
A translational study from Achilles tendinosis to cyclooxygenase
84
The endocannabinoid system: A translational study from Achilles tendinosis to cyclooxygenase Emmelie Björklund Department of Pharmacology and Clinical neuroscience Umeå 2014
Transcript of A translational study from Achilles tendinosis to cyclooxygenase

The endocannabinoid system: A translational study from Achilles tendinosis to cyclooxygenase
Emmelie Björklund
Department of Pharmacology and Clinical neuroscience Umeå 2014

Responsible publisher under swedish law: the Dean of the Medical Faculty This work is protected by the Swedish Copyright Legislation (Act 1960:729) All previously published papers were reproduced with permission from the publisher ISBN: 978-91-7601-089-1 ISSN: 0346-6612 New series no: 1663 Elektronisk version tillgänglig på http://umu.diva-portal.org/ Tryck/Printed by: Print & Media Umeå, Sweden 2014

“If we knew what it was we were doing, it would not be called research, would it?” Albert Einstein

i
Table of contents
Original papers ............................................................................................................... iii Abstract ............................................................................................................................. iv Populärvetenskaplig sammanfattning ................................................................... vi Abbrevations ................................................................................................................ viii Introduction ..................................................................................................................... 1 The endocannabinoid system ........................................................................................................................ 1 Cannabinoid receptors ..................................................................................................................................... 2 Non-‐cannabinoid receptors targeted by AEA ........................................................................................ 6 Transient receptor potential vanilloid 1 (TRPV1) .................................................................... 6 Peroxisome proliferator-‐activated receptors ............................................................................. 6
Synthesis of endocannabinoids ..................................................................................................................... 7 NAEs .............................................................................................................................................................. 7 2-‐arachidonoylglycerol ......................................................................................................................... 9
The cellular processing of endocannabinoids ..................................................................................... 10 Degradation of AEA .............................................................................................................................. 12 Other enzymes responsible for endocannabinoid inactivation ........................................ 13 Degradation of 2-‐AG ............................................................................................................................. 14 Other enzymes responsible for 2-‐AG metabolism .................................................................. 15 The importance of the COX-‐2 pathway in endocannabinoid signalling ........................ 15
Endocannabinoids and pain ....................................................................................................................... 16 Inhibition of endocannabinoid hydrolysis as a therapeutic target for the treatment of pain ......................................................................................................................................................... 20 Endocannabinoid system in human pain states ...................................................................... 21 Chronic pain in the human Achilles tendon ............................................................................... 22
Aims of the thesis ......................................................................................................... 23 Methodological considerations ............................................................................... 24 Subjects (Paper I) ............................................................................................................................................ 24 Achilles tendinosis patients .............................................................................................................. 24 Controls ..................................................................................................................................................... 25
Ethics (Paper I) ................................................................................................................................................. 26 Sampling, fixation and sectioning (Paper I) ........................................................................................ 26 Immunofluorescence and control staining (Paper I) ....................................................................... 27 Hematoxylin-‐eosin staining ........................................................................................................................ 27 Antibodies (Paper I) ....................................................................................................................................... 28 Assay for FAAH and MGL activities (Paper II, III, IV, V) .................................................................. 29 Cell culture (Paper II, III, IV) ....................................................................................................................... 32 Uptake assay of [3H]AEA, [3H]2-‐AG and [3H]PEA (Paper II, III, IV) ........................................... 33 Binding to CB1 receptors (Paper II) ......................................................................................................... 34

ii
RNA extraction, cDNA synthesis and PCR (Paper IV) ...................................................................... 34 Statistics .............................................................................................................................................................. 36 Mann-‐Whitney U test ........................................................................................................................... 36 One-‐way and two-‐way ANOVA ........................................................................................................ 36 pI50 and IC50 ............................................................................................................................................. 36
Results ............................................................................................................................. 37 CB1 receptor immunoreactivity in normal Achilles tendon and Achilles tendinosis (Paper 1) ............................................................................................................................................................................. 37 Comparison of tenocyte CB1IR in normal Achilles tendon with Achilles tendinosis ....................................................................................................................................................................... 37
Inhibition of FAAH and COX by Flu-‐AM1 and Nap-‐AM1 (Paper II) ........................................... 39 Inhibition of cellular uptake of AEA by ketoconazole (Paper III) .............................................. 40 Inhibitory effect of ketoconazole upon FAAH activity .......................................................... 40
Expression of FAAH mRNA in AT-‐1, RBL2H3, C6 and P19 cells (Paper IV) ............................ 42 Activity of FAAH and accumulation of [3H]AEA in RBL2H3, C6, AT-‐1 and P19 cells (Paper IV) ........................................................................................................................................................................... 43 Expression and activity of FABP5 in RBL2H3, C6, AT-‐1 and P19 cells (Paper IV) .............. 43 Screening of selected compounds for inhibition of MGL using the spectrophotometric NPA assay (Paper V) ....................................................................................................................................... 44 Troglitazone and N-‐arachidonoyl dopamine as inhibitors of MGL ................................. 44
Discussion ...................................................................................................................... 47 Overall comments ............................................................................................................................................ 47 Achilles tendinosis ........................................................................................................................................... 47 Inhibitors of endocannabinoid metabolism and uptake ................................................................ 49 Future perspectives ......................................................................................................................................... 52
Conclusions .................................................................................................................... 53 Acknowledgements ..................................................................................................... 54 References ...................................................................................................................... 57

iii
Original papers
The present thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I. Emmelie Björklund, Sture Forsgren, Håkan Alfredson, Christopher J. Fowler. Increased expression of cannabinoid CB1 receptors in Achilles Tendinosis. PLoS ONE. 2011 September; 6 (11): e24731
II. Mariateresa Cipriano, Emmelie Björklund, Alan A. Wilson, Cenzo Congiu, Valentina Onnis, Christopher J. Fowler. Inhibition of fatty acid amide hydrolase and cyclooxygenase by the N-(3-methylpyridin-2-yl)amide derivates of flurbiprofen and naproxen. European Journal of Pharmacology. 2013 October; 720 (2013): 383-390
III. Emmelie Björklund, Therése Larsson, Stig Jacobsson, Christopher J. Fowler. Ketoconazole inhibits the cellular uptake of anandamide via inhibition of FAAH at pharmacologically relevant concentrations. PLoS ONE. 2014 January 9 (14): e87542
IV. Emmelie Björklund, Anders Blomqvist, Joel Hedlin, Emma Persson, Christopher J. Fowler. Involvement of fatty acid amide hydrolase and fatty acid binding protein 5 in the uptake of anandamide by cell lines with different levels of fatty acid amide hydrolase expression: a pharmacological study. Submitted
V. Emmelie Björklund, Erika Norén, Johanna Nilsson, Christopher J. Fowler. Inhibition of monoacylglycerol lipase by troglitazone, N-arachidonyl dopamine and the irreversible inhibitor JZL184: comparison of two different assays. British Journal of Pharmacology. 2010 December; 161 (7): 1512-1526

iv
Abstract
The endogenous cannabinoids anandamide (arachidonoyl ethanolamide, AEA) and
2-arachidonoyl glycerol (2-AG) exert their effect by activating cannabinoid
receptors (CB). These receptors mediate a broad range of physiological functions
such as beneficial effects in pain and inflammation, although little is known about
the expression of CB receptors in human pain conditions. AEA and 2-AG are short-
lived molecules due to their rapid cellular accumulation and metabolism. The
enzymes primarily responsible for their degradation are fatty acid amide hydrolase
(FAAH) for AEA and monoacylglycerol lipase (MGL) for 2-AG. Inhibition of
endocannabinoid metabolism is a potential approach for drug development, and
there is a need for the identification of novel compounds with inhibitory effects
upon FAAH and MGL.
In Paper I of this thesis, the expression of CB1 receptors in human Achilles tendon
was examined. We found expression of CB1 receptors in tenocytes, blood vessel
wall as well as in the perineurium of the nerve. A semi-quantitative analysis showed
an increase of CB1 receptors in painful human Achilles tendinosis.
In papers II and III, termination of AEA signalling was investigated via inhibition of
FAAH. In Paper II, Flu-AM1, an analogue of flurbiprofen, was investigated. The
compound inhibited both FAAH and the oxygenation of 2-AG by cyclooxygenase-2.
In Paper III the antifungal compound ketoconazole was shown to inhibit the cellular
uptake of AEA in HepG2, CaCo-2 and C6 cell lines in a manner consistent with
inhibition of FAAH.
The role of FAAH in gating the cellular accumulation of AEA was investigated in
Paper IV. FAAH has been shown to control the concentration gradient of AEA
across the plasmamembrane in RBL2H3 cells, whereas no such effect is seen in
other FAAH-expressing cell lines. To determine whether this effect is assay
dependent or due to intrinsic differences between the cell lines, we assayed four cell
lines with different levels of FAAH expression using the same methodology. We

v
found that the sensitivity of FAAH uptake inhibition was not dependent on the
expression level of FAAH, suggesting that factors other than FAAH are important
for uptake.
Paper V is focused on the inhibition of MGL. Prior to this study no selective
inhibitors of the enzyme had been described. Thus, we screened a number of
compounds for their inhibitory effect on MGL. Troglitazone was found to be an
inhibitor of MGL, although its potency was dependent upon the enzyme assay used.

vi
Populärvetenskaplig sammanfattning
Trots att cannabis har använts för berusning och i medicinska syften under hela
mänsklighetens historia dröjde det enda till 60-talet innan den primära psykoaktiva
komponenten i cannabis, Δ9-tetrahydrocannabinol (THC) upptäcktes. Trettio år
senare, i början av 90-talet lyckades man identifiera de proteinmolekyler i kroppen
till vilka THC kan binda och aktivera. Dessa proteinmolekyler kallar man för
cannabinoidreceptor 1 och 2 (CB1 och CB2). Upptäckten av receptorer i kroppen för
växtbaserade substanser ledde forskningen vidare och 1992-5 upptäcktes kroppsegna
cannabinoider (endocannabinoider) som endogena ligander för CB receptorer. De
två mest studerade endocannabinoiderna är anandamide (AEA) och 2-
arakidonoylglycerol (2-AG). Endocannabinoider bildas vid olika sjukdomstillstånd,
inklusive smärta och inflammation, men eftersom kroppens celler snabbt tar upp och
sedan bryter ner dem är deras effekt kortvarig.
Det är idag oklart hur dessa signaleringsmolekyler tas upp av cellen. Än så länge
finns det inget identifierat transportprotein som står för förflyttningen över
cellmembranet men det finns en rad hypoteser om hur endocannabinoider
transporteras från utsidan av cellen till de metaboliserande enzymerna. De mest
studerade enzymerna som bryter ner endocannabinoider är fettsyraamidhydrolas
(FAAH) och monoacylglycerollipas (MGL). Då man i en rad olika djurmodeller har
påvisat en smärtlindrande effekt av cannabinoidsignalering ökar intresset för att
farmakologiskt modifiera detta system och på så sätt få en ökad signalering.
Användning av cannabinoider begränsas på grund av psykogena effekter men
teoretisk skulle blockering av metabolismen av de kroppsegna cannabinoiderna leda
till terapeutisk effekt utan biverkan på centrala nervsystemet.
Den mesta forskningen som är gjord på endocannabinoidsystemet vid smärttillstånd
är utförd i gnagare vilket gör att man inte helt känner till hur detta system är uttryckt
hos människor. Inledningsvis studerade vi därför uttrycket av cannabinoidreceptorer
i mänsklig frisk hälsena och jämförde detta mot uttrycket hos patienter med
smärtande hälsenor, så kallad Akilles tendinos. Vi fann att uttrycket skilde sig

vii
mellan dessa grupper och det gav oss en grund till att systemet är förändrat även i
mänskliga smärttillstånd.
I de följande studierna använde vi oss av odlade celler och enzymextrakt för att
undersöka olika substansers verkan på FAAH och MGL. Baserat på diskussionen
ovan om att hämning av dessa enzymer potentiellt kan öka
endocannabinoidsignaleringen och ge positiva terapeutiska effekter mot smärta var
ett delsyfte med denna avhandling att finna substanser som har just denna
hämmande effekt. Det finns FAAH-hämmande substanser som är under klinisk
prövning men utfallet av dessa har varit svagt. Det finns därför ett behov av nya
substanser som har denna FAAH-hämmande effekt. I studie II och III undersökte vi
substanser som används eller har används kliniskt, alternativt substanser som har
syntetiserats baserat på klinisk verksamma substanser och fann två substanser som
var verksamma som FAAH-hämmare. Förutom studierna kring hämning av
nedbrytning utredde vi i studie IV FAAHs inverkan på upptaget av anandamide i
olika celltyper. Processen kring cellens upptag av anandamide är omdiskuterat och
resultaten skiljer sig mellan olika laboratorier och celltyper. Vi ville därför utreda
huruvida detta beror på skillnad i metodologi eller hos de olika cellernas egenskaper.
Vad gäller MGL så fanns det inga hämmande substanser som är selektiva mot det
enzymet när vi startade arbetet med denna avhandling. I och med att behovet av
sådana substanser är stort undersökte vi en rad substansers förmåga att påverka
MGL och fann två lovande substanser som kan tjänstgöra som mall för framtida
struktur-aktivitetssambands studier.
Sammanfattningsvis så visar resultaten från dessa studier att
endocannabinoidsystemet är ändrat i smärttillstånd hos människan. De visar även
prov på substanser som potentiellt skulle kunna utgöra grund för nya FAAH- och
MLG hämmande läkemedel.

viii
Abbrevations
2-AG 2-arachidonoylglycerol
2-OG 2-oleoylglycerol
ABHD6/12 abhydrolase domain-containing protein 6 and 12
AEA anandamide, arachidonoyl ethanolamide
AM404 N-(4-hydroxyphenyl)arachidonylamide (uptake inhibitor)
BSA bovine serum albumin
CB cannabinoid
CB1 cannabinoid receptor type 1
CB2 cannabinoid receptor type 2
CB1IR CB receptor immunoreactivity
COX cyclooxygenase
DAG diacylglycerol
DAGL diacylglycerol lipase
DRG dorsal root ganglion
FAAH fatty acid amide hydrolase
FABP fatty acid binding protein
FLAT FAAH-like anandamide transporter
JZL184 4-nitrophenyl-4-(di-benzo[d][1,3]dioxol-5-
yl(hydroxy)methyl)piperidine-1-carboxylate (MGL inhibitor)
LOX lipoxygenase
MGL monoacylglycerol lipase
NAE N-acetylethanolamine
NAPE N-acylphosphatidylethanolamine
NAPE-PLD N-acylphosphatidylethanolamine phospholipase D
PEA palmitoylethanolamide
PPAR peroxisome proliferator-activated receptors
THC Δ9-tetrahydrocannabinol
TRPV1 transient receptor potential vanilloid type 1
URB597 cyclohexylcarbamic acid 39- carbamoylbiphenyl-3-yl ester (FAAH
inhibitor)

ix

1
Introduction
The endocannabinoid system
Extracts from the plant Cannabis sativa have been used for many centuries both for
medicinal and for recreational purposes. The main psychoactive ingredient of
cannabis is Δ9-tetrahydrocannabinol (THC). Initially, the term “cannabinoid” was
used to indicate a structure with similarity to THC, However, the definition has
evolved to include compounds that interact with cannabinoid receptors (see below).
Most of the biological effects of THC and synthetic cannabinoids are mediated
through specific G-protein-coupled receptors, named cannabinoid receptors. At
present, there are two characterized cannabinoid receptors, CB1 and CB2, which
were cloned in 1990 and 1993 (Matsuda et al., 1990; Munro et al., 1993) Signalling
through cannabinoid receptors results in a broad repertoire of systemic responses
such as the beneficial effects of analgesia and inflammation, appetite regulation,
relief of spasticity in multiple sclerosis as well as decreased intestinal motility, and,
of course, the psychoactive effects sought after by recreational users of cannabis
(Howlett et al., 2002).
The discovery of cannabinoid receptors led to the identification of endogenous
ligands, called endocannabinoids. In 1992 the first endocannabinoid to be
discovered was anandamide (N-arachidonoylethanolamine, AEA) (Devane et al.,
1992). A few years later, 2-arachidonoylglycerol (2-AG) was identified as an
agonist of cannabinoid receptors (Mechoulam et al., 1995; Sugiura et al., 1995).
These are the most well-studied endocannabinoids although other compounds have
been proposed to be endocannabinoids such as 2-arachidonyl-glycerol ether (noladin
ether) (Hanuš et al., 2001) and the unsaturated fatty acid ethanolamides
docosahexaenoyl ethanolamide (DHEA) and docosatetraenoyl ethanolamide (DEA)
(Hanuš et al., 1993).

2
Briefly, the endocannabinoid system comprises CB1 and CB2, the endogenous
ligands and enzymes responsible for biosynthesis and degradation. These are
considered in more detail below.
Cannabinoid receptors
CB1 receptors were first identified in rat brain (Devane et al., 1988) and later cloned
in both rat (Devane et al., 1988; Matsuda et al., 1990) and human (Gérard et al.,
1991). CB2 was discovered shortly after in the human promyelocytic leukemic cell
line HL60 (Munro et al., 1993). The two receptors belong to the seven
transmembrane domain family of G-protein-coupled receptors.
CB1 receptors are abundantly expressed in the central nervous system (CNS). A high
expression of CB1 receptors is seen in, e.g. cerebral cortex, hippocampus,
hypothalamus, basal ganglia and cerebellum (Glass et al., 1997; Herkenham et al.,
1991). CB1 receptors are also expressed in lower levels of the brain stem, spinal cord
and in peripheral tissues such as the reproductive system, gastrointestinal tract, heart
and vasculature (Bonz et al., 2003; Liu et al., 2000; Ruiz-Llorente et al., 2003;
Wright et al., 2005). The distribution of CB1 receptor expression correlates well
with the known physiological effects of cannabinoids, e.g. modulation of cognition,
memory, motor function and analgesia (Pertwee et al., 2010). Some of the
physiological functions of peripheral CB1 receptors are listed in Table 1.
Studies have shown that in the brain, CB1 receptors are mainly located
presynaptically (Katona et al., 1999). Endocannabinoids are generally considered as
retrograde signals due to their synthesis and release into the synaptic cleft following
elevated levels of intracellular calcium in postsynaptic neurons (Alger et al., 2011).
They activate CB1 receptors on the presynaptic membrane and supress inhibitory or
excitatory neurotransmitter release (see Fig. 1).

3
Table 1. Examples of physiological functions mediated by CB1 receptors.
Tissue Agonist effect Reference
Vasculature induces hypotension (Szekeres et al., 2012)
Prostate gland inhibits contraction of the gland (Ruiz-Llorente et al., 2003)
Testis supresses hormone secretion (Wenger et al., 2001)
Urinary tract increases urine output (Sofia et al., 1977)
Uterus affects implantation of embryo (Paria et al., 1998)
Intestinal tract depresses gastrointestinal motility,
delays gastric emptying
(Pertwee, 2001)
CB1 receptors undergo constitutive internalization and following activation of
receptors at the plasma membrane, they are internalized via endocytosis. Receptors
are trafficked through the recycling endosomal pathway back to the plasma
membrane (Leterrier et al., 2004). It should be noted, however, that functional CB1
receptors are also expressed in intracellular compartments (Brailoiu et al., 2011;
Rozenfeld et al., 2008) Endosomes containing CB1 receptors are believed to result
from constitutive endocytosis.
The CB2 receptor was initially identified in a human promyelocytic leukemic cell
line (Munro et al., 1993). CB2 is often described as a peripheral receptor, mainly
expressed in immune tissues, such as spleen, tonsils and immune cells (B-cells and
natural killer cells) (Galiègue et al., 1995). When activated, CB2 receptors can
modulate immune cell migration and cytokine release (Pertwee et al., 2010).
However, there is some evidence that CB2 receptors have a limited distribution
within the CNS and may be involved in memory consolidation (García-Gutiérrez et
al., 2013).

4
Fig. 1. Activation of postsynaptic metabotropic glutamate receptors (mGluR) leads to
release of intracellular Ca2+ and Ca2+ influx, triggering biosynthesis of endocannabinoids
(see section below). The endocannabinoid (usually 2-AG) is released and presynaptic CB1
receptors are activated following retrograde diffusion leading to inhibition of Ca2+ channels.
Decreased intracellular Ca2+ levels leads to reduced inhibitory or excitatory neurotransmitter
release from the presynaptic terminal.
Upon stimulation both CB1 and CB2 receptors can inhibit adenylyl cyclase and
activate mitogen-activated protein kinase by signalling through Gi/o proteins (see
Fig. 1). CB1 receptors can also mediate increase of potassium current and inhibit
calcium channel activity (Pertwee et al., 2010). Examples of known ligands to
cannabinoid receptors are listed in Table 2.
In recent years functional studies have suggested that both endogenous and synthetic
cannabinoids have effects independently of CB1 and CB2 receptors. In some cases,
the target receptors are part of other receptor families (see below), whereas in others,
putative additional cannabinoid receptors have been suggested. The most studied of
the latter is the orphan G-protein receptor GPR55 (Ryberg et al., 2007). However
there is conflicting evidence regarding the ligands with which this receptor interact
mGluR Ca2+ influx
Lipid precursor
eCB
K+ influx
CB1
Ca2+ influx
Neurotransmitter
ATP cAMP AC
Gi/0
Presynaptic neuron
Postsynaptic neuron
5
and it is not yet clear whether or not GPR55 should be classified as a novel
cannabinoid receptor (Pertwee et al., 2010).
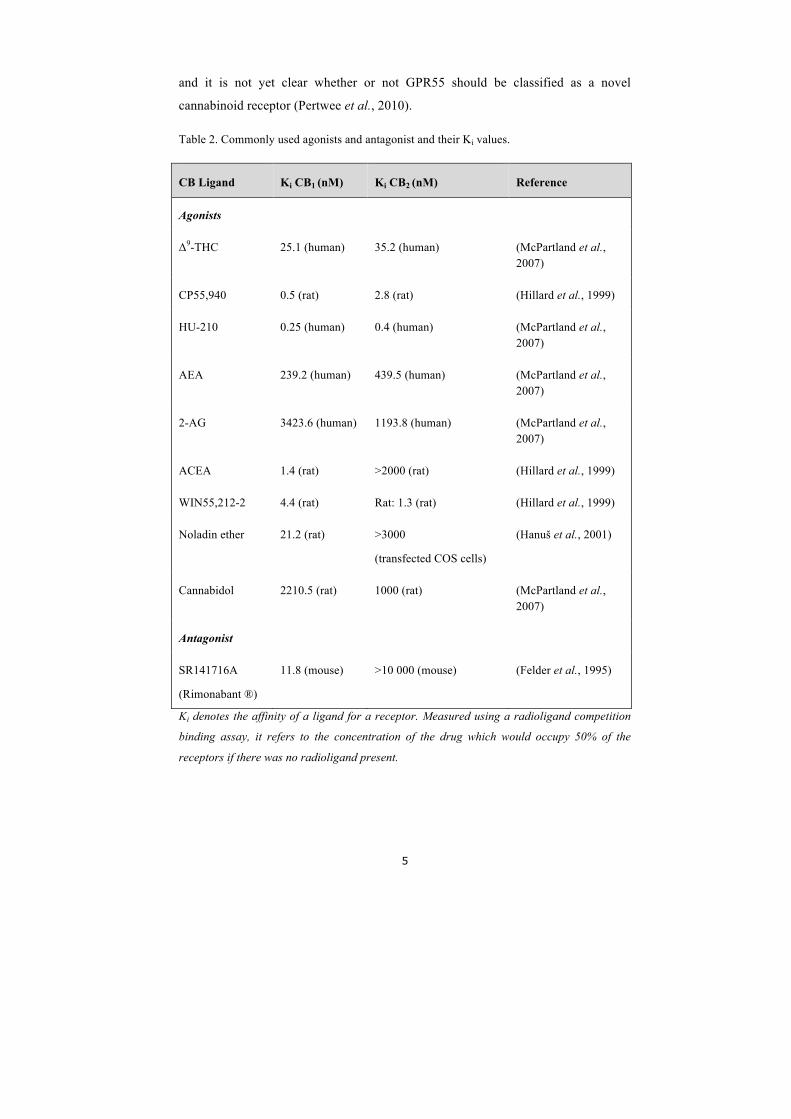
Table 2. Commonly used agonists and antagonist and their Ki values.
CB Ligand Ki CB1 (nM) Ki CB2 (nM) Reference
Agonists
Δ9-THC 25.1 (human) 35.2 (human) (McPartland et al., 2007)
CP55,940 0.5 (rat) 2.8 (rat) (Hillard et al., 1999)
HU-210 0.25 (human) 0.4 (human) (McPartland et al., 2007)
AEA 239.2 (human) 439.5 (human) (McPartland et al., 2007)
2-AG 3423.6 (human) 1193.8 (human) (McPartland et al., 2007)
ACEA 1.4 (rat) >2000 (rat) (Hillard et al., 1999)
WIN55,212-2 4.4 (rat) Rat: 1.3 (rat) (Hillard et al., 1999)
Noladin ether 21.2 (rat) >3000
(transfected COS cells)
(Hanuš et al., 2001)
Cannabidol 2210.5 (rat) 1000 (rat) (McPartland et al., 2007)
Antagonist
SR141716A
(Rimonabant ®)
11.8 (mouse) >10 000 (mouse) (Felder et al., 1995)
Ki denotes the affinity of a ligand for a receptor. Measured using a radioligand competition
binding assay, it refers to the concentration of the drug which would occupy 50% of the
receptors if there was no radioligand present.

6
Non-cannabinoid receptors targeted by AEA
Transient receptor potential vanilloid 1 (TRPV1)
It is now generally accepted that the endogenous CB1/CB2 receptor agonist AEA and
certain of its analogues are agonists for TRPV1 receptor (Howlett et al., 2002;
Zygmunt et al., 1999). TRPV1 is part of a family of transient receptor potential
channels. It is activated by a range of stimuli such as heat, low pH and compounds
including capsaicin, the main pungent ingredient of chilli pepper (Caterina et al.,
1997; Tominaga et al., 1998). The human receptor was cloned in 2000 and is most
highly expressed in the dorsal root ganglion (DRG) (Hayes et al., 2000). Although
the vanilloid receptor is a molecular target for AEA the affinity towards this receptor
is lower than that for CB1 (Ross, 2003). However, inflammatory mediators can
increase both the potency and efficacy of AEA (Singh Tahim et al., 2005),
suggesting a pro-nociceptive effect rather than an anti-nociceptive effect of AEA in
pathological situations. The endocannabinoid 2-AG is a partial agonist at this
receptor (Pertwee et al., 2010), and sufficient 2-AG is synthesised in response to the
appropriate receptor stimulation (see section on endocannabinoid synthesis below)
to activate TRPV1 receptors expressed in HEK293 cells (Zygmunt et al., 2013).
Peroxisome proliferator-activated receptors
Peroxisome proliferator activated receptors are ligand-activated transcription factors
that belong to the nuclear receptor family with different fatty acids as classical
agonists (Pertwee et al., 2010). There are three isoforms of PPARs; α, β/δ and γ.
PPAR-α is expressed in liver, kidney, muscle and fat (Desvergne et al., 1999).
PPAR-γ is highly expressed in intestine and adipose tissue (Braissant et al., 1996).
Studies have reported that AEA can activate PPAR-α and PPAR-γ (Bouaboula et al.,
2005; Sun et al., 2006) while 2-AG activates PPAR-γ and PPAR-β/δ (Pertwee et al.,
2010; Rockwell et al., 2006).
Other targets for AEA include some types of opioid, dopamine, and muscarinic
acetylcholine receptors as well as a variety of ion channels (Hampson et al., 1998;
Kimura et al., 1998; Lagalwar et al., 1999; Pertwee et al., 2010).

7
Synthesis of endocannabinoids
Under normal conditions the tissue concentrations of AEA are low. Thus, for
example, in the mouse brain, a concentration of AEA of 13.6 ± 3.2 pmol/g,
representing 1.3% of the total content of the N-acylethanolamines (the class of lipids
to which AEA belongs) in the brain (Degn et al., 2007). In contrast, the
concentration of 2-AG is much higher (12 ± 1 nmol/g) (Degn et al., 2007), although
this represents the total concentration of this lipid, which is not only an
endocannabinoid but also a metabolic intermediate. Interstitial levels of basal AEA
and 2-AG in the nucleus accumbens are less divergent. In vivo microdialysis of this
region in C57/BL6 mice gave AEA and 2-AG concentrations in the microdialysate
of ~0.6 and ~4.5 nM, respectively. Selective blockade of the hydrolysis of 2-AG
(discussed below) increased 2-AG levels in the microdialysate without affecting
AEA levels (Long et al., 2009). AEA levels are not always low with respect to other
N-acylethanolamines. In the periimplantation mouse uterus, for example, AEA is the
most predominant of this class of lipids (Schmid et al., 1997).
Due to their highly lipophilic structure they are not stored in intracellular vesicles
but are synthesized on demand (Freund et al., 2003). Examples of stimuli that
trigger this response are listed in Table 3.
NAEs
AEA is a member of the N-acetylethanolamine (NAE) family, a large group of
bioactive lipids that also includes compounds such as N-oleoylethanolamine (OEA)
and N-palmitoylethanomamine (PEA). The latter two are not considered as
endocannabinoids since they lack affinity towards cannabinoid receptors. However,
they activate TRPV1 and PPARα and produce a range of biological effects such as
appetite suppression and analgesia (Fu et al., 2003; Lo Verme et al., 2005; Movahed
et al., 2005; Rodríguez de Fonseca et al., 2001).

8
Table 3. Examples of stimuli affecting endocannabinoid production.
Stimulus triggering endocannabinoid synthesis Reference
G-protein receptors coupled to phospholipase C
e.g. Metabotropic glutamate receptors
Subtype 1 (mGluR1) and 5 (mGluR5)
Histamine H1 receptors
(Maejima et al., 2001)
(Ohno-Shosaku et al., 2002)
(Zygmunt et al., 2013)
NMDA-receptors (Ohno-Shosaku et al., 2007)
Formalin (Walker et al., 1999)
Inflammation (Rettori et al., 2012)
Lipopolysaccharide (Hu et al., 2012)
Nerve injury
Neuropathic pain
Closed head injury (increased 2-AG levels)
Concussive head trauma (increased AEA levels)
Stroke
Parkinson's disease
Multiple Sclerosis
(Petrosino et al., 2007) (Mitrirattanakul et al., 2006)
(Panikashvili et al., 2001) (Hansen et al., 2001) (Schäbitz et al., 2002)
(Naccarato et al., 2010) (Pisani et al., 2005)
(Eljaschewitsch et al., 2006)
Several different pathways have been suggested to contribute to the synthesis of
NAEs from their corresponding N-acylphosphatidylethanolamine (NAPE)
precursors, which exist as a minor component of membrane phospholipids. The
most well studied pathway is a two-step enzymatic reaction involving NAPE-
phospholipase D (NAPE-PLD). The first step is the transfer of a fatty acyl chain

9
from the sn-1 position of glycerophospholipids to phosphatidylethanolamine,
catalyzed by Ca2+ dependent N-acyltransferase (Ca-NAT) (Ueda et al., 2010). The
second step comprises Ca2+-sensitive NAPE-PLD which catalyses the hydrolysis of
NAPE to produce the NAE (Okamoto et al., 2004). Even though NAEs are
produced on demand, NAPE-PLD seems to be kept in a constitutively active form
(Muccioli, 2010). A Ca2+-independent NAT, named iNAT has been cloned (Jin et
al., 2009). This enzyme is reported to be capable of forming NAPE. However, it is
unclear whether or not iNAT contributes to the biosynthesis of NAPE and NAEs in
vivo.
Two other pathways have been described. The first, which mainly has been
characterized in macrophages and mouse brain, is the two-step process involving
cleavage of NAPE by phospholipase C to yield phosphoanandamide. This is then
dephosphorylated by phosphatases to yield AEA (Liu et al., 2006). In the second
pathway, NAPE is first hydrolysed to lyso-NAPE by an enzyme with phospholipase
2 activity. NAE is then released from lyso-NAPE by a lysophospholipase D-like
enzyme (Natarajan et al., 1983).
2-arachidonoylglycerol
2-AG, similar to AEA, is produced in a stimulus-dependent fashion. Increased
intracellular Ca2+ triggers synthesis but the synthetic pathways differs (Bisogno et
al., 1997b; Kondo et al., 1998). 2-AG can be synthesized in a two-step reaction.
Diacylglycerol (DAG) is generated from phosphatidylinositol (PI), a minor
component of membrane phospholipids, by phospholipase C (PLC). DAG is then
hydrolysed by a diacylglycerol lipase (DAGL) to yield 2-AG. Inhibition of these
enzymes results in decreased 2-AG levels. In DAGL-alpha knockout mice the levels
of 2-AG are reduced by up to 80% in brain and spinal cord and about 60% in liver.
Depletion of the other subtype of DAGL, DAGL-beta, yields a 50% reduction of 2-
AG levels in brain but no difference in spinal cord (Gao et al., 2010). These lowered
concentrations of 2-AG result in a loss of retrograde endocannabinoid signalling in
the hippocampus of mice (Gao et al., 2010). The key intermediate DAG can also be

10
produced from phosphatidic acid by a phosphatidic acid hydrolase. This represents
an alternative pathway to DAG production (Muccioli, 2010).
A second pathway for 2-AG production involves 2-arachidonoyl-
lysophosphatidylinositol (lyso-PI) as intermediate. Phosphatidylinositol-preferring
phospholipase A1 produces the lyso-PI intermediate from PI. Secondly, a
lysophosphatidylinositol-selective phospholipase C generates 2-AG in a Ca2+-
independent manner in rat brain (Ueda et al., 1993). The relevance of this pathway
to generate 2-AG compared to the PLC-DAGL cascade is less clear. In addition,
DAG and 2-AG are intermediates in several pathways, one being arachidonic acid
release. It is likely, therefore, that not all the pathways leading to 2-AG are actually
involved in physiological endocannabinoid signalling (Muccioli, 2010).
Additionally, there are studies suggesting that all 2-AG is not necessarily produced
on demand but that there is a pool that is pre-synthesised and stored until needed
(Min et al., 2010; Zhang et al., 2011). However, this requires further study, and new
reports support the idea that on demand 2-AG biosynthesis is required for retrograde
endocannabinoid signalling (Hashimotodani et al., 2013).
The cellular processing of endocannabinoids
Termination of endocannabinoid signalling occurs through an uptake mechanism
followed primarily by enzymatic hydrolysis. The mechanism(s) responsible for
cellular uptake has not yet been clarified. There are studies of endocannabinoid
uptake showing a time- and temperature dependency, saturability but also an ATP-
and sodium independency (Chicca et al., 2012; Di Marzo et al., 1994; Hillard et al.,
1997). In addition, the fact that AEA analogues such as AM404 inhibit the uptake
process indicates the presence of a membrane transporter (Beltramo et al., 1997).
All these characteristics are consistent with a facilitated transport mechanism.
Nonetheless, at the time of writing of this thesis, a plasma membrane transporter
protein has still not been cloned.

11
If AEA uptake is driven by passive transport, e.g. in absence of a transporter protein,
the uptake will cease once the extracellular and intracellular levels of AEA reach
equilibrium. However, this equilibrium can be affected both by sequestration of the
intracellular AEA, such has been described in lipid droplets (Kaczocha et al., 2010;
Oddi et al., 2008), and/or by intracellular metabolism of endocannabinoids (the
degradation of endocannabinoids are described in more detail in the section below).
Fatty acid amide hydrolase (FAAH) is the key enzyme responsible for the
hydrolysis of AEA (Cravatt et al., 1996; Deutsch et al., 1993). At present, there are
clearly disagreements in the literature concerning the importance of FAAH in
controlling the cellular uptake of AEA. In FAAH-containing neuroblastoma (N18),
glioma (C6) and rat basophilic leukaemia cells, the net uptake of AEA is decreased
in the presence of FAAH inhibitors (Day et al., 2001; Deutsch et al., 2001).
However, compounds such as AM404, which are structurally similar to AEA,
decrease AEA uptake in cells lacking FAAH (Fegley et al., 2004; Ligresti et al.,
2004; Ortega-Gutiérrez et al., 2004). One explanation for these differences is that
they are due to cellular differences, but it might also be a matter of methodological
artefacts. The cellular accumulation of 2-AG is not dependent upon its subsequent
metabolism in RBL2H3, AT-1 and PC3 cells, but may gate the uptake in Neuro-2a
cells (Fowler et al., 2008).
Due to their highly lipophilic nature, endocannabinoids require intracellular
transporters to carry them throughout the cytoplasm of the cell to their catabolic
enzymes and/or intracellular targets (the AEA-binding site of the TRPV1 receptor,
for example, is located on the intracellular face of the receptor) once they have
crossed the plasma membrane. Several intracellular AEA carrier proteins have been
proposed including fatty acid binding protein (FABP), heat shock protein 70,
albumin and FAAH-like anandamide transporter (FLAT) (Bojesen et al., 2003; Fu et
al., 2012; Kaczocha et al., 2009; Oddi et al., 2009), although such a role of FLAT
has been questioned (Leung et al., 2013). In an effort to reduce the cellular uptake of
AEA, blockade of intracellular carrier proteins is in theory an alternative to FAAH
inhibitors. Recently, Berger et al. identified SB-FI-26 as a novel inhibitor of
FABP5. SB-FI-26 reduces FABP-mediated AEA uptake in HeLa cells and produces

12
antinociceptive and anti-inflammatory effects in mice (Berger et al., 2012).
Recently, ARN272 was found to block AEA binding to FLAT selectively. Systemic
administration of this compound in mice caused a dose-dependent reduction of
formalin-induced pain behaviour. In addition, it produced anti-inflammatory and
anti-hyperalgesic effects when injected intraplantarally (Fu et al., 2012).
Degradation of AEA
N-Acylethanolamines are hydrolysed to free fatty acids and ethanolamines. The key
enzyme in this process is the intracellular enzyme fatty acid amide hydrolase
(FAAH) (Cravatt et al., 1996; Deutsch et al., 1993). FAAH is a 63kDa membrane-
bound serine hydrolyse enzyme distributed widely throughout the body. Examples
of tissues expressing FAAH is brain, liver, testis, uterus and spleen (Bobrov et al.,
2000; Cravatt et al., 1996; Deutsch et al., 1993; Maccarrone et al., 2000; Watanabe
et al., 1998). In the brain, the distribution of FAAH and CB1 receptors are
complementary, CB1 is principally located presynaptically in contrast to the FAAH,
which is postsynaptic (Egertová et al., 1998). FAAH has a wide substrate specificity
and is capable of metabolising not only AEA but also other fatty acid amides such as
PEA and oleamide as well as 2-AG (Bisogno et al., 1997a; Cravatt et al., 1996;
Goparaju et al., 1999; Lang et al., 1999; Maccarrone et al., 1998; Tiger et al., 2000).
In 2006, an isoenzyme of FAAH, referred to as FAAH-2, with ~20% sequence
identity at amino acid level was found to be expressed in humans but not rodents
(Wei et al., 2006).
Early studies of FAAH demonstrated that it is inhibited by non-selective compounds
such as phenylmethylsulfonyl fluoride (Deutsch et al., 1993) and compounds
structurally related to the substrates of FAAH such as arachidoloyltrifluoromethyl
ketones (Boger et al., 1999; Koutek et al., 1994). The carbamate-type inhibitor
URB597 is frequently used as a selective FAAH inhibitor (Kathuria et al., 2003). In
rodents, URB597 elevates the AEA levels, induces antidepressant-like effects,
reduces blood pressure, analgesia and reduces inflammation (Adamczyk et al., 2008;
Bátkai et al., 2004; Fegley et al., 2005; Holt et al., 2005; Jayamanne et al., 2006;
Kathuria et al., 2003). There are FAAH inhibitors in Phase I and Phase II clinical

13
trials. PF-04457845, which possesses antinociceptive effect in rodents (Ahn et al.,
2011), has recently been investigated in a randomized, placebo-controlled clinical
trial of patients with osteoarthritis of the knee. Despite increasing levels of four
NAEs in the plasma at the doses used, it did not have an analgesic effect (Huggins et
al., 2012). One possible explanation is at least in part to cyclooxygenase-2 (COX-2)
metabolism of AEA, i.e. that the increased AEA resulting from the FAAH inhibition
is rerouted along the COX-2 metabolic pathway (see section below).
Other enzymes responsible for endocannabinoid inactivation
Apart from FAAH, AEA can also be metabolised by several other enzymes (see Fig.
2). COX-2, which is expressed during inflammation, converts AEA to several
prostaglandin ethanolamides (PG-EAs) (Kozak et al., 2002a; Yu et al., 1997). The
major metabolites from LOX- oxidation are hydroxylated derivatives of AEA. The
lipooxygenase (LOX) derivate 12-hydroxyanandamide is capable of binding to the
CB1 receptor with twice the affinity that of AEA (Hampson et al., 1995).
Cytochrome P450 oxygenases metabolises arachidonic acid to form
epoxyeicosatrienoic acids (EETs) and hydroxyeicosatetraenoic acid (HETEs) but
also to metabolise AEA in both mouse and human (Bornheim et al., 1995; Snider et
al., 2007). One of the many P450-derived metabolites of AEA, 5,6-EET-EA has
been show to bind and functionally activate recombinant human CB2 receptor
(Snider et al., 2009).
Fig. 2. The main degrading enzymes and products of AEA. EA, Ethanolamine; AA,
arachi-donic acid; PG-EA, prostaglandin ethanolamide; HPETE, hydroper-
oxyeicosatetraenoic acid; HETE, hydroxyeicosatetraenoic acid; EET, epoxyeicosatrienoic
acid
Anandamide(
FAAH( COX.2( LOX( P450(
AA(EA( PG.EA( HPETE( HETE(EET(

14
Degradation of 2-AG
The main hydrolysing enzyme converting 2-AG to glycerol and arachidonic acid is
monoacylglycerol lipase (MGL). This enzyme is a 33kDa protein originally cloned
and purified in adipose tissue (Karlsson et al., 1997). Later in 2002, northern blot
and in situ hybridization analyses revealed that MGL mRNA is heterogeneously
expressed in the rat brain, with highest levels in regions where CB1 receptors are
also present (hippocampus, cortex, anterior thalamus and cerebellum) (Dinh et al.,
2002). Like CB1 receptors, it is predominantly localized to presynaptic axon
terminals (Gulyas et al., 2004). Its primary mechanism is inactivation of 2-AG
(Dinh et al., 2002) and functional proteomic approaches have been used to explore
different 2-AG hydrolases in mouse brain membrane homogenates. Under the
conditions used, approximately 85% of brain 2-AG is hydrolysed by MGL, the
remaining 15% is mostly catalysed by the serine hydrolases ABHD6 and ABHD12
(Blankman et al., 2007). MGL has been described to be both cytosolic- and
membrane bound (Sakurada et al., 1981) with a pH optimum of ~8 (Tornqvist et al.,
1976). MGL is expressed in a number of tissues including adipose tissue, adrenal
gland, ovary, heart, brain, spleen, lung, liver, skeletal muscle, kidney and testis
(Karlsson et al., 1997).
At the time when this thesis work was started, selective inhibitors of MGL were not
available. Early studies suggested that MGL activity is sensitive to general serine
hydrolase inhibitors such as phenylmethanesulfonyl fluoride (PMSF), arachidonoyl
trifluoromethylketone, and hexadecysulfonylfluoride (Ghafouri et al., 2004; Saario
et al., 2004), compounds also inhibiting FAAH. It has also been implied that MGL
is inhibited by non-specific sulfhydryl agents, including p-chloromercuribenzoic
acid and N- ethlymaleimide (Sakurada et al., 1981; Tornqvist et al., 1976). The
carbamate compound URB602 was initially reported as the first selective inhibitor
of MGL (Hohmann et al., 2005). However its selectivity has been a matter of debate
since it has been shown to be equally potent against FAAH in vitro (Vandevoorde et
al., 2007). In 2009, JZL184, a novel highly potent selective MGL inhibitor was
reported (Long et al., 2009). When administrated to mice, JZL184 raised brain 2-
AG levels by eight-fold without altering AEA levels. In agreement with previous

15
studies (Blankman et al., 2007), brain membranes maintained a residual of ~15% 2-
AG hydrolysis activity even at the highest concentrations of JZL184 tested (Long et
al., 2009). JZL184 showed to exhibit analgesic properties in different pain assays
including the acetic acid writhing test of visceral pain, tail-immersion test of acute
thermal pain sensation and the formalin test of noxious chemical pain. The CB1
antagonist Rimonabant blocked these effects. However, in contrast to FAAH
inhibitors JZL184 induced hypothermia and hypomotility but not catalepsy (Long et
al., 2009).
Other enzymes responsible for 2-AG metabolism
As with AEA, 2-AG can also be metabolised by FAAH, COX-2, LOX and CYP
(Goparaju et al., 1998; Guindon et al., 2008; Hu et al., 2008; Kozak et al., 2002a;
Kozak et al., 2002b; Kozak et al., 2002c). As mentioned previously, Blankman et al.
confirmed that MGL is the key enzyme (~85% of hydrolysis) responsible for
metabolism of 2-AG in mouse brain whereas ABHD6 and 12 is responsible for
about 4% and 9% respectively. It is suggested that based on their distinct cellular
and/or subcellular localization, MGL, ABHD6 and 12 regulate distinct pools of 2-
AG in the brain. While MGL is a soluble enzyme that associates with membranes,
ABHD6 and ABHD12 are integral membrane enzymes (ABHD6 facing the
cytoplasm and ABHD12 the extracellular compartments of the cell) (Blankman et
al., 2007).
The physiological and pathophysiological roles of ABHD6 and 12 have not been
well examined but mutations of the ABHD12 gene cause the human
neurodegenerative disease PHARC (polyneuropathy, hearing loss, ataxia, retinitis
pigmentosa, and cataract) (Fiskerstrand et al., 2010). Recently ABHD6 has also
been shown to control 2-AG accumulation in Neuro2A cells, which lack MGL (Hsu
et al., 2012).
The importance of the COX-2 pathway in endocannabinoid signalling
As mentioned above, COX-2 converts AEA to several prostaglandin ethanolamides
(PG-EAs). This pathway, first demonstrated in vitro by (Yu et al., 1997) is of

16
importance in vivo, particularly under inflammatory conditions (Duggan et al., 2011;
Gatta et al., 2012), or in the absence of FAAH following a priming dose of AEA
(Weber et al., 2004). Indirect evidence for the importance of COX-2 as an
endocannabinoid metabolic enzyme has been obtained in studies of AEA uptake by
the mouse brain in vivo: inhibition of COX-2 resulted in higher AEA uptake and
stability (Glaser et al., 2010). AEA and 2-AG levels are also higher in the brains of
COX-2 knockout mice compared to wild type animals (Hermanson et al., 2013).
Inhibitors of COX-2 has also been shown to potentiate retrograde signalling in
hippocampal pyramidal cells, e.g. activate presynaptic CB1 receptors and transiently
reduce GABAergic transmission, a process termed depolarization induced
suppression of inhibition (Kim et al., 2004). Recently, an indomethacin analogue,
LM-4131, has been identified as a substrate-selective COX-2 inhibitor, inhibiting
the oxygenation of AEA and 2-AG, but not arachidonic acid, by this enzyme form.
LM-4131 increased AEA levels without affecting central or peripheral
prostaglandins. It also had effect on 2-AG levels, albeit more modest. Furthermore,
LM-4131 treatment did not further increase AEA and 2-AG levels in COX-2
knockout mice over and above the effects produced by the genetic deletion per se. In
line with its substrate selectivity, LM-4131 did not affect levels of arachidonic acid
or prostaglandin levels in the brain, in contrast to indomethacin, which increased
arachidonic acid levels and decreased prostaglandin levels as expected for a general
inhibition of COX-2. Other N-acylethanolamines such as palmitoylethanolamine,
were not affected. The authors also found that LM-4131 decreased anxiety-like
behaviours in a Rimonabant-sensitive manner, suggesting that the augmentation of
endocannabinoid signalling by the compound in vivo is sufficient to produce
behavioural effects (Hermanson et al., 2013).
Endocannabinoids and pain
According to the International Association for the Study of Pain (IASP), pain is
defined as an unpleasant sensory and emotional experience associated with actual or
potential tissue damage, or described in terms of such damage. IASP have further
classified pain in to different categories. Nociceptive pain is described as pain that

17
arises from actual or threatened damage to non-neural tissue and is due to the
activation of nociceptors, whereas neuropathic pain is caused by a lesion or disease
of the somatosensory nervous system. Pain resulting from tissue inflammation is
referred to as inflammatory pain. Inflammation gives rise to peripheral sensitization,
which is defined as increased responsiveness of nociceptive neurons to their normal
input, and/or recruitment of a response to normally sub-threshold inputs. Pain that
persists more than 3 months is defined as chronic pain (www.iasp-pain.org).
Chronic pain is common and affects quality of life negatively (Breivik et al., 2006).
Studies using experimental animals show that the anti-nociceptive effects of
cannabinoids are not only centrally mediated, but that spinal and peripheral
cannabinoid receptors are involved (Guindon et al., 2009). Support for supraspinal
sites for the analgesic action of cannabinoids was found in studies in which synthetic
CB receptor agonists were injected into different brain regions, including the
periaqueductal grey (PAG) (Lichtman et al., 1996; Martin et al., 1995), thalamus
(Martin et al., 1996) and amygdala (Marsicano et al., 2002; Martin et al., 1999)
among others, in rat. These studies suggest that cannabinoids might act in the
midbrain to produce antinociception under physiological conditions. In 1999,
Walker et al. found that electric stimulation of the dorsolateral PAG produced
antinociception in the tail-flick test and increased levels of endogenous AEA in this
area as measured by microdialysis. This analgesic effect was blocked by
Rimonabant (Walker et al., 1999). Intraplantar administration of formalin was also
shown to increase levels of endogenous AEA in the dorsolateral PAG (Walker et al.,
1999). Endocannabinoid levels are altered in specific brain regions following nerve
injury. For example, injury of the sciatic nerve increases the levels of AEA and 2-
AG in the PAG as well as in the spinal cord (Petrosino et al., 2007).
A number of animal studies have demonstrated that cannabinoids act at the spinal
level to modulate pain. Intrathecal administration of cannabinoids produces
antinociception and supresses nociceptive neuronal activity (Hohmann et al., 1998;
Smith et al., 1992; Welch et al., 1995; Yaksh, 1981). Additionally, the CB2-selective
agonist AM1241 supresses C-fibre-evoked responses of dorsal horn neurons in rats

18
in the presence of inflammation (Nackley et al., 2004). Also, CB1 receptors are
observed to be up-regulated in the spinal cord following nerve injury (Lim et al.,
2003; Sagar et al., 2005). Non-opioid, stress-induced analgesia increases 2-AG but
not AEA levels in the lumbar spinal cord. Spinal administration of inhibitors of
endocannabinoid hydrolysis (URB597 for FAAH) and (URB602 for MGL)
enhanced stress-induced analgesia through a CB1 mediated mechanism (Suplita et
al., 2006). Spinal cord levels of AEA and 2-AG are increased following cisplatin-
induced peripheral neuropathy. Also, inhibition of FAAH (URB937, URB597) and
MGL (JZL184) suppresses cisplatin-evoked mechanical and cold allodynia
(Guindon et al., 2013).
Evidence for peripheral involvement of cannabinoids in the modulation of pain has
been presented in numerous animal pain models. Peripheral, but not systemic
administration of AEA inhibits oedema, capsaicin-evoked plasma extravasation into
the hind paw and carrageenan-induced thermal induction of hyperalgesia. Peripheral
administration also reduces hyperalgesia after its development via interaction with
CB1 receptors, as revealed by using the CB1 antagonist Rimonabant (Richardson et
al., 1998). In the formalin-evoked pain model AEA prevents pain when injected
locally (intraplantar), an effect which was blocked by Rimonabant. A further
experiment showed that 94% of the injected AEA remained associated with the
injected paw. This indicates that AEA inhibits nociception after formalin injection
by activating peripheral CB1 receptors located on sensory neurons involved in pain
transmission (Calignano et al., 1998). In inflamed paw following carrageean-
induced inflammation a decrease in FAAH activity is seen compared to the non-
inflamed mice. Also intraperitoneal injections of the FAAH inhibitor URB597
reduce oedema formation (Holt et al., 2005). Intraplantar administration of AEA and
ibuprofen have antinociceptive effects in rat paw formalin test. The compounds
showed synergistic effects that were completely antagonised by the CB1 antagonist
AM251 (Guindon et al., 2006).
The peripheral contribution of endocannabinoid-mediated analgesia has been
investigated further by generation of transgenic mice lacking CB1 receptors in

19
nociceptors, preserving expression in the spinal neurons, brain and all other organs.
These genetically modified mice shows that specific loss of CB1 in nociceptors leads
to reduced response to noxious heat, reduced response thresholds to mechanical
stimuli and greater responses to intraplantar injections of capsaicin and formalin
(Agarwal et al., 2007). Additionally, low doses of a peripherally applied synthetic
cannabinoid reduced inflammatory and neuropathic pain, an effect that was nearly
completely lost on nociceptor-specific deletion of CB1 receptors (Agarwal et al.,
2007). Following the induction of neuropathy, e.g. by spinal nerve ligation, both
AEA and 2-AG are increased only in the ipsilateral DRG (Mitrirattanakul et al.,
2006). FAAH has been found in the rat DRG, spinal cord and peripheral nerve tissue
(Lever et al., 2009). An increase of FAAH in the ipsilateral DRG occurred after
spinal nerve lesion but not after chronic inflammation of the rat hind paw 2 d after
injection of complete Freund's adjuvant (Lever et al., 2009). This reveals the
location of FAAH in neural tissue involved in peripheral nociception and provides
targets for manipulation of the endocannabinoid system for the treatment of pain.
As mentioned earlier, AEA can also activate the TRPV1 receptor (Zygmunt et al.,
1999). N-arachidonoyl-5-hydroxytryptamine (AA-5-HT), a compound with a “dual”
ability to inhibit the fatty acid amide hydrolase (FAAH) and to antagonize the
TRPV1 receptors shows strong analgesic activity after systemic administration in
acute or chronic pain models in rodents (Maione et al., 2007). Intra-periaqueductal
grey (PAG) administration of the compound significantly increased basal levels of
2-AG and OEA (which activates TRPV1 receptors) but not those of AEA. Injection
of AA-5-HT also produced anti-nociceptive effects in the formalin model of pain, an
effect erased by co-injection by either AM251 (a CB1 antagonist) or I-RTX (a
TRPV1 antagonist) (de Novellis et al., 2008). In 2013, Zygmunt et al. demonstrated
that 2-AG and 1-AG activate the TRPV1 receptor. Additionally, the MGL inhibitor
JZL184 produced a TRPV1-dependent anti-nociceptive effect in the first phase of
the mouse formalin test (Zygmunt et al., 2013).

20
Inhibition of endocannabinoid hydrolysis as a therapeutic target for the treatment
of pain
Although AEA binds to CB1 receptors and has been implicated in the suppression of
pain, its rapid degradation by FAAH is a challenge in investigation the physiological
functions of this endocannabinoid. There is evidence that FAAH knockout mice
have a 15-fold increase of AEA levels in brain and display reduced pain sensation
that is reversed by Rimonabant (Cravatt et al., 2001). In a mouse collagen-induced
arthritis (CIA) model, FAAH knockout mice displayed decreased severity of CIA
and associated hyperalgesia (Kinsey et al., 2011). However, as mentioned in a
previous section, the irreversible FAAH inhibitor PF-004457845 has been
investigated in a randomized, placebo-controlled clinical trial of patients with
osteoarthritis of the knee. Although elevation of AEA plasma levels were seen, this
inhibitor did not produce significant analgesia in the patient population investigated
(Huggins et al., 2012). One explanation for this might be the choice of outcome
measures (Rice et al., 2008). Another explanation is that AEA uses metabolic
pathways other than FAAH in the presence of an FAAH inhibitor. AEA and 2-AG
are also substrates of COX-2 (see above) and FAAH inhibitors have been shown to
give synergistic analgesic interactions together with non-steroidal anti-inflammatory
drugs in experimental animals (Naidu et al., 2009; Sasso et al., 2012). Further, the
FAAH inhibition reduced the gastrointestinal disturbances produced by the non-
steroidal anti-inflammatory drugs (Naidu et al., 2009; Sasso et al., 2012).
Most of the research examining the role of endocannabinoid catabolic enzymes in
nociception has focused on FAAH, largely due to the lack of a selective MGL
inhibitor. At the start of the present thesis, the only compound available was
URB602 (Hohmann et al., 2005), the selectivity of which had been questioned
(Vandevoorde et al., 2007). The development of JZL184 (Long et al., 2009) opened
up for experiments to evaluate the role of 2-AG in pain perception. JZL184 when
administered acutely increases 2-AG brain levels, without altering AEA brain levels
(Long et al., 2009). Systemic administration of JZL184 has been demonstrated to
reduce nociceptive responses in several different animal models including tail
withdrawal, formalin, acetic acid stretching tests and chronic constriction injury

21
model of neuropathic pain in mice (Kinsey et al., 2009; Long et al., 2009).
Intraplantar injection of JZL184 produces antinociception in the formalin test and in
the capsaicin model of nociception (Guindon et al., 2011; Spradley et al., 2010).
Additionally, JZL184 significantly inhibits inflammatory pain in the carrageenan
assay and more specifically, JZL184 attenuated the development of paw oedema and
mechanical allodynia. The compound also reversed oedema and allodynia when
administered after carrageenan (Ghosh et al., 2013).
There are drugs used in the clinic, such as Sativex®, that affect the cannabinoid
system. However, compounds with one primary mechanism of action can have
additional biological targets, including components of the endocannabinoid system.
One interesting approach in the design of novel inhibitors of endocannabinoid
metabolism is the use of clinically known compounds as a template. This has an
advantage that clinical data is available, at least for the starting compound. An
example of this is ibuprofen, which has a primary action to inhibit COX, but which
has been demonstrated to also inhibit FAAH (Fowler et al., 1997). Moreover,
studies have shown that paracetamol (acetaminoprofen) can be metabolised in the
brain into the anandamide uptake inhibitor AM404 in a FAAH-dependent manner
(Högestätt et al., 2005). Ibuprofen and paracetamol analogues with low to sub-
micromolar potency have been identified (De Wael et al., 2010; Holt et al., 2007;
Onnis et al., 2010; Patel et al., 2013) and one of these, the N-(3-methylpyridin-2-
yl)amide derivative of ibuprofen, combines an FAAH inhibitory effect with a
substrate-selective inhibition of COX-2 (Fowler et al., 2013).
Endocannabinoid system in human pain states
Current data from experimental animals, summarised above, suggest that the
endocannabinoid system is dysfunctional in pain states. As mentioned earlier, there
is evidence for this in animal pain models but at the time when this thesis was
started little was, and still is, known about the situation in human pain. In 2008 it
was reported that CB1 receptor expression in pancreatic nerves was negatively
correlated to pain symptoms of patients with pancreatic cancer (Michalski et al.,
2008). One year later, in 2009, it was stated that patients with complex regional pain

22
syndrome show higher AEA plasma concentrations compared to age- and sex-
matched controls (Kaufmann et al., 2009). Additionally, a study suggests that
increased CB1 receptor immunoreactive nerve fibres may be related to bladder pain
in painful bladder syndrome (Mukerji et al., 2010). A negative correlation of CB2
mRNA in human spinal cord is seen in patients with joint chondropathy (Burston et
al., 2013).
Chronic pain in the human Achilles tendon
The Achilles tendon is the strongest tendon in the body (O'Brien, 1992). When there
is chronic pain, swelling and impaired function in the Achilles tendon, the condition
is referred to as tendinopathy (Khan et al., 1999). If patients with tendon pain,
swelling and impaired function also demonstrate structural tissue changes, the
condition is termed tendinosis (Alfredson et al., 2005). Biopsies of symptomatic
tendons show changes in the appearance of the tenocytes, such as rounded nuclei
and a less spindle-shaped appearance, hypercellularity, neovascularization,
degeneration and disordered arrangement of collagen fibres (Khan et al., 1999;
Åström et al., 1995). There are no signs of classic inflammation in chronic
tendinosis, i.e. presence of inflammatory cells and elevated prostaglandin levels
(Alfredson et al., 1999; Khan et al., 1999). The aetiology of Achilles tendinopathy is
still unclear although several molecular candidates have been identified and
proposed as mediators of the pain in Achilles tendinosis (Riley, 2008). Given the
ubiquity of the endocannabinoid system and its role in pain, a dysregulation of the
endocannabinoid system might also occur in Achilles tendinosis.

23
Aims of the thesis
It is known that the endocannabinoid system plays an important role in the control
of pain and that activation of the cannabinoid receptors shows clinical utility in a
variety of pain states. However, there are many gaps in the knowledge about the
situation in human pain syndromes. Thus, nothing is known about the potential
involvement of CB1 receptors in Achilles tendinosis. There is also a need for a better
understanding of how the endocannabinoid system can be modulated
pharmacologically in order to strengthen existing signalling patterns. One approach
is to investigate compounds that are already in clinical use, to determine whether
they inhibit endocannabinoid metabolism as an additional effect. Such an approach
can then form the basis of structure-activity studies designed to optimise the
endocannabinoid component of the drug in question. An example of this is
ibuprofen, which in addition to their effects upon COX, inhibits FAAH (Fowler et
al., 1997). The N-(3-methylpyridin-2-yl)amide analogue of ibuprofen is 2-3 orders
of magnitude more potent as an inhibitor of FAAH but retains the COX inhibitory
properties of the parent compound (Holt et al., 2007). The aims of the thesis are as
follows:
Paper I: To evaluate if CB1 receptors are expressed in Achilles tendons, and
whether this expression is anomalous in Achilles tendinosis.
Paper II: To determine whether the N-(3-methylpyridin-2-yl)amide analogue of
flurbiprofen has greater potency than the corresponding ibuprofen analogue as an
FAAH inhibitor, and whether it shows a substrate-selective inhibition of COX-2.
Paper III: To determine whether, and how, the antifungal agent ketoconazole
inhibits the cellular uptake of AEA at pharmacologically relevant concentrations.
Paper IV: To determine whether the expression level of FAAH is an absolute
determinant of the sensitivity of AEA uptake to FAAH inhibition.
Paper V: To determine whether compounds inhibiting the activity of MGL can be
identified from a screen of a library of drugs and biologically active compounds.

24
Methodological considerations
During the work of this thesis, several methods have been used. The details of each
method are given in the original papers and are summarized in this section.
Subjects (Paper I)
The subjects participating in the study were from a group of patients suffering from
Achilles tendinosis or healthy controls. In total, samples from 24 individuals; 11
males and 13 females (Table 4) were analysed. All participants were healthy, free
from medication, non-smokers and choosed to be included in the research program
on voluntary basis.
Achilles tendinosis patients
The Achilles tendinosis group consisted of 17 patients suffering from chronic
painful mid-portion Achilles tendinosis verified by ultrasonography and clinical
examination.
Inclusion criteria for this group were:
o Pain in the Achilles tendon for more than 3 months
o Clinical symptoms: tender thickening in the Achilles tendon mid-portion
and ultrasound verified tendinosis changes corresponding to the region with
clinical findings
Exclusion criteria were:
o Diseases or injuries causing radiating pain in the lower limb
o Smokers
o Acute or chronic inflammatory diseases
There were and 9 females (aged 47, 52, 53, 55, 59, 61, 61, 68 and 75 years) and 8
males (ages 28, 28, 29, 36, 53, 58, 60 and 70 years) in this group.

25
Controls
This group included 7 individuals (4 females, aged 21, 47, 47 and 47 years; 3 males
ages 39, 39 and 46 years) with no history of pain symptoms from their Achilles
tendons. Ultrasonography showed normal tendons.
Inclusion criteria for this group were:
o No diseases or injuries affecting the lower extremities
o Non-smokers
o No ongoing or previous pain in the Achilles tendon
o Normal findings on ultrasonography
o Good health and free from medication
Table 4. Overview of subjects for the study. M/F: Male/Female, Age: Mean age
Subjects 24
M/F 11/13
Age (range) 48 (21-70)
Tendinosis 17
M/F 8/9
Age (range) 51 (28-70)
Controls 7
M/F 3 / 4
Age (range) 41 (21-47)

26
Ethics (Paper I)
The Committee of Ethics at the Faculty of Medicine, Umeå University and the
Regional Ethical Review Board in Umeå approved the study. All participants read
an explanatory statement and received a verbal summary of the project before they
gave verbal consent to participate in the research. All procedures followed the
principles of the Declaration of Helsinki.
Sampling, fixation and sectioning (Paper I)
All biopsies were taken during surgical treatment and under strict sterile condition.
In the tendinosis group, tendon tissue (macroscopically abnormal) was taken
through a longitudinal incision lateral to the tendon mid-portion from the ventral
part of the Achilles tendon. The samples were taken from different depths of the
tendon and were approximately 2 mm wide and 1–5 mm long. Biopsies from the
control group (same size as from tendinosis patients) were carefully taken from the
dorsal part of the tendon using a longitudinal plain incision under local anaesthesia.
The dorsal part of the tendon was chosen for ethical and practical reasons.
The samples were fixed overnight at 4 °C in a solution of 4 % formaldehyde in 0.1
M phosphate buffer, pH 7.0 and were thoroughly washed in Tyrode’s solution
containing 10% sucrose. The samples were then mounted on thin cardboard in OCT
embedding medium and frozen at -80 °C until sectioning.
The samples were cut in a series of sections (7 µm thick) using a cryostat. The
sections were mounted on slides, pre-coated with Crome Alum Gelatin solution,
dried and thereafter used for immunohistochemistry. Reference tissues (human
colonic and rat dorsal root ganglion tissue), which had been fixed and handled in the
same way as the tendon samples were examined in parallel.

27
Immunofluorescence and control staining (Paper I)
Briefly, sections were initially treated with acid potassium permanganate in order to
enhance the visualization of specific immunofluorescence reaction sites (Hansson et
al., 1995) prior to incubation for 20 min in a 1 % solution of Triton X-100. After
rinsing three times in PBS the sections were incubated in 5 % normal swine serum
supplemented with 0.1 % bovine serum albumin (BSA) for 15 min in a humid
environment in room temperature. The samples were thereafter incubated with the
primary antibody for 60 min at 37 °C in a humid environment. Prior to a 30 min
incubation at 37 °C with the secondary antibody the samples were again incubated
with normal swine serum. This antibody corresponded to tetramethylrhodamine
isothiocyanate (TRITC)-conjugated swine antirabbit IgG.
All sections were evaluated for the intensity of CB1 receptor immunoreactivity in the
normal and Achilles tendinosis tendons by two independent investigators (this
author and Prof. Sture Forsgren). Tenocytes were scored for immunoreactive
intensity (0–3 where 0 is absent and 3 is high), and the average value was taken.
To confirm the validity of the method, parallel staining on reference tissue (human
colon and dorsal root ganglia) were performed. For control purposes, the primary
CB1 antibody was pre-absorbed overnight at 4 °C with its immunogenic peptide
(20–100 mg/ml; ab50542; Abcam, Cambridge, UK) prior to incubation on the
sections. As an additional control, the primary antibody was substituted with
PBS/BSA. The PGP9.5 (protein gene product 9.5) (see Table 5) antibody for
staining nerve fibres is a well-used antibody in the laboratory and in numerous
studies by others (Andersson et al., 2007; Bjur et al., 2005).
Hematoxylin-eosin staining
To outline tissue morphology, hematoxylin-eosin staining of parallel sections to
those processed for immunohistochemistry was performed. Hematoxylin gives a
blue colour to basophilic structures such as cell nuclei and ribosomes whereas eosin
28
colours eosinophilic structures such as intra- and extracellular proteins in different
shades of red.
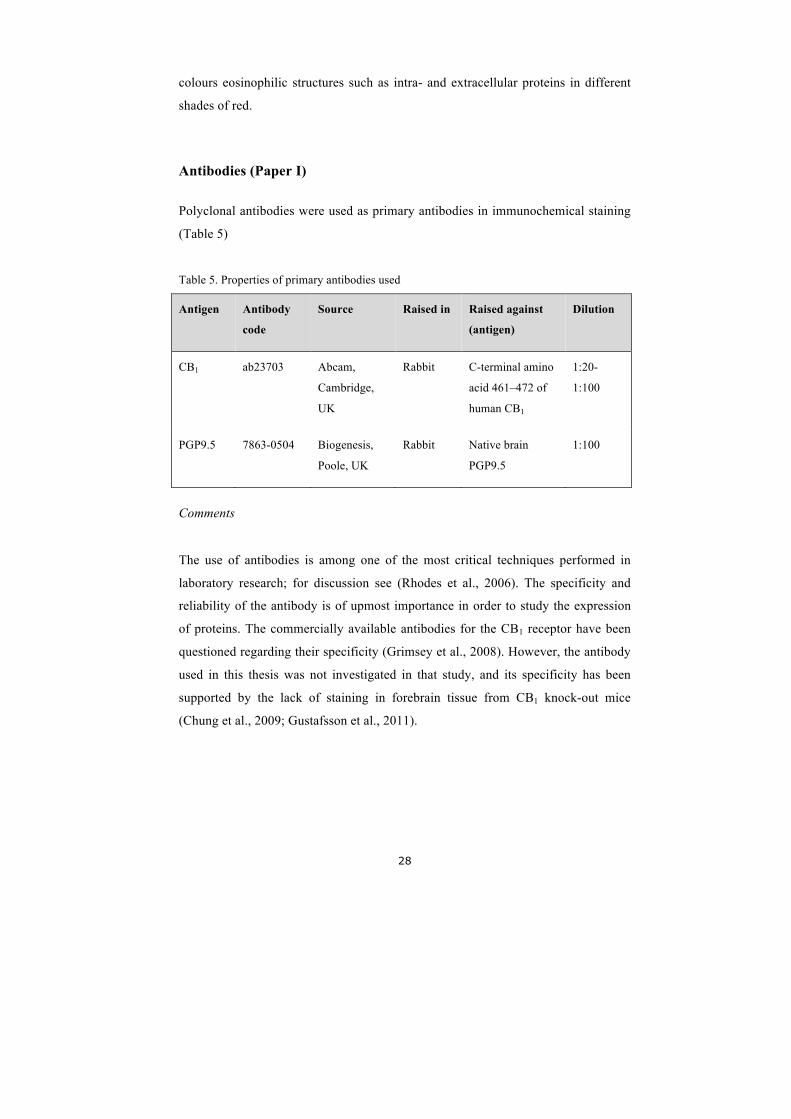
Antibodies (Paper I)
Polyclonal antibodies were used as primary antibodies in immunochemical staining
(Table 5)
Table 5. Properties of primary antibodies used
Antigen Antibody
code
Source Raised in Raised against
(antigen)
Dilution
CB1 ab23703 Abcam,
Cambridge,
UK
Rabbit C-terminal amino
acid 461–472 of
human CB1
1:20-
1:100
PGP9.5 7863-0504 Biogenesis,
Poole, UK
Rabbit Native brain
PGP9.5
1:100
Comments
The use of antibodies is among one of the most critical techniques performed in
laboratory research; for discussion see (Rhodes et al., 2006). The specificity and
reliability of the antibody is of upmost importance in order to study the expression
of proteins. The commercially available antibodies for the CB1 receptor have been
questioned regarding their specificity (Grimsey et al., 2008). However, the antibody
used in this thesis was not investigated in that study, and its specificity has been
supported by the lack of staining in forebrain tissue from CB1 knock-out mice
(Chung et al., 2009; Gustafsson et al., 2011).

29
Assay for FAAH and MGL activities (Paper II, III, IV, V)
The assays used for the activity of either FAAH or MGL are based on the
measurement of [3H]ethanolamine formed from hydrolysis of [3H]AEA and
[3H]glycerol formed from [3H]2-OG hydrolysis, respectively (Dinh et al., 2002;
Omeir et al., 1995). The original assay described by Omeir et al., 1995 used
chloroform:methanol extraction for separation of the water-soluble molecules
(products) from lipophilic molecules (substrates). In the present form, the
hydrophilic products ([3H]ethanolamine and [3H]glycerol) were separated from the
lipophilic substrates ([3H]AEA and [3H]2-OG) by addition of charcoal:HCl (Boldrup
et al., 2004).
For FAAH assays, the homogenates used were prepared from adult Sprague-Dawley
or Wistar rats. Frozen brains (minus cerebellum) were thawed, homogenized and
thereafter centrifuged at 4 °C. The pellets were resuspended in buffer followed by
recentrifugation and a second resuspension in buffer. After incubation at 37 °C, in
order to hydrolyse all endogenous FAAH substrates, the pellets were again
centrifuged, resuspended in 50 mM Tris-HCl buffer (pH 7.4) and frozen at -80 °C in
aliquots until used.
For cell homogenates, cells were grown in culturing flasks. Cells were washed two
times with ice-cold PBS prior to collection using a rubber policeman. Cells were
centrifuged, resuspended in 10 mM Tris-HCl buffer and stored at -80 °C until use.
The protein concentration was determined using the Bradford assay with BSA as
standard (Bradford, 1976).
For MGL assays, homogenates of adult Sprague-Dawley or Wistar rats cerebella
were prepared in 50 mM sodium phosphate buffer (pH 8.0) containing 0.32 M
sucrose and centrifuged for one hour at 4 °C. The supernatants (cytosolic fraction)
were saved and the pellets (membrane fraction) were resuspended in 50 mM sodium
phosphate buffer (pH 8.0). The protein concentration of all homogenates was

30
determined by the method of Lowry or Bradford using bovine serum albumin as
standard (Bradford, 1976; Lowry et al., 1951).
For both the FAAH and the MGL assay, enzyme (homogenates or cytosolic
fraction) were pretreated with test compound prior to addition of [3H]AEA (labelled
in the ethanolamine part) or [3H]2-OG (labelled in the glycerol part). The samples
were incubated for 10 min at 37 °C and the reaction was then stopped by addition of
active charcoal:HCl, vortex mixing of the tubes and placing them on ice. The tubes
were then centrifuged and aliquots of the aqueous phase (containing the hydrophilic
[3H]EA and [3H]glycerol products) were transferred into scintillation vials and
counted with scintillation spectroscopy with quench correction. The lipophilic AEA
molecule remains bound to the charcoal. Blanks contained assay buffer instead of
homogenate solution.
For experiments using intact cells, [3H]AEA and [3H]2-AG hydrolysis were
measured as described by Paylor et al., 2006. However a slightly modified protocol
was used. Briefly, cells were seeded in a 24-well plate and grown over night. Wells
were washed with warm assay buffer (Krebs-Ringer HEPES (KRH) buffer, pH 7.4),
preincubated with test compound and then incubated with [3H]AEA (labelled in the
ethanolamine part) or [3H]2-AG (labelled in the glycerol part). To stop the reaction,
plates were put on ice and a mixture of active charcoal:HCl was added. The plates
were left at room temperature for approximately 30 min. prior to centrifugation.
Aliquots of the upper aqueous phase (containing the hydrophilic [3H]EA and
[3H]glycerol products) were transferred to scintillation vials and assayed for tritium
content by liquid scintillation spectroscopy with quench correction. The lipophilic
AEA molecule remains bound to the charcoal.
The spectrophotometric assay for MGL used in this thesis was developed by
Muccioli et al., 2008. Briefly, human recombinant MGL (either clear lysates or
enzyme further purified through a nickel column) in 10 mM Tris–HCl, 1 mM
EDTA, pH 7.4 and selected test compounds were added to a 96-well microtiter
plate. The reaction was started by rapid addition of the substrate 4-nitrophenyl

31
acetate (NPA). Blanks contained buffer alone. The absorbance was measured at 405
nm after 0 min (to rule out effects of the compounds per se on the absorbance) and
at least two 20 min intervals thereafter using a Thermomax Microplate Reader.
Comments
A number of different assays for FAAH, including chromatographic separation of
substrate, spectrophotometric assays with p-nitroanilide substrates, coupled assays
and simple organic separation of radiolabelled product from radiolabelled substrates,
have been described (Boldrup et al., 2004). The principle of the assay was originally
described by Omeir et al., 1995 and Dinh et al., 2002, although these authors used a
chloroform/methanol extraction procedure. The use of charcoal to separate the
metabolites of AEA (Boldrup et al., 2004) avoids exposure to potentially hazardous
organic solvents.
One advantage of this assay is that it is simple and easy to perform. However, the
observed activity of FAAH is highly pH-dependent, and the same can be true for its
inhibition (Holt et al., 2001; Paylor et al., 2006). Throughout the experiments of this
thesis, an assay pH of 7.4 has been used in order to measure activity under
physiological conditions. However, in Paper III a pH of 9 was used to be as close to
the pH optimum for FAAH as possible.
As discussed in the introduction, FAAH and MGL are not the only enzymes
involved in the degradation of AEA and MGL. Oxidative enzymes might also
contribute to activity, at least in theory. However, oxidation of AEA by COX or
LOX generates lipophilic products that will not be extracted in the water phase
measured in our assays (Patrignani et al., 2005; Ueda et al., 1995). Evidence of
FAAH as the major contributing enzyme for the metabolism of AEA have been
shown in studies where mice lacking FAAH possess a 50-100 fold reduced rate of
AEA hydrolysis (Cravatt et al., 2001). Futhermore, the FAAH isoenzyme FAAH-2,
with a pH optimum of 8.0, is not as effective as FAAH-1 in the hydrolysis of AEA.
In addition, it is not expressed in rodents (Wei et al., 2006). As mentioned earlier,

32
brain 2-AG can be metabolised not only by MGL but also by ABHD6 and 12. In
Paper V we found that, using rat cerebellar cytosolic fractions and the charcoal assay
to separate the products, the selective MGL inhibitor JZL184 inhibited the
hydrolysis of 2-OG by ~90%, suggesting that the involvement of other enzymes is
minor, consistent with the literature (Blankman et al., 2007).
The number of available MGL inhibitors is limited, mostly due to the lack of rapid
and accurate pharmacological assays for the enzyme. The spectrophotometric assay
using 4-nitrophenylacetate (4-NPA) as a substrate simplifies the screening for novel
inhibitors of the enzyme. Since 4-NPA is a substrate for many esterases, human
recombinant MGL was used instead of cytosol fraction of brain homogenates.
Throughout the experiments reported in this thesis, the 20 min. time point was used
to ensure that initial activities were measured but at the same time allow for
sufficient product formation. Given that the MGL used is a recombinant his-tagged
enzyme, the assumption is made that the recombinant enzyme behaves in an
identical manner to the natural enzyme.
Cell culture (Paper II, III, IV)
RBL2H3 rat basophilic leukaemia cells (ATCC® CRL-2256™), C6 rat glioma cells
(ATCC® CCL-107™), SH-SY5Y human neuroblastoma cells (ATCC® CRL-
2266™), P19 mouse teratocarcinoma (ATCC® CRL-1825™), R3327 AT-1 rat
prostate cancer cells (kind gift from Prof. Anders Bergh, Umeå University), HepG2
human liver hepatocellular carcinoma cells (ATCC® HB-8065™), CaCo-2 human
epithelial colorectal adenocarcinoma cells (ATCC® HTB-37™) and PC-3 human
prostate epithelial cells were used (ATCC® CRL-1435™). All cells were grown in
75 cm2 culturing flasks at 37 °C with 5% CO2 in humidified atmospheric pressure.
Comments
RBL2H3 cells were the first peripheral cell line investigated in detail for AEA
uptake (Bisogno et al., 1997a; Rakhshan et al., 2000). C6 cells are commonly used

33
in neurobiological research and are also widely used when investigating the cellular
uptake of AEA. The well-characterized accumulation of AEA in C6 (and RBL2H3)
cells makes is easier to compare findings between laboratories (Deutsch et al., 2001;
Ligresti et al., 2004). All cells chosen for this thesis are well characterised regarding
the endocannabinoid system, and have been reported to express endocannabinoid
components such as FAAH. In contrast, PC-3 cells do not express this enzyme
(Endsley et al., 2008; Ligresti et al., 2003; Pasquariello et al., 2009; Thors et al.,
2007; Wu et al., 2010) which makes them useful as a negative control for the
FAAH-involvement in AEA uptake.
Uptake assay of [3H]AEA, [3H]2-AG and [3H]PEA (Paper II, III, IV)
The assay for [3H]AEA, [3H]2-AG and [3H]PEA accumulation for adherent cells
was originally described by (Rakhshan et al., 2000) and later modified by (Sandberg
et al., 2005). Cells were plated in 24-well plates and incubated over night. The cells
were washed with warm assay buffer (Krebs-Ringer HEPES (KRH) buffer, pH 7.4)
prior to preincubation with selected compounds or vehicle. To start the reaction,
[3H]AEA, [3H]2-AG (both labelled in the arachidonate part of the molecule) or
[3H]PEA (labelled in the palmitoyl part of the molecule) were added to the wells.
Placing the plates on ice stopped the reaction. The wells were then washed three
times with ice-cold assay buffer containing 1% bovine serum albumin (BSA).
Finally, 0.2 M NaOH was added and the plates were incubated at 75 °C to solubilise
cellular material. Aliquots were transferred to scintillation vials and the tritium
content was measured by liquid scintillation spectroscopy with quench correction.
Comments
As discussed in the introduction, the mechanism of uptake of AEA is a matter of
debate. Also, the specific conditions used for AEA uptake assays have not been
standardized and may be a confounding issue for the analysis of the data.

34
Throughout the experiments of this thesis we have used BSA to stabilize AEA in the
buffer. BSA is known to bind to AEA and thereby prevent its binding to plastics
(Bojesen et al., 2003). However, the use of BSA in assay media is discussed widely
among researchers (Glaser et al., 2005). It was reported that addition of BSA to the
transport media greatly reduced AEA uptake (Di Marzo et al., 1994), although this
is presumably because it decreases the amount of free AEA available for uptake,
rather than directly affecting the uptake process itself. For discussion see Thors et
al., 2006. On the other hand, BSA decreases non-specific binding of AEA to plastic
wells, which is an advantage (Karlsson et al., 2004). In all the assays the BSA
concentration never exceeded 0.1%.
Binding to CB1 receptors (Paper II)
The protocol of Thomas et al., 1998 was used with minor modifications. The
binding of [3H]CP55,940, a CB1 receptor agonist (Hillard et al., 1999) were
undertaken in flat-bottomed 96-well plates using rat brain (without the cerebellum)
homogenates. The homogenates were prepared as described above. Non-specific
binding was determined in the presence of 10 µM CP55,940. Samples were
incubated for 90 min. at 37 °C and the bound radioactivity was separated from
unbound by vacuum filtration through pre-wetted FilterMAT filters using a cell
harvester. The tritium retained by the filters was measured by liquid scintillation
spectroscopy with quench correction.
RNA extraction, cDNA synthesis and PCR (Paper IV)
Total RNA was extracted using the miRNeasy Kit (Qiagen, Hilden, Germany, Cat.
No. 217004) following the manufacturer’s instructions. Briefly, cells were washed
and lysed in the recommended volume of QIAzol Lysis Reagent. Rat prefrontal
cortex was lysed in the same buffer using a homogenisator and RNA was extracted
as described above. The RNA concentration was determined by measuring the
absorbance at 260 nm using a NanoDrop spectrophotometer.

35
cDNA was synthesized using the High.Capacity cDNA Reverse Transcription kit
(Applied Biosystems, Foster City, CA, USA). RNA was diluted with RT buffer
(keeps the enzyme stable), RT random primers (bind to mRNA in order for reverse
transcription to occur), dNTP mix (for cDNA synthesis), multiscribe transcriptase
(RNA dependent-DNA polymerase, MuLV), RNAse inhibitor together with
nuclease-free water. The thermal cycler conditions used were an initial step of 10
min. at 25 °C followed by 120 min. at 37 °C, 85 s at 85 °C and finally cooled to 4
°C.
For detection of rat and mouse faah and fabp5 mRNA, samples of equal cDNA
concentrations were amplified in a polymerase chain reactions (PCR) using a Taq
PCR Core kit (Qiagen, Hilden, Germany, Cat. No. 201223). The primers used were:
forward rat faah forward 5’-GTGCTGAGCGAAGTGTGGACC-3’, reverse 5’-
GGGCCTGGGACAGCTGAGTCT-3’, rat fabp5 forward 5’- CGACCGTGTTTTC
TTGCACC-3’, reverse 5’-TGGCATTGTTCATGACGCAC-3’, mouse faah forward
5’-GTGGTGCTAACCCCCATGCTGG-3’, reverse 5’-TCCACCTCCCGCATGAA
CCGCAGACA-3’, mouse fabp5 forward 5’-GACGGTCTGCACCTTCCAAG-3’,
reverse 5’-CAGGATGACGAGGAAGCCC-3’. Samples were amplified with the
following conditions; an initial denaturation step at 94 °C for 2 min., followed by 35
cycles with denaturing at 94 °C for 40 s, annealing at 60 °C for 40 s and elongation
at 72 °C for 60 s, followed by a final elongation step at 72 °C for 3 min. The PCR
products were separated electrophoretically on a 1.5 % agarose gel. The expected
sizes of the fragments were: rat FAAH 382 bp, rat FABP5 192 bp, mouse FAAH
302 bp, and mouse FABP5 176 bp.
Other methodologies
Other methodologies in the papers include: synthesis of racemic Flu-AM1, Flu-
AM2, Nap-AM1 and the in vivo interaction of Flu-AM1 with rat brain FAAH,
performed at the University of Cagliari, Italy and the Centre for Addiction and
Mental Health, Toronto, Toronto, Canada, respectively; sampling and fixation of
human and animal material for Paper I, undertaken at the Section for Anatomy,
Department of Integrative Medical Biology and at the Sports Medicine Unit,

36
Department of Surgical and Perioperative Sciences, Umeå University, Sweden;
assays of COX inhibition in Paper II, performed by Ph. D. Mariateresa Cipriano at
our laboratory. For details of these methodologies, the reader is referred to the
individual papers.
Statistics
With the exception of analyses conducted using the R computer programme (paper
IV), all ll statistical calculations were undertaken using the computer software
GraphPad Prism 5 or 6 for the Macintosh (GraphPad Software Inc., San Diego, CA,
USA). Statistical significance was set to p<0.05.
Mann-Whitney U test
The non-parametric test is used for comparison of two independent groups. This test
was applied in Paper I.
One-way and two-way ANOVA
The one-way ANOVA (analysis of variance) test compares whether the mean of
three or more samples differ significantly, and the two-way ANOVA tests the
influence of different independent variables upon a dependent variable. Upon
significance, Dunnett's multiple comparison test was undertaken to compare every
mean with a control mean or Šídák’s multiple comparison test to compare selected
means with each other. These tests were used in papers II, III and IV.
pI50 and IC50
IC50 is defined as the molar concentration of a substance needed to produce 50 % of
maximum possible inhibition. pI50 is the negative logarithm of the IC50 value. The
pI50 and IC50 were calculated using the built-in equation log(inhibitor) vs. response
from the data expressed as % of control. The top was set to 100 and the bottom to
either 0 or allowed to float. For comparison between the curves Akaike’s
Informative Criteria was used. These calculations were used in papers II, III and V.

37
Results
CB1 receptor immunoreactivity in normal Achilles tendon and Achilles
tendinosis (Paper 1)
CB1 receptor immunoreactivity (CB1IR) was seen in normal Achilles tendon as well
as in Achilles tendinosis (Fig. 3). In the normal Achilles tendon, tendon cells
(tenocytes) had the characteristic appearance of being elongated, and CB1IR was
seen along the length of the cells (Fig. 3A). In the case for Achilles tendinosis,
CB1IR appeared in a granular pattern, which was seen especially in the abnormally-
formed tenocytes, e.g. rounded/swollen tenocytes and in tenocytes with a wavy
appearance, which is characteristic for Achilles tendinosis (Fig. 3B).
CB1IR in the form of fine pointed reactions was also seen in the walls of small blood
vessels (confirmed by haematoxylin-eosin staining). CB1IR was also seen in the
perineurium of nerve fascicles (demarcated with PGP9.5 immunoreactivity) but not
within the interior of nerve fascicles. CB1IR in blood vessels and nerve structures
was seen especially in the tendinosis samples (Fig. 3C, D).
As a positive control, CB1IR was also investigated in human colon, where distinct
reactivity was seen in both cells the mucosal, submucosal and the epithelial layers
and in dorsal root ganglia where CB1IR was seen as granular, intracellular reactions
(Paper I, Fig. 1c, e) No immunostaining was seen when the antibody was
preabsorbed with the corresponding peptide (Paper I, Fig. 1b, d, f and Fig. 2b).
Comparison of tenocyte CB1IR in normal Achilles tendon with Achilles tendinosis
A semi-quantitative analysis was performed based on the scoring of the CB1IR
intensity in tenocytes from patients either having pain-free Achilles tendon or from
patients suffering from Achilles tendinosis. Analysis of the data indicated that CB1
receptor expression was significantly upregulated in Achilles tendinosis (p<0.05)
(Fig 4). The tendon from the control with the highest CB1IR showed characteristic
tendinosis pathology, although the individual was asymptomatic at the time of the

38
biopsy. This makes the categorisation of this sample as a control questionable, and
when it was excluded from the data the significance level increased to p<0.005.
Fig. 3. Immunofluorescence for CB1 in normal tendon (A) and Achilles tendinosis (B-D).
Panels show sections processed for CB1 immunostaining. Immunoreactivity (white arrows) is
seen in normal tendon tissue (A), Achilles tendinosis tendon (B), small blood vessels (C) and
a nerve fascicle (D). Original magnification x40 (A), x63 (B, C, D).
Fig. 4. CB1IR scores for biopsies from control patients and patients with Achilles
tendinosis. Boxplot of CB1IR scores from control patients (normal pain-free Achilles tendon,
n=7) vs. Achilles tendinosis patients (n=17). *p=<0.05, two-tailed Mann-Whitney U test.
Values are median of the individual scoring made by the two investigators. Whiskers
represents min. to max. values.
0
1
2
3
CB
1IR
sco
re
*
Control Ach tendinosis
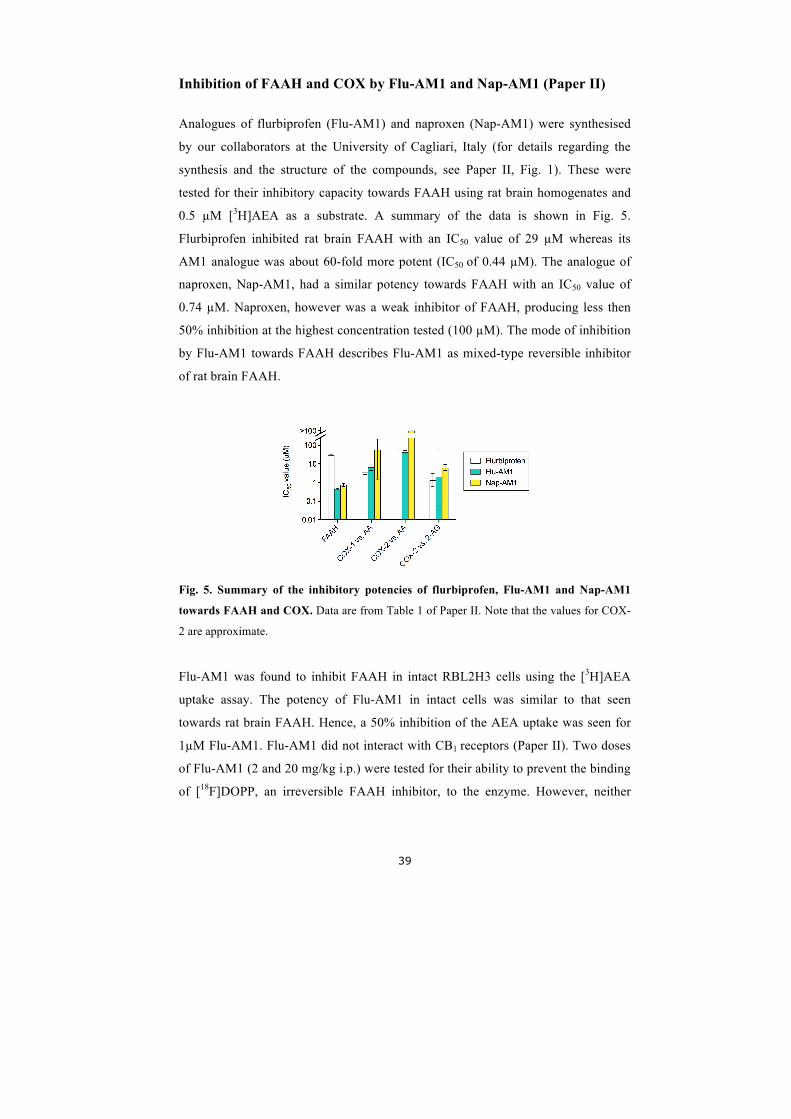
39
Inhibition of FAAH and COX by Flu-AM1 and Nap-AM1 (Paper II)
Analogues of flurbiprofen (Flu-AM1) and naproxen (Nap-AM1) were synthesised
by our collaborators at the University of Cagliari, Italy (for details regarding the
synthesis and the structure of the compounds, see Paper II, Fig. 1). These were
tested for their inhibitory capacity towards FAAH using rat brain homogenates and
0.5 µM [3H]AEA as a substrate. A summary of the data is shown in Fig. 5.
Flurbiprofen inhibited rat brain FAAH with an IC50 value of 29 µM whereas its
AM1 analogue was about 60-fold more potent (IC50 of 0.44 µM). The analogue of
naproxen, Nap-AM1, had a similar potency towards FAAH with an IC50 value of
0.74 µM. Naproxen, however was a weak inhibitor of FAAH, producing less then
50% inhibition at the highest concentration tested (100 µM). The mode of inhibition
by Flu-AM1 towards FAAH describes Flu-AM1 as mixed-type reversible inhibitor
of rat brain FAAH.
Fig. 5. Summary of the inhibitory potencies of flurbiprofen, Flu-AM1 and Nap-AM1
towards FAAH and COX. Data are from Table 1 of Paper II. Note that the values for COX-
2 are approximate.
Flu-AM1 was found to inhibit FAAH in intact RBL2H3 cells using the [3H]AEA
uptake assay. The potency of Flu-AM1 in intact cells was similar to that seen
towards rat brain FAAH. Hence, a 50% inhibition of the AEA uptake was seen for
1µM Flu-AM1. Flu-AM1 did not interact with CB1 receptors (Paper II). Two doses
of Flu-AM1 (2 and 20 mg/kg i.p.) were tested for their ability to prevent the binding
of [18F]DOPP, an irreversible FAAH inhibitor, to the enzyme. However, neither

40
dose affected the binding profile of this imaging agent measured ex vivo (Fig. 5 of
Paper II).
From the COX assay performed by Ph. D. Mariateresa Cipriano, it was found that
Flu-AM1 is a potent inhibitor of the COX-1 catalysed cyclooxygenation of
arachidonic acid. COX-2 was less affected by Flu-AM1 when arachidonic acid was
used as a substrate. However, when using 2-AG as a substrate, COX-2 catalysed
oxygenation was greatly reduced. A similar pattern was seen for flurbiprofen. Nap-
AM1, on the other hand, was a poor inhibitor of both COX isoforms (Fig. 5).
Inhibition of cellular uptake of AEA by ketoconazole (Paper III)
The antifungal agent ketoconazole was found to inhibit the cellular uptake of AEA
in HepG2 and CaCo-2 cells with IC50 values of 17 µM and 18 µM respectively (Fig.
6). The compound also reduced the retention of AEA by the wells, i.e. the binding of
AEA to plastic. However, experiments using different incubation times revealed that
ketoconazole significantly reduced the rate of cellular uptake of AEA by HepG2
cells, while the retention to the wells alone was low over time.
Inhibitory effect of ketoconazole upon FAAH activity
URB597 produced a large reduction of AEA accumulation in the uptake assay with
both HepG2 and CaCo-2 cells, demonstrating the importance of FAAH in
preserving the gradient across the plasma membrane for these cells. Further
investigation of the inhibitory effect of FAAH by ketoconazole was undertaken
where the inhibition produced by ketoconazole was not additive to that produced by
URB597 alone (Fig. 7). This indicates that these compounds act on the same
pathway. This was further validated using PC-3 cells, which lack FAAH. In this cell
line ketoconazole produced a modest inhibition of AEA uptake (Fig. 7).
Experiments using HepG2 and CaCo-2 lysates revealed an inhibitory effect of
ketoconazole upon the hydrolysis of AEA.

41
Fig. 6. The inhibitory effect upon the uptake of [3H]AEA by ketoconazole in CaCo-2
cells, HepG2 cells, PC-3 cells and wells alone. Cells (or wells) were preincubated with
ketoconazole for 10 min followed by addition of [3H]AEA (100 nM) and an additional
incubation time for 4 min. Shown are means ± s.e.m., n=3-9.
Fig. 7. The effect of ketoconazole upon [3H]AEA uptake in HepG2 cells in combination
with URB597. Cells were preincubated with ketoconazole in combination with URB597
followed by incubation with [3H]AEA. Shown are means ± s.e.m., n=3.
0 0.1 1 10 0 0.1 1 10 0 0.1 1 10 0 0.1 1 100
300
600
900
[URB597] (nM)
[3 H]A
EA u
ptak
e(fm
ol p
er w
ell)
0 1 10 100[Ketoconazole] (µM)
0.1 1 10 1000
25
50
75
100
125
Concentration (µM)
[3 H]A
EA a
ccum
ulat
ion
(% o
f con
trol)
CaCo-2HepG2PC-3Wells

42
Expression of FAAH mRNA in AT-1, RBL2H3, C6 and P19 cells (Paper
IV)
The role of FAAH in gating AEA uptake was further explored in Paper IV. faah
mRNA expression in three rat cell lines, one mouse cell line together with rat brain,
as a positive control, was investigated using RT-PCR. Specific primers were
designed to amplify a PCR product corresponding to the +4 to +385 region of the
coding sequence of the rat faah gene. The PCR analysis displayed the presence of a
band in agreement with the expected fragment size of 382 bp for rat faah and 302
for mouse faah. Equal amount of cDNA was added to the reaction and the relative
faah expression levels were found to be RBL2H3 > C6 >AT-1 > P19 cells (Fig. 8).
There was no evidence of a band with the predicted molecular weight of FLAT (178
bp), a finding consistent with the study of Leung et al. (2013). FLAT resembles the
faah gene except that it lacks a 204 bp segment encoding amino acid residues 9-76
(Fu et al., 2012).
Fig. 8. RT-PCR analysis of mRNA expression of faah. Expression of faah mRNA was
determined in rat brain lysate (lane 2), and in AT-1 (lane 3), C6 (lane 4) RBL2H3 (lane 5) and
P19 (lane 6) cells using RT-PCR. Lane 1 and 7 are a 100 bp marker and the arrows show the
expected size of rat FAAH (382 bp) and mouse FAAH (302 bp) products. Total RNA was
isolated and reverse transcribed into cDNA. Samples (n=4) were pooled. Note that the
photograph has been inverted to show the bands more clearly.

43
Activity of FAAH and accumulation of [3H]AEA in RBL2H3, C6, AT-1
and P19 cells (Paper IV)
The FAAH activity in intact cells (RBL2H3, C6, AT-1 and P19) was measured by
following the accumulation of tritiated product after incubation of the cells with 100
nM [3H]AEA. C6 showed the highest rate of hydrolysis with an activity of 3.3±0.66
pmol/well/10 min. RBL2H3, AT-1 and P19 hydrolysed the substrate with activities
of 2.1±0.16, 0.31±0.09 and 0.7±0.09 pmol/well/10 min.
The cellular accumulation of [3H]AEA was measured in all four cell lines (C6,
RBL2H3, AT-1 and P19) in the absence and presence of URB597. A significant
decrease in uptake was seen for all cell lines tested. Despite differences in the rates
of uptake in the absence of URB597, a rather similar residual uptake in all cells was
seen in its presence (Paper IV, fig 1 and 2).
We also compared Compound 33, known to be an inhibitor of FAAH (Onnis et al.,
2010), for its ability to inhibit the hydrolysis of AEA in intact AT-1, C6 and
RBL2H3 cells in relation to its capability of affecting uptake of AEA in the same
cells. We found a clear relationship between % inhibition of AEA metabolism and
uptake in all of the three cell lines. However, the slopes of the regression lines were
different. This indicates that a given degree of FAAH inhibition may not lead to the
same degree of inhibition of AEA uptake for different cells despite similarities in
FAAH expression (Paper IV, Fig 3 and 4).
Expression and activity of FABP5 in RBL2H3, C6, AT-1 and P19 cells
(Paper IV)
AEA is shuttled to FAAH for hydrolysis by transporting proteins. The diffences
seen between the cells might be due to different expression levels of such carriers.
Therefore, we examined the expression and function of FABP5, an intracellular
carrier protein of AEA (Kaczocha et al., 2009), using the FABP5 inhibitor SB-FI-26
(Berger et al., 2012). The PCR revealed a band, which correlates well with the

44
expected size of the fragment of 192 bp for rat FABP5 mRNA and 179 bp for mouse
FABP5 mRNA (Paper IV, Fig 5a). The cells show similar expression levels of
FABP5. Further, in all cells tested 50 µM SB-FI-26 significantly decreased the
accumulation of AEA (Paper IV, Fig 5b-f). Thus, the differences between the cells
cannot be ascribed to differences in expression of FABP5, although other carriers
may still be involved.
Screening of selected compounds for inhibition of MGL using the
spectrophotometric NPA assay (Paper V)
Initial experiments were undertaken by co-authors Norén and Nilsson to characterise
the assay with respect to enzyme concentrations and incubation times used, pH
optima, sensitivity to inhibition by 2-AG and 2-OG, and maximum tolerable solvent
concentrations (Norén & Nilsson, undergraduate thesis project, 15 hp, Autumn term
2008). The screening included 96 compounds known to interact with the CB system
or have other actions upon targets in the brain, NSAIDs or naturally occurring
compounds. Selected compounds and their hMGL activity are listed in Paper V,
Table 1. In the initial screen, compounds were tested at two different concentrations
(3 and 10 µM). A compound producing >60% inhibition was classified as a “hit”.
Further characterisation of six of the “hits” was performed by dose-response curves
over the range of 0.1-10 µM (Paper V, Fig. 1a). The pI50 values (IC50 values in
brackets) were for troglitazone, CP55,940, N-arachidonoyl dopamine and AM404
5.95±0.04 (1.1µM), 5.31±0.07 (4.9 µM), 6.11±0.08 (0.78 µM) and 5.53±0.09 (3.1
µM) respectively. There was a large variability in the observed inhibition at the
highest concentration for WIN55,212-2 and JWH015 yielding difficulties to
determine their IC50 values.
Troglitazone and N-arachidonoyl dopamine as inhibitors of MGL
The PPARγ ligand troglitazone was compared to other PPARγ ligands toward their
ability to inhibit hMGL. The potency order was troglitazone > ciglitazone >
rosiglitazone > 15-deoxy-Δ12,14-prostaglandin J2 ≈ CAY10415 > CAY10514 (Paper
V, Fig. 1b).

45
The inhibition of hMGL with troglitazone showed no time-dependency indicating
that the inhibition is reversible (Fig. 9a). However, additional experiment indicates
that this reversibility is not rapid. Saturation curves for NPA incubated with 0 and
0.75 µM troglitazone gave Km values of 0.17 and 0.70 mM respectively (Fig. 9b),
assuming a competitive mode of action.
Both troglitazone and the endogenous TRPV1 ligand N-arachidonoyl dopamine
were investigated in two assay systems in order to determine if the inhibition of
MGL was assay, substrate and/or species dependent. N-arachidonoyl dopamine
inhibited the hydrolysis by the hMGL of NPA and 2-OG with IC50 values in the low
µM range (0.78 µM and 2.2 µM respectively). However, the compound was less
potent as an inhibitor of the hydrolysis of 2-OG by rat cytosol (IC50 value of 20 µM)
(Fig. 10a). For troglitazone there was a large inter-assay variability. The compound
inhibited purified hMGL with a threefold lower potency then seen for the lysate in
the NPA assay. Yet, in the radiochemical assay troglitazone was a weak inhibitor of
both rat cytosol and hMGL with 2-OG as substrate (Fig. 10b).
Different BSA concentrations in the two assays were found to affect the observed
inhibitory properties of troglitazone. In the NPA assay, the inhibition of hMGL
produced by both troglitazone and N-arachidonoyl dopamine were greater in the
absence of BSA. Also, when assayed radiochemically, N-arachidonoyl dopamine
inhibited hMGL lysate better without BSA in the buffer compared to the presence of
BSA. Troglitazone was also a better inhibitor of hMGL lysate in the radiochemically
assay in the absence of BSA, although its potency was still lower then that seen in
the NPA assay.

46
Fig. 9. Mode of inhibition by troglitazone upon hydrolysis of 4-NPA by hMGL. In panel
A, lysates were preincubated with troglitazone at room temperature prior to addition of 4-
NPA (0.25mM). In panel B, no preincubation was used prior to addition of 4-NPA (0.08-0.64
mM). Shown are means ± s.e.m., n=3 except for the data in Panel B with 0.32 mM NPA,
where n=2. To illustrate the competitive nature of the inhibition, a double reciprocal plot of
the mean data with 0 and 0.75 mM troglitazone is inserted in Panel B
Fig. 10. The potencies of N-arachidonoyl dopamine (A) and troglitazone (B) towards
MGL possessed in different assays. No preincubation phase were used prior to addition of
substrate in any of the assay system used. Shown are mean ± s.e.m., n=3-6.

47
Discussion
Overall comments
The studies included in this thesis provides fruitful novel information about the
endocannabinoid system in a human pain state, e.g. Achilles tendinosis as well as a
better understanding on how to pharmacologically modify this system. The results
demonstrate that CB1 receptors can be observed immunohistochemically in Achilles
tenocytes and that their immunochemical staining intensity in increased in
tendinosis. This is consistent with the suggestion of a dysregulated endocannabinoid
system in a painful human condition. As a consequence of this, an enhanced
knowledge on how to modulate this system in a pharmacological manner would be
beneficial. The findings presented here delineate new openings for the development
of novel inhibitors of endocannabinoid metabolism.
Achilles tendinosis
Prior to Paper I, as discussed in the introduction, little was known about the
endocannabinoid system in human pain. Likewise, the Achilles tendon is the
strongest tendon in the body but the reasons behind the pain experienced in Achilles
tendinosis remains unclear. Based on this gap in our knowledge, we investigated the
expression of CB1 receptors in Achilles tendon tissue using biopsy samples from
patients suffering from Achilles tendinosis and from individuals having
asymptomatic pain free tendons. The result of the semi-quantitative analysis,
performed by two independent blinded researchers, implied an up-regulation of CB1
receptor expression in Achilles tendinosis. We found CB1IR along the length of
tenocytes in normal tendons. For tenocytes having a characteristic tendinosis
morphology, i.e. rounded/swollen tenocytes and tenocytes with a wavy appearance,
the CB1IR had a granular intracellular pattern. The presence of intracellular
reactions can also be seen in the literature for other cells in the body (Bridges et al.,
2003). In 2001 Brailoiu et al., 2011 showed that AEA can activate intracellular CB1

48
receptors and thereby provided evidence that these intracellular receptors are
functional.
Recently, Dean et al., 2013 have critically reviewed the reliability of published
articles in the tendinosis research field. They conducted a systematic review of the
scientific literature where inclusion criteria and methods of analysis were specified
in advance. The search, selection of studies and data analysis was performed by two
individuals. The methodological quality of the selected papers was assessed using a
10-point scale where a point was awarded for each of the following criteria:
inclusion/exclusion criteria clearly described; study population clearly described;
study population representative of those in clinical practice; control group clearly
described; sampling method clearly described; live study subjects; live control
subjects; quantitative method or semi-quantitative method using minimum of two
independent observers; reliability and/or validity of methods described; and study
limitations addressed (Dean et al., 2013). Our study attained a high score (9/10),
although it was felt that the study limitations should have been further discussed.
Hence, these are addressed below.
Our control group consisted of healthy asymptomatic patients (as opposed to
cadaveric controls or patients with Achilles tendinosis rupture). Although this is a
strength - the tendon rupture patients might not yet show symptoms of tendinosis
albeit the tendon tissue is somehow impaired which leads to the rupture - the use of
healthy controls is a study limitation in terms of the size of the control group.
Further, for ethical reasons, the tendon samples taken from the controls were from
the dorsal part of the tendon. Another limitation of the study is that CB1
immunoreactivity alone does not tell us whether or not CB1 receptors are functional
or active. One way of investigating this would be the study of CB1-mediated
receptor signalling in primary tenocyte cultures. In collaboration with the research
group of Ph. D. Patrick Danielsson (Department of Integrative Medical Biology,
Section of Anatomy, Umeå University), we have tried to evaluate this possibility,
but we were unable to obtain robust data in the time frame available. Additionally,
although immuneoreactivity gives information about protein expression, it does not

49
provide data concerning the underlying regulation accounting for observed
differences, e.g. regulation at the level of mRNA expression or post-translationally.
Moreover, small or local changes in endocannabinoid signalling would be difficult
to detect. Although the latter question is beyond the scope of our paper, it would be
of clear interest to investigate local endocannabinoid levels using, for example the
microdialysis technique. An additional approach, which we initiated, was to evaluate
whether the expression of the endocannabinoid metabolising enzyme FAAH was
altered in Achilles tendinosis. However, we were not able to find a robust antibody
with which to assess immunoreactivity.
Inhibitors of endocannabinoid metabolism and uptake
Termination of endocannabinoid signalling is known to occur through cellular
uptake followed by hydrolysis by metabolising enzymes. Although the catabolic
process is well defined, the mechanism(s) of cellular uptake has not yet been
clarified. A number of inhibitors of AEA uptake and AEA metabolism have been
developed, most of them with reported effect both in vitro and in vivo. Inhibitors of
FAAH are in phase I and II clinical trials, but so far no compounds have
successfully reached the market. When this thesis was started, no selective MGL
inhibitors had been disclosed. The irreversible inhibitor of MGL, JZL184, was
reported in 2009 but there are issues of tolerance with this compound (Schlosburg et
al., 2010). Our goal was to identify novel compounds, preferably inhibiting
endocannabinoid metabolism in a reversible manner. Our approach was to
investigate compounds that are already in clinical use and elucidate whether
inhibition of endocannabinoid metabolism is an additional effect either per se, or a
result of chemical modification of the compound in question.
A previous article from our lab revealed the inhibitory effect of ibuprofen on FAAH,
in addition to COX (Fowler et al., 1997). Furthermore, its N-(3-methylpyridin-2-
yl)amide analogue proved to be 2-3 orders of magnitude more potent as an FAAH
inhibitor but with preserved effect on COX (Holt et al., 2007). Mice treated with
FAAH inhibitors together with compounds inhibiting COX had a lower incidence of

50
gastric haemorrhages (Naidu et al., 2009). Based on these results, in Paper II we
synthesised the analogue of flurbiprofen, Flu-AM1, in order to investigate its ability
to inhibit FAAH and COX. We found that Flu-AM1 inhibited FAAH with preserved
effect upon COX. By using RBL2H3 cells, in which FAAH has been shown to gate
AEA uptake (Deutsch et al., 2001), we found that Flu-AM1 had the ability to cross
membranes and inhibit FAAH but it did not affect brain FAAH. This raises the
question as to whether or not the concentrations tested were too low, or the
compound has a limited capacity to cross the blood-brain barrier. The answer to this
was beyond the scope of this project. Nonetheless, identification of novel dual-
action compounds is clearly an area worth pursuing, given the synergistic effects of
FAAH inhibitors and NSAIDs in preclinical pain models (Naidu et al., 2009).
In line with the concept of finding novel actions of established compounds, in Paper
III we investigated whether or not the antifungal compound ketoconazole affects
AEA uptake. Ketoconazole has previously been shown to interact with CYP3A4
(Sai et al., 2000) and lipooxygenase (Beetens et al., 1986), two enzymes that can
metabolise AEA. Ketoconazole inhibited AEA uptake in two FAAH-containing cell
lines, namely HepG2 and CaCo-2, in a manner consistent with inhibition of FAAH.
This is supported by the fact that ketoconazole had no effect upon the accumulation
of AEA in PC-3 cells, which have low expression of FAAH. In addition we found
that the inhibitory effects of ketoconazole and the FAAH inhibitor URB597 upon
AEA uptake were not additive suggesting that the two compounds act on the same
pathway.
As pointed out above, due to the lack of selective MGL inhibitors, more research
about the activity of the enzyme and potential inhibitors is needed. As already
discussed regarding ketoconazole, our basic idea was to investigate a range of
compounds that are either currently, or have been previously, in clinical use and/or
that are known to have some kind of activity towards the endocannabinoid system.
We used the assay of Muccioli et al., 2008, which allows the screening of a larger
number of compounds than usually possible in our radiochemical assay. The “hits”
from this preliminary screen were then further evaluated. We found that the PPARγ

51
ligand troglitazone (Willson et al., 1996) inhibited MGL in the low micromolar
range but also that there was a large inter-assay difference. Troglitazone was
withdrawn from the market due to hepatotoxicity and related ligands such as
rosiglitazone are associated with cardiovascular events, but it may be possible to use
troglitazone as a starting point for novel MGL inhibitors devoid of these issues.
Taken together, by screening compounds that are, or previously have been, in
clinical use for their FAAH and MGL inhibitory properties gives us good templates
with which to design novel FAAH and MGL inhibitors. By synthesising analogues
of a promising compound, it may be possible to optimise effects upon the
endocannabinoid system while retaining (or not, as appropriate) its effects on other
systems.
As mentioned in the introduction, the uptake process of AEA in to cells is a matter
of debate. FAAH activity has been shown to gate AEA uptake in RBL2H3 cells but
in other FAAH-expressing cells, the effect is not seen (Deutsch et al., 2001). This
raises the question as to whether or not the degree of FAAH-sensitivity is the result
of methodological differences in different laboratories, or if it reflects the properties
of the cells themselves. To answer this we investigated the effect of FAAH
inhibition upon AEA accumulation in four cell lines. We found that RBL2H3 and
C6 cells, two cell lines having roughly similar levels of faah gene expression, had
different sensitivities to a given level of FAAH inhibition. This implies that the
reported differences in FAAH sensitivities of AEA uptake in the literature are not
due to methodological differences alone but to the properties of the cells themselves.
One suggestion might be that different cells may take up and accumulate AEA for
different purposes (Hillard et al., 2005), e.g. as a requirement for cell signalling or
as a source for the production of arachidonic acid, and in consequence utilise
different transport mechanisms. The presence of intracellular AEA transporting
proteins might also contribute to the differences in FAAH sensitivity between the
cells, although variations in FABP5 expression can be ruled out.

52
Future perspectives
Future directions of this work could be:
• Examine whether or not the CB1 receptors in Achilles tendinosis are
functionally active, for example by investigating the signalling of the CB1
receptors in fresh tissue from tendinosis patients.
• Determine the strength of local endocannabinoid signalling in the normal
and pathological Achilles tendon. Ideally, this could be undertaken by
measuring local endocannabinoid levels, but a more indirect measure
would be the determination of expression levels of the endocannabinoid
synthetic and degradative enzymes, or by investigating the activation of
downstream targets of CB1.
• In vitro and in vivo experiments with analogues of ketoconazole,
troglitazone and Flu-AM1 optimised for inhibition of FAAH, MGL and
FAAH/COX-2, respectively.

53
Conclusions
CB1 receptors are expressed in tenocytes of Achilles tendon. In painful Achilles
tendinosis, expression is up-regulated compared to normal, healthy tendons.
The analogue of flurbiprofen, Flu-AM1, shows promising potency as a dual-action
compound effecting both FAAH and COX.
Ketoconazole, an antifungal compound with inhibitory effect on enzymes in the
CYP family, can also inhibit the cellular uptake of AEA primarily due to effects
upon FAAH.
Cells with broadly similar levels of FAAH-expression show different sensitivities to
FAAH inhibition upon AEA uptake.
Troglitazone inhibits MGL in a manner dependent upon the enzyme assay used.

54
Acknowledgements
Finally I can say, I’m done! The work presented in this thesis is mine and I’m alone
responsible for all possible faults and errors. Anyhow, the work would not have
been possible without the help and support from the people around me. Most of
them work, or have worked, at the Department of Pharmacology and Clinical
Neuroscience, Umeå University and I’m so lucky to have such talented people by
my side.
First of all, I would like to give special thanks to my great supervisor Christopher
Fowler. Your ability to come up with ideas, interpret data combined with an
incredible speedy way of writing papers is really impressive. You have always been
inspiring and supportive to me. Thank you for your encouragement and for giving
me opportunities that I could never dream of. I’m forever grateful that I had the
chance to work with you!
My co-supervisor Stig Jacobsson, thank you for believing in me. You have always
been there and helped me whenever I needed. No matter if the question is personal,
scientific or concerns technology issues, you always have the answer. You are an
important figure at the department. Thank you for bringing joy to the people around
you.
My second co-supervisor Sture Forsgren at the department of Integrative Medical
Biology, Umeå University, thank you for guiding me in the world of tendons. You
are a true inspiration!
Gunnar Tiger, thank you for giving me a memorable first day as a biomedical
student on the course of pharmacology. You are the reason for my great interest in
the field! Your personality is one of a kind and has given me many laughs.
The time I’ve spent at the lab became much more funny thanks to the former and
present PhD-student at the department. First of all, “double-doctor” Sofia
Gustafsson, you are amazing. You have given me so many joyful moments, I really

55
miss you at the department. One day I will become a “double-doctor” just like you!
Dina Popova, Jessica Karlsson and Linda Gabrielsson, thanks for the pleasant
time both at work but also outside of the lab. I wish you the best of luck on your
way to the PhD degree. Jessica, now that Dina is at home taking care of her cute
little baby girl, you are the “överstedoktorand”. Never forget that ;) And Linda, I
feel safe leaving the place now that we have a new “bodensare” in the house. Stand
up for Boden!
I would also like to thank the former post-docs Linus Plym Forshell, Marialuisa
Rojo, Antonio Rodríguez and Mariateresa Cipriano for bringing some
international feeling to the lab. Mariateresa, you gave the word “fika” a whole new
meaning :)
Thanks also to the rest of the staff at the department. Mona Svensson and Staffan
Tavelin, even though I haven’t had a chance to work with you for that long, you are
really an add-on to the staff and to the great atmosphere at the department. I would
like to give a special thank to Eva Hallin and Agneta Walleskog. Eva, your
helpfulness at the lab, all from ordering, finding and cleaning stuff to
methodological question were indispensably. You were my teammate! Agneta,
thank you for providing me excellent help with administrative questions, and of
course, for your great personality. Olov Nilsson, your social skills and your way of
telling anecdotes never makes the coffee room quiet and boring. Jonathan
Gilthorpe, thank you for your valuable feedback and linguistic help with the writing
of this thesis.
There are also people outside the lab who are as important for making this thesis
possible. Maria Lestander, what would I do without you? You are my nearest and
dearest friend. We have known each other for more than 25 years and you have
always stood by my side. Remember when we were kids and saw the two elder
ladies with walkers? We said that was going to be us one day. You know time
passes, we’re soon there! ;)

56
Micaela Wiik, no one knows me as you do. We share so many great memories. You
always put a smile on my face and make me do stuff I never though I would do. I
just wish we lived closer but I know you are just a phone call away. Therese
Eriksson, thank you for being who you are. Your friendship means the world to me.
Lisa Holmström, David Jonsson, Caroline Lithner, Joel Weinmar, Sandra
Högqvist, Maria Solvang, Josip and Ana Marusic, you make (and made) my life
here in Umeå much more memorable and eventful.
Eva Levén and Tina Lundquist, we have known each other for so long. Even
though we live far away and we don’t see each other that often you are still close
and valuable friends to me. Matilda Lundström, you are as a sister to me. We share
something no one can understand. I love you.
Last but definitively not least I would like to thank my family. My mother Gunvor,
father Bengt-Arne, brother Marcus and of course the best and cutest niece Nova,
words can’t express what you mean to me. Thank you for giving me the best support
and love I can ever imagine. This journey would not been possible if it weren’t for
all of you! “Puss och kram!”
For those of you who are not mentioned by name, I just want to say that I haven’t
forgotten you. You are just as important!

57
References
Adamczyk P, Golda A, McCreary AC, Filip M, Przegalinski E (2008). Activation of endocannabinoid transmission induces antidepressant-like effects in rats. J Physiol Pharmacol 59(2): 217-228.
Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, et al. (2007). Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci 10(7): 870-879.
Ahn K, Smith SE, Liimatta MB, Beidler D, Sadagopan N, Dudley DT, et al. (2011). Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther 338(1): 114-124.
Alfredson H, Thorsen K, Lorentzon R (1999). In situ microdialysis in tendon tissue: high levels of glutamate, but not prostaglandin E2 in chronic Achilles tendon pain. Knee surgery, sports traumatology, arthroscopy : official journal of the ESSKA 7(6): 378-381.
Alfredson H, Öhberg L (2005). Neovascularisation in chronic painful patellar tendinosis--promising results after sclerosing neovessels outside the tendon challenge the need for surgery. Knee surgery, sports traumatology, arthroscopy : official journal of the ESSKA 13(2): 74-80.
Alger BE, Kim J (2011). Supply and demand for endocannabinoids. Trends Neurosci 34(6): 304-315.
Andersson G, Danielson P, Alfredson H, Forsgren S (2007). Nerve-related characteristics of ventral paratendinous tissue in chronic Achilles tendinosis. Knee surgery, sports traumatology, arthroscopy : official journal of the ESSKA 15(10): 1272-1279.
Bátkai S, Pacher P, Osei-Hyiaman D, Radaeva S, Liu J, Harvey-White J, et al. (2004). Endocannabinoids acting at cannabinoid-1 receptors regulate cardiovascular function in hypertension. Circulation 110(14): 1996-2002.
Beetens J, Loots W, Somers Y, Coene M, De Clerck F (1986). Ketoconazole inhibits the biosynthesis of leukotrienes in vitro and in vivo. Biochem Pharmacol 35(6): 883-891.
Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997). Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277(5329): 1094-1097.
Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, et al. (2012). Targeting fatty acid binding protein (FABP) anandamide transporters - a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One 7(12): e50968.
Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V (1997a). Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem 272(6): 3315-3323.
Bisogno T, Sepe N, Melck D, Maurelli S, De Petrocellis L, Di Marzo V (1997b). Biosynthesis, release and degradation of the novel endogenous cannabimimetic metabolite 2-arachidonoylglycerol in mouse neuroblastoma cells. Biochem J 322 (Pt 2): 671-677.

58
Bjur D, Alfredson H, Forsgren S (2005). The innervation pattern of the human Achilles tendon: studies of the normal and tendinosis tendon with markers for general and sensory innervation. Cell Tissue Res 320(1): 201-206.
Blankman JL, Simon GM, Cravatt BF (2007). A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14(12): 1347-1356.
Bobrov MY, Shevchenko VP, Yudushkin IA, Rogov SI, Remov MN, Fomina-Ageeva EV, et al. (2000). Hydrolysis of anandamide and eicosapentaenoic acid ethanolamide in mouse splenocytes. Biochemistry (Mosc) 65(5): 615-619.
Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, et al. (1999). Trifluoromethyl ketone inhibitors of fatty acid amide hydrolase: a probe of structural and conformational features contributing to inhibition. Bioorg Med Chem Lett 9(2): 265-270.
Bojesen IN, Hansen HS (2003). Binding of anandamide to bovine serum albumin. J Lipid Res 44(9): 1790-1794.
Boldrup L, Wilson SJ, Barbier AJ, Fowler CJ (2004). A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J Biochem Biophys Methods 60(2): 171-177.
Bonz A, Laser M, Küllmer S, Kniesch S, Babin-Ebell J, Popp V, et al. (2003). Cannabinoids acting on CB1 receptors decrease contractile performance in human atrial muscle. J Cardiovasc Pharmacol 41(4): 657-664.
Bornheim LM, Kim KY, Chen B, Correia MA (1995). Microsomal cytochrome P450-mediated liver and brain anandamide metabolism. Biochem Pharmacol 50(5): 677-686.
Bouaboula M, Hilairet S, Marchand J, Fajas L, Le Fur G, Casellas P (2005). Anandamide induced PPARgamma transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur J Pharmacol 517(3): 174-181.
Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
Brailoiu GC, Oprea TI, Zhao P, Abood ME, Brailoiu E (2011). Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J Biol Chem 286(33): 29166-29174.
Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W (1996). Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 137(1): 354-366.
Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D (2006). Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain 10(4): 287-333.
Bridges D, Rice AS, Egertova M, Elphick MR, Winter J, Michael GJ (2003). Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience 119(3): 803-812.
Burston JJ, Sagar DR, Shao P, Bai M, King E, Brailsford L, et al. (2013). Cannabinoid CB2 Receptors Regulate Central Sensitization and Pain Responses Associated with Osteoarthritis of the Knee Joint. PLoS One 8(11): e80440.
Calignano A, La Rana G, Giuffrida A, Piomelli D (1998). Control of pain initiation by endogenous cannabinoids. Nature 394(6690): 277-281.

59
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389(6653): 816-824.
Chicca A, Marazzi J, Nicolussi S, Gertsch J (2012). Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem 287(41): 34660-34682.
Chung SC, Hammarsten P, Josefsson A, Stattin P, Granfors T, Egevad L, et al. (2009). A high cannabinoid CB(1) receptor immunoreactivity is associated with disease severity and outcome in prostate cancer. Eur J Cancer 45(1): 174-182.
Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, et al. (2001). Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A 98(16): 9371-9376.
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384(6604): 83-87.
Day TA, Rakhshan F, Deutsch DG, Barker EL (2001). Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol Pharmacol 59(6): 1369-1375.
de Novellis V, Palazzo E, Rossi F, De Petrocellis L, Petrosino S, Guida F, et al. (2008). The analgesic effect of N-arachidonoyl-serotonin, a FAAH inhibitor and TRPV1 receptor antagonist, associated with changes in rostral ventromedial medulla and locus coeruleus cell activity in rats. Neuropharmacology 55(7): 1105-1113.
De Wael F, Muccioli GG, Lambert DM, Sergent T, Schneider YJ, Rees JF, et al. (2010). Chemistry around imidazopyrazine and ibuprofen: discovery of novel fatty acid amide hydrolase (FAAH) inhibitors. Eur J Med Chem 45(9): 3564-3574.
Dean BJ, Franklin SL, Carr AJ (2013). The peripheral neuronal phenotype is important in the pathogenesis of painful human tendinopathy: a systematic review. Clinical orthopaedics and related research 471(9): 3036-3046.
Degn M, Lambertsen KL, Petersen G, Meldgaard M, Artmann A, Clausen BH, et al. (2007). Changes in brain levels of N-acylethanolamines and 2-arachidonoylglycerol in focal cerebral ischemia in mice. J Neurochem 103(5): 1907-1916.
Desvergne B, Wahli W (1999). Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 20(5): 649-688.
Deutsch DG, Chin SA (1993). Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46(5): 791-796.
Deutsch DG, Glaser ST, Howell JM, Kunz JS, Puffenbarger RA, Hillard CJ, et al. (2001). The cellular uptake of anandamide is coupled to its breakdown by fatty-acid amide hydrolase. J Biol Chem 276(10): 6967-6973.
Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS, Howlett AC (1988). Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34(5): 605-613.
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258(5090): 1946-1949.

60
Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372(6507): 686-691.
Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. (2002). Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A 99(16): 10819-10824.
Duggan KC, Hermanson DJ, Musee J, Prusakiewicz JJ, Scheib JL, Carter BD, et al. (2011). (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chem Biol 7(11): 803-809.
Egertová M, Giang DK, Cravatt BF, Elphick MR (1998). A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci 265(1410): 2081-2085.
Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, et al. (2006). The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49(1): 67-79.
Endsley MP, Thill R, Choudhry I, Williams CL, Kajdacsy-Balla A, Campbell WB, et al. (2008). Expression and function of fatty acid amide hydrolase in prostate cancer. International journal of cancer. Journal international du cancer 123(6): 1318-1326.
Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, et al. (2005). Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3'-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther 313(1): 352-358.
Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, et al. (2004). Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci U S A 101(23): 8756-8761.
Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, et al. (1995). Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48(3): 443-450.
Fiskerstrand T, H'Mida-Ben Brahim D, Johansson S, M'Zahem A, Haukanes BI, Drouot N, et al. (2010). Mutations in ABHD12 cause the neurodegenerative disease PHARC: An inborn error of endocannabinoid metabolism. Am J Hum Genet 87(3): 410-417.
Fowler CJ, Bjorklund E, Lichtman AH, Naidu PS, Congiu C, Onnis V (2013). Inhibitory properties of ibuprofen and its amide analogues towards the hydrolysis and cyclooxygenation of the endocannabinoid anandamide. J Enzyme Inhib Med Chem 28(1): 172-182.
Fowler CJ, Ghafouri N (2008). Does the hydrolysis of 2-arachidonoylglycerol regulate its cellular uptake? Pharmacol Res 58(1): 72-76.
Fowler CJ, Tiger G, Stenstrom A (1997). Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J Pharmacol Exp Ther 283(2): 729-734.
Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83(3): 1017-1066.
Fu J, Bottegoni G, Sasso O, Bertorelli R, Rocchia W, Masetti M, et al. (2012). A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci 15(1): 64-69.

61
Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, et al. (2003). Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 425(6953): 90-93.
Galiègue S, Mary S, Marchand J, Dussossoy D, CarriÈre D, Carayon P, et al. (1995). Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232(1): 54-61.
Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, et al. (2010). Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30(6): 2017-2024.
García-Gutiérrez MS, Ortega-Álvaro A, Busquets-García A, Pérez-Ortiz JM, Caltana L, Ricatti MJ, et al. (2013). Synaptic plasticity alterations associated with memory impairment induced by deletion of CB2 cannabinoid receptors. Neuropharmacology 73: 388-396.
Gatta L, Piscitelli F, Giordano C, Boccella S, Lichtman A, Maione S, et al. (2012). Discovery of prostamide F2alpha and its role in inflammatory pain and dorsal horn nociceptive neuron hyperexcitability. PLoS One 7(2): e31111.
Gérard CM, Mollereau C, Vassart G, Parmentier M (1991). Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J 279 ( Pt 1): 129-134.
Ghafouri N, Tiger G, Razdan RK, Mahadevan A, Pertwee RG, Martin BR, et al. (2004). Inhibition of monoacylglycerol lipase and fatty acid amide hydrolase by analogues of 2-arachidonoylglycerol. Br J Pharmacol 143(6): 774-784.
Ghosh S, Wise LE, Chen Y, Gujjar R, Mahadevan A, Cravatt BF, et al. (2013). The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci 92(8-9): 498-505.
Glaser ST, Kaczocha M (2010). Cyclooxygenase-2 mediates anandamide metabolism in the mouse brain. J Pharmacol Exp Ther 335(2): 380-388.
Glaser ST, Kaczocha M, Deutsch DG (2005). Anandamide transport: a critical review. Life Sci 77(14): 1584-1604.
Glass M, Dragunow M, Faull RL (1997). Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 77(2): 299-318.
Goparaju SK, Ueda N, Taniguchi K, Yamamoto S (1999). Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem Pharmacol 57(4): 417-423.
Goparaju SK, Ueda N, Yamaguchi H, Yamamoto S (1998). Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett 422(1): 69-73.
Grimsey NL, Goodfellow CE, Scotter EL, Dowie MJ, Glass M, Graham ES (2008). Specific detection of CB1 receptors; cannabinoid CB1 receptor antibodies are not all created equal! J Neurosci Methods 171(1): 78-86.
Guindon J, De Léan A, Beaulieu P (2006). Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain 121(1-2): 85-93.

62
Guindon J, Guijarro A, Piomelli D, Hohmann AG (2011). Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol 163(7): 1464-1478.
Guindon J, Hohmann AG (2009). The endocannabinoid system and pain. CNS Neurol Disord Drug Targets 8(6): 403-421.
Guindon J, Hohmann AG (2008). A physiological role for endocannabinoid-derived products of cyclooxygenase-2-mediated oxidative metabolism. Br J Pharmacol 153(7): 1341-1343.
Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG (2013). Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol Res 67(1): 94-109.
Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, et al. (2004). Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci 20(2): 441-458.
Gustafsson SB, Palmqvist R, Henriksson ML, Dahlin AM, Edin S, Jacobsson SO, et al. (2011). High tumour cannabinoid CB1 receptor immunoreactivity negatively impacts disease-specific survival in stage II microsatellite stable colorectal cancer. PLoS One 6(8): e23003.
Hampson AJ, Bornheim LM, Scanziani M, Yost CS, Gray AT, Hansen BM, et al. (1998). Dual effects of anandamide on NMDA receptor-mediated responses and neurotransmission. J Neurochem 70(2): 671-676.
Hampson AJ, Hill WA, Zan-Phillips M, Makriyannis A, Leung E, Eglen RM, et al. (1995). Anandamide hydroxylation by brain lipoxygenase:metabolite structures and potencies at the cannabinoid receptor. Biochim Biophys Acta 1259(2): 173-179.
Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, et al. (2001). Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem 78(6): 1415-1427.
Hansson M, Forsgren S (1995). Immunoreactive atrial and brain natriuretic peptides are co-localized in Purkinje fibres but not in the innervation of the bovine heart conduction system. Histochem J 27(3): 222-230.
Hanuš L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, et al. (2001). 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci U S A 98(7): 3662-3665.
Hanuš L, Gopher A, Almog S, Mechoulam R (1993). Two new unsaturated fatty acid ethanolamides in brain that bind to the cannabinoid receptor. J Med Chem 36(20): 3032-3034.
Hashimotodani Y, Ohno-Shosaku T, Tanimura A, Kita Y, Sano Y, Shimizu T, et al. (2013). Acute inhibition of diacylglycerol lipase blocks endocannabinoid-mediated retrograde synaptic suppression: evidence for on-demand biosynthesis of 2-arachidonoylglycerol. J Physiol.
Hayes P, Meadows HJ, Gunthorpe MJ, Harries MH, Duckworth DM, Cairns W, et al. (2000). Cloning and functional expression of a human orthologue of rat vanilloid receptor-1. Pain 88(2): 205-215.
Herkenham M, Lynn AB, de Costa BR, Richfield EK (1991). Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res 547(2): 267-274.

63
Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, et al. (2013). Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci 16(9): 1291-1298.
Hillard C, Jarrahian A (2005). Accumulation of anandamide: evidence for cellular diversity. Neuropharmacology 48(8): 1072-1078.
Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997). Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem 69(2): 631-638.
Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, et al. (1999). Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther 289(3): 1427-1433.
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. (2005). An endocannabinoid mechanism for stress-induced analgesia. Nature 435(7045): 1108-1112.
Hohmann AG, Tsou K, Walker JM (1998). Cannabinoid modulation of wide dynamic range neurons in the lumbar dorsal horn of the rat by spinally administered WIN55,212-2. Neuroscience letters 257(3): 119-122.
Holt S, Comelli F, Costa B, Fowler CJ (2005). Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol 146(3): 467-476.
Holt S, Nilsson J, Omeir R, Tiger G, Fowler CJ (2001). Effects of pH on the inhibition of fatty acid amidohydrolase by ibuprofen. Br J Pharmacol 133(4): 513-520.
Holt S, Paylor B, Boldrup L, Alajakku K, Vandevoorde S, Sundstrom A, et al. (2007). Inhibition of fatty acid amide hydrolase, a key endocannabinoid metabolizing enzyme, by analogues of ibuprofen and indomethacin. Eur J Pharmacol 565(1-3): 26-36.
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. (2002). International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54(2): 161-202.
Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF (2012). DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol 8(12): 999-1007.
Hu H, Ho W, Mackie K, Pittman QJ, Sharkey KA (2012). Brain CB(1) receptor expression following lipopolysaccharide-induced inflammation. Neuroscience 227: 211-222.
Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM (2008). Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol 153(7): 1538-1549.
Huggins JP, Smart TS, Langman S, Taylor L, Young T (2012). An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153(9): 1837-1846.
Högestätt ED, Jönsson BA, Ermund A, Andersson DA, Björk H, Alexander JP, et al. (2005). Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem 280(36): 31405-31412.

64
Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW (2006). Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol 147(3): 281-288.
Jin XH, Uyama T, Wang J, Okamoto Y, Tonai T, Ueda N (2009). cDNA cloning and characterization of human and mouse Ca(2+)-independent phosphatidylethanolamine N-acyltransferases. Biochim Biophys Acta 1791(1): 32-38.
Kaczocha M, Glaser ST, Chae J, Brown DA, Deutsch DG (2010). Lipid droplets are novel sites of N-acylethanolamine inactivation by fatty acid amide hydrolase-2. J Biol Chem 285(4): 2796-2806.
Kaczocha M, Glaser ST, Deutsch DG (2009). Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci U S A 106(15): 6375-6380.
Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C (1997). cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem 272(43): 27218-27223.
Karlsson M, Påhlsson C, Fowler CJ (2004). Reversible, temperature-dependent, and AM404-inhibitable adsorption of anandamide to cell culture wells as a confounding factor in release experiments. Eur J Pharm Sci 22(2-3): 181-189.
Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. (2003). Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9(1): 76-81.
Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, et al. (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19(11): 4544-4558.
Kaufmann I, Hauer D, Huge V, Vogeser M, Campolongo P, Chouker A, et al. (2009). Enhanced anandamide plasma levels in patients with complex regional pain syndrome following traumatic injury: a preliminary report. European surgical research. Europaische chirurgische Forschung. Recherches chirurgicales europeennes 43(4): 325-329.
Khan KM, Cook JL, Bonar F, Harcourt P, Astrom M (1999). Histopathology of common tendinopathies. Update and implications for clinical management. Sports medicine 27(6): 393-408.
Kim J, Alger BE (2004). Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci 7(7): 697-698.
Kimura T, Ohta T, Watanabe K, Yoshimura H, Yamamoto I (1998). Anandamide, an endogenous cannabinoid receptor ligand, also interacts with 5-hydroxytryptamine (5-HT) receptor. Biol Pharm Bull 21(3): 224-226.
Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, et al. (2009). Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther 330(3): 902-910.
Kinsey SG, Naidu PS, Cravatt BF, Dudley DT, Lichtman AH (2011). Fatty acid amide hydrolase blockade attenuates the development of collagen-induced arthritis and related thermal hyperalgesia in mice. Pharmacology, biochemistry, and behavior 99(4): 718-725.
Kondo S, Kondo H, Nakane S, Kodaka T, Tokumura A, Waku K, et al. (1998). 2-Arachidonoylglycerol, an endogenous cannabinoid receptor agonist: identification as one of the major species of monoacylglycerols in various rat tissues, and evidence for its

65
generation through CA2+-dependent and -independent mechanisms. FEBS Lett 429(2): 152-156.
Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, et al. (1994). Inhibitors of arachidonoyl ethanolamide hydrolysis. J Biol Chem 269(37): 22937-22940.
Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, et al. (2002a). Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem 277(47): 44877-44885.
Kozak KR, Gupta RA, Moody JS, Ji C, Boeglin WE, DuBois RN, et al. (2002b). 15-Lipoxygenase metabolism of 2-arachidonylglycerol. Generation of a peroxisome proliferator-activated receptor alpha agonist. J Biol Chem 277(26): 23278-23286.
Kozak KR, Marnett LJ (2002c). Oxidative metabolism of endocannabinoids. Prostaglandins Leukot Essent Fatty Acids 66(2-3): 211-220.
Lagalwar S, Bordayo EZ, Hoffmann KL, Fawcett JR, Frey WH, 2nd (1999). Anandamides inhibit binding to the muscarinic acetylcholine receptor. J Mol Neurosci 13(1-2): 55-61.
Lang W, Qin C, Lin S, Khanolkar AD, Goutopoulos A, Fan P, et al. (1999). Substrate specificity and stereoselectivity of rat brain microsomal anandamide amidohydrolase. J Med Chem 42(5): 896-902.
Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z (2004). Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem 279(34): 36013-36021.
Leung K, Elmes MW, Glaser ST, Deutsch DG, Kaczocha M (2013). Role of FAAH-Like Anandamide Transporter in Anandamide Inactivation. PLoS One 8(11): e79355.
Lever IJ, Robinson M, Cibelli M, Paule C, Santha P, Yee L, et al. (2009). Localization of the endocannabinoid-degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. J Neurosci 29(12): 3766-3780.
Lichtman AH, Cook SA, Martin BR (1996). Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther 276(2): 585-593.
Ligresti A, Bisogno T, Matias I, De Petrocellis L, Cascio MG, Cosenza V, et al. (2003). Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 125(3): 677-687.
Ligresti A, Morera E, Van Der Stelt M, Monory K, Lutz B, Ortar G, et al. (2004). Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem J 380(Pt 1): 265-272.
Lim G, Sung B, Ji RR, Mao J (2003). Upregulation of spinal cannabinoid-1-receptors following nerve injury enhances the effects of Win 55,212-2 on neuropathic pain behaviors in rats. Pain 105(1-2): 275-283.
Liu J, Gao B, Mirshahi F, Sanyal AJ, Khanolkar AD, Makriyannis A, et al. (2000). Functional CB1 cannabinoid receptors in human vascular endothelial cells. Biochem J 346 Pt 3: 835-840.
Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, et al. (2006). A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A 103(36): 13345-13350.

66
Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. (2005). The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 67(1): 15-19.
Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. (2009). Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5(1): 37-44.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951). Protein measurement with the Folin phenol reagent. J Biol Chem 193(1): 265-275.
Maccarrone M, De Felici M, Bari M, Klinger F, Siracusa G, Finazzi-Agro A (2000). Down-regulation of anandamide hydrolase in mouse uterus by sex hormones. Eur J Biochem 267(10): 2991-2997.
Maccarrone M, van der Stelt M, Rossi A, Veldink GA, Vliegenthart JFG, Agrò AF (1998). Anandamide hydrolysis by human cells in culture and brain. J Biol Chem 273(48): 32332-32339.
Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001). Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31(3): 463-475.
Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, et al. (2007). Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol 150(6): 766-781.
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418(6897): 530-534.
Martin WJ, Coffin PO, Attias E, Balinsky M, Tsou K, Walker JM (1999). Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Res 822(1-2): 237-242.
Martin WJ, Hohmann AG, Walker JM (1996). Suppression of noxious stimulus-evoked activity in the ventral posterolateral nucleus of the thalamus by a cannabinoid agonist: correlation between electrophysiological and antinociceptive effects. J Neurosci 16(20): 6601-6611.
Martin WJ, Patrick SL, Coffin PO, Tsou K, Walker JM (1995). An examination of the central sites of action of cannabinoid-induced antinociception in the rat. Life Sci 56(23-24): 2103-2109.
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346(6284): 561-564.
McPartland JM, Glass M, Pertwee RG (2007). Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol 152(5): 583-593.
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. (1995). Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50(1): 83-90.
Michalski CW, Oti FE, Erkan M, Sauliunaite D, Bergmann F, Pacher P, et al. (2008). Cannabinoids in pancreatic cancer: correlation with survival and pain. International journal of cancer. Journal international du cancer 122(4): 742-750.

67
Min R, Di Marzo V, Mansvelder HD (2010). DAG lipase involvement in depolarization-induced suppression of inhibition: does endocannabinoid biosynthesis always meet the demand? Neuroscientist 16(6): 608-613.
Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, et al. (2006). Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 126(1-3): 102-114.
Movahed P, Jonsson BA, Birnir B, Wingstrand JA, Jorgensen TD, Ermund A, et al. (2005). Endogenous unsaturated C18 N-acylethanolamines are vanilloid receptor (TRPV1) agonists. J Biol Chem 280(46): 38496-38504.
Muccioli GG (2010). Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov Today 15(11-12): 474-483.
Muccioli GG, Labar G, Lambert DM (2008). CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. Chembiochem 9(16): 2704-2710.
Mukerji G, Yiangou Y, Agarwal SK, Anand P (2010). Increased cannabinoid receptor 1-immunoreactive nerve fibers in overactive and painful bladder disorders and their correlation with symptoms. Urology 75(6): 1514 e1515-1520.
Munro S, Thomas KL, Abu-Shaar M (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature 365(6441): 61-65.
Naccarato M, Pizzuti D, Petrosino S, Simonetto M, Ferigo L, Grandi FC, et al. (2010). Possible Anandamide and Palmitoylethanolamide involvement in human stroke. Lipids Health Dis 9: 47.
Nackley AG, Zvonok AM, Makriyannis A, Hohmann AG (2004). Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absence and presence of inflammation. Journal of neurophysiology 92(6): 3562-3574.
Naidu PS, Booker L, Cravatt BF, Lichtman AH (2009). Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther 329(1): 48-56.
Natarajan V, Schmid PC, Reddy PV, Zuzarte-Augustin ML, Schmid HH (1983). Biosynthesis of N-acylethanolamine phospholipids by dog brain preparations. J Neurochem 41(5): 1303-1312.
O'Brien M (1992). Functional anatomy and physiology of tendons. Clinics in sports medicine 11(3): 505-520.
Oddi S, Fezza F, Pasquariello N, D'Agostino A, Catanzaro G, De Simone C, et al. (2009). Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol 16(6): 624-632.
Oddi S, Fezza F, Pasquariello N, De Simone C, Rapino C, Dainese E, et al. (2008). Evidence for the intracellular accumulation of anandamide in adiposomes. Cellular and molecular life sciences : CMLS 65(5): 840-850.
Ohno-Shosaku T, Hashimotodani Y, Ano M, Takeda S, Tsubokawa H, Kano M (2007). Endocannabinoid signalling triggered by NMDA receptor-mediated calcium entry into rat hippocampal neurons. J Physiol 584(Pt 2): 407-418.
Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M (2002). Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci 15(6): 953-961.

68
Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N (2004). Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279(7): 5298-5305.
Omeir RL, Chin S, Hong Y, Ahern DG, Deutsch DG (1995). Arachidonoyl ethanolamide-[1,2-14C] as a substrate for anandamide amidase. Life Sci 56(23-24): 1999-2005.
Onnis V, Congiu C, Bjorklund E, Hempel F, Soderstrom E, Fowler CJ (2010). Synthesis and evaluation of paracetamol esters as novel fatty acid amide hydrolase inhibitors. J Med Chem 53(5): 2286-2298.
Ortega-Gutiérrez S, Hawkins EG, Viso A, López-Rodríguez ML, Cravatt BF (2004). Comparison of anandamide transport in FAAH wild-type and knockout neurons: evidence for contributions by both FAAH and the CB1 receptor to anandamide uptake. Biochemistry 43(25): 8184-8190.
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, et al. (2001). An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413(6855): 527-531.
Paria BC, Ma W, Andrenyak DM, Schmid PC, Schmid HHO, Moody DE, et al. (1998). Effects of cannabinoids on preimplantation mouse embryo development and implantation are mediated by brain-type cannabinoid receptors. Biol Reprod 58(6): 1490-1495.
Pasquariello N, Catanzaro G, Marzano V, Amadio D, Barcaroli D, Oddi S, et al. (2009). Characterization of the endocannabinoid system in human neuronal cells and proteomic analysis of anandamide-induced apoptosis. J Biol Chem 284(43): 29413-29426.
Patel JZ, Parkkari T, Laitinen T, Kaczor AA, Saario SM, Savinainen JR, et al. (2013). Chiral 1,3,4-Oxadiazol-2-ones as Highly Selective FAAH Inhibitors. J Med Chem 56(21): 8484-8496.
Patrignani P, Tacconelli S, Sciulli MG, Capone ML (2005). New insights into COX-2 biology and inhibition. Brain Res Brain Res Rev 48(2): 352-359.
Paylor B, Holt S, Fowler CJ (2006). The potency of the fatty acid amide hydrolase inhibitor URB597 is dependent upon the assay pH. Pharmacol Res 54(6): 481-485.
Pertwee RG (2001). Cannabinoids and the gastrointestinal tract. Gut 48(6): 859-867.
Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev 62(4): 588-631.
Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S, et al. (2007). Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology 52(2): 415-422.
Pisani A, Fezza F, Galati S, Battista N, Napolitano S, Finazzi-Agrò A, et al. (2005). High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson's disease patients. Ann Neurol 57(5): 777-779.
Rakhshan F, Day TA, Blakely RD, Barker EL (2000). Carrier-mediated uptake of the endogenous cannabinoid anandamide in RBL-2H3 cells. J Pharmacol Exp Ther 292(3): 960-967.
Rettori E, De Laurentiis A, Zorrilla Zubilete M, Rettori V, Elverdin JC (2012). Anti-inflammatory effect of the endocannabinoid anandamide in experimental periodontitis and stress in the rat. Neuroimmunomodulation 19(5): 293-303.

69
Rhodes KJ, Trimmer JS (2006). Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J Neurosci 26(31): 8017-8020.
Rice ASC, Cimino-Brown D, Eisenach JC, Kontinen VK, Lacroix-Fralish ML, Machin I, et al. (2008). Animal models and the prediction of efficacy in clinical trials of analgesic drugs: a critical appraisal and call for uniform reporting standards. Pain 139(2): 243-247.
Richardson JD, Kilo S, Hargreaves KM (1998). Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain 75(1): 111-119.
Riley G (2008). Tendinopathy--from basic science to treatment. Nature clinical practice. Rheumatology 4(2): 82-89.
Rockwell CE, Snider NT, Thompson JT, Vanden Heuvel JP, Kaminski NE (2006). Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor gamma independently of cannabinoid receptors 1 and 2. Mol Pharmacol 70(1): 101-111.
Rodríguez de Fonseca F, Navarro M, Gómez R, Escuredo L, Nava F, Fu J, et al. (2001). An anorexic lipid mediator regulated by feeding. Nature 414(6860): 209-212.
Ross RA (2003). Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140(5): 790-801.
Rozenfeld R, Devi LA (2008). Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. Faseb J 22(7): 2311-2322.
Ruiz-Llorente L, Sánchez MG, Carmena MJ, Prieto JC, Sánchez-Chapado M, Izquierdo A, et al. (2003). Expression of functionally active cannabinoid receptor CB1 in the human prostate gland. Prostate 54(2): 95-102.
Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, et al. (2007). The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152(7): 1092-1101.
Saario SM, Savinainen JR, Laitinen JT, Järvinen T, Niemi R (2004). Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem Pharmacol 67(7): 1381-1387.
Sagar DR, Kelly S, Millns PJ, O'Shaughnessey CT, Kendall DA, Chapman V (2005). Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci 22(2): 371-379.
Sai Y, Dai R, Yang T, Krausz K, Gonzalez F, Gelboin H, et al. (2000). Assessment of specificity of eight chemical inhibitors using cDNA-expressed cytochromes P450. Xenobiotica 30(4): 327-343.
Sakurada T, Noma A (1981). Subcellular localization and some properties of monoacylglycerol lipase in rat adipocytes. J Biochem 90(5): 1413-1419.
Sandberg A, Fowler CJ (2005). Measurement of saturable and non-saturable components of anandamide uptake into P19 embryonic carcinoma cells in the presence of fatty acid-free bovine serum albumin. Chem Phys Lipids 134(2): 131-139.
Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, et al. (2012). Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res 65(5): 553-563.

70
Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, et al. (2010). Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13(9): 1113-1119.
Schmid PC, Paria BC, Krebsbach RJ, Schmid HHO, Dey SK (1997). Changes in anandamide levels in mouse uterus are associated with uterine receptivity for embryo implantation. Proc Natl Acad Sci U S A 94(8): 4188-4192.
Schäbitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, et al. (2002). Release of fatty acid amides in a patient with hemispheric stroke: a microdialysis study. Stroke 33(8): 2112-2114.
Singh Tahim A, Sántha P, Nagy I (2005). Inflammatory mediators convert anandamide into a potent activator of the vanilloid type 1 transient receptor potential receptor in nociceptive primary sensory neurons. Neuroscience 136(2): 539-548.
Smith PB, Martin BR (1992). Spinal mechanisms of delta 9-tetrahydrocannabinol-induced analgesia. Brain Res 578(1-2): 8-12.
Snider NT, Kornilov AM, Kent UM, Hollenberg PF (2007). Anandamide metabolism by human liver and kidney microsomal cytochrome p450 enzymes to form hydroxyeicosatetraenoic and epoxyeicosatrienoic acid ethanolamides. J Pharmacol Exp Ther 321(2): 590-597.
Snider NT, Nast JA, Tesmer LA, Hollenberg PF (2009). A cytochrome P450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol Pharmacol 75(4): 965-972.
Sofia RD, Knobloch LC, Harakal JJ, Erikson DJ (1977). Comparative diuretic activity of delta9-tetrahydrocannabinol, cannabidiol, cannabinol and hydrochlorothiazide in the rat. Arch Int Pharmacodyn Ther 225(1): 77-87.
Spradley JM, Guindon J, Hohmann AG (2010). Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol Res 62(3): 249-258.
Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. (1995). 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun 215(1): 89-97.
Sun Y, Alexander SPH, Kendall DA, Bennett AJ (2006). Cannabinoids and PPARalpha signalling. Biochem Soc Trans 34(Pt 6): 1095-1097.
Suplita RL, 2nd, Gutierrez T, Fegley D, Piomelli D, Hohmann AG (2006). Endocannabinoids at the spinal level regulate, but do not mediate, nonopioid stress-induced analgesia. Neuropharmacology 50(3): 372-379.
Szekeres M, Nádasy GL, Turu G, Soltész-Katona E, Tóth ZE, Balla A, et al. (2012). Angiotensin II induces vascular endocannabinoid release, which attenuates its vasoconstrictor effect via CB1 cannabinoid receptors. J Biol Chem 287(37): 31540-31550.
Thomas BF, Gilliam AF, Burch DF, Roche MJ, Seltzman HH (1998). Comparative receptor binding analyses of cannabinoid agonists and antagonists. J Pharmacol Exp Ther 285(1): 285-292.
Thors L, Eriksson J, Fowler CJ (2007). Inhibition of the cellular uptake of anandamide by genistein and its analogue daidzein in cells with different levels of fatty acid amide hydrolase-driven uptake. Br J Pharmacol 152(5): 744-750.

71
Thors L, Fowler CJ (2006). Is there a temperature-dependent uptake of anandamide into cells? Br J Pharmacol 149(1): 73-81.
Tiger G, Stenström A, Fowler CJ (2000). Pharmacological properties of rat brain fatty acid amidohydrolase in different subcellular fractions using palmitoylethanolamide as substrate. Biochem Pharmacol 59(6): 647-653.
Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, et al. (1998). The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21(3): 531-543.
Tornqvist H, Belfrage P (1976). Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J Biol Chem 251(3): 813-819.
Ueda H, Kobayashi T, Kishimoto M, Tsutsumi T, Okuyama H (1993). A possible pathway of phosphoinositide metabolism through EDTA-insensitive phospholipase A1 followed by lysophosphoinositide-specific phospholipase C in rat brain. J Neurochem 61(5): 1874-1881.
Ueda N, Tsuboi K, Uyama T (2010). Enzymological studies on the biosynthesis of N-acylethanolamines. Biochim Biophys Acta 1801(12): 1274-1285.
Ueda N, Yamamoto K, Yamamoto S, Tokunaga T, Shirakawa E, Shinkai H, et al. (1995). Lipoxygenase-catalyzed oxygenation of arachidonylethanolamide, a cannabinoid receptor agonist. Biochim Biophys Acta 1254(2): 127-134.
Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ (2007). Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol 150(2): 186-191.
Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC (1999). Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A 96(21): 12198-12203.
Watanabe K, Ogi H, Nakamura S, Kayano Y, Matsunaga T, Yoshimura H, et al. (1998). Distribution and characterization of anandamide amidohydrolase in mouse brain and liver. Life Sci 62(14): 1223-1229.
Weber A, Ni J, Ling KH, Acheampong A, Tang-Liu DD, Burk R, et al. (2004). Formation of prostamides from anandamide in FAAH knockout mice analyzed by HPLC with tandem mass spectrometry. J Lipid Res 45(4): 757-763.
Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF (2006). A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 281(48): 36569-36578.
Welch SP, Thomas C, Patrick GS (1995). Modulation of cannabinoid-induced antinociception after intracerebroventricular versus intrathecal administration to mice: possible mechanisms for interaction with morphine. J Pharmacol Exp Ther 272(1): 310-321.
Wenger T, Ledent C, Csernus V, Gerendai I (2001). The central cannabinoid receptor inactivation suppresses endocrine reproductive functions. Biochem Biophys Res Commun 284(2): 363-368.
Willson T, Cobb J, Cowan D, Wiethe R, Correa I, Prakash S, et al. (1996). The structure-activity relationship between peroxisome proliferator-activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones. J Med Chem 39(3): 665-668.

72
Wright K, Rooney N, Feeney M, Tate J, Robertson D, Welham M, et al. (2005). Differential expression of cannabinoid receptors in the human colon: cannabinoids promote epithelial wound healing. Gastroenterology 129(2): 437-453.
Wu WJ, Yang Q, Cao QF, Zhang YW, Xia YJ, Hu XW, et al. (2010). [Membrane cholesterol mediates the endocannabinoids-anandamide affection on HepG2 cells]. Zhonghua Gan Zang Bing Za Zhi 18(3): 204-208.
Yaksh TL (1981). The antinociceptive effects of intrathecally administered levonantradol and desacetyllevonantradol in the rat. Journal of clinical pharmacology 21(8-9 Suppl): 334S-340S.
Yu M, Ives D, Ramesha CS (1997). Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem 272(34): 21181-21186.
Zhang L, Wang M, Bisogno T, Di Marzo V, Alger BE (2011). Endocannabinoids generated by Ca2+ or by metabotropic glutamate receptors appear to arise from different pools of diacylglycerol lipase. PLoS One 6(1): e16305.
Zygmunt PM, Ermund A, Movahed P, Andersson DA, Simonsen C, Jönsson BAG, et al. (2013). Monoacylglycerols Activate TRPV1 - A Link between Phospholipase C and TRPV1. PLoS One 8(12): e81618.
Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgard M, Di Marzo V, et al. (1999). Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400(6743): 452-457.
Åström M, Rausing A (1995). Chronic Achilles tendinopathy. A survey of surgical and histopathologic findings. Clinical orthopaedics and related research(316): 151-164.


















