DENNIS JOHNQALEXANDER - Spiral: Home · cellular protein synthesis in infected chick embryo cells...
248
Royal Postgraduate Medical School Library REFERENCE COPY NOT to be taken away CVO STUDIES U4TU NEWCASTLE DISEASE VIRUS by DENNIS JOHNQALEXANDER Department of Virology Royal Postgraduate Medical School London. Ph.D. Thesis University of London 1971
Transcript of DENNIS JOHNQALEXANDER - Spiral: Home · cellular protein synthesis in infected chick embryo cells...

Royal Postgraduate Medical School Library
REFERENCE COPY
NOT to be taken away
CVO STUDIES U4TU NEWCASTLE DISEASE VIRUS
by
DENNIS JOHNQALEXANDER
Department of Virology
Royal Postgraduate Medical School
London.
Ph.D. Thesis
University of London
1971

ABSTRACT
The object of this work was to investigate the cause of the
differences of virulence between Newcastle disease virus (NDV) strains,
with particular reference to the role of virus proteins.
Methods for the purification of NAV using zonal rotors are
described.
Separation of purified virus by acrylamide gel discelectrophoresis
after sodium dodecylsulphate (SDS) treatment revealed four structural
polypeptides which were similar in migration and proportion for all
strains. The molecular weights of the three major polypeptides were:
88,000, 58,000 and 42,000; haemagglutinin, ribonucleoprotein and
neuraminidase were associated respectively with these polypeptides.
The minor polypeptide, molecular weight) 200,000, was not associated
with any known component or property of the virus.
The four structural proteins and tWo other 'non-structural'
proteins were detectable in infected chick cells. Strain Herts
induced two other non-structural proteins but later than 10HR,after
infection. Virus induced proteins were detectable earliest after
infection with virulent strains.
Neither the biochethical properties of neuraminidase nor the
amount per virion showed any relationship to virulence. The cell-
associated titre of neuraminidase in chorioallantoic membranes 22 HR
after infection was directly related to the virulence of the infecting
strain.
The extent of cell fusion and haemadaorption, and inhibition of
cellular protein synthesis in infected chick embryo cells were related
to the virulence of the infecting strain. All three required
protein synthesis in the first four hours of infection.

Congo red gave some protection of chick embryos against low
but lethal, doses of virulent strains. Very high doses of avirulent
virus showed greatly increased lethality to chick embryos in the
presence of congo red.
The cumulative production of RNA in infected cells treated
with actinomycin D was directly related to the virulence of the
infecting strain.
It is concluded that the virulence of NDV strains is a result
of the rate of production of one or more virus product in infected
cells.

ACKNOWLEDGEMENTS
This work was aided by grants from the Agricultural Research
Council and Action for the Crippled Child (Polio Research Fund).
I am indebted to Dr. Peter Reeve for his constant help, encouragement
and guidance throughout the course of this work and the preparation of
this thesis.
I would also like to thank: Professor A.P. Waterson for his
advice and encouragement; Mr. W.H. Allan for supplying the virus
strains used in this work and his helpful advice and discussions;
Mr. Peter Lister for his help and advice particularly during the
preparation of the figures; Miss Gill Pope and Miss Joy Pacey for
their help, and all those who during the course of this work have
offered advice and criticism.

CONTENTS
PAGE
I REVIEW AND INTRODUCTION
A. History 1
B. Morphology structure and chemistry 3
1. The virion 3
2. The substructures 3
a) Ribonucleoprotein 3
b) The envelope 5
C. Classification of NDV - a myxovirus 8
D. Replication of NDV 12
1. Adsorption 12
2. Penetration 12
3. Eclipse phase 13
a) General events 13
b) Types of NDV-specified RNA produced during infection 13
c) Role of complementary RNA 14
d) NDV-specified RNA-dependent RNA polymerise 15
4. Virus maturation and release 16
E. Virulence - range, and methods of assessment 18
P. Strain differences and virulence 21
Virulence definition 21

Foreword 22
1. Morphology and virulence 23
2. Serology 23
3. Biophysical properties 24
4. Cytopathogenecity 24
a) Haemadsorption 25
b) Syncytial formation - cell fusion from within 25
c) Plaque formation 26
d) Plaque size 26
e) Lysosome damage 28
f) Syncytial formation - cell fusion from without 28
3. Biological activities 30
a) Haemagglutinin 30
b) Neuraminidase 31
6. Virus multiplication 32
a) Growth rates 32
b) Growth site 35
7. Effects on cell metabolism 36
a) Protein and RNA synthesis 37
b) DNA synthesis 38
c) Lipid synthesis 39
8. Interferon and virulence 39
a) Interferon sensitivity 39
b) Interferon production 39
G. Effect of congo red on NDV infection 42
H. Virus specified proteins in NDV-infected cells 43

J. Purification of NDV 45
K. Objects of investigation 47
II MATERIALS AND METHODS 48
A. Reagents and equipment 48
1. Reagents 48
2. Equipment 50
B. Virus strains and virus techniques 52
1. Strains 52
2. Growth of virus 53
C. Cell culture techniques 54
1. Preparation of chick embryo fibroblast cells 54
2. Infection of cell monolayers 54
3. Staining cell monolayers 55
a) Giemsa - May GrUnwald 55
b) Acridine orange 55
D. Biological assays 57
1. Haemagglutinin 57
2. Infectivity 57
3. Plaque assay 57
4. Neuraminidase assay 57
5. Protein assay 58
6. Lactate dehydrogenase assay 58
7. Estimation of haemadsorption 58
8. Estimation of cell fusion 59

a) Size of syncytia 59
b) 7 polykaryocytosis 59
9. Estimation of cell-associated neuraminidase activity 59
E. Virus disruption 60
1. Sodium dodecyl sulphate-2-mercaptoethanol 60
2. Wean 20 at pH 10 60
3. Sodium demqcholate 61
F. Radioactive isotope counting techniques 62
1. Scintillation counter 62
2. Scintillation fluids 62
3. Estimation of uptake of radioactive isotope into coverelip cell. cultures 62
4. Estimation of radioactivity in gel slices 63
5._ Quench curve 63
6. Radioactive virus 65
G. Polyacrylamide gel discelectrophoresis 66
1. Preparation of 7.57. gels 66
2. Running 67
3. Staining 67
4. Estimation of polypeptide molecular weights 68
H. Double labelling techniques 69
J. Density gradient centrifugation - virus purification 71

A. Virus
III RESULTS
72 purification and biophysical properties
1. Linear sucrose gradient 72
2. Linear tartrate gradient 72
3. Association of viral properties to peaks 72
4. Discontinuous gradients 76
5. Recentrifugation of HA peaks 76
6. Nature of major and minor HA peaks 80
7. Separation under varying conditions 80
8. Estimation of virus purity 80
9. Behaviour of different strains of NDV on density gradients 84
10. Discussion of results 87
B. The structural proteins of NDV 89
1. Polyacrylamide gel discelectrophoresis after disruption by SDS 89
2. Polypeptide patterns of different strains 89
3. Radioactive labelled virus 89
4. Relative proportions of the polypeptides 95
5. Estimation of the molecular weight of the polypeptides 97
6. Identification of NDV polypeptides 97
a) Nucleocapsid polypeptide 97
b) Neuraminidase and haemagglutinin 100
C. Neuraminidase studies 108
1. Relationship of neuraminidase to HA and infectivity 108

2. Kinetic studies of virus-bound neuraminidase
108
3. Heat stability of virus-bound neuraminidase
113
4. Effect of Tween 20 at pH 10
113
5. Neuraminidase activity in infected chorioallantoic membranes
118
D. Cell fusion (from within) and haemadsorption
120
I. Haemadsorption
120
2. Cell fusion (from within)
127
3. Effect of inhibition of protein synthesis on virus induced cell fusion and haemadsorption
133
4. Effect
1. Effect
2. Effect
3. Effect
F. Effect of
E. Inhibition
of time of addition of FPA on cell fusion and haemadsorption
of cellular protein synthesis
of infection with different strains on cellular protein synthesis
of p-fluorophenylalanine on virus induced inhibition of cell protein synthesis
of time of addition of FPA on virus induced inhibition of cell protein synthesis
congo red on the mortality of eggs infected with NDV
133
137
137
139
139
142
G. The synthesis of NDV induced proteins 148
H. RNA production of infected cells in the presence of actinomycin D 157

IV DISCUSSION 159
A. Structural proteins 159
B. Neuraminidase studies 162
C. Cell fusion and haemadsorption 164
1. Haemadsorption 164
2. Cell fusion 164
3, Effect of inhibition of protein synthesis on cell fusion and_haemadsorption 166
D. Inhibition of cellular protein synthesis 167
E. Effect of congo red on the mortality of eggs infected with NDV 168
F. The synthesis of NDV induced proteins 169
G. Production of NDV specified RNA 172
H. General discussion 173
V CONCLUSIONS 176
VI SUGGESTIONS FOR NEITHER WORK 176
VII REFERENCES 178

I REVIEW AND INTRODUCTION
A. HISTORY
Newcastle disease was first recorded on the island of Java in
1926 (Kranevald, 1926) but first recognised as a virus infection
after a later -outbreak on a farm near Newcastle-on-Tyne (Doyle,
1927). These early outbreaks showed incubation periods from 4-11
days; respiratory and nervous signs (twitching and paralysis) as
well as general malaise were the moat apparent symptoms and mortality
rates were as high as 98-100%. Because of the high mortality the
early outbreaks in England were self-limiting. However in 1933 and
again in 1947 there were outbreaks of equal virulence. A milder
form of the disease also appeared in 1947 and has been endemic in
England since that time.
In 1944 it was recognised that a disease of chickens that had
prevailed in California for almost nine years called "chicken flu"
or "pneumoencephalitis" (Beach, 1943) was, in fact, Newcastle disease
(Beach, 1944). The delay in recognition was due to the entirely
different clinical features of the disease to those usually presented
by Newcastle disease. The disease was far milder than any form of
Newcastle disease known at that time, the respiratory and neurologi-
cal symptoms were notably mild and the mortality rate was often less
than 15%.
Newcastle disease is now known to be present in virtually every
country in the world (Levine, 1964) although in some countries, 2141.
Ireland and Australia the strains of virus present are particularly
avirulent. Bankowaki (1961) and McFerran (1966, unpublished) have
1

isolated virus strains of this kind which are symptomless in adult
birds, infection prior to isolation is detectable only by haema -
gglutination inhibition tests on the fowls serum.
In England since the early outbreaks the environment of the
chicken has greatly changed. Intensive rearing methods used in the
'broiler house' system haimeant that large flocks of many tens of
thousands of birds are kept. These flocks are indoors with the
birds in very close proximity to each other which gives ideal con-
ditions for infection to spread amongst a flock. It has been the
policy in EnglanU. since 1965 to control the endemic disease by
vaccination with inactivated virus; until Chi* time a policy of
slaughter, which is still maintained in Scotland. had been practiced.
Although several outbreaks have occurred since the introduction of
vaccination these were never extensive epidemics and the disease
VAS generally considered, perhaps too complacently, Co be under con-
trol.
In the autumn of 1970 a new outbreak of Newcastle disease
occurred in Essex which proceeded to spread throughout England as
an epidemic of unprecedented proportions, by February 1971 over
12,000,000 birds bad died. The virus unlike other strains of high
virulence is pneumotropl(ic rather than neurotropgic and the chick-
ens appear to die of respiratory malfunction (Allan, personal
communication). Conventional vaccination has had little success
in preventing the spread of the disease and in January 1971 the
Ministry of Agriculture gave permission for vaccination with live
virus in the form of the vaccinal strain B1. The effectiveness
of vaccination with this virus in controlling the present outbreak
has yet to be seen.
2

B. MORPHOLOGY STRUCTURE AND CHEMISTRY
1) The Virion
Viewed in the electron microscope using negative staining
techniquesI NDV populations can be seen to be grossly pleomorphic
(Schafer, Schramm and Traub, 1949), while individual virions have
no rigid structure and frequently appear disrupted on the grid.
The upper limits of the greatest dimensions of the NDV virion are
120-130 nm. (Waterson and Cruickshank, 1963)
Chemical analysis of pure NOV virions has shown the following
composition by weight: 677. protein, 1% RNA, 24% lipid, 77. carbo-
hydrate (Cunha et al, 1947). The viral RNA is surrounded by
protein to form a helical nucleocapsid (Kingsbury and Darlington,
1968). This nucleocapsid is itself contained inside a lipoprotein
membrane of host origin (Cruickshank, 1964) from which project
numerous regular spikes (Horne et al, 1960). The neuraminidase and
haemagglutinin activities are also associated with the envelope.
2) The substructures
a) Ribonucleoprotein (RNP)
Treatment of the virus with ether was shown to release the inner
component, the RNP (Schafer and Rott, 1959). The RNP of NDV is 17-
18 nm. wide has a central bole 4-5 nm. in diameter and a periodi-
city of 5 am. along the axis of the helix (Horne and Waterson, 1960).
Release of the RNP by ether tends to produce pieces of variable
length but osmotic lysis of NDV infected rhesus monkey kidney
cells gave a value of 1.06p for the intact length (Compans and
Choppin, 1967) and from deoxycholate treated virions 1.3-1.4p
(Kingsbury and Darlington, 1968).
3

RNP contains 9.3% RNA (Rott, Waterson and Reda, 1963) which
can be freed of the protein by treatment with sodium dodecylsul-
phate solution (Kingsbury and Darlington, 1968). Estimates of the
M.W. of the RNA from NDV vary from 5.8 x 106 (Nakajima and Obara,
1967) to 7.5 x 106 (Duesberg and Robinson, 1965). The sedimenta-
tion coefficient of the RNA in 0.05M Nadi, 0.01M sodium acetate
0.5% SDS was 49.2S increasing to 57S in 0.1M Nadi 0.01M Tris/HC1
and 0.001M EDTA (Adams, 1965, Duesberg and Robinson, 1965, Kingsbury,
1966). A low molecular weight component (36-4S) isolated was
assumed to be a degradation product of the 57S RNA. The isolated
RNA was easily degraded by RNase, suggesting it is entirely single
stranded RNA (Nakajima and Obara, 1967). Newcastle disease virus
has therefore a genome which is several times larger than that of
any other single strand RNA virus (except Sendai) which implies afar
greater genetic information content.
High molecular weight RNA isolates from the Beaudette C
strain of NDV examined by Kingsbury (1966) failed to show any sign
of infectivity in chick embryo monolayers, although isolated
Western equine encephalomyelitis virus RNA used as a control under
the same conditions did. However, infectivity was assessed by
ability to form plaques which in the NDV system is not necessarily
indicative of infection (Table 1).
The protein of the RNP has recently been shown by polyacryl-
amide gel electrophoresis techniques in the presence of SDS to con-
sist of a single polypeptide subunit (Evans and Kingsbury, 1969,
'bishop Cheyne and White, 1969 and Bikel and Duesberg, 1969).
Evans and Kingsbury (1969) and Bikel and Duesberg (1969) report
the molecular weight of the polypeptide sub unit as 62,000 daltons

while Haslam et al using a similar procedure quote 54,000 daltons
and estimate that the amount of thlsprotein per virion is 45% of
the total virion protein. All!these estimations of molecular
weight used the technique of Shapiro, vinuela and Maizel (1967),
and should be regarded only as approximations.
b) The envelope
Difficulties have arisen in recent years over the nomencla-
ture of myxovirus sub units. Originally haemagglutinin referred
to the rosette-like structures produced by ether treatment which
retained both haemagglutinating activity and neuraminidase activity
(Waterson, 1964). In the last few years subunits showing haema-
gglutinin activity have bean separated from those with neuramini-
dase activity (at least with influenza virus). The subunit possess-
ing haemagglutinin activity has been called haemagglutinin as well.
In this review haemagglutinin refers to the subunit protein which
possesses haemagglutinating activity and nothing else.
The envelope consists of protein, lipid and carbohydrate.
It is apparently derived from host cell membranes which have been
modified by the incorporation of virus specified material.
(Cruickshank, 1964, Hoyle, 1954). The envelope has an outer cover-
ing of spike-like projections 8O long, 10-158 wide and spaced 10-
1008 apart over the envelope surface. The spikes in this and
other mymoviruses are associated with the haemagglutinin activity
of the virion (Waterson, 1964, Biddle, 1968).
Because of the ease with which influenza can be degraded with
biologically active subunits, compared with NDV, moat studies of
myxovirus substructure have been with that virus (Laver, 1963, 1964,
Laver and Kilbourne, 1966, Laver and Valentine, 1969, Drzenfek,
5

Frank and Rott, 1968) and little has been done to isolate pure
biologically active subunits from NDV. Drzeniek and Rott (1963)
however treated purified, freeze-dried virus with n-butanol and
trypsin and extracted a non-haemagglutinating neuraminidase-con-
taining component which had a molecular weight of 200,000. It
was, luggested that this was a single molecule of neuraminidase,
however the component still induced neutralizing and haemagglutin-
ation inhibition antibody when given intravenously to rabbits
(Rott, 1964).
A study has been made of the biochemical properties of the
neuraminidases of two strains of NDV, Italian and Beaudette C and
it has been shown that although they are alike in their biochemical
properties, (Rott, Drzeniek, Saber and Reichert, 1966, Drzeniek,
Seto and Rott, 1966) NDV neuraminidases show significantly
different characteristics from most influenza neuraminidases.
Drzeniek (1967) has also shown that NDV neuraminidase is unlike
Vibrio cholerae neuraminidase, in particular NDV cleaves only 2-3'
linked sialolactose whereas Vibrio cholerae neuraminidase cleaves
both 2-3' and 2-6' linkages.
The actual relationships between haemagglutinin and neuramini-
dase and their structural positions on the envelope are not clear.
There is strong evidence•in the influenza system that haemagglutinin
and neuraminidase are proteins of separate entity (Drzeniek, Frank
and Rott, 1968, Laver and Valentine, 1969). However much of the
evidence comes from experiments employing recombinants of strains
which either have no neuraminidase activity or no haemagglutinin
activity. These do not necessarily lack the other protein but may
possess only an inactive form. The methods of disrupting the virion
6

to bring about separation of the biological activities are
usually very harsh (SDS treatment or_proteolytic enzyme action
in conjunction with a solvent) and could merely be separating
polypeptides of a single protein. Rott (1964) has suggested that
NDV neuraminidase may be a monovalent part of a polyvalent haema-
gglutinin.
Disc electrophoresis of NDV on acrylamide gels after SDS
treatment has revealed three major protein bands and several onts
smaller/(Evans and Kingsbury, 1969, Haslam, Cheyne and White,
1969, Bikel and Duesberg, 1969). Of the three major bands the
middle one is without doubt the RNP polypeptide while the other
two bands remain to be satisfactorily identified. The slowest
moving band M.W. 80,-90,000 has been associated with haemagglutinin
activity while the fastest moving band M.W. 38,-40,000 with neura-
minidase (Haslai et al, 1969). However in neither case was the
protein isolated and purified which are the ideal conditions for
this type of identification.
Several other biological activities associated with the OrAti
envelopey/haemolytic activity, ATPase and ADPase have been found
in preparatioo3 of NDV, these are now considered to be of host-
cell origin. (Neurath and Sokol, 1963, Neurath, 1965, Allison,
1967).
7

C. CLASSIFICATION OF NDV - A NWOVIRUS
Andreves, Bang and Burner (1955) suggested the name myxovirus
to include influenza. A, B, C, fowl plague, Newcastle disease
and mumps virus. All these viruses had distinct interactions with
mucoproteins (haemagglutination, and elution due to neuraminidase)
and the prefix myxo- from the Greek myna meaning nasal secretion
or slime, and hence mucus, was particularly apt.
The development of phosphotungstenic acid negative staining
showed all the myxoviruses to be morphologically similar, consist-
ing of a coiled helix surrounded by an envelope covered with spike-
like projections. At about the same time the group as a whole
were also shown to be RNA viruses. However, measles, distemper,
rinderpest and respiratory syncytial viruses were all shown to be
similar in morphology possessing spiked envelopes and helical RNP,
and although only measles showed haemagglutination activity and
none possessed nouraminidase they were added to the group (Virus
Sub-Committee of the International Nomenclature Committee, 1963).
Lwoff, Horne and Tournier (1962) proposed that viruses should be
classified by the following characteristics 1) Nucleic acid,
DNA or RNA?; 2) Symmetry of nucleocapsid; 3) Presence or
absence of an envelope surrounding the nucleocapsid; 4) The
diameter of the nucleocapsid or the number of capsomers. The
myxovirus group were therefore: 1) RNA 2) Helical 3) Envel-
oped. But on the basis of the diameter of the nucleocapsid the
myxovlruses could be divided into two groups (Waterson, 1962):
The influenza group with an inner helix diameter of 9-10 mm,
including the influenzas and fowl plague. The NDV group including

NDV, mumps, the parainfluenzas, measles, rinderpest and distemper virlAse-%
all of which have a helix diameter of 17-18 nm. (Fig. 1)
This method of classification presents some problems however as
viruses such as rabies would fall into one or other of the groups
although morphologically as well as in other respects they are
fundamentally different.
It has since been suggested (Waterson and Almeida, 1966) that
a further distinction of more significance than the size of the
nucleocapsid should be made in the myxovirus group.- This is to
separate those viruses showing an affinity fsti'mucoproteins, i.e.
the myxophilic 'true myxoviruses' which possess neuraminidase and
those which do not, "pseudomyxoviruses". The scheme suggested by
Waterson and Almeida is outlined in Figures 1 and 2 . In the
original scheme bovine parainfluenza 3 virus was classified as a
pseudomyxovirus of NDV morphology but it has since been shown to
possess neuraminidase (Drzenialt,, B8gel and Rott, 1967) and so
should be included in the true myxovirus group.
9

FIGURE
The Lwoff-Horna•Tournier Classification of Myxovirusos.
10
Virus
RNA
Helical
Enveloped
170-180 a
NOV Rabios 90A
Mumps VSV
Parsinfluanzas Avion orythro-
Measles -myeloblastosis 42A
Rindarpott
DistanTer
Characteristic:
Nucleic acids
Symmetry:
Envelops{
()isolator of nuclaocapsid:
O
90-100 A
InfIvonzas
Fowl plague

FIGURE 2
The OT-thitarson-Almoida Classification of Myxoviruaeo.
11
Characteristic:
Nucleic acid:
Symmetry:
Envelopes
Myxovirus morphology
Virus
RNA
F-- Helical
F-- Enveloped
Uthor compound helical viruses so. Rabies, Viltit Avian orythromyolo--blastosis
Myxophily:
True myxoviruses
Size of nucloocapsids
influon2se NOV
Pseudomyxoviruses
'NOV morphology' 'Influmorpenza hology'
1(11s1as •••
ParninflunnmOindorpnnt
Diatnmpor

D. REPLICATION OF NDV
In many instances the details of NDV replication are not Fo be-
specifically known and are assumed/analogous to other myxoviruses.
In the following account, unless otherwise specified, all ref-
erences are to experiments with NDV.
1. Adsorption
Presumably adsorption of NDV to cells is mediated by haema-
gglutinin attachment to specific cell sites. After treatment of
cells with Vibrio 'cholerae neuraminidase cells fail tolfuse, become
infected, and red cells fail to agglutinate.on inoculation with
NDV, because of the destruction of the cell-receptors. (Kohn, 1964,
Waterson, 1968).
The actual mode of attachment seems to be controlled by
electrostatic forces for while it is impossible to adsorb virus
particles in isotonic glucose medium free,. of ions, adsorption can
he achieved by the addition of electrolytes, divalent cations
being the most efficient (Rott and Scholtissek, 1967).
2. Penetration
&makes de St. Groth (1948) proposed that myxoviruses entered
host cells by active engulfment (viropexis) of whole virus by the
cells. Musegay and Weibel (1962) enhanced this theory by showing
that particles of intact NDV virions can be observed in the cyto-
plasm as early as 30 minutes after inoculation of chick embryo
cells. It is postulated that liberation of RNP then occurs. Hoyle
(1962) has shown in in vitro studies that normal cytoplasmic par-
ticles of the chorioallantoic membrane engulf virus particles
causing disintegration and release of RN?. This release of RNP
12

has been shown to occur in cells with 32
P labelled influenza virus
(Hoyle and Frisch-Niggemeyer, 1955). The release of the RNP and
the uncoating of the viral RNA all appear to be carried out by
enzymes associated with lysosomes (Ralph, 1969).
3. Eclipse Phase
a) General Events
Because NDV cuts off cell macromolecule metabolism only very
slowly, if at all, study of many of the events during the eclipse
phase, in particular viral RNA production, would be extremely
difficult if NDV was not relatively insensitive to actinomycin D
(Kingsbury, 1962).
The synthesis of viral RNA starts between 2-4 hours after
infection, reaching a maximum at around 7 hours. The actual site
of RNA replication in the cell is in some dispute. Wheelock (1963)
claimed the cytoplasm was the site of NDV RNA synthesis. However
Bukrinskaya, Burdulea and Vorkunova (1966) point out that unless
the labelled pulse is brief the label is found throughout the
cytoplasm; whereas with short pulses of labelled precursor they
were able to show, by autoradiography, that RNA appears to replicate
in the nucleolus and migrate to the cytoplasm. More recently
Brett and Robinson (1967) have presented further evidence for a
cytoplasmic site, however, they have sampled somewhat later after
infection and may have missed an early nuclear site.
Between 3-4 hours after infection RNP, haemagglutinin and
neuraminidase are produced in the cytoplasm and shortly afterwards
infective virus can be detected.
b) Types of NDV-specified RNA produced during infection
It has been shown that of the RNA produced in NDV infected cells
13

80% of that produced between 3-5 hours and 90% of RNA produced
at 7 hours is complementary (-)RNA (Kingsbury, 1970) although some
(+) RNA is always produced (Kingsbury, 1966b, Brett and Robinson,
1967, Blair and Robinson, 1968). Some of the (+) RNA sediments
at the same rate as RNA from virions (570 but there is also more
slowly sedimenting (+) RNA which may be free or associated with
(-) RNA (Brett and Robinson, 1967). It is not sure in these
experiments however how much (+) RNA is derived from nucleocap-
aid or cell associated virions. Evidence generally suggests that
there is no pool of viral genomes although work by Reeve and Poste
(1970) suggests genome RNA may be used as a messenger.
The NDV specified complementary RNA is heterogenous in sedi-
mentation behaviour producing peaks'at 188, 228 and 358 and some
57S RNA (Brett and Robinson, 1967, Blair and Robinson, 1968).
Brett and Robinson (1967) showed that 50% of the NDV genome
is made RNAse resistant when annealed with (-) 183 RNA. Since 18S
RNA would probably have a molecular weight one sixth of that of
the NDV genome this work suggests that 188 (-)RNA may be hetero-
genous with respect to nucleotide sequence.
c) Role of complementary RNA
In other non-myxoviruses it is generally accepted that com-
plementary RNA functions as a template for (+) RNA production,
the viral strand being replaced in the double-stranded complex as
a new strand is initiated. In NDV infections the most logical can-
didate for this function is a 57S(-)RNA, i.e. one of the same size
as the genome. However this does not account for the vast majority
of the (-) RNA which has insufficient information to fulfil this
function. Similarly although small amounts of 35S and 57S RNAs
14

resist RNase digestion, there is no clear evidence of RNase-
resistant double-stranded viral RNA replication intermediates
(Brett and Robinson, 1967).
An alternative hypothesis is that the (-) RNAs are messenger
RNA. The fact that most of the RNA associated with the polyriba.
somas is of the (-) RNA type supports this hypothesis (Brett and
Robinson, 1967, Blair and Robinson, 1968, Kingsbury, 1970), although
the presence in the polyribosomes may be fortuitous.
Schafer, Pieter and Schneider (1967) by showing viral antigens
are produced in cells even after viral RNA synthesis is inhibited
have proposed that NDV (+) RNA functions as the sole messenger.
Work by Reeve and Poste (1970) has strengthened this opinion while
Ralph (1969) suggests that both (-) RNA and (+) RNA may serve as
messengers at different stages of the infection by NDV.
d) NDV-specified RNA dependent RNA-polymerase
Although it is a fairly obvious speculation that a NDV-
specified RNA-polymerase is produced in infected cells there is
currently only one report of such enzymii activity. The enzyme
detected by Scholtissek and Rott (1969) was not found in noninfected
cells and was not inhibited by actinomycin D. The enzyMe was
first detectable 3 hours after infection, the peak activity was
reached after 5 hours. Predictably it was shown by nearest neighbour
analysis and hybridization experiments that the vast majority if
not all the RNA produced in vitro by the enzyme was (-) RNA.
However no information vas recorded as to the sedimentation coeffic-
ient of this RNA. The RNA polymerase activity was detected in
the cytoplasmic extract of infected cells but there is no indication
as to whether or not the nuclear fraction was tested for activity.
15

4. Virus maturation and release
However much the myxovirus subgroups differ from one another
in other respectsi the maturation of the virion at or near the cell
surface seems to be common to all the myxoviruses (Chanock and
Coates, 1964). For this reason it has been generally accepted
that the mode of release for all the myxoviruses is very similar.
Mature NDV production is detectable about 4 hours after
infection, with cytopathic strains production ceases after 10,42
hours but with non-cytopathic strains virus production may con-
tinue somewhat longer. At about the same time as mature virus is
first detectedi Narcus (1962) has shown that infected cells become W4V-
able to adsorb red cells and suggested/this was brought about by
the incorporation of viral heemagglutinin into the cell membrane
prior to virus release: Furthermore Rott (1964) has shown that
prior to release iviral nucleocapsid can be seen to accumulate
beneath the cell surface of NDV infected cells.
Using SV5 virus (Compans et al, 1966) it has been shorn that
the nucleocapsid becomes aligned under the cell membrane and is
slowly budded off to form a released virus particle, part of the
infected cell membrane becoming the viral envelope. It is not
clear, however, whether the nucleoprotein aligns and then cell sur-
face modification occurs or vice versa.
Seto and Rott (1966) have demonstrated that neuraminidase
plays an important part in fowl plague virus (FPV) release. When
neuraminidase specific antiserum was added to FPV infected mono-
layers no HA, infective virus or neuraminidase activity was
released from the cells, although the normal amount of complement
fixing antigen and at least one third of the normal infectivity
16

was detectable associated with the cells. These findings have been
confirmed morphologically by electron microscopy of infected cells
after treatment with specific antiserum (Seto and Chang, 1969).
It has been estimated that the release time of NDV. from chick
embryo lung epithelium cells is one hour (Rubin, Franklin and
Bamda, 1957).
17

E. VIRULENCE - RANGE, AND METHODS OF ASSESSMENT
Although Newcastle disease has remained endemic in this
country since the 1940's, until the present outbreak, highly vims
lent outbreaks similar to those of the 1920's were rare. Occasional
outbreaks causing high mortality were generally self limiting or
controlled by slaughtering the infected fowls. Because of these
conditions of low virulence endemic virus and occasional virulent
outbreaksi many strains of NDV have now been isolated and they show
a continuous spectrum between the two limits of virulence.
Three methods have been universally accepted for the
assessment of virulence. They are detailed in Methods for the
examination of poultry biologics (1963).
a) Mean death time in embryonated eggs (MDT)
The mean death time is the average time in which eggs inocu-
lated with one minimum lethal dose of virus are killed.
b) Intracerebral pathogenicity in chicks
The intracerebral pathogenicity index (ICPI) is estimated
from time taken for one-day-old chicks to die or show disease symp-
toms after intracerebral inoculation of virus. The results are
based on a scoring system in which the maximum index possible is
2 (100% mortality in one day) and the minimum is 0 (no recorded
symptoms after 8 days).
c) Intravenous pathogenicity index (IVPI)
This is the least used method. It is very similar to the
ICPI method but 6-week-old chicks are used and are injected intra- ,
venously,/maximum score is 3.
Table 1 shows the MDT, ICPI and IVPI of all the strains used
18
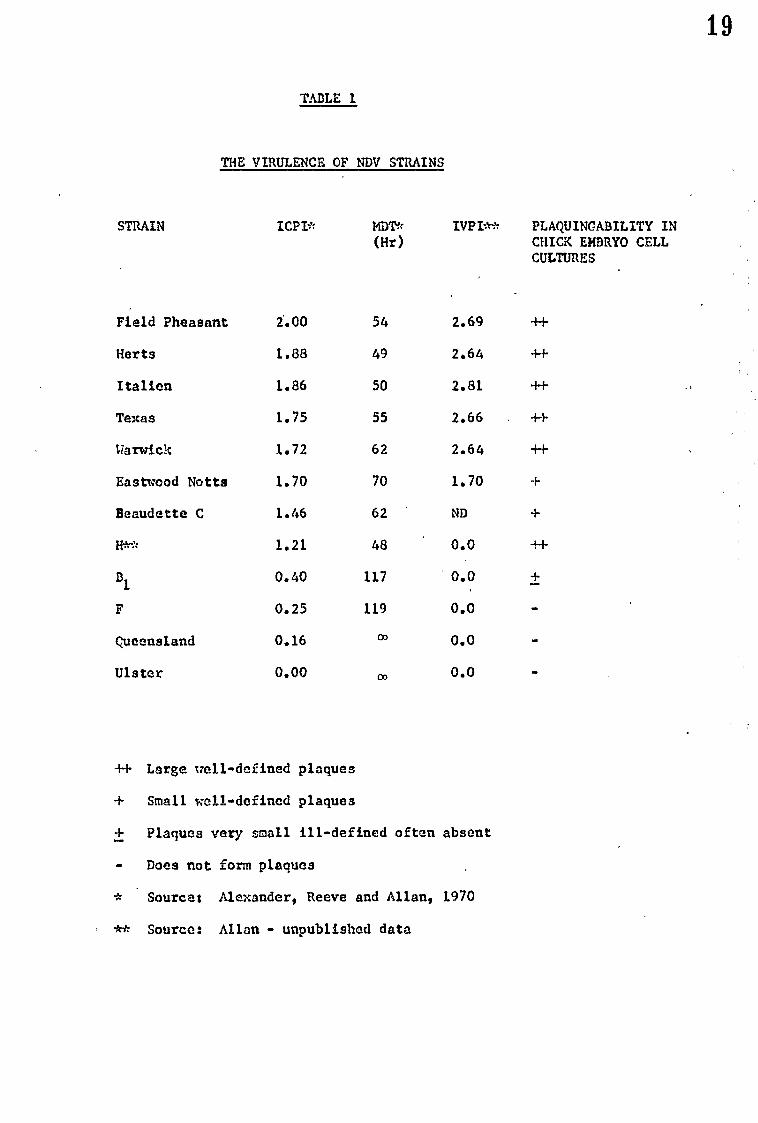
TABLE 1
THE VIRULENCE OF NDV STRAINS
STRAIN ICPI* MDT* IVPI** PLAQUINCABILITY IN (Hr) CHICK EMBRYO CELL
CULTURES
Field Pheasant 2.00 54 2.69 +I-
Herts 1.88 49 2.64 ++
Italian 1.86 50 2.81 -H-
Texas 1.75 55 2.66 +I-
Warwick 1.72 62 2.64 -t-i-
Eastwood Notts 1.70 70 1.70 +
Beaudette C 1.46 62 ND +
it." 1.21 48 0.0 ++
B1
0.40 117 ' 0.0 ±
F 0.25 119 0.0
Queensland 0.16 m 0.0
Ulster 0.00 co 0.0
++ Large well-defined plaques
+ Small -till-defined plaques
+ Plaques very small ill-defined often absent
• Does not form plaques
* Source: Alexander, Reeve and Allan, 1970
** Source: Allan - unpublished data
19

in this study. The large range of virulence can be seen. Hanson
and Brandly (1955) grouped the strains of NDV according to the
MDT:-
Vologenic strains MDT 40-60 hours
Nesogenic strains MDT 60-90 hours
Lentogenic strains EDT > 90 hours
The divisions are purely arbitrary but are worth mentioning as the
terminology is still often used.
Other properties can also be used as a guide or a measure of
virulence. For example, Schloer and Hanson (1968) have shown that
plaque size is directly related to virulence. The ability to
plaque at all is also indicative of virulence and the growl diff-
erencesof plaguing ability are also shown in Table 1.
It is worth noting that although the different measures of
virulence run parallel or inversely paral1t there are strains
whose relative virulence varies with the method used. Strain 11
for example shows low virulence in chicks and yet produces the
lowest EDT in eggs and the largest plaques in chick embryo fibroblasts
of all strains tested. This may indicate a difference in the
cause of virulence from one system to another.
20

F. STRAIN DIFFERENCES AND VIRULENCE
Virulence Definition
'The capacity of a micro-organism to kill is covered by the
term virulence, and virulence may be determined by toxigenicity,
invasiveness, or many other factors and is qualified by the host
in which it is measured.' (Wilson and Miles, 1964)
21

Foreword
'In any study of virulence in the NDV system it is particularly
important that the number and range of virulence of the strains
used are carefully selected: In the past often only two strains
have been studied, a virulent and a less virulent. Because of
this strain differences have sometimes been put forward as caus-
ing a difference in virulence but which are merely differences
between the two strains used. It is particularly unfortunate that
in many of the studies Beaudette C has been used as the strain
of lower virulence (Rott, Reda and Schafer, 1962, 1963, Schafer
and Rott, 1959, Drzeniek, Seto and Rott, 1966, Wilson, 1968 etc).
Beaudette C is not an avirulent strain being classed as mesogenic
by Hanson and Brandly (1955) but in fact is considerably virulent
in eggs,(see Table 1). Beaudette C is also a particularly atyp-
ical strain as it is not a natural isolate but a laboratory mutant
isolated solely for its heat stability (Granoff, 1959).
To reach any conclusions as to the cause of the wide range of
virulence in the NDV system it is necessary to study a reasonably
large number of strains which cover a wide range of virulence
from all parts of the spectrum. The converse, however, is not
necessarily true and it is safe to say that any one property is
not the cause of difference in virulence if one virulent and one
avirulent strain are identical in that property, providing the
strains are frankly virulent and frankly avirulent.
22

1) Morphology and virulence
The pleomorphic nature of NDV virus makes morphological
comparisons between strains difficult. Waterson and Cruickshank
(1963) compared the morphologyi before and after ether treatment)
of eight strains of varying virulence. There was some variation,
though of no relation to virulence, in the ease of ether disrup-
tionibut morphologically the whole virion, the diirupted particles,
end the RNP were similar for all the strains tested. Rott, Bade
and Schafer (1962) reported that up to 187. of their preparation
of strain Beaudette C had no RNP while strain Italian preparations
had only 27. empty particles. Since only two strains were examined
further work is necessary to show if there is any relationship
with virulence or not.
2) Serology
Serological studies of the surface antigens of NDV strains
using a serum neutralisation test (Upton, Hanson and Brandly, 1953)
showed up to 105-fold differences between strains. However, the
differences shown in no way related to the virulence of the strains.
The procedure used by Upton et al (1953) was later discredited
and using the now accepted procedure for serum neutralisation reac-
tions and comparing haemagglutination inhibition it has been shown
that no significant differences exist between the most virulent
and avirulent strains. (Waterson, Pennington and Allan, 1967).
Pennington (1967) using more sensitive techniques, and examin-
ing the internal nucleocapsid antigen as well as the external anti-
gens, showed only minor serological differences between strains
which in no way correlated with virulence.
The word strain then, when applied to the NDV system, is used
23

to mean a stable well characterised line of virus, and does not
imply a serologically distinguishable type.
3. Biophysical properties
Strains that differ as widely in virulence as those of HDV
may well differ in the constituents of the virion or in the ratio
of these constituents. The variation may be sufficient to give
a different bouyant density to the virions of the strains.
Roisman and Roan&-.(1961) have shown just such a difference between
two strains of herpes simplex virus. cletd showed different
cytopathic effects but otherwise appeared identical. However,
they could be separated on caesium chloride gradients. [T-he strains
were both grown under identical conditions on the same cell lice]
Stenback and Durand (1963) showed that NDV populations of
strain California behaved as if they contained infectious particles
with a wide range of density. By using a wide range of cells for
virus growth it was shown that the density in caesium chloride
was host controlled. The average densities in various avian cells
was between 1.219-1.221 whereas in mammalian cells 1.236-1.242.
The host control is not totally unexpected as the envelope of
HDV which must greatly affect the bouyant density is generally
accepted as being a modified piece of cell membrane (Cruickshank,
1964). This however raises the possibility that avirulent NDV
strains may have a different density than virulent strains due
to their reported maturation at different sites in the cell
(Bang, 1953).
4. Cytopathogenicity
During NDV infections there are several different kinds of
cytopathic effect and often only one particular effect has been
24

studied by any one author. For this reason this section is
divided into six parts although this is in no way intended to
suggest different mechanisms are involved or that the effects are
not different manifestations of the same event.
a) Haemadsorption
The ability of red cells to adsorb to the surface of infected
cells is the first evidence of cytopathic effect seen in NDV
infections. Marcus (1962) has shown that haemadsorption can be
detected as early as 4-6 hours in his HeLa or chick embryo cell
systems. He suggests that this cell surface modification is
brought about by the incorporation of newly synthesized haemagglu-
tinin into the membrane. Using a system in which single cells
could be studied he show& that, following normaf- growth curve kin-
etics, the amount of haemadsorption increased from a single BBC
per cell until the whole cell surface wts covered by adsorbed red
blood cells. Very little work has been carried out comparing the
ability of strains of NDV to induce the phenomenon but Bankowski
(1964) him shown that the less virulent strains tend to take longer
to show haemadsorption in monolayers.
b) Syncytial formation - cell fusion from within
When cells are infected with a cytopathic strain of NDV,
syncytia can first be noticed at about eight hours after infection
(Brett and Gallaher, 1969) and gradually increase in size, as well
as in numbers, by the fusion of more cells until cell death occurs.
There is no evidence that the size of single syncytia which may
contain 3-200 or more nuclei or the overall formation in a monolayer
has any relationship with strain virulence. However it has been
25

shown that the avirulent strain B1 fails to form syncytia
(Bankowski, 1964).
c) Plaque formation
If low doses of virus are applied to a cell sheeti between
24-72 hours/discrete:lesions (plaques) can be seen in the sheets
particularly after staining with neutral red. Neutral red is
adsorbed into the lysosomes of active cells hence normal cells
appear red in colour while dead or dying cells fail to take up
neutral red. Not all strains of NDV are capable of forming plaques.
Under certain conditions it has been noticed, with viruses
other than NDV, that groups of infected cells take up more neutral
red than surrounding uninfected cells and give the appearance of
'negative' or 'red' plaques (Bonifas and Schlesinger, 1959). Thiry
(1963) showed that similar red-plaque-forming mutants occurred
spontaneously in wild-type NDV populations and could be induced
using nitrous acid or hydroxylamine as mutagens. Thiry further
showed that cell sheets recovered completely from infections by
red plaque mutants but were destroyed by white mutants.
Bankowski (1964) has studied the cytopathology of five strains
of NDV of differing virulence and recorded the cytopathic occur-
ences over 72 hours, his work is summarised in Table 2.
d) Plaque size
Schloer and Hanson (1968) showed that of the 14 representa-
tive strains of NDV they studied only the velogenic and mesogenic *rains
formed plaques. Furthermore it was shown that of the strains that
formed plaques i the frankly virulent strains produced much larger
plaques than the mesogenic strains, and, as a general trend the
larger plaques were formed by the more virulent strains. Kohn and
26

TIME OBSERVED HOURS p.i.
TABLE 2
CYTOPATHIC EFFECTS OF DIFFERENT STRAINS OF NDV
SOURCE: BANKOUSKI (1964)
CPE/STRAINS
TEXAS
CAL 11914
TCND
A
B1
2
6
I0
24
48
72
•••
•••
WO
OM. IMO
Small syncytid formed, showing haemadsorption
Cell sheet destroyed
Cell sheet destroyed
As for Texas
Large syncytia and massive haemadsorption at edges of discrete plaques
As for Texas
As for Texas
As for CAL 11914
As for Texas
Very small syncytia and microscopic plaques some haem-adsorption
Destroyed 501 of cell sheet. Remaining cells show evidence of small plaques
Some cell death and occasional red cell attachment but no syncytid or plaques even when very high multiplicities used
Cells: Chick embryo
Inoculum: 100 TCID 50 per Leighton tube

Fuchs (1969) however, using eight strains of NDV were unable to
show more than a general tendency for plaque size to vary with
virulence. There is though no doubt that the very virulent strains
produce largo well defined plaques, the more moderately virulent
strains smaller, less well-defined plaques and the avirulent strains
no plaque at all.
e) Lysosome damage
Allison and Sande cin(1963) suggested that in virus infections
release of lysosomal enzymes may well contribute to cytopathic
effect. Further work, with viruses which varied in their cytopathic
effect including red and white plaque mutants of NDV, (Allison
and Mallucci, 1965) has shown that there are apparently two stages
in lysosomal damage:
i) Lysosomal membranes become permeable but do not Nr
release enzymes. This is demonstrable by the increased
uptake of neutral red into the lysosomes giving rise to
red plaques. The cell may recover from this state as it
does with the red plaque mutants of NDV.
ii) Lysosomal enzymes are released into the cytoplasm
due to further increase in permeability of the membranes.
The cells round up and uptake of neutral red decreases,
resulting in the formation of white plaques.
Work with other viruses has tended to confirm these original
findings (Allison, 1967, Poste, 1910) and lysosome damage is now
considered as almost certainly a cause of cell fusion if not total
cytopathogenicity.
f) Syncytial formation - cell fusion from without
If animal cells are treated with large concentrations (> 100
28

KID50/cell) of NDV, cell fusion occurs and polykaryocytes contain-
ing 3-150 nuclei/cell may be formed (Kohn, 1965). The fusion
begins within an hour and reaches a maximum within 2-4 hours. The
extent of cell fusion varies with the input multiplicity. Brett
and Gallaher (1969) have shown that this is a quite separate phen-
omenon from that of cell fusion after infection (fusion from
within). Fusion from within requires low inoculli of infectious
virus and protein synthesis while fusion from without needs neither
infectious virus nor protein synthesis for induction. They have
also shown that strains of NDV which induce fusion from within
are not necessarily good at inducing fusion from without and that
strains that are particularly good at fusion from without do not
necessarily produce fusion by infection at all.
Kohn (1965) attempted to demonstrate the viral component
responsible for fusion from without. Destruction of the cell recep-
tors with Vibrio &holerae neuraminidase stopped cell fusion so it
is necessary for the virus to become attached to the cells. However
since phospholipases were the most active in destroying cell fusion
ability Kohn concluded that the integrity of the phospholipids of
the viral membrane was the main requirement for the formation of
polykaryocytee.
Kohn and Fuchs (1969) studied eight strains of NDV and showed
that the ability to fuse cells from without varied enormously from
strain to strain but there was no correlation whatsoever with this
ability and virulence.
29

5) Biological Activities
In addition to cytopathogenicity NDV viriona have several
other biological activities associated with them: haemagglutina-
tion,neuraminidase, haemolysin, ADPase and ATPase. It is a
short step to argue that the differences in virulence may be due
to greater or smaller amounts, or greater or lesser activity of
one or more of these active units.
Only HA and neuraminidase of the activities listed above
appear to be integral parts of the virus structure; the others
being of host cell origin, perhaps derived from cellular lysosomes
(Neurath, 1964, Allison, 1967). However, Kohn and Fuchs (1969)
have shown that the variation in haemolysin activity has no rela-
tionship with the variation of virulence of the eight strains they
tested.
a) Haemagglutinin
Although the evidence is somewhat confused it appears that
viral haemagglutinin activity is in no way related to",,virulence.
Studies with unpurified virus are limited in their use since Rott,
Reda and Schafer (1962, 1963) have shown that with Beaudette 'C 187.
of HA present in their preparations was due to empty virus particles
whereas with Italien the figure was only 2%. Purification is
evidently necessary so that intact vArions only are examined.
Kohn and Fuchs (1969) using 8 strains of purified virus showed that
although the ratio of HA to infectivity varied it did not correlate
with the differences in virulence. Schafer and Rott (1959) reported-
that treatment of virus with ether produced a large fall in HA
activity with strain Italien whereas Beaudette C retained its
activity after treatment. Pennington (1967) examined many more
30

strains and showed that the loss or retention of HA activity after
treatment with ether is independent of virulence. Kohn and Fuchs
(1969) have also shown that the differences in heat lability of
HA is not related to virulence.
b) Neuraminidase
Of all the activities associated with the virion the most
likely candidate to cause the differences in virulence is neura-.
minidase. It seems highly likely that the production of neuramini-
dase in any quantity in the cell would lead to cell damage due to
the effect - on the mucoproteins in the cell membranes, Scholtissek,
Becht and Drzeniek (1967) have in fact shown that the cytopatho-
genicity of influenza virus was related to the presence of neura-
minidase activity.
Drzeniek, Seto and Rott (1966) have shown that the neuramtni-
dases of Italian and Beaudette C are identical in serological and
chemical properties, these strains are close in virulence but assum-
ing their findings applied to all strains then any variation of
neuraminidasa activity that correlated with virulence_ would be
quantitative rather than qualitative. Mussgay (1960) comparing
strains Italian (virulent) and B1 (avirulent) showed that the rate
of elution from fowl erythrocytes was far greater with Italian.
Howevar,it is difficult to make any conclueiona as to the relation-
ship with virulence from an experiment using only two strains.
It should be borne in mind that even if the quantity or activity
of the virus subunits do not vary from strain to strain as part of
the virion, the intracellular production may be unrelated to the
amount per virion. The possibility of this is discussed elsewhere
in this review (see virus multiplication growth studies section).
31

6. Virus multiplication
a) Growth rates
In any study with growth rates and virulence it is important
that 'one-step' growth curves should be examined. Using a one
step growth curve secondary adsorption, infection and inactivation
will not necessarily effect the experiment and that in fact the
growth cycle in one cell is being measured although with many
duplications. It is also important that both intracellular and
extracellUlar (or released) virus is measured as cell death may
be due to failure to release virus or due to damage at virus
release. For example Compans et al (1966) showed that BHK21-F
cells infected with' SV5 virus accumulated viral ribonucleoprotein
and disintegrated after extensive fusion although little mature
virus Was released. In contrast primary rhesus monkey kidney cells
infected with SV5 did not accumulate RNP, released infective virus,
but were not destroyed.
Studies with NDV have usually been confined to two strains,
one virulent and one avirulent. Liii and Bang (1953) compared the
growth of a virulent strain (CG179) and the lentogenic B1 in the
allantois of fertile eggs. The released virus reached a titre of
109 rt,50/mtin 26-28 hours with both strains but a further 10-fold
increase took 56 houri with the vaccine strain and only seven with
the virulent. Drain (1969) found similar results with B1 and Texas
(sea Table 3) but in neither of these cases were one step growth
curves used.
Using chick-embryo fibroblast monolayers and 100 /050/cell (i.e.
one step) Drain 1969 showed that in both intracellular and extracell-
ular production strains Texas and B1 were vary similar but events
32

TABLE 3
GROWTH OF VIRUS IN EGGS
Release of infectious virus into the allantoic cavity after low
infection (100 ID50 per egg)
Titre attained
Time taken (Hrs) ID50/m1
8 3 . 10
3 9. 10
9 8 . 10
10 9
1 10 0
Texas
24-25
35
47
CG 179
26
33
B1
28
40
72
B1
28
84
:Drain (19690
Liu S. Bang (1953)
6)9

with 81 tended to occur later (approximately one hour behind). In
a similar system Reeve and Waterson (1970) showed that the aviru-
lent strains Queensland and Ulster had growth cycles which showed
only minor differences to that of Italian and concluded that the
gross differences in cytopathogenicity between the two avirulent
and virulent Italian were in no way related to the differences in
the kinetics of growth in fibroblasts.
Hirsagay (1960) had earlier made the opposite concluaions
after studying Italian and F, reporting that the difference in
cytopathogenicity of the two strains was probably due to the slower
release of strain F. The differences noted by Mussgay were small,
however, and seem unlikely to account for the large difference in
the plaque sire. The crucial point is whether or not the avirulent
viruses produce plaques when a similar titre of virus to the viru-
lent strains is reached. Pennington (1967) studied strains F,
Queensland and Ulster which did not produce plaques in his system:
even after six days there was no sign of plaques although the titre
of released virus exceeded 10 10*ID50/ml which was greater than
the titre of released virus with virulent strains at the time
plaques were apparent.
The distinction,between those that form plaques and those
that do not is an important one and leads to speculation that there
are two groups which vary in virulence but by- different mechanisms:
1) The plaque formers which within themselves vary in
virulence due to the speed of growth but which eventually all
kill eggs and chickens and which if left a sufficient time would
(it Is postulated) all have a similarlmaximum plaque size.
2) The non-plaque formers which do not form plaques at all
34

and do not differ merely in growth rate.
It is important to remember in the study of growth rates that
with some strains up to 18% of released virus (as measured by
BA) may be present as non-infective virus (Rott et al, 1963) it is
therefore imperative that HA should be measured in conjunction
with infectivity. Fortunately the strains discussed above have
similar levels of non-infective particles.
Pennington (1967) showed that 40 hours after infection the
ratio of the HA titre of homogenated CAM to the HA titre of the
allantoic fluid was much higher for virulent strains than avirulent
strains; eight strains of varying virulence were tested and a
marked correlation was evident between the ratios and the MDT
in eggs. Reeve, Rosemblum and Alexander (1970) have made a more
detailed study of growth of different strains in eggs. Growth
curve studies of both CAM homogenate and allantoic fluid of infected
eggs led them to suggest that there were two discrete groups of
strains: 1) Virulent strains which kill chick embryos rapidly
and accumulate all the major viral components in the chorioallantoic
membrane to titres higher than in the allantoic fluid, and 2) aviru-
lent strains which kill embryos slowly or not at all and produce
similar extracellular antigen titres to virulent strains but in
which the intracellular accumulation of virus antigen is limited.
b) Growth site
The actual site of multiplication has also been implicated in
the differences in virulence of NDV strains. Kaman and Bang (1951)
compared the rates of increase of two strains CG179 (virulent) and
B (avirulent) in various tissues after intramuscular inoculation of
chickens. Both strains increased at the same rate in the external
9 r J

tissues, i.e. the blood, lungs, rectum and spleen, but the viru-
lent strain showed an accelerated growth rate in the brain. The
titre of the virulent strain was in fact 2000 times greater than
that of the avirulent strain after 24 hours. However the aviru-
lent strain eventually reached a higher titre in the brain though
much later. There was no brain damage or chicken death with the
avirulent virus which suggests that speed of growth, rather than
the titre reached is significant.
Bang (1953) also showed differences in the site of growth on
a cellular level by electron microscopy on thin-sections of infec-
ted Chorio-allantoic membranes. The avirulent strain (B1) showed
no damage to the cells and mature virus particles appeared to be
released from microvilli formed at the cell surface. The virulent
strain (C0179) however caused gross cellular damage and disorder
and appeared to mature within the cytoplasm. No other evidence
has been recorded to confirm the suggestion by Bang that this may
be the direct cause of the difference in virulence.
7. Effects on cell metabolism
Many viruses Which cause microscopically or grossly visible
cytopathic effects also inhibit the synthesis of cellular macro-
molecules (Martin and Kerr, 1968). The effects and causes of the
cut off of cell macromolecule synthesis have been studied in detail
for many viruses and although Bablanian, Eggers and'Tamm (1965)
have shown that poliovirus-induced switch-off of cell metabolism
was not responsible for the cytopathic effect of that virus it is
not certain to what extent other virus systems are affected.
36

a) Protein and RNA synthesis
Protein and RNA synthesis are closely linked and in general
have been examined together in studies involving NDV. Inhibition
of cell protein synthesis and RNA synthesis by NDV, using the
virulent Hickman strain, was shown by Wheelock and Tem (1961).
Using HeLa cells it was shown that 53k hours after infection
there was no incorporation of radioactive labelled precursors into
proteins. RNA synthesis appears to be more gradually inhibited;
by hour 10 a decrease of 40% of precursor incorporation had occurred.
One important feature of this work was that protein synthesis cut-
off occurred long before the gross cytopathic effect was apparent,
showing that it was unlikely that cell protein synthesis was
affected merely as a result of cell damage as Rott (personal communi-
cation) has suggested.
Bolognesi and Wilson (1966) using strain Texas G.B. with chick
embryo fibroblasts showed that cell protein synthesis inhibition
was initiated five hours after infection and by nine hours had
reached 85% inhibition.iiing the RNA inhibitor azauridine viral
protein synthesis cut-off could be prevented if the inhibitor was
added at the time of infection but not if added after three hours.
These, and other results with actinomycin D and cycloheximide indi-
cated that proteins synthesised during early NDV replication were
responsible for the inhibition of protein synthesis and the synthe-
sis of these proteins required the prior synthesis of RNA.
Scholtissek and Rott(1965)reported that with strain Italien and
chick embryo fibroblasts inhibition of cell protein synthesis was
reversible and infected cells recovered after ten hours. Wilson
37

(1962) using strains Texas G.B. and Beaudetta C was unable to
show a similar recovery.
Wilson (1962), using Texas G.B. (virulent) and Beaudette C
(mesogenic, - but see note at beginning of section) has made the
only attempt to compare two strains of differing virulence to see
if their ability to cut off cell protein and RNA synthesis varied.
Both strains inhibited protein synthesis to a similar level but
experiments with metabolic inhibitors suggested that different
mechanisms were involved. RNA synthesis inhibition by Texas was
marked (more than 807. inhibition at 10 hours) and shown by use
of cycloheximide and azauridine to involve the synthesis of inhibi-
tory proteins coded by the virus genome. Beaudette C shoved very
little inhibition of RNA synthesis at all, in fact a slight increase
was recorded presumably due to viral RNA synthesis. This however
may not be associated with virulence as Scholtissek and Rott (1965)
showed that strain Italian (highly virulent) produced a similar
effect in their system.
b) DNA synthesis
Wheelock and Teimn (1961) showed that using the virulent
Hickman strain in a HeLa cell system DNA synthesis became almost
completely inhibited by NDV within five hours of infection. Further
work with synchronized and unsynchronized,L cell cultures using
strain Hickman has lead to a greater understanding of the mechan-
ism of DNA inhibition by NDV (Ensminger and TamM, 1970a, 1970b).
Cellular template DNA retains its structure after NDV infection
so that it is unlikely that degradation of template DNA causes
DNA synthesis inhibition. Similarly synthesis and polymerization
enzymes of precursor deoxyribonucleotides are unaffected. However
38

it has been shown that maintenance of DNA synthesis requires con-
tinual protein synthesis in uninfected cells (Ensminger and Tamm,
1970b). Ensminger and Tamm postulate, therefore, that NDV inhibits
cellular DNA synthesis secondary to the inhibition of protein
synthesis and that viral inhibition of protein synthesis affects
a process required for the initiation of DNA synthesis upon new
sites of replication.
c) Lipid' synthesis
Drain (1969) extensively studied the effect on 14C-acetate
incorporation into lipid of infection by strains Texas and B1
(avirulent). Very little difference was observed, both strains
produced gradual suppression of lipid metabolism to as low as
309. by 20 hours after infection.
8. Interferon and virulence
a) Interferon sensitivity
Sensitivity to interferon could obviously be a factor affecting
the virulence of a virus, and it is known that in some virus systems
the less virulent strains are more sensitive to interferon than the
virulent strains (Penner, 1968). However, Baron (1964) has shown
that nine strains of NDV of widely varying virulence are relatively
insensitive to interferon and that interferon sensitivity is unlikely
to have much significance in the variation of virulence between
NDV strains.
b) Interferon production
It has been shown that in cells infected with Semliki Forest
virus (Pinter, 1964), vesicular stomatitis virus (Wagner et al, 1963),
foot and mouth disease virus (Sellers, 1963, 1964) and herpes simp-
39

lex virus (Aurelian and &Oxman, 1965) in the system studied the
lower the virulence the greater the yield of interferon. Fenner
(1968) has suggested that the lack of interferon production in
virulent strains is due to the rapid cut off of cell macromolecule
synthesis by the virulent strains compared to.the'less virulent
,strains. Gandhi and Burke (1970) drew similar conclusions from
' their work with NDV. However Thacore and YOungner (1970) using
strain Harts and a mutant from persistently - infected L cells
showed that in L cells interferon production still occurs although
shut off of cell synthesis is far more complete than in chick
'embryo cells which do not produce interferon on'infection. They
suggest that the defect in induction of interferon synthesis in
chick embryo cells infected with Harts strain of NDV is not rela-
ted to the general shut off of host cell synthesis but to the fail-
ure of a more specific event. Thiry (1963) has shown using 'red'
mutants that in the NDV system the interferon production of cells
in vitro is inversely proportional to the virulence of the infec-
ting strain. Baron (1964) has similar findings with infected
fertile chicken eggs but shows that little if any interferon is
produced with the truly lentogenic strains B1 and LaSota.
Lomniczi (1970a) has studied the in vivo production of inter-
feron in chicks infected with NDV and has shown that virulent
strains are more potent inducers of interferon than avirulent
strains, these findings in fact agree with those of Baron (1964)
as the avirulent strains used were Blb LaSota and F. In addition
Lomnicsi has shown that induction of maximum interferon was uni-
formly at the second hour after infection with avirulent strains
40

whereas with virulent strains the maximum was considerably later
and varied with strain. Looniest (1970b) also showed a different
mechanism was involved,since damage to the viral nucleic acid
affected the interferon inducing ability of the virulent strains
whereas damage to the viral protein affected the interferon induc-
ing ability of the avirulent strains.
41

G. EFFECT OF CONGO RED ON NDV INFECTIONS
Finkelstein (1961) showed that congo red, trypan blue and
other azo dyes protected eggs against low but lethal doses of viru-
lent NDV. Further to this Becht and Dreeniek (1960 have shown
that neuraminidase activity in vitro is inhibited by congo red and
that no infective virus is released from infected chick embryo
cells in the presence of this dye.
Since it has been shown that neuraminidase plays an important
part in myxovirus release (Seto and Rott, 1966, Seto and Chang,
1969; discussed above) it seems likely that Finkelstein's observa-
tions were due to the azo dye inhibiting neuraminidase which pre-
vented release of infective particles. This in turn would mean
that only a sublethal number of cells in the CAH were infected and
subsequently killed.
If congo red does act merely baj prevention of release then,
if sufficiently high doses are used, virulent virus should be unaff-
ected in lethality to eggs.
It has been suggested (Reeve et al, 1970) that the accumula-
tion of virus antigens in infected cells results in the death of
these cells and eventually the death of the infected egg. If
congo red prevents virus release but not virus production in infec-
ted cells it could be postulated that avirulent viruses would
accumulate in and therefore kill cells treated with congo red.
Similarly very high doses of avirulent strains would be lethal to
eggs in the presence of congo red.
42

H. VIRUS-SPECIFIED PROTEINS IN NDV-INFECTED CELLS
When a virus particle infects a cell several events may take
place: uncoating, cessation of cell metabolism, synthesis of
viral nucleic acid, synthesis of virus structural proteins, assem-
bly of proteins,.cytopathic effect and, with NDV, cell fusion and
haemadsorption. Any one of these may require the synthesis of a
separate virus-specified enzyme or other non structural proteins.
A method for investigating the proteins synthesised during
viral infections was reported by Summers, Maisel and Darnall (1965).
This method consisted of radioactive labelling infected cells with
an amino acid in the presence of actinomycin D, to prevent cell
protein synthesis, extracting the proteins using sodium dodecyl
sulphate, and separating them by polYacrylamide disc electrophor-
esis. In this way the proteins of the cell are separated accord-
ing to their molecular Weights and form discrete bands on the gel.
The,gel was then sliced and the radioactivity present in each band -
measured. By this method Summers at al (1965) have shown that 14
virus-specified proteins are made after infection with poliovirus,
while only 4 are seen if pure virus is extracted and similarly
treated. 'Hay, Skehel and Burke (1968) have modified this method
so that host Cell metabolism need not necessarily be stopped. In
their method infected cells are labelled with 3H-amino acid while
uninfected cells are labelled with 14C-amino acid, the uninfected
and infected cultures are harvested, mixed and then, after extrac-
tion and disc electrophoresis, sliced and counted. The ratio of
the 3H dpm to the 14C dpm is highest in bands which are virus-
43

specified since these proteins are not present in uninfected cells.
Hay at al (1968) have shown by this method that Semliki Forest
virus grown in chick embryo cell monolayers formed six virus-
specific proteins, only two of which were present in similarly
extracted pure virus. This refinement of the method is partic-
ularly useful for studies with NDV, because although NDV is insen-
sitive to actinomycin D added after infection it has been reported
to be affected by the addition of actinomycin D to cultures before
infection (Kingsbury, 1962, Granoff and Kingsbury, 1964, Brett
and Robinson, 1967). Addition of actinomycin D at the time of
infection would mean that analysis of virus-specified proteins
produced immediately after infection would be masked by cellular
protein synthesis still occurring, unless the double labelling
technique is used.
Virus-specified proteins may be of great importance in the
NDV system since it can be postulated that one particular protein
(or proteins) which is responsible for cytopathogenicity and cell
death is present only in virulent strains. The time at which
specific proteins are produced during NDV infections can also be
monitored by this method and may prove significant to the virulence
of the infecting strain.
44

3. PURIFICATION OF NDV
Purification of NDV, and large RNA viruses in general, poses
several special problems, mainly because the virions have many
physical and chemical properties similar to those of cellular mem-
branes and because of the great range in size of individual par-
ticles.
Adsorption and elution from erythrocytes (Ada and Perry, 1956,
Hoyle, Horne and Waterson, 1961) has been used for myzovirus puri-
fication but the main disadvantage of this procedure is the intro-
duction of red cell debris into the virus preparation.
The surface properties of myzoviruses have also been used to
separate and purify virus by column chromatography using, alumin..
ium phosphate-silica gel (Prommhagen and Knight, 1959), calcium
phosphate (Taverns, Marshall and Fulton, 1958), or ion exchange
cellulose derivatives (Wilson, 1962, Nicoll, Retail and Colobert,
1964).
Commonly, virus is partially purified and concentrated by
differential centrifugation, consisting basically of low speed
centrifugation to remove large fragments of cellular material and
high speed centrifugation to collect the virus. However signifi-
cant amounts of cellular material remain in the virus pellet. •
Ammonium sulphate precipitation has also been used as an initial ,
concentration step (Minocha, Consign and Eisenstark, 1968) but is
less desirable since it is known to cause loss of infection in sev-
eral viruses (Robinson and Duesberg, 1968).
Centrifugation in density gradients separates components on
the basis of their buoyant density (isopycnic separation) or rate
45

of migration which may be determined by size and shape (rate zonal
separation). Gradients of heavy metal salts such as caesium chlor-
ide have been used to purify NDV (Minocha et al, 1968, Stenback and
Durand, 1963) but with high loss of infectivity. Potassium tar-
trate (McCrea, Epstein and Barry, 1961) and sucrose (Duesberg and
Robinson, 1963) gradients have produced far superior yields of infec-
tious virus.
The introduction of specially designed 'zonal' rotors has
enabled large volumes of viral suspension to be purified to a high
level by a fairly simple method (Fox at al, 1967, Fox et al, 1968).
The major disadvantage of density gradient centrifugation is
that the buoyant density of the myxoviruses range from 1.15-1.25
g/ml which is exactly the range of density of most cell membranes.
This may result in difficulties in purifying highly contaminated
suspensions of tissue culture-grown virus.
In this study due to the availability of zonal rotorsi density
gradient centrifugation was chosen for the purification of virus.
Most of the virus to be purifiedkis egg grown and does not suffer
from gross contamination with cell debris.
46

K. OBJECTS OF INVESTIGATION
It is the object of this investigation to obtain a fuller
understanding of the broad spectrum of virulence seen with diff-
erent strains of NDV.
The major part of this work deals with the proteins of NDV:
the structural proteins, their activity, quantity and the number
present in the virion; an investigation into the virus-specified
proteins and the time that they are produced.
An attempt has also been made to clarify past results which
have been confusing or contradictory. Consequently the phenomena
of cell fusion and haemadaorption, cell protein synthesis inhibi-
tion and the growth of strains of NDV in eggs and cell culture
under certain conditions have all been studied with respect to
variation in strain virulence.
47

II MATERIALS AND METHODS
A. REAGENTS AND EQUIPMENT
1) Reagents
The following materials were obtained from the manufacturers
specified below. A11 other materials wore normal laboratory reag-
ents of 'Analar' quality unless otherwise stated.
Congo Red (for microscopical staining); sodium dodecylsulphate
(specially pure): B.D.H. Chemicals Ltd., Poole, England.
Actinomycin D (STAT..PACK); Nauramine Lactose (from beef colos-
trum): Cal -biochem Ltd., Los Angeles, U.S.A.
Acridine Orange, Gismos Solution, May Grftwald solution, Tween
20; G.T. Gurr Ltd., London England
Buffer Tablets pH6.8: Hopkins and Williams Ltd., Chadvell Heath,
England.
Crystamycin (Sodium penicillin G + streptomycin): Glaxo Labora-
tories Ltd., Greenford, England.
Trypsin solution (sterile, freeze-dried); 4.4% sodium bicarbon-
ate; Eagle's HER medium 10 x strength; Tryptose phosphate broth:
Wellcome Reagents Ltd., Beckenham, England.
Foetal calf serum; Flow Laboratories Ltd., Irvine, Scotland.
Ianagar No. 2; Earle solution (Earle's saline): Oxoid Ltd.,
London, England.
Sephadex G-200: Pharmacia, Uppsala, Sweden.
The following radioisotopes were all obtained from The Radio-
chemical Centre, Amersham, England:
L-Leucine 4,5-T; 19.7-23.0 C/MM (3H-Leucine)
48

L-Leucine-C14(u) 344mC/MM (14C-leucine)
Utidine -5 -T, 17.5-26.5 C/mM (311-uridine)
D -Clucosamine-1 -C14 hydrochloride, 45mC/mM (14C-glucosamine)
L-Arginine -5 -T monohydrochloride, 500 mC/mM (3H-arginine)
n-Hexadecane 1,2, -H3, 200pC/g (3H-hexadecane)
n -Hexadocane -1 -C14 1.10pC/g (14C -hexadecane)
Fetuin
Fetuin was prepared from foetal calf serum by the method of
Graham (1961).
Phosphate Buffered Saline (PBS)
PBS was made from tablets of phosphate buffered saline 'A'
(Dulbecco and Vogt, 1954) obtained from Oxoid Ltd., London.
Coll Culture Media
i) Growth medium
Growth medium was prepared with sterile distilled water to
contain:
10% v/v Eagle's BHIC 10 x strength
10% v/v Foetal calf serum
2% v/v Sodium bicarbonate (4.4% solution)
107. v/v Tryptose phosphate broth
Penicillin and streptomycin to a final concentration of 200
units and 200pg respectively.
ii) Maintenance medium
Maintenance medium was prepared in the same way as growth
medium with the following exceptions.
Foetal calf serum 2%
Tryptose phosphate broth 5%
49

Chicken Red Blood Cells
Chickens ware bled from a wing vein into Alsever's solution
(sodium citrate 2.25g, citric acid 0.8g, glucose 2.2g and water to
100 ml.). The blood cells were then washed three times and suspen-
ded in PBS. The 'packed cell' concentration was calculated using an
haematocrit tube, centrifuging at 3,000 r.p.m. for 20 minuntes in
a M.S.E. 'minor' centrifuge.
2) Equipment
a) Centrifuges
The following centrifuges were used: M.S.E. superapeed 65,
M.S.E. 'minor', M.S.E. 6L; Measuring and Scientific Equip-
ment Ltd., London, England. Sorvall superspeed RC2-B, Ivan Sorvall
Inc., Conneticut, U.S.A.
b) Spectrophotometers
The following spectrophotometers were used for Optical density
measurements: SP600 series 2, SP500 series 2, SP8000; Unice&
Instruments Ltd., Cambridge, England.
LKB Uvicord /I; LKB Produkter AB, Stockholm, Sweden.
c) Densitometers
Photographs of stained gels were scanned using the SP590
electrophoresis strip scanning accessory to the SP500.
Unstained and stained gels were scanned,sed-using the gel-
scanning attachment to a Hilger-Gilford Reaction Kinetics Spectro-
photometer.
d) Fraction collector
In all experiments involving fraction collecting an Ultrorac
7000 automatic fraction collector (LKB Produkter AB, Stockholm,
Sweden) was used.
n Ju

a) Microscopes
A Leitz Orthoplan microscope (Ernst Leitz, Wetzlar, Germany)
was used for most microscopy and photomicroscopy.
A Nikon inverted microscope (PEN Optics Ltd, London, England)
was used for cell culture work.
51

B. VIRUS STRAINS AND VIRUS TECHNIgpES
1) Strains
The following strains were used in this investigation, their
virulence for chickens, chick embryos and ability to form plaques in
chick embryo cell monolayers is given in Table 1.
Harts 33 (Herts)*, isolated at Weybridge in 1933 (Dobson, 1939).
Field Pheasant*, isolated at Weybridge in 1962 from pheasants,
(Allan, personal communication).
Texas C.B. (Texas)* isolated in 1948 near Austen, Texas, U.S.A.
(Boney, 1951).
Warwick* isolated in Warwickshire in 1966 (Allan, personal
communication).
Italian**, isolated in Italy at some date before 1949 (Schafer,
Schramm and Traub, 1949).
Eastwood Notts*, isolated in England in 1966 (Allan, personal
communication).
Beaudette C**, this strain is a heat stable variant isolated
from the Beaudette strain of NDV by Granoff (1959). The parent
strain was isolated in the U.S.A. before 1946 (Granoff, 1964).
'11 1 * isolated in the U.S.A. (Iyer and Dobson, 1940).
B1* accidently isolated as a supposed contaminant of an infec-
tious bronchitis virus culture (Hitchner, 1948).
F*, isolated in England in 1949 (Asplin, 1952).
Queensland V4 (Queensland)* isolated in 1966 by Rylie in
Brisbane, Australia (Allan, personal communication).
Ulster*** isolated in Northern Ireland in 1966 from the faeces
of apparently healthy chickens by NcFerran (personal communication).
52

Seed viruses were received from:
*W.H. Allan, Ministry of Agriculture, Fisheries and Food,
Central Veterinary Laboratories, Weybridge, Surrey, England.
**R. Rott, Institute of Virology, Giessen, West Germany.
***J.B. NePerran, Ministry of Agriculture for Ireland, Belfast,
Northern Ireland.
2) Growth of virus
Stock virus and all other virus, i.e. radioactive virus, virus
for treatment with detergent and testing neuraminidase activity,
was grown in eggs.
Fertile hens eggs were obtained from Poyndon Farm Ltd., Goffs
Oak, Hertfordshire, and incubated at 37°C for 9-11 days before use.
Seed virt) or infected allantoic fluid (0.121 of a 10-2 dilu-
tion) was inoculated into the allantoic cavity and the eggs incu-
bated nt 37°C. After 42 hours, with virulent strains, or 72 hours
with avirulent strains, the eggs were chilled at 4°C for several
hours and the allantoic fluid was harvested.
After low speed centrifugation (1000) to remove red blood
cells and any cell debris, virus was either stored as infective ellen-
tole fluid or centrifuged at 40,0001 and resuspended as a more con-
centrated suspension before storing.
Virus suspensions were stored overnight at 4°C or for longer
periods in a -70°C deep freeze.
53

C. CELL CULTURE TECHNIQUES
1) Preparation of chick embryo fibroblast cultures
The heads, legs, guts and viscera of 10-12 day old chick
embryos were removed and the bodies forced through a 10m1 syringe
into sterile PBS. The macerated embryos were washed three times
in PBS and then 100m1 of 0.27w/v trypsin in PBS was added for
every 12 embryos used. The mixture was then stirred at 37°C for
30 minutes after which 10% v/v calf serum was added to inhibit the
trypsin. The suspension obtained was then filtered through a coarse
wire mash strainer and centrifuged for 15 minutes at 1300g. The
supernatant obtained was discarded and the cells resuspended in
growth medium. After passing the cells through a sintered glass
filter they were diluted with 0.17w/v trypan blue in PBS and coun-
ted using a Neubauer counting chamber.
Cells were usually adjusted to 2 x 106/ml for seeding but
sometimes when confluent monolayers were not required concentrations
of 0.6-0.8 x 106 cells/mi were used.
Leighton tube coverslips were seeded with 1.5m1 of 2 x 106/ml,
5cm diameter plastic Petri dishes received 4m1 and 14 cm glass die.-
hes received 2$al to give confluent monolayers in 48 hours.
All cell cultures were incubated at 37°C. Culture vessels
which are not air tight were incubated in an incubator continuously
flushed with a CO2/Air mixture giving a final concentration of 57.
carbon dioxide.
2) Infection of cell monolavers
The required dilution of infected allantoic fluid was made in
maintenance medium and a sufficient volume to cover the surface of
54

the monolayer was used. After adsorption at 37°C for one hour
cells were washed twice with warm PBS or maintenance medium and
fresh maintenance medium added.
The following multiplicities were used:
Experiment Multiplicity
Cell fusion and haemadsorption
10 EID50 /cell
Cell fusion and haenadsorption in
the presence of inhibitors and all
other experiments involving meta-
bolic inhibitors
50 EID50/cell
Double-labelling experiments
50-100 EID50/cell
LDS experiments
50 BID50 /cell
3) Staining call monolayers
a) Giemsa--May GrUnwald
A modification of the method of Jacobson and Webb (1952) was
used. After washing with PBS,coverslip-monolayers to be stained
were fixed in methanol for for 15-30 minutes. The coverslips were
then ibeed in pH6.8 buffer solution containing 5% v/v Giemea solu-
tion and 5% v/m, May GrUnwald solution and left for 20-25 minutes.
The coverslips were then washed once in tap-water and twice in
pH6.8 buffer. After drying under infra red lamps the coverslips
were mounted in DePex (G.T. Gurr Ltd.) for microscopy.
b) Acridine orange
To stain cells with acridine orange the method of Alwyn (1968)
was used.
Michaelis' buffer (Michaelis, 1931) pH4.2 was prepared:
Sodium acetate 9.7g
55

Sodium barbiturate 14.7g
Distilled water to 500m1
For use this buffer was diluted:
Michaelis' Buffer 50m1
Sodium chloride 8.574w/v 20m1
UC1 120m1
Distilled water 60m1
Coverslip cell cultures were immersed in acridine orange 0.017.w/v
In diluted buffer for 10.20 minutes. They were then washed three
times in diluted buffer and mounted in 8%v/v glycerol. Cultures
were then examined under the Leitz microscope using fluorescent light
transmission filter 5mm. B12 and suppression filter K510 or 1530.
In cells stained with acridine orange RUA appeared red while DNA
appeared yellow-green.
Acridine orange solution is extremely susceptible to light and
was therefore stored in a brown glass bottle covered in silver foil
at 4°C.

D. BIOLOGICAL ASSAYS
1) Haemagmlutinin assay
Haemagglutinin (HA) activity vas measured in standard plastic
plates in 0.2m1. volumes. PBS was used as a diluent with 1% chicken
red blood calls.
2) Infectivity
Infectivity for chick embryos was measured by inoculating 0.1m1.
volumes of serially diluted virus suspensions into the allantoic
cavity of 9 or 10-day old fertile chicken eggs. Eggs were candled
daily and after 7 days the allantoic fluid of surviving eggs was
tested for the presence of haemagglutinin. The 50% egg infective
dose (BID50) per ml. was estimated by the Kaerber method.
3) Plaque assay
Primary chick fibroblast monolayers in 5cm diameter plastic
Petri dishes were infected with ten-fold dilutions of virus in PBS
containing 2% foetal calf serum. After adsorption, for one hour, the
inoculum was removed and 4m1 of 0.97.w/v /onager in maintenance media
was added. Dishes were stained with neutral red 0.1%w/v in PBS and
examined for plaques three days after infection.
4) Neuraminidase assay
Neuramimidase (mucopolysaccharide N-acetylneuraminyl hydrolase,
BC.3.2.1.18.) activity was estimated by measuring the amount of N-
acetyl neuraminic acid (NANA) released from a suitable substrate.
The solution to be tested (0.1m1) was incubated with 0.1m1
substrate and 0.3m1 buffer for 10 minutes at 37°C. The NANA rele-
ased was estimated by the method of Aminoff (1961).
The substrate used was either bovine neuramine lactose (vhere,
57

specified) 200pg per test, or the equivalent amount of fetuin.
NANA solutions were used as standards and enzyme activity was
expressed as pg of NANA released.
The buffer was, unless otherwise specified, 0.1H sodium acetate-
acetic acid buffer, pH5.2 for fetuin or pH5.4 for neuramine lactose.
5) Protein assay
The method of Lowry et al (1951) was used for the estimation
of proteins. The final volume was 6.5m1 and optical density (OD) was
read at a wavelength of 600nm. Bovine serum albumin was used as
a standard.
6) Lactate dehydrogenase activity
Lactate dehydrogenase (LDH) activity was estimated by the
spectrophotometric method described by Kornberg (1955).
7) Estimation of haemadsorption
Coverslip cell cultures were washed three times with ice cold
PBS at the specified times after infection and then received 0.5m1
of a 0.5%v/v suspension of chicken red blood cells. After 15-20
minutes at 4°C, unadsorbed red blood cells were removed by three
further washes with ice-cold PBS. The cultures were then fixed in
methanol, stained with May-GrUnwald.4iemsa, and the number of red
blood cells adsorbed to the cells counted.
The amount of haemadsorption was expressed as the number of
red blood cells adsorbed per cell or per unit area. In experiments
with metabolic inhibitors the number of red blood cells per unit
area in monolayers treated with inhibitor was expressed as a per-
centage of the number in the same area of inhibitor free cultures,
measured 8 hours after infection.
58

8) Estimation of cell fusion
Cell fusion was estimated by one of two methods:
a) Size of syncytia
The size of syncytia WAS estimated from coverslip monolayers
of chick embryo cells stained with Giensa-May GrUnwald 12 hours
after infection. Size was expressed as the average number of
nuclei seen in syncytia at that time.
b) 7. polykaryocytosis
The extent of cell fusion was estimated from confluent cover-
slip monolayers stained with Giemsa-May Grunwald 15 hours after
infection. The results are expressed as the number of nuclei invol-
ved in syncytia in a given area, i.e. one field of the microscope,
as a percentage of the total number of nuclei in that area.
In experiments with metabolic inhibitors the % polykaryocy-
tosis with treated cells is expressed as 7. of the polykaryocytosis
in inhibitor free infected-cultures
9) Estimation of call-associated neuraminidase activity
For each strain tested 1 x 109 EID50 were inoculated into the
allantoic cavity of 10-day-old eggs. After 22 hours incubation
at 37°C the chorioallantoic membrane was removed from each egg,
washed three times in ice-cold PBS and stored at -70°C until assayed.
The membranes were then thawed, and blended in homogenizers with
motor driven Teflon pestles in 0.0114 sodium acetate-acetic acid
buffer, 05.2. The homogenate was then assayed for neuraminidase
activity and protein.
J r

B. VIRUS DISRUPTION
1) Sodium dodecyl sulphate (SDS) 2-marcaptoethanol
Virus disruption prior to acrylamide gel electrophoresis
was done by suspending pelleted virus in 17. w/v sodium dodecyl
sulphate (SDS) and 1% w/v mercaptoethanol (Shapiro, Vinuela and
Maisel, 1967) or by making suspensions this concentration by addi-
tion of 107. w/v SDS-mercaptoethanol. The mixture clarified immed-
iately but was heated for 30-60 minutes at 37°C. No difference was
seen in the SDS-gel pattern of disrupted virus if 17. w/v dithio-
threitol (DTT) was used instead of mercaptoethanol (Gordon, 1969),
the mixture was heated at 100°C for 1 minute - (Maisel, White and
Scharff, 1968) or if 0.5M urea was present.
When run in parallel with double-labelled cell extract, i.e.
at the same time but not in the same tubes, virus was disrupted by
the method of Summers, Maizell and Darnell (1965, also sae below),
however no difference in gel patterns was seen when compared to ord-
inary SDS disrupted virus gels.
2) Tween 20 at pH10
The method used to disrupt virus with Tween 20 was that of
Webster and Darlington (1969). Virus was suspended 17v/v Tween 20
in 0.02M bicarbonate buffer, 010, The mixture was held at 37°C
overnight and intact virus removed by centrifugation at 45,000g for
30 minutes.
Tween 20 was separated from the supernatant and partial separa-
tion of neuraminidase and HA activities was obtained by separation
on a 0200 sophadex column.. A column of Sephadex G200 was made in a
60

K25/100 Sephadex glass column (Pharmacia, Uppsala, Sweden). The
length of the column was 35cm and the width 2.5cm. The samples were
applied to the bottom and elution with PBS was upwards using an
LKB peristaltic pump run at 10ml/hr., 3 ml. fractions were collected.
The column was kept at 4°C.
3) Sodium deoxycholate
To disrupt virus with sodium deoxycholate a modification of
the method of Kingsbury and Darlington (1966) was used. Virus was
pelleted by centrifugation at 40,0008 for 40 minutes and resuspen-
ded to a concentration of 1mg/m1 of protein in 0.01M deoxycholate.
RNP was separated from the disrupted virus membrane by sucrose den-
sity gradient centrifugation. The gradient was made 5-407.w/v
sucrose in 0.01M DOC and centrifugation was in a 3 x 23m1 swingout
rotor at 4°C for IA hours at 60,000g.
61

F. RADIOACTIVE ISOTOPE COUNTING TECHNIQUES
1) Scintillation counter
The counter used to count all labelled materials vas an ABAC
S1.40 liquid scintillation spectrometer: Intertechnique Ltd.,
Portslade, England.
2) Scintillation fluids
a) Non aqueous samples
Non aqueous samples, including dried coverslip monolayers and
dried glassfibre filters, were counted in 15 or 10m1 of scintillation
fluid prepared:
Butyl-PBD (Intertechnique Ltd.) 8.0g
Toluene (Scintillation grade) to 1000m1
b) Aqueous samples, including dissolved polyacrylamide gel
fractions were counted in 15m1 of scintillation fluid prepared:
Butyl-PBD. 8.0g
Naphthelene (Intertechnique Ltd.) 80.0g
Thixotropic gal (Cab-O-Sil, Packard Instruments Ltd.,
Wembley, London) 40.0g
Made up to 1000m1 with a 2-ethoxyethanol 40%v/v:
Toluene 60%v/v mixture.
Cab-O-Sil is used to keep any precipitate that may form evenly
distributed throughout the scintillation fluid (Ott et al, 1959).
3) Estimation of uptake of radioactive isotope into coverslip cell
cultures
To estimate the amount of uptake of radioactive labelled pre-
cursor into macromolecules in coverslip cell cultures the following
62

method was employed.
After removal of the media containing the radioactive precur-
sor cultures were washed twice in ice cold PBS and three times in
ice cold 5Zv/v trichloracetic acid (TCA). After a further wash in
acetone the coverslips were air-dried and then transferred to 15m1
toluene butyl PBD scintillation fluid for estimation of radioactivity.
Maintenance media containing 1.0µC/ml of 311.4eucine was used
to pulse label coverslip call cultures for 30 or 60 minutes in
experiments involving protein synthesis.
In RNA-uptake experiments 1.0µC/ml in maintenance media vas
used, in the presence of 5µg/m1 Actinomycin D but accumulation of
counts was measured.
4) Estimation of radioactivity_ in Bel slices
Polyacrylamide gels containing radioactive labelled polypep-
tides were fractionated using a Mickle gel slicer (Joyceuloebl Ltd.,
London, England) into slices 1mm thick. Each slice vas then diss-
olved in "100 volumes" hydrogen peroxide by incubating for 1-2
hours at 90°C or 60°C overnight (Young and Fulhorst, 1965). This
treatment also decolorises gels previously stained with amid*
black. Scintillation fluid (15m1, of 2 -Othoxyethanol-toluane4ab-
0-sil) was then added to the dissolved gel and the radioactivity
counted. Usually the vials were held at 4°C for a few hours before
counting as this facilitated the removal of air bubbles by gentle
shaking.
5) Quench curve
The estimation of radioactivity due to 14C and 311 in a double
labelled sample using liquid scintillation spectrometry poses sav-
63

eral problems. The energy spectra of disintegration of 14C and 3H
overlap in the machine so that without some method of resolution it
is not possible to distinguish the CPM contributed by each isotope.
Furthe4 many substances, in this case dissolved acrylamide gel,
reduce the counting efficiency of, or 'quench' these isotopes
causing an increase in the overlapping of the spectra. The degree
of quench relates to the concentration of the quenching material.
To separate 14C counts from 3H counts in a double labelled
sample a quench curve (i.e. the relationship between efficiency of
counting and the amount of quench) was constructed. This was done
using the external standard ratio method described in the Intertech-
nique Handbook for the ADAC SL4O spectrometer which approximates
the counting efficiency/quench relationship to a third order poly-
nomial curve. This enables the machine to compute the cross-
contribution of 14C activity to the 3H spectrum and calculate the
disintegrations per minute of each sample.
Standard radioactive solutions were made using 14C-hexadecane
and 3H-hexadecane. Quench material was polyacrylamide gel dissolved
in hydrogen peroxide.
Occasionally dissolved fractions of gel formed a fine precipi-
tate on addition of scintillation fluid. However because Cab-06-
sil keeps fine precipitates evenly distributed throughout the
scintillation fluid the quench curve could be constructed using
the 'external standard ratio' method.
The curve constructed gave correct disintegrations per minute
(DPM) of double labelled standards to within ±1% of the known
radioactivity.
64

6) Radioactive virus
Radioactive virus was grown in an identical manner to unlabelled
virus except that prior to Infection radioactive precursor was
inoculated into each egg. Usually 5pC 14C-labelled precursor or
10pC 38-labelled precursor were added in 0.1m1 of PBS to each egg.
65

G. POLYACRYLAHIDE GEL DISCELECTROPHORES/8
The method used is basically a modification of that of Ornstein
(1964) and Davis (1964).
The apparatus used was the standard 'Shandon' equipment
(Shandon Scientific Co. Ltd., London,England) except that glass
running tubes were either: 0.7cm internal diameter, 8.5cm length;
or 0.5cm internal diameter, 7.5cm length.
1) Preparation of 7.5% eels
The following stock solutions were made and stored at 4°C.
a) Buffer
Tris (hydroxymethyl) methylamine (Tris) 36.6g
N,N,N ,N1:Tetramethylethylenediamine (TED) 0.23ma
IN Hydrochloric acid to p88.9 approximately 48ml
Distilled water to 100m1
b) Acrylamids
30g
Distilled water to
100m1
c) N,Hr-methylenebisecrylamide (BIS)
cP5g
Distilled water to 100m1
To prepare 7.3% acrylamide gels the above solutions were mixed:
a) one part,
b) two parts
c) one part
Distilled water four parts
The mixture was deearated by vacuum and made ' 0.6%v/v with ammon-
ium persulphate solution (7%w/v), shaken gently in a circular motion
so that reiteration did not occur. Glass tubes, suitably stoppered
with rubber or plastic caps, were then filled with mixture to lcm
from the top of the tube. A distilled water overlay was then
66

added with sufficient care to avoid disturbing the interphase. A
syringe was used and the needle pressed against the side of the
tube to avoid such mixing. The water overlay serves two purposes;
it excludes air, which prevents polymerisation, and produces a flat
interphase. The gels were then allowed to polymerize, this took
10-20 minutes at room temperature.
2) Running
Protein samples to be run were prepared in the same buffer as
the reservoir buffer but at 1/10 concentration, which helped sharpen
the bands. Samples were made 10%v/v with 60%w/w sucrose and 107.
v/v with 0.001%w/v Bromophenol blue or 0.001%w/v Phenol red and
layered carefully onto the surface of the gels through the reservoir
buffer with a 'Hamilton' syringe.
A current of 1mA per tube was applied for 15 minutes followed
by a current of 4mA per tube until the marker had migrated a pre-
determined distance, usually 4.5cm. Migration was from the cathode
(-) to the anode(+), i.e. top to bottom in this case.
The reservoir buffer in all the experiments described was
made:
Tris 10.0g
Disodiumethylendiaminetetra-acetic acid (EDTA) 1.0g
adjusted to pH9.2 with saturated boric acid solution and made up
to 1000m1 with distilled water. In experiments in which proteins
treated with SDS were run the reservoir buffer contained 0.17w/v
SDS.
3) Staining
Gels were removed from the tubes by rleming with a needle while
7%w/v acetic acid was injected between the gel and the glass. They
67

were then fixed and stained overnight with 1%w/v amido black in 7%
acetic acid. Excess stain was removed in one of three ways:
a) Repeated washing and soaking in 7% acetic acid.
b) Repeated washing and soaking in 20%v/v ethanol in
7% w/v acetic acid at 60°C.
c) Transverse destaining using a "Transverse Disc Destainer"
(Shandon Ltd.).
Unstained gels for densitometer scanning were fixed in 5%w/v
TCA and stored at 4°C.
4) Estimation of nolypentide molecular veizht
The method of Shapiro, Vinuela and Maizel (1967) vas used,
except that 7.5% acrylamide gals were used.
Marker proteins were all obtained as "Non enzymic protein
molecular weight markers" from Hann Research Laboratories Inc., New
York, U.S.A.
68

H. DOUBLE LABELLING TECHNIQUE
The method described below is a modification of the methods of
Summers, Maizel and Darnell (1965) and Hay, Skehel and Burke (1968).
Chick embryo cell monolayers were grown in 14cm glass Petri
dishes and half the total dishes were infected with 50-100 E/D50/
cell of the strain under investigation. After adsorption for one
hour the cells were washed twice with PBS at 37°C and 20m1 main-
tenance media containing actinomycin D was added. Actinomycin D
was used at a concentration of 5pg/ml for samples up to the sixth
hour and 1pg/ml for later samples. At the times specified cells
were washed twice with warm Earle's saline (Earle 1959) containing
2%v/v foetal calf serum and the following amount of radioactive
amino acid added:
a) To infected cells,
10m1 of Earle's saline + 27 foetal calf serum, containing
10µC/ml 3H-leucine.
b) To uninfected calls,
10m1 of Earle's saline + 2% foetal calf serum, containing
_ -. 2.5pC/m1 14 u Leucine.
After a one hour pulse, cultures were washed twice with ice cold
PBS and then once with ice cold hypotonic buffer (magnesium chloride
103M, Tris/HC1 10-3M pH8.3, 2-mercaptoethanol (0.1%v/v) 1.4 x
10-214). The cells were scraped from the Petri dishes into one ml
of hypotonic buffer and the infected 3H-leucine labelled cells
mixed with the uninfected 14C-leucine labelled cells.
The cell mixture was made 107.v/v with glacial acetic acid,
69

0.5M with urea and 17.w/v with SDS. The mixture was then incubated
at 37°C for 30-60 minutes and dialysed overnight against 2000-3000
volumes of 1/10 TEB pR 9.2, 0.17. SDS, 0.1% 2-mercaptosthanol and
0.5M urea, at room temperature.
The dialysate was decanted from any insoluble material remain-
ing and adjusted to 2mg protein per ml. Samples of 0.1m1 were then
analysed by electrophoresis on 7.57. acrylamide gels. At the end
of a run the gels were stained in amido black and then sliced, diss-
olved in hydrogen peroxide and radioactivity estimated. The radio-
activity was expressed as a ratio of 311 DEM to 14C DPM. Because
4 times the concentration of 3H-leucine compared to 14C-leucine was
used ratios were divided by 4.
70

J. DENSITY GRADIENT CENTRIFUGATION - VIRUS PURIFICATION
Preliminary purification and concentration of virus was done
as described above. Depending on the quantity of virus to be puri-
fied one of the following rotors for the MSE 65 centrifuge was
used for further purification on density gradients: B XIV zonal
rotor, D XV zonal rotor, 3 x 65m1 swing out, 3 x 23m1 swing out and
3 x 10m1 swing out.
Linear sucrose or tartrate gradients were prepared using the
relevant size MSE gradient mixer (M.S.E. Ltd., London, England).
Tubes were pierced using an MSE tube piercer and all fractions
collected on the LKB ultroi-4c fraction collector. Ultraviolet
adsorption was measured on the LKB uvicord.
Sucrose and potassium tartrate solutions in 0.01M Tris/HC1
buffer pH7.4 wore made as w/w solutions in accordance with the
Handbook of Chemistry and Physics (Weast, 1968).
The refractive index of solutions was measured using a refrac-
tometer (Hilger Watts Ltd., London, England) at 20°C. Densities
were calculated from the refractive index and are expressed as g/ml
at 20°C.
Radioactivity in fractions after density centrifugation of virus
containing 3H-uridine was measured after precipitation on to glass
fibre filters with ice cold 5% TCA, washing each filter twice with
ice cold TCA and drying the filters under infra red lamps before
transferring them to scintillation fluid for counting.
71

/II RESULTS
A. VIRUS PURIFICATION AND BIOPHYSICAL PROPERTIES
1) Linear sucrose gradient
A typical result of centrifuging strain Italian on a linear
sucrose gradient using a B XIV zonal rotor is shown in Figure 3.
There are two major protein peaks, one is found at the top of the
gradient while the other is associated with the major haamagglu-
tinin peak at a density of 1.19g/ml. Another minor peak of HA
activity can be seen at a density of 1.12-1.13g/m1 while some HA
remains on top of the gradient.
2) Linear tartrate gradient
The separation of virus (strain Italian) on a linear potassium
tartrate gradient using a BXIV zonal rotor is shown in Figure 4.
The over all appearance is similar to the results on a linear sucr-
ose gradient, however, the major HA peak appears at a density of
1.22g/m1 while the minor HA peak is found at 1.16g/ml.
3) Association of viral prozerties to peaks
Virus (strain Italian) labelled with 3H-uridine was prepared
and run on a linear sucrose gradient in a 3 x 23m1 swing out rotor.
Figure 5 shows that an acid-insoluble radioactivity peak and infec-
tivity are identifiable with the major HA peak at a density of 1.19
g/ml. The minor HA peak shows little infectivity and appears to
possess no RNA. These results confirm that the whole virus is
found at a density of 1.190m1 in sucrose. Similar experiments
showed that the peak at 1.22g/m1 in potassium tartrate is mature virus.
Electron microscopy confirmed these finding* typical intact virus
72

Ha
em
ag
glu
tin
sn
pe
r m
l x
10
00
M.
M:
M.
M-
12."
2.1
Euo 01
10-
7
Z" '"11111,1111 muumuui
•
FIGURE 3
73
.1 06
4 e 12 16 20 24 2. 32 36 40 44 45 12 66 60 64
Fraction number (10 ml each)
Purification of strain Italian on a linear sucrose gradient;
BXIV rotor, centrifugation: 40,000rpm for 90min.
Histogram is the reciprocal of HA titre.
• solution density.
protein.

.. r .
200
HA
100
C a)
12 .w•. ♦......
..... • ............ • ........... A
............ • .........
.....
•
40"•••••%/4/
.........
FIGURE 4
74
30
40
Fraction (10m1)
Purification of strain Italian on a linear potassium tartrate
gradient; OXIV rotor, centrifugation: 45,000rpm for 16hr.
Histogram is the reciprocal of the HA titre.
,••

0 c) I-x
E a-C.)
- s
- a
-4
20-'
18-
18-, o o -
E 12- - 10
1.24 .-.
3
2 4 8 8 10 12
FIGURE 5
Fraction number
Purification of strain Italian on a linear sucrose gradient;
3 X 23m1. awing-out rotor, centrifugation: 30,000rpm for 90min.
o solution density.
• infectivity. _
• CPM due to 3H-uridine uptake into TCA-insoluble material.
75

could be seen at the higher density while the minor HA peak revealed
what appeared to be fragments of virus or modified cell membrane.
4) Discontinuous gradients
Experiments were carried out to investigate the possibility
of using the simpler technique of discontinuous gradients. Succ-
essful separation of virus was obtained using gradients made from
sucrose solutions of the following densities: 1.04, 1.16, 1.26.
and 1.288/m1 in the ratio of 1:2:1:eto fill'. These gradients
possess two 'steps' in which the density changes rapidly (Figure
6) because of this the virus peak is much sharper and appears in
fewer fractions. If centrifugation was continued for long periods,
i.e. 12-18 hours or discontinuous gradients were left for several
hours before use, mixing of the bands occurred and shallower steps
in the gradient were obtained. With potassium tartrate solutions
discontinuous gradients could not be obtained: evidently the lack
of viscosity of tartrate permitted wady mixing so that almost linear
gradients were formed.
5) Recentrifugation of HA peaks
The occurrence of two haemagglutinin peaks, one of which was
possibly virus fragments could be caused by degradation of the virus
during the purification procedure. To discover if this was so
material from fractions corresponding to the two major HA peaks on
a sucrose gradient was sedimented and recentrifuged on sucrose
gradients. The single peak obtained from the whole virus at density
1.19g/m1 (Figure 7) suggests that the procedure does not degrade
intact virions. Recentrifugation of the lower density HA peak
showed that some whole virus is also associated with the fragments
(Figure 8).
76

1.22
1.20
1.18
E 1.10
0
1•10
munc 6
77
40
0 0 0
30
20
5
0 0
10
am ro 10,0,,000644//0/0/ Cd7 ALW71,
4 8 12 18 20 24 28 32 38 40 44 48
Fraction number (icimi each)
X
Purification of strain Italian on a discontinuouo sucroao
gradient; 3XIV rotor, Contrifugations 40,00Orpm for DOmin.
Histogram is tho reciprocal of the HA titre.

1 20
E ......
CO
118
1.10
1 22
1.10 H
aem
agg
luti
nin
per
ml x
10
00
4 8 12 18 20 24 28 32 38 40
FIGURE 7
78
Fraction number (to ml each)
Recentrifugation or major HA peak obtained from a gradient
similar to that seen in Fig. 3. EIXIV rotor, sucrose gradient
centrifugation: 40,000rpm for 90min.

FIGURE 8
79
5 10 15 20
Fraction
Rocentrifugotion of minor HA peak obtained from a gradient
similar to that seen in Fig. 3. 3 X 23m1. ewing*out rotor.
centrifugations 35od0Orpm for 180min., sucrose gradient.
I 30
E I 20
); 1 C 0 0
110
1
- 15
- 10 N
9
X
< I
- 5

6) Nature of major and minor HA peaks
Lack of RNA and infectivity, together with electron microscopic
evidence all suggest that the minor HA peek found at a density of
1.13 when strain Italian is run on a sucrose gradient consists of
virus fragments or modified cell membrane fragments. When whole
NDV is disrupted with SDS polyscrylemide gel discelectrophoresis
yields 3 major bands (see below). When samples from the major HA
peak and minor HA peak were treated in this manner, the major HA
peak showed 3 major bands while the minor HA peak showed only 2
bands the middle band being absent (Figure 9).
7) Separation undpr varying conditions
Whole virus banded at its isopycnic density after very short
runs, as brief as 45 minutes at 45,000 r.p.m., or long runs, 18
hours at 45,000 r.p.m., on linear or discontinuous gradients (Table
4). This is an important point, because virus is so easily sedi-
mented to its isopycnic density short runs are preferable for virus
purification since under these conditions virus is subjected to
both isopycnic and rate zonal separation. Cell fragments which may
have the same density as virus but a slower sedimentation rate are
prevented from contaminating the virus. This'is less desirable if
contaminating proteins of greater density than the virus are present,
but results have shown that these are minimal. Where protein peaks
have appeared at densities greater than the virus peak electron
microscopy has shown them to consist of RNP fragments.
8) Estimation of virus purity
Estimation of purity poses a considerable problem. Purifica-
tion can be monitored by measuring HA per mg protein at each step
(Table 5). By this method it can be seen that virus after zonal
80

81
A B
g in HA
nt.
d H p k • 1 nd '. di ru t d 1 1 SO -
d
b •
- H
H p •

TABLE 4
Densities at which haemagglutinin peaks occurred on sucrose gradients
under different conditions. Virus: Italien.
Length of run R.P.M. Density Type of Gradient (Hours) (BXIV rotor) ( g/ml at 20°C)
HA Peaks
I II
45,000 1.19 1.13 Discontinuous
1. 45,000 1.19 1.13 Linear
11/2 45,000 1.18 1.14 Discontinuous
2 45,000 1.185 1.13 Discontinuous
3 47,000 1.18 1.12 Linear
14 30,000 1.19 1.12 Linear '
18 30,000 1.195 1.13 Linear
15 47,000 1.18 1.13 Discontinuous
82

TABLE 5
Purification of NDV strain Italian
83
HA/ml Protein ,ug/m1
HA per mg protein
5,120 2,000 2,560
163,840 2,600 63,000
40,960 50 819,200
Sample
Original allantoic fluid
Concentrate from allantoic fluid
Virus after zonal centrifugation

centrifugation is 320-fold purer than infected allantoic fluid;
however this does not show that no contaminating proteins are pres-
ent. Purification can also be monitored by the use of polyacryla..
mide gel discelectrophoresis. Whole virus will not enter 7.5%
acrylamide gels while smaller soluble proteins will, figure 10
shows therefore that after sonal centrifugation virus is free of
soluble proteins. Bands of 1 -2pg of cytochrmhs C can be seen quite
distinctly on similar gels. Soluble proteins too large to enter
the gel could be present but this is unlikely since purified virus
passed through both a sephadex G200 column and a DEAE DE32 (Whatman
Ltd) column as a single protein peak.
9) Behaviour of different strains of NDV on density gradients
The buoyant densities of the whole virus and the minor HA peak
of seven different strains of NDV which differ in virulence was
measured on gradients of sucrose, potassium tartrate and caesium
chloride. Very little variation was seen amongst the whole virus
of the different strains on sucrose, the range was 1.19-1.20g/m1
and caesium chloride, the range was 1.20-1.21; potassium tartrate
gradients gave more heterogenous results the range was 1.20-1•.22
g/ml but this was not considered significant as there is no appar-
ent trend if compared with virulence (Table 6). The high density
of virus on potassium tartrate and caesium chloride gradients is
presumed due to the uptake of these low molecular weight salts into
the virion while this does not occur with sucrose.
The minor HA peak was more heterogamous, having a range of 1.13-
1.16g/ml on sucrose and 1.12-1.168/m1 on tartrate. These ranges are
no greater than have been recorded in different preparations of any
one strain and should be expected if this band is fragments of virus
84

85
(1
411
Purification of NtV. The fallowing were diluted so that
0.1m1. layered on to polyacrylemide gels contained 2094 of
proteinta) Original infected allantoic fluid. b) Allantoic
fluid after low speed centrifugation. c) Pellet obtained after
high speed centrifugation. d) Supernatant obtained after high
speed centrifugation. a) Virus suspension after zonal cent-
.rifugation.

86 TABLE 6
The buoyant densities of the major and minor haemagglutinin peaks of different strains of NDV.
Strain Density g/ml at 20°C in:
Sucrose
I II
Sodium tartrate Caesium chloride
Harts (BHK) 1.19 1.15 1.21 1.12 ND
Herts 1.19 1.14 1.20 1.14 1.20
Italien 1.19 1.13 1.22 1.16 1.21
Texas ND ND 1.20 ND 1.20
Beaudette C 1.19 1.16 1.22 1.16 1.21
B1 1.20 1.15 1.215 1.15 1.21
Queensland 1.20 1.16 1.21 1.14 1.21
Ulster 1.19 1.14 1.20 1.13 ND
Herts (BHK) was strain Herts grown in suspended BHK cells.
All other strains were grown in eggs.
ND Not determined.
I vs Whole virus peak.
II ==, Minor HA peak.

or modified cell membranes. There is little difference between
the lamyant density of the minor peak in sucrose and tartrate as
there is not complete membrane present. In caesium chloride grad-
ient experiments already purified virus was used.
10) Discussion of results
Satisfactory separation of NDV from large volumes of infected
allantoic fluid vas obtained using either sucrose or potassium
tartrate gradients in zonal rotors. Discontinuous sucrose gradients
simplified the technique without affecting the purification.
Separation of smaller volumes of virus on gradients made in
tubes in Ewing out rotors was less successful. However using the
following procedure pure virus could be readily obtained: 1) After
concentration virus was centrifuged through potassium tartrate sol-
ution at a density of 1.16g/m1 onto a sucrose cushion at a density
of 1.24 using a Sorvall centrifuge at 40,000A for 2 hours. 2) The
band at the interphase was collected and after dilution was placed
on a linear gradient of 0-60% sucrose and spun at 22,000 r.p.m. in
the 3 x 65m1 rotor for 2 hours. In this way satisfactorily puri-
fied virus could be obtained.
Since the initial experiments gradients of potassium tartrate
were seldom used. Generally, purified virus was needed for treat-
ment with SDS and the presence of potassium causes precipitation
of this detergent.
From the results it can be seen that virulent strains do not
differ significantly in bubyant density from avirulent strains.
Stonback and Durand (1963) showed that the density of the virion
was host controlled and some variation might have been expected as
87

Bang (1953) reported that avirulent and virulent strains differ
in their site of maturity within the cell. However if such diff-
erences exist they may not be large enough. to detect using these
techniques. In this respect NDV is unlike the herpes simplex virus
system in which it has been shown that strains differing in viru-
lence differ in buoyant density (Holzman and Roane, 1961).
88

B. THE STRUCTURAL PROTEINS OF NEWCASTLE DISEASE VIRUS
1) Polyacrylamide gel discelectrophoresis after disruption by SDS
Discelectrophoresis on 7.5% polyacrylamide gels in the presence
of 0.1% SDS after disruption of purified virus with 1% SDS and 17
mercaptoethanol reveals three major polypeptide bands, I, II, III
and one minor band, A, after staining with amidoblack (Fig. 11,
photograph). Occasionally other minor bands were seen B, C, and
IIa (Fig. 11, diagram).
2) Polypeptide patterns of different strains
The polypeptide patterns seen with all the strains tested were
essentially identical. Three major and one minor bands were seen
consistently. The migrations of these bands as percent migration
of bromophenol blue or phenol red markers are shown in Table 7.
The values for Beaudette C (a) and Beaudette C (b) are the extremes
recorded with this strain, while the migration of the bands of
the other strains is an average of at least three gels. It can
be seen that the variation from strain to strain is no greater
than may be recorded using the same strain.
3) Radioactive labelled virus
Labelling virus with radioactive amino acids confirms the
peptide nature of the bands seen in stained gels, since radioactivity
appeared'in peaks corresponding by migration to the stained bands,
Figures 12, 13 and 14 show that all four strains tested have the
three major bands seen in stained gels and although the minor band
A is hardly above background it may be distinguished as a minor peak.
Figure 12 shows analysis of two strains, Texas and Beaudette C,
labelled with 14C and 3H-leucine respectively, after electrophoresis
on the same gel, it can be seen that all peaks have exactly tha came
migrations.
89

FIGURE 11
90
A
B
C
ila
Bromophenol blue
NA structural proteins.
The photograph of the gel shows typical separation of NA
polypeptides (Herts) after polyacrylamide gel electrophoresis
in the presence of 505 after TOS-mercaptoethanol disruption.
Staining: Amido black.
The diagram shows thu minor bands occasionally seen and
their migration in relation to the major polypeptide bands.
..0,011.1710P4,Trwt.c. Origin

TABLE 7
Migration of the polypeptide bands of different strains of NDV
on 7.57 polyacrylamide gels.
Strain Band
Distance migrated 7. bromophenol blue
Herts 8.5 45.0 59.0 70.0
Texas 8.5 48.0 62.0 72.0
*Beaudette C (a) 8.7 50.0 63.0 73.0
*Beaudette C (b) 9.5 45.0 60.0 69.0
B1 9.5 45.0 60.0 69.0
Queensland 9.3 46.5 61.0 70.0
Distance migrated as 7. phenol red
Warwick 16.5 53.0 69.0 73.0
Beaudette C 15.0 50.5 69.5 78.0
F 14.5 51.0 70.0 76.5
Queensland 10.5 48.0 66.0 76.0
Ulster 13.5 53.0 69.0 76.0
* Extremes recorded for this strain. The other fesults are the average
of at least three runs.
91
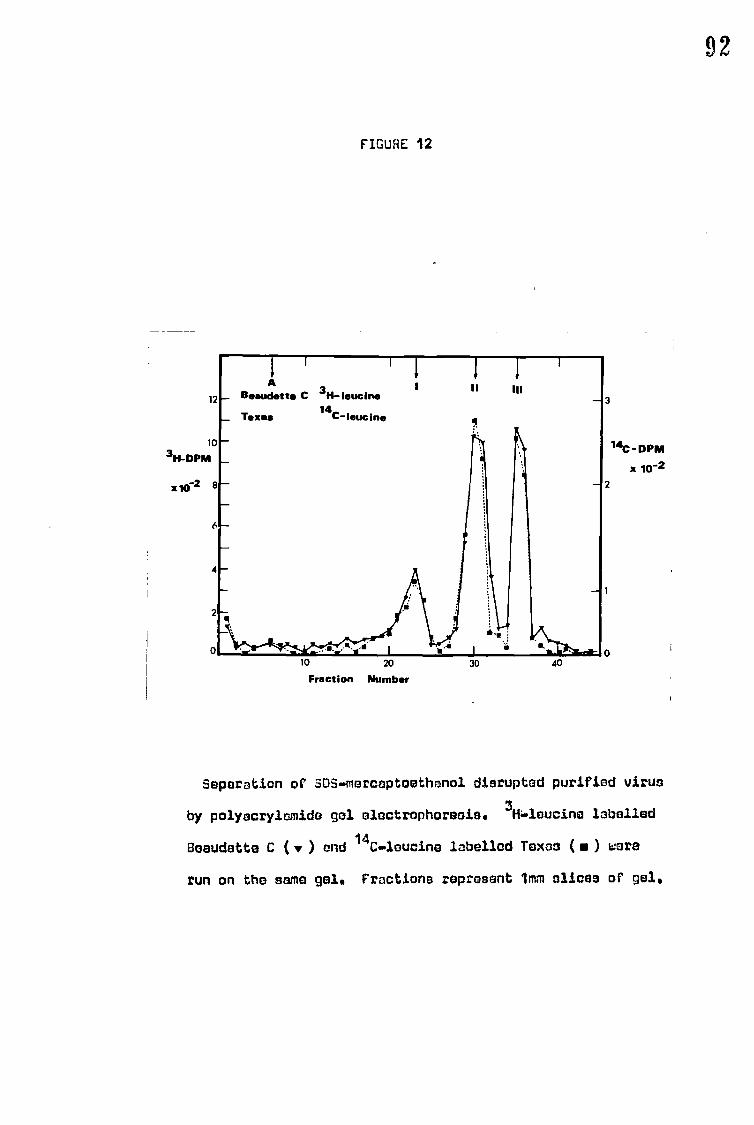
3
141C -DPM x 10-2
2
11 111 A
Beaudette C 3H- !suckle
Texas 14
C-leucine
12
10 3H-DPM
x102
..._ ..a...
10 20 30
Fraction Number 40
FIGURE 12
92
Separation of SOS.mereaptoethanol disrupted purified virus
by polyacrylemida gal electrophoreois. 3H6pleucine labelled
Beaudette C (v) and 14C.leucine labelled Texas ( • ) ware
run on the same gel. Fractions represent 1mm nlicss of gel.
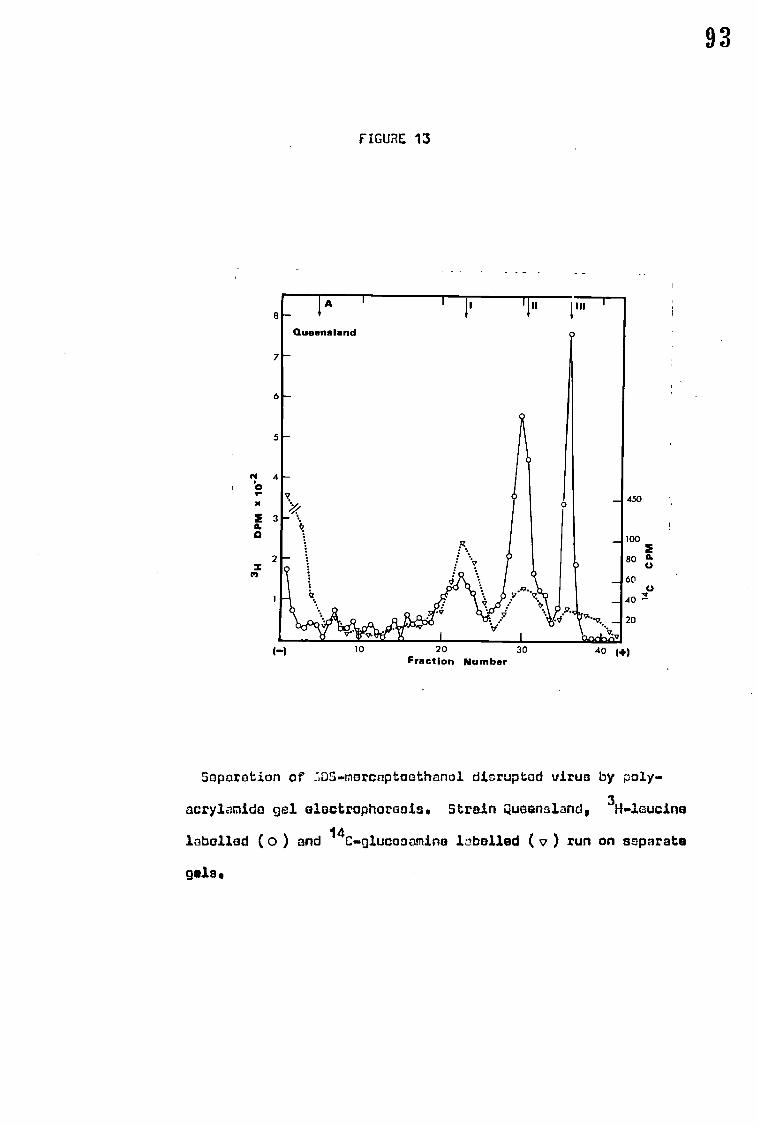
FIGURE 13
93
8 I A II
land
7
6 —
5 —
N 4 oc,
2 3 a.
2 X
0
0
450
100
80 IL C.)
60
40
20
1-1 10 20 30 Fraction Number
40 (+)
Saparation of LDS-morcaptoothanol disrupted virus by poly-
acrylamide gel electrophoraais. Strain Queensland, 3H-loucine
labelled (o) and 14C-glucosamina libelled ( v ) run on separate
gels.

FIGURE 14
94
1
1
1
Herts 3H- arginine
200
DPM
100
(-) 10 20
30
40
1+)
Fraction Number
Savration of 0.5-mercvtosthanol disrupted purified virus
by pelyecrylamicio gel olectrehoresis. 3H-arginine inbollnd
Herts.

4) Relative proportions of the polypeptides
The proportion of the total protein found in any one band can be
estimated either by measuring the amount of radioactivity in any
band after labelling or by measuring the area under the curves of
a stained or unstained densitometer trace of 600 nm or 280 nm
wavelength respectively. Within the limitations of the method
the results are remarkably consistent for all the methods of
estimation used (Table 8). The average values are: I - 21%,
II • 44%, III - 307., and A • 2.5%. The greatest variation was with
polypeptide II which ranged from 40.57. (Harts stained trace) to 46.07.
(Queensland, 3H-leucine), however the figures of, 44.67. for Texas
and 41.6% for Beaudette C suggest that this is more likely to
be an experimental variation than a real difference and is certainly
not related to virulence. In comparison with the results of
Haslam et al (1969) polypeptide III shows a much higher proportion,
307. compared to 217.,while the others are similar.
Table 8 also shows the molecular ratios of the polypeptides,
these indicate the relative ratio of the molecules of each poly-
peptides in the virion. The strains tested are remarkably similar
in:this respect. Labelling with 3H-arginine indicates that
polypeptide I is particularly low in this amino acid.
Virus (strain Queensland) was also grown in the presence of
radioactive glucosamine as this specifically labels the glycoproteina
(Klenk, Caliguri and Choppin, 1970; Burge and Strauss, 1970).
Although counts were low due to the low specific activity of the
available isotope the results show that there is a peak of activity
at the position of polypeptide I which indicates that this is a
glycoprotein (Fig. 13). Similarly there is a somewhat smaller
peak at polypeptide II which suggests that this is also a glycoprotein,
while although there is a small peak at the position of polypeptide
III it seems unlikely that this is of sufficient significance to
demonstrate that polypeptide III is a glycoprotein.
95

96 TABLE 8
Relative proportions of NDV polypeptidea
Strain 7. Total yrotein !lethod
Molecular ratio*
III A Rest
V-8 VB1/66 18 45 21 5 11 Haslam et al
(avirulent) '0.42 1.54 1.0 (1969)
Texas 21.0 44.6 30.5 2.7 1.2 14C-leucine 0.33 1.06 1.0 0.019 -
Beaudette C 21.2 41.6 31.0 2.8 3.5 3H-leucine
0.32 0.97 1.0 0.018 -
Queensland 19.1 46.0 29.7 2.5 2.7 3H-leucine
0.31 1.12 1.0 0.018 -
Herts 21.0 40.5 30.6 7.9 Amido black
0.33 0.96 1.0 stained trace
Herts 21.6 40.5 32.9 5.2 Unstained trace
0.31 0.90 1.0
Herts 12.0 45.0 31.0 12,0 3H-arginine
0.18 1.05 1.0
k The molecular ratios were calculated by dividing the percenta3e of virus
protein by the molecular weight of the polypeptide. The resultant ratios
were then divided by the value for peak III so that the results may be
compared.

97 5) Estimation of molecular weight of the polypeptides
Treatment of proteins with SDS produces a rod-like particle the
length of which varies with the molecular weight of the protein in the
complex (Reynold and Tanford, 1970). Because of this property tha
distance of protein migration on polyacrylamide gels in the presence of
SDS is a direct consequence of the molecular weight of the protein. In
fact migration shows a straight line relationship to the log of the
molecular weight (Shapiro, Vifluela and Maizel, 1967).
This method has been used to estimate the molecular weights of the
NDV polypeptides (Fig. 15). The results obtained are shown in Table 9
together with the molecular weights reported by other authors using a
similar technique. The range from experiment to experiment is considerable -
84-90,000 for pblypeptide I, 56-60,000 for II, 40-42,000 for III - however
the figures obtained are a useful approximation. The average molecular
weight for each polypeptide compares favoUrablY' with the average of the
previously reported values although these have even greater ranges.
Polypeptides in the region of polypeptide A have a large molecular
weight of 200,000 or more which suggests that this may be due to incomplete
dissociation of virus or reeggregation of polypeptides. The occasional
bands B, 160,000 and C, 120-140,000 may be produced in a similar way. The
other occasional band, tIe, which has a molecular weight of 50,000 appears
to correspond to the polypeptide in this position obtained from
nucleocapsid isolated after tryptic digestion of infected cells
(Mountcastle et al 1970).
6) Identification of NDV polypeptides
a) Nucleocapsid polypeptide.
Treatment of pure virus, strain Harts, with deoxycholate disrupts the
virus membrane and releases RNP. RNP can then be separated from the
disrupted membrane by rate zonal centrifugation on a sucrose gradient

Queensland
15 2x BSA
MW 10
X10 4 8
2 x0VALBUMIN
0 IH.L )
BSA
4
6 HO
1 \ \\Sf\
ii OVALBUMIN
ill PEPSIN
2
FIGURE 15
98
30 40 50 60
70
80
Migration 7. Bromophenol blue
Estimation of the molecular weights of NOV polypeptidas
by polyscrylamide gel electrophoresis. 5DS-merceptoethanol
disrupted virus (e.g. Queensland) was run in parallel with
the following marker proteins of known molecular weight*
H or L = heavy or light chain ofp.globulin (human); OSA m
bovine serum albumin; ovalbumin; pepsin. Each gel wee run
with a bromophenol blue marker.

99
TABLE 9
The molecular weights of NDV polypeptides
Strain
Polypeptide Source
I II III
M.W. X103
Beaudette C 100 62 45 Bikel cC Duesberg
(1969)
Beaudette C 90 62 42 Evans S. Kingsbury
(1969)
V-8 VRI/66 80 54 38 Haslam et al (1970)
Herta 85 40 58
Herts 84 56 42'
Herts 95 60 42
Beaudette C 90 57 42
Oueensland 88 57 42
AVERAGE 88 58 42

100 containing deoxycholate (Kingsbury and Darlington 1968). Figure 16
shows the separation of RNP on a sucrose gradient.
The RNP obtained was further purified by separation on a caesium
chloride gradient, banding at a density of 1.30 g/ml. After dialysis
against PBS and centrifugation the resuspended pellet was shown to
consist of RNP by its characteristic spectrum from wavelength 240 nm
to 320 nm (Fig. 17) which is identical to that shown by Kingsbury
and Darlington (1968).
Treatment of the purified RNP with SDS separates the nucleocapaid
polypeptide from the RNA. Analysis of SDS-treated isolated RNP by
polyacrylamide gel electrophoresis showed a single polypeptide which
corresponded to polypeptide II of the whole virus, having a migration of
59.57. of that of bromophenol blue compared to 60.67. of the migration of
polypeptide II of whole virus (Table 10).
b) Neuraminidase and haemagglutinin
It has been shown that treatment of NDV with TWeen 20 at pH10
releases the HA and neuraminidase without destroying their activities,
although HA activity is prevented in the presence of A'ween 20 (Vebster
- • •-•:-• end Darlington 1969).
The supernatant obtained after centrifugation of purified virus
(Ulster) treated with Teen 20, was applied to a 0200-sephadex column
and the subsequent fractions collected were assayed for neuraminidase and
HA activity (Fig. 18). Separation was not complete since no fractions
contained haemagglutinin activity with no contaminating neuraminidase.
However some fractions showed neuraminidase activity but no detectable HA.
Fractions 27-31 were pooled as a 'HA pool' and fractions 35-37 as a
'neuraminidase pool'.
The two pools were precipitated with 57. TCA and after treatment with
SDS and mercaptoethanol, the two pools, the pellet and supernatant from
the Tween 20 treatment and original virus were each analysed by poly-
acrylamide gel electrophoresis. Table 11 shows the area under the curve

t30 — 15
— Ei w
too at
I - 5
5 V IS
fraCtX,C11nIll
101
FIGURE: 15
Separation of RN!) from other virus matoria/ by oucr000--
dooxycholato 7,ralibnt contriFugatiun. Mathod is daseritsd
in toxt. Vireo: Herta. Migration from right to left.
Histogram in tha reciprocal of the HA titre.

102
FIGURE 17
03
..\.,..,..„ ..... ...
OD 0.2.1
0. .
0.0 . ■ 240 -260 280 300
nm. wavelength
Spectrum of isolatcd riMP.

103 TABLE 10
Identification of nucleocapsid polypeptide
Band 7 Migration of bromophenol blue marker
Gel 1 (virus)
Gel 2 (RNP)
10.0 mls
I 45.2
Mb
II 60.6
59.5
III 70.0 ••

—50
4 4 Z5.
10
—40
!
—30 ri;
4
20
,-10
WAD VOLUME
60
50 ►
40 E
a 30
0. 2
20
10
104
F/GURE 18
70
10 20 30 40 50
Fraction Number
Separation of virus (Ulster) subunits on a G200 sophadex
column after disruption with Teflon 20 at pH 10. The methods
used are described in the text.
0 Neuraminidase activity (pg NANA released).
■ Reciprocal of the HA titre.
Protein, pg.

105
of the peaks after a densitometer trace of the stained gels. Figure 19
shows the densitometer trace of a photograph of the five gels. The gel
of the neuraminidase pool, which had no detectable HA activity, had
only one band discernable by eye although the densitometer detected
another minor peak. The major peak of the neuraminidase pool had the
same migration as peak III in the whole virus gel, while the minor peak
migrated to the same position as band I of the whole virus. The 'HA
pool' which also had neuraminidase activity showed two peaks, corresponding
to I and III of the whole virus, the larger peak corresponding to I.
These results suggest that band III of the whole virus is the
polypeptide associated with neuraminidase activity while band I is
associated with the HA activity.
Other methods used to separate Tween 20 - released PA and
neuraminidase, sucrose density gradient centrifugation and DEAF:
chromatography produced similar results, complete separation of Hal and
neuraminidase was never obtained.
It is interesting to note that polypeptide I had a slightly lower
migration rate when treated with SDS in the presence of Tween 20 while
on the removal of Tween 20 it reverts to its former rate (Fig. 19).

1c
TABLE 11
Separation of haemagglutinin and neuraminidase
Area under curve (mm2) of densitometer trace
I II III REST
Virus (200dug) 997 1917 1450 374
Pellet 861 3817 1541 1282
Supernatant 1334 1570 1383 665
HA pool (50)ug) 573 - 495 -
N pool (20/ug) 8 - 142
N pool* 177 - 1055 OD
* Deflection across trace 0.2 OD. All others 1.0 OD.
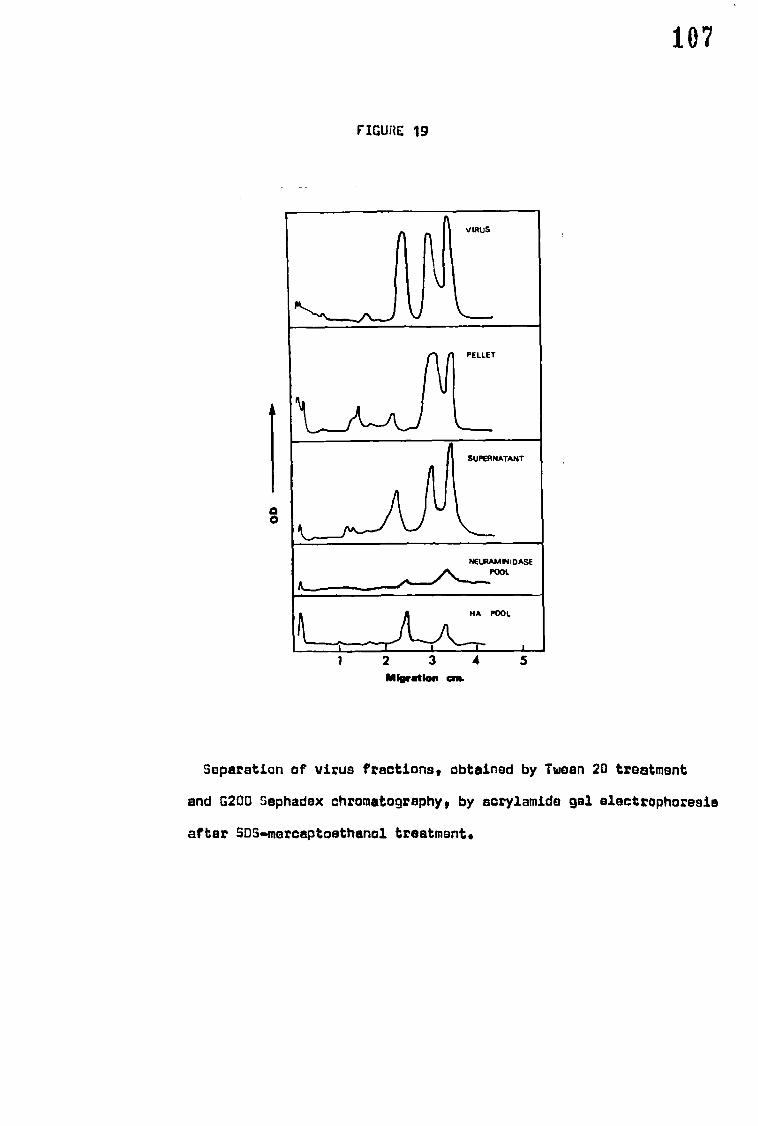
107
FIGURE 19
ik"•-•N___"_,, ki
vows
PELLET
%....__,_...
SUPERNATANT
NEURAM IN I DASE ...........",... POOL
kr
HA POOL
IL.1I 1` 1 2 3 4
5
Migration au.
Separation of virus fractions, obtained by Tween 20 treatment
and G200 Sephadex chromatography, by acrylamide gel alectrophoresie
after SDS-mercaptoethanol treatment.
0 0

108 C. NEURAMINIDASE STUDIES
1) Relationship of neuraminidase to HA and infectivity
In order to determine if any strains possessed more neuraminidase -
than others the neuraminidase activity was compared with HA titre
(Table 12). HA was taken to be a measure of viral surface area and
preferred to a particle count because of the pleomorphism of the virus.
The results show that the ratio of neuraminidase activity to HA units
is generally constant, except for strains Beaudette C and, to a lesser
extent, Ulster which have considerably lower ratios.
The relationship between HA, neuraminidase and infectivity was
also investigated. The HA/EID50 and the neuraminidase /EID50 ratios,
again with the exceptions of Beaudette C and Ulster, even after allowing
for their high HA titres, were very similar for all strains (Table 13).
2) Kinetic studies of virus-bound neuraminidase
The amount of NANA released after 10 minutes was plotted against the
concentration of substrate used. All eight strains examined gave kinetics
curves essentially identical to that of strain Herts (Fig. 10).
The Michaelis-Menten constants (Km) were obtained by Lineweaver-Burk
plots (Fig. 20). All the strains had a Km of approximately 1 x 1e3M
(Table 14).
With fetdin as substrate the optimum pH of the neuraminidase of the
different strains, though varying slightly from experiment to experiment,
always fell in the range pH5.1-5.3 (Table 15) and was considered to be
the same for all the strains tested. Typical curves of pH effect with
fetitn as a substrate are shown in Figs. 21A and 21B. A second minor peak

109 TABLE 12
Relationship between haemagglutinin and neuraminidase activity
Strain Neuraminidase activity/ml
HA/ml Neuraminidase/ HA x 10 3
Field Pheasant 38.0 1500 25.4
Herts 360.0 10240 35.0
Texas 87.5 2560 34.1
WaiWick 84.5 2560 33.0
Eastwood Notts 171.0 5120 33.3
Beaudette C 271.0 20480 13.3
Al 176.0 5120 34.4
F 74.5 2560 28.1
(ueensland 172.5 5120 33.6
Ulster 845.0 40960 20.6

110
TABLE 13
Relationship between infectivity, haemagglutinin and neuraminidase activity
Strain HA
EID50 x 10-7
Neuraminidase activity (pH 6.0)
EID 50 x 109
Herts 2.9 8:5
Texas 3.9 7.3
Eastwood Notts 3.6 7.5
Beaudette C 19.6 22.2
B1 2.4 5.2
7 2.3 10.3
Ulster 10.1 14.2

60
20
0
111
FIGURE 20
Neuramine lactose (pmoles) 0 I 01 03
OA
1/s
Kinetics of the virus hound neuraminidaso of strain Herts.
Standard enzyme essay procedure was used except that the
concentration of neuramine lactose ues as above, In the
Linewoaver•Burk plot the units of Ve and 1/v Ewer moles/ml•

1:1.2 TABLE 14
Michaelis-Menten constant of neuraminidase at pH 5.4
Strain Km
Field Pheasant 1.0 x 10-3 M
Herts 1.0 x 10-3 M
Italien 0.9 x 10-3 M
Texas 1.0 x 10-3 M
Beaudette C 1.0 x 10-3 M
F 0.9 x 10-3 M
nueensland 0.9 x 10-3 M
Ulster 1.0 x 10-3 M

113
at pH6.3 was seen with all the strains tested except strain F. If
neuramine lactose was used as the substrate the pH optimum for all the
strains was raised slightly to pH5.4 (Fig. 21B), the second minor peak
was far more obvious although it still occurred at pH6.3. In these
experiments sodium acetate buffer was usually used for pH values from
4.1-5.5, phosphate buffer for pH5.6-7.2. If sodium acid maleate
buffer was used from pH5.1 to pH7.2 with both fetbin and neuramine lactose
the two peaks were present but with neuramine lactose the "minor" peak
was so much greater that it became the larger of the two (Fig. 21C and
21D).
In Table 15 the neuraminidase activity at physiological pH-:(07.2)
is shown as a percentage of the activity at the optimum pH. The strains
tested were generally in the range of 50-707. and although differences in
the strains were consistently found there was no relationship with
virulence.
3) Heat stability of virus-bound neuraminidase
The heat stability of virus-bound neuraminidase was estimated by
incubating virus suspensions at 56°C and measuring the remaining
neuraminidase activity at intervals. Within 20 minutes the neuraminidase
activity had been destroyed in all the strains tested except Beaudette C,
which still had 307 of the original activity (Fig. 22).
4) Effect of Tween 20 at pHIO
Treatment of virus with 17. v/v Tween 20 at pH10 caused the release
of neuraminidase with all the strains tested; between 70-100% of the
remaining activity was present as free neuraminidase that could not be
sedimented (Table 16). The total yield after treatment varied considerably
from strain to strain. With strains Herts and Beaudette C there was an
807. loss of neuraminidase activity while with Field Pheasant there was an
increase of nearly 407..

114
TABLE 15
The effect of pH on neuraminidase activity using fetuin as a substrate
Strain pH optimum pH of minor % of optimum peak of activity neuraminidase
activity at pH 7.2
Herts 5.1 - 5.2 6.3 53.4
Texas 5.1 - 5.2 6.3 60.0
Beaudette C 5.2 - 5.3 6.1 69.0
F 5.2 - 5.3 No minor peak 62.5
Ulster 5.1 - 5.2 6.3 49.7

I
a
Neu
ram
insd
ase
12
12
0
115
FIGURE 21
5
7 5
7
p H
The affect of pH on virus-bound neureminidaso undor different
conditions,
The normal assay procedure was used with the following
variations: Asistrain: Harts; buffers pH 4'2 - 5'5, O'lrl
sodium acetate-acetic acid buffer, pH 5'6 • 7'21 OiM phosphate
buffer; substrata: fetuin. 8.strain: Harts; buffer: acetate/
phosphate; substrate: neuramine lactose. C.strain: Ulster;
buffer: acetate/phosphate ( ); 042M sodium acid malaite
buffer (o )1 substrates fetuin. 0-strain: Herta; buffer:
sodium acid meloate; substrates neuramine lactose.

Neu
ram
inid
ase
0
to
1 00
80
50
40
20
116
FIGURE 22
0
5
10
15
20
Exposure t i me ( min )
rioA stability of virus-bound n,:uraminidnso ;-2t 560 C. t
tha spocifi,:d Urns 0•1m1 of purifi2d virus suspension was
added to N3m1 of ice-cold buffer, to stop further heat in-
activation, and then assayed as described in the text.
• Harts
• Texas
• Oeaudette C
O Queeneland
O Ulster

117 TABLE 16
Effect of Tween 20 (1% v/v pH 10) on virus-bound neuraminidase
Strain
Field Pheasant
7. activity of total control activity
control treated
Supernatant 10.0 125.0
pellet 90.0 13.3
Herts
supernatant 3.0 16.7
pellet 97.0 0.0
Texas
supernatant 4.1 79.6
pellet 95.9 8.5
Beaudette C
supernatant 10.0 20.0
pellet 90.0 2.2
Queensland
supernatant 8.3 92.5
pellet 91.7 12.1
Ulster
supernatant 1.7 70.0
pellet 98.3 7.6
'Control' samples were incubated overnight at 37°C with bicarbonate
buffer pH 10. For 'treated' samples the buffer contained 1% v/v Tween 20.
All samples were spun at 45,000 xg for 60 min, the pellets resuspended to
volume and the solutions tested for neuraminidase activity.

118
5) Neuraminidase activity in infected chorioallantoic membranes
The levels of neuraminidase per/mg. protein in the chorioallantoic
membranes:- of eggs infected with different strains appear to be related
to the virulence of the infecting strain, (Table 17). The plaque-
forming strains, i.e. Eastwood Notts and the more virulent strains,
show a linear relationship between cell associated neuraminidase and
MDT. The less virulent strains show less marked differences in
neuraminidase levels, presumably because there is very little
accumulation of the enzyme and much of the activity recorded is due
to whole virus particles associated with the membrane.

119
TABLE 17
Cell associated neuraminidase levels in infected chorioallantoic membranes
22 hrs after infection.
Strain Neuraminidase activity/mg protein
MDT (hours)
Herts 11.1 49
Texas 7.3 55
Warwick 5.9 62
Beaudette C 4.6 62
Eastwood Notts 2.8 70
B1 1.5 117
F i_16 119
Queensland 0.83 00
Ulster 0.5 co

120 D. CELL FUSION (FROM WITHIN) AND HAEMADSORPTION
Haemadsorotion
The adsorption of chick red blood cells to chick embryo fibroblast
cells infected with NDV was first seen 4-6 hours after infection. Figure
23 shows typical early haemadsorption to cells infected with strain
Texas. The *virulent strains, in contrast, show very little haemadsorption
until 8 hours or more after infection. RBC that become attached to
cells infected with virulent virus a few hours after infection always
attach at a central area of the cells (Fig. 23), whereas with avirulent
strains haemadsorption *Bays appears' at the cell extremities (Fig. 24).
The frequency with which this difference occurs suggests that this is
not merely the same effect viewed from a different angle. The virulent
strains examineds Herts, Warwick, Texas and Beaudette C; show
haemadsorption which increases exponentially until about 12 hours after
infection, by which time cells were almost totally covered by RBC and
syncytia had appeared. Massive cell fusion and haemadsorption caused
by Herts 16 hours after infection can be seen in Figure 25.
A comparison of haemadsorption by cells infected with Texas and
Ulster 22 hours after infection is shown in Figures 26 and 27. Infection
by strains F, Queensland and Ulster always produced far less
haemadsorption than cells infected with virulent strains, rarely
exceeding an average of 10 RBC/cell even after 36 hours.
The number of RBC attached per cell at different time after
infection was also estimated (Fig. 28). Haemadsorption occurcat a
much greater rate and is more extensive after infection with virulent
strains than with avirulent while Beaudette C which is often classified
as mesogenic appears to be intermediate to the two levels.

121
FIGURE 23
Haemadeorption by chick embryo coils 4hr. after infection
with strain Texas. Stains May GrUnwald•Giemsa, Magnifications
X1800.

FIGURE 24
•
a
Hasmnisorption by chiek embryo calls 10hr. after infection
with strain F. Stain: May GrUnwald-Giemsa. Magnification X1300.
r J

123
FIGURE 25
Haemedaorption by chick embryo cells 16hr. after infection
with strain Herts. Steins Noy Granwald.Giemse.Magnification X750.

FIGURE 26
Heemedsorption by chick embryo cells 22hr, after infection
with strain Texas. Stein: acridine orange. MegnificationiX440.

1 Jr
FI2URE 27
Hsanadsorption by chick embryo cells 22hr. after infection
with strain Ulster. Stainsacridine orange, Magnification X530.

cv 9 a 8
7 6 5
30
20 O
10
0
a)
E
ery
th
rocy
tes
• .0"
• •
•
5
-0-
••••• .60.•
"act—
1 6
FIGURE 28
4 8 12 16 20 24 28 32 36
Hr after inoculation
Haemadsorption by chick embryo cells infected with different
strains of NOV. At least 50 infect ,:d culls were observed for
each point.
• Harts
• Texas
• Warwick
• Beaudette C
0 r
• Queensland
A ulster

19 7 Difficulty was experienced in estimating attached RBC at 12 hours
or more after infection with virulent strains, because of the large numbers
of RBC sndi-the cell fusion that had occurred. However by seeding cultures
with low concentrations of chick embryo cells, infecting before
confluent monolayers were formed and by counting each nucleus in a
syncytium as one cell this problem was to some extent overcome.
No haemadsorption was seen in uninfected cells (Fig. 29).
2) Cell fusion (from within)
The ability of NDV strains to form syncytia in infected cultures
coincided directly with their ability to form plaques (Table 18). The
avirulent strains tested never formed plaques or syncytia, with the
exception of strain F which occasionally formed small syncytia. Cell
fusion was generally in evidence by 8 hours after infection although
with strains Herts and Texas small syncytia could be seen somewhat
earlier. Large syncytia containing up to 20 nuclei could be seen in
Hertz infected cell cultures by 14 hours (Fig. 30) although with other
virulent strains fusion to this extent took longer. Figure 31 shows a
large syncytium formed by Warwick 22 hours after infection. By 24 hours
after infection with Harts or Texas massive cell fusion had occurred
and the monolayer was almost completely fused into a single syncytium
(Fig. 32).
The size of syncytia formed appeared to vary from strain to strain
and, by counting the number of nuclei in each polykaryocyte at 12 hours
after infection the size was shown to be directly related to the virulence
of the infecting strain (Table 18). Occasionally a few binucleate cells
were present in uninfected cell monolayers, but no syncytia were ever
seen (Fig. 29).

1w8
FIGURE 29
Uninfected chick embryo calls. :twin: ley :rdnweld-Giemsa.
.Magoinciltions X430.

129
TABLE 18
Comparison of the ability to form plaques and syncytia in chick embryo
cell cultures infected with different strains of NDV.
Strain Presence of:
Plaques
Syncytia Average number of nuclei per syncytium
Herts ++ 16
Texas ++ + 15
Warwick -H- + 10
Beaudette C + + 8
4
rueensland
Ulster
The numbers of nuclei per syncytium were estimated from counts on at
least 50 syncytia 12 bra after infection.
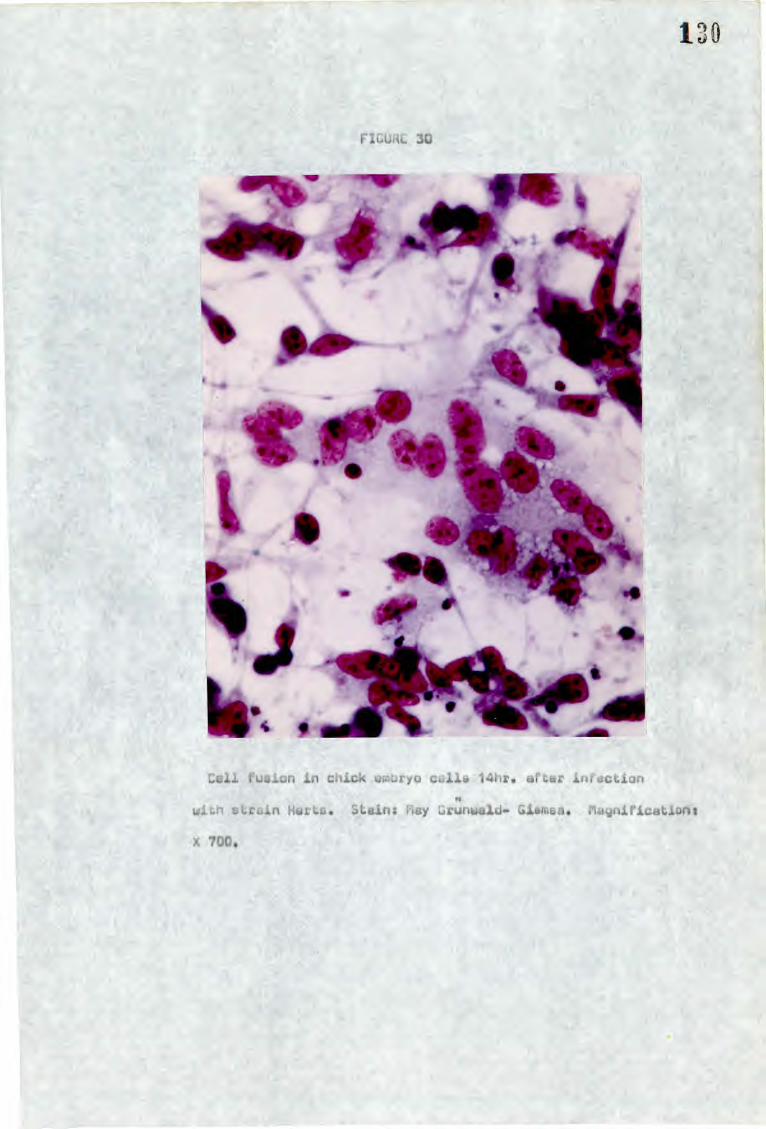
190
FIGUiiL 30
Cell fusion in chick wiouryo calls 14hr. afGar infection
wiLh strcin Hares. Stains flay Grunwald- Glsmsa. Magnifications
X 700.

Syncytium soon in chick etriryo call culture 22hr. after
infection with strain Uaruick. Stain: r:y Grunwald-Ciemsa.
Magnification: X 700
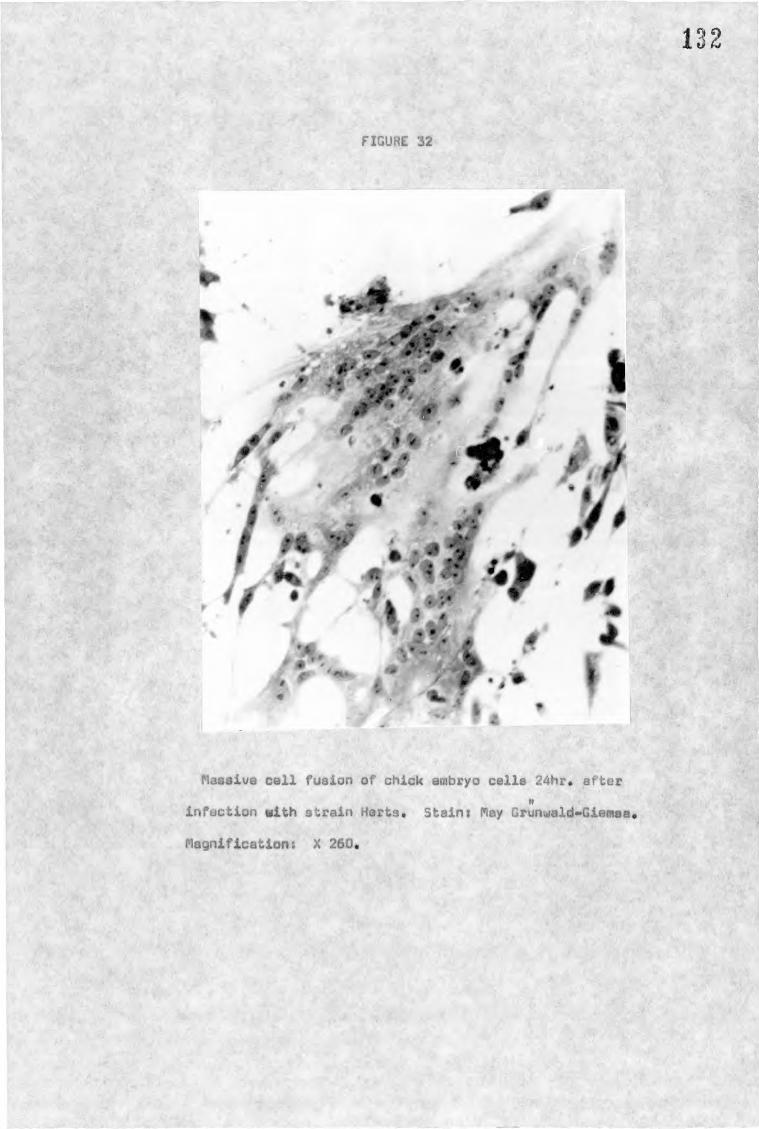
*12111 ", *41".
•
(11
9
•
132
FIGURE 32
Massive cell fusion of chick embryo cells 24hr. after If
infection with strain Herts. Stain: May Grunwald..Giemsa*
Magnification: X 260.

133 3) Effect of inhibition of protein synthesis on virus induced
cell fusion and haemadsorption
The effect on haemadsorption and cell fusion (measured at 8 and
15 hours post infection respectively) of different concentrations of
2.-fluorophenylalanine (FPA) after infection by strain Herts is shown in
Figure 33. Fluorophenylalanine inhibits protein synthesis by replacing
phenylalanine to form non-functional proteins, 400,ug/m1 FPA inhibited
the level of both haemadsorption and cell fusion to as little as 10%
of that in untreated cells.
Cycloheximide also inhibits protein synthesis, with this drug
however the inhibition is due to interference to the messenger RNA
• ribosome complex (Mahler and Cordes, 1966). Cell protein synthesis
in uninfected cells, haemadsorption and cell fusion are all inhibited
to the same extent by cycloheximide (Fig. 34).
The inference from these experiments is that after infection
protein synthesis of a functional protein, or proteins, must occur
if NDV-induced cell fusion or haemadsorption are to subsequently
develop.
4) The effect of time of addition of FPA on cell fusion and haemadsorption
Fluorophenylalanine (500 pg/ml) was added to chick embryo cell
cultures at different times after infection with strain Herts, and the
effect on cell fusion and haemadsorption (measured at 15 and 8 hours after
infection respectively) was estimated (Fig. 35).
Both cell fusion and haemadsorption were almott completely inhibited
if the FPA was added within 2 hours of infection, but were unaffected if
added later than 4 hours after infection. This suggests that the protein
or proteins that are necessary for these phenomena must be synthesised
within 3 hours of infection.
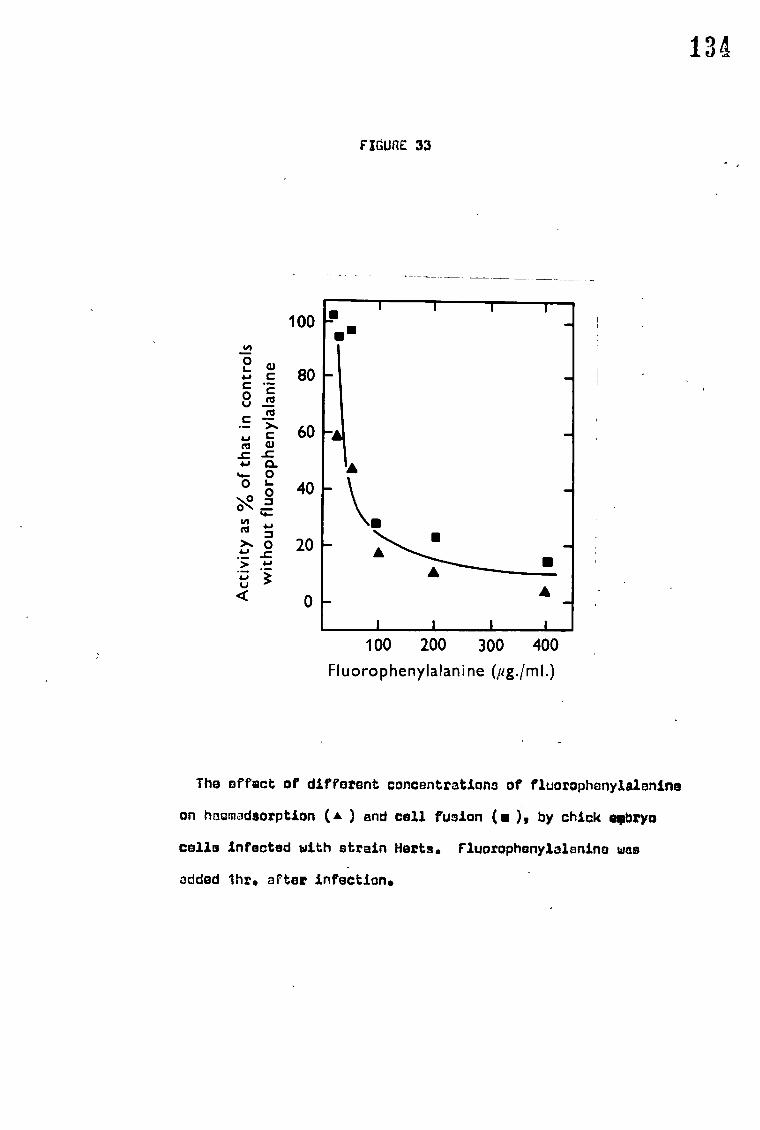
• •
• A
• • •
134
FIGURE 33
100
0
0
80
a 60 4.) CU nd
_C -C 4-0 CL
0 0
0 40 4-
0 ld
0 20
<r) 0
100 200 300 400
Fluorophenylalanine (fig./m1.)
The effect of different concentrations of fluorophenylalanine
on haemadsorption (A ) and cell fusion (m), by chick splay*
cells infected with strain Herts. Fluorophenylalanine was
added ihr. after infection.

135
FIGURE 34
100
15
lEj • 80
C W
rt E _c • R 60 (1.)
O -c O
*,;°
40 5 0
> _c • . _
4 14C
0
20
5
10 15
20
Cycloheximide /mI.)
Effect of difforsnt concontrationl of cyclohoxinido on protein
synthesis in uninfoctad chick embryo coils ( • ) and on hr!en-
adsorption ( A) and cell fusion (is ) in chick embryo cells
infected with strain Herts. Cycloheximide was added lhr.
altar infection. Protein synthesis W33 measured in uninfected
cells 3hr. after the addition of cycloheximide by the uptake
of 3144suoino into en acid-insoluble fraction.

136
FIGURE 35 -
0
C 0
C
0
r3
0 • 80 C
ed
• 60 _c 0
0 40
0
20
0
0 4 8 12
16 Hr after infection
Effect of ceding fluerophanylelanine (5031pgim1) at different
iron after infection on heemadeorption (A) and cell fusion
(a) in chick embryo cells infoctod with strain Herts. The
points indicate the times after infection at which fluorc»
phenylelenine was added.

137 E. INHIBITION OF CELLULAR PROTEIN SYNTHESIS
1) Effect of infection with different strains on cellular protein synthesis
The effect of different strains of NDV on cellular protein synthesis
was measured by the ability of chick embryo cells to incorporate' 3H-leucine
in the trichloracetic acid - insoluble material after a 30 minute pulse.
In Figure 36 incorporation of radioactive label is expressed as per cent
of incorporation into uninfected cells. The virulent strains Herts,
Warwick, H, Texas, Field Pheasant, and Beaudette C all caused marked
inhibition of protein synthesis, 50% at 6-8 hours after infection and
as much as 90% by 12 hours. The vaccine' strain F inhibited prOfein
synthesis by 40% but this level was not reached until 18 hours after
infection. The avirulent strains Queensland and Ulster had very little
effect on protein synthesis.
The virulent strains also cause a marked cytopathic effect in chick cells
and there is a possibility that inhibition of protein synthesis is due
solely to the death of cells. Disruption of the cell membranes and
lysis of the cell is associated with cell death. The degree of cell
death can,. therefore, be measured by the release of a substance, normally
found solely in the cells, into the supernatant. The enzyme lactate
dehydrogenase (LDH) is such a substance and the measurement of release
of LDH activity after infection with Herts, Texas, Queensland and F was
measured and expressed as a percentage of total cellular lactate
dehydrogenase activity. A slow loss of LDH activity was recorded
during the first 16 hours after infection with all the strains. After
this time a rapid increase was seen only in cells infected with virulent
strains. These results suggest that gross cellular damage induced by
virus infection occurs after 16 hours and could not, therefore, account
for cell protein synthesis inhibition which occurs much earlier.

O 0 00 • • 100
80
3 • 60
1 40
20
0
19 8
1
I 9 6
V
40
:4 • 20
g 0 120
0
100 U
• 80
o ▪ 60V
40
20 0
,
FIGURE 36
1 I I -
I-
0- 0
I I i HERTS
•-•
o
I
o
I
o-'
1 I 1 1 1 TEXAS
o
0
I I I I I H -
_
...:
...:
-
- WARWICK.• BEAUDETTE C FIELD PHEASANT
-
-
_
00-0/
1 1
QUEENSLAND
0
0/
/ 0
1 1 1 1 1 1
ULSTER
1 1 1 1
ea............ F\,...:
-,
0-0
,0''
0.0/ I I I I 4 8 12 16 20 24 4 8 12 16 20 24
Hr after infection 4 8 12 16 20 24
Protoin Synthosis inhibition by different strains of NOV.
Protein synthesis in infected ombryo cells wen measured by
tho uptake of 3H-leucine into a TCA insoluble fraction and
axpressed us incorporation into uninfected calls (40.
The releaso of LDH ftorn chick embryo cells infect©d with WV
strains (o) is oxprossed as 5 total collulnr LDH.

139 2) Effect of p-fluorophenylalanine on virus induced inhibition of
cell protein synthesis
Two strains were examined - Harts and H - to study the effect of
FPA on virus induced cell protein inhibition. One hour after infection
FPA at various concentrations was added to cell cultures. Seven hours
later cultures were pulsed for 1 hour with 3H-leucine and incorporation
into a TCA-insoluble fraction was measured. At this time control cells
with no FPA show 60% inhibition of cell protein synthesis. The results
obtained (Fig. 37) show that FPA decreases the amount of cell protein
synthesis inhibition, in the presence of 400 ug/m1 FPA cell protein
synthesis is inhibited by only 107. These results indicate that like
cell fusion and haemadsorption protein synthesis is necessary for the
virus induced inhibition of cell protein synthesis.
3) Effect of time of addition of FPA on virus induced inhibition
of cell protein synthesis
Cells were infected with strain Herts and at intervals after infection
500 ug/ml FPA was added. At 10 hours after infection the cultures
were pulsed for 1 hour with 3H-leucine and the extent to which cellular
protein synthesis had been inhibited was estimated (Fig. 38).
The addition of FPA within 3 hours of infection completely reversed
the inhibition of cell protein synthesis but the later the FPA was
added after 3 hours the nearer the level of inhibition approached 707.
which was the amount of inhibition obtained with infected cella in the
absence of FPA. These results suggest that a protein, or proteins, ,
synthesised within 3 hours of infection is responsible for. the
inhibition of cell protein synthesis.

-
FIGURE 37
140
100
ii7; 80
60 •_
40 0
•_ 20
:E
100 200 300 400 Fluorophenylalanine (pg./ml.)
Prevention by fluorophonylplanine of tho inhibition of
cellular protein synthesis in chick embryo cells by strains
Horto (•) and H (0). Fluorophsnylnlenine was added ihr.
after infection to control and infected cultures: 7hr. later
cultures were pulsed for lhr. with 3H-loucine and incorporation
into an acid-insolublo fraction wee measured.
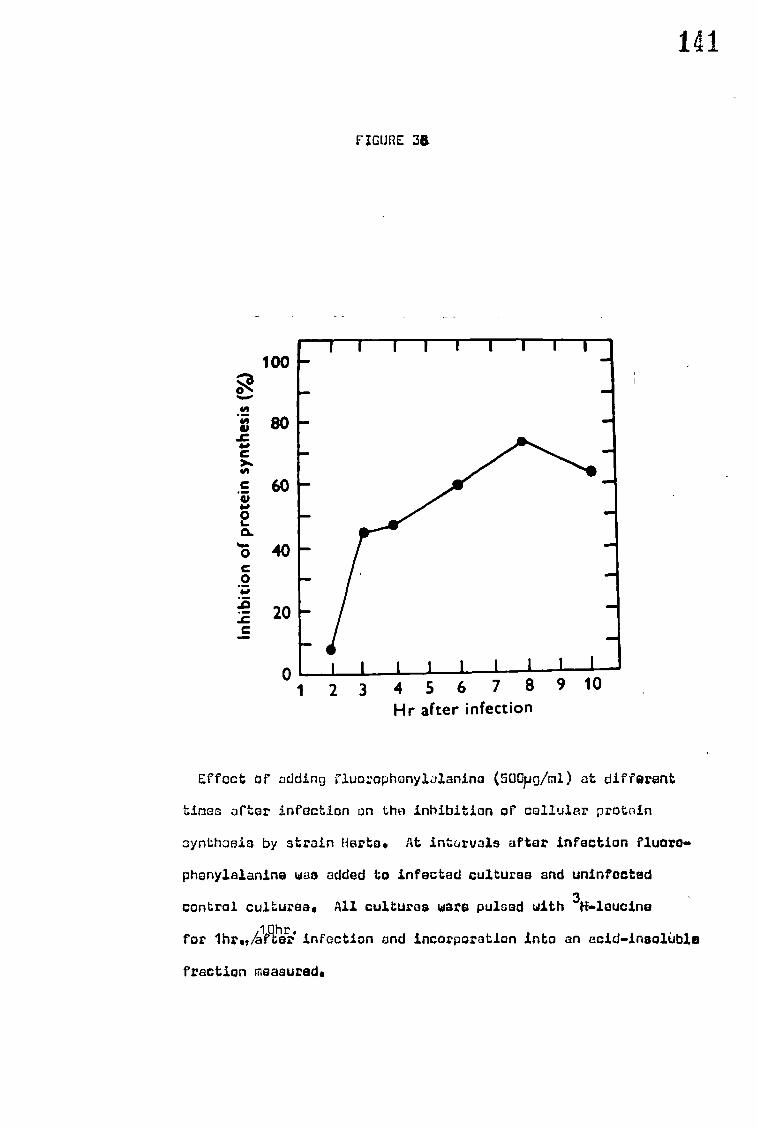
141
FIGURE 38
100 0
•V go .c
C 60
15 0 CL
40 C 0
20 C
1 2 3 4 5 6 7 8 9 10 Hr after infection
Effect of adding fluorophonylalanina (50Gpg/m1) at different
time; after infection on the inhibition of collular protein
synthosis by strain Herta. At intervals after infection fluoro-
phanylalanine was added to infected cultures and uninfected
control cultures. All cultures ware pulsed with 31-6.1eucina
for ihr"agicTr. infection and incorporation into an acid-insoluble
fraction measured.

142 F. EFFECT OF CONGO RED ON THE MORTALITY OF EGGS INFECTED WITH NDV
The effect of congo red on the virulence in eggs of 5 strains:
Herta, Texas, B1, Queensland and F, was tested. Each of the virus
strains was inoculated into the allantois of 10-day-old eggs as a
high dose, . 5 x 108 EID50, or as a low dose, 1-50 EID50. Congo red
was inoculated immediately prior to infection. The mortality of
uninfected eggs treated with conga red was not greater than 57. at
168 hours after inoculation.
Up to 72 hours after infection treatment with congo red gave
considerable protection to eggs infected with low doses of the two
virulent strains (Fig. 39). In later hours, however, the mortality
reaches a similar level in both treated and untreated eggs. Initial
protection suggests that prevention of release does not enable
infection of sufficient cells to cause the death of the chick embryo.
High mortality at all, although greatly delayed, is presumably due
to infective particles being released by death and Lysis of originally
infected cells and eventual infection of sufficient cells to cause
death of the embryo.
Congo red has very little effect on the death of eggs infected
with high doses of virulent virus (Fig. 40). This is predictable
as high doses of virus would ensure sufficient\cells are infected and
killed to bring about death of the embryo even without release of
virus particles.
Eggs infected with low doses of strain B1 and Ulster in the
presence of congo red showed very little variation from infected eggs
without congo red (Fig. 41). Eggs infected with Queensland in the
presence of congo red however produced up to 407. mortality within 96
hours. There is no apparent explanation for this although repeated
experiments produced similar results.

143
High doses of avirulent (or low virulence) virus produced high
mortality of eggs in the presence of congo reds 80-907. in 96 hours
and 1007. in 168 hours compared to 20-307. at 96 hours without congo
red (Fig. 42). This suggests that the prevention of release of
virus can bring about the death of an embryo if sufficient numbers
of cells are infected, even if the virus strain is normally
avirulent.
It should be noted that embryo mortality in the presence of
congo red occurs at a much higher rate after virulent infections
than with avirulent infections (Figs. 40 and 42).

60
cs-P N 0 0 u, 20
0 >- 1- = 4 o-. cc
2 0100
60--
20 1 96
1 HE RTS
I fl NI' 24 48 54 72
HR. AFTER INFECTION
100
i 1 FIELD PHEASANT
144
FIGURE 39
Virulent_virus: low inoculum
Virus w-s inoculated at the following levels: Herta 50 EIDro
per egg; Field Pheasant 12E/050 per egg. Congo red (1mg in
0•25m1) uns added immedistely prior to infection (closed
histogram) control eggs received 0•25m1 sterile distilled
water prior to infedtion(open histogram). NR: not racorded.

X.5
FIGURZ 40
100
60
o 20 w 0
LL
' *Tr I- 100 O cr
2
60
20
HERTS.
FIELD PHEASANT
24 40 48 54 72
96
HR. AFTER INFECTION
Virulent virus: hi:-41 inoculum
Virulent virus was inoculated at the following levels:
Harts 2+5 x 109 EID50 per egg; Field Pheasant Gx 100 CID50 per egg. Congo red (1mg in 0•25m1) W13 added immediately
prior to infection (closed histogrcm), Control eggs received
0,25m1 of distilled water prior to infection (open histogram).
Mn: not recorded,
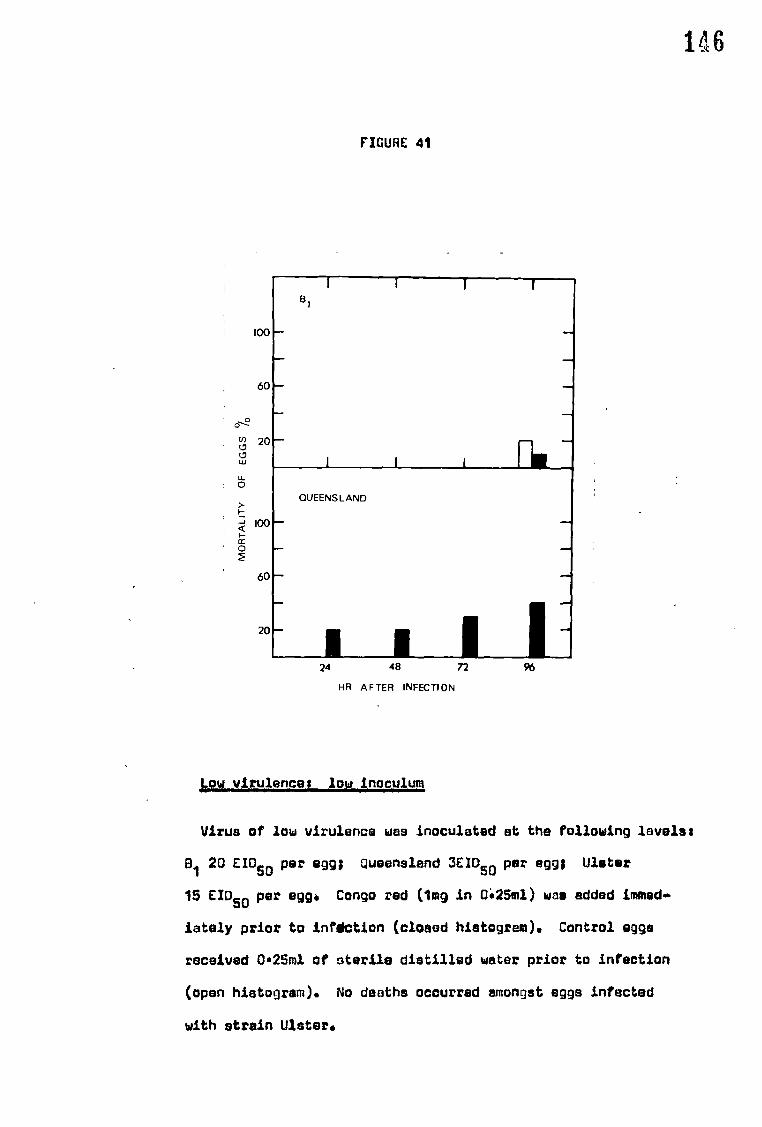
146
FIGURE 41
0-12
100
60
20
81
I I I H.
U. 0
M
OR
TA
LIT
Y
100
60
QUEENSLAND
20
24 48 72
96
HR AFTER INFECTION
Low virulence: low inoculum
Virus of low virulence was inoculated at the following levels:
B1 20 EIDSO per egg; Queensland 3EI050 per egg; Ulster
15 EID 50 per egg. Congo red (1mg in 0:25m1) was added immed..
iately prior to infdction (closed histogram). Control eggs
received 0.25m1 of sterile distilled water prior to infection
(open histogram). No deaths occurred amongst eggs infected
with strain Ulster.
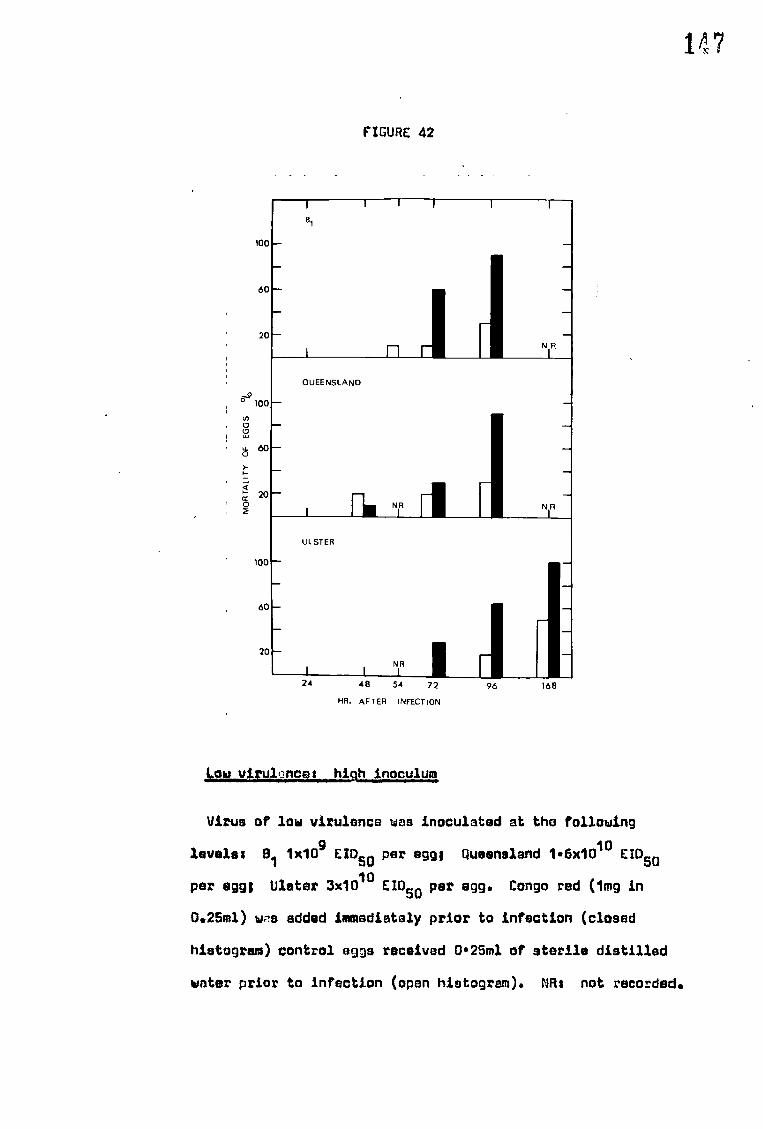
B1
mo—
60 -
1
-
20
QUEENSLAND
c.t) LI — I,
R. NR rl fl N R
ULSTER
MO
RTA
LITY OF 60-
20 -
100 -
60 -
20 - NR
147
FIGURE 42
24 48 54 72 96
168
HR. AF TER INFECTION
'ow viyulonces high inoculum
N R
Virus of low virulence was inoculated at the following
levels; 81 1x109 EIDSO per egg; Queensland 1•6x1010 EID50
per egg; Ulster 3x1010 EIDSO per egg. Congo red (1mg in
0.25m1) was added immediately prior to infection (closed
histogram) control eggs received 0625m1 of sterile distilled
water prior to infection (open histogram). NRs not recorded.

148
G. THE SYNTHESIS OF NDV-INDUCED PROTEINS
The synthesis of virus-induced proteins in NDV-infected cells
could be detected iising the double labelling technique described
in the methods section. The extracted proteins gave the pattern
seen in Figure 43 after polyacrylamide gel electrophoresis and
staining with amido black. Since total cell protein has been
separated on the gel there are very many bands, so many in fact
that it would be impossible to distinguish the extra bands induced
by infection with virus and demonstrates the necessity for labelling
the virus-induced proteins.
All proteins in uninfected cells labelled with 3H-leucina have
complementary proteins in 14C-leucine labelled uninfected cells.
The graph in figure 43 shows the ratio of 3H to "C DPH in a sliced
gel after electrophoresis of a double-labelled uninfected cell
preparation, as expected there are no major peaks and the proteins
show an almost constant ratio of 3 H/14 C. In infected cells, where
virus-induced proteins labelled with 3H have no complementary 14C
labelled proteins in uninfected cells, obvious peaks of 3H/14C ratios
can be seen (Figs. 44-48).
By comparison with egg-grown purified virus run in parallel with
the double-labelled cell extracts it can be seen that the three major
structural proteins of NDV could be detected from the second hour
(1-2 HR) in cells infected with Herta and Field Pheasant but not
until the 4th hour (3-4 HR) with strain Queensland. In the second
hour structural proteins I and III were detectable in Beaudette C
infected cells but protein II was not, although it appeared in the
4th hour.

149
FIGURE 43
3H/ 14c 2
UNINFECTED CELLS
•••••••1
1
••••••V••••••W1/4.••••••••••••%...vii....
10 20 30 40
50
Migration(mm.)
Polyacrylamide gel electrophoresis of polypeptides extracted
from 3H-lewcine and 14C-leucine uninfected cells. Cells wore
pulse-labelled for one hour.
The photograph shows a typical pattern obtained after
electrophoresis and staining pulypeptides extracted from chick
embryo cells whether infected or not.

1 1 1 1 A
—
.—
I I
/ I I[
?I
i
I
1 1 HERTS III
—
—
—.
I I
—
I I I
FIELD PHEASANT
—
I I BEAUDETTE C
1--
.— .... I 1 I I 1
QUEENSLAND
...- —.
1.4.011VPHII4844••••NMIORResemeeseVeliP0 ....., I I I 1
A I Ix
— • • •
—
—
I%
III mum) • —
—
—
gym I
2 3H/14c
2
B CPM 6
x10-2 4
0
3
ail .14 C 2
150
FIGURE 44
10 20 30 40 50 Migration(mm)
Induced proteins I-2hr. aftor infection.
Polyacrillamide gel electrophorosie of polypeptides extracted
from 3H-leucine labelled infected cells end 14C-leucine
labelled uninfected cells. Celle were pulee4abelled one
hour after infection for one hour. Purified 3H-loucine lab.
oiled virus (strain Queensland) was similarly extracted and
run in parallel for comparison (o ).

4c
3
2
BEAUDETTE C
3IY 2 1— 14c
151
FIGURE 45
QUEENSLAND
2 —311/ 'MC
1 —
1 10 2 3
114 5
Migration (mm.)
Induced proteins 3.'4hr. after infection.
Pelyacrylamide gel electrophoresis of polypaptides from
3H-leucine labelled infected cells and 14C-laucine labelled
uninfected cells. Cells ware pulse-labelled 3hr. after
infection for one hour. Similarly extracted purified virus
(strain Harts) was run in parallel, the migration of the
structural polypeptides detected by amido black staining are
indicated.
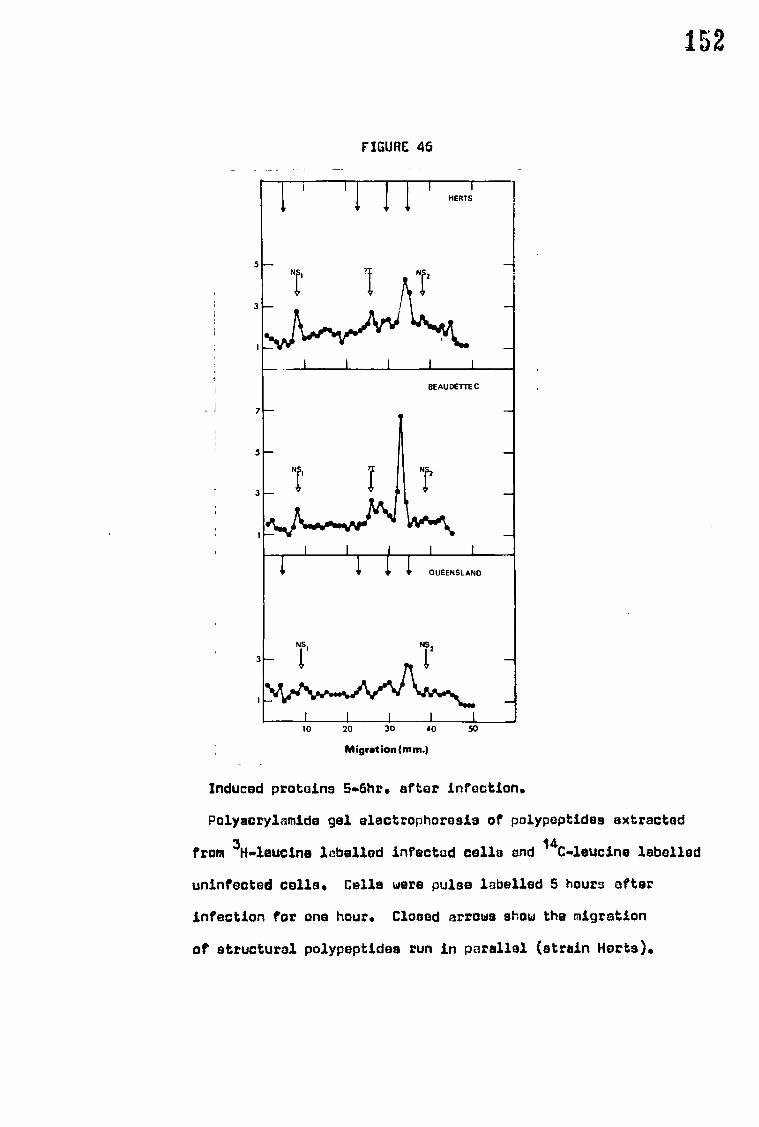
3
I 1 1 i 10 20 30 40 50
7
5
3
1
I
QUEENSLAND
152
FIGURE 46
HERTS
5
3
i
t
1
1 BEAUDETTE C
Migration (mm.)
Induced proteins 5.6hr. after infection.
Polyacrylamide gel electrophoresis of polypeptides extracted
from 3H.leucine labelled infected cells and 14C.leucine labelled
uninfected cells. Celle were pulse labelled 5 hours after
infection for one hour. Closed arrows show the migration
of structural polypeptides run in parallel (strain Herta).

I I I i 1 10 20 30 40 50
Migration(mm.)
153
FIGURE 47
Induced proteins 8-10hr. after infection.
Polyecrylamide gel electrophoresis of polypeptides extracted
from 3H.mleucine labelled infected calls and 14C-leucine
labelled uninfected cells, Celia were pulse labelled 8hr.
after infection (strain BeaudetteC) or 9hr. after infection
(strains Herta and Queensland) for one hour. Closed arrows
show the migration of structural polypeptides run in parallel
(strain Hefts).
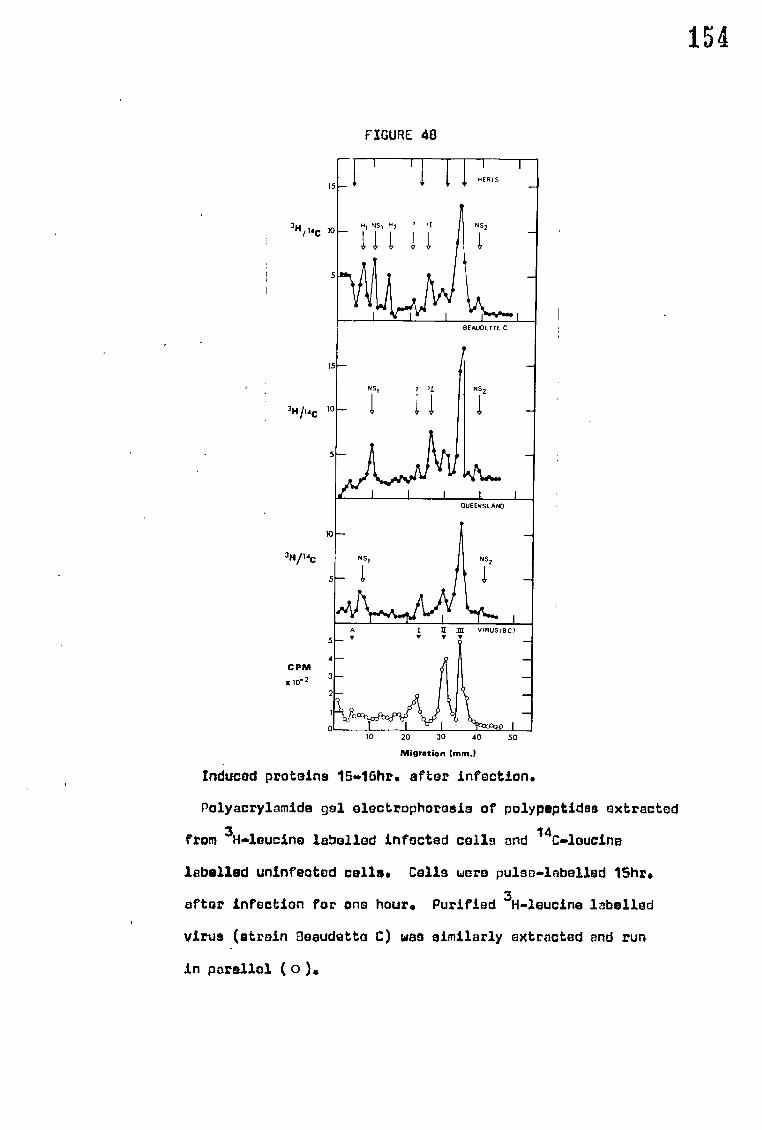
,_.
1
•
ii, NSI
/1,
I
H2 '
/ 11
I M)111011ra%!
'I
I
1 I HERTS
-
f
NS2 -
1
I-
NS, 1 'I
/ i 1
BEAuDETTE C
NS2
-.
I I
NS,
- 1 1. 1
4.1j\rAWL1J1‘.14(
OUEENSLAND
-.
NS2
--
1.... I A I II ICE
• • VIRUS IBC)
31-1/ 14c t
314114c I
3H14c
10
5
4
2
0
CPM
ato-2
154
FIGURE 48
10 20 30 40
50
Migration (mm.)
Induced proteins 15.46hrs after infection.
Polyacrylamide gel electrophoresis of polypeptidss extracted
from 3H4eucine labelled infected cella and 14Cibleucine
labelled uninfected cells. Cells wore pulse-labelled 15hrs
after infection for one hour. Purified 3H-leucine labelled
virus (strain 3eaudetto C) was similarly extracted and run
in parallel ( o ).

155 The structural protein 1 which had a corresponding protein in
Queensland infected cells seems to be represented in Beaudette C and
Herta infections by protein '?I', although this protein_migrated further
than protein I of egg grown pure virus preparations of either of the
strains run on parallel gels (Figs. 44-48). This assumption is
further complicated however by the occasional appearance in some gels
of these strains of a peak showing the same migration as polypeptide
(Fig. 44 1-2 HR Beaudette C, Fig. 48).
Structural protein A which is not always discernable in gels
of pure radioactive labelled virus can be seen in all three of the
strains in the later samples.
Two non-structural proteins are produced which are common to
all three strains. The most prominent of these, NS, which has a
molecular weight of approximately 180,000 is first seen in all the
strains in the 6th hour (5-6HR'Fig. 46). The production of NS, in
Queensland infected cells is at a maximum at the 11th hour when it is
the most dominant peak (Fig. 47). The other non-structural protein,
NS2, has a molecular weight of approximately 36-38,000. The peak
produced by N52 was always very small, however, it is unlikely to be
adventitious as it was present consistently from the 4th hour.
Herts infected cells produced two non-structural proteins which
were not produced in cells infected with the other two strains.
Protein H1 was first'seen 9-10 HR.after infection (Fig. 47) and has
an approximate molecular weight of 200,000. in addition another protein,
H2' was detected in the 16th hour which has an approximate molecular
weight of 140,000 (Fig. 48 Herta 15-16). Smaller peaks at the same
migration as H2 were seen earlier in infection but were inconsistent
in both appearance and migration.
A summary of the results and the proteins induced in infected
cells is given in Table 19.

Strain and times seen
Protein
All strains and pure virus
All Queensland gels except 1-2 HR and in pure virus preparations virus
In all Herts and Beaudette C gels but not in pure virus preparations
In all'gels except 1-2 HR
III
A
I
NS1 In all gels after 3-4 HR. Not in pure virus preparations
Non-structural 180,000
If NS2 36-38,000 In most gels but not in pure virus preparations
111 200,000 Detectable in Harts 9-10 HR and Hertsl5-16 HR only
II H2 140,000 In Herts 15-16 HR only
156
TABLE 19
Summary of the induced proteins seen during NDV infections
Type Approximate molecular weight
Structural ) 200,000
Structural 88,000
Structural?? 78,000
Structural 58,000
11 42,000

157
H. RNA PRODUCTION OF INFECTED CELLS IN THE PRESENCE OF ACTINOUYCIN D
The production of RNA in chick embryo coverslip cell-cultures
infected with different strains of NDV was measured by the incorporation
of 3H-uridine into an acid insoluble fraction in the presence of 5 ug/ml
actinomycin D. Under these conditions only the production of virus
specified RNA is measured. The results show a remarkable correlation
between the amount of RNA produced and the virulence of the five strains
tested (Fig. 49). Strain Herts shows 2357. more uptake of 3H-uridine
than Queensland after 12HR, while Texas and Beaudette C show respectively
1857. and 1357. more at that time.
RNA production also appears to begin somewhat earlier with the
virulent strains than the avirulent.
The drop in RNA production seen in the more virulent strains,
notably Herts, appears to be due to loss of cells from the monolayers
as a consequence of the cytopatbic effect of the virus.

NDV RPM production
Chick embryo cells HE RTS
TEXAS
ULSTER
QUEENSL AND
CONTROL 0
...
BEAUDE TTE C
158
FIGURE 49
300
2 a. U
200 a
E 0
100
2
4
a
10
12
Hr after infection
NDV RNA production in chick embryo cells.
The accumulative uptake of 3H-uridine in the presence of
actinomycin 0 (Speml) into an acid-insoluble fraction was
measured at hourly intervals after infection of chick embryo
cell coverslip cultures. Results are expressed as a % of
Queenalanctrifacted cells after 12hr. (1001As approx. 3,000cpm.).

19 IV DISCUSSION
The object of this work was to investigate the wide range of
virulence seen between strains of NDV. Most of the sections of the
work studied have been previously suggested as, or shown in preliminary
experiments to be, likely candidates for the causitive agent of the
differences in virulence.
A. STRUCTURAL PROTEINS
Since the demonstrations by Ornstein (1964) and Davis (1964)
of the advantages of polyacrylamide gel discelectrophoresis over
other techniques used for protein separation this method has been
increasingly used in investigations into the structure of virions and
production of virus proteins in infected cells. In particular,
acrylamide gel electrophoresis has been used in the presence of SDS
after disruption of virions or treatment of other proteins with this
detergent, since under these conditions migration of proteins is directly
related to their molecular weights (Shapiro, Vifluela d Maize', 1967).
Although acrylamide gel electrophoresis of disrupted virus after
treatment with SDS is a powerful tool in the investigation of virus-
structural-proteins there are certain drawbacks to the method that
should be considered. The most obvious criticism is that more than
one polypeptide may be represented in any one band. There may also
be one or more components of the virion which are of particular importance
but present in such low amounts as to be undetectable by this method.
The three major and one minor polypeptide bands consistently seen in
this study should, therefore, be regarded only as the minimum number
of polypeptides likely to be of viral origin.
The exact correspondence of the peaks of radioactivity in gels of
labelled virus to the stained bands demonstrates that the polypeptides

160
are specifically induced by the virus, incorporation into the virions
of proteins made before infection would mean that stained bands of these
proteins would not show a corresponding radioactive peak. However,
there is no information as to whether or not the proteins are coded
for directly by the virus genane or are produced by the cell as a
response to infection. The results show (with the considerations
mentioned ) that the NDV virion consists of three major polypeptides
I, II and III and one minor polypeptide, A.
The validity of polypeptide II as a single or multiple polypeptide
is of particular interest. In this work, that of Evans and Kingsbury
(1969), Haslam et al (1969), Bikel and Duesberg (1969), Mountcastle
et al (1970) and Mountcastle, Compans and Choppin (1971), it has been
shown that isolated RNP migrates as a single band to the position of
polypeptide II of whole virus and it has been assumed that the RNP
polypeptide is the only polypeptide in this position. However
Mountcastle et al (1970) have also shown that the polypeptide of RNP
isolated from a tryptic digest of cells infected with NDV migrates with
a molecular weight of 47,000 to the position of polypeptide IIa in this
study. A band at this position was also reported as an occasional band
by most of the other authors. Presumably the partial digestion of the
RNP by trypsin is sometimes mimicked by SDS disruption causing the
appearance of the occasional band IIa. Another problem is the apparent
uptake of labelled glucosamine into polypeptide II - the band identified
as the RNP polypeptide. Using fluorescent antibody techniques RNP is
first detected in the cytoplasm of infected cells close to the nucleus
(Rott and Schiffer 1964, Rott and Scholtissek, 1967) and would seem
unlikely to be a glycoprotein, as glycoproteins are usually associated
with cell surface membranes. Mountcastle et al (1971) also show activity

161 at the same migration as polypeptide II, identified as RNP, after
labelling with radioactive glucosamine, however) they report that there
is no uptake of labelled glucosamine detectable in isolated RNP and
suggest that the results obtained are due to another polypeptide showing
identical migration to polypeptide II. If this is the case it is
remarkable that in the attempts to separate and isolate the sub units of
NDV both in this study and that of Haslam et al (1969) no further evidence
of this protein has appeared.
To identify a viral subunit that unit should,ideallyt be isolated
and purified. The lack of success in the studies undertaken so far, i.e.
this study and that of Haslam et al (1969) and Evans and Kingsbury (1969)
in separating the neuraminidase and haemagglutinin sub units has prevented
the absolute confirmation of the suggestions that polypeptide I is associated
with haemagglutinin, and polypeptide III with neuraminidase. However,
although different methods of disruption and analysis were used, similar
conclusions as to the nature of the polypeptides were reached in each
study, although Evans and Kingsbury (1969) do•:not associate neuraminidase
with polypeptide III. The fact that polypeptide I is a glycoprotein
gives more significance to the possibility that this band is HA since it is
known that haemagglutinin originates at the cell surface membrane
(Mott and Scholtissek, 1967). Failure to bring about separation of the
viral sub units in a biologically active form may well be a warning against
the general assumption that the activities are associated with separate
proteins, it is arguable that both activities could be associated with
one protein or with different polypeptides of the same protein.
The absence of any significant difference in the number, molecular
weight or proportion of the polypeptides, seen in this study, from one
strain to another suggests that differences in virulence are due to
differences that occur in the production of the virus between the
moment of entry and the moment of release.

162
B. NEURAMINIDASE STUDIES
None of the properties of NDV neuraminidase examined show any great
variation from strain to strain. Unlike influenza virus (Rafelson et al,
1963) it appears that different NDV strains possess neuraminidases which
are essentially identical in their biochemical properties.
On assessment of physical characteristics Beaudette C appears to be
the most atypical strain. The heat resistance shown by Beaudette C
neuraminidase is probably due to the protection of the intact virion since
Drzeniek et al (1966) have shown that neuraminidase isolated from Beaudette
C virus is just as heat labile as neuraminidase from the other strains.
Beaudette C has twice as much haemagglutinin as other strains which could
account for the protection of the neuraminidase, although estimations of
the proportion of polypeptide I is no greater in Beaudette C than other
strains. Rott, Reda and Schafer (1962) have shown that a large fraction of
the virus in their Beaudette C suspensions was devoid of ribonucleoprotein.
From the HA/EID50 ratios which are, in fact, estimations of non-infectious
particles, this does not appear to be a general characteristic of all the
strains, although Ulster also seems to have a high HA/EID50 ratio.
It is difficult to assess the significance of the differences in the
effect of Tween 20 on the neuraminidase of the different strains.
Presumably the enzyme of those strains showing a low yield of released 6tRel
neuraminidase after Tween 20 treatment is/more intimately associated with
the lipid membrane of. the virus, or that their enzyme is different in
some way and rendered inactive in the presence of Tween 20.
The pH optimum of NDV neuraminidase is lower than that recorded by
Drzeniek et al (1966) for Italien and Beaudette C. However this is
probably due to variation in the substrate, it has been shown by
Rafelson et al (1963) that pH optima of neuraminidase differ with
substrate. The observation of two peaks of enzyme activity has been
previously recorded by Drzeniek, Bagel and Rott (1967) with the neuraminidase
of bovine parainfluenza 3 virus. Since in both the substrates used the

163
NANA is not specifically attached i the two peaks could be produced by the
availability of substrate varying with pH. However, Drzeniek (1967) using
2-3' and 2-6' linked sialolactose, has shown that NDV neuraminidase,
unlike Vibrio cholerae neuraminidase cleaves only 2-3' linkages and that
- the failure to cleave 2-6' linkages i* unaffected by pH changes.
Alternatively the enzyme may exist in more than one form, with different
pH optima.
The relationship between virulence and cell-associated neuraminidase
levels has been noted previously by Reeve et al (1970) who have shown
that all major virus antigens accumulate in the chorioallantoic membrane
of eggs infected with virulent strains but not in eggs infected with
avirulent strains of NDV. Accumulation of neuraminidase and other antigens
in virulent strains infers that there will be a far more extensive
modification of the cell membrane than with avirulent strains and this
alone could cause cell death. However, it is also possible that cell
death could be brought about by generalised disruption of cell membrane
caused by the accumulating neuraminidase, or by the action of neuraminidase
at a specific site or sites in the cell (Bang 1953, Allison and
Mallucci, 1965).

164
C. CELL FUSION AND HAEMADSORPTION
1. Haemadsorntion
Bang (1953) reported that avirulent stains were produced at the cell
surface on microvilli while virulent strains appeared to originate more
within the cytoplasm. This may well account for the observation in this
study that intial haemadsorption to cells infected with avirulent strains
is at the extremities of the cell but closer to the nucleus with virulent
strains.
Banhowski (1964) had previously shown that cells infected with
avirulent strains of NDV took longer to show haemadsorption than cells
infected with virulent virus, in the present study it is demonstrated that
haemadsorption is a direct consequence of the virulence of the infecting
strain, at least on the basis of gross virulence, i.e. velogenic
mesogenic and lentogenic strains. The extensive modification of cell
membrane, demonstrated by haemadsorption, could in itself be responsible
for the death of cells and ultimately the virulence of the infecting
strain. It is particularly noticeable that avirulent strains appear
never to reach the levels of haemadsorption attained by virulent strains,
increase in red cell attachment is negligible between 16-36 HR after
infection by avirulent strains. The differences in haemadsorption could
be due solely to a difference in rate of virus production but this does
not explain the evidence which suggests that virus is released at the
same rate to similar levels from all strains (Reeve and Waterson, 1970,
Liu and Bang, 1953, Pennington, 1967). It seems likely that differences
in haemadsorption are related to the accumulation of virus antigen recorded
by Reeve et al (1970). However this too may be the result of a different
rate of production.
2. Cell fusion
Cell fusion in chick embryo cells was also shown to be directly
related to the virulence of the infecting strain, the most virulent

165
Mit.• 0...40 J.
strains producing the largest polykaryocytes. Similarly only strains
which produce plaques showed cell fusion to any great extent, which
may suggest that cell fusion is an initial stage in plaque forma.
The relationship of cell fusion to virulence has been reported with
other viruses (Plowright, 1962, Bodon and Greczi, 1966, Ilahmias and
Dawdle, 1968) but the reason why virulent virus and not avirulent should
induce cell fusion and the mechanism by which they cause it, remains
obscure. At the moment there are two main theories for the mechanism
of cell fusion (for review see Postei 1970). The first (Ejercito,
Kieff d Roizman, 1968) postulates that maturation and accumulation of
virus-products at the cell surface causes gross changes in the cell
membrane which result in cell fusion. In the NDV system it is known
that accumulation of virus products is important with respect to virus
virulence and the vast haemadsorption to cells infected with virulent
strains is evidence of the massive cell modification that occurs.
However Poste (1970) disagrees with this theory on the basis that some
viruses that mature at the cell surface do not fuse cells. This is
exactly the case in the NDV system and yet only the strains that
accumulate, irus components and drastically modify the cell surface
fuse cells. The other theory, credited to Allison (1967), is
basically that modification of the cell surface glycoproteins which
must occur before cell fusion can take place is brought about by the
release of lysosomal enzymes at the cell surface (for review see Poste,
1970). This is supported by the findings of Allison and Mallucci
(1965), using NDV, they have reported that lysosomal damage and release
of enzymes occurs in virulent but-not avirulent infections. However
this explanation merely shifts the question one step since there is
no explanation of why virulent virus infections cause lysosomal
damage.

166
3. EFFECT OF INHIBITION OF PROTEIN SYNTHESIS ON CELL FUSION AND HAEMADSORPTION
The results obtained of the effect on haemadsorption and cell
fusion of FPA and cycloheximide suggest that not only is protein
synthesis necessary for the manifestation of these phenomena but that
inhibition after 4 hours post infection has little effect on their
production. This suggests that the proteins responsible for both cell
fusion and haemadsorption are produced in sufficient quantities in the
first 4 Hitt° render further synthesis of these,proteins unnecessary
for maximum occurrence of cell fusion and haemadsorption. Or, in the
case of haemadsorption, that sufficient protein (i.e. haemagglutinin)
is produced in the first 4 hours to produce maximum modification of
the cell membrane.
The inhibition of cell fusion by inhibition of protein synthesis
has previously been reported with other viruses. Kaku and Kamshbraa
(1964) reported the inhibition of cell fusion in vaccinia infected L-cells
in the presence of puromycin and Falk* (1965, 1967) showed that fusion
by harps* simplex virus of rabbit kidney cells was prevented by the presence
of cycloheximide and FPA but not'chloramphenicol.
The nature of the proteins or protein that are responsible for
cell fusion by NDV is not blear. The fact that high multiplicities of
virus can fuse cells without protein synthesis (cell fusion from without)
and the inhibition of both types of cell fusion with NDV specific
antiserum (Brett and Gallaher, 1969) suggests that a structural protein
may be implicated by its action at the cell surface. Although it is
difficult to visualise this in terms of the 'lysosome labilisation'
theory of cell fusion already discussed. In fact the failure of
cells to fuse in the presence of specific antisera is excellent
evidence that the maturation of the virus at the cell surface is
important for cell fusion.

167 D. INHIBITION OF CELLULAR PROTEIN SYNTHESIS
The results show that inhibition of cell protein synthesis by NDV
strains was directly related to the virulence of the infecting strain.
The results of LDH release from infected cells suggest that the inhibition
of cell protein synthesis is not merely a consequence of cytopathic
effect, this conclusion was also reached by Wheelock and Tamm (1961)
using Hela cells.
Scholtissek and Rott (1965) reported that after 10 hours chick
embryo fibroblast cells recovered from the initial inhibition of protein
synthesis induced by strain Italien. There is no evidence, in this
investigation of any such recovery after inhibition by any of the
strains used. Similarly Wilson (1968) using Texas and Beaudette C
failed to show recovery.
Inhibition of cell protein synthesis by other viruses has also
been recorded (Martin and Kerr, 1968). In particular the picornaviruses
and vaccinia virus show inhibition (Martin and Kerr, 1968, Bablaniin,
1970) which is particularly rapid (within 3 HR of infection) and seems
unlikely to be comparable to the slower inhibition by NDV virulent strains.
The experiments using FPA to inhibit functional protein synthesis
demonstrate that a protein (or proteins) synthesised in the first 3 HR
after infection is responsible for the inhibition of cell protein synthesis.
This infers that the genom6 of avirulent strains is unable to code for
a protein (or proteins) with this ability.
It is unlikely that the inhibition of cell protein synthesis, or of
all macromolecule synthesis, is alone responsible for the difference
in virulence and cytopathogenicity between strains of NDV. Neither
puromycin, which inhibits protein synthesis, or actinomycin, which
prevents RNA synthesis cause the death of Kreb's ascites or Hela cells
in less than 24 HR (Martin and Kerr, 1968); while control chick fibroblast
or BHK cells treated with actinomycin D azauridine or FPA showed no
sign of cytopathology at times after treatment when similarly treated
but infected cells show gross cytopathic effects.

I
E. EFFECT OF CONGO RED ON THE MORTALITY OF EGGS INFECTED WITH NDV
The results obtained are a remarkable vindication of the hypothesis
outlined in the introduction ( see review section). It must be concluded
from the results that the prevention of release of virus can bring about
the death of an embryo, if a sufficient number of cells are infected,
even if the virus strain is normally avirulent. This agrees with the
conclusion of Reeve et al (1970) that the accumulation of viral antigen,
which is presumably related to the rate of growth since they suggest
virus is released at the same rate, is directly related to virulence.
This is particularly supported by the speed with which high doses of
virulent virus in the presence of congo red reached 1007. mortality
(48-54 HR after infection) compared with high doses of avirulent
virus in the presence of congo red (96-168 HR).

169 F. THE SYNTHESIS OF NDV-INDUCED PROTEINS
Using the double labelling technique described, altogether six
proteins were seen common to all strains, if it is assumed that protein
'71' seen in gels from cells infected with Herts and Beaudette C
Corresponds to protein I seen in Queensland infected cells and pure
disrupted virus of all strains. Of the six protefns,A, I, II and III
correspond to polypeptides seen in pure virion preparations and can
be regarded as structural proteins. The major interest of these
proteins is the time at which they were produced after infection. The
virulent strains show all three major peaks HR after infection
while Queensland shows no obvious production until 3-4 HR. This is
further evidence that virulent strains are produced at a faster rate.
The two other polypeptide peaks NS„cand NS2 could not be identified
with structural proteins, NS2 was produced in very small amounts while
the production of NS1 even exceeded the production of the virus
proteins on one occasion. If NS1 and NS2 are the only two non-structural
proteins produced, at least in avirulent'infections, then one at
least would be expected to be an RNA-polymerise. NS1 was not detected
until the,6th hour which seems late for RNA-polymerase production
although experiments show that virus RNA production is not detectable
until 4-6 HR after infection (Results section H, also Brett and
Robinson, 1971). If NS1, NS2, or both, are responsible for RNA-
polymerase activity it is strange that production of these polypeptides
continue after the 'fall off' of RNA production. However the
apparent decline in RNA production seen in this study, and those of
Brett and Robinson (1967, 1971) with virulent strains may be due to
cytopathic effect:.

170 The significance of the induced polypeptides H1 and H2 which are
only detectable in Herts infections after the 10th hour, is difficult
to assess as they appear too late to have any relevance to the
differences in the virulence of the strains. One possibility is that
immediately prior to cell death, protein metabolism is sufficiently
disrupted to cause uptake of labelled amino acid into some cell
proteins even in the presence of actinomycin D.
The molecular weight of the RNA genome of NDV has been estimated
as approximately 6 x 10L6 daltons (Nakajima and Obara, 1967, Duesberg,
1968) which, using an average nucleotide molecular weight and an average
amino acid molecular weight, could code for protein of approximate
M.W. 800,000. The total estimated molecular weights of the four
structural proteins and the two non-structural NS1 and NS2 is 605,000
or 660,000 if the structural glycoprotein of molecular weight 56,000
identified by Mountcastle et al (1971) is included. This means that while
there is sufficient genetic information in the NOV genome to code for
both the structural and non-structural proteins detected in this study
it is unlikely that H1 and H2 are in fact coded for by the NDV genome.t,
It should be noted here that recent work (Huang, Baltimore and
Brett, 1971) has suggested that there is an RNA dependent RNA-polymerase
in the virions of NDV. This polymerase has such low activity that
it seems unlikely to be present in sufficient quantity to produce a
band on acrylamide gels, similarly early RNA production due to this
polymerase is unlikely to be detected by the method used in this study
to measure RNA production in cells. No evidence has been presented to
show that the virion polymerase is not a polymerase that is present in
cells infected with NDV late in infections and taken into the virion
particles with RNP at release. However if the virion polymerase is a
discrete protein it may account for polypeptides NS1 or NS2 or if
undetected by the double-labelling technique should have a molecular
weight not greater than 140,000.

171 Work with the effect of protein inhibitors on haemadsorption, cell
fusion and protein synthesis inhibition suggested a protein (or
proteins) necessary for these effects was produced in the first 4
hours after infection. Of the proteins induced by NDV only the structural
proteins and perhaps NS2 were detectable within 4 HR of infection.

172 G. PRODUCTION OF NDV-SPECIFIED RNA
The production of NDV RNA in cells offers further evidence that
the rate of formation of the virus products is important to the
virulence of the infecting strain. Most of the RNA produced, 80-90%
in Beaudette C infections, is in fact complementary to the parental,
strand (Kingsbury, 1970) so that production of RNA is not necessarily
a measure of virion production unless parental RNA production is directly
related to complementary RNA production.
Schafer, Pister and Schneider (1967) demonstrated the production
of viral antigens in the absence of RNA synthesis and proposed that the
NDV input strand of RNA, (+) RNA, acted as the sole messenger for virus
protein production. Reeve and co-workers (Reeve and Poste, 1971,
Reeve et al, 1971) have shown that even in the presence of azauridine,
which inhibits RNA synthesis, cell fusion and haemadsorption still
occur, findings that support those of Schafer et al (1967). Other
authors support the hypothesis that RNA complementary to the virus genome,
(-) RNA, acts as messenger RNA (Brett and Robinson, 1967, Blair and Robinson,
1968, Kingsbury, 1970) while Ralph (1969) has proposed that at
different stages of infection both (+) RNA and (-) RNA may act as
messenger RNA. in this present investigation increase in RNA production
due to NDV infection was not detected earlier than 4 HR after infection.
Structural proteins however were detected some time earlier than this,
1-2 HR after infection in the case of the virulent strains, which
supporta the hypothesis that, initially at least, the NDV genome
acts as messenger RNA. Of course RNA production before the 4 HR may
be merely too small tallye detected by the method used.

173 H. GENERAL DISCUSSION
Table 20 summarises the relationship of the various parts of this
work to virulence. Most of the phenomena that relate to virulence are
associated with the rate of production of virus. Cell fusion and
protein synthesis cut off appear to have no such relationship. The
theories on the cause of cell fusion have been previously discussed
but if massive cell modification by the virus could be responsible
then presumably this would relate to the rate of growth of the infecting
strain. Although protein synthesis inhibition by NDV and other viruses
has been thoroughly investigated little is known of the actual mechanisms
involved (Martin and Kerr, 1968) for this reason it is difficult to
relate protein synthesis inhibition in NDV to any other aspect of
virus infection.
The results obtained showing the effect of congo red on virulence,
in addition to the accumulation of neuraminidase and the other virus
antigens (Reeve et al, 1970), suggests that accumulation is responsible
for the death of NDV-infected cells and eggs. However this does not
explain why, if virus is produced at a faster rate,during infection by
virulent strains, it is not merely released at a faster rate and
accumulation prevented. Unless, of course, the release of virus is
controlled by the cell and not the virus. This may be so, Compans
et al (1966) showed that BHK 21-F cells infected with SV5 accumulated
viral RNP, released very little mature virus and were eventually killed,
while , primary rhesus monkey kidney (MK) cells, infected with the same
virus, released infective virus to high titres, did not accumulate
RNP and were not destroyed. Further to this, studies of NDV growth
rates have shown no differences in released virus titres that can be
related to virulence (Reeve and Waterson, 1970, Pennington, 1967,
Drain, 1969, Reeve et al, 1970.)

174 15
If the inference/that the rate of release of virus particles is
constant or approximately so, while the rate of productiOn varies
with strain,two states of infection could occur:
i) Infection in which the rate of viral production is less than
or equal'to the rate of virus release, i.e. the strictly
avirulent strains Queensland and Ulster.
ii) Infections in which the rate of production exceeds the rate
of release, causing accumulation of viral products and eventual
cell death. The virulent strains would be in this category
and the degree of virulence vary with the amount the rate
of production of viral antigens exceeded the rate of virus
release. So that virus production in Harts infections would
be far in excess of virus release while B1 (which forms
plaques occasionally and can kill eggs, see Table 1, also
Drain, 1969) would have a virus production rate only slightly
above the rate of release.

1 5 Table 20
Summary of the relationship to virulence of the properties investigated
Related to virulence
Not related to virulence
Accumulation of neuraminidase. Biophysical properties.
The extent of cell fusion
Biochemical properties of
of infected chick cells. neuraminidase.
The extent of haemadsorption Structural proteins.
to infected chick cells.
Inhibition of cellular Induced proteins up to 9 HR after
protein synthesis after infection. (After this time Herts
infection. infected cells induced two unique
protein; the relevance of which is
lleath of eggs after high discussed in the text).
doses of virus in the
presence of congo red.
Time of appearance of induced
proteins in infected chick cells.
ProductiOn of RNA in infected
chick cells treated with
Actinomycin D.
It has also been demonstrated that high doses of avirulent virus
show much higher lethality to eggs in the presence of congo red.

176
V. CONCLUSIONS
The relationship to virulence of the properties investigated are
summarised in Table 20. It is concluded from these results and previous
work by other authors:
1. Death of cells infected with NDV is a consequence of
accumulation of virus products in the cell.
2. Accumulation is related to the rate of production of the virus
components, therefore variations in rate cause variations in
virulence.
VI. SUGGESTIONS FOR FURTHER WORK
This work suggests that the rate of production of the virus components
in cells is directly related to the virulence of the infecting strain.
The most obvious step in the continuation of this work is therefore
to determine why production should be at a faster rate with one strain
than another. Since the cell's mechanism is used for the translation
of viral messenger-RNA it can be assumed that the limit to virus protein
production, which appears from this work to be responsible for cyto-
pathogenecity, is the production of viral messenger-RNA. Since RNA
production also appears to be related to virulence it would be significant
to study the NDV specified RNA-dependent RNA polymerase made during
NDV infections (Scholtissek and Rott, 1969) for differences between
strains with respect to both the rate of production by the polymerase
and the species of RNA produced.
In this study proteins produced during infections with either
virulent or avirulent strains have been shon to be similar. However
only extracts of whole cells have been examined, it is suggested that
sub-cellular fractions should be examined using similar techniques
to determine the exact site of virus specified protein production.
The hypothesis is that virulent strains may be able to modify membranes

177
that the avirulent strains cannot. Bang (1951) has shown that while
virulent strains appeared to develop within the cytoplasm avirulent
strains were formed only at the cell surface. The theory that lysosome
damage may be responsible for the initial cytopathic effect - cell
fusion, adds significance to such a study.
One of the more fundamentally important considerations in this
thesis is that the parental ANA may alone code for virus proteins.
Although Reeve and Poste .(1971) and Schafer et al (1967) both reached
the same conclusion using different techniques to measure viral
protein production in both cases the inhibitor 6-azauridine was used.
Similar experiments using a wide range of RNA inhibitors are now
necessary to confirm the results obtained with 6-azauridine. Double'
labelling experiments could be run in conjunction with experiments in
which RNA production is inhibited to determine whether all the virus-
induced proteins can be coded for by the input strand.

178
VII REFERENCES
Ada, G.L. and Perry, B.T. (1956) Journal of General Microbiology
14, 623.
Adams, W.R. (1965) Federation Proceedings Abstracts 24, 159.
Allison, A. (1967) Perspectives in Virology 5, 29.
Allison, A.C. and Mallucci, L. (1965) Journal of Experimental
Medicine 121, 463. ••••••••••••
Allison, A.C. and Sandecin, K. (1963) Journal of Experimental
Medicine 117, 879.
Alwyn, J. (1968) Ph.D. Thesis, University of London.
Aminoff, D. (1961) Biochemical Journal 81, 384.
Andrewes,C.H., Bang, F.B. and Burnett, F.M. (1955) Virology, 1, 176.
Asplin, F.D. (1952) Veterinary Record 64, 245.
Aurelian, L. and Roizman, B. (1965) Journal of Molecular Biology
11, 539.

179
Bablanian, R. (1970) Journal of General Virology 6, 221.
Bablanian, R., Eggers, H.J. and Tam, I. (1965) Virology, 26, 100.
Bang, F.B. (1953) Bulletin of the Johr Hopkins Hospital 92, 291.
Bankowski, R.A. (1961) Research in Veterinary Science 2, 193.
Baron, S. (1964) In Newcastle Disease Virus. An Evolving Pathogen,
p205. Ed. by Hanson, R.P. Madison, University of Wisconsin Press.
Beach, J.R. (1943) Proceedings of the 46th Annual Meeting of the
United States Livestock Sanitary Association. p203.
Beach, J.R. (1944) Science 100, 361.
Becht, H. and Drzeniek, R. (1968) Journal of General Virology 2, 261.
Biddle, F. (1968) Journal of General Virology 2, 19.
Bikel, I. and Duesberg, P.H. (1969) Journal of Virology 4, 388.
Blair, C.D. and Robinson, W.S. (1968) Virology 35, 537. •••••••••
Bodo% L. and Grecci, E. (1966) Acta microbiologica Academiae
scientiarum hungaricae 13, 185.
Bolognesi, D.P. and Wilson, D.E. (1966) Journal of Bacteriology 91, 1896.
Boney, W.A. (1951) South Wales Veterinarian 5, 19.

180
Bonifas, V. and Schlesinger, R.W. (1959) Federation Proceedings 18, 560.
Bratt, M.A. and Gallaher, W.R. (1969) Proceedings of the National
Academy of Sciences of the United States of America 64, 536.
Bratt, M.A. and Robinson, W.S. (1967) Journal of Molecular Biology 23, 1.
Bratty M.A. and Robinson, W.S. (1971) Journal of General Virology 10, 139. 111110•11•11
:Iukrinskaya, A.G., Burdolea, 0., and Vorkunova, G.K. (1966)
Proceedings of the Society for Experimental Biology and Medicine 123, 236. .1.111.1.111.1••
Burge, B.W. and Strauss, J.H. (1970) Journal of Molecular Biology 47, 449.
-Chenock, R.M. and Coates, H.V. (1964) In Newcastle Disease Virus.
An Evolving Pathogen. p279. Ed. by Hanson, R.P. Madison: University
of Wisconsin Press.
Compans, R.W. and Choppin, P.W. (1967) Virology 33, 344.
Compans, R.W., Holmes, K.W., Dales, S. and Choppin, (1966)
Virology, 30, 411.
Cruickshank, J.G. (1964) -Im-Cellular Biology of Myxovirus Infections:
Ciba Foundation $ymposium. p5. Ed. by Wolatenholme, G.E.W. and Knight,
3. London: Churchill Ltd.
Cunha, R., Weil, M.L., Beard, D., Taylor, A.R., Sharp, D.J. and Beard,
J.W. (1947) Journal of Immunology 55, 69.

181
Davis, B.J. (1964) Annals of the New York Academy of Sciences 121, 404.
Dobson, N. (1939) Proceedings of the 7th World's Poultry Congress,
Cleveland. p250.
Doyle, T.M. (1927) Journal of Comparative Pathology and Therapeutics
40, 144.
Drain, J.E. (1969) Ph,D. Thesis, University of London.
Drzeniek, R. (1967) Biochemical and Biophysical Research Communications
26, 631.
Drzeniek, R. and Rott,11. (1963) Zeitschrift fUr Naturforschung 18 b, 1127.
Drzeniek, R., Bogel, K. and Rott,1.R. (1967) Vi*ology 31, 725.
Drzeniek, R., Frank, H. and Rott,'R. (1968) Virology 36, 703.
Drzeniek, R., Seto, J.T. and Rott,,R. (1966) Biochimica et biophysics
acta 128, 547.
Duesberg, P.H. (1968) Proceedings of the National Academy of Sciences
of the United States of America 60, 1511. .101111.1111
Duesberg, P.H. and Robinson, W.S. (1965) Proceedings of the National
Academy of Sciences of the United States of America 54, 794.
Dulbecco, R. and Vogt, M. (1954) Journal of Experimental Medicine 99, 167.

182
Earle, H. (1959) Science 130, 432.
Ejercitot P.M., Kiefft E.D. and Roizman, B. (1968) Journal of
General Virology 2, 357.
Ensmingert W.D. and Tammt I. (1970 a) Virology 40, 152.
Ensminger, W.D. and Tammt I. (1970 b) Journal of Virology 5, 672.
Evans, M.J. and Kingsbury, D.W. (1969) Virology 37, 597.
Falket D. (1965) Archly fUr die gesamte Virusforschung 15, 38.
Falket D. (1967) Zeitachrift fUr Medical Mikrobiologie and
Lnmunoforschung 153, 175.
Fazekas de at. Grothp S. (1948) Nature, London 162, 294.
Fenner, F. (1968) In the Biology of Animal Viruses vol 1, p393.
New York, Academic Press.
Finkelstein, R.A. (1961) Journal of Immunology 87, 707. •••••=1•.
Finter, N.B. (1964) Journal of Hygiene, Cambridge 62, 337.
Fox, S.M., Birnie, G.D., Martin, E.M. and Sonnabend, J.A. (1967)
Journal of General Virology 1, 577.
Fox, S.M., Birnie, C.D., Harvey, D.R., Martin, E.M. and Sonnabend, J.A. (1968)
Journal of General Virology 2, 455.

183
Frommhagen, L.H. and Knight, C.A. (1959) Virology, 13, 448.
Gandhi, S.S. and Burke, D.C. (1970) Journal of General Virology 6, 95.
Gordon, A.H. (1969) Electrophoresis of proteins on Polyacrylamide and'
Starch Gels. p27. North Holland Publishing Co., Amsterdam-London.
Graham, E.R.B. (1961) Australian Journal of Science 24, 140.
Granoff, A. (1.959) Virology 9, 636.
Granoff, A. (1964) In Newcastle Disease Virus. An Evolving Pathogen,
p147. Ed. by Hanson, R.P. Madison: University of Wisconsin Press.
Granoff, A. and Kingsbury, D.W. (1964) In Cellular Biology of Wxovirus
Infections: Ciba Foundation Symposium, p96. Ed. by Wostenholme, G.E.W.
and Knight, J. London: Churchill.
Hanson, R.P. and Brandly, C.A. (1955) Science 122* 756. ••••••=1•1•Mr
Haslam, E.A., Cheyne, I.M. and White, D.O. (1969) Virology 39, 118.
Hay, A.J., Skehel, J.J. and Burke, D.C. (1968) Journal of General
Virology 3, 175.
Hitchner, S.B. and Johnson, E.P. (1948) Veterinary Medicine 43, 525. 4••••••••
Horne, R.W. and Waterson, A.P. (1960) Journal of Molecular Biology 2, 75.

18
Home, R.W., Waterson, A.P., Wildy, P. and Farnham, A.E. (1960)
Virology 11, 79.
Hoyle, L. (1954) Journal of Hygiene, Cambridge 52, 180.
Hoyle, L. (1962) Cold Spring Harbour Symposium on 9gantitative
Biology 17, 113.
Hoyle, L. and Frisch-Niggemeyer, W. (1955) Journal of Hygiene, Cambridge
53, 474.
Hoyle, L., Horne, R.W.' and Waterson, A.P. (1961) Virology 13, 448. •••••••••
Huang, A.S., Baltimore, D. and Bratt, M.A. (1971) Journal of Virology
7, 389.
Iyer, S.G. and Dobson, N. (1940) Veterinary Rpcords 52, 889.
Jacobson, W. and Webb, M. (1952) Experimental Cell Research 3, 163.
Karzon, D.T. and Bang, F.B. (1951) Journal of Experimental Medicine
93, 267.
Kingsbury, D.W. (1962) Biochemical and Biophysical Research Communications
9, 156.
Kingsbury, D.W. (1966 a) Journal of Molecular Biology 18, 195.
Kingsbury, D.W. (1966 b) Journal of Molecular Biology 18, 204.

185
Kingsbury, D.W. (1966 b) Journal of Molecular Biology 18, 204.
Kingsbury, D.W. (1970) Progress in Medical Virology 12, 49.
Kingsbury, D.W. and Darlington, R.W. (1968) Journal of Virology 2, 248.
Klenk, H.-D., Caliguri, L.A. and Choppin, P.W. (1970) Virology 42, 473. sim••••••
Kohn, A. (1965) Virology 26, 228.
Kohn, A. and Fuchs, P. (1969) Journal of Virology 3, 539.
Kornberg, A. (1955) In Methods in Enzymology, vol 40 p441. Ed. by
Colwick, S.P. and Kaplan, N.O., New York: Academic Press.
Kraneveld, P.C. (1926) Nederlandisch-Indische Bladen Voor Dierganeeskiende
38, 448.
Laver, W.G. (1963) Virology 20, 251.
Laver, W.G. (1964) Journal of Molecular Biology 9, 109.
Laver, W.G. and Kilbourne, E.D. (1966) Virology 30, 493.
Laver, W.G. and Valentine, R.C. (1969) Virology 88, 105.
Levine, P.P. (1964) In Newcastle Disease Virus. An Evolving_ Pathogen,
p65. Ed. by Hanson, R.P. Madison: University of WisCorisin Press.

186
Liu, C. and Bang, F.B. (1953) Journal of Immunology 70, 538.
Lominiczi, B. (1970 a) Archie fUr die Resents Virusforschun& 30, 159.
Lominiczi, B. (1970 b) Archie fUr die gesante Virusforschunik 30, 167.
Lowry, 0.H., Roseborough, N.J., Farr, A.L. and Randall, R.J: (1951)
Journal of Biological Chemistry 193, 265.
Lwoff, A., Horne, R.W. and Tournier, P. (1962) Cold Spring Harbour
ymposium on Quantitative Biology 27, 51; 111.11111.
Mahler,.H.R. and Cordes, E.H. (1966) Biological Chemistry.
New York and London: Harper and Row.
Waal, J.V., White, D.O. and Scharff, M.D. (1968) Virology, 36, 115.
Marcus, P.I. (1962) Cold Spring Harbour Symposium on Quantitative
Biology 17, 351.
Martin, E.M. and Kerr, I.M. (1968) Symposium of the Society of
General Microbiology 18, 15.
McCrea, J.F., Epstein, B.S. and Barry, R.D. (1961) Nature, London 189, 220.
Methods for the examination of poultry biologics (1963) Publication
No. 1038 National Academy of Sciences, National Research Council,
Washington, D.C., Uf.S.A.

187
Michaelis, L. (1931) Biochemische Zeitschrift 234, 139.
Minocha, H.C., Consiglio R.A. and Eisenstark, A. (1968) American
Journal of Veterinary Research 29, 877. ••••••••
Mountcastle, W.E., Compans, R.W. and Choppin, P.W. (1971)
Journal of Virology, 7, 47.
Mountcastle, W.E., Compans, R.W., CaliguirifiL.A. and Choppin,
(1970) Journal of Virology 6, 677.
Mussgay, M. (1960) Zentralblatt fUr Bakteriologie Parasitenkunde
Infections - kronkheiten and hygiene 4, 437.
Mussgay, M. and Weibel, J. (1962) Virology 16, 506.
Nahmias, A.J. and Dawdle, W.R. (1968) Progress in Medical Virology 10, 110.
Nakajima, H. and Obara, J. (1967) Archie fUr die gesamte Virusforschung.
20, 287.
Neurath, A.R. (1964) Acta Virologica (Prague). Sr 154.
Neurath, A.R. and Sokol, F. (1963) Zeitschrift fUr Naturforschung
18 b, 1050.
Nicoll, J., BetaiL, G. and Colobert, L. (1964) Annales de 1'Institut
PasteWr 107, 192.

188
Ornstein, L. (1964) Annals of the New York Academy of Sciences 121, 321.
Ott, D.G., Richmond, C.R., Trijillo, T.Y. and Foreman, H. (1959)
Nucleonics 17, 106.
Pennington, T.H. (1967) Ph.D. Thesis, University of London.
Plowright, W. (1962) Annals of the New York Academy of Sciences 101, 548.
Poste, G. (1970) Advances in Virus Research 16, 303.'
Rafelson, M.E., Schneir, M. and Wilson, V. (1963) Archives of
Biochemistry and Biophysics 103, 424.
Ralph, R.K. (1969) Advances in Virus Research 151 _115. 1101111.11•.•
Reeve, P. and Poste, G. (1971) Nature, New Biology 229, 157.
Reeve, P. and Waterson, A.P. (1970) Microbios 2, 5.
Reeve, P., Rosenblum, M. and Alexander, D.J. (1970) Journal of
Hygiene, Cambridge 68, 61. •=.11••••
Reeve, P., Alexander, D.J., Pope, G. and Poste, G. (1971) Journal of
General Virology 11, 25.
Reynolds, J.A. and Tanford, C. (1970) Journal of Biological Chemistry
245, 5161.

189
Robinson, W.S. and Duesberg, P.M. (1968) In Molecular Basis of
Virology, p255. Ed. by Fraenkl-Conrat, H. New York: Reinhold.
Roizman, P. and Roane, P.R. (1961) Virology 15, 75. 4111m11•••11
Rott, R. (1964) In Cellular Biology of Myxovirus Infections: Ciba
Foundation Symposium, p175. Ed. by Wolatenholme, G.E.W. and Knight,
3. London: Churchill.
Rott, R. (1964) In Newcastle Disease Virus. An Evolving Pathogen,
p147. Ed. by Hanson, R.P. Madison: University of Wisconsin Press.
Rott, R. and Schafer, W. (1964) In Cellular Biology of Myxovirus
Infections: Ciba Foundation Symposium, p27. Ed. by Wolstenholme, G.E.W.
and Knight, 3. London: Churchill.
Rott, R. and Scholtissek, C. (1967) Modern Trends in Medical Virology 1, 25.
Rott, R., Drzeniek, R., Saber, M.S. and Reichert, E. (1966) Archie fUr
die &esamte Virusforsch 19, 273.
Rott, R., Reda, I.M. and Schafer, W. (1962) Virology 16, 207.
Rott, R., Reda, I.M. and Schafer, W. (1963) Zeitschrift fUr
Naturforschuns 18, 118.
Rott, R., Waterson, A.P. and Reda, I.M. (1963) Virology 21, 663.
Rubin, H., Franklin, R.M., and Bamba, M.W. (1957) Vijology 3, 587.

190
Schafer, W. and Rott, R. (1959) Zeitschrift fUr Naturforschung 14 b, 629. ••••••••
Schlfer, W., Pister, L. and Schneider, R. (1967) Zeitschrift fUr
Naturforschuilg 22 b, 1319.
Schafer, W., Schramm, G. and Traub, E. (1949) Zeitschrift fUr
Naturforechung 4, 157.
Schider, G. and Hanson, R.P. (1968) Journal of Virology 2, 40.
Scholtissek, C. and Rote, R. (1965) Nature, London 206, 729.
Scholtissek, C. and Rott, R. (1969) Journal of General Virology 4, 565.
Scholtissek, C., Becht, H. and Drzeniek, R. (1967) Journal of
General Virology 1, 219.
Sellers, R.F. (1963) Nature 198, 1228. •••••••••••
Sellers, R.F. (1964) Journal of Immunology 93, 6.
Seto, J.T. and Chang, F.S. (1969) Journal of Virology 4, 58.
Seto, J.T. and Rott, R. (1966) Virology 30, 731.
Shapiro, A.L., Vfnuela, E. and Dietzel, J.V. (1967) Biochemical and
Biophysical Research Communications 28, 815.

101
Stenback, W.A. and Durand, D.P. (1963) Virology 20, 545.
Summers, D.F., Maizel, J.V. and Darnell, J.E. (1965) Proceedings
of the National Academy of Sciences of the United States of America 54, 505.
Taverne, J., Marshall, J.H. and Fulton, F. (1958) Journal of General
Virology, 19, 451.
Thacore, H. and Youngner, J.S. (1970) Journal of Virology 6, 42.
Thiry, L. (1963) Virology 19, 225.
Upton, E., Hanson, R.P. and Brandly, c:.A. Proceedings of the Society
for Experimental Biology and Medicine 84, 691.
Wagner, R.R., Levy, A.H., Snyder, R.M., RatcIiff, G.A. and Hyatt, D.F.
(1963) Journal of Immunology 91, 112. 4111M1mil•
Waterson, A.P. (1962) Nature, London 193, 1163. 41••••••••••••••
Waterson, A.P. (1964) In Newcastle Disease Virus. An Evolving Pathogen.
Ed. by Hanson, R.P. Madison: University of Wisconsin Press.
Waterson, A.P. and Almeida, J.D. (1966) Nature 210, 1138.
Waterson, A.P. and Cruickshank, J.G. (1963) Zeitschrift fiir NaturforschunR
186, 114.

1.92
Waterson, A.P., Pennington, T.H. and Allan, N.H. (1967) British
Medical Bulletin 23, 138.
Weast, R.C. (1968) Ed. of The Handbook of Chemistry and Physics 4th Edition.
The Chemical Rubber Company, Cleveland, Ohio, U.S.A.
Webster, R.O. and Darlington, R.W. (1969) Journal of,Virology 4, 182.
Wheelock, E.F. (1963) Proceedings of the Society for Experimental
Biology, New York 114, 56.
Wheelock, E.F. and Tamm, I. .(1961) Journal of Experimental Medicine 114, 617.
Wilson, D.E. (1961) Journal of Bacteriology 84, 295.
Wilson, D.E. (1968) Journal of Virology 2, 1.
Wilson, G.S. and Miles, A.A. (1964) Editorss Topley and Wilson's
Principles of Bacteriology and Immunity. London: Edward Arnold.
Young, R.W. and Fulhorst, H.W. (1965) Analytical Biochemistry 11, 389.

Reprinted from MICROBIOS

I sopycnic separation of Newcastle disease virus on sucrose gradients using zonal ultracentrifuge rotors
Peter Reeve and Dennis J. Alexander
Department of Virology. Royal Postgraduate Medical School Hammersmith Hospital London W12 Great Britain
Abstract
H ighly purified preparations of Newcastle disease virus were obtained from allantoic fluids following centrifugation on sucrose or potassium tartrate gradients in an MSE BXIV zonal ultracentrifuge rotor.
Introduction
The use of zonal rotors for the purification of viruses is well established. For example, Fox and others have described the use of both continuous flow and batch type rotors for separating Semliki Forest virus and polyoma virus from cell culture fluids (Fox et al., 1967; Fox et al., i 968). Our studies on the properties of strains of Newcastle disease virus (NDV) required relatively large amounts of highly purified virus. This paper describes the isopycnic separation of NDV from allantoic fluids using both sucrose and potassium tartrate gradients; we also describe a simple discontinuous gradient method which should be applic-able to other systems.
Materials and methods
Viruses
The following strains of NDV were used • Herts 33 (Herts), received from W. H Allan, Ministry of Agriculture, Fisheries and Food, Central Veterinary Laboratory, Weybridge, Surrey, England; isolated at Weybridge in 1933 by Dobson (1939). Italien, received from Professor R Rott, Institute of Virology, Giessen, Ger-many; isolated in Italy (Schafer, Schramm and Traub, 1949). Ulster, received from J. B. McFerran, Ministry of Agriculture for Ireland, Belfast, Northern Ireland; isolated in 1966 in Northern Ireland from the faeces of apparently healthy chickens.
Virus culture Batches of 200-500 ten-day-old fertile eggs were inoculated by the allantoic route with about to5 ° egg infectious doses (EID„) per embryo. Forty eight hr later the eggs were chilled for about 4 hr and the allantoic fluids harvested.
Virus (strain Herts 33) grown in BHK21 cells was obtained from Dr M. Berry, Glaxo Laboratories, Greenford, Middlesex, England. Virus containing 3H-uridine was obtained by inoculating each chick embryo with too µc uridine-5-T (purchased from the Radiochemical Centre, Amersham, Buckinghamshire, England) : acid-insoluble radioactivity was determined using a Packard 300 Tricarb Liquid Scintillation Spectrometer after precipitation onto cellulose acetate membrane filters with ice cold 5% (w/v) trichloracetic acid.
53 Microbios 1970 5 53-58

24cT
22C, A
E120
!so 0
4 8 12 16 20 24 36 03 36 40 44 46 50 58 GO 54
20
0 o 18- 0
le-
c 12-
Z 10—..
n 6-
126
1 24
1 22
1 20
116
1 16
1
0
110
1 08
Virus purification Virus from infected allantoic fluid or tissue culture medium was centrifuged at 40,000 x g for 3o min and resuspended to i / 1 oo original volume in phosphate buffered saline (PBS) at pH 7.4 before centrifuging in a sucrose or potassium tartrate gradient in an MSE BXIV titanium zonal rotor at 40,000 rpm
00,000 x g) using an MSE 65 ultracentrifuge. Fractions were collected using an LKB Ultrarac fraction collector after measuring ultraviolet absorption at 259 nm in an LKB Uvicord II. When material, e.g. radioactive virus, was scarce, samples were centrifuged at 30,000 rpm ( too,000 x g) in sucrose or potassium tartrate gradients in 23 ml tubes using a swing-out rotor.
Sucrose and potassium tartrate solutions (w/w) containing (poi M tris pH 7.4, were made using the tables given in the Handbook of Chemistry and Physics (Weast, 1968) : densities were estimated from the refractive index of the solutions using the same tables.
Protein concentrations Protein concentrations were estimated by Lowry's method (1951) using crystal-line bovine serum albumin as a standard. Haemagglutinin was titrated using t•o% chicken erythrocytes in o • 5 M NaCl. Tests were done in plastic trays and the last dilution giving complete agglutination was taken as the end point.
Results
When virus was centrifuged for 6o—go min at 100,0w X g on continuous gradients made using an MSE gradient mixing device two major peaks absorb-ing ultraviolet light were seen. The viral haemagglutinin was distributed throughout the gradient, but a major peak could be seen, as well as two smaller peaks at lesser densities (Figure 1). Similar results were obtained with gradients made using io% to 6o% (w/w) sucrose or i ocy„ to 40% (w/w) potassium tartrate. The result was the same whether centrifugation continued for 6o min,
Fraction number in ml each)
Figure 1 Purification of NOV strain Italien on a sucrose gradient BkIV titanium rotor centrifuged 40,000 rpm for 90 min protein N—N solution density Histogram reciprocal of HA titre
54 Microbios P Reeve and D J Alexander

8 12 II 20 24 28 32 36 40
Den
sit
y 9
/m
1
1 22
120
118
ire
114
1 12
110
0 0
a 0.
2 4 6 8 10 12
Fraction number
Figure 2 Purification of NDV, strain Italien on a sucrose gradient Twenty three nil tubes in a swing out rotor 30 000 rpm 90 min 0-0 solution density .--110 infectivity PFU per ml III—IIII 3 H radioactivity Histogram reciprocal of HA titre
go min or 18 hr. Examination of material sedimented by subsequent high speed centrifugation from the major haemagglutinin peaks found at densities of about i •o4; I • I 2 and 1 • 18 g per ml using a Philips EM 30o electron microscope re-vealed typical intact virus particles at the highest density but only fragments at lower densities. Gradient centrifugation performed using tubes and a swing-out rotor showed that virus infectivity and radioactivity of 3H-labelled virus was associated with the peak of highest density (Figure 2).
In order to determine whether virus preparations were originally heterogene-ous or had been degraded during purification, material from fractions corres-ponding to the three major haemagglutinin peaks was sedimented and recentri-fuged in sucrose or potassium tartrate gradients. Single peaks were obtained at the same densities as those from which the material had originally been obtained, suggesting that the purification procedure had not degraded previously intact particles (Figure 3).
Fraction number (to mt each)
Figure 3 Centrifugation of NDV purified by zonal centrifugation on sucrose gradient BXIV titanium rotor, 40 000 rpm for 90 min iiii—ip solution density Histogram reciprocal of HA titre
55 Zonal centrifugation of NDV

0
O O •— 30
E 20
O
160 ,3,
40
4 6 12 16 20 24 26 32 36 40 44 4
1 22
1 20
1 16
1 16
1 14
1 12 E 0
1 10
Discontinuous gradients
Discontinuous gradients were made to obtain a sharper separation of intact NDV particles from host material and non-infective haemagglutinating frag-ments. Gradients were made by filling the zonal rotors with three separate sucrose or potassium tartrate solutions with densities of about 1.04, 1-16 and I •26 g per ml. Using sucrose, gradients were obtained with two 'steps' at which the density changed rapidly. After centrifugation for go min at 4°C, virus banded on such gradients to give two or three sharp peaks of haemagglutinin. Infectivity was associated with a major peak isopycnic with sucrose at densities of about i •i8 g per ml and this was easily separated from other fractions which only contained haemagglutinating virus fragments (Figure 4). If centrifugation was continued for 18 hr, or gradients were allowed to equilibrate by centrifuging at 2500 rpm for 1-2 hr before use, more mixing occurred between the sucrose solutions and less sharp changes of density in the subsequent gradients were seen. With potassium tartrate solution discontinuous gradients could not be obtained: evidently the lack of viscosity of potassium tartrate permitted ready mixing so that almost linear gradients were obtained.
Fraction number ( lornl each)
Figure 4 Purification of NDV on a discontinuous sucrose gradient— BXIV titanium rotor 40 000 rpm for 00 min 0-0 solution density Histogram reciprocal of HA tine
Estimation of virus purity
The protein concentration of the fractions was compared with the titres of haemagglutinin Estimates of purity were also obtained by subjecting virus sus-pension to electrophoresis on 7-5% polyacrylamide gels in a Shandon disc electrophoresis apparatus run at a constant current of I or 4 mA. Infected allantoic fluid contained about 1-2 mg protein per ml and as many as 5 bands were seen on polyacrylamide gels stained with 1% amido black in 7% acetic acid (Table 1, Figure 5a).
Approximately go% non-viral protein was removed after sedimenting virus from infected fluid but such virus still produced several bands on polyacrylamide
56 Microbios P Reeve and D J Alexander

Table 1 Purification of NDV, strain Italien, from allantoic fluid. After centrifuging at 20,000 rpm for 30 min to sediment virus the concentrate obtained was centrifuged on a sucrose gradient in a BXIV zonal rotor for 90 min at 45,000 rpm.
Haemagglutinin per ml
Protein Ag per ml
Haemagglutinin per mg protein
Original allantoic fluid 5,120 2,000 2,560
Concentrate from allantoic fluid 163,840 2,600 63,000
Virus after zonal centrifugation 40,960 50 819,200
gels indicating the presence of several contaminating soluble proteins (Figure 5c), but after zonal centrifugation virus suspensions were obtained containing about ro5 HNU per mg protein which did not produce adventitious bands following polyacrylamide gel electrophoresis (Table t, Figure 5e). Tests showed that less than zpo eg bovine serum albumin could be detected by polyacrylamide gel electrophoresis so that virus suspensions purified by zonal centrifugation were substantially freed of contaminating soluble host proteins, but of course, host protein may well have remained closely adsorbed to virus particles.
Figure 5 Polyacrylamide gel electrophoresis of NDV infected allantoic fluid. About 200 Ag protein was layered onto 7.5% polyacrylamide gels before electrophoresis at 4 mA pH 9.2. (a) Original infected allantoic fluid. (b) Allantoic fluid after low speed centrifugation. (c) Pellet obtained after high speed centrifugation. (d) Supernatant obtained after high speed centrifugation. (e) Virus suspension after zonal centrifugation.
Discussion
Satisfactory isopycnic separation of NDV from infected allantoic fluids was obtained using either sucrose or potassium tartrate gradients. Duesberg and Robinson (1967) stated that `NDV could not be banded on sucrose' since sufficiently high densities could not be obtained. However, using solutions made on a weight for weight basis, as given in the Handbook of Chemistry and Physics ( 1968), sucrose solutions can readily be obtained with densities of up to 1.28 g per ml and this is sufficiently high to permit isopycnic banding of NDV, whose mean density is about i •18. Sucrose permitted discontinuous gradients to be made without the use of a gradient mixing machine. With such gradients it was possible to separate readily complete infectious NDV virions from the heterogeneous suspension obtained from infected allantoic fluids. Virus grown in
57 Zonal centrifugation of NDV

cell culture proved difficult to purify, because large amounts of contaminating host protein always remained associated with virus suspensions. No differences were seen in the behaviour of the different strains and there were no differences in the mean densities of the purified virus even though these strains exhibit great differences in virulence for chick embryos and cell cultures (Waterson, Penning-ton and Allan, 1967). Further, no morphological differences were seen when negatively stained preparations of the isolated virions were examined in the electron microscope.
Acknowledgements
We thank Mr J. Hall and Miss Joy Pacey for excellent technical assistance, Mrs J. D. Almeida for electron microscopy and Professor A P Waterson for his encouragement and help with the manuscript This work was aided by grants from the Wellcome Trust and the Agricultural Research Council
References
Fox S. M , Birnie G D., Martin E M. and Sonnabend J A. 1967 Isolation of polyoma virus and Semliki Forest virus from tissue culture fluid by continuous flow zonal ultracentrifugation J gem Virol 1 577
Fox S M , Birnie G D , Harvey D R.. Martin E M. and Sonnabend J A. 1968 The use of batch-type zonal ultracentrifuge rotors for the isolation and purification of viruses J gen Viral 2 455
Dobson N 1939 Newcastle disease Proc 7th World s Poultry Cong Cleveland. 250
Lowry 0. H , Rosebrough N. J , Farr A L and Randall R. J 1951 Protein estimation with the Folin phenol reagent J biol Chem 193 265
Schafer W , Schramm G and Traub E 1949 Untersuchungen uber das Virus der atypischen Geflugelpest Z Naturforschung 46157
Weast R C 1968 Editor-in-Chief of The Handbook of Chemistry and Physics 4th edition 1967-8 The Chemical Rubber Company Cleveland Ohio U S A
Accepted 2 December 1969
58 Microbios P Reeve and D J Alexander

THE FACULTY PRESS 88 Regent Street Cambridge England

Reprinted from MICROBIOS

Characterization and biological properties of the neuraminidase of strains of Newcastle disease virus which differ in virulence
Dennis J Alexander, Peter Reeve and 'W. H Allan
Department of Virology Royal Postgraduate Medical School London W 12. Great Britain and 'Ministry of Agriculture Fisheries and Food Central Veterinary Laboratories New Haw Weybridge Surrey, Great Britain
Abstract
Different strains of NDV vary widely in virulence and cytopathogenicity The biological and biochemical characteristics of the virus-bound neuraminidase of a representative number of NDV strains were examined All the strains were found to have essentially identical neuraminidase in vitro However, the levels of cell-associated neuraminidase in the chono-allantoic membrane 22 hrafterinfection appeared to be related to thevirulenceof the infecting strain.
Introduction
The enzyme neuraminidase (mucopolysaccharidc N-acetylneuraminyl hydro-lase EC 3 2.1. I 8) has been found to be distributed among many tissues, micro-organisms and, in particular, the myxoviruses In general the study of myxo-virus neuraminidase has been concerned mainly with influenza virus, and the characteristics, structure and morphology of influenza virus neuraminidase have been extensively studied (Rafelson, Schneir and Wilson, 1963; Drzeniek, Seto and Rott, 1966; Drzeniek, Frank and Rott, 1968, Kendal, Biddle and Belyavin, 1968; Paniker, 1968).
Different strains of Newcastle disease virus (NDV) show a continuous spectrum of differing virulence, as measured by lethality in chickens or eggs (Waterson, Pennington and Allan, 1967), and cytopathogenicity (Schloer and Hanson, 1968). This system is therefore particularly useful in studies of the relationship between viral structure and activity and virulence or cytopatho-genicity.
Waterson et al. (1967) have suggested that difference in virulence of the different strains of NDV could in some way be related to the neuraminidase activity. This suggestion was given more weight by the work of Scholtissek, Becht and Drzeniek (1967), who showed that cytopathogenicity of influenza virus was related to neuraminidase activity. Furthermore it has been shown by Kohn and Fuchs (1969) that other properties of the NDV virion are not re-lated to virulence.
Drzeniek et al. (1966), in a preliminary study, characterized the neuramini-dase, both virus-bound and released, of two strains of NDV (Italien and Beau-dette C) and showed that they were alike in their properties. In this work we have limited our studies to virus-bound neuraminidase but have examined a larger number of strains of much greater variation in virulence and cyto-pathogenicity, and included other biological properties of the NDV neura-minidase.
155 Microbios 1970 6 155-165

Materials and methods
Viruses
The following strains of NDV were used: Field Pheasant — isolated at the Central Veterinary Laboratories, Weybridge, Great Britain, 1962. Herts 33 (Herts) — Dobson (1939). Italien — Schafer, Schramm and Traub (1949). Texas G.B. (Texas) — isolated near Austin, Texas, U.S.A., 1948. Warwick — isolated in Great Britain, 1966. Eastwood Notts — isolated in Great Britain, 1966. Queensland V4 (Queensland) — isolated in Brisbane, Australia, 1966. Ulster — received from J. B. McFerran, Ministry of Agriculture for Northern Ireland, Belfast, and isolated in 1966. B1 — Hitchner and Johnson (1948) . F — Asplin (1952). Bcaudette C — Granoff (1959). Virus was grown in the allantois of io-day-old fertile hen's eggs. The allantoic fluid was harvested, centrifuged at low speed to remove cell debris, and the virus sedimented at 45,000 x g for 45 min. The virus was partially purified (> 95% of protein removed) by sedimentation through 32% (w/w) sucrose onto a 6o% (w/w) sucrose cushion. In experiments involving haemagglutinin and infectivity titrations, infected allantoic fluid was used without further treatment.
Measurement of virulence and ability to form plaques
The methods used for the estimation of intracerebral pathogenicity index (ICPI) in chickens, and mean death time in eggs (MDT) have been described in detail before (Methodsfor the examination of poultry biologics, 1963).
Primary chick fibroblast monolayers, overlaid with 0.9% (w/v) nutrient agar and incubated at 37°C, were examined for plaques 3 days after infection with virus.
Titrations of haemagglutinating activity and infectivity
Haemagglutinin (HA) was measured in standard plastic plates in 0•2 ml
volumes, phosphate buffered saline pH 7.2 (PBS) was used as diluent with I% (w/v) chicken red blood cells.
Infectivity for chick embryos was estimated by inoculating o•i ml volumes of serially diluted virus suspensions into the allantoic cavity of 9- or o-day-old fertile chicken eggs. Eggs were candled daily and after 7 days the allantoic fluid of surviving eggs was tested for the presence of hacmagglutinin The 5o% egg infective dose (EID„) per ml was estimated by the Kaerber method.
Neuraminidase assay
Neurammidase activity was estimated by measuring the amount of N-acetyl neuraminic acid (NANA) released from a suitable substrate The solution to be tested (0.1 ml) was incubated with o•1 ml substrate and o.3 ml of buffer for io min at 37°C. The NANA released was estimated by the method of Aminoff
156 Microbios D J. Alexander, P Reeve and W H Allan

(1961). The substrate used was either bovine neuramme lactose (Calbiochem Ltd) 200 jig per test, or the equivalent amount of fetuin prepared from foetal calf serum (Graham, 1961).
Enzyme activity was expressed as jig of NANA released.
Release of neuraminidase activity by Tween 20 at pH 10.0
Virus was sedimented at 45,00o x g for 45 min and resuspended in 1% v/v Tween 20 in bicarbonate buffer pH to•o (Webster and Darlington, 1969) and incubated at 37°C overnight. The mixture was then centrifuged at 45,000 x g for 6o min, the pellet resuspended to volume and both suspensions tested for neuraminidase activity In each case a control was performed under identical conditions but was incubated in bicarbonate buffer which did not contain Tween 20.
Estimation of cell-associated neuraminidase activity
For each strain tested i x 109 EID„ were inoculated into the allantoic cavity of o-day-old eggs. After 22 hr incubation at 37°C the chorioallantoic membrane
was removed from each egg, washed three times in ice cold PBS and stored at — 7o°C until assayed. The membranes were thawed and blended in homogeni-zers with motor driven Teflon pestles in sodium acetate buffer (pH 5.2). The homogenate was then assayed for neuraminidase activity and protein was estimated by the method of Lowry et al. (1951).
Results
Virulence and cytopathogenicity
The strains used covered a wide range of virulence, whether measured by intracerebral pathogenicity index in chickens or mean death time in eggs
Table 1 Virulence and cytopathogenicity of different NIDV strains
Strain ICPI MDT Plaguing ability
Field Pheasant 2 00 54 + + Herts 1 88 49 + +
Italian 1 86 50 + + Texas 1 75 55 + + Warwick 1 72 62 + + Eastwood Notts 1 70 70 Beaudette C 1 46 62 + B, 040 117 ± F 0 25 119 — Queensland 0 16 ix, — Ulster 0 00 ci, —
+ + Large well-defined plaques + Small well-defined plaques ± Plaques very small, ill defined and often absent — Does not form plaques ICPI Intracerebral pathogenicity index MDT Mean death time in eggs in hours
1 57 N DV virus-bound neuraminidase

(Table 1). The two methods of estimation generally run inversely parallel, Beaudette C and Field Pheasant being exceptions.
Ability to form plaques in chicken embryo cell sheets, although only the gross differences were measured, varied with virulence.
Relationship of neuraminidase to HA and infectivity
In order to determine if any strains possessed more neuraminidase than others the neuraminidase activity was compared with HA titre (Table 2a). HA was taken to be a measure of viral surface area and preferred to a particle count because of the pleomorphism of the virus. The results show that the ratio of neuraminidase activity to HA units is generally constant, except for strains Beaudette C and, to a lesser extent, Ulster which have considerably lower ratios.
Table 2a Relationship between haemagglutinin and neuraminidase activity
Strain Neuramimdase activity/m1 HA/ml
Neuraminidase HA x 10-3
Field Pheasant 38 0 1500 25 4
Hurts 360 0 10240 35 0
Texas 87 5 2560 34 1
Warwick 84 5 2560 33 0
Eastwood Notts 171 0 5120 33 3
Beaudette C 271 0 20480 13 3
B, 176 0 5120 34 4
F 74 5 2560 28 1
Queensland 172 5 5120 33 6
Ulster 845 0 40960 20 6
The relationship between HA, neuraminidase and infectivity was also investigated. The HA/EID50 and the neuraminidase/EID„ ratios, again with the exceptions of Beaudette C and Ulster, even after allowing for their high HA titres, were very similar for all strains (Table 2b).
Table 2b Relationship between infectivity, haemagglutinin and neuraminidase activity
Strain HA Neuraminidase activity (pH 6 0)
EID„ x 10-7 ElDso x 10-9
Harts 2 9 85
Texas 39 73
Eastwood Notts 36 75
Beaudette C 196 22 2
24 52
F 23 103
Ulster 101 142
158 Microbios D J Alexander, P Reeve and W. H. Allan

0 12
42 0 04 z a z
Kinetic studies of virus-bound neuraminidase
The amount of NANA released after to min was plotted against the concentra-tion of substrate used. All eight strains examined gave kinetic curves essentially Identical to that of strain Herts (Figure 1 ). The Michaelis-Menten constants
0 10
0 20
0 30 0 40
Neuramine lactose [}moles )
Figure 1 Kinetics of the virus-bound neuraminidase of strain Herts Standard enzyme assay procedure was used except that the concentration of neuramine lactose was as above, pH was 5 4 with 0 1 M sodium acetate-acetic acid buffer
(Km) were obtained by Lineweaver-Burk plots. All the strains had a Km of approximately i x io-3 m (Table 3).
With fetuin as substrate the optimum pH of the neuraminidase of the different strains, though varying slightly from experiment to experiment, always fell in the range pH 5-1-5.3 (Table 4) and was considered to be the same for all the strains tested. Typical curves of pH effect with fetuin as a substrate are shown in Figures 2a and 2c. A second minor peak at pH 6.3 was seen with all the strains tested except strain F. If neuramine lactose was used as the substrate the pH
Table 3 Michaelis- Menton constant of neuramindase at pH 5 4
Strain Km
Field Pheasant 1 0.10-3M Harts 10x10-3M Italien 0 9.10-3M Texas 1 0 x 10-3M Beaudette C 1 0.10-3M
F 09x10-'M Queensland 0 9.10-3M Ulster 1 0.10-3M
159 N DV virus-bound neuraminidase
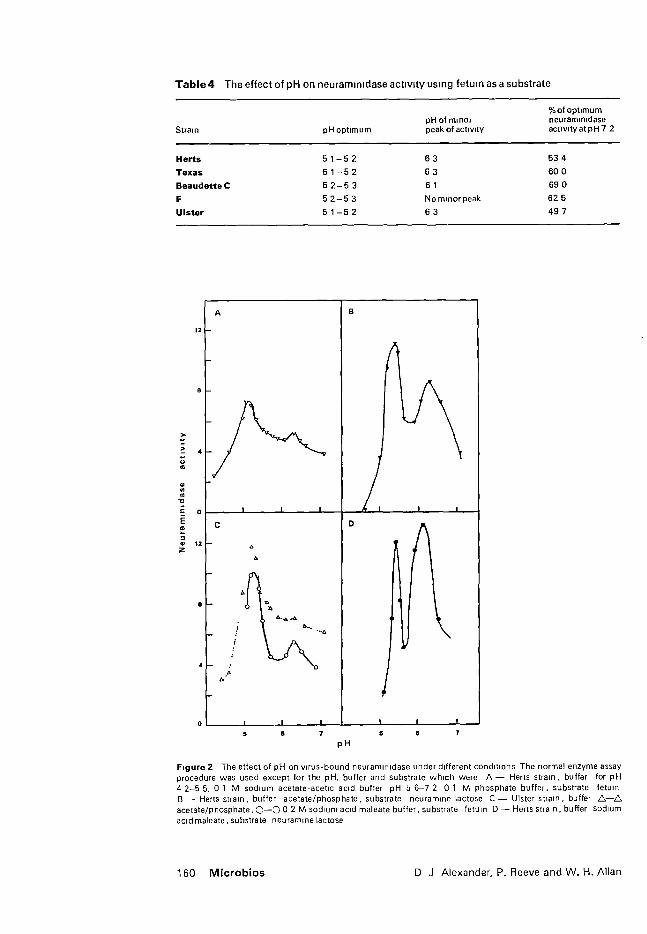
12
a
O
12
Neura
mmid
ase
a
4
0
Table4 The effect of pH on neuraminidase activity using fetuin as a substrate
%of optimum pH of minor neuraminidase
Strain pH optimum peak of activity activity at pH 7 2
Harts 5 1-5 2 63 534 Texas 5 1-5 2 6 3 60 0 Beaudette C 5 2-5 3 6 1 69 0 F 5 2-5 3 No minor peak 62 5
Ulster 5 1-5 2 6 3 49 7
A B
- ..,, A----
- - 1 \.
5 7 a a r
p H
Figure 2 The effect of pH on virus-bound neuraminidase under different conditions The normal enzyme assay procedure was used except for the pH. buffer and substrate which were A — Herts strain, buffer for pH 4 2-5 5, 0 1 M sodium acetate-acetic acid buffer pH 5 6-7 2 0 1 M phosphate buffer, substrate tot= B — Herts strain, buffer acetate/phosphate , substrate neuramine lactose C — Ulster strain, buffer L—A acetate/phosphate .0-0 0 2 M sodium acid maleate buffer, substrate fetu in D — Herts strain, buffer sodium acid maleate , substrate neuramine lactose
160 Microbios D J Alexander, P. Reeve and W. H. Allan

s-?
Neura
min
ida
se
0
0
S
S
100
60
4
6
20
optimum for all the strains was raised slightly to pH 5.4 (Figure 2b), the second minor peak was far more obvious although it still occurred at pH 6.3. In these experiments sodium acetate buffer was usually used for pH values from 4.1 to 5.5, phosphate buffer for pH 5.6-7'2. If sodium acid maleate buffer was used from pH 5.1 to pH 7.2 with both fetuin and neuramine lactose the two peaks were present but with neuramine lactose the 'minor' peak was so much greater that it became the larger of the two (Figures 2c and 2d).
In Table 4 the neuraminidase activity at physiological pH (pH 7.2) is shown as a percentage of the activity at the optimum pH. The strains tested were generally in the range of 50-7o% and although differences in the strains were consistently found there was no relationship with virulence
Heat stability of virus-bound neuraminidase
The heat stability of virus-bound neuraminidase was estimated by incubating virus suspensions at 56°C and measuring the remaining neuraminidase activity at intervals. Within 20 min the neuraminidase activity had been destroyed in all the strains tested except Beaudette C, which still had 3o% of the original activity (Figure 3).
0
5 10 15
20
Exposure time I miri I
Figure 3 Heat stability of virus-bound neuraminidase of different strains of NOV at 56°C At specified times 0 1 ml of enzyme was added to 0 3 ml of ice cold buffer to stop further heat inactivation . 0-0 Herts: III—E Texas, A—A Beaudette C .E1-0 Queensland ,0-0 Ulster.
161 N DV virus-bound neuraminidase
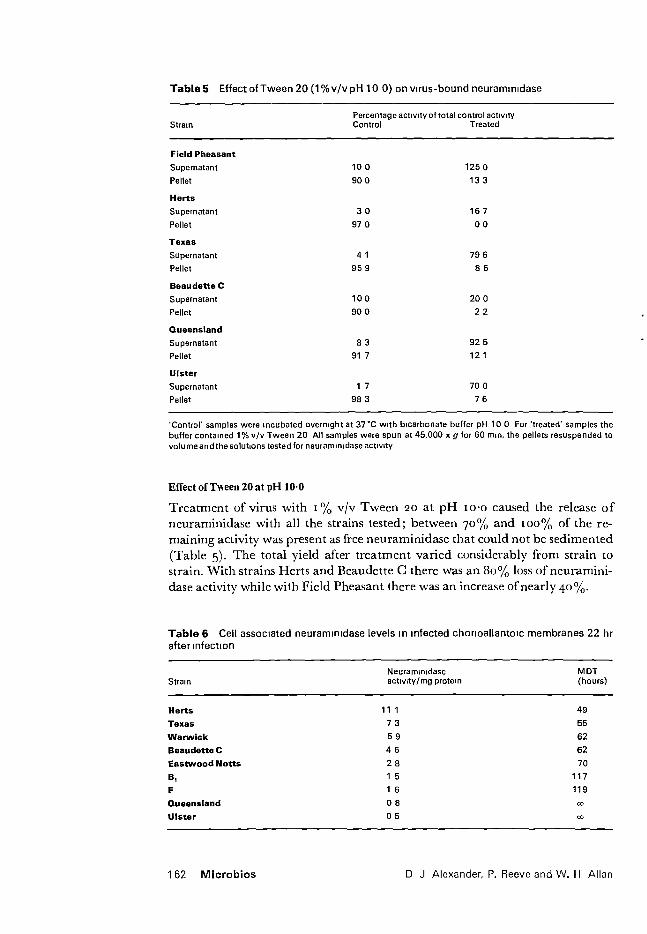
Table 5 Effect of Tween 20 (1% v/v pH 10 0) on virus-bound neuraminidase
Strain Percentage activity of total control activity Control Treated
Field Pheasant Supernatant 10 0 125 0 Pellet 90 0 13 3
Hens Supernatant 3 0 16 7
Pellet 97 0 0 0
Texas Supernatant 4 1 79 6
Pellet 95 9 8 5
Beaudette C Supernatant 10 0 20 0
Pellet 90 0 2 2
Queensland Supernatant 8 3 92 5
Pellet 91 7 12 1
Ulster Supernatant 1 7 70 0
Pellet 98 3 7 6
'Control' samples were incubated overnight at 37°C with bicarbonate buffer pH 10 0 For 'treated' samples the buffer contained 1% v/v Tween 20 All samples were spun at 45,000 x g for 60 min, the pellets resuspended to volume and the solutions tested for neuraminidase activity
Effect of Tneen 20 at pH 10.0
Treatment of virus with 1 % v/v Tween 20 at pH to-o caused the release of neuraminidase with all the strains tested; between 7o% and t00% of the re-maining activity was present as free neuraminidase that could not be sedimented (Table 5). The total yield after treatment varied considerably from strain to strain. With strains Herts and Beaudette C there was an 8o% loss of neuramini-dase activity while with Field Pheasant there was an increase of nearly 40%.
Table 6 Cell associated neuraminidase levels in infected chorioallantoic membranes 22 hr after infection
Strain Neuraminidase activity/mg protein
MDT (hours)
Harts 11 1 49
Texas 7 3 55
Warwick 5 9 62
Beaudette C 4 6 62
Eastwood Notts 2 8 70
B, 1 5 117
F 1 6 119 Queensland 0 8 m
Ulster 0 5 co
162 Microbios D J Alexander, P. Reeve and W. H Allan

Neuraminidase activity in infected chorioallantoic membranes
The levels of neuraminidase per mg protein in the chorioallantoic membranes of eggs infected with different strains appear to be related to the virulence of the infecting strain (Table 6). The plaque-forming strains, i.e. Eastwood Notts and the more virulent strains, show a linear relationship between cell associated neuraminidase and MDT. The less virulent strains show less marked differ-ences in neuraminidase levels, presumably because there is very little accumula-tion of the enzyme and much of the activity recorded is due to whole virus particles associated with the membrane.
Discussion
None of the properties of NDV neuraminidase examined show any great variation from strain to strain. Unlike influenza virus (Rafelson et al., 1963) it appears that different NDV strains possess neuraminidases which are essentially identical in their biochemical properties.
On assessment of physical characteristics Beaudette C appears to be the most atypical strain. The heat resistance shown by Beaudette C neuraminidase is probably due to the protection of the intact virion since Drzeniek et al. (1966) have shown that neuraminidase isolated from Beaudette C virus is just as heat labile as neuraminidase from the other strains. Beaudette C has twice as much hacmagglutinin as other strains which could account for the protection of the neuraminidase. Rott, Reda and Schafer (1962) have shown that a large fraction of the virus in their Beaudette C suspensions was devoid of ribonucleo-protein. From the HA/EID„ ratios which arc, in fact, estimation of non-infectious particles this does not appear to be a general characteristic of all the strains although Ulster also seems to have a high HA/EID5O ratio.
It is difficult to assess the significance of the differences in the effect of Tween 20 on the neuraminidase of the different strains. Presumably the enzyme of those strains showing a low yield of released neuraminidase after Tween 20 treatment is more intimately associated with the lipid membrane of the virus, or that their enzyme is different in some way and rendered inactive in the presence of Tween 20.
The pH optimum of NDV neuraminidase is lower than that recorded by Drzeniek et al. (1966) for Italien and Beaudette C. However, this is probably due to variation in the substrate. It has been shown by Raffelson et al. (1963) that pH optima of neuraminidase differ with substrate. The observation of two peaks of enzyme activity has been previously recorded by Drzeniek, Bogel and Rott (1967) with the neuraminidase of bovine parainfluenza 3 virus. Since in both the substrates we have used, the NANA is not specifically attached the two peaks could be produced by the availability of substrate varying with pH. However, Drzeniek (1967) using 2-3' and 2-6' linked sialolactose, has shown that NDV neuraminidase, unlike Vibrio cholerae neuraminidase cleaves only 2-3' linkages and that the failure to cleave 2-6' linkages is unaffected by pH changes. Alternatively the enzyme may exist in more than one form, with different pH optima.
163 NDV virus-bound neuraminidase

The relationship between virulence and cell-associated neuraminidase levels has been noted previously by Reeve, Rosenblum and Alexander (1970) who have shown that all the major virus antigens accumulate in the chorioallantoic membrane of eggs infected with virulent strains but not in eggs infected with avirulent strains of NDV. Accumulation of neuraminidase and other viral antigens in virulent strains infers that there will be a far more extensive modifi-cation of the cell membrane than with avirulent strains and this alone could cause cell death. However, it is also possible that cell death could be brought about by generalized disruption of cell membranes caused by the accumulating neuraminidase, or by the action of neuraminidase at a specific site or sites in the cell (Bang, 1953; Allison and Mallucci, 1965).
Acknowledgements
D J Alexander is supported by the Agricultural Research Council to whom we are in-debted for a grant for technical assistance and supplies This research was aided by grants from the Wellcome Trust and Action for the Crippled Child (Polio Research Fund) We are most grateful to Professor A P Waterson for his help and encouragement
References
Allison A. C. and Mallucci L. 1965 Histochemical studies of lysosomes and lysosomal enzymes in virus infected cell cultures J. exp med.121 463
Aminoff D 1961 Methods for the quantitative estimation of NANA and their application to hydrolysates of sialomucoids Biochem J 81 384
Asplin F. D 1952 Immunisation against Newcastle disease with a virus of low virulence (strain F) and observa- tions on sub-clinical infection in partially resistant fowls Vet Roc, 64 245
Bang F. B. 1953 The development of Newcastle disease virus in cells of the chorioallantoic membrane as studied by thin section Bull Johns Hopkins Hosp 92 309
Dobson N.1939 Newcastle Disease Proc 7th Worlds Poultry Cong Cleveland p 250
Drzeniek R 1967 Differences in splitting capacity of virus and V cholerae neurammidase on sialic acid type substrates Blochem Blophys Res Comm 26 631
Drzeniek R , Bogel K and Rott R 1967 On the classification of bovine parainfluenza 3 virus Virology31 725
Drzeniek R , Frank H and Rott R 1968 Electron microscopy of purified influenza neuraminidase Virology 36 703 Drzeniek R , Seto J T and Rott R 1966 Characterisation of neuraminidases from myxoviruses Biochem biophys Acta 128 547
Graham E R. B. 1961 Some aspects of the carbohydrate moiety of fetuin Australian J So 24 140
Granoff A 1959 Studies on mixed infection with Newcastle disease virus Virology9 636
Hitchner S. B and Johnson E. P. 1948 A virus of low virulence for immunizing fowls against Newcastle disease (avian pneumoencephalitis) Vet Med 43 525
Kendal A P , Biddle F. and Belyavin G 1968 Influenza virus neuraminidase and the viral surface Biochem blophys Acta 165 419
Kohn A and Fuchs P 1969 Cell fusion by various strains of NOV and their virulence J Virol 3 539
Lowry 0. H., Rosebrough N J , Farr A. L and Randall R J 1951 Protein measurement with the Foil n phenol reagent J .61°1 Chem 193 265
Methods for the examination of poultry biologics 1963 Publication No 1038 National Academy of Sciences— National Research Council, Washington DC USA
Paniker C K. J 1968 Serological relationships between the neuraminidases of influenza viruses J gen Virol 2 385 Rafelson M E , Schneir M , Wilson V 1963 Studies on the neuraminidase of influenza virus II Additional properties of the enzyme from the Asian and PR8 strain Arch. Biochem Biophys 103 424
Reeve P , Rosenblum M and Alexander D J 1970 Growth in chorioallantoic membranes of strains of Newcastle disease virus of differing virulence J Hyg 68 61
Rott R , Reda I M. and Schafer W 1 962 Isolation and characterisation of haemagglutinating non infectious particles produced during multiplication of Newcastle disease virus (NDV) Virology16 207
164 Microbios D. J Alexander, P. Reeve and W. H. Allan

Schafer W.. Schramm G. and Traub E. 1949 Untersuchungen uber das Virus der atypischen Geflugelpest Z Naturforsch 46 157
Schloer G. and Hanson R. P. 1968 Relationship of plaque size and virulence for chickens of 14 representative Newcastle disease virus strains J.14rol 2 40
Scholtissek C , Becht H. and Drzeniek R. 1967 Biochemical studies on the cytopathic effect of influenza virus J.gen.Virol 1 219. Waterson A. P , Pennington T. H and Allan W. H. 1967 Virulence in Newcastle disease virus A preliminary study Br.med.Bul1,23 138
Webster R. G and Darlington R W 1969 Disruption of myxoviruses with Tween 20 and isolation of biologically active haemagglutinin and neurammidase subunits J Oro, 4 182
Accepted 7 April 1970
165 N DV virus-bound neuraminidase

THE FACULTY PRESS 88 Regent Street Cambridge England .

Reprinted from CYTOBIOS

Plaque formation, cell fusion and haemadsorption by Newcastle disease virus
Peter Reeve and Dennis J. Alexander
Department of Virology. Royal Postgraduate Medical School Hammersmith Hospital London W12, Great Britain
Abstract
Only those strains of Newcastle disease virus (NDV) which Induced syncytia in chicic embryo fibroblasts formed plaques. Haemadsorption also was greater with plaque forming strains than with other strains, suggesting a more extensive modification of the cell surface.
The myxoviruses, which include Newcastle disease virus (NDV), are assembled at the cell surface, which they modify in such a way that erythrocytes can be adsorbed by the cells (haemadsorption). Cells infected with NDV are also known to fuse to form syncytia. We have examined strains of NDV grown in chick embryo fibroblasts to test the hypothesis that plaque formation depends on polykaryocytosis, which is itself determined by the extent of host cell modifica-tion. Virus pools were prepared from NDV-infected allantoic fluid of to-day chick embryos and stored at — 70°C until required.
The following strains were used, Herts 33 (Dobson, 1939), Texas G. B., Warwick, Beaudette C (Granoff, 1959), 'F' (Asplin, 1952), Queensland and Ulster. Of these the first four produced plaques in chick embryo fibroblast monolayers, whereas the other three did not. Although strains Queensland and Ulster failed to form plaques they grew well in chick embryo fibroblasts, re-leasing high titres of infectious particles (Reeve and Waterson, 197o). Poly-karyocytosis was looked for in monolayers infected with a multiplicity of about o egg infectious doses (EID„) per cell, incubated at 37°C and stained with
Giemsa 12 hr and 20 hr respectively after infection. Syncytia were seen after in-fection with strains Herts, Texas G.B., Warwick and Beaudette C (Table t). A few small syncytia were seen following infection with strain `F but these were absent in cultures infected with Queensland or Ulster. Syncytia varied in size, as judged by the number of nuclei seen 12 hr after infection; Herts and Texas G.B. inducing the largest and Beaudette C the smallest (Table ). Haemadsorption was estimated by adding i % chicken erythrocytes in phos-phate buffered saline (PBS) at pH 7.2 to cultures washed three times with o•15M NaCl. After 15 min at +4°C unadsorbed erythrocytes were removed by three further washes with saline. Cultures were then stained with Giemsa and the number of erythrocytes adsorbed to individual cells counted. Uninfected cultures failed to adsorb erythrocytes. With strains Herts, Texas G B., Warwick and Beaudette C, haemadsorption was seen 4-6 hr after infection, the number of erythrocytes adsorbed to individual cells increasing exponentially until about 12 hr after infection, when infected cells were covered with erythrocytes, syn-cytia began to appear and accurate counts were difficult. With strains `F', Queensland and Ulster, haemadsorption was always less than with the virulent
55 Cytobios 1970 5 55-57

a 10
9 6 5
3
Num
ber o
f ery
thro
cyte
s
Table 1 Comparison of the ability to form plaques and syncytia in chick embryo fibroblast cell cultures infected with strains of NDV. Number of nuclei per syncytium estimated from counts on at least 50 syncytia, 12 hr after infection
Strain Ability to form plaques Presence of syncytia Average number of nuclei per syncytium
Herts 33 + + 16 Texas GB + + 15 Warwick + + 10 Beaudette C + + 8
Queensland
—
— , 4
0 Ulster — — 0
strains, only about 8 erythrocytes adsorbed to individual cells 36 hr after in-fection (Figure t) ; occasionally cells or syncytia which had adsorbed larger numhers of erythrocytes were seen
Our strains show a direct relationship between plaque formation and the presence of syncytia, and it is known that the size of the plaques induced by strains of NM' varies, being directly related to their virulence for chick embryos (Schloer and Hanson, 1968). Our results suggest differences in the size of syncytia may determine plaque size. Roizman (1962) has shown that alteration of the cell surface is a prerequisite for polykaryocytosis, and described variants of herpes simplex virus which showed a correlation between plaque size and ability to form syncytia. We suggest that modification of the cell surface beyond a certain point induces cell fusion; with virulent strains this point is reached rapidly in all cells infected; with avirulent strains this point is rarely or never
fibro
bla
st
30 • ,
20 / • •
7' - /A
F 71 •
o - • I • %• 2. ❑ ii o
• II" , 0 • 0
1 o
0 1 /b I
2 °
4 8 12 16 20 24 28 32 36
Hr after inoculation
Figure 1 Haemadsorption by chick embryo fibroblasts infected with strains of N DV The figures given are mean values for 2-3 experiments for each strain, at least 50 infected cells were observed for each point • Herts 33 • Texas G B A Warwick VBeaudette C 0 F' ❑ Queensland L Ulster
56 Cytobios P. Reeve and D J Alexander

reached. The presence of haemagglutinin at the cell surface may by itself cause sufficient alteration to induce cell fusion. HoN‘ever, it is equally likely that this may only be a secondary phenomenon occurring in parallel with other cellular changes which are responsible for cell fusion. Poste (1969) has suggested that cellular lysosomes are always damaged before cell fusion occurs regardless of the nature of the agent inducing fusion, and Allison and Mallucci (1965) have reported changes in the lysosomes of cells infected with NDV. They compared two strains of this virus. A highly cytopathic strain caused irreversible destruc-tion of lysosomes, release of enzymes and consequent cell damage, while a less cytopathic strain induced only transient lysosomal damage
Kohn and Fuchs (1969) showed that the cell-fusing abilities of NDV were not related to virulence, findings at first sight at variance with ours. Their methods involved the use of high multiplicities of virus, most of the cell surface being affected by virus during adsorption so that cell fusion was seen within 3 hr, before the formation of new virus.
Acknowledgements
We thank W H Allen for supplying virus strains and Professor A P Waterson for help The work was aided by grants from the Wellcome Trust and the Agricultural Research Council
References Asphn F D 1952 Immunization against Newcastle disease with a virus of low virulence (strain F) and observa-tions on subclinical infection in partially resistant fowls Vet Rec 64 245 Dobson N 1939 Newcastle Disease Proc 7th World's Poultry Cong Cleveland 250 Granoff A 1 959 Studies on mixed infection with Newcastle disease virus Virology 9 248 Kohn A and Fuchs P 1969 Cell fusion by various strains of Newcastle disease virus and their virulence J Virol 3 539
Reeve P and Waterson A P 1970 The growth cycle of avirulent strains of Newcastle disease virus Microbtos 5 5-9 Roizman B 1962 Polykaroycytosis Cold Sprang Harb Symp Quant Saul 27 327 Poste G H 1969 A study of virus-induced polykaryocytosis Ph D thesis University of Bristol Schloer G M and Hanson R P 1968 Relationship of plaque size and virulence for chickens of 14 represent-ative Newcastle disease virus strains J Vuol 2 40
Accepted 10 December 1969
57 NDV, cell fusion and haemadsorption

THE FACULTY PRESS 88 Regent Street Cambridge England

J. Hyg., Crank (1970), 68, 61 61 Printed in Great Britain
Growth in chick chorioallantoic membranes of strains of Newcastle disease virus of differing virulence
BY P. REEVE, MARGARET ROSENBLUM AND
D. J. ALEXANDER Department of Virology, Royal Postgraduate Medical School,
London, W.12
(Received 31 July 1969)
SUMMARY The growth of eight strains of Newcastle disease virus in chick embryo chorio-
allantoic membranes was studied by comparing, at different times after infection, the amounts of haemagglutinin released into the allantoic fluid (extracellular haemagglutinin) with that associated with the membrane (cell-associated haemag-glutinin). The virulence of the strains examined differed in that some killed chick embryos more rapidly than others. All strains released similar amounts of extra-cellular haemagglutinin and maximum titres were achieved about 12 hr. after infection. With virulent strains cell-associated haemagglutinin titres increased exponentially until the death of the host and maximum titres were much higher than those of extracellular haemagglutinin. With avirulent strains cell-associated haemagglutinin titres increased exponentially for only a limited time and titres were always lower than the titres of extracellular haemagglutinin.
Similar results were obtained when the titres of neuraminidase and viral ribo-nucleoprotein were measured during the growth of two virulent and two avirulent strains. Virulence appears to be associated with the continued intracellular accumulation of viral antigens.
INTRODUCTION Newcastle disease virus (NDV) has been suggested as a suitable virus for the
study of virulence (Waterson, Pennington & Allan, 1967). Many strains are available and their virulence, defined here as the ability to cause disease or death in a host, ranges from highly virulent strains, which are extremely pathogenic for chickens, kill chick embryos rapidly and destroy infected chick embryo fibroblast cells in culture, to strains isolated from apparently normal chickens which are not cytopathic and kill chick embryos infrequently or not at all. Virulence can be measured accurately and reproducibly in chick embryos and young chickens, and all strains grow readily in the chick embryo chorioallantoic membrane and release high titres of infectious virus into the allantoic fluid. They can, therefore, be cul-tured in quantity. Virus titres can be accurately and easily measured in terms of infectivity, haemagglutinin and neuraminidase activity. Besides this, NDV is not only an important animal pathogen in its own right, but is biologically akin to some important pathogens of man and other vertebrates, including mumps, the

62 P. REEVE, MARGARET ROSENBLUM AND D. J. ALEXANDER
para-influenzas, measles and distemper viruses : studies with NDV are thus relevant in a wide context.
Studies on the isolated virion show a surprising uniformity among strains : all those so far examined under the electron microscope look alike (Waterson & Cruickshank, 1963) and no major serological differences have been seen using serum neutralization or haemagglutination-inhibition (HAI) tests (W. H. Allan, unpublished observations) or in gel diffusion studies (Pennington, 1967).
Strains which differ in virulence for the chick embryo also differ, in parallel with this, in cytopathogenicity. Schloer & Hanson (1968) have shown, for example, that the most virulent strains for chick embryos produce the largest plaques on chick embryo fibroblast monolayers. This suggests that it is in the intracellular stages of virus development that strain differences might be found.
T. H. Pennington (unpublished observations) has reported differences in the quantity of haemagglutinin found in the chorioallantoic membranes of chick embryos infected with different strains of NDV, although with all strains sub-stantially similar quantities of haemagglutinin (HA) were released into the allantoic fluid. However, these findings were based on samples taken, for all strains, 40 hr. after inoculation of the chick embryos: thus no information was available on the rates at which the haemagglutinin was produced. We have, there-fore, measured the production of haemagglutinin in chorioallantoic membranes of chick embryos throughout one cycle of growth of several strains of NDV. We have compared these results with the titres of haemagglutinin released into the allantoic fluid and we have also measured the rates at which neuraminidase (Mucopoly-saccharide N-acetylneuraminylhydrolase) and viral ribonucleoprotein are pro-duced.
METHODS Viruses
The following strains of NDV were used: Herts 33 (Herts), received from W. H. Allan, Ministry of Agriculture, Fisheries
and Food, Central Veterinary Laboratory, Weybridge, Surrey. Isolated at Wey-bridge in 1933 by Dobson (1939).
Italien, received from Professor R. Rott, Institute of Virology, Giessen, Ger-many. Isolated in Italy at some date before 1949 (Schafer, Schramm & Traub, 1949).
Texas GB, (Texas), received from W. H. Allan; isolated in 1948 near Austin, Texas, U.S.A.
Field Pheasant, isolated at Weybridge in 1962 from pheasants; clinically more virulent than Herts 33 (W. H. Allan, personal communication).
Beaudette C, received from Professor R. Rott. This is a heat stable variant isolated from the Beaudette strain by Granoff (1959).
Strain F, received from W. H. Allan, isolated in England by Asplin in 1949 (Asplin, 1952).
Queensland V 4 (Queensland), received from W. H. Allan, isolated in 1966 by Rylie, in Brisbane, Australia (W. H. Allan, personal communication).

Growth of NDV in chick chorioallantoic membranes 63
Ulster, received from J. B. McFerran, Ministry of Agriculture for Ireland, Belfast. Isolated in 1966 in Northern Ireland from the faeces passed by healthy chickens.
The viruses were grown in the allantois of 10-day-old chick embryos incubated at 37° C., and virus-infected allantoic fluids were stored at — 70° C.
Growth cycles in chick embryo chorioallantoic membranes For each experiment about 50 10-day-old chick embryos were inoculated with
0.5 ml. of infected allantoic fluid containing about 109 Egg m50, and incubated at 37° C. At intervals thereafter three chick embryos were harvested and the allantoic fluid pooled. The chorioallantoic membranes were pooled after three washes with ice cold saline (0.15m). All samples were stored at — 70° C. until assayed.
Allantoic membranes were thawed and blended in homogenizers with motor-driven Teflon pestles to give 50 % (w/v) suspensions in phosphate buffered saline, pH 7.2 (PBS).
Haemagglutination titrations Plastic plates were used with 0.2 ml. volumes and 1 % (v/v) chicken erythro-
cytes; titres are expressed as the reciprocal of the last dilution giving complete agglutination.
Neuraminidase assays Sample volumes of 0.1 ml. were incubated at 37° C. for 10 min. with 0.1 ml.
fetuin and 0.3 ml. 0.2m-KH2PO4-Na2HPO4 buffer (pH 6.0). Fetuin was prepared from foetal calf serum by the method of Graham (1961). The N-acetyl neuraminic acid (NANA) released was assayed by the method of Aminoff (1961). Results are expressed as pg. NANA liberated per ml. of sample.
Incorporation of 3H-uridine The allantoic fluid of each egg at the start of the growth cycle was inoculated
with 100 pc. 3H-uridine 5-T (purchased from the Radiochemical Centre, Amer-sham).
The estimation of 311-uridine incorporated into ribonuclease-insensitive acid-insoluble material was based on the method of Plagemann (1968). For each sample,
ml. was diluted with 0.5 ml. 0.01 M sodium deoxycholate, 0.1 ml. ribonuclease (100 pg. per ml. in PBS) and incubated at 37° C. for 30 min.; 2.0 ml. 1 N perchloric acid was added and, after centrifugation at 1000 g for 10 min., the precipitate was washed three times with ice cold 5 % (w/v) trichloracetic acid. The precipitate was dissolved by adding 0.5 ml. 1 N sodium hydroxide and incubating at 60° C. for 30 min.: 0.2 ml. of this solution was added to 10 ml. of scintillation fluid con-taining 8.0 g. butyl PBD (Ciba Ltd), 80.0 g. naphthalene, 400 ml. oxitol (ICI Ltd) and 600 ml. toluene. Radioactivity was measured using an ABAC SL 40 Liquid Scintillation Spectrometer (Inter-technique Ltd) with an efficiency of about 35 %.

64 P. REEVE, MARGARET ROSENBLITM AND D. J. ALEXANDER
RESULTS
The growth of seven strains of NDV in chick embryo chorioallantoic membranes was examined by measuring the amount of haemagglutinin released into the allantoic fluid (extracellular haemagglutinin) and that still associated with the allantoic membrane (cell-associated haemagglutinin). The strains examined differed in virulence in that some killed chick embryos more quickly than others ; Table 1 shows the mean death time (MDT) of chick embryos inoculated with one minimum lethal dose. Herts was the most virulent strain examined, with an MDT of 49 hr. Neither Queensland nor Ulster consistently killed chick embryos, many of which survived even high inocula of these strains. With all strains an increase in haemagglutinin was first detected in the allantoic fluid about 4 hr. after inoculation, and thereafter titres increased exponentially until 12-14 hr. after inoculation (Fig. 1).
Table 1. Relative virulence of Newcastle disease virus strains (from Waterson, Pennington & Allan, 1967)
Mean death time Intracerebral Strain (hr.) index
Herts 49 1.88 Itahen 50 1.86 Texas 50 F80 Beaudette C 62 1.48 F 168 0.25 Queensland oo 0.25 Ulster co 0.00
Mean death time is the average time in which eggs inoculated with one minimum lethal dose of virus are killed. The intracerebral index is estimated from the time taken for chicks to die after intracerebral inoculation. The results are based on a scoring system in which the maximum index is 2 (100 % mortality in one day) and the minimum is 0 (no recorded symptoms after 8 days).
All strains produced approximately the same levels of extracellular haemagglutinin but marked differences were apparent when haemagglutinin associated with the allantoic membranes was assayed. With strains Herts, Italien and Beaudette C the production of cell-associated haemagglutinin continued exponentially until at least 20 hr. after inoculation and only diminished when at least 50 % of the chick embryos were already dead (Table 2). Cell-associated haemagglutinin titres of these virulent strains greatly exceeded those measured in the allantoic fluid at the end of the growth cycle.
Strains Queensland, Ulster and F, on the other hand, produced only low levels of cell-associated haemagglutinin and the rate of production appeared to diminish after approximately 12 hr. The titres of cell-associated haemagglutinin in these avirulent strains never exceeded those measured in the allantoic fluid.
All chick embryos inoculated with the strains Herts, Italien and Beaudette C had died after approximately 30 hr.; of the less virulent strains only F con-sistently killed the majority of chick embryos (90 % dead within 96 hr.) whereas

Growth of NDV in chick chorioallantoic membranes 65
50 % of chick embryos inoculated with Queensland and Ulster survived for seven days after inoculation (Table 2).
Four strains (Herts, Texas, Queensland and Ulster) were examined in greater detail to see if other structural antigens making up the NDV virions, viz. neur-aminidase and ribonucleoprotein, also accumulated in the allantoic membranes infected with virulent virus but not in those infected with avirulent strains. The results obtained supported this possibility: 3H-uridine incorporated into ribo-nuclease-resistant, acid-insoluble material and neuraminidase titres both increased exponentially in allantoic membranes infected with the virulent strains Herts and
— Herts.
— •
• —
•
•
F
• to,. Le
• • ••
oo o
00 — Field Pheasant
• o
_ • •
_
Ulster
o o o o
— Italien
• —
Queensland •
• • ••
o _
...
.
LA; V
• 111111111111111
_ Beaudette C •
• — .
• • • o
—
0
• •
iiiiiiiiiiilill
2 6 10 14 18 22 26 30
2 6 10 14 18 22 26 30 Hours after infection
Fig. 1. The comparison of cell-associated (0-0) and extracellular (•—•) haemagglutination titres during the growth of seven different strains of NDV in eggs.
5 tr VG 68
Reci
proc
al H
A t
itre
10,000
1,000
100
10
0 10,000
1,000
100
10
1,000
100
10
10,000
1,000
100
10

66 P. REEVE, MARGARET ROSENBLUM AND D. J. ALEXANDER
Texas until the embryos died (Fig. 2). We have assumed that the 311-labelled product is either viral RNA or ribonucleoprotein: both double-stranded RNA (Plagemann, 1968) and ribonucleoprotein (Kingsbury & Darlington, 1968) resist digestion with ribonuclease whereas single-stranded (cellular) RNA would be destroyed. In allantoic membranes infected with the avirulent strain Queensland, titres of neuraminidase and ribonuclease-resistant, acid-insoluble 3H-labelled material increased exponentially only for the first 14-16 hr. after infection and then levelled off; with strain Ulster the exponential phase extended rather longer but levelled off within 24 hr. after infection (Fig. 3).
Table 2. Percentage death of chick embryos in eggs inoculated with different strains of Newcastle disease virus
Tune after
inocula- tion (hr.)
Strains of NDV
Herts Italien Beau- dette C F
Queens- land Ulster
22 0 0 0 0 0 0 24 7.5 — 0 0 0 0 26 15 59 9 0 0 0 28 34 78 56 0 0 0 29 — 100 — 0 0 0 30 44 100 0 0 0 32 100 0 0 0 48 0 0 0 69 57 7 20 74 70 7 20 89 97 20 27 96 100 23 30
114 36 43 138 40 53 142 57 53
DISCUSSION
The results obtained show that some strains of Newcastle disease virus had higher titres of viral components associated with the chorioallantoic membrane than with the allantoic fluid, suggesting two discrete groups of strains; (1) Virulent strains which kill chick embryos rapidly and accumulate all the major viral components in the chorioallantoic membrane to titres higher than in the allantoic fluid, and (2) avirulent strains which kill embryos slowly or not at all and produce similar extracellular antigen titres to virulent strains but in which the intracellular accumulation of virus antigen is limited.
Our results show that virulence is associated with the continued exponential production of cell-associated viral antigens without concomitant release of mature virions, a process terminated only by the death of the chick embryo.
Newcastle disease virus, like other myxoviruses, is assembled at the cell surface and the envelope may consist of or contain cell membrane (Waterson, 1968). The modification of the cell membrane presumably has a drastic effect on the cell

(a) (a) •
0
. . .
0 o •
.o 0
0 0
. . . . . .
(b) (b) •
•
o
„6....0...............").
o
(c)
o
• o
(c) • •
o
10,000 Rec
ipro
cal
HA t
itre
1,000
100
10
1,000
E
C O
L.) 100
25
100
E
10 ou
1
Growth of NDV in chick chorioallantoic membranes 67
Texas Herts
2 6 10 14 18 22 26 2 6 10 14 18 22 26 Hours after infection
Fig. 2. Comparison of the extracellular (.-.) and cell-associated (0-0) major antigens of two virulent strains of NDV during growth in eggs. (a) Haemagglutinm. (b) Material incorporating 3H-uridino, in an acid-insoluble RNase-resistant fraction. (c) Nouraminidase, as ug. NANA released.
permeability and metabolism. It could therefore be postulated that the strains of virus producing the most intracellular haemagglutinin would be the most virulent. The loss of ability to carry out cellular repair would enhance this effect and Wilson (1968) has shown that infection with the strain Texas GB, which is highly virulent, inhibits host cell protein synthesis in chick embryo fibroblasts.
Compans et al. (1966) in a morphological study showed that BHK21-F cells infected with SV5 virus accumulated viral ribonucleoprotein and disintegrated after extensive fusion although little mature virus was released. In contrast, primary rhesus monkey kidney (MK) cells infected with SV5 did not accumulate RNP, released infective virus, but were not destroyed. Virulence in their system
5-z
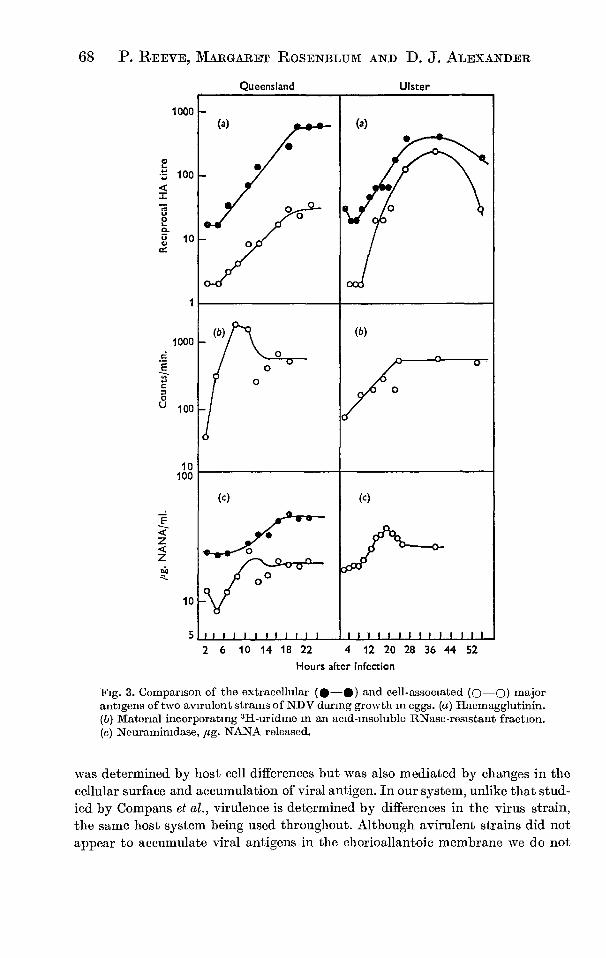
68 P. REEVE, MARGARET ROSENBLUM AND D. J. ALEXANDER
Queensland
Ulster
1000
1000 E
O
100
10 100
0
10
5
(a)
—
. • •
•
o • .
•
•
o
(a)
0 • •
•••
•
. .
.
(b) —
.
.
o
0 (b)
/o 0
(c)
_
II 1
O
i i l
•
(c)
.• '. •
..• ..
Ill I I
.
l 1 i I
.
i I I i I
O O
III II I 2 6 10 14 18 22 4 12 20 28 36 44 52
Hours after infection
Fig. 3. Comparison of the extracellular (0-0) and cell-associated (Q—Q) major antigens of two avirulent strains of NDV during growth in eggs. (a) Haemagglutinin. (b) Material incorporating 31-1-uridine in an acid-insoluble RNase-resistant fraction. (c) Neuramimdase, pg. NANA released.
was determined by host cell differences but was also mediated by changes in the cellular surface and accumulation of viral antigen. In our system, unlike that stud-ied by Compans et al., virulence is determined by differences in the virus strain, the same host system being used throughout. Although avirulent strains did not appear to accumulate viral antigens in the chorioallantoic membrane we do not

Growth of N DV in chick chorioallantoic membranes 69
know if all the cells were infected. Fewer cells may have been infected by avirulent strains than by virulent strains, but since extracellular virus titres are similar with all strains this would require cells infected with avirulent viruses to release relatively more virus into the allantoic fluid than cells infected with virulent virus.
If the same number of cells is infected the difference between strains may be that the intracellular production of viral antigen is limited in cells infected with avirulent strains either by some host cell response or some limiting factor in the virus antigen production mechanism.
Margaret Rosenblum was a visiting student assistant supported by the Polio Research Fund (A.C.C.). D. J. Alexander is supported by the Agricultural Research Council. This research was aided by a grant from the Wellcome Trust. We are indebted to the British Egg Marketing Board for a grant for the eggs used. We acknowledge the technical assistance of Miss Joy Pacey and John Hall and we are most grateful to Professor A. P. Waterson for his help and encouragement.
REFERENCES
AMINOFF, D. (1961). Methods for quantitative estimation of N-acetylneurammic acid and their application to hydrolysates of sialomucoids. Biochemical Journal 81, 384.
ASvLIN, F. D. (1952). Immumsation against Newcastle disease with a virus of low virulence (strain F) and observations on sub-clinical infection in partially resistant fowls. Veterinary Record 64, 245.
COMPANS, R. W., HOLMES, KATHRYN V., DALES, S. & CFIOPPIN, P. W. (1966). An electron microscopic study of moderate and virulent virus-cell interactions of the paramfluenza virus SV5. Virology 30, 411.
DOBSON, N. (1939). Newcastle Disease. Proceedings of the 7th TVorld's Poultry Congress, Cleveland, p. 250.
GRAHAM, E. R. B. (1961). Some aspects of the structure of the carbohydrate moiety of fetuin. Australian Journal of Science 24, 140.
GRANOFF, A. (1959). Studies on mixed infection with Newcastle disease virus. Virology 9, 636. KINGSBURY, D. W. & DARLINGTON, R. W. (1968). Isolation and properties of Newcastle
disease virus nucleocapsid. Journal of Virology 2, 248. PENNINGTON, T. H. (1967). Studies with Newcastle disease virus. Ph.D. Thesis, University of
London. PLAGEMANN, P. W. (1968). Mengovirus replication in Novikoff rat hepatoma and mouse L
cells: Effects on synthesis of host-cell macromolecules and virus specific synthesis of ribonucleic acid. Journal of Virology 2, 461.
ScHAFER, W., SCHRAMM, G. & TRAUB, E. (1949). Untersuchungen uber das Virus der atyp-ischen Geflugelpest. Zeitschrift fur Naturforschung 46, 157.
SCHLOER, G. M. & HANsoN, R. P. (1968). Relationship of plaque size and virulence for chickens of 14 representative Newcastle disease virus strains. Journal of Virology 2, 40.
WATERSON, A. P. (1968). Introduction to Animal Virology. 2nd ed., Cambridge University Press.
WATERSON, A. P. & CRUICKSHANK, J. G. (1963). The effect of ether on Newcastle disease virus: a morphological study of eight strains. Zeitschrift fur Naturforschung 18B, 114.
WATERSON, A. P., PENNINGTON, T. H. & ALLAN, W. H. (1967). Virulence in Newcastle disease virus. A preliminary study. British Medical Bulletin 23 (2), 138.
WILSON, D. E. (1968). Inhibition of host-cell protein and RNA synthesis by Newcastle disease virus. Journal of Virology 2, 1.

J. gen. Viral. 0970, II, 25-34 25 Printed in Great Britain
Studies on the Cytopathic Effects of Newcastle Disease Virus: Metabolic Requirements
By P. REEVE, D. J. ALEXANDER, GILLIAN POPE AND G. POSTE
Department of Virology, Royal Postgraduate Medical School, Du Cane Road, London, W12, England
(Accepted 17 December 1970)
SUMMARY
Nine strains of Newcastle disease virus were examined for their ability to inhibit cellular protein synthesis and to cause cell fusion. Inhibition of cellular protein synthesis was confined to infection with virulent strains (HERTS, WARWICK, TEXAS, `H', FIELD PHEASANT) and the mesogenic strain BEAUDETTE C. No inhibition of synthesis was recorded with avirulent strains (ULSTER, F, QUEENSLAND). Inhibition of cellular protein synthesis required virus protein synthesis within 3 hr of infection and could be inhibited by parafluorophenylalanine and Congo red. Cell fusion and haemadsorption by the various Newcastle disease virus strains also required virus-induced protein synthesis and were inhibited by cycloheximide, parafluorophenyl-alanine and Congo red. However, neither virus-induced inhibition of cellular protein synthesis nor cell fusion required new virus RNA synthesis, since azauridine did not affect these processes. The importance of virus-induced proteins in the inhibition of protein sythesis and cell fusion is discussed.
INTRODUCTION
Previous studies (Reeve & Alexander, 1970; Reeve & Poste, 1971) have shown that the formation of polykaryocytes by cell fusion is the initial cytopathic effect induced by New-castle disease virus (NDV) grown in chick embryo cells. These studies on NDV also showed that there was a direct relation between virulence and the capacity of strains to cause cytopathic effect. In parallel with cytopathic effect many viruses also produce profound alterations in cellular metabolism, particularly by the inhibition of the synthesis of host cell macromolecules (for review, see Roizman & Spear, 1969). However, only limited informa-tion is available on the effect of NDV on host cell macromolecular synthesis. Wilson (1968) found that the virulent strain TEXAS inhibited host cell RNA and protein synthesis, while the mesogenic strain BEAUDETTE c inhibited only protein synthesis. The inhibition of host cell protein synthesis induced by NDV occurs relatively late in the virus growth cycle, in contrast to the 'early' inhibition induced by the picorna-, adeno- and poxviruses (Martin & Kerr, 1968). The significance of NDV-induced inhibition of cellular macromolecular synthesis is not clear, nor is it known whether this inhibition is related to the virulence of the infecting strain. Several strains of NDV of distinct virulence have therefore been examined for their ability to inhibit cellular protein synthesis. Experiments were also done with selective metabolic inhibitors to test whether the ability of NDV to inhibit protein synthesis and to cause cytopathic effect were linked through a common mechanism.

26 P. REEVE AND OTHERS
METHODS
Virus strains and cell culture. The cell culture techniques and most of the virus strains have been described (Reeve & Waterson, 1970; Reeve, Rosenblum & Alexander, 1970). Strains H' and WARWICK were obtained from W. H. Allan Central Veterinary Laboratories, Wey-
bridge, Surrey. All experiments were done on coverslip cell cultures. Chick embryo cell monolayers were inoculated with 0.5 ml. virus-infected allantoic fluid diluted in maintenance medium (Eagle's BH K supplemented with 2 % foetal calf serum, Burroughs Wellcome Ltd., Beckenham, Kent) to provide a multiplicity of infection of 5o infective particles/cell. After adsorption at 37° for 1 hr cultures were washed three times with pre-warmed phosphate-buffered saline (PBS) to remove any remaining virus and fresh maintenance medium was added with metabolic inhibitors or radioisotopes as appropriate. Cultures were then in-cubated at 37° in the stationary position.
Metabolic inhibitors. Congo red, azauridme, cycloheximide and p-fluorophenylalanine (FPA) were obtained from Koch—Light Laboratories, Colnbrook, England and used in cell culture media as freshly prepared solutions at the appropriate concentrations.
Radioisotopes. [3H]L-leucine 4,5-T (19.7 c/mM) and [3H]uridine 5-T (26.5 c/mm) were obtained from the Radiochemical Centre, Amersham, Buckinghamshire.
Incorporation of radioisotopes. The rates of protein and RNA synthesis were estimated by measuring the incorporation of radioactive precursors into trichloroacetic acid-insoluble material. Maintenance medium containing 1-0 pc/ml. isotope was used to pulse coverslip cell cultures for 3o or 6o min. The coverslips were then washed once with ice-cold PBS, four times with ice-cold 5 % (w/v) trichloracetic acid and once in acetone. The coverslips were air-dried, and the acid-insoluble radioactivity estimated in an Intertechnique ABAC SL 4o liquid scintillation spectrometer using toluene containing 8-0 g./1. butyl PBD as scintillation fluid.
Estimation of cell fusion as % polykaryocytosis. The extent of cell fusion produced by virus strains was estimated 15 hr after infection from counts on stained coverslip cell cultures (Reeve & Poste, 1971).
Estimation of haemadsorption. Coverslip cell cultures were washed three times with ice-cold PBS 8 hr after infection and then received 0.5 ml. of a o•5 % (v/v) suspension of chicken erythrocytes. After 15 to 20 min. at 4°, unadsorbed erythrocytes were removed by three further washes with ice-cold PBS. The cultures were then fixed in methanol, stained with May—Griinwald Giemsa (Jacobson & Webb, 1952), and the number of erythrocytes adsorbed to the cells was counted. The amount of haemadsorption is expressed in terms of the number of erythrocytes in cultures treated with metabolic inhibitors expressed as a percentage of the number in inhibitor free cultures. Uninfected chick embryo cell cultures did not adsorb erythrocytes.
Estimation of cellular permeability. The permeability of infected cells was estimated by assaying the amount of lactate dehydrogenase released from the cells into the culture medium. For this assay, cell cultures were grown in 15 cm. glass Petri dishes and the lactate dehydrogenase released was estimated spectrophotometrically (Kornberg, 1955) and ex-pressed as a percentage of the total lactate dehydrogenase present.

4 8 12 16 20 24 4 8 12 16 20 24 Hr after infection
NDV cytopathic effect 27
1 1 1 1 1 1 HERTS
—
_
—
_
0
—WARWICK
1 1 1 1 1 1 TEXAS
0
1 1 1 1 1 H —
—
iiri\\e\e
_
v\\—BEAUDETTE C FIELD PHEASANT —
—
QUEENSLAND
illArNr7--". 1:1
— 13/
a/ — 0'
00-0/ 111111
ULSTER
VA's...7A
111111
F sre......._-\\::
—
0-0 ,Nn/ — i..i .,Ei .-. ,
❑
no/1 1 1 1
Fig t Incorporation of [31-I]leucine into trichloracetic acid-insoluble fractions of chick embryo cells infected with different strains of NDV (•—•). The results are expressed as of the incorporation into uninfected control cells. The release of lactate dehydrogenase from chick embryo cells infected with NDV strains (—E1), is expressed as a percentage of the total cellular lactate dehydrogenase.
0 4 8 12 16 20 24
100
80
60
40
20
0
120
100
80
60
40
20
0
100
80
60 Cou
nts
/min
. as
% c
ontr
ol. R
ele
ased
lact
ate d
ehyd
rogen
ase
as
% to
tal ce
llula
r LD
H
40
20

2 0 " c O E
E 1. 1,4 5
0
>-o
100
80
60
40
20
0
• •
A
• A
(b) —
•
• Inhi
bitio
n o
f p
rote
in s
yn
thes
is (
%) 100
80
60
40
20
(a) —
■
AM
28 P. REEVE AND OTHERS
RESULTS
Relation between virulence of the infecting strain and inhibition of cellular protein synthesis
The virulent Newcastle disease virus strains HERTS, WARWICK, `LI', TEXAS and FIELD PHEASANT and the mesogenic strain BEAUDETTE c all caused marked inhibition of protein synthesis, as judged by the ability of the infected cells to incorporate [3H]-leucine in tri-chloroacetic acid-insoluble material after a 3o min. pulse when compared with the uninfected control cells (Fig. I). All of these strains inhibited cellular protein synthesis by 5o% at 6 to 8 hr after infection, at which time new infective virus appeared in the medium and 5o % of the final yield of mature virus had been synthesized (Reeve & Waterson, 1970). The avirulent vaccine strain F inhibited cellular protein synthesis by about 40 %, but this was not detected until 18 hr after infection. In contrast, the avirulent strains QUEENSLAND and ULSTER did not inhibit cellular protein synthesis.
100 200 300 400 100 200 300 400
Fluorophenylalanme Gig /ml )
Fluorophenylalanine (ig /ml )
Fig 2 (a). Prevention by p-fluorophenylalanine of the inhibition of cellular protein synthesis in chick embryo cells by strains HERTS (0-110) and 're (II—E).Fluorophenylalanine was added' hr after infection to control and infected cultures 7 hr later cultures were pulsed for 1 hr with PHI. leucine and incorporation into an acid insoluble fraction was measured The incorporation of isotope by infected cultures was compared with that of corresponding uninfected control cultures in the presence of fluorophenylalanine and results are expressed as % inhibition of incorporation (b) The effect of different concentrations of fluorophenylalanine on haemadsorption (•—A) and cell fusion (•—•), by chick embryo cells infected with strain HERTS. i hr after infection fluoro-phenylalanine was added, haemadsorption and cell fusion were measured 8 and 15 hr after infection, respectively. The results arc expressed as °/;) of the amount of haemadsorption or cell fusion in infected cultures without fluorophenylalanine
Relation between inhibition of cellular protein synthesis, cytopathic effect and cell damage
All the strains which inhibited cellular protein synthesis induced extensive cell fusion and damage. Cytopathic effect, in the form of cell fusion, was first seen 6 to 8 hr after infection at the time when 5o % inhibition of cellular protein synthesis was recorded. Inhibition of cellular protein synthesis might then be merely a non-specific effect secondary to cell damage and to the development of cytopathic effect. We therefore compared the rate at which inhibition of protein synthesis occurred with the rate at which the enzyme lactate dehydro-genase was lost from the cell through the damaged plasma membrane. With two virulent

NDV cytopathic effect 29
strains, HERTS and TEXAS and the avirulent QUEENSLAND and F strains, a slow loss of lactate dehydrogenase was recorded during the first i6 hr after infection. After this a rapid increase in lactate dehydrogenase release, indicative of extensive cellular damage, was seen only in cells infected with the virulent strains (Fig. 1). These results suggest that gross cellular damage produced by virus replication occurred at 16 to 24 hr after infection and too late to account for the marked inhibition of protein synthesis found 6 to 8 hr after infection.
Effect of inhibitors of protein synthesis on virus-induced inhibition of protein synthesis, cell fusion and haemadsorption
From these experiments we concluded that inhibition of cellular protein synthesis was related directly to the virulence of the infecting strain of NDV. Other properties of the virus which are related directly to its virulence include the ability to produce cell fusion (Reeve & Poste, 197o) and haemadsorption (Reeve & Alexander, 1970). We therefore examined these three properties to determine whether there was a common requirement for protein synthesis.
Cell cultures were infected with NDV strains in the presence of the following metabolic inhibitors and the inhibition of cellular protein synthesis, extent of cell fusion and the degree of haemadsorption measured.
100
0-
80 at" _c
60 C)
0 o_ 40 0 C 0
_r3 20 C C
0
Act
ivit
y as
% o
f th
at in c
ontr
ols w
itho
ut C
ongo
Red
100
80
60
40
20
0
20 40 60
80
Congo Red (pg /ml )
20 40 60 80
Congo Red (pg Iml)
Fig. 3 (a). Prevention by Congo red of the inhibition of cellular protein synthesis in chick embryo cells by strain HERTS. Congo red was added to control and infected cultures 1 hr after infection. Cultures were pulsed 7 hr later fort hr with [31-1]leucine and incorporation into acid-insoluble material measured The incorporation of isotope by infected cultures was compared with that of corresponding uninfected control cultures in the presence of Congo red and results are expressed as
inhibition of incorporation observed. (b). Effect of different concentrations of Congo red on haemadsorption (♦—A) and cell fusion (■—■) by chick embryo cells infected with strain BER-rs. Congo red was added i hr after infection, and haemadsorption and cell fusion measured 8 and 15 hr after infection, respectively The results are expressed as of the amount of haemadsorp- tion or cell fusion in infected cultures without Congo red.
p-Fluorophenylalanine, which inhibits protein synthesis by replacing phenylalanine to form non-functional proteins, prevented the inhibition of cellular protein synthesis by strains HERTS and `1-1' (Fig. 2a) and also prevented cell fusion and haemadsorption by strain HERTS (Fig. 2h).
Congo red. Becht & Drzeniek (1968) described the inhibition of NDV replication by this agent. Doses of io pg./ml. or greater inhibited the synthesis of virus proteins, haemagglutinin
3 vig II
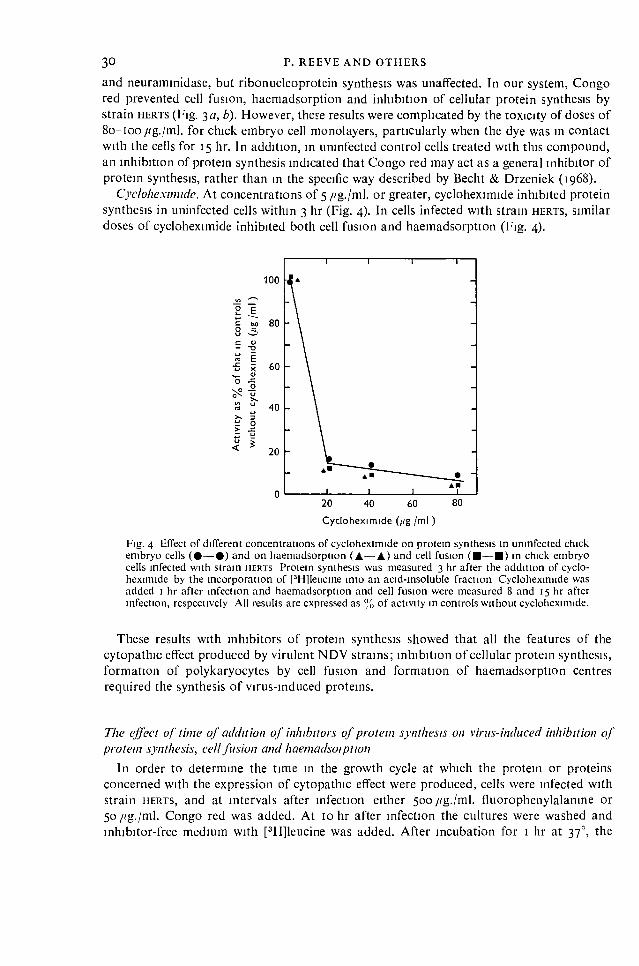
30 P. REEVE AND OTHERS
and neuraminidase, but ribonucleoprotein synthesis was unaffected. In our system, Congo red prevented cell fusion, haemadsorption and inhibition of cellular protein synthesis by strain HERTS (Fig. 3a, b). However, these results were complicated by the toxicity of doses of 8o—too //g./ml. for chick embryo cell monolayers, particularly when the dye was in contact with the cells for 15 hr. In addition, in uninfected control cells treated with this compound, an inhibition of protein synthesis indicated that Congo red may act as a general inhibitor of protein synthesis, rather than in the specific way described by Becht & Drzeniek (1968).
Cycloheximide. At concentrations of 5 fig./ml. or greater, cycloheximide inhibited protein synthesis in uninfected cells within 3 hr (Fig. 4). In cells infected with strain HERTS, similar doses of cycloheximide inhibited both cell fusion and haemadsorption (Fig. 4).
100
80
c coi — -0
60
O ON-2
40 0 _c
•Lt.' 20
0 20 40 60
80
Cycloheximide (Fig /ml )
Fig. 4 Effect of different concentrations of cycloheximide on protein synthesis in uninfected chick embryo cells (•—•) and on haemadsorption (♦—A) and cell fusion (■—■) in chick embryo cells infected with strain HERTS Protein synthesis was measured 3 hr after the addition of cyclo-heximide by the incorporation of PH]leneine into an acid-insoluble fraction Cycloheximide was added i hr after infection and haemadsorption and cell fusion were measured 8 and 15 hr after infection, respectively All results are expressed as °,„' of activity in controls without cycloheximide.
These results with inhibitors of protein synthesis showed that all the features of the cytopathic effect produced by virulent NDV strains; inhibition of cellular protein synthesis, formation of polykaryocytes by cell fusion and formation of haemadsorption centres required the synthesis of virus-induced proteins.
The effect of time of addition of inhibitors of protein synthesis on virus-induced inhibition of protein synthesis, cell fusion and haemadsorption
In order to determine the time in the growth cycle at which the protein or proteins concerned with the expression of cytopathic effect were produced, cells were infected with strain HERTS, and at intervals after infection either 500 fig./ml. fluorophenylalanine or 50 pg./mi. Congo red was added. At To hr after infection the cultures were washed and inhibitor-free medium with [3H]leucine was added. After incubation for i hr at 37°, the

1 2 3 4 5 6 7 8 9 10 Hr after infection
0 4 8 12
16 Hr after infection
100 ..ts3
r, • 80 _c E'
• 60
O
'15 40 C O
_E 20
0
100
o c 80 C E
8 ,"=
c 60 ,3 .c 13_
"F, L'2 O 40 4;. 0 _
> 20
0
NDV cytopathic effect 31
incorporation of the isotope was measured and the extent to which cellular protein synthesis had been inhibited by virus infection was estimated.
The addition of either inhibitor 2 hr after infection prevented the inhibition of cellular protein synthesis by the virus, but after that time the inhibition increased (Fig. 5). Thus, it appears that protein synthesis is required within 3 hr of infection for virus-induced inhibition of cellular protein synthesis to occur. A concentration of 500 €g.Jml. fluorophenylalanine prevented cell fusion and haemadsorption by strain HFRTS when added within 2 hr of infection, but not when added over 4 hr after infection (Fig. 6). Experiments with strains ' and WARWICK gave similar results.
We conclude that protein synthesis must occur very early in the growth cycle for cyto-pathic effect to occur, and that the protein or proteins implicated in this cytopathic effect must be synthesized within 3 hr of infection.
Fig 5 Fig. 6
Fig. 5. Effect of adding fluorophenylalanine (5oo pg ml , 0-0) or Congo red (5o pg , •—•) at different times after infection on the degree of inhibition of cellular protein synthesis by strain HERTS At intervals after infection fluorophenylalanine or Congo red was added to infected cultures and uninfected control cultures All cultures were pulsed with [ 31-illeticine for i hr at to hr after infection and incorporation into an acid-insoluble fraction was measured The results are expressed as % inhibition of protein synthesis calculated from the amount of incorporation of [3H]leucine by uninfected cultures The points indicate the times after infection at which inhibitor was added Fig 6. Effect of adding fluorophenylalanine (500 pg ml) at different times after infection on haemadsorption ( A.—•) and cell fusion (•—•) in chick embryo cells infected with strain HERTS. The points indicate the times after infection at which fluorophenylalanine was added. Haemad-sorption and cell fusion were estimated 8 and 15 hr after infection, respectively The results are expressed as of those for infected cultures without fluorophenylalanine
Effect of inhibition of RNA synthesis on cytopathic effect Since the protein synthesis associated with the development of cytopathic effect occurred
early in the growth cycle, it suggested that the RNA of the infecting particle, rather than newly synthesized RNA, was used directly for coding this synthesis. To test this hypothesis, azauridine was used to block RNA synthesis. Wilson & Lo Gerfo (1964) showed that azauri-dine inhibited the synthesis of virus and cellular RNA to the same extent. In our system, azauridine at 90 pg./ml. inhibited cellular RNA synthesis by over 50 %, as shown by the
3-2

32 P. REEVE AND OTHERS
incorporation of [3H]uridine into uninfected cells. At woo itg./ml. or greater cellular RNA synthesis was inhibited by at least 90 % (Fig. 7). However, cell fusion and haemadsorption in cells infected with strain HERTS were unaffected by up to 3000 azauridine (Fig. 7).
Activit
y as
% o
f th
at in c
on
tro
ls w
ithou
t aza
uri
din
e
1 2
3 Azauridine (mg /ml )
Fig 7. Effect of different concentrations of azauridine on RNA synthesis in uninfected chick embryo cells (•—s) and on haemadsorption (A—A) and cell fusion (■—■) in chick embryo cells infected with strain HERTS RNA synthesis was measured by the incorporation of [3H]uridine into an acid-insoluble fraction 3 hr after the addition of azauridme. Azaurichne was added 1 hr after infection and haemadsorption and cell fusion measured 8 and 15 hr after infection, respectively. Results are expressed as °,10 of activity in cultures without azauridine.
DISCUSSION
Our results show that the ability of Newcastle disease virus strains to inhibit cellular protein synthesis is related directly to the virulence of the infecting strain. Inhibition of cellular protein synthesis by NDV strains occurred fairly late in the growth cycle. A 5o % inhibition was recorded 6 to 8 hr after infection, by which time 5o % of the final yield of mature virus had already been synthesized (Reeve & Waterson, 1970). It is therefore unlikely that the inhibition produced by NDV is comparable with the rapid inhibition seen with the picornaviruses and vaccinia virus (Martin & Kerr, 1968; Bablanian, 1970). However, since extensive leakage of lactate dehydrogenase from infected cells was not seen until some hr after inhibition of protein synthesis, it is unlikely that inhibition results merely from gross cellular damage, but must be due to an early event induced by virus. The results obtained using inhibitors showed that some protein synthesis was required early in the virus growth cycle if cellular protein synthesis was to be inhibited subsequently. The other manifestations of virus infection, haemadsorption and cell fusion, were also dependent upon early protein synthesis.
A requirement for protein synthesis during virus-induced cell fusion has been demonstrated with other viruses. In vaccinia virus-infected L cells, puromycin inhibited the formation of polykaryocytes by cell fusion (Kaku & Kamahora, 1964). Falke (1965, 1967) found that

NDV cytopathic effect 33 cycloheximide and fluorophenylalanine, but not chloramphenicol, prevented the fusion of rabbit kidney cells by herpes simplex virus. More recently, Bratt & Gallaher (1969) showed that cycloheximide prevented cell fusion in chick embryo cells infected with the VICTORIA
strain of NDV. It is not clear whether the proteins involved in cell fusion by NDV are structural com-
ponents of the virus particle or how they act to induce fusion. The labilization of lysosomes is known to be important in producing the cell surface changes necessary for fusion (Allison, 1967; Poste, 1970; Poste & Allison, 1971), and the possibility must be considered that the virus-induced proteins involved in fusion act on the lysosome system. There are indications from the other virus—cell systems that virus-induced proteins are responsible for specific labilization of lysosomes (Amako & Dales, 1967; Thacore & Wolff, 1968; Guskey, Smith & Wolff, 1970). Furthermore, Allison & Mallucci (1965) showed that virulent strains of NDV produced extensive and permanent damage to lysosomes, while avirulent strains induced only transient and reversible damage.
Although our results do not permit a strict distinction between the cellular events involved in the inhibition of protein synthesis and those in cell fusion, it is unlikely that both are mediated by the same mechanism, even though the same proteins may be implicated.
Newcastle disease virus induces the synthesis of both RNA complementary to that of the virus particle, — RNA, and replicas of the virus genome, + RNA. In the first 7 hr of infection of chick embryo cells by BEAUDETTE c, up to 90 % of virus RNA is negative-stranded (Kings-bury, 1970). The functions of the different RNAs are not clear: Kingsbury (1966) and Bratt & Robinson (1967) suggested that the —RNA acts as messenger for some or all virus proteins. However, Schafer, Pister & Schneider (1967) showed that virus antigens could be synthesized in chick embryo cells infected with BEAUDETTE C in the presence of azauridine, and concluded that parental + RNA acted as messenger. Since both virus-induced cell fusion and haemadsorption were unaffected by azauridine, our results support the suggestion of Schafer et al. (1967) that parental RNA can act as messenger. Long & Burke (1970), using u.v. irradiation to inhibit RNA synthesis, also concluded that the parental strand of fowl plague RNA acted directly as messenger for the proteins involved in the inhibition of host cell protein synthesis. Recent studies have also shown that some virus-induced proteins were synthesized in Sendai virus infected cells using the RNA of the infecting particle as mess-enger (Mekler, Shlyankevitch & Shevliaghyn, 1970). It is important to confirm, therefore, whether the nucleic acid of the virus particle can act directly as a messenger for the proteins involved in cytopathogenicity, since our work implies that the synthesis of nucleic acid and the subsequent assembly and release of mature particles may be irrelevant to the expression of virus-induced cell damage.
This research was aided by grants from the Wellcome Trust, The Agricultural Research Council and Action for the Crippled Child (Polio Research Fund). D. A. was supported by the Agricultural Research Council, G. P. by the Cancer Research Campaign and P. R. by the Wellcome Trust. We are most grateful to Professor A. P. Waterson for his help and encouragement throughout this work and in the preparation of the paper.

34
P. REEVE AND OTHERS
REFERENCES
ALLISON, A c (1967). Lysosomes in virus infected cells Perspectives in Virology 5, 29 ALLISON, A c & MALLUCCI, L (1965) Histochemical Studies of lysosomes and lysosomal enzymes in virus-
infected cell cultures. Journal of Ever 'menial Medicine 12i, 463. AMAKO, K & DALES, S (1967) Cytopathology of mengovirus infection. I Relationship between cellular
disintegration and virulence. Virology 32, 201 BABLAN1AN, R (1970). Studies on the mechanism of vaccinia virus cytopathic effects. effect of inhibitors of
RNA and protein synthesis on early virus-induced cell damage. Journal of General Virology 6, 221 BECHT, H & DRZENIEK, R. (1968) The effect of azo dyes on myxovirus neuraminidase and on virus multi-
plication Journal of General Vuology 2, 261. BRATT, M. A. S. GALLAHER, W R (1969) Preliminary analysis of the requirements for fusion from within and
fusion from without by Newcastle disease virus. Proceedings of the National Academy of Sciences of the United States of Amer ca 64, 536
BRATT, M A & ROBINSON, W. S. (1967). RNA synthesis in cells infected with Newcastle disease virus Journal of Molecular Biology 23, 1.
GUSKEY, L. E , SMITH, P. C. & WOLFF, D A. (197o). Patterns of cytopathology and lysosomal enzyme release in poliovirus-infected HEp-2 cells treated with either 2-(a-hydroxybenzy1)-Benzimiclazole or guanidine HCl Jour nal of General Vuology 6, 151.
FALKE, D. (1965). Untersuchungen uber die Beziehungen zwischen Riesenzellbildung und Infektiostat von Herpes simplex virus. Al chi'. fur die gesanite Vousforschting 15, 38.
FALKE, D. (1967) Ca + Histidtne und als Faktoren bei der Riesenzellibildung durch das Herpes-virus hommis. Zettschi ift fur Medical Mikrobiologle und Immunoforschung 153, 175.
JACOBSON, NV & WEBB, M. (1952). Two types of nucleic acid Experimental Cell Research 3. 163 KAKU, KAMAHORA, T. (1964). Giant cell formation in L cells infected with active vaccinia virus. Biken's
Journal 6, 299 KINGSBURY, D. W. (1966) Newcastle disease virus RNA. II. Preferential synthesis of RNA complementary to
parental viral RNA in chick embryo cells Jour nal of Molecular Biology 18, 204. KINGSBURY, D w. (1970). Replication and function of myxovirus ribonucleic acids Pi ogress in Medical
Vu ology 12, 49. KORNBERG, A (1955) Lactic dehydrogenase of muscle In Methods in Enzymology, Vol 1, Ed by S. P
Colwick and N 0. Kaplan. p. 441 New York Academic Press LONG, NV F. & BURKE, D. C (1970) The effect of infection with fowl plague virus on protein synthesis in chick
embryo cells Journal of General Vuology 6, I. MARTIN, E Ni & KERR, I. M. (1968) Virus-induced changes in host-cell macromolecular synthesis Symposia of
the Society for General Microbiology 18, 15. MEKLER, L B , SHLYANKEVICH, M. A. & SHEVLIAGHYN, V J. (1970) Sendai virus RNA as messenger RNA
determining the synthesis of early virus specific proteins Ai cluv fur die gesamte Vti usjor seining 3o, 309 POSTE, G. (1970) Virus-induced polykaryocytosis and the mechanism of cell fusion Advances in Virus
Reseal ch 16, 303 POSTE, G & ALLISON, A C (1971). The membrane fusion reaction a theory Journal of Theoretical Biology
(In the Press ) REEVE, P & ALEXANDER, D. J. (1970) Plaque formation, cell fusion and haemadsorption by Newcastle disease
virus Cytobtos 2, 55 REEVE, P & POSTE, G. (1971). Studies on the cytopathogemcity of Newcastle disease virus Relation between
virulence, polykaryocytosis and plaque size Jour nal of General Virology II, 17 REEVE, P. & WATERSON, A P (1970). The growth cycle of avirulent strains of Newcastle disease virus Ma., obios
2, 5. REEVE, P , ROSENBLUM, 51 & ALEXANDER, D J (Iwo) Growth in chick chorioallantoic membranes of strains of
Newcastle disease virus of differing virulence. Journal of Hygiene, Cambridge 68, 6i ROIZMAN, B & SPEAR, P. G (1969). Macromolecular biosynthesis in animal cells infected with cytolytic viruses.
On lent Topics in Developmental Biology 4, 79 SCHAFER, W PISTER, L & SCHNEIDER, R. (1967). Analyse des Vermehrungsmechanismus des Newcastle disease
virus (NDV) mit Hilfe verschiedener Inhibitoren Zeitsclu ift fur Naturforschung 22b, 1319 THACORE, H. & WOLFF, D A (1968) Activation of isolated lysosomes by poliovirus-infected cell extracts
Nature, London 218, 1063. WILSON, D. E (1968) Inhibition of host cell protein and ribonucleic acid synthesis by Newcastle disease virus
Joni nal of Virology 2, I WILSON, D E & LO GERFO, P (1964) Inhibition of RNA synthesis in Newcastle disease virus-infected cells by
puromycin and 6-azauridine. Journal of Bacteriology 88, 1550
(Received 24 July 1970)