
Enhancement of TGF-b Signaling Responses by...
13
Molecular and Cellular Pathobiology Enhancement of TGF-b Signaling Responses by the E3 Ubiquitin Ligase Arkadia Provides Tumor Suppression in Colorectal Cancer Vikas Sharma 1 , Anna G. Antonacopoulou 2 , Shinya Tanaka 1 , Alexios A. Panoutsopoulos 1 , Vasiliki Bravou 3 , Haralabos P. Kalofonos 2 , and Vasso Episkopou 1 Abstract TGF-b signaling provides tumor protection against colorectal cancer (CRC). Mechanisms that support its tumor-suppressive properties remain unclear. The ubiquitin ligase Arkadia/RNF111 enhances TGF-b signaling responses by targeting repressors of the pathway for degradation. The corepressors SnoN/Ski, critical substrates of Arkadia, complex with the activated TGF-b signaling effectors Smad2/3 (pSmad2/3) on the promoters of target genes and block their transcription. Arkadia degrades this complex including pSmad2/3 and unblocks the promoter. Here, we report that Arkadia is expressed highly in the mouse colonic epithelium. Heterozygous Akd þ/ mice are normal but express less Arkadia. This leads to reduced expression of several TGF-b target genes, suggesting that normal levels of Arkadia are required for efficient signaling responses. Critically, Akd þ/ mice exhibit increased susceptibility to azoxymethane/dextran sodium sulfate carcinogen–induced CRC, as they develop four-fold more tumors than wild-type mice. Akd þ/ tumors also exhibit a more aggressive pathology, higher proliferation index, and reduced cytostasis. Therefore, Arkadia functions as a tumor suppressor whose peak expression is required to suppress CRC development and progression. The accumulation of nuclear SnoN and pSmad2, along with the downregulation of TGF-b target genes observed in Akd þ/ colon and tumors, suggest that tumor-suppressing properties of Arkadia are mediated by its ability to derepress TGF-b signaling. Consistent with this likelihood, we identified mutations in primary colorectal tumors from human patients that reduce Arkadia function and are associated with the accumulation of nuclear SNON. Collectively, our findings reveal that Arkadia enhances TGF-b signaling responses and supports its tumor-suppressing properties in CRC. Cancer Res; 71(20); 6438–49. Ó2011 AACR. Introduction Colorectal cancer (CRC) is one of the most common malig- nancies worldwide and accounts for over 600,000 deaths each year (1). Mutations that inactivate TGF-b signaling compo- nents are associated with CRC, consistent with a role of this pathway in tumor suppression (2, 3). Specifically, mutation or deletion of the receptor genes TGFbR1 and TGFbR2, and the effector genes SMAD2, SMAD3, and SMAD4 of the pathway, have been reported in CRC (2, 4). Germ line mutations of SMAD4 are also associated with juvenile polyposis in the colon (5). In addition, mouse models in which these core signal transduction components are inactivated result in an increased susceptibility to develop adenocarcinomas under oncogenic stress (3). However, analyses of tumor samples have revealed that the TGF-b pathway also exhibits oncogenic properties in CRC, as it promotes survival, invasion, and metastasis (6). Mechanisms that impair TGF-b–mediated tumor suppression but do not abolish signaling may allow TGF-b–mediated metastasis to occur more frequently later in tumorigenesis. Regulators of such mechanisms could be used as early markers for susceptibility and prognosis and help in both the choice of drug treatment and in the development of novel anticancer agents. The TGF-b ligands, including TGF-b-1/2/3, Nodal and Activin, bind to specific cell surface receptors and activate, via phosphorylation, the Smad2/3 effectors (pSmad2/3). pSmads complex with Smad4 and translocate to the nucleus where they function as transcription factors (7, 8). Within the constantly regenerating colonic epithelium, the normal func- tion of the TGF-b pathway is to maintain homeostasis by counteracting proliferation and promoting differentiation and apoptosis (9). TGF-b factors specify cell fate and differentiation Authors' Affiliations: 1 Department of Experimental Medicine, Imperial College, Hammersmith Hospital Campus, London, United Kingdom; 2 Divi- sion of Oncology, Department of Medicine, University of Patras Medical School, Patras; and 3 Department of Anatomy-Histology-Embryology, Medical School, University of Patras, Rio, Greece Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). Corresponding Authors: Vasso Episkopou, Imperial College, Hammer- smith Hospital Campus, Burlington Danes 4th Floor, Du Cane Road, London W12 0NN, United Kingdom. Phone: 44-(0)20-7594-6587; Fax: 44-(0)20-7594-6548; E-mail: [email protected] and Vasiliki Bravou, Department of Anatomy-Histology-Embryology, Medical School, University of Patras, 26500, Rio, Greece; E-mail: [email protected] doi: 10.1158/0008-5472.CAN-11-1645 Ó2011 American Association for Cancer Research. Cancer Research Cancer Res; 71(20) October 15, 2011 6438 Cancer Research. on September 16, 2018. © 2011 American Association for cancerres.aacrjournals.org Downloaded from
Transcript of Enhancement of TGF-b Signaling Responses by...

Molecular and Cellular Pathobiology
Enhancement of TGF-b Signaling Responses by the E3Ubiquitin Ligase Arkadia Provides Tumor Suppression inColorectal Cancer
Vikas Sharma1, Anna G. Antonacopoulou2, Shinya Tanaka1, Alexios A. Panoutsopoulos1, Vasiliki Bravou3,Haralabos P. Kalofonos2, and Vasso Episkopou1
AbstractTGF-b signaling provides tumor protection against colorectal cancer (CRC). Mechanisms that support its
tumor-suppressive properties remain unclear. The ubiquitin ligase Arkadia/RNF111 enhances TGF-b signalingresponses by targeting repressors of the pathway for degradation. The corepressors SnoN/Ski, critical substratesof Arkadia, complex with the activated TGF-b signaling effectors Smad2/3 (pSmad2/3) on the promoters of targetgenes and block their transcription. Arkadia degrades this complex including pSmad2/3 and unblocks thepromoter. Here, we report thatArkadia is expressed highly in themouse colonic epithelium.HeterozygousAkdþ/�
mice are normal but express less Arkadia. This leads to reduced expression of several TGF-b target genes,suggesting that normal levels of Arkadia are required for efficient signaling responses. Critically, Akdþ/� miceexhibit increased susceptibility to azoxymethane/dextran sodium sulfate carcinogen–induced CRC, as theydevelop four-fold more tumors than wild-type mice. Akdþ/� tumors also exhibit a more aggressive pathology,higher proliferation index, and reduced cytostasis. Therefore, Arkadia functions as a tumor suppressor whosepeak expression is required to suppress CRC development and progression. The accumulation of nuclear SnoNand pSmad2, alongwith the downregulation of TGF-b target genes observed inAkdþ/� colon and tumors, suggestthat tumor-suppressing properties of Arkadia aremediated by its ability to derepress TGF-b signaling. Consistentwith this likelihood, we identified mutations in primary colorectal tumors from human patients that reduceArkadia function and are associated with the accumulation of nuclear SNON. Collectively, our findings reveal thatArkadia enhances TGF-b signaling responses and supports its tumor-suppressing properties in CRC. Cancer Res;71(20); 6438–49. �2011 AACR.
Introduction
Colorectal cancer (CRC) is one of the most common malig-nancies worldwide and accounts for over 600,000 deaths eachyear (1). Mutations that inactivate TGF-b signaling compo-nents are associated with CRC, consistent with a role of thispathway in tumor suppression (2, 3). Specifically, mutation ordeletion of the receptor genes TGFbR1 and TGFbR2, and theeffector genes SMAD2, SMAD3, and SMAD4 of the pathway,
have been reported in CRC (2, 4). Germ line mutations ofSMAD4 are also associated with juvenile polyposis in the colon(5). In addition, mouse models in which these core signaltransduction components are inactivated result in anincreased susceptibility to develop adenocarcinomas underoncogenic stress (3). However, analyses of tumor samples haverevealed that the TGF-b pathway also exhibits oncogenicproperties in CRC, as it promotes survival, invasion, andmetastasis (6). Mechanisms that impair TGF-b–mediatedtumor suppression but do not abolish signaling may allowTGF-b–mediated metastasis to occur more frequently later intumorigenesis. Regulators of such mechanisms could be usedas early markers for susceptibility and prognosis and help inboth the choice of drug treatment and in the development ofnovel anticancer agents.
The TGF-b ligands, including TGF-b-1/2/3, Nodal andActivin, bind to specific cell surface receptors and activate,via phosphorylation, the Smad2/3 effectors (pSmad2/3).pSmads complex with Smad4 and translocate to the nucleuswhere they function as transcription factors (7, 8). Within theconstantly regenerating colonic epithelium, the normal func-tion of the TGF-b pathway is to maintain homeostasis bycounteracting proliferation and promoting differentiation andapoptosis (9). TGF-b factors specify cell fate and differentiation
Authors' Affiliations: 1Department of Experimental Medicine, ImperialCollege, Hammersmith Hospital Campus, London, United Kingdom; 2Divi-sion of Oncology, Department of Medicine, University of Patras MedicalSchool, Patras; and 3Department of Anatomy-Histology-Embryology,Medical School, University of Patras, Rio, Greece
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Authors: Vasso Episkopou, Imperial College, Hammer-smith Hospital Campus, Burlington Danes 4th Floor, Du Cane Road,London W12 0NN, United Kingdom. Phone: 44-(0)20-7594-6587; Fax:44-(0)20-7594-6548; E-mail: [email protected] and VasilikiBravou, Department of Anatomy-Histology-Embryology, Medical School,University of Patras, 26500, Rio, Greece; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-11-1645
�2011 American Association for Cancer Research.
CancerResearch
Cancer Res; 71(20) October 15, 20116438
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

in a dose-dependent fashion during development (10). How-ever, reduction of TGF-b signaling in the adult colonic epithe-lium and its involvement in CRC is not clear.TGF-b signaling is tightly regulated by both extracellular and
intracellular mechanisms. Key intracellular regulators includethe inhibitors Smad6/7, which mediate the degradation ofreceptors and interfere with the phosphorylation of effectorSmads, and the nuclear corepressors SnoN and Ski, whichdirectly interact with the effector Smad proteins and recruit acorepressor complex containing histone deacetylase andnuclear receptor corepressor to targeted gene promoters toblock expression (11). Gene amplification and overexpressionof SMAD7 and SNON/SKI proteins have also been linked toCRC, attributed to loss of TGF-b signaling (12, 13). We previ-ously showed that Arkadia, a nuclear RING domain E3 ubiqui-tin ligase, is a positive regulator of the TGF-b/Nodal signalingbranch. It mediates the ubiquitin–proteasome degradation ofall the abovementioned negative regulators of the pathway andtherefore constitutes a potent "derepressor" that enhancesTGF-b target gene transcription (14–17). Degradation ofSnoN/Ski by Arkadia depends upon their specific interactionwith pSmad2/3 (15, 18, 19). As Arkadia also interacts anddegrades pSmad2/3, its function results in clearing the pro-moters from used/blocked effectors, thereby allowing fresheffectors to bind and activate transcription (20). Consistentwith this, absence of Arkadia results in increased levels ofstable SnoN/Ski and pSmad2/3, but as these are together in acomplex, the promoters of target genes are occupied by stable,repressed pSmad2/3 leading to repression (20). Arkadia isbroadly expressed in the mouse embryo and its absence leadsto loss of a subset of Nodal signaling responses necessary forthe development of anterior/head structures, which are alsolost with the genetic reduction of Nodal (20–22). However,whether Arkadia enhances TGF-b signaling responses in theadult colonic epithelium and how this affects CRC develop-ment is unknown.Using a deep sequencing screen of human Arkadia (AKD)
mRNA from tumors of patients with CRC, we identified soma-tic mutations that reduce AKD function. We showed thatreduction in Arkadia levels increased susceptibility to developCRC in a mouse model and showed that this mechanisminvolves increased stability of SnoN and pSmad2 and a reduc-tion of TGF-b–mediated target gene transcription. Collective-ly, our data reveal that Arkadia is required for peak efficiency ofa subset of TGF-b transcriptional responses in the colonicepithelium and in colorectal tumors and thereby supports thetumor-suppressive arm of this pathway.
Materials and Methods
Deep sequencingTotal RNA was extracted from formalin-fixed, paraffin-
embedded tumor and adjacent normal tissue sections aspreviously described (23) and reverse transcribed. Five to 6overlapping PCR amplifications spanning the 300-bp C-termi-nus of AKD were conducted per patient/tissue type; eachrepresenting an "amplicon library" (primer sequences in Sup-plementary Section). Preparation of DNA-carrying beads was
conducted as previously described (24). Beads were purifiedand loaded onto a 16-gasket picotiter plate for high-through-put pyrosequencing using the GS20 454-sequencer (Roche).Each amplicon library yielded an average of 4,000 sequencingreads. Data were analyzed as previously described (24). TheEnsembl entry ENST00000380504 was used as the referencesequence for AKD (corresponding to protein Q6ZNA4-1).
Plasmid construction and cell culture assaysSite-directed mutagenesis to generate the different point
mutations was conducted on a full-length human AKD cDNAclone (Invitrogen; ID: 30336202; see Supplementary Sectionfor primer details). Fragments were subsequently PCRamplified and cloned in-frame into the expression plasmidpTriex2-GFP as XhoI-digested products (20). Transfectionsand luciferase assays were conducted in HEK293T cells(Cancer Research UK) as previously described (15). Forimmunoblotting, cells were harvested 24 hours posttrans-fection and lysates were analyzed for green fluorescentproteins (GFP). The HEK293T cell line was authenticatedin July 2011 by the Health Protection Agency using shorttandem repeat multiloci genotyping.
ImmunohistochemistryImmunohistochemistry (IHC) was conducted as previously
described (25) using the following rabbit polyclonal antibodies:Arkadia (1:100; LifeSpan BioSciences, Inc.); SnoN, H-317 (1:80;Santa Cruz); and pSmad2 (1:50; Millipore). The ABC–DABdetection system was used (ABC detection kit, Pierce; DABsubstrate, Thermo Scientific). Blocking solution was usedinstead of primary antibodies for negative controls. Immuno-reactivity was graded on a scale of 0 to 3 (0, negative; 1, weak; 2,moderate; and 3, strong) according to intensity of staining andpercentage of immunopositive cells as previously described(26). All sections were counterstained with hematoxylin.
ImmunoblottingExperiments were carried out as described previously (20),
using the following rabbit polyclonal antibodies: GFP (1:1,000;Invitrogen); pSmad2 (1:500; Cell Signaling); SnoN, H-317(1:5,000; Santa Cruz); p21, C-19 (1:200; Santa Cruz); andHistoneH3 (H3), ab-1791 (1:10,000; Abcam). The following monoclonalantibodies were used: rabbit Smad2 (1:1000, Epitomics); mouseproliferating cell nuclear antigen (Pcna), PC-10 (1:3000, SantaCruz), and mouse active b-catenin (anti-b-catn), 8E7 (1:500,Millipore). Densitometric quantification of bands was con-ducted using ImageJ software.
Colorectal tumor induction protocol and histologicanalysis
Twenty-week old wild-type and Akdþ/� mice in a 129SVccinbred genetic background were injected with a single intra-peritoneal injection of the carcinogen azoxymethane (AOM; 7.4mg/kg; Sigma). After one week, mice were subjected to 2%dextran sodium sulfate (DSS; molecular weight: 36,000–50,000;MP Biomedical) in their drinking water for a period of 5 days.Two more cycles of the 5-day DSS treatment were given, eachseparated by a 16-day period on normal drinkingwater. Twelve
Arkadia Functions as a Tumor Suppressor in Colorectal Cancer
www.aacrjournals.org Cancer Res; 71(20) October 15, 2011 6439
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

weeks after the final DSS treatment, mice were sacrificed andtheir colons analyzed for tumors. This protocol was repeatedusing mice in a 129SVcc/CD1 hybrid background. Tumorcounts were made under a dissecting microscope. Colonswere fixed in 4% paraformaldehyde and paraffin embedded.X-gal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside)staining was conducted as previously described (21). Sec-tions of 4 mm thickness were either hematoxylin and eosinstained or used in IHC. Histologic analysis was conductedfor all wild-type tumors and at least 3 representative tumorsper Akdþ/� mouse.
Statistical analysisStatistical analysis was conducted using the SPSS for Win-
dows, release 12.0 (SPSS Inc.). x2 analysis was used to test thesignificance of differences in immunoreactivity scores or his-topathologic parameters between wild-type and Akdþ/�
tumors. All other data were analyzed using the Student t testand presented as mean � SEM. P< 0.05 was considered assignificant and denoted with a single asterisk. P� 0.01 and P�0.001 were denoted with 2 and 3 asterisks, respectively.
Results
Arkadia mutations in selected human colorectal tumorswith high SNON
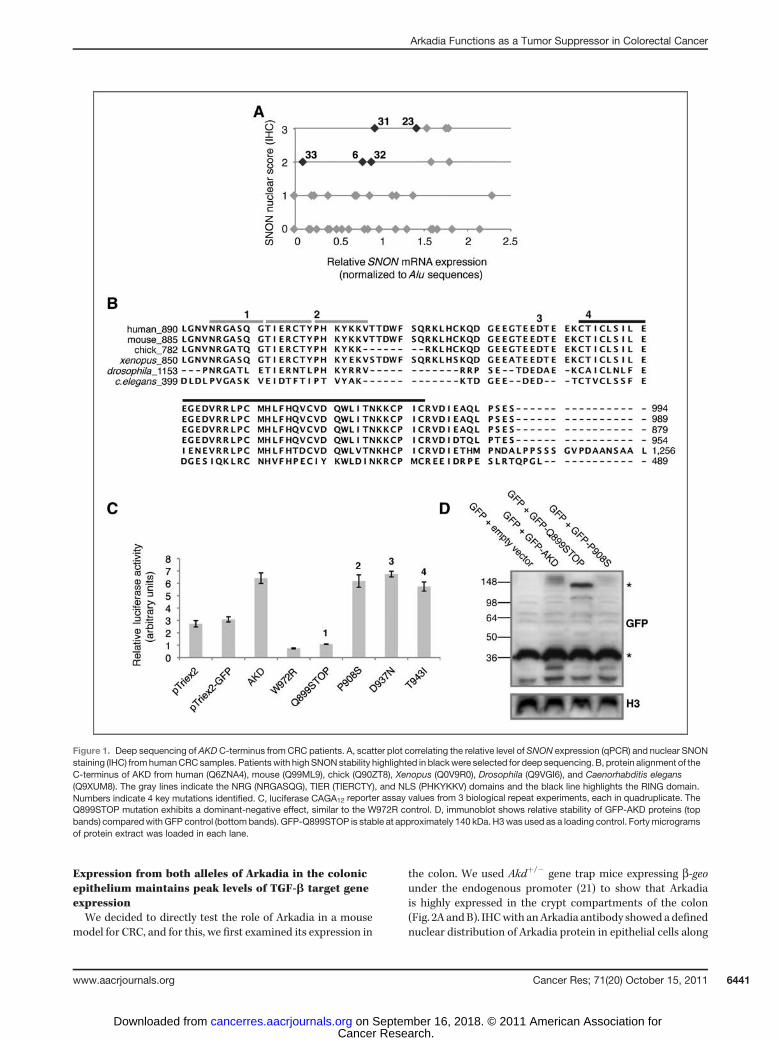
We recently described a cohort of primary human CRCs, ofwhich 83 of 87 (95.4%) overexpressed SNON (26). NuclearSNON expression was identified in 42 of 87 (48.3%) of thesetumors and, importantly, this correlatedwith advancing tumorgrade. Interestingly, we showed that SNON accumulation inthese tumors was not a result of elevated SNON mRNA,suggesting increased protein stability. Because SNON is asubstrate of AKD, itself a nuclear protein, it is possible thatinactivating mutations in AKD may account, in part, for thisstable nuclear SNON phenotype. To test this possibility, weconducted a deep sequencing screen for mutations in AKDmRNA extracted fromhumanCRCparaffin-embedded tumors.We selected 5 CRC patients with tumors displaying the highestlevels of nuclear SNON protein but a relatively low level ofSNON mRNA expression (Fig. 1A; ref. 26). Normal expressionlevels of AKD mRNA were present in these samples (data notshown).
We hypothesized that mutations which inactivate the ubi-quitin ligase activity of AKD, but do not disrupt substratebinding, would protect the substrates from ubiquitination andsubsequent degradation and therefore prevent wild-type AKDfunction. The domains that are required for the activity of AKDare located in the highly conservedC-terminal 100 amino acids.This region comprises the NRG, which is at the end of thedomain required for substrate (pSMAD2 and SNON/SKI) rec-ognition, followed by a conserved TIER domain (of unknownfunction), a nuclear localization signal (NLS), and a RINGdomain, required for the ubiquitin ligase activity (20). Inacti-vation of the RING domain or deletion of the TIER domain inArkadia converts its function to dominant-negative, as shownin luciferase reporter experiments carried out in HEK293Tcells using the SMAD-dependent reporter CAGA12-Luc
(Supplementary Fig. S1). Interestingly, the COSMIC databasehas catalogued 2 different point mutations at the C-terminal ofAKD from ovarian cancer (27). This supported our hypothesisthat mutations in the C-terminus are more likely to affect AKDfunction and led us to focus the mRNA sequencing screen onthe last 300 bp of AKD.
The paraffin-embedded tumor sections that we used forour sequencing analysis comprised a heterogeneous popu-lation of cells including both tumorigenic epithelial cells andnontumorigenic stromal cells such as fibroblasts and inflam-matory cells. In addition, not all cells exhibited an accumu-lation of nuclear SNON and therefore a potential mutation inAKD. Furthermore, as AKD mutations may arise from only 1allele in these cells, they are likely to be represented at a lowfrequency in the sample. To determine the true frequency ofsuch mutations in the mRNA (cDNA), we applied the GS20system to achieve thousands of bidirectional sequencingreads for the C-terminal of AKD from each sample (24).Mutations were identified in 2 of the 5 patients (patients 6and 32; Supplementary Table S1). Flowgram analysis wasconducted to validate these mutations (Supplementary Fig.S2). Table 1 summarizes 4 of these mutations identified inthe screen, each corresponding to a conserved amino acidresidue (Fig. 1B).
We tested the functionality of each of these mutations inHEK293T cells using the CAGA12-Luc reporter assay (Fig.1C). Overexpression of GFP-tagged AKD carrying the P908S,D937N, or T943I mutation enhanced reporter expression to asimilar level as that of wild-type GFP-AKD (which is >2-foldabove endogenous signaling, as assessed by overexpressionof empty vector controls). In contrast, the Q899STOP mutantreduced signaling 3-fold compared with controls (Fig. 1C),suggesting that it exhibits a dominant-negative functionsimilar to the control RING-W972R mutant AKD. Consistentwith our hypothesis, this mutation introduces a STOP codonat the end of the substrate recognition domain, resulting in atruncated protein lacking the enzymatic RING domain. TheP908S mutation was observed at the highest frequency(9.94%). As this is positioned within the NLS of AKD, weexamined the localization of its GFP-tagged form inHEK293T transfections but found it to be nuclear likewild-type AKD (data not shown).
We have previously shown that deletion of the RING orNRG stabilizes the normally unstable wild-type Arkadia(20). By coexpressing a GFP control plasmid with eachGFP-tagged AKD mutant in HEK293T cells, we observedan equal abundance of short GFP in all lysates followingimmunoblotting. However, only the GFP-Q899STOP AKDmutant gave a larger stable and visible band (Fig. 1D). Thestabilization of Q899STOP AKD most likely contributes toits ability to strongly suppress wild-type AKD function whenthis mutation occurs in 1 allele. Considering that only aproportion of tumor cells displayed stabilized nuclearSNON (26), combined with the basis of screening for sin-gle-allele mutations in a heterogeneous tumor sample, theobserved frequency of 2.76% for the Q899STOP mutationhas a good association to be causal to the accumulation ofSNON.
Sharma et al.
Cancer Res; 71(20) October 15, 2011 Cancer Research6440
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from
Expression from both alleles of Arkadia in the colonicepithelium maintains peak levels of TGF-b target geneexpressionWe decided to directly test the role of Arkadia in a mouse
model for CRC, and for this, we first examined its expression in
the colon. We used Akdþ/� gene trap mice expressing b-geounder the endogenous promoter (21) to show that Arkadiais highly expressed in the crypt compartments of the colon(Fig. 2A andB). IHCwith anArkadia antibody showed a definednuclear distribution of Arkadia protein in epithelial cells along
Figure 1. Deep sequencing of AKDC-terminus from CRC patients. A, scatter plot correlating the relative level of SNON expression (qPCR) and nuclear SNONstaining (IHC) fromhumanCRCsamples. Patientswith highSNONstability highlighted in blackwere selected for deep sequencing. B, protein alignment of theC-terminus of AKD from human (Q6ZNA4), mouse (Q99ML9), chick (Q90ZT8), Xenopus (Q0V9R0), Drosophila (Q9VGI6), and Caenorhabditis elegans(Q9XUM8). The gray lines indicate the NRG (NRGASQG), TIER (TIERCTY), and NLS (PHKYKKV) domains and the black line highlights the RING domain.Numbers indicate 4 key mutations identified. C, luciferase CAGA12 reporter assay values from 3 biological repeat experiments, each in quadruplicate. TheQ899STOP mutation exhibits a dominant-negative effect, similar to the W972R control. D, immunoblot shows relative stability of GFP-AKD proteins (topbands) comparedwith GFP control (bottom bands). GFP-Q899STOP is stable at approximately 140 kDa. H3was used as a loading control. Fortymicrogramsof protein extract was loaded in each lane.
Arkadia Functions as a Tumor Suppressor in Colorectal Cancer
www.aacrjournals.org Cancer Res; 71(20) October 15, 2011 6441
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

the crypt (Fig. 2C). Bothmethods showedweaker expression inthe surface epithelium. These expression data suggest a role forArkadia in the colonic epithelium, where TGF-b is involved inmaintaining tissue homeostasis.
The gene trap insertion terminates transcription of the Akdallele before the first exon (21) and quantitative PCR (qPCR)confirmed that Akdþ/� colons express half the level of Arkadiacompared with wild-type colons (Fig. 2D). Analysis of TGF-btarget gene expression revealed significant downregulationof genes such as p15INK4b, Pai-1 (Serpine1), Smad7, and Tmepai(Pmepa1) in the Akdþ/� colon, whereas targets, such as SnoNand p21WAF, showed no difference (Fig. 2E). Immunoblottingrevealed higher levels of pSmad2 in Akdþ/� than in wild-typecolon (Fig. 2F). We also conducted IHC for SnoN and observedhigher levels in Akdþ/� than in wild-type colon (Fig. 2G and H).These observations are consistent with the known molecularfunction of Arkadia to degrade SnoN–Smad complexes andenhance TGF-b target gene expression (14, 15, 20), as it does inembryos, embryonic stem, and other cell lines (14, 20).
Interestingly, the above data suggest that even a reductionof Arkadia function by the loss of 1 wild-type allele isassociated with the reduction of TGF-b signaling responses,which was not known before. Multipotent stem cells resideat the base of the crypts and give rise to proliferatingdaughter epithelial cells that migrate upward before finallyacquiring a differentiated epithelial phenotype at the surfacewith TGF-b signaling counteracting proliferation and main-taining homeostasis (28). Notably, the increase of SnoN inthe Akdþ/� colon occurs predominantly in the more differ-entiated cells of the surface epithelium (compare Fig. 2G andH). This suggests that the downregulation of TGF-b signalingresponses occurs within the epithelium and most likelyimpairs the maintenance of balance between proliferationand differentiation.
Akdþ/� mice exhibit increased susceptibility to CRCTo determine whether loss of 1 allele of Arkadia increases
susceptibility to develop tumors, we induced CRC usingthe AOM/DSS method (29, 30) in age-matched wild-type andAkdþ/� mice in 2 different genetic backgrounds (129SVccinbred and 129SVcc/CD1 hybrid) under a specific pathogen-free environment (Fig. 3A). Azoxymethane is a potent carcin-ogen that reproduces the activating mutations in b-cateninand KRAS (31, 32) and the inactivating mutations in adeno-matous polyposis coli (APC) that are observed in human CRC(33, 34). Crucially, 5 of 12 (42%) wild-type mice did not developany tumors throughout the duration of the protocol, whereas
all Akdþ/� mice developed tumors (Fig. 3B). Akdþ/� micedeveloped almost 4-fold the number of colorectal tumors aswild-type mice in a 129SVcc background (number of Akdþ/�
tumors, mean � SEM ¼ 7.6 � 1.0; P ¼ 0.0001; Fig. 3C, left).We found very similar results in the 129SVcc/CD1 background,with a near 3-fold increase in tumor multiplicity in Akdþ/�
mice (P ¼ 0.0053, Fig. 3C, right). These results show thatAkdþ/� mice are more susceptible to carcinogenesis and thatwild-type levels of Arkadia provide a protection against CRC.This supports the hypothesis that mutations that simplyreduce AKD function in patients may contribute to the devel-opment of CRC.
Enhanced tumor progression in Akdþ/� miceTo characterize the tumor pathology from wild-type and
Akdþ/� mice, all 23 tumors from wild-type mice and a total of24 tumors from Akdþ/�mice from a 129SVcc backgroundweresubjected to histopathologic analysis. The lesions identified inthe colon of treatedmice included tubular adenomas with low-or high-grade dysplasia, intramucosal carcinomas, and inva-sive carcinomas (Table 2). Although the majority of tumorsfrom wild-type mice developed to intramucosal carcinomas, asignificant proportion of tumors (13%) did not develop beyondadenomas with high-grade dysplasia (Fig. 3D–F). In contrast,all (24 of 24) Akdþ/� tumors analyzed developed to intramu-cosal carcinomas (Fig. 3G), with 1 Akdþ/� mouse even har-boring an invasive carcinoma (Fig. 3H). We also observed avessel invasion within a carcinoma from another Akdþ/�
mouse (Fig. 3I).The above analysis shows that lesions progress more rapidly
through the adenoma–carcinoma sequence in Akdþ/� mice,whichmay explainwhy a high proportion of wild-typemice didnot develop any gross tumors. Statistical analysis confirmedthat loss of 1 allele of Akd significantly correlated with theprogression of lesions from adenoma to intramucosal andinvasive carcinomas (P ¼ 0.03; Table 2).
Tumors from Akdþ/� mice exhibit reduced TGF-b–mediated cytostasis and increased proliferation
Wenext examined the tumors at amolecular level to identifythe mechanisms involved in their enhanced progression. Weanalyzed the expression of various genes by conductingimmunoblot and qPCR analysis on proteins and RNA extractedfrom 6 wild-type (T1–T6) and 6 Akdþ/� (TA–TF) tumors(Fig. 4A–C). Using the proliferationmarker Pcna, we found thattumors from Akdþ/� mice (hereafter termed Akdþ/� tumors)exhibited significantly higher levels of proliferation than
Table 1. Summary of the Q899STOP, P908S, D937N, and T943I mutations of AKD identified in the deepsequencing screen
Mutation Codon change Amino acid change Total frequency, % (reads) Patient
1 CAG>TAG Q899STOP 2.76 (4,167) 62 CCA>TCA P908S 9.94 (4,167) 63 GAC>AAC D937N 4.30 (3,833) 324 ACT>ATT T943I 1.59 (3,833),1.76 (5,282) 32,6
Sharma et al.
Cancer Res; 71(20) October 15, 2011 Cancer Research6442
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

tumors from wild-type mice (hereafter termed wild-typetumors; Fig. 4A).As activation of Wnt/b-catenin signaling is critically
involved in the proliferation of the regenerating colonic epi-thelium and in CRC, we examined whether differences in itsactivation underpinned this hyperproliferation. An antibodythat specifically detects the active nuclear form of b-catenin(35) showed no difference between Akdþ/� and wild-typetumors (Fig. 4A). Furthermore, the expression of the Wnt/
b-catenin target oncogene c-Myc was also not significantlydifferent between Akdþ/� and wild-type tumors (Fig. 4C).These data suggest that an alternative mechanism underliesthe more aggressive pathology observed in Akdþ/� tumors.
As reduction in cytostasis can also lead to hyperproliferationand as this critically depends on TGF-b signaling in the colonicepithelium, we examined the levels of the p21WAF cyclin-dependent kinase inhibitor in tumors (36). Indeed, this factorwas significantly lower in all 6Akdþ/� tumors and in 2 of 6wild-
Figure 2. Expression of Arkadia inthemouse colon. A andB,wild-type(wt) (A) and Akdþ/� (B) colonsections stained with X-gal. C, anti-Arkadia IHC in wild-type colon. Dand E, mRNA expression of Akd (D)and 5 TGF-b target genes (E) in wtand Akdþ/� colon tissue asdetermined by qPCR. F,immunoblot with pSmad2 antibodyand H3 loading control. G and H,expression of SnoN in (G) wild-typeand (H) Akdþ/� colon tissue.Elevated SnoN staining is observedin Akdþ/� colons, particularly in thedifferentiated epithelial cells at thesurface of the crypts. All images areshown at 40� magnification; scalebars, 25 mm.
Arkadia Functions as a Tumor Suppressor in Colorectal Cancer
www.aacrjournals.org Cancer Res; 71(20) October 15, 2011 6443
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

type tumors (Fig. 4B). However, we did not observe a corre-sponding decrease in mRNA expression of p21WAF in Akdþ/�
tumors (Fig. 4C), suggesting a possible reduction in p21WAF
protein stability in these tumors. In contrast, transcription ofthe cytostatic target p15INK4b was significantly reduced inAkdþ/� tumors (Fig. 4C). This reduction in p15INK4b expressionpreexists in the colon of untreated Akdþ/� mice (Fig. 2E).Together, the data suggest that reduced cytostasis is respon-sible for the hyperproliferation and enhanced progression ofAkdþ/� tumors.
Furthermore, examination of the levels of Arkadia's sub-strate SnoN showed that it is elevated in tumors and adjacentnormal colon of Akdþ/�mice compared to wild-typemice (Fig.4A and B). SnoN mRNA expression, however, was not signif-icantly different between Akdþ/� and wild-type tumors (Fig.4C), suggesting an increased stability of the SnoN protein in
Akdþ/� tumors. Consistent with the molecular function ofArkadia, pSmad2, which complexes with SnoN and is alsodegraded by Arkadia, was also increased in Akdþ/� tumors(Fig. 4A and B). As the expression of the TGF-b target genePAI-1, like p15INK4b, was also reduced in Akdþ/� tumors(Fig. 4C), collectively, the above data strongly suggest thatrepression of the pathway is increased because of the reductionor loss of Arkadia from cells of Akdþ/� tumors.
Enhanced tumor progression in Akdþ/� mice isassociated with reduction and not loss of Arkadiafunction
To address the distribution and level of TGF-b pathwayrepression in tumors, we conducted IHC to visualize SnoN andpSmad2 proteins in 6 Akdþ/� tumors and 4 wild-type tumors.We found that pSmad2 stainingwas stronger inAkdþ/� tumors
Figure 3. AOM/DSS induction ofCRC in wild-type (wt) and Akdþ/�
mice. A, macroscopic view ofrepresentative X-gal–stainedmouse colons (129SVccbackground) at end of protocol(distal end at top; bar, 1 cm). B,percentage of mice that developedtumors. C, mean number of tumorsobserved from wild-type (n ¼ 12)and Akdþ/� (n ¼ 7) mice in a129SVcc background and wild-type (n¼ 6) and Akdþ/� (n¼ 6) micein a 129SVcc/CD1 background. D–I, hematoxylin and eosin staining ofcolons and tumors from treatedwild-type and Akdþ/� mice. Wild-type: D, normal colon; E, adenomawith severe dysplasia; and F,intramucosal carcinoma. Akdþ/�:G, intramucosal carcinoma withmucus production; H, invasiveadenocarcinoma; and I, vesselinvasion. Black arrowheads marksites of invasion. D–H, 2.5�magnification; scale bars, 250 mm.I, 20� magnification; scale bars,50 mm.
Sharma et al.
Cancer Res; 71(20) October 15, 2011 Cancer Research6444
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

(score 2 or 3 in 4 of 6 tumors) than in wild-type tumors (score 1in 4 of 4 tumors; Fig. 5A and B). This difference, however, didnot reach statistical significance, most likely because pSmad2levels also depend on the amount of active ligand that eachtumor expresses, which may vary between tumors. There wasno difference in the cellular localization of pSmad2 inwild-typeand Akdþ/� tumors, as both were nuclear. In contrast, SnoNlevels in Akdþ/� tumors were classified as either moderate(score 2 in 2 of 6 tumors) or strong (score 3 in 4 of 6 tumors),whereas expression in wild-type tumors was frequently weak(score 1 in 3 of 4 tumors). This difference was statisticallysignificant (x2; P ¼ 0.036). Notably, SnoN cellular localizationwas cytoplasmic inwild-type tumors (3 of 4 tumors) and largelynuclear in Akdþ/� tumors (Fig. 5C and D). Both SnoN andpSmad2were found to benuclear in the cells that constitute thelining of the tubular structures of the adenocarcinoma fromAkdþ/� mice, suggesting increased repression of TGF-b sig-naling in these cells (Fig. 5B and D).To examine whether complete loss of Arkadia expression is
associated with the enhanced progression of Akdþ/� tumors,we conducted qPCR on wild-type and Akdþ/� tumors andfound that Akdþ/� tumors consistently express Akd but at onlyhalf the level of wild-type tumors (Fig. 4C). To determinewhether this reduction is an average expression of all tumorcells rather than complete loss in a subset of cells, we con-ducted IHC for Arkadia in tumors (this antibody is raisedagainst the C-terminus of AKD and is expected to recognize
functional Arkadia proteins). We observed the presence ofnuclear Arkadia protein in almost every cell lining the tubularstructures of tumors from both genotypes (Fig. 5E and F), evenin the most aggressive Akdþ/� tumors (Fig. 5F shows Arkadiastaining on the same invasive adenocarcinoma shown in Fig.3H). Therefore, even in the most advanced tumors, Arkadia isnot lost and this therefore cannot account for the enhancedgrowth of Akdþ/� tumors. Collectively, the above data suggestthat reduction of Arkadia rather than loss is responsible for theenhanced tumor progression.
Our findings also mirror reports that Smad4 and Tgfbr1haploinsufficiency promotes CRC development in a geneticbackground carrying Apc mutations (37–40), which alonecauses adenomas (37, 41). Together, these studies emphasizethat gene dosage of important components of the TGF-bpathway acts as key determinants in CRC susceptibility andsupports the hypothesis that the tumor-suppressing propertiesof this pathway depend on its peak levels. Furthermore, TGF-bsignaling responses and cytostasis are reduced inAkdþ/�mice,suggesting that Arkadia's tumor-suppressing properties inboth the normal colonic epithelium and colorectal tumors aremediated by the enhancement of this pathway.
Discussion
TGF-b signaling exerts an antitumorigenic function in thecolonic epithelium, whereas in advanced tumors, it can
Table 2. Histopathology of wild-type and Akdþ/� colorectal tumors
Mouse Tumors Tumors analyzed Adenomas Intramucosal carcinomas Invasive carcinomas
wt_1 5 5 — 5 —
wt_2 1 1 — 1 —
wt_3 2 2 — 2 —
wt_4 3 3 1 2 —
wt_5 3 3 — 3 —
wt_6 — — n/a n/a n/awt_7 — — n/a n/a n/awt_8 — — n/a n/a n/awt_9 — — n/a n/a n/awt_10 — — n/a n/a n/awt_11 6 6 2 4 —
wt_12 3 3 — 3 —
Total 23 23 3 (13%) 20 (87%) —
Akdþ/�_1 10 3 — 3 —
Akdþ/�_2 9 3 — 3VI —
Akdþ/�_3 9 3 — 2 1Akdþ/�_4 3 3 — 3 —
Akdþ/�_5 5 3 — 3 —
Akdþ/�_6 7 4 — 4 —
Akdþ/�_7 10 5 — 5 —
Total 53 24 — 23 (96%) 1 (4%)
NOTE: Tumors fromall wild-type (n¼ 12) and allAkdþ/� (n¼ 7)mice (129SVccbackground)were subjected to histopathologic analysis.Colons were examined along the entire proximal-distal axis.Abbreviations: n/a, not applicable; VI, vessel invasion (found in 1 tumor).
Arkadia Functions as a Tumor Suppressor in Colorectal Cancer
www.aacrjournals.org Cancer Res; 71(20) October 15, 2011 6445
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

promote metastasis and progression. What regulates thesedifferent downstream TGF-b signaling effects remains largelyunknown. In this study, we provide evidence that the E3ubiquitin ligase Arkadia, whose major function is to enhanceTGF-b signaling by degrading negative regulators of the path-way, exhibits tumor-suppressing properties in CRC (Fig. 3and Table 2). We found that 2 wild-type alleles of Arkadia arerequired to maintain sufficiently low levels of these negativeregulators (and therefore a high activity of the TGF-b pathway)and lower the susceptibility for CRC development and pro-gression under oncogenic stress in mice (Figs. 4 and 5). We didnot find evidence of complete loss of Arkadia in invasivetumors from Akdþ/�mice, suggesting that the tumor-suppres-sing properties of TGF-b signaling depend on enhanced down-stream responses. Furthermore, we screened for mutationsthat lower AKD function in patients with CRC exhibitingstabilization of SNON (26), a key repressor and substrate forAKD ubiquitination and degradation. We found several pointmutations in the C-terminal functional domains of AKD thatmay account for the nuclear accumulation of SNON (Fig. 1and Table 1). As this is associated with amore advanced tumorgrade, AKD emerges as a likely tumor suppressor in CRC.
In the regenerating colonic epithelium, homeostasis isthought to be maintained by the Wnt/b-catenin pathwaythat promotes proliferation of epithelial cells (and thereforeexhibits oncogenic potential) and the TGF-b pathway,which promotes differentiation, cytostasis, and apoptosis(tumor-suppressive properties; ref. 9). Mutations in Wnt/b-catenin signaling, such as the constitutive activation ofb-catenin or the inactivation of APC that regulates b-cate-nin activity, lead to hyperproliferation and adenoma for-mation. However, for progression to CRC, it is clear thatthere is a requirement for cooperation with other pathways(3, 28, 42). Mutations that inactivate TGF-b signaling havebeen shown to contribute to CRC progression (37–39, 41).However, whereas activation of Wnt/b-catenin can be gen-erated in a single hit (i.e., 1 allele of either APC or b-catenin),inactivation of a component of the TGF-b pathway usuallyhas to occur in both alleles in the same cell. This is a veryrare event and most likely occurs late after the accumula-tion of mutations that increase genomic instability. Geneamplification of negative regulators of the TGF-b pathway,such as SMAD6/7 and SNON/SKI, has been found in CRC(12, 13). However, amplification rarely occurs early enough
Figure 4. Immunoblot analysis ofwild-type (wt) and Akdþ/� tumors.A, immunoblotting with SnoN,pSmad2, active b-catenin (b-catn),and Pcna. N, treated adjacentnormal colon tissue; T,representative tumor samples fromwild-type (T1–T3) and Akdþ/�(TA–TC) mice. H3 was used as aloading control. Protein levelsnormalized to H3 are shown in the 3charts. B, analysis of p21WAF,SnoN, and pSmad2 protein levels in6 wild-type (T1–T6) and 6 Akdþ/�
(TA–TF) tumors. Ten micrograms ofprotein extract was loaded in eachlane for all gels. Because of theenhanced stability of pSmad2 inAkdþ/� tumors, we also observedmore stable levels of total Smad2.C, mRNA expression levels ofvarious genes (as indicated) in theabove analyzed tumors, asdetermined by qPCR. Average foldchanges are shown in the charts onthe right.
Sharma et al.
Cancer Res; 71(20) October 15, 2011 Cancer Research6446
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

to correspond to a tumor-promoting event, nor have singlemutations leading to the overexpression of these factorsbeen described.In contrast, as Arkadia mediates the degradation of all the
above negative regulators, and not only that of SNON, thesimple reduction of its function by pointmutationsmay lead torepression of TGF-b signaling responses and constitute anearly tumor-promoting event. This is supported in this study bythe identification of several missense mutations in AKD fromCRC patients with stabilized nuclear SNON. One mutation, inparticular Q899STOP, introduces a stop codon at the C-ter-minus that eliminates the final 100 amino acids harboring theubiquitin ligase activity, but preserves the substrate recogni-tion domains (Fig. 1B). This truncated AKD is more stable (Fig.1D), binds to the substrates, and protects them from ubiqui-tination by wild-type AKD (Fig. 1C). In fact, a mutation thatintroduces a stop codon anywhere in the C-terminus betweenresidues 889 and 978 (essential for enzymatic activity; Fig. 1B)would be expected to disrupt AKD function and exhibit adominant-negative effect. Other missense mutations withinthis highly conserved regionmay also inactivate the enzymaticactivity of Arkadia and result in a dominant-negative effect.The COSMIC database of somatic mutations in cancer reports2 missense mutations in AKD (R904S and K915I), both in
heterozygosity from primary ovarian tumors (27). The R904Smutation is located in the center of the TIER domain (Fig. 1B).Deletion of the TIER domain results in a dominant-negativeformof Arkadia (Supplementary Fig. S1), suggesting that R904SAKD may also act in this fashion. Therefore, each AKD alleleexhibits a large region sensitive to single-hit mutations, whichcould act as an early event in the progression of adenomas toCRC and perhaps in other tumors that depend onTGF-b tumorsuppression.
In our carcinogenesis model, both wild-type and Akdþ/�
tumors displayed a similar level of nuclear b-catenin. How-ever, only Akdþ/� tumors showed nuclear accumulation ofSnoN and pSmad2 (Fig. 5), suggesting that repression ofTGF-b signaling by SnoN, and most likely Ski (not examinedin this study), is associated with susceptibility to CRCdevelopment and increased progression. In human CRCs,we also reported a correlation between nuclear SNON/SKIaccumulation and nuclear b-catenin (26). This suggests thattumors with Wnt/b-catenin activation, combined withmutations that increase SNON, lead to a more aggressiveCRC pathology. Therefore, as mutations inactivating just 1allele of AKD can lead to stable nuclear SNON, combinationwith nuclear b-catenin may provide an early prognosticmarker.
Figure 5. IHC for SnoN, pSmad2,and Arkadia in wild-type (wt) andAkdþ/� tumors. Representativesections of adenocarcinomasstained with anti-pSmad2: (A) wild-type and (B) Akdþ/�; anti-SnoN: (C)wild-type and (D) Akdþ/�; andanti-Arkadia: (E) wild-type and (F)Akdþ/�. All images are shown at40� magnification; scale bars,25 mm.
A
C
E F
B
D
Arkadia Functions as a Tumor Suppressor in Colorectal Cancer
www.aacrjournals.org Cancer Res; 71(20) October 15, 2011 6447
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

In this study, we have shown a strong association betweensusceptibility to CRC and repression of TGF-b signalingresponses caused by loss of 1 allele of Arkadia in mice anda dominant-negative functioning mutant of AKD from ahuman CRC patient. Interestingly, in Akdþ/� colons andtumors, expression of the TGF-b target genes p21WAF andSnoN are not obviously affected, whereas p15INK4b, Pai-1,Tmepai, and Smad7 are reduced (Figs. 2E and 4C). Similarly,in Akd-null embryos and embryonic stem cells, not all TGF-b/Nodal target genes show equally reduced expression levels(20). Most likely SnoN/Ski do not repress all target genepromoters, suggesting that their targets are involved in thetumor-suppressing function of TGF-b signaling. As muta-tions in AKD do not completely inactivate all TGF-b signal-ing responses, this will increase the risk of TGF-b–mediatedmetastasis later, which most likely involves a differentsubset of target genes. Collectively, our data suggest thatsomatic mutations in a single AKD allele that reduce itsfunction could act as an early tumor-promoting event inhuman CRC development and progression.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Drs. Laurence Game, Mick Jones, and Nikolaos Karoulias,Nathalie Lambie and Frederique Maheo for assistance with the deep sequencingexperiments; Steve Bottoms and Dr. Tammy Kalber for help with histology; andDr. Jes�us Gil and Prof. Robin-Lovell Badge for critical reading of the manuscript.
Grant Support
The study was supported by Cancer Research UK grant number: A9066; MRCCore Funding; Greek Ministry of Education. Financial support was provided byCancer Research UK (V. Sharma), Greek Ministry of Education (A.G. Antona-copoulou, V. Bravou, and H.P. Kalofonos), and Medical Research Council(S. Tanaka, A.A. Panoutsopoulos, and V. Episkopou).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received May 12, 2011; revised July 21, 2011; accepted August 19, 2011;published online October 13, 2011.
References1. Gellad ZF, Provenzale D. Colorectal cancer: national and international
perspective on the burden of disease and public health impact.Gastroenterology 2010;138:2177–90.
2. Levy L, Hill CS. Alterations in components of the TGF-beta superfamilysignaling pathways in human cancer. Cytokine Growth Factor Rev2006;17:41–58.
3. Xu Y, Pasche B. TGF-beta signaling alterations and susceptibility tocolorectal cancer. Hum Mol Genet 2007;16:R14–20.
4. Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectalcancer: genetics of development and metastasis. J Gastroenterol2006;41:185–92.
5. Howe JR, Roth S, Ringold JC, Summers RW, Jarvinen HJ, Sistonen P,et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis.Science 1998;280:1086–8.
6. IkushimaH,Miyazono K. TGFbeta signalling: a complex web in cancerprogression. Nat Rev Cancer 2010;10:415–24.
7. Massague J, Seoane J, Wotton D. Smad transcription factors. GenesDev 2005;19:2783–810.
8. Feng XH, Derynck R. Specificity and versatility in tgf-beta signalingthrough Smads. Annu Rev Cell Dev Biol 2005;21:659–93.
9. Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal devel-opment and cancer. Annu Rev Cell Dev Biol 2004;20:695–723.
10. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic devel-opment and homeostasis. Dev Cell 2009;16:329–43.
11. Moustakas A, Heldin CH. The regulation of TGFbeta signal transduc-tion. Development 2009;136:3699–714.
12. Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L, et al.SMAD7 is a prognostic marker in patients with colorectal cancer. Int JCancer 2003;104:446–9.
13. Deheuninck J, LuoK.Ski andSnoN, potent negative regulators of TGF-beta signaling. Cell Res 2009;19:47–57.
14. Nagano Y, Mavrakis KJ, Lee KL, Fujii T, Koinuma D, Sase H, et al.Arkadia induces degradation of SnoN and c-Ski to enhance trans-forming growth factor-beta signaling. J Biol Chem 2007;282:20492–501.
15. Levy L, Howell M, Das D, Harkin S, Episkopou V, Hill CS. Arkadiaactivates Smad3/Smad4-dependent transcription by triggeringsignal-induced SnoN degradation. Mol Cell Biol 2007;27:6068–83.
16. Koinuma D, Shinozaki M, Komuro A, Goto K, Saitoh M, Hanyu A, et al.Arkadia amplifies TGF-beta superfamily signalling through degrada-tion of Smad7. EMBO J 2003;22:6458–70.
17. Liu W, Rui H, Wang J, Lin S, He Y, Chen M, et al. Axin is a scaffoldprotein in TGF-beta signaling that promotes degradation of Smad7 byArkadia. EMBO J 2006;25:1646–58.
18. LeScolanE,ZhuQ,WangL,BandyopadhyayA, JavelaudD,Mauviel A,et al. Transforming growth factor-beta suppresses the ability of Ski toinhibit tumor metastasis by inducing its degradation. Cancer Res2008;68:3277–85.
19. Nagano Y, Koinuma D, Miyazawa K, Miyazono K. Context-dependentregulation of the expression of c-Ski protein by Arkadia in humancancer cells. J Biochem 2010;147:545–54.
20. Mavrakis KJ, Andrew RL, Lee KL, Petropoulou C, Dixon JE, Nava-ratnam N, et al. Arkadia enhances Nodal/TGF-beta signaling bycoupling phospho-Smad2/3 activity and turnover. PLoS Biol2007;5:e67.
21. Episkopou V, Arkell R, Timmons PM, Walsh JJ, Andrew RL, Swan D.Induction of the mammalian node requires Arkadia function in theextraembryonic lineages. Nature 2001;410:825–30.
22. Niederlander C, Walsh JJ, Episkopou V, Jones CM. Arkadia enhancesnodal-related signalling to induce mesendoderm. Nature 2001;410:830–4.
23. Taron M, Rosell R, Felip E, Mendez P, Souglakos J, Ronco MS, et al.BRCA1mRNA expression levels as an indicator of chemoresistance inlung cancer. Hum Mol Genet 2004;13:2443–9.
24. ThomasRK, Nickerson E, Simons JF, Janne PA, Tengs T, Yuza Y, et al.Sensitive mutation detection in heterogeneous cancer specimens bymassively parallel picoliter reactor sequencing. Nat Med 2006;12:852–5.
25. Bravou V, Klironomos G, Papadaki E, Taraviras S, Varakis J. ILK over-expression in human colon cancer progression correlates with acti-vationof beta-catenin, down-regulationof E-cadherin andactivationofthe Akt-FKHR pathway. J Pathol 2006;208:91–9.
26. Bravou V, Antonacopoulou A, Papadaki H, Floratou K, StavropoulosM, Episkopou V, et al. TGF-beta repressors SnoN and Ski are impli-cated in human colorectal carcinogenesis. Cell Oncol 2009;31:41–51.
27. Wellcome Trust Sanger Institute. Available from: http://www.sanger.ac.uk/search?db¼cosmic&t¼RNF111 [cited 2011 Sept 20].
28. Clevers H. Wnt/beta-catenin signaling in development and disease.Cell 2006;127:469–80.
29. Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. Theforkhead box M1 transcription factor contributes to the develop-ment and growth of mouse colorectal cancer. Gastroenterology2007;132:1420–31.
Sharma et al.
Cancer Res; 71(20) October 15, 2011 Cancer Research6448
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

30. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novelinflammation-related mouse colon carcinogenesis model induced byazoxymethane and dextran sodium sulfate. Cancer Sci 2003;94:965–73.
31. Erdman SH, Wu HD, Hixson LJ, Ahnen DJ, Gerner EW. Assessment ofmutations in Ki-ras and p53 in colon cancers from azoxymethane- anddimethylhydrazine-treated rats. Mol Carcinog 1997;19:137–44.
32. Takahashi M, Nakatsugi S, Sugimura T, Wakabayashi K. Frequentmutations of the beta-catenin gene in mouse colon tumors induced byazoxymethane. Carcinogenesis 2000;21:1117–20.
33. Mollersen L, Paulsen JE, Alexander J. Loss of heterozygosity andnonsense mutation in Apc in azoxymethane-induced colonic tumoursin min mice. Anticancer Res 2004;24:2595–9.
34. Maltzman T, Whittington J, Driggers L, Stephens J, Ahnen D. AOM-induced mouse colon tumors do not express full-length APC protein.Carcinogenesis 1997;18:2435–9.
35. van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wntsignaling controls the phosphorylation status of beta-catenin. J BiolChem 2002;277:17901–5.
36. Moustakas A, Kardassis D. Regulation of the human p21/WAF1/Cip1promoter in hepatic cells by functional interactions between Sp1 andSmad family members. Proc Natl Acad Sci U S A 1998;95:6733–8.
37. Alberici P, Jagmohan-Changur S, De Pater E, VanDer ValkM, Smits R,Hohenstein P, et al. Smad4 haploinsufficiency in mouse models forintestinal cancer. Oncogene 2006;25:1841–51.
38. Hohenstein P, Molenaar L, Elsinga J,Morreau H, van der Klift H, StruijkA, et al. Serrated adenomas and mixed polyposis caused by a spliceacceptor deletion in the mouse Smad4 gene. Genes ChromosomesCancer 2003;36:273–82.
39. Taketo MM, Takaku K. Gastrointestinal tumorigenesis in Smad4(Dpc4) mutant mice. Hum Cell 2000;13:85–95.
40. Zeng Q, Phukan S, Xu Y, Sadim M, Rosman DS, Pennison M, et al.Tgfbr1 haploinsufficiency is a potent modifier of colorectal cancerdevelopment. Cancer Res 2009;69:678–86.
41. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM.Intestinal tumorigenesis in compound mutant mice of both Dpc4(Smad4) and Apc genes. Cell 1998;92:645–56.
42. Massague J. TGFbeta in cancer. Cell 2008;134:215–30.
Arkadia Functions as a Tumor Suppressor in Colorectal Cancer
www.aacrjournals.org Cancer Res; 71(20) October 15, 2011 6449
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from

2011;71:6438-6449. Cancer Res Vikas Sharma, Anna G. Antonacopoulou, Shinya Tanaka, et al. Colorectal CancerUbiquitin Ligase Arkadia Provides Tumor Suppression in
Signaling Responses by the E3βEnhancement of TGF-
Updated version
http://cancerres.aacrjournals.org/content/71/20/6438
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2011/10/11/0008-5472.CAN-11-1645.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/71/20/6438.full#ref-list-1
This article cites 41 articles, 11 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/71/20/6438.full#related-urls
This article has been cited by 5 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/71/20/6438To request permission to re-use all or part of this article, use this link
Cancer Research. on September 16, 2018. © 2011 American Association forcancerres.aacrjournals.org Downloaded from












![Research Paper TGF-β and NF-κB signaling pathway crosstalk ... · expression via a Smad3-mediated signaling pathway [22]. Increased levels of TGF-β have been reported in several](https://static.fdocuments.net/doc/165x107/5dd0c05cd6be591ccb6284f7/research-paper-tgf-and-nf-b-signaling-pathway-crosstalk-expression-via.jpg)





