In varieties of chemical reactions, oxidation reactions...
57
Chapter 1 INTRODUCTION
Transcript of In varieties of chemical reactions, oxidation reactions...

Chapter 1
INTRODUCTION

1
1.1 Reaction Kinetics and Mechanism
Chemical Kinetics is a part of science of dynamics. It plays a key
role in elucidation of reaction mechanism. It deals with the rate of
chemical reactions, at various conditions such as concentration,
temperature, influencing the rate of reaction, and the explanation of all
the rates in terms of the reaction mechanism. The course of the products
of the reaction is controlled by the relative rates of several competing
reactions, which in turn depend on several factors. Studies on the
mechanism of a reaction are conveniently made by following the
quantitative variation of the rate under the influence of varying
conditions of concentrations and temperature.
Chemical kinetics, with this view point, can be contrasted with
thermodynamics which provides a static view point. Thermodynamics
deals only in the initial and a final state of a system, but time is not one
of the thermodynamic variables. Kinetics aims fundamentally at the
details of the process in which a system converts from one state to
another and the time required for the transformation. Hence, chemical
kinetics provides information about the rate of reaction on possible
pathways, by which the reactants are transformed into products. Thus,
the fundamental of objective of the study of the kinetics of chemical
reaction is to unfold the mysteries of chemical processes.
In chemical equilibria, only the initial and final stages of the
reactions are considered, while thermodynamics describes the energy
relations between the reactants and products.1 In chemical kinetics,2 the

2
rate and mechanism of a chemical reaction is to determine the overall
stoichiometry of the reaction and to identify any side‐reaction. This
study involves the determination of the effect of changes in the
concentrations of reactant and product species. One reason for studying
the rates of the reaction is to predict how quickly a reaction mixture
approaches equilibrium. Another is that the study of reaction rates leads
to an understanding of the mechanism of a reaction and analysis into a
sequence of elementary steps.
Many reactions occur in a series of steps, each of which involves
only one or two molecules. An elementary reaction may be denoted by
its chemical equation without displaying the physical state of the
species. The slowest elementary reaction, designated as the rate
determining step, controls the rate of the overall reaction. The rate
determining step is not just the slowest step, it must be slow and be a
crucial gateway for the formation of products.
The number of molecules coming together to react in an
elementary reaction is termed the molecularity of that reaction. In a
unimolecular reaction, a single molecule shakes itself apart or its atoms
into a new arrangement. In a bimolecular reaction, two molecules
collide, and exchange energy, or undergo some other kind of changes.
The number of molecules taking part in the rate‐determining step is
termed the order of the overall reaction and it is theoretically, equal to
the sum of the powers of concentration terms involved in the overall

3
rate law. Accordingly, the reactions are designated as first, second, third
or higher‐order reactions.
Many gas‐phase reactions and liquid‐phase polymerization
reactions are chain reactions, reactions in which an intermediate
produced in one step generates another reactive intermediate in the
subsequent step(s). The intermediates responsible for the propagation
of a chain reaction (propagation steps) are called chain carriers. The
chain carriers may be radicals, ions or even neutrons. The first chain
carrier is formed in the initiation step of the chain reaction. The chain
carrier might attack a product molecule formed earlier in the reaction.
Because this attack decreases the net rate of formation of product, it is
called a retardation step. Elementary reactions, in which the chain
carriers combine and end the chain are called termination steps. A chain
reaction often leads to a complicated rate‐law. The complexity of the
rate law suggests that a complicated mechanism is involved.
1.1.1 Techniques to study reaction rates
A wide variety of experimental techniques involving both physical
and chemical methods are available to investigate many types of
chemical reactions. In all this techniques, decreasing concentration of
products or corresponding changes in any physical property is measured
at various time intervals during reaction in progress.
For reactions in solution, the mechanism is formulated by the
determination of different kinetic parameters, the most important
among them being the order of reaction with respect to the different

4
reactants, effect of concentration of the catalyst (if any), ionic strength3,
solvent4, dielectric constant5 of the medium and temperature2.
Determination of the stoichiometry of the reaction, detection and
estimation of products and effect of substituents on the reaction rate
are also important factors, which throw considerable light on the
mechanism of the reaction and confirm the rate‐determining step.
Further, the isolation of intermediates and identification of their
structure and use of isotopic methods6 have also proved to be of great
significance in elucidating reaction mechanism.
The selection of a typical method depends on the nature of the
species involved and how rapidly their concentration change. For
reactions that are relatively slow, conductometric, potentiometric,
optical methods, polarimetry and spectrophotometry are used. For
reactions in which one or more of the products are gases, the reaction
rate determination involves monitoring pressure.
Reactions in solutions involving ionic species may be studied by
monitoring their conductivity. The change in EMF of an electrochemical
reaction as given by the Nernst equation, can be followed
potentiometrically. Spectrophotometry measurements of the intensity
of absorption in a particular spectral region are widely used to monitor
concentration. This technique is particularly useful when one substance
in the reaction mixture has a characteristic absorption in a conveniently
accessible region of the spectrum. Reactions that involve a change in the

5
concentrations of H+ ions may be studied by monitoring the pH of the
solution with a glass electrode.
Other methods of the monitoring the composition of the reaction
mixture include the detection of fluorescence and phosphorescence,
mass spectrometry, gas chromatography and magnetic resonance (both
NMR and ESR). Polarimetric method, the observation of the optical
activity of a reaction mixture, is occasionally employed. In the quenching
method, the reaction is stopped after being allowed to proceed for a
certain time and the composition is analyzed. The entire reaction
mixture may be quenched either by sudden cooling or by adding it to a
large volume of the solvent. This method is applicable to reactions that
are slow enough so that there is little reaction during the time it takes to
quench the reaction mixture. Fast reactions which have half lives of few
minutes are studied by special techniques such as stopped flow
methods, relaxation methods, shock tubes, flash photolysis etc.
1.1.2 Theories of reaction rates
There are two well known theories2 of reaction rates: (i) collision
theory and (ii) activated complex theory. Collision theory suggests that
the reactant molecules collide before they react. When molecules
possessing energy equal to or greater than the energy of activation for
this reaction collide and give activated complex. The activated complex
decomposes to products. The expression for rate constant is written as
RTaEeZZfk /−== (1)

6
where Z is the total number of molecules colliding per cc per sec and f is
the fraction of molecules which are in activated state. This theory gives a
simple and clear picture of the mechanism and predicts the values of the
rate constant satisfactorily for reactions, which involve relatively simple
molecules if the activation energy is known.
Improvement to the collision theory is made by considering two
factors. (i) All collisions do not yield the products, only collisions, known
as effective collisions, possessing energy sufficient enough to break the
bonds in the reacting molecules lead to the formation of products and
(ii) the colliding molecules must have proper orientation for the reaction
to occur. If the molecules approach directly there will be effective
collision. If the molecule collide in a grazing manner or in poor
orientation, the collision cannot be effective.
These shortcomings are overcome in the Absolute Reaction Rate
Theory, simply designated as ARRT. This theory attempts to treat the
reaction rates from thermodynamic considerations. It is assumed that an
equilibrium is established between the reactants and the activated
complex. The activated complex then disproportionates at a certain rate
to give products. It is this rate that determines the overall rate of the
reaction. The difference between the energy of the reactant and the
activated complex is the energy of activation Ea for the forward reaction.
The rate of the reaction for the process
A+ B X‡ may be written as
=/= KhTk k
(2)

7
where k is Boltzmann constant, h is Planck’s constant and K≠ is the
equilibrium constant, which is expressed in terms of ∆G≠, the increase in
Gibb’s free energy in the passage from the initial state to the activated
state. The result is
RTGehTk /k =/Δ−= (3)
If the free energy of activation is expressed in terms of entropy and heat
of activation is ∆H≠ ‐T∆S≠, the equation becomes
RTHRS eehTk
=/=/ Δ−Δ=k
(4)
The entropy and enthalpy of activation can be calculated from the
experimental values of rate constant and activation energy. Based on the
Arrhenius theory Ea can be evaluated by determining the rate constant of
the reaction at different temperatures and plotting a graph of log k
versus 1/T. In most of the simple cases, the plot will be straight line with
a negative slope. Then
(5) 1)987.1303.2( −××−= molcalslopeEa
The values of ∆H≠, ∆S≠ and ∆G≠ can be calculated graphically from
the slope and intercept of log (k/T) versus 1/T plots (Eyring plots). The
sign of the ∆S≠ value thus obtained tells an useful information, that is, a
negative sign means that the activated complex is more ordered than
the reactants and a positive sign means that the activated complex is less
ordered than the reactant.
Solvent effects provide important information regarding (i) the
nature of the reacting species in the rate determining step and (ii)
structure of the activated complex. For ionic reactions, polar solvents

8
seem to be the best mediums. Bronsted7 has given a relation between
the reaction rate constant k and the ionic strength (I) in a reaction
involving ions of charges ZA and ZB as,
21
2loglog IZZkk BAo α+= (6)
Here α , a constant for aqueous solution is 0.5 at 298 K and ko is the rate
constant at zero ionic strength. According to this equation, a plot of log k
versus I1/2 is a straight line with a slope, equal to (2αZA ZB). The validity
of this equation has been experimentally tested.
The value of the slope will be zero, positive or negative depending
on the nature of charge on the reacting species. If one of the reactants is
neutral, the slope will be zero, showing that the rate constant is
independent of ionic strength of the medium. However, more detailed
treatment of the effects of ionic strength on reactions between ions and
neutral molecules indicates that there is a small ionic strength effect. If
the reaction involves ions of like charges in the rate determining step,
the rate constant will increase with the increase in I, but will decrease if
the ions are of unlike charges. The extent of variation depends on the
magnitude of ZAZB.
The rate of a reaction in solution is varied with changes in
pressure, that is, the proportion of products at equilibrium is decreased
or increased by pressure according as the volume change is positive or
negative. The van’t Hoff equation that relates pressure and the rate is
given as
RTV
pk
T
=/Δ−=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂
∂ ln (7)

9
This equation means that the rate constant of a reaction increases
with increasing pressure if ∆V‡, volume of activation is negative, i.e., the
activated state has a smaller volume than the initial state. Conversely,
pressure has an adverse effect on the rates if there is a volume decrease
when the activated complex is formed. The integration of this equation
results in
PRTVkk o
=/Δ−= lnln (8)
which helps in the determination of volume of activation from the slope
of the zero intercept line obtained by plotting log (k/ko) against P. Based
on the values of ∆V‡, the reactions are classified into three broad classes
viz., slow reactions (reaction between ions of same sign), normal
reactions (replacement of negative ions) and fast reactions (reactions
between ions of opposite sign).
Enzyme kinetics, the study of the effect of enzymes on the rates
of reactions, is also important. This kind of analysis leads to the
elucidation of the mechanisms of enzyme‐catalysed reactions. The
steady‐state approximation and graphical analyses of data in the form of
Michaelis‐Menten kinetics can reveal detailed information about these
reactions and how they are affected by inhibitors and coenzymes.
Enzyme‐catalysed reactions may also show a more complex
temperature‐dependence because raising the temperature may provoke
conformational changes and even denaturation and degradation that
lower the effectiveness of the enzymes. The net rate of formation of the
intermediate complex is the difference between the rates of its

10
formation and its decay. An intermediate is any species that does not
appear in the overall reaction, but which has been invoked in the
mechanism.
There are also many specialized theories as Lindemann theory,
Kassel‐Rice‐Ramsberger theory, Hinshelwood theory and Slater theory to
deal with unimolecular reactions and Marcus theory to deal with
electron transfer reactions.
1.1.3 Structure reactivity relationships
A number of quantitative relationships have been proposed in
connection with the effects of substituents on the rate constant of
reactions. One of the best known and the most useful of these is an
equation proposed by Hammett8 which relates equilibrium and rate
constants for the reactions of meta and para substituted benzene
derivatives. The equation applies to series of aromatic compounds
having the same reaction centre. An example of this is a group of
substituted benzoic esters. According to the Hammett relationship, a
rate or equilibrium constant for reaction of any of these compound is
related to the value for the unsubstituted compound in terms of two
parameters ρ and σ. In the case of rate constant, the relationship is
ρσ+= okk loglog (9)
where ko is the rate constant for the parent compound. For equilibrium
constants
ρσ+= oKK loglog (10)

11
Of these two constants, σ depends only on the substituent, while ρ is a
reaction constant, which varies with the reaction and external conditions
such as solvent. Hammett’s equation applies quite accurately to large
number of rate and equilibrium constants, and is therefore of significant
value for predicting such constants from a small number of values of ρ
and σ. Substituents with negative σ values attract electrons more weakly
than hydrogen. Reactions with positive ρ values are accelerated by
electron withdrawal from the benzene ring, whereas those with negative
ρ values are retarded by electron withdrawal.
Excellent monographs are published dealing with the importance
of Hammett equation and its modifications. In recent years, the trend of
correlation analysis is directed towards the separation of inductive,
resonance and steric effects. It was observed earlier that in the study of
correlation analysis, Hammett’s σ substituent constants are not
sufficient to explain the behavior of certain substituents at para
positions where strong resonance interaction between the substituent
and developing charge is possible. This results in the postulation of σ+
value by Brown and Okomoto9 for the groups capable of electron
donation by resonance in electrophilic aromatic substitution reactions.
Similarly, to account for the resonance interaction between electron
withdrawing groups like –NO2, ‐CN, ‐COOH, ‐COOR in the reactions
involving production of negative charge, exalted σ− values were
required.9 The discovery of these equations resulted in the first
refinement of Hammett equation through duality of substituent

12
constants. The values of σ− have been applied extensively to the
correlation of data for aniline and phenol derivatives, carbanions and
radicals and these values have also been applied to nucleophilic aromatic
substitutions. Brown’s equation involving σ+ has been applied
extensively to solvolysis, aldehyde reactions and most notably to
electrophilic aromatic substitution.
The use of σ+ and σ− greatly extends the range of applicability of
Hammett equation. There are, however, certain situations in which the
use of σ is unsatisfactory, but the use of σ+ or σ− appears to be
appropriate for the effect of cross conjugation. To overcome this
difficulty, a sliding scale of σ values was proposed by Yukawa and
Tsunoy.10 However, the approach of the Japanese workers has been
subjected to much criticism and particularly has been found to be
unsuitable for two step process.
Taft and Lewis11 first suggested that inductive and resonance
effects could be quantitatively separated, through the eqs. (11) and (12).
RIm σασσ += (11)
RIp ασσσ += (12)
The inductive effect, given by σI, is assumed to operate equally from the
meta and para positions. The resonance effect given by σR, contributing
to σM indirectly is the relay co‐efficient (α< 1). The importance of the
separation of parameters into σI and σR type contributions is that it
suggests the possibility of a dual substituent parameter equation for a
reaction series through the equation of the form

13
RRIIokk σρσρ +=)/(log (13)
Taft12 has made efforts to evaluate quantitatively, the
inductive and resonance contributions to Hammett values assuming that
(a) inductive and resonance effects are additive (b) inductive effects are
equal in meta and para positions and (c) the resonance effect in the
meta position is smaller than in the para position by a constant fraction.
He put forward an equation,
so Ekk δσρ += **)/(log (14)
This is of the same form as Hammett equation, but another
term has been added. The polar parameter σ* is a measure of the polar
effect of a substituent while ρ* measures the reaction sensitivity to the
polar effect. Similarly Es is a measure of the polar effect introduced by
the presence of a substituent while δ measures the sensitivity of this
reaction to the steric effect. Taft made use of the hydrolysis of ortho
substituted benzoate esters as a model reaction, catalyzed both by acids
and bases. To evaluate steric constants, the polar effects in the acid
catalyzed hydrolysis reaction are assumed to be unimportant. Equation
(26) is then reduced to:
Eskk o δ=)/log( (15)
By setting δ =1 for this reaction, we obtain the values of Es which can be
tabulated.
If log k gives a linear plot against σ*, it implies that steric effects
do not cause rate differences. If the first plot is not linear, one may try
plotting log k versus Es. If this is linear, equation (15) applies and we

14
conclude that electric effects are not important. If neither of these gives
a linear plot, one can try (log k/ko – Es) versus σ*. Linearity in this plot
suggests that both steric and electronic effects play a key role in
determining the rate and that δ = 1. If even this is not linear, it is possible
to try with equation (14) to get a good correlation of the data. If a
correlation is expected, deviation of points from the correlation may
indicate a change in the mechanism.
To deal with the influence of –R and +R substituents respectively
on reactions that are more or less electron‐demanding than the
ionization of benzoic acid, Yukawa and Tsuno13 and Yasioka14 formulated
equations (16) and (17), known as Yukawa‐Tsuno equations.
)(loglog ++Δ++= Ro rkk σσρ (16)
)(loglog −−Δ++= Ro rkk σσρ (17)
where = σ+− σ and = σ−− σ, r± gives a measure of the extend
to which cross conjugation of substituents with reaction centers
stabilizes the transition sate or product relative to the initial state. The r+
in eq. (16) can have values varying from 0 to unity and values greater
than one are also possible for r− in eq. (17).
+Δ Rσ −Δ Rσ
1.1.4 Iso‐kinetic relationships
Variation in rate within a reaction series may be caused by
changes in either enthalphy or entropy of activation or both. Four
categories can be recognized:

15
i) Changes in rate are caused by changes in ΔH‡ when ΔS‡ is
substantially constant. Many reaction series that follow the
Hammett ρσ relationship fall within this category.
ii) Changes in rate are caused chiefly by changes in ΔS‡ when ΔH‡ is
substantially constant.
iii) Changes in rate are caused by random changes in both ΔH‡ and ΔS‡
iv) Changes in rate are caused by changes in both ΔH‡ and ΔS‡, but
these quantities vary in a parallel fashion.
The Hammett equation is applicable to a reaction series in which either
ΔS‡ is constant or in which the variation in ΔS‡ is linearly related15 to
changes in ΔH‡. Leffler16 modified the Hammett equation to get an iso‐
kinetic equation correlating ΔH‡ and ΔS‡ as
(18) =/=/ Δ+Δ=Δ SHH o β
which holds good for a particular temperature. The validity of the
relation can be tested graphically by plotting ΔH‡ versus ΔS‡. A linear
relationship between entropies and enthalpies of activation is taken as
evidence to the point that all the reactions in a series proceed by the
same mechanism.17 The slope of such a plot has units of absolute
temperature and it is called the iso‐kinetic temperature (β) at which all
reactions in the series proceed at the same rate. The phenomenon is
sometimes called the compensation effect because it implies that
enthalphy variations throughout the series of reactions are exactly
compensated by entropy changes. According to Petersen, in order for
the observed iso‐kinetic relationship to be valid,18 the range of observed

16
ΔH‡ (ΔΔH‡) must be twice the maximum possible error (δ) in ΔH‡ i.e.
ΔΔH‡ > 2δ.
A graphical method for finding the iso‐kinetic temperature has
been suggested by Exner19 when the rate constants are measured at two
different temperatures. A plot of log k(T2) versus log k(T1) is called Exner
plot. The linearity of this plot indicates that the reactions in the selected
series proceed with the same mechanism. The isokinetic temperature β
can be calculated by employing the simple equation
12 loglog kbak += (19)
where k2 and k1 are the rate constants at the temperatures T2 and T1
respectively and with T2 > T1. The isokinetic temperature β can be
evaluated from the expression
bTT
bT−
−=
)/()1(
21
1β (20)
where b is the slope of the Exner plot. The existence of isokinetic
relationship and linear free energy relations is of considerable
importance and they are valuable tools for chemists for mechanistic
investigation when used as supporting evidence along with other types
of data.
1.2 Oxidation Reactions – An Overview
In varieties of chemical conversions, oxidation reactions have a
prominent position. Generally, oxidation is defined in two ways; (i) loss
of electrons and (ii) increase in oxidation number. Oxidation of an
organic molecule usually corresponds to increasing in oxygen content or

17
to decreasing its hydrogen content. Oxidation of an organic compound
may be more broadly defined as a reaction that increases its content of
any element more electronegative than carbon. Of course, when an
organic compound is oxidized, something else – the oxidizing agent –
must be reduced. Most oxidations in organic chemistry involve a gain of
oxygen and / or a loss of hydrogen (Lavoisier’s original definition of
oxidation). The concept of loss / gain of electron(s) cannot be directly
applied to organic reactions, since in most organic reactions direct
electron transfer does not take place.
Oxidation in organic chemistry can be precisely defined as the
conversion of the functional group in a molecule from a low oxidation
state category to a higher one. The conversion of functional groups
serves as a tool for the study of rate and mechanism of chemical
reactions.
Oxidation reactions are classified20 into the following five groups
depending on the type of bond changes involved:
1. Elimination of Hydrogen
2. Oxidations Involving Cleavage of C‐C Bonds
3. Reactions involving replacement of hydrogen by oxygen
4. Reactions in which oxygen is added to the substrate
5. Oxidative coupling
The important prerequisite for an oxidant to be useful are its
mildness, versatility, selectivity and operational simplicity. A variety of
oxidants have been used as mild and selective oxidizing reagents in

18
synthetic organic chemistry. A number of reports on the oxidation of
organic compounds by different oxidants are available in the literature.
Chromium(VI) containing compounds, with heterocyclic bases,
like pyridinium chlorochromate,21,22 2,2’‐bipyridinium chlorochromate,23
pyridinium bromochromate,24 pyridinium fluorochromate,25 quinolinium
fluorochromate,26 quinolinium bromochromate,27 imidazolium fluoro‐
chromate28 and benzyltrimethylammonium fluorochromate29 have been
developed to improve the selectivity of oxidation of organic compounds.
Metal ion oxidants such as ammonium metavanadate,30
KMnO4,31,32 superoxochromium(III) ion,33 hexacyanoferrate(III),34 and
Ce(IV)35 have been employed as oxidizing agents to carry out the
oxidation of various organic compounds. Peroxo compounds like
peroxodisulphate,36 peroxomonosulphate,37 periodate38, bromate39,
perborate40, and organic peroxides41 are economically and
environmentally preferred42 for oxidation of organic compounds than
the conventional metal ion oxidants. But, under ordinary conditions,
oxidation by these reagents is slow and requires various metal ions as
catalysts.43 Nowadays the N‐halo reagents are increasingly used as
oxidants for the oxidation of various organic compounds. As the present
study employs N‐bromophthalimide and N‐chlorosaccharin as oxidants,
a detailed literature review pertaining to works employing N‐halo
compounds as oxidants is furnished in the following sections.

19
1.3 N‐Halo Compounds as Oxidants
Synthetic methodology, the building block of organic synthesis,
continuously seeks for new reagents, better reaction conditions, and
more efficient and selective methods. In this regard, a large group of
compounds entitled N‐halo reagents are widely used in fine organic
synthesis. The application of N‐halo compounds in the field of organic
synthesis is very wide, such as oxidation reactions, deprotection and
protection of different functional groups, halogenation of saturated and
unsaturated compounds, acylation of alcohols, phenols, amines or thiols,
epoxidation of alkenes, aziridination and finding more and more
applications.44 The N‐halo reagents include N‐chloro, ‐bromo, and ‐iodo
derivatives of amines, amides, imides, urea, saccharins, sulphonamides,
sulphonimides, etc. The different N‐halo reagents available for synthesis
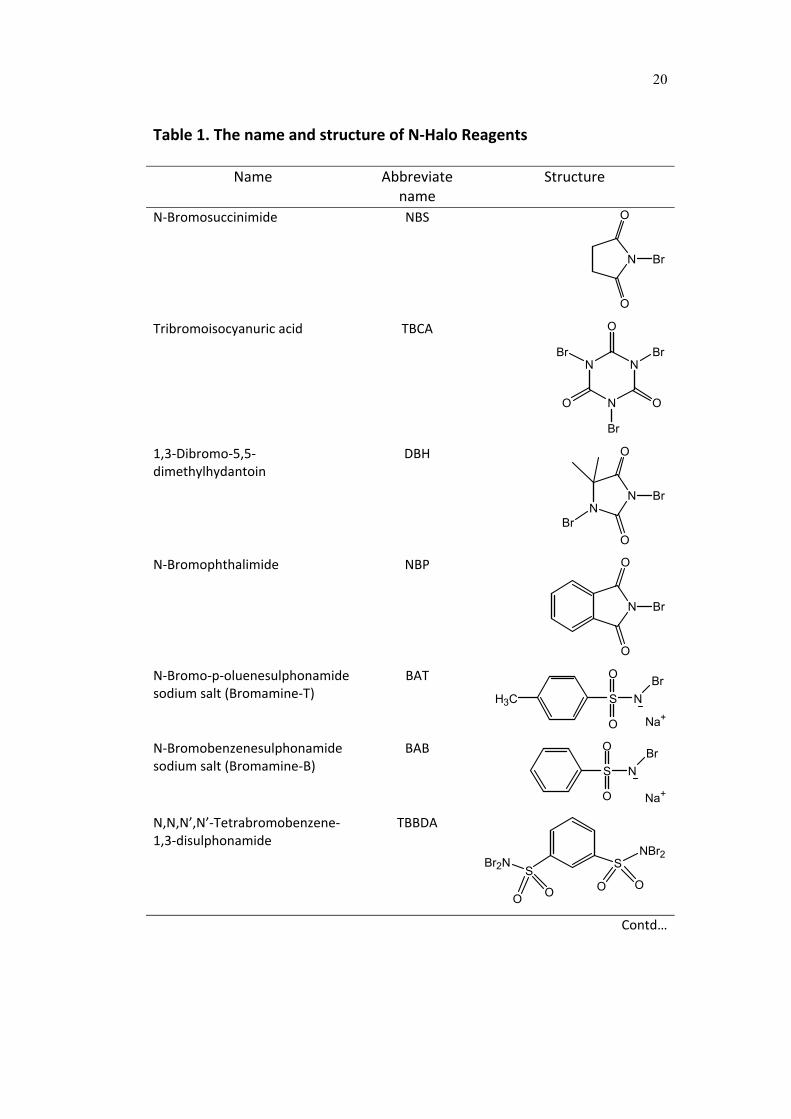
and oxidation of organic compounds are listed in Table 1.
The specific feature of N‐halo reagents is the high activity of the
N–X bond and various modes of its splitting. Depending on the
conditions, a number of highly reactive intermediates can be formed
including halogen radicals, halogen cations, halogen anions, N‐radicals,
N‐cations, N‐anions, etc. Consequently, N‐halo reagents have the
potential to promote important reactions in synthetic and natural
products chemistry. Since the N‐halo compounds exhibit appreciable
stability both in acid and alkaline mediums, it is probably for this reason
that these reagents have frequently been used as redox titrants45 in
analytical chemistry.
20
Table 1. The name and structure of N‐Halo Reagents
Name Abbreviate name
Structure
N‐Bromosuccinimide NBS
N
O
O
Br
Tribromoisocyanuric acid TBCA
N
N
N
O
O O
Br
Br
Br
1,3‐Dibromo‐5,5‐dimethylhydantoin
DBH
NN
O
O
Br
Br
N‐Bromophthalimide NBP
N
O
O
Br
N‐Bromo‐p‐oluenesulphonamide sodium salt (Bromamine‐T)
BAT
H3C S
O
O
NBr
Na+
N‐Bromobenzenesulphonamide sodium salt (Bromamine‐B)
BAB
S
O
O
NBr
Na+
N,N,N’,N’‐Tetrabromobenzene‐1,3‐disulphonamide
TBBDA
SS
NBr2Br2N
O O O O
Contd…
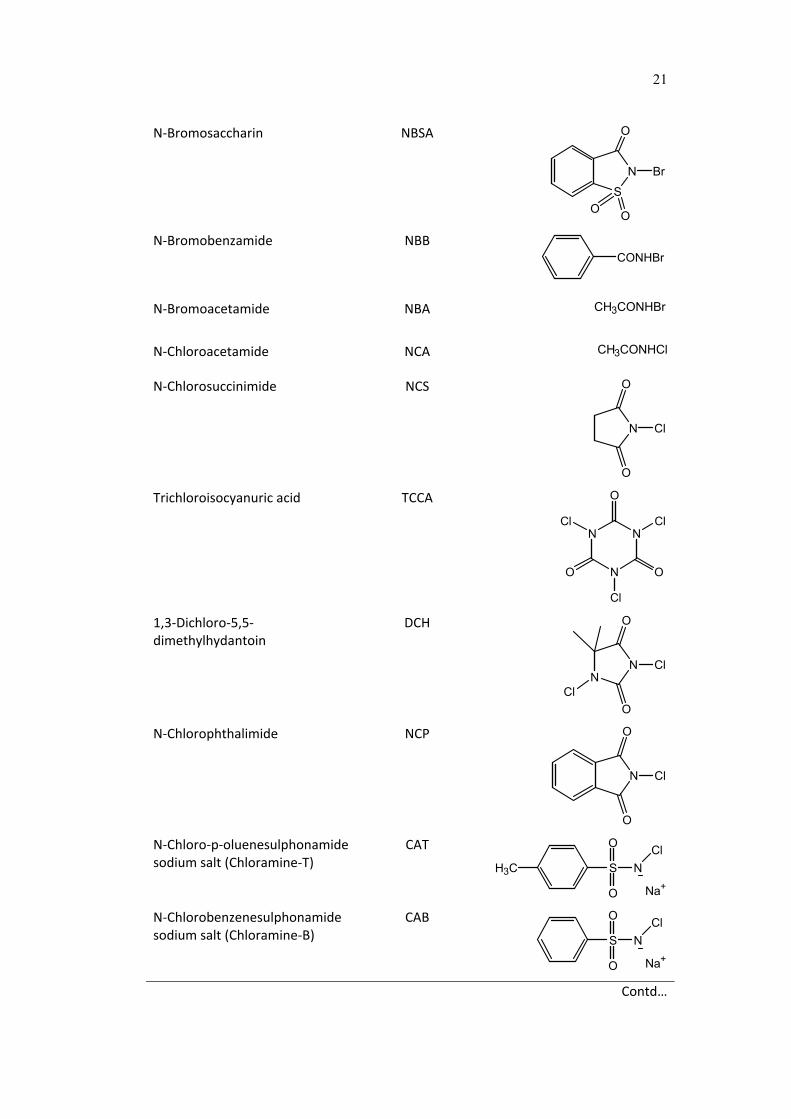
21
N‐Bromosaccharin NBSA
S
N
O
O
Br
O
N‐Bromobenzamide NBB CONHBr
N‐Bromoacetamide NBA CH3CONHBr
N‐Chloroacetamide NCA CH3CONHCl
N‐Chlorosuccinimide NCS
N
O
O
Cl
Trichloroisocyanuric acid TCCA
N
N
N
O
O O
Cl
Cl
Cl
1,3‐Dichloro‐5,5‐dimethylhydantoin
DCH
NN
O
O
Cl
Cl
N‐Chlorophthalimide NCP
N
O
O
Cl
N‐Chloro‐p‐oluenesulphonamide sodium salt (Chloramine‐T)
CAT
H3C S
O
O
NCl
Na+
N‐Chlorobenzenesulphonamide sodium salt (Chloramine‐B)
CAB
S
O
O
NCl
Na+
Contd…

22
N‐Chlorosaccharin NCSA
S
N
O
O
Cl
O
Trichloromelamine TCM
N
N
N
NHClClHN
NHCl
N‐Chloronicotinimide NCN
N
NHCl
O
N‐Fluoro‐2,4‐dinitroimidazole NFDNI
NN
NO2
O2N
F
N‐Iodosuccinimide NIS
N
O
O
I
N‐Iodophthalimide NIP
N
O
O
I
N‐Iodosaccharin NISA
S
N
O
O
I
O
Aqueous solutions of halogens have a strong oxidizing character.
Different species can be responsible for such oxidizing character
depending on the acidity of the medium. Some of the possible

23
equilibria46,47 existing between the oxidizing species in the aqueous
solution of halogens are:
X2(g) X2(aq)
X2(aq) + 2H2O(l) HOX(aq) + H3O+(aq) + X−
(aq)
X2(aq) + 2OH−(aq) XO−
(aq) + H2O(l) + X−(aq)
X2(aq) X+(aq) + X
−(aq)
X2(aq) + H2O(l) H2OX+(aq) + X−
(aq)
HOX(aq) + H2O(l) XO−(aq) + H3O+
(aq)
3XO−(aq) XO3
−(aq) + 2X−
(aq)
X2(aq) + X−(aq) X3
−(aq)
These oxidant species react readily with N‐compounds to give the
corresponding N‐halo compounds. The oxidation products depend on
the ratio [X2(aq)]/[N‐compounds], and on the acidity of the medium. The
possible reactive species48 of N‐halo compounds in acid solution are >NX,
HOX, >N+HX or H2O+X and the reactive species in alkaline solution are
>NX, HOX and OX−.
A review of literature shows that N‐halo compounds such as NCS,
NBS, NCN, NBB, NBSA, NCSA, CAT, BAT, CAB, BAB etc. are commonly
used for the oxidation of various organic compounds such as alcohols,
aldehydes, amino acids, keto acids, sulphides, etc.
N‐Halo succinimides are sources of positive halogens and these
reagents have been exploited as oxidant for a variety of substances in
both acidic and alkaline mediums.49‐56 Kinetics of oxidation of L‐arginine

24
by NBS in the presence of mercury(II) acetate in perchloric acid medium
have been studied49 with a view to elucidate the mechanism of the
reaction and to identify the active species of the oxidant in acid medium.
A suitable mechanism in consistent with the kinetic results has been
suggested (Scheme 1).
H2C
H2CCO
NH+Br
OC
H2C
H2CCO
NBr
OC
+ H+
H2C
H2CCO
NBr
OC
+R CH COOH
+NH3
Complex (C)
C
HOOC
C
R
NH +H2C
H2CCO
NH
OC
+ HBr + H+
HOOC
C
R
NH + H2O RCHO + NH3 + CO2
k1
k2
kslow
fast
Scheme1
Kinetics of oxidation of aromatic aldehydes by NCS in the presence of
HClO4 and NaCl in aqueous acetic acid medium has been reported.55 The
observed rate of oxidation is first‐order with respect to [NCS], [H+] and
[Cl−]. The order with respect to aldehyde is always zero. The product of
oxidation is the corresponding acid. Arrhenius and the activation
parameters have been calculated. Based on the kinetic results a suitable
mechanism has been proposed (Scheme 2).

25
NCS + H+ NCSH+
NCSH+ + Cl− Succinimide + Cl2
Cl2 + H2O HOCl + HCl
HOCl + X−C6H4CHO X−C6H4COCl + H2O
X-C6H4COCl + H2O X−C6H4COOH + HClfast
fast
fast
slowk1
fast
K
Scheme 2
In the kinetic study56 of the reaction between iodide ion and NCS, it has
been found that the reaction is first‐order in [NCS], [I−] and [H+] and it
follows a general‐base catalysis pathway. A mechanism involving a
transfer of Cl+ from NCS to the iodide ion has been proposed (Scheme 3).
H2C
H2C C
N
C
Cl
O
O
ICl
ClOH
H2C
H2CC
N
CO
O
I−
OH−
+
Scheme 3
N‐Halosulphonamides have been widely used in kinetic studies as
oxidants both in acid as well as in alkaline mediums. Some of the
reagents of this category, which have been effectively used in organic
transformations, are Chloramine‐T,57‐62 Bromamine‐T,63 Chloramine‐B64
and Bromamine‐B.65 The kinetic investigation of the oxidation of glycine
by chloramine‐T has been made62b in the presence of an anionic
surfactant, sodium dodecyl sulphate at 313 K. The rate of the reaction
shows first‐order dependence on CAT and fractional‐order dependence
on glycine, according to eq. (21).

26
2CH3.C6H4SO2N.NaCl + CH2.NH2.COOH → 2CH3.C6H4SO2NH2 + HCN +
CO2 + 2NaCl (21)
The kinetics of oxidation of ethanolamines by bromamine‐B in
alkaline buffer medium (pH 8.7 – 12.2) have been studied.65 Michaelis‐
Menten type of kinetics has been observed. The formation and
decomposition constants of ethanolamine‐BAB complex have been
evaluated. TBBDA has been used as effective oxidizing agent for the
conversion of urazoles and bis‐urazoles to the corresponding
triazolinediones under mild and heterogeneous conditions.66 TBBDA and
some other poly‐N‐bromo sulphonamide reagents have been used67 for
the oxidation of 1,3,5‐trisubstituted pyrazolines to their corresponding
pyrazoles in solvent‐free conditions and of primary and secondary
alcohols to corresponding carbonyl compounds in the presence of
DMSO.
DBH has also been used for the efficient oxidation of mono and
bis‐urazoles both in solution and under solvent‐free conditions.68a The
oxidation of 1,3,5‐trisubstituted pyrazolines to the corresponding
pyrazoles using DBH under both heterogeneous and solvent‐free
conditions has been carried out.68b,c Walters et. al. have studied the use
of DBH for the oxidation of hydroxylamines to gem‐halonitro compounds
in the presence of ozone.68d
TCCA has been used69 for the enantioselective epoxidation of
chalcones, enones and alkenes in aqueous acetone and for the oxidation
of urazoles and bis‐urazoles to their thiazolinediones under both

27
heterogeneous and solvent‐free conditions. An interesting application of
TCCA has been reported70 by Heiagl et. al. in the conversion of α‐amino
acids into nitriles by oxidative decarboxylation in water or methanol in
the presence of pyridine. Using TCCA, efficient oxidation of primary
alcohols to carboxylic acids or methyl esters has been carried out71 at
room temperature and in acetone/water or in dichloromethane (eq. 22).
OH OMe
O
TCCACH3OHCH2Cl2 (22)
N‐Haloacetamide72, N‐halobenzamide73,74 and N‐chloronicotinamide75
have been used as oxidizing and halogenating agents for a large number
of compounds.
Kinetics of oxidation of few α‐amino acids by NCN in aqueous
acetic acid medium in presence of HCl has been investigated75b. The
observed rate of oxidation is first‐order in both [NCN] and [HCl]. A small
increase in rate is observed with increase in [amino acid]. The
corresponding aldehydes, ammonia and CO2 have been identified as the
oxidation products. Molecular chlorine has been postulated as the
reactive oxidizing species (Scheme 4).
Scheme 4
NCN + H+ + Cl− Nicotinamide + Cl 2
Cl2 + Amino acid complex
complex products
k1
k−1
k−2
k2
k3
slow

28
The literature pertaining to N‐bromophthalimide and N‐chlorosaccharin,
which are the oxidizing agents in the present study, has been dealt
elaborately in the following sections.
1.3.1 N‐Halosaccharin as oxidant
N‐Chlorosaccharin has been introduced as an oxidimetric titrant
for use in aqueous acetic acid medium.45 It is very stable in solid state
and its solution in anhydrous acetic acid has better keeping qualities
than those of most other oxidants of similar type.
The kinetic study of oxidation of benzaldehyde and o‐chloro‐
benzaldehyde with NCSA in aqueous acetic acid and perchloric acid
mediums has been reported.76 The reaction is first‐order each with
respect to oxidant and substrate. Both the reactions are acid‐catalyzed.
The thermodynamic parameters have been evaluated. It has been
suggested that the hydrated form of the substrate is readily oxidized to
acid with H2O+Cl species of NCSA (Scheme 5).
Scheme 5
XC6H4CHO + H2OK1
XC6H4 C OH
OH
H
NCSA + H2O Saccharin + HOClK2
HOCl + H+ H2O+ClK3
XC6H4 C OH
OH
H
+ H2O+Cl XC6H4 CO
OH
H
H O
H
Cl
XC6H4 C OH
O
+ HCl + H3O+
slowk

29
The kinetic studies of NCSA oxidation of acetaldehyde and
propionaldehyde in aqueous acetic acid medium have been reported77
by Khan et. al. The reactions follow identical kinetics being first‐order in
NCSA and one to zero order with respect to substrate and [H+]. A positive
effect on the oxidation rate is observed for solvents whereas saccharin
exhibits a negative effect. A suitable mechanism consistent with the
experimental results has been proposed.
The kinetics of oxidation of keto acids by NCSA in aqueous acetic
acid medium in the presence of perchloric acid has been investigated.78
The observed rate of oxidation is first‐order each in [keto acid], [NCSA]
and [H+]. The main product is the corresponding carboxylic acid.
Hypochlorous acidium ion (H2O+Cl) has been postulated as the reactive
oxidizing species. A suitable mechanism, supported by substituent‐ and
temperature‐effect studies, has been proposed (Scheme 6).
NCSA + H3O+ H2O+Cl + SaccharinK1
C CH2CH2COOH
O
+ H3O+K2 C CH2CH2COOH
+OH
+ H2O
C CH2CH2COOH
+OH
+ H2OK3 C CHCH2COOH
OH
+ H3O+
C CHCH2COOH
OH
H2O+Cl
C CHCH2COOH
OH
H2OCl
fast
C OH
O
+ other products
k
slow
Scheme 6

30
A mechanistic approach of oxidation of some primary alcohols by
NCSA has been made79 in different solvents by Tiwari et. al. The
reactions are first‐order in NCSA and HClO4 and the order varies from
one to zero in substrate. The oxidation is catalyzed by acid and retarded
by added saccharin. A positive effect of solvents establishes a positive
ion – dipole interaction. Various thermodynamic parameters have been
computed. The reactivity increases in the order MeOH > EtOH > PrOH >
BuOH. A rate degradation of hypochlorite with deprotonation has been
proposed.
The kinetic and mechanistic considerations of the degradative
oxidation of substituted mandelic acids by NCSA in aqueous acetic acid
medium have been reported.80 The reaction rate is a direct function of
both [oxidant] and [substrate] in the lower concentration region but
tends towards zero order at higher concentrations. A retarding trend of
H+ ions, solvent composition and saccharin on the system has been
observed.
Mechanism of oxidation of some active methylene compounds,
CH3COCH2CO2Et and CH2(COOEt)2 by NCSA in aqueous acetic acid
medium has been studied.81 The reaction is first‐order with respect to
both [oxidant] and [substrate]. The reaction is acid‐catalyzed. The effects
of variation of solvent and ionic strength on the reaction have also been
examined. The reaction rate is retarded by the addition of saccharin. The
stoichiometry of the oxidation is 1:2 which shows the formation of a
transitory complex between the enolic form of the substrate and the

31
oxidant. A plausible mechanism with a suitable rate law has been
envisaged.
NCSA oxidation of 2‐alkanones has been investigated82 in aqueous
acetic acid and perchloric acid mediums. The reaction exhibits first‐order
dependence in the oxidant. The order of the reaction with respect to
substrate and perchloric acid varies from one to zero. The reaction rate
is retarded by the addition of saccharin and increasing percentage of
binary mixture of acetic acid and water, whereas the primary salt effect
shows slightly increasing trend in rate. The proposed complex‐formation‐
mechanism involves H2O+Cl as oxidizing species (Scheme 7).
Scheme 7
HOCl + H+ H2O+ClK2
NCSA + H2O HOCl + SaccharinK1
R C CH2R
OH+
R C CHR
OH
R C CHR
OH
+ H2O+Clk
slowR C CHR
O
+ H2O + HCl
R C CHR
O
+ H2Ofast
R C CH
O
R
OH
+ H+
NCSA has been shown to undergo an electrophilic Ritter‐type
reaction with alkenes in acetonitrile.83 N‐Halosaccharins have been
used84 for regioselective cleavage of epoxides into vicinal halohydrins
and dihalides in the presence of triphenyl phosphine (eq. 23).
O
PhONCSA (or) NBSA1% aq. CH3CN
PhOCH2CH(OH)CH2X (23)

32
Sanchez and Fumarola have reported85 an efficient method for benzylic
and α‐carbonylic bromination using NBSA under mild conditions (eq. 24). CH3 CH2Br
(i) NBSA, 5% Benzoyl peroxide
(ii) dark, 80 oC, 2h(24)
NBSA has been successfully used86 for chemoselective oxidation of thiols
to their corresponding disulphides in CH2Cl2 under microwave irradiation
in high yields (eq. 25). NBSA has been applied87 as an efficient reagent
for the oxidative cleavage of oximes to the corresponding aldehydes and
ketones under microwave irradiation with reasonable yields by Khazaei
et. al. They have also reported88 the above transformation with NBSA in
water and acetone as solvent at room temperature or by conventional
heating (eq. 26).
(25)R SHNBSA
CH2Cl2R S S R
NOH
R2R1
NBSA
Acetone / H2O
O
R2R1
(26)
1.3.2 N‐Bromophthalimide as oxidant
N‐Bromophthalimide acts as a moderate oxidant with a redox
potential of 1.09 V.89 The reaction of NBP with acetophenone has been
studied90 in presence of excess of mercuric acetate in aqueous acetic
acid medium. The reaction is first‐order in [NBP] and fractional order in
[acetophenone]. Variation of phthalimide, mercuric acetate and ionic
strength shows an insignificant effect on reaction rate. The

33
stoichiometric ratio of substrate:oxidant is 1:2 and the products are
benzoic acid and formaldehyde. Hammett plot of log k versus σ+ gives a
ρ+ value of −0.52 and Exner’s plot gives an isokinetic temperature of
263 K. Thermodynamic and activation parameters have been evaluated
and a mechanism consistent with the kinetic data has been proposed
(Scheme 8).
Scheme 8
fastPh C CH3
O+ NBP
K1
Adduct
Adductk
slow NH
O
O
+ Intermediate
Intermediate + NBP + H2OPh C OH
O+ NH
O
O
+ CH2O + HBr
The oxidative kinetics of some para‐substituted benzaldehydes
has been carried out with NBP in presence of excess of mercuric acetate
in aqueous acetic acid medium.91 Results of detailed kinetic effects, viz.,
solvent, temperature, concentration and salt effects, support the
Michaelis‐Menten type of mechanism. The stoichiometric ratio of
NBP:aldehyde is 1:1 and the product of the reaction is benzoic acid.
Thermodynamic and activation parameters have been determined.
Based on substituent‐effect studies and other kinetic observations, a
suitable mechanism has been proposed (Scheme 9).

34
C6H5 C H
O
+ H2O C6H5CH(OH)2
C6H5CH(OH)2 + N
O
O
Brfast
K1
HOC
O
C6H5 H
H
N
O
O
Br
Adduct
Adductk
slowPh C OH
O+ NH
O
O
+ HBr
Scheme 9
The oxidation of the dipeptide, glycylglycine, has been carried out92 with
NBP in presence of mercuric acetate. The study reveals a first‐order
dependence on [NBP] and fractional order dependence on glycylglycine.
Michaelis‐Menten type mechanism has been proposed with the support
of thermodynamic parameters (Scheme 10).
N
O
O
Br + H2N CH2 CONHCH2COOHfast
K1Adduct
Adductk
slowHN CH CONHCH2COOH + NH
O
O
+ HBr
HN CH CONHCH2COOHH2Ofast
O CH CONHCH2COOH + NH3
Scheme 10
Potentiometric and visual end‐point titrations with NBP and NBSA
have been reported93 for the determination of vitamin C in various
pharmaceutical preparations. During this titration, ascorbic acid is
oxidized to dehydroascorbic acid. Also, NBP and NBSA have been used
for the determination of some sulpha drugs and carbohydrates, on
account of their stability and reactivity.94 These two halo reagents are

35
reported to be better brominating agents in the estimation of para‐
aminobenzoic acid in pharmaceutical preparations.95,96
The kinetics of the oxidation of aspirin by NBP, BAT and NBS have
been studied97 in aqueous perchloric acid at 303 K. The oxidation
reaction follows identical kinetics with first‐order in [oxidant], fractional
order in [aspirin], and inverse fractional order in [H+]. Under identical
experimental conditions, the extent of oxidation with different oxidizing
agents is in the order, NBS > BAT > NBP. Based on the solvent isotope
and temperature effect studies, a suitable mechanism has been
proposed.
The kinetics of oxidation of glycine by NBP has been studied98 in
the presence of an anionic surfactant, sodium dodecyl sulphate in acidic
medium at 303 K. The reaction is first‐order in [NBP] and fractional order
in [glycine] and [H+]. The addition of phthalimide has no significant effect
on the reaction rate. The main oxidation product is found to be HCN. The
various activation parameters have been computed. A mechanism well
suited to the kinetic results has been proposed.
The oxidation of α‐hydroxy acids, namely mandelic acid, lactic
acid, malic acid, benzilic acid and atrolatic acid with NBP to give the
corresponding carbonyl compounds has been carried out99 in order to
ascertain whether the alcoholic OH or the carboxylic OH is involved in
the oxidative decarboxylation process. The effect of pH on reaction rate
and primary kinetic isotope effect establish that the alcoholic OH gets
oxidized. The reactive species is acyl or alkyl hypobromite at higher or

36
lower pH, respectively. The NBP and NBS oxidations of α‐hydroxy acids
are found to be well correlated.
NBP has been found100 to be an efficient and selective reagent for
the mild oxidative cleavage of oximes to yield the corresponding
carbonyl compounds in good to excellent yields (eq. 27). NBP has been
used for the facile oxidation of thiols to symmetrical disulphides in a
mixture of acetone‐water under microwave irradiation.101 Both aromatic
and aliphatic thiols are selectively oxidized in good to excellent yields
(eq. 28). Reaction of substituted benzene rings with NBP under neutral
conditions gives the corresponding bromo derivatives with a preference
for the formation of para over the ortho isomers102a (eq. 29). NBP has
also been used for the bromination of some deoxyhexoses.102b
NOH
R2R1
NBP
Acetone / H2O
O
R2R1
(27)(R = aromatic or aliphatic)
(28)R SHNBP
Acetone / waterR S S R (R = aromatic or aliphatic)
R R
(29)NBP
Et2O
Br
(R = OMe, NHAc, NEt2, OH, CONH2)
The present investigation deals with the kinetics of oxidation of
(arylthio)acetic acids and diaryl sulphides with NCSA and NBP. Therefore,
some of the previous works relating to the oxidation of organic sulphur

37
compounds with N‐halo reagents and other oxidants are mentioned and
discussed hereunder.
1.4 Oxidation of Organic Sulphur Compounds
The oxidation of sulphides to sulphoxides is of significant
importance in organic chemistry both for fundamental research and for a
wide range of applications.103 The synthesis of sulphoxides has been
reported for the first time by Marcker in 1865 and since then, a number
of methods have been developed for the conversion of sulphides into
sulphoxides.
1.4.1 Studies of oxidation of (arylthio)acetic acids
(Phenylthio)acetic acid (PTAA) and (substituted phenyl‐
thio)acetic acids were found to possess many biological activities and act
as herbicides,104 systemic fungicides,105 pesticides,106 and are used to
activate the growth of certain plants.106,107 Certain (phenylthio)acetic
acids were found to be of great use as precursors in the biosynthesis of a
penicillin.108 The degree of dissociation and the ionization of several
(substituted phenylthio)acetic acids have been measured at various
experimental conditions and solvent systems.109‐111 From the comparison
of Hammett ρ value of (phenylthio)acetic acids (0.30) with the ρ values
of β‐phenylpropionic acids (0.24) and phenoxyacetic acids (0.23) it is
clear that thio group transmits inductive effect more effectively than
methylene or oxo group111 and the order of dissociation constant109 is
phenoxyacetic acid < PTAA < β‐phenylpropionic acid.

38
Srinivasan and Pitchumani112 have reported the substituent
effects on the 1H NMR spectra of (substituted phenylthio)acetic acids.
The chemical shifts of the methylene protons (in Hz) in C6H5XCH2COOH,
(X = O, S, SO or SO2) are S: 329, SO: 339, SO2: 367 and O: 421. This is in
accordance with the electronegativity order (S < SO < SO2 < O) and also
consistent with the acidities of the acid. PTAA undergoes a one electron
reduction polarographically113 to give PhS− and acetic acid. The half wave
potential for the reduction is 2.425 V. Photoinduced decarboxylation of
PTAA to methyl phenyl sulphide was photosensitized by benzo‐
phenone,114,115 aromatic ketones,116 aromatic nitro compounds117 and
heterocyclic compounds.118
The oxidation of (phenylthio)acetic acid is interesting and only
very few reports on the kinetics of oxidation of (phenylthio)acetic acids
are available. Sodium metaperiodate,119 hydrogen peroxide,120
microsomal fractions of rat liver homogenates,121 sodium‐N‐chloro‐
benzenesulphonamide,122 iodobenzene dichloride123 and potassium
peroxodisulphate124 oxidize PTAA and yield phenylsulphinylacetic acid.
Potassium bromate oxidizes PTAA solution containing potassium
bromide and hydrochloric acid by absorbing two equivalents of bromine
rapidly forming sulphoxide.125
The kinetics of oxidation of PTAA by peroxomonophosphoric
acid in 10% aqueous acetonitrile126 is first‐order each in oxidant and the
pH dependence of the rate was rationalized in terms of different species
of peroxomonophosphoric acid. Kinetics of oxidation of PTAA with the

39
isoelectronic and isostructural peroxoanions viz., peroxodisulphate127
and peroxodiphosphate128 ions were extensively studied by Srinivasan
and Pitchumani. Both oxidants follow second order kinetics, first‐order in
each reactant. The reaction is accelerated by electron withdrawing
substituents and a good correlation exists between the rate constants
and σ+/σ− constants. A mechanism involving a bimolecular nucleophilic
displacement of the sulphur on the peroxoanion in the rate‐determining
step has been proposed.
The kinetics of chloramines‐T (CAT) oxidation of PTAA129 in
alkaline medium indicates first‐order dependence in [CAT] and [PTAA]
and a near inverse first order dependence in [OH−]. The oxidation rate
considerably decreases with increase of pH. The reaction exhibits a
positive salt effect and added p‐toluenesulphonamide increases the rate.
The oxidation has been shown to proceed via two paths, the major path
involving CAT as the main oxidizing species and the other involving
hypochlorite ion.
Rajagopal130 has studied the kinetics of Cr(VI) and picolonic acid
catalyzed Cr(VI) oxidation of PTAA in 50% aqueous acetic acid. The
mechanism of Cr(VI) oxidation envisages the formation of a cation
radical intermediate in the rate‐determining step. This cation radical
intermediate may attack the Cr=O bond of Cr(V) to form a complex
which on solvolysis yields sulphoxide. Picolinic acid catalyzed oxidation
also proceeds via the sulphur cation radical as a result of the interaction
between PTAA and active cyclic intermediate formed between chromic

40
acid and picolinic acid. The reaction is first‐order each in substrate,
oxidant and picolinic acid. The kinetics of oxidation of PTAA by
phenyliodosodiacetate (PIA) has been investigated130 in aqueous
acetonitrile. The reaction is first‐order each in PIA and PTAA. A
mechanism involving the reversible formation of an iodine (III)‐
sulphonium ion intermediate complex, followed by its decomposition
has been proposed for this oxidation. The reactivity data well correlated
with Hammett σ constants and gave the reaction constant −1.35.
Gurumurthy et al.131a have studied the kinetics of oxidation of
several (phenylthio)acetic acids by ceric ammonium nitrate in the
presence of perchloric acid spectrophotometrically in 50% (v/v) aqueous
acetic acid. The order with respect to Ce(IV) is one and with respect to
(phenylthio)acetic acid is found to be 0.8. A linear plot of 1/kobs versus
1/[substrate] with an intercept on the rate axis suggests the formation of
an equilibrium complex between the reactants prior to the rate‐
determining step. The added acrylonitrile retards the reaction rate
considerably suggesting that the oxidation process may involve a free
radical mechanism. A good correlation is found to exist between log k1.8
and Hammett σ constants.
The kinetics of oxidation of several substituted ethyl
phenylthioacetates by Bromamine‐B has been studied131b in 50% (v/v)
aqueous ethanol medium. Two mechanisms (Schemes 11a and b) have
been proposed. In the presence of Hg(II), rate = k[C1][H2O+Br] and in the
absence, rate = k[S][H2O+Br].

41
BAB + H2O HOBr + C6H5SO2NH2
HOBr + H+ H2O+Br
Hg(II) + S C1
C1 + H2O+Br SO + 2H+ + Br−
Scheme 11a [in the presence of Hg(II)]
slow
BAB + H2O HOBr + C6H5SO2NH2
HOBr + H+ H2O+Br
+ H2O+Br SO + 2H+ + Br−
Scheme 11b [in the absence of Hg(II)]
S slow
Kabilan et al.132 have studied the kinetics of oxidation of
(phenylthio)acetic acid by pyridinium fluorochromate in aqueous acetic
acid medium. A Michaelis‐Menten type of kinetics is observed. A
mechanism involving the formation of phenylsulphinylacetic acid in a
slow step has been proposed.
Thenraja et al.133 have followed the kinetics of oxidation of alkyl
aryl sulphides, diphenyl sulphide and (arylthio)acetic acids by N‐chloro‐
succinimide in 75% acetonitrile‐25% water mixture in the presence of
perchloric acid. The oxidation has been found to be first‐order both in
NCS and substrate. While the rate of oxidation of methyl phenyl sulphide
or diphenyl sulphide increases with [H+], that of PTAA decreases. NCS
and its protonated species have been proposed as the active oxidizing
species in the oxidation of sulphides and NCS itself as the active species
in the case of (arylthio)acetic acids. Structure‐Reactivity correlations in

42
the two cases yield ρ values of −3.33 and −2.73 respectively, providing
evidence for the formation of chlorosulphonium ion intermediate.
Read et al.134 have investigated the kinetics and mechanism of the
oxidation of (phenylthio)acetic acid and thiodiglycolic acid by potassium
ferrate under pseudo and non‐pseudo first‐order conditions. (Phenyl‐
thio)acetic acid is oxidized to the sulphone and Fe(II) within 300 seconds.
Above a pH value of 8.7 the kinetics is first‐order in [H+], whereas at
lower pH values the kinetics is independent of [H+]. The possible
mechanism involves a reaction between protonated ferrate and the
(phenylthio) acetic acid as the rate‐determining step.
The kinetics of oxidation of phenylsulphanylacetate ions by
Bromamine‐T have been studied at three temperatures in aqueous KOH
by Srinivasan et al.135 The reaction follows overall second‐order kinetics,
first‐order in each reactant. The Hammett correlation is excellent giving
a high ρ value of −2.42. A mechanism involving an attack of BrO− on
phenylsulphanylacetate ion in the rate determining step has been
proposed. Mechanistic investigations of the oxidation of phenyl‐
sulphanylacetate ions by potassium hexacyanoferrate(III) in aqueous
NaOH were done by Srinivasan and Subramaniam.136 The oxidation
follows first‐order kinetics in each of the oxidant, substrate and OH− at
constant ionic strength. While the added radical scavenger, acrylamide
enhances the rate, potassium hexacyanoferrate(II) retards it. Rate
studies with substituted phenylsulphanylacetate ions give an excellent
Hammett correlation with a positive reaction constant (ρ = 1.31). On the

43
basis of the kinetic evidence, a mechanism which involves an initial
reversible proton abstraction followed by another reversible electron
transfer step has been postulated.
1.4.2 Studies of oxidation of sulphides and sulphoxides
Harville et al. have reported137 that treatment of aliphatic and
aromatic sulphides with NCS or NBS in a large volume of anhydrous
methanol yields the corresponding sulphoxide in excellent yields. The
probable mechanism for the formation of sulphoxides in this system is
considered to parallel that suggested by Oae and coworkers138 involving
the initial formation of an intermediate halosulphonium compound
which reacts with the excess methanol to yield an alkoxysulphonium salt.
Dialkyl and alkyl aryl sulphides oxidized with NBS in aqueous medium
undergo C–S bond cleavage, but aromatic sulphides are oxidized to the
sulphoxides under the same reaction conditions. Oxidation of sulphides
with NBS has been carried out139 using anhydrous solvents at various
temperatures. It has been shown that certain aromatic sulphides can be
oxidized to sulphoxides in 70% dioxane‐water by NBS.
The kinetics of oxidation of several substituted phenyl methyl
sulphides by N‐chloroacetamide yielding sulphoxides have been
studied140 in acidic aqueous acetonitrile medium. The reaction displays
first‐order dependence each in [sulphide], [NCA] and [H+]. The reaction
rate is not influenced by the addition of acetamide, mercuric acetate and
acrylamide. Hammett correlation yields a ρ value of −3.29 establishing a
polar mechanism involving the rate‐limiting formation of a

44
chlorosulphonium cation by the electrophilic attack of a protonated NCA
on the sulphur. The formation of a halosulphonium cation is also the
rate‐limiting step in the oxidation of sulphides by NCS.141
The kinetic and mechanistic study of oxidation of sulphides with
N‐bromoacetamide (NBA) in the presence of Hg(II) salts suggests142 that
both NBA and sulphide form complexes with Hg(II) ions and that these
complexes participate in the rate‐determining step. The formation of a
halosulphonium cation which hydrolyses to sulphoxide has been
discussed. It has also been concluded that in the oxidation of sulphides
with N‐chloroamides, the presence of Hg(II) is not necessary.143
Chowdhury et al. have studied144 the kinetics of oxidation of
sulphides by N‐bromobenzamide to yield the corresponding sulphoxides.
The reaction is first‐order with respect to NBB, sulphide and hydrogen
ion. Protonated NBB has been postulated as the reactive oxidizing
species. Reactivity of the sulphides towards NBB has been analyzed using
multiparametric correlation equations. A mechanism involving formation
of a halogenosulphonium cation in the rate‐determining step has been
proposed (Scheme 12).
RCONHX + H+ RCON+H2X
R1SR2 +
R2
SR1
XRCON+H2X + RCONH2
slow
R2
SR1
X + H2Ofast
R2
SR1
OH + HX
R2
SR1
OHfast
R2
SR1
O + H+
Scheme 12

45
The oxidation with NBB does not require the presence of Hg(II) as a
bromine scavenger, in contrast to the NBA‐mediated oxidation of
sulphides.145
A simple and highly selective oxidation of sulphides to sulphoxides
by NBS catalyzed by β‐cyclodextrin in water has been reported.146 A
series of sulphides are oxidized selectively at room temperature in
excellent yields. This reaction proceeds without overoxidation to
sulphones under mild conditions using water as a solvent. Oxidation of
alkyl aryl sulphides by NBS in the presence of mercuric acetate
exhibits147 a clean second‐order kinetics, first‐order in each reactant.
Change in ionic strength and variation of added succinimide and
mercuric acetate concentrations have no effect on the rate. The reaction
affords a negative ρ value (−2.0). Addition of H+ catalyzes the reaction.
These results are argued in favour of a mechanism involving the rate‐
limiting electrophilic attack of NBS in neutral medium (and of NBS as well
as NBSH+ in acid medium) on the sulphide sulphur resulting in the
formation of sulphonium ion which undergoes fast hydrolysis to
sulphoxide.
A kinetic study of the oxidation of aryl methyl and diaryl sulphides
by NCS in a mixed acetonitrile – water solvent containing 0.001 M
perchloric acid has revealed133 that NCS and its protonated form are the
oxidizing species (Scheme 13).

46
Scheme 13
Z Cl + H+
slow
H Z Cl
Z Cl + R1SR2
R2
SR1
Cl Zδ+ δ− fast
R2
SR1
Cl + Z−
slow+ R1SR2H Z Cl
R2
SR1
Cl + ZH
R2
SR1
Cl + H2Ofast
R2
SR1
O + HCl + H+
Z− + H+ ZHfast
Kinetics of osmium(VIII) catalyzed oxidation of methyl phenyl
sulphide by Bromamine‐B in alkaline medium has been followed148 in 1:1
t‐butanol – water solvent. The product is methyl phenyl sulphoxide and a
suitable mechanism has been proposed. The osmium(VIII)‐catalyzed BAB
oxidation of some diaryl and dimethyl sulphoxides to sulphones in a
strongly alkaline (pH ~11.5) t‐butanol‐water medium has been
investigated.149 The kinetics of oxidation of methyl phenyl sulphoxide by
CAT in the presence of HCl (0.05 ‐ 0.08 M) and NaCl (0.4 ‐ 0.7 M) has
been studied150 at 10 oC. The oxidation of phenyl methyl sulphoxides by
CAT has been carried out151 in buffered ethanol ‐ water (1:1 v/v, pH =
7.0). A possible mechanism involving three rate‐controlling steps has
been proposed with the derivation of mixed‐order rate law.
Mahadevappa and others have reported152 RNHCl as the oxidizing
species in the Os(VIII)‐catalyzed oxidation of diphenyl sulphoxide by CAT
in alkaline medium.

47
Lee et al.153 have reported that the oxidation of sulphides and
sulphoxides by permanganate in anhydrous acetone solution is catalyzed
by Lewis acids such as FeCl3, ZnCl2 and HgCl2. The reaction is first‐order
each in permanganate and Lewis acid. But the order with respect to
sulphide is first‐order at low concentration and zero‐order at high
concentration of sulphide. Hammett analysis of the rate constants for
the oxidation of a series of substituted thioanisoles gives a negative ρ
value indicating an electron deficient transition state. Based on the
kinetic results a mechanism involving the rate‐limiting step of ligand
formation i.e., the attachment of the sulphide to manganese by a
coordinate covalent bond has been proposed. Similar kinetic results are
observed with the oxidation of sulphoxides also and hence a mechanism
analogous to the one proposed for the oxidation of sulphides has been
suggested (Scheme 14).
MeS
PhO +
OMn
O OZnCl2
O MeS
PhO
OMn
O OZnCl2
O
MeS
Ph
OMn
O OZnCl2
OO
MeS
Ph
O
O+ MnO3ZnCl2
−
Scheme 14
The Cr(VI) oxidation of alkyl aryl and diphenyl sulphides and
sulphoxides has been studied in aqueous acetic acid and in aqueous
acetonitrile by Srinivasan and others.154,155 Baliah and Satyanarayana156

48
have postulated the formulation of a cation radical intermediate in the
rate‐determinig step for Cr(VI) oxidation of aryl methyl sulphoxides. This
cation radical intermediate may attack the Cr=O bond of Cr(VI) or Cr(V)
species to form a complex which on solvolysis yields sulphone. An
analogous mechanism has been proposed by Srinivasan et ai.155 in the
Cr(VI) oxidation of diphenyl sulphoxides.
The kinetics and mechanistic aspects of Ru(III) catalyzed
oxidation of several diaryl, dialkyl, and alkyl aryl sulphoxides with HSO5−
have been reported.157 The reaction exhibits first order dependence each
in oxidant and reductant in the absence of Ru(III). If Ru(III) catalyzes the
reaction a mixed order dependence on the concentration of Ru complex
is observed. A mechanism involving nucleophilic attack of sulphoxide
sulphur on Ru=O has been proposed (Scheme 15).
Scheme 15
Ru(III) + HSO5−
RuV = O + HSO4−
RSOR'
R'S
RRuV O
O
δ+ δ−Products
Brovo et al. have explored158 the use of bromine as a catalyst in
the oxidation of sulphides to sulphoxides with H2O2 in CH2Cl2/H2O and
found that the oxidation in this system is strongly dependent on the
structure of the sulphides. Both alkyl and aryl sulphides can be efficiently
oxidized159,160 to the sulphoxides using NaBrO3 in combination with
NH4Cl in aqueous acetonitrile in the presence of Mg(HSO4)2.

49
Iodosobenzene (PhIO) as an efficient oxidant of alkyl aryl
sulphides to the corresponding sulphoxides has been described161 by
Kannan et al. This oxidation may be carried out in a suspension of
acetonitrile or in the solid state. Systematic studies of kinetics of
oxidation of alkyl aryl, dialkyl and diphenyl sulphides to the
corresponding sulphoxides with PhIO catalyzed by MnIII(salen),162,163
CrIII(salen)164 and FeIII(salen)165 complexes (salen = N,N’‐ethylenebis‐
(salicylideneaminato)) in acetonitrile medium have been reported. Single
electron transfer, SN2 and Michaelis‐Menten type mechanisms have
been envisaged for the above three systems, respectively. The oxidation
of various sulphides to sulphoxides with PhIO – (salen)CrIII system has
been reported166 by Kim et al.
The oxidation of various para‐substituted methyl phenyl
sulphoxides with several substituted oxo(salen)manganese(V) complexes
has been reported by Chellamani et al.167 The reaction follows an over all
second‐order kinetics, first‐order each in sulphoxide and oxo(salen)‐
manganese(V)complex. The less nucleophilic sulphoxides are more
sensitive to substituent effect compared to the corresponding sulphides.
These results are interpreted with a SN2 mechanism (Scheme 16).
(Salen)MnV
O
+ ArSOMe (Salen)MnV
O SOAr
Me
(Salen)MnIII + ArSO2Me
slow
Scheme 16

50
Oxidation of the diaryl sulphides to the corresponding sulphoxides
ith phenyliodoso diacetate (PIA), or poly(diacetoxyiodo)styrene (PDAIS)
in the of KBr in water has been reported168 In the oxidation
of diaryl sulphides with PDAIS, however, the major product is the
sulphone, whilst that in the case of PIA is the sulphoxide. The kinetics of
oxidation of several substituted alkyl aryl sulphides by PIA have been
investi
actions involving a heterolytic splitting of O–O bond, which is
promo
w
presence
gated169 in acetonitrile‐water mixture. The reaction is fractional
order in [sulphide] and first‐order in [oxidant]. Hammett and Taft
correlation analyses have been carried out. A mechanism involving a pre‐
equilibrium between oxidant and sulphide has been proposed.
Venkatachalapathy et al. have demonstrated170 the application of
clays as supports for tetrabutylammonium periodate in the oxidation of
sulphides to sulphoxides. Kim et al. have investigated171 the periodic acid
hydrate (H5IO6) in the presence of FeCl3 as a mild and highly selective
oxidant for the oxidation of sulphides to sulphoxides in acetonitrile
medium.
Hydrogen peroxide and alkyl peroxides have been widely used as
oxidant, solely or in combination with other reagents as catalysts, for the
oxidation of sulphides to sulphoxides. Modena and others have
studied172 the oxidation of organic sulphides by H2O2 and other organic
peroxides. They have found that these reactions are electrophilic oxygen
transfer re
ted by a concerted inter‐ or intra‐molecular proton transfer. Silica
gel and alumina mediate the TBHP (t‐butyl hydroperoxide) oxidation of

51
sulphides and sulphoxides.4 Ravikumar t al. have reported17 the
transformation of various sulphides into sulphoxides by H2O2 in
hexafluoro‐2‐propanol as the solvent.
The oxidation of dialkyl and diphenyl sulphides by pyridinium
chlorochromate155 is found to be catalyzed by organic acids and a
Michaelis‐Menten behaviour has been reported. Pyridinium flouro‐
chromate also oxidizes several aryl methyl, alkyl phenyl, dialkyl and
diphenyl sulphides to the corresponding sulphoxides and a rate‐
determining electrophilic oxygen trans
1 e 3
fer from PFC to the sulphide has
been
nic effect (ρ = ‐3.3) and also supported by the
observ
proposed.174 The oxidation of several sulphides by quinolinium
bromochromate has been found27b to result in the formation of the
corresponding sulphoxides. A mechanism involving the formation of a
sulphurane intermediate in the slow step has been proposed. Very
recently it has been reported that the oxidation of organic sulphides by
morpholinium chlorochromate proceeds through a sulphonium cation
intermediate.175
Ce(IV)‐catalysed autooxidation of sulphdes has been described176a
by Riley et al. A zwitter ion R2S+OO− has been envisaged as the probable
intermediate. But, for the oxidation of dilakyl, alkyl aryl and diaryl
sulphides with Ce(IV) carried out in the absence of oxygen,176b an
electron transfer mechanism has been proposed on the basis of
observed electro
ation that alkyl aryl sulphides are significantly more reactive than
dialkyl sulphides and that the reaction rate is slowed by added Ce(III).

52
Lead tetraacetate (LTA) has been found177 to oxidize various organic
sulphides to sulphoxides. In this work, Banerji has reported that this
oxidation is catalyzed by H+ and susceptible to changes in the solvent
composition. A mechanism involving a nucleophilic attack of sulphide on
lead in LTA in the rate‐determining step has been postulated
(Scheme 17).
S1
2+ Pb(OAc)3 - OAc
R
R
slowS
1
2
+R
RPb(OAc)3 + OAc-
SR
R2
+1OH + OAc- S
R1
R2
O + AcOH
Scheme 17
S1
2
+R
RPb(OAc)3 S
1
2
+R
ROH Ac2O Pb(OAc)2+ ++ AcOH
Kinetic studies of oxidation of organic sulphur compounds using several
other oxidants such as selenonic acids,178 Zn(MnO4)2,179
peroxomonosulphate ion37b etc. are also found in large numbers in the
literature.
1.5 Scope of the Present Investigation
Kinetic study is an expanding field of research in chemistry with a
to industrially o
thodologies, to improve the selectivity or to increase the
focuses on the oxidation of
scope of developing newer reagents which can be used for the
conversion of substrates commercially, r biologically
important products. The new method is to make easy the hurdles in the
existing me
yield of the product. The present study

53
(arylth
p
lity both in acid and alkaline
mediu
ns, deprotection and protection of different
functio
io)aceic acids and diaryl sulphides using the N‐halo compounds, N‐
bromo thalimide and N‐chlorosaccharin.
The role of N‐halo compounds in the synthetic field is very wide.
The diverse nature of the chemistry of these compounds is due to their
ability to act as sources of halonium cations, hypohalite species and
nitrogen anions, which act both as bases and nucleophiles.180 The N‐halo
compounds are considered to be disinfectants and antiseptic.72e The N‐
halo compounds have appreciable stabi
ms. The electronegativities of halogens, except fluorine, are less
than that of nitrogen. Hence, they acquire a positive charge when linked
with nitrogen. The electronegativity of nitrogen is further enhanced by
linking it with certain electron‐withdrawing groups. The N–X bond in N‐
halo compounds is fairly strong electrophilic, since halogen leaves as X+
ion in reactions.181
From the literature works cited in the previous sections it
becomes clear that the N‐halo compounds, commonly used in studies of
oxidation of organic substrates such as alcohols, aldehydes, amino acids,
keto acids, sulphides, etc., are NCS, NBS, NCN, CAT, BAB, NBB, NBSA,
NCSA, NBP, etc. Various organic functional group transformations such
as oxidation reactio
nal groups, halogenations of saturated and unsaturated
compounds, acylation of alcohols, phenols, amines or thiols, epoxidation
of alkenes, aziridination etc. have been carried out182,183 using NBP and
NCSA. These two reagents have also been used94 as oxidimetric reagents

54
for the estimation of different organic compounds. Yet, there is scant
information in the literature about the kinetics and the mechanistic
aspects of these re ctions. Although the works n the kinetics of
oxidation of organic sulphur compounds, particularly, (arylthio)acetic
acids and diaryl sulphides with NCS, NBS, NCA and NBB are abound,
those with NCSA and NBP are not traceable. All these aspects are taken
into consideration while choosing NCSA and NBP as oxidants in the
present study.
Oxidation reactions, capable of converting sulphides to
sulphoxides and then to sulphones are perhaps useful in the
detoxification of harmful and poisonous substances like nerve agents
and mustard gas.184 Organic sulphoxides are also useful synthetic
intermediates for the construction of various chemical and biological
systems and so
a o
me play key roles in the activity of some enzymes.185,186
They o t h p s
i
n
t t r
ften play an importan role as t era eutic agent such as anti‐
ulcer (proton pump nhibitor),187 antibacterial, antifungal, anti‐
atherosclerotic,188 anthelmintic,189 anti‐hypertensive,190 and cardiotonic
agents191 as well as psychoto ics192 and vasodilators.193 Insecticides,
which are sulphoxides are commercially manufactured by oxidizing
sulphides with H2O2 and the conversion of penicillins to their S‐oxides is
commercially important for cephalosporin derivatives.194 The oxidation
of sulphides is he mos straight‐forward method fo the synthesis of
sulphoxides. The conditions of the reaction, that is, time, temperature
and the relative amount of oxidants, have to be controlled to avoid

55
forming side‐products of the oxidation. In view of these aspects, organic
sulphides are chosen as substrates for the present investigation. The
experiments have been focused to explore the following aspects.
(1) The order of the reaction with respect to [oxidant], [substrate]
and [H+]
(3) The effect of ionic strength of the medium on the reaction
(2) The effect of dielectric constant of the reaction medium on the
reaction
rate
product of the oxidant, phthalimide or saccharin
n
and diaryl sulphides, the methods of kinetic
presented in Chapter 2. Chapters 3 and 4 describe the results obtained in
the kinetic studies of oxidation of several para‐substituted
limide (NBP)
and N‐chlorosaccharin (NCSA), respectively.
(4) The effects of added substances, viz., free‐radical scavenger and
the reduced
(5) The effect of substituents on the phenyl ring of the substarte on
the reactio
(6) Determination of reaction constant and isokinetic temperature
(7) Determination of activation parameters for the reaction, and
(8) Elucidation of a suitable mechanism for the reaction.
The experimental aspects of the study including the preparation
of (arylthio)acetic acids
measurements, product analysis and the tables of kinetic runs are
(phenylthio)acetic acids (listed below) with N‐Bromophtha
56
(Arylthio)acetic acids (p‐XC6H4SCH2COOH)
S CH2COOHX
X =
H : Phenylth o)acetic acid (H‐ )
OCH3 : (4‐Methoxyphenylthio)acetic acid (OMe‐S)
CH3 : (4‐Methylphenylthio)acetic aci
( i S
d (Me‐S)
F : (4
Cl : (4‐Chlorophenylthio)acetic acid (Cl‐S)
NO2 : (4‐Nitrophenylthio)acetic acid (NO2‐S)
a er obtained in etic studies of
oxidation se ide with
N‐chlorosa h
4SC6H
X
H
hyldiphenyl sulphide (Me‐S)
Cl : 4‐Chlor S)
NO2 : 4‐ (NO2‐S)
‐Fluorophenylthio)acetic acid (F‐S)
Ch pt 5 describes the results the kin
of veral para‐substituted diaryl sulph s (listed below)
cc arin.
Diaryl sulphides (p‐XC6H 5)
SX
=
: Diphenyl sulphide (H‐S)
OCH3 : 4‐Methoxydiphenyl sulphide (OMe‐S)
CH3 : 4‐Met
odiphenyl sulphide (Cl‐
Nitrodiphenyl sulphide