
Core/Shell Nanospheres, Hollow Capsules, and...
15
Core/Shell Nanospheres, Hollow Capsules, and Bottles Gang Zhang Kai Zhang Jilin University, Changchun, People’s Republic of China Yi Yu Chinese Academy of Sciences, Changchun, People’s Republic of China Bai Yang Jilin University, Changchun, People’s Republic of China INTRODUCTION The design and synthesis of nanoscale materials is im- portant in the fabrication of advanced devices for optics, electronics, and biotechnology. [1–6] Over the past decade, considerable effort has been devoted to the design and controlled fabrication of nanostructured materials with functional properties. The interest in nanoscale materials comes from the fact that their properties (optical, elec- trical, or chemical, etc.) are the functions of their size, composition, and structural order. Colloidal particles represent attractive building blocks from which to create ordered and complex materials. They are also of wide- spread interest in chemical engineering, biological, and pharmaceutical applications. [7] In biotechnology, the encapsulation and delivery of proteins and DNA into cells has led to the implementation of intracellular medicinal therapies such as gene therapy. [8,9] Comple- tion of the human genome project has ensured the former, leaving the synthesis of encapsulation and delivery materials as perhaps the single most important challenge in intracellular medicinal therapies. Recently, core-shell particles and micro- or nanosized capsules have received considerable attentions for their techno- logical importance in many fields. [10–25] There have been many efforts to fabricate core-shell colloidal materials with tailored structural, surface, and optical proper- ties. [26–28] The creation of core-shell colloidal particles is also of interest from a fundamental viewpoint, especially in the areas of colloid and interface science. They can be utilized as model systems to investigate factors govern- ing colloidal interactions and stabilization and to gain valuable information on the properties of concentrated dispersions. [29,30] OVERVIEW The synthesis of core-shell particles typically involves tailoring the surface properties of particles, often accom- plished by coating or encapsulating them within a shell of a preferred material. The shell can alter the charge, functionality, and reactivity of the surface, and can en- hance the stability and dispersibility of the colloidal core. Optical, magnetic, or catalytic functions may be readily imparted to the dispersed colloidal matter depending on the properties of the coating. Encapsulating colloids in a shell of different composition may also protect the core from extraneous chemical and physical changes. [31,32] Core-shell particles often exhibit improved physical and chemical properties over their single-component counter- parts and, hence, are potentially useful in a broader range of applications. Optimization of the surface characteristics of particles through coating processes is also of primary importance for the successful application of composite particles. Recent methods offer new alternatives for the controlled synthesis of novel, stable, and functional core- shell type materials. An important extension of core-shell particles is the subsequent removal of the core by either thermal or chemical means (selective etching with a solvent or cal- cination in air), forming hollow spheres. A variety of procedures currently used to fabricate a wide range of stable hollow capsules of various compositions have been reported. [33] Hollow capsules of nanometer to micrometer dimensions constitute an important class of materials that are employed in various technological applications, such as encapsulate agents for delivery of cosmetic, drug, ca- talysis, and protecting sensitive agents. They may also provide some immediate advantages over their solid Dekker Encyclopedia of Nanoscience and Nanotechnology 865 DOI: 10.1081/E-ENN 120013629 Copyright D 2004 by Marcel Dekker, Inc. All rights reserved.
Transcript of Core/Shell Nanospheres, Hollow Capsules, and...

Core/Shell Nanospheres, Hollow Capsules,and Bottles
Gang ZhangKai ZhangJilin University, Changchun, People’s Republic of China
Yi YuChinese Academy of Sciences, Changchun, People’s Republic of China
Bai YangJilin University, Changchun, People’s Republic of China
INTRODUCTION
The design and synthesis of nanoscale materials is im-
portant in the fabrication of advanced devices for optics,
electronics, and biotechnology.[1–6] Over the past decade,
considerable effort has been devoted to the design and
controlled fabrication of nanostructured materials with
functional properties. The interest in nanoscale materials
comes from the fact that their properties (optical, elec-
trical, or chemical, etc.) are the functions of their size,
composition, and structural order. Colloidal particles
represent attractive building blocks from which to create
ordered and complex materials. They are also of wide-
spread interest in chemical engineering, biological, and
pharmaceutical applications.[7] In biotechnology, the
encapsulation and delivery of proteins and DNA into
cells has led to the implementation of intracellular
medicinal therapies such as gene therapy.[8,9] Comple-
tion of the human genome project has ensured the
former, leaving the synthesis of encapsulation and
delivery materials as perhaps the single most important
challenge in intracellular medicinal therapies. Recently,
core-shell particles and micro- or nanosized capsules
have received considerable attentions for their techno-
logical importance in many fields.[10–25] There have been
many efforts to fabricate core-shell colloidal materials
with tailored structural, surface, and optical proper-
ties.[26–28] The creation of core-shell colloidal particles is
also of interest from a fundamental viewpoint, especially
in the areas of colloid and interface science. They can be
utilized as model systems to investigate factors govern-
ing colloidal interactions and stabilization and to gain
valuable information on the properties of concentrated
dispersions.[29,30]
OVERVIEW
The synthesis of core-shell particles typically involves
tailoring the surface properties of particles, often accom-
plished by coating or encapsulating them within a shell
of a preferred material. The shell can alter the charge,
functionality, and reactivity of the surface, and can en-
hance the stability and dispersibility of the colloidal core.
Optical, magnetic, or catalytic functions may be readily
imparted to the dispersed colloidal matter depending on
the properties of the coating. Encapsulating colloids in a
shell of different composition may also protect the core
from extraneous chemical and physical changes.[31,32]
Core-shell particles often exhibit improved physical and
chemical properties over their single-component counter-
parts and, hence, are potentially useful in a broader range
of applications. Optimization of the surface characteristics
of particles through coating processes is also of primary
importance for the successful application of composite
particles. Recent methods offer new alternatives for the
controlled synthesis of novel, stable, and functional core-
shell type materials.
An important extension of core-shell particles is the
subsequent removal of the core by either thermal or
chemical means (selective etching with a solvent or cal-
cination in air), forming hollow spheres. A variety of
procedures currently used to fabricate a wide range of
stable hollow capsules of various compositions have been
reported.[33] Hollow capsules of nanometer to micrometer
dimensions constitute an important class of materials that
are employed in various technological applications, such
as encapsulate agents for delivery of cosmetic, drug, ca-
talysis, and protecting sensitive agents. They may also
provide some immediate advantages over their solid
Dekker Encyclopedia of Nanoscience and Nanotechnology 865
DOI: 10.1081/E-ENN 120013629
Copyright D 2004 by Marcel Dekker, Inc. All rights reserved.

ORDER REPRINTS
counterparts because of their relatively low densities and
as fillers with low dielectric constant in electronic com-
ponents. Using various chemical and physicochemical
methods nowadays routinely produces hollow capsules
comprising polymer, glass, metal, and ceramic.
As particular examples, hollow spheres with mesopor-
ous wall have been synthesized from gel composite;[34,35]
however, large entities such as macromolecules usually
cannot penetrate such microspheres. It would be desirable
to leave a hole on the shell surface for transporting var-
ious molecules. Lin and coworkers synthesized a vesicular
hollow microspheres that possess a pair of holes of
submicron size on exactly opposite sides.[36] Recently, our
group obtained the nanobottles through the removal of
template functional polymer and silica cores through
programmed calcination at high temperature and solution
etching, respectively. Because there is an opening on the
hollow cavity of silica nanobottle, it can afford both a
channel for transmission and a container for storage. So
the nanobottles can be used as an extremely small con-
tainer for encapsulation, as well as a nanosized carrier and
reactor for catalysis and microreaction. Furthermore, the
encapsulation of rare earth complex in the nanobottles re-
veals a potential application for nanotechnique.
This article provides an overview of the various
methods used to synthesize core-shell particles, hollow
capsules, and bottles in the nanometer to the micrometer
size range, detailing early and very recent developments
in the above area.
NANOSIZED CORE-SHELL SPHERES
Nanosized Core-Shell Sphereswith Polymer Shell
Polymer-coated spheres offer interesting prospects in a
broad spectrum of applications, ranging from catalysis to
additives and dyes, where they are exploited in the
manufacture of cosmetics, inks, and paints. The synthetic
routes that have been developed in order to produce
polymer-coated spheres fall into two main classes:
polymerization at the sphere surface or adsorption onto
the spheres. Hofman-Caris has comprehensively reviewed
the processes used to obtain spheres that consist of an
inorganic core and a polymer shell through polymeriza-
tion and chemical coupling procedures prior to 1994.[28]
We will deal with more recent strategies used to coat
spheres with polymers, polymerization approaches, and
the self-assembly of polymers from solution.
A number of polymerization-based methods have been
employed to produce spheres that consist of solid cores
coated with a shell of polymeric materials. These include
monomer adsorption onto spheres followed by subse-
quent polymerization,[37–42] heterocoagulation–polymeri-
zation,[43] and emulsion polymerization.[28,44] In the first
approach, one of the most frequently employed to achieve
polymer coatings on solid spheres, the polymerization
reaction can be catalyzed either by an initiator to promote
the process or by the colloidal spheres themselves.
Atom transfer radical polymerization (ATRP) is a
versatile technique, which offers several advantages over
other polymerization routes including control over mo-
lecular weight and molecular weight distribution.[45,46]
Also, the polymers can be end-functionalized or block
copolymerized upon the addition of other monomers.[46]
Not only does this feature offer tailorability of the
polymer coating with a variety of compositions and
functionalities, but also this feature may be important in
biomedical applications to modify the polymer shell with
biological moieties for specific cellular interactions.
ATRP has been able to form PMMA and PS shells on
silica nanoparticles,[45] and provides magnetic core-shell
nanospheres with size <15 nm. Magnetic studies show
a decrease in coercivity, which is consistent with the
reduction of magnetic surface anisotropy upon polymer
coating. Certainly the magnetic core of these core-shell
nanospheres can be selected, depending upon the desired
super paramagnetic properties for specific applications
such as in data storage and MRI contrast enhance-
ment.[47] Moreover, the resulting core-shell nanospheres
are within the biological size restrictions and may poten-
tially be modified for a particular biospecificity.
Matijevic et al. reported the coating of aluminum
hydrous oxide-modified silica spheres with poly(divinyl-
benzene) (PDVB) layers by pretreatment of the inorganic
cores with coupling agents such as 4-vinylpyridine or 1-
vinyl-2-pyrrolidone, followed by subsequent admixing of
divinylbenzene and a radical initiator.[37] Polymer layers
of poly(vinylbenzyl chloride) (PVBC), copolymers of
PDVB–PVBC, and double shells of PDVB and PVBC
were also synthesized around inorganic spheres using a
similar approach.[38]
The use of electrochemical or soluble initiators can be
eliminated by utilizing catalytically active cores to ef-
fect the polymerization of monomers adsorbed on the
surface of spheres. This approach was employed to obtain
poly(pyrrole) coatings on a range of inorganic cores by
using the active sites on the metal oxide surfaces to initiate
the polymerization of pyrrole.[39] Hematite, silica-modi-
fied hematite, and cerium(IV) oxide (CeO2) were coated
with poly(pyrrole) by exposing the inorganic cores to the
polymerization medium of pyrrole in an ethanol/water
mixture and heating to 100�C.
Moller et al. presented a work directed toward the
formation of core-shell particles consisting of a nanocrys-
tal of Au surrounded by a shell of conducting polymer,
e.g., polypyrrole.[48] Because of the different chemical
866 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
nature of the two materials, charge transfer might be
expected at the interface and the optical functions of both
materials should be drastically different from the
corresponding bulk materials.[49] Preliminary experiments
using solutions of tetrachloroauric acid (HAuCl4) and
pyrrole, without diblock copolymer, have demonstrated
the formation of PPY and Au. However, in this case it was
not possible to prevent the macroscopic segregation of the
polymer and the metal phase. Macroscopic segregation
can be prevented efficiently if the composite particles are
formed within the cores of the micelles of a diblock
copolymer. Fig. 1 shows the micrograph recorded after
annealing the film at 130�C. Uniform Au particles with a
diameter of 7 nm formed within the micelles. The figure
indicates that the originally indistinct ultrasmall clusters
of elementary gold coalesced upon treatment above the
glass transition temperature of both blocks. When the
same annealing procedure was applied to an Au-loaded
micellar film that had not been treated with pyrrole, larger
Au particles were formed and the micellar organization
was destroyed. Thus the presence of polypyrrole or
pyrrole oligomers is essential to yield a single Au particle
within each spherical microdomain.
Methods to coat a polymer shell with a controllable
thickness on magnetic nanoparticles may aid in the
development of ordered arrays of magnetic nanoparti-
cles. The formation of polymeric shells is essential for
biomedical applications of magnetic nanoparticles such as
magnetic targeting drug delivery and magnetic resonance
imaging (MRI) contrast enhancement. Many methods
usually create micrometer-sized magnetic polymer
spheres, which are large for in vivo applications.[50] A
less than 20-nm size has been suggested for the efficient
diffusion of nanoparticles through tissue in MRI applica-
tions.[51] An emulsion polymerization of poly(methyl
methacrylate) (PMMA) on �10-nm core of mixed-phase
iron oxides has made improvement; the particle size is
still >130 nm.[52,53] Polystyrene (PS) is easy to synthesize
for testing various strategies of coating nanoparticles
with polymer shells. Zhang et al. reported the formation of
magnetic MnFe2O4 PS nanoparticles using ATRP yield-
ing a core-shell nanoparticle with size <15 nm.[54] Most
polymer-coating studies on magnetic nanoparticles form
the nanoparticle core (typically Fe, Fe2O3, or Fe3O4) at the
same time as that of polymerization.[55] The MnFe2O4
nanoparticles as the magnetic core were separately
prepared by a reverse micelle microemulsion proce-
dure.[56] Polymerization initiators are chemically attached
onto the surface of nanoparticles. The modified nanopar-
ticles are then used as macro-initiators in the subsequent
polymerization reaction. This approach provides great
flexibility in the selection of magnetic core. Conse-
quently, magnetic tunability can be introduced into these
core-shell nanosphere systems to achieve the desired
super paramagnetic response.[57]
Inspired by the nanosized, amphiphilic core-shell
structure of lipoproteins, shell cross-linked nanoparticles
with a hydrophobic core, contained within a hydrogel
network, were prepared by the self-assembly of amphi-
philic block copolymers followed by intramicellar cross-
linking between the polymeric chains located within the
shell.[58] The control over size, shape, and composition of
these nanoparticles holds great potential in drug delivery
applications.[59,60] Intramicellar cross-linking of the poly-
mer chains within the shells of polystyrene-b-poly(acrylic
acid) micelles by reaction with difunctional poly(ethylene
oxide) afforded unimolecular amphiphilic core-shell
nanospheres (50 nm hydrodynamic radius).[61]
The controlled release of polymer chains from the core
by adjusting the cross-link density of the shell opens the
possibilities of designing polymeric nanoparticles with
specific shell permeabilities, capable of delivery of large
guests. This approach may provide a solution to some
of the delivery problems posed by biologically active
molecules, such as peptides and proteins, genes and
Fig. 1 TEM micrograph of a colloidal polymer film ([PY]/
[HAuCl4]=3.0) after annealing at 130�C for 140 min, exhibiting
7 nm wide Au clusters in each micelle encapsulated by PPY.
(From Ref. [48].)
Core/Shell Nanospheres, Hollow Capsules, and Bottles 867

ORDER REPRINTS
oligonucleotides. The results of this study also provide
a foundation for better understanding of the porosity of
the cross-linked shell.[62] This represents a methodology
to probe the permeability of nanoscopic membranes and
a means for applications in the controlled release of
macromolecular species.
Our group described a flexible method for preparing
monodisperse silica–PS core-shell microspheres. In this
method, silica nanoparticles grafted with 3-(trimethoxy-
silyl) propyl methacrylate (MPS) was employed in an
emulsion polymerization as seeds. The thickness of the
shells could be changed through varying the amount of
monomer and emulsifier. The monodispersity and dia-
meters of the core-shell microspheres were found to
depend on the size of grafted silica nanoparticles and the
concentration of emulsifier.
The monodisperse silica microspheres with average
radii ranging from 35 to 90 nm were prepared in ethanol
according to the Stober method[63] at ambient tempera-
ture. In order to obtain a functionalized surface, MPS
with C C bond was added and reacted with the Si–OH
group on the surface of the silica by hydrolysis. Mono-
disperse silica–polymer core-shell microspheres were ob-
tained through emulsion polymerization of styrene (St)
or methyl methacrylate (MMA), while grafted silica
particles dispersed in ethanol, which acts as ‘‘seeds’’ in
the polymerization process.[64]
Fig. 2 shows the TEM images of the resulting silica–
PMMA (left) and silica–PS (right) core-shell micro-
spheres. The spherical particles show obvious core-shell
structures, light shells (PMMA or PS) coat the dark
grafted silica microspheres cores, and over about 90%
of these core-shell microspheres have only one single
core. The average radius of the monodisperse core-shell
microspheres varies from 45 to 150 nm for silica–PMMA
and from 80 to 210 nm for silica–PS, which have been
confirmed by the TEM.
A series of TEM images of core-shell microspheres
prepared by increasing the amount of styrene (St) prove
that the grafted silica nanoparticles act as ‘‘seeds’’ in the
emulsion polymerization.[64] The ‘‘raspberry’’ morpholo-
gy of core-shell microspheres was seen, and it was clearly
visible that the surfaces of shells became smoother and the
shells thickened with increasing the amount of monomer;
the core-shell microspheres were still monodisperse.
Nanosized Core-Shell Spheres withInorganic and Composite Shell
Various procedures have been employed in the fabrication
of inorganic/hybrid coatings on particles, allowing a broad
range of materials with different properties to be prepared.
The specific methods of solid-core inorganic/hybrid-shell
sphere preparation can be classified into two general
Fig. 2 TEM images of silica-PMMA (left), silica-PS core-shell spheres (right). (From Ref. [64].)
868 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
categories: precipitation and surface reactions, and the
controlled deposition of preformed inorganic colloids.
Previous investigations have demonstrated that poly-
meric and inorganic particles dispersed in aqueous
solutions can be coated with layers of various inorganic
materials either by precipitation of the coating materials
onto the cores or by direct surface reactions utilizing spe-
cific functional groups on the cores to induce coat-
ing.[17,27,31,65–76] The inorganic coatings prepared using
these approaches include silica,[17,27,31,65–73] yttrium basic
carbonate, titania,[74–76] and polyelectrolytes onto particles
via the layer- by-layer ( LbL) and LB techniques. Early
work focused on the coating of titania microparticles with
silica layers; however, significant particle clumping and
coalescence took place during silica deposition. Using the
precipitation method, in which the coating material is
precipitated directly onto the core, Ohmori and Matijevic
optimized the coating conditions and coated spindle-
shaped hematite (a-Fe2O3) particles with silica layers by
hydrolysis of the alkoxide tetraethoxysilane (TEOS) in
2-propanol.[31,65,77] Uniform silica coatings on individual
a-Fe2O3 particles were obtained when the kinetics of the
TEOS hydrolysis was properly controlled. Dispersions of
uniform submicrometer spherical particles consisting of
silica cores and yttria coatings, as well as yttria cores with
silica coatings, were also prepared by a similar method.
Electrostatic interactions between nanoparticles and
larger particles via solution self-assembly have been
widely exploited to prepare core-shell materials.[66,78–82]
Homola et al. reported the coating of g-Fe2O3 particles
with preformed smaller silica particles by combining
the particle mixtures under conditions where the two
types of particles are oppositely charged. This resulted in
better dispersion and less aggregation of the magnetic
particles. Similarly, nanosized silica was deposited on a
range of larger inorganic particles, thus forming a pro-
tective layer. Nanocomposite multilayers can be assem-
bled on particle surfaces by using the LbL method based
on colloidal templates.
Lu et al. described a sol–gel approach for the coating of
super paramagnetic iron oxide nanoparticles with uniform
shells of amorphous silica.[83] The coating process has
been successfully applied to particles contained in a com-
mercial ferrofluid and those synthesized through a wet
chemical process. The thickness of the silica coating could
be conveniently controlled in the range of 2–100 nm by
changing the concentration of the sol–gel solution. Fluo-
rescent dyes could also be incorporated into these silica
shells through a covalent coupling between these organic
dyes and the sol–gel precursor. Also, they and Liz-Marzan
et al. demonstrated that gold nanoparticles could be di-
rectly coated with uniform shells of amorphous silica
using a sol–gel process (Fig. 3).[27,83,84] The thickness of
such a conformal coating could be changed from tens to
several hundred nanometers by controlling the concentra-
tion of TEOS precursor or the deposition time. The po-
tential use of these spherical, core-shell colloids in fabri-
cating photonic devices has been illustrated with two
examples: photonic crystals and plasmonic waveguides.
These demonstrations suggest that Au–SiO2 core-shell
particles with well-controlled sizes are promising building
blocks for nanoscale integrated optics, in which the di-
mensions of structures for guiding and modulating pho-
tons will no longer be limited by the wavelength of light.
Stable colloidal core-shell particles consisting of a PS
core and a titania coating were prepared in one step by the
hydrolysis of a titanium alkoxide in the presence of a
cationic PS latex.[85] Although this study used PS as a
core, it should be possible to replace it with other poly-
mer colloids that can be given cationic surface groups or
with negatively charged particles that can be made pos-
itive by coating with a polyelectrolyte. This results in
unusually smooth and uniform titania shells that can be
made as thin as a few nanometers. This is attributed to
a very rapid collection of the negatively charged titania
oligomers by the positively charged surfaces. The coat-
ings are very smooth and uniform and can be varied in
thickness from just a few nanometers to at least 50 nm.
Thicker coatings should also be possible but only through
a multistep seeded growth process. The coated spheres
have the same monodispersity as the starting latex, allow-
ing them to form colloidal crystals.
Fig. 3 (A) TEM image of gold nanoparticles with an average diameter of 50 nm. (B,C) TEM images of such gold nanoparticles after
their surfaces had been coated with amorphous silica shells of �20 and �80 nm in thickness, respectively. (From Ref. [83].)
Core/Shell Nanospheres, Hollow Capsules, and Bottles 869

ORDER REPRINTS
Novel fine polymer particles containing ultrafine Pd,
Pt, or Rh metal dispersed on the core-shell-type sphere
were prepared by the emulsifier-free emulsion polymer-
ization, followed by the addition of a mixture of Ln(NO3)3
and NaH2PO4.[86] Rogach et al. and Caruso et al. report
on the fabrication of 3-D colloidal photonic crystals by the
self-organization of submicrometer-sized PS latex spheres
covered via the consecutive electrostatic adsorption of
charged polyelectrolytes and luminescent semiconductor
nanocrystals (Fig. 1).[82] CdTe and CdTe(S) nanocrys-
tals,[87] capped on the surface with different thiols and
with sizes ranging from 2.5 to 5 nm, have been prepared
by a wet chemical route.[88] They show a pronounced size
dependence on the position of their electronic transitions
and luminescence maxima due to the quantum confine-
ment effect. Relatively narrow and reasonably strong
‘‘excitonic’’ luminescence occurs close to the onset of
absorption and is tunable between 530 and 680 nm.
Highly monodispersed CdSe–CdS core-shell nanopar-
ticles have been prepared by a novel route involving
thermolysis in TOPO in a one-pot synthesis.[89] This
route is a simple and convenient route to produce rea-
sonable quantities of high-quality, monodispersed core-
shell nanoparticles. The precursors are easy to synthesize
and store and give high yields of TOPO-capped quan-
tum dots.
Submicrometer-sized anionic PS latexes have been
coated with uniform layers of iron compounds by aging, at
elevated temperature. Dispersions of the polymer colloid
in the presence of aqueous solutions of ferric chloride,
urea, hydrochloric acid, and polyvinylpyrrolidone have
been produced.[25] The thickness of the deposited layers
could be altered by suitable adjustment of the reactant
concentrations, and they could also be increased by further
aging of the coated particles in the presence of aqueous
solutions of ferric chloride. Hollow colloidal spheres of
iron compounds were obtained by calcinations of the so-
coated PS latexes at elevated temperature in air. Different
chemical compositions of hollow colloidal spheres were
obtained by heating them in hydrogen.
HOLLOW CAPSULES AND NANOBOTTLES
Hollow spheres are useful in a variety of areas. They can
be used in catalysis, delivery of drugs, development of
artificial cells, or protection of biologically active agents
(such as proteins, enzymes, or DNAs). Hollow spheres
may also provide some immediate advantages over their
solid counterparts because of their relatively low densi-
ties. In a typical procedure, hollow spheres are prepared
by the removal of the ‘‘cores’’ (via selective etching with
a solvent or calcination in air) from core-shell structure
nanospheres. There are a variety of methods currently
used to fabricate a wide range of stable, hollow spheres of
various compositions. These methods include nozzle
reactor systems,[90–92] emulsion/phase separation techni-
ques coupled with sol–gel processing,[73,93,94] sacrificial
core procedures,[77,95–97] and LbL technique (consecu-
tively assembling inorganic nanoparticles and polymer
onto colloids and subsequently removing the templated
colloid).[1,10–15,98–102] There have been some successful
examples for the preparation of different kinds of hol-
low microsphere materials (such as silica,[12,16–21,80] zir-
conium[16] hydrous oxide, yttrium compounds,[15,22,23]
titania,[75,99–101] copper compounds,[102] zeolite,[103] and
magnetic nanoparticles.[17,24,25,104]) Yin et al. synthesized
Fig. 4 TEM and SEM (inset) of hollow palladium spheres.
(From Ref. [105].)
870 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
mesoscopic hollow microspheres of ceramic materials
with functionalized interior surfaces.[96] Kim et al. fab-
ricated hollow palladium microspheres and successfully
applied them to the recyclable heterogeneous catalyst for
Suzuki coupling reactions (Fig. 4).[105] Caruso and cow-
orkers prepared many kinds of inorganic and hybrid
hollow spheres (SiO2, TiO2, Fe3O4, luminescent polyelec-
trolyte, etc.) by consecutively assembling inorganic nano-
particles and polymer onto colloids (LbL technology) and
subsequently removing the templated colloid (Fig. 5).[106]
Polymer Hollow Capsules and Nanobottles
Recent advances in supramolecular chemistry have given
chemists unprecedented control over the composition and
shape of nanoscopic objects. An example of such
development is the synthesis of nanometer-sized organic
hollow spheres, which can find numerous applications in
drug delivery/targeting, extraction and as nanoreactors.
Sun et al. described a new strategy for synthesizing
nanometer-scale organic hollow spheres using Au colloids
as templates.[107] The whole structure is held together
by S–S bonds. Oxidation of gold nanoparticles protected
by thiolated bicyclodextrin molecules leads to the forma-
tion of water-soluble polycyclodextrin nanocapsules held
together by S–S bonds. They are currently working on
broadening the described strategy to other substrates/
templates and probing the encapsulation properties of the
hollow spheres.
Marinakos et al. described new methods for synthesiz-
ing nanometer-sized hollow capsules of poly(pyrrole),
poly(N-methylpyrrole), and polyalkenes.[41,42] These
methods utilized nanometer-sized gold particles as tem-
plates from which to grow polymer shells. Dissolution of
the template particles yielded structurally intact hollow
polymer capsules with interior volume and shell thickness
governed by the diameter of the template particle and the
polymerization time, respectively. Moreover, they showed
that alkanethiols were encapsulated in the hollow polymer
core by attaching them to the gold template particles prior
to polymerization and particle etching, and small mole-
cule diffusion rates through the pyrrole-based polymer
capsules depended on polymer oxidation state. They also
described a method for converting alkylthiolate mono-
layers on gold particles into hollow polymer capsules.[108]
The synthetic design of the tripodal ligand provides the
potential to ultimately control the functionality present on
the surface of the particle as well as that present internally.
Marinakos et al. show that small molecules and
enzymes can be trapped inside poly(pyrrole), poly(N-
methylpyrrole), and poly-(3-methylthiophene) capsules
synthesized using the gold particle template method.[1]
Diffusion coefficients of small molecules through the
capsule shell were found to vary by almost 3 orders of
magnitude depending on the polymer, polymer oxidation
state, and counter anion incorporated during polymer
synthesis. A small molecule (anthraquinone) and an en-
zyme (horseradish peroxidase) were trapped inside hol-
low capsules by attaching them to the template particle
prior to polymerization and particle etching. A thin
poly(pyrrole) shell protected the enzyme two times longer
in neat toluene compared to unencapsulated enzyme.
Finally, the potential for using conductive polymer nano-
particles for intracellular delivery or diagnostics was
examined by administering particle suspensions to 3T3
murine fibroblasts. Particles ranging in size from 25 to
100 nm were engulfed by fibroblasts without compromis-
ing cell viability.[1]
Hollow polymer spheres synthesized from a vesicle-
directed polymerization can be dried and redispersed
Fig. 5 Illustration of procedures for preparing inorganic and hybrid hollow spheres. The scheme is shown for PS latex particles. (From
Ref. [106].)
Core/Shell Nanospheres, Hollow Capsules, and Bottles 871

ORDER REPRINTS
in water using a variety of nonionic ethoxylated alcohol
surfactants as stabilizers.[109] The final dispersions consist
of both polymer shells and surfactant micelles, which
remain together in colloidal suspension for at least several
months. Small-angle neutron scattering (SANS) is used
to measure the polymer shell thickness (6.3 nm) and core
radius (56 nm) of the surfactant-stabilized hollow poly-
mer spheres in the presence of surfactant micelles.
Hollow polymer microsphere latexes were synthesized
according to polymer–polymer core-shell emulsion poly-
merization then removing the core by selective sol-
vents.[41] Kamata et al. have demonstrated a practical
route to the facile synthesis of spherical hollow colloids of
PBzMA that contained movable cores of Au nanoparticles
(Fig. 6).[110] This procedure should be extendable to many
other systems that involve the use of different combi-
nations of materials for the core and the shell. These core-
shell colloids may also find use as building blocks to
form colloidal crystals with photonic band gap proper-
ties different from those of conventional core-shell or
hollow particles.
Water-soluble polyelectrolyte nanocapsules as pH-
sensitive nanocontainers can be synthesized by vesicular
or emulsion polymerization via core-shell latexes.[111]
These particles show a reversible pH and ionic strength-
dependent swelling transition causing a considerable
increase (decrease) of their radius. During this transition,
gated pores are opened (closed) in the spherical polymer
shells, which enable free molecular exchange between the
interior of the hollow sphere and the bulk medium. This
pH-switchable control of the permeability of the poly-
electrolyte envelopes can be used to trigger the release
of encapsulated materials from their central cavity.
Inorganic Hollow Capsules and Nanobottles
Previous studies have provided successful procedures for
the preparation of composite particles consisting of in-
organic and organic cores covered with shells of other
inorganic materials by controlled surface precipitation
processes.[85,112–114] Such composite particles may be
useful in many applications because the properties (mag-
netic, optical, electric, adsorptive, etc.) of these particles
can be altered by appropriate coatings. Other studies have
also shown that these procedures can be used for the
preparation of polymer particles covered with yttrium,
zirconium, iron, and titanium compounds by controlled
surface precipitation processes, which makes it possible
to extend the use of these colloids to different areas of
high technology.
Owing to their lower density, large specific surface
area, and optical properties, hollow particles have been of
interest as fillers, coatings, catalysts, capsule agents, etc.
In a novel approach, it was shown that hollow inorganic
colloidal spheres of narrow size distribution could be
obtained by thermal decomposition of the polymer core of
PS particles coated with yttrium, zirconium, iron, and
titanium compounds. Kawahashi and Shiho described
the application of these processes to other systems. Thus,
under certain conditions, copper compounds can be de-
posited uniformly on PS latexes by precipitation using
Fig. 6 (A,B) Backscattering SEM and (C,D) TEM images of Au–SiO2–PBzMA particles before (A,C) and after (B,D) HF etching. The
polymerization time was 4 hr, and the polymer shell was �22 nm thick. (E,F) TEM images of Au–Air–PBzMA particles synthesized
using different polymerization times: (E) 3 hr and (F) 6 hr. The polymer shells were �2 and �32 nm in thickness, respectively. (From
Ref. [110].)
872 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
solutions of the corresponding salts in the presence of
urea. Hollow metallic copper and copper oxide particles
of a narrow size distribution can be obtained by calci-
nation of particles coated in this manner at elevated tem-
peratures in nitrogen and air, respectively.[102]
Calcination of sulfate-stabilized PS latexes coated with
nanoparticle/polymer multilayers results in the production
of hollow silica spheres.[11,84] The calcination process
removes the organic matter (the colloidal core and bridg-
ing polymer) during heating to 450�C, as confirmed by
thermogravimetric analysis.
Hollow spheres of zeolite b and silicalite-1 with
different sizes were fabricated efficiently and convenient-
ly through LbL self-assembly of nanozeolite–polymer
multilayers on PS latex, coupled with the removal of the
core by calcination.[103] The pH and ionic strength of the
colloidal solution, crystal size of nanozeolites, and size of
PS latex templates are factors affecting the fabrication of
hollow zeolite spheres. Hollow spheres of other zeolites
such as ZSM-5 and TS-1 have also been successfully
fabricated in the same manner. Currently, the application
of these novel materials in catalysis, separation and
delivery systems is in progress in our laboratory.
Fowler et al. prepared hollow silica microspheres in
high yields by a one-step facile synthesis under ambient
conditions.[34] By controlling the rate of TEOS hydrolysis
specifically at the droplet/water interface, intact micro-
spheres with uniform wall thickness and thermal stability
can be routinely synthesized. The procedure can be
readily extended to the synthesis of organo-functionalized
silica shells, microspheres with encapsulated organic
pigment, and hollow silica capsules with submicrometer
dimensions. Such materials could have a wide range of
uses in diverse materials applications. And they reported
the facile synthesis of thermally stable hollow spherical
shells with ordered mesoporous walls, approximately 20
nm or less in thickness. The structures were synthesized at
room temperature by hydrolysis and condensation of
TEOS in an aqueous solution of cetyltrimethyl ammonium
bromide (CTABr), which was subjected to rapid quench-
ing by dilution followed by pH neutralization after an
induction period.[35]
Novel fine polymer particles containing ultrafine Pd
particles dispersed on the surface of core-shell [core,
poly(styrene-co-acrylic acid); shell, PrPO4]-type micro-
spheres were prepared by the emulsifier-free emulsion
polymerization of styrene with acrylic acid followed by
the addition of PdCl2 and a mixture of Pr(NO3)3 and
NaH2PO2. Pyrolysis of the resulting polymer particles at
900�C provides organic polymer-free hollow capsules
composed of Pd metal and PrPO4.[115]
Most work in this area has been focused on the
development of synthetic methodologies. Very little
attention has been directed toward the functionalization
of the interiors of these hollow particles. In addition, there
are only limited sets of reports that address the diffusion
of chemical reagents across the shells of hollow particles.
Yin et al. described a method based on template-directed
synthesis for generating ceramic hollow spheres whose
interior surfaces could be functionalized with the pre-
specified, nanoscopic objects.[96] The templates involved
in this process were crystalline lattices of monodispersed
polymer beads whose surfaces had been derivatized with
functional objects such as nanoparticles, quantum dots, or
other nanoscale objects. These nanoscopic objects were
left behind on the interior surfaces of the hollow spheres
when the templates were selectively removed through
etching or calcination (Fig. 7).
On the other hand, nanosized hollow inorganic spheres
with a hole in the wall (denoted as nanobottle) had been
successfully prepared from the assembly of functional
polymer nanosphere with tetraethoxysilane or tetrabutyl
titanate, coupled with the removal of the cores by
programmed calcination. Cross-linked polymer nano-
Fig. 7 Schematic outline of the experimental procedure. The
polymer template could be either dissolved with a solvent or
burnt out through calcination at elevated temperatures. (From
Ref. [96].)
Core/Shell Nanospheres, Hollow Capsules, and Bottles 873

ORDER REPRINTS
spheres with quaternary ammonium species on the surface
were synthesized using an emulsifier-free emulsion
copolymerization. The polymerization and purification
were carried out according to a published procedure,[116]
and cross-linked polymer nanospheres with a uniform
size of about 45 nm were obtained. As-synthesized silica-
coated polymer nanospheres were hydrothermally pre-
pared from chemical assembly of TEOS with the
functional polymer nanospheres.
After calcination at 550�C, the polymer template was
removed and hollow silica spheres were obtained (named
as silica nanobottles). Fig. 8 shows the TEM images of
functional polymer nanospheres from emulsion polymer-
ization process, as-synthesized silica microspheres, and
calcined hollow silica samples. After the self-assembly of
the silica gel with the functional polymer nanospheres, the
as-synthesized silica microspheres also show a very
uniform size at 52–55 nm (B), which are nearly 10 nm
thicker than the polymer nanospheres.[117–120] Calcina-
tion of the as-synthesized silica spheres results in the
complete removal of the polymer nanospheres, forming
hollow nanospheres with the size of 50–53 nm (C). As
shown in Fig. 8D, a hole with the size of 5–8 nm can be
seen on the surface of some hollow silica microspheres.
These results may suggest that the holes on the silica
hollow microspheres are formed in the following steps:
Calcination at 550�C leads to decomposition of polymer
nanospheres to smaller gas molecules, which have high
pressure in the closed hollow microspheres. Then the
gaseous molecules with high pressure break through
the shells of the hollow microspheres, and the hole in
the silica hollow microsphere is formed (scheme as Fig. 9).
Therefore these silica hollow microspheres with the hole
are referred to as silica nanobottles.[121]
The AFM observation of functional polymer nano-
spheres, as-synthesized silica microspheres, and calcined
silica samples were carried out. Similar to the TEM
images, the functional polymer nanospheres have a uni-
form size of 52–56 nm and as-synthesized silica micro-
spheres show a bigger size of 58–62 nm (not shown here).
In addition, it can be seen clearly in Fig. 10 that the
surface of every shell contains one hole of 9–12 nm in
Fig. 8 TEM images of (A) polymer spheres, (B) silica spheres before calcination, (C) hollow silica spheres after calcinations, and (D)
magnification of silica nanobottles. (From Ref. [121].)
Fig. 9 The procedure for preparation of silica nanobottles. (From Ref. [121].)
874 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
diameter. Obviously, the sample size characterized by
AFM is slightly larger than that by TEM technique,
which could be explained by assuming that the probe
does not follow the microsphere’s surface precisely be-
cause of the blunt scanning tip. Interestingly, the AFM
images also show that there are holes on the hollow
microspheres and each hollow microsphere possesses
only one hole, which is in good agreement with the
images observed by TEM. These results further con-
firmed that the sample is a kind of silica nanobottles.
The nitrogen adsorption–desorption isotherms of silica
nanobottles and uncalcined silica-coated polymer micro-
spheres are well measured. The comparisons of adsorp-
tion results suggest that the calcined sample is a kind of
opening hollow nanosphere (nanobottles). The relatively
larger pore volume of silica nanobottles may be poten-
tially useful for the encapsulation of functional com-
pounds in the silica nanobottles.
Composite Capsules
Hollow inorganic–organic composite spheres can be
obtained by selection of a solvent that decomposes the
templated core but leaves the polymer bridging the
nanoparticles in the shell. The choice of solvent depends
on the type of core employed; for example, acidic or
dimethyl sulfoxide solutions cause the removal of MF
polymer latex core templates, tetrahydrofuran the removal
of some PS cores, and highly oxidizing solutions decom-
pose proteinaceous cores.
Similar to the pure polymer shells, the nanoparticle/
polymer multiplayer shell assembled onto MF particles
obtained upon decomposition of the MF core by acid
assumes a rather flat confirmation on the substrate when
dried.[84] Confocal microscopy images of the hollow
composite microspheres again show that the shells often
maintain their spherical shape in solution. Interestingly,
permeating the nanoparticle/polymer shell still readily
expels the oligomers produced as a result of decomposing
the MF particles. Higher magnification TEM reveals that
the shell is composed of nanoparticles embedded in the
polymer matrix.
Nanoparticle/polymer-coated biocolloids (gluteralde-
hyde-fixed echinocytes) can also be utilized for the
production of composite hollow structures. The template
has a jagged and highly structured surface. After the
removal of the core by exposure to deproteinizer, hollow
composite silica/polymer capsules are obtained. Unlike
the polymer or nanoparticle/polymer shells produced by
the removal of MF-templated cores by acid solutions,
these hollow structures mimic the original shape, includ-
ing the secondary structure (spikes) of the templates, and
do not significantly spread-out on the surface when dried.
This is most probably due to the gelation of the silica
particles as a result of the decomposing solution. SEM
experiments confirmed that these structures were hollow.
ENCAPSULATION OF RARE EARTHCOMPLEX IN NANOBOTTLES
The abovementioned silica bottles are nanosized materials
and there is a hole on the surface of it, which may be
useful for further encapsulations. Rare earth (RE) complex
Eu(TTA)3(TPPO)2 (TTA: 1-(2-thenoyl)-3,3,3-trifluorace-
tate, TPPO: triphenyl phosphineoxide) was selected as a
Fig. 10 AFM height and amplitude images of silica nanobottles. (From Ref. [121].) (View this art in color at www.dekker.com).
Core/Shell Nanospheres, Hollow Capsules, and Bottles 875
ORDER REPRINTS
guest molecule. After the modification of silica nanobot-
tles with APTES (NH2–(CH2)3 Si(OC2H5)3),[122] the RE
complex was mixed with the silica nanobottles in
chloroform, followed by filtering and washing with
chloroform until the filter liquors gave no luminescence
under UV radiation.
After RE complex encapsulation, the SEM images of
silica nanobottles give most like morphology as before
encapsulation, indicating that the silica nanobottles still
remained after the encapsulation of RE complex. Further-
more, the encapsulation of Eu(TTA)3(TPPO)2 in nano-
bottles was characterized by energy dispersive X-ray
analysis (EDX). The results indicate that RE complex still
exists in silica nanobottle samples after careful washing.
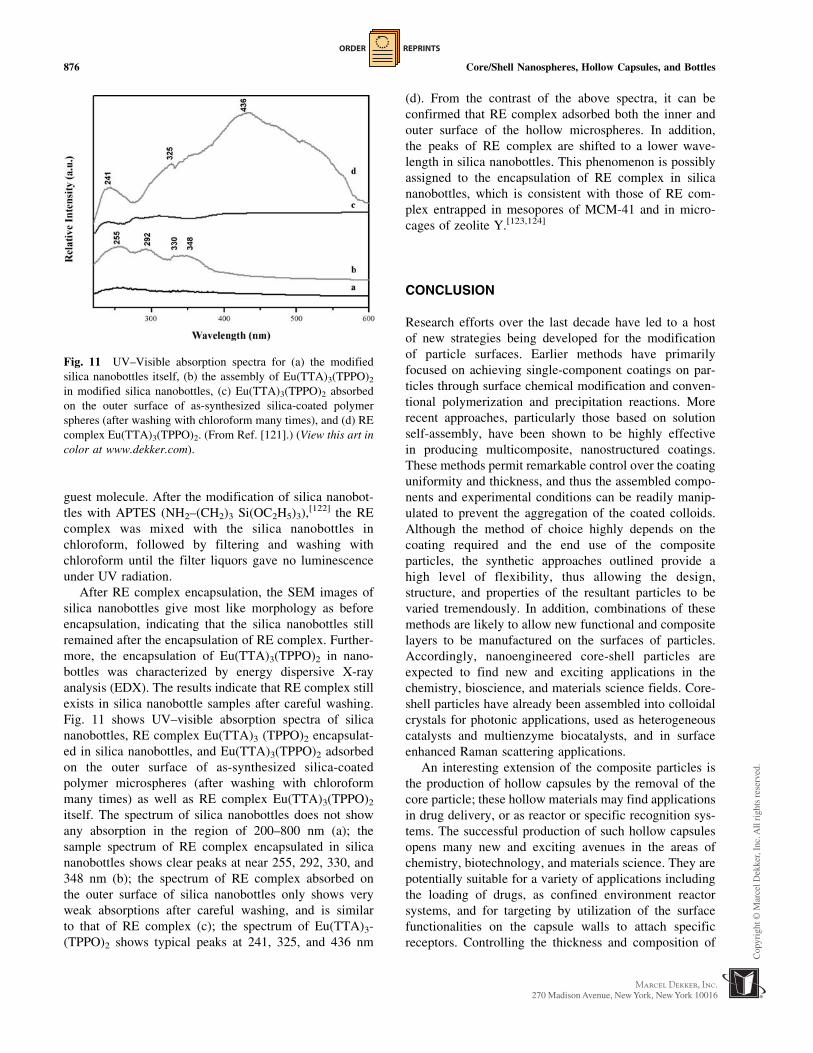
Fig. 11 shows UV–visible absorption spectra of silica
nanobottles, RE complex Eu(TTA)3 (TPPO)2 encapsulat-
ed in silica nanobottles, and Eu(TTA)3(TPPO)2 adsorbed
on the outer surface of as-synthesized silica-coated
polymer microspheres (after washing with chloroform
many times) as well as RE complex Eu(TTA)3(TPPO)2
itself. The spectrum of silica nanobottles does not show
any absorption in the region of 200–800 nm (a); the
sample spectrum of RE complex encapsulated in silica
nanobottles shows clear peaks at near 255, 292, 330, and
348 nm (b); the spectrum of RE complex absorbed on
the outer surface of silica nanobottles only shows very
weak absorptions after careful washing, and is similar
to that of RE complex (c); the spectrum of Eu(TTA)3-
(TPPO)2 shows typical peaks at 241, 325, and 436 nm
(d). From the contrast of the above spectra, it can be
confirmed that RE complex adsorbed both the inner and
outer surface of the hollow microspheres. In addition,
the peaks of RE complex are shifted to a lower wave-
length in silica nanobottles. This phenomenon is possibly
assigned to the encapsulation of RE complex in silica
nanobottles, which is consistent with those of RE com-
plex entrapped in mesopores of MCM-41 and in micro-
cages of zeolite Y.[123,124]
CONCLUSION
Research efforts over the last decade have led to a host
of new strategies being developed for the modification
of particle surfaces. Earlier methods have primarily
focused on achieving single-component coatings on par-
ticles through surface chemical modification and conven-
tional polymerization and precipitation reactions. More
recent approaches, particularly those based on solution
self-assembly, have been shown to be highly effective
in producing multicomposite, nanostructured coatings.
These methods permit remarkable control over the coating
uniformity and thickness, and thus the assembled compo-
nents and experimental conditions can be readily manip-
ulated to prevent the aggregation of the coated colloids.
Although the method of choice highly depends on the
coating required and the end use of the composite
particles, the synthetic approaches outlined provide a
high level of flexibility, thus allowing the design,
structure, and properties of the resultant particles to be
varied tremendously. In addition, combinations of these
methods are likely to allow new functional and composite
layers to be manufactured on the surfaces of particles.
Accordingly, nanoengineered core-shell particles are
expected to find new and exciting applications in the
chemistry, bioscience, and materials science fields. Core-
shell particles have already been assembled into colloidal
crystals for photonic applications, used as heterogeneous
catalysts and multienzyme biocatalysts, and in surface
enhanced Raman scattering applications.
An interesting extension of the composite particles is
the production of hollow capsules by the removal of the
core particle; these hollow materials may find applications
in drug delivery, or as reactor or specific recognition sys-
tems. The successful production of such hollow capsules
opens many new and exciting avenues in the areas of
chemistry, biotechnology, and materials science. They are
potentially suitable for a variety of applications including
the loading of drugs, as confined environment reactor
systems, and for targeting by utilization of the surface
functionalities on the capsule walls to attach specific
receptors. Controlling the thickness and composition of
Fig. 11 UV–Visible absorption spectra for (a) the modified
silica nanobottles itself, (b) the assembly of Eu(TTA)3(TPPO)2
in modified silica nanobottles, (c) Eu(TTA)3(TPPO)2 absorbed
on the outer surface of as-synthesized silica-coated polymer
spheres (after washing with chloroform many times), and (d) RE
complex Eu(TTA)3(TPPO)2. (From Ref. [121].) (View this art in
color at www.dekker.com).
876 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
the capsule walls should allow selective and switchable
permeation for the encapsulation and release of various
substances. The use of cross-linkable, pH- or temperature-
sensitive polymers as capsule wall constituents are attrac-
tive candidates for controlling and varying the permeabil-
ity, while the incorporation of specific reactive groups
inside the capsule walls would allow specific chemistry to
be carried out in these systems. Coupling of biospecies to
the surfaces of the capsules through functional groups
would provide biofunctionalized capsules.
Some experiments demonstrate that it is possible to
coat the outer and inner surfaces of hollow polymer
capsules with phospholipid bilayers. The polymer cap-
sules are permeable to small low molecular weight dyes,
but not to polyelectrolytes with molecular weights greater
than 4000 or molecules larger than 5–10 nm in diameter.
The phospholipid coating reduces the permeability to
small organic dyes. The precipitation of small organic dye
molecules inside polymer capsules has been achieved, as
has the solubilization of various organic solvents. Func-
tional biomolecules have also been encapsulated at a very
high loading capacity in polymer capsules; these systems
are expected to be used in enzyme catalysis applications.
The coating technique is currently being extended to
inorganic templates to create novel hollow capsules of
nanometer size and to emulsion-based systems. A lumi-
nescent RE complex is successfully encapsulated in silica
nanobottles, showing this material has potential nano-
technology applications.
ACKNOWLEDGMENTS
This work was supported by NSFC (29925412) and the
Major State Basic Research Development Program
(G2000078102).
REFERENCES
1. Marinakos, S.M.; Anderson, M.F.; Ryan, J.A.;
Martin, L.D.; Feldheim, D.L. J. Phys. Chem., B
2001, 105, 8872.
2. Hostetler, M.J.; Templeton, A.C.; Murray, R.W.
Langmuir 1999, 15, 3782.
3. Foss, C.A., Jr.; Tierney, M.J.; Martin, C.R. J. Phys.
Chem. 1992, 96, 9001.
4. Sandrock, M.L.; Pibel, C.D.; Geiger, F.M.; Foss,
C.A., Jr. J. Phys. Chem., B 1999, 103, 2668.
5. Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storh-
off, J. J. Nat. 1996, 382, 607.
6. Freeman, R.G.; Grabar, K.C.; Allison, K.J.; Bright,
R.M.; Davis, J.A.; Guthrie, A.P.; Hommer, M.B.;
Jackson, M.A.; Smith, P.C.; Walter, D.G.; Natan, M.
J. Sci. 1995, 267, 1629.
7. Caruso, F. Adv. Mater. 2001, 13, 11.
8. Bateman, A.R.; Harrington, K.J.; Melcher, A.A.
Expert Opin. Investig. Drugs 2000, 9, 2799.
9. Garcia-Blanco, M.A.; Puttaraju, M.; Mansfield,
S.G.; Mitchell, L.G. Gene Ther. Reg. 2000, 1, 141.
10. Caruso, F. Chem. Eur. J. 2000, 6, 413.
11. Caruso, F.; Caruso, R.A.; Mohwald, H. Science
1998, 282, 1111.
12. Giersig, M.; Ung, T.; Liz-Marzan, L.M.; Mulva-
ney, P. Adv. Mater. 1997, 9, 570.
13. Philipse, A.P.; van Bruggen, M.P.B.; Pathmama-
noharan, C. Langmuir 1994, 10, 92.
14. Chang, S.Y.; Liu, L.; Asher, S.A. J. Am. Chem.
Soc. 1994, 116, 6739.
15. Ung, T.; Liz-Marzan, L.M.; Mulvaney, P. Lang-
muir 1998, 14, 3740.
16. Kawahashi, N.; Persson, C.; Matijevic, E. J. Mater.
Chem. 1991, 1, 577.
17. Kawahashi, N.; Matijevic, E. J. Colloid Interface
Sci. 1991, 143, 103.
18. Bamnolker, H.; Nitzan, B.; Gura, S.; Margel, S. J.
Mater. Sci. Lett. 1997, 16, 1412.
19. Walsh, D.; Mann, S. Nature 1995, 377, 320.
20. Garg, A.; Matijevic, E. J. Colloid Interface Sci.
1988, 126, 243.
21. Correa-Duarte, M.A.; Giersig, M.; Liz-Marzan,
L.M. Chem. Phys. Lett. 1998, 286, 497.
22. Kawahashi, N.; Matijevic, E. J. Colloid Interface
Sci. 1990, 138, 534.
23. Gieshe, H.; Matijevic, E. J. Mater. Res. 1994, 9, 436.
24. Shiho, H.; Manabe, Y.; Kawahashi, N. J. Mater.
Chem. 2000, 10, 333.
25. Shiho, H.; Kawahashi, N. J. Colloid Interface Sci.
2000, 226, 91.
26. Davies, R.; Schurr, G.A.; Meenan, P.; Nelson,
R.D.; Bergna, H.E.; Brevett, C.A.S.; Goldbaum,
R.H. Adv. Mater. 1998, 10, 1264.
27. Liz-Marzan, L.M.; Giersig, M.; Mulvaney, P.
Langmuir 1996, 12, 4329.
28. Hofman-Caris, C.H.M. New J. Chem. 1994, 18,1087.
29. Antelmi, D.A.; Spalla, O. Langmuir 1999, 15,7478.
30. Bartsch, E.; Frenz, V.; Baschnagel, J.; Schaertl, W.;
Silescu, H. J. Chem. Phys. 1997, 106, 3743.
31. Ohmori, M.; Matijevic, E. J. Colloid Interface Sci.
1993, 160, 288.
32. Goia, D.V.; Matijevic, E. New J. Chem. 1998,1203.
33. Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davis,
S.A.; Mohwald, H. Angew. Chem., Int. Ed. Engl.
1998, 37, 2201.
Core/Shell Nanospheres, Hollow Capsules, and Bottles 877

ORDER REPRINTS
34. Fowler, C.E.; Khushalani, D.; Mann, S. J. Mater.
Chem. 2001, 11, 1968.
35. Fowler, C.E.; Khushalani, D.; Mann, S. Chem.
Commun. 2001, 2028.
36. Lin, H.P.; Mou, C.Y.; Liu, S.B.; Tang, C.Y. Chem.
Commun. 2001, 1970.
37. Oyama, H.T.; Sprycha, R.; Xie, Y.; Partch, R.E.;
Matijevic, E. J. Colloid Interface Sci. 1993, 160, 298.
38. Sprycha, R.; Oyama, H.T.; Zelenev, A.; Matijevic,
E. Colloid Polym. Sci. 1995, 273, 693.
39. Partch, R.; Gangolli, S.G.; Matijevic, E.; Cai, W.;
Arajs, S. J. Colloid Interface Sci. 1991, 144, 27.
40. Marinakos, S.M.; Brousseau, L.C.; Jones, A.;
Feldheim, D.L. Chem. Mater. 1998, 10, 1214.
41. Marinakos, S.M.; Shultz, D.A.; Feldheim, D.L.
Adv. Mater. 1999, 11, 34.
42. Marinakos, S.M.; Novak, J.P.; Brousseau, L.C.;
House, A.B.; Edeki, E.M.; Feldhaus, J.C.; Feld-
heim, D.L. J. Am. Chem. Soc. 1999, 121, 8518.
43. Ottewill, R.H.; Schofield, A.B.; Waters, J.A.;
Williams, N.S. J. Colloid Polym. Sci. 1997, 275, 274.
44. Quaroni, L.; Chumanov, G. J. Am. Chem. Soc.
1999, 121, 10642.
45. Von Werne, T.; Patten, T.E. J. Am. Chem. Soc.
2001, 123, 7497.
46. Patten, T.E.; Matyjaszewski, K. Adv. Mater. 1998,10, 901.
47. Sun, S.; Murray, C.B.; Weller, D.; Folks, D.;
Moser, A. Science 2000, 287, 1989.
48. Selvan, S.T.; Spatz, J.P.; Klok, H.A.; Moller, M.
Adv. Mater. 1998, 10, 132.
49. Godovski, D.Y. Adv. Polym. Sci. 1995, 119, 79.
50. Go’mez-Lopera, S.A.; Plaza, R.C.; Delgado, A.W.
J. Colloid Interface Sci. 2001, 240, 40.
51. Portet, D.; Denizot, B.; Rump, E.; Lejeune, J.J.;
Jallot, P. J. Colloid Interface Sci. 2001, 238, 37.
52. Xu, X.; Friedman, G.; Humfeld, K.D.; Majetich,
S.A.; Asher, S.A. Adv. Mater. 2001, 13, 1681.
53. Xu, X.; Friedman, G.; Humfeld, K.D.; Majetich,
S.A.; Asher, S.A. Chem. Mater. 2002, 14, 1249.
54. Vestal, C.R.; Zhang, Z.J. J. Am. Chem. Soc. 2002,124, 14312.
55. Srikanth, H.; Hajndl, R.; Chirinos, C.; Sanders, J.;
Sampath, A.; Sudarshan, T.S. Appl. Phys. Lett.
2001, 79, 3503.
56. Liu, C.; Zou, B.; Rondinone, A.J.; Zhang, Z.J. J.
Phys. Chem., B 2000, 104, 1141.
57. Liu, C.; Zou, B.; Rondinone, A.J.; Zhang, Z.J. J.
Am. Chem. Soc. 2000, 122, 6263.
58. Huang, H.; Kowalewski, T.; Remsen, E.E.;
Gertzmann, R.; Wooley, K.L. J. Am. Chem.
Soc. 1997, 119, 11653.
59. Butun, V.; Billingham, N.C.; Armes, S.P. J. Am.
Chem. Soc. 1998, 120, 12135.
60. Zhang, Q.; Remsen, E.E.; Wooley, K.L. J. Am.
Chem. Soc. 2000, 122, 3642.
61. Huang, H.; Remsen, E.E.; Wooley, K.L. Chem.
Commun. 1998, 1415.
62. Murthy, K.S.; Ma, Q.; Clark, C.G.; Remsen, E.E.;
Wooley, K.L. Chem. Commun. 2001, 773.
63. Stober, W.; Fink, A. J. Colloid Interface Sci. 1968,26, 62.
64. Zhang, K.; Chen, H.; Chen, X.; Chen, Z.; Cui, Z.;
Yang, B. Macromol. Mater. Eng. 2003, 288, 380.
65. Ohmori, M.; Matijevic, E. J. Colloid Interface Sci.
1992, 150, 594.
66. Liz-Marzan, L.M.; Giersig, M.; Mulvaney, P.
Chem. Commun. 1996, 731.
67. Dokoutchaev, A.; James, J.T.; Koene, S.C.; Pathak,
S.; Prakash, G.K.S.; Thompson, M.E. Chem.
Mater. 1999, 11, 2389.
68. Liu, Q.; Xu, Z.; Finch, J.A.; Egerton, R. Chem.
Mater. 1998, 10, 3936.
69. Liz-Marzan, L.M.; Philipse, A.P. J. Colloid Inter-
face Sci. 1995, 176, 459.
70. Ung, T.; Liz-Marzan, L.M.; Mulvaney, P. J. Phys.
Chem., B 1999, 103, 6770.
71. Bruggen, M.P.B. Langmuir 1998, 14, 2245.
72. Hall, S.R.; Davis, S.A.; Mann, S. Langmuir 2000,16, 1454.
73. Hardikar, V.V.; Matijevic, E. J. Colloid Interface
Sci. 2000, 221, 133.
74. Hanprasopwattana, A.; Srinivasan, S.; Sault, A.G.;
Datye, A.K. Langmuir 1996, 12, 3173.
75. Guo, X.C.; Dong, P. Langmuir 1999, 15, 5535.
76. Pastoriza-Santos, I.; Koktysh, D.S.; Mamedov,
A.A.; Giersig, M.; Kotov, N.A.; Liz-Marzan,
L.M. Langmuir 2000, 16, 2731.
77. Stober, W.; Fink, A.; Bohn, E. J. Colloid Interface
Sci. 1968, 26, 62.
78. Keller, S.W.; Johnson, S.A.; Brigham, E.S.;
Yonemoto, E.H.; Mallouk, T.E. J. Am. Chem.
Soc. 1995, 117, 12879.
79. Caruso, F.; Lichtenfeld, H.; Giersig, M.; Mohwald,
H. J. Am. Chem. Soc. 1998, 120, 8523.
80. Caruso, F.; Mohwald, H. Langmuir 1999, 15, 8276.
81. Caruso, F.; Susha, A.S.; Giersig, M.; Moehwald, H.
Adv. Mater. 1999, 11, 950.
82. Rogach, A.; Susha, A.; Caruso, F.; Sukhorukov, G.;
Kornowski, A.; Kershaw, S.; Mohwald, H.; Eych-
muller, A.; Weller, H. Adv. Mater. 2000, 12, 333.
83. Lu, Y.; Yin, Y.; Mayers, B.T.; Xia, Y. Nano Lett.
2002, 2, 183.
84. Caruso, F.; Caruso, R.A.; Mohwald, H. Chem.
Mater. 1999, 11, 3309.
85. Imhof, A. Langmuir 2001, 17, 3579.
86. Tamai, H.; Ikeya, T.; Nishiyama, F.; Yasuda, H. J.
Mater. Sci. 2000, 35, 4945.
878 Core/Shell Nanospheres, Hollow Capsules, and Bottles

ORDER REPRINTS
87. Susha, A.S.; Caruso, F.; Rogach, A.L.; Sukhor-
ukov, G.B.; Kornowski, A.; Mohwald, H.; Giersig,
M.; Eychmller, A.; Weller, H. Colloids Surf., A
2000, 163, 39.
88. Gao, M.; Kirstein, S.; Mohwald, H.; Rogach, A.L.;
Kornowski, A.; Eychmller, A.; Weller, H. J. Phys.
Chem., B 1998, 102, 8360.
89. Revaprasadu, N.; Malik, M.A.; O’Brien, P.; Wake-
field, G. Chem. Commun. 1999, 1573.
90. Lu, Y.; Fan, H.; Stump, A.; Ward, T.L.; Rieker, T.;
Brinker, C. J. Nat. 1999, 398, 223.
91. Bruinsma, P.J.; Kim, A.Y.; Liu, J.; Baskaran, S.
Chem. Mater. 1997, 9, 2507.
92. Iida, M.; Sasaki, T.; Watanabe, M. Chem. Mater.
1998, 10, 3780.
93. Pekarek, K.J.; Jacob, J.S.; Mathiowitz, E. Nature
1994, 367, 258.
94. Liu, J.G.; Wilcox, D.L. J. Mater. Res. 1995, 10, 84.
95. Hubert, D.H.W.; Jung, M.; Frederik, P.M.; Bow-
mans, P.H.H.; Meuldijk, J.; German, A.L. Adv.
Mater. 2000, 12, 1286.
96. Yin, Y.D.; Lu, Y.; Gates, B.; Xia, Y.N. Chem.
Mater. 2001, 13, 1146.
97. Rhodes, K.H.; Davis, S.A.; Caruso, F.; Zhang, B.;
Mann, S. Chem. Mater. 2000, 12, 2832.
98. Decher, G. Science 1997, 277, 1232.
99. Zhong, Z.; Yin, Y.; Gates, B.; Xia, Y.N. Adv.
Mater. 2000, 12, 206.
100. Caruso, R.A.; Susha, A.; Caruso, F. Chem. Mater.
2001, 13, 400.
101. Shiho, H.; Kawahashi, N. J. Colloid Polym. Sci.
2000, 278, 270.
102. Kawahashi, N.; Shiho, H. J. Mater. Chem. 2000,10, 2294.
103. Wang, X.D.; Yang, W.L.; Tang, Y.; Wang, Y.J.;
Fu, S.K.; Gao, Z. Chem. Commun. 2000, 2161.
104. Caruso, F.; Spasova, M.; Susha, A.; Giersig, M.;
Caruso, R.A. Chem. Mater. 2001, 13, 109.
105. Kim, S.W.; Kim, M.; Lee, W.Y.; Hyeon, T. J. Am.
Chem. Soc. 2002, 124, 7642.
106. Caruso, F.; Caruso, R.A.; Mohwald, H. Science
1998, 282, 6.
107. Sun, L.; Crooks, R.M.; Chechik, V. Chem.
Commun. 2001, 359.
108. Wu, M.; O’Neill, S.A.; BrousseauIII, L.C.;
McConnell, W.; Shultz, D.A.; Feldheim, D.L.;
Linderman, R. J. Chem. Commun. 2000, 775.
109. McKelvey, C.A.; Kaler1, E.W. J. Colloid Interface
Sci. 2001, 245, 68.
110. Kamata, K.; Lu, Y.; Xia, Y.N. J. Am. Chem. Soc.
2003, 125, 2384.
111. Sauer, M.; Streich, D.; Meier, W. Adv. Mater.
2001, 13, 1649.
112. Garg, A.; Matijevic, E. Langmuir 1988, 4, 38.
113. Aiken, B.; Matijevic, E. J. Colloid Interface Sci.
1988, 126, 645.
114. Furusawa, K.; Anzai, C. Colloids Surf. 1992, 63, 103.
115. Tamai, H.; Ikeya, T.; Yasuda, H. J. Colloid
Interface Sci. 1999, 218, 217.
116. Chen, X.; Cui, Z.; Chen, Z.; Zhang, K.; Lu, G.;
Zhang, G.; Yang, B. Polymer 2002, 43, 4147.
117. Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz,
M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.;
Olson, D.H.; Sheppard, E.W.; McCullen, S.B.;
Higgins, J.B.; Schlenker, J.L. J. Am. Chem. Soc.
1992, 114, 10834.
118. Zhao, D.Y.; Feng, J.L.; Huo, Q.S.; Melosh, N.;
Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D.
Science 1998, 279, 548.
119. Xiao, F.S.; Han, Y.; Yu, Y.; Meng, X.J.; Yang, M.;
Wu, S. J. Am. Chem. Soc. 2002, 124, 888.
120. Zhang, Z.T.; Han, Y.; Xiao, F.S.; Qiu, S.L.; Zhu, L.;
Wang, R.W.; Yu, Y.; Zhang, Z.; Zou, B.S.; Wang,
Y.Q.; Sun, H.P.; Zhao, D.Y.; Wei, Y. J. Am. Chem.
Soc. 2001, 123, 5014.
121. Zhang, G.; Yu, Y.; Chen, X.; Han, Y.; Di, Y.;
Yang, B.; Xiao, F.; Shen, J. J. Colloid Interface
Sci. 2003, 263, 467.
122. Liu, C.J.; Li, S.G.; Pang, W.Q.; Che, C.M. Chem.
Commun. 1997, 65.
123. Xu, Q.H.; Li, L.S.; Li, B.; Yu, J.H.; Xu, R.R.
Microporous Mesoporous Mater. 2000, 38, 351.
124. Rose, I.L.V.; Serra, O.A.; Nassa, E.J. Lumin 1997,72, 532.
Core/Shell Nanospheres, Hollow Capsules, and Bottles 879


















