
DẪN XUẤT HALOGEN 1.1. Dẫn xuất monohalogen 1.1.1. Cấu tạo ...
101
Hóa hữu cơ 2 1 CHƯƠNG I: DẪN XUẤT HALOGEN 1.1. Dẫn xuất monohalogen 1.1.1. Cấu tạo và gọi tên Dẫn xuất monohalogen là những hợp chất có công thức chung R-X (trong đó: R – : C n H 2n + 2 – 2a’ – 1 – với n: số cacbon của R, a’ = số π + số vòng của R). Ví dụ: (CH 3 ) 3 CX : tert-butylhalogenua Cl Phenyl clorua Theo IUPAC: xem X là nhóm thế gọi tên theo hydrocacbon tương ứng. Ví dụ: Thông dụng: Đọc như este vô cơ Gốc hydrocacbon + halogenua Ví dụ: Benzyl clorua CH 2 CH CH 2 Allyl clorua CH 2 Cl Cl 1.1.2. Tính chất vật lý Các dẫn xuất halogen có các tính chất chung sau đây: - Nhiệt độ sôi các dẫn xuất halogen cao hơn hydrocacbon tương ứng do phân tử có phân cực lớn. - Nhiệt độ sôi tùy thuộc vào halogen tương ứng: dẫn xuất Flo có t 0 s thấp nhất và cao nhất là dẫn xuất Iot - Do không có liên kết hydro liên phân tử với H 2 O nên dẫn xuất halogen không tan trong nước nhưng lại tan tốt trong các dung môi hữu cơ. - Các dẫn xuất của I, Br và polyclo có tỉ trọng lớn hơn d nước . 1.1.3. Các phương pháp điều chế (1)Từ các Hydrocacbon tương ứng: - Từ ankan: dùng phản ứng thế theo cơ chế gốc tự do S R , tương tự đối với gốc ankyl gắn vào vòng thơm. - Từ hydrocacbon thơm: dùng phản ứng thế theo cơ chế S E - Từ các hydrocacbon không no: dùng phản ứng cộng A E ở các nối π C=C, riêng cộng vào vòng benzen dùng phản ứng cộng A R (2)Từ ancol: dùng SOCl 2 , PX 3 , PX 5 , HX R – OH + SOCl 2 → R – X + SO 2 + HX R – OH + PX 3 → R – X + POX 3 + HX R – OH + HX → R – X + H 2 O (3)Từ andehyt và xeton: dùng PX 5 Clobenzen H 3 C CH Cl CH 2 CH 3 2-clobutan Cl
Transcript of DẪN XUẤT HALOGEN 1.1. Dẫn xuất monohalogen 1.1.1. Cấu tạo ...

Hóa hữu cơ 2
1
CHƯƠNG I: DẪN XUẤT HALOGEN
1.1. Dẫn xuất monohalogen 1.1.1. Cấu tạo và gọi tên Dẫn xuất monohalogen là những hợp chất có công thức chung R-X (trong đó: R – : CnH2n + 2 – 2a’ – 1 – với n: số cacbon của R, a’ = số π + số vòng của R). Ví dụ: (CH3)3CX : tert-butylhalogenua
Cl
Phenyl clorua
Theo IUPAC: xem X là nhóm thế gọi tên theo hydrocacbon tương ứng. Ví dụ:
Thông dụng: Đọc như este vô cơ Gốc hydrocacbon + halogenua Ví dụ:
Benzyl cloruaCH2 CH CH2 Allyl clorua CH2ClCl
1.1.2. Tính chất vật lý Các dẫn xuất halogen có các tính chất chung sau đây: - Nhiệt độ sôi các dẫn xuất halogen cao hơn hydrocacbon tương ứng do phân tử có phân cực lớn. - Nhiệt độ sôi tùy thuộc vào halogen tương ứng: dẫn xuất Flo có t0s thấp nhất và cao nhất là dẫn xuất Iot - Do không có liên kết hydro liên phân tử với H2O nên dẫn xuất halogen không tan trong nước nhưng lại tan tốt trong các dung môi hữu cơ. - Các dẫn xuất của I, Br và polyclo có tỉ trọng lớn hơn dnước. 1.1.3. Các phương pháp điều chế (1)Từ các Hydrocacbon tương ứng:
- Từ ankan: dùng phản ứng thế theo cơ chế gốc tự do SR, tương tự đối với gốc ankyl gắn vào vòng thơm.
- Từ hydrocacbon thơm: dùng phản ứng thế theo cơ chế SE - Từ các hydrocacbon không no: dùng phản ứng cộng AE ở các nối π C=C,
riêng cộng vào vòng benzen dùng phản ứng cộng AR (2)Từ ancol: dùng SOCl2, PX3, PX5, HX R – OH + SOCl2 → R – X + SO2 + HX R – OH + PX3 → R – X + POX3 + HX R – OH + HX → R – X + H2O (3)Từ andehyt và xeton: dùng PX5
ClobenzenH3C CH
Cl
CH2 CH3 2-clobutan Cl

Hóa hữu cơ 2
2
C O + PX5 CX2 + POX3
(4)Thay thế halogen lẫn nhau:
(X: Cl, Br) Các phương pháp (2), (3), (4) là các phương pháp tổng hợp
1.1.4. Tính chất hóa học Do X có độ âm điện lớn hơn C, do đó liên kết C – X phân cực mạnh, X có thể được thay thế bởi một tác nhân có tính bazơ mạnh hơn X. Tác
nhân bazơ có cặp điện tử tự do hay giàu electron sẽ tấn công vào trung tâm dương điện của phân tử. Phản ứng loại này được gọi là phản ứng thế ái nhân (SN). Ngoài ra, R – X còn tham gia phản ứng tách loại H – X. 1.1.4.1. Phản ứng thế nucleophin SN: Ta có thể mô tả phản ứng thế nucleophin ở nguyên tử cacbon no bằng sơ đồ tổng quát như sau:
Y + R X R Y + X Trong đó X là nhóm được thay thế, có tính chất hút electron, là tác nhân nucleophin. Nhiều phản ứng quan trọng trong hóa hữu cơ thuộc loại SN.
- Chuyển hóa ankyl halogenua hoặc arylsunfonat ankyl (trong đó X: Cl, Br, I, OSO3Ar)
Tùy vào tác nhân nucleophin mà ta có các sản phẩm khác nhau:
Y + R X
ancol (Y : OH, H2O)
ete (Y : RO, ROH)
este (Y : RCOO, RCOOH)
- Ankyl hóa amin bằng ankyl halogenua hoặc arylsunfonat ankyl - Ankyl hóa amin bằng ankyl halogenua hoặc arylsunfonat ankyl - Chuyển hóa ancol thành dẫn xuất halogen nhờ tác dụng của HX, PX3, PX5. - Chuyển hóa ancol thành ete trong môi trường axit. - Phân cắt ete nhờ tác dụng của axit HX
Y: + XOH ROH
HOH → R – OH + HX O ROR' + XR + XCN R C N + XI RI
R - X + I R - I + X
R X
Y
R SO
OO Ar

Hóa hữu cơ 2
3
R' C C R C C R' + X v.v... 1.1.4.2. Phản ứng thế nucleophin SN2 (thế nucleophin lưỡng phân tử) a. Cơ chế phản ứng SN2 - Đặc điểm cơ bản của SN2 là hình thành phức hoạt động hay là trạng thái chuyển tiếp trong quá trình phản ứng, không tạo ra sản phẩm trung gian. Khi tác nhân nucleophin đến gần chất phản ứng, liên kết mới giữa C và nhóm Y được hình thành đồng thời với sự yếu đi và tách ra của liên kết cũ giữa C và nhóm X. Như vậy cả hai thành phần: tác nhân Y- và chất phản ứng RX đều tham gia vào giai
đoạn tạo ra trạng thái chuyển
tiếp.
Thí dụ: CH3Br tác dụng với dung dịch NaOH
+H3C Br OH
H
HH
BrHO
H
H
HHO + Br
- Bậc động học của phản ứng là 2: v = k.[CH3Br].[OH-] b. Giản đồ năng lượng của phản ứng SN2
c. Hóa học lập thể SN2: phản ứng xảy ra với sự nghịch đảo cấu hình Ví dụ: R(-)-2-bromoctan tác dụng với NaOH thu được S(-)-octan-2-ol (cấu hình R → S).
Y + R X Y X
R R Y + X
Toạ độ phản ứng
Trạng thái chuyển E

Hóa hữu cơ 2
4
HC6H13
H3CBr + NaOH H
C6H13
CH3
HO + NaBr
Sự nghịch đảo cấu hình đưa đến kết luận: trong phản ứng SN2 tác nhân
nucleophin tấn công từ phía ngược với nhóm halogen. d. Phản ứng phụ kém theo SN2 là phản ứng E2
H3C (CH2)5 CH
Br
CH3 + H3C CH2 OSN2
E2
H3C (CH2)5 CH
OC2H5
CH3
H3C (CH2)4 CH CH CH3
1.1.4.2. Phản ứng thế nucleophin SN1 a. Cơ chế phản ứng SN1 phản ứng trải qua 2 giai đoạn:
C
CH3
Br
CH3
H3C C
CH3
CH3
H3C + Br (1)
C
CH3
CH3
H3C + OHnhanh C
CH3
CH3
OH
H3C (2)
Bậc của phản ứng là bậc 1: nếu giai đoạn 2 thật nhanh so với giai đoạn đầu,
vận tốc của phản ứng chỉ tùy thuộc vào chất tác dụng: v = k.[(CH3)3C Br] b. Giản đồ năng lượng của phản ứng SN1
c. Hóa học lập thể SN1 Ta phân biệt các trường hợp sau:
E
Toạ độ phản ứng
Ion hoá tổ hợp Ion
C XC Y
C X
C

Hóa hữu cơ 2
5
- Trường hợp cacbocation hoàn toàn tự do: Nếu thực hiện trong dung môi có khả năng phân ly tốt như: HCOOH,
C2H5OH + H2O, CH3COCH3 + H2O, H2O. Sau khi được ion hóa để cho ra cặp ion, cặp ion bị dung môi hóa và tách ra hẳn. Trong điều kiện này, cacbocation phẳng và đối xứng, sự tấn công của nhóm Y- vào hai bên xảy ra với xác xuất như nhau. Như vậy nếu chất R – X có tính đối quang thì sau phản ứng kết quả thu được biến thể raxemic.
Y C Y
YC
Y C
- Trường hợp cacbocation dưới dạng cặp ion: Đối với dung môi không có khả năng phân ly lớn (ví dụ CH3COOH chẳng
hạn), cacbocation sinh ra dưới dạng cặp ion, lúc đó một bên cacbocation bị nhóm X- cản trở, chất nucleophin tấn công về phía đối diện, tạo ra nghịch đảo cấu hình một phần.
C
Y
Trong thực tế phản ứng SN1 có thể cho ra cả một “phổ” kết quả khác nhau từ
phía ưu tiên raxemic hóa đến phía ưu tiên quay cấu hình. Mức độ raxemic hóa sẽ tăng lên theo độ bền của cacbocation và độ trơ hóa
học của môi trường với cacbocation. Nếu không tạp được cacbocation phẳng thì không xảy ra phản ứng SN1được. Ví dụ: không cho phản ứng SN1được
d. Phản ứng phụ kèm theo SN1 Phản ứng SN1 có tạo ra cacbocation nên những phản ứng phụ có thể xảy ra
như phản ứng khử, chuyển vị. 1.1.4.3. Những yếu tố ảnh hưởng đến phản ứng thế nucleophin Có thể xảy ra trường hợp ưu tiên SN1, SN2 hay cả hai với vận tốc nhất định phụ thuộc vào:
hướng tấn công ưu thế
Br

Hóa hữu cơ 2
6
1.1.4.3.1. Ảnh hưởng bởi gốc R R ảnh hưởng chính đến cơ chế thế SN
- Nếu gốc R là ankyl bậc 1 SN2 xảy ra dễ dàng - Nếu gốc R là aralkyl, ankyl bậc cao SN1 xảy ra dễ dàng - Nếu gốc R là allyl, aryl metyl xảy ra đồng thời SN1 và SN2 - Nếu gốc R là vinyl hay phenyl khó xảy ra SN1 lẫn SN2 - Cacbocation càng bền thì phản ứng xảy ra theo cơ chế SN1 càng thuận lợi, cụ thể ta chú ý từng gốc: + Gốc ankyl: bậc của Cacbon mang X càng cao thì khả năng tham gia SN2 càng giảm, trong khi đó khả năng tham gia SN1 càng tăng.
C
R
R'
R''
X CH
R
R'
X CH2R X
SN1 SN2
+ Gốc allyl và aryl metyl:
(cacbocation bền nhờ + C cho nên SN1 tốt) Mặt khác SN2 của allyl cũng dễ dàng vì trạng thái chuyển tiếp tương ứng
được ổn định nhờ sự xen phủ giữa orbital π của nối đôi với orbital của trạng thái chuyển tiếp.
CCH
H
H
CH
H
X
Y
Do đó trong điều kiện này yếu tố phân ly của dung môi, tính ái nhân của dung môi sẽ rất quan trọng.
Đối với gốc benzyl cũng xảy ra theo hai hướng SN1 và SN2, cách giải thích tương tự như đối với gốc allyl. Thực nghiệm đã chứng minh rằng khả năng SN1 thay đổi như sau:
Allyl > benzyl > R3C – X > R2CH – X > RCH2 – X > CH3 – X 1.1.4.3.2. Ảnh hưởng của nhóm bị thay thế (nhóm xuất ra – X) - Khả năng phản ứng:
CH2 CH CH2 X CH2 CH CH2

Hóa hữu cơ 2
7
- I : 3- Br : 1- Cl : 0,02- F : 0,0001
- OH2 : 1- OH : 10-10
O N
O
O
: 0,01
- Đối với R – X thì: I > Br > Cl >> F , nguyên nhân là do độ dài liên kết C-I là lớn nhất và sự phân cực hóa ở I là lớn nhất. - Các nhóm – OH phản ứng kém nhất, do đó bình thường không tách ra ion – OH được, chính vì thế ancol chỉ được este hóa trong môi trường axit và ete chỉ được phân cắt trong môi trường axit. 1.1.4.3.3. Ảnh hưởng của tác nhân nucleophin (nhóm nhập – Y) Chỉ có phản ứng SN2 mới phụ thuộc vào tác nhân nucleophin. Giữa tính nucleophin và tính bazơ thường có tính “song song”. Những bazơ mạnh như: C2H5O-, HO- cũng đồng thời có tính nucleophin mạnh, nhưng không phải bao giờ cũng vậy.
a) Tác nhân nucleophin mang điện tích âm có lực nucleophin lớn hơn axit liên hợp của nó, phù hợp với trật tự về lực bazơ HO- > HOH; NH2
- > NH3 b) Đối với tác nhân nucleophin có cấu tạo tương tự nhau và ứng với các
nguyên tố trong chu kì nhỏ, sự biến thiên về lực nucleophin song song với lực bazơ NH2
- > RO- > HO- > R2NH > ArO- > NH3 > F- > H2O c) Đối với các nguyên tố trong cùng một phân nhóm: lực ái nhân tăng từ
trên xuống theo chiều tằn của ban kính nguyên tử và độ phân cực hóa, trái với trật tự lực bazơ I- > Br- > Cl- > F- HS- > HO- ; C2H5S- > C2H5O-
d) Đối với tác nhân nucleophin có nhóm thế, khả năng phản ứng giảm khi sự án ngữ không gian tăng lên dù lực bazơ tăng hay giảm
1.1.4.3.4. Ảnh hưởng của dung môi SN1 hay SN2 chỉ là lý tưởng do ta bỏ qua dung môi, thực tế, không có sự tương tác của dung môi phản ứng không xảy ra. Ví dụ: (C6H5)3CCl + CH3OH trong benzen với v=k[(C6H5)3CCl].[CH3OH] Trong phenol: v= k[(C6H5)3CCl].[CH3OH].[C6H5OH] mặc dù C6H5OH không có mặt trong phương trình tỉ lượng của phản ứng. Đó là vì CH3OH và C6H5OH đã tham gia. Quá trình tạo ra trạng thái chuyển tiếp
C6H5
C6H5 C6H5
ClO
H3C
HH O CH3

Hóa hữu cơ 2
8
Và ảnh hưởng của dung môi đến phản ứng thế mucleophin có thể hiểu như sau:
a) Những dung môi có cả hai tính chất nucleophin và electrophin như H2O, rượu, amin thì: - solvat hóa cả anion (và những phân tử có dư electron) do chúng có H linh động - solvat hóa cả anion (và phân tử thiếu electron) do chúng còn dư cặp electron.
Đối với phản ứng SN quan trọng là sự solvat hóa, anion bị tách ra bằng con đường tạo liên kết H.
R X H OR' Thực nghiệm cho thấy khả năng làm ổn định anion:
HCOOH > C6H5OH > R’OH Do đó: phản ứng SN2 trong rượu nhưng SN1 trong HCOOH. b) Những dung môi có tính nucleophin và electrophin như ROH, axeton...
không có khả năng tạo liên kết H với anion nên chỉ thuận lợi cho sự solvat hóa cation.
1.1.4.4. Phản ứng thế SN ở vòng benzen - Các dẫn xuất halogen gắn trực tiếp vào vòng benzen tham gia phản ứng thế SN rất khó khăn. Điều này được giải thích là do hiệu ứng p-π làm cho liên kết C-X kém phân cực.
X
- Cơ chế: giống như cơ chế thế SE (phần hydrocacbon thơm) chỉ khác phức σ là ion âm - Dẫn xuất nếu có các nhóm thế hút e mạnh như: NO2, SO3H, - CHO, - COOH, -CN gắn với vị trí octo, para đối với X- thì phản ứng SN xảy ra dễ dàng. Giải thích dựa vào cơ chế và độ bền của phức σ. 1.1.4.5. Phản ứng tách Hydrohalogenua của dẫn xuất halogen - Khi đun nóng RX với kiềm + C2H5OH thường xảy ra:
RX
-HX
SN
C C + H2O + X
ROH + X - Về mức độ tách: RI > RBr > RCl > RF
1.1.4.5.1. Tách E1 a. Cơ chế
X

Hóa hữu cơ 2
9
Tương tự SN1, E1 là phản ứng hai giai đoạn, sản phẩm trung gian là R+. Nếu không ghi vai trò rất quan trọng của dung môi ta có sơ đồ:
H C C X H C C X
H C C C C H
Ví dụ:
(CH3)3CBr- Br
(CH3)3CH2O
-H3O(CH3)2C CH2
b. Nhận xét - Cơ chế tách đơn phân tử E1 tương tự cơ chế SN1 về giai đoạn chậm tạo ra
cacbocation, nhưng ở giai đoạn sau thì khác về hướng của phản ứng. Trong phản ứng E1, cacbocation tách prôton tạo ra sản phẩm chưa no. Phản ứng xảy ra theo hướng nhóm X bị tách ra cùng với nguyên tử -hidro ở cacbon bậc cao nhất và tạo ra nối đôi có nhiều nhóm thế nhất (qui tắc Zaixep), nghĩa là tạo ra cacbocation bền.
- Cấu trúc cacbocation của chất phản ứng, tác nhân: + Tốc độ phản ứng phụ thuộc vào độ bền của cacbocation. Cacbocation càng
bền, tốc độ phản ứng càng cao. Nhóm thế có hiệu ứng +I, +C ở vị trí đối với trung tâm phản ứng làm ổn
định cacbocation, làm tăng tốc độ phản ứng: (CH3)3CBr > (CH3)2CHBr > CH3CH2Br > CH3Br (C6H5)3CBr > (C6H5)2CHBr > C6H5CH2Br > CH3Br
Như vậy phản ứng xảy ra dễ dàng với dẫn xuất bậc ba để hình thành cacbocation bậc ba. + Phản ứng tách E1 thường dùng cho các hợp chất ancol trong môi trường axit mạnh để tổng hợp anken qua cacbocation hơn là các dẫn xuất halogen hay sunfonat cho hỗn hợp sản phẩm phức tạp:
OH
H3PO4 H2O
85% + Tác nhân: không có tính bazơ hoặc bazơ rất yếu thuận lợi cho E1.
- Ảnh hưởng của nhóm đi ra: Tốc độ phản ứng tách phụ thuộc vào độ bền liên kết C – X. Liên kết càng kém bền, phản ứng xảy ra càng dễ. Tốc độ phản ứng:
Chậm
Nhanh
Nhanh

Hóa hữu cơ 2
10
R – F << R – Cl < R – Br < R – I - Động học của phản ứng: Phản ứng E1 là phản ứng bậc 1 vì vận tốc của phản ứng chỉ phụ thuộc nồng độ của chất phản ứng chứ không phụ thuộc vào nồng độ tác nhân nucleophin. - Ảnh hưởng lập thể
Về phương diện hoá học lập thể, các phản ứng E1 trong dung dịch không có tính đặc thù như phản ứng E2, vì cacbocation trung gian sinh ra có cấu trúc phẳng, do đó sự tách không phụ thuộc vào cấu hình của phân tử ban đầu. Tuy vậy nếu nhóm X chưa rời khỏi nguyên tử C mang điện dương một khoảng cách đủ xa mà đã xảy ra sự tách proton hoặc nếu trong phản ứng chỉ tạo ra những cặp ion trung gian, thì sự tách theo kiểu trans sẽ chiếm ưu thế hơn. Ví dụ: Trong thực tế, ở những điều kiện của phản ứng E1, từ hai đồng phân cis và trans của 1-metyl-2-cloxyclohexan đều tạo ra 1-metylxyclohexen là chính, cùng với 3-metylxyclohexen, nhưng từ đồng phân cis ( tách kiểu trans) tỷ lệ 1-metylxclohexen cao hơn:
H
CH3
H
H
CH3
Cl
H
CH3
H
H
Cl
CIS-
TRANS-
CH3
1-Metylxyclohexen
H
CH3
3-Metylxiclohexen
- Sự chuyển vị Phản ứng E1 còn có sự chuyển vị của cacbocation như ở SN1, biểu hiện
trong những phản ứng xúc tác axit. Chẳng hạn, phản ứng tách của 3 – metyl – 2 – butanol không phải cho 2 mà 3 sản phẩm:
CH3 C C CH3
CH3
H H
OH
CH3 C C CH2
CH3
H
H
CH3 C C CH3
CH3 H
H C C CH2
H
CH3
CH3
3-metyl-2-butanol
2-metyl-2-buten 3-metyl-1-buten 2-metyl-1-buten Bởi vì có quá trình chuyển vị hidrua từ cation bậc 2 tới bậc 3:

Hóa hữu cơ 2
11
CH3 C C CH3
CH3
H H
CH3 C C CH3
CH3
H
H
Cation 3-metyl-2-butyl cation 2-metyl-2-butyl Theo cơ chế chung sau:
CH3 C C CH3
CH3
H H
OH
3-metyl-2-butanol
HCH3 C C CH3
CH3
H OH2
H
- H2O
CH3 C C CH3
CH3
H
H
CH3 C C CH2
CH3
Ha
H
3-metyl-2-butanol
Hb
CH3 C C CH3
CH3 H
CH3 C C CH2
CH3
H
H
- Ha
- Hb
Hx CH2 C C
CH3
CH3
H
Hy
CH3 C C CH3
CH3 H
- Hy
- Hx CH2 C
CH3
CH2CH3 6.3.3.1.1.4.5.2. Tách E2 a. Cơ chế
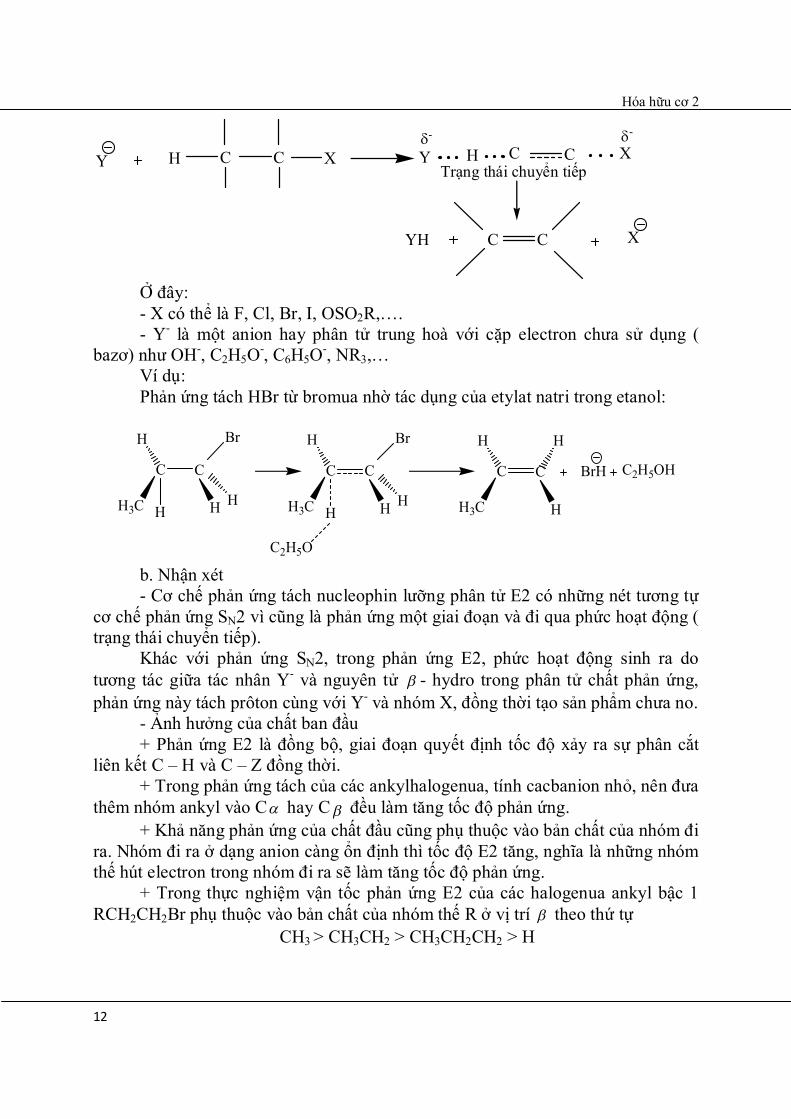
Hóa hữu cơ 2
12
Y H C C X Y H C C X
YH C C X
- -
Ở đây: - X có thể là F, Cl, Br, I, OSO2R,…. - Y- là một anion hay phân tử trung hoà với cặp electron chưa sử dụng ( bazơ) như OH-, C2H5O-, C6H5O-, NR3,… Ví dụ:
Phản ứng tách HBr từ bromua nhờ tác dụng của etylat natri trong etanol:
C C
H
H3C H
Br
HH
C C
H
H3C H
Br
HH
C2H5O
C C
H
H3C H
H
BrH C2H5OH
b. Nhận xét - Cơ chế phản ứng tách nucleophin lưỡng phân tử E2 có những nét tương tự cơ chế phản ứng SN2 vì cũng là phản ứng một giai đoạn và đi qua phức hoạt động ( trạng thái chuyển tiếp).
Khác với phản ứng SN2, trong phản ứng E2, phức hoạt động sinh ra do tương tác giữa tác nhân Y- và nguyên tử - hydro trong phân tử chất phản ứng, phản ứng này tách prôton cùng với Y- và nhóm X, đồng thời tạo sản phẩm chưa no. - Ảnh hưởng của chất ban đầu + Phản ứng E2 là đồng bộ, giai đoạn quyết định tốc độ xảy ra sự phân cắt liên kết C – H và C – Z đồng thời. + Trong phản ứng tách của các ankylhalogenua, tính cacbanion nhỏ, nên đưa thêm nhóm ankyl vào C hay C đều làm tăng tốc độ phản ứng. + Khả năng phản ứng của chất đầu cũng phụ thuộc vào bản chất của nhóm đi ra. Nhóm đi ra ở dạng anion càng ổn định thì tốc độ E2 tăng, nghĩa là những nhóm thế hút electron trong nhóm đi ra sẽ làm tăng tốc độ phản ứng.
+ Trong thực nghiệm vận tốc phản ứng E2 của các halogenua ankyl bậc 1 RCH2CH2Br phụ thuộc vào bản chất của nhóm thế R ở vị trí theo thứ tự
CH3 > CH3CH2 > CH3CH2CH2 > H
Trạng thái chuyển tiếp

Hóa hữu cơ 2
13
+ Khi đưa một nhóm ankyl vào vị trí hoặc của halogenua ankyl bậc 2 hoặc bậc 3 ta cũng thấy vận tốc phản ứng E2 tăng lên. - Ảnh hưởng của tác nhân nucleophin Phản ứng E2 tỷ lệ với nồng độ của bazơ trong phương trình tốc độ, mặt khác E2 cũng rất nhạy với tính bazơ của tác nhân. Tính bazơ của tác nhân lớn, tốc độ phản ứng càng tăng. Và thấy rằng trong phản ứng E2 cần dùng bazơ mạnh hay dùng tác nhân KOH/C2H5OH. - Động học của phản ứng Phản ứng E2 là phản ứng bậc 2 vì vận tốc phản ứng thường phụ thuộc cả hai thành phần tham gia phản ứng. - Ảnh hưởng của dung môi: Dung môi càng phân cực thì làm giảm tốc độ phản ứng. - Ảnh hưởng lập thể + Về mặt lý thuyết, nhóm X có thể bị tách ra cùng nguyên tử - hydro ở cùng phía ( tách kiểu cis hay là kiểu syn) hoặc khác phía với nó ( tách kiểu trans hay là kiểu anti).
C C
H XH
X
C C
H
X H
X
- Thực tế chứng tỏ phản ứng E2 xảy ra theo kiểu trans. Tính đặc thù lập thể
này có nhiều nguyên nhân: + Một là, nếu so sánh năng lượng của các trạng thái chuyển tiếp thì trạng thái ứng với sự tách kiểu trans ổn định hơn trạng thái ứng với sự tách kiểu cis, vì không có sự đẩy nhau giữa các nhóm C…H…Y và C…X + Hai là, sự tạo thành các obitan sẽ thuận lợi hơnnếu phản ứng tách xảy ra theo kiểu trans. Ta có thể phát biểu một qui luật chung như sau: Sự tách lưỡng phân tử E2 chỉ xảy ra dễ dàng khi nào một trung tâm tham gia phản ứng ( H – C – C – X ) nằm trong một mặt phẳng, nghĩa là các nhóm bị tách ở vị trí trans (hay anti) đối với nhau. - Phản ứng E2 ở các hợp chất vòng cũng chạy theo kiểu trans. - Tuy nhiên, quy luật tách kiểu trans chỉ áp dụng được cho các hình thể (cấu dạng) có những nhóm bị tách ở vị trí trans kiểu trục. Các hình thể trans (e,e) có những nhóm bị tách ở vị trí biên không tham gia phản ứng tách lưỡng phân tử, vì bốn trung tâm H – C – C – X không nằm trong một mặt phẳng. Các đồng phân cis (a,e) hoặc (e,a) chỉ phản ứng rất chậm hoặc không phản ứng.
- Sự cạnh tranh E1 và E2
Hợp chất chưa no Hợp chất no Hợp chất chưa no Hợp chất no TÁCH KIỂU CIS TÁCH KIỂU TRANS

Hóa hữu cơ 2
14
+ E1: Ưu tiên tách ở cacbon bậc cao nhất và tạo ra nối đôi có nhiều nhóm thế nhất. ( Tách theo qui tắc Zaixep). Ví dụ:
CH3 CH
Cl
CH2 CH3KOH/C2H5OH
CH2 CH CH2 CH3
E1 + E2: Trong phản ứng E2, nếu X không mang điện dương và không có án
ngữ không gian ( Cl, Br…) thì sẽ tách ra cùng với H ở C bậc cao nhất ( Quy tắc Zaixep), nếu X mang điện tích dương và kém hoạt động hoặc rất cồng kềnh (+NR3, +SR2…) thì sẽ tách ở cacbon bậc thấp.( Tách theo qui tắc hôpman). Như vậy, quy tắc Hôpman ngược với quy tắc Zaixep và chủ yếu áp dụng cho phản ứng E2. Ví dụ:
(CH3)2CH CH CH3
NMe3
HO (CH3)2CHCH CH2
Quy tắc Zaixep: HX được tách ra do X và H gắn với C bậc cao kế bên C mang X. Điều này được giải thích là do anken sinh ra bền do có hiệu ứng siêu liên hợp.
1.1.4.6. Phản ứng với kim loại a) Tác dụng với Na
b) Tác dụng với Zn c) Tác dụng với Mg Có thể có phản ứng phụ 1.1.5. Giới thiệu các hợp chất R-X thông dụng 1.1.5.1. CH3Br Là chất khí ở điều kiện thường, độc, dùng để xông hơi, chống mối mọt ở tàu biển, kho lương thực Ơgenol + CH3Cl + CH3Br: chất dẫn dụ côn trùng 1.1.5.2. vinyl clorua 1.1.5.3. C6H5Cl Từ phenyl clorua tạo ra DDT 1.1.5.4. CHCl3 (clorofoc) Là chất lỏng, thường làm dung môi cho các hợp chất hữu cơ khác để chiết các hợp chất lấy từ thực vật, các ankaloit, steroit, chất béo và còn dùng làm thuốc mê. 1.1.5.5. CHI3 (Iodofoc) 1.1.5.6. CF2Cl2 (Freon 12 hay Frigen) CF2Cl2 có tnc = -1550C, ts = -300C, không cháy. 1.1.5.7. CF2=CF2

Hóa hữu cơ 2
15
Teflon có tính chất quý giá chịu được khoảng nhiệt độ từ -730C đến +2600C, không tan trong bất kỳ dung môi nào, không tác dụng với bazơ. Nên teflon được dùng để phủ lên bề mặt kim loại chống rỉ, công nghiệp chế tạo máy móc, điện...

Hóa hữu cơ 2
16
CHƯƠNG II: HỢP CHẤT CƠ NGUYÊN TỐ
2.1. Định nghĩa Hợp chất cơ nguyên tố là hợp chất hữu cơ mà trong đó nguyên tử nguyên tố Cacbon liên kết với các nguyên tố khác, gọi là cơ kim nếu là kim loại và cơ phi kim nếu là phi kim 2.2. Hợp chất cơ kim 2.2.1. Phân loại Có hai loại: + Cơ kim đơn giản: chỉ kim loại liên kết trực tiếp với Cacbon Ví dụ: LiCH3; CH3-Zn-CH3; CH3Na + Cơ kim hỗn tạp: kim loại M ngoài việc liên kết với Cacbon còn liên kết với các nguyên tố khác thường là Halogen. Ví dụ: C2H5MgCl 2.2.2. Gọi tên - Đối với cơ kim đơn giản: Tên gốc R + tên kim loại - Đối với cơ kim hỗn tạp: Tên gốc R + tên kim loại + tên nguyên tố khác Ví dụ: C2H5MgCl: metyl magie clorua 2.2.3. Cấu tạo Phụ thuộc vào bản chất của liên kết C – M: - Khi khối lượng mol nguyên tử M tăng, liên kết càng phân cực và không bền. :Hợp chất cơ nguyên tố mạnh (C6H5)4Pb: Hợp chất mang tính cộng hóa trị nên phản ứng kém. Quan trọng nhất là cơ Mg hỗn tạp R-Mg-X 2.2.4. Điều chế 2.2.4.1. Hợp chất cơ nguyên tố kim loại kiềm R – M - Từ R – X (R là ankyl hay aryl) R – X + M → R – M + MX (1) R – M + R – X → R – R + MX (2) Cần chú ý: dùng dung môi trơ vì R – M hoạt tính mạnh với các dung môi có H linh động; thường xảy ra đến phản ứng (2) tạo R – R nếu M là Na còn với các kim loại khác thường dừng lại ở phản ứng (1). - Từ hợp chất cơ thủy ngân R – Hg – R + M → 2R – M + Hg - Từ ankin-1 R C C H + NaNH2 R C C Na + NH3 2.2.4.2. Hợp chất cơ nguyên tố kim loại kiềm thổ (khảo sát cơ Mg) - Từ R – X R – X + Mg → R – Mg – X
H C C Na

Hóa hữu cơ 2
17
- Từ hợp chất có H linh động
R C C H + RMgX R C C MgX + RH 2.2.5. Tính chất hóa học 2.2.5.1. Hợp chất cơ nguyên tố kim loại kiềm R – M - Phản ứng thế H bằng kim loại kiềm ở R – M
CH2Cl+ CH3Na
CH2Na+ CH4
R C C H + R C C Na + CH4CH3Na - Phản ứng trao đổi thành cơ kim mới
R X
+ R' Li R Li + R' X - Phản ứng cộng vào hợp chất không no
R CH CH2 + C4H9Li R CH CH2
Li
C4H9
Loại phản ứng này được dùng điều chế cao su tổng hợp
+ C4H9LiCH2 CH CH CH2 CH2 CH CH CH2
C4H9
Li
CH2 CH CH CH2
CH2
C C
CH2
C4H9
H H
CH2
C CH H
CH2Li
2.2.5.2. Hợp chất cơ nguyên tố kim loại kiềm thổ Trong môi trường ete khan R – MgX tồn tại như sau:
2R MgX R Mg
X
R
Mg X R Mg R + MgX2
Thông thường hay viết dưới dạng R – Mg – X
Do khả năng phân cực R – Mg tốt nên các phản ứng đặc trưng bao gồm:
- Phản ứng như là một bazơ (với các hợp chất có H linh động) R – Mg – X + H – A → RH↑ + A – Mg – X
R Mg
X
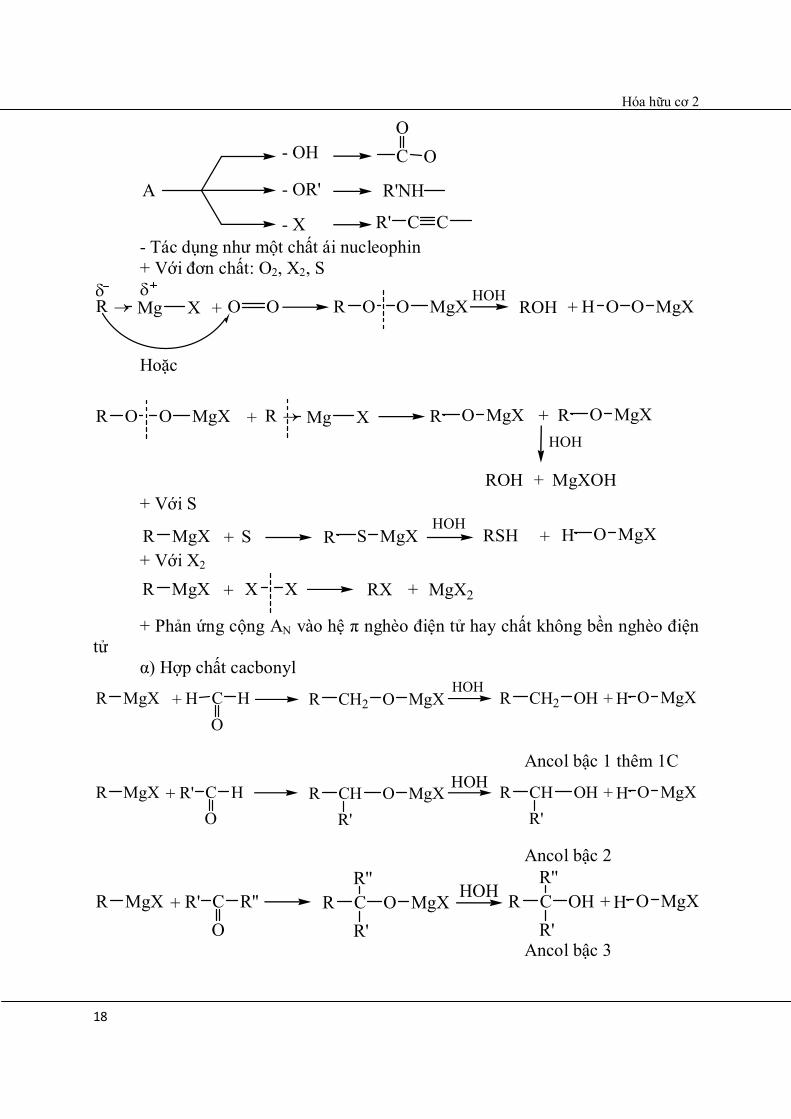
Hóa hữu cơ 2
18
A
- OH
- OR'
- X
C OO
R'NH
R' C C - Tác dụng như một chất ái nucleophin + Với đơn chất: O2, X2, S
R Mg
X + O O O OR MgXHOH
ROH + O OH MgX
Hoặc
O OR MgX + R Mg X OR MgX + OR MgXHOH
ROH + MgXOH + Với S
MgXR + S SR MgXHOH
RSH + OH MgX
+ Với X2
MgXR + X X RX + MgX2
+ Phản ứng cộng AN vào hệ π nghèo điện tử hay chất không bền nghèo điện tử α) Hợp chất cacbonyl
MgXR + CO
HH CH2R O MgXHOH
CH2R OH + OH MgX
Ancol bậc 1 thêm 1C
MgXR + CO
HR' CHR O MgXHOH CHR OH + OH MgX
R' R' Ancol bậc 2
MgXR + CO
R''R' CR O MgX HOH CR OH + OH MgXR' R'
R'' R''
Ancol bậc 3

Hóa hữu cơ 2
19
MgXR + CO
OR' R'' CR OOMgX
R'R'' HOH CR O
OH
R'R'' + OH MgX
CO
RR' + R''OHMgXR
CROMgX
R'RCR
OH
R'R
HOH+OH MgX
Các cloruaaxit, anhydrit axit và RCONH2 tương tự như este về cơ chế phản ứng. Cho đến nay vẫn chưa cơ chế nào được chứng minh đầy đủ, nhưng phản ứng theo cơ chế vòng được nhiều người công nhận nhất.
MgXR'+ OH MgX
CO
RR + MgX2R CO
RR
. . .
. . . Mg XR'
MgX
R'
CR O MgXR'
R
HOHCR OH +R'
R
β) Tác dụng vào các trung tâm nghèo điện tử - Với anken oxit
HOHMgXR + CH2H2C
O
HOMgX
+
+CH2R CH2 OMgX CH2R CH2 OH
HOMgX
++
MgXR + CH2CH
O
R' CH2R CH OMgX
R'
HOH CH2R CH OH
R' Tấn công vào C ít chướng ngại lập thể - Phản ứng với CO2
R Mg
X + C OH3O+
+ HO MgXO CO
R O MgX CO
R O H
- Phản ứng với hợp chất nitrin

Hóa hữu cơ 2
20
+ HO MgXR MgX + C NR' CR
R' N MgXHOH
CR
R' NH
CO
R R' + NH3
(Xetimin)
Các andimin và xetinin dễ bị thủy phân. - Phản ứng với hợp chất RX
R' MgX + XR R' R + MgX2
2.3. Hợp chất cơ phi kim (khảo sát cơ P) 2.3.1. Phân loại Chia làm hai loại: - Loại 1: P liên kết trực tiếp với Cacbon. Tương tự như NH3, ta có PH3 (hợp chất tiêu biểu đầu tiên), do H được thay thế bởi các gốc hydrocacbon ta có photphin bậc I, II, III. Các hợp chất photphin dễ bị oxi hóa hơn rất nhiều lần so với hợp chất của NH3, thành các oxit như: RH2P=O, R2HP=O, R3P=O. Loại hợp chất quan trọng khác của loại này là axit hữu cơ của P
PRH
OHO P
HO
HO
RO PR
R
OHO
- Loại 2: Các sản phẩm của axit vô cơ của P, trong đó P không liên kết trực tiếp với C mà qua các nguyên tố khác như O, S, N. 2.3.2. Gọi tên 2.3.3. Điều chế 2.3.3.1. Điều chế cơ photpho loại 1 - Ankyl hóa các photphin kim loại
PH3 + Na PH2Na + 1/2H2ete khan
PH2Na + R' X
SN
R'-PH2 + NaX
Nếu tiếp tục ta thu được các photphin bậc 2, 3 - Từ clorua photphin + Khử hóa bằng [H]
PRCl
Cl+ 4[H] PH2R + 2HCl

Hóa hữu cơ 2
21
+ Khử hóa bằng [H]
PRCl
Cl+ 2H2O PR
OH
OH+ 2HCl
PR
Cl
R+ H2O PR
OH
R+ HCl
- Cộng photphin vào anken PH3CHC6H13 CH2 + CHC6H13 CH3
PH2 - Tổng hợp bằng cơ Mg
R Mg
X + PCl
Cl
Cl
PR RR
+3 3MgXCl
2.3.3.2. Điều chế cơ photpho loại 2
- Từ clorua photphin
PR
Cl
Cl+ PR
OC2H5
OC2H5
+ 2NaCl2Na OC2H5
Este này bị chuyển vị khi tác dụng với RX như sau:
PRO
O+
C2H5
C2H5R' X PR
OOC2H5 2NaCl
R'+
Tương tự như vậy ta có thể điều chế các hợp chất còn lại
P
Cl
Cl
Cl
+ 3 R' O Na P
O
O
O R'
R'
R'
3NaCl+
POO
O R'
R'R'
R1X+ P
O
O
O R'
R'R1 R'X+
Các este này dễ bị thủy phân tạo thành các axit tương ứng:

Hóa hữu cơ 2
22
P
O
O
O C2H5
C2H5R + 2C2H5OH+ 2HOH H+P
O
OH
OH
R
PRO
OC2H5
R+ HOH H+
PRO
OHR
+ C2H5OH
2.3.4. Tính chất hóa học - Các hợp chất photphin dễ bị oxi hóa RPH2 + [O] → RH2PO3 R2PH + [O] → R2HPO3 R3P + [O] → R3PO - Photphin bậc 3 tác dụng với R – X tạo ra muối photphonin
PR
R
R
+ R' X PR
R
R
R' X
2.3.5. Ứng dụng của cơ photpho Hợp chất cơ Photpho có hoạt tính sinh học cao nhất đó là axit của cơ P có hóa trị 5. Số lượng cơ P tổng hợp được có hoạt tính sinh học cao trên 50 chất. Hợp chất cơ photpho là chất độc đối với sâu bọ, côn trùng, động vật máu nóng kể cả con người. Nguyên nhân là do cơ P làm tê liệt men Cholinestera (kí hiệu ChE) đóng vai trò quan trọng trong cơ thể người.
N CH2
CH3
H3C O CO
CH3ChE NH CH2
CH3
H3C OH CH3COOH+
Axetyl cholin
PO
O
O R'
R'RChE-H + PO
OOR'
R'R
ChEH-ROH
PO
OOR'
R'
ChE
P
O
OOR'
R'
ECh
Do tính độc, cơ P được chia ra hai hướng ứng dụng: - Hướng 1: làm chất độc chiến tranh

Hóa hữu cơ 2
23
+ Tabun: Đimetyl aminoetyl xyanphotphat (Trilon-83), kí hiệu 6A
PO
CNOC2H5
(H3C)2N
+ Xarin: Este isopropyl của Floruametyl photphinic (Trilon- 46), kí hiệu 6B
PO
F O CHH3C
CH3
CH3
+ Xoman: Florua của este phinacolic của axit metyl photphinic
P
O
F
O CH3C
CH3
CH3
C CH3
CH3
OH + Chất V:
PO
RO S CH2
H3C
CH2 NCH3
CH3 Mức độ độc hai:
Tabun: 1 Xarin: 10 Xoman: 30 Chất V: 2000 - Hướng 2: làm thuốc diệt cỏ, sâu bọ Vonphatoc:
PO S O
O
H3C
CH3
NO2 Dipteret:
PHO
O
CHCH3
CH3
H3C
Thiophot:
PO O
O
CH2 NO2H3C
O C2H5 Malathion:

Hóa hữu cơ 2
24
PO S
S
CH2H3C
O C2H5
CH
CH2COOC2H5
COOC2H5
Mecatophot:
PO CH2
S
CH2H3C CH2 S
OCH2H3C
C2H5

Hóa hữu cơ 2
25
CHƯƠNG III: DẪN XUẤT HYDROXI CỦA HYDROCACBON
A. ANCOL 3.1. Cấu tạo, phân loại, danh pháp 3.1.1. Cấu tạo Là những hợp chất có công thức tổng quát R(OH)n, trong đó nhóm – OH gắn vào cacbon no. 3.1.2. Phân loại - Theo số nhóm chức ancol: ancol đơn chức và ancol đa chức - Theo bậc ancol (khái niệm bậc ancol) - Theo bản chất gốc R 3.1.3. Danh pháp 3.1.3.1. Tên quốc tế (IUPAC) Tên loại hydrocacbon tương ứng - số chỉ vị trí nhóm chức – OL CH3 – OH: metanol
H3C CH2 CCH3
HCH2 CH2 OH 2-metylpentan-1-ol:
CH3
C CHC2H5
CH2 CH2 OH 4-metyl-3-etylpent-4-en-1-ol:H2C
3.1.3.2. Tên thông thường Ancol + Tên gốc hydrocacbon + ic
CH CH2 OH Ancol allylic:H2C
H3C CH2 CCH3
CH3
OH Ancol tecpentylic:
3.1.3.3. Tên gọi cacbinol Xuất phát từ tên gọi của CH3 – OH là cacbinol (C6H5)3C – OH: Triphenyl cacbinol (CH3)2CH(OH): Đimetyl cacbinol 3.1.3.4. Tên đặc biệt
:H2C CH2
OH OHEtylen glycol
:CH CH2
OH OHGlyxerinH2C
OH 3.2. Tính chất vật lý

Hóa hữu cơ 2
26
Thủy phân Men rượu
Tinh bột
Các ancol chủ yếu tồn tại ở dạng lỏng, kể cả CH3OH. Điều này được giải thích là do phân tử ancol có liên kết hydro liên phân tử.
H OR
. . . H OR
. . . H OR
. . .
Khả năng hòa tan trong nước tốt do tạo liên kết H liên phân tử với nước
H OR
. . . H OH
. . . H OR
. . .
Thông thường C1 – C5 tan tốt trong nước. Phổ hồng ngoại của – O–H: - νOH riêng biệt = 3620 cm-1 - νOH có liên kết H = 3350 cm-1 Nguyên nhân sự giảm tần số dao động phụ thuộc vào năng lượng liên kết Hydro. Liên kết hydro càng tăng sự giảm tần số dao động càng nhiều. 3.3. Điều chế 3.3.1. Các phương pháp công nghiệp 3.3.1.1. Hydrat hóa các anken
CH RH2C + HOHH2SO4 90 - 98%
CH3 CH
OH
R
Phản ứng tuân theo quy tắc Macconhicop. Phản ứng trái Macconhicop trong trường hợp dùng Hidrobo hóa.
CH RH2C1) B2H6
CH2 CH2 R2) HCOOH
OH 3.3.1.2. Lên men và thủy phân cacbohydrat C12H22O11 (Saccarozo + Mantozo)
(C6H10O5)n
Xenlulozo
C6H12O6 C2H5OH + CO2
Men Clostridum acetobutylium n-C4H9OH (60%) + C2H5OH (20%) + aceton
3.3.1.3. Các phương pháp đặc biệt tổng hợp metanol CO + 2H2 → CH3OH 2CH4 + O2 → 2CH3OH
3.3.2. Các phương pháp tổng hợp trong phòng thí nghiệm 3.3.2.1. Phương pháp Grinha
- Điều chế ancol bậc 1
Tinh bột

Hóa hữu cơ 2
27
R Mg X +
O2 R OMgXHOH
R OH
HCHO R CH2 OMgXHOH
R CH2OH
OR CH2 CH2 OMgX
HOHR CH2CH2OH
- Điều chế ancol bậc 2
R Mg X +
R CHHOH
CH2CH
O
R'CHO OMgXR'
R CH OHR'
R' R CH2 CH R'OH
R' là ankyl - Điều chế ancol bậc 3
R Mg X +
R C OHR'
R' CO
R''
R''R' C
OO C2H5 R C OH
R'
R 3.3.2.2. Phương pháp khử hóa các hợp chất cacbonyl, axit cacboxylic và dẫn xuất
C O
[H]CH OH
Chất khử có thể là H2/Ni hay LiAlH4 Đối với H2/Ni bên cạnh khử hóa nhóm cacbonyl thành ancol còn có thế khử hóa liên kết π nên cần khống chế điều kiện phản ứng. RCOOH thông thường khử khó, thường là dẫn xuất của chúng
CO
R X[H] CH2 OHR
X có thể là – OH, - OR’, - Cl... Thường dùng LiAlH4 để khử axit cacboxylic thành R – CH2 – OH; đối với este thì dùng Na/C2H5OH. Ngoài ra để điều chế ROH người ta còn dùng các phương pháp: thủy phân RX, thủy phân este vô cơ và hữu cơ. 3.3.3. Tính chất hóa học
Sự phân cực của liên kết O – C, O – H tốt dẫn đến momen lưỡng cực của phân tử tăng.
H3C
O
H
1,2 Ao
1,5 Ao
1050

Hóa hữu cơ 2
28
- Dễ tham gia phản ứng dị ly - Đứt liên kết – O – H: phản ứng thế H bởi kim loại, phản ứng este hóa - Đứt liên kết – O – C: phản ứng este hóa vô cơ với axit vô cơ mạnh - Ngoài ra còn có phản ứng tại gốc CxHy
3.3.3.1. Phản ứng tạo ancolat ROH + Na → RONa + 1/2H2 ROH có tính axit cực kì yếu, ancolat dễ bị thủy phân
RONa + H2 → ROH + NaOH 3.3.3.2. Phản ứng ete hóa
R OH + ROH ROR + H2O
H2SO4
1400C phản ứng xảy ra theo cơ chế SN:
R OH + H+ HOR
H
SN1
SN2
ROR + H2O + H+
3.3.3.3. Phản ứng este hóa hữu cơ
C OH + R'OH + H2OH2SO4
T0CO
R C OR'
O
R
Các dẫn xuất axit:
C X
O
R C O
O
R CO
R
Cơ chế:
C OH + H+
O
R C OHR
OH
R'OHC OH
OH
RO R'H
C OH2
OH
R
O R'- H2O
C
OH
R O R' + H2O
C OR'
O
R

Hóa hữu cơ 2
29
Không bền
Lưu ý:
C O + H+ C O H C OHOxoni
3.3.3.4. Phản ứng este hóa vô cơ
OHR + HX XR + H2O
Cơ chế:
OHR + H+ OH2R XR + H2O+ X-
Đối với HX: Mức độ phản ứng của HI > HBr > HCl >>> HF Do HCl khó phản ứng nên thực tế dùng xúc tác ZnCl2. Dung dịch ZnCl2 + HCl được gọi với tên: thuốc thử Lucas.
OHR + ZnCl2 ORH
ZnCl2 R+ ZnCl2+ OH+ Cl-
RClSN1 : Vì với thuốc thử Lucas phản ứng xảy ra theo cơ chế SN1 nên: rượu bậc 3
(xảy ra tức khắc), rượu bậc 2 ( chậm sau vài phút), rượu bậc 1 (chỉ tác dụng wor nhiệt độ cao).
Để tạo ra dẫn xuất halogen còn có các tác nhân khác như PCl5, PCl3, PBr3,... hay P đỏ + X2 tương ứng.
OHR + PCl33 ClR + H3PO33Piridin
Ngoài ra còn dùng SOCl2 (Tionyl clorua)
OHR + SOCl2 ClRPiridin
ClSO
OR + SO2
3.3.3.5. Phản ứng đehyđrat hóa
CC
H OH
(1)H2SO4 t0 > 1700
(2) Al2O3 t0 = 400-8000CC + H2O
Đối với Al2O3, cơ chế phản ứng như sau:
+CH OH
R'
CH2R Al2O3 CH O
R'
CH2R
H
Al2O3

Hóa hữu cơ 2
30
Chuyển vị
Etách > E thế = 1,5 – 2,0kcal/mol
+
H2O
HO Al2O3 CH
R'
CH2R- H+
CH R'CHR + H+
Al2O3 +
Xúc tác H2SO4 đậm đặc:
CH OH
R'
CHR
R1+ H+
CH OH2
R'
CHR
R1
CH
R'
CHR
R1
CH R'CHR
R1
CH R'CR
R1
CH R'CR
R1
Xúc tác H2SO4 đậm đặc có sự tranh chấp của hai phản ứng:
- Thế SN (ete hóa) - Tách E (tách H2O)
3.3.3.6. Phản ứng oxy hóa + Oxy hoá monancol thành hợp chất cacbonyl
CHOH C = O
Sự oxy hoá các ancol bậc một và bậc hai bằng pemanganat kali xảy ra rất chậm trong dung dịch trung tính, nhưng thường lại rất nhanh trong dung dịch bazơ và axit mạnh. Khảo sát phản ứng oxy hoá (C6H5)2CHOH trong dung dịch trung tính và bazơ, người ta thấy: V = k [alcol][MnO4
-][OH-] hiệu ứng đồng vị ở 250C khi oxy hoá (C6H5)2CDOH có giá trị bằng 6,6; benzophenon sinh ra không chứa oxy từ pemanganat kali. Từ các dữ kiện đó người ta đề nghị cơ chế phản ứng như sau:
(-)
(C6H5)2CHOH + HO (C6H5)2CHO + H2O
(C6H5)2CHO + MnO4 (C6H5)2C = O + HMnO4
HMnO4 + MnO4 + HO 2MnO4 + H2Onhanh
(-)
(-)
(-)
(-) (-)2- 2-
2-
chậm

Hóa hữu cơ 2
31
Khảo sát phản ứng oxy hoá ancol isopropylic bằng axit cromic người ta thấy tốc độ phản ứng trong dung dịch axit axetic lớn hơn trong dung dịch axit vô cơ với cùng giá trị về H0; khi thay thế nguyên tử α-hydro bằng đơteri ở 250C tốc độ giảm 6,6 lần, trong khi đó thay thế các nguyên tử - hydro bằng đơteri hầu như không làm thay đổi tốc độ phản ứng: (CH3)2 CH - OH (CH3)2 CDOH (CD3)2 CHOH Từ các dữ kiện người ta đề nghị cơ chế phản ứng như sau:
(CH3)2 CHOH + HCrO4 + 2H (CH3)2CHOCrO3H2 + H2O(+)(-) (+)
CH3
C C
O - CrO3H2 CH3
CH3
= O + BH + HCrO3(+) (+)
(Cr )VL
(+)
CH3 H
: B
(Cr )IV
Sau đó: (+)
3Cr Cr + 2CrIIIIV IVH
Hoặc:
(+)
3Cr + Cr + 2CrIV IV
Cr + (CH3)2CHOH Cr + (CH3)2 C = OIIIV
V
-H + Oxy hoá cắt mạch 1,2 – điol Các 1,2 – điol có đặc tính khác và monoalcol ở chỗ chúng bị oxy hoá bởi
axit peiođic và tetraxetat chì làm đứt liên kết
- C - OH
C - OH
- C - OH
C - OH+
- COH - COH -
Khi oxy hoá bằng HIO4 phản ứng đi qua một este vòng trung gian:
- C - OH
C - OH+ HIO4
- C - O
C - OIO3H
- C - OH
C - OH+ HIO3
HIO4 hoặc
Pb(CH3COO)4
Tốc độ: 1,0 0,16 1,0

Hóa hữu cơ 2
32
Những thí nghiệm dùng 18O đã cho thấy rằng nguyên tử oxy cacbonyl ở sản phẩm cũng chính là nguyên tử oxy –glycol.
Cơ chế tạo este vòng trung gian của phản ứng trên đã được xác nhận bằng các dữ kiện động học và quang phổ, dẫn chứng đặc biệt là các glycol với hai nhóm trans-OH thí dụ trans-9,10-đecalinđiol không phản ứng với axit peiođic.
OH
OH
+ HIO4
Khi oxy hoá 1,2 -điolbằng Pb (CH3COO)4 người ta cũng có nhiều dữ kiện
cho thấy phản ứng xảy ra theo cơ chế vòng. Thí dụ phản ứng có bậc một nối đôi với mỗi chất phản ứng giảm đi, cis-điol bị oxy hoá nhanh hơn trans-điol tương ứng…Cơ chế vòng như sau:
- C - OH
C - OH+ Pb ( CH3COO)4 Pb(CH3COO)2
- C - OH
C - OH+-2CH3COOH Pb(CH3COO)2
- C - O
C - O
Tuy nhiên, có một số trans-1,2-điol lại dễ phản ứng hơn dạng cis tương ứng. Ví dụ trường hợp 9,10 –đihyđrophenantren-9,10 -điol dạng trans phản ứng nhanh hơn dạng cis 9,4 lần, trans 9,10 –đecalinđiol không phản ứng với HIO4 nhưng lại bị oxy hoá bởi Pb(CH3COO)4. Như vậy, có thể ở đây phản ứng đã xảy ra theo cơ chế không vòng.
Pb(CH3COO)2-CH3COOH -2CH3COOH
OH
OH O
O
Pb(CH3COO)2
O
O
+ Pb(CH3COO)2
1,2-điol cũng có thể bị oxy hoá cắt mạch bởi axit cromic và phản ứng cũng chạy theo cơ chế este vòng trung gian.
- Oxy hoá anđehit và xeton + Oxy hoá bằng tác nhân vô cơ: Các hợp chất cacbonyl, nhất là alđehyl có thể bị oxy hoá bởi các hợp chất
oxy hoá khác nhau như MnVII, CrVI, Cu'', AgI, CeIV… Nghiên cứu phản ứng oxy hoá anđehit thơm bằng KMnO4 trong dung dịch
trung tính người ta thấy rằng phản ứng có bậc một đối với anđehit cũng như đối với pemanganat, hiệu ứng đồng vị đơteri bằng 7, nếu dùng KMn18O4, 18O sẽ xuất hiện trong thành phần phân tử của axit thơm sinh ra, các nhóm thế ở vòng thơm chỉ có ảnh hưởng yếu đến tốc độ phản ứng (ρ = -0,25).
Ta có thể mô tả cơ chế phản ứng oxy hoá đó bằng sơ đồ sau:

Hóa hữu cơ 2
33
ArC + MnO4 + H Ar - C OMnO3(-)
H H
OHO(+)
Ar - C - O - MnO3 ArCOOH + BH + MnO3
H
OH(+)(+)
: B
Phản ứng oxy hoá anđehit bằng axit cromic có nhiều nét tương tự phản ứng oxy hoá alcol cũng bằng tác nhân đó. Cơ chế phản ứng như sau:
RC - H + HCrO4 + H RC - H RC + HCrO3 + H3O:OH2
OH
O
O OH
O- CrO3H
(-) (+) (+)
Trong trường hợp dùng C6H5CDO người ta thấy hiệu ứng đồng vị bằng 4,3, điều đó chứng tỏ giai đoạn phân cắt liên kết cacbon-hyđro quyết định tốc độ chung của phản ứng.
Giữa hai phản ứng oxy hoá anđehit và oxy hoá alcol bằng axit crômic có hai điểm khác nhau đáng kể. Một là, các nhóm thế hút electron xúc tiến sự oxy hoá anđehit (ρ = +1,02) nhưng lại làm giảm nhẹ tốc độ oxy hoá alcol (ρ = -1,0). Hai là thay thế dung môi nước bằng axit axetic không làm tăng mạnh tốc độ oxy hoá anđehit như đã thấy khi oxy hoá alcol. Khác với anđehit, xeton tương đối khó bị oxy hoá bởi pemangant kali và axit cromic. Tuy vậy các xetôn vòng dẽ dàng bị oxy hoá thành axit đicacboxylic.
Ví dụ:
O COOHCOOHCr2O3
hay HNO3
Có thể các xeton đã tác dụng dưới dạng enol tạo ra este vô cơ trung gian, sau đó este này bị thuỷ phân:
chậm
Axit ađipic

Hóa hữu cơ 2
34
(-)
- C - C - - C = C - - C - C -
O H OH OH OCrO3 H2(+)
(+)
- C - C - - C - C -
- H(+)
O OH O OCrO3 H2
H2CrO4
Đáng chú ý là nhóm α-metylen của xeton cũng như anđehit có thể bị oxy hoá thành cacbonyl thứ hai nhờ tác dụng của oxyt selen:
- C - CH2 - + SeO2 - C - C - + Se +H2O-H2O
O
AeOH
O O
Trong phản ứng trên, tác nhân oxy hoá trực tiếp có lẽ là axit selenơ: axit này tác dụng với liên kết π của dạng enol theo sơ đồ:
- C - CH2 - - C = CH - - C = CH -H2SeO3
-H2OO OH O - SeO2H
- C - C - - C - CH -
O O O O - SeOH
- Se- H2O
3.4. Giới thiệu các ancol tiêu biểu 3.4.1. Ancol metylic: CH3OH 3.4.1.1. Điều chế - Chưng cất khan gỗ
-
-
3.4.1.2. Tính chất, ứng dụng - chất lỏng, dễ cháy, độc (có thể gây mù mắt, chết người)
+CO H2200 atm, 3000
CH3OH (xt: MnO, Cr2O3, ZnO)
+2CH4 O2p, xt, t0
2CH3OH
Oxy hoá tiếp thuỷ phân

Hóa hữu cơ 2
35
- Trùng hợp HCHO, đimetyl sunfat: tác nhân metyl hóa, CH3OH là dung môi trong công nghiệp, pha vào xăng động cơ phản lực. Lưu ý: CH3OH + CaCl2 → CaCl2.4CH3OH tinh thể: do đó không dùng CaCl2 làm khan CH3OH được. 3.4.2. Ancol etylic: C2H5OH 3.4.2.1. Điều chế - Phản ứng lên men: C6H12O6 → 2C2H5OH + CO2
- Hydrat hóa etylen:
H2C CH2 + H2OP = 80atm, xt: H3PO4, SiO2
3000CH3C CH2 OH
3.4.2.2. Tính chất - ở áp suất thường, nhiệt độ sôi của rượu etylic: 78,30C. - Dung dịch rượu với 95,57% rượu và 4,43% nước tạo thành hỗn hợp đẳng phí có nhiệt độ sôi: 78,150C, do vậy không tách C2H5OH ra khỏi nước bằng chưng cất thường được. Muốn tách, trước tiên cho CaO vào nhiều lần nhằm nâng nồng độ cồn lên 960C. Sau đó cho tiếp Mg kim loại vào hay RONa và đun nhiều giờ được cồn 99,50 hoặc có thể dùng C6H6 để phá hỗn hợp đẳng phí. 3.4.2.3. Ứng dụng - Pha rượu - Là dung môi hữu cơ tốt: pha sơn, vecni, pha vào hương liệu, nước hoa, mỹ phẩm. - Trong y học dùng để sát trùng. - Nguyên liệu tổng hợp ete, este... 3.4.3. Một số ancol không no 3.4.3.1. Ancol allylic Điều chế:
H2C CH CH3 + Cl25000C
H2C CH CH2 Cl-HCl
-OH-H2C CH CH2 OH
3.4.3.2. Các ancol không no có trong hợp chất thiên nhiên Xitronelol (tinh dầu sả)
C CH CH2H3C
CH3
CH2 CH CH2
CH3
CH2 OH
Geraniol (tinh dầu hoa hồng)
C CH CH2H3C
CH3
CH2 C CH
CH3
CH2 OH
Bombicol (do bướm cái tiết ra có mùi thơm thu hút bướm đực)
Hóa hữu cơ 2
36
C C
CH2
H
C C
H (CH2)8 CH2
CH2H3C
H
H
OH
3.4.4. Glycol
1,2: -glycol
1,3: -glycol
1,4: - glycol Xét etylen glycol: 3.4.4.1. Điều chế - Thủy phân dẫn xuất Halogen:
H2C CH2
ClCl
H2C CH2
OHOH
+ 2H2O + OH-+ 2HCl
- Khử hóa các este:
R O CO
(CH2)n CO
O R[H]
Na/C2H5OHH2C
OH(CH2)n CH2
OH+ 2ROH
- Thủy phân các anken oxyt:
H2C CH2
O
+ H2O H2C CH2
OHOH
+ H+
3.4.4.2. Tính chất - Tính chất của glycol tương tự như R – OH. Dùng các α-glycol phản ứng được với Cu(OH)2 tạo ra phức xanh thẫm. Đây là phản ứng đặc trưng để nhận biết α-glycol.
+2 CH2 CH2
OHOH
Cu(OH)2
CH2
CH2 O
O
Cu O
CH2
CH2
O
+ 2H2O
3.4.4.3. Ứng dụng 3.4.5. Glyxerol 3.4.5.1. Điều chế - Thủy phân chất béo:
(RCOO)3C3H5 + 3NaOH 3RCOONa + C3H5(OH)3 - Từ propilen:

Hóa hữu cơ 2
37
Hyđroxi benzen
H2C CH CH3 + Cl25000C
H2C CH CH2 Cl-HCl
+ Cl2 + H2OH2C CH CH2
ClCl OH+ OH-
H2C CH CH2
OHOH OH
- Từ acrolein: CH2 = CH – CHO + H2O2
CH2
CH
CHO
H2C CH CH3 H2C CH CH2
OHOH OH
H2C CH CHO
OH OH
+ H2O2 + H2/Ni
3.4.5.2. Tính chất Tính chất của glyxerol tương tự như R – OH. Dùng các α-glycol phản ứng được với Cu(OH)2 tạo ra phức xanh thẫm. Đây là phản ứng đặc trưng để nhận biết glyxerol. 3.4.5.3. Ứng dụng - Trong công nghiệp thuộc da - Pha vào kem đánh răng - Tạo thuốc nổ TNG, sơn epoxit B. PHENOL 3.1. Cấu tạo, phân loại, danh pháp 3.1.1. Cấu tạo Là những hợp chất có công thức tổng quát Ar(OH)n, trong đó nhóm – OH gắn vào benzen. 3.1.2. Danh pháp - Chất đơn giản nhất là:
OH
Phenol
Axit phenic - Phenol của naphtalen là naphtol Phenol của antraxen là antranol Phenol của phenantren là phenantrol

Hóa hữu cơ 2
38
OH
phenol
OH
Clp-clophenol
OH
-naphtol
OH
1-hydroxi naphtalenantranol-1
HO
phenantranol-5
OH
Catechol
OHOH
OH
Rezoxinol
OH
RezoxinolOH
OH CH3
Crezol Lưu ý cách đánh số hợp chất vòng: - Hệ vòng giáp thẳng:
12
345
6
78
1,4,5,8: α ; còn lại: β
12
345
6
78 9
10 1,4,5,8: α ; 2,3,6,7: β; còn lại: µ - Hệ vòng gãy gấp:
12
34
56
7
8
910
3.2. Tính chất vật lý Phenol ở điều kiện thường là chất rắn, tnc = 410C, ts = 1820C, để lâu trong
không khí chuyển sang màu hồng, do có sự biến đổi như sau:
1,2-đihiđroxibenzen 1,3-đihiđroxibenzen 1,4-đihiđroxibenzen

Hóa hữu cơ 2
39
Hầu như không phản ứng
OH O
Tạo ra chất có sự liên hợp như trên nên có màu. 3.3. Tính chất hóa học
O H
3.3.1. Các phản ứng thể hiện tính axit 3.3.1.1. Phản ứng với dung dịch bazơ ArOH + NaOH → ArONa + ½ H2 3.3.1.2. Phản ứng tạo ete (phản ứng Williamson)
Ar O Na + R X Ar O R + NaXSN
Ar O H + H2C N N Ar O CH3 + N2
3.3.1.3. Phản ứng tạo este –chuyển vị Frizơ
+Ar O H
RCOOH
R C ClO
(RCO)2O
R C OO
Ar
Khi thực hiện phản ứng este hóa phenol với xúc tác AlCl3 (hay bo triflorua, ZnCl2), sẽ xuất hiện chuyển vị axyl đến vị trí orto, para của nhân thơm – gọi là chuyển vị Frizơ, dùng để điều chế các Hyđroxi xeton thơm.
Cơ chế chuyển vị Frizơ:

Hóa hữu cơ 2
40
O
CO
R
+ AlCl3O C
OR
AlCl3
O AlCl3+
C
O
R
SE
OAlCl3
H
CO
R
OAlCl2
CO
R
H2O, T0
OH
CO
R + AlCl3
3.3.2. Các phản ứng ở nhân thơm Do – OH là nhóm thế loại 1 nên ưu tiên thế vào vị trí octo và para; khả năng thế cũng tốt hơn so với benzen. 3.3.2.1. Phản ứng với X2
OH
+ 3Br2
OHBr
Br
Br
+ 3HBr
3.3.2.2. Phản ứng nitro hóa Phản ứng được ngay với cả dung dich HNO3 20%. Đối với đung dịch HNO3 đậm đặc:
OH
+ 3HNO3
OHNO2
NO2
O2N
+ 3H2O
Axit picric Các đồng phân o, p- nitrophenol có thể tách nhau ra bằng phương pháp
chưng cất lôi cuốn hơi nước do đồng phân octo tạo ra phức Chelat nên dễ bị lôi cuốn hơi nước hơn.

Hóa hữu cơ 2
41
O
N
H
O
O
.. .
3.3.2.3. Phản ứng sunfo hóa
OH
H2SO4
OH
OH
SO3H
SO3H
OHSO3H
SO3H
HO3S
Axit phenol-2,4,6-trisunfonic
3.3.2.4. Phản ứng ankyl hóa (phản ứng Fridencraft)
OH
+ 2RX
OH OH
+ AlCl3R
R
+2 + 2HX
3.3.2.5. Phản ứng axyl hóa - Trực tiếp:
OH
+ R C ClO
OH
C
O
R
OH
C
O
R
+
- Gián tiếp: tạo ra este – chuyển vị Frizơ

Hóa hữu cơ 2
42
+ R C XO
OH
CO
RO C
OROH
AlCl3 250
1650 OH
C
O
R
3.3.2.6. Phản ứng ghép đôi với muối điazoni
+ R N N
OH
Cl
OH
N N R+
OH
N N R
3.3.2.7. Phản ứng KONBE O Na
+ O C O1250C, 7atm
OH
C O NaO
OH
COOHH+
3.3.2.8. Phản ứng RAIMO - TIMAN
+
OH
CHCl3 + NaOH
OHCHO
+ 2HCl + NaCl
Cơ chế:

Hóa hữu cơ 2
43
OH + HCClCl
ClC
ClCl
Cl+ H2O
CClCl
ClCl C
Cl
Cl+
OH
CCl
Cl
OHCH
Cl
Cl
OHCH
Cl
ClHOH
OHCHO
+ 2HCl
3.3.2.9. Phản ứng với HCHO
OH
HCHOH+/OH-
OH
CH2OH C6H5OH
OH
CH2 OH
C6H5OH
HCHO
OH
CH2 OHH2C....
CH2
....
CH2
OH CH2
CH2
....
Xúc tác axit:
+
OH
H C H
O
H+ H C H
OH+ SE
OH
CH2 OH
OH
CH2 OH

Hóa hữu cơ 2
44
Quá trình được lặp đi lặp lại, thế SE và ngưng tụ liên tục tạo ra polyme. Xúc tác OH -:
O
+H C H
O
+
O
CH2 OH
n 3.3.3. Phản ứng oxi hóa phenol
OH O
[O]
Các gốc này có thể đime hóa hoặc bị oxi hóa khử dị hóa, tùy thuộc bản chất gốc R gắn vào vòng benzen nên cho màu khác nhau. 3.3.3.1. Đime hóa
O
2 OO
3.3.3.2. Oxi hóa khử dị hóa O
2
CH3
O
CH2
+
OH
CH3 3.4. Giới thiệu các phenol thông dụng 3.4.1. Phenol Là nguyên liệu rất quan trọng để sản xuất: - Nhựa dẻo phenolfomanđehyt - axit salixilic - chất màu - dược phẩm - sợi nilon - nhựa epoxit - C6Cl5(OH): chất bảo quản gỗ, chống mối mọt - Thuốc diệt cỏ - chất 2,4-D; 2,4,5-T

Hóa hữu cơ 2
45
OH
OH-
O
+ Cl CH2 COOH
O CH2 COOH O CH2 COOH
Cl
ClO CH2 COOH
Cl
ClCl
3.4.2. Crezol
OHCH3
- Thu hồi do chưng cất nhựa than đá - nguyên liệu để sản xuất chất dẻo phenolfomanđehyt 3.4.3. Điphenyl 3.4.3.1. Catechol
OHCl
HOH
1900C
OHOH
a) Phản ứng đặc trưng tạo vòng
O
O
Na
Na+
Cl C Cl
O
-2NaClO
OC O + 2NaCl
O
O
Na
Na+
Cl
CH2
Cl O
OCH2 + 2NaCl
b) Phản ứng khử ion Ag+
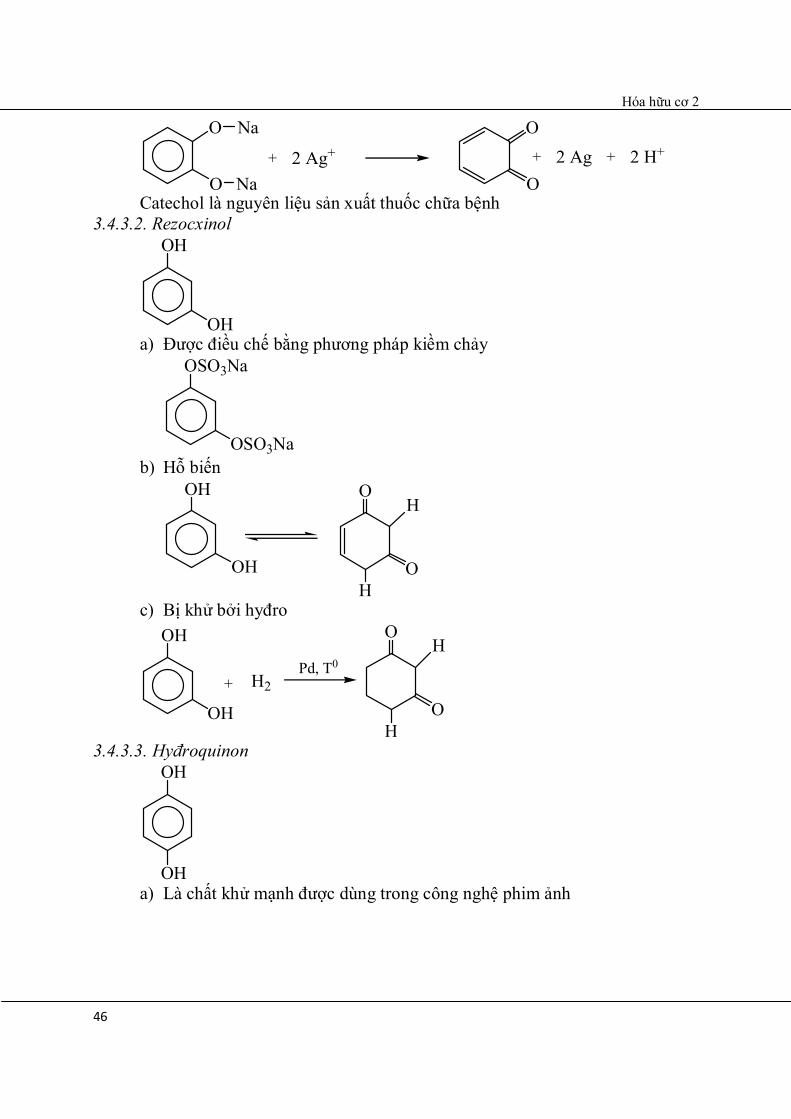
Hóa hữu cơ 2
46
O
O
Na
Na+
O
O2 Ag+ + 2 Ag + 2 H+
Catechol là nguyên liệu sản xuất thuốc chữa bệnh 3.4.3.2. Rezocxinol
OH
OH a) Được điều chế bằng phương pháp kiềm chảy
OSO3Na
OSO3Na b) Hỗ biến
OH
OH
O
O
H
H c) Bị khử bởi hyđro
+
OH
OH
O
O
H
H
H2Pd, T0
3.4.3.3. Hyđroquinon
OH
OH a) Là chất khử mạnh được dùng trong công nghệ phim ảnh
Hóa hữu cơ 2
47
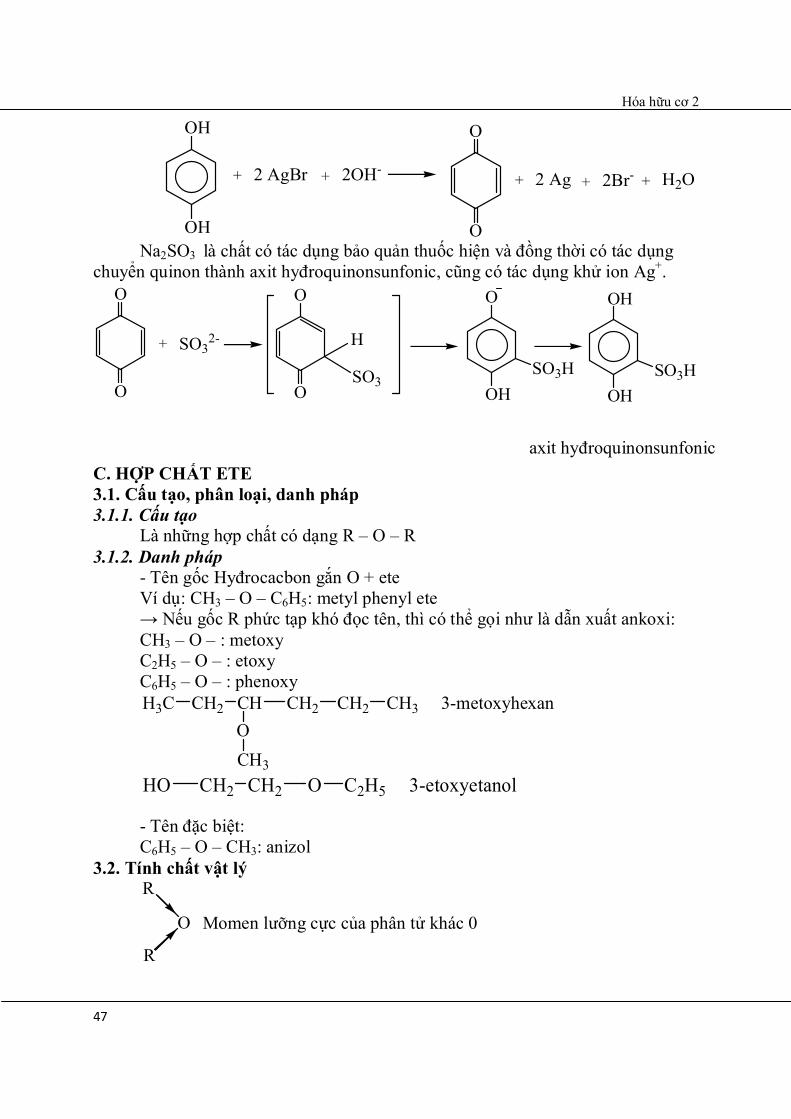
axit hyđroquinonsunfonic
+
OH
OH
2 AgBr + 2OH-
O
O
+ 2 Ag + 2Br- + H2O
Na2SO3 là chất có tác dụng bảo quản thuốc hiện và đồng thời có tác dụng chuyển quinon thành axit hyđroquinonsunfonic, cũng có tác dụng khử ion Ag+.
O
O
+ SO32-
O
O
H
SO3
O
OHSO3H
OH
OHSO3H
C. HỢP CHẤT ETE 3.1. Cấu tạo, phân loại, danh pháp 3.1.1. Cấu tạo Là những hợp chất có dạng R – O – R 3.1.2. Danh pháp - Tên gốc Hyđrocacbon gắn O + ete Ví dụ: CH3 – O – C6H5: metyl phenyl ete → Nếu gốc R phức tạp khó đọc tên, thì có thể gọi như là dẫn xuất ankoxi: CH3 – O – : metoxy C2H5 – O – : etoxy C6H5 – O – : phenoxy
H3C CH2 CHO
CH2 CH2 CH3
CH3
3-metoxyhexan
CH2 CH2 O C2H5HO 3-etoxyetanol
- Tên đặc biệt: C6H5 – O – CH3: anizol
3.2. Tính chất vật lý
O
R
R
Momen lưỡng cực của phân tử khác 0

Hóa hữu cơ 2
48
- Không có liên kết Hyđro như rượu nên ts< trượu - Có liên kết liên phân tử với H2O nên tan tốt trong nước. 3.3. Điều chế 3.3.1. Phản ứng tách nước từ 2 nhóm – OH của ancol
R O H + H O RH2SO4, 1400C
R O R + H2O
H2C CH2
O
+ H2OH2C CH2
OHOH
H2SO4, 1400C
Cơ chế (Mevai):
+ H+R O H R O H
Hion oxoni
R OHR O R
H
+ H2O
R O R + H+
Đối với phenol, khả năng đứt liên kết – C – OH khó do đó không điều chế Ar – O – Ar bằng phương pháp này. 3.3.2. Phản ứng Williamson
R O Na + R X R O R + NaXSN
Phản ứng này dùng để điều chế Ar – O – Ar và R – O – R’.
Ar O Na +
SN
I Ar O Ar + NaI
Tất nhiên đối với Cl – Ar hay Br – Ar phản ứng chỉ xảy ra đối với vòng benzen có các nhóm thế hút điện tử nhất là gắn vào ví trí octo hay para đối với – X. Ví dụ:
C6H5 O CH2 NO2 (A)
+(1) (A)O2N Cl Ar O Na
(2) OCH2O2N Na + Cl
Phản ứng trên không dùng với dẫn xuất halogen bậc 3 vì dẫn xuất halogen bậc 3 dễ tham gia tách loại ở nhiệt độ cao.

Hóa hữu cơ 2
49
H3C CH2 Br + C
CH3
CH3
CH3
ONa H3C CH2 C
CH3
CH3
CH3
O
H3C CH2 ONa + (CH3)3CCl 3.3.3. Các phương pháp khác H2C CH OH + CH2CHHO X H2C CH O CH2CH
CH2 CH2 OH2ClH2SO4,T0C
CH2 CH2 OCl CH2 CH2 ClKOH, T0cao
CH2 CH O CH CH2 H2C CH O R X H2C CH OH + R OH
xt: RONa, T0C
HC CH + R OH 3.4. Tính chất hóa học
Do trên O còn cặp electron tự do nên tính chất hóa học cơ bản là tính nucleophin và tính bazơ. 3.4.1. Tính bazơ - Với HX:
R O R + HX R O R
H
X
ion oxoni - Với axit Lewis:
R O R + BF3 R O R
BF3
3.4.2. Phân cắt bởi axit
R O R + H+ R O R
H
X-
(1) SN1(2) SN2
R OH + RX
3.4.3. Phản ứng chuyển vị Claizen Khi ở nhiệt độ 2000C, allyl phenyl ete chuyển gần như hoàn toàn về o-allyl phenol. Cơ chế:

Hóa hữu cơ 2
50
Chuyển dịch H+
OCH2
CHH2C
H T0C
OCH2
CHCH2
HOH
CH2
CHCH2
H
1414 14
Nếu các vị trị octo bị chiếm chỗ thì chuyển vị về para.
O CH2 CH CH2RR
T0C
O
CH2CHCH2
RR
OHRR
CH2 CH CH2 3.5. Giới thiệu riêng các ete 3.5.1. Đietyl ete: C2H5 – O – C2H5 - Chất lỏng không màu có mùi đặc trưng, nhẹ, dễ cháy, dễ bay hơi. - Làm dung môi, chất gây mê trong y học - Để lâu dễ bị oxi hóa nên khi chưng cất dễ nổ - Đietyl ete tuyệt đối thu được bằng cách chưng cất ete + H2SO4 và sau đó đựng trong bình kín có chứa Na kim loại. 3.5.2. Etylen oxyt (oxiran, epoxit) - Điều chế:
H2C CH2
O
+ 1/2 O2H2C CH2Ag, 2500C
C C
OH
+ X2C C
X
+ OH-C C
OHOH
- H2OC C
O Oxi hóa bằng peraxit:
+ RCOOOHC C C C
O
+ RCOOH

Hóa hữu cơ 2
51
là chất có hoạt tính mạnh nên dễ tham gia phản ứng mở vòng. Phản ứng mở vòng có thể xúc tác bằng H+.
C C
O
+ H+
C C
OH
+ Z-
C C
ZOH
Tác nhân mở vòng là: HOH; R – OH; HX; RMgX 3.5.3. Các ete khác - Metyl phenyl ete (Anizol)
O CH3
- Anetol (tinh dầu quả hồi)
O CH3
HC CH CH3 - Gaiacol (tổng hợp chất vani)
OH
O CH3
- Euganol (1-hyđroxi-2-metoxy-4-allylbenzen)
Thành phần chủ yếu của tinh dầu cây đinh hương và hương nhu.
- Veretrol Veretrol dùng để tổng hợp ancaloit papaverin.
C C
O
OH
HC CH CH3
O CH3
O
O CH3
CH3

Hóa hữu cơ 2
52
Chất lignin của gỗ cũng được tổng hợp trên cơ sở các metyl phenyl ete, do đó mà khi chưng cất than gỗ các ancol metylic sinh ra.

Hóa hữu cơ 2
53
CHƯƠNG IV: HỢP CHẤT CACBONYL
(ANĐEHYT VÀ XETON) 4.1. Cấu tạo, phân loại, danh pháp 4.1.1. Cấu tạo Hợp chất cacbonyl (hợp chất oxo) là những hợp chất hữu cơ trong công thức có nhóm hóa trị II .
- Nếu là anđehyt: C
O
H
- Nếu là xeton:
C
O Các góc gần bằng 1200, phân cực mạnh tại liên kết C = O
4.1.2. Danh pháp 4.1.2.1.Đối với anđehyt a. Tên IUPAC - Tên hyđrocacbon tương ứng + al CH3 – CH2 – CHO: propanal - Nếu có đồng phân thì đánh số mạch chính với C số 1 là – CHO
CH3 CHCH3
CH2 CHO 3-metylbutanal1234
CH3 CH
CH3
C C CHO12345
4-metylpent-2-in-1-al
b. Tên thông thường Đọc từ tên thông thường của axit cacboxylic tương ứng
CH3 – CHO: anđehyt axetic HCHO: anđehyt fomic Chú ý: Trường hợp phức tạp xem – CHO là nhóm thế (Focmyl) C6H5 – CHO: Focmylbenzen (benzanđehyt)
C O
C OR
R

Hóa hữu cơ 2
54
12
345
6
78 9
10
CHO
9-focmylantraxen
Ngoài ra, C = O còn được đọc là nhóm oxo OHC – CHO: 1,2-đioxoetan
OHC CO
CH2 CH21 2 3 4 5
CHO 1,2,5-trioxopentan
4.1.2.2.Đối với xeton
a. Tên IUPAC - Tên hyđrocacbon tương ứng + on
CH3 CO
CH2 CH21 2 3 4 5
CH3 pentan-2-on
b. Tên thông thường
Tên các gốc hyđrocacbon gắn với nhóm oxo + xeton
CH3 CO
CH2 CH21 2 3 4 5
CH3 metyl n-propyl xeton
Để đọc xeton phức tạp: Ví dụ:
C CH3
Oaxetylbenzen hay benzoylmetan
CO
benzoylxyclopentan
Ngoài ra, C = O còn được đọc là nhóm oxo (tương tự như trường hợp
anđehyt). 4.2. Tính chất vật lý - Do nhóm cacbonyl phân cực tốt nên so với ete, dẫn xuất halogen, hydrocacbon có phân tử lượng tương tự, anđehyt và xeton có nhiệt độ sôi cao hơn. - Tuy nhiên, do không có liên kết Hyđro nên có nhiệt độ sôi nhỏ hơn rượu. Nhưng vẫn tạo được liên kết Hyđro với nước nên tan tốt trong nước. 4.3. Điều chế 4.3.1. Đi từ hyđrocacbon 4.3.1.1. Oxi hóa ankan, anken, ankin

Hóa hữu cơ 2
55
CH4 + O2
600-7000C
NOHCHO + H2O
CH3Ar + H2O+ [O]CrO2Cl2 CHOAr
O3CC +H3O+
OC + OC
1/2O2CH2H2C +PbCl2 +CuCl2 CH3CHO
4.3.1.2. Hyđrat hóa ankin 4.3.1.3. Hyđrofomyl hóa anken
CC + H2+ COCo(CO)4
t0, PCHOC
4.3.2. Đi từ hyđrocacbon 4.3.2.1. Thủy phân dẫn xuất halogen
CCR
R X
X+ 2OH- RC
O
R + 2X- + 2H2O
4.3.2.2. Oxi hóa, thủy phân monohalogen
CH2R ClH2O, MnO2
CHOR 4.3.3. Đi từ ancol Đehyđro hóa hay oxi hóa monoancol - Trong công nghiệp
CH2R OHCu, T0
CHOR + H2
RC
O
RRCH
OH
RCu, T0
+ H2
- Trong phòng thí nghiệm
RC
O
R
CH2R OH CHOR + H2
RCH
OH
R + H2
[O]
[O]
(1) CrO3(2) K2Cr2O7
4.3.4. Đi từ dẫn xuất của axit cacboxylic
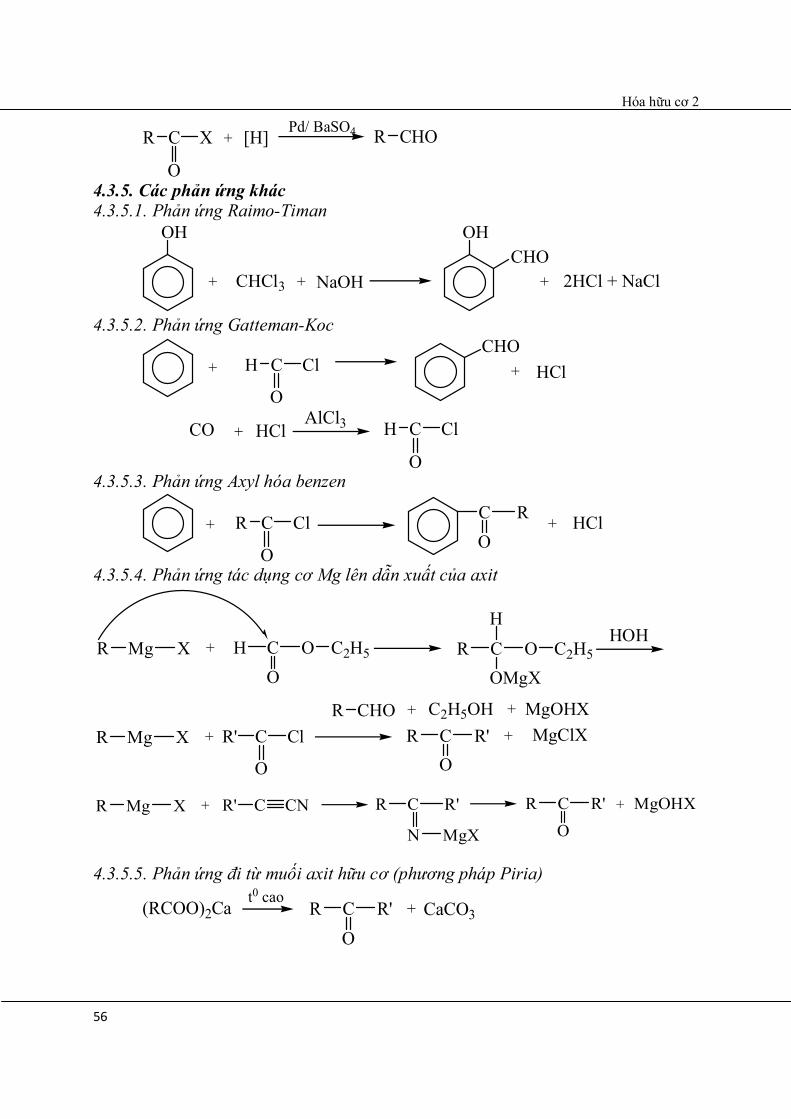
Hóa hữu cơ 2
56
[H] Pd/ BaSO4CR X
O
+ CHOR
4.3.5. Các phản ứng khác 4.3.5.1. Phản ứng Raimo-Timan
+
OH
CHCl3 + NaOH
OHCHO
+ 2HCl + NaCl
4.3.5.2. Phản ứng Gatteman-Koc
HCl+ CH Cl
O
CHO+
CO + HClAlCl3 CH Cl
O 4.3.5.3. Phản ứng Axyl hóa benzen
HCl+ CR Cl
O
C +RO
4.3.5.4. Phản ứng tác dụng cơ Mg lên dẫn xuất của axit
MgR X + OCO
H C2H5 OCOMgX
R C2H5
HHOH
CHOR + C2H5OH + MgOHX MgR X + CR' Cl
O
R'CO
R + MgClX
MgOHX+MgR X + CR' CN CR
N
R'
MgX
R'CO
R
4.3.5.5. Phản ứng đi từ muối axit hữu cơ (phương pháp Piria)
(RCOO)2Ca t0 caoR'C
OR + CaCO3

Hóa hữu cơ 2
57
OCR
C OCa
O
O
t0 cao
Có thể nhiệt phân RCOOH (phương pháp Sabachie).
2RCOOHMnO2
300-4000CRC
OR + CO2 + H2O
4.3. Tính chất hóa học 4.3.1. Phản ứng cộng AN điển hình 4.3.1.1. Cơ chế phản ứng cộng AN điển hình
R'
C OR-+
+ H Y-+
R'C
OHR
Y
HY là: H – OH, H – O – C2H5, H – CN, H – SO3Na XY là: R – MgX, – CH = CH – Na
R'
C OR-+
+ H Y-+
R'
C
O
R Y
R'
C
OH
R Y
4.3.1.2. Các yếu tố ảnh hưởng đến cộng AN Gốc R càng nhỏ và rút điện tử càng mạnh thì AN càng tốt. Hệ quả: - R – CHO không vòng thì H – CHO tốt nhất rồi đến mạch C nhỏ hơn. - R – CHO có vòng và R – CHO không vòng thì R – CHO không vòng tốt hơn. - xeton kém thua R – CHO. - xeton thấp tốt hơn xeton cao. 4.3.1.3. Hóa học lập thể của cộng AN
Có cấu trúc phẳng nên phản ứng cộng AN có tính đặc thù. Tuy nhiên, nếu nhóm liên kết với một trung tâm bất đối xứng, lúc bấy giờ, phản ứng cộng sẽ tạo ra hai đồng phân quang học không đối quang (Treo
và eritro). Để dự đoán được đồng phân nào ưu thế ta dùng quy tắc Cram: - Nếu 3 nhóm thế ở nguyên tử C bất đối đính với C=O có kích thước khác
C O

Hóa hữu cơ 2
58
nhau. Kí hiệu L: lớn, N: nhỏ, Tb: trung bình thì: tác nhân nucleophin sẽ tấn công vào nhóm cacbonyl từ phía ít bị án ngữ không gian nhất.
L
Tb N
HYR
O
RY
Tb N
L
OH
Ví dụ:
C6H5CH3
HY C6H5
CH3
O
CH3
C6H5
CH3OH
H(1) Ar - Mg - X
(2) H3O+
H
OH
C6H5 CH3
H
CH3C6H5
4.3.1.4. Các phản ứng cộng AN điển hình a. Cộng với H2O
+OC HOH OHC
OH b. Cộng với R’ – OH
+R'CO
R (K) OHR1 OC
OH
R
R'
R1 OC
O
R
R'
R1
R1Semiaxetal Axetal
c. Cộng HCN
+
O
C HCN CNC
OH d. Cộng NaHSO3
R C
O
C
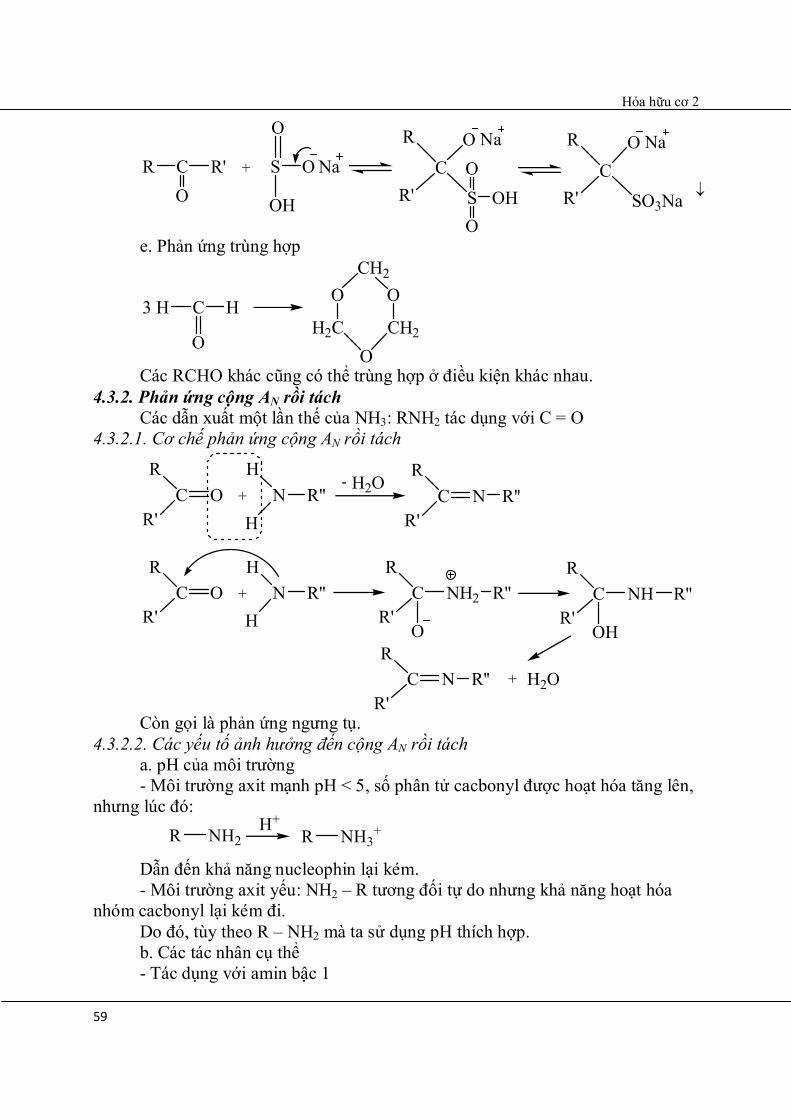
Hóa hữu cơ 2
59
+R'CO
R OSO
C
S
R
R'
O
OH
NaNa
O
OOH
OC
SO3Na
R
R'
Na
e. Phản ứng trùng hợp
O
C H3 HO
H2CO
CH2
OCH2
Các RCHO khác cũng có thể trùng hợp ở điều kiện khác nhau.
4.3.2. Phản ứng cộng AN rồi tách Các dẫn xuất một lần thế của NH3: RNH2 tác dụng với C = O 4.3.2.1. Cơ chế phản ứng cộng AN rồi tách
OCR'
R+ R''NH
HH2O-
NCR'
RR''
+ H2O
OCR'
R+ R''NH
HNH2C
R'
RR''
O
NHCR'
RR''
OH
NCR'
RR''
Còn gọi là phản ứng ngưng tụ. 4.3.2.2. Các yếu tố ảnh hưởng đến cộng AN rồi tách
a. pH của môi trường - Môi trường axit mạnh pH < 5, số phân tử cacbonyl được hoạt hóa tăng lên,
nhưng lúc đó: Dẫn đến khả năng nucleophin lại kém. - Môi trường axit yếu: NH2 – R tương đối tự do nhưng khả năng hoạt hóa
nhóm cacbonyl lại kém đi. Do đó, tùy theo R – NH2 mà ta sử dụng pH thích hợp. b. Các tác nhân cụ thể - Tác dụng với amin bậc 1
R NH2H+
R NH3+

Hóa hữu cơ 2
60
Dễ bị trime hóa (nếu R thơm không có g/đ này
+ H2O+C HO
H2N CH N
Bazơ Ship R – CH = N – R’ thường kém bền dễ thủy phân trong môi trường axit cho ra anđehyt và amin tương ứng. Tuy nhiên Ar – CH = N – R’ lại bền.
- Tác dụng với Hyđroxy Amin H2N – OH
OCR'
R+ + H2OOHH2N NCR OH
R'Andoxim (xetoxim)
- Tác dụng với Hyđrazin
OCR'
R+ + H2ONH2H2N NC
RNH2
R' Hydrazon
NCR
R'N C
R
R'
OCR'R
Azin Đối với xeton việc tạo ra xetozin khó khăn hơn. Do đó, đối với xeton thường
dừng lại ở Hyđrazon. Các hyđrazon và azin rắn kết tinh dễ bị thủy phân trong môi trường axit tạo ra lại nhóm cacbonyl, do vậy dùng phản ứng với hyđrazin để nhận biết nhóm cacbonyl.
Với các dẫn xuất hyđrazin là C6H5 – NH – NH2, p-nitro phenyl hyđrazin, 2,4-đinitrophenyl hyđrazin, tạo ra các dẫn xuất Hyđrazon tương ứng có các tính chất: rắn kết tinh, dễ bị thủy phân trong môi trường axit cho ra cacbonyl ban đầu là phản ứng đặc trưng để nhận biết nhóm cacbonyl.
- Tác dụng với NH3
CHOR + NH3 CHROH
NH2
NHCHR
CH
HNCH
NH
CHNH
R
R
R
Đặc biệt, HCHO tác dụng với NH3 cho ra urotropin (C6H12N4)

Hóa hữu cơ 2
61
Hexogen (xiclonit): chất nổ mạnh, dùng trong thế chiến thứ II
6 HCHO + 4 NH3 → C6H12N4 + 6 H2O
NH2C CH2
CH2
NN
N
CH2CH2
CH2
3HNO3
N N
N
NO2
NO2
O2N+ 3HCHO + NH3
- Tác dụng với PCl5
OCR'
R+ P
ClCl
Cl
Cl
Cl
ClC
OPCl4
R
R'
ClC
Cl
R
R'
+ POCl3
4.3.2.3. Phản ứng với các hợp chất có nhóm – CH2 – linh động
- Những hợp chất có Hα linh động thì hiệu ứng – C
C
H
H
- C
- Phản ứng này được tiến hành như sau a. Phản ứng andol hóa Phản ứng chủ yếu xảy ra với R – CHO
OC +-+
C
H
H
C
O
H C C CHO
OH
Ví dụ: CH3CHO + H CH2 CHO H3C CH CH2 CHO
OH b. Sản phẩm andol sinh ra nếu bị tách nước (phản ứng Croton hóa)
H3C CH CH CHO
OH H
- H2O H3C CH CH CHO
c. Hợp chất crotonic tiếp tục tác dụng với hợp phần metylen (phản ứng
Maicơn)
Anđehyt crotonic

Hóa hữu cơ 2
62
Cơ chế E1cb Cb: cacbanion
H3C CH CH CHO + H C C
H
H
O
H H3C CH CH2 CHOCH2 CHO
- Cơ chế phản ứng: phản ứng có thể xúc tác bằng H+ hay OH- a. Xúc tác bằng OH-
OH- + H C C
H
H
O
H (Ar) CH2 C
O
H (Ar) + H2O CH2 C
O
H (Ar)-
-
R CO
H
CH CH2 C
O
H (Ar)R
O
H2O CH CH2 C
O
HR
OH b. Xúc tác bằng H+
+R C
O
H H+ R CH OH
Nếu là xeton: R C
O
R' + H+ R C
OH
R' R C
OH
R' + H+
Enol sẽ cộng vào anđehyt đã được hoạt hóa, phản ứng cộng bị tách H+ thành β – hyđroxyxeton
+Ar CH OH CH2 C
OH
R' Ar CH
OH
CH2 C R'
OH
Ar CH
OH
CH2 C R'
O
+ H+
β – hyđroxyxeton sinh ra bị tách nước:
+ OH-Ar CH
OH
CH C Ar
OH
Ar CH
OH
CH2 C Ar
O
Ar CH CH C Ar
O
+ OH-

Hóa hữu cơ 2
63
Trong môi trường axit, β – hyđroxyxeton bị enol hóa cho ra sản phẩm cuối cùng.
+ H+Ar CH
OH
CH C Ar
OH
Ar CH
OH
CH C Ar
OH
Ar CH CH C Ar
OH
H
OH2
- H2OAr CH CH C Ar
O Các phản ứng xảy ra giữa nhóm cacbonyl và hợp phần metylen: - anđehyt với RCHO: tất nhiên phải có một chất có Hα. - RCHO + xeton: xeton có hợp phần metylen linh động. - xeton + xeton: nói chung xeton kém vì lí do: Hα kém linh động và bị án
ngữ không gian. - RCHO và dẫn xuất RCOOH. Phản ứng Peckin - Benzanđehyt hay anđehyt thơm khác nhất là có nhóm hút electron gắn vào
vòng benzen, khi cho tác dụng với anđehyt axit, xúc tác CH3COONa ta thu được α,β-không no.
+C6H5 CHOCH3 C
O
OCO
CH3
CH3COONaC6H5 CH CH COOH
Axit xinamic
CH3COONa: vai trò xúc tác bazơ. - Cơ chế phản ứng
H+
- H2O
+CH3COO- H CH2 CO O CH3 CH2 CO O CH3 + CH3COOH
CO
H
CHO
CH2 CO O CH3
CHOH
CH2 C O COCH3
OCH CH C O COCH3
O
H2O
CH CH COOH
Chú ý: Anđehyt dãy béo không tham gia vào phản ứng Peckin. Phản ứng Knuevenagen Anđehyt thơm và béo khi có xúc tác bazơ tham gia Hα của axit malonic và
các hợp chất có nhóm metylen khác như: CH3 – CN, CH3 – NO2...ngưng tụ croton.

Hóa hữu cơ 2
64
+CO
HCOOH
CH2
COOH - H2O CH C
COOH
- CO2
CH CH COOH
COOH
Phản ứng benzoin hóa Ar – CHO và thơm khác khi có mặt CN- tham gia phản ứng tương tự andol
về hình thức.
+ CN-Ar C
O
H ArC
O
H
Ar C
OH
C
O
Ar
Cơ chế:
CN-+CO
H C H
C6H5 C
OH
CH
O
C6H5
O
C N
C
OH
C N
CO
H
CN
C6H5 C
O
CH
OH
C6H5
CNC6H5 C
O
CH
OH
C6H5
4.3.2.4. Phản ứng khử hóa và oxi hóa 4.3.2.4.1. Phản ứng khử tạo andol
+ H2
RC O
RCH OH
xt: Ni/Pd
Mức độ phản ứng kém hơn vào nối đôi C = C

Hóa hữu cơ 2
65
+ H2C C CH2 CH2xt: Ni/Pd
H = -30Kcal/mol
C O + H2 CH OH H = -12Kcal/mol
Chất khử tốt là LiAlH4, AlH4- ≡ H-AlH3
Cơ chế:
C O + LiAlH4+ -
C O
H
Li + AlH3
+ H3O+
C OH
H
+ Li
4.3.2.4.2. Phản ứng khử tạo hyđrocacbon
C O + [H] CH2 + ......
a. Phương pháp Clemensen
C O CH2Zn + Hg + HCl
b. Phương pháp Kizne – Vonfo Được dùng khi gốc R không bền trong môi trường axit.
C O CH2N NH2 N NH2C2H5ONa CH2 + N2
4.3.2.4.3. Phản ứng oxi hóa tạo – COOH a. Tác nhân oxi hóa có thể là: - Thuốc thử Tolen (AgNO3 + NH3 dư) - Thuốc thử Felinh (phức Cu2+ với ion tactrat) - Thuốc thử Beneđic (phức Cu2+ với ion citrat) - Dung dịch K2Cr2O7 - Dung dịch KMnO4 b. Cơ chế oxi hóa bằng KMnO4
KMnO4 +H+H+ MnO4
-

Hóa hữu cơ 2
66
Phản ứng Baiơ – Viligiơ
Với HMnO4
+ H+ MnO4-R C
O
H + R C OMnO3
OH
HR C OH
O
+ HMnO3
c. Cơ chế oxi hóa bằng H2CrO4
+ H+ HCrO4-R C
O
H + R C OCrO3H
OH
HR C OH
O
+ HCrO3
Với xeton chỉ bị oxi hóa khi đun nóng mạnh với các chất oxi hóa rất mạnh, lúc đó:
+CH2 CR CH2
OR
(1)(2)(1)
(2)
CH2R COOH R' COOH
+CH2R' COOH R COOH
Nếu chất oxi hóa là peraxit thì:
R CO
R'R1 COOOH
R CO
O R' hay R' CO
O R
c. Phản ứng oxi hóa khử dị hóa Phản ứng Cannizaro:
OH-Ar C
O
H + Ar C
O
H Ar COO- + Ar CH2 OH
Cơ chế:
OH-+CO
H C O
H
OH
C6H5 CO
H
Ar COO- + Ar CH2 OHAr C
O
OH + Ar CH2 O
4.3.2.4.4. Phản ứng ở gốc R
a. Halogen hóa ở Hα

Hóa hữu cơ 2
67
+HC
H
H
C
O
H X2 (Cl2 >Br2 > I2) C CHO
X
+ HX
Qúa trình enol hóa xúc tác bằng H+ hay OH-. Nếu dùng môi trường OH- R C
OCH3 + 3X2 + 3NaOH R C
OCX3 + 3NaX + 3H2O
NaOH
R CO
ONa + CHX3
Trong thực tế: - Halogenfoc: CHCl3, CHBr3, CHI3 được điều chế từ axeton, ancol etylic - CHI3 kết tủa vàng sáng, ít tan trong nước, có mùi đặc trưng dùng nhận biết nhóm cacbonyl có dạng: R C
OCH3
b. Phản ứng thế SE ở nhân thơm
+CO
H H2SO4
CHO
SO3H 4.4. Giới thiệu các hợp chất điển hình 4.4.1. HCHO HCHO có lượng nhỏ khi đốt cháy không hoàn toàn nhiều chất hữu cơ như gỗ, đường, than, hyđrocacbon...Vì vậy trong khói bếp có một ít HCHO làm cho khói có tính sát trùng. Dung dịch 40% HCHO trong nước gọi là focmon hay fomalin. HCHO là chất khí, có mùi xốc. Do tính độc đối với vi khuẩn, do đó HCHO làm chất sát trùng, tẩy uế. HCHO làm biến tính protit, biến protit thành chất đàn hồi nên được dùng trong việc chuẩn bị các mẫu giải phẩu. HCHO còn được dùng trong ngành công nghiệp nhựa tổng hợp (nhựa Bakelit, nhựa cacbanoit, ure fomanđehyt), dùng trong công nghiệp tổng hợp phẩm nhuộm. 4.4.2. CH3CHO CH3CHO được điều chế từ axetylen, etylen và ancol etylic. Là nguyên liệu để điều chế CH3COOH, etyl axetat.

Hóa hữu cơ 2
68
Những R – CHO béo có Hα không tham gia phản ứng Cannizaro vì trong dung dịch OH- phản ứng andol chiếm ưu thế, sẽ tham gia phản ứng oxi hóa-khử lẫn nhau tạo thành este nếu có xúc tác nhôm ancolat hoặc Titan ancolat (tức là không phải bazơ mạnh). 4.4.3. Axeton Điều chế trong công nghiệp: - Cất khan gỗ - Nhiệt phân Canxi axetat - Đề hydro hóa ancol bậc 2 tương ứng - hyđrat hóa propylen trong công nghiệp dầu mỏ - Oxi hóa Cumen - Lên men gluxit nhờ vi khuẩn Baccilus acetobutylicus Axeton là nguyên liệu quan trọng tổng hợp các chất hữu cơ như clorofoc, iodofoc, metyl metacrylic... 4.5. Hợp chất Policacbonyl 4.5.1. Hợp chất đicacbonyl 4.5.1.1. Hợp chất 1,2-đicacbonyl a. Điều chế - Oxi hóa nhóm – CH2 – của monocacbonyl
CH3 CHO
+ SeO2 OHC CHO + H2O + SeH3C C
OCH3 + SeO2 H3C C
OCHO
+ H2O + Se
H3C C
OCH2 CH3 + SeO2 H3C C
OC CH3
O+ H2O + Se
- Oxim hóa nhóm α-metylen của monocacbonyl sau đó thủy phân
H3C CO
CH2 H + HO N O H3C CO
CH2 N O
H3C CO
CH N OH + H2O H3C CO
C HO
NH2 OH+
- Oxi hóa hay đehyđro hóa α-glycol hoặc α-hyđroxyxeton
H2C CH2
OHOH
Cu, t0 OHC CHO + 2H2
H3C CHOH
C HO
Cu, t0 H3C CO
C HO
+ H2
b. Tính chất hóa học

Hóa hữu cơ 2
69
Do ảnh hưởng của hai nhóm cacbonyl kề nhau nên hợp chất 1,2-đicacbonyl phản ứng rất cao đặc biệt OHC – CHO (glioxal).
Trừ các hợp chất đicacbonyl thơm như điphenylglioxal, còn lại các hợp chất 1,2-đicacbonyl tham gia các phản ứng như monocacboxyl (cộng), andol hóa, ngưng tụ...
Ngoài ra chúng còn có các phản ứng riêng đặc trưng cho hai nhóm cacbonyl liền nhau.
- Phản ứng với H2O2 làm đứt liên kết giữa hai nhóm – C = O
H3C CO
C CH3
O
+ HOOH 2H3C CO
OH
- Phản ứng khử một nhóm cacbonyl tạo thành hyđroxyxeton
C6H5 CO
C C6H5
O
+[H] C6H5 CHOH
- Phản ứng Cannizaro nội phân tử tạo thành α-hyđroxyaxit, phản ứng này chỉ xảy ra đối với đianđehyt hay anđehyt xeton.
CH3 CHO + H2O CH2OH COOHOH-
H3C CO
C HO
+ H2OOH-
H3C CHOH
C OHO
- Phản ứng ngưng tụ Croton thành quinon.
H3C
CH3
O
O
CH3
H3C
O OOH-
H3C
HO OH
CH3O
O
- H2OH3C
CH3O
O
Andol 2,5-dimetylbenzoquinon - Chuyển vị benzyl: khi đun nóng điphenylglioxal trong môi trường kiềm sẽ
chuyển vị một nhóm C6H5 – tạo thành axit α-hiđroxi điphenylaxetic (axit benzilic) Cơ chế:
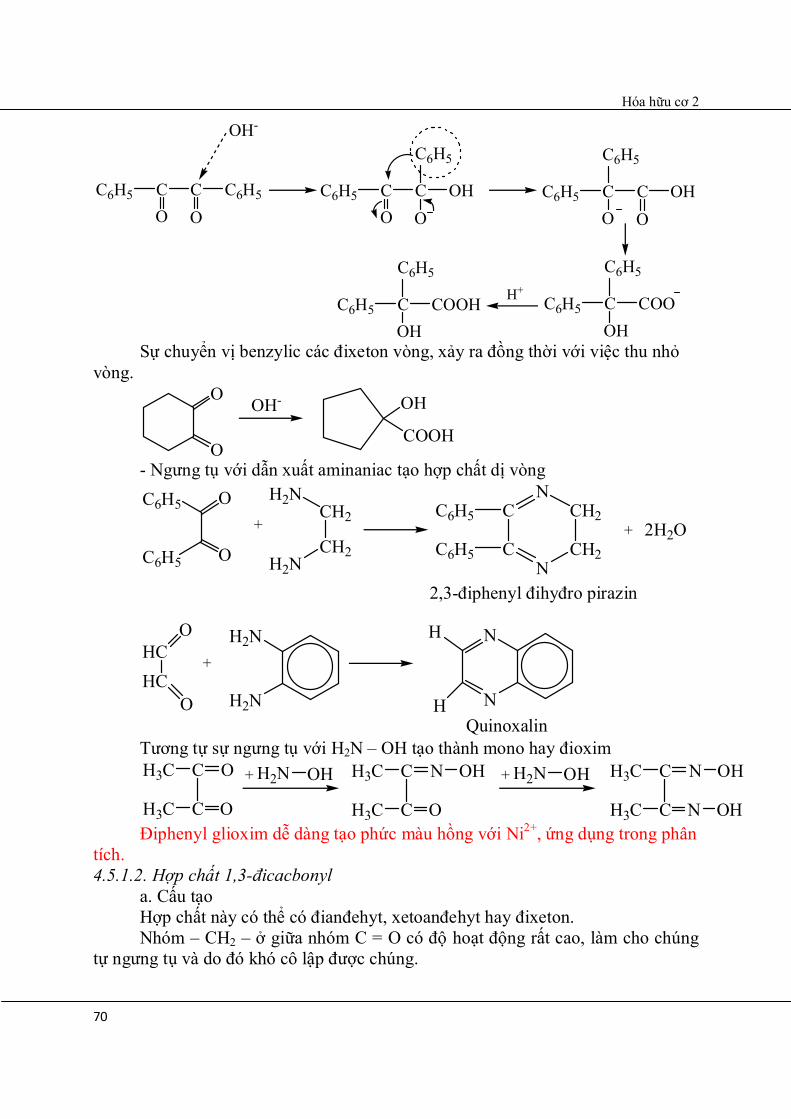
Hóa hữu cơ 2
70
2,3-điphenyl đihyđro pirazin
C6H5 CO
C C6H5
O
OH-
C6H5 CO
C OHO
C6H5
C6H5 CO
C OHO
C6H5
C6H5 COH
COO
C6H5
H+C6H5 C
OHCOOH
C6H5
Sự chuyển vị benzylic các đixeton vòng, xảy ra đồng thời với việc thu nhỏ
vòng. O
O
OH- OH
COOH
- Ngưng tụ với dẫn xuất aminaniac tạo hợp chất dị vòng
+ 2H2OC6H5
C6H5 O
O
+
H2NCH2
CH2H2N
C
CN
CH2
CH2
NC6H5
C6H5
HCHC
O
O
+
H2N
H2N
N
N
H
HQuinoxalin
Tương tự sự ngưng tụ với H2N – OH tạo thành mono hay đioxim
C
C O
O
+H3C
H3C
H2N OH
C
C N
O
H3C
H3C
OH + H2N OH
C
C N
N
H3C
H3C
OH
OH Điphenyl glioxim dễ dàng tạo phức màu hồng với Ni2+, ứng dụng trong phân tích. 4.5.1.2. Hợp chất 1,3-đicacbonyl a. Cấu tạo Hợp chất này có thể có đianđehyt, xetoanđehyt hay đixeton. Nhóm – CH2 – ở giữa nhóm C = O có độ hoạt động rất cao, làm cho chúng tự ngưng tụ và do đó khó cô lập được chúng.

Hóa hữu cơ 2
71
Sản phẩm O-ankyl
Sản phẩm C-ankyl
CH3 C
O
CH2 C CH3
O
CH3 C
OH
CH C CH3
Oenol 85%
Trong khi đó:
CH3 C
O
CH3
2,5x10-4%
CH3 C
OH
CH2
Vì dạng enol của axetylaxeton xuất hiện sự liên hợp và có liên kết hyđro nội phân tử.
C
OH
O
CC
H
CH3H3C
hayC
OH
O
CC
H
CH3H3C
b. Tính chất hóa học Nhờ sự tồn tại đồng thời của hai dạng enol và axetylaxeton nên tính chất hóa
học cũng rất phong phú và đặc trưng. - Phản ứng ankyl hóa α) Tác dụng được với kim loại kiềm CH3 C
OH
CH C CH3
O
+ Na CH3 C
ONa
CH C CH3
O
+ 1/2H2
β) Các enolat kiềm tác dụng với R – X CH3 C
O
CH C CH3
O Na+
+ R X CH3 C
O
CH C CH3
O R
+ NaX
CH3 C
O
CH C CH3
O Na+
+ R X CH3 C
O
CH C CH3
O
R
Thật ra: enolat Ag thì dễ tạo ra O-ankyl hơn Còn enolat kiềm thì dễ tạo ra C-ankyl. - Phản ứng ngưng tụ với hợp chất N tạo thành dị vòng
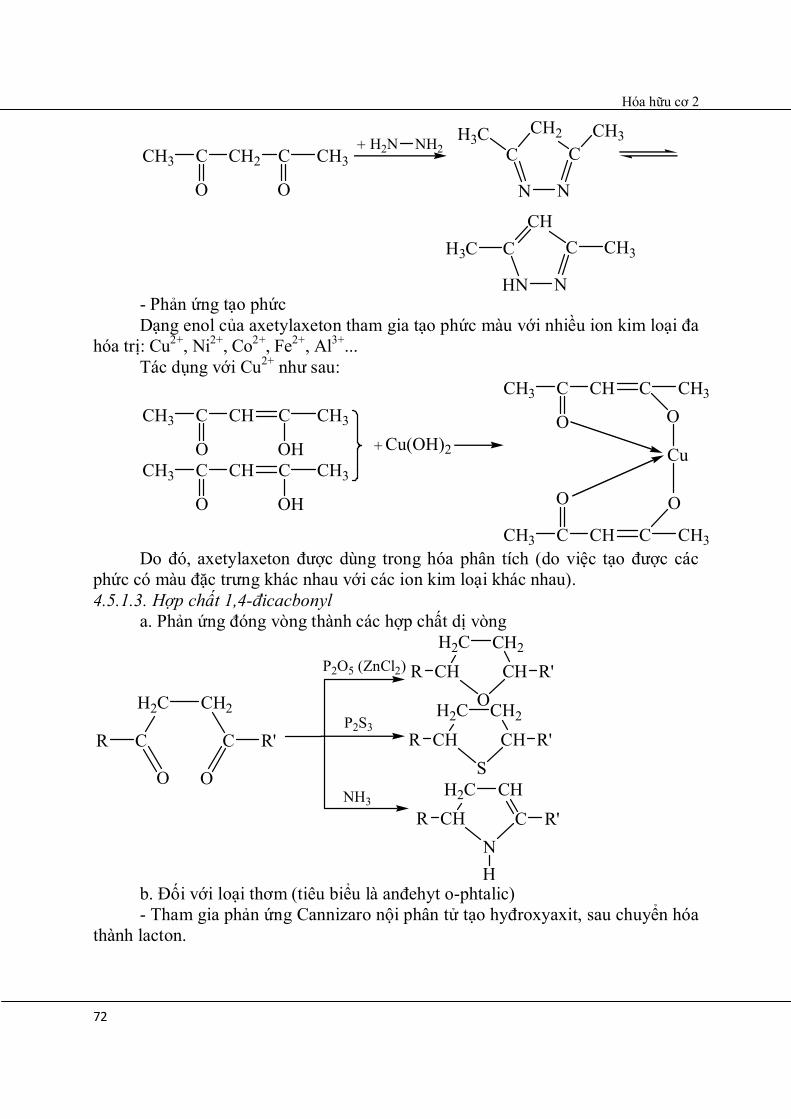
Hóa hữu cơ 2
72
CH3 C
O
CH2 C CH3
O
+ H2N NH2
N N
CCH2
CCH3H3C
HN N
CCH
C CH3H3C
- Phản ứng tạo phức Dạng enol của axetylaxeton tham gia tạo phức màu với nhiều ion kim loại đa
hóa trị: Cu2+, Ni2+, Co2+, Fe2+, Al3+... Tác dụng với Cu2+ như sau:
CH3 C
O
CH C CH3
OHCH3 C
O
CH C CH3
OH
+ Cu(OH)2
CH3 C
O
CH C CH3
O
Cu
CH3 C
O
CH C CH3
O
Do đó, axetylaxeton được dùng trong hóa phân tích (do việc tạo được các phức có màu đặc trưng khác nhau với các ion kim loại khác nhau). 4.5.1.3. Hợp chất 1,4-đicacbonyl a. Phản ứng đóng vòng thành các hợp chất dị vòng
O O
C
CH2H2C
CR R'
P2O5 (ZnCl2)
P2S3
NH3
OCH
CH2H2CCHR R'
SCH
CH2H2CCHR R'
NC
CHH2CCHR R'
H b. Đối với loại thơm (tiêu biểu là anđehyt o-phtalic) - Tham gia phản ứng Cannizaro nội phân tử tạo hyđroxyaxit, sau chuyển hóa thành lacton.
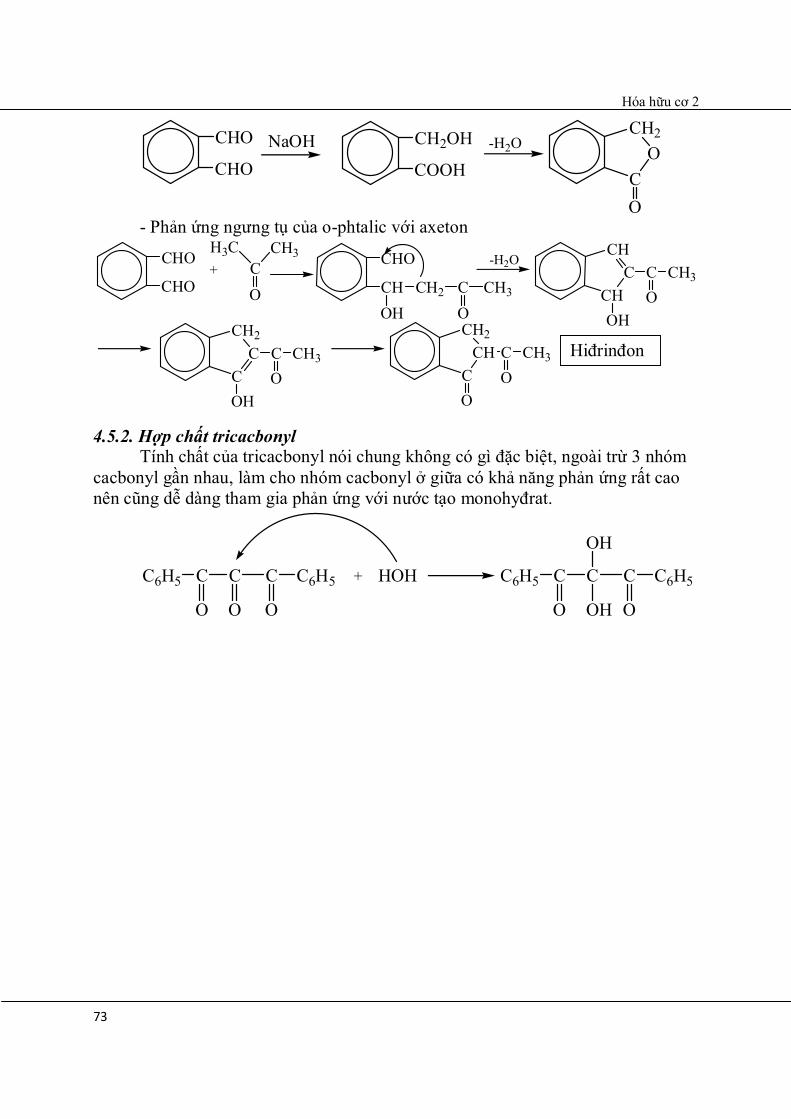
Hóa hữu cơ 2
73
CHO
CHONaOH CH2OH
COOH-H2O
CO
CH2
O - Phản ứng ngưng tụ của o-phtalic với axeton
CHO
CHO+
OC
CH3H3CCHO
CH CH2
OHCO
CH3
-H2O
CHC
CH
OH
CO
CH3
CC
CH2
OH
CO
CH3C
CHCH2
O
CO
CH3
4.5.2. Hợp chất tricacbonyl Tính chất của tricacbonyl nói chung không có gì đặc biệt, ngoài trừ 3 nhóm cacbonyl gần nhau, làm cho nhóm cacbonyl ở giữa có khả năng phản ứng rất cao nên cũng dễ dàng tham gia phản ứng với nước tạo monohyđrat.
C6H5 C
O
C C C6H5
O
+
O
HOH C6H5 C
O
C C C6H5
OOH
OH
Hiđrinđon

Hóa hữu cơ 2
74
Axit thơm
CHƯƠNG V: AXIT CACBOXYLIC
5.1. Cấu tạo, phân loại, danh pháp 5.1.1. Cấu tạo Là những hợp chất hữu cơ có nhóm – COOH đính vào gốc hyđrocacbon. 5.1.2. Phân loại - Theo số nhóm chức -COOH: axit monocacbonxylic và axit đicacboxylic - Theo bản chất gốc R: axit cacboxylic no và axit cacboxylic thơm 5.1.3. Danh pháp 5.1.3.1. Tên IUPAC Axit + tên hyđrocacbon tương ứng(kể cả C của axit) + oic Ví dụ: HCOOH: axit metanoic
CH3 CHCH3
CH2 COOH axit 3-metylbutanoic1234
5.1.3.2. Tên gọi khác Đối với gốc R quá phức tạp, trong đó, axit cacboxylic được gọi là dẫn xuất của thế nguyên tử Hyđro của hyđrocacbon bằng nhóm cacboxyl. Axit + tên hyđrocacbon tương ứng + cacboxylic Ví dụ: CH3COOH: axit metancacboxylic 5.1.3.3. Tên thông thường Tên thông thường liên quan đến nguồn gốc tìm ra axit. Ví dụ: axit fomic, axit axetic, axit stearic. 5.1.3.4. Tên hợp lý Lấy CH3COOH làm chuẩn, những axit khác thế Hα bằng gốc R. C6H5 – CH2 – COOH: axit phenylaxetic 5.2. Điều chế 5.2.1. Oxi hóa các hợp chất hữu cơ có bậc oxi hóa thấp hơn ở Cacbon - Ankan:
Ankan O2, xt: Mn Axit - Anken bị oxi hóa mạnh tai liên kết C=C tạo ra 2 axit
CnH2n+1
[O]
- Ancol bậc 1 hay anđehyt khi bị oxi hóa cho ra axit cacboxylic tương ứng (giữ nguyên mạch) - Ancol bậc 2 hay xeton khó bị oxi hóa, đứt mạch cacbon cho ra axit cacboxylic.

Hóa hữu cơ 2
75
Các dẫn xuất axit
5.2.2. Thủy phân các dẫn xuất axit và dẫn xuất axit octofomic
R CO
X
R CO
O C R'O
R CO
O R'
R CO
NH2
R C N
+ HOH R CO
OHxt: H+
OH-
Các dẫn xuất của axit octofomic là:
R CX3
R C
OR'
OR'OR'
R CX
XX
Thủy phân các dẫn xuất này, ví dụ:
R C
OR'
OR'OR'
+ 3HOH R C
OH
OHOH
+ 3R'OH
- HOH
R CO
OH
5.2.3. Đưa thêm nhóm cacboxyl vào phân tử chất hữu cơ 5.2.3.1. Cacboxyl hóa cơ kim Đây là phương pháp quan trọng điều chế axit cacboxylic trong phòng thí nghiệm và đặc biệt điều chế axit có động vị C14 trong nhóm – COOH (dùng 14CO2).
R Mg X + CO2 R CO
OMgX dd HCl R CO
OH + MgX
Cl
5.2.3.2. Cacboxyl hóa ancolat kim loại
+NaOH CO 120-1300CP = 6-8atm
HCOONa

Hóa hữu cơ 2
76
+C2H5ONa CO C2H5COONa
5.2.3.2. Tổng hợp tăng mạch cacbon các axit monocacboxylic a. Dùng este của axit malonic (tổng hợp malonic)
CH2
COOC2H5
COOC2H5
Na hay C2H5ONaCH
COOC2H5
COOC2H5
NaR X
CHCOOC2H5
COOC2H5
R H+CH
COOH
COOHR
t0 cao CH2 COOHR + CO2
b. Phản ứng ngưng tụ este (phản ứng Claizen) Khi cho tác dụng Na hay RONa, 2 phân tử este có Hα linh động sẽ ngưng tụ nhau, gọi là ngưng tụ Claizen.
R C
O
O R' + H CH
R1
COOR''Na hay C2H5ONa
C CH
R1
COOR''R
O+ R'OH
H+
C CH
R1
COOHR
O Cơ chế:
R'O + CH2 C
O
O R''H R'OH + CH2 C
O
O R''
R CO
O R1
SN
CH2 C
O
O R''CO
O
RR1
CH2 C
O
O R''C
O
R
c. Cacboxyl hóa anken:

Hóa hữu cơ 2
77
+CH2 CH2(1)Ni(CO)4
(2)HOHCH2 C
O
OHH3CCO
d. Điazo hóa clorua axit – Tổng hợp Acnơ-Aixtơ
+ClCO
R 2CH2N2 CHN2CO
R + CH3Cl + N2
Ag2O, T>1000C- N2
CCHR O + N2HOHCH2 C
O
OHR
Cơ chế: Cl
CO
R + CH2 N N
C CH
H
NR
O
Cl
N C CH NR
O
N
CH2 N N HCl CH3 N N CH3Cl + N2
CH N N
CO
R Ag+, H2O
- N2
CH2 COOHR
Vì:
CH N N
C
O
R Ag+, H2O
- N2
C CHRO
C CHO R
C CHO R HOH C CHO R
OH 5.3. Tính chất vật lý Taọ liên kết hyđro liên phân tử mạnh hơn của rượu và dạng đime
R CO
O H
H
O
OC R
ΔHđime hóa = -14kcal/mol Bên cạnh dạng đime là dạng polyme

Hóa hữu cơ 2
78
R
COO
HR
COO
HR
COO
H Tan được trong nước do tạo được liên kết hyđro với nước, C1 → C3 tan gần như vô hạn trong nước. 5.4. Tính chất hóa học Nhận xét cấu tạo
C
O
O H
Có thể xảy ra các phản ứng sau: - Phản ứng đứt liên kết – O – H - Phản ứng đứt liên kết – C – OH - Phản ứng Đecacboxxyl hóa - Phản ứng ở gốc R 5.4.1. Phản ứng đứt liên kết – O – H Sự phân li trong nước RCOOH + HOH RCOO- + H3O+
Bảng so sánh tính axit các loại
Tính axit của các ancol
Tính axit của phenol Tính axit của axit cacboxylic aliphatic
Tính axit của các axit có vòng thơm
- Axit rất yếu:
ROH RO- + H+
Mức độ phân ly phụ thuộc vào bản chất của gốc hydrocacbon R.
- Độ phân ly của ancol no như CH3OH, C2H5OH,..yếu hơn nước. Nguyên nhân là ancol có nhóm ankyl thể hiện hiệu ứng +I. Chính hiệu ứng này đã làm tăng
- Phenol cũng như enol (axit nhóm II) nên tính axit của phenol luôn luôn mạnh hơn ở ancol no và nước
- tiôphenol (C6H5SH) có tính axit mạnh hơn thiôancol (C2H5SH), C6H5OH < C6H5SH (?)
- Các nhóm thế khác nhau trong vòng benzen của phenol làm thay đổi tính axit một cách rõ rệt. Các nhóm hút electron làm tăng tính axit còn các nhóm đẩy electron làm giảm tính axit.
- Axit cacboxylic (thuộc nhóm II) là những axit hữu cơ điển hình, có đại diện đơn giản nhất là HCOOH. Khi thay thế nguyên tử H-C trong HCOOH bằng các gốc ankyl (thể hiện hiệu ứng +I), tính axit yếu đi rõ rệt.
Ví dụ: HCOOH (pKa = 3,75) > CH3COOH (pKa = 4,76).
Tuy nhiên nếu tăng thêm chiều dài mạch n-ankyl thì pKa thay đổi rất ít (nguyên nhân có thể là ảnh hưởng +I ở đây không phải là yếu tố duy nhất làm thay đổi tính axit của nhóm COOH mà còn những yếu tố khác như hiệu ứng siêu liên hợp, tương tác giữa nhóm cacboxyl với nguyên tử H nào đó trong một số
- Khi thay thế nguyên tử H trong HCOOH bằng gốc phenyl, tính axit giảm đi vì gốc phenyl một mặt thể hiện hiệu ứng -I, mặt khác còn thể hiện hiệu ứng +C.
- Khi thay thế nguyên tử H trong CH3COOH bằng gốc phenyl thì được axit phenylaxetic có tính axit mạnh hơn axit axetic vì gốc phenyl chủ yếu chỉ thể hiện hiệu ứng -I.
Ví dụ:
C6H5CH2 - CH2 - COOH < C6H5CH2COOH.
Chú ý: C6H5COOH >

Hóa hữu cơ 2
79
cường mật độ electron ở nguyên tử oxy trong ROH và làm giảm độ bền vững của anion RO- (so với OH-
).
- C2H5OH < C2H5SH (?)
- Khi thay thế nguyên tử H trong nhóm CH3 của metanol bằng những nhóm nguyên tử khác nhau sẽ làm thay đổi tính axit của ancol.
+ Các nhóm ankyl làm giảm tính axit (vì có hiệu ứng +I).
+ Halogen, metoxyl và các nhóm -I khác sẽ làm tăng tính axit.
- Số nhóm hydroxyl trong ancol tăng lên (polyol) thì tính axit cũng tăng tuy không nhiều.
- Chú ý: Tính axit của các enol. Enol thuộc nhóm axit II, trong đó ngoài sự phân cực cảm ứng trong phân tử còn có sự liên
- Các nhóm thế ở vị trí octô đối với nhóm OH gây ảnh hưởng không gian không lớn lắm so với ảnh hưởng như thế đối với NO2 hoặc COOH.
Ví dụ:
OH
NO2
OH
NO2
OH
NO2
H3C CH3
H3C CH3
pKa 7,16 8,24 7,21
- So sánh:
OH
NO2
OH
OCH3
OH
CH3
OH
NH2
> > >
- Xét về hiệu ứng điện tử thì para có ảnh hưởng mạnh hơn so với octô, meta. Nhưng xét về hiệu ứng không gian thì octô có ảnh hưởng mạnh hơn p-, m- (tuy nhiên ảnh hưởng không lớn).
gốc hydrocacbon,v.v...
- Thay thế các nguyên tử H trong axit axetic bằng các nhóm metyl rõ ràng có làm giảm tính axit vì không những hiệu ứng +I tăng lên mà cả hiệu ứng không gian án ngữ quá trình sonvat hoá anion cũng tăng.
- Thay thế H bằng những nhóm thế có ảnh hưởng -I thì tính axit tăng, càng nhiều nhóm thế -I, tính axit càng tăng nhanh.
Ví dụ:
a, O2N - CH2 - COOH > CH3SO2 - CH2 - COOH > CH3O - CH2 - COOH > HO - CH2 - COOH.
b, Cl3C - COOH > F - CH2 - COOH > Cl - CH2 - COOH > Br - CH2 - COOH.
- Các nhóm thế ở những vị trí càng ở xa vị trí gây ảnh hưởng càng yếu do sự tắt nhanh hiệu ứng khi mạch liên kết kéo dài.
Ví dụ:
F3C - COOH > F3C - CH2COOH > F3C - CH2 - CH2COOH.
- Những axit cacboxylic chưa no nói chung có tính axit mạnh hơn axit no tương ứng vì các nhóm C=C có hiệu ứng -I. Vị trí của nối đôi trong gốc hydrocacbon có khi cũng gây ảnh hưởng lớn đến tính axit của nhóm COOH. Nhóm C=C càng ở gần nhóm COOH thì tính axit càng tăng, trừ trường hợp nối đôi ở vị trí
, là vị trí thuận lợi cho sự liên hợp giữa nhóm C=C với nhóm C=O. Hiệu ứng này (+C) gây ảnh hưởng ngược lại hiệu ứng -I. Do đó axit cacboxylic , - chưa no
C6H5CH2COOH? (theo lý thuyết về hiệu ứng là ngược lại:
C O
O H-C
+C-I
+C
CH2
C
O
O H
-C
+C
-I
<
- Khi đưa các nhóm thế vào gốc phenyl trong phân tử các axit trên, tính axit sẽ biến đổi tuỳ theo bản chất và vị trí của nhóm thế trong nhân thơm và về mức độ ảnh hưởng của nhóm thế còn phụ thuộc cả vào cấu tạo của mắt xích trung gian giữa gốc phenyl và nhóm cacboxyl.
- Ảnh hưởng của các nhóm thế ở vị trí mêta và para:
+ Ankyl: luôn làm giảm tính axit.
+ Các nhóm metoxy và hydroxy ở vị trí meta (-I) nên làm tăng tính axit, nhưng ở vị trí para chúng có thể phát huy đầy đủ ảnh hưởng +C (cùng với -I) do đó độ phân ly của axit giảm.
+ Các halogen ở bất kỳ vị trí nào cũng có hiệu ứng -I mạnh hơn +C, nhất là ở mêta nên chúng luôn làm tăng độ phân ly của axit, khi ấy đồng phân meta phân ly mạnh hơn đồng phân para.
+ Nhóm NO2- là nhóm
Hóa hữu cơ 2
80
hợp giữa cặp electron vơi electron n của nguyên tử O làm giảm mật độ electron của nguyên tử O. Chính vì vậy mà tính axit của enol cao hơn của ancol no, nhất là khi trong gốc R có nhóm cacboxyl liên hợp với nối đôi.
Ví dụ:
CH3 C
O
CH C
OH
không phải là axit mạnh nhất trong dãy các axit chưa no.
Ví dụ:
CH3 - CH=CH - CH2 - COOH > CH2=CH - CH2 - CH2 - COOH > CH3 - CH2 - CH = CH - COOH.
- Giữa hai đồng phân cis - trans của axit cacboxylic , - chưa no, đồng phân cis thường phân ly mạnh hơn. Nguyên nhân của hiện tượng khác biệt này là do những nhóm ankyl và aryl trong đồng phân cis ở gần nhóm COOH nên đã gây ảnh hưởng tương tự ảnh hưởng octô.
- Liên kết C≡C dù ở vị trí , cũng làm tăng mạnh tính axit của nhóm cacboxyl một cách đột ngột do nguyên tử C ở trạng thái lai hoá sp thể hiện hiệu ứng -I lớn hơn các nguyên tử C khác, mặt khác trong số hai liên kết của nối ba chỉ có một trực tiếp tham gia liên hợp với -CO-, còn một nữa vì có mặt phẳng thẳng góc với mặt phẳng của nhóm -CO- nên không có tương tác đó.
- Các axit đicacboxylic phân ly theo hai nấc, ở nấc thứ nhất có ảnh hưởng của nhóm COOH làm tăng tính axit, còn ở nấc thứ hai có ảnh hưởng của nhóm mang điện âm COO- gây tác dụng ngược lại. Trong đại đa số trường hợp các ảnh hưởng đó chủ yếu là ảnh hưởng cảm ứng ( I )
Ví dụ:
a, HOOC - COOH > HOOC - CH2 - COOH > HOOC - CH2 - CH2 - COOH
b, HOOC - CH2 - CH2 - COOH <
hút e- mạnh cả về -C và -I nên các axit có nhóm NO2 ở vị trí para bao giờ cũng phân ly mạnh hơn mêta và tất cả đều mạnh hơn các axit tương uớng có chứa halogen trong nhân thơm.
- Các nhóm thế ở vị trí octô gây ảnh hưởng rất đặc biệt đến tính axit của axit benzoic. Bất kể nhóm thế thuộc loại nào ít nhiều cũng đều làm tăng độ phân ly của axit do hiệu ứng octo. Khi kéo dài quãng cách giữa gốc phenyl với nhóm COOH bằng cách đưa thêm các nhóm trung gian như CH2, CH2 - CH2 vào thì hiệu ứng octô không còn rõ nữa.
- Xét về hiệu ứng điện tử thì para có ảnh hưởng mạnh hơn so với octo, meta. Nhưng xét về hiệu ứng không gian thì octô mạnh hơn p-, m-. (Nếu ion COO- có khả năng bền hoá bởi hiệu ứng o- như liên kết hidro thì làm tăng tính axit, nếu nhóm thế o- gây hiệu ứng không gian loại một thì làm tăng tính axit?)

Hóa hữu cơ 2
81
HOOC - CH=CH - COOH.
5.4.2. Các phản ứng cộng và thế của nhóm cacbonyl Phần lớn các phản ứng cộng vào nhóm cacbonyl rồi đứt liên kết – C – OH tạo ra các dẫn xuất sẽ học kĩ hơn ở phần điều chế dẫn xuất axit từ RCOOH. ở đây, chỉ đề cấp phản ứng cộng AN thuần túy vào nhóm cacbonyl. 5.4.2.1. Phản ứng cộng với HOH
R CO
O H H O H+ R COHOH
OHoctohydrat
5.4.2.2. Phản ứng khử hóa Không khử hóa bằng H2 và các chất khử thông thường khác mà bằng LiAlH4.
R COOH
(1) LiAlH4
(2) H+ R CH2OH
Cơ chế: Thực chất của phản ứng là phản ứng cộng AN của LiAlH4 +R COOH LiAlH4 R COOLi + AlH3 + H2
+
LiAlH4
R CO
O Li Li AlH3 .H- R CO
O Li
H
Li+ AlH3
HOH/H+
R CHOH
OH + 2LiOHR CHO + H2O
R CH2 O LiHOH/H+
R CH2 OH 5.4.2.3. Phản ứng đecacboxyl hóa R COOH CO2 + RH Thực tế người ta dùng:
R CO
O Na + NaOHCaO, t0cao
RHNa2CO3 +
Như vậy phản ứng đêcacboxyl hóa no không có nhóm thế hút electron khó xảy ra. Phản ứng càng dễ dàng nếu như vị trí α có những nhóm thế hút electron tốt như – NH2; - C≡N; -CO-.

Hóa hữu cơ 2
82
Ví dụ:
H3C CO
CH2 CO
OH
H3C CO
CH3 + CO2
5.4.2.4. Phản ứng ở gốc R 5.4.2.4.1. Phản ứng oxi hóa Nói chung nhóm – COOH trơ đối với tác nhân oxi hóa (trừ HCOOH) nhưng gốc ankyl có thể bị oxi hóa tạo thành axit oxocacboxylic.
R CH2 COOH + SeO2 R C COOH
O
+ Se + H2O
Trong quá trình sinh học, sự oxi hóa nhờ xúc tác men lại xảy ra ở cả vị trí β.
CH2 CH2 COOH + SeO2 + Se + H2OR
C CH2 COOHR
O
men
5.4.2.4.2. Phản ứng halogenua ở gốc ankyl Có thể theo cơ chế gốc hay ion tùy theo điều kiện phản ứng. Halogen hóa bằng xúc tác P (phương pháp Hen-Fonhac-Zelinxki), theo cơ chế thế sau: tạo ra axit α-halogencacboxylic. 2P + 3Br2 2PBr3 - Tạo dẫn xuất halogen axit R CH2 C
OOH + PBr3 R CH2 C
OBr + P
OBr + HBr
- Enol hóa, xúc tác H+
R CH CO
Br R CH COH
Br
HH+
- Enol tác dụng với Br2
R CH COH
Br+ Br2 R CH C
OBr
Br+ HBr
R CH C
OBr
Br+ R CH2 COOH R CH COOH
Br
R CH2 CO
Br+
Phản ứng clo hóa axit , điều kiện hν tuân theo cơ chế gốc tự do. Đối với clo chủ yếu chỉ có β và γ, một lượng nhỏ ở α.

Hóa hữu cơ 2
83
CH2 CH2 COOH + Cl2H3C
h
CH2 CH COOHH3CCl
5%
CH CH2 COOHH3CCl
64%
CH2 CH2 COOHH2C 31%Cl
5.4.2.4.3. Phản ứng halogenua ở nhân thơm Do nhóm – COOH hút e tốt nên khi gắn trực tiếp vào vòng benzen nên thế SE ở vị trí meta và khó hơn bình thường.
COOH
+ HNO3, H2SO4
COOH
NO2 Sự phản hoạt hóa nhân thơm do nhóm – COOH gây ra làm cho vòng benzen không tham gia phản ứng Friđenhaft. 5.5. Giới thiệu riêng 5.5.1. Axit fomic (HCOOH) Axit fomic có rải rác trong thiên nhiên: trong cơ thể động, thực vật. Vào thế kỷ 17, người ta tìm thấy trong loài kiến Fomicarufai. Có thể điều chế từ: - Thủy phân CHCl3 - Axit H-CN tác dụng với nước - oxi hóa CH3-OH - Nhiệt phân axit oxalic, xúc tác là glyxerin
CO + NaOH HCOONa H+HCOOH
Trong công nghiệp ứng dụng trong công nghiệp dêt, da và thực phẩm. 5.5.2. Axit axetic (CH3COOH) Được điều từ thời thượng cổ, trong dấm ăn (3 – 6% là CH3COOH), được điều chế đậm đặc mới từ 200 năm lại đây. Ban đầu đi từ chưng cất than gỗ cho ra hỗn hợp CH3COOH, CH3OH, CH3COCH3. Tiếp tục cho hỗn hợp tác dụng với Ca(OH)2 thu được muối canxi axetat, tác dụng với dung dịch axit cho ra axit axetic. Ngày nay, trong công nghiệp:
HC CH + H2OHg2+
CH3CHOO2, Mn2+
CH3COOH
n-butan O2, t0 = 1800C
p = 70atmCH3COOH

Hóa hữu cơ 2
84
Axit axetic nguyên chất là chất lỏng, mùi xốc, ts = +1180C, tđ đ = +160C ở dạng tinh thể như nước đá (gọi là axit axetic nước đá, hay axit axetic kết tinh). Axit axetic được ứng dụng làm dấm, tổng hợp các chất thơm, axeton, muối axetat. Axetat natri là chất háo nước, tác nhân ngưng tụ, sản xuất axit anhyđrit axetic. Axetat nhôm là chất cầm màu vì trong nước nó tạo ra muối bazơ tương tự như sắt axetat. 5.5.3. Các axit không no Là các axit có nối đôi, nối ba ở gốc R, phần lớn, các axit không no có chứa nối đôi ở vị trí liên hợp (vị trí α, β) đối với nhóm cacbonyl. 5.5.3. 1.Axit anylic CH2=CH-COOH Là chất lỏng, mùi xốc, ts = 1400C, được điều chế bằng cách: - Thủy phân anylonitrin: H2C CH CN + 2H2O H2C CH COOH + NH3 - Cho photgen tác dụng với etylen rồi sau đó thủy phân
H2C CH COOHH2C CH H + Cl C ClO
AlCl3 CH2 C ClO
CH2Cl + H2O
- Phương pháp Reppe
HC CH + CONi(CO)4 H2C CH COOH
5.5.3. 2.Axit metacrylic CH2=C(CH3)-COOH Là chất lỏng, ts = 1600C, được điều chế bằng cách: - Từ axeton
H3C C CH3O
+ HCN H3C C CH3OH
CN
-
H2O
H2C C COOHCH3
H3C C CH3OH
OH
H2O
+ H2C C COOHCH3
H3C CH COOHCH3
Br2 H3C C COOHCH3
Br- HBr

Hóa hữu cơ 2
85
CHƯƠNG VI: DẪN XUẤT CỦA AXIT CACBOXYLIC
6.1. Mở đầu Khi thay thế nhóm – OH của axit cacboxylic bằng những nhóm thế khác nhau ta dựa vào dẫn xuất ở nhóm chức của axit cacboxylic.
R C OH
OR C Z
O Tùy theo nhóm Z, ta có:
R C Halogen
OHalogenua axit
R C OO
C RO
R C OO
R' Este
R C NO
R'(H)Amit
R''(H)
R C NHO
HydrazitNH2
R C NHO
Axit hydroxamicOH
R C N3O
Azit
Dẫn xuất của axit còn có cả những sản phẩm thế nguyên tử O trong nhóm cacbonyl và nhóm – OH nữa. Ví du: Nitrin R – C ≡ N Trong phạm vi giáo trình này ta chỉ nghiên cứu các dẫn xuất thông dụng như: halogenua axit, anhyđrit axit, este, amit. 6.2. Tính chất chung Thế SN ở C không no nhóm cacbonyl. 6.2.1. Sơ đồ chung
ZR C YO
+ R C YO
ZYR C Z
O+
6.2.2. Đặc điểm - Tương tự như SN ở C no nhưng dễ dàng hơn vì:
Anhyđrit axit

Hóa hữu cơ 2
86
C O+
nên C dương hơn, cấu tạo phẳng không bị chướng ngại lập thể nhiều như SN ở C tứ diện. Giai đoạn đầu của phản ứng tạo ra trạng thái chuyển tiếp như cộng AN vào anđehyt hay xeton. Giai đoạn tiếp theo thay vì bền hóa sản phẩn bằng cách kết hợp proton lại xảy ra sự táchđi nhóm Y. - Phản ứng SN ở C = O có thể xúc tác bằng H+ hay OH-. + Trong môi trường axit: axit proton hóa nhóm cacbonyl làm giảm năng lượng hoạt hóa Ea của phản ứng.
R C YO
+ H+ R C Y
OH
ZR C Y
O
ZYR C Z
O+
+ Trong môi trường bazơ: chính OH-
là tác nhân nucleophin mạnh trực tiếp tấn công vào nhóm cacbonyl.
R C ZO
R C OHO
Z+ OH- OHR C Z
O+
- Khả năng phản ứng: R C Halogen
O> R Halogen
R C NH2
O> R NH2
R C OR'
O> R OR'
R C Halogen
O> (RCO)2O > R C OH
O> R C OR'
O> R C NH2
O6.3. Este 6.3.1. Định nghĩa Este là sản phẩm thế nhóm – OH của axit cacboxylic R – COOH bằng nhóm ankoxi: OR’ của ancol hay nhóm Ar-O của phenol. 6.3.2. Danh pháp 6.3.2.1. Tên IUPAC Tên gốc R của rượu hay phenol + tên gốc axit theo IUPAC Ví dụ: CH3COOCH3 : metyl etanoat 6.3.2.2. Tên thông thường Tên gốc R của rượu hay phenol + tên gốc axit theo tên thông thường Ví dụ: CH3COOCH3 : metyl axetat

Hóa hữu cơ 2
87
6.3.3. Điều chế 6.3.3.1. Từ axit RCOOH tác dụng với R’OH (ancol)
+ +R C OR'
OR COOH R' OH
H+
H2O
6.3.3.2. Từ clorua axit tác dụng với ArOH (hay ancol)
R C Cl
O+ R' OH +R C OR'
OHCl
6.3.3.3. Từ anhyđrit axit tác dụng với ArOH (hay ancol) Tương tự như trên 6.3.3.4. Từ axit tác dụng với điazometan
+R C OO
H CH2 N N
CH2 N N t0C CH2 + N2
R C OO
CH3 + N2
6.3.3.5. Từ xeten tác dụng với ArOH (hay ancol)
+CH2 C O R' O H H3C C OO
R'
6.3.3.6. Từ muối natri và muối bạc
RCOO-NaRCOO-Ag
+ R'I +R C OR' AgI
6.3.3.7. Từ hợp chất nitrin
+R C N R' O H R C ONH
R'
6.3.4. Tính chất vật lí Nói chung este không có liên kết hyđro nhưng phân tử phân cực nên ts < ts axit tương ứng, đều nhẹ hơn nước và ít tan trong nước. Các este có M nhỏ thường ở dạng lỏng và có mùi thơm trái cây, còn este có M lớn là chất rắn. 6.3.5. Tính chất hóa học 6.3.5.1. Phản ứng ở nhóm cacbonyl (thế SN) a. Thủy phân - Xúc tác H+
+R C O
OR' H+ R C O
OHR' hay R C O
OR'
H

Hóa hữu cơ 2
88
Biến thế Raxemic nếu SN1
+ R C OO
R'H
H OH
R C OO
R'H
OH2
R CO
OH2 + R'OH
R C OO
H + H+
+
-
R'OH+
H OH
R C OOH
R' R C OOH
R'OH2
R C OOH
R'OH
H
R'OH
R C
OH
OH
- H+R C OO
H H+ +
- Xúc tác OH-
R C OO
H R'OHO- + R C OO
R' R C OO
R'
OH+
R C OO
R'OH+
Phản ứng thủy phân este là lưỡng phân tử SN2, phân tử este bị phân cắt ở liên kết:
Thực nghiệm chứng minh bằng các dữ kiện sau: + Dùng phương pháp hóa lập thể
HO-+C6H5 C OO
CC2H5
CH3
H(1) (2)
(1)C6H5 C O
O (1)+ HO C
C2H5
CH3
H(2)(2)
C6H5 C OO
+ HO CC2H5
CH3
HOHC
C2H5H3CH+
R C O
O
R'

Hóa hữu cơ 2
89
p-metoxi benzhyđrylphtalat (quang hoạt)
p-metoxi benzhyđrol (raxemic)
Muốn xảy ra kiểu (1) thì rượu thu được là sec-butylic giữ nguyên cấu hình vì các liên kết với C* được giữ nguyên. Nếu xảy ra theo kiểu (2) thì ancol
thu được là sec-butylic thay đổi cấu hình hoàn toàn (nếu SN2) hoặc tạo ra biến thể raxemic (nếu SN1).
Thực nghiệm cho thấy, ancol thu được giữ nguyên cấu hình, tức là phân cắt theo kiểu (1). + Dùng phương pháp đồng vị đánh dấu
HO-CH2 C OO
C2H5H3C18
+ CH2 COOH3C + OHC2H518
Các yếu tố ảnh hưởng đến sự thủy phân este: - R của axit càng cồng kềnh, phản ứng càng kém - R của axit càng hút electron, phản ứng càng tốt Cơ chế SN2 ở trên phổ biến đối với este thông thường. Trong trường hợp thông thường, người ta còn gặp cơ chế liên kết ankyl-oxi đơn phân tử.
R C O
O
R'
Muốn thế cation R’+ sinh ra phải có độ bền cao. Ví dụ 1:
H O H+H3C O CH
OC
OC OH
O
H3C O CH
H O H
H3C O CH
OH Ví dụ 2:
CH3 C OO
C18CH3
CH3
CH3
H2O CCH3
H3CCH3
18H2O
CCH3
CH3
CH3
HO18
Qua 2 ví dụ trên chứng tỏ thủy phân (1) và (2) là đứt liên kết theo kiểu (2).
b. Phản ứng trao đổi este Khi đun nóng este với ancol, xúc tác H+ hay RONa sẽ xảy ra phản ứng trao đổi este.

Hóa hữu cơ 2
90
Ancol bậc 3
R C O
OR1
H++ OHR2 R C O
OR2 + OHR1
Cơ chế trao đổi este trong môi trường axit tương tự như cơ chế thủy phân
este trong môi trường axit, tương tự đối với môi trường kiềm. Tức là thế SN2 nhưng có điều phản ứng thuận nghịch và R2OH phải có tính nucleophin tốt thì cân bằng dễ xảy ra theo chiều thuận.
c. Phản ứng với NH3 và dẫn xuất của amoniac (cơ chế SN2) OHR'+ NH3 RCONH2 +
+ R'NH2 RCONHR OHR'+
+ R1NHR2 R CO
N R1
R2
+ H2N NH2 R CO
NH NH2 OHR'+
+ H2N OH R CO
NH OH OHR'+
R C OO
R'
d. Phản ứng với cơ kim tạo ra ancol bậc 3
OHR'+
R C OO
R' + R1 Mg X+ -
R C OOMgX
R'H2O
R C OOH
R'OH
+ MgOHX
R C R1O
e. Phản ứng khử hóa
+
RCH2 OH
R C OO
R' Li AlH3 .H- R CO
OR'
H
Li
R C HO
LiAlH4
+ R'OH
R C OO
R'(1) Na/ancol
(2)H2/xt: Cu cromitRCH2 OH + R'OH
6.3.5.2. Phản ứng ở nhóm metylen linh động a. Phản ứng ngưng tụ este (ngưng tụ Claizen) Khi có xúc tác Na hay RONa, t0C các este có ít nhất Hα của nhóm metylen sẽ ngưng tụ tạo ra este của axit β-xetocacboxyl.

Hóa hữu cơ 2
91
Bền do cộng hưởng
+ Na hay C2H5ONaR C
O
O C2H5 H CH
R'
C
O
O C2H5 CH
R'
C
O
O C2H5C
O
R
Cơ chế: tương tự cơ chế cộng andol
+RO Na CH2 C
O
O C2H5H(1)
ROH + Na CH2 C
O
O C2H5
+CH2 C
O
O C2H5 CH3 C
O
O C2H5 CH3 C
O
O C2H5
CH2 COOC2H5
CH2 C
O
O C2H5C
O
H3C
- Nếu 2 este đều có HX → hỗn hợp các sản phẩm - Nếu 1 este có HX → 1 sản phẩm - Không có HX → không có phản ứng trên * Este cũng có thể ngưng tụ với xeton → β-đixeton
C2H5ONaCH3 C
O
O C2H5
+
CH2 CO
H CH3 C
O
O C2H5
CH2 COAr
CH2 C
O
O ArC
O
H3C OC2H5
b. Phản ứng ankyl hóa este của axit malonic
(Tổng hợp malonic)
CH2
COOR
COOR
Na hay C2H5ONaCH
COOR
COOR
R' X CHCOOR
COORR'
H+CH
COOH
COOHR'
t0 caoCH2 COOHR' + CO2

Hóa hữu cơ 2
92
6.4. Halogenua Axit 6.4.1. Danh pháp 6.4.1.1. Tên IUPAC Tên axit quốc tế bỏ ic thêm yl
Ví dụ: Axit propanoic → propanoyl clorua 6.4.1.2. Tên thông thường
tương tự Axit propionic → propionyl clorua Như vậy tên gồm 2 phần: tên gốc axit gọi là axyl và tên halogen → axyl
halogenua 6.4.2. Điều chế 6.4.2.1.Clorua axit được điều chế như sau
+R C
O
O H PCl5 R C
O
Cl + HCl + POCl3 (1)
+R C
O
O H SOCl2 R C
O
Cl + HCl + SO2 (2)
+R C
O
O H SO2Cl2 2R C
O
Cl + Na2SO4 (3)
+R C
O
O H COCl2 R C
O
Cl + HCl + CO2 (4)
Để điều chế CH3COCl, trong phòng thí nghiệm dùng phản ứng (1), (2). Trong công nghiệp, dùng phản ứng (3), (4). PCl5 là tác nhân phản ứng rất mạnh và dùng để điều chế clorua của điaxit, polyaxit. 6.4.2.2.Phản ứng của axit với clorua axit khác
+R COOH CC
O
O
ClCl
R C
O
Cl + CC
O
O
O + HCl
Điều chế RCOBr hay RCOI tương tự, đối với RCOF dùng KHF2 hay HF. 6.4.3. Tính chất vật lý RCOX thấp là chất lỏng, mùi xốc, ts < ts axit tương ứng. 6.4.4. Tính chất hóa học RCOX hoạt động mạnh do X hút e (-I) tốt. Ngoài ra còn xảy ra ở Hα, tuy nhiên ít thấy vì nguyên tử X gắn với nhóm cacbonyl còn hoạt động mạnh hơn nhiều. 6.4.4.1. Phản ứng axyl hóa bằng halogenua axit

Hóa hữu cơ 2
93
R CO
N R1
R2
R C XO
+ HOH RCOOH+ HOR' RCOOR'+ HOAr RCOOAr+ Na+-OOCR' O C
ORC
OR
+ NH3 RCONH2
+ R1NHR2
+ Na+N3- R C
ON3
6.4.4.2. Tác dụng với cơ kim
+R C ClO
R'MgXR C R'
OR C
R'
OH
R' Với cơ kim thì RCOF > RCOCl > RCOBr ( -F có –I tốt và kích thước nhỏ). Điều này trái với quy luật R – F < R – Cl < R – Br. 6.4.4.3. Khử dẫn xuất axit RCOOH không bị khử chỉ trừ LiAlH4 (hay H- khác) nhưng dẫn xuất axit thì có thể bị khử thành ancol nhờ các chất khử khác. Cụ thể:
+R C ClO
H2Pd, t0C R CHO + HCl
+R C ClO
H2Pd(S),1250C R CHO + HCl
Toluen
Phương pháp Rozenmun cho S vào để giảm hoạt tính xúc tác Pd. 6.4.4.4. Phản ứng ở gốc R Thế Hα tương tự như axit
+ + HClH3C C ClO
Cl2 H2C C ClOCl
6.4.5. Giới thiệu riêng các RCOX 6.4.5.1. Axetyl clorua Là tác nhân khơi mào cho cơ chế gốc vì tạo ra các gốc tự do có hoạt tính mạnh:

Hóa hữu cơ 2
94
+H3C C ClO
Na2O2 H3C C O
O
2 O CO
CH3 + 2NaCl
t0C
CH3 + CH3COO 6.4.5.2. Benzoyl clorua Điều chế bằng cách:
+C6H5 CCl3 H2OH2SO4 C6H5 COCl + 2HCl
C6H5COCl là tác nhân benzoyl hóa của nhiều hợp chất: amin, phenol... 6.5. Anhyđrit Axit 6.5.1. Danh pháp 6.5.1.1. Tên IUPAC Anhyđrit + tên axit theo IUPAC 6.4.1.2. Tên thông thường
tương tự Anhyđrit + tên axit thông thường
6.5.2. Điều chế 6.5.2.1.Tách nước từ các axit cacboxylic
2 R COOHP2O5, t0C R C O
O
CO
R + H2O
Đối với đicacboxylic chỉ cần tiến hành ở nhiệt độ cao:
H2OCC
O
O
O +CC
O
O
OHOH
t0C
6.5.2.2. Axyl hóa muối RCOONa
+ NaCl+R' C O
O
Na R C Cl
O
R' C O
O
CO
R
Phương pháp này xảy ra theo cơ chế SN2 vào cacbonyl, điều chế được anđehyt hỗn tạp. 6.5.2.3. Các phương pháp khác Để điều chế R – COOCOCH3 cho RCOOH tác dụng với xeten (CH2=C=O).

Hóa hữu cơ 2
95
Đối với anhyđrit hỗn tạp như H – COOCOR được điều chế từ HCOOH tác dụng với R – COOCO – R, đó là tác nhân focmyl hóa thay cho anhyđrit fomic không bền để điều chế được este khác của axit fomic. 6.5.3. Tính chất vật lí 6.5.4. Tính chất hóa học Nói chung tương tự như RCOCl: anhyđrit tham gia các phản ứng ở nhóm cacbonyl. - Khử bằng LiAlH4 cho ra ancol bậc 1 - phản ứng FriđenCraft - Phản ứng với các tác nhân giàu electron như:
+
R CO
N R1
R2
+ HOH 2RCOOH+ HOR' RCOOR'+ HOAr RCOOAr
+ NH3 RCONH2
+ R1NHR2
RCOOH+ RCOOH
+ RCOOH+ RCOOH
- Phản ứng thế ở R (phản ứng Peckin)
C6H5 CHO H3C C O
O
CO
CH3+CH3COONa
C6H5 CH CH COOHAxit xinamic
Với anhydrit propionic, xúc tác Natri propionat phản ứng xảy ra tương tự. Ta có:
C6H5 CHO + (CH3CH2CO)2OCH3CH2COONa
C6H5 CH CCH3
COOH
Axit -metyl xinamic Cơ chế:
CH2 CO OCOCH3+CH3COO- Na+
H
CH2 CO OCOCH3 CH3COOH
CO
H
C6H5 CH CH2 COOCOCH3
OC6H5 CH CH2 COOCOCH3
OH-HOH
C6H5 CH CH COOCOCH3 Chú ý: andehyt béo không tham gia phản ứng Peckin

Hóa hữu cơ 2
96
6.5.5. Giới thiệu riêng 6.5.5.1. Andehyt axetic - Trong công nghiệp:
+ (CH3CO)2OCH3COOH CH2 C O - Tác nhân axetyl hóa (gắn nhóm metoxy) tốt hơn CH3COCl 6.5.5.2. Anhyđrit phtalic
CH3
CH3 [O] CC
O
O
O
O2 CC
O
O
O
ROHCC
O
O
O +CC
O
O
OROH
t0C ROH CC
O
O
OROR
+CC
O
O
O NH3lanh C
C
O
O
NH2
COOHamit
t0C+
CC
O
O
O NH3CC
O
O
NH + H2O
imit

Hóa hữu cơ 2
97
Amit bậc 1
Amit bậc 2
Amit bậc 3
+CC
O
O
OArOH
CC
O
O
O
OH
+
H
OH
SEZnCl2 khan
CO
HO
OH
C O
Phenolphtalein
Chất chỉ thị màu axit – bazơ 6.5. Amit Khi axyl hóa (RCO-) vào NH3 có thể cho 3 sản phẩm:
R C
O
NH2
R C
O
NH C
O
R
R C
O 3
N
Chỉ có amit bậc một mới có ý nghĩa trong thực tế
6.5.1. Danh pháp Lấy từ axit bỏ vị ngữ “ic” thay bằng “amit”. - Tên quốc tế (IUPAC): tên axit IUPAC bỏ vĩ ngữ “ic” thay bằng “amit”.
CH2 C
O
NH2H3C propanoamit
- Tên thông thường: propionamit
6.5.2. Điều chế 6.5.2.1. Dẫn xuất khác của axit
+ NH3
R C
O
X
R C
O
O C
O
R
R C
O
O R'

Hóa hữu cơ 2
98
(nếu có amit thế, thay NH3 bằng amin bậc 1,2) 6.5.2.2. Từ muối amoni
RCOONH42200C
R C
O
NH2 + H2O
6.5.2.3. Thủy phân không hoàn toàn R – CN (hoàn toàn được RCOOH)
+ H2OR C N R C
O
NH2
6.5.2. Tính chất
- Trừ focmamit ở dạng lỏng, còn lại đều là chất rắn kết tinh, không màu. - Amit thấp tan tốt trong nước vì có liên kết H liên phân tử với H2O, nếu
amit còn H – N. 6.5.2.1. Amit là hợp chất lưỡng tính
+ Tính bazơ rất yếu:
R C
O
NH2
Dẫn đến chỉ tác dụng với axit mạnh
+ H2O+ HClR C
O
NH2 R C
O
NH3 Cl + HClR C
O
NH2
+ Tính axit rất yếu: Do có +C làm cho H linh động nhưng vỗn cảu NH nên yếu.
R C
O
NH H
Chỉ tác dụng với kim loại mạnh (Na/NH3)
+ H2OR C
O
NH2 + NaOH R C
O
NH Na
6.5.2.2. Các phản ứng quan trọng 6.5.2.2.1. Thủy phân
+ H+ hay OH-
R C
O
NH2 + H2O R COOH + NH3
Khả năng thủy phân kém hơn este. 6.5.2.2.2. Phản ứng khử tạo ra amin:
R C
O
NH2 + H2 R CH2 NH2
6.5.2.2.3. Tách nước

Hóa hữu cơ 2
99
Chuyển vị R qua gắn với N và giải phóng Br−
R C
O
NH2 R C NP2O5, t = 2000C
+ H2O
6.5.2.2.4. Phản ứng đặc biệt Hopman (thoái biến Hopman):
Khi cho Br2 (hay NaBrO) hay NaClO trong dung dịch kiềm tác dụng amit tạo ra amin (kém hơn 1 C so với axit).
R C
O
NH2 + Br2 R NH2OH-
Cơ chế: Tính chất axit thứ nhất:
R C
O
NH H OH-R C
O
NH + H2O
(thể hiện tính axit của amit)
+R C
O
NH Br Br+ -
R C
O
NH Br + Br
Tác dụng với Brδ+ Tính chất axit thứ hai:
CO2
OH- H2OR C
O
N Br +
H
R C
O
N Br +
CO N R + Br
OH H
CO N ROH2
CHO N ROH
CHO NH RO
R NH2 +
6.5.3. Giới thiệu riêng 6.5.3.1. Focmamit
Chất lỏng không màu, điều chế từ:
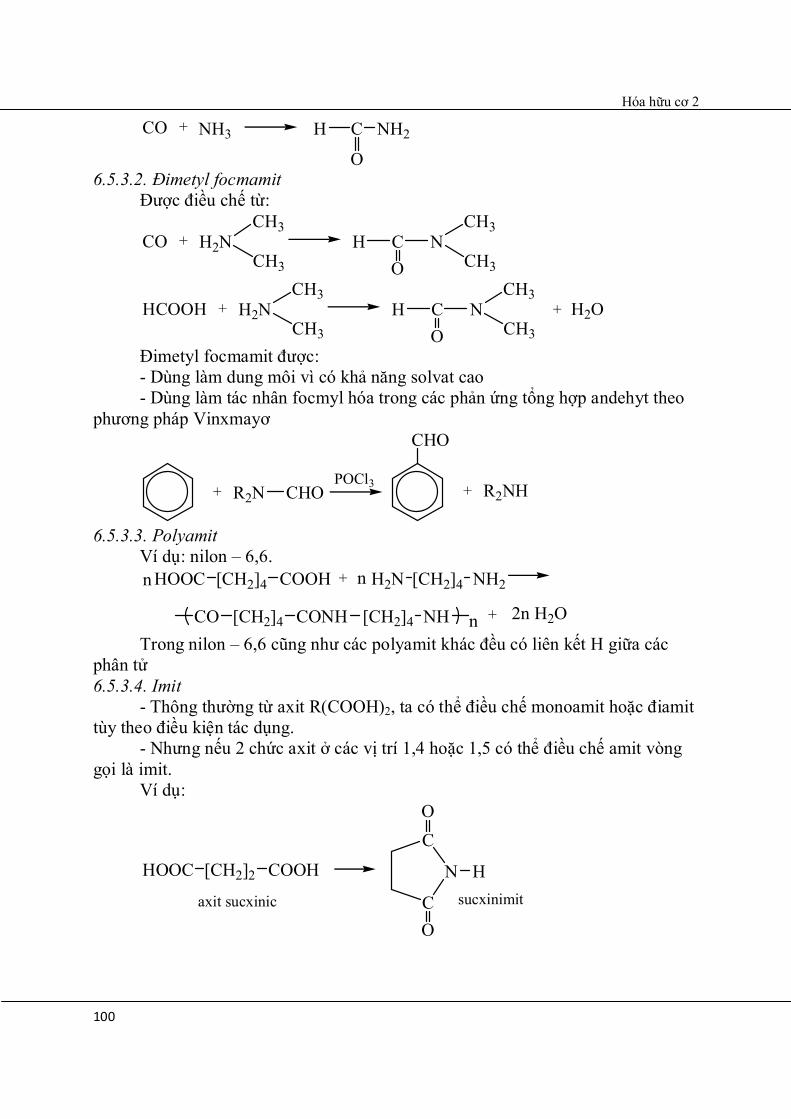
Hóa hữu cơ 2
100
+CO NH3 H C
O
NH2
6.5.3.2. Đimetyl focmamit Được điều chế từ:
+CO H2NCH3
CH3
NCH3
CH3
CHO
H2O++HCOOH H2NCH3
CH3
NCH3
CH3
CHO
Đimetyl focmamit được: - Dùng làm dung môi vì có khả năng solvat cao - Dùng làm tác nhân focmyl hóa trong các phản ứng tổng hợp andehyt theo
phương pháp Vinxmayơ
+ R2N CHOPOCl3
CHO
+ R2NH
6.5.3.3. Polyamit Ví dụ: nilon – 6,6.
+
H2O
[CH2]4 COOHHOOCn n [CH2]4 NH2H2N
[CH2]4 CONHCO [CH2]4 NH n + 2n
Trong nilon – 6,6 cũng như các polyamit khác đều có liên kết H giữa các phân tử 6.5.3.4. Imit
- Thông thường từ axit R(COOH)2, ta có thể điều chế monoamit hoặc điamit tùy theo điều kiện tác dụng.
- Nhưng nếu 2 chức axit ở các vị trí 1,4 hoặc 1,5 có thể điều chế amit vòng gọi là imit.
Ví dụ:
[CH2]2 COOHHOOC
C
N
CO
O
Haxit sucxinic sucxinimit

Hóa hữu cơ 2
101
Saccarin
CHHOOC CH COOHC
N
CO
O
Hmaleimitaxit maleic
COOHCOOH C
NCO
O
H
axit phtalic axit phtalimit
Các imit có tính axit hơn RCONH2 và dễ bị thủy phân trong môi trường kiềm.
CC
O
O
ONaNH2
NaOH
CN
CO
O
HH+ C
C
O
O
OHNH2
Imit của axit o-sunfobenzoic có tên là saccarin là một loại đường ngọt gấp
500 lần đường saccarozơ, được tổng hợp theo sơ đồ sau:
NaOH
CH3 OH S ClO
OSE
SO
OCl
NH3
SO
ONH2
o-Toluen sunfoclorua o-Toluen sunfamit+ KMnO4
SO
ONH2
CH3 CH3
COOH- H2O
SN
CO
O
HO
SO2
NNaCO


















