
EXPERIENCES WITH SICKLE CELL DISEASE SCREENING AND …
111
EXPERIENCES WITH SICKLE CELL DISEASE SCREENING AND DIAGNOSIS
Transcript of EXPERIENCES WITH SICKLE CELL DISEASE SCREENING AND …

EXPERIENCES WITH SICKLE CELL DISEASE SCREENING AND DIAGNOSIS

USING PARENTAL EXPERIENCES WITH SICKLE CELL DISEASE SCREENING
AND DIAGNOSIS TO GUIDE HEALTH SCIENCE EDUCATION:
A SYSTEMATIC REVIEW AND INTEGRATIVE QUALITATIVE
META-SYNTHESIS
BY
SIMONE GRIFFITH, B.HSc.
A THESIS
SUBMITTED TO THE DEPARTMENT OF HEALTH SCIENCE EDUCATION
AND THE SCHOOL OF GRADUATE STUDIES
OF MCMASTER UNIVERSITY
IN PARTIAL FULFILMENT OF THE REQUIREMENTS
FOR THE DEGREE OF
MASTER OF SCIENCE
McMaster University © Copyright by Simone Griffith, April 2021

ii
MASTER OF SCIENCE (2021) McMaster University
(Department of Health Science Education) Hamilton, Ontario
TITLE: Using Parental Experiences with Sickle Cell Disease
Screening and Diagnosis to Guide Health Science Education:
A Systematic Review and Integrative Qualitative
Meta-Synthesis
AUTHOR: Simone Griffith, B.HSc.
SUPERVISOR: Dr. Meredith Vanstone
NUMBER OF PAGES: xiii, 110

iii
Abstract
Sickle cell disease (SCD) is a chronic, lifelong, often debilitating, inherited disorder that
can affect every organ system. Affected individuals often experience repetitive pain
crises, multiple hospitalizations and a diminished quality of life. Many people at risk for
SCD are unaware of their sickle cell carrier status and surprisingly health care providers’
knowledge of SCD is limited. Research literature focuses mainly on management of
clinical manifestations of the disease. This systematic review and integrative qualitative
meta-synthesis aims to capture parents’ perspectives on the screening process and
diagnosis of SCD or sickle cell trait (SCT). Information generated by this review will be
helpful in contributing to the development or enhancement of guidelines and protocols in
SCD and SCT management for health care providers and health care educators.
.

iv
Acknowledgements
This work would not have been possible without the support of McMaster Health
Science Education staff members and Marija Radomirovic, who liaised with the Graduate
Department on several occasions on my behalf.
I am grateful to Dr. Lawrence Grierson, whose support facilitated my ability to
complete this program. The members of my Thesis Committee graciously gave of their
time, too often on short notice, and provided critical and positive feedback, all while
navigating scholastic challenges during the COVID pandemic. They offered kind words
of encouragement and gently guided me.
I would especially like to thank Dr. Meredith Vanstone, who exemplified the
epitome of kindness and never ending patience. Through her sage guidance, she
mentored me and gave me courage to believe that I could complete my Master’s thesis
while I maintained a fulltime job in the health care sector. She always greeted me with a
smile and she never made me feel as though I was too much work. I am not sure that my
words can truly capture the depth of my gratitude.
And my family, who I have most certainly neglected in the months preceding my
Master’s thesis defence. My mother, Cecile, has been invaluable, loving me and
supporting me as always, and my son, Kyle, always the cheerleader. I am fortunate to
have their support now and always.

v
Table of Contents
Abstract ........................................................................................................................... iii
Acknowledgements ........................................................................................................ iv
Table of Contents ............................................................................................................ v
List of Tables .................................................................................................................. ix
List of Figures .................................................................................................................. x
List of Diagrams ............................................................................................................. xi
List of Abbreviations .................................................................................................... xii
Declaration of Academic Achievement ...................................................................... xiii
1. Introduction ............................................................................................................ 14
2. Clinical Background .............................................................................................. 17
Etiology of Sickle Cell Disease ....................................................................... 18
Types of Sickle Cell Disease ........................................................................... 18
Sickle Cell Trait ....................................................................................... 18
Inheritance Pattern of Sickle Cell Disease ............................................. 20
Sequelae of Sickle Cell Disease ............................................................... 21
3. Lifespan, Treatment and Cure ............................................................................. 22
Lifespan............................................................................................................ 22
Treatment ........................................................................................................ 23
Cure .................................................................................................................. 23
Haematopoietic Stem Cell Transplant ................................................... 23
Gene Therapy ........................................................................................... 24
4. Screening and Diagnosis of Sickle Cell Disease and Sickle Cell Trait .............. 24
Haemoglobin Electrophoresis ........................................................................ 24
Sickle Cell Solubility Test .............................................................................. 24
Newborn Screen .............................................................................................. 24
Genetic Counseling for Pre-marital or Pre-conception Screening ............. 25
Genetic Counseling during the Antenatal Period ........................................ 25
Chorionic Villus Sampling ...................................................................... 25

vi
Amniocentesis ........................................................................................... 26
Author’s Professional Experience with Sickle Cell
Disease Screening ............................................................................................ 26
5. Method .................................................................................................................... 27
Methodology .................................................................................................... 27
Study Design .................................................................................................... 28
Data Extraction ............................................................................................... 29
Data Analysis ................................................................................................... 29
6. Results ..................................................................................................................... 37
Antenatal Screening for Sickle Cell
Disease (Haemoglobinopathy) ....................................................................... 39
Informed Choice to Screen and Reasons to Accept
Sickle Cell Disease (Haemoglobinopathy) Screening ........................... 39
Awareness and Education of Sickle Cell
Disease or Sickle Cell Trait ..................................................................... 41
Timing of Sickle Cell Disease (Haemoglobinopathy) Screening,
Planning of Pregnancy and Termination of Pregnancy ....................... 42
Responsibility for Sickle Cell Disease (Haemoglobinopathy)
Screening .................................................................................................. 43
Family Influence on Sickle Cell Disease
(Haemoglobinopathy) Screening ............................................................ 44
Role of Religion in Sickle Cell Disease
(Haemoglobinopathy) Screening ............................................................ 45
Sickle Cell Trait Diagnosis ............................................................................. 45
Awareness and Education of Sickle Cell Trait...................................... 46
Antenatal Screening ......................................................................... 46
Newborn Screening ........................................................................... 46
Disclosure of Sickle Cell Trait Results ................................................... 47
Communication of Sickle Cell Trait Results ......................................... 48

vii
Emotional Response to Sickle Cell Trait Diagnosis .............................. 49
Sharing of Sickle Cell Trait Results and Family Support.................... 49
Paternity ................................................................................................... 50
Cascade Testing ....................................................................................... 50
Sickle Cell Disease Diagnosis ......................................................................... 51
Awareness and Education of Sickle Cell Disease .................................. 51
Communication of Sickle Cell Disease Results ..................................... 52
Emotional Response to Sickle Cell Disease Diagnosis .......................... 53
Child’s Future .......................................................................................... 53
Implications for Future Pregnancy Planning and
Parental Relationships............................................................................. 54
Sharing Sickle Cell Disease Diagnosis and Support ............................. 54
7. Discussion ............................................................................................................... 55
8. Interpretation of Results ....................................................................................... 57
Lived Experience of People with Sickle Cell Disease .................................. 57
Informed Choice ............................................................................................. 58
Role of the Health Care Provider and the Responsibility of the
Education System ............................................................................................ 61
Role of the Health Care Provider ........................................................... 61
Role of the Health Care Education System ........................................... 62
Individual Screening versus Population Based Screening
for Sickle Cell Disease..................................................................................... 63
Antenatal Screening for Sickle Cell Disease .......................................... 63
Population Based Screening for Sickle Cell Disease ............................. 64
Racism in Healthcare and Research ............................................................. 65
Reflexivity ........................................................................................................ 69
Personal Experience of a Midwife ................................................................. 69
Experience of a Black Nurse .......................................................................... 71
9. Recommendations .................................................................................................. 72

viii
Implicit Bias ..................................................................................................... 72
Increase Funding for Sickle Cell Disease Research ..................................... 73
Improve Sickle Cell Disease Health Education ............................................ 74
Hidden Curriculum ........................................................................................ 75
Informed Choice Discussion and Communication ...................................... 76
Sickle Cell Disease Patient Directed
Health Care Provider Education ................................................................... 77
10. Limitations .............................................................................................................. 78
11. Conclusions ............................................................................................................. 79
12. References ............................................................................................................... 80
13. Appendix A Sickle Cell Search Strategy ......................................................... 88
14 Appendix B Characteristics of Excluded Studies ......................................... 102
15. Appendix C Newborn Screen for Sickle Cell Disease in Canadian ............ 109
16. Appendix D Sickle Cell Trait Disclosure
(countries listed in the studies reviewed) ................................. 110

ix
List of Tables
Table 1. Sickle Cell Disease Variations .............................................................. 19
Table 2. Inclusion and Exclusion Criteria ......................................................... 28
Table 3. Characteristics of Included Studies ..................................................... 31
Table 4. Themes Identified by the Authors of the Studies Reviewed .............. 34
Table 5. Sickle Cell Search Strategy ................................................................... 88
Table 6. Characteristics of Excluded Studies .................................................. 102
Table 7. Newborn Screen for Sickle Cell Disease in Canada ......................... 109
Table 8. Sickle Cell Trait Disclosure (by countries listed in the studies) ...... 110

x
List of Figures
Figure 1. Systematic Literature Search PRISMA Diagram .............................. 30

xi
List of Diagrams
Diagram 1. Prevalence of Sickle Cell Disease Worldwide ..................................... 16
Diagram 2. Inheritance Pattern of Sickle Cell Anaemia
and Sickle Cell Trait.............................................................................. 20

xii
List of Abbreviations
CF Cystic Fibrosis
GPs General Practitioners
HbP Haemoglobinopathies
HbA Normal Haemoglobin
HbS Haemoglobin S (Sickle Haemoglobin)
HSCT Haematopoietic Stem Cell Transplant
MV Meredith Vanstone
NBS Newborn screen
NSAID Non Steroidal Anti-Inflammatory Drug
PCP Primary Care Provider
SC Sickle Cell
SCD Sickle Cell Disease
SCT Sickle Cell Trait
SG Simone Griffith
T Thalassemia
UK United Kingdom
USA United States of America

xiii
Declaration of Academic Achievement
Simone Griffith completed this research work independently, with scholarly guidance
from her supervisor (Dr. Meredith Vanstone) and committee members (Dr. Madeleine
Verhovsek and Dr. Melissa Kimber). She received instrumental support from research
assistants to sort the references for the systematic review and integrative qualitative meta-
synthesis.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
14
Introduction
Sickle cell disease (SCD) is an inherited blood disorder that affects millions of
people worldwide (Diagram 1) (1-4). The disease is chronic, lifelong and can be
debilitating. It has the potential to affect virtually every organ system in the body;
potential consequences include - acute chest syndrome (chest pain, cough and fever),
stroke, chronic anaemia, leg ulcers, dactylitis (inflammation of the hands), gallstones,
kidney damage, bacterial infections and early death (1, 2). The disease not only impacts
the quality of life of affected individuals, from frequent hospital admissions to higher
rates of absence from school or work, but also the quality of life of the immediate family
(5). Family caregivers of sickle cell patients often experience job loss, decreased earning
potential and mental and physical fatigue (5).
SCD is the most commonly inherited group of blood disorders (6, 7). Mutations on
the gene encoding the haemoglobin subunit β result in the change of the structure of the
haemoglobin molecule (3, 6, 8). Sickle haemoglobin (HbS) carriers oxygen in the same
way as normal haemoglobin (HbA) but there can be reduced oxygen delivery to tissues
when there is sickle vaso-occlusion (blockage of post-capillary venules, causing reduced
blood flow through the capillary beds) (1, 7, 8). SCD is an autosomal recessive disorder
– affected individuals inherit two copies of the abnormal gene, one from each parent.
Drug therapy and blood transfusions are the treatment of choice to manage SCD.
Haematopoietic stem cell transplantation (stem cells from bone marrow or blood) is the

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
15
only curative option for SCD, however, it carries a risk of complications such as
infection, graft rejection and death (8, 9). The lack of available matched donors makes
this therapy out of reach for the majority of patients with SCD (9). Gene therapy as a
curative option for SCD is on the horizon – multiple gene therapy strategies are in clinical
trials (9). Eighty to ninety percent of children born with SCD in high income countries
will survive to adulthood, nonetheless, medical complications of SCD often lead to a
shortened lifespan of 54-65 years (10, 11). Without early diagnosis of SCD and
comprehensive medical care, children with SCD in low income countries and Africa may
die before the age of five years (2, 11, 12). SCD is a global public health issue – 75% of
the SCD population live in sub-Saharan Africa where inadequate health infrastructure,
limited access to drug treatment, and poor nutrition leads to a high incidence of morbidity
and mortality (2).
SCT is the carrier state for SCD. A normal haemoglobin gene is inherited from one
parent and a sickle haemoglobin gene in inherited from the other parent (6-8, 11). Most
people are unaware of their SCT carrier status (1, 2). SCT typically presents as a benign
condition, however, venous thromboembolism (blood clots) and renal complications can
occur at higher rates, and other complications can occur under periods of extreme
exercise in affected individuals (13, 14).
It is believed that 3,000 to 5,000 people in Canada have SCD (4). SCD is most
prevalent in people of Black African, South Asian, Mediterranean (Italian, Greek,
Turkish), and Middle Eastern descent – regions endemic for malaria (11).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
16
Diagram 1. Prevalence of Sickle Cell Disease Worldwide
As the demographic of the population of Canada continues to become more ethnically
and racially diverse, the incidence of SCD will continue to increase, as will the burden on
the Canadian healthcare system (16). Aside from a set of national consensus
recommendations for the care of patients with SCD in Canada (4), no nationally based
strategies, policies or registries exist for guidance on the management of SCD in Canada
(16). Health care providers, educational institutions and researchers are left to create
policies and research at their discretion.
For many parents, the diagnosis of their child’s SCD or sickle cell carrier status
through newborn screen (NBS), will also act as the first diagnosis, albeit indirect, of their
own sickle cell carrier status (17, 18). Research literature reveals that while many people
at risk for SCD may be familiar with the disease, 25% – 50% of them will be unaware of
Not Alone in Sickle Cell (15)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
17
their own sickle cell carrier status (19-23). The psychological and physical demands of
caring for a child with SCD can be devastating – from upheaval of the family dynamics,
parental exhaustion of balancing multiple hospital admissions and medical appointments
to financial challenges associated with absences from their job (5). There is value in
understanding parents’ lived experience with SCD and SCT given their important role in
the identification, management and access to treatments for SCD and SCT for their
children, respectively.
This systematic review and integrative qualitative meta-synthesis aims to
consolidate what is known of SCD and SCT from the perspective of parents, it will also
generate recommendations on aspects of the health care system that function well and
those that do not function well and identify any gaps that exist. Information generated by
this review will be helpful in contributing to the development or enhancement of
guidelines and protocols in SCD and SCT management for health care providers and
health care educators.
Clinical Background
Research has shown that while people may be familiar with term SCD they are not
familiar with the details of the disease (19). Rudimentary information on SCD is
provided to enhance the reader’s ability to appreciate the complexity of the SCD and
dispel misconceptions of the disease.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
18
Etiology of Sickle Cell Disease
SCD is an inherited disease of the blood affecting the formation of haemoglobin.
Haemoglobin is a protein in the blood that carries oxygen from the lungs to rest of the
body (1, 6, 11). A single point mutation on the sixth codon of the beta-globin chain on
chromosome 11, causes a permanent change of the haemoglobin structure, creating a new
and abnormal haemoglobin gene (designated haemoglobin S - HbS). Mutations can also
occur in different locations of the alpha globin and beta globin chain, which causes the
creation of different or variant forms of haemoglobin genes (1, 6, 11).
Types of Sickle Cell Disease
Sickle cell disease: results from the inheritance of two abnormal haemoglobin genes
(Diagram 2). Any combination of one abnormal haemoglobin S gene paired with another
variant (abnormal) haemoglobin gene results in SCD. Sickle cell anaemia, the most
prevalent and well known type of SCD, results from the inheritance of two abnormal
haemoglobin S genes (3, 7, 12). Other SCD variants are described in Table 1.
Sickle cell trait: is characterized by the inheritance of one normal haemoglobin
gene and one abnormal haemoglobin S gene (Table 1.) (1, 3, 6, 11). SCT is typically an
asymptomatic and benign condition, however, in rare instances and under extreme
conditions, such as excessive exercise, people affected with SCT can experience health
concerns (13, 14). It is not uncommon for people affected with SCT to be unaware of

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
19
their carrier status. Furthermore, there remains confusion or misunderstanding of the
genetic significance of SCD among people who know they are sickle cell carriers (13,
20). These potential parents may then be surprised to have a child affected with SCD (13,
20).
Table 1. Sickle Cell Disease Variations
Sickle Cell Disease Haemoglobin
Variants
Prevalence Disease
Manifestation
Sickle Cell Anaemia HbSS Most common
form of sickle cell
disease
Severe symptoms
Sickle Cell Beta
Thalassemia +
HbSβ+ Less common Less severe
symptoms than
sickle cell anaemia
Sickle Cell Beta-zero
Thalassemia 0
HbSβ0 Less common Severe symptoms
Sickle Cell C HbSC Less common Less severe
symptoms than
sickle cell anaemia
Sickle Cell D HbSD Rare Rare, almost no
symptoms
Sickle Cell E HbSE Rare Rare, almost no
symptoms
Sickle Cell Arab HbSO Rare Rare, almost no
symptoms
Ware et al (2017) (8)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
20
Diagram 2. Inheritance Pattern of Sickle Cell Anaemia and Sickle Cell Trait
Inheritance Pattern of Sickle Cell Disease
If both parents have SCD, all their children will also have SCD.
If both parents have SCT, in each conception there is a 25% chance that their child will have
SCD.
If both parents have SCT, in each conception there is 50% chance that their child will have
SCT.
If both parents have SCT, in each conception there is a 25% chance that their child will have
normal haemoglobin.
If one parent has SCT and one parent has SCD, in each conception there is a 50% that their
child will have SCD.
If one parent has SCT and one parent has SCD, in each conception there is a 50% that their
child will have SCT (if the SCD consists of a haemoglobin S gene and a variant haemoglobin
gene, in each conception there is a 25% chance their child will have SCT and a 25% chance
their child will have a haemoglobin variant trait).
If one parent has SCT and one parent has normal haemoglobin, in each conception there is a
50% chance that their child will have normal haemoglobin.
If one parent has SCT and one parent has normal haemoglobin, in each conception there is a
50% chance that their child will have SCT (1, 6).
Microsoft Library

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
21
Sequelae of Sickle Cell Disease
Normal red blood cells are flexible, round in shape and travel easily through blood
vessels (squeezing and changing shape) as they transport oxygen through the body (3, 6).
Conditions of deoxygenation (oxygen molecules detach from the haemoglobin molecule)
typically have no significant affect on normal haemoglobin molecules. However,
deoxygenation of abnormal haemoglobin S molecules causes them to link together,
leading to a change in the shape of the red blood cell (they assume a sickle shape, denoted
by the notation of HbS) (1, 2, 7). The abnormal HbS red blood cells experience an
episode of sickling. Sickled shaped red blood cells transport fewer oxygen molecules, are
more fragile, rigid and less flexible and become jammed in the blood vessels because they
cannot flow through blood vessels easily. Episodes of sickling deprive organs of vital
oxygen which leads to periods of pain that are often termed pain crises. Repeated cycles
of sickling, shorten the lifespan of the fragile sickle cell red blood cell to an average of 17
days versus 90 days for a normal red blood cell (1, 2, 7).
Chronic anaemia, fatigue, lethargy and frequent pain crises are the hallmark
symptoms of SCD (6-8). Every organ system is subject to injury and permanent damage
in SCD. The severity of symptoms and damage to organs in SCD is largely dependent
upon the type of variant haemoglobin gene(s) inherited.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
22
Lifespan, Treatment and Cure
Lifespan
The quality of life and life expectancy of people affected with SCD vary widely
depending on country of birth, age of diagnosis, age at initiation of drug treatment, and
access to care (1, 11, 24). In high income countries, more than 90% of infants born with
SCD will survive to adulthood. Early diagnosis of SCD by NBS and greater access to
drug therapy and comprehensive medical care, reduce the incidence of childhood
morbidity and mortality (2, 11, 24). Clinical manifestations of SCD can vary from person
to person and within the types SCD (6). Some patients will experience many symptoms
while other patients will experience few symptoms over their lifetime (6). Adherence to
prophylactic drug regimens and vaccination schedules, and general health routines affect
the quality of life and life span of SCD patients. However, the lifespan of people affected
with SCD is generally reduced by 15 to 24 years as compared to people not affected with
SCD (25).
Infants born in low income or low resource countries, as in African countries, where
NBS is not routine and there is limited access to drug treatment, or low resources of drug
treatment early in life, may only have a 10%-50% survival rate to the age of five years
(12, 24). Low rates of early detection in infancy and early childhood and limited access
to prophylactic penicillin and hydroxyurea, limit the lifespan to 15-20 years for children
who survive beyond the age of five years in low income countries (11, 12).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
23
Treatment
• Prophylactic penicillin (preventative), initiated by three months of age, is used
for management of SCD in children and reduces the frequency of infections.
• Hydroxyurea, is an oral medication that is used to prevent anaemia and reduce
the incidence of pain crises and other acute and chronic complications.
• L-glutamine is an oral powder used to help reduce the frequency of pain crises
• Crizanlizumab is an intravenous drug that is used to help reduce the frequency of
pain crises.
• Morphine is an opioid drug used to help relieve pain. Other opioid and/or
nonsteroidal anti-inflammatory drug (NSAID) analgesics are also among the
available options for pain treatment.
• Voxelotor is an oral drug that is used to improve anaemia.
• Blood transfusions are frequent and commonly used to manage severe and
chronic anaemia associated with the SCD.
Cure
Haematopoietic stem cell transplantation (HSCT): is the only curative treatment
for SCD. It is expensive, with a risk of toxicity, and health concerns post transplant,
including early death. HSCT is often reserved for cases in which repeated blood
transfusions are no longer feasible (4, 6). Periods of sickling can lead to progressive
organ and tissue damage as SCD patients age. Organ dysfunction can increase risks of

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
24
organ toxicity, morbidity and transplant-related mortality exponentially and for this
reason, HSCT is often reserved for children younger than 17 years (4, 6).
Gene therapy: is a relatively new curative option in the clinical trial phase (9).
Screening and Diagnosis of Sickle Cell Disease and Sickle Cell Trait
Screening for SCD can be offered to women and men pre-maritally, pre-conception,
and antenatally. The United Kingdom (UK) offers universal antenatal screening for SCD
for women at risk for SCD (26, 27). Antenatal screening for SCD in Canada, the USA,
and the Netherlands is offered at the discretion of the health care provider (26). Infants
can be screened via the NBS within a few days of birth. In other instances, infants may
not be offered screening or diagnosis for SCD until symptoms manifest.
Haemoglobin electrophoresis: a blood test used to identify normal and abnormal
haemoglobins. This test is able to diagnose all forms of SCD and SCT (8).
Sickle cell solubility test (commonly called SICKLEDEX®): a blood test used to
identify the presence of haemoglobin S. The test is unable to detect other abnormal
haemoglobins and can only be used as a screening tool (8). Further diagnostic tests need
to be run when haemoglobin S is identified. The absence of haemoglobin S can be used
to exclude SCD or SCT only. This test is not reliable in the neonatal period.
Newborn screen: a blood test used on newborn infants to assess the probable
presence of disorders (genetic, metabolic, blood or hormone related) that may not be

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
25
apparent immediately after birth. The disorders that can be screened are not uniform and
vary by jurisdiction. Canada does not have a national universal newborn screening
program – each province and territory operates its own newborn screening program.
Currently only eight provinces and one territory offer universal screening for SCD
(Alberta, British Columbia, New Brunswick, Newfoundland and Labrador, Nova Scotia,
Ontario, Prince Edward Island, Quebec and the Yukon) (28).
Genetic counseling for pre-marital or pre-conception: can be offered to couples if
each parent carries one sickle cell gene. These couples have a one in four chance of baby
with SCD (Diagram 2). A genetic counselor may enquire of the family history of each
couple and offer pre-implantation screening of embryos, which can identify embryos that
may carry SCD or SCT.
Genetic counseling during the antenatal period: can be offered to parents when
each parent screens positive for the sickle cell gene during pregnancy. Prenatal diagnosis
for the fetus (unborn baby) can be offered in early pregnancy to determine whether the
fetus has SCD.
• Chorionic villus sampling: is performed between 10 and 12 weeks of pregnancy
and involves obtaining a sample of the placental tissue (8).
o The procedure can be performed transcervical – a catheter is inserted through
the cervix into the placenta to obtain tissue.
o The procedure can be performed transabdominal - a needle is inserted
through the abdomen and the uterus into the placenta to obtain tissue

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
26
• Amniocentesis: is performed between 15 and 20 weeks of pregnancy and
involves the insertion of a thin needle through the abdomen and the uterus into
the amniotic fluid to obtain a small sample of amniotic fluid.
Chorionic villus sampling and amniocentesis carry a risk of infection, miscarriage,
preterm labour and limb defects.
Author’s Professional Experience with Sickle Cell Disease Screening
In my practice as a midwife, I have chosen to offer antenatal screening for
haemoglobinopathies, which encompasses SCD, to all my clients irrespective of their
ethnicity as anyone of any ethnicity can have SCT (6). I also have encouraged all my
colleagues to offer haemoglobinopathy screening to all their clients. The midwifery
profession is prefaced on the belief of empowering and supporting women during
pregnancy and I believe I have an opportunity and obligation to educate all my clients
about SCD. While I have not maintained accurate statics, during 17 years of midwifery
practice, the vast majority of my clients and their partners have either never heard of SCD
or they are familiar with SCD, but unaware of their sickle cell carrier status. Through this
screening practice, many of my clients have learned of their sickle cell or thalassemia
carrier status for the first time. It is important to be aware that the sickle solubility test is
the only test that can be ordered within a midwife’s scope of practice in Ontario. Only
one diagnostic laboratory processes haemoglobin electrophoresis tests ordered by
midwives.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
27
Method
Methodology
This systematic review and integrative qualitative meta-synthesis uses Sandelowski
and Barosso’s methodology (29). Integrative qualitative meta-synthesis is also known as
qualitative research integration. It aims to summarize the findings of multiple studies
with two aims: first, this technique aggregates results to reflect the range of findings
across studies while still retaining original meaning. Second, it produces a new
integrative interpretation by comparing and contrasting findings across studies.
Integrative qualitative meta-synthesis is the appropriate synthesis methodology for
the current project because it balances description and interpretation, focusing on
understanding what has been said about a topic and offering new understanding by
looking at the cumulative evidence. This approach is more consistent with systematic
review methodology than many other qualitative synthesis approaches because it allows a
priori definition of the research question and search strategy, as opposed to using a
progressively iterating research question with a more flexible search strategy (29). This
methodology focuses on the synthesis of qualitative findings, which is congruent with my
exploratory research question about experiences.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
28
Study Design
This systematic review and integrative qualitative meta-synthesis answers the
research question - What are parents' experiences with sickle cell disease screening and
diagnosis?
Table 2. Inclusion and Exclusion Criteria
Search terms were developed based on truncations of the words sickle cell, sickle
cell trait, anaemia, and combined with a validated search mega-filter developed and
validated to identify qualitative research (30) (Appendix A). Wild card and Boolean
Search Operators were used to capture the most relevant studies. A total of seven
databases were searched, which included OVID Medline Epub Ahead of Print, In-Process
& Other Non-Indexed Citations, Ovid MEDLINE® Daily and Ovid MEDLINE® 1946 to
Present, Embase (1974-2019), PsychINFO, AMED, OVID Emcare, and CINAHL. The
Inclusion
Exclusion
English language, full text articles
Published, peer reviewed, research
Primary qualitative empirical research (using any
descriptive or interpretive qualitative methodology,
including the qualitative component of mixed-
methodology)
Studies involving the experiences or perspectives of
people who have sickle cell disease or trait and/or
their parents
Studies published after 2006
Quantitative
Not empirical
Not English
Not patient or parent
perspective
Not peer reviewed
Not primary
Not sickle cell
Animal study
No abstract
Plant Study
Relevant but not eligible
Sickle cell is secondary focus
Studies published before 2006

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
29
search process began 2019 June 18 and was complete 2019 June 28. No date limits were
applied. Backward citation chaining of the included studies supplemented the database
searches.
Article screening was completed in duplicate independently by SG and research
assistants, who resolved disagreements through discussion. Eligible articles included
English-language primary qualitative research studies addressing the experiences of
parents or intended parents relevant to screening and diagnosis of SCD or SCT (Table 2).
Data Extraction
Each study was carefully reviewed and the country of origin, number of
participants, and study design was recorded in an excel spreadsheet. Themes identified
in the original study that represented the experiences of the participants were extracted
and analyzed using the process detailed below.
Data Analysis
Following the analytic instructions of qualitative meta-synthesis (29), we aimed for
findings which produced both a comprehensive summary of the original study articles, as
well as an interpretation of the meaning of this information from the perspective of
parents. This was achieved through multiple rounds of coding, following an adapted
version of the staged coding process of Grounded Theory (31). First, descriptive codes
were used to summarize key points in each article. Next, codes were gathered together

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
30
with similar ideas into categories. The third stage of coding involved the application of
interpretative ideas, another pass through the data to recognize relevant data excerpts, and
then a re-combination of the categories. Analysis was led by SG, who discussed
emerging analytic ideas with MV.
Figure 1. Systematic Literature Search PRISMA Diagram
12,615 references retrieved through
database searching
6,994 references after duplicates removed
(134 additional duplicates removed)
203 primary eligible qualitative research
87 mixed methods
43 grey
333 potentially relevant to research
43 eligible studies
Title/abstract review (full text review if
necessary) of articles to assess eligibility
[6661 removed] (exclusion category; i.e. not
primary, not sickle cell, etc)
Title/Abstract screening for exclusion criteria
5463 (Quantitative)
249 (Not empirical)
7 (Not English)
108 (Not patient or parent perspective)
53 (Not peer reviewed)
236 (Not primary)
193 (Not sickle cell)
67 (Animal study)
4 (No abstract)
5 (Plant study)
33 (Relevant but not eligible)
109 (Sickle cell is secondary focus)
Total excluded: 6661
Full text screening for relevance to [RQ]
[290 removed] (exclusion category; i.e. not
primary, not sickle cell, etc)
13 included - hand search
of references
14 studies included

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
31
Table 3. Characteristics of Included Studies
Author(s) Title Country Topic Style Participants
Biwott, P.J. et
al (2017)
Disclosure of
Sickle Cell Disease
Results to
Parents/Guardians
Participating In a
Research at a
Hemato-Oncology
Clinic in Eldoret
Kenya
Kenya Screening
of children
Interviews
and
questionnaire
46 parents,
guardians,
health care
providers
Bruce, A.A.,
et al. (2018)
A complex
interface: Exploring
sickle cell disease
from a parent's
perspective, after
moving from Sub-
Saharan Africa to
North America.
Canada Screening
of children
Interviews 12 parents
Chudleigh, J.,
et al. (2016)
Parents'
Experiences of
Receiving the
Initial Positive
Newborn Screening
(NBS) Result for
Cystic Fibrosis and
Sickle Cell Disease
UK Newborn
screening
Interviews 22 parents
Collins, J. L.,
et al. (2013)
Factors that
influence parents'
experiences with
results disclosure
after newborn
screening identifies
genetic carrier
status for cystic
fibrosis or sickle
cell
hemoglobinopathy
USA Newborn
screening
Interview
and
questionnaire
270 parents
Inclusion
Exclusion
English language, full text articles
Published, peer reviewed, research
Primary qualitative empirical research (using any
descriptive or interpretive qualitative methodology,
including the qualitative component of mixed-
methodology)
Studies involving the experiences or perspectives of people
who have sickle cell disease or trait and/or their parents
Studies published after 2006
Not empirical
Not English
Animal Studies
Not patient or parent perspective
Not peer reviewed
Not primary
Not sickle cell
No abstract
Plant Study
Quantitative
Relevant but not eligible
Sickle cell is secondary focus
Studies published before 2006

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
32
Table 3. cont. Characteristics of Included Studies
Author(s) Title Country Topic Style Participants
Holtkamp,
K.C.A, et al.
(2018)
Experiences of a
High-Risk
Population with
Prenatal
Hemoglobinopathy
Carrier Screening
in a Primary Care
Setting: a
Qualitative Study
Netherlands Antenatal
screening
Interviews 26
expectant
women
La Pean, A., et
al. (2012)
A qualitative
secondary
evaluation of
statewide follow-up
interviews for
abnormal newborn
screening results
for cystic fibrosis
and sickle cell
hemoglobinopathy
USA Newborn
screening
Interviews 195 parents
Lebensburger,
J.D., et al.
(2015)
Understanding and
improving health
education among
first-time parents of
infants with sickle
cell anemia in
Alabama: A mixed
methods approach
USA Newborn
screening
Interviews
and
questionnaire
8 parents
Locock, L.
and Kai, J.
(2008)
Parents'
experiences of
universal screening
for haemoglobin
disorders:
implications for
practice in a new
genetics era
UK Antenatal
and
newborn
screening
Interviews
39 parents
Miller, F.A.,
et al. (2010)
Understanding
sickle cell carrier
status identified
through newborn
screening: a
qualitative study
Canada Newborn
screening
Interviews 6 parents, 8
community
based
advocates,
42 health
care
providers

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
33
Table 3. cont. Characteristics of Included Studies
Author(s) Title Country Topic Style Participants
Reed, K.
(2009)
'It's them faulty
genes again':
women, men, and
the gendered nature
of genetic
responsibility in
prenatal blood
screening
UK Antenatal
screening
Interviews 48 parents
Reed, K.
(2011)
'He's the dad isn't
he?' Gender, race
and the politics of
prenatal screening
UK Antenatal
screening
Interviews
and focus
groups
48 parents
Tsianakas, V.,
et al. (2012)
Offering antenatal
sickle cell and
thalassaemia
screening to
pregnant women in
primary care: a
qualitative study of
women's
experiences and
expectations of
participation
UK Antenatal
screening
Interviews 21
expectant
women
Ulph, F., et al.
(2011)
Familial influences
on antenatal and
newborn
haemoglobinopathy
screening
UK Antenatal
and
newborn
screening
Interviews 37 parents
Ulph, F., et al.
(2015)
Parents' responses
to receiving sickle
cell or cystic
fibrosis carrier
results for their
child following
newborn screening
UK Newborn
screening
Interviews 67 parents

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
34
Table 4. Themes as identified by the Authors of the Studies Reviewed
Author Title Themes
Biwott, P.J.
et al (2017)
Disclosure of Sickle Cell Disease
Results to Parents / Guardians
Participating In a Research at a
Hemato-Oncology Clinic in Eldoret
Kenya
• How the parent or guardian found
out about the illness
• Communication process
• The person communicating sickle
cell disease information
• Expectations on the results
• Perception of health care providers
on the disclosure process
• Adequacy of disclosure
• Adequacy of time used during
disclosure
• Feelings after disclosure
• Perception of health care providers
on the adequacy of information
disclosed and psychological impact
• Tests used to screen sickle cell
disease
• Ethics of the disclosure
Bruce, A.A.,
et al. (2018)
A complex interface: Exploring sickle
cell disease from a parent's
perspective, after moving from Sub-
Saharan Africa to North America.
• Parents recalling cases about SCD in
their communities in Africa
• Shock
• Acceptance
• Thinking about the future
• Self-reliance
• Guarded trust
Chudleigh,
J., et al.
(2016)
Parents' Experiences of Receiving the
Initial Positive Newborn Screening
(NBS) Result for Cystic Fibrosis and
Sickle Cell Disease
• Prior knowledge of the condition
(CF and SCD) and NBS
• Receiving the initial positive NBS
result
• Reactions to the positive NBS result
• Impact of the screening process on
parental relationships
• Future support strategies

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
35
Table 4 cont. Themes as identified by the Authors of the Studies Reviewed
Author(s) Title Themes
Collins, J. L., et
al. (2013)
Factors that influence parents'
experiences with results disclosure
after newborn screening identifies
genetic carrier status for cystic
fibrosis or sickle cell
hemoglobinopathy
• Specific content messages that
influenced parents’ experiences
• PCP traits that influenced parents’
experiences and reactions
• Aspects of the setting that
influenced parents
• Positive and negative reactions
reported by parents
• Associations between factors and
reactions
Holtkamp,
K.C.A, et al.
(2018)
Experiences of a High-Risk
Population with Prenatal
Hemoglobinopathy Carrier
Screening in a Primary Care Setting:
a Qualitative Study
• Familiarity with HbP’s and carrier
screening
• HbP carrier screening: reasons to
accept or decline testing
• multistep process of decision
making
• perceived information overload
during counseling
La Pean, A., et
al. (2012)
A qualitative secondary evaluation
of statewide follow-up interviews for
abnormal newborn screening results
for cystic fibrosis and sickle cell
hemoglobinopathy
• Parents’ opinions about the
Project’s follow-up interview
• Reasons why parents found the
Project’s follow-up interview
beneficial
• Parents’ emotional reactions to the
Project’s follow-up interview
Lebensburger,
J.D., et al.
(2015)
Understanding and Improving Health
Education among First-Time Parents
of Infants With Sickle Cell Anemia
in Alabama: A Mixed Methods
Approach
• Parental fear of sickle cell anemia
and trust in sources of health
information
• Trust in sources of health
information
Locock, L. and
Kai, J. (2008)
Parents' experiences of universal
screening for haemoglobin disorders:
implications for practice in a new
genetics era
• Being informed about screening
• Timing of screening tests
• Understanding carrier status and
genetic risk
• Communication of carrier results
and advice about options
• Religious influences on individual
decision making

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
36
Table 4 cont. Themes as identified by the Authors of the Studies Reviewed
Author(s) Title Themes
Miller, FA., et
al. (2010)
Understanding sickle cell carrier
status identified through newborn
screening: A qualitative study
• Uncertainty
• Dissonant claims of clinical
significance
• Health-care providers’ equivocation
Reed, K. (2009) 'It's them faulty genes again':
women, men and the gendered
nature of genetic responsibility in
prenatal blood screening
• Women and ‘embodied’
responsibility
• ‘It’s them faulty genes again’:
women and accountability
Reed, K. (2011) 'He's the dad isn't he?' Gender, race
and the politics of prenatal screening
• ‘He’s the dad isn’t he?’ BME men
and the positive result
• Whiteness, and men’s participation
• Ethnicity, men and screening
professionals
Tsianakas, V.,
et al. (2012)
Offering antenatal sickle cell and
thalassaemia screening to pregnant
women in primary care: a qualitative
study of women's experiences and
expectations of participation
• The perceived benefits of early
screening
• Satisfaction and expectations of
being involved in decision making
• The need for information
Ulph, F., et al.
(2011)
Familial influences on antenatal and
newborn haemoglobinopathy
screening
• Family providing knowledge base
• Family involvement in decisions
• The control of information within
families
• Intergenerational control
• Facilitating the messengers
Ulph, F., et al.
(2015)
Parents' responses to receiving sickle
cell or cystic fibrosis carrier results
for their child following newborn
screening
• Impact of knowing carrier results
• Effect of process of communication
• Considering ‘cascade’ testing
within the family
• Sharing carrier information with
extended family

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
37
Results
Fourteen studies were reviewed for this paper, of which only three focused
exclusively on SCD or SCT (32-34). Of the remaining 11 studies, it was not always
possible to accurately discern whether the participants’ comments related to experiences
with screening for SCD, cystic fibrosis (CF) or thalassemia. By combining all these
conditions, important nuances related to experiences of particular conditions (e.g., impact
of race or stigma of an invisible chronic pain condition) may have been missed by the
original researchers. Given that the current work was a secondary analysis, it was not
possible to re-design the studies and so the current findings are limited by these existing
design features.
From an initial body of 12, 615 references, the full text of 43 articles were
reviewed. Thirteen eligible studies were identified (Figure 1). The reference section of
each of the included studies (n=13) was hand searched. One additional study resulted
from this process, for a total of 14 included studies.
Of the 14 studies analysed for this systematic review and integrative qualitative
meta-synthesis, six focused on antenatal screening for SCT, four studies focused on SCT
diagnosis from the NBS and four studies focused on SCD diagnosis from the NBS. There
were no studies addressing the opportunity for screening before an individual reached
reproductive age, married, or planning to conceive a pregnancy.
These 14 studies were conducted in Africa (n=1), Canada (n=2), the Netherlands
(n=1), the UK (n=7) and the USA (n=3) and used a variety of methodologies including

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
38
grounded theory and focused ethnographies. A total of 876 participants were included
across the studies - 766 women, 108 men and two participants of unspecified gender.
Women constituted 87% of the study population. The participants in the study were
described by the authors as: Black, Black-Caribbean, Black African, White, Asian,
Latino, Middle Eastern, Chinese, and Mixed-race ethnicities. The terms SCD, SCT,
haemoglobinopathy and haemoglobinopathies were used variably in the studies - a
reflection of the topic addressed in different studies – and these terms are reflected in the
results section of this paper.
The UK employs universal antenatal screening of women at risk for SCD or any
haemoglobinopathy, whereas in Canada, the Netherlands and the USA, antenatal
screening is performed at the discretion of the health care provider (26, 27). Conversely,
the UK offers newborn screening as a choice, while eight Canadian provinces and one
territory, the Netherlands and the USA employ universal NBS to detect SCD (26-28). No
formal SCD screening systems exist in Africa, although pilot studies for SCD screening
have been conducted in several African countries (35).
SCD screening of adults serves the purpose of detecting the sickle cell gene
affording prospective parents the opportunity for pre-conception planning, prenatal
diagnosis or termination of a pregnancy should the fetus have SCD or SCT. The primary
purpose of the NBS is the identification of infants who have SCD. SCT detection from
the NBS is an incidental finding (23, 34). The results have been divided into the themes -
screening for SCT antenatally, and SCD and SCT screening post delivery (via the NBS)
to capture the differences in parents’ experiences with SCD screening.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
39
Antenatal Screening for Sickle Cell Disease (Haemoglobinopathy)
Analysis of the studies revealed that experiences of screening for SCD
(haemoglobinopathies) related to six themes: informed choice to screen and reasons to
accept SCD (haemoglobinopathy) screening, awareness and education of SCD or SCT,
timing of SCD (haemoglobinopathy) screening and planning of pregnancy and
termination of pregnancy, responsibility for SCD (haemoglobinopathy) screening, family
influence on SCD (haemoglobinopathy) screening and the role of religion in SCD
(haemoglobinopathy) screening.
Informed Choice to Screen and Reasons to Accept Sickle Cell Disease
(Haemoglobinopathy) Screening
Women’s reasons for accepting screening varied. Most women in the studies
accepted screening when it was offered to them and expressed satisfaction with their
choice to be screened for SCT or a haemoglobinopathy (26, 27, 36). Some women felt
their doctor provided a clear explanation of the purpose for screening and the significance
of inheritance patterns of SCD and offered them time to accept or decline screening (27,
36); others reported feeling poorly informed of screening options and uncertain of the
risks of SCD (36).
Women who expressed dissatisfaction with the information they received, reported
receiving only a pamphlet or leaflet regarding haemoglobinopathies (36). Tsianakas et al
reported that some women wanted more information from their doctor: “At the initial

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
40
consultations with their GPs, women were given a leaflet about SC and T [sickle cell and
thalassemia] to take home and read. However, a number of women explained that in
addition to that, GPs should have spent more time explaining the conditions face-to-face.”
(36, pg. 120).
Some women reported they were not offered a choice to screen or felt they were
encouraged or led by their doctor or midwife to accept screening (26, 36). Other women,
although given the choice to screen, felt incapable of making an educated decision, opting
instead to defer to the doctor or midwife “who is the expert” (26, 27, 36). These women
expressed complete trust in the health care provider and deemed them to be more
knowledgeable about SCD.
The ease of the test - one extra blood test added to the standard tests done at the
initial doctor or midwifery antenatal visit - was the impetus for a few women to accept
screening (26). Holtkamp et al found that: “Women argued that screening is performed
simultaneously with other blood tests, and thus entails only one extra sample of blood.”
(26 pg. 639). Along this same line of reasoning, some of the women who agreed to be
screened never gave thought to the consequences of having a positive screen for SCT
(26).
Women for whom English was a second language expressed dissatisfaction,
confusion or misunderstanding with the screening process. One woman’s request for an
interpreter went unfulfilled while another woman complained of lack of educational
literature in her native language (36).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
41
Men constituted a small proportion of study participants. Reed reported that many
of the men of colour in her study were familiar with SCD or haemoglobinopathies, all of
whom accepted antenatal screening (38). Only half of the White men accepted screening.
Some of the White men who declined to be screened were surprised they were offered
screening, because they did not think they, as White men, were at risk for a
haemoglobinopathy (38). Holtkamp et al also reported that some of the White men in
their study did not expect the need to be screened (26).
Awareness and Education of Sickle Cell Disease or Sickle Cell Trait
Knowledge or lack thereof and understanding/misunderstanding of SCD risk
factors, as well as related health concerns of the disease were inextricably linked to
women’s choices to accept or decline screening for SCD (haemoglobinopathies). Some
women were aware of SCD or haemoglobinopathies, but they did not understand the
hereditary nature of the disease when they accepted SCD (haemoglobinopathy) screening.
Others, who were aware of SCD and its hereditary consequences, still declined antenatal
screening on the premise the test results would only provide information of their own
sickle cell carrier status and that “carrier screening would not provide certainty about the
condition of their unborn child.” (26, pg. 640). These women deferred to the NBS of the
baby instead (26, 27).
With respect to risk factors, some women thought, mistakenly, that their advanced
age was a risk factor for SCD (haemoglobinopathy) (36).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
42
Several women reported they were unfamiliar with SCD or haemoglobinopathies.
Other women suggested they were more aware of Zika disease than SCD suggesting the
healthcare system did not provide enough information about haemoglobinopathies (26).
A few White women were surprised to be offered antenatal screening for SCD
(haemoglobinopathy) (26).
Only a few women declined antenatal screening for SCD (haemoglobinopathy).
Holtkamp et al found that women used lack of family history of a haemoglobinopathy as
a reason to decline screening: “One woman argued that there was no need for
haemoglobinopathy carrier screening as haemoglobinopathies did not run in her family
and that she already has healthy children” (26, pg. 639). Other women chose to decline
screening on the belief they were healthy (26, 27) and other women declined screening on
the basis that their partners had been screened previously and were told that they did not
have a haemoglobinopathy (36).
Timing of Sickle Cell Disease (Haemoglobinopathy) Screening, Planning of Pregnancy
and Termination of Pregnancy
Women overall were happy with the opportunity to be screened, however many
questioned the timing of screening.
Some women reported feeling overwhelmed when SCD (haemoglobinopathy)
screening was offered during the initial antenatal visit for a pregnancy. They reported too

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
43
much information was provided at one time making it difficult to process screening
options and make a decision at one visit (26). Many of these women were either
unfamiliar with SCD or had not appreciated the hereditary risks or aspects of SCD
(haemoglobinopathies) (26). Women offered suggestions of a separate appointment time
to discuss SCD (haemoglobinopathy) screening as a solution to minimize excessive
information at the first antenatal visit. Yet other women felt capable to process the
information provided to them at the initial antenatal visit for that pregnancy (26, 27).
Tsianakas et al found that some women believed waiting to offer SCD
(haemoglobinopathy) screening during the pregnancy was not ideal: “Some women said
SC&T [sickle cell and thalassemia screening] should be done pre-conceptually in order
for parents to be aware of their carrier status before conceiving.” (36, pg. 119). Many
women suggested that SCD (haemoglobinopathy) screening should be offered pre-
maritally, preconception, prenatally or as early as possible in pregnancy to afford women
or couples the time to make decisions about marriage partners, conception, prenatal
diagnosis or termination of a pregnancy (26, 27, 36).
Responsibility for Sickle Cell Disease (Haemoglobinopathy) Screening
Women reported, in two studies, they felt they bore the majority of the burden of
the choice to screen (36, 37) since “the screening was happening within their body” (37,
pg. 350). They reported feeling a connection to the baby and responsibility for the baby’s
well being (36, 37).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
44
Some of the women who were involved in relationships with men who were at risk
for SCD (haemoglobinopathy), found it difficult to persuade their partner to be screened
(27). Locock et al found that: “Some men felt sure they were not carriers because they
were fit and healthy, and told their partners they did not need to be screened.” (27, pg.
164). A complicating factor of partner screening was hesitancy related to fetal paternity
(27); other women declined partner screening on the basis their partner had previous
carrier screening (26).
The men who participated in screening for SCD articulated an increased sense of
responsibility for the well being of their baby as the reason they chose to screen (38).
Family Influence on Sickle Cell Disease (Haemoglobinopathy) Screening
One study focused on the role of family members in the decision process of
antenatal screening (39). One woman deferred decision making to her family stating that
her partner, family and friends discouraged her from screening for SCD (39).
Other women opted to control or limit the influence their family had on screening
by selectively choosing which aspects of the screening process to share with their family.
Some women chose not to disclose they had learned they were at risk for SCD, while
others chose not to disclose they had been screened (39). Many “participants felt that
when information was conveyed to family so was a right to be involved.” (39, pg. 367).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
45
Role of Religion in Sickle Cell Disease (Haemoglobinopathy) Screening
Women of strong Christian and Muslim faiths framed reasons to decline or accept
screening around their belief in God. Some women believed that their willingness or need
to accept any child precluded their ability to accept screening. They “described a sense of
resignation to God’s plan.” (27, pg. 165)
On the contrary, other women described the belief that early antenatal screening and
detection could allow for prenatal diagnosis of the baby. Some women of both Christian
and Muslim faiths stated that termination of an affected baby was a possibility provided it
occurred early in the pregnancy (27).
Sickle Cell Trait Diagnosis
Some studies differentiated between screening and diagnosis of SCT. SCT is not a
disease. It results from the inheritance of one normal haemoglobin gene and one sickle
cell gene. Most people with SCT will have no symptoms and may be unaware of they
carry the sickle cell gene (1-3).
Analysis of this group of studies revealed that experiences of disclosure of SCT
results related to seven themes: awareness and education of SCT, disclosure of SCT
results, communication of SCT results, emotional response to SCT diagnosis, sharing of
SCT results and family support, paternity, and cascade testing. Two studies focused on

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
46
antenatal SCT (haemoglobinopathy) screening and four studies focused on newborn
screening.
Awareness and Education of Sickle Cell Trait
Antenatal screening
Women who screened positive for SCT in pregnancy needed to make follow-up
decisions. For one woman the decision was simple as her partner willingly chose to be
screened (38). Women in another study were unprepared for a positive SCT results
despite willingly accepting antenatal screening for SCD. They were unsure of the
consequence of SCT for them or their unborn baby (36).
Newborn Screening
Several studies described the existence of SCT screening in population based
newborn screening (17, 18, 34, 40). In these studies, it was common for parents to be
unaware of their own sickle cell carrier status prior to the NBS.
The vast majority of families in the studies reviewed were unfamiliar with SCT and
all wanted to learn more about sickle cell carrier status, in particular, information on the
impact sickle cell carrier status would have on the health of their baby (17, 18, 27, 34).
Some parents reported they were not aware their child had been screened for SCD or they
reported they did not understand the purpose of the NBS.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
47
Often parents wanted more information on the inheritance pattern of SCT, in
particular, they wanted to know which parent carried the sickle cell gene (18). This was
particularly relevant to parents who did not know they were at risk for SCT, such as a
White couple who were confused by the SCT results of their baby (27).
Some parents misunderstood the role haemoglobin or blood played in SCT. Many
reported hearing that their child had a problem associated with their blood. Parents
interpreted this to mean their baby had bad blood (27).
Disclosure of Sickle Cell Trait Results
Disclosure of SCT detected by newborn screening differed by jurisdiction. SCT
diagnosis is habitually disclosed to parents in the UK and the USA, but not every
province or territory in Canada (17, 18, 27, 34).
All but one parent reported gratitude for knowledge of their child’s sickle cell
carrier status (18, 34, 39). Some parents found identification of sickle cell carrier status a
helpful tool to plan for the child’s future. Parents who did not receive disclosure of sickle
cell carrier status, argued for continued disclosure of sickle cell carrier status (34).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
48
Communication of Sickle Cell Trait Results
Parents received SCT (carrier) results by letter, phone call or in person. A few
parents reported they had not received any results and others had no recollection of being
informed that their baby was being screened for SCD (27).
Almost all parents preferred to receive positive SCT results in person, by a
practitioner who had knowledge of SCD and SCT, and preferably known to them (18, 27,
34). They emphasized the importance of being able to speak directly with a health care
provider about the meaning and implication of results and for this reason they preferred
not to receive results via the telephone, or voicemail and on Fridays. They reported
messages left on a Friday provided little opportunity to speak with a health care provider
before the week ended. Some parents reported receiving a letter alerting them of the need
to call their doctor for disclosure of the SCT test results. None of these parents reported
liking this method of results disclosure (27).
Parents reported they had greater understanding of SCT when health care providers
were patient, used non-medical terms and took time to explain in detail SCT and the
implications of sickle cell carrier status. Several parents also reported additional written
material enhanced their comprehension of SCT (18, 39).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
49
Emotional Response to Sickle Cell Trait Diagnosis
Parents experienced a range of emotions when first learning of their child’s SCT
diagnosis. The detail provided to parents and the manner in which their health care
provider delivered test results contributed to a parent’s positive or negative emotional
state (40). Even parents who reported good understanding of SCT or parents who had
personal experience of SCT experienced negative emotional response upon learning their
child had SCT (34); emotional responses included shock, surprise, sadness, and anxiety
(17, 39). Many parents reported feeling scared and worried for their child’s future and
health; this was the case even if parents were made aware that SCT is considered benign
(39). Other parents reported feeling reassured, calm, relieved, confident, and comfortable
after their health care provider carefully explained the details of SCT, which emphasizes
the important role health care providers have in providing information to contextualize
test results (27).
Sharing of Sickle Cell Trait Results and Family Support
Parents’ decision to share the SCT results for their baby seemed to be linked to their
own experience with SCD or family history of SCD (39). Families for whom SCD or
SCT had evoked negative experiences, criticism, or pity, seemed less willing to openly
share their child’s sickle cell carrier status (39). This pattern was more common in
families of African descent or African immigrants. Parents reported a desire to avoid
negative comments, pity, or shunning behaviour (39).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
50
Some families found that SCT information was too difficult to explain to family
members (39). Others preferred to remain secretive and one woman was very upset when
she learned that her SCT results had been shared with her sister without her consent (39).
The need for secrecy was not expressed by all parents. Some parents believed it
important to share the SCT results while others had no concern regarding informing
family members of their child’s results (27, 40). Families who had positive experiences
with sharing the results of their baby’s screen were also more likely to receive emotional
family support (39). Sometimes the parents turned to their families for support when they
first received the SCT results (39).
Paternity
It is not uncommon for many people to be unaware of their own sickle cell carrier
status and issues of paternity were more common when the SCT diagnosis was
unexpected. A White mother expressed gratitude that her White husband did not question
the paternity of their child upon learning that her child had SCT. She stated she associated
SCT as a disease that affects the Black race (27).
Cascade Testing
Upon learning of their child’s sickle cell carrier status, some parents expressed a
desire to know their own sickle cell carrier status and that of their other children (40).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
51
This pattern of one diagnosis leading to many other tests is referred by Ulph et al as
“cascade testing” (40). Some parents expressed frustration and worry when they were
unable to have immediate family members tested. Yet other families reported that conflict
arose between families when other relatives chose not to pursue their own sickle cell
carrier status (40).
Sickle Cell Disease Diagnosis
Some studies focused on the diagnosis of SCD, rather than SCT (32, 33, 41, 42).
Analysis of the studies revealed that experiences of disclosure of SCD results related to
six themes: awareness and education of SCD, communication of SCD results, emotional
response to SCD diagnosis, child’s future, implication for future pregnancy planning and
parental relationship, and sharing SCD diagnosis and support. In North America children
who have SCD are most likely to be identified at birth. It is not uncommon, however, for
many African children to be identified in early childhood. Two studies were based on
NBS results, one study was based on SCD screening of children in Kenya, and one study
was based on SCD screening of children who had immigrated to Canada from sub-
Saharan African countries.
Awareness and Education of Sickle Cell Disease
Many of the parents who lived in African or had recently immigrated to Canada
from Africa had witnessed children with symptoms characteristic of SCD. It is not

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
52
uncommon for children living in Africa to be misdiagnosed with malaria. Most of these
parents did not anticipate a positive SCD diagnosis (32, 33). Some parents reported they
were not aware their child had been screened or reported they did not understand the
purpose of the NBS.
Many of the parents knew of family members or friends who had SCD. However, a
few parents had no prior experience with SCD (41, 42). Most parents were unaware of
their own sickle cell carrier status (32, 33).
All the parents expressed a desire to learn more information about SCD and the
inheritance pattern of SCD. Some parents reported their health care provider was very
informative of SCD and provided additional literature which they found helpful (42).
Other parents reported relying on the internet, books or pamphlets to fill the gap left by
health care providers (42).
Communication of Sickle Cell Disease Results
Many of the parents received results in person by a doctor or health care provider
(32, 33), while some parents received a letter informing them a health care provider
would deliver the newborn screen results in person at their home. Parents reported a
preference for in person disclosure of SCD diagnosis.
Parents also reported that some doctors or health care providers appeared rushed,
had too many patients or had busy clinics that did not afford the time required to discuss

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
53
the SCD results in detail (32). The parents in this group did not feel they had the
opportunity to ask questions or learn enough about SCD. Some of these parents reported
relying on family, friends or the internet to learn more about SCD when they perceived
the doctors did not answer their questions (42).
Other parents also revealed lack of privacy or confidentiality during the disclosure
process (32).
Emotional Response to Sickle Cell Disease Diagnosis
Almost all parents expressed profound reactions to the SCD diagnosis. Most parents
expressed surprise and shock despite witnessing many unwell children in their hometown
or village (32, 33). The results were at variance with their expectations (32).
Emotions expressed by many parents ranged from shock, disbelief, sadness, anger,
and devastation to fear, guilt, blame and regret (32, 33, 41, 42). Many parents expressed
difficulty reconciling their child’s diagnosis and some expressed regret for having a child
(41).
Child’s Future
All parents reported feeling consumed with worry for their child’s future. Many
wanted to know how to recognize when their child was ill or in pain, other parents
worried they would not know how to take care of their child and many were fearful their

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
54
child would die young (32, 33, 41, 42). Parents also expressed concern their child would
not find a partner as an adult.
Parents who had family members with SCD reflected on their memories of
relatives’ experiences of being labeled a drug addict or drug seeker when they sought
morphine treatment to manage painful sickle cell crises (32).
Implication for Future Pregnancy Planning and Parental Relationships
Parents expressed regret for not knowing their own sickle cell carrier status. They
reflected that prior knowledge (pre-marital or preconception) of their sickle cell carrier
status would likely have influenced their choice of partner or provided more options for
pregnancy planning and prenatal diagnosis (32, 41, 42).
Other parents contemplated the option of not staying with their partner or choosing
not to have more children (33).
Sharing Sickle Cell Disease Diagnosis and Support
Parents often chose not to disclose their child’s SCD diagnosis to family or friends.
Many parents expressed fear their child would be treated differently than other children
(33, 41). They also anticipated being shunned or pitied. Parents stated they did not want
friends and family speaking poorly of them in their absence. Many parents desired for
their children to be treated the same as children who did not have SCD (33, 41).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
55
Parents’ choice to remain secretive regarding their child’s diagnosis ultimately
created a feeling of isolation and lack of support (32). Some parents reported that they
trusted only their health care provider and one or two close family members (32).
It is clear from the results that study participants would have benefited from prior
knowledge of their own sickle cell carrier status.
Discussion
This systematic review and integrative qualitative meta-synthesis described the
perspective of parents’ experiences with SCD screening and diagnosis. The predominant
themes from the 14 studies reviewed were; awareness and education of SCD or SCT,
informed choice, communication between health care provider and parents, emotional
impact of a positive diagnosis of SCD or SCT, and stigma and shame.
Across all of the studies reviewed, it was apparent that many of the parents were
unfamiliar with SCD or SCT, and of the parents who knew of SCD or SCT, only a
handful were aware of their own sickle cell carrier status. Futhermore, there was general
misunderstanding of the health implications of SCD and SCT or that SCD and SCT are
inherited, not communicable diseases (26, 27, 36, 40, 41, 42). While most parents
accepted the offer to be screened for SCD or SCT, it was evident that not all parents
perceived they had a choice to decline screening (27). Additionally, of the parents whose
infants screened positive for SCD or SCT from the NBS, most failed to recall any
discussion or offer of the NBS (26, 27, 36, 40, 41, 42).

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
56
Communication between health care providers and parents was a very important
theme in all fourteen studies during the antenatal SCD screening process or disclosure of
positive SCD or SCT results. Consistently in all of the studies, parents who perceived
their health care provider as informative, thorough and clear, either during the screening
process or disclosure of positive SCD or SCT results, overall reported more positive
experiences (17, 18, 26, 27, 41, 42). Conversly parents who perceived their health care
provider as uninformative, or unclear or not helpful during the SCD screening process
and disclosure of results, reported more negative experiences, particularly when screening
results were positive for SCD or SCT (17, 18, 26, 27, 41, 42).
Almost all of the parents of children who were diagnosed with SCD, reported
experiencing a range of negative emotions ranging from disbelief to guilt to despair (32,
33, 41, 42). For many parents, the manner in which positive results were disclosed was
imporant, with parents overwhelming preferring to be informed in person or by telephone
rather than by letter (18, 26, 32, 42) .
Many of the parents of children diagnosed with SCD reported a need or desire to
hide their child’s positive SCD results from their family for fear of being shunned, or
treated differently by their family.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
57
Interpretation of Results
Lived Experience of People with Sickle Cell Disease
The studies reviewed in this paper captured parents’ experiences with the screening
process for SCD. An appreciation of the impact of SCD on affected individuals and their
caregivers and family is important. SCD is a chronic, lifelong and often debilitating
disease. Children living with SCD in high income countries have many appointments
with family doctors, SCD clinics (where available) and often have high absenteeism from
school. Parents or family caregivers often are required to be absent from work or need to
change their career to facilitate the care of a child affected with SCD (5). The lived
experience of a young man with SCD and a parent of young man with SCD are captured
on the Centers for Disease Control and Prevention website:
One young man named Lance, reflected on his experience living with SCD:
“The news altered my family dynamic in a major way. My mom, a nurse at the
time, lost her job because she needed to stay home and take care of me. Her
career goals were gone. My parents also had a separation period because of the
hardships.” (43)
Lance also shared his relationship with his siblings:
“Every time I got sick and had to go to the hospital, they [my sister and brother]
went to our grandmother’s house. To them, I received special treatment because
I needed it and they got less time and attention from my parents. To me, my
parents’ focus was always on keeping me healthy, not on my goals and dreams.
My parents always saw me as the sick child. But my siblings, they let them
spread their wings.” (43)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
58
A parent of a child affected with SCD shared her views on the manner in which
SCD impacted her life and that of her child:
“I finally met a man who was on track to be older than the age [doctors] gave me
for how long my son would live. When he talked to us, he didn’t talk to us from
a sense of hopelessness. He talked to us about living a life, enjoying life, and the
reality that we would have challenges, but how we view those challenges could
not take away from the life that we were still meant to live. It was a level of
hope that I had not allowed myself to have up to that point. Up until then, all I
thought about was that sickle cell was a disease that was going to kill my child,
and it was devastating. That encounter changed the course of my life and my
son’s life. I truly believe that my son is still alive today because of that
conversation.” (42)
These testaments highlight the psychological impact that SCD has on individuals
affected with SCD and their families and the value and importance of SCD screening. In
essence, SCD is a preventable disease as tools exist to detect carriers of SCT. The
incidence of SCD could be reduced or eliminated by the intentional choice of a life
partner or donor sperm that does not carry the sickle cell gene, or parents’ choice not to
procreate, or the option of prenatal diagnosis and/or pre-implantation testing (available in
high income countries) (46).
Informed Choice
The screening process for SCD in most countries is optional, whether offered
universally or at the discretion of a health care provider, prospective parents have the
choice to opt in or opt out of the screening process. The health care provider is
responsible for offering the opportunity to be screened. Informed choice is the

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
59
cornerstone of the screening process. The terms informed consent and informed choice
are often used interchangeably, even in medical literature, but they are fundamentally two
different processes. Informed consent is the choice to accept the test(s) that have been
recommended by a clinician. Informed choice involves a deliberate decision based on
relevant knowledge and values (44). The clinician does not seek to recommend or
encourage any particular course of action, and counsels with the aim of providing
information and facilitating an autonomous choice from the patient (45). Informed choice
is the foundation of prenatal screening in Canada and many countries (45). Martineau et
al describe informed choice as:
“..an informed choice to undergo screening occurs when an individual has a
positive attitude towards undergoing a test, and has relevant knowledge about
the test and undergoes it. An informed choice to decline a test occurs when an
individual holds a negative attitude towards undergoing the test, has relevant
knowledge about the test and does not undergo it.” (44)
Within the context of this review, the health care providers were responsible for not
only offering SCD screening (either antenatally or post delivery through the newborn
screening process) but were also responsible to ensure parents fully comprehended the
purpose of the SCD screen, and the subsequent consequences and follow up options
should they or their baby screen positive for SCD or SCT. Thus, in order for women or
parents to make an informed decision to accept or decline SCD, do they need to fully
understand SCD?
The challenge is that unlike other routine tests offered during pregnancy, for
example, diabetes screening, blood tests or screening for Down Syndrome, that may be

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
60
familiar to women, either from experience with a previous pregnancy or through
experiences of friends and family, SCD is not well known or understood (20, 21). This
was true for many of the women in the studies reviewed, particularly for women who
were not Black, who reported they were not familiar with SCD or haemoglobinopathy
screening (26, 36). Many women accepted the offer of antenatal SCD screening but
stated they did not fully understand the purpose of the SCD screen, while others reported
that the antenatal SCD screen was not presented as a choice or they perceived their health
care provider wanted them to screen for SCD, and consequently agreed to be screened
(26, 27, 36). Essentially, these women exercised informed consent rather than informed
choice (44).
Of the parents whose infant was diagnosed with SCD or SCT from the NBS, most
could not recall being informed or offered the NBS. This was true whether the NBS was
offered as a choice (opt-in program), as in the UK, or performed routinely (opt-out
program) as in Canada and the USA (17, 26, 27, 41, 42).
Brown et al propose that women make informed choices to accept or decline
antenatal SCT screening when they are offered screening at appropriate time during their
pregnancy, understand the screen and the consequences of the screen (46). Conversely,
women make an uninformed choice to accept or decline an antenatal SCD screen when
the test is offered at an appropriate time during pregnancy, but they may not understand
the screen or the consequences of not screening (46). Some of the participants in the
studies reviewed, expressed regret for accepting the SCD screen once they learned they
screened positive for SCT, as they had not contemplated the need for follow up

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
61
testing (26). While other women’s choice to decline SCT could be interpreted as an
uninformed choice, particularly when they stated that “sickle cell disease doesn’t run in
my family” or “I don’t feel as though I have sickle cell disease” (26, pg. 639).
The question remains when screening tests are offered for health conditions for
which most patients will be unfamiliar or are misunderstood, who bears the responsibility
of educating patients so that they may make an informed decision rather than uninformed
decision to accept or decline the screen?
Role of the Health Care Provider and the Responsibility of Education System
Role of the Health Care Provider
Family doctors, nurse practitioners and midwives are well positioned to offer
antenatal SCD screening to pregnant women, as they will often see pregnant women
within the first trimester of pregnancy. Conducting a SCD screen in the first trimester
affords women who screen positive for SCT more reproductive options, including
prenatal diagnosis, partner screening and early termination of their pregnancy should the
fetus screen positive for SCD (26). However, in my experience as a midwife, the majority
of my clients who are at risk for SCT, enter care unaware of their sickle cell carrier status
and they often report they have little or no knowledge about SCD. Literature suggests
that health care providers may have a basic knowledge of SCD but many of them do not
believe they have sufficient knowledge to counsel patients effectively on SCD, SCT or
the genetic inheritance pattern of the disease (47-50). In order to facilitate informed

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
62
choice for antenatal SCD screening, health care providers need to be equipped to explain
SCD to their patients and answer relevant questions about SCD (49). In the studies
reviewed, some parents expressed they felt that their health care providers offered good
counsel on SCD, while others reported feeling confused and unsure of the value of SCD
screening (26, 27, 36).
Role of the Health Care Education System
Given that SCD affects millions of people worldwide and causes recurrent
morbidity, shortened life span and negatively impacts the quality of life of affected
people, do health care education systems have an obligation to arm practitioners with the
knowledgeable and capability to educate, inform and screen their patients? I argue that
every health care provider who treats patients should be able to counsel their own
patients. Most medical school, nursing and midwifery curricula fall short on teaching
students about SCD (51). SCD modules may be offered in post graduate medical
haematology residency programmes with focus on disease presentation and management,
but a more generalist approach is required, to ensure that all health care providers
understand the epidemiology, presentation, and treatment of SCD.
Vaso-occlusive crises (pain crises) are often characterized by periods of
excruciating pain that may persist for several weeks (52-54). Morphine and/or fentanyl
are the standard drugs used to manage the pain crisis. It is very well documented that
physicians and nurses are more likely to doubt SCD patients’ claims of pain, leading

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
63
many patients to be under medicated, labeled as drug addicts and stigmatized (52-54).
There is evidence that the stigmatizing medical language such as “narcotic dependant”,
“frequent flyer in the ER department” “insisting their pain is still a 10” is frequently
written in SCD patients’ medical charts (53). Comments such as these, when read by and
verbally relayed to health care providers, can lead to them to acquire implicit bias toward
SCD patients (53, 54). It behooves the medical and nursing academia to make more
effort to reduce and eliminate racist, harmful and stigmatizing language.
SCD may be addressed briefly and at a basic level in midwifery education
programme, in large part because midwives do not care for sickle cell patients, however,
midwives still have the capacity and responsibility, at the very least, to ensure clients
understand SCD when offering SCD screening.
Individual Screening versus Population Based Screening for Sickle Cell Disease
Antenatal Screening for Sickle Cell Disease
In the UK SCD screening is offered universally to women who are a higher risk for
SCD, that is, women of Black African, South Asian, Mediterranean or Middle Eastern
descent. In Canada and the USA, antenatal SCD screening is offered as individual
screening (26, 27). In Canada and the USA, each couple or woman is screened based on
criteria commonly associated with SCD – ethnicity of the parents and values of routine
blood work (mean corpuscular volume) (55, 56). Individual screening aims to offer
autonomy to the patient and reduce the ethical dilemma that can occur with population

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
64
based screened (57). However, individual screening for SCD leads to the potential of not
identifying people who may carry the SCT.
Population Based Screening for Sickle Cell Disease
Screening all people for SCD would aim to identify people who carry the sickle cell
gene irrespective of ethnicity, thus affording more reproductive options, such as choosing
not to conceive, choosing a partner who does not have SCT or engage in prenatal
diagnosis (54, 57). In the context of the studies reviewed for this paper, all the women in
the studies conducted in the UK and the Netherlands were offered antenatal screening,
which functioned as a form of population based screening (26, 36, 37-39). Universal
newborn screening in the USA functions as a population-based screening program as all
infants in the USA are screened for SCD (unless a parent declines the NBS). All infants
who carry the sickle cell gene are identified, however, there remained a challenge with
communication of SCT results in the studies reviewed (17, 18, 40).
The universal newborn screening program in Canada falls short of functioning as
population based screening for SCD on two fronts; 1) SCD is not included in the NBS in
all provinces or territories and 2) only four of the provinces and one territory that include
SCD in the NBS, disclose SCT results to parents. Parents in Ontario have the option to
request their child’s NBS results. It is unclear how many parents and primary care health
care providers are aware of this option or how many parents request their child’s results.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
65
It is important to understand that a positive SCT result from a NBS indicates at least
one parent must carry the sickle cell gene (Diagram 2) (1-3). Considering that most
people are unaware of their sickle cell carrier status, and most health care providers are
not screening their patients for SCD (18, 19, 21, 26), I find it difficult to reconcile how a
government institution can justify wilfully with holding medical information that has the
potential to upend a parent’s future. One can argue that the SCT results can be released
upon request, but most parents and health care providers are unaware of the option to
request SCT results (56).
Racism in Health Care and Research
Comparing SCD education, research, treatment and screening to those for genetic
conditions with comparable consequences and hereditary patterns reveals some stark
inequities.
Many people affected with sickle cell anaemia report they feel they are treated
differently, judged and stigmatized, in particular when seeking pain relief during a pain
crisis (51).
The term racism is often difficult for many people to accept. It often conjures
images of slavery, name calling, hatred or prejudice most commonly depicted as
unjustified acts committed by the majority race toward a minority race (58). However, it
is even more difficult to accept the term racism when paired with the term healthcare –
health care providers are expected to take care of all people equally. Yet numerous

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
66
studies have confirmed that racism is not only alive a well in healthcare, but it is
institutionalized (59-61). Jones defines institutional racism as “differential access to the
goods, services and opportunities of society by race” (58). Racism in healthcare should
not be seen as the blatantly obvious, segregating actions practiced in the 1950s and 1960s,
but in the manner in which people are presented or not represented in medicine. Terms
such as ‘pink’, and ‘blue’ are still used in medical textbooks today to describe skin tone.
In the context of the studies reviewed for this thesis, I acknowledge that it may be
more difficult to align racism with the screening process for SCD since many of the acts
attributed to racism against people with SCD are missing – no denial of anaelgeisc drugs,
no reference to the study participants as drug addicts or drug seeking and the study
participants ethnically diverse. However, it is problematic that although the World Health
Organization (WHO) declared in 2006 that SCD is a significant global public health
concern and recommended the implementation of counselling, screening and raising
public awareness of SCD (62), parents and some health care providers in the studies
reviewed still reported having little knowledge and understanding of SCD (26).
Furthermore, although the American College of Gynecologists (55) and the Society of
Obstetricians and Gynecologists of Canada (56) recommend offering SCD screening in
early pregnancy to women and their partners who are at risk for SCD, many obstetricians
fail to meet or follow these guidelines (63). The lack of parental and health care provider
knowledge of SCD represent significant barriers to care (64), potentially leading to
missed opportunities for parents or prospective parents to be screened prior to pregnancy,
ultimately limiting parents’ reproductive options (63). Little to no public awareness of

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
67
SCD and limited or brief education modules on SCD for health care providers contribute
to the knowledge deficit of SCD (47-49, 65).
Dr. James Herrick first identified the peculiar, sickled shape red blood cells of his
medical student in 1910 (1). A century later, much progress has been made from
identifying the gene for many haemoglobin variants and development of medications to
improved management of SCD. Yet in comparison to the progress made in the
management and screening for Tay-Sachs disease and CF, SCD has a long way to go. In
the USA disease-specific drug development has favoured CF (four versus one drug
approval for CF and SCD respectively) (52).
Tay-Sachs disease is a rare, lethal, neurodegenerative, autosomal recessive disease
that is prevalent in people of Ashkenazi Jewish descent (66). A voluntary population
based screening program for Tay-Sachs disease began in the 1970’s and to date more than
1.4 million people have been screened for Tay-Sachs disease worldwide (66). The
incidence rate of Tay-Sachs was reduced by more than 90% by 2000 (66). Montreal
implemented a high school based voluntary population screening program for Tay-Sachs
disease from 1973 to 1993, resulting in a reduction rate of the disease by 90%-95% (67).
Tay-Sachs disease is included in the newborn screening program nationally in Canada.
CF is a rare, autosomal recessive disease most prevalent in people of Northern
European ancestry, affecting the respiratory and digestive system (68). Voluntary
population based screening has been offered for CF since 1989 in many countries,
including Canada. CF disease is included in the newborn screening program nationally in

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
68
Canada. Additionally, CF is offered as a pre-conception screening in all provinces in
Canada (68).
There are no population based screening programs for SCD that can compare with
population based screening programs for Tay-Sachs disease or CF. In fact, a search of
most medical databases for the term ‘population based screening and sickle cell disease’
produces no specific articles. The research in SCD is still focused on disease
management, treatment and screening. A study by Farooq et al, found that more federal
funding was provided for CF as compared with SCD in the USA (52).
The SCD birth rate worldwide is estimated at more than 300,000 annually with
numbers projected to be 400,000 annually by 2050 (69). Conversely, the incidence CF is
declining worldwide and the survival rate of CF patients is increasing to a median
lifespan of 50 years (70). Early diagnosis of CF by the newborn screen, genetic
counselling for prospective parents, prenatal diagnosis and new advances in medications,
and treatment of CF, account for significant improvements in the management of CF (70).
SCD is estimated to affect 3,000 – 5,000 people in Canada (4) in comparison to 4,000
people affected with CF (70). It is difficult not to compare the SCD statistics with CF
statistics and question whether similar, nationally based strategies for SCD, such as
population based screening, national universal newborn screening for SCD and national
universal disclosure for SCT could positively impact the course of SCD in Canada.
At the writing of this thesis, a very well known and popular National Hockey
League player donned custom hockey skates and a stick bearing the SickKids’ logo, in an

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
69
effort to raise public awareness and money for CF and CF research. A description of CF
disease was featured prominently on several social media platforms. I could find no
similar public awareness campaigns for SCD current or past.
It is problematic that SCD receives far less focus and funding despite the burden of
the disease on patients, their families and the health care system. It is worthy to note that
SCD and SCT is the most common genetic condition identified in the newborn screening
program (71).
Reflexivity
Personal Experience of a Midwife
I have practiced midwifery for 17 years, during which time I have routinely offered
antenatal haemoglobinopathy screening, which encompasses screening for SCD and
thalassemia, to all my clients. I have also encouraged all my colleagues to follow suit. I
genuinely believed I was well informed on SCD – I understood the inheritance pattern of
SCD, and I knew that SCD and SCT trait was prevalent amongst people of Black African
descent (1). I prided myself in delving deeper and taking the time to elucidate the ethnic
background of all my clients, even when they appeared White.
Through the process of conducting this thesis, reviewing the studies for this thesis,
and being guided by a haematologist, one of my committee members, I realise that my
knowledge of SCD was inadequate. I did not know that people of Italian, Greek, South

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
70
Asian, and Middle Eastern descent, are at risk for SCD (1-4). I believed they were only at
risk for thalassemia. I also did not know that an incidental finding of SCT on the NBS is
only routinely disclosed to health care providers in some provinces in Canada (Table 7),
and therefore I was not aware that parents can request the SCT results from the NBS (71).
I am very embarrassed.
As I read the studies for this thesis, I recognized that I may not have offered SCD
and newborn screening as an informed choice discussion to my clients. I now recognize
my own failure to participate in informed choice discussions about SCD screening (44-
48). I should clarify that informed choice discussion is one of the three tenants of
midwifery care – of all health care providers surely, I should excel at informed choice
discussion? For all other tests that I offer in the antenatal period I do excel at conducting
an informed choice discussion (44, 45).
The problem is that my own strongly held beliefs, as a Black midwife, that all
people at risk should know their sickle cell carrier status, and my own personal
experience of being screened for SCD as an adult, have clouded my perspective. I have
been as guilty as some of the health care providers in the studies in this review. I did not
always explain the consequences of a positive SCT result – further follow-up testing for
the father of the baby, potential referral to a Prenatal Diagnostic Clinic in Hamilton. I
also did not thoroughly review the NBS with my clients during the antenatal period.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
71
I have since changed my habits – I endeavour to engage in an informed choice
discussion for antenatal screening for SCD and NBS. I am still trying to reconcile my
own knowledge deficit of SCD.
Experience of a Black Nurse
My adult son works is a registered nurse at a hospital. He relayed to me his
experience with one of his patients - a young, Black man, age 20, who has SCD. He
reported to me that his patient was in the hospital for pain crisis that required
management by intravenous hydromorphone and fentanyl. It was not uncommon for his
patient to be hospitalized for a period of four weeks, during which time he always
receives opioid medication for pain relief. Two things stood out. My son reported that
none of his White, nurse colleagues believed the severity of this Black patient’s reports of
pain. The nurses allegedly assumed that his ability to ambulate in his room or to the
bathroom, was proof that his pain was not severe. They all believed he was drug seeking
and used the hospital to access opioid drugs. Unfortunately, intravenous opioids, are the
only drug of choice to manage sickle cell pain crisis. My son is unaware whether his
patient has been offered palliative care as an option to manage SCD.
This young, Black patient reported he was thrilled to have a Black, male nurse, a
first experience for him. The patient reportedly stated that none of the nurses nor
physicians believed the severity of his pain, and he reportedly expressed no one

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
72
comprehends his experience with pain crisis or SCD. My son’s patient’s experience
highlights the discrimination, labeling and stigma associated with SCD.
Recommendations
I believe that there is no simple solution to resolving the issues with SCD screening
and diagnosis. A multistep, interdisciplinary approach is necessary. I will outline some
options.
Implicit bias: it is important to acknowledge and embrace that the terms racism in
conjunction with the term healthcare cannot be easily unpacked. It is easier to identify
explicit bias, that is, overt expressions of prejudice and negative racial stereotypes (72),
than implicit bias, unconscious negative ethnic and racial stereotypes and attitudes (72,
73). Studies have shown that it is not uncommon for health care providers to have
unconscious negative attitudes toward Black people, while having positive attitudes to
White people (73). The challenge is that implicit bias too often negatively affects the
quality of care delivered to Black people – for example they are less likely to be referred
to specialists, are offered fewer clinical tests, their pain is poorly managed, and they are
more likely to be perceived as being less cooperative, less compliant and combative (72,
73).
Implicit racial and ethnic bias training courses have shown promise at improving
health care providers’ recognition of their own biases (74-76) and should be included in
the curriculum of all health care programs as well as a requirement of ongoing

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
73
certification for health care providers in practice, much like the Accessibility for
Ontarians with Disabilities Act (AODA) training that is required for all employers and
employees in Ontario (77). It should be noted that some health care learners and health
care providers may resist or deny their own implicit bias (78).
Increase funding for sickle cell disease research: in the era of evidence based
medicine, medical research is often used to guide medical procedures, protocols and
policies. Health care education in turn, is influenced by updated and newly developed
procedures, protocols and policies (79, 80). SCD research continues to be underfunded
compared to funding for other inherited disorders such as CF and Tay-Sachs disease (52).
Farooq et al, found that despite SCD being three times as prevalent as CF in the USA,
both diseases received a similar amount of federal government research funding between
2008 and 2018 (52). Unfortunately, there are no comparable Canadian based studies.
There is a dire need for more SCD specific research, specifically Canadian based
SCD research. Additionally, 75% of people affected with SCD live in Africa where most
African countries lack the infrastructure and resources to conduct scientific studies, let
alone studies that would meet the rigour expected by Western world standards (69).
However, I recognize a caveat - the people for whom SCD and SCT is more prevalent are
of Black African descent. Black people are underrepresented in research and they also
tend to have distrust in medical research (79). It would be beneficial to determine ways
to engage Black sickle cell patients to participate in more research – research that could

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
74
potentially lead to better disease management. It is beyond the scope of this thesis to
address this in detail - this may be a topic is appropriate for PhD.
I initially wanted to focus on parents' experiences with SCD diagnosis for my
Master’s thesis, but after a thorough and detailed search, only three studies specific to
SCD met the criteria, of which only one was Canadian. I had to expand my topic to
include the screening process for SCD and diagnosis for SCT. To compound this
problem, of the 14 studies that met eligibility criteria for this review, only two were
conducted in Canada (33, 34) and 11 studies focused on SCD or SCT and cystic fibrosis
or SCD or SCT and thalassemia (17, 18, 26, 27, 36-41). This trend was also reflected in
the references of the studies - only a small percentage of the references were dedicated to
SCD or SCT only. There is need for more sickle cell specific research to guide health care
practices, policies, protocols, pharmaceutical advancement and curative treatments.
Improve sickle cell disease health care education: many studies have identified that
health care providers and health care learners, have poor knowledge and understanding of
SCD, which in turn affects their management of sickle cell patients (19, 21, 80). SCD is
still a relatively rare disease and it is likely that most health care providers will have
limited experience treating SCD patients. Furthermore, SCD is rarely included in the core
curriculum of health care programs, which could represent a missed opportunity to study
the disease in depth (51).
The studies reviewed for this thesis showed that many of the study participants had
little knowledge of SCD and they also expressed dissatisfaction with the SCD information

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
75
provided to them by their health care providers (26, 27, 36). Given that the majority of
health care providers who offer sickle cell disease screening and disclose positive results
are not hematologists, it would be beneficial for all non-specialty health care providers to
improve their knowledge of SCD (as well as their communication skills) thus improving
the care of their patients or clients (63).
It is also important to remember that not all sickle cell patients will have access to
SCD specialty clinics, as SCD clinics in Hamilton, or Toronto. Their care may be
managed by a health care provider who may have limited knowledge and understanding
of SCD (47-49). SCD education modules and SCD conferences have been shown to help
improve baseline knowledge of SCD, however it is unclear whether improved knowledge
is sustained (47, 50, 51, 80, 81). More research is warranted.
One study has shown that a 90 hour, three month distance course on SCD, increased
the knowledge of health care providers (81). An elective course designed by Bulgin et al
shows promise to address the knowledge gaps for SCD in core curricula (51).
There is also value in incorporating the lived experience of SCD patients and their
caregivers to guide health care providers and enhance SCD curricula (5, 82).
Hidden curriculum: there is a difference between health care pedagogy and the
clinical teaching environment. Given that SCD education is limited in most health care
programs, health care learners would likely need to learn about SCD management from
observation during clinical rotations or clinical placements (83). Stigmatizing and
labelling language is often used to describe sickle cell patients who seek medical care

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
76
when experiencing a pain crisis (53). The patient’s pain is usually managed with
intravenous opioid medication, and it is not uncommon for health care staff to discredit a
patient’s pain which can lead to poor management of their pain (53). Stigmatizing
language such as “frequent flyer in the ER department” “insisting their pain is still a 10”
is often used and documented in a patient’s chart, which can lead to the transmission of
bias in the medical records (53). The health care learner may acquire habits that have the
potential to become incorporated in to their practice and the cycle of stigma and labeling
could repeat once the health care learner becomes the teacher.
Health care programs should endeavour to remember that the clinical observation
may serve as a stronger and more permanent teaching tool that traditional literature based
pedagogy (84). Health care staff need to acknowledge the stigma and bias inherent in the
health care setting and appreciate the emotional and physical impact on the patient, as
some sickle cell patients develop and aversion and fear of the health care providers and
have been known to delay seeking treatment (53, 79).
Informed choice discussion and communication: the parents in this systematic
review and integrative qualitative meta-synthesis generally accepted the offer to be
screened but expressed surprise if they or their children were diagnosed with SCD or
SCT. It would appear that some the parents did not fully understand purpose for SCD
screening, the impact of the disease or the hereditary pattern of SCD (26, 27). The
parents’ comments in this review are supported by literature that reveals that knowledge
and understanding of SCD is limited (19, 20). A review by Asnani et al, revealed that

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
77
intervention strategies were successful at improving patient knowledge of SCD that were
sustained for a period of time (62). This lends support that a public health education
program could be beneficial for improving the knowledge of general public on SCD.
Some parents’ failure to fully comprehend the consequences of SCD screening may
also suggest that they provided inform consent for SCD screening rather than making an
informed choice decision (26, 44, 46). It would be beneficial for health care providers to
have a deeper understanding of the difference between informed consent and informed
choice (44-46), a reminder of the power dynamic of the clinician-patient dyad and the
challenges some patients may face making decisions for their health management (85).
Sickle cell disease patient directed health care provider education: the voices of
the SCD patients and caregivers of SCD patients need to be included in the development
of SCD curricula for health care learners and health care providers (5, 82). Policy
statements made only from the perspective the health care provider risks distancing and
alienating SCD patients. However, change can only occur if policy stakeholders, health
care providers, health care educators and health care learners are willing to address their
implicit biases and engage in reflexive behaviour (72-76).
It is evident from the parents’ comments in the studies in this review that health
care provider’s method of communication, whether in offering SCD screening or the
disclosure of results, is very important. Parents’ expressed more positive experiences –
feeling informed, better understanding of test results, decreased stress and anxiety – when

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
78
they perceived their health care provider as thorough, caring, patient and knowledgeable
of SCD (17, 18, 26, 32, 36, 42).
The studies revealed that poor communication from health care providers often had
a negative impact on experience with SCD screening and the diagnosis process of SCD
and SCT (17, 18, 26, 32, 36, 42). It would be beneficial for health care providers to
engage in education programs/courses that focus on the value of good communication,
the disadvantages to using complex and technical medical terminology and an
appreciation of patients’ reliance on internet based medical information (80). Health care
providers should also be reminded or taught the true value of reflexivity.
Limitations
Many of the parents in the studies reviewed, live in the USA. Unlike Canada, the
Netherlands, the UK, the USA does not have a universal health care system. It is possible
that some of the parents had to pay for their infant’s NBS and subsequent follow-up
appointments with doctors, potentially adding to a parent’s emotional and financial
burden. It is possible that not all the parents’ comments may be applicable to the
Canadian population. Parents of two of the studies were from Africa, where SCD is more
prevalent and SCD screening and newborn screening is not offered routinely. Once
again, these parents’ experiences may not relate well to parents who were raised or lived
in Canada, the UK or the USA.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
79
Conclusions
This systematic review and integrative qualitative meta-synthesis explored parental
experiences with screening for SCD as a tool to guide health science education. Parents
overall accepted screening for SCD and appreciated learning of their own sickle cell
carrier status and that of their child. However, it is clear that many parents did not fully
understand the significance of SCD, or its inheritance pattern. Additionally, it is clear that
health care providers need to ensure they practice an informed choice discussion rather
than informed consent for SCD screening. Health care providers also need to be mindful
of the manner in which diagnostic results are delivered to patients. In particular Canada
requires a nationally based SCD strategy and network. Finally, a better follow-up system
and future reproductive options should be in place for parents and infants who have SCT.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
80
References
1 Kato, G.J., Piel, F.B., Reid, C.D., Gaston, M.H., Ohene-Frempong, K., Krishnamurti,
L., Smith, W.R., Panepinto, J.A., Weatherall, D.J., Costa, F.F., Vichinsky, E.P.
(2018). Sickle Cell Disease. Nature Reviews. Disease Primers, 4:18010: 1-22
2 Mburu, J., Odame, I (2019). Sickle cell disease: Reducing the global disease burden.
International Journal of Laboratory Hematology. 41(Suppl 1): 82-88
3 Pinto, V.M., Balocco, M., Quintino, S., Forni, G.L. (2019). Sickle cell disease: a
review for the internist. Internal and Emergency Medicine. 14(7):1051-1064
4 Verhovsek, M., Ewurabena, S. (2014) Consensus Statement on the Care of Patients
with Sickle Cell Disease in Canada. The Canadian Haemoglobinopathy Association.
Retrieved from https://www.canhaem.org/wp-content/uploads/2018/05/Sickle-Cell-
Consensus.pdf#:~:text=The%20Consensus%20Statement%20on%20the%20Care%2
0of%20Patients,Hemoglobinopathy%20Association%20%28CanHaem%29%20repre
sents%20the%20first-ever%20national%20guidance
5 Abu Ali, R.M., Abedl Razeq, N.M. (2017). The Lived Experience of Parents of
Children with Sickle Cell Disease: A Qualitative Study. Open Journal of Nursing.
7(11): 1348-1364
6 Inusa, B.P.D., Hsu, L.L., Kohli, N.J., Patel, A., Ominu-Evbota, K., Anie, K.A.,
Atoyebi, W. (2019). Sickle Cell Disease – Genetics, Pathophysiology, Clinical
Presentation and Treatment. International Journal of Neonatal Screening. 5(2):20-34
7 Piel, F.B., Steinberg, M.H., Rees, D.C. (2017) Sickle Cell Disease. The New England
Journal of Medicine. 376 (16):1561-1573
8 Ware, RE., de Montalembert, M., Tshilolo, L., Abboud, M.R. (2017). Sickle cell
disease. The Lancet, 390(10091):311-323
9 Leonard, A., Tisdale, J., Abraham, A. (2020). Curative options for sickle-cell
disease: haploidentical stem cell transplant or gene therapy? British Journal of
Haematology. 189 (3):408-423
10 Chaturvedi, S., DeBaun, M.R. (2018). Evolution of sickle cell disease from a life-
threatening disease of children to a chronic disease of adults: The last 40 years.
American Journal of Hematology. 91(1):5-14

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
81
11 Houwing, M.E., de Pagter P.J., van Beers E.J., Biemond B.J., Rettenbacher, E.,
Rijneveld A.W., Schols E.M., Philipsen J.N.J., Tamminga R.Y.J., Fijn van Draat, K.,
Nur E., Cnossen, M.H. (2019). Sickle cell disease: Clinical presentation and
management of a global health challenge. Blood Reviews. 37(100580)
12 Macharia, A.W., Mochamah, G., Uyoga, S., Ndila, C.M., Nyutu, G., Makale, J.,
Tendwa, M., Nyatichi, E., Ojal, J., Shebe, M., Awuondo, K.O., Mturi, N., Peshu, N.,
Tsofa, B., Scott, J.A.G., Maitland, K., Williams, T.N. (2018). The clinical
epidemiology of sickle cell anemia in Africa. American Journal of Hematology.
93(3):363-370
13 Pecker, L.H., Naik, R.P. (2018). The current state of sickle cell trait: implications for
reproductive and genetic counseling. Blood. 132(22):2231-2338
14 Xu, J.Z., Thein, S.L. (2019). The carrier state for sickle cell disease is not completely
harmless. Haematologica. 104(6):1106-1111
15 People affected by sickle cell disease (n.d.) Not Alone in Sickle Cell. Retrieved
from https://www.notaloneinsicklecell.com/Global-Impact-Of-SCD/
16 Sickle Cell Disease Association of Canada. The Need for a National Strategy for
Sickle Cell Disease (SCD). Retrieved from
http://www.sicklecelldisease.ca/pdf/SCDAC-National-Strategy-Document-PartI-
April-28th.pdf
17 Collins, J. L., La Pean, A., O’Tool, F., Eskra, K.L., Roedl, S.J. (2013). Factors that
influence parents' experiences with results disclosure after newborn screening
identifies genetic carrier status for cystic fibrosis or sickle cell hemoglobinopathy.
Patient Education and Counseling. 90(3): 378-385
18 La Pean, A., Collins, J.L., Christopher, S.A., Eskra, K.L., Roedl, J.S., Tluczek, A.,
Farrell, M.H. (2012). A Qualitative Secondary Evaluation of Statewide Follow-up
Interviews for Abnormal Newborn Screening Results for Cystic Fibrosis and Sickle
Cell Hemoglobinopathy. Genetics in Medicine. 14(2):207-214
19 Acharya, K., Lang, C.W., Ross, L. A Pilot Study to Explore Knowledge, Attitudes
and Beliefs about Sickle Cell Trait and Disease. (2009). Journal of the National
Medical Association. 101(11):1163-1172
20 Aderotoye-Oni, S., Diaku-Akinwumi, I.N., Adeniran, A., Falase, B. (2018).
Unprepared and Misinformed Parents of Children with Sickle Cell Disease: Time to
Rethink Awareness Campaigns. Cureus. 10(12):e3806-e3821.

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
82
21 Boyd, J.H. Watkins, A.R., Price, C.L., Fleming, F., DeBaun, M.R. (2005).
Inadequate community knowledge about sickle cell disease among African-American
women. Journal of the National Medical Association. 97(1): 62-67
22 Gustafson, S.L., Getting, E.A., Watt-Morse, M., Krishnamurti, L. (2007). Health
beliefs among African-American women regarding genetic testing and counseling for
sickle cell disease. Genetics in Medicine. 9(5):303-310
23 Mayo-Gamble, T.L., Barnes, P.A., Cunningham Erves, J., Middlestadt, S.E., Lin,
H.C. (2018) ‘It means everyone should know their status’: exploring lay conceptions
of sickle cell trait and sickle cell trait screening among African Americans within
middle reproductive age. Ethnicity and Health. 23(7):813-829
24 de Montalembert, M., Tshilolo, L., Allali, S. (2019). Sickle cell disease: a
comprehensive program of care from birth. Hematology. The American Society of
Hematology Education Program. 2019(1):490-495
25 Lubeck, D., Agodoa, I., Bhakta, N., Danese, M., Pappu, K., Howard, R., Gleeson,
M., Halperin, M., Lanzkron, S. (2019) Estimated Life Expectancy and Income of
Patients With Sickle Cell Disease Compared With Those Without Sickle Cell
Disease. JAMA Network Open. 2(11):e1915374-e1915387
26 Holtkamp, K.C.A., Lakeman, P., Hader, H., Jans, S.MJ.P., Hoenderdos, M., Playfair,
H.A.M., Cornel, M.C., Peters, M., Hennenman, L. (2018). Experiences of a High-
Risk Population with Prenatal Hemoglobinopathy Carrier Screening in a Primary
Care Setting: a Qualitative Study. Journal of Genetic Counseling. 27(3):635-646
27 Locock, L., Kai, J. (2008). Parents' experiences of universal screening for
haemoglobin disorders: implications for practice in a new genetics era. The British
Journal of General Practice. 58(548):161-168
28 El-Haj, N., Hoppe, C.C. Newborn Screening for SCD in the USA and Canada.
(2018). International Journal of Neonatal Screening. 4(4):36-45
29 Sandelowski, M., Barroso, J. (2007). Handbook for Synthesizing Qualitative
Research. New York, NY. Springer Publishing Company, Inc.
30 Dejuan., Giacomini, N., Simeonov, D., Smith, A. (2016) Finding qualitative research
evidence for health technology assessment. Qualitative Health Research.
26(10):1301-1317
31 Charmatz, K. Second Edition (2014). Constructing Grounded Theory. Los Angeles.
Sage

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
83
32 Biwott, P.J., Njiru, D., Naanyu, V. (2017). Disclosure of Sickle Cell Disease Results
to Parents/Guardians Participating In Research at a Hemato-Oncology Clinic in
Eldoret Kenya. Journal of Nursing and Health Science. 6(3):8-18
33 Bruce, A.,Witol, A., Alvadj-Korenic, T., Mayan, M., Greenslade, H., Plaha, M.,
Venner, M.A. (2018). A complex interface: Exploring sickle cell disease from a
parent's perspective, after moving from Sub-Saharan Africa to North America.
Pediatric Hematology and Oncology. 35(7-8):373-384.
34 Miller, F. A., Paynter, M., Hayeems, R.Z., Little, J., Carroll, J.C., Wilson, B.J.,
Allanson, J., Butautas, J.P., Chakraborty, P. (2010). Understanding sickle cell carrier
status identified through newborn screening: A qualitative study. European Journal
of Human Genetics. 18(3):303-308
35 Hsu, L., Nnodu, O.E., Brown, B.J., Tluway, F., King, S., Dogara, L.G., Patil, C.,
Shevkoplyas, S., Lettre, G., Cooper, R.S., Gordeuk, V.R., Tayo, B.O. (2018). White
Paper: Pathways to Progress in Newborn Screening for Sickle Cell Disease in Sub-
Saharan Africa. Journal of Tropical Disease and Public Health. 6(2):260-279
36 Tsianakas, V., Atkin, K., Calnan, M.W., Dormandy, D., Marteau, TM. (2012).
Offering antenatal sickle cell and thalassaemia screening to pregnant women in
primary care: a qualitative study of women's experiences and expectations of
participation. Health Expectations. 15(2):115-125
37 Reed, K. (2009). 'It's them faulty genes again': women, men and the gendered nature
of genetic responsibility in prenatal blood screening. Sociology of Health and Illness.
31(3):343-359
38 Reed, K. (2011). 'He's the dad isn't he?' Gender, race and the politics of prenatal
screening. Ethnicity and Health. 16(4-5):327-341
39 Ulph, F., Cullinan, T., Quershi, N., Kai, J. (2011). Familial influences on antenatal
and newborn haemoglobinopathy screening. Ethnicity and Health. 16(4-5):361-375
40 Ulph, F., Cullinan, T., Qureshi, N, Kai, J. (2015). Parents' responses to receiving
sickle cell or cystic fibrosis carrier results for their child following newborn
screening. European Journal of Human Genetics. 23(4):459-465
41 Chudleigh, J., Buckingham, S., Dignan, J., O’Driscoll, S., Johnson, K., Rees, D.,
Wyatt, H., Metcalfe, A. (2016). Parents' Experiences of Receiving the Initial Positive
Newborn Screening (NBS) Result for Cystic Fibrosis and Sickle Cell Disease.
Journal of Genetic Counseling. 25(6):1215-1226

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
84
42 Lebensburger, J. D., Grosse, S.D., Altice, J.L., Thierry, J.M., Ivankova, N.V. (2015).
Understanding and Improving Health Education Among First-Time Parents of
Infants With Sickle Cell Anemia in Alabama: A Mixed Methods Approach. Journal
of Pediatric Hematology/Oncology. 37(1):35-42
43 Centers for Disease Control and Prevention. Caregivers and Sickle Cell Disease.
Retrieved at: https://www.cdc.gov/ncbddd/sicklecell/features/sickle-cell-
caregivers.html
44 Marteau, T.M., Dormandy, E., Michie, S. (2001). A measure of informed choice.
Health Expectations. 4(2):99-108
45 Majid, U., Kandasamy, S., Farrah, K., Vanstone, M. (2019). Women’s preferences
and experiences of cervical cancer screening in rural and remote areas: a systematic
review and qualitative meta-synthesis. Rural and Remote Health. 19(4):5190-5200
46 Brown, K., Dormandy, E., Reid, E., Gulliford, M., Marteau, T. (2011). Impact on
informed choice of offering antenatal sickle cell and thalassemia screening in
primary care: a randomized trial. Journal of Medical Screening. 18(2):65-75
47 Aboagye, S., Torto, M., Asah-Opoku, K. Nuamah, M.A., Oppong, S.A., Samba, A.
(2019). Sickle Cell Education: A Survey of Antenatal Health Care Givers. American
Journal of Tropical Medicine and Hygiene. 101(3):684-688
48 De, D. (2006). How Well Does Midwifery Education Enable Professionals to Work
With Families and Individuals Affected by Sickle Cell and Thalassaemia? Research
Policy and Planning. 24(2):121-133
49 Jans, S.M.P.J., de Jonge, A., Henneman, L., Cornel, M.C., Lagro-Janssen, A.L.M.
(2012). Attitudes of general practitioners and midwives towards ethnicity-based
haemoglobinopathy carrier screening. European Journal of Human Genetics.
20(11):1112-1117
50 Rolfe, V., Fowler, M., Dyson S.M. (2011). Sickle cell in the university curriculum: a
survey assessing demand for open-access educational materials in a constructed
community of interest. Diversity and Equality in Health and Care. 8(4):239-249
51 Bulgin, D., Tanabe, P., Asnani, M., Royal, C.D.M. (2019). Twelve tips for teaching a
comprehensive disease-focused course with a global perspective: A sickle cell
disease example. Medical Teacher. 41(3):275-281
52 Farooq, F., Mogayzel, P.J., Lanzkron, S., Haywood,C., Strouse, J.J. (2020).
Comparison of US Federal and Foundation Funding of Research for Sickle Cell
Disease and Cystic Fibrosis and Factors Associated with Research Productivity.
JAMA Network Open. 3(3): e201737-e201749

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
85
53 Goddu, A.P., O’Connor, K.J., Lanzkron, S., Saheed, M.O., Saha, S., Peek, M.E.,
Haywood, C., Beach, C. (2019). Do Words Matter? Stigmatizing Language and the
Transmission of Bias in the Medical Record. General Internal Medicine. 33(5):685-
691
54 Lee, L., Smith-Whitley, K., Banks, S., Puckrein, G. (2019). Reducing Health Care
Disparities in Sickle Cell Disease: A Review. Tropical Review. 13(6):599-607
55 American College of Obstetricians and Gynecologist. Retrieved at
https://www.acog.org/womens-health/faqs/carrier-screening-for-hemoglobinopathies
56 Langois, S., Ford, J.C., Chitayat, D. (2008). Carrier screening for thalassemia and
hemoglobinopathies in Canada. Journal of Obstetrics and Gynaecology of Canada.
30(10):950-959
57 Zwahlena, M., Lowa, N., Borisch, B., Egger, M., Künzli, N., Obrist, R., Paccaud, F.,
Zybach, U., Probst-Hensch, N.M. (2010). Population-based screening – the difficulty
of how to do more good than harm and how to achieve it. Swiss Medical Weekly.
140:w13061
58 Jones, P.C. (2000). Levels of Racism: A Theoretic Framework and a Gardner’s Tale.
American Journal of Public Health. 90(8):1212-1215
59 Power-Hays, A., McGann, P.T. (2020). When Actions Speak Louder Than Words –
Racism and Sickle Cell Disease. The New England Journal of Medicine.
383(20):1902-1903
60 Dryden, O.S., Nnorom, O. (2021). Time to dismantle systemic anti-Black racism in
medicine in Canada. Canadian Medical Association Journal. 193(2):E55-E57
61 Mahabir, D.F., O’Campo, P., Lofters, A., Shankardass, K., Salmon, C., Muntaner, C.
(2021). Experiences of everyday racism in Toronto’s health care system: a concept
mapping study. International Journal for Equity in Health. 20(1):74-88
62 World Health Organization 59th World Health Assembly (2006). Retrieved at
https://lapps.who.int./gb/ebwha/pdf_files/WHA59_REC1/e/WHA59_2006_REC1-
en.pdf?us=1
63 Azonobi, I.C., Anderson, B.L., Byams, V.R., Grant, A.M., Schulkin, J. (2014).
Obstetrician-Gynecologists’ knowledge of sickle cell disease screening and
management. BMC Pregnancy and Childbirth. 14(1):356-360
64 Brennan-Cook, J., Bonnabeau, E., Aponte, R., Augustin, C., Tanabe, P. (2018).
Barriers to Care for Persons with Sickle Cell Disease: The Case Manager’s
Opportunity to Improve Patient Outcomes. Professional Case Management.
23(4):213-219

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
86
65 Gomes, L.M.X., Vieira, M.M., Reis, T.C., Barbosa, T.L.A., Caldeira, A.P. (2011).
Knowledge of family health program practitioners in Brazil about sickle cell disease:
a descriptive, cross-sectional study. BMC Family Practice. 12:89-95
66 Kaback, M.M. (2000). Population-based genetic screening for reproductive
counseling: the Tay-Sachs disease model. European Journal of Pediatrics. 159(Suppl
3): S192-S195
67 Mitchell J.J, Capua, A., Clow, C., Scriver, C.R. (1996). Twenty-Year Outcome
Analysis of Genetic Screening Programs for Tay-Sachs and B-Thalassemia Disease
Carriers in High Schools. American Journal of Genetics. 59(4):793-798
68 Ioannou, L., Mclaren, B.J., Massie, J., Lewis, S., Metcalfe, S.A, Forrest, L.,
Delatycki, M.B. (2014). Population-based carrier screening for cystic fibrosis: a
systematic review of 23 years of research. Genetics in Medicine. 16(3):207-216
69 Thein, M.S., Thein, S.L. (2016). World Sickle Cell Day 2016: A time for appraisal.
Indian Journal of Medical Research. 143(6):678-681
70 Scotet, V., L’Hostis, C., Ferec, C. (2020). The Changing Epidemiology of Cystic
Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes.
11(6):589-601
71 Ontario Newborn Screen. Retrieved at https://www.newbornscreening.on.ca/
72 Burgess, D., van Ryn, M., Dovidio, J., Saha, S. (2017). Reducing Racial Bias Among
Health Care Providers: Lessons from Social-Cognitive Psychology. Journal of
General Internal Medicine. 22(6):882-887
73 Hall, W.J., Chapman, M.V., Lee, K.M., Merino, Y.M., Thomas, T.W., Payne, B.K.,
Eng, E., Day, S.H., Coyne-Beasley, T. (2015). Implicit Racial/Ethnic Bias Among
Health Care Professionals and Its Influence on Health Care Outcomes: A Systematic
Review. American Journal of Public Health. 105(12):e60-e76
74 Marcelin, J.R., Siraj, D.S, Victor, R., Kotadia, S., Maldonado, Y.A. (2019). The
Impact of Unconscious Bias in Healthcare: How to Recognize and Mitigate It. The
Journal of Infectious Diseases. 220(Suppl 2): S62-S73
75 Zeidan, A.J., Khatri, U.G., Aysola, J., Shofer, F.S., Mamtani, M., Scott, K.R.,
Conlon, L.W., Lopez, B.L. (2019). Implicit Bias Education and Emergency Medicine
Training: Step One? Awareness. AEM Education and Training. 3(1):81-85
76 Sherman, M.D., Ricco, J., Nelson, S.C., Nezhad, S.J., Prasad, S. (2019). Implicit Bias
Training in Residency Program: Aiming for Enduring Effects. Family Medicine.
51(8):677-681

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
87
77 Accessibility for Ontarians with Disability Act. (2018). AODA Training
Requirements: Who Needs It and Why? Retrieved at https://www.aoda.ca/aoda-
training-requirements-who-needs-it-and-why/
78 Gonzalez, C.M., Deno, M.L., Kintzer, D., Marantz, P.R., Lypson, M.L, McKee, M.D.
(2019). A Qualitative Study of New York Medical Student Views on Implicit Bias
Training Instruction: Implications for Curriculum Development. Journal of General
Internal Medicine. 34(5):692-698
79 Omondi, N.A., Stickney Ferguson, S.E., Majhail, N.S., Denzen, E.M., Buchanan,
G.R., Haight, A.E., Labotka, R.J., Rizzo, J.D., Murphy, E.A. (2013). Barriers to
Hematopoietic Cell Transplantation Clinical Trial Participation of African-American
and Black Youth with Sickle Cell Disease and Their Parents. Journal of Pediatric
Hematological Oncology. 35(4):289-298
80 Asnani, M.R., Quimby, K.R., Bennett, N.R., Francis, D.K. (2016). Interventions for
patients and caregivers to improve knowledge of sickle cell disease and recognition
of its related complications. Cochrane Database of Systematic Reviews. 10(10):1-57
81 Santos Diniz, K.K., Pagano, A.S., Pinheiro, A.P., Afonso Reis, I., Pinheiro Junior,
K.G., de Carvalho Torres, H. (2019). Knowledge of professional healthcare providers
about sickle cell disease: Impact of a distance education course. Hematology,
Transfusion and Cell Therapy. 41(1):62-68
82 Burnes, D.P.R., Antle, B.J., Williams, C.C., Cook, L. (2008). Mothers Raising
Children with Sickle Cell Disease at the Intersection of Race, Gender and Illness
Stigma. Health and Social Work. 33(3):211-220
83 Turners, S., Krebs, E., Axtell, S. (2002). The Hidden Curriculum in Multicultural
Medical Education: The Role of Case Examples. Academic Medicine. 77(3):209-216
84 Browning, D.M., Meyer, E.C., Truog, R.D., Solomon, M.Z. (2007). Difficult
Conversations in Health Care: Cultivating Relational Learning to Address the Hidden
Curriculum. Academic Medicine. 82(9):905-913
85 Woolf, S.H., Chan, E.C.Y., Harris, R., Sheridan, S.L., Braddock 3rd, C.H., Kaplan,
R.M., Krist, A., O’Connor, A.M., Tunis, S. (2005). Promoting Informed Choice:
Transforming Health Care to Dispense Knowledge for Decision Making. Annals of
Internal Medicine. 143(4):293-300
86 Sickle Cell Trait (n.d.). Sickle Cell Awareness Group of Ontario. Retrieved at
https://sicklecellanemia.ca/?s=sickle+cell+trait

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
88
Appendix A
Table 5. Sickle Cell June 2019 Search Strategy
Search date: June 18, 2019
Databases searched:
OVID Medline Epub Ahead of Print, In-Process & Other Non-Indexed Citations,
Ovid MEDLINE® Daily and Ovid MEDLINE® 1946 to Present: 3054 hits
Embase (1974-2019): 5744 hits
PsychINFO: 787 hits
AMED: 14 hits
OVID Emcare: 1120 hits
CINAHL: 624 hits
Web of Science: 542 hits
6994 references after duplicates removed
Database: OVID Medline Epub Ahead of Print, In-Process & Other Non-Indexed
Citations, Ovid MEDLINE® Daily and Ovid MEDLINE® 1946 to Present
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
89
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
Aug;26(10):1307-
17.
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 Limit 42 to (English language and humans)
44 TS=exp Anemia, Sickle Cell Intervention terms
45 TS=exp Since Cell Trait
46 TS=sickle cell.mp. Key word
47 #44 OR #45 OR #46 This line combines
the intervention
concepts and key
word

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
90
48 #43 AND #47 Combine
Intervention and
filter
Database: PsychINFO (1987-2019)
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
Aug;26(10):1307-
17.
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
91
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 TS=sickle cell disease Intervention term
44 TS=sickle cell.mp. Key word
45 #43 OR #44 This line combines
the intervention
concepts and key
word
46 #42 AND #45 Combine
Intervention and
filter

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
92
Database: Embase (1974-2019)
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
Aug;26(10):1307-
17.
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
93
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 Limit 42 to (English language and humans)
44 TS=sickle cell Intervention terms
45 TS=((sickle cell anemia) OR (hemoglobin sc disease) OR
(hemoglobin sd disease) OR (sickle cell beta thalassemia)
or (sickle cell crisis) OR (sickle cell trait)
46 TS=sickle cell.mp. Key word
47 #44 OR #45 OR #46 This line combines
the intervention
concepts and key
word
48 #43 AND #47 Combine
Intervention and
filter

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
94
Database: AMED (1985-2019)
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
Aug;26(10):1307-
17.
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
95
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 TS=anemia, sickle cell Intervention term
44 TS=sickle cell.mp. Key word
45 #43 OR #44 This line combines
the intervention
concepts and key
word
46 #42 AND #45 Combine
Intervention and
filter

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
96
Database: OVID Emcare
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
Aug;26(10):1307-
17.
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
97
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 Limit 42 to (English language and humans)
44 TS=(sickle cell) or (sickle cell anemia) or (sickle cell beta
thalassemia) or (sickle cell crisis)
Intervention terms
45 TS=sickle cell.mp. Key word
46 #44 OR #45 This line combines
the intervention
concepts and key
word
47 #43 AND #46 Combine
Intervention and
filter

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
98
Database: CINAHL
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
Aug;26(10):1307-
17.
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
99
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 TS=anemia, sickle cell Intervention terms
44 TS=sickle cell trait
45 TS=sickle cell.mp. Key word
46 #43 OR #44 OR #45 This line combines
the intervention
concepts and key
word
47 #42 AND #46 Combine
Intervention and
filter

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
100
Database: Web of Science (Social Science Citation Index)
Line
#
Search Notes
1 TS=interview*
Hybrid Filter: ISI
Web of Science,
Social Science
Citation Index
Would recommend
citing in
methods/report:
DeJean D,
Giacomini M,
Simeonov D,
Smith A. Finding
Qualitative
Research
Evidence for
Health Technology
Assessment. Qual
Health Res. 2016
Aug;26(10):1307-
17.
2 TS=(theme*)
3 TS=(thematic analysis)
4 TS=qualitative
5 TS=nursing research methodology
6 TS=questionnaire
7 TS=(ethnograph*)
8 TS= (ethnonursing)
9 TS=(ethnological research)
10 TS=(phenomenol*)
11 TS=(grounded theor*) OR TS=(grounded stud*) OR
TS=(grounded research) OR TS=(grounded analys?s)
12 TS=(life stor*) OR TS=(women's stor*)
13 TS=(emic) OR TS=(etic) OR TS=(hermeneutic) OR
TS=(heuristic) OR TS=(semiotic) OR TS=(data saturat*)
OR TS=(participant observ*)
14 TS=(social construct*) OR TS=(postmodern*) OR
TS=(post structural*) OR TS=(feminis*) OR
TS=(interpret*)
15 TS=(action research) OR TS=(co-operative inquir*)
16 TS=(humanistic) OR TS=(existential) OR
TS=(experiential) OR TS=(paradigm*)
17 TS=(field stud*) OR TS=(field research)
18 TS=(human science)
19 TS=(biographical method*)
20 TS=(theoretical sampl*)
21 TS=(purposive sampl*)
22 TS=(open-ended account*) OR TS=(unstructured account)
OR TS=(narrative*) OR TS=(text*)
23 TS=(life world) OR TS=(conversation analys?s) OR
TS=(theoretical saturation)
24 TS=(lived experience*) OR TS=(life experience*)
25 TS=(cluster sampl*)
26 TS=observational method*
27 TS=(content analysis)
28 TS=(constant comparative)
29 TS=(discourse analys?s) or TS =(discurs* analys?s)
30 TS=(narrative analys?s)
31 TS=(heidegger*)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
101
32 TS=(colaizzi*)
33 TS=(spiegelberg*)
34 TS=(van manen*)
35 TS=(van kaam*)
36 TS=(merleau ponty*)
37 TS=(husserl*)
38 TS=(foucault*)
39 TS=(corbin*)
40 TS=(strauss*)
41 TS=(glaser*)
42 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR
#9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR
#16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR
#23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 OR
#30 OR #31 OR #32 OR #33 OR #34 OR #35 OR #36 OR
#37 OR #38 OR #39 OR #40 OR #41
This line combines
all the qualitative
filter keywords
43 TS=sickle cell.mp. Key word
44 #42 AND #43 Combine
Intervention and
filter

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
102
Appendix B
Table 6. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Acharya, K.,
et al. (2009)
A Pilot Study to
Explore
Knowledge,
Attitudes, and
Beliefs about-
Sickle Cell Trait
and Disease
USA Opinions
about sickle
cell trait
Interviews
and
surveys
Not about
parental
experience
with
screening
Ahmad, N.Y.,
Farrell, MH
(2014)
Linguistic
markers of
emotion in
mothers of sickle
cell carrier
infants: What are
they and what do
they mean?
USA Diagnosis of
carrier status
from
newborn
screening
Interviews
and
surveys
Results
presented
quantitatively
Alexander, S.,
et al. (2017)
Knowledge of and
attitudes toward
heel prick
screening for
sickle cell disease
in Saint Lucia
Jamaica Knowledge
of newborn
screening
Interviews
and survey
Not about
parental
experience
with
screening
Atkin, K., et
al. (1998)
Screening and
counseling for
sickle cell
disorders and
thalassaemia: the
experience of
parents and health
professionals
UK Screening Interviews Prior to 2006
Dennis-Antwi,
J. A., et al.
(2011)
I can die today, I
can die
tomorrow': Lay
perceptions of
sickle cell disease
in Kumasi, Ghana
at a point of
transition
Africa Fathers
perspective
of their
child’s sickle
cell disease
Interviews Not about
experience
with the
screening
process

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
103
Table 6. cont. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Dormandy, E.,
et al. (2007)
Development of a
measure of
informed choice
suitable for use in
low literacy
populations
UK Informed
choice
Interviews
and
surveys
Quantitative
Dyson, S. M.,
et al. (2007)
Ethnicity
questions and
antenatal
screening for
sickle
cell/thalassaemia
(EQUANS) in
England:
observation and
interview study
UK Antenatal
barriers to
screening
Interviews Not about
parental
experience of
screening for
sickle cell
disease
Guedes, C.
(2012)
Reproductive
decisions and
newborn
screening: The
perspective of
female caregivers
of children with
sickle cell disease
Brazil Mothers of
children
with sickle
cell disease
perception
of screening
Interviews Not English
Hill, M., et al.
(2014)
Client views and
attitudes to non-
invasive prenatal
diagnosis for
sickle cell
disease,
thalassaemia and
cystic fibrosis.
USA Opinion of
genetic
testing
Interviews Not about
parental
experience of
screening for
sickle cell
disease
Hill, S. A.
(1994)
Motherhood and
the obfuscation of
Medical
Knowledge: The
Case of Sickle
Cell Disease
USA Screening of
know sickle
cell disease
mothers
Interviews Prior to 2006

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
104
Table 6. cont. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Kai, J., et al.
(2009)
Communication
of carrier status
information
following
universal
newborn
screening for
sickle cell
disorders and
cystic fibrosis:
qualitative study
of experience and
practice
UK
Disclosure
of carrier
status
Interviews Not primary
Lang, C. W.,
et al. (2009)
Maternal
knowledge and
attitudes about
newborn
screening for
sickle cell disease
and cystic fibrosis
USA Knowledge
of Newborn
screen
Interview
and
surveys
Quantitative
Lawrence,
R.H.,
Bediako, M.S.
(2012)
Social and
behavioral
implications of
the NCAA-
mandated sickle
cell trait testing
policy
USA Screening of
athletes
Interview
and
surveys
Abstract
Lebensburger,
J.D., et al.
(2012)
The process of
acquiring health
education for first
time parents of an
infant with sickle
cell disease
USA Health
education
for parents
with sickle
cell child
Interviews
and
surveys
Poster
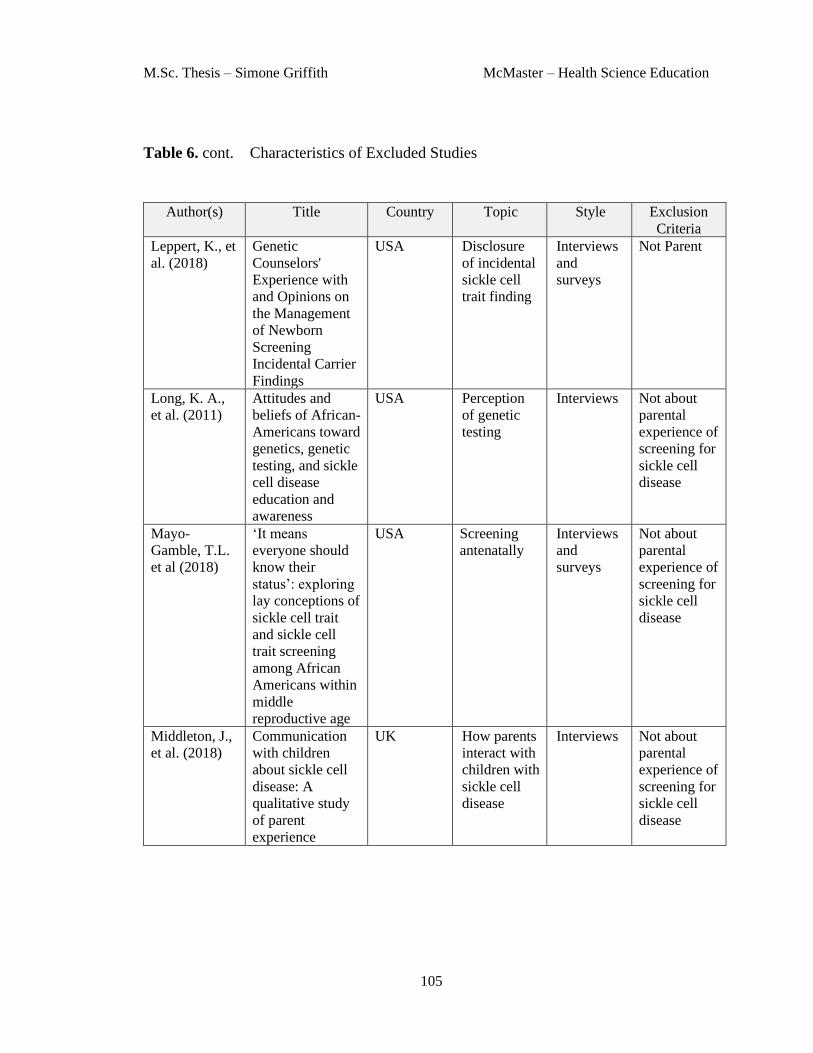
M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
105
Table 6. cont. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Leppert, K., et
al. (2018)
Genetic
Counselors'
Experience with
and Opinions on
the Management
of Newborn
Screening
Incidental Carrier
Findings
USA Disclosure
of incidental
sickle cell
trait finding
Interviews
and
surveys
Not Parent
Long, K. A.,
et al. (2011)
Attitudes and
beliefs of African-
Americans toward
genetics, genetic
testing, and sickle
cell disease
education and
awareness
USA Perception
of genetic
testing
Interviews Not about
parental
experience of
screening for
sickle cell
disease
Mayo-
Gamble, T.L.
et al (2018)
‘It means
everyone should
know their
status’: exploring
lay conceptions of
sickle cell trait
and sickle cell
trait screening
among African
Americans within
middle
reproductive age
USA Screening
antenatally
Interviews
and
surveys
Not about
parental
experience of
screening for
sickle cell
disease
Middleton, J.,
et al. (2018)
Communication
with children
about sickle cell
disease: A
qualitative study
of parent
experience
UK How parents
interact with
children with
sickle cell
disease
Interviews Not about
parental
experience of
screening for
sickle cell
disease

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
106
Table 6. cont. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Moody, L., et
al. (2017)
Healthcare
professionals' and
parents'
experiences of the
confirmatory
testing period: a
qualitative study
of the UK
expanded
newborn
screening pilot.
UK Experience
with positive
newborn
screen -
Interviews Not sickle
cell
Noke, M. and
Ulph, F.
(2014)
Young adults'
pre-existing
knowledge of
cystic fibrosis and
sickle cell
diseases:
implications for
newborn
screening
UK Knowledge
of newborn
screening
Interviews Not about the
experience of
screening for
sickle cell
disease
Noke, M., et
al. (2016)
A qualitative
study to explore
how professionals
in the United
Kingdom make
decisions to test
children for a
sickle cell carrier
status
UK Health care
professionals
opinion on
screening
Interviews Not parent
O'Connor, S.,
et al. (2014)
Attitudes among
healthcare
providers and
patients
diagnosed with
sickle cell
disease.
USA Diagnosis -
Experience
of diagnosis
of sickle cell
disease –
young adults
Interviews Not parent

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
107
Table 6. cont. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Ross, P. T.
(2015)
Motivations of
women with
sickle cell disease
for asking their
partners to
undergo genetic
testing
USA Partner
testing for
sickle cell
disease
Interviews Not about the
experience of
screening for
sickle cell
disease
Shook, L. M.,
et al. (2011)
Preferences for
genetic
counseling and
educational
materials for
African
immigrant
families with a
child diagnosed
with sickle cell
disease
UK Diagnosis –
African
Immigrants
experience
Interviews Not parent
Skirton, H., et
al. (2015)
An easy test but a
hard decision:
Ethical issues
concerning non-
invasive prenatal
testing for
autosomal
recessive
disorders
UK Antenatal
screening
Interviews Not sickle
cell
Stewart, K. A.
(2008)
An examination
of African
American college
students'
knowledge and
attitudes
regarding sickle
cell disease and
sickle cell disease
carrier testing: A
mixed methods
study
USA Screening –
knowledge
about
screening
Interviews Not peer
reviewed

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
108
Table 6. cont. Characteristics of Excluded Studies
Author(s) Title Country Topic Style Exclusion
Criteria
Treadwell, M.
J., et al.
(2006)
Using qualitative
and quantitative
strategies to
evaluate
knowledge and
perceptions about
sickle cell disease
and sickle cell
trait
USA Knowledge
of sickle
disease –
indirectly
related to
screening
Interviews Not about the
experience of
screening for
sickle cell
disease
Ulph, F., et al.
(2014)
Informing
children of their
newborn
screening carrier
result for sickle
cell or cystic
fibrosis:
qualitative study
of parents'
intentions, views
and support needs
UK Disclosure
of sickle cell
status to
children
Interviews Not about the
experience of
screening for
sickle cell
disease
Warren, N. S.,
et al. (1982)
Newborn
screening for
hemoglobinopathi
es in New York
State: Experience
of physicians and
parents of
affected children
USA Screening
and
diagnosis -
Interviews Prior to 2006
Wright, S. W.,
et al. (1994)
Screening for
sickle-cell trait in
the emergency
department."
USA Screening -
Antenatal/Pr
e-conceptual
screening of
me and
women in an
ER
Interviews Prior to 2006

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
109
Appendix C
Table 7. Newborn Screen for Sickle Cell Disease in Canada
Province Sickle Cell Disease
included in the
Newborn Screen
Routine Disclosure
of Sickle Cell Trait
Results
Alberta Yes Yes
British Columba Yes No (disclosed only
upon request)
Manitoba No N/A
New Brunswick Yes Yes
Newfoundland Yes Yes
North West Territories No N/A
Nova Scotia Yes Yes
Nunavut No N/A
Ontario Yes No (disclosed only
upon request)
Prince Edward Island Yes Yes
Quebec Yes No (disclosed only
upon request)
Saskatchewan No N/A
Yukon Yes Yes
Sickle Cell Awareness Group of Ontario (86)

M.Sc. Thesis – Simone Griffith McMaster – Health Science Education
110
Appendix D
Table 8. Sickle Cell Trait Disclosure Practice (countries listed in the studies reviewed)
Country
(in studies
reviewed)
Antenatal Screening Sickle Cell Disease included
on Newborn Screen
Disclosure of SCT
finding
Canada Offered at health
care provider’s
discretion
Universal screening offered
in 8 provinces and one
territory2. Opt-out system3
Alberta, New
Brunswick,
Newfoundland and
Labrador, Nova
Scotia, Prince
Edward Island and
the Yukon
Netherlands Universal Screening
(for study only)
National Universal
screening
Not reported to
parents
United
Kingdom
Universal screening
offered for women
at risk1
Offered as a choice. Opt-in 4 Reported to parents.
No national reporting
standard
United States of
America
Offered at health
care provider’s
discretion
National Universal
screening (50 states). Opt-
out system3
Reported to parents.
No national reporting
standard
1 Includes women of African, Black Caribbean, Mediterranean, Middle Eastern or South
Asian descent (26)
2 Offered in Alberta, British Columbia, New Brunswick, Newfoundland and Labrador, Nova
Scotia, Ontario, Prince Edward Island, Quebec and the Yukon (28)
3 Opt-out system – Infants will be tested automatically unless parent(s) decline(s). A refusal
form required to be signed (28)
4 Opt-in system – Parent(s) is/are offered newborn screen. Consent required (27)


















