus souvent la consé-quence d’une diminution des apports avec mobilisationdes réserves énergétiques et utilisation de la massemaigre musculaire à des fins énergétiques. L’existencede besoins augmentés et (ou) de pertes accrues accélèrela vitesse de survenue de la dénutrition et en modifiel’expression clinique dans le sens d’une dénutrition àprédominance protéique.
Dénutrition par réduction des apports
Il existe 2 formes de dénutrition, l’une globale, protéino-énergétique ou marasme, d’évolution chronique, l’autreà prédominance protéique ou kwashiorkor, d’évolutionsubaiguë. Dans les pays occidentaux, la carence d’ap-port « isolée » la plus caractéristique est représentée parl’anorexie mentale non compliquée : elle entraîne unedénutrition de type « marasme ». La réduction desapports conduit à une érosion de la masse maigre et àune diminution de la masse grasse, d’autant plus rapideque la carence est importante. En cas de jeûne total(grève de la faim), la synthèse accrue de corps céto-niques supplée partiellement aux besoins des tissusgluco-dépendants, notamment du cerveau et de la moelleosseuse.Les situations caractérisées essentiellement par la réductiondes apports, dites anorexiantes, se rencontrent dans :• de nombreuses pathologies chroniquestelles que les
syndromes algiques, dépressifs, de maldigestion ou demalabsorption, la cirrhose, l’alcoolisme, le sida, lescancers, la bronchopneumopathie, l’insuffisance rénale… ;
• comme conséquence de thérapeutiques lourdes tellesla chimiothérapie, la radiothérapie ou la chirurgie ;
• chez les personnes âgées et dans le quart monde :isolement social, denture, polymédication, escarres,insuffisance de ressources…
Dénutrition par hypermétabolisme
L’hypermétabolisme est une caractéristique des étatsd’agression : brûlure, polytraumatisme, infection,syndromes inflammatoires, suite de chirurgie lourde…
Il y a dénutrition lorsque le bilan protéique, influencénégativement par un bilan énergétique négatif, devientinsuffisant pour répondre aux besoins métaboliques del’organisme. Elle s’accompagne d’une diminution de lamasse maigre (MM), notamment de la masse dite « cellulaire active », de la masse musculaire et d’unealtération de fonctions physiologiques notamment musculaire, immunitaire, de cicatrisation et de la vie de relation ou psychique. Ces altérations dépendent plus de la réduction nette des ingesta, si la durée estsupérieure à 7 jours, que de la masse proprement dite ;elles peuvent donc être observées de façon précoce aucours de la dénutrition.
DénutritionSignes cliniques et biologiques, traitement
PR Luc CYNOBER 1, DR Pascal CRENN 2, PR Bernard MESSING 2,3
1. Service de biochimie et président du Comité de liaison alimentation nutrition (CLAN), L’Hôtel-Dieu, 75181 Paris Cedex 04.2. Service d’hépato-gastro-entérologie et d’assistance nutrititionnelle, hôpital Lariboisière.3. Président du CLAN, hôpital Lariboisière, 75475 Paris Cedex 10.
• Un dépistage systématique de la dénutrition,quel qu’en soit le degré, chez des patients hospitalisés et dans la communauté, est nécessaire car sa prévalence est respectivementde l’ordre de 30 % et 10 %. Elle est majoréechez les personnes âgées.
• La dénutrition résulte d’un bilan protéino-énergétique négatif. Dans la majorité des cas,une réduction absolue ou relative des ingesta estretrouvée ; elle est isolée ou associée à une aug-mentation des besoins dans tous les états trau-matiques, infectieux et inflammatoires et (ou) une augmentation des pertes, notammentdans les syndromes de malabsorption et les brûlures.
• Le traitement de la dénutrition repose donc sur des apports nutritionnels adaptés et suffisants sous forme soit de supplémentsoraux, soit d’une nutrition artificielle entéraleou parentérale ; le mode entéral doit être utiliséde façon préférentielle au mode parentéral.
• L’efficacité de la renutrition doit être vérifiée :elle est plus facilement obtenue lorsque ses complications sont prévenues et maintenuesà un taux minimal par un programme d’assurance qualité sous la responsabilité d’un Comité de liaison pour la nutrition et l’alimentation (CLAN).
Points Forts à comprendre

La dénutrition est alors plus protéique que calorique etla perte de masse musculaire, accélérée par l’immobi-lisation, est de survenue étonnamment rapide. L’hyper-métabolisme est relayé par une réaction immuno-neuro-endocrine (tableau I). Il a pour but de satisfairel’augmentation des besoins énergétiques secondaire à laredistribution de la synthèse protéique, notamment versles protéines de l’inflammation, l’hématopoïèse et laréparation des tissus lésés. Le bilan protéique négatif estdû au déséquilibre entre la synthèse protéique et celle,plus forte, de la protéolyse. L’utilisation des acides aminés à des fins néoglucogéniques entraîne une azoturie qui peut atteindre 20 g/j, soit un équivalent protéique de 125 g (20 x 6,25), soit l’équivalent de 500 g de masse musculaire. Ces situations hyper-métaboliques entraînent également une consommationaccrue de certains acides aminés, de minéraux et demicronutriments (vitamines et oligo-éléments).
Clinique de la dénutrition
Interrogatoire
L’anamnèse pondérale (données déclaratives) semi-récente (3 à 6 derniers mois) et récente (2 dernièressemaines) précisant le poids de référence personnel pré-morbide, ainsi que le recueil du niveau semi-quantitatif(médical) récent des ingesta (normaux, réduits, nuls)sont des données clés faisant partie de l’évaluation del’état nutritionnel (tableau II). Il est également nécessairede disposer de l’enquête alimentaire quantitative diététique en macronutriments, jugée sur la prise oraledes 3 à 7 derniers jours. Une modification de l’activitéphysique et des signes de carence en minéraux et enmicronutriments (crampes, paresthésies, douleursosseuses, etc.) sont recherchés.
Poids et taille
Le poids (en kg, mesuré avec une balance de qualitémédicale) et la taille (en cm) sont notés. Le degréd’amaigrissement, non volontaire, est alors exprimé enpourcentage du poids usuel. Toute perte de poids égale à 5 % est significative ; elle traduit une dénutrition modérée et sévère lorsqu’elle atteint respectivement10 % et 20 %. L’existence d’œdèmes surestime le poidset sous-estime le degré de dénutrition. Même en leurabsence, il existe une augmentation de l’eau extra-cellulaire, parallèle au degré de sévérité de la dénutrition chronique. Le poids (en kg) divisé par la taille (en m2) constituel’indice de masse corporelle (IMC) de Quetelet devaleurs normales comprises entre 20 et 25 kg/m2.Associé à un amaigrissement, un indice de masse corpo-relle entre 20 et 18,5 traduit un risque de dénutrition ;entre 18,5 et 16 une dénutrition modérée ; entre 16 et 13 une dénutrition sévère et en dessous de 13 une dénutrition grave avec augmentation significative durisque de décès à court terme (figure). Chez le vieillard,la taille peut être difficile à déterminer : on peut utiliser àla place de celle-ci la hauteur du genou ou l’enverguresterno-digitale ; chez l’enfant, la courbe de croissanceest un bon marqueur de l’état nutritionnel.
Examen clinique
Il permet de reconnaître plusieurs facteurs.• Le risque de dénutritionpeut être évoqué en présenced’une pathologie chronique, devant une réduction desingesta O 50 % des besoins et O 7 j. Dans ce cas,la perte de poids involontaire et récente peut ne pasdépasser 5 à 10 % du poids usuel.• Dans la dénutrition « établie »,l’index clinique leplus utile qui permet d’évaluer l’état nutritionnel estcelui dit « de Detsky ». Il s’agit d’un index subjectif clinique global qui classe les patients en 3 catégoriesd’état nutritionnel (normal, dénutri sévère et état inter-médiaire ou dénutrition modérée). Il associe les para-
D É N U T R I T I O N
1594 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Facteur Variations Principales de synthèse actions résultantes
Hormones
Cortisol protéolyse musculaireefflux musculaire
des acides aminésGlucagon captation hépatique
des acides aminésglycogénolyse,
néoglucogenèseCatécholamines lipolyse,
glycogénolyseInsuline Insulinorésistance :
hyperglycémie,lipolyse,
hypertriglycéridémie,synthèses protéiques
Hormone Résistance à l’action de croissance de la GH : IGF1,
synthèses protéiques
Médiateurs
Cytokines Anorexigènepro-inflammatoires activation de l’axe(tumour necrosis hypothalamo-factor α, hypophyso-surrénalinterleukines 1 et 6) synthèse des protéines
de l’inflammationresynthèse hépatique
d’acides grasChémokines mobilisation(dont interleukine 8) des cellules immunitaires
(polynucléaires)Prostaglandines, peroxydation lipidique leucotriènes génération de radicaux
libres
➔➔
➔
➔
➔
➔
➔
➔➔
➔
➔
➔➔
➔
➔
➔
➔
➔
➔
➔
➔
Médiation de la réponseà l’agression
TABLEAU I

mètres décrits dans le tableau II. Cet index a, aprèsapprentissage, une bonne reproductibilité entre observa-teurs indépendants pour le diagnostic de ces 3 classes ; il a une meilleure valeur prédictive positive et négativede complications postopératoires que des variables biologiques nutritionnelles, isolées ou associées à l’anthro-pométrie. L’examen cardiaque, neurologique, cutanéo-muqueux et des phanères (perte des cheveux, onglesstriés, desquamation, hyperpigmentation cutanée, folli-culite, chéilite, muqueuses [gencives hémorragiques…])et l’état dentaire apportent des arguments pour descarences minérales, en vitamines ou oligo-éléments.Des œdèmes et une hépatomégalie molle (stéatose) sont 2 signes cardinaux d’une dénutrition protéique prédominante et leur absence, en présence d’un amai-grissement notable, est en faveur d’une dénutrition àprédominance énergétique.
Complications de la dénutrition
Une dénutrition significative est responsable d’une morbidité et d’une mortalité accrues, indépendammentdu diagnostic étiologique de la dénutrition. Cela se traduitpar une prolongation du séjour hospitalier, source d’uneaugmentation des dépenses de santé :– défaut de cicatrisation avec l’absence de prise de
greffe de peau (brûlé), des fistules… ;– immunodépression avec des infections plus fréquentes
et plus sévères majorant la dénutrition ;– défaillance musculaire respiratoire, cardiomyopathie,
retard de la vidange gastrique, état grabataire avecescarres… ;
– altération des fonctions intellectuelles, irritabilité,dépression ;
– aménorrhée, hypothermie, hypoglycémie, acro-syndrome… ;
– et, selon la cause de la dénutrition, les carences miné-rales ou en micronutriments peuvent être au 1er plan :iléus paralytique dû à une hypokaliémie, maladie deGayet-Wernicke à cause du déficit en vitamine B1,pancytopénie due au déficit en folates, acrodermatiteentéropathique liée à un déficit sévère en zinc, neuro-pathie périphérique et (ou) centrale due aux carencesvitaminiques (B1, B12), fractures pathologiques pardéficit prolongé en calcium et en vitamine D…
Diagnostic différentiel
Il faut attirer l’attention sur 2 pièges diagnostiques :– les faux positifs ; en effet, la maigreur dite constitu-
tionnelle n’est pas synonyme de dénutrition car l’indicede masse corporelle est certes bas, compris entre 20 et 16, mais il n’y a ni amaigrissement, ni réductionpatente des ingesta, ni signes carentiels objectifs ;
– les faux négatifs sont plus nombreux, car l’indice demasse corporelle peut rester supérieur à 20 et l’amai-grissement, non volontaire (musculaire), peut êtremasqué par la persistance d’une masse adipeuse sous-cutanée normale ou augmentée.
Endocrinologie - Métabolisme - Nutrition
1595L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Histoire
1 / Perte de poids des 6 derniers mois (% poids de forme)
❑ < 5 %❑ 6 à 10 %❑ > 10 %Évolution pondérale des 2 dernières semaines :
❑ prise de poids❑ poids stable❑ perte de poids
2 / Modification de la prise alimentaire (versus ingesta habituel)
❑ pas de changement❑ modification (depuis combien de semaines : ................)Type : ❑ diminution nette
❑ jeûne
3 / Symptômes digestifs d’une durée > 2 semaines
❑ aucun❑ nausées❑ vomissements❑ diarrhée❑ anorexie
4 / Capacités fonctionnelles
❑ normales❑ perturbées : durée : (.......... semaines)Type : ❑ travaille presque normalement
❑ garde quelques activités❑ reste au lit le plus souvent
5. Degré d’agression : dépense énergétique attendue
❑ normale❑ peu augmentée❑ très augmentée
Normal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0Dénutrition modérée . . . . . . . . . . . . . . . . . . . . . . . . . . . 1Dénutrition nette . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2Dénutrition sévère . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Perte de masse grasse (plis cutanés quadricipital, tricipital,préthoracique)Perte musculaire (quadriceps, fessiers, deltoïde)Œdèmes (prétibial, chevilles, lombes)
A / État nutritionnel normal (absence de symptômes)B / Dénutrition modérée ou potentielle (intermédiaire entre A et C)C / Dénutrition sévère (forte présence de symptômes)
Index dit de Detsky* (d’après J Parent Enteral Nutr 1987 ; 11 : 8-13).
Examen clinique
Classement (3 stades)
Évaluation clinique subjective globale de l’état nutritionnel*
TABLEAU II

Composition corporelle
Des mesures de la composition corporelle peuventobjectiver une diminution de la masse musculaire et de la masse grasse (MG) au cours de la dénutrition :en pratique clinique, elles sont surtout utiles à évaluerl’efficacité d’une renutrition par des mesures séquentielles.Il faut remarquer que la masse musculaire comporteenviron 75 % d’eau. Ce secteur se subdivise en eauxextra- et intracellulaire dont le rapport usuel est de 2 tiers/1 tiers et dont l’estimation est possible au lit dumalade par impédancemétrie. La masse grasse est estimée par la mesure des 4 plis cutanés (bicipital,tricipital, sus-scapulaire et supra-iliaque) et la massemusculaire par la mesure de la circonférence musculairebrachiale (CMB) ou la créatininurie.
Signes biologiques
Protéines marqueurs de l’état nutritionnel
L’albumine, la transthyrétine (TTR ou préalbumine), latransferrine et la retinol binding protein(RBP) sont desprotéines exclusivement synthétisées par le foie. Leursynthèse est dépendante de l’état nutritionnel, leursconcentrations circulantes diminuant lors d’une dénutri-tion et augmentant à nouveau lors de la renutrition. Leursensibilité à la dénutrition, et surtout à la renutrition, estd’autant meilleure que leur demi-vie est courte (48 hpour la transthyrétine, 20 j pour l’albumine). Cependant,les syndromes inflammatoires, les maladies hépatiqueset les modifications des secteurs hydriques de l’organismepeuvent faire varier ces marqueurs. Ces protéines marqueurs de l’état nutritionnel voient leur synthèseinhibée par les cytokines, dont les sécrétions sont augmentées lors de processus inflammatoires, même enl’absence de dénutrition. Ainsi, le dosage d’une protéinemarqueur de l’état nutritionnel doit être systématiquementassocié à celui d’une protéine de la réaction inflammatoire.En pratique clinique, il faut retenir :– l’albuminémie, marqueur pronostique global dont la
diminution (< 35 g/L) fait passer le degré de dénutritionde modéré à sévère ;
– la transthyrétine est un marqueur d’anabolisme quiindique ce déficit pour un seuil < 0,2 g/L. La trans-thyrétine augmente dès le 5e j d’une renutrition efficace ;
– une protéine de la réaction inflammatoire telle quel’ α1-glycoprotéine acide (orosomucoïde) et surtout laprotéine C réactive qui est un très bon marqueur del’existence d’un syndrome inflammatoire ;
– il est utile d’analyser la créatininurie selon, bienentendu, la fonction rénale, mais aussi à l’aune de lamasse musculaire. Elle diminue parallèlement audegré d’amyotrophie.
Le dosage de la transferrine n’a aucun intérêt car l’existence fréquente d’une anémie ferriprive et (ou)d’une inflammation contrebalance ses variations secon-daires à la dénutrition.
Bilan azoté
La détermination du bilan azoté (différence entre l’azoteabsorbé ou perfusé et l’azote excrété) est utile au coursdes états cataboliques. Schématiquement, la balance estpositive en cas de rétention azotée, négative en situationde perte nette d’azote, par carence d’apport et (ou) parpertes accrues. Sa détermination est, en pratique clinique,difficile (erreurs dans la mesure des entrées et des sorties). Elle peut contribuer, avec la détermination de latransthyrétine, à l’évaluation de l’efficacité de la renutrition.Le dosage de l’azote est pratiqué sur les urines et, en casde diarrhée par malabsorption, sur les selles (recueilcomplet de 3 j). Il est réalisé par la méthode de Kjeldahlou par chimioluminescence. À défaut, on dose l’uréemais cela conduit à des erreurs liées à la variabilité despertes azotées sous forme d’ammoniaque.
Immunité
La lymphopénie (< 1 200/mm3) est un marqueur trèssimple mais non spécifique de la dénutrition. Les testscutanés d’immunité cellulaire (Multitest) sont étroitementliés à l’état nutritionnel, avec hypoergie ou anergie, quis’améliorent lors de la renutrition.
Statut en minéraux et en micronutriments
La détermination des concentrations plasmatiques desminéraux (calcium, phosphore, magnésium) et, plusrarement, celle des oligo-éléments (fer, zinc, sélénium)et des vitamines (25-OH-D3, B12, acide folique…) sontpratiquées selon l’orientation clinique. En situation dedésordres hydro-électrolytiques et de pertes réduites ouaccrues, les bilans sodé et potassique doivent être mesurés.
Traitement
Principes et bénéfices attendus
La prise en charge diagnostique et thérapeutique dupatient dénutri doit être faite par une équipe de nutrition(CLAN) qui seule peut obtenir les meilleurs résultats,c’est-à-dire un taux minimal d’iatrogénicité et unmeilleur rapport efficacité-coût. Le traitement de ladénutrition est complémentaire du traitement de lamaladie causale dont il améliore le pronostic en enréduisant la morbi-mortalité. Le diagnostic et le traitement précoces de la dénutrition (avant le 5e j d’hospitalisation) réduisent la durée du séjour hospitalieret de la convalescence. La correction des désordreshydro-électrolytiques et en minéraux est une urgencethérapeutique ; celle-ci augmente l’efficacité de la renutrition, de même que son caractère « complet »incluant électrolytes, vitamines et oligo-éléments, et cequelle que soit la durée du traitement par nutrition artificielle. La prescription du traitement de la dénutritionpar nutrition artificielle et sa surveillance doivent être
D É N U T R I T I O N
1596 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

Ainsi, le bénéfice initial de la renutrition ne se juge passur le gain de poids qui, s’il dépasse 250-300 g/j, traduitune rétention hydrosodée. Les techniques de nutritionartificielle ne doivent pas contraindre à l’immobilisationdu patient et l’on doit encourager une reprise prudente etprogressive de l’activité physique avec l’aide d’unekinésithérapie adaptée.
Indications
Une assistance nutritive est formellement indiquée chezle patient ayant une dénutrition sévère définie par l’unde ces facteurs :– une perte de poids, involontaire et récente, dans les
3 à 6 mois qui précèdent, égale à 20 % du poids deréférence usuel antérieur (donnée déclarative) ; ou,maigreur constitutionnelle exclue :
– un indice de masse corporelle o 16 ;– un stade « C » à l’évaluation clinique subjective
globale de Detsky. Le patient ayant une dénutrition modérée associée à untaux d’albumine sérique de 35g/L doit aussi bénéficierd’une assistance nutritive. Ce « stade modéré » est défini par l’un des paramètres suivants :– une perte de poids, involontaire et récente dans les 3 à
6 mois qui précèdent, de 10 à 19 % du poids usuel ;– un indice de masse corporelle égal à 18,5 ; – un stade « B » à l’évaluation clinique subjective
globale de Detsky.Une assistance nutritive peut être aussi indiquée chez lepatient à risque de dénutrition. Ce risque est difficile àquantifier ; il semble raisonnable de poser l’indicationen présence de l’association des 3 valeurs « seuils » suivantes :– une perte de poids, involontaire et récente, de 5 à 9 %
du poids usuel ;– une situation médicale « anorexiante », quelle qu’elle
soit ;– une transthyrétinémie inférieure à 200 mg/L.
Méthodes d’assistance nutritive
1. Suppléments diététiques oraux (Renutryl, Fortimel…)
Ils sont indiqués en 1re intention en cas d’ingesta baschez les patients à risque de dénutrition ou ayant unedénutrition modérée. Le niveau calorique des supplé-ments est habituellement limité à 1 000 kcal/j. Ils sontprescrits pour une durée de 1 mois, renouvelable unefois et, en règle générale, ils ne s’accompagnent pas dela réduction des ingesta spontanés.
2. Nutrition artificielle Elle est indiquée dès lors que le malade ne peut pas ouplus s’alimenter suffisamment :– en préopératoire avec une dénutrition sévère avant une
chirurgie lourde programmée (l’assistance nutritive estpoursuivie en postopératoire) ;
faites par un CLAN sur documents spécifiques, préétablis,adaptés à la catégorie de patients traités. Normonutritionprotéino-énergétique et nutrition complète préviennentle « syndrome de renutrition », parfois mortel, secondai-re aux déficits en nutriments, notamment en phosphore,magnésium, potassium, sélénium, zinc et vitamines.Une renutrition bien conduite doit être efficace en 1 à 2semaines avec améliorations fonctionnelles, musculaires,immunitaires et psychologiques, bilan azoté positif,augmentation de la transthyrétine et bilan hydrosodééquilibré ou négatif (hyperdiurèse et fonte des œdèmes).
Endocrinologie - Métabolisme - Nutrition
1597L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Classification de l’état nutritionnel selon l’indice demasse corporelle [IMC = poids (kg)/taille (m)2].
Obésité sévère
IMC
40
30
25
20
18,5
16
13
Poids (kg)pour une taillede 1,70 m
115
85
70
55
53
46
37,5
Obésité
Surcharge pondérale
Zone de normalité
Risque de dénutrition
Dénutrition modérée
Dénutrition sévère
Risque de décès
Une courbe en U relie le risque de décès à l’indice de masse corporelle,le risque le plus faible étant compris entre 20 et 25 (zone de normalité).

– en postopératoire et en réanimation, chez les maladesà risque de dénutrition ou ayant une dénutrition modérée ; la nutrition entérale précoce diminue la fréquence des infections ;
– en présence d’une insuffisance intestinale aiguë tran-sitoire (2 semaines) ou prolongée (malabsorptionsévère de cause médicale ou chirurgicale), la nutritionparentérale est indiquée.
3. Détermination des besoinsLes besoins énergétiques peuvent être calculés à l’aidede la formule de Harris et Benedict qui prend en comptele sexe, la taille, le poids et l’âge. Les valeurs obtenuesdoivent être corrigées pour tenir compte du niveaud’agression :x 1,1 en période postopératoire,x 1,1 à 1,3 en cas de fractures multiples,x 1,3 à 1,6 dans lesinfections sévères,x 1,5 à 2,1 chez le brûlé, en fonctionde la surface corporelle lésée. Cependant, leur applicationaboutit souvent à une surestimation des dépenses éner-gétiques vraies. Les besoins énergétiques peuvent êtremesurés par la calorimétrie indirecte qui permet d’approcher les dépenses réelles du malade. Cetteméthode repose sur la détermination de la consom-mation d’oxygène et de la production de gaz carbonique.De réalisation relativement simple, elle nécessite cependant des précautions méthodologiques. La dépense énergétique de repos est ainsi d’environ 30 kcal/kg/24 h chez l’homme et 25 kcal/kg/24 h chezla femme. Elle est réduite dans les états de dénutritionchronique et chez les malades normométaboliques sédatés sous ventilation mécanique ou hypothermiques.Elle est augmentée chez les malades agressés, d’un facteur variable selon la maladie (v. ci-dessus). En pratique, la majorité des patients peut être correctementnourrie avec 1 600 à 2 400 kcal/j.Les besoins azotés se situent entre 150 et 350 mg d’azote/kg/j. D’un point de vue qualitatif, on utilise desprotéines de bonne valeur nutritionnelle (nutrition enté-rale) ou des mélanges d’acides aminés reproduisant lacomposition des protéines de valeur nutritionnelle élevée (nutrition parentérale). Certains acides aminés,tels que la glutamine (Dipeptiven), l’arginine (Impact,Hyperamine) et l’alpha-cétoglutarate d’ornithine, pré-curseur des 2 précédents (Ornicétil, Cétornan) possèdentdes propriétés pharmacologiques (sur le métabolismeprotéique, la cicatrisation, l’immunité) lorsqu’ils sontapportés en quantités importantes (de l’ordre de 10 à 30 g/j). Leur utilisation est justifiée chez les patientssévèrement dénutris et (ou) cataboliques, lorsqu’unenutrition conventionnelle est inefficace. Parce que l’accrétion azotée a un coût énergétique, lerapport entre apports caloriques et apports azotés doitêtre optimal : 1 g d’azote pour 200 calories glucido-lipidiques chez les malades dénutris chroniques et 1 pour 100 à 1 pour 125 chez les patients agressés.L’administration d’eau et d’électrolytes doit être adaptéeà la pathologie. Les apports en sodium doivent être limitésà 3 mmol/kg/j. Au contraire, les besoins en potassiumsont augmentés (de l’ordre de 6 mmol/g d’azote) car cet
élément est nécessaire à l’utilisation du glucose et del’azote. Les besoins en phosphore sont élevés chez lesmalades agressés. Ceux en magnésium sont augmentésdans les entéropathies. Les apports en vitamines etoligo-éléments doivent être systématiques.
4. Nutrition entéraleLa voie entérale doit être utilisée dès lors que l’état anatomique et fonctionnel du tube digestif le permet. Eneffet, pour une efficacité nutritionnelle identique, elleest moins onéreuse et plus sûre que la voie parentérale.Cette voie d’abord digestive implique l’utilisation d’unesonde, mise en place le plus en amont possible afin detirer profit au mieux de la fonctionnalité digestive :– la sonde naso-gastrique est la méthode la plus adaptée
du fait de sa simplicité ;– la sonde naso-duodénale ou naso-jéjunale est moins
bien tolérée mais permet une nutrition entérale en casde gastroparésie ou de pancréatite aiguë.
– pour la gastrostomie perendoscopique, les indicationspréférentielles de ce geste réalisable sous anesthésielocale sont les cancers des voies aérodigestives supérieures et les troubles de déglutition ;
– la jéjunostomie chirurgicale.• Modalités d’administration :elle se fait de manièrecontinue sur le nycthémère au moyen d’une pompe péri-staltique. Le débit initial est augmenté toutes les 24 à 48 hen fonction de la tolérance digestive.La plupart des produits sont iso-osmolaires et contien-nent 1 kcal/mL. On préfère les produits polymériques(nutriments non dégradés : protéines, polysaccharides,triglycérides à chaînes longues : Normoréal, Nutrison…).Lorsque les capacités intestinales de digestion-absorptionsont réduites (entéropathies), on utilise des produitssemi-élémentaires (peptides, dextrines, triglycérides àchaîne moyenne) [Réabilan].• Parmi les complications mécaniques,les plus fré-quentes sont les déplacements secondaires de la sonde.Leur prévention passe par sa mise en place par un personnel entraîné et la vérification régulière du bonpositionnement de son extrémité. Les obstructions sontprévenues par son rinçage régulier.• Les régurgitations avec risque d’inhalationsont unautre type de complications. La pneumopathie d’inhala-tion est la complication la plus grave. Sa fréquence estaugmentée dans les maladies neurologiques et est diminuée en position semi-assise.• L’intolérance digestiven’est pas rare, surtout chez lepatient agressé chez qui la motricité gastro-intestinaleest altérée. Les nausées et vomissements (10 à 20 % despatients) imposent l’arrêt ou la modification de la tech-nique de nutrition. Des ballonnements et des crampesabdominales peuvent survenir. La diarrhée est une com-plication fréquente (40 % des patients), en rapport avecl’osmolalité des produits, la vitesse d’infusion (elle doitêtre inférieure à 180-240 mL/h), une infection nosoco-miale favorisée par une antibiothérapie.• Certaines complications sont liées à la sonde ;ils’agit des œsophagites et de l’érosion de l’aile du nez.
D É N U T R I T I O N
1598 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

– la survenue d’une fièvre chez un malade porteur d’uncathéter central doit faire systématiquement suspecterson infection et réaliser une hémoculture périphériqueet sur le cathéter ;
– hépatiques (stéatose, cholestase) ;– osseuses (en cas de nutrition parentérale au long cours).
5. Nutrition parentéraleSes indications sont les contre-indications et impossibilitésde la nutrition entérale : sténose digestive haute, occlu-sion intestinale chronique, colites sévères (notammentmaladie de Crohn), syndromes de malabsorption chro-niques sévères (insuffisance intestinale) qui répondent àune indication au long cours et est réalisée le plus souvent au domicile du patient.• La voie d’abord vasculaire(nutrition par veine péri-phérique) est réservée à une nutrition de courte durée (< 2 à 3 semaines) et suppose un capital veineux suffisant. L’osmolarité des solutions ne doit pas dépasser800 mOsm/L.Dans la nutrition par voie centrale, le cathéter est placédans la veine sous-clavière ou dans la veine jugulaireinterne.Dans tous les cas, la nutrition parentérale nécessite descathéters biocompatibles et inertes.• En ce qui concerne les modalités d’administration,on peut utiliser des flacons séparés pour chaque macro-nutriment (acides aminés, glucides, lipides) et perfuséssimultanément en Y. Il existe des poches où les 3 macro-nutriments sont prémélangés (mélanges ternaires :Vitrimix…) ou doivent l’être extemporanément (pochesbi- ou tri-compartimentées ; Aminomix, Clinomel).Chez les malades agressés, on préfère une perfusion 24 hsur 24 ; sinon, une nutrition cyclique nocturne, sur 12 h à 16 h sur 24, est la plus adaptée.Les besoins énergétiques sont couverts par un apportmixte glucido-lipidique. Les glucides sont apportés sousforme de glucose qui fournit 4 kcal/g. L’apport est compris entre 3 et 4 g/kg/j. Les émulsions lipidiques (9 kcal/g) sont administrées à la dose de 1 à 2 g/kg/j,perfusées sur au moins 6 h, idéalement sur 12 à 16 h.Les émulsions lipidiques contiennent :– des triglycérides à chaînes longues (TCL) [Intralipide] ;
on préfère les émulsions à 20 % ;– un mélange équilibré triglycérides à chaînes longues/
triglycérides à chaînes moyennes (TCM) [Médialipide].Les triglycérides à chaînes moyennes présententl’avantage de pénétrer dans les mitochondries sansrecourir à un transporteur, la carnitine, qui pourraitêtre déficient chez les malades agressés ;
– les émulsions à base d’huile d’olive (Clinoléic) sontriches en acide oléique et en vitamine E, leur conférantdes propriétés antioxydantes.
De nombreuses solutions d’acides aminés cristallisésexistent. La plupart reproduisent la composition de protéines de référence (Vintène, Vamine, etc.). Certainessont plus riches en acides aminés non essentiels(Nutrilamine).• Les complicationspouvant survenir sont de différentsordres :– acidose, désordres métaboliques et autres désordres
hydro-électrolytiques. Leur surveillance nécessite lamesure régulière de l’ionogramme sanguin et urinaireet de la glycémie ;
– thrombose veineuse sur cathéter ;
Endocrinologie - Métabolisme - Nutrition
1599L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• La dénutrition est responsable d’une morbiditéet d’une mortalité accrues avec prolongation du séjour hospitalier.
• La dénutrition, qu’elle soit sévère ou modérée,entraîne des anomalies fonctionnelles qui sontresponsables de complications propres,et notamment d’une augmentation de la fréquenceet de la sévérité des infections.
• Son diagnostic repose sur un amaigrissementnon volontaire, supérieur à 5 % du poids usuel,parfois considérable, touchant préférentiellementla masse musculaire, associé à une réductiondes ingesta. Un indice de masse corporelle inférieur à 20, à condition de le distinguer de la maigreur, est un bon argument diagnostique;cependant un indice de masse corporelle normal ou fort peut masquer une dénutritionmême sévère.
• Les méthodes de renutrition par nutrition artificielle font appel soit aux complémentsoraux, soit à la nutrition entérale et, en casd’échec ou d’impossibilité de celle-ci, à la nutritionparentérale. Ces traitements doivent obéir à des référentiels qui, sous l’égide des comitésde liaison pour la nutrition et l’alimentation,en garantissent la sécurité et l’efficacité.
• Le traitement précoce de la dénutrition estcomplémentaire du traitement de la maladiecausale ; il améliore le pronostic en réduisant la morbi-mortalité, la durée du séjour hospitalieret la qualité de vie.
Points Forts à retenir
Carences nutritionnelles : étiologies et dépistage. Paris : INSERM,1999.
Cynober L, Aussel C. Exploration de l’état nutritionnel.Collection Explorations fonctionnelles humaines. Cachan : Édi-tions Médicales Internationales, 1998.
Ricour, Ghisolfi J, Putet G, Goulet O. Traité de nutrition pédiatrique.Paris : Maloine, 1993.
Leverve X, Cosnes J, Erny Ph, Hasselmann M. Traité de nutritionartificielle de l’adulte. Paris : Mariette Guena, 1998.
POUR EN SAVOIR PLUS

Endocrinologie - Métabolisme - NutritionB 330
1473L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
animaux de diabètes auto-immuns et les rares étudeshistologiques humaines suggèrent que l’évolution se faiten 3 phases :– le déclenchement se caractériserait par une présentation
macrophagique d’auto-antigènes pancréatiques, qu’uneapoptose initiale des cellules bêta pourrait favoriser ;
– le développement d’une insulite se caractérise par uneinfiltration de lymphocytes CD4 et CD8 autour desîlots, avec peu ou pas de destruction des cellules bêta(« péri-insulite ») et par une production de cytokinesinflammatoires, interleukine 2 (IL2), interféron γ(IFN γ), TNF α (tumor necrosis factor α) ;
– la phase terminale se caractérise par une prédominancedes CD8 (cytotoxiques). La destruction des cellulesbêta est secondaire à leur apoptose. L’origine de celle-ciest probablement multifactorielle, liée entre autres àl’expression de Fas à leur surface (favorisée par lescytokines inflammatoires), les lymphocytes T activésexprimant Fas ligand.
La réaction immunitaire cellulaire spécifique pour différents antigènes pancréatiques témoigne de leurimportance pathogénique. En particulier, la suppressionpar transgenèse de l’expression de la protéine GAD(glutamic acid decarboxylase), au niveau des cellulesbêta des souris diabétiques NOD, permet de prévenir lediabète. De plus, les îlots comportant des cellules bêtan’exprimant pas GAD, transplantés chez les sourisNOD, ne subissent pas l’attaque auto-immune, contrai-rement aux îlots contenant des cellules bêta normales.La protéine GAD apparaît donc comme un antigènemajeur dans le déroulement de la réaction immunitairechez la souris NOD. La démonstration de son rôle prépondérant n’a pas encore été faite chez l’homme.Au cours de cette réaction d’immunité cellulaire, sontproduits des auto-anticorps dirigés contre certains anti-gènes pancréatiques. Ces auto-anticorps n’ont pas eneux-mêmes de rôle pathogène mais sont des marqueursfiables du déroulement du processus auto-immun. Ilssont essentiellement au nombre de 4 : les anticorps anti-îlots (islet cell antibody, ICA) présents chez 60 à 80 % despatients au début du diabète ; les anticorps anti-GAD présents chez 80 % des patients présentant un diabète detype 1 ; les auto-anticorps anti-insuline retrouvés surtoutchez l’enfant ; les anticorps anti-IA2 dirigés contre unetyrosine phosphatase membranaire, présents chez 50 à75 % des patients présentant un diabète de type 1.
Épidémiologie
Le diabète de type 1 touche environ 10 % de l’ensemblede la population diabétique, soit en France 150 000 per-sonnes. Il survient à tout âge, mais surtout avant l’âge de 20 ans, avec un pic de fréquence vers 12 ans.L’incidence avant l’âge de 15 ans en France est de 7 pour 100 000. Il existe un important gradient nord-sud,puisque l’incidence en Finlande est de 42 pour 100 000. Ce gradient nord-sud s’expliquerait par des raisons génétiques imparfaitement connues et par desfacteurs d’environnement totalement inconnus, bien queles virus fassent figure de suspects numéro un. Enfin, ilsemble exister une augmentation de l’incidence dans lemonde d’environ 3 % par an, sans que l’on en connaissela raison.
Physiopathologie
Processus auto-immun
Le diabète de type 1 est dû dans l’immense majorité descas à une destruction auto-immune des cellules bêta dupancréas, qui se déroule à bas bruit pendant plusieursannées avant le début du diabète. L’étude des modèles
Diabète insulinodépendantÉtiologie, physiopathologie, diagnostic, complications, traitement
PR André GRIMALDI, DR Agnès HARTEMANN-HEURTIER
Service de diabétologie et métabolisme, groupe hospitalier La Pitié-La Salpêtrière, 75651 Paris Cedex 13.
• Le diabète de type 1 est dû à une destructiondes cellules bêta du pancréas, dans la majoritédes cas d’origine auto-immune.
• Le diabète apparaît lorsque plus de 80 % des cellules bêta sont détruites. Il représentedonc la « cicatrice » métabolique d’une maladieimmunitaire.
• Son traitement repose sur l’insulinothérapievisant à remplacer l’insulinosécrétion physiologique. Le but du traitement est d’obtenir un équilibre glycémique moyenaussi proche que possible de la normale pour éviter les complications de micro- et de macroangiopathie.
Points Forts à comprendre

Le dosage des anticorps anti-GAD, anti-IA2 et anti-insuline fait appel à des techniques radio-immunologiqueset tend à suppléer le dosage des anticorps anti-îlots parimmunofluorescence sur coupes de pancréas humain,dans le dépistage des patients à risque de développer undiabète. Le risque de survenue de diabète augmente avecle taux et le nombre des anticorps présentés par le patient.
Terrain génétique de susceptibilité
Cette insulite prédiabétique survient sur un terrain génétique prédisposé, mais il s’agit d’une susceptibilitéfaible puisque lorsque la mère est diabétique insulino-dépendante, le risque pour l’enfant est de 2 à 3 %, lorsquele père est diabétique insulinodépendant le risque est de 4 à 5 %. Lorsqu’il existe des frères et sœurs, le risque est de 5 % et lorsqu’il s’agit de jumeaux univitellins, laconcordance n’est que de 30 à 40 %. Finalement, on neretrouve une hérédité familiale de diabète de type 1 chezun nouveau diabétique qu’une fois sur 10.Il s’agit en réalité d’une susceptibilité plurigénique avecau moins une dizaine de gènes en cause. Le principal(rendant compte de 40 à 50 % de la susceptibilité géné-tique) se situe sur le chromosome 6 au niveau des gènesdu système HLA de classe 2 avec un risque relatif de 3 à 5 lorsqu’il existe un antigène HLA DR3 ou DR4. Le risque relatif atteint 20 à 40 lorsque les 2 antigènesDR3 et DR4 sont associés (l’association DR3 DR4 estfréquente dans la population diabétique et exceptionnelledans la population non diabétique). Ainsi, le risque pourdes frères et des sœurs doit être précisé en fonctiond’une identité HLA avec le sujet diabétique. Le risqueest de 15 % lorsque les frères ou sœurs ont les 2 haplo-types HLA en commun avec le parent diabétique. Iln’est que de 7 % lorsqu’ils n’ont qu’un seul haplotypeen commun et il est inférieur à 1 % lorsque les 2 haplo-types sont différents. La caractérisation moléculaire desgènes des molécules de classe 2 a permis d’identifier ungrand nombre d’allèles nouveaux. Les anciennes spéci-ficités DR et DQ définies sérologiquement ont été divisées en sous-types dont certains sont associés au dia-bète de type 1, le risque relatif de certains allèles DQétant supérieur à celui obtenu pour DR. Ainsi, une fortesusceptibilité est apportée par les haplotypes DR3 DQB1 02 01 et DR4 DQ B1 03 02. À l’inverse, une forteprotection est conférée par l’haplotype DR 15 DQ B1 06 02.Le mécanisme expliquant le lien entre ces différentshaplotypes HLA et la survenue du diabète auto-immun,reste en partie inconnu. On a identifié plusieurs régions contenant des gènes deprédisposition, en particulier dans les régions prochesdu gène de l’insuline sur le chromosome 11, proches durécepteur de l’IGF1 sur le chromosome 15. Certainsgènes de susceptibilité pourraient être communs auxdiabètes de types 1 et 2, expliquant l’augmentation del’hérédité du diabète de type 2 retrouvée chez les sujetsdiabétiques de type 1.
Facteurs déclenchants
Des facteurs d’environnement sont probablement à l’origine du déclenchement du processus auto-immuni-taire, qu’il s’agisse de facteurs nutritionnels, toxiques ouviraux. Des faits cliniques tels que la présence d’un diabète insulinodépendant en cas de rubéole congénitale(environ 20 %) et des faits expérimentaux observés chezl’animal font suspecter un rôle essentiel des virus (oreillons,Coxsackie B4, cytomégalovirus, hépatite B…). Lesvirus pourraient intervenir de multiples façons :– certains virus pourraient présenter un mimétisme anti-
génique avec des protéines de cellules bêta (il existeune séquence peptique commune entre le virusCoxsackie et la GAD par exemple) ;
– l’infection virale pourrait être responsable de la sécré-tion de cytokines, en particulier d’interféron γ, entraînantune expression anormale des antigènes de classe II,avec présentation d’auto-antigènes pancréatiques auxrécepteurs des lymphocytes T CD4, et une surexpressiondes antigènes de classe I, accélérant le processus de destruction par les lymphocytes cytotoxiques CD8 ;
– les virus pourraient également participer à l’inductiond’une apoptose des cellules B (par des cytokinesinflammatoires) initiant le processus auto-immunitaire ;
– enfin, l’infection virale pourrait rompre la toléranceimmunitaire en activant une insulite quiescente ou enrompant l’équilibre entre les lymphocytes TH1 (orientantla réaction immunitaire vers l’immunité cellulaire),les lymphocytes TH2 (orientant la réaction immunitairevers l’immunité humorale), c’est-à-dire en levant lasuppression de la réaction auto-immune cellulaire.
Conséquences pour le clinicien
On peut retenir 4 messages essentiels.Le risque génétique est faible (voir :Pour approfondir 1).Il ne s’agit pas de la transmission d’un gène pathologiqueresponsable par lui-même de l’apparition de la maladie,mais seulement de la transmission plurigénique d’unesusceptibilité, l’apparition de la maladie étant déterminéepar des facteurs d’environnement.La maladie immunologique évolue à bas bruit pendantdes années avant l’apparition du diabète. Les infectionset chocs psychologiques précédant de quelques semainesou de quelques mois l’apparition du diabète, souventincriminés par les malades ou leur entourage, ne peuventdonc jouer qu’un rôle de révélateur.On connaît mal l’histoire naturelle de l’insulite pancréatiquechez l’homme. Cependant, il semble que la destructiondes cellules bêta ne soit pas linéaire, mais connaisse uneaccélération finale, si bien que la préservation du capitalinsulinosécrétoire restant lors de la découverte du diabètenécessite la mise en place d’une insulinothérapie optimaleen urgence.On peut aujourd’hui dépister simplement l’insulite pré-diabétique grâce à un dosage des anticorps anti-GAD.Cependant, dans la mesure où nous ne disposons pasd’une thérapeutique préventive validée, il ne saurait être
D I A B È T E I N S U L I N O D É P E N D A N T
1474 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

– un diabète de type 2 avec surpoids, et souvent une fortehérédité familiale, débutant par un syndrome cardinalavec cétose. Cette situation se rencontre particulièrementchez les Africains et les Indiens. La négativité desauto-anticorps permet d’éliminer un diabète de type 1.L’évolution de ce type de diabète, ni 1 ni 2, se fait en 3 temps : début initial cétosique nécessitant le recourstransitoire à l’insulinothérapie, puis équilibration dudiabète par hypoglycémiants oraux évoluant secondai-rement en quelques années vers l’insulinorequérance.Ce diabète associerait une insulinorésistance et undéfaut insulinosécrétoire secondaire à une diminutionde la masse cellulaire bêta d’origine génétique.
À l’inverse, devant une hyperglycémie modérée survenantchez une personne de moins de 20 ans dans un contextefamilial évocateur d’hérédité autosomique dominante, ilfaut suspecter un diabète MODY (maturity-onset dia-betes of the youth). De même, si le diabète est d’héréditématernelle et s’accompagne de maladies évocatrices(surdité, rétinopathie dégénérative non diabétique, déficitmusculaire, pseudo-accident vasculaire cérébral…), ondoit penser à un diabète mitochondrial. Ces diabètesgénétiques bénéficient aujourd’hui d’un diagnosticmoléculaire.
Traitement
Schéma d’insulinothérapie
Le traitement du diabète insulinodépendant repose surl’insulinothérapie visant à reproduire le mieux possiblel’insulinosécrétion physiologique grâce aux injectionssous-cutanées d’insuline ou à une perfusion continuesous-cutanée par pompe portable, permettant de réaliserun schéma dit « basal-prandial ». En effet, l’insulino-sécrétion physiologique comporte une insulinosécrétionde base continue, persistante, y compris après plusieursjours de jeûne, à laquelle viennent s’ajouter des picsinsulinosécrétoires adaptés à la quantité de glucidesingérés lors des repas. L’insulinothérapie basale estassurée soit par le débit de base de la pompe, soit par 2 injections d’insuline semi-lente (NPH) ou lente.L’insulinothérapie prandiale est assurée par des injectionsd’insuline rapide (Insuline Actrapid, Umuline rapide,Insuman rapide, Orgasuline) ou par des analogues del’insuline rapide (Insuline Lispro, Insuline Aspart)injectées avant chaque repas.La dose d’insuline basale est d’environ 0,35 unités/kg,sachant qu’il s’agit d’une dose moyenne, variable d’unpatient à l’autre et souvent plus faible chez les patientsminces ayant une activité physique importante ou endébut de diabète lorsqu’il persiste une insulinosécrétionrésiduelle endogène (phase dite de « lune de miel »). Lesdoses d’insuline basale changent peu au cours de l’année.Les malades doivent être éduqués pour adapter les dosesd’insuline basale en fonction des résultats des contrôlesdes glycémies capillaires de la semaine ou des 2 semainesprécédentes.
question d’un dépistage systématique. Néanmoins,beaucoup de diabétologues conseillent ce dépistage dansles fratries de sujets diabétiques pour au moins 2 raisons.Un dépistage positif permettrait au sujet prédiabétiquede participer à un protocole de recherche d’immuno-thérapie préventive. L’administration d’insuline par voieorale serait susceptible d’induire une tolérance immunitaireen favorisant la réponse TH2 au détriment de la réponseTH1 et en stimulant le développement de lymphocytes Tprotecteurs sécrétant du TGFβ (transforming growthfactor β) au niveau des îlots pancréatiques eux-mêmes. À défaut, le dépistage permettrait une surveillance rapprochée, autorisant la mise en route d’une insulino-thérapie avant la décompensation cétosique du diabète,le risque d’éclosion d’un diabète dans un délai d’un anétant pratiquement de 100 % chez les frères et sœursd’un patient diabétique insulinodépendant présentant desanticorps anti-GAD et une glycémie à jeun à plusieursreprises supérieure à 1,10 g/L.
Clinique
Le début clinique du diabète de type 1 est le plus souventrapide, marqué par l’apparition soudaine d’un syndromecardinal associant polyurie, soif et amaigrissementcontrastant avec une polyphagie. Souvent, ce syndromecardinal s’enrichit d’une grande fatigue en particulier àl’effort, de douleurs musculaires et de troubles de la vueà type d’hypermétropie secondaire aux perturbationsosmotiques du cristallin. Le diagnostic est confirmé parun simple dosage de la glycémie ; s’il n’est pas porté àce stade, l’évolution se fait vers l’acidocétose avecdéshydratation, nausées, vomissements, douleurs abdo-minales et surtout polypnée. Une mesure de la glycémiecapillaire et une recherche de la cétonurie suffisent àconfirmer le diagnostic.Devant un tableau clinique typique, il faut instituer sanstarder le traitement par insulinothérapie. Point n’estbesoin de confirmer le diagnostic par des examens com-plémentaires. Ni la détermination du phénotype HLA nile dosage des auto-anticorps ou de l’insulinémie ne sontnécessaires. Seuls peuvent être justifiés les examenscomplémentaires, guidés par la clinique, à la recherched’une éventuelle cause déclenchante telle qu’une infection. Le dosage des auto-anticorps anti-GAD peut en revancheêtre utile lorsqu’il existe une atypie clinique, en particulierdans les 2 situations suivantes :– une hyperglycémie modérée sans cétose pouvant évoquer
un diabète non insulinodépendant mais dont on est« surpris » par la survenue chez une personne de moinsde 40 ans, ou par l’absence d’obésité, ou par l’associationà d’autres maladies auto-immunes d’organes (vitiligo,dysthyroïdie, maladie de Biermer…). La présence d’anti-corps anti-GAD permet alors d’évoquer un diabète detype 1 d’évolution lente (LADA), représentant 15 à20 % des diabètes non insulinodépendants survenantavant40 ans, surtout en l’absence de surpoids et évoluant en quelques années vers l’insulinorequérance ;
Endocrinologie - Métabolisme - Nutrition
1475L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

Les doses d’insuline rapide ou ultrarapide injectéesavant les repas doivent être calculées en fonction :– des apports glucidiques et protidiques prandiaux ; on
considère que pour 10 g de glucides, il faut 2 unitésd’insuline rapide au petit déjeuner, 1 unité au déjeunerde midi, 1 unité et demie au dîner, et que pour 20 g deprotides, il faut ajouter 1 unité d’insuline (ces valeursindicatives doivent être adaptées individuellement) ;
– de l’activité physique prévue dans les heures qui suiventle repas (si celle-ci est importante, la dose d’insulinepeut être divisée par 2 voire supprimée) ;
– des résultats glycémiques des jours précédents, dansla période horaire correspondant à la durée d’efficacitéde l’insuline injectée ;
– de la glycémie instantanée, le malade réalisant ainsiun véritable « correctif thérapeutique », diminuant ladose d’insuline de 1 ou 2 unités si la glycémie estinférieure à 1,20g/L ou 0,80 g/L, l’augmentant de 1 à 2 unités si elle est supérieure à 1,60 ou 2 g/L.L’adaptation de la dose d’insuline en fonction de laglycémie instantanée reste controversée.
Éducation thérapeutique
Pour réaliser une telle adaptation thérapeutique, le maladedoit mesurer sa glycémie capillaire 3 à 4 fois par jour àl’aide d’un stylo auto-piqueur permettant d’obtenir defaçon quasi indolore une goutte de sang au bout dudoigt. Il doit connaître le délai, le pic et la durée d’actionde ses insulines (voir :Pour approfondir 2). Enfin, il doitavoir une formation diététique pour évaluer la quantitéde glucides ingérée et connaître les équivalences (voir :Pour approfondir 3). Le régime du diabétique insulinodépendant est donc unrégime équilibré, adapté en fonction de l’âge et desbesoins énergétiques, apportant environ 50 % de caloriessous forme glucidique, 35 % sous forme lipidique, 15 %de protéines. Les seuls glucides interdits en dehors del’hypoglycémie sont les glucides d’absorption rapideessentiellement en raison de leurs propriétés physiquesliquides (sodas). Les autres glucides y compris les glucidessimples, tels que la saccharose ou le fructose, ne sont pasinterdits aux sujets diabétiques insulinodépendants, àcondition que le malade adapte en conséquence les dosesd’insuline. Pour limiter le risque athérogène, il est conseilléde diminuer les graisses saturées d’origine animale (endehors des poissons) et d’augmenter les graisses mono-insaturées (huile d’olive, huile d’arachide, colza, avocat).Trois verres de vin par jour sont autorisés (l’alcool necontient pas de sucre ; seuls les vins doux, les vins cuitset la bière contiennent des glucides). Il est déconseilléaux sujets diabétiques de fumer et une aide doit être systématiquement proposée pour le sevrage tabagique.
Prévention et traitement de l’hypoglycémie
Le but du traitement est d’éviter les complications dégé-nératives sévères du diabète survenant à long terme,grâce à un équilibre glycémique aussi bon que possible,
tout en permettant une qualité de vie quotidienne accep-table. En effet, une hémoglobine glycosylée (HbA1c)autour de 7,5 % (soit une moyenne glycémique autourde 1,60 g/L) n’est obtenue qu’au prix de 2 à 3 hypo-glycémies diurnes par semaine. Abaisser le seuil d’hémo-globine glycosylée au-dessous de 7,5% pour se rapprocherde la normale, augmente proportionnellement le risqued’hypoglycémie. Des hypoglycémies modérées sympto-matiques, permettant un resucrage immédiat sont pratiquement sans conséquence, mais la répétition deshypoglycémies peut émousser certains des symptômesd’alerte neurovégétatifs conduisant à des hypoglycémiessévères non perçues par le malade, pouvant entraînerdes comas répétés. Le risque hypoglycémique doit doncêtre évalué pour chaque malade et la prescription d’uneinsulinothérapie doit aller de pair avec une éducation surla prévention et le traitement de l’hypoglycémie (voir :Pour approfondir 4, 5 et 6).
D I A B È T E I N S U L I N O D É P E N D A N T
1476 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
HbA1c = 6% 1,20 g/L
Pour chaque pourcent d’HbA1C, on ajoute 0,30 g/L soit❑ HbA1c = 7 % 1,50 g/L❑ HbA1c = 8 % 1,80 g/L❑ HbA1c = 9 % 2,10 g/L
* En dehors de la grossesse, pour une technique HPLC (normale 4,8 ± 0,4 %).
Correspondance entre les valeursd’HbA1c et la glycémie moyenne
des 2 mois précédant le prélèvement*
TABLEAU
Vingt à 30 % des sujets diabétiques insulinodépendantsprésentent chaque année une hypoglycémie sévère (néces-sitant l’aide d’une tierce personne) ; 10 % présentent uncoma hypoglycémique ou une crise comitiale provoquéepar l’hypoglycémie ; moins de 1% des diabétiques décèdentd’un coma hypoglycémique, mais le pourcentage est plusélevé s’il s’agit de sujets diabétiques insulinodépendantsvivant seuls et sentant mal les hypoglycémies ou de per-sonnes diabétiques ayant une insuffisance rénale terminaleou encore présentant une intoxication alcoolique.
Symptômes de l’hypoglycémie
Il existe 2 types de symptômes.
1. Symptômes neurovégétatifsIl s’agit de palpitations, de tremblements, de sueurs palmaires, de sueurs diffuses, d’une pilo-érection, depâleur et de fringales.
2. Signes de neuroglycopénieIl s’agit d’un ralentissement intellectuel, de troubles dela vue (diplopie, perte de la vision des couleurs, perte de la vision de profondeur du champ ou, au contraire,

chaque repas, ce que ne permettent pas les mélangespréparés d’insulines dites biphasiques (insuline Mixtardou Umuline Profil…).L’obtention d’un bon équilibre glycémique nécessite lapratique quotidienne de l’autocontrôle glycémique 3 à 4 fois par jour. La fréquence des autocontrôles dépendde la possibilité pour le malade d’en tirer des conclusionsthérapeutiques, en particulier pour l’adaptation de sesdoses d’insuline, mais aussi pour le délai entre l’injectionet le repas. Les schémas d’insulinothérapie optimisée« basal-prandial », en particulier ceux comprenant uneinjection d’insuline ultrarapide, nécessitent la pratiquede l’adaptation thérapeutique immédiate en fonction desrésultats. Le tenue d’un carnet de surveillance diabétiqueest un outil utile au malade pour l’adaptation de son traitement dans l’intervalle des consultations médicales.Le carnet permet rétrospectivement de discuter avec lemédecin des décisions thérapeutiques prises par le patient.Devant un mauvais équilibre du diabète, en particulierune instabilité glycémique, il importe de rechercher sys-tématiquement l’existence de lipo-hypertrophies auniveau des bras et des cuisses. Ces lipo-hypertrophies,dues à l’absence de variation de point de piqûre, perturbentla résorption de l’insuline. Il est donc important que lespatients soient informés de la nécessité de varier lespoints d’injection, même s’il est conseillé de garder lesmêmes territoires aux mêmes heures d’injection (parexemple : le ventre le matin, car la résorption accéléréed’insuline rapide permet de mieux contrôler le pichyperglycémique suivant le petit déjeuner, les bras lemidi en raison de leur commodité et les cuisses le soirpour assurer une résorption plus lente des insulines retard).La survenue d’hypoglycémies est évidemment une limiteimportante du traitement par injections d’insuline sous-cutanée. Néanmoins, la plupart des sujets diabétiquestolèrent fort bien les hypoglycémies et parfois même« trop bien », quand ils n’en perçoivent plus les symp-tômes d’alerte. Par contre, un petit nombre de sujets diabétiques (5 à 10 %) ont une peur panique de l’hypo-glycémie avec une sensation de malaise dès que la glycémieatteint 1,20 g/L voire plus, entraînant des resucragesintempestifs et une sous-insulinisation. Ces patients ontune hémoglobine glycosylée autour de 9 %, comportantun risque de complications dégénératives sévères.D’autres patient(e)s (5 à 10 %) associent à cette peurpanique de l’hypoglycémie une peur phobique de laprise de poids. Ils maintiennent un « poids idéal » grâce à une sous-insulinisation délibérée assurant uneglycosurie élevée dont témoigne l’hémoglobine glycosyléedépassant souvent 11 %. Les poussées hyperglycémiquesentraînées par les grignotages, les accès compulsifs etles crises de boulimie sont responsables de cures de diurèse osmotique entraînées par la glycosurie massiveévitant le recours aux laxatifs ou aux vomissements pro-voqués. L’existence de ces troubles du comportementalimentaire sévères associés au diabète comportent unrisque majeur d’évolution vers les complications sévèreset justifient une double prise en charge diabétologique etpsychologique.
éloignement des objets, points brillants devant les yeux,flou visuel…), de troubles de la parole, de troubles del’équilibre, de mouvements anormaux, de convulsions,de crises d’épilepsie, de troubles du comportement, d’unsyndrome confusionnel, de troubles de l’humeur (plussouvent de tristesse ou d’angoisse que de jovialité oud’euphorie), d’une confusion ou d’un coma.
Patients diabétiques à haut risque d’hypoglycémie sévère
L’hypoglycémie sévère (coma, convulsions, hypoglycé-mies nécessitant le recours à une tierce personne) survient principalement chez :– les patients surdosés en insuline avec des doses totales
supérieures à 1 U/kg/j ou des doses d’insuline basalessupérieures à 0,40 U/kg/j ;
– les sujets diabétiques qui ont déjà fait plusieurs hypo-glycémies sévères ;
– les enfants de moins de 7 ans ;– les personnes âgées de plus de 70 ans ;– les patients présentant une intoxication alcoolique ou
même une alcoolisation aiguë ;– les patients ayant une pancréatectomie ou une pan-
créatite chronique calcifiante ;– les patients ne percevant plus les symptômes d’alerte
neurovégétatifs de l’hypoglycémie ; – les patientes enceintes chez lesquelles on cherche une
normalisation de l’hémoglobine glycosylée avec unrisque accru d’hypoglycémie sévère lors des premiersmois de grossesse ;
– les patients ayant un déficit hormonal hypophysaireou surrénal associé au diabète insulinodépendant ;
– classiquement, les malades traités par bêtaboquants.En réalité, les bêtabloquants ne suppriment que lespalpitations et non les autres symptômes neurovégétatifsd’alerte. En revanche, en réduisant le débit cardiaquelors de l’hypoglycémie, ils diminuent de ce fait le débitsanguin glucosé cérébral et majorent la neuroglycopénie.
Obstacles au bon équilibre du diabèteinsulinodépendant
Environ la moitié des sujets diabétiques insulinodépendantsadultes obtiennent des valeurs d’hémoglobine glycosyléesouhaitées inférieures à 7,5 %. Cinquante pour cent parcontre n’atteignent pas cet objectif et ont un risque decomplications sévères de microangiopathie. Il convientd’analyser méthodiquement les obstacles rencontrés parles malades.Un schéma insulinique inadéquat, avec en particulierdes insulines retard trop courtes, ne couvre pas la find’après-midi ou la fin de nuit. Il peut alors être utile dedécaler les insulines retard par exemple en reportantl’insuline retard (neutral protamine hagedorn, NPH) dudîner au coucher et (ou) d’ajouter une injection d’insulineretard le midi. Les injections d’insuline doivent permettrede séparer les « insulines basales » et les « insulinesprandiales », avec une injection d’insuline rapide avant
Endocrinologie - Métabolisme - Nutrition
1477L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

Le trouble psychologique le plus fréquent à l’origine dumauvais équilibre du diabète est la « pseudo-acceptation »de la maladie ou plus exactement le refus conscientd’être diabétique que manifestent 15 à 20 % des sujetsdiabétiques. Ces patients présentent un clivage durableentre le « moi non diabétique » et le « moi diabétique »réduit au strict temps nécessaire pour réaliser lescontraintes minimales, le plus souvent 1 ou 2 injectionsavec refus de la 3e injection et des autocontrôles glycé-miques et évitement des hypoglycémies, moins en raisonde leur désagrément symptomatique qu’à cause du rappelinopportun du diabète qu’elles imposent. Ces patientscachent fréquemment leur diabète, parfois même à leurentourage proche, et affirment volontiers une force decaractère particulière, se traduisant notamment par uneactivité intense parfois même dangereuse sur le planmédical quand elle se fait sans règle de sécurité diabéto-logique. Il s’agit alors véritablement de conduites àrisque lors d’activités professionnelles ou sportives.Reste un groupe de patients diabétiques insulinodépendants(15 à 20 %) acceptant leur maladie mais ayant du mal àgérer quotidiennement les contraintes des traitementspour des raisons émotionnelles : manque de confianceen soi, tendance dépressive, dépendance vis-à-vis desautres, difficultés de programmation des activités de lajournée et de projection dans l’avenir, fatalisme… Cespatients peuvent être aidés par une prise en charge rapprochée avec des consultations tous les 15 jours outous les mois (voir :Pour approfondir 7).
Complications dégénérativesLes complications dégénératives du diabète permettentde distinguer la microangiopathie d’une part, la macro-angiopathie d’autre part.
Microangiopathie diabétique
L’hyperglycémie chronique est le seul facteur causal dela microangiopathie (rétinopathie, glomérulopathie,neuropathie diabétiques, qui sont fréquemment associées,formant la classique triopathie diabétique). Il existe uneforte corrélation entre d’une part la survenue de cescomplications et d’autre part la durée du diabète et leniveau d’hyperglycémie. Un point en plus ou en moinsd’hémoglobine glycosylée (ce qui correspond à unediminution ou une augmentation de la glycémie moyennede 0,30 g/L) entraîne une augmentation ou une diminutiondu risque d’apparition ou d’aggravation de la micro-angiopathie diabétique d’environ 30 %. L’hyperglycémie chronique est responsable d’unemicroangiopathie fonctionnelle caractérisée par uneaugmentation du débit, de la pression et de la perméabilitécapillaires, une perte de l’autorégulation hémodynamiqueentraînant un retentissement sur la microcirculation dela pression artérielle systémique et une tendance thrombo-gène secondaire, notamment une augmentation du facteurVon Willebrand, du fibrinogène et du PAI1 (plasmino-gen activator inhibitor1).
1. Rétinopathie diabétiqueLa rétinopathie diabétique se développe à bas bruit, sansque le malade perçoive pendant longtemps de symptôme.La baisse de l’acuité visuelle témoigne de lésions trèsavancées. Il est donc essentiel que tout patient diabétiquereçoive une éducation sur le dépistage des lésions réti-niennes par un examen systématique annuel du fondd’œil à la recherche de microanévrismes. L’angio-graphie rétinienne n’est utile que si l’examen soigneuxdu fond d’œil montre des anomalies évolutives. La rétinopathie diabétique se développe sur 2 modesévolutifs fréquemment associés : l’ischémie et l’œdème.Les hémorragies intrarétiniennes témoignent de l’ischémie,surtout lorsqu’elles sont nombreuses et étendues, les territoires non perfusés vus à l’angiographie, les nodulescotonneux témoignant d’une obstruction artériolaire, lesanomalies de calibre veineux (veines tortueuses etboucles veineuses), les néovaisseaux intrarétiniens (ano-malies microvasculaires intrarétiniennes), puis prérétiniens et notamment prépapillaires provoquant ledéveloppement d’une fibrose gliale tirant sur la rétine etfinissant par la décoller. La rétinopathie proliférantetouche environ 50 à 60 % des sujets diabétiques insulino-dépendants et 25 à 30 % des diabétiques non insulino-dépendants après 20 ans d’évolution du diabète. L’œdèmepeut être responsable d’exsudats durs, prédominant aupôle postérieur. La maculopathie œdémateuse est unedes causes de perte de l’acuité visuelle du sujet diabétique.Cet œdème maculaire peut s’associer à une ischémiemaculaire définie par un doublement de la surface de lazone avasculaire centrale. L’œdème maculaire affecteenviron 30 % des sujets diabétiques insulinodépendantsaprès 20 ans d’évolution du diabète. Sa prévalencedépend en fait de la sévérité de la rétinopathie diabétique,l’œdème maculaire étant beaucoup plus fréquent au coursdes rétinopathies proliférantes (voir :Pour approfondir 8). Le traitement de la rétinopathie diabétique suppose uneéquilibration glycémique aussi parfaite que possible,une baisse de la pression artérielle au-dessous de 130/80 mmHg. Le bénéfice des traitements par anti-plaquettaires est mineur. Le traitement par photocoagulationau rayon laser a 2 indications : une photocoagulationpanrétinienne est indiquée lorsqu’il existe une rétino-pathie proliférante débutante, la seconde indication de laphotocoagulation au laser est la maculopathie œdémateuse.
2. Neuropathie diabétiqueLa neuropathie diabétique est fréquente. Si l’on retientdes critères cliniques, on estime sa prévalence entre 25 et50 % chez les sujets diabétiques dont la maladie évoluedepuis plus de 20 ans. Elle est bien corrélée à l’équilibreglycémique, mais l’âge est un facteur essentiel de suscep-tibilité, la majorité des neuropathies diabétiques survenantaprès l’âge de 50 ans. On distingue les mononeuropathieset mononeuropathies multiples (10 à 15 % des neuro-pathies diabétiques) et les polyneuropathies diabétiquesbeaucoup plus fréquentes (80 à 85 %). • Les mononeuropathies et mononeuropathies multiplesse traduisent essentiellement par des signes moteursdéficitaires, des douleurs évocatrices par leur exacerbation
D I A B È T E I N S U L I N O D É P E N D A N T
1478 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

Les manifestations urogénitales :l’éjaculation rétrogradeest cause d’infertilité. L’impuissance atteindrait 30 %des sujets diabétiques. Multifactorielle, elle est rarementdue à une obstruction artérielle et ne semble pas plusfréquemment endocrinienne chez le sujet diabétique quechez le non diabétique. Elle peut être exclusivementd’origine psychique et est souvent aggravée par de nom-breux médicaments dont pratiquement tous les antihyper-tenseurs, les fibrates, les antihistaminiques H2, les médi-caments à visée neuropsychique. Elle a surtout 2 causes :d’une part, la fibrose pénienne en particulier des corpscaverneux, d’autre part la neuropathie autonome.L’atteinte vésicale est fréquente, mise en évidence parune altération de la débitmétrie urinaire. Les formes évi-dentes sont rares et correspondent à des stades évolués,compliqués d’infection ou de rétention. En effet, lerisque majeur est celui de la rétention d’urine, responsabled’une infection avec reflux vésico-urétéral menaçant lehaut appareil. L’échographie post-mictionnelle, examenanodin, permet d’en apprécier l’importance (de façonfiable au-dessus de 100 mL) et de vérifier l’absence deretentissement sur les voies urinaires. Une explorationurodynamique spécialisée s’impose pour décider de laconduite thérapeutique.Les manifestations digestives peuvent intéresser tout letube digestif, mais 2 types de manifestations sont plusfréquentes : d’une part, la gastroparésie avec achlorhy-drie gastrique qui peut être responsable d’hypoglycémiespost-prandialesd’horaire inhabituel (les formes sévèrespeuvent être améliorées par une prescription d’érythro-mycine, agoniste de la motiline), d’autre part, la diarrhée,comportant 10 à 30 selles par jour, impérieuses, survenantsouvent après les repas et parfois la nuit ou à l’occasiond’une hypoglycémie et s’accompagnant dans 50 % descas d’une incontinence anale, survenant par poussées dequelques jours à quelques semaines. Dans tous les cas, ilimporte d’éliminer une diarrhée provoquée par la prisede metformine (Glucophage ; Stagid ; Glucinan), unediarrhée avec stéatorrhée due à une pancréatite chronique,une diarrhée secondaire à une hyperthyroïdie associéeau diabète, ou encore une diarrhée due à une maladiecœliaque qu’évoquerait l’existence de troubles demalabsorption.La dysautonomie diabétique peut encore être responsablede troubles pupillaires avec exceptionnellement un signed’Argyll Robertson, de déficit endocrinien notammentd’un hyporéninisme hypo-aldostéronisme parfois res-ponsable d’une hyperkaliémie avec acidose tubulaire.Enfin, elle participe aux troubles trophiques, dominéspar les maux perforants plantaires et l’ostéo-arthropathiediabétique.
3. Néphropathie diabétique La glomérulopathie diabétique complique 25 à 30 % desdiabètes insulinodépendants. La principale manifestationde la glomérulopathie diabétique est l’augmentation del’albuminurie que l’on peut aujourd’hui dépister préco-cement grâce aux dosages immunologiques. L’élévationprogressive de la pression artérielle va de pair avec l’élé-vation de l’albuminurie. Pour confirmer le diagnostic, il
nocturne. Elles comprennent des cruralgies, remarquablespar l’absence de syndrome rachidien et l’atteinte volontierspluriradiculaire, des méralgies paresthésiques. Les nerfsdes membres supérieurs sont moins souvent touchés. L’atteinte du nerf oculomoteur est parmi les plus fréquentes.Un tiers des paralysies oculomotrices seraient d’originediabétique, le III et le VI sont plus souvent touchés quele IV. La paralysie est souvent précédée pendant quelquesjours de douleurs vives. L’atteinte du III épargne enrègle les fibres plus superficielles du III intrinsèque. Laréactivité pupillaire est donc normale. Une mydriaseassociée à une paralysie du III doit en effet faire recherchersystématiquement un anévrisme de la carotide interneou une tumeur, par tomodensitométrie ou imagerie parrésonance magnétique.Exceptionnellement, le thorax et l’abdomen peuventêtre touchés avec des mono- ou polyradiculopathies res-ponsables de douleurs thoraciques uni- ou bilatéralesparfois abdominales. L’amyotrophie diabétique proximale pseudomyopathiqueest rare. Elle intéresse les racines, en particulier lespsoas et quadriceps amyotrophiés et douloureux à lapalpation. Son évolution est le plus souvent favorable. • Les polyneuropathies diabétiquessont beaucoup plusfréquentes. Leur topographie est habituellement distale,bilatérale et symétrique, le plus souvent en chaussette,plus rarement en gant, exceptionnellement thoraco-abdominale. Les manifestations subjectives sont de 2 ordres : le plus souvent il s’agit de paresthésies et dedysesthésies mais parfois de douleurs volontiers exa-cerbées la nuit, intolérables avec sensations d’écrasementou de brûlures continues ou fulgurantes. Leur intensitépeut être telle qu’elle provoque une dépression réaction-nelle avec anorexie, amaigrissement sévère, permettantd’individualiser une forme cachectique pseudo-néopla-sique. L’examen neurologique trouve une abolition desréflexes achilléens et parfois rotuliens, une altération dela sensibilité profonde, des troubles de la sensibilitésuperficielle, tactile, thermique et douloureuse. L’anesthésie à la douleur joue un rôle essentiel dans lapathogénie des ulcérations trophiques des pieds. En per-dant la sensibilité à la douleur, le malade diabétique perden effet le moyen fondamental de la protection des pieds.L’électromyogramme de la neuropathie diabétique révèleun ralentissement des vitesses de conduction nerveuseainsi qu’une diminution de l’amplitude des potentielsd’action des nerfs sensitifs, puis moteurs. C’est un examenle plus souvent inutile pour le diagnostic et la surveillancede la neuropathie diabétique. Son indication relève doncdu spécialiste.• La neuropathie végétativecomporte plusieurs typesde manifestations.Les manifestations cardiovasculaires et sudorales :ladénervation cardiaque, mise en évidence par l’étude dela variation de la fréquence cardiaque lors de la respirationprofonde de l’épreuve de Valsalva et du passage de laposition couchée à la position debout, comporte un risquede mort subite. L’hypotension orthostatique témoigned’une dénervation sympathique périphérique intéressantles membres inférieurs et le territoire splanchnique.
Endocrinologie - Métabolisme - Nutrition
1479L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

suffit de vérifier l’absence d’autre maladie uro-néphrolo-gique (l’ECBU est normal, la taille des reins est normale,l’échographie ne révèle pas d’anomalie des voies urinaires)et de s’assurer de l’existence d’une rétinopathie diabétique,le plus souvent sévère. L’absence de rétinopathie diabétiquedoit en effet amener à réviser le diagnostic et en tout casà demander l’avis d’un néphrologue qui décide de l’op-portunité éventuelle d’une ponction-biopsie rénale.L’évolution se fait en 5 stades (voir :Pour approfondir 9).Au stade de « néphropathie incipiens », la clairance glo-mérulaire n’est pas normale mais augmentée, pouvantatteindre 150 à 200 mL/min. Elle commence à décroîtrelorsque la micro-albuminurie dépasse 70 mg/24 h et estencore normale lorsque la néphropathie patente apparaît(albuminurie supérieure à 300 mg/24 h). Le traitement de la néphropathie diabétique repose sur :– une équilibration du diabète avec une hémoglobine
glycosylée inférieure à 8%, connaissant le risque d’hypo-glycémie sévère chez ces patients ;
– un traitement antihypertenseur visant à obtenir unepression artérielle inférieure à 130/80 mmHg ;
– même en l’absence d’hypertension artérielle, uneprescription d’inhibiteur de l’enzyme de conversionde l’angiotensine censé diminuer la pression hydrosta-tique intraglomérulaire ;
– un régime hypoprotidique modéré, autour de 0,8 g/kg/j;– la correction d’une hyperlipidémie ;– l’arrêt d’une intoxication tabagique.Le traitement des facteurs de risque cardiovasculairesest essentiel. En effet, la néphropathie diabétique est responsable d’une angiopathie maligne associant micro-angiopathie sévère, athérosclérose accélérée, responsabled’accidents vasculaires cérébraux, d’infarctus du myocardeet surtout d’artérite des membres inférieurs. La recherched’une ischémie myocardique silencieuse et une explorationpar écho-doppler des troncs supra-aortiques et des artèresdes membres inférieurs sont indispensables pour un traitement suffisamment précoce des lésions d’athérome. L’infection urinaire n’est pas plus fréquente chez lessujets diabétiques bien équilibrés que chez les non dia-bétiques. En revanche, l’infection urinaire est plus sévèrechez les diabétiques, justifiant un dépistage systématiqueà l’aide des bandelettes dépistant leucocyturie et nitrites.En cas de positivité, il convient de demander un examencytobactériologique urinaire avec antibiogramme etd’entreprendre un traitement prolongé pendant 8 jours.Les traitements des infections urinaires et des uropathiesobstructives ont rendu la nécrose papillaire rare aujourd’hui. Quant à la néphropathie due aux produits de contrasteiodés, elle n’est pas à proprement parler favorisée par lediabète, mais par l’insuffisance rénale. Elle doit être sys-tématiquement prévenue par des protocoles d’hydratation(apport per os d’eau de Vichy ou perfusion de sérumphysiologique) des sujets diabétiques avant l’injectionde produits de contraste iodés. Elle impose un contrôlede la diurèse et une mesure de la créatininémie 48 haprès l’examen. La prise de metformine (Glucophage,Stagid, Glucinan) doit être impérativement suspenduedans les 48 h qui précèdent et qui suivent l’examen.
Macroangiopathie diabétique
L’athérosclérose est devenue la principale cause dedécès des patients diabétiques. Le diabète entraîne unrisque relatif d’athérosclérose de 1,5 à 2 pour les accidentsvasculaires cérébraux, de 2 à 4 pour l’insuffisance coro-naire, de 5 à 10 pour l’artériopathie des membres inférieurs. Il comporte un risque relatif plus élevé pourla femme diabétique qui perd en partie son avantage surl’homme avant l’âge de la ménopause. Cependant, lacorrélation entre l’hyperglycémie et la morbi-mortalitécardiovasculaire est moins importante que celle entrel’hyperglycémie et la microangiopathie diabétique. Pourun point en plus d’hémoglobine glycosylée, le risqued’événement cardiovasculaire n’augmente dans les 10 ansque de 10%. Le risque absolu dépend donc essentiellementde l’association aux autres facteurs de risque : hypertensionartérielle, augmentation du LDL cholestérol (low densi-ty lipoprotein), modifications qualitatives du LDL cho-lestérol avec augmentation des LDL petites et denses,diminution du HDL cholestérol (high density lipopro-tein), tabagisme… L’importance du risque vasculaireabsolu chez le sujet diabétique insulinodépendantconduit donc à proposer des seuils d’intervention théra-peutique plus stricts à partir de 140/80 mmHg pour lapression artérielle, de 1,30 g/L pour le LDL cholestérol.Les complications de l’athérosclérose ont une mortalitéglobalement double de celle du sujet non diabétique.Les accidents vasculaires cérébraux sont plus rarementhémorragiques chez le diabétique en dépit de l’augmen-tation de la fréquence de l’hypertension artérielle. Enrevanche, les micro-infarctus responsables de lacunessemblent plus fréquents.L’ischémie myocardique est 2 à 3 fois plus souventindolore chez le sujet diabétique. L’infarctus du myocardeest ainsi très souvent indolore, bien que rarementasymptomatique. Il faut y penser systématiquementdevant la survenue soudaine de symptômes par ailleursinexpliqués : troubles digestifs et parfois douleurs épi-gastriques, dyspnée d’effort, asthénie en particulier àl’effort, troubles du rythme cardiaque, embolie et parfoissimple déséquilibre inexpliqué du diabète ou baisse dela pression artérielle. La mortalité après infarctus dumyocarde est double à 1 mois, 1 an et 5 ans par rapportau sujet non diabétique. Cette surmortalité tient essen-tiellement à la fréquence de l’insuffisance cardiaqueséquellaire, en particulier chez la femme diabétique obèse.L’artériopathie des membres inférieurs se révèle parfoispar une claudication intermittente avec sa douleurconstrictive en étau, imposant l’arrêt de la marche. Enréalité, 5 fois sur 6, cette douleur fait défaut en raison de la coexistence d’une neuropathie diabétique. Outrel’association fréquente à une neuropathie responsabledu caractère indolore de l’ischémie, l’artériopathie desmembres inférieurs du malade diabétique est caractériséepar sa topographie : dans un tiers des cas elle est proximale,bien corrélée aux facteurs de risque classiques (hyper-tension artérielle, hyperlipidémie, tabagisme), dans untiers des cas elle est distale (corrélée à l’hyperglycémie),
D I A B È T E I N S U L I N O D É P E N D A N T
1480 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

La lésion la plus banale est le mal perforant plantaire,secondaire à une callosité ou à un durillon sous lespoints d’appui (tête des métatarsiens, talon, styloïde du5e méta), l’hyperkératose indolore en raison de la neuro-pathie forme l’équivalent d’une pierre dure, blessant letissu sous-cutané, provoquant la formation d’une petitepoche séro-hématique. Le liquide sous tension lors del’appui dissèque les tissus, formant une véritable chambrede décollement. L’hyperkératose sèche se fendille,ouvrant la voie à la surinfection avec constitution d’unvéritable abcès sous-cutané, qui peut fuser vers l’os oules parties molles ou s’ouvrir à la peau, formant le classique mal perforant entouré de sa couronne d’hyper-kératose. Le mal perforant n’est donc ni plus ni moinsqu’un « durillon qui a mal tourné » parce qu’il était indolore. Sa prévention repose donc sur l’abrasionmécanique de la kératose et le graissage quotidien despieds pour éviter sa récidive.Le pied de Charcot correspond à une ostéodystrophienerveuse avec effondrement du sommet de l’arche internedu pied, secondaire à une ostéonécrose du scaphoïde et(ou) du premier cunéiforme et (ou) de la base du premier méta, provoquant une dislocation secondaire de l’articulation de Lisfranc. Il constitue donc un piedplat avec élargissement du coup de pied et saillie dubord interne du pied. Il s’accompagne d’une dénervationsympathique avec des shunts artérioveineux, des poulsbondissants, un pied chaud et sec, initialement pseudo-inflammatoire. ■
siégeant en dessous du genou et dans un tiers des caselle est globale, proximale et distale. Par chance, mêmelorsque l’artérite est distale sous-poplitée, l’artèrepédieuse reste le plus souvent perméable. La palpationd’un pouls pédieux n’élimine donc en rien l’existenced’une artérite sévère des axes jambiers et sus-jacents,mais il est sûrement l’un des meilleurs arguments pronostiques de l’artérite diabétique. En effet, cette persistance permet de réaliser des pontages distaux (utilisant la veine saphène interne dévalvulée in situ ouinversée) dans le cadre d’un sauvetage de membrenécessité par une gangrène du pied.L’artériopathie des membres inférieurs se révèle en effettrop souvent par un trouble trophique avec début de gan-grène secondaire à un traumatisme minime (frottementdans la chaussure, ongle mal taillé blessant l’orteil voisin,ongle incarné, mycose interdigitale, absence de protectiondes talons lors de l’alitement prolongé…). La survenued’un tel trouble trophique avec nécrose ischémiqueimpose toujours l’hospitalisation du patient pour explo-rations vasculaires (écho-doppler, mesure de la pressiontranscutanée en oxygène, artériographie) qui permettentune décision thérapeutique de sauvetage.En effet, la gangrène, même limitée, n’est jamais secondaireà une microangiopathie diabétique. Elle témoigne toujoursd’une atteinte des artères musculaires, même s’il s’agitd’artères de petit calibre et elle doit donc bénéficier, àchaque fois que cela est possible, d’une revascularisation.Un geste d’amputation a minima fait sans explorationvasculaire risque de ne jamais cicatriser et d’entraînerune aggravation secondaire de l’ischémie avec amputationmajeure.
Complications du pied diabétique
On comptabilise environ 5 000 amputations par an duesau diabète, dont 5 à 10 % surviennent chez les sujets diabétiques de type 1. On estime que 50 % de ces ampu-tations pourraient être évitées. Il est donc indispensablede reconnaître les diabétiques à risque podologique,c’est-à-dire les diabétiques ayant perdu la sensibilité à ladouleur au niveau des pieds ou les diabétiques ayant uneartériopathie des membres inférieurs. Tout diabétique doit donc bénéficier d’un dépistageannuel du risque podologique. Si un risque est mis enévidence (existence d’une neuropathie avec diminutionde la sensibilité à la douleur et [ou] d’une artérite desmembres inférieurs) l’examen des pieds et des chaussuresdoit être fait à chaque consultation. Le sujet diabétique à risque podologique doit bénéficier d’une éducationspécialisée pour la protection des pieds vis-à-vis desagents agressifs (chaussures, troubles statiques, hyper-kératose, troubles unguéaux, mycose interdigitale, cori-cides, objets divers blessants…).Lorsqu’un risque podologique est dépisté, il faut s’assurerque le malade peut voir et toucher ses pieds et qu’il peutassurer lui-même les soins de prévention. Si ce n’est pasle cas, il faut s’enquérir de la tierce personne, en particulierdans l’entourage, susceptible de le faire.
Endocrinologie - Métabolisme - Nutrition
1481L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• La prise en charge du diabète de type 1 a connu des progrès spectaculaires grâce à l’autocontrôle glycémique, à l’améliorationdes techniques d’injection d’insuline et à l’éducation thérapeutique des patients leur permettant de réaliser une insulinothéra-pie plus physiologique, adaptée à leur mode devie et à leur alimentation, qui ne diffère plusguère de celle conseillée pour les non-diabé-tiques.
• La surveillance ophtalmologique annuelle systématique et si nécessaire le traitement par photocoagulation au laser doivent permettre de faire disparaître la cécité des complications du diabète de type 1. L’effortprincipal porte actuellement sur la préventionde la néphropathie diabétique patente,complication la plus redoutable de la maladie.
• L’avenir appartient à la prévention du diabètede type 1 par l’immunothérapie de l’insuliteprédiabétique et à la guérison du diabète de type 1 par la mise au point de pancréas
Points Forts à retenir
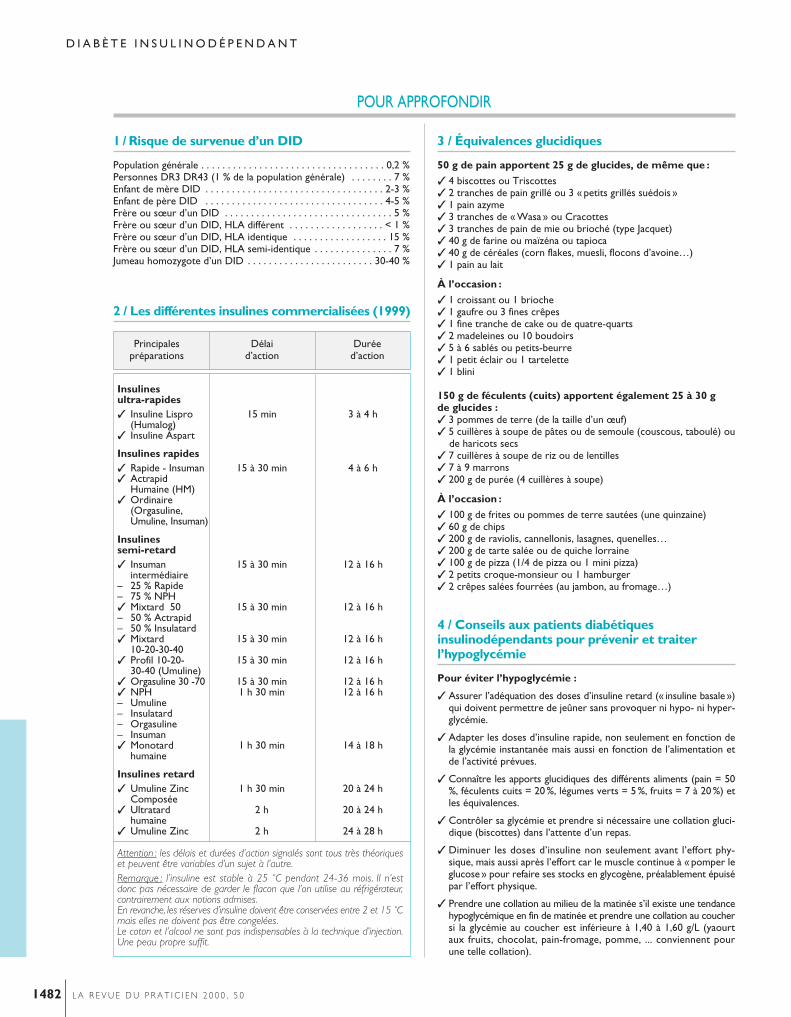
D I A B È T E I N S U L I N O D É P E N D A N T
1482 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
1 / Risque de survenue d’un DID
Population générale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 0,2 %Personnes DR3 DR43 (1 % de la population générale) . . . . . . . . 7 %Enfant de mère DID . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2-3 %Enfant de père DID . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4-5 %Frère ou sœur d’un DID . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 %Frère ou sœur d’un DID, HLA différent . . . . . . . . . . . . . . . . . . < 1 %Frère ou sœur d’un DID, HLA identique . . . . . . . . . . . . . . . . . . 15 %Frère ou sœur d’un DID, HLA semi-identique . . . . . . . . . . . . . . . 7 %Jumeau homozygote d’un DID . . . . . . . . . . . . . . . . . . . . . . . . 30-40 %
2 / Les différentes insulines commercialisées (1999)
3 / Équivalences glucidiques
50 g de pain apportent 25 g de glucides, de même que :
✓ 4 biscottes ou Triscottes✓ 2 tranches de pain grillé ou 3 « petits grillés suédois »✓ 1 pain azyme✓ 3 tranches de « Wasa » ou Cracottes✓ 3 tranches de pain de mie ou brioché (type Jacquet)✓ 40 g de farine ou maïzéna ou tapioca✓ 40 g de céréales (corn flakes, muesli, flocons d’avoine…)✓ 1 pain au lait
À l’occasion :
✓ 1 croissant ou 1 brioche✓ 1 gaufre ou 3 fines crêpes✓ 1 fine tranche de cake ou de quatre-quarts✓ 2 madeleines ou 10 boudoirs✓ 5 à 6 sablés ou petits-beurre✓ 1 petit éclair ou 1 tartelette✓ 1 blini
150 g de féculents (cuits) apportent également 25 à 30 g de glucides :✓ 3 pommes de terre (de la taille d’un œuf)✓ 5 cuillères à soupe de pâtes ou de semoule (couscous, taboulé) ou
de haricots secs✓ 7 cuillères à soupe de riz ou de lentilles✓ 7 à 9 marrons✓ 200 g de purée (4 cuillères à soupe)
À l’occasion :
✓ 100 g de frites ou pommes de terre sautées (une quinzaine)✓ 60 g de chips✓ 200 g de raviolis, cannellonis, lasagnes, quenelles…✓ 200 g de tarte salée ou de quiche lorraine✓ 100 g de pizza (1/4 de pizza ou 1 mini pizza)✓ 2 petits croque-monsieur ou 1 hamburger✓ 2 crêpes salées fourrées (au jambon, au fromage…)
4 / Conseils aux patients diabétiques insulinodépendants pour prévenir et traiter l’hypoglycémie
Pour éviter l’hypoglycémie :
✓ Assurer l’adéquation des doses d’insuline retard (« insuline basale »)qui doivent permettre de jeûner sans provoquer ni hypo- ni hyper-glycémie.
✓ Adapter les doses d’insuline rapide, non seulement en fonction dela glycémie instantanée mais aussi en fonction de l’alimentation etde l’activité prévues.
✓ Connaître les apports glucidiques des différents aliments (pain = 50%, féculents cuits = 20 %, légumes verts = 5 %, fruits = 7 à 20 %) etles équivalences.
✓ Contrôler sa glycémie et prendre si nécessaire une collation gluci-dique (biscottes) dans l’attente d’un repas.
✓ Diminuer les doses d’insuline non seulement avant l’effort phy-sique, mais aussi après l’effort car le muscle continue à « pomper le glucose » pour refaire ses stocks en glycogène, préalablement épuisépar l’effort physique.
✓ Prendre une collation au milieu de la matinée s’il existe une tendancehypoglycémique en fin de matinée et prendre une collation au couchersi la glycémie au coucher est inférieure à 1,40 à 1,60 g/L (yaourtaux fruits, chocolat, pain-fromage, pomme, ... conviennent pourune telle collation).
POUR APPROFONDIR
Principales préparations
Délai d’action
Durée d’action
Insulines ultra-rapides✓ Insuline Lispro 15 min 3 à 4 h
(Humalog) ✓ Insuline Aspart
Insulines rapides✓ Rapide - Insuman 15 à 30 min 4 à 6 h✓ Actrapid
Humaine (HM)✓ Ordinaire
(Orgasuline, Umuline, Insuman)
Insulines semi-retard✓ Insuman 15 à 30 min 12 à 16 h
intermédiaire– 25 % Rapide– 75 % NPH✓ Mixtard 50 15 à 30 min 12 à 16 h– 50 % Actrapid– 50 % Insulatard✓ Mixtard 15 à 30 min 12 à 16 h
10-20-30-40✓ Profil 10-20- 15 à 30 min 12 à 16 h
30-40 (Umuline)✓ Orgasuline 30 -70 15 à 30 min 12 à 16 h✓ NPH 1 h 30 min 12 à 16 h– Umuline– Insulatard– Orgasuline– Insuman✓ Monotard 1 h 30 min 14 à 18 h
humaine
Insulines retard✓ Umuline Zinc 1 h 30 min 20 à 24 h
Composée✓ Ultratard 2 h 20 à 24 h
humaine✓ Umuline Zinc 2 h 24 à 28 h
Attention : les délais et durées d’action signalés sont tous très théoriqueset peuvent être variables d’un sujet à l’autre.Remarque : l’insuline est stable à 25 ˚C pendant 24-36 mois. Il n’est donc pas nécessaire de garder le flacon que l’on utilise au réfrigérateur,contrairement aux notions admises.En revanche, les réserves d’insuline doivent être conservées entre 2 et 15 ˚Cmais elles ne doivent pas être congelées.Le coton et l’alcool ne sont pas indispensables à la technique d’injection.Une peau propre suffit.

Endocrinologie - Métabolisme - Nutrition
1483L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
✓ Il est parfois nécessaire de recommander de faire l’injection d’insulinerapide du midi après le repas et non avant, s’il existe un risque d’hypo-glycémie au cours du trajet ou de la file d’attente à la cantine ou au self.
✓ Avoir toujours sur soi 3 sucres dans la poche, à prendre immédia-tement en cas de malaise (l’application de cette recommandationdoit être vérifiée à chaque consultation).
✓ Contrôler sa glycémie au bout du doigt avant de conduire un véhicu-le et avoir à portée de main 3 sucres.
✓ En cas de malaise hypoglycémique, prendre immédiatement 3 mor-ceaux de sucre ou un jus de fruit ou 3 cuillerées à café de confiture ;on conseillera au diabétique de ne mesurer sa glycémie au bout dudoigt qu’après ce premier resucrage ; si la glycémie est inférieure à0,40 g/L, il prendra une deuxième dose de resucrage identique à lapremière, puis il recontrôlera sa glycémie au bout du doigt 10 à 15 minaprès le second resucrage.
✓ En effet, le sentiment de malaise persiste longtemps après que laglycémie est redevenue normale ou élevée (ce sentiment de malaisehypoglycémique persistant conduit souvent le malade à un excèsde resucrage).
✓ Analyser, après correction du malaise, les causes de l’hypoglycémieet éventuellement adapter les doses d’insuline correspondantespour éviter la récidive de l’hypoglycémie le lendemain à la mêmeheure (par contre, le plus souvent, il ne faut pas diminuer les dosesd’insuline dans les heures qui suivent le malaise hypoglycémiquecar le resucrage et la contre-régulation hormonale sont souvent res-ponsables d’un rebond hyperglycémique et d’une insulinorésistance prolongée).
✓ Avoir chez soi, sur son lieu de travail, sur son lieu de résidencesecondaire ou de loisirs, du Glucagon gardé au frais au bas du réfri-gérateur, non périmé (en ayant eu soin d’informer et de formerune tierce personne à l’injection de Glucagon à faire immédiate-ment en cas de coma ou d’impossibilité de resucrage per os ; lerenouvellement du Glucagon périmé doit être l’occasion pour letiers en question de manipuler « à blanc » seringue, Glucagon etsolvant).
5 / Prévention des hypoglycémies
Pour prévenir les hypoglycémies, des collations, entre les repas, peu-vent parfois être nécessaires, notamment à 22 h, si la glycémie est infé-rieure ou égale à 1,60 g/L.Les exemples de collations d’assimilation « lente » (peu hyper-glycémiante) ci-dessous apportent 15 à 20 g de glucides (c’est-à-direl’équivalent de 3 à 4 morceaux de sucre).
« Sur place »
✓ 2 biscottes beurrées✓ 30 g de pain avec du fromage✓ 1 mousse au chocolat (100 g)✓ 1 crème caramel (type Flanby)✓ 1 part de riz ou de semoule au lait (100 g)✓ 20 à 25 g de céréales dans du lait ou du yaourt✓ 1 esquimau✓ 2 boules de glace✓ 1/6 de pizza (60 g)
À « emporter »
✓ 1 fruit moyen✓ 2 à 3 fruits secs (dattes, pruneaux…)✓ 1 madeleine✓ 3 à 4 biscuits secs (sablés, petits-beurre…)✓ 4 à 6 carrés de chocolat✓ 1 barre chocolatée (25 à 30 g)✓ 2 bâtonnets de Craquinettes
✓ 1 briquette de lait chocolaté✓ 5 marrons✓ 1barre de céréales (type Jump ou Grany)✓ 1 petit croissant✓ 1 brioche ou 1 pain au lait✓ 1 crêpe
6 / Traitement des hypoglycémies
En cas d’hypoglycémie, prendre immédiatement un sucre « rapide » (très hyperglycémiant) :✓ 3 carrés de sucre (n˚ 4)✓ 2 c. à soupe de miel ou de confiture✓ 1 verre de soda ou de jus de fruit (15 cL)✓ 3 à 4 tablettes de dextrose (glucose) : Vitagermine, Nergi-sport✓ 1 pâte de fruit (30 g)
7 / Comportements liés aux phases d’acceptationd’une maladie
Phase du travail de deuil de l’état de santé antérieur que doit effectuer toutnouveau diabétique insulinodépendant, d’après Anne Lacroix, psychologue,division d’enseignement pour les maladies chroniques, Pr. J.-P. Assal, Hôpitalcantonal universitaire de Genève.
POUR APPROFONDIR (SUITE)
Patient ExempleComportement
habitueldu praticien
Attitude adéquate
ChocSurpris à angoissé Je ne réalise Donne le maximum Soutenir
pas très bien d’instructions Aider le patientà se retrouver
DénégationDétaché Il y a des maladies Persuasif Instaurer un climatBanalise plus graves de confiance
Chercher en quoile patient se sentmenacé
RévolteAgressif C’est la faute Se sent attaqué Chercher l’objetRevendicatif de si… Juge le patient de sa révolte
caractérielMarchandagePlus collaborant Je n’accepterai pas Irrité Négocier sur desManipulateur une 2e injection Remis en question points secondairesDépressionTriste Je réalise que je Peu attentif Renforcer l’écouteMéditatif me suis servi de active, susciter
mon diabète pour… un projet d’avenirAcceptationTranquille Je vis avec Gratifié RenforcerCollaborant et non pas malgré la formation
mon diabète personnalisée du patient
RésignationPassif Je m’en remets Dévoué Eviter Docile à vous Docteur la chronicisation
iatrogéniquePseudo-acceptationRefuse Je refuse d’être Impuissant Tenter consciemment un handicapé Menaçant de rejoindrede se sentir le niveau malade émotionnel

D I A B È T E I N S U L I N O D É P E N D A N T
1484 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
8 / Classification simplifiée de la rétinopathie diabétique
a Pas de rétinopathie
b Rétinopathie diabétique non proliférante : minime (microanévrismes sansdiffusion de fluorescéine et/ou dilatation veineuse) ; modérée ; sévè-re
c Rétinopathie préproliférante
d Rétinopathie proliférante : débutante, modérée, sévère
e Maculopathie : ischémique, œdémateuse focale, œdémateuse diffu-se cystoïde, œdémateuse diffuse non cystoïde
9 / Les 5 stades de la néphropathie diabétique
Stade I : néphropathie fonctionnelle
✓ Augmentation de la taille des reins et du volume glomérulaire
✓ Augmentation de la filtration glomérulaire de 20 à 40 %
✓ Tension artérielle normale
✓ Albuminurie normale
Stade II : lésions rénales histologiques sans traduction clinique
Stade III : néphropathie incipiens
✓ Augmentation de la filtration glomérulaire
✓ Augmentation de l’albuminurie entre 20 et 200 µg/min ou entre 30 et300 mg/24 h (croissance annuelle de 20 à 50 %)
✓ Augmentation annuelle de la pression artérielle de 3 à 4 mmHg(micro HTA)
Stade IV : néphropathie clinique
✓ Dépôts mésangiaux nodulaires ou diffus
✓ Hyalinose artériolaire (touchant les artères glomérulaires afférenteet efférente)
✓ Diminution de la filtration glomérulaire
✓ Protéinurie croissante
✓ Hypertension artérielle (> 140/90 mmHg)
Stade V : insuffisance rénale terminale
✓ Obstructions glomérulaires
✓ Filtration glomérulaire < 10 mL/min
✓ Hypertension artérielle volodépendante
POUR APPROFONDIR (SUITE)
Grimaldi A, Cornet P, Masseboeuf F, Popelier M, Sachon C. Guidepratique du diabète. Collection Médiguides. Le Généraliste. GazMed (Paris), 1997.
Lacroix A, Assal JP. L’éducation thérapeutique des patients.Nouvelles approches de la maladie chronique. Collection « Éduca-tion du patient » dirigée par Jean-François d’Ivernois. Paris : Vigot,1998.
POUR EN SAVOIR PLUS

Endocrinologie-Métabolisme-NutritionB 331
629L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
Circonstances de découverteLe diabète non insulinodépendant peut être découvertdans différentes circonstances sachant que, dans cettepathologie, l’hyperglycémie est susceptible d’évoluersilencieusement pendant des années. Ces circonstances dediagnostic sont un bilan systématique, un syndromepolyuro-polydipsique, l’existence d’une complication,alors que le diabète était méconnu, soit une compli-cation dégénérative de type rétinopathie, neuropathie ounéphropathie ou une complication de macro-angiopathie.Enfin, la dernière circonstance de découverte est dans lecadre du dépistage des sujets à risque. En effet, plusieursfacteurs prédictifs de diabète non insulinodépendantsont identifiés : des antécédents familiaux de diabètenon insulinodépendant, une obésité androïde, pour lesfemmes, des enfants de poids de naissance supérieur à 4 kg , et le fait d’avoir déjà eu une glycémie supérieureou égale à 2 g/L à un moment donné de son existence.
ÉpidémiologieEn France, la prévalence du diabète non insulinodépen-dant dans la population générale est estimée à 2 % envi-ron. Cette approximation s’explique par le faible nombrede données épidémiologiques fiables et l’absence d’unepolitique de dépistage systématique. Aux États-Unis, laprévalence du diabète non insulinodépendant est estimée àenviron 6 % dans la population âgée de plus de 40 ans et àplus de 20 % dans celle de plus de 60 ans. Les diabétiquesont un risque coronaire 3 à 4 fois plus important que celuide la population générale. Aux États-Unis comme enSuède, le diabète est responsable de plus de 50 % desamputations non traumatiques, de 15 % des cécités et de16 à 35 % des insuffisances rénales au stade de la dialyse(70 à 90 % des diabétiques en dialyse étant non insulino-dépendants). La prévalence du diabète augmente avecl’âge. Elle était par exemple de 2,4 % de 20 à 44 ans, de8,5 % de 45 à 54 ans, de 12,8 % de 55 à 64 ans, et de 17,7 %au-delà dans l’étude d’une population américaine. On a pucalculer ainsi que, au-delà de 60 ans, le risque de dévelop-per un diabète est égal à environ 30 fois celui des sujetsâgés de 20 à 30 ans. Ainsi, l’âge est un facteur de risque de
Critères diagnostiques du diabète non insulinodépendant ou diabète de type 2Selon les critères de l’Organisation mondiale de la santé,le diabète se définissait par une glycémie supérieure à 7,8 mmol/L (1,4 g/L) de plasma veineux à jeun à 2reprises, et à 11,1 mmol/L (2 g/L) 2 heures après inges-tion orale de 75 grammes de glucose. Ces critères vien-nent d’être modifiés, et les seuils de définition du diabèteont été abaissés et ramenés à une glycémie à jeun supé-rieure ou égale à 7 mmol/L (1,26 g/L) à 2 reprises et (ou)une glycémie 2 heures après charge orale de 75 g de glu-cose supérieure ou égale à 11,1 mmol/L (2g/L). La glycémie à jeun normale est inférieure à 6,1 mmol/L(1,10 g/L).Les personnes ayant une glycémie à jeun entre 1,10 et 1,25 g/L présentent une hyperglycémie modérée à jeun,qui apparaît comme l’équivalent de l’intolérance au glucose.L’homologation de ces nouveaux critères risque de pro-voquer l’abandon de l’hyperglycémie provoquée parvoie orale. Il est en effet préconisé d’utiliser la glycémieà jeun comme critère diagnostique de première intention,dans un souci de standardisation et de simplification.
Diabète non insulinodépendantÉpidémiologie, étiologie, physiopathologie, diagnostic, complications, traitement
PR Denis RACCAH, DR Blandine JANAND-DELENNE, PR Philippe VAGUE
Service de nutrition-maladies métaboliques-endocrinologie, hôpital de la Timone, 13385 Marseille cedex 05.
• Maladie très hétérogène qui débuterait par une insulinorésistance qui pourrait être génétique, acquise ou les deux à la fois.
• Les complications sont fréquentes à l’origined’une morbidité et mortalité non négligeable.
• Dans la forme commune, il existe une obésitéassociée. Le premier principe du traitement est d’améliorer l’insulinorésistance.La prise en charge diététique est à la base de la thérapeutique. Dans un second temps,le recours aux médicaments agissant sur l’insulinorésistance voire surl’insulinosécrétion serait utile.
Points Forts à comprendre

diabète non insulinodépendant dont il faut tenir comptedans les enquêtes de prévalence. La prévalence du diabète non insulinodépendant ajustée pour l’âge est plusélevée aux États-Unis dans la population noire, et plusencore dans les minorités d’origine hispanique, que dansla population blanche. Elle est maximale dans une ethnieparticulière : les indiens Pima. Il est intéressant de noterque d’une façon globale, la prévalence du diabète chez lesmigrants tend à rejoindre celle de la population d’accueil,ce qui suggère l’importance de l’environnement. En fait,les variations de prévalence du diabète non insulinodé-pendant observées dans les populations migrantes et dansles pays ayant récemment modifié leur mode de vie,tiennent en grande partie aux modifications de l’alimen-tation, à l’augmentation de la prévalence de l’obésité et à l’acquisition d’un mode de vie sédentaire.La composante génétique reste également un point trèsimportant. Le cas particulier des diabètes de type MODY(Maturity Onset Diabetes of the Young), observés chez dessujets jeunes dont l’étude des antécédents familiauxindique une transmission autosomique dominante, montrebien l’implication de facteurs génétiques dans cette caté-gorie de diabète, certes peu fréquente. Dans les formesplus communes de diabète non insulinodépendant, l’exis-tence d’un terrain génétique de susceptibilité est suggéréepar la forte agrégation des cas familiaux. Le risque de dia-bète est d’environ 40 % chez des apparentés au premierdegré de sujets diabétiques. La fréquence d’antécédentsfamiliaux de diabète non insulinodépendant chez les sujetsatteints est néanmoins influencée par l’âge de survenue dudiabète. En effet, dans plusieurs études, l’existence d’anté-cédents familiaux de diabète est environ 2 fois plus fréquente, lorsque la maladie est observée avant 45 ou 50 ans, que lorsqu’elle est observée à un âge plus avancé.
Physiopathologie
1. Formes monogéniques
Les études génétiques du diabète non insulinodépendantont montré qu’il existait de nombreuses familles où lediabète se transmettait de manière mendélienne. Dans cesformes monogéniques, environ 5 à 15 % des diabètes, unemutation d’un seul gène est suffisante pour entraîner unehyperglycémie. Les facteurs environnementaux intervien-nent alors pour en moduler l’expression clinique.L’interrogatoire familial, allié aux techniques de biologiemoléculaire, permet aujourd’hui un diagnostic génétiqued’une proportion importante de ces cas familiaux. Lesdiabètes monogéniques ont un début souvent précoceavec une forte pénétrance. Cela a permis d’étudier degrands arbres généalogiques, comportant de nombreuxmembres atteints, et d’identifier certains gènes respon-sables des troubles de l’homéostasie glycémique. Le diabète de type MODY est la forme monogénique dediabète non insulinodépendant la plus fréquente et saprévalence est d’environ 5 % des diabètes non insulino-dépendants. Il s’agit d’un diabète à début précoce (avantl’âge de 25 ans) présentant une transmission autosomique
dominante à forte pénétrance (près de 90 %). Le phénotypeMODY est caractérisé par une hyperglycémie chronique,d’origine non auto-immune, qui s’aggrave avec le temps etentraîne l’apparition de complications dégénératives.Biologiquement, les patients MODY ont en général desconcentrations normales ou basses d’insuline, malgré leurhyperglycémie chronique, témoignant d’une anomalie pri-mitive de l’insulinosécrétion. Cette observation a conduit àcentrer l’étude génétique des diabètes non insulino-dépendants à début précoce, sur l’étude des gènes candidatsdont les produits jouent un rôle dans la régulation de l’insulinosécrétion en réponse au glucose. Ainsi, uneliaison génétique a été démontrée entre le MODY 2 et lelocus de la glucokinase sur le bras court du chromosome 7. Une quarantaine de mutations disséminées sur la majeurepartie des régions codantes de ce gène, ont été identifiéeschez des patients MODY. On peut actuellement estimer à2 % la proportion de patients caucasiens atteints de diabète non insulinodépendant, qui présentent des muta-tions du gène de la glucokinase, soit environ 30 000 personnes en France. La glucokinase est une enzyme clédu contrôle du métabolisme intracellulaire du glucosedans les cellules insulinosécrétoires du pancréas endocrineet dans les hépatocytes. La phosphorylation du glucose englucose-6-phosphate, catalysée par la glucokinase dansces tissus, est en effet la première étape du métabolismeintracellulaire du glucose. Dans les cellules β du pan-créas, le métabolisme du glucose et l’insulinosécrétionsont fortement dépendants de l’activité de l’enzyme et ladiminution de l’activité enzymatique de la glucokinaseest associée à une diminution du flux glycolytique pourun niveau glycémique donné. Les mutations du gène de laglucokinase ne sont pas les seules trouvées dans leMODY. En effet, le locus MODY 1, localisé en 1992 surle chromosome 20q, a été récemment identifié commecelui du gène codant HNF-4α (hepatocyte nuclear factor-4α), un facteur de transcription appartenant à lasuper-famille des récepteurs aux stéroïdes. La nature duligand endogène d’HNF-4α, s’il existe, ainsi que lesmécanismes génétiques et physiopathologiques de lamaladie restent méconnus. Le troisième gène du MODY,localisé en 1995 sur le chromosome 12 q, vient d’êtreégalement identifié. Il s’agit du gène codant HNF-1α, unfacteur de transcription homéotique exprimé dans le foie,dans le pancréas endocrine et dans d’autres organes. Celocus MODY 3 est responsable d’à peu près 25 à 30 %des cas de diabètes non insulinodépendants à débuts pré-coces. À la différence de la glucokinase, HNF-1α est responsable d’un diabète sévère de début post-pubertaire,rapidement évolutif et associé à des complications micro-angiopathiques précoces, rétiniennes et rénales. Environ30 % des sujets atteints évoluent vers un diabète insulino-requérant.Enfin, la prédominance de la transmission du diabète noninsulinodépendant par les mères diabétiques, et la présencede familles de diabétiques à hérédité purement maternel-le, ont conduit plusieurs équipes à étudier le rôle du gènemitochondrial dans la susceptibilité génétique au diabètenon insulinodépendant. Une mutation ponctuelle de l’acide
D I A B È T E N O N I N S U L I N O D É P E N D A N T
630 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9

Hypothèses concernant l’étiologie du diabète non insulinodépendant.1
dépendant se caractérise par une diminution de la fonctiondes cellules bêta du pancréas et de l’insulinosécrétioncompensatrice. Cette perte de fonction est probablementgénétiquement programmée, et aggravée par le phénomènede glucotoxicité et lipotoxicité.Il semble donc que la première anomalie à l’origine dudiabète non insulinodépendant soit une baisse d’efficacitéde l’insuline en tant que facteur d’utilisation du glucose,et principalement au niveau du muscle squelettique. Eneffet, l’usage de techniques variées, en particulier leclamp euglycémique hyperinsulinémique, a permis demettre en évidence un état de résistance à l’insuline chezles patients diabétiques non insulino-dépendants. En fait,80 % des patients diabétiques non insulinodépendantsprésentent une obésité dite androïde, c’est-à-dire carac-térisée par un excès de graisse viscérale. Cette obésité vis-cérale est associée à une résistance périphérique à l’actionde l’insuline. Pour expliquer ce lien, une des hypothèsesest que le tissu graisseux viscéral présente une grandeactivité métabolique avec lipolyse accrue et libérationexagérée d’acides gras libres dans le système porte. Cesacides gras libres favoriseraient l’insulinorésistance auniveau hépatique par une diminution de la clairance del’insuline, stimulation de la néoglucogenèse et inhibitionde la glycogénolyse ; et au niveau musculaire, par inhibitioncompétitive de la captation du glucose. Chez les sujetsobèses androïdes et diabétiques non insulinodépendants,l’oxydation musculaire du glucose et le stockage sousforme de glycogène sont réduits de 40 à 50 % lors declamps euglycémiques hyperinsulinémiques. Cependant, tous les sujets présentant une obésité viscéra-le ne deviennent pas intolérants au glucose ou diabétiquesnon insulinodépendants. En effet, tant que les cellules βpancréatiques de ces sujets peuvent répondre de façonadaptée et proportionnelle à l’insulinorésistance, en aug-mentant l’insulinosécrétion, la tolérance au glucose peutrester normale. Une deuxième anomalie doit donc néces-sairement coexister avec l’insulinorésistance pour expliquerle développement d’un diabète non insulinodépendant :
désoxyribonucléique mitochondrial, située au niveau dela séquence codant l’acide ribonucléique de transfert de laleucine (mutation en position 32-43), a été découvertedans des familles de diabétiques. Cette mutation en coségré-gation avec un diabète non insulinodépendant et une hypo-acousie neurosensorielle, a été retrouvée dans 2 % desfamilles de diabétiques non insulinodépendants d’unecohorte française. La mutation 32-43 est la même anoma-lie que celle auparavant identifiée dans le syndromeMELAS (Myopathy Encephalopathy Lactic Acidosis andStroke). Les mitochondries mutées sont apparemmentnon fonctionnelles et une concentration importante deformes anormales dans un tissu donné en réduirait lafonctionnalité, comme par exemple la sécrétion d’insuline.Ce type de diabète débute souvent chez des adultesjeunes, chez lesquels il peut conduire à l’insulinodépen-dance en quelques années. Les mécanismes physiopatho-logiques conduisant au diabète non insulinodépendant et au diabète insulinorequérant dans ce syndrome sontprobablement complexes et multifactoriels ; des défautsde production d’insuline, une toxicité du glucose et uneinsulinorésistance pouvant y être associés.
2. Physiopathologie du diabète non insulinodépendant commun
L’histoire naturelle du diabète non insulinodépendantcommun apparaît sur la figure. C’est une maladie trèshétérogène qui ne peut s’expliquer par une physio-pathologie unique. Elle débuterait par une insulino-résistance qui pourrait être génétique, acquise, ou les 2 àla fois. Si le fonctionnement des cellules β du pancréas estnormal, une hyperinsulinémie compensatrice se met enplace pour permettre le maintien d’une homéostasie glucidique normale. Il existe en effet une insulino-résistance et une hyperinsulinémie chez les prédiabé-tiques ayant une tolérance normale au glucose, et ce, bienavant le début d’un diabète non insulinodépendant. Latransition de l’état prédiabétique au diabète non insulino-
Endocrinologie-Métabolisme-Nutrition
631L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
Compensation de l’insulinorésistanceTolérance normale au glucose
Diabète non insulinodépendantInsulinorésistance
Hyperproduction de glucose par le foieDiminution de l’insulinosécrétion
Insulinorésistance HyperinsulinémieGénétique
Acquise
AcquiseGlucotoxicitéLipotoxicité
Génétique Déficience des cellules β

une dysfonction de l’insulinosécrétion. Dès lors qu’ilexiste une insulinopénie relative au regard de l’insulinoré-sistance, la tolérance au glucose est altérée et peut condui-re à un diabète non insulinodépendant patent. Cette ano-malie de l’insulinosécrétion a probablement des origines génétiques, mais certainement polygéniques, etnon encore clairement identifiées. Cependant, des fac-teurs aggravants peuvent intervenir, avec en particulierl’accumulation de triglycérides dans le pancréas secon-dairement à l’augmentation chronique de la concentrationdes acides gras et des triglycérides dans le plasma,conduisant au phénomène de lipotoxicité ; le phénomènede glucotoxicité, avec une diminution de l’insulino-sécrétion en condition d’hyperglycémie chronique; etenfin la découverte de dépôts d’amyline dans les cellulesβ du pancréas des patients diabétiques non insulino-dépendants.En fait, la forme commune de diabète non insulino-dépendant peut être considérée comme la conséquence de l’excès de graisse viscérale et l’insulinorésistanceassociée. Le diabète non insulinodépendant n’est pas laseule conséquence de cette insulinorésistance et de l’hyperinsulinémie compensatrice qui en résulte. En effet,d’autres anomalies métaboliques ont été associées, etregroupées sous le nom de syndrome plurimétabolique ousyndrome X. Parmi ces anomalies, une dyslipidémie estcouramment retrouvée, avec une élévation des VLDL(Very Low Density Lipoprotein) lipoparticules véhiculantprincipalement les triglycérides dans le sang, et une diminution du taux de HDL (High Density Lipoprotein)qui assurent principalement le transport inverse du choles-térol de la périphérie, en particulier des parois artérielles,vers le foie où il est catabolisé. L’hyperinsulinémie chro-nique, en particulier portale, stimule la synthèse hépa-tique des VLDL, et explique l’élévation du taux de trigly-cérides plasmatiques. Par ailleurs, les 2 principalessources de HDL sont d’une part la production hépatiquesous la forme de particules discoïdes comportant desphospholipides et des apoprotéines A1 et A2, et d’autrepart, la libération de composants de surface des chylomi-crons et des VLDL au cours de l’hydrolyse des trigly-cérides de ces lipoprotéines par la lipoprotéine lipase. Dans le syndrome d’insulinorésistance avec hyper-insulinémie, il existe une diminution d’activité de la lipoprotéine lipase et donc une diminution d’une dessources de production des HDL. Par ailleurs, l’anomaliedu métabolisme des lipoprotéines aboutit à la productionde LDL (Low Density Lipoprotein) petites et denses, etparticulièrement oxydables.Une troisième anomalie liée à l’insulinorésistance et à l’hyperinsulinémie est une augmentation de la pression artérielle. En effet, lorsque l’on compare des sujetstémoins et des sujets atteints d’une hypertension artérielleessentielle, appariés par l’âge et le poids, les sujets hypertendus présentent une résistance à l’action de l’insu-line lors de clamps euglycémiques hyperinsulinémiquespar rapport aux sujets témoins. Le lien entre hyper-insulinémie et hypertension artérielle n’est pas clairementétabli. Il pourrait s’agir de liens génétiques ou de liens
physiologiques, connaissant l’action de l’insuline auniveau du système nerveux sympathique, et au niveau detransporteurs ioniques impliqués dans la genèse de l’hypertension artérielle. Enfin, une dernière anomalie aété décrite, à savoir une hypofibrinolyse. La fibrinolyseest un mécanisme physiologique de défense contre lathrombose. En effet, lors d’une brèche endothéliale, lesphénomènes de la coagulation aboutissent à la formationd’un caillot de fibrine insoluble, pour réparer la lésion. Lafibrinolyse se met en route quasiment simultanément afinde dégrader ce caillot de fibrine insoluble en produits dedégradation de la fibrine solubles. L’enzyme clé de lafibrinolyse est la plasmine, qui provient d’un pro-enzymeinactif, le plasminogène. Le plasminogène est susceptibled’être activé par l’activateur tissulaire du plasminogène(tPA) lui-même fortement inhibé par l’inhibiteur de l’activateur du plasminogène 1 (PAI-1). Le PAI-1 estdonc un régulateur inhibiteur puissant de la fibrinolyse,puisqu’il agit à l’origine de cette cascade de réactions. Or,dans l’obésité viscérale avec insulinorésistance, les tauxplasmatiques de PAI-1 sont élevés, avec donc une tendan-ce à l’hypofibrinolyse, et à l’accumulation intravasculairede fibrine. Ce syndrome plurimétabolique associant diabète noninsulinodépendant, dyslipidémie, augmentation de lapression artérielle et hypofibrinolyse, explique la grandeprévalence de morbi-mortalité cardiovasculaire chez lespatients présentant ces anomalies.
Complications
Le diabète non insulinodépendant est encore trop souventconsidéré comme une pathologie bénigne. Pourtant sescomplications sont fréquentes, à l’origine d’une morbi-dité et mortalité non négligeables, mais également d’uncoût économique élevé. Le risque cardiovasculaire parmacro-angiopathie est majeur dans la population qui enest atteinte. En effet, comme nous l’avons vu précé-demment, les patients diabétiques de type 2 ont souventdes anomalies métaboliques associées, qui représententdes facteurs de risque cardiovasculaire et majorent la prévalence de la morbi-mortalité par athérothrombose.Par ailleurs, le patient diabétique non insulinodépendant,soumis à une hyperglycémie chronique, est susceptible dedévelopper aussi des atteintes spécifiques du diabète liéesà une micro-angiopathie, à savoir une néphropathie, unerétinopathie et une neuropathie périphérique. Au cours de 20 dernières années, le nombre de patientsdiabétiques devant subir une épuration extrarénale a net-tement augmenté dans les pays industrialisés. En France,le pourcentage de diabétiques dans cette population estpassé de 7 à 13 % entre 1982 et 1992. Cette augmentationest la conséquence d’un accroissement de la prévalencede la maladie dans la population générale qui vieillit,mais également d’une meilleure prise en charge de lacoronaropathie qui permet aux patients diabétiques noninsulinodépendants d’atteindre le stade de l’insuffisancerénale terminale. Il existe certainement des facteurs
D I A B È T E N O N I N S U L I N O D É P E N D A N T
632 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9

génétiques impliqués dans l’étiopathogénie de la néphro-pathie diabétique. La physiopathologie de la néphro-pathie dans le diabète non insulinodépendant comporteencore de nombreuses incertitudes. L’hypertension arté-rielle y joue un rôle tout à fait particulier. Plus de 50 %des diabétiques non insulinodépendants sont hypertendusau moment du diagnostic, parfois depuis plusieursannées ; et cette hypertension artérielle a déjà pu engen-drer des lésions rénales auxquelles se superposent ultérieurement les lésions de glomérulopathie diabétique.On manque encore d’un marqueur précoce de la néphro-pathie dans le diabète non insulinodépendant. En effet, lamicro-albuminurie n’a pas la même signification dans les2 formes de diabète. Alors qu’une micro-albuminuriepathologique entre 30 et 300 mg/24 heures, à 2 ou 3recueils urinaires, représente un signe précoce de néphro-pathie chez le diabétique insulinodépendant ou diabète de type 1, chez le diabétique non insulinodépendant, sasignification est plus complexe. Elle ne traduit pas forcé-ment une altération de la fonction rénale et elle ne préditpas forcément le développement vers la macroprotéinurieet l’insuffisance rénale. Elle traduit surtout un risque cardiovasculaire accru. En effet, une étude prospectivesur 8 années comparant 153 diabétiques non insulino-dépendants micro-albuminuriques à autant de diabétiquesnon insulinodépendants normo-albuminuriques, appariéspar l’âge, le sexe et la durée du diabète, retrouvent uneélévation du risque de décès par maladies cardiovasculairesdès que la micro-albuminurie dépasse 10,5 µg/min. L’excrétion pathologique d’albumine traduirait dans cettepopulation une souffrance endothéliale diffuse et non uni-quement localisée au glomérule rénal. La recherche d’unecoronaropathie silencieuse est donc indiquée chez toutpatient diabétique non insulinodépendant présentant unemicro-albuminurie pathologique. Le diagnostic denéphropathie diabétique n’est pas si facile, puisque lesétudes histologiques retrouvent fréquemment des lésionsd’une autre étiologie que le diabète. La biopsie rénalen’est pas systématique. Un faisceau d’arguments est enfaveur du diagnostic : une micro-albuminurie augmentéeou une protéinurie en l’absence d’autres anomalies duculot urinaire et la présence d’une autre complicationdégénérative du diabète, comme une rétinopathie ou uneneuropathie périphérique. La rétinopathie diabétique reste la première cause de cécitéavant l’âge de 50 ans dans les pays industrialisés. Après20 ans d’évolution, 60 % des diabétiques non insulino-dépendants présentent une rétinopathie diabétique. Elleest d’autant plus fréquente que le diabète est ancien et maléquilibré. Il existe une relation parallèle entre l’équilibreglycémique et l’incidence ou la progression de la rétino-pathie. Le dépistage de cette complication s’effectue par unfond d’œil après dilatation pupillaire et une angiographierétinienne en fluorescence. Le diabète non insulino-dépendant pouvant évoluer depuis plusieurs années avantsa découverte, le fond d’œil doit être systématique dès lediagnostic de diabète posé. Enfin, la neuropathie peut affecter aussi bien le systèmenerveux périphérique que le système nerveux autonome.
Endocrinologie-Métabolisme-Nutrition
633L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
La neuropathie est liée à la durée d’évolution du diabèteet à la qualité du contrôle glycémique. les signes cliniquesles plus fréquents sont la polyneuropathie symétrique etdistale, et surtout, de façon plus fréquente que dans le diabète insulinodépendant, des multinévrites ou mononé-vrites intéressant en particulier les nerfs oculomoteurs. La neuropathie autonome cardiaque tient une place particulière par sa fréquence et sa gravité. Elle associeune hypotension orthostatique, des modifications de larégulation tensionnelle, une tachycardie permanente etune mauvaise adaptation cardiovasculaire à l’effort. Ellepourrait expliquer le caractère silencieux de l’ischémiemyocardique et serait à l’origine de mort subite chez lepatient diabétique.
Traitement
1. Lutte contre l’insulinorésistance
Rappelons que dans la forme commune de diabète noninsulinodépendant, il existe le plus souvent une obésitéassociée et en particulier, une obésité viscérale.L’insulinorésistance existe donc toujours dans cesconditions, et le premier principe du traitement estl’amélioration de la sensibilité à l’insuline, en particu-lier au niveau musculaire. La prise en charge diététiqueest à la base de la thérapeutique du diabète non insulino-dépendant, avec pour objectif, non seulement l’amélio-ration de l’équilibre glycémique, mais aussi le contrôledes autres facteurs de risque cardiovasculaire. Il s’agit leplus souvent d’une restriction calorique qui permetd’obtenir une perte de poids moyenne de l’ordre de 3 kgpar mois, et qui correspond en général à une réductionde l’apport calorique de 30 % par rapport aux dépensesénergétiques globales. Ces dernières peuvent être esti-mées à partir de tableaux qui tiennent compte de l’âge,du sexe, du poids et de l’activité physique. L’apport alimentaire doit respecter un équilibre entre glucides,lipides et protéines. Le pourcentage stable de caloriesprotidiques est voisin de 15 à 20 % de la ration éner-gétique totale. La ration glucidique peut être compriseentre 40 et 55 %, en favorisant les glucides complexes, àdigestion et absorption lentes. Choisir la premièreoption revient à porter le pourcentage des lipides à 45 %de l’apport calorique total et choisir la deuxième consis-te à réduire les calories lipidiques à 30 %. En fait, touteune série de positions intermédiaires sont possibles, laplus fréquente étant 45 % de glucides, 20 % de graissesmono-insaturées, avec un rapport acides gras mono-insaturés/acides gras polyinsaturés /acides gras saturéségal à 2/1/1. Le choix est conditionné par le poids, laconcentration plasmatique des triglycérides, du HDLcholestérol et de l’activité physique. Associée à cesmesures diététiques, la pratique d’un exercice physiqueadapté à chaque patient doit être préconisée. En effet,l’exercice physique améliore la sensibilité musculaire àl’insuline. Les conditions dans lesquelles il doit être pratiqué commencent à être définies : il doit avoir lieu

de 3 à 5 fois par semaine pour avoir un effet métaboliqueprolongé; son intensité doit atteindre 70 % de la VO2maximale ; enfin, sa durée doit varier entre 20 et 50 min. La pratique de l’exercice physique n’est pas sansrisque et doit être réalisée sous surveillance médicale.Diverses complications sont possibles : cardiovasculaireschez un malade coronarien méconnu, hémorragiques surune rétinopathie proliférante méconnue, et orthopédiques.Le troisième moyen d’améliorer la sensibilité à l’insulineest la prescription de biguanides, et en particulier de met-formine, seul composant commercialisé en France. La présence d’insuline est nécessaire à l’action de la met-formine qui n’est pas un stimulant de la sécrétion d’insuline. Son site d’action est le foie, où elle diminue lanéoglucogenèse hépatique à partir du lactate de l’alanineet du glycérol. Cette action semble largement expliquer ladiminution de la production hépatique de glucose. Uneffet inhibiteur sur la glycogénolyse a aussi été évoqué.Par ailleurs, la metformine agit aussi au niveau musculai-re en augmentant l’utilisation du glucose, et en stimulantprincipalement son métabolisme non oxydatif, c’est-à-dire son stockage sous forme de glycogène musculaire.Cet effet semble passer par la stimulation de la trans-location des transporteurs GLUT 4 du glucose.Il existe d’autres molécules susceptibles d’améliorer lasensibilité à l’insuline. Le benfluorex semble augmenterl’utilisation musculaire du glucose, en stimulant sonmétabolisme oxydatif. Par ailleurs, la famille des thiazolidinediones avec commechef de file la troglitazone, améliore la sensibilité muscu-laire à l’insuline, par un mécanisme d’action différent. En effet, ces molécules se lient spécifiquement à des récepteurs nucléaires de la famille PPAR-γ (peroxysome proliferator activated receptor gamma).L’activation de PPAR-γ entraînerait une diminution de l’insulinorésistance par régulation de l’expression desgènes impliqués dans l’action de l’insuline, principale-ment au niveau du tissu adipeux. Les études cliniquesavec ces produits sont actuellement ralenties à caused’une potentielle toxicité hépatique.
2. Inhibiteurs des α-glucosidases
L’absorption intestinale de l’amidon et des disaccharidesrequiert l’action d’α-glucosidases qui hydrolysent lesliaisons α-glucosides, situées sur la bordure en brosse descellules épithéliales de l’intestin. Des composés présen-tant une homologie structurale avec les saccharides del’alimentation ont été isolés et présentent une activitéd’inhibition compétitive des α-glucosidases : l’acarboseet le miglitol. Ces molécules ralentissent la digestion desglucides. Leur absorption a donc lieu plus dans l’iléonque dans le jéjunum. L’apparition de glucose dans le sangaprès un repas est donc retardée et étalée dans le temps, cequi diminue la glycémie post-prandiale. Cependant, lamaldigestion et la malabsorption des oligosaccharidesdans l’intestin grêle, peuvent favoriser la croissance bactérienne colique, avec comme corollaire, des effetsindésirables digestifs, comme flatulences et diarrhées.
3. Lutte contre l’anomalie de l’insulinosécrétion
Les sulfamides hypoglycémiants sont une classe de médi-caments présentant la capacité de stimuler l’insulino-sécrétion par les cellules β pancréatiques. En effet, ilsagissent directement sur les canaux potassiques adénosi-ne triphosphate dépendants situés dans les membranes deces cellules. Il existe plusieurs types de sulfamides hypo-glycémiants, de durée de vie et de puissance d’action dif-férentes.L’insulinothérapie exogène peut enfin être envisagée dansla thérapeutique du diabète non insulinodépendant, soiten cas de contre-indication aux antidiabétiques oraux soitau moment d’un événement intercurrent aigu, tel qu’unecomplication métabolique, une intervention chirurgicaleoù l’insulinothérapie sera généralement transitoire, soit,enfin, du fait de l’histoire naturelle du diabète non insuli-nodépendant, avec au bout d’un certain nombre d’annéesd’évolution, une insulinopénie vraie. ■
D I A B È T E N O N I N S U L I N O D É P E N D A N T
634 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 9 , 4 9
• Le diabète non insulinodépendant est le type de diabète le plus fréquent. Il touche environ 2 % de la population française.
• Il se définit comme une glycémie à jeunsupérieure ou égale à 1,26 g/l.
• Il est le plus souvent associé à une obésité de type androïde, et s’accompagne d’autresanomalies métaboliques, regroupées sous le nom de syndrome plurimétabolique ou syndrome X.
• La physiopathologie de la forme commune de ce type de diabète repose sur 2 anomalies.D’une part une résistance périphérique à l’insuline, en particulier au niveaumusculaire, et d’autre part, une anomalie de l’insulinosécrétion.
• Les complications de ce diabète sont de 2 types : une macro-angiopathie avec maladies cardiovasculaires touchant les artères de moyen et gros calibres,et une micro-angiopathie spécifique de l’hyperglycémie chronique.
• Les principes du traitement reposent d’une part, sur la lutte contrel’insulinorésistance par la perte de poids,l’exercice physique, les biguanidesprincipalement ; d’autre part,le ralentissement de la digestion et de l’absorption des glucides de l’alimentation, et enfin la stimulation de l’insulinosécrétion par les sulfamideshypoglycémiants, voire même uneinsulinothérapie exogène.
Points Forts à retenir

Endocrinologie – Métabolisme – NutritionA 58
2163L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Dans d’autres cas, ce sont des signes cliniques quiconduisent le médecin à palper la région cervicale (douleur, gêne, signes de compression locorégionale,signes en faveur d’une hypothyroïdie ou d’une hyper-thyroïdie).Parfois, le goitre est découvert sur une imagerie deman-dée pour une pathologie autre (échographie cervicale,radiographie pulmonaire montrant une opacité se proje-tant dans le médiastin antérieur et supérieur, ou bientomodensitométrie thoracique).
Éléments cliniques d’orientation
1. Éléments anamnestiques
Ils permettent de préciser l’ancienneté du goitre, l’évo-lution (progression, stabilisation), la notion de radio-thérapie cervicale dans l’enfance (susceptible d’induireun cancer thyroïdien), les traitements suivis, en particulierceux contenant de l’iode ou le lithium, l’origine géo-graphique, ethnique (régions de carence iodée), lesmaladies thyroïdiennes dans la famille.
2. Caractéristiques du goitreL’examen de la glande thyroïde est le temps essentielpermettant l’orientation diagnostique. Il comprend l’ins-pection, la palpation et l’auscultation. L’inspection permetde visualiser le goitre chez les sujets maigres ou en casde goitre volumineux. On recherche aussi des signes decompression veineuse, une turgescence des jugulairesou encore une circulation veineuse collatérale. Pour lapalpation, l’examinateur se met derrière le patient assis,et place ses doigts dans la région thyroïdienne. Le caractèremobile de la glande thyroïde est confirmé en demandantau patient de déglutir. La palpation des aires ganglionnairescervicales complète l’examen. L’ensemble des résultatsretrouvé peut être consigné sur un schéma et les caracté-ristiques suivantes seront précisées : taille – mesure dupérimètre cervical – ; consistance – molle, souple, élas-tique, ferme, dure, pierreuse, caractère douloureux,symétrie, nodules associés, mobilité à la déglutition,adénopathies jugulo-carotidiennes, prétrachéales, sus-claviculaires, spinales et sous-maxillaires, thrill ougoitre soufflant.
Orientation diagnostique devant un goitre
Définition
Le goitre est une augmentation diffuse ou localisée de laglande thyroïde, consécutive soit à une augmentation dunombre des cellules épithéliales et des follicules thyroï-diens (goitre diffus sporadique ou endémique), soit àune infiltration de la glande thyroïde par un processusinflammatoire, auto-immun ou néoplasique. On distingueaussi le goitre parenchymateux ferme, constitué depetites vésicules pauvres en colloïde à l’opposé dugoitre colloïde, rénitent, de consistance molle car constituéde vésicules dilatées remplies de colloïde.
Circonstances de découverte
Le goitre peut être découvert fortuitement par le patientlui-même, son entourage ou bien par le médecin lorsd’une palpation systématique de la région cervicale.
Goitre diffus et nodule thyroïdien Orientation diagnostique
DR Sophie VENAULT, PR Vincent ROHMER
Service d’endocrinologie-nutrition, médecine C, CHU,Angers Cedex 01.
• La découverte d’un goitre ou d’un nodule thyroïdien est un motif fréquent de consultationen endocrinologie.
• L’orientation étiologique dépend des donnéesfournies par l’anamnèse, l’examen clinique,les dosages hormonaux et les explorations à visée morphologique : échographie et scintigraphie.
• Dans le cas des nodules thyroïdiens palpables,la cytoponction est l’examen essentiel à pratiquer. La démarche diagnostique vise ensuite à choisir au mieux l’indication thérapeutique, à savoir : soit simple surveillance,soit traitement médical ou chirurgical.
Points Forts à comprendre

3. Examen généralIl recherche :• des signes d’hyperthyroïdie ou d’hypothyroïdie ;• des signes de compression locorégionale :dyspnée
inspiratoire par compression trachéale, dysphagie parrefoulement ou compression œsophagienne, dysphonie,voix bitonale par atteinte récurrentielle, syndromecave supérieur avec œdème en pèlerine par compres-sion de la veine cave supérieure ;
• une altération de l’état général,la notion de diarrhéeou de bouffées vasomotrices faciales ;
• une anomalie de la pression artérielle.
Examens biologiques
Le dosage de la TSH est indispensable dans tous les cas.Il est complété par un dosage de la thyroxine libre (T4 libre) et des anticorps antithyroïdiens (anticorpsanti-thyroperoxydases, anti-thyroglobuline) et anticorpsanti-récepteurs de la TSH en fonction du contexte clinique. Un dosage de l’iodémie ou de l’iodurie peutêtre demandé si l’on suspecte une surcharge iodée. Encas de goitre nodulaire, un dosage de la calcitonine estproposé.
Examens à visée morphologique
1. ÉchographieC’est un examen simple, non traumatique et peu coûteux.Des coupes transversales et longitudinales de la glandethyroïde sont réalisées. Cet examen permet de donnerles mensurations et le volume thyroïdien. En moyenne,la hauteur varie de 4 à 6 cm, la largeur et l’épaisseur de1,5 à 2,5 cm. Le caractère homogène ou non et l’écho-génicité du goitre doivent être précisés, de même la pré-sence éventuelle de nodule infraclinique ou de calcifica-tions. La présence d’adénopathie est recherchée et notéedans le compte rendu.
2. Scintigraphie au technétium ou à l’iode 123 Elle garde uniquement 2 indications dans le cadre desgoitres non nodulaires : le goitre avec une hyperthyroïdieet le goitre plongeant. Dans le premier cas, le but estd’apprécier le pourcentage de captation du traceur et lecaractère homogène ou non de la fixation. Dans le secondcas, l’intérêt est de confirmer le caractère plongeant ouendothoracique du goitre. Dans tous les cas, cet examenest contre-indiqué en cas de grossesse, et réalisé endébut de cycle chez la femme si une contraception efficace n’est pas suivie. Si une surcharge iodée est sus-pectée ou si le patient est déjà traité par des hormonesthyroïdiennes, il n’y a aucun intérêt, en normothyroïdie,à réaliser cet examen car la fixation sera nulle.
3. Radiographie thoracique Elle détecte le caractère plongeant du goitre sous laforme d’une opacité médiastinale supérieure. Cet élémentne peut pas être visualisé par l’échographie. L’aspect dela trachée, déviée ou rétrécie, est à observer.
4. Radiographie de la trachéeElle permet d’évaluer s’il existe un rétrécissement de la trachée par le goitre. Il sera parfois visualisé des calcifications. Mais cet examen n’est pas indispensable.
5. Scanner cervico-thoracique Réalisé avec injection d’iode, il est demandé en cas degoitre volumineux plongeant ou totalement endothora-cique, compressif, en préopératoire. Il vise à préciser lesrapports de la glande thyroïde avec les organes de voisi-nage. Si une scintigraphie thyroïdienne est nécessaire,cet examen est réalisé au préalable, l’injection d’ioderendant la scintigraphie ininterprétable. Le produit decontraste iodé peut induire, rarement, un trouble fonctionnel thyroïdien.
Diagnostic étiologique et conduite à tenir
1. Goitre diffus avec euthyroïdie
• Le goitre simplese définit comme une augmentationdu volume de la thyroïde non liée à un processus inflam-matoire ou néoplasique et associée à une euthyroïdie. Ilsurvient préférentiellement chez la femme, et l’on parlede goitre sporadique lorsqu’il touche moins de 10 % dela population d’une même région. Il est en général devolume modéré, souple, homogène, indolore, et débuteen période péripubertaire. En échographie, le goitreapparaît homogène. Il peut évoluer vers une augmentationprogressive du volume avec, éventuellement, formationde nodule(s), signes compressifs, rarement, dysfonc-tionnement thyroïdien (fig. 1).L’augmentation de l’incidence du goitre simple au coursde la grossesse est connue. Ainsi, dans une étude récente,il a été montré par une étude échographique de la glandethyroïde, une augmentation du volume thyroïdien allantde 20 à 130 % chez plus de 80 % des parturientes et celade façon continue tout au long de la grossesse. Ces goitresdisparaissent en général dans les 6 mois qui suivent l’accouchement.• Le goitre exclusivement endothoraciqueévoque unetumeur du médiastin antéro-supérieur. Mais la scintigraphieprécédant l’examen tomodensitométrique thoraciquepermet en général d’en faire le diagnostic. Il peut s’ac-compagner de signes compressifs.• Le goitre de cause iatrogéniquesans trouble fonctionnelavéré est le plus souvent lié à la prise de lithium.• La thyroïdite de Hashimotoà sa phase initiale peutentraîner un goitre diffus sans dysthyroïdie.• Le goitre endémiquesurvient dans une zone danslaquelle plus de 10 % de la population des enfants âgésde 6 à 12 ans ont un goitre. Les besoins journaliers eniode sont de 150 µg chez l’adulte, 90 à 120 µg chez lenourrisson et l’enfant. En 1992, la carence iodée affectaitencore 140 millions d’habitants, 97 millions d’entre euxavaient un goitre. L’origine géographique et le caractèrefamilial du goitre orientent le diagnostic. La préventionpasse par l’iodation du sel notamment ou de l’eau de
G O I T R E D I F F U S E T N O D U L E T H Y R O Ï D I E N
2164 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

• La surcharge iodéeassocie une notion de prise médicamenteuse (médicament le plus souvent iodé) ; un goitre ancien, diffus, une hyperthyroïdie marquée.La scintigraphie est blanche, l’iodémie est élevée s’ils’agit d’une hyperthyroïdie induite par l’iode.• La thyroïdite silencieuseou indolore est représentéeprincipalement par la thyroïdite du post-partum. Elle est caractérisée par la survenue d’une hyperthyroïdiemodérée, d’une durée de 2 à 8 semaines, survenant 1 à 3 mois suivant l’accouchement, associée à un goitreisolé. Puis, survient une phase d’hypothyroïdie versle 3e et le 6e mois du post-partum avec un retour versl’euthyroïdie au 10e mois. Devant une hyperthyroïdiesurvenant dans le post-partum, le premier diagnostic àévoquer est celui de thyroïdite du post-partum (70 à80 % des cas), il s’agit plus rarement d’une maladie deBasedow (10 à 15 % des cas). En cas de doute diagnos-tique, une scintigraphie pourra être demandée. Ellemontre une captation nulle ou diminuée. La suspensionde l’allaitement pendant 7 jours après la scintigraphieest nécessaire.• L’hyperplasie toxique de la thyroïdeassocie un goitrediffus, vasculaire sans ophtalmopathie et sans stigmated’auto-immunité et ayant un caractère familial. La thyroidstimulating hormone(TSH) est effondrée. Une mutationactivatrice germinale du récepteur de la TSH en est lacause.• L’adénome hypophysaire à TSHassocie un goitrediffus, homogène avec des signes de thyrotoxicose. Laprésence d’un syndrome hypophysaire associé peut rétablir le diagnostic mais surtout le profil hormonal quiassocie une élévation de la T4 libre avec une TSH nor-male ou augmentée.
boisson. Actuellement, l’apport d’iode en France se faitsurtout par le lait via l’utilisation de la Bétadine pournettoyer les cuves dans les industries laitières et le sel.Le manioc, s’il est surconsommé, est également responsablede goitres endémiques dans certaines régions du monde.• La thyroïdite de Riedel,rare, est caractérisée par ungoitre de consistance dure, et les signes de compressionssont au premier plan.
2. Goitre diffus avec une hyperthyroïdie• La maladie de Basedow est une maladie auto-immunede la femme jeune, faisant souvent suite à un choc psy-choaffectif. Elle associe typiquement : un goitre diffus,symétrique, homogène, non nodulaire, non douloureux,de consistance élastique, soufflant ; une exophtalmie ;des signes de thyrotoxicose francs.Le goitre est homogène en échographie et l’image scin-tigraphique est symétrique, bilatérale et homogène. Lesanticorps anti-récepteurs de la TSH sont positifs dans lamajorité des cas.• La thyroïdite de Hashimoto(forme appelée hashi-toxicosis) peut être responsable à la phase initiale d’unehyperthyroïdie.• La thyroïdite subaiguë de De Quervainsurvient defaçon saisonnière, sous forme d’épidémie, dans un contexted’infection virale. La clinique est d’emblée évocatrice etassocie : un goitre récent, ferme, irrégulier et surtoutdouloureux rendant la palpation difficile,un syndromefébrile et un syndrome inflammatoire ; des signes dethyrotoxicoses modérés et transitoires.Le goitre est très hypoéchogène et hétérogène en écho-graphie et la captation en scintigraphie est nulle en l’absence de surcharge iodée.
Endocrinologie – Métabolisme – Nutrition
2165L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Diagnostic d’un goitre diffus.1
Goitre diffus
Dosage de la TSH
Hyperthyroïdie
Maladie de BasedowThyroïdie de de Quervainorigine médicamenteuse
HashitoxicoseThyroïdite du post-partum
Hypothyroïdie
Thyroïdite de Hashimotocarence iodée
Thyroïdite du post-partumorigine médicamenteuse
Euthyroïdie
Goitre simpleGoitre endémique
Thyroïdite de Hashimotoorigine médicamanteuse

• Le syndrome de résistance partielle aux hormonesthyroïdiennesassocie : des signes de thyrotoxicose, ungoitre, dans un contexte familial, se rencontrant dans lescas de résistance hypophysaire aux hormones thyroï-diennes. Dans ce cas, la thyroxine libre est augmentée etla TSH est normale ou augmentée.
3. Goitre diffus avec une hypothyroïdie• La thyroïdite chronique de Hashimoto est une affectionauto-immune qui survient le plus souvent chez la femme,entre 30 et 60 ans et réalise typiquement un goitreferme, symétrique, indolore, d’apparition progressive,parfois bosselé, parfois associé à des adénopathies jugulo-carotidiennes. Initialement, le patient peut être euthyroï-dien, ou plus rarement hyperthyroïdien (hashitoxicosis)mais l’évolution se fait vers l’hypothyroïdie. En écho-graphie, le goitre apparaît hétérogène, hypoéchogène,avec parfois des plages pseudo-nodulaires. Les anticorpsantithyroïdiens sont très positifs. La scintigraphie, enrègle générale inutile à présent, montre une image hété-rogène, en damier, parfois avec des plages plus fixantes,pouvant donner un aspect de pseudo-nodule chaud.• La thyroïdite du post-partumpeut être responsabled’une hypothyroïdie.• Le goitre endémique par carence iodéepeut être responsable d’une hypothyroïdie.• Le goitre lié à un trouble de l’hormonogenèse àrévélation tardive,non reconnu dans l’enfance, ce quiest exceptionnel à présent.• Parmi les causes médicamenteuses :antithyroïdiensde synthèse, iode (amiodarone surtout), lithium, iode 131,l’interféron ou d’autres cytokines.
Orientation diagnostique devant un nodule thyroïdien
Généralités
Le nodule thyroïdien est défini cliniquement commeune tuméfaction localisée de la glande thyroïde. Il estfréquent, sa prévalence chez l’adulte est variable selonles moyens utilisés pour l’identifier : 2,5 à 4 % clinique-ment, 27 à 51 % en échographie, plus de 50 % sur desséries autopsiques ; de plus, l’incidence croît avec l’âge.Devant un nodule thyroïdien, la question souvent poséepar le patient est de savoir s’il est bénin ou malin. Desarguments épidémiologiques doivent permettre de rassurer le patient : 90 % des nodules thyroïdiens exploréssont bénins, l’incidence annuelle du cancer thyroïdienest faible (2,5 pour 100 000 habitants), le pronostic descancers thyroïdiens différenciés est bon ; cependant,chez le sujet de sexe masculin, ou ayant des antécédentsd’irradiation cervicale dans l’enfance, ou chez l’enfant,une attention particulière devra être portée compte tenude la plus grande fréquence des cancers par rapport à lapopulation générale.
La préoccupation du clinicien devant un nodule thyroïdienest de savoir aussi s’il s’accompagne ou non d’unehyperthyroïdie.
Circonstances de découverte
Le nodule peut être découvert lors d’une palpation systématique effectuée par le patient lui-même ou lemédecin, ou bien devant des signes d’appel : un dys-fonctionnement thyroïdien, une douleur, des signescompressifs, ou bien encore, des signes évocateurs d’un carcinome médullaire de la thyroïde (flush et diarrhée).Une douleur d’apparition subite permettant de découvrirune tuméfaction thyroïdienne oriente soit vers un kystehémorragique, soit vers une nécrose partielle d’un nodulesolide. Elle est à distinguer de la douleur de la thyroïditesubaiguë de De Quervain plus diffuse et accompagnéede signes inflammatoires, ou de l’exceptionnel abcèsthyroïdien.Parfois, c’est dans un contexte de dépistage devant desantécédents familiaux de cancer thyroïdien ou de nodulethyroïdien ou de néoplasie endocrinienne multiple detype 2. Enfin, c’est aussi au cours d’une imagerie cervicaleprescrite pour un autre motif que le nodule est découvert.
Examen clinique
Le nodule thyroïdien est perçu sous la forme d’unehypertrophie arrondie et localisée dont on précise lesiège, la consistance, les dimensions, la sensibilité, lescontours, la mobilité. L’exploration des aires ganglion-naires satellites est systématique.L’examen général ainsi que la recherche de signes enfaveur d’une hyperthyroïdie ou d’une hypothyroïdie etde signes de compression complètent le bilan.
Examens paracliniques
• Le dosage de la TSH détermine l’état de la fonctionthyroïdienne.• Le dosage de la calcitonineest discuté mais devraitêtre réalisé devant tout nodule thyroïdien (voir :Pourapprofondir 1).• L’échographie thyroïdienneest prescrite ; il est préci-sé le nombre, la taille, la localisation, le caractère bienlimité ou non, l’échogénicité du nodule et la présenced’un halo hypoéchogène entourant la totalité du nodule(en faveur du caractère plutôt bénin). La recherched’adénopathies non palpables est importante.• La scintigraphie à l’iode 123est intéressante, unique-ment s’il existe une hyperthyroïdie clinique ou bien sim-plement une TSH basse afin de rechercher un noduletoxique ou prétoxique (voir :Pour approfondir 2). Lesprécautions d’emploi et les contre-indications sont lesmêmes que celles citées précédemment.• La cytoponctiondu nodule est indispensable (voir :Pour approfondir 3).
G O I T R E D I F F U S E T N O D U L E T H Y R O Ï D I E N
2166 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

Éléments d’orientation
Un faisceau d’arguments cliniques et paracliniques vapermettre de répondre aux 2 questions posées : s’agit-ild’un nodule toxique? s’agit-il d’un cancer ? (fig. 2).• En faveur d’un nodule toxique,on retient : les signescliniques d’hyperthyroïdie ; une TSH effondrée ; unehyperfixation en scintigraphie unilatérale, en regard dunodule, avec extinction partielle ou complète du reste du parenchyme ; dans tous les cas, une cytoponction est réalisée car la présence d’un cancer thyroïdien ausein d’un nodule chaud est rare mais existe (4 % danscertaines séries).• Les éléments suivants peuvent orienter vers un cancer :antécédents familiaux de cancer thyroïdien ; associationsparticulières : polypose colique, syndrome de Cowden ;irradiation du cou ; les caractéristiques cliniques dunodule : augmentation rapide de la taille, consistancedure, de surface irrégulière, le caractère fixé, la présencede signes de compression, la présence d’adénopathiessuspectes (l’augmentation progressive de taille d’unnodule au sein d’une thyroïdite lymphocytaire chro-nique chez un adulte de plus de 60 ans peut évoquer un lymphome) ; une augmentation de la calcitonineoriente vers un carcinome médullaire de la thyroïde ; enéchographie, le caractère anéchogène en totalité ouhyperéchogène du nodule est rassurant, de même la présence d’un halo hypoéchogène entourant la totalitédu nodule, les nodules cancéreux étant le plus souventhypoéchogènes ou hétérogènes. La présence de micro-calcifications n’a aucune valeur d’orientation ; néan-moins, la présence d’un piqueté de microcalcificationspeut évoquer un carcinome papillaire. Les limitesimprécises d’un nodule sont aussi suspectes sans êtrecaractéristiques. Des adénopathies hypoéchogènes detaille supérieure à 1 cm sont suspectes ; la cytoponctiona une sensibilité de 90 % et une spécificité de 70 à 80 %pour le diagnostic de cancer. ■
Endocrinologie – Métabolisme – Nutrition
2167L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• Le goitre diffus est une augmentation de volume de l’ensemble de la glande thyroïde.L’orientation diagnostique dépend des caractéristiques cliniques du goitre et du statut thyroïdien. Les examens complémentaires essentiels sont le dosage de la TSH et l’échographie. Si une hyperthyroïdieest associée, une scintigraphie complète les explorations.
• La prévalence du nodule thyroïdien est élevéesurtout si l’on utilise l’échographie commemoyen de dépistage (prévalence de 50 %). Ainsi, seuls les nodules cliniquement palpables,ou de taille supérieure à 1 cm, devront donnerlieu à des explorations complémentaires,en dehors des cas où il existe des signes échographiques suspects (limites irrégulières,microcalcifications, adénopathies non résolutives) ou des antécédents familiauxnotables (carcinome médullaire de la thyroïde,carcinome papillaire touchant déjà plusieursmembres de la famille). La stratégie diagnostique consiste à sélectionner les nodulesconduisant à un traitement spécifique :les adénomes toxiques et les carcinomes thyroïdiens en sachant que 90 % des nodulessont bénins. Pour cela, la clinique est indispensable. Le dosage de la TSH et de la calcitonine est demandé, ainsi qu’une échographie thyroïdienne. La cytoponction à l’aiguille fine est le tempsessentiel dans la prise en charge du nodule en routine. La scintigraphie thyroïdienne a peu d’intérêt en l’absence d’hyperthyroïdie.
Points Forts à retenir
Diagnostic d’un nodule thyroïdien palpable.2
Nodule thyroïdien palpable
Dosage de la TSH – Échographie
Hyperthyroïdie
Scintigraphie
Nodule toxiqueGoitre multinodulaire toxique
Euthyroïdie
Calcitonine élevéeCarcinome médullaire
de la thyroïdeMalinBénin
DouteuxIninterprétable
Cytoponction

Endocrinologie - Métabolisme - NutritionB 341
1701L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Étiologie
La sécrétion de cortisol s’intègre dans un ensemblefonctionnel : l’axe corticotrope (fig. 1). La corticolibérine(CRH) et la vasopressine (AVP) d’origine hypothalamiquestimulent la synthèse de la pro-opiomélanocortine(POMC) dans les cellules corticotropes anté-hypophysaireset sa maturation donnant naissance à différents peptidesdont la corticotrophine (ACTH), la lipotrophine (LPH),etc. La corticotrophine exerce un effet trophique sur lescorticosurrénales ; stimule la synthèse et la sécrétion decortisol, de déhydro-épiandrostérone (ou DHEA, andro-gène) et de minéralocorticoïdes (désoxycorticostéroneou DOC notamment). La sécrétion de corticotrophine etde cortisol suit un rythme nycthéméral : leur concentrationplasmatique est minimale vers 0 h, augmente progressi-vement en seconde partie de nuit, atteint son maximumen fin de nuit puis décroît progressivement durant lajournée. L’activité de l’axe corticotrope est asservie parun rétrocontrôle négatif : l’augmentation de la cortisolémieinhibe la synthèse et la libération de la corticolibérine etde la corticotrophine. La sécrétion excessive et chronique de cortisol (hyper-cortisolisme) est responsable du syndrome de Cushingendogène. La prise chronique de corticoïdes de synthèsepeut être à l’origine d’un syndrome de Cushing exogène.D’un point de vue physiopathologique, il existe 2 catégoriesde causes d’hypercortisolisme (ou syndrome de Cushingendogène) (fig. 1, tableau I) :
Hypercortisolisme de l’adulteÉtiologie, diagnostic
PR Antoine TABARIN
Département d’endocrinologie, diabétologie et maladies métaboliques, CHU de Bordeaux ; hôpital du Haut-Levêque, 33604 Pessac, Cedex.
• La différenciation physiopathologique entre les hypercortisolismes secondaires à une sécrétion inappropriée de corticotrophine(ou ACTH-dépendants) et les hypercortisolismesindépendants de la sécrétion de corticotrophine(ou d’origine surrénalienne).
• L’origine hypophysaire ou non hypophysaire(ectopique) de la sécrétion de corticotrophinedans l’hypercortisolisme ACTH-dépendant.
• Le diagnostic différentiel entre maladie de Cushing et sécrétion ectopique d’ACTH parfois très délicat et qui repose sur un faisceaud’arguments.
Points Forts à comprendre
Physiopathologie et classification du syndrome de Cushing.1
Adénome/carcinomesurrénalien
Hyperplasie macronodulaireprimitive
CRH AVP
ACTHCortisol
Normal
AVPCRH
Cortisol
ACTH
Maladie de Cushing Sécrétion ectopique d'ACTH
Cortisol
ACTH
AVPCRH
Syndrome de Cushing ACTH-dépendant
AVPCRH
CortisolACTH
AVPCRH
Cortisol
ACTH
Syndrome de Cushing ACTH-indépendant

– l’hypercortisolisme ACTH-indépendant : la sécrétionsurrénalienne est autonome et l’hypercortisolisme,par rétrocontrôle, supprime la sécrétion de corticotrophinedont la concentration circulante est effondrée. Unetumeur surrénalienne unilatérale bénigne (adénomecortisolique) ou plus rarement maligne (carcinomeprimitif ou corticosurrénalome malin) est à l’originede l’hypercortisolisme (tableau I). Rarement, les 2 surrénales sécrètent en excès le cortisol (hyperplasiemacronodulaire ou dysplasie corticotrophine-indé-pendantes). Dans certains cas, l’hyperplasie macrono-dulaire est secondaire à une expression illicite derécepteurs dans la corticosurrénale comme le récepteurdu GIP (gastric inhibitory peptide), peptide sécrété par le tube digestif lors de l’alimentation, et qui stimule alors de manière aberrante la sécrétion de cortisol ;
– l’hypercortisolisme ACRTH-dépendant : les surré-nales sont stimulées par une sécrétion excessive etinappropriée de corticotrophine. La corticotrophinepeut être d’origine eutopique et sécrétée par un adénomedéveloppé à partir des cellules corticotropes hypo-physaires, c’est la maladie de Cushing (tableau I).Plus rarement, la corticotrophine est d’origine ectopique,produite par une tumeur neuro-endocrine non hypo-physaire. Cette sécrétion ectopique de corticotrophineest responsable d’un syndrome de Cushing paranéo-plasique. Le syndrome de Cushing paranéoplasiquepar sécrétion ectopique exclusive de corticolibérineest exceptionnel.
Diagnostic
Il s’articule en plusieurs étapes dont la chronologie doitêtre respectée. Il faut successivement :– évoquer l’hypercortisolisme cliniquement en recherchant
les symptômes les plus spécifiques ;
– dépister biologiquement l’hypercortisolisme avec desinvestigations simples puis le confirmer lorsque ledépistage est positif ;
– éliminer les diagnostics différentiels de l’hypercortiso-lisme (principalement les hypercortisolismes fonc-tionnels) ;
– déterminer l’étiologie de l’hypercortisolisme, préambuleindispensable à un traitement adapté.
Du fait de la fréquence des lésions surrénaliennes ethypophysaires non fonctionnelles et asymptomatiquesdans la population générale, le diagnostic positifd’hypercortisolisme repose exclusivement sur des donnéescliniques et biologiques. L’imagerie médicale n’a deplace qu’une fois le diagnostic de syndrome de Cushingendogène acquis.
Sémiologie clinique
Certains symptômes cliniques d’hypercortisolisme (obésité,hypertension, diabète sucré, dépression) sont fréquemmentrencontrés dans la population générale. Il est donc capitalde rechercher les symptômes les plus spécifiques : cesont ceux engendrés par les effets cataboliques et anti-anaboliques du cortisol sur le métabolisme protidique.
1. Anomalies morphologiques Ces anomalies permettent d’évoquer le diagnostic dèsl’inspection (fig. 2 et 3).Elles sont acquises et peuvent être différenciées desaspects constitutionnels en examinant des photographiesanciennes des patients.• La prise pondéraleest généralement modérée (unedizaine de kg), survient en l’absence de modificationdes habitudes alimentaires et résiste à la restriction calorique. Surtout, elle présente une topographie parti-culière, facio-tronculaire : le visage devient arrondi,bouffi, les creux sus-claviculaires se comblent, l’accu-mulation adipeuse au niveau de la nuque provoque unaspect en « bosse de bison ».
H Y P E R C O R T I S O L I S M E D E L ’ A D U L T E
1702 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Étiologie Prévalence relative (%)
Syndrome de Cushing 80ACTH-dépendant❑ maladie de Cushing 65-70❑ sécrétion ectopique d’ACTH 10-15❑ sécrétion ectopique de CRH < 1
Syndrome de Cushing 20ACTH-indépendant❑ adénome cortisolique 15❑ carcinome primitif 5❑ hyperplasie et dysplasies primitives 1-2
Prévalence relative des causes d’hypercortisolisme
TABLEAU I
Faciès bouffi et érythrosique caractéristique du syndromede Cushing.
2
• Symptômes d’hyperandrogénie :ils peuvent êtreassociés, se limitant généralement à un hirsutismemodéré, une séborrhée et une acné séborrhéique du visage.
2. Autres symptômes • Ostéopénie et ostéoporosesont évocatrices avantl’âge de 50 ans. Souvent asymptomatiques ou pauci-symptomatiques (lombalgies d’horaire mécanique), ellespeuvent être objectivées par ostéodensimétrie. Des fractures pathologiques, costales ou vertébrales peuventsurvenir chez les patients âgés ou quand l’hypercortiso-lisme est intense.• Troubles gonadiques :spanioménorrhée voire amé-norrhée secondaire, muette (sans bouffées de chaleur)chez la femme ; baisse de la libido et impuissance chezl’homme. • Hypertension artérielle :elle est le plus souventmodérée mais concourt à la morbidité cardiovasculairedu syndrome de Cushing. • Troubles psychiatriques :à type d’irritabilité, anxiété,insomnie nocturne et tendance dépressive fluctuant en intensité. Un tableau psychiatrique aigu à type deconfusion mentale ou psychose hallucinatoire est rarement rencontré.
3. Anomalies biologiques non spécifiquesCes anomalies peuvent renforcer la suspicion clinique.• Intolérance aux hydrates de carbone et diabètesucré : ils sont secondaires à l’insulinorésistance engendrées par l’hypercortisolisme et sont associés àl’augmentation modérée du taux de triglycérides et decholestérol. • Plus rarement, la numération formule sanguine(NFS) peut objectiver une hyperleucocytose à poly-nucléaires neutrophiles avec relative lymphopénie.• Une alcalose hypokaliémiqueest rencontrée danscertaines causes d’hypercortisolisme (carcinomes sur-rénaux, sécrétion ectopique de corticotrophine) oulorsque celui-ci est intense.
4. Présentations cliniques particulièresIl est nécessaire de connaître diverses formes :• paucisymptomatiquescorrespondant à des formes de
début, à un hypercorticisme modéré ou intermittent.On évoque l’hypercortisolisme devant une ostéoporosene faisant pas la preuve de son étiologie, devant undiabète sucré de présentation atypique ainsi quedevant toute tumeur cortico-surrénalienne de découvertefortuite (« incidentalome ») ;
• enrichies, secondaires à certaines causes. Citons les formes virilisantes (aménorrhée, hirsutisme marqué, raucité de la voix, golfes fronto-temporaux,clitoridomégalie), témoins d’une hypersécrétion d’androgènes et orientant vers un carcinome surrénalien ;les formes cachectiques avec des signes de catabolismeprotidique intense et éventuellement mélanodermie(témoin d’une hypersécrétion de peptides cortico-tropes) évoquant une sécrétion ectopique de cortico-trophine.
• Conséquences morphologiquesdes effets cataboliqueset anti-anaboliques du cortisol : elles ont un intérêt diagnostique particulier :– l’amyotrophie prédomine au niveau des ceintures et
de la sangle abdominale. Elle peut être responsabled’une fatigabilité à la marche et lors de la montéed’escaliers voire confiner le patient au lit. Elle est par-fois visible au niveau de la face antérieure des cuisseset contraste avec l’adiposité facio-tronculaire et l’abdomen protubérant par relâchement de la sangleabdominale. Parfois plus discrète et il faudra larechercher par la palpation du quadriceps crural et lamanœuvre du tabouret ;
– l’atrophie cutanée et sous-cutanée est responsabled’une lenteur à la cicatrisation retrouvée à l’inter-rogatoire. La peau au niveau de la face dorsale des mains est amincie (en « feuille de papier à cigarette ») ;
– la fragilité cutanéo-capillaire est responsable d’ecchy-moses survenant au moindre choc (crête tibiale, dosde la main, avant-bras) ;
– des vergetures cutanées sont caractéristiques : larges,pourpres, orientées horizontalement sur les flancs et àla racine des membres ou à disposition radiaire dans larégion mammaire et périombilicale ;
– enfin, la peau du visage est érythrosique, avec destélangiectasies liées à l’atrophie de l’épiderme.
Endocrinologie - Métabolisme - Nutrition
1703L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Vergetures pourpres caractéristiques du syndrome deCushing.
3

Diagnostic biologique d’hypercortisolisme
Les anomalies biologiques dans l’hypercortisolismesont quantitatives et qualitatives.
1. Anomalies quantitatives Elles mettent en évidence la sécrétion excessive de cortisol. • La concentration du cortisol plasmatiquematinal,maximale chez les sujets sains, est peu discriminante.La cortisolémie vespérale (23-24 h), physiologiquementminimale, est par contre très discriminante mais nécessiteune hospitalisation. Le cortisol salivaire, étroitementcorrélé au cortisol libre plasmatique, peut être dosé dansdes prélèvements vespéraux réalisés en ambulatoire.• La mesure du cortisol libre urinaire (CLU) des 24 h(fraction du cortisol excrété sans être métabolisé) estl’examen de choix qui permet d’apprécier la quantité decortisol produit sur l’ensemble du nycthémère. Cet examen fondamental se heurte à 2 écueils principaux :– la difficulté d’un recueil adéquat des urines de 24 h. Il
est indispensable d’expliquer précisément au patientles aspects pratiques du recueil et d’associer systéma-tiquement le dosage de la créatininurie (constante permettant d’apprécier la qualité du recueil). Desrecueils urinaires sur une durée plus limitée (urines de la nuit par exemple) ont été proposés ;
– les fluctuations spontanées de la sécrétion dans l’hy-percortisolisme. Il est donc impératif de prélever lesurines pendant plusieurs jours.
2. Anomalies qualitatives• Rupture du rythme nycthéméral de sécrétion du cortisol : elle peut être mise en évidence en réalisant desprélèvements veineux diurnes et nocturnes. Cette inves-tigation ne se conçoit donc qu’en hospitalisation afind’éviter les augmentations de la cortisolémie liées austress de la ponction veineuse (pose d’un cathéter) ou àun sommeil perturbé.• Perte du freinage physiologique par les glucocorti-coïdes exogènes :elle est étudiée avec un corticoïde desynthèse, la dexaméthasone (qui n’est pas « reconnue »par les dosages du cortisol). Plusieurs modalités defreinage peuvent être proposées :– le freinage « minute » est la modalité la plus simple et
est réalisable en ambulatoire : la cortisolémie matinaleest déterminée après la prise de 1 mg de dexaméthasonela veille vers 23 h. L’hypercortisolisme doit être évoquélorsque la cortisolémie est supérieure à 100 nmol/L(3,6 mg /100 mL) ;
– le test de freinage « faible » (souvent appelé freinage« standard ») utilise 0,5 mg de dexaméthasone toutesles 6 h (2 mg/j) pendant 2 j et se juge sur la diminutiondu cortisol libre urinaire (normale < 10 mg/j) et (ou) de lacortisolémie (normale < 50 nmol/L ou 1,8 mg /100 mL).
Des faux positifs peuvent survenir lors de la prise d’inducteurs enzymatiques (rifampicine, phénobarbital,diphénylhydantoïne, etc.) qui accélèrent la clairance dela dexaméthasone, d’œstrogènes (contraceptifs oraux)qui augmentent la production de la protéine porteuse du
cortisol (transcortine) et entraînent une élévation artifi-cielle de la cortisolémie ; en cas d’affection intercurrente,de dépression ou de stress intenses. Le freinage «standard»est plus spécifique (moins de faux positifs) que le freinage« minute ». Ce dernier est donc plutôt utilisé en premièreintention pour le dépistage de l’hypercortisolisme et lefreinage standard pour sa confirmation.
3. Stratégie d’exploration paracliniqueElle dépend du degré de vraisemblance clinique de l’hypercortisolisme :– lorsque le diagnostic est hypothétique, on peut recourir
dans un premier temps à un dépistage ambulatoire. Enl’absence de prise médicamenteuse susceptible d’inter-férer, on réalise préférentiellement un test de freinage« minute » à la dexaméthasone. Dans les autres cas, onaura recours à la mesure du cortisol libre urinaire et dela créatininurie sur 24 h voire sur les urines de la nuitlorsque le patient n’est pas très discipliné ;
– lorsque l’hypercortisolisme est cliniquement très vrai-semblable, une hospitalisation pour la mesure du cortisol libre urinaire des 24 h pendant plusieurs joursconsécutifs. Un cortisol libre urinaire supérieur à 4 fois la normale affirme le diagnostic. Lorsqu’il estmoins franchement élevé, le test de freinage standardet la cortisolémie vespérale permettent de trancher.
Dans de rares cas, l’hypercorticisme alterne avec des périodes d’eucorticisme pouvant durer plusieurssemaines voire mois (Cushing intermittent). Il faut évoquercette possibilité lorsque la clinique contraste avec unebiologie normale ou lorsque des symptômes s’amendentspontanément puis réapparaissent. Dans ce cas, on s’aideradu dosage du cortisol salivaire en demandant aux patientsde recueillir quotidiennement quelques millilitres de saliveau lever et au coucher pendant plusieurs semaines.
4. Diagnostic différentiel• L’obésité, qui cliniquement ne s’accompagne pas designes cliniques cataboliques, n’entraîne généralementpas d’élévation du cortisol libre urinaire. • Le syndrome de Cushing iatrogénique par priseocculte de corticoïdes :la fonction corticotrope est freinée(cortisol plasmatique et cortisol libre urinaire effondrés)et contraste avec la symptomatologie clinique. Le profilpsychiatrique des patients et la recherche de corticoïdesde synthèse circulants par spectrographie de masseconfirment le diagnostic.• Les hypercortisolismes fonctionnels :les dépressionssévères activent l’axe corticotrope et entraînent une élévation modérée du cortisol libre urinaire et (ou) untest de freinage « minute » négatif. Cela peut être à l’origine de problèmes diagnostiques chez les sujetsdépressifs et (ou) éthyliques, lorsqu’il existe une sympto-matologie clinique compatible avec l’hypercortisolisme(pseudo-syndrome de Cushing). Le diagnostic reposesur un faisceau d’arguments cliniques (absence designes cataboliques) et biologiques parmi lesquels lacortisolémie à minuit est particulièrement discriminante.L’épreuve du temps avec la réévaluation clinique et
H Y P E R C O R T I S O L I S M E D E L ’ A D U L T E
1704 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

biologique des patients à distance, éventuellement aprèsmise en route d’un traitement psychotrope adapté ouaprès sevrage éthylique, permet souvent de trancher.
Diagnostic étiologique de l’hypercorticisme
La première étape de l’enquête étiologique consiste àétablir si l’hypercorticisme dépend ou non de la cortico-trophine.
1. Étude de l’ACTH-dépendance de l’hypercorticismeElle repose sur le dosage de la corticotrophine plasmatiquepar méthode immunoradiométrique (IRMA,immuno-radiometric assay), très sensible et permettant de mesurerde faibles concentrations de corticotrophine. Néanmoins,cette molécule est fragile et requiert des précautions particulières de recueil et de conservation des échantillonssanguins. Plusieurs prélèvements pour le dosage du cortisolet de corticotrophine seront réalisés en fin d’après-midiou la nuit en milieu spécialisé. Une concentration effondréede corticotrophine (< 5 pg/mL) signe l’hypercorticismeACTH-indépendant. Une concentration de corticotrophineconservée (> 15 pg/mL) signe l’hypercorticisme ACTH-dépendant.
2. Diagnostic du syndrome de Cushing ACTH-indépendantLa 1reétape est de réaliser une imagerie surrénalienne (scan-ner en coupes fines ou imagerie par résonance magnétique). • Dans la majorité des cas, une tumeur unilatérale estvisualisée et la surrénale controlatérale est atrophique(fig. 4). Il s’agit d’un adénome cortisolique ou d’un carci-nome primitif. Le diagnostic est souvent aisé en cas depetite lésion (< 5 cm) homogène évoquant un adénomeou de volumineuse tumeur hétérogène et métastatiqueévoquant un carcinome. Devant une volumineuse tumeursans essaimage décelable, la distinction entre adénomeet carcinome de bas grade de malignité est parfois délicateet repose sur un faisceau d’arguments (tableau II) parmilesquels l’imagerie occupe une place de choix. • De volumineuses masses polylobées bilatérales sontrarement mises en évidence :il s’agit d’une hyperplasiesurrénalienne macronodulaire.• Exceptionnellement, les surrénales apparaissentnormales ou discrètement hypertrophiques.Deux entitésrares peuvent être à l’origine du syndrome de Cushingde l’adulte jeune et se rencontrent généralement chezl’enfant et l’adolescent :– la dysplasie surrénalienne micronodulaire (ou pigmentaire)
survient dans un contexte sporadique ou familial ets’intègre parfois dans un syndrome de Carney (lentiginosecutanée, myxomes cardiaques et autres tumeurs…) ;
– un syndrome de Mac Cune-Albright (taches cutanéescafé au lait, dysplasie des os plats, endocrinopathiesvariées). L’hypercorticisme découle de l’activationspontanée du récepteur de la corticotrophine par unemutation intéressant les protéines membranaires Gcouplant le récepteur avec l’adénylate-cyclase.
1705L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Scanner surrénalien.A – Aspect fin du jambage des surrénales normales (flèches).B – Lésion hypodense de 3 cm gauche caractéristique d’unadénome (flèche).C – Volumineuse lésion (6 cm) hétérogène caractéristiqued’un carcinome surrénalien (étoile). Notez l’aspect fin de lasurrénale droite (flèche).VC veine cave inférieure ; RE : rein ; A : aorte ; RA : rate.
4
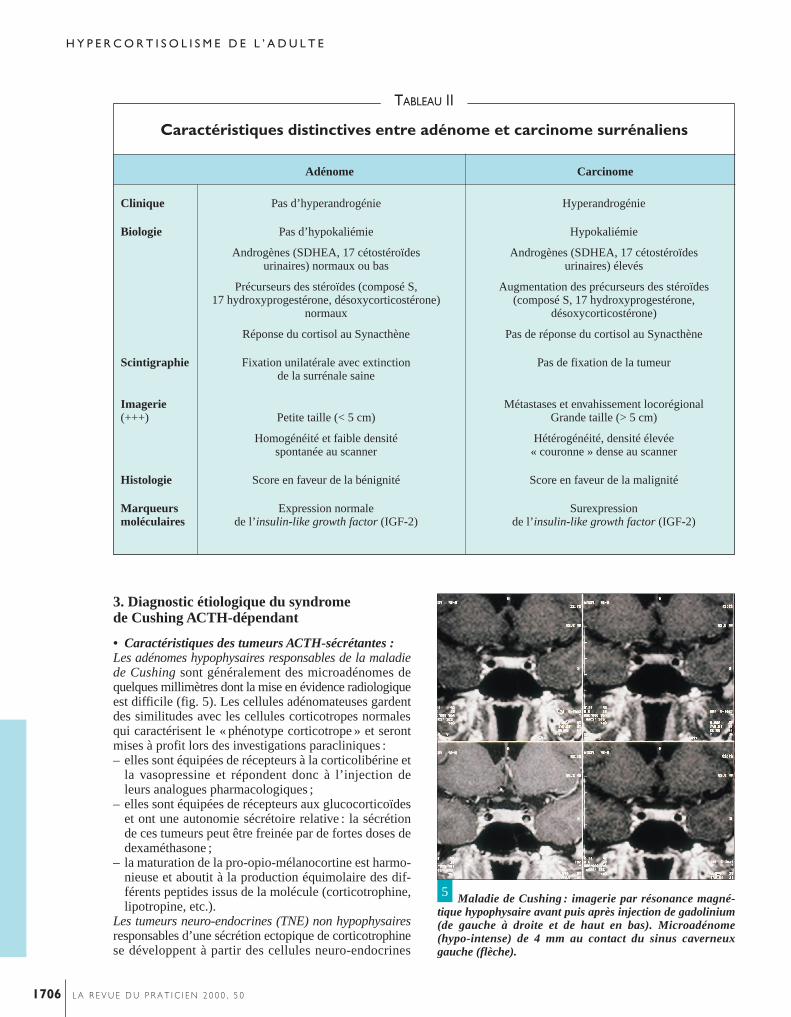
3. Diagnostic étiologique du syndrome de Cushing ACTH-dépendant
• Caractéristiques des tumeurs ACTH-sécrétantes :Les adénomes hypophysaires responsables de la maladiede Cushingsont généralement des microadénomes dequelques millimètres dont la mise en évidence radiologiqueest difficile (fig. 5). Les cellules adénomateuses gardentdes similitudes avec les cellules corticotropes normalesqui caractérisent le « phénotype corticotrope » et serontmises à profit lors des investigations paracliniques :– elles sont équipées de récepteurs à la corticolibérine et
la vasopressine et répondent donc à l’injection deleurs analogues pharmacologiques ;
– elles sont équipées de récepteurs aux glucocorticoïdeset ont une autonomie sécrétoire relative : la sécrétionde ces tumeurs peut être freinée par de fortes doses dedexaméthasone ;
– la maturation de la pro-opio-mélanocortine est harmo-nieuse et aboutit à la production équimolaire des dif-férents peptides issus de la molécule (corticotrophine,lipotropine, etc.).
Les tumeurs neuro-endocrines (TNE) non hypophysairesresponsables d’une sécrétion ectopique de corticotrophinese développent à partir des cellules neuro-endocrines
H Y P E R C O R T I S O L I S M E D E L ’ A D U L T E
1706 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Adénome Carcinome
Clinique Pas d’hyperandrogénie Hyperandrogénie
Biologie Pas d’hypokaliémie Hypokaliémie
Androgènes (SDHEA, 17 cétostéroïdes Androgènes (SDHEA, 17 cétostéroïdes urinaires) normaux ou bas urinaires) élevés
Précurseurs des stéroïdes (composé S, Augmentation des précurseurs des stéroïdes17 hydroxyprogestérone, désoxycorticostérone) (composé S, 17 hydroxyprogestérone,
normaux désoxycorticostérone)
Réponse du cortisol au Synacthène Pas de réponse du cortisol au Synacthène
Scintigraphie Fixation unilatérale avec extinction Pas de fixation de la tumeurde la surrénale saine
Imagerie Métastases et envahissement locorégional(+++) Petite taille (< 5 cm) Grande taille (> 5 cm)
Homogénéité et faible densité Hétérogénéité, densité élevéespontanée au scanner « couronne » dense au scanner
Histologie Score en faveur de la bénignité Score en faveur de la malignité
Marqueurs Expression normale Surexpressionmoléculaires de l’insulin-like growth factor (IGF-2) de l’insulin-like growth factor (IGF-2)
Caractéristiques distinctives entre adénome et carcinome surrénaliens
TABLEAU II
Maladie de Cushing : imagerie par résonance magné-tique hypophysaire avant puis après injection de gadolinium(de gauche à droite et de haut en bas). Microadénome (hypo-intense) de 4 mm au contact du sinus caverneuxgauche (flèche).
5
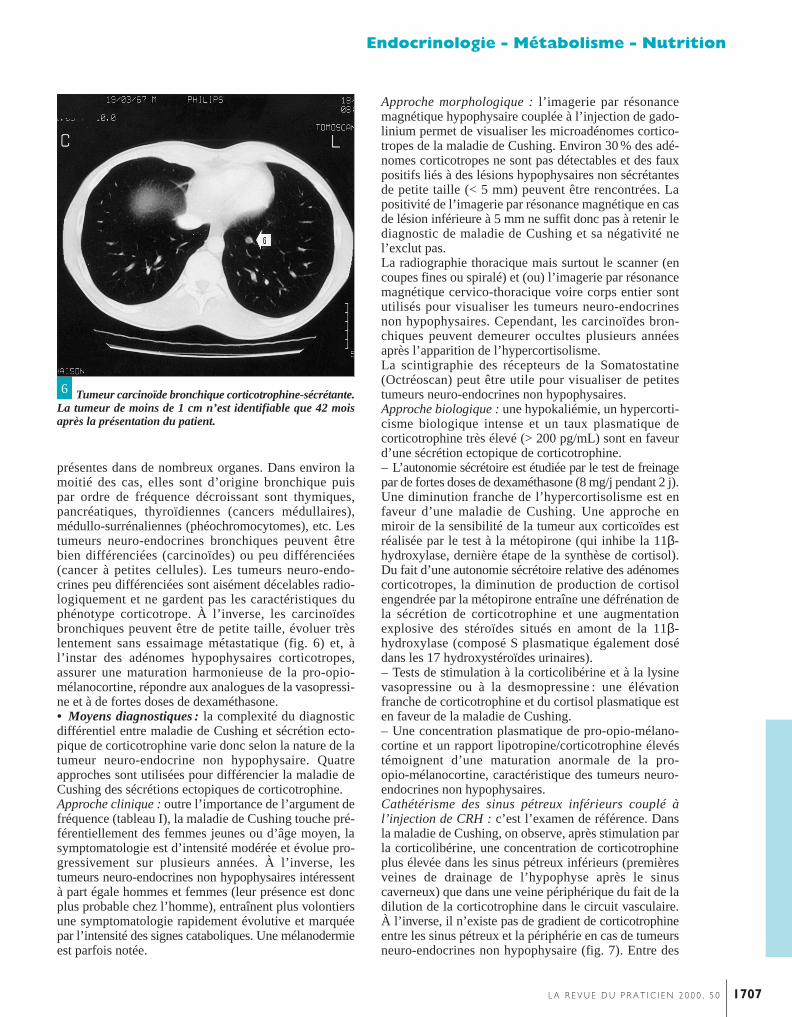
Approche morphologique :l’imagerie par résonancemagnétique hypophysaire couplée à l’injection de gado-linium permet de visualiser les microadénomes cortico-tropes de la maladie de Cushing. Environ 30 % des adé-nomes corticotropes ne sont pas détectables et des fauxpositifs liés à des lésions hypophysaires non sécrétantesde petite taille (< 5 mm) peuvent être rencontrées. Lapositivité de l’imagerie par résonance magnétique en casde lésion inférieure à 5 mm ne suffit donc pas à retenir lediagnostic de maladie de Cushing et sa négativité nel’exclut pas.La radiographie thoracique mais surtout le scanner (encoupes fines ou spiralé) et (ou) l’imagerie par résonancemagnétique cervico-thoracique voire corps entier sontutilisés pour visualiser les tumeurs neuro-endocrinesnon hypophysaires. Cependant, les carcinoïdes bron-chiques peuvent demeurer occultes plusieurs annéesaprès l’apparition de l’hypercortisolisme.La scintigraphie des récepteurs de la Somatostatine(Octréoscan) peut être utile pour visualiser de petitestumeurs neuro-endocrines non hypophysaires.Approche biologique :une hypokaliémie, un hypercorti-cisme biologique intense et un taux plasmatique de corticotrophine très élevé (> 200 pg/mL) sont en faveurd’une sécrétion ectopique de corticotrophine.– L’autonomie sécrétoire est étudiée par le test de freinagepar de fortes doses de dexaméthasone (8 mg/j pendant 2 j).Une diminution franche de l’hypercortisolisme est enfaveur d’une maladie de Cushing. Une approche enmiroir de la sensibilité de la tumeur aux corticoïdes estréalisée par le test à la métopirone (qui inhibe la 11β-hydroxylase, dernière étape de la synthèse de cortisol).Du fait d’une autonomie sécrétoire relative des adénomescorticotropes, la diminution de production de cortisolengendrée par la métopirone entraîne une défrénation dela sécrétion de corticotrophine et une augmentationexplosive des stéroïdes situés en amont de la 11β-hydroxylase (composé S plasmatique également dosédans les 17 hydroxystéroïdes urinaires). – Tests de stimulation à la corticolibérine et à la lysinevasopressine ou à la desmopressine : une élévationfranche de corticotrophine et du cortisol plasmatique esten faveur de la maladie de Cushing. – Une concentration plasmatique de pro-opio-mélano-cortine et un rapport lipotropine/corticotrophine élevéstémoignent d’une maturation anormale de la pro-opio-mélanocortine, caractéristique des tumeurs neuro-endocrines non hypophysaires.Cathétérisme des sinus pétreux inférieurs couplé à l’injection de CRH :c’est l’examen de référence. Dansla maladie de Cushing, on observe, après stimulation parla corticolibérine, une concentration de corticotrophineplus élevée dans les sinus pétreux inférieurs (premièresveines de drainage de l’hypophyse après le sinus caverneux) que dans une veine périphérique du fait de ladilution de la corticotrophine dans le circuit vasculaire.À l’inverse, il n’existe pas de gradient de corticotrophineentre les sinus pétreux et la périphérie en cas de tumeursneuro-endocrines non hypophysaire (fig. 7). Entre des
présentes dans de nombreux organes. Dans environ lamoitié des cas, elles sont d’origine bronchique puis par ordre de fréquence décroissant sont thymiques,pancréatiques, thyroïdiennes (cancers médullaires),médullo-surrénaliennes (phéochromocytomes), etc. Lestumeurs neuro-endocrines bronchiques peuvent êtrebien différenciées (carcinoïdes) ou peu différenciées(cancer à petites cellules). Les tumeurs neuro-endo-crines peu différenciées sont aisément décelables radio-logiquement et ne gardent pas les caractéristiques duphénotype corticotrope. À l’inverse, les carcinoïdesbronchiques peuvent être de petite taille, évoluer trèslentement sans essaimage métastatique (fig. 6) et, àl’instar des adénomes hypophysaires corticotropes,assurer une maturation harmonieuse de la pro-opio-mélanocortine, répondre aux analogues de la vasopressi-ne et à de fortes doses de dexaméthasone. • Moyens diagnostiques :la complexité du diagnosticdifférentiel entre maladie de Cushing et sécrétion ecto-pique de corticotrophine varie donc selon la nature de latumeur neuro-endocrine non hypophysaire. Quatreapproches sont utilisées pour différencier la maladie deCushing des sécrétions ectopiques de corticotrophine.Approche clinique :outre l’importance de l’argument defréquence (tableau I), la maladie de Cushing touche pré-férentiellement des femmes jeunes ou d’âge moyen, lasymptomatologie est d’intensité modérée et évolue pro-gressivement sur plusieurs années. À l’inverse, lestumeurs neuro-endocrines non hypophysaires intéressentà part égale hommes et femmes (leur présence est doncplus probable chez l’homme), entraînent plus volontiersune symptomatologie rapidement évolutive et marquéepar l’intensité des signes cataboliques. Une mélanodermieest parfois notée.
Endocrinologie - Métabolisme - Nutrition
1707L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Tumeur carcinoïde bronchique corticotrophine-sécrétante.La tumeur de moins de 1 cm n’est identifiable que 42 moisaprès la présentation du patient.
6

mains entraînées, cet examen invasif est réalisable dansla majorité des cas et n’est qu’exceptionnellement grevéd’effets indésirables.• Démarche diagnostique :le diagnostic étiologique del’hypercorticisme ACTH-dépendant repose sur un faisceaud’arguments mais il n’existe pas d’algorithme diagnostiquefaisant l’unanimité (le test à la Métopirone par exempleest fondamental pour certains mais abandonné pard’autres).Pour la plupart des équipes, l’investigation paracliniquecomporte initialement :– une imagerie par résonance magnétique hypophysaire
(du fait de l’argument de fréquence de la maladie deCushing) et une radiographie pulmonaire lue par unradiologue averti ;
– une kaliémie, un test de freinage fort à la dexamétha-sone voire un test à la corticolibérine.
Au terme de ce premier bilan, 3 situations sont possibles :– l’ensemble de ces arguments convergent vers une
maladie de Cushing, l’intervention chirurgicale etl’anatomopathologie confirmeront le diagnostic ;
– l’ensemble de ces arguments convergent vers unesécrétion ectopique de corticotrophine, la recherchebiologique (métanéphrines pour les phéochromocytomes,calcitonine pour les carcinomes médullaires thyroïdiens)et morphologique (scanner/imagerie par résonancemagnétique) d’une sécrétion ectopique de corticotrophines’impose. Elle est positive : l’intervention chirurgicaleet l’anatomopathologie confirmeront le diagnostic ;
– le diagnostic reste en suspens : la tumeur corticotro-phine-sécrétante n’est pas visualisée ou une lésionhypophysaire de très petite taille (< 5 mm) est vue mais
H Y P E R C O R T I S O L I S M E D E L ’ A D U L T E
1708 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• Le diagnostic positif d’hypercortisolisme repose exclusivement sur des données cliniqueset biologiques. L’imagerie médicale (surrénalienneet hypophysaire) n’a de place qu’une fois le diagnostic de syndrome de Cushing endogèneacquis.
• L’importance diagnostique des signes cliniquesde catabolisme protidique.
• Le test de freinage «minute» à la dexaméthasonedans le dépistage ambulatoire de l’hyper-cortisolisme.
• L’intérêt (et les difficultés pratiques) du cortisollibre urinaire des 24 h.
• Le dosage de la corticotrophine pour guider les investigations une fois le diagnostic d’hypercortisolisme acquis.
• La difficulté de mise en évidence morphologiquedes tumeurs corticotrophine-sécrétantes à l’origine de l’hypercortisolisme.
• Le diagnostic différentiel entre maladie de Cushing et sécrétion ectopique de corticotrophine qui repose sur un faisceaud’arguments biologiques et morphologiques.
Points Forts à retenir
Tabarin A. Diagnostic du syndrome de Cushing. Méd ThérEndocrino1999 ; 1 : 55-74.
POUR EN SAVOIR PLUS
n’emporte pas la conviction et (ou)s’accompagne d’arguments biologiquesdiscordants. Dans ce cas, le cathété-risme des sinus pétreux couplé à l’in-jection de corticolibérine est l’examende référence. Il peut être précédé ousuivi d’un Octréoscan pour tenter devisualiser une tumeur neuro-endocrinenon hypophysaire. Au terme de cesinvestigations, il est souvent possiblede poser le diagnostic de maladie deCushing ou de sécrétion ectopique decorticotrophine sans nécessairementvisualiser la tumeur. Dans ce cas,c’est l’exploration chirurgicale hypo-physaire (en cas de cathétérisme évo-quant une maladie de Cushing) ou lasurveillance morphologique rigoureuseaprès traitement symptomatique del’hypercortisolisme qui permettrontle diagnostic. ■
Cathétérisme des sinus pétreux (SPI) dans la maladie de Cushing. La concen-tration de corticotrophine est nettement plus élevée dans le sinus pétreux (tête deflèche) gauche (3 420 pg/mL) que dans une veine périphérique (85 pg/mL).Encadré : sinus caverneux ; étoile : loge pituitaire.
7
85

1353L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
B 336
Physiopathologie
1. Lipides totauxLes lipides circulants les plus importants comprennent lecholestérol (libre et estérifié), les triglycérides (TG), lesphospholipides et acides gras libres (AGL).
2. LipoprotéinesLes lipides ne sont pas hydrosolubles. Dans le plasma, ilscirculent associés à des protéines [appelées apolipopro-téines (Apo)] sous forme de complexes macromolécu-laires : les lipoprotéines.Il en existe 4 principales séparées par ultracentrifugationet par leur migration en électrophorèse. Ce sont les chylo-microns, les VLDL (very low density lipoprotein), les LDL(low density lipoprotein) et les HDL (high density lipo-protein) (tableau II).
3. Métabolisme des lipoprotéinesUne partie des triglycérides exogènes (alimentaires) esthydrolysée dans l’estomac. Dans le duodénum, les acides
HyperlipoprotéinémiesÉpidémiologie, étiologie, physiopathologie, diagnostic, traitement
Dr Rita CHADAREVIANDr Pascale COHEN-PRESBERGPr Éric BRUCKERTPr Gérard TURPINService d’encrinologie-métabolisme (Professeur Turpin), hôpital Pitié-Salpêtrière,75651 Paris cedex 13.
Les hyperlipoprotéinémies correspondent à uneaugmentation du taux des lipoprotéinescirculantes. Il s’agit d’un groupe de pathologieshétérogènes classées en 6 phénotypes en fonctiondu type de lipoprotéine(s) dont la concentrationplasmatique est augmentée.Elles peuvent être d’origine primitive (génétique)et/ou secondaire (autre pathologie, facteursnutritionnels...).Le diagnostic de l’anomalie lipidique repose surun dosage plasmatique réalisé après 12 heures de jeûne, en situation métabolique stable et préalablement à tout traitement.L’élévation du cholestérol total et du LDL-C est correlée aux complications cardiovasculairesischémiques. Inversement, leur diminutionthérapeutique entraîne une diminution de lamorbidité et de la mortalité cardiovasculaire. Ceci implique un dépistage précoce, un diagnosticprécis et un traitement adéquat.
Points Forts à comprendre gras à chaîne courte ou moyenne (moins de 12 atomes decarbone) sont absorbés directement via le système portalet le foie, puis passent dans la circulation générale.
Au niveau de l’entérocyte, les triglycérides peuvent alorsêtre resynthétisés à partir des acides gras libres et s’asso-cient avec les apolipoprotéines synthétisées dans lamuqueuse intestinale pour former les chylomicrons.
Les triglycérides des chylomicrons sont hydrolysés par lalipoprotéine lipase (LPL) en présence de son cofacteur,l’apolipoprotéine CII. Les acides gras sont captés essen-tiellement par les tissus adipeux et musculaires. Les chy-lomicrons appauvris en triglycérides sont appelés chylo-microns « remnants » et sont captés par un récepteurhépatique reconnaissant l’ApoE.
Les triglycérides sont progressivement hydrolysés par lalipoprotéine lipase. Les VLDL s’appauvrissent donc en tri-glycérides et s’enrichissent en cholestérol pour former lesIDL (intermediary density lipoprotein). Les IDL peuventêtre captées par le foie (récepteur des remnants) ou trans-formées en LDL toujours sous l’action de la lipoprotéinelipase. L’ApoE est codée par un système codominant à3 allèles sur un même locus (e2, e3, e4). Six phénotypessont donc possibles ; l’allèle e3 est le plus fréquent. L’ApoEse lie beaucoup moins au récepteur hépatique que l’ApoEou E4.
Environ 70 % du cholestérol est transporté par les LDL quipermettent sa distribution aux cellules de l’organisme. LesLDL se fixent sur le récepteur Apo B/E (décrit par Brownet Goldstein) présent sur les membranes cellulaires de l’or-ganisme. Au total, 70 % des récepteurs aux LDL sont situésau niveau du foie. La liaison des LDL à leur récepteur peutêtre alétérée et entraîner un allongement de leur demi-vieplasmatique. Les LDL subissent alors des modifications àtype d’oxydation, de glycosylation, etc. Elles passent dansl’espace sous-endothélial et sont captées par les macro-phages par le biais d’un récepteur « éboueur » appelé sca-venger. Les macrophages se transforment alors en cellulesspumeuses par enrichissement en cholestérol. Il s’agit d’unphénomène majeur dans le processus d’athérosclérose.
Les HDL sont essentiellement synthétisées par le foie etaccessoirement par l’intestin. Elles assurent le « transportreverse » du cholestérol des tissus périphériques vers le foiepermettant ainsi son élimination par les voies biliaires.

1354 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R L I P O P R O T É Ï N É M I E S
Étiologie et épidémiologiedes hyperlipoprotéinémies
Les hyperlipoprotéinémies correspondent à une augmen-tation des lipides circulants et de leurs apolipoprotéines(fig. 1). Plus de 2 % de la population générale est affectéepar ces pathologies, et un certain nombre de sujets ne sontpas dépistés. Les hyperlipoprotéinémies sont le plus sou-vent primaires mais une hyperlipidémie secondaire seratoujours recherchée.
Hyperlipoprotéinémies primaires
La détermination de la fraction lipidique augmentée per-met de typer l’hyperlipoprotéinémie. La classification inter-nationale de Fredrickson repose sur les données de l’élec-trophorèse des lipoprotéines. Six phénotypes sont décrits,correspondant à l’augmentation isolée ou non des chylo-microns, LDL, VLDL et IDL.
1. L’hypercholestérolémie pure (type IIa)
• Forme monogénique– Épidémiologie et aspects génétiques :. transmission autosomique dominante monogénique ;. trois gradations : la forme mineure où le cholestérol totalest situé entre 2,4 et 4 g/L ; la forme sévère où le cholesté-rol total est voisin de 4 g/L, qui correspond le plus souventà la forme hétérozygote de la xanthomatose tendineusehypercholestérolémique familiale (fréquence 1 cas/500) ;la forme majeure où le cholestérol total est situé entre 6 et12 g/L, qui correspond à la forme homozygote de la xan-thomatose tendineuse hypercholestérolémique familiale(fréquence 1 cas/1 million).– Les deux principaux pour mécanismes sont :. mutation du gène codant le récepteur des LDL (plusieursmutations ont été décrites) ; le récepteur peut être soit absent(de 0 à 50 % selon que la forme est homo- ou hétérozy-gote), soit anormal ;
CT cholestérol total
TG triglycérides
AGL acides gras libres
LDLC low density lipoprotein cholesterol
HDL C high density lipoprotein cholesterol
VLDL very low density lipoprotein
IDL intermediar density lipoprotein
Apo Apolipoprotéine
Migration Composition Principalesélectrophorèse moyenne en lipides apolipoprotéines
Chylomicrons absents à jeun TG 90 %-CT 3 % Apo A1-Apo B 48-Apo CII-Apo E
VDL pré-β TG 60 %-CT 20 % Apo B100-Apo CII-Apo E
LDL β TG 10 %-CT 45 % Apo B100
IDL absentes à jeun TG 50 %-CT 50 % Apo B100-Apo E
HDL α TG 5 %-CT 20 % Apo A1-Apo A2
Type de l’hyperlipidémie Athérome Pancréatite Fréquence de l’hyperlipidémie
Types I et V non oui rare
Type IV peut-être oui fréquente
Type IIb oui non fréquente
Type IIa oui non fréquente
Type III oui non rare
Liste des abréviations
TABLEAU I
Migration normale des lipoprotéines à l’électrophorèseet composition
TABLEAU II
Principales complications observéesen fonction du type d’hyperlipidémie
TABLEAU III

Classification physiopathologique des hyperlipoprotéïné-mies.
1
1355L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
. mutation de l’apoprotéine B100 (en général 3 500e acideaminé).• La forme polygénique, caractérisée par des antécédentsfamiliaux variables, en rapport avec des petites anomaliesgénétiques révélées ou aggravées par des erreurs diété-tiques. Une alimentation trop riche en cholestérol etgraisses saturées peut entraîner une déficience fonction-nelle des récepteurs.
2. Hyperlipidémie mixte ou combinée
• Hyperlipidémie de type IIb– Aspects génétiques : transmission autosomique domi-nante, mais le phénotype peut varier chez les apparentés,en fonction de l’âge, des conditions diététiques, du sexedes individus. La même anomalie génétique peut se pré-senter comme une hypercholestérolémie pure ou une hyper-triglycéridémie isolée.– Mécanisme : augmentation de la synthèse hépatique del’apo B100.• Hyperlipidémie de type III.– Épidémiologie : moins de 1 % des hyperlipoprotéinémies,1/10 000 sujets.– Aspects génétiques : transmission autosomique récessivele plus souvent.– Mécanismes : accumulation plasmatique de chylomicronsremnants et d’IDL par défaut de captation hépatique. Ledéveloppement de cette dyslipidémie nécessite deux condi-tions :. une anomalie de l’ApoE : 90 % des patients ont un phé-notype homozygote de l’ApoE E2/E2 de l’affinité de liai-son de l’ApoE2 à son récepteur est inférieure à 1 % parrapport à l’affinité de liaison des Apo E3 et E4 qui est de100 % ;. un facteur associé génétique ou environnemental (sur-poids par exemple) responsable d’une augmentation de lasynthèse des VLDL.
Ainsi, le phénotype E2/E2 seul ne suffit pas à entraîner unehyperlipidémie de type III. En effet, seulement 1 % despatients ayant le phénotype E2/E2 vont développer un typeIII.
3. Hypertriglycéridémie
• Hypertriglycéridémie pure de type IV– Hypertriglycéridémie alcoolo-glucido-pondéro-dépen-dante :. aspects génétiques : non identifiés,. mécanisme : la surcharge pondérale, la consommationd’alcool et l’insulinorésistance augmentent la synthèsehépatique en VLDL et une baisse de leur catabolisme.– Hypertriglycéridémie familiale :. épidémiologie : environ 10 % des patients hyperlipidé-miques,. aspects génétiques : transmission autosomique dominante.• Hypertriglycéridémie de type– Épidémiologie : exceptionnelle, 1 cas sur 1 million, moinsde 1 % des hyperlipoprotéinémies.– Aspects génétiques : mutation du gène de la lipoprotéinelipase, transmission autosomique récessive.– Mécanisme : défaut d’activité de la lipoprotéine lipaselié à une anomalie de l’enzyme ou de son activateur phy-siologique (ApoCII) entraînant une accumulation de chy-lomicrons.Cette hypertriglycéridémie est dépendante des graisses exo-gènes (alimentaires).• Hypertriglycéridémie de type V– Épidémiologie : également exceptionnelle, moins de 1 %des hyperlipoprotéinémies.– Mécanisme : double surcharge en chylomicrons et VLDLavec une double dépendance aux graisses et aux sucres. Leplus souvent, les sujets ayant une hyperlipidémie de typeIV avec une forte augmentation des VLDL peuvent tran-sitoirement avoir un phénotype V du fait de la saturationdes possibilités de lipolyse de la lipoprotéine lipase entraî-nant une accumulation de chylomicrons.
Hyperlipoprotéinémies secondaires
Elles peuvent être découvertes dans le bilan de l’affectioncausale dont elles peuvent modifier le pronostic, cardio-vasculaire en particulier.Elles doivent systématiquement être recherchées devanttoute hyperlipoprotéinémie en apparence primaire.
1. Diabète
• Diabète de type 1, insulino-dépendant (DID) : la carenceinsulinique majeure entraîne une diminution de l’activitéde la lipoprotéine lipase et donc une hypetriglycéridémie.Cependant, l’hypertriglycéridémie majeure reste rare, ycompris dans le cadre d’un diabète insulino-dépendantdécompensé.• Diabète de type II, non insulino-dépendant (DNID) :augmentation de la synthèse des VLDL par augmentationdu flux portal de substrats (acides gras libres et glucose).
Chylomicron
Type I
Type IVType II
Type II a
Type II b = type IV et II a Type V = type I et IV
Déficit en LPLou Apo CII
Hypersynthèse des VLDLouDiminution du catabolismedes VLDL Hyperproduction
de VLDLet
Défaut ApoE
Hypersynthèse des LDLouDiminution du catabolismedes LDL
Foie
VLDL
LDL
Classification physiopathologiquedes hyperlipoprotéinémies
iDL
Celluleperiphé-
rique
Tube digestifTube digestif

1356 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R L I P O P R O T É Ï N É M I E S
L’hyperinsulinisme pourrait potentialiser cette augmenta-tion de synthèse des VLDL. L’hypertriglycéridémie modé-rée est fréquente chez ces patients, surtout s’il existe unesurcharge pondérale.
2. HypothyroïdieDiminution de la synthèse mais surtout du catabolisme deslipoprotéines. Responsable d’une hyperlipoprotéinémie detype IIa ou IIb avec HDL-C élevé.
3. Syndrome néphrotiqueAugmentation de synthèse hépatique des VLDL secon-dairement à l’hypoalbuminémie. Responsable d’une hyper-lipidémie mixte.
4. Insuffisance rénaleAccumulation de remnants de VLDL, augmentation desVLDL, baisse des HDL et enrichissement en triglycéridesdes LDL. Responsable d’une hypertriglycéridémie.
5. CholestaseL’obstruction biliaire intra- ou extrahépatique, quelle quesoit l’étiologie, entraîne l’apparition de lipoprotéines spé-cifiques appelées lipoprotéines X.Elle est responsable d’une hypercholestérolémie.
6. SidaIl est fréquemment associé à une hypertriglycéridémie dephysiopathologie complexe.
7. Hyperlipidémies iatrogéniques– Œstrogènes par voie orale : parfois responsables d’unehypertriglycéridémie avec HDL-C élevé.– Progestatifs dérivés de la nor-testostérone (nor-stéroïdes) :responsables d’une diminution du HDL-C.– Corticoïdes : responsables d’une hypertriglycéridémieavec HDL-C élevé.– Rétinoïdes : responsables d’une hyperlipidémie combi-née.– Antihypertenseurs : certains diurétiques et les β-bloquantssans activité sympathomimétique intrinsèque sont respon-sables d’une hypertriglycéridémie modérée avec souventdiminution du HDL-C.– Autres : antiprotéases, ciclosporine…
Diagnostic
Démarche diagnostiqueLors d’une consultation pour hyperlipidémie, elle doit com-prendre systématiquement :• un interrogatoire précisele mode de découverte (lorsd’un examen systématique ou lors d’une complication car-diovasculaire), les taux des paramètres lipidiques, lesrégimes prescrits, les traitements antérieurement reçus ainsique leur tolérance et leur efficacité éventuelle, les facteursde risque cardiovasculaires associés (obésité, tabac, hyper-tension artérielle, diabète sucré) et l’existence d’antécé-
dents ou de signes actuels d’angor, infarctus du myocarde,claudication intermittente, accident vasculaire cérébral ;• une analyse des antécédents familiaux d’hyperlipidé-mie et d’affections cardiovasculaires ;• un examen clinique pour évaluer le retentissement vas-culaire de l’hyperlipidémie et rechercher des dépôts lipi-diques extravasculaires ;• l’analyse critique du bilan biologique qui doit être pra-tiqué après 12 heures de jeûne. Les Références médicalesopposables (RMO) recommandent lors du premier bilanun dosage du cholestérol total (CT) et des triglycérides ;chez un homme de moins de 50 ans et chez une femme nonménopausée, asymptomatique, il n’est pas recommandé derecontrôler ces dosages avant un délai de 5 ans. En casd’anomalie d’un ou des deux paramètres, on complètera lebilan par l’examen de l’aspect du sérum à jeun et le dosagedu HDL-C (par précipitation). Le LDL-C sera calculé selonla formule de Friedewald (les paramètres sont en g/L) : LDL= CT – HDL – TG/5 (formule valable uniquement si lestriglycérides sont inférieurs à 4 g/L et en l’absence d’IDL).Le diagnostic précis de l’hyperlipidémie repose sur undosage fiable à confirmer par un deuxième bilan. L’élec-trophorèse des lipoprotéines est parfois utile ;• la recherche d’une cause secondaire ou iatrogéniqued’hyperlipidémie par des examens clinique et paraclinique.
Hypercholestérolémie pure (type IIa)1. Présentation cliniqueLe tableau clinique dépend essentiellement du taux de cho-lestérolémie :– l’arc cornéen est un arc blanchâtre complet ou non, situéà la périphérie de la cornée, évocateur d’hypercholestéro-lémie modérée surtout chez les sujets avant 50 ans ;– le xanthélasma est un dépôt jaunâtre à la partie internedes paupières supérieures et inférieures ;– les xanthomes tendineux sont des épaississements ou desnodules visibles et (ou) palpables (lorsque le cholestéroltotal est supérieur à 4 g/L) sur les tendons extenseurs desdoigts des mains et les tendons d’Achille (pouvant entraî-ner une tendinite du tendon d’Achille) ; ils apparaissentvers l’âge de 20-30 ans ;– les xanthomes cutanés plans ou tubéreux peuvent appa-raître dans l’enfance et sont spécifiques de la forme homo-zygote.
2. Signes biologiquesAprès 12 heures de jeûne, le sérum est clair ; il y a une aug-mentation du cholestérol total par augmentation du LDL-C.Les triglycérides sont normaux. Le HDL-C est normal oubas.
3. PronosticL’augmentation du LDL-C est associée de façon indiscu-table à un risque athérogène accru, en particulier au niveaucoronaire. Toutes les gradations de type IIa sont concer-nées. L’augmentation du niveau de risque est corrélée autaux de cholestérolémie : plus les taux de cholestérol totalet LDL-C sont élevés, plus les accidents coronaires seront

1357L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
de survenue précoce. Dans la forme homozygote, en l’ab-sence de traitement adéquat, le décès par infarctus du myo-carde survient habituellement avant l’âge de 20 ans.
4. Hypercholestérolémies secondairesIl faut rechercher devant une hypercholestérolémie : unehypothyroïdie, une cholestase et un syndrome néphrotique.
Hyperlipidémie mixte (type IIb ou III)Il s’agit d’une élévation du cholestérol total et des trigly-cérides. Le taux de HDL-C est le plus souvent abaissé (dufait de l’hypertriglycéridémie).
1. Présentation clinique• L’hyperlipidémie de type III (dysbêtalipoprotéinémie)est dépistée après l’âge de 20 ans et est évoquée clinique-ment par la présence de dépôts jaune orangé des plis pal-maires et de xanthomes tubéreux. Cependant, leur absencen’exclut pas le diagnostic. Les atteintes vasculaires sontfréquentes et souvent multiples (coronaires, carotides,fémorales).• L’hyperlipidémie de type IIb est athérogène et peut êtreévoquée devant un arc cornéen ou un xanthélasma. Ellecorrespond dans certains cas à l’hyperlipidémie familialecombinée, hautement athérogène où les différents membresd’une même famille présentent des phénotypes différents.
2. Signes biologiquesLe sérum est opalescent à jeun.• Dans l’hyperlipidémie de type III : l’élévation du cho-lestérol total et des triglycérides se fait dans des propor-tions similaires, entre 3 et 5 g/L. Dans ce cas, l’électro-phorèse des lipoprotéines est très utile car elle peut montrerune bande anormale caractéristique : la broad bêta lipo-protéine qui correspond aux IDL. L’étude du péhnotype del’ApoE peut contribuer au diagnostic (classiquement, phé-notype E2/E2).• Dans l’hyperlipidémie de type IIb : élévation du choles-térol total et LDL-C et des tryglicérides dans des propor-tions variables d’un jour à l’autre, l’apoprotéine B est aug-mentée. L’électrophorèse des lipoprotéines montre uneélévation des β-lipoprotéines (LDL) et des préβ-lipopro-téines (VLDL).
Hypertriglycéridémie pure (type IV)1. Présentation cliniqueL’hypertriglycéridémie est le plus souvent asymptoma-tique. Elle peut être associée à une obésité androïde, untrouble du métabolisme glucidique, et une hypertensionartérielle, l’ensemble de ces anomalies s’intégrant dans lesyndrome X de Reaven. Sous l’influence d’une consom-mation importante d’alcool et/ou d’une décompensationd’un diabète préalablement connu ou pas et/ou d’un écartde régime, l’hyperlipidémie peut s’amplifier en type IVmajeur (triglycérides supérieurs à 10 g/L). On peut alorsobserver une hépatomégalie par stéatose, une xanthoma-tose éruptive (papules centrées d’une lésion jaunâtre), uneasthénie et une somnolence post-prandiale. La complica-
tion majeure, dans ce cas, est la pancréatite aiguë qui peutd’ailleurs constituer le mode de révélation de cette dysli-pidémie.
2. Signes biologiquesLe sérum est trouble à jeun. Les triglycérides sont élevés(supérieurs à 1,5 g/L) mais extrêmement variables d’un jourà l’autre. Le cholestérol total est normal ou élevé (par élé-vation du cholestérol contenu dans les VLDL), le HDL-Cest abaissé. Sous traitement, la décroissance des triglycé-rides est plus rapide que celle du cholestérol total. Celaexplique que si le bilan est réalisé quelques jours aprèsl’épisode aigu, on peut observer un taux de cholestérol totalsupérieur à celui des triglycérides.
3. Hypertriglycéridémies secondairesDevant une hypertriglycéridémie, il faut doser la glycémieà jeun pour rechercher un diabète sucré. Il faut rechercheraussi une insuffisance rénale chronique, une prise d’alcoolou d’œstrogènes par voie orale (dans ces deux derniers cas,le HDL-C est normal ou élevé malgré l’élévation des tri-glycérides).
Hyperchylomicronémie (type I ou V)1. Présentation cliniqueL’hyperlipidémie de type I est révélée dans l’enfance pardes épisodes itératifs de douleurs abdominales, ou de pan-créatites aiguës (si triglycérides supérieurs à 10 g/L), oupar une canthomatose éruptive. Elle doit être prise encharge dans un service spécialisé.L’hyperlipidémie detype V associe les anomalies du type I et du type IV.
2. Signes biologiquesLe sérum à jeun est lactescent. Après 24 h de décantationà 4 °C, le sérum est clair avec un surnageant crémeux dansle type I ou opalescent avec un surnageant crémeux dansle type V. Le bilan confirme l’hypertriglycéridémie parfoistrès élevée avec un cholestérol total normal. L’électropho-rèse des lipoprotéines retrouve une hyperchylomicronémieisolée (type I) ou associée à une augmentation des pré-β-lipoprotéines (type V).
Traitement
Preuves apportées par les études de préventionLes études de prévention, qu’elles soient primaires ousecondaires, ont démontré clairement l’efficacité du trai-tement hypolipémiant sur la baisse de la morbidité et de lamortalité cardiovasculaires via la baisse du cholestérol totalet LDL-C. Les études récentes effectuées avec les statinesont démontré en plus une diminution significative de lamortalité globale et que le bénéfice thérapeutique peut êtretrès précoce, avant même le 6e mois qui suit l’intaurationdu traitement. C’est à partir de ces études qu’ont été défi-nies les valeurs seuils d’instauration du traitement diété-tique ou médicamenteux et la valeur cible de LDL-C à

1358 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R L I P O P R O T É Ï N É M I E S
atteindre. Les valeurs à partir desquelles un traitement serainstauré ainsi que les valeurs cibles souhaitables dépendentdu type de prévention (primaire ou secondaire), de l’âgedu patient et des facteurs de risque associés.
1. Régime hypocholestérolémiantIl est conseillé de :– diminuer les apports en cholestérol (cholestérol alimen-taire, apports inférieurs à 300 mg/j) ; ces conseils se tra-duisent en pratique pour le patient par une diminution dela consommation de viande rouge, charcuterie, abats, fro-mage, œufs, au profit du maintien ou de l’augmentation dela consommation de poisson (surtout gras), de viandeblanche, volaille, légumes, légumineuse et huiles végé-tales ;– diminuer la consommation quotidienne en lipides dontl’apport ne doit pas dépasser 30-35 % de la ration caloriquequotidienne ;– surtout diminuer les apports en graisses saturées et lesremplacer par des graisses insaturées (produites à partir decertains végétaux : huile ou margarine au tournesol, huilede maïs, huile de colza, huile d’olive).
2. Régime hypotriglycéridémiantCompte tenu du caractère athérogène et thrombogène desgraisses saturées, il est important chez ces patients d’enlimiter l’apport.Il faut plus particulièrement axer les conseils en fonctionde la dépendance alimentaire de l’hypertriglycéridémie :– suppression de l’alcool dans la forme alcoolo-dépen-dante ;– limitation draconienne des hydrates de carbone à indexglycémique élevé dans la forme glucido-dépendante (fruits,boissons sucrées, glaces, confiserie, etc.) ;– limitation calorique en cas de surpoids.
Traitements médicamenteuxLes principaux modes d’action des hypolipédimants sontrésumés dans la figure 2.
1. Résines échangeuses d’ionsLa colestyramine (Questran) à la dose de 4 à 24 g/j entraînela diminution du LDL-C. Les inconvénients sont les effetssecondaires qui sont atténués par l’augmentation progres-sive de la posologie (constipation, ballonnements, nausées,changement du goût) et le risque d’interférence médica-menteuse par diminution de l’absorption des médicamentspris simultanément. Le Questran doit être pris à distancedes autres médicaments. Il faut en particulier que les digi-taliques et les antivitamines K soient pris au minimum1 h 30 à 2 h avant le Questran. Une élévation modérée destriglycérides est parfois notée.
2. Statines ou inhibiteurs de l’HMG Co-A(hydroxy-3-méthylglutaryl coenzyme A)réductaseLa simvastatine (Zocor et Lodalès à la dose de 5 à 40 mg/j),la pravastatine (Élisor ou Vasten à la dose de 10 à 40 mg/j),la fluvastatine (Fractal et lescol à la dose de 20 à 80 mg/j),
l’atorvastatine (Tahor 10 à 80 mg/j). Ces médicaments,seuls ou en combinaison avec les résines, diminuent signi-ficativement le taux de cholestérol total et cela de façondose-dépendante (le LDL-C diminue d’environ 30-50 %)et diminuent modérément les triglycérides. Ces produitssont efficaces en prise unique et doivent être pris au repasdu soir. Ils sont contre-indiqués chez l’enfant, la femmeenceinte ou susceptible de l’être, et en cas d’insuffisancehépatique ou rénale sévère. La tolérance clinique est bonne.La surveillance du traitement comprend un dosage sys-tématique des transaminases (ASAT et ALAT) et des CPK(créatine phosphokinases) tous les 4 à 6 mois et plus pré-cocement en cas de douleur musculaire. Le traitement doitêtre interrompu si les transaminases sont à plus de trois foisla normale ou si les CPK sont à plus de cinq fois la normale.
3. Dérivés des fibrates
Ils permettent l’abaissement des VLDL et des triglycéridesd’environ 50 %, la modification favorable de la taille et dela structure des LDL, la réduction de leur taux d’environ10 à 15 % et, enfin, l’augmentation du HDL-C d’environ10 %. Tous peuvent être donnés en une seule prise, le soir :ciprofibrate (Lipanor à la dose de 100 mg/j), bézafibrate(Befizal à la dose de 400 à 600 mg/j) et fénofibrate (Lipan-thyl 67 ou 200 micronisé ou Secalip 300). En pratique cou-rante, ce sont des médicaments bien tolérés ; les effetssecondaires sont rares : impuissance sexuelle de physiopa-thologie inconnue, élévation des transaminases et des CPKet peut-être lithiase biliaire. Leur liaison aux protéinessériques est importante, d’où risque d’interaction médica-menteuse, en particulier avec les antivitamines K. Ils sontcontre-indiqués en cas d’insuffisance hépatique ou rénale,chez l’enfant et la femme enceinte ou susceptible de l’être.
Principaux modes d’action des hypolipémiants et indica-tions en fonction du type d’hyperlipoprotéïnémie.
2
Fibrates
Type II aII b
IIIIVV
Type II a
Type II aII b
VLDL
Principaux modes d’action des hypolipémiantset indications en fonction du type d’hyperlipoprotéinémie
FOIE
ApoB100
HMG Co-AReductase
Acidesbiliaires
Bile
AGL
Cholestérol
LDL
Statines
Résines
-
+
-
+

1359L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Endocrinologie - Métabolisme - Nutrition
Scandanivian Simvastatin Survival Study Group. Randomised trialof cholesterol lowering in 4 444 patients with coronary heartdisease. Lancet 1994 ; 344 : 1383-9.Shepherd J, Cobbe SM, Ford I et al. West of Scotland coronaryprevention study group. Prevention of coronary heart diseasewith pravastatin in men with hypercholesterolemia. N Engl J Med1995 ; 333 : 1301-7.Turpin G. Pourquoi, quand, comment traiter les dyslipoprotéi-némies ? Édition 1997.
POUR EN SAVOIR PLUS
POUR APPROFONDIR
– L’hypercholestérolémie est un facteur de risque cardiovasculaire par-faitement établi, indépendant de tous les autres facteurs. Cependant, ilpersiste des controverses sur le caractère indépendant ou pas de l’hyper-triglycéridémie, essentiellement du fait de la très forte interaction (tri-glycérides-HDL C. Il s’agirait plutôt d’un marqueur de risque.– La lipoprotéine (a) : Lp(a) est une lipoprotéine dont la partie protéiqueest constituée d’ApoB100 et d’Apo(a). Le rôle athérogène est médié parl’ApoB qui permet l’accumulation de la Lp(a) dans la paroi artérielle. Lerôle thrombogène est lié à l’analogie de structure de l’Apo(a) et du plas-minogène entraînant une inhibition de la fibrinolyse. Son taux est déter-miné génétiquement et ne varie pratiquement pas au cours de la vie (onnote une augmentation chez la femme après la ménopause). De plus, iln’est sensible ni à la diététique ni aux traitements médicamenteux.
4. Huiles de poissonÀ base d’acides gras oméga 3 polyinsaturés (Maxepa 4 à 6capsules par jour) elles ont un effet hypotriglycéridémiant àfortes doses et ont des propriétés antiagrégeantes. Elles per-mettent une baisse de la triglycéridémie d’environ 25%, pro-bablement via une inhibition de la synthèse, cet effet étantdose-dépendant. Elles ont également un effet favorable surla coagulation et l’agrégation plaquettaire. Des événementsindésirables bénins peuvent survenir, à type d’éructations,flatulence, vomissements, troubles du transit.
5. LDL aphérèsesElles permettent l’épuration des LDL ; il s’agit d’un trai-tement extrêmement efficace mais très lourd et coûteux.
Indications thérapeutiquesL’instauration d’un traitement diététique ou médicamenteuxest réalisée après le diagnostic précis de l’anomalie lipidiquequi nécessite 2 à 3 dosages à quelques semaines d’intervalle.Les indications doivent être bien étudiées car une fois le trai-tement prescrit, il doit être poursuivi à vie. La décision demettre en place un traitement médicamenteux est fondéeessentiellement sur le taux de LDL-C. L’efficacité et la tolé-rance du traitement seront surveillées tous les 6 mois envi-ron. Il faut, bien entendu, veiller à la lutte contre les autresfacteurs de risque, en particulier le tabagisme, et encouragerle patient à pratiquer une activité physique régulière.
1. Type IIaLa diététique est la première étape et doit être poursuiviemême si un traitement médicamenteux est instauré. Le pre-mier médicament qui peut être utilisé est une résine ou unestatine.Les fibrates sont intéressants, même dans les hypercho-lestérolémies pures en raison de leur coût moindre.L’association résine-statine est synergique. L’associationstatines-fibrates est classiquement déconseillée en raisondu risque potentiellement accru de myolyse.Enfin, les LDL aphérèses sont à réserver aux patientsatteints d’hyperlipémie de type IIa homozygote.
2. Type IVLa diététique est primordiale et suffit le plus souvent à nor-maliser l’anomalie lipidique. La valeur cible de la trigly-céridémie est inférieure à 2 g/L ou même à 1,50 g/L en pri-vilégiant la diététique. Si celle-ci ne permet pas d’atteindrela valeur cible préconisée, il faut alors évaluer soigneuse-ment le risque global de l’hyperlipidémie (hypoHDLémieinférieure à 0,35 g/L ou diabète associés) et si nécessaireajouter un médicament [fibrates et (ou) des huiles de pois-son]. Lors d’un passage en type IV majeur, le patient est àrisque de pancréatite. La diétérique stricte et le sevrage del’alcool sont indispensables. L’hospitalisation est justifiéeen cas de douleur abdominales. En cas de diabète décom-pensé associé, l’insulinothérapie entraîne une diminutionrapide des triglycérides.
3. Types IIb ou IIISchématiquement, les patients doivent bénéficier desmesures mentionnées dans les 2 paragraphes précédents
pour le choix du médicament et la diététique. Le traitementde choix est représenté par les dérivés des fibrates surtoutsi les triglycérides sont très élevés avec un HDL-C bas.Si le dépassement des triglycérides est modéré, les statinespeuvent être utilisées isolément.
4. Type IIl nécessite une prise en charge spécialisée ; le régime pres-crit est sévère, très pauvre en graisses (moins de 10 % dela ration quotidienne) afin de limiter l’hypertriglycéridé-mie et le risque de pancréatite aiguë secondaire. Ce régimenécessite des compléments lipidiques représentés par lesacides gras à chaîne moyenne et une supplémentation envitamines liposolubles. ■
• Augmentation du cholestérol total et du LDL-cholestérol : facteurs de risque cardiovasculaireayant le statut de facteur causal. La diminutionthérapeutique de ces paramètres est bénéfique surla morbidité et la mortalité cardiovasculaires.• Penser à éliminer une hyperlipidémie secondaire.• Plusieurs dosages nécessaires sans traitementpour bien typer l’hyperlipoprotéinémie.• Distinguer les hypercholestérolémies des hypertriglycéridémies car traitement et complications différents : athérosclérose et pancréatite respectivement.• La valeur seuil pour l’instauration du traitementmédicamenteux est fonction du niveau de risquecardiovasculaire individuel.• La diététique est fondamentale et constitue la première étape de la prise en chargethérapeutique.
Points Forts à retenir

HyperthyroïdieÉtiologie, physiopathologie, diagnostic, évolution, traitement
PR JEAN-LOUIS WEMEAUClinique endocrinologique Marc-Linquette, CHU, 59037 Lille cedex
Étiopathogénie
Les manifestations de la thyrotoxicose s’expliquent parl’effet des hormones thyroïdiennes qui augmentent laproduction énergétique et la consommation en oxygène,et accélèrent les différents métabolismes.
Maladie de Basedow
Elle est définie comme un goitre diffus d’apparitionrécente, responsable d’hyperthyroïdie, et habituellementassociée à une ophtalmopathie œdémateuse.Elle résulte de la production par les lymphocytes intra-thyroïdiens d’immunoglobulines thyréostimulantes.Elle survient sur un terrain génétiquement prédisposé(particulièrement dans les groupes tissulaires HLA A1B8 et DR3 chez les caucasiens) où s’expriment à la sur-face des thyrocytes les antigènes tissulaires de classe II.L’ophtalmopathie procède également d’un mécanismeauto-immun mais sa nature est encore mal précisée.On rapproche de la maladie de Basedow les goitresbasedowifiés qui surviennent sur un goitre préexistant.
Nodule toxique
Le nodule toxique de Plummer est une hypertrophielocalisée, autonome, hyperfonctionnelle, extinctive vis-à-vis du reste du parenchyme thyroïdien. La présenta-tion est donc celle d’un nodule solitaire avec thyrotoxi-cose. La majorité de ces formations s’explique parl’apparition de mutations somatiques du récepteur de laTSH (voir: pour approfondir 1).On en rapproche le goitre multinodulaire hétérogènesecondairement toxique où s’individualisent au seind’un goitre une ou plusieurs formations nodulaires.Certaines d’entre elles s’avèrent hyperfonctionnelles etresponsables d’hyperthyroïdie, d’autres sont isofixantesou hypofixantes vis-à-vis des isotopes radioactifs.
Hyperthyroïdies induites par l’iodesur glande saine ou pathologique
Le mécanisme des hyperthyroïdies induites par l’iodeest imparfaitement compris. Elles s’observent surtoutdans les régions de carence iodée relative qui favorise laconstitution au sein du parenchyme thyroïdien de foyersd’hyperplasie dont l’hyperactivité se révèle à la faveurde la disponibilité accrue en iode. On évoque aussi desphénomènes de thyroïdite iodée conduisant à la destruc-tion des structures vésiculaires et à la libération dumatériel hormonal. Très rarement , elles s’expliquentpar l’induction d’une auto-immunité spécifique (JodBasedow).
Hyperthyroïdie des thyroïdites
Des inflammations du parenchyme thyroïdien sont sus-ceptibles d’altérer la structure vésiculaire, de libérer lecontenu intrathyroïdien en iode, thyroglobuline et enhormones, ce qui détermine une phase thyrotoxiqueordinairement transitoire, spontanément résolutive enquelques semaines. Une phase d’hypothyroïdie peut lui suc-céder traduisant l’inhibition fonctionnelle des cellules vési-culaires.On peut observer cette évolution diphasique surtout dans lesthyroïdites subaiguës de De Quervain, ( réactionnelles à desaffections virales), dans les thyroïdites dites silencieuses ouindolores (thyroïdites lymphocytaires d’origine auto-immu-ne que l’on observe surtout dans les semaines ou les moissuivant un accouchement).
Endocrinologie-Métabolisme-NutritionB 337
1377L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
• L’hyperthyroïdie désigne l’hyperfonctionnementthyroïdien. Elle accroît la productiondes hormones thyroïdiennes dont la conséquenceest la thyrotoxicose.• Cette situation est fréquente, affectant 1 à 2 %de la population et prédomine largement dans lesexe féminin (sex ratio 1/8). Elle est responsabled’un inconfort général et sa méconnaissanceexpose à des complications, notammentcardiaques et osseuses. Elle est liée à de trèsnombreuses affections dont la reconnaissanceconditionne le pronostic et les choixthérapeutiques.• Il n’y a pas de traitement idéal deshyperthyroïdies : antithyroïdiens de synthèse,chirurgie, dose thérapeutique d’iode 131 ontchacun un certain nombre d’avantages etd’inconvénients. Il est important d’en connaîtreles règles de prescription et de surveillance.
Points Forts à comprendre

Ces situations sont à distinguer des hyperthyroïdiesconstatées au cours des thyroïdites de Hashimoto(hashitoxicoses) occasionnées, comme les maladies deBasedow, par la production d’immunoglobulines thy-réostimulantes.
Hyperthyroïdie non auto-immune familiale
Rarement des formes familiales d’hyperthyroïdie résultentde goitres diffus toxiques sans atteinte oculaire, et sansanticorps stimulants, sans infiltrat lymphoplasmocytairethyroïdien. Elles sont liées à des mutations germinalesdu récepteur de la TSH (voir:pour approfondir 1).
Hyperthyroïdie de la grossesse et vomis-sements gravidiques
Des signes thyrotoxiques peuvent être constatés aucours du premier trimestre de la grossesse du fait despropriétés thyréostimulantes de l’hCG placentaire. Cettesituation revêt une particulière intensité au cours desvomissements incoercibles de la grossesse.
Hyperthyroïdies centralesDans ces situations très rares, l’hyperthyroïdie résulte d’unsyndrome de sécrétion inappropriée de TSH, avec desconcentrations de TSH accrues ou paradoxalement nor-males coïncidant avec l’hyperhormonémie thyroïdienne.Elles sont liées soit à un adénome hypophysaire développéaux dépens des cellules thyréotropes de l’antéhypophysequi produisent préférentiellement la chaîne a-spécifique,soit à un défaut de rétrocontrôle des hormones thyroï-diennes au niveau de l’antéhypophyse qui conduisent à laproduction excessive d’une TSH normale.
Hyperthyroïdies liées aux tumeursLes cas rapportés d’hyperthyroïdies au cours de cancersviscéraux relèvent d’associations.
En revanche existent authentiquement des hyperthyroï-dies résultant de môles hydatiformes ou de choriocarci-nomes, car les tumeurs placentaires produisent l’hCG enexcès.On a décrit aussi des hyperthyroïdies relevant de la pré-sence au sein des ovaires d’un tissu thyroïdien différen-cié (goitre ovarien toxique).
Thyrotoxicoses factices
Elles ne sont pas au sens propre responsables d’unehyperthyroïdie, car l’état thyrotoxique lié à la prise sou-vent clandestine d’hormones thyroïdiennes, met aurepos le parenchyme thyroïdien qui est atrophique etnon fonctionnel.
Diagnostic
Diagnostic positif
Évoquée cliniquement, la thyrotoxicose sera confirméepar les examens biologiques.
1. Signes cliniques• Dans la forme complète, le diagnostic n’est pas dou-teux : amaigrissement rapide contrastant avec un appétitconservé pouvant conduire à une véritable cachexie ;asthénie musculaire avec amyotrophie, notamment desceintures, responsable dans les formes sévères d’un han-dicap moteur (signe du tabouret de Froment) et de modi-fications du timbre de la voix (assourdie et voilée) ;signes de dysrégulation thermique : thermophobie,hypersudation, élévation thermique discrète, polydipsie.L’aspect de la « main basedowienne » chaude et moite,avec chaleur irradiée, est très évocateur ; éréthisme car-diovasculaire : tachycardie permanente, avec poulsvibrant, palpitations, dyspnée d’effort, bruits du cœurrapides et éclatants avec possibilité de souffle et de ryth-
1378 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R T H Y R O Ï D I E
- Maladie de Basedow - Hyperthyroïdies induites par l’iode
- Nodule toxique - Thyroïdite subaigue- Goitres secondairement toxiques - Thyroïdite silencieuse- Hashitoxicose - Thyrotoxicose factice- Hyperthyroïdies centrales - Métastases hyperfonctionnelles- Hyperthyroïdies gravidiques - Goitre ovarien toxique- Hyperthyroïdies auto-immunes familiale- Hyperthyroïdies des tumeurs placentaires
* L’étude de la fixation cervicale de l’iode radioactif n’a aucun interët dans l’affirmation de l’hyperthyroïdie qui est le fait
des dosages hormonaux. En revanche, elle peut s’avérer précieuse pour obtenir des précisions sur le type étiologique.
• Hyperthyroïdies avec fixation accrue • Hyperthyroïdies avec fixation diminuée
Fixation de l’iode radioactif *
TABLEAU

me pseudomitral, augmentation de la pression artériellesystolique et des constantes cardio-artérielles (vitessecirculatoire, débit cardiaque) ; diarrhée ou disparitiond’une constipation ancienne ; tremblement fin, rapide,régulier des extrémités, apparaissant au maintien desattitudes, parfois accompagné d’une impression de tré-mulation intérieure ; nervosité, agitation, instabilité del’humeur ; troubles trophiques des ongles, des cheveux ;troubles génitaux (irrégularité menstruelle, hypoménor-rhée, anovulation) ; gynécomastie.• Dans les formes frustes, la symptomatologie thyro-toxique est dissociée, réduite à quelques signes diverse-ment associés : tachycardie, petit tremblement, thermo-phobie, sudation, diarrhée, amaigrissement discret....• Dans les formes trompeuses,la symptomatologieprend le masque :- d’une affection cardiovasculaire (cardiothyréose) extra-systoles ; crises de tachycardie paroxystique ; accès deflutter ou de fibrillation auriculaire ; tachyarythmiecomplète par fibrillation auriculaire ; asystolie (particu-lière par l’absence de cardiopathie, la coexistence del’arythmie complète, la relative rareté des accidentsthrombo-emboliques (ce qui ne doit pas dispenser du traite-ment anticoagulant) ; les valeurs paradoxalement normalesde la vitesse circulatoire et du débit cardiaque ; la résistan-ce au traitement digitalique et la sensibilité élective autraitement antithyroïdien, exceptionnellement angor etbloc auriculo-ventriculaire ;– de signes paradoxaux avec prise de poids souvent avecaménorrhée chez la femme jeune, anorexie chez le sujet âgé; – d’une anomalie cutanée avec prurit ;– d’une affection digestive en raison de la diarrhée et del’amaigrissement ;– d’une affection musculaire ou neuropsychiatrique :myopathie pseudoparalytique ou pseudo-myasthénique,troubles paresthésiques ou paralytiques, modificationsdu comportement, états anxiodépressifs, délirants ouconfusionnels ;– d’une affection osseuse : douleurs osseuses et tasse-ments vertébraux liés à l’ostéopénie ;– d’une infection sévère : crise aiguë thyrotoxique avechyperthermie et déshydratation.Quelle que soit la présentation clinique de la thyrotoxi-cose, la confirmation du diagnostic sera le fait des exa-mens biologiques.
2. Explorations complémentaires
• Le retentissement périphériquede l’hyperhormoné-mie est confirmé par la mise en évidence d’une augmen-tation du métabolisme basal et le raccourcissement dutemps de contraction et de demi-relaxation de l’achilléo-réflexogramme qui sont des examens désuets ; le cholestéroldiminué (< 1,50 g/L), l’augmentation de la gamma-GT, desphosphatases alcalines, de l’ostéocalcine, de l’enzyme deconversion de l’angiotensine I, de la ferritine, de lasex bindingprotein, la tendance à l’hypercalcémie (Ca > 100 mg/L), à lamicrocystose (VGM < 82 µm3), à la leucopénie.
• Les modifications de l’état hormonalse traduisenttypiquement par l’augmentation des concentrations deT4 et de T3 totales (ou mieux de leurs formes libres FT4et FT 3) et la diminution de TSH. Dans la majorité dessituations le rapport T3/T4 est accru. Parfois, elles serésument à une augmentation de la FT3 (hyperthyroï-dies à T3, surtout dans les formes débutantes de la mala-die, chez les sujets jeunes et en situation de carenceiodée), hyperthyroïdies à T4, surtout en surcharge iodée,chez le sujet âgé ou débilité); ou simplement à un abais-sement isolé de TSH, sans accroissement de T4 et T3(formes frustes ou de tout début). Des valeurs paradoxa-lement normales ou accrues de TSH, inappropriées àl’hyperhormonémie thyroïdienne caractérisent leshyperthyroïdies centrales (adénome thyréotrope, résis-tance aux hormones thyroïdiennes).
Diagnostic différentiel
1. Sur le plan clinique
On pourrait évoquer à tort l’hyperthyroïdie chez l’éthyliqueen phase aiguë ou de sevrage, dans le phéochromocytomedu fait de l’amaigrissement, la tachycardie, la nervosité quiconfèrent au patient l’aspect pseudobasedowien ; cependantl’hypertension artérielle est systolo-diastolique, il n’y a pasde goitre ni de diarrhée et les catécholamines sont aug-mentées ; dans le syndrome d’hyperexcitabilité desbêtarécepteurs, avec notamment tachycardie, mais sansanomalie hormonale. La symptomatologie se réduitsous bêtabloquants.
2. Sur le plan hormonal
Une augmentation isolée de T3 libre (avec souvent T4basse) s’observe dans certains goitres par carence iodée(goitres à T3) ; une hyperthyroxinémie sans hyperthy-roïdie s’observe dans les maladies psychiatriques, enaltitude, sous amphétamines, propranolol à fortes doseset amiodarone; les anticorps anti-T3 et anti-T4 peuventartificiellement élever les dosages de T4 et T3 (dosagespar double anticorps) ; un abaissement isolé de TSH estcompatible avec une hyperthyroïdie fruste débutantemais peut aussi s’observer dans la grossesse, dans lesimprégnations glucocorticoïdes et dopaminergiquesmassives et récentes et en pathologie psychiatrique.
Diagnostic étiologique
La recherche des causes d’ hyperthyroïdie se fonde surun certain nombre d’arguments :- présence d’un goitre diffus ou nodulaire, ancien ourécent ;- recherche de signes oculaires, notion de prises médica-menteuses, notamment riches en iode ; recherche desanticorps antithyroïdiens : anti-thyroglobuline (anti-Tg),anti-thyroperoxydase (anti-TPO) et surtout anti-récep-teur de TSH (TBI Ab ou PA2) ; dosage de l’iodémie, de
Endocrinologie-Métabolisme-Nutrition
1379L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8

l’iodurie des 24 h, ou du rapport iodurie/créatininuriesur un échantillon d’urines, étude de la fixation de l’iode123 par la thyroïde.
1. Maladie de Basedow
C’est la variété la plus classique, la plus répandue et cli-niquement la plus impressionnante de toutes les hyper-thyroïdies. Elle survient fréquemment dans un contextefamilial de thyropathie, et avec prédilection chez lafemme jeune, mais n’épargne pas l’enfant, l’homme etle sujet plus âgé. Elle peut même être observée transitoi-rement chez le nouveau-né du fait du passage transpla-centaire des immunoglobulines thyréostimulantes.• Le débutde l’affection est parfois insidieux et progres-sif, parfois brutal. Son déclenchement au décours d’unchoc émotionnel ou d’un épisode de la vie génitale(puberté, grossesse, ménopause) n’est pas rare.• À la phase d’état, souvent l’aspect général du patientau regard fixe et brillant, aux yeux saillants, à la base ducou légèrement tuméfiée et se plaignant de sudations, desoif vive, de palpitations, d’asthénie et d’amaigrisse-ment est suffisamment évocateur pour permettre d’em-blée le diagnostic.À l’analyse on reconnaît :- Un goitre d’apparition récente, diffus mais parfoisasymétrique, ferme, indolore, vasculaire avec érythèmecervical, thrill et souffle systolique ou systolodiastoliqueà renforcement systolique, isolé, sans symptomatologiecompressive, sans adénopathie satellite.- Une ophtalmopathie est habituelle, mais contingenteet d’intensité variable, éventuellement responsable designes d’irritation conjonctivale (picotements, larmoie-ment, photophobie) et se complique parfois de diplopie,elle est habituellement bilatérale mais parfois asymé-trique, voire strictement unilatérale. Elle est constituée àdes degrés divers :✎d’un élément palpébro-rétractile avec élargissement dela fente palpébrale (signe de Dalrymple) créant unaspect de fausse exophtalmie, rareté du clignement, asy-nergie oculo-palpébrale dans le regard vers le bas (signede Von Graefe), oculofrontale dans le regard vers le haut(signe de Sainton) ; .d’une ophtalmopathie œdémateuse avec exophtalmievraie par protrusion des globes oculaires, axile et réduc-tible mesurable à l’exophtalmomètre. Dans les formesimportantes, on peut observer les fibres d’insertion desmuscles droits externes sur le globe oculaire (signe deBonamour), œdème et pigmentation des paupières (signe deJellinek), œdème conjonctival (chémosis), œdème desmuscles oculomoteurs déterminant une réduction de lamobilité des globes (prédominant souvent sur l’élévation) etun défaut de convergence (signe de Moebius).L’ensemble de ces signes confèrent au regard sa fixité, sonaspect tragique.- Le syndrome de thyrotoxicose est l’expression de l’in-flation hormonale, il est typique, discret ou trompeur.- Les signes associéssont inconstants, et témoignentd’anomalies auto-immunes associées :
. dermopathie basedowienne, constituée de nodules etde placards fermes, indolores, de coloration beige, infil-trant le derme avec élargissement des pores en peaud’orange et développement de longs poils noirs (aspectde peau de porc), localisée à la face antéro-externe desjambes (myxœdème prétibial), ou plus diffuse d’aspect élé-phantiasique sur les membres inférieurs, ou affectant le troncet les bras, parfois précédée de simples œdèmes inflamma-toires des jambes, en dehors de toute hyposystolie ; . vitiligo ; . périarthrite scapulo-humérale ;. hippocratisme digital ; . association à d’autres maladies auto-immunes : diabètesucré, insuffisance surrénale, anémie hémolytique,maladie rhumatoïde….• L’évolution s’effectue par poussées, ordinairementécourtées par le traitement. Elle est parfois émaillée decomplications liées à la thyrotoxicose ou l’ophtalmopa-thie.• Le diagnostic d’hyperthyroïdie est confirmé par lamesure de FT3, FT4 et de TSH.Y a-t-il lieu d’effectuer plus spécifiquement d’autresexamens ?– La présence d’anticorps anti-récepteur de TSH estnotée dans plus de 90 % des cas. Elle constitue un argu-ment en faveur de l’origine basedowienne de l’hyper-thyroïdie et de son évolutivité. On observe aussi souventdes anticorps anti-Tg et (ou) anti-TPO à des titres modé-rés (voir: pour approfondir 2).– La fixation de l’iode 123 est en principe augmentée,sauf si la maladie s’associe à une surcharge iodée. L’étudede la fixation de l’iode 131 n’est utile que si est envisagé untraitement radio-isotopique pour le calcul de la dose.– La cartographie thyroïdienne révèle une fixation diffuse ethomogène de l’isotope au sein de la glande hypertrophiée.Elle est particulièrement indiquée s’il existe une indurationlocalisée ou une formation nodulaire à la recherche d’unfoyer d’hypofixation qui sera ponctionné pour étude cytolo-gique.– En échographie, le parenchyme est globalement hypo-échogène et hypervasculaire. Cet examen est utile pour pré-ciser la signification de formations nodulaires associées.– Les formes sévères d’atteinte oculaire peuvent êtreévaluées par l’exploration fonctionnelle visuelle, latomodensitométrie (elle confirme l’absence de massetumorale, apprécie quantitativement l’épaisseur desmuscles oculomoteurs et l’exophtalmie par la mesure del’indice oculo-orbitaire) ou la résonance magnétiquenucléaire. Une fixation de l’analogue retard de la soma-tostatine marqué par l’indium (octréoscan orbitaire)s’observe dans les formes évolutives.
2. Nodule toxique de Plummer
Adénome autonome, hypersécrétant et extinctif vis-à-vis du reste du parenchyme thyroïdien, il s’observe àtous les âges mais reste plus fréquent dans le sexe fémi-nin. Il complique parfois l’évolution d’un noduledemeuré jusque-là normofonctionnel.
1380 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R T H Y R O I D I E

On constate un nodule thyroïdien : hyperthyroïdie loca-lisée d’une partie d’un lobe, parfois de tout un lobe oude l’isthme de la thyroïde, à limites précises, non hyper-vasculaire. Le reste du parenchyme thyroïdien n’est pasperçu ; un syndrome de thyrotoxicose complet, fruste outrompeur ; l’absence de signes oculaires œdémateux. Seulsun éclat du regard, une discrète rétraction palpébrale liés àl’hyperhormonémie peuvent être parfois notés.Les dosages hormonaux ayant confirmé l’hyperthyroï-die, l’examen fondamental du diagnostic est constituépar la scintigraphie thyroïdienne : la cartographie clas-sique par balayage (au technétium 99m ou mieux à l’io-de 123) révèle une fixation élective au niveau du nodulepalpé. Le reste du parenchyme thyroïdien est éteint (etseulement visualisé par l’enregistrement prolongé à lagamma-caméra, ou par l’échographie).
3. Goitres diffus secondairement toxiques
L’hyperthyroïdie survient sur un goitre préexistant.Le goitre est diffus, parfois sensiblement homogène,plus souvent irrégulier et bosselé, déformé par la présen-ce de formations nodulaires. On recherchera un retentis-sement compressif veineux, récurrentiel, trachéal ; unprolongement endothoracique (radiographies de face etde profil, éventuellement complétées par la tomodensi-tométrie ou la résonance magnétique nucléaire si l’onveut éviter l’administration d’un agent de contrasteradiographique iodé) ; des adénopathies.Les signes thyrotoxiques sont typiques, frustes ou trom-peurs.La présence d’une ophtalmopathie basedowienne estpossible, mais inhabituelle.Les dosages des anticorps anti-récepteurs de TSH, lacartographie thyroïdienne au technétium, ou mieux, àl’iode 123, éventuellement complétée par l’examencytologique et échographique des nodules suspects quipermettront de préciser si l’hyperthyroïdie relève d’ungoitre « basedowifié » (par stimulation diffuse du paren-chyme thyroïdien par les anticorps stimulants), ou plussouvent d’un goitre multinodulaire hétérogène secon-dairement toxique (par autonomisation d’une ou plu-sieurs formations nodulaires).
4. Hyperthyroïdies induites par l’iodesur glande saine ou pathologique
Chacune des variétés précédentes d’hyperthyroïdie peutêtre révélée par l’introduction d’iode en excès dans l’or-ganisme. Mais il existe aussi d’authentiques dysfonc-tions thyroïdiennes purement iatrogéniques apparaissantchez les patients porteurs d’un goitre simple ou mêmede thyroïdes apparemment saines, à la faveur de prisesmédicamenteuses iodées (antitussifs, antidiarrhéiques,amiodarone), d’agents de contraste iodés ou de prépara-tions alimentaires riches en iode.Elles sont ordinairement caractérisées par un tableau dethyrotoxicose pure, sans signe oculaire, une fixation
basse de l’iode 123, une augmentation de l’iodurie, l’ab-sence d’anticorps antithyroïdiens stimulants, une évolu-tion ordinairement spontanément régressive en quelquessemaines ou quelques mois, parallèlement à l’élimina-tion de la surcharge iodée, parfois suivie d’une phasetransitoire d’hypothyroïdie.Aucune de ces caractéristiques n’a cependant de valeurformelle, notamment en cas de pathologie thyroïdienneprécessive. Il n’est pas exceptionnel, après amiodarone,d’observer des évolutions sévères et prolongées.On peut en rapprocher les hyperthyroïdies survenantaprès prise de cytokines (interféron).
5. Hyperthyroïdies des thyroïdites
Une phase thyrotoxique peut s’observer :• au début des thyroïdites subaiguës de De Quervain,typiquement précession d’un syndrome infectieux d’al-lure grippale, puis apparition de douleurs cervicalesantérieures avec parfois otalgies, hyperthermie, hyper-trophie thyroïdienne électivement douloureuse. L’étatthyrotoxique est souvent discret, parfois méconnu,authentifié par l’exploration hormonale ; en phase aiguë,la vitesse de sédimentation est très augmentée, les anti-corps antithyroïdiens sont absents ou présents à des tauxfaibles et dissociés, le captage de l’iode 123 est effon-dré. La symptomatologie régresse spontanément enquelques semaines, parfois suivie d’une phase d’hypo-thyroïdie ;• au cours des thyroïdites d’Hashimoto (hashitoxicoses),le goitre est très ferme, d’une consistance comparable àcelle du suif ou du caoutchouc, l’ophtalmopathie base-dowienne est possible. La vitesse de sédimentation estaugmentée, les taux des anticorps anti-Tg et anti-TPOsont très élevés et on détecte des anticorps anti-récep-teurs de TSH, la fixation de l’iode 123 est accrue.L’hyperthyroïdie d’intensité variable nécessite un traite-ment anti-thyroïdien, mais celui-ci devra tenir compted’une évolution possible de la maladie vers l’hypothy-roïdie en quelques mois ou années ;• au début les thyroïdites silencieusessont respon-sables d’un état thyrotoxique discret avec hypocaptationthyroïdienne, spontanément régressif en quelques semaines etqui peut faire place à une hypothyroïdie, le plus souvent transi-toire également. Le taux des anticorps antithyroïdiens estélevé, le goitre est indolore, siège d’une thyroïdite lymphocy-taire. Cette expression particulière des thyroïdites auto-immunes peut survenir spontanément, mais s’observe avecune particulière fréquence dans le post-partum (5 % des gros-sesses).
6. Hyperthyroïdies auto-immunes familiales
Se différenciant des formes familiales de maladie de Basedow,de rares hyperthyroïdies familiales surviennent avec une égalefréquence dans les deux sexes, en l'absence d’atteinte oculaire.Il n’y a pas d’anticorps détectables et on peut caractériser desmutations ponctuelles du récepteur deTSH.
Endocrinologie-Métabolisme-Nutrition
1381L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8

7. Hyperthyroïdies gravidiques et vomissementsde la grossesse
Une baisse isolée de TSH est constatée chez environ 10 % des femmes enceintes au cours du premier tri-mestre. Dans certaines formes plus sévères, et notam-ment chez les femmes atteintes de vomissements incoer-cibles de la grossesse, l’hormonémie thyroïdienne estaccrue et l’on observe des signes thyrotoxiques.
8. Hyperthyroïdies centrales
Elles sont très rares, parfois longtemps méconnues, etse caractérisent par un goitre diffus, parfois tardive-ment remanié par l’apparition de nodules, un étatthyrotoxique souvent discret, l’absence de signesoculaires.L’hyperhormonémie thyroïdienne coïncide avec desvaleurs paradoxalement normales ou hautes de TSH,l’absence d’anticorps anti-récepteur de TSH, une fixa-tion accrue de l’iode 123.Elles peuvent résulter soit d’un adénome thyréotrope del’antéhypophyse : typiquement le rapport chaîne a /TSH est accru, la TSH est non stimulable par TRH etun adénome hypophysaire (le plus souvent un macroa-dénome, rarement un microadénome) est détectable parles radiographies de selle turcique, l’étude tomodensito-métrique et (ou) la résonance magnétique nucléaire ;soit d’un syndrome de résistance hypophysaire sélectiveou préférentielle aux hormones thyroïdiennes : typique-ment le rapport chaîne a /TSH est normal, la TSHrépond à la stimulation par TRH et l’enquête morpholo-gique est négative.
9. Hyperthyroïdies tumorales
On considère, en règle générale, que les hyperthyroïdiesconstatées au cours des cancers digestifs, bronchiques,thymiques relèvent, non pas de syndromes paranéopla-siques, mais de simples associations.En revanche, on peut observer rarement des hyperthy-roïdies liées : à des cancers thyroïdiens sécrétants, nodu-laires ou plus diffus ; à des métastases fonctionnelles(osseuses, pulmonaires) d’un cancer thyroïdien différen-cié antérieurement opéré ; à des môles hydatiformes oudes choriocarcinomes placentaires, producteurs de b-hCG ; à des goitres ovariens toxiques.
10. Thyrotoxicoses factices
Elles ne résultent pas d’une hyperactivité thyroïdiennemais de la prise souvent clandestine d’hormones thyroï-diennes ou de leurs dérivés cataboliques (acide triiodo-thyroacétique) par des femmes dans le but de maigrir.Elles réalisent un état de thyrotoxicose très pure, sansgoitre et sans atteinte oculaire. La thyroglobuline circu-lante est indétectable et le captage de l’iode 123 esteffondré.
Évolution
Certaines hyperthyroïdies peuvent spontanément régres-ser (thyroïdites, prise d’iode), d’autres évoluer par pous-sées (maladie de Basedow) ou s’aggraver progressive-ment.Les hyperthyroïdies sont des situations inconfortablesqui peuvent déterminer à la longue des complicationsparfois dramatiques.
1. État thyrotoxique
• Complications cardiovasculaires: arythmie complète,asystolie basedowienne, angor.• Complications musculaires : myopathies avec pseudo-paralysies (à différencier des formes s’associant auxparalysies périodiques).• Complications neurologiques: névrites des membres,polyradiculonévrites.• Complications psychiatriques(psychothyréoses) : agita-tion anxieuse, bouffées délirantes, états confusionnels.• Complications osseuses: ostéose hyperthyroïdienneavec tassements vertébraux, fractures. Elles prédomi-nent chez les femmes âgées et sont plus dépendantes del’ancienneté de l’hyperthyroïdie que de son intensité.Elles peuvent être dépistées précocement par l’étudedensitométrique osseuse, et révéleraient des signes his-tomorphométriques spécifiques.• Complications hépatiques: syndrome rétentionnelavec ictère.• Crise aiguë thyrotoxique : c’est une urgence endocri-nologique traditionnellement favorisée par la thyroïdec-tomie ou l’administration intempestive d’iode 131appliquée sans précaution, ou une infection intercurren-te. La présentation est sévère : hyperthermie importante,déshydratation, amaigrissement rapide, défaillance car-diaque, agitation anxieuse (forme sthénique) ou apathieextrême (forme asthénique), troubles métaboliques avechémoconcentration et déplétion potassée. L’évolutionvers le coma et la mort, parfois hâtée par des hémorra-gies, des accidents thrombotiques ou une infection estpossible.• Cachexie.
3. Ophtalmopathie
Environ 3 % des patients avec maladie de Basedowconstituent une atteinte oculaire sévère justifiant desexplorations et une thérapeutique spécifiques :• troubles oculomoteursavec diplopie permanentedépendant de l’atteinte musculaire ;• conjonctivites, kératites, ulcérations cornéennespou-vant conduire à la panophtalmie;• atteinte du nerf optiqueavec altération de la vision descouleurs, amblyopie ;• exophtalmie maligne : complication classique des thy-roïdectomies et des administrations intempestives d’iode131. Elles réalisent une véritable subluxation de l’œil avec
1382 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R T H Y R O Ï D I E

exophtalmie non réductible, perte de l’oculomotricité,baisse de la vision t risque d’ulcération sévère.
TraitementLe but du traitement des hyperthyroïdies est de réduirel’hyperfonctionnement thyroïdien et ses conséquences,et d’en prévenir les récidives.
Méthodes
1. Thérapeutiques symptomatiques
• Repos.• Bêtabloquants : ils atténuent les effets périphériquesdes hormones thyroïdiennes sans modifier la productionthyroïdienne ; l’effet est rapide sur le tremblement et latachycardie ; surtout prescrits sous forme de propranolol(Avlocardyl) qui réduit de plus la conversion de T4 enT3 ; en respectant les contre-indications classiques : sur-tout asthme, bloc auriculo-ventriculaire, diabète traitépar insuline ou sulfamides ; en cas d’arythmie : seméfier des risques de décompensation hémodynamique(intérêt de l’association aux digitaliques) et des acci-dents thrombo-emboliques (intérêt des anticoagulants).
2. Traitement médical
• Les antithyroïdiens de synthèse(ATS) réduisent laproduction thyroïdienne en inhibant l’oxydation et l’or-ganification de l’iode ; ils diminuent aussi la productiondes immunoglobulines thyréostimulantes. Ils sont pres-crits sous forme de carbimazole (Néo-Mercazole, com-primés à 5 et 20 mg), de benzylthiouracile (Basdène,comprimés à 5 mg) ou éventuellement de propylthioura-cile (PTU, disponible dans les pharmacies hospita-lières). La posologie d'attaque varie entre 15 et 60 mgpar jour. Les cas d’intolérance cutanée, digestive, hépa-tique sont rares, mais le risque de leuconeutropénietoxo-allergique justifie la surveillance régulière del’hémogramme, tout particulièrement durant les deux pre-miers mois de la prescription et lors de sa réintroduction. Laréduction de l’hyperthyroïdie est obtenue en 2 à 6 semaines et l’efficacité du traitement se juge sur l’étatclinique, les dosages hormonaux, éventuellement le tauxdes TBII.• L’iode minéralen excès bloque transitoirement l’organifi-cation (effet Wolff-Chaikoff). Il est prescrit sous forme desolution de Lugol Fort (I2 2 g; IK 4 g, eau qsq 40 g), 30 à 60gouttes par jour, surtout en préparation à la chirurgie.• Le lithium bloque transitoirement la protéolyse de la thy-roglobuline et peut ainsi être mis à profit en préparation à lachirurgie, en cas de leucopénie, d’intolérance à l’iode, et enassociation aux antithyroïdiens de synthèse (ATS) lorsqu'ilest nécessaire de réduire très rapidement une hyperthy-roïdie.
3. Traitement chirurgical
• Exérèse large(thyroïdectomie des 19/20e ou thyroï-dectomie totale) pour goitre diffus basedowien ou pluri-
nodulaire, ou lobectomie-isthmectomie pour noduletoxique.• Toujours après réduction soigneuse de l’hyperfonc-tionnement thyroïdien par les ATSpuis, éventuelle-ment, préparation par la solution de Lugol Fort ou lelithium.• Les risques de paralysie récurrentielle et d’atteinteparathyroïdienneexistent, mais sont mineurs si l’inter-vention est confiée à un chirurgien spécialisé.• C’est un traitement radicalqui assure la guérison desnodules toxiques ; après thyroïdectomie large pourmaladie de Basedow, les récidives d’hyperthyroïdie sontrares ; l’hypothyroïdie précoce ou retardée est possible,toutefois aisément compensable par l’hormonothérapiesubstitutive.
4. Traitement radio-isotopique
• Administration d’une activité thérapeutique ordinai-rement de 200 à 400 Mbq, calculée à partir du volumedu goitre, de la courbe de fixation.• Contre-indiqué en cas de grossesse, mais aussi de thy-rotoxicose ou d’ophtalmopathie sévères ; on l’évitehabituellement chez le sujet jeune et en âge de procréa-tion.• La tolérance est bonne ; l’exacerbation initiale de lathyrotoxicose est possible, souvent discrète, son rôledans l’aggravation des signes oculaires est discuté.• À terme, le risque est celui de l’hypothyroïdie(celle-cis’observe dans plus de la moitié des cas, 10 ans aprèsl’application du traitement radio-isotopique de la mala-die de Basedow).
Indications
Elles sont difficilement codifiables, fonction de l’âgedes patients, des caractéristiques du goitre, de l’impor-tance de la thyrotoxicose, de la participation oculaire etdes habitudes des thérapeutes.
1. Maladie de Basedow
• Cas habituel : femme jeune avec ophtalmopathiemodérée:– repos, antithyroïdiens de synthèse, éventuellementassociés au propranolol ;– le traitement d’attaque est, soit poursuivi à fortesdoses (20 à 60 mg/ j) en association avec l’hormonothé-rapie substitutive, soit poursuivi seul à une posologieprogressivement réduite, adaptée à l’état clinique et hor-monal. La durée habituelle du traitement est de 18 mois.La normalité du taux des anticorps anti-récepteurs deTSH et de la thyroglobuline circulante, en fin de traite-ment, constitue un élément favorable, mais ne permetpas d’affirmer la guérison définitive ;– en cas de reprise évolutive à l’arrêt du traitement ou derechute à distance, la reprise du traitement initial estpossible, mais on peut aussi envisager l’application d’untraitement radical : soit la thyroïdectomie (surtout chezle sujet jeune et en cas de volumineux goitre), soit
Endocrinologie-Métabolisme-Nutrition
1383L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8

l’application d’une dose thérapeutique d’iode 131 (chezle sujet plus âgé et en l’absence d’atteinte oculaireimportante).
• Cas particuliers:
– goitre volumineux de fixation hétérogène : chirurgieaprès réduction médicale de l’hyperfonctionnement thy-roïdien ;– atteinte cardiaque : traitement radical, le plus souventradio-isotopique, et à défaut chirurgical après réductionmédicale de hyperthyroïdie ;– sujet âgé : indication d’emblée de l’iode radioactif ;– femme enceinte : antithyroïdiens de synthèse, notam-ment un thiouracile, en recherchant la posologie minima-le. Le parfait équilibre hormonal maternel et l’atténuationdes stigmates d’auto-immunité spécifique représentent lameilleure prévention des dysfonctions néonatales ;– Basedow néonatal : bêtabloquant ou Lugol ou ATS enattendant la résolution spontanée ;– enfant et adolescent : traitement prolongé encadrant lapuberté ;– crise aiguë thyrotoxique : propranolol à fortes doses,éventuellement par voie veineuse, antithyroïdiens de syn-thèse à fortes doses, rééquilibration hydro-électrolytiqueavec notamment recharge potassée, éventuellement plas-maphérèse ;– ophtalmopathies sévères : du fait de la rétraction palpé-brale : collyre à la guanéthidine ; du fait de l’atteinteconjonctivale, cornéenne : collyres antiseptiques, antibio-tiques, corticoïdes, voire tarsorraphie ; du fait de l’exoph-talmie et des troubles oculomoteurs ; corticothérapie pré-coce à fortes posologies (1 mg/kg/jour) durant 3 à 6 mois,éventuellement télécobaltothérapie, ciclosporine, plas-maphérèse, intervention décompressive (OGURA) ouréimplantations musculaires au stade de fibroses ;– myxœdème prétibial : corticoïdes sous pansementsocclusifs.
2. Nodule toxiqueC’est une indication à un traitement radical :• soit chirurgie, après réduction médicale de l’hyperthy-roïdie, surtout chez le sujet jeune, en cas de nodule trèsvolumineux ou hétérogène ;• soit administration d’iode 131, notamment chez le sujetplus âgé, en cas d’atteinte cardiaque.
3. Goitres secondairement toxiques
En fonction des caractéristiques cliniques du goitre et ducontexte, discuter l’opportunité :• soit d’un traitement chirurgicalaprès réduction médi-cale de l’hyperthyroïdie ;• soit d’un traitement radio-isotopique si les conditionsde captation l’autorisent ;• soit du maintien d’un traitement antithyroïdienaulong cours, avec surveillance de la morphologie dugoitre.
4. Hyperthyroïdies induites par l’iode
En règle générale, le retour à l’euthyroïdie est spontané-ment obtenu, éventuellement sous couvert de bêtablo-quants.Certaines formes sévères et prolongées, notammentaprès amiodarone, font envisager un traitement soit parles antithyroïdiens de synthèse à fortes doses, seuls ouassociés au perchlorate de potassium, soit par la cortico-thérapie (surtout en cas de thyroïdite iodée). Le recoursaux plasmaphérèses, ou la chirurgie, est également envi-sageable.
5. Hyperthyroïdies des thyroïdites
• Thyroïdites subaiguës: bêtabloquants, anti-inflamma-toires.• Hashitoxicoses: antithyroïdiens de synthèse.• Thyroïdites silencieuses: bêtabloquants.
6. Hyperthyroïdies auto-immunes familiales
On peut discuter l’opportunité du traitement médical aulong cours par l’iode 131 et surtout de la thyroïdectomietotale.
7. Hyperthyroïdies de la grossesse
On peut proposer un traitement bêtabloquant, ou anti-thyroïdien transitoirement dans les formes sévères.
8. Hyperthyroïdies centrales
• Adénome thyrotrope: exérèse sélective, éventuelle-ment complétée par la télécobaltothérapie, les analoguesretard de la somatostatine.• Résistance aux hormones thyroïdiennes: analogueretard de la somatostatine, D-thyroxine ou acide triiodo-thyroacétique.
9. Hyperthyroïdies des tumeurs
• Traitement étiologiquechaque fois qu’il est possible.• Iode 131pour les métastases des tumeurs.
10. Thyroïdites factices
Tenter de convaincre le patient de la nécessité de l’arrêtde la prise hormonale. ■
1384 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P E R T H Y R O I D I E

Endocrinologie-Métabolisme-Nutrition
1385L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
POUR APPROFONDIR
1 / 1 Le récepteur de la TSH-R-TSH• C’est une protéine de 764 acides aminés qui comprend :– un domaine extracellulaire de 394 amino-acides qui constitue le sitede liaison de la TSH (ou les anticorps anti-récepteur de TSH) ;– 7 domaines membranaires de 266 acides aminés ;– un domaine intracellulaire de 83 amino-acides qui possèdent dessites potentiels de phosphorylation.• Le récepteur de la TSH appartient à la superfamille des récepteursaux protéines G. Il possède une forte homologie avec ceux de FSH,LH, hCG.• Le récepteur de la TSH est couplé à l’adénylcyclase par l’intermé-diaire de la protéine Gs : l’activité de l’unité effectrice productriced’AMPc favorise le développement de la cellule vésiculaire et activela biosynthèse des hormones thyroïdiennes. Le récepteur de la TSHstimule aussi la voie de la phospholipase C (production de diacylgly-cérol et d’inositol phosphates) régulant les processus d’iodation ausein de la thyroglobuline.• L’activation du récepteur de la TSH est obtenue :– par la TSH : en physiologie ;– par les anticorps anti-récepteurs de TSH : dans la maladie deBasedow et le goitre basedowifié. On ne connaît guère les sites antigé-niques impliqués dans l’apparition des anticorps spécifiques ;– par la b-HCG : grossesse, tumeur placentaire ;– par le développement de mutations constitutives de R-TSH qui sesituent surtout dans le domaine transmembranaire (portion terminalede la 3e boucle du 6e domaine transmembranaire). Les récepteursmutés activent l’adénylcyclase en l’absence de liguant. Les mutationspeuvent être :. somatiques : nodules toxiques,. germinales : hyperthyroïdie diffuse non auto-immune familiale
2 /Les auto-anticorps anti-récepteursde TSH
• Ils apparaissent spontanément mais préférentiellement dans le sexeféminin et dans des familles prédisposées aux thyropathies et auxmaladies auto-immunes.• Ce sont les IgG qui franchissent la barrière placentaire (d’où le trans-fert d’hyperthyroïdie in utero).• Ils déterminent l’accroissement du volume thyroïdien, l’hyperfec-tionnement de la glande. Leur implication dans la pathogénie dessignes oculaires de la maladie de Basedow est plus incertaine et nonexclusive.• Ils peuvent être détectés soit par technique de déplacement (thyroidbinding inhibiting immunoglobulin : TBI Ab, immunoglobulines inhi-bant la liaison de la TSH à son récepteur qui prennent en compte à lafois les anticorps bloquants et stimulants), soit par technique de géné-ration d’AMPc sur des cellules thyroïdiennes en culture (thyroid sti-mulating immunoglobulin : TSI ou TS Ab)
• L’hyperthyroïdie n’est pas une maladie,mais un état d’hyperfonctionnement thyroïdienqui résulte de causes diverses qu’il importe de préciser.• L’atteinte oculaire œdémateuse est spécifiquede la maladie de Basedow et des goitresbasedowifiés.• « Les basedowiens souffrent et meurent du cœur. »• La baisse de TSH constitue l’indice le plusprécoce des hyperfonctionnements primitivementthyroïdiens.• Les auto-anticorps anti-récepteurs de TSHconstituent un marqueur diagnostique et pronostique de la maladie de Basedow.• Les antithyroïdiens de synthèse ne permettentpas la guérison du nodule toxique et du goitremultinodulaire secondairement toxique.• Les antithyroïdiens de synthèse nécessitentune surveillance hématologique.• L’iode 131 est à manier avec prudence en cas d’atteinte oculaire importante.• La chirurgie de l’hyperthyroïdie ne s’envisagequ’après réduction médicamenteuse de l’hyperthyroïdie.
Points Forts à retenir
Leclere J, Orgiazzi J , Rousset B , Schlienger JL , Wemeau JL .La thyroïde. Expansion Scientifique Française, 1992.de Roux N . Mutation du récepteur de TSH et hyperthyroïdie :Métabolismes, Hormones, Nutrition. 1997 ; 1 : 7-15.J Clerc. Traitement de l’hyperthyroïdie par l’iode 131.Métabolismes, Hormones, Nutrition. 1997 ; 1 : 40-7.
POUR EN SAVOIR PLUS

HypoglycémieOrientation diagnostique et conduite à tenir en situation d’urgence avec la posologiemédicamenteuse
PR Gérard SLAMAService de diabétologie, l’Hôtel-Dieu de-Paris, 75181 Paris cedex 04
Les hypoglycémies sont un syndrome clinique dontles manifestations sont liées à un taux anormale-
ment bas de la glycémie. Leur diagnostic passe doncimpérativement, à un moment ou à un autre, par la miseen évidence d’une glycémie dosée dans des conditionsparfaites au laboratoire, mesurée en dessous de 0,50 get, plus caractéristique, en dessous de 0,30 g/l.Une hypoglycémie sévère pouvant mettre en jeu le pro-nostic vital ou fonctionnel (accident vasculaire céré-bral), le diagnostic doit être rapide et le traitementimmédiat : il s’agit d’une urgence diagnostique et théra-peutique.Schématiquement, deux circonstances diagnostiquespeuvent se rencontrer : ou bien on assiste à un accidenthypoglycémique, ou bien c’est rétrospectivement que lediagnostic doit être évoqué.
Accident paroxystique évocateur d’hypoglycémie
Manifestations polymorphes :
1.Manifestations évocatrices • Soit mineures: sensation de malaise avec asthénie brutale,sensation de fringale, tremblements, tachycardie,palpitations,sueurs.• Soit majeures : coma, qui fait facilement évoquer unehypoglycémie quand, à une altération brusque de l’état deconscience s’associent convulsions, contractures avectrismus,Babinski bilatéral,hyperréflectivité ostéotendineuse,chez un patient mouillé de sueurs.
2. Manifestations aspécifiques:
Elles peuvent donner :• des signes neurologiques focalisés, crise d’épilepsieBravais-Jacksonienne, hémiparésie, monoplégie, paralysiefaciale;• des troubles psychiatriques paroxystiques, par exemplesyndrome confusionnel simulant une ivresse,colère clastiqueou comportement bizarre;• un coma profond, calme, aréflexique sans sueur peut êtreégalement une hypoglycémie;3. Manifestations trompeusesIl peut s’agir d’hypertension artérielle paroxystique ; crisede tachycardie ; d’une douleur d’angine de poitrine.
Endocrinologie-Métabolisme-NutritionA 59
1821L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
• Le diagnostic de certitude d’une hypoglycémiepasse par la constatation d’un tauxanormalement bas de la glycémie,sur un prélèvement fait dans des conditionstechniques parfaites (tube adéquat,acheminement immédiat) et dosé dans unlaboratoire.• On commence à parler d’hypoglycémiebiologiquement quand la glycémie est en dessousde 0,50 g ; à partir de 0,30 g, dans les conditionsde prélèvement évoquées ci-dessus,l’hypoglycémie est indiscutable.• La détermination de la glycémie capillaire est très utile mais ne constitue, à elle seule,qu’une orientation diagnostique et non pas la preuve de l’hypoglycémie.• Les signes cliniques ne sont en rien spécifiquesde l’hypoglycémie ; ils constituent le motif du prélèvement sanguin.• L’interrogatoire minutieux est, en revanche,le meilleur guide vers la découverte d’uneétiologie, une fois le diagnostic d’hypoglycémieétabli par la biologie.• Tout signe ou symptôme neurologique et (ou)psychiatrique, surtout d’installation brutale, peutêtre dû à une hypoglycémie; dans le doute,ne pas s’abstenir; traiter immédiatement comme une hypoglycémie ne peut en aucun cas êtredangereux ; faire précéder si possible letraitement par une détermination de la glycémiecapillaire utilisant un dispositif en bon état de marche et (ou) des bandelettes réactives non périmées et conservées de façon adéquate.• Les hypoglycémies survenant après les repas ne sont jamais graves (sauf patient diabétiquetraité par sulfamides hypoglycémiants).• Les hypoglycémies survenant au jeûne,spontanément (non iatrogèniques), évoquent une cause sérieuse, voire grave.• Les causes les plus fréquentes sont :– les diabètes ;– l’alcool et les médicaments ;– les hypoglycémies postprandiales fonctionnelles
Points Forts à comprendre

En fait, rien n’étant caractéristique dans ce tableau, lediagnostic ne peut naître que d’une suspicion systéma-tique de l’hypoglycémie devant toute manifestationparoxystique particulièrement neurologique ou psychia-trique.Il faut alors :• Si l’état d’agitation du patient et les conditionslocales le permettent, au mieux donc, faire une prise desang immédiate pour dosage au laboratoire de la glycé-mie sur plasma veineux avant tout traitement ; recom-mander au laboratoire de garder le plasma non utilisécongelé.• Là encore si les conditions locales le permettent,c’est-à-dire si l’on dispose du matériel, apprécier la gly-cémie capillaire à l’aide de bandelettes réactives (àcondition que le lecteur éventuel soit bien entretenu, lesbandelettes réactives non périmées et conservées dansdes conditions adéquates) : une valeur appréciée basse,en dessous de 0,50 g par exemple est, une bonne orien-tation du diagnostic d’hypoglycémie mais ne peutconstituer une preuve absolue au diagnostic.• De toute façon, et sans attendre les résultats des pré-lèvements envoyés au laboratoire, il faut traiter cetteéventuelle hypoglycémie :– si le patient est suffisamment conscient, c’est-à-dires’il n’y a pas de risque de fausse route, par l’administra-tion orale d’une boisson sucrée, par exemple 20 cL d’unjus de fruits, ou l’équivalent de 20 g de saccharose, soiten solution, soit en morceaux (4 carrés trempés dans del’eau, et disposés entre la joue et les dents s’il y a tris-mus) ;– si le patient est inconscient, le traitement le plusimmédiatement efficace est l’administration intravei-neuse d’une vingtaine de grammes de glucose, soit 60 mL de sérum glucosé à 30 % en 1 à 3 min ; le réveil estquasi immédiat, il faut alors profiter du réveil du patientpour lui administrer un glucide par voie orale, assurantainsi une administration de 20 à 25 g de glucides. Cetteinjection intraveineuse n’est parfois pas possible si lepatient est très agité. C’est dans ce cas seulement (ou sion a la certitude qu’il s’agit d’un patient diabétique trai-té à l’insuline) que l’on a recours à une injection intra-musculaire ou sous-cutanée, si nécessaire même à tra-vers les vêtements, de 1 mg de glucagon. Il faut savoir
attendre souvent une bonne dizaine de minutes pour enmesurer l’efficacité, quasi constante s’il s’agit biend’une hypoglycémie : cette injection peut être inefficaceen cas d’hypoglycémie alcoolique ; elle peut entraînerune amélioration très transitoire suivie d’une nouvellehypoglycémie profonde si cette dernière est liée à laprise de sulfamides hypoglycémiants ou en cas detumeur bêta-pancréatique (effet insulino-sécréteur duglucagon) : pour ces raisons, il est de bonne règle defaire suivre l’injection de glucagon par une prise alimen-taire d’une quinzaine de grammes de glucides dès que lepatient est en mesure de déglutir.La réponse au traitement est un argument importantmais non suffisant au diagnostic, quand il entraîne uneamélioration franche et rapide des signes cliniques.L’absence de réponse n’exclut pas que l’accident initialait pu être une hypoglycémie (coma post-hypoglycé-mique par œdème cérébral ou séquelle neurologiquedurable compliquent parfois des hypoglycémies ayantduré de nombreuses heures).Une fois la conscience retrouvée, une surveillance desglycémies capillaires permet de décider de la nécessitéde poursuivre le resucrage par voie orale, voire misesous perfusion immédiate de sérum glucosé à 10 % si ona la conviction que l’hypoglycémie est liée à une sur-charge importante en sulfamides hypoglycémiants :l’hypoglycémie dans ce cas-là peut récidiver pendantplus de 48 h.C’est alors que, secondairement, on pourra disposer desrésultats de la glycémie dosée au laboratoire : seule laconstatation d’un chiffre nettement inférieur aux normespour la technique de dosage utilisée, peut affirmer lediagnostic avec certitude. Encore faut-il savoir que cetteglycémie peut être trouvée faussement basse :– parce que le prélèvement a été recueilli sur un tubeinadéquat ne comportant pas de bloqueur de la glycoly-se, et laissé à la température ambiante trop longtemps ;– parce qu’il existe une leucémie (un grand nombre deglobules blancs consomme le glucose).À condition d’une attitude rigoureuse quand on assisteaux manifestations paroxystiques, le diagnostic d’hypo-glycémie est donc fait avec certitude.On n’assiste pas à l’hypoglycémie
1822 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P O G L Y C É M I E
Symptômes liés à l’hypoglycémie TABLEAU I
Dus… à une neuroglycopénie Dus à la riposte neurovégétative adrénergique et cholinergique
Ralentissement mental palpitations sensation de froid
Troubles de la vue + diplopie (= signe musculaire) tremblements chair de poule, frissons
Lenteur verbale sueurs pâleur
Mouvements anormaux pâleur faim
Troubles neurologiques et psychiatriques

A distance de l’accident paroxystique
C’est également à ce stade que le médecin se trouveconfronté après coup à la plainte d’un patient qui décritun accident neurologique et (ou) psychiatrique aigu, ouun malaise.Il est dès lors essentiel de retrouver une cause à cettehypoglycémie. Schématiquement, l’interrogatoireminutieux du patient et de son entourage permet dedécrire 3 situations possibles : on apprend que le patientest diabétique ; le patient prend certains médicaments ;le malaise est apparemment survenu spontanément.
Patient diabétique connu et traité
C’est le cas le plus fréquent des hypoglycémies authen-tiques.• Il peut s’agir, d’un diabétique traité à l’insuline : toutdiabétique traité à l’insuline a fait ou fera au moins unefois dans sa vie, le plus souvent maintes fois, l’expérien-ce d’un malaise ou d’un coma hypoglycémique.Ces hypoglycémies sont le plus souvent la conséquenced’une erreur évidente dans l’administration de la dosed'insuline : dose trop importante, erreur d’insuline àinjecter, existence de lipodystrophies, injection endovei-neuse accidentelle, changement de type d’insuline (ana-logue), erreur diététique (consommation insuffisante deglucides, répartition inadéquate des apports glucidiques)ou effort physique inhabituel sans que des mesuresappropriées pour éviter l’hypoglycémie aient été prises.Dans tous les cas, l’information diabétologique dupatient doit être renforcée, le patient est le plus souventrenvoyé vers son médecin habituel une fois l’hypoglycé-mie passée sans qu’il soit nécessaire d’hospitaliser lepatient.• Il peut s’agir d’un patient diabétique traité avec desantidiabétiques oraux: les sulfamides antidiabétiquespeuvent entraîner des hypoglycémies extrêmementgraves, récidivantes après resucrage, éventuellementmortelles. Ces hypoglycémies sont toujours la consé-quence d’une erreur thérapeutique grossière : traitementoral d’un diabétique ne nécessitant au plus que quelquesmesures diététiques ; utilisation de sulfamides à duréed’action trop longue ou allongée par l’existence négli-gée d’une insuffisance rénale ou hépatique suivant leurmétabolisme ; utilisation de produits trop puissants, àposologie trop forte ; prescription de produits , tels quecertains sulfamides antibactériens, anti-inflammatoiresnon stéroïdiens (AINS), kétoconazole qui potentialisentl’effet hypoglycémiant des sulfamides.La meilleure prévention des hypoglycémies graves soussulfamides est la diminution des posologies au strictminimum et le dépistage, à l'interrogatoire, des hypogly-cémies mineures survenant en fin d’après-midi ou ladétection de glycémies en dessous de 0,90 g à 18-19 h.• Les biguanides et les inhibiteurs des a_- glucosidasesseuls ne donnent jamais d’hypoglycémie, mais peuventla provoquer, associés au jeûne et (ou) à l’alcool.
Médicaments hypoglycémiants
Dans la 2e situation clinique schématique l’interrogatoireapprend que le patient (diabétique, ou non diabétique) a prisdes médicaments qui peuvent occasionnellement entraînerdes hypoglycémies : de nombreuses drogues peuvent entraî-ner des hypoglycémies. Parmi les plus importantes : l’al-cool, surtout s’il est associé au jeûne chez l’adulte mais éga-lement chez l’enfant et le très jeune enfant ; le diagnostic estsouvent méconnu ; les salicylées, surtout quand leur admi-nistration est massive et qu’il existe une insuffisance rénaleou une dénutrition ; le propoxyphène, la clonidine, lesbêtabloquants à dose toxique.
Hypoglycémie spontanée
Enfin, troisième éventualité, il s’agit d’une hypoglycé-mie probablement spontanée. L’interrogatoire est, làencore, l’élément clé de l’orientation du diagnostic : ildoit être minutieux. Il permet en effet de faire la distinc-tion fondamentale entre :– les hypoglycémies survenant en période de jeûne ouaprès un effort musculaire : peuvent être très sévères etrelèvent de causes souvent graves ;– les hypoglycémies survenant après les repas : ceshypoglycémies ne sont jamais graves, elles sont ditesfonctionnelles.
1. Hypoglycémies spontanées survenant au jeûneElles ont comme caractéristique de faire alterner desmanifestations neurologiques ou psychiatriquesmineures et des manifestations majeures dont elles ontpratiquement l’exclusivité. Ces manifestations neurolo-giques ou psychiatriques surviennent loin des repas, sur-tout le matin avant le petit déjeuner, parfois déclenchéespar un effort physique ou l’omission d’un repas (le dia-gnostic d’hypoglycémie de jeûne peut très exceptionnel-lement être porté, en l’absence de toute manifestationclinique, par la constatation fortuite puis contrôlée d’uneglycémie très anormalement basse le matin à jeun). Ceshypoglycémies associent de façon caractéristique la triadede Wipple : accident neuropsychique survenant à jeun,réversible sous hydrates de carbone, et constatation d’uneglycémie inférieure ou égale à 0,50 g.Comme la connaissance d’une hypoglycémie de jeûneconduit à des décisions thérapeutiques majeures, l’exa-men clinique et l’exploration fonctionnelle conduisantau diagnostic étiologique doivent être menés avec unetrès grande rigueur.• Parfois l’examen clinique est pratiquement suffisantau diagnostic étiologique : c’est le cas le moins fré-quent.Il peut s’agir :– d’un panhypopituitarisme évident avec l’associationd’un hypogonadisme ou d’une aménorrhée avec dépig-mentation et dépilation, d’une hypothyroïdie souventmodérée, d’une insuffisance surrénale dont l’hypoglycé-mie est l’une des manifestations ;
Endocrinologie-Métabolisme-Nutrition
1823L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8

– l’examen clinique peut retrouver également des argu-ments en faveur d’une maladie d’Addison, avec sa pig-mentation particulière, son asthénie, son hypotensionartérielle.Le traitement de ces insuffisances hormonales suffit àfaire disparaître les hypoglycémies.– L’examen clinique peut également trouver une grossetumeur à l’examen du thorax, de l’abdomen, de la régionlombaire ou du petit bassin. Ces tumeurs mésenchyma-teuses ne sauraient échapper à un examen clinique etradiologique simple, car il s’agit de tumeurs volontiersvolumineuses. Le dosage des facteurs de croissance,dans des laboratoires hautement spécialisés, confirme-ront le diagnostic et le mécanisme de l’hypoglycémie :mise en évidence d’un taux anormalement élevé deIGF2.– Le diagnostic est cliniquement aisé chez un patientatteint d’insuffisance hépatique grave : cirrhose au stadeultime, hépatite fulminante virale ou toxique, cancersecondaire évolué, certaines intoxications (amanitephalloïde, tétrachlorure de carbone). Le traitement deces hypoglycémies est symptomatique et ne modifie pasle pronostic désastreux à court terme.On est parfois également confronté à un patient atteintde sida, très dénutri, au stade ultime de sa maladie et oùl’étiologie de l’hypoglycémie peut être multifactorielle.• Le plus souvent l’examen clinique n’apporte aucunélément notable: cette absence de signes ou de symp-tômes oriente en soi vers une hypoglycémie organiquepar hyperinsulinisme et tumeur ß-langheransienne.– Le diagnostic d’hypoglycémie par hyperinsulinismeendogène repose sur la mise en évidence simultanéed’une glycémie franchement basse, dosée au laboratoireau-dessous de 0,30 g, et d’un hyperinsulinisme. C’est lacause la plus fréquente des hypoglycémies organiques.Le diagnostic erre parfois depuis de très nombreusesannées avant d’être posé. Cela, joint à l’absorption répé-tée d’aliments pour lutter contre les malaises hypoglycé-miques, entraîne parfois une prise de poids notable. Lapreuve de l’origine se fait au mieux par le dosage de la gly-cémie et de l’insulinémie au moment d’un accident (d’oùl'intérêt d’avoir demandé au laboratoire de conserver unplasma congelé lors de ce premier accident), ou sur plu-sieurs prélèvements faits le matin à jeun. Sinon le patientsera hospitalisé en service spécialisé pour y subir une épreu-ve de jeûne, exploration dangereuse si elle n’est pas menéedans un environnement adéquat.– Une hypoglycémie franche en dessous de 0,30 g / Lest habituellement observée dans les 24 premières heures,exceptionnellement au-delà de la mise au jeûne du patient.L’épreuve ne doit en aucun cas être stoppée sur la seuleconstatation de symptômes cliniques, mais sur la constata-tion d’une glycémie significativement basse (< 0,30 g / L)dont le résultat doit être obtenu immédiatement du labora-toire. La preuve de l’hyperinsulinisme est fondée sur laconjonction d’une glycémie basse et d’une insulinémie éle-vée ou simplement dosable sur un même prélèvement. Cethyperinsulinisme peut être dû :
. à une tumeur β-langerhansienne, bénigne ou maligne :le peptide C est augmenté en même temps que l’insuli-némie et que la glycémie est basse ;. peut être lié à l’absorption subreptice de sulfamides
hypoglycémiants : la situation est indiscernable du casprécédent (cf. infra)..à une injection subreptice d’insuline par le patient lui-
même, ou quelqu’un de son entourage : dans ce cas làl’insulinémie est élevée mais le peptide C est indosable.Peut coexister un taux modérément élevé d’anticorpsanti-insuline provoqué par les injections répétées d’in-suline.Il existe des cas rarissimes de maladies auto-immunesavec anticorps anti-insuline spontanés : la glycémie estbasse, l'insulinémie extrêmement élevée, mais il existedes taux très élevés d’anticorps anti-insuline.– Dans l’immense majorité des cas donc, la constatationd’une glycémie basse et d’une insulinémie élevéetémoigne d’une sécrétion endogène par une tumeur b- pancréatique. Il s’agit donc là d’un diagnostic avanttout biologique qu’on recherchera secondairement lesarguments anatomiques de la tumeur : 2 examens delocalisation de la tumeur pancréatique, anodins et depratique courante, confirment le diagnostic (quand les 2examens sont concordants), et vont guider un acte chi-rurgical éventuel :. L’échographie pancréatique par fibroscopie gastro-
duodénale : c’est un examen sensible et spécifique quine peut être pratiqué que par des personnes entraînées ;. L’angioscanner hélicoïdal qui permet, en couplant un
scanner hélicoïdal à une injection intraveineuse de pro-duit de contraste, de détecter des tumeurs du pancréas dequelques millimètres.. Un 3e examen, surtout indiqué si les 2 précédents ne don-nent pas des résultats concordants, doit être pratiqué : l’arté-riographie cœliaque et mésentérique, pratiquée à conditionqu’un acte chirurgical ne soit pas de toute façon exclu.. Ces examens, couplés également à l’échographie
hépatique, permettent de dépister des métastases gan-glionnaires ou du pédicule hépatique, ou des métastasesintrahépatiques.• Conduite thérapeutique devant un hyperinsulinismeendogène: en attendant les résultats des examens biolo-giques et d’imagerie, un traitement symptomatique pardiététique apportant une quantité supérieure à 250 gd’hydrates de carbone par jour, répartie tout au long dunycthémère, et si nécessaire en milieu de nuit, associé àdu Diazoxide, 100 à 300 mg/j , peut être prescrit.Le diagnostic fait, la tumeur localisée, une discussionchirurgicale va être faite qui doit tenir compte de l’âge,du risque opératoire et des données de l’imagerie.La démonstration d’une tumeur langerhansienne, qui estplus fréquemment un adénome (85 % des cas) qu’un can-cer, conduit le plus souvent à proposer un acte chirurgicalradical programmé sans urgence. L’intervention chirurgica-le, faite par un chirurgien entraîné et habitué à cette patholo-gie, sera également guidée par l’échographie pancréatiquepéropératoire qui permet de localiser des tumeurs multiples.
1824 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P O G L Y C É M I E
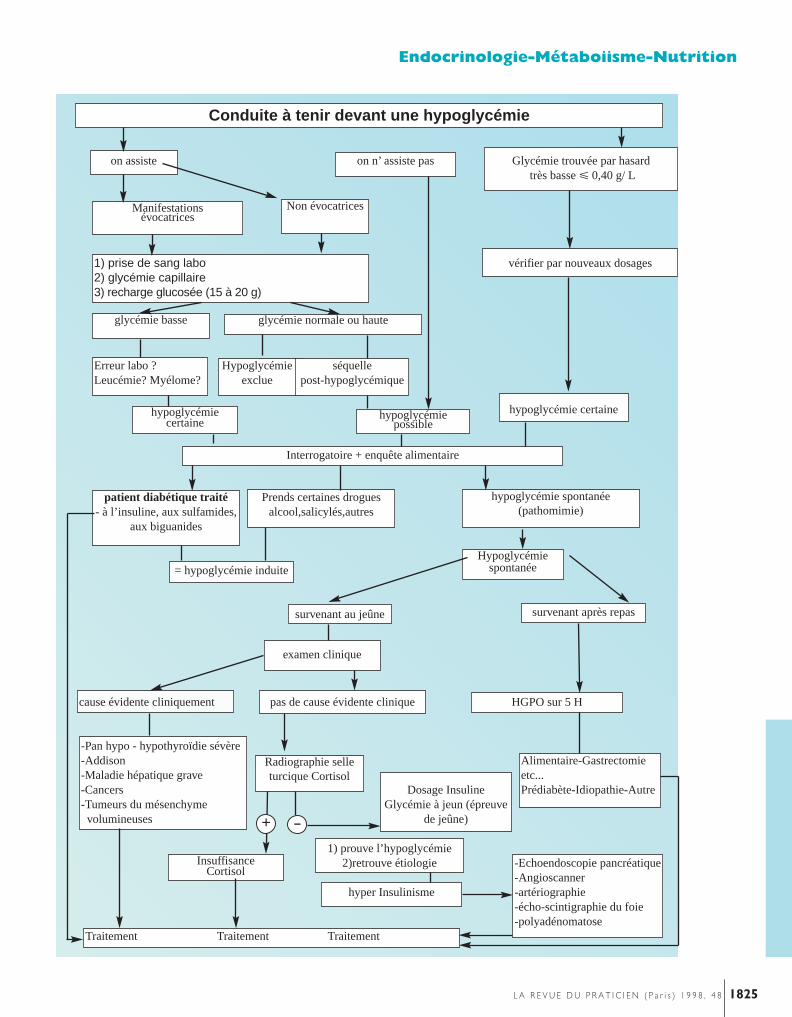
Endocrinologie-Métaboiisme-Nutrition
1825L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
Conduite à tenir devant une hypoglycémie
Manifestationsévocatrices
Non évocatrices
vérifier par nouveaux dosages1) prise de sang labo2) glycémie capillaire3) recharge glucosée (15 à 20 g)
on assiste on n’ assiste pas Glycémie trouvée par hasardtrès basse o 0,40 g/ L
glycémie basse glycémie normale ou haute
Erreur labo ?Leucémie? Myélome?
Hypoglycémie exclue
séquelle post-hypoglycémique
hypoglycémiecertaine
hypoglycémie possible
hypoglycémie certaine
Interrogatoire + enquête alimentaire
patient diabétique traité - à l’insuline, aux sulfamides,
aux biguanides
Prends certaines droguesalcool,salicylés,autres
hypoglycémie spontanée (pathomimie)
cause évidente cliniquement
examen clinique
HGPO sur 5 Hpas de cause évidente clinique
= hypoglycémie induiteHypoglycémie
spontanée
survenant au jeûne survenant après repas
-Pan hypo - hypothyroïdie sévère-Addison-Maladie hépatique grave-Cancers-Tumeurs du mésenchyme volumineuses
Radiographie selleturcique Cortisol
Dosage InsulineGlycémie à jeun (épreuve
de jeûne)
Alimentaire-Gastrectomieetc...Prédiabète-Idiopathie-Autre
1) prouve l’hypoglycémie2)retrouve étiologieInsuffisance
Cortisol
hyper Insulinisme
-Echoendoscopie pancréatique-Angioscanner-artériographie-écho-scintigraphie du foie-polyadénomatose
Traitement Traitement Traitement
+ -

1826 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P O G L Y C É M I E
Le geste chirurgical peut être une énucléation de la tumeurou une pancréatectomie partielle.La découverte d’une tumeur langerhansienne doitconduire à rechercher d’autres adénomes : parathyroï-diens, hypophysaires, thyroïdiens, surrénaliens.• Autres causes d’hypoglycémies survenant au jeûne :outre les tumeurs mésenchymateuses que nous avonsvues, il peut s’agir d’une insuffisance surrénale lente,dont l’hypoglycémie peut être le seul symptôme : ledosage de la cortisolémie, des épreuves de stimulationau Synacthène, voire un text à la métopyrone en servicespécialisé, permettent de faire le diagnostic.– Il peut s’agir également d’une insuffisance surrénaled’origine haute non évidente cliniquement, l’explora-tion radiologique de la région hypothalamo-hypophyai-re, peut s’imposer en cas de doute.– Enfin il peut s’agir d’une hypoglycémie induite parl’auto-administration de substances hypoglycémiantesdans un but de pathomimie. Ce diagnostic est à recher-cher systématiquement, particulièrement quand lepatient a des rapports avec le milieu médical ou a un dia-bétique dans sa famille. Le diagnostic, très rare, restetrès difficile à faire. Il repose avant tout sur la mise enévidence du produit dans l’environnement immédiat dupatient, conviction difficile à obtenir, et le dosage san-guin spécifique du produit ainsi suspecté.
2. Hypoglycémies post-alimentaires,dites fonctionnelles
Elles ne mettent jamais en jeu le pronostic vital. Ce sontle plus souvent des hypoglycémies modérées, ne s’ac-compagnant que très exceptionnellement, sinon jamais,d’accident neurologique profond.Leur diagnostic est souvent porté par excès.– Ces hypoglycémies surviennent 2 à 4 h après un repas,en fin de matinée ou en fin d’après-midi, survenant sur-tout après les repas très riches en glucides. Le meilleurcritère diagnostique est le dosage de la glycémie aumoment du « malaise » : il est rarement possible. Le dia-gnostic repose alors sur la pratique d’une hyperglycémieprovoquée orale sur 5 heures, avec des prélèvements dedemi-heure en demi-heure. Le diagnostic d’hypoglycé-mie post-alimentaire peut être posé si le test reproduitles symptômes habituellement reproduits par le patientau moment où une glycémie o 0,50 g est dosée (25 %des sujets normaux, souvent jeunes, ne se plaignantd’aucun symptôme, ont une glycémie o 0,50 g au coursd’une hyperglycémie provoquée orale systématique).L’épreuve de jeûne est parfaitement supportée. Troistypes d’hypoglycémie post-alimentaire peuvent être dis-tingués ;– les hypoglycémies liées à un trouble de la cinétique dela digestion des glucides (gastrectomie, gastro-entéro-stomie, vagotomie, pyloroplastie).– hypoglycémies liées à un trouble de la cinétique del’insulinosécrétion ; hypoglycémies post-alimentairesdes obèses ayant des antécédents familiaux de diabète ;
– hypoglycémies post-alimentaires idiopathiques, sou-vent chez des jeunes femmes extrêmement minces etégalement anxieuses ;Le traitement de ces hypoglycémies post-alimentairespasse par la réduction de l’absorption de quantités tropimportantes de glucides en une seule fois, et éventuelle-ment de l’utilisation en seconde intention d’un inhibi-teur des α− glucosidases (acarbose, miglitol).
3. Il existe des cas exceptionnels d’hypoglycémie
– Hypoglycémie par pathologie auto-immune avec pré-sence d’anticorps anti-récepteurs à l’insuline : ces anti-corps sont stimulants. L’insulinémie est basse, à la diffé-rence du cas évoqué plus haut d’hypoglycémieauto-immune par anticorps anti-insuline.– Perfusion massive prolongée de solutés glucosés intra-veineux, stoppés trop brutalement.– Exercice physique mené jusqu’à épuisement (mara-thon, course cycliste, etc.).– Dénutrition pré-mortem (cancer, sida, grève de lafaim…).Les hypoglycémies de l’enfant sont un chapitre à partqui n’est pas évoqué ici. ■
• Les signes cliniques ne sont en rien spécifiques.• Les causes les plus fréquentes sont les diabètes,;l’alcool et les médicaments; les hypoglycémiespost prandiales.
Points Forts à retenir
Pinsky MR, Dhainaut J.F. Pathophysiologic foundations of criti-cal care. Williams and Wilkins.
POUR EN SAVOIR PLUS

HypothyroïdieEtiologie, physiopathologie, diagnostic, évolution, traitementPR Jean-Claude VALCKEService de médecine interne II, hôpital Boucicaut, 75730 Paris cedex 15
Étiologie
Les étiologies de l’hypothyroïdie seront envisagées suc-cessivement chez l’adulte et chez l’enfant.
Chez l’adulte
1. Hypothyroïdies primaires
● L’atrophie : c’est une cause fréquente (40 %) survenantpréférentiellement chez la femme post-ménopausée de 50 à55 ans, mais parfois plus jeune. Le diagnostic est assuré parl’absence de goitre et même l’atrophie du corps thyroïde.● La maladie d’Hashimoto: c’est l’expression la plus fré-quente et la plus typique des thyroïdites auto-immunes.L’élément clinique essentiel est le Goitre, diffus, régulierou finement bosselé, ferme ou dur (consistance du caout-chouc) indolore. L’hypothyroïdie apparaît en cours d’évolu-tion dans 50 % des cas. Le diagnostic est confirmé par la présence d’anticorps anti-thyroïdiens à titre élevé (anti-
thyroglobuline, anti-thyroperoxydase). Elle peut être asso-ciée à une maladie d’Addison d’origine auto-immune,constituant le syndrome de Schmidt.● D’autres thyroïditespeuvent plus rarement évoluer versune hypothyroïdie souvent transitoire : thyroïdites subai-guës, thyroïdites du post-partum.● Les hypothyroïdies iatrogènes, à forte incidenceactuelle, où l’on distinguera : les hypothyroïdies post-thérapeutiques : thyroïdectomie pour cancer ou maladie deBasedow ; séquelles d’un traitement par l’iode radio-actif(IRA) ; radiothérapie pour cancer du larynx, de l’œsophageou maladie de Hodgkin ; et les hypothyroïdies médicamen-teuses par prise d’antithyroïdien de synthèse (ATS) ; delithium, surtout par saturation iodée (Amiodarone et pro-duits de contraste) ; il s’agit d’une hypothyroïdie modéréeavec goitre ferme ; iodémie et iodurie sont élevées ; il existeune fixation précoce du radio-iode et une image scintigra-phique correcte peut être obtenue dans les 30 à 60 premièresminutes ; le test au perchlorate est positif mettant en éviden-ce le bloc de l’organification. Enfin, les traitements parinterféron peuvent être compliqués par une hypothyroïdie.● Une destruction du corps thyroïde par un processus infil-tratif, sarcoïdose, amylose, néoplasme ou lymphome diffus,thyroïdite fibrosante de Riedel, est plus rare.
2. Hypothyroïdies centrales :
Elles sont liées à un déficit hypothalamique ou hypophysai-re acquis par tumeur (craniopharyngiome, adénome), irra-diation, chirurgie hypophysaire, maladie de Sheehan,hémochromatose, granulome, (sarcoïdose). Le déficit enhormone thyréotrope (TSH) est associé à d’autres déficitshormonaux hypophysaires.
Chez l’enfant les hypothyroïdies sontcongénitales ou acquises.
1. Les hypothyroïdies primaires
● Congénitales: ce sont les dysgénésies thyroïdiennes :ectopie thyroïdienne et athyréose ; le diagnostic est assurépar la scintigraphie ; et les troubles de l’hormono-synthèseplus rares .● Acquises: il s’agit le plus souvent d’une thyroïditeauto-immune attestée par la présence d’auto-anticorpsthyroïdiens à titre élevé ; plus rarement, d’une carence
Endocrinologie-Métabolisme-NutritionB 338
2157L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
• L’hypothyroïdie est l’expression clinique et biologique de la carence en hormonesthyroïdiennes (HT) .• Les hormones thyroïdiennes exercent leurseffets physiologiques sur de nombreux tissus et métabolismes ; de ce fait, leur carences’exprime de façon polymorphe, peu spécifiquedans ses stades initiaux .• Cependant, la qualité de l’explorationfonctionnelle thyroïdienne actuelle permet un diagnostic précoce et ainsi d’éviter les formes évoluées et compliquées,en particulier le coma hypothyroïdien .• L’hypothyroïdie résulte soit d’une altérationfonctionnelle ou organique du corps thyroïde(CT) lui-même : ce sont les hypothyroïdiesprimaires ou périphériques (95 % des étiologies); soit d’un déficit de la commande hypothalamo-hypophysaire :ce sont les hypothyroïdies secondaires ou centrales (5 % des causes).
Points Forts à comprendre

en iode : celle-ci entraîne avant tout un goitre, rarementcompliqué dans nos régions d’une hypothyroïdie ;exceptionnellement enfin, d’une séquelle d’irradiationcervicale ou d’une maladie de surcharge : thalassémie,cystinose...
2. Hypothyroïdies centrales● Congénitales: elles sont liées à un déficit en TSH, fai-sant partie d’une insuffisance antéhypophysaire plusglobale incluant presque toujours l’hormone de crois-sance. ● Acquises: ce sont les causes tumorales de la régionhypothalamo-hypophysaire, en particulier le craniopha-ryngiome à rechercher en priorité par tomodensitomé-trie-imagerie par résonance magnétique. Plus rarement,elles sont la séquelle d’une irradiation crânienne ou d’unprocessus infiltratif (histiocytose).
Physiopathologie
De l’Hypothyroïdie
La carence en hormones thyroïdiennes a de multiplesconséquences métaboliques et tissulaires.
1. Métaboliques
Les principales sont la diminution de la consommationd’O2 et de la production de chaleur centrale ; la diminu-tion de la synthèse et de la dégradation protéique ; la diminution de la dégradation du cholestérol et aug-mentation de la concentration des LDL apoprotéines ;l’augmentation des triglycérides ; enfin la diminution del’absorption intestinale des glucides et de la néogluco-genèse.
2. Tissulaires Elles sont ubiquitaires et intéressent : le tissu sous-cuta-né infiltré par le dépôt de glyco-aminoglycans (GAG) ;le système cardiovasculaire où la diminution des effetschronotrope et inotrope positifs des hormones thyroï-diennes conduit à une diminution du débit cardiaque ;l’hypertension artérielle n’est probablement qu’uneassociation ; l'athérogenèse est accélérée par les modifi-cations lipidiques ; les poumons où la carence en hor-mones thyroïdiennes a des effets musculaires périphé-riques et nerveux central, aboutissant à unehypoventilation avec hypoxie-hypercapnie ; les musclesstriés, avec ralentissement de la contraction et de larelaxation musculaire ; le système nerveux central avecralentissement de l’activité ; le système digestif, avechypopéristaltisme œsophagien, gastrique, intestinal etcolique ; l'hématopoïèse avec diminution de l'érythro-poïèse ; les autres systèmes hormonaux : diminution ducatabolisme du cortisol ; diminution de l’hormone decroissance et de sa réponse à divers stimuli ; diminutionde la clearance de l’eau libre en partie liée à une hypersé-crétion d’hormone antidiurétique ; augmentation de la
prolactine. Chez le nouveau-né et l’enfant, la carence enhormones thyroïdiennes a en outre deux conséquencesredoutables : l’altération du développement et de lamaturation du système nerveux central entraînant retardmental et diverses anomalies neurologiques ; le retard dela maturation osseuse et la présence d’une dysgénésieépiphysaire qui ont pour conséquence un retard de crois-sance. Plus accessoirement, un retard pubertaire. Lesmodifications enzymatiques hépatiques expliquent l’ic-tère prolongé néonatal, lié à leur retard de maturation.
Des diverses causes
1. Atrophie idiopathique
De mécanisme longtemps mystérieux, est probablementle stade ultime d’une thyroïdite auto-immune atrophiante.
2. Thyroïdites auto-immunes Leur mécanisme est incomplètement élucidé mais attestépar de nombreux arguments : prédisposition génétique,anomalie de l’immunorégulation avec présence d'auto-anticorps antithyroïdiens à titre élevé et infiltration lym-phocytaire du corps thyroïde.
3. Hypothyroïdies iatrogèniquesElles sont pour la plupart de mécanisme évident : anti-thyroïdiens de synthèse, destruction chirurgicale ou pariode radioactif du corps thyroïde etc. L’iode inhibe lasynthèse des hormones thyroïdiennes et la protéolyse,donc la libération des hormones thyroïdiennes mais chezle sujet normal, il y a un phénomène d’échappement.
4. Déficit marqué en iodeDans certains pays, il entraîne une insuffisance de pro-duction hormonale.
Diagnostic
La sévérité et la durée du déficit hormonal déterminentde nombreuses formes cliniques. De plus, les signesrévélateurs de même que l’expression clinique desformes patentes sont différents chez l’adulte et l’enfant etseront considérés séparément.
Formes cliniques de l’hypothyroïdie de l’adulte
1. Hypothyroïdie fruste ou débutanteLes signes habituels sont : apathie et diminution de l’acti-vité, asthénie globale avec lenteur intellectuelle, frilosité etsécheresse de la peau, crampes, bouffissure des paupièreset du visage, gain de poids, constipation, syndromedépressif.
2. Forme typique, patente
Elle associe les signes suivants à caractère acquis.● Le syndrome cutanéo-muqueuxqui comporte une
2158 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P O T H Y R O Ï D I E

infiltration de la peau par le myxœdème ou faux œdème,élastique, ne prenant pas le godet, épaississant les pau-pières, le dos des mains et des pieds, les doigts et orteilsd’aspect boudiné, les creux sus-claviculaires, lesmuqueuses avec macroglossie, raucité de la voix, hypo-acousie, ronflements nocturnes. La peau est sèche, épais-sie et froide. Les phanères sont altérées : cheveux raréfiés,secs et cassants ; ongles striés et cassants, dépilation axilo-pubienne et de la queue du sourcil, signe classique. Dansl’ensemble, ce syndrome donne un aspect caractéristique àla face, bouffie, en pleine lune, inexpressive, à la pâleurcireuse. Il rend compte de la prise de poids modérée, diffu-se, sans jamais d’obésité marquée.● Signes neuropsychiques: le maître mot est ralentisse-ment : ralentissement psychique avec indifférence à sonétat, somnolence, apathie, diminution de l’attention, de lamémoire, de l’idéation ; ralentissement moteur : asthénied’effort, gestes rares et lents. Outre ce ralentissement psy-chomoteur, on peut observer : des paresthésies dans le ter-ritoire du nerf médian témoin d’un syndrome du canal car-pien ; des troubles du comportement intermittents :bouffées de colère , agitation paradoxale avec délire hallu-cinatoire, dépression, une polyneuropathie sensitivo-motrice diffuse plus rarement, enfin, un syndrome cérébel-leux exceptionnel.● Signes musculaires: fréquents, il comportent, lescrampes, l’enraidissement et la lenteur de décontraction.La myopathie thyroïdienne lorsqu’elle est présente déter-mine soit une amyotrophie, soit une myopathie hypertro-phique pseudomyotonique.● Signes cardiovasculaires : une dyspnée d’effort peutdominer le tableau clinique ; les bruits du coeur sont assour-dis, la bradycardie est fréquente, la pression artérielle estnormale ou un peu élevée. Les signes sont surtout paracli-niques : cardiomégalie, peu battante à la radio, témoin del’infiltration myocardiaque et de l’épanchement péricar-dique ; électrocardiogramme avec bas voltage, T plat ouinversé ; l’échocardiographie précise ces données myocar-diques et péricardiques. ● Signes digestifs: il s’agit d’anorexie, nausées, dysphagie; la constipation est le symptôme le plus net ; à un stadetrès évolué, mégacôlon et iléus paralytique peuvent s’ob-server ; une ascite, riche en protéine, est présente dans 4 %des cas.● Le syndrome d’hypométabolismecomporte, outre l’as-thénie physique, intellectuelle et sexuelle, une frilositéinconstante et une hypothermie modérée.● Les signes génitaux: chez la femme jeune, on observefréquemment des méno-métrorragies, des cycles anovula-toires avec hypofécondité voire stérilité ou un syndromed’aménorrhée galactorrhée par hyperprolactinémie, enfindes avortements précoces répétés. Chez l’homme, le signele plus fréquent est l’impuissance.
3. Formes compliquées de l’hypothyroïdie de l’adulteEn l’absence de traitement, l’évolution spontanée conduitprogressivement aux complications.
● Coeur myxœdémateux: les 2 complications cardiaquespossibles sont l’insuffisance cardiaque rare, de réalité dis-cutée en l’absence de maladie cardiaque sous-jacente, cau-sée par une cardiomyopathie hypothyroïdienne ou par unépanchement péricardique abondant et l’insuffisance coro-narienne : plus fréquente chez l’hypothyroïdien que chezle sujet euthyroïdien de mêmes âge et sexe ; conséquenced’une accélération (probable mais non prouvée) de l’athé-rosclérose coronarienne au cours de l’hypothyroïdie pro-longée ; masquée par l’hypométabolisme et la diminutiondes besoins en O2 : les manifestations angineuses sontrares en état d’hypothyroïdie ; démasquée, notion essen-tielle, sous l’effet du traitement et du retour à l’euthyroïdie; à rechercher donc avec soin avant le début du traitement.● Le coma myxœdémateux: expression ultime d’unehypothyroïdie négligée, c’est une complication majeuresouvent mortelle, malgré le traitement. Cliniquement,c’est l’installation progressive d’un coma plus au moinsprofond, sans signe de localisation, aréflexique, avec sou-vent hyperprotéinorachie, associé dans un quart des cas àdes crises convulsives généralisées ; et aux signes clas-siques accentués : bradycardie, bradypnée, infiltrationcutanée et surtout hypothermie, excellent signe ; biologi-quement : hyponatrémie, hypokaliémie, hypoglycémie,anémie, hypoxie et acidose respiratoire (par hypoventila-tion alvéolaire, encombrement, macroglossie) ; les fac-teurs déclenchants sont à connaître : froid ; infections,stress chirurgical ou traumatique, hémorragie, accidentvasculaire cérébral(parfois au premier plan, il peut faireméconnaître le coma hypothyroïdien), médicaments(sédatifs, barbituriques, opiacés).
Formes cliniques de l’hypothyroïdiede l’enfant
1. Chez le nouveau-né et nourrisson
L’hypothyroïdie est le plus souvent diagnostiquée par undépistage systématique au 5e jour par le dosage de TSH surdu sang capillaire. Les signes évocateurs en période néo-natale sont prolongation de l’ictère néonatal, hypotonieaxiale, fontanelle postérieure large, hypothermie ; dans lespremières semaines de la vie, difficulté à boire, macro-glossie, constipation, ballonnement abdominal avec her-nie, raucité du cri, enfant trop sage et somnolent ; en l’ab-sence de traitement et dans les premiers mois, on constateune insuffisance de croissance staturale avec croissancepondérale normale ; un retard d’âge osseux et surtout unretard psychomoteur irréversible.
2. Chez l’enfantLe tableau clinique dépend de l’importance de l’hypothy-roïdie. Le signe le plus évocateur est le ralentissement de lavitesse de croissance staturale, surtout s’il est associé à uneprise pondérale. Les autres signes sont : asthénie, constipa-tion, frilosité, diminution des performances scolaires. Laprésence d’un goitre dépend de l’étiologie de l’hypothyroï-die. L’âge osseux est inférieur à l’âge chronologique.
Endocrinologie-Métabolisme-Nutrition
2159L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8

Diagnostic biologique
Étant donné les difficultés du diagnostic clinique desformes précoces, les examens de laboratoire peuventêtre largement utilisés.
1. Dosages hormonaux Le diagnostic de l’hypothyroïdie primaire est extrême-ment simple :● Dans la situation typique: l’abaissement de la T4libre caractérise l’hypothyroïdie (T4 est le principal pro-duit de sécrétion du corps thyroïde) ; le taux de T3 amoins d’intérêt car il peut être encore normal alors quela T4 est abaissée ; il est fréquemment abaissé chez dessujets euthyroïdiens mais atteints d’une maladie nonthyroïdienne aiguë ou chronique (infection aiguë, cir-rhose, insuffisance rénale, dénutrition etc.). C’est le syn-drome de basse T3 isolé sans hypofonction thyroïdien-ne. L’élévation du taux de TSH plasmatique est le test leplus sensible du diagnostic d’hypothyroïdie primaire.● Autres possibilités: au début de l’hyposécrétion thy-roïdienne, les taux plasmatiques de T4 et de T3 peuventêtre encore dans la fourchette de la zone normale, com-pensés par une hypersécrétion de TSH dont le taux estisolément élevé. Le test à la TRH n’a plus de véritabled’intérêt dans l’hypothyroïdie primaire, compte tenu del’extrême sensibilité du dosage de TSH.● L’hypothyroïdie secondaireest caractérisé par un tauxde T4 libre abaissé, un taux de TSH bas ou normal (sans“ riposte ” à l’abaissement de T4). Un test à la TRH per-met schématiquement de distinguer si l’hypothyroïdieest d’origine hypophysaire ou hypothalamique.
2. Retentissement hormonal ● Au niveau hypophysaire,on constate une hyperpro-lactinémie dans 75 % des cas, pouvant entraîner un syn-drome aménorrhée-galactorrhée qui peut être révélateurde l’hypothyroïdie. ● Au niveau corticosurrénalien, cortisolémie et cortisollibre urinaire (FLU) sont normaux ; cependant, le tauxde sécrétion du cortisol peut être diminué dans 50 % descas dans les hypothyroïdies profondes et prolongées,réversible sous traitement. L’existence d’une insuffisan-ce corticosurrénalienne organique associée devra detoute façon être recherchée.
3. Autres anomalies biologiques Elles témoignent du retentissement périphérique de la
carence en hormones thyroïdiennes. Quelquefois révéla-trices, elles sont cependant de valeur diagnostiquemédiocre. Il s’agit d’une hypercholestérolémie et moinssouvent d’une hypertriglycéridémie ; d’une anémie pré-sente dans 30 à 60 % des cas, soit normocytaire, normo-chrome, arégénérative par hypoplasie médullaire, soitmacrocytaire par diminution de la vitamine B12 et desfolates et exceptionnellement par association à une ané-mie de Biermer, soit microcytaire hypochrome par dimi-nution de l’absorption du fer et présence de ménomé-
trorragies. Les enzymes musculaires CPKMM sont éle-vées, témoin de la myopathie ; une hyponatrémie dedilution s’observe dans les formes sévères. Les enzymeshépatiques telle la lactico-déshydrogénase, les ASAT etALAT sont fréquemment élevées.
4. Explorations à visée étiologique Elles seront demandées en fonction du contexte :dosage d’anticorps antithyroïdiens, scintigraphie et cap-tation d’iode radioactif, iodémie et iodurie, recherched’autres déficits hormonaux, notamment hypophysaires,échographie thyroïdienne.
Traitement
Son but est de restaurer les concentrations tissulairesadéquates d’hormones thyroïdiennes pour supprimer lesanomalies cliniques et biologiques. Il est rarement étio-logique : suppression d’un médicament, d’une surchargeiodée, ablation d’une tumeur hypophysaire ; traitementd’une sarcoïdose. Il s’agit le plus souvent d’un traite-ment substitutif à vie qu’il faudra faire accepter par lepatient.
Les hormones synthétiques sontles seules utilisées
La lévothyroxine (l-T4) se présente sous forme degouttes pédiatriques (1 goutte = 5 µg) ; de comprimés à25, 75, 100 et 150 µg. La L-triiodo-thyronine (l-T3) seprésente sous forme de comprimés de 25 µg. De façongénérale, la l-T3, hormone immédiatement active estpeu utilisée à cause de sa demi-vie courte nécessitantdes prises répétées et provoquant de petits signes d’into-lérance cardiovasculaire chez le sujet âgé. Elle est parcontre indiquée dans des situations particulières : sevra-ge ou traitement rapide. La l-T4 est préférable car elle sefixe plus sur les protéines vectrices ; son relargage estlent et sa demi-vie longue ; elle se comporte comme unepro-hormone de T3 produite à partir du réservoir de T4.
Posologies
● Chez l’adulte jeune, elle est de 1,5 à 2,5 µg/kg/j, c’est-à-dire 150 et 200 µg par jour (quelques patients ont besoinde doses supérieures à 200 µg par jour car l’absorptionvarie d’un sujet à l’autre). La posologie diminue avecl’âge.● Chez l’enfant,la forme en goutte est utilisée. La poso-logie est de 8 µg/kg par jour durant le premier trimestrede vie puis les besoins diminuent pour atteindre 5 µg/kg/jour vers l’âge de 2 ans.
Modalités d’administration :
Les produits sont à prendre en une fois, à la mêmeheure, avant les repas. La posologie initiale sera déter-minée en fonction de l’âge, de l’ancienneté et la sévérité
2160 L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
H Y P O T H Y R O Ï D I E

de l’hypothyroïdie, de l’existence d’une atteinte cardio-vasculaire.
Surveillance sous traitement
Les meilleurs éléments de surveillance sont d’abord cli-niques avec initialement perte de poids et polyurie ; dispa-rition complète de tous les signes. On vérifiera l’absencede surdosage (palpitations, nervosité, et à la longue risqued’ostéopénie) ; on vérifiera aussi la tolérance cardiaqueclinique et électrique. Chez l’enfant, la surveillance se faitsur l’évolution de la croissance staturo-pondérale et sur lafréquence cardiaque. Puis, biologiques : dans l’hypothy-roïdie primaire, le taux de TSH est l’élément essentiel àdoser 3 à 5 semaines après la dernière modification desposologies : il doit être dans la zone de normalité. La T3 etla T4 doivent être normalisées (la T4 libre est souvent unpeu élevée). Chez le sujet âgé, la normalisation de T4 et T3suffit, celle de TSH n’est pas toujours nécessaire. Dansl’hypothyroïdie secondaire, seuls les dosages de T3 et T4sont utilisables.
Cas particuliers
1. Insuffisance coronaire
L’existence reconnue d’une insuffisance coronarienneimpose que la mise en route du traitement soit faite enmilieu hospitalier : correction d’une éventuelle anémiepuis traitement substitutif très progressif associé à desmesures cardiologiques spécifiques.
2. Insuffisance thyroïdienne secondaire
Il faudra rechercher l’existence d’une insuffisance cor-ticotrope associée et la traiter avant l'hypothyroïdie pouréviter une insuffisance surrénale aiguë. Dans le doute,on adjoindra 30 à 50 mg d’hydrocortisone par jour à lathyroxine.
3. Le coma myxœdémateux
C’est une urgence à traiter en milieu hospitalier. On n’at-tendra pas les résultats des dosages hormonaux. Dès lesprélèvements sanguins faits, on utilisera les mesuressuivantes : L-T4 : 200 à 500 µg IV lente ou IM le premierjour ; hydrocortisone : 200 à 300 mg IV toutes les 3 à 4heures ; oxygénation et parfois assistance respiratoire.■
Endocrinologie-Métabolisme-Nutrition
2161L A R E V U E D U P R A T I C I E N ( P a r i s ) 1 9 9 8 , 4 8
• L’hypothyroïdie peut être méconnue du fait de son polymorphisme clinique : de ce fait, lasymptomatologie initiale doit être toujoursprésente à l’esprit .• Le diagnostic hormonal est aisé. Chez lenouveau-né, le dépistage systématique permetd’éviter un grave et irréversibledysfonctionnement intellectuel et neurologique et plus tard un nanisme dysharmonieux. • Dans tous les cas, il faut préciser la cause del’hypothyroïdie, parfois directement curable ;• Le traitement, le plus souvent substitutif à vie,est simple mais doit toujours prendre en compteune observance médiocre, le risque d’unedécompensation coronarienne chez le sujet âgé,plus rarement le risque d’une insuffisancesurrénale aiguë. Il permet d’éviter unecomplication rare mais redoutable : le comahypothyroïdien.
Points Forts à retenir

Endocrinologie - Métabolisme - NutritionB 342
1805L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
Insuffisance surrénale aiguë d’origine surrénale
1. L’insuffisance surrénale chronique est connue et traitéeIl s’agit d’une maladie d’Addison quelle qu’en soitl’origine (auto-immune le plus souvent), et l’on doitimpérativement rechercher un facteur déclenchant ladécompensation aiguë :– arrêt inopiné du traitement par le patient lui-même ;– régime désodé ;– effort physique majeur ;– perte de sel : vomissements, diarrhée, sueurs abon-dantes dans un contexte de chaleur excessive ;– syndrome infectieux (grippe souvent) ;– traumatisme ;– chirurgie ;– grossesse évolutive ;– stress de toute nature ;– origine médicamenteuse : diurétiques, laxatifs, opiacés,sédatifs et médicaments inducteurs enzymatiques[rifampicine (Rifadine), phénobarbital (Gardénal), phényl-hydantoïne (Di-Hydan)].Cette situation est évitable par la prévention et l’éduca-tion du patient.Les troubles de l’hormonosynthèse surrénale (bloc en 21 hydroxylase) sont connus depuis l’enfance, traitéset peuvent donner dans les mêmes circonstances uneinsuffisance surrénale aiguë. Chez l’adulte, ces blocsenzymatiques surrénaux ne sont jamais découverts dansce contexte.
2. L’insuffisance surrénale chronique est méconnueElle doit être suspectée par l’interrogatoire à larecherche d’une symptomatologie évocatrice d’insuffi-sance surrénale lente pouvant évoluer depuis des moisvoire des années et par l’examen clinique qui retrouveune mélanodermie importante. La décompensationaiguë de la maladie latente se fait le plus souvent à l’oc-casion d’un des facteurs déclenchants décrits ci-dessus.L’origine en est le plus souvent auto-immune, confirméepar la recherche d’anticorps anti-surrénaux, plus rare-ment d’origine tuberculeuse.
Étiologie
L’insuffisance surrénale aiguë (ISA) peut être d’originebasse, surrénale ou haute : insuffisance corticotrope.L’insuffisance surrénale aiguë d’origine basse induit un double déficit hormonal : cortisol et aldostérone,secondaire à une atteinte des deux glandes surrénales.L’insuffisance corticotrope, d’origine hypothalamo-hypophysaire, n’entraîne qu’un déficit de production decortisol, les glandes surrénales étant normales et assu-rant la production d’aldostérone.Sur le plan étiologique, il existe 4 circonstances cliniques différentes à l’origine d’une insuffisance surrénale aiguë.
Insuffisance surrénaleaiguë de l’adulte Étiologie, diagnostic, prévention, conduite à tenir en situation d’urgence avec la posologiemédicamenteuse
PR Sylvie ARLOT
Service d’endocrinologie, hôpital Sud, 80054 Amiens Cedex.
• L’insuffisance surrénale aiguë est une situation rare, mais fatale en l’absencede traitement d’urgence. Elle survient le plussouvent dans un contexte de maladie d’Addisonconnue où un facteur déclenchant est négligé. Il s’agit alors d’un double déficit hormonal en cortisol et en aldostérone à l’origine des signes cliniques et biologiques.
• L’insuffisance surrénale aiguë peut être d’origine haute : insuffisance corticotrope. Le déficit ne porte que sur la production de cortisol. La symptomatologie est pluspauvre. Celle-ci survient le plus souvent après sevrage d’une corticothérapie,circonstance également évitable par la prévention.
• En situation de stress, le patient doit adapterson traitement. Cela justifie la prévention par l’éducation du patient et de son entourage.Le traitement doit être débuté en urgence dès la suspicion clinique du diagnostic.
Points Forts à comprendre

3. L’insuffisance surrénale aiguë est brutale(lésions bilatérales aiguës des 2 surrénales)
• Hémorragies et (ou) hématomes bilatéraux des sur-rénales :ils sont rares et gravissimes, souvent liés à untrouble de l’hémostase :– coagulation intravasculaire disséminée (CIVD) ;– thrombopénie à l’héparine ;– traitement anticoagulant (lors de l’institution d’untraitement par antivitamines K) ;– lupus ;– syndrome des antiphospholipides ;– maladie thrombotique (cancers, hémopathies).Exceptionnellement, ils peuvent survenir dans uncontexte traumatique : traumatisme thoraco-abdominalou crânien sévère.• Les métastases bilatérales des surrénalessont fré-quentes au cours de l’évolution des pathologies cancé-reuses. Elles sont le plus souvent asymptomatiques maispeuvent parfois se manifester sous forme d’une insuffi-sance surrénale aiguë.• Certaines infections opportunistesau cours d’un sidaavéré peuvent se localiser au niveau des surrénales :mycose, cytomégalovirus…
Insuffisance surrénale aiguë d’originehypothalamo-hypophysaire : insuffisancecorticotrope
• L’insuffisance corticotrope aiguëpeut être d’originehypophysaire : 1re manifestation d’un panhypopitui-tarisme, ancien et méconnu, quelle qu’en soit la cause(tumeur hypophysaire, syndrome de Sheehan…). Ilexiste habituellement un facteur déclenchant qui aggravela carence en cortisol et induit une insuffisance cortico-trope aiguë : tous les types de stress ou d’affectionsintercurrentes. Les autres signes de panhypopituitarisme,en particulier de déficit gonadotrope, sont faciles àretrouver par l’interrogatoire. Mais l’insuffisance cortico-trope peut être récente dans certaines pathologies hypo-physaires comme l’apoplexie hypophysaire. Elle peutêtre également isolée, seule manifestation du déficithypophysaire.Après chirurgie hypophysaire pour un macro-adénome,le risque est faible, car le traitement substitutif est misen œuvre d’emblée.• Sevrage d’une corticothérapie au long cours :l’insuf-fisance surrénale aiguë apparaît lors d’un arrêt brutal de lacorticothérapie ou lors d’une dégression trop rapide desdoses de corticoïdes. Elle peut également apparaître àdistance du sevrage, lors d’une affection intercurrente.En pratique, lorsqu’un sevrage d’une corticothérapie aulong cours est programmé, il convient de diminuer lesdoses de corticoïdes jusqu’à l’équivalent de 30 mg/jd’hydrocortisone (environ 7 mg/j de prednisone,Cortancyl), de les remplacer par l’hydrocortisone à laposologie de 30 mg/j et de proposer une alimentationnormosodée. Une surveillance clinique et hormonale estnécessaire pour envisager le sevrage définitif.
• Les cause iatrogéniqueslors du traitement d’un syn-drome de Cushing : lors d’un traitement médical par anti-cortisoliques de synthèse [aminoglutéthimide, Orimétène(risque immédiat), op’DDD, Mitotane (après une quinzainede jours de traitement), kétoconazole, Nizoral, à fortesdoses (après quelques jours)], un traitement par hydro-cortisone doit systématiquement être associé.Après traitement chirurgical d’un syndrome de Cushingtel que l’ablation d’une tumeur surrénalienne unilatéralesécrétante, la surrénalectomie totale bilatérale ou l’adé-nomectomie hypophysaire dans une maladie deCushing, l’insuffisance corticotrope est classique parinertie de l’axe corticotrope. Un traitement par hydro-cortisone doit systématiquement être institué dès l’actechirurgical.
DiagnosticLe diagnostic doit être évoqué dès l’examen cliniqueafin de démarrer le traitement d’urgence. Les signes bio-logiques viendront étayer l’hypothèse diagnostique, quiest confirmée définitivement par les dosages hormonauxreçus ultérieurement.
Signes cliniques
Ils s’installent rapidement en quelques heures, d’embléeou après une phase prodromique parfois longue(quelques mois voire années) d’insuffisance surrénalelente (tableau I).Quatre signes majeurs caractérisent le tableau d’insuffi-sance surrénale aiguë.
1. Signes digestifsNausées, vomissements, diarrhée parfois cholériformeévoluent dans un contexte de douleurs épigastriques ouabdominales diffuses. L’examen clinique abdominal estnormal. Le tableau peut évoquer un aspect pseudo-chirurgical.
2. Troubles psychiquesIls sont variables. L’asthénie évolue vers une adynamieextrême puis un coma. Parfois un tableau psychiatriquepeut apparaître sous forme d’agitation, de délire ou deconfusion. L’examen neurologique ne retrouve aucunsigne de localisation.
3. Troubles tensionnelsLa tension artérielle s’abaisse jusqu’au collapsus, avec unpouls petit, filant et rapide. Les extrémités sont froides.
4. Perte de poids Elle est intense, accompagnée d’une déshydratationextracellulaire majeure : un pli cutané et une hypotoniedes globes oculaires sont constants.
5. Autres troublesDes douleurs diffuses sont fréquentes : myalgies,arthralgies et céphalées. Une hyperthermie peut exister,sans signes infectieux.
I N S U F F I S A N C E S U R R É N A L E A I G U Ë D E L ’ A D U L T E
1806 L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9

Dans l’insuffisance corticotrope, les signes biologiquesliés au déficit minéralocorticoïde sont absents, seulel’hypoglycémie est présente. Parfois, une hyponatrémiede dilution est observée.
Diagnostic hormonal
Dès la suspicion du diagnostic d’insuffisance surrénaleaiguë, il faut effectuer des prélèvements sanguins pourdosages plasmatiques du cortisol, de l’ACTH (adreno-corticotrophic hormone), de la rénine et de l’aldostérone.Il ne faut pas attendre les résultats de ces examens pourtraiter. Les résultats confirment définitivement le diagnostic devant des taux effondrés de cortisol et d’aldostérone, et des taux élevés d’ACTH et de rénine.Dans l’insuffisance corticotrope, le taux plasmatique decortisol est bas, le taux d’ACTH normal ou bas.
Prévention
La prévention repose sur l’éducation du patient dèsqu’un diagnostic d’insuffisance surrénale est posé. Ellepermet ainsi d’éviter ce type de décompensation. Lesprincipes sont les suivants :– information du patient et de son entourage ;– alimentation normalement sodée ;– proscrire les régimes sans sel, les diurétiques, les laxatifs ;– en cas de stress ou d’affection intercurrente, doublervoire tripler les doses quotidiennes d’hydrocortisone ;– en cas de troubles digestifs, utiliser la voie intramuscu-laire pour l’injection d’hémisuccinate d’hydrocortisone ;– prévenir l’anesthésiste en cas d’intervention chirurgicale.L’ensemble de ces principes doit être inscrit sur la carted’insuffisant surrénal donnée au patient. Sur cette carte,doivent également figurer les noms de son médecin trai-tant et de son spécialiste ainsi que le traitement habituel.
À ce stade, le diagnostic d’insuffisance surrénale aiguëdoit être évoqué, après avoir recherché une mélanodermieou une phase prodromique d’insuffisance surrénale lenteet un facteur déclenchant. Dès lors le traitement doit êtreinstitué.S’il s’agit d’une insuffisance corticotrope, les signes cli-niques sont pauvres : asthénie, anorexie, douleursvagues à type de céphalées ou d’arthralgies, sensationde malaise général. Les hypoglycémies cliniques sontrares, de type organique. Ces signes cliniques peuventêtre évocateurs dans un contexte de sevrage de cortico-thérapie, d’autant qu’ils contrastent avec un faciès évo-cateur de syndrome de Cushing.
Signes biologiques
Le bilan biologique doit être fait d’urgence, en mêmetemps que débute le traitement. Ce bilan doit comporter :– dans le sang : numération formule sanguine (NFS), glycé-mie, ionogramme, urée, créatinine, protides et gaz du sang ;– dans les urines : ionogramme sur un échantillon.Ces examens biologiques (tableau II) donnent les résultatssuivants :• dans le sang,la natrémie est basse, parfois inférieureà 120 mmol/L ; l’hyperkaliémie peut être menaçanteimpliquant la réalisation d’un électrocardiogramme(ECG), la chlorémie est abaissée. Les gaz du sang met-tent en évidence une acidose d’origine métabolique.L’hypoglycémie est constante, majorant les signes psy-chiques. La numération formule sanguine montre unehyperéosinophilie classique, peu spécifique. Les signesd’insuffisance rénale fonctionnelle sont constants(déshydratation extracellulaire) : élévation des tauxd’urée, de protides et de l’hématocrite ;• dans les urines,la natriurèse est élevée par fuite sodéeet la kaliurèse est basse, témoignant du déficit en aldo-stérone (en l’absence de prise de diurétiques « épar-gneurs » de potassium).
Endocrinologie - Métabolisme - Nutrition
1807L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
Signes digestifs
Asthénie
Hypotension artérielle
Amaigrissement
Mélanodermie
Tableau pseudo-chirurgical
Adynamie, coma
Collapsus
Déshydratation extracellulaire
Mélanodermie
Insuffisance surrénale lente
Insuffisancesurrénale aiguë
Évolution des signes cliniques de l’insuffisance surrénale lente
méconnue vers l’insuffisancesurrénale aiguë
TABLEAU I
Natrémie
Kaliémie
Chlorémie
Bicarbonates
Glycémie
Urée
Créatinine
Natriurèse
Kaliurèse
Sang Urines
Signes biologiques de l’insuffisance surrénale aiguë
TABLEAU II

Conduite à tenir en situation d’urgence avec la posologie médicamenteuse
Le pronostic vital est mis en jeu à court terme. Celanécessite, si besoin est (gravité du tableau clinique, éloi-gnement du centre hospitalier), une prise en charge àdomicile sous forme d’une injection intraveineuse de100 mg d’hémisuccinate d’hydrocortisone et une hospi-talisation en milieu spécialisé pour mise en place dutraitement. Le traitement comporte 2 volets essentiels :corriger les désordres hydro-électrolytiques et compenserle déficit hormonal.
Mise en condition du patient
Une voie d’abord veineuse périphérique ou centrale,selon la gravité clinique, est mise en place. Les soins denursage seront prescrits avec une prévention des complications de décubitus par héparine de bas poidsmoléculaire (Lovenox). Au niveau des examens complémentaires, on réalise des prélèvements bactério-logiques systématiques ainsi qu’un électrocardiogramme.Un test de grossesse doit être demandé s’il s’agit d’unefemme jeune.
Recharge volémique et sodée
S’il existe un collapsus, une perfusion de solutés macro-moléculaires est mise en place sous contrôle de la pres-sion veineuse centrale.La réhydratation comporte une perfusion de 4 à 6 L desérum glucosé à 10 % pour les premières 24 heures : 2 à3 L pendant les 6 premières heures, 2 à 3 L dans les 18heures suivantes, avec 4 à 6 g de chlorure de sodium parlitre sans adjonction de potassium.
Traitement hormonal
Si l’injection intraveineuse de 100 mg d’hémisuccinated’hydrocortisone n’a pas été faite à domicile, un bolusde 100 mg doit être injecté par voie veineuse et relayépar 300 mg par 24 heures d’hémisuccinate d’hydrocorti-sone également par voie veineuse, à la seringue élec-trique, pour les 24 premières heures.
Traitement du facteur déclenchant
Le traitement antibiotique est quasi systématique aprèsles prélèvements bactériologiques. Une grossesse doitêtre systématiquement recherchée chez une femmejeune.
Éléments de surveillance
La surveillance clinique implique de noter : l’état deconscience ; le pouls ; la pression artérielle ; la fréquencerespiratoire ; la température ; le poids ; la diurèse.
On y associe la surveillance biologique des iono-grammes sanguins et urinaires, de la glycémie et destaux d’urée et de créatinine plasmatiques, toutes les 3 heures au début.L’électrocardiogramme doit également être surveillé.
Évolution
L’amélioration clinique est rapide, en moins de 24 h. La posologie d’hémisuccinate d’hydrocortisone par voie veineuse à la seringue électrique peut être réduite à 200 mg/24 h le lendemain, puis 100 mg/24 h et dégression progressive jusqu’à la posologie d’entretiende 30 à 40 mg d’hydrocortisone. Cette dégression estfaite par voie orale dès que l’état du patient le permet.La fludrocortisone est introduite dès que la voie orale estpossible à raison de 50 µg/j le matin.L’éducation du patient est systématiquement revue avantsa sortie. ■
I N S U F F I S A N C E S U R R É N A L E A I G U Ë D E L ’ A D U L T E
1808 L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
• Le diagnostic d’une insuffisance surrénaleaiguë est un diagnostic clinique, qui doit impliquer la mise en route du traitement d’urgence. Le plus souvent, il s’agit d’uneinsuffisance surrénale d’origine basse, connueou non, qui décompense dans un contexted’agression psychique, physique…
• Les signes biologiques confortent l’hypothèsediagnostique clinique de carence hormonale :hypoglycémie, hyponatrémie avec fuite sodéeurinaire et hyperkaliémie.
• Le traitement comporte 2 éléments essentiels : compenser la perte hydro-électrolytique et le déficit hormonal.
• L’insuffisance corticotrope est de diagnosticplus difficile. Elle se voit le plus souvent après sevrage brutal et non compensé d’une corticothérapie au long cours, facilementretrouvée par l’interrogatoire et la présence de signes de syndrome de Cushing iatrogénique.
• La prévention par l’éducation du patient est primordiale pour éviter la récidive.
Points Forts à retenir
Bertagna X. Corticothérapie et fonction surrénalienne. Éditionstechniques. Encycl Med Chir (Paris, France). Endocrinologie-Nutrition, 1990 ; 10015 A20, 12 : 5 p.
Mosnier-Pudar H, Paoli V, Luton JP. Insuffisances surrénales. Éditions techniques. Encycl Med Chir (Paris, France).Endocrinologie-Nutrition, 1991 ; 10015 A10 : 14 p.
POUR EN SAVOIR PLUS

Endocrinologie - Métabolisme - NutritionB 343
2257L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
La sécrétion d’aldostérone, indépendante de la cortico-trophine, est soumise au contrôle exercé par l’angio-tensine II (résultant de l’activation du système rénine-angiotensine) et le potassium circulant.L’atteinte directe des 2 glandes surrénales par un processuspathologique (insuffisance surrénale lente primitive oumaladie d’Addison) entraîne un déficit global en cortico-stéroïdes (cortisol, aldostérone et androgènes) et s’ac-compagne d’une élévation réactionnelle du taux plasma-tique de corticotrophinepar absence de rétrocontrôlenégatif du cortisol sur l’hypophyse. L’augmentation de laconcentration plasmatique de corticotrophine est alorsresponsable d’une stimulation de la production de mélaninepar les mélanocytes cutanés (mélanodermie). À l’inverse,l’insuffisance surrénale peut résulter d’un déficit en cor-ticotrophine (insuffisance corticotrope). Il s’agit alorsd’une insuffisance surrénale lente secondaire dont l’origine peut être fonctionnelle ou organique. Dans cecas, la sécrétion des minéralocorticoïdes est conservée. Le tableau clinique de l’insuffisance surrénale, quirésulte des déficits stéroïdiens et du taux plasmatique decorticotrophine, est par conséquent variable, selon l’origine(primitive ou secondaire) de l’atteinte surrénale.
Étiologie
Insuffisances surrénales lentes primitives(maladie d’Addison)
1. Insuffisance corticosurrénale auto-immuneC’est, actuellement, la première cause d’insuffisancesurrénale lente primitive. Elle peut être isolée ou, plusrarement, associée à d’autres affections auto-immunesendocriniennes (maladie de Basedow, thyroïdite deHashimoto, diabète insulinodépendant, insuffisancegonadique, hypoparathyroïdie) ou non endocriniennes(à titre d’exemples : maladie de Biermer, vitiligo, poly-arthrite rhumatoïde). Deux grands types de polyendo-crinopathies auto-immunes ont été décrits. Le type Iassocie une hypoparathyroïdie, une insuffisance surrénalelente primitive et une candidose cutanéo-muqueuse.Le type II comporte une insuffisance surrénale lente primitive, une maladie thyroïdienne auto-immune et undiabète insulino-dépendant.
Les insuffisances surrénales lentes (ISL) sont définies parun déficit de sécrétion des hormones corticosurrénaliennes(glucocorticoïdes, minéralocorticoïdes et androgènessurrénaliens) d’installation progressive. Elles peuventêtre primitives par atteinte directe des glandes surrénalesou secondaires à un déficit en corticotrophine (ACTH).Dans tous les cas, elles exposent au risque d’insuffisan-ce surrénale aiguë (ISA), menaçant le pronostic vital, etdont il faut prévenir l’apparition.
PhysiopathologieChacune des trois couches cellulaires (ou zones) du cortex surrénalien est spécialisée dans la synthèse et lasécrétion d’un type de stéroïde. La zone gloméruléesécrète les minéralocorticoïdes (aldostérone), la zone fas-ciculée produit les glucocorticoïdes (cortisol) et la zoneréticulée libère les androgènes surrénaux. L’activitésécrétoire des zones fasciculée et réticulée est contrôléepar l’axe hypothalamo-hypophysaire (fig. 1). L’hypo-thalamus libère la corticolibérine (ou corticotrophin-releasing hormone, CRH) qui stimule la sécrétion anté-hypophysaire de corticotrophine. Celle-ci, libérée dans lacirculation générale, va elle-même stimuler la productionde cortisol et d’androgènes par le cortex surrénal. La cor-tisolémie exerce en retour un rétrocontrôle négatif sur lessécrétions de corticotrophine et de CRH.
Insuffisance surrénalelente de l’adulteÉtiologie, diagnostic, traitement
PR Hervé LEFEBVRE
Service d’endocrinologie et maladies métaboliques, hôpital de Boisguillaume, CHU de Rouen, 76031 Rouen Cedex
• Les signes cliniques et biologiques de l’insuffisance surrénale lente résultent des déficits en gluco- et minéralocorticoïdes.
• La présence d’une mélanodermie, témoin de l’élévation du taux sanguin d’ACTH (adrenocorticorticotropic hormone),permet d’affirmer l’origine primitive d’une insuffisance surrénale lente.
• La fonction minéralocorticoïde est préservée aucours de l’insuffisance surrénale lente secondaire.
• Le traitement substitutif de l’insuffisance surrénale lente (hydrocortisone) a pour but de mimer la sécrétion physiologique de cortisol.
Points Forts à comprendre

2. Tuberculose
Environ 20 % des insuffisances surrénales lentes primitives sont dues à une tuberculose. Le bacille deKoch atteint les glandes surrénales par voie sanguine.Les lésions initialement caséeuses évoluent vers l’atrophie puis la calcification dans 1 cas sur 2.
3. AdrénomyéloneuropathieIl s’agit d’une affection de cause génétique caractériséepar une accumulation d’acides gras à longue chaînedans les tissus surrénal et nerveux. Elle se révèle chezl’adulte jeune par une insuffisance surrénale lente primitive associée à des signes neurologiques défici-taires d’origine médullaire.
4. Infections mycotiquesL’histoplasmose, la coccidioïdomycose et la cryptococ-cose peuvent être à l’origine d’une insuffisance surrénalelente primitive, en particulier dans le cadre d’un syndromede l’immunodéficience acquise (cf. infra).
5. Syndrome de l’immunodéficience acquise (sida)Les infections opportunistes (mycobactérioses atypiques,cytomégalovirus, cryptococcoses, toxoplasmoses) peuvententraîner des destructions des glandes surrénales chezenviron 5 % des patients atteints de sida.
6. Métastases surrénales bilatéralesElles peuvent être responsables d’insuffisance surrénalelente primitive, notamment en cas de cancer primitif dupoumon, de l’estomac, du côlon, de mélanome malin etde lymphomes.
7. Causes iatrogéniquesL’insuffisance surrénale lente primitive peut être laconséquence d’un traitement anticortisolique (Op’DDD[mitotane], kétoconazole…) ou d’une surrénalectomiebilatérale chez un patient traité pour un hypercorticisme.
Insuffisances surrénales lentes secondaires
Les insuffisances surrénales lentes secondaires peuventêtre isolées ou s’intégrer dans le cadre d’une insuffisanceantéhypophysaire globale. Leurs principales causes sontiatrogéniques ou organiques.
1. Iatrogéniques ou fonctionnellesC’est la plus fréquente des causes d’insuffisance surrénalelente secondaire isolée. Les traitements prolongés parcorticoïdes de synthèse entraînent une inertie de l’axecorticotrope par inhibition des sécrétions de CRH et de
I N S U F F I S A N C E S U R R É N A L E L E N T E D E L ’ A D U L T E
2258 L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
Représentation schématique de la régulation de l’activité sécrétoire du cortex surrénal. La corticotrophine stimule lessécrétions de cortisol et d’androgènes alors que la production d’aldostérone est contrôlée par l’angiotensine II et le potassium.En retour, le cortisol rétro-inhibe les sécrétions de CRH et de corticotrophine et l’augmentation de la volémie induite par l’aldostérone freine la sécrétion de rénine.
1
Rénine
Angiotensine I
Angiotensine II
Volémie
Aldostérone
K+
Cortisol
Androgènes
ACTH
Antéhypophyse
CRH
Hypothalamus
Glomérulée
Fasciculée
Réticulée
Cortex surrénalien
+
+
+
+ +–
–
–

En dehors de l’insuffisance surrénale aiguë, ils se limitent à une anorexie globale et à une constipation. Laprésence de nausées, douleurs abdominales et diarrhéeannonce la décompensation. • Amaigrissement : il est constamment présent et souvent modéré. Son mécanisme est multifactoriel :perte hydrosodée consécutive au déficit en minéralo-corticoïdes, diminution de l’apport calorique lié à l’anorexie, réduction de l’anabolisme protidique secon-daire à la carence androgénique.• Autres symptômes :le déficit en glucocorticoïdes peutêtre responsable de signes d’hypoglycémie (sueurs, frin-gale, céphalée, faiblesse générale…) en période dejeûne. Des arthralgies et des myalgies (crampes voire contrac-tures) ont également été décrites.
2. Insuffisance surrénale lente secondaire(insuffisance corticotrope)Cette affection comporte beaucoup de signes cliniqueset biologiques communs avec ceux de l’insuffisance sur-rénale lente primitive (tableau I). Néanmoins, l’absence
corticotrophine (rétrocontrôle négatif). Cette inertie corti-cotropen’est que très lentement réversible après arrêt dela corticothérapie. Par conséquent, toute diminutionrapide ou interruption brutale d’une corticothérapie anti-inflammatoire prolongée ne laisse pas le temps nécessaireà la normalisation de la sécrétion de corticotrophine etexpose à un risque élevé d’insuffisance surrénale aiguë.
2. OrganiquesConsécutives à un processus pathologique du complexehypothalamo-hypophysaire, les insuffisances surrénaleslentes secondaires organiques s’associent le plus souventà des déficits touchant les autres axes hypophysaires.Leurs causes sont superposables à celles de l’insuffisanceantéhypophysaire qui fait l’objet d’une question spécifique. Lorsqu’un adénome hypophysaire est en cause, letableau clinique peut également comporter un syndrometumoral hypophysaire associant céphalées et anomaliesdu champ visuel.
Diagnostic positif
Signes cliniques
Leur installation est souvent très insidieuse.
1. Insuffisance surrénale lente primitive (maladie d’Addison)• Asthénie :conséquence du déficit en cortisol, elle estle signe le plus précoce. Absente au lever, elle apparaîten cours de journée pour atteindre un maximum le soiret lors des efforts. Elle est classiquement physique(musculaire), psychique et sexuelle, et va peu à peus’aggraver.• Mélanodermie :c’est un élément fondamental du diagnostic. Elle est le reflet de l’augmentation du tauxplasmatique de corticotrophine. Sa présence témoigne del’origine primitivement surrénale de l’insuffisance. Sescaractères sémiologiques permettent de l’identifier. Elle prédomine au niveau des régions découvertes touten se distinguant du hâle solaire par son hétérogénéité ;des zones normalement pigmentées (mamelons) ; deszones de flexion et (ou) de frottement (plis palmaires,coudes, ceinture, encolure) ; des cicatrices. Les muqueuses peuvent être le siège de taches ardoiséeslocalisées principalement à la face interne des lèvres etdes joues.• Hypotension artérielle :elle est la conséquence del’hypovolémie liée au déficit en minéralocorticoïdes eten cortisol. En dehors des épisodes de décompensationaiguë, elle est modérée et s’exprime essentiellement parune hypotension orthostatique. La déplétion hydrosodéepeut également entraîner une accélération du pouls etune appétence marquée pour le sel. • Troubles digestifs :ils relèvent de la carence en minéralocorticoïdes responsable de perturbations deséchanges ioniques à travers les muqueuses digestives.
Endocrinologie - Métabolisme - Nutrition
2259L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
Communs aux insuffisances surrénales primitives et secondaires
❑ Asthénie❑ Anorexie, amaigrissement❑ Hypotension orthostatique❑ Troubles digestifs❑ Hypoglycémie❑ Anémie, lymphocytose, éosinophilie❑ Cortisolémie basse à l’état basal et insuffisamment
réactive lors du test au Synacthène
Orientant vers une insuffisance surrénale primitive
❑ Mélanodermie❑ Pathologie auto-immune associée❑ Hyponatrémie de déplétion❑ Hyperkaliémie❑ Corticotrophine élevée
Orientant vers une insuffisance surrénale secondaire
❑ Pâleur cutanée❑ Signes d’insuffisance antéhypophysaire❑ Syndrôme tumoral hypophysaire : céphalées,
altérations du champ visuel❑ Hyponatrémie de dilution❑ Kaliémie normale❑ Corticotrophine plasmatique normale ou basse
Signes cliniques et biologiquesdes insuffisances surrénales lentes
TABLEAU

de mélanodermie (la sécrétion de corticotrophine est icidiminuée) est un élément fondamental qui doit faire évo-quer une origine hypophysaire à l’insuffisance surrénalelente. En outre, le tableau clinique comporte fréquem-ment des signes d’autres déficits hypophysaires et (ou) unsyndrometumoral hypophysaire (tableau I).
Signes biologiques
1. Non spécifiques• Au cours de l’insuffisance surrénale lente primitive,les perturbations électrolytiques liées à l’hypo-aldosté-ronisme, hyponatrémie de déplétion et hyperkaliémie,sont souvent modérées voire absentes en dehors despoussées d’insuffisance surrénale aiguë. L’hypocortiso-lisme peut se traduire par une diminution de la glycémieà jeun et des anomalies de la numération formule san-guine : lymphocytose modérée, hyperéosinophilie, ané-mie normochrome et normocytaire. L’anémie relève à lafois de la carence en glucocorticoïdes et du déficit enandrogènes. • Au cours de l’insuffisance surrénale lente secondaire,la fonction minéralocorticoïde est ici respectée. Les examens biologiques courants ne montrent donc pasd’hyperkaliémie. La présence d’une hyponatrémie estpossible mais son mécanisme relève d’une hémodilutionque l’on peut opposer à l’hyponatrémie de déplétionrencontrée au cours de l’insuffisance surrénale lente primitive.
2. SpécifiquesLe diagnostic d’insuffisance surrénale lente devra êtreconfirmé par des dosages hormonaux spécifiques.Lorsque les contextes clinique et biologique fontcraindre une décompensation aiguë imminente (hypo-tension, fièvre, douleurs abdominales, vomissements,diarrhée, perturbations électrolytiques franches), leséchantillons sanguins destinés à ces dosages doivent êtreprélevés rapidement et le traitement doit être débutésans attendre les résultats.• Signes communs aux 2 types d’insuffisance surréna-le lente :en dehors de tout contexte d’urgence, le dia-gnostic positif de l’insuffisance surrénale lente, quelqu’en soit le mécanisme, repose en premier lieu sur lamise en évidence d’un taux abaissé de cortisol plasma-tique à 8 h,horaire qui correspond au pic physiologique dela sécrétion de cette hormone ou sur les résultats d’unestimulation par le Synacthène (β 1-24 ACTH). Ce testconsiste à mesurer la cortisolémie à l’état basal, puis 30 et 60 min après l’administration intraveineuse (ouintramusculaire) de 250 µg de Synacthène. Au cours del’insuffisance surrénale lente, la cortisolémie, spontané-ment abaissée, s’élève insuffisamment après injectionde Synacthène (pic sécrétoire normalement supérieur à600 nmol/L). Ce profil de réponse, logique lorsque l’in-suffisance surrénale lente est primitive (le tissu surrénalest alors détruit par un processus pathologique et ne peutrépondre à la stimulation), s’observe également au cours
de l’insuffisance surrénale lente secondaire, en raison del’atrophie surrénale consécutive au déficit prolongé encorticotrophine endogène. • Signes permettant de préciser le mécanisme (primitifou secondaire) de l’insuffisance surrénale lente :ladétermination du niveau de l’atteinte surrénale repose surle dosage de corticotrophine plasmatique à 8 h.En cas d’insuffisance surrénale lente primitive, le tauxplasmatique de corticotrophine est constamment supé-rieur aux valeurs normales.Lorsque l’hypocortisolisme est lié à une insuffisancecorticotrope, le taux plasmatique de corticotrophine estabaissé ou paradoxalement normal, contrastant alorsavec l’abaissement du taux de cortisol.Certains tests dynamiques peuvent être utiles. Le test àla métopirone et le test de l’hypoglycémie insuliniquesont formellement contre-indiqués en cas d’insuffisancesurrénale lente primitive (corticotrophine plasmatique debase élevée) en raison du risque élevé de décompensa-tion aiguë (et de mort subite) qu’ils font courir aupatient. En revanche, ils peuvent être pratiqués, enmilieu spécialisé et en dehors de tout contexte de menacede décompensation aiguë, lorsque le diagnostic étiolo-gique s’oriente vers une insuffisance surrénale lentesecondaire (corticotrophine plasmatique de base non éle-vée). Dans ce contexte, un test de stimulation de cortico-trophine par la CRH (ou une association CRH-lysinevasopressine) peut également être employé pour distinguerles atteintes hypothalamiques (réponse positive avec élé-vation de la corticotrophine ) des atteintes hypophysaires(absence de réponse de la corticotrophine lors du test).La détermination du mécanisme, primitif ou secondaire,de l’insuffisance surrénale lente doit s’aider de l’explo-ration de la fonction minéralocorticoïde. Cette explora-tion est impérative car un éventuel déficit en minéralo-corticoïdes devra obligatoirement être substitué. Au cours de l’insuffisance surrénale lente primitive, cedéficit est affirmé à l’aide d’un dosage de la réninémie(ou de l’activité rénine plasmatique) qui montre unchiffre élevé contrastant avec un taux sanguin d’aldo-stérone abaissé ou à la limite inférieure de la normale. En cas d’insuffisance surrénale lente secondaire, lesconcentrations plasmatiques de rénine et d’aldostéronesont normales.Les signes biologiques communs et respectifs des insuf-fisances surrénales lentes primitive et secondaire sontrésumés dans le tableau.
Diagnostic étiologique
Au cours de l’insuffisance surrénale lenteprimitive
Le diagnostic étiologique de l’atteinte surrénale reposealors sur la réalisation d’une tomodensitométrie abdo-minale avec des coupes centrées sur les aires surrénales.Plusieurs cas peuvent être envisagés selon l’aspect desglandes surrénales.
I N S U F F I S A N C E S U R R É N A L E L E N T E D E L ’ A D U L T E
2260 L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
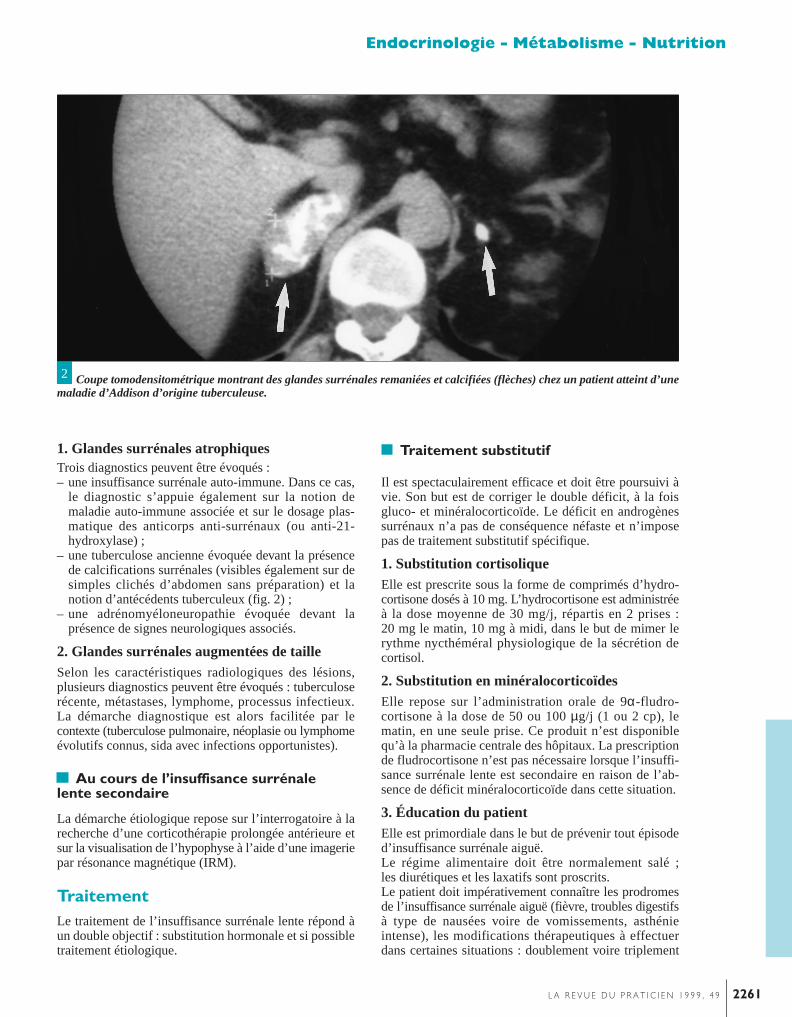
Traitement substitutif
Il est spectaculairement efficace et doit être poursuivi àvie. Son but est de corriger le double déficit, à la foisgluco- et minéralocorticoïde. Le déficit en androgènessurrénaux n’a pas de conséquence néfaste et n’imposepas de traitement substitutif spécifique.
1. Substitution cortisolique Elle est prescrite sous la forme de comprimés d’hydro-cortisone dosés à 10 mg. L’hydrocortisone est administréeà la dose moyenne de 30 mg/j, répartis en 2 prises :20 mg le matin, 10 mg à midi, dans le but de mimer lerythme nycthéméral physiologique de la sécrétion decortisol.
2. Substitution en minéralocorticoïdes Elle repose sur l’administration orale de 9α-fludro-cortisone à la dose de 50 ou 100 µg/j (1 ou 2 cp), lematin, en une seule prise. Ce produit n’est disponiblequ’à la pharmacie centrale des hôpitaux. La prescriptionde fludrocortisone n’est pas nécessaire lorsque l’insuffi-sance surrénale lente est secondaire en raison de l’ab-sence de déficit minéralocorticoïde dans cette situation.
3. Éducation du patient Elle est primordiale dans le but de prévenir tout épisoded’insuffisance surrénale aiguë.Le régime alimentaire doit être normalement salé ; les diurétiques et les laxatifs sont proscrits. Le patient doit impérativement connaître les prodromesde l’insuffisance surrénale aiguë (fièvre, troubles digestifsà type de nausées voire de vomissements, asthénieintense), les modifications thérapeutiques à effectuerdans certaines situations : doublement voire triplement
1. Glandes surrénales atrophiquesTrois diagnostics peuvent être évoqués :– une insuffisance surrénale auto-immune. Dans ce cas,
le diagnostic s’appuie également sur la notion demaladie auto-immune associée et sur le dosage plas-matique des anticorps anti-surrénaux (ou anti-21-hydroxylase) ;
– une tuberculose ancienne évoquée devant la présencede calcifications surrénales (visibles également sur desimples clichés d’abdomen sans préparation) et lanotion d’antécédents tuberculeux (fig. 2) ;
– une adrénomyéloneuropathie évoquée devant la présence de signes neurologiques associés.
2. Glandes surrénales augmentées de tailleSelon les caractéristiques radiologiques des lésions,plusieurs diagnostics peuvent être évoqués : tuberculoserécente, métastases, lymphome, processus infectieux.La démarche diagnostique est alors facilitée par lecontexte (tuberculose pulmonaire, néoplasie ou lymphomeévolutifs connus, sida avec infections opportunistes).
Au cours de l’insuffisance surrénale lente secondaire
La démarche étiologique repose sur l’interrogatoire à larecherche d’une corticothérapie prolongée antérieure etsur la visualisation de l’hypophyse à l’aide d’une imageriepar résonance magnétique (IRM).
TraitementLe traitement de l’insuffisance surrénale lente répond àun double objectif : substitution hormonale et si possibletraitement étiologique.
2261L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
Coupe tomodensitométrique montrant des glandes surrénales remaniées et calcifiées (flèches) chez un patient atteint d’unemaladie d’Addison d’origine tuberculeuse.
2
Endocrinologie - Métabolisme - Nutrition

de la dose d’hydrocortisone en cas de grande chaleur,de stress aigu, d’infection ou de traumatisme. Le port permanent d’une carte mentionnant l’insuffisancesurrénale lente ainsi que son traitement est impératif. Letraitement ne doit jamais être interrompu, et le patientdoit disposer d’une ampoule d’hydrocortisone (stockée àdomicile au réfrigérateur) à administrer par voie intramusculaire en cas d’intolérance digestive, qu’ellesoit ou non annonciatrice d’insuffisance surrénale aiguë.
4. Interventions chirurgicales Elles font l’objet d’une préparation particulière :doublement de la dose d’hydrocortisone la veille de l’intervention, administration parentérale de 50 à 100 mg d’hydrocortisone le matin de l’intervention etmise en place d’une perfusion d’hydrocortisone pendantl’intervention.
5. Grossesse La posologie de l’hydrocortisone devra être augmentéeau cours du 1er trimestre, en raison des nausées fréquentes à cette période de la gestation et lorsde l’accouchement.
6. Surveillance du traitement substitutif L’adaptation des doses de gluco- et minéralocorticoïdessera fonction des données cliniques et de l’ionogrammesanguin : disparition de l’asthénie et de la mélanodermie,normalisation du poids, de la pression artérielle, de lanatrémie et de la kaliémie. Un surdosage en hydro-cortisone peut être responsable d’une prise pondérale,d’une fragilité cutanée et faire courir un risque de démi-néralisation osseuse. Enfin, il faut souligner que lesdosages hormonaux (cortisolémie et corticotrophineplasmatique) n’ont aucun intérêt dans ce cadre.
Traitement étiologique
Le traitement causal de l’insuffisance surrénale lente(traitement antituberculeux, antimycotique…) estréalisé lorsqu’il est possible.
Complications
La complication majeure de l’insuffisance surrénalelente est l’insuffisance surrénale aiguë (se reporter à laquestion spécifique du programme de l’internat). Ils’agit d’une urgence thérapeutique qui met en jeu le pronostic vital. L’insuffisance surrénale aiguë est le plus souvent secon-daire à une insuffisance surrénale lente méconnue, nontraitée ou insuffisamment traitée dans une situation destress, d’agression physique (déshydratation, interven-tion chirurgicale, infections) ou psychique. Elle peutégalement être favorisée par une erreur thérapeutiquecomme un régime sans sel, un traitement diurétique oulaxatif, ou encore un arrêt inopiné de l’hormonothérapiesubstitutive.
En dehors de ces situations, l’insuffisance surrénaleaiguë peut être inaugurale, qu’elle soit primitive etconsécutive à une hémorragie bilatérale des surrénaleslors d’un traitement anticoagulant, d’une septicémie oud’un syndrome des anticorps anti-phospholipides (ils’agit alors d’une nécrose hémorragique secondaire à unethrombose bilatérale des artères surrénales) ; ou qu’ellesoit secondaire, à l’occasion d’une apoplexie hypo-physaire, d’une nécrose hémorragique du post-partumou de la cure chirurgicale d’une maladie de Cushing. Après une phase prodromique marquée par les signescliniques cités plus haut, le tableau clinique se complèteensuite avec des douleurs abdominales pouvant parfoismimer une urgence chirurgicale, une grande déshydrata-tion à la fois extra- et intracellulaire, une hypotensionartérielle et des troubles de la conscience. En l’absenced’un traitement rapide, l’évolution spontanée se fait versle collapsus cardiovasculaire et la mort.Les examens biologiques et hormonaux dont il ne fautpas attendre les résultats pour traiter le patient mettenten évidence une hyponatrémie notable (inférieure à 130 mmol/L parfois 120), une hyperkaliémie modérée,une acidose métabolique et une hypoglycémie. La cortiso-lémie de base est constamment effondrée.Le traitement d’une insuffisance surrénale aiguë doitêtre réalisé en urgence, en milieu spécialisé. La prise encharge thérapeutique comprend d’une part, le traitementsymptomatique avec réhydratation, apport sodé puisglucosé et, d’autre part, l’administration d’hémisuccinated’hydrocortisone et éventuellement d’acétate de désoxy-corticostérone (minéralocorticoïde) par voie parentérale.Le facteur déclenchant doit également être traité lorsquecela est possible.Enfin, le meilleur traitement de l’insuffisance surrénaleaiguë est préventif. Dans cette optique, la reprise del’éducation du patient est indispensable après chaquedécompensation. ■
I N S U F F I S A N C E S U R R É N A L E L E N T E D E L ’ A D U L T E
2262 L A R E V U E D U P R A T I C I E N 1 9 9 9 , 4 9
• L’insuffisance surrénale lente est un diagnosticauquel il faut penser au risque d’identifier la maladie lors d’une décompensation aiguëpotentiellement létale.
• Le diagnostic positif repose sur la mesure de la cortisolémie à 8 h ou après stimulationpar le Synacthène.
• la détermination du mécanisme primitif ou secondaire de l’insuffisance surrénale lenterepose sur la mesure du taux de corticotrophine plasmatique à 8 h.
• Le traitement substitutif en glucocorticoïdes et éventuellement en minéralocorticoïdes ne doit jamais être interrompu.
• L’éducation du patient est primordiale pour prévenir toute décompensation aiguë.
Points Forts à retenir

Endocrinologie - Métabolisme - NutritionB 333
549L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
dans les pays du Pacifique et de l’océan Indien(Mélanésie, Polynésie et Micronésie). Dans les pays duSud-Est asiatique, d’Afrique et de l’Est méditerranéen,nous ne disposons pas d’étude représentative de bonnequalité pour apprécier la prévalence de l’obésité.
2. Évolution dans le tempsLa prévalence de l’obésité augmente de façon alarmantedans les pays développés, mais également dans les paysen cours d’industrialisation, comme la Chine. Le phéno-mène est particulièrement grave aux États-Unis. Cela estobservé dans tous les groupes ethniques et dans les 2 sexes. La prévalence de l’obésité est passée de 12 à19,7 % chez les hommes et de 14,8 à 24,7 % pour lesfemmes, entre 1980 et 1990. En Europe, la prévalencede l’obésité a augmenté de 10 à 40 % dans la majoritédes pays, au cours des 10 à 15 dernières années.
Facteurs de risque de l’obésité
1. Différences en fonction du sexe La composition corporelle varie en fonction du sexe.Une femme présente une masse grasse plus importantequ’un homme de même poids et de même taille, soit respectivement 20 à 25 % et 15 à 20 % de la masse corporelle chez l’adulte jeune. Globalement, la prévalence de l’obésité tend à être plusimportante chez la femme que chez l’homme dans laplupart des études, et notamment en Europe et auxÉtats-Unis.
2. Effets de l’âgeL’augmentation de la prévalence de l’obésité sembleencore plus nette chez les sujets jeunes que chez lesadultes. Cependant, les index pondéraux augmententavec l’âge dans les 2 sexes, mais proportionnellement demanière plus importante chez la femme et ce jusqu’à 60 ans. Après 60 ans, la prévalence de l’obésité diminue.La composition corporelle change aussi avec l’âge, avecune diminution de la masse maigre et une augmentationde la masse grasse. Le vieillissement affecte aussi larépartition du tissu adipeux, avec prédominance du tissugraisseux dans la partie supérieure du corps.
3. Différences raciales et ethniques La prévalence de l’obésité est plus importante chez lesfemmes de race noire que chez les femmes de raceblanche, quel que soit l’âge. Par exemple, 48,6 % desfemmes de race noire ont un excès pondéral contre 33,2%des femmes de race blanche aux États-Unis.
Épidémiologie
Prévalence de l’obésité
1. Selon la zone géographiqueL’obésité existe désormais dans tous les pays du monde.La prévalence, particulièrement élevée dans certainspays industrialisés, augmente aussi dans les pays en voiede développement. L’obésité semble peu fréquente enAfrique et en Asie, mais cela n’est vrai que dans leszones rurales, car la maladie se développe dans leszones urbaines. En Europe, la prévalence de l’obésité estestimée entre 10 et 20 % chez les hommes et 10 à 25 %chez les femmes. Toutefois, les résultats varient consi-dérablement selon les pays et les régions. La prévalencede l’obésité est la plus forte en Lituanie et la plus faibleen Suède. Elle est particulièrement élevée (20 à 45 %)chez les femmes des pays européens du Sud, commel’Espagne ou le Portugal, et de l’Est, comme la Pologneet l’ex-URSS. En France, la prévalence de l’obésité est de 6 % dans lesdeux sexes. Les données d’une étude réalisée aux États-Unis entre 1988 et 1991 ont montré qu’environ 20 % deshommes et 25 % des femmes sont obèses. Les chiffresobservés au Canada sont un peu plus faibles : 15 % deshommes et des femmes sont obèses. Au Brésil, seul payslatino-américain pour lequel on dispose d’une étudenationale représentative, l’obésité affecte 6 % deshommes et 13% des femmes. La prévalence de la maladieest particulièrement élevée dans les pays des Caraïbes.Dans les pays du Pacifique ouest, en Australie et enNouvelle-Zélande, 9,3 % des hommes et 11,1 % desfemmes sont obèses. L’obésité est aussi très fréquente
ObésitéÉpidémiologie, diagnostic, complications
PR Denis RACCAH
Service de nutrition, maladies métaboliques, endocrinologie, hôpital de la Timone, 13385 Marseille Cedex 05.
• L’obésité est une maladie complexe,tant pour ses formes cliniques, ses facteurs physiopathologiques, sa sémiologie et ses conséquences pathologiques.
• L’augmentation de la prévalence et de l’incidence de l’obésité, en fonction du changement du mode de vie et de la modernisation, constitue à l’évidence un problème préoccupant de santé publiquedans les pays industrialisés et dans beaucoup de pays en voie de développement.
Points Forts à comprendre

4. Facteurs génétiques et environnementaux
• Des facteurs génétiquessont impliqués dans la genèse de l’obésité. Il est admis que l’obésité est une maladie polygénique, à forte composante environ-nementale. On ne connaît actuellement que quelquesgènes de susceptibilité. Seule la mutation ponctuelle du récepteur bêta-adrénergique a fait l’objet d’étudesdans différentes populations, bien que son rôle étio-pathogénique soit controversé : sa prévalence, qui variede 4 à 19 % selon les pays, est la même dans la population générale et chez les sujets obèses. Lesindiens Pima, population qui présente une prévalenceparticulièrement élevée d’obésité et de diabète de type2, font exception, car la prévalence de la mutation atteint30 %. • Les facteurs environnementaux ont joué un rôleconsidérable dans l’augmentation de l’incidence del’obésité au cours des 10 dernières années, car les seulsfacteurs génétiques ne peuvent expliquer une évolutionaussi rapide. Paradoxalement, les apports énergétiquesont diminué, ce qui suggère que l’augmentation de lasédentarité ou d’autres modifications du mode de viesont en cause.
4. Facteurs socio-économiques Dans les pays développés, l’obésité est plus fréquentedans les classes sociales défavorisées et notamment chezles femmes. Ainsi, dans une étude anglaise, l’indice demasse corporelle moyen des femmes ayant un faibleniveau socio-économique est plus élevé de 2 points quecelui des femmes plus favorisées. La profession nesemble pas jouer de rôle direct. À l’inverse, l’obésitéconcerne plutôt les classes aisées dans les pays en voiede développement.
Diagnostic
L’évaluation de l’obésité repose sur l’analyse de 2 para-mètres qui jouent un rôle indépendant par rapport auxcomplications de la maladie : l’excès de masse grasse etla répartition du tissu adipeux.
Excès de masse grasse
L’obésité peut être définie comme un excès de massegrasse susceptible d’avoir un effet néfaste sur la santé.L’indice de masse corporelle ou indice de Quetelet est leplus utilisé actuellement pour définir l’obésité. Il corres-pond au rapport du poids en kilogrammes au carré de lataille en mètre carré. Il est donc facile à calculer etsimple. L’indice de masse corporelle est fortement corrélé à la masse grasse (r = 0,7 à 0,8). On définit lasurcharge pondérale par un indice de masse corporellecompris entre 25 et 29,9 kg/m2 et l’obésité par un indicede masse corporelle supérieur ou égal à 30 kg/m2. Cettedéfinition présente 3 avantages :
– c’est une référence internationale et elle permet decomparer les études menées dans différents pays ;
– elle a une signification pronostique vis-à-vis desrisques de la maladie ;
– elle est valable chez l’adulte quels que soit le sexe etl’âge.
L’utilisation de l’indice de masse corporelle pour définirl’obésité a néanmoins des limites, car cet indice ne tientpas compte de la composition corporelle qui peut êtredifférente pour un même indice de masse corporelle,notamment en fonction de l’âge, du sexe et de l’activitéphysique. Par exemple, une valeur élevée de l’indice demasse corporelle chez un travailleur de force ou un spor-tif de haut niveau, correspond à une masse musculaireimportante et non pas à un excès de tissu adipeux.
Répartition du tissu adipeux
C’est Jean Vague qui avait décrit dès 1947 le caractèrebipolaire des obésités. Il est maintenant convenu de dis-tinguer d’un côté l’obésité androïde, caractérisée parune répartition du tissu graisseux à la partie supérieuredu corps et en particulier abdominale et viscérale ; et del’autre l’obésité gynoïde, caractérisée par une répartitiondu tissu graisseux à la partie inférieure du corps et enparticulier au niveau des hanches et des membres inférieurs.La mesure des circonférences au niveau de la taille etdes hanches permet de calculer le rapport taille surhanche. L’obésité androïde est définie chez l’homme parun rapport taille sur hanche supérieur à 1 et chez lafemme par un rapport supérieur à 0,85. La mesure dutour de taille serait un meilleur indice, car il est mieuxrelié que le rapport taille sur hanche au risque de comor-bidité. La valeur seuil pour laquelle la comorbidité aug-mente est voisine de 100 cm.
Complications
Mortalité totale
Les grandes études épidémiologiques ont démontré lesfaits suivants.Il existe dans les 2 sexes une relation curvilinéaire entrel’indice de masse corporelle et le risque de mortalitétotale. Cette courbe a une forme de U ou de J, la surmortalitédes sujets maigres pouvant être attribuée schématiquementaux cancers et celle des plus corpulents aux maladiescardiovasculaires. L’indice de masse corporelle optimal,correspondant à une mortalité minimale, est situé entre19 et 25.L’obésité sévère est associée à un doublement du risquerelatif de mortalité totale.L’obésité abdominale est probablement la forme cliniquela plus associée à cet excès de mortalité. En effet, lessujets obèses androïdes semblent surtout prédisposésaux complications cardiovasculaires.
O B É S I T É
550 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

sujets atteints d’une hypertension artérielle essentielle,appariés par l’âge et le poids, les sujets hypertendus présentent une résistance à l’action de l’insuline lors declamps euglycémiques hyperinsulinémiques par rapportaux sujets témoins. La nature de ce lien reste à préciser.Certains arguments sont en faveur d’un lien purementgénétique. En effet, dans quelques ethnies (MexicainsAméricains), les sujets avec antécédents familiaux dediabète de type 2 ont des insulinémies plus élevées etune fréquence d’hypertension artérielle plus importante,que ceux sans antécédents familiaux de diabète de type 2.Mais d’autres arguments sont en faveur d’un lien physiologique entre hyperinsulinémie et hypertensionartérielle, faisant intervenir l’activité de la pompe àsodium proton, la natriurèse et le tonus sympathique.
4. HypofibrinolyseLa fibrinolyse est un mécanisme physiologique dedéfense contre la thrombose. En effet, lors d’une brècheendothéliale, les phénomènes de la coagulation aboutissentà la formation d’un caillot de fibrine insoluble pourréparer la lésion. La fibrinolyse se met en route quasimentsimultanément afin de dégrader ce caillot de fibrineinsoluble en produits de dégradation de la fibrinesoluble. L’enzyme clef de la fibrinolyse est la plasmine,qui provient d’une pro-enzyme inactive, le plasminogène.Le plasminogène est susceptible d’être activé par l’activateur tissulaire du plasminogène (tPA), lui-mêmefortement inhibé par l’inhibiteur de l’activateur du plasmi-nogène 1 (PAI-1). Le PAI-1 est donc un régulateur inhi-biteur puissant de la fibrinolyse, puisqu’il agit à l’originede cette cascade de réactions. Or, dans l’obésité viscéraleavec insulinorésistance, les taux plasmatiques de PAI-1sont élevés avec une tendance à l’hypofibrinolyse et àl’accumulation intravasculaire de fibrine.Ces 4 complications métaboliques de l’obésité viscérale,regroupées sous le nom de syndrome plurimétabolique,expliquent la grande prévalence de maladies cardio-vasculaires chez ces patients. Les mécanismes pouvantexpliquer le lien entre excès de graisse viscérale et insulinorésistance sont débattus. Cependant, l’hypothèsemétabolique retenue est que le tissu graisseux viscéralpossède une grande activité métabolique, avec, en parti-culier, une lipolyse accrue et une libération exagéréed’acides gras libres dans le système porte. Ces acidesgras libres favoriseraient l’insulinorésistance au niveauhépatique, par diminution de la clairance de l’insuline,stimulation de la néoglucogenèse et inhibition de la glycolyse; et au niveau musculaire, par inhibition compétitive de la captation du glucose.
Pathologies cardiaques
L’obésité est en tant que telle un facteur de risque d’hyper-trophie ventriculaire gauche et d’insuffisance cardiaque.L’obésité accroît le travail cardiaque et l’augmentationdes pressions de remplissage du ventricule entraîne unehypertrophie de type excentrique et donc une dilatationdes cavités. La mort subite est 3 à 6 fois plus fréquente
Complications métaboliques
Ces complications sont spécifiques de l’obésité androïdeet (ou) viscérale. En effet, l’excès de graisse viscérales’accompagne d’une résistance à l’insuline, principalementau niveau musculaire. Cette particularité est à l’origined’un syndrome appelé «syndrome d’insulino-résistance»,ou « syndrome plurimétabolique » ou « syndrome X ».Ce syndrome associe 4 anomalies métaboliques :– un diabète de type 2 ou diabète non insulinodépendant ;– une dyslipidémie ;– une hypertension artérielle ;– une hypofibrinolyse.
1. Diabète de type 2L’excès de graisse viscérale s’accompagne d’une résistanceà l’insuline. Tant que les cellules bêtapancréatiques sontcapables de compenser exactement cette résistance àl’insuline par une hypersécrétion insulinique, la glycémieest équilibrée. Il faut donc que coexiste une 2e anomaliepour expliquer le développement d’un diabète non insulino-dépendant, c’est une anomalie de l’insulinosécrétionavec insulinopénie relative par rapport à l’insulino-résistance. Dès que la capacité insulinosécrétoire pan-créatique ne compense pas exactement la résistance àl’insuline, la glycémie s’élève, provoquant dans un 1er temps une hyperglycémie à jeun modérée, puis unvrai diabète de type 2.
2. Dyslipidémie L’hyperinsulinémie chronique qui accompagne l’insulino-résistance est à l’origine d’une dyslipidémie courammentretrouvée, avec une élévation des VLDL (very low densitylipoprotein), lipoparticules véhiculant principalementles triglycérides dans le sang, et une diminution du tauxde HDL (high density lipoprotein), qui assure le transportinverse du cholestérol de la périphérie, en particulier desparois artérielles, vers le foie où il est catabolisé. Eneffet, l’hyperinsulinémie endogène s’accompagne d’unehyperinsulinémie portale, qui stimule la synthèse hépatiquedes VLDL et explique l’élévation du taux de triglycéridesplasmatiques. Par ailleurs, les 2 principales sources deHDL sont d’une part la production hépatique sous laforme de particules discoïdes comportant des phospho-lipides et des apoprotéines A1 et A2 et, d’autre part, lalibération de composantes de surface des chylomicronset des VLDL, au cours de l’hydrolyse des triglycéridesde ces lipoprotéines par la lipoprotéine lipase. Dans lesyndrome d’insulinorésistance avec hyperinsulinémie, ilexiste une diminution de l’activité de la lipoprotéinelipase et donc une diminution d’une des sources de pro-duction des HDL. Enfin, l’anomalie du métabolisme delipoprotéines aboutit à la production de LDL (low densitylipoprotein) petites et denses et particulièrement oxydables.
3. Hypertension artérielle Les travaux du groupe de Ferranini ont mis en évidencele lien entre hypertension artérielle et insulinorésistance.En effet, lorsqu’on compare des sujets témoins et des
Endocrinologie - Métabolisme - Nutrition
551L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0

chez les sujets obèses en fonction de l’âge et du sexe.Les troubles du rythme ventriculaire associés à l’hyper-trophie ventriculaire gauche en sont responsables :tachycardie et fibrillation.
Complications bronchopulmonaires
Les complications respiratoires des obésités comportentdes altérations de la mécanique ventilatoire, un syndromerestrictif, des modifications des échanges gazeux etconduisent à 2 syndromes particuliers qui peuventmettre en cause le pronostic vital. Le syndrome d’hypo-ventilation alvéolaire est responsable d’une hypoxémieet d’une hypercapnie. Le syndrome d’apnée du sommeilse manifeste par des apnées, une hypersomnolence diurne, des troubles neuropsychiques, des céphalées etune ronchopathie.
Complications rhumatologiques
L’obésité joue probablement un rôle déclenchant ouaggravant de nombreuses affections dégénératives de l’ap-pareil locomoteur, telles que l’arthrose et, en particulier, lagonarthrose et la coxarthrose. Les douleurs rachidiennessont fréquentes et l’ensemble de ces anomalies favorise la sédentarité et l’inactivité physique de ces patients.
Cancers
L’incidence de certains cancers est augmentée chez lessujets obèses. Ce sont surtout des cancers dépendant deshormones : chez la femme, endomètre, ovaires et seinsaprès la ménopause, et chez l’homme, prostate; et cancersdigestifs (côlon, rectum et vésicule biliaire).
Maladies hépatobiliaires et digestives
La lithiase vésiculaire est beaucoup plus fréquente chezles sujets obèses que chez les sujets de poids normal, enparticulier dans le sexe féminin. La stéatose hépatiquecentrolobaire ou diffuse est fréquente chez l’obèse.Cette stéatose facilement diagnostiquée par l’échographiepeut être responsable d’anomalies mineures du bilanhépatique, comme une augmentation des gamma-glutamyl-transférases (γ GT) et une cytolyse modérée. ■
O B É S I T É
552 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• L’obésité est définie comme un excès de masse grasse, quantifiée par l’indice de masse corporelle (IMC), correspondant au rapport du poids en kilogrammes au carréde la taille en mètre carré.
• La surcharge pondérale est définie par un indice de masse corporelle compris entre 25 et 29,9, alors que l’obésité est définiepar un indice de masse corporelle supérieur ou égal à 30.
• La répartition corporelle du tissu graisseux,appréciée par le rapport taille/hanche ou le tour de taille est un 2e paramètre à prendre en compte, particulièrement vis-à-visdu risque de complications. En effet, l’obésitédite androïde est associée spécifiquement au risque de complications métaboliques.
• La prévalence de l’obésité est en augmentation dans les pays industrialisés et en voie de développement. Les facteurs environnementaux jouent un rôle considérabledans l’augmentation de la prévalence de l’obésitéet ces facteurs interviennent sur des terrainsgénétiques favorisants.
• Parmi les complications de l’obésité, il faut retenir une surmortalité totale ; des complicationsmétaboliques spécifiques de l’obésité androïderegroupées sous le nom de syndrome pluri-métabolique associant un diabète de type 2,une dyslipidémie, une hypertension artérielle et une hypofibrinolyse ; des pathologies cardiaques, à type d’hypertrophie ventriculairegauche et insuffisance cardiaque ; des complications bronchopulmonaires avec en particulier le syndrome d’apnée du sommeil ;des complications rhumatologiques, en particulierarthrose du genou et des hanches ; certains cancers dépendant des hormones ; des maladieshépatobiliaires et digestives à type de lithiasevésiculaire et stéatose hépatique.
Points Forts à retenir

Endocrinologie - Métabolisme - NutritionA 57
791L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Clinique
Le diabète insipide peut en effet survenir à tous les âges,en proportion sensiblement égale entre sexes féminin oumasculin.L’apparition du syndrome polyuro-polydipsique estvolontiers rapide, si ce n’est brutale, notamment enfonction de l’étiologie. Néanmoins, le début peut être,plus rarement, progressif voire insidieux.De même, en fonction de l’intensité de la polyurie, lasoif est impérieuse, éprouvante, entraînant anxiété etfatigue, car maintenue durant la nuit et perturbant lesommeil. La persistance nocturne du syndrome polyuro-polydipsique est un signe en faveur d’une organicité.Dans la plupart des cas de diabète insipide modéré, lesyndrome polyuro-polydipsique est toutefois assez bientoléré si les boissons arrivent à compenser la diurèse. Iln’existe pas, dans ces conditions, de signes de déshydra-tation et notamment le poids reste stable.Quand existe une perturbation de la vigilance ou de lasoif associée, le diabète insipide peut entraîner unedéshydratation intracellulaire, puis globale, voire uncollapsus cardiovasculaire. Lorsque le syndrome poly-uro-polydipsique est ancien, intense, prolongé et malcontrôlé, il peut être à l’origine d’une dilatation globaledes voies urinaires, particulièrement chez l’enfant,l’urétéro-hydronéphrose étant éventuellement respon-sable d’un diabète insipide néphrogénique surajouté.Par ailleurs, un diabète insipide central peut ne pas êtreapparent dans 2 circonstances : lors d’une carence encortisol primitivement surrénale ou secondaire à unhypopituitarisme corticotrope, le diabète insipide estalors révélé par le traitement hormonal surrénal substitutif ; lors d’une atteinte conjointe du centre de lasoif (adipsie ou oligodipsie responsable d’une hyper-natrémie neurogène).
BiologieLes examens doivent démontrer 2 choses.
1. Le syndrome polyuro-polydipsique est hypotoniqueLa densité urinaire est inférieure à 1 005, l’osmolalitéurinaire est inférieure à 200 mOsm/kg d’eau et la clairance de l’eau libre (CH2O) nettement positive.
Diagnostic positif
Définition
Le syndrome polyuro-polydipsique hypotonique du diabète insipide est défini par l’excrétion anormalementimportante d’urines diluées, atteignant ou dépassant 50 mL/kg de poids en régime de boissons libres chezl’adulte, soit une diurèse égale ou supérieure à 3,5 L/24 hde densité urinaire (DU) inférieure à 1 005, d’osmolalité(OsmU) inférieure à 200 mOsm/kg d’eau et avec uneclairance de l’eau libre calculée nettement positive. Lesnormes du nourrisson et du jeune enfant sont moins biendéfinies, probablement plus élevées que chez l’adulte,de l’ordre de 75 à 100 mL/kg de poids. La diurèse peutêtre relativement modérée (4 à 5 L/24 h) ou parfoisénorme (15 à 30 L/24 h) chez l’adulte et dépassant le poids corporel chez le nourrisson. Parallèlement àl’intensité de la diurèse, les urines sont à peine ou pas du tout colorées.
Syndrome polyuro-polydipsiqueOrientation diagnostique
PR Jean LEFEBVRE, DR Marie-Christine VANTYGHEM
Service d’endocrinologie et maladies métaboliques, clinique Marc-Linquette, CHRU, 59037 Lille Cedex.
• Un syndrome polyuro-polydipsique (SPP) est secondaire soit à une polyurie primaire,hyper- ou hypotonique, soit à une polydipsieprimaire (PP) organique ou fonctionnelle par dérèglement de la soif ou par potomanie.
• Le diabète insipide central (DIC) est le prototypede syndrome polyuro-polydipsique hypotoniquesensible à l’hormone antidiurétique (HAD).
• Le syndrome polyuro-polydipsique secondaireà une potomanie est plus fréquent que le diabèteinsipide central.
• Le diabète insipide central est plus souventacquis que congénital ou apparemment idiopathique.
• Il peut être masqué par une adipsie ou par un hypocortisolisme et alors révélé par le traitement substitutif.
• Un diabète insipide (DI) peut être d’origine centrale ou néphrogénique (DIN),soit congénitale, soit acquise.
Points Forts à comprendre

2. Le syndrome polyuro-polydipsique est dû à une carence en hormone antidiurétique
• Dosage de la vasopressine dans le sang ou les urines :l’intérêt est plus théorique que pratique. Si ce dosage estinutile en routine, il est parfois indispensable lorsd’épreuves dynamiques, nécessaires dans certaines circonstances cliniques et en milieu spécialisé.• Épreuve de restriction hydrique (RH) :c’est lemeilleur examen pour mettre en évidence un diabèteinsipide, avec injection de desmopressine (1-déamino-8-D-AVP) en fin d’épreuve pour apprécier la sensibilité dutubule rénal à la vasopressine ; la restriction hydrique esttoutefois une épreuve non dénuée de risques et doit êtreobligatoirement réalisée sous stricte surveillance, enmilieu endocrinologique spécialisé, en raison de risquesde déshydratation rapide ; les résultats normaux etpathologiques figurent sur le tableau I (v. infra).
D’autres épreuves dynamiques, anciennes ou plusrécentes, comme la mesure de l’excrétion urinaire del’aquaporine 2, sont beaucoup moins utilisées car parfois dangereuses ou, en réalité, peu informatives.Reste parfois l’essai de traitement à la desmopressine(Minirin) par voie nasale ou orale, utile pour le diagnostic des formes partielles de diabète insipidecentral ou diabète insipide néphrogénique et de poly-dipsie primaire psychogène. Il consiste à administrer10 à 20 mg de desmopressine par 24 h en régime deboisson libre. Cependant, comme tous les tests dyna-miques, il nécessite une surveillance clinique particu-lièrement attentive car, au cours d’une potomanie, il ya un risque réel d’hyponatrémie sévère et d’intoxi-cation par l’eau.
Explorations morphologiques
1. Examen du fond d’œil (FO) et du champ visuel (CV)
Il est nécessaire, notamment lors d’un hypopituitarismeantérieur ou d’une atteinte du centre de la soif associée(v. supra).
2. Radiographie du crâneDe face et de profil, centrée sur la selle turcique, ellepeut donner de précieux renseignements, notammentlors de la présence de calcifications, pour l’orientationdu diagnostic étiologique (fig. 1).
S Y N D R O M E P O L Y U R O - P O L Y D I P S I Q U E
792 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Osmolarité urinaire (mOsm/kg d’eau) DiagnosticEn fin de restriction Après
hydrique desmopressine
> 700
< 250
< 250
250-700
> 700
> 500
< 250
< 700
Sujet normal
Diabète insipidecentral
Diabète insipidenéphrogénique
Diabète insipidecentral partiel
Diabète insipidenéphrogéniquepartiel
Potomanie
Interprétation de l’épreuvede restriction hydrique
TABLEAU I
• Le syndrome polyuro-polydipsique est sensible àl’injection de desmopressine :c’est le cas pour le dia-bète insipide central partiel ou complet. La restrictionhydrique sans dosage conjoint de la vasopressine plasmatique (AVPp) permet d’explorer valablement les syndromes polyuro-polydipsiques hypotoniques dansplus de 80 % des cas. Néanmoins, la mesure de la vaso-pressine plasmatique sous stimulation osmotique par lesérum salé hypertonique à 5 %, sous contrôle strict enmilieu spécialisé, peut informer au cours du diabèteinsipide partiel car elle permet d’apprécier l’insuffisance de sécrétion de vasopressine par rapportau stimulus osmotique. Cette épreuve permet égale-ment d’évaluer le seuil de déclenchement de la soif et de libération de vasopressine.
Radiographie du crâne de profil.Calcifications suprahypophysaires et agrandissement de laselle turcique évocatrice d’un craniopharyngiome.
1

à une potomanie
Il est plus fréquent que le diabète insipide central.L’excès d’eau ingérée entraîne une polyurie par unmécanisme direct, lié à l’augmentation du flux tubulairerénal, associée à une diminution du gradient corticopa-pillaire et indirect, par inflation des liquides extracellu-laires (LEC) qui inhibe la vasopressine et provoque undiabète insipide induit.
Diagnostic différentiel
Polyuries primaires hypertoniques
La densité urinaire supérieure à 1 015 et la mesure del’osmolalité urinaire permettent de les reconnaître faci-lement. L’osmolalité urinaire est en effet très nettementsupérieure à 300 mOsm/kg d’eau ainsi qu’à l’osmolalité plasmatique (OsmP) mesurée conjointe-ment ou calculée. La CH2O est en conséquence négative.Ces syndromes polyuro-polydipsiques hypertoniquessont essentiellement dus à la glycosurie d’un diabètesucré déséquilibré, à une polyurie uréique et (ou) sodéeaprès une insuffisance rénale aiguë ou subaiguë, ouune uropathie obstructive et (ou) une levée d’obstaclesur les voies urinaires.
3. Imagerie par résonance magnétique
L’exploration morphologique hypothalamo-hypophy-saire est réalisée au mieux par l’imagerie par résonancemagnétique (IRM), nettement plus performante quel’examen tomodensitométrique (scanner). Elle estnécessaire dès que le diagnostic de syndrome polyuro-polydipsique hypotonique est fait de façon à orienter larecherche d’une étiologie. La présence ou l’absence del’hypersignal spontané de la post-hypophyse en séquencepondérée T1 lors de l’imagerie est en effet précieuse.Dans ces conditions, l’hypersignal est présent chez 90 à100 % des sujets normaux avec toutefois un déclin pro-gressif en fonction de l’âge.Réserve faite des données précédentes, la disparition del’hypersignal T1 de la post-hypophyse est un argumentde poids en faveur du diagnostic de diabète insipide central, très vraisemblablement en rapport avec unedéplétion de la vasopressine stockée par les granulesneurosécrétoires, sauf dans certaines formes familialespar anomalies des osmorécepteurs sans déficit de vaso-pressine et au tout début d’un diabète insipide centralidiopathique. L’absence d’hypersignal post-hypophysairene permet toutefois pas d’exclure un diabète insipidenéphrogénique responsable d’une déshydratation chro-nique prolongée.
4. Apport de l’imagerie par résonance magnétique
Il est surtout remarquable pour la mise en évidenced’une éventuelle tumeur de la région neuro-hypothala-mo-hypophysaire, d’un épaississement isolé de la tigepituitaire, évoquant un processus infiltratif tumoral ouinflammatoire, ou d’une section de tige post-trauma-tique (fig. 2), parfois suivie d’une reconstitution d’unlobe postérieur ectopique.L’enregistrement électroencéphalographique (EEG) estutile lors d’un syndrome polyuro-polydipsique secon-daire à un trouble de la soif, potomanie ou surtout dipso-manie qui peut être un équivalent d’une comitialitéméconnue.
Physiopathologie du syndrome polyuro-polydipsique hypotonique
1. Diabète insipide central complet
Il ne se produit qu’après destruction de plus de 85 %des neurones sécrétant de la vasopressine au niveau desnoyaux supra-optiques et paraventriculaires. La lésiondoit en plus être haut située, au niveau hypothalamiqueou du tractus supra-optico-post-hypophysaire, pourprovoquer un diabète insipide permanent. Beaucoupplus exceptionnellement, le diabète insipide centralrelève d’une anomalie de l’osmorégulation ou d’uneanomalie congénitale du gène de la propressophysine.
2. Syndrome polyuro-polydipsique secondaire
Endocrinologie - Métabolisme - Nutrition
793L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Imagerie par résonance magnétique coupe coronale T1.Petite image hyperintense visible juste sous le chiasmaoptique correspondant à une post-hypophyse rétractée vers lehaut. La tige pituitaire n’est pas visible en dessous d’elle ;l’antéhypophyse est normalement située dans la selle turcique.
2

Polyuries primaires hypotoniques
Dans un contexte neurochirurgical ou traumatique, unepolyurie hypotonique peut être due à l’élimination phy-siologique d’une surcharge préalable en solutés hypoto-niques ou isotoniques glucosés. La natrémie est alorsvolontiers basse ou normale. Si l’apport hydrique estmaintenu, la polyurie est entretenue et évoque un diabèteinsipide. L’utilisation de desmopressine à titre diagnos-tique est toutefois déconseillée en raison du risque d’hyponatrémie. Si la réduction des apports ne modifiepas la polyurie hypotonique, inférieure à 200 mOsm/kgd’eau, et si la natrémie est supérieure à 145 mmol/L, lediagnostic de diabète insipide est assuré.En dehors d’un tel contexte et d’un diagnostic formel dediabète insipide central pitressino-sensible, les syndromespolyuro-polydipsiques par polyuries primaires hypoto-niques peuvent être classés en 2 grandes catégories :syndromes polyuro-polydipsiques hypotoniques pitres-sino-sensibles ; syndromes polyuro-polydipsiqueshypotoniques pitressino-résistants.
1. Syndromes polyuro-polydipsiques hypotoniques pitressino-sensiblesIls ont 2 origines différentes, lésionnelles ou fonction-nelles.• Polydipsies primaires lésionnelles :elles sont rares etsecondaires à une atteinte du centre de la soif par unelésion hypothalamique organique dont les étiologiessont identiques à celles du diabète insipide central (v. infra).• Polydipsies primaires fonctionnelles :elles corres-pondent soit à une dipsomanie, soit beaucoup plus fré-quemment à une potomanie dont le diagnostic est diffi-cile car la polydipsie induit un diabète insipide qui atoutes les caractéristiques d’un authentique diabète insi-pide central (v. supra).Le syndrome polyuro-polydipsique hypotonique s’ins-talle progressivement sur un terrain psychologique per-turbé, parfois de façon intermittente, rythmé par l’évolu-tion de la psychopathie. Dans de telles conditionsparticulièrement difficiles, une restriction hydrique pro-longée avec injection de desmopressine la plus tardivepossible peut faire le diagnostic qui reste toutefois aléa-toire et nécessite alors le recours éventuel à un décondi-tionnement progressif sous surveillance spécialisée.
2. Syndromes polyuro-polydipsiques hypotoniques pitressino-résistantsIls correspondent aux diabètes insipides néphrogé-niques, caractérisés par l’absence d’augmentation del’osmolalité urinaire au cours de la restriction hydrique,l’inefficacité de la desmopressine injectée en fin de res-triction et des taux de vasopressine plasmatique nor-maux ou élevés à ce moment-là. Ce dernier dosage estsouvent utile dans les diabètes insipides néphrogéniquespartiels.Ces diabètes insipides néphrogéniques sont soit congé-nitaux et familiaux, soit acquis (tableau II).
• Formes congénitales et familiales :les diabètes insi-pides néphrogéniques de ce type sont rares, d’apparitionnéonatale contrairement aux diabètes insipides centrauxfamiliaux plus tardivement révélés. Ils comportentactuellement 2 types d’anomalie génétique :– le diabète insipide néphrogénique par mutation dugène du récepteur V2 de la vasopressine, à transmissionrécessive liée à l’X. Le gène est situé dans la brancheq28 du bras long du chromosome X comme le gène durécepteur V2 de la vasopressine ;– le diabète insipide néphrogénique par mutation dugène de l’aquaporine 2, beaucoup plus rare, à transmis-sion autosomique récessive.• Formes acquises :elles sont, pour la plupart, liées àune pathologie rénale, en l’occurrence une néphropathietubulo-interstitielle chronique, primitive ou secondaire àdes maladies systémiques, ainsi qu’à des perturbationsmétaboliques ou à une origine iatrogénique (tableau II).
Diagnostic étiologique
En matière de syndromes polyuro-polydipsiques hypo-toniques pitressino-sensibles d’origine centrale ou de diabète insipide central, comme pour les diabètes insi-pides néphrogéniques, ces diabètes insipides centrauxsont soit congénitaux et familiaux, soit acquis et alorssecondaires à diverses étiologies (tableau III).
S Y N D R O M E P O L Y U R O - P O L Y D I P S I Q U E
794 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
1 / Formes congénitales et familiales
2 / Formes acquises
❑ Atteintes rénales :néphropathies tubulaires aiguës ou chroniques après uropathies malformatives ou obstructives,polykystoses, maladies kystiques médullaires,ou après transplantation rénale
❑ Maladies systémiques variées :myélome multiple, amylose, sarcoïdose, drépanocytose,syndrome de Sjögren et syndrome de Fanconi
❑ Perturbations métaboliques :hypercalcémie, hypokaliémie, carence d’apport protéique, régime désodé strict prolongé
❑ Origine iatrogénique :lithium, amphotéricine B, vinblastine, anesthésie au méthoxyflurane
Étiologie des diabètes insipides néphrogéniques
TABLEAU II

Dans ce cas, le diabète insipide central survient rapide-ment et se poursuit durant 4 à 5 jours puis est suivi d’unerémission entre le 5e et le 7e jour, et parfois même d’unhyper-vasopressinisme, puis d’une réapparition dumême diabète, cette fois assez souvent définitif. Cemode évolutif est plus fréquent lors d’une interventionpar voie haute, sous-frontale. Le caractère définitif outransitoire du diabète insipide central dépend essentiel-lement du niveau de l’atteinte de la tige hypophysaire :plus elle est haute, proche des noyaux supra-optiques etparaventriculaires, plus le diabète risque d’être définitif.Il est complet ou partiel en fonction de l’étendue de lalésion et par conséquent du nombre de neurones fonc-tionnels persistant.
2. Lésions neuro-hypothalamiquesLe diabète insipide est beaucoup plus rare quand latumeur responsable n’a pas d’extension suprasellaire etpeut être aussi masqué lors d’un hypopituitarisme corti-cotrope associé.• Primitives : les craniopharyngiomes sont la cause laplus fréquente, notamment chez l’enfant où il occupe le2e rang, après le germinome. C’est une tumeur bénigne,à croissance lente, à l’origine d’un syndrome tumoraléventuel et d’un hypopituitarisme antérieur associé,responsable d’un diabète insipide central dans environ15 % des cas en raison de son développement supra-sellaire souvent important. La présence de calcificationsvisibles à la radiographie standard est un bon argumentdiagnostique (fig. 2).Le germinome est aussi une tumeur prédominant chezl’enfant et l’adolescent, responsable d’un diabète insipidecentral à début souvent brutal, assez souvent associé àdes signes de compression chiasmatique et à un hypopi-tuitarisme antérieur. La cytologie du liquide céphalo-rachidien (LCR) ainsi que le dosage de la βhCG (humanchorionic gonadiotrophin) dans le liquide céphalorachi-dien sont très utiles au diagnostic et permettent d’éviterla biopsie dans la mesure du possible. C’est en effet unetumeur maligne, mais très radio-sensible. Le diabèteinsipide central est d’une fréquence remarquable, dansprès de 90 % des cas. L’exploration morphologique parimagerie par résonance magnétique est indispensable.L’élargissement de la tige pituitaire et la disparition del’hypersignal post-hypophysaire normal sont des signesprécoces.Des tumeurs diverses peuvent causer un diabète insipidecentral. Il survient lors d’adénomes invasifs à dévelop-pement suprasellaire et peut être masqué par une insuffi-sance corticotrope.Les tumeurs à cellules granuleuses ou choristomes sontdes tumeurs bénignes exceptionnelles de la post-hypo-physe, à croissance très lente, apparaissant volontiersaprès 40 ans et révélées par un diabète insipide associé à un syndrome tumoral suprasellaire. Ces tumeurs,très vascularisées, apparaissent homogènes et trèscontrastées en imagerie par résonance magnétique.Un lymphome hypophysaire peut très exceptionnel-lement être révélé par diabète insipide central.
Formes congénitales et familiales
Les diabètes insipides centraux de ce type sont encoretrès rares. Le diabète insipide central par mutation dugène de la vasopressine est autosomique dominant etapparaît entre 6 mois et 6 ans, puis s’aggrave progressi-vement. Ces diabètes insipides centraux sont pitresso-sensibles et la vasopressine circulante est très générale-ment indétectable. En imagerie par résonancemagnétique, l’hypersignal post-hypophysaire est sou-vent conservé. Le diabète insipide est isolé, sans déficitantéhypophysaire associé. Le diabète insipide centralfamilial est donc une affection autosomique dominantesecondaire à une mutation hétérozygote du gène de lavasopressine atteignant la partie codant le peptide signalou celle qui code la neurophysine II. Il existe aussi un seul cas connu de forme récessive liée à l’X. Le second type de diabète insipide central familialappartient au syndrome de Wolfram ou DIDMOAD(Diabetes Insipidus, Diabetes Mellitus, Optic Atrophia,Deafness). Ce syndrome est à transmission autosomiquerécessive et pourrait être dû à une mutation de l’ADNmitochondrial, mais il apparaît hétérogène sur le plangénotypique.
Formes acquises
1. Post-traumatiques et postopératoiresLe diabète insipide central peut y être transitoire, notam-ment après intervention intrasellaire par voie basse, oupermanent par atteintes hautes hypothalamiques ou de latige pituitaire et éventuellement d’évolution triphasique.
Endocrinologie - Métabolisme - Nutrition
795L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
1 / Formes congénitales et familiales
2 / Formes acquises
❑ Traumatismes cranio-cérébraux et chirurgie hypophysaire
❑ Lésions neuro-hypothalamiques :– primitives : craniopharyngiome, germinome, diverses ;– secondaires : métastases hypophysaires
❑ Maladies systémiques : sarcoïdose, histiocytose X
❑ Maladies infectieuses
❑ Maladies auto-immunes : hypophysite lymphocytaire
❑ Diabète insipide de la grossesse
❑ Idiopathiques
Étiologie des diabètes insipides centraux
TABLEAU III
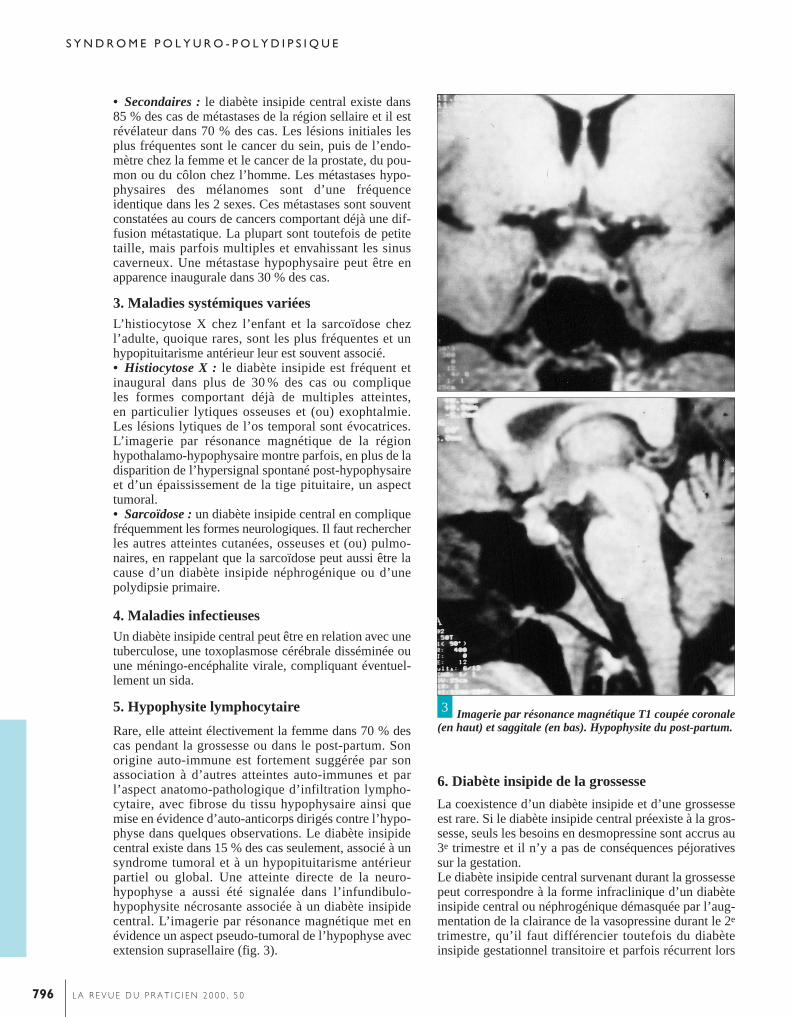
• Secondaires :le diabète insipide central existe dans85 % des cas de métastases de la région sellaire et il estrévélateur dans 70 % des cas. Les lésions initiales lesplus fréquentes sont le cancer du sein, puis de l’endo-mètre chez la femme et le cancer de la prostate, du pou-mon ou du côlon chez l’homme. Les métastases hypo-physaires des mélanomes sont d’une fréquenceidentique dans les 2 sexes. Ces métastases sont souventconstatées au cours de cancers comportant déjà une dif-fusion métastatique. La plupart sont toutefois de petitetaille, mais parfois multiples et envahissant les sinuscaverneux. Une métastase hypophysaire peut être enapparence inaugurale dans 30 % des cas.
3. Maladies systémiques variéesL’histiocytose X chez l’enfant et la sarcoïdose chezl’adulte, quoique rares, sont les plus fréquentes et unhypopituitarisme antérieur leur est souvent associé.• Histiocytose X :le diabète insipide est fréquent etinaugural dans plus de 30 % des cas ou complique les formes comportant déjà de multiples atteintes,en particulier lytiques osseuses et (ou) exophtalmie. Les lésions lytiques de l’os temporal sont évocatrices.L’imagerie par résonance magnétique de la région hypothalamo-hypophysaire montre parfois, en plus de ladisparition de l’hypersignal spontané post-hypophysaireet d’un épaississement de la tige pituitaire, un aspecttumoral. • Sarcoïdose :un diabète insipide central en compliquefréquemment les formes neurologiques. Il faut rechercherles autres atteintes cutanées, osseuses et (ou) pulmo-naires, en rappelant que la sarcoïdose peut aussi être lacause d’un diabète insipide néphrogénique ou d’unepolydipsie primaire.
4. Maladies infectieusesUn diabète insipide central peut être en relation avec unetuberculose, une toxoplasmose cérébrale disséminée ouune méningo-encéphalite virale, compliquant éventuel-lement un sida.
5. Hypophysite lymphocytaire
Rare, elle atteint électivement la femme dans 70 % descas pendant la grossesse ou dans le post-partum. Sonorigine auto-immune est fortement suggérée par sonassociation à d’autres atteintes auto-immunes et parl’aspect anatomo-pathologique d’infiltration lympho-cytaire, avec fibrose du tissu hypophysaire ainsi quemise en évidence d’auto-anticorps dirigés contre l’hypo-physe dans quelques observations. Le diabète insipidecentral existe dans 15 % des cas seulement, associé à unsyndrome tumoral et à un hypopituitarisme antérieurpartiel ou global. Une atteinte directe de la neuro-hypophyse a aussi été signalée dans l’infundibulo-hypophysite nécrosante associée à un diabète insipidecentral. L’imagerie par résonance magnétique met enévidence un aspect pseudo-tumoral de l’hypophyse avecextension suprasellaire (fig. 3).
6. Diabète insipide de la grossesse
La coexistence d’un diabète insipide et d’une grossesseest rare. Si le diabète insipide central préexiste à la gros-sesse, seuls les besoins en desmopressine sont accrus au3e trimestre et il n’y a pas de conséquences péjorativessur la gestation.Le diabète insipide central survenant durant la grossessepeut correspondre à la forme infraclinique d’un diabèteinsipide central ou néphrogénique démasquée par l’aug-mentation de la clairance de la vasopressine durant le 2e
trimestre, qu’il faut différencier toutefois du diabèteinsipide gestationnel transitoire et parfois récurrent lors
S Y N D R O M E P O L Y U R O - P O L Y D I P S I Q U E
796 L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
Imagerie par résonance magnétique T1 coupée coronale(en haut) et saggitale (en bas). Hypophysite du post-partum.
3

• chlorpropamide :100 mg/j à adapter, mais l’effethypoglycémiant est redoutable, en particulier chezl’enfant et en cas d’hypopituitarisme antérieur associé ;
• diurétiques thiazidiques et clofibrate :les posologiessont très variables et difficiles à manier, ces produitspouvant provoquer des incidents ;
• carbamazépine :la posologie est comprise entre 200 et600 mg/24 h, en général la carbamazépine est trèsbien tolérée et c’est certainement le meilleur traite-ment oral en cas de diabète insipide central partiel.■
d’une nouvelle grossesse, en rapport avec une augmen-tation de la vasopressinase associée ou non à une ano-malie du fonctionnement hépatique.
7. Diabète insipide central idiopathiqueLes progrès réalisés dans l’exploration morphologiquehypophysaire au cours du diabète insipide central ontpermis sans nul doute de diminuer sa prévalence dans laforme idiopathique. On doit y ajouter les données obte-nues grâce aux progrès de la génétique moléculaire donton peut espérer de nouvelles avancées, diminuant d’au-tant ce cadre d’attente.
Traitement
Desmopressine
La 1-déamino-8-D-AVP ou desmopressine, quelquefoisécrite DDAVP, est un analogue structural de synthèse del’hormone antidiurétique ou vasopressine. Ce médicamentde choix, commercialisé sous le nom de Minirin (desmo-pressine) a une stabilité et un pouvoir antidiurétiqueaccrus par rapport à l’hormone native, alors que son activité pressive est pratiquement négligeable. L’activitéantidiurétique est variable en fonction de la posologie etde la forme pharmacologique utilisée. Sur ce plan, ladesmopressine existe actuellement sous 4 formes :• injectable :ampoule de 1 mL dosée à 4 µg ; 2 µg/12 h
suffisent en général pour traiter un diabète insipidepitresso-sensible chez un adulte ;
• nasale, en spray :10 µg par pulvérisation ;• nasale par « Rhinyl » gradué :10 à 20 µg/24 h chez
l’adulte ;• comprimés par voie orale :0,1 à 0,2 mg par comprimé.
Traitement du diabète insipide central complet
• En contexte post-neurochirurgical ou post-trauma-tique : outre la réhydratation, on prescrit la desmopres-sine injectable : (1 à 4 µg/24 h) sous surveillance biolo-gique attentive de la natrémie.• En dehors du contexte d’urgence,la prescription estla suivante :– des comprimés à 0,1 mg 2 fois par 24 h, à adapter en
fonction de la diurèse fractionnée ;– la posologie, chez l’adulte, est comprise entre 0,1 et
0,2 mg, 3 fois par 24 h.• La forme nasaleest utilisée si l’équilibre n’est pasobtenu par voie orale.
Traitement du diabète insipide central partiel
Il touche 60 à 80 % des patients. La desmopressine peutêtre utilisée, mais à posologie plus faible et adaptée.D’autres traitements ont été proposés :
Endocrinologie - Métabolisme - Nutrition
797L A R E V U E D U P R A T I C I E N 2 0 0 0 , 5 0
• Les syndromes polyuro-polydipsiques sont,de façon générale, assez fréquents et surtoutde nature et d’étiologies multiples. Après avoir éliminé les syndromes polyuro-polydipsique hypertoniques, de diagnosticsimple et répondant à des causes la plupart du temps évidentes, la difficulté essentielleconsiste non pas à reconnaître les formes complètes, quasiment caricaturales des diabètesinsipides centraux et néphrogéniques, mais à faire le diagnostic des formes partielles et des formes masquées par une atteinte hypophysaire antérieure ou une polydipsie psychogène primitive susceptible d’induire un véritable diabète insipide.
• L’examen clinique est tout à fait fondamentalpour orienter au mieux le diagnostic. L’épreuve de restriction hydrique, réalisée soussurveillance spécialisée et dans de bonnes conditions techniques, reste la pièce maîtressede l’identification des formes partielles ou intriquées, voire modifiées par des traite-ments inadéquats.
Points Forts à retenir
Decanter C, Hober C, Hamon M, Laffite JJ, Lefebvre J, VantyghemMC. Diabète insipide révélateur d’une métastase hypophysaired’un carcinome bronchique. Ann Endocrinol (Paris) 1996 ; 57 :411-7.
Lefebvre J, Vantyghem MC, Hober C. Physiologie de l’hormoneantidiurétique. Encycl Med Chir (Elsevier, Paris), Endocrinologie-Nutrition, 10-024-A10, 1996, 5 pages.
Lefebvre J, Vantyghem MC, Hober C. Hypovasopressinismes.Encycl Med Chir (Elsevier, Paris), Endocrinologie-Nutrition, 10-024-A30, 1996, 6 pages.
Vantyghem MC, Hober C, Bouthors AS, Cappoen JP, Monnier JC,Lefebvre J. Stratégie diagnostique et thérapeutique devant un diabète insipide de la grossesse et du post-partum. Rev FrEndocrinol Clin 1993 ; 34 : 379-86.
Vantyghem MC, Hober C, Lefebvre J. Diabètes insipides congé-nitaux. Acquisitions récentes en génétique moléculaire. PresseMed 1996 ; 25 : 299-303.
Vantyghem MC, Douillard C, Evrard A, Lefebvre J. Relations entrel’axe corticotrope et l’arginine vasopressine en pathologie
POUR EN SAVOIR PLUS


















