
Characterization of the complex formation of 1,6-anhydro-β-maltose and potassium ions using NMR...
6
Characterization of the complex formation of 1,6-anhydro-b-maltose and potassium ions using NMR spectroscopy and single-crystal X-ray crystallography Takashi Fujimoto a , Takayuki Kato a , Yosuke Usui a , Osamu Kamo b , Kazuo Furihata c , Koji Tsubono b , Toshiyo Kato b , Tomoya Machinami a , Mitsuru Tashiro a,⇑ a Department of Chemistry, School of Sciences and Engineering, Meisei University, Hodokubo, Hino, Tokyo 191-8506, Japan b Solution & Marketing Division, JEOL RESONANCE Inc., Musashino, Akishima, Tokyo 196-8558, Japan c Division of Agriculture and Agricultural Life Sciences, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan article info Article history: Received 29 April 2011 Received in revised form 11 June 2011 Accepted 14 June 2011 Keywords: 1,6-Anhydro-b-maltose Potassium Complex formation 39 K NMR spectroscopy Single-crystal X-ray crystallography abstract The formation of a complex between 1,6-anhydro-b-maltose and potassium ions was characterized using 1 H, 13 C and 39 K NMR spectroscopy and single-crystal X-ray crystallography. In the NMR study, the spin- lattice relaxation times (T 1 ) of C1, C3, C5, C6, and C5 0 significantly decreased in the presence of potassium ions, and 39 K-T 1 also decreased in the presence of 1,6-anhydro-b-maltose, indicating complex formation. In a crystal, both 8- and 9-coordination structures, corresponding to the distorted capped pentagonal bipyramidal structure and the capped hexagonal bipyramidal structure, respectively, were identified. A potassium ion was positioned in the center of each bipyramidal structure. Ó 2011 Elsevier Ltd. All rights reserved. 1. Introduction The complex formation abilities of carbohydrates with cations have been used in the separation of sugar mixtures and the isola- tion of single tautomeric forms in solution. Sugars can be separated on columns of cation-exchange resins using water as the eluent. 1,2 For instance, D-glucose and D-fructose were separated using a Ca 2+ column, 3 and the a and b anomers of D-allose (both as pyranose and furanose forms) were isolated owing to the differences in their abilities to coordinate with cations. 4 It is well known that the hy- droxyl groups of carbohydrates form complexes in solution with monovalent cations. 5,6 Electrophoretic studies have provided infor- mation on the abilities of alkali metal ions to form complexes with polyhydroxy compounds. The migration rate of 1,6-anhydro-b-D- glucopyranose in the presence of metal ions was extremely higher than that alone, indicating complex formation. 6 Our group has been studying the complex formation of alkali metal ions and 1,6-anhydro sugars, 7,8 which were prepared from the corresponding free sugars through intramolecular dehydration. In past studies, the complexation of 1,6-anhydro-b-maltose with rubidium, that of 1,6-anhydro-b-D-glucopyranose with rubidium 9 and that of 1,6-anhydro-b-maltotriose with potassium 10 were characterized using NMR spectroscopy. Besides these studies, interactions between sucrose and various metal ions were investi- gated by Rondeau et al. using mid-infrared and 13 C NMR spectros- copy. 11,12 The crystal structure of the complex formed by two 1,6- anhydro-b-maltoses with a peroxide and two sodium ions, [Na 2 (1,6-anhydro-b-maltose) 2 (H 2 O) 3 ]O 2 , was determined by sin- gle-crystal X-ray crystallography. 13 The O 2 moiety, 1.496(2) Å for the O–O distance in the complex, was identified as a discrete per- oxide dianion, whereas two sodium and two carbohydrate mole- cules constituted a binuclear complex counter cation. The O 2 moiety had to be present as the peroxide dianion to maintain the charge balance. The complex containing the discrete peroxide dianion proved to be a useful oxidizing agent, which successfully oxidized the a,b-unsaturated ketone to epoxy-ketone. For the pur- pose of elucidating the mechanism of 1,6-anhydro sugar-forming complexes with alkali metal ions, we have investigated the com- plex formation of 1,6-anhydro-b-maltose (Fig. 1) with potassium ions using 1 H, 13 C and 39 K NMR spectroscopy and single-crystal X-ray crystallography. Analytical methods to selectively monitor potassium ions in the solution state are rather limited. In this re- search, 39 K NMR spectroscopy was used as a valuable method to selectively observe 39 K nuclei in solution. 0008-6215/$ - see front matter Ó 2011 Elsevier Ltd. All rights reserved. doi:10.1016/j.carres.2011.06.017 ⇑ Corresponding author. Tel.: +81 42 591 5597; fax: +81 42 591 7331. E-mail address: [email protected] (M. Tashiro). Carbohydrate Research 346 (2011) 1991–1996 Contents lists available at ScienceDirect Carbohydrate Research journal homepage: www.elsevier.com/locate/carres
-
Upload
takashi-fujimoto -
Category
Documents
-
view
213 -
download
1
Transcript of Characterization of the complex formation of 1,6-anhydro-β-maltose and potassium ions using NMR...

Carbohydrate Research 346 (2011) 1991–1996
Contents lists available at ScienceDirect
Carbohydrate Research
journal homepage: www.elsevier .com/locate /carres
Characterization of the complex formation of 1,6-anhydro-b-maltoseand potassium ions using NMR spectroscopy and single-crystal X-raycrystallography
Takashi Fujimoto a, Takayuki Kato a, Yosuke Usui a, Osamu Kamo b, Kazuo Furihata c, Koji Tsubono b,Toshiyo Kato b, Tomoya Machinami a, Mitsuru Tashiro a,⇑a Department of Chemistry, School of Sciences and Engineering, Meisei University, Hodokubo, Hino, Tokyo 191-8506, Japanb Solution & Marketing Division, JEOL RESONANCE Inc., Musashino, Akishima, Tokyo 196-8558, Japanc Division of Agriculture and Agricultural Life Sciences, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan
a r t i c l e i n f o
Article history:Received 29 April 2011Received in revised form 11 June 2011Accepted 14 June 2011
Keywords:1,6-Anhydro-b-maltosePotassiumComplex formation39K NMR spectroscopySingle-crystal X-ray crystallography
0008-6215/$ - see front matter � 2011 Elsevier Ltd. Adoi:10.1016/j.carres.2011.06.017
⇑ Corresponding author. Tel.: +81 42 591 5597; faxE-mail address: [email protected] (M. T
a b s t r a c t
The formation of a complex between 1,6-anhydro-b-maltose and potassium ions was characterized using1H, 13C and 39K NMR spectroscopy and single-crystal X-ray crystallography. In the NMR study, the spin-lattice relaxation times (T1) of C1, C3, C5, C6, and C50 significantly decreased in the presence of potassiumions, and 39K-T1 also decreased in the presence of 1,6-anhydro-b-maltose, indicating complex formation.In a crystal, both 8- and 9-coordination structures, corresponding to the distorted capped pentagonalbipyramidal structure and the capped hexagonal bipyramidal structure, respectively, were identified. Apotassium ion was positioned in the center of each bipyramidal structure.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
The complex formation abilities of carbohydrates with cationshave been used in the separation of sugar mixtures and the isola-tion of single tautomeric forms in solution. Sugars can be separatedon columns of cation-exchange resins using water as the eluent.1,2
For instance, D-glucose and D-fructose were separated using a Ca2+
column,3 and the a and b anomers of D-allose (both as pyranoseand furanose forms) were isolated owing to the differences in theirabilities to coordinate with cations.4 It is well known that the hy-droxyl groups of carbohydrates form complexes in solution withmonovalent cations.5,6 Electrophoretic studies have provided infor-mation on the abilities of alkali metal ions to form complexes withpolyhydroxy compounds. The migration rate of 1,6-anhydro-b-D-glucopyranose in the presence of metal ions was extremely higherthan that alone, indicating complex formation.6
Our group has been studying the complex formation of alkalimetal ions and 1,6-anhydro sugars,7,8 which were prepared fromthe corresponding free sugars through intramolecular dehydration.In past studies, the complexation of 1,6-anhydro-b-maltose with
ll rights reserved.
: +81 42 591 7331.ashiro).
rubidium, that of 1,6-anhydro-b-D-glucopyranose with rubidium9
and that of 1,6-anhydro-b-maltotriose with potassium10 werecharacterized using NMR spectroscopy. Besides these studies,interactions between sucrose and various metal ions were investi-gated by Rondeau et al. using mid-infrared and 13C NMR spectros-copy.11,12 The crystal structure of the complex formed by two 1,6-anhydro-b-maltoses with a peroxide and two sodium ions,[Na2(1,6-anhydro-b-maltose)2(H2O)3]O2, was determined by sin-gle-crystal X-ray crystallography.13 The O2 moiety, 1.496(2) Å forthe O–O distance in the complex, was identified as a discrete per-oxide dianion, whereas two sodium and two carbohydrate mole-cules constituted a binuclear complex counter cation. The O2
moiety had to be present as the peroxide dianion to maintain thecharge balance. The complex containing the discrete peroxidedianion proved to be a useful oxidizing agent, which successfullyoxidized the a,b-unsaturated ketone to epoxy-ketone. For the pur-pose of elucidating the mechanism of 1,6-anhydro sugar-formingcomplexes with alkali metal ions, we have investigated the com-plex formation of 1,6-anhydro-b-maltose (Fig. 1) with potassiumions using 1H, 13C and 39K NMR spectroscopy and single-crystalX-ray crystallography. Analytical methods to selectively monitorpotassium ions in the solution state are rather limited. In this re-search, 39K NMR spectroscopy was used as a valuable method toselectively observe 39K nuclei in solution.
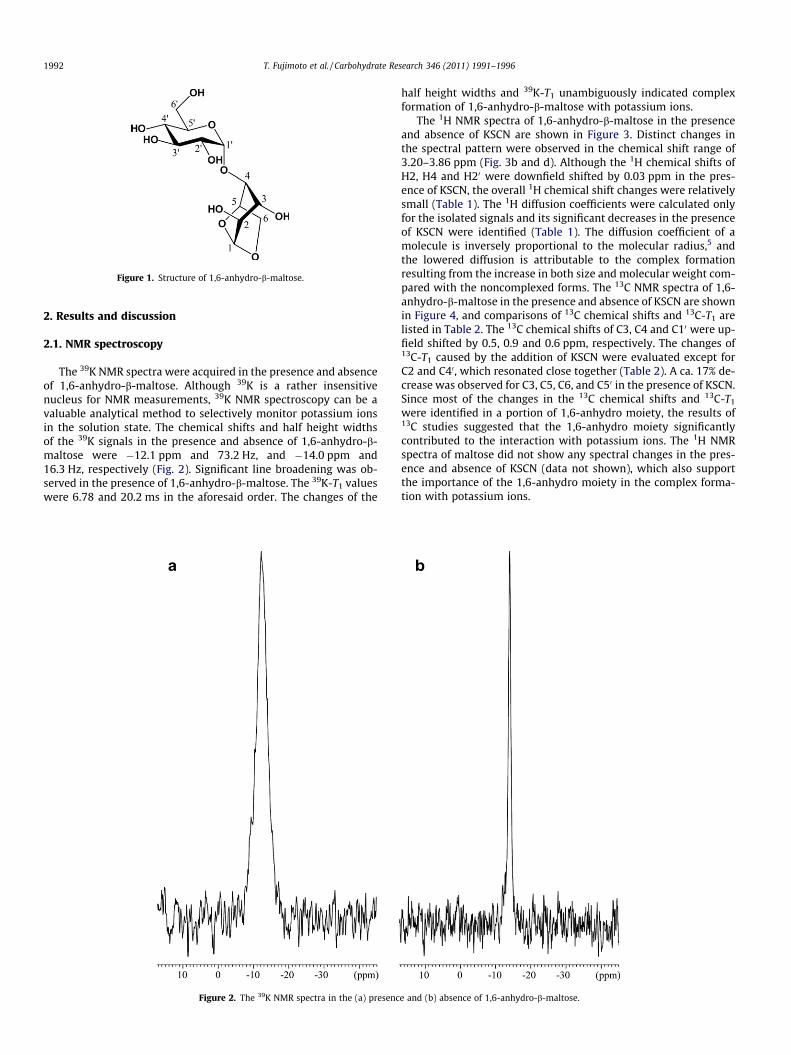
Figure 1. Structure of 1,6-anhydro-b-maltose.
1992 T. Fujimoto et al. / Carbohydrate Research 346 (2011) 1991–1996
2. Results and discussion
2.1. NMR spectroscopy
The 39K NMR spectra were acquired in the presence and absenceof 1,6-anhydro-b-maltose. Although 39K is a rather insensitivenucleus for NMR measurements, 39K NMR spectroscopy can be avaluable analytical method to selectively monitor potassium ionsin the solution state. The chemical shifts and half height widthsof the 39K signals in the presence and absence of 1,6-anhydro-b-maltose were �12.1 ppm and 73.2 Hz, and �14.0 ppm and16.3 Hz, respectively (Fig. 2). Significant line broadening was ob-served in the presence of 1,6-anhydro-b-maltose. The 39K-T1 valueswere 6.78 and 20.2 ms in the aforesaid order. The changes of the
Figure 2. The 39K NMR spectra in the (a) presenc
half height widths and 39K-T1 unambiguously indicated complexformation of 1,6-anhydro-b-maltose with potassium ions.
The 1H NMR spectra of 1,6-anhydro-b-maltose in the presenceand absence of KSCN are shown in Figure 3. Distinct changes inthe spectral pattern were observed in the chemical shift range of3.20–3.86 ppm (Fig. 3b and d). Although the 1H chemical shifts ofH2, H4 and H20 were downfield shifted by 0.03 ppm in the pres-ence of KSCN, the overall 1H chemical shift changes were relativelysmall (Table 1). The 1H diffusion coefficients were calculated onlyfor the isolated signals and its significant decreases in the presenceof KSCN were identified (Table 1). The diffusion coefficient of amolecule is inversely proportional to the molecular radius,5 andthe lowered diffusion is attributable to the complex formationresulting from the increase in both size and molecular weight com-pared with the noncomplexed forms. The 13C NMR spectra of 1,6-anhydro-b-maltose in the presence and absence of KSCN are shownin Figure 4, and comparisons of 13C chemical shifts and 13C-T1 arelisted in Table 2. The 13C chemical shifts of C3, C4 and C10 were up-field shifted by 0.5, 0.9 and 0.6 ppm, respectively. The changes of13C-T1 caused by the addition of KSCN were evaluated except forC2 and C40, which resonated close together (Table 2). A ca. 17% de-crease was observed for C3, C5, C6, and C50 in the presence of KSCN.Since most of the changes in the 13C chemical shifts and 13C-T1
were identified in a portion of 1,6-anhydro moiety, the results of13C studies suggested that the 1,6-anhydro moiety significantlycontributed to the interaction with potassium ions. The 1H NMRspectra of maltose did not show any spectral changes in the pres-ence and absence of KSCN (data not shown), which also supportthe importance of the 1,6-anhydro moiety in the complex forma-tion with potassium ions.
e and (b) absence of 1,6-anhydro-b-maltose.
Figure 3. The 1H NMR spectra of 1,6-anhydro-b-maltose in the (a, b) presence and (c, d) absence of KSCN. (b, d) Expanded spectra are presented to show the changes of thespectral patterns in the region of 3.20–4.10 ppm.
Table 1Comparison of the 1H chemical shifts and 1H diffusion coefficients (D) of 1,6-anhydro-b-maltose in the presence and absence of KSCN
d1H (ppm) 1H-D (�10�10 m2/s)
Free +KSCN D Free +KSCN Ratio
H1 5.28 5.30 0.02 6.66 ± 0.05 5.39 ± 0.08 0.81H2 3.39 3.42 0.03H3 3.73 3.74 0.01 5.05 ± 0.05H4 3.60 3.63 0.03 6.66 ± 0.04H5 4.65 4.63 �0.02 6.59 ± 0.03 5.14 ± 0.02 0.78H6 4.08/3.65 4.10/3.67 0.02/0.02 6.74 ± 0.06 5.45 ± 0.05 0.81H10 5.00 5.02 0.02 6.82 ± 0.08 5.22 ± 0.05 0.77H20 3.38 3.41 0.03H30 3.69 3.68 �0.01H40 3.26 3.27 0.01 6.79 ± 0.06H50 3.71 3.72 0.01H60 3.84/3.65 3.83/3.65 0.01/0.01 6.75 ± 0.06 5.08 ± 0.03 0.75
T. Fujimoto et al. / Carbohydrate Research 346 (2011) 1991–1996 1993
For the purpose of observing conformational changes in1,6-anhydro-b-maltose in complexation with potassium ions, 2DNOESY spectra and selective J-resolved HMBC14 spectra wereacquired. The notable differences could not be identified in com-parison of 2D NOESY spectra in the presence and absence of KSCN(data not shown). The coupling constants of 3JCH around theglycosylic bond were measured using the selective J-resolvedHMBC experiment. The 3JCH of H10 and C4 were measured to be5.42 and 5.17 Hz in the presence and absence of KSCN, respec-tively. These results indicated that the stereochemical relation-ship between H10 and C4 was close to gauche in both samplesolutions, and that significant conformational change was not in-duced in 1,6-anhydro-b-maltose by binding with potassium ions.
2.2. Single-crystal X-ray crystallography
In the crystal structure as determined by X-ray crystallography,both 8- and 9-coordination forms were identified as shown in Fig-ure 5. The 8-coordination structure, which can be defined as thedistorted capped pentagonal bipyramidal structure, comprisedtwo 1,6-anhydro-b-maltoses (designated as anhydro sugars A andB), a thiocyanate ion, an MeOH molecule, and a potassium ion(Fig. 5a and b). A potassium ion was positioned in the center ofthe bipyramidal structure, where two 1,6-anhydro-b-maltoses,possessing identical conformation, surrounded a potassium ion.The distances comprising 8-coordination form around a potassiumion are listed in Table 3. The distances between a potassium ionand all oxygen atoms involved in the 8-coordination structurewere in the range of 2.72–2.98 Å. The pentagon comprised O10,O20 and O5 of anhydro sugar A and O3 and O6 of anhydro sugarB. The O2 of anhydro sugar A and a sulfur atom of a thiocyanateion corresponded to the peak of each pyramid, although the dis-tance between a potassium ion and a thiocyanate ion (3.86 Å)was considered to be long. The oxygen atom of MeOH was posi-tioned as capping for the pyramid, involving a sulfur atom of thethiocyanate ion as a peak.
The 9-coordination structure, which can be defined as thecapped hexagonal bipyramidal structure, comprised three 1,6-anhydro-b-maltoses (designated as anhydro sugars A, B, and C), athiocyanate ion, and a potassium ion (Fig. 5c and d). Although apotassium ion was positioned in the center of the bipyramidalstructure as observed in the 8-coordination structure, three1,6-anhydro-b-maltoses, possessing identical conformations,surrounded a potassium ion. The hexagon comprised O10, O20,and O5 of anhydro sugar A, O3, and O6 of anhydro sugar B andO50 of anhydro sugar C. The O60 of anhydro sugar C and a sulfur

Figure 4. The 13C NMR spectra of 1,6-anhydro-b-maltose in the (a, b) presence and (c, d) absence of KSCN. (b, d) Expanded spectra are presented to show the changes of thespectral patterns in the region of 63.0–78.0 ppm.
Table 2Comparison of the 13C chemical shifts and 13C-T1 of 1,6-anhydro-b-maltose in thepresence and absence of KSCN
d13C (ppm) 13C-T1 (s)
Free +KSCN D Free +KSCN Ratio
C1 103.5 103.4 0.1 0.81 ± 0.01 0.70 ± 0.02 0.86C2 72.3 72.0 0.3 0.80 ± 0.01C3 71.8 71.3 0.5 0.84 ± 0.02 0.70 ± 0.03 0.83C4 78.2 77.3 0.9 0.77 ± 0.02 0.72 ± 0.03 0.94C5 76.9 76.9 0.0 0.72 ± 0.02 0.60 ± 0.02 0.83C6 66.6 66.5 0.1 0.46 ± 0.01 0.38 ± 0.02 0.83C10 99.6 99.0 0.6 0.73 ± 0.01 0.67 ± 0.01 0.92C20 73.6 73.5 0.1 0.71 ± 0.01 0.64 ± 0.02 0.90C30 75.0 75.0 0.0 0.75 ± 0.01 0.67 ± 0.02 0.89C40 72.0 71.9 0.1 0.74 ± 0.01C50 74.5 74.4 0.1 0.74 ± 0.01 0.62 ± 0.02 0.84C60 62.8 62.8 0.0 0.42 ± 0.01 0.39 ± 0.02 0.93
1994 T. Fujimoto et al. / Carbohydrate Research 346 (2011) 1991–1996
atom of a thiocyanate ion corresponded to the peak of each pyra-mid, and O2 of anhydro sugar A was positioned as capping forthe pyramid, involving O60 of anhydro sugar C as a peak. Thedistances comprising the 9-coordination structure around a potas-sium ion are listed in Table 3. The distances between a potassiumion and all oxygen atoms comprising the 9-coordination structurewere in the range of 2.69–3.51 Å. In these distances, O3 of anhydrosugar B and O50 of anhydro sugar C were distantly positioned froma potassium ion for 3.51 and 3.30 Å, respectively, and rest of theatoms were positioned within 2.94 Å.
In 8- and 9-coordination structures, O2, O5, O10 and O20 ofanhydro sugar A and O3 and O6 of anhydro sugar B commonly con-tributed to forming the bipyramidal structures (Fig. 5b and d). Fourof these six oxygen atoms, O2, O3, O5 and O6 were involved in the1,6-anhydro moiety. From the results of the 13C-T1 measurements,
13C-T1 of C1, C3, C5, C6, and C50 were shown to be significantly de-creased (Table 2). The C1, C3, C5, and C6 are positioned adjacent tothe above four oxygen atoms, which could be the reason for affect-ing its 13C-T1. The C50 is positioned adjacent to O50, which was in-volved in the hexagon of the 9-coordination structure (Fig. 5c andd). The 13C NMR studies indicated the contribution of 1,6-anhydromoiety in forming complex with potassium ions, which was alsosupported by the single-crystal X-ray structure.
In conclusion, complex formation of 1,6-anhydro-b-maltose andpotassium ions has been elucidated using 1H, 13C and 39K NMRspectroscopy and single-crystal X-ray crystallography. In solution,1H diffusion coefficients and 39K-T1 unambiguously indicated com-plex formation, and 13C NMR studies indicated the importance ofthe 1,6-anhydro moiety in the complex formation, which was alsosupported in the analysis of the X-ray crystal structure.
3. Experimental
3.1. NMR spectroscopy
Conventional 1H and 13C NMR spectra were acquired on a Var-ian 600 spectrometer using 250 lL sample solutions of 30 mM 1,6-anhydro-b-maltose in the presence and absence of 30 mM KSCNand 30 mM KSCN alone in 99.9% CD3OD in Shigemi tubes at30 �C. The 39K NMR spectra were acquired on a JEOL ECS400 spec-trometer equipped with a multitunable probe. The 1H and 13Cassignments were carried out using the conventional 1D and 2DNMR techniques, and the mixing time of 2D NOESY was set to be500 ms. The selective J-resolved HMBC experiment14 was carriedout to measure the long-range coupling constants of 3JCH aroundthe glycosylic bond. The experimental parameters of the selectiveJ-resolved HMBC experiment were data size in t1 = 256 complexpoints; data size in t2 = 1024; spectral width in f1 = 14000 Hz;

Figure 5. The crystal structure of 1,6-anhydro-b-maltose forming an inclusion complex with a potassium ion. (a) 8-Coordination structure and (b) its schematic drawing. (c)9-Coordination structure and (d) its schematic drawing. In (a) and (c), close contacts between 1,6-anhydro-b-maltoses and a potassium ion are shown by dashed lines, andoutlines of each coordination structure, corresponding to the schematic drawings of (b) and (d), are shown by solid lines.
Table 3Distances (Å) in the 8- and 9-coordination forms around a potassium ion
8-Coordination 9-Coordination
Atoms Distances Atoms Distances
K� � �O2(A)a 2.98 K� � �O2(A) 2.72K� � �O3(B) 2.78 K� � �O3(B) 3.51K� � �O5(A) 2.74 K� � �O5(A) 2.83K� � �O6(B) 2.74 K� � �O6(B) 2.81K� � �O10(A) 2.81 K� � �O10(A) 2.94K� � �O20(A) 2.85 K� � �O20(A) 2.77K� � �MeOH 2.72 K� � �O50(C) 3.30K� � �SCN� 3.86 K� � �O60(C) 2.69
K� � �SCN� 3.27
a ‘A’ means anhydro sugar A defined in Figure 5.
Table 4Crystal and experimental data
Formula: C27H43K2N2O21S2
Formula weight: 873.95Crystal system: monoclinicSpace group: P 2yb Z = 2Crystal dimensions (mm) = 0.20 � 0.15 � 0.15Temperature = 123 Ka = 9.784(2) Åb = 11.919(3) Åc = 16.041(4) ÅV = 1867.1(8) Å3
Dcalcd = 1.556 g/cm3
No. of reflections = 218752hmax = 60.0� with Mo KaR = 0.0515Max. shift/error in final cycle 0.001Maximum peak in final diff. map 0.88 e�/Å3
Minimum peak in final diff. map �0.73 e�/Å3
Measurements R-AXIS RAPID (Rigaku, 2000)Program system Crystal structure 4.0Structure determination SIR200415
Refinement Full-matrix least-squares on F2
T. Fujimoto et al. / Carbohydrate Research 346 (2011) 1991–1996 1995
f2 = 2500 Hz; number of transients per increment = 32; recycletime = 1.5 s; the scaling factor, n = 25. The 13C-T1 measurementswere carried out six times using the inversion recovery method.The experimental parameters were number of scans = 1024, re-cycle time = 30 s. The pulse intervals were 16 steps. The measure-ments of 1H diffusion coefficients were carried out six times. Theexperimental parameters were number of scans = 32; recycletime = 28 s. The gradient amplitudes ranged from 2 to 30 G/cm in16 steps. The 39K-T1 measurements were carried out using theinversion recovery method. The experimental parameters werenumber of scans = 4000; pulse repetition time = 0.9 s. The pulseintervals were ten steps. The baseline corrections were applied be-fore calculation. The 13C and 39K-T1 were calculated using the lin-ear least-squares analysis, and the half height widths of 39Ksignals were calculated using the peak separation programequipped in the JEOL Delta NMR software.
3.2. Single-crystal X-ray crystallography
Equal molar amounts of 1,6-anhydro-b-maltose (324 mg) andKSCN (97.1 mg) were dissolved in 3 mL of MeOH, and the solutionwas kept at room temperature for 48 h. After filtration, a colorlessprism, mp 161–162 �C, having approximate dimensions of0.20 � 0.15 � 0.15 mm was mounted in a loop. All measurementswere carried out using a Rigaku RAXIS RAPID imaging plate area

1996 T. Fujimoto et al. / Carbohydrate Research 346 (2011) 1991–1996
detector with graphite monochromated Mo Ka radiation at 123 K.The crystal and experimental data are given in Table 4.
4. Supplementary data
Crystallographic data, excluding structure factors, have beendeposited with the Cambridge Crystallographic Data Centre as sup-plementary publication with CCDC No. 816367. Copies of the datacan be obtained free of charge on application with the Director,CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223336 033; e-mail: [email protected]).
Acknowledgments
This study was supported by an Ito Science Foundation and aGrant-in-Aid for Scientific Research (No. 21550092 for M.T.) fromthe Ministry of Education, Culture, Sports, Science, andTechnology.
References
1. Angyal, S. J. Adv. Carbohydr. Chem. Biochem. 1989, 47, 1–43.2. Gyurcsik, B.; Nagy, L. Coord. Chem. 2000, 203, 81–149.3. Barker, P. E.; Thawait, S. Chem. Ind. (London) 1983, 817–821.4. Angyal, S. J.; Bethell, G. S.; Beveridge, R. J. Carbohydr. Res. 1979, 73, 9–18.5. Diaz, M. D.; Berger, S. Carbohydr. Res. 2000, 329, 1–5.6. Stilbs, P.; Johnson, C. S., Jr. Anal. Chem. 1994, 66, 211–215.7. Schuerch, C. Adv. Carbohydr. Chem. Biochem. 1981, 39, 157–212.8. Tanaka, T.; Huang, W. C.; Noguchi, M.; Kobayashi, A.; Shoda, S. Tetrahedron Lett.
2009, 50, 2154–2157.9. Fujimoto, T.; Sakurai, S.; Tsutsui, A.; Furihata, K.; Machinami, T.; Tashiro, M.
Anal. Sci. 2005, 21, 1245–1247.10. Kato, T.; Tsubono, K.; Kamo, O.; Kato, T.; Furihata, K.; Fujimoto, T.; Machinami,
T.; Tashiro, M. Magn. Reson. Chem. 2009, 47, 948–952.11. Rondeau, P.; Sers, S.; Jhurry, D.; Cadet, F. Appl. Spectrosc. 2003, 57, 466–472.12. Rondeau, P.; Jhurry, D.; Sers, S.; Cadet, F. Appl. Spectrosc. 2004, 58, 816–822.13. Kato, T.; Fujimoto, T.; Tsutsui, A.; Tashiro, M.; Mitsutsuka, Y.; Machinami, T.
Chem. Lett. 2010, 39, 136–137.14. Furihata, K.; Tashiro, M.; Seto, H. Magn. Reson. Chem. 2009, 47, 814–818.15. Burla, M. C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G. L.; De Caro,
L.; Giacovazzo, C.; Polidori, G.; Spagna, R. J. Appl. Crystallogr. 2005, 38, 381–388.


















