
environmentally friendly corrosion inhibitors for amine based co2 ...
176
ENVIRONMENTALLY-FRIENDLY CORROSION INHIBITORS FOR THE AMINE-BASED CO 2 ABSORPTION PROCESS A Thesis Submitted to the Faculty of Graduate Studies and Research In Partial Fulfillment of the Requirements for the Degree of Master of Applied Science in Process Systems Engineering University of Regina By Sureshkumar Srinivasan Regina, Saskatchewan November, 2012 Copyright © 2012: Sureshkumar Srinivasan
-
Upload
duongkhuong -
Category
Documents
-
view
244 -
download
2
Transcript of environmentally friendly corrosion inhibitors for amine based co2 ...

ENVIRONMENTALLY-FRIENDLY CORROSION INHIBITORS FOR THE
AMINE-BASED CO2 ABSORPTION PROCESS
A Thesis
Submitted to the Faculty of Graduate Studies and Research
In Partial Fulfillment of the Requirements for the
Degree of Master of Applied Science in
Process Systems Engineering
University of Regina
By
Sureshkumar Srinivasan
Regina, Saskatchewan
November, 2012
Copyright © 2012: Sureshkumar Srinivasan

UNIVERSITY OF REGINA
FACULTY OF GRADUATE STUDIES AND RESEARCH
SUPERVISORY AND EXAMINING COMMITTEE
Sureshkumar Srinivasan, candidate for the degree of Master of Applied Science in Process Systems Engineering, has presented a thesis titled, Environmentally-Friendly Corrosion Inhibitors for the Amine-Based CO2 Absorption Process, in an oral examination held on November 13, 2012. The following committee members have found the thesis acceptable in form and content, and that the candidate demonstrated satisfactory knowledge of the subject material. External Examiner: Dr. Farshid Torabi, Petroleum Systems Engineering
Supervisor: Dr. Amornvadee Veawab, Process Systems Engineering
Committee Member: Dr. Amr Henni, Process Systems ENgineering
Committee Member: Dr. Adisorn Aroonwilas, Industrial Systems Engineering
Chair of Defense: Dr. Doug Durst, Faculty of Social Work *Not present at defense

i
ABSTRACT
Corrosion in an amine-based carbon dioxide (CO2) absorption process is one of
the most serious operational problems affecting both plant safety and economics.
Corrosion inhibitors are widely applied for corrosion control, mainly due to their
adaptability. However, the most effective corrosion inhibitors are generally toxic or
heavy metal based, which makes their handling and disposal difficult and expensive.
Owing to increasing environmental regulations, the search for an environmentally-
friendly corrosion inhibitor is more relevant now than ever before. In this work, a pool of
environmentally-friendly corrosion inhibitors for the CO2 absorption process was
identified based on the principles of hard and soft acids and bases (HSAB), toxicity
properties, and quantum chemical analysis. Eight compounds were experimentally tested
using electrochemical techniques. The experiments were carried out to evaluate inhibition
performance on carbon steel in 5.0 kmol/m3 monoethanolamine (MEA) solution at 80
oC
and 0.55 mol/mol CO2 loading. The effects of corrosion inhibitor concentration and
process contaminants (i.e., formate and chloride) on inhibition performance were also
studied.
The results show that the tested corrosion inhibitors reduced the corrosion rate
from 4.27 mmpy (uninhibited) to 0.35 to 2.50 mmpy (i.e., corrosion inhibition
efficiencies were in the range of 20 to 92%). The highest corrosion inhibition efficiency
was obtained for sodium thiosulfate, which was 92% in the absence of chloride and
formate. 2-aminobenzene sulfonic acid, 3-aminobenzene sulfonic acid, 4-aminobenzene
sulfonic acid, sulfapyridine, and sulfolane showed corrosion inhibition efficiencies in the

ii
range of 85 to 90%. Sulfanilamide and thiosalicylic acid were removed from the
screening tests due to their performance and incompatibility with the solution,
respectively. The inhibition efficiency of sodium thiosulfate and sulfolane was not
affected by the presence of chloride and formate. However, the inhibition efficiency of 3-
aminobenzene sulfonic acid and sulfapyridine deteriorated in the presence of chloride.
Those of 2-aminobenzene sulfonic acid, 3-aminobenzene sulfonic acid, and 4-
aminobenzene sulfonic acid were reduced in the presence of formate.
Based on the electrochemical results, only four compounds, namely 4-
aminobenzene sulfonic acid, sulfapyridine, sulfolane, and sodium thiosulfate were further
tested using weight loss techniques for 28 days. Despite their promise in electrochemical
tests, sodium thiosulfate and 4-aminobenzene sulfonic acid did not perform well in
longer-duration tests. Sulfapyridine and sulfolane, on the other hand, were found to be
effective.

iii
ACKNOWLEDGEMENTS
First, I would like to express my earnest gratefulness to my mentor,
Dr. Amornvadee Veawab, for providing me with constant guidance, support, direction,
freedom, and encouragement both professionally and personally throughout my research
career to help me successfully complete my research work. I would also like to thank
Dr. Adisorn Aroonwilas for his help in setting up my experiments and invaluable
suggestions for my research. I would also like to extend my thanks to the Faculty of
Graduate Studies and Research at the University of Regina and Natural Sciences and
Engineering Research Council (NSERC) for their financial support.
I take this opportunity to express my heartfelt gratitude to my parents, Srinivasan
and Adilakshmi, my brother Sathish, and my fiancé Soundarya for their unconditional
love and understanding through the dips and peaks of my life. I would also like to express
my gratefulness to my dear friend (Late) Hariprakash and his parents Ramamoorthy and
Uma Rani for their love and support.
I would like to extend special thanks to my dearest friends Pathi, Ameer, Prakash,
Avinash, Neelu, Ranga, Rengu, Ganesh, Balaji, Ezhiyl, Sridhar, Mani, and my colleagues
for their support and motivation.

iv
TABLE OF CONTENTS
ABSTRACT i
ACKNOWLEDGEMENT iii
TABLE OF CONTENTS iv
LIST OF TABLES viii
LIST OF FIGURES ix
NOMENCLATURE xvii
1. INTRODUCTION 1
1.1 Carbon capture from industrial waste gas 1
1.2 Corrosion and its impacts 4
1.3 Corrosion inhibitors 11
1.4 Research Motivation 17
1.5 Research Objectives and Scope 21
2. FUNDAMENTALS OF CORROSION AND CORROSION 24
INHIBITION
2.1 Thermodynamic aspects of corrosion 24
2.1.1 Origin of Electrode potential 24
2.1.2 Electrode Processes 25
2.1.3 Concept of Mixed Potential 27
2.1.4 Free Energy - Electrode potential Relationship 28

v
2.2 Kinetics of Corrosion 28
2.2.1 Faraday’s law 30
2.2.2 Polarization 31
2.2.2.1 Activation polarization 34
2.2.2.2 Concentration polarization 35
2.2.2.3 Combined polarization 37
2.3 Passivity 38
2.4 Corrosion characterization techniques 39
2.4.1 Tafel extrapolation 39
2.4.2 Potentiodynamic cyclic polarization 41
2.4.3 Electrochemical impedance spectroscopy 41
2.5 Corrosion control techniques – Corrosion inhibitors 47
2.5.1 Anodic Inhibitors 48
2.5.2 Cathodic Inhibitors 48
2.5.3 Film forming inhibitors 50
2.6 Selection of corrosion inhibitors 50
2.7 Quantum chemical analysis of corrosion inhibitors 52
3. SELECTION AND TESTING OF CORROSION INHIBITORS 55
3.1 Selection of tested corrosion inhibitors 55
3.1.1 Selection of compounds 55
3.1.2 Toxicity evaluation 58
3.1.3 Quantum chemical analysis 62

vi
3.2 Corrosion testing 66
3.2.1 Electrochemical experiments 66
3.2.1.1 Experimental setup 66
3.2.1.2 Specimen preparation 68
3.2.1.3 Solution preparation 68
3.2.1.4 Experimental procedure 70
3.2.1.5 Validation of experimental setup 73
3.2.1.6 Data analysis 75
3.2.2 Weight loss experiments 76
3.2.2.1 Experimental setup 76
3.2.2.2 Specimen preparation 78
3.2.2.3 Solution preparation 78
3.2.2.4 Experimental procedure 78
3.2.2.5 Weight loss analysis 80
3.2.2.6 Surface analysis 82
4. RESULTS AND DISCUSSION 83
4.1 Electrochemical tests 83
4.1.1 Corrosion behaviour of uninhibited MEA systems 83
4.1.2 Corrosion behaviour of inhibited MEA systems 92
4.1.2.1 2-aminobenzenesulfonic acid 92
4.1.2.2 3-aminobenzenesulfonic acid 97

vii
4.1.2.3 4-aminobenzenesulfonic acid 102
4.1.2.4 Sulfapyridine 107
4.1.2.5 Sulfanilamide 111
4.1.2.6 Sulfolane 113
4.1.2.7 Thiosalicylic acid 116
4.1.2.8 Sodium thiosulfate 120
4.1.3 Comparison of corrosion inhibition performance of 126
different inhibitors
4.2 Weight loss tests 130
4.2.1 Corrosion behaviour of uninhibited MEA systems 130
4.2.2 Corrosion behaviour of inhibited MEA systems 134
5. CONCLUSIONS AND FUTURE WORK 143
5.1 Conclusions 143
5.2 Recommendations for future work 144
REFERENCES 146

viii
LIST OF TABLES
Table 1.1 Summary of plant experience on corrosion in gas treating plants 6
Table 1.2 Summary of corrosion inhibitors in gas treating plants 12
Table 1.3 Ecological information of patented corrosion inhibitors 19
Table 3.1 List of selected compounds 56
Table 3.2 Toxicity of absorbents 60
Table 3.3 Ecological information of selected corrosion inhibitors 61
Table 3.4 Quantum chemical parameters for selected compounds 64
Table 3.5 Summary of chemicals used in corrosion experiments 72
Table 4.1 Summary of experimental parameters and conditions 84
Table 4.2 Summary of electrochemical experimental Results 86
Table 4.3 Summary of weight loss experimental results 131

ix
LIST OF FIGURES
Figure 1.1 Process flow diagram for the amine-based CO2 absorption 3
process
Figure 2.1 Corrosion of Iron in deaerated hydochloric acid solution 26
Figure 2.2 Evans Diagram for mixed potential 29
Figure 2.3 A typical electrochemical cell a) Daniel Cell b) Polarization 33
behaviour of Daniell cell
Figure 2.4 Types of polarization a) Activation polarization 36
(b) Concentration polarization (c) Mixed polarization
Figure 2.5 Active passive transition behaviour of a metal 40
Figure 2.6 A typical Tafel plot 42
Figure 2.7 Typical potentiodynamic cyclic polarization curves 43
(a) No pitting (b) Pitting
Figure 2.8 Impedance analyses for a corroding metal surface without 45
diffusion control (a) Equivalent circuit for a corroding
metal surface b) Nyquist plot for the equivalent circuit
Figure 2.9 Impedance analyses for a corroding metal surface with 46
diffusion control (a) Equivalent circuit for a corroding
metal surface (b) Nyquist plot for the equivalent circuit
Figure 2.10 Types of inhibitors (a) Anodic inhibitors (b) Cathodic 49
inhibitors (c) Film forming inhibitors

x
Figure 3.1 Trends of different quantum chemical parameters 65
(a) Highest occupied molecular orbital energy (EHOMO)
(b) Energy gap (∆E) (c) Dipole moment (µ) (d) Fraction
of electron transferred (∆N) (e) Charge of the sulfur atom (Zs)
Figure 3.2 Experimental setup for electrochemical corrosion testing 67
Figure 3.3 A sketch of electrochemical testing specimens 69
Figure 3.4 Chittick’s apparatus for CO2 loading measurement 71
Figure 3.5 Validation of experimental setup and procedure (a) Validation 74
of potentiodynamic polarization using ASTM G5-94 (2004)
(b) Validation of impedance measurement using ASTM
G106-89 (2010)
Figure 3.6 A schematic diagram of the experimental setup for corrosion 77
weight loss testing
Figure 3.7 A sketch of the weight loss testing specimen 79
Figure 3.8 Estimation of weight loss of the tested specimen (ASTM G1-90) 81
Figure 4.1 Corrosion behaviour of uninhibited MEA solutions (5.0 kmol/m3 85
MEA, 80oC, 0.55 mol/mol CO2 loading and no process
contaminant) (a) Polarization behaviour (b) Impedance behaviour
Figure 4.2 Corrosion behaviour of uninhibited MEA solutions with and 90
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading) (a) Comparison of corrosion rate
(b) Polarization behaviour (c) Comparison of polarization
resistance (d) Impedance behaviour

xi
Figure 4.3 Corrosion behaviour of ‘2-aminobenzene sulfonic acid’ inhibited 93
MEA solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2
loading, no process contaminant) (a) Comparison of corrosion
rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance
(e) Impedance behaviour
Figure 4.4 Corrosion behaviour of inhibited MEA solutions with and 96
Without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 1000 ppm 2-aminobenzene
sulfonic acid) (a) Comparison of corrosion rate (b) Comparison
of inhibition efficiencies (c) Polarization behaviour
(d) Comparison of polarization resistance (e) Impedance
behaviour
Figure 4.5 Corrosion behaviour of ‘3-aminobenzene sulfonic acid’ 98
inhibited MEA solutions (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, no process contaminant)
(a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour
(d) Comparison of polarization resistance
(e) Impedance behaviour

xii
Figure 4.6 Corrosion behaviour of inhibited MEA solutions with and 100
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 1000 ppm 3-aminobenzene
sulfonic acid) (a) Comparison of corrosion rate
(b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance
(e) Impedance behaviour
Figure 4.7 Pitting tendency of 3-aminobenzene sulfonic acid in presence of 101
chloride (a) Cyclic polarization curve indicating tendency for
pitting (b) SEM images showing pitted areas (5.0 kmol/m3
MEA, 80oC, 0.55 mol/mol CO2 loading, 1000 ppm
3-aminobenzene sulfonic acid and 10000 ppm chloride)
Figure 4.8 Corrosion behaviour of ‘4-aminobenzene sulfonic acid’ inhibited 103
MEA solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2
loading, no process contaminant) (a) Comparison of corrosion
rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance
(e) Impedance behaviour
Figure 4.9 Corrosion behaviour of inhibited MEA solutions with and 106
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 1000 ppm 4-aminobenzene
sulfonic acid) (a) Comparison of corrosion rate (b) Comparison
of inhibition efficiencies (c) Polarization behaviour

xiii
(d) Comparison of polarization resistance
(e) Impedance behaviour
Figure 4.10 Corrosion behaviour of ‘sulfapyridine’ inhibited MEA solutions 108
(5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process
contaminant) (a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour (d) Comparison of
polarization resistance (e) Impedance behaviour
Figure 4.11 Corrosion behaviour of inhibited MEA solutions with and 110
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 2000 ppm sulfapyridine)
(a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour
(d) Comparison of polarization resistance
(e) Impedance behaviour
Figure 4.12 Corrosion behaviour of ‘sulfanilamide’ inhibited MEA solutions 112
(5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process
contaminant) (a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour (d) Comparison
of polarization resistance (e) Impedance behaviour
Figure 4.13 Corrosion behaviour of ‘sulfolane’ inhibited MEA solutions 114
(5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process
contaminant) (a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour

xiv
(d) Comparison of polarization resistance (e) Impedance
behaviour
Figure 4.14 Corrosion behaviour of inhibited MEA solutions with and 117
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 2000 ppm sulfolane) (a) Comparison
of corrosion rate (b) Comparison of inhibition efficiencies
(c) Polarization behaviour (d) Comparison of polarization
resistance (e) Impedance behaviour
Figure 4.15 Corrosion behaviour of ‘thiosalicylic acid’ inhibited MEA 119
Solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2
loading, no process contaminant) (a) Comparison of corrosion
rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance
(e) Impedance behaviour
Figure 4.16 Corrosion behaviour of ‘sodium thiosulfate’ inhibited MEA 121
solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading,
no process contaminant) (a) Comparison of corrosion rate (b)
Comparison of inhibition efficiencies (c) Comparison of
polarization resistance (d) Impedance behaviour
Figure 4.17 (a) Polarization behaviour of ‘sodium thiosulfate’ inhibited MEA 122
solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading,
no process contaminant) (b) Working electrode after stable open
circuit potential in presence of sodium thiosulfate (5.0 kmol/m3

xv
MEA, 80oC, 0.55 mol/mol CO2 loading and 1000 ppm sodium
thiosulfate)
Figure 4.18 Corrosion behaviour of inhibited MEA solutions with and 124
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 1000 ppm sodium thiosulfate)
(a) Comparison of corrosion rate (b) Comparison of inhibition
efficiencies (c) Polarization behaviour (d) Comparison of
polarization resistance (e) Impedance behaviour
Figure 4.19 Corrosion behaviour of inhibited MEA solutions with and 127
without process contaminants (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading) (a) Comparison of corrosion
rate (b) Comparison of polarization resistance (c) Comparison
of inhibition efficiencies
Figure 4.20 Comparison of corrosion rates of inhibited MEA solutions 132
(5.0 kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading,
no process contaminant)
Figure 4.21 Surface analysis of tested specimen after 28 days – (a) SEM 133
images (500X Magnification) (b) EDS spectra (c) XRD spectra
(5.0 kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading,
uninhibited)
Figure 4.22 Surface analysis of fresh CS1018 specimen (a) SEM image 135
(Magnification – 500 X) (b) EDS analysis with Wt% of the
element

xvi
Figure 4.23 Comparison of corrosion rates of inhibited MEA solutions 136
(5.0 kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading, no
process contaminant)
Figure 4.24 Surface analysis of tested specimen after 28 days – (a) SEM 137
images (500X Magnification) (b) EDS spectra (c) XRD spectra
(5 kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading,
1000 ppm 4-aminobenzene sulfonic acid)
Figure 4.25 Surface analysis of tested specimen after 28 days – (a) SEM 139
images (500X Magnification) (b) EDS spectra (c) XRD spectra
(5kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading, 2000
ppm sulfapyridine)
Figure 4.26 Surface analysis of tested specimen after 28 days – (a) SEM 140
images (500X Magnification) (b) EDS spectra (c) XRD spectra
(5 kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading,
2000 ppm sulfolane)
Figure 4.27 Surface analysis of tested specimen after 28 days – (a) SEM 141
images (500X Magnification) (b) EDS spectra (c) XRD spectra
(5 kmol/m3 MEA, 80
oC and 0.55 mol/mol CO2 loading, 1000 ppm
sodium thiosulfate)

xvii
NOMENCLATURE
a Atomic weight (g/mol)
ap Activity of products
ar Activity of reactants
A Electron affinity (eV)
AC Alternating current
AMP 2-Amino-2-methyl-1-propanol
ASTM American Society for Testing and Materials
C Capacitance (farad)
CAA Clean Air Act
CCS Carbon capture and storage
Cdl Double layer capacitance (μF/cm2)
CE Counter electrode
CEPA Canadian Environmental Protection Act
CR Corrosion rate (mmpy)
CS Carbon steel
CWA Clean Water Act
oC Degree Centigrade
Di Diffusion coefficient
D Density (g/cm3)
DC Direct current
DEA Diethanolamine

xviii
DGA Diglycolamine
DIPA Diisopropanolamine
E Electrode potential (V)
Eo Standard electrode potential (V)
Eb Breakdown potential or pitting potential (V)
Ecorr Corrosion potential (V)
EDS Energy-dispersive X-ray spectroscopy
EHOMO Highest occupied molecular orbital energy (eV)
ELUMO Lowest unoccupied molecular orbital energy (eV)
EIS Electrochemical Impedance Spectroscopy
EPA Environmental Protection Agency
Epp Primary passivation potential (V)
Erev Equilibrium potential (or Reversible potential) (V)
Erp Repassivation potential (V)
∆E Energy gap (eV)
f Frequency (Hz)
F Faraday’s constant (96,500 coulombs per mole)
GHG Greenhouse gas
ΔG Free energy change
HSAB Hard and soft acids and bases
ΔH Enthalpy change
ia Anodic current density (A/cm2)
ic Cathodic current density (A/cm2)

xix
icorr Corrosion current density (A/cm2)
icrit Critical current density (A/cm2)
iL Limiting current density (A/cm2)
io Equilibrium exchange current density (A/cm2)
ipass Passivation current density (A/cm2)
I Ionization potential (eV)
ICDD International Centre for Diffraction Data
IPCC Intergovernmental Panel for Climatic Change
LC50 Lethal concentration (dose large enough to kill 50% of sample animals
under test)
mmpy Millimetre per year
MDEA Methyldiethanolamine
MEA Monoethanolamine
MS Mild carbon steel
n Number of electrons per atom of the species involved in the reaction
n
Hardness (eV)
ΔN Fraction of electrons transferred
OCP Open circuit potential
pow Partition in octanol / water
PAR Princeton Applied Research
PLONOR Poses little or no risk
PM6 Parameterized model number 6
R Gas constant (JK-1
mol-1
)

xx
RE Reference electrode
RP Polarization resistance (ohm cm2)
RS Solution resistance (ohm cm2)
SCC Stress corrosion cracking
SEM Scanning electron microscopy
SS Stainless steel
ΔS Change in entropy
T Absolute temperature (oC)
TEA Triethanolamine
W Warburg impedance (ohm cm2)
WE Working electrode
Wt% Weight percent
XRD X-ray Powder Diffraction
Z Impedance (ohm cm2)
Z' Real impedance (ohm cm2)
Z" Imaginary impedance (ohm cm2)
Zs Charge on the sulfur atom

xxi
Greek Letters:
βa Anodic Tafel slope (mV/decade of current density)
βc Cathodic Tafel slope (mV/decade of current density)
Ƞa Activation polarization (V)
Ƞc Concentration polarization (V)
Ƞdiss Dissolution overpotential (V)
Ƞredn Reduction overpotential (V)
ȠT Total polarization (V)
θ Phase angle (degree)
µ Dipole moment (debye)
χ Electronegativity (eV)
ω Angular frequency

1
1. INTRODUCTION
1.1 Carbon capture from industrial waste gas
The Intergovernmental Panel for Climatic Change (IPCC) has reported that
between 1995 and 2006, eleven out of twelve years were the warmest in the instrumental
record of global surface temperature [IPCC1, 2007]. Melting of glaciers and continual
increases in sea level are the direct effects of global warming. This is mainly attributed to
the increase in the atmospheric concentrations of greenhouse gases (GHGs) in recent
times, which is evident from the fact that GHG emissions now are 70% higher than their
value in the 1970s [IPCC1, 2007]. Particularly, carbon dioxide (CO2) is the most
significant greenhouse gas as its emissions have increased by 80% in the same time
frame, and CO2 represented 77% of the total anthropogenic GHG emissions in 2004
[IPCC1, 2007]. Coal-fired power plants, natural gas processing plants, and manufacturing
industries such as cement, ammonia, and steel plants are some of the major sources of
CO2 emissions [IPCC2, 2005]. Among the above, coal-fired power plants assume specific
importance as they typically contribute to approximately 30% of the total CO2 emissions
[Aaron and Tsouris, 2005].
Carbon capture and storage (CCS) is a technology used to remove CO2 from
industrial flue gas especially from power plants where it can effect a gross reduction of
CO2 emissions by approximately 85 - 95% [IPCC2, 2005]. The CO2 removal can be
accomplished by a number of processes such as membrane separation, adsorption onto
solids, and absorption into liquids. However, the latter is most commonly used for gas
treating applications [Astarita et al., 1983]. The industrial separation of CO2 for natural

2
gas processing and ammonia manufacture by absorption into liquid is a mature
technology and has been successfully used for many decades. However, adaptation of this
technology for flue gas treatment began only in the 1980s [Kittel et al., 2009]. For
example, IMC Global Inc (previously North American chemicals), in Trona, USA,
features a CO2 capture unit that is used to sequester CO2 from flue gas from a coal-fired
power generation plant that started operation in 1978 and is still functioning. Similarly,
Indo-Gulf Corporation, a fertilizer industry in India, features CO2 capture from flue gas
of the ammonia reformer unit that has been operational since 1988 capturing 150 tonnes
CO2/day. Bellingham Cogeneration facility, Massachusetts, USA, produces food grade
CO2 by treating 300 tonnes/day of CO2 from flue gas emitted from an electricity
generation plant since 1991. Sumitomo Chemicals, Japan, treats flue gas generated from
onsite boilers and coal/oil boilers since 1994 with a capacity of 150 tonnes CO2/day. As
illustrated by the above examples of successful and continuous adaptation of this
technology for the past three decades, it is clearly discernible that the flue gas treatment
using absorption into liquid is viewed as a promising technology.
In a typical CO2 absorption process as illustrated in Figure 1.1, a flue gas stream
containing CO2 enters the absorber from the bottom and interacts counter-currently with
down flowing chemical solvent entering from top. CO2 reacts with the solvent and is
absorbed, rendering the gas stream with permissible levels of CO2, and the treated gas
leaves the absorber top. The CO2 loaded rich solvent leaving the bottom of the absorber
passes through the rich-lean heat exchanger where it is preheated and then enters the
regenerator from the top where, on application of heat in the form of steam, the solvent is
stripped of CO2, and the lean solvent is recycled back into the absorber after being cooled
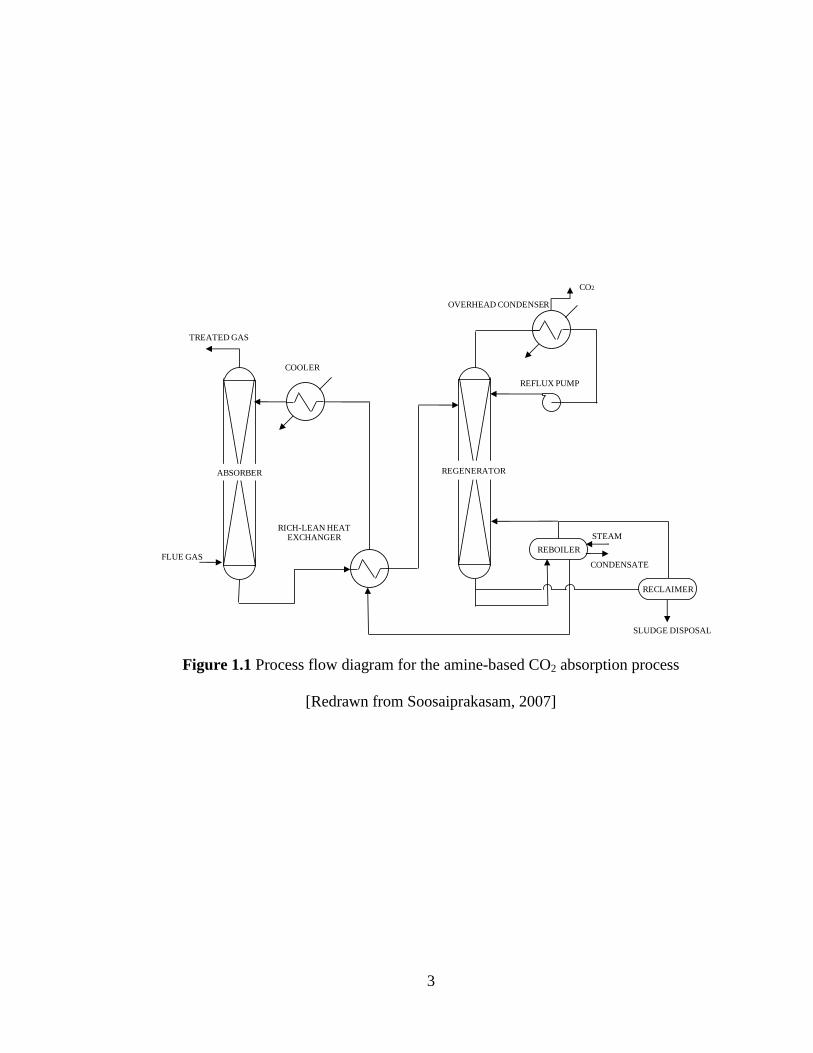
3
Figure 1.1 Process flow diagram for the amine-based CO2 absorption process
[Redrawn from Soosaiprakasam, 2007]
ABSORBER
TREATED GAS
COOLER
RICH-LEAN HEAT EXCHANGER
FLUE GAS
STEAM
CONDENSATE
OVERHEAD CONDENSER
CO2
REFLUX PUMP
REGENERATOR
REBOILER
RECLAIMER
SLUDGE DISPOSAL

4
down to the required operating temperature. A portion of lean solvent is withdrawn at the
reclaimer where it is heated and the vapour mixture containing amine and CO2 are
reintroduced into the regenerator. From the bottom of the reclaimer, a sludge containing
insoluble salts and other chemicals are obtained which is removed for waste handling.
The vapor mixture containing CO2 and water vapor leaves the regenerator and enters the
overhead condenser where most of the water vapor is condensed and recycled back to the
regenerator and the concentrated CO2 leaves the overhead condenser [Aroonwilas, 1996].
A wide range of absorption solvents have been used for CO2 absorption
processes, among which, aqueous alkanolomine-based solvents are the most widely used
absorbents. Alkanolamines can be classified into three categories, namely, primary,
secondary, and tertiary amines. Monoethanolamine (MEA) and diglycolamine (DGA)
belong to the primary type whereas diethanolamine (DEA) and diisopropanolamine
(DIPA) are the secondary type. Methyldiethanolamine (MDEA) and triethanolamine
(TEA) are examples of tertiary amines. In general, primary amines have high reaction
rates with CO2, followed by secondary amines and tertiary amines, respectively [Veawab,
2000]. Since, for flue gas applications, CO2 partial pressures are low and the gas flow rate
is extremely high compared to natural gas processing, the absorption rate has to be
correspondingly faster. With this consideration, MEA shows promise and could well be
the first available solvent absorbent for this application [Kittel et al., 2009; Kittel et al.,
2010].

5
1.2 Corrosion and its impacts
A typical CO2 absorption process can have a number of factors that can cause
operational difficulties, but corrosion is the chief influencing factor from an economic
perspective [Kohl and Nielson, 1997]. Corrosion can greatly influence both economics
and safety associated with the CO2 absorption process. The economic losses are caused
by unplanned downtime, production losses, and reduced equipment life or safety issues
such as injury or death of plant personnel [Dupart et al., 1993]. A summary of plant
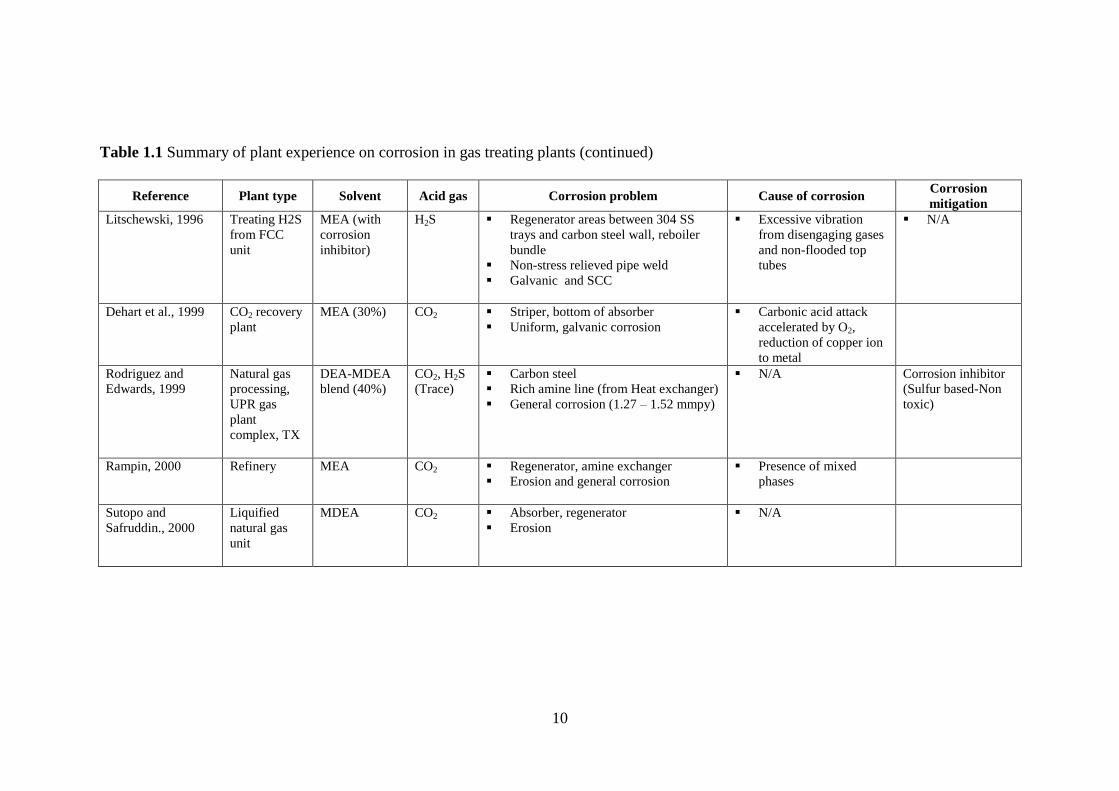
experiences on corrosion in the CO2 absorption process is given in Table 1.1.
From Table 1.1, it can be observed that the absorber bottom, regenerator, heat
exchanger, and associated piping and valves are areas susceptible to severe corrosion.
Both general (uniform) and localized corrosion were observed in CO2 absorption plants.
Localized corrosion such as erosion corrosion due to the presence of foreign particles in
the circulating solution and pitting corrosion are reported to occur in addition to galvanic
corrosion, stress corrosion cracking (SCC), and intergranular corrosion. Acid gas flashing
on walls, high lean loading, high solution velocities, the presence of particulate
contaminants, coupling of dissimilar alloys, and improper metal stress treatment are some
of the reported causes of corrosion. Corrosion mitigation measures include use of
corrosion inhibitors, design measures to reduce acid gas flashing, and replacement of
carbon steel with corrosion resistant alloys in the heat exchanger and regenerator areas
(trays and valves) in some cases.

6
Table 1.1 Summary of plant experience on corrosion in gas treating plants
Reference Plant type Solvent Acid gas Corrosion problem Cause of corrosion Corrosion
mitigation
Dingman et al.,
1966
Sour gas
treating plant
MEA CO2 and
H2S
Rich-lean heat exchanger, solution
letdown valve, piping downstream
letdown valve, upper portion of
regenerator
Erosion corrosion
Flashing of acid gas
from hot surface
High solution velocity
Change in direction of
fluid flow
Contamination with
solids such as iron
oxide, iron sulfide, mill
scale and sand
N/A
Smith and
Younger., 1972
Twenty-four
Sour gas
treating plants
in western
Canada
DEA CO2 and
H2s
Erosion corrosion
- Rich lean heat exchanger
- Regenerator
- Reboiler-vapor line and letdown
- Rich solution piping
Stress corrosion cracking
- Stainless steel heat exchanger
Contamination of
foreign particles in
circulating solution
High solution velocity
(5.5 ft/s)
Insufficient liquid level
over tight tube spacing
in reboiler and heat
exchanger
Chloride ion evolved
from gasket material
used between plates
N/A
Heisler and Weiss,
1975
Natural gas
treating plant,
Aderklaa,
Austria
MEA CO2 and
H2S
Tray type regenerator including wall
internal, downcomer, circumferential
joint, weld seam and joint, and tray
Uniform, pitting, erosion
Cavity in vapor flash N/A
Schmeal et al., 1978 Sour gas
treating plant
Sulfinol
(DIPA +
sulfolane)
CO2 and
H2S
Absorber below 5th
tray
Pitting
Acid gas flashing N/A
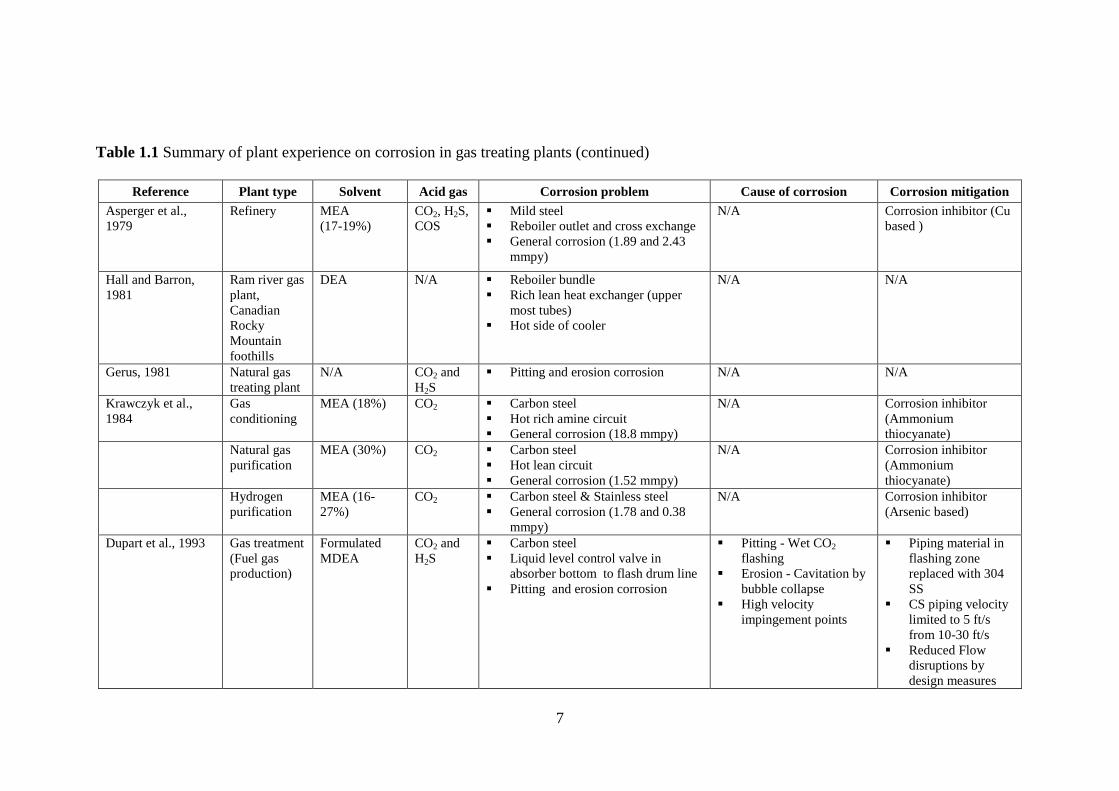
7
Table 1.1 Summary of plant experience on corrosion in gas treating plants (continued)
Reference Plant type Solvent Acid gas Corrosion problem Cause of corrosion Corrosion mitigation
Asperger et al.,
1979
Refinery MEA
(17-19%)
CO2, H2S,
COS
Mild steel
Reboiler outlet and cross exchange
General corrosion (1.89 and 2.43
mmpy)
N/A Corrosion inhibitor (Cu
based )
Hall and Barron,
1981
Ram river gas
plant,
Canadian
Rocky
Mountain
foothills
DEA N/A Reboiler bundle
Rich lean heat exchanger (upper
most tubes)
Hot side of cooler
N/A N/A
Gerus, 1981 Natural gas
treating plant
N/A CO2 and
H2S
Pitting and erosion corrosion N/A N/A
Krawczyk et al.,
1984
Gas
conditioning
MEA (18%) CO2 Carbon steel
Hot rich amine circuit
General corrosion (18.8 mmpy)
N/A Corrosion inhibitor
(Ammonium
thiocyanate)
Natural gas
purification
MEA (30%) CO2 Carbon steel
Hot lean circuit
General corrosion (1.52 mmpy)
N/A Corrosion inhibitor
(Ammonium
thiocyanate)
Hydrogen
purification
MEA (16-
27%)
CO2 Carbon steel & Stainless steel
General corrosion (1.78 and 0.38
mmpy)
N/A Corrosion inhibitor
(Arsenic based)
Dupart et al., 1993 Gas treatment
(Fuel gas
production)
Formulated
MDEA
CO2 and
H2S
Carbon steel
Liquid level control valve in
absorber bottom to flash drum line
Pitting and erosion corrosion
Pitting - Wet CO2
flashing
Erosion - Cavitation by
bubble collapse
High velocity
impingement points
Piping material in
flashing zone
replaced with 304
SS
CS piping velocity
limited to 5 ft/s
from 10-30 ft/s
Reduced Flow
disruptions by
design measures
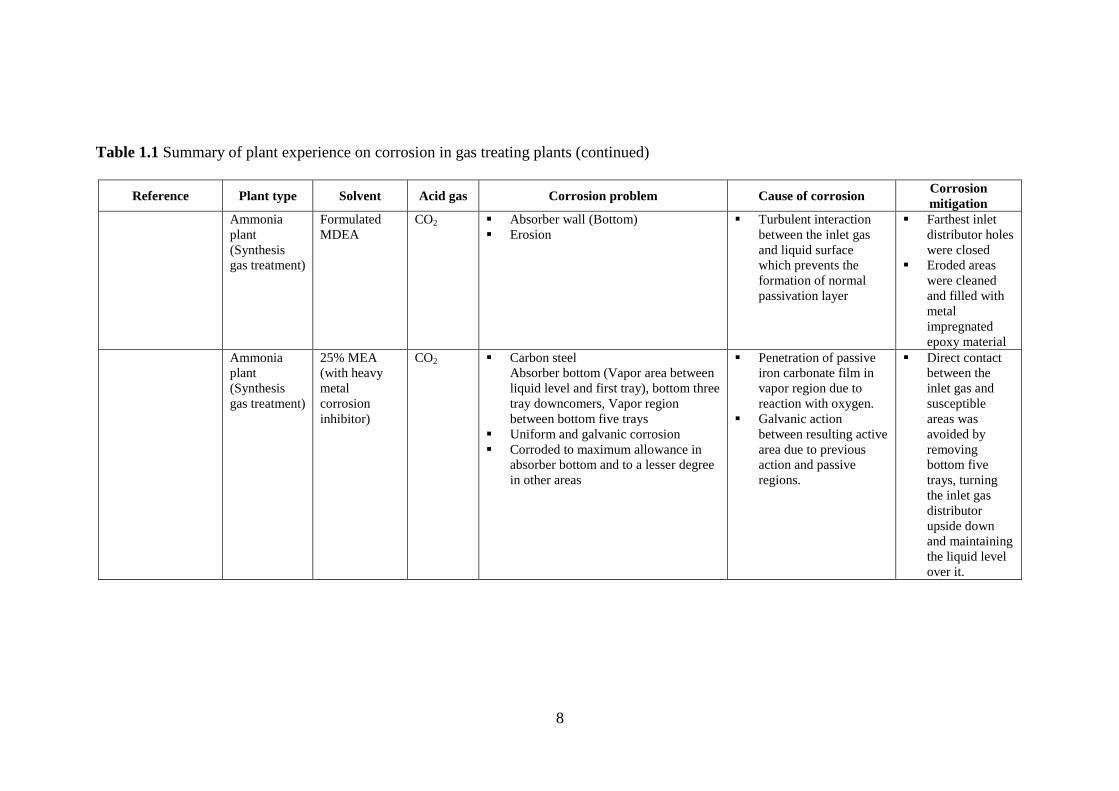
8
Table 1.1 Summary of plant experience on corrosion in gas treating plants (continued)
Reference Plant type Solvent Acid gas Corrosion problem Cause of corrosion Corrosion
mitigation
Ammonia
plant
(Synthesis
gas treatment)
Formulated
MDEA
CO2 Absorber wall (Bottom)
Erosion
Turbulent interaction
between the inlet gas
and liquid surface
which prevents the
formation of normal
passivation layer
Farthest inlet
distributor holes
were closed
Eroded areas
were cleaned
and filled with
metal
impregnated
epoxy material
Ammonia
plant
(Synthesis
gas treatment)
25% MEA
(with heavy
metal
corrosion
inhibitor)
CO2 Carbon steel
Absorber bottom (Vapor area between
liquid level and first tray), bottom three
tray downcomers, Vapor region
between bottom five trays
Uniform and galvanic corrosion
Corroded to maximum allowance in
absorber bottom and to a lesser degree
in other areas
Penetration of passive
iron carbonate film in
vapor region due to
reaction with oxygen.
Galvanic action
between resulting active
area due to previous
action and passive
regions.
Direct contact
between the
inlet gas and
susceptible
areas was
avoided by
removing
bottom five
trays, turning
the inlet gas
distributor
upside down
and maintaining
the liquid level
over it.
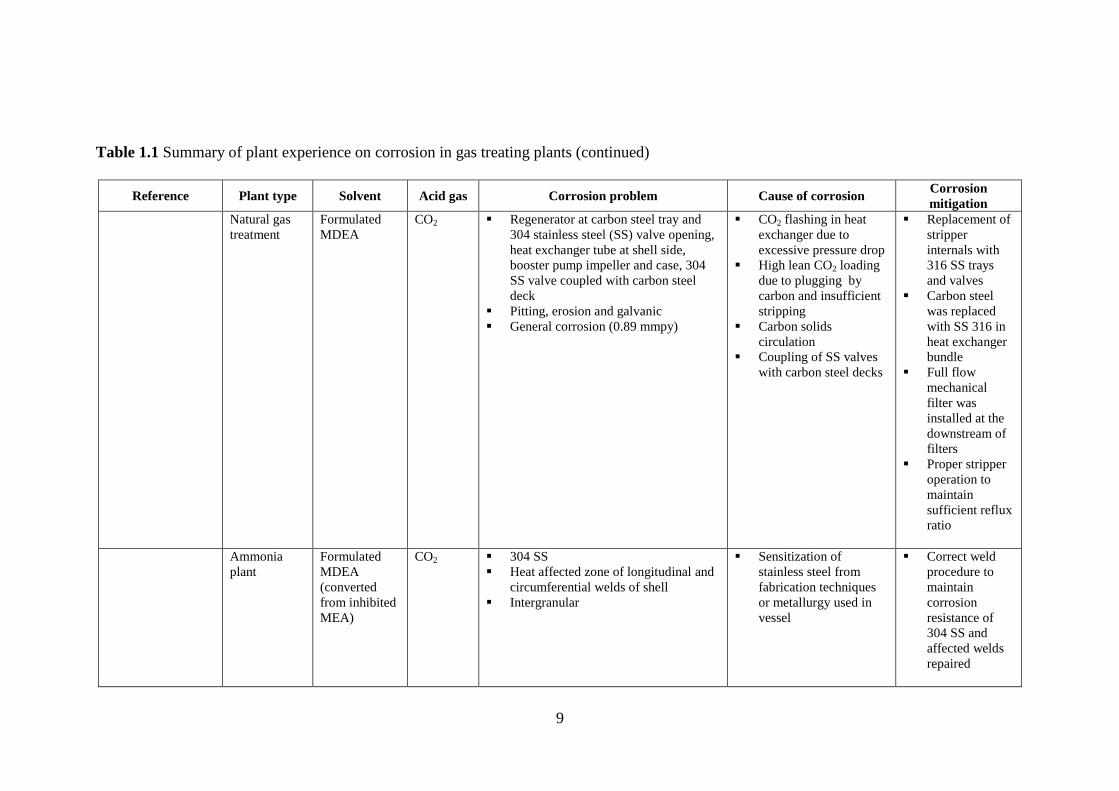
9
Table 1.1 Summary of plant experience on corrosion in gas treating plants (continued)
Reference Plant type Solvent Acid gas Corrosion problem Cause of corrosion Corrosion
mitigation
Natural gas
treatment
Formulated
MDEA
CO2 Regenerator at carbon steel tray and
304 stainless steel (SS) valve opening,
heat exchanger tube at shell side,
booster pump impeller and case, 304
SS valve coupled with carbon steel
deck
Pitting, erosion and galvanic
General corrosion (0.89 mmpy)
CO2 flashing in heat
exchanger due to
excessive pressure drop
High lean CO2 loading
due to plugging by
carbon and insufficient
stripping
Carbon solids
circulation
Coupling of SS valves
with carbon steel decks
Replacement of
stripper
internals with
316 SS trays
and valves
Carbon steel
was replaced
with SS 316 in
heat exchanger
bundle
Full flow
mechanical
filter was
installed at the
downstream of
filters
Proper stripper
operation to
maintain
sufficient reflux
ratio
Ammonia
plant
Formulated
MDEA
(converted
from inhibited
MEA)
CO2 304 SS
Heat affected zone of longitudinal and
circumferential welds of shell
Intergranular
Sensitization of
stainless steel from
fabrication techniques
or metallurgy used in
vessel
Correct weld
procedure to
maintain
corrosion
resistance of
304 SS and
affected welds
repaired
10
Table 1.1 Summary of plant experience on corrosion in gas treating plants (continued)
Reference Plant type Solvent Acid gas Corrosion problem Cause of corrosion Corrosion
mitigation
Litschewski, 1996 Treating H2S
from FCC
unit
MEA (with
corrosion
inhibitor)
H2S Regenerator areas between 304 SS
trays and carbon steel wall, reboiler
bundle
Non-stress relieved pipe weld
Galvanic and SCC
Excessive vibration
from disengaging gases
and non-flooded top
tubes
N/A
Dehart et al., 1999 CO2 recovery
plant
MEA (30%) CO2 Striper, bottom of absorber
Uniform, galvanic corrosion
Carbonic acid attack
accelerated by O2,
reduction of copper ion
to metal
Rodriguez and
Edwards, 1999
Natural gas
processing,
UPR gas
plant
complex, TX
DEA-MDEA
blend (40%)
CO2, H2S
(Trace)
Carbon steel
Rich amine line (from Heat exchanger)
General corrosion (1.27 – 1.52 mmpy)
N/A Corrosion inhibitor
(Sulfur based-Non
toxic)
Rampin, 2000 Refinery MEA CO2 Regenerator, amine exchanger
Erosion and general corrosion
Presence of mixed
phases
Sutopo and
Safruddin., 2000
Liquified
natural gas
unit
MDEA CO2 Absorber, regenerator
Erosion
N/A

11
1.3 Corrosion inhibitors
There are many alternative approaches to mitigating corrosion in CO2
absorption plants, such as proper equipment and process design, use of corrosion
resistant materials, side stream removal of particulate matters from amine solution,
and use of corrosion inhibitors. Among these, the use of corrosion inhibitors is
considered the most economical, mainly because it requires no major process
modification [Kohl and Nielson, 1997; Dupart et al., 1993; Veawab, 2000]. From the
plant experiences detailed in Table 1.1, it can be seen that despite the usage of
corrosion inhibitors, corrosion can still occur due to certain design problems such as
coupling of dissimilar metals and acid gas flashing in selected areas [Dupart et al.,
1993]. Hence, corrosion inhibitors in combination with one or all of the above
approaches have to be deployed for effective corrosion reduction. This thesis work
focused mainly on corrosion inhibitors.
A corrosion inhibitor is defined as a chemical substance that, when added in
small concentrations to the fluid phase of a corroding environment, are capable of
retarding corrosion by interacting either with the metal surface or the environment
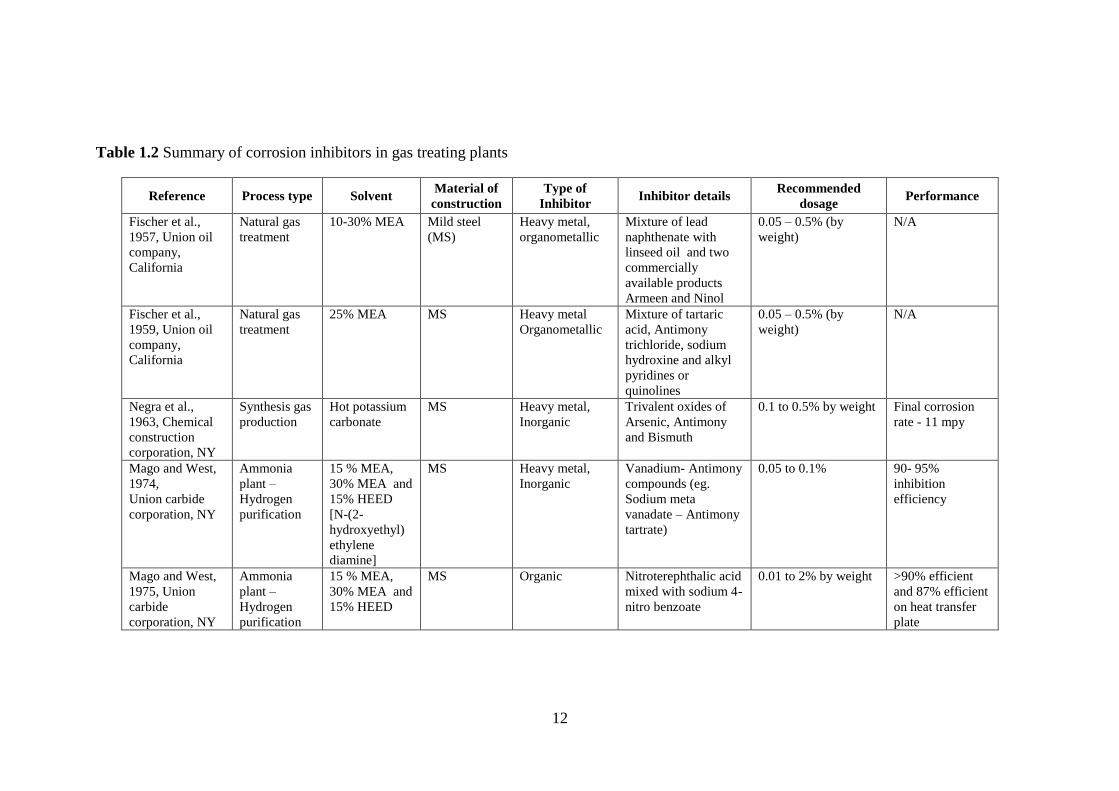
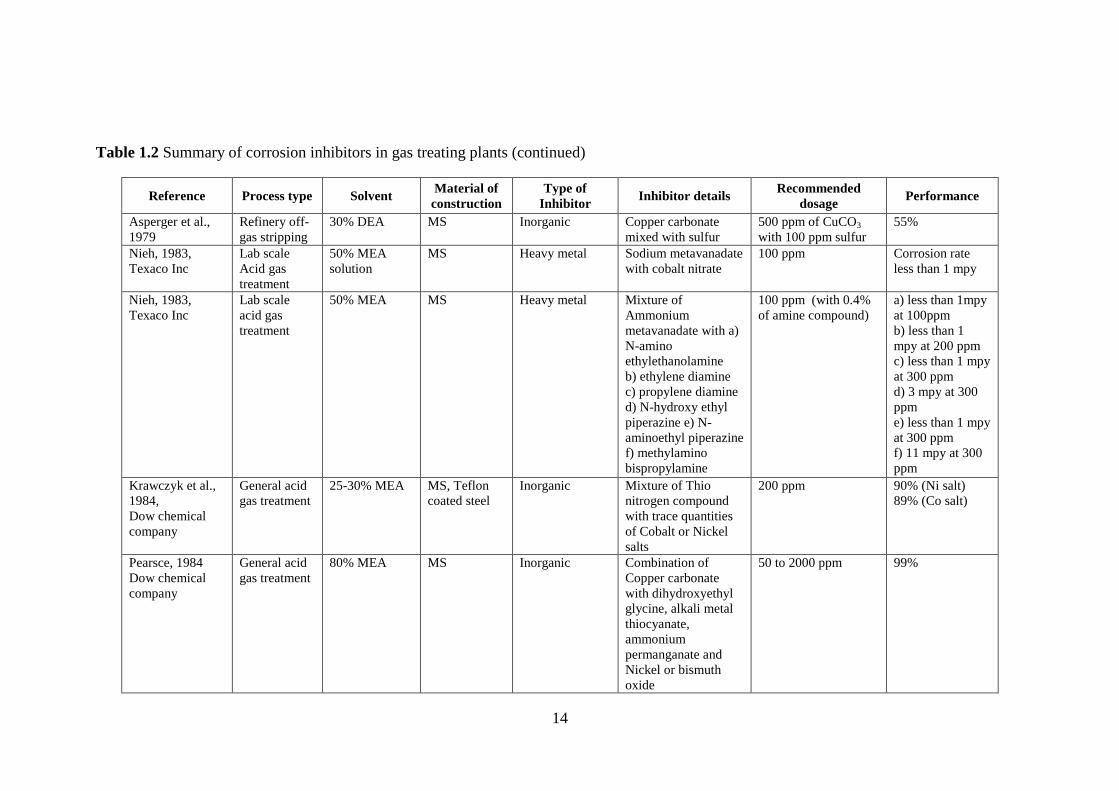
[Sastri, 2001]. A wide array of corrosion inhibitors were tested and patented for gas
treating applications over the past fifty years (Table 1.2), but the most effective ones
are based on heavy metals such as arsenic and vanadium [Dupart et al., 1993]. For a
period of around two decades, beginning in 1957, several heavy metal inhibitors such
as lead-based, antimony and bismuth-based, arsenic, vanadium, and tin-based
compounds were tested and patented. Despite being very effective inhibitors, their
usage was restricted because they are toxic inorganic compounds, which makes their
disposal difficult and costly [Kohl and Nielson, 1997]. As a result, a shift in trend
towards development of environmentally-friendly corrosion inhibitors was inevitable.
12
Table 1.2 Summary of corrosion inhibitors in gas treating plants
Reference Process type Solvent Material of
construction
Type of
Inhibitor Inhibitor details
Recommended
dosage Performance
Fischer et al.,
1957, Union oil
company,
California
Natural gas
treatment
10-30% MEA Mild steel
(MS)
Heavy metal,
organometallic
Mixture of lead
naphthenate with
linseed oil and two
commercially
available products
Armeen and Ninol
0.05 – 0.5% (by
weight)
N/A
Fischer et al.,
1959, Union oil
company,
California
Natural gas
treatment
25% MEA MS Heavy metal
Organometallic
Mixture of tartaric
acid, Antimony
trichloride, sodium
hydroxine and alkyl
pyridines or
quinolines
0.05 – 0.5% (by
weight)
N/A
Negra et al.,
1963, Chemical
construction
corporation, NY
Synthesis gas
production
Hot potassium
carbonate
MS Heavy metal,
Inorganic
Trivalent oxides of
Arsenic, Antimony
and Bismuth
0.1 to 0.5% by weight Final corrosion
rate - 11 mpy
Mago and West,
1974,
Union carbide
corporation, NY
Ammonia
plant –
Hydrogen
purification
15 % MEA,
30% MEA and
15% HEED
[N-(2-
hydroxyethyl)
ethylene
diamine]
MS Heavy metal,
Inorganic
Vanadium- Antimony
compounds (eg.
Sodium meta
vanadate – Antimony
tartrate)
0.05 to 0.1% 90- 95%
inhibition
efficiency
Mago and West,
1975, Union
carbide
corporation, NY
Ammonia
plant –
Hydrogen
purification
15 % MEA,
30% MEA and
15% HEED
MS Organic Nitroterephthalic acid
mixed with sodium 4-
nitro benzoate
0.01 to 2% by weight >90% efficient
and 87% efficient
on heat transfer
plate

13
Table 1.2 Summary of corrosion inhibitors in gas treating plants (continued)
Reference Process type Solvent Material of
construction
Type of
Inhibitor Inhibitor details
Recommended
dosage Performance
Mago, 1976,
Union carbide
corporation, NY
Lab scale
acid gas
treatment
Hot potassium
carbonate (with
5%
bicarbonate)
MS (Cold
rolled)
Heavy metal,
Inorganic
a)Vanadium
compounds
b) Antimony
compounds
c) Combination of
above
0.01 to 2% a) -400%
(aggravated
corrosion)
b) -70%
(aggravated
corrosion)
c) 84-90%
Mago and West,
1976,
Union carbide
corporation, NY
Ammonia
plant –
Hydrogen
purification
30% MEA,
30% 1:1
MEA:HEED
MS Heavy metal,
Inorganic
Stannous tartarate 0.01 to 2% 80-95%
Davidson et al.,
1978, Dow
chemical
company
Natural or
synthesis gas
treatment
(pilot plant)
30% MEA MS Inorganic, Reaction product of
copper and sulfur
yielding compounds
with
Monoethanolamine
20 – 20000 ppm
(0.002 – 2%)
N/A
Asperger and
Clouse, 1978,
Dow chemical
company
Natural or
synthesis gas
treatment
30% MEA MS Organic Tetradecyl
polyalkylpyridinium
bromide with
polyethylene
polyamine
100 ppm 91%
Clouse and
Asperger, 1978,
Dow chemical
company
Lab scale
Acid gas
treatment
30% MEA MS Organic +
Inorganic
Tetradecyl
polyalkylpyridinium
bromide with thio
urea and cobalt
acetate
50 ppm (with10-50
ppm cobalt acetate)
96%
Clouse and
Asperger, 1978,
Dow chemical
company
Lab scale
Acid gas
treatment
30% MEA MS Organic Tetradecyl
polyalkylpyridinium
with a) Thio urea
b) Thiocyanate or c)
Thionicotinamide
50 ppm a) 77%
b) 91%
c) 92%
14
Table 1.2 Summary of corrosion inhibitors in gas treating plants (continued)
Reference Process type Solvent Material of
construction
Type of
Inhibitor Inhibitor details
Recommended
dosage Performance
Asperger et al.,
1979
Refinery off-
gas stripping
30% DEA MS Inorganic Copper carbonate
mixed with sulfur
500 ppm of CuCO3
with 100 ppm sulfur
55%
Nieh, 1983,
Texaco Inc
Lab scale
Acid gas
treatment
50% MEA
solution
MS Heavy metal Sodium metavanadate
with cobalt nitrate
100 ppm Corrosion rate
less than 1 mpy
Nieh, 1983,
Texaco Inc
Lab scale
acid gas
treatment
50% MEA MS Heavy metal Mixture of
Ammonium
metavanadate with a)
N-amino
ethylethanolamine
b) ethylene diamine
c) propylene diamine
d) N-hydroxy ethyl
piperazine e) N-
aminoethyl piperazine
f) methylamino
bispropylamine
100 ppm (with 0.4%
of amine compound)
a) less than 1mpy
at 100ppm
b) less than 1
mpy at 200 ppm
c) less than 1 mpy
at 300 ppm
d) 3 mpy at 300
ppm
e) less than 1 mpy
at 300 ppm
f) 11 mpy at 300
ppm
Krawczyk et al.,
1984,
Dow chemical
company
General acid
gas treatment
25-30% MEA MS, Teflon
coated steel
Inorganic Mixture of Thio
nitrogen compound
with trace quantities
of Cobalt or Nickel
salts
200 ppm 90% (Ni salt)
89% (Co salt)
Pearsce, 1984
Dow chemical
company
General acid
gas treatment
80% MEA MS Inorganic Combination of
Copper carbonate
with dihydroxyethyl
glycine, alkali metal
thiocyanate,
ammonium
permanganate and
Nickel or bismuth
oxide
50 to 2000 ppm 99%

15
Table 1.2 Summary of corrosion inhibitors in gas treating plants (continued)
Reference Process type Solvent Material of
construction
Type of
Inhibitor Inhibitor details
Recommended
dosage Performance
Dupart et al.,
1984,
Dow chemical
Company
Natural &
synthesis gas
treatment
30% MEA
solution
MS, Stainless
steel (SS)
type 304, type
316 and
Monel
Inorganic Combination of
Ammonium
thiocyanate, Bismuth
citrate and Nickel
sulfate
Greater than 50 ppm
of thiocyanate (with
Bismuth citrate 1-100
ppm)
93.6% efficiency
Jones and Alkire,
1985,
Standard oil
company
Natural &
synthesis gas
treatment
20 – 60% DEA MS Organic-
Inorganic
Dodecylbenzyl
chloride with
alkylpyridine still
bottoms and Nickel
acetate
2000 ppm (with 35
ppm Ni compound)
Maximum
efficiency of
93.1%
Henson et al.,
1986,
Dow chemical
company
Refinery gas
conditioning
30% MEA Carbon steel
(CS)
Organic-
Inorganic
Mixture of
Aminoethyl
piperazine-
Formaldehyde-
thiourea polymer and
Nickel sulfate
5000 ppm As close as 100%
reported
Trevino, 1987,
Hylsa, S.A
Gas
processing
25 – 30%
MEA
CS Inorganic Mixture of cupric
oxide and Zinc sulfate
with bronze pieces (to
maintain Copper and
Zinc concentration)
Copper - 1-500ppm
Zinc – 100-500 ppm
80% efficiency at
regenerator top
and 90% at
absorber bottom
Sekine et al.,
1992, Dept. of
Industrial
chemistry,
University of
Tokyo
General acid
gas treatment
23% hot
potassium
carbonate
MS and SS Organic Combination of
following a)2-
Aminothiophenol
(ATP) b) 1-
Hydroxyethylidene
bisphosphonic acid
(HEDP) and c)
Diethanolamine
(DEA)
a) 10 ppm
b) 100ppm
c) 3%
Maximum
efficiency of 92%
for SS41 and
95% for SS104

16
Table 1.2 Summary of corrosion inhibitors in gas treating plants (continued)
Reference Process
type Solvent
Material of
construction
Type of
Inhibitor Inhibitor details
Recommended
dosage Performance
Minevski and
Labmbousy, 1998,
BetzDearborn Inc
General
acid gas
treatment
18% MEA (140
ppm sulfuric acid,
150 ppm oxalic
acid, 240 ppm
formic acid and 30
ppm sodium
chloride)
MS Organic 1,6 Hexanedithiol
with cyclohexanethiol
or decanethiol or
dodecane thiol is used
25 – 100 ppm (with
less than 1% by
weight of thiol)
Maximum
efficiency of
around 90% when
only CO2 is
present and 75%
when H2S is also
present.
Minevski, 2000,
BetzDearborn Inc
General
acid gas
treatment
18% MEA (140
ppm sulfuric acid,
150 ppm oxalic
acid, 240 ppm
formic acid and 30
ppm sodium
chloride)
MS Organic Combination of
Thiomorpholine with
Phthalic acid
500 – 2000 ppm Maximum
effiency of about
92%
Veawab et al., 2001,
University of Regina
Industrial
gas
treatment
3M MEA CS Organic a)Amine-based
b) Sulfoxide based
c) Carboxylic acid
based
a) 1000 ppm
b) 1000 ppm
c) 5000 ppm
a) around 75%
b) over 75%
c)98%
Veldman and Trahan,
2001, Coastal
Chemical Co
Natural
gas plant
and
Refinery
Hydrogen
treatment
50% MDEA
solution
MS and SS Heavy metal Sodium molybdate in
presence of
hydroquinone,
ethylketoxime and
diethylhydroxyl
amine
3.5% by weight Over 99%
Chang et al., 2008,
GE Betz Inc
General
acid gas
treatment
25% MEA and
30% DEA
CS Organic Polythia ether
compounds
10-20 ppm 96% for DEA and
99% for MEA
Soosaiprakasam and
Veawab, 2009,
University of Regina
General
acid gas
treatment
MEA (5M, 7M
and 9M)
CS Inorganic Copper carbonate 250 ppm Above 80%

17
New corrosion inhibitors were initially relatively less toxic either because of
the choice of the chemical compounds or by combination of heavy metals with less
toxic compounds. Davidson et al., 1978, registered a patent for the claim of copper
and sulfur-based chemical compounds as corrosion inhibitors that were less toxic than
heavy metal corrosion inhibitors. Copper carbonate in combination or alone has been
suggested a potential corrosion inhibitor since 1979 [Asperger et al., 1979; Pearsce,
1984; Trevino, 1987; Soosaiprakasam and Veawab, 2009]. Many organic compounds
such as pyridinium-based, piperazine-based, thiophenol- and thiol-based, and amine
and carboxylic acid-based compounds were also reported with comparable inhibition
efficiencies of around 90%. Inorganic inhibitors were generally used in the
concentration range of 20 – 2000 ppm whereas organic inhibitors were in the range of
100 – 20000 ppm. During the initial development period, only inhibition efficiency
was of primary concern. However, increased environmental awareness has led to the
choice of corrosion inhibitors that are not only effective but also eco-friendly.
1.4 Research motivation
Disposal of toxic chemicals has resulted in significant damage to human and
environmental health and, based on those experiences, environmental awareness has
seen tremendous growth in the last few decades. As a result, a number of initiatives
were taken across the world. For instance, in the United States, for a period of over
100 years, since the late 1800s, only 20 environmental laws were passed. However, in
the few decades that followed, over 120 environmental regulatory laws were set in
place. Consequently, the cost of compliance with those environmental regulations
through waste treatment, control, and disposal were high and has been estimated to be
in the range of 100-150 billion USD per year for the affected industries [Anastas and

18
Williamson, 1998; Doble and Kruthiventi, 2007]. In Canada, usage of toxic
substances is regulated by the Canadian Environmental Protection Act (CEPA). For
example, inorganic arsenic and cadmium compounds are classified as carcinogenic
and considered toxic and were listed as CEPA 1999 Schedule-I compounds, and as a
result, their usage was banned. In the USA, the Environmental Protection Agency
(EPA) regulates the usage of chemicals through the Clean Water Act (CWA) and
Clean Air Act (CAA). In Europe, an environmental regulatory mechanism OSPAR
was established by fifteen Northeast Atlantic nations by unifying their policies in the
1972 Oslo convention against waste dumping to protect the marine environment. This
was later broadened to cover land-based pollutant sources and offshore industries in
the Paris Convention of 1974 [OSPAR, 2011]. As per the guidelines set by OSPAR
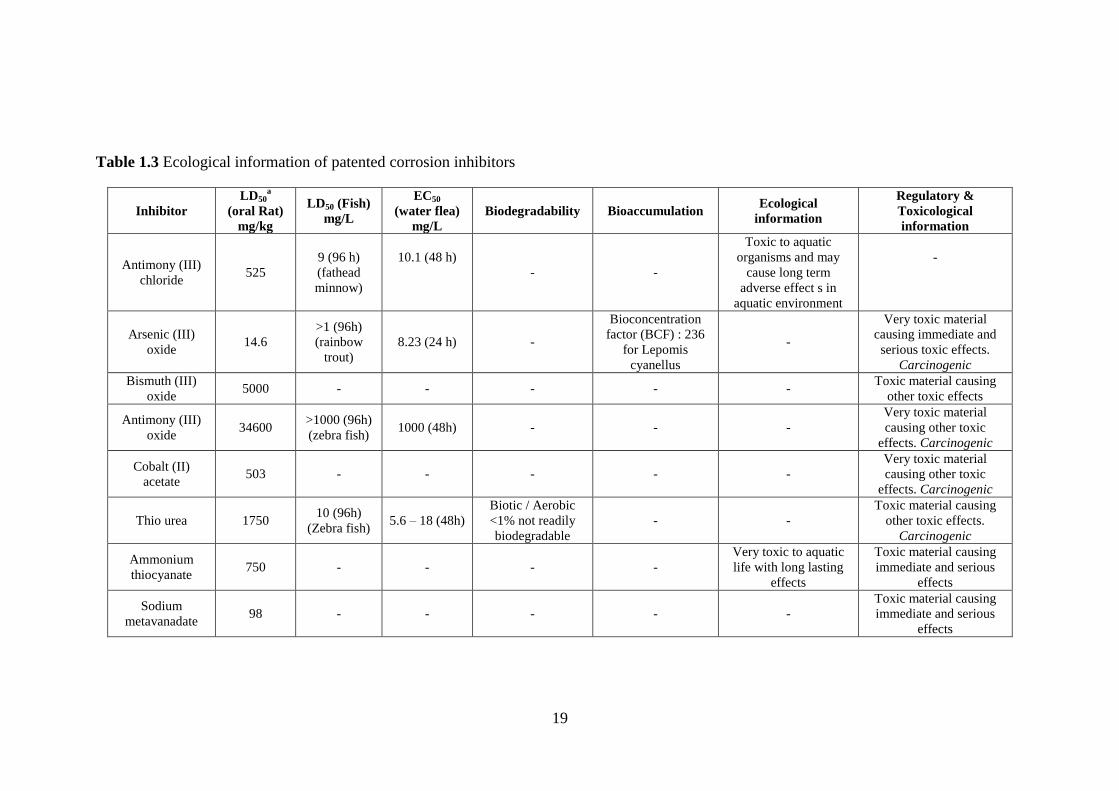
for environmentally-friendly chemicals, for a chemical to be listed in PLONOR
(poses little or no risk), two out of three of the following requirements has to be
satisfied with its biodegradability being superior to 20% in 28 days: a)
Biodegradability (>60% in 28 days), b) Toxicity [Lethal concentration (LC50) or
Effective concentration (EC50)] > 1mg/L for inorganic species and (LC50 or EC50 >
10mg/L for organic species) where LC50 or EC50 is the dose large enough to kill 50%
of sample animals under test; and c) Bioaccumulation ( logpow < 3 where pow is the
partition in octanol/water)
Based on the above OSPAR guidelines, it can be observed from Table 1.3 that
most corrosion inhibitors that are used, tested, and patented are non-environmentally
friendly. For instance, Antimony (III) oxide, arsenic oxide, cobalt acetate, thiourea,
aniline, pyridine, and vanadium compounds are toxic and carcinogenic. Hence, there
is a need to develop an environmentally-friendly corrosion inhibitor that can replace
the present highly toxic ones and also provide comparable inhibition efficiencies.
19
Table 1.3 Ecological information of patented corrosion inhibitors
Inhibitor
LD50a
(oral Rat)
mg/kg
LD50 (Fish)
mg/L
EC50
(water flea)
mg/L
Biodegradability Bioaccumulation Ecological
information
Regulatory &
Toxicological
information
Antimony (III)
chloride 525
9 (96 h)
(fathead
minnow)
10.1 (48 h)
- -
Toxic to aquatic
organisms and may
cause long term
adverse effect s in
aquatic environment
-
Arsenic (III)
oxide 14.6
>1 (96h)
(rainbow
trout)
8.23 (24 h) -
Bioconcentration
factor (BCF) : 236
for Lepomis
cyanellus
-
Very toxic material
causing immediate and
serious toxic effects.
Carcinogenic
Bismuth (III)
oxide 5000 - - - - -
Toxic material causing
other toxic effects
Antimony (III)
oxide 34600
>1000 (96h)
(zebra fish) 1000 (48h) - - -
Very toxic material
causing other toxic
effects. Carcinogenic
Cobalt (II)
acetate 503 - - - - -
Very toxic material
causing other toxic
effects. Carcinogenic
Thio urea 1750 10 (96h)
(Zebra fish) 5.6 – 18 (48h)
Biotic / Aerobic
<1% not readily
biodegradable
- -
Toxic material causing
other toxic effects.
Carcinogenic
Ammonium
thiocyanate 750 - - - -
Very toxic to aquatic
life with long lasting
effects
Toxic material causing
immediate and serious
effects
Sodium
metavanadate 98 - - - - -
Toxic material causing
immediate and serious
effects
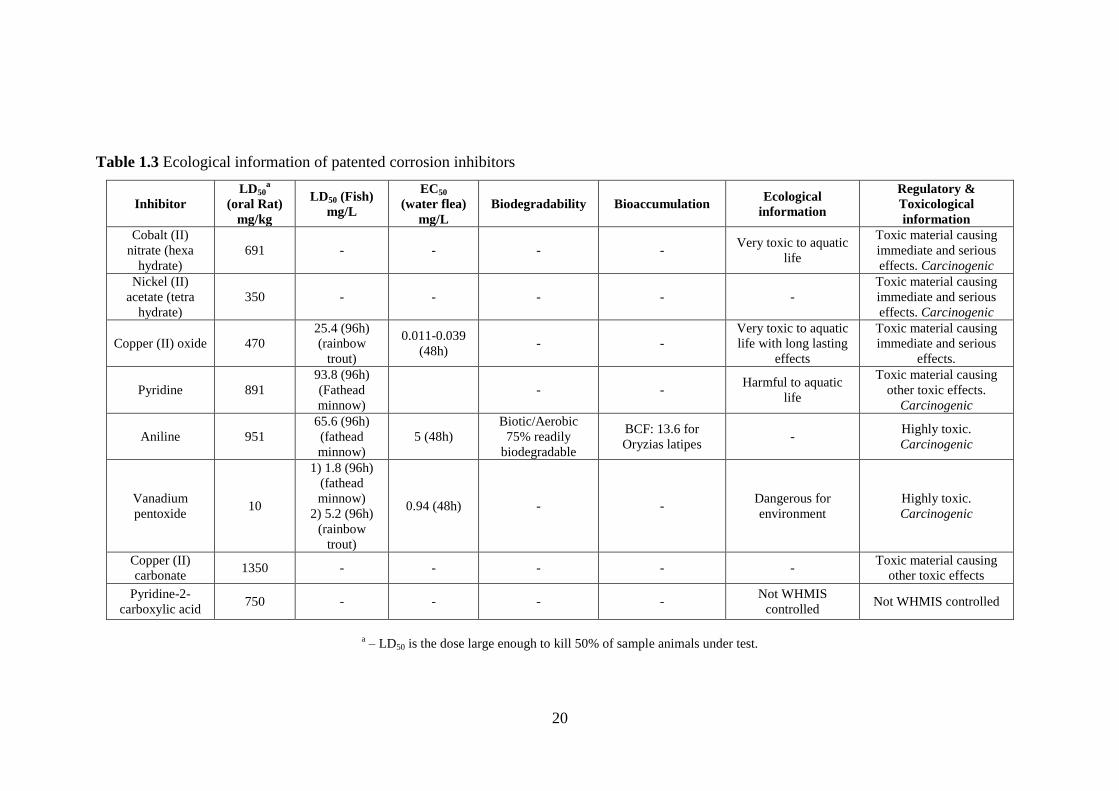
20
Table 1.3 Ecological information of patented corrosion inhibitors
Inhibitor
LD50a
(oral Rat)
mg/kg
LD50 (Fish)
mg/L
EC50
(water flea)
mg/L
Biodegradability Bioaccumulation Ecological
information
Regulatory &
Toxicological
information
Cobalt (II)
nitrate (hexa
hydrate)
691 - - - - Very toxic to aquatic
life
Toxic material causing
immediate and serious
effects. Carcinogenic
Nickel (II)
acetate (tetra
hydrate)
350 - - - - -
Toxic material causing
immediate and serious
effects. Carcinogenic
Copper (II) oxide 470
25.4 (96h)
(rainbow
trout)
0.011-0.039
(48h) - -
Very toxic to aquatic
life with long lasting
effects
Toxic material causing
immediate and serious
effects.
Pyridine 891
93.8 (96h)
(Fathead
minnow)
- - Harmful to aquatic
life
Toxic material causing
other toxic effects.
Carcinogenic
Aniline 951
65.6 (96h)
(fathead
minnow)
5 (48h)
Biotic/Aerobic
75% readily
biodegradable
BCF: 13.6 for
Oryzias latipes -
Highly toxic.
Carcinogenic
Vanadium
pentoxide 10
1) 1.8 (96h)
(fathead
minnow)
2) 5.2 (96h)
(rainbow
trout)
0.94 (48h) - - Dangerous for
environment
Highly toxic.
Carcinogenic
Copper (II)
carbonate 1350 - - - - -
Toxic material causing
other toxic effects
Pyridine-2-
carboxylic acid 750 - - - -
Not WHMIS
controlled Not WHMIS controlled
a – LD50 is the dose large enough to kill 50% of sample animals under test.

Many previous works have attempted to search for a suitable green corrosion
inhibitor for absorption-based gas treating plants. In general, it was observed that organic
inhibitors are much more environmentally friendly than inorganic inhibitors [Veawab et
al, 2001]. Asperger and Clouse [1978] have patented two different organic corrosion
inhibitors based on polyalkyl pyridinium. Minevsky and Lambousy [1998] have reported
a corrosion inhibitor based on organic thiol and dithiols. Minevsky [2001] has also
reported a thiomorpholine-based corrosion inhibitor. Similarly, Veawab et al. [2001]
have reported eight organic inhibitors based on carboxylic acid, sulfoxide, and amines
that are all reportedly less toxic than the conventional vanadium-based inhibitors and
comparably efficient. All these suggest that searching for a potential corrosion inhibitor
that is environmentally friendly as well as efficient compared to conventional corrosion
inhibitors has been a continuous process, but no compound yet has emerged as a
satisfactory candidate for replacement of conventional corrosion inhibitors.
1.5 Research objectives and scope
The most effective corrosion inhibitors patented are not eco-friendly, as can be
seen in Table 1.3. This leads to an objective to search for an environmentally-friendly
corrosion inhibitor with comparable inhibition performance that can replace conventional
highly toxic corrosion inhibitors. To achieve this objective, this work was implemented
through 4 tasks:
i. Selection of chemical compounds for testing as corrosion inhibitors
On understanding of chemistry, it is possible to select a chemical
compound that can best interact with the metal (surface) to be protected. On that

22
basis, in this work, thirteen organic compounds were selected and studied. That
also includes the structural isomers of some of the selected compounds that have
the same functional groups and reaction centers as those of the parent compound.
ii. Analysis of the inhibitor for eco-friendliness
Since the primary focus of this work is to search for an environmentally-
friendly corrosion inhibitor, ecological analysis assumes primary importance.
Selected compounds were evaluated based on their toxicity values in terms of
LD50 (Lethal dosage to kill 50% of sample animals under test) and other available
ecological data.
iii. Theoretical analysis of the inhibitor for performance
Quantum chemical studies are extensively used in corrosion inhibitor
development mainly because it can provide a predictive capability of corrosion
inhibition performance of different compounds. In this work, an attempt was
made to evaluate compounds on that basis.
iv. Experimental testing, Evaluation, and Recommendation
Final shortlisted compounds were subjected to experimental analysis to
evaluate the corrosion inhibition performance of these compounds and understand
the mechanisms of interaction. Corrosion inhibitors were evaluated based on their
effect on corrosion rate of carbon steel, which is a common material of
construction in amine-based CO2 capture plants. Inhibitors were
electrochemically tested at different concentrations ranging from as low as 250
ppm to 10,000 ppm. Also, the effect of the presence of possible process
contaminants on corrosion inhibition performance was studied. Their performance

23
was evaluated based on various experimentally-obtained parameters such as
corrosion rate, polarization resistance, corrosion current, and inhibition
efficiencies. Weight loss testing was carried out to corroborate the results
obtained from the electrochemical tests.
Based on the above results, those compounds that manifested good potential for corrosion
inhibition were recommended for further study.

24
2. FUNDAMENTALS OF CORROSION AND CORROSION INHIBITION
Knowledge of fundamentals of corrosion is imperative to understanding the
mechanism of corrosion based on which a congruent selection of corrosion preventive
method can be made. It is also useful in the prognosis of corrosion behaviour of different
metals under variegated conditions. In order to reasonably understand the process of
corrosion, it is necessary to understand the thermodynamics and associated kinetics of the
corroding system under investigation.
2.1 Thermodynamic aspects of corrosion
Thermodynamic principles can be used to ascertain the driving force and
spontaneous direction for any reaction. In general, driving force is characterized as the
balance between the effect of change in energy (enthalpy, ∆H) and the effect of change in
thermodynamic probability (entropy, ∆S). Free energy change (∆G) is used to quantify
the above and at constant temperature can be expressed as
∆G = ∆H - T∆S (2.1)
where T is the absolute temperature. Only those reactions that lower the energy of the
system can be spontaneous, which implies that the free energy change will be negative
for a spontaneous reaction [ASM Handbook (13), 1987].
2.1.1 Origin of electrode potential
Corrosion of metals, especially in aqueous environments, is electrochemical in
nature involving at least two electrochemical reactions occurring concurrently on the

25
metal surface. The corroding metal surface is the electrode and the aqueous medium,
which acts as an ionic conductor, is the electrolyte. Thus, when an electrode is brought in
contact with the electrolyte, a discontinuity is introduced, and due to the anisotropic
forces, the properties of the solution at the interface become different from those of the
bulk solution. Due to the reactions occurring at the metal surface, the
electrode/electrolyte interface is electrified and results in a double layer formation (i.e.,
separation of charges between metal (electrons) and solution (ions)). Various phenomena,
such as interaction of ions with water molecules, adsorption of ions on electrodes, and
diffusion influence, the double layer properties. The characteristic feature of the double
layer is that it gives rise to a potential difference between the metal side and solution side
of the interface leading to the definition of electrode potential [Bockris and Reddy, 2000].
2.1.2 Electrode processes
Electrochemical reactions are considered a special case of chemical reaction that
involves two simultaneous half-cell reactions, oxidation and reduction, with
corresponding half-cell electrode potentials. Oxidation occurs at the anode and is
characterized by the removal of electrons from an atom, whereas reduction occurs at the
cathode and is characterized by the addition of electrons to an atom. As an example,
corrosion of iron in deaerated hydrochloric acid is depicted as a simplest case scenario in
Figure 2.1 where dissolution of iron generates two electrons through oxidation, which are
then consumed by the reduction of hydrogen ions forming molecular hydrogen. The
corresponding partial or half cell reactions of oxidation and reduction are as follows:

26
Figure 2.1 Corrosion of iron in deaerated hydrochloric acid solution (Redrawn
from Soosaiprakasam, 2007)
Fe2+
Fe2+
Fe2+
Fe2+
H2
H+
H+
H+
H+
H+
H+
H+
e-
Anode Fe → Fe2+ + 2 e-
Cathode 2H+ + 2e- → H2
Iron Solution

27
Oxidation (Anodic): Fe Fe2+
+ 2e (2.2)
Reduction (Cathodic): 2H+ + 2e H2 (2.3)
As can be seen from Equations (2.2 and 2.3), oxidation and reduction have to occur at the
same rate, or, otherwise, there will be a net accumulation or deficiency of charge in the
electrode, which is not possible. Each reaction has a characteristic half-cell potential, and
the difference is termed as the electrode potential, which can be expressed as follows:
Eo = EA + EC (2.4)
where Eo is the standard electrode potential corresponding to unit activity of reactants and
products at 298 K, EA and EC are half-cell anodic and cathodic potentials, respectively,
measured with reference to the standard hydrogen electrode (SHE), which is arbitrarily
assigned a value of zero volts. Any change in standard potential in response to the
changes in conditions such as concentration and temperature can be determined using the
Nernst equation:
r
p
oa
a
nF
RTEE ln (2.5)
where n is the number of electrons per atom of the species involved in the reaction, F is
the charge per mole of electrons (96480 C/mol), R is the gas constant, T is the
temperature, and ap and ar are the activity of the products and reactants, respectively.
[ASM Handbook (13), 1987; Veawab, 2000]
2.1.3 Concept of mixed potential
Though the potential for cathodic and anodic reactions are characteristic and
different, when occurring simultaneously on the same metal surface, they tend to drift
away from their corresponding equilibrium values and establish a combined potential

28
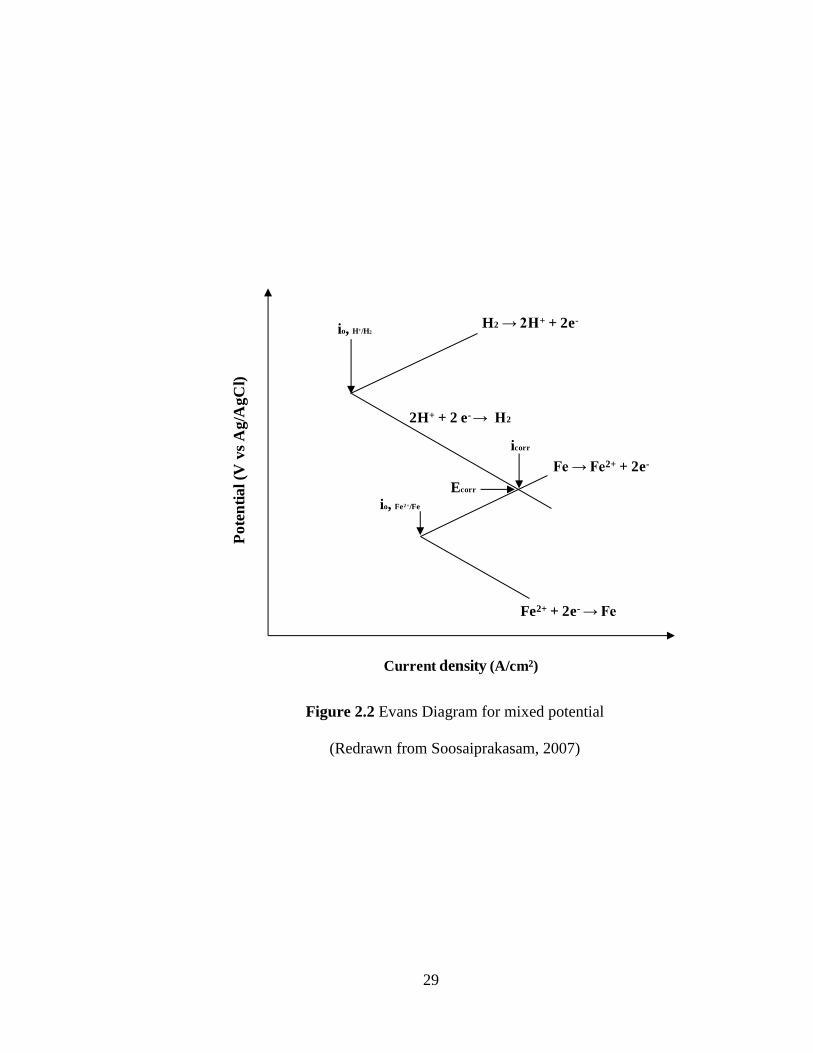
called the mixed or corrosion potential (Ecorr). The concept of mixed potential for iron
dissolution in deaerated hydrochloric acid is shown in Figure 2.2. The reduction process
need not be as simple as in the considered case but can be more complicated, involving
more than one reaction in addition to hydrogen evolution, such as oxygen reduction in the
case of aerated solutions [Fontana, 1986].
2.1.4 Free energy - electrode potential relationship
The change in free energy associated with an electrochemical reaction can be
expressed in terms of potential by the following expression:
∆G = − nFE (2.6)
where n is the number of electrons per atom of the species involved in the reaction, F is
the charge of 1 mol of electrons (96480 C/mol), and E is the electrode reduction
potential. As a convention, positive potential is associated with the spontaneous reaction
and, hence, the negative sign is used in Equation (2.6).
2.2 Kinetics of corrosion
Thermodynamics can provide a framework of possibility for different corrosion
reactions to occur. However, only based on kinetics, it is possible to elucidate those
reactions that will primarily occur and the rate at which they occur among the reactions
that are thermodynamically possible. For example, aluminum displays a predicative
thermodynamic tendency to react but is limited by its slow kinetics, which renders it
more resistant than other metals that are innately less reactive in certain environments.
29
Figure 2.2 Evans Diagram for mixed potential
(Redrawn from Soosaiprakasam, 2007)
H2 → 2H+ + 2e-
2H+ + 2 e- → H2
io, H+/H2
icorr
Ecorr
Fe → Fe2+ + 2e-
Fe2+ + 2e- → Fe
io, Fe2+/Fe
Current density (A/cm2)
Pote
nti
al (V
vs
Ag/A
gC
l)

30
2.2.1 Faraday’s law
A metal undergoing corrosion can be viewed as analogous to a short circuited
energy-producing electrochemical cell wherein the energy is produced by consumption of
reactants to form corrosion products. To be consistent with the principle of conservation
of mass, the amount of corrosion products formed has to be equal to the amount of the
reactants consumed. Also, for an electrochemical reaction, the electrons liberated by
anodic reaction have to be consumed by the cathodic reaction at the same rate, which
makes it possible to express corrosion in terms of electrochemical current. Hence, it can
be stated that the current flowing from an anodic reaction will be equal and opposite to
the current flowing into the cathodic reaction. This current can be used as an indicator of
rate of corrosion and can be correlated to the amount of material corroded by Faraday’s
law.
nF
Itam (2.7)
where m is the weight of the metal corroded (g), I is the current passed (A), t is time (s), a
is the atomic weight of the metal (g/mol), n is the number of electrons transferred, and F
is the charge per mole of electron. If multiple cathodic and anodic reactions can take
place, the corrosion current represents the sum of component partial currents. Also,
though, the anodic and cathodic currents have to be equal in magnitude, the
corresponding anodic and cathodic areas need not be equal. Hence, the respective current
density, which is clearly a function of the surface area of the corroding metal, can be
different. So when the anodic area of the corroding metal is almost equal to the cathodic
area, the corrosion is uniform. However, when the anodic area of the corroding metal is

31
relatively much smaller than the cathodic area, the nature of the corrosion will be
localized [ASM Handbook (13a), 1987].
Dividing Equation (2.7) by the surface area of the corroding metal (Ae) and
rearranging the yields, the following correlation is found between corrosion rate and
current density:
Corrosion rate (CR) = nF
ai
tA
m corr
e
(2.8)
where icorr
eA
Irepresents the corrosion current density.
2.2.2 Polarization
Even though corrosion is seldom an equilibrium process, it is essential to
understand the equilibrium state properties in order to analyze its non-equilibrium
behaviour. For any reaction at equilibrium, the rate of forward reaction is equal to the rate
of reverse reaction, and as a consequence, there is no net reaction but only an exchange
reaction rate. Exchange reaction rate can be readily expressed using exchange current
density (io), which can be defined as the current density at equilibrium corresponding to
the equal forward and reverse reactions at the electrode. For example, consider the
reversible hydrogen evolution reaction:
2H+ + 2e H2 (2.9)
For this reaction, the correlation between exchange reaction rate and current
density can be deduced from Faraday’s law:
nF
airr o
redoxid (2.10)

32
where io is the exchange current density and roxid and rred are the oxidation and reduction
rates at equilibrium, respectively.
A characteristic feature of io is its dependency on the nature of the surface
antithetical to the thermodynamic parameter, the potential. Thus, when a net current is
involved in an electrode process, the equilibrium is disturbed and causes a potential
change that is dependent on the direction and magnitude of the current. This potential
change, as a result of a net current, is called polarization and can be measured in volts.
The concept of polarization can be better understood by illustration using an
electrochemical cell (Daniell cell) as depicted in Figure 2.3a.
A typical cell consists of zinc in zinc sulfate solution and copper in copper sulfate
solution with the electrodes connected to a variable resistance (R), a voltmeter (V), and
an ammeter (A). From the polarization diagram (Figure 2.3b), it can be noted that under
the condition of no current, potential difference corresponds to the difference in the
respective thermodynamic potentials of zinc and copper, which is approximately 1 V. As
the current starts flowing, electrodes are polarized towards each other and, hence, a
constant decrease in the potential difference is observed. When the electrodes are short
circuited, maximum current flows and the potential difference approaches zero.
Polarization of zinc traces the path abc and that of copper is def in Figure 2.3b. At point
b, polarization of zinc is given by (b-a) and similarly for copper at point e; polarization is
given by (e-d), and this deviation from the equilibrium value is known as overpotential
(Ƞ). The potential difference is b-e corresponding to a current I1. Under a short circuited
condition, the potential difference is at its least value and is equal to the product of
current (Imax) and the electrolytic resistance (Re).

33
(a)
(b)
Figure 2.3 A typical electrochemical cell (Redrawn from Revie and Uhlig, 2008)
(a) Daniell Cell (b) Polarization behaviour of Daniell cell
V
A
Cu Zn
R
CuSO4 ZnSO4
log current
Po
ten
tia
l
φcorr
φCu
φZn
Imax Re
d
e
f
c
b
I1
a
Imax

34
Imax is indicative of the equivalent deposition rate of copper and, more importantly, the
equivalent corrosion rate for Zinc. As illustrated above, the corrosion process is
analogous to a short circuited electrolytic cell where the current Imax is given as the
corrosion current, Icorr, and the measured potential is given as the corrosion potential,
Ecorr, which represents the mixed potential of polarized anodes and cathodes. For
corrosion studies, alternatively, an external source can be used to polarize the metal
surface either in the anodic or cathodic direction, and the response of the system can be
evaluated. Cathodic polarization is characterized by the supply of electrons to the metal
rendering the metal negative in potential with respect to its equilibrium potential.
Conversely, for anodic polarization, electrons are withdrawn from the metal resulting in
an electron deficiency and, thereby, shifting the surface potential towards more positive
values [Revie and Uhlig, 2008]. Polarization can be sorted into three types based upon its
causes, and these are, namely, activation, concentration, and combined polarization.
2.2.2.1 Activation polarization
When one of the reactions occurring at the interface controls the net rate of an
electrochemical process, then the reaction is said to be under activation polarization. For
example, the hydrogen evolution reaction occurring on the metal surface in Equation
(2.3), in a simplest case scenario, can involve a number of steps such as adsorption of H+
ions onto the surface followed by electron transfer resulting in reduction of H+ to H, and,
finally, the hydrogen atoms coalescing to form a bubble of hydrogen gas. If one of the
above steps is the slowest and, hence, the rate determining step, then the system is

35
activation controlled. For activation polarization, the overpotential can be correlated to
the rate of oxidation or reduction using the Tafel equation as follows:
Ƞa = ± β
oi
ilog (2.11)
where Ƞa is the activation polarization, i is the current density corresponding to the rate of
oxidation or reduction, io is the equilibrium exchange current density, and β is the Tafel
constant, which ranges between 0.03 and 0.2. As can be observed from Figure 2.4a, Ƞa is
linearly related to the log current density with a slope of β.
2.2.2.2 Concentration polarization
When the rate of an electrochemical reaction at the interface is controlled by the
bulk solution properties, such as diffusion of the ionic species to the interface, then the
condition is known as concentration polarization and is generally pronounced in the case
of dilute solutions with very low concentrations of the reducible species. It is important to
emphasize that this condition is generally observed only in cathodic polarization but is
not applicable to anodic polarization owing to the extremely vast availability of metal
atoms for dissolution. In the case of hydrogen evolution, a sufficiently high reduction rate
causes a depletion of hydrogen ions in the proximity of the interface and leads to a
limiting rate, which will be determined by the diffusion of ions from the bulk to the
interface. This condition is illustrated in Figure 2.4b. For an electrode only under
concentration polarization, the correlation between overpotential and reduction rate is
expressed using Equation 2.12.

36
(a)
(b)
(c)
Figure 2.4 Types of polarization (a) Activation polarization (b) Concentration
polarization (c) Combined polarization
-0.3
-0.2
-0.1
0
0.1
0.2
0.001 0.01 0.1 1 10 100
-0.3
-0.2
-0.1
0
0.1
0.2
0.001 0.01 0.1 1 10 100
io,H2/H+
log i
Po
lari
zati
on
,Ƞ (
V)
Anodic slope = βa
Cathodic slope = βc
Acti
ve
No
ble
H2 → 2H+ + 2e-
2H+ + 2 e- → H2
0
iL
log i
(+)
(-)
Ƞco
nc
0
io
log i
(+)
(-)
Po
ten
tial
Ƞconc
ȠactȠT = Ƞa + Ƞc

37
Ƞc =
Li
i
nF
RT1log3.2 (2.12)
where Ƞc is the concentration polarization, i is the current density corresponding to the
rate of reduction, and Li is the limiting diffusion current density, which can be expressed
as a function of concentration of the reacting ions (CB) as follows:
x
nFCDi Bi
L (2.13)
where Di is the diffusion coefficient of the reacting ions and x is the thickness of the
diffusion layer. Environmental variables such as solution velocity, concentration, and
temperature are directly related to the limiting diffusion current density and, hence, the
net reaction rate [Fontana, 1986].
2.2.2.3 Combined polarization
Usually the combination of activation and concentration polarization is likely to
occur at an electrode where, at low reaction rates, the system is under activation
polarization and at higher rates, concentration polarization takes control. Hence, the total
polarization of the system can be expressed as follows:
ȠT = Ƞa + Ƞc (2.14)
Figure 2.4c represents the combined polarization curve. For anodic dissolution, Ƞc
can be eliminated for the reasons mentioned earlier, and, hence, the correlation between
the overpotential and current can be expressed as follows:
Ƞdiss = β
oi
ilog (2.15)

38
However, for reduction reactions, the effect of concentration has to be accounted
for in the overall expression of the kinetics, which is the combination of expressions for
activation and concentration polarization.
Ƞredn = - β
oi
ilog +
Li
i
nF
RT1log3.2 (2.16)
where Ƞdiss and Ƞredn represent the dissolution and reduction overpotentials, respectively.
[Fontana, 1986]
2.3 Passivity
Passivity is a complex phenomenon occurring on the metal surface under certain
specific oxidizing conditions that renders the metal surface corrosion resistant due to the
formation of a thin protective film in the dimensions of nanometers. This phenomenon is
known as the active passive transition tendency. Fortunately, some of the extensively
used engineering materials such as iron, nickel, chromium, and their alloys display this
tendency to become relatively inert to chemical reactivity with increasing potential and
anodic polarization and are characterized by a reduction of corrosion rate by an order of
up to 106
times in this passive state. It is generally considered a special case of activation
polarization due to the formation of a surface film that is stable over a significant range of
oxidizing conditions, and it is eventually destroyed at much stronger oxidizing conditions
[Jones, 1992].
There are a number of disparate theories applied in an attempt to explain the
source of passivity with some of the suggested reasons being allotropic modifications,
adsorbed oxygen, adsorbed hydroxyl ions, and bulk oxide. However, there exists no
precise understanding of the exact nature of this barrier. Figure 2.5 illustrates the

39
corrosion current density of an active passive metal as a function of potential. At low
values, corrosion rate increases with the potential until primary passivation potential (Epp)
is reached, beyond which the passive film is stabilized and the corrosion current density
is reduced to a lower value of ipass and remains almost constant. At much higher potential
(Eb), the passive film yields a transpassive state, and the current density increases again
with the potential. Environmental parameters such as solution velocity and oxidizer
concentration can either increase or not affect the corrosion rate, depending on the nature
of the metal and environment, and are generally complex in nature. An increase in
temperature, however, has been reported to increase the corrosion rate [Fontana, 1986].
2.4 Corrosion characterization techniques
2.4.1 Tafel extrapolation
Corrosion rate determination using Tafel extrapolation was initially carried out to
corroborate the validity of the mixed potential theory using the cathodic and anodic
polarization data [Fontana, 1986]. It was shown earlier, in Equation (2.11), that the
overpotential is linearly related to the logarithmic value of corrosion current density.
However, when a current is applied externally, such as, for example, cathodic current in
the case of cathodic polarization studies, at low values of current, the polarization curve
is non-linear due to the presence of a finite anodic current. However, at higher values of
current, the curve becomes linear as the corresponding anodic current is negligible, and
the opposite is observed during the measurement of anodic polarization. In actual
experiments, the polarization curve becomes linear at approximately 50 mV more active
or noble than the corrosion potential.

40
Figure 2.5 Active-passive transition behaviour of a metal (Redrawn from Jones, 1992)
Current density (A/cm2)
Active
Passive
Transpassive
icritipass
Eb
Epp
Ecorr
Po
ten
tial
(V
)

41
This linear portion of the curve is known as the Tafel region and can be extrapolated to
determine the corresponding Ecorr and Icorr, as illustrated in Figure 2.6.
2.4.2 Potentiodynamic cyclic polarization
A potentiodynamic polarization is different from the Tafel technique in that the
electrodes are polarized to more anodic values of potential, past the active area, allowing
one to study the corrosion characteristics. As discussed in section 2.3, some metals
exhibit active-passive transition behaviour as the potential is increased to more anodic
values. After the breakdown of the passive film in the transpassive zone, polarization is
reversed by changing the potential towards the cathodic direction as the curve traces back
to Ecorr. The resultant cyclic polarization curve can be used to predict the tendency of the
metal to undergo localized corrosion such as pitting. As shown in Figure 2.7a, pitting is
unlikely when the reverse curve lies to the left of the forward curve (negative hysteresis)
and pitting is likely to occur if the reverse curve lies to the right, as shown in Figure 2.7b
(positive hysteresis). Also if the repassivation potential is closer to the pitting potential,
then the metal is capable of repairing the passive layer to protect its surface
[Soosaiprakasam, 2007].
2.4.3 Electrochemical impedance spectroscopy
Impedance analysis relies on the frequency dependent response of a corroding
system to provide useful insights to the mechanistic details of a corrosion process, which
may not be available from potentiodynamic polarization analysis and are extensively
useful in corrosion inhibitor evaluation. Some of the common electrode processes, such

42
Figure 2.6 A typical Tafel plot (Redrawn from Srinivasan, 2006)
Ecorr
Icorr
Log current density
Pote
nti
al Anodic
Cathodic

43
(a)
(b)
Figure 2.7 A typical potentiodynamic cyclic polarization curve
(a) No pitting (b) Pitting
log i (A/cm2)
Active
Passive
Transpassive
icritipass
Eb
Epp
Ecorr
Epass
Erp
Negative
Po
ten
tial
(V
)
log i (A/cm2)
Active
Passive
Transpassive
icritipass
Eb
Epp
Ecorr
Epass
Erp
Positive
Po
ten
tial
(V
vs
Ag
/Ag
Cl)

44
as slow electrode kinetics, slow preceding chemical reactions, and diffusion, can all
impede the flow of an AC current, causing a time lag and phase shift (θ), and these are
analogous to resistors and capacitors in the AC circuit, which can be interpreted based on
equivalent circuit models of electrode/electrolyte interface. Electrochemical impedance
can be expressed as the ratio of applied alternating potential V (t) and the time-dependent
current response I (t) as follows:
Z (ω) = )(
)(
tI
tV (2.17)
V (t) = Vo sin ωt (2.18)
I (t) = Io (sin ωt + θ) (2.19)
where ω is the angular frequency and θ is the phase angle between V (t) and I (t). The
impedance includes real component Z (ω) and imaginary component Z (ω), which are
plotted against each other in a Nyquist plot used to express the impedance behaviour of a
system.
Figure 2.8a represents a simple equivalent circuit model for an electrochemical
interface undergoing corrosion without any diffusion control, which includes solution
resistance (RS), double layer capacitance (Cdl), and polarization resistance (RP). At high
frequency measurements, impedance of the capacitor is extremely low, and, hence, the
total impedance represents the solution resistance (RS) alone. However, at the low
frequency end, the impedance of the capacitor approaches infinity, and, hence, the total
impedance is the sum of solution and polarization resistance (RP). The Nyquist plot for
the above circuit is presented in Figure 2.8b. The semicircle represents the capacitive
loop due to charge transfer kinetics at the interface. Figure 2.9a represents the equivalent
circuit model in the presence of diffusion control, which includes an additional
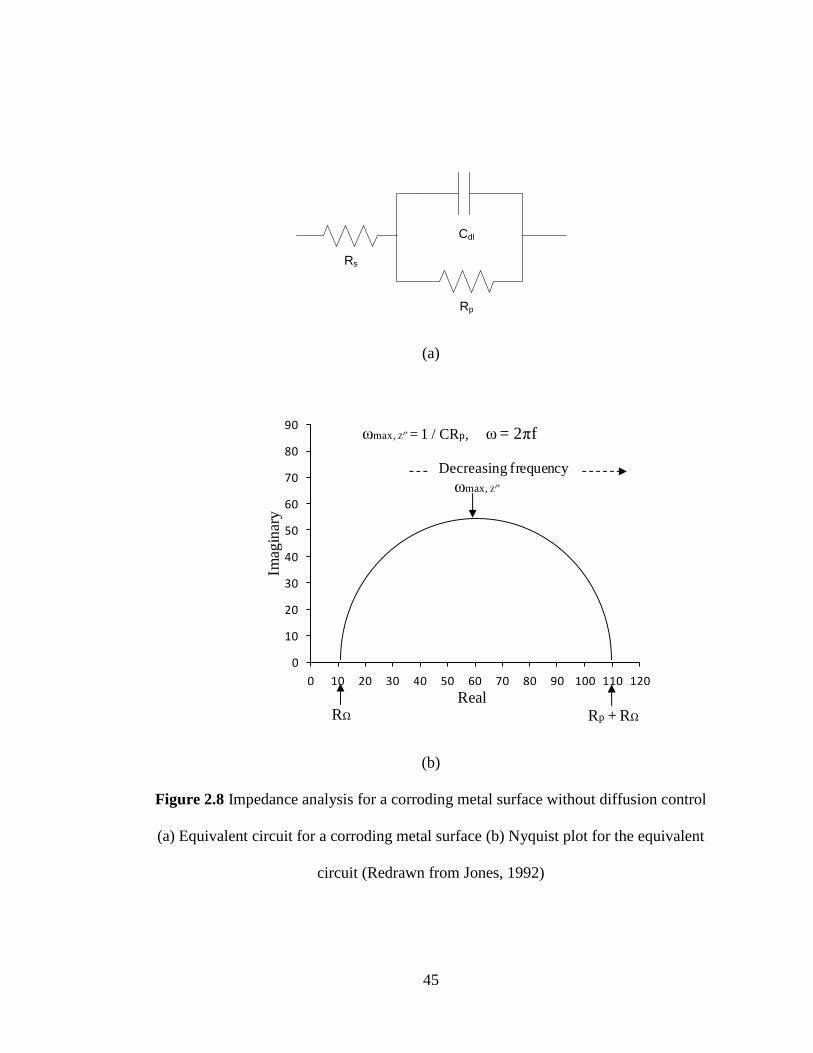
45
(a)
(b)
Figure 2.8 Impedance analysis for a corroding metal surface without diffusion control
(a) Equivalent circuit for a corroding metal surface (b) Nyquist plot for the equivalent
circuit (Redrawn from Jones, 1992)
Rp
Cdl
Rs
0
10
20
30
40
50
60
70
80
90
0 10 20 30 40 50 60 70 80 90 100 110 120
0
10
20
30
40
50
60
70
80
90
0 10 20 30 40 50 60 70 80 90 100 110 120
RΩ Rp + RΩ
Real
Imag
inar
y
Decreasing frequency
ωmax, Z'''
ωmax, Z''' = 1 / CRp, ω = 2πf
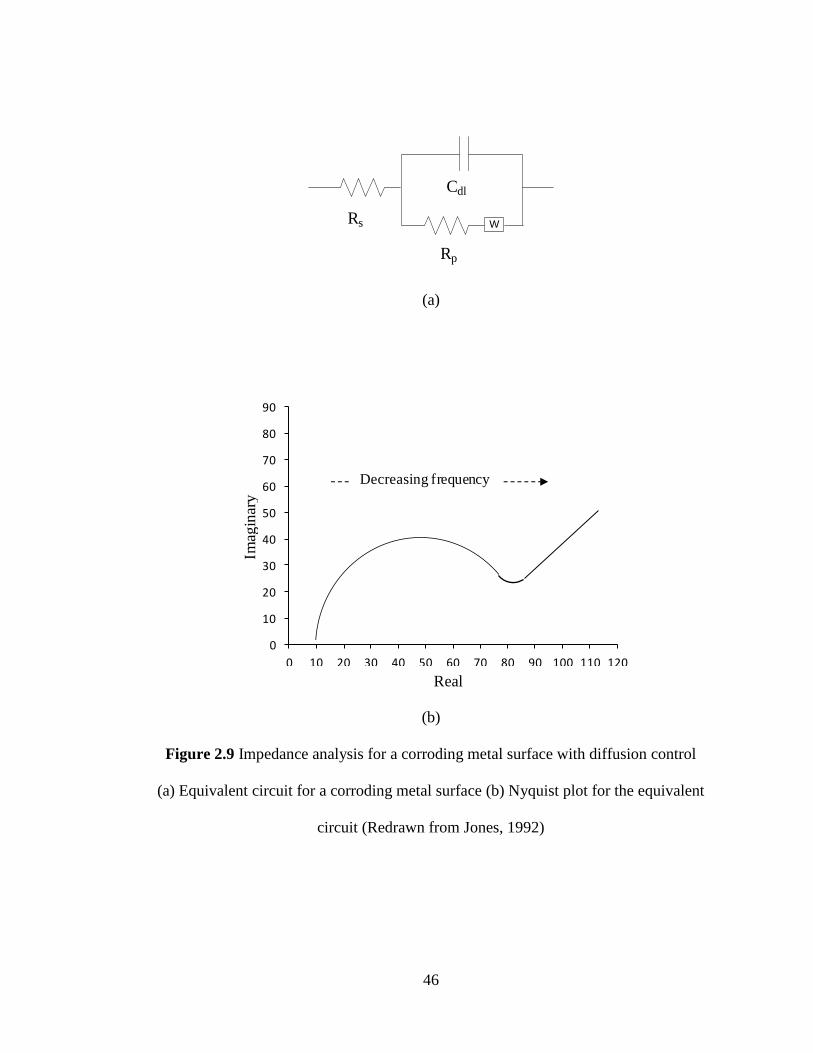
46
(a)
(b)
Figure 2.9 Impedance analysis for a corroding metal surface with diffusion control
(a) Equivalent circuit for a corroding metal surface (b) Nyquist plot for the equivalent
circuit (Redrawn from Jones, 1992)
Rp
Cdl
Rs
W
Rp
Rs
Cdl
Real
Imag
inar
y
0
10
20
30
40
50
60
70
80
90
0 10 20 30 40 50 60 70 80 90 100 110 120
0
10
20
30
40
50
60
70
80
90
0 10 20 30 40 50 60 70 80 90 100 110 120
Decreasing frequency

47
component of resistance in series with the polarization resistance, called Warburg
impedance. The low frequency end of the Nyquist plot is characterized by linearity
(diffusion tail) as shown in Figure 2.9b. This condition is frequently observed when a
surface film is formed over the metal surface, which limits the diffusion of corrosive
species across the interface.
Despite the advantages of the EIS techniques, it has some drawbacks such as the
complexity and ambiguity in the data interpretation at low frequency measurements
where it tends to approach the DC measurements. Nevertheless, impedance analysis can
provide some unique mechanistic information that cannot be obtained using DC
techniques.
2.5 Corrosion control techniques – Corrosion inhibitors
Corrosion inhibitors are chemicals that are capable of reacting either with the
metallic surface or the environment and reduce corrosion. Usage of corrosion inhibitors
has been one of the commonly employed methods to mitigate corrosion, and, hence, a
myriad of compounds have been reported to act as corrosion inhibitors by different
mechanisms. Corrosion inhibitors usually function by either increasing anodic or
cathodic polarization behaviour, by restricting the movement of ions to the metal surface,
or by increasing the electrical resistance of the surface to be protected. They can be
classified based on their mechanism, chemistry, or application. The most common
approach to classifying inhibitors is by their mechanism, and, as such, they can be
categorized as anodic, cathodic, and film forming inhibitors.

48
2.5.1 Anodic inhibitors
Anodic inhibitors or passivators are the most effective and commonly used type,
and they function by shifting the corrosion potential to more anodic values, as shown in
Figure 2.10a. This forces the metal to its passive region. They can either contain
oxidizing ions such as chromate or nitrite that can effectively passivate steel even without
the presence of oxygen, or they can contain non-oxidizing ions such as phosphate or
molybdate that can also passivate steel but only in the presence of oxygen. The
concentration of the inhibitor is a critical factor since a deficiency can cause localized
pitting corrosion. Hence, it is important to monitor the concentration of the inhibitor on a
regular basis.
2.5.2 Cathodic inhibitors
Cathodic inhibitors typically interfere with the cathodic reactions of corrosion by
either directly slowing down the reaction or by formation of surface deposits that limit
the diffusion. They are generally characterized by shifting the corrosion potential to more
cathodic values, as depicted in Figure 2.10b, and are considered safer than anodic
inhibitors as they do not pose risks of pitting corrosion. They can function by three
different mechanisms, namely, cathodic poisoning, precipitation, and oxygen scavenging.
Inhibitors based on arsenic and antimony function by making the recombination and
discharge of hydrogen difficult, thereby reducing the rate of cathodic reaction and, hence,
the net corrosion rate. Inhibitors based on zinc and calcium function by precipitating in
the form of oxides on the metal surface. Finally, oxygen scavengers such as sulfite and
bisulfite ions function by specifically reacting with oxygen.
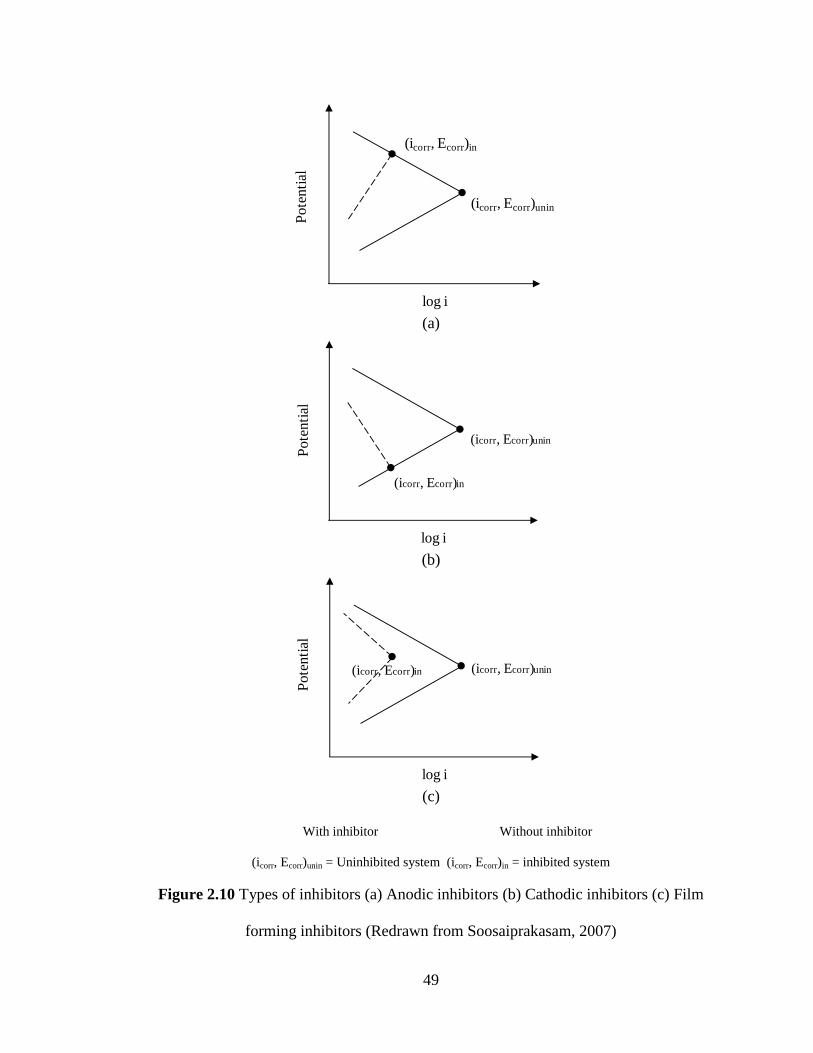
49
(a)
(b)
(c)
With inhibitor Without inhibitor
(icorr, Ecorr)unin = Uninhibited system (icorr, Ecorr)in = inhibited system
Figure 2.10 Types of inhibitors (a) Anodic inhibitors (b) Cathodic inhibitors (c) Film
forming inhibitors (Redrawn from Soosaiprakasam, 2007)
log iP
ote
nti
al
(icorr, Ecorr)unin
(icorr, Ecorr)in
log i
Pote
nti
al
(icorr, Ecorr)unin
(icorr, Ecorr)in
log i
Po
ten
tial
(icorr, Ecorr)unin(icorr, Ecorr)in

50
2.5.3 Film forming inhibitors
This category of inhibitors is primarily constituted by organic compounds that are
capable of forming a hydrophobic film on the corroding metal surface that can act as a
physical barrier. When present in adequate levels of concentration, they tend to interact
with the entire metal surface and both anodic and cathodic effects might be observed as
shown in Figure 2.10c. Film formation is clearly based on adsorption, and, consequently,
the environmental parameters such as temperature and pressure can be critical. Chemical
composition, molecular structure, and the strength of the adsorption bond formed with the
metal surface are some of the factors that can influence the effectiveness of an organic
inhibitor. They can either be cationic in nature, such as amines, or anionic as in the case
of sulfolane, and depending on the residual charge of the metal, they will be adsorbed
[NACE1, 2012]. Generally, when organic inhibitors are introduced in a corroding system,
they alter the properties of the double layer by adsorbing at the metal-solution interface,
and the reaction can be summarized as follows:
M (nH2O) ads + I(s) MIads + nH2O (2.20)
Corrosion inhibitor molecules present in the solution displace the water molecules
adsorbed on the metal surface and are preferentially adsorbed.
2.6 Selection of corrosion inhibitors
In the process of development of a corrosion inhibitor, selection of appropriate
chemical compounds is the most crucial step. The selection of inhibitors has mostly been
empirical and based on the macroscopic understanding of physico-chemical properties
only. However, many recent works emphasize the importance of understanding the

51
compounds at a molecular level to comprehend their inhibition properties [Öğretir et al
(1999); Sastri and Perumareddi (1997)]. This assumes specific importance because
corrosion inhibition is generally considered to involve electron transfer between the
inhibitor and the metal in either direction. Consequently, factors such as availability of π
electrons due to the presence of unsaturated bonds, as in aromatic compounds and the
presence of reactive functional groups with the ability to donate electrons, which are
considered to facilitate electron transfer from the inhibitor to the metal. Properties of the
donor atom of the functional group, such as electron density and polarizability, can also
influence the inhibition characteristics of the inhibitor. Hence, it is an enhanced
understanding of the properties of the chemical compounds at a molecular level is
essential, in addition to general empirical knowledge, to render the selection process
more scientific.
The hard and soft acids and bases (HSAB) principle is a tool that can be
extensively useful in the process of design and selection of a corrosion inhibitor [Sastri,
2001]. As per the HSAB principle, every chemical compound can be classified into a
hard, soft, or borderline category of acid or base depending upon the compund’s
molecular parameters such as electronegativity, polarizability, size, and hardness
(calculated from ionization potential and electron affinity). For instance, when the
polarizability of a compound is low, it signifies hard characteristics, and higher values
signify soft characteristics, whereas the converse is true for electronegativity. Also
according to HSAB theory, homogenous interactions among the species are greatly
favoured, which implies that hard acids tend to form complexes with hard bases, whereas
soft acids tend to form complexes with soft bases and borderline bases can interact with

52
both soft and hard bases. Thus, it is important to classify the metal to be protected based
on the HSAB principle so that the potential corrosion inhibitors can be chosen
accordingly.
2.7 Quantum chemical analysis of corrosion inhibitors
Quantum chemical methods for corrosion inhibition studies, though they cannot
be used to directly predict corrosion in an absolute sense, can still be used to obtain
useful qualitative to semi-quantitative information to understand the corrosion inhibition
process. Generally, corrosion inhibition studies from a quantum chemical perspective
cannot be rigorously achieved due to the numerous parameters associated with the
process and the enormously complex interaction between them [Öğretir et al., 1999].
Some of the commonly reported quantum chemical indices that are correlated with
inhibition efficiencies are Highest occupied molecular orbital energy (EHOMO), Energy
gap (∆E) between highest occupied molecular orbital energy and lowest unoccupied
molecular orbital energy (EHOMO - ELUMO), Dipole moment (µ), and Charge on the donor
atom (Z) [Chakrabarti (1984); Sastri (2001); Khalil (2003); Öğretir et al (1999)]. EHOMO
is associated with the electron donating ability of a molecule, and, hence, higher values
indicate better inhibition efficiency. Similarly, ELUMO represents the ability of a molecule
to accept electrons, and, consequently, lower values of ∆E can cause higher inhibition
efficiency. For µ, lower values will favour accumulation of inhibitor in the surface layer,
leading to better inhibition, and in case of Z, the higher the value, the better the corrosion
inhibition [Khaled and Hackerman, 2003]. In some works, the fraction of electrons (∆N)

53
transferred from the inhibitor to the metal was also reported to have a direct correlation
with the corrosion inhibition performance [Sastri and Perumareddi, 1997].
∆N can be calculated based on the correlations below. As per Koopman’s
theorem, EHOMO and ELUMO are related to ionization potential (I) and electron affinity (A)
as follows:
- EHOMO = I (2.21)
- ELUMO = A (2.22)
Mulliken electronegativity (χ) and Absolute hardness (ň) can be approximated by
Equations (2.23 and 2.24), respectively.
2
AI (2.23)
2
AIn
(2.24)
∆N can be calculated using Equation (2.25), which has a direct relation with the
corrosion inhibition. For iron as bulk metal, a theoretical electronegativity value of 7.0 is
used, and absolute hardness is considered to be zero.
inhibitorMetal
inhibitorMetal
nN
n
(2.25)
These indices are used to analyze the effect of different substituents on the
corrosion inhibition characteristics of a particular parent organic compound. Öğretir et al
[1999] have used EHOMO, ∆E and µ to evaluate the performance of different pyridine
derivatives compared to the parent compound. In addition to the indices used in the
previous work, Sastri and Perumareddi [1997] have also used ∆N to evaluate the
corrosion inhibition behaviour of substituted pyridines and ethane compounds.

54
In another work by the same authors EHOMO, ∆E and charge on the nitrogen atom
(Zn) were correlated with the corrosion inhibition performance [Sastri and Perumareddi,
1994].

55
3. SELECTION AND TESTING OF CORROSION INHIBITORS
3.1 Selection of tested corrosion inhibitors
3.1.1 Selection of compounds
The principle of hard and soft acids and bases (HSAB) was used for theoretical
selection of corrosion inhibitors to be tested. For the case of amine-based CO2 absorption
processes, the most commonly used material of construction is carbon steel and the
general corrosion products was reported to be iron carbonate (FeCO3) [Kohl and Nielson,
1997, Hamah-Ali et al, 2011]. In FeCO3, iron is in a divalent (Fe2+
) state, which is
classified as a borderline acid [Sastri, 2001]. Therefore, the preferred choice of inhibitor
must possess borderline basic characteristics in order to best protect carbon steel
predominantly in a divalent oxidation state.
Also based on the general empirical understanding of organic compounds used as
corrosion inhibitors, sulfur-containing compounds were reported to be more effective due
to their better electron donating ability than nitrogen or oxygen [Sastri (2001);
Hackerman and Makrides (1954); Khaled and Hackerman (2003)]. Theoretical and
empirical considerations, as illustrated above, were used in the selection of inhibitors.
A total of thirteen compounds were chosen in which ten compounds were selected
based on the HSAB principle, including aniline- and pyridine-based compounds, and the
other three compounds were based on general empirical considerations. The list of
compounds chosen is presented in Table 3.1. Broadly, the selected inhibitors can be
sorted into three categories, namely, aniline-based, pyridine-based, and other compounds.
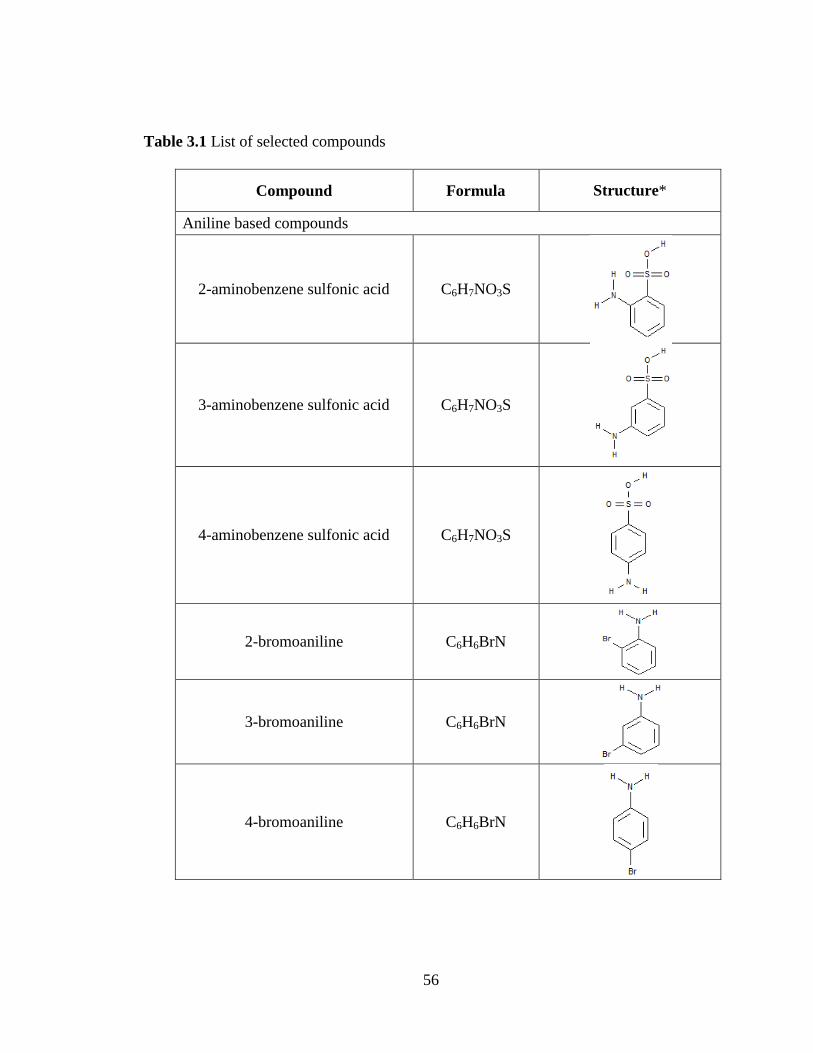
56
Table 3.1 List of selected compounds
Compound Formula Structure*
Aniline based compounds
2-aminobenzene sulfonic acid C6H7NO3S
3-aminobenzene sulfonic acid C6H7NO3S
4-aminobenzene sulfonic acid C6H7NO3S
2-bromoaniline C6H6BrN
3-bromoaniline C6H6BrN
4-bromoaniline C6H6BrN

57
Table 3.1 List of selected compounds (continued)
Compound Formula Structure*
Sulfapyridine C11H11N3O2S
Sulfanilamide C6H8N2O2S
Pyridine based compounds
2-bromopyridine C5H4BrN
3-bromopyridine C5H4BrN
Other compounds
Sulfolane C4H8O2S
Thiosalicylic acid C7H6O2S
Sodium thiosulfate Na2S2O3
* Molecular structures redrawn from PubChem database [PubChem1]

58
Eight substituted compounds of aniline were chosen due to their borderline
characteristics that enable them to interact with the metal surface effectively. Ortho,
para, and meta substituted aminobenzene sulfonic acids contain a sulfur (S) and an
oxygen (O) donor atom in addition to a nitrogen (N) atom that can act as a reaction center
for adsorption. Similarly, ortho, para, and meta substituted bromoanilines were chosen
since the natural borderline characteristics of the aniline will be enhanced by a borderline
substituent Br-. Sulfanilamide is also a substituted compound of aniline with one S and
two N and O reaction centers. Sulfpyridine is a derivative of sulfanilamide with a
pyridine ring attached to the nitrogen atom.
Substituted pyridine compounds also display borderline characteristics besides
being reported as corrosion inhibitors and, hence, are ideal choices for corrosion
inhibitors, especially the bromine (Br-) substituted compounds such as 2-bromopyridine
and 3-bromopyridine where Br- can enhance the borderline characteristics.
Sulfolane, a heterocyclic compound with a sulfonyl reaction center and
thiosalicylic acid with one S and two O reaction centers were the other compounds
chosen. Sodium thiosulfate, a process contaminant in the amine-based CO2 absorption
process, when tested for its effect in the preliminary experiments, caused a drastic
reduction in corrosion rate. Hence, sodium thiosulfate was also tested along with other
organic inhibitors.
3.1.2 Toxicity evaluation
The preliminary selection was followed by a screening process that used the
values of toxicity of the compounds as the screening criteria. The specific criteria is that

59
the corrosion inhibitors must not be more toxic than the absorbents used in the CO2
capture process (i.e., their lethal dosage (LD50) values must be equal or greater than those
of the absorbents). Although DEA is the most toxic absorbent among those listed in
Table 3.2, the toxicity of MEA was set as the basis for screening inhibitors. This is
because MEA is the most prevalent benchmark solvent for absorbents used in carbon
capture applications. Thus, the cut-point toxicity is LD50 ORAL Rat < 1720 mg/kg.
Ecological information on the selected organic compounds is compiled in Table
3.3. It was not possible to obtain all the details of ecological information for all the
chemical compounds considered; however, based on the available details, it was possible
to eliminate certain compounds that are explicitly not safe and do not meet the screening
criteria. Generally, bromine compounds are considered suspected carcinogens, and
among the Br- substituted compounds selected, only the toxicity values of 4-bromoaniline
and 2-bromopyridine were available and neither met the screening criteria. It can also be
observed that in the material safety data sheets (MSDS) for these compounds, the
ecological properties of the parent compound and some of its derivatives for which the
data is available were considered representative of a whole group of compounds. In line
with that, the toxicity values of those compounds for which the data is available were
taken as a guideline for assessing compounds that are structural isomers of the same
compound.
Also, hazard rating for each compound was taken as a guideline to define the
toxicity of the material. Due to their toxicity and suspected carcinogenic nature, the five
Br- substituted compounds were eliminated from further analysis.
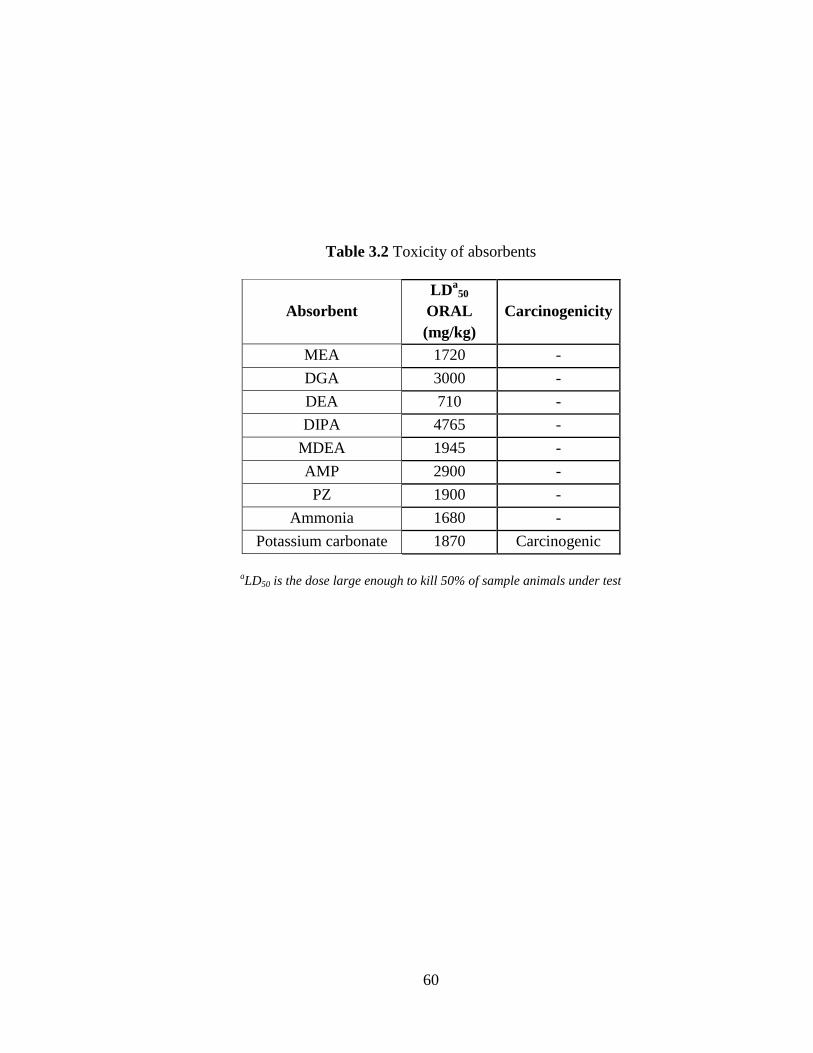
60
Table 3.2 Toxicity of absorbents
Absorbent
LDa
50
ORAL
(mg/kg)
Carcinogenicity
MEA 1720 -
DGA 3000 -
DEA 710 -
DIPA 4765 -
MDEA 1945 -
AMP 2900 -
PZ 1900 -
Ammonia 1680 -
Potassium carbonate 1870 Carcinogenic
aLD50 is the dose large enough to kill 50% of sample animals under test
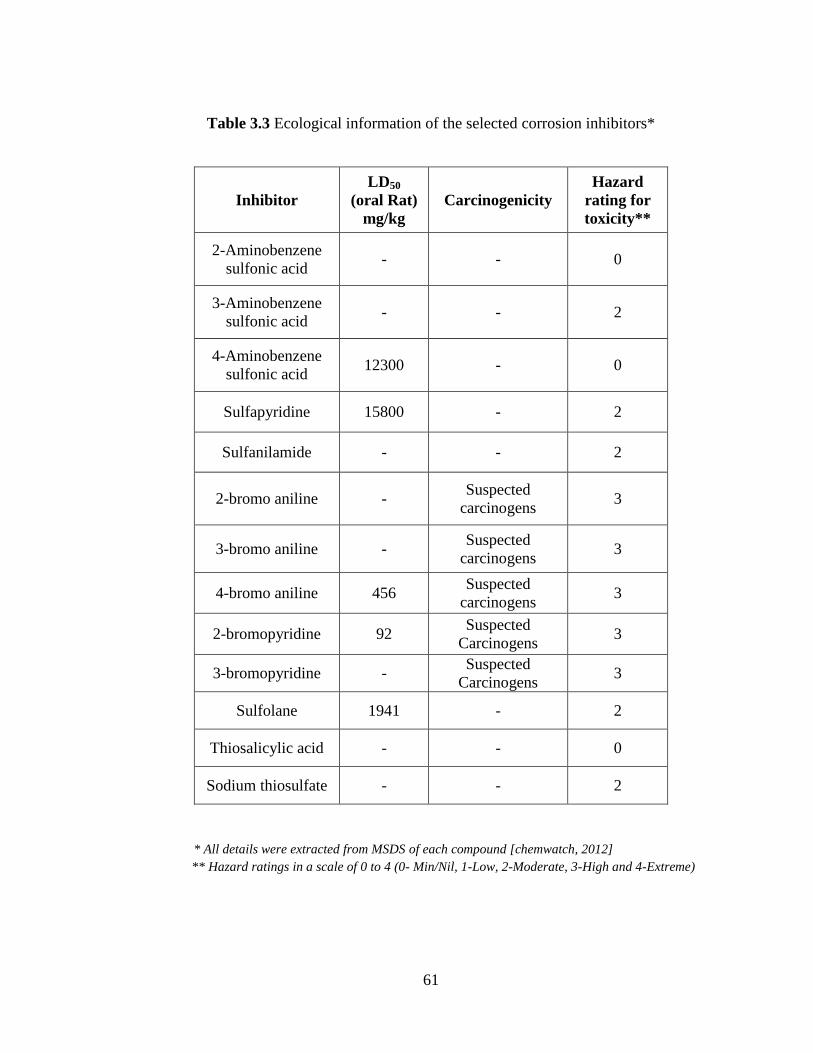
61
Table 3.3 Ecological information of the selected corrosion inhibitors*
Inhibitor
LD50
(oral Rat)
mg/kg
Carcinogenicity
Hazard
rating for
toxicity**
2-Aminobenzene
sulfonic acid - - 0
3-Aminobenzene
sulfonic acid - - 2
4-Aminobenzene
sulfonic acid 12300 - 0
Sulfapyridine 15800 - 2
Sulfanilamide - - 2
2-bromo aniline - Suspected
carcinogens 3
3-bromo aniline - Suspected
carcinogens 3
4-bromo aniline 456 Suspected
carcinogens 3
2-bromopyridine 92 Suspected
Carcinogens 3
3-bromopyridine - Suspected
Carcinogens 3
Sulfolane 1941 - 2
Thiosalicylic acid - - 0
Sodium thiosulfate - - 2
* All details were extracted from MSDS of each compound [chemwatch, 2012]
** Hazard ratings in a scale of 0 to 4 (0- Min/Nil, 1-Low, 2-Moderate, 3-High and 4-Extreme)

62
Only eight compounds were selected for further analysis, namely 2-aminobenzene
sulfonic acid, 3-aminobenzene sulfonic acid, 4-aminobenzene sulfonic acid,
sulfapyridine, sulfanilamide, sulfolane, thiosalicylic acid, and sodium thiosulfate.
3.1.3 Quantum chemical analysis
Quantum chemical analysis was carried out to determine the quantum chemical
parameters such as highest occupied molecular orbital energy (EHOMO), energy gap (∆E)
between highest occupied molecular orbital energy and lowest unoccupied molecular
orbital energy (EHOMO - ELUMO), dipole moment (µ), fraction of electron transferred (∆N),
and charge of the donor atom (Z). EHOMO, ∆N and Z have a direct relation with inhibition
efficiencies whereas ∆E and µ have an inverse relation [Chakrabarti (1984); Sastri
(2001); Khalil (2003); Khaled and Hackerman (2003); Sastri and Perumareddi (1997)].
Generally, these indices are used to analyze the effect of different substituents on
the corrosion inhibition characteristics of a particular parent organic compound.
However, for this study, of the eight compounds selected, only seven compounds were
organic. Among them, only five compounds were derivatives of one parent compound,
aniline, and, hence, could be evaluated comparatively. The quantum chemical parameters
for the other two compounds that were distinctly different from the rest were also
determined. The only commonality among all the selected organic compounds is the
presence of a sulfur atom that can act as the adsorption center. Hence, the charge of the
sulfur atom (Zs) and ∆N were used to compare all the compounds studied.
Semi-empirical quantum chemistry calculations package MOPAC2007 was used
to perform PM-6 (Parameterization) calculations from which the quantum chemical

63
parameters for selected corrosion inhibitors were obtained by specifying the molecular
geometry of each compound [PubChem1, 2011]. Values of EHOMO, ∆E, and µ can be
directly obtained from the software, whereas the values of ∆N and Zs were obtained by
further analysis and are presented in Table 3.4. Trends of different parameters are
presented in Figure 3.1.
In Figures 3.1a, 3.1b, and 3.1c, five aniline derivatives are compared, where
sulfapyridine has the highest EHOMO value and, hence, better than the other compounds.
However, in terms of ∆E and µ, 3-aminobenzene sulfonic acid and 2-aminobenzene
sulfonic acid were better than other compounds, respectively. In the case of ∆N and Zs
(Figures 3.1d and 3.1e), sulfapyridine was better than all the other compounds. From the
comparisons based on different parameters, 2-aminobenzene sulfonic acid, 3-
aminobenzene sulfonic acid, and sulfapyridine are potential corrosion inhibitors.

64
Table 3.4 Quantum chemical parameters for selected compounds
Compound EHOMO
(eV)
ΔE
(eV)
µ
(debye) ∆N Zs
2-aminobenzene sulfonic acid -9.17 8.61 4.64 0.25 2.54
3-aminobenzene sulfonic acid -9.10 8.29 5.19 0.25 2.45
4-aminobenzene sulfonic acid -9.45 8.85 8.58 0.22 2.48
Sulfanilamide -9.18 8.89 6.50 0.25 2.52
Sulfapyridine -9.01 8.78 7.96 0.27 2.56
thiosalicylic acid -9.27 7.82 2.73 0.21 -0.18
Sulfolane -10.82 2.44 5.88 0.18 2.44
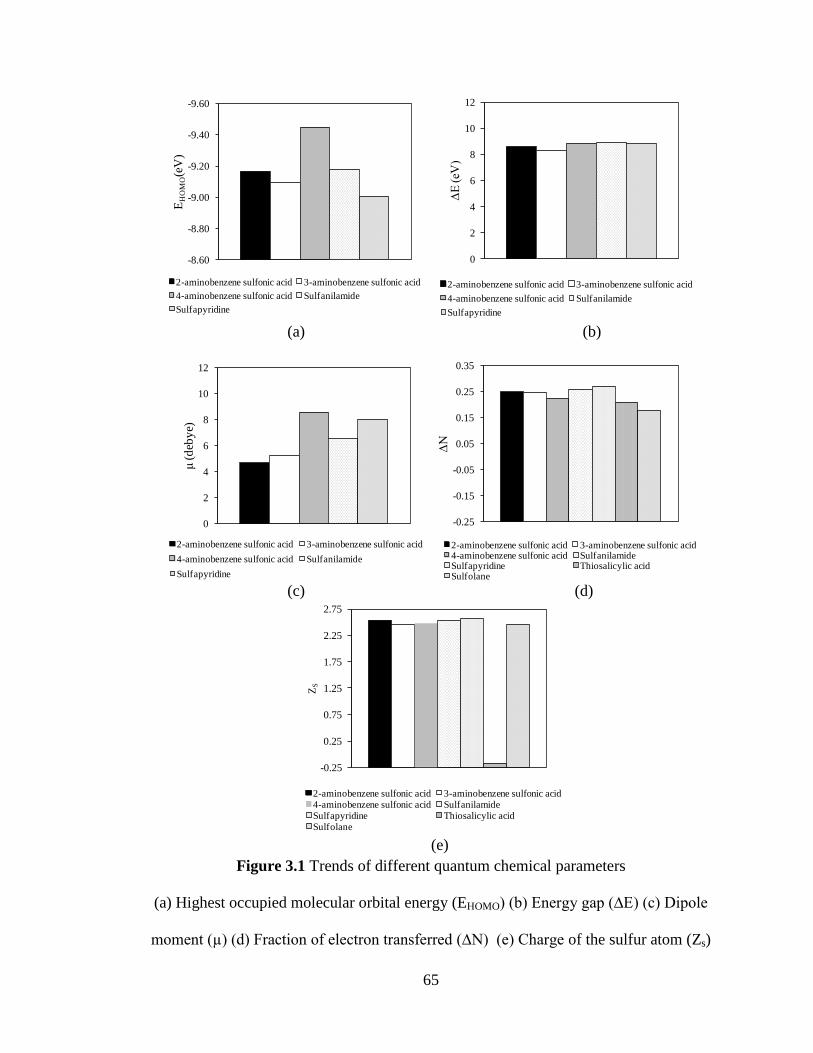
65
(a) (b)
(c) (d)
(e)
Figure 3.1 Trends of different quantum chemical parameters
(a) Highest occupied molecular orbital energy (EHOMO) (b) Energy gap (∆E) (c) Dipole
moment (µ) (d) Fraction of electron transferred (∆N) (e) Charge of the sulfur atom (Zs)
-9.60
-9.40
-9.20
-9.00
-8.80
-8.60
EH
OM
O(e
V)
2-aminobenzene sulfonic acid 3-aminobenzene sulfonic acid
4-aminobenzene sulfonic acid Sulfanilamide
Sulfapyridine
0
2
4
6
8
10
12
∆E
(eV
)
2-aminobenzene sulfonic acid 3-aminobenzene sulfonic acid
4-aminobenzene sulfonic acid Sulfanilamide
Sulfapyridine
0
2
4
6
8
10
12
μ(d
eby
e)
2-aminobenzene sulfonic acid 3-aminobenzene sulfonic acid
4-aminobenzene sulfonic acid Sulfanilamide
Sulfapyridine
-0.25
-0.15
-0.05
0.05
0.15
0.25
0.35
∆N
2-aminobenzene sulfonic acid 3-aminobenzene sulfonic acid4-aminobenzene sulfonic acid SulfanilamideSulfapyridine Thiosalicylic acidSulfolane
-0.25
0.25
0.75
1.25
1.75
2.25
2.75
ZS
2-aminobenzene sulfonic acid 3-aminobenzene sulfonic acid4-aminobenzene sulfonic acid SulfanilamideSulfapyridine Thiosalicylic acidSulfolane

66
3.2 Corrosion testing
In this work, two types of corrosion experiments were conducted in order to
evaluate the performance of corrosion inhibitors (i.e., electrochemical and weight loss
testing). The details of the experimental setup, procedure, and associated analysis are
described in the following sections.
3.2.1 Electrochemical experiments
3.2.1.1 Experimental setup
Figure 3.2 illustrates the experimental setup used for the electrochemical testing
in this work. It comprises a number of components as follows:
i) 100 ml double walled microcell (Model 636 ring disc electrode (RDE)
assembly, PAR, USA), which is a three electrode assembly with a cylindrical
working electrode at the centre and a silver/silver chloride (Ag/AgCl)
reference electrode and a platinum counter electrode on either side
ii) A water bath with a temperature controller to maintain the temperature of the
corrosion cell at a required temperature by circulating hot water through the
outer jacket of the corrosion cell
iii) A CO2 and N2 supply set consisting of a CO2 and N2 cylinder with a gas
regulator and a flow meter, each in series
iv) A water cooled condenser to avoid evaporation losses from the corrosion cell
v) A potentiostat (PAR 263A, Princeton Applied Research, USA) interfaced
with an impedance system (Model 5210 Lock-in amplifier)
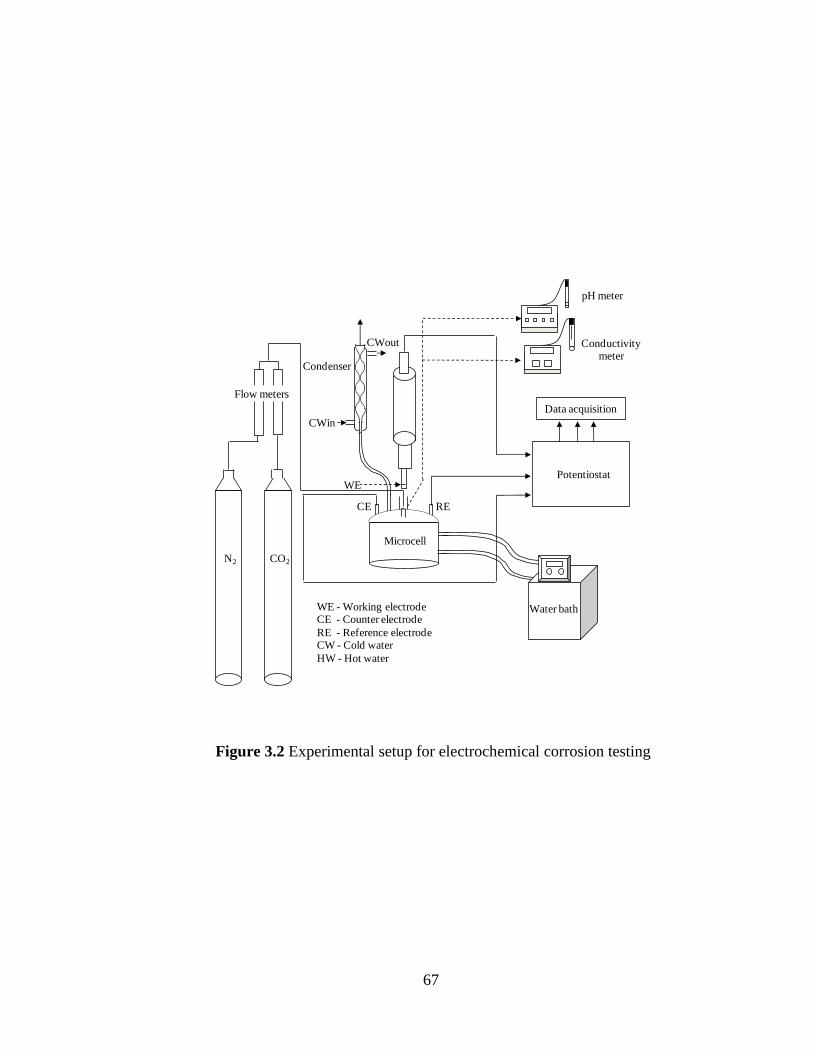
67
Figure 3.2 Experimental setup for electrochemical corrosion testing
Potentiostat
Data acquisition
N2 CO2
Flow meters
Condenser
CWin
WE - Working electrode CE - Counter electrode
RE - Reference electrode CW - Cold water
HW - Hot water
CE RE
CWout
WE
Conductivity meter
pH meter
Microcell
Water bath

68
vi) Powercorr (Version 2.53) software to record and analyze the results by
interfacing with a PC
vii) A pH meter (Oakton pH510 series)
viii) A conductivity meter (YSI 3200 conductivity instrument)
3.2.1.2 Specimen preparation
Carbon steel (CS 1018 with the composition of 0.175% carbon, 0.75%
manganese, and balance iron) was chosen as the working electrode (specimen) due to its
common use as construction material in amine-based CO2 capture plants [Dupart et al.,
1993]. The specimens were cylindrical with dimensions of 0.8 cm in height, 1.2 cm
outside diameter, and 0.6 cm central hole, as shown in Figure 3.3. The surface of each
specimen was prepared by wet grinding with 600 grit silicon carbide papers using
deionized water, being degreased with high purity methanol, rinsed with deionized water,
and then dried as per ASTM G1-90 (1999) before being introduced into the solution for
testing.
3.2.1.3 Solution preparation
Monoethanolamine (MEA) was chosen in this work as the absorption solvent
since it has been used as the benchmark in both demonstration and commercial CO2
capture plants. Its concentration was fixed at 5.0 kmol/m3 or 30% by weight to represent
the common strength of the service MEA solution in practice [Kohl and Nielson, 1997].
The aqueous solution of MEA was prepared from 99% reagent grade MEA and deionized
water.

69
Figure 3.3 A sketch of electrochemical testing specimens
0.6 cm
1.2 cm
0.8 cm

70
The solution was saturated with CO2 with a CO2 loading of 0.55±0.05 mol CO2 /
mol MEA. To these final solutions, a measured weight of tested corrosion inhibitor was
added. Inhibitor concentrations (250-10000 ppm) were chosen to be within the range of
the literature values (Table 1.2). The same procedure was repeated for different corrosion
inhibitors tested. In some tests, process contaminants, namely sodium chloride (NaCl)
and formic acid (CH2O2), with concentrations of 10,000 ppm were added to the test
solution for the study of process contaminant effects on the inhibitor performance. To
determine the MEA solution concentration, a measured volume of the solution sample
was titrated with hydrochloric acid (HCl) using methyl orange indicator. For every drop
of HCl added, a corresponding volume of CO2 was liberated. Its volume was measured
using a Chittick’s apparatus, which was attached to the titration setup (Figure 3.4). Based
on the volume of CO2 liberated, the CO2 loading of the solution was determined. A
summary of the chemicals used for the experiments are presented in Table 3.5.
3.2.1.4 Experimental procedure
The experiment began with charging the prepared MEA solution into the
corrosion cell. The corrosion cell was then fitted with a condenser to prevent evaporation
loss of the test solution, purged with gaseous CO2 to maintain the CO2 saturated loading
of the solution, and connected to a heater circulator to maintain the temperature of
solution at 80oC. After the solution temperature reached 80
oC, solution samples were
taken for analysis of MEA concentration and CO2 loading. The conductivity and pH of
the tested solution were directly measured from the corrosion cell. The corrosion cell was
assembled with a working electrode (specimen), a counter electrode, and a reference

71
Figure 3.4 Chittick’s apparatus for CO2 loading measurement
1 1
1 11
11
Burette (50 ml)
Sample
Conical Flask
Levelling bulb
Graduated gas measuring tube
Stopper
Glass T-tube
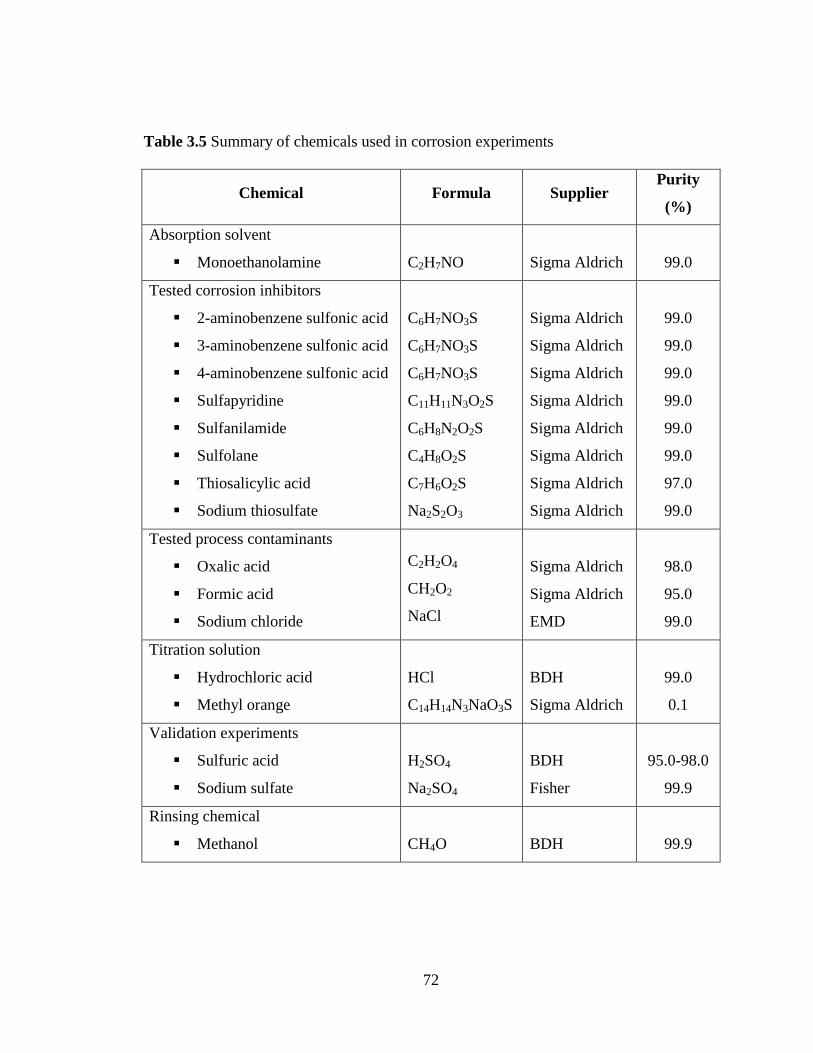
72
Table 3.5 Summary of chemicals used in corrosion experiments
Chemical Formula Supplier Purity
(%)
Absorption solvent
Monoethanolamine
C2H7NO
Sigma Aldrich
99.0
Tested corrosion inhibitors
2-aminobenzene sulfonic acid
3-aminobenzene sulfonic acid
4-aminobenzene sulfonic acid
Sulfapyridine
Sulfanilamide
Sulfolane
Thiosalicylic acid
Sodium thiosulfate
C6H7NO3S
C6H7NO3S
C6H7NO3S
C11H11N3O2S
C6H8N2O2S
C4H8O2S
C7H6O2S
Na2S2O3
Sigma Aldrich
Sigma Aldrich
Sigma Aldrich
Sigma Aldrich
Sigma Aldrich
Sigma Aldrich
Sigma Aldrich
Sigma Aldrich
99.0
99.0
99.0
99.0
99.0
99.0
97.0
99.0
Tested process contaminants
Oxalic acid
Formic acid
Sodium chloride
C2H2O4
CH2O2
NaCl
Sigma Aldrich
Sigma Aldrich
EMD
98.0
95.0
99.0
Titration solution
Hydrochloric acid
Methyl orange
HCl
C14H14N3NaO3S
BDH
Sigma Aldrich
99.0
0.1
Validation experiments
Sulfuric acid
Sodium sulfate
H2SO4
Na2SO4
BDH
Fisher
95.0-98.0
99.9
Rinsing chemical
Methanol
CH4O
BDH
99.9

73
electrode and then connected to the potentiostat. The open circuit potentials (OCP) were
recorded as a function of time until reaching a steady state value (±1 mV for 300
seconds). After the steady state OCP value was obtained, impedance analysis was carried
out. The AC amplitude of 10 mV was applied over a frequency range of 10 kHz to 10
mHz. The corresponding impedance values, resolved into imaginary and real
components, were measured as a function of frequency. Since the excitation potential was
a very small magnitude, there was a minimum system perturbation OCP and the system
returned to its steady state OCP in 5 to 10 minutes.
After the impedance measurement, cyclic potentiodynamic polarization scans
were carried out at a scan rate of 10 mV/min. On the completion of each experiment,
solution samples were taken again for measuring MEA concentration and CO2 loading of
solutions. The values of conductivity and pH of the solution were measured and recorded.
3.2.1.5 Validation of experimental setup
The experimental setup and procedure for potentiodynamic polarization was
validated using ASTM G5-94 (Reapproved 2004) preceding actual testing for corrosion
inhibitors. The working electrode used for this purpose was stainless steel type 430
(SS430), which was prepared by wet polishing of specimen with 600 grit silicon carbide
papers. The procedure involved anodic polarization of the SS430 specimen in 1.0 N
sulfuric acid (H2SO4) solution at 30oC. The corrosion cell was purged with nitrogen (N2)
at a flow rate of 150 cm3/min for 30 minutes prior to the polarization experiment.
Resulting polarization curves were compared with the ASTM reference band. As
illustrated in Figure 3.5a, the polarization curve produced from this work lies within the

74
(a)
(b)
Figure 3.5 Validation of experimental setup and procedure
(a) Validation of potentiodynamic polarization using ASTM G5-94 (2004) (b) Validation
of impedance measurement using ASTM G106-89 (2010)
-0.800
-0.400
0.000
0.400
0.800
1.200
1.600
0.1 1 10 100 1000 10000 100000
Po
ten
tial
(V
) v
s A
g/A
gC
l
Current density (µA/cm2)
ASTM- maximum
This work
ASTM- minimum
0
15
30
45
60
75
0 40 80 120 160 200
-Z"
(ohm
cm
2)
Z' (ohm cm2 )
ASTM maximum
This work
ASTM minimum

75
ASTM standard curves, thereby validating our experimental setup and polarization
measuring procedure.
Similarly, the experimental setup and procedure for impedance measurements
were validated using ASTM G106-89 (Reapproved 2010) where SS430 specimen was
used as the working electrode and prepared similar to ASTM G5-94. The corrosion cell
was purged with N2 at a flow of 150 cm3/min for 60 minutes prior to the experiment and
was continued throughout the experiment. The procedure involved the impedance
analysis of SS430 specimen in 0.495 M sodium sulfate (Na2SO4) solution containing
0.005 M H2SO4 at 30oC. The resulting Nyquist plot was compared with the ASTM
reference band. Figure 3.5b shows that the plot produced from this work conforms to the
ASTM band, thereby validating the experimental setup and impedance measuring
procedure.
3.2.1.6 Data analysis
The Tafel extrapolation technique, taking into account only ±250 mV from the
corrosion potential (Ecorr) of the potentiodynamic polarization curves, was used to
estimate the corrosion current density (icorr) from which corrosion rate (CR) could be
determined using Equation (3.1).
D
1027.3 3
n
aiCR corr
(3.1)
where CR is the corrosion rate in millimeters per year (mmpy), icorr is the corrosion
current density in µA/cm2, a is the atomic weight, n is the number of electrons, and D is
the density of specimen in g/cm3. Inhibition efficiency (IE) can be calculated from the

76
corrosion rates obtained for uninhibited inhibited conditions using the following
expression:
duninhibite
inhibitedduninhibite
CR
CRCRIE 100(%)
(3.2)
3.2.2 Weight loss experiments
3.2.2.1 Experimental setup
The experimental setup used for weight loss corrosion testing is illustrated in
Figure 3.6, and it included the following components:
i) A two liter double walled cylindrical glass corrosion cell attached to a metal
lid equipped with provisions for a thermometer, a gas dispenser, a condenser,
and central glass specimen hooks.
ii) A thermometer for monitoring the solution temperature
iii) A water-cooled condenser for minimizing solution vaporization loss
iv) A water bath with a temperature controller for maintaining the solution
temperature at a required temperature by circulating hot water through the
outer jacket of the corrosion cell
v) A CO2 supply set consisting of a CO2 cylinder with a gas regulator and flow
meter in series
vi) Glass specimen hooks with provisions for mounting four metal specimens at
one time.
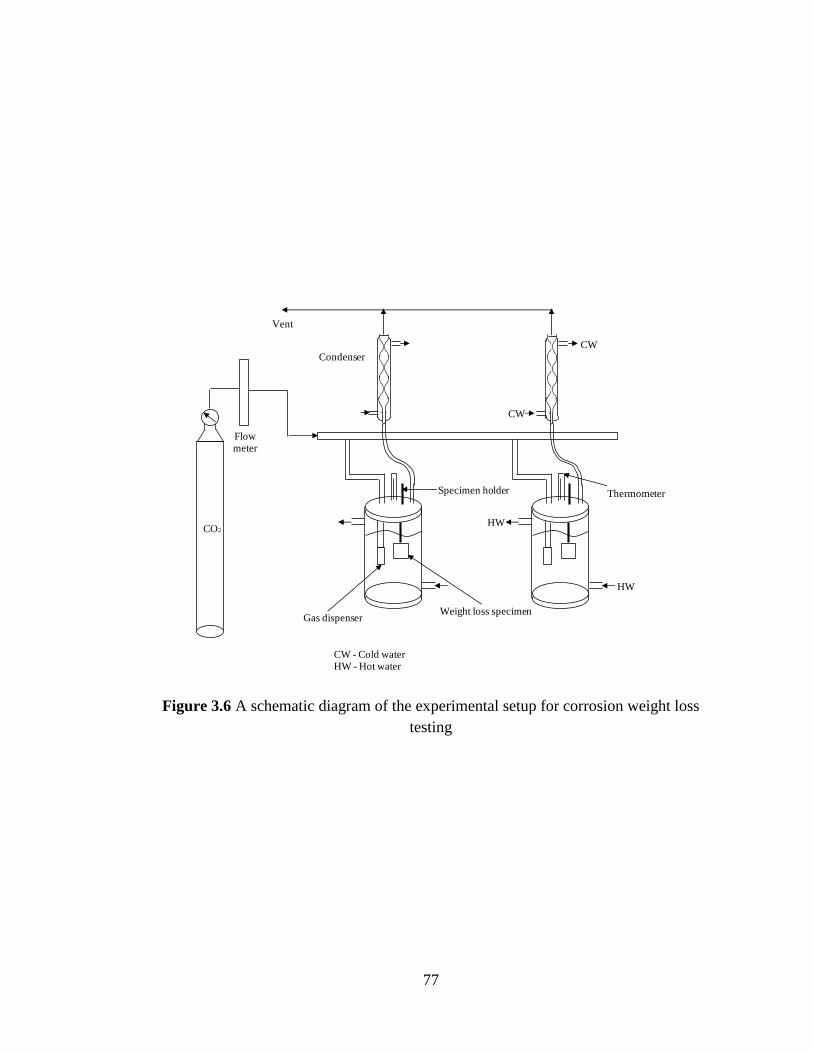
77
Figure 3.6 A schematic diagram of the experimental setup for corrosion weight loss
testing
Flowmeter
CO2
Vent
Condenser
Weight loss specimenGas dispenser
ThermometerSpecimen holder
HW
HW
CW
CW
CW - Cold water HW - Hot water

78
3.2.2.2 Specimen preparation
Carbon steel 1018 (CS1018) specimens were used for the reasons discussed in
section 3.2.1.2. The specimens were rectangular coupons with dimensions of 2.5 cm in
height, 2.5 cm in width, and 0.3 cm thickness, as shown in Figure 3.7. They were surface
finished with 600 grit silicon papers, rinsed with high purity methanol followed by
deionized water, and finally dried before being introduced into the test solution.
3.2.2.3 Solution preparation
The solution preparation for weight loss testing follows the same procedure as
detailed in section 3.2.1.3 except for the volume of test solution, which was two liters in
this case. The solution was preloaded with CO2 until the saturation point and corrosion
inhibitor with a specific concentration was added as required.
3.2.2.4 Experimental procedure
The test solution was transferred to the corrosion cell. The prepared specimens
were weighed using a microbalance with an accuracy of ±0.10 mg, and then mounted
onto the glass specimen hooks fitted to the center of the metal lid. The specimens were
immersed in the test solution, and the metal lid was fastened to the corrosion cell. The
gaseous CO2 was purged continuously throughout the experiment to maintain the CO2
saturation in the test solution. Hot water was circulated in the outer jacket of the
corrosion cell to elevate the temperature of the solution to the required condition. The
solution concentration, CO2 loading, and solution temperature were regularly monitored
throughout the experiments. Owing to its relatively long testing time, the test solution
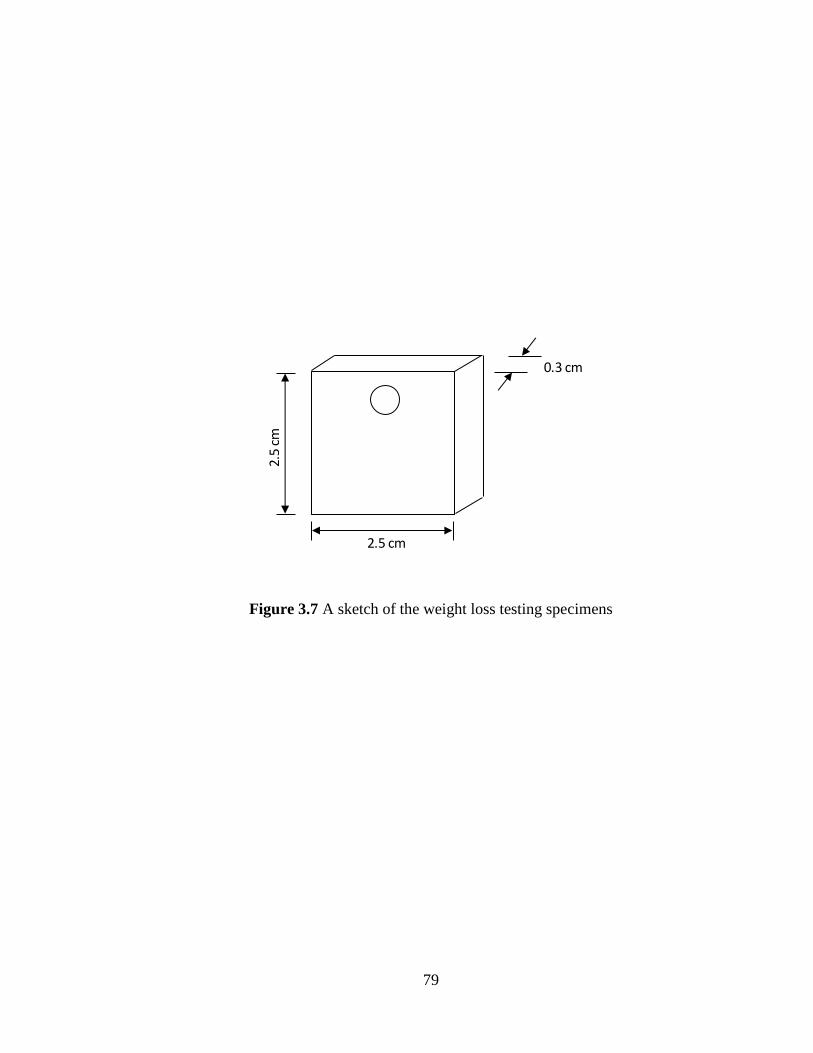
79
Figure 3.7 A sketch of the weight loss testing specimens
0.3 cm
2.5 cm
2.5
cm

80
required replenishment by addition of the calculated amounts of water at regular intervals
to account for the water loss by evaporation. The specimens were removed from the
corrosion cell at specific intervals of time. From the weight loss of these specimens,
corrosion rate was obtained.
3.2.2.5 Weight loss analysis
The corrosion products were removed from the tested specimens as per ASTM
standard G1-90 (1999). The tested specimens were cleaned mechanically by
simultaneous washing in running tap water and brushing with a non-metallic bristle to
remove the loose corrosion product and the weight loss compared to the initial weight of
the specimen was recorded. Then, the specimens were chemically cleaned by immersion
in a solution of concentrated hydrochloric acid (HCl) inhibited with 20 g antimony
trioxide (Sb2O3) and 50 g stannous chloride (SnCl2) for one minute. The mechanical and
chemical cleaning cycles were repeated, and the weight loss after each cycle was
determined. Determined weight loss was plotted against the number of cycles as shown
in Figure 3.8. The curve AB corresponds to the weight loss due to the removal of
corrosion product from the metal surface. As the corrosion product is removed, the bare
metal is exposed during the chemical cleaning and the measured weight loss in the
specimen became constant as shown in the curve BC. The point of intersection between
AB and BC corresponds to the weight loss due to corrosion from which the corrosion rate
can be calculated using Equation (3.3).
D
TA
WKCR
(3.3)

81
Figure 3.8 Estimation of weight loss of the tested specimen (ASTM G1-90)
0.36
0.37
0.37
0.38
0.38
0.39
0.39
0.40
0 2 4 6
Wei
gh
t lo
ss
Number of cleaning cycles
B
C
A

82
where CR is the corrosion rate in mmpy, K is the constant with a value of 8.76 x 104, W is
the weight loss in g, A is the area of the metal specimen in cm2, T is the time of exposure
in hours, and D is the density of the specimen in g/cm3.
3.2.2.6 Surface analysis
The weight loss specimen exposed to the longest duration of testing was subjected
to the surface analytical techniques including X-ray Diffraction analysis (XRD), Energy-
dispersive X-ray Spectroscopy (EDS), and Scanning Electron Microscopy (SEM) to
understand and characterize the corrosion products for different conditions tested. The
XRD analysis was carried out to characterize any crystalline phases in the corrosion
products. A Bruker Discover diffractometer (nickel - filtered Cu K - 0.154056 nm as a
radiation source) was used for analysis, and reference data from the International Centre
for Diffraction Data (ICDD) was used for identifying the phases. For the SEM analysis, a
JSM – 5600 (JEOL, USA) microscope was used for obtaining the images at different
magnifications. The EDS analysis was carried out using an EDAX Genesis 7000 model,
which was used for elemental analysis of the specimen surface. Following by surface
analysis, the specimens were subjected to a cleaning procedure as detailed in Section
3.2.2.5 to determine the weight loss and, in turn, the corrosion rate.

83
4. RESULTS AND DISCUSSION
The inhibition performance of the selected corrosion inhibitors was evaluated
using two corrosion testing techniques, namely electrochemical and weight loss testing.
The electrochemical testing provides an insight into the behaviour of corrosion and
corrosion inhibition while the weight loss testing provides information of corrosion
magnitudes in longer periods of exposure time than the electrochemical testing. Details of
results from both tests are given below.
4.1 Electrochemical tests
Corrosion characteristics of uninhibited and inhibited systems were studied by
evaluating the corrosion behaviour of carbon steel (CS1018) at different experimental
conditions as shown in Table 4.1. Electrochemical tests employed two techniques,
namely DC - cyclic potentiodynamic polarization and AC - impedance analysis.
4.1.1 Corrosion behaviour of uninhibited MEA systems
a) Absence of process contaminants
From the cyclic potentiodynamic polarization curve (Figure 4.1a), it can be
observed that the carbon steel exhibits active-passive behaviour in the uninhibited MEA
system (5.0 kmol/m3 MEA at 80
oC and 0.55 mol/mol CO2). Under these experimental
conditions, the metal undergoes active corrosion at a rate of 4.27 mmpy (Table 4.2).
Negative hysteresis (reverse curve lies left of the forward curve) suggests no pitting
corrosion tendency (Figure 4.1a).
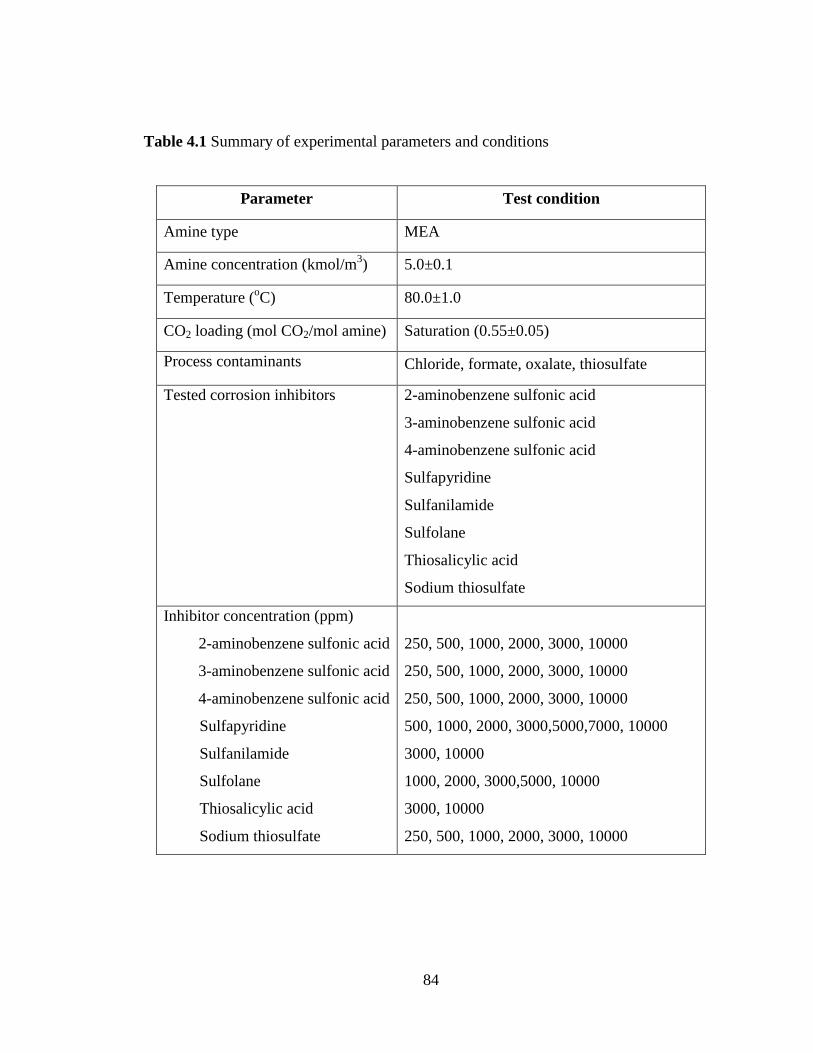
84
Table 4.1 Summary of experimental parameters and conditions
Parameter Test condition
Amine type MEA
Amine concentration (kmol/m3) 5.0±0.1
Temperature (oC) 80.0±1.0
CO2 loading (mol CO2/mol amine) Saturation (0.55±0.05)
Process contaminants Chloride, formate, oxalate, thiosulfate
Tested corrosion inhibitors 2-aminobenzene sulfonic acid
3-aminobenzene sulfonic acid
4-aminobenzene sulfonic acid
Sulfapyridine
Sulfanilamide
Sulfolane
Thiosalicylic acid
Sodium thiosulfate
Inhibitor concentration (ppm)
2-aminobenzene sulfonic acid
3-aminobenzene sulfonic acid
4-aminobenzene sulfonic acid
Sulfapyridine
Sulfanilamide
Sulfolane
Thiosalicylic acid
Sodium thiosulfate
250, 500, 1000, 2000, 3000, 10000
250, 500, 1000, 2000, 3000, 10000
250, 500, 1000, 2000, 3000, 10000
500, 1000, 2000, 3000,5000,7000, 10000
3000, 10000
1000, 2000, 3000,5000, 10000
3000, 10000
250, 500, 1000, 2000, 3000, 10000
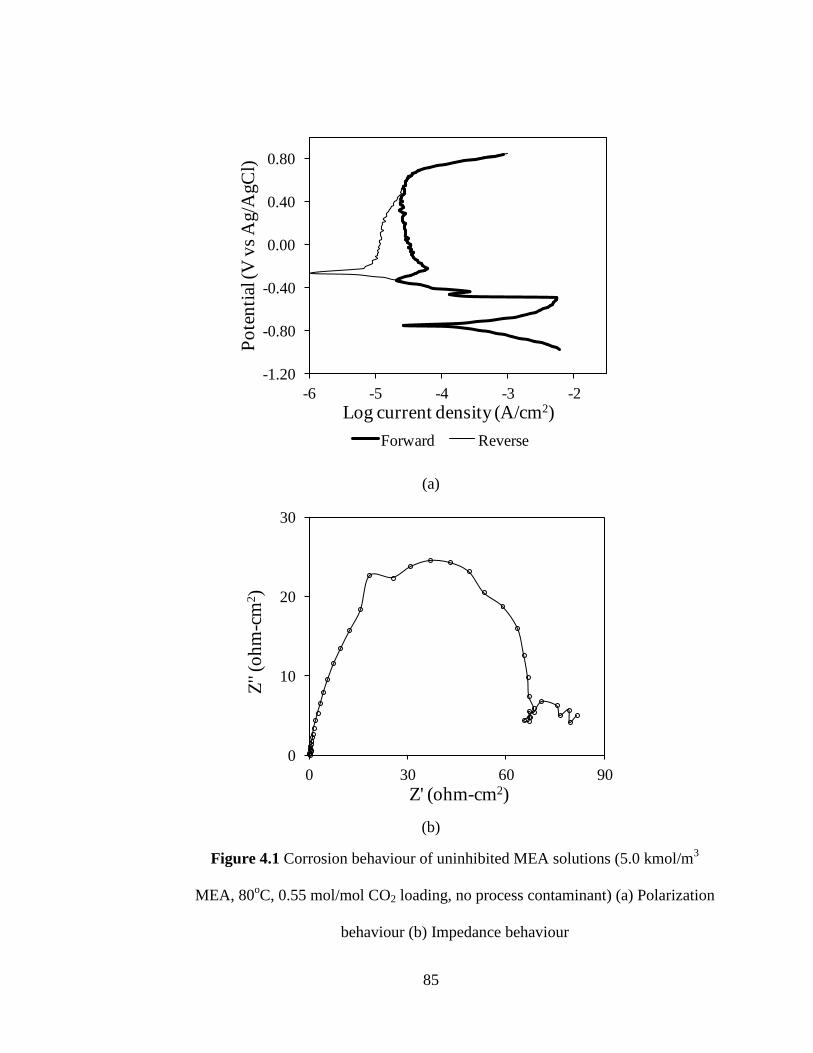
85
(a)
(b)
Figure 4.1 Corrosion behaviour of uninhibited MEA solutions (5.0 kmol/m3
MEA, 80oC, 0.55 mol/mol CO2 loading, no process contaminant) (a) Polarization
behaviour (b) Impedance behaviour
-1.20
-0.80
-0.40
0.00
0.40
0.80
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
Forward Reverse
0
10
20
30
0 30 60 90
Z''
(oh
m-c
m2)
Z' (ohm-cm2)

86
Table 4.2 Summary of electrochemical experimental results
Experimental Condition pH σ
(mS/cm)
Ecorr
(mV vs
Ag/AgCl)
icorr
(μA/cm2)
βc
(mV/
decade)
βa
(mV/
decade)
Rp
(ohm-cm2)
CR
(mmpy)
IE
(%)
Pitting
tendency
No corrosion inhibitor :
5.0 kmol/m3 MEA, 80oC,
α=0.55
- 8.15±0.03 42.55±1.75 -751.92±0.70 364.24±1.19 88.84±11.20 146.08±27.1 72.04±0.40 4.27±0.01 - No
10000 ppm
thiosulfate 8.78±0.05 39.44±1.20 -703.05±0.61 20.33±0.73 98.82±3.10 135.71±6.40 995.19±25.01 0.26±0.01 - No
10000 ppm
oxalate 8.24±0.02 43.24±2.60 -773.31±5.44 66.89±0.60 133.10±0.10 77.60±2.60 172.50±8.60 0.78±0.01 - No
10000 ppm
formate 7.98±0.03 50.67±1.60 -756.42±1.23 45.03±1.39 83.64±8.62 159.30±11.26 73.10±11.12 5.29±0.02 - No
10000 ppm
chloride 8.36±0.04 45.20±2.20 -750.34±0.74 344.37±2.15 71.60±3.40 120.90±6.30 86.84±2.40 4.04±0.03 - No
2-aminobenzene sulfonic
acid :
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
250 ppm 8.22±0.01 42.08±1.80 -787.91±0.71 47.02±0.83 123.73±2.20 63.58±1.30 390.21±6.90 0.55±0.01 87 No
500 ppm 8.30±0.04 41.40±1.40 -773.40±3.14 48.68±1.26 128.42±1.60 66.36±4.56 359.44±10.36 0.57±0.01 87 No
1000 ppm 8.37±0.02 40.06±2.10 -786.44±3.90 46.36±1.21 127.40±0.52 71.20±3.24 417.84±4.10 0.54±0.01 87 No
2000 ppm 8.48±0.02 38.87±1.20 -786.71±0.74 39.40±2.40 119.40±1.04 65.86±2.64 401.16±21.54 0.46±0.00 89 No
3000 ppm 8.60±0.01 37.95±0.80 -778.93±4.30 47.02±1.74 129.14±3.00 63.54±2.42 349.76±12.73 0.55±0.00 87 No
10000 ppm 8.80±0.06 36.60±2.40 -753.92±5.84 138.08±1.27 121.42±2.60 73.10±8.26 133.54±11.40 1.62±0.01 62 No
2-aminobenzene sulfonic
acid (1000 ppm) :
5.0 kmol/m3 MEA, 80oC,
α=0.55
10000 ppm
formate 8.20±0.02 47.78±3.40 -742.36±8.24 52.65±0.50 90.74±8.46 127.73±2.12 84.70±3.26 5.70±0.01 -34 No
10000 ppm
chloride 8.47±0.02 46.95±2.90 -762.84±2.80 486.76±1.42 175.50±1.88 86.93±4.78 380.57±5.60 0.62±0.00 86 No
3-aminobenzene sulfonic
acid:
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
250 ppm 8.21±0.02 41.98±2.20 -740.56±7.20 41.39±1.16 116.50±1.70 55.23±2.03 391.74±8.90 0.49±0.01 89 No
500 ppm 8.27±0.01 41.63±1.60 -766.08±5.84 40.73±0.41 122.82±8.24 67.94±5.02 427.51±12.40 0.48±0.01 89 No
1000 ppm 8.35±0.03 40.72±3.10 -764.12±13.80 40.73±3.18 110.30±6.81 62.36±2.73 368.94±21.02 0.48±0.04 89±1 No
2000 ppm 8.47±0.02 40.10±1.80 -768.61±2.42 41.72±3.77 120.10±16.22 69.42±8.50 387.02±24.01 0.49±0.04 89±1 No
3000 ppm 8.63±0.01 39.31±1.50 -760.60±3.84 42.05±2.05 122.64±0.14 66.06±1.18 407.20±3.51 0.49±0.02 89±1 No
10000 ppm 8.71±0.01 37.91±2.30 -732.42±5.82 324.83±1.41 120.86±2.83 172.40±7.80 88.24±2.83 3.81±0.02 11±1 No
3-aminobenzene sulfonic
acid (1000 ppm) :
5.0 kmol/m3 MEA, 80oC,
α=0.55
10000 ppm
formate 7.92±0.04 49.28±1.70 -747.81±6.23 453.64±1.85 199.40±3.12 141.12±0.80 105.42±13.30 5.31±0.02 -24±1 No
10000 ppm
chloride 8.45±0.03 44.02±2.10 -772.11±4.78 140.73±1.06 162.90±4.20 104.78±5.23 87.90±9.78 1.65±0.01 61 Yes

87
Table 4.2 Summary of electrochemical experimental results (continued)
Experimental Condition pH σ
(mS/cm)
Ecorr
(mV vs
Ag/AgCl)
icorr
(μA/cm2)
βc
(mV/
decade)
βa
(mV/
decade)
Rp
(ohm-cm2)
CR
(mmpy)
IE
(%)
Pitting
tendency
4-aminobenzene sulfonic
acid :
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
250 ppm 8.21±0.02 42.30±2.30 -766.61±0.30 47.68±2.95 132.74±3.90 71.60±3.34 400.94±7.40 0.56±0.04 87±1 No
500 ppm 8.26±0.01 41.69±2.60 -769.45±2.54 45.36±0.26 130.56±4.12 70.84±8.91 459.90±24.04 0.54±0.02 88 No
1000 ppm 8.35±0.02 41.44±1.80 -786.20±6.80 38.41±1.46 126.45±2.04 60.10±6.10 440.90±9.31 0.45±0.02 90 No
2000 ppm 8.42±0.01 40.86±2.10 -791.56±9.72 35.10±5.62 115.21±6.70 69.22±13.31 419.18±0.94 0.41±0.07 90±2 No
3000 ppm 8.54±0.01 39.90±1.60 -779.47±5.04 32.68±2.60 99.20±13.45 53.10±5.94 355.16±3.80 0.38±0.03 91±1 No
10000 ppm 8.70±0.03 37.82±3.20 -736.67±2.48 321.19±2.68 85.60±9.05 108.70±12.32 99.14±4.20 3.77±0.03 12±1 No
4-aminobenzene sulfonic
acid (1000 ppm) :
5.0 kmol/m3 MEA, 80oC,
α=0.55
10000 ppm
formate 8.24±0.02 50.02±4.10 -748.30±3.42 463.58±2.09 122.22±8.65 149.91±8.30 86.63±6.70 5.45±0.02 -28 No
10000 ppm
chloride 8.45±0.03 46.04±1.20 -776.51±1.50 39.74±0.83 121.8±10.0 62.44±3.10 385.24±8.90 0.46±0.03 89 No
Sulfapyridine :
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
500 ppm 8.25±0.03 42.45±1.80 -748.72±2.12 364.24±0.79 99.50±2.62 142.94±1.23 86.30±5.92 4.26±0.01 0 No
1000 ppm 8.29±0.01 42.38±1.40 -746.44±8.64 261.26±2.75 151.16±1.83 106.41±3.24 104.54±12.10 3.07±0.03 28±1 No
2000 ppm 8.42±0.01 41.02±3.10 -783.82±6.89 37.42±2.19 126.92±4.90 69.30±5.78 468.68±9.72 0.44±0.03 90±1 No
3000 ppm 8.58±0.02 39.39±2.60 -796.17±5.43 35.43±1.69 118.60±5.92 63.30±11.12 396.78±6.14 0.42±0.02 90±1 No
5000 ppm 8.63±0.03 38.27±1.70 -783.27±4.82 326.49±1.08 122.53±9.18 65.74±13.23 444.70±4.28 0.38±0.01 91 No
7000 ppm 8.71±0.04 37.65±3.30 -784.67±3.69 34.77±3.16 122.65±4.70 59.91±6.41 407.66±3.20 0.41±0.03 90±1 No
10000 ppm 8.76±0.04 36.41±4.10 -786.21±9.12 28.21±4.01 121.50±9.92 64.26±8.14 527.91±1.20 0.33±0.05 92±1 No
Sulfapyridine (2000 ppm) :
5.0 kmol/m3 MEA, 80oC,
α=0.55
10000 ppm
formate 8.25±0.01 47.32±2.60 -778.44±7.43 44.37±2.68 135.36±4.12 58.66±10.12 358.50±9.45 0.52±0.03 88±1 No
10000 ppm
chloride 8.45±0.03 48.59±1.10 -741.95±12.12 37.09±2.53 107.40±3.87 165.56±7.96 90.92±4.20 4.34±0.03 -2±1 No
Sulfanilamide:
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
3000 ppm 8.16±0.01 42.12±1.80 -767.61±8.20 212.91±1.52 148.64±8.70 97.34±6.40 103.60±6.80 2.50±0.02 42±1 No
10000 ppm 8.25±0.03 41.69±2.40 -744.82±4.60 291.06±2.07 76.42±10.40 131.32±8.12 84.30±4.80 3.41±0.02 20±1 No
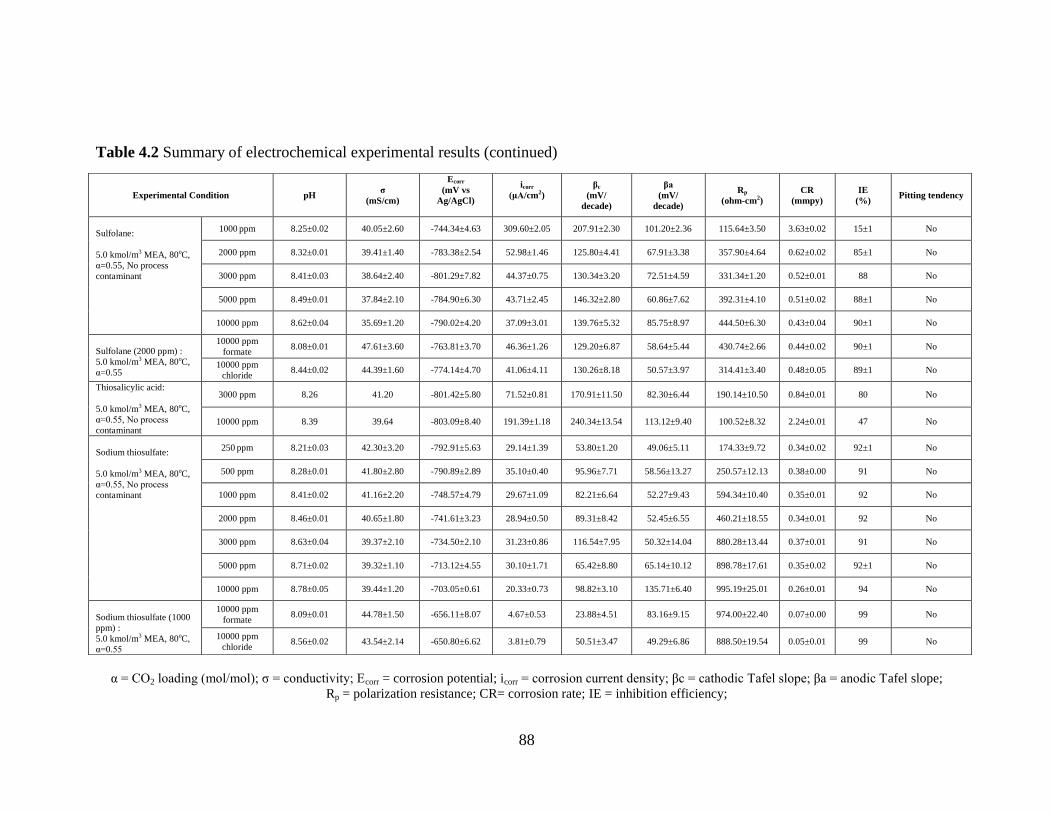
88
Table 4.2 Summary of electrochemical experimental results (continued)
α = CO2 loading (mol/mol); σ = conductivity; Ecorr = corrosion potential; icorr = corrosion current density; βc = cathodic Tafel slope; βa = anodic Tafel slope;
Rp = polarization resistance; CR= corrosion rate; IE = inhibition efficiency;
Experimental Condition pH σ
(mS/cm)
Ecorr
(mV vs
Ag/AgCl)
icorr
(μA/cm2)
βc
(mV/
decade)
βa
(mV/
decade)
Rp
(ohm-cm2)
CR
(mmpy)
IE
(%) Pitting tendency
Sulfolane:
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
1000 ppm 8.25±0.02 40.05±2.60 -744.34±4.63 309.60±2.05 207.91±2.30 101.20±2.36 115.64±3.50 3.63±0.02 15±1 No
2000 ppm 8.32±0.01 39.41±1.40 -783.38±2.54 52.98±1.46 125.80±4.41 67.91±3.38 357.90±4.64 0.62±0.02 85±1 No
3000 ppm 8.41±0.03 38.64±2.40 -801.29±7.82 44.37±0.75 130.34±3.20 72.51±4.59 331.34±1.20 0.52±0.01 88 No
5000 ppm 8.49±0.01 37.84±2.10 -784.90±6.30 43.71±2.45 146.32±2.80 60.86±7.62 392.31±4.10 0.51±0.02 88±1 No
10000 ppm 8.62±0.04 35.69±1.20 -790.02±4.20 37.09±3.01 139.76±5.32 85.75±8.97 444.50±6.30 0.43±0.04 90±1 No
Sulfolane (2000 ppm) :
5.0 kmol/m3 MEA, 80oC,
α=0.55
10000 ppm
formate 8.08±0.01 47.61±3.60 -763.81±3.70 46.36±1.26 129.20±6.87 58.64±5.44 430.74±2.66 0.44±0.02 90±1 No
10000 ppm
chloride 8.44±0.02 44.39±1.60 -774.14±4.70 41.06±4.11 130.26±8.18 50.57±3.97 314.41±3.40 0.48±0.05 89±1 No
Thiosalicylic acid:
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
3000 ppm 8.26 41.20 -801.42±5.80 71.52±0.81 170.91±11.50 82.30±6.44 190.14±10.50 0.84±0.01 80 No
10000 ppm 8.39 39.64 -803.09±8.40 191.39±1.18 240.34±13.54 113.12±9.40 100.52±8.32 2.24±0.01 47 No
Sodium thiosulfate:
5.0 kmol/m3 MEA, 80oC,
α=0.55, No process
contaminant
250 ppm 8.21±0.03 42.30±3.20 -792.91±5.63 29.14±1.39 53.80±1.20 49.06±5.11 174.33±9.72 0.34±0.02 92±1 No
500 ppm 8.28±0.01 41.80±2.80 -790.89±2.89 35.10±0.40 95.96±7.71 58.56±13.27 250.57±12.13 0.38±0.00 91 No
1000 ppm 8.41±0.02 41.16±2.20 -748.57±4.79 29.67±1.09 82.21±6.64 52.27±9.43 594.34±10.40 0.35±0.01 92 No
2000 ppm 8.46±0.01 40.65±1.80 -741.61±3.23 28.94±0.50 89.31±8.42 52.45±6.55 460.21±18.55 0.34±0.01 92 No
3000 ppm 8.63±0.04 39.37±2.10 -734.50±2.10 31.23±0.86 116.54±7.95 50.32±14.04 880.28±13.44 0.37±0.01 91 No
5000 ppm 8.71±0.02 39.32±1.10 -713.12±4.55 30.10±1.71 65.42±8.80 65.14±10.12 898.78±17.61 0.35±0.02 92±1 No
10000 ppm 8.78±0.05 39.44±1.20 -703.05±0.61 20.33±0.73 98.82±3.10 135.71±6.40 995.19±25.01 0.26±0.01 94 No
Sodium thiosulfate (1000
ppm) :
5.0 kmol/m3 MEA, 80oC,
α=0.55
10000 ppm
formate 8.09±0.01 44.78±1.50 -656.11±8.07 4.67±0.53 23.88±4.51 83.16±9.15 974.00±22.40 0.07±0.00 99 No
10000 ppm
chloride 8.56±0.02 43.54±2.14 -650.80±6.62 3.81±0.79 50.51±3.47 49.29±6.86 888.50±19.54 0.05±0.01 99 No

89
From the impedance analysis (Figure 4.1b), polarization resistance (RP) can be
obtained from the diameter of the semicircle that can be used as an indicator of the
corrosion behaviour of a system in addition to corrosion rate. For the uninhibited MEA
system in the absence of process contaminants, an RP value of 72 ohm-cm2 was obtained.
The impedance spectrum showed a semicircle, characteristic of a capacitive loop due to
charge transfer kinetics at the interface (Figure 4.1b).
b) Presence of process contaminants
The effect of process contaminants, namely chloride, formate, oxalate, and
thiosulfate on corrosion behaviour of the uninhibited MEA system (5.0 kmol/m3 MEA at
80oC and 0.55 mol/mol CO2 loading) were examined. Results (Figure 4.2a) show that the
corrosion rate was increased in the presence of formate but decreased in presence of the
chloride, oxalate, and thiosulfate. This might be due the significant reduction of the
solution pH in the presence of formate. From the corresponding potentiodynamic
polarization curves obtained for each case (Figure 4.2b), it can be observed that in the
presence of oxalate and thiosulfate, both cathodic and anodic current densities were lower
than the ‘No contaminant’ condition, and, hence, there was a reduction in corrosion rate.
In the presence of chloride and formate, the anodic current densities were lower than the
‘No contaminant’ condition, but the cathodic current densities were higher in the case of
formate and almost unchanged in the case of chloride. This change in polarization might
be due to the change in the composition of the metal-solution interfacial double layer in
the presence of process contaminants. The change in Tafel slopes (Table 4.2) in the
presence and absence of contaminants also suggests a change in corrosion mechanism.
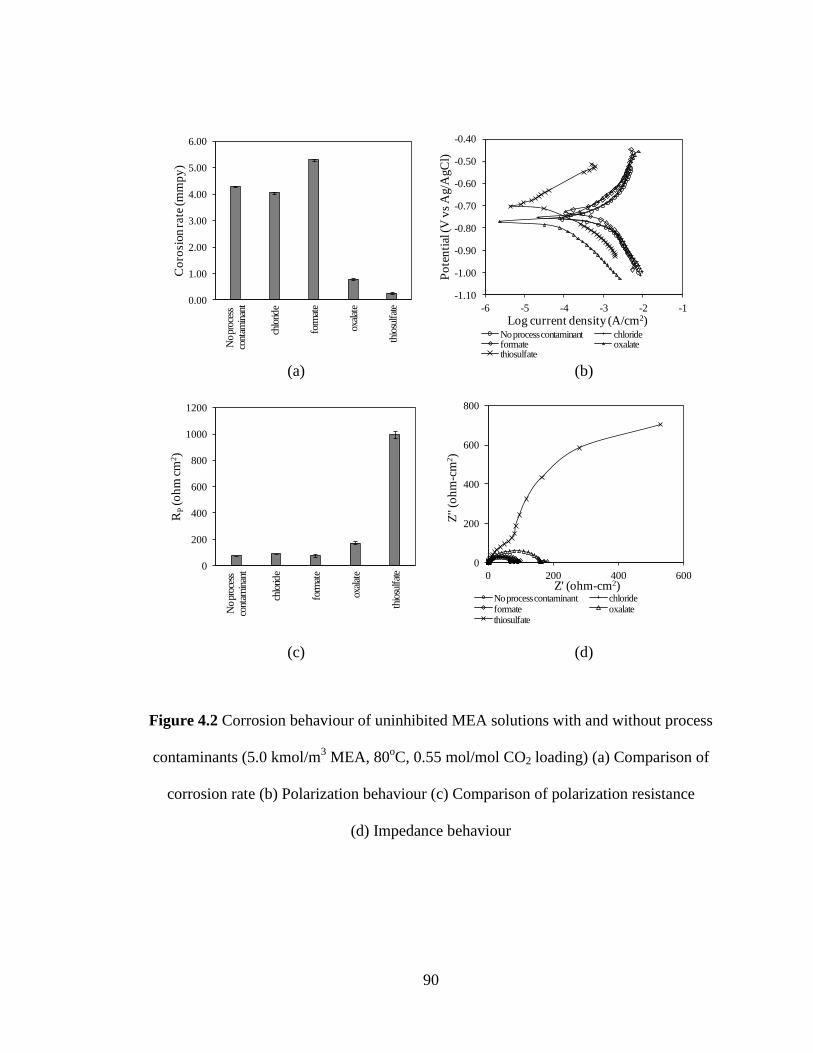
90
(a) (b)
(c) (d)
Figure 4.2 Corrosion behaviour of uninhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading) (a) Comparison of
corrosion rate (b) Polarization behaviour (c) Comparison of polarization resistance
(d) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
6.00
No
proc
ess
cont
amin
ant
chlo
ride
form
ate
oxal
ate
thio
sulf
ate
Co
rosi
on r
ate
(m
mp
y)
-1.10
-1.00
-0.90
-0.80
-0.70
-0.60
-0.50
-0.40
-6 -5 -4 -3 -2 -1
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)No process contaminant chlorideformate oxalatethiosulfate
0
200
400
600
800
1000
1200
No
proc
ess
cont
amin
ant
chlo
ride
form
ate
oxal
ate
thio
sulf
ate
RP
(oh
m c
m2)
0
200
400
600
800
0 200 400 600
Z''
(oh
m-c
m2)
Z' (ohm-cm2)No process contaminant chlorideformate oxalatethiosulfate

91
Pitting tendency was not induced by the presence of process contaminants.
Polarization resistance (RP) values obtained from impedance analysis in the
absence and presence of different process contaminants can also be used to explain the
change in corrosion behaviour of the system. From Figure 4.2c, it can be observed that
the RP values in the presence of chloride and formate were comparable to the ‘No
contaminant’ result. In the case of oxalate and thiosulfate, RP values were higher,
representing a reduced corrosivity of the system (Figure 4.2c). The impedance behaviour
of thiosulfate was different from the other process contaminants. An impedance spectrum
corresponding to the ‘No contaminant’ condition and the presence of chloride, formate,
and oxalate showed a semicircle, characteristic of a capacitive loop due to charge transfer
kinetics at the interface (Figure 4.2d). In the case of thiosulfate, in addition to the
semicircle, the low frequency end (right end of X axis) of the spectrum was characterized
by linearity (diffusion tail). This might be due to the thin black film formed over the
metal surface by the adsorption of thiosulfate molecules (S2O32-
) limiting the diffusion of
corrosive species across the interface.
Based on the above results, formate, which increased the corrosion rate of the
uninhibited MEA system, was chosen to be tested for the effect on corrosion inhibition
performance of selected inhibitors. Though chloride does not increase the corrosion rate,
it can potentially cause localized pitting corrosion, and, hence, it is important to study the
effect of chloride on the performance of inhibitors. Consequently, in order to evaluate the
performance of the inhibitors, only the effect of formate and chloride were studied.
Oxalate was not tested due to its reduction of corrosion rate in uninhibited MEA systems,
and thiosulfate was tested for its inhibition performance.

92
4.1.2 Corrosion behaviour of inhibited MEA systems
The selected corrosion inhibitors (Table 4.1) were tested at different
concentrations. Changes in the corrosion behaviour of carbon steel in different inhibited
MEA systems (5.0 kmol/m3 MEA at 80
oC and 0.55 mol/mol CO2 loading) compared to
the uninhibited MEA systems were studied. The effect of process contaminants (chloride
and formate) on the performance of different corrosion inhibitors was also studied.
4.1.2.1 2-aminobenzene sulfonic acid
a) Absence of process contaminants
Corrosion inhibition performance of 2-aminobenzene sulfonic acid was tested as a
function of corrosion inhibitor concentration. From Figure 4.3a, it can be observed that
the corrosion rate of carbon steel in the 2-aminobenzene sulfonic acid inhibited MEA
system was significantly reduced (0.46 – 0.57 mmpy) compared to the uninhibited
corrosion rate of 4.27 mmpy, when the inhibitor concentration was in the range of 250 to
3000 ppm. However at 10,000 ppm, corrosion rate increased to 1.62 mmpy. Corrosion
inhibition efficiency was in the range of 87–89 % for the inhibitor concentration of 250 –
3000 ppm, which dropped to 62 % at 10,000 ppm (Figure 4.3b).
From the potentiodynamic polarization curves for the inhibited MEA systems
(Figure 4.3c), it can be observed that even though the metal undergoes active corrosion,
the cathodic current densities were lower than the uninhibited condition while the anodic
current densities were almost unchanged. This suggests that the corrosion inhibition was
cathodic in nature and was due to the preferential adsorption of inhibitor molecules onto
the cathodic sites of the metal surface.

93
(a) (b)
(c) (d)
(e)
Figure 4.3 Corrosion behaviour of ‘2-aminobenzene sulfonic acid’ inhibited MEA
solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant)
(a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
1.50
3.00
4.50
0 250 500 1000 2000 3000 10000
Co
rosi
on r
ate
(m
mp
y)
Inhibitor concentration (ppm)
2-aminobenzene sulfonic acid
0%
25%
50%
75%
100%
250 500 1000 2000 3000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm10000 ppm
0
100
200
300
400
500
0 250 500 1000 2000 3000 10000
RP
(oh
m-c
m2)
Inhibitor concentration (ppm)
2-aminobenzene sulfonic acid
0
30
60
90
120
150
180
0 100 200 300 400 500
Z''
(oh
m-c
m2)
Z' (ohm-cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm10000 ppm

94
The change in Tafel slopes of the inhibited potentiodynamic polarization curves
compared to the uninhibited condition also suggested a change in corrosion mechanism
(Table 4.2). No pitting tendency was induced by the presence of corrosion inhibitor at
different concentrations.
Polarization resistance (RP) values obtained from the impedance analysis of
inhibited MEA systems at different corrosion inhibitor concentrations can also be used to
understand the effect of concentration on corrosion inhibition performance. As can be
observed from Figure 4.3d, when the corrosion inhibitor concentrations were in the range
of 250 to 3000 ppm, higher RP (350 – 418 ohm-cm2) was observed, while at 10,000 ppm,
a drop in RP (134 ohm-cm2) was observed compared to the uninhibited condition (72
ohm-cm2). Impedance analysis (Figure 4.3e) of inhibited MEA systems at different
inhibitor concentrations traced a semicircle (a capacitive loop due to charge transfer
kinetics) similar to that of the uninhibited system, suggesting the absence of any passive
film. Thus, the higher RP values obtained in the case of inhibited systems can be
attributed to the adsorption of inhibitor molecules on to the metal surface. The
deterioration of inhibition performance at higher inhibitor concentrations might be due to
the increase in the attractive lateral interactions in the adsorbed layer of molecules on the
metal surface [Vračar and Dražić, 2002].
As no specific trend could be established between inhibitor concentration and
corrosion rate or the RP, an inhibitor concentration of 1000 ppm was chosen to test the
effect of process contaminants on corrosion inhibition performance of 2-aminobenzene
sulfonic acid.

95
b) Presence of process contaminants
Inhibition performance of 2-aminobenzene sulfonic acid (1000 ppm) was almost
unaffected by the presence of chloride but was detrimentally affected by the presence of
formate. It can be observed that the corrosion rate in the presence of chloride (4.04
mmpy) was virtually unchanged compared to the ‘No contaminant’ results (Figure 4.4a).
However, in the presence of formate (5.29 mmpy), the corrosion rate was higher than the
uninhibited corrosion rates both with and without formate. It is more apparent from
Figure 4.4b, where the presence of formate was characterized by an aggravation (-33 %)
of corrosion. From the potentiodynamic polarization behaviour (Figure 4.4c), it can be
observed that in the case of chloride, both cathodic and anodic current densities were
slightly lower than the ‘No contaminant’ condition, but in case of formate, though the
anodic current densities were comparable, the cathodic current densities were much
higher than the ‘No contaminant’ condition. This suggests that the adsorption of inhibitor
molecules on the metal surface was disrupted and possibly displaced by the formate ions
from the contaminant. No pitting tendency was induced by the presence of the process
contaminants.
RP values obtained from the impedance analysis of the 2-aminobenzene sulfonic
acid (1000 ppm) inhibited MEA systems were also used to understand the effect of
process contaminants on the corrosion behaviour of the system. From Figure 4.4d, it can
be observed that in the presence of chloride, the RP was slightly lower, but in the
presence of formate, the RP was almost as low as the uninhibited system. From the
impedance analysis, it can be observed that a semicircular loop characteristic of charge
transfer kinetics was obtained, suggesting the absence of any passive film (Figure 4.4e).

96
(a) (b)
(c) (d)
(e)
Figure 4.4 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, 1000 ppm 2-
aminobenzene sulfonic acid) (a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour (d) Comparison of polarization
resistance (e) Impedance behaviour
0.00
2.00
4.00
6.00
No process contaminant
chloride formate
Co
rosi
on r
ate
(m
mp
y)
Uninhibited Inhibited
-50%
-25%
0%
25%
50%
75%
100%
No process contaminant
chloride formate
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-7 -6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
No process contaminant chloride formate
0
150
300
450
No process contaminant
chloride formate
RP
(oh
m-c
m2)
Uninhibited Inhibited
0
30
60
90
120
150
180
0 100 200 300 400
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
No process contaminant chloride formate

97
4.1.2.2 3-aminobenzenesulfonic acid
a) Absence of process contaminants
The corrosion rates of carbon steel in the 3-aminobenzene sulfonic acid inhibited
MEA systems (5.0 kmol/m3 MEA at 80
oC, 0.55 mol/mol CO2 loading and no process
contaminant) were found to be in the range of 0.48 to 0.49 mmpy when the corrosion
inhibitor concentration was in the range of 250 to 3000 ppm. The corrosion rate increased
to a value of 3.81 mmpy when this inhibitor was increased to 10,000 ppm (Figure 4.5a).
Corrosion inhibition efficiency was 89% when the corrosion inhibitor concentration was
in the range of 250 – 3000 ppm, which then dropped to 11% at 10,000 ppm (Figure 4.5b).
It can be observed from the potentiodynamic polarization behaviour (Figure 4.5c) that the
metal was in an active state. Hence, the decrease in corrosion rate might be due to the
adsorption of inhibitor molecules onto the metal surface. When the corrosion inhibitor
concentration was in the range of 250-3000 ppm, the cathodic current densities were
lower than the uninhibited condition. This suggests that the cathodic reactions were
impeded by the adsorption of inhibitor molecules onto the metal surface. However, at a
corrosion inhibitor concentration of 10,000 ppm, the cathodic current densities were
closer to the uninhibited values and were only slightly lower. This suggests that at this
concentration, the adsorption of inhibitor molecules to the metal surface is affected. This
might be due to the increased lateral attractions in the adsorbed layer. No pitting
tendency was induced by the presence of corrosion inhibitor at different concentrations.
Polarization resistance (RP) was in the range of 369 to 428 ohm-cm2
when the
inhibitor concentration was 250 to 3000 ppm, but dropped to 88 ohm-cm2 at 10000 ppm
(Figure 4.5d). From the impedance analysis (Figure 4.5e) in the presence of corrosion
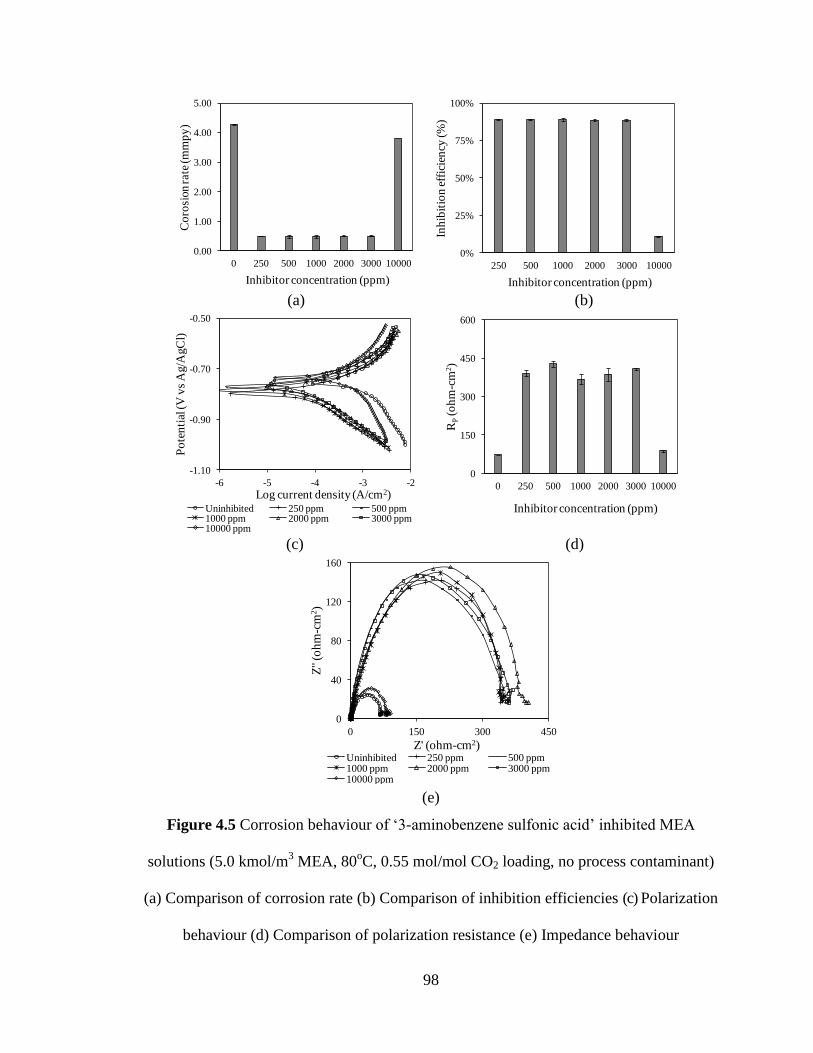
98
(a) (b)
(c) (d)
(e)
Figure 4.5 Corrosion behaviour of ‘3-aminobenzene sulfonic acid’ inhibited MEA
solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant)
(a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
0 250 500 1000 2000 3000 10000
Co
rosi
on r
ate
(m
mp
y)
Inhibitor concentration (ppm)
0%
25%
50%
75%
100%
250 500 1000 2000 3000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
3-aminobenzene sulfonic acid
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm10000 ppm
0
150
300
450
600
0 250 500 1000 2000 3000 10000
RP
(oh
m-c
m2)
Inhibitor concentration (ppm)
0
40
80
120
160
0 150 300 450
Z''
(oh
m-c
m2)
Z' (ohm-cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm10000 ppm

99
inhibitor, semicircular loops with larger diameters than the uninhibited condition could be
observed. Absence of a diffusion tail suggests that no passive film was formed. Thus, the
increase in RP might be due to the adsorption of inhibitor molecules onto the metal
surface. As no trend could be observed between the corrosion inhibitor concentration and
corrosion rate or RP, 1000 ppm was chosen to test the effects of process contaminants.
b) Presence of process contaminants
The presence of process contaminants (chloride or formate) had a deteriorating
effect on corrosion inhibition performance of 3-aminobenzene sulfonic acid (1000 ppm).
The corrosion rate in the presence of chloride increased to 1.65 mmpy from 0.48 mmpy
in the absence of any contaminant. In the case of formate, the corrosion rate was 5.31
mmpy, which was higher than the uninhibited corrosion rate both in the presence and
absence of formate (Figure 4.6a). The corrosion inhibition efficiency of 3-aminobenzene
sulfonic acid in the presence of chloride dropped to 62% compared to the ‘No
contaminant’ condition (89%). In the presence of formate, an efficiency of -24% was
observed, suggesting that the inhibitor actually aggravated the corrosion (Figure 4.6b).
From the potentiodynamic polarization analysis (Figure 4.6c), it can be observed that the
metal underwent active corrosion, and the cathodic current densities were characterized
by higher values in the presence of contaminant than the ‘No contaminant’ condition.
This might be due to the disruption of adsorption of corrosion inhibitor molecules onto
the metal surface in the presence of process contaminants, and this effect is more
pronounced in the presence of formate than chloride. From the cyclic polarization
analysis (Figure 4.7a), positive hysteresis (reverse curve lies to the right of the forward

100
(a) (b)
(c) (d)
(e)
Figure 4.6 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, 1000 ppm 3-
aminobenzene sulfonic acid) (a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour (d) Comparison of polarization
resistance (e) Impedance behaviour
0.00
2.00
4.00
6.00
No process contaminant
chloride formate
Co
rosi
on r
ate
(m
mp
y)
Uninhibited Inhibited
-50%
-25%
0%
25%
50%
75%
100%
No process contaminant
chloride formate
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-7 -6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
No process contaminant chloride formate
0
200
400
No process contaminant
chloride formate
RP
(oh
m-c
m2)
Uninhibited Inhibited
0
30
60
90
120
150
180
0 100 200 300 400
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
No process contaminant formate chloride
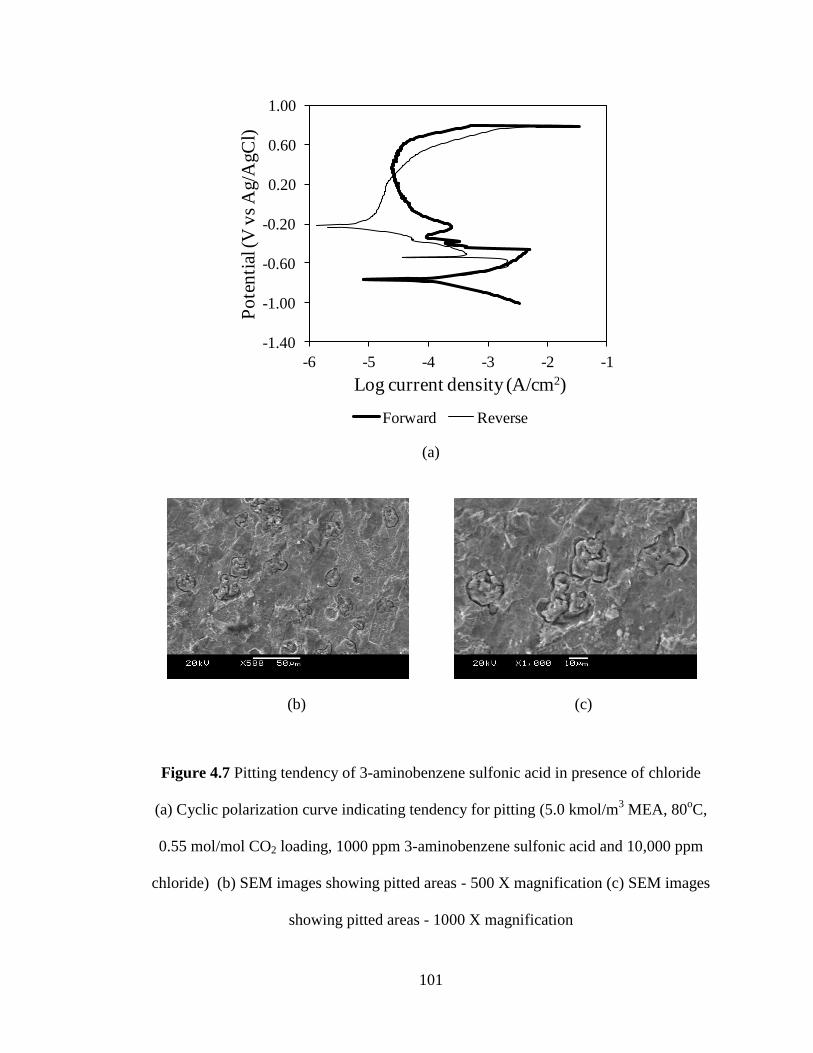
101
(a)
(b) (c)
Figure 4.7 Pitting tendency of 3-aminobenzene sulfonic acid in presence of chloride
(a) Cyclic polarization curve indicating tendency for pitting (5.0 kmol/m3 MEA, 80
oC,
0.55 mol/mol CO2 loading, 1000 ppm 3-aminobenzene sulfonic acid and 10,000 ppm
chloride) (b) SEM images showing pitted areas - 500 X magnification (c) SEM images
showing pitted areas - 1000 X magnification
-1.40
-1.00
-0.60
-0.20
0.20
0.60
1.00
-6 -5 -4 -3 -2 -1
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
Forward Reverse

102
curve) was observed in the presence of chloride, suggesting the possibility of pitting
corrosion. SEM analysis of the working electrode after the experiment confirmed the
occurrence of pitting corrosion (Figure 4.7b and Figure 4.7c).
Polarization resistances (RP) obtained from the impedance analysis corroborated
the observation from the potentiodynamic polarization analyses. The deterioration of
corrosion inhibition performance in the presence of process contaminants was
characterized by a drop in RP to the value of 88 ohm-cm2 in the presence of chloride and
105 ohm-cm2 in the case of formate from 369 ohm-cm
2 for the ‘No contaminant’
condition (Figure 4.6d). From the impedance analysis (Figure 4.6e), the semicircular loop
obtained was characteristic of a dissolution process taking place at the metal/solution
interface. The lower RP values in the presence of process contaminants might be
attributed to the interference in the adsorption of corrosion inhibitor onto the metal
surface by the chloride or formate ions.
4.1.2.3 4-aminobenzenesulfonic acid
a) Absence of process contaminants
Corrosion rates of carbon steel in 4-aminobenzene sulfonic acid inhibited MEA
systems (5.0 kmol/m3 MEA at 80
oC, 0.55 mol/mol CO2 loading and no process
contaminant) were in the range of 0.38 to 0.56 mmpy when the corrosion inhibitor
concentrations were 250 to 3000 ppm but increased to 3.77 mmpy at 10,000 ppm (Figure
4.8a). Corrosion inhibition efficiencies were in the range of 87 to 91% when the inhibitor
concentration was 250 to 3000 ppm, which dropped to 12% at 10,000 ppm (Figure 4.8b).
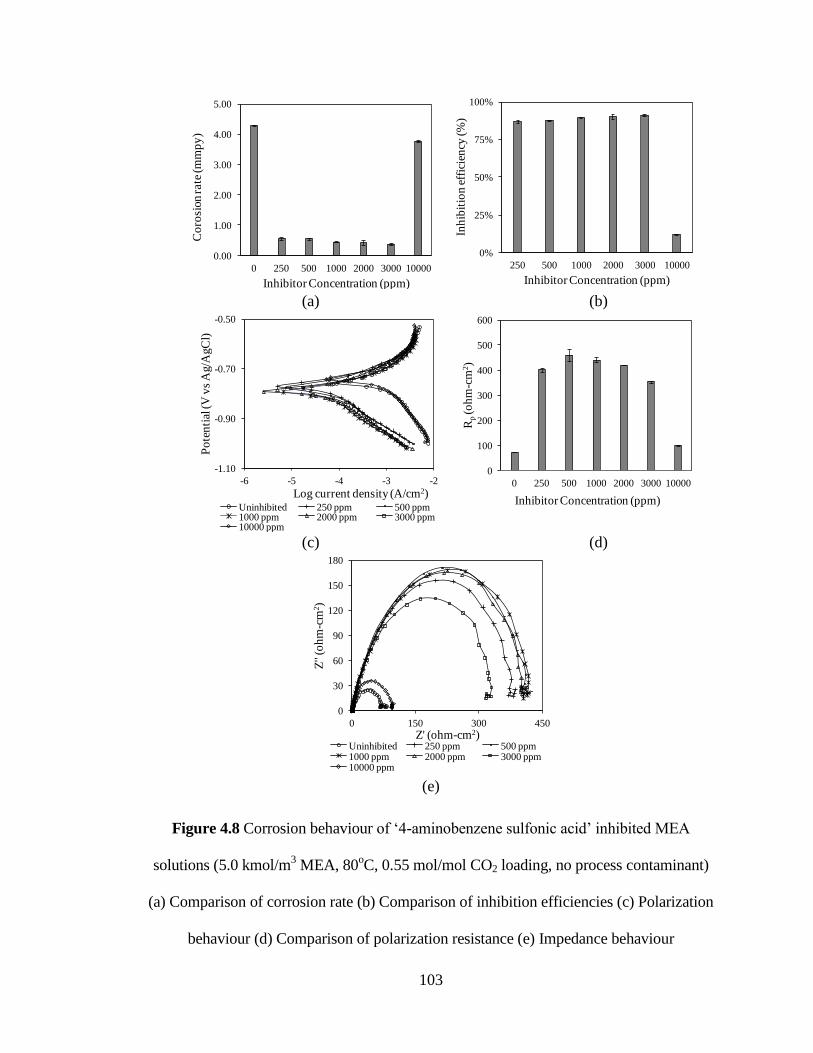
103
(a) (b)
(c) (d)
(e)
Figure 4.8 Corrosion behaviour of ‘4-aminobenzene sulfonic acid’ inhibited MEA
solutions (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant)
(a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
0 250 500 1000 2000 3000 10000
Co
rosi
on r
ate
(m
mp
y)
Inhibitor Concentration (ppm)
4-aminobenzene sulfonic acid
0%
25%
50%
75%
100%
250 500 1000 2000 3000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor Concentration (ppm)
4-aminobenzene sulfonic acid
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm10000 ppm
0
100
200
300
400
500
600
0 250 500 1000 2000 3000 10000
Rp(o
hm
-cm
2)
Inhibitor Concentration (ppm)
4-aminobenzene sulfonic acid
0
30
60
90
120
150
180
0 150 300 450
Z''
(oh
m-c
m2)
Z' (ohm-cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm10000 ppm

104
From the potentiodynamic polarization curves (Figure 4.8c), it can be observed that even
though the metal undergoes active corrosion, lower corrosion rates were obtained in the
presence of corrosion inhibitor. This can be attributed to the adsorption of inhibitor
molecules onto the metal surface. At the corrosion inhibitor concentrations of 250 - 3000
ppm, the cathodic current densities were lower than the uninhibited cathodic current
densities suggesting that the cathodic reactions were highly impeded by the adsorption of
corrosion inhibitor. No pitting tendency was induced by the presence of corrosion
inhibitor at any concentration.
Polarization resistances (RP) obtained from the impedance analysis at different
concentrations of corrosion inhibitor corroborated the inferences from the
potentiodynamic polarization analysis. At the inhibitor concentration of 250 - 3000 ppm,
RP values obtained were in the range of 355 - 460 ohm-cm2, which dropped to 99 ohm-
cm2 at 10,000 ppm (Figure 4.8d). The deterioration of performance at higher inhibitor
concentrations might be due to the increase in the attractive lateral interactions in the
adsorbed layer of molecules on the metal surface [Vračar and Dražić, 2002]. Impedance
analysis yielded a semicircle characteristic of charge transfer kinetics at the metal-
solution interface, suggesting that no passive layer might be present (Figure 4.8e).
Hence, the increase in RP value might be due to the adsorption of inhibitor molecules
onto the metal surface. As no specific trend between corrosion inhibitor concentration
and corrosion rate or RP could be observed, a corrosion inhibitor concentration of 1000
ppm was chosen to test the effect of process contaminants on inhibition performance of
4-aminobenzene sulfonic acid.

105
b) Presence of process contaminants
Corrosion inhibition performance of 4-aminobenzene sulfonic acid (1000 ppm)
was almost unaffected by the presence of chloride but deteriorated in the presence of
formate. While the corrosion rate in the presence of chloride was 0.46 mmpy, the
corrosion rate in the presence of formate was 5.45 mmpy compared to the corrosion rate
in the absence of any contaminant, which was 0.45 mmpy (Figure 4.9a). Corrosion
inhibition efficiency in the presence of chloride was 89% and in the presence of formate
was -28%, implying that the corrosion rate of the system was increased by the inhibitor in
the presence of formate (Figure 4.9b). From the potentiodynamic polarization behaviour
(Figure 4.9c), it can be observed that the metal underwent active corrosion, and in the
presence of chloride, the anodic and cathodic current densities were virtually the same as
those of the ‘No contaminant’ condition, but in the presence of formate, the cathodic
current densities were much higher in comparison. An increase in corrosion rate in the
presence of formate might be caused by the interference of formate ions in the adsorption
of inhibitor molecules onto the metal surface. No pitting tendency was present in either
case.
While the polarization resistance (RP) in the presence of chloride was slightly
lower (385 ohm-cm2) than the no contaminant condition (441 ohm-cm
2), RP in the
presence of formate was drastically lower (87 ohm-cm2) (Figure 4.9d). From the
impedance analysis (Figure 4.9e), it can be observed that a semicircular loop
characteristic of charge transfer kinetics was obtained. The lower values of RP in the
presence of formate might be due to the interference of formate ions in the adsorption of
inhibitor molecules onto the metal surface.

106
(a) (b)
(c) (d)
(e)
Figure 4.9 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, 1000 ppm 4-
aminobenzene sulfonic acid) (a) Comparison of corrosion rate (b) Comparison of
inhibition efficiencies (c) Polarization behaviour (d) Comparison of polarization
resistance (e) Impedance behaviour
0.00
2.00
4.00
6.00
No process contaminant
chloride formate
Co
rosi
on r
ate
(m
mp
y)
Uninhibited Inhibited
-50%
-25%
0%
25%
50%
75%
100%
No process contaminant
chloride formate
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-7 -6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
No process contaminant chloride formate
0
100
200
300
400
500
No process contaminant
chloride formate
RP
(oh
m-c
m2)
Uninhibited Inhibited
0
30
60
90
120
150
180
0 150 300 450
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
No process contaminant chloride formate

107
4.1.2.4 Sulfapyridine
a) Absence of process contaminants
Corrosion rates of carbon steel in sulfapyridine inhibited MEA systems (5.0
kmol/m3 MEA at 80
oC, 0.55 mol/mol CO2 loading and no process contaminant) were in
the range of 0.33-0.44 mmpy when the inhibitor concentrations were in the range of
2000-10,000 ppm. While the corrosion rate was slightly lower at an inhibitor
concentration of 1000 ppm (3.07 mmpy), at 500 ppm (4.26 mmpy), the corrosion rate
was almost as high as the uninhibited corrosion rate (4.27 mmpy) (Figure 4.10a).
Corrosion inhibition efficiencies were in the range of 90–91% when the inhibitor
concentrations were in the range of 2000-10,000 ppm (Figure 4.10b). At inhibitor
concentrations of 500 and 1000 ppm, the corrosion inhibition efficiencies were 0 and
28%, respectively, suggesting that the corrosion inhibitor was not effective below a
concentration of 2000 ppm. However, no relationship was observed between corrosion
inhibitor concentration and corrosion rate above 2000 ppm (Figure 4.10a). From the
potentiodynamic polarization behaviour (Figure 4.10c), it can be observed that the anodic
current densities were almost comparable for different corrosion inhibitor concentrations,
but the cathodic current densities were lower in the range of 2000-10,000 ppm and
proximate to the uninhibited result at 500 and 1000 ppm. From the potentiodynamic
polarization behaviour (Figure 4.10c), it can be observed that the metal was in an active
state, and, hence, the lower corrosion rate might be due to the adsorption of inhibitor onto
the metal surface. No pitting tendency was induced due to the presence of corrosion
inhibitor at any concentration.

108
(a) (b)
(c) (d)
(e)
Figure 4.10 Corrosion behaviour of ‘sulfapyridine’ inhibited MEA solutions (5.0 kmol/m3
MEA, 80oC, 0.55 mol/mol CO2 loading, no process contaminant) (a) Comparison of
corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization behaviour
(d) Comparison of polarization resistance (e) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
Co
rosi
on r
ate
(m
mp
y)
Inhibitor concentration (ppm)
sulfapyridine
0%
25%
50%
75%
100%
500 1000 2000 3000 5000 7000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
sulfapyridine
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)Uninhibited 500 ppm 1000 ppm2000 ppm 3000 ppm 5000 ppm7000 ppm 10000 ppm
0
100
200
300
400
500
600
RP
(oh
m-c
m2)
Inhibitor concentration (ppm)
sulfapyridine
0
40
80
120
160
200
0 200 400 600
Z''
(oh
m-c
m2)
Z' (ohm-cm2)Uninhibited 500 ppm 1000 ppm2000 ppm 3000 ppm 5000 ppm7000 ppm 10000 ppm

109
Polarization resistances (RP) obtained from the impedance analysis of the
sulfapyridine inhibited MEA systems at different concentrations of corrosion inhibitor
reinforced the inferences from the potentiodynamic polarization analysis. The results in
Figure 4.10d show that the RP values were in the range of 397 – 528 ohm-cm2 when the
corrosion inhibitor concentrations were 2000-10,000 ppm and dropped to 86 and 105
ohm-cm2 at 500 and 1000 ppm, respectively. From the impedance analysis (Figure
4.10e), the semicircular loop characteristic of charge transfer kinetics at the interface
suggests the possibility that no passive layer was present. Hence, the higher RP values
obtained might be due to the adsorption of inhibitor onto the metal surface.
Based on the potentiodynamic polarization and impedance analysis, it can be
inferred that the corrosion inhibitor does not function effectively below 2000 ppm.
Hence, 2000 ppm was chosen as the concentration of corrosion inhibitor to test the effect
of process contaminants on the corrosion inhibition performance of sulfapyridine.
b) Presence of process contaminants
The inhibition performance of sulfapyridine was detrimentally affected by the
presence of chloride but virtually unaffected by the presence of formate. In the presence
of chloride, the corrosion rate of carbon steel in sulfapyridine inhibited MEA solution
(5.0 kmol/m3 MEA at 80
oC and 0.55 mol/mol CO2 loading) was 4.34 mmpy, and in
presence of formate, it was 0.52 mmpy (Figure 4.11a). While in the presence of formate,
the corrosion inhibition efficiency was slightly reduced to 88% compared to the ‘No
contaminant’ condition of 90%, and it was -2% in the presence of chloride (Figure
4.11b).

110
(a) (b)
(c) (d)
(e)
Figure 4.11 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, 2000 ppm
sulfapyridine) (a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies
(c) Polarization behaviour (d) Comparison of polarization resistance (e) Impedance
behaviour
0.00
2.00
4.00
6.00
No process contaminant
chloride formate
Co
rosi
on r
ate
(m
mp
y)
Uninhibited Inhibited
-25%
0%
25%
50%
75%
100%
No process contaminant
chloride formate
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-7 -6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
No process contaminant formate chloride
0
150
300
450
600
No process contaminant
chloride formate
RP
(oh
m-c
m2)
Uninhibited Inhibited
0
40
80
120
160
200
0 100 200 300 400 500
Z' (
oh
m-c
m2)
Z' (ohm-cm2)No process contaminant formate chloride

111
From the potentiodynamic polarization analysis (Figure 4.11c), it can be observed that in
the presence of chloride, both the cathodic and anodic current densities were higher than
the ‘No contaminant’ condition but were almost comparable in the case of formate. This
suggests that the adsorption of inhibitor onto the metal surface was drastically affected by
the presence of chloride. No pitting tendency was induced by the presence of process
contaminants.
From Figure 4.11d, it can be observed that in the presence of formate, RP was
lower than the ‘No contaminant’ condition, but in the presence of chloride, RP was almost
as low as in the uninhibited condition. From the impedance analysis in the presence of
chloride and formate (Figure 4.11e), a semicircular loop was obtained, which is
characteristic of a capacitive loop due to charge transfer kinetics at the interface. Thus,
the reduction in the RP value in the presence of process contaminants suggests that the
adsorption of sulfapyridine molecules onto the metal surface was disrupted, and the effect
was more pronounced in the presence of chloride.
4.1.2.5 Sulfanilamide (Absence of process contaminants)
The corrosion rates of carbon steel in sulfanilamide inhibited MEA systems (5.0
kmol/m3 MEA at 80
oC, 0.55 mol/mol CO2 loading and no process contaminant) were
2.50 mmpy and 3.41 mmpy (Figure 4.12a) when the corrosion inhibitor concentrations
were 3000 and 10,000 ppm, respectively, with corresponding corrosion inhibition
efficiencies of 42% and 20% (Figure 4.12b). From the potentiodynamic polarization
analyses (Figure 4.12c), it can be observed that the metal underwent active corrosion.

112
(a) (b)
(c) (d)
(e)
Figure 4.12 Corrosion behaviour of ‘sulfanilamide’ inhibited MEA solutions (5.0
kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant) (a)
Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
0 3000 10000
Co
rosi
on r
ate
(m
mp
y)
Inhibitor Concentration (ppm)
Sulfanilamide
0%
25%
50%
75%
100%
3000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor Concentration (ppm)
Sulfanilamide
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l
Log current density (A/cm2)
uninhibited 3000 ppm 10000 ppm
0
60
120
0 3000 10000
Rp(o
hm
-cm
2)
Inhibitor Concentration (ppm)
Sulfanilamide
0
20
40
0 60 120
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
uninhibited 3000 ppm 10000 ppm

113
Compared to the uninhibited condition, the cathodic current densities were lower in the
case of 3000 ppm and almost similar in the case of 10,000 ppm (Figure 4.12c). No pitting
tendency was induced by the presence of inhibitor at any concentration.
The polarization resistances (RP) obtained from the impedance analysis were 104
and 84 ohm-cm2 at corrosion inhibitor concentrations of 3000 and 10,000 ppm,
respectively, compared to the uninhibited RP value of 72 ohm-cm2 (Figure 4.12d).
Impedance analysis in the presence of inhibitor at 3000 and 10,000 ppm (Figure 4.12e)
traced a semicircle (a capacitive loop due to charge transfer kinetics) similar to that of the
uninhibited system, suggesting that no passive film might be present. Thus, the small
increase in the RP value in the presence of inhibitor might be due to the adsorption of
inhibitor molecules onto the metal surface, but as the corrosion inhibition efficiencies at
the measured concentrations were relatively poor in comparison with other inhibitors,
sulfanilamide was not tested further for the effect of its concentration and the effect of
process contaminants.
4.1.2.6 Sulfolane
a) Absence of process contaminants
Corrosion rates of carbon steel in sulfolane inhibited MEA system (5 kmol/m3
MEA at 80oC, 0.55 mol/mol CO2 loading and no process contaminant) were in the range
of 0.43 – 0.62 mmpy when the inhibitor concentrations were in the range of 2000-10,000
ppm (Figure 4.13a). At an inhibitor concentration of 1000 ppm, the corrosion rate was
3.63 mmpy, which is only slightly lower than the uninhibited corrosion rate of 4.27
mmpy. Corrosion inhibition efficiency was in the range of 85 – 90 % when the corrosion

114
(a) (b)
(c) (d)
(e)
Figure 4.13 Corrosion behaviour of ‘sulfolane’ inhibited MEA solutions (5.0 kmol/m3
MEA, 80oC, 0.55 mol/mol CO2 loading, no process contaminant) (a) Comparison of
corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization behaviour (d)
Comparison of polarization resistance (e) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
0 1000 2000 3000 5000 10000
Co
rosi
on r
ate
(m
mp
y)
Inhibitor concentration (ppm)
sulfolane
0%
25%
50%
75%
100%
1000 2000 3000 5000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
sulfolane
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
Uninhibited 1000 ppm 2000 ppm
3000 ppm 5000 ppm 10000 ppm
0
100
200
300
400
500
0 1000 2000 3000 5000 10000
RP
(oh
m-c
m2)
Inhibitor concentration (ppm)
sulfolane
0
30
60
90
120
150
180
0 150 300 450
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
Uninhibited 1000 ppm 2000 ppm3000 ppm 5000 ppm 10000 ppm

115
inhibitor concentrations were in the range of 2000-10,000 ppm, which dropped to 15% at
1000 ppm (Figure 4.13b). This suggested that the minimum concentration required for
effective corrosion inhibition was 2000 ppm. However, no specific trend was observed
between corrosion rate and concentration of corrosion inhibitor above 2000 ppm. From
the potentiodynamic polarization behaviour (Figure 4.13c), it can be observed that when
the corrosion inhibitor concentration was 1000 ppm, the cathodic and anodic current
densities were close to the uninhibited condition, but at higher concentrations (2000 -
10000 ppm), the cathodic current densities were lower. This suggests that the presence of
corrosion inhibitor impeded the cathodic reaction when the inhibitor concentration was
2000 – 10,000 ppm. As the metal was in an active state, the decrease in corrosion rate
might be due to the adsorption of sulfolane molecules onto the metal surface. No pitting
tendency was induced by the presence of sulfolane at any concentration.
Polarization resistances (RP) obtained from the impedance analysis reinforced the
observation from the potentiodynamic polarization analysis. When the concentration of
corrosion inhibitors was in the range of 2000 – 10,000 ppm, higher RP values (331– 445
ohm-cm2) were obtained, but at 1000 ppm, it dropped to 116 ohm-cm
2. An approximate
linear trend between concentration of corrosion inhibitor and RP value was observed
(Figure 4.13d). From the impedance analysis (Figure 4.13e), the semicircular loop
characteristic of charge transfer kinetics at the metal/solution interface suggests that no
passive layer might be present. Hence, the increase in RP might be due to the adsorption
of inhibitor molecules onto the metal surface. An inhibitor concentration of 2000 ppm,
below which the corrosion inhibition was not effective, was chosen for testing the effect
of process contaminants on the inhibition performance of sulfolane.

116
b) Presence of process contaminants
The inhibition performance of sulfolane was not affected by the presence of
chloride or formate. The corrosion rate was 0.48 mmpy in the presence of chloride and
0.44 mmpy in the presence of formate, which was better than the corrosion rate for the
‘No contaminant’ condition, which was 0.62 mmpy (Figure 4.14a). Consequently, the
inhibition efficiencies were marginally better in the presence of chloride and formate
compared to the ‘No contaminant’ condition (Figure 4.14b). From the potentiodynamic
polarization analysis (Figure 4.14c), it was observed that anodic and cathodic current
densities in the presence of process contaminants were slightly lower than the anodic and
cathodic current densities in the absence of the process contaminants. No pitting tendency
was induced by the presence of process contaminants.
From Figure 4.14d, it can be observed that the polarization resistance (RP)
obtained from the impedance analysis was slightly lower in the presence of chloride (314
ohm-cm2) and was higher in the presence of formate (431 ohm-cm
2) compared to the ‘No
process contaminant’ condition (358 ohm-cm2). The semicircular loop obtained from the
impedance analysis (4.14e) represents a capacitive loop due to the charge transfer
kinetics at the interface. Thus, the adsorption of inhibitor molecules onto the metal
surface might be slightly affected by the presence of chloride but was enhanced by the
presence of formate.
4.1.2.7 Thiosalicylic acid (Absence of process contaminants)
Corrosion rates of carbon steel in ‘thiosalicylic acid’ inhibited MEA systems (5.0
kmol/m3 MEA at 80
oC, 0.55 mol/mol CO2 loading and no process contaminant) were

117
(a) (b)
(a) (d)
(e)
Figure 4.14 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, 2000 ppm sulfolane)
(a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
2.00
4.00
6.00
No process contaminant
chloride formate
Co
rosi
on r
ate
(m
mp
y)
Uninhibited Inhibited
0%
25%
50%
75%
100%
No process contaminant
chloride formate
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
No process contaminant chloride formate
0
150
300
450
No process contaminant
chloride formate
RP
(oh
m-c
m2)
Uninhibited Inhibited
0
30
60
90
120
150
180
0 100 200 300 400 500
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
No process contaminant chloride formate

118
0.84 mmpy and 2.24 mmpy when the corrosion inhibitor concentrations were 3000 and
10,000 ppm, respectively (Figure 4.15a). The corresponding corrosion inhibition
efficiencies were 80% and 47%, respectively (Figure 4.15b). From the potentiodynamic
polarization analyses (Figure 4.15c), it was observed that relative to the uninhibited
condition, the anodic current densities were almost comparable, but the cathodic current
densities were lower for both corrosion inhibitor concentrations.
Polarization resistances (RP) from the impedance analysis were determined to be
101 and 190 ohm-cm2 for the corrosion inhibitor concentrations of 3000 and 10,000 ppm,
respectively, compared to the uninhibited RP value of 72 ohm-cm2 (Figure 4.15d). At the
corrosion inhibitor concentrations of 3000 and 10,000 ppm, the impedance analyses
(Figure 4.15e) yielded a semicircle characteristic of a capacitive loop due to charge
transfer kinetics at the interface. This suggests that no passive layer was present on the
metal surface. Thus, the higher RP values obtained can be attributed to the adsorption of
thiosalicylic acid molecules onto the metal surface.
In the course of the experiment, the test solution containing thiosalicylic acid
turned black at both 3000 and 10,000 ppm, which might be due to the incompatibility of
the inhibitor with the solution. This suggested that the use of thiosalicylic acid as a
corrosion inhibitor might cause other complications in the CO2 absorption process. Thus,
thiosalicylic acid was not tested further at other concentrations and for the effect of
process contaminants.
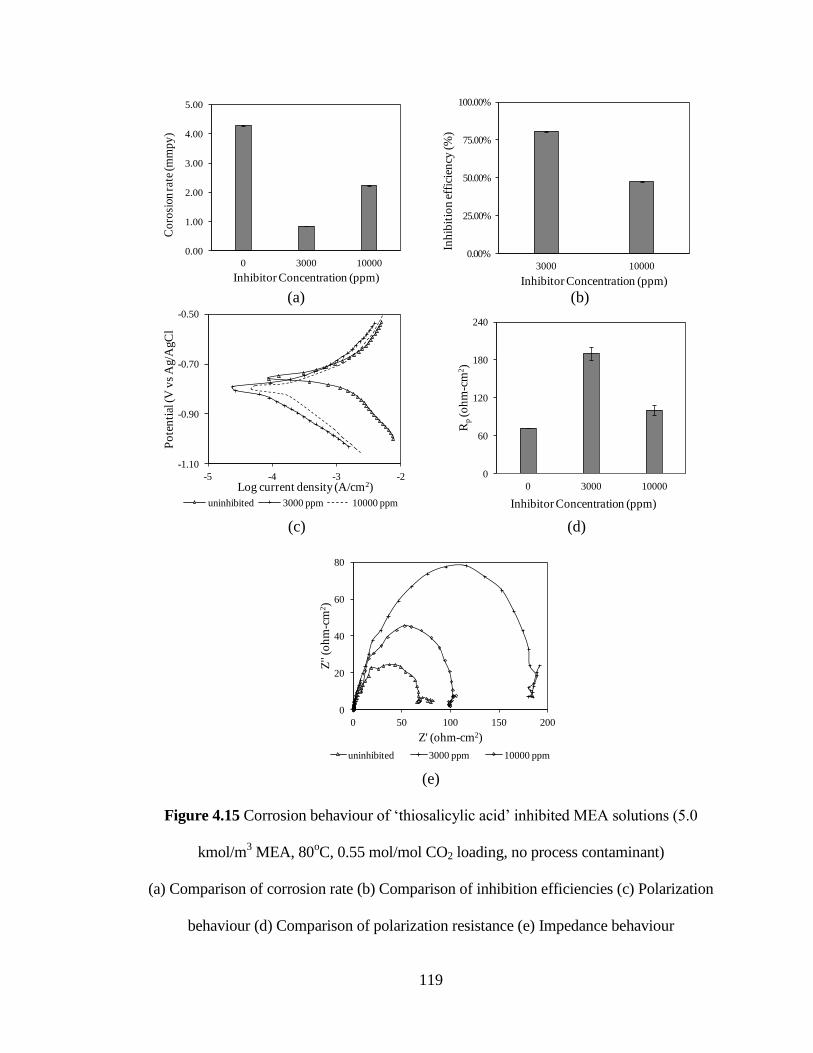
119
(a) (b)
(c) (d)
(e)
Figure 4.15 Corrosion behaviour of ‘thiosalicylic acid’ inhibited MEA solutions (5.0
kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant)
(a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c) Polarization
behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
1.00
2.00
3.00
4.00
5.00
0 3000 10000
Co
rosi
on r
ate
(m
mp
y)
Inhibitor Concentration (ppm)
Thiosalicylic acid
0.00%
25.00%
50.00%
75.00%
100.00%
3000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor Concentration (ppm)
-1.10
-0.90
-0.70
-0.50
-5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l
Log current density (A/cm2)
uninhibited 3000 ppm 10000 ppm
0
60
120
180
240
0 3000 10000
Rp(o
hm
-cm
2)
Inhibitor Concentration (ppm)
Thiosalicylic acid
0
20
40
60
80
0 50 100 150 200
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
uninhibited 3000 ppm 10000 ppm

120
4.1.2.8 Sodium thiosulfate
a) Absence of process contaminants
Corrosion rates of carbon steel in the sodium thiosulfate inhibited MEA systems
(5.0 kmol/m3 MEA at 80
oC, 0.55 mol/mol CO2 loading and no process contaminant) were
in the range of 0.26 - 0.38 mmpy when the corrosion inhibitor concentrations were 250 –
10,000 ppm (Figure 4.16a) with corresponding corrosion inhibition efficiencies of 91-
94% (Figure 4.16b). From the potentiodynamic polarization analysis (Figure 4.17a), two
different behaviours, depending on the concentration of corrosion inhibitor, were
observed. When the corrosion inhibitor concentration was 250 or 500 ppm, the metal was
in an active state. The anodic current densities were higher and the cathodic current
densities were lower than the uninhibited condition. However, at higher corrosion
inhibitor concentrations (1000 – 10,000 ppm), the metal showed passivation tendency
(Figure 4.17a), but the passive layer was weak and unstable. From the anodic polarization
curves, it can be observed that as the initial passive layer broke down, an increase in
anodic current densities was observed. This was followed by a repassivation and lower
current densities, and this cycle repeated until a stable passive layer was formed.
Formation of a thin black film over the metal surface (Figure 4.17b) was observed, which
might be due to the adsorption of thiosulfate (S2O32-
) ions. The corrosion inhibition was
anodic in nature. Though no trend between corrosion rate and corrosion inhibitor
concentration was found, the passive behaviour was observed only at 1000 ppm and
above. No pitting tendency was induced by the presence of inhibitor at any concentration.
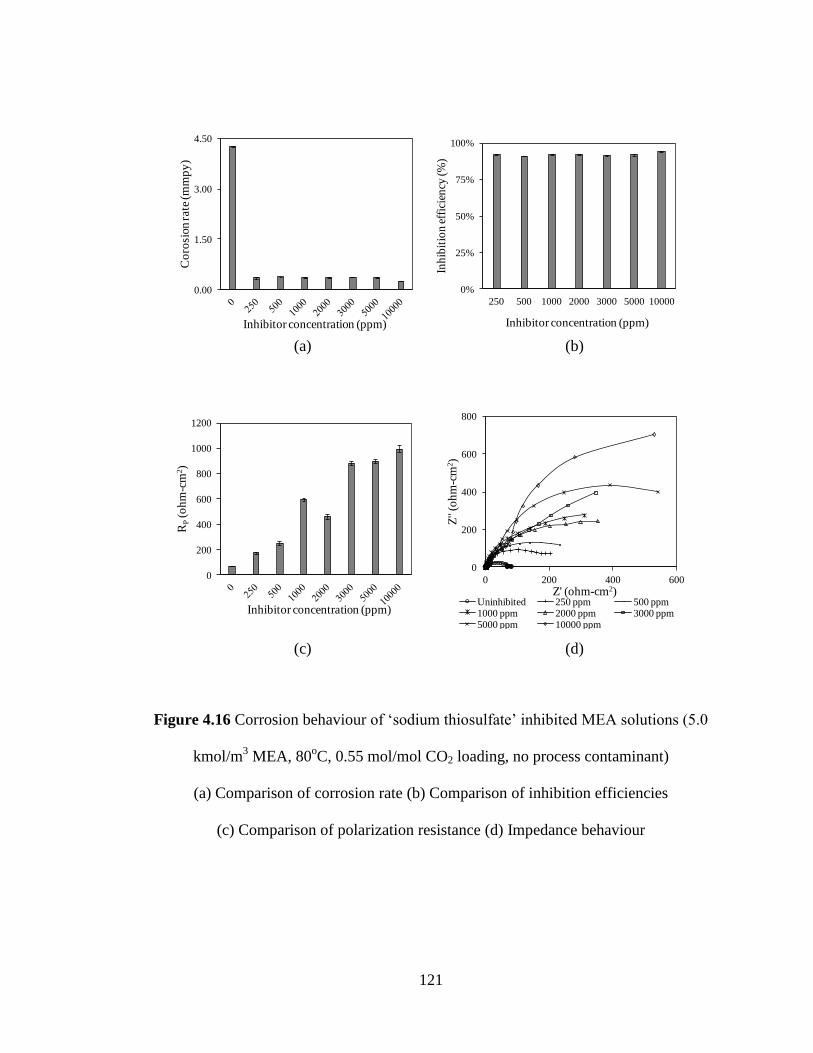
121
(a) (b)
(c) (d)
Figure 4.16 Corrosion behaviour of ‘sodium thiosulfate’ inhibited MEA solutions (5.0
kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant)
(a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies
(c) Comparison of polarization resistance (d) Impedance behaviour
0.00
1.50
3.00
4.50
Co
rosi
on r
ate
(m
mp
y)
Inhibitor concentration (ppm)
thiosulfate
0%
25%
50%
75%
100%
250 500 1000 2000 3000 5000 10000
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
thiosulfate
0
200
400
600
800
1000
1200
RP
(oh
m-c
m2)
Inhibitor concentration (ppm)thiosulfate
0
200
400
600
800
0 200 400 600
Z''
(oh
m-c
m2)
Z' (ohm-cm2)Uninhibited 250 ppm 500 ppm1000 ppm 2000 ppm 3000 ppm5000 ppm 10000 ppm

122
(a)
(b)
Figure 4.17 (a) Polarization behaviour of ‘sodium thiosulfate’ inhibited MEA solutions
(5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, no process contaminant)
(b) Working electrode after stable open circuit potential in presence of sodium thiosulfate
(5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading and 1000 ppm sodium thiosulfate)
(Original in colour)
-1.20
-0.90
-0.60
-0.30
0.00
0.30
0.60
0.90
-7 -6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
0 ppm 250 ppm 500 ppm
1000 ppm 2000 ppm 3000ppm

123
Polarization resistances (RP) obtained from the impedance analysis of the sodium
thiosulfate inhibited MEA systems at different concentrations of corrosion inhibitor were
in the range of 174–995 ohm-cm2 (Figure 4.16c). An approximately direct relationship
between RP and concentration of corrosion inhibitor could be observed (Figure 4.16c). It
can be observed from the impedance behaviour (Figure 4.16d) that, in addition to a
semicircular loop characteristic of charge transfer kinetics at the interface, the low
frequency end (right end of X axis) of the spectrum was marked by linearity (diffusion
tail) suggesting a passive film formation over the metal surface. This might be due to the
adsorption of thiosulfate molecules (S2O32-
), which limited the diffusion of corrosive
species across the interface. At a corrosion inhibitor concentration of 250 or 500 ppm, it
can be observed that the diffusion tail was not as pronounced (Figure 4.16d) as in higher
concentrations (1000 – 10,000 ppm). The minimum inhibitor concentration at which the
metal displayed passive tendency was 1000 ppm, and it was chosen to test the effect of
process contaminants on the inhibition performance of sodium thiosulfate.
b) Presence of process contaminants
In the presence of chloride and formate, the corrosion rate of sodium thiosulfate
inhibited MEA system dropped to a value of 0.05 and 0.07 mmpy compared to the ‘No
contaminant’ corrosion rate of 0.35 mmpy (Figure 4.18a). Corrosion inhibition efficiency
in the presence of chloride and formate was 99% (Figure 4.18b). However, from the
potentiodynamic polarization behaviour (Figure 4.18c), it can be observed that even
though the metal tended to passivate at lower anodic current densities compared to the
‘No contaminant’ condition, the passive layer was highly unstable.

124
(a) (b)
(c) (d)
(e)
Figure 4.18 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading, 1000 ppm sodium
thiosulfate) (a) Comparison of corrosion rate (b) Comparison of inhibition efficiencies (c)
Polarization behaviour (d) Comparison of polarization resistance (e) Impedance behaviour
0.00
2.00
4.00
6.00
No process contaminant
chloride formate
Co
rosi
on r
ate
(m
mp
y)
Uninhibited Inhibited
0%
25%
50%
75%
100%
No process contaminant
chloride formate
Inh
ibit
ion
eff
icie
ncy
(%
)
Inhibitor concentration (ppm)
-1.20
-0.90
-0.60
-0.30
0.00
0.30
0.60
0.90
-7 -6 -5 -4 -3 -2
Po
ten
tial
(V
vs
Ag/A
gC
l)
Log current density (A/cm2)
No process contaminant chloride formate
0
200
400
600
800
1000
No process contaminant
chloride formate
RP
(oh
m-c
m2)
Uninhibited Inhibited
0
100
200
300
400
0 100 200 300 400 500
Z''
(oh
m-c
m2)
Z' (ohm-cm2)
No contaminant formate chloride

125
As the initial passive layer broke down, an increase in anodic current densities was
observed, which was followed by repassivation. This cycle repeated without the
formation of a stable passive layer throughout the anodic polarization, suggesting that the
passive layer might not be protective in the presence of process contaminant. No pitting
tendency was induced by the presence of process contaminants.
Polarization resistances (RP) obtained from the impedance analysis in the
presence of chloride and formate were 889 and 974 ohm-cm2, respectively, which was
higher than the ‘No contaminant’ condition (594 ohm-cm2) (Figure 4.18d). This was in
agreement with the lower corrosion rates obtained from potentiodynamic polarization
analysis. Impedance behaviour (Figure 4.18e) in the presence and absence of the process
contaminants was similar in terms of trends characterized by the presence of a diffusion
tail suggesting that the metal was in a passive state. The unstable nature of the passive
layer in the presence of process contaminants could not be predicted, mainly because the
impedance analysis is carried out at the equilibrium potential of the metal unlike
potentiodynamic polarization.

126
4.1.3 Comparison of corrosion inhibition performance of different inhibitors
Corrosion inhibition performance of different corrosion inhibitors was compared
based on the corrosion rates obtained for each case (Figure 4.19a). Corrosion rates at the
inhibitor concentration at which the effect of process contaminants was tested in each
case were chosen as the basis for comparison. In the case of sulfanilamide and
thiosalicylic acid, for which the effects of process contaminants were not tested, the
corrosion inhibitor concentration corresponding to the lowest corrosion rate was chosen.
In the absence of process contaminants, corrosion rates for different corrosion
inhibitors were in the range of 0.35 to 2.50 mmpy compared to the uninhibited corrosion
rate of 4.27 mmpy. The lowest corrosion rate was observed for sodium thiosulfate and the
highest corrosion rate was obtained for sulfanilamide. In the absence of process
contaminants, the corrosion rate decreased in the following order: sulfanilamide >
thiosalicylic acid > sulfolane > 2-aminobenzene sulfonic acid > 3-aminobenzene sulfonic
acid > 4-aminobenzene sulfonic acid > sulfapyridine > sodium thiosulfate.
In presence of chloride, the corrosion rate of 3-amino benzene sulfonic acid
increased to 1.65 mmpy from 0.48 mmpy, whereas in case of sulfapyridine, the corrosion
rate increased to 4.34 mmpy from 0.44 mmpy. For 4-aminobenzene sulfonic acid,
sulfolane and sodium thiosulfate, the corrosion rate was lower than the ‘No contaminant’
condition, and in the case of 2-aminobenzene sulfonic acid, the corrosion rate was
slightly higher. In the presence of chloride, the corrosion rate decreased in the following
order: sulfapyridine > 3-aminobenzene sulfonic acid > 2-aminobenzene sulfonic acid >
sulfolane > 4-aminobenzene sulfonic acid > sodium thiosulfate.

127
(a) (b)
(c)
Figure 4.19 Corrosion behaviour of inhibited MEA solutions with and without process
contaminants (5.0 kmol/m3 MEA, 80
oC, 0.55 mol/mol CO2 loading) (a) Comparison of
corrosion rate (b) Comparison of polarization resistance (c) Comparison of inhibition
efficiencies
0.00
1.00
2.00
3.00
4.00
5.00
6.00
2-a
min
ob
enze
ne
sulf
on
ic a
cid
3-a
min
ob
enze
ne
sulf
on
ic a
cid
4-a
min
ob
enze
ne
sulf
on
ic a
cid
sulf
ap
yri
din
e
sulf
an
ilam
ide
sulf
ola
ne
thio
sali
cyli
c aci
d
thio
sulf
ate
Co
rosi
on r
ate
(m
mp
y)
No process contaminant chloride formate
0
200
400
600
800
1000
2-a
min
ob
enze
ne
sulf
on
ic a
cid
3-a
min
ob
enze
ne
sulf
on
ic a
cid
4-a
min
ob
enze
ne
sulf
on
ic a
cid
sulf
ap
yri
din
e
sulf
an
ilam
ide
sulf
ola
ne
thio
sali
cyli
c aci
d
thio
sulf
ate
RP
(oh
m-c
m2)
No process contaminant chloride formate
-40%
-20%
0%
20%
40%
60%
80%
100%
Inh
ibit
ion
eff
icie
ncy
(%
)
No process contaminant chloride formate

128
In the presence of formate, 2-aminobenzene sulfonic acid, 3-aminobenzene
sulfonic acid, and 4-aminobenzene sulfonic acid had increased corrosion rates higher than
the uninhibited condition. For sulfapyridine, the corrosion rate was slightly higher than
the ‘No contaminant’ condition, whereas in the case of sulfolane and sodium thiosulfate,
lower corrosion rates were observed. In the presence of formate, the corrosion rate
decreased in the following order: 2-aminobenzene sulfonic acid > 3-aminobenzene
sulfonic acid > 4-aminobenzene sulfonic acid > sulfapyridine > sulfolane > sodium
thiosulfate.
Similarly, polarization resistances (RP) obtained from impedance analysis for
different corrosion inhibitors can also be used to compare the corrosion inhibition
performance (Figure 4.19b). The results were in agreement with the inferences of the
corrosion rate comparison. In the absence of process contaminants, RP decreases in the
following order: sodium thiosulfate > sulfapyridine > 4-aminobenzene sulfonic acid > 2-
aminobenzene sulfonic acid > 3-aminobenzene sulfonic acid > sulfolane > thiosalicylic
acid > sulfanilamide. In presence of chloride, RP decreased in the following order:
sodium thiosulfate > 4-aminobenzene sulfonic acid > 2-aminobenzene sulfonic acid >
sulfolane > sulfapyridine > 3-aminobenzene sulfonic acid. In the presence of formate,
the RP decreased in the following order: sodium thiosulfate > sulfolane > sulfapyridine >
3-aminobenzene sulfonic acid > 4-aminobenzene sulfonic acid > 2-aminobenzene
sulfonic acid. From Figure 4.19c, it can be observed that in the absence of process
contaminants, the corrosion inhibition efficiencies were in the range of 42 to 92%. The
lowest corrosion inhibition efficiency was obtained for sulfanilamide, whereas the
highest efficiency was observed in the case of sodium thiosulfate.

129
In the presence of chloride, sulfapyridine displayed a corrosion inhibition
efficiency of -2%, suggesting that the corrosion inhibitor slightly aggravated the
corrosivity of the MEA system. In the presence of formate, 2-aminobenzene sulfonic
acid, 3-aminobenzene sulfonic acid, and 4-aminobenzene sulfonic acid showed corrosion
inhibition efficiencies of -34%, -25%, and -28%, respectively. Sulfolane and sodium
thiosulfate showed an increase in the corrosion inhibition efficiency compared to the ‘No
contaminant’ condition. In the absence of process contaminants, corrosion inhibition
efficiency decreased in the following order: sodium thiosulfate > sulfapyridine > 4-
aminobenzene sulfonic acid > 3-aminobenzene sulfonic acid > 2-aminobenzene sulfonic
acid > sulfolane > thiosalicylic acid > sulfanilamide. In the presence of formate, the
corrosion inhibition efficiency decreased in the following order: sodium thiosulfate >
sulfolane > sulfapyridine > 3-aminobenzene sulfonic acid > 4-aminobenzene sulfonic
acid > 2-aminobenzene sulfonic acid. In the presence of chloride, the corrosion inhibition
efficiency decreased in the following order: sodium thiosulfate > 4-aminobenzene
sulfonic acid > sulfolane > 2-aminobenzene sulfonic acid > 3-aminobenzene sulfonic acid
> sulfapyridine.
The effect of process contaminants on corrosion inhibition performance of sodium
thiosulfate was peculiar, as it was not apparent from the corrosion rate, RP, and corrosion
inhibition efficiencies. In the presence of sodium thiosulfate, a passive film was formed
over the metal surface and the stability of the passive film was affected by the process
contaminants (formate and chloride) as discussed in section 4.1.2.8 (b). Based on the
comparison of corrosion inhibition performances of different corrosion inhibitors, the

130
following compounds were considered for the weight loss testing: 4-aminobenzene
sulfonic acid, sulfapyridine, sulfolane, and sodium thiosulfate.
4.2 Weight loss tests
From the weight loss values of the carbon steel specimens withdrawn at regular
time intervals from the weight loss tests, the corrosion rates were determined. In order to
analyze the nature of the corrosion product formed in each case, the specimens tested for
the longest duration (28 days) were subjected to surface analytical testing such as
Scanning Electron Microscopy (SEM), Energy Dispersive X-Ray Spectroscopy (EDS),
and X-Ray Diffraction (XRD).
4.2.1 Corrosion behaviour of uninhibited MEA systems
The corrosion rate of carbon steel in uninhibited MEA solution (5.0 kmol/m3
MEA, 80oC, 0.55 mol/mol CO2 loading and No process contaminant) after 28 days was
0.50 mmpy (Table 4.3). From Figure 4.20, for the uninhibited condition, it can be
observed that a steep increase in corrosion rate after 7 days was followed by a gradual
increase until it reached a steady value after 21 days (0.50 mmpy). The final corrosion
rate of 0.50 mmpy was lower than the electrochemical corrosion rate of carbon steel in an
uninhibited MEA system, which was 4.27 mmpy. This might be due to the formation of a
stable corrosion product as a result of longer exposure time, which might act as a barrier
between the metal and the solution. From the SEM images (Figure 4.21a), a uniform
corrosion product formed over the metal surface can be clearly seen. From the EDS
analysis (4.21b), a significant increase in the amounts of oxygen and carbon in the tested

131
Table 4.3 Summary of weight loss experimental results
Experimental
condition
Corrosion rate (mmpy) I.E (after
28 days)
(%)
pH σ
(mS/cm) 7 days 14 days 21 days 28 days
Uninhibited 0.39 0.46 0.50 0.50 - 8.34 38.97
4-aminobenzene
sulfonic acid - 0.69 0.68 0.77 -54.81 8.38 39.09
Sulfapyridine 0.24 0.18 0.19 0.23 54.72 8.33 42.30
Sulfolane 0.34 0.30 0.28 0.28 44.26 8.32 41.27
Thiosulfate 0.44 0.43 0.48 0.45 10.62 8.31 42.66
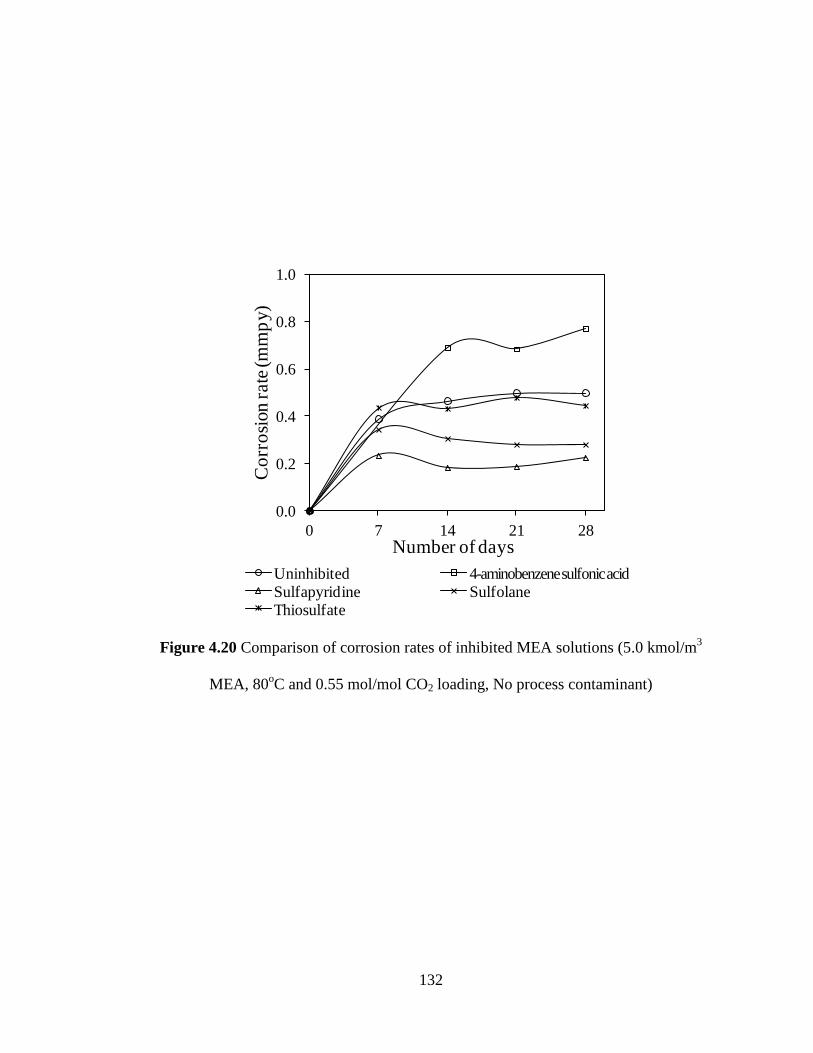
132
Figure 4.20 Comparison of corrosion rates of inhibited MEA solutions (5.0 kmol/m
3
MEA, 80oC and 0.55 mol/mol CO2 loading, No process contaminant)
0.0
0.2
0.4
0.6
0.8
1.0
0 7 14 21 28
Co
rro
sion
rate
(m
mp
y)
Number of days
Uninhibited 4-aminobenzene sulfonic acid
Sulfapyridine SulfolaneThiosulfate
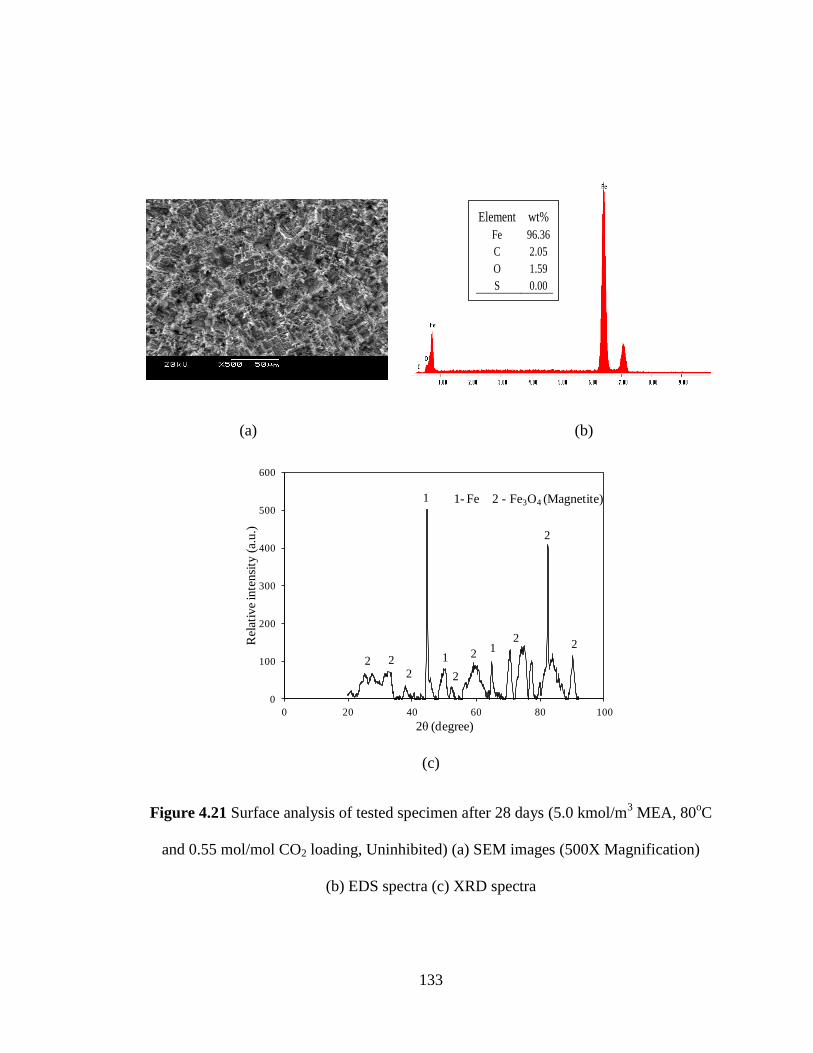
133
(a) (b)
(c)
Figure 4.21 Surface analysis of tested specimen after 28 days (5.0 kmol/m3 MEA, 80
oC
and 0.55 mol/mol CO2 loading, Uninhibited) (a) SEM images (500X Magnification)
(b) EDS spectra (c) XRD spectra
Element wt%
Fe 96.36
C 2.05
O 1.59
S 0.00
0
100
200
300
400
500
600
0 20 40 60 80 100
Rel
ativ
e in
tensi
ty (
a.u.)
2θ (degree)
2 22
1
11
2
2
2
2
2
1- Fe 2 - Fe3O4 (Magnetite)

134
specimens compared to the fresh specimen could be observed (Figure 4.22). This might
be due to the formation of corrosion products based on carbon and oxygen. In the XRD
analysis (Figure 4.22c), the peaks obtained were broader, suggesting that the corrosion
product might be poorly crystalline. Hence, different peaks lying in proximity could
possibly have merged together to form a single broad peak, making it difficult to analyze.
Comparing the results with the reference data (ICDD) for different compounds, the
corrosion product was characterized mainly to be in the form of Fe3O4 (magnetite). In
addition to Fe3O4, peaks corresponding to iron (Fe) could also be seen, which suggests
that the surface layer is less protective. Additionally, the increase in the percent weight
of carbon suggests that the surface might also contain amorphous iron carbonate
(FeCO3).
4.2.2 Corrosion behaviour of inhibited MEA systems
For 4-aminobenzene sulfonic acid, it can be observed (Table 4.3) that the
corrosion rate after 28 days of weight loss testing was 0.77 mmpy corresponding to an
aggravation of corrosion by about 55% (Figure 4.20). From electrochemical testing, it
was observed that the corrosion inhibitor actually increased the corrosion rate in the
presence of formate (Figure 4.9). Solution degradation products such as formate could be
formed in long-term tests and might interfere with the adsorption of the inhibitor onto the
metal surface and expose the metal. From SEM images (Figure 4.24a), it can be observed
that the uniform corrosion products over the metal surface were similar in appearance and
distribution to the uninhibited result (Figure 4.21a). From EDS analysis (Figure 4.24b),
larger quantities of oxygen and carbon than in the uninhibited condition were observed,
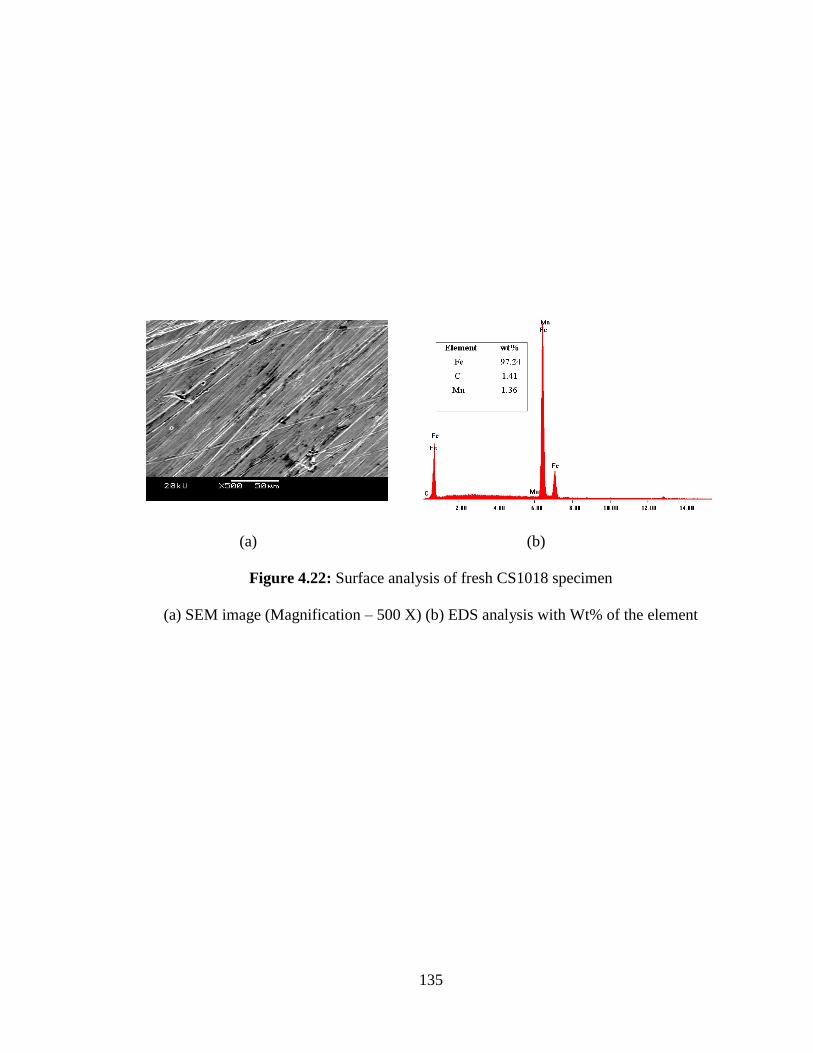
135
(a) (b)
Figure 4.22: Surface analysis of fresh CS1018 specimen
(a) SEM image (Magnification – 500 X) (b) EDS analysis with Wt% of the element
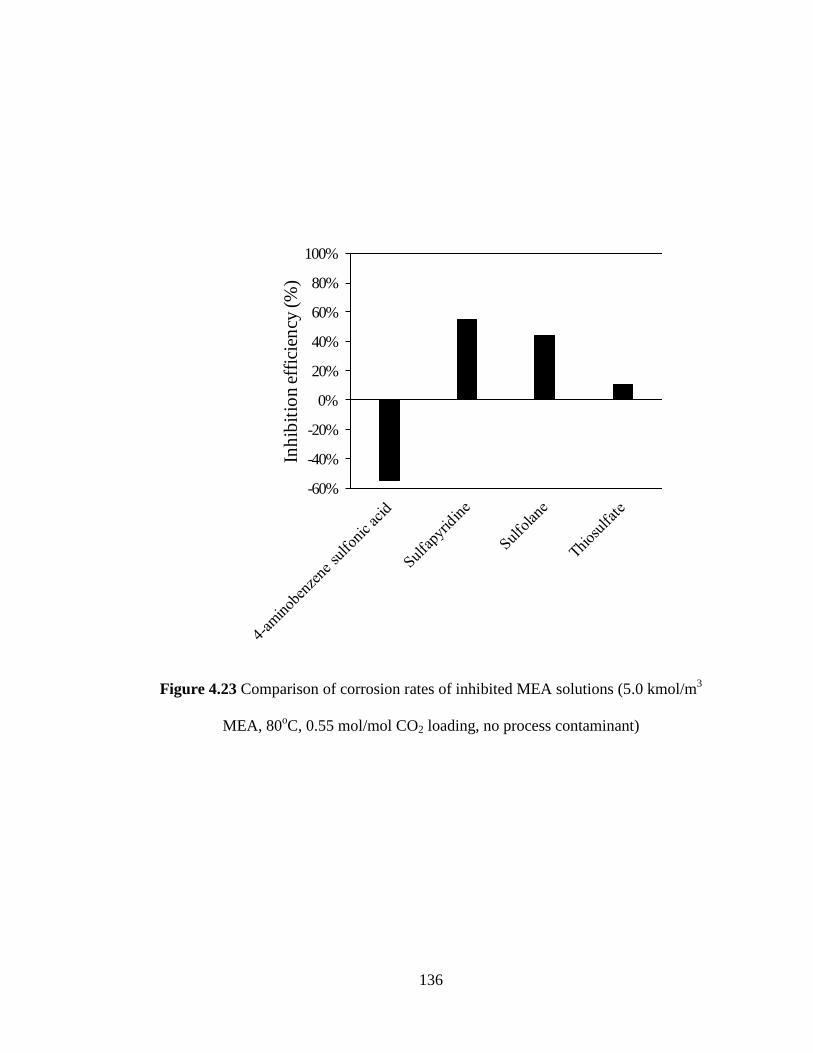
136
Figure 4.23 Comparison of corrosion rates of inhibited MEA solutions (5.0 kmol/m3
MEA, 80oC, 0.55 mol/mol CO2 loading, no process contaminant)
-60%
-40%
-20%
0%
20%
40%
60%
80%
100%
Inh
ibit
ion
eff
icie
ncy
(%
)
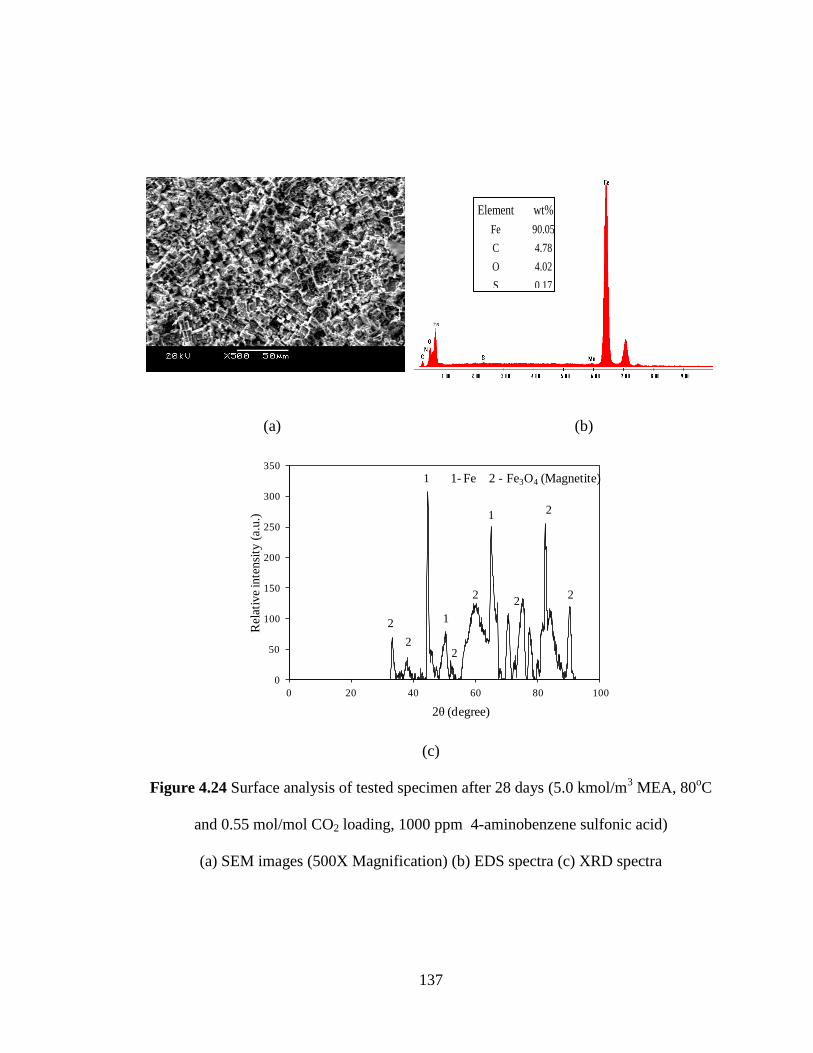
137
(a) (b)
(c)
Figure 4.24 Surface analysis of tested specimen after 28 days (5.0 kmol/m3 MEA, 80
oC
and 0.55 mol/mol CO2 loading, 1000 ppm 4-aminobenzene sulfonic acid)
(a) SEM images (500X Magnification) (b) EDS spectra (c) XRD spectra
Element wt%
Fe 90.05
C 4.78
O 4.02
S 0.17
Element wt%
Fe 90.05
C 4.78
O 4.02
S 0.17
0
50
100
150
200
250
300
350
0 20 40 60 80 100
Rel
ativ
e in
ten
sity
(a.
u.)
2θ (degree)
2
2
1
1
2
2
1
22
2
1- Fe 2 - Fe3O4 (Magnetite)

138
indicating a larger amount of corrosion product formed. Increase in corrosion rate with
the number of days suggests that the corrosion product formed was not protective. This
was also confirmed by the XRD results (Figure 4.24c), where peaks corresponding to Fe
were characterized in addition to the primary corrosion product Fe3O4.
In the case of sulfapyridine, the final corrosion rate was 0.23 mmpy,
corresponding to a final inhibition efficiency of 55% (Figure 4.23). On visual observation
of the specimen, a tenacious surface layer was seen. From SEM images (Figure 4.25a), it
can be seen that, in this case, a non-uniformly distributed corrosion product was formed.
Increase in quantities of both oxygen and carbon was observed from EDS analysis
(Figure 4.25b) and from XRD spectra (Figure 4.25c). Fe3O4 was characterized to be the
primary component of the corrosion product, but Fe was also present.
In the case of sulfolane, the corrosion rate was initially high, which then dropped
and reached a steady value corresponding to a final inhibition efficiency of around 44%
(Figure 4.22). Visual examination of the specimen suggested that the surface was covered
by a tenacious surface layer. As can be seen from the SEM image (Figure 4.26a), the
surface was not covered with a densely porous and uniform corrosion product as in the
previous cases but was characterized by an intact protective layer on the surface. The
amount of carbon and oxygen on the metal surface based on EDS analysis (Figure 4.26b)
was the least in comparison with other inhibitors. Fe3O4 was characterized to be the main
component of the corrosion product from XRD analysis (Figure 4.26c) along with the
presence of Fe.
In the case of thiosulfate, loose corrosion product could be visually observed
during the removal of the specimens. From SEM images (Figure 4.27a), two distinct
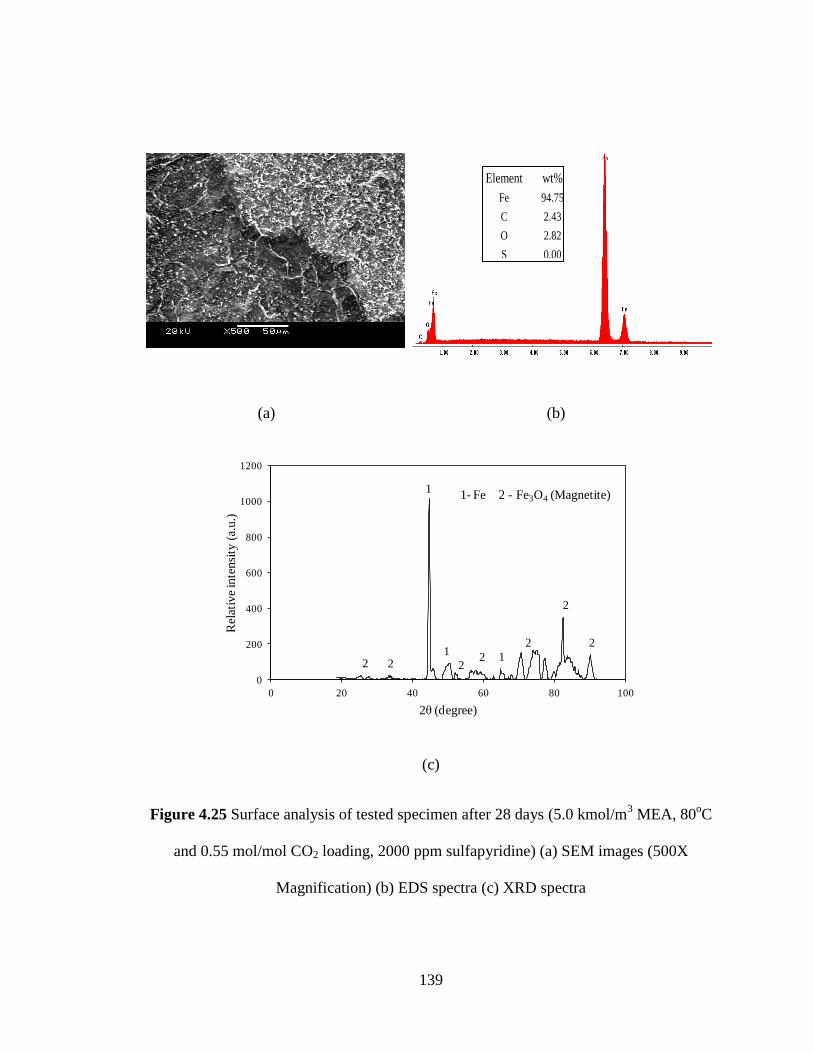
139
(a) (b)
(c)
Figure 4.25 Surface analysis of tested specimen after 28 days (5.0 kmol/m3 MEA, 80
oC
and 0.55 mol/mol CO2 loading, 2000 ppm sulfapyridine) (a) SEM images (500X
Magnification) (b) EDS spectra (c) XRD spectra
Element wt%
Fe 94.75
C 2.43
O 2.82
S 0.00
Element wt%
Fe 94.75
C 2.43
O 2.82
S 0.00
0
200
400
600
800
1000
1200
0 20 40 60 80 100
Rel
ativ
e in
ten
sity
(a.
u.)
2θ (degree)
2 2
1
1
2
2
12
2
2
1- Fe 2 - Fe3O4 (Magnetite)

140
(a) (b)
(c)
Figure 4.26 Surface analysis of tested specimen after 28 days (5.0 kmol/m3 MEA, 80
oC
and 0.55 mol/mol CO2 loading, 2000 ppm sulfolane) (a) SEM images (500X
Magnification) (b) EDS spectra (c) XRD spectra
Element wt%
Fe 96.98
C 1.68
O 1.26
S 0.10
Element wt%
Fe 96.98
C 1.68
O 1.26
S 0.10
0
50
100
150
200
250
300
350
400
0 20 40 60 80 100
Rel
ativ
e in
ten
sity
(a.
u.)
2θ (degree)
2
2
1
1
1
2
2
2
2
1- Fe 2 - Fe3O4 (Magnetite)
2

141
(a) (b)
(c)
Figure 4.27 Surface analysis of tested specimen after 28 days (5.0 kmol/m3 MEA, 80
oC
and 0.55 mol/mol CO2 loading, 1000 ppm sodium thiosulfate) (a) SEM images (500X
Magnification) (b) EDS spectra (c) XRD spectra
Element wt%
Fe 64.68
C 1.83
O 4.72
S 28.77
Element wt%
Fe 64.68
C 1.83
O 4.72
S 28.77
-20
30
80
130
180
230
280
330
380
430
0 20 40 60 80 100
Rel
ativ
e in
tensi
ty (
a.u.)
2θ (degree)
22
2
1
1
1
22
2
2
2
1- Fe 2 - Fe3O4 (Magnetite)

142
types of corrosion products were visible; one was the loose and bulk corrosion product on
the outer surface and the other was a relatively fine and intact corrosion product beneath.
A large quantity of sulfur (29%) was characterized by EDS analysis (Figure 4.27b),
suggesting the presence of a sulfur-based corrosion product on the surface. Sodium
thiosulfate can potentially decompose into hydrogen sulfide (H2S) at the tested
conditions, which implies that the corrosion product could be speculated to be iron
sulfide based on the EDS results. However, based on the XRD results (Figure 4.27c),
Fe3O4 was characterized to be the primary component of the surface layer, along with the
presence of Fe. Thus, iron sulfide might be present in amorphous phase. Corrosion
product was less protective in nature as the corrosion inhibition efficiency was only
around 10%. This can be explained based on its polarization behaviour in the presence of
formate and chloride. In long-term testing, the solution degradation products such as
formate that could be formed in the solution might have caused the destabilization of
passive film formed, leading to an increase in corrosion rate.
In summary, sulfapyridine and sulfolane were found to be the best-performing
corrosion inhibitors among the selected compounds. While sulfolane would be stable in
the presence of both formate and chloride, the performance of sulfapyridine might be
affected in the presence of chloride. Sodium thiosulfate was not effective in long-term
protection, whereas 4-aminobenzene sulfonic acid showed increased corrosion rate in its
presence. Iron oxide (Fe3O4) was the primary corrosion product. Increase in the amounts
of carbon and oxygen in all cases and large quantities of sulfur in the case of thiosulfate
suggest that other non-crystalline compounds such as iron carbonate (FeCO3) or iron
sulfide (FeS) might also be present.

143
5. CONCLUSIONS AND FUTURE WORK
5.1 Conclusions
Eight environmentally-friendly corrosion inhibitors were successfully chosen
based on the principle of hard and soft acids and bases (HSAB), toxicity, and quantum
chemistry for the evaluation of their corrosion inhibition performance. The important
findings of the performance evaluation are as follows:
2-aminobenzene sulfonic acid is effective, with an inhibition efficiency of 87–89% at
a concentration range of 250-3000 ppm. However, its performance can deteriorate in
the presence of formate. Corrosion inhibition is due to the adsorption of inhibitor onto
the metal surface.
3-aminobenzene sulfonic acid performs well, with an inhibition efficiency of 88-89%
at a concentration range of 250-3000 ppm. The inhibition efficiency can deteriorate in
the presence of chloride and formate. Corrosion inhibition is due to adsorption of
inhibitor on metal surface.
4-aminobenzene sulfonic acid is effective, with an inhibition efficiency of 87-91% at
a concentration range of 250-3000 ppm. Its performance can be reduced in the
presence of formate and becomes ineffective in long-term exposure. Corrosion
inhibition is due to adsorption of inhibitor onto the metal surface.
Sulfapyridine performs well, with an inhibition efficiency of 90-92% when its
concentration is at 2000 ppm or greater, but its performance can be deteriorated by
chloride. The inhibitor performance of sulfapyridine can be maintained in long-term
exposure.

144
Sulfanilamide is not an effective corrosion inhibitor, as its inhibition efficiency is 20-
42%.
Sulfolane is an effective corrosion inhibitor when its concentration is at least 2000
ppm. Its inhibition performance is unaffected by the presence of chloride and formate,
and it can be maintained during long-term exposure. Corrosion inhibition is due to the
adsorption of inhibitor on the metal surface.
Thiosalicylic acid is not an effective corrosion inhibitor, as its efficiency is 47-80%.
The presence of thiosalicylic acid causes color change of the MEA solution.
Sodium thiosulfate is an effective corrosion inhibitor in short-term exposure with an
inhibition efficiency of 91-94% at a concentration range of 250-10,000 ppm, but it
becomes ineffective in long-term. This is due to the instability of the passive film
formed. Its performance can be deteriorated by the presence of chloride and formate.
Sulfolane and sulfapyridine show promise as potential corrosion inhibitors due to
their inhibition performance in both short-term and long-term exposure and are
recommended for further evaluation.
5.1 Recommendations for future work
The corrosion inhibitors identified from this work can be further tested for their
effectiveness in the MEA-based CO2 absorption process under a wider range of operating
conditions. Further tests can include parametric effects on corrosion inhibition
performance by the presence of dissolved oxygen, solution velocity, solution temperature
(up to 120oC), and the presence of other process contaminants (sulfite, bicine, and
acetate). Flow loop tests can be conducted to simulate the effect of real flow conditions in

145
the plant on corrosion inhibition. The inhibitors that show promise in the above
mentioned tests can be evaluated for their effectiveness in other amine-based CO2
absorption processes, such as diethanolamine (DEA), 2-amino-2-methyl-1-propanol
(AMP), and piperazine (PZ).

146
REFERENCES
Aaron, D., Tsouris, C., (2005), Separation of CO2 from flue gas: a review, Separation
Science and Technology, 40, 321-348.
Anastas. P. T., Williamson. T. C., (1998), Green Chemistry: Frontiers in benign
chemical syntheses and processes, Oxford University Press, NY, USA.
Aroonwilas, A., (1996), High efficiency structured packing for CO2 absorption using 2-
Amino-2-methyl-2-propanol (AMP), M.A.Sc. Thesis, University of Regina,
Regina, Saskatchewan, Canada.
Asperger, R.G., Clouse, R.C., (1978), Quarternary salt-polyamine inhibitor for sour gas
conditioning solutions, US Patent 4100099.
Asperger, R.G., Krawczyk, L.S., Oakes, B.D., (1979), Method and composition for
inhibiting the corrosion of ferrous metals, US Patent 4143119.
Astarita. G., Savage. D. W., Bisio. A., (1983), Gas treating with chemical solvents,
John Wiley Publications, NY, USA.
ASM Handbook: Volume 13A – Corrosion: Fundamentals, Testing and Protection,
(2003), ASM International, OH, USA.
ASTM Standard G1-03, (2003), Standard practice for preparing, cleaning and
evaluating corrosion test specimens. In Annual Book of ASTM Standards;
American Society of Testing and Materials: West Conshohocken, PA.
ASTM Standard G106 – 89 (Reapproved 2010), Standard practice for verification of
algorithm and equipment for electrochemical impedance measurements, In
Annual Book of ASTM Standards; American Society of Testing and Materials:
West Conshohocken, PA.

147
ASTM Standard G5-94 (Reapproved 2004), Standard reference test method for making
potentiostatic and potentiodynamic anodic polarization measurements, In
Annual Book of ASTM Standards; American Society of Testing and Materials:
West Conshohocken, PA.
Bockris, J.O’M., Reddy, A.K.N., (2000), Modern Electrochemistry:Volume 2,
Springer, NY, USA.
Chakrabarti, A., (1984), Quantum chemical study of the corrosion inhibition of mild
steel in 6 percent (wt/wt) HCl by means of cyanoguanidine derivatives, Br.
Corros. J., 19, 124–126.
Chang, Z., Minevski, L., (2008), Polythiaether compounds and their use as corrosion
inhibitors, US Patent 7335794 B2
Clouse, R.C., Asperger, R.G., (1978), Cobalt-containing inhibitor for sour gas
conditioning solutions, US Patent 4100100.
Clouse, R.C., Asperger, R.G., (1978), Inhibitors for gas conditioning solutions, US
Patent 4102804.
Davidson, J.R., Friedli, H.R., (1978), Method and composition for inhibiting the
corrosion of metals, US Patent 4071470
DeHart, T. R., Hansen, D.A., Mariz, C. L., McCullough, J. G., (1999), Solving
corrosion problems at the Nea Bellingham Massachusetts carbon dioxide
recovery plant, Corrosion 99; NACE International, Paper No. 264.
Dingman, J. C., Allen, D. L., Moore, T. F., (1966), Minimize corrosion in MEA units,
Hydrocarbon Processing, 285-290.

148
Doble, M., Kruthiventi. A.K., (2007), Green Chemistry and Engineering, Academic
press, MA, USA.
Dupart, M.S., Bacon, T.R., Edwards, D.J., (1993), Understanding corrosion in
alkanolamine gas treating plants, Part 1 & 2, Hydrocarbon Processing, 93, 75-
80.
Dupart, M.S., Oakes, B.D., Cringle, D.C., (1984) Method and composition for reducing
corrosion in the removal of acidic gases from gaseous mixtures, US Patent
4446119.
Fischer, P.W., (1957), Corrosion prevention in gas recovery systems, US Patent
2776870.
Fischer, P.W., (1959), Method of gas purification utilizing an amin solution and an
anti-corrosion agent, US Patent 2869978.
Fontana, M. G., (1986), Corrosion Engineering, (3rd ed.), New York: McGraw-Hill.
Gerus, B.R.D., (1981), Detection and mitigation of weight loss corrosion in sour gas
gathering systems. In H2S corrosion in Oil & Gas Production – A Compilation
of classic papers, National Association of Corrosion Engineers: Houston, TX,
USA, 888-903.
Hackerman, N., Makrides, A.C., (1954), Action of polar organic inhibitors in acid
dissolution of metals, Ing. Eng. Chem., 46 (3), 523-527.
Hall, W. D., Barron, J. G., (1981), Solving gas treating problems – A different
approach, Proceedings of Gas Conditioning conference, 31st Annual, University
of Oklahoma, C1-C13.

149
Hamah-Ali, B., Ali, B. S., Yusoff, R., Aroua, M. K., (2011), Corrosion of carbon steel
in aqueous carbonated solution of MEA/ [bmim] [DCA], Int. J. Electrochem.
Sci., 6, 181-198.
Heisler, L., Weiss, I.H., (1975), Operating Experience at Aderklaa with Alkanolamine
Gas Treating Plants for Sour Natural Gas Sweetening, Proceedings of Gas
Conditioning conference, 25th
Annual, University of Oklahoma, H1-H22.
Hensen, E.R., Tipton, T.M., Courtwright, J.G., (1986), Corrosoin inhibitors for
Alkanolamines, US Patent 4595723.
IPCC1 (2007), IPCC Fourth Assessment Report (AR4), Climate Change 2007:
Synthesis Report, Retrieved from www.ipcc.ch on February 2012.
IPCC2 (2005), IPCC special report on CO2 capture and storage, Retrieved from
www.ipcc-wg3.de on February 2012.
Jones, D. A., (1992), Principles and Prevention of Corrosion, New York: Macmillan
Publishing Company.
Jones, L.W., Alkire, J.D., (1985), Corrosion inhibitors for amine gas sweetening
systems, US Patent 4541946.
Khaled, K.F., Hackerman, N., (2003), Investigation of the inhibitive effect of ortho-
substituted anilines on corrosion of iron in 1 M HCl solutions, Electrochimica
Acta, 48 (19), 2715-2723
Khalil, N., (2003), Quantum chemical approach of corrosion inhibition, Electrochim.
Acta., 48 (18), 2635–2640.

150
Kittel, J., Fleury, E., Vuillemin, B., Gonzalez, S., Ropital, F., Oltra, R., (2010),
Corrosion in alkanolamine used for acid gas removal: From natural gas
processing to CO2 capture, Materials and Corrosion, 63 (3), 223-230.
Kittel, J., Idem, R., Gelowitz, D., Tontiwachwuthikul, P., Parrain, G., Bonneau, A.,
(2009), Corrosion in MEA units for CO2 capture: pilot plant studies, Energy
Procedia, 1, 791-797.
Kohl, A. L., Nielsen, R. B., (1997), Gas Purification, (5thed.). Houston, Texas: Gulf
Publishing Co.
Krawczyk, L. S., Martin, C. W., Pearce, R. L., (1984), Inhibitors for Acid gas
conditioning solutions, US Patent 4431563.
Litschewski, M.J., (1996), More experiences with corrosion and fouling in a refinery
amine system, Corrosion 96; NACE International annual conference and
exposition, Paper No. 391.
Mago, B.F., West, C.W., (1974), Antimony-Vanadium corrosion inhibitors for
alkanolamine gas treating system, US Patent 3808140.
Mago, B.F., West, C.W., (1975), Nitrosubstituted Aromatic acid corrosion inhibitors
for alkanolamine gas treating system, US Patent 3896044.
Mago, B.F., (1976), Corrosion inhibition of aqueous potassium carbonate gas treating
systems, US Patent 3951844.
Mago, B.F., West, C.W., (1976), Corrosion Inhibitors for alkanolamines gas treating
system, US Patent 3959170.
Minevski, L.V., (2000), Corrosion inhibitor for alkanolamine units, US Patent
6036888.

151
Minevski, L.V., Lambousy, A.L., (1998) Corrosion inhibitor for alkanolamine units,
US Patent 5843299.
NACE1, (2012), Retrieved from www.nace.org on April 2012.
Negra, J.S., McCloskey, J.W., (1963) Corrosion inhibiting, US Patent 3087778
Nieh, E.C.Y., (1983), Vanadium-Cobalt corrosion inhibitor system for sour gas
conditioning solutions, US Patent 4371450.
Nieh, E.C.Y., (1983), Vanadium-amine corrosion inhibitor system for sour gas
conditioning solutions, US Patent 4372873.
OSPAR, (2011), Retrieved from www.ospar.org on March 2011.
Öğretir, C., Mihçi, B., Bereket, G., (1999), Quantum chemical studies of some pyridine
derivatives as corrosion inhibitors, Journal of Molecular Structure (Theochem),
488 (1-3), 223-231.
Pearce, R.L., (1984), Process for removal of carbon dioxide from industrial gases, US
Patent 4440731.
PubChem1, (2011), Retrieved from http://pubchem.ncbi.nlm.nih.gov/ on Jan 2011
Rampin, P., (2000), Amine units: results of a survey on structural reliability,
Proceedings of International Conference on Corrosion in refinery,
petrochemical and power generation plants, May 18-19, Venezia.
Revie, R.W., Uhlig, H.H., (2008), Corrosion and Corrosion control, 3rd
edition, John
Wiley & Sons, Inc. Hoboken, New Jersey.
Rodriguez, E.F., Edwards, M.A., (1999), The use of organic inhibitor to control
corrosion in alkanolamine units processing gas containing CO2, Corrosion 99;
NACE International, Paper No. 260.

152
Sastri, V. S., (2001), Corrosion Inhibitors: Principles and Applications, West Sussex,
England: John Wiley & Sons Ltd.
Sastri, V.S., Perumareddi, J.R., (1994), Selection of corrosion Inhibitors for use in sour
media, Corrosion, 50 (6), 433-437.
Sastri, V.S., Perumareddi, J.R., (1997), Molecular orbital theoretical studies of some
organic corrosion inhibitors, 53 (8), 617-622.
Schmeal, W. R., MacNab, A. J., Rhodes, P. R., (1978), Corrosion in amine/sour gas
treating contactors, Chemical Engineering Progress, 74(3), 37-42.
Sekine, I., Shimode, T., Yuasa. M., Takaoka. K., (1992), Corrosion inhibition of
structural steels in the carbon dioxide absorption process by organic inhibitor
composed of 2-aminothiophenol, (1-hydroxyethylidene)bis(phosphonic acid),
and diethanolamine, Ind. Eng. Chem. Res., 31(1), 434-439.
Smith, R. F., Younger, A. H., (1972), Operating experiences of Canadian
diethanolamine plants, Proceedings of Gas Conditioning conference, 22nd
Annual, University of Oklahoma, E1-E17.
Soosaiprakasam, I. R., (2007), Corrosion inhibition performance of copper carbonate in
CO2 absorption process using aqueous solution of monoethanolamine, M.A. Sc.
Thesis, University of Regina, Regina, Saskatchewan, Canada.
Soosaiprakasam, I. R., Veawab, A., (2009), Corrosion inhibition performance of copper
carbonate in MEA-CO2 capture unit, Energy Procedia, 1 (1), 225-229.
Srinivasan, P., (2006), Synergistic effect of multi heat-stable salts on corrosion of
carbon steel in amine treating plants, M.A. Sc. Thesis, University of Regina,
Regina, Saskatchewan, Canada.

153
Sutopo., Safruddin. R., (2000), Twenty years of experience in controlling corrosion in
amine unit, Badak LNG plant, Corrosion 2000; NACE International, Paper No.
497.
Treviňo, J.A.V., (1987), Corrosion inhibitor for CO2 absorption process using
Alkanolamines, US Patent 4714597.
Veawab, A., (2000), Corrosion and corrosion control in CO2 absorption process using
aqueous amine solutions, Ph.D. Thesis, University of Regina, Regina,
Saskatchewan, Canada.
Veawab, A., Tontiwachwuthikul, P., Chakma, A., (2001), Investigation of Low-Toxic
corrosion inhibitors for CO2 separation process using aqueous MEA solvent.
Ind. Eng. Chem. Res., 40(22), 4771-4777.
Veldman, R.R., Trahan, D.O., (2001), Gas treating solution corrosion inhibitor, US
Patent 6299836 B1
Vračar, Lj. M., and Dražić, D.M., (2002), Adsorption and corrosion inhibitive
properties of some organic molecules on iron electrode in sulfuric acid.
Corrosion Science, 44 (8), 1669-1680.


















