
Benign and Intermediate Fibroblastic/Myofibroblastic ... foreninger/Den... · Fibromatosis colli...
19
1 Benign and Intermediate Fibroblastic/Myofibroblastic Lesions Rita Alaggio Fibroblastic/myofibroblastic tumors Fibroblastic/myofibroblastic tumors account for about 12% of all pediatric soft tissue tumors. The WHO classification recognizes 3 prognostic categories: Benign tumors, tumors with intermediate prognosis (either locally aggressive or only rarely metastasizing) and malignant tumors. The majority of myofibroblastic proliferations is represented by benign or reactive/pseudo-neoplastic lesions; the remaining have an intermediate prognosis. Malignant myofibroblastic tumors are very rare in children. The histologic variation of Fibroblastic/myofibroblastic tumours, may be sometimes subtle even between tumors with different clinical behavior and render the differential diagnosis challenging. This review will focus on benign and intermediate myofibroblastic lesions. Table 1 summarizes the fibroblastic-myofibroblastic tumors more frequent in pediatric age. Table 1 FIBROBLASTIC / MYOFIBROBLASTIC TUMOURS Pseudo-sarcomatous proliferations Nodular fasciitis Cranial Fasciitis Myositis ossificans/fibro-osseous pseudo-tumour of digits Benign Lesions Fibromas Fibroma of tendon sheath Gardner fibroma Fibromatoses (Juvenile type) Fibrous hamartoma of infancy Myofibroma / Myofibromatosis Fibromatosis colli Juvenile hyaline fibromatosis/Inclusion body fibromatosis Calcifying aponeurotic fibroma Lesions with intermediate prognosis (locally aggressive) Fibromatoses (Juvenile type) Lipofibromatosis Fibromatoses (adult type) Palmar-plantar fibromatosis Desmoid-type fibromatoses Lesions with intermediate prognosis (rarely metastasizing) Inflammatory myofibroblastic tumour Low-grade myofibroblastic sarcoma Infantile fibrosarcoma Pseudosarcomas
Transcript of Benign and Intermediate Fibroblastic/Myofibroblastic ... foreninger/Den... · Fibromatosis colli...

1
Benign and Intermediate Fibroblastic/Myofibroblastic Lesions
Rita Alaggio
Fibroblastic/myofibroblastic tumors
Fibroblastic/myofibroblastic tumors account for about 12% of all pediatric soft tissue
tumors. The WHO classification recognizes 3 prognostic categories: Benign tumors,
tumors with intermediate prognosis (either locally aggressive or only rarely
metastasizing) and malignant tumors. The majority of myofibroblastic proliferations is
represented by benign or reactive/pseudo-neoplastic lesions; the remaining have an
intermediate prognosis. Malignant myofibroblastic tumors are very rare in children. The
histologic variation of Fibroblastic/myofibroblastic tumours, may be sometimes subtle
even between tumors with different clinical behavior and render the differential
diagnosis challenging.
This review will focus on benign and intermediate myofibroblastic lesions.
Table 1 summarizes the fibroblastic-myofibroblastic tumors more frequent in pediatric
age.
Table 1 FIBROBLASTIC / MYOFIBROBLASTIC TUMOURS
Pseudo-sarcomatous proliferations Nodular fasciitis Cranial Fasciitis Myositis ossificans/fibro-osseous pseudo-tumour of digits
Benign Lesions Fibromas
Fibroma of tendon sheath Gardner fibroma Fibromatoses (Juvenile type)
Fibrous hamartoma of infancy Myofibroma / Myofibromatosis Fibromatosis colli Juvenile hyaline fibromatosis/Inclusion body fibromatosis Calcifying aponeurotic fibroma
Lesions with intermediate prognosis (locally aggressive) Fibromatoses (Juvenile type)
Lipofibromatosis Fibromatoses (adult type) Palmar-plantar fibromatosis Desmoid-type fibromatoses
Lesions with intermediate prognosis (rarely metastasizing) Inflammatory myofibroblastic tumour Low-grade myofibroblastic sarcoma Infantile fibrosarcoma
Pseudosarcomas

2
Nodular fasciitis
Nodular fasciitis is the most common pseudo-sarcomatous lesion in adults. It is rare in
children younger than 5 years, although infants and small children may be rarely
affected. There is a slight predominance in males. It may involve any site, including
head and neck, trunk and extremities. Nodules are located in deep subcutaneous tissue,
muscle and fascia, but dermal variants are also reported. A previous trauma may be
identified in less than 15% of cases.
Macroscopically nodular fasciitis is a small non-encapsulated, nodule, usually less than
3.0 cm in greatest diameter with a cut surface, varying from myxoid to firm and
leathery.
The histological features show variations according to the phase of the process (table 2).
Early lesions show a prominent myxoid matrix with spindle-stellate cells simulating
fibroblasts grown in tissue culture. Extravasated red blood cells, scattered lymphocytes
and plasma-cells are common.
In the florid phase, there is a higher cellularity, with myofibroblasts and ganglion-like
mesenchymal cells in short fascicles and sheets intermingled with collagen bundles.
Occasionally a zonation pattern may be observed, with a myxoid or hyalinized central
area and a densely cellular periphery. In late stage lesions myxoid matrix is minimal,
replaced by collagen with hyaline, keloid-like appearance. Broad fascicles of
myofibroblasts display a storiform pattern. Microcystic spaces containing mucoid
material may be found. Mitoses are more frequent in recent lesions, but are never
atypical. Cartilagineous metaplasia, osteoid, calcification, and heterotopic ossification
may be occasionally found.
Immunohistochemically myofibroblasts are reactive for vimentin, muscle-specific actin,
smooth muscle actin, and calponin, whereas h-caldesmon is negative. CD68 stains
histiocytes and rare osteoclast-like giant cells.
Unusual variants of nodular fasciitis include intra-articular and intravascular nodular
fasciitis. They closely resemble the common form of nodular fasciitis.
The differential diagnosis of nodular fasciitis includes a wide spectrum of benign and
malignant lesions (table 2). Nodular fasciitis is treated by surgical excision. In case of
incomplete resection, the lesion may spontaneously regress. Recurrences are extremely

3
rare and may be related to the proliferation of reactive myofibroblasts present in the
residual lesion in response to the trauma of resection.
Table 2: differential diagnosis of Nodular Fasciitis
Histology Differential Diagnosis Diagnostic clues
Early lesion: Abundant myxoid matrix Fibroblasts in “cell culture-like” pattern Red cells, lymphocytes plasmacells Florid lesion:
High cellularity Occasional zonation (hyalinized center and cellular periphery) Older lesions: Abundant keloid-like collagen
Malignant peripheral nerve sheath tumor
Neurofibroma
Lipoblastoma
myxoid liposarcoma:
myxofibrosarcoma:
Myofibromatosis
Spindle cell sarcomas (embryonal rhabdomyosarcoma, fibrosarcoma, inflammatory myofibroblastic tumor)
Desmoid Fibromatosis:
Keloid
Inflammatory myofibroblastic tumor
cellular atypia, weavy nucley, necrosis, Antoni A and B areas.
absent inflammatory infiltrate and red cells, S100+ cells
lipoblasts, organoid pattern with collagen septa
lipoblasts, typical vascular network
cellular atypia, atypical mitoses
spindle cells in whorls, inflammatory infiltrate or extravased red cells not prominent
Larger lesions, deeply located, cytologic atypia, necrosis
Long fascicles, scattered mast cells, IHC: beta-catenin+
larger lesions, deeply located, ALK+ in some case
Fibromas
Gardner fibroma
Gardner fibroma, has been recognized by the 2002 World Health Organization
classification of soft tissue tumors as a distinctive benign lesion, typically associated
with desmoids and familial adenomatous polyposis coli or Gardner syndrome. A family
history of APC is found in 70% of patients and in 10% other members of family have a
history of desmoid fibromatosis.
It has a predilection for the first 2 decades of life, and is located in superficial and deep
soft tissues of the paraspinal region, back, chest wall, flank, head and neck, and
extremities. A desmoid fibromatosis may be found in continuity with the Gardner
fibroma at time of biopsy or may develop subsequently in about 50% of cases.
Macroscopically, it is a poorly circumscribed, plaque-like lesion with a rubbery, white
cut-surface.
Microscopically it is extremely hypocellular, densely collagenized, with scattered
spindle cells separated by clefts from the eosinophilic collagen fibers. At the periphery
the lesion is infiltrative with focal entrapment of fat, peripheral nerve fibers, and blood
vessels. Sparse mast cells may be present.
The spindled cells are positive for vimentin and CD34, but negative for muscle- specific
actin, smooth muscle actin, desmin, estrogen receptor, and progesterone receptor

4
proteins. Nuclear beta-catenin reactivity is variable and can be positive in up to two-
thirds of cases. It may be negative in patients with APC mutation.
Nuchal- type fibroma typical of young adults is a lesion strictly related to Gardner
Fibroma. It may be associated with diabetes mellitus and shows an increased number of
small nerve bundles, absent in Gardner fibroma. Its presence in children may be
associated with FAP.
Gardner fibroma is a benign lesion, whether it is a malformation or a true neoplasm
representing a juvenile form of desmoid fibromatosis is yet to be determined. The
correct diagnsis is important because it may represent a sentinel event and should alert
clinicians to the possibility of an underlying Gardner Syndrome or FAP.
Fibromatoses
Fibromatoses represent an unique group of fibroblastic myofibroblastic lesions
accounting for over the 90% of fibroblastic-myofibroblastic tumors in children.They
include different clinico-pathologic entities, some typical of children (juvenile
fibromatoses), others occurring in adults and children (adult fibromatoses).
The juvenile fibromatoses are generally benign and some of them may have a
spontaneous regression. Some juvenile fibromatoses such as Juvenile hyaline
fibromatosis and gingival fibromatosis are hereditary.
The adult fibromatoses include superficial (palmar / plantar) and Desmoid-type
fibromatoses, which are considered as intermediate, locally aggressive lesions in 2002
WHO classification. In table 3 clinical features of fibromatoses are reported. We will
focus on the most common types.

5
Table 3: Clinical features of Juvenile and Adult (*)Fibromatoses
Age M/F Site Prognosis Treatment
Myofibromatosis Solitary multicentric Generalized
Birth or later Congenital Congenital
3/1 1/2
Skin, soft tissue Skin, soft tissue, bone Skin, soft tissue, bone, viscera
Favorable Favorable Progression Death (75%)
Surgery, possible regression Surgery, possible regression Chemotherapy
Fibromatosis colli
Congenital Male predominance
Sternocleidomastoid muscle, right, may be bilateral
Self-limiting Physical therapy
Infantile digital fibroma/inclusion body fibromatosis
Infants/older children
No sex predilection
40-60% multiple dorso-lateral pprtion of digits,
Recurrence in xx%
Surgical resection
Fibrous Hamartoma of Infancy
Infants-2yr 1/1 Axilla, groin, upper arms, upper trunk, external genitalia
favourable Surgery
Lipofibromatosis Infants-2 yrs 2:1 distal extremities, trunk and head
Recurrence or persistent growth in 72%
Surgical resection
Juvenile nasopharingeal fibroma
10-20 yr male posterior nasal cavity ethmoid and maxillary sinuses
recurrence rate: 68%. Rare spontaneous regression.
Surgery, pharmacotherapy with anti-androgens. Embolization or chemotherapy.
Juvenile hyaline fibromatosis Juvenile Infantile systemic
First few years of life Infants
No sex predilection
Skin lesions: hands, scalp, ears, nose. Large subcutaneous tumors. Gingival hypertrophy
visceral involvement
progressive joint contractures osteopenia lethal
Interferon 2 alpha
Calcifying aponeurotic fibroma
0-64 yr Slight male predominance
Hands (fingers and palms), feet, and wrist. Proximal extremities and back occasionally involved
50% recurrence.. surgical excision
Palmar/plantar Fibromatosis*
Adults (10% in children between 2-12 yr)
1/2
plantar, less frequently palmar.
Recurrence: 83% Surgery reserved to lesions not responding to pharmacologic therapies and to early contractures in proximal inter-phalangeal joints of the hands
Desmoid-type fibromatosis*
Adults (20% in children between 0-19 yr)
1/1 Extremities, trunk, head, neck,shoulder, hip
Local growth, Relapse 20%
Surgery, CT/RT, Estrogen antagonists, Imatinib
Juvenile Fibromatoses Infantile myofibroma/myofibromatosis Infantile myofibroma or myofibromatosis is the most common type of fibromatosis in
infancy and childhood.
It occurs in 3 clinical forms: solitary, multicentric, and generalized. (table)The term
myofibromatosis is reserved to the last two forms. Solitary and multicentric IM are the
most common forms and manifest as isolated or multiple nodules localized in the skin,
soft tissues and bone. The most frequent regions involved are head and neck, followed
by trunk and extremities.

6
Generalized IM shows concomitant lesions in lung, heart, gastrointestinal tract or even
central nervous system (Table 3).
Macroscopically myofibromas are nodules of variable size, with an average greatest
diameter of 1.5 cm, but large lesions may occur. Skin lesions are well circumscribed,
whereas those deeply located tend to have less defined margins. The nodules have firm
consistency and white-gray color, occasionally pinky, with cysts or central yellow
necrotic areas, which can be prominent in larger lesions. Scattered calcifications are also
found.
Histologically myofibromas/myofibromatosis are nodular proliferation variously
composed of myoid, elongated cells showing eosinophilic cytoplasm, nuclei with finely
dispersed chromatine and one or two small nucleoli and more primitive elongated-
polygonal cells with larger nuclei, uniform chromatine and amphophilic cytoplasms.
Their appearance varies according to the stage of the lesion.
Immature lesions are highly cellular and the primitive elongated-polygonal cells are
predominant. The cells are arranged in sheets or short fascicle, and only focal areas
show slightly more mature myofibroblasts. At the periphery the margins may be poorly
circumscribed or infiltrative. Delicate irregularly branching blood vessels in a
hemangiopericytomatous pattern may be present. In the past, such cases were regarded
as "infantile hemangiopericytoma", but they are now recognized as a variant of infantile
myofibromatosis.
A zonal microscopic appearance may be found in some cases charcaterized by immature
cells in a hemangiopericytomatous pattern in the center of lesion and myoid cells
arranged in short interlacing fascicles interspersed in a variable collagen or myxoid
matrix or forming whorls at the periphery. In the adjacent non-lesional tissue, peripheral
satellite nodules with a whorled pattern may be found. Focal perivascular and
intravascular proliferation of immature myofibroblastic and primitive mesenchymal cells
can give a false impression of vascular invasion.
Mitoses are rare, but occasianally are frequent. Necrosis may be extensive, with
associated dystrophic calcifications.

7
Immunohistochemical staining of the tumor cells reveals alpha-smooth muscle actin
strong reactivity in the myoid component. S100 protein, epithelial membrane antigen,
keratin and desmin are negative.
Cytogenetic analyses or molecular genetics of infantile myofibroma/myofibromatosis
have demonstrated nonspecific findings with chromosome 8 abnormalities and
chromosome 6 deletions.
An autosomal dominant pattern of inheritance with variable penetrance has been
observed in rare cases.
The differential diagnosis includes other fibromatoses, particularly desmoid
fibromatosis, nodular fasciitis, myofibroblastic sarcoma (Table 4).
Solitary and multicentric myofibroma/myofibromatosis are frequently a self-healing
process, with spontaneous regression of nodules. Surgical excision is the treatment of
choice in those lesions that do not regress. Myofibromas with a destructive growth in
non-resectable sites may be responsive to chemotherapy. Generalized
myofibromatosis does not regress spontaneously and visceral involvement may be
fatal in about 70% of cases.
Fibrous hamartoma of infancy
Fibrous hamartoma is a subdermal fibromatous tumor of infants, generally
diagnosed within the first 2 years of life, with nearly 20% detected at birth. There is a
male prevalence, without familial or syndromic association. It commonly involves the
axillary and inguinal regions, upper arms, upper trunk and external genital areas.
Extremities are rarely involved. Fibrous hamartoma of infancy is generally a solitary
subcutaneous mass.
Grossly, it is poorly demarcated from the surrounding adipose tissue and has a pale,
gray-white fibrotic cut surface with focal soft yellow areas resembling fat.
The triphasic histologic pattern composed of primitive small rounded or stellate cells in
nests and whorls, mature fibrous tissue with collagen bands and islands of adipose tissue
is virtually diagnostic of fibrous hamartoma.
Morphologic variations include a prominent fat or collagen myxoid component,
simulating respectively a lipoblastoma or a neurofibroma. Immunohistochemistry may
be helpful in such cases, demonstrating their fibroblastic-myofibroblastic nature. In fact

8
the primitive mesenchymal cells express only vimentin, whereas in the fibroblastic
component smooth muscle actin and desmin may be positive. CD34 is generally limited
to blood vessels, but primitive mesenchymal cells may be occasionally positive. A
reciprocal translocation t(2;3)(q31;q21) and a t(6;12;8)(q25;q24.3;q13) have been
reported in 2 cases.
Fibrous Hamartoma of infancy is treated with complete surgical excision. The
recurrence rate is 15%, mostly related to an incomplete resection. Spontaneous
regression has not been reported.
Lipofibromatosis
Lipofibromatosis has been described by Fetsch et al. in 2000.
In the past, lesions with morphologic features of lipofibromatosis had been variously
classified as infantile fibromatosis, fibrous hamartoma of infancy; calcifying aponeurotic
fibroma and lipoblastoma. Clinically lipofibromatosis manifests as a slow-growing
mass, arising in the upper and lower distal extremities and, less frequently, in the trunk
and head of infants and children. Up to 25% of cases are congenital. Males are affected
more often than females. Macroscopically the lesions are poorly demarcated, of variable
size, from 2 cm to 7 in maximum dimension, of firm consistency, with a gritty,
yellowish or tan–white cut surface.
Histologically, lipofibromatosis is composed of abundant adipose tissue, with thick
fibrous septa containing fascicles of fibroblasts embedded in a variable amount of
collagen. Focal myxoid changes may be present. Both the adipose tissue and the spindle
cell components are integral to the neoplastic process.
Uni-vacuolated cells in small aggregates may be found at the periphery of adipose
lobules, adjacent to the fibrous septa. These cells are pseudo-lipoblasts, probably
representing adipocytes with regressive changes or cells in transitional stage between
fibroblast and adipocyte. Lipoblasts are not found. At the periphery, the lesions are
poorly circumscribed with evidence of fibro-fatty tissue, infiltrating and entrapping the
adjacent structures. Mitoses are rare. Cytologic atypia is absent. The spindle cell
component shows a positive staining for CD99, CD34, alpha-smooth muscle actin.
BCL-2, S-100 protein, muscle specific actin and EMA may be occasionally expressed.

9
One case has been reported with a complex translocation t(4;9;6). A similar region in
chromosome 6 is involved in a complex translocation found in a fibrous hamartoma of
infancy raising the possibility of a common pathogenetic link related to 6q
rearrangement.
The differential diagnosis of lipofibromatosis includes fibrous hamartoma of infancy,
lipoblastoma, calcifying aponeurotic fibroma, desmoid-type fibromatosis, and
fibrolipoma (table 4).
Surgical resection is the elective treatment for lipofibromatosis. Recurrence or persistent
growth has occurred in 72% of reported cases. Congenital onset, male sex, hands and
feet location, incomplete surgery, and a high mitotic activity are associated with a risk of
recurrence.
Adult Fibromatoses in children and adolescents
Desmoid fibromatosis
Desmoid tumors are monoclonal fibroblastic-myofibroblastic proliferations occurring
throughout the body, generally in association with the deep aponeurotic tissue. They
account for up to 60% of fibrous tumors in childhood, with an age range from the birth
to 19 years. Up to 30% occur in the first year of life, with a peak around 4.5 years.
Congenital cases have also been reported. In pediatric series a male predominance has
been observed.
Desmoid fibromatosis can be sporadic or arise in the setting of Familial Adenomatous
Polyposis (FAP) or the Gardner’s variant of the syndrome.
In children desmoid fibromatosis may involve extremities, trunk, head and neck,
shoulder and hip. Abdomen, retroperitoneum, breast and spermatic cord are rarely
involved. It is a slow growing, mass with an infiltrative growth with involvement of the
adjacent structures, without systemic symptoms. Desmoid tumors arising in Gardner
fibroma may be painful and have a rapid growth.
On gross examination, desmoid fibromatosis is a firm, gray-white mass with a
glistening, whorled cut surface. Hemorrhagic foci or necrosis are absent. The lesion may

10
be apparently well circumscribed, but at a closer inspection, subtle trabeculae or wisps
dissect the surrounding tissues.
Histologically the lesions vary in cellularity and amount of collagen matrix. Uniform
spindle cells and subtle collagen bundles are arranged in long fascicles or sheets with
thin-walled blood vessels parallel to the fascicles. Morphologic variations include
lesions extensively collagenized or prominently myxoid lesions. Nuclear hypercromasia,
cellular atypia and pleomorphism are absent. Mitoses are rare. Mast cells may be
present, especially around blood vessels. At the periphery the tumors show a typical
infiltrative pattern, with subtle bands of collagen or cells in fascicles dissecting adipose
tissue or entrapping muscle fibers. Recurrent lesions show identical morphological
features. The extensive collagenization related to the scarring process may render
difficult the differential diagnosis with a recurrence and the evaluation of surgical
margins.
Immunostains confirm the fibroblastic-myofibroblastic nature of desmoid fibromatoses,
showing positive staining for vimentin and variable expression of muscle specific actin,
desmin and smooth muscle actin. S100 protein and cytokeratins are negative.
Cytogenetic investigation of desmoids in FAP has clarified the key role played by beta-
catenin. Beta catenin in mesenchymal tissues is important as a transcriptional activator
involved in the promotion of cell proliferation. In physiological conditions its cytosolic
levels are under control of adenomatosis polyposis coli (APC) complex, which
phosphorylates four critical aminoacids determining its proteosomal degradation. The
portion of beta-catenin involved in the phosphorylation is encoded by exon 3 of
CTNNB1 gene. Any interruption of phosphorylation results in the accumulation of beta-
catenin in the cytoplasm and its migration to the nucleus with a permanen activation of
genes involved in cell proliferation. In FAP-related desmoids germ-line mutations in
APC gene inhibit its ability to induce the phosphorilation of beta catenin. In sporadic
desmoids three different mutations resulting in a non-phosphorylated active beta-catenin
have been identified. The most frequent mutations in CTNNB1 gene involve codons 41
(41A) and 45 (45F and 45P). The mutated form of beta-catenin shows a positive nuclear

11
immunostaing.
In a series of 42 pediatric desmoids Niero et al identified a mutation in APC gene in
17%, CTNNB1 in 64% and no mutations in 19%. A positive nuclear staining for beta-
catenin was present in about 85% of cases.
Some studies indicate that increased nuclear expression of beta-catenin, especially if
associated to p53 positivity may be predictive of a high recurrence rate, in other studies,
this correlation has not emerged and has been found a significant association between
CTNNB1 45F mutation and local recurrence.
The differential diagnosis includes a wide spectrum of benign and malignant
myofibroblastic proliferations.
The natural history of desmoid is that of a slow growing neoplasm, only locally
aggressive. It is unclear why some tumors continue to grow; others remain stable and
occasionally spontaneously regress. The therapeutic approach should take into account
the patient’s characteristics, the site and the observed aggressivity. Surgery remains the
mainstay for resectable sporadic desmoid tumors. The goal should be a radical resection
with negative margins, in order to reduce the risk of local recurrences. However, the
influence of positive margins on the local relapse is still debated, hence a mutilating
surgery should be avoided. In recent years the multi-modal approach including
radiotherapy and chemotherapy has contributed to the decrease of 5-year recurrence rate
to 20%. Mortality from locally aggressive desmoid is less than 10%. Estrogen
antagonists have been proven to be effective. Imatinib mesylate, a tyrosine-kinase
inhibitor, targeting c-kit and PDGFR have been recently used in a clinical trial. The
good clinical response reported in some cases is probably related to other mechanisms,
not involving c-kit and PGDFR. Clinical features associated with a higher risk of
recurrence include younger age, mesenteric lesions and occurrence in the setting of a
FAP or Gardner syndrome.
12
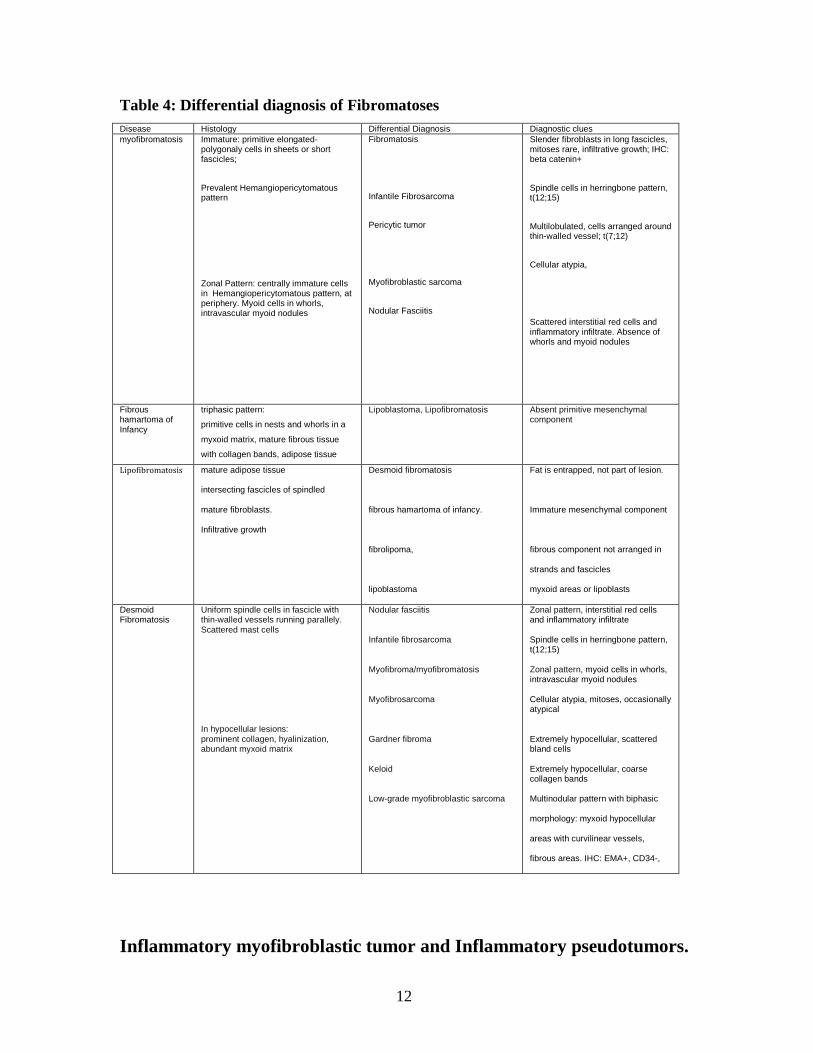
Table 4: Differential diagnosis of Fibromatoses
Disease Histology Differential Diagnosis Diagnostic clues
myofibromatosis
Immature: primitive elongated-polygonaly cells in sheets or short fascicles;
Prevalent Hemangiopericytomatous pattern
Zonal Pattern: centrally immature cells in Hemangiopericytomatous pattern, at periphery. Myoid cells in whorls, intravascular myoid nodules
Fibromatosis
Infantile Fibrosarcoma
Pericytic tumor
Myofibroblastic sarcoma
Nodular Fasciitis
Slender fibroblasts in long fascicles, mitoses rare, infiltrative growth; IHC: beta catenin+
Spindle cells in herringbone pattern, t(12;15)
Multilobulated, cells arranged around thin-walled vessel; t(7;12)
Cellular atypia,
Scattered interstitial red cells and inflammatory infiltrate. Absence of whorls and myoid nodules
Fibrous hamartoma of Infancy
triphasic pattern:
primitive cells in nests and whorls in a
myxoid matrix, mature fibrous tissue
with collagen bands, adipose tissue
Lipoblastoma, Lipofibromatosis Absent primitive mesenchymal component
Lipofibromatosis mature adipose tissue
intersecting fascicles of spindled
mature fibroblasts.
Infiltrative growth
Desmoid fibromatosis
fibrous hamartoma of infancy.
fibrolipoma,
lipoblastoma
Fat is entrapped, not part of lesion.
Immature mesenchymal component
fibrous component not arranged in
strands and fascicles
myxoid areas or lipoblasts
Desmoid Fibromatosis
Uniform spindle cells in fascicle with thin-walled vessels running parallely. Scattered mast cells In hypocellular lesions: prominent collagen, hyalinization, abundant myxoid matrix
Nodular fasciitis Infantile fibrosarcoma Myofibroma/myofibromatosis Myofibrosarcoma Gardner fibroma Keloid Low-grade myofibroblastic sarcoma
Zonal pattern, interstitial red cells and inflammatory infiltrate Spindle cells in herringbone pattern, t(12;15) Zonal pattern, myoid cells in whorls, intravascular myoid nodules Cellular atypia, mitoses, occasionally atypical Extremely hypocellular, scattered bland cells Extremely hypocellular, coarse collagen bands Multinodular pattern with biphasic
morphology: myxoid hypocellular
areas with curvilinear vessels,
fibrous areas. IHC: EMA+, CD34-,
Inflammatory myofibroblastic tumor and Inflammatory pseudotumors.

13
The generic term “inflammatory pseudotumor” includes reactive or infectious processes,
such as spindle cell nodules of genito-urinary tract, mycobacterial pseudotumor and
pseudotumors of lymph node and neoplastic clonal lesions such as the inflammatory
myofibroblastic tumor and follicular dendritic tumors of spleen, in the past diagnosed as
splenic pseudotumors.
In recent years a new group of inflammatory pseudotumors has emerged, including the
idiopathic fibro-sclerosing lesions related to a new syndrome with elevated serum levels
of IgG4.
Inflammatory myofibroblastic tumor.
IMT have been observed mainly in children and young adults, but can arise also in older
adults. The most frequent anatomic sites are the mesentery, omentum, retroperitoneum,
abdominal soft tissues, lung, mediastinum, liver, and head and neck.
In the current World Health Organization classification of soft tissue and bone tumors,
inflammatory myofibroblastic tumors are regarded as a neoplasm of intermediate
biologic potential with tendency for local recurrence and very rare metastases
A palpable mass may be the clinical presentation, but in up to a third of patients it is
discovered after the occurrence of an inflammatory syndrome associated with alteration
of laboratory exams (microcytic hypochromic anemia, thrombocytosis, polyclonal
hyperglobulinemia).
Macroscopically, inflammatory myofibroblastic tumors are nodular, circumscribed
lesions, with a firm consistency, and may reach a large size. Intra-abdominal IMTs are
generally larger, frequently multinodular, non-capsulated, adherent to and sometimes
infiltrating the intestinal wall. On cross-section, the surface is whitish with firm, fleshy,
or myxoid areas.
Histologically IMT are composed of long or intermediate-sized myofibroblasts with
eosinophilic-amphophilic cytoplasm and indistinct cellular borders, showing variable
cellularity and pattern of growth, intermixed with a prominent inflammatory infiltrate
composed of plasma cells, lymphocytes, and eosinophils. Coffin et al identified three
main morphologic patterns: fasciitis-like, fibrohistiocytoma-like and desmoid-like.
In the fasciitis-like pattern interlacing bundles of spindle cells or haphazardly arranged
plump to polygonal myofibroblasts are embedded in an abundant myxoid matrix

14
containing a mixed inflammatory infiltrate A prominent network of small dilated vessels
and typical ganglion-like cells with evident nucleoli may be also present.
The fibrohistiocytoma-like pattern is more cellular with spindle cells in fascicles or in a
storiform pattern alternating with collagenized or myxoid areas. Scattered ganglion like
cells may be seen.
The desmoid-type pattern is hypocellular, with a rich dense collagenous matrix and
sparse spindle cells, intermixed with a discrete inflammatory infiltrate.
A rare variant of round cell inflammatory myofibroblastic tumor has been recently
described. It is characterized by non-cohesive round to polygonal cells with eccentric,
vesicular nuclei, prominent nucleoli and abundant eosinophilic cytoplasm.
Scattered foci of necrosis, calcifications and osseous metaplasia may be occasionally
found in inflammatory myofibroblastic tumors. Mitotic activity is generally low with
less than 2 mitoses per 10 HPF, and atypical mitoses are rare. Bizarre cells, large
atypical myofibroblasts may be seen.
Immunohistochemistry shows strong reactivity for vimentin and variable staining from
focal to diffuse for smooth muscle actin, muscle specific actin and desmin. Focal
positivity for CD68 may be found. Follicular dendritic cell markers, CD21, CD23 are
negative, whereas clusterin may be occasionally positive. Approximately one-third of
cases show focal keratin reactivity.
ALK-1 staining is detected in approximately half of inflammatory myofibroblastic
tumors and correlates with the presence of rearrangements of the gene. ALK is a
receptor tyrosine-kinase gene located on chromosome 2p23. When rearranged, it results
in a protein persistently activated. In inflammatory myofibroblastic tumors a variety of
fusion gene partners have been identified, including ATIC, CARS, TPM3, TMP4, TPM3
and TPM4, CLTC, RANBP2 and SEC31L genes, each one characteristically associated
with a peculiar pattern of immunostaining.
ALK gene rearrangements are more frequent in pediatric inflammatory myofibroblastic
tumors and in those arising in abdomen.
The differential diagnosis is extensive and varies according to the predominant
histologic pattern.
The recurrence rate varies according to the anatomical site. Extra-pulmonary lesions

15
tend to recur more frequently, with a relapse rate of 25%. Distant metastases are rare,
occurring in less than 5% of cases, mostly in lung and brain, followed by liver and bone.
Tumor size, cellularity and histologic features do not appear to influence the clinical
behavior, even if some studies suggest that cellular atypia and prominent myxoid stroma
may be associated with an aggressive course. However cytologic atypia is frequently
found in IMT with a favorable prognosis. Aneuploidy may indicate a more aggressive
potential. There is no evidence of a prognostic significance of ALK expression.
Interestingly, intra-abdominal tumors, which are those with higher recurrence rate, tend
to show a prominent myxoid background, higher cytologic atypia and more frequent
ALK gene rearrangements. ALK negative tumors, on the other side, appear to have a
higher risk of metastasis. An aggressive behavior has been reporre in round cell IMT.
The fw tumors investigated showed an ALK- RANBP2 fusion gene.
IgG4 related fibro-sclerosing lesions (CASE 2 SLIDE SEMINAR)
Sclerosing mesenteritis, idiopathic retroperitoneal fibrosis, sclerosing mediastinitis, and
orbital inflammatory pseudo-tumor are lesions, which histologically can mimic
inflammatory myofibroblastic tumor.
After the report by Hamano et al in 2001 of elevated serum level of immunoglobulin G4
(IgG4) in patients affected by autoimmune sclerosing pancreatitis, there has been a
growing evidence that iper-IgG4 and sclerosing lesions affecting various organs,
represent an unique disease with different clinical manifestations. A recent study
reported the association of increased IgG4-positive plasma cells also in plasma cell-rich
pulmonary inflammatory pseudotumors.
Iper-IgG4 sclerosing diseases occur mainly in adults, nonetheless, retroperitoneal
fibrosis, autoimmune pancreatitis and orbital inflammatory pseudotumor have been
reported also in children.
Grossly, these processes form ill-defined masses often encasing adjacent structures.
Histologically there is a prominent fibrosclerosis extremely hypocellular, with a minimal
spindle cell component. A lymphoplasmacytic infiltrate is present with evidence of
lymphoid follicles. The fibrosis surrounds the parenchymal structures, such as pancreatic
16
acini, with atrophy or loss. Obstructive phlebitis may be helpful in the differential
diagnosis from IMT.
The criteria to diagnose an IgG4-related disease on the basis of the degree of IgG4
positive plasma cells have not been clarified yet and the ratio IgG4+/IgG+ plasma cells
more than 0.1 is not reliable to discriminate from IMT (Coffin et al 2010).
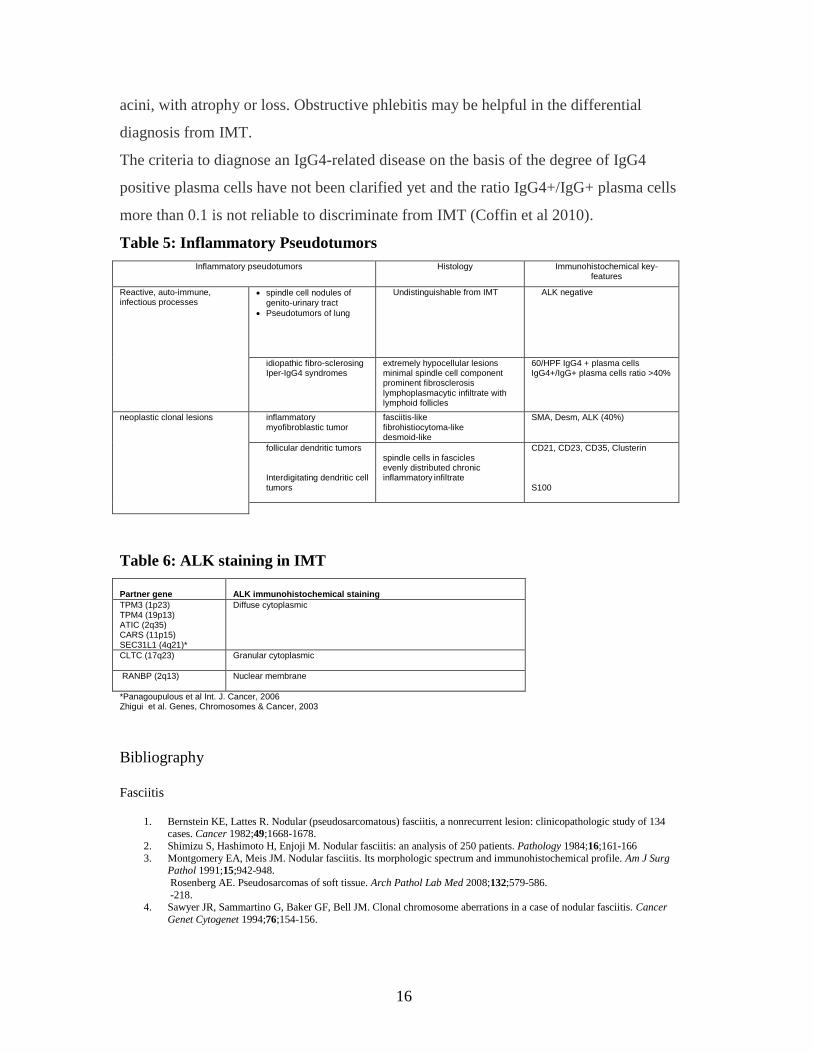
Table 5: Inflammatory Pseudotumors
Inflammatory pseudotumors Histology Immunohistochemical key-features
Reactive, auto-immune, infectious processes
spindle cell nodules of genito-urinary tract
Pseudotumors of lung
Undistinguishable from IMT ALK negative
idiopathic fibro-sclerosing Iper-IgG4 syndromes
extremely hypocellular lesions minimal spindle cell component prominent fibrosclerosis lymphoplasmacytic infiltrate with lymphoid follicles
60/HPF IgG4 + plasma cells IgG4+/IgG+ plasma cells ratio >40%
neoplastic clonal lesions inflammatory myofibroblastic tumor
fasciitis-like fibrohistiocytoma-like desmoid-like
SMA, Desm, ALK (40%)
follicular dendritic tumors Interdigitating dendritic cell tumors
spindle cells in fascicles evenly distributed chronic inflammatory infiltrate
CD21, CD23, CD35, Clusterin
S100
Table 6: ALK staining in IMT
Partner gene
ALK immunohistochemical staining
TPM3 (1p23) TPM4 (19p13) ATIC (2q35) CARS (11p15) SEC31L1 (4q21)*
Diffuse cytoplasmic
CLTC (17q23)
Granular cytoplasmic
RANBP (2q13) Nuclear membrane
*Panagoupulous et al Int. J. Cancer, 2006 Zhigui et al. Genes, Chromosomes & Cancer, 2003
Bibliography
Fasciitis
1. Bernstein KE, Lattes R. Nodular (pseudosarcomatous) fasciitis, a nonrecurrent lesion: clinicopathologic study of 134 cases. Cancer 1982;49;1668-1678.
2. Shimizu S, Hashimoto H, Enjoji M. Nodular fasciitis: an analysis of 250 patients. Pathology 1984;16;161-166
3. Montgomery EA, Meis JM. Nodular fasciitis. Its morphologic spectrum and immunohistochemical profile. Am J Surg Pathol 1991;15;942-948.
Rosenberg AE. Pseudosarcomas of soft tissue. Arch Pathol Lab Med 2008;132;579-586.
-218. 4. Sawyer JR, Sammartino G, Baker GF, Bell JM. Clonal chromosome aberrations in a case of nodular fasciitis. Cancer
Genet Cytogenet 1994;76;154-156.

17
5. Meng GZ, Zhang HY, Zhang Z, Wei B, Bu H. Myofibroblastic sarcoma vs nodular fasciitis: a comparative study of
chromosomal imbalances. Am J Clin Pathol 2009;131;701-709.
Fibromatoses 6. Mackenzie DH. The fibromatoses: a clinicopathological concept. Br Med J 1972;4;277-281.
7. Allen PW. The fibromatoses: a clinicopathologic classification based on 140 cases. Am J Surg Pathol 1977;1;255-270. 8. Stout AP. Juvenile fibromatoses. Cancer 1954;7;953-978.
9. Chung EB, Enzinger FM. Infantile myofibromatosis. Cancer 1981;48;1807-1818.
10. Coffin CM, Neilson KA, Ingels S, Frank-Gerszberg R, Dehner LP. Congenital generalized myofibromatosis: a disseminated angiocentric myofibromatosis. Pediatr Pathol Lab Med 1995;15;571-587.
11. Mentzel T, Calonje E, Nascimento AG, Fletcher CD. Infantile hemangiopericytoma versus infantile myofibromatosis.
Study of a series suggesting a continuous spectrum of infantile myofibroblastic lesions. Am J Surg Pathol 1994;18;922-930.
12. Stenman G, Nadal N, Persson S, Gunterberg B, Angervall L. del(6)(q12q15) as the sole cytogenetic anomaly in a case of
solitary infantile myofibromatosis. Oncol Rep 1999;6;1101-1104. 13. Arcangeli F, Calista D. Congenital myofibromatosis in two siblings. Eur J Dermatol 2006;16;181-183.
14. Zand DJ, Huff D, Everman D et al. Autosomal dominant inheritance of infantile myofibromatosis. Am J Med Genet A
2004;126A;261-266.
15. Enzinger FM. Fibrous Hamartoma of Infancy. Cancer 1965;18;241-248.
16. Dickey GE, Sotelo-Avila C. Fibrous hamartoma of infancy: current review. Pediatr Dev Pathol 1999;2;236-243.
17. Paller AS, Gonzalez-Crussi F, Sherman JO. Fibrous hamartoma of infancy. Eight additional cases and a review of the literature. Arch Dermatol 1989;125;88-91.
18. Sotelo-Avila C, Bale PM. Subdermal fibrous hamartoma of infancy: pathology of 40 cases and differential diagnosis.
Pediatr Pathol 1994;14;39-52. 19. Fletcher CD, Powell G, van Noorden S, McKee PH. Fibrous hamartoma of infancy: a histochemical and
immunohistochemical study. Histopathology 1988;12;65-74. 20. Carretto E, Dall'Igna P, Alaggio R et al. Fibrous hamartoma of infancy: an Italian multi-institutional experience. J Am
Acad Dermatol 2006;54;800-803.
21. Popek EJ, Montgomery EA, Fourcroy JL. Fibrous hamartoma of infancy in the genital region: findings in 15 cases. J Urol 1994;152;990-993.
22. Lakshminarayanan R, Konia T, Welborn J. Fibrous hamartoma of infancy: a case report with associated cytogenetic
findings. Arch Pathol Lab Med 2005;129;520-522. 23. Rougemont AL, Fetni R, Murthy S, Fournet JC. A complex translocation (6;12;8)(q25;q24.3;q13) in a fibrous hamartoma
of infancy. Cancer Genet Cytogenet 2006;171;115-118.
24. Fetsch JF, Miettinen M, Laskin WB, Michal M, Enzinger FM. A clinicopathologic study of 45 pediatric soft tissue tumors with an admixture of adipose tissue and fibroblastic elements, and a proposal for classification as lipofibromatosis. Am J
Surg Pathol 2000;24;1491-1500.
25. Herrmann BW, Dehner LP, Forsen JW, Jr. Lipofibromatosis presenting as a pediatric neck mass. Int J Pediatr Otorhinolaryngol 2004;68;1545-1549.
26. Alman BA, Pajerski ME, Diaz-Cano S, Corboy K, Wolfe HJ. Aggressive fibromatosis (desmoid tumor) is a monoclonal
disorder. Diagn Mol Pathol 1997;6;98-101. 27. Reitamo JJ, Scheinin TM, Hayry P. The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the
desmoid tumor. Am J Surg 1986;151;230-237.
28. Ayala AG, Ro JY, Goepfert H, Cangir A, Khorsand J, Flake G. Desmoid fibromatosis: a clinicopathologic study of 25 children. Semin Diagn Pathol 1986;3;138-150.
29. Coffin CM, Dehner LP. Fibroblastic-myofibroblastic tumors in children and adolescents: a clinicopathologic study of 108
examples in 103 patients. Pediatr Pathol 1991;11;569-588. 30. Gebert C, Hardes J, Kersting C et al. Expression of beta-catenin and p53 are prognostic factors in deep aggressive
fibromatosis. Histopathology 2007;50;491-497.
31. Knudsen AL, Bulow S. Desmoid tumour in familial adenomatous polyposis. A review of literature. Fam Cancer 2001;1;111-119.
32. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 2006;101;385-398.
Meazza C, Bisogno G, Gronchi A et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 2010;116;233-240.
33. Lazar AJ, Tuvin D, Hajibashi S et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local
recurrence in sporadic desmoid tumors. Am J Pathol 2008;173;1518-1527. 34. Lefevre JH, Parc Y, Kerneis S et al. Risk factors for development of desmoid tumours in familial adenomatous polyposis.
Br J Surg 2008;95;1136-1139.
35. Lips DJ, Barker N, Clevers H, Hennipman A. The role of APC and beta-catenin in the aetiology of aggressive fibromatosis (desmoid tumors). Eur J Surg Oncol 2009;35;3-10.
Lazar AJ, Hajibashi S, Lev D. Desmoid tumor: from surgical extirpation to molecular dissection. Curr Opin Oncol
2009;21;352-359. 36. Bhattacharya B, Dilworth HP, Iacobuzio-Donahue C et al. Nuclear beta-catenin expression distinguishes deep
fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol 2005;29;653-
659. 37. Thway K, Gibson S, Ramsay A, Sebire NJ. Beta-catenin expression in pediatric fibroblastic and myofibroblastic lesions:
a study of 100 cases. Pediatr Dev Pathol 2009;12;292-296.
38. Sharma A, Ngan BY, Sandor GK, Campisi P, Forte V. Pediatric aggressive fibromatosis of the head and neck: a 20-year retrospective review. J Pediatr Surg 2008;43;1596-1604.

18
39. Deyrup AT, Tretiakova M, Montag AG. Estrogen receptor-beta expression in extraabdominal fibromatoses: an analysis of
40 cases. Cancer 2006;106;208-213. 40. Hornick JL, Fletcher CD. Immunohistochemical staining for KIT (CD117) in soft tissue sarcomas is very limited in
distribution. Am J Clin Pathol 2002;117;188-193.
41. Matono H, Oda Y, Nakamori M et al. Correlation between beta-catenin widespread nuclear expression and matrix metalloproteinase-7 overexpression in sporadic desmoid tumors. Hum Pathol 2008;39;1802-1808.
42. Francis WP, Zippel D, Mack LA et al. Desmoids: a revelation in biology and treatment. Ann Surg Oncol 2009;16;1650-
1654. 43. Stoeckle E, Coindre JM, Longy M et al. A critical analysis of treatment strategies in desmoid tumours: a review of a
series of 106 cases. Eur J Surg Oncol 2009;35;129-134.
Inflammatory Myofibroblastic Tumor and Inflammatory Pseudotumors 44. Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol 2008;61;428-437.
45. Pettinato G, Manivel JC, Insabato L, De Chiara A, Petrella G. Plasma cell granuloma (inflammatory pseudotumor) of the
breast. Am J Clin Pathol 1988;90;627-632. 46. Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory
pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995;19;859-872.
47. Coffin CM, Dehner LP, Meis-Kindblom JM. Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: an historical review with differential diagnostic considerations. Semin Diagn Pathol 1998;15;102-110.
48. Coffin CM, Humphrey PA, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor: a clinical and pathological
survey. Semin Diagn Pathol 1998;15;85-101. 49. Meis-Kindblom JM, Kjellstrom C, Kindblom LG. Inflammatory fibrosarcoma: update, reappraisal, and perspective on its
place in the spectrum of inflammatory myofibroblastic tumors. Semin Diagn Pathol 1998;15;133-143.
50. Chen ST, Lee JC. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: combination of distinct morphologic, immunohistochemical, and genetic features. Hum Pathol 2008;39;1854-1858.
51. Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic,
and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 2007;31;509-520.
52. Alaggio R, Cecchetto G, Bisogno G et al. Inflammatory myofibroblastic tumors in childhood: a report from the Italian
Cooperative Group studies. Cancer 2010;116;216-226. 53. Cook JR, Dehner LP, Collins MH et al. Anaplastic lymphoma kinase (ALK) expression in the inflammatory
myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol 2001;25;1364-1371.
Tsuzuki T, Magi-Galluzzi C, Epstein JI. ALK-1 expression in inflammatory myofibroblastic tumor of the urinary bladder. Am J Surg Pathol 2004;28;1609-1614.
54. Bridge JA, Kanamori M, Ma Z et al. Fusion of the ALK gene to the clathrin heavy chain gene, CLTC, in inflammatory
myofibroblastic tumor. Am J Pathol 2001;159;411-415. 55. Sirvent N, Hawkins AL, Moeglin D et al. ALK probe rearrangement in a t(2;11;2)(p23;p15;q31) translocation found in a
prenatal myofibroblastic fibrous lesion: toward a molecular definition of an inflammatory myofibroblastic tumor family?
Genes Chromosomes Cancer 2001;31;85-90. 56. Debelenko LV, Arthur DC, Pack SD, Helman LJ, Schrump DS, Tsokos M. Identification of CARS-ALK fusion in
primary and metastatic lesions of an inflammatory myofibroblastic tumor. Lab Invest 2003;83;1255-1265.
57. Ma Z, Hill DA, Collins MH et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2003;37;98-105.
58. Chun YS, Wang L, Nascimento AG, Moir CR, Rodeberg DA. Pediatric inflammatory myofibroblastic tumor: anaplastic
lymphoma kinase (ALK) expression and prognosis. Pediatr Blood Cancer 2005;45;796-801. 59. Cessna MH, Zhou H, Sanger WG et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its
mesenchymal mimics: a study of 135 cases. Mod Pathol 2002;15;931-938.
60. Soriano AO, Thompson MA, Admirand JH et al. Follicular dendritic cell sarcoma: a report of 14 cases and a review of the literature. Am J Hematol 2007;82;725-728.
61. Pileri SA, Grogan TM, Harris NL et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical
approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002;41;1-29.
62. Hussong JW, Brown M, Perkins SL, Dehner LP, Coffin CM. Comparison of DNA ploidy, histologic, and immunohistochemical findings with clinical outcome in inflammatory myofibroblastic tumors. Mod Pathol 1999;12;279-
286.
63. Yamamoto H, Oda Y, Saito T et al. p53 Mutation and MDM2 amplification in inflammatory myofibroblastic tumours.
Histopathology 2003;42;431-439.
64. Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and
chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol 2001;14;569-576.
Autoimmune Pancreatitis and other sclerosing diseases hyper-IgG4 related 65. Hamano H, Kawa S, Horiuchi A et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J
Med 2001;344;732-738.
66. Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med 2006;4;23.
67. Kamisawa T, Funata N, Hayashi Y et al. A new clinicopathological entity of IgG4-related autoimmune disease. J
Gastroenterol 2003;38;982-984. 68. Zen Y, Kitagawa S, Minato H et al. IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of
the lung. Hum Pathol 2005;36;710-717.
69. Miller OF, Smith LJ, Ferrara EX, McAleer IM, Kaplan GW. Presentation of idiopathic retroperitoneal fibrosis in the pediatric population. J Pediatr Surg 2003;38;1685-1688.

19
70. Blejter J, Weller S, Pace R, Cusumano H, Giambini D. Autoimmune pancreatitis: an adolescent case and review of
literature. J Pediatr Surg 2008;43;1368-1372. 71. Zamboni G, Luttges J, Capelli P et al. Histopathological features of diagnostic and clinical relevance in autoimmune
pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004;445;552-563.
Kloppel G, Sipos B, Zamboni G, Kojima M, Morohoshi T. Autoimmune pancreatitis: histo- and immunopathological features. J Gastroenterol 2007;42 Suppl 18;28-31.
72. Zhang L, Notohara K, Levy MJ, Chari ST, Smyrk TC. IgG4-positive plasma cell infiltration in the diagnosis of
autoimmune pancreatitis. Mod Pathol 2007;20;23-28. 73. Dhall D, Suriawinata AA, Tang LH, Shia J, Klimstra DS. Use of immunohistochemistry for IgG4 in the distinction of
autoimmune pancreatitis from peritumoral pancreatitis. Hum Pathol 2010.
74. Yamamoto H, Yamaguchi H, Aishima S et al. Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol 2009;33;1330-1340.


















