Innate Inmune Response to H3N2 an H1N1
11
Innate immune response to H3N2 and H1N1 influenza virus infection in a human lung organ culture model Wenxin Wu a, ⁎, J. Leland Booth a , Elizabeth S. Duggan a , Shuhua Wu a,d , Krupa B. Patel a , K. Mark Coggeshall c , Jordan P. Metcalf a,b a Pulmonary and Critical Care Division, Department of Medicine, University of Oklahoma Health Sciences Center, RM 425, RP1, 800 N. Research Pkwy., Oklahoma City, OK 73104, USA b Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA c Programs in Immunology and Cancer, Oklahoma Medical Research Foundation, Oklahoma City, OK 73104, USA d Department of Emergency and Critical Care, the Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215004, P.R. China abstract article info Article history: Received 12 March 2009 Returned to author for revision 9 April 2009 Accepted 12 October 2009 Available online 12 November 2009 Keywords: Influenza virus Lung Cytokine Chemokine MAPK IP-10 MIP-1α We studied cytokine responses to influenza virus PR8 (H1N1) and Oklahoma/309/06 (OK/06, H3N2) in a novel human lung tissue model. Exposure of the model to influenza virus rapidly activated the mitogen- activated protein kinase signaling (MAPK) pathways ERK, p38 and JNK. In addition, RNase protection assay demonstrated the induction of several cytokine and chemokine mRNAs by virus. This finding was reflected at the translational level as IL-6, MCP-1, MIP-1α/β, IL-8 and IP-10 proteins were induced as determined by ELISA. Immunohistochemistry for IP-10 and MIP-1α revealed that alveolar epithelial cells and macrophages were the source of these two cytokines. Taken together, both PR8 and OK/06 cause similar induction of cytokines in human lung, although OK/06 is less effective at inducing the chemokines MCP-1 and IL-8. This human organ culture model should thus provide a relevant platform to study the biological responses of human lung to influenza virus infection. Published by Elsevier Inc. Introduction In the United States, influenza annually infects 5–20% of the population, hospitalizes 200,000, and kills 36,000. Thus, it is a leading cause of death (Izurieta et al., 2000; Thompson et al., 2004). The devastating Spanish influenza A virus infected about a third of the world's population and killed 40 million people during the pandemic of 1918. Following replication in the superficial cells of the respiratory tract, influenza virus causes a spectrum of acute clinical syndromes that can progress to a fatal outcome. The clinical syndromes include asymptomatic infection and primary viral or secondary bacterial pneumonia. Therefore, although the virus infects the entire respira- tory tract, morbidity and mortality is associated with lower respiratory tract involvement (Hers, 1966; Hers et al., 1958; Hers and Mulder, 1961). Human influenza virus belongs to the orthomyxoviridae family, which consists of four genera: influenza A, B, and C virus, and Thogovirus. Only influenza virus A and B are pathogenic among human beings. Influenza A viruses are further subdivided into subtypes based on the antigenicity of two transmembrane glycoproteins, hemagglu- tinin (HA) and neuraminidase (NA). There are 16 HA and 9 NA subtypes in influenza virus A. Viruses with HA types H1–H3 and NA types N1 and N2 are found in humans. Epithelial cells are the primary site of viral replication for influenza, although monocytes/macrophages and other leukocytes can also be infected (Ronni et al., 1997). Influenza virus specific antigen has been found in type 1 and type 2 alveolar epithelial cells, as well as in alveolar macrophages. Viruses initiate infection by binding of the viral HA to sialic acid on the cell surface and enter the cells by receptor-mediated endocytosis. Once inside the cells, influenza virus shuts off host cell protein synthesis and replicates in a fast and efficient way. This process results in host cell apoptosis or death by cytolysis. However, the host cells respond in several ways to limit viral spreading. The most significant response is production of cytokines and chemokines by epithelial cells and leukocytes via activation of multiple transcriptional and posttranslational systems (Julkunen et al., 2000). Cytokines are extracellular signal proteins that stimulate adjacent and distant cells to activate host antiviral defense. Chemokines are low molecular weight chemoattractant cytokines which bind to their specific receptors in leukocytes, recruit inflammatory cells to the site of infection, and activate innate immune responses (Baggiolini, 1998). Cytokines are released from lung epithelial cell lines during influenza virus infection. These include type 1 IFN (Ronni et al., 1997), IL-6 and the chemokines Virology 396 (2010) 178–188 ⁎ Corresponding author. Fax: +1 405 271 5440. E-mail address: [email protected] (W. Wu). 0042-6822/$ – see front matter. Published by Elsevier Inc. doi:10.1016/j.virol.2009.10.016 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/yviro
Transcript of Innate Inmune Response to H3N2 an H1N1

Virology 396 (2010) 178–188
Contents lists available at ScienceDirect
Virology
j ourna l homepage: www.e lsev ie r.com/ locate /yv i ro
Innate immune response to H3N2 and H1N1 influenza virus infection in a humanlung organ culture model
Wenxin Wu a,⁎, J. Leland Booth a, Elizabeth S. Duggan a, Shuhua Wu a,d, Krupa B. Patel a,K. Mark Coggeshall c, Jordan P. Metcalf a,b
a Pulmonary and Critical Care Division, Department of Medicine, University of Oklahoma Health Sciences Center, RM 425, RP1, 800 N. Research Pkwy., Oklahoma City, OK 73104, USAb Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USAc Programs in Immunology and Cancer, Oklahoma Medical Research Foundation, Oklahoma City, OK 73104, USAd Department of Emergency and Critical Care, the Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215004, P.R. China
⁎ Corresponding author. Fax: +1 405 271 5440.E-mail address: [email protected] (W. Wu).
0042-6822/$ – see front matter. Published by Elsevierdoi:10.1016/j.virol.2009.10.016
a b s t r a c t
a r t i c l e i n f oArticle history:Received 12 March 2009Returned to author for revision 9 April 2009Accepted 12 October 2009Available online 12 November 2009
Keywords:Influenza virusLungCytokineChemokineMAPKIP-10MIP-1α
We studied cytokine responses to influenza virus PR8 (H1N1) and Oklahoma/309/06 (OK/06, H3N2) in anovel human lung tissue model. Exposure of the model to influenza virus rapidly activated the mitogen-activated protein kinase signaling (MAPK) pathways ERK, p38 and JNK. In addition, RNase protection assaydemonstrated the induction of several cytokine and chemokine mRNAs by virus. This finding was reflected atthe translational level as IL-6, MCP-1, MIP-1α/β, IL-8 and IP-10 proteins were induced as determined byELISA. Immunohistochemistry for IP-10 and MIP-1α revealed that alveolar epithelial cells and macrophageswere the source of these two cytokines. Taken together, both PR8 and OK/06 cause similar induction ofcytokines in human lung, although OK/06 is less effective at inducing the chemokines MCP-1 and IL-8. Thishuman organ culture model should thus provide a relevant platform to study the biological responses ofhuman lung to influenza virus infection.
Published by Elsevier Inc.
Introduction
In the United States, influenza annually infects 5–20% of thepopulation, hospitalizes 200,000, and kills 36,000. Thus, it is a leadingcause of death (Izurieta et al., 2000; Thompson et al., 2004). Thedevastating Spanish influenza A virus infected about a third of theworld's population and killed 40 million people during the pandemicof 1918.
Following replication in the superficial cells of the respiratorytract, influenza virus causes a spectrum of acute clinical syndromesthat can progress to a fatal outcome. The clinical syndromes includeasymptomatic infection and primary viral or secondary bacterialpneumonia. Therefore, although the virus infects the entire respira-tory tract, morbidity and mortality is associated with lowerrespiratory tract involvement (Hers, 1966; Hers et al., 1958; Hersand Mulder, 1961).
Human influenza virus belongs to the orthomyxoviridae family,which consists of four genera: influenza A, B, and C virus, andThogovirus. Only influenza virus A and B are pathogenic amonghumanbeings. Influenza A viruses are further subdivided into subtypes based
Inc.
on the antigenicity of two transmembrane glycoproteins, hemagglu-tinin (HA) and neuraminidase (NA). There are 16 HA and 9 NAsubtypes in influenza virus A. Viruses with HA types H1–H3 and NAtypes N1 and N2 are found in humans.
Epithelial cells are the primary site of viral replication for influenza,although monocytes/macrophages and other leukocytes can also beinfected (Ronni et al., 1997). Influenza virus specific antigen has beenfound in type 1 and type 2 alveolar epithelial cells, aswell as in alveolarmacrophages. Viruses initiate infection by binding of the viral HA tosialic acid on the cell surface and enter the cells by receptor-mediatedendocytosis. Once inside the cells, influenza virus shuts off host cellprotein synthesis and replicates in a fast and efficientway. This processresults in host cell apoptosis or death by cytolysis. However, the hostcells respond in several ways to limit viral spreading. The mostsignificant response is production of cytokines and chemokines byepithelial cells and leukocytes via activation ofmultiple transcriptionaland posttranslational systems (Julkunen et al., 2000). Cytokines areextracellular signal proteins that stimulate adjacent anddistant cells toactivate host antiviral defense. Chemokines are low molecular weightchemoattractant cytokines which bind to their specific receptors inleukocytes, recruit inflammatory cells to the site of infection, andactivate innate immune responses (Baggiolini, 1998). Cytokines arereleased from lung epithelial cell lines during influenza virus infection.These include type 1 IFN (Ronni et al., 1997), IL-6 and the chemokines
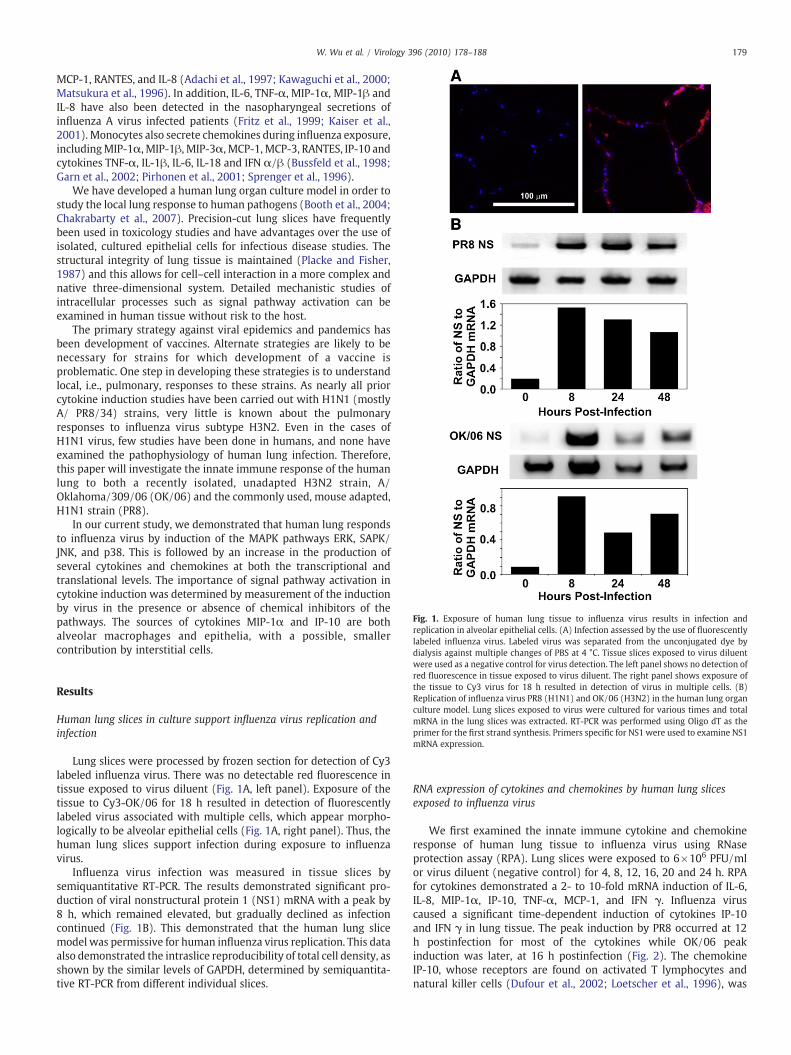
Fig. 1. Exposure of human lung tissue to influenza virus results in infection andreplication in alveolar epithelial cells. (A) Infection assessed by the use of fluorescentlylabeled influenza virus. Labeled virus was separated from the unconjugated dye bydialysis against multiple changes of PBS at 4 °C. Tissue slices exposed to virus diluent
179W. Wu et al. / Virology 396 (2010) 178–188
MCP-1, RANTES, and IL-8 (Adachi et al., 1997; Kawaguchi et al., 2000;Matsukura et al., 1996). In addition, IL-6, TNF-α, MIP-1α, MIP-1β andIL-8 have also been detected in the nasopharyngeal secretions ofinfluenza A virus infected patients (Fritz et al., 1999; Kaiser et al.,2001). Monocytes also secrete chemokines during influenza exposure,includingMIP-1α, MIP-1β, MIP-3α, MCP-1, MCP-3, RANTES, IP-10 andcytokines TNF-α, IL-1β, IL-6, IL-18 and IFN α/β (Bussfeld et al., 1998;Garn et al., 2002; Pirhonen et al., 2001; Sprenger et al., 1996).
We have developed a human lung organ culture model in order tostudy the local lung response to human pathogens (Booth et al., 2004;Chakrabarty et al., 2007). Precision-cut lung slices have frequentlybeen used in toxicology studies and have advantages over the use ofisolated, cultured epithelial cells for infectious disease studies. Thestructural integrity of lung tissue is maintained (Placke and Fisher,1987) and this allows for cell–cell interaction in a more complex andnative three-dimensional system. Detailed mechanistic studies ofintracellular processes such as signal pathway activation can beexamined in human tissue without risk to the host.
The primary strategy against viral epidemics and pandemics hasbeen development of vaccines. Alternate strategies are likely to benecessary for strains for which development of a vaccine isproblematic. One step in developing these strategies is to understandlocal, i.e., pulmonary, responses to these strains. As nearly all priorcytokine induction studies have been carried out with H1N1 (mostlyA/ PR8/34) strains, very little is known about the pulmonaryresponses to influenza virus subtype H3N2. Even in the cases ofH1N1 virus, few studies have been done in humans, and none haveexamined the pathophysiology of human lung infection. Therefore,this paper will investigate the innate immune response of the humanlung to both a recently isolated, unadapted H3N2 strain, A/Oklahoma/309/06 (OK/06) and the commonly used, mouse adapted,H1N1 strain (PR8).
In our current study, we demonstrated that human lung respondsto influenza virus by induction of the MAPK pathways ERK, SAPK/JNK, and p38. This is followed by an increase in the production ofseveral cytokines and chemokines at both the transcriptional andtranslational levels. The importance of signal pathway activation incytokine induction was determined by measurement of the inductionby virus in the presence or absence of chemical inhibitors of thepathways. The sources of cytokines MIP-1α and IP-10 are bothalveolar macrophages and epithelia, with a possible, smallercontribution by interstitial cells.
were used as a negative control for virus detection. The left panel shows no detection ofred fluorescence in tissue exposed to virus diluent. The right panel shows exposure ofthe tissue to Cy3 virus for 18 h resulted in detection of virus in multiple cells. (B)Replication of influenza virus PR8 (H1N1) and OK/06 (H3N2) in the human lung organculture model. Lung slices exposed to virus were cultured for various times and totalmRNA in the lung slices was extracted. RT-PCR was performed using Oligo dT as theprimer for the first strand synthesis. Primers specific for NS1 were used to examine NS1mRNA expression.
Results
Human lung slices in culture support influenza virus replication andinfection
Lung slices were processed by frozen section for detection of Cy3labeled influenza virus. There was no detectable red fluorescence intissue exposed to virus diluent (Fig. 1A, left panel). Exposure of thetissue to Cy3-OK/06 for 18 h resulted in detection of fluorescentlylabeled virus associated with multiple cells, which appear morpho-logically to be alveolar epithelial cells (Fig. 1A, right panel). Thus, thehuman lung slices support infection during exposure to influenzavirus.
Influenza virus infection was measured in tissue slices bysemiquantitative RT-PCR. The results demonstrated significant pro-duction of viral nonstructural protein 1 (NS1) mRNA with a peak by8 h, which remained elevated, but gradually declined as infectioncontinued (Fig. 1B). This demonstrated that the human lung slicemodel was permissive for human influenza virus replication. This dataalso demonstrated the intraslice reproducibility of total cell density, asshown by the similar levels of GAPDH, determined by semiquantita-tive RT-PCR from different individual slices.
RNA expression of cytokines and chemokines by human lung slicesexposed to influenza virus
We first examined the innate immune cytokine and chemokineresponse of human lung tissue to influenza virus using RNaseprotection assay (RPA). Lung slices were exposed to 6×106 PFU/mlor virus diluent (negative control) for 4, 8, 12, 16, 20 and 24 h. RPAfor cytokines demonstrated a 2- to 10-fold mRNA induction of IL-6,IL-8, MIP-1α, IP-10, TNF-α, MCP-1, and IFN γ. Influenza viruscaused a significant time-dependent induction of cytokines IP-10and IFN γ in lung tissue. The peak induction by PR8 occurred at 12h postinfection for most of the cytokines while OK/06 peakinduction was later, at 16 h postinfection (Fig. 2). The chemokineIP-10, whose receptors are found on activated T lymphocytes andnatural killer cells (Dufour et al., 2002; Loetscher et al., 1996), was

Fig. 2. Cytokine and chemokine mRNA response of human lung to influenza virusinfection. Human lung tissue slices (3 slices per data point) were exposed to 6×106
PFU/ml of influenza virus PR8 (A) and OK/06 (B). Equal volumes of virus diluent wereused as a negative control. mRNA expression levels were determined using a customcytokine template (see Materials and methods). Fold increase was determined bynormalization for levels of housekeeping genes present in each sample.
Fig. 3. Influenza virus stimulates chemokine and cytokine release in human lung. Foreach data point, multiple lung slices were exposed to 6×106 PFU/ml of influenza virusPR8 and OK/06 and allowed to incubate at 37 °C, 5% CO2 for the indicated periods.Virus diluent was used as a negative control, and PMA (100 ng/ml) was used as apositive control. Chemokine and cytokine protein levels were determined by ELISA onlung slice supernatants. Data are expressed as the means±SEM from three separatelung slice donor experiments. Statistical significance was determined by ANOVA.Means were compared to data from the negative control group. ⁎Pb0.05; ⁎⁎Pb0.01;NS represents no significant difference.
180 W. Wu et al. / Virology 396 (2010) 178–188
the most highly induced cytokine in terms of mRNA fold increase.As to the patterns of cytokines induced, there was no discernabledifference in induction for the two viral strains. Thus, RPA resultssuggest a broad cytokine immune response and also a monocyteand neutrophil chemokine response to influenza virus exposure byhuman lung.
Induction of cytokines in human lung tissue infected with influenza virus
RPA data indicated influenza induction of mRNA levels of severalcytokines and chemokines. To confirm that this increase in mRNAwasreflected at the level of translation, we tested for the correspondingcytokine protein in supernatants of lung slices exposed to influenzavirus for 6, 20, 30 and 40 h using ELISA. Human lung slices were mocktreated with virus diluent, treated with 6×106 PFU/ml of influenzavirus or treatedwith PMA (100 ng/ml) as a positive control. Consistentwith the RPA results, we saw an increase in the cytokines IL-6 and thechemokines IL-8, MCP-1, IP-10 and MIP-1α/β with virus exposure(Fig. 3). Specifically, therewas a peak fold increase over background inPR8-induced cytokine and chemokine levels of IL-6, IP-10, IL-8,MCP-1,and MIP-1α/β of 16, 75, 2, 12 and 50, respectively. MIP-1α/βmaximum levels were seen at 20 h, IL-8 at 30 h while the maximumincrease in IL-6, IP-10 and MCP-1 were seen at 40 h. At all the timestested, MCP-1 was stimulated to a greater extent than that seen withthe positive control, 100 ng/ml of PMA (Fig. 3C). The most stimulatedcytokine was IP-10, 75-fold over mock, which is consistent with theRPA result showing IP-10 is the most highly induced cytokine at the

Fig. 4. Kinetics of ERK1/2, SAPK/JNK and p38 phosphorylation by influenza virus inhuman lung slices. Human lung tissue slices were exposed to 6×106 PFU/ml ofinfluenza virus PR8 and OK/06 for the times indicated. Lung lysates were prepared, andlevels of total and phosphorylated ERK1/2 (A, B, and C), p38 (D, E, and F), and SAPK/JNK(G, H, and I) were assessed by Western blot analysis. PMA (100 ng/ml) was used aspositive controls, and virus diluent was used as a negative control. Western blots wereprobed with antibody specific for phosphorylated ERK1/2 (A), p38 (D), or SAPK/JNK(G) are shown. Identically prepared blots were also probed with pan-anti-kinaseantibody against ERK1/2 (B), p38 (E), or SAPK/JNK (H). Activation as determined bythe ratio of phosphorylated kinase/total kinase for ERK1/2 (C), p38 (F), and SAPK/JNK(I) is graphed as shown.
181W. Wu et al. / Virology 396 (2010) 178–188
level of transcription. PMA is not a strong inducer for IP-10, whichsuggests that the human lung innate response involving IP-10 isspecific to virus (Fig. 3E). Although we saw a significant induction ofIFN-γ mRNA in the RPA, IFN-γ protein levels were low (less than100 pg/ml) in lung slice supernatants, and undetectable in lung tissueextracts (data not shown). However, as IP-10 (10 kDa interferon-gamma-induced protein) is secreted by several cell types in responseto IFN-γ (Luster et al., 1985), induction of IP-10 suggests that IFN-γinduction may have occurred in our system despite the ELISA results.For the most part, PR8 and OK/06 caused similar induction of most ofthe cytokines tested except MCP-1 and IL-8. OK/06 more weaklyinduces MCP-1 than PR8, at two of the four times tested. For IL-8 induction, OK/06 also appears to induce IL-8 less readily than PR8,but this only reached statistical significance at 30 h after infection.These results show that the induction of cytokine and chemokinegenes, as determined by RPA, is consistently reflected at the proteinlevel. As such, both influenza virus strains induced a broad innateimmune chemokine and cytokine response in human lung.
Induction of MAPK signaling pathways in human lung slices exposed toinfluenza virus
To determine the role of signal pathway activation in cytokine andchemokine induction by influenza virus in human lung, we nextstudied the kinetics of virus-induced activation of the MAPK signalingcascades. We assessed ERK, SAPK/JNK, and p38 activation as exhibitedby phosphorylation in human lung infected with virus (6×106 PFU/ml). Stimulation with PMA (100 ng/ml) for 1 h served as a positivecontrol for phosphorylation, and mock-infected, unstimulated lysateswere prepared at 1 and 8 h as a negative control. At various times afterinfection, lung lysates were prepared, and total and phosphorylatedERK, SAPK/JNK, and p38 levels were assessed by Western blotanalysis. Membranes were also probed for total ERK, p38 and JNKMAPK as a control for protein loading (Rodriguez-Viciana et al., 2006;Williams et al., 2001). Phosphorylation was assessed by determiningthe ratio of phosphorylated (Figs. 4A, D and G) to total kinase (Figs. 4B,E and H), and the resultant activities were graphed (Figs. 4C, F and I).The ratio corrects for variations in the amount of sample proteinloaded onto SDS–PAGE gels. These results revealed that ERK, SAPK/JNK, and p38 were all activated by either, or both, of the influenzavirus strains in the human lung organ culture model.
For PR8, p38 and JNK were activated and were greatest at the lasttime point measured (8 h). Activation of p38 and SAPK/JNK at thattime was 2.7- and 1.9-fold, respectively, over mock-infected controls.PR8-induced ERK phosphorylation exhibited a constant, moderateincrease over the negative control at all times tested, ranging from1.1- to 1.5-fold. For OK/06, ERK phosphorylation also exhibited aconstant, moderate increase over the negative control at most timestested, ranging from 1.2- to 1.7-fold. With this strain, in contrast toPR8, activation of p38 occurred soon after infection, and declinedthereafter. Also, in contrast to PR8, no induction of JNK was evidentduring the experiment. These results indicate that exposure toinfluenza virus elicits a modest increase in ERK, SAPK/JNK, and p38activation which precedes cytokine mRNA induction and depends onthe influenza virus strain used.
Requirement for activation of multiple signaling pathways in cytokineinduction by influenza virus
The findings described above demonstrated that the MAPKsignaling pathways are activated when human lung slices are exposedto influenza virus. We next sought to determine whether theactivation of these signaling pathways is essential for the inductionof cytokines. Lung slices were preincubated for 4 h in medium with50 μMof the ERK pathway inhibitor U0126, 0.5 μMof the p38 pathwayinhibitor SB203580, or 0.5 μM of the SAPK/JNK pathway inhibitor
SP600125. For human lung, these doses were sufficient to inhibitinduction of their corresponding signaling pathways by PMA/LPS. Acombination of the three inhibitors was also used. The inhibitorsremained in the medium throughout the duration of the experiment,and slices that were not treated with inhibitors were exposed to aninhibitor solvent (DMSO) as an additional control. The lung sliceswere incubated for 20 h either with virus (6×106 PFU/ml) or withPMA (100 ng/ml) prior to collection of the supernatants for cytokineand chemokine determination.

182 W. Wu et al. / Virology 396 (2010) 178–188
The data revealed differences in the requirements for signalingpathway activation for the induction of specific cytokines by influenzavirus (Fig. 5). With the exception of virus induction of IL-8 and MIP-
Fig. 5. Inhibition of influenza virus induction of cytokines and chemokines by signalpathway inhibitors. Human lung tissue slices were preincubated with 0.5 μM ofSB203580, 0.5 μM of SP600125 and 50 μM of U0126, or all three together (mixture) orinhibitor solvent (Mock), and the concentrations of these reagents was maintainedthroughout. Slices were then exposed to virus diluent, 6×106 PFU/ml of influenza virusPR8 and OK/06, or PMA (100 ng/ml) for 20 h prior to the measurement of cytokinesand chemokines by ELISA. The data are expressed as the means±SEM of fourexperiments, and three tissue slices were used per experiment. Statistical significancewas determined by ANOVA. Means were compared to data from the virus-infectedcontrol group without inhibitors. ⁎Pb0.05; ⁎⁎Pb0.01.
1α/β, addition of any of the three inhibitors diminished cytokine andchemokine induction by influenza virus. In the case of MIP-1α/β,inhibition of the p38 pathway individually did not alter induction ofthis chemokine by virus. For all cytokines and chemokines tested,inhibition of all three pathways appeared to enhance blockade ofvirus-mediated cytokine and chemokine induction.
OK/06 was more sensitive to the p38 inhibitor, SB203580, for IL-6induction than PR8, but not for induction of MIP-1α/β, MCP-1, IP-10and IL-8. PR8-induced MCP-1 and IP-10 were p38 dependent whileMIP-1α/β induction was likely independent of activation of thispathway. The different sensitivities of the two strains to inhibitorswere not likely due to inhibitor-caused viral replication defects sinceNS1 gene expression of both strains was not affected in inhibitortreated tissue (data not shown).
Cellular source of cytokine induction by influenza virus
To determine the lung cellular elements that participate in thelung innate immune cytokine response to influenza virus, weperformed immunohistochemistry on virus-exposed lung slices.Lung slices were exposed to virus at 6×106 PFU/ml or virus buffersfor 24 h in the presence of Brefeldin A to enhance detection ofcytokines. The slices were then processed for immunohistochemistryfor detection of influenza virus nucleoprotein (NP) and the cytokinesMIP-1α and IP-10. Macrophages were also detected by using an anti-CD 68 polyclonal antibody (De Groot et al., 1997; Noorman et al.,1997). Tissues exposed to virus diluent were used to demonstratebasal cytokine and chemokine detection. An additional negativecontrol was performed for MIP-1α and IP-10 detection by using thesame staining protocol but with the MIP-1α or IP-10 primaryantibody omitted. There was minimal background immunofluores-cence in the absence of MIP-1α and IP-10 primary antibodies (Figs. S1and S2). MIP-1α and IP-10 detection were significantly enhanced byinfluenza virus infection (Figs. 6 and 7, panels C). BothMIP-1α and IP-10 were detected in both epithelial cells and CD 68+ alveolarmacrophages (Figs. 6 and 7, panels F). There were also scatteredinterstitial cells that also stained positive for MIP-1α and IP-10. Theresults indicate that both lung epithelia and alveolar macrophagescontribute to the innate immune response through induction ofcytokines. Additional interstitial cells may also contribute to thisresponse. There is no obvious cell specificity difference for cytokineexpression between the two influenza strains.
Discussion
Host innate immunity is the first line of protection againstinfection by virus and is essential in local control of invadingmicrobes. The innate immune system is composed of macrophages,neutrophils, natural killer cells and dendritic cells which play crucialroles in the initiation and subsequent direction the of adaptiveimmune response, as well as in promoting localized inflammation andproducing cytokines that recruit additional leukocytes to the site ofinfection. However, unrestrained or excessive stimulation of theinnate immune response can be harmful. The recent study fromKobasa et al. using reconstructed 1918 influenza virus in macaquesrevealed that the unprecedented lethality of the 1918 pandemic virusis linked to an aberrant innate immune response (Kobasa et al., 2007).Similar analysis of mice infected with the reconstructed 1918influenza virus also demonstrated an increased and acceleratedactivation of host immune response genes associated with severepulmonary pathology (Kash et al., 2006). With increased mobility ofpotentially infected subjects due to modern travel, concern has beenraised about the introduction of highly pathogenic avian influenzaviruses into humans and the possibility, if adaptation of the virusenables person-to-person spread, of a resultant worldwide pandemic.Therefore, a comprehensive understanding of the human innate

Fig. 6. Cellular source of MIP-1α induction by influenza virus in human lung. Lung slices were exposed to 6×106 PFU/ml of influenza virus PR8 and OK/06 or virus diluent for 24 h inthe presence of BFA to enhance detection of cytokines. The slices were then processed for immunohistochemistry for detection of the chemokine MIP-1α using goat polyclonalantibodies, viral nucleoprotein (NP) using rabbit polyclonal antibody and macrophages using an anti-CD 68 polyclonal antibody. Panels E are brightfield images that demonstratethat lung architecture is preserved during the experiment. The rest of the panels are fluorescent images that demonstrate nuclei (panels A, blue), NP (panels B, red), MIP-1α (panelsC, green) and macrophages (panels D, cyan). Panels F are overlays of the fluorescent images and demonstrate that the primary cellular sources of the cytokines are alveolarmacrophages (arrows) and epithelial cells. Some interstitial cells are also positive for MIP-1α. The bar represents 100 μm.
183W. Wu et al. / Virology 396 (2010) 178–188
immune response to influenza virus is important. Moreover, under-standing the contribution of host immune responses to virulentinfluenza virus infections is an important starting point for theidentification of prognostic indicators and the development of novelantiviral therapies.
Our current study examines the interaction of influenza virus withhuman lung tissue. Influenza virus induces a strong pro-inflammatorycytokine and a monocyte and lymphocyte chemokine response inhuman lung tissue, as shown by our RPA and ELISA results (Figs. 2
and 3). Specifically, this response includes induction of the cytokinesIL-6 and IFN-γ, the monocyte chemokines MIP-1 α/β and MCP-1, theneutrophil chemokine IL-8, and the lymphocyte chemokine IP-10.Immunohistochemistry data clearly shows that MIP-1α and IP-10 areinduced not only in macrophages, as is traditionally thought, but alsoin alveolar epithelial cells. This discovery suggested epithelial cellsmay play more important roles in contributing to the pathogenesis ofairway inflammation caused by influenza virus infection. Prior to thedata presented in this work, only IL-6, IL-8, MCP-1, Eotaxin and

Fig. 7. Cellular source of IP-10 induction by influenza virus. Lung slices were exposed to 6×106 PFU/ml of influenza virus PR8 and OK/06 or virus diluents for 24 h in the presence ofBFA to enhance detection of cytokines. The slices were then processed for immunohistochemistry for detection of the chemokine IP-10 using goat polyclonal antibodies, viral NPusing rabbit polyclonal antibody and macrophages using an anti-CD 68 polyclonal antibody. Panels E are brightfield images that demonstrate that lung architecture is preservedduring the experiment. The rest of the panels are fluorescent images that demonstrate nuclei (Panels A, blue), NP (Panels B, red), IP-10 (Panels C, green) andmacrophages (Panels D,cyan). Panels F are overlays of the fluorescent images and demonstrate that the primary cellular sources of the cytokines are alveolarmacrophages (arrows) and epithelial cells. Someinterstitial cells are also positive for IP-10. The bar represents 100 μm.
184 W. Wu et al. / Virology 396 (2010) 178–188
RANTES have been shown to be induced by influenza in isolatedhuman epithelial cells, or cell lines in culture (Adachi et al., 1997; Choiand Jacoby, 1992; Kawaguchi et al., 2000; Matsukura et al., 1996).Macrophages and dendritic cells have been the traditional focal pointsfor study of the initial innate immune response to influenza virusinfection and are, of course, important in the response to thepathogen. However, the lung epithelium is the first site of exposurefor virus and contains a large, 80 m2 surface area for viral contact.Thus, due to the sheer number of the cells involved, and ourdemonstration that these cells respond to the virus with elaboration
of cytokine, and chemokines, the contribution of these cells to theinnate immune response is likely significant.
Our human lung organ culture model, unlike artificial modelsusing differentiated cell lines, reproduces the normal lung architec-ture as it maintains the three dimensional structure present in nativetissue. Also, current cell culture models lack the diversity of cell typesfound in normal lung. Thus, complex interactions of the different celltypes in the native lung are not accurately modeled in cultured celllines, but are closely replicated and reproduced structurally in ourmodel. It is likely that the diverse cytokine and chemokine responses

185W. Wu et al. / Virology 396 (2010) 178–188
to influenza presented herein more accurately describe the actuallung innate immune response to the viruses.
One limitation of our study is that some of the lung tissue used wasfrom patients who were undergoing resection for lung cancer. Thus,they are older, frequently suffer from COPD, and usually have previouscigarette smoke exposure. These patient-specific factors have variableeffects on cytokine responses to stimuli. However, the overallresponses, though not the magnitude or exact timing of the cytokineresponses, are frequently comparable (Gaschler et al., 2008; Hackettet al., 2008). We have found that in the general group of patients fromwhom we have obtained tissues there are consistent cytokineresponses to stimuli not only from slice to slice, but also from donorto donor. Thus, we believe that although the magnitude of theresponses might be different, the overall characteristics of theresponses are likely similar to those that occur in patients affectedwith influenza virus. Furthermore, we included in the inhibitorexperiment both tissues from surgeries and from National DiseaseResearch Interchange (NDRI). The overall cytokine responses of thelung from resection are comparable to a healthy human lung (Fig. 5 vs.Fig. 3 at 20 h after infection).
The initial phase of influenza virus infection is characterized byneutrophilic infiltration followed by a mixed monocytic/neutrophilicinfiltration from the peripheral blood across the endo-/epithelialbarrier into the alveolar air space (Toms et al., 1977). Later, theairways are filled with exudates containing monocytes and lympho-cytes (Haesebrouck et al., 1985; Van Reeth, 2000). Induction of theneutrophil chemotaxin IL-8 is consistent with the finding ofneutrophil infiltration in the initial phase. The induction of monocytechemotaxins MIP-1α/β and MCP-1 is consistent with monocyteinfiltration that occurs later in the course of infection. Finally, therobust expression of the lymphocyte chemotaxin IP-10 is likelyimportant in recruiting lymphocytes that are prominent in the airwayat latter stages of disease, and participate in the adaptive immuneresponse to influenza. It is important to recognize that strictcorrelation of the autopsy findings with our present study is limitedby the fact that autopsy studies represent findings of the terminalstages of influenza infection, while this current study examines virus-host interactions throughout the course of the illness.
The elucidation of intracellular signaling pathways that areactivated by influenza A virus infection is important for theunderstanding of both viral replication strategies and host defensemechanisms. Here, we show that productive infection of lung slicesresults in a moderate activation of all the subgroup of MAPK signalingpathways, p38, ERK and JNK. Inhibition experiments showed thatindividual pathways are important in induction of specific cytokinesand chemokines. However, as addition of all three inhibitors togetherprovided additional suppression of the cytokine and chemokineresponses, it is likely that multiple pathways play a role in cytokineand chemokine induction by influenza virus. Our data extends studiesthat show that p38 MAP kinase and JNK regulates RANTES productionby bronchial epithelial cells induced by influenza virus A. Also p38MAP kinase has been identified as important for hyperinduction ofTNF-α expression in human macrophages due to the avian H5N1influenza virus (Kujime et al., 2000; Lee et al., 2005). Ludwig et al.have also demonstrated that influenza virus-induced activation of JNKand AP-1 appears to be part of the innate antiviral response of theMDCK and U937 cells (Ludwig et al., 2001). This is the first report thatall the three MAPK signaling pathways are involved in cytokineinduction by influenza virus in cultured human lung tissue.
We chose to include an H3N2 influenza virus recently isolatedfrom the community in our study. The emergence of H3N2 influenzavirus strains as a major seasonal pathogen is a major public healthconcern. This is because vaccination and antiviral therapy are themainstays of planning against yearly influenza outbreaks, and H3N2vaccine production is problematic. Most human H3N2 influenzaviruses isolated after 1992 have lost the ability to agglutinate chicken
red blood cells and only bind to human or guinea pig erythrocytes(Gulati et al., 2005; Nobusawa et al., 2000). These viruses growwell inMDCK cells, but they are difficult to adapt to egg culture, which hascaused vaccine shortages. H3N2 viruses account for over 90% of thestrains that are adamantane resistant, which has created additionalpublic health issues (MMWR Journal Article, 2006). Understandingthe pathogenesis of H3N2 infection in order to develop noveltherapies is particularly important. Our use of both the H1N1 andH3N2 strains will ensure that the results can be correlated withresponses studied using adapted strains in other models, and with theresponses that occur when the general public is exposed to non-adapted influenza. In isolated human type II alveolar epithelial cells(ATII), both H1N1 and H3N2 viruses induced release of pro-inflammatory cytokines such as IL-6, IL-8, RANTES, MCP-1, and MIP-1β. Although both viruses have similar ability to infect and replicate inATIIs, the wild-type strain PR8 is a stronger inducer of chemokinesand cytokines than A/Phil/82 (a H3N2 reassortant PR/8 virus, Wanget al., 2009). Furthermore, since the ferret is considered the mostreproducible model similar to human in terms of pathological changesand cytokine responses to influenza (Smith and Sweet, 1988), analysisof the local ferret immune response to human influenza isolates of theH1N1 and H3N2 subtypes showed that cytokine responses aresubtype-independent (Svitek et al., 2008). In A549 cells (a humantype II alveolar epithelial adenocarcinoma cell line), influenza AH1N1-induced cytokinemRNA expressionwas detectable at 24 h afterinfection, while H3N2 infection-induced mRNA expression after 12h of infection. Although influenza A H1N1 virus induced somewhatlower chemokine mRNA expression compared to H3N2 or influenza Bviruses, the chemokine protein induction was nearly equal for all theinfluenza viruses tested (Veckman et al., 2006). In this study, despitethe large difference in these strains in terms of growth in egg culturesystems and adamantane resistance, we found that there were onlysubtle differences in terms of the innate immune cytokine response toH1N1 and H3N2 in human lung.
One research group studied the induction of the MAPK signalcascade by two seasonal human influenza A viruses, A/HK/218449/06 (H3N2) and A/HK/218847/06 (H1N1). Infection with H3N2 virusresulted in substantially increased activation of ERK signalingcompared to that caused by H1N1 (Marjuki et al., 2007). Our dataagree with earlier results in primary macrophages that only modestERK and p38 signaling was induced during H1N1 infection (Lee et al.,2005). Furthermore, modest JNK activation was also observed in ourmodel by H1N1.
Taken together, our results indicate that the human lung respondsto infection by the influenza virus by producing a robust cytokine andchemokine response which recruits neutrophils, monocytes andlymphocytes to participate in the innate immune response. We alsodemonstrate that the production of the pro-inflammatory cytokinesand chemokines are causally related to the activation of the signalingpathways ERK, JNK, and p38. This is the first description of the initialhuman lung innate immune response to influenza virus in a humanorgan culture model.
Materials and methods
Preparation of influenza virus stock
The viruses used in this study include A/Oklahoma/309/06, aWisconsin/05 like H3N2 isolate, and A/PR/34, H1N1 virus. A/Oklahoma/309/06 was isolated in the University of Oklahoma HealthSciences Center clinical microbiology laboratory in 2006. The viruseshave been passaged in Madin–Darby canine kidney (MDCK) cells.MDCK cells were cultured in supplemented Dulbecco's modifiedEagle's medium (DMEM). Viruses were grown in MDCK cells inDMEM/F12 with ITS+ (BD Biosciences, Franklin Lakes, NJ) andtrypsin (Liu and Air, 1993), harvested at 72 h postinfection and titered

186 W. Wu et al. / Virology 396 (2010) 178–188
by plaque assay in MDCK cells. There was no detectable endotoxin inthe final viral preparations used in the experiments as determined bylimulus amebocyte lysate assay (Cambrex, Walkersville, MD). Thelower limit of detection of this assay is 0.1 EU/ml or approximately20 pg/ml LPS.
Lung explant culture
Human lung tissue was obtained from patients undergoing lungresection for cancer in accordance with protocols approved by theInstitutional Review Boards of the University of Oklahoma, VeteransAdministration Hospital, Baptist-Integris Hospital, St. Anthony'sHospital, and Mercy Health Center, all of Oklahoma City, OK. Onlytissue that did not contain tumor was used for experiments. Forexperiments onMAPK inhibition, lung tissue fromNDRI was also usedfor comparison with tissue from surgeries. The tumor free lung tissuewas transported on ice in sterile Phosphate buffered saline (PBS)containing 200 μg gentamicin/ml, 100 U penicillin/ml, 100 μgstreptomycin/ml, and 2.5 μg amphotericin B/ml (PBS+antibiotics)and processed immediately. The subsegmental bronchi of lung tissuewith intact outer pleura were isolated and cannulated with a flexibleTeflon catheter, and the lung segments were gently inflatedwith 37 °Clung slice medium (LSM) containing 1.5% low-melting–low-gellingagarose (BioWhittaker, Rockland, ME). LSM consisted of minimalessential medium (Sigma Chemical Co., St. Louis, MO) supplementedwith 1.0 μg of bovine insulin/ml, 0.1 μg of hydrocortisone/ml, 0.1 μg ofretinyl acetate/ml, 200 μg of gentamicin/ml, 100 U of penicillin/ml,100 μg of streptomycin/ml, and 1.25 μg of amphotericin B/ml. Afterthe agarose inflated lung was solidified, 1 cm diameter tissue cores ofnon-emphysematous tissue were prepared and sliced into 500 μmthick sections using a Krumdieck Tissue Slicer (Alabama Research andDevelopment, Munford, AL). During slicing, the cores were sub-merged in chilled PBS + antibiotics. Each slice was placed in 0.5 ml ofLSM in a single well of a 24-well plate, then placed in a humidifiedincubator at 37 °C in 5% CO2. The LSMwas replaced prior to subjectingthe slices to the experimental treatments.
Infection of human lung slices with influenza virus
After overnight incubation of the lung slices, the culture mediumwas replaced with fresh LSM. For each data point, three lung sliceswere each exposed to 6×106 PFU/ml of influenza virus PR8 and OK/06, and allowed to incubate at 37 °C, 5% CO2 for the indicated periods.Virus diluent was used as a negative control, and PMA (100 ng/ml)was used as a positive control. Following stimulation for varioustimes, media supernatants were harvested and stored at−20 °C priorto ELISA.
OK/06 H3N2 virus was reacted with Cy3 dye in 0.1 M sodiumcarbonate buffer (pH 9.3) for 30 min at room temperature withoccasional mixing according to the manufacturer's protocol forlabeling proteins. The labeled virus was then dialyzed in PBScontaining 0.6 mM CaCl2 and 0.5 mM MgCl2 (CaMg-PBS ) for 72 hwith three buffer changes to remove unlabeled dye. The tissue slicesexposed to virus-free diluent were used as a negative control forinfluenza virus detection.
RNA preparation and RNase protection assay (RPA)
Lung slices were harvested by homogenization in TRIzol Reagent(Invitrogen, Carlsbad, CA), and the total RNAwas isolated according tothemanufacturer's protocol using glycogen (20mg/ml) as the carrier.Triplicate slices yielded 8–10 μg total RNA, 6 μg of which were used fora single RPA reaction.
Relative gene expression was determined with the RiboQuantMulti-Probe RNase Protection Assay system (BD Biosciences). Acustom cytokine set was used containing probes for TNF-α, IL 12/
p35, IP-10, IFN-γ, MIP-1α, MCP-1, IL-8 and IL-6. The template setcontained probes for ribosomal protein (L32) and GAPDH to use fornormalization of RNA loading. Labeled riboprobe was made with theIn vitro Transcription Kit (BD Biosciences) and (α-32P) UTP. The RPAKit (BD Biosciences) was used for hybridization of the probe with thetarget RNA in the samples, and for digestion of unpaired transcripts.Additional controls included a sample containing yeast total RNA, asample with the custom template control RNA, and one withunprotected probe. The resulting mRNA duplexes were separated ona standard 50 cm long, 0.4 mm thick polyacrylamide gel. The gel wasdried and imaged using a phosphorimager (Molecular Dynamics,Sunnyvale, CA). The image was analyzed with ImageQuant 5.0software (Molecular Dynamics) using the volume quantitationmethod with histogram peak background subtraction. The identityof each protected band in a sample lane was determined from theposition of the bands in the unprotected probe lane. Fold increase foreach RNA species over control samples prepared at the same timepoints was determined after correction for loading using the L32 andGAPDH standards.
Signal pathway inhibition and cytokine and chemokine proteindetermination by ELISA
After overnight incubation of the lung slices, the culture mediumwas replaced with fresh LSM. Lung slices were exposed to 6×106
PFU/ml of influenza virus PR8 and OK/06 in triplicate wells of a 24-well plate and allowed to incubate at 37 °C for 24 h. Virus diluent wasused as a negative control, and PMA (100 ng/ml) was used as apositive control.
To determine the effect of inhibition of the ERK, p38, and JNKsignaling pathways on cytokine induction, the specific inhibitorsU0126, SB203580 and SP600125 (Calbiochem, San Diego, CA) wereused, respectively. Lung slices were pre-incubated with 0.5 μM ofSB203580, 0.5 μM of SP600125 and 50 μM of U0126 for 4 h at 37 °C.These optimized doses were determined using dose range experi-ments in lung slices stimulated with PMA/LPS. The medium wasreplaced with LSM, and the lung slices were exposed to 6×106 PFU/ml of influenza virus PR8 and OK/06 for 20 h at 37 °C. The finalconcentration of the inhibitors was maintained throughout theexperiment. PMA (100 ng/ml) was used as a positive control, andmock-infected slices were exposed to the same concentration of theinhibitor solvent, dimethyl sulfoxide (DMSO), as present when allthree inhibitors were used. After incubation, the supernatants werecollected, centrifuged at 10,000×g for 2 min, transferred to a newtube and stored at −20 °C.
Cytokine ELISA's were performed using anti-cytokine monoclonalprimary antibodies and biotinylated anti-cytokine polyclonal second-ary antibodies (R&D Systems, Minneapolis, MN). As the MIP-1αmonoclonal antibody crossreacts with MIP-1β, the results wereexpressed as MIP-1α/β levels. Plates were developed using the TMBreagent (BD Biosciences).
Signaling pathway kinase assay
Human lung slices were maintained overnight at 37 °C, 5% CO2 in0.5 ml LSM and the mediumwas replaced prior to stimulation. Four toeight slices per condition were used for MAPK family assays. Sliceswere stimulated with either 6×106 PFU/ml of influenza virus or PMA.Mock-infected, negative control slices were exposed to an equivalentvolume of virus-free diluent. After incubation at 37 °C, 5% CO2 for theindicated times, the slices were harvested and homogenized in 100 μlcold RIPA lysis buffer. Clarified lung slice homogenates containing 20–30 μg protein were heat denatured in SDS–PAGE sample buffer. Thesamples were separated on a 10% SDS–PAGE gel and then electropho-retically transferred to polyvinylidene fluoride (PVDF)membranes. Todetect activated, phosphorylated ERK, p38 or SAPK/JNK, the

187W. Wu et al. / Virology 396 (2010) 178–188
membranes were blocked overnight in 5% powdered milk in Tris-buffered saline (TBS) and then immunoblotted with specific affinity-purified, rabbit polyclonal antibodies (Cell Signaling Technology,Beverly, MA). Identically prepared membranes were probed witheither rabbit polyclonal anti-ERK, anti-p38 or anti-SAPK/JNK anti-bodies that recognized both phosphorylated and non-phosphorylatedforms of the signaling proteins (Cell Signaling Technology). Themembranes were developed with horseradish peroxidase-conjugatedgoat anti-rabbit IgG (Cell Signaling Technology) and chemilumines-cent reagents (Pierce Biotechnology, Rockford, IL). The developedmembranes were exposed to X-ray film, and digital scans of the filmwere quantified using ImageQuant software (BD/MolecularDynamics,Bedford, MA).
Cytokine immunohistochemistry on lung tissue explants
To examine which cell types in the lung tissue produced IP-10 andMIP-1α after influenza infection, lung slices were exposed to 6×106
PFU/ml of influenza virus or virus diluent in fresh LSM and incubatedat 37 °C for 24 h. Brefeldin A (L C Laboratories, Wofford, MA) wasadded at a concentration of 5 μg/ml to block protein export in orderto enhance cytokine detection. Following the incubation, the lungslices were fixed with 4% paraformaldehyde in PBS at roomtemperature for 30 min and were then imbedded in paraffin.Sections (3–5 μm) were mounted on glass slides and immunoprobedovernight at 4 °C with a goat anti-human polyclonal antibody for IP-10 (R&D Systems) or MIP-1α (Abcam, Cambridge, MA), a polyclonalrabbit anti-NP serum (Zhang and Air, 1994) and an anti-CD 68monoclonal antibody (Dakocytomation, Carpinteria, CA). Afterwashing, the sections were probed with a donkey anti-goatsecondary antibody conjugated to Alexa Fluor 350, a donkey anti-rabbit secondary antibody conjugated to Alexa Fluor 546 and adonkey anti-mouse secondary antibody conjugated to Alexa Fluor647, and the cell nuclei were stained with SYTOX green (all fromMolecular Probes). Transmitted light and fluorescent microscopyimages were obtained using a Leica SP2 MP Confocal microscope.
Statistical analysis
Where applicable, the data have been expressed as the means±standard error of the mean (SEM). Statistical significance wasdetermined by one-way ANOVA with Student-Newman-Keuls posthoc correction for multiple comparisons. Significance was consideredas Pb0.05.
Acknowledgments
We thank Dr. Gillian Air for providing us the influenza virus strainsand the polyclonal rabbit anti-NP serum. The research described inthis work was partially supported by a Clinical Innovator Award ofFlight Attendant Medical Research Institute (to W.W.) and by theNational Institute of Allergy and Infectious Diseases, project 1U19AI62629 (to J.P.M. and K.M.C.).
We acknowledge the kind assistance of the Departments ofPathology of the Veterans Administration Hospital, the University ofOklahoma Medical Center, Baptist-Integris Hospital, St. Anthony'sHospital, and Mercy Health Center, all of Oklahoma City, OK. We alsoacknowledge the assistance and expertise of Julie Maier of theOklahoma Medical Research Foundation Imaging Analysis corefacility.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.virol.2009.10.016.
References
Adachi, M., Matsukura, S., Tokunaga, H., Kokubu, F., 1997. Expression of cytokines onhuman bronchial epithelial cells induced by influenza virus A. Int. Arch. AllergyImmunol. 113 (1-3), 307–311.
Baggiolini, M., 1998. Chemokines and leukocyte traffic. Nature 392 (6676), 565–568.Booth, J.L., Coggeshall, K.M., Gordon, B.E., Metcalf, J.P., 2004. Adenovirus type 7 induces
interleukin-8 in a lung slice model and requires activation of Erk. J. Virol. 78 (8),4156–4164.
Bussfeld, D., Kaufmann, A., Meyer, R.G., Gemsa, D., Sprenger, H., 1998. Differentialmononuclear leukocyte attracting chemokine production after stimulation withactive and inactivated influenza A virus. Cell. Immunol. 186 (1), 1–7.
Chakrabarty, K., Wu, W., Booth, J.L., Duggan, E.S., Nagle, N.N., Coggeshall, K.M., Metcalf,J.P., 2007. Human lung innate immune response to Bacillus anthracis sporeinfection. Infect. Immun. 75 (8), 3729–3738 electronic publication 2007 May 3721.
Choi, A.M., Jacoby, D.B., 1992. Influenza virus A infection induces interleukin-8 geneexpression in human airway epithelial cells. FEBS Lett. 309 (3), 327–329.
De Groot, C.J., Ruuls, S.R., Theeuwes, J.W., Dijkstra, C.D., Van der Valk, P., 1997.Immunocytochemical characterization of the expression of inducible and consti-tutive isoforms of nitric oxide synthase in demyelinating multiple sclerosis lesions.J. Neuropathol. Exp. Neurol. 56 (1), 10–20.
Dufour, J.H., Dziejman, M., Liu, M.T., Leung, J.H., Lane, T.E., Luster, A.D., 2002. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10in effector T cell generation and trafficking. J. Immunol. 168 (7), 3195–3204.
Fritz, R.S., Hayden, F.G., Calfee, D.P., Cass, L.M., Peng, A.W., Alvord, W.G., Strober, W.,Straus, S.E., 1999. Nasal cytokine and chemokine responses in experimentalinfluenza A virus infection: results of a placebo-controlled trial of intravenouszanamivir treatment. J. Infect. Dis. 180 (3), 586–593.
Garn, H., Schmidt, A., Grau, V., Stumpf, S., Kaufmann, A., Becker, M., Gemsa, D., Siese, A.,2002. IL-24 is expressed by rat and human macrophages. Immunobiology 205 (3),321–334.
Gaschler, G.J., Zavitz, C.C., Bauer, C.M., Skrtic, M., Lindahl, M., Robbins, C.S., Chen, B.,Stampfli, M.R., 2008. Cigarette smoke exposure attenuates cytokine production bymouse alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 38 (2), 218–226electronic publication 2007 Sep 2013.
Gulati, U., Wu, W., Gulati, S., Kumari, K., Waner, J.L., Air, G.M., 2005. Mismatchedhemagglutinin and neuraminidase specificities in recent human H3N2 influenzaviruses. Virology 339 (1), 12–20.
Hackett, T.L., Holloway, R., Holgate, S.T., Warner, J.A., 2008. Dynamics of pro-inflammatory and anti-inflammatory cytokine release during acute inflammationin chronic obstructive pulmonary disease: an ex vivo study. Respir. Res. 9, 47.
Haesebrouck, F., Biront, P., Pensaert, M.B., Leunen, J., 1985. Epizootics of respiratorytract disease in swine in Belgium due to H3N2 influenza virus and experimentalreproduction of disease. Am. J. Vet. Res. 46 (9), 1926–1928.
Hers, J.F., 1966. Disturbances of the ciliated epithelium due to influenza virus. Am. Rev.Respir. Dis. 93 ((3), Suppl.), 162–177.
Hers, J.F., Mulder, J., 1961. Broad aspects of the pathology and pathogenesis of humaninfluenza. Am. Rev. Respir. Dis. 83 (2)Pt(2), 84–97.
Hers, J.F., Masurel, N., Mulder, J., 1958. Bacteriology and histopathology of therespiratory tract and lungs in fatal Asian influenza. Lancet 2 (7057), 1141–1143.
Izurieta, H.S., Thompson, W.W., Kramarz, P., Shay, D.K., Davis, R.L., DeStefano, F., Black,S., Shinefield, H., Fukuda, K., 2000. Influenza and the rates of hospitalization forrespiratory disease among infants and young children.[see comment]. N. Eng. J.Med. 342 (4), 232–239.
Julkunen, I., Melen, K., Nyqvist, M., Pirhonen, J., Sareneva, T., Matikainen, S., 2000.Inflammatory responses in influenza A virus infection. Vaccine 19 (Suppl. 1),S32–37.
Kaiser, L., Fritz, R.S., Straus, S.E., Gubareva, L., Hayden, F.G., 2001. Symptompathogenesis during acute influenza: interleukin-6 and other cytokine responses.J. Med. Virol. 64 (3), 262–268.
Kash, J.C., Tumpey, T.M., Proll, S.C., Carter, V., Perwitasari, O., Thomas, M.J., Basler, C.F.,Palese, P., Taubenberger, J.K., Garcia-Sastre, A., Swayne, D.E., Katze, M.G., 2006.Genomic analysis of increased host immune and cell death responses induced by1918 influenza virus. Nature 443 (7111), 578–581 electronic publication 2006 Sep2027.
Kawaguchi, M., Kokubu, F., Kuga, H., Tomita, T., Matsukura, S., Kadokura, M., Adachi, M.,2000. Expression of eotaxin by normal airway epithelial cells after influenza virus Ainfection. Int. Arch. Allergy Immunol. 122 (Suppl. 1), 44–49.
Kobasa, D., Jones, S.M., Shinya, K., Kash, J.C., Copps, J., Ebihara, H., Hatta, Y., Kim, J.H.,Halfmann, P., Hatta, M., Feldmann, F., Alimonti, J.B., Fernando, L., Li, Y., Katze, M.G.,Feldmann, H., Kawaoka, Y., 2007. Aberrant innate immune response in lethalinfection of macaques with the 1918 influenza virus. Nature 445 (7125), 319–323.
Kujime, K., Hashimoto, S., Gon, Y., Shimizu, K., Horie, T., 2000. p38 mitogen-activatedprotein kinase and c-jun-NH2-terminal kinase regulate RANTES production byinfluenza virus-infected human bronchial epithelial cells. J. Immunol. 164 (6),3222–3228.
Lee, D.C., Cheung, C.Y., Law, A.H., Mok, C.K., Peiris, M., Lau, A.S., 2005. p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factoralpha expression in response to avian influenza virus H5N1. J. Virol. 79 (16),10147–10154.
Liu, C., Air, G.M., 1993. Selection and characterization of a neuraminidase-minus mutantof influenza virus and its rescue by cloned neuraminidase genes. Virology 194 (1),403–407.
Loetscher, M., Gerber, B., Loetscher, P., Jones, S.A., Piali, L., Clark-Lewis, I., Baggiolini, M.,Moser, B., 1996. Chemokine receptor specific for IP10 and mig: structure, function,and expression in activated T-lymphocytes. J. Exp. Med. 184 (3), 963–969.

188 W. Wu et al. / Virology 396 (2010) 178–188
Ludwig, S., Ehrhardt, C., Neumeier, E.R., Kracht, M., Rapp, U.R., Pleschka, S., 2001.Influenza virus-induced AP-1-dependent gene expression requires activation of theJNK signaling pathway. J. Biol. Chem. 276 (24), 10990–10998.
Luster, A.D., Unkeless, J.C., Ravetch, J.V., 1985. Gamma-interferon transcriptionallyregulates an early-response gene containing homology to platelet proteins. Nature315 (6021), 672–676.
Marjuki, H., Yen, H.L., Franks, J., Webster, R.G., Pleschka, S., Hoffmann, E., 2007. Higherpolymerase activity of a human influenza virus enhances activation of thehemagglutinin-induced Raf/MEK/ERK signal cascade. Virol. J. 4, 134.
Matsukura, S., Kokubu, F., Noda, H., Tokunaga, H., Adachi, M., 1996. Expression of IL-6,IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced byinfluenza virus A. J. Allergy Clin. Immunol. 98 (6 Pt 1), 1080–1087.
MMWR Journal Article, 2006. High levels of adamantane resistance among influenza A(H3N2) viruses and interim guidelines for use of antiviral agents–United States,2005-06 influenza season. MMWR, Morb. Mortal. Wkly. Rep. 55 (2), 44–46.
Nobusawa, E., Ishihara, H., Morishita, T., Sato, K., Nakajima, K., 2000. Change in receptor-binding specificity of recent human influenza A viruses (H3N2): a single amino acidchange in hemagglutinin altered its recognition of sialyloligosaccharides. Virology278 (2), 587–596.
Noorman, F., Braat, E.A., Barrett-Bergshoeff, M., Barbe, E., van Leeuwen, A., Lindeman, J.,Rijken, D.C., 1997. Monoclonal antibodies against the human mannose receptor asa specific marker in flow cytometry and immunohistochemistry for macrophages.J. Leukoc. Biol. 61 (1), 63–72.
Pirhonen, J., Sareneva, T., Julkunen, I., Matikainen, S., 2001. Virus infection inducesproteolytic processing of IL-18 in human macrophages via caspase-1 and caspase-3activation. Eur. J. Immunol. 31 (3), 726–733.
Placke, M.E., Fisher, G.L., 1987. Adult peripheral lung organ culture–a model forrespiratory tract toxicology. Toxicol. Appl. Pharmacol. 90 (2), 284–298.
Rodriguez-Viciana, P., Tetsu, O., Tidyman, W.E., Estep, A.L., Conger, B.A., Cruz, M.S.,McCormick, F., Rauen, K.A., 2006. Germline mutations in genes within the MAPKpathway cause cardio-facio-cutaneous syndrome. Science 311 (5765), 1287–1290electronic publication 2006 Jan 1226.
Ronni, T., Matikainen, S., Sareneva, T., Melen, K., Pirhonen, J., Keskinen, P., Julkunen, I.,
1997. Regulation of IFN-alpha/beta, MxA, 2′,5′-oligoadenylate synthetase, and HLAgene expression in influenza A-infected human lung epithelial cells. J. Immunol.158 (5), 2363–2374.
Smith, H., Sweet, C., 1988. Lessons for human influenza from pathogenicity studies withferrets. Rev. Infect. Dis. 10 (1), 56–75.
Sprenger, H., Meyer, R.G., Kaufmann, A., Bussfeld, D., Rischkowsky, E., Gemsa, D., 1996.Selective induction of monocyte and not neutrophil-attracting chemokines afterinfluenza A virus infection. J. Exp. Med. 184 (3), 1191–1196.
Svitek, N., Rudd, P.A., Obojes, K., Pillet, S., von Messling, V., 2008. Severe seasonalinfluenza in ferrets correlates with reduced interferon and increased IL-6 induction.Virology 376 (1), 53–59.
Thompson, W.W., Shay, D.K., Weintraub, E., Brammer, L., Bridges, C.B., Cox, N.J., Fukuda,K., 2004. Influenza-associated hospitalizations in the United States. JAMA 292 (11),1333–1340.
Toms, G.L., Davies, J.A., Woodward, C.G., Sweet, C., Smith, H., 1977. The relation ofpyrexia and nasal inflammatory response to virus levels in nasal washings of ferretsinfected with influenza viruses of differing virulence. Br. J. Exp. Pathol. 58 (4),444–458.
Van Reeth, K., 2000. Cytokines in the pathogenesis of influenza. Vet. Microbiol. 74 (1-2),109–116.
Veckman, V., Osterlund, P., Fagerlund, R., Melen, K., Matikainen, S., Julkunen, I., 2006.TNF-alpha and IFN-alpha enhance influenza-A-virus-induced chemokine geneexpression in human A549 lung epithelial cells. Virology 345 (1), 96–104 electronicpublication 2005 Oct 2025.
Wang, J., Oberley-Deegan, R., Wang, S., Nikrad, M., Funk, C.J., Hartshorn, K.L., Mason, R.J.,2009. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J. Immunol. 182 (3), 1296–1304.
Williams, J.A., Su, H.S., Bernards, A., Field, J., Sehgal, A., 2001. A circadian output inDrosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science 293 (5538),2251–2256.
Zhang, H., Air, G.M., 1994. Expression of functional influenza virus A polymeraseproteins and template from cloned cDNAS in recombinant vaccinia virus infectedcells. Biochem. Biophys. Res. Commun. 200 (1), 95–101.