
Bax and Bak Are Required for Apoptosis Induction by ... · intake of cruciferous vegetables may be...
10
Bax and Bak Are Required for Apoptosis Induction by Sulforaphane, a Cruciferous Vegetable–Derived Cancer Chemopreventive Agent Sunga Choi and Shivendra V. Singh Department of Pharmacology and University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania Abstract Sulforaphane, a constituent of many edible cruciferous vegetables, including broccoli, effectively suppresses prolifer- ation of cancer cells in culture and in vivo by causing apoptosis induction, but the sequence of events leading to cell death is poorly defined. Here, we show that multidomain proapoptotic Bcl-2 family members Bax and Bak play a critical role in apoptosis induction by sulforaphane. This conclusion is based on the following observations: (a ) sulforaphane treatment caused a dose- and time-dependent increase in the protein levels of both Bax and Bak and conformational change and mitochondrial translocation of Bax in SV40-transformed mouse embryonic fibroblasts (MEF) derived from wild-type mice to trigger cytosolic release of apoptogenic molecules (cytochrome c and Smac/DIABLO), activation of caspase-9 and caspase-3, and ultimately cell death; (b) MEFs derived from Bax or Bak knockout mice resisted cell death by sulforaphane, and (c ) MEFs derived from Bax and Bak double knockout mice exhibited even greater protection against sulforaphane-induced cytochrome c release, caspase activation, and apoptosis compared with wild-type or single knockout cells. Interestingly, sulforaphane treatment also caused a dose- and time-dependent increase in the protein level of Apaf-1 in wild-type, Bax / , and Bak / MEFs but not in double knockout, suggesting that Bax and Bak might regulate sulforaphane-mediated induction of Apaf- 1 protein. A marked decline in the protein level of X-linked inhibitor of apoptosis on treatment with sulforaphane was also observed. Thus, it is reasonable to postulate that sulforaphane-induced apoptosis is amplified by a decrease in X-linked inhibitor of apoptosis level, which functions to block cell death by inhibiting activities of caspases. In conclusion, the results of the present study indicate that Bax and Bak proteins play a critical role in initiation of cell death by sulforaphane. (Cancer Res 2005; 65(5): 2035-43) Introduction Epidemiologic data continue to support the premise that dietary intake of cruciferous vegetables may be protective against the risk of various types of cancers (1–4). Laboratory studies suggest that cancer-protective effect of cruciferous vegetables may be due to isothiocyanates that occur as thioglucoside conjugate (glucosino- lates) in a variety of edible plants, including broccoli, cabbage, watercress, etc. (5–8). Isothiocyanates are generated due to hydrolysis of corresponding glucosinolates through catalytic mediation of myrosinase, which is released on damage of the plant cells during processing (e.g., cutting or chewing) of cruciferous vegetables (7). Isothiocyanates, including phenethyl- isothiocyanate and benzyl-isothiocyanate, have been shown to offer significant protection against cancer in animal models induced by a variety of chemical carcinogens (5, 6, 8–13). Sulforaphane [1-isothiocyanato-4-(methylsulfinyl)-butane; CH 3 -SO-(CH 2 ) 4 -N=C=S] is a naturally occurring member of the isothiocyanate family of cancer chemopreventive agents that has attracted particular attention due to its potent anticancer effects (14–19). For example, sulforaphane was shown to offer statistically significant protection against 9,10-dimethyl-1,2-benzanthracene- induced mammary tumorigenesis in rats (15). Interestingly, sulforaphane exhibited bactericidal activity against clinical isolates as well as antibiotic-resistant strains of Helicobacter pylori and inhibited benzo[a ]pyrene-induced forestomach cancer in mice (18). Eradication of H. pylori in human gastric xenografts implanted in nude mice on sulforaphane administration was also documented (19). Sulforaphane as well as its N -acetylcysteine conjugate given during the postinitiation period significantly inhibited azoxyme- thane-induced colonic aberrant crypt foci formation in rats (17). The mechanism by which sulforaphane inhibits chemically induced cancers is believed to involve impairment of carcinogen metabolism due to inhibition of cytochrome P450–dependent monooxygenases and/or induction of phase II detoxification enzymes, such as glutathione transferase (reviewed in refs. 5, 6, 8). More recent studies, including those from our laboratory, have indicated that sulforaphane can suppress proliferation of cancer cells in culture and in vivo by inhibiting cell cycle progression and/ or causing apoptosis induction (20–29). Growth inhibition, cell cycle arrest, and/or apoptosis induction by sulforaphane has been documented in human colon, leukemia, medulloblastoma, and prostate cancer cells (20–29). Gamet-Payrastre et al. (20) were the first to show that treatment of HT29 human colon cancer cells with sulforaphane resulted in G 2 -M-phase cell cycle arrest and apoptosis induction. Although the mechanism of cell cycle block was not thoroughly examined by these investigators, the sulforaphane- induced apoptosis in HT29 cells was associated with induction of Bax protein expression and cytosolic release of cytochrome c (20). Recent studies from our laboratory have offered novel insights into the mechanism of cell cycle block by sulforaphane (29). Using PC-3 human prostate cancer cells as a model, we showed that sulforaphane-induced G 2 -M-phase cell cycle arrest was associated with rapid and sustained activation of checkpoint kinase 2, which promoted Ser 216 phosphorylation of cell division cycle 25C leading to its translocation from nucleus to the cytosol (29). The net result of these effects was accumulation of Tyr 15 -phosphorylated (inactive) cyclin-dependent kinase 1 (29), which together with the B-type cyclins plays an important role in regulation of G 2 -M progression (30). Although significant progress has been made toward our understanding of the signal transduction pathways Requests for reprints: Shivendra V. Singh, University of Pittsburgh, Hillman Cancer Center Research Pavilion, Suite 2.32A, 5117 Centre Avenue, Pittsburgh, PA 15213. Phone: 412-623-3263; Fax: 412-623-7828; E-mail: [email protected]. I2005 American Association for Cancer Research. www.aacrjournals.org 2035 Cancer Res 2005; 65: (5). March 1, 2005 Research Article Research. on May 22, 2020. © 2005 American Association for Cancer cancerres.aacrjournals.org Downloaded from
Transcript of Bax and Bak Are Required for Apoptosis Induction by ... · intake of cruciferous vegetables may be...

Bax and Bak Are Required for Apoptosis Induction by Sulforaphane,
a Cruciferous Vegetable–Derived Cancer Chemopreventive Agent
Sunga Choi and Shivendra V. Singh
Department of Pharmacology and University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine,Pittsburgh, Pennsylvania
Abstract
Sulforaphane, a constituent of many edible cruciferousvegetables, including broccoli, effectively suppresses prolifer-ation of cancer cells in culture and in vivo by causingapoptosis induction, but the sequence of events leading to celldeath is poorly defined. Here, we show that multidomainproapoptotic Bcl-2 family members Bax and Bak play acritical role in apoptosis induction by sulforaphane. Thisconclusion is based on the following observations: (a)sulforaphane treatment caused a dose- and time-dependentincrease in the protein levels of both Bax and Bak andconformational change and mitochondrial translocation ofBax in SV40-transformed mouse embryonic fibroblasts (MEF)derived from wild-type mice to trigger cytosolic release ofapoptogenic molecules (cytochrome c and Smac/DIABLO),activation of caspase-9 and caspase-3, and ultimately celldeath; (b) MEFs derived from Bax or Bak knockout miceresisted cell death by sulforaphane, and (c) MEFs derivedfrom Bax and Bak double knockout mice exhibited evengreater protection against sulforaphane-induced cytochromec release, caspase activation, and apoptosis compared withwild-type or single knockout cells. Interestingly, sulforaphanetreatment also caused a dose- and time-dependent increase inthe protein level of Apaf-1 in wild-type, Bax��/����/����/��, and Bak��/����/����/��
MEFs but not in double knockout, suggesting that Bax andBak might regulate sulforaphane-mediated induction of Apaf-1 protein. A marked decline in the protein level of X-linkedinhibitor of apoptosis on treatment with sulforaphane wasalso observed. Thus, it is reasonable to postulate thatsulforaphane-induced apoptosis is amplified by a decreasein X-linked inhibitor of apoptosis level, which functions toblock cell death by inhibiting activities of caspases. Inconclusion, the results of the present study indicate thatBax and Bak proteins play a critical role in initiation of celldeath by sulforaphane. (Cancer Res 2005; 65(5): 2035-43)
Introduction
Epidemiologic data continue to support the premise that dietaryintake of cruciferous vegetables may be protective against the riskof various types of cancers (1–4). Laboratory studies suggest thatcancer-protective effect of cruciferous vegetables may be due toisothiocyanates that occur as thioglucoside conjugate (glucosino-lates) in a variety of edible plants, including broccoli, cabbage,watercress, etc. (5–8). Isothiocyanates are generated due tohydrolysis of corresponding glucosinolates through catalytic
mediation of myrosinase, which is released on damage of theplant cells during processing (e.g., cutting or chewing) ofcruciferous vegetables (7). Isothiocyanates, including phenethyl-isothiocyanate and benzyl-isothiocyanate, have been shown to offersignificant protection against cancer in animal models induced bya variety of chemical carcinogens (5, 6, 8–13).Sulforaphane [1-isothiocyanato-4-(methylsulfinyl)-butane;
CH3-SO-(CH2)4-N=C=S] is a naturally occurring member of theisothiocyanate family of cancer chemopreventive agents that hasattracted particular attention due to its potent anticancer effects(14–19). For example, sulforaphane was shown to offer statisticallysignificant protection against 9,10-dimethyl-1,2-benzanthracene-induced mammary tumorigenesis in rats (15). Interestingly,sulforaphane exhibited bactericidal activity against clinical isolatesas well as antibiotic-resistant strains of Helicobacter pylori andinhibited benzo[a]pyrene-induced forestomach cancer in mice(18). Eradication of H. pylori in human gastric xenografts implantedin nude mice on sulforaphane administration was also documented(19). Sulforaphane as well as its N-acetylcysteine conjugate givenduring the postinitiation period significantly inhibited azoxyme-thane-induced colonic aberrant crypt foci formation in rats (17). Themechanism by which sulforaphane inhibits chemically inducedcancers is believed to involve impairment of carcinogen metabolismdue to inhibition of cytochrome P450–dependent monooxygenasesand/or induction of phase II detoxification enzymes, such asglutathione transferase (reviewed in refs. 5, 6, 8).More recent studies, including those from our laboratory, have
indicated that sulforaphane can suppress proliferation of cancercells in culture and in vivo by inhibiting cell cycle progression and/or causing apoptosis induction (20–29). Growth inhibition, cellcycle arrest, and/or apoptosis induction by sulforaphane has beendocumented in human colon, leukemia, medulloblastoma, andprostate cancer cells (20–29). Gamet-Payrastre et al. (20) were thefirst to show that treatment of HT29 human colon cancer cells withsulforaphane resulted in G2-M-phase cell cycle arrest and apoptosisinduction. Although the mechanism of cell cycle block was notthoroughly examined by these investigators, the sulforaphane-induced apoptosis in HT29 cells was associated with induction ofBax protein expression and cytosolic release of cytochrome c (20).Recent studies from our laboratory have offered novel insights intothe mechanism of cell cycle block by sulforaphane (29). Using PC-3human prostate cancer cells as a model, we showed thatsulforaphane-induced G2-M-phase cell cycle arrest was associatedwith rapid and sustained activation of checkpoint kinase 2, whichpromoted Ser216 phosphorylation of cell division cycle 25C leadingto its translocation from nucleus to the cytosol (29). The net resultof these effects was accumulation of Tyr15-phosphorylated(inactive) cyclin-dependent kinase 1 (29), which together with theB-type cyclins plays an important role in regulation of G2-Mprogression (30). Although significant progress has been madetoward our understanding of the signal transduction pathways
Requests for reprints: Shivendra V. Singh, University of Pittsburgh, HillmanCancer Center Research Pavilion, Suite 2.32A, 5117 Centre Avenue, Pittsburgh, PA15213. Phone: 412-623-3263; Fax: 412-623-7828; E-mail: [email protected].
I2005 American Association for Cancer Research.
www.aacrjournals.org 2035 Cancer Res 2005; 65: (5). March 1, 2005
Research Article
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

responsible for inhibition of cell cycle progression (20, 24–27, 29),the mechanism of sulforaphane-induced apoptosis is poorlycharacterized. For example, apoptosis induction by sulforaphanein different cellular systems is associated with induction of Baxprotein expression (20, 21, 25, 28), yet studies that couldexperimentally test the role of this protein in cell death are lacking.In the present study, we used SV40-transformed mouse
embryonic fibroblasts (MEFs) derived from wild-type and Baxand/or Bak knockout mice (31) to gain insights into the role ofthese multidomain proapoptotic Bcl-2 family members in celldeath caused by sulforaphane. The present study indicates thatboth Bax and Bak are essential for apoptosis induction bysulforaphane and that sulforaphane treatment causes activationand mitochondrial translocation of Bax to trigger cytosolic releaseof apoptogenic molecules.
Materials and Methods
Reagents. Sulforaphane (>99% pure) was purchased from LKT
Laboratories (St. Paul, MN). Tissue culture medium, penicillin-streptomycinantibiotic mixture, and fetal bovine serum were from Life Technologies
(Grand Island, NY). Propidium iodide and 4V,6-diamidino-2-phenylindole
(DAPI) were from Sigma (St. Louis, MO), RNase A was from Promega
(Madison, WI), and the kit for quantitation of cytoplasmic histone-associated DNA fragmentation was from Roche Diagnostics (Mannheim,
Germany). Antibodies against Bax (clone N-20), Bak (clone G-23), Smac/
DIABLO (clone V-17), and Apaf-1 (clone H-324) were from Santa Cruz
Biotechnology (Santa Cruz, CA). Antibodies against caspase-3, caspase-9,and poly(ADP-ribose)polymerase (PARP) were from Cell Signaling Tech-
nology (Beverly, MA). Antibodies against cytochrome c and X-linked
inhibitor of apoptosis (XIAP) were from BD PharMingen (San Diego, CA).Anti-actin antibody was from Oncogene Research Products (Boston, MA).
Anti-Bax monoclonal antibody 6A7 that recognizes an epitope on the NH2
terminus (between amino acids 12 and 24) of conformationally changed
(activated) Bax protein was purchased from BD PharMingen.Cell Culture and Cell Survival Assay. Primary MEFs derived from wild-
type, Bax knockout (Bax�/�), Bak knockout (Bak�/�), and Bax-Bak double
knockout (DKO) mice and immortalized by transfection with a plasmid
containing SV40 genomic DNA were generously provided by Dr. Stanley J.Korsmeyer (Dana-Farber Cancer Institute, Boston, MA; ref. 31). MEFs were
maintained in DMEM supplemented with 10% (v/v) heat-inactivated fetal
bovine serum, 0.1 mmol/L nonessential amino acids, 0.1 Amol/L2-mercaptoethanol, and antibiotics. Normal human bronchial epithelial
cell line BEAS2B and normal prostate epithelial cell line PrEC were cultured
in LHC-9 medium (Biosource, Camarillo, CA) and PrEBM medium
(Cambrex, Rockland, ME), respectively. Human lung cancer cell lineH1299 was maintained in DMEM containing 10% FCS and antibiotics.
Human prostate cancer cell line LNCaP was cultured in RPMI 1640
supplemented with 10% FCS, 10 mmol/L HEPES, 1 mmol/L sodium
pyruvate, 0.2% glucose, and antibiotics. Each line was maintained in ahumidified atmosphere of 95% air and 5% CO2 at 37jC. The effect of
sulforaphane treatment on cell viability was determined by trypan blue dye
exclusion assay. Briefly, the desired MEF line (5 � 103) was plated in six-wellplates and allowed to attach overnight. The medium was replaced with
fresh complete medium containing different concentrations of sulfora-
phane, and incubation was continued for 24 hours at 37jC. Stock solution
of sulforaphane was prepared in DMSO, and an equal volume of DMSO( final concentration 0.1%) was added to controls. At the end of the
incubation, both floating and adherent cells were collected and suspended
in PBS. The cells were then mixed with 0.4% trypan blue solution, and live
(unstained) and dead (stained) cells were counted under an invertedmicroscope.
Apoptosis Assays. Apoptosis induction in sulforaphane-treated MEFs,
normal epithelial cells (BEAS2B or PrEC), and cancer cells (H1299 or
LNCaP) was assessed by analysis of cytoplasmic histone-associated DNA
fragmentation using a kit from Roche Diagnostics according to themanufacturer’s instructions or flow cytometric analysis of cells with
sub-G0/G1 DNA content following staining with propidium iodide as
described by us previously (28, 29) or microscopic analysis of apoptotic
cells with condensed nuclei following staining with DAPI as describedpreviously (32).
Immunoblotting. MEFs were treated with the desired concentrations
of sulforaphane or DMSO (control) as described above and lysed as
reported by us previously (28). The cell lysate was cleared bycentrifugation at 14,000 � g for 15 minutes. Supernatant proteins were
resolved by 12.5% SDS-PAGE and transferred onto polyvinylidene
difluoride membrane. After blocking with 5% nonfat dry milk in TBS
containing 0.05% Tween 20, the membrane was incubated with thedesired primary antibody for 1 hour at room temperature. The
membrane was then treated with appropriate secondary antibody, and
the immunoreactive bands were visualized using enhanced chemilumi-nescence method. Each membrane was stripped and reprobed with anti-
actin antibody to normalize for differences in protein loading. Change in
protein level was assessed by densitometric scanning of the immuno-
reactive bands followed by correction for actin loading control.Immunohistochemistry for Localization of Cytochrome c . MEFs
were cultured on coverslips and treated with 30 Amol/L sulforaphane or
DMSO (control) for 8 hours. The MEFs were then washed with PBS and
stained for 1 hour at 37jC with 100 nmol/L mitochondria-specific dyeMitoTracker Red (Molecular Probes, Eugene, OR). After washing with
PBS, MEFs were fixed with 4% paraformaldehyde and permeabilized with
0.1% Triton X-100. The MEFs were incubated with normal goat serum(1:20 dilution, Sigma) in PBS for 45 minutes. Subsequently, the MEFs
were treated with anti–cytochrome c antibody (1:200 dilution) for 2 hours,
washed with PBS, and incubated with Alexa Fluor 488–conjugated
secondary antibody (1:1,000 dilution, Molecular Probes) for 1 hour. Afterwashing, cells were treated with DAPI (1 Ag/mL) for 5 minutes to stain
DNA. The cells were visualized under a Leica DC300F fluorescence
microscope.
Analysis of Bax Conformation Change. Wild-type MEFs were treatedwith 40 Amol/L sulforaphane for 4, 12, or 24 hours and lysed using a
solution containing 10 mmol/L HEPES (pH 7.4), 150 mmol/L NaCl,
1% CHAPS, and protease inhibitor cocktail. Aliquots containing 1 mglysate protein in 0.5 mL lysis buffer were incubated overnight at 4jCwith 2 Ag anti-Bax monoclonal antibody 6A7. Protein G-agarose beads
(40 AL, Santa Cruz Biotechnology) were then added to each sample, and
the incubation was continued for 2 hours at 4jC. The immunoprecipi-tated complexes were washed thrice with lysis buffer and subjected to
SDS-PAGE followed by immunoblotting using polyclonal anti-Bax
antibody.
Immunohistochemistry for Localization of Bax. Wild-type MEFs werecultured on coverslips and treated with sulforaphane (40 Amol/L) or DMSO
for 12 hours. After staining with MitoTracker Red, MEFs were incubated
with anti-Bax antibody (1:1,000 dilution) for 2 hours followed by incubation
with Alexa Fluor 488–conjugated secondary antibody (1:1,000 dilution) for 1hour. MEFs were washed with PBS, stained with DAPI, and examined under
a fluorescence microscope.
Statistical Analysis. One-way ANOVA was used to test the significanceof differences in measured variables between control and treated groups
followed by Bonferroni’s test for multiple comparisons. Statistical
significance was determined at the 0.05 level.
Results
Sulforaphane-Induced Apoptosis in Wild-type MEFs WasAssociated with Induction of Bax and Bak Proteins. Previousstudies have shown that apoptosis induction by sulforaphane indifferent cellular systems is associated with induction of Baxprotein expression (20, 21, 25, 28), but the role of this protein in celldeath has not been experimentally verified. In the present study, weused MEFs derived from wild-type, Bax knockout (Bax�/�), Bak
Cancer Research
Cancer Res 2005; 65: (5). March 1, 2005 2036 www.aacrjournals.org
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

knockout (Bak�/�), and Bax and Bak DKO mice immortalized bytransfection with a plasmid containing SV40 genomic DNA (31) toinvestigate the role of these proteins in apoptosis induction bysulforaphane. First, we determined the sensitivity of wild-typeMEFs to cell killing and apoptosis induction by sulforaphane toassess suitability of these cells for our work. The effect ofsulforaphane treatment on viability of wild-type MEFs was
determined by trypan blue dye exclusion assay (Fig. 1A). Similarto other cellular systems, the viability of wild-type MEFs wasreduced significantly in the presence of sulforaphane in aconcentration-dependent manner (Fig. 1A). Next, we addressedthe question whether reduced viability of wild-type MEFs in thepresence of sulforaphane was due to apoptosis induction.Apoptosis was assessed by analysis of cytoplasmic histone-associated DNA fragmentation, which has emerged as a sensitivemethod for detection of apoptotic cell death. As can be seen inFig. 1B , a 24-hour treatment of wild-type MEFs with sulfor-aphane resulted in a concentration-dependent and statisticallysignificant increase in cytoplasmic histone-associated DNAfragmentation. For instance, the DNA fragmentation in wild-type MEFs treated with 20 and 40 Amol/L sulforaphane wasincreased by f6- and 8-fold, respectively, compared with control(Fig. 1B). These results indicated that reduced viability of wild-type MEFs in the presence of sulforaphane was indeed due toapoptosis induction.We raised the question whether sulforaphane-induced cell death
in wild-type MEFs was associated with induction of Bax and/or Bakprotein expression. We explored this possibility by determining theeffect of sulforaphane treatment on levels of Bax and Bak proteinsby immunoblotting, and representative immunoblots are shown inFig. 1C . Treatment of wild-type MEFs with sulforaphane resulted ina concentration- and time-dependent increase in the protein levelsof both Bax and Bak. For example, a 24-hour treatment of wild-typeMEFs with 20, 30, and 40 Amol/L sulforaphane caused an f42%,80%, and 155% increase in Bax protein level, respectively, whencompared with control (Fig. 1C). In time course experiments using40 Amol/L sulforaphane, the induction of Bax and Bak proteins wasevident as early as 4 to 8 hours after treatment and increasedgradually with increasing exposure time. Collectively, these resultsindicated that sulforaphane-mediated apoptosis in wild-type MEFswas associated with induction of Bax and Bak proteins.Next, we raised the question whether sulforaphane-induced
apoptosis is selective for cancer cells, which is a highly desirablefeature of potential chemopreventive agents. We addressed thisquestion by determining the effect of sulforaphane treatment oncytoplasmic histone-associated DNA fragmentation in BEAS2Bnormal bronchial epithelial cell line, PrEC normal prostate
epithelial cell line, H1299 human lung cancer cells, and LNCaPhuman prostate cancer cells. As can be seen in Fig. 1D and E , bothcancer cells (H1299 and LNCaP) were significantly more sensitiveto sulforaphane-induced cytoplasmic histone-associated DNAfragmentation compared with BEAS2B and PrEC normal epithelialcells. These results indicated that normal epithelial cells were
significantly more resistant to apoptosis induction by sulforaphanecompared with cancer cells.Bax and Bak DKO MEFs Were Resistant to Cell Death by
Sulforaphane. We compared sensitivities of Bax and/or Bakknockout MEFs toward sulforaphane-induced apoptosis todetermine the contribution of these proteins in cell deathcaused by this agent. The wild-type MEFs were included in theanalysis for direct comparison. Apoptosis induction bysulforaphane was assessed by flow cytometric analysis ofsubdiploid cells, which is a characteristic feature of cellsundergoing apoptosis. As shown in Fig. 2A , a 24-hour treatmentof wild-type MEFs with 30 Amol/L sulforaphane caused a >30-fold increase in percentage of subdiploid cells compared withDMSO-treated control. The sulforaphane-mediated increase insubdiploid fraction was much less pronounced in Bax�/� and
Figure 1. Sulforaphane (SFN )-induced cell death in wld-type MEFs wasassociated with induction of Bax and Bak protein expression. A, effect ofsulforaphane treatment on viability of wild-type MEFs determined by trypan bluedye exclusion assay following a 24-hour exposure to DMSO (control) or differentconcentrations of sulforaphane. Columns, % of control and mean (n = 3); bars,SE. *, P < 0.05, significantly different compared with control by one-way ANOVA.Experiment was repeated twice, and results were comparable. B, effect ofsulforaphane treatment on apoptosis induction in wild-type MEFs assessed byquantitation of cytoplasmic histone-associated DNA fragmentation. Wild-typeMEFs were plated, allowed to attach by overnight incubation, exposed to DMSO(control) or different concentrations of sulforaphane for 24 hours, and processedfor analysis of DNA fragmentation. Experiment was repeated twice in duplicate.Columns, mean (n = 4); bars, SE. *, P < 0.05, significantly different comparedwith control by one-way ANOVA. C, immunoblotting for Bax and Bak usinglysates from wild-type MEFs treated with sulforaphane for the indicatedconcentrations and time periods. Blots were stripped and reprobed with anti-actinantibody to normalize for equal protein loading. Immunoblotting for each proteinwas done at least twice, and results were comparable. D, effect of sulforaphanetreatment on cytoplasmic histone-associated DNA fragmentation in BEAS2Bnormal human bronchial epithelial and H1299 human lung cancer cells. Cellswere treated with DMSO (control) or desired concentration of sulforaphane for 24hours. Columns, mean (n = 3); bars, SE. *, P < 0.05, significantly differentcompared with DMSO-treated control by one-way ANOVA. E, effect ofsulforaphane treatment on cytoplasmic histone-associated DNA fragmentation inPrEC normal prostate epithelial and LNCaP human prostate cancer cells. Cellswere treated with DMSO (control) or desired concentration of sulforaphane for 24hours. Columns, mean (n = 3); bars, SE. *, P < 0.05, significantly differentcompared with DMSO-treated control by one-way ANOVA.
Role of Bax and Bak in Sulforaphane-Induced Apoptosis
www.aacrjournals.org 2037 Cancer Res 2005; 65: (5). March 1, 2005
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
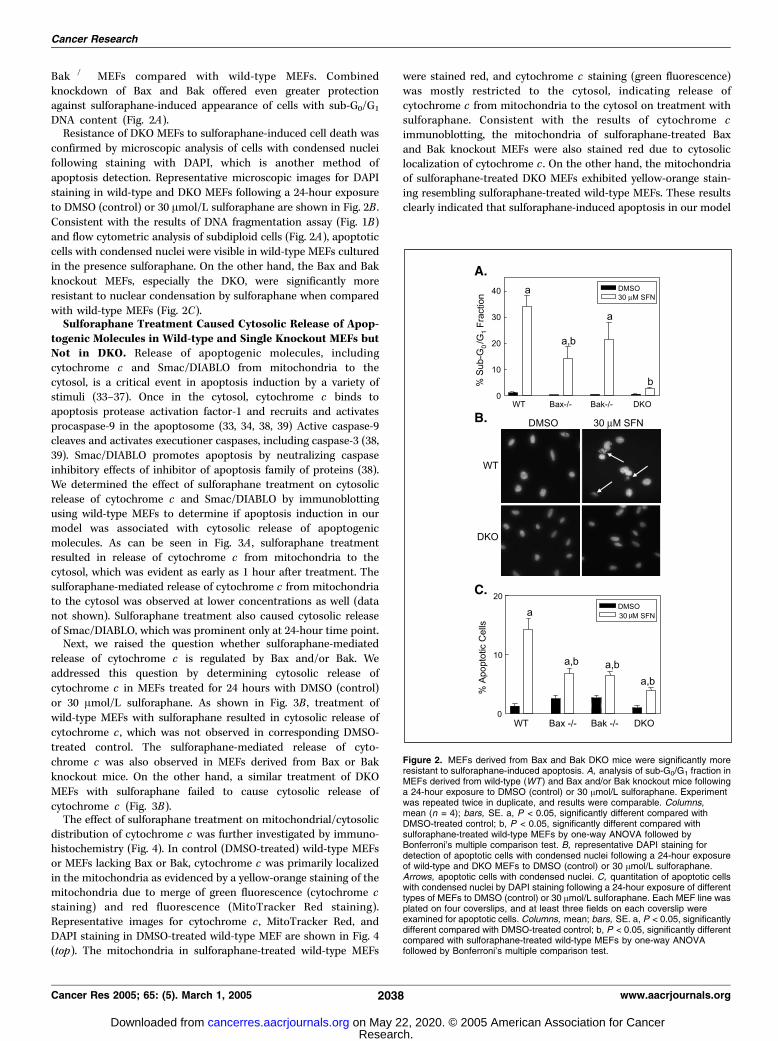
Bak�/� MEFs compared with wild-type MEFs. Combinedknockdown of Bax and Bak offered even greater protectionagainst sulforaphane-induced appearance of cells with sub-G0/G1
DNA content (Fig. 2A).Resistance of DKO MEFs to sulforaphane-induced cell death was
confirmed by microscopic analysis of cells with condensed nucleifollowing staining with DAPI, which is another method ofapoptosis detection. Representative microscopic images for DAPIstaining in wild-type and DKO MEFs following a 24-hour exposureto DMSO (control) or 30 Amol/L sulforaphane are shown in Fig. 2B .Consistent with the results of DNA fragmentation assay (Fig. 1B)and flow cytometric analysis of subdiploid cells (Fig. 2A), apoptoticcells with condensed nuclei were visible in wild-type MEFs culturedin the presence sulforaphane. On the other hand, the Bax and Bakknockout MEFs, especially the DKO, were significantly moreresistant to nuclear condensation by sulforaphane when comparedwith wild-type MEFs (Fig. 2C).Sulforaphane Treatment Caused Cytosolic Release of Apop-
togenic Molecules in Wild-type and Single Knockout MEFs butNot in DKO. Release of apoptogenic molecules, includingcytochrome c and Smac/DIABLO from mitochondria to thecytosol, is a critical event in apoptosis induction by a variety ofstimuli (33–37). Once in the cytosol, cytochrome c binds toapoptosis protease activation factor-1 and recruits and activatesprocaspase-9 in the apoptosome (33, 34, 38, 39) Active caspase-9cleaves and activates executioner caspases, including caspase-3 (38,39). Smac/DIABLO promotes apoptosis by neutralizing caspaseinhibitory effects of inhibitor of apoptosis family of proteins (38).We determined the effect of sulforaphane treatment on cytosolicrelease of cytochrome c and Smac/DIABLO by immunoblottingusing wild-type MEFs to determine if apoptosis induction in ourmodel was associated with cytosolic release of apoptogenicmolecules. As can be seen in Fig. 3A , sulforaphane treatmentresulted in release of cytochrome c from mitochondria to thecytosol, which was evident as early as 1 hour after treatment. Thesulforaphane-mediated release of cytochrome c from mitochondriato the cytosol was observed at lower concentrations as well (datanot shown). Sulforaphane treatment also caused cytosolic releaseof Smac/DIABLO, which was prominent only at 24-hour time point.Next, we raised the question whether sulforaphane-mediated
release of cytochrome c is regulated by Bax and/or Bak. Weaddressed this question by determining cytosolic release ofcytochrome c in MEFs treated for 24 hours with DMSO (control)or 30 Amol/L sulforaphane. As shown in Fig. 3B , treatment ofwild-type MEFs with sulforaphane resulted in cytosolic release ofcytochrome c , which was not observed in corresponding DMSO-treated control. The sulforaphane-mediated release of cyto-chrome c was also observed in MEFs derived from Bax or Bakknockout mice. On the other hand, a similar treatment of DKOMEFs with sulforaphane failed to cause cytosolic release ofcytochrome c (Fig. 3B).The effect of sulforaphane treatment on mitochondrial/cytosolic
distribution of cytochrome c was further investigated by immuno-histochemistry (Fig. 4). In control (DMSO-treated) wild-type MEFsor MEFs lacking Bax or Bak, cytochrome c was primarily localizedin the mitochondria as evidenced by a yellow-orange staining of themitochondria due to merge of green fluorescence (cytochrome cstaining) and red fluorescence (MitoTracker Red staining).Representative images for cytochrome c , MitoTracker Red, andDAPI staining in DMSO-treated wild-type MEF are shown in Fig. 4(top). The mitochondria in sulforaphane-treated wild-type MEFs
were stained red, and cytochrome c staining (green fluorescence)was mostly restricted to the cytosol, indicating release ofcytochrome c from mitochondria to the cytosol on treatment withsulforaphane. Consistent with the results of cytochrome cimmunoblotting, the mitochondria of sulforaphane-treated Baxand Bak knockout MEFs were also stained red due to cytosoliclocalization of cytochrome c . On the other hand, the mitochondriaof sulforaphane-treated DKO MEFs exhibited yellow-orange stain-ing resembling sulforaphane-treated wild-type MEFs. These resultsclearly indicated that sulforaphane-induced apoptosis in our model
Figure 2. MEFs derived from Bax and Bak DKO mice were significantly moreresistant to sulforaphane-induced apoptosis. A, analysis of sub-G0/G1 fraction inMEFs derived from wild-type (WT ) and Bax and/or Bak knockout mice followinga 24-hour exposure to DMSO (control) or 30 Amol/L sulforaphane. Experimentwas repeated twice in duplicate, and results were comparable. Columns,mean (n = 4); bars, SE. a, P < 0.05, significantly different compared withDMSO-treated control; b, P < 0.05, significantly different compared withsulforaphane-treated wild-type MEFs by one-way ANOVA followed byBonferroni’s multiple comparison test. B, representative DAPI staining fordetection of apoptotic cells with condensed nuclei following a 24-hour exposureof wild-type and DKO MEFs to DMSO (control) or 30 Amol/L sulforaphane.Arrows, apoptotic cells with condensed nuclei. C, quantitation of apoptotic cellswith condensed nuclei by DAPI staining following a 24-hour exposure of differenttypes of MEFs to DMSO (control) or 30 Amol/L sulforaphane. Each MEF line wasplated on four coverslips, and at least three fields on each coverslip wereexamined for apoptotic cells. Columns, mean; bars, SE. a, P < 0.05, significantlydifferent compared with DMSO-treated control; b, P < 0.05, significantly differentcompared with sulforaphane-treated wild-type MEFs by one-way ANOVAfollowed by Bonferroni’s multiple comparison test.
Cancer Research
Cancer Res 2005; 65: (5). March 1, 2005 2038 www.aacrjournals.org
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

was triggered by cytosolic release of cytochrome c and that both
Bax and Bak were required for this effect.Sulforaphane Treatment Caused Cleavage of Caspases in
Wild-type MEFs but Not in DKO MEFs. Caspases are aspartate-
specific cysteine proteases that exist as latent zymogens (38, 39).
On activation by apoptotic stimuli, caspases can systematically
cleave key cellular proteins, including DNA repair enzyme PARP
(38, 39). The mitochondria-mediated caspase cascade (also known
as intrinsic pathway) involves mitochondrial membrane perme-
ability change that triggers release of cytochrome c and other
proapoptotic molecules from mitochondria to the cytosol (38–40).
Because cytochrome c release was observed in sulforaphane-
treated wild-type MEFs, we determined the effect of sulforaphane
treatment on proteolytic cleavage of caspase-9 and caspase-3 by
immunoblotting using wild-type MEFs. As can be seen in Fig. 5A ,
sulforaphane treatment caused proteolytic cleavage of both
caspase-9 and caspase-3 in a concentration-dependent manner.
In time course experiments using 40 Amol/L sulforaphane,
proteolytic cleavage of caspase-9 was evident as early as 2 hours
after treatment, whereas caspase-3 cleavage was not observed until
12 hours (Fig. 5B). Activation of caspase-3 leads to cleavage and
inactivation of key cellular proteins, including the DNA repair
enzyme PARP. We therefore determined the effect of sulforaphane
treatment on cleavage of PARP to confirm caspase-3 activation. As
can be seen in Fig. 5C , PARP cleavage was observed in
sulforaphane-treated wild-type MEFs.The effect of sulforaphane treatment (30 Amol/L, 24 hours) on
cleavage of caspase-3 and PARP was compared using wild-type andBax and/or Bak knockout MEFs (Fig. 5D). Treatment of wild-typeMEFs as well as the MEFs derived from Bax or Bak knockout micewith sulforaphane resulted in proteolytic cleavage of caspase-3,which was barely seen in DKO MEFs (Fig. 5D). Consistent with
these results, sulforaphane-induced cleavage of PARP was muchmore pronounced in wild-type MEFs and MEFs derived from Baxor Bak single knockout mice when compared with DKO. Somecleavage of PARP in the DKO MEFs is expected becausesulforaphane is able to induce residual cell death in these cells(Fig. 2). These results indicate involvement of a nonmitochondrialcomponent in sulforaphane-induced apoptosis in DKO.Sulforaphane Treatment Caused Induction of Apaf-1 and
Down-regulation of XIAP Protein Expression. Activation ofcaspase-9 is regulated by Apaf-1, which facilitates recruitment ofprocaspase-9 to the apoptosome (37–40). The XIAP proteininhibits activity of caspases (41). To determine possible involve-ment of Apaf-1 and XIAP in apoptosis induction by sulforaphane,we determined its effect on levels of above proteins byimmunoblotting using wild-type MEFs. As shown in Fig. 6A , a24-hour treatment of wild-type MEFs with sulforaphane resulted ina dose-dependent induction of Apaf-1 protein expression. Thesulforaphane-mediated increase in Apaf-1 protein level was evidentas early as 8 hours after treatment (data not shown). Interestingly,the sulforaphane-treated wild-type MEFs also exhibited a concen-tration-dependent reduction in XIAP protein level, especially athigher concentrations of sulforaphane (Fig. 6A). Next, we raised thequestion whether sulforaphane-mediated change in Apaf-1 or XIAPprotein levels was regulated by Bax or Bak. As can be seen inFig. 6B , the sulforaphane-mediated induction of Apaf-1 proteinexpression was observed in wild-type and Bax and Bak singleknockout MEFs but not in the MEFs derived from DKO mice. Onthe other hand, the sulforaphane-mediated decline in XIAP proteinlevel was observed in wild-type as well as Bax and Bak knockoutMEFs, including DKO (Fig. 6B). These results suggested thatsulforaphane-mediated induction of Apaf-1 protein might beregulated by Bax and Bak proteins.Sulforaphane Treatment Caused Mitochondrial Transloca-
tion of Bax. In normal cells, the Bax protein exists in an inactiveform in the cytosol but can be induced to change conformationand translocate to the mitochondria in response to certainapoptotic stimuli (42, 43). The activated Bax protein oligomerizeson the outer mitochondrial membrane and induces release ofapoptogenic molecules to the cytoplasm (42, 43). Becausecytosolic release of cytochrome c was observed in sulfora-phane-treated wild-type MEFs, we raised the question whethersulforaphane treatment caused activation of Bax to initiate thecell death process. We tested this possibility by two different butcomplementary approaches. First, we determined whethersulforaphane treatment causes conformational change of Bax.The conformational change was assessed by immunoprecipita-tion of Bax using a monoclonal antibody (6A7) that recognizesan epitope at the NH2 terminus of the protein, which becomesexposed only after a change in conformation of Bax. Theimmunoprecipitated complex was then subjected to SDS-PAGEfollowed by immunoblotting using anti-Bax polyclonal antibody.As can be seen in Fig. 6C , sulforaphane treatment caused a changein conformation of Bax that was evident as early as 4 hours afterexposure and gradually increased with increasing exposure time.Activation of Bax on treatment with sulforaphane was confirmedby immunohistochemistry using wild-type MEFs (Fig. 6D). InMEFs treated for 12 hours with DMSO (control), the mitochondriawere stained red and Bax immunostaining was generally restrictedto the cytosol. On the other hand, the mitochondria in sulfor-aphane-treated MEFs were stained yellow-orange due to merge ofgreen fluorescence (Bax immunostaining) and red fluorescence
Figure 3. Sulforaphane treatment caused release of apoptogenic molecules tothe cytosol in wild-type, Bax knockout, and Bak knockout MEFs but not in DKO.A, immunoblotting for cytochrome c and Smac/DIABLO using cytosolic fractionsfrom wild-type MEFs treated with 40 Amol/L sulforaphane for the indicated timeperiods. B, immunoblotting for cytochrome c in cytosolic fractions prepared fromMEFs following a 24-hour exposure to DMSO (control) or 30 Amol/Lsulforaphane. Blots were stripped and reprobed with anti-actin antibody tonormalize for equal protein loading. Immunoblotting for each protein was done atleast twice, and results were comparable.
Role of Bax and Bak in Sulforaphane-Induced Apoptosis
www.aacrjournals.org 2039 Cancer Res 2005; 65: (5). March 1, 2005
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

(MitoTracker Red staining), indicating translocation of Bax fromcytosol to the mitochondria. To the best of our knowledge, our studyis the first published report to indicate Bax activation insulforaphane-treated cells.
Discussion
Evidence is accumulating to indicate that sulforaphane not onlyoffers protection against chemically induced cancer in animalmodels (15, 17, 18) but also suppresses proliferation of cancer cells inculture as well as in vivo (20–29). Studies have indicated thatinhibitory effect of sulforaphane against proliferation of culturedcancer cells is attributable to cell cycle block as well as apoptosisinduction (20–29). A novel mechanism of chemoprevention bysulforaphane involving inhibition of histone deacetylase was alsosuggested recently (26). Our own work has revealed that sulfor-aphane treatment causes apoptosis in PC-3 human prostate cancercells in association with activation of caspases (28). Despite theseadvances, however, the mechanism of sulforaphane-induced apo-ptosis remains poorly defined. For instance, the signaling pathwaysupstream of caspase activation in sulforaphane-induced cell deathare not characterized. The present study provides experimentalevidence to indicate that Bcl-2 family proapoptotic proteins Bax andBak play a critical role in regulation of cell death by sulforaphane.
The Bcl-2 family proteins have emerged as critical regulators of
the mitochondria-mediated apoptosis by functioning as either
promoters (e.g., Bax and Bak) or inhibitors (e.g., Bcl-2 and Bcl-xL) of
the cell death process (44–48). Antiapoptotic Bcl-2 family members
(Bcl-2 and Bcl-xL) possess four conserved BH domains (BH1-BH4)
and mainly prevent the release of apoptogenic molecules
(e.g., cytochrome c) from mitochondria to the cytosol by forming
heterodimer with proapoptotic proteins, such as Bax (44–48). The
proapoptotic Bcl-2 family proteins, which can be subdivided into
the Bax subfamily of multidomain proteins (e.g., Bax and Bak) or
BH3-only subfamily (e.g., Bid and Bim), induce mitochondrial
membrane permeabilization and release of apoptogenic molecules
from mitochondria to the cytosol (44–48). Previous studies,
including those from our laboratory, indicated that sulforaphane-
induced apoptosis in different cellular systems was associated with
induction of Bax protein expression (20, 21, 25, 26). The present
study was designed to experimentally test the role of Bax and Bak
in apoptosis induction by sulforaphane using SV40-transformed
MEFs derived from Bax and/or Bak knockout mice. Data presented
herein indicate that both Bax and Bak are required for apoptosis
induction by sulforaphane. This conclusion is based on the
following observations: (a) sulforaphane treatment causes a dose-
and time-dependent increase in protein levels of both Bax and Bak
Figure 4. Sulforaphane treatment causedtranslocation of cytochrome c frommitochondria to the cytosol in wild-type MEFsbut not in DKO. Immunohistochemistryfor analysis of cytochrome c localization inwild-type MEFs and in Bax- and/orBak-deficient MEFs following 8-hourexposure to DMSO (data for wild-type MEFsare shown) or 30 Amol/L sulforaphane.Green, red , and blue fluorescence,staining for cytochrome c , mitochondria,and nucleus, respectively. Images weremerged to detect mitochondrial/cytosolicdistribution of cytochrome c .
Cancer Research
Cancer Res 2005; 65: (5). March 1, 2005 2040 www.aacrjournals.org
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

in wild-type MEFs, (b) the MEFs derived from Bax and Bakknockout mice exhibit increased resistance to apoptosis inductionby sulforaphane when compared with wild-type MEFs, and (c) theprotection against sulforaphane-induced apoptosis in DKO is
greater than in cells lacking either Bax or Bak. Consistent with
these results, sulforaphane treatment causes release of apoptogenic
molecules and caspase-3 activation in wild-type MEFs but not in
DKO. Our data also suggest that Bax and Bak proteins may have
overlapping functions because the single knockout cells exhibited
significant apoptosis in response to treatment with sulforaphane.
Thus, sulforaphane resembles various other agents that requireboth Bax and Bak proteins to initiate the cell death, including
Figure 5. Sulforaphane treatment caused cleavage of caspase-9 and caspase-3.A, immunoblotting for cleavage of caspase-9 and caspase-3 using lysates fromwild-type MEFs treated for 24 hours with DMSO (control) or indicatedconcentrations of sulforaphane. B, immunoblotting for cleavage of caspase-9 andcaspase-3 using lysates from wild-type MEFs treated with 40 Amol/L sulforaphanefor the indicated time periods. C, immunoblotting for PARP cleavage using lysatesfrom wild-type MEFs treated with indicated concentrations of sulforaphane forspecified time intervals. D, immunoblotting for cleavage of caspase-3 and PARPusing lysates from different MEFs treated for 24 hours with DMSO (control) or30 Amol/L sulforaphane. Blots were stripped and reprobed with anti-actin antibodyto normalize for equal protein loading. Immunoblotting for each protein was done atleast twice, and results were comparable.
Figure 6. Sulforaphane treatment caused induction of Apaf-1, down-regulationof XIAP protein, and conformational change and mitochondrial translocation ofBax in wild-type MEFs. A, immunoblotting for Apaf-1 and XIAP proteins usinglysates from wild-type MEFs treated for 24 hours with DMSO (control) orindicated concentrations of sulforaphane. B, immunoblotting for Apaf-1 and XIAPproteins using lysates from different MEFs treated for 24 hours with DMSO(control) or 30 Amol/L sulforaphane. Blots were stripped and reprobed withanti-actin antibody to normalize for equal protein loading. Immunoblotting foreach protein was done at least twice, and results were comparable. C, analysisof conformational change of Bax using lysates from wild-type MEFs treated withsulforaphane for the indicated time periods. Bax protein was immunoprecipitatedfrom equal amounts of lysate proteins using anti-Bax monoclonal antibody 6A7.Immunoprecipitated complexes were subjected to SDS-PAGE followed byimmunoblotting using anti-Bax polyclonal antibody. D, immunohistochemistry foranalysis of Bax localization. Wild-type MEFs were treated for 12 hours withDMSO or 40 Amol/L sulforaphane. Cells were then stained with anti-Bax (greenfluorescence), MitoTracker Red (red fluorescence ), and DAPI (bluefluorescence). Merged images are shown, which indicate yellow-orange stainingof mitochondria in sulforaphane-treated MEFs due to merge of green and redfluorescence . Mitochondria in DMSO-treated control were stained red .
Role of Bax and Bak in Sulforaphane-Induced Apoptosis
www.aacrjournals.org 2041 Cancer Res 2005; 65: (5). March 1, 2005
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

tunicamycin, staurosporine, etoposide, and tumor necrosis factor–related apoptosis-inducing ligand (31, 49). The kinetics ofsulforaphane-mediated induction of Bax and Bak proteins wascomparable, suggesting that these proteins may have commonregulatory mechanism. However, further studies are needed todetermine the mechanism by which sulforaphane treatment causesinduction of Bax or Bak protein expression.In normal cells, the Bax protein exists in an inactive form
mainly in the cytosol but can be induced to change conformationand translocate to the mitochondria in response to certainapoptotic stimuli (42, 43). The conformationally changed Baxprotein oligomerizes on the outer mitochondrial membraneand induces release of apoptogenic molecules to the cytoplasm(42, 43). Recent studies have indicated that microtubule-damagingagents cause Bax activation to trigger the cell death (50). Becausesulforaphane was shown recently to disrupt tubulin polymeriza-tion (25), we reasoned that apoptosis induction by thisphytochemical may be due to Bax activation. The results of thepresent study indicate that sulforaphane treatment indeed causesconformational change and mitochondrial translocation of Bax.To the best of our knowledge, the present study is the firstpublished report to show Bax activation in sulforaphane-inducedcell death.Apaf-1 is a critical regulator of mitochondria-mediated
activation of caspase-9, whereas XIAP functions to inhibitcaspases (38–41). The present study reveals that sulforaphanetreatment causes an increase in protein level of Apaf-1 and adecrease in XIAP protein level. Thus, it is reasonable to postulatethat sulforaphane-mediated activation of caspases and apoptosisin our model is probably amplified by induction of Apaf-1 anddown-regulation of XIAP. It is interesting to note that sulfor-aphane-mediated induction of Apaf-1 was not observed in MEFslacking both Bax and Bak. These results suggest that Bax and Bakmay regulate sulforaphane-mediated induction of Apaf-1, butadditional studies are needed to systematically explore thispossibility. Notably, the sulforaphane-mediated decline in XIAPprotein level was not influenced by the presence or absence of Baxand/or Bak.
Cell growth inhibition and apoptosis induction by sulfora-phane in SV40-transformed MEFs (present study) as well as inother cells, including colon, Jurkat T leukemia, and prostatecancer cells, have been observed at 10 to 40 Amol/L concen-trations (20–29). A fundamental question, which remainsunanswered, is whether the micromolar concentrations ofsulforaphane needed to trigger cell death are achievable inhumans. Although the answer to this question awaits pharma-cokinetic data in humans using pure sulforaphane, thepharmacokinetic variables for sulforaphane were determinedrecently in rats after oral dosing (50 Amol; ref. 51). Sulforaphanewas detectable in the plasma after 1 hour, peaked around 20Amol/L at 4 hours after dosing, and declined with a half-life off2.2 hours (51). The isothiocyanates, including sulforaphane, aremainly excreted as thiol conjugates in the urine (51–53).Interestingly, the thiol conjugates of sulforaphane retain chemo-preventive activity (8, 12, 13). Thus, it is highly likely that theconcentrations of sulforaphane needed to cause cell death maybe achievable.In conclusion, the results of the present study indicate that
sulforaphane treatment causes induction of Bax and Bak proteinexpression and conformational change and mitochondrialtranslocation of Bax to trigger the release of apoptogenicmolecules from mitochondria to the cytosol leading to activationof caspases and cell death. The sulforaphane-mediated caspaseactivation is probably amplified due to induction of Apaf-1 anddown-regulation of XIAP protein. Furthermore, we provideexperimental evidence to indicate that both Bax and Bak areessential for sulforaphane-induced cell death.
Acknowledgments
Received 10/7/2004; revised 11/24/2004; accepted 12/21/2004.Grant support: USPHS grants CA101753 and CA076348 awarded by the National
Cancer Institute.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Yan Zeng for technical assistance and Dr. Stanley Korsmeyer for thegenerous gift of MEFs.
References1. Verhoeven DT, Goldbohm RA, van Poppel G,Verhagen H, van den Brandt PA. Epidemiologicalstudies on Brassica vegetables and cancer risk. CancerEpidemiol Biomarkers Prev 1996;5:733–48.
2. Cohen JH, Kristal AR, Stanford JL. Fruit and vegetableintakes and prostate cancer risk. J Natl Cancer Inst2000;92:61–8.
3. Zhang SM, Hunter DJ, Rosner BA, et al. Intakes offruits, vegetables, and related nutrients and the risk ofnon-Hodgkin’s lymphoma among women. CancerEpidemiol Biomarkers Prev 2000;9:477–85.
4. Ambrosone CB, McCann SE, Freudenheim JL,Marshall JR, Zhang Y, Shields PG. Breast cancer riskin premenopausal women is inversely associated withconsumption of broccoli, a source of isothiocyanates,but is not modified by GST genotype. J Nutr2004;134:1134–8.
5. Hecht SS. Inhibition of carcinogenesis by isothiocya-nates. Drug Metab Rev 2000;32:395–411.
6. Talalay P, Fahey JW. Phytochemicals from cruciferousplants protect against cancer by modulating carcinogenmetabolism. J Nutr 2001;131:3027–33s.
7. Fahey JW, Zalcmann AT, Talalay P. The chemicaldiversity and distribution of glucosinolates andisothiocyanates among plants. Phytochemistry 2001;56:5–51.
8. Conaway CC, Yang YM, Chung FL. Isothiocyanatesas cancer chemopreventive agents: their biologicalactivities and metabolism in rodents and humans.Curr Drug Metab 2002;3:233–55.
9. Wattenberg LW. Inhibition of carcinogenic effects ofpolycyclic hydrocarbons by benzyl isothiocyanate andrelated compounds. J Natl Cancer Inst 1977;58:395–8.
10. Stoner GD, Morrissey DT, Heur YH, Daniel EM,Galati AJ, Wagner SA. Inhibitory effects of phenethylisothiocyanate on N -nitrosobenzylmethylamine carci-nogenesis in the rat esophagus. Cancer Res 1991;51:2063–8.
11. Morse MA, Wang CX, Stoner GD, et al. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-inducedDNA adduct formation and tumorigenicity in the lung ofF344 rats by dietary phenethyl isothiocyanate. CancerRes 1989;49:549–53.
12. Jiao D, Smith TJ, Yang CS, et al. Chemopreventiveactivity of thiol conjugates of isothiocyanates for lungtumorigenesis. Carcinogenesis 1997;18:2143–7.
13. Yang YM, Conaway CC, Chiao JW, et al. Inhibition ofbenzo(a)pyrene-induced lung tumorigenesis in A/Jmice by dietary N -acetylcysteine conjugates of benzyland phenethyl isothiocyanates during the postinitiationphase is associated with activation of mitogen-activatedprotein kinases and p53 activity and induction ofapoptosis. Cancer Res 2002;62:2–7.
14. Zhang Y, Talalay P, Cho CG, Posner GH. A majorinducer of anticarcinogenic protective enzymes frombroccoli: isolation and elucidation of structure. ProcNatl Acad Sci U S A 1992;89:2399–403.
15. Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P.Anticarcinogenic activities of sulforaphane andstructurally related synthetic norbornyl isothiocyanates.Proc Natl Acad Sci U S A 1994;91:3147–50.
16. Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: anexceptionally rich source of inducers of enzymes thatprotect against chemical carcinogens. Proc Natl AcadSci U S A 1997;94:10367–72.
17. Chung FL, Conaway CC, Rao CV, Reddy BS. Chemo-prevention of colonic aberrant crypt foci in Fischer ratsby sulforaphane and phenethyl isothiocyanate. Carci-nogenesis 2000;21:2287–91.
18. Fahey JW, Haristoy X, Dolan PM, et al. Sulforaphaneinhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and preventsbenzo[a ]pyrene-induced stomach tumors. Proc NatlAcad Sci U S A 2002;99:7610–5.
19. Haristoy X, Angioi-Duprez K, Duprez A, Lozniewski A.Efficacy of sulforaphane in eradicating Helicobacterpylori human gastric xenografts implanted in nudemice. Antimicrob Agents Chemother 2003;47:3982–4.
20. Gamet-Payrastre L, Li P, Lumeau S, et al. Sulfor-aphane, a naturally occurring isothiocyanate, induces
Cancer Research
Cancer Res 2005; 65: (5). March 1, 2005 2042 www.aacrjournals.org
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

cell cycle arrest and apoptosis in HT29 human coloncancer cells. Cancer Res 2000;60:1426–33.
21. Fimognari C, Nusse M, Cesari R, Iori R, Cantelli-Forti G,Hrelia P. Growth inhibition, cell-cycle arrest andapoptosis in human T-cell leukemia by the isothiocyanatesulforaphane. Carcinogenesis 2002;23:581–6.
22. Misiewicz I, Skupinska K, Kasprzycka-Guttman T.Sulforaphane and 2-oxohexyl isothiocyanate induce cellgrowth arrest and apoptosis in L-1210 leukemia andME-18 melanoma cells. Oncol Rep 2003;10:2045–50.
23. Gingras D, Gendron M, Boivin D, Moghrabi A,Theoret Y, Beliveau R. Induction of medulloblastomacell apoptosis by sulforaphane, a dietary anticarcinogenfrom Brassica vegetables. Cancer Lett 2004;203:35–43.
24. Wang L, Liu D, Ahmed T, Chung FL, Conaway CC,Chiao JW. Targeting cell cycle machinery as a molecularmechanism of sulforaphane in prostate cancer preven-tion. Int J Oncol 2004;24:187–92.
25. Jackson SJT, Singletary KW. Sulforaphane: a naturallyoccurring mammary carcinoma mitotic inhibitor,which disrupts tubulin polymerization. Carcinogenesis2004;25:219–27.
26. Myzak MC, Karplus PA, Chung FL, Dashwood RH. Anovel mechanism of chemoprotection by sulforaphane:inhibition of histone deacetylase. Cancer Res 2004;64:5767–74.
27. Parnaud G, Li P, Cassar G, et al. Mechanism ofsulforaphane-induced cell cycle arrest and apoptosis inhuman colon cancer cells. Nutr Cancer 2004;48:198–206.
28. Singh AV, Xiao D, Lew KL, Dhir R, Singh SV.Sulforaphane induces caspase-mediated apoptosis incultured PC-3 human prostate cancer cells and retardsgrowth of PC-3 xenografts in vivo . Carcinogenesis2004;25:83–90.
29. Singh SV, Herman-Antosiewicz A, Singh AV, et al.Sulforaphane-induced G2-M phase cell cycle arrestinvolves checkpoint kinase 2 mediated phosphorylationof Cdc25C. J Biol Chem 2004;279:25813–22.
30. Hartwell LH, Kastan MB. Cell cycle control andcancer. Science 1994;266:1821–8.
31. Wei MC, Zong WX, Cheng EH, et al. Proapoptotic Baxand Bak: a requisite gateway to mitochondrial dysfunc-tion and death. Science 2001;292:727–30.
32. Xiao D, Choi S, Johnson DE, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cellsinvolves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation ofBcl-2. Oncogene 2004;23:5594–606.
33. Green DR, Reed JC. Mitochondria and apoptosis.Science 1998;281:1309–12.
34. Hengartner MO. The biochemistry of apoptosis.Nature 2000;407:770–6.
35. Liu X, Kim C, Yang J, Jemmerson R, Wang X.Induction of apoptotic program in cell-free extracts:requirement for dATP and cytochrome c . Cell1996;86:147–57.
36. Susin SA, Lorenzo HK, Zamzami N, et al. Molecularcharacterization of mitochondrial apoptosis-inducingfactors. Nature 1999;397:441–6.
37. Du C, Fang M, Li Y, Li L, Wang X. Smac, amitochondrial protein that promotes cytochrome c -dependent caspase activation by eliminating IAPinhibition. Cell 2000;102:33–42.
38. Thornberry N, Lazebnick Y. Caspases: enemieswithin. Science 1998;281:1312–6.
39. Wolf BB, Green DR. Suicidal tendencies: apoptoticcell death by caspase family proteinases. J Biol Chem1999;274:20049–52.
40. Verhagen AM, Ekert PG, Pakusch M, et al. Identifi-cation of DIABLO, a mammalian protein that promotesapoptosis by binding to and antagonizing IAP proteins.Cell 2000;102:43–53.
41. Salvesen GS, Duckett CS. IAP proteins: blocking theroad to death’s door. Nat Rev Mol Cell Biol 2002;3:401–10.
42. Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG,Youle RJ. Movement of Bax from the cytosol tomitochondria during apoptosis. J Cell Biol 1997;139:1281–92.
43. Yamaguchi H, Wang HG. The protein kinase PKB/Aktregulates cell survival and apoptosis by inhibiting Baxconformational change. Oncogene 2001;20:7779–86.
44. Hockenbery D, Nunez G, Milliman C, Schreiber RD,Korsmeyer SJ. Bcl-2 is an inner mitochondrial mem-brane protein that blocks programmed cell death.Nature 1990;348:334–6.
45. Reed JC. Bcl-2 family proteins: regulators of apopto-sis and chemoresistance in hematologic malignancies.Semin Hematol 1997;34:9–19.
46. Chao DT, Korsmeyer SJ. Bcl-2 family: regulators ofcell death. Annu Rev Immunol 1998;16:395–419.
47. Adams JM, Cory S. The Bcl-2 protein family: arbitersof cell survival. Science 1998;281:1322–6.
48. Martinou JC, Green DR. Breaking the mitochondrialbarrier. Nat Rev Mol Cell Biol 2001;2:63–7.
49. Kandasamy K, Srinivasula SM, Alnemri ES, et al.Involvement of proapoptotic molecules Bax and Bak intumor necrosis factor-related apoptosis-inducing ligand(TRAIL)-induced mitochondrial disruption and apopto-sis: differential regulation of cytochrome c and Smac/DIABLO release. Cancer Res 2003;63:1712–21.
50. Yamaguchi H, Chen J, Bhalla K, Wang HG.Regulation of Bax activation and apoptotic responseto microtubule-damaging agents by p53 transcription-dependent and -independent pathways. J Biol Chem2004;279:39431–7.
51. Hu R, Hebbar V, Kim BR, et al. In vivo pharmaco-kinetics and regulation of gene expression profiles forisothiocyanate sulforaphane in the rat. J Pharmacol ExpTher 2004;310:263–71.
52. Liebes L, Conaway CC, Hochster H, et al. High-performance liquid chromatography-based determina-tion of total isothiocyanate levels in human plasma:application to studies with 2-phenethyl isothiocyanate.Anal Biochem 2001;291:279–89.
53. Ji Y, Morris ME. Determination of phenethyl iso-thiocyanate in human plasma and urine by ammoniaderivatization and liquid chromatography-tandemmass spectrometry. Anal Biochem 2003;323:39–47.
Role of Bax and Bak in Sulforaphane-Induced Apoptosis
www.aacrjournals.org 2043 Cancer Res 2005; 65: (5). March 1, 2005
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

2005;65:2035-2043. Cancer Res Sunga Choi and Shivendra V. Singh Chemopreventive Agent
Derived Cancer−Sulforaphane, a Cruciferous Vegetable Bax and Bak Are Required for Apoptosis Induction by
Updated version
http://cancerres.aacrjournals.org/content/65/5/2035
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/65/5/2035.full#ref-list-1
This article cites 52 articles, 24 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/5/2035.full#related-urls
This article has been cited by 18 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/65/5/2035To request permission to re-use all or part of this article, use this link
Research. on May 22, 2020. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from


















![5[1]. Cruciferous Vegetables(1)](https://static.fdocuments.net/doc/165x107/577d2a701a28ab4e1ea92c6b/51-cruciferous-vegetables1.jpg)