
BONES AND MUSCLES ROBERTO D. PADUA JR., MD, DPSP DEPARTMENT OF PATHOLOGY FATIMA COLLEGE OF MEDICINE.
112
BONES AND MUSCLES BONES AND MUSCLES ROBERTO D. PADUA JR., MD, DPSP ROBERTO D. PADUA JR., MD, DPSP DEPARTMENT OF PATHOLOGY DEPARTMENT OF PATHOLOGY FATIMA COLLEGE OF MEDICINE FATIMA COLLEGE OF MEDICINE
-
Upload
gabriella-joseph -
Category
Documents
-
view
216 -
download
1
Transcript of BONES AND MUSCLES ROBERTO D. PADUA JR., MD, DPSP DEPARTMENT OF PATHOLOGY FATIMA COLLEGE OF MEDICINE.

BONES AND MUSCLESBONES AND MUSCLES
ROBERTO D. PADUA JR., MD, DPSPROBERTO D. PADUA JR., MD, DPSPDEPARTMENT OF PATHOLOGYDEPARTMENT OF PATHOLOGYFATIMA COLLEGE OF MEDICINEFATIMA COLLEGE OF MEDICINE

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• CHONDRODYSPLASIAS• Abnormalities in the size and shape of bones• Disproportionate shortness in stature• Named after the part of the bone affected• Other names refer to the appearance of the bone
– Diastrophic (twisted)– Thanatophoric (death-bearing)– Metatropic (changing)
• Family history of disease is obligatory• Radiologic appearance can be confused with other
metabolic bone diseases• Serum levels of biochemical markers are normal• Bone is well mineralized

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 1. ACHONDROPLASIA• Most common cause of disproportionately
short stature• 1 of 40,000 live births, autosomal dominant• Head is large, frontal region is protuberant,
nasal bridge is depressed• Lordosis and lumbar kyphosis are present• Anteroposterior flattening of the pelvic inlet• Failure of normal endochondral ossification at
the level of the proliferating and maturing cartilage

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 2. ACHONDROGENESIS• Affected infants are either stillborn or do not survive
the immediate neonatal period• 2 syndromes
– a) Achondrogenesis I (Parenti-Fraccaro)• Associated with congenital heart defects• No ossification in the skull & vertebral bodies
– b) Achondrogenesis II (Langer-Saldino)• Shortened limbs & disproportionately large head• Underdeveloped ossification centers in the vertebral
bodies and pelvis• Epiphyseal cartilage is lobulated with increased
vascularity• Completely disorganized endochondral ossification of
the growth plate and there is no column formation

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 3. THANATOPHORIC DYSPLASIA• Infants are either stillborn or die of respiratory
distress during the neonatal period• Pattern of inheritance is unknown• Length of trunk is normal but the head is large with
cranio-facial disproportion• Common CVS and CNS anomalies• Pronounced platyspondyly of the lumbar vertebrae
with an inverted U appearance • Curvature of femurs with medial and lateral spikes at
their lower ends; short, flared ribs• Endochondral ossification is disrupted at the growth
plate, no regular column formation

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 4. CHONDROECTODERMAL DYSPLASIA• Also known as Ellis-van Creveld syndrome• Short limb dwarfism• Consanguity is an important factor in the
etiology of the disease• Clinical presentation:
• Narrowing of rib cage• Congenital heart disease• Ectodermal abnormalities• Acromegalic micromelia (shortening of the
distal segment of the limb

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 5. ASPHYXIATING THORACIC DYSPLASIA
• Jeune’s syndrome• Narrowing of the chest and immobility• Stippled epiphyses and chondroplasia
punctata are striking features on x-ray

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 6. OSTEOPETROSIS• Marble bone disease• Defective osteoclast function that impairs
skeletal resorption• Primary spongiosa persists during adult life
• Increased incidence of parental consanguity• Early symptom is malformation of mastoid
and paranasal sinuses• Pathognomonic histologic finding is the
failure of osteoclast to resorb skeletal tissue

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 7. PROGRESSIVE DIAPHYSEAL DYSPLASIA
• Camurati-Engelman disease• Rare autosomal dominant disorder• Formation of new bone at both the
periosteal and endosteal surfaces

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 8. ENDOSTEAL HYPEROSTOSIS• Van Buchem disease• Autosomal dominant/recessive disorder• Progressive enlargement of mandible during
puberty• Radiographic feature: dense and
homogenous diaphyseal cortex with narrowing of the medullary canal

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 9. OSTEOPOIKILOSIS• Presence of numerous foci of sclerosis in
cancellous bone (“spotted bones”)• Autosomal dominant disorder• Bone changes are asymptomatic• Found incidentally on radiographs

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 10. OSTEOPATHIA STRIATA• Characterized by linear striations at the ends of
long bones and in the ilium• X-ray shows gracile linear striations in the
cancellous region of the skeleton• Autosomal dominant trait

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 11. MELORHEOSTOSIS• Characterized by hyperostosis of the limb
bones• X-ray : likened to appearance of melted wax
that is dripped down the side of the candle• Typical histologic finding is endosteal
thickening

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 12. PACHYDERMOPERIOSTOSIS• Hypertrophic osteoarthropathy• Characterized by clubbing of digits,
hyperhidrosis, thickening of skin around the face and forehead and periosteal new bone formation in the distal limbs
• Inherited as autosomal dominant trait• Men>women• X-ray shows thickening and sclerosis of the
distal portions of the tubular bones

SKELETAL DEVELOPMENTAL AND SKELETAL DEVELOPMENTAL AND GENETIC DISORDERSGENETIC DISORDERS
• 13. OSTEOGENESIS IMPERFECTA• “Brittle bone disease”• Hereditary disorder involving defects in the synthesis
or structure of collagen type I• Cardinal features : osteopenia associated with
recurrent fracture and skeletal deformity• Biochemical findings : increased AP, increased level
of hydroxyproline, hypercalciuria• Histology : abnormal skeletal matrix; cartilaginous
bars formed by vascular invasion of the metaphyses do not become envelop by bones; cortical bone is almost non-existent

METABOLIC BONE DISEASESMETABOLIC BONE DISEASES
• 1. OSTEOPOROSIS• Loss of normally minerralized bone• Diagnosed clinically with non-invasive radiographic
techniques that measures bone density• Changes in the bone :
• Structurally weak• Loss of trabecular bone• Enlargement of the medullary space• Cortical porosity• Reduction in cortical thickness

METABOLIC BONE DISEASESMETABOLIC BONE DISEASES
• 2. RENAL OSTEODYSTROPHY• Seen in patients with advanced renal failure• Clinical presentations :
• Bone pain (most common), spontaneous fractures, aseptic necrosis of hip, myopathy
• Laboratory findings :• Low levels of 1;25(OH)2D3, hyperphosphatemia,
hypocalcemia, alterations in the secretion or activity of PTH
• X-ray : • Subperiosteal erosions, patchy osteosclerosis
(“rugger jersey” appearance of thoracic vertebral spine on lateral views, “salt and pepper” appearance of skull, slipped epiphyses

METABOLIC BONE DISEASESMETABOLIC BONE DISEASES
• 3. OSTEOMALACIA• Defective mineralization of the trabecular
and cortical bone matrix• Associated with decreased serum calcium
phosphate product• A common complication of chronic renal
failure in adults• Secondary to Vitamin D deficiency
• Histologically characterized by excessive quantities of osteoid because of the failed matrix calcification despite continued matrix synthesis by the osteoblasts

METABOLIC BONE DISEASESMETABOLIC BONE DISEASES
• 4. RICKETS• Defective mineralization of the epiphyseal growth
plate cartilage• Clinical features : craniotabes, frontal bossing,
rachitic rosary, pectus excavatum, “Harrison’s groove”, thoracic kyphosis, rachitic potbelly, genu varum/genu valgum
• Histologic appearance : • Rachitic growth plate is wide and irregular• Columnar rearrangement of the hypertrophic
chondrocyts is lost• Zone of provisional calcification disappears• Cartilage extends deep into the metaphyses

TRAUMATRAUMA
• FRACTURE REPAIR• Blastema wound closure scar formation• Initial repair tissue formed is called a CALLUS which is
composed of fibrous tissue, woven bone and cartilage• 3 phases of fracture healing :
• A) inflammatory phase• B) reparative phase = orderly removal and replacement of
immature woven bone by cartilage differentiation• C) modeling phase = realignment & mechanical shaping of
the bone and callus; restoration of the medullary cavity and bone marrow
• Complications of fracture healing :• A) nonunions• B) fibrous union

INFLAMMATORY BONE DISORDERSINFLAMMATORY BONE DISORDERS
• OSTEOMYELITIS• Classified according to several factors
• 1. its duration = acute, subacute or chronic• 2. nature of the exudate = hemorrhagic,
purulent, or nonsuppurative• 3. its location = bone, periosteum, or epiphyses• 4. etiologic agent = Staphylococcus, Tb, etc.
• Histologically, inflammatory cells are seen• Loss of normal marrow architecture• Hematopoietic elements and fat are replaced
by leukocytic infiltrates

INFLAMMATORY BONE DISEASESINFLAMMATORY BONE DISEASES
• OSTEOMYELITIS…..• “Chronic sclerosing osteomyelitis of Garre”
• A chronic form of osteomyelitis with findings of dense, scarred bone and few clinical symptoms without any abscess formation.
• “Brodies abscess”• Osteomyelitis sharply limited to one side with
formation of abscess cavity surrounded by a rim of sclerotic bone.
• Causes:• Coagulase (+) Staph. Aureus (60-90%)• Strep, Pneumococcus, E. coli, Klebsiella,
Salmonella, Bacteroides

INFLAMMATORY BONE DISEASESINFLAMMATORY BONE DISEASES
• OSTEOMYELITIS…..• Causes :
– Tuberculosis• Spread hematogenously• Characteristic lesion : Chronic caseating
granulomatous inflammation which often involves the subchondral part of the joint. Sequestrum forms in the subchondral bone and articular cartilage resulting in a “kissing sequestrum”.
• Pott’s disease – Tb of the spine

OSTEOMYELITISOSTEOMYELITIS
X-RAYGROSS : UPPER FEMUR

INFLAMMATORY BONE DISEASESINFLAMMATORY BONE DISEASES
• SARCOIDOSIS• Noncaseating granulomatous process• Manifest as small lytic and sclerotic foci in
the bones of the hand• Large areas of destruction are not typically
found

INFLAMMATORY BONE DISEASESINFLAMMATORY BONE DISEASES
• PAGET’S DISEASE OF BONE (OSTEITIS DEFORMANS)
• A chronic osteolytic and osteosclerotic disease of uncertain cause
• May involve one or more bones• Presents with pain, skeletal deformities, and
occasionally sarcomatous transformation• Usually affects 3% of white population over 40
y/o• Incidence increases with age; men>women• Most patients are asymptomatic (80-90%)

INFLAMMATORY BONE DISEASESINFLAMMATORY BONE DISEASES
• PAGET’S DISEASE OF BONE…..• Common skeletal sites of involvement are the sacrum, spine,
pelvis, skull, femur, clavicle, tibia, ribs, and humerus• Histopathology:
• Normal marrow is replaced by a richly vascular, loose fibrous connective tissue
• Isolated clusters of inflammatory cells may be seen
• Osteoclasts aggregate on the existing bone trabeculae and within the cortex
• Innumerable small, irregularly shaped bone fragments (mosaic pattern)
• Grossly resembles the gritty but brittle texture of pumice or lava rock

INFLAMMATORY BONE DISEASESINFLAMMATORY BONE DISEASES
• PAGET’S DISEASE OF BONE….• X-RAY : flocculant, radiopaque deposit
likened to cotton wool. Pelvis is the most common site of involvement.
• Elevated AP and osteocalcin level• Elevated urinary excretion of
hydroxyproline, pyridinoline and deoxypyridinoline
• Malignant transformation are also observed• Osteosarcomas• Fibrosarcomas• Giant cell malignant fibrous histiocytoma

PAGET’S DISEASE OF BONEPAGET’S DISEASE OF BONE
X-RAY OF TIBIA SHOWING BONE DESTRUCTION AND BONE FORMATION
EARLY CHANGES SHOWING PROMINENT OSTEOCLASTIC ACTIVITY

DEGENERATIVE DISEASES OF DEGENERATIVE DISEASES OF BONEBONE
• OSTEONECROSIS• Infarction of bone typically involving the femoral
head• 3 generic categories = postfracture, idiopathic, and
renal transplant associated• Also known as “avascular necrosis of bone”• Earliest histologic changes are death of the bone and
the surrounding hematopoietic & fatty marrow• X-ray : “Crescent sign” , a separation of fracture cleft
forms between the impacted fragments and the overlying subchondral plate. Increased density within the necrotic bone.

BONE TUMORSBONE TUMORS
• Most malignant tumors arise de novo• Benign bone lesions that predispose to the
development of skeletal malignancies• Paget’s disease, chondromatosis,
osteochondromatosis, fibrous dysplasia, and osteofibrous dysplasia
• Five basic parameters in the diagnosis of bone tumors
• Age of the patient• Bone involved• Specific area within the bone• Radiographic appearance• Microscopic appearance

BONE FORMING TUMORSBONE FORMING TUMORS
• 1. OSTEOMA• Seen almost exclusively in the flat bones of skull
and face• Microscopically: composed of dense, mature,
predominantly lamellar bone• Benign• Associated with Gardner’s syndrome

BONE-FORMING TUMORSBONE-FORMING TUMORS
• 2. OSTEOID OSTEOMA• Benign neoplasm seen in patients between 10 and 30 y/o• 2:1 male-female ratio• Intense pain is the most prominent symptom• Reported in practically every bone, most are centered in the
cortex (85%), spongiosa (13%), or subperiosteal region (2%)• X-ray: typical finding is a radiolucent nidus that is seldom
larger than 1.5 cm and may or may not contain a dense center. This nidus is surrounded by a peripheral sclerotic reaction.
• Microscopic: sharply delineated central nidus composed of more or less calcified osteoid lined by plump osteoblast and growing within vascularized connective tissue, without evidence of inflammation.

OSTEOID OSTEOMAOSTEOID OSTEOMA
X-RAY GROSS MICROSCOPIC

BONE-FORMING TUMORSBONE-FORMING TUMORS
• 2. OSTEOBLASTOMA• Benign osteoblastoma, giant osteoid osteoma• Closely related to osteoid osteoma both microscopically and
ultrastructurally• It has a larger size of the nidus, absence of surrounding area
of reactive bone formation, and the lack of intense pain• A cartilaginous matrix is present in some cases• Most cases arise in the spongiosa of the bone involving the
spine or major bones of the lower extremity• Osteomalacia can be seen as a complication

BONE-FORMING TUMORSBONE-FORMING TUMORS
• 3. OSTEOSARCOMA• The most frequent primary malignant tumor,
exclusive of hematopoietic malignancy• Usually occurs in patients between 10 and 25
years of age and is rare in pre-school children• Another peak age incidence occurs after the age of
40, in association with other disorders• Most osteosarcomas arise de novo, but others
arise within the context of a preexisting condition• Paget’s disease, radiation exposure, chemotherapy,
preexisting benign bone lesions, foreign bodies, trauma

BONE-FORMING TUMORSBONE-FORMING TUMORS
• OSTEOSARCOMA…..• Located in the metaphyseal area of long bones,
particularly the lower end of femur, upper end of the tibia, and the upper end of the humerus
• Large majority arise within the medullary cavity from which they extend into the cortex
• Gross appearance varies depending on the relative amounts of bone, cartilage, cellular stroma and vessels bony hard to cystic, friable, and hemorrhagic
• From its usual origin in the metaphysis of a long bone, the tumor may spread along the marrow cavity, invade the adjacent cortex, or elevate or perforate the periosteum (Codman’s triangle)

BONE-FORMING TUMORSBONE-FORMING TUMORS
• OSTEOSARCOMA…..• Extend into the soft tissues, extend into the
epiphysis, extend into the joint space, form satellite nodules independent from the main tumor mass proximal to the primary lesion (“skip metastases”), metastasize through the blood stream to distant sites particularly the lung.

BONE-FORMING TUMORSBONE-FORMING TUMORS
• OSTEOSARCOMA…..• Microscopic features
• May destroy preexisting bone trabeculae or grow around them in an appositional fashion
• Key feature is the presence of osteoid and or bone produced directly by tumor cells without interposition of cartilage
• Osteoblastic areas are often mixed with fibroblastic and chondroblastic foci
• Tumor cells may grow in diffuse, nesting or pseudopapillary arrangements

BONE-FORMING TUMORSBONE-FORMING TUMORS
• OSTEOSARCOMA…..• OS cells usually exhibit strong AP activity,
regardless of their appearance• Ultrastructurally, tumor cells resemble normal
osteoblasts• Consistently expresses Vimentin• In some cases, they are positive for smooth
muscle actin, desmin, EMA, S-100 protein• Osteonectin, osteocalcin, osteopontin bone
morphogenetic protein and bone GLA protein have been identified immunohistochemically

BONE-FORMING TUMORSBONE-FORMING TUMORS
• OSTEOSARCOMA…..• Microscopic variants
• Telangiectatic• Small cell • Fibrohistiocytic• Anaplastic• Well-differentiated intramedullary• Others = parosteal (juxtacortical), periosteal

OSTEOSARCOMAOSTEOSARCOMA
GROSS SHOWING SKIP METASTASIS
MICROSCOPIC APPEARANCE

OSTEOSARCOMAOSTEOSARCOMA
TELANGIECTATIC VARIANT OF OSTEOSARCOMA

OSTEOSARCOMAOSTEOSARCOMA
JUXTACORTICAL OSTEOSARCOMA

BONE-FORMING TUMORSBONE-FORMING TUMORS
• OSTEOSARCOMA…..• Diagnosis: characteristic radiographic appearance,
open biopsy, needle biopsy, FNAB, frozen section.• Therapy: amputation or disarticulation. At present,
limb-sparing procedures coupled with other therapeutic modalities.
• Prognosis: • Poor : presence of Paget’s disease, multifocal OS,
chondroblastic type, Telangiectatic variant, elevated AP, low postchemotherapy tumor necrosis, loss of heterozygosity of the RB gene, HER2/neu expression, expression of P-glycoprotein

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
• 1. CHONDROMA• A common benign cartilaginous tumor that occurs most
frequently in the small bones of the hands and feet, particularly the proximal phalanges
• 30% are multiple• Microscopically, they are composed of mature hyaline
cartilage. Foci of myxoid degeneration, calcification, and endochondral ossification are common
• Enchondromas begins in the spongiosa of the diaphysis from which they expand and thin out the cortex
• Lesions with predominantly unilateral distribution are referred to as Ollier’s disease
• Its association with soft tissue hemangiomas is known as Maffucci’s syndrome

CHONDROMACHONDROMA
X-RAY GROSS
MICROSCOPIC

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
• 2. OSTEOCHONDROMA• Most frequent benign tumor• Usually asymptomatic, but may lead to deformity or interfere with
the function of adjacent structures such as tendons and blood vessels
• Most common locations are metaphyses of the lower femur, upper tibia, upper humerus and pelvis
• Average age of onset is 10 y/o, majority appears before the age of 20
• Average greatest diameter is 4 cm but may reach 10 cm or more• A cap of cartilage covered by fibrous membrane continous with the
periosteum of the adjacent bone• Microscopically, the cells resemble those of normal hyaline
cartilage. Eosinophilic, PAS-(+) inclusions may be seen in the cytoplasm. The bulk of the lesion is composed of mature bone trabeculae located beneath the cartilaginous cap and containing normal bone marrow.

OSTEOCHONDROMAOSTEOCHONDROMA
GROSS, CUT SECTION MICROSCOPIC

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
• 3. CHONDROBLASTOMA• Occurs predominantly in males under 20 y/o• Usually arises in the epiphyseal end of long bones before the
epiphyseal cartilage has disappeared, particularly in the distal end of femur, proximal end of humerus, and proximal end of tibia
• X-ray: tumor is fairly well delimited and contains areas of rarefaction
• Microscopic:• the basic tumor cell is an embryonic chondroblast with
only a limited capacity for the production of cartilaginous matrix.
• Presence of occasional scattered giant cells

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
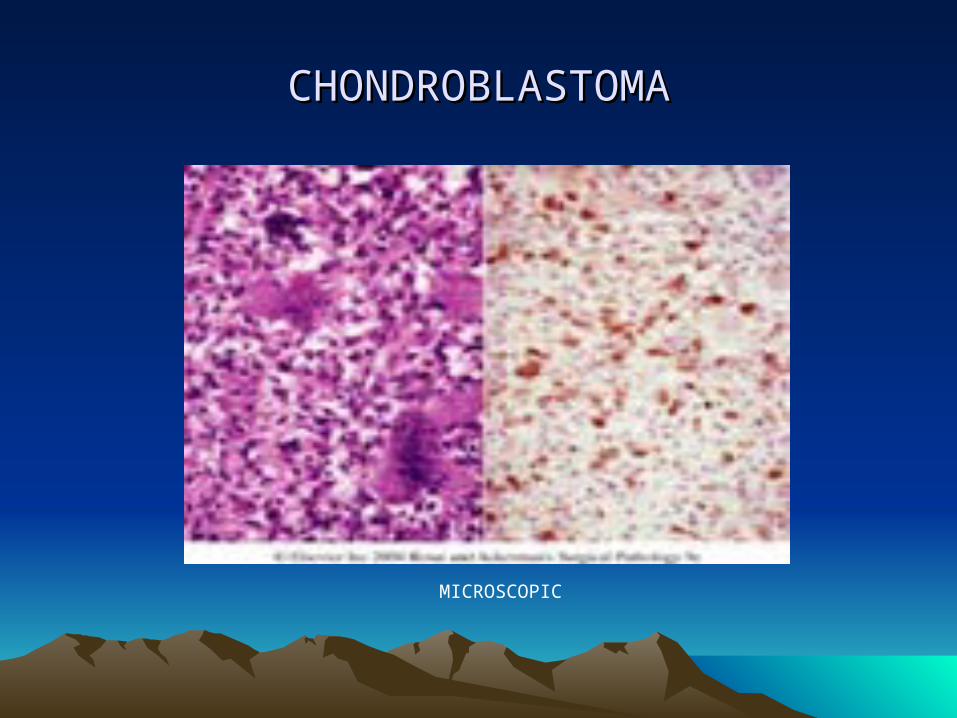
• CHONDROBLASTOMA…..• Microscopic:
• Cells are usually polyhedral with round to indented nuclei. Reticulin fibers surround each individual cell.
• Presence of small zones of focal calcification (“chicken wire”)
• Diagnosis can be made by fine needle aspiration which will show neoplastic chondroblast, multinucleated osteoclast-like giant cells, and chondroid myxoid fragments
• Treatment is by curettement with bone grafting

CHONDROBLASTOMACHONDROBLASTOMA
X-RAY GROSS
CHONDROBLASTOMACHONDROBLASTOMA
MICROSCOPIC

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
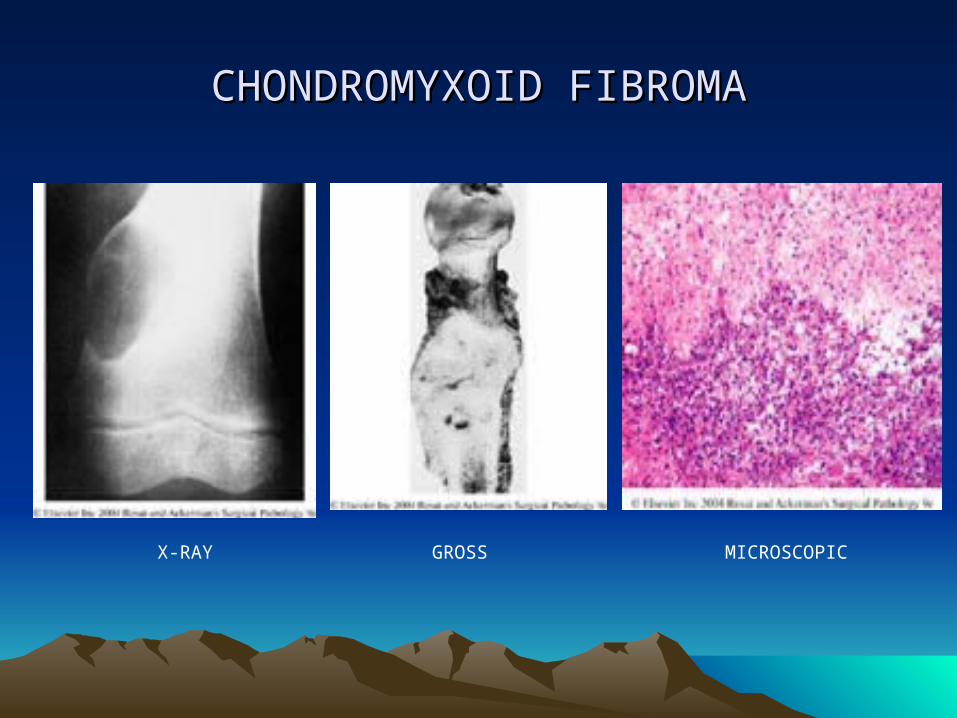
• 4. CHONDROMYXOID FIBROMA• An unusual benign tumor• Usually occurs in long bones of young adults• Radiographically, it is sharply defined and may attain a large
size• Grossly, it is solid and yellowish white or tan, replaces bone
and thins the cortex.• Microscopically, shows hypocellular lobules with a
chondromyxoid appearance separated by intersecting bands of fibroblast-like spindle cells and osteoclasts
• Strong positivity to S-100 protein• Treatment is by curettage with a recurrence rate of 25%
CHONDROMYXOID FIBROMACHONDROMYXOID FIBROMA
X-RAY GROSS MICROSCOPIC

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
• 5. CHONDROSARCOMA• A malignant tumor of cartilage-forming tissues• Divided into conventional and variants• Conventional chondrosarcoma can be
• Central = located in the medullary cavity, usually of flat or long bone. X-ray show osteolytic lesion with splotchy calcification with ill-defined margins, fusiform thickening of the shaft, and perforation of the cortex
• Peripheral = may arise de novo or from the cartilaginous cap of a preexisting osteochondroma
• Juxtacortical (periosteal) = involves the shaft of a long bone characterized by a cartilaginous lobular pattern with areas of splotchy calcification and endochondral ossification

CARTILAGE-FORMING TUMORSCARTILAGE-FORMING TUMORS
• CHONDROSARCOMA…..• Microscopically, there is production of cartilaginous
matrix and the lack of direct bone formation by the tumor cells
• Soft tissue implantation following biopsy is a well known complication
• Chondrosarcoma variants• Clear cell chondrosarcoma• Myxoid chondrosarcoma• Dedifferentiated chondrosarcoma• Mesenchymal chondrosarcoma

CHONDROSARCOMACHONDROSARCOMA
GROSS APPEARANCES OF CHONDROSARCOMA

CHONDROSARCOMACHONDROSARCOMA
X-RAY, FEMUR

CHONDROSARCOMACHONDROSARCOMA
WELL-DIFFERENTIATED CLEAR CELL VARIANT

GIANT CELL TUMORGIANT CELL TUMOR
• Osteoclastoma• Patients are over 20 years of age• More common in women then men• More frequently in Oriental than Western countries• Classic location is epiphysis of long bone• Affects more commonly the lower end of femur, upper
end of tibia, and lower end of the radius. It also occurs in the humerus, fibula, and skull particularly the sphenoid bone.
• Multicentricity has been reported particularly in young patients and in the small bones of hands and feet

GIANT CELL TUMORGIANT CELL TUMOR
• X-ray: • The typical appearance is that of an entirely lytic,
expansile lesion in the epiphysis, usually without peripheral bone sclerosis, or periosteal reaction
• Gross:• The size of the tumor varies; when large, it may be
associated with a pathologic fracture• The cut surface is solid and tan or light brown,
traversed by fibrous trabeculae, and often contains hemorrhagic areas
• The cortex is thinned, but periosteal new bone formation is rare

GIANT CELL TUMORGIANT CELL TUMOR
• Microscopic:• Two main components are stromal cells and
giant cells• The giant cells are usually large and have over
twenty or thirty nuclei, most of then are arranged toward the center. – They resemble osteoclasts at all levels: ultrastructurally,
enzyme histochemically and immunohistochemical– Result of fusion of circulating monocytes that have
been recruited into the lesion• Possible mechanisms: 1. Autocrine or paracrine loop mediated by
transforming growth factor beta

GIANT CELL TUMORGIANT CELL TUMOR
• Microscopic…..• Possible mechanisms….
2. Production of osteoprotegerin ligand (a factor essential for osteoclastogenesis)
3. Expression of the ligand for RANK (receptor activator of nuclear factor Kappa B)
• Mononuclear stromal cells is the only proliferating element in the lesion and the one exhibiting atypia in the rare cytologically malignant examples of this tumor
• These changes may be focal, hence a thorough sampling is required
• Produces type I & III collagen and has receptors for PTH

GIANT CELL TUMORGIANT CELL TUMOR
X-RAY GROSS APPEARANCE

GIANT CELL TUMORGIANT CELL TUMOR
MICROSCOPIC APPEARANCE

GIANT CELL TUMORGIANT CELL TUMOR
• Frequent positivity for S-100 protein• Many benign lesions with giant cells have
been diagnosed as giant cell tumor in the past– A diagnosis of a lesion other than GCT should
be favored if:• 1. patient is a child• 2. lesion is located in the metaphysis or diaphysis
of a long bone• 3. lesion is multiple• 4. lesion is located in the vertebrae, jaw, or bones
of the hands or feet

GIANT CELL TUMORGIANT CELL TUMOR
• Treatment : – Surgical = consists of curettage with
bone grafting or en bloc resection with allograft or artificial material
– Use of radiation therapy should be reserved only for cases in which surgical removal is impossible

MARROW TUMORSMARROW TUMORS
• 1. EWING’S SARCOMA/PRIMITIVE NEUROECTODERMAL TUMOR (PNET)
• Undifferentiated type of bone sarcoma in children
• Related to the neoplasm originally described in the soft tissues as primitive (peripheral) neuroectodermal tumor
• Usually seen in patients between the ages of 5 and 20 years
• Clinically, the tumor may simulate OM because of pain, fever, and leukocytosis

EWING’S/PNETEWING’S/PNET
• Occurs most often in long bones (femur, tibia, humerus, and fibula) and in the bones of the pelvis, rib, vertebrae, mandible and clavicle
• It generally arises in the medullary canal of the shaft, from which it permeates the cortex and invade the tissues
• Can present clinically as a soft tissue neoplasm with a normal appearance of the underlying bone on plain x-ray films
• X-ray : cortical thickening and widening of the medullary canal. With progression of the lesion, reactive periosteal bone may be deposited in layers parallel to the cortex (“onion-skin” appearance) or at right angle to it (“sun-ray” appearance)

EWING’S SARCOMA/PNETEWING’S SARCOMA/PNET
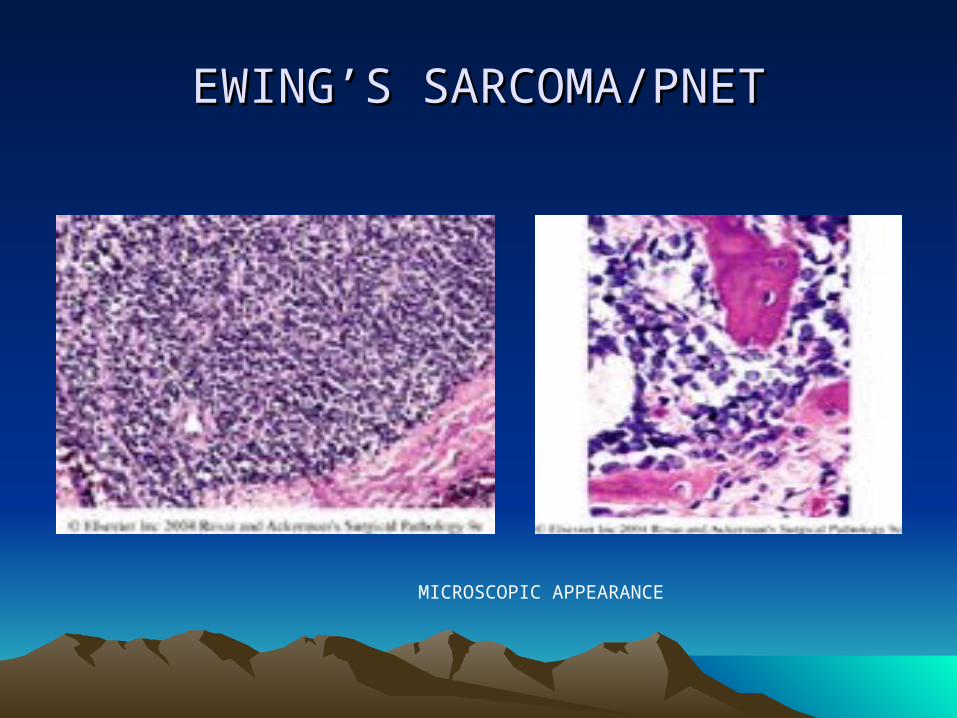
• Microscopic :• Consists of solid sheets of cells divided into irregular
masses by fibrous bands• Individual cells are small and uniform• The cells outline are indistinct, resulting in a
“syncitial” appearance• The nuclei are round, with frequent indentations,
small nucleoli and variable but usually brisk mitotic activity
• There is well developed vascular network• Pseudorosettes and rosettes arrangement of cells
may be seen

EWING’S SARCOMA/PNETEWING’S SARCOMA/PNET
X-RAY GROSS APPEARANCE
EWING’S SARCOMA/PNETEWING’S SARCOMA/PNET
MICROSCOPIC APPEARANCE

EWING’S SARCOMA/PNETEWING’S SARCOMA/PNET
• Cells contains large amounts of cytoplasmic glycogen --- (+) PAS
• Ultrastructurally, a few dense core granules will be found either in the cell cytoplasm or in cell prolongations
• Immunohistochemically, positive for vimentin, LMW keratin, NSE, protein gene product 9.5, Leu7, and neurofilaments
• Over 95% of cases show a reciprocal translocation 11;22 (q24;q12), which results in the fusion of EWS gene with FLI or ERG genes

EWING’S SARCOMA/PNETEWING’S SARCOMA/PNET
• Metastatic spread is to the lungs and pleura, other bones (particularly the skull), CNS, and (rarely) regional LN
• About 25% of the patients have multiple bone and/or visceral lesions at the time of presentation
• Treatment:• Combination of high-dose irradiation and
multidrug chemotherapy– sometimes combined with limited surgery

MARROW TUMORSMARROW TUMORS
• 2. MALIGNANT LYMPHOMA• Can involve the skeletal system primarily or as a
manifestation of a systemic disease• LARGE CELL LYMPHOMA
• More common in adults than in children• 60% of cases occurring in patients over 30 y/o• No sex predilection• Most cases involve the diaphysis or metaphysis og
long bones or vertebrae producing patchy cortical and medullary destruction associated with minimal to moderate periosteal reaction
• The tumor is pinkish gray and granular, frequently extends into the soft tissues and invades the muscle

MALIGNANT LYMPHOMAMALIGNANT LYMPHOMA
• LARGE CELL LYMPHOMA…..• Radiographically, a combination of bone
production and bone destruction often involves a wide area of a long bone
• Microscopically, the appearance is similar to that of the large cell lymphoma in nodal and other extranodal sites, some cases are accompanied by prominent fibrosis
• The 5-year survival rate for localized B-cell lymphoma of bone has ranged from 30-60%
• The stage of the disease is the single most important prognostic determinator

MALIGNANT LYMPHOMAMALIGNANT LYMPHOMA
X-RAY MICROSCOPIC APPEARANCE

MALIGNANT LYMPHOMAMALIGNANT LYMPHOMA
• HODGKIN’S LYMPHOMA• Produces radiographically detectable bone lesions
in approximately 15% of the patients• Involvement is multifocal in about 60% of cases,
most frequent sites being vertebrae, pelvis, ribs, sternum, and femur
• Osseous lesions are often asymptomatic and in half of the cases are not demonstrable radiographically

MALIGNANT LYMPHOMAMALIGNANT LYMPHOMA
• Anaplastic large cell lymphoma
• Burkitt’s lymphoma
• Lymphoblastic lymphoma

ACUTE LEUKEMIAACUTE LEUKEMIA
• Associated with radiographic abnormalities in the skeletal system in 70-90% of cases
• Destructive bone lesions are extremely rare in the chronic leukemias

VASCULAR TUMORSVASCULAR TUMORS
• 1. HEMANGIOMA• Often seen in the vertebrae as an incidental post-mortem
finding• The most common locations are the skull, vertebrae, and jaw• Cut section has a currant jelly appearnce• Microscopically, there is a thick-walled lattice-like pattern of
endothelial lined cavernous spaces filled with blood• Multiple hemangiomas are mainly seen in children and are
associated in about half of the cases with cutaneous, soft tissue, or visceral hemangiomas

VASCULAR TUMORSVASCULAR TUMORS
• 2. MASSIVE OSTEOLYSIS• Gorham’s disease• Not a vascular neoplasm• Has microscopic similarities with skeletal
angiomatosis• It has a destructive character• It results in reabsorption of a whole bone or
several bones and the filling of the residual spaces by a heavy vascularized fibrous tissue

VASCULAR TUMORSVASCULAR TUMORS
• 3. LYMPHANGIOMAS• Most cases are multiple and associated with soft
tissue tumors of similar appearance
• 4. GLOMUS TUMOR• May erode the underlying bone
• 5. HEMANGIOPERICYTOMA• Can present as a primary bone lesion, most
common location is the pelvis

VASCULAR TUMORSVASCULAR TUMORS
• 6. EPITHELIOID HEMANGIOENDOTHELIOMA
• A borderline type of vascular neoplasm characterized microscopically by the presence epithelial- or histiocyte-like endothelial cells with abundant acidophilic and often vacuolated cytoplasm, large vesicular nucleus, modest atypia, scanty mitotic activity, inconspicous or absent anastomosing channels, recent and old hemorrhage and an inconstant but sometimes prominent inflammatory component rich in eosinophils

VASCULAR TUMORSVASCULAR TUMORS
• 7. ANGIOSARCOMA• Malignant hemangioendothelioma,
hemangioendothelial sarcoma• Exhibits obvious atypia of the tumor cells,
formation of solid areas alternating with others with anastomosing vascular channels, and foci of necrosis and hemorrhage
• Multicentricity is common• Distant metastasis are common, particularly lungs

VASCULAR TUMORSVASCULAR TUMORS
CAVRNOUS HEMANGIOMA OF BONE

VASCULAR TUMORSVASCULAR TUMORS
EPITHELIOID HEMANGIOENDOTHELIOMA

METASTATIC TUMORSMETASTATIC TUMORS
• In most cases the lesions are multiple• More than 80% arises from the breast, lung,
prostate, thyroid, or kidney• These metastases can be accompanied by
visceral deposits or represent the only apparent site of dissemination
• Soft tissue sarcomas rarely metastasize to the bones except embryonal rhabdomyosarcoma in children
• They are usually osteolytic but maybe osteoblastic or mixed

Metastatic tumorsMetastatic tumors
• The mechanism is thought to be the production of bone growth factors by tumor cells, such as TGF-beta, fibroblast growth factor, and bone morphogenetic proteins
• Symptoms is usually pain• Treatment is relief of pain and to prevent
fracture of weight-bearing bones

TUMORLIKE LESIONSTUMORLIKE LESIONS
• 1. SOLITARY BONE CYST• Unicameral bone cyst• Usually occur in long bones, most often in the
upper portion of the shaft of the humerus and femur
• Also seen in the short bones, calcaneus• Mostly affects males and are seen in patients
under 20 years• Usually are advanced when first seen, most are
centered in the metaphysis and they migrate away from the epiphyseal line

TUMORLIKE LESIONSTUMORLIKE LESIONS
• SOLITARY BONE CYST….• The cysts contains a clear or yellow fluid that is
lined by a smooth fibrous membrane• Maybe hemorrhagic if previous fracture occurred• Microscopic: well-vascularized connective tissue,
hemosiderin and cholesterol clefts are frequent• Treatment of choice is curettement and
replacement of the cyst with bone chips

SOLITARY BONE CYSTSOLITARY BONE CYST
X-RAY GROSS APPEARANCE

TUMORLIKE LESIONSTUMORLIKE LESIONS
• 2. ANEURYSMAL BONE CYST• Usually seen in patients between 10 and 20 years
of age• More common in females• Occurs mainly in the vertebrae and flat bones but
can also arise in the shaft of long bones• Multiple involvement is common in the vertebral
lesions• X-ray: shows eccentric expansion of the bone with
erosion and destruction of the cortex and a small area of periosteal new bone formation

TUMORLIKE LESIONSTUMORLIKE LESIONS
• ANEURYSMAL BONE CYST….• GROSS: it forms a spongy hemorrhagic mass
covered by a thin shell of reactive bone, which may extend into the soft tissue
• Microscopic: • show large spaces filled with blood• They do not contain endothelial lining but are rather
delimited by cells with similar features to fibroblast, myofibroblasts, and histiocytes
• A row of osteoclasts is often seen immediately beneath the surface
• There is significant deposition of generated calcifying fibromyxoid tissue.

TUMORLIKE LESIONSTUMORLIKE LESIONS
• ANEURYSMAL BONE CYST….• Pathogenesis is still unknown• In a few cases, the lesion is preceded by trauma with fracture
or subperiosteal hematoma• It may also arise in some preexisting bone lesion as a result
of changed hemodynamics• Insulin-like growth factor-I may play in its pathogenesis• Ddx: chondroblastoma, GCT, fibrous dysplasia, nonossifying
fibroma, osteoblastoma, chondrosarcoma• Treatment : en bloc resection or curettage with bone grafting

ANEURYSMAL BONE CYSTANEURYSMAL BONE CYST
X-RAY GROSS APPEARANCE

ANEURYSMAL BONE CYSTANEURYSMAL BONE CYST
MICROSCOPIC APPEARANCE

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA• Most common soft tissue sarcoma of childhood
and adolescence• Usually appears before the age of 20• Commonly occurs in the head and neck or
genitourinary tract, extremities• Cytogenic abnormalities
• t(2;13)(q35;q14)• t(1;13)(q36;14)

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA• In the more common translocation, t(2;13), the
PAX3 gene on chromosome 2 fuses with the FKHR gene on chromosome 13
• PAX3 gene functions upstream of genes that control muscle differentiation
• Pathogenesis of tumor involves dysregulation of muscle differentiation by the chimeric PAX3-FKHR protein

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA – MORPHOLOGY• EMBRYONAL • ALVEOLAR• PLEOMORPHIC*** Diagnostic cell is the RHABDOMYOBLAST
= contains eccentric eosinophilic granular cytoplasm rich in thick and thin filaments = may be round or elongated (tadpole or strap
cells= Ultrastructurally, contain sarcomeres = Immunohistochemically, they stain with antibodies to the myogenic markers
desmin, MYOD1 and myogenin

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA – MORPHOLOGY
• EMBRYONAL RHABDOMYOSARCOMA• Most common type, accounting to 60%• Includes Sarcoma Botryoides• Occurs in children under 10 years of age• Typically arises in the nasal cavity, orbit, middle
ear, prostate and paratesticular region• Allelic loss of chromosome 11p15.5 as its major
genomic abnormality

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA – MORPHOLOGY
• EMBRYONAL RHABDOMYOSARCOMA• Grossly,they present as a soft gray infiltrative
mass• Microscopically, the tumor cells mimic skeletal
muscle cells at various stages of embryogenesis and consist of sheets of both malignant round and spindled cells in a variably myxoid stroma
• Sarcoma botryoides grows in a polypoid fashion, producing the appearance of a cluster of grapes protruding into a hollow structure such as the bladder or vagina

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA – MORPHOLOGY
• ALVEOLAR RHABDOMYOSARCOMA• Most common in the early and mid-adolescence
and usually arises in the deep musculature of the extremities
• Histologically, the tumor is traversed by a network of fibrous septae that divide the cells into clusters or aggregates; as the central cells degenerate and drop out, resembles pulmonary alveolae
• Tumor cells are moderate in size and have little cytoplasm

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA – MORPHOLOGY
• ALVEOLAR RHABDOMYOSARCOMA• Cells with cross-striations are identified in about
25% of cases• Cytogenetic studies show a t(2;13) or t(1;13)
chromosomal translocations

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA – MORPHOLOGY
• PLEOMORPHIC RHABDOMYOSARCOMA• Characterized by numerous large, sometimes
multinucleated, bizarre eosinophilic tumor cells• This variant is rare• Arises in the deep soft tissue of adults • Resemble malignant fibrous histiocytoma
histologically

TUMORS OF SKELETAL MUSCLESTUMORS OF SKELETAL MUSCLES
• RHABDOMYOSARCOMA • Usually treated with a combination of surgey and
chemotherapy with or without radiation• Histologic variant and location of the tumor
influence survival• Sarcoma botryoides have the best prognosis,
followed by embryonal, pleomorphic, and alveolar variants
• Overall prognosis for children is good = 65%; less for adults

TUMORS OF SMOOTH MUSCLETUMORS OF SMOOTH MUSCLE
• LEIOMYOMA• Benign smooth muscle tumor, commonly arises in
the uterus• May also arise in the erector pili muscles found in
the skin, nipples, scrotum and labia and less in the deep soft tissues
• Multiple lesions is thought to be hereditary and transmitted as an autosomal dominant trait
• Occur in adolescence and early adult life

TUMORS OF SMOOTH MUSCLETUMORS OF SMOOTH MUSCLE
• LEIOMYOMA• Tumors are usually not larger than 1 to 2 cm in
greatest dimension • Composed of fascicles of spindle cells that tend to
intersect each other at right angles• Tumor cells have blunt-ended, elongated nuclei
and show minimal atypia and few mitotic figures• Treatment is surgical

TUMORS OF SMOOTH MUSCLETUMORS OF SMOOTH MUSCLE
• LEIOMYOSARCOMA• Account for 10 to 20% of soft tissue sarcomas• Occurs in adults, women>men• Most develop in the skin and deep soft tissues of
the extremities and retroperitoneum• Present as painful, firm masses• Retroperitoneal tumors may be large and bulky
and cause abdominal symptoms

TUMORS OF SMOOTH MUSCLETUMORS OF SMOOTH MUSCLE
• LEIOMYOSARCOMA• Histologically, characterized by malignant spindle
cells that have cigar-shaped nuclei arranged in interweaving fascicles
• Immunologically, they stain with antibodies to vimentin, actin, smooth muscle actin and desmin
• Treatment depends on the size, location and grade of the tumor



















