
Crystal Structures of a Multifunctional …Crystal Structures of a Multifunctional...
15
Crystal Structures of a Multifunctional Triterpene/Flavonoid Glycosyltransferase from Medicago truncatula Hui Shao, 1 Xianzhi He, 1 Lahoucine Achnine, Jack W. Blount, Richard A. Dixon, and Xiaoqiang Wang 2 Plant Biology Division, Samuel Roberts Noble Foundation, Ardmore, Oklahoma 73401 Glycosylation is a ubiquitous reaction controlling the bioactivity and storage of plant natural products. Glycosylation of small molecules is catalyzed by a superfamily of glycosyltransferases (GTs) in most plant species studied to date. We present crystal structures of the UDP flavonoid/triterpene GT UGT71G1 from Medicago truncatula bound to UDP or UDP- glucose. The structures reveal the key residues involved in the recognition of donor substrate and, by comparison with other GT structures, suggest His-22 as the catalytic base and Asp-121 as a key residue that may assist deprotonation of the acceptor by forming an electron transfer chain with the catalytic base. Mutagenesis confirmed the roles of these key residues in donor substrate binding and enzyme activity. Our results provide an initial structural basis for understanding the complex substrate specificity and regiospecificity underlying the glycosylation of plant natural products and other small molecules. This information will direct future attempts to engineer bioactive compounds in crop plants to improve plant, animal, and human health and to facilitate the rational design of GTs to improve the storage and stability of novel engineered bioactive compounds. INTRODUCTION Glycosylation is quantitatively the most significant reaction on earth. It is catalyzed by a superfamily of enzymes, the glycosyl- transferases (GTs), which have been classified into >70 families (Campbell et al., 1997) (http://afmb.cnrs-mrs.fr/CAZY/index. html). Family 1 GT enzymes, the UDP glycosyltransferases (UGTs), transfer UDP-activated sugar moieties to specific ac- ceptor molecules. Twenty-seven UGTs have been identified in the human genome (de Wildt et al., 1999; Li et al., 2001); these play central roles in the metabolism and detoxification of foreign chemicals such as carcinogens and hydrophobic drugs. By contrast, plants contain a very large number of UGTs that are involved in the glycosylation of natural products (Vogt and Jones, 2000; Jones and Vogt, 2001) and contain a highly conserved consensus signature sequence called the PSPG motif (Hughes and Hughes, 1994; Paquette et al., 2003). Plants collectively synthesize >200,000 natural products, and a significant pro- portion of them are glycosylated. Consistent with this chemical complexity, 117 putative UGT genes have been identified in the genome of Arabidopsis thaliana (Ross et al., 2001). Likewise, >100 UGTs are predicted in the model legume Medicago truncatula based on an analysis of currently available EST and genomic sequences (Achnine et al., 2005). Glycosylation is generally seen as a mechanism for the de- toxification of xenobiotics or for facilitating the storage of bio- active molecules in plant vacuoles (Sandermann, 1992; Paquette et al., 2003). However, in some cases, glycosylated plant natural products exhibit higher bioactivity than their corresponding aglycones, as in the case of the triterpene saponins (Osbourn, 2003). UGT71G1 was recently identified in M. truncatula as an enzyme functionally involved in the biosynthesis of saponins, although the enzyme exhibits more favorable kinetics with the flavonoid quercetin (Achnine et al., 2005). Triterpene glycosides have allelopathic, antimicrobial, insecticidal, and antiherbivory activities for plant protection (Osbourn et al., 1996; Small, 1996; Oleszek et al., 1999; Papadopoulou et al., 1999). They also have pharmacological, anticholesterolemic, anticancer, adjuvant, and hemolytic activities (Behboudi et al., 1999; Oh et al., 2000; Haridas et al., 2001), act on the cardiovascular, nervous, and digestive systems, and are used in sneezing powders, emetics, and cough syrups to facilitate expectoration (Dennehy, 2001). The saponins in M. truncatula are glycosides of the five triterpene aglycones soyasapogenol B, soyasapogenol E, medicagenic acid, hederagenin, and bayogenin (Huhman and Sumner, 2002). These aglycones share similar structures, and UGT71G1 recog- nizes both medicagenic acid and hederagenin as acceptors (Figure 1) for the sugar moiety delivered from UDP-glucose. It is not yet clear which positions on the triterpene scaffold are glycosylated by UGT71G1; O-glycosylation at positions 3 and 28 is most common for this class of compound, but C-glycosylation of plant natural products has also been observed, and these products are of particular pharmaceutical interest (Clemens et al., 2004). Understanding the structural basis for the glycosylation of triterpene natural products will facilitate future metabolic engi- neering of these compounds to improve the health of plants (resistance to disease and herbivory), animals (via improved forage quality), and humans (through drug discovery). 1 These authors contributed equally to this work. 2 To whom correspondence should be addressed. E-mail xwang@ noble.org; fax 580-224-6692. The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Xiaoqiang Wang ([email protected]). Article, publication date, and citation information can be found at www.plantcell.org/cgi/doi/10.1105/tpc.105.035055. The Plant Cell, Vol. 17, 3141–3154, November 2005, www.plantcell.org ª 2005 American Society of Plant Biologists
Transcript of Crystal Structures of a Multifunctional …Crystal Structures of a Multifunctional...

Crystal Structures of a Multifunctional Triterpene/FlavonoidGlycosyltransferase from Medicago truncatula
Hui Shao,1 Xianzhi He,1 Lahoucine Achnine, Jack W. Blount, Richard A. Dixon, and Xiaoqiang Wang2
Plant Biology Division, Samuel Roberts Noble Foundation, Ardmore, Oklahoma 73401
Glycosylation is a ubiquitous reaction controlling the bioactivity and storage of plant natural products. Glycosylation of
small molecules is catalyzed by a superfamily of glycosyltransferases (GTs) in most plant species studied to date. We
present crystal structures of the UDP flavonoid/triterpene GT UGT71G1 from Medicago truncatula bound to UDP or UDP-
glucose. The structures reveal the key residues involved in the recognition of donor substrate and, by comparison with other
GT structures, suggest His-22 as the catalytic base and Asp-121 as a key residue that may assist deprotonation of the
acceptor by forming an electron transfer chain with the catalytic base. Mutagenesis confirmed the roles of these key
residues in donor substrate binding and enzyme activity. Our results provide an initial structural basis for understanding the
complex substrate specificity and regiospecificity underlying the glycosylation of plant natural products and other small
molecules. This information will direct future attempts to engineer bioactive compounds in crop plants to improve plant,
animal, and human health and to facilitate the rational design of GTs to improve the storage and stability of novel engineered
bioactive compounds.
INTRODUCTION
Glycosylation is quantitatively the most significant reaction on
earth. It is catalyzed by a superfamily of enzymes, the glycosyl-
transferases (GTs), which have been classified into >70 families
(Campbell et al., 1997) (http://afmb.cnrs-mrs.fr/CAZY/index.
html). Family 1 GT enzymes, the UDP glycosyltransferases
(UGTs), transfer UDP-activated sugar moieties to specific ac-
ceptor molecules. Twenty-seven UGTs have been identified in
the human genome (de Wildt et al., 1999; Li et al., 2001); these
play central roles in the metabolism and detoxification of foreign
chemicals such as carcinogens and hydrophobic drugs. By
contrast, plants contain a very large number of UGTs that are
involved in the glycosylation of natural products (Vogt and Jones,
2000; Jones and Vogt, 2001) and contain a highly conserved
consensus signature sequence called the PSPG motif (Hughes
and Hughes, 1994; Paquette et al., 2003). Plants collectively
synthesize >200,000 natural products, and a significant pro-
portion of them are glycosylated. Consistent with this chemical
complexity, 117 putative UGT genes have been identified in the
genome of Arabidopsis thaliana (Ross et al., 2001). Likewise,
>100 UGTs are predicted in the model legume Medicago
truncatula based on an analysis of currently available EST and
genomic sequences (Achnine et al., 2005).
Glycosylation is generally seen as a mechanism for the de-
toxification of xenobiotics or for facilitating the storage of bio-
activemolecules in plant vacuoles (Sandermann, 1992; Paquette
et al., 2003). However, in some cases, glycosylated plant natural
products exhibit higher bioactivity than their corresponding
aglycones, as in the case of the triterpene saponins (Osbourn,
2003). UGT71G1 was recently identified in M. truncatula as an
enzyme functionally involved in the biosynthesis of saponins,
although the enzyme exhibits more favorable kinetics with the
flavonoid quercetin (Achnine et al., 2005). Triterpene glycosides
have allelopathic, antimicrobial, insecticidal, and antiherbivory
activities for plant protection (Osbourn et al., 1996; Small, 1996;
Oleszek et al., 1999; Papadopoulou et al., 1999). They also have
pharmacological, anticholesterolemic, anticancer, adjuvant, and
hemolytic activities (Behboudi et al., 1999; Oh et al., 2000;
Haridas et al., 2001), act on the cardiovascular, nervous, and
digestive systems, and are used in sneezing powders, emetics,
and cough syrups to facilitate expectoration (Dennehy, 2001).
The saponins inM. truncatula are glycosides of the five triterpene
aglycones soyasapogenol B, soyasapogenol E, medicagenic
acid, hederagenin, and bayogenin (Huhman and Sumner, 2002).
These aglycones share similar structures, and UGT71G1 recog-
nizes both medicagenic acid and hederagenin as acceptors
(Figure 1) for the sugar moiety delivered from UDP-glucose. It is
not yet clear which positions on the triterpene scaffold are
glycosylated by UGT71G1;O-glycosylation at positions 3 and 28
is most common for this class of compound, butC-glycosylation
of plant natural products has also been observed, and these
products areof particular pharmaceutical interest (Clemenset al.,
2004). Understanding the structural basis for the glycosylation of
triterpene natural products will facilitate future metabolic engi-
neering of these compounds to improve the health of plants
(resistance to disease and herbivory), animals (via improved
forage quality), and humans (through drug discovery).
1 These authors contributed equally to this work.2 To whom correspondence should be addressed. E-mail [email protected]; fax 580-224-6692.The author responsible for distribution of materials integral to thefindings presented in this article in accordance with the policy describedin the Instructions for Authors (www.plantcell.org) is: Xiaoqiang Wang([email protected]).Article, publication date, and citation information can be found atwww.plantcell.org/cgi/doi/10.1105/tpc.105.035055.
The Plant Cell, Vol. 17, 3141–3154, November 2005, www.plantcell.orgª 2005 American Society of Plant Biologists

To date, 17 members of the GT superfamily (but none from
plants) have been analyzed structurally (Goldsmith et al., 1989;
Vrielink et al., 1994; Charnock and Davies, 1999; Gastinel
et al., 1999, 2001; Ha et al., 2000; Pedersen et al., 2000, 2003;
Unligil et al., 2000; Persson et al., 2001; Gibbons et al., 2002;
Gibson et al., 2002; Buschiazzo et al., 2004; Fritz et al., 2004;
Lobsanov et al., 2004; Mulichak et al., 2004), and these struc-
tures fall into two different topologies/folds, GT-A and GT-B
(Breton et al., 2002; Hu and Walker, 2002; Coutinho et al., 2003).
Plant UGTs have been predicted to be similar to the bacterial GT
family 1 TDP-epi-vancosaminyltransferasesGtfA, GtfB, andGtfD
from Amycolatopsis orientalis (Mulichak et al., 2001, 2003, 2004;
Hans et al., 2004; Bowles et al., 2005), which belong to the GT-B
fold comprising twoRossmann-like domains. However, bacterial
Gtfs share only ;10% sequence identity with plant UGTs,
making it very difficult to model plant glycosylation processes
without knowledge of plant UGT crystal structures.
Here, we report the structures of UGT71G1 fromM. truncatula
bound to UDP or UDP-glucose. These plant UGT crystal struc-
tures reveal the detailed interactions between the enzyme and its
donor substrate as well as the roles of the UGT signature motif in
substrate recognition and the catalytic activity of the enzyme.
Further mutational and structural analyses, and comparisons
with other GT structures, will provide a basis for understanding
the complex and often overlapping substrate specificity and
regiospecificity within the plant UGT superfamily.
RESULTS
Overall Structure
The three-dimensional structure of UGT71G1 fromM. truncatula
bound with a UDP molecule was determined using the multi-
wavelength anomalous dispersion (MAD) method with a crystal
of Se-Met–substituted enzyme and refined to 2.0-A resolution
with a native data set. Data collection, phasing, and refinement
statistics are presented in Table 1.
The structure of UGT71G1 consists of two N- and C-terminal
domains with similar Rossmann-type folds and, as predicted,
belongs to the GT-B fold (Figures 2 and 3). The N-terminal
domain contains a central seven-stranded parallel b sheet
flanked by eight a helices on both sides and a small two-
stranded b sheet. The C-terminal domain contains a six-
stranded b sheet flanked by eight a helices. The two domains
pack very tightly and form a deep cleft with a UDP molecule
bound.
The asymmetric unit contains two independent UGT71G1
molecules related by a noncrystallographic twofold axis. These
two molecules are very similar to each other, with root mean
square deviations (RMSD) of 1.04 and 1.52 A for 456 Ca atoms
and all atoms (residues 8 to 463), respectively. However, small
differences were observed between the two molecules, espe-
cially in the region near the UDP binding site in the cleft between
the N- and C-terminal domains, which included the C-terminal
part of helix Na1 (around residue Ile-20); loop Nb2-Na2 (around
residuePro-50),which is highlymobilewith highB factors (;80 A2)
and poorly defined electron density in both molecules A and B;
and the loop linker between the N- and C-terminal domains
(around residue Asn-255), the electron density of which was
poorly defined inmolecule B. The helix Na1 (P18 toH36) and loop
Nb2-Na2 and the following helix Na2 (K48 to Q66) in molecule B
shift away slightly from the C-terminal domain, with the largest
movement, 2.6 A, for the Ca atom of Leu-23, which is adjacent to
the key catalytic residue His-22 (see below). This suggests that
the flexibility in the cleft region between the two subunits allows
for movement to occur during substrate binding and catalysis.
Comparison with Other Members of the GT Superfamily
There are three known structures of family 1 GTs, all for bacterial
vancomycin UDP glucosyltransferase Gtfs. Superimposing the
structures of plant UGT71G1 and bacterial GtfD revealed strong
structural similarity, with an RMSD of 2.23 A for 242 Ca atoms,
although the sequence identity is very low (;10%) (Figure 3). The
largest difference between these two enzymes was observed
in the region between the helix Na5 and the strand Nb6 (;155
to 210) in their N-terminal domains. A two-stranded b sheet
is present in this region of the UGT71G1 structure, resembling
an arm flipped to the opposite side near the helix Ca5 of the
C-terminal domain, compared with the corresponding region in
the GtfD structure. This region forms one side of the substrate
(acceptor molecule) binding pocket, and this large difference in
Figure 1. Structures of Three Substrates of M. truncatula UGT71G1.
The known or predicted glycosylation sites are labeled with numbers: quercetin (3, 5, 7, 39, and 49), medicagenic acid (3 or 28), and hederagenin (3
or 28).
3142 The Plant Cell

conformation/folding changes the shape and size of the binding
pocket. The comparison also shows that the similarity between
the C-terminal domains of UGT71G1 and GtfD (RMSD ¼ 2.13 A
for 137 Ca atoms) is higher than that between their N-terminal
domains (RMSD ¼ 2.34 A for 120 Ca atoms), presumably
because the C-terminal domains recognize similar donor mole-
cules but the N-terminal domains recognize quite different
acceptor molecules. The UGT71G1 structure is more compact
in its N-terminal domain, presumably because its substrates,
such as medicagenic acid and quercetin, are considerably
smaller than the acceptor substrate desvancosaminyl vancomy-
cin (DVV) of GtfD.
Comparison was also performed between the UGT71G1
structure and those of other GTs with the GT-B fold. Escherichia
coliMurG (Coutinho et al., 2003) is closely related to UGT71G1 in
the overall GT classification. Superimpositions between portions
of the structures of UGT71G1 and MurG gave RMSD values of
2.27 A (199 Ca atoms of the overall structures), 2.23 A (95 Ca
atoms in the N-terminal domains), and 1.95 A (110 Ca atoms in
the C-terminal domains).
Trehalose-6-phosphate synthetase OtsA, T4 bacteriophage
b-glucosyltransferase (BGT), and glycogen synthase are rela-
tively distant from UGT71G1 in classification, although they use
the same UDP-sugar or, in the case of glycogen synthase, the
related ADP-sugar. Superimposing the overall structures of
UGT71G1 and OtsA, which have very little sequence identity,
gave an RMSD of 2.21 A (158 Ca atoms). Superimposing their
N- and C-terminal domains gave RMSD values of 2.17 A (96 Ca
atoms) and 2.27 A (70 Ca atoms), respectively; the N-terminal
rather than the C-terminal domain is more similar to the corre-
sponding domain of UGT71G1, in contrast with the situation with
UGT71G1 and GtfD or MurG. Similarly, superimposing the
N- and C-terminal domains of UGT71G1 and BGT gave RMSD
values of 2.28 A (79 Ca atoms) and 2.29 A (70 Ca atoms),
respectively.
Interactions between Enzyme and Donor Ligands
Although UDP-galactose (a nondonor) and quercetin were in-
cluded for cocrystallization with UGT71G1, the only substrate
electron density observed was for the UDP moiety of UDP-
galactose (Figure 4A) as a result of possible hydrolysis. This is
similar to the situation with the bacterial GtfD structure, in which
only TDP was observed, although GtfD was cocrystallized with
TDP-glucose (Mulichak et al., 2004). Soaking crystals of
UGT71G1 with UDP-glucose resulted in a weak electron density
map for the glucose portion of the molecule in the enzyme
structure at 2.6 A (Figure 4B). The B factor of the glucose moiety
is higher than that of the UDP moiety, indicating either flexibility
or low occupancy of the sugar attributable to the possible
hydrolysis of UDP-glucose.
UDP or UDP-glucose is almost completely buried in a long,
narrow channel mainly within the C-terminal domain of the
enzyme. In the 2.0-A structure of UGT71G1 complexed with
UDP, the enzyme interacts with UDP mainly through residues in
the UGT signature PSPG motif (residues 339 to 382) (Figure 4C,
Table 2). The uracil ring of UDP interacts through hydrogen
bonds between the N3 and O4 atoms of the ring and the main
chain oxygen and nitrogen atoms of Ala-340 and also forms
parallel p-stacking interactions with the indole ring of Trp-339.
Table 1. Data Collection, Phasing, and Refinement Statistics
Data Collection and Phasing Statistics
Native (þ UDP) Se peak (þ UDP) Se inflection (þ UDP) Se remote (þ UDP) Complex (þ UPG)
Resolution (A) 2.0 2.4 2.4 2.4 2.6
Wavelength (A) 1.5418 0.9797 0.98 0.9393 1.5418
Unique reflections 62,825 36,121 35,553 34,461 29,545
Completeness (%) 98.2 (97.7) 94.4 (81.8) 97.0 (85.0) 92.4 (78.4) 99.8 (99.7)
Rsyma (%) 4.1 (27.7) 8.2 (26.0) 5.7 (25.2) 6.0 (28.6) 5.9 (44.2)
I/s (I) 13.7 (2.7) 16.5 (2.7) 16 (2.6) 16.1 (3.1) 12.1 (2.3)
Figure of merit 0.44
Refinement Statistics
Enzyme-UDP Enzyme-UDP glucose (UPG)
R factor (%) 18.7 20.5
Rfree (%) 22.9 28.0
Number of protein atoms 7204 7204
Number of solvent atoms 728 17
Number of ligand atoms 50 (two UDP molecules) 72 (two UDP-glucose molecules)
Average B factors (A2) 31.8 56.3
RMSD from ideal values
Bond length (A) 0.0057 0.0070
Bond angle (8) 1.27 1.29
Numbers in parentheses are for the highest-resolution shell.a Rsym ¼ Shkl jI � <I>j/SI, where I is the observed intensity and <I> is the average intensity from observations of symmetry-related reflections. A subset
of the data (8%) was excluded from the refinement and used to calculate the free R value (Rfree). R factor ¼ SkFoj � jFck/SjFoj.
UDP Glycosyltransferase Structure 3143

The ribose ring of UDP makes contacts with the enzyme through
hydrogen bonds between its O2* and O3* atoms and the OE1
and OE2 atoms of Glu-365 and between its O2* atom and the
NE2 atom of Gln-342. The a-phosphate forms hydrogen bonds
through its O1A atom and the ND2 atom of Asn-361, its O2A
atom and the OG and N atoms of Ser-362, and its O3A atom and
the NE2 atom of His-357. The b-phosphate forms hydrogen
bonds through its O3B oxygen and the OH atom of Tyr-379 and
the NE2 atom of His-357 and between its O2B oxygen and the
OG and N atoms of Ser-285, the only interacting residue outside
of the UGT signature motif region.
Interactions between the enzyme and the glucose moiety of
the donor are observed in the 2.6-A structure of UGT71G1 in its
complexwith UDP-glucose (Figure 4C, Table 2). TheO29 andO39
atoms of the glucose ring form hydrogen bonds with the NE2
atom of Gln-382 and the OE1 atom of Glu-381. The O49 atom of
the glucose ring contacts both the OE2 atom of Glu-381 and the
N atom of Trp-360. On the other hand, several interactions
observed between UDP and the enzyme are not present in the
model of UDP-glucose bound to the enzyme. A small rotation
and shift in the position of the b-phosphate of UDP-glucose
weakens the interaction of the O2B and O3B atoms of
b-phosphate and the O3A atom of a-phosphate with protein
residues.
In UGT71G1 binding UDP-glucose, GtfD binding TDP, treha-
lose-6-phosphate synthetase OtsA binding UDP, MurG binding
UDP-GlcNAc, and T4 bacteriophage BGT and its mutant D100A
binding UDP or UDP-glucose, the sugar donor molecules are
observed in a similar region of the enzymes (Gibson et al., 2002;
Hu et al., 2003; Lariviere et al., 2003; Mulichak et al., 2004). The
PSPG motif of UGT71G1 and the corresponding sugar binding
regions in these different GTs share a very similar conformation,
a/b/a/b, even though the sequence identities are very low and
the detailed interactions with the sugar donors also vary. Super-
impositions show that the UGT signature motif of UGT71G1 and
the corresponding regions of GtfD and MurG share higher
structural similarity, indicative of similar binding modes with
sugar donors, and differ from those for OtsA and BGT, in which
the first b strands of their a/b/a/bmotifs are long and the second
b strands cannot interact with sugar donors (Figure 5).
The Acceptor Binding Site
A very narrow, deep pocket is observed adjacent to the UDP
binding site, mainly consisting of residues from the N-terminal
domain (Figure 6), which would appear to be the binding pocket
for acceptor molecules. The location of this pocket is similar to
that of the substrate binding site observed in the structure ofGtfD
bound with DVV and TDP (Mulichak et al., 2004). In the GtfD
structure, Asp-13 is close to both DVV and TDP and has been
proposed as the general base for the catalysis of glycosyl
transfer. His-22 occupies the corresponding position in
UGT71G1 and is 4.7 A from the C19 atom of UDP-glucose, as
shown in the structure of UGT71G1 (molecule A) complexed with
UDP-glucose. Close to it (;3 A), there is an acidic residue (Asp-
121) that may form an electron transfer chain to His-22 to help
deprotonate acceptor molecules. The sequence alignment of
multiple plant UGTs (Figure 7) shows that corresponding His and
Asp residues are conserved in most plant UGTs.
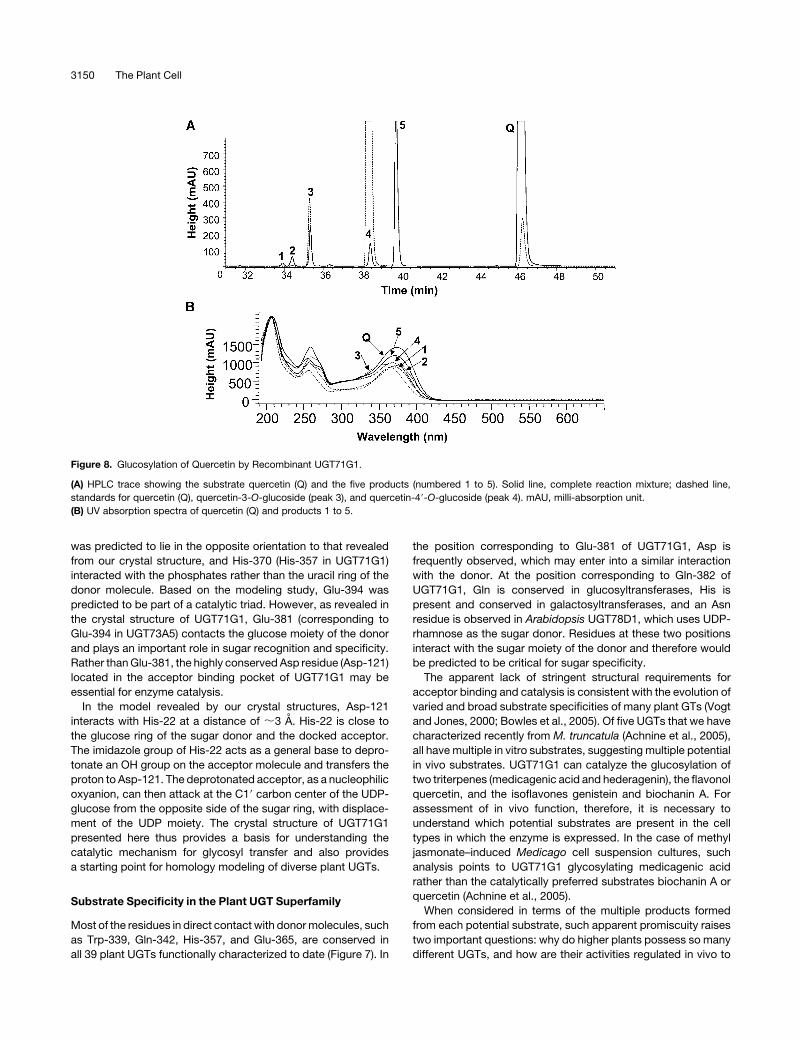
There are several potential O-glycosylation sites (five hydroxyl
groups) on the quercetin molecule, and multiple products are
formed after incubation of quercetin and UDP-glucose with
UGT71G1 (Figure 8A). All five products from the in vitro in-
cubation of recombinant UGT71G1 with UDP-glucose and
quercetin exhibited a similar UV spectrum to that of quercetin
(Figure 8B), had identicalmolecular ionmasses (462.5 to 462.9D)
consistent with quercetin monoglucoside structures, and dis-
appeared after incubation with b-glucosidase (data not shown).
This indicates that, in spite of the production of one favored
product (peak 5 in Figure 8A, identified as quercetin-39-O-
glucoside), UGT71G1 is capable of glucose transfer to each of
the five hydroxyl groups on the quercetin molecule. To probe the
structural basis of the apparently promiscuous acceptor binding
to UGT71G1, molecular docking was performed using the
Figure 2. Ribbon Diagram of the Structure of UGT71G1 with Bound UDP.
The N- and C-terminal domains are shown in orange and green, with the
secondary structures and the N and C termini labeled. The a helices and
b strands in the N- and C-terminal domains are numbered separately.
The UDP molecule is shown as a ball-and-stick model colored by atom
type (nitrogen, blue; carbon, yellow; oxygen, red; phosphorus, green).
Figures 2, 3B, 4, 5, and 6 were prepared with MOLSCRIPT (Kraulis, 1991)
and RASTER3D (Merritt and Bacon, 1997).
3144 The Plant Cell
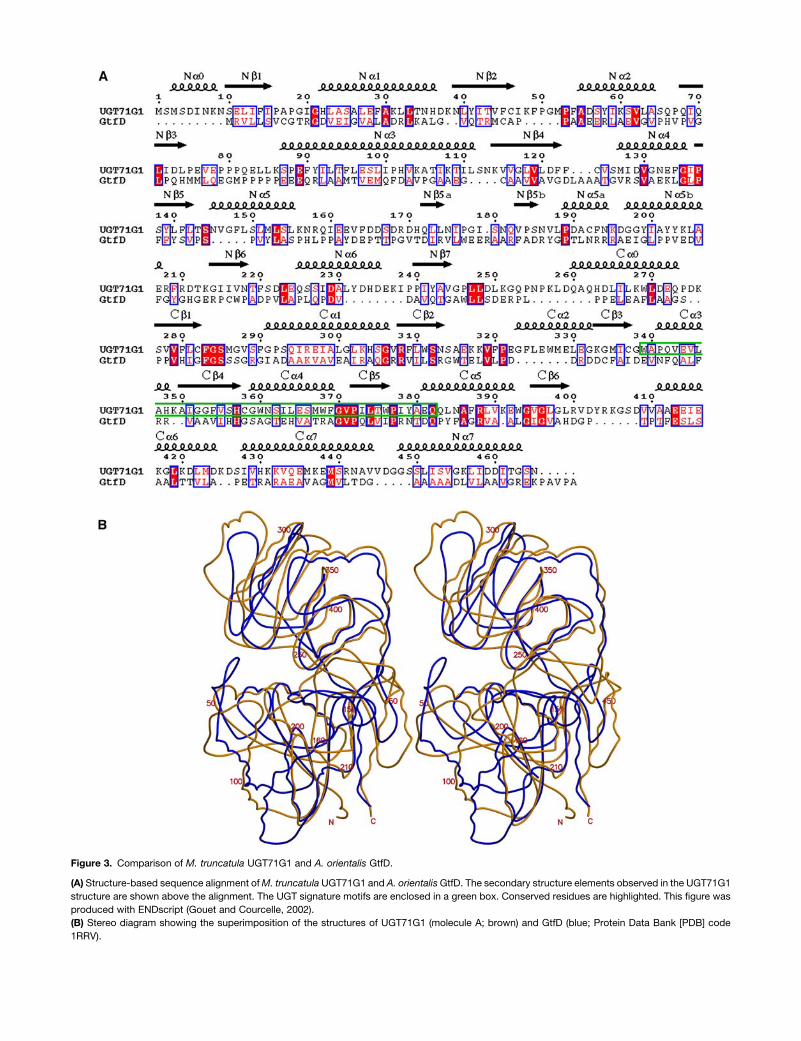
Figure 3. Comparison of M. truncatula UGT71G1 and A. orientalis GtfD.
(A) Structure-based sequence alignment ofM. truncatula UGT71G1 and A. orientalisGtfD. The secondary structure elements observed in the UGT71G1
structure are shown above the alignment. The UGT signature motifs are enclosed in a green box. Conserved residues are highlighted. This figure was
produced with ENDscript (Gouet and Courcelle, 2002).
(B) Stereo diagram showing the superimposition of the structures of UGT71G1 (molecule A; brown) and GtfD (blue; Protein Data Bank [PDB] code
1RRV).

software GOLD. Consistent with the multiple product specificity
with quercetin and the fact that quercetinwas not observed in the
structurewhen included in the crystallization liquid or soaked into
crystals, all five hydroxyl groups of quercetin could be docked
close to His-22, which lies in the middle of the potential acceptor
channel. Three aromatic residues, Phe-148, Phe-122, and Tyr-
202, and an acidic residue, Asp-190, are located at one end of
the channel; the other end is relatively larger, containing two
aromatic residues, Phe-49 and Phe-54, and two Mets, Met-52
and Met-286. An aromatic residue, Phe-123, and an acidic
residue, Glu-89, are in the middle of the channel, opposite UDP-
glucose. The pocket is much longer and larger than quercetin,
and quercetin may be fit in easily, with any of its hydroxyl groups
located ;3 A from the NE2 atom of His-22.
The docking in the active site of the hydroxyl groups at
positions 3 or 39 of quercetin gives fitness scores slightly higher
than for the other three hydroxyls, and also more potential
interactions between quercetin and the protein and donor. In the
model with the 39-OH docked in the active site (Figure 6A),
hydrogen bonds may form between the 3-OH of quercetin and
the O1B atom of UDP-glucose and between the 5-OH of
quercetin and the OG atom of Ser-285.
The triterpene substrates hederagenin and medicagenic acid
are more hydrophobic and significantly larger than quercetin.
Compared with the docking results with quercetin, docking with
medicagenic acid and hederagenin gave lower fitness scores
and weaker interaction with the enzyme, consistent with pre-
viously reported kinetic data (Achnine et al., 2005). Themolecular
docking study for hederagenin and medicagenic acid also
suggested that O-glycosylation at position 3 would be preferred
over other positions. In the model docked with hederagenin
(Figure 6B) or medicagenic acid, the OH group at position 3
points to the NE2 atom of His-22 (;3.2 A) and is located just
above the C19 atom of UDP-glucose (;3.8 A). Similar to the
proposed general mechanism of other inverting GTs (Unligil and
Rini, 2000), His-22 would act as an effective general base to
deprotonate the OH group of the acceptor molecule, then the
newly formed nucleophilic oxyanionwould attack the C19 carbon
of the UDP-glucose directly to displace the UDP moiety. The
carboxyl groups at position 28 of the triterpene acceptors may
hydrogen bond with the main chain oxygen atom of Gly-51. The
residues Pro-18, Phe-49, Pro-53, Phe-54, Leu-85, Pro-88, Ile-92,
and Phe-123 provide a hydrophobic environment for the accep-
tor substrates. A solution of the model for docking the carboxyl
oxygen atom at position 28 of hederagenin to the active site had
a slightly lower fitness score than that for the 3-hydroxyl. In the
former case, the hydroxyl group at position 3 may interact with
the main chain oxygen atom of Gly-51.
Mutagenesis of UGT71G1
To further explore the roles of the potential key residues
identified above, structure-directed point mutants of UGT71G1
were constructed, targeting residues involved in direct contact
with the sugar donor, the proposed general base His-22, and its
partner, Asp-121. Substitution of His-22 with Ala (H22A) led to
the complete loss of enzyme activity, which is consistent with the
proposed role of His-22 as the general base. Substitution of Asp-
121 with Ala or Asn resulted in no detectable enzyme activity.
This is similar to the situation with trypsin, for which the mutant
D102N (in the catalytic triad) showed a large loss in activity
(Sprang et al., 1987). Also similar to the case of trypsin, Asp-121
in UGT71G1 is nearly buried and forms a hydrogen bond with
a catalytic His residue. Our results demonstrate that Asp-121
and its proper interaction with His-22 are essential for enzyme
activity. In themodel docked with acceptors, Asp-121 is not able
to contact the acceptor molecules directly. This finding suggests
that Asp-121 may play an important role in assisting His-22 by
initialization or activation of the reaction through electron trans-
fer. The E381A mutant also showed no detectable activity,
demonstrating the key role of Glu-381 in recognition of the sugar
donor substrate because the side chain of Glu-381 forms
hydrogen bonds with the O39 and O49 atoms of the glucose
ring of the donor (Figure 4C). By contrast, mutant A340F retained
Figure 4. Donor Molecules and Their Interactions with UGT71G1.
(A) A jFobsj � jFcalcj electron density omit map of bound UDP contoured
at 1.5 s is superimposed on a ball-and-stick model of the UDP molecule.
(B) A jFobsj � jFcalcj electron density omit map of bound UDP-glucose
contoured at 1.5 s is superimposed on a ball-and-stick model of the
UDP-glucose molecule.
(C) Stereo diagram showing interactions between bound UDP-glucose
and UGT71G1 side chains. The structure of UDP-glucose is shown as
a ball-and-stick model. Hydrogen bonding interactions are indicated by
dashed lines. Interactions observed only between UDP(-galactose) and
UGT71G1 in the 2.0-A structure are indicated by green dashed lines.
3146 The Plant Cell
activity similar to that of the wild-type enzyme reported pre-
viously (Achnine et al., 2005). Ala-340 forms hydrogen bond
interactions with the uracil ring of the donor through its main
chain oxygen and nitrogen atoms; therefore, such bonds will still
be supplied by Phe.
DISCUSSION
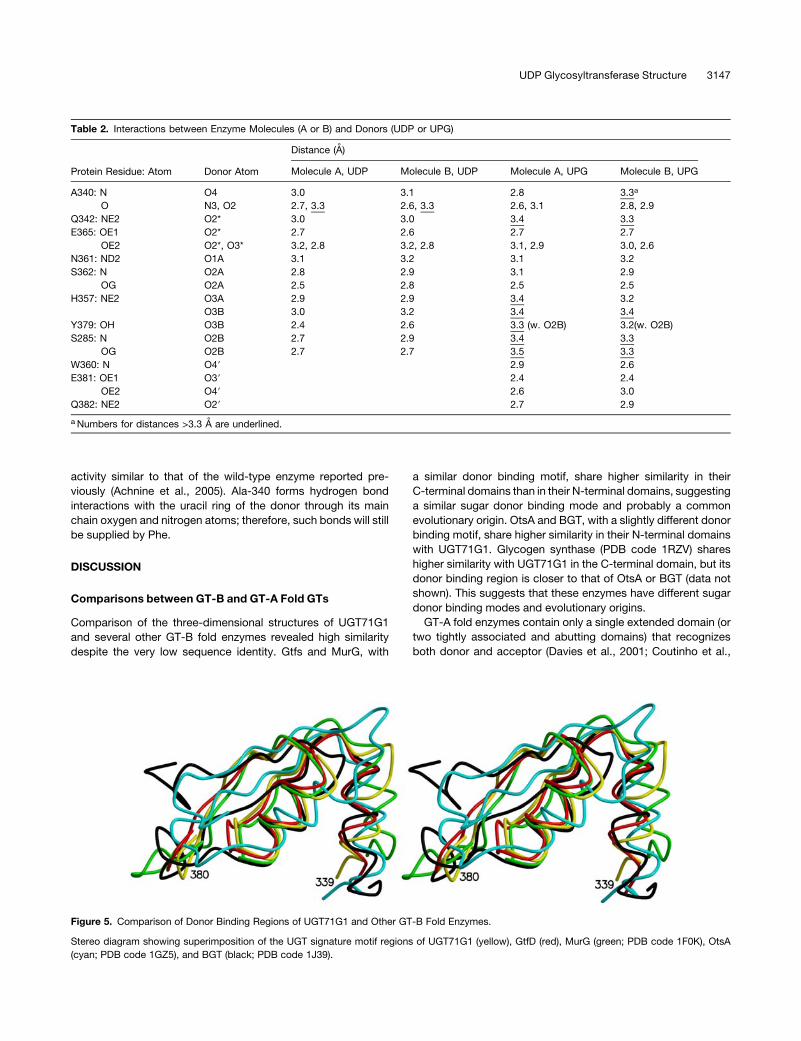
Comparisons between GT-B and GT-A Fold GTs
Comparison of the three-dimensional structures of UGT71G1
and several other GT-B fold enzymes revealed high similarity
despite the very low sequence identity. Gtfs and MurG, with
a similar donor binding motif, share higher similarity in their
C-terminal domains than in their N-terminal domains, suggesting
a similar sugar donor binding mode and probably a common
evolutionary origin. OtsA and BGT, with a slightly different donor
binding motif, share higher similarity in their N-terminal domains
with UGT71G1. Glycogen synthase (PDB code 1RZV) shares
higher similarity with UGT71G1 in the C-terminal domain, but its
donor binding region is closer to that of OtsA or BGT (data not
shown). This suggests that these enzymes have different sugar
donor binding modes and evolutionary origins.
GT-A fold enzymes contain only a single extended domain (or
two tightly associated and abutting domains) that recognizes
both donor and acceptor (Davies et al., 2001; Coutinho et al.,
Table 2. Interactions between Enzyme Molecules (A or B) and Donors (UDP or UPG)
Protein Residue: Atom Donor Atom
Distance (A)
Molecule A, UDP Molecule B, UDP Molecule A, UPG Molecule B, UPG
A340: N O4 3.0 3.1 2.8 3.3a
O N3, O2 2.7, 3.3 2.6, 3.3 2.6, 3.1 2.8, 2.9
Q342: NE2 O2* 3.0 3.0 3.4 3.3
E365: OE1 O2* 2.7 2.6 2.7 2.7
OE2 O2*, O3* 3.2, 2.8 3.2, 2.8 3.1, 2.9 3.0, 2.6
N361: ND2 O1A 3.1 3.2 3.1 3.2
S362: N O2A 2.8 2.9 3.1 2.9
OG O2A 2.5 2.8 2.5 2.5
H357: NE2 O3A 2.9 2.9 3.4 3.2
O3B 3.0 3.2 3.4 3.4
Y379: OH O3B 2.4 2.6 3.3 (w. O2B) 3.2(w. O2B)
S285: N O2B 2.7 2.9 3.4 3.3
OG O2B 2.7 2.7 3.5 3.3
W360: N O49 2.9 2.6
E381: OE1 O39 2.4 2.4
OE2 O49 2.6 3.0
Q382: NE2 O29 2.7 2.9
a Numbers for distances >3.3 A are underlined.
Figure 5. Comparison of Donor Binding Regions of UGT71G1 and Other GT-B Fold Enzymes.
Stereo diagram showing superimposition of the UGT signature motif regions of UGT71G1 (yellow), GtfD (red), MurG (green; PDB code 1F0K), OtsA
(cyan; PDB code 1GZ5), and BGT (black; PDB code 1J39).
UDP Glycosyltransferase Structure 3147

2003). The structures of GT-A fold enzymes and the N- and
C-terminal domains of GT-B fold enzymes are similar to each
other. Superimposition between a GT-A enzyme, human b1,
3-glucuronyltransferase I (family 43; PDB code 1FGG) (Pedersen
et al., 2000), and each domain of UGT71G1 gaveRMSDvalues of
2.3 A for 80 Ca atoms in the N-terminal domain and 1.96 A for 40
Ca atoms in the C-terminal domain. This finding suggests that
GT-A and GT-B enzymes are evolutionarily related. During
evolution, two domain enzymes were probably produced, with
one domain recognizing similar donors and another adapting to
divergent acceptors in the natural environment.
Substrate Binding and Catalysis by UGT71G1
Kubo et al. (2004) reported that a single mutation, H374Q, in
Aralia cordata UDP-galactose:anthocyanin galactosyltransfer-
ase (corresponding to Gln-382 in UGT71G1) switched the pre-
ferred donor from UDP-galactose to UDP-glucose and
suggested that the last residue, Gln or His, in this signature motif
would define the sugar donor specificity. However, the Scutel-
laria baicalensisUDP-glucose:flavonoid glucosyltransferasemu-
tant Q382H does not acquire galactosyltransferase activity,
suggesting that other residues might also play roles in sugar
recognition and specificity in plant UGTs. In the structure of
UGT71G1 bound with UDP-glucose, the O29 and O39 atoms of
the glucose ring of the donor form hydrogen bonds with the NE2
atom of Gln-382 and the OE1 atom of Glu-381, respectively. The
O49 atom of the glucose ring contacts both the OE2 atom of Glu-
381 and the N atom of Trp-360. If a UDP-galactose is placed in
the position of the UDP-glucose in the structure of UGT71G1, the
O49 atomof galactose points away from theOE2atomofGlu-381
and can no longer form a hydrogen bond. This suggests that Glu-
381 may be a key residue for sugar recognition and specificity.
A structural model for catalysis by plant UGTs has been
proposed based on a modeling and mutagenesis study of
UGT73A5, a betanidin glucosyltransferase from Dorotheanthus
bellidiformis (Hans et al., 2004). However, the low identity
(;14%) between UGT73A5 and the bacterial GtfB used as the
basis for the homology modeling made it difficult to model
regions with insertion or deletion of amino acid residues. Nev-
ertheless, His-22 was identified as a key residue for enzyme
activity, although it was oriented differently from that shown in
the crystal structure of UGT71G1, such that the acceptor binding
pocket was predicted to be in a different location from that
suggested by our docking results. The position of the binding
pocket for the sugar donor was predicted correctly, but the sugar
Figure 6. The Putative Acceptor Binding Pocket.
Stereo diagram showing quercetin (A) and hederagenin (B) docked into the proposed binding pockets. Quercetin, hederagenin, and UDP-glucose are
shown as ball-and-stick models. Some protein residues in the acceptor binding pocket are labeled and shown in cyan as bond models. Distances (A)
between the OH group of acceptors and the atom NE2 of His-22 or the atom C19 of UDP-glucose are labeled and indicated with dashed lines.
3148 The Plant Cell

Figure 7. Comparison of the Donor and Acceptor Regions of Functionally Characterized Plant UGTs.
Sequence alignment of 39 plant UGTs, showing the acceptor binding region (first three fragments: 1 to 55, 81 to 95, and 117 to 151) and the donor
binding region (last two fragments: 282 to 286 and 339 to 382). Identical residues are highlighted, and similar residues are enclosed in boxes. Residues
His-22 and Asp-121 are marked with asterisks. The alignment was performed using ClustalX (Thompson et al., 1997). The plant UGTs includeMedicago
truncatula UGT71G1 and UGT73K1; Allium cepa UGT73G1 and UGT73J1; Aralia cordata GaT; Bellis perennis UGAT; Brassica napus SGT1;
Catharanthus roseus UGT2; Citrus maxima 1,2RhaT; Crocus sativus UGTCs2; Dorotheanthus bellidiformis UGT73A5; Gentiana triflora 39GT;
Glycyrrhiza echinata UGT73F1; Nicotiana tabacum GT1a, GT2, and GT3; Phaseolus lunatus ZOG1; Phaseolus vulgaris ZOX1; Sorghum bicolor
UGT85B1; Stevia rebaudiana UGT74G1, UGT76G1, and UGT85C2; and Zea mays cisZOC1 and cisZOG2. All others are from Arabidopsis
thaliana.
was predicted to lie in the opposite orientation to that revealed
from our crystal structure, and His-370 (His-357 in UGT71G1)
interacted with the phosphates rather than the uracil ring of the
donor molecule. Based on the modeling study, Glu-394 was
predicted to be part of a catalytic triad. However, as revealed in
the crystal structure of UGT71G1, Glu-381 (corresponding to
Glu-394 in UGT73A5) contacts the glucose moiety of the donor
and plays an important role in sugar recognition and specificity.
Rather thanGlu-381, the highly conserved Asp residue (Asp-121)
located in the acceptor binding pocket of UGT71G1 may be
essential for enzyme catalysis.
In the model revealed by our crystal structures, Asp-121
interacts with His-22 at a distance of ;3 A. His-22 is close to
the glucose ring of the sugar donor and the docked acceptor.
The imidazole group of His-22 acts as a general base to depro-
tonate an OH group on the acceptor molecule and transfers the
proton to Asp-121. The deprotonated acceptor, as a nucleophilic
oxyanion, can then attack at the C19 carbon center of the UDP-
glucose from the opposite side of the sugar ring, with displace-
ment of the UDP moiety. The crystal structure of UGT71G1
presented here thus provides a basis for understanding the
catalytic mechanism for glycosyl transfer and also provides
a starting point for homology modeling of diverse plant UGTs.
Substrate Specificity in the Plant UGT Superfamily
Most of the residues in direct contact with donormolecules, such
as Trp-339, Gln-342, His-357, and Glu-365, are conserved in
all 39 plant UGTs functionally characterized to date (Figure 7). In
the position corresponding to Glu-381 of UGT71G1, Asp is
frequently observed, which may enter into a similar interaction
with the donor. At the position corresponding to Gln-382 of
UGT71G1, Gln is conserved in glucosyltransferases, His is
present and conserved in galactosyltransferases, and an Asn
residue is observed in Arabidopsis UGT78D1, which uses UDP-
rhamnose as the sugar donor. Residues at these two positions
interact with the sugar moiety of the donor and therefore would
be predicted to be critical for sugar specificity.
The apparent lack of stringent structural requirements for
acceptor binding and catalysis is consistent with the evolution of
varied and broad substrate specificities of many plant GTs (Vogt
and Jones, 2000; Bowles et al., 2005). Of five UGTs that we have
characterized recently from M. truncatula (Achnine et al., 2005),
all have multiple in vitro substrates, suggesting multiple potential
in vivo substrates. UGT71G1 can catalyze the glucosylation of
two triterpenes (medicagenic acid and hederagenin), the flavonol
quercetin, and the isoflavones genistein and biochanin A. For
assessment of in vivo function, therefore, it is necessary to
understand which potential substrates are present in the cell
types in which the enzyme is expressed. In the case of methyl
jasmonate–induced Medicago cell suspension cultures, such
analysis points to UGT71G1 glycosylating medicagenic acid
rather than the catalytically preferred substrates biochanin A or
quercetin (Achnine et al., 2005).
When considered in terms of the multiple products formed
from each potential substrate, such apparent promiscuity raises
two important questions: why do higher plants possess so many
different UGTs, and how are their activities regulated in vivo to
Figure 8. Glucosylation of Quercetin by Recombinant UGT71G1.
(A) HPLC trace showing the substrate quercetin (Q) and the five products (numbered 1 to 5). Solid line, complete reaction mixture; dashed line,
standards for quercetin (Q), quercetin-3-O-glucoside (peak 3), and quercetin-49-O-glucoside (peak 4). mAU, milli-absorption unit.
(B) UV absorption spectra of quercetin (Q) and products 1 to 5.
3150 The Plant Cell

allow for the production of a more limited number of products
than suggested by their in vitro substrate/product specificities?
The importance of UGTs for the detoxification of xenobiotics
(allelochemicals before the human introduction of synthetic
herbicides and pesticides) may have driven the evolution of
new forms of the enzyme differing in residues around the
acceptor binding pocket.
It has been suggested that UGTsmay be associated in enzyme
complexes with the early enzymes of the pathway in which they
are involved, and such metabolic channeling would essentially
limit a particular GT to a specific pathway irrespective of the
enzyme’s in vitro substrate specificity (Jorgensen et al., 2005).
The presence and/or nature of physical interactions between any
UGT and a related biosynthetic enzyme remain unknown. The
structures described in this work provide a basis for the explo-
ration of such interactions.
METHODS
Expression and Purification of UGT71G1
The UGT71G1 cDNA fromMedicago truncatula (Achnine et al., 2005) was
cloned into the pET28a expression vector (Novagen) with a hexahistidine
tag and a thrombin cleavage site. Escherichia coli BL21(DE3) cells
transformed with the plasmid were grown at 378C in Luria-Bertani
medium containing 50 mg/mL kanamycin until A600 ¼ 0.6 to 0.8. Cultures
were induced with 0.5 mM isopropyl 1-thio-b-galactopyranoside and
grown overnight at 168C. Cells were pelleted and resuspended in lysis
buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10 mM imidazole, and
10 mM b-mercaptoethanol). After lysis with a French press and centri-
fugation at 12,000 rpm at 48C for 20min, Ni2þ-nitrilotriacetic acid agarose
was added to the supernatant containing the target proteins. After
incubation for 40 to 60 min, the mixture was transferred into a disposable
column and washed extensively with lysis buffer (;50 column volumes).
The His-tagged proteins were eluted with elution buffer (50 mM Tris-HCl,
pH 8.0, 500 mM NaCl, 250 mM imidazole, and 10 mM b-mercaptoetha-
nol). Incubationwith biotinylated thrombin during overnight dialysis at 48C
against 20 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 10 mM b-mercap-
toethanol cleaved the N-terminal His tag. Dialyzed proteins were in-
cubated with streptavidin agarose and Ni2þ-nitrilotriacetic acid agarose
to remove thrombin and the cleaved His tag, respectively. The proteins
were further purified on Resource Q and Superdex-200 gel filtration
columns (AmershamPharmacia Biotech) and concentrated to 5mg/mL in
10 mM NaCl, 10 mM Tris-HCl, pH 7.5, and 5 mM b-mercaptoethanol.
Se-Met–substituted UGT71G1 was prepared by expressing the re-
combinant protein in E. coli B834(DE3) cells (Novagen) grown in M9
minimal medium supplemented with Se-Met (Doublie, 1997). The pro-
cedure for expression and purification was similar to the procedure
described above for the native protein.
Crystallization and Data Collection
Crystals of UGT71G1 were grown from hanging drops by the vapor
diffusion method. UGT71G1 protein at a concentration of 5 mg/mL was
mixed with 5 mM UDP-galactose and 5 mM quercetin at a 2:1 (v/v) ratio,
then mixed with an equal volume of reservoir solution containing 40%
(w/v) polyethylene glycol 3350, 0.2 M ammonium acetate, and 0.1 M
sodium citrate, pH 5.6. The mixture was equilibrated over the reservoir
solution at 208C. Crystals typically grew over 2 to 5 d with dimensions of
0.1 3 0.1 3 0.2 mm.
Before data collection, the crystals were flash-cooled to �1808C. Data
from a native protein crystal were measured to 2.0 A with a Raxis IVþþ
image plate detector and a RU3H x-ray source. The crystal belonged to
space group P21 (a ¼ 53.7 A, b ¼ 90.6 A, c ¼ 101.8 A, b ¼ 102.68). There
were two molecules per crystallographic asymmetric unit, with 47%
solvent content and a Matthews coefficient VM of 2.6 A3/D. A 2.4-A MAD
data set from a Se-Met derivative crystal, using three wavelengths, was
collected at the Gulf Coast Protein Crystallography Consortium beamline
using a MAR CCD detector.
Crystals of UGT71G1 complexed with UDP-glucose were obtained by
soaking the crystals described above with mother liquid containing
saturated UDP-glucose. Five changes of this solution were made during
the 3-d soaking period, the last being a few hours before freezing the
crystal for data collection. A 2.6-A diffraction data set was collected with
a Raxis IVþþ image plate detector and a RU3H x-ray source.
All data sets were processed using denzo and scalepack or HKL2000
(Otwinowski and Minor, 1997).
Structure Determination and Refinement
The structure of UGT71G1 was determined using the MAD method. The
MAD data (40 to 2.4 A) were analyzed with SOLVE (Terwilliger and
Berendzen, 1999), and 20 of 24 Se sites were located, yielding an overall
figure of merit of 0.44. The program RESOLVE (Terwilliger, 2000) was
used for electron density modification, phase extension to 2.0 A by
combining with the native data, and automated model building. Interac-
tive model building and crystallographic refinement were performed
using the programs O (Jones et al., 1991) and CNS (Brunger et al., 1998),
respectively. A bulk solvent correction was applied. B factors were
refined individually for the structure of UGT71G1 complexed with UDP at
2.0 A, and grouped B factor refinement was performed for the structure of
UGT71G1 complexed with UDP-glucose at 2.6 A. Water molecules were
added with Arp/wArp (Lamzin et al., 2001) and checked manually for
inclusion. In themodels, the first two and seven amino acid residues in the
N terminus were not observed inmolecules A andB, respectively, and the
last two residues in the C terminus were disordered in both molecules.
The program PROCHECK (Laskowski et al., 1993) was used to check
themodels. All backbone f-c torsion angles are within allowed regions of
the Ramachandran plot.
Molecular Docking
The automated docking program GOLD was used to dock acceptors into
the UGT71G1 active site (Jones et al., 1997). Default genetic algorithm
parameters for controlling the operation of the docking process were
used. All docking calculations were restricted to the predicted binding
pocket by defining the active site with residue His-22. For quercetin,
medicagenic acid, and hederagenin, there are several potential glyco-
sylation sites, and a distance constraint was used to define each potential
site for calculation. GOLDscore was used to identify the lowest energy
docking results. The hydrogen bonds and van der Waals contacts
between ligands and enzyme were analyzed to identify the optimal
binding mode.
Mutagenesis and Enzyme Assay
Site-directed mutants of UGT71G1 were constructed using the Quik-
Change strategy (Stratagene). The mutant proteins were expressed and
purified using the same procedures described above for native protein
except for the thrombin cleavage. Enzyme assay was performed essen-
tially according to a reported method (Lim et al., 2003). Each GT activity
assaymix (200mL) contains 2mg of recombinant protein, 50mMTris-HCl,
pH 7.0, 14 mM b-mercaptoethanol, 5 mM UDP-glucose, and 1 mM
quercetin. For mutants, increasing concentrations of substrates (2 mM
quercetin and 10 mMUDP-glucose, or 5 mM quercetin and 25 mMUDP-
glucose) and proteins (4 mg) were also used. The reaction was performed
UDP Glycosyltransferase Structure 3151

at 308C for 1 h and stoppedby the addition of 20mL of trichloroacetic acid,
quick-frozen, and stored at �208C before HPLC analysis with a Waters
Spherisorb 5u ODS2 C18 reverse-phase column (250 3 4.6 mm) on an
AgilentHP1100HPLCdevice equippedwith anautosampler, a quaternary
pump, and a diode array detector. Elution was as described previously
(Achnine et al., 2005).
Accession Numbers
The coordinates and structure factors for the structures of UGT71G1 in
complexes with UDP and UDP-glucose have been deposited to the PDB
with codes 2ACV and 2ACW, respectively. The plant UGTs included in the
alignment (Figure 7) and their accession numbers are as follows:
Medicago truncatula UGT71G1 (accession number AAW56092) and
UGT73K1 (AAW56091); Allium cepa UGT73G1 (AAP88406) and
UGT73J1 (AAP88407); Aralia cordata GaT (BAD06514); Bellis perennis
UGAT (BAD77944); Brassica napus SGT1 (AAF98390); Catharanthus
roseus UGT2 (BAD29722); Citrus maxima 1,2RhaT (AAL06646); Crocus
sativus UGTCs2 (AAP94878); Dorotheanthus bellidiformis UGT73A5
(CAB56231); Gentiana triflora 39GT (BAC54092); Glycyrrhiza echinata
UGT73F1 (BAC78438); Nicotiana tabacum GT1a (BAB60721), GT2
(BAB88935), and GT3 (BAB88934); Phaseolus lunatus ZOG1
(AAD04166); Phaseolus vulgaris ZOX1 (AAD51778); Sorghum bicolor
UGT85B1 (AAF17077); Stevia rebaudiana UGT74G1 (AAR06920),
UGT76G1 (AAR06912), and UGT85C2 (AAR06916); Zea mays cisZOC1
(AAK53551) and cisZOG2 (AAL92460); and Arabidopsis thaliana
UGT71C1 (AAC35226), UGT72B1 (AAB61023), UGT72E2 (BAA97275),
UGT73C5 (AAD20156), UGT73C6 (AAD20155), UGT74B1 (AAC00570),
UGT74F2 (AAB64024), UGT84A2 (BAB02351), UGT84A1 (CAB10326),
UGT84B1 (AAB87119), UGT76C1 (BAB10792), UGT76C2 (AAN28835),
UGT78D1 (AAF19756), UGT85A1 (AAF18537), and UGT91B1
(BAA98174).
ACKNOWLEDGMENTS
We thank S. Liu for initial protein purification and crystallization
screening, J. Lin for technical assistance, L.W. Sumner, Z. Lei, and D.
Huhman for mass spectrometric analysis, N. Duke (Structural Biology
Center) and I. Koshelev (Industrial Macromolecular Crystallography
Association–Collaborative Access Team) at the Advanced Photon
Source (Argonne National Laboratory) for assistance with data collection
of initial crystals, H. Bellamy and D. Neau of the Gulf Coast Protein
Crystallography Consortium beamline of the Center for Advanced
Microstructures and Devices (Louisiana State University) for help with
Se-Met MAD data collection, and T.M.T. Hall and P. Xu for critical
reading of the manuscript. This work was supported by National
Science Foundation Grant 0416883 and the Samuel Roberts Noble
Foundation.
Received June 8, 2005; revised August 21, 2005; accepted September
19, 2005; published October 7, 2005.
REFERENCES
Achnine, L., Huhman, D.V., Farag, M.A., Sumner, L.W., Blount, J.W.,
and Dixon, R.A. (2005). Genomics-based selection and functional
characterization of triterpene glycosyltransferases from the model
legume Medicago truncatula. Plant J. 41, 875–887.
Behboudi, S., Morein, B., and Villacres-Eriksson, M.C. (1999). Quil-
laja saponin formulations that stimulate proinflammatory cytokines
elicit a potent acquired cell-mediated immunity. Scand. J. Immunol.
50, 371–377.
Bowles, D., Isayenkova, J., Lim, E., and Poppenberger, B. (2005).
Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant
Biol. 8, 254–263.
Breton, C., Heissigerova, H., Jeanneau, C., Moravcova, J., and
Imberty, A. (2002). Comparative aspects of glycosyltransferases.
Biochem. Soc. Symp. 69, 23–32.
Brunger, A.T., et al. (1998). Crystallography & NMR system: A new
software suite for macromolecular structure determination. Acta
Crystallogr. D Biol. Crystallogr. 54, 905–921.
Buschiazzo, A., Ugalde, J.E., Guerin, M.E., Shepard, W., Ugalde,
R.A., and Alzari, P.M. (2004). Crystal structure of glycogen synthase:
Homologous enzymes catalyze glycogen synthesis and degradation.
EMBO J. 23, 3196–3205.
Campbell, J.A., Davies, G.J., Bulone, V., and Henrissat, B. (1997). A
classification of nucleotide-diphospho-sugar glycosyltransferases
based on amino acid sequence similarities. Biochem. J. 326,
929–939.
Charnock, S.J., and Davies, G.J. (1999). Structure of the nucleotide-
diphospho-sugar transferase, SpsA from Bacillus subtilis, in native
and nucleotide-complexed forms. Biochemistry 38, 6380–6385.
Clemens, D., Dirk, H., Sven-Eric, W., Koji, I., Monika, W., Ursula,
V.M., Jon, S.T., and Andreas, B. (2004). The glycosyltransferase
UrdGT2 catalyzes both C- and O-glycosidic sugar transfers. Angew.
Chem. Int. Ed. Engl. 43, 2962–2965.
Coutinho, P.M., Deleury, E., Davies, G.J., and Henrissat, B. (2003).
An evolving hierarchical family classification for glycosyltransferases.
J. Mol. Biol. 328, 307–317.
Davies, G.J., Charnock, S.J., and Henrissat, B. (2001). The enzymatic
synthesis of glycosidic bonds: ‘‘Glycosynthases’’ and glycosyltrans-
ferases. Trends Glycosci. Glycotechnol. 13, 105–120.
Dennehy, C. (2001). Botanicals in cardiovascular health. Clin. Obstet.
Gynecol. 44, 814–823.
de Wildt, S.N., Kearns, G.L., Leeder, J.S., and van den Anker, J.N.
(1999). Glucuronidation in humans. Pharmacogenetic and develop-
mental aspects. Clin. Pharmacokinet. 36, 439–452.
Doublie, S. (1997). Preparation of selenomethionyl proteins for phase
determination. Methods Enzymol. 276, 523–530.
Fritz, T.A., Hurley, J.H., Trinh, L.B., Shiloach, J., and Tabak, L.A.
(2004). The beginnings of mucin biosynthesis: The crystal structure of
UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-
T1. Proc. Natl. Acad. Sci. USA 101, 15307–15312.
Gastinel, L.N., Bignon, C., Misra, A.K., Hindsgaul, O., Shaper, J.H.,
and Joziasse, D.H. (2001). Bovine alpha1,3-galactosyltransferase
catalytic domain structure and its relationship with ABO histo-blood
group and glycosphingolipid glycosyltransferases. EMBO J. 20,
638–649.
Gastinel, L.N., Cambillau, C., and Bourne, Y. (1999). Crystal struc-
tures of the bovine beta4-galactosyltransferase catalytic domain
and its complex with uridine diphosphogalactose. EMBO J. 18,
3546–3557.
Gibbons, B.J., Roach, P.J., and Hurley, T.D. (2002). Crystal structure
of the autocatalytic initiator of glycogen biosynthesis, glycogenin.
J. Mol. Biol. 319, 463–477.
Gibson, R.P., Turkenburg, J.P., Charnock, S.J., Lloyd, R., and
Davies, G.J. (2002). Insights into trehalose synthesis provided by
the structure of the retaining glucosyltransferase OtsA. Chem. Biol. 9,
1337–1346.
Goldsmith, E.J., Sprang, S.R., Hamlin, R., Xuong, N.H., and Fletterick,
R.J. (1989). Domain separation in the activation of glycogen phos-
phorylase a. Science 245, 528–532.
Gouet, P., and Courcelle, E. (2002). ENDscript: A workflow with web
interface to display sequence and structure information. Bioinformat-
ics 18, 767–768.
3152 The Plant Cell

Ha, S., Walker, D., Shi, Y., and Walker, S. (2000). The 1.9 A crystal
structure of Escherichia coli MurG, a membrane-associated glycosyl-
transferase involved in peptidoglycan biosynthesis. Protein Sci. 9,
1045–1052.
Hans, J., Brandt, W., and Vogt, T. (2004). Site-directed mutagenesis
and protein 3D-homology modelling suggest a catalytic mechanism
for UDP-glucose-dependent betanidin 5-O-glucosyltransferase from
Dorotheanthus bellidiformis. Plant J. 39, 319–333.
Haridas, V., Higuchi, M., Jayatilake, G.S., Bailey, D., Mujoo, K.,
Blake, M.E., Arntzen, C.J., and Gutterman, J.U. (2001). Avicins:
Triterpenoid saponins from Acacia victoriae (Bentham) induce apo-
ptosis by mitochondrial perturbation. Proc. Natl. Acad. Sci. USA 98,
5821–5826.
Hu, Y., Chen, L., Ha, S., Gross, B., Falcone, B., Walker, D.,
Mokhtarzadeh, M., and Walker, S. (2003). Crystal structure of the
MurG:UDP-GlcNAc complex reveals common structural principles of
a superfamily of glycosyltransferases. Proc. Natl. Acad. Sci. USA 100,
845–849.
Hu, Y., and Walker, S. (2002). Remarkable structural similarities
between diverse glycosyltransferases. Chem. Biol. 9, 1287–1296.
Hughes, J., and Hughes, M.A. (1994). Multiple secondary plant product
UDP-glucose glucosyltransferase genes expressed in cassava (Man-
ihot esculenta Crantz) cotyledons. DNA Seq. 5, 41–49.
Huhman, D.V., and Sumner, L.W. (2002). Metabolic profiling of
saponins in Medicago sativa and Medicago truncatula using HPLC
coupled to an electrospray ion-trap mass spectrometer. Phytochem-
istry 59, 347–360.
Jones, G., Willett, P., Glen, R.C., Leach, A.R., and Taylor, R. (1997).
Development and validation of a genetic algorithm for flexible dock-
ing. J. Mol. Biol. 267, 727–748.
Jones, P., and Vogt, T. (2001). Glycosyltransferases in secondary
plant metabolism: Tranquilizers and stimulant controllers. Planta 213,
164–174.
Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. (1991).
Improved methods for the building of protein models in electron
density maps and the location of errors in these models. Acta
Crystallogr. A47, 110–119.
Jorgensen, K., Rasmussed, A.V., Morant, M., Nielsen, A.H.,
Bjarnholt, N., Zagrobelny, M., Bak, S., and Moller, B.L. (2005).
Metabolon formation and metabolic channeling in the biosynthesis of
plant natural products. Curr. Opin. Plant Biol. 8, 280–291.
Kraulis, P.J. (1991). MOLSCRIPT: A program to produce both detailed
and schematic plots of protein structures. J. Appl. Crystallogr. 24,
946–950.
Kubo, A., Arai, Y., Nagashima, S., and Yoshikawa, T. (2004).
Alteration of sugar donor specificities of plant glycosyltransferases
by a single point mutation. Arch. Biochem. Biophys. 429, 198–203.
Lamzin, V.S., Perrakis, A., and Wilson, K.S. (2001). The ARP/WARP
suite for automated construction and refinement of protein models.
In International Tables for Crystallography, M.G. Rossmann and
E. Arnold, eds (Dordrecht, The Netherlands: Kluwer Academic Pub-
lishers), pp. 720–722.
Lariviere, L., Gueguen-Chaignon, V., and Morera, S. (2003). Crystal
structures of the T4 phage beta-glucosyltransferase and the D100A
mutant in complex with UDP-glucose: Glucose binding and identifi-
cation of the catalytic base for a direct displacement mechanism.
J. Mol. Biol. 330, 1077–1086.
Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M.
(1993). PROCHECK—A program to check the stereochemical quality
of protein structures. J. Appl. Crystallogr. 26, 283–291.
Li, Y., Baldauf, S., Lim, E.K., and Bowles, D.J. (2001). Phylogenetic
analysis of the UDP-glycosyltransferase multigene family of Arabi-
dopsis thaliana. J. Biol. Chem. 276, 4338–4343.
Lim, E., Baldauf, S., Li, Y., Elias, L., Worrall, D., Spencer, S.P.,
Jackson, R.G., Taguchi, G., Ross, J., and Bowles, D.J. (2003).
Evolution of substrate recognition across a multigene family of
glycosyltransferases in Arabidopsis. Glycobiology 13, 139–145.
Lobsanov, Y.D., Romero, P.A., Sleno, B., Yu, B., Yip, P., Herscovics,
A., and Howell, P.L. (2004). Structure of Kre2p/Mnt1p: A yeast
alpha1,2-mannosyltransferase involved in mannoprotein biosynthesis.
J. Biol. Chem. 279, 17921–17931.
Merritt, E.A., and Bacon, D.J. (1997). Raster3D: Photorealistic molec-
ular graphics. In Methods in Enzymology, C.W.J. Carter and R.M.
Sweet, eds (New York: Academic Press), pp. 505–524.
Mulichak, A.M., Losey, H.C., Lu, W., Wawrzak, Z., Walsh, C.T., and
Garavito, R.M. (2003). Structure of the TDP-epi-vancosaminyltrans-
ferase GtfA from the chloroeremomycin biosynthetic pathway. Proc.
Natl. Acad. Sci. USA 100, 9238–9243.
Mulichak, A.M., Losey, H.C., Walsh, C.T., and Garavito, R.M. (2001).
Structure of the UDP-glucosyltransferase GtfB that modifies the
heptapeptide aglycone in the biosynthesis of vancomycin group
antibiotics. Structure 9, 547–557.
Mulichak, A.M., Lu, W., Losey, H.C., Walsh, C.T., and Garavito, R.M.
(2004). Crystal structure of vancosaminyltransferase GtfD from the
vancomycin biosynthetic pathway: Interactions with acceptor and
nucleotide ligands. Biochemistry 43, 5170–5180.
Oh, S.R., Kinjo, J., Shii, T., Ikeda, T., Nohara, T., Ahn, K.S., Kim, J.H.,
and Lee, H.K. (2000). Effects of triterpenoids from Pueraria lobata on
immunohemolysis: Beta-D-glucuronic acid plays an active role in
anticomplementary activity in vitro. Planta Med. 66, 506–510.
Oleszek, W., Junkuszew, M., and Stochmal, A. (1999). Determination
and toxicity of saponins from Amaranthus cruentus seeds. J. Agric.
Food Chem. 47, 3685–3687.
Osbourn, A.E. (2003). Molecules of interest. Saponins in cereals.
Phytochemistry 62, 1–4.
Osbourn, A.E., Bowyer, P., and Daniels, M.J. (1996). Saponin
detoxification by plant pathogenic fungi. Adv. Exp. Med. Biol. 404,
547–555.
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction
data collected in oscillation mode. In Methods in Enzymology, C.W.L.
Carter and R.M. Sweet, eds (New York: Academic Press), pp.
307–326.
Papadopoulou, K., Melton, R.E., Leggett, M., Daniels, M.J., and
Osbourn, A.E. (1999). Compromised disease resistance in saponin-
deficient plants. Proc. Natl. Acad. Sci. USA 96, 12923–12928.
Paquette, S., Moller, B.L., and Bak, S. (2003). On the origin of family 1
plant glycosyltransferases. Phytochemistry 62, 399–413.
Pedersen, L.C., Dong, J., Taniguchi, F., Kitagawa, H., Krahn, J.M.,
Pedersen, L.G., Sugahara, K., and Negishi, M. (2003). Crystal
structure of an alpha 1,4-N-acetylhexosaminyltransferase (EXTL2),
a member of the exostosin gene family involved in heparan sulfate
biosynthesis. J. Biol. Chem. 278, 14420–14428.
Pedersen, L.C., Tsuchida, K., Kitagawa, H., Sugahara, K., Darden,
T.A., and Negishi, M. (2000). Heparan/chondroitin sulfate biosynthe-
sis: Structure and mechanism of human glucuronyltransferase I. J.
Biol. Chem. 275, 34580–34585.
Persson, K., Ly, H.D., Dieckelmann, M., Wakarchuk, W.W., Withers,
S.G., and Strynadka, N.C. (2001). Crystal structure of the retaining
galactosyltransferase LgtC from Neisseria meningitidis in complex
with donor and acceptor sugar analogs. Nat. Struct. Biol. 8, 166–175.
Ross, J., Li, Y., Lim, E., and Bowles, D.J. (2001). Higher plant
glycosyltransferases. Genome Biol. 2, 3004.1–3004.6.
Sandermann, H., Jr. (1992). Plant metabolism of xenobiotics. Trends
Biochem. Sci. 17, 82–84.
Small, E. (1996). Adaptations to herbivory in alfalfa (Medicago sativa).
Can. J. Bot. 74, 807–822.
UDP Glycosyltransferase Structure 3153

Sprang, S., Standing, T., Fletterick, R.J., Stroud, R.M., Finer-Moore,
J., Xuong, N.H., Hamlin, R., Rutter, W.J., and Craik, C.S. (1987).
The three-dimensional structure of Asn102 mutant of trypsin: role of
Asp102 in serine protease catalysis. Science 237, 905–909.
Terwilliger, T.C. (2000). Maximum likelihood density modification. Acta
Crystallogr. D Biol. Crystallogr. 56, 965–972.
Terwilliger, T.C., and Berendzen, J. (1999). Automated MAD and
MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55,
849–861.
Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., and
Higgins, D.G. (1997). The ClustalX windows interface: Flexible strat-
egies for multiple sequence alignment aided by quality analysis tools.
Nucleic Acids Res. 24, 4876–4882.
Unligil, U.M., and Rini, J.M. (2000). Glylcosyltransferase structure and
mechanism. Curr. Opin. Struct. Biol. 10, 510–517.
Unligil, U.M., Zhou, S., Yuwaraj, S., Sarkar, M., Schachter, H., and
Rini, J.M. (2000). X-ray crystal structure of rabbit N-acetylglucosa-
minyltransferase I: Catalytic mechanism and a new protein super-
family. EMBO J. 19, 5269–5280.
Vogt, T., and Jones, P. (2000). Glycosyltransferases in plant natural
product synthesis: Characterization of a supergene family. Trends
Plant Sci. 5, 380–386.
Vrielink, A., Ruger, W., Driessen, H.P., and Freemont, P.S. (1994).
Crystal structure of the DNA modifying enzyme b-glucosyltransferase
in the presence and absence of the substrate uridine diphosphoglu-
cose. EMBO J. 13, 3413–3422.
3154 The Plant Cell

DOI 10.1105/tpc.105.035055; originally published online October 7, 2005; 2005;17;3141-3154Plant Cell
Hui Shao, Xianzhi He, Lahoucine Achnine, Jack W. Blount, Richard A. Dixon and Xiaoqiang Wangtruncatula
MedicagoCrystal Structures of a Multifunctional Triterpene/Flavonoid Glycosyltransferase from
This information is current as of February 25, 2020
References /content/17/11/3141.full.html#ref-list-1
This article cites 62 articles, 17 of which can be accessed free at:
Permissions https://www.copyright.com/ccc/openurl.do?sid=pd_hw1532298X&issn=1532298X&WT.mc_id=pd_hw1532298X
eTOCs http://www.plantcell.org/cgi/alerts/ctmain
Sign up for eTOCs at:
CiteTrack Alerts http://www.plantcell.org/cgi/alerts/ctmain
Sign up for CiteTrack Alerts at:
Subscription Information http://www.aspb.org/publications/subscriptions.cfm
is available at:Plant Physiology and The Plant CellSubscription Information for
ADVANCING THE SCIENCE OF PLANT BIOLOGY © American Society of Plant Biologists


















