
DNA Functional Groups Required for Formation of Open Complexes ...

DEVELOPMENT OF FUNCTIONAL DNA-BASED SENSORS AND
INVESTIGATIONS INTO THEIR MECHANISM
BY
NANDINI NAGRAJ
DISSERTATION
Submitted in partial fulfillment of the requirements
for the degree of Doctor of Philosophy in Chemistry
in the Graduate College of the
University of Illinois at Urbana-Champaign, 2010
Urbana, Illinois
Doctoral Committee:
Professor Yi Lu, Chair
Associate Professor Scott Silverman
Professor Kenneth Suslick
Professor Jonathan Sweedler

ii
ABSTRACT
The discovery that nucleic acids could perform functional roles in addition to being
genetic materials carriers opened doors to a new paradigm in nucleic acid chemistry. Catalytic
DNA molecules also known as deoxyribozymes or DNAzymes were first isolated in 1994
through an in vitro selection procedure and have since been engineered and isolated to perform
various functions that include both RNA and DNA cleavage and ligation. The 8-17 DNAzyme is
an RNA-cleaving DNAzyme that has shown high selectivity for Pb2+
under different selection
conditions. It has been explored extensively in terms of its applications for bio-sensing as well as
for exploring its mechanism from a more fundamental perspective.
A critical barrier of DNA-based sensors for practical applications, such as environmental
monitoring, is their highly variable sensing performance with changing temperatures, due to the
reliance of sensor design on temperature-dependent hybridization. In Chapter 2, this issue has
been addressed through the introduction of mismatches in the DNA hybridization arms of this
Pb2+
-specific 8-17 DNAzyme and these fluorescent sensors can resist temperature-dependent
variations from 4 °C to 30 °C. The strategy of using mismatches to tune the temperature
dependence is a novel and inexpensive method that can be applied in other nucleic acid sensors
for either metal ions or other molecular targets.
Currently there is no structure, (either X-ray or NMR) available for the 8-17 DNAzyme.
Hence, understanding its mechanism has posed a challenge, particularly in regard to the high
selectivity of Pb2+
for this DNAzyme. In Chapter 3, the systematic activity, folding and structural
studies of the 8-17 DNAzyme with both monovalent and divalent metal ions has been carried
out. The results obtained suggest a clear trend between the folding and activity of all the metal

iii
ions studied, the lower the activity, the lesser the folding and vice-versa. Structural studies based
on CD and folding studies based on FRET have demonstrated that Pb2+
behaves in a manner that
is different from other metal ions and hence it is hypothesized that the 8-17 DNAzyme may have
a specific binding pocket for Pb2+
.
The possibility that the 8-17 DNAzyme might have a metal ion binding pocket for Pb2+
has been investigated in Chapter 4, through systematic phosphorothioate (PS) modifications on
the backbone of the DNAzyme. Kinetic assays with the PS modified bases have shown that there
are specific bases on the enzyme strand which are important for activity mainly in the presence
of Pb2+
. The activities of the identical PS modified enzymes are, however, not significantly
altered in the presence of Mg2+
and Cd2+
. 31
P NMR has been used as an additional tool to directly
visualize the backbone phosphates since a single PS modification shifts the signal of the
phosphate downfield by ~50 ppm. These results, in conjunction, have led to the identification of
a proposed metal ion binding site, specifically a potential Pb2+
-binding site for the 8-17
DNAzyme.
The starting point towards the development of a successful functional nucleic acid-based
sensor is its isolation and this is done through in vitro selection. In vitro selection to isolate
DNAzymes for Hg2+
and Cd2+
and the use of negative selections to overcome Pb2+
interference
at various stages of selection has been described in Chapter 5. Chapter 6 describes the structure-
switching strategy to isolate DNA aptamers specific for endotoxins. It is anticipated that the
results obtained from the current study and future characterizations will lead to the development
of functional DNA sensors for endotoxins.

iv
Dedicated to my family who have given me their unwavering love and support through
everything and who have been the driving force through all the ups and downs.

v
ACKNOWLEDGEMENTS
I would like to firstly acknowledge my research advisor, Prof. Yi Lu for guiding me
through this journey and giving me the freedom to experiment and learn, and providing
encouragement when everything seemed to be stumbling along the way. I also would like to
thank my committee members Prof. Scott Silverman, Prof. Ken Suslick and Prof. Jonathan
Sweedler for their helpful suggestions, particularly during my preliminary examination. Their
suggestions and comments added great value to my research and thesis. I would also like to
particularly acknowledge the guidance and suggestions provided by Prof. Scott Silverman and
his group members, Dr. Elizabeth Pratico and Dr. Dana Baum during trouble-shooting in vitro
selection procedures. I also want to thank my collaborator from the Illinois Sustainable
technologies Center, Jennifer Deluhery for working with me on the selection of endotoxin
aptamers and bringing both her enthusiasm and passion on the table.
The secretaries in the IMP office, Connie Knight, Beth Myler, Teresa Struss and Sandy
Pijanowski, have really made life extremely smooth for graduate students with their reminders
and virtual take-over of all the formalities, and they have really kept us well-fed as well with
their wonderful treats and treat-days every month. I also want to acknowledge Dot Gordon who
has been a great resource and „go-to‟ person for graduate students and Joyce Beasley for all their
help over the years.
The Lu lab has been like family and many relationships, both professional and personal
that have been formed here will most certainly last for a long time. I want to specially thank Dr.
Juewen Liu for pioneering various projects and for great intellectual advice. I also want to thank
Dr. Hee-Kyung Kim and Dr. Debapriya Mazumdar for their collaboration on the biochemical

vi
and biophysical characterization of the 8-17 DNAzyme with monovalent metal ions project and
thank them as well for valued discussions and for long-lasting support and friendships. I also
want to thank Eric Null for all his help on 31
P NMR and Seyed-Fakhreddin Torabi for help with
the activity assays related to the PS project. I also want to thank undergraduates Stephanie
Sterling and Jenny Wu for their help. I want to specially thank Tian Lan and Hannah Ihms for
being cubicle buddies and great sources of selection discussions. I also want to thank Weichen
Xu, Natasha Yeung and Masha Savelieff for their friendship over the years in the Lu lab. I also
want to thank other past and present members of both the protein and DNA lab for sharing their
research-related sorrows and joys and for being great people to work with.
Finally, I want to thank my family and other friends without whom I wouldn‟t be here
today. My parents Appa and Amma, have given me a lot of freedom and have always motivated
me to do my best and leave the rest, and encouraged me every step of the way and been my rock
of emotional strength and support . My sister Aarts who has also provided me constant emotional
support through the years and has always amazed me with her courage and persistence. My
parents-in-law, Ma and Baba have encouraged me constantly to be focused and attain my goals.
My friends outside of lab, Madhu and Sunni, Gagan and Sukhi, Devi and Inder, Deepa and
Adarsh, Preeti and Sandeep, Vikas and Deepti, Sagar and Shweta and Sima have been a lot of
fun to hang out with and enjoy time with outside of lab. I finally want to specially thank my
husband, Indraneel who has been very supportive of me and my ambitions and shared all the ups
and downs, and the joys and the frustrations of my graduate school life and has always been
there for me. I humbly acknowledge the Blessings and Grace of Bhagawan Sri Sathya Sai Baba
whose love is the constant in my life.

vii
TABLE OF CONTENTS
1 Introduction ....................................................................................................................... 1
1.1 Nucleic acids .......................................................................................................... 1 1.1.1 Functional nucleic acids ...................................................................................... 1
1.2 Role of metal ion cofactors in catalysis by functional nucleic acid enzymes ... 6 1.2.1 Catalysis based on electrostatic charge screening ............................................... 7 1.2.2 Direct involvement of metal ions in catalysis ..................................................... 8 1.2.3 Indirect involvement of metal ions in catalysis ................................................. 10
1.3 Characterization of functional nucleic acid enzymes ...................................... 10 1.3.1 Biochemical characterization ............................................................................ 10 1.3.2 Biophysical characterization ............................................................................. 12
1.3.3 Crystallographic studies .................................................................................... 12
1.4 Bio-sensing Applications of functional nucleic acids ....................................... 15 1.4.1 Fluorescence-based sensing with DNAzymes .................................................. 15 1.4.2 Fluorescence-based sensing with aptamers ....................................................... 16
1.4.3 Fluorescence-based sensing using aptamers and nucleic acid enzymes ........... 17
1.5 Research focus of the thesis ................................................................................ 18
1.6 References ............................................................................................................ 19
2 Low Temperature-Resistant Lead (Pb2+
) Sensing Based on a Fluorescent Catalytic
Beacon Sensor.................................................................................................................. 31
2.1 Introduction ......................................................................................................... 31 2.1.1 Importance of Pb
2+ sensing in water ................................................................. 31
2.1.2 Current methods of Pb2+
detection .................................................................... 32
2.1.3 Use of DNAzymes for Pb2+
detection in water ................................................. 33 2.1.4 Research goals ................................................................................................... 35
2.2 Materials and methods ....................................................................................... 36 2.2.1 Materials ............................................................................................................ 36 2.2.2 Sensor preparation ............................................................................................. 36
2.2.3 Fluorescence measurements and calculations ................................................... 37
2.3 Results and discussion ........................................................................................ 38 2.3.1 Temperature dependence of the original sensor (or RT Sensor) ....................... 38 2.3.2 Strategies to modulate temperature dependence of the sensor .......................... 40 2.3.3 The MM1 sensor and its temperature dependence ............................................ 42

viii
2.3.4 The MM2 sensor and its temperature dependence ............................................ 43
2.3.5 Selectivity and sensitivity of the MM1 sensor .................................................. 45 2.3.6 Role of location of the mismatch ...................................................................... 47 2.3.7 Introduction of dual mismatches ....................................................................... 49
2.4 Conclusions .......................................................................................................... 49
2.5 References ............................................................................................................ 51
3 Effect of Metal Ions on the Global Folding, Activity and Structure of the 8-17
DNAzyme ......................................................................................................................... 54
3.1 Introduction ......................................................................................................... 54 3.1.1 Metal ion-dependent catalysis in nucleic acid enzymes .................................... 55 3.1.2 Metal ion-dependent folding ............................................................................. 56
3.1.3 Research goals ................................................................................................... 58
3.2 Materials and methods ....................................................................................... 59 3.2.1 Materials ............................................................................................................ 59 3.2.2 Kinetic gel-based activity assays ....................................................................... 59
3.2.3 FRET experiments ............................................................................................. 60 3.2.4 CD experiments ................................................................................................. 63
3.3 Results .................................................................................................................. 64 3.3.1 Activity-based on kinetic assays ....................................................................... 64 3.3.2 Folding studies using FRET .............................................................................. 68
3.3.3 Monitoring structural changes using CD .......................................................... 75
3.4 Discussion............................................................................................................. 80 3.4.1 Comparison of monovalent ion-dependent activities between DNAzymes and
ribozymes……………… .................................................................................. 80
3.4.2 Correlation between ionic radii and folding ...................................................... 82 3.4.3 Correlation between activity and folding .......................................................... 83
3.4.4 Electrostatic and site-specific interaction .......................................................... 84 3.4.5 CD evidence for Z-DNA formation .................................................................. 85
3.5 Conclusions .......................................................................................................... 86
3.6 References ............................................................................................................ 87
4 Investigating Potential Metal Ion Binding Sites of the 8-17 DNAzyme through
Phosphorothioate Modifications and 31
P NMR ............................................................ 93
4.1 Introduction ......................................................................................................... 93

ix
4.1.1 Insights into the mechanism of the 8-17 DNAzyme ......................................... 93
4.1.2 Phosphorothioate (or PS) modifications ........................................................... 95 4.1.3 Research goals ................................................................................................... 97
4.2 Materials and methods ....................................................................................... 98 4.2.1 Materials ............................................................................................................ 98 4.2.2 Kinetic assays .................................................................................................... 98 4.2.3
31P NMR studies .............................................................................................. 100
4.3 Results ................................................................................................................ 102 4.3.1 Dependence of the 8-17 DNAzyme activity on position of PS modification. 102
4.3.2 Probing metal Binding based on 31
P NMR ..................................................... 107
4.4 Discussion........................................................................................................... 117 4.4.1 Effect of PS modifications on AGC terminal loop based on kinetic assays ... 117 4.4.2 Effect of PS modifications at bases on the TCGAA loop based on kinetic
assays………… ............................................................................................... 119 4.4.3 Effect of PS modifications on the substrate strand based on kinetic activity
assays……………………. .............................................................................. 120 4.4.4 31
P NMR studies of the 8-17 DNAzyme ......................................................... 120
4.4.5 Implications for metal ion binding and activity .............................................. 123
4.5 Conclusions ........................................................................................................ 126
4.6 References .......................................................................................................... 128
5 In Vitro Selection of DNAzymes Specific for the Detection of Mercury (Hg2+
) and
Cadmium (Cd2+
) ............................................................................................................ 131
5.1 Introduction ....................................................................................................... 131 5.1.1 Deoxyribozymes (or DNAzymes) and ribozymes .......................................... 131
5.1.2 In vitro selection .............................................................................................. 132 5.1.3 Metal-dependent DNAzymes that cleave RNA .............................................. 134
5.1.4 Mercury contamination and toxicity ............................................................... 134 5.1.5 Cadmium toxicity and EPA Maximum Contamination Limit (MCL) ............ 135
5.1.6 Research focus ................................................................................................. 136
5.2 Materials and methods ..................................................................................... 138 5.2.1 Materials .......................................................................................................... 138
5.2.2 Template and primer design ............................................................................ 139 5.2.3 In vitro selection and PCR protocols ............................................................... 141 5.2.4 Real time PCR measurements ......................................................................... 146 5.2.5 Gel-based activity assays ................................................................................ 148
5.2.6 Cloning and sequencing .................................................................................. 149

x
5.3 Results and discussion ...................................................................................... 150 5.3.1 In vitro selection of a Hg
2+- specific DNAzyme ............................................. 150
5.3.2 In vitro selection of Cd2+
-specific DNAzymes ............................................... 156
5.4 Conclusions ........................................................................................................ 162
5.5 Future Directions .............................................................................................. 163
5.6 References .......................................................................................................... 164
6 In vitro selection of a DNA Aptamer specific for Endotoxins ................................... 170
6.1 Introduction ....................................................................................................... 170 6.1.1 Endotoxins and their impact on health ............................................................ 170
6.1.2 Current methods of detection and need for improved detection methods ....... 171
6.1.3 Aptamers and their use as biosensors .............................................................. 173 6.1.4 Advantages of using aptamers for detection ................................................... 174
6.1.5 Research goals ................................................................................................. 175
6.2 Materials and methods ..................................................................................... 176 6.2.1 Materials .......................................................................................................... 176
6.2.2 Generation of the initial pool .......................................................................... 176 6.2.3 Optimization of selection conditions and in vitro selection ............................ 179
6.2.4 Regeneration of the pool after selection .......................................................... 181
6.3 Results and discussion ...................................................................................... 182 6.3.1 Design of DNA library .................................................................................... 182 6.3.2 Selection strategy and parallel selections ........................................................ 184
6.4 Conclusions ........................................................................................................ 193
6.5 Future directions ............................................................................................... 194
6.6 References .......................................................................................................... 195

1
1 Introduction
1.1 Nucleic acids
Nucleic acids are biopolymers that are named according to their existence in the nucleus
of a cell. The monomers of these polymers are called nucleotides and each nucleotide consists of
three parts, a heterocyclic base which could either be a purine or a pyrimidine, a pentose sugar,
namely ribose or 2-deoxyribose, and a phosphate group. DNA or „deoxyribonucleic acid‟ and
RNA or „ribonucleic acid‟ are the two types of nucleic acids found in nature and they differ from
each other based on the nature of the pentose sugar (2-deoxyribose in DNA and ribose in RNA),
and a single base (thymine (T) in DNA and uracil (U) in RNA). The three bases common to both
RNA and DNA are adenine (A), guanine (G) and cytosine (C). Another difference that is seen in
biological systems between RNA and DNA is that the former is usually single-stranded while the
latter adopts a double-stranded helical structure with the two strands being held together by two
hydrogen bonds between A and T, and three hydrogen bonds between G and C, respectively. In
terms of function, DNA is responsible for carrying genetic information and transcribing it into
RNA, which in turn is translated into proteins, constituting what is thought to be the central
dogma of molecular biology.1
1.1.1 Functional nucleic acids
Discoveries made over the last thirty years have demonstrated that in addition to being
genetic material carriers, nucleic acids can perform active roles in binding and catalysis, leading
to the term „functional nucleic acids‟.2-5
This finding has opened doors to a new research
paradigm in nucleic acid chemistry and biology.

2
1.1.1.1 Catalytic RNA molecules or ribozymes
RNA-based functional nucleic acids that can facilitate catalysis are called ribozymes.6-9
The proposal of nucleic acids to perform catalysis and other biochemical activities through the
formation of three-dimensional shapes and structures was initially suggested by Spiegelman et
al. in the 1960s.10
However, it was decades later that the first example of a naturally occurring
ribozyme was discovered, in the early-80s by Cech and co-workers, in the intron of an RNA
transcript of Tetrahymena thermophilia, that could catalyze a transesterification reaction.2 This
discovery and others thereafter led to the speculation of an „RNA world‟ wherein the RNA was
involved in carrying out all biochemical reactions of the cell in the absence of any protein
machinery.11-15
Subsequently, over the last few decades, experiments have demonstrated that
non-natural ribozymes can be isolated in the laboratory through a selection procedure called
SELEX (Systematic Evolution of Ligands through Exponential Enrichment),16
for a wide variety
and range of catalytic functions, further adding ground to the theory of an „RNA world‟.
Identification of the earliest examples of artificial ribozymes and RNA aptamers (discussed in
the next section) was performed using the in vitro or SELEX selection process, in relevance to
the isolation of aptamers by Tuerk and Gold in 1990.16
However, it is currently used
interchangeably in regard to the isolation of catalytic RNA or DNA as well. A detailed
description of in vitro selection is discussed in chapter 5.
One of the first artificial ribozymes was discovered by Bartel and Szostak, which showed
RNA ligation activity.17
Later on, ribozymes that were capable of catalyzing a variety of other
reactions including RNA cleavage,18-21
RNA capping,22-24
Diels-Alder reactions,25-28
Michael
addition,29
aminoacylation30-32
and porphyrin metalation33
were also isolated. Ribozymes have

3
been broadly classified into two categories, based upon the identity of the nucleophile that
attacks the phosphodiester linkage.34-37
The „small‟ self-cleaving ribozymes include the
hammerhead,38-40
hairpin,41-43
hepatitis delta virus (HDV),44-46
and leadzyme47-49
ribozymes, and
in these systems, the 2′-OH of the ribose moiety attacks its own 3′-phosphodiester linkage. In
„large‟ ribozymes, such as the Group I50-52
and Group II introns,53-56
Neurospora VS,57-59
and
RNaseP systems,60, 61
the mechanism involves an exogenous nucleophile, such as water or the 2′
or 3′-OH of another nucleotide.
1.1.1.2 Aptamers
The speculation of an „RNA world‟ also led to the idea that RNA molecules could form
complex three-dimensional structures capable of binding molecules.13
Indeed arguments in favor
of this theory became more substantial when RNA molecules that showed binding toward small
organic dyes were first discovered by Ellington and Szostak in 1990.3 They also coined the term,
„aptamers‟, which was derived from the Latin word „aptus‟, meaning „to fit‟. Since then, both
RNA and DNA aptamers have been isolated for numerous functions and applications using the
SELEX method.62-64
In fact, a therapeutic aptamer that is directed against the vascular
endothelial growth factor (VEGF), called Macugen® (pegaptanib sodium) has been clinically
approved by the U.S. FDA (Food and Drug Administration) for the treatment of age-related
macular degeneration (AMD) and diabetic retinopathy,65-67
and is the first example of an
aptamer-based drug. An online database that provides information about the list of targets for
which aptamers have been isolated against has been created by Ellington and co-workers.68
In
addition, partial lists of aptamers that bind a variety of targets have also been documented.5, 63

4
Additional evidence that the primitive world was based on RNA came from more recent
discoveries that RNA motifs could behave as specific regulatory switches called riboswitches,
using feedback regulatory loops in the absence of any helper proteins and control their own
expression.69-71
These riboswitches are structured elements typically found in the 5' untranslated
regions of mRNAs, where they regulate gene expression by binding to small metabolites.72
It is
believed that through the evolution process, they formed structured and highly selective
receptors for these small drug-like metabolite molecules. Therefore, riboswitches could have
significant applications as antibacterial drug targets.73-78
1.1.1.3 Catalytic DNA molecules or DNAzymes
DNAzymes or catalytic DNA molecules on the other hand, have not been known to occur
in nature. Due to the absence of the 2'-hydroxyl group in DNA as compared to RNA, it has been
thought that isolation of functionally active DNA molecules would pose a much greater
challenge than functional RNA. However, through the isolation of a small RNA-cleaving
DNAzyme, Breaker and Joyce demonstrated in 1994 that it was indeed possible to isolate
catalytic DNA molecules as well.4 Widespread efforts have been since made for the isolation of
these molecules in laboratories through a combinatorial biology technique known as in vitro
selection, also referred to as SELEX as mentioned in the previous section for various
applications.79-95
DNA is more stable toward hydrolysis in comparison with RNA, much easier to
chemically synthesize and is also easily amenable to chemical modifications that could be
potentially useful for promoting applications.5 The repertoire of reactions catalyzed by
DNAzymes is shown in Table 1.1.5 The 8-17 and the 10-23 DNAzymes are two of the most
widely studied RNA-cleaving deoxyribozymes isolated using in vitro selection.

5
1.1.1.4 The 8-17 DNAzyme
A small RNA-cleaving DNAzyme called the 8-17 DNAzyme has been the focus of
various studies by the Lu lab and other researchers in terms of its applications as well as its
mechanistic aspects.96-120
It specifically possesses a very small catalytic core (in comparison to
other RNA-cleaving DNAzymes), has a relatively fast reaction rate and can catalyze the RNA-
cleavage reaction with both DNA as well as RNA substrates. Interestingly, variants of this
catalytic motif have been isolated under various selection conditions and by different research
laboratories in the presence of different metal-ion co-factors such as 10 mM Mg2+
,116
0.5 mM
Mg2+
/50 mM histidine,121, 122
0.1 mM Zn2+
,123
7.5 mM Mg2+
/7.5 mM Mn2+
,111
and 50 µM
Cu2+
/7.5 mM Mg2+
/7.5 mM Mn2+
.114
In one instance the same motif was selected in the absence
of any metal ion co-factor as well.124
The three common variants obtained include the original 8-
17 motif selected by Santoro and Joyce in 1997,116
the Mg5 variant isolated by Faulhammer and
Famulok in 1997121, 122
and the 17E variant selected by the Lu lab in 2000.123
The utility of the 8-17 DNAzyme has been explored through a variety of applications that
include in vitro bio-sensing,106, 125-129
logic gates and DNA-based programming,130, 131
proofreading and error-removal,132, 133
directing the outcome of selections134
and as gene control
and antiviral agents.101, 117

6
Table 1.1 Examples of DNAzymes isolated by in vitro selection. This table has been updated
from Cao et al.5 and the PhD thesis of former group member, Dr. Juewen Liu.
Reaction Cofactor kmax (min-1
)
kcat/kuncat Ref.
RNA cleavage Pb2+
1 105 4
Mg2+
0.01 105
135
Ca2+
0.1 104
121
Mg2+
10 >105
116
None 0.01 108
124
L-histidine 0.2 106
136
Zn2+
~40 >105
123
*Zn2+
~4 137
Mg2+
1.7 138
**None 0.044 139
Co2+
7 140
Cd2+
, Mn2+
, Ni2+
~1 141
UO22+
~1.2 142
DNA cleavage Cu2+
0.2 >106
79, 81
DNA hydrolysis Mn2+
, Zn2+
2.7 h-1
1012
143
RNA ligation Mn2+
~2.2 >106
144
Mg2+
0.5 105
145
Mg2+
0.013 1.9×104
146
Mg2+
0.1 450 118
Zn2+
0.5 1.7×104
147
DNA ligation Cu2+
or Zn2+
0.07 105
148
Mn2+
10-4
>105
149
DNA phosphorylation Ca2+
0.01 109
150
DNA depurination IO4-
151
DNA adenylation Cu2+
0.003 >1010
152
Thymine dimer cleavage None 4.5 2.5×104
153
Phosphoramidate bond cleavage Mg2+
~5×10-4
>103
154
N-glycosylation Ca2+
0.5 106
155
Porphyrin metallation None 1.3 103
156
Carbon-carbon bond formation Ca2+
3.0 4×105
157
*imidazole-modified DNA, **imidazole-,amine-modified DNA
1.2 Role of metal ion cofactors in catalysis by functional nucleic acid enzymes
Nucleic acids are essentially biopolymers with a negatively-charged phosphate backbone;
hence, metal ions are essential in balancing and stabilizing their functionally important

7
conformations.158, 159
It is interesting to note that despite their limited number of chemical
functionalities, nucleic acid and metal ion interactions are considerably complex and have been
extensively studied by researchers for a variety of reasons ranging from fundamental
advancement of inorganic chemistry and structural biology to practical utility in pharmaceutical
and biotechnological industries. For instance, an important area of research involves the role of
metal ions in mechanisms catalyzed by ribozymes, which could play a key role in the origin of
life as hypothesized by the theory of the „RNA world‟.159-161
A more practical application of
metal-nucleic acid interactions is based on the binding of platinum complexes to DNA leading to
the development of anticancer drugs, the most famous being cisplatin and its analogs.162-165
Most
ribozymes and DNAzymes are dependent on metal ion co-factors for both their structure and
function, although there are instances of artificial ribozymes or DNAzymes, isolated in vitro in
the absence of any metal ion.124, 166
In general, the role of metal ions in nucleic acid enzymes can
be broadly divided into four categories depending on the nature of their interactions: non-specific
charge-screening based on neutralizing the negatively-charged nucleic acid phosphate backbone;
specific binding to well-defined sites on the nucleic acid characterized by a concentration of
negative electrostatic potential; inner-sphere binding wherein the metal ion interacts directly with
the nucleic acid ligands, and outer-sphere binding wherein metal ions interact with the nucleic
acid via water molecules.159, 161, 167
1.2.1 Catalysis-based on electrostatic charge screening
Electrostatic effects are used by many enzymes to indirectly activate functional groups at
the active site to thereby facilitate catalysis. Divalent metal ions are particularly useful since they
have a high charge density and relatively well-defined coordination geometry.167
These features

8
have been shown to induce and perturb the acidity of various nucleobases. For instance, a Mg2+
in the vicinity of the guanosine N7 nitrogen will change the pKa of N1.168
This is an important
interaction in the hairpin ribozyme where an (N1)-deprotonated site on an active site guanosine
is implicated in catalysis.169, 170
Another aspect of electrostatic screening by metal ions is demonstrated by the fact that
several small ribozymes that include the hairpin, hammerhead and VS ribozymes can in fact
react and show activity at molar concentrations of monovalent metal ions.46, 171-174
The hepatitis
delta virus (HDV) ribozyme displays an absolute requirement for divalent metal ions in
functional and/or structural roles.173, 175
However, in addition to Co(NH3)63+
, polyamines and
aminoglycoside antibiotics can replace Mg2+
ions to induce cleavage activity by the hairpin
ribozyme.176
This observation is rather interesting since it suggests that many of these ribozymes
need not contain any specific sites for binding of the divalent metal ions, rather the metal ions are
required only for structure formation and therefore might play a minimal role in function.171
1.2.2 Direct involvement of metal ions in catalysis
A metal ion that is required for catalytic function and is directly involved in catalysis
(also referred to as inner-sphere binding or coordination) can participate in several ways: a) as a
general acid catalyst, where a proton from a metal bound water stabilizes a developing negative
charge on the leaving group; b) a general base catalyst where a metal coordinated hydroxide
abstracts the proton from the 2-OH; c) as a Lewis acid catalyst that stabilizes the leaving group;
d) a Lewis acid that promotes deprotonation of the attacking nucleophile, or as e) an electrophilic
catalyst that increases the electrophilicity of the phosphorus atom in the cleaved phosphodiester
bond, as shown in Figure 1.1.177

9
Figure 1.1 Potential roles of catalytic metal ions in RNA cleavage. Metal ions and metal bound
water molecules are proposed to facilitate (a) general acid, (b) general base, (c-d) Lewis acid or
(e) electrophilic catalysis in reactions catalyzed by ribozymes(or DNAzymes). "Figure has been
adapted from Takagi, Y.; Warashina, M.; Stec, W. J.; Yoshinari, K.; Taira, K. Recent advances
in the elucidation of the mechanisms of action of ribozymes. Nucleic Acids Res. 2001, 29, 1815-
1834. (http://nar.oxfordjournals.org/cgi/content/abstract/29/9/1815) Reprinted by permission of
Oxford University Press."
The generally accepted mechanism for the hammerhead reaction is a single metal ion
mechanism in which the hydroxide ion form of Mg2+
acts as a general base to deprotonate the 2'-
OH.178, 179
A two-metal ion mechanism has been implicated for the group I intron, in which one
metal ion acts as general acid in deprotonating the 2'-hydroxyl and another metal ion acts as a
general base in stabilizing the leaving 5'-oxygen.51, 180, 181
The crystal structure of the group I
intron revealed a highly coordinated cluster of two Mg2+
ions, where five of the water ligands of
one of the Mg2+
ions are replaced with oxygen atoms from the RNA. 180, 181

10
1.2.3 Indirect involvement of metal ions in catalysis
Catalysis by several enzymes has also been demonstrated to take place in the presence of
metal ion co-factors that are completely hydrated and participate in catalysis through this
hydrated form (i.e., outer-sphere/ligand-mediated interactions). This was demonstrated in the
hairpin ribozyme wherein replacement of Mg2+
by the coordinatively saturated and
substitutionally inert metal complex, Co(NH3)63+
, activity was still observed.182
Similar results
coupled with analysis of the Mg2+
-dependence of an acyl-transferase ribozyme demonstrated that
Mg2+
indeed participated via outer-sphere interactions through its fully hydrated form,
Mg(H2O)62+
.183
1.3 Characterization of functional nucleic acid enzymes
1.3.1 Biochemical characterization
Various kinetic and thermodynamic studies have been carried out to elucidate the
mechanism of both ribozymes as well as DNAzymes. For instance, the phosphodiester cleavage
reaction of the 8-17 DNAzyme was proposed to adopt the same mechanism as the small self-
cleaving ribozymes19, 57, 184, 185
and the 10-23 DNAzyme.116, 120
The 2-OH group at the cleavage
site acts as a nucleophile that attacks the scissile phosphorus, forming in a penta-coordinated
phosphate intermediate, followed by elimination of the 5-oxygen as shown in Figure 1.2. In
addition to this transesterification step, Pb2+
catalyzes the subsequent hydrolysis of the 2,3-
cyclic phosphate.120
The identical two-step mechanism has been observed in the Pb2+
-dependent
ribozyme (leadzyme), while no evidence of catalytic hydrolysis of the 2, 3-cyclic phosphate
was found in the Pb2+
-dependent cleavage product of yeast tRNAphe
.19, 186

11
Figure 1.2 Proposed reaction mechanism of the hydrolytic cleavage reaction catalyzed by small
ribozymes and DNAzymes. The transesterfication step occurs in the presence of Pb2+
, Zn2+
, and
Mg2+
, but further hydrolysis of the 2, 3-cyclic phosphate occurs only in the presence of Pb2+
.
The figure was adapted from a former group member, Dr. Juewen Liu‟s PhD thesis.
Phosphorothioate modifications have been used extensively in ribozymes to map catalytic
metal ion binding sites.187
In these experiments, one of the non-bridging oxygen atoms (a hard
ligand) in the nucleic acid backbone is replaced with a sulfur atom (a soft ligand), resulting in
one of two possible stereoisomers. Sulfur substitutions have been used to identify possible direct
metal-coordination sites to specific oxygen atoms in the Group I52, 188
and Group II introns.53
The
absence of a thio affect on the activity of the hairpin ribozyme was taken as evidence that no
GO
O
OH OP
AO
O OH
5'-ACTCACTAT
GAAGAGATG-3'
GAAGAGATG-3'
GO
OH5'-ACTCACTAT A
O
O O
P
O O-
+
5'-ACTCACTAT
O-
O-
OP
AO
O OH
Pb2+, H2O

12
direct coordination of a Mg2+
ion with any non-bridging oxygen atoms at the cleavage site is
necessary for activity.182, 189
1.3.2 Biophysical characterization
Fluorescence spectroscopy, specifically FRET or Fluorescence Resonance Energy
Transfer, has been used for studying the relationship between structure and function of
ribozymes9 and the 8-17 DNAzyme.
97, 99, 100, 112 Ion-induced folding studies of the hammerhead
ribozyme using FRET revealed that the ribozyme undergoes a well-defined two-stage folding
process induced by the sequential binding of two Mg2+
ions with the second structural transition,
corresponding to the formation of the catalytic domain of the ribozyme.40, 190, 191
The folding has
been shown to have a close relationship to catalytic activity of the ribozyme.192
Global folding of
the hairpin ribozyme has also been well characterized and shown to have a good correlation
between the extent of ion-induced folding and cleavage activity.193
This method has also been
used to observe conformational changes that take place in the 8-17 DNAzyme in the presence of
different metal ion co-factors.
1.3.3 Crystallographic studies
X-ray crystal structures for a variety of ribozymes have been obtained that include the
hammerhead,194
leadzyme,48, 195
hairpin,196
HDV,44
RNA-polymerase ribozyme197
and Group I
intron ribozymes.198
It is important to note, however, that the crystal structure may not always
correspond to the solution structure of the active species or reaction intermediates, and therefore
solution NMR experiments are also needed to support mechanistic hypotheses based on crystal
structures.199, 200

13
A 3Å crystal structure of the RNA-polymerase ribozyme is shown in Figure 1.3.197
The
authors proposed a mechanism for this class I ligase based on their structure and the mechanism
of model proteinaceous enzymes, whereby the substrate α-phosphate and backbone phosphates
of A29 and C30 jointly bind a catalytic Mg
2+, thereby activating the primer 3'-hydroxyl for
nucleophilic attack and stabilizing the transition state. They further suggested the presence of a
second metal ion complexed with an NTP (Nucleoside triphosphate) that would
remain
coordinated by oxygens on the β- and γ-phosphates, helping to stabilize the transition state
through the development of a negative charge on the pyrophosphate leaving group.
197
Despite the number of ribozyme crystal structures, crystallographic data for DNAzymes
however, continues to remain elusive. The 10-23 DNAzyme was crystallized as a dimer;
however, the observed 2:2 stoichiometry of the enzyme and substrate strands was inconsistent
with that observed in single-turnover kinetic studies,201
suggesting that the structure obtained did
not represent the active enzyme-substrate complex. Hence, until the crystallization of active
forms of DNAzymes is achieved, biophysical and biochemical methods are the tools at hand
used for better understanding the mechanism of this class of functional nucleic acid enzymes.

14
Figure 1.3 Crystal Structure of the RNA-polymerase ribozyme A) Secondary structure and
reaction scheme of a ligase variant with decreased Mg2+
dependence B) Revised secondary
structure of the crystallization construct C) Ribbon representation of ligase structure, as if
peering into the active site (yellow) and ligation junction (red). (D) Top-down view, relative to
(C). "Figure from Shechner, D. M.; Grant, R. A.; Bagby, S. C.; Koldobskaya, Y.; Piccirilli, J. A.;
Bartel, D. P. Crystal structure of the catalytic core of an RNA-polymerase ribozyme. Science
2009, 326, 1271-1275. (http://www.sciencemag.org/cgi/content/full/326/5957/1271) Reprinted
with permission from AAAS."

15
1.4 Bio-sensing applications of functional nucleic acids
Functional nucleic acids are well suited for use in the development of sensors for the
specific targets that they have been selected against by SELEX,16
since the desired selectivity
and specificity for the target of choice can be engineered during their evolution. In fact, the
binding ability of aptamers is often times comparable to that of antibodies and in certain cases
they have been shown to out-perform the latter against the same target.202, 203
In contrast to
antibodies, aptamers can be chemically synthesized and can be targeted against small molecule
targets. The use of nucleic acid enzymes to sense their respective metal ion co-factors has also
gained considerable ground over the past decade with the development of several DNAzyme-
based metal ion detection techniques. Moreover, the ability to convert either functional nucleic
acid target binding or metal-ion cleavage events into signal transduction events using techniques
such as fluorescence, color, electrochemical detection, magnetic resonance imaging, surface
acoustic wave-based detection, microfabricated cantilever-based detection, mass spectrometry
and surface plasmon resonance-based detection, provides an opportunity to perform on-site and
real-time detection of their specific targets.5 Some fluorescent methods for detection based on
both functional nucleic acid metal ion and molecular targets are briefly discussed below.
1.4.1 Fluorescence-based sensing with DNAzymes
Several functional nucleic acids have been labeled with fluorophores and quenchers and
converted into fluorescent sensors due to the sensitivity afforded by this detection method.5, 107
A
common strategy utilized in sensing applications of RNA-cleaving DNAzymes is based on the
catalytic beacon method. An example of this strategy is the 39E DNAzyme that can specifically
detect UO22+
.142
The sensor is assembled by hybridizing the enzyme strand containing a single

16
quencher and a substrate strand containing an RNA base that undergoes cleavage, a fluorophore
and quencher. In the absence of the metal ion, the fluorescence is quenched due to proximity of
the fluorophore with the quencher. In the presence of the metal ion however, cleavage of the
RNA base takes place, leading to the release of the substrate arm containing the fluorophore and
an overall increase in the fluorescence intensity (~15 times in this case). The detection limit for
UO22+
based on this method was found to be 45 pM, which was considerably lower than the U.S.
EPA (Environment Protection Agency) defined limit of 130 nM.204
In addition, this sensor also
demonstrated more than one million-fold selectivity over the other metal ions tested. Sensors
based on this method have also been developed for Pb2+
and Hg2+
.105, 125, 129, 204, 205
In addition, a
number of other methods based on fluorescence, specifically with the fluorescently labeled 8-17
DNAzyme for Pb2+
detection, have been explored that include its immobilization onto
surfaces,126, 127
and assays in micro- and nano-fluidic sensing platforms with improvements in
overall sensitivity or ease of use.128, 206
New DNAzymes with fluorescently modified nucleotides
were isolated by Li and co-workers140, 141
and utilized directly for metal sensing.98, 115
1.4.2 Fluorescence-based sensing with aptamers
A general approach for converting aptamers into fluorescent sensors is the aptamer
beacon approach.5 This technique is based on classic molecular beacon technology wherein the 5'
and 3' ends of the DNA (or RNA) are extended to form a hairpin loop and one end of this hairpin
is labeled with a fluorophore and the other end is labeled with a quencher.207-209
In the absence of
the target, the fluorescence of the fluorophore is quenched. The presence of the target leads to
disruption of the hairpin, leading to an increase in the fluorescence intensity. Another approach
involves using the change in secondary structure of the aptamer upon target binding, called

17
„structure-switching signaling aptamers‟ and was reported by Nutiu and co-workers and
demonstrated in the detection of ATP.210-213
Label-free sensing of molecular targets based on
fluorescence methods has also been demonstrated either through intercalation of fluorescent dyes
or by a design, wherein, aptamers for dyes such as malachite green (MG) which when unbound,
exhibits low fluorescence intensity, but when bound, shows significant increase in the
fluorescence intensity.214
This idea is the basis for a new label-free sensor design for the
detection of adenosine. In this instance, the MG aptamer strand is and the adenosine aptamer are
held together by a bridging DNA strand that is complementary to both aptamer strands. This
aptamer-bridging DNA system prevents MG from binding to the MG aptamer, resulting in a low
background fluorescence of MG in the absence of adenosine. However, addition of the target
(adenosine) causes binding of the adenosine aptamer, thereby weakening the hybridization of the
MG aptamer strand with the bridging strand, making it possible for MG to bind the free aptamer
and exhibit an increase in the overall fluorescence intensity. Since this design is based purely on
nucleic acid hybridization, it can be generally applied to other aptamers for the label-free
detection of a broad range of analytes. Aptamers for cellular targets have also been developed
based on fluorescence methods.215
1.4.3 Fluorescence-based sensing using aptamers and nucleic acid enzymes
Sensors that combine the metal ion detection of nucleic acid enzymes with the target-
binding capability of aptamers have been constructed as well and these are termed allosteric
DNA/RNA aptazymes.62, 216, 217
For instance, Rueda and Walter created a theophylline aptazyme
based biosensor by combining the hammerhead ribozyme with a theophylline aptamer and
labeling the ends of the hammerhead substrate arm with a FRET pair.218
In the absence of

18
theophylline, energy transfer is seen between the FRET pair due to their proximity; however, in
the presence of the target, substrate cleavage and disassociation takes place leading to an overall
decrease in the FRET efficiency. An immobilized ligation aptazyme has been used for sensing
ATP through the ligation of a short fluorophore-labeled DNA with a long circular DNA template
in the presence of the target, leading to an increased fluorescence signal.219
Hence, DNAzymes, aptamers and aptazymes are useful target recognition elements that
can be combined with appropriate signal transduction elements for a variety of sensing
applications.
1.5 Research focus of the thesis
The 8-17 DNAzyme, a small RNA-cleaving DNAzyme has been explored extensively in
terms of its applications for bio-sensing as well as for exploring its mechanism from a more
fundamental perspective. The first part of this thesis focuses on both these aspects of the 8-17
DNAzyme. Chapter 2 describes a strategy to increase the utility of a fluorescent Pb2+
sensor
based on the 8-17 DNAzyme, at different temperatures ranging from 4 °C to 30 °C through the
introduction of mismatches on the enzyme strand. Chapters 3 and 4 describe our efforts in
gaining a more fundamental perspective into the role played by various monovalent and divalent
metal ions on the overall activity, folding and structure of the 8-17 DNAzyme and the use of
phosphorothioate modifications to identify functional phosphates and a potential Pb2+
-binding
site in the 8-17 DNAzyme, respectively. Gel-based kinetic assays, bulk FRET assays, circular
dichroism (CD) studies and 31
P NMR are the techniques employed to carry out these studies.
The starting point for the development of new bio-sensors based on functional nucleic
acids is their isolation, which is achieved using the in vitro selection or SELEX method.

19
Chapter 5 focuses on our efforts to use in vitro selection to identify DNAzymes specific for
Hg2+
and Cd2+
. Negative selections to overcome Pb2+
interference at various stages of selection
are described as well. Chapter 6 focuses on our efforts in carrying out in vitro selection to
isolate structure-switching signaling aptamers that can specifically detect endotoxins. The
aptamers obtained based on this strategy can be directly converted into sensors in the future for
endotoxins. A parallel positive selection with KDO or 2-keto-3-deoxyoctulosonic acid, a small
molecule that is common to the endotoxins expressed by various strains of gram negative
bacteria as a strategy to increase the applicability of the aptamers selected is also discussed.
1.6 References
1. Alberts, B.; Alexander, J.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P., Molecular Biology
of the Cell, 4th Edition. 2004; p 2000.
2. Kruger, K.; Grabowski, P. J.; Zaug, A. J.; Sands, J.; Gottschling, D. E.; Cech, T. R. Self-
splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening
sequence of Tetrahymena. Cell 1982, 31, 147-157.
3. Ellington, A. D.; Szostak, J. W. In vitro selection of RNA molecules that bind specific
ligands. Nature 1990, 346, 818-822.
4. Breaker, R. R.; Joyce, G. F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223-
229.
5. Liu, J.; Cao, Z.; Lu, Y. Functional nucleic acid sensors. Chem. Rev. 2009, 109, 1948-
1998.
6. Doherty, E. A.; Doudna, J. A. Ribozyme structures and mechanisms. Annu. Rev.
Biochem. 2000, 69, 597-615.
7. Doudna, J. A.; Cech, T. R. The chemical repertoire of natural ribozymes. Nature 2002,
418, 222-228.
8. Doudna, J. A.; Lorsch, J. R. Ribozyme catalysis: not different, just worse. Nat. Struct.
Mol. Biol. 2005, 12, 395-402.
9. Lilley, D. M. Structure, folding and mechanisms of ribozymes. Curr. Opin. Struct. Biol.
2005, 15, 313-323.
10. Mills, D. R.; Peterson, R. L.; Spiegelman, S. An extracellular Darwinian experiment with
a self-duplicating nucleic acid molecule. Proc. Natl. Acad. Sci. U. S. A. 1967, 58, 217-
224.
11. Gilbert, W.; Marchionni, M.; McKnight, G. On the antiquity of introns. Cell 1986, 46,
151-153.
12. Gilbert, W. Evolution of antibodies. The road not taken. Nature 1986, 320, 485-486.

20
13. Benner, S. A.; Ellington, A. D.; Tauer, A. Modern metabolism as a palimpsest of the
RNA world. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 7054-7058.
14. Benner, S. A.; Ellington, A. D. Evolution and structural theory: the frontier between
chemistry and biology. Bioorg. Chem. Front. 1990, 1, 1-70.
15. Benner, S. A.; Burgstaller, P.; Battersby, T. R.; Jurczyk, S. Did the RNA world exploit an
expanded genetic alphabet? Cold Spring Harbor Monogr. Ser. 1999, 37, 163-181.
16. Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA
ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505-510.
17. Bartel, D. P.; Szostak, J. W. Isolation of new ribozymes from a large pool of random
sequences. Science 1993, 261, 1411-1418.
18. Pan, T.; Uhlenbeck, O. C. In vitro selection of RNAs that undergo autolytic cleavage
with Pb2+. Biochemistry 1992, 31, 3887-3895.
19. Pan, T.; Uhlenbeck, O. C. A small metalloribozyme with a two-step mechanism. Nature
1992, 358, 560-563.
20. Williams, K. P.; Ciafre, S.; Tocchini-Valentini, G. P. Selection of novel Mg2+
-dependent
self-cleaving ribozymes. EMBO J. 1995, 14, 4551-4557.
21. Jayasena, V. K.; Gold, L. In vitro selection of self-cleaving RNAs with a low pH
optimum. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10612-10617.
22. Chapman, K. B.; Szostak, J. W. Isolation of a ribozyme with 5'-5' ligase activity. Chem.
Biol. 1995, 2, 325-333.
23. Huang, F.; Yang, Z.; Yarus, M. RNA enzymes with two small-molecule substrates.
Chem. Biol. 1998, 5, 669-678.
24. Huang, F.; Yarus, M. 5'-RNA self-capping from guanosine diphosphate. Biochemistry
1997, 36, 6557-6563.
25. Morris, K. N.; Tarasow, T. M.; Julin, C. M.; Simons, S. L.; Hilvert, D.; Gold, L.
Enrichment for RNA molecules that bind a Diels-Alder transition state analog. Proc.
Natl. Acad. Sci. U. S. A. 1994, 91, 13028-13032.
26. Tarasow, T. M.; Tarasow, S. L.; Eaton, B. E. RNA-catalyzed carbon-carbon bond
formation. Nature 1997, 389, 54-57.
27. Seelig, B.; Jaschke, A. A small catalytic RNA motif with Diels-Alderase activity. Chem.
Biol. 1999, 6, 167-176.
28. Agresti, J. J.; Kelly, B. T.; Jaschke, A.; Griffiths, A. D. Selection of ribozymes that
catalyse multiple-turnover Diels-Alder cycloadditions by using in vitro
compartmentalization. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16170-16175.
29. Sengle, G.; Eisenfuhr, A.; Arora, P. S.; Nowick, J. S.; Famulok, M. Novel RNA catalysts
for the Michael reaction. Chem. Biol. 2001, 8, 459-473.
30. Illangasekare, M.; Sanchez, G.; Nickles, T.; Yarus, M. Aminoacyl-RNA synthesis
catalyzed by an RNA. Science 1995, 267, 643-647.
31. Illangasekare, M.; Yarus, M. A tiny RNA that catalyzes both aminoacyl-RNA and
peptidyl-RNA synthesis. RNA 1999, 5, 1482-1489.
32. Yarus, M.; Illangasekare, M. Aminoacyl-tRNA synthetases and self-acylating ribozymes.
Cold Spring Harbor Monogr. Ser. 1999, 37, 183-196.
33. Conn, M. M.; Prudent, J. R.; Schultz, P. G. Porphyrin Metalation Catalyzed by a Small
RNA Molecule. J. Am. Chem. Soc. 1996, 118, 7012-7013.

21
34. Cochrane, J. C.; Strobel, S. A. Catalytic strategies of self-cleaving ribozymes. Acc. Chem.
Res. 2008, 41, 1027-1035.
35. Narlikar, G. J.; Herschlag, D. Mechanistic aspects of enzymatic catalysis: lessons from
comparison of RNA and protein enzymes. Annu. Rev. Biochem. 1997, 66, 19-59.
36. Scott, W. G. Ribozymes. Curr. Opin. Struct. Biol. 2007, 17, 280-286.
37. Sigel, R. K.; Pyle, A. M. Lanthanide ions as probes for metal ions in the structure and
catalytic mechanism of ribozymes. Met. Ions Biol. Syst. 2003, 40, 477-512.
38. Eckstein, F. The hammerhead ribozyme. Biochem. Soc. Trans. 1996, 24, 601-604.
39. Bassi, G. S.; Mollegaard, N. E.; Murchie, A. I.; Lilley, D. M. RNA folding and
misfolding of the hammerhead ribozyme. Biochemistry 1999, 38, 3345-3354.
40. Hammann, C.; Lilley, D. M. Folding and activity of the hammerhead ribozyme.
ChemBioChem 2002, 3, 690-700.
41. Lilley, D. M. Folding and catalysis by the hairpin ribozyme. FEBS Lett. 1999, 452, 26-
30.
42. Fedor, M. J. Structure and function of the hairpin ribozyme. J. Mol. Biol. 2000, 297, 269-
291.
43. Wilson, T. J.; Nahas, M.; Ha, T.; Lilley, D. M. Folding and catalysis of the hairpin
ribozyme. Biochem. Soc. Trans. 2005, 33, 461-465.
44. Ferre-D'Amare, A. R.; Zhou, K.; Doudna, J. A. Crystal structure of a hepatitis delta virus
ribozyme. Nature 1998, 395, 567-574.
45. Shih, I. H.; Been, M. D. Catalytic strategies of the hepatitis delta virus ribozymes. Annu.
Rev. Biochem. 2002, 71, 887-917.
46. Ke, A.; Ding, F.; Batchelor, J. D.; Doudna, J. A. Structural roles of monovalent cations in
the HDV ribozyme. Structure 2007, 15, 281-287.
47. Westhof, E.; Hermann, T. Leadzyme RNA catalysis. Nat. Struct. Biol. 1999, 6, 208-209.
48. Wedekind, J. E.; McKay, D. B. Crystal structure of the leadzyme at 1.8 A resolution:
metal ion binding and the implications for catalytic mechanism and allo site ion
regulation. Biochemistry 2003, 42, 9554-9563.
49. Hoogstraten, C. G.; Wank, J. R.; Pardi, A. Active site dynamics in the lead-dependent
ribozyme. Biochemistry 2000, 39, 9951-9958.
50. Michel, F.; Ellington, A. D.; Couture, S.; Szostak, J. W. Phylogenetic and genetic
evidence for base-triples in the catalytic domain of group I introns. Nature 1990, 347,
578-580.
51. Woodson, S. A. Structure and assembly of group I introns. Curr. Opin. Struct. Biol. 2005,
15, 324-330.
52. Forconi, M.; Lee, J.; Lee, J. K.; Piccirilli, J. A.; Herschlag, D. Functional identification of
ligands for a catalytic metal ion in group I introns. Biochemistry 2008, 47, 6883-6894.
53. Michels, W. J., Jr.; Pyle, A. M. Conversion of a group II intron into a new multiple-
turnover ribozyme that selectively cleaves oligonucleotides: elucidation of reaction
mechanism and structure/function relationships. Biochemistry 1995, 34, 2965-2977.
54. Sigel, R. K.; Sashital, D. G.; Abramovitz, D. L.; Palmer, A. G.; Butcher, S. E.; Pyle, A.
M. Solution structure of domain 5 of a group II intron ribozyme reveals a new RNA
motif. Nat. Struct. Mol. Biol. 2004, 11, 187-192.

22
55. Sigel, R. K. O. Group II intron ribozymes and metal ions - a delicate relationship. Eur. J.
Inorg. Chem. 2005, 2281-2292.
56. Erat, M. C.; Sigel, R. K. Divalent metal ions tune the self-splicing reaction of the yeast
mitochondrial group II intron Sc.ai5gamma. J. Biol. Inorg. Chem. 2008, 13, 1025-1036.
57. Saville, B. J.; Collins, R. A. A site-specific self-cleavage reaction performed by a novel
RNA in Neurospora mitochondria. Cell 1990, 61, 685-696.
58. Beattie, T. L.; Olive, J. E.; Collins, R. A. A secondary-structure model for the self-
cleaving region of Neurospora VS RNA. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 4686-
4690.
59. Rastogi, T.; Beattie, T. L.; Olive, J. E.; Collins, R. A. A long-range pseudoknot is
required for activity of the Neurospora VS ribozyme. EMBO J. 1996, 15, 2820-2825.
60. Frank, D. N.; Pace, N. R. Ribonuclease P: unity and diversity in a tRNA processing
ribozyme. Annu. Rev. Biochem. 1998, 67, 153-180.
61. Kazantsev, A. V.; Krivenko, A. A.; Pace, N. R. Mapping metal-binding sites in the
catalytic domain of bacterial RNase P RNA. RNA 2009, 15, 266-276.
62. Famulok, M.; Hartig, J. S.; Mayer, G. Functional aptamers and aptazymes in
biotechnology, diagnostics, and therapy. Chem. Rev. 2007, 107, 3715-3743.
63. Klussmann, S., The Aptamer Handbook: Functional Oligonucleotides and Their
Applications. Wiley-VCH: Weinheim, 2006; p 490.
64. Lee, Y. J.; Lee, S.-W. In vitro selection of cancer-specific RNA aptamers. J. Microbiol.
Biotechnol. 2006, 16, 1149-1153.
65. Ng, E. W.; Shima, D. T.; Calias, P.; Cunningham, E. T., Jr.; Guyer, D. R.; Adamis, A. P.
Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug
Discov. 2006, 5, 123-132.
66. Ulrich, H.; Trujillo, C. A.; Nery, A. A.; Alves, J. M.; Majumder, P.; Resende, R. R.;
Martins, A. H. DNA and RNA aptamers: from tools for basic research towards
therapeutic applications. Comb. Chem. High Throughput Screen. 2006, 9, 619-632.
67. Majumder, P.; Gomes, K. N.; Ulrich, H. Aptamers: from bench side research towards
patented molecules with therapeutic applications. Expert Opin. Ther. Pat. 2009, 19,
1603-1613.
68. Lee, J. F.; Hesselberth, J. R.; Meyers, L. A.; Ellington, A. D. Aptamer database. Nucleic
Acids Res. 2004, 32, D95-100.
69. Mandal, M.; Boese, B.; Barrick, J. E.; Winkler, W. C.; Breaker, R. R. Riboswitches
control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell
2003, 113, 577-586.
70. Mandal, M.; Breaker, R. R. Gene regulation by riboswitches. Nat. Rev. Mol. Cell Biol.
2004, 5, 451-463.
71. Sullenger, B. A. Riboswitches--to kill or save the messenger. N. Engl. J. Med. 2004, 351,
2759-2760.
72. Edwards, T. E.; Klein, D. J.; Ferre-D'Amare, A. R. Riboswitches: small-molecule
recognition by gene regulatory RNAs. Curr. Opin. Struct. Biol. 2007, 17, 273-279.
73. Tucker, B. J.; Breaker, R. R. Riboswitches as versatile gene control elements. Curr. Opin.
Struct. Biol. 2005, 15, 342-348.

23
74. Sashital, D. G.; Butcher, S. E. Flipping off the riboswitch: RNA structures that control
gene expression. ACS Chem. Biol. 2006, 1, 341-345.
75. Topp, S.; Gallivan, J. P. Guiding bacteria with small molecules and RNA. J. Am. Chem.
Soc. 2007, 129, 6807-6811.
76. Cochrane, J. C.; Strobel, S. A. Riboswitch effectors as protein enzyme cofactors. RNA
2008, 14, 993-1002.
77. Topp, S.; Gallivan, J. P. Emerging applications of riboswitches in chemical biology. ACS
Chem. Biol. 2010, 5, 139-148.
78. Yamauchi, T.; Miyoshi, D.; Kubodera, T.; Ban, M.; Nishimura, A.; Sugimoto, N.
Riboswitches for enhancing target gene expression in eukaryotes. ChemBioChem 2008,
9, 1040-1043.
79. Carmi, N.; Shultz, L. A.; Breaker, R. R. In vitro selection of self-cleaving DNAs. Chem.
Biol. 1996, 3, 1039-1046.
80. Breaker, R. R. DNA enzymes. Nat. Biotechnol. 1997, 15, 427-431.
81. Carmi, N.; Balkhi, S. R.; Breaker, R. R. Cleaving DNA with DNA. Proc. Natl. Acad. Sci.
U. S. A. 1998, 95, 2233-2237.
82. Kurz, M.; Breaker, R. R. In vitro selection of nucleic acid enzymes. Curr. Top.
Microbiol. Immunol. 1999, 243, 137-158.
83. Li, Y.; Breaker, R. R. Deoxyribozymes: new players in the ancient game of biocatalysis.
Curr. Opin. Struct. Biol. 1999, 9, 315-323.
84. Sen, D.; Geyer, C. R. DNA enzymes. Curr. Opin. Chem. Biol. 1998, 2, 680-687.
85. Jaschke, A. Artificial ribozymes and deoxyribozymes. Curr. Opin. Struct. Biol. 2001, 11,
321-326.
86. Peracchi, A. DNA catalysis: potential, limitations, open questions. ChemBioChem 2005,
6, 1316-1322.
87. Silverman, S. K. Deoxyribozymes: DNA catalysts for bioorganic chemistry. Org. Biomol.
Chem. 2004, 2, 2701-2706.
88. Silverman, S. K. Breaking up is easy to do (if you're a DNA enzyme that cleaves RNA).
Chem. Biol. 2004, 11, 7-8.
89. Achenbach, J. C.; Chiuman, W.; Cruz, R. P.; Li, Y. DNAzymes: from creation in vitro to
application in vivo. Curr. Pharm. Biotechnol. 2004, 5, 321-336.
90. Silverman, S. K. In vitro selection, characterization, and application of deoxyribozymes
that cleave RNA. Nucleic Acids Res. 2005, 33, 6151-6163.
91. Hobartner, C.; Silverman, S. K. Recent advances in DNA catalysis. Biopolymers 2007,
87, 279-292.
92. Baum, D. A.; Silverman, S. K. Deoxyribozymes: useful DNA catalysts in vitro and in
vivo. Cell. Mol. Life Sci. 2008, 65, 2156-2174.
93. Willner, I.; Shlyahovsky, B.; Zayats, M.; Willner, B. DNAzymes for sensing,
nanobiotechnology and logic gate applications. Chem. Soc. Rev. 2008, 37, 1153-1165.
94. Silverman, S. K. Deoxyribozymes: selection design and serendipity in the development
of DNA catalysts. Acc. Chem. Res. 2009, 42, 1521-1531.
95. Schlosser, K.; Li, Y. Biologically inspired synthetic enzymes made from DNA. Chem.
Biol. 2009, 16, 311-322.

24
96. Schlosser, K.; Lam, J. C.; Li, Y. Characterization of long RNA-cleaving deoxyribozymes
with short catalytic cores: the effect of excess sequence elements on the outcome of in
vitro selection. Nucleic Acids Res. 2006, 34, 2445-2454.
97. Kim, H. K.; Rasnik, I.; Liu, J.; Ha, T.; Lu, Y. Dissecting metal ion-dependent folding and
catalysis of a single DNAzyme. Nat. Chem. Biol. 2007, 3, 763-768.
98. Shen, Y.; Mackey, G.; Rupcich, N.; Gloster, D.; Chiuman, W.; Li, Y.; Brennan, J. D.
Entrapment of fluorescence signaling DNA enzymes in sol-gel-derived materials for
metal ion sensing. Anal. Chem. 2007, 79, 3494-3503.
99. Kim, H. K.; Liu, J.; Li, J.; Nagraj, N.; Li, M.; Pavot, C. M.; Lu, Y. Metal-dependent
global folding and activity of the 8-17 DNAzyme studied by fluorescence resonance
energy transfer. J. Am. Chem. Soc. 2007, 129, 6896-6902.
100. Mazumdar, D.; Nagraj, N.; Kim, H. K.; Meng, X.; Brown, A. K.; Sun, Q.; Li, W.; Lu, Y.
Activity, Folding and Z-DNA Formation of the 8-17 DNAzyme in the Presence of
Monovalent Ions. J. Am. Chem. Soc. 2009, 131, 5506-5515.
101. Liu, Y.; Sen, D. Light-regulated catalysis by an RNA-cleaving deoxyribozyme. J. Mol.
Biol. 2004, 341, 887-892.
102. Liu, Y.; Sen, D. A contact photo-cross-linking investigation of the active site of the 8-17
deoxyribozyme. J. Mol. Biol. 2008, 381, 845-859.
103. Liu, Y.; Sen, D. Local rather than global folding enables the lead-dependent activity of
the 8-17 deoxyribozyme: evidence from contact photo-crosslinking. J. Mol. Biol. 2010,
395, 234-241.
104. Liu, J.; Lu, Y. FRET study of a trifluorophore-labeled DNAzyme. J. Am. Chem. Soc.
2002, 124, 15208-15216.
105. Liu, J.; Lu, Y. Improving fluorescent DNAzyme biosensors by combining inter- and
intramolecular quenchers. Anal. Chem. 2003, 75, 6666-6672.
106. Liu, J.; Lu, Y. Accelerated color change of gold nanoparticles assembled by DNAzymes
for simple and fast colorimetric Pb2+ detection. J. Am. Chem. Soc. 2004, 126, 12298-
12305.
107. Liu, J.; Lu, Y. Fluorescent DNAzyme biosensors for metal ions based on catalytic
molecular beacons. Methods Mol. Biol. 2006, 335, 275-288.
108. Peracchi, A.; Bonaccio, M.; Clerici, M. A mutational analysis of the 8-17 deoxyribozyme
core. J. Mol. Biol. 2005, 352, 783-794.
109. Schlosser, K.; Gu, J.; Sule, L.; Li, Y. Sequence-function relationships provide new
insight into the cleavage site selectivity of the 8-17 RNA-cleaving deoxyribozyme.
Nucleic Acids Res. 2008, 36, 1472-1481.
110. Bonaccio, M.; Credali, A.; Peracchi, A. Kinetic and thermodynamic characterization of
the RNA-cleaving 8-17 deoxyribozyme. Nucleic Acids Res. 2004, 32, 916-925.
111. Cruz, R. P.; Withers, J. B.; Li, Y. Dinucleotide junction cleavage versatility of 8-17
deoxyribozyme. Chem. Biol. 2004, 11, 57-67.
112. Lee, N. K.; Koh, H. R.; Han, K. Y.; Kim, S. K. Folding of 8-17 deoxyribozyme studied
by three-color alternating-laser excitation of single molecules. J. Am. Chem. Soc. 2007,
129, 15526-15534.

25
113. Xiang, Y.; Tong, A.; Lu, Y. Abasic site-containing DNAzyme and aptamer for label-free
fluorescent detection of Pb(2+) and adenosine with high sensitivity, selectivity, and
tunable dynamic range. J. Am. Chem. Soc. 2009, 131, 15352-15357.
114. Schlosser, K.; Li, Y. Tracing sequence diversity change of RNA-cleaving
deoxyribozymes under increasing selection pressure during in vitro selection.
Biochemistry 2004, 43, 9695-9707.
115. Chiuman, W.; Li, Y. Efficient signaling platforms built from a small catalytic DNA and
doubly labeled fluorogenic substrates. Nucleic Acids Res. 2007, 35, 401-405.
116. Santoro, S. W.; Joyce, G. F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl.
Acad. Sci. U. S. A. 1997, 94, 4262-4266.
117. Chakraborti, S.; Banerjea, A. C. Inhibition of HIV-1 gene expression by novel DNA
enzymes targeted to cleave HIV-1 TAR RNA: potential effectiveness against all HIV-1
isolates. Mol. Ther. 2003, 7, 817-826.
118. Flynn-Charlebois, A.; Prior, T. K.; Hoadley, K. A.; Silverman, S. K. In vitro evolution of
an RNA-cleaving DNA enzyme into an RNA ligase switches the selectivity from 3'-5' to
2'-5'. J. Am. Chem. Soc. 2003, 125, 5346-5350.
119. Wang, H.; Kim, Y.; Liu, H.; Zhu, Z.; Bamrungsap, S.; Tan, W. Engineering a
unimolecular DNA-catalytic probe for single lead ion monitoring. J. Am. Chem. Soc.
2009, 131, 8221-8226.
120. Brown, A. K.; Li, J.; Pavot, C. M.; Lu, Y. A lead-dependent DNAzyme with a two-step
mechanism. Biochemistry 2003, 42, 7152-7161.
121. Faulhammer, D.; Famulok, M. The Ca2+
ion as a cofactor for a novel RNA-cleaving
deoxyribozyme. Angew. Chem. Int. Ed. 1996, 35, 2837-2841.
122. Faulhammer, D.; Famulok, M. Characterization and divalent metal-ion dependence of in
vitro selected deoxyribozymes which cleave DNA/RNA chimeric oligonucleotides. J.
Mol. Biol. 1997, 269, 188-202.
123. Li, J.; Zheng, W.; Kwon, A. H.; Lu, Y. In vitro selection and characterization of a highly
efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000, 28,
481-488.
124. Geyer, C. R.; Sen, D. Evidence for the metal-cofactor independence of an RNA
phosphodiester-cleaving DNA enzyme. Chem. Biol. 1997, 4, 579-593.
125. Li, J.; Lu, Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J.
Am. Chem. Soc. 2000, 122, 10466-10467.
126. Swearingen, C. B.; Wernette, D. P.; Cropek, D. M.; Lu, Y.; Sweedler, J. V.; Bohn, P. W.
Immobilization of a catalytic DNA molecular beacon on Au for Pb(II) detection. Anal.
Chem. 2005, 77, 442-448.
127. Wernette, D. P.; Mead, C.; Bohn, P. W.; Lu, Y. Surface immobilization of catalytic
beacons based on ratiometric fluorescent DNAzyme sensors: a systematic study.
Langmuir 2007, 23, 9513-9521.
128. Wernette, D. P.; Swearingen, C. B.; Cropek, D. M.; Lu, Y.; Sweedler, J. V.; Bohn, P. W.
Incorporation of a DNAzyme into Au-coated nanocapillary array membranes with an
internal standard for Pb(ii) sensing. Analyst 2006, 131, 41-47.
129. Nagraj, N.; Liu, J.; Sterling, S.; Wu, J.; Lu, Y. DNAzyme catalytic beacon sensors that
resist temperature-dependent variations. Chem. Commun. 2009, 4103-4105.

26
130. Macdonald, J.; Stefanovic, D.; Stojanovic, M. N. Solution-phase molecular-scale
computation with deoxyribozyme-based logic gates and fluorescent readouts. Methods
Mol. Biol. 2006, 335, 343-363.
131. Stojanovic, M. N.; Mitchell, T. E.; Stefanovic, D. Deoxyribozyme-based logic gates. J.
Am. Chem. Soc. 2002, 124, 3555-3561.
132. Liu, J.; Wernette, D. P.; Lu, Y. Proofreading and error removal in a nanomaterial
assembly. Angew. Chem. Int. Ed. 2005, 44, 7290-7293.
133. Lu, Y.; Liu, J. Smart nanomaterials inspired by biology: dynamic assembly of error-free
nanomaterials in response to multiple chemical and biological stimuli. Acc. Chem. Res.
2007, 40, 315-323.
134. Wang, Y.; Silverman, S. K. Directing the outcome of deoxyribozyme selections to favor
native 3'-5' RNA ligation. Biochemistry 2005, 44, 3017-3023.
135. Breaker, R. R.; Joyce, G. F. A DNA enzyme with Mg(2+)-dependent RNA
phosphoesterase activity. Chem. Biol. 1995, 2, 655-660.
136. Roth, A.; Breaker, R. R. An amino acid as a cofactor for a catalytic polynucleotide. Proc.
Natl. Acad. Sci. U. S. A. 1998, 95, 6027-6031.
137. Santoro, S. W.; Joyce, G. F.; Sakthivel, K.; Gramatikova, S.; Barbas, C. F., 3rd RNA
cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc.
2000, 122, 2433-2439.
138. Feldman, A. R.; Sen, D. A new and efficient DNA enzyme for the sequence-specific
cleavage of RNA. J. Mol. Biol. 2001, 313, 283-294.
139. Perrin, D. M.; Garestier, T.; Helene, C. Bridging the gap between proteins and nucleic
acids: a metal-independent RNAseA mimic with two protein-like functionalities. J. Am.
Chem. Soc. 2001, 123, 1556-1563.
140. Mei, S. H.; Liu, Z.; Brennan, J. D.; Li, Y. An efficient RNA-cleaving DNA enzyme that
synchronizes catalysis with fluorescence signaling. J. Am. Chem. Soc. 2003, 125, 412-
420.
141. Liu, Z.; Mei, S. H.; Brennan, J. D.; Li, Y. Assemblage of signaling DNA enzymes with
intriguing metal-ion specificities and pH dependences. J. Am. Chem. Soc. 2003, 125,
7539-7545.
142. Liu, J.; Brown, A. K.; Meng, X.; Cropek, D. M.; Istok, J. D.; Watson, D. B.; Lu, Y. A
catalytic beacon sensor for uranium with parts-per-trillion sensitivity and millionfold
selectivity. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2056-2061.
143. Chandra, M.; Sachdeva, A.; Silverman, S. K. DNA-catalyzed sequence-specific
hydrolysis of DNA. Nat. Chem. Biol. 2009, 5, 718-720.
144. Wang, Y.; Silverman, S. K. Deoxyribozymes that synthesize branched and lariat RNA. J.
Am. Chem. Soc. 2003, 125, 6880-6881.
145. Coppins, R. L.; Silverman, S. K. A DNA enzyme that mimics the first step of RNA
splicing. Nat. Struct. Mol. Biol. 2004, 11, 270-274.
146. Flynn-Charlebois, A.; Wang, Y.; Prior, T. K.; Rashid, I.; Hoadley, K. A.; Coppins, R. L.;
Wolf, A. C.; Silverman, S. K. Deoxyribozymes with 2'-5' RNA ligase activity. J. Am.
Chem. Soc. 2003, 125, 2444-2454.

27
147. Hoadley, K. A.; Purtha, W. E.; Wolf, A. C.; Flynn-Charlebois, A.; Silverman, S. K.
Zn2+-dependent deoxyribozymes that form natural and unnatural RNA linkages.
Biochemistry 2005, 44, 9217-9231.
148. Cuenoud, B.; Szostak, J. W. A DNA metalloenzyme with DNA ligase activity. Nature
1995, 375, 611-614.
149. Sreedhara, A.; Li, Y.; Breaker, R. R. Ligating DNA with DNA. J. Am. Chem. Soc. 2004,
126, 3454-3460.
150. Li, Y.; Breaker, R. R. Phosphorylating DNA with DNA. Proc. Natl. Acad. Sci. U. S. A.
1999, 96, 2746-2751.
151. Hobartner, C.; Pradeepkumar, P. I.; Silverman, S. K. Site-selective depurination by a
periodate-dependent deoxyribozyme. Chem. Commun. 2007, 2255-2257.
152. Li, Y.; Liu, Y.; Breaker, R. R. Capping DNA with DNA. Biochemistry 2000, 39, 3106-
3114.
153. Chinnapen, D. J.; Sen, D. A deoxyribozyme that harnesses light to repair thymine dimers
in DNA. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 65-69.
154. Burmeister, J.; von Kiedrowski, G.; Ellington, A. D. Cofactor-assisted self-cleavage in
DNA libraries with a 3'-'5'-phosphoramidate bond. Angew.Chem. Int. Ed. 1997, 36, 1321-
1324.
155. Sheppard, T. L.; Ordoukhanian, P.; Joyce, G. F. A DNA enzyme with N-glycosylase
activity. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 7802-7807.
156. Li, Y.; Sen, D. A catalytic DNA for porphyrin metallation. Nat. Struct. Biol. 1996, 3,
743-747.
157. Chandra, M.; Silverman, S. K. DNA and RNA can be equally efficient catalysts for
carbon-carbon bond formation. J. Am. Chem. Soc. 2008, 130, 2936-2937.
158. Kazakov, S. A.; Hecht, S. M., Nucleic Acid-Metal Ion Interactions. In Encyclopedia of
Inorganic Chemistry, King, R. B., Ed. Wiley: Chichester, 2005; Vol. VI, p 3690.
159. Sigel, R. K.; Pyle, A. M. Alternative roles for metal ions in enzyme catalysis and the
implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97-113.
160. Chen, X.; Li, N.; Ellington, A. D. Ribozyme catalysis of metabolism in the RNA world.
Chem. Biodivers. 2007, 4, 633-655.
161. Freisinger, E.; Sigel, R. K. O. From nucleotides to ribozymes-A comparison of their
metal ion binding properties. Coord. Chem. Rev. 2007, 251, 1834-1851.
162. Erkkila, K. E.; Odom, D. T.; Barton, J. K. Recognition and reaction of
metallointercalators with DNA. Chem. Rev. 1999, 99, 2777-2796.
163. Reedijk, J. New clues for platinum antitumor chemistry: kinetically controlled metal
binding to DNA. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3611-3616.
164. Jamieson, E. R.; Lippard, S. J. Structure, Recognition, and Processing of Cisplatin-DNA
Adducts. Chem. Rev. 1999, 99, 2467-2498.
165. Zhang, C. X.; Lippard, S. J. New metal complexes as potential therapeutics. Curr. Opin.
Chem. Biol. 2003, 7, 481-489.
166. Schnabl, J.; Sigel, R. K. Controlling ribozyme activity by metal ions. Curr. Opin. Chem.
Biol., doi:10.1016/j.cbpa.2009.1011.1024
167. Sigel, R. K. Intimate Relationships between Metal Ions and Nucleic Acids. Angew.
Chem. Int. Ed. 2007, 46, 654-656.

28
168. Lippert, B., Alterations of nucleobase pKa values upon metal coordination: Origins and
consequences. John Wiley & Sons: 2005; Vol. 54, p 385-447.
169. Wilson, T. J.; Ouellet, J.; Zhao, Z. Y.; Harusawa, S.; Araki, L.; Kurihara, T.; Lilley, D.
M. Nucleobase catalysis in the hairpin ribozyme. RNA 2006, 12, 980-987.
170. Zhao, Z. Y.; McLeod, A.; Harusawa, S.; Araki, L.; Yamaguchi, M.; Kurihara, T.; Lilley,
D. M. Nucleobase participation in ribozyme catalysis. J. Am. Chem. Soc. 2005, 127,
5026-5027.
171. Murray, J. B.; Seyhan, A. A.; Walter, N. G.; Burke, J. M.; Scott, W. G. The hammerhead,
hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem.
Biol. 1998, 5, 587-595.
172. O'Rear, J. L.; Wang, S.; Feig, A. L.; Beigelman, L.; Uhlenbeck, O. C.; Herschlag, D.
Comparison of the hammerhead cleavage reactions stimulated by monovalent and
divalent cations. RNA 2001, 7, 537-545.
173. Perrotta, A. T.; Been, M. D. HDV ribozyme activity in monovalent cations. Biochemistry
2006, 45, 11357-11365.
174. Curtis, E. A.; Bartel, D. P. The hammerhead cleavage reaction in monovalent cations.
RNA 2001, 7, 546-552.
175. Perrotta, A. T.; Been, M. D. A single nucleotide linked to a switch in metal ion reactivity
preference in the HDV ribozymes. Biochemistry 2007, 46, 5124-5130.
176. Earnshaw, D. J.; Gait, M. J. Hairpin ribozyme cleavage catalyzed by aminoglycoside
antibiotics and the polyamine spermine in the absence of metal ions. Nucleic Acids Res
1998, 26, 5551-5561.
177. Takagi, Y.; Warashina, M.; Stec, W. J.; Yoshinari, K.; Taira, K. Recent advances in the
elucidation of the mechanisms of action of ribozymes. Nucleic Acids Res. 2001, 29,
1815-1834.
178. Vogt, M.; Lahiri, S.; Hoogstraten, C. G.; Britt, R. D.; DeRose, V. J. Coordination
environment of a site-bound metal ion in the hammerhead ribozyme determined by 15N
and 2H ESEEM spectroscopy. J. Am. Chem. Soc. 2006, 128, 16764-16770.
179. Murray, J. B.; Scott, W. G. Does a single metal ion bridge the A-9 and scissile phosphate
groups in the catalytically active hammerhead ribozyme structure? J. Mol. Biol. 2000,
296, 33-41.
180. Stahley, M. R.; Strobel, S. A. Structural evidence for a two-metal-ion mechanism of
group I intron splicing. Science 2005, 309, 1587-1590.
181. Adams, P. L.; Stahley, M. R.; Kosek, A. B.; Wang, J.; Strobel, S. A. Crystal structure of a
self-splicing group I intron with both exons. Nature 2004, 430, 45-50.
182. Young, K. J.; Gill, F.; Grasby, J. A. Metal ions play a passive role in the hairpin
ribozyme catalysed reaction. Nucleic Acids Res. 1997, 25, 3760-3766.
183. Suga, H.; Cowan, J. A.; Szostak, J. W. Unusual metal ion catalysis in an acyl-transferase
ribozyme. Biochemistry 1998, 37, 10118-10125.
184. Hampel, A.; Tritz, R. RNA catalytic properties of the minimum (-)sTRSV sequence.
Biochemistry 1989, 28, 4929-4933.
185. Thill, G.; Blumenfeld, M.; Lescure, F.; Vasseur, M. Self-cleavage of a 71 nucleotide-long
ribozyme derived from hepatitis delta virus genomic RNA. Nucleic Acids Res. 1991, 19,
6519-6525.

29
186. Uhlenbeck, O. C. A small catalytic oligoribonucleotide. Nature 1987, 328, 596-600.
187. Koizumi, M.; Ohtsuka, E. Effects of phosphorothioate and 2-amino groups in
hammerhead ribozymes on cleavage rates and Mg2+ binding. Biochemistry 1991, 30,
5145-5150.
188. Szewczak, A. A.; Kosek, A. B.; Piccirilli, J. A.; Strobel, S. A. Identification of an active
site ligand for a group I ribozyme catalytic metal ion. Biochemistry 2002, 41, 2516-2525.
189. Nesbitt, S.; Hegg, L. A.; Fedor, M. J. An unusual pH-independent and metal-ion-
independent mechanism for hairpin ribozyme catalysis. Chem. Biol. 1997, 4, 619-630.
190. Singh, K. K.; Parwaresch, R.; Krupp, G. Rapid kinetic characterization of hammerhead
ribozymes by real-time monitoring of fluorescence resonance energy transfer (FRET).
RNA 1999, 5, 1348-1356.
191. Bassi, G. S.; Murchie, A. I.; Walter, F.; Clegg, R. M.; Lilley, D. M. Ion-induced folding
of the hammerhead ribozyme: a fluorescence resonance energy transfer study. EMBO J.
1997, 16, 7481-7489.
192. Penedo, J. C.; Wilson, T. J.; Jayasena, S. D.; Khvorova, A.; Lilley, D. M. Folding of the
natural hammerhead ribozyme is enhanced by interaction of auxiliary elements. RNA
2004, 10, 880-888.
193. Wilson, T. J.; Lilley, D. M. Metal ion binding and the folding of the hairpin ribozyme.
RNA 2002, 8, 587-600.
194. Wedekind, J. E.; McKay, D. B. Crystallographic structures of the hammerhead ribozyme:
relationship to ribozyme folding and catalysis. Annu. Rev. Biophys. Biomol. Struct. 1998,
27, 475-502.
195. Wedekind, J. E.; McKay, D. B. Crystal structure of a lead-dependent ribozyme revealing
metal binding sites relevant to catalysis. Nat. Struct. Biol. 1999, 6, 261-268.
196. Alam, S.; Grum-Tokars, V.; Krucinska, J.; Kundracik, M. L.; Wedekind, J. E.
Conformational heterogeneity at position U37 of an all-RNA hairpin ribozyme with
implications for metal binding and the catalytic structure of the S-turn. Biochemistry
2005, 44, 14396-14408.
197. Shechner, D. M.; Grant, R. A.; Bagby, S. C.; Koldobskaya, Y.; Piccirilli, J. A.; Bartel, D.
P. Crystal structure of the catalytic core of an RNA-polymerase ribozyme. Science 2009,
326, 1271-1275.
198. Golden, B. L.; Gooding, A. R.; Podell, E. R.; Cech, T. R. A preorganized active site in
the crystal structure of the Tetrahymena ribozyme. Science 1998, 282, 259-264.
199. Furtig, B.; Richter, C.; Wohnert, J.; Schwalbe, H. NMR spectroscopy of RNA.
ChemBioChem 2003, 4, 936-962.
200. DeRose, V. J. Two decades of RNA catalysis. Chem. Biol. 2002, 9, 961-969.
201. Nowakowski, J.; Shim, P. J.; Prasad, G. S.; Stout, C. D.; Joyce, G. F. Crystal structure of
an 82-nucleotide RNA-DNA complex formed by the 10-23 DNA enzyme. Nat. Struct.
Biol. 1999, 6, 151-156.
202. O'Sullivan, C. K. Aptasensors--the future of biosensing? Anal. Bioanal. Chem. 2002, 372,
44-48.
203. Bunka, D. H.; Stockley, P. G. Aptamers come of age - at last. Nat. Rev. Microbiol. 2006,
4, 588-596.

30
204. Liu, J.; Lu, Y. Rational design of "turn-on" allosteric DNAzyme catalytic beacons for
aqueous mercury ions with ultrahigh sensitivity and selectivity. Angew. Chem. Int. Ed.
2007, 46, 7587-7590.
205. Wang, Z.; Heon Lee, J.; Lu, Y. Highly sensitive "turn-on" fluorescent sensor for Hg2+
in
aqueous solution based on structure-switching DNA. Chem. Commun. 2008, 6005-6007.
206. Dalavoy, T. S.; Wernette, D. P.; Gong, M.; Sweedler, J. V.; Lu, Y.; Flachsbart, B. R.;
Shannon, M. A.; Bohn, P. W.; Cropek, D. M. Immobilization of DNAzyme catalytic
beacons on PMMA for Pb2+ detection. Lab Chip 2008, 8, 786-793.
207. Tan, L.; Li, Y.; Drake, T. J.; Moroz, L.; Wang, K.; Li, J.; Munteanu, A.; Chaoyong, J. Y.;
Martinez, K.; Tan, W. Molecular beacons for bioanalytical applications. Analyst 2005,
130, 1002-1005.
208. Kim, Y.; Sohn, D.; Tan, W. Molecular beacons in biomedical detection and clinical
diagnosis. Int. J. Clin. Exp. Pathol. 2008, 1, 105-116.
209. Li, J.; Cao, Z. C.; Tang, Z.; Wang, K.; Tan, W. Molecular beacons for protein-DNA
interaction studies. Methods Mol. Biol. 2008, 429, 209-224.
210. Nutiu, R.; Li, Y. Structure-switching signaling aptamers. J. Am. Chem. Soc. 2003, 125,
4771-4778.
211. Nutiu, R.; Li, Y. Structure-switching signaling aptamers: transducing molecular
recognition into fluorescence signaling. Chem. Eur. J. 2004, 10, 1868-1876.
212. Nutiu, R.; Li, Y. Aptamers with fluorescence-signaling properties. Methods 2005, 37, 16-
25.
213. Nutiu, R.; Li, Y. In vitro selection of structure-switching signaling aptamers. Angew.
Chem. Int. Ed. 2005, 44, 1061-1065.
214. Xu, W.; Lu, Y. Label-free fluorescent aptamer sensor based on regulation of malachite
green fluorescence. Anal. Chem. 2010, 82, 574-578.
215. Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G. C.; Cheng, J.; Lu, Y. Reversible cell-
specific drug delivery with aptamer-functionalized liposomes. Angew. Chem. Int. Ed.
2009, 48, 6494-6498.
216. Famulok, M. Allosteric aptamers and aptazymes as probes for screening approaches.
Curr. Opin. Mol. Ther. 2005, 7, 137-143.
217. Ogawa, A.; Maeda, M. Aptazyme-based riboswitches as label-free and detector-free
sensors for cofactors. Bioorg. Med. Chem. Lett. 2007, 17, 3156-3160.
218. Rueda, D.; Bokinsky, G.; Rhodes, M. M.; Rust, M. J.; Zhuang, X.; Walter, N. G. Single-
molecule enzymology of RNA: essential functional groups impact catalysis from a
distance. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 10066-10071.
219. Cho, E. J.; Yang, L.; Levy, M.; Ellington, A. D. Using a deoxyribozyme ligase and
rolling circle amplification to detect a non-nucleic acid analyte, ATP. J. Am. Chem. Soc.
2005, 127, 2022-2023.

31
2 Low Temperature-Resistant Lead (Pb2+
) Sensing Based on a Fluorescent
Catalytic Beacon Sensor
Note and Acknowledgments: This work was done in collaboration with Dr. Juewen Liu
and undergraduates Stephanie Sterling and Jenny Wu. This chapter is the basis of a published
manuscript, “DNAzyme Catalytic Beacon Sensors that Resist Temperature-dependent
Variations” Nandini Nagraj, Juewen Liu, Stephanie Sterling, Jenny Wu and Yi Lu Chem.
Commun., 2009, 27, 4103-4105.
2.1 Introduction
2.1.1 Importance of Pb2+
sensing in water
Metal ions play a critical role in most of the everyday processes of life, from biological
and functional roles, to those in industry and medicine. However, several heavy metal ions,
previously used for some of the afore-mentioned applications, have been shown to have long-
term toxic effects on humans. Some of the most common metals responsible for “heavy-metal
poisoning” include lead, mercury, arsenic, cadmium, barium, thallium and antimony.
(http://extoxnet.orst.edu/faqs/safedrink/metals.htm) Detection of these trace contaminants is
therefore particularly important in shared natural resources including soil and water. Lead (Pb2+
)
is a non-essential, detrimental trace element that has a ubiquitous presence in water resources,
through its use over centuries in lead vessels and more recently in paints, pipes and as additives
in gasoline. Some of the chronic toxicity attributed to Pb2+
includes renal malfunction, anemia,
gout and nervous system disorders, including inhibition of brain development in children and

32
fetuses.1-3
Children in particular are much more susceptible to the toxic effects of Pb2+
in
comparison with adults and long-term exposure to non-toxic levels of Pb2+
is known to cause
neurodegeneration and learning disabilities in children.4 The Environmental Protection Agency
(EPA) recommended Maximum Contaminant Level (MCL) of Pb2+
in water is 15 ppb or 72 nM
(http://www.epa.gov/lead/).
2.1.2 Current methods of Pb2+
detection
Standard laboratory detection techniques used for the detection of Pb2+
include the ICP-
MS or Inductively Coupled Plasma-Mass Spectrometry method5-8
and the AAS or Atomic
Absorption Spectrometry methods.9-12
These instrumental methods exhibit very high selectivity
and sensitivity toward the target metal ion, in this instance Pb2+
, with detection limits in the 0.1-1
ppt range.13
Despite this level of sensitivity and accuracy, one disadvantage of these techniques
is that they cannot be used for real-time and on-site measurements. The detection analysis is
restricted to specific laboratory locations and skilled technicians.
Various electrochemical and optical sensors have also been developed, but many of these
sensors suffer from interferences and are also more expensive.14-16
A reversible fluorescent
chemosensor for Pb2+
based on its chelation by rhodamine B has also been synthesized and
characterized and the selectivity of this sensor is about 200 times greater for Pb2+
than for Zn2+
.17
However, sensing in this case is carried out in 100 % acetonitrile rather than in water. Portable
Pb2+
detection kits based on the color change upon Pb2+
chelation by sodium rhodizonate18-20
as
well as X-ray fluorescence have also been developed, however, the former suffers from
interferences due to pigmentation while the latter, although highly quantitative, requires
expensive instrumentation and a radioactive source for generation of the characteristic X-rays

33
from Pb2+
. The antibody approach has also been developed by Blake and co-workers21
who
generated monoclonal antibodies against Pb2+
-bound KLH (keyhole limpet hemocyanin) via a
bifunctional chelator, and one of the antibodies secreted bound the Pb2+
- chelator complex but
not the metal-free chelator.21
Detection of Pb2+
based on the ELISA (Enzyme-Linked
ImmunoSorbent Assay) method using this antibody was 56 nM, with minimal cross-reactivity
toward other divalent and trivalent metal ions. While this method showed clear utility for
commercial Pb2+
detection purposes, the ELISA assays are generally expensive due to the cost
associated with generation of the monoclonal antibodies.22, 23
2.1.3 Use of DNAzymes for Pb2+
detection in water
Functional DNA molecules, specifically deoxyribozymes or DNAzymes have been
isolated for specific targets through an in vitro selection method24
which was specifically
referred to as SELEX (Systematic Evolution of Ligands through EXponential Enrichment) in
relevance to the isolation of aptamers.25
A previous member of the Lu lab isolated a DNAzyme
called 17E through this in vitro selection process.26
Its core was identical to the 8-17 DNAzyme
motifs, and it catalyzed the cleavage of a single RNA base on the substrate strand in the presence
of Pb2+
with high specificity and selectivity over other metal ions. This DNAzyme was also
converted into a fluorescent sensor for Pb2+
through the introduction of a fluorophore/quencher
pair on the substrate/enzyme arms respectively.27
In the absence of the divalent metal ion (M2+
),
the fluorescence of the fluorophore (TAMRA) was quenched due to its proximity to the enzyme
strand containing the quencher molecule (DABCYL). The presence of the M2+
however, caused
the cleavage of the single RNA base on the substrate strand followed by release of the substrate
strand containing the fluorophore. This is lead to a significant increase of the fluorescent

34
intensity over the background as the quencher was no longer in close proximity to the
fluorophore. The detection limit achieved using this method was 10 nM and the selectivity for
Pb2+
over other M2+
ions tested was at least 80-fold. This method of using a DNAzyme-based
catalytic beacon sensor for the fluorescent detection of Pb2+
was significant because this was an
example of a relatively simple and environmentally-benign platform based on DNA for Pb2+
detection in water.27
A drawback of this method, however, was significant background
fluorescence intensity in the absence of any M2+
. This was overcome through the introduction of
a second quencher on the substrate strand to aid in both intramolecular and intermolecular
quenching.28
Immobilization of this DNAzyme on a gold surface further lowered its detection
limit to 1 nM due to the extremely low background fluorescence achieved through successive
washing of the excess DNA.29, 30
Sensor-regeneration and long-term storage could also be
achieved through this method.31
The use of micro- and nano-fluidic devices for Pb2+
detection
through the use of the 8-17 DNAzyme has also been demonstrated.32, 33
The microfluidic device
was able to achieve Pb2+
sensing through the use of < 1 nL of DNA solution,32
while a voltage-
controlled nanofluidic device could detect 11 nM Pb2+
through the use of an interconnecting
nanocapillary array.33
Electrochemical sensors based on the 8-17 DNAzyme have also been
developed and show high sensitivity towards Pb2+
, with detection limits as low as 1 nM.34, 35
Colorimetric sensors for Pb2+
have also developed through the functionalization of gold
nanoparticles with the 17E DNAzyme.36-39
Sensing is based on the color change that occurs due
to disassembly of the nanoparticles in the presence of Pb2+
, wherein a color change from blue to
red is observed. A lateral-flow dipstick for Pb2+
detection in paint has also been recently
developed which has a detection of limit of 0.5 µM and has been successfully used to

35
demonstrate Pb2+
detection in paint samples collected from real homes, in accordance with the
federally-defined threshold for paint to be classified as lead-based.40
The nanoparticles in this
case are conjugated to the DNAzyme and used as labels on the DNA and the color on the test
strips is due to accumulation of gold nanoparticles at the appropriate zones of the device.
2.1.4 Research goals
Despite the great promise and huge potentials of these DNAzyme sensors, there is a
major unsolved issue that prevents their widespread application. Since DNA hybridization and
dehybridization play a critical role in almost all DNAzyme sensor designs, the sensor
performance is vulnerable to temperature variations. This makes simple on-site and real-time
detection difficult, particularly since temperature variations commonly occur between different
locations and times of the day. Implementing temperature control units or calibrations increases
both the cost and complexity of the sensing operation. Therefore, it is highly desirable to design
functional DNA sensors whose performance withstands temperature variations in the practical
range. In this chapter, a novel strategy of using mismatches in the DNAzyme to tune and control
its temperature stability so that the catalytic beacon exhibits similar fluorescence signals at
temperatures between 4 °C and 30 °C is demonstrated. In the process, the importance of the
nature and the position of the mismatch are also shown and therefore, the strategy demonstrated
here can be applied to other nucleic acid-based sensors as well.

36
2.2 Materials and methods
2.2.1 Materials
All oligonucleotides were synthesized and HPLC-purified by Integrated DNA
Technologies Inc. (Coralville, Iowa). The substrate strand was labeled with FAM (fluorescein)
on the 5' end and a blackhole quencher (BHQ1®
) on the 3' end while all the different enzyme
strands used were labeled with a DABCYL quencher on the 3' end. The sequences of all the
enzyme and substrate strands used in this study are shown in Table 2.1. Ultrapure HEPES-
sodium salt, Trizma® acetate and PIPES were purchased from Sigma-Aldrich or USB
Corporation while ultrapure hydrochloric acid, puratronic® sodium chloride and puratronic
® lead
acetate were purchased from Alfa-Aesar. The latter was used for preparation of lead stock
solutions in 0.5% of acetic acid. Buffers were brought up to pH using ultrapure hydrochloric
acid. Stock solutions of buffers were treated with Chelex 100 beads to chelate any metal ion as
an added level of precaution, to prevent any contamination.
2.2.2 Sensor preparation
The enzyme substrate complex was prepared with 1 µM each of +5_17E (A16.7-G) or
+5_17E (T16.5-C) and +5_17S in either 50 mM HEPES buffer, pH 7.2 or a mixed buffer of 50
mM Tris acetate/PIPES at pH 6.8, both containing 100 mM of NaCl. The sample was heated to
80 °C for 3 min and cooled to 23 °C over 45 min for annealing the enzyme and substrate strands.
Sensors made in the mixed buffer solution at pH 6.8 could be air-dried in small aliquots and
stored at room temperature for over one month without significant loss of activity. The sensors

37
were then rehydrated in 50 mM HEPES, pH 7.2 containing 100 mM NaCl and used for
fluorescence measurements.
2.2.3 Fluorescence measurements and calculations
Fluorescence experiments were carried out on a Fluromax-2 fluorimeter (HORIBA Jobin
Yvon inc., Edison, NJ) using the Constant Wavelength Analysis (CWA) mode. The temperature
of the sample was adjusted using a temperature controller connected to the fluorimeter. (LSI-
3751 from Wavelength Electronics) The performance of the sensors was monitored by studying
the overall change in the fluorescence intensity at 520 nm, at 10 sec. time intervals upon Pb2+
addition (Figure 2.1b). The initial rate of fluorescent increase, attributable to release of the
substrate arm was measured by plotting the change in fluorescence intensity for 16 seconds, at a
2 sec. time interval, after Pb2+
addition using the equation: y = y0 + Vfluox; where Vfluo is the
initial rate of release of the substrate arm.

38
Table 2.1 Sequences of all the oligonucleotides used in the current study. The base underlined
indicates the position of the mismatch used.
2.3 Results and discussions
2.3.1 Temperature dependence of the original sensor (or RT Sensor)
The temperature-dependence of the +5_17E DNAzyme catalytic beacon sensor (Figure
2.1a) was followed in real-time using fluorescence spectroscopy. The fluorescence increase over
the background was monitored at 520 nm, the emission wavelength of FAM, as a function of
time. At 25 °C, a dramatic increase in fluorescence intensity was observed upon addition of 2
µM of Pb2+
. In contrast, when the same experiment was carried out at 4 °C, negligible increase
was observed (Figure 2.1c). The initial rate of substrate release of the sensor was further studied
between 4 °C and 30 °C (Figure 2.2a) and while the increase was very slow at 4 °C, it became
faster at 15 °C, and reached the maximum at ~ 25 °C, after which the rate started to drop again.
Name Sequence (5' 3')
RT: +5_17E (Dy) ACA GAC ATC TCT TCT CCG AGC CGG TCG AAA TAG TGA GT-DABCYL
MM1:+5_17E(T16.5-C) ACA GAC ATC TCT TCT CCG AGC CGG TCG AAA TAG CGA GT-DABCYL
MM2:+5_17E(A16.7-G) ACA GAC ATC TCT TCT CCG AGC CGG TCG AAA TAG TGG GT-DABCYL
+5_17E(G16.4-T) ACA GAC ATC TCT TCT CCG AGC CGG TCG AAA TAG TTA GT-DABCYL
+5_17E(T2.4 -C) ACA GAC ATC TCC TCT CCG AGC CGG TCG AAA TAG TGA GT-DABCYL
DMM1:+5_17E (T16.5-C,
A16.7-G)
ACA GAC ATC TCT TCT CCG AGC CGG TCG AAA TAG CGG GT-DABCYL
DMM2: +5_17E ( T2.4-C,
A16.7-G)
ACA GAC ATC TCC TCT CCG AGC CGG TCG AAA TAG TGG GT-DABCYL
+5_17E-1(Dy) ACA GAC ATC TCT TCT CCG AGC CGG TCG AAA TAG TG GT-DABCYL
+5_17S FAM- ACT CAC TAT rA GGA AGA GAT GTC TGT -BHQ_1
+5_17S-1(FAM) FAM- AC CAC TAT rA GGA AGA GAT GTC TGT -BHQ_1

39
Since this sensor exhibited high activity at ~ 25 °C, it was referred to as the RT (“room
temperature”) sensor. The variance of pH of 50 mM HEPES with 100 mM NaCl buffer used was
< 0.4% between 4-30 °C and therefore, did not contribute to the temperature dependence of the
sensor. For the efficient functioning of the catalytic beacon sensor, a fine balance had to be
struck between the high stability of the substrate/enzyme strands for obtaining low background
fluorescence in the absence of Pb2+
, and low stability of the product/enzyme strands for fast
product release and high fluorescence increase in the presence of Pb2+
. For the RT sensor, the
stability of the product/enzyme strand was so high at 4 °C that the product was not released even
after Pb2+
-dependent cleavage. Increasing the temperature provided sufficient energy to facilitate
the release of the product strand. Higher rates of fluorescence increase were observed at 15 °C
and 25 °C. Above 25 °C, however, the stability of the substrate/enzyme complex gradually
started to decrease, causing dehybridization and high fluorescence background before Pb2+
addition. Thereby, the rate of florescence increase upon Pb2+
addition was slightly lowered again
at 30 °C.

40
Figure 2.1 a) Predicted secondary structure of the fluorescent 17E DNAzyme Pb2+
sensor. F
represents the fluorophore, FAM, Q1 is the quencher BHQ-1® and Q2 is the DABCYL
quencher. The single RNA base on the substrate arm is denoted by rA; b) Schematic
representation of the catalytic beacon sensor; c) Kinetics of fluorescence increase of RT (sensor
that contains no mismatch), over the background at 25 °C (in red) and 4 °C, (in blue) with 2 µM
Pb2+
in 50 mM HEPES with 100 mM NaCl at pH 7.2. Pb2+
is added at the 20 sec time point.
2.3.2 Strategies to modulate temperature dependence of the sensor
There are three different strategies that can be employed to change the overall release
kinetics of the substrate arm from the enzyme-substrate complex. First, the total number of bases
on the enzyme-substrate complex can be varied to reduce the overall melting temperature. The
original 17E DNAzyme sensor used for the detection of Pb2+
in fact had only nine base-pairs on
both the releasing and the non-releasing arm of the enzyme-substrate complex. However, this

41
complex was efficient only at 4 °C. At 25 °C, there was considerable dehybridization of the
complex, resulting in significant background fluorescence and hence, minimum fluorescence
increase over the background. Moreover, if this strategy had to be employed, various
enzyme/substrate strands of different lengths would have to be used for activity at different
temperatures, thereby leading to an overall increase in the cost involved. The second strategy that
could be employed would be to vary the total NaCl concentration since the overall stability of the
complex, its melting temperature, and its release-kinetics depends on the same. The drawback of
this method is that a minimum amount of NaCl is required to maintain the overall secondary
structure of the DNAzyme-substrate complex. Hence, the variance in temperature due to addition
of NaCl can only be unidirectional in terms of temperature. The third strategy that could be
utilized would be to introduce mismatches on the enzyme-substrate complex. The substrate
strand has an RNA base, a fluorophore, and a quencher modification, and is more expensive than
the enzyme strand. Therefore, the introduction of mismatches on the DNAzyme would be
beneficial since this strand that has only a single quencher modification. If only the enzyme
strand is modified, these enzyme strands containing different mismatches can be used with the
same substrate strand. Previous studies have shown that there is a considerable change in the
dissociation kinetics and the melting temperatures of oligonucleotides that are perfectly matched
versus oligonucleotides with stable mismatches versus those containing unstable mismatches.41,
42 The trend of stability of internal mismatches in DNA, based on the free energy change (ΔG°37
(kcal/mol)) values at pH 7.0 in 1 M NaCl, calculated using the nearest-neighbor model, follows
the order G·T ≥ G·A > C·T > A·C. 43-48

42
2.3.3 The MM1 sensor and its temperature dependence
In order to tune the stability of the system and improve performance at lower
temperatures, we proposed the introduction of the least stable A·C mismatch in the middle of the
releasing arm of the enzyme strand 17E (T16.5-C) (called MM1 sensor, Figure 2.2a). Such a
mismatch may allow considerable destabilization and thus, release of the product even at low
temperatures. The MM1 sensor with the mismatch showed a much higher increase of
fluorescence intensity at 4 °C in comparison to that of RT sensor (Figure 2.2b). The rates in
fluorescence increase were then plotted as a function of temperature (Figure 2.4a). While the
MM1 sensor showed a high signal response at 4 °C upon Pb2+
addition, such a response
decreased with increasing temperature from 4 °C to 30 °C. Since the temperature-dependent
fluorescence changes of RT and MM1 sensors were almost opposite to each other, we
investigated the possibility of mixing the two sensors at different ratios to develop a sensor
system, whose signal is relatively independent of temperature. Interestingly, at a ratio of
4:1(MM1:RT), the fluorescence signal response to Pb2+
addition remains almost constant
between 4 °C to 30 °C (Figure 2.4a).

43
Figure 2.2 a) Predicted secondary structure of the fluorescent 17E (T16.5-C) or MM1 DNAzyme
Pb2+
sensor. F represents the fluorophore, FAM, Q1 is the quencher BHQ-1® and Q2 is the
DABCYL quencher. The single RNA base on the substrate arm is denoted by rA ; b) Kinetics of
fluorescence increase of MM1 over the background at 25 °C (in red) and 4 °C, (in blue) with 2
µM Pb2+
in 50 mM HEPES with 100 mM NaCl at pH 7.2. Pb2+
is added at the 20 sec. time point.
2.3.4 The MM2 sensor and its temperature dependence
Having developed a catalytic beacon sensor system that was relatively independent of
temperature using a mixture of two sensors with opposite temperature dependence, we wanted to
investigate whether there could be an even simpler system consisting of only a single sensor.
Hence, instead of the complete A·C mismatch as in MM1, we tested 17E (A16.7-G) (called MM2
sensor, Figure 2.3a) that contained a G·T wobble pair. This wobble pair is less stable than a G-C

44
Watson-Crick pair, but more stable than the A·C mismatch.10
The wobble pair may be stable
enough to allow hybridization of the substrate and enzyme strand even at higher temperatures
while exerting enough destabilization to allow release of the product arm at low temperature.
Indeed, this MM2 sensor system displayed a high increase in the fluorescence intensity at both 4
°C and 25 °C (Figure 2.3b) and upon measuring the initial rate, a relatively constant response
between 4-30 °C was observed (Figure 2.4b).
Figure 2.3 a) Predicted secondary structure of the fluorescent 17E (A16.7-G) or MM2 DNAzyme
Pb2+
sensor. F represents the fluorophore, FAM, Q1 is the quencher BHQ-1® and Q2 is the
DABCYL quencher. The single RNA base on the substrate arm is denoted by rA ; b) Kinetics of
fluorescence increase of MM1 over the background at 25 °C (in red) and 4 °C, (in blue) with 2
µM Pb2+
in 50 mM HEPES with 100 mM NaCl at pH 7.2. Pb2+
is added at the 20 sec. time point.

45
Figure 2.4 Temperature-dependent initial rate of substrate release of a) RT, (in red) MM1, (in
black) and (4:1) MM1: RT (in blue) upon addition of 2 µM Pb2+
and b) MM2 (in green) upon
addition of 750 nM and 500 nM Pb2+
; RT (in red) upon addition of 750 nM Pb2+
in 50 mM
HEPES with 100 mM NaCl at pH 7.2.
2.3.5 Selectivity and sensitivity of the MM1 sensor
To ensure that the development of the temperature-independent catalytic beacon sensor
did not compromise the sensitivity and selectivity, signal responses of the MM2 sensor with
different metal ions was studied. All competing metal ions tested showed negligible response
with the sensor at both 4 °C and 25 °C, suggesting excellent selectivity (Figure 2.5). The
sensitivity of the MM2 sensor was also tested and the detection limits for Pb2+
at 4 °C and 25 °C
were determined to be 50 nM and 20 nM, respectively (Figure 2.6). Since the MCL for Pb2+
in
drinking water is defined by the US Environment Protection Agency to be 72 nM and the limit
levels for Pb2+
in other media such as paint, dust and soil are even higher than 72 nM, the
temperature-independent DNAzyme catalytic beacons described here are well suited for
monitoring Pb2+
in the environment.

46
Figure 2.5 Selectivity of the MM2 sensor. Kinetics of fluorescence increase relative to
background at 6 minutes after incubation with a) 1 µM of M2+
ions at 4 °C and at b) 500 nM of
M2+
ions at 25 °C, in buffer containing 50 mM HEPES with 100 mM NaCl at pH 7.2.
Figure 2.6 Sensitivity of the MM2 sensor. a) Kinetics of fluorescence increase relative to the
background, with varying concentrations of Pb2+
at 25 °C. b) Variation of the initial rate with
Pb2+
concentrations at 4°C. The inset presents the linear range in submicromolar concentrations
of Pb2+
. The rates were measured in buffer containing 50 mM HEPES, 100 mM NaCl at pH 7.2.

47
2.3.6 Role of location of the mismatch
The location of the mismatch was an important factor in determining the overall
temperature-dependence of the sensor, in addition to the actual nature of the mismatch. To test
the importance of the location of the mismatch, several control sensors were designed and their
temperature-dependence was studied.
TGTCTGTAGAGAAGGrATATCACTCA
ACAGACATCTCTTCT ATAGTGAGTCC
CGAG C
GGT C
GA
A
5'
5' 3'
3'F
Q2
Q1
T
a
Figure 2.7 a) Predicted secondary structure of the lead sensor showing the position of the
mismatch 17E (G16.6-T) b) Normalized fluorescence response and c) temperature dependence of
the sensor with 2 µM Pb2+
in 50 mM HEPES at pH 7.2 with 100 mM NaCl.

48
A sensor containing a complete T·C mismatch on the releasing arm of the enzyme was
studied (Figure 2.7a) and this sensor exhibited behavior similar to MM1.(Figure 2.7b,c) On the
other hand, a sensor containing an A·C mismatch on the non-releasing arm of the enzyme
showed a response similar to the original sensor, RT. (Figure 2.8a-c) This confirmed that the use
of the same mismatch on the non-releasing arm of the enzyme gave results similar to the original
RT sensor, since it was not involved in the release of the substrate arm that dictated the overall
sensor behavior.
TGTCTGTAGAGAAGGrATATCACTCA
ACAGACATCTCTTCT ATAGTGAGTCC
CGAG C
GGT C
GA
A
5'
5' 3'
3'F
Q2
Q1
C
a
Figure 2.8 a) Predicted secondary structure of the Pb2+
sensor showing the position of the
mismatch 17E (T2.4- C) b) Normalized fluorescence response and c) temperature dependence of
the sensor with 2 µM Pb2+
in 50 mM HEPES at pH 7.2 with 100 mM NaCl.

49
2.3.7 Introduction of dual mismatches
The fluorescence response of sensors containing dual mismatches was also studied
(Figure 2.9). The first dual mismatch sensor, DMM1 containing the same two mismatches as the
MM1 and MM2 sensors (A·G and T·C) on the releasing arm showed a fluorescence response
similar to MM1 albeit lower, owing to its considerable destabilization. The second dual
mismatch sensor, DMM2 on the other hand, containing one mismatch on the releasing arm
(A·G), and one on the non-releasing arm (T·C), showed a response similar to MM2. The overall
response, therefore, was dictated by the mismatch on the releasing arm of the substrate, which in
this case was identical to MM2. This further confirms that the sensor behavior was dependent on
the nature as well as the location of the mismatch either on the releasing or non-releasing arm.
2.4 Conclusions
The work done in this chapter demonstrates the use of the intrinsic lowered stability of
mismatches to develop catalytic beacon Pb2+
sensors whose performance is independent of
temperatures between 4-30 °C. In the process, it has been shown that the A·C mismatch and G·T
wobble pair can both be utilized, albeit with different strategies, in achieving the same goal. Free
of the need for temperature calibration, such a temperature-independent sensor system can now
find even wider applications for the on-site and real-time detection of Pb2+
in the environment.
The methodology developed in this study can also be applied for designing other nucleic acid
sensors that resist temperature-dependent variations.

50
TGTCTGTAGAGAAGGrATATCACTCA
ACAGACATCTCTTCT ATAGTGAGTCC
CGAG C
GGT C
GA
A
5'
5' 3'
3'F
Q2
Q1
GC
a
TGTCTGTAGAGAAGGrATATCACTCA
ACAGACATCTCTTCT ATAGTGAGTCC
CGAG C
GGT C
GA
A
5'
5' 3'
3'F
Q2
Q1
GC
b
Figure 2.9 a) Predicted secondary structure of the lead sensors a) DMM1, showing the position
of the mismatches 17E (T16.5-C, A16.7-G) and b) DMM2, showing the position of the mismatches
17E (T2.4-C, A16.7-G). c) Normalized fluorescence response of the sensors with 2 µM Pb2+
in 50
mM HEPES at pH 7.2 with 100 mM NaCl.

51
2.5 References
1. Needleman, H. L., Human Lead Exposure. CRC Press: Boca Raton, 1992; p 290.
2. Needleman, H. L. Clair Patterson and Robert kehoe: two views of lead toxicity.
Environ.Res. 1998, 78, 79-85.
3. Martin, R. B.; King, R. B., Metal Ion Toxicity. In Encyclopedia of Inorganic Chemistry,
King, R. B., Ed. Wiley: Chichester, 2007; Vol. 4, p 2185.
4. Healey, N. Lead toxicity, vulnerable subpopulations and emergency preparedness.
Radiat. Prot. Dosim. 2009, 134, 143-151.
5. Al-Rashdan, A.; Heitkemper, D.; Caruso, J. A. Lead speciation by HPLC-ICP-AES and
HPLC-ICP-MS. J. Chromatogr. Sci. 1991, 29, 98-102.
6. Schutz, A.; Bergdahl, I. A.; Ekholm, A.; Skerfving, S. Measurement by ICP-MS of lead
in plasma and whole blood of lead workers and controls. Occup. Environ. Med. 1996, 53,
736-740.
7. Widmer, C. R.; Krahenbuhl, U.; Kramers, J.; Tobler, L. Lead isotope measurements on
aerosol samples with ICP-MS. Fresenius J. Anal. Chem. 2000, 366, 171-173.
8. Ettler, V.; Mihaljevic, M.; Komarek, M. ICP-MS measurements of lead isotopic ratios in
soils heavily contaminated by lead smelting: tracing the sources of pollution. Anal.
Bioanal. Chem. 2004, 378, 311-317.
9. Naghmush, A. M.; Pyrzynska, K.; Trojanowicz, M. Flame AAS determination of lead in
water with flow-injection preconcentration and speciation using functionalized cellulose
sorbent. Talanta 1995, 42, 851-860.
10. Novotny, K.; Turzikova, A.; Komarek, J. Speciation of copper, lead and cadmium in
aquatic systems by circulating dialysis combined with flame AAS. Fresenius J. Anal.
Chem. 2000, 366, 209-212.
11. Bravo-Sanchez, L. R.; San Vicente de la Riva, B.; Costa-Fernandez, J. M.; Pereiro, R.;
Sanz-Medel, A. Determination of lead and mercury in sea water by preconcentration in a
flow injection system followed by atomic absorption spectrometry detection. Talanta
2001, 55, 1071-1078.
12. Burguera, J. L.; Burguera, M. Determination of lead in biological materials by
microwave-assisted mineralization and flow injection electrothermal atomic absorption
spectrometry. J. Anal. At. Spectrom. 1993, 8, 235-241.
13. Yebra-Biurrun, M. C.; Moreno-Cid Barinaga, A. Literature survey of on-line
spectroscopic methods for lead determination in environmental solid samples.
Chemosphere 2002, 48, 511-518.
14. Herdan, J.; Feeney, R.; Kounaves, S. P.; Flannery, A. F.; Storment, C. W.; Kovacs, G. T.
A.; Darling, R. B. Field Evaluation of an Electrochemical Probe for in Situ Screening of
Heavy Metals in Groundwater. Environ. Sci. Technol. 1998, 32, 131-136.
15. Reeder, G. S.; Heineman, W. R. Electrochemical characterization of a microfabricated
thick-film carbon sensor for trace determination of lead. Sens. Actuators, B 1998, B52,
58-64.
16. Wilson, R.; Schiffrin, D. J.; Luff, B. J.; Wilkinson, J. S. Optoelectrochemical sensor for
lead based on electrochemically assisted solvent extraction. Sens. Actuators, B 2000, B63,
115-121.

52
17. Kwon, J. Y.; Jang, Y. J.; Lee, Y. J.; Kim, K. M.; Seo, M. S.; Nam, W.; Yoon, J. A highly
selective fluorescent chemosensor for Pb2+
. J. Am. Chem. Soc. 2005, 127, 10107-10111.
18. Feigl, F.; Suter, H. A. Analytical use of sodium rhodizonate. Ind. Eng. Chem., Anal. Ed.
1942, 14, 840-842.
19. Feigl, F.; Braile, N. Analysis by spot reactions. III. Use of sodium rhodizonate in the
analysis of mineral products and alloys. Chemist-Analyst 1943, 32, 52-59.
20. Ashley, K.; Fischbach, T. J.; Song, R. Evaluation of a chemical spot-test kit for the
detection of airborne particulate lead in the workplace. Am. Ind. Hyg. Assoc. J. 1996, 57,
161-165.
21. Khosraviani, M.; Blake, R. C., 2nd; Pavlov, A. R.; Lorbach, S. C.; Yu, H.; Delehanty, J.
B.; Brechbiel, M. W.; Blake, D. A. Binding properties of a monoclonal antibody directed
toward lead-chelate complexes. Bioconjug. Chem. 2000, 11, 267-277.
22. Blake, D. A.; Jones, R. M.; Blake, R. C., 2nd; Pavlov, A. R.; Darwish, I. A.; Yu, H.
Antibody-based sensors for heavy metal ions. Biosens. Bioelectron. 2001, 16, 799-809.
23. Zhu, X.; Hu, B.; Lou, Y.; Xu, L.; Yang, F.; Yu, H.; Blake, D. A.; Liu, F. Characterization
of monoclonal antibodies for lead-chelate complexes: applications in antibody-based
assays. J. Agric. Food Chem. 2007, 55, 4993-4998.
24. Breaker, R. R.; Joyce, G. F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223-
229.
25. Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA
ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505-510.
26. Li, J.; Zheng, W.; Kwon, A. H.; Lu, Y. In vitro selection and characterization of a highly
efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000, 28,
481-488.
27. Li, J.; Lu, Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J.
Am. Chem. Soc. 2000, 122, 10466-10467.
28. Liu, J.; Lu, Y. Improving fluorescent DNAzyme biosensors by combining inter- and
intramolecular quenchers. Anal. Chem. 2003, 75, 6666-6672.
29. Swearingen, C. B.; Wernette, D. P.; Cropek, D. M.; Lu, Y.; Sweedler, J. V.; Bohn, P. W.
Immobilization of a catalytic DNA molecular beacon on Au for Pb(II) detection. Anal.
Chem. 2005, 77, 442-448.
30. Wernette, D. P.; Mead, C.; Bohn, P. W.; Lu, Y. Surface immobilization of catalytic
beacons based on ratiometric fluorescent DNAzyme sensors: a systematic study.
Langmuir 2007, 23, 9513-9521.
31. Wernette, D. P.; Swearingen, C. B.; Cropek, D. M.; Lu, Y.; Sweedler, J. V.; Bohn, P. W.
Incorporation of a DNAzyme into Au-coated nanocapillary array membranes with an
internal standard for Pb(ii) sensing. Analyst 2006, 131, 41-47.
32. Dalavoy, T. S.; Wernette, D. P.; Gong, M.; Sweedler, J. V.; Lu, Y.; Flachsbart, B. R.;
Shannon, M. A.; Bohn, P. W.; Cropek, D. M. Immobilization of DNAzyme catalytic
beacons on PMMA for Pb2+
detection. Lab Chip 2008, 8, 786-793.
33. Chang, I. H.; Tulock, J. J.; Liu, J.; Kim, W. S.; Cannon, D. M., Jr.; Lu, Y.; Bohn, P. W.;
Sweedler, J. V.; Cropek, D. M. Miniaturized lead sensor based on lead-specific
DNAzyme in a nanocapillary interconnected microfluidic device. Environ. Sci. Technol.
2005, 39, 3756-3761.

53
34. Xiao, Y.; Rowe, A. A.; Plaxco, K. W. Electrochemical detection of parts-per-billion lead
via an electrode-bound DNAzyme assembly. J. Am. Chem. Soc. 2007, 129, 262-263.
35. Shen, L.; Chen, Z.; Li, Y.; He, S.; Xie, S.; Xu, X.; Liang, Z.; Meng, X.; Li, Q.; Zhu, Z.;
Li, M.; Le, X. C.; Shao, Y. Electrochemical DNAzyme sensor for lead based on
amplification of DNA-Au bio-bar codes. Anal. Chem. 2008, 80, 6323-6328.
36. Liu, J.; Lu, Y. A colorimetric lead biosensor using DNAzyme-directed assembly of gold
nanoparticles. J. Am. Chem. Soc. 2003, 125, 6642-6643.
37. Liu, J.; Lu, Y. Accelerated color change of gold nanoparticles assembled by DNAzymes
for simple and fast colorimetric Pb2+
detection. J. Am. Chem. Soc. 2004, 126, 12298-
12305.
38. Liu, J.; Lu, Y. Colorimetric biosensors based on DNAzyme-assembled gold
nanoparticles. J. Fluoresc. 2004, 14, 343-354.
39. Wei, H.; Li, B.; Li, J.; Dong, S.; Wang, E. DNAzyme-based colorimetric sensing of lead
(Pb2+
) using unmodified gold nanoparticle probes. Nanotechnology 2008, 19,
095501/095501-095501/095505.
40. Mazumdar, D.; Liu, J.; Lu, G.; Zhou, J.; Lu, Y. Easy-to-use dipstick tests for detection of
lead in paints using non-cross-linked gold nanoparticle–DNAzyme conjugates. Chem.
Commun. 2010, DOI: 10.1039/b917772h.
41. Aboul-ela, F.; Koh, D.; Tinoco, I., Jr.; Martin, F. H. Base-base mismatches.
Thermodynamics of double helix formation for dCA3XA3G + dCT3YT3G (X, Y =
A,C,G,T). Nucleic Acids Res. 1985, 13, 4811-4824.
42. Ikuta, S.; Takagi, K.; Wallace, R. B.; Itakura, K. Dissociation kinetics of 19 base paired
oligonucleotide-DNA duplexes containing different single mismatched base pairs.
Nucleic Acids Res. 1987, 15, 797-811.
43. Allawi, H. T.; SantaLucia, J., Jr. Thermodynamics and NMR of internal G.T mismatches
in DNA. Biochemistry 1997, 36, 10581-10594.
44. Allawi, H. T.; SantaLucia, J., Jr. NMR solution structure of a DNA dodecamer containing
single G.T mismatches. Nucleic Acids Res. 1998, 26, 4925-4934.
45. Allawi, H. T.; SantaLucia, J., Jr. Nearest-neighbor thermodynamics of internal A.C
mismatches in DNA: sequence dependence and pH effects. Biochemistry 1998, 37, 9435-
9444.
46. Allawi, H. T.; SantaLucia, J., Jr. Thermodynamics of internal C.T mismatches in DNA.
Nucleic Acids Res. 1998, 26, 2694-2701.
47. Allawi, H. T.; SantaLucia, J., Jr. Nearest neighbor thermodynamic parameters for internal
G.A mismatches in DNA. Biochemistry 1998, 37, 2170-2179.
48. Riccelli, P. V.; Vallone, P. M.; Kashin, I.; Faldasz, B. D.; Lane, M. J.; Benight, A. S.
Thermodynamic, spectroscopic, and equilibrium binding studies of DNA sequence
context effects in six 22-base pair deoxyoligonucleotides. Biochemistry 1999, 38, 11197-
11208.

54
3 Effect of Metal Ions on the Global Folding, Activity and Structure of the
8-17 DNAzyme
Note and acknowledgments: This work was done in direct collaboration with Dr.
Debapriya Mazumdar and Dr. Hee-Kyung Kim. We thank Professor Martin Gruebele at the
University of Illinois for the use of their CD instrument. We also thank Krishnarjun Sarkar,
Apratim Dhar and Sharlene Denos for assistance with the CD measurements. The material in this
chapter is the basis for two published manuscripts- „Reproduced in part‟ with permission from:
1) “Activity, folding and Z-DNA formation of the 8-17 DNAzyme in the presence of monovalent
ions.” Debapriya Mazumdar, Nandini Nagraj, Hee-Kyung Kim, Xiangli Meng, Andrea
K. Brown, Qian Sun, Wei Li and Yi Lu J. Am. Chem. Soc. 2009, 131, 5506-5515
(http://pubs.acs.org/doi/abs/10.1021/ja8082939); and 2) “Metal-Dependent Global Folding and
Activity of the 8-17 DNAzyme Studied by Fluorescence Resonance Energy Transfer.” Hee-
Kyung Kim, Juewen Liu, Jing Li, Nandini Nagraj, Mingxi Li, Caroline M.-B. Pavot and Yi Lu.
J. Am. Chem. Soc. 2007, 129, 6896-6902 (http://pubs.acs.org/doi/abs/10.1021/ja0712625) -
Copyright 2009 and 2007, American Chemical Society.
3.1 Introduction
Metal ions play an integral role in the catalysis facilitated by most nucleic acid enzymes
and their roles and possible modes of effecting catalysis have been discussed previously. Most
nucleic acid enzymes are efficient catalysts in the presence of divalent metal ion co-factors that
have been proposed to play both structural and functional roles, i.e., they can function to stabilize

55
the active structure of the enzyme and can also be directly involved in active-site chemistry.
While divalent metal-dependent catalysis is the most common mode of activation in enzyme
catalysis, monovalent metal ion-dependent catalysis has also been observed, and has been
insightful for understanding their mechanisms.1-10
3.1.1 Metal ion-dependent catalysis in nucleic acid enzymes
3.1.1.1 Monovalent metal ion-dependent activity of ribozymes
Studies with naturally occurring ribozymes such as the hairpin,11-15
hammerhead,16, 17
hepatitis delta virus (HDV),18
Varkud satellite (VS),1 and glmS ribozymes
19 have demonstrated
that these enzymes can promote catalysis in the presence of molar concentrations of monovalent
ions, such as Li+, Na
+, and K
+. In many cases, the observed rate constants (kobs) in the presence
of monovalent ions approach that of the divalent metal ion catalyzed reaction, although a higher
higher concentration of monovalent ions is required. For instance, the hammerhead ribozyme
displays monovalent ion-dependent activity (kobs with 4 M Li+: 0.29 min
-1) that is within 10-fold
of divalent metal ion-dependent activity (kobs with 10 mM Mg2+
: 2.2 min-1
).16
The high
concentration of monovalent ions required for catalysis suggests that these ribozymes do not
contain specific binding sites for alkali metal ions. Instead, it is likely that at high concentrations,
monovalent ions can induce the same active conformation as divalent metal ions by electrostatic
stabilization of the negative charge. Thermodynamic and chemical probing studies by Jaeger et
al. have established that similar RNA structures can form in 10 mM Mg2+
and 1 M Na+, that
molar concentrations of NaCl can readily substitute for the millimolar concentrations of Mg2+
that is generally used for RNA tertiary structure formation.20

56
It may also be possible that catalysis can be supported by monovalent ions occupying the
same active site regions as divalent ions, although catalytic site binding of monovalent ions has
not been reported in the crystal structure of any of these small ribozymes. However, in the crystal
structure of the large intact group I intron (which catalyzes trans-esterification using a two-metal
ion mechanism), one of the two catalytic metal binding sites was occupied by K+.21
3.1.1.2 Monovalent ion-dependent catalysis in DNAzymes
While a number of studies have been reported on ribozyme catalysis in the presence of
monovalent ions alone, there have not been similar reports on the monovalent ion-dependent
activity of catalytically proficient DNAzymes that have been selected in the presence of divalent
ions. In contrast, Geyer et al. reported the in vitro selection for an RNA-cleaving DNAzyme
which was carried out in the presence of Na+ without any divalent metal ions in the buffer.
22 This
resulted in DNAzymes that were active without any divalent metal ions and catalysis was
supported by monovalent ions only. Interestingly, the addition of divalent metal ions did not
change the catalytic rate significantly. Thus, this DNAzyme may be fundamentally different
from the naturally occurring ribozymes and other in vitro selected DNAzymes that utilize
divalent metal ions as their most efficient co-factor. Therefore, it is important to study the effects
of monovalent ions on DNAzymes that are active in the presence of divalent metal ions.
3.1.2 Metal ion-dependent folding
Metal ion-dependent folding can play a critical role in metalloenzyme function.
Understanding the relationship between folding and enzymatic reactions is critical in obtaining
deeper insights into the enzyme mechanism, which in turn may allow the design of better

57
enzymes for practical applications. Toward this goal, a number of studies have been carried out
with protein metalloenzymes, which show that an active-site metal-dependent global folding may
precede enzymatic reaction in some cases, while such a folding is not required in others.23
3.1.2.1 Metal ion-dependent folding of ribozymes
Metal ion-specific folding and misfolding has been previously shown to have close
relation with functions of ribozymes,24-26
including the hairpin,27
hammerhead,28
VS,29
group I
intron,30, 31
and RNase P.32-34
The hammerhead ribozyme was shown to misfold due to
competitive binding of Mg2+
and Co(NH3)63+
, and existence of two distinct reaction pathways
promoted by divalent or non-divalent (especially Co(NH3)63+
) metal ions was proposed.28
The
group I intron and RNase P have been shown to form the active site only in the presence of
divalent metal ions.30, 33, 35, 36
Kinetics in folding of RNase P was very sensitive to different group
IIA metal ions.34, 35
All these studies indicated that metal ion properties such as size and
hydration energy affected the structural reorganization of the ribozymes studied.
Monovalent metal ion-dependent folding of small ribozymes has also been studied.
Interestingly, these ions have been shown to induce similar active conformations as divalent
metal ions.27, 37-41
These findings suggest that ribozymes require metal ions mainly for
electrostatic or structural stabilization, while for chemical catalysis; their role is minimal or
absent. The conditions in these studies were not conducted under physiological conditions.
3.1.2.2 Metal ion-dependent folding of DNAzymes
The 8-17 DNAzyme has also been the subject of a few folding studies.42-46
It was
observed that both Mg2+
and Zn2+
induced folding of the 8-17 DNAzyme into a compact

58
conformation prior to the cleavage reaction,43, 44
similar to what was observed with ribozymes.
However, the presence of Pb2+
, which was the most efficient co-factor of the DNAzyme, did not
show any evidence for folding of the DNAzyme.43, 44
Uni-directional folding and cleavage of the
8-17 DNAzyme was also observed in single-molecule FRET assays.44
These observations led to
the proposal that the DNAzyme could catalyze the cleavage reaction with different reaction
pathways depending on the nature of the metal ions used. Since a conformational change was
also not observed prior to cleavage in the presence of Pb2+
, it was proposed that the DNAzyme
might have been pre-arranged to accept Pb2+
.44
3.1.3 Research goals
Detailed investigations into the relationship between the activities and folding of several
ribozymes in the presence of both monovalent and divalent metal ions have been carried out.
These studies have provided critical insights into the extent to which different cations alter the
dynamics, stabilization via folding and the catalytic function of these nucleic acid enzymes. The
8-17 DNAzyme, that has been discussed in chapter 1, has been converted into both
fluorescent,47-51
and colorimetric Pb2+
sensors52-55
by various members of the Lu lab, owing to its
highest activity in the presence of Pb2+
.56
Therefore, there is significant interest in understanding
the relationship that exists between metal ion dependent folding and activity of the 8-17
DNAzyme in the presence of both divalent and monovalent metal ions. Moreover, monovalent
ion-dependence of the 8-17 DNAzyme has not been reported previously and this investigation
would also help design more selective sensors because monovalent ions are often present during
sensing applications. Hence, our goal was to systematically investigate the metal ion-dependent
catalysis and folding of the 8-17 DNAzyme.

59
Herein, we report the gel-based activity assays and folding studies on the 8-17
DNAzyme in the presence of monovalent ions (group IA). In addition, we also report folding
studies in the presence of some divalent metal ions belonging to group IIA and the d-block, for
which activity data has been previously reported.56
Circular dichroism (CD) has also been used
to probe structural changes of the 8-17 DNAzyme in the presence of monovalent ions and Mg2+
,
Zn2+
, and Pb2+
. Relations between metal ion size, folding efficiency, and activity have been
examined, and the role of metal ions in catalysis of the 8-17 DNAzyme is discussed.
3.2 Materials and Methods
3.2.1 Materials
All the oligonucleotides (HPLC-purified) were purchased from Integrated DNA
Technologies Inc., Coralville, IA. All metal salts were obtained from Alfa-Aesar and were of
Puratronic® grade, 99.999 % pure.
3.2.2 Kinetic gel-based activity assays
Activity assays of the 8-17 DNAzyme with various M+ metal ions were carried out in a
reaction buffer containing 50 mM Na-HEPES and 25 mM EDTA, (to prevent any M2+
dependent
activity) at pH 7.5. Since concentrated stock solutions of 5 M metal ions were prepared in the
reaction buffer, the pH of the buffer was no longer maintained. The pH was then re-adjusted
using concentrated NaOH to 7.5 using a glass pH electrode (Orion) and further verified using pH
paper before every assay. The substrate 17S was 5´ labeled with [-32
P]-dATP (GE Healthcare)
using T4 kinase from Invitrogen. The sample solution containing the enzyme (5 × 5 µM) and
32P-labeled substrate (5 × 1 nM) was denatured by heating the mixture in a water bath at 90
oC

60
for 3 min and annealed by gradual cooling to room temperature over 30 min. The reaction was
initiated by adding one volume of enzyme-substrate mixture (5 × concentration) into four
volumes of 5 M metal solution at room temperature, such that the final concentrations of the
enzyme, substrate, and metal ion were 5 µM, 1 nM, and 4 M, respectively. The reaction was
stopped at specific time points by adding a 2.5 µL aliquot of the reaction mixture into 50 µL of
stop buffer containing 8 M urea + 50 mM EDTA + 0.05 % bromophenol blue + 0.05 % xylene
cyanol. Such a large volume (20 ×) of stop solution was used for these experiments in
comparison to that routinely used with assays containing M2+
ions, as this would stop the
reaction by diluting the ion concentration significantly rather than chelation of the metal ion by
EDTA. The uncleaved substrate and the cleaved product were separated on a denaturing 20 %
polyacrylamide gel, imaged on a Molecular Dynamics Storm 430 phosphorimager and quantified
using Image Quant software. The % product at time „t‟ was calculated by taking the ratio of the
5'-cleaved product to the total of the cleaved product and the uncleaved substrate, after
subtraction from the background radioactivity on the gel. Kinetic plots were created using Sigma
Plot and fit to the equation:
% Pt = % P0 + % P∞ (1-e-kt
)
wherein, % P0 is the initial amount of product (at t = 0), % Pt is the amount of product at
time t and % P∞ is the amount of product formed at the endpoint of the reaction ( at t = ∞ ) and k
= kobs , the observed rate of cleavage.
3.2.3 FRET experiments
To study metal induced folding, 10 µM each of fluorophore-labeled substrate and
enzyme, called Cy3-17DS and Cy5-17E, respectively (Figure 3.1b) were annealed in 50 mM Na-

61
HEPES acetate buffer (pH 7.5) by heating the sample at 85 C for 5 min and subsequently
cooling it to 4 C over 2 h. The annealed product was purified with a native 16% polyacrylamide
gel at 4 C.42, 57
The band containing the annealed DNAzyme was located by inspection, cut out,
crushed, and subsequently recovered by soaking the crushed gel bits in 50 mM Na-HEPES
acetate, pH 7.5. All the procedures were carried out at 4 C to avoid dissociation of the
DNAzyme complex. The sample concentration was determined by measuring its absorption at
260 nm and the final concentration was diluted to <100 nM in the same buffer. At this
concentration, the fluorophores were dilute enough to avoid inner-filter effects.58, 59
Concentrated
metal ion solutions in 50 mM Na-HEPES acetate, pH 7.5 were titrated into the sample to initiate
folding.
Steady-state fluorescence emission spectra were recorded on a Fluromax-2 fluorometer
(HORIBA Jobin Yvon Inc., NJ). Polarization artifacts were avoided by setting the polarizers
under ‘magic angle conditions’.58
Fluorescence measurements were carried out at 10 C to avoid
denaturation of the DNAzyme complex.
The FRET efficiency (EFRET) was calculated using the (ratio)A method.43
A detailed
description of the (ratio)A method can be found in literature.59
Briefly, for a FRET pair, when the
donor (D) is excited, the nearby (10-100 Å) acceptor (A) can receive energy from the donor with
efficiency, (ratio)A expressed as:59
where is the donor-to-acceptor distance. is defined by
Å6.
D is the quantum yield of the donor and
2 is the
66
0
6
0
RR
RE
R 0R
642236
0 )(10785.8 vJR D

62
orientation factor for dipole coupling. If both the donor and acceptor can rotate freely during the
excited state lifetime of the donor, 2 has the average value of 2/3.
60 is the refractive index of
the media. J(v) is the overlap integral of the fluorescence spectrum of the donor and the
absorption spectrum of the acceptor.
In the method, the FRET spectrum, (at , the acceptor may have
some absorbance also), is fitted to the sum of the two components. The first component is the
emission spectrum of the sample excited at (only the acceptor absorbs at ).
The second component is a singly labeled donor emission spectrum excited at .
,
where and α are the two weighting factors for the two components. Therefore,
,
where is the extinction coefficient of the fluorophore and is the fraction of DNA
labeled with donor. When donor labeling efficiency is 100%, equals one. can then be
calculated from the above equation. Here, was 513 nm and was 648 nm.
The EFRET values were plotted against metal concentration [M] and fit to the following
equation using Origin software, in order to determine the apparent dissociation constant (Kd ) for
folding:
Aratio)( ),( D
exemF D
ex
),( A
exemF A
ex A
ex
),( D
exem
DF D
ex
),(),()(),( D
exem
DA
exemA
D
exem FFratioF
Aratio)(
)(
)(
)(
)(
),(
),(),()(
A
ex
A
D
ex
D
A
ex
A
D
ex
A
A
exem
D
exem
DD
exemA Ed
F
FFratio
d
d E
D
ex A
ex
EFRET = EFRET (0) + C [M]
Kd + [M]EFRET = EFRET (0) +
C [M]
Kd + [M]
C [M]
Kd + [M]

63
3.2.4 CD experiments
To perform the CD studies, a non-cleavable substrate analog, called 17DS was used
where the single ribo-A of 17S was replaced by a deoxy-A (Figure 3.1a). 17E and 17DS (200
µM each) was annealed in 50 mM Na-HEPES acetate buffer (pH 7.5) by heating the sample at
85 C for 5 min and subsequently cooling it to room temperature over 1.5 h and then to 4 C for
~ 1-2 h. CD spectra were recorded on a JASCO J-515 spectropolarimeter in a 1 mm path length
cuvette using 500 µL of sample volume. Each spectrum was recorded over a wavelength range
of 340-200 nm using a scan speed of 50 nm/sec and a 0.5 sec response time. The final
concentration of DNA was diluted to 40 µM for all the experiments. By using a short path length
cuvette and a high DNA concentration, buffer absorbance due to Na-HEPES was minimal. (A
blank spectrum recorded with Na-HEPES buffer showed minimal noise in the 240-220 nm
region under these conditions, whereas if a 1 cm cuvette with 4 µM DNA was used, significant
noise was seen at that region). Divalent metal ions were titrated by incremental addition of
concentrated metal solution, such that the total volume increased by < 5%. In the case of
monovalent ion titrations, a large volume of stock monovalent solution (5 M) had to be added
and therefore the DNA concentration was kept constant by adding concentrated DNA stock upon
metal addition.To monitor the formation of Z-DNA, the CD signal at 294 nm (CD294) was
plotted against metal concentration [M] using Origin software and fit to the following equation to
obtain Kd from CD data. Since the value of the CD signal changed from positive to negative with
change in metal concentration, a constant was added to all the data points before curve fitting.
CD294 = C [M]
Kd + [M]CD294 =
C [M]
Kd + [M]
C [M]
Kd + [M]

64
Figure 3.1 Predicted secondary structures of the 8-17 DNAzyme constructs. a) Non-extended
and non-modified 8-17 DNAzyme used for activity assays. The substrate containing a single
RNA base, rA (indicated in red), at the cleavage site is called 17S and the enzyme is called 17E.
For the CD studies, the ribonucleotide (rA) at the cleavage site on the substrate was replaced
with a deoxyribonucleotide (A) to render it non-cleavable, and this was called 17DS. b) Modified
8-17 DNAzyme used for FRET study. The end of each arm is extended by two base pairs for
thermal stabilization and the rA was replaced with A. Substrate (Cy3-17DS) and enzyme (Cy5-
17E) were modified with Cy3 and Cy5 fluorophores, respectively to form a FRET pair.
3.3 Results
3.3.1 Activity based on kinetic assays
Since monovalent metal ion-dependent activity assays were performed with a modified
protocol starting with 5 × concentration of enzyme-substrate mixture (as compared to 2 ×
concentration for divalent metal ion-dependent reaction), we first ensured that this difference did
not cause any difference in the cleavage rate by performing a Zn2+
dependent assay starting at
both conditions (Figure 3.2).

65
Time (mins)
0 20 40 60 80 100
% c
leava
ge
0
20
40
60
80
100
2X Enz-sub
5X Enz-sub
Figure 3.2 Kinetic plot for Zn2+
dependent 8-17 DNAzyme reaction. The open triangles
represent reactions initiated with 2 × concentration of the enzyme – substrate complex and the
closed circles represent reactions initiated with 5 × concentration of the enzyme – substrate
complex.
The activity of the 8-17 DNAzyme was measured in the presence of alkali metal ions
(Li+, Na
+, K
+, Rb
+, and Cs
+) or NH4
+, each at 4 M concentration. All the assays were carried out
in 50 mM HEPES-acetate buffer, pH 7.5 which contained 25 mM EDTA. Our results showed
that if EDTA was not present in the buffer, trace amounts of divalent metal contaminants present
in the buffer caused significant cleavage after ~ 2 hours. Thus, it was essential to include EDTA
in the buffer.

66
Figure 3.3 Representative gels for the 8-17 DNAzyme reaction in the presence of 4 M
monovalent ions (Li+, Na
+, K
+, Rb
+, Cs
+, and NH4
+) or 80 mM Co(NH3)6
3+ in 50 mM Na-
HEPES, 25 mM EDTA, pH 7.5. The left panels are reactions carried out in the presence of both
enzyme and substrate, and the right panels are the control reactions carried out in the absence of
enzyme. S and P indicate the position of uncleaved substrate and product bands, respectively.

67
Figure 3.3 depicts representative gels for the activity assays performed using 4 M
monovalent ions or 80 mM [Co(NH3)6]3+
. The total time for which the reaction was carried out
varied between 45-55 h. The left panels show the substrate cleavage in the presence of
DNAzyme (called enzymatic cleavage), while the right panels display substrate cleavage in the
absence of the DNAzyme (called background cleavage) under identical conditions. In the
presence of Li+ and NH4
+, the intensity of product band can be seen increasing with time when
DNAzyme is present, suggesting the 8-17 DNAzyme is active with these two monovalent ions.
A small amount of cleavage is also observed for Na+ (< 10% in 50 h) over the background
cleavage. In the case of the other monovalent ions (K+, Rb
+, and Cs
+), no significant difference
was observed between enzymatic cleavage and background cleavage after ~50 h. The percent
cleavage is plotted vs. time in Figure 3.4. The observed rate constant (kobs) for 4 M Li+ and NH4
+
are 7 × 10-4
and 1 ×10-3
min-1
, respectively (calculated as an average of three independent trials).
This rate constant is at least 4-5 orders of magnitude lower than the estimated rate constants at
pH 7.5 with the most effective divalent metal co-factors, 100 µM Pb2+
(estimated kobs ~220 min-
1) and 10 mM Zn
2+ (estimated kobs ~50 min
-1) and ~3 orders of magnitude lower than that with 10
mM Mg2+
(kobs ~1 min-1
).56
However, the rate constant with monovalent ions still represent a rate
enhancement of ~107
over the spontaneous rate of cleavage for the dinucleotide junction A-rA.22
In addition to monovalent ions, the exchange inert complex [Co(NH3)6]3+
was also used
to probe if direct metal coordination is required for catalysis in ribozymes.1, 11, 16
We tested the
activity of the 8-17 DNAzyme in the presence of 80 mM [Co(NH3)6]3+
, and did not observe any
noticeable rate enhancement in enzymatic cleavage over background cleavage (Figure 3.3,
bottom panels).

68
Figure 3.4 Kinetic trace for the 8-17 DNAzyme reaction in the presence of 4 M metal chloride
solutions in 50 mM Na-HEPES and 25 mM EDTA (pH 7.5). The observed rate constants (kobs)
that are reported are an average of three independent experiments.
3.3.2 Folding studies using FRET
Metal ion-dependent folding of the 8-17 DNAzyme was probed by FRET. FRET can
provide information on the relative distance between the arms of the DNAzyme when labeled
with a fluorescent donor/acceptor pair (Figure 3.1b). In order to carry out the FRET studies, a
non-cleavable analog of the substrate, where the single RNA base was replaced by a DNA base,
was used. Additionally, the DNAzyme complex was extended by two base pairs on both ends to
increase the stability of the complex for FRET studies. The enzyme was labeled on the 5´ end
with the acceptor fluorophore, Cy5 and the substrate was labeled on the 5´ end with the donor
fluorophore, Cy3.
Figure 3.5a shows the typical FRET spectra for monovalent ions. The samples were
excited at 513 nm, and emission spectra were collected from 530 to 700 nm. There are two main

69
emission peaks: the peak at ~562 nm is from the donor (Cy3), and the acceptor (Cy5) peak at
~663 nm can be attributed to energy transfer from the donor. As the concentration of the metal
ion was increased, there was a decrease in the intensity of the donor peak and an increase in the
intensity of the acceptor peak. A considerable dilution effect on the fluorescence signal was
observed due to the large volume increase upon addition of a large amount of monovalent ion
solution. Thus, anti-correlated fluorescence intensity changes of the donor and acceptor were not
observed directly. However, the use of the (ratio)A method took into account the dilution effect
and an increase in EFRET was observed as the concentrations of the ions were increased.59
EFRET
changes in the presence of monovalent metal ions or NH4+
were obtained as a function of their
concentrations (Figure 3.5b). Interestingly, in contrast to activity assays where activity was
observed only in the presence of Li+, NH4
+ and, to a lesser extent with Na
+; folding was seen
with all the monovalent ions investigated. The Kd values obtained can be placed into two groups:
a stronger Kd for Li+, Na
+, and NH4
+ induced folding (0.228, 0.212 and 0.218 M, respectively)
and a weaker Kd for Rb+ and Cs
+ induced folding (0.359 and 0.344 M, respectively).

70
Figure 3.5 Monovalent ion-dependent FRET studies. a) Typical fluorescence spectra of the 8-17
DNAzyme construct used to probe FRET in the presence of monovalent ions. b) Monovalent
ion-dependent EFRET changes in the 8-17 DNAzyme. The Kd values reported are an average of
two independent measurements.
For comparison to folding in the presence of monovalent ions, divalent metal ion-
dependent folding was also studied with select transition and alkaline earth metal ions. Figure
3.6a shows the plot of EFRET values vs. metal concentration for Zn2+
and Cd2+
. The folding in the
presence of these transition metal ions requires a much lower concentration of metal ions, with
Kd values of 3.10 and 3.06 µM for Zn2+
and Cd2+
, respectively. The alkaline earth metal ion-
dependent EFRET showed two distinct kinds of folding. In the presence of Mg2+
and Ca2+
, higher
saturated EFRET states (EFRET ~0.66) were accompanied by stronger Kd values of 0.717 and 0.700
mM, respectively. This is in comparison to Sr2+
and Ba2+
(EFRET ~0.58) which observed Kd
values of 1.09 and 1.06 mM, respectively (Figure 3.6b). In general, the Kd values obtained for all
of the divalent metal ions studied are much stronger than those obtained with monovalent metal

71
ions, indicating that a higher concentration of monovalent ions is required for folding of the
DNAzyme.
Figure 3.6 Divalent metal ion-dependent EFRET changes in the 8-17 DNAzyme. Dependence of
EFRET on the concentration of a) transition metal ions b) alkaline earth metal ions. The Kd values
reported are an average of two independent measurements.
3.3.2.1 Mg2+
and Zn2+
titrations in different Na+ concentrations
From the FRET results with the series of di- and monovalent ions described, the
following questions were raised. Firstly, “Is the folding induced by electrostatic interaction
between metal ions and DNA or by site-specific interaction?” Secondly, “Are the folded state(s)
induced by monovalent ions identical with that induced by divalent ions?” In order to examine
these questions, we carried out additional FRET measurements, wherein, titrations were carried
out with Mg2+
and Zn2+
in the presence of different background concentrations of Na+ in the
range of 30-500 mM. Higher background Na+ concentration will screen more electrostatic
interactions between metal ions and DNA. Any trend in folding behavior changes could also

72
possibly provide us an insight into site-specific binding interactions of divalent metal ions and
folded structures.
The DNA sample was prepared in 30 mM HEPES-Na (pH 7.5) buffer instead of 50
mM, and corresponding Na+ background was prepared by adding small volume (2-47 µl) of 5M
NaCl in 50 mM HEPES-Na (pH 7.5). Mg2+
-dependent folding is shown in Figure 3.7 and
summarized in Table 3.1. The binding constants, Kd for folding linearly increased as the
background Na+ concentration increased. The Hill coefficient (n) values also increased gradually
to ~1. The final EFRET values were the same over the entire range of Na+ conditions used,
indicating that the DNAzyme folded into the same structure finally in the presence of Na+ and
Mg2+
. Zn2+
-dependent folding is shown Figure 3.8 and summarized in Table 3.1. Similar to the
Mg2+
-dependent folding, an increase in Kd was observed with an increase in the Na+
concentration. However, there was no clear trend seen with the Hill coefficient values and the
final EFRET values were all different in contrast with the Mg2+
-dependent folding.

73
0 15 300.2
0.3
0.4
0.5
0.6
30 mM Na
Kd = 0.412 ± 0.078 mM
n = 0.58 ± 0.04
0 15 300.2
0.3
0.4
0.5
0.6
50 mM Na
Kd = 0.557 ± 0.065 mM
n = 0.76 ± 0.07
0 15 300.2
0.3
0.4
0.5
0.6
EF
RE
T
100 mM Na
Kd = 1.21 ± 0.106 mM
n = 0.83 ± 0.06
0 15 300.2
0.3
0.4
0.5
0.6
200 mM Na
Kd = 2.13 ± 0.15 mM
n = 0.98 ± 0.07
0 15 300.2
0.3
0.4
0.5
0.6
[MgCl2] (mM)
500 mM Na
Kd = 7.97 ± 1.32 mM
n = 0.92 ± 0.15
0 15 30
0.3
0.4
0.5
0.6
30 mM Na
50 mM Na
100 mM Na
200 mM Na
500 mM Na
[MgCl2] (mM)
Figure 3.7 Mg2+
-dependent folding in the presence of different background Na+ concentrations
from 30-500 mM. Kd was obtained by fitting the plot with a single binding curve, y = y0 + a
x/(Kd+x), a Hill coefficient, n, was obtained by fitting the plot with a Hill plot, y = y0 +Vmax
xn/(K
n+x
n).

74
0 100 200 300 400 5000.2
0.3
0.4
0.5
0.6
30 mM Na
Kd = 0.78 ± 0.17 M
n = 0.70 ± 0.11
0 100 200 300 400 5000.2
0.3
0.4
0.5
0.6
50 mM Na
Kd = 1.55 ± 0.25 M
n = 0.78 ± 0.09
0 100 200 300 400 5000.2
0.3
0.4
0.5
0.6
EF
RE
T
100 mM Na
Kd = 3.91 ± 0.62 M
n = 0.86 ± 0.11
0 100 200 300 400 5000.2
0.3
0.4
0.5
0.6
200 mM Na
Kd = 7.80 ± 1.30 M
n = 0.78 ± 0.11
0 100 200 300 400 5000.2
0.3
0.4
0.5
0.6
[ZnCl2] (M))
500 mM Na
Kd = 22.8 ± 4.8 M
n = 0.61 ± 0.13
0 100 200 300 400 5000.2
0.3
0.4
0.5
0.6
30 mM Na
50 mM Na
100 mM Na
200 mM Na
500 mM Na
[ZnCl2] (M))
Figure 3.8 Zn2+
-dependent folding in the presence of different background Na+ concentrations
from 30-500 mM. Kd was obtained by fitting the plot with a single binding curve, y = y0 + a
x/(Kd+x), a Hill coefficient, n, was obtained by fitting the plot with a Hill plot, y = y0 +Vmax
xn/(K
n+x
n).
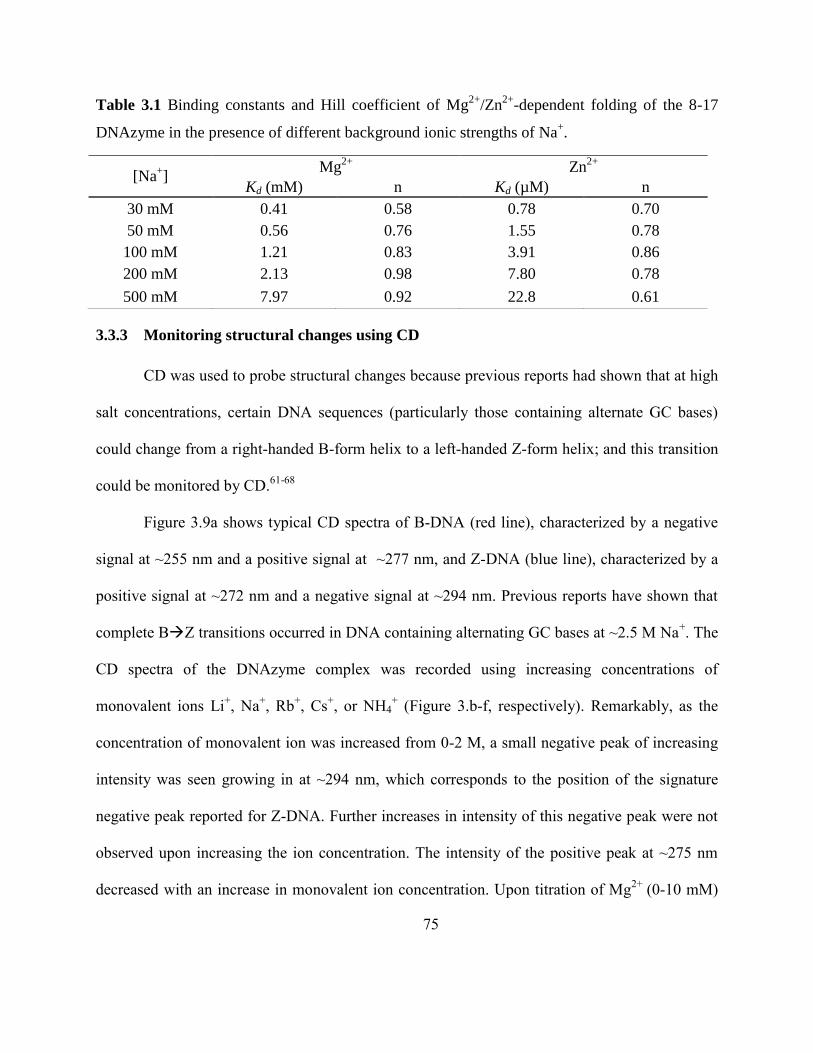
75
Table 3.1 Binding constants and Hill coefficient of Mg2+
/Zn2+
-dependent folding of the 8-17
DNAzyme in the presence of different background ionic strengths of Na+.
3.3.3 Monitoring structural changes using CD
CD was used to probe structural changes because previous reports had shown that at high
salt concentrations, certain DNA sequences (particularly those containing alternate GC bases)
could change from a right-handed B-form helix to a left-handed Z-form helix; and this transition
could be monitored by CD.61-68
Figure 3.9a shows typical CD spectra of B-DNA (red line), characterized by a negative
signal at ~255 nm and a positive signal at ~277 nm, and Z-DNA (blue line), characterized by a
positive signal at ~272 nm and a negative signal at ~294 nm. Previous reports have shown that
complete BZ transitions occurred in DNA containing alternating GC bases at ~2.5 M Na+. The
CD spectra of the DNAzyme complex was recorded using increasing concentrations of
monovalent ions Li+, Na
+, Rb
+, Cs
+, or NH4
+ (Figure 3.b-f, respectively). Remarkably, as the
concentration of monovalent ion was increased from 0-2 M, a small negative peak of increasing
intensity was seen growing in at ~294 nm, which corresponds to the position of the signature
negative peak reported for Z-DNA. Further increases in intensity of this negative peak were not
observed upon increasing the ion concentration. The intensity of the positive peak at ~275 nm
decreased with an increase in monovalent ion concentration. Upon titration of Mg2+
(0-10 mM)
[Na+]
Mg2+
Zn2+
Kd (mM) n Kd (µM) n
30 mM 0.41 0.58 0.78 0.70
50 mM 0.56 0.76 1.55 0.78
100 mM 1.21 0.83 3.91 0.86
200 mM 2.13 0.98 7.80 0.78
500 mM 7.97 0.92 22.8 0.61

76
and Zn2+
(0-300 µM), the same effect was observed (Figure 3.9g, h respectively). In contrast,
when Pb2+
was added (0-380 µM), we did not observe any negative peak at this wavelength
(Figure 3.9i).
Since the 8-17 DNAzyme does not contain a sequence that is known to form Z-DNA, it is
expected that complete BZ conversion does not take place. It is possible that a small fraction
of the DNAzyme converts to the Z-form or that a particular tract in the DNAzyme sequence can
promote the formation of Z-DNA. Two different control experiments were performed in order to
test if these results were typical for the 8-17 construct. First, the substrate (17DS) was hybridized
with a fully complementary sequence to form a DNA duplex and the CD spectra were recorded
with increasing concentrations of Zn2+
(Figure 3.10a), because Zn2+
showed the maximum
increase in the 294 nm peak. No changes in the spectra can be seen up to 340 µM of Zn2+.
Furthermore, since Z-DNA formation is mainly seen in sequences that contain alternate GCs, the
GC rich stem of the enzyme emerged as a starting point to test the effect of sequence elements on
the formation of Z-DNA.
Next, the effect of mutation on the three base pair stem in the catalytic core of the 8-17
DNAzyme was examined, as this region is rich in GC’s which are known to form Z-DNA. A
single GC base pair of the stem was replaced with an AT pair (enzyme is called
17S(C4A,G10C)), and the CD spectra were recorded with Zn2+
(Figure 3.10b) Remarkably, even
with this small change in the sequence, we did not observe a negative peak growing in at 294 nm
with a Zn2+
concentration of 940 µM. These results suggest that the stem region of the 8-17
DNAzyme may contain the sequence that leads to the structural changes observed by CD.
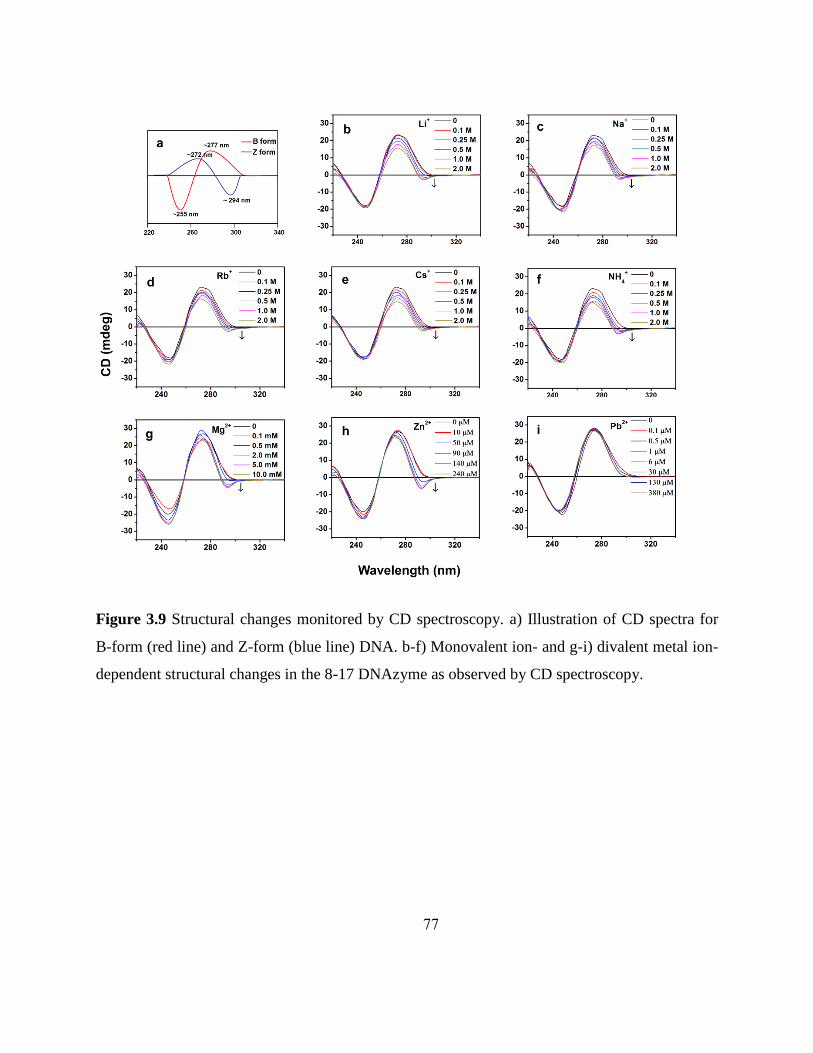
77
Figure 3.9 Structural changes monitored by CD spectroscopy. a) Illustration of CD spectra for
B-form (red line) and Z-form (blue line) DNA. b-f) Monovalent ion- and g-i) divalent metal ion-
dependent structural changes in the 8-17 DNAzyme as observed by CD spectroscopy.

78
Figure 3.10 Predicted secondary structures (top) and CD spectra in the presence of Zn2+
(bottom) for two controls: a) duplex DNA where substrate, 17DS was hybridized with a fully
complementary sequence, comp-17DS instead of the enzyme. b) Enzyme modified to replace
one GC pair in the stem with an AT pair (depicted in blue), called 17E (C4A, G10C).
In order to quantify the metal dependence, the CD signal at 294 nm was plotted against
metal concentration for the monovalent ions (Figure 3.11a), Mg2+
(Figure 3.11b) or Zn2+
(Figure
3.11c). A clear transition from positive intensity to increasing intensity of a negative peak was
observed. The Kd’s (from CD) was determined to be 0.313, 0.235, 0.269, 0.236, 0.149 M for Li+,
Na+, Rb
+, Cs
+, and NH4
+, respectively. These Kd values (from CD) are in a similar range as the
Kd of folding obtained from FRET studies (0.212 – 0.359 M for monovalent ions). The stronger
Kd’s of the divalent metal ions (Mg2+
: 1.13 mM and Zn2+
: 55.6 µM) are also comparable to the
Kd values obtained using FRET (Mg2+
: 0.71 mM and Zn2+
: 3.10 µM). These results indicate that
the metal-dependent global folding, monitored using FRET, is closely related to the structural

79
changes that are observed by CD, suggesting that the formation of Z-DNA may play an
important role for activity, except in the case of Pb2+
.
Figure 3.11 CD signal at 294 nm vs. metal concentration for a) monovalent ions, b) Mg2+
, or c)
Zn2+
. Note: Since the CD signal at 295 nm transitioned from a positive to negative value during
the metal-dependent titrations, we added a constant value of -3.5 to all the recorded CD (mdeg)
signals, so that all the data points could be brought to a single data quadrant for data fitting to
obtain the Kd values.

80
3.4 Discussion
3.4.1 Comparison of monovalent ion-dependent activities between DNAzymes and
ribozymes
It was shown previously that several ribozymes that showed activity in the presence of
divalent metal ions under physiological conditions were also active in the presence of a very high
concentration of monovalent cations.1, 37
Since the 8-17 DNAzyme shares a similar secondary
structure to the hammerhead ribozyme, such as the presence of a bulged three-way junction at
the catalytic core, as well as ribonucleotide phosphodiester transfer activity, we compared the
kobs for the 8-17 DNAzyme in the presence of 4 M monovalent ions to that reported for the
hammerhead ribozyme (Table 3.2). In the case of the 8-17 DNAzyme, the kobs values obtained
with the monovalent ions were much lower than those obtained for the hammerhead ribozyme.
For example, for the hammerhead ribozyme, the 4 M Li+ induced activity is only ~10-fold less
than the 10 mM Mg2+
induced activity, indicating that high concentrations of monovalent ion can
replace divalent ions in catalysis. This result led to the hypothesis that molar concentrations of
monovalent ions could substitute for millimolar concentrations of Mg2+
during catalysis by the
hammerhead ribozyme, indicating primarily the structural contributions of metal ions to
catalysis.1, 16, 17
In contrast, the 8-17 DNAzyme activity in the presence of 4 M monovalent ions
(Li+ and NH4
+) was ~1000-fold lower than that in the presence of 10 mM Mg
2+, and ~200,000-
fold slower than the estimated rate constant in the presence of 100 µM Pb2+
. Therefore, our
studies indicate that through the in vitro selection process, the 8-17 DNAzyme has a more
stringent requirement for divalent metal ions to obtain high activity as compared to the naturally
occurring ribozymes.69
Another major difference between the two nucleic acid enzymes is the

81
[Co(NH3)6]3+
-dependent activities. While [Co(NH3)6]3+
alone can support hammerhead ribozyme
activity,70
its role in 8-17 DNAzyme activity is not detectable, suggesting that outer-sphere metal
coordination is not likely to operate, at least in the presence of [Co(NH3)6]3+
.
Table 3.2 Comparison of observed rate constants (kobs) for the 8-17 DNAzyme, the hammerhead
ribozyme, and the G3 DNAzyme in the presence of selected metal ions or Co(NH3)63+
under
single turn-over conditions. aData from the present study in 50 mM HEPES and 25 mM EDTA
(pH 7.5). bData taken from Ref.
56 in 50 mM HEPES (pH 7.0) for the Mg
2+ assay and 50 mM
MES (pH 6.0) for the Pb2+
assay. *The Pb2+
-dependent cleavage was too fast to be measured at
higher pH. However, from the pH-dependent rate profile, it is estimated that the rate at pH 7.5 is
~220 min-1
. cData taken from Ref.
71 in 50 mM MES (pH 6.0). **The Zn
2+-dependent cleavage
was too fast to be measured at higher pH. However, from the pH-dependent rate profile, it is
estimated that the rate at pH 7.5 is ~50 min-1
. dData taken from Ref.
16 in 50 mM Tris (pH 8.0) for
the Mg2+
dependent assay and in 50 mM Tris and 25 mM EDTA (pH 8.0) for the M+ assays.
eData taken from Ref.
22 in 50 mM HEPES and 1 mM EDTA (pH 7.0). „N.D.‟ denotes not
detectable and „–‟ denotes not reported.
We also compared our results using the 8-17 DNAzyme to that of the G3 DNAzyme
(Table 3.2) isolated by Geyer et al. using in vitro selection in the absence of any divalent metal
kobs (min-1
)
Li+ Na
+ NH4+ Mg
2+ Zn2+ Pb
2+ Co(NH3)63
+ 8-17
DNAzyme 7.0×10
-4a
(4 M)
~10-5a
(4 M)
1.0×10-3a
(4 M)
0.9b
(10
mM)
1.3c
[**50.0]
(10 mM)
6.5b
[*220.0]
(100
µM)
N.Da
(80 mM)
hammerhead
ribozyme 2.9×10
-1d
(4 M)
7.5×10-3d
(4 M)
1.4×10-2d
(4 M)
2.2d
(10 mM)
- - 7.1×10-3d
(100 mM)
G3
DNAzyme 3.0×10
-3e
(500 mM)
4.0×10-3e
(500 mM)
4. ×10-3e
(500 mM)
N.D.e
(20 mM)
- - N.D.e
(100 mM)

82
co-factors.22
In contrast to the hammerhead ribozyme and the 8-17 DNAzyme, the G3 DNAzyme
displayed no activity in the presence of divalent metal ions and is a prototype monovalent ion-
dependent DNAzyme.22
These DNAzymes utilize the monovalent metal ion more efficiently as
they achieve similar kobs (~10-3
min-1
) at a much lower concentration of monovalent ion (500 mM
as opposed to the 4 M concentration required by the 8-17 DNAzyme). These results indicate that
laboratory selected DNAzymes may differ from naturally occurring ribozymes in their metal
dependence because their active sites can evolve to have more stringent discrimination between
different types of metal ions, albeit serendipitously during the in vitro selection process since the
selection is carried out in an excess of monovalent metal ions.
3.4.2 Correlation between ionic radii and folding
The Kd values obtained by FRET for different metal ions were plotted as a function of Z
(charge)/r (ionic radii) (Figure 3.12a). It can be seen that the monovalent ions which have the
largest ionic radii, and thus the lowest charge density, have the weakest Kd (~100 mM), whereas
the divalent transition metal ions have the highest Z/r ratio and the strongest Kd (~10-3
mM). It
was previously reported that small ions are more effective than large ions in condensing DNA
and stabilizing the tertiary conformation of ribozymes.72-79
Woodson and co-workers interpreted
the greater stability of RNA with small metal ions in terms of electrostatic interactions only.76-78
They discussed two factors for small, high charge density cations to be better in RNA folding.
First, these can approach the RNA more closely, resulting in stronger attractive Coulomb
interactions. Second, the excluded volume of each ion is smaller, which lowers the entropic
penalty for confining the ions to a small volume around the RNA.72, 75-78
A similar observation in

83
this study in relation to folding and ion sizes suggests that the observed folding is at least
partially due to electrostatic interactions between ions and the DNAzyme.
Figure 3.12 a) Relation between Z/r and Kd for monovalent and divalent ions. b) Relation
between kobs and Kd for monovalent and divalent ions.
3.4.3 Correlation between activity and folding
In order to examine the relationship between folding and activity of the DNAzyme, we
have co-plotted the Kd of folding from FRET and kobs from the cleavage reaction in Figure 3.12b.
The activity of the 8-17 DNAzyme is dependent on metal cofactors with the following order of
activity; Pb2+
>> Zn2+
>> Cd2+
>> Mg2+
~ Ca2+
> Ba2+
~ Sr2+
>> ~ NH4+
~ Li+ >> Na
+. We found
close correlation between folding and activity, (i.e., stronger binding affinity in folding results in
higher activity), except in the case of the Pb2+
ion, which does not induce folding.43, 44
These
correlations strongly suggest metal ion dependent folding plays an important role in the catalytic
function of DNAzymes.

84
3.4.4 Electrostatic and site-specific interaction
Two of the four binding modes of metal ions to nucleic acids include electrostatic
charge screening and site-specific inner sphere interactions. Electrostatic interactions neutralize
negative charges on phosphate backbones by a diffuse cloud of ions and are important in the
conformational transitions of nucleic acids, and thus metal ions generally play a critical role in
mediating structural alterations.80-83
Metal ions can also bind nucleic acids through inner-sphere
interaction with specific ligands on the nucleic acid, forming a defined local geometry. The
binding will be more specific to certain metal ions, and usually divalent metal ions are involved
in this interaction.5, 6
The electrostatic interaction between metal ions and nucleic acids may be
able to explain the relations between ion size, folding, and activity in some extent. However, the
difference between different groups of metal ions cannot be merely simply explained by on this
basis. In order to gain deeper insight into the nature of the metal-DNA interaction and further the
role of the metal ions on the DNAzyme catalysis, we carried out divalent metal-dependent FRET
measurements in different ionic strength background Na+ concentrations.
Mg2+
-dependent folding of the 8-17 DNAzyme was studied in the presence of different
background Na+ concentrations. Increases in the Kd values and a Hill coefficient „n‟ for folding
were observed as the background sodium concentrations increased. Anti-cooperative binding of
Mg2+
with a Hill coefficient n<1 was observed in a low ionic strength background Na+ ions,
which is typical for diffuse binding of Mg2+
ions to nucleic acids.84, 85
This suggests that charge
screening by positive charged ions is required for the folding of the DNAzyme. At higher ionic
strength background Na+ ions such as ≥ 200 mM of Na
+, non-cooperative binding of Mg
2+ was
observed with a Hill coefficient n close to 1. Under these conditions, the overall charge on the

85
DNA is significantly reduced by the diffuse binding of Na+ ions, and the electrostatic
interactions between Mg2+
and DNA will be relatively weak.86, 87
Further, induced folding is
probably by site-specific binding of a Mg2+
ion to the DNA. Almost identical results were
previously reported for a four-way RNA junction derived by complementing the loops of the
hairpin ribozyme.27, 81
It was interpreted that there is a single critical Mg2+
ion bound at the four-
way junction, but that effective folding also requires a significant ionic strength to provide
general charge neutralization. Initial EFRET value increased as background ionic strength
increased, but the final EFRET did not change, indicating that the DNAzyme was pre-folded by the
background Na+ and further folded by Mg
2+ ions. These observations further support the idea
that folding of the DNAzyme is induced from a combination of both electrostatic interactions
and site-specific binding. The Hill coefficient n of Zn2+
-dependent folding was ~ 06-0.9. In
contrast with what was observed with the Mg2+
-dependent folding study, there was no systematic
change in the Hill coefficient values of Zn2+
-dependent folding in different background ionic
strengths of Na+ ions. Another difference from the Mg
2+-dependence case is that slightly
different final EFRET values were observed depending on background ionic strength of Na+,
implying that different folding pathways may be induced by Na+ from that by Zn
2+.
3.4.5 CD evidence for Z-DNA formation
Since FRET experiments provide information only on global folding, but not on
structural changes, we sought to use CD to search for any structural changes in the presence of
different metal ions. We found very interesting evidence of a small percentage of Z-DNA
formation, evidenced by the increase in the ~294 nm peak with increasing concentration of metal
ions. It is not surprising that the percentage of Z-DNA formation in the 8-17 DNAzyme is small

86
because it contains only a small portion of alternate GC bases (in the stem region) that are known
to form Z-DNA.61-68
CD has previously been used to monitor structural changes in the hammerhead
ribozyme,88-90
HDV ribozyme,91
hairpin ribozyme,92
lead ribozyme,93
RNA subunit of RNase
P,94, 95
10-23 DNAzyme,96
Dk5 DNAzyme,97
and a Ca2+
- dependent DNAzyme;98
however, to
the best of our knowledge, this is the first study where metal-dependent Z-DNA formation has
been reported among DNAzymes and ribozymes. More importantly, since the Kd obtained from
the CD studies are similar to those obtained from the FRET studies and activity assays, these
results suggest that both metal ion-dependent folding and Z-DNA formation play important roles
in monovalent ion- and divalent metal ion-dependent activities. The only exception is Pb2+
, in the
presence of which, no folding or Z-DNA formation was observed, indicating Pb2+
-dependent
enzymatic function is based upon a completely different mechanism from those of monovalent
metal ions and other divalent metal ions.
3.5 Conclusions
Metal ion-dependent activity and folding of the 8-17 DNAzyme was examined. In the
presence of 4 M Li+ or NH4
+, ~1000-fold lower and an estimated ~200,000-fold lower activity
was observed when compared to 10 mM Mg2+
and 100 µM Pb2+
, respectively, indicating high
selectivity of the DNAzyme for divalent metal ions. This contrasted sharply to that seen in the
hammerhead ribozyme where the activity with 4 M Li+ was only ~10 fold lower than that of
Mg2+
. Clear trends between the ionic radii of the metal ions, effective in folding and activity
were been observed, suggesting that, with the exception of Pb2+
, the most active metal ion,
electrostatic interactions between metal ions and DNA are important in the folding and activity

87
of the 8-17 DNAzyme. Structural changes of the 8-17 DNAzyme in the presence of various
metal ions was monitored by CD and these assays provided interesting evidence for Z-DNA
formation in the presence of monovalent ions, Mg2+
and Zn2+
, but not in the case of Pb2+
. This
provided further proof that Pb2+
acts in a different manner as opposed to other active metal ions.
To the best of our knowledge, this is the first report of Z-DNA formation in ribozymes and
DNAzymes. Further investigation would be required to gain insight into the sequence elements
that maybe responsible for the formation of Z-DNA.
3.6 References
1. Murray, J. B.; Seyhan, A. A.; Walter, N. G.; Burke, J. M.; Scott, W. G. The hammerhead,
hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem.
Biol. 1998, 5, 587-595.
2. DeRose, V. J. Metal ion binding to catalytic RNA molecules. Curr. Opin. Struct. Biol.
2003, 13, 317-324.
3. Sigel, R. K. O. Group II intron ribozymes and metal ions - a delicate relationship. Eur. J.
Inorg. Chem. 2005, 2281-2292.
4. Freisinger, E.; Sigel, R. K. O. From nucleotides to ribozymes-A comparison of their
metal ion binding properties. Coord. Chem. Rev. 2007, 251, 1834-1851.
5. Sigel, R. K. Intimate Relationships between Metal Ions and Nucleic Acids. Angew.
Chem. Int. Ed. 2007, 46, 654-656.
6. Sigel, R. K.; Pyle, A. M. Alternative roles for metal ions in enzyme catalysis and the
implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97-113.
7. Schnabl, J.; Sigel, R. K. Controlling ribozyme activity by metal ions. Curr. Opin. Chem.
Biol. 2010.
8. Woodson, S. A. Metal ions and RNA folding: a highly charged topic with a dynamic
future. Curr. Opin. Chem. Biol. 2005, 9, 104-109.
9. Suelter, C. H. Enzymes activated by monovalent cations. Science 1970, 168, 789-795.
10. Suelter, C. H. Monovalent cations in enzymes-catalyzed reactions. Met. Ions Biol. Syst.
1974, 3, 201-251.
11. Hampel, A.; Cowan, J. A. A unique mechanism for RNA catalysis: the role of metal
cofactors in hairpin ribozyme cleavage. Chem. Biol. 1997, 4, 513-517.
12. Nesbitt, S.; Hegg, L. A.; Fedor, M. J. An unusual pH-independent and metal-ion-
independent mechanism for hairpin ribozyme catalysis. Chem. Biol. 1997, 4, 619-630.
13. Young, K. J.; Gill, F.; Grasby, J. A. Metal ions play a passive role in the hairpin
ribozyme catalysed reaction. Nucleic Acids Res. 1997, 25, 3760-3766.

88
14. Earnshaw, D. J.; Gait, M. J. Hairpin ribozyme cleavage catalyzed by aminoglycoside
antibiotics and the polyamine spermine in the absence of metal ions. Nucleic Acids Res.
1998, 26, 5551-5561.
15. Seyhan, A. A.; Amaral, J.; Burke, J. M. Intracellular RNA cleavage by the hairpin
ribozyme. Nucleic Acids Res. 1998, 26, 3494-3504.
16. Curtis, E. A.; Bartel, D. P. The hammerhead cleavage reaction in monovalent cations.
RNA 2001, 7, 546-552.
17. O'Rear, J. L.; Wang, S.; Feig, A. L.; Beigelman, L.; Uhlenbeck, O. C.; Herschlag, D.
Comparison of the hammerhead cleavage reactions stimulated by monovalent and
divalent cations. RNA 2001, 7, 537-545.
18. Perrotta, A. T.; Been, M. D. HDV ribozyme activity in monovalent cations. Biochemistry
2006, 45, 11357-11365.
19. Roth, A.; Nahvi, A.; Lee, M.; Jona, I.; Breaker, R. R. Characteristics of the glmS
ribozyme suggest only structural roles for divalent metal ions. RNA 2006, 12, 607-619.
20. Jaeger, J. A.; Zuker, M.; Turner, D. H. Melting and chemical modification of a cyclized
self-splicing group I intron: similarity of structures in 1 M Na+, in 10 mM Mg2+, and in
the presence of substrate. Biochemistry 1990, 29, 10147-10158.
21. Adams, P. L.; Stahley, M. R.; Kosek, A. B.; Wang, J.; Strobel, S. A. Crystal structure of a
self-splicing group I intron with both exons. Nature 2004, 430, 45-50.
22. Geyer, C. R.; Sen, D. Evidence for the metal-cofactor independence of an RNA
phosphodiester-cleaving DNA enzyme. Chem. Biol. 1997, 4, 579-593.
23. Bren, K. L.; Pecoraro, V. L.; Gray, H. B. Metalloprotein folding. Inorg. Chem. 2004, 43,
7894-7896.
24. Lilley, D. M. Structure, folding and catalysis of the small nucleolytic ribozymes. Curr.
Opin. Struct. Biol. 1999, 9, 330-338.
25. Hanna, R.; Doudna, J. A. Metal ions in ribozyme folding and catalysis. Curr. Opin.
Chem. Biol. 2000, 4, 166-170.
26. Sosnick, T. R.; Pan, T. RNA folding: models and perspectives. Curr. Opin. Struct. Biol.
2003, 13, 309-316.
27. Wilson, T. J.; Lilley, D. M. Metal ion binding and the folding of the hairpin ribozyme.
RNA 2002, 8, 587-600.
28. Hammann, C.; Lilley, D. M. Folding and activity of the hammerhead ribozyme.
ChemBioChem 2002, 3, 690-700.
29. Lafontaine, D. A.; Norman, D. G.; Lilley, D. M. The global structure of the VS ribozyme.
EMBO J. 2002, 21, 2461-2471.
30. Stahley, M. R.; Strobel, S. A. Structural evidence for a two-metal-ion mechanism of
group I intron splicing. Science 2005, 309, 1587-1590.
31. Stahley, M. R.; Strobel, S. A. RNA splicing: group I intron crystal structures reveal the
basis of splice site selection and metal ion catalysis. Curr. Opin. Struct. Biol. 2006, 16,
319-326.
32. Qu, X.; Smith, G. J.; Lee, K. T.; Sosnick, T. R.; Pan, T.; Scherer, N. F. Single-molecule
nonequilibrium periodic Mg2+-concentration jump experiments reveal details of the early
folding pathways of a large RNA. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6602-6607.

89
33. Baird, N. J.; Fang, X. W.; Srividya, N.; Pan, T.; Sosnick, T. R. Folding of a universal
ribozyme: the ribonuclease P RNA. Q. Rev. Biophys. 2007, 40, 113-161.
34. Kazantsev, A. V.; Krivenko, A. A.; Pace, N. R. Mapping metal-binding sites in the
catalytic domain of bacterial RNase P RNA. RNA 2009, 15, 266-276.
35. Beebe, J. A.; Kurz, J. C.; Fierke, C. A. Magnesium ions are required by Bacillus subtilis
ribonuclease P RNA for both binding and cleaving precursor tRNAAsp. Biochemistry
1996, 35, 10493-10505.
36. Fang, X. W.; Pan, T.; Sosnick, T. R. Mg2+
-dependent folding of a large ribozyme without
kinetic traps. Nat. Struct. Biol. 1999, 6, 1091-1095.
37. Basu, S.; Rambo, R. P.; Strauss-Soukup, J.; Cate, J. H.; Ferre-D'Amare, A. R.; Strobel, S.
A.; Doudna, J. A. A specific monovalent metal ion integral to the AA platform of the
RNA tetraloop receptor. Nat. Struct. Biol. 1998, 5, 986-992.
38. Takamoto, K.; He, Q.; Morris, S.; Chance, M. R.; Brenowitz, M. Monovalent cations
mediate formation of native tertiary structure of the Tetrahymena thermophila ribozyme.
Nat. Struct. Biol. 2002, 9, 928-933.
39. Penedo, J. C.; Wilson, T. J.; Jayasena, S. D.; Khvorova, A.; Lilley, D. M. Folding of the
natural hammerhead ribozyme is enhanced by interaction of auxiliary elements. RNA
2004, 10, 880-888.
40. Shcherbakova, I.; Gupta, S.; Chance, M. R.; Brenowitz, M. Monovalent ion-mediated
folding of the Tetrahymena thermophila ribozyme. J. Mol. Biol. 2004, 342, 1431-1442.
41. Jiang, Y. F.; Xiao, M.; Yin, P.; Zhang, Y. Monovalent cations use multiple mechanisms
to resolve ribozyme misfolding. RNA 2006, 12, 561-566.
42. Liu, J.; Lu, Y. FRET study of a trifluorophore-labeled DNAzyme. J. Am. Chem. Soc.
2002, 124, 15208-15216.
43. Kim, H. K.; Liu, J.; Li, J.; Nagraj, N.; Li, M.; Pavot, C. M.; Lu, Y. Metal-dependent
global folding and activity of the 8-17 DNAzyme studied by fluorescence resonance
energy transfer. J. Am. Chem. Soc. 2007, 129, 6896-6902.
44. Kim, H. K.; Rasnik, I.; Liu, J.; Ha, T.; Lu, Y. Dissecting metal ion-dependent folding and
catalysis of a single DNAzyme. Nat. Chem. Biol. 2007, 3, 763-768.
45. Lee, N. K.; Koh, H. R.; Han, K. Y.; Kim, S. K. Folding of 8-17 deoxyribozyme studied
by three-color alternating-laser excitation of single molecules. J. Am. Chem. Soc. 2007,
129, 15526-15534.
46. Liu, Y.; Sen, D. Local rather than global folding enables the lead-dependent activity of
the 8-17 deoxyribozyme: evidence from contact photo-crosslinking. J. Mol. Biol. 2010,
395, 234-241.
47. Li, J.; Lu, Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J.
Am. Chem. Soc. 2000, 122, 10466-10467.
48. Liu, J.; Lu, Y. Improving fluorescent DNAzyme biosensors by combining inter- and
intramolecular quenchers. Anal Chem 2003, 75, 6666-6672.
49. Nagraj, N.; Liu, J.; Sterling, S.; Wu, J.; Lu, Y. DNAzyme catalytic beacon sensors that
resist temperature-dependent variations. Chem. Commun. 2009, 4103-4105.
50. Wernette, D. P.; Mead, C.; Bohn, P. W.; Lu, Y. Surface immobilization of catalytic
beacons based on ratiometric fluorescent DNAzyme sensors: a systematic study.
Langmuir 2007, 23, 9513-9521.

90
51. Xiang, Y.; Tong, A.; Lu, Y. Abasic site-containing DNAzyme and aptamer for label-free
fluorescent detection of Pb(2+) and adenosine with high sensitivity, selectivity, and
tunable dynamic range. J. Am. Chem. Soc. 2009, 131, 15352-15357.
52. Liu, J.; Lu, Y. A colorimetric lead biosensor using DNAzyme-directed assembly of gold
nanoparticles. J. Am. Chem. Soc. 2003, 125, 6642-6643.
53. Liu, J.; Lu, Y. Colorimetric biosensors based on DNAzyme-assembled gold
nanoparticles. J. Fluoresc. 2004, 14, 343-354.
54. Liu, J.; Lu, Y. Accelerated color change of gold nanoparticles assembled by DNAzymes
for simple and fast colorimetric Pb2+ detection. J. Am. Chem. Soc. 2004, 126, 12298-
12305.
55. Liu, J.; Lu, Y. Optimization of a Pb2+-Directed Gold Nanoparticle/DNAzyme Assembly
and Its Application as a Colorimetric Biosensor for Pb2+. Chem. Mater. 2004, 16, 3231-
3238.
56. Brown, A. K.; Li, J.; Pavot, C. M.; Lu, Y. A lead-dependent DNAzyme with a two-step
mechanism. Biochemistry 2003, 42, 7152-7161.
57. Liu, J.; Lu, Y. Multi-fluorophore fluorescence resonance energy transfer for probing
nucleic acids structure and folding. Methods Mol. Biol. 2006, 335, 257-271.
58. Lakowicz, J. R., Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum:
New York, 1999.
59. Clegg, R. M. Fluorescence resonance energy transfer and nucleic acids. Methods
Enzymol. 1992, 211, 353-388.
60. Van der Meer, B. W. Kappa-squared: from nuisance to new sense. Rev. Mol. Biotechnol.
2002, 82, 181-196.
61. Hernandez, B.; Baumruk, V.; Gouyette, C.; Ghomi, M. Thermal stability, structural
features, and B-to-Z transition in DNA tetraloop hairpins as determined by optical
spectroscopy in d(CG)(3)T(4)(CG)(3) and d(CG)(3)A(4)(CG)(3) oligodeoxynucleotides.
Biopolymers 2005, 78, 21-34.
62. Benight, A. S.; Wang, Y. S.; Amaratunga, M.; Chattopadhyaya, R.; Henderson, J.;
Hanlon, S.; Ikuta, S. Conformation and dynamics of a left-handed Z-DNA hairpin:
studies of d(CGCGCGTTTTCGCGCG) in solution. Biochemistry 1989, 28, 3323-3332.
63. Chattopadhyaya, R.; Ikuta, S.; Grzeskowiak, K.; Dickerson, R. E. X-ray structure of a
DNA hairpin molecule. Nature 1988, 334, 175-179.
64. Xodo, L. E.; Manzini, G.; Quadrifoglio, F.; van der Marel, G. A.; van Boom, J. H. The B-
Z conformational transition in folded oligodeoxynucleotides: loop size and stability of Z-
hairpins. Biochemistry 1988, 27, 6327-6331.
65. Wang, Y.; Thomas, G. A.; Peticolas, W. L. Sequence dependence of the B to Z transition
in crystals and aqueous NaCl solutions for deoxyoligonucleotides containing all four
canonical DNA bases. Biochemistry 1987, 26, 5178-5186.
66. Germann, M. W.; Schoenwaelder, K. H.; van de Sande, J. H. Right- and left-handed (Z)
helical conformations of the hairpin d(C-G)5T4(C-G)5 monomer and dimer.
Biochemistry 1985, 24, 5698-5702.
67. Thamann, T. J.; Lord, R. C.; Wang, A. H.; Rich, A. The high salt form of poly(dG-
dC).poly(dG-dC) is left-handed Z-DNA: Raman spectra of crystals and solutions. Nucleic
Acids Res. 1981, 9, 5443-5457.

91
68. Pohl, F. M.; Jovin, T. M. Salt-induced co-operative conformational change of a synthetic
DNA: equilibrium and kinetic studies with poly (dG-dC). J. Mol. Biol. 1972, 67, 375-
396.
69. Weinstein, L. B.; Jones, B. C.; Cosstick, R.; Cech, T. R. A second catalytic metal ion in
group I ribozyme. Nature 1997, 388, 805-808.
70. Seyhan, A. A.; Burke, J. M. Mg2+
-independent hairpin ribozyme catalysis in hydrated
RNA films. RNA 2000, 6, 189-198.
71. Li, J.; Zheng, W.; Kwon, A. H.; Lu, Y. In vitro selection and characterization of a highly
efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000, 28,
481-488.
72. Rouzina, I.; Bloomfield, V. A. Influence of Ligand Spatial Organization on Competitive
Electrostatic Binding to DNA. J. Phys. Chem. 1996, 100, 4305-4313.
73. Plum, G. E.; Arscott, P. G.; Bloomfield, V. A. Condensation of DNA by trivalent cations.
2. Effects of cation structure. Biopolymers 1990, 30, 631-643.
74. Trend, B. L.; Knoll, D. A.; Ueno, M.; Evans, D. F.; Bloomfield, V. A. Cation radius
effects on the helix-coil transition of DNA. Cryptates and other large cations. Biophys. J.
1990, 57, 829-834.
75. Stigter, D.; Dill, K. A. Binding of ionic ligands to polyelectrolytes. Biophys. J. 1996, 71,
2064-2074.
76. Heilman-Miller, S. L.; Thirumalai, D.; Woodson, S. A. Role of counterion condensation
in folding of the Tetrahymena ribozyme. I. Equilibrium stabilization by cations. J. Mol.
Biol. 2001, 306, 1157-1166.
77. Heilman-Miller, S. L.; Pan, J.; Thirumalai, D.; Woodson, S. A. Role of counterion
condensation in folding of the Tetrahymena ribozyme. II. Counterion-dependence of
folding kinetics. J. Mol. Biol. 2001, 309, 57-68.
78. Koculi, E.; Hyeon, C.; Thirumalai, D.; Woodson, S. A. Charge density of divalent metal
cations determines RNA stability. J. Am. Chem. Soc. 2007, 129, 2676-2682.
79. Bai, Y.; Greenfeld, M.; Travers, K. J.; Chu, V. B.; Lipfert, J.; Doniach, S.; Herschlag, D.
Quantitative and comprehensive decomposition of the ion atmosphere around nucleic
acids. J. Am. Chem. Soc. 2007, 129, 14981-14988.
80. Clegg, R. M.; Murchie, A. I.; Lilley, D. M. The four-way DNA junction: a fluorescence
resonance energy transfer study. Braz. J. Med. Biol. Res. 1993, 26, 405-416.
81. Lilley, D. M.; Clegg, R. M. The structure of the four-way junction in DNA. Annu. Rev.
Biophys. Biomol. Struct. 1993, 22, 299-328.
82. Misra, V. K.; Draper, D. E. On the role of magnesium ions in RNA stability. Biopolymers
1998, 48, 113-135.
83. Misra, V. K.; Draper, D. E. A thermodynamic framework for Mg2+
binding to RNA.
Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12456-12461.
84. Krakauer, H. The binding of Mg2+
ions to polyadenylate, polyuridylate, and their
complexes. Biopolymers 1971, 10, 2459-2490.
85. Owczarzy, R.; Moreira, B. G.; You, Y.; Behlke, M. A.; Walder, J. A. Predicting stability
of DNA duplexes in solutions containing magnesium and monovalent cations.
Biochemistry 2008, 47, 5336-5353.

92
86. Bukhman, Y. V.; Draper, D. E. Affinities and selectivities of divalent cation binding sites
within an RNA tertiary structure. J. Mol. Biol. 1997, 273, 1020-1031.
87. Peracchi, A. Preferential activation of the 8-17 deoxyribozyme by Ca2+
ions. Evidence
for the identity of 8-17 with the catalytic domain of the Mg5 deoxyribozyme. J. Biol.
Chem. 2000, 275, 11693-11697.
88. Horton, T. E.; DeRose, V. J. Cobalt hexammine inhibition of the hammerhead ribozyme.
Biochemistry 2000, 39, 11408-11416.
89. Mikulecky, P. J.; Feig, A. L. Cold denaturation of the hammerhead ribozyme. J. Am.
Chem. Soc. 2002, 124, 890-891.
90. Sakamoto, T.; Kim, M. H.; Kurihara, Y.; Sasaki, N.; Noguchi, T.; Katahira, M.; Uesugi,
S. Properties of a hammerhead ribozyme with deletion of stem II. J. Biochem. 1997, 121,
288-294.
91. Tinsley, R. A.; Harris, D. A.; Walter, N. G. Magnesium dependence of the amplified
conformational switch in the trans-acting hepatitis delta virus ribozyme. Biochemistry
2004, 43, 8935-8945.
92. Komatsu, Y.; Kumagai, I.; Ohtsuka, E. Investigation of the recognition of an important
uridine in an internal loop of a hairpin ribozyme prepared using post-synthetically
modified oligonucleotides. Nucleic Acids Res. 1999, 27, 4314-4323.
93. Kim, M. H.; Sugiyama, T.; Kanagawa, M.; Tanaka, Y.; Nagaoka, M.; Nishimura, Y.;
Kohno, T.; Kurihara, Y.; Katahira, M.; Uesugi, S. Effects of metal ions on activity and
structure of a lead ribozyme. Nucleic Acids Symp.Ser. 1996, 35, 133-134.
94. Pan, T.; Sosnick, T. R. Intermediates and kinetic traps in the folding of a large ribozyme
revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity.
Nat. Struct. Biol. 1997, 4, 931-938.
95. Fang, X. W.; Golden, B. L.; Littrell, K.; Shelton, V.; Thiyagarajan, P.; Pan, T.; Sosnick,
T. R. The thermodynamic origin of the stability of a thermophilic ribozyme. Proc. Natl.
Acad. Sci. U. S. A. 2001, 98, 4355-4360.
96. Ota, N.; Warashina, M.; Hirano, K.; Hatanaka, K.; Taira, K. Effects of helical structures
formed by the binding arms of DNAzymes and their substrates on catalytic activity.
Nucleic Acids Res. 1998, 26, 3385-3391.
97. McManus, S. A.; Li, Y. A deoxyribozyme with a novel guanine quartet-helix pseudoknot
structure. J. Mol. Biol. 2008, 375, 960-968.
98. Okumoto, Y.; Tanabe, Y.; Sugimoto, N. Factors that contribute to efficient catalytic
activity of a small Ca2+
-dependent deoxyribozyme in relation to its RNA cleavage
function. Biochemistry 2003, 42, 2158-2165.

93
4 Investigating Potential Metal Ion Binding Sites of the 8-17 DNAzyme
through Phosphorothioate Modifications and 31
P NMR
Notes and Acknowledgements: The work in this chapter was done in collaboration with
Eric Null (31
P NMR) and Seyed-Fakhreddin Torabi. The authors also thank Dr. Feng Lin for his
assistance with 31
P NMR experiments. This work is the basis of an unpublished manuscript,
“Investigating potential metal-binding sites of the 8-17 DNAzyme through phosphorothioate
modifications and 31
P NMR.” Nandini Nagraj, Eric Null, Seyed-Fakhreddin Torabi and Yi Lu. In
preparation.
4.1 Introduction
4.1.1 Insights into the mechanism of the 8-17 DNAzyme
Extensive characterization studies of the 8-17 DNAzyme have been previously carried
out based on various biochemical probing methods.1-7
Like most ribozymes and DNAzymes, the
8-17 DNAzyme depends on divalent metal ions for activity. The DNAzyme activity is the
highest in the presence of Pb2+
, following the order: Pb2+
>> Zn2+
>> Mn2+
≈ Co2+
> Ni2+
> Mg2+
≈ Ca2+
> Sr2+
≈ Ba2+
, in spite of the fact that the 8-17 motifs were selected under varying
conditions and with metal ions other than Pb2+
.2, 5, 8
This DNAzyme is a highly efficient enzyme
with its maximum reaction rate estimated to be ~ 220 min-1
in the presence of 200 µM Pb2+
at
physiological pH (7.5), which is among the highest in DNAzymes/ribozymes.2
Systematic modifications of bases through the introduction of non-standard nucleotides
in the catalytic core of the 8-17 DNAzyme (Figure 4.1) have been carried out and the activity of
these modified DNAzymes have been calculated through kinetic assays.4, 5
The results of these

94
studies correlated well with the original in vitro selection results obtained by Santoro and Joyce
and the impact of mutations on the overall activity of the DNAzyme was postulated.9 Photo
cross-linking studies have also been undertaken by Liu and Sen to investigate the cross-linking
patterns and proximity between different bases with the scissile site of the substrate.7 This has
been done by systematic replacement of each of these bases with the thio- and halogen-
substituted base analogues, followed by photochemical activation to yield cross-links within the
enzyme-substrate complex, in the presence of 10 mM Mg2+
. A model of the active site of the 8-
17 DNAzyme has been generated based on their data which the authors suggest is generally
representative of the pre-folded active site in the presence of both lower and higher magnesium
concentrations.7 In chapter 4, the effect of various monovalent and divalent ions on the activity,
global folding and structure formation of the 8-17 DNAzyme has been discussed. It was
observed from both bulk and single-molecule FRET studies that Pb2+
, the most efficient co-
factor of the 8-17 DNAzyme, did not cause any folding of the enzyme-substrate complex before
cleavage activity.10, 11
In addition, no structural change from the right-handed B-form to the Z-
form was observed with Pb2+
, whereas, in the presence of sufficient concentrations of other
divalent and monovalent metal ions, the 8-17 DNAzyme folded to form a more compact
structure prior to activity.12
CD measurements also indicated a structural change from the B-form
to the Z-form in the presence of sufficient concentrations of these metal ions. This led to the
proposition that the 8-17 DNAzyme might form a special pocket to accept Pb2+
or was
structurally pre-organized through directed evolution to accept, and catalytically utilize Pb2+
.

95
T2.1
5'
3'
G1.1rN
A6
G7 C8
C13
G14
A15W
12
A15.05'
3'
Bulge Loop
Terminal Loop
Cleavage Site
W = A or T
N = A, G, C or T
Figure 4.1 Predicted secondary structure of the trans-cleaving 8-17 DNAzyme showing the
conserved residues in blue and indicating positions of the nucleotides based on the numbering
system proposed by Peracchi et al.1, 5
The bold lines represent nucleotides on the helical regions
while the thin lines represent Watson-Crick base pairs across these helices.
4.1.2 Phosphorothioate (or PS) modifications
Replacement of the P-O bond of a phosphate group by a P-S bond (Figure 4.2) is
referred to as a „phosphorothioate (or PS) modification‟.13, 14
This represents a rather
conservative elemental replacement in terms of the overall structure of the phosphate, since the
P-S bond is only 0.3 Å longer than the P-O bond. However, there is a significant difference in
terms of reactivity toward different metal ions since the negative charge in the case of P-S bond
is localized to a greater extent on the more polarizable sulfur. Therefore, divalent metal ions that
have been traditionally classified as being „hard‟ such as the alkaline earth metals, prefer binding
to the phosphate oxygen, while metal ions, such as Cd2+
, that have been traditionally classified as
being „soft‟ prefer binding to the sulfur. There are „borderline‟ metal ions that can bind both
oxygen and sulfur and these include Pb2+
, Co2+
, Zn2+
and Mn2+
.

96
Figure 4.2 Schematic representation of a phosphate to a phosphorothioate modification on the
DNA backbone.
4.1.2.1 Identification of metal-binding sites in ribozymes using PS modifications
Site-specific phosphorothioate labeling on the phosphate backbone of ribozymes has been
used extensively to predict important structural and functional sites relevant to catalysis.15-19
Metal-specificity switch or phosphorothioate interference experiments have been used to probe
metal ion binding of ribozymes, on the basis of differing affinities of metal ions toward oxygen
and sulfur.20-22
According to the HSAB (Hard Soft Acid Base theory), a reduction in the cleavage
of a „soft‟ thio-substituted substrate (of the ribozyme) should be observed in the presence of a
„hard‟ Mg2+
cation (the thio effect) and restoration of normal cleavage rate of a sulfur-containing
substrate should occur in the presence of a thiophilic cation such as Cd2+
(the rescue effect). This
rescue in the activity of the ribozyme has been generally accepted to be evidence of functional
metal-binding to the site of substitution. The mechanistic aspect of ribozymes that include the
group I and II introns,23-25
the RNA subunit of RNase P,26, 27
and the hammerhead,28-36
have been

97
explained to a greater degree through the analysis of these types of interactions. Although
interpretation of these results has been complicated, and quite often debated,37-39
due to the
ability of divalent metal ions to cause structural and steric effects on the ribozyme; overall
comparisons of the crystal structure of ribozymes obtained correlate well with the metal-binding
sites observed using phosphorothioate modifications. Results obtained based on this technique
have also been complemented using additional probing techniques such as 31
P NMR, EPR and
ENDOR of phosphorothioate modified ribozymes to determine metal-binding sites.33, 34, 39-42
Mapping of the functional phosphate groups in the catalytic core of the 10-23 DNAzyme
has also been carried out recently.43
These experiments suggested the both oxygens at the P5
phosphate position and the proRP oxygen at the P9 position played a role in metal coordination.
In addition, the authors found that oxygen at the C6 position of G6 contributed to metal ion
binding and that interaction was essential for the catalytic activity of the10-23 DNAzyme.43
4.1.3 Research goals
Over the last decade, considerable attention has been directed towards both isolating as
well as understanding the mechanism of laboratory-evolved DNAzymes for various applications.
Researchers have also spent a lot of effort in elucidating the role of metal ions as well as
nucleotide bases in the activity and folding of the 8-17 DNAzyme.10-12, 44, 45
However, obtaining
a crystal structure for this DNAzyme (or a functional structure for any DNAzyme to date) has
thus far proved to be extremely challenging. Phosphorothioate modifications on the backbone of
ribozymes have proved to be very insightful in better elucidating their mechanism and
crystallographic data obtained for several ribozymes correlates well with the results obtained
through this kind of mapping for identification of functional metal ion binding sites. The use of

98
31P NMR by DeRose and co-workers,
29, 35 has been a valuable tool in directly viewing the
implications of metal ion addition on specific phosphates of the hammerhead ribozyme and
correlations with the binding of the metal ion. This chapter details our efforts in identifying
functional phosphates on the backbone of the 8-17 DNAzyme, in the presence of Pb2+
, through
single phosphorothioate modifications on previously identified conserved residues using both
kinetic assays and 31
P NMR. This is the first systematic study elucidating the role of the
phosphates as potential metal ion binding sites on the 8-17 DNAzyme and it is anticipated that
the results obtained during this study, in addition to the various biophysical and biochemical
characterizations will give us a better mechanistic understanding of this DNAzyme.
4.2 Materials and methods
4.2.1 Materials
All oligonucleotides were obtained standard desalted from Integrated DNA Technologies
Inc., Coralville, IA and purified by PAGE (Poly-Acrylamide Gel Electrophoresis) for kinetic
assays. The oligonucleotides containing the PS modifications were obtained as racemic mixtures,
containing both the RP and SP stereoisomers, and were used without any further separation.
Oligonucleotides for 31
P NMR were used as obtained. All metal salts used were obtained from
Alfa-Aesar and were of Puratronic® grade, 99.999 % pure. [γ-
32P]-deoxyadenosine triphosphate
(dATP) was obtained from Perkin Elmer and T4 kinase was obtained from Invitrogen.
4.2.2 Kinetic assays
The substrate oligonucleotide (17S) was 5'-radiolabeled with [γ-32
P]-dATP using T4
kinase from Invitrogen, purified on a C18 Sep-Pak® cartridge and stored at -20 °C. All kinetic

99
assays were carried out under single-turnover conditions with 5 µM enzyme and 0.5 -1.5 nM
substrate in a buffer containing 50 mM MES-HCl at pH 6.0, and 100 mM NaCl. The sample
solution containing the enzyme and substrate in buffer (Figure 4.3a) was denatured at 95°C in a
water bath and then annealed by gradual cooling to room temperature over ~ 45 minutes. Upon
annealing the solution, a 5 µL aliquot was added to 25 µL of a „stop solution‟ containing 8 M
urea, 1 × TBE and 0.5 % each bromophenol blue and xylene cyanol in a 96-well plate. This was
the „zero-point‟ control before the initiation of the reaction. Thereafter, appropriate volumes of
metal solutions were added to the sample to initiate the reaction and 5 µL aliquots were taken at
suitable time-intervals and added to the stop solution to quench the reaction on a 96-well plate.
The final concentrations of Pb2+
and Zn2+
used for the assays was 100 µM, while 10 mM Mg2+
,
Cd2+
, Mn2+
and 2.5 mM each of Cu2+
and Cu+ were used. The uncleaved substrate and the
cleaved product were separated on a denaturing 20 % polyacrylamide gel, imaged on a
Molecular Dynamics Storm 430 phosphorimager and quantified using Image-Quant software.
The % product at time „t‟ was calculated by taking the ratio of the 5'-cleaved product to the total
of the cleaved product and the uncleaved substrate, after subtraction from the background
radioactivity on the gel. Kinetic plots were created using Origin 8.0 software and fit to the
equation,
% Pt = % P0 + % P∞ (1-e-kt
)
wherein, % P0 is the initial amount of product (at time, t = 0), % P∞ is the amount of
product formed at the endpoint of the reaction ( at t = ∞ ), % Pt is the amount of product at time t
and k = kobs, the observed rate constant.

100
4.2.3 31P NMR studies
For the 31
P NMR studies the concentrations of the enzyme and substrate (Figure 4.3b)
were maintained at 669 µM each in 50 mM MES-Na at pH 6.0 in 10 % D2O. Samples were
prepared by first dialyzing equal amounts of the enzyme and substrate strands in buffer
containing 50 mM MES-Na at pH 6.0 against the same buffer, followed by lyophilization. The
samples were then re-dissolved in 10 % D2O, and then denatured at 55 °C in a water bath
followed by gradual cooling to room temperature to ensure complete hybridization. 31
P NMR
spectra were collected at 242.8 MHz on a Varian UNITY INOVA spectrometer with a 5 mm
AutoTuneX probe. Temperature was held constant at 25 °C and samples were referenced using
an internal coaxial tube containing either 5 % 5'-thymidine monophosphate (4.15 ppm) or
trimethyl phosphate (3.70 ppm) in D2O. Metal ion titrations were carried out at room temperature
by removing the sample from the NMR tube and adding either a 100 mM or 200 mM M2+
solution as appropriate, with constant magnetic stirring. Samples containing Pb2+
were chloride-
free to minimize precipitation. Samples were allowed to equilibrate for ten minutes before the
start of acquisition.

101
CATCTCTTCT2.1 A
16.1TGGTGAGT
5'
3'
GTAGAGAAGG1.1rAT
17.1ATCACTCA
C9
A6
G7 C8
G10
G11
C13
G14
A15
T12
A15.0
5'
3'
C3C
4G
5
Cleavage Sitea
17S
17E
CATCTCTTCT2.1 A
16.1TGGTGAGT
5'
3'
GTAGAGAAGG1.1
A T
17.1ATCACTCA
C9
A6
G7 C8
G10
G11
C13
G14
A15
T12
A15.0
5'
3'
C3C
4G
5
Non-cleavable substrate
17DS
b
17E
CATCTCTTCTCCGAGCCGGTCGAAATGGTGAGT
5'
3'
GTAGAGAAGAGGCTCGGCCAGCTTTATCACTCA
5'
3'
c
Duplex control with 17EG14PS
Figure 4.3 Predicted secondary structures of a) 17ES DNAzyme-substrate complex used for the
kinetic assays b) 17EDS DNAzyme-substrate complex containing a non-cleavable base at the
cleavage site, used for 31
P NMR studies. The position of the PS modifications used for both the
kinetic and NMR studies is indicated in Table 5.1. c) Duplex control used for 31
P NMR that
contains a completely complementary sequence to the 17EG14PS enzyme strand.

102
4.3 Results
4.3.1 Dependence of the 8-17 DNAzyme activity on position of PS modification
The roles of the phosphates on several bases of the 8-17 DNAzyme, implicated
previously in catalysis, were probed using phosphorothioate modifications. A single replacement
of the P-O bond by P-S was made on each of the individual bases implicated in catalysis, and the
rate of cleavage upon modification was systematically studied. The thio effect, which was
representative of the extent of perturbation upon a PS modification, was calculated in the
presence of Pb2+
and rates of the cleavage reaction were also calculated in the presence of a
range of soft metal ions that included Cd2+
, Mn2+
, Cu+, borderline soft namely Zn
2+, and also in
the presence of Mg2+
which is classified as a hard metal ion. Activity assays were also carried
out in the presence of Cu2+
as a control for the Cu+ studies.
4.3.1.1 Activity of modified DNAzymes with Pb2+
As shown in Figure 4.4a, the DNAzymes with PS modifications at positions C13 and G14
showed negligible activity and therefore, maximum perturbation with 100 µM Pb2+
. In
comparison, the PS modifications on positions A6 and A16.1 did not result in significant
perturbation and the kinetic rate constants obtained were almost the same as that obtained with
the unmodified 17E DNAzyme. Modifications at positions G7, T2.1 A15 and C8 did lead to some
perturbation and an overall loss in activity in comparison with the unmodified 17E DNAzyme by
approximately 1-2 orders of magnitude respectively. However, the overall activity was still
quantifiable in comparison with enzymes containing modifications at positions C13 and G14. In
fact, a ratio of the rate constants of the unmodified 17E DNAzyme to PS-modified enzyme,

103
known as the thio effect (shown in Table 4.1), range from ~1.0 in the case of A6 and A16.1
positions (highlighted in a red box in Table 4.1), to intermediate values between 20 and 60 in the
case of A15,T2.1 and G7( highlighted in blue) ; to extremely perturbed with thio effects of ~265
and unquantifiable in the case of C8, C13 and G14 ( highlighted in green) respectively. Any PS
modification at the cleavage site, namely the RNA base on the substrate strand, resulted in a
complete loss of activity. Further, any substitution on G1.1 as well as T17.0, both neighboring the
RNA cleavage site on the substrate, also resulted in a complete loss of activity with Pb2+
, Cd2+
and Mg2+
(Table 4.1) .
4.3.1.2 Activity of modified enzymes with other metal ions
Interestingly, when the kinetic assays of the DNAzyme with any PS modification on the
enzyme strand were performed with 10 mM Mg2+
and 10 mM Cd2+
, which are at the two
extremes of the hard-soft metal ions scale, no significant difference in kobs values were observed
in comparison with the unmodified 17E DNAzyme as shown in figure 4.4b and 4.4c. (Initial
experiments with 1 mM Mg2+
and 1 mM Cd2+
after ~24 hours did not yield any activity even
with the unmodified 17ES DNAzyme and hence a concentration of 10 mM was chosen). The
thio effects (shown in Table 4.1) ranged from about ~ 1-2.5 with 10 mM Cd2+
and from ~ 1-3.5
with 10 mM Mg2+
, reflecting that many of the single PS substitutions on the 17E DNAzyme
perturbed the activity with 100 µM Pb2+
to a much greater extent, than with the other two metal
ions. Results obtained with 10 mM Mn2+
also reflected the same trend when the activity of the
most perturbed PS-modified bases, C13 (kobs = 0.093 min-1
) and G14 ( kobs = 0.072 min-1
) as well
as the 17E ( kobs = 0.086 min-1
) were carried out (Figure 4.5a). Kinetic assays with another
„soft‟ metal ion namely 2.5 mM Cu+ did not yield any activity with 17E, 17EC13PS as well as

104
17EG14PS. Since Cu+ may be gradually oxidized in water or buffer to Cu
2+ over the period of the
activity assay; as a control, the assays were also carried out with 2.5 mM Cu2+
(Figure 4.5b).
However, no activity was seen with both forms of copper. Higher concentrations of Cu+ in a 10
% acetonitrile and buffer mixture led to significant precipitation issues and therefore could not
be used. Since Zn2+
is also known as a „borderline‟ metal ion with regard to the HSAB theory,
the kinetic assays of the most perturbed bases namely 17EC8PS, 17EC13PS and 17EG14PS were
also carried out with 100 µM Zn2+
, (concentration at which 17E also shows activity with Zn2+
with kobs = 0.073 min-1
) and the resulting trends observed were surprisingly similar to that seen
with Pb2+
. The PS-modification at these positions caused significant perturbations and no
detectable activity was measured after 2 hours. To ensure the similarity in the trend observed, the
activity assays were also carried out with A16.1PS and A6PS with 100 µM Zn2+
, (which were non-
perturbed with 100 µM Pb2+
) and the rate constants measured were 0.064 min-1
and 0.030 min-1
with a thio effect of 1.1 and 2.4 respectively (Figure 4.5c).

105
Figure 4.4 Kinetic traces of the 17E variants containing single PS modifications on the enzyme
strand with a) 100 µM Pb2+
, b) 10 mM Cd2+
and c) 10 mM Mg2+
under single turn-over
conditions in buffer containing 50 mM MES , 100 mM NaCl at pH 6.0.

106
Table 4.1 List of observed rate constants (kobs) for the phosphorothioate modified 17E variants
under single-turnover conditions.
Name of the 17E
Variant
With 100 µM Pb2+
With 10 mM Cd2+
With 10 mM Mg2+
kobs
(min-1
) a
Thio
effect d
kobs
(min-1
) b
Thio
effect
kobs
(min-1
) c
Thio
effect
Unmodified 17E 5.3 2.6 x 10-2
1.2 x 10-2
17EA6PS 3.9 1.4 1.2 x 10-2
2.2 4.7 x 10-3
2.6
17EA16.1PS 3.6 1.5 2.5 x 10-2
1.0 1.2 x10-2
1.0
17EG7PS 0.25 21 2.0 x 10-2
1.3 4.7 x 10-3
2.6
17ET2.1PS 0.15 35 1.3 x 10-2
2.0 3.7 x 10-3
3.2
17EA15PS 0.08 66 1.1 x 10-2
2.4 3.8 x 10-3
3.2
17EC8PS ~ 0.02 ~265 2.0 x 10-2
1.3 6.0 x 10-3
2.0
17EC13PS N.D - 2.1 x 10-2
1.2 9.4 x 10-3
1.3
17EG14PS N.D - 2.1 x 10-2
1.2 3.9 x 10-3
3.1
17SrAPS N.D - N.D - N.D -
17SrG1.1PS N.D - N.D - N.D -
17ST17.1PS N.D - N.D - N.D -
a Data shown is the mean value obtained from three trials.
b, c Data shown is the average
value obtained after two trials. All the assays were carried out in 50 mM MES containing 100
mM NaCl at pH 6.0. d The thio effect is the ratio of the kobs of the unmodified 17E to the kobs
value of the PS-modified variant. “N.D” denotes “not detectable” (at time-intervals similar to
activity of the unmodified 17E) and “–”denotes “not calculated.”

107
Figure 4.5 Kinetic traces of some of the 17E variants containing single PS modifications on the
enzyme strand with a) 2.5 mM Cu+ in 10 % acetonitrile and 2.5 mM Cu
2+ as a control in buffer,
b) 10 mM Mn2+
and c) 100 µM Zn2+
under single turn-over conditions in buffer containing 50
mM MES , 100 mM NaCl at pH 6.0.
4.3.2 Probing metal binding based on 31
P NMR
Enzymes 17EC13PS and 17EG14PS containing individual PS modifications were chosen
for 31
P NMR studies based on the afore-mentioned kinetic gel-based activity assays, which
indicated significant perturbation on the overall activity of these modified enzymes upon Pb2+
addition. On the other hand the 31
P NMR characterization of the enzyme non-cleavable substrate

108
complex containing a phosphorothioate modification at position A6 was also performed, since
the resulting modified enzyme showed activity that was comparable to the unmodified 17E with
100 µM Pb2+
. The 31
P NMR characterization of these modified enzymes was also carried out
with Cd2+
, which did not show any significant change in its overall kinetic activity upon PS
modification, as reflected in the thio effect values (Table 4.1).
Figure 4.6a shows the 31
P NMR spectra of the three PS-modified 17E enzymes with
17DS (non-cleavable substrate) in the absence of any metal ion. The two, well-resolved peaks of
the 17A6PS correspond to the two phosphorothioate diastereomers, RP and SP which are in slow
exchange. The 31
P NMR of 17EC13PS does indicate the presence of the two isomers; however,
the peaks observed are much broader and indicate a relatively faster conformational change
between the two isomers. In the case of the 17EG14PS only one peak is observed indicating that
the two isomers are undergoing rapid exchange to coalesce to the single feature observed. Upon
reducing the temperature from 25 °C to 4 °C, (Figure 4.6b) the single peak of the 17EG14PS at
55 ppm, starts resolving into two phosphorothioate features, corresponding to the RP and SP
isomers.

109
Figure 4.6 a) 31
P NMR of the three individual 17E enzymes containing single PS modifications
+ 17DS (non-cleavable substrate), in the absence of any metal ion in 50 mM MES-Na, at pH 6.0
at 25 °C. b) Effect of temperature on the 31
P NMR of 17EG14PS + 17DS, in the absence of any
metal ion. All measurements were carried out at pH 6.0 in 50 mM MES-Na and 10 % D2O.
4.3.2.1 Effect of Pb2+
addition on the 31
P NMR spectra
Addition of increasing (0-5) equivalents of Pb2+
to the 17EG14PS + 17DS complex,
results in an upfield shift of the phosphorothioate peak (Figure 4.7a). Previous studies with
ribozymes have indicated that metal ion addition, specifically Cd2+
addition to a
phosphorothioate-modified hammerhead ribozyme results in a significant change in the chemical
shift, moving upfield due to the covalency of the Cd2+
-sulfur bond. Similar experiments
performed with 17EC13PS + 17DS, results in upfield shifts of both phosphorothioate features
upon addition of Pb2+
, although the change in the chemical shift change is smaller in comparison
with that seen at the 17EG14PS position (Figure 4.7b). Interestingly, upon addition of Pb2+
to
17EA6PS + 17DS, the two phosphorothioate peaks are shifted to a small extent, in opposite

110
directions, the upfield peak shifts slightly upfield while the downfield peak shifts slightly
downfield. (Figure 4.7c) Such an observation was also made upon titration of Cd2+
to the PS-
modified cleavage site of a minimal hammerhead ribozyme wherein the Rp diastereomer shifted
upfield, indicating direct Cd2+
-sulfur binding, whereas the Sp isomer shifted downfield, thereby
indicating a conformational change either in addition to metal-binding or in place of the same.
Moreover, the shifts observed with A6PS are much smaller in comparison to the shifts observed
with 17EC13PS and 17EG14PS, indicative of minimal metal-interactions (Table 4.2).
An important control experiment was also performed wherein the 31
P NMR spectrum of
a duplex DNA was collected, with the enzyme strand identical to the 17EG14PS and the
“substrate strand” which was fully complementary to the same (as opposed to the partially
complementary 17DS), was titrated with increasing equivalents of Pb2+
. The peak shift in this
case, showed minimal changes even with 5 equivalents of Pb2+
and was < 0.4 ppm, thereby
indicating that the phosphorothioate modification on this duplex DNA was not a site of Pb2+
-
binding (Figure 4.7d). This also implied that simply having a phosphorothioate modification on a
duplex DNA did not lead to significant changes in the chemical shift upon Pb2+
-addition, rather,
significant changes in the chemical shifts were observed only when the metal ion bound to
specific modified bases, important for its activity or caused an overall conformational change of
the complex in addition to binding.
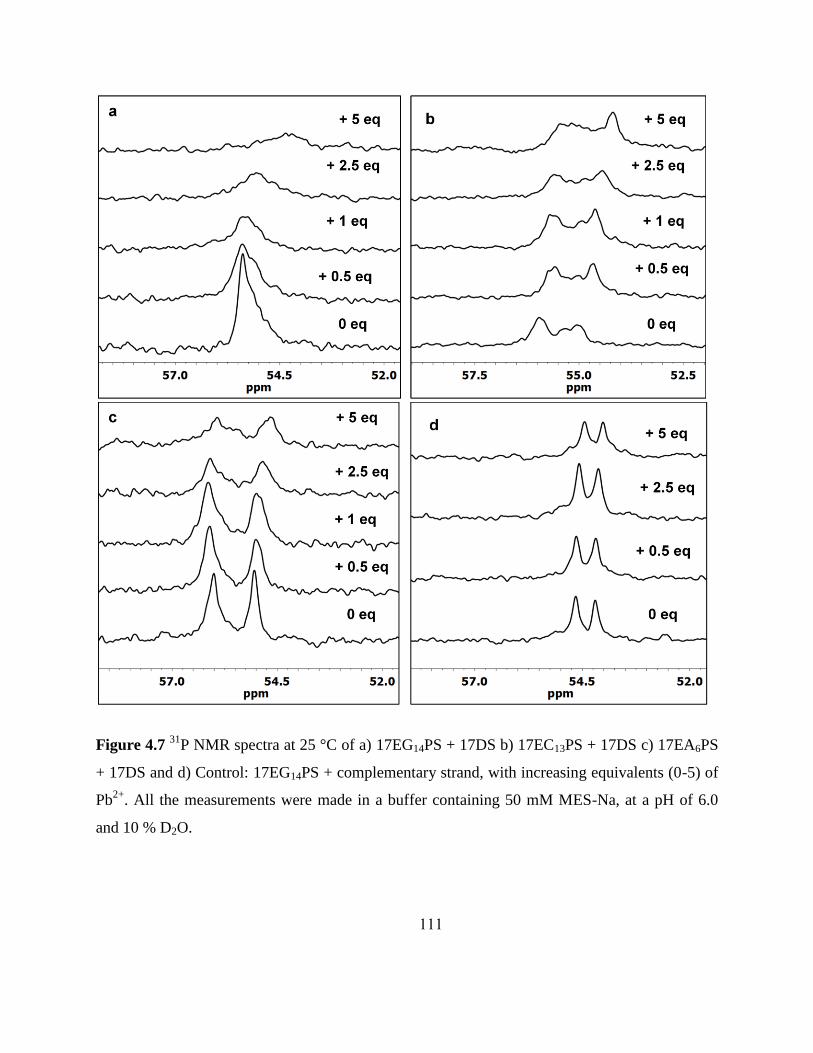
111
Figure 4.7 31
P NMR spectra at 25 °C of a) 17EG14PS + 17DS b) 17EC13PS + 17DS c) 17EA6PS
+ 17DS and d) Control: 17EG14PS + complementary strand, with increasing equivalents (0-5) of
Pb2+
. All the measurements were made in a buffer containing 50 mM MES-Na, at a pH of 6.0
and 10 % D2O.

112
4.3.2.2 Effect of Cd2+
addition on the 31
P NMR
The addition of Cd2+
to the 17EG14PS + 17DS complex results in an upfield shift of the
phosphorothioate peak while with 17EC13PS + 17DS upfield shifts of both phosphorothioate
features are again observed similar to that seen with Pb2+
addition. Addition of Cd2+
to 17EA6PS
+ 17DS also results in movement of both peaks in opposite directions as observed with Pb2+
(Figure 4.8a-c). However, there is a clear difference in terms of the extent of movement of the
chemical shift of the upfield peak between Pb2+
and Cd2+
. All the three PS-modified enzymes
show almost identical shifts upon addition of 10 equivalents of Cd2+
, with the exception of the
downfield peak of 17EA6PS, which can be attributed to either a conformational change
accompanying metal-binding as mentioned previously or due to the binding of the metal ion to
the oxygen of a phosphorothioate. On the other hand, the change in chemical shifts upon Pb2+
addition is very close with 17EC13PS and 17EG14PS, whereas with the 17EA6PS, the shift of the
upfield peak is almost one-third smaller in comparison (Table 4.2 and 4.3). A significant
broadening of the peaks is also observed. The peak shift with the control duplex DNA is also
minimal upon addition of Cd2+
, again confirming that the Cd2+
also specifically binds functional
phosphorothioates on the DNA backbone rather than binding any modified phosphorothioate.

113
Figure 4.8 31
P NMR spectra at 25 °C of a) 17EG14PS + 17DS b) 17EC13PS + 17DS and c)
17EA6PS + 17DS with increasing equivalents (0-10) of Cd2+
. All measurements were made in a
buffer containing 50 mM MES-Na, at a pH of 6.0 and 10 % D2O.

114
4.3.2.3 31P NMR of PS modifications on the substrate strand
The site of cleavage on the substrate strand (17S) was replaced by a non-cleavable DNA
base (17DS) for all the NMR experiments as shown in Figure 4.3b. A PS modification was also
made at the cleavage site of this 17DS, referred to as 17DSAPS. Any modification at this
cleavage site leads to a complete loss of catalytic activity with both Pb2+
and Cd2+
. The 31
P NMR
of this modified enzyme-substrate complex showed two extremely well-resolved peaks in the
absence of any metal ion, indicating that the two diastereomers were undergoing a slow
exchange at 25 °C and were conformationally locked in comparison to 17EG14PS + 17DS. Upon
addition of Pb2+
, both peaks shifted very minimally upfield, while upon addition of up to 5
equivalents of Cd2+
, there was again minimal movement of the peaks. However, upon addition of
10 equivalents of Cd2+
, a significant broadening of the peaks was observed accompanied by a
shift of ~0.4 and ~0.5 ppm for the downfield and the upfield peak respectively. Data with Pb2+
was not collected at 10 equivalents due to precipitation issues (Figure 4.9 and Tables 4.2, 4.3).
Figure 4.9 31
P NMR spectra of a) 17E + 17DASPS with increasing equivalents of a) Pb2+
and b)
Cd2+
. All the samples were in 50 mM MES-Na, at a pH of 6.0 and 10 % D2O.
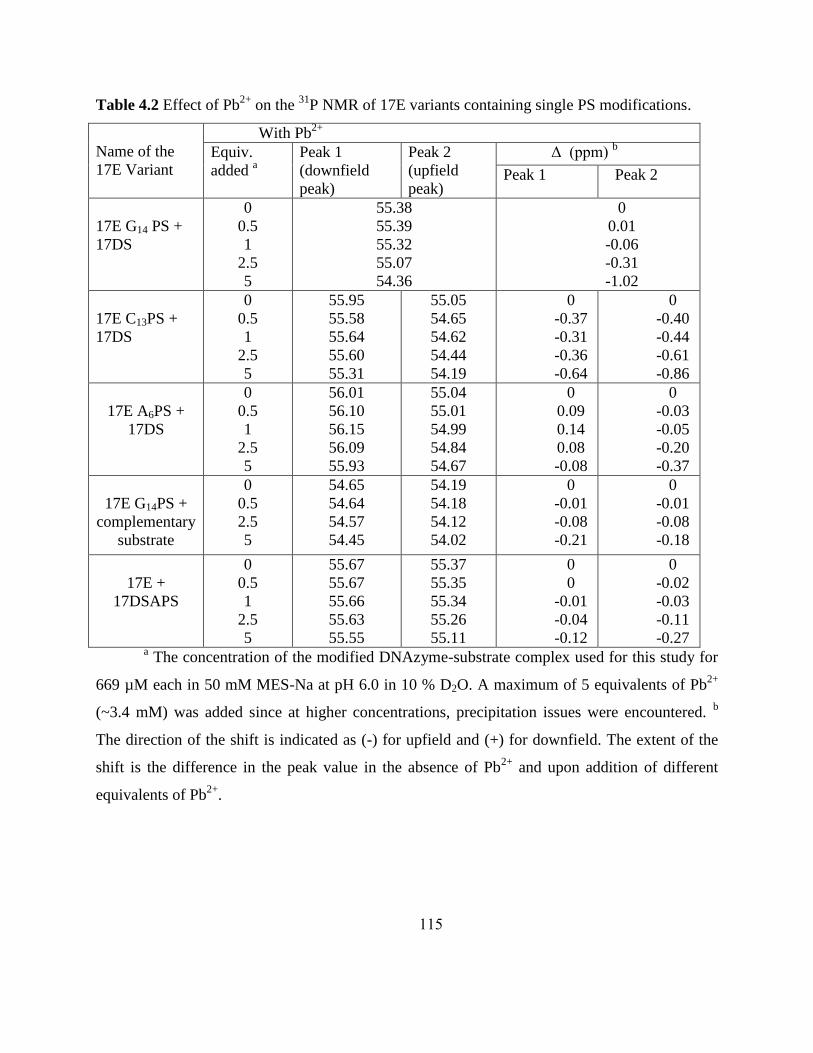
115
Table 4.2 Effect of Pb2+
on the 31
P NMR of 17E variants containing single PS modifications.
Name of the
17E Variant
With Pb2+
Equiv.
added a
Peak 1
(downfield
peak)
Peak 2
(upfield
peak)
Δ (ppm) b
Peak 1 Peak 2
17E G14 PS +
17DS
0
0.5
1
2.5
5
55.38
55.39
55.32
55.07
54.36
0
0.01
-0.06
-0.31
-1.02
17E C13PS +
17DS
0
0.5
1
2.5
5
55.95
55.58
55.64
55.60
55.31
55.05
54.65
54.62
54.44
54.19
0
-0.37
-0.31
-0.36
-0.64
0
-0.40
-0.44
-0.61
-0.86
17E A6PS +
17DS
0
0.5
1
2.5
5
56.01
56.10
56.15
56.09
55.93
55.04
55.01
54.99
54.84
54.67
0
0.09
0.14
0.08
-0.08
0
-0.03
-0.05
-0.20
-0.37
17E G14PS +
complementary
substrate
0
0.5
2.5
5
54.65
54.64
54.57
54.45
54.19
54.18
54.12
54.02
0
-0.01
-0.08
-0.21
0
-0.01
-0.08
-0.18
17E +
17DSAPS
0
0.5
1
2.5
5
55.67
55.67
55.66
55.63
55.55
55.37
55.35
55.34
55.26
55.11
0
0
-0.01
-0.04
-0.12
0
-0.02
-0.03
-0.11
-0.27 a The concentration of the modified DNAzyme-substrate complex used for this study for
669 µM each in 50 mM MES-Na at pH 6.0 in 10 % D2O. A maximum of 5 equivalents of Pb2+
(~3.4 mM) was added since at higher concentrations, precipitation issues were encountered. b
The direction of the shift is indicated as (-) for upfield and (+) for downfield. The extent of the
shift is the difference in the peak value in the absence of Pb2+
and upon addition of different
equivalents of Pb2+
.

116
Table 4.3 Effect of Cd2+
on the 31
P NMR of 17E variants containing single PS modifications.
Name of the
17E Variant
With Cd2+
Equiv.
added a
Peak 1
(downfield
peak)
Peak 2
(upfield
peak)
Δ (ppm) b
Peak 1 Peak 2
17E G14 PS +
17DS
0
0.5
2.5
5
10
55.4
55.12
54.97
54.40
N.D
0
-0.28
-0.43
-0.99
N.D
17E C13PS +
17DS
0
0.5
2.5
5
10
55.95
55.62
55.63
55.54
55.20
55.05
54.38
54.41
54.37
54.14
0
-0.33
-0.32
-0.42
-0.75
0
-0.68
-0.64
-0.67
-0.92
17E A6PS +
17DS
0
0.5
2.5
5
10
56.00
56.01
56.14
56.17
56.20
55.03
54.85
54.80
54.34
54.20
0
0.01
0.14
0.17
0.20
0
-0.18
-0.23
-0.69
-0.83
17E G14PS +
complementary
substrate
0
0.5
2.5
5
54.66
54.65
54.63
54.58
54.20
54.20
54.19
54.14
0
-0.01
-0.03
-0.08
0
0.00
-0.02
-0.06
17E +
17DSAPS
0
1
2.5
5
10
55.67
55.66
55.64
55.58
55.29
55.36
55.35
55.29
55.18
54.86
0
-0.01
-0.03
-0.09
-0.37
0
-0.01
-0.06
-0.02
-0.49 a The concentration of the modified DNAzyme-substrate complex used for this study was
669 µM each in 50 mM MES-Na at pH 6.0 in 10 % D2O. A maximum of 10 equivalents of Cd2+
(~6.7 mM) was used as at this concentration significant broadening issues were encountered. b
The direction of the shift is indicated as (-) for upfield and (+) for downfield. The extent of the
shift is the difference in the peak value in the absence of Cd2+
and upon addition of different
equivalents of Cd2+
.

117
4.4 Discussion
While significant efforts have been made in postulating the role of the metal ions
responsible for activity, identifying binding sites on the actual enzyme-substrate complex is still
a challenge, particularly, in the absence of either an X-ray crystal structure or an NMR structure
for the 8-17 DNAzyme. Even more challenging is the fact that the most effective co-factor of
this DNAzyme is Pb2+
, which is a spectroscopically silent metal ion. Hence, characterization
techniques such as UV-Vis and EPR, which are routinely used to identify metal ion binding sites
of metalloproteins, are, in the case of this nucleic acid enzyme, very challenging to use without
prior modifications. The use of phosphorothioate modifications on the phosphate backbone of
nucleic acid enzymes, therefore, is a simple but effective technique that has been extensively
utilized to identify and confirm potential metal ion binding sites of various ribozymes as
discussed previously.
The results obtained in the current study signify the roles that phosphates play in metal
ion binding, particularly with Pb2+
, thereby, influencing the overall catalytic activity of the 8-17
DNAzyme and providing a snapshot of a potential „active metal ion binding site‟.
4.4.1 Effect of PS modifications on AGC terminal loop based on kinetic assays
Table 4.1 shows that under single-turnover conditions, the effect of PS modifications on
the overall kobs values with A6, G7 and C8 with 100 µM Pb2+
varies with the position of the actual
modification. Interestingly, this thio effect with Pb2+
follows the order: A6 < G7 < C8, with kobs
for A6 being virtually unchanged in comparison with the unmodified enzyme, while kobs for G7
lowered by an order of magnitude and that for C8 lowered by approximately two-fold. The
original in vitro selection study on the 8-17 DNAzyme has indicated that bases A6, G7 and C8 on

118
the terminal loop are strictly conserved and are critical for the function of the DNAzyme.46
Mutagenesis studies have also indicated that any mutations on this loop results in a > 50-fold
loss in the catalytic activity.1, 2, 4, 5
Residues A6 and G7 are thought to be involved in close interaction with other portions of
the enzyme-substrate complex through hydrogen-bond interactions and any replacement by base
analogues leads to a significant decrease in catalytic activity. Mutations on residue C8 on the
other hand lead to a five- to ten-fold decrease in the overall catalytic activity. Cross-linking
studies have indicated that the contact point of the AGC loop is the actual cleavage site of the
substrate rather than any other portion of the enzyme-substrate complex including the bulge loop
or the helical stem.7 Folding studies have also indicated that bases A6 and G7 are important
elements for the overall folding of the 8-17 DNAzyme.45
This reversal in the trend of activity of
the A6 residue with Pb2+
upon PS modification, in comparison with the mutational studies with
base analogues, suggests that the phosphate backbone of this base is not involved in Pb2+
-
binding. The reduction of the kobs value by an order of magnitude upon PS modification at G7,
suggests that the phosphate backbone of this particular base maybe involved in weak binding
towards Pb2+
. However, the thio effect of ~265, in the case of PS-modified C8 suggests that the
phosphate backbone of this particular residue is more likely to be involved in Pb2+
-binding.
Our current studies lead us to believe that bases A6 and G7 do indeed perform an
important role in the overall structure adopted by the 8-17 DNAzyme for its catalytic activity
rather than being involved in metal-binding. The phosphate backbone of C8 on the other hand
seems to be important for Pb2+
binding and is functional for overall activity of the DNAzyme.
On the other hand the thio effect values obtained with both 10 mM Cd2+
and 10 mM Mg2+

119
suggest that the binding of these metal ions at any of these three phosphates is not functionally
important. Instead, it could be either postulated that the binding of these metal ions to the PS-
modified positions is not responsible for activity, or that there is no specific binding to any
modified base at all. Rather, the presence of either Mg2+
or Cd2+
only facilitate the formation of
an active structure before catalysis and helps in stabilizing the overall electrostatic interactions.
4.4.2 Effect of PS modifications at bases on the TCGAA loop based on kinetic assays
Single PS modifications at positions C13 and G14 (Table 4.1) resulted in a significant loss
in the catalytic activity of the 8-17 DNAzyme with 100 µM Pb2+
. In fact, the kobs values of the
PS-modified enzymes with 100 µM Pb2+
could not be estimated during the course of one hour
while the kobs for the unmodified 17E enzyme is calculated to be ~ 5.3 min-1
under identical
conditions. This significant difference in activity suggests that the phosphate groups of these
bases play an important role in the catalytic activity of the 8-17 DNAzyme with implications for
metal ion binding, specifically with either Pb2+
or Zn2+
( since Zn2+
also exhibits similar behavior
relative to the unmodified enzyme).
Previous mutagenesis studies have indicated that replacement of the C13 base by 5-bromo
cytosine residue resulted in only a four-fold drop in activity as compared to a forty-fold drop in
activity as expected for a cytosine group that performed a general acid-base catalyst role at
neutral pH.5 This excluded the role of this base as a general acid-base catalyst. Replacement of
the G14 base by other base analogues suggested the importance of the 2-amino group for activity.
However, when these mutant G14 enzymes were tested with various metal ions, there was no
preference for any metal ion seen suggesting that G14 did not bind to any crucial metal ion. Also,
while the functional importance of the 2-amino group of G14 was observed, there was no clear

120
evidence for a direct involvement of the base in catalysis.5 Our results, however, suggest that the
phosphate group of both C13 and G14 bases do indeed show a significant preference in terms of
activity with different metal ions, thereby indicating that the nucleotide itself is not involved in
metal ion binding, rather the phosphate group of the base is involved in metal ion binding,
specifically binding with Pb2+
and Zn2+
.
4.4.3 Effect of PS modifications on the substrate strand based on kinetic activity assays
Any PS modification on the substrate strand lead to a complete loss in activity of the
enzyme with either Pb2+
, Cd2+
or Mg2+
.This could be interpreted to mean either that phosphates
at positions G1.1, T17.1 or at the cleavage site, rAPS, are functional for activity or that there is a
disruption of the active conformation of the enzyme-substrate complex responsible for catalytic
activity, due to the PS replacement.
4.4.4 31P NMR studies of the 8-17 DNAzyme
31P NMR investigations of the modified 8-17 DNAzymes, 17EA6PS, 17EC13PS and
17EG14PS have been carried out with both Pb2+
as well as Cd2+
, which have exhibited different
activities with these enzymes based on the kinetic activity assay studies. Most of the modified
enzyme-substrate complexes studied have shown the presence of the two diastereomers formed
upon a single PS modification on the phosphate, namely RP and SP, shown by the presence of
two peaks in the NMR spectra. However, in this study, the two isomers have not been resolved
since the aim of this study was to primarily identify functional phosphates involved metal ion
binding sites, rather than the specific spatial arrangement of the ligands bound to the metal ion.

121
This will be, however, attempted in the future when additional information is available either
through X-ray crystallography or structural NMR studies.
The 31
P NMR spectrum of 17EG14PS shows only a single peak in the absence of any
metal ion, indicating the conformational flexibility of this site and the fast exchange of the two
diastereomers RP and SP. The addition of either Pb2+
or Cd2+
causes a significant shift in the
chemical shift and also broadens the peak indicating that there indeed is metal binding that takes
place to the sulfur at this particular position (Figures 4.7a and 4.8a). However, the binding in the
case of Pb2+
is functional for activity, while the binding in the case of Cd2+
is not functional as
shown from the kinetic activity assays. This is confirmed by the fact that the 31
P NMR spectrum
of 17EG14PS with a complementary DNA strand (Figure 4.3c) in place of the substrate strand
(17DS) with Pb2+
shows 2 peaks, both of which are shifted by ~0.2 ppm with Pb2+
in comparison
with the ~1.00 ppm shift seen with 17EG14PS with 17DS. The binding of Cd2+
to the 17EG14PS
with complementary substrate also results in a change in the chemical shift value of both the
peaks; however, the extent of the shift is ~ half that of what is seen with the 17EG14PS
with17DS. This indicates that Pb2+
binding to the 17EG14PS site is present and is functional for
activity while binding for Cd2+
is present, while not important or essential for function.
In the absence of any metal ion the 31
P NMR spectra of both 17EC13PS and 17EA6PS,
with 17DS, appear to be conformationally restrained and resolved clearly into the two
diastereomers, RP and SP. Upon addition of either Pb2+
or Cd2+
ions, there is a significant shift in
the position of both the peaks with the PS modified C13 enzyme (Table 4.2 and 4.3). However, in
comparison with the highly perturbed G14PS and C13PS sites, the shift with the A6PS modified
enzyme is much lower with Pb2+
, with the downfield peak barely shifting with increasing Pb2+

122
equivalents while the upfield peak shifting about ~0.37 ppm. However, the shift of the A6PS
modified enzyme with Cd2+
is significant in the case of the upfield peak, similar to that seen in
the C13PS modified enzyme indicating the lack of preference of Cd2+
towards the position of the
phosphate in comparison with Pb2+
which exhibits more specific binding to particular
phosphates.
The relatively small change (~ 0.2-0.3 ppm) in the chemical shift of the PS-modified
substrate strand, 17DSAPS with unmodified 17E, in the presence of both Pb2+
and Cd2+
suggests
that the phosphates at this position are less likely to be involved in direct metal- ion binding. It is
more likely that the replacement of the PS in place of the PO bond at this position results in the
disruption of the active site conformation, responsible for catalytic activity Therefore, these
observations suggest a role for the functional groups of the nucleobases on the substrate arm
being directly involved in metal ion binding. Insights obtained from photo cross-linking studies
also indicate that there are three major contacts seen between the two nucleotides flanking the
scissile phosphodiester site of the substrate and the DNAzyme strand: firstly, with the
DNAzyme‟s T2.1 residue, which is a part of the functionally important G1.1T2.1 wobble pair,
secondly with C13, G14 and A15 of the WCGAA bulge loop and finally weak contacts with the A6
residue of the AGC terminal loop.7 The role of the T17.1 has not been implicated as being
important for function several mutational studies previously carried out. Hence, these combined
results suggest an extremely important role for the bases at the site of the cleavage as well as the
phosphates of the bases as being responsible for maintaining an active-site conformation of the
enzyme-substrate complex rather than being directly involved in metal ion binding.

123
4.4.5 Implications for metal ion binding and activity
The insights obtained from this study implicate some important phosphates in the
catalytic core forming a specific metal-binding pocket for Pb2+
thereby facilitating activity.
Based on the previously discussed results, these bases are the C8, C13 and G14 on the enzyme
strand. Importantly, our current kinetic studies have been carried out with racemic mixtures of
the PS-modified oligonucleotides, and hence, we cannot distinguish binding between the two
stereoisomers namely the Pro-RP and the Pro-SP of the phosphate as is shown in Figure 4.10.
Hence, Pb2+
binding to either of the two stereoisomeric oxygens might be envisioned. This
active-site binding pocket may be responsible for the efficient catalysis of the 8-17 DNAzyme
observed in the presence of Pb2+
. It is interesting to note that Zn2+
also perturbs the C8, C13, G14
phosphate modifications to the same extent as does Pb2+
implying that these phosphates may also
play an important role, and facilitate function in the presence of Zn2+
as well. However, recent
unpublished results (personal communication, courtesy Eric Null) based on 1H NMR studies
indicate that peaks at certain positions shift similarly for Pb2+
and Zn2+
while some are different.
It is likely that Zn2+
is causing mostly rearrangement and does show not direct binding to some
imino protons of the nucleobases, while Pb2+
doesn‟t need the rearrangement and hence causes
minimal shifts. Hence, the 8-17 DNAzyme exhibits a lowered activity for Zn2+
in comparison
with Pb2+
; however it is still higher than all the other divalent metal-ion cofactors, indicating that
the proposed binding site pocket might play a role in facilitating function with Zn2+
, albeit lower
than Pb2+
. Previous studies have shown that sequence-variations of the 8-17 DNAzyme seem to
affect the activities of different metal ions that include Ca2+
, Mg2+
, Pb2+
and Zn2+
in a similar

124
manner, suggesting that the specificity of the sequence is independent of the metal ions used in
the reaction.5
On the other hand, it has been proposed that there are multiple reaction pathways of the
8-17 DNAzyme depending on the nature of metal ion used.10-12
Since the reaction pathway of the
8-17 DNAzyme does not undergo folding with Pb2+
before cleavage activity, it is thought that
the DNAzyme maybe prearranged to accept Pb2+
and may have a ligand binding pocket, like that
seen in the case of the GlmS or the group I ribozyme.12, 21, 47
Moreover, since this behavior of
Pb2+
is in contrast with that observed with Zn2+
and Mg2+
it is also thought that Pb2+
could
occupy a binding site different from these metal ions, or bind to a similar site with a different
mode so that no significant global conformational change occurs. Our current kinetic assays
coupled with the 31
P NMR data with Pb2+
, suggest that the original phosphates at the PS-
modified positions, are potentially a part of an active-site binding pocket that is evolved through
the directed evolution process to accept and bind Pb2+
specifically. Since activity is significantly
affected upon PS modification on the substrate arm, whereas no binding is seen based on 31
P
NMR, the functional groups of the bases are thought to be directly involved in for metal ion
binding (Figure 4.10). Our kinetic results suggest that binding with Zn2+
also may take place at
the same active-site, but as indicated by the FRET studies the mechanism maybe different, and
may involve the folding-step (for easy accessibility to the active-site or to cross a potential
energy barrier) prior to the active-site binding step, thereby, leading to the lowered activity of the
8-17 DNAzyme complex for Zn2+
in comparison with Pb2+
. Also, a two-step mechanism for the
8-17 DNAzyme has also been demonstrated with Pb2+
(whereas only a single step with Zn2+
and
Mg2+
) and it has been proposed that to complete this two-step mechanism, a specific, three-

125
dimensional structure that forms a Pb2+
-binding site may be required,2 further adding value to
our current results.
The NMR studies also indicate that binding to the PS-modified bases does occur in the
presence of Cd2+
. However, this occurs at all PS-modified positions studied indicating the lack of
specificity for a particular site with Cd2+
. The kinetic studies further suggest that the PS
modifications at different positions on the DNAzyme catalytic core do not affect the overall
activity of the 8-17DNAzyme in the presence of either Mg2+
or Cd2+
, implying that these metal
ions either form weak contacts with the phosphates at these positions, but metal ion binding at
these positions may not be crucial for activity of the enzyme. Or that there could be just non-
specific binding that occurs at these phosphate positions, which is more structural in nature than
functional. Structural CD studies on the 8-17 DNAzyme have also indicated that there is a
transition observed from the right-handed B helical form to the left-handed Z helical form in the
presence of various monovalent metal ions as well as Zn2+
and Mg2+
, but no such transition is
observed with Pb2+
,12
potentially indicating the easy access that Pb2+
has for the active-site in
comparison with Zn2+
.

126
G1.1
rA
Pb2+
X
O
C13O
O
PO
O G14
O
O
O
P O
O
O
OOC8
O
PO
O
O
Pro-RP or
Pro-SP ?
Pro-RP or
Pro-SP ?
Pro-SP or
Pro-RP ?
X = H2O?
Figure 4.10 Proposed binding sites for Pb2+
on the 8-17 DNAzyme based on phosphorothioate
modifications and 31
P NMR. The bases shown in red indicate the scissile cleavage site and the
conserved „G‟ next to the cleavage site on the substrate strand, while the bases shown in green
refer to the most perturbed bases on the enzyme strand respectively. Based on our current kinetic
studies with racemic mixtures of PS-modified oligonucleotides, it is envisioned that Pb2+
might
bind either of the two stereo-isomeric oxygens of the phosphates, namely either pro-RP or pro-SP.
4.5 Conclusions
In the current study, the role of phosphates in catalysis carried out by the 8-17 DNAzyme
has been investigated through the systematic replacement of single P-O bonds on the phosphate
backbone by P-S modifications on conserved bases shown previously to be important based on
mutational and cross-linking studies. We have also used 31
P NMR to observe the dependence of

127
metal-binding toward phosphorothioate modifications. Bases that were previously identified as
being important sites on the catalytic core of the 17E DNAzyme were chosen for replacement
with individual P-S bonds in place of P-O bonds on the phosphate backbone, and the resulting
enzymes were tested with different metal ions that included the most effective co-factors of the
DNAzyme, namely Pb2+
, the borderline Zn2+
ion as well as soft metal ions such as Cd2+
, Mn2+
and Cu+ as well as the hard Mg
2+ ion. Cu
2+ was used a control for the Cu
+ experiments. In kinetic
assays, some of the PS-modified enzymes studied were perturbed significantly over the
unmodified enzyme in the presence of 100 µM Pb2+
and Zn2+
, thereby, suggesting the
importance of the phosphates at these positions for metal-binding. Whereas, no significant
differences in terms of activity of the PS-modified 17E DNAzyme over the unmodified enzyme
have been observed with other metal ions including Cd2+
, Mg2+
, Mn2+
, Cu2+
and Cu+ suggesting
no specific preference for different phosphates of the enzyme. Any PS modification on the
substrate strand, including the single RNA base at the cleavage site, as well nucleotides flanking
either side of it, namely G1.1 belonging to the G·T wobble pair and T17.1, resulted in a total loss of
activity with Pb2+
, Cd2+
and Mg2+
. However the 31
P NMR with PS modification at the cleavage
site (17DSAPS+ 17E) did not show a significant shift in the peak position upon Pb2+
- addition,
and relatively minimal shifts with Cd2+
. This suggested that binding of the metal ion to the
phosphate of the RNA base was not crucial for activity; rather the PS replacement might have
resulted in a disruption of the active-site conformation, thereby suggesting a role for the
functional groups of the nucleobases in metal ion binding. .
Hence, in this study, we demonstrated the use of phosphorothioate modifications to
identify functional phosphates of the 8-17 DNAzyme that could be involved in metal ion

128
binding. 31
P NMR has been used as an additional tool to directly visualize the backbone
phosphates since a single PS modification shifts the signal of the phosphate downfield by by ~50
ppm. These results, in conjunction, have led to the identification of a proposed metal ion,
specifically Pb2+
-binding site for the 8-17 DNAzyme.
4.6 References
1. Peracchi, A. Preferential activation of the 8-17 deoxyribozyme by Ca(2+) ions. Evidence
for the identity of 8-17 with the catalytic domain of the Mg5 deoxyribozyme. J. Biol.
Chem. 2000, 275, 11693-11697.
2. Brown, A. K.; Li, J.; Pavot, C. M.; Lu, Y. A lead-dependent DNAzyme with a two-step
mechanism. Biochemistry 2003, 42, 7152-7161.
3. Cruz, R. P.; Withers, J. B.; Li, Y. Dinucleotide junction cleavage versatility of 8-17
deoxyribozyme. Chem. Biol. 2004, 11, 57-67.
4. Bonaccio, M.; Credali, A.; Peracchi, A. Kinetic and thermodynamic characterization of
the RNA-cleaving 8-17 deoxyribozyme. Nucleic Acids Res. 2004, 32, 916-925.
5. Peracchi, A.; Bonaccio, M.; Clerici, M. A mutational analysis of the 8-17 deoxyribozyme
core. J. Mol. Biol. 2005, 352, 783-794.
6. Schlosser, K.; Gu, J.; Sule, L.; Li, Y. Sequence-function relationships provide new
insight into the cleavage site selectivity of the 8-17 RNA-cleaving deoxyribozyme.
Nucleic Acids Res. 2008, 36, 1472-1481.
7. Liu, Y.; Sen, D. A contact photo-cross-linking investigation of the active site of the 8-17
deoxyribozyme. J. Mol. Biol. 2008, 381, 845-859.
8. Li, J.; Zheng, W.; Kwon, A. H.; Lu, Y. In vitro selection and characterization of a highly
efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000, 28,
481-488.
9. Santoro, S. W.; Joyce, G. F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl.
Acad. Sci. U. S. A. 1997, 94, 4262-4266.
10. Kim, H. K.; Liu, J.; Li, J.; Nagraj, N.; Li, M.; Pavot, C. M.; Lu, Y. Metal-dependent
global folding and activity of the 8-17 DNAzyme studied by fluorescence resonance
energy transfer. J. Am. Chem. Soc. 2007, 129, 6896-6902.
11. Kim, H. K.; Rasnik, I.; Liu, J.; Ha, T.; Lu, Y. Dissecting metal ion-dependent folding and
catalysis of a single DNAzyme. Nat. Chem. Biol. 2007, 3, 763-768.
12. Mazumdar, D.; Nagraj, N.; Kim, H. K.; Meng, X.; Brown, A. K.; Sun, Q.; Li, W.; Lu, Y.
Activity, Folding and Z-DNA Formation of the 8-17 DNAzyme in the Presence of
Monovalent Ions. J. Am. Chem. Soc. 2009, 131, 5506-5515.
13. Eckstein, F. Nucleoside phosphorothioates. J. Am. Chem. Soc. 1970, 92, 4718-4723.
14. Matzura, H.; Eckstein, F. A polyribonucleotide containing alternation P=O and P=S
linkages. Eur. J. Biochem. 1968, 3, 448-452.

129
15. Eckstein, F. Nucleoside phosphorothioates - tools for the investigation of enzyme
mechanisms. Phosphorus Chem.Directed Biol., Lect. Int. Symp. 1980, 125-127.
16. Eckstein, F. Nucleoside phosphorothioates for the study of enzyme mechanisms. ACS
Symp. Ser. 1981, 171, 99-101.
17. Eckstein, F. Nucleoside phosphorothioates. Annu. Rev. Biochem. 1985, 54, 367-402.
18. Olsen, D. B.; Sayers, J. R.; Kotzorek, G.; Eckstein, F. Interaction of restriction
endonucleases with phosphorothioate-containing DNA. Nucleosides Nucleotides 1991,
10, 665-667.
19. Eckstein, F. Developments in RNA chemistry, a personal view. Biochimie 2002, 84, 841-
848.
20. Basu, S.; Strobel, S. A. Thiophilic metal ion rescue of phosphorothioate interference
within the Tetrahymena ribozyme P4-P6 domain. RNA 1999, 5, 1399-1407.
21. Cernak, P.; Madix, R. A.; Kuo, L. Y.; Lehman, N. Accommodation of Ca(II) ions for
catalytic activity by a group I ribozyme. J. Inorg. Biochem. 2008, 102, 1495-1506.
22. Hunsicker, L. M.; DeRose, V. J. Activities and relative affinities of divalent metals in
unmodified and phosphorothioate-substituted hammerhead ribozymes. J. Inorg. Biochem.
2000, 80, 271-281.
23. Herschlag, D.; Khosla, M. Comparison of pH dependencies of the Tetrahymena ribozyme
reactions with RNA 2'-substituted and phosphorothioate substrates reveals a rate-limiting
conformational step. Biochemistry 1994, 33, 5291-5297.
24. Lambowitz, A. M.; Caprara, M. G.; Zimmerly, S.; Perlman, P. S. Group I and group II
ribozymes as RNPs: clues to the past and guides to the future. Cold Spring Harbor
Monogr.Ser. 1999, 37, 451-485.
25. Pyle, A. M. Catalytic reaction mechanisms and structural features of group II intron
ribozymes. Nucleic Acids Mol. Biol. 1996, 10, 75-107.
26. Crary, S. M.; Kurz, J. C.; Fierke, C. A. Specific phosphorothioate substitutions probe the
active site of Bacillus subtilis ribonuclease P. RNA 2002, 8, 933-947.
27. Sun, L.; Harris, M. E. Evidence that binding of C5 protein to P RNA enhances ribozyme
catalysis by influencing active site metal ion affinity. RNA 2007, 13, 1505-1515.
28. Osborne, E. M.; Schaak, J. E.; Derose, V. J. Characterization of a native hammerhead
ribozyme derived from schistosomes. RNA 2005, 11, 187-196.
29. Osborne, E. M.; Ward, W. L.; Ruehle, M. Z.; DeRose, V. J. The identity of the
nucleophile substitution may influence metal interactions with the cleavage site of the
minimal hammerhead ribozyme. Biochemistry 2009, 48, 10654-10664.
30. Peracchi, A.; Beigelman, L.; Scott, E. C.; Uhlenbeck, O. C.; Herschlag, D. Involvement
of a specific metal ion in the transition of the hammerhead ribozyme to its catalytic
conformation. J. Biol. Chem. 1997, 272, 26822-26826.
31. Ruffner, D. E.; Uhlenbeck, O. C. Thiophosphate interference experiments locate
phosphates important for the hammerhead RNA self-cleavage reaction. Nucleic Acids
Res. 1990, 18, 6025-6029.
32. Knoll, R.; Bald, R.; Furste, J. P. Complete identification of nonbridging phosphate
oxygens involved in hammerhead cleavage. RNA 1997, 3, 132-140.

130
33. Horton, T. E.; Maderia, M.; DeRose, V. J. Impact of phosphorothioate substitutions on
the thermodynamic stability of an RNA GAAA tetraloop: an unexpected stabilization.
Biochemistry 2000, 39, 8201-8207.
34. Maderia, M.; Horton, T. E.; DeRose, V. J. Metal interactions with a GAAA RNA
tetraloop characterized by 31P NMR and phosphorothioate substitutions. Biochemistry
2000, 39, 8193-8200.
35. Maderia, M.; Hunsicker, L. M.; DeRose, V. J. Metal-phosphate interactions in the
hammerhead ribozyme observed by 31P NMR and phosphorothioate substitutions.
Biochemistry 2000, 39, 12113-12120.
36. Kumar, P. K. R.; Zhou, D. M.; Yoshinari, K.; Taira, K. Mechanistic studies on
hammerhead ribozymes. Nucleic Acids Mol. Biol. 1996, 10, 217-230.
37. Scott, E. C.; Uhlenbeck, O. C. A re-investigation of the thio effect at the hammerhead
cleavage site. Nucleic Acids Res. 1999, 27, 479-484.
38. Zhou, D.-M.; Kumar, P. K. R.; Zhang, L.-H.; Taira, K. Ribozyme mechanism revisited:
Evidence against direct coordination of a Mg2+
ion with the pro-R oxygen of the scissile
phosphate in the transition state of a hammerhead ribozyme-catalyzed reaction. J. Am.
Chem. Soc. 1996, 118, 8969-8970.
39. Suzumura, K.; Takagi, Y.; Orita, M.; Taira, K. NMR-based reappraisal of the
coordination of a metal ion at the pro-Rp oxygen of the A9/G10.1 site in a hammerhead
ribozyme. J. Am. Chem. Soc. 2004, 126, 15504-15511.
40. Horton, T. E.; Clardy, D. R.; DeRose, V. J. Electron paramagnetic resonance
spectroscopic measurement of Mn2+
binding affinities to the hammerhead ribozyme and
correlation with cleavage activity. Biochemistry 1998, 37, 18094-18101.
41. Morrissey, S. R.; Horton, T. E.; DeRose, V. J. Mn2+
Sites in the Hammerhead Ribozyme
Investigated by EPR and Continuous-Wave Q-band ENDOR Spectroscopies. J. Am.
Chem. Soc. 2000, 122, 3473-3481.
42. Vogt, M.; Lahiri, S.; Hoogstraten, C. G.; Britt, R. D.; DeRose, V. J. Coordination
environment of a site-bound metal ion in the hammerhead ribozyme determined by 15N
and 2H ESEEM spectroscopy. J. Am. Chem. Soc. 2006, 128, 16764-16770.
43. Nawrot, B.; Widera, K.; Wojcik, M.; Rebowska, B.; Nowak, G.; Stec, W. J. Mapping of
the functional phosphate groups in the catalytic core of deoxyribozyme 10-23. FEBS J.
2007, 274, 1062-1072.
44. Kim, H. K.; Li, J.; Nagraj, N.; Lu, Y. Probing metal binding in the 8-17 DNAzyme by
TbIII luminescence spectroscopy. Chem. Eur. J. 2008, 14, 8696-8703.
45. Lee, N. K.; Koh, H. R.; Han, K. Y.; Kim, S. K. Folding of 8-17 deoxyribozyme studied
by three-color alternating-laser excitation of single molecules. J. Am. Chem. Soc. 2007,
129, 15526-15534.
46. Faulhammer, D.; Famulok, M. Characterization and divalent metal-ion dependence of in
vitro selected deoxyribozymes which cleave DNA/RNA chimeric oligonucleotides. J.
Mol. Biol. 1997, 269, 188-202.
47. Lim, J.; Grove, B. C.; Roth, A.; Breaker, R. R. Characteristics of ligand recognition by a
glmS self-cleaving ribozyme. Angew. Chem. Int. Ed. 2006, 45, 6689-6693.

131
5 In Vitro Selection of DNAzymes Specific for the Detection of Mercury
(Hg2+
) and Cadmium (Cd2+
)
Note and Acknowledgements: The in vitro selection of DNAzymes specific for Cd2+
was done in collaboration with Dr. Debapriya Mazumdar and is being continued by Hannah
Ihms. The random pools used in both the selections were designed in collaboration with Dr.
Geng Lu and Dr. Debapriya Mazumdar. I wish to thank Professor Scott Silverman for valuable
suggestions during the design of the random pool and during my preliminary examination, and
Silverman laboratory members (particularly Dr. Elizabeth D. Pratico and Dr. Dana A. Baum) for
helpful suggestions while troubleshooting experimental procedures.
5.1 Introduction
5.1.1 Deoxyribozymes (or DNAzymes) and ribozymes
The terms deoxyribozymes or DNAzymes and ribozymes have been coined to denote
DNA and RNA molecules that have catalytic activity respectively.1-5
Ribozymes have been
implicated in biochemical processes that include protein translation and RNA splicing.6 These
RNA enzymes are also believed to have played an important catalytic role in primitive earth.7
Naturally occurring ribozymes were first discovered in the early „80s by Cech and co-workers in
the intron of an RNA transcript,6 and in the RNA component of the RNase P complex that is
involved in the maturation of pre-tRNAs.8, 9
The discovery of ribozymes appears to be a critical
component and connection to the concept of an „RNA world‟; wherein the majority of catalytic
functions were performed by RNA molecules since proteins were non-existent.7, 10-14
RNA
molecules termed as aptamers15
were also shown to form tertiary structures and bind to

132
molecular targets in 1990 through the combinatorial technique (discussed in Chapter 1) known as
SELEX (Systematic Evolution of Ligands by Exponential Enrichment) or in vitro selection.16
This technique was also useful in eventually producing both artificial ribozymes17, 18
as well as
DNAzymes, the latter of which have not been known to occur naturally thus far. The first report
of a DNAzyme was in 1994 by Breaker and Joyce,19
who isolated DNA molecules that could
specifically catalyze the cleavage of a specific RNA linkage within the nucleic acid strand in the
presence of Pb2+
ions. Various classes of DNAzymes have since been isolated through the in
vitro selection technique, that include RNA ligation, RNA lariat formation, DNA
phosphorylation, DNA ligation, oxidative DNA cleavage and porphyrin metallation.4, 5, 20-27
5.1.2 In vitro selection
The starting point of this method is the use of large populations (1013
-1016
) of random
DNA or RNA sequences that are subsequently iteratively subjected to selection pressure to attain
either a desired catalytic or binding function.25, 28-31
After every round of selection, sequences
that display the desired activity are separated from the other DNA/RNA molecules commonly by
either gel electrophoresis or column-based separations. Once the desired sequences have been
identified, they are amplified using the polymerase chain reaction (PCR). After several rounds of
selection the „winner‟ sequence exhibiting highest selectivity and sensitivity is identified and
characterized. This combinatorial technique, therefore, allows the screening of very large
libraries through these iterative selection-amplification cycles.
5.1.2.1 Important considerations during the selection process
The key factors that therefore govern any selection and its success include the length of
the random region incorporated into the pool, the selection pressure introduced, the conditions at

133
which selection is carried out and finally the parameter that is monitored for activity.25, 32
Those
selections that involve the catalysis of complex reactions generally start with pools containing
longer random regions due to their inherent capability to fold into functional conformations.
However, difficulties can arise from having a long pool sequence. Large pools could misfold
due to interactions of extraneous regions with functional regions within the sequence. Small but
efficient motifs in larger copy numbers than larger motifs could increase in relative abundance
during the early rounds of selections when the selective pressure is less stringent. Hence, a fine
balance needs to be struck between the length of the pool and the probability of the evolution of
a functionally active sequence. The number of possible sequences for a random region
containing „n‟ number of bases is 4n and successful in vitro selections have been carried out with
random region ranges from < 30 to > 200 nucleotides.25, 33
For a system containing 40 random
nucleotides, 440
or 1024
possible sequences exist, while 1036
sequences are possible for a 60 base
random domain. During the generation of the initial pool, ~ 0.01-10 nmol template is used and
hence the typical starting random pool contains between 1013
-1016
sequences. Although only a
small amount of the sequence space is sampled (~10-10
coverage), the number of successful
selections carried out demonstrate the presence of functional sequences within the space
explored. In addition, some variations in sequence will be permissible leading to redundancy in
sequences that adopt a particular structure or function. In order to increase the sampling of
sequence space, or the set of all possible sequences for a given number of random nucleotides,
reselection is often performed. Reselection following the initial selection uses a degenerate
template derived from the most active generation of the selection to introduce a high degree of
diversity which may lead to the isolation of more active or specific sequences that vary from the

134
template by a relatively small number of bases. Further diversity can also be introduced by in
vitro evolution, via mutagenesis, such as the use of error-prone PCR.
5.1.3 Metal-dependent DNAzymes that cleave RNA
Many classes of DNAzymes that have been isolated thus far using the in vitro selection
method require the presence of a divalent metal co-factor and display metal-dependent catalytic
activities (also shown in Chapter 1, Table 1.1). Two of the most common motifs isolated
amongst DNAzymes that cleave RNA are the „10-23‟ and „8-17‟ DNAzymes.33
The 10-23
DNAzyme performs the RNA cleavage at a pyrimdine-purine junction in the presence of
divalent metal ions that include Mg2+
and Mn2+
.34, 35
Its utility has been mainly explored in
relevance to therapeutic applications. The 8-17 DNAzyme, on the other hand, has been isolated
by several different research groups under various selection conditions in the presence of
different divalent metal ions and in one instance, even in the absence of a metal ion.36-39
The
highest activity of this DNAzyme has been observed with Pb2+
and it has subsequently been
converted into both a colorimetric and fluorescence sensor for lead.40-49
DNAzymes which show
activity in the presence of UO2+
,50
Zn2+
,38
Cu2+
,51
Co2+
,52
and Mn2+ 26
have also been isolated and
many of these DNAzymes have also been converted to sense their specific metal ion co-
factors.53-56
5.1.4 Mercury contamination and toxicity
Mercury is a well-known water contaminant. Like several other environmental
pollutants, it undergoes bioaccumulation, whereby the rate of accumulation of mercury in
humans and other organisms is much more rapid, than its elimination. Biomagnification is the
incremental increase in concentration of a contaminant at each level of a food chain. This

135
phenomenon occurs because the food source for organisms higher on the food chain is
progressively more concentrated in mercury and other contaminants, thus magnifying
bioaccumulation rates at the top of the food chain (http://water.usgs.gov/wid/FS_216-
95/FS_216-95.pdf). The majority of mercury in the atmosphere is in the inorganic form (Hg2+
),
which in water enters complex cycles wherein it can convert from one form to other.
Methylmercury is one of the forms commonly produced, and it is extremely toxic. The health
effects of mercury include the impairing of speech, hearing, vision and gait. Methylmercury
poisoning in the Minamata bay in 1956 lead to hundreds of deaths, and caused severe nervous
system disorders leading to permanent brain damage.57-60
The EPA-recommended MCL
(Maximum Contamination Level) of mercury in water is 2 ppb.
(http://www.epa.gov/ogwdw000/contaminants/dw_contamfs/mercury.html)
5.1.5 Cadmium toxicity and EPA Maximum Contamination Limit (MCL)
Cadmium is a toxic metal that can induce cytotoxicity, chromosomal aberrations, DNA-
strand breakage and mutagenicity in mammalian cells.61, 62
Animal studies have shown that
cadmium can increase the incidence of leukemia, as well as testicular interstitial-cell tumors and
prostrate tumors. Cadmium is particularly dangerous as it can interact with both DNA and
proteins.63
It can inhibit DNA replication and DNA repair pathways. Since it is more thiophilic
than zinc, it can interfere with zinc binding in proteins that form metal – enzyme complexes.
Cadmium is used mainly in Ni-Cd storage batteries, electroplating of various metals and as a
component in different alloys. Major industrial releases of cadmium are due to waste streams and
leaching of landfills. In particular, cadmium can be released into drinking water from the
corrosion of some galvanized plumbing and water main pipe materials. Dust, fumes and waste

136
from these industries are the main sources of cadmium contamination in the environment.
Workers in these industries have the highest potential exposure from airborne dust and fumes.
Waste from these industries can also increase cadmium concentration in surface waters and
crops.64, 65
The MCL for cadmium in water has been set at 5 ppb by the EPA
(http://www.epa.gov/OGWDW/contaminants/dw_contamfs/cadmium.html).
5.1.5.1 Interaction of cadmium with nucleic acids
Cadmium is more likely to interact with the bases of nucleic acids than with the
phosphates on the backbone. It has been reported that Cd2+
possibly coordinates to the N7 of
guanine and adenine. Various synthetic models are being investigated to find out more about the
nature of cadmium binding.66, 67
Recently Zivarts et al.68
reported an engineered allosteric
ribozyme that is activated by a number of divalent metal ions including Cd2+
. However they also
report Cd2+
to have an inhibitory effect on the activity at a concentration greater than 1 mM,
possibly due to non-specific binding of the metal to RNA, causing disruption of structure and
function. Cd2+
has also been used in mechanistic studies, to determine the interaction of metals
with ribozymes. The substitution of phosphate oxygen with sulfur to form phosphorothioate is a
common tool used in mechanistic studies to determine the importance of particular oxygens in
forming the catalytic metal binding site. Cd2+
has been found to rescue catalytic activity from
deleterious effects of such substitution. 69-73
5.1.6 Research focus
Several techniques currently exist for the detection of both Hg2+
and Cd2+
, including
atomic absorption spectroscopy, gas chromatography, ICP-MS, atomic fluorescence

137
spectroscopy, ICP-atomic emission spectroscopy and reverse-phase HPLC, all of which can
provide limits of detection at the ppb level.74-79
However, all of these techniques require
elaborate and time-consuming sample preparation as well as analysis. Considerable efforts are
therefore devoted to develop sensors that are capable of on-site monitoring without the use of
sophisticated instrumentation. Recently Hg2+
sensors based on oligonucleotides with poly-T
templates have been developed.80
Molecular beacons,80, 81
gold nanoparticles82
and allosteric
DNAzymes83
have all been developed based on this poly-T design.
DNAzymes provide a stable metal-sensing platform and we have previously
demonstrated the use of the 8-17 system for effective Pb2+
sensing in water and paints.43, 84
The
need for expensive instrumentation and elaborate analysis is eliminated with the use of the
colorimetric lead sensor and therefore, effective on-site sensing can be achieved. DNA molecules
are also preferred for sensing applications because of their high stability in comparison with
RNA, as well as their solubility in aqueous solutions.55
The denaturation-renaturation cycle of
DNA can also be carried out multiple times without any significant loss of binding or catalytic
activities. The use of negative selection in the combinatorial in vitro technique also minimizes
interference caused due to other divalent metal ions with similar physical and chemical
properties and hence very high selectivity can be achieved.52
The iterative rounds of selection
also lead to the isolation of highly specific DNA sequences.25
The research focus of this project was initially the in vitro selection of a DNAzyme
specific for Hg2+
. However, during the period of selection, a molecular beacon sensor approach
based on DNA containing poly-T‟s was developed in the Lu lab for the detection of Hg2+
, and
the detection limit based on this method was found to be 3.2 nM.81
This sensor also exhibited a

138
very high selectivity toward Hg2+
over other metal ions tested. Since such a good sensor for Hg2+
based on DNA was already developed in the Lu lab, the author‟s focus and efforts were then
directed towards the isolation of a DNAzyme specific for Cd2+
(which was an on-going effort in
the lab) which could eventually be developed as a biosensor for the real-time and on-site
detection of Cd2+
in environmental monitoring and clinical toxicology.
5.2 Materials and Methods
5.2.1 Materials
HPLC-purified template and primers were purchased from Integrated DNA
Technologies, Inc., and were used without any further purification. Mercury perchlorate
(puratronic®, 99.995 %) and Cadmium chloride.xH2O (x~2.5) (puratronic
®, 99.995 %) were
purchased from Alfa Aesar. HEPES, monosodium salt, was purchased from Sigma-Aldrich while
sodium perchlorate was purchased from Fisher Scientific. All buffers and metal solutions were
made in Millipore water. The selection buffer was made as a 2 × solution and contained 100 mM
HEPES, 500 mM sodium perchlorate at pH 7.0. Ultrapure nitric acid (from Alfa-Aesar) was used
for adjustment of the pH. The concentrated mercury metal-stock solution contained 50 mM
mercury perchlorate in water while the cadmium-stock solution contained 100 mM cadmium
chloride in water with a few drops of HCl (~0.05 %). Subsequent dilutions in Millipore water
were made to obtain 2 × metal solutions of appropriate concentrations. Taq polymerase with
PCR buffers (5U/L) and dNTPs (10 mM) was purchased from Invitrogen. [32
P]-
deoxyadenosine triphosphate (dATP) and [P]-dATP were purchased initially from GE
Healthcare and later from Perkin Elmer. The original TA cloning® kit was purchased from
Invitrogen.

139
5.2.2 Template and primer design
The same template and primers were used for both the Hg2+
and Cd2+
selections. The
design of the pool was initiated by Dr. Geng Lu and modified by Dr. Debapriya Mazumdar with
inputs from the author (Figure 5.1). The pool consisted of a 50-nucleotide (N50) random region
flanked by two primer-binding regions. This pool has 4-5 base pairs flanking the random region
and a 4-nucleotide loop that facilitates this base pairing. There were 3 primers used for extension
and amplification and were denoted as P2, P3 and P4. Sequence of template and primers used is
shown in Table 5.1.
Figure 5.1 Schematic representation of the expected secondary structure of the random pool
used for the in vitro selections of both Hg2+
and Cd2+
. rN represents the single RNA base while
N50 represents the 50-nucleotide random region.
N50
rN

140
Table 5.1 Sequences of the oligonucleotides used for the in vitro selection. The random pool
generated after PCR is also shown.
Name Sequence of Oligonucleotide (from 5' 3')
Template CTCGGATCCATACCCTGAAN50CAACGAGGGTTGTCTGTAGATCTTCAGGTGATCTAG
P2 GAATCACCTACTAGATCACCTGAAGATCrTACAGACAACCCTCG
P3 GAATCACCTACTAGATCACCTGAAGATrC
P4 (AAC)12-Sp-C18-CTCGGATCCATACCC
Random
pool
GAATCACCTACTAGATCACCTGAAGATrCGACAGACAACCCTCGTTGTCN50TTCAGGGT
ATGGATCCGAG
The oligo-analyzer program was used to analyze the secondary structure, shown in Figure
5.2, and the thermodynamic parameters of the initial pool at 23 °C, and 250 mM Na+. The
template and primers were also checked for the formation of hairpins, self-dimers and hetero
dimers using the oligo-analyzer on the website of Integrated DNA Technologies Inc.
(http://scitools.idtdna.com/analyzer/oligocalc.asp) using the defaults settings (50 mM salt, 0.25
μM DNA). This presence of these structures could compromise the efficiency of the desired PCR
reactions between the template and the primers or lead to amplification of unwanted DNA that
could contaminate the selection. The base-pairing between the template and primers was also
checked to ensure the absence of mispriming.

141
Figure 5.2 Predicted secondary structure of the random pool used for in vitro selection.
5.2.3 In vitro selection and PCR protocols
The in vitro selection method used was based on the scheme originally developed by
Breaker and Joyce.15
However gel-based separations based on the methods of Williams and
Bartel64
were used in place of the column-based separation. The initial pool used for both the
Hg2+
and Cd2+
selections was identical. It contained a randomized N50 region flanked by 2
primer-binding regions, one containing the single RNA base which serves as a cleavage site. The
P4 primer contained a hexaethyleneglycol spacer (Spacer-18, from Integrated DNA

142
Technologies) which served as a polymerization termination site, followed by (AAC) 12, which
gave rise to two strands of unequal lengths (Figure 5.3).
5.2.3.1 Initial pool preparation
The initial pool was generated by template-directed extension, followed by PCR
amplification. The extension was carried out in 20 centrifuge tubes with 0.1 µM of DNA
template and 0.2 M primer P3 (for incorporation of the RNA base), 0.2 mM dNTP mix, 1 ×
PCR buffer and 1.5 mM MgCl2; the total volume in each tube being 1.05 mL. 1.0 L of Taq
polymerase was added to each of the tubes at 85°C and 7 cycles of PCR were carried out. To
each of the tubes containing the extension product, 13.4 L of a solution containing 1 × PCR
buffer, 0.2 mM dNTP mix, 1.5 mM MgCl2, 5.7 M P4 (for introducing the spacer and
termination site), 2.1 M P3 and 2 L of [-32
P]-dATP were added.7 additional cycles of PCR
were carried out and all the amplification products were pooled together. The PCR products were
then neutralized with 10 % of a 3 M sodium acetate solution, at pH 5.2, and precipitated using
2.5 × volume of ethanol. The samples were stored at -80 °C for 1 hour, then centrifuged,
decanted and lyophilized.
5.2.3.2 32P-labeled gel-based separations and ethanol precipitations
The lyophilized samples were dissolved in water and an equal amount of loading buffer
was added. This loading buffer also called as the „stop buffer‟ contained 8 M urea, 50 mM
EDTA and 1 × TBE (Tris, Boric acid, EDTA). The reaction products were purified using a 10 %
denaturing PAGE gel, with the use of 1 × TBE as the running buffer. The DNA was run on the
gel alongside markers corresponding to the cleaved (88-mer) and uncleaved (118-mer) pool. The
gel was then covered with a plastic wrap and exposed to an X-ray film because the DNA was

143
now radioactively labeled and „hot‟. The film was then developed and the band corresponding to
the 118-mer marker on the gel was extracted with a 10 mM Tris, 1 mM EDTA, and 300 mM
sodium perchlorate solution (soak solution). The solution was centrifuged 2-3 times to remove
any gel bits and was ethanol precipitated using the above-mentioned procedure.
5.2.3.3 Selection procedure
With Hg2+
: The lyophilized pools after the initial pool generation (and subsequent PCR
amplifications) were dissolved in 2 × selection buffer and incubated with the desired 2 ×
Hg(ClO4)2 solution. The selection was gradually made stringent by varying the reaction time.
The reaction was stopped by the addition of an equal amount of the stop buffer. The selection
conditions that include the concentrations of Hg2+
and the incubation times used for selection are
shown in Table 5.2. Purification was carried out on a 10 % gel alongside the markers used
earlier. This time the cleaved band corresponding to the lower (88-mer) marker was cut out,
extracted using the soak solution, ethanol precipitated and lyophilized.
With Cd2+
: The lyophilized pools were dissolved in 2 × selection buffer and incubated
with the desired 2 × cadmium chloride solution or 2 × lead acetate solution (for the first 2 rounds
of strategy 1). The concentration of Pb2+
used for the first 2 rounds of negative selection was 100
M (1 ×) and this was followed by selection with 100 M of Cd2+
for rounds 3-5. For strategy 2,
the pool was incubated with 100 M Cd2+
starting from round 1. The selection was gradually
made more stringent by varying the reaction time (decreasing from 5 h in round 1 to ~3.5 hours
in round 5) and was stopped by the addition of an equal amount of stop buffer. Purification was
carried out on a 10 % gel alongside the markers used earlier. This time the cleaved band

144
corresponding to the lower (88-mer) marker was cut out, extracted using the soak solution,
ethanol precipitated and lyophilized.
Figure 5.3 In vitro selection scheme adapted from former group member, Dr. Debapriya
Mazumdar‟s thesis.
(cleavage products)
Template directed extension
rAP3
Template
random region
N50
PCR amplification of initial pool
STOP
rA
rA
P4
STOP
rA
STOP
rA
rA
STOPSTOP
rA
STOP
Gel purification of random pool
M2+
Cleavage
rA
P2
(uncleaved)
Gel purification of cleavage product
PCR extension of cleavage product
PCR extension and amplification

145
5.2.3.4 PCR extension and amplification
Half of the lyophilized sample was used for PCR extension and amplification while the
other half was stored for future use. This DNA sample was extended using 1 × PCR buffer, 1.5
mM MgCl2, 0.2 mM dNTPs, 0.4 μM P2 primer (for extension) and 0.4 μM P4 primer. 1 L Taq
polymerase was added to the mixture at 80°C and 10 cycles of amplification were carried out. 10
mL of this PCR product was used for PCR 2 which contained 0.2 mM dNTPs, 1 × PCR buffer,
1.5 mM MgCl2, 0.5 μM P3 primer, 0.25 μM P4 primer and 1 μL α-P32
ATP. 1 L of Taq
polymerase was again added at 80 °C and 20 cycles of PCR amplification were carried out. The
extended and amplified product after PCR 2 was again purified by 10 % PAGE gel
electrophoresis. The sample was again run against the markers. The band corresponding to the
upper (118-mer) marker was cut out, extracted using the soak solution, ethanol precipitated and
then lyophilized (Figure 5.4). The extended and amplified DNA was then used for the next round
of selection.

146
Figure 5.4 a) Representative X-ray film image exposed to a 10 % PAGE gel following PCR
after round 1 of selection with 5 mM Hg2+
. The bands on the left indicate the marker while the
bands on the right indicate the PCR products obtained. The band boxed in blue, corresponds to
the 118-mer marker, is cut out crushed and soaked overnight for DNA extraction. b) Image
following in vitro selection after round 7 with 100 µM Hg2+
and 500 µM Hg2+
, alongside marker
(in center).The lower bands boxed in blue, correspond to the 88-mer marker and are cut out ,
crushed and soaked overnight.
5.2.4 Real-time PCR measurements
Real-time PCR measurements were carried out in a DNA Engine Opticon™ System from
MJ Research. The instrument was kindly provided to the Lu lab from Dan Cropek, CERL.
DyNAmo™ SYBR® Green real-time qPCR kits were purchased from Finnzymes. Real-time
PCR was done in low-profile PCR strip tubes. 8 * 50 L (each) reactions were carried out. The
PCR mixture contained 25 L of 2 × master mix, 1.75 L of primer P4, 2.5 L P3 primer, 5 L
of PCR 1 product (after round 6) and 15.75 L of water. PCR conditions were maintained to be
identical to the regular PCR 2 conditions used for selection. This real-time PCR was used to
determine whether the PCR conditions used were optimized to the correct number of
amplification cycles and that there was no over-amplification, mispriming or unspecific
amplification taking place. Figure 5.5 shows the results of the real-time PCR data. SYBR® Green
a
118
88
b
118
88

147
is a dye that binds only to double-stranded DNA and it is the fluorescence of this particular dye
that is monitored during the real-time PCR.
Figure 5.5 Real-time PCR data showing the increase in fluorescence intensity of SYBR® Green
during 20 cycles of amplification.
The PCR protocol initially created by Dr. Jing Li and later modified by others including
the author was used, and 20 cycles of amplification were carried out. Negative controls that did
not contain either the pool DNA from PCR 1 or the primers did not show any increase in the
fluorescence intensity after 20 rounds of amplification. The amplification cycles that were used
appear to be the correct number, as good amplification was seen starting from round 10 and a
steady fluorescence from round 12. Also the single peak seen from the melting curve data (data
not shown) indicated that there was only one type of duplex DNA formed. Hence, the PCR
conditions used were optimal for good extension and amplification.

148
5.2.5 Gel-based activity assays
Activity assays were performed after various rounds of selection, in order to determine
the cleavage activity of the selected pool. The P3 primer was radioactively labeled with [32
P]-
dATP using T4 kinase from Invitrogen and was called P3*. The labeling mixture contained 1
mM P3, 1 × forward reaction buffer, 2.5 L of T4 kinase and 3 L of [-32
P]-dATP, the total
volume being 20 L. This was incubated at 37 °C for 1.5 h.
10 L of the DNA pool after PCR-1 of the specific round, „y‟, was labeled by carrying
out PCR 2 with 0.5 M P3*, 0.1 M P3, 0.05 M P4, 1 × PCR buffer, 0.06 mM dNTP mix and
1.5 mM MgCl2. Taq polymerase was added at 80 °C and 20 cycles of PCR 2 were carried out.
The DNA pool was purified using 10 % PAGE (as described earlier) and the band corresponding
to the upper (118-mer) marker was cut out and extracted with soak solution. The DNA was
purified with a C-18 Sep-Pak desalting column (from Waters) and then lyophilized. This DNA
pool was called the round y PCR 2* pool (where the asterisk denotes a gamma-labeled, and thus
quantitatively-labeled, pool).
The round y PCR 2* population was dissolved in 2 × selection buffer and denatured at 95
°C for 3 min. and annealed by cooling at room temperature for around 30 min. The reaction was
initiated by the addition of an equal volume of 2 × metal solution to the DNA in 2 × selection
buffer. At predetermined time points, 10 L aliquots of the reaction mixture were withdrawn and
added to 20 L of stop solution containing 50 mM EDTA, 8 M Urea, 1 × TBE, 0.05 % xylene
cyanol and 0.05 % bromophenol blue. The uncleaved and cleaved pools were separated on a 20
% PAGE gel (32 W, 2.5h). The gel was then wrapped and exposed to a phosphorimager cassette.
The cassette was then imaged using a Molecular Dynamics Storm 430 Phosphorimager (from

149
Amersham Biosciences) and the fraction of cleavage was calculated using Image Quant software
(Molecular Dynamics). Kinetic curves were plotted using Sigma plot and fit to the equation:
% Pt = % P0 + % P (1-e-kt
)
where % P0 is the initial percent product (t = 0), % P is the % product at the end point of the
reaction (t = ) ,% Pt is the % product at time t, and k is kobs is the rate of cleavage.
5.2.6 Cloning and sequencing
Cloning was carried out with PCR products after round 7 (primers used did not contain
the ribonucleotide and the stop sequence) using the original TA Cloning® kit (Invitrogen). DNA
was inserted into vectors by the TA cloning reaction and the vector was transformed into E.Coli
competent cells via heat-shock. Approximately 50 µL of transformed cells were plated on LB
plates containing ampicillin and incubated at 37 °C overnight. Individual white clones that grew
on the plates were picked using pipette tips (~ 96 clones selected) and grown in separate tubes
containing 5 mL LB media containing 40 µg/mL ampicillin at 37 C overnight. DNA was
extracted using QIAprep Spin Miniprep kits (Qiagen) and eluted into water. The concentration of
8 randomly selected clones were determined by measuring the absorbance (A260 × 50 =
concentration in ng/µL). The concentration was adjusted to ~ 140 ng/µL and the clones were
submitted for sequencing to the W.M. Keck Sequencing facility, University of Illinois at Urbana
Champaign.

150
5.3 Results and discussion
5.3.1 In vitro selection of a Hg2+
- specific DNAzyme
Seven rounds of selection were carried out according to the protocols mentioned in the
materials and methods (Section 5.2.3.3). These rounds are summarized as Trial-1 in Table 5.2.
The Hg2+
concentration was gradually reduced from 5 mM in round 1 to 2 mM in round 7,
thereby introducing stringency in the selection procedure. Incubation times were also reduced to
further increase stringency during the selection process, from 5 h in round 1 to 2.5 h in round 7.
After every round of selection, no lower band (length of 88-mer) corresponding to the cleaved
DNA was observed on a gel after the selection. This could have probably been due to low
populations of DNA selective for Hg2+
or also due to the fact that mercury at millimolar
concentrations could have had some inhibitory effect on the DNA.
Activity assays were carried out after rounds 5, 6 and 7. Assays done after round 6 in the
presence of 5 and 2 mM Hg2+
, 5 mM Cd2+
and Zn2+
, 500 M Pb2+
indicated that the pool was
active only in the presence of Pb2+
. (Figure 5.8a) Activity assays performed with 500 M Hg2+
after round 6 also did not show any cleavage indicating the bias of the pool for lower
concentrations of Pb2+
rather than even 5 mM of Hg2+
. (Figure 5.8b) A second trial selection was
then started, wherein after round 3 the concentration of Hg2+
used was lowered to bias the pool
for greater specificity toward Hg2+
. An activity assay performed after round 6 with 50 M Pb2+
and 100 M of Pb2+
after 3h, showed about 50 % cleavage and 30 % cleavage respectively
(Figure 5.8c and Table 5.3). However, with 2 M, 500 M and 100 M Hg2+
no activity was
seen even after 3 h. Hence, at round 6, there was more Pb2+
-dependence rather than Hg2+
-
dependence, indicating a bias of the pool toward Pb2+
over Hg2+
. Hence, a negative selection

151
with 500 M Pb2+
was performed at round 7 and only those sequences that did not show any
activity were extracted from the gel and retained for further selections. These sequences are
shown in Figure 5.8d; in this case the upper band corresponds to the 118-mer marker. A round of
negative selection with 500 M Pb2+
was followed by a round of positive selection with 500 M
(Figure 5.8e) or 100 M Hg2+
. An activity assay carried out following these two rounds of
selection showed no cleavage with either 500 Mor 100MPb2+
after ~2.5 h. However, no
activity was seen with 500 Mand100MHg2+
as well. Upon discussion of this issue during
the course of the author‟s preliminary examinations, suggestions were made to start selection
with a much lowered concentration of Hg2+
. This was necessary because the goal of the selection
was to isolate a DNAzyme with very high specificity towards Hg2+
, the EPA-recommended
MCL of which was 10 nM. Hence, further trial selections with an initial concentration of 1 mM
Hg2+
and a parallel selection with 100 µM Hg2+
starting from round 1 to round 3 were carried
out. However, the selections were discontinued after round 3 of this third trial, since around the
same time a highly selective and sensitive DNA sensor for Hg2+
had been developed by another
member of the Lu lab. This sensor was based on a fluorescent molecular beacon approach, and
had a detection limit of 3.2 nM for Hg2+
.

152
Table 5.2 Summary of metal concentrations and incubation time of the pool with metal ions for
various rounds of in vitro selection with Hg2+
.
Trial-1 Round [Metal ion] Incubation
Time (h)
Trial-2
Round
[Metal ion] Incubation
Time (h) 1,2,3,4 Hg
2+ 5 mM 5 1,2,3 Hg
2+ 5 mM 5
5 Hg2+
5 mM 4 4,5 Hg2+
500M 3
6 Hg2+
2 mM 3 6,7 Hg2+
500M 2.5, 2
7 Hg2+
500M, 100
M
3 7(negative
selection) Pb
2+ 500M 2.5
Table 5.3 Results of activity assays after Trial-2, round 6 of Hg2+
selection.
5.3.1.1 Problems encountered during actual selection procedure
Several attempts of trial-1, rounds 4-7 were carried out. During the attempts it was
observed that after round 6, the band on the gel after PCR extension and amplification was very
faint. The number of amplification cycles was then increased from 20 to 30, however some non-
specific amplification was observed. The number of amplification cycles was then reduced back
to 20, but this time fresh -32
P was used for labeling and all solutions were re-made. This time
clear bands after extension and amplification were observed. After round 6, activity assays were
also carried out initially. However, it was observed that several bands (even uncleaved DNA)
disappeared on the gel, as shown in Figure 5.6. There appeared to be a random appearance and
disappearance that could not be due to actual Hg2+
-dependent activity of the DNAzymes. Hence,
Metal concentration % Cleavage
0 min
% Cleavage
30 min
% Cleavage
120 min
% Cleavage
180 min Hg
2+-2 mM 0.76 3.51 2.05 1.78
Hg2+
-500 M 3.39 3.38 3.42 2.97
Hg2+
-100 M 2.11 3.65 3.06 2.42
Pb2+
- 500 M 2.21 22.87 47.82 52.19
Pb2+
- 100M 3.78 7.11 12.34 31.74

153
as a first step to overcome this problem, all solutions including buffers, gel stocks as well as
filters for Millipore water were discarded and made fresh. Rounds 5 and 6 of selection were
again carried out and then the activity assay after round 6 was again carried out. All the bands
were present as expected and no unusual appearance or disappearance of bands was observed
(Figure 5.7).
Figure 5.6 Gel image showing the activity assay after trial-1, round 6 of Hg2+
selection with
disappearing bands
5.3.1.2 Challenges overall with Hg2+
selection
The behavior of the Hg2+
with nucleic acids differs from that of other transition metals.
The Hg2+
ion has been shown to bind very strongly to all of the four nucleosides of DNA with
the logarithm of the stability constants following the order guanosine (4.00) < adenosine (4.25) <
cytidine (9.50) < thymidine (10.60).85
Also, Hg2+
has been shown to facilitate the formation and
stabilization of T-T mismatches of DNA duplexes and this property has been used to detect the
ion. The endo- and exocyclic amine DNA nucleobase functionalities as well as the purinyl C8
position have been reported as suitable binding sites for Hg2+
.86
In fact, the in vitro selection of a
DNAzyme specific for Hg2+
has been designed, wherein the intrinsic affinity of imidazole for
Hg2+
has been utilized to generate non-natural nucleotides dUaa
and dAim
that have been
incorporated into the DNA pool for selection.87
The self-cleaving DNAzyme shows rate constant
2mM 500 µM 100 µMPb2+Hg2+
500 µM 100 µM
0 min
3 h
Uncleaved
Cleaved

154
kobs of 0.037 ± 0.002 min-1
in the presence of 5 µM Hg2+
and shows minimal cleavage in the
presence of comparable concentrations of other metal ions tested. In this instance, the authors
hypothesize that based on the thermodynamics, the DNAzyme 10-13 utilizes the imidazole
residues to chelate Hg2+
cations, rather than making use of the T-Hg2+
-T dimeric interaction seen
in other systems. Hence, till date, the introduction of specific Hg2+
binding strategies in nucleic
acids has proved to be more useful for selection as well as detection strategies. This ion has been
reported to interrupt helices and to facilitate non-canonical base pairings.88
In addition, Hg2+
facilitates base stacking in d(TpT) and d(TpA) dimers, an effect that is highly sequence-
dependent and has not been confirmed in longer DNA strands.88
Mercuration, or covalent
modification of nucleobases can also occur at physiological temperatures.89, 90
So, during the
actual in vitro selection process, it is highly possible that Hg2+
may bind to different functional
units of the DNA and may disrupt the formation of structures that may be essential for the
cleavage activity of RNA that is monitored. All of the afore-mentioned factors contribute to the
extremely challenging prospect as well as the importance of design elements involved in
identifying non-modified, natural DNAzymes that are specific for Hg2+
.

155
Figure 5.7 Gel images showing activity assay results after a, b) round 6 of trial-1 of Hg2+
selection c) round 6 of trial-2 of Hg2+
selection and e) round 7 of trial-2 of Hg2+
selection. d) X-
ray film image exposed to a 10 % gel showing results after round 7 of negative selection with
Pb2+
, of trial-2 of the Hg2+
selection.
Hg2+
5 mM 2 mM 5 mM 5 mM 500 µM
Cd2+ Zn2+ Pb2+
0 min
3 ha
Hg2+
500 µM
Uncleaved
Cleaved
100 µM 500 µM 100 µM0 min
3 h
Pb2+
b
0 min
c100 µM500 µM500 µM 100 µM2 mM
Pb2+Hg2+
3 h
d
118
88
500 µM 100 µM 500 µM 100 µM
Hg2+ Pb2+
2.5 h
e
0 min

156
5.3.2 In vitro selection of Cd2+
-specific DNAzymes
Seven rounds of in vitro selection with Cd2+
were carried out according to the protocol
described in section 5.2.3.3. Earlier selections to isolate DNAzymes specific for Cd2+
with the
current pool had been carried out by former group member, Dr. Debapriya Mazumdar, who
carried out 10 rounds of selection (results published in her PhD thesis). These results indicated
that although the DNA pool after ten rounds of selection showed activity with Cd2+
, Pb2+
-specific
sequences were still obtained as shown in Figure 5.8. In order to bias the random pool away from
specificity toward Pb2+
and also to increase the specificity toward Cd2+
, two parallel selection
strategies were designed and carried out with Cd2+
. For the first strategy, the initial 2 rounds
consisted of carrying out negative selection with 100 M of Pb2+
for 5 hours. These 2 rounds of
negative selection were incorporated so as to remove any sequences that were specific for Pb2+
early on in selection itself. Perrin and co-workers have shown that sequences selective for Hg2+
with high specificity and selectivity could be obtained, through the incorporation of negative
selection rounds during their initial selection rounds. Hence, the introduction of these two
negative selection rounds should be able to remove any Pb2+
specificity that had been previously
observed after several selection rounds. For the subsequent selection rounds, 100 M of Cd2+
was used and the time was varied from 4.5 hours to 3 hours. For strategy 2, the random pool was
incubated with 100 M of Cd2+
till round 4 and no negative selection rounds were introduced.
Stringency was incorporated into the selection by gradually reducing the incubation time from 5
h to 3 h (Table 5.4). The introduction of these parallel selection strategies would give us an
advantage of comparing sequences with negative selection in the initial selection rounds versus
that in later selection rounds. “Would selectivity toward Cd2+
be increased” and “Would

157
selectivity toward Pb2+
be decreased” were the questions that these selection strategies sought to
address. The concentration of Cd2+
for both selection strategies was also significantly lower than
the 5 mM metal concentration the Lu lab usually used for initial rounds of selection. This
strategy was incorporated to increase the sensitivity for lowered Cd2+
concentrations as well as to
obtain sequences with a lowered detection limit since the EPA MCL for Cd2+
was 5 ppb which is
~ 44 nM. Starting with 100 M Cd2+
and gradually lowering the concentration to around 100 nM
would give us an increased probability of finding sequences with a higher sensitivity for the nM-
M range of Cd2+
detection. However, after 7 rounds of parallel selection, no bands
corresponding to a cleavage reaction after the in vitro selection was observed. (A representative
gel after in vitro selection is shown in Figure 5.9a). This could probably be due to the fact that
the enrichment upon 7 rounds of selection did not yield a significant population of DNA that
could actually be visualized on a gel and hence the overall numbers of sequences specific for
activity in the presence of Cd2+
were low in number. For the first strategy, since a negative
selection with Pb2+
was incorporated into the first two rounds of selection, the overall population
of the DNA pool would be expected to be considerably less. This is because activity assays
based on previous selections indicated that the pool did have a good activity with Pb2+
. Removal
of these Pb2+
-specific sequences probably lead to an overall loss in the total number of sequences
present in the pool.

158
Table 5.4 Summary of metal concentrations and incubation time of the pool with metal ions for
various rounds of in vitro selection with Cd2+
.
Strategy-1
Round
[Metal ion] Incubation
Time (h)
Trial-2
Round
[Metal ion] Incubation
Time (h) 1,2(negative
selection) Pb
2+ 100 M 5 1,2,3 Cd
2+ 100 M 5
3 Cd2+
100 M 4 4,5 Cd2+
100 M 3
4,5,6 Cd2+
100 M 3 6,7 Cd2+
100 M 2.5, 2
7,8 Cd2+
100,50 M 3, 2 7(negative
selection) Pb
2+ 100 M 2.5
Figure 5.8 Activity assay gel image from round 6 of selection with Cd2+
with pool containing
rCT at the cleavage site adapted from the thesis of Dr. Debapriya Mazumdar. “NM” stands for
the control with no metal added.
5.3.2.1 Problems encountered during actual selection procedure
Several trials of rounds 4-7 (based on strategies 1 and 2) were carried out repeatedly
since there were instances were no bands were seen on the gel after PCR or selection. Moreover,
during the trials it was also observed that after round 4 and 5, the band on the gel after extension
and amplification was not seen. The number of amplification cycles had already been increased
5 15 35 60 131 180 326 35 131 326 35 131 326Zn Co Ni Mn Ca NM
5 mM, 326 minCd, 5mM Cd, 1mM Pb, 500 uM
5 15 35 60 131 180 326 35 131 326 35 131 326Zn Co Ni Mn Ca NM
5 mM, 326 minCd, 5mM Cd, 1mM Pb, 500 uM

159
from 20 to 30 since strategy 1 had a smaller number of DNA sequences after the initial rounds of
negative Pb2+
selection. Fresh -32
P was also used for labeling and all PCR solutions were made
fresh. However, no bands were seen again (data not shown). Then the concentration of dNTPs
was lowered by 1/10, which subsequently resulted in the appearance of bands again. Hence, the
incorporation of labeled ATP posed a problem when the concentration of template was lowered
during progressive selection rounds and therefore lowered dNTP concentrations needed to be
used accordingly.
Another significant problem that was encountered during the course of selection was that
at the end of rounds 5-7, 10 % gels after either PCR 2 or PCR 2* (see section 5.2.5) amplification
indicated a band that corresponded to the position of the lower cleaved marker as shown in
Figure 5.9b and 5.9c respectively (88-mer). This was extremely surprising, since at this stage
after either PCR 2 or PCR 2*, the pool should have been regenerated and should have appeared at
the position corresponding to the uncleaved marker (118-mer). Something seemed to have been
cleaving the entire pool present. While the population of DNA corresponding to this uncleaved
pool at the position of the 118-mer marker was significant enough to extract from the gel and
proceed with future rounds of selection, the amount of the band after PCR 2* was almost
insignificant and hence activity assays could not be performed. In order to address this issue, all
the PCR buffers were replaced. Further, the purity of the primers used for amplification was
tested using mass spectrometry (ESI-MS) and this did not reveal anything amiss. Another
member of the Lu lab, Hannah Ihms, who was also working on the Cd2+
selection encountered
identical problems when she used primers that she had ordered, identical to the primers that the
author was using and also encountered similar problems. Upon subsequent re-ordering of the

160
primers and PCR, the same problems were again seen. Hence, to view the overall pool, after
round 7 the DNA pool was subject to PCR, followed by transformation, cloning and sequencing.
However, the results of sequencing indicated that the vectors were all self-ligated and no insert
corresponding to the round 7 pool was present. This was rather unexpected since cloning with a
positive control yielded blue colonies while the colonies carrying the insert were white in color
(according to the manufacturer‟s protocol). Hence, either the sequencing had failed completely
or there was a problem with the insertion of the round 7 pool in the vector since the EcoR1 sites
flanking the insert site were missing in the sequenced DNA.
Figure 5.9 Representative X-ray film images exposed to 10 % gels after a) in vitro selection b)
after PCR 2 regeneration of the DNA pools upon in vitro selection and c) after PCR 2*
regeneration of the pool upon selection using a [γ-32
P]-dATP labeled primer for activity assays.
The central lane in all the gels is a marker containing DNA of lengths 118, corresponding to the
upper uncleaved DNA pool and 88, corresponding to the lower cleaved DNA pool.
a
118
88
Uncleaved
Cleaved
118
88
bc
118
88
Cleaved
Pool

161
5.3.2.2 Comparison with previous Cd2+
selections
Two independent in vitro selections, which differed only at the dinucleotide junction at
the cleavage site, were performed previously to isolate a Cd2+
-dependent DNAzyme
(unpublished results based on former group member, Dr.Debapriya Mazumdar‟s PhD thesis).
The first selection with rA-G at the cleavage site yielded robust Cd2+
-dependent DNAzyme
populations limited by their poor metal-ion specificity (the pool was active with Pb2+
, Zn2+
Co2+
,
Ni2+
and Mn2+
). This specificity could not be improved over three rounds of negative selection.
These results indicated that there might have been a number of sequences in the pool that had a
high activity in the presence of a number of different transition metal ions, and their binding site
could not discriminate between these ions. Another in vitro selection with a pool that had rC-T at
the cleavage site was performed based on reports by Yingfu Li and co-workers39
about the
influence of pool dinucleotide junctions on pool specificity. They found that the dinucleotide
junctions containing rN-G yielded a high percentage of sequences containing the 8-17 motif.
(The 8-17 motif is a small catalytic motif that is very efficient in catalyzing RNA cleavage but is
not highly selective amongst many divalent transition metal ions). On the other hand, they found
that dinucleotide junctions formed by rN-T yielded a low percentage of 8-17 containing
sequences. When rC-T was previously employed as the cleavage site (unpublished results
obtained from the thesis of by Dr. Debapriya Mazumdar), a Cd2+
-dependent population with
specificity over other divalent metal ions such as, Zn2+
Co2+
, Ni2+
, Mn
2+, Hg
2+, Ca
2+ and Mg
2+
was obtained, indicating that the choice of the cleavage site was an important factor for obtaining
specificity. Pb2+
-dependent cleavage with observed rate constants similar to Cd2+
-dependent
cleavage was still observed, and thus this selection failed to provide specificity over Pb2+
. In

162
addition, the pool was not sensitive and required 1 mM Cd2+
for activity, which is not practical
for biosensing applications. Two strategies were therefore adopted in performing the current in
vitro selection study. First, the initial concentrations of Cd2+
used were lowered to 100 µM,
which is about an order of magnitude lower than the previously used 5 mM initial Cd2+
concentration. This increased the sensitivity of the pool so that by the end of the selection it was
comparable to the concentrations needed for bio-sensing applications. Negative selection with
Pb2+
in this case was performed at a later round. Secondly, a parallel selection was performed
with the anticipation that negative selections with Pb2+
during the initial rounds of selection
would significantly lower the bias of the entire pool towards it, albeit at the expense of losing
some DNA populations specific towards Cd2+
. These strategies also provided a means to
compare the arrangement of the negative selection steps during the selection procedure on the
overall sequences of DNA isolated and whether the timing of negative selection played an
important role in isolating a functional DNAzyme.
5.4 Conclusions
Efforts toward the isolation of DNAzymes specific for Hg2+
and Cd2+
using in vitro
selection is reported in this chapter. Selection conditions including the concentration of metal
ions, the choice of buffer and pH conditions as well as the introduction of counter selections or
negative selections are critical for the selection of functional DNAzymes. Selections that have
been carried out with both Hg2+
and Cd2+
demonstrate the importance of negative selection for
removing populations of DNA that are Pb2+
-specific. Moreover, the initial concentration of the
metal ions, especially Cd2+
, has also been significantly lowered with the view that DNA
sequences with higher specificity can be obtained if stringency is introduced in the beginning of

163
selection. No functional DNAzymes that were Hg2+
-specific have obtained at the end of the
various rounds of selection carried out, reiterating the key challenges that Hg2+
poses in terms of
in vitro selection due to its known interactions with different components of DNA. Further
selection rounds were, however, discontinued due to the development of a functional Hg2+
sensor. This Hg2+
sensor was based on the highly sensitive and selective interaction of Hg2+
with
T-bases on DNA. While selections with Cd2+
are still being continued, the presence of significant
PCR artifacts indicates the possibility that there might be some inherent cleavage that takes place
during the PCR in several sequences based on the current pool design and this non-robust
population may have arisen over the course of selections and may compete with the actual
functional sequences. It is anticipated that the results discussed indicate the importance of
negative selection in isolating DNA populations that show metal-ion specificity and that a
method for engineering sensitivity into the selection procedure is to lower the initial
concentration of the target used.
5.5 Future directions
The DNA pools obtained from the two selection strategies will be cloned and sequenced
again to compare and contrast the sequences. Whether the use of negative selection during initial
rounds of selection is an important factor versus that in later rounds of selection will be
compared. This will allow us to determine whether small differences during the sequence of in
vitro selection are critical to the selectivity and sensitivity of the final sequences obtained for the
same target. In vitro selections are also being continued with an 80% degenerate pool that is
based on a clone previously isolated, to test if random mutations introduced into the sequence
can improve sensitivity. The final goal of this project is to obtain a DNAzyme sequence that is

164
highly active and specific for Cd2+
, and develop that into a sensor. Biochemical characterization
of the DNAzyme will be carried out to have a better understanding about the sequence and
structural requirements for activity and specificity.
5.6 References
1. Breaker, R. R. DNA enzymes. Nat. Biotechnol. 1997, 15, 427-431.
2. Li, Y.; Breaker, R. R. Deoxyribozymes: new players in the ancient game of biocatalysis.
Curr. Opin. Struct. Biol. 1999, 9, 315-323.
3. Jaschke, A. Artificial ribozymes and deoxyribozymes. Curr. Opin. Struct. Biol. 2001, 11,
321-326.
4. Silverman, S. K. Catalytic DNA (deoxyribozymes) for synthetic applications-current
abilities and future prospects. Chem. Commun. 2008, 3467-3485.
5. Baum, D. A.; Silverman, S. K. Deoxyribozymes: useful DNA catalysts in vitro and in
vivo. Cell Mol. Life Sci. 2008, 65, 2156-2174.
6. Kruger, K.; Grabowski, P. J.; Zaug, A. J.; Sands, J.; Gottschling, D. E.; Cech, T. R. Self-
splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening
sequence of Tetrahymena. Cell 1982, 31, 147-157.
7. Cech, T. R. The efficiency and versatility of catalytic RNA: implications for an RNA
world. Gene 1993, 135, 33-36.
8. Gardiner, K.; Pace, N. R. RNase P of Bacillus subtilis has a RNA component. J. Biol.
Chem. 1980, 255, 7507-7509.
9. Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of
ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849-857.
10. Cech, T. R. RNA. Fishing for fresh catalysts. Nature 1993, 365, 204-205.
11. Cech, T. R. Catalytic RNA: structure and mechanism. Biochem. Soc. Trans. 1993, 21,
229-234.
12. Cech, T. R. Ribozymes, the first 20 years. Biochem. Soc. Trans. 2002, 30, 1162-1166.
13. Gilbert, W.; De Souza, S. J., Introns and the RNA World. 1999; Vol. 37, p 221.
14. Roy, S. W.; Nosaka, M.; de Souza, S. J.; Gilbert, W. Centripetal modules and ancient
introns. Gene 1999, 238, 85-91.
15. Ellington, A. D.; Szostak, J. W. In vitro selection of RNA molecules that bind specific
ligands. Nature 1990, 346, 818-822.
16. Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA
ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505-510.
17. Bartel, D. P.; Szostak, J. W. Isolation of new ribozymes from a large pool of random
sequences [see comment]. Science 1993, 261, 1411-1418.
18. DeRose, V. J. Two decades of RNA catalysis. Chem. Biol. 2002, 9, 961-969.
19. Breaker, R. R.; Joyce, G. F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223-
229.
20. Chandra, M.; Silverman, S. K. DNA and RNA can be equally efficient catalysts for
carbon-carbon bond formation. J. Am. Chem. Soc. 2008, 130, 2936-2937.

165
21. Gerdt, J. P.; Miduturu, C. V.; Silverman, S. K. Selective stabilization of natively folded
RNA structure by DNA constraints. J. Am. Chem. Soc. 2008, 130, 14920-14921.
22. Hobartner, C.; Pradeepkumar, P. I.; Silverman, S. K. Site-selective depurination by a
periodate-dependent deoxyribozyme. Chem. Commun. 2007, 2255-2257.
23. Hobartner, C.; Silverman, S. K. Recent advances in DNA catalysis. Biopolymers 2007,
87, 279-292.
24. Pradeepkumar, P. I.; Hobartner, C.; Baum, D. A.; Silverman, S. K. DNA-catalyzed
formation of nucleopeptide linkages. Angew. Chem. Int. Ed. 2008, 47, 1753-1757.
25. Silverman, S. K. In vitro selection, characterization, and application of deoxyribozymes
that cleave RNA. Nucleic Acids Res. 2005, 33, 6151-6163.
26. Wang, Y.; Silverman, S. K. Deoxyribozymes that synthesize branched and lariat RNA. J.
Am. Chem. Soc. 2003, 125, 6880-6881.
27. Schlosser, K.; Li, Y. Biologically inspired synthetic enzymes made from DNA. Chem.
Biol. 2009, 16, 311-322.
28. Klug, S. J.; Famulok, M. All you wanted to know about SELEX. Mol. Biol. Rep. 1994,
20, 97-107.
29. Wilson, D. S.; Szostak, J. W. In vitro selection of functional nucleic acids. Annu. Rev.
Biochem. 1999, 68, 611-647.
30. Sen, D. DNA selection and amplification. Compr. Nat. Prod. Chem. 1999, 7, 615-641.
31. Sen, D.; Geyer, C. R. DNA enzymes. Curr. Opin. Chem. Biol. 1998, 2, 680-687.
32. Jhaveri, S. D.; Hirao, I.; Bell, S.; Uphoff, K. W.; Ellington, A. D. Landscapes for
molecular evolution: lessons from in vitro selection experiments with nucleic acids.
Annu. Rep. Comb. Chem. Mol. Diversity 1997, 1, 169-191.
33. Klussmann, S., The Aptamer Handbook: Functional Oligonucleotides and Their
Applications. 2006; p 490.
34. Santoro, S. W.; Joyce, G. F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl.
Acad. Sci. U. S. A. 1997, 94, 4262-4266.
35. Joyce, G. F. RNA cleavage by the 10-23 DNA enzyme. Methods Enzymol. 2001, 341,
503-517.
36. Faulhammer, D.; Famulok, M. The Ca2+
ion as a cofactor for a novel RNA-cleaving
deoxyribozyme. Angew. Chem. Int. Ed. 1996, 35, 2837-2841.
37. Geyer, C. R.; Sen, D. Evidence for the metal-cofactor independence of an RNA
phosphodiester-cleaving DNA enzyme. Chem. Biol. 1997, 4, 579-593.
38. Li, J.; Zheng, W.; Kwon, A. H.; Lu, Y. In vitro selection and characterization of a highly
efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000, 28,
481-488.
39. Cruz, R. P.; Withers, J. B.; Li, Y. Dinucleotide junction cleavage versatility of 8-17
deoxyribozyme. Chem. Biol. 2004, 11, 57-67.
40. Li, J.; Lu, Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J.
Am. Chem. Soc. 2000, 122, 10466-10467.
41. Liu, J.; Lu, Y. Improving fluorescent DNAzyme biosensors by combining inter- and
intramolecular quenchers. Anal. Chem. 2003, 75, 6666-6672.
42. Liu, J.; Lu, Y. A colorimetric lead biosensor using DNAzyme-directed assembly of gold
nanoparticles. J. Am. Chem. Soc. 2003, 125, 6642-6643.

166
43. Liu, J.; Lu, Y. Colorimetric biosensors based on DNAzyme-assembled gold
nanoparticles. J. Fluoresc. 2004, 14, 343-354.
44. Liu, J.; Lu, Y. Accelerated color change of gold nanoparticles assembled by DNAzymes
for simple and fast colorimetric Pb2+
detection. J. Am. Chem. Soc. 2004, 126, 12298-
12305.
45. Swearingen, C. B.; Wernette, D. P.; Cropek, D. M.; Lu, Y.; Sweedler, J. V.; Bohn, P. W.
Immobilization of a catalytic DNA molecular beacon on Au for Pb(II) detection. Anal.
Chem. 2005, 77, 442-448.
46. Wernette, D. P.; Swearingen, C. B.; Cropek, D. M.; Lu, Y.; Sweedler, J. V.; Bohn, P. W.
Incorporation of a DNAzyme into Au-coated nanocapillary array membranes with an
internal standard for Pb(ii) sensing. Analyst 2006, 131, 41-47.
47. Wernette, D. P.; Mead, C.; Bohn, P. W.; Lu, Y. Surface immobilization of catalytic
beacons based on ratiometric fluorescent DNAzyme sensors: a systematic study.
Langmuir 2007, 23, 9513-9521.
48. Dalavoy, T. S.; Wernette, D. P.; Gong, M.; Sweedler, J. V.; Lu, Y.; Flachsbart, B. R.;
Shannon, M. A.; Bohn, P. W.; Cropek, D. M. Immobilization of DNAzyme catalytic
beacons on PMMA for Pb2+
detection. Lab Chip 2008, 8, 786-793.
49. Nagraj, N.; Liu, J.; Sterling, S.; Wu, J.; Lu, Y. DNAzyme catalytic beacon sensors that
resist temperature-dependent variations. Chem. Commun. 2009, 4103-4105.
50. Liu, J.; Brown, A. K.; Meng, X.; Cropek, D. M.; Istok, J. D.; Watson, D. B.; Lu, Y. A
catalytic beacon sensor for uranium with parts-per-trillion sensitivity and millionfold
selectivity. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 2056-2061.
51. Carmi, N.; Breaker, R. R. Characterization of a DNA-cleaving deoxyribozyme. Bioorg.
Med. Chem. 2001, 9, 2589-2600.
52. Bruesehoff, P. J.; Li, J.; Augustine, A. J., 3rd; Lu, Y. Improving metal ion specificity
during in vitro selection of catalytic DNA. Comb. Chem. High Throughput Screen. 2002,
5, 327-335.
53. Liu, J.; Lu, Y. A DNAzyme catalytic beacon sensor for paramagnetic Cu2+
ions in
aqueous solution with high sensitivity and selectivity. J. Am. Chem. Soc. 2007, 129,
9838-9839.
54. Wang, Z.; Lu, Y. Functional DNA directed assembly of nanomaterials for biosensing. J.
Mater. Chem. 2009, 19, 1788-1798.
55. Liu, J.; Cao, Z.; Lu, Y. Functional nucleic acid sensors. Chem. Rev. 2009, 109, 1948-
1998.
56. Lu, Y.; Liu, J. Functional DNA nanotechnology: emerging applications of DNAzymes
and aptamers. Curr. Opin. Biotechnol. 2006, 17, 580-588.
57. Clarkson, T. W. Human toxicology of mercury. J. Trace Elem. Exp. Med. 1998, 11, 303-
317.
58. Clarkson, T. W.; Magos, L.; Myers, G. J. The toxicology of mercury - current exposures
and clinical manifestations. N. Engl. J. Med. 2003, 349, 1731-1737.
59. Harris, H. H.; Pickering, I. J.; George, G. N. The chemical form of mercury in fish.
Science 2003, 301, 1203.

167
60. Tchounwou, P. B.; Ayensu, W. K.; Ninashvili, N.; Sutton, D. Environmental exposure to
mercury and its toxicopathologic implications for public health. Environ. Toxicol. 2003,
18, 149-175.
61. Ochi, T.; Ohsawa, M. Induction of 6-thioguanine-resistant mutants and single-strand
scission of DNA by cadmium chloride in cultured Chinese hamster cells. Mutat. Res.
1983, 111, 69-78.
62. Ochi, T.; Takayanagi, M.; Ohsawa, M. Cadmium-induced DNA single-strand scissions
and their repair in cultured Chinese hamster cells. Toxicol. Lett. 1983, 18, 177-183.
63. Nordberg, G. Cadmium and human health: A perspective based on recent studies in
China. J. Trace Elem. Exp. Med. 2003, 16, 307-319.
64. Satarug, S.; Baker, J. R.; Urbenjapol, S.; Haswell-Elkins, M.; Reilly, P. E.; Williams, D.
J.; Moore, M. R. A global perspective on cadmium pollution and toxicity in non-
occupationally exposed population. Toxicol. Lett. 2003, 137, 65-83.
65. Jarup, L.; Akesson, A. Current status of cadmium as an environmental health problem.
Toxicol. Appl. Pharmacol. 2009, 238, 201-208.
66. Ochoa, P. A.; Rodriguez-Tapiador, M. I.; Alexandre, S. S.; Pastor, C.; Zamora, F.
Structural models for the interaction of Cd(II) with DNA: trans-[Cd(9-RGH-
N7)2(H2O)4]2+
. J. Inorg. Biochem. 2005, 99, 1540-1547.
67. Buchko, G. W.; Hess, N. J.; Kennedy, M. A. Cadmium mutagenicity and human
nucleotide excision repair protein XPA: CD, EXAFS and (1)H/(15)N-NMR
spectroscopic studies on the zinc(II)- and cadmium(II)-associated minimal DNA-binding
domain (M98-F219). Carcinogenesis 2000, 21, 1051-1057.
68. Zivarts, M.; Liu, Y.; Breaker, R. R. Engineered allosteric ribozymes that respond to
specific divalent metal ions. Nucleic Acids Res. 2005, 33, 622-631.
69. Wang, S.; Karbstein, K.; Peracchi, A.; Beigelman, L.; Herschlag, D. Identification of the
hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious
effect of a cleavage site phosphorothioate. Biochemistry 1999, 38, 14363-14378.
70. Maderia, M.; Hunsicker, L. M.; DeRose, V. J. Metal-phosphate interactions in the
hammerhead ribozyme observed by 31P NMR and phosphorothioate substitutions.
Biochemistry 2000, 39, 12113-12120.
71. Suzumura, K.; Takagi, Y.; Orita, M.; Taira, K. NMR-based reappraisal of the
coordination of a metal ion at the pro-Rp oxygen of the A9/G10.1 site in a hammerhead
ribozyme. J. Am. Chem. Soc. 2004, 126, 15504-15511.
72. Forconi, M.; Lee, J.; Lee, J. K.; Piccirilli, J. A.; Herschlag, D. Functional identification of
ligands for a catalytic metal ion in group I introns. Biochemistry 2008, 47, 6883-6894.
73. Osborne, E. M.; Ward, W. L.; Ruehle, M. Z.; DeRose, V. J. The identity of the
nucleophile substitution may influence metal interactions with the cleavage site of the
minimal hammerhead ribozyme. Biochemistry 2009, 48, 10654-10664.
74. Hu, Q.; Yang, G.; Zhao, Y.; Yin, J. Determination of copper, nickel, cobalt, silver, lead,
cadmium, and mercury ions in water by solid-phase extraction and the RP-HPLC with
UV-Vis detection. Anal. Bioanal. Chem. 2003, 375, 831-835.
75. Paull, B.; Twohill, E.; Bashir, W. Determination of trace cadmium in environmental
water samples using ion-interaction reversed-phase liquid chromatography with
fluorescence detection. J. Chromatogr. A 2000, 877, 123-132.

168
76. Bravo-Sanchez, L. R.; de la Riva, B. S.; Costa-Fernandez, J. M.; Pereiro, R.; Sanz-Medel,
A. Determination of lead and mercury in sea water by preconcentration in a flow
injection system followed by atomic absorption spectrometry detection. Talanta 2001,
55, 1071-1078.
77. Matusiewicz, H.; Kopras, M.; Sturgeon, R. E. Determination of cadmium in
environmental samples by hydride generation with in situ concentration and atomic
absorption detection. Analyst 1997, 122, 331-336.
78. Sunaga, H.; Kobayashi, E.; Shimojo, N.; Suzuki, K. T. Detection of sulfur-containing
compounds in control and cadmium-exposed rat organs by high-performance liquid
chromatography--vacuum-ultraviolet inductively coupled plasma-atomic emission
spectrometry (HPLC-ICP). Anal. Biochem. 1987, 160, 160-168.
79. Krupp, E. M.; Milne, B. F.; Mestrot, A.; Meharg, A. A.; Feldmann, J. Investigation into
mercury bound to biothiols: structural identification using ESI-ion-trap MS and
introduction of a method for their HPLC separation with simultaneous detection by ICP-
MS and ESI-MS. Anal. Bioanal. Chem. 2008, 390, 1753-1764.
80. Ono, A.; Togashi, H. Highly selective oligonucleotide-based sensor for mercury(II) in
aqueous solutions. Angew. Chem. Int. Ed. 2004, 43, 4300-4302.
81. Wang, Z.; Heon Lee, J.; Lu, Y. Highly sensitive "turn-on" fluorescent sensor for Hg2+
in
aqueous solution based on structure-switching DNA. Chem. Commun. 2008, 6005-6007.
82. Lee, J. S.; Han, M. S.; Mirkin, C. A. Colorimetric detection of mercuric ion (Hg2+
) in
aqueous media using DNA-functionalized gold nanoparticles. Angew. Chem. Int. Ed.
2007, 46, 4093-4096.
83. Liu, J.; Lu, Y. Rational design of "turn-on" allosteric DNAzyme catalytic beacons for
aqueous mercury ions with ultrahigh sensitivity and selectivity. Angew. Chem. Int. Ed.
2007, 46, 7587-7590.
84. Mazumdar, D.; Liu, J.; Lu, Y., Functional nucleic acid-directed assembly of
nanomaterials and their applications as colorimetric and fluorescent sensors for trace
contaminants in water. In Nanotechnology Applications for Clean Water, 2009; pp 427-
446.
85. Kazakov, S. A.; Hecht, S. M.; King, R. B., Nucleic Acid-Metal Ion Interactions. In
Encyclopedia of Inorganic Chemistry, Wiley: Chichester, 2005; Vol. VI, p 3690.
86. Onyido, I.; Norris, A. R.; Buncel, E. Biomolecule--mercury interactions: modalities of
DNA base--mercury binding mechanisms. Remediation strategies. Chem. Rev. 2004, 104,
5911-5929.
87. Hollenstein, M.; Hipolito, C.; Lam, C.; Dietrich, D.; Perrin, D. M. A highly selective
DNAzyme sensor for mercuric ions. Angew. Chem. Int. Ed. 2008, 47, 4346-4350.
88. Sponer, J.; Sabat, M.; Burda, J. V.; Leszczynski, J.; Hobza, P. Interaction of the Adenine-
Thymine Watson-Crick and Adenine-Adenine Reverse-Hoogsteen DNA Base Pairs with
Hydrated Group IIa (Mg2+, Ca2+, Sr2+, Ba2+) and IIb (Zn2+, Cd2+, Hg2+) Metal
Cations: Absence of the Base Pair Stabilization by Metal-Induced Polarization Effects. J.
Phys. Chem. B 1999, 103, 2528-2534.
89. Dale, R. M.; Martin, E.; Livingston, D. C.; Ward, D. C. Direct covalent mercuration of
nucleotides and polynucleotides. Biochemistry 1975, 14, 2447-2457.

169
90. Dale, R. M.; Ward, D. C. Mercurated polynucleotides: new probes for hybridization and
selective polymer fractionation. Biochemistry 1975, 14, 2458-2469.

170
6 In vitro selection of a DNA Aptamer specific for Endotoxins
Notes and Acknowledgements: The work presented in this chapter was done in
collaboration with Jennifer Deluhery from the Illinois Sustainable Technology Center. I would
like to thank Tian Lan for helpful discussions and suggestions during the course of the selection
and Seyed-Fakhreddin Torabi who will continue to work on this project in the future. I would
also like to thank the Illinois Sustainable Technology Center for their funding.
6.1 Introduction
6.1.1 Endotoxins and their impact on health
Endotoxins are potentially toxic natural compounds that are found in the outer cell
membrane of various gram-negative bacteria. These compounds are historically also known as
LPS (LipoPolySaccharides) and consist of a polysaccharide chain and a lipid moiety known as
Lipid A (Figure 6.1) which anchors the entire molecule in the cell wall of the bacteria.1 In fact,
endotoxins are in large part responsible for the dramatic clinical manifestations of infections with
pathogenic gram-negative bacteria, such as Neisseria meningitidis, the pathogen that causes
meningitis. These toxins are released by the bacteria either during growth or upon cell lysis and
have been found to produce a number of physiological effects including decreased spirometry,
pulmonary inflammation, fever, septic shock and hyperimmune responses.2-4
Humans can be
exposed to endotoxins either through ingestion and inhalation of gram negative bacteria in
medical environments or through intravenous injections, medical implants, dialysis and surgery.
These toxins are also common in petroleum products and in metal fabrication environments
wherein they are transported in the form of aerosols. Many ailments including skin rashes,

171
malaise, fevers and respiratory distress have been attributed to endotoxin exposure from
petroleum products.5 With proposed health-based occupational exposure limits of 50 ng/mL and
increased symptoms at 50 ng/mL exposure levels, workers in above mentioned environments are
at high risk of developing fever as well as respiratory and skin related ailments.6 Chronic
exposure would also greatly increase the risk and severity of these symptoms. Hence, detection
of endotoxins in medical as well several industrial environments is essential.
O-Specific ChainOuter
Core
Inner
CoreLipid A
Polysaccharide Lipid
a)
CO2-
CO2-
CO2-
Kdo
Kdo
Kdo
GluN heptose
D-
glucoseD-
galactoseD-
glucose
D-
galactose
heptose heptose
PO4-
PO4-
PO4-PO
4-
D-
GluN
D-
GluN
Polysaccharides Lipid
b)
Figure 6.1 a) General architecture of endotoxins b) Structure of endotoxins; wherein, GluN:
acetylglucosamine; Etn: ethanolamine; Kdo (or KDO): 2-keto-3-deoxyoctulosonic acid.
6.1.2 Current methods of detection and need for improved detection methods
The current USP (United States Pharmacopeia) and FDA (Food and Drug
Administration) standard is the Limulus Amebocyte Lysate (LAL) gel-clot method.7 LAL is an

172
aqueous extract of blood cells from the horseshoe crab, which causes gel formation upon
addition to a sample containing endotoxin. Additional techniques that include kinetic and
endpoint chromogenic and turbidimetric methods are also based on the LAL. Each method has
different sensitivity ranges and interferences and therefore applicability is extended to various
situations. Owing to the diversity and differences across the population of horseshoe crabs,
different sensitivity levels of LAL are available, thereby allowing quantitative analysis of a
sample rather than a simple pass-fail method. The turbidimetric method utilizes the same
reaction that occurs in the gel-clot method; however, plates and tube readers are now used to
measure the turbidity of the sample. The turbidity of the solution increases upon clotting of the
sample. A measurement of the amount of endotoxin present in the sample is possible by
measuring the rate of the increase in turbidity. For the chromogenic methods, a modification to
the basic LAL extract has been made, thereby allowing for the release of a chromophore in the
presence of endotoxin. The rate of change in optical density of the chromophore is an indication
of the amount of endotoxin in the sample.7 The current testing limits utilizing materials available
from the Associates of Cape Cod Inc. are 0.03 ng/mL, 0.001 ng/mL and 0.005 ng/mL for the gel-
clot, turbidimetric and chromogenic methods respectively.8, 9
The main drawback of these
methods in an industrial environment is the large number of contaminants and interferences
present.9 In metalworking fluids, high pH, high salt content and surfactants are common. All of
these factors interfere with the performance of the LAL test. In addition to sample matrix issues,
there are other problems associated with these methods. Both of the kinetics-based chromogenic
and turbidimetric methods require the use of expensive equipments for detection. For example, a
microplate reader or a tube reader is needed for measurements. Trained technicians and analysts

173
would be required for data analysis and processing. In gel-clot, the gel in a positive result can
easily be broken by poor sample handling. Pretreatment of samples before actual endotoxin
detection using LAL is a time consuming prerequisite to overcome assay inhibition or
enhancement. The approach used to overcoming the inhibition/enhancement is dilution of the
sample, reducing the ability to detect low concentrations of LPS in sample with high levels of
contaminants.9 Gas chromatography (GC) has also been used for endotoxin detection.
10 This
method focuses on the detection of 3-hydroxy fatty acids for the measurement of endotoxins in a
sample. One of the advantages of using GC is that, identification of the type of bacteria present
in the sample is possible by analysis of the types of fatty acids present.11
However, this method
again requires the use of expensive instrumentation as well as experienced and qualified
technicians for data processing and analysis.
6.1.3 Aptamers and their use as biosensors
The discovery that nucleic acids were more than just carriers of genetic information and
could, in fact, bind molecular targets and even perform catalysis in the presence of metal
cofactors, led to the idea of using these molecules as biosensors and even as drug delivery
agents.12-17
DNA and RNA molecules that selectively bound to molecular targets were
discovered in the early 1990s and were given the name “aptamers”; which stemmed from the
Latin word, aptus, the past participle of „to fit‟.18
Aptamers can be isolated in the laboratory
through an in vitro selection procedure also known as SELEX (Systematic Evolution of Ligands
by Exponential Enrichment),19
which is described in detail in chapters 1 and 5. Typically every
round of selection takes an average of two days to complete (total number of rounds of selection
vary from 5-18) and aptamers with high selectivity and sensitivity can be obtained through the

174
introduction of stringency during the selection procedure.20
Aptamers that bind a wide variety of
analytes with high affinity and selectivity have since been discovered.21
The range of analytes
varies from small organic molecules (e.g., cocaine, adenosine) and antibiotics (e.g., vancomycin,
kanamycin) to complex and large entities, such as viral particles, prion proteins and even whole
cells.22-25
Based on the experience, results and success obtained with such a wide range of
analytes, theoretically, aptamers can be generated to bind to essentially, any target molecule.
6.1.4 Advantages of using aptamers for detection
6.1.4.1 The use of a combinatorial approach
One of the main advantages of using aptamers is the fact that they can be isolated
through in vitro selection for endotoxins with both high affinity and selectivity. Selection is
carried out in the absence of animals and is both time and cost effective relative to the antibody
approach. The binding affinities between many aptamers and their targets are very high. For
instance, the Kd value obtained for the binding between a protein, namely, platelet derived
growth factor β-chain and its DNA aptamer was found to be 0.1 nM,26, 27
while that for
ethanolamine (which is the smallest molecule for which an aptamer has been obtained) and its
DNA aptamer was found to be 6 nM.28
6.1.4.2 Stability of DNA
Under physiological conditions, DNA is nearly 1,000-fold more stable against hydrolysis
than proteins (such as antibodies) and nearly 100,000-fold more stable than RNA. Unlike
antibodies, DNA aptamers can typically be denatured and renatured many times without losing
their binding. Also, they can be stored under denaturing conditions, and their catalytic activity is

175
restored when permissive incubation conditions are re-established. Due to these characteristics,
DNA aptamers have a much longer shelf life than antibodies or RNA aptamers and are
particularly suitable for field applications.13
6.1.4.3 Capability for practical sensing applications
Aptamers have become „ideal‟ diagnostic agents owing to their high affinity and
selectivity.13, 29-32
Their sensing applications are predominantly based on target induced
conformational changes, which can be detected by methods that include fluorescence
quenching,33
excimer formation,34
electrochemical signal generation35
and a micro-fabricated
cantilever36
technique. Another detection method makes use of the visible color change induced
by aptamer-mediated disassembly of DNA-functionalized gold nanoparticles.37
This principle
has also been used for the creation of a lateral flow detection method for the analysis of
adenosine and cocaine on a dipstick.38, 39
6.1.5 Research goals
The goal of this project is to use in vitro selection to isolate DNA aptamers that can bind
endotoxins with high affinity and selectivity. The long-term goal of this project would be to
convert the aptamers obtained into sensors for endotoxin detection in standard conditions of
metalworking industrial fluids, for on-site and real-time detection.
This chapter describes our efforts in designing a strategy for the in vitro selection of an
aptamer specific for endotoxins and generalizing the type of endotoxins detected through a
parallel selection with KDO (2-keto-3-deoxyoctulosonic acid) (Figure 6.6), a sugar unit that is
common to most endotoxins produced by different strains of bacteria. We are currently at a

176
stage wherein the DNA sequences isolated show activity toward both of our targets of choice.
The next step of cloning and sequencing the pool will be carried out in the weeks following the
submission of this dissertation and hence is beyond the scope of discussion of the current
chapter. The biochemical characterizations and optimization of the DNA aptamers isolated will
help identify specific and conserved sequences responsible for activity.
6.2 Materials and methods
6.2.1 Materials
The selected endotoxin target, Control Standard Endotoxin (or CSE) was obtained from
the Associates of Cape Cod Inc. while the endotoxins used for other studies was obtained from
Fisher Scientific. All DNA sequences (Table 6.1) were obtained standard desalted from
Integrated DNA technologies Inc. (Coralville, IA) and the template was purified by 10% PAGE.
Buffers were prepared using ultrapure chemicals from Sigma-Aldrich. Platinum® Taq DNA
polymerase (5 U/μL) and PCR grade dNTPs (10 mM mix) were purchased from Invitrogen. [-
32P]-dATP and [-
32P]-ATP (250 μCi) were purchased from Perkin Elmer.
6.2.2 Generation of the initial pool
The initial pool used for selection was checked for unwanted secondary structure and
mispriming using the oligo-analyzer program (http://scitools.idtdna.com/analyzer/oligocalc.asp)
from Integrated DNA Technologies and then generated using PCR (Figure 6.2).

177
Figure 6.2 Predicted secondary structure of the random pool used for in vitro selection.
Table 6.1 Sequences of DNA oligonucleotides for in vitro selection and aptamers previously
isolated for LPS.
Name Sequences (from 5' 3')
Capture-
BDNA Biotin-TACCGCAAAAAAAAA-GCA TATGA GCTAC CGTAG
Random
Pool CCTGCCACGCTCCGCAAGCTTN15CTACGGTAGCTCATATGCN25TAAGCTTGGCAC
CCGCATCGT P1 GCG GAG CGT GGC AGG
P2 ACG ATG CGG GTG CCA AGC TTrA
P3 CCTGCCACGCTCCG
19LPS GGCGTCCACTCCCAGCCGCTCACTAGTTTCTGCGTGGGTGA
100
marker ATA GAC TAT GTG CGA TAG TAC GTG TAC GAT ATC GAT CGG ATT ATA GCT
AGC TGG ACT TAC GAT CGG TTA GCT ATA ATC GCT ACG TAC GTA ACT GAG
ACT A 79
marker ATA GTG ATA TCG CCT AGA CTA CGT AGC TAC ATT CGG CTA GCT AAG AGC
TCG TAG CTA TAC GCA TAT CGT GCT AGC TCC T

178
Different PCR conditions were tested including variation of the concentrations of
template and primers used as well as the number of amplification rounds. Based on the results
obtained from these initial optimizations the following PCR conditions were chosen. Ten PCR
reactions were prepared by mixing 100 µL of the 10 × PCR buffer, 100 µL of 2 mM dNTPs, 10
µL of the 18 µM template DNA, 75 µL each of 10 µM P2 and P3, 20 µL of 50 mM MgCl2, and
600 µL of Millipore H2O. This mixture was then divided into ten tubes and in one of the tubes 2
µL of [-32
P]-dATP, the final volume in each tube being 99 µL. The PCR reactions were
initiated by the addition of 1 µL of Platinum®
Taq DNA polymerase to each tube. The PCR
conditions used are shown in Table 6.2. The PCR products obtained were then hydrolyzed with
10 µL of 6 M NaOH by heating at 90 °C for 10 minutes followed by neutralization with 10 µL of
6 M HCl at room temperature and purified by 10 % PAGE against markers containing 100-mer
DNA, corresponding to the uncleaved pool and 79-mer DNA corresponding to the cleaved pool.
The gel was then exposed to an X-ray film for a few minutes and the film was developed using
an X-ray developer to reveal the location of the 32
P-labeled DNA bands. The upper band
(corresponding to the uncleaved pool) was cut out and was crushed and soaked in ~ 1.0 mL soak
solution (10 mM Tris pH 7.5, 1 mM EDTA, 300 mM NaCl, pH 7.5). This was placed on a rocker
for ~15 h, following which the gel bits were removed from solution using a Biorad Polyprep
column. DNA was precipitated by adding 10 % volume of 3 M sodium acetate at a pH of 5.3,
250 % volume of 100 % ethanol and chilling in the -80 C freezer for at least ~1.5 h. This was
followed by centrifugation (SS-34 rotor) at 15,000 rpm for 30 min at -4 C, which resulted in a
white DNA pellet at the bottom of the tubes. The supernatant was discarded and the DNA was

179
lyophilized overnight for complete drying. This purified DNA pool was used for the initial round
of selection.
Table 6.2 PCR conditions used for amplification of random pool after every round of selection.
Reagent Initial
Concentration
Target
Concentration
Amount Added
(L)
Millipore DI water 51
dNTP 2 mM 200 µM 10
PCR Buffer 10 × 1 × 10
MgCl2 50 mM 1 mM 2
Primer 2rna 10 µM 5 pmol 7.5
Primer 3 10 µM 5 pmol 7.5
DNA Template 18 µM 20 pmol 1
Platinum Taq DNA 1
6.2.3 Optimization of selection conditions and in vitro selection
The control standard endotoxin (CSE) used for selection was initially dissolved in
different buffers at pH values ranging from 6.0 (in MES) to 7.5( in HEPES) ; containing varying
concentrations of divalent (Mg2+
) and monovalent metal (K+ and Na
+) ions. This was done to
ensure that no precipitation was seen since it is known that high concentrations of metal ions
may cause the CSE to precipitate. The 1 × selection buffer (SB) that was finally used for the
selection based on our tests for precipitation with the endotoxin was: 50 mM HEPES-Na at pH
7.35 containing 50 mM NaCl, 5 mM KCl and 5 mM MgCl2. A stock of 5 × concentration was
always made to minimize any errors caused by weighing and then diluted to the appropriate 1 ×
concentration. Gel-clot assays were also carried out using the afore-mentioned LAL method at
the facilities of the Illinois Sustainable Technology Center, to ensure that concentration of CSE
(1.25 µg/mL) used for selection was significant. An exact determination of the final
concentration in terms of molarity for endotoxins in general is a challenge since the exact

180
molecular weight of the endotoxins is usually not listed, rather the amount in a vial for sale, is
listed in terms of endotoxin units or EU. Therefore, the concentrations used for the selections are
expressed in units of µg/mL rather than molarity.
For the first round of selection, the purified DNA (after PCR, PAGE purification and
ethanol precipitation) was dissolved in 38 µL of 1 × SB. Then 30 µL of this DNA pool was
added to a mixture of 5 µL of 500 µM capture-BDNA, and 1.5 µL each of 500 µM P1 and P2
and denatured in a water bath at 85 °C. For every subsequent round of selection, the same
procedure was followed except that, 5 µL of 50 µM capture B-DNA and 1.5 µL each of 50 µM
P1 and P2 were used, since the population of unbound DNA reduced significantly after the initial
round of selection. The mixture was then annealed over a period of 1 hour by cooling the DNA
mixture in the water bath to room temperature. This DNA pool was subsequently loaded onto an
avidin column, ( ~300 µL bed volume; total volume loaded 600 µL) which was previously
washed with 3 mL of PBS buffer and equilibrated with 3 mL of the 1 × SB. The pool was then
incubated at room temperature for 1 hour and then washed with 5 mL of 1 × SB and collected as
10 x 500 µL fractions to remove the unbound DNA. After another hour, the column was again
washed with 1 mL of 1xSB and 2 x 500 µL fractions were collected to determine the
background. To this column, (final volume~500 µL) 10 µL of 125 µg/mL CSE corresponding to
a final concentration of 1.25 µg/mL was added and incubation was allowed to proceed for 1 hour
at room temperature. Elution was carried out with 2 mL of 1 × SB containing 1.94 µg/mL CSE,
and 4 x 500 µL fractions were collected. The radiation count of each fraction was determined by
liquid scintillation counting (LSC), for which, 100 µL of the sample was dissolved in 4.9 mL of
LSC fluid. The ratio of the radioactivity of DNA eluted by the target, specifically elution

181
fraction 1, since most of the DNA was eluted in this fraction, to that eluted by the selection
buffer alone i.e. the average of the background fractions 1 and 2, was defined as the switching
activity. After every round of selection, this switching activity was measured and this measure
was an indication of the selection progress after each round.
The selection procedure for the control selection with ATP and positive parallel with
KDO was identical to that described with CSE, except that the concentration of ATP for
selection was maintained at 500 mM while the concentration of KDO used for selection was
maintained to be the same as that for the CSE i.e. 1.25 µg/mL. The in vitro selection with CSE
was carried out for the first seven rounds after which a duplicate PCR round was carried out with
the round 7 elution fraction and 2 parallel selections with CSE and KDO were carried out
starting from round 8 under almost identical conditions simultaneously. The incubation time till
round 16 of selection for both CSE and KDO, and for ATP till round 11 was maintained at 1
hour. At round 17 of selection with CSE and KDO, the incubation time was lowered to 10 min to
increase stringency during the selection process.
6.2.4 Regeneration of the pool after selection
After each round of selection, 10 µL of solution from elution fraction 1 (with the highest
switching activity) was amplified by PCR. The PCR protocol used was identical to the
previously mentioned pool generation protocol. The PCR reaction mixture in this case contained
10 µL of elution fraction 1, 10 µL of 10 PCR buffer, 10 µL of 2 mM dNTPs, 7.5 µL each of
primers P2 and P3, 2 µL of 50 mM MgCl2 , 1 µL of [α-32
P] dATP and 1 µL of Platinum® Taq
DNA polymerase. The amplification product was then hydrolyzed using 10 µL of 6 M NaOH
and then neutralized with 10 µL of 6 M HCl and purified by 10% PAGE, followed by extraction

182
and ethanol precipitation as described in section 6.2.2. This purified DNA pool was used for
subsequent rounds of selection.
biotin avidin-coated bead
P1 P2
R
R
1. Incubation
2. Elution
PCR1. NaOH
2. PAGE
annealing
5'5'
5'5'
5'
5'5'
5'
5'5'
5'
5'
5'5'
5'
5'
5'
annealing 1
2
34
5
P1
P2
RP3
Capture BDNA Random regions of
pool
Figure 6.3 In vitro selection scheme of structure-switching aptamers. Figure has been adapted
and modified from Nutiu et al.40
6.3 Results and discussion
6.3.1 Design of DNA library
The design of the DNA library and combinatorial selection was based on the structure-
switching strategy originally developed by Nutiu and Li.40-43
The DNA library consisted of two
random regions, N15 and N25 respectively (Figure 6.3), which were flanked by 2 primer binding
arms and separated by a 16 base-pair fixed region. The central fixed region was designed to be
complementary to an antisense capture oligonucleotide (referred to as „capture BDNA‟) that was

183
biotinylated at its 5' end and this was optimized to be 18-bases long for minimum background
(data not shown). Hybridization of this oligonucleotide to the DNA library would then allow
indirect immobilization of the latter onto an avidin column. Two other short oligonucleotides, P1
and P2 that were complementary to the two flanking primer binding regions, were introduced to
prevent any tertiary interactions between the two arms. The immobilized DNA assembly was
then incubated with the target toxin, followed by elution. Only those DNA sequences that were
able to bind to the toxin and disrupt the capture BDNA base-pairing would elute from the
column. These eluted DNA molecules were then collected, PCR amplified. During PCR, the
reverse primer (P3) was designed to contain a ribonucleotide (R) at its 3' end, so that the
chimeric antisense strand generated during PCR amplification was prone to NaOH cleavage. The
two strands could therefore be separated by PAGE (Poly Acrylamide Gel Electrophoresis)
because of the difference in length. The upper, uncleaved band containing the desired DNA
sequences was extracted from the gel and purified (Figure 6.4). The purified DNA was again
incubated with the capture oligonucleotide, P1 and P2 as well as the avidin beads for further
rounds of selection.

184
Figure 6.4 Representative X-ray film image exposed to 10 % gel after PCR amplification
followed by hydrolysis and neutralization. The upper uncleaved pools, corresponding to the 100-
mer marker in the central lane, were extracted and used for subsequent selection rounds.
This strategy avoided the immobilization of the target onto a solid support which was
particularly useful for the selection of endotoxin since it was a lipopolysaccharide molecule with
several functional moieties and immobilizing this target onto a column would prove to be
challenging. Another advantage was that the ATP aptamers isolated using this method were
directly converted into fluorescent sensors, without significant changes in the overall design
strategy which was advantageous since the long-term goal of the current project was to obtain
sensors based on the DNA aptamers isolated.
6.3.2 Selection strategy and parallel selections
A control selection with ATP was initially carried out to primarily allow the collaborator
to get familiar with DNA handling, radiation, gel-based purification and PCR and afforded the
author to get familiar with the specific structure-switching method of selection and optimize
selection conditions. A parallel selection with endotoxins was then started after two rounds of
selection with ATP. At round 8 of selection with the CSE, the pool was again regenerated using
PCR and an additional selection with KDO, which was a positive control, was performed. The
100-mer
79-mer

185
selection was continued with ATP for about 11 rounds, and for the CSE for about 16 rounds after
which it was discovered that there was an error with the pH of the selection buffer used. Hence,
we went back to round 6 of the selection with the CSE, at which the pH of the selection buffer
was definitely at 7.3 and re-started selection. The control selection with ATP was however not
pursued any further, since at this stage, both members involved with the project were sufficiently
familiar with the selection methodology. Moreover, parallel selections with KDO (the more
conserved part of the CSE across different strains of bacteria) and the CSE were being
simultaneously pursued.
6.3.2.1 Selection with ATP as a control
The in vitro selection process was started using ATP as a control due to the fact that it is
a relatively small molecule, for which numerous aptamers have been isolated in the past, using
both the immobilization as well as the structure-switching method.40, 44-46
The initial optimization
conditions for PCR as well as the length of the capture oligonucleotides, also known as capture
BDNA was carried out prior to the ATP control selection. We were able to complete 11 rounds
of selection, using 500 mM of ATP as the target concentration and an incubation of one hour.
After approximately 11 rounds, no increase in the switching activity was obtained and this was in
contrast to the results obtained by Nutiu and Li40
as well as a control selection carried out by
Tian Lan in the Lu lab (unpublished results). It was then discovered during the process of
trouble-shooting and questioning, that the pH of the selection buffer after round 11 was not
maintained at 7.3. Hence, all the buffers were remade and the focus was maintained on the
selection of the CSE and the parallel KDO selection as discussed in the next section. The control
ATP selection was discontinued at round 11 (Figure 6.5).

186
6.3.2.2 Selection with CSE (control standard endotoxin) and KDO
KDO (2-keto-3-deoxyoctulosonic acid) shown in Figure 6.6 is unique in the sense that it
is expressed on most of the different types of CSE produced by various strains of gram negative
bacteria. A positive selection with KDO was also carried out to compare as well as ensure that
the sequence obtained upon selection with CSE could be used against a wide number of gram-
negative bacterial strains rather than just the specific strain(from E.coli) that we used for
selection.
Figure 6.5 Progression of in vitro selection of DNA pool with ATP. The pH of the selection
buffer used in this selection from round 6-11 was found to be higher than the expected 7.5.
Hence, the selection after round 11 was discontinued.

187
Figure 6.6 Structure of KDO (2-keto-3-deoxyoctulosonic acid)
Figure 6.7 Progression of in vitro selection of DNA pool with CSE or control standard
endotoxin and KDO. The pH of the selection buffer used in this selection from round 6-16 (CSE
and KDO) was found to be higher than the expected 7.5. Hence, the selection was restarted from
round 6.
The concentration of both CSE and KDO used for the selection was kept at 1.25 µg/mL
while the elution contained 1.93 µg/mL, to ensure that switching into the solution took place.

188
The incubation time for the first 16 rounds used was one hour. As mentioned in the previous
section, during trouble-shooting of the control selection with ATP, it was discovered that the pH
of the selection buffer was not maintained at pH 7.3 (Figure 6.7). Hence, the selection was
restarted from round 6 with both CSE and KDO using the above mentioned selection conditions,
however, with the right selection buffer at a pH of 7.3. At round 16 of the selection with CSE, an
increase in the switching activity from about 1 to ~ 2.5 was obtained, while that with KDO was
found to be ~ 4. To test the sensitivity of the pool, the incubation time at round 17 was lowered
to 10 min. This increased stringency in round 17 lowered the activity of the KDO to about 1.5,
while the activity with CSE was maintained at ~2.4. Hence, the sensitivity of the DNA pool with
KDO on the other hand, shows a significant drop with the increased stringency in the pool
suggesting therefore that further selection with lowered KDO incubation times need to be carried
out to obtain a more sensitive DNA population. The progress of selection and the comparison of
the switching activity of KDO and the CSE are shown in Figure 6.8.

189
Figure 6.8 The selection progress and comparison of the switching activity of CSE (control
standard endotoxin) and KDO in 50 mM HEPES-Na at pH 7.35 containing 50 mM NaCl, 5 mM
KCl and 5 mM MgCl2.
6.3.2.3 Binding assays to evaluate LPS (Lipopolysaccharide) aptamers discovered
previously
During the course of the CSE selection, a paper by Chen et al.47
was published which
describes the isolation of DNA aptamers that showed binding towards LPS. Their end goal was
to isolate aptamers that could be used for the treatment of sepsis caused by LPS (or endotoxins).
These aptamers were isolated through SELEX based on the nitro-cellulose binding method and
the progress of selection was monitored by purely measuring the total number of LSC (liquid

190
scintillation counts) counts, CPM or counts per minute rather than a ratio of the counts of the
sample over the background. A control with BSA (Bovine Serum Albumin) was however carried
out at every round of selection. When aptamers isolated based on this method with the tightest
binding activity (18 LPS and 19 LPS) were injected into mice, which were previously infected
with LPS. There was a decrease in their infection, thereby indicating that the aptamers probably
did bind to the LPS (Figure 6.9a-c).
Based on these results we ordered the identical aptamer sequences as described in their
paper and tried to carry out the nitrocellulose binding assays with our target CSE and KDO,
using the same buffer and target concentrations as mentioned. The background radiation in this
method was extremely high and hence, it was really hard to quantify. We therefore chose to use
the gel-shift assay to study binding between the aptamers and the CSE produced by E.Coli that
we used for our selections.

191
Figure 6.9 Relative binding rate of ssDNA and LPS. (A) CPM of residual radioactive nucleic
acids on nitrocellulose membrane after screening. (B) Electrophoregram of screened ssDNA. (C)
Aptamer 19 inhibited LPS-induced endotoxin shock in mice. C57BL/6 mice were injected with
LPS via caudal vein (1 mg/mouse, n = 20), or with LPS and 10 μmol aptamer 19 (n = 20) or with
LPS and 10 μmol aptamer 18 (n = 20). The mice surviving 12 h, 24 h, and 72 h after injection
were counted, and the survival rate was calculated (%). "Reprinted from Biophysical and
Biochemical Research Communications, 382, Wen, A. Q.; Yang, Q. W.; Li, J. C.; Lv, F. L.;
Zhong, Q.; Chen, C. Y, „A novel lipopolysaccharide-antagonizing aptamer protects mice against
endotoxemia‟, 140-144, 2009, with permission from Elsevier."
C

192
We initially studied the binding activity of the 19 LPS aptamer, which showed the
highest binding affinity toward LPS, using a control which was 10 pmol DNA in binding buffer
with either no target, or with our strain of LPS at a concentration of 100 ng/mL CSE and also
with KDO at a concentration of 100 ng/mL. All samples were incubated with or without target at
37 °C for 1 h and run on a native gel for 8.5 h. The concentrations of DNA and target used were
identical to that described in the paper. However, we did not see any binding with both our CSE
and the KDO as shown in comparison with a control containing only a DNA pool in the absence
of any target (Figure 6.10a). This could probably be due to either the fact that we used a different
strain of LPS or because we used a different method of studying the binding activity, i.e. the
native gel-shift assay rather than the nitrocellulose binding. Hence, the same strain of endotoxin
or LPS, namely the LPS (0111B4) was purchased from Sigma-Aldrich and used for studying the
binding. However, again no gel-shift was observed at the above-described concentrations;
wherein activity had been seen according to the paper (Figure 6.10b). Based on our results using
the gel-shift assay, we were unable to detect any binding between 19 LPS aptamer and the both
strains of LPS used as well as the KDO. This was surprising since the mice that were injected
with the 19 LPS aptamer as described by the authors did show a greater survival rate as
compared to the control. However, no additional method of binding of the DNA aptamer to the
target was demonstrated and it seems highly probable that aptamer binding to the injected LPS in
the mice, may have not been the reason for higher survival rate. Even if it were the case, no
additional supporting method was described in the current paper. Hence, it was decided that we
would continue to carry out selection of DNA aptamers specific for both CSE and KDO.
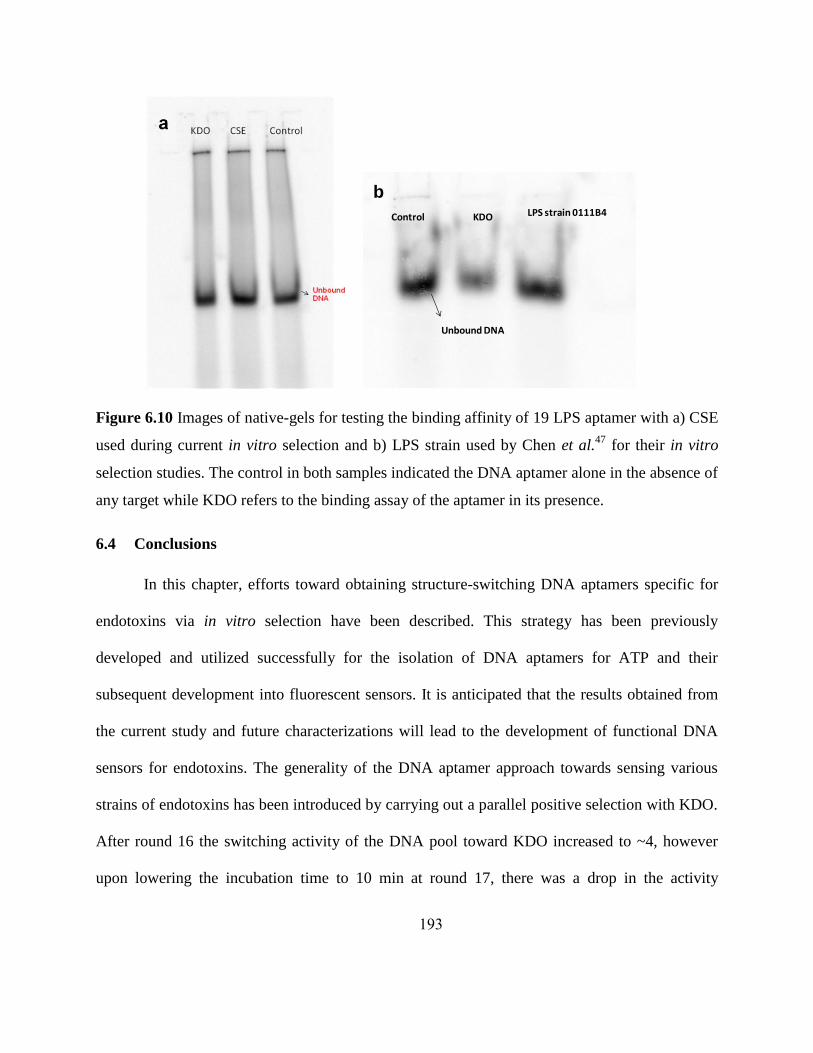
193
Figure 6.10 Images of native-gels for testing the binding affinity of 19 LPS aptamer with a) CSE
used during current in vitro selection and b) LPS strain used by Chen et al.47
for their in vitro
selection studies. The control in both samples indicated the DNA aptamer alone in the absence of
any target while KDO refers to the binding assay of the aptamer in its presence.
6.4 Conclusions
In this chapter, efforts toward obtaining structure-switching DNA aptamers specific for
endotoxins via in vitro selection have been described. This strategy has been previously
developed and utilized successfully for the isolation of DNA aptamers for ATP and their
subsequent development into fluorescent sensors. It is anticipated that the results obtained from
the current study and future characterizations will lead to the development of functional DNA
sensors for endotoxins. The generality of the DNA aptamer approach towards sensing various
strains of endotoxins has been introduced by carrying out a parallel positive selection with KDO.
After round 16 the switching activity of the DNA pool toward KDO increased to ~4, however
upon lowering the incubation time to 10 min at round 17, there was a drop in the activity
Control LPS strain 0111B4KDO
Unbound DNA
b

194
observed implying that a few further rounds of selection would have to be carried out to obtain
sequences with greater sensitivity. The switching activity of the CSE on the other hand has
remained steady at ~2.4 over rounds 16 and 17 of selection; thereby implying that the overall
sensitivity toward the target did not change with incubation time and hence binding was strong.
However, biochemical characterizations and binding assays would have to be carried out to
determine whether the activity of the aptamers was significant enough to be observed during
sensing applications.
Determination of the binding efficiency of a previously isolated LPS aptamer (19 LPS
aptamer) using native gel-shift assays has also been reported in this chapter and these results
indicate the absence of any significant binding with both CSE as well as the LPS strain used in
their studies. While the assays carried out in this current chapter are not conclusive to point that
the 19 LPS aptamer indeed does not show binding, it does stress the importance of carrying out
complementary binding assays in addition to that used during in vitro selection, for greater
applicability and validation purposes.
6.5 Future directions
Specificity of the DNA aptamer toward the CSE in the presence of other contaminants
and interfering species is an extremely important consideration while designing the in vitro
selection strategy. There exists a high possibility that a DNA aptamer that binds with one toxin
may cross-react with another contaminant that is closely related in structure. Hence, efforts are
currently underway to determine the efficiency of the random DNA pool after round 16, to detect
CSE in an industrial metal-networking fluid mixture. Previously, metal-ion selectivity of
DNAzymes has been eliminated through the introduction of a „negative selection‟ strategy,48

195
wherein, undesired analytes are used during a selection step in the place of the desired target.
The DNA aptamers that are capable of functioning in the presence of the contaminants are
discarded and only those specific toward the target (CSE) are retained and used for further
selection rounds. Negative selection will be carried out if the random pool suffers from
interferences in industrial fluids.
Cloning, sequencing and biochemical characterization of the pool (at the round that
highest activity is seen) will also be undertaken and complementary binding assays will also be
performed to ensure activity of the pool toward the target is not merely an artifact or accidental.
6.6 References
1. Raetz, C. R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002,
71, 635-700.
2. Petsch, D.; Beeskow, T. C.; Anspach, F. B.; Deckwer, W. D. Membrane adsorbers for
selective removal of bacterial endotoxin. J. Chromatogr. B Biomed. Sci. Appl. 1997, 693,
79-91.
3. Douglas, H.; Rossmoore, H. W.; Passman, F. J.; Rossmoore, L. A. Evaluation of
endotoxin-biocide interaction by the Limulus amebocyte lysate assay. Dev. Ind.
Microbiol. 1990, 31, 221-224.
4. Rossmoore, L. A.; Rossmoore, H. W. Metalworking fluid microbiology. Manuf. Eng.
Mater. Process. 1994, 41, 247-271.
5. Hill, E. C. The current state of the art on chemical and physical antimicrobial measures
[on metalworking fluids]. Schmierst. Metallbearb., Int. Kolloq.,3rd
1982, 2, 82.81-82.84.
6. Svrcek, C.; Smith, D. W. Cyanobacteria toxins and the current state of knowledge on
water treatment options: A review. J. Environ. Eng. Sci. 2004, 3, 155-185.
7. Iwanaga, S. The limulus clotting reaction. Curr. Opin. Immunol. 1993, 5, 74-82.
8. Obata, T.; Nomura, M.; Kase, Y.; Sasaki, H.; Shirasawa, Y. Early detection of the
Limulus amebocyte lysate reaction evoked by endotoxins. Anal. Biochem. 2008, 373,
281-286.
9. Bohrer, D.; Horner, R.; do Nascimento, P. C.; Adaime, M.; Pereira, M. E.; Martins, A. F.;
Hartz, S. A. Interference in the Limulus amebocyte lysate assay for endotoxin
determination in peritoneal dialysis fluids and concentrates for hemodialysis. J. Pharm.
Biomed. Anal. 2001, 26, 811-818.
10. Mielniczuk, Z.; Mielniczuk, E.; Larsson, L. Gas chromatography-mass spectrometry
methods for analysis of 2- and 3-hydroxylated fatty acids: application for endotoxin
measurement. J. Microbiol. Methods 1993, 17, 91-102.

196
11. Rybka, J.; Gamian, A. Determination of endotoxin by the measurement of the acetylated
methyl glycoside derivative of Kdo with gas-liquid chromatography-mass spectrometry.
J. Microbiol. Methods 2006, 64, 171-184.
12. Song, S.; Wang, L.; Li, J.; Fan, C.; Zhao, J. Aptamer-based biosensors. TrAC, Trends
Anal. Chem. 2008, 27, 108-117.
13. Liu, J.; Cao, Z.; Lu, Y. Functional nucleic acid sensors. Chem. Rev. 2009, 109, 1948-
1998.
14. Li, J.; Lu, Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J.
Am. Chem. Soc. 2000, 122, 10466-10467.
15. Levy-Nissenbaum, E.; Radovic-Moreno, A. F.; Wang, A. Z.; Langer, R.; Farokhzad, O.
C. Nanotechnology and aptamers: applications in drug delivery. Trends Biotechnol. 2008,
26, 442-449.
16. Potyrailo, R. A.; Conrad, R. C.; Ellington, A. D.; Hieftje, G. M. Adapting selected
nucleic acid ligands (aptamers) to biosensors. Anal. Chem. 1998, 70, 3419-3425.
17. Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G. C.; Cheng, J.; Lu, Y. Reversible cell-
specific drug delivery with aptamer-functionalized liposomes. Angew. Chem. Int. Ed.
2009, 48, 6494-6498.
18. Ellington, A. D.; Szostak, J. W. In vitro selection of RNA molecules that bind specific
ligands. Nature 1990, 346, 818-822.
19. Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA
ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505-510.
20. Hall, B.; Micheletti, J. M.; Satya, P.; Ogle, K.; Pollard, J.; Ellington, A. D. Design,
synthesis, and amplification of DNA pools for in vitro selection. Curr. Protoc. Mol. Biol.
2009, Chapter 24, Unit 24 22.
21. Mayer, G. The chemical biology of aptamers. Angew. Chem. Int. Ed. 2009, 48, 2672-
2689.
22. Klussmann, S., The Aptamer Handbook: Functional Oligonucleotides and Their
Applications. Wiley-VCH: Weinheim, 2006; p 490.
23. Famulok, M.; Hartig, J. S.; Mayer, G. Functional aptamers and aptazymes in
biotechnology, diagnostics, and therapy. Chem. Rev. 2007, 107, 3715-3743.
24. Ulrich, H.; Trujillo, C. A.; Nery, A. A.; Alves, J. M.; Majumder, P.; Resende, R. R.;
Martins, A. H. DNA and RNA aptamers: from tools for basic research towards
therapeutic applications. Comb. Chem. High Throughput Screen. 2006, 9, 619-632.
25. Majumder, P.; Gomes, K. N.; Ulrich, H. Aptamers: from bench side research towards
patented molecules with therapeutic applications. Expert Opin. Ther. Pat. 2009, 19,
1603-1613.
26. Jellinek, D.; Lynott, C. K.; Rifkin, D. B.; Janjic, N. High-affinity RNA ligands to basic
fibroblast growth factor inhibit receptor binding. Proc. Natl. Acad. Sci. U. S. A. 1993, 90,
11227-11231.
27. Jellinek, D.; Green, L. S.; Bell, C.; Janjic, N. Inhibition of receptor binding by high-
affinity RNA ligands to vascular endothelial growth factor. Biochemistry 1994, 33,
10450-10456.
28. Mann, D.; Reinemann, C.; Stoltenburg, R.; Strehlitz, B. In vitro selection of DNA
aptamers binding ethanolamine. Biochem. Biophys. Res. Commun. 2005, 338, 1928-1934.

197
29. Mok, W.; Li, Y. Recent Progress in Nucleic Acid Aptamer-Based Biosensors and
Bioassays. Sensors 2008, 8, 7050-7084.
30. Kim, Y.; Sohn, D.; Tan, W. Molecular beacons in biomedical detection and clinical
diagnosis. Int. J. Clin. Exp. Pathol. 2008, 1, 105-116.
31. Tan, L.; Li, Y.; Drake, T. J.; Moroz, L.; Wang, K.; Li, J.; Munteanu, A.; Chaoyong, J. Y.;
Martinez, K.; Tan, W. Molecular beacons for bioanalytical applications. Analyst 2005,
130, 1002-1005.
32. Lu, Y.; Liu, J. Functional DNA nanotechnology: emerging applications of DNAzymes
and aptamers. Curr. Opin. Biotechnol. 2006, 17, 580-588.
33. Cao, Z.; Suljak, S. W.; Tan, W. Molecular beacon aptamers for protein monitoring in
real-time and in homogeneous solutions. Current Proteomics 2005, 2, 31-40.
34. Conlon, P.; Yang, C. J.; Wu, Y.; Chen, Y.; Martinez, K.; Kim, Y.; Stevens, N.; Marti, A.
A.; Jockusch, S.; Turro, N. J.; Tan, W. Pyrene excimer signaling molecular beacons for
probing nucleic acids. J. Am. Chem. Soc. 2008, 130, 336-342.
35. Zuo, X.; Xiao, Y.; Plaxco, K. W. High specificity, electrochemical sandwich assays
based on single aptamer sequences and suitable for the direct detection of small-molecule
targets in blood and other complex matrices. J. Am. Chem. Soc. 2009, 131, 6944-6945.
36. Savran, C. A.; Knudsen, S. M.; Ellington, A. D.; Manalis, S. R. Micromechanical
detection of proteins using aptamer-based receptor molecules. Anal. Chem. 2004, 76,
3194-3198.
37. Zhao, W.; Chiuman, W.; Lam, J. C.; McManus, S. A.; Chen, W.; Cui, Y.; Pelton, R.;
Brook, M. A.; Li, Y. DNA aptamer folding on gold nanoparticles: from colloid chemistry
to biosensors. J. Am. Chem. Soc. 2008, 130, 3610-3618.
38. Liu, J.; Mazumdar, D.; Lu, Y. A simple and sensitive "dipstick" test in serum based on
lateral flow separation of aptamer-linked nanostructures. Angew. Chem. Int. Ed. 2006, 45,
7955-7959.
39. Liu, J.; Lu, Y. Preparation of aptamer-linked gold nanoparticle purple aggregates for
colorimetric sensing of analytes. Nat. Protoc. 2006, 1, 246-252.
40. Nutiu, R.; Li, Y. In vitro selection of structure-switching signaling aptamers. Angew.
Chem. Int. Ed. 2005, 44, 1061-1065.
41. Nutiu, R.; Li, Y. Structure-switching signaling aptamers. J. Am. Chem. Soc. 2003, 125,
4771-4778.
42. Nutiu, R.; Yu, J. M.; Li, Y. Signaling aptamers for monitoring enzymatic activity and for
inhibitor screening. ChemBioChem 2004, 5, 1139-1144.
43. Nutiu, R.; Li, Y. Aptamers with fluorescence-signaling properties. Methods 2005, 37, 16-
25.
44. Huizenga, D. E.; Szostak, J. W. A DNA aptamer that binds adenosine and ATP.
Biochemistry 1995, 34, 656-665.
45. Jhaveri, S.; Rajendran, M.; Ellington, A. D. In vitro selection of signaling aptamers. Nat.
Biotechnol. 2000, 18, 1293-1297.
46. Vaish, N. K.; Larralde, R.; Fraley, A. W.; Szostak, J. W.; McLaughlin, L. W. A novel,
modification-dependent ATP-binding aptamer selected from an RNA library
incorporating a cationic functionality. Biochemistry 2003, 42, 8842-8851.

198
47. Wen, A. Q.; Yang, Q. W.; Li, J. C.; Lv, F. L.; Zhong, Q.; Chen, C. Y. A novel
lipopolysaccharide-antagonizing aptamer protects mice against endotoxemia. Biochem.
Biophys. Res. Commun. 2009, 382, 140-144.
48. Bruesehoff, P. J.; Li, J.; Augustine, A. J., 3rd; Lu, Y. Improving metal ion specificity
during in vitro selection of catalytic DNA. Comb. Chem. High Throughput Screening
2002, 5, 327-335.


















