
薬物乱用・依存状況の 実態把握と 薬物依存症者の …...薬物乱用・依存状況の 実態把握と 薬物依存症者の 社会復帰に向けた 支援に関する研究

ヒューマトロープヒューマトロープヒューマトロープヒューマトロープ C6mgC6mgC6mgC6mg ヒューマトロープヒューマトロープヒューマトロープヒューマトロープ C12mgC12mgC12mgC12mg
2.7 臨臨臨臨 床床床床 概概概概 要要要要
日本日本日本日本イーライリリーイーライリリーイーライリリーイーライリリー株式会社株式会社株式会社株式会社

ヒューマトロープ CTD(2.7 臨床概要)
2.7 臨床概要 ························································································································ 1
2.7.1 生物薬剤学及び関連する分析法の概要 ································································· 1
2.7.2 臨床薬理の概要······································································································ 2
2.7.3 臨床的有効性の概要 ······························································································ 3
2.7.3.1 背景及び概観 ···································································································· 3
(1) プラセボ対照二重盲検比較試験(K01A試験)············································· 3
(2) 長期臨床試験(K02A試験) ·········································································· 4
(3) 外国臨床試験(参考資料) ············································································ 4
2.7.3.2 個々の試験結果の要約 ····················································································· 7
(1) プラセボ対照二重盲検比較試験(K01A試験)············································· 7
(2) 長期臨床試験(K02A試験) ········································································ 16
(3) 外国臨床試験(参考資料) ·········································································· 23
2.7.3.3 全試験を通しての結果の比較と解析 ····························································· 25
2.7.3.3.1 試験対象集団 ······························································································ 25
(1) 国内臨床試験 ································································································ 25
(2) 外国臨床試験(参考資料) ·········································································· 27
2.7.3.3.2 全有効性試験の結果の比較検討································································· 28
(1) 有効性の主要評価(LBMの変化) ······························································ 28
(2) 有効性の主要評価に関連する外国臨床成績(参考資料) ··························· 30
(3) 有効性の副次評価 ························································································· 34
(4) その他の有効性評価(血清脂質の変化) ···················································· 48
2.7.3.3.3 部分集団における結果の比較 ···································································· 57
(1) 性別、発症時期 ······························································································ 57
(2) 高齢者における臨床効果(外国臨床成績;参考資料)······························· 59
2.7.3.4 推奨用法・用量に関する臨床情報の解析 ······················································ 60
(1) 投与開始用量(0.021 mg/kg/週)及び最大用量(0.084 mg/kg/週)の設
定根拠 ··········································································································· 61
(2) 血清 IGF-I濃度を指標とした用量調整法の設定根拠 ··································· 64
(3) 1日投与量の上限·························································································· 72
(4) 投与量の性差に関する考察 ·········································································· 72
2.7.3.5 効果の持続、耐薬性 ······················································································· 75
2.7.4 臨床的安全性の概要 ···························································································· 79
2.7.4.1 医薬品への曝露 ······························································································ 79
2.7.4.1.1 総括的安全性評価計画及び安全性試験の記述 ··········································· 79
2.7.4.1.2 全般的な曝露状況······················································································· 79
(1) 国内臨床試験における曝露(K01A試験及び K02A試験)························· 79
(2) 外国臨床試験における曝露(E003/4試験、E005/6試験、GDED 試験
及び T002試験)(参考資料) ···································································· 81
2.7.4.1.3 治験対象集団の人口統計学的特性及びその他の特性 ································ 83

ヒューマトロープ CTD(2.7 臨床概要)
(1) 国内臨床試験 ································································································ 83
(2) 外国臨床試験(参考資料) ·········································································· 85
2.7.4.2 有害事象 ········································································································· 88
2.7.4.2.1 有害事象の解析 ·························································································· 88
(1) 国内 AGHD被験者における安全性の検討 ··················································· 88
(2) 外国 AGHD被験者における安全性の検討(参考資料) ··························· 117
2.7.4.2.1.1 比較的よく見られる有害事象 ·························································· 122
(1) 国内臨床試験で比較的よく見られた有害事象の重症度別での検討 ·········· 122
(2) 国内臨床試験で比較的よく見られた有害事象の投与期間での検討 ·········· 125
(3) 国内臨床試験で比較的よく見られた有害事象の投与量での検討 ·············· 128
2.7.4.2.1.2 死 亡 ······························································································ 129
(1) 国内臨床試験 ······························································································ 129
(2) 外国臨床試験(参考資料) ········································································ 129
2.7.4.2.1.3 その他の重篤な有害事象 ································································· 131
(1) 国内臨床試験で認められた、その他の重篤な有害事象····························· 131
(2) 外国臨床試験で認められた、その他の重篤な有害事象(参考資料)······· 133
2.7.4.2.1.4 その他の重要な有害事象 ································································· 138
(1) 国内臨床試験 ······························································································ 138
(2) 外国臨床試験(参考資料) ········································································ 141
2.7.4.2.1.5 器官別又は症候群別有害事象の解析 ··············································· 143
(1) 国内臨床試験 ······························································································ 143
(2) 外国臨床試験(参考資料) ········································································ 147
2.7.4.2.2 個別有害事象の文章による説明 ······················································ 149
2.7.4.3 臨床検査値の評価························································································· 149
(1) 国内臨床試験 ······························································································ 149
(2) 外国臨床試験(参考資料) ········································································ 166
2.7.4.4 バイタルサイン、身体的所見及び安全性に関連する他の観察項目 ············ 168
(1) 国内臨床試験 ······························································································ 168
(2) 外国臨床試験(参考資料) ········································································ 177
2.7.4.5 特別な患者集団及び状況下における安全性 ················································ 181
2.7.4.5.1 内因性要因································································································ 181
(1) 男女別及び発症時期別の有害事象 ····························································· 181
(2) 年齢別の有害事象 ······················································································· 184
2.7.4.5.2 外因性要因································································································ 191
2.7.4.5.3 薬物相互作用 ···························································································· 191
2.7.4.5.4 妊娠及び授乳時の使用 ············································································· 191
2.7.4.5.5 過量投与 ··································································································· 191
2.7.4.5.6 薬物乱用 ··································································································· 191
2.7.4.5.7 離脱症状及び反跳現象 ············································································· 191

ヒューマトロープ CTD(2.7 臨床概要)
2.7.4.5.8 自動車運転及び機械操作に対する影響又は精神機能の障害 ··················· 192
2.7.4.6 市販後データ ································································································ 193
(1) GDDQ調査 ································································································· 193
(2) 定期的安全性最新報告················································································ 208
2.7.5 参考文献············································································································· 220
2.7.6 個々の試験のまとめ ·························································································· 222
(1) 臨床試験一覧表··························································································· 222
(2) 個々の試験の概要 ······················································································· 225

ヒューマトロープ CTD(2.7 臨床概要) Page 1
1
2.7 臨床概要臨床概要臨床概要臨床概要 2.7.1 生物薬剤学及び関連する分析法の概要
今回の申請は新効能医薬品としての一部変更承認申請であり、新たに追加すべきデータはな
いため、本項は該当しない。

ヒューマトロープ CTD(2.7 臨床概要) Page 2
2
2.7.2 臨床薬理の概要
ヒトでの薬物動態については、初回申請(下垂体性小人症)及びターナー症候群における低
身長の効能追加の申請時の資料(1987年 11月及び 1990年 9月申請)で提出した。それ以降、
19 年に外国臨床データの外挿の可能性を考慮して、日本人と白人における薬物動態の民族間
差を検討するための臨床薬物動態試験( GDFR試験*)を実施した。その結果、日本人
と白人とで薬物動態は類似していた。これは既に提出資料に記載されている薬物動態に民族間
差はないという考察と一致していた。しかしながら、本申請は国内臨床データのみを評価資料
としており、かつ日本人における薬物動態に関する資料は、既提出資料で十分であると判断し
た。
* 新薬承認情報提供時に置き換えた

ヒューマトロープ CTD(2.7 臨床概要) Page 3
3
2.7.3 臨床的有効性の概要
2.7.3.1 背景及び概観
成人成長ホルモン分泌不全症(Adult Growth Hormone Deficiency;以下 AGHD)に対する国
内臨床第Ⅲ相試験の開始に先立ち、20 年 月 日に第Ⅱ相試験終了後治験相談を行った。
その後、医薬品副作用被害救済・研究振興調査機構から得られた助言を踏まえ、LY137998(以
下、本剤)の有効性を評価するための臨床試験計画を立案した。
「
」との助言を踏まえ、
国内第Ⅲ相試験では、プラセボ対照二重盲検比較試験(K01A試験)を実施した。また、AGHD
患者に対する GH補充療法は長期にわたることが予想されるため、プラセボ対照二重盲検比較
試験終了後に、同一被験者を対象に、継続して投与し、長期投与における有効性を検討するた
め長期臨床試験(K02A試験)を実施した。
なお、外国で実施された臨床試験(E003/4試験、E005/6試験、GDED試験、T002試験)は、
有効性評価の参考資料とした。
(1) プラセボ対照二重盲検比較試験(K01A試験)
20 年 月 日の治験相談において、「
」との医薬品副作用被害救済・研究振興調
査機構の見解を得た。
本治験は AGHDに対する GH補充療法の有効性及び安全性を、プラセボを対照として検討し
た多施設共同二重盲検比較試験である。20 年 月から 20 年 月まで AGHD患者 65例を
対象に、本剤又はプラセボを二重盲検下で 24 週間投与し、有効性を検証するとともに安全性
を評価した。
対象患者として、18歳以上 64歳未満(同意取得時)の日本人男女で、AGHDを発症しうる
病因を経験し、GH分泌刺激試験において GH頂値が 3 ng/mL未満である AGHD患者を選択し
た。
目標症例数については、外国臨床成績(GDED試験)を参考に、主要評価項目である除脂肪
体重(Lean Body Mass;以下 LBM)の有意な変化を検出できる症例数を算出した。その結果、
58例を最大の解析対象集団の症例数とした。また、小児期発症型の患者数を確保するため、58
例中小児期発症型の症例数を 18例以上とした。更に投与開始までの脱落率を 15%と仮定し、
最終的に目標組入れ症例数を 68例とした。
組み入れられた症例は、登録センターにより登録票の内容から組入れ基準を満たしているこ
とを確認された後、AGHDの発症時期(成人期・小児期)を層別因子として、性別、年齢及び
施設を因子とした層別確率化最小化法に基づき、各治療群に割り付けられた。
投与期間は 24週間とし、本剤を投与した群においては、1週間に体重(kg)当たり、ソマト
ロピン(遺伝子組換え)として規定する用量を、6~7回に分けて皮下投与した。用量は最初の
4週間、次の 8週間及び最後の 12週間を、それぞれ 0.021 mg/kg/週、0.042 mg/kg/週及び 0.084
mg/kg/週とした。なお、プラセボを投与した群においては LY群と同一液量のプラセボを投与
した。

ヒューマトロープ CTD(2.7 臨床概要) Page 4
4
本治験への参加の同意があり、組入れ基準をすべて満たし、治療群を割り付けられた被験者
のうち、1 回以上の治験薬投与が行われた被験者の集団を最大の解析対象集団と定義し、有効
性の評価に用いた。また、主要評価項目は LBMの変化率とした。主要目的は、本剤投与群の
投与 24週目におけるベースラインからの LBMの変化率がプラセボ群に比べて、有意に大きい
ことを検証することである。なお、治療群間における比較には Studentの t検定を用い、両群の
母平均値が等しいという帰無仮説に対し、両群の母平均値が異なるという対立仮説を検定した。
また、投与 24 週目におけるベースラインからのインスリン様成長因子-I(Insulin-like Growth
Factor-I;以下 IGF-I)濃度、インスリン様成長因子結合蛋白質-3(Insulin-like Growth Factor Binding
Protein-3;以下 IGFBP-3)濃度、QOL及び血清脂質の変化を治療群間で比較した。
本治験では、主要な解析において LOCFを用いることとし、試験実施計画書にその旨を規定
した。これは以下の理由によるものである。
本治験は最大の解析集団を有効性の解析対象集団とした。LBMの解析において、投与後 24
週のデータが欠損の場合、変化率を計算することができず、早期に中止された症例は解析から
除かざるを得なくなる。この場合、実際の解析の対象となる集団と最大の解析対象集団との間
に乖離が生じることとなる。このことを回避するため、欠損データを何らかの方法で補填する
こととした。補填の方法は、通常よく使われている LOCFを採用した。その際、外国臨床試験
の成績から、早期に中止した場合には、LBMの大きな変化は見込めないため、LOCFを用いる
ことにより実薬群に有利になることはないと判断した。
(2) 長期臨床試験(K02A試験)
本試験では、20 年 月から、先行するプラセボ対照二重盲検比較試験(K01A試験)に参
加した AGHD患者を対象に、非盲検無対照試験で本剤の 48週間の投与を行い、長期投与にお
ける有効性を評価した。
先行する K01A試験において、本剤が投与された患者に対しては、継続投与により、LBMの
変化が維持されることを確認した。また、プラセボが投与された患者に対しては、長期試験期
間と二重盲検比較期間の LBMの変化率を比較した。更に IGF-I濃度、IGFBP-3濃度、QOL及
び血清脂質の本剤投与前後の変化も併せて評価した。
投与期間は 48週間とした。用量は、治験開始から、1週間に体重(kg)あたり、0.021 mgを
6~7回に分けて 8週間皮下投与した。その後は、前回来院時の血清 IGF-I濃度に基づいて、0.021
~0.084 mg/kg/週の範囲内で、適宜投与量を増減した。この際、「IGF-I濃度の年齢別男女別基
準範囲」を参照し、血清 IGF-I濃度が-1.96SD以上、+1.96SD以下に維持されるように調整を
行った。このように各患者の血清 IGF-I濃度に基づき投与量を調整する方法は、GRSコンセン
サス・ガイドライン、外国臨床試験成績及び外国における使用経験を参考にしたものである。
また、本治験では、K01A試験と同様の理由により、主要な解析において、LOCFを用いた。
(3) 外国臨床試験(参考資料)
1) 成人期発症型の AGHD患者に対する GH補充療法の有効性評価(E003試験、E004試験)
19 年から、欧州 6ヵ国 7施設において、成人期発症型の AGHD患者を対象に GH補充療法
の有効性を検討した。本試験の成績は欧米における承認申請に用いられた。

ヒューマトロープ CTD(2.7 臨床概要) Page 5
5
有効性の主要評価項目は、皮下脂肪厚、生体インピーダンス法で測定した体組成値〔LBM、
体脂肪量(Fat Mass;以下 FM)、体脂肪率〕、Nottingham Health Profile(以下、NHP)を用い
た QOLスコア、血清脂質濃度(総コレステロール値、HDLコレステロール値、LDLコレステ
ロール値、VLDLコレステロール値、中性脂肪、アポリポ蛋白質 A-I及び B)とした。また、
副次評価項目は、血清 IGF-I濃度、血清 IGFBP-3濃度、血清オステオカルシン濃度、尿中ピリ
ジノリン濃度、尿中デオキシピリジノリン濃度及び日常生活動作とした。
1 ヵ月の単盲検プラセボ投与観察期から開始し、6 ヵ月のプラセボ対照二重盲検比較期間の
後、非盲検下で 12ヵ月本剤を投与した。投与量は、最初の 4週間を 6.25 µg /kg/日とし、その
後 12.5 µg /kg/日に増量した。12ヵ月の非盲検期では、最初の 4週間を 6.25 µg /kg/日に減量し
た後、12.5 µg /kg/日に増量して、11ヵ月間投与を継続した。
E003試験と E004試験は、同一内容の実施計画により実施したため、E003/4試験として併合
した結果を示した。
2) 小児期発症型の AGHD患者に対する GH補充療法の有効性評価(E005試験、E006試験)
19 年から、欧州 6ヵ国 8施設において、小児期発症型の AGHD患者を対象に GH補充療法
の有効性を検討した。本試験の成績は欧米における本剤の承認申請に用いられた。
有効性の主要評価項目は、皮下脂肪厚、生体インピーダンス法で測定した体組成値(LBM、
FM、体脂肪率)、NHPを用いた QOLスコア、血清脂質濃度(総コレステロール値、HDLコ
レステロール値、LDLコレステロール値、VLDLコレステロール値、中性脂肪、アポリポ蛋白
質 A-I及び B)とした。また、副次評価項目は、血清 IGF-I濃度、血清 IGFBP-3濃度、血清オ
ステオカルシン濃度、尿中ピリジノリン濃度、尿中デオキシピリジノリン濃度及び日常生活動
作とした。
1 ヵ月の単盲検プラセボ投与観察期から開始し、6 ヵ月のプラセボ対照二重盲検比較期間の
後、非盲検下で 12ヵ月本剤を投与した。投与量は、最初の 4週間を 6.25 µg /kg/日とし、その
後 12.5 µg /kg/日に増量した。12ヵ月の非盲検期では、最初の 4週間を 6.25 µg /kg/日に減量し
た後、12.5 µg /kg/日に増量して、11ヵ月間投与を継続した。
E005試験と E006試験は、同一内容の実施計画により実施したため、E005/6試験として、併
合した結果を示した。
3) AGHD患者に対する GHの投与量別の有効性を評価した市販後臨床試験(GDED試験)
19 ~19 年、欧州及びカナダ 13ヵ国 56施設において、AGHD患者を対象に、GHの低用
量(3.0 µg/kg/日→6.0 µg/kg/日)を投与した群と高用量(6.0 µg/kg/日→12.0 µg/kg/日;既承認用
量)を投与した群の有効性を、無作為化非盲検並行群試験で比較した。本試験では、成人期発
症型及び小児期発症型の AGHD患者のいずれも対象とした。
投与期間は 6ヵ月に設定した。有効性の主要評価項目は、LBMの変化率とした。また、副次
評価項目は、FM、体脂肪率、皮下脂肪厚、BMI、ウエスト/ヒップ比、QOL(Questions on Life
Satisfaction;以下、QLS)、血清 IGF-I濃度、血清 IGFBP-3濃度及び血中脂質とした。低用量
群の投与量は、最初の 3ヵ月を 3.0 µg/kg/日、次の 3ヵ月を 6.0 µg/kg/日とした。一方、高用量
群の投与量は、最初の 3ヵ月を 6.0 µg/kg/日、次の 3ヵ月を 12.0 µg/kg/日とした。

ヒューマトロープ CTD(2.7 臨床概要) Page 6
6
4) AGHD患者を対象として 2種類の用法・用量を検討した市販後臨床試験(T002試験)
19 ~20 年、欧米 6ヵ国 61施設において、AGHD患者を対象に、用量の決定方法が異な
る 2群間における有効性の成績を、無作為化非盲検並行群試験で比較した。本試験では、成人
期発症型及び小児期発症型の AGHD患者のいずれも対象とした。
有効性の主要評価項目は、FM のベースラインからの変化率とした。また、副次評価項目は
QOL(QLS-H)、LBM、血中脂質、血清 IGF-I濃度とした。用量の決定方法の違いにより、無
作為に 2群に割り付けた。最初の 8週間は 200 µg/日を投与し、その後 8週間ごとに、血清 IGF-I
濃度及び臨床症候に基づいて用量を調整する患者群を ID群とした。一方、最初の 16週間、次
の 8週間及び最後の 8週間の用量をそれぞれ、4.0 µg/kg/日、8.0 µg/kg/日及び 12.0 µg/kg/日とす
る群を FD群とした。ただし、FD群も血清 IGF-I濃度が規定した数値以上に上昇したときは減
量することとした。

ヒューマトロープ CTD(2.7 臨床概要) Page 7
7
2.7.3.2 個々の試験結果の要約
(1) プラセボ対照二重盲検比較試験(K01A試験)
試験方法の概略を表 1に示した。
表 1 試験方法の概略(K01A試験)
項 目 内 容
治験題目 成人成長ホルモン分泌不全症患者における LY137998[ソマトロピン(遺伝子組換え)]のプラセボ対照二重盲検比較試験
治験の目的 成人成長ホルモン分泌不全症患者において、実薬投与群の投与 24週目におけるベースラインからの全身 LBMの変化率がプラセボ投与群に比し有意に大きいことを検証する。
治験の種類 プラセボ対照、多施設共同、無作為化、並行群間、二重盲検比較試験 対象被験者 成人成長ホルモン分泌不全症 選択基準 本治験に組み入れられるには、以下のすべての基準を満たす必要がある。
(1) 小児期(18歳未満)又は成人期(18歳以上)に、成人成長ホルモン分泌不全症を発症し得る、以下のいずれかの病因を経験した者(ただし、小児低身長に対する治療を行った患者の場合、成長速度が 1.0 cm/年以下になっていること) 1) 下垂体又はその周辺の腫瘍の治療を受け、寛解した者(この場合、当該治療完了後、本治験参加への同意取得までに 2年以上経過している者を対象とする)
2) 下垂体又はその周辺を損傷し得る外傷を受けた者 3) シーハン症候群患者 4) トルコ鞍空虚の認められる者 5) 小児期に特発性又は先天性の成長ホルモン分泌不全症であると診断された者
(2) 成長ホルモン分泌刺激試験*において GH頂値が 3.0 ng/mL未満である者 *インスリン負荷試験を行う。ただし、インスリン負荷試験禁忌の患者(心電図異常、虚血性心疾患の既往歴/現病歴又は痙攣性の障害がある等)又はインスリン負荷試験の経験が少ない 60歳以上の患者で、インスリン負荷試験が困難と判断される場合はアルギニン負荷試験又はグルカゴン負荷試験を行う。
(3) 他の下垂体ホルモンが欠乏しており治療が必要とされる場合、少なくとも本治験参加への同意取得前 3ヵ月の間、必要なホルモンの補充療法を受けている者 ただし、性腺刺激ホルモン欠損又は卵巣疾患により女性ホルモンの補充療法を受ける必要のある女性の場合は、以下の者を本治験の対象とする。 1) 本治験参加への同意取得時における年齢が 40歳未満の場合:少なくとも同意取得前 3ヵ月の間、当該療法を受けている者
2) 本治験参加への同意取得時における年齢が 40歳以上 55歳未満の場合:少なくとも同意取得前 3ヵ月の間、当該療法実施の有無に変更がない者
3) 本治験参加への同意取得時における年齢が 55歳以上の場合:少なくとも同意取得前 3ヵ月の間、当該療法を受けていない者
(4) 本治験参加への同意取得時に 18歳以上 64歳未満の日本人男女 除外基準 以下の基準のいずれかに該当する者は本治験の対象から除外する。
<既往症・合併症> (1) 高度の心機能障害、肺機能障害、腎機能障害、肝機能障害の認められる者 (2) 高度の精神疾患の認められる者 (3) 悪性腫瘍の認められる者 (4) 末端肥大症の既往のある者 (5) 良性頭蓋内圧亢進の認められる者 (6) 増殖性網膜障害の認められる者 (7) 治療を行っても十分に血圧コントロールができない高血圧患者 (8) 糖尿病患者 (9) 高度の神経筋疾患の認められる者 (10) 染色体性又は遺伝性の異常(malformation)(成長ホルモン分泌不全症の病因となる
遺伝障害を除く)の認められる者 <薬剤投与> (11) 本治験参加への同意取得前 3ヵ月以内に食欲抑制剤又は抗うつ薬の投与を受けた者

ヒューマトロープ CTD(2.7 臨床概要) Page 8
8
項 目 内 容 (12) 本治験参加への同意取得前3ヵ月以内に長期使用の目的で補充量以外の用量で副腎皮
質ステロイドの投与を受けている者、又は性腺ステロイド補充療法以外に本治験参加への同意取得前3ヵ月以内に長期使用の目的で同化ステロイドの投与を受けている者(上記薬剤の補充療法を受けている患者で、患者の状態により必要と判断された結果、一時的に用量を適宜増減した場合は本基準に該当しない)
(13) 本治験参加への同意取得前4ヵ月以内に厚生労働省の承認を得ていない薬剤の投与を受けた者
(14) 本治験参加への同意取得前 6ヵ月以内に成長ホルモンの投与を受けた者 <その他> (15) 治験責任医師、治験分担医師、治験協力者、治験依頼者の社員及びそれらの配偶者、
両親、子供、兄弟姉妹(血縁関係及び法律上の関係の双方を含む) (16) 妊婦、授乳中又は妊娠している可能性のある女性、もしくは予定の治験期間中に妊娠
を希望している女性 (17) 金属片、ギブス包帯等、二重エネルギーX線吸収法(Dual Energy X-ray Absorptiometry、
以下 DEXA法)による検査を妨害することが明らかな体内・体外器具を装着している者
(18) 本治験の実施計画(投与スケジュール、来院等)の遵守が困難である者 (19) その他、治験責任医師又は治験分担医師が本治験の対象として適当でないと判断した
者 目標症例数 最大の解析対象集団対象例として、58例(LY137998投与群及びプラセボ投与群各 29例)。
そのうち、小児期発症型を 18例以上と定める。以上に投与開始までの脱落 15%を考慮し、組入れ症例数として 68例とする。
使用薬剤 LY137998 ソマトロピン(遺伝子組換え)6.56 mg 専用注入器装着時、6 mgまで使用できる プラセボ製剤 アミノ酢酸及びマンニトールを主体とするプラセボ薬剤
用法・用量 0.021 mg/kg/週を開始用量として皮下投与し、0.084 mg/kg/週まで漸増する。プラセボ投与群においては LY137998投与群と同一液量のプラセボを投与する。但し、体重を基に算出される 1日投与量が 1mgを超える場合は、体重に関わらず 1mg/日を投与する。同一の副作用が 2週間以上継続して認められた場合、治験責任医師又は治験分担医師が規定の 25-50%減量の投与の必要性を判断する。
投与期間 24週間 併用禁止薬 食欲抑制剤
抗うつ薬 厚生労働省の承認を得ていない他の薬剤 他の成長ホルモン製剤

ヒューマトロープ CTD(2.7 臨床概要) Page 9
9
項 目 内 容
観察・検査・評価項目及び時期
来院 同意取得時
スクリーニング時
登録時投与開始直前
投与4週目
投与8週目
投与 12週目
投与 24週目 中止時
規定検査日 ─ ─ -28日目 1日目 29日目 57日目 85日目 169日目 ─
範囲(日) ─ ─ ±7 ─ ±7 ±7 ±7 ±14 ─
同意取得 X
患者背景 X
成長ホルモン 分泌刺激試験
X
臨床検査 X X X X X X
血圧 X X X X X X
身長 X
体重 X X X X X X LBM X X X QOL (SF36、QLS) X X X X
投与量 X X X X X
投与状況 X X X X X
合併症及び 有害事象
X X X X X X X X X
観察・検査等の項目
併用薬 併用療法
X X X X X X X X X
主要評価項目 各被験者の投与 24週目における LBMのベースラインからの変化率 解析方法 主要評価項目:LBM
各被験者の投与 24週目における LBMのベースラインからの変化率につき、治療群ごとに要約統計量を算出し、治療群間における比較検定を行う。比較には Studentの t検定を用い、両群の母平均値が等しいという帰無仮説に対し、両群の母平均値が異なるという対立仮説を検定し、この検定を本治験の主要解析とする。ただし、有意水準は両側 5%とする。また、両群母平均値の差の推定値と信頼係数両側 95%の信頼区間を示す。変化率の分布に正規性が仮定出来ない場合は、Studentの t検定ではなくWilcoxon検定を行うとともに優越確率差の信頼係数両側 95%の信頼区間を求める。検定により有意差が認められた場合には、成長ホルモン分泌不全症の発症時期についての質的交互作用の有無を検討する。 成長ホルモン分泌不全症の発症時期、及び発症時期と性別により層別した部分集団において、上記と同様な解析を行う。 安全性の解析 (1) 有害事象
各治療群について、投与後観測された有害事象の発現例数及び発現件数を、重症度及び因果関係別に集計する。重症度分布の集計には、同一被験者に重症度の異なる同一事象が発現した場合、重症度の最も高い事象のみを用いる。 各治療群について、投与後観測された有害事象の発現例数及び発現件数を 4週間ごとに算出し、発現頻度の経時推移グラフを作成する。
(2) 臨床検査値及び血圧 各治療群について、来院ごとに要約統計量を算出すること及び経時推移グラフを作成することにより経時推移を要約する。 各治療群について、投与開始直前における観測値を投与前値、投与 24週目における観測値を投与後値とした、投与前後の比較を行う。ただし、中止された被験者については中止時の観測値を投与後値とする。 各治療群について、正常範囲を用いて各観測値を異常高値、正常、異常低値に分類し、投与前値と投与後値の交差分類表を作成する。また、異常変動の発現例数を重症度及び因果関係別に集計する。
治験調整医師

ヒューマトロープ CTD(2.7 臨床概要) Page 10
10
項 目 内 容
治験 アドバイザー
外部治験 アドバイザー
代表施設名 及び施設数
計 25施設
治験期間 20 年 月~20 年 月
1) 被験者の内訳
被験者の内訳を図 1に示した。本治験は 25施設で実施し、23施設から 69名の被験者が参加
した。そのうち、スクリーニング検査で不適格とされた 1例、登録時に不適格とされた 2例及
び同意を撤回した 1例、計 4例を除く 65例が登録され、LY群に 33例、プラセボ群に 32例が
割り付けられた。
65例に本剤又はプラセボが投与された。LY群では、完了例は 31例であった。また、2例が
有害事象により治験を中止した。一方、プラセボ群では、完了例は 29 例であった。また、中
止例は 3例で、有害事象による中止、治験責任医師の判断による中止及び必要な観察・測定が
困難になったことによる中止が、それぞれ 1例であった。
図 1 被験者の内訳(K01A試験)
*1:スクリーニング時不適格(1)、登録時不適格(2)、同意撤回(1) *2:中止(2){有害事象(2)}*3:中止(3){有害事象(1)、医師の判断(1)、継続的実施の困難(1)}
非組入れ例(4)*1
未投与例(0)
完了例(31) 未完了例(2)*2
LY 群(33)
完了例(29) 未完了例 (3)*3
プラセボ群(32)
投与例(65)
組入れ例(65)
同意取得例(69)

ヒューマトロープ CTD(2.7 臨床概要) Page 11
11
2) 患者背景
本剤又はプラセボが投与された 65例のうち、1例は組入れ基準を満たしていなかったことが
治験終了後に判明したため、評価対象例から除外した。その結果、64例を有効性評価対象例と
した。
有効性評価対象例の患者背景を表 2に示した。
表 2 患者背景(K01A試験)
項 目 LY群 (N= 33)
プラセボ群 (N= 31) p値*1
平均±標準偏差 37.2±13.9 39.0±13.8 0.600
18~19 2 (6.1%) 1 (3.2%)
20~29 10 (30.3%) 9 (29.0%)
30~39 8 (24.2%) 8 (25.8%) 同意時年齢
40~63 13 (39.4%) 13 (41.9%)
0.955
男 性 16 (48.5%) 15 (48.4%) 性 別
女 性 17 (51.5%) 16 (51.6%) 0.994
成人期発症型 14 (42.4%) 13 (41.9%) 発症時期
小児期発症型 19 (57.6%) 18 (58.1%) 0.968
特発性又は先天性 6 (18.2%) 9 (29.0%)
腫 瘍 14 (42.4%) 9 (29.0%)
腫瘍に対する治療 9 (27.3%) 10 (32.3%)
シーハン症候群 2 (6.1%) 2 (6.5%)
トルコ鞍空虚 1 (3.0%) 1 (3.2%)
成長ホルモン分泌不全の病因
外 傷 1 (3.0%) 0 (0.0%)
0.749
GH単独分泌不全症 0 (0.0%) 1 (3.2%) 病 態
複合型下垂体機能低下症 33 (100.0%) 30 (96.8%) 0.298
LBM (kg;平均±標準偏差) 40.52±9.70 38.99±10.19 0.413
IGF-I (ng/mL;平均±標準偏差) 65±46 73±49 0.563
IGF-I SDS*2 (平均±標準偏差) -2.42±1.85 -2.03±1.58 0.643
*1 年齢、LBM、IGF-I:Wilcoxon検定、カテゴリカル項目:カイ 2乗検定 *2 IGF-I Standard Deviation Score
割付因子である年齢、性別及び発症時期を含む主な患者背景並びにベースライン時の有効性
評価項目について、LY群とプラセボ群との間に不均衡は認められなかった。
組み入れられた症例の平均年齢は、LY群で 37.2歳、プラセボ群で 39.0歳であった。性別は、
LY群で男性 16例及び女性 17例であった。一方、プラセボ群では男性 15例及び女性 16例であっ
た。成人期発症型及び小児期発症型の症例数は、LY群でそれぞれ 14例及び 19例、プラセボ
群で 13例及び 18例であった。成長ホルモン分泌不全の病因は、LY群では、腫瘍が 14例(42.4%)、
腫瘍に対する治療が 9例(27.3%)、特発性又は先天性が 6例(18.2%)の順であった。プラセ
ボ群においてもこれらの頻度が高く、腫瘍が 9例(29.0%)、腫瘍に対する治療が 10例(32.3%)、
特発性又は先天性が 9例(29.0%)であった。病態については、プラセボ群の成人期発症型の 1
例を除く全例が複合型であった。また、IGF-I SDSのベースラインについては、LY群の 21例
及びプラセボ群の 16例は基準範囲以下であった。

ヒューマトロープ CTD(2.7 臨床概要) Page 12
12
3) LBMの変化
本剤及びプラセボ投与前後における LBMの変化を表 3に示した。LY群の LBMは増加し、
ベースラインからの平均変化率は 4.7%であり、投与前後で統計学的有意差が認められた
(p<0.001)。一方、プラセボ群では、ベースラインからの平均変化率は-0.5%であった。
表 3 LBMの変化
項目 投与群 来院 例数 平均 標準偏差 最大値 中央値 最小値 p値*2
0週 33 40.52 9.702 59.80 38.18 28.43 - 24週 31 42.18 9.609 62.94 40.19 30.47 -
24週 LOCF 32 41.94 9.546 62.94 38.91 30.47 - LY群
変化*1 32 4.7 3.88 15.6 5.0 -2.4 <0.001
0週 31 38.99 10.186 67.89 35.46 26.12 -
24週 29 38.27 9.717 65.55 34.24 26.94 -
24週 LOCF 29 38.27 9.717 65.55 34.24 26.94 -
LBM (kg)
プラセボ群
変化*1 29 -0.5 4.10 9.2 -0.7 -6.5 0.519
*1 0週から 24週 LOCFまでの変化率(%) *2 対応のある t検定(ベースライン vs 24週 LOCF)
4) IGF-I及び IGFBP-3の変化
LY群及びプラセボ群における血清 IGF-I濃度と血清 IGFBP-3濃度の分布の変化を表 4に示
した。
LY群の血清 IGF-I濃度は投与前には 33例中 21例が基準範囲より低く、12例が基準範囲内
の値を示したが、本剤投与後 24週時には 31例中 22例が基準範囲内の値を示し、6例が基準範
囲超の高値を示し、3例が基準範囲より低かった。一方、プラセボ群では、投与前には 31例中
17例が基準範囲より低かったが、本剤投与後 24週時においても 29例中 17例が基準範囲より
低かった。
LY群の血清 IGFBP-3濃度は投与前には 33例中 8例が基準範囲内の値を示し、1例が基準範
囲超の高値を示し、24例が基準範囲より低かったが、本剤投与後 24週時には 31例中 22例が
基準範囲内の値を示し、5例が基準範囲超の高値を示し、4例が基準範囲より低かった。一方、
プラセボ群では、投与前には 31例中 18例が基準範囲より低かったが、本剤投与後 24週時に
おいても 29例中 18例が基準範囲より低かった。
表 4 血清 IGF-I濃度と血清 IGFBP-3濃度の分布の変化
血清 IGF-I濃度 血清 IGFBP-3濃度 投与群 血清濃度
0週 24週 0週 24週
基準範囲超 0 6 1 5
基準範囲 12 22 8 22 LY群
基準範囲未満 21 3 24 4
基準範囲超 0 0 1 0
基準範囲 14 12 12 11 プラセボ群
基準範囲未満 17 17 18 18

ヒューマトロープ CTD(2.7 臨床概要) Page 13
13
表 5、表 6に、K01A試験の血清 IGF-I濃度及び血清 IGFBP-3濃度のシフトテーブルを示した。
表 5に示すとおり、LY投与群の 6症例が 24週時点で血清 IGF-I濃度が高値を示した。その
うち、5症例は投与開始前には基準範囲内、残る 1症例は基準範囲以下であった。
表 5 血清 IGF-I濃度のシフトテーブル
投与群 24週
0週 基準範囲未満 基準範囲 基準範囲超
基準範囲超 0 0 0
基準範囲 0 6 5 LY群
基準範囲未満 3 16 1
基準範囲超 0 0 0
基準範囲 1 12 0 プラセボ群
基準範囲未満 16 0 0
表 6に示すとおり、LY投与群の 5症例が 24週時点で血清 IGFBP-3濃度が高値を示した。そ
のうち、1 症例は投与開始前で基準範囲を超えており、3 症例は投与開始前には基準範囲内、
残る 1症例は基準範囲以下であった。
表 6 血清 IGFBP-3濃度のシフトテーブル
24週
0週 基準範囲未満 基準範囲 基準範囲超
基準範囲超 0 0 1
基準範囲 0 4 3 LY群
基準範囲未満 4 18 1
基準範囲超 0 0 0
基準範囲 1 10 0 プラセボ群
基準範囲未満 17 1 0
5) LBM、IGF-I、IGFBP-3変化の群間比較
LBMの変化率並びに IGF-I、IGF-I SDS、IGFBP-3及び IGFBP-3 SDSの変化量についての群間
比較を表 7に示した。
LBMの平均変化率は、LY群及びプラセボ群で、それぞれ 4.7%及び-0.5%であり、LY群がプ
ラセボ群より統計学的に有意に大きかった(p<0.001)。また、IGF-I、IGF-I SDS、IGFBP-3及
び IGFBP-3 SDSの変化量のいずれについても、LY群がプラセボ群より大きく、統計学的有意
差が認められた(p<0.001)。

ヒューマトロープ CTD(2.7 臨床概要) Page 14
14
表 7 LBM、IGF-I、IGFBP-3の変化の群間比較
LY群 プラセボ群 差(LY-プラセボ) 項目*1
平均±標準偏差 平均±標準偏差 p値*2 LBM (%) 4.7±3.9 -0.5±4.1 <0.001 IGF-I (ng/mL) 175±108 -8±25 <0.001 IGF-I SDS 3.26±1.85 -0.18±0.61 <0.001 IGFBP-3 (µg/mL) 1.3±0.8 -0.0±0.4 <0.001 IGFBP-3 SDS 3.48±2.45 -0.31±1.40 <0.001
*1 0週から 24週 LOCFまでの変化量〔LBMは変化率(%)〕 *2 LBM:対応のない t検定、IGF-I、IGFBP-3:2標本のWilcoxon検定
6) QOLの評価
QOLは、QOL質問票「MOS Short-Form 36-Item Health Survey(以下、SF-36)」及び「Questions
on Life Satisfaction(以下、QLS)」で評価した。
(a) SF-36 本剤又はプラセボ投与前後における SF-36の変化を表 8に示した。なお、QOLが改善したと
きは、正方向に変化する。
LY群では、8項目中 6項目の平均得点が上昇し、2項目の平均得点が低下した。いずれの項
目においても、本剤投与前後における得点の変化は、統計学的に有意ではなかった。
プラセボ群では、8 項目中 3 項目の平均得点が上昇し、5 項目の平均得点が低下した。いず
れの項目においても、プラセボ投与前後における得点の変化は、統計学的に有意ではなかった。
下位尺度の 8 項目すべてについて、LY 群とプラセボ群の変化量には、有意差が認められな
かった。
表 8 SF-36下位尺度得点の変化量の群間比較
LY群 プラセボ群 差(LY-プラセボ) 項目*1
平均±標準偏差 平均±標準偏差 p値*2
PF:身体機能 2.0±9.2 2.1±12.4 0.675
RP:日常役割機能(身体) 0.0±20.9 -6.1±27.4 0.410
BP:体の痛み -3.1±22.2 -3.4±25.2 0.618
GH:全体的健康感 1.2±10.2 3.1±12.6 0.605
VT:活力 1.6±19.4 0.7±15.6 0.754
SF:社会生活機能 5.1±23.7 -0.8±19.9 0.597
RE:日常役割機能(精神) 2.9±18.8 -5.0±33.7 0.238
MH:心の健康 2.8±17.0 -3.9±18.9 0.323
*1 0週から 24週 LOCFまでの変化量 *2 2標本のWilcoxon検定
(b) QLS a) QLSの信頼性及び妥当性
QLS質問票は、AGHDに疾患特異的な質問票として、外国で開発され、その信頼性及び妥当
性が確認されている。本質問票を国内で用いるにあたって、日本語版の信頼性及び妥当性を検

ヒューマトロープ CTD(2.7 臨床概要) Page 15
15
討した。
QLS質問票の信頼性及び妥当性の検討のため、本質問票を用いた外国における結果との比較
を含めた計量心理学的検討を行った。QLSの 3つの尺度、一般事項(QLS-A)、健康(QLS-G)、
ホルモンの不足(QLS-H)について、Cronbach’s α係数及び部分全体相関の検討による内的整
合性(等質性)の確認と、再テスト法による信頼性の検討を行った。更に、ホルモンの不足
(QLS-H)に関しては、尺度の治療効果への感度及び SF-36との相関性の面から妥当性の検討
を行った。
これらの結果を海外における結果と比較したところ、すべての尺度において十分な部分全体
相関と Cronbach’s α係数による高い内的整合性(QLS-A:0.83、QLS-G:0.90、QLS-H:0.89)
が示された。また、QLS-Hと SF-36の 8項目の下位尺度との相関性の検討では、社会生活機能
を除き、ある程度の相関性が認められた。しかし、再テスト法による信頼性は海外に比して低
かった(QLS-A:0.69、QLS-G:0.76、QLS-H:0.66)。また、ホルモンの不足(QLS-H)の尺
度の治療効果への感度も十分でなかった。
結論として、本治験で用いた疾患特異的 QLS質問票の信頼性及び妥当性を確認するには至ら
なかった。
b) QLSの変化(参考成績)
本剤及びプラセボ投与前後における QLSの変化を表 9に示した。なお、QLS質問票の信頼
性及び妥当性を確認するには至らなかったため、参考成績とした。
表 9 QLSの変化量の群間比較
LY群 プラセボ群 差(LY-プラセボ) 項目*1
平均±標準偏差 平均±標準偏差 p値*2
QLS-A:一般事項 2.2±23.2 0.9±18.3 0.892
QLS-G:健康 3.6±30.2 3.5±35.3 0.941
QLS-H:ホルモンの不足 0.1±61.3 -10.5±30.5 0.164
*1 0週から 24週 LOCFまでの変化量 *2 2標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 16
16
(2) 長期臨床試験(K02A試験)
試験方法の概略を表 10に示した。
表 10 試験方法の概略(K02A試験)
項 目 内 容
治験課題名 成人成長ホルモン分泌不全症患者における LY137998[ソマトロピン(遺伝子組換え)]の長期臨床試験
治験の目的 LY137998による成人成長ホルモン分泌不全症患者に対する 48週間の治療における安全性を評価する。
治験の種類 多施設共同、無対照試験 対象被験者 成人成長ホルモン分泌不全症 選択基準 本治験に組み入れられるには、以下のすべての基準を満たす必要がある。
(1) 他の下垂体ホルモンが欠乏しており治療が必要とされる場合、少なくとも本治験参加への同意取得前 3ヵ月の間、必要なホルモンの補充療法を受けている者 ただし、性腺刺激ホルモン欠損又は卵巣疾患により女性ホルモンの補充療法を受ける必要のある女性の場合は、以下の者を本治験の対象とする。 1) 本治験参加への同意取得時における年齢が 40歳未満の場合:少なくとも同意取得前
3ヵ月の間、当該療法を受けている者 2) 本治験参加への同意取得時における年齢が 40歳以上 56歳未満の場合:当該療法実施の有無にかかわらず、少なくとも同意取得前 3ヵ月の間、当該療法実施の有無に変更がない者
3) 本治験参加への同意取得時における年齢が 56歳以上の場合:少なくとも同意取得前3ヵ月の間、当該療法を受けていない者
(2) 先行試験において少なくとも 12週間治験薬の投与を行い、先行試験の投与 12週目来院時における検査・観察が行われた者
除外基準 以下の基準のいずれかに該当する者は治験から除外する。 <既往症・合併症> (1) 高度の心機能障害、肺機能障害、腎機能障害、肝機能障害の認められる者 (2) 高度の精神疾患の認められる者 (3) 悪性腫瘍が認められる者 (4) 良性頭蓋内圧亢進の認められる者 (5) 増殖性網膜障害の認められる者 (6) 治療を行っても十分に血圧コントロールができない高血圧患者 (7) 糖尿病患者 (8) 高度の神経筋疾患の認められる者 <薬剤投与> (9) 本治験参加への同意取得前 3ヵ月以内に食欲抑制剤又は抗うつ薬の投与を受けた者 (10) 本治験参加への同意取得前 3ヵ月以内に長期使用の目的で補充量以外の用量で副腎皮質
ステロイドの投与を受けている者、又は性腺ステロイド補充療法以外に本治験参加への同意取得前 3ヵ月以内に長期使用の目的で同化ステロイドの投与を受けている者(上記薬剤の補充療法を受けている患者で、患者の状態により必要と判断された結果、一時的に用量を適宜増減した場合は本基準に該当しない)
(11) 本治験参加への同意取得前 4ヵ月以内に厚生労働省の承認を得ていない薬剤の投与を受けた者(LY137998を除く)
<その他> (12) 治験責任医師、治験分担医師、治験協力者、治験依頼者の社員及びそれらの配偶者、両
親、子供、兄弟姉妹(血縁関係及び法律上の関係の双方を含む) (13) 妊婦、授乳中又は妊娠している可能性のある女性、もしくは予定の治験期間中に妊娠を
希望している女性 (14) 金属片、ギブス包帯等、二重エネルギーX線吸収法(Dual Energy X-ray Absorptiometry、
以下 DEXA法)による検査を妨害することが明らかな体内・体外器具を装着している者(15) 本治験の実施計画(投与スケジュール、来院等)の遵守が困難である者 (16) その他、治験責任医師又は治験分担医師が本治験の対象として適当でないと判断した者

ヒューマトロープ CTD(2.7 臨床概要) Page 17
17
項 目 内 容
目標症例数及び設定根拠
成人成長ホルモン分泌不全症患者における LY137998[ソマトロピン(遺伝子組換え)]のプラセボ対照二重盲検比較試験(治験実施計画書番号:B9R-JE-K01A)で対象とされた成人成長ホルモン分泌不全症患者のうち、治験責任医師又は治験分担医師によって安全性の観点から長期投与への移行が問題ないと判断され、かつ長期投与移行の同意が得られた患者が対象となるため、症例数設定は行わない。
使用薬剤 LY137998 ソマトロピン(遺伝子組換え)6.56 mg 専用注入器装着時、6 mgまで使用できる
用法・用量 0.021 mg/kg/週にて 8週間皮下投与し、それ以降は前回来院時の血清 IGF-I濃度に基づき、0.021~0.084 mg/kg/週の範囲内で適宜投与量を増減する。但し、体重を基に算出される 1日投与量が 1 mgを超える場合は、体重に関わらず 1 mg/日を投与する。また、同一の副作用が 2週間以上継続して認められた場合、治験責任医師又は治験分担医師は規定の 25-50%減量の投与の必要性を判断する。
投与期間 48週間 併用禁止薬 食欲抑制剤
抗うつ薬 厚生労働省の承認を得ていない他の薬剤 他の成長ホルモン製剤
観察・検査・評価項目及び時期
来院 同意取得時
投与開始直前
投与4 週目
投与8 週目
投与12週目
投与16週目
投与20週目
投与 24 週目
投与 36 週目
投与48 週目
中止時
規定検査日 ─1 日目
29日目
57日目
85日目
113日目
141日目
169 日目
253 日目
337 日目 ─
範囲(日) ─ ─ ±7 ±7 ±7 ±7 ±7 ±7 ±14 ±14 ─
同意取得 X
臨床検査 X X X X X X X X X X
血圧 X X X X X X X X X X
体重 X X X X X X X X X X LBM X X X X QOL (SF36、QLS) X X X X
投与量 X X X X X X X X X
投与状況 X X X X X X X X X
合併症及び 有害事象
X X X X X X X X X X
観察・検査等の項目
併用薬 併用療法
X X X X X X X X X X
主要評価項目 48週間の治療における安全性 解析方法 有効性の解析
(1) 先行試験において実薬を投与した群 1) LBM 投与開始直前、投与 24週目及び 48週目の観測値の要約統計量及び信頼係数両側 95%の信頼区間を算出すること、並びに投与開始直前から投与 48週目までの観測値の経時推移グラフを作成することにより、先行試験における投与開始から投与 24週目までに得られた LBM増加が維持されていることを確認する。
2) IGF-I、IGFBP-3 投与開始直前から投与 24週目及び 48週目までの観測値の要約統計量及び信頼係数両側 95%の信頼区間を算出すること、並びに投与開始直前から投与 48週目までの観測値の経時推移グラフを作成し、観測値が正常範囲内に維持されることを確認する。また、IGF-Iと IGFBP-3の相関関係を評価する。

ヒューマトロープ CTD(2.7 臨床概要) Page 18
18
項 目 内 容
3) QOL(SF-36、QLS) 投与開始直前、投与 24週目及び 48週目の QOL評価を要約し、先行試験における投与開始から投与 24週目までに得られた QOL改善が維持されていることを確認する。
(2) 先行試験においてプラセボを投与した群 1) LBM 各被験者の投与 24週目及び 48週目における観測値の投与開始直前観測値からの変化率につき、要約統計量及び信頼係数両側 95%の信頼区間を算出し、先行試験時の投与 24週目における投与開始直前値からの LBMの変化率と比較する。
2) IGF-I、IGFBP-3 各被験者の投与 24週目及び 48週目における観測値の投与開始直前からの変化につき、要約統計量及び信頼係数両側 95%の信頼区間を算出し、先行試験時の投与 24週目における投与開始直前からの変化と比較する。また、IGF-Iと IGFBP-3の相関関係を評価する。
3) QOL(SF-36、QLS) 各被験者の投与 24週目及び 48週目における QOL評価の投与開始直前からの変化につき要約し、先行試験時の投与 24週目における投与開始直前からの QOL変化と比較する。
安全性の解析 先行試験時に実薬を投与された群の 48週間及び先行試験時も加えた 72週間(24+48週間)、並びにプラセボを投与された群の 48週間の 3つについて以下の解析を行う。ただし、ベースラインを、先行試験時にプラセボを投与した群は投与開始直前とし、実薬を投与された群は先行試験時の投与開始直前とする。 (1) 有害事象
投与後観測された有害事象の発現例数及び発現件数を、重症度及び因果関係別に集計する。重症度分布の集計には、同一被験者に重症度の異なる同一事象が発現した場合、重症度の最も高い事象のみを用いる。 投与後観測された有害事象の発現例数及び発現件数を 4週間ごとに算出し、発現頻度の経時推移グラフを作成する。
(2) 臨床検査値及び血圧 来院ごとに要約統計量を算出すること及び経時推移グラフを作成することにより経時推移を要約する。 ベースラインを投与前値とし、投与 48週目における観測値を投与後値とし、投与前後の比較を行う。ただし、中止された被験者については中止時の観測値を投与後値とする。正常範囲を用いて各観測値を異常高値、正常、異常低値に分類し、投与前値と投与後値の交差分類表を作成する。また、異常変動の発現例数を重症度及び因果関係別に集計する。
治験調整医師
治験 アドバイザー
外部治験 アドバイザー
代表施設名及び施設数
計 25施設
治験期間 20 年 月~20 年 月

ヒューマトロープ CTD(2.7 臨床概要) Page 19
19
1) 被験者の内訳
被験者の内訳を図 2に示した。本治験は 25施設で実施した。組み入れられた 60例すべてに
本剤が投与された。K01A試験で LY群に割り付けられ、K02A試験においても、引き続いて本
剤の投与を 48週間受けた患者群を LY/LY48w群と表記した。また、K01A試験でプラセボ群
に割り付けられ、K02A試験で初めて、本剤の投与を 48週間受けた患者群をプラセボ/LY48w
群と表記した。
LY/LY48w群では 30例が 48週目の来院を完了し、1例が有害事象により治験を中止した。
一方、プラセボ/LY48w群では 27例が 48週目の来院を完了し、1例が組入れ基準違反(先行
試験開始時からの併用禁止薬の使用)のため、更に 1例が治験の継続的実施が困難となった(最
終観察時に来院せず)ため、計 2例が治験を中止した。
有効性の評価は、組入れ基準違反のため治験を中止した 1例を除いて、59例(LY/LY48w
群:31例、プラセボ/LY48w群:28例)を対象に行った。
*1: 中止(1)[有害事象(1)]*2: 中止(2)[併用禁止治療(1), 治験の継続的実施困難(1)]
非組み入れ例(0)
未投与例(0)
完了例(30) 未完了例(1)*1
LY/LY48W群(31)
完了例(27) 未完了例(2)*2
プラセボ/LY48W群(29)
投与例(60)
組み入れ例(60)
同意取得例(60)
図 2 被験者の内訳(K02A試験)
2) 患者背景
有効性評価対象例 59 例の主な患者背景を表 11 に示した。平均年齢(同意取得時)は、LY
/LY48w群で 36.2歳、プラセボ/LY48w群で 39.6歳であった。性別は、LY/LY48w群で男
性 15例及び女性 16例であった。一方、プラセボ/LY48w群では男性 14例及び女性 14例であっ
た。成人期発症型と小児期発症型の症例数は、LY/LY48w群で、それぞれ 12例及び 19例、
プラセボ/LY48w群でそれぞれ 11例及び 17例であった。成長ホルモン分泌不全の病因は、LY
/LY48w群では、腫瘍が 13例(41.9%)、腫瘍に対する治療が 9例(29.0%)、特発性又は先
天性が 6例(19.4%)の順であった。プラセボ/LY48w群においても、これらの頻度が高く、
腫瘍が 7例(25.0%)、腫瘍に対する治療が 9例(32.1%)、特発性又は先天性が 9例(32.1%)
であった。病態については、プラセボ/LY48w 群の成人期発症型 1 例を除く全例が複合型で
あった。

ヒューマトロープ CTD(2.7 臨床概要) Page 20
20
表 11 患者背景(K02A試験)
項 目 LY/LY48w群 (N=31)
プラセボ/LY48w群(N=28)
平均±標準偏差 36.2±13.3 39.6±13.7 18~19 2 (6.5%) 1 (3.6%)
20~29 10 (32.3%) 8 (28.6%)
30~39 8 (25.8%) 7 (25.0%) 同意時年齢
40~64 11 (35.5%) 12 (42.9%)
男 性 15 (48.4%) 14 (50.0%) 性 別
女 性 16 (51.6%) 14 (50.0%)
成人期発症型 12 (38.7%) 11 (39.3%) 発症時期
小児期発症型 19 (61.3%) 17 (60.7%)
特発性又は先天性 6 (19.4%) 9 (32.1%)
腫 瘍 13 (41.9%) 7 (25.0%)
腫瘍に対する治療 9 (29.0%) 9 (32.1%)
シーハン症候群 1 (3.2%) 2 (7.1%)
トルコ鞍空虚 1 (3.2%) 1 (3.6%)
成長ホルモン分泌不全の病因
外 傷 1 (3.2%) 0 (0.0%)
GH単独分泌不全症 0 (0.0%) 1 (3.6%) 病 態
複合型下垂体機能低下症 31 (100.0%) 27 (96.4%) LBM (kg) 平均±標準偏差 42.18±9.61 38.60±9.73
IGF-I (ng/mL) 平均±標準偏差 243±114 61±39 IGF-I SDS 平均±標準偏差 0.82±2.28 -2.33±1.42
3) LBMの変化
K02A試験の LBMの変化を表 12に示した。
LY/LY48w群において、K02A試験開始時から 24週時までの LBMの平均変化率は 0.1%で
あった。また、K02A試験開始時から 48週時までの LBMの平均変化率は 1.2%であり、K01A
試験で認められた LBMの変化は、本治験 48週後においても維持されていた。
プラセボ/LY48w群において、本剤投与開始から 24週時までの LBMの平均変化率は 3.4%、
48週時までの LBMの平均変化率は 4.5%であり、投与前後で統計学的に有意であった(表 12
及び表 13、いずれも p<0.001)。また、先行試験期間中(プラセボ投与)の変化と比較しても、
有意に大きかった(表 13、24週変化率:p=0.009、48週変化率:p=0.001)。

ヒューマトロープ CTD(2.7 臨床概要) Page 21
21
表 12 LBMの変化
項目 投与群 来院 例数 平均 標準偏差 最大値 中央値 最小値
0週 31 42.18 9.609 62.94 40.19 30.47
24週 28 42.76 10.874 64.00 41.05 28.90
24週 LOCF 31 42.38 10.545 64.00 39.57 28.90
48週 29 43.16 10.664 66.35 40.64 30.32
48週 LOCF 31 42.81 10.641 66.35 40.64 28.90
24週変化率 (%) 31 0.1 4.68 14.1 0.3 -7.1
LY/LY48w群
48週変化率(%) 31 1.2 4.91 12.3 0.5 -6.5
0週 28 38.60 9.729 65.55 35.84 26.94
24週 26 39.95 10.378 66.23 35.79 25.93
24週 LOCF 28 39.89 10.010 66.23 36.43 25.93
48週 26 40.43 9.552 65.45 37.31 26.20
48週 LOCF 28 40.16 9.467 65.45 37.31 26.20
24週変化率 (%) 28 3.4 4.79 13.8 3.3 -7.3
LBM (kg)
プラセボ/LY48w群
48週変化率 (%) 28 4.5 5.25 15.9 4.2 -4.3
表 13 プラセボ/LY群 48wにおける LBM変化率の先行試験との比較
試験番号 例数 平均値 標準偏差 最大値 中央値 最小値 検定*3
p値 検定*4
p値
K01A試験 24週 28 -0.6 4.12 9.2 -1.1 -6.5 0.436 -
K02A試験 24週*1 28 3.4 4.79 13.8 3.3 -7.3 <0.001 0.009
K02A試験 48週*2 28 4.5 5.25 15.9 4.2 -4.3 <0.001 0.001
*10週から 24週 LOCFまでの変化率(%) *2 0週から 48週 LOCFまでの変化率(%) *3 帰無仮説:変化率=0、対応のある t検定 *4 帰無仮説:K01A試験と K02A試験の変化率が等しい、対応のある t検定
4) IGF-I及び IGFBP-3の変化
K02A試験開始後の血清 IGF-I濃度と血清 IGFBP-3濃度の分布の変化を表 14に示した。LY
/LY48w群において、血清 IGF-I濃度は K02A試験開始時には、31例中 22例が基準範囲内の
値を示し、6例が基準範囲超の高値を示し、3例が基準範囲より低かったが、K02A試験開始後
24週時には 29例中 24例が基準範囲内の値を示し、5例が基準範囲超の高値を示した。K02A
試験開始後 48週時には 30例中 27例が基準範囲内の値を示し、3例が基準範囲超の高値を示し
た。一方、プラセボ/LY48w群では、血清 IGF-I濃度は K02A試験開始時には、28例中 11例
が基準範囲内の値を示し、17例が基準範囲より低かったが、本剤投与後 24週時では、27例中
20例が基準範囲内の値を示し、4例が基準範囲超の高値を示し、3例が基準範囲より低い値を
示した。本剤投与後 48週時では、27例中 22例が基準範囲内の値を示し、3例が基準範囲超の
高値を示し、2例が基準範囲より低い値を示した。
LY/LY48w群において、血清 IGFBP-3濃度は、K02A試験開始時には、31例中 22例が基準
範囲内の値を示し、5例が基準範囲超の高値を示し、4例が基準範囲より低かったが、K02A試

ヒューマトロープ CTD(2.7 臨床概要) Page 22
22
験開始後 24週時では、29例中 22例が基準範囲内の値を示し、4例が基準範囲超の高値を示し、
3例が基準範囲より低い値を示した。K02A試験開始後 48週時では、30例中 21例が基準範囲
内の値を示し、7 例が基準範囲超の高値を示し、2 例が基準範囲より低い値を示した。一方、
プラセボ/LY48w群では、血清 IGFBP-3濃度は、K02A試験開始時には、28例中 10例が基準
範囲内の値を示し、18例が基準範囲より低かったが K02A試験開始後 24週時では、27例中 18
例が基準範囲内の値を示し、3 例が基準範囲超の高値を示し、6 例が基準範囲より低かった。
K02A試験開始後 48週時では、27例中 17例が基準範囲内の値を示し、4例が基準範囲超の高
値を示し、6例が基準範囲より低い値を示した。
表 14 血清 IGF-I濃度と血清 IGFBP-3濃度の分布の変化
血清 IGF-I濃度 血清 IGFBP-3濃度 投与群 血清濃度
0週 24週 48週 0週 24週 48週
基準範囲超 6 5 3 5 4 7
基準範囲 22 24 27 22 22 21 LY/LY48w群
基準範囲未満 3 0 0 4 3 2
基準範囲超 0 4 3 0 3 4
基準範囲 11 20 22 10 18 17 プラセボ/LY48w群
基準範囲未満 17 3 2 18 6 6
5) SF-36を用いた QOLの評価
K02A 試験において、K01A 試験から引き続き本剤が 24 週間投与された結果、LY/LY48w
群では、4項目の平均得点が上昇し、4項目の平均得点が低下した(表 15)。その後、48週間
まで投与された結果、5 項目の平均得点が上昇し、3 項目の平均得点が低下した。一方、プラ
セボ/LY48w群では、K02A試験投与開始後から本剤が 24週間投与された結果、6項目の平均
得点が上昇し、1項目の平均得点が低下した。その後、48週間投与された結果、すべての項目
で平均得点が低下した。いずれの項目においても、本剤投与前後の変化は、統計学的に有意で
はなく、先行試験でのプラセボ投与期間中の平均変化量との間に統計学的有意差は認められな
かった。

ヒューマトロープ CTD(2.7 臨床概要) Page 23
23
表 15 SF-36の変化量
24週投与時*1 48週投与時*2
LY/LY48w群 プラセボ/LY48w群 LY/LY 48w群 プラセボ/LY 48w群項目
平均値±標準偏差 平均値±標準偏差 平均値±標準偏差 平均値±標準偏差
PF:身体機能 -1.2 ± 9.7 (29)
0.4 ± 6.6 (27)
0.2 ± 7.5 (30)
-0.8 ± 8.2 (28)
RP:日常役割機能(身体) 3.0 ± 18.2 (31)
2.5 ± 24.8 (28)
5.0 ± 20.1 (31)
-1.7 ± 16.9 (28)
BP:体の痛み -1.2 ± 18.7 (31)
-2.6 ± 30.0 (28)
-2.2 ± 20.5 (31)
-4.4 ± 26.4 (28)
GH:全体的健康感 2.3 ± 10.8 (29)
1.0±13.3 (28)
0.7 ± 15.9 (31)
-0.2 ± 13.9 (28)
VT:活力 0.9 ± 18.1 (29)
0.7 ± 18.8 (28)
3.0 ± 14.5 (31)
-4.5 ± 19.6 (28)
SF:社会生活機能 -2.0 ± 20.2 (31)
0.0 ± 25.2 (28)
-8.1 ± 25.5 (31)
-9.8 ± 25.3 (28)
RE:日常役割機能(精神) 1.1 ± 18.6 (31)
6.8 ± 26.2 (28)
1.6 ± 23.4 (31)
-0.9 ± 28.1 (28)
MH:心の健康 -0.5 ± 19.8 (29)
0.7 ± 15.0 (28)
-3.7 ± 18.7 (31)
-5.1 ± 19.7 (28)
*1 0週から 24週 LOCFまでの変化量 *2 0週から 48週 LOCFまでの変化量
(3) 外国臨床試験(参考資料)
1) 成人期発症型の AGHD患者に対する GH補充療法の有効性評価(E003/4試験)
102例が組み入れられ、そのうち 4例がプラセボ投与観察期において中止されたため、98例
が LY群又はプラセボ群に無作為割り付けされた。割り付けられた全症例(98例)の平均年齢
は 43.5歳で、男性患者数及び女性患者数は、それぞれ 62例及び 36例であった。年齢、性別な
どの患者背景に、両群間で有意な差はなかった。
LY群に割り付けられた 52例のうち、完了例は二重盲検比較期間及び長期投与期間において、
それぞれ 49例及び 44例であった。また、プラセボ群に割付けられた 46例のうち、完了例は
二重盲検比較期間及び長期投与期間において、それぞれ 44例及び 40例であった。
6ヵ月のプラセボ対照二重盲検比較期間において、LY群で LBMの増加が認められた。LBM
が平均 2.59 kg増加し、ベースラインと比較して、統計学的有意差が認められた。一方、プラ
セボ群の変化は平均-0.22 kgであった。また、LY群とプラセボ群との LBMの変化量の差は、
統計学的に有意であった。
投与 18ヵ月後の結果から、LY群における LBMは、ベースラインから投与 18ヵ月後までに
平均 2.07 kg増加し、投与後 6ヵ月時に認められた LBMの増加は維持されていた。
2) 小児期発症型の AGHD患者に対する GH補充療法の有効性評価(E005/6試験)
71 例が組み入れられ、そのうち 4 例がプラセボ投与観察期において中止されたため、67 例
が LY群又はプラセボ群に無作為割り付けされた。割り付けられた全症例の平均年齢は 28.4歳
で、男性患者数及び女性患者数は、それぞれ 49例及び 18例であった。年齢、性別などの患者
背景に、両群間で有意な差はなかった。
LY群に割り付けられた 32例のうち完了例は、二重盲検比較期間及び長期投与期間において、

ヒューマトロープ CTD(2.7 臨床概要) Page 24
24
それぞれ 28例及び 22例であった。また、プラセボ群に割付けられた 35例のうち、完了例は
二重盲検比較期間及び長期投与期間において、それぞれ 30例及び 26例であった。
6ヵ月のプラセボ対照二重盲検比較期間において、LY群で LBMの増加が認められた。LBM
が平均 3.68 kg増加し、ベースラインと比較して、統計学的有意差が認められた。一方、プラ
セボ群の変化は平均-1.91 kgであった。また、LY群とプラセボ群との LBMの変化量の差は、
統計学的に有意であった。
投与 18ヵ月後の結果から、LY群における LBMは、ベースラインから投与 18ヵ月後までに
平均 3.58 kg増加し、投与後 6ヵ月時に認められた LBMの増加は維持されていた。
3) AGHD患者に対する GHの投与量別の有効性を評価した市販後臨床試験(GDED試験)
595例が組み入れられ、低用量群(3.0 µg/kg/日→6.0 µg/kg/日)に 302例及び高用量群(6.0 µg/kg/
日→12.0 µg/kg/日;既承認用量)に 293例が割り付けられた。低用量群及び高用量群の平均年
齢は、それぞれ 43.0歳及び 42.9歳であった。年齢、性別及び発症時期などの患者背景に、両
群間で有意な差はなかった。
低用量群の 302例中 274例及び高用量群の 293例中 246例が 6ヵ月の試験期間を完了した。
ベースラインから投与 3ヵ月後までの LBMの平均変化率は、低用量群及び高用量群で、そ
れぞれ 2.43%及び 3.58%であり、いずれも有意な変化が認められた。また、用量群間の差は、
統計学的に有意であった。
ベースラインから投与 6ヵ月後までの LBMの平均変化率は、低用量群及び高用量群で、そ
れぞれ 4.38%及び 5.21%であり、いずれも有意な変化が認められた。しかし、用量群間には統
計学的有意差は認められなかった。
4) AGHD患者を対象として 2種類の用法用量を検討した市販後臨床試験(T002試験)
387例が組み入れられ、ID群に 187例及び FD群に 200例が、無作為に割り付けられた。全
症例の平均年齢は 46.7歳で、男性患者数及び女性患者数は、それぞれ 224例及び 163例であっ
た。また、79.8%の症例が成人期発症型の AGHD 患者であった。両群の背景因子については、
年齢、男女の分布、民族構成及び発症時期の分布並びに身長、体重、全身 FM 及び血清 IGF-I
濃度の平均の全項目において大きな差はなかった。FD群の 26例及び ID群の 29例が投与を中
止した。
主要評価項目であるベースライン及び投与終了時(32週間投与後)の FMの測定が適正に実
施された 281例を対象に有効性を評価した。その結果、FD群及び ID群における FMの平均減
少率は、それぞれ 10.9%及び 7.9%で、いずれの投与群においても投与開始時より有意に減少し
ていた。

ヒューマトロープ CTD(2.7 臨床概要) Page 25
25
2.7.3.3 全試験を通しての結果の比較と解析
2.7.3.3.1 試験対象集団
(1) 国内臨床試験
各投与群の表記については、図 3に示すように、K01A試験において、本剤が投与された患
者群を「LY群」、プラセボが投与された患者群を「プラセボ群」と表記した。次に、K02A試
験において、K01A 試験から引き続いて本剤が投与された患者群の K02A 試験期間中の成績
(K01A 試験投与開始時から起算して、24~72 週までの成績)を示す際、「LY/LY48w 群」
と表記した。一方、K02A試験において、本剤が初めて 48週間投与された患者群の K02A試験
期間中の成績を示す際、「プラセボ/LY48w群」と表記した。更に、K01A試験及び K02A試
験を通じて、本剤が 72週間投与された患者群の K01A試験開始時から投与後 72週時(K02A
試験投与後 48週時)までの成績を示す際、「LY72w群」と表記した。なお、LY72w群及び LY
/LY48w 群のベースラインは K01A 試験開始時とし、プラセボ/LY48w 群のベースラインは
K02A試験開始時とした。
用量漸増
プラセボ
0週 4週 72週
0.021*
0.042*
0.084*
*:mg/kg/週
用法用量
試験番号
群の名称
LY群
LY72w群
12週 32週
K01A試験 K02A試験
0.021*
投与方法
群の名称
投与方法
48週24週
血 清 I G F – I 濃 度 に よ る 用 量 調 整
血 清 I G F – I 濃 度 に よ る 用 量 調 整
LY/LY48w群
プラセボ/LY48w群プラセボ群
図 3 国内臨床試験における投与群の設定
国内臨床試験に参加した AGHD患者の患者背景を表 16に示した。
K01A試験には 65例が組み入れられ、発症時期(成人期発症型・小児期発症型)を層別因子
として、性別、年齢及び施設を因子とした層別確率化最小化法に基づき、33例が LY群に、ま

ヒューマトロープ CTD(2.7 臨床概要) Page 26
26
た 32例がプラセボ群に割り付けられた。組入れ基準を充足していなかったことが、K02A試験
期間中に判明したプラセボ群 1例を除いて、64例を有効性評価対象例とした。
64例のうち男性は 31例(48.4%)、女性は 33例(51.6%)であり、平均年齢は 38.1歳であっ
た。発症時期では、成人期発症型が 27例(42.2%)、小児期発症型が 37例(57.8%)であった。
また、BMIの平均値は、24.0であった。AGHDの病因については、器質性が 49例、特発性又
は先天性が 15例であった。64例中成人期発症型の 1例が GH単独分泌不全症で、他の 63例は
複合型下垂体機能低下症であった。
表 16 患者背景(K01A/K02A試験)
投与群 LY群* (N=33)
プラセボ群 (N=31)
プラセボ/ LY48w群
(N=28) 年齢 (平均値±標準偏差) 37.2±13.9 39.0±13.8 39.6±13.7
男性 (例数) 16 15 14 性別
女性 (例数) 17 16 14
成人期発症型 (例数) 14 13 11 発症 時期 小児期発症型 (例数) 19 18 17
GH分泌刺激試験 (ng/mL) (中央値、最小値~最大値)
0.1 0.1~2.4
0.2 0.1~2.7
0.2 0.1~2.7
器質性 (例数) 27 22 19 AGHDの 病因 特発性又は先天性 (例数) 6 9 9
GH単独分泌不全症 (例数) 0 1 1 病態
複合型下垂体機能低下症 (例数) 33 30 27
BMI (平均値±標準偏差) 24.6±4.5 23.3±4.3 22.9±3.6
* LY72w群も同一患者であるため背景は同一である。
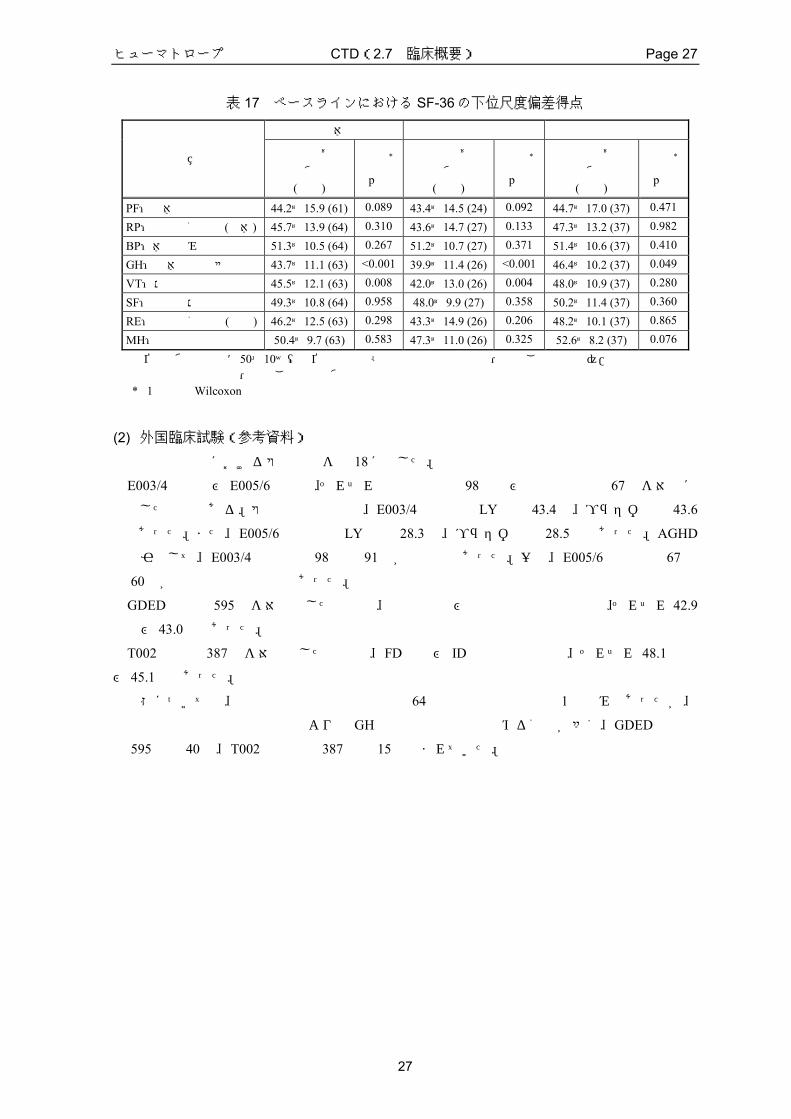
K01A試験に参加したAGHD患者のベースラインにおける SF-36の下位尺度偏差得点を表 17
に示した。また、AGHD 患者を発症時期別に分け、それぞれの下位尺度偏差得点も同様に示し
た。なお、QOLの評価に偏差得点を用いたのは、AGHD患者の QOLスコアと国民標準値との
対比を容易にするためである。偏差得点の算出には、年齢・性別の影響を排除するため、年齢・
性別ごとの標準値を用いた。この偏差得点において、国民標準値は 50点であり、1標準偏差の
増減は 10点の増減として表わされる。
AGHD患者全体では、ベースラインにおいて下位尺度 8項目中 6項目の平均偏差得点が国民
標準値(50点)より低く、そのうち 2項目は、統計学的に有意に低かった(「全体的健康感」:
p<0.001、「活力」:p=0.008)。このことから、K01A試験に参加した AGHD患者の QOLは、
ベースラインにおいて、国民標準値より低いことが示された。
次に、発症時期別にベースラインにおける偏差得点を検討したところ、いずれの下位尺度に
ついても、成人期発症型の方が小児期発症型より、平均偏差得点が低かった。成人期発症型に
おいて、平均偏差得点が 45 点以下であった下位尺度は、「全体的健康感」、「活力」、「日
常役割機能(精神)」、「身体機能」及び「日常役割機能(身体)」であり、国民標準値(50
点)より統計学的に有意に低かった下位尺度は「全体的健康感」(p<0.001)及び「活力」(p=0.004)
であった。一方、小児期発症型において、平均偏差得点が 45点以下であった下位尺度は、「身
体機能」のみであった。
ヒューマトロープ CTD(2.7 臨床概要) Page 27
27
表 17 ベースラインにおける SF-36の下位尺度偏差得点
全体 成人期発症型 小児期発症型
項 目 平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
PF:身体機能 44.2±15.9 (61) 0.089 43.4±14.5 (24) 0.092 44.7±17.0 (37) 0.471
RP:日常役割機能(身体) 45.7±13.9 (64) 0.310 43.6±14.7 (27) 0.133 47.3±13.2 (37) 0.982
BP:体の痛み 51.3±10.5 (64) 0.267 51.2±10.7 (27) 0.371 51.4±10.6 (37) 0.410
GH:全体的健康感 43.7±11.1 (63) <0.001 39.9±11.4 (26) <0.001 46.4±10.2 (37) 0.049
VT:活力 45.5±12.1 (63) 0.008 42.0±13.0 (26) 0.004 48.0±10.9 (37) 0.280
SF:社会生活機能 49.3±10.8 (64) 0.958 48.0±9.9 (27) 0.358 50.2±11.4 (37) 0.360
RE:日常役割機能(精神) 46.2±12.5 (63) 0.298 43.3±14.9 (26) 0.206 48.2±10.1 (37) 0.865
MH:心の健康 50.4±9.7 (63) 0.583 47.3±11.0 (26) 0.325 52.6±8.2 (37) 0.076
下位尺度偏差得点=50+10×(下位尺度得点-日本人健康人の年齢・性別ごとの平均)/ 日本人健康人の年齢・性別ごとの標準偏差値 * 1標本のWilcoxon検定
(2) 外国臨床試験(参考資料)
外国臨床試験における患者背景を表 18に示した。
E003/4試験及び E005/6試験は、それぞれ成人期発症型 98例及び小児期発症型 67例を対象に
実施した試験である。患者の平均年齢は、E003/4試験では LY群で 43.4歳、プラセボ群で 43.6
歳であった。また、E005/6試験では LY群で 28.3歳、プラセボ群で 28.5歳であった。AGHD
の病因として、E003/4試験では 98例中 91例が器質性であった。一方、E005/6試験では 67例
中 60例が特発性又は先天性であった。
GDED試験は 595例を対象とした試験で、高用量群及び低用量群の平均年齢は、それぞれ 42.9
歳及び 43.0歳であった。
T002試験は 387例を対象とした試験で、FD群及び ID群の平均年齢は、それぞれ 48.1歳及
び 45.1歳であった。
病態については、単独型は国内臨床試験では 64例中成人期発症型の 1例のみであったが、
外国臨床試験では国内臨床試験よりも GH単独分泌不全症の占める割合が多く、GDED試験で
は 595例中 40例、T002試験では 387例中 15例含まれていた。

ヒューマトロープ CTD(2.7 臨床概要) Page 28
28
表 18 外国臨床試験における患者背景
E003/4試験 E005/6試験 GDED試験 T002試験 試験番号
LY群 プラセボ群
LY群プラセボ群
低用量群*1 高用量群*2 FD群 ID群*3
年齢 (平均値±標準偏差) 43.4 43.6 28.3 28.5 43.03± 13.93
42.87± 14.98
48.08±13.24
45.13±14.07
男性 (例数)
36 26 26 23 174 185 115 109 性別
女性 (例数)
16 20 6 12 128 108 85 78
成人期発症型 (例数)
52 46 - - 204 199 162 147 発症 時期 小児期発症型
(例数) - - 32 35 98 93 38 40
器質性 (例数)
91 7 236 233 - - AGHDの病因 特発性又は先天性
(例数) 7 60 65 60 - -
GH単独分泌不全症 (例数)
- - - - 20 20 6 9
病態 複合型 下垂体機能低下症
(例数) - - - - 281 273 194 177
*1 低用量群中、1例の病因及び病態のデータなし *2 高用量群中、1例の発症時期のデータなし *3 ID群中、1例の病態のデータなし
2.7.3.3.2 全有効性試験の結果の比較検討
国内で実施した K01A試験及び K02A試験は、同一患者を対象に実施したため 2試験の成績
を併せて有効性を評価した。
本項では、外国臨床試験で得られた成績についても、参考資料として示した。
(1) 有効性の主要評価(LBMの変化)
国内臨床試験では、LBM の変化率を有効性の主要評価項目とした。本剤又はプラセボが投
与された患者群におけるベースラインからの LBMの変化率を表 19及び表 20に示した。
K01A試験において、LY 群のベースラインから投与後 24週時までの LBMの平均変化率は
4.7%であり、投与前後で統計学的有意差が認められた(p<0.001)。一方、プラセボ群におけ
る平均変化率は-0.5%であった。また、この時点までの変化率について、LY群とプラセボ群と
の差は、統計学的に有意であった(p<0.001)(表 19)。
K01A試験及び K02A試験を通じ、本剤が 72週間投与された患者群(LY72w群)におけるベー
スラインから投与後 48週時(K02A試験投与開始後 24週時)までのLBMの平均変化率は 4.7%、
投与後 72週時(K02A試験投与開始後 48週時)までは 5.8%であった。いずれの場合も投与前
後で統計学的有意差が認められた(p<0.001)。このことから、K01A試験において認められた
LBMの増加は、投与後 72週時においても維持されていることが明らかとなった。K02A試験
におけるプラセボ/LY48w 群のベースライン(K02A 試験投与開始時)から投与後 24 週時ま

ヒューマトロープ CTD(2.7 臨床概要) Page 29
29
での LBMの平均変化率は 3.4%であり、投与後 48週時までは 4.5%であった。いずれの場合も
投与前後で統計学的有意差が認められた(p<0.001)(表 20)。
表 19 LBMの変化率(投与 24週間)
LBM変化率:(%)
投与期間(週) 24
投与群 例数 平均値±標準偏差検定*1
p値 検定*2 p値
LY群 32 4.7 ± 3.9 <0.001
プラセボ群 29 -0.5 ± 4.1 0.519<0.001
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 対応のある t検定 *2 対応のない t検定
表 20 LBMの変化率(投与 48週間)
LBM変化率:(%) 投与期間(週)
24 48 72
投与群 例数 平均値±標準偏差
検定*
p値 平均値± 標準偏差
検定*
p値 平均値± 標準偏差
検定*
p値
LY72w群 32 4.7 ± 3.9 <0.001 4.7 ± 3.9 <0.001 5.8 ± 4.7 <0.001
プラセボ/LY 48w群 28 3.4 ± 4.8 <0.001 4.5 ± 5.2 <0.001 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 対応のある t検定
本剤又はプラセボが投与された患者群におけるベースラインからの体重の変化を表 21 及び
表 22に示した。
LY群及びプラセボ群のベースラインから投与後 24週時までの体重の平均変化率は、それぞ
れ 0.4%及び 0.2%で、いずれも有意な変化は認められなかった(表 21)。LY72w群においても、
ベースラインから投与後 48週時までの体重の平均変化率は 0.8%、投与後 72週時までは 1.3%
であり、統計学的に有意な変化は認められなかった。また、プラセボ/LY48w群のベースライ
ンから投与後 24週時までの体重の平均変化率は-1.0%、投与後 48週時までは-0.4%であり、統
計学的に有意な変化は認められなかった(表 22)。
これらの結果から、本剤が投与された群において、LBM が増加したにもかかわらず体重が
変化してしていないこと、及び骨塩量は 72週間では顕著に変化しないことから、FMが減少し
たと考えられた。更に、LBMの増加及び FMの減少から、体組成が改善したと判断した。
なお、骨塩量は 72週間では顕著に変化しないと考えた理由は以下のとおりである。
AGHD患者における骨への影響を検討した外国臨床試験の成績では、本剤の 2年間投与によ
り、骨塩量はベースラインの約 2.1 kgから有意な増加を示したが、その変化量は約 0.2 kgであっ
た 1。また、2 年間の GH 投与を行った外国臨床試験の成績から、骨塩量はベースラインの約
2.7 kgから有意な増加を示したが、その変化量は約 0.1 kgであった 2。それぞれの臨床試験にお
ける患者の平均体重が約 62 kg及び約 80 kgであったことを考慮すると、全体重に対する骨塩

ヒューマトロープ CTD(2.7 臨床概要) Page 30
30
量の変化量は、非常に小さいと考えられた。
以上のことより 72週間における骨塩量の変化は顕著ではないと考えた。
なお、日本人において骨塩量は全体重のうち、おおむね 2 kg前後を占めているが 3、上記の
とおり GH投与による骨塩量の変化は、もともと骨塩量が全体重に比較し小さいことから、全
体重と LBMから体脂肪量を評価する際に考慮すべきほど大きい変化ではないと判断した。
表 21 体重の推移及び変化率(投与 24週間)
体 重 0 24 投与期(週)
実測値(kg) 実測値(kg) 変化率(%)
投与群 例数 平均値± 標準偏差
平均値± 標準偏差
平均値± 標準偏差
検定*
p値
LY群 33 65.9 ± 15.2 66.3 ± 15.7 0.4 ± 3.4 0.521
プラセボ群 31 61.6 ± 15.2 61.6 ± 14.8 0.2 ± 3.6 0.807
投与開始後 24週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
表 22 体重の推移及び変化率(投与 48週間)
体 重
0 24 48 72 投与期(週)
実測値 (kg)
実測値 (kg)
変化率 (%)
実測値 (kg)
変化率 (%)
実測値 (kg)
変化率 (%)
投与群 例数 平均値±標準偏差
平均値± 標準偏差
平均値±標準偏差
検定*
p値 平均値± 標準偏差
平均値±標準偏差
検定*
p値平均値± 標準偏差
平均値±標準偏差
検定*
p値
LY72w群 33 65.9±15.2 66.3±15.7 0.4±3.4 0.521 66.7±16.8 0.8±4.0 0.312 67.1±17.3 1.3±4.5 0.158
プラセボ/LY48w群
28 60.6±13.8 59.9±13.4 -1.0±4.0 0.077 60.2±13.4 -0.4±3.7 0.521 - - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
(2) 有効性の主要評価に関連する外国臨床成績(参考資料)
1) LBMの変化
外国臨床試験で得られた LBMの変化に関する成績を表 23にまとめ、ベースラインからの変
化量(kg)又は変化率(%)で示した。
いずれの試験においても、本剤が投与された患者群では LBMが増加し、ベースラインと比
較して、統計学的有意差が認められた。また、48週間以上の長期投与においても、LBMのベー
スラインからの増加は維持されていた。なお、プラセボ群では有意な変化は認められなかった。
E003/4試験及び E005/6試験における、LY群とプラセボ群との LBMの変化量の差は統計学
的に有意であった。
GDED試験及び T002試験におけるベースラインから投与後 24週時又は 48週時までの平均
変化率は、4.38~5.21%であり、国内臨床試験の LY群の平均変化率 4.7%と同程度であった。

ヒューマトロープ CTD(2.7 臨床概要) Page 31
31
表 23 LBMの変化量(kg)又は変化率(%)(外国臨床試験)
LBMの変化率(%)又は変化量(kg):平均値±標準偏差 投与期間(週) 試験番号 投与群 例数
12 24 32 48 72 LY群 46 - 2.59±8.43 kg*
- 1.61±9.11 kg 2.07±10.88 kg
プラセボ群 45 - -0.22±5.50 kg - - - E003/4試験
プラセボ/LY群 43 - - - 2.60±5.41 kg* - LY群 31 - 3.68±4.16 kg**
- 3.20±4.56 kg* 3.58±5.09 kg**
プラセボ群 34 - -1.91±5.76 kg - - - E005/6試験 プラセボ/LY群 30 - - - 5.18±4.35 kg** -
高用量群 235 3.58±4.69%** 5.21±5.99%** - - - GDED試験
低用量群 232 2.43±4.33%** 4.38±5.34%** - - - ID群 140 - - 4.70±6.22%** - -
T002試験 FD群 141 - - 5.01±5.99%** - -
0週からの変化量又は変化率
*:p<0.05、**:p<0.001
2) FMの変化(外国臨床成績)
外国臨床試験で得られた FMの変化に関する成績を表 24にまとめ、ベースラインからの変
化量(kg)又は変化率(%)で示した。
いずれの試験においても、本剤が投与された患者群では、ベースラインから投与後 12~32
週までに、FM が顕著に減少した。また、48 週間以上の長期投与において、FM はベースライ
ンからは減少していたが、投与後 24 週時までの変化ほど顕著ではなかった。なお、プラセボ
群では有意な変化は認められなかった。
表 24 FMの変化量(kg)又は変化率(%)(外国臨床試験)
FMの変化率 (%)又は変化量 (kg) :平均値±標準偏差 投与期間(週) 試験番号 投与群 例数
12 24 32 48 72 LY群 46 - -3.27±9.60 kg*
- -0.84±10.38 kg -0.62±11.35 kg
プラセボ群 45 - 0.56±6.18 kg - - - E003/4試験
プラセボ/LY群 43 - - - -3.69±5.29 kg** - LY群 31 - -3.33±4.64 kg**
- -2.48±4.67 kg* -1.67±5.79 kg
プラセボ群 34 - 2.23±6.48 kg - - - E005/6試験 プラセボ/LY群 30 - - - -4.71±5.40 kg** -
高用量群 235 -5.53±8.64%** -9.45±12.07%**- - -
GDED試験 低用量群 232 -2.81±7.81%** -6.35±9.42%** - - -
ID群 140 - - -7.91±11.93%**- -
T002試験 FD群 141 - - -10.9±11.54%** - -
0週からの変化量又は変化率
*:p<0.05、**:p<0.001
3) 皮下脂肪厚の推移(外国臨床成績)
外国臨床試験において、本剤又はプラセボが投与された患者群における皮下脂肪厚の推移を
表 25に示した。
*
**
**
**
*
***
*

ヒューマトロープ CTD(2.7 臨床概要) Page 32
32
いずれの試験においても、本剤投与後 24 週時には皮下脂肪厚は減少し、ベースライン値と
の差は、いずれも統計学的に有意であった。また、E003/4 試験及び E005/6 試験において、投
与後 24 週時までの、本剤投与群とプラセボ投与群との皮下脂肪厚の変化量の差は、いずれも
統計学的に有意であった。一方、プラセボが投与された患者においては、皮下脂肪厚の有意な
変化は認められなかった。
表 25 皮下脂肪厚の推移(外国臨床試験)
皮下脂肪厚(mm):平均値±標準偏差(例数) 投与期間(週) 試験番号 投与群
0 8 12 16 24 32 48 72
LY群 83.80± 30.61 (46)
- - - 71.25±21.31**
(44) -
68.35± 22.69**
(42)
68.68±21.68**
(40)
プラセボ群 92.96± 34.72 (46)
- - - 88.83±27.88 (43)
- - - E003/4試験
プラセボ/LY群
88.83± 27.88 (43)
- - - 81.36±28.81**
(42) -
78.03± 24.01**
(40) -
LY群 83.17± 39.32 (31)
- - - 69.83±29.33*
(26) -
67.85± 25.83**
(24)
69.03±31.77*
(20)
プラセボ群 85.08± 34.85 (35)
- - - 81.30±30.11 (32)
- - - E005/6試験
プラセボ/LY群
81.30± 30.11 (32)
- - - 68.05±31.52*
(28) -
66.85± 27.26**
(24) -
高用量群 77.61± 28.48 (289)
- 75.93±29.77**
(289) -
73.90±28.78**
(289) - - -
GDED試験
低用量群 79.53± 26.73 (295)
- 77.20±26.43**
(295) -
76.22±25.79**
(295) - - -
ID群 81.46± 28.15 (178)
80.92±28.57**
(173) -
78.73±28.96**
(165)
78.14±29.29**
(158)
76.05± 29.71**
(156) - -
T002試験
FD群 77.52± 28.19 (194)
76.08±28.09**
(184) -
74.28±28.95**
(178)
71.73±29.25**
(176)
70.52± 29.20**
(172) - -
*:p<0.05、**:p<0.001
4) 運動能力(Exercise Capacity)の変化(外国臨床成績)
E003/4試験において、一部の施設で運動能力が測定された。対象となった 20例の患者は、6ヵ
月間、本剤又はプラセボの投与を受けた成人期発症型の AGHD患者であった。
表 26~表 32に、最大酸素消費量、最大換気量、無酸素性作業閾値、最大運動負荷量、体重
あたりの最大運動負荷量、LBMあたりの最大運動負荷量及び運動時間の推移を示した。
本剤が投与された患者群では、最大酸素消費量、最大換気量、無酸素性作業閾値及び運動時
間は、いずれの項目も増加し、ベースラインとの差は統計学的に有意であった。一方、プラセ
ボが投与された患者群では、いずれの項目についても、統計学的に有意な差は認められなかっ
た。
最大運動負荷量は、本剤が投与された患者群で増加し、ベースラインとの差は統計学的に有
意であった。また、体重あたりの最大運動負荷量も、本剤が投与された患者群で増加し、ベー
*
*

ヒューマトロープ CTD(2.7 臨床概要) Page 33
33
スラインとの差は統計学的に有意であった。しかし、本剤が投与された患者群において、LBM
が投与前後で 52 kgから 56.6 kgに増加し、ベースラインとの差は統計学的に有意であったにも
かかわらず、LBMあたりの最大運動負荷量は増加せず、統計学的に有意な差は認められなかっ
た。なお、プラセボ群では投与前後の LBMの変化に統計学的に有意な差は認められなかった
(47 kg→49.9 kg)。このことから、最大運動負荷量の増加は LBMの増加によるものである可
能性が高いと考えられた。
表 26 最大酸素消費量(VO2 max)の推移(E003/4試験)
VO2 max(L/分):平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 1.9±0.2 2.6±0.18*
プラセボ群 10 1.9±0.18 2.2±0.18
*:p<0.01
表 27 最大換気量(Maximal Ventilation)の推移(E003/4試験)
Maximal ventilation(L/分):平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 53.4±6.3 59.4±6.0*
プラセボ群 10 50.5±5.3 53.9±5.7
*:p<0.05
表 28 無酸素性作業閾値(Anaerobic Threshold/kg BW)の推移(E003/4試験)
Anaerobic threshold/kg BW(mL/分・kg):平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 16.9±1.8 20.4±1.7*
プラセボ群 10 16.04±1.0 18.4±1.2
*:p<0.01
表 29 最大運動負荷量(Maximal Power Output)の推移(E003/4試験)
Maximal power output(Watts):平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 192.5±13.5 227.5±11.5*
プラセボ群 10 187.5±16.8 192.5±14.9
*:p<0.01

ヒューマトロープ CTD(2.7 臨床概要) Page 34
34
表 30 体重あたりの最大運動負荷量(Maximal power output BW)の推移(E003/4試験)
Maximal power output BW(Watts/kg):平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 2.2±0.17 2.4±0.18*
プラセボ群 10 2.1±0.19 2.1±0.13
*:p<0.01
表 31 LBMあたりの最大運動負荷量(Maximal power output/lean body mass)の推移
(E003/4/試験)
Maximal power output/lean body mass(Watts/kg): 平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 3.7±0.3 3.5±0.3
プラセボ群 10 3.8±0.2 3.8±0.1
表 32 運動時間(Exercise time)の推移(E003/4/試験)
Exercise time(分):平均値±標準誤差
投与期間(週) 投与群 例数
0 24
LY群 10 6.35±0.6 6.77±0.5*
プラセボ群 10 6.05±0.7 6.17±0.6
*:p<0.05
(3) 有効性の副次評価
1) 血清 IGF-I濃度の推移
(a) 国内臨床試験における成績
国内臨床試験において、本剤又はプラセボが投与された患者群における血清 IGF-I濃度の推
移を表 33及び 表 34に示した。なお、K02A試験においては、血清 IGF-I濃度が基準範囲内に
入るように本剤の投与量を調整した。
LY群では、平均血清 IGF-I濃度の上昇が投与後 4週時から認められ、投与量の増加に伴い、
投与後 24週時まで上昇した。一方、プラセボ群では、平均 IGF-I濃度の平均値は変化しなかっ
た。
LY72w群では、投与後 24週時まで上昇した平均血清 IGF-I濃度は投与後 28週時に一時低下
したが、これは K02A試験開始から投与方法が変更され低用量(0.021mg/kg/週)が投与された
ためと考えられた。その後、投与量の増量に伴い、平均血清 IGF-I濃度は上昇した。プラセボ
/LY48w群では、平均血清 IGF-I濃度の上昇が投与後 4週時から認められた。

ヒューマトロープ CTD(2.7 臨床概要) Page 35
35
表 33 血清 IGF-I濃度の推移(投与 24週間)
血清 IGF-I濃度:ng/mL 投与期(週) 0 4 8 12 24
LY群 65 ± 46 (33)
153 ± 87 (33)
217 ± 109 (32)
181 ± 110 (32)
243 ± 114 (31)
プラセボ群 73 ± 49 (31)
69 ± 48 (30)
70 ± 47 (28)
67 ± 44 (29)
63 ± 39 (29)
平均値±標準偏差(例数)
表 34 血清 IGF-I濃度の推移(投与 72週間)
血清 IGF-I濃度:ng/mL 投与期間(週)
0 4 8 12 16 20 24 28 32 36 40 44 48 60 72
LY72w群 (ng/mL)
65± 46
(33)
153±87
(33)
217± 109 (32)
181± 110 (32)
- - 243±114(31)
148±84
(31)
147±89
(31)
170±81
(31)
184± 74
(30)
187± 86
(30)
217± 71
(29)
203±76
(30)
206±77
(30)
プラセボ/ LY48w群(ng/mL)
61± 39
(28)
133±82
(28)
140± 73
(28)
169± 86
(28)
178±87
(28)
181±79
(28)
195±92
(27)- -
213±90
(28)- -
191± 78
(27)
平均値±標準偏差(例数)
次に、本剤又はプラセボが投与された患者群における IGF-I SDSの推移を表 35及び 表 36に
示した。
LY群の投与開始時における IGF-I SDSの平均値は、基準範囲(-1.96SDS以上、1.96SDS以下)
よりも低かったが、投与後 4週時には基準範囲内の値を示し、投与後 24週時まで基準範囲内
で維持された。一方、プラセボ群では、IGF-I SDSの平均値は変化しなかった。
LY72w群では、投与後 24週時まで上昇した IGF-I SDSの平均値は、K02A試験から低用量が
投与されたため、投与後 28週時に低下したが、基準範囲内の値を示した。その後、投与後 72
週時まで IGF-I SDSの平均値は基準範囲内で維持された。プラセボ/LY48w群において、投与
開始時における IGF-I SDSの平均値は基準範囲よりも低かったが、投与後 4週時には基準範囲
内の値を示し、投与後 48週時まで維持された。
表 35 IGF-I SDSの推移(投与 24週間)
IGF-I SDS 投与期(週) 0 4 8 12 24
LY群 -2.42 ± 1.85 (33)
-0.58 ± 2.13 (33)
0.40 ± 2.37 (32)
-0.09 ± 2.43 (32)
0.82 ± 2.28 (31)
プラセボ群 -2.03 ± 1.58 (31)
-2.13 ± 1.66 (30)
-2.05 ± 1.59 (28)
-2.16 ± 1.57 (29)
-2.26 ± 1.44 (29)
平均値±標準偏差(例数)

ヒューマトロープ CTD(2.7 臨床概要) Page 36
36
表 36 IGF-I SDSの推移(投与 72週間) IGF-I SDS 投与期間
(週) 0 4 8 12 16 20 24 28 32 36 40 44 48 60 72
LY72w群 -2.42± 1.85 (33)
-0.58± 2.13 (33)
0.40± 2.37 (32)
-0.09± 2.43 (32)
- - 0.82±2.28(31)
-0.74±1.95(31)
-0.73±2.04(31)
-0.34±2.00(31)
0.11± 1.31 (30)
0.18± 1.56 (30)
0.65± 1.11 (29)
0.48±1.29(30)
0.51±1.25(30)
プラセボ/ LY48w群
-2.33± 1.42 (28)
-0.81± 2.02 (28)
-0.59± 1.81 (28)
-0.12± 1.66 (28)
0.05±1.56(28)
0.07±1.46(28)
0.32±1.70(27)
- - 0.67±1.65(28)
- - 0.38± 1.67 (27)
平均値±標準偏差(例数)
次に、各患者の血清 IGF-I濃度を基準範囲超、基準範囲内及び基準範囲未満に分類し、分布
の推移を表 37及び表 38に示した。
LY群では、投与開始時に基準範囲未満の症例は 33例中 21例であったが、投与後 24週時で
は、基準範囲未満の症例は 3例まで減少し、基準範囲内の値を示した症例は 22例、基準範囲
超の高値を示した症例は 6例であった。一方、プラセボ群では、投与開始時に基準範囲未満の
症例は 31例中 17例であり、投与後 24週時においても 29例中 17例が基準範囲未満であった。
LY72w群の投与後 24週時において、基準範囲未満の症例は 31例中 3例であったが、K02A
試験から低用量が投与されたため、投与後 28週時において基準範囲未満の症例は 7例となっ
た。その後、投与後 72 週時では、基準範囲未満の症例は認められず、基準範囲内の値を示し
た症例は 27例、基準範囲超の高値を示した症例は 3例であった。プラセボ/LY48w群の本剤
投与開始時では、28例中 17例が基準範囲未満の低い値を示していたが、投与後 48週時におい
ては基準範囲未満の症例は 2例に減少し、基準範囲内の値を示した症例は 22例、基準範囲超
の高値を示した症例は 3例であった。
以上、本剤が投与された患者群では、血清 IGF-I濃度及び IGF-I SDSは上昇した。一方、プ
ラセボが投与された患者群では血清 IGF-I濃度及び IGF-I SDSは変化しなかった。
表 37 血清 IGF-I濃度の分布の推移(投与 24週間)
投与期間(週) 0 4 8 12 24
基準範囲超 0 2 8 7 6
基準範囲 12 23 20 19 22 LY群 (例数)
基準範囲未満 21 8 4 6 3
基準範囲超 0 0 0 0 0
基準範囲 14 13 13 11 12 プラセボ群 (例数)
基準範囲未満 17 17 15 18 17
表 38 血清 IGF-I濃度の分布の推移(投与 72週間)
投与期間(週) 0 4 8 12 16 20 24 28 32 36 40 44 48 60 72
基準範囲超 0 2 8 7 - - 6 2 3 3 2 3 5 3 3
基準範囲 12 23 20 19 - - 22 22 19 24 26 24 24 26 27LY72w群 (例数)
基準範囲未満 21 8 4 6 - - 3 7 9 4 2 3 0 1 0
基準範囲超 0 2 2 2 4 1 4 - - 5 - - 3
基準範囲 11 17 21 21 20 24 20 - - 20 - - 22 プラセボ/LY48w群 (例数) 基準範囲未満 17 9 5 5 4 3 3 - - 3 - - 2

ヒューマトロープ CTD(2.7 臨床概要) Page 37
37
(b) 外国臨床試験における成績(参考資料)
外国臨床試験において、本剤又はプラセボが投与された患者群における血清 IGF-I濃度及び
IGF-I SDSの推移を表 39及び表 40に示した。いずれの試験においても、国内臨床成績と同様
に、本剤が投与された患者群では、血清 IGF-I濃度は上昇した。一方、プラセボが投与された
患者群では、血清 IGF-I濃度は変化しなかった。
表 39 血清 IGF-I濃度の推移(外国臨床試験)
血清 IGF-I濃度:平均値±標準偏差(例数) 投与期間(週)
試験番号 (単位) 投与群
0 8 12 16 24 32 48 72
LY群 73.35± 40.14 (46)
- - - 223.35±113.47
(43) -
249.28± 101.99
(43)
242.51±108.44
(39)
プラセボ群 70.28± 31.64 (46)
- - - 72.21±35.07 (43)
- - - E003/4試験 (ng/mL)
プラセボ/LY群
72.21± 35.07 (43)
- - - 245.95±108.47
(42) -
239.62± 101.14
(39) -
LY群 60.71± 69.50 (32)
- - - 195.18±146.98
(28) -
224.92± 147.73
(24)
214.67±131.07
(21)
プラセボ群 54.39± 45.66 (34)
- - - 55.33±43.16 (33)
- - - E005/6試験 (ng/mL)
プラセボ/LY群
55.33± 43.16 (33)
- - - 231.65±134.35
(26) -
210.19± 137.89
(26) -
高用量群 64.52± 43.72 (286)
- 162.48±
94.76 (286)
- 207.26±130.92(286)
- - - GDED試験
(ng/mL) 低用量群
61.01± 40. 11 (298)
- 127.34±
72.09 (298)
- 162.06±
93.91 (298)
- - -
ID群 78.681± 45.978 (181)
136.057±69.144(181)
- 167.334±
74.973(172)
180.045±75.362(166)
187.387± 73.346 (159)
- - T002試験 (ng/mL)
FD群 77.131± 44.922 (193)
157.904±77.336(191)
- 161.106±
81.741(188)
197.374±88.151(180)
190.515± 94.304 (177)
- -
表 40 IGF-I SDSの推移(外国臨床試験)
IGF-I SDS:平均値±標準偏差(例数) 投与期間(週) 試験番号 投与群
0 8 12 16 24 32
高用量群 -4.30±2.75 (286) - -1.30±2.54
(286) - -0.67±2.80 (286) -
GDED試験 低用量群 -4.41±2.92
(298) - -1.94±2.58 (298) - -1.20±2.39
(298) -
ID群 -3.096±2.130 (180)
-1.248±1.656(180) - -0.564±1.593
(172) -0.317±1.462
(165) -0.161±1.483
(159) T002試験
FD群 -3.043±2.033 (193)
-0.770±1.731(191) - -0.659±1.599
(188) -0.010±1.553
(180) -0.177±1.699
(177)

ヒューマトロープ CTD(2.7 臨床概要) Page 38
38
2) 血清 IGFBP-3濃度の推移
(a) 国内臨床試験における成績
国内臨床試験において、本剤又はプラセボが投与された患者群における血清 IGFBP-3濃度の
推移を表 41及び表 42に示した。
LY群では、平均血清 IGFBP-3濃度の上昇が投与後 4週時から認められ、投与量の増加に伴
い、投与後 24週まで上昇した。一方、プラセボ群では、平均血清 IGFBP-3濃度は変化しなかっ
た。
LY72w群では、K02A試験の投与後 24週時まで上昇した平均血清 IGFBP-3濃度は、K02A試
験の開始時には低用量が投与されたため、投与後 28 週時に低下したが、その後投与量の増加
に伴い上昇した。血清 IGFBP-3濃度の推移は、血清 IGF-I濃度の推移と一致していた。プラセ
ボ/LY48w群では、投与後 4週時以降から平均血清 IGFBP-3濃度の上昇が認められた。
表 41 血清 IGFBP-3濃度の推移(投与 24週間)
血清 IGFBP-3濃度:µg/mL 投与期(週) 0 4 8 12 24
LY群 2.0 ± 1.0 (33)
2.7 ± 1.2 (33)
3.1 ± 1.0 (32)
2.9 ± 1.2 (32)
3.3 ± 1.0 (31)
プラセボ群 2.1 ± 1.0 (31)
2.1 ± 1.2 (30)
2.0 ± 1.0 (28)
1.9 ± 1.0 (29)
1.9 ± 1.0 (29)
平均値±標準偏差(例数)
表 42 血清 IGFBP-3濃度の推移(投与 72週間)
血清 IGBP-3濃度:µg/mL 投与期間(週) 0 4 8 12 16 20 24 28 32 36 40 44 48 60 72
LY72w群 (µg/mL)
2.0± 1.0 (33)
2.7±1.2 (33)
3.1± 1.0 (32)
2.9± 1.2 (32) - -
3.3±1.0 (31)
2.6±1.0 (31)
2.7±1.1 (31)
2.9±1.1 (31)
3.2±1.1 (30)
3.2± 1.1 (30)
3.5± 1.0 (29)
3.3±0.9 (30)
3.5±1.0(30)
プラセボ/LY48w群(µg/mL)
1.9± 1.0 (28)
2.6±1.2 (28)
2.7± 1.1 (28)
2.9± 1.1 (28)
2.9±1.1 (28)
2.9±1.0 (28)
3.2±1.1 (27)
- - 3.2±
1.0 (28)
- - 3.3±
1.1 (27)
平均値±標準偏差(例数)
(b) 外国臨床試験における成績(参考資料)
外国臨床試験において、本剤又はプラセボが投与された患者群における血清 IGFBP-3濃度の
推移を表 43 に示した。いずれの試験においても、国内臨床成績と同様に、本剤が投与された
患者群では、血清 IGFBP-3 濃度は上昇した。一方、プラセボが投与された患者群では血清
IGFBP-3濃度は変化しなかった。

ヒューマトロープ CTD(2.7 臨床概要) Page 39
39
表 43 血清 IGFBP-3濃度の推移(外国臨床試験)
血清 IGFBP-3濃度:平均値±標準偏差(例数) 投与期間(週)
試験番号(単位) 投与群
0 8 12 16 24 32 48 72
LY群 2474.81±
992.89 (46)
- - - 3528.93±1113.05
(43) - -
3488.82±1053.31
(39)
プラセボ群 2317.20±
929.37 (46)
- - - 2276.81±
742.01 (43)
- - - E003/4試験 (ng/mL)
プラセボ/LY群
2276.81± 742.01
(43) - - - - -
3530.05±880.71
(37) -
LY群 1562.50± 1041.93
(32) - - -
2737.21±1030.40
(28) - -
3555.49±1255.47
(20)
プラセボ群 1595.83±
970.19 (35)
- - - 1560.00±1036.44
(33) - - - E005/6試験
(ng/mL)
プラセボ/LY群
1560.00± 1036.44
(33) - - - - -
3312.56±1342.69
(25) -
高用量群 2.06±1.01 (286) - 3.00±1.19
(286) - 3.27±1.24(286) - - - GDED試験
(µg/L) 低用量群 2.01±1.03 (298) - 2.79±1.15
(298) - 3.04±1.12(298) - - -
ID群 2.18± 0.90 (186)
2.94± 1.03 (181)
- 3.25± 1.01 (172)
3.19± 1.01 (165)
3.20± 0.88 (159)
- - T002試験(µg/L)
FD群 2.16± 0.93 (198)
3.03± 1.01 (191)
- 3.05± 0.96 (188)
3.35± 1.05 (180)
3.20± 0.93 (177)
- -
3) QOLの評価
(a) 国内臨床試験における成績
a) SF-36を用いた QOLの評価
① 全患者を対象とした SF-36の評価
国内臨床試験において、本剤又はプラセボが投与された患者群における SF-36の推移を表 44
に示した。
LY群では、ベースラインから投与後 24週時までに、6項目の平均偏差得点が上昇し、2項
目が低下した。いずれの下位尺度においても、本剤投与前後における偏差得点の変化は、統計
学的に有意ではなかった。プラセボ群では、3 項目の平均偏差得点が上昇し、5 項目が低下し
た。いずれの下位尺度においても、プラセボ投与前後における偏差得点の変化は、統計学的に
有意ではなかった。また、いずれの下位尺度においても、LY 群とプラセボ群との偏差得点の
変化量の差は、統計学的に有意ではなかった。
次に、K01A試験及び K02A試験を併合し、本剤が投与された期間における SF-36の推移を
表 45に示した。
LY72w群では、ベースラインから投与後 48週時までに、7項目の平均偏差得点が上昇し、1
項目が低下した。また、ベースラインから投与後 72週時までに、5項目の平均偏差得点が上昇
し、3 項目が低下した。しかしながら、いずれの下位尺度においても、本剤投与前後における
偏差得点の変化は、統計学的に有意ではなかった。

ヒューマトロープ CTD(2.7 臨床概要) Page 40
40
プラセボ/LY48w群では、本剤投与が開始されたベースライン(K02A試験投与開始時)か
ら投与後 48週時までに、2項目の平均偏差得点が上昇し、1項目は変化なく、5項目が低下し
たが、いずれの下位尺度においても、本剤投与前後における偏差得点の変化は、統計学的に有
意ではなかった。
表 44 QOL(SF-36)の推移(投与 24週間)
投与期間(週) 0 24
項 目 投与群 平均値± 標準偏差
(例数)
平均値± 標準偏差
(例数)
検定*1
p値 検定*2
p値
LY群 43.9±18.8 (31) 44.8±23.6 (31) 0.670 PF:身体機能
プラセボ群 44.5±12.6 (30) 46.1±11.3 (30) 0.703 0.726
LY群 44.1±15.7 (33) 43.6±18.6 (32) 0.964 RP:日常役割機能 (身体)
プラセボ群 47.5±11.7 (31) 42.0±20.4 (30) 0.258 0.398
LY群 50.7±11.7 (33) 49.2±11.3 (32) 0.408 BP:体の痛み
プラセボ群 51.9±9.3 (31) 50.1±13.1 (30) 0.617 0.659
LY群 44.6±12.1 (33) 45.5±11.6 (32) 0.835 GH:全体的健康感
プラセボ群 42.7±10.1 (30) 45.3±9.1 (30) 0.403 0.664
LY群 47.1±13.4 (33) 48.0±11.1 (32) 0.873 VT:活力
プラセボ群 43.7±10.3 (30) 44.3±12.3 (30) 0.705 0.783
LY群 48.4±11.6 (33) 50.6±9.5 (32) 0.423 SF:社会生活機能
プラセボ群 50.3±9.9 (31) 49.7±10.3 (30) 0.980 0.695
LY群 44.6±12.5 (32) 46.5±12.7 (32) 0.178 RE:日常役割機能 (精神)
プラセボ群 47.9±12.4 (31) 44.4±14.6 (30) 0.435 0.209
LY群 50.0±11.3 (33) 51.5±10.2 (32) 0.623 MH:心の健康
プラセボ群 50.9±7.8 (30) 48.9±10.8 (30) 0.413 0.398
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 41
41
表 45 QOL(SF-36)の推移(投与 72週間)
投与期間(週) 0 24 48 72
投与群 項 目 平均値±標準偏差
(例数)
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
PF: 身体機能
43.9±18.8(31)
44.8±23.6 (31) 0.670 45.1±23.9
(32) 0.363 45.2±25.6 (32) 0.050
RP: 日常役割機能(身体)
44.1±15.7(33)
43.6±18.6 (32) 0.964 45.2±18.7
(32) 0.337 46.9±13.7 (32) 0.116
BP: 体の痛み
50.7±11.7(33)
49.2±11.3 (32) 0.408 48.5±12.8
(32) 0.357 48.1±11.2 (32) 0.208
GH: 全体的健康感
44.6±12.1(33)
45.5±11.6 (32) 0.835 46.8±12.7
(32) 0.272 46.0±12.8 (32) 0.850
VT: 活力
47.1±13.4(33)
48.0±11.1 (32) 0.873 48.2±12.7
(32) 0.916 49.4±12.1 (32) 0.293
SF: 社会生活機能
48.4±11.6(33)
50.6±9.5 (32) 0.423 49.5±12.3
(32) 0.435 46.5±14.2 (32) 0.265
RE: 日常役割機能(精神)
44.6±12.5(32)
46.5±12.7 (32) 0.178 46.5±13.2
(32) 0.134 46.8±14.3 (32) 0.098
LY72w群
MH: 心の健康
50.0±11.3(33)
51.5±10.2 (32) 0.623 51.2±12.3
(32) 0.956 49.4±11.7 (32) 0.598
PF: 身体機能
47.3±8.7 (28)
47.3±10.9 (27) 0.471 47.0±10.8
(28) 0.918 - -
RP: 日常役割機能(身体)
45.7±13.2(28)
47.5±12.6 (28) 0.738 45.7±13.9
(28) 0.844 - -
BP: 体の痛み
51.4±9.8 (28)
50.4±9.0 (28) 0.700 49.5±12.0
(28) 0.452 - -
GH: 全体的健康感
45.6±9.3 (28)
46.3±9.6 (28) 0.532 45.9±10.3
(28) 0.771 - -
VT: 活力
45.3±12.0(28)
45.8±10.4 (28) 0.826 43.0±12.6
(28) 0.221 - -
SF: 社会生活機能
49.1±10.4(28)
49.3±11.1 (28) 0.900 44.3±13.6
(28) 0.065 - -
RE: 日常役割機能(精神)
46.5±12.5(28)
50.4±8.5 (28) 0.130 46.6±12.6
(28) 0.978 - -
プラセボ/LY48w群
MH: 心の健康
49.9±10.5(28)
50.4±8.3 (28) 0.742 47.2±10.7
(28) 0.185 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
② 発症時期別の SF-36の評価
ベースラインにおける下位尺度偏差得点が、成人期発症型の方が小児期発症型より低かった
ことから(「2.7.3.3.1 試験対象集団」参照)、成人期発症型及び小児期発症型の QOLの推移
を検討した。
K01A 試験において、本剤又はプラセボが投与された成人期発症型の患者群における SF-36
の推移を表 46に示した。
LY群において、7項目の平均偏差得点が上昇し、1項目が低下したが、いずれの下位尺度も
ベースラインとの差は統計学的に有意ではなかった。プラセボ群においては、5 項目の平均偏
差得点が上昇し、3 項目が低下したが、いずれの下位尺度もベースラインとの差は統計学的に
有意ではなかった。「体の痛み」において、LY 群の方がプラセボ群に比して有意に低下して

ヒューマトロープ CTD(2.7 臨床概要) Page 42
42
いた(p=0.037)。
次に、K01A試験及び K02A試験を併合し、本剤が投与された期間における成人期発症型患
者の SF-36の推移を表 47に示した。
LY72w 群において、ベースラインから投与後 48週時にかけて、7項目の平均偏差得点が上
昇し、1 項目が低下した。平均偏差得点が上昇した下位尺度の中で、ベースラインとの差が統
計学的に有意であったのは、「日常役割機能(身体)」(p=0.027)と「日常役割機能(精神)」
(p=0.006)の 2項目であった。これらの変化は、投与後 72週時も同じ傾向であり、投与後 72
週時において平均偏差得点が上昇した下位尺度の中で、ベースラインとの差が統計学的に有意
であったのは、「身体機能」(p=0.012)、「日常役割機能(身体)」(p=0.039)及び「日常
役割機能(精神)」(p=0.003)であった。
一方、プラセボ/LY48w 群においては、ベースラインから投与後 48週時にかけて、7項目
の平均偏差得点が低下し、「社会生活機能」はベースラインとの差は統計学的に有意(p=0.047)
であった。
表 46 成人期発症型における QOL(SF-36)の推移(投与 24週間)
投与期間(週) 0 24
項 目 投与群 平均値± 標準偏差
(例数)
平均値± 標準偏差
(例数)
検定*1
p値 検定*2
p値
LY群 44.7±14.5 (12) 49.8±11.2 (13) 0.094 PF:身体機能
プラセボ群 42.0±15.0 (12) 47.9±9.2 (12) 0.234 0.925
LY群 43.3±14.1 (14) 46.8±13.4 (13) 0.203 RP:日常役割機能(身体)
プラセボ群 43.9±15.8 (13) 42.1±15.6 (12) 0.938 0.330
LY群 51.9±11.2 (14) 45.7±10.1 (13) 0.064 BP:体の痛み
プラセボ群 50.5±10.4 (13) 51.4±12.7 (12) 0.688 0.037
LY群 43.1±12.0 (14) 45.1±10.3 (13) 0.533 GH:全体的健康感
プラセボ群 36.2±9.9 (12) 43.7±7.9 (12) 0.064 0.324
LY群 44.5±13.7 (14) 46.9±10.5 (13) 0.569 VT:活力
プラセボ群 39.0±12.1 (12) 42.1±11.7 (12) 0.420 0.931
LY群 46.6±10.8 (14) 51.5±7.4 (13) 0.195 SF:社会生活機能
プラセボ群 49.6±9.0 (13) 48.8±11.2 (12) 0.945 0.333
LY群 42.3±13.8 (13) 45.0±12.8 (13) 0.320 RE:日常役割機能 (精神)
プラセボ群 44.3±16.5 (13) 43.7±14.2 (12) 1.000 0.458
LY群 47.2±13.7 (14) 48.6±13.2 (13) 0.945 MH:心の健康
プラセボ群 47.5±7.2 (12) 49.3±10.1 (12) 0.695 0.954
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 43
43
表 47 成人期発症型における QOL(SF-36)の推移(投与 72週間)
投与期間(週) 0 24 48 72
投与群 項 目 平均値± 標準偏差
(例数)
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
PF: 身体機能
44.7±14.5 (12)
49.8±11.2 (13) 0.094 48.7±17.8
(13) 0.359 49.2±12.9 (13) 0.012
RP: 日常役割機能(身体)
43.3±14.1 (14)
46.8±13.4 (13) 0.203 49.0±14.5
(13) 0.027 47.0±15.0 (13) 0.039
BP: 体の痛み
51.9±11.2 (14)
45.7±10.1 (13) 0.064 47.4±12.6
(13) 0.266 48.0±11.1 (13) 0.275
GH: 全体的健康感
43.1±12.0 (14)
45.1±10.3 (13) 0.533 47.5±10.2
(13) 0.214 46.5±11.2 (13) 0.365
VT: 活力
44.5±13.7 (14)
46.9±10.5 (13) 0.569 48.7±12.7
(13) 0.569 48.8±12.5 (13) 0.160
SF: 社会生活機能
46.6±10.8 (14)
51.5±7.4 (13) 0.195 50.9±10.1
(13) 0.156 48.3±12.1 (13) 0.496
RE: 日常役割機能(精神)
42.3±13.8 (13)
45.0±12.8 (13) 0.320 46.9±14.2
(13) 0.006 47.8±13.9 (13) 0.003
LY72w群
MH: 心の健康
47.2±13.7 (14)
48.6±13.2 (13) 0.945 49.7±14.4
(13) 0.970 49.7±13.1 (13) 0.638
PF: 身体機能
47.5±9.6 (11)
47.2±8.4 (10) 0.750 46.1±11.6
(11) 1.000 - -
RP: 日常役割機能(身体)
44.1±14.7 (11)
45.6±11.8 (11) 1.000 41.3±16.6
(11) 0.438 - -
BP: 体の痛み
50.5±12.8 (11)
48.0±10.5 (11) 0.734 48.5±13.3
(11) 0.563 - -
GH: 全体的健康感
44.2±8.1 (11)
44.9±11.3 (11) 0.898 44.2±13.2
(11) 0.982 - -
VT: 活力
43.6±11.0 (11)
43.5±11.9 (11) 0.844 39.9±17.4
(11) 0.413 - -
SF: 社会生活機能
48.1±11.4 (11)
41.5±12.2 (11) 0.078 40.2±16.7
(11) 0.047 - -
RE: 日常役割機能(精神)
45.6±13.2 (11)
46.1±9.9 (11) 0.938 42.3±16.1
(11) 0.844 - -
プラセボ/LY48w群
MH: 心の健康
50.3±9.9 (11)
49.1±5.6 (11) 0.637 43.0±13.9
(11) 0.083 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
K01A 試験において、本剤又はプラセボが投与された小児期発症型の患者群における SF-36
の推移を表 48に示した。
LY群において、4項目の平均偏差得点が上昇し、4項目が低下したが、いずれの下位尺度も
ベースラインとの差は統計学的に有意ではなかった。プラセボ群においては、8 項目すべての
平均偏差得点が低下したが、いずれの下位尺度もベースラインとの差は統計学的に有意ではな
かった。LY 群とプラセボ群との変化量の差については、いずれの下位尺度においても、統計
学的に有意ではなかった。
次に、K01A試験及び K02A試験を併合し、本剤が投与された期間における小児期発症型患
者の SF-36の推移を表 49に示した。
LY72w 群において、ベースラインから投与後 48週時にかけて、3項目の平均偏差得点が上

ヒューマトロープ CTD(2.7 臨床概要) Page 44
44
昇し、5 項目が低下したが、いずれの下位尺度も、ベースラインとの差は統計学的に有意では
なかった。また、ベースラインから投与後 72週時までに、2項目の平均偏差得点が上昇し、1
項目が変化なく、5項目が低下した。「社会生活機能」を除き、いずれの下位尺度においても、
投与前後における偏差得点の差は統計学的に有意ではなかった。
プラセボ/LY48w 群において、ベースラインから投与後 48週時にかけて、5 項目の平均偏
差得点が上昇し、3 項目が低下した。いずれの下位尺度においても、投与前後における偏差得
点の差は統計学的に有意ではなかった。
表 48 小児期発症型における QOL(SF-36)の推移(投与 24週間;K01A試験)
投与期間(週) 0 24
項 目 投与群 平均値± 標準偏差
(例数)
平均値± 標準偏差
(例数)
検定*1
p値 検定*2
p値
LY群 43.4±21.5 (19) 41.2±29.4 (18) 0.414 PF:身体機能
プラセボ群 46.1±10.8 (18) 44.9±12.6 (18) 0.836 0.607
LY群 44.8±17.1 (19) 41.4±21.6 (19) 0.320 RP:日常役割機能(身体)
プラセボ群 50.1±6.8 (18) 41.9±23.4 (18) 0.250 0.759
LY群 49.9±12.2 (19) 51.6±11.8 (19) 0.700 BP:体の痛み
プラセボ群 52.9±8.6 (18) 49.2±13.7 (18) 0.233 0.217
LY群 45.8±12.3 (19) 45.7±12.7 (19) 0.796 GH:全体的健康感
プラセボ群 47.1±7.7 (18) 46.4±9.9 (18) 0.492 0.818
LY群 49.1±13.3 (19) 48.6±11.7 (19) 0.681 VT:活力
プラセボ群 46.8±7.8 (18) 45.8±12.8 (18) 0.709 0.988
LY群 49.6±12.2 (19) 50.0±10.8 (19) 0.570 SF:社会生活機能
プラセボ群 50.9±10.7 (18) 50.2±9.9 (18) 1.000 0.703
LY群 46.1±11.7 (19) 47.4±12.8 (19) 0.377 RE:日常役割機能(精神)
プラセボ群 50.5±7.8 (18) 44.8±15.2 (18) 0.370 0.390
LY群 52.0±8.9 (19) 53.5±7.4 (19) 0.423 MH:心の健康
プラセボ群 53.3±7.5 (18) 48.7±11.6 (18) 0.147 0.140
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 45
45
表 49 小児期発症型における QOL(SF-36)の推移(投与 72週間)
投与期間(週) 0 24 48 72
投与群 項 目 平均値± 標準偏差
(例数)
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
平均値± 標準偏差
(例数)
検定*
p値
PF: 身体機能
43.4±21.5 (19)
41.2±29.4 (18) 0.414 42.6±27.5
(19) 0.934 42.5±31.5 (19) 0.516
RP: 日常役割機能(身体)
44.8±17.1 (19)
41.4±21.6 (19) 0.320 42.7±21.1
(19) 0.754 46.8±13.1 (19) 0.483
BP: 体の痛み
49.9±12.2 (19)
51.6±11.8 (19) 0.700 49.3±13.1
(19) 0.970 48.2±11.5 (19) 0.542
GH: 全体的健康感
45.8±12.3 (19)
45.7±12.7 (19) 0.796 46.2±14.5
(19) 0.686 45.7±14.1 (19) 0.417
VT: 活力
49.1±13.3 (19)
48.6±11.7 (19) 0.681 47.9±13.1
(19) 0.711 49.9±12.2 (19) 0.755
SF: 社会生活機能
49.6±12.2 (19)
50.0±10.8 (19) 0.570 48.6±13.8
(19) 0.625 45.3±15.7 (19) 0.034
RE: 日常役割機能(精神)
46.1±11.7 (19)
47.4±12.8 (19) 0.377 46.2±12.8
(19) 0.783 46.1±14.9 (19) 0.953
LY72w群
MH: 心の健康
52.0±8.9 (19)
53.5±7.4 (19) 0.423 52.2±11.0
(19) 0.930 49.1±11.0 (19) 0.280
PF: 身体機能
47.1±8.3 (17)
47.3±12.4 (17) 0.490 47.5±10.5
(17) 0.818 - -
RP: 日常役割機能(身体)
46.7±12.5 (17)
48.7±13.3 (17) 0.505 48.5±11.5
(17) 0.747 - -
BP: 体の痛み
51.9±7.6 (17)
51.9±7.8 (17) 0.970 50.1±11.4
(17) 0.761 - -
GH: 全体的健康感
46.4±10.2 (17)
47.1±8.6 (17) 0.553 46.9±8.2
(17) 0.486 - -
VT: 活力
46.4±12.8 (17)
47.3±9.5 (17) 0.868 45.0±8.3
(17) 0.385 - -
SF: 社会生活機能
49.8±10.0 (17)
54.4±6.8 (17) 0.135 46.9±10.9
(17) 0.455 - -
RE: 日常役割機能(精神)
47.0±12.5 (17)
53.2±6.4 (17) 0.067 49.3±9.2
(17) 0.749 - -
プラセボ/LY48w群
MH: 心の健康
49.6±11.2 (17)
51.2±9.8 (17) 0.497 49.9±7.3
(17) 0.946 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
国内臨床試験における、SF-36を用いた AGHD患者の QOLの評価において、成人期発症型
と小児期発症型に差が見られたことについて考察した。
成人期発症型及び小児期発症型の AGHD患者におけるベースラインの下位偏差得点は、発症
時期により異なり、全般的に成人期発症型の方が低かった(表 17)。その理由として、成人期
発症型では健康な時期を経験しており、健康であった状態と現状を比較することができ、その
違いを十分認識しているのに対し、小児期発症型では小児期より GH分泌不全を発症し、正常
な状態を経験していないため、自分自身の病態に対する認識が薄いことが挙げられる。そのた
め、特に自分自身の QOL が低下しているとの認識が薄い小児期発症型の患者において、疾患
特異的ではない SF-36の質問票を用いて QOLを評価することは困難であり、小児期発症型の
患者のベースラインにおいて、QOLスコアの低下として現れていなかったものと考えられた。

ヒューマトロープ CTD(2.7 臨床概要) Page 46
46
さらに、小児期発症型に対する GH投与の治療効果を検出することも困難であった。
一般的に、小児期発症型の患者では、成長障害などの治療のために小児期に GH投与を受け
ていることが多い。K01A試験においても小児期発症型の患者 37例のうち 31例が小児期に GH
投与を受けていた。これらの被験者における小児期の GH投与終了から、本治験での GH投与
までの期間を集計したところ、261日~6706日(平均±SD:3174日±1997日)と個人差が非
常に大きかった。そこで、この GH未投与期間が QOLのベースラインの点数や治療後の QOL
の変化に関係していないかを検討したところ、特に相関を認めなかった。
一方、Vahlらによって、小児期発症型の QOL評価に関する報告がなされた 4。この報告によ
ると、GHを投与している小児期発症型の AGHD患者を対象として、そのまま GH投与を継続
した群及び、いったん GH投与を中止し 12ヵ月間プラセボ投与を実施した後に、再び GH投与
を行った群において、General Health Questionnaireを用いて QOLを評価した。その結果、12ヵ
月の GH又はプラセボ投与を終えた時点では、両群の QOLスコアにベースラインからの変化
は認められなかったが、プラセボ投与後 GH投与を再開した群において、QOLの改善が認めら
れた。
このことから、Vahlらの報告は GHを投与しない期間が単一かつ 12ヵ月と短く、国内臨床
試験で GHを投与しない期間に非常な差があった小児期発症型患者での成績との比較は困難で
あり、さらに使用している QOL 質問票が異なることを考慮する必要があるものの、国内にお
いても他の適切な QOLの評価方法を採用すれば、小児期発症型の AGHD患者を対象にしても、
QOLスコアの変化は検出できると考えられる。
成人期発症型の投与後 48及び 72週時に有意な上昇が認められた「日常役割機能(身体)」
と「日常役割機能(精神)」の 2項目は、日常生活において、被験者が有職者もしくは主婦な
ど、明確な役割を担っている場合に変化が認められる項目である。一般的に成人期発症型の場
合、小児期発症型に比べて、職業を有している AGHD患者が多いため、結果に差が出たと考え
られた。また、同様に LY72w群において投与後 72週時に有意な上昇が認められた「身体機能」
については、外国臨床試験(E003/4試験)でも運動機能の向上が認められているように、本試
験においてもスコアの上昇となって反映された可能性が考えられる。
しかしながら、プラセボ/LY48w群では、これらの項目に関し LY72w群で認められたよう
な有意な上昇は認められず、「社会生活機能」については統計学的に有意な低下が認められた。
LY72w群とプラセボ/LY48w群では、LBM、IGF-I、血清脂質及び体重等に対する GH補充療
法の効果は両群間で差は認められていない。相違点としては、プラセボ/LY48w群では本剤投
与前に 24週間のプラセボ投与期間があったことがあげられるが、両群間の QOLスコアの推移
に差が認められた原因は現時点では不明である。
また、小児期発症型の LY72w 群で有意な低下が認められた「社会生活機能」については、
成人期発症型のプラセボ/LY48w群においても認められている。しかし、これらの群において
特に有害事象が増加しているわけではなく、成人期発症型の LY72w 群では逆に上昇している
こと、及び外国臨床試験ではこのような傾向は見られておらず、その原因は現時点では不明で
ある。

ヒューマトロープ CTD(2.7 臨床概要) Page 47
47
b) QLSを用いた QOLの評価(参考成績)
K01A試験において、疾患特異的 QLS質問票の信頼性及び妥当性を確認するには至らなかっ
たため、参考成績とした。
LY72w群、プラセボ群、プラセボ/LY48w群におけるQLSの各項目の推移を表 50に示した。
表 50 QLSの推移
QLS:平均値±標準偏差(例数) 投与期間(週) 項 目 投与群
0 24 48 72
LY72w群 20.7±73.3 (32)
20.8±56.1 (32)
34.7±54.7 (32)
37.5±63.2(32)
プラセボ群 28.7±51.0 (30)
16.8±46.2 (30) - - QLS-H:ホルモンの不足
プラセボ/LY48w群 18.8±47.2 (28)
25.7±44.5 (28)
36.8±55.5 (28) -
LY72w群 19.4±33.3 (32)
23.3±31.3 (31)
24.7±36.3 (32)
22.6±35.4(32)
プラセボ群 20.4±36.9 (28)
21.4±31.9 (30) - - QLS-A:一般事項
プラセボ/LY48w群 22.9±32.6 (28)
22.4±33.3 (28)
23.6±39.2 (28) -
LY72w群 31.6±47.0 (32)
34.2±45.4 (32)
36.1±50.5 (32)
33.4±44.7(32)
プラセボ群 26.3±37.4 (29)
28.9±44.0 (30) - - QLS-G:健康
プラセボ/LY48w群 31.8±44.1 (28)
33.1±39.4 (28)
32.0±42.0 (28) -
(b) 外国臨床試験における成績(参考資料)
GDED試験及び T002試験で得られた QLSの成績を表 51に示した。
GDED試験において、投与後 12週時及び 24週時の QLSが評価された。その結果、いずれの
時点においても、ベースラインと比較して統計学的有意差が認められ、QOLが改善したことが
確認された。
T002試験において、投与後 32週時の QLSが評価された。その結果、ベースラインと比較し
て、統計学的有意差が認められ、QOLが改善したことが確認された。

ヒューマトロープ CTD(2.7 臨床概要) Page 48
48
表 51 QLSの推移(外国臨床試験)
QLS:平均値±標準偏差(例数) 投与期間(週) 試験番号 項 目 投与群
0 12 24 32
高用量群 27.31±39.52 (148)
40.78±35.13** (148)
45.66±35.97 ** (148) - QLS-H:
ホルモンの不足 低用量群 26.84±34.95
(150) 41.84±33.15 **
(150) 48.84±34.55 **
(150) -
高用量群 45.23±42.01 (150)
52.49±35.67* (150)
59.16±37.50** (150) - QLS-A:
一般事項 低用量群 47.51±36.01
(147) 60.44±33.37**
(147) 64.21±34.41**
(147) -
高用量群 38.59±49.63 (149)
50.11±45.26**(149)
59.68±46.80** (149) -
GDED試験
QLS-G: 健康
低用量群 38.09±49.85 (149)
57.57±41.33**(149)
64.58±40.93** (149) -
ID群 27.209±52.261(174) - - 58.045±47.654**
(174) QLS-H: ホルモンの不足
FD群 29.178±44.792(186) - - 55.511±46.193**
(186)
ID群 46.860±37.756(175) - - 62.998±36.323**
(175) QLS-A: 一般事項
FD群 52.091±34.797(187) - - 65.053±36.161**
(187)
ID群 44.462±50.151(174) - - 68.381±47.944**
(174)
T002試験
QLS-G: 健康
FD群 44.275±45.690(189) - - 70.090±46.308**
(189)
*:p<0.05、**:p<0.001
(4) その他の有効性評価(血清脂質の変化)
1) 国内臨床試験における成績
国内臨床試験において、本剤又はプラセボが投与された患者群における血清脂質(総コレス
テロール値、LDLコレステロール値)の推移を表 52~表 60に示した。
(a) 総コレステロール値の変化
LY群において、総コレステロール値は低下し、ベースラインとの差は統計学的に有意であっ
た(p=0.025、表 52)。一方、プラセボ群では総コレステロール値の低下は認められなかった。
また、LY 群とプラセボ群との変化量の差は統計学的に有意であった(p=0.036)。この結果、
24週間の GH補充療法による総コレステロール値の低下が確認された。
次に、K01A試験及び K02A試験を併合し、本剤が投与された期間における患者群の総コレ
ステロール値の推移を表 53に示した。
LY72w群では、投与 24週時に低下した総コレステロール値は投与後 48週時に上昇したが、
ベースラインよりは低かった。ベースラインと投与後 48 週時との差は統計学的に有意ではな
かった。その後、投与後 48週時から投与後 72週時にかけて、総コレステロール値は低下した。
ベースラインと投与後 72週時との差は、統計学的に有意(p=0.042)であった。プラセボ/LY48w
群では、ベースラインから投与後 24 週時までに、総コレステロール値は低下し、ベースライ

ヒューマトロープ CTD(2.7 臨床概要) Page 49
49
ンとの差は統計学的に有意(p=0.011)であった。投与後 24週時から投与後 48週時にかけて、
総コレステロール値は上昇したが、ベースラインよりは低かった。ベースラインと投与後 48
週時までの差は、統計学的に有意ではなかった。
表 52 総コレステロール値の推移(投与 24週間)
総コレステロール(mg/dL) 投与期(週)
0 24
試験番号 投与群 例数 平均値±標準偏差 平均値±標準偏差 検定*1
p値 検定*2
p値
LY群 33 220±43 206±38 0.025 K01A試験
プラセボ群 31 209±39 216±44 0.585 0.036
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定
表 53 総コレステロール値の推移(投与 72週間)
総コレステロール(mg/dL) 投与期(週)
0 24 48 72
投与群 例数 平均値± 標準偏差
平均値± 標準偏差
検定*
p値 平均値± 標準偏差
検定* p値
平均値± 標準偏差
検定*
p値
LY72w群 33 220 ± 43 206 ± 38 0.025 215 ± 44 0.115 213 ± 37 0.042
プラセボ/ LY48w群
28 210 ± 42 193 ± 38 0.011 199 ± 38 0.103 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
続いて、ベースラインの総コレステロール値が異常高値(220 mg/dL以上)、正常(150 mg/dL
以上、219 mg/dL以下)及び異常低値(149 mg/dL以下)であった患者群に分類して総コレステ
ロール値の推移を検討した。
K01A試験における結果を表 54に示した。
LY 群ではベースラインが異常高値であった患者群においてのみ、総コレステロール値は低
下し、ベースラインとの差は統計学的に有意(p=0.006)であった。一方、プラセボ群では総コ
レステロール値の有意な低下は認められなかった。また、ベースラインの総コレステロール値
が異常高値の患者群における、LY群とプラセボ群との変化量の差は統計学的に有意(p=0.027)
であった。
次に、K01A試験及び K02A試験を併合した時の結果を表 55に示した。
ベースラインが異常高値であった LY72w群では、投与後 24週時に低下した総コレステロー
ル値は 48週時に上昇したが、ベースラインよりは低かった。ベースラインと投与後 48週時の
差は統計学的に有意(p=0.040)であった。その後、投与後 48週時から投与後 72週時にかけて
総コレステロール値は低下し、ベースラインとの差は統計学的に有意(p=0.005)であった。
ベースラインが異常高値であったプラセボ/LY48w群でも、ベースラインから投与後 24週
時にかけて総コレステロール値は低下し、ベースラインとの差は統計学的に有意(p=0.025)で

ヒューマトロープ CTD(2.7 臨床概要) Page 50
50
あった。その後、投与後 24週時から投与後 48週時にかけて総コレステロール値は上昇したが、
ベースラインよりは低かった。ベースラインと投与後 48 週時との差は、統計学的に有意では
なかった。
ベースラインの総コレステロール値が正常及び異常低値であった LY72w 群及びプラセボ/
LY48w群では、いずれの群においても有意な変化は認められなかった。
これらの結果より、GH は総コレステロール値が異常高値である患者に顕著に作用して、総
コレステロール値を低下させることが示唆された。
表 54 ベースライン値別の総コレステロール値の推移(投与 24週間)
総コレステロール(mg/dL) 投与期(週)
0 24
ベースライン 投与群 例数 平均値± 標準偏差
平均値± 標準偏差
検定*1 p値
検定*2 p値
LY群 14 261±28 227±40 0.006 異常高値
プラセボ群 14 243±16 236±32 0.463 0.027
LY群 18 193±17 191±30 0.726 正 常
プラセボ群 13 193±17 210±44 0.250 0.179
LY群 1 141 190 1.000 異常低値
プラセボ群 4 141±7 166±48 0.375 0.724
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定
表 55 ベースライン値別の総コレステロール値の推移(投与 72週間)
総コレステロール(mg/dL) 投与期(週)
0 24 48 72
ベースライン 投与群 例数 平均値±標準偏差
平均値±標準偏差
検定*
p値 平均値±標準偏差
検定* p値
平均値±標準偏差
検定*
p値
LY72w群 14 261±28 227±40 0.006 247±43 0.040 240±29 0.005
異常高値 プラセボ/ LY48w群
12 251±24 219±31 0.025 226±29 0.062 - -
LY72w群 18 193±17 191±30 0.726 193±28 0.907 191±29 0.502
正常 プラセボ/ LY48w群
14 187±14 182±23 0.366 185±24 0.761 - -
LY72w群 1 141 190 1.000 164 1.000 222 1.000
異常低値 プラセボ/ LY48w群
2 133±11 118±16 1.000 129±5 1.000 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 51
51
LY72w 群において投与 24 週後から 48 週後の総コレステロール値が上昇したため、LY/
LY48w群における総コレステロール値の変動を検討した(表 56)。
ベースライン値が異常高値であった患者における投与後 24週時と 48週時の総コレステロー
ル値の差は統計学的に有意であった。一方、ベースライン値が正常値又は異常低値であった患
者における投与後 24週時と 48週時の総コレステロール値に有意な差はなかった。
表 56 LY/LY48w群の投与後 24週から 48週における
ベースライン値別の総コレステロール値の推移
投与期(週) 総コレステロール(mg/dL)
24 48 ベースライン 例数
平均値±標準偏差 平均値±標準偏差
検定* p値
異常高値 14 227±40 247±43 0.037
正常値 16 190±31 193±29 0.553
異常低値 1 190 164 1.000
投与開始後 24週及び 48週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
(b) LDLコレステロール値の変化
K01A試験において、本剤又はプラセボが投与された患者群における LDLコレステロール値
の推移を表 57に示した。
LY群及びプラセボ群において、LDLコレステロール値のベースラインと投与後 24週時の差
は、統計学的に有意ではなかったが、LY 群とプラセボ群との変化量の差は統計学的に有意で
あった(p=0.040)。
この結果、24週間の GH補充療法による LDLコレステロール値の低下が示唆された。
次に、K01A試験及び K02A試験を併合し、本剤が投与された期間における患者群の LDLコ
レステロール値の推移を表 58に示した。
LY72w群では、LDLコレステロール値はベースラインから投与後 72週時にかけてわずかに
低下したが、統計学的に有意な変化ではなかった。プラセボ/LY48w 群においては、LDL コ
レステロール値は投与後 24週から 48週時にかけて低下し、ベースラインから投与後 24週及
び 48週時までの変化はいずれも統計学的に有意(p=0.013 、p=0.032)であった。
表 57 LDLコレステロール値の推移(投与 24週間)
LDLコレステロール(mg/dL) 投与期(週)
0 24
試験番号 投与群 例数 平均値±標準偏差 平均値±標準偏差 検定*1
p値 検定*2
p値
LY群 33 128±38 120±34 0.151 K01A試験
プラセボ群 31 123±39 131±36 0.138 0.040
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 52
52
表 58 LDLコレステロール値の推移(投与 72週間)
LDLコレステロール(mg/dL) 投与期(週)
0 24 48 72
投与群 例数 平均値± 標準偏差
平均値± 標準偏差
検定*
p値 平均値± 標準偏差
検定*
p値 平均値± 標準偏差
検定*
p値
LY72w群 33 128 ± 38 120 ± 34 0.151 127 ± 41 0.780 126 ± 35 0.272
プラセボ/ LY48w群
28 127 ± 34 114 ± 36 0.013 116 ± 38 0.032 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
続いて、ベースラインの LDLコレステロール値が異常高値(140 mg/dL以上)、正常(70 mg/dL
以上 139 mg/dL以下)及び異常低値(69 mg/dL以下)であった患者群に分類して LDLコレス
テロール値の推移を検討した。
K01A試験における結果を表 59に示した。
ベースラインが異常高値であった LY群において、LDLコレステロール値は低下し、ベース
ラインとの差は統計学的に有意であった(p=0.008)。一方、プラセボ群では LDLコレステロー
ル値の有意な低下は認められなかった。なお、LY 群とプラセボ群の変化量との変化量の差は
統計学的に有意ではなかった。
次に、K01A試験及び K02A試験を併合した結果を表 60に示した。
ベースラインが異常高値であった LY72w群では、投与後 24週時に低下した LDLコレステ
ロール値は 48週時に上昇したが、ベースラインよりは低かった。ベースラインと投与後 48週
時の差は統計学的に有意ではなかった。その後、投与後 48週時から投与後 72週時にかけて LDL
コレステロール値は低下し、ベースラインと投与後 72週時との差は統計学的に有意(p=0.012)
であった。
ベースラインが異常高値であったプラセボ/LY48w群でも、投与後 24週から 48週時にかけ
て LDLコレステロール値は低下し、ベースラインから投与後 24週及び 48週時までの変化はい
ずれも統計学的に有意(p=0.020 、p=0.029)であった。投与後 24週時から投与後 48週時にか
けて、LDLコレステロール値はわずかに上昇したが、ベースラインより低かった。
ベースラインの LDLコレステロール値が正常又は異常低値であった LY72w群及びプラセボ
/LY48w群におけるLDLコレステロール値は、いずれも統計学的に有意な変化ではなかった。
以上、LDLコレステロール値の変化は総コレステロール値の変化と同様に、GHは異常高値
である患者に顕著に作用して、LDLコレステロール値を低下させることが示唆された。

ヒューマトロープ CTD(2.7 臨床概要) Page 53
53
表 59 ベースライン値別の LDLコレステロール値の推移(投与 24週間)
LDLコレステロール(mg/dL) 投与期(週) 0 24
ベースライン 投与群 例数平均値± 標準偏差
平均値± 標準偏差
検定*1
p値 検定*2
p値
LY群 9 177 ± 28 148 ± 26 0.008 異常高値
プラセボ群 10 169 ± 21 165 ± 27 0.695 0.094
LY群 24 109 ± 21 110 ± 30 0.835 正常
プラセボ群 20 103 ± 19 118 ± 23 0.017 0.083
LY群 - - - - 異常低値
プラセボ群 1 46 48 1.000 -
投与開始後 24週のデータが欠測の場合、LOCFにより補填した *1 1標本のWilcoxon検定 *2 2標本のWilcoxon検定
表 60 ベースライン値別の LDLコレステロール値の推移(投与 72週間)
LDLコレステロール(mg/dL) 投与期(週)
0 24 48 72
ベースライン 投与群 例数 平均値±標準偏差
平均値±標準偏差
検定*
p値 平均値±標準偏差
検定* p値
平均値±標準偏差
検定*
p値
LY72w群 9 177 ± 28 148 ± 26 0.008 166 ± 42 0.164 154 ± 28 0.012
異常高値 プラセボ/ LY48w群
10 162 ± 20 140 ± 27 0.020 142 ± 40 0.029 - -
LY72w群 24 109 ± 21 110 ± 30 0.835 112 ± 29 0.598 116 ± 32 0.648
正常 プラセボ/ LY48w群
17 111 ± 17 103 ± 30 0.207 103 ± 29 0.178 - -
LY72w群 - - - - - - - - 異常低値
プラセボ/ LY48w群
1 48 42 1.000 74 1.000 - -
投与開始後 24週、48週及び 72週のデータが欠測の場合、LOCFにより補填した * 1標本のWilcoxon検定
(c) 血清脂質の変動に関する考察
K01A試験及び K02A試験を通じて、本剤を 72週間あるいは 48週間投与された患者では、
特に、本剤投与前の総コレステロール値あるいは LDL コレステロール値が異常高値の患者に
おいて、ベースラインと比較して統計学的に有意な総コレステロール値あるいは LDL コレス
テロール値の低下が認められた。ただし、LY72w 群及びプラセボ/LY48w 群のいずれの群に
おいても本剤の投与開始後 24週時に低下した総コレステロール及び LDLコレステロール値は、
投与開始後 48週時点で若干上昇し、LY72w群では投与後 72週時にまた低下するという推移を
示した。
コレステロール代謝に関して、GHは、肝臓の LDL受容体数を増加させることにより、コレ

ヒューマトロープ CTD(2.7 臨床概要) Page 54
54
ステロールに富んだ血中 LDL及び Intermediate Density Lipoprotein (以下、IDL)、レムナント
を肝臓に取り込み、血中コレステロール値を低下させる 5。肝臓に取り込まれたコレステロー
ルは、GHによって活性化される Cholesterol 7α-Hydroxylaseによって、胆汁酸に代謝され、胆
管を経て消化管から体外へ排泄される。一方、GH は脂肪組織等で脂肪を分解し、中性脂肪
(Triglyceride; 以下 TG)から遊離脂肪酸(Free Fatty Acid; 以下、FFA)を産生する。多量に血
中に放出された FFAは、肝臓に取り込まれ、GHの働きで肝内 TG産生に使用され、また同時
に GH作用により、アポリポ蛋白 B(ApoB)産生も促進される。その結果、コレステロールと
TGに富む Very Low Density Lipoprotein (以下、VLDL)が肝臓から血中へ放出され、血中コレ
ステロール値は上昇する。VLDL は、リポプロテインリパーゼ(Lipoprotein Lipase)や肝トリ
グリセリドリパーゼによってレムナントや IDL、更に LDLに代謝され、再び肝臓や末梢組織の
LDL受容体に結合して、細胞内に取り込まれる。
このように、GH は単に血中コレステロール値を低下させる物質ではなく、コレステロール
代謝のいくつものステップに関与し、全体としてコレステロール代謝を活性化する、あるいは
代謝回転を早くするホルモンであると言える。
さらに脂質代謝そのものは、他の体内物質や患者のライフスタイル 6、食事の内容などによ
り影響を受けることから、総コレステロール値及び LDL コレステロール値の短期間の変動を
説明することは困難であると考えられる。
GH投与による総コレステロール値及び LDLコレステロール値の低下が、ベースライン値が
異常高値であった AGHD患者のみに認められることは他にも報告があり 7, 8、これは GH補充
療法によって、GH の欠乏に起因する臨床症候が改善したことを意味しているものと考えられ
る。一方、総コレステロール値及び LDL コレステロール値のベースライン値が正常であった
患者では、血清脂質に異常は見られなくても他に様々な臨床症候を有しており、GH を投与す
ることによって、それらの臨床症候を改善する必要があることから、それらの AGHD患者に対
しても、GH投与は実施すべきであると考えられる。
2) 外国臨床試験における成績(参考資料)
外国臨床試験において、本剤又はプラセボが投与された患者群における総コレステロール値
及び LDLコレステロール値の推移を表 61及び表 62に示した。
総コレステロール値については、E005/6試験の LY群及びプラセボ/LY群並びに GDED試
験の低用量群を除く本剤投与群において、投与後 24週時又は投与後 32週時に有意な低下が認
められた。LDLコレステロール値については、E005/6試験の LY群及びプラセボ/LY群を除
く本剤投与群において、投与後 24週時又は 32週時に有意な低下が認められた。

ヒューマトロープ CTD(2.7 臨床概要) Page 55
55
表 61 総コレステロール値の推移(外国臨床試験)
総コレステロール:平均値±標準偏差(例数) 投与期間(週)
試験番号(単位) 投与群
0 24 32 48 72
LY群 237.85±
52.68 (46)
221.70± 42.34*
(43) -
240.00± 42.37 (38)
232.20± 35.31 (40)
プラセボ群 245.18±
51.72 (45)
243.60± 54.22 (42)
- - - E003/4試験 (mg/dL)
プラセボ/LY群 243.60±
54.22 (42)
235.22± 47.24*
(37) -
246.36± 46.66 (39)
-
LY群 211.27±
45.10 (30)
201.19± 46.99 (27)
- 205.36±
39.13 (25)
205.57± 46.76 (21)
プラセボ群 220.47±
61.45 (32)
223.30± 55.90 (30)
- - - E005/6試験 (mg/dL)
プラセボ/LY群 223.30±
55.90 (30)
224.41± 60.23 (27)
- 211.24± 49.82*
(25) -
高用量群 5.79± 1.36 (289)
5.54± 1.30** (289)
- - - GDED試験
(mmol/L) 低用量群
5.76± 1.34 (297)
5.63± 1.21 (297)
- - -
ID群 219.87±
44.84 (180)
- 214.53± 42.67* (180)
- - T002試験 (mg/dL)
FD群 217.25±
47.04 (191)
- 207.75± 43.98 **
(191) - -
*:p<0.05、**:p<0.001
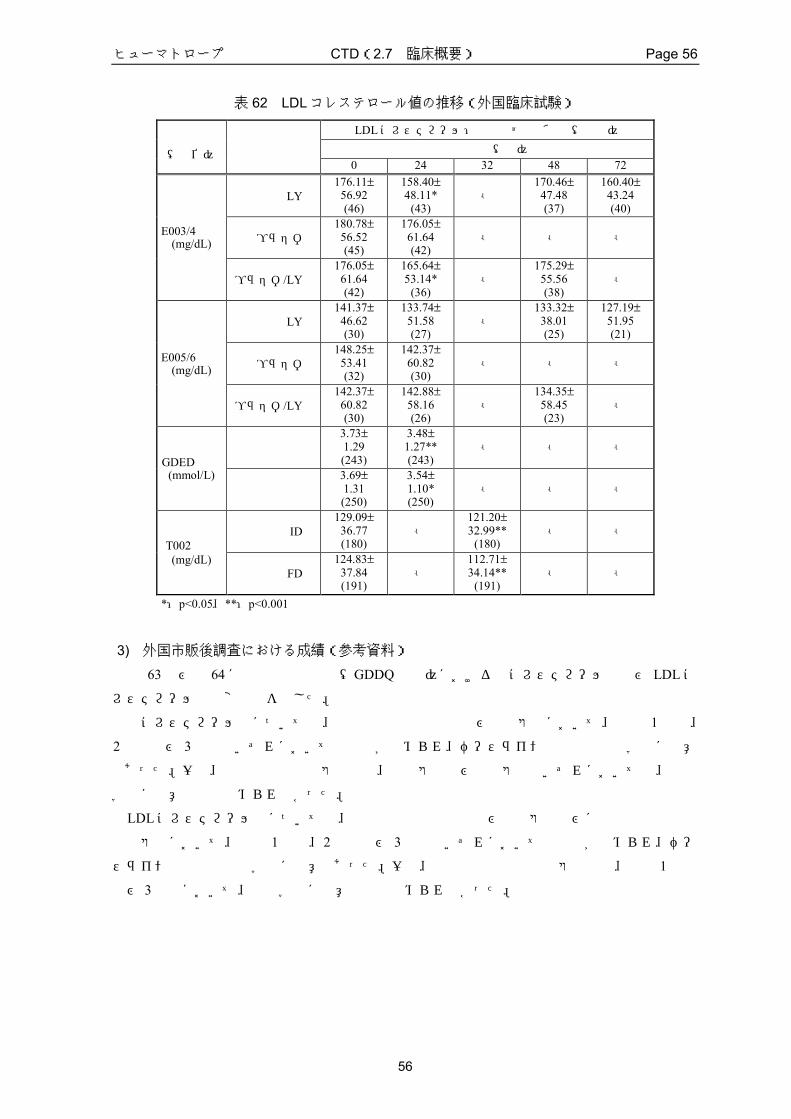
ヒューマトロープ CTD(2.7 臨床概要) Page 56
56
表 62 LDLコレステロール値の推移(外国臨床試験)
LDLコレステロール:平均値±標準偏差(例数) 投与期間(週)
試験番号(単位) 投与群
0 24 32 48 72
LY群 176.11±
56.92 (46)
158.40± 48.11*
(43) -
170.46± 47.48 (37)
160.40± 43.24 (40)
プラセボ群 180.78±
56.52 (45)
176.05± 61.64 (42)
- - - E003/4試験 (mg/dL)
プラセボ/LY群 176.05±
61.64 (42)
165.64± 53.14*
(36) -
175.29± 55.56 (38)
-
LY群 141.37±
46.62 (30)
133.74± 51.58 (27)
- 133.32±
38.01 (25)
127.19± 51.95 (21)
プラセボ群 148.25±
53.41 (32)
142.37± 60.82 (30)
- - - E005/6試験 (mg/dL)
プラセボ/LY群 142.37±
60.82 (30)
142.88± 58.16 (26)
- 134.35±
58.45 (23)
-
高用量群 3.73± 1.29 (243)
3.48± 1.27** (243)
- - - GDED試験
(mmol/L) 低用量群
3.69± 1.31 (250)
3.54± 1.10* (250)
- - -
ID群 129.09±
36.77 (180)
- 121.20± 32.99**
(180) - -
T002試験 (mg/dL)
FD群 124.83±
37.84 (191)
- 112.71± 34.14**
(191) - -
*:p<0.05、**:p<0.001
3) 外国市販後調査における成績(参考資料)
表 63及び表 64に外国市販後調査(GDDQ調査)における総コレステロール値及び LDLコ
レステロール値の変化量を示した。
総コレステロール値については、成人期発症型の男性及び女性患者において、投与 1 年後、
2年後及び 3年後のいずれにおいても低下が認められ、ベースラインとの差は統計学的に有意
であった。一方、小児期発症型の患者では、男性患者及び女性患者のいずれにおいても、統計
学的に有意な差は認められなかった。
LDLコレステロール値については、成人期発症型の男性及び女性患者並びに小児期発症型の
男性患者において、投与 1年後、2年後及び 3年後のいずれにおいても低下が認められ、ベー
スラインとの差は統計学的に有意であった。一方、小児期発症型の女性患者では、投与 1年後
及び 3年後において、統計学的に有意な差は認められなかった。

ヒューマトロープ CTD(2.7 臨床概要) Page 57
57
表 63 総コレステロール値の変化量(GDDQ調査)
総コレステロール値(mmol/L):平均値±標準偏差(例数) 投与期(年)
1 2 3
発症時期 性別平均値± 標準偏差
検定 p値
平均値± 標準偏差
検定 p値
平均値± 標準偏差
検定 p値
男性 -0.24±1.09 (265) <0.001 -0.35±1.24
(189) <0.001 -0.30±1.21 (124) 0.007
成人期 発症型
女性 -0.16±1.20 (258) 0.029 -0.15±1.12
(170) 0.012 -0.12±1.38 (91) 0.008
男性 -0.15±0.81 (77) 0.116 -0.14±0.76
(59) 0.149 -0.25±0.84 (33) 0.090
小児期 発症型
女性 0.04±1.04 (56) 0.753 -0.17±0.90
(52) 0.185 -0.13±0.99 (25) 0.519
表 64 LDLコレステロール値の変化量(GDDQ調査)
LDLコレステロール値(mmol/L):平均値±標準偏差(例数) 投与期(年)
1 2 3
発症時期 性別平均値± 標準偏差
検定 p値
平均値± 標準偏差
検定 p値
平均値± 標準偏差
検定 p値
男性 -0.33±0.83 (222) <0.001 -0.40±1.11
(164) <0.001 -0.42±1.05 (110) <0.001
成人期 発症型
女性 -0.20±0.75 (219) <0.001 -0.31±0.85
(140) <0.001 -0.33±0.88 (72) 0.002
男性 -0.20±0.76 (63) 0.036 -0.30±0.74
(50) 0.006 -0.39±1.00 (28) 0.050
小児期 発症型
女性 -0.17±0.83 (47) 0.162 -0.29±0.93
(45) 0.042 -0.29±0.86 (23) 0.120
2.7.3.3.3 部分集団における結果の比較
LBMの変化率における治療と各種因子(性別、発症時期、測定機器)との交互作用を、K01A
試験の結果を用いて検討した。更に、高齢者における有効性について、外国臨床試験成績を用
いて検討した。
(1) 性別、発症時期
K01A試験での性別及び発症時期により層別した部分集団における LBMの変化率を表 65及
び表 66に示した。
いずれの部分集団においても、LY群における LBMの平均変化率は、プラセボ群の平均変化
率より大きく、治療とこれらの因子との間に質的な交互作用は認められなかった。
次に、治療とこれらの因子との量的交互作用を分散分析で検討した(表 67)。モデルには、
これらの因子及び治療との交互作用のほかに、LBMの測定機器を因子に加えた。有意水準 15%
において有意であった交互作用は、「治療×発症時期」であった(p=0.088)。
そのため、発症時期の各部分集団(成人期発症型、小児期発症型)において、LBM の変化
率を、LY群とプラセボ群で比較した。その結果、LY群とプラセボ群との LBMの変化率の差

ヒューマトロープ CTD(2.7 臨床概要) Page 58
58
は、どちらの発症時期においても、統計学的に有意であった(成人期発症型:p<0.001、小児期
発症型:p=0.014)。
以上より、治療と発症時期の間に量的な交互作用が示唆されたが、LY 群とプラセボ群との
LBMの変化率の差は両部分集団において有意であり、どちらの発症時期においても、GH補充
療法が有効であると考えられた。
表 65 男女別の LBMの変化率
LBM変化率(%) 投与期間(週)
24
性別 投与群 例数 平均値±標準偏差
LY群 15 3.8±4.2 男性
プラセボ群 14 -0.8±4.2
LY群 17 5.5±3.5 女性
プラセボ群 15 -0.2±4.2
投与開始後 24週のデータが欠測の場合、LOCFにより補填した
表 66 発症時期別の LBMの変化率
LBM変化率(%) 投与期間(週)
24
発症時期 投与群 例数 平均値±標準偏差検定*
p値
LY群 13 5.5±3.2 成人期発症型
プラセボ群 12 -2.0±3.8 <0.001
LY群 19 4.2±4.3 小児期発症型
プラセボ群 17 0.6±4.1 0.014
投与開始後 24週のデータが欠測の場合、LOCFにより補填した * 対応のない t検定
表 67 LBMの変化率における交互作用の検討
要 因 自由度 SS MS F値 p値
治 療 1 243.34 243.34 16.75 <0.001
発症時期 1 42.29 42.29 2.91 0.095
治療×発症時期 1 44.01 44.01 3.03 0.088
性 別 1 33.49 33.49 2.31 0.136
治療×性別 1 2.19 2.19 0.15 0.700
測定機器 5 168.50 33.70 2.32 0.058
治療×測定機器 4 18.32 4.58 0.32 0.866

ヒューマトロープ CTD(2.7 臨床概要) Page 59
59
(2) 高齢者における臨床効果(外国臨床成績;参考資料)
65 歳以上の高齢者を含む外国臨床試験(GDED 試験、T002 試験)におけるベースラインか
らの LBMの変化率を表 68に示した。
GDED試験では、低用量(3.0 µg/kg/日→6.0 µg/kg/日)を投与された高齢者では、投与後 12
週時及び 24週時のいずれの時点においても、LBMの有意な増加は認められなかった。一方、
高用量(6.0 µg/kg/日→12.0 µg/kg/日;既承認用量)を投与された高齢者では、投与後 12週時及
び 24週時のいずれの時点においても、LBMの有意な増加が認められた。
T002試験では、高齢者では ID群及び FD群のいずれにおいても LBMの有意な増加が認めら
れた。
以上、GDED 試験の低用量群を除いたすべての投与群において、LBM の有意な増加が認め
られた。
表 68 高齢者及び非高齢者における LBMの変化率(GDED試験、T002試験)
LBM変化率(%):平均値±標準偏差(例数)
投与期間(週) 試験番号 高齢者又は非高齢者 投与群
12 24 32
高用量群 5.035±4.364 (18) ** 7.146±9.511 (18) * - 高齢者
低用量群 -0.010±3.492 (10) 0.180±5.524 (10) -
高用量群 3.905±4.818 (192) ** 5.616±5.635 (195) ** - GDED試験
非高齢者 低用量群 2.726±4.436 (207) ** 4.789±5.295 (212) ** -
ID群 - - 4.955±3.139 (9) * 高齢者
FD群 - - 3.221±4.016 (11) *
ID群 - - 4.685±6.127 (93) **T002試験
非高齢者 FD群 - - 5.427±5.975 (93) **
0週からの変化率
*:p<0.05、**:p<0.001

ヒューマトロープ CTD(2.7 臨床概要) Page 60
60
2.7.3.4 推奨用法・用量に関する臨床情報の解析
AGHDの効能・効果に対し推奨される用法・用量は以下のとおりである。
[推奨用法・用量]
通常開始用量として、1週間に体重 kg当たり、ソマトロピン(遺伝子組換え)として
0.021 mgを 6~7回に分けて皮下に注射する。患者の臨床症状に応じて 1週間に体重
kg当たり 0.084 mgを上限として漸増し、1週間に 6~7回に分けて皮下に注射する。
なお、投与量は副作用の発現及び血清 IGF-I濃度に応じて適宜増減する。ただし、1
日量として 1 mgを超えないこと。
国内臨床試験成績、GRS コンセンサス・ガイドライン及び海外臨床試験成績を基に推奨用
法・用量を設定した根拠を以下に述べる。図 4にその概念図を示した。*
図 4 臨床推奨用量の設定根拠
* 新薬承認情報提供時に置き換えた
小児における成長ホルモン
分泌不全症の治療用量
(1980年代後半~)
E003/4試験*
E005/6試験*
GDED試験*
K01A試験*
K02A試験*
推奨用量
0.021 ~ 0.042*
0.14 ~ 0.175*
臨床症候及びIGF-Iによる用量調整
0.021 ~ 0.084*
0.021 ~ 0.084*
(臨床症候及びIGF-Iによる用量調整)
ただし、1日1mgを超えないこと
国内臨床試験(2001年)
海外臨床試験(1992
海外市販後臨床試験(1997年)
GRS
コンセンサスガイドライン
(1997年)
低用量より投与開始
0.04375 ~ 0.0875*
低用量 高用量
* : 単位はmg/kg/週
0.042 ~ 0.084*
T002試験*
海外市販後臨床試験(1998年)
臨床症状と
血中IGF-I濃度を
指標とした用量調整
1.4 ~ 5.6 mg/週
0.028 ~ 0.084*
臨床症状と
IGF-Iによる用量調整 1mg/日を超えない
1日投与量の
上限

ヒューマトロープ CTD(2.7 臨床概要) Page 61
61
(1) 投与開始用量(0.021 mg/kg/週)及び最大用量(0.084 mg/kg/週)の設定根拠
1) 背景(E003/4試験、E005/6試験、GDED試験、GRSコンセンサス・ガイドライン)
AGHDに対する研究が開始された当初、小児における GH分泌不全性低身長症の承認用量で
ある 0.175 mg/kg/週前後の投与量が選択された 9-11。その結果、体組成の改善が認められた。し
かし、GH の有する抗ナトリウム利尿作用に起因する浮腫や関節痛といった投与初期に発現し
やすい副作用が多く認められた。また、GH の分泌能は加齢に伴い減少することが知られてい
る。以上のことより、AGHDに対する至適投与量は小児における低身長症の治療に用いられる
投与量よりも低いと考えられた。
その後、AGHDに対する研究が更に進められ、低身長症の治療に用いられる 0.175 mg/kg/週
の 1/4である 0.04375 mg/kg/週を投与開始用量とし、最大用量を 0.0875 mg/kg/週とした臨床試
験(E003/4試験及び E005/6試験)が実施された。なお、欧米ではこれら 4試験の成績を基に
承認申請が行われ、「成人成長ホルモン分泌不全症」として承認されている。
実地診療下において AGHD 患者に対する GH 補充療法が実施されるようになった後、The
Growth Hormone Research Society(以下、GRS)より「AGHDの診断と治療のためのコンセンサ
ス・ガイドライン」が公表された。GRS コンセンサス・ガイドラインでは AGHD の治療に関
する以下の事項について合意がなされている。
• 投与の目的は治療による利益を最大化し、副作用を最小化することである。
• 低用量(0.15~0.3 mg/日*)より投与を開始することを推奨する。
• 用量変更の間隔は 1ヵ月以上あけることが望ましい。 *:体重 60 kgの患者の場合、0.0175~0.035 mg/kg/週に相当する。
GRSコンセンサス・ガイドラインを踏まえ、E003/4試験及び E005/6試験よりも投与開始用
量が低用量である 0.021 mg/kg/週から投与開始し 0.042 mg/kg/週まで増量する低用量群と 0.042
mg/kg/週から投与開始し 0.084 mg/kg/週まで増量する高用量群の比較試験(GDED 試験)が実
施された。その結果、有効性の主要評価項目である投与開始 3ヵ月間の LBM増加は、両群で
認められたが、低用量群よりも高用量群で有効であった。しかし、高用量群は低用量群よりも
有害事象が多く見られた。したがって、安全性と有効性のバランスを考慮すると、低用量(0.021
mg/kg/週)から投与を開始することが望ましいと考えられた。
2) 国内臨床試験
GRSコンセンサス・ガイドライン及び外国臨床試験成績などから得られた知見から、国内臨
床試験では開始用量を 0.021 mg/kg/週とし、0.084 mg/kg/週を上限として増量できる投与方法を
設定した。特に K01A試験では 0.021 mg/kg/週から 0.084 mg/kg/週まで漸増する方法(以下、用
量漸増法)を用いた。
(a) 有効性に関する臨床情報の解析
K01A試験における、本剤及びプラセボ投与 24週前後の LBMの平均変化率を「2.7.3.2 個々
の試験結果の要約」の表 3に示した。

ヒューマトロープ CTD(2.7 臨床概要) Page 62
62
LY群の LBMの平均変化率は 4.7%であり、ベースラインとの間に統計学的有意差が認めら
れた(p<0.001)。プラセボ群の平均変化率は-0.5%であり、有意な変化ではなかった。また、
LY群の LBMの変化率は、プラセボ群と比較して、統計学的に有意に大きかった(p<0.001)。
以上、開始用量を 0.021 mg/kg/週とし 0.084 mg/kg/週を上限として、用量を増量する方法は有
効であることが確認された。
(b) 安全性に関する臨床情報の解析
K01A試験において認められた有害事象の要約を表 69に示した。
表 69 有害事象の要約(K01A試験)
投与群 区分*1 LY群
(N=33) プラセボ群 (N=31)
p値*2
死亡 0 0.0% 0 0.0% NA
重篤な有害事象 1 1 3.0% 4 1 3.2% 0.964
有害事象による中止*3 1 1 3.0% 1 1 3.2% 0.964
有害事象による減量*3 6 6 18.2% 1 1 3.2% 0.055
有害事象 173 28 84.8% 170 26 83.9% 0.914
副作用 38 14 42.4% 11 7 22.6% 0.091
*1 件数、例数、発現率%、 *2 カイ 2乗検定、 *3 重篤な有害事象例を除く
LY 群、プラセボ群とも死亡例は認められなかったが、重篤な有害事象が 1 例ずつ認められ
た(LY 群:脳梗塞、プラセボ群:交通事故)。また、有害事象による投与中止例及び指示投
与量が減量された例は、LY群でそれぞれ 1例と 6例、プラセボ群で 1例ずつ認められた。有
害事象の発現率について、LY群とプラセボ群との間に、統計学的有意差は認められなかった。

ヒューマトロープ CTD(2.7 臨床概要) Page 63
63
次に、有害事象の一覧表を表 70に示した。
表 70 LY群において 4例以上発現した有害事象
投与群 器官別*1
有害事象 LY群 (N=33)
プラセボ群 (N=31)
p値*2
呼吸器、胸郭および縦隔障害 56 17 51.5% 63 22 71.0% 0.111
鼻咽頭炎 18 11 33.3% 27 17 54.8% 0.083
咳嗽 9 7 21.2% 11 6 19.4% 0.854
鼻漏 8 7 21.2% 7 6 19.4% 0.854
咽喉頭疼痛 8 6 18.2% 8 5 16.1% 0.828
上気道の炎症 8 5 15.2% 5 4 12.9% 0.796
全身障害および投与局所様態 26 15 45.5% 28 16 51.6% 0.622
発熱 9 7 21.2% 12 12 38.7% 0.126
浮腫NOS 4 4 12.1% 2 2 6.5% 0.437
筋骨格系および結合組織障害 16 13 39.4% 7 4 12.9% 0.016
関節痛 7 6 18.2% 4 4 12.9% 0.561
神経系障害 14 9 27.3% 8 6 19.4% 0.455
頭痛 7 6 18.2% 4 4 12.9% 0.561
皮膚および皮下組織障害 11 9 27.3% 13 10 32.3% 0.663
胃腸障害 19 8 24.2% 19 12 38.7% 0.212
感染症および寄生虫症 7 5 15.2% 6 6 19.4% 0.656
臨床検査 7 5 15.2% 6 3 9.7% 0.508
代謝および栄養障害 5 4 12.1% 5 5 16.1% 0.645
*1 MedDRA(J) 5.1(件数、例数、発現率%) *2 カイ 2乗検定
個別の有害事象の発現率について、LY 群とプラセボ群との間に統計学的有意差が認められ
た有害事象はなかった。
次に、有害事象の器官分類別発現率を検討した結果、「筋骨格系および結合組織障害」につ
いてのみ、LY群がプラセボ群と比較して、有意に発現率が高かった(p=0.016)。なお、「筋
骨格系および結合組織障害」に分類される有害事象には、関節痛などが含まれる。関節痛は本
剤の抗ナトリウム利尿作用に起因し発現することが知られており、予測される事象であり、投
与量を調整することにより対処しうると判断した。
以上、開始用量を 0.021 mg/kg/週とし、0.084 mg/kg/週を上限とする用量設定は安全であるこ
とが確認された。
3) 投与開始用量(0.021 mg/kg/週)及び最大用量(0.084 mg/kg/週)に関する結論
有効性及び安全性の観点から総合的に判断し、開始用量を 0.021 mg/kg/週とし、0.084 mg/kg/
週を上限として増量する方法が適切であることが確認された。

ヒューマトロープ CTD(2.7 臨床概要) Page 64
64
(2) 血清 IGF-I濃度を指標とした用量調整法の設定根拠
1) 背景(T002試験、GRSコンセンサス・ガイドライン)
AGHDに対する治療が開始された当初、投与量の調整は GHに起因する副作用が発現した場
合のみ行われていた。その後、GH に対する感受性及び必要補充量は個々の患者で異なること
が分かり、副作用発現時のみ投与量を調整することでは不十分と考えられた。
一般的にホルモン補充療法においては、補充したホルモンの血中濃度をモニタリングしなが
ら投与量を調整することが多いが、皮下投与された GH は投与 24 時間後にはすでに血中から
消失し、毎日連続投与を行っても血中濃度は定常状態に達することはない。GH の薬理作用に
は直接作用と IGF-Iを介する間接作用があり、さらに GHを皮下投与した時の IGF-Iの血中消
失速度は緩やかで、GHの連続投与後 4週間で IGF-Iの血中濃度は定常状態に達することが報告
されている 12。このことから、GHの補充量が適切であることを測る指標の一つとして血清 IGF-I
濃度が広く用いられるようになった。その結果、患者の臨床症候及び血清 IGF-I濃度を指標と
した用量調整法の重要性が認識されてきた。
GH 依存性生化学マーカーとして、IGF-I以外に IGFBP-3、ALS(acid-labile subunit)が知ら
れているが、De Boerらの報告では、AGHD患者 46例にGH補充療法を 1年間実施した結果 IGF-I、
IGFBP-3、ALSの 3項目ともGH投与量に相関して血中濃度の平均値が上昇したものの、IGFBP-3
及び ALS濃度の変動はすべて基準範囲内であり、基準範囲を超えて変動したのは IGF-Iのみで
あった。また、被験者数でみると、IGF-Iの基準範囲を超えた症例は 46例中 19例であったの
に対し、IGFBP-3及び ALSではそれぞれ 2例及び 8例であった 13。
GRSコンセンサス・ガイドラインでは、GH補充療法における用量調整方法に関して、以下
の指針が示されている。
• GH投与に対する感受性は、個人間でバラツキがあることが知られており、特に、高齢者
は感受性が高い。
• 過量投与を避けるため、血清 IGF-I濃度を用量設定に利用し、血清 IGF-I濃度を年齢別に
定められた基準範囲に維持する必要がある。なお、投与開始初期には 1~2ヵ月ごとに血
清 IGF-I濃度を測定する必要がある。
• GH作用の生化学マーカーとしては、血清 IGF-I濃度が唯一信頼できる。
• 血清 IGF-I濃度は年齢別の基準範囲が設定されている。
GRSコンセンサス・ガイドラインの指針を踏まえ、体重当たりの用量によって、投与量を段
階的に増量した群(FD群)(0.028~0.084 mg/kg/週)と、血清 IGF-I濃度及び臨床症状に基づ
き、個々の患者ごとに投与量を調整した群(ID群)(1.4~5.6 mg/週)における 32週間の無作
為化並行群間比較試験(T002試験)を実施した。有効性について、主要評価項目である投与 8ヵ
月間における脂肪量変化率は、いずれの投与群においても投与開始時より有意に減少していた
(FD群:10.9±11.5%、ID群:7.9±11.9%)。安全性については、末梢性浮腫の発現率につい
てのみ、投与群間に有意差が認められた(FD群:16.5%、ID群:9.1%)。以上の結果から、ID
群は忍容性の面でわずかに FD群より優っていると見られた。

ヒューマトロープ CTD(2.7 臨床概要) Page 65
65
以上より、GH 補充療法の際には患者の臨床症候及び血清 IGF-I 濃度を指標とした用量調整
法が重要であると考えられた。
2) 国内臨床試験
GRSコンセンサス・ガイドラインの用量調整に関する指針及び T002試験結果から得られた
知見から、K02A 試験では、患者の臨床症候及び血清 IGF-I 濃度を指標とした用量調整法(以
下、用量調整法)を設定した。
(a) 投与量と血清 IGF-I濃度に関する考察
LY群、LY/LY48w群及びプラセボ/LY48w群における投与量の推移を表 71に示した。用
量漸増法を用いた LY群の投与 12週時、投与 24週時の平均投与量はそれぞれ 0.041 mg/kg/週、
0.078 mg/kg/週であった。一方、用量調整法を用いた LY/LY48w群の投与 36週時(K02A試験
投与 12週時)、投与 48週時及び投与 72週時の平均投与量はそれぞれ 0.032 mg/kg/週、0.052
mg/kg/週及び 0.050mg/kg/週であり、プラセボ/LY48w群の投与 12週時(K02A試験投与 12週
時)、投与 24週時及び投与 48週時の平均投与量はそれぞれ 0.034 mg/kg/週、0.049 mg/kg/週及
び 0.049 mg/kg/週であった。
以上の通り、用量調整法を用いた場合の投与開始から投与 24週までの投与量増量の推移は、
用量漸増法を用いた場合に比べ緩やかであり、その後投与 72 週時まで投与量はほとんど変化
はなく、維持されていた。

ヒューマトロープ CTD(2.7 臨床概要) Page 66
66
表 71 投与量の推移の要約統計量
投与群 来院 例数 平均値 標準偏差 最大値 中央値 最小値
4週 33 0.021 0.0005 0.022 0.021 0.020
8週 32 0.042 0.0004 0.043 0.042 0.041
12週 32 0.041 0.0043 0.043 0.042 0.021 LY群
24週 31 0.078 0.0153 0.085 0.084 0.021
28週 31 0.021 0.0005 0.022 0.021 0.020
32週 31 0.021 0.0005 0.022 0.021 0.020
36週 30 0.032 0.0100 0.043 0.037 0.010
40週 30 0.039 0.0177 0.084 0.041 0.010
44週 30 0.046 0.0235 0.084 0.041 0.011
48週 29 0.052 0.0236 0.084 0.042 0.011
60週 30 0.050 0.0243 0.084 0.042 0.010
LY/LY48w群
72週 30 0.050 0.0243 0.085 0.042 0.010
4週 28 0.021 0.0006 0.022 0.021 0.020
8週 28 0.021 0.0006 0.022 0.021 0.020
12週 28 0.034 0.0142 0.084 0.041 0.020
16週 28 0.039 0.0197 0.085 0.041 0.020
20週 28 0.044 0.0218 0.085 0.042 0.020
24週 27 0.049 0.0230 0.084 0.043 0.020
36週 28 0.049 0.0245 0.085 0.043 0.020
プラセボ/LY48w群
48週 27 0.049 0.0256 0.084 0.042 0.020
投与量(mg/kg/週)を示す
次に、K01A試験及び K02A試験における IGF-I SDSの分布の推移を「2.7.3.3.2 血清 IGF-I
濃度の分布の推移」の表 37及び表 72に示した。また、用量調整法を用いた K02A試験におけ
る IGF-I SDSの推移を図 5に、K01A試験及び K02A試験における本剤投与後の IGF-I SDSの推
移を表 73に示した。
LY群の投与開始時における IGF-I SDS基準値未満の症例は 33例中 21例であり、投与 24週
時には 3例となった。投与方法を用量漸増法から用量調整法に変更し、更に 48週の本剤投与
がなされた LY/LY48w群の投与 72週時において、基準値未満の値を示した症例は認められな
かった(表 72)。用量調整法を用いたプラセボ/LY48w群の投与開始時において、IGF-I SDS
が基準値未満の症例は、28例中 17例であり、投与 48週時には 2例に減少した(表 72)。以上
の結果から、投与方法に関係なく、GH補充療法によって必要量の GH補充が行われたと判断
した。
更に、IGF-I SDSの基準範囲を超える症例について検討した。LY群の用量漸増法による投与
24週時において基準範囲を超えた症例は 6例であり、その最大値は 6.81と基準範囲を大きく
超えていた。投与 24週以降、投与方法を用量調整法に変更した LY/LY48w群の投与 72週時
において基準範囲を超えた症例は 3例であり、その最大値は 2.82であった。用量調整法を用い
たプラセボ/LY48w群の投与 48週時において基準範囲を超えた症例は 3例であり、その最大
値は 4.23であった(表 73)。
用量漸増法を用いた場合、血清 IGF-I濃度が基準範囲を大きく超える症例がみられたことか

ヒューマトロープ CTD(2.7 臨床概要) Page 67
67
ら、これらの症例において過量投与が懸念された。一方、用量調整法を用いた場合、臨床症候
や血清 IGF-I濃度を指標として投与量を調整した結果、投与量増量の推移は用量漸増法に比べ
緩やかであり、血清 IGF-I濃度が基準範囲を大きく超える症例はなかったことから、過量投与
がなされず、適切量の GHが補充されたと判断した。
以上の結果から、臨床症候及び血清 IGF-I濃度を指標とした用量調整法は過量投与を避け、
適切量の GHを補充する最適の用法であると判断した。
-----:血清 IGF-I濃度基準範囲
図 5 IGF-I SDSの推移
表 72 血清 IGF-I 濃度の推移
投与期間(週) 0 24 28 32 36 40 44 48 60 72
基準範囲超 0 6 2 3 3 2 3 5 3 3
基準範囲 12 22 22 19 24 26 24 24 26 27 LY/LY48w群 (例数)
基準範囲未満 21 3 7 9 4 2 3 0 1 0
投与期間(週) - 0 4 8 12 16 20 24 36 48
基準範囲超 - 0 2 2 2 4 1 4 5 3
基準範囲 - 11 17 21 21 20 24 20 20 22 プラセボ/LY48w群
(例数)
基準範囲未満 - 17 9 5 5 4 3 3 3 2
週
LY/LY48w群 プラセボ/LY48w群

ヒューマトロープ CTD(2.7 臨床概要) Page 68
68
表 73 IGF-I SDS の推移の要約統計量
投与群 来院 例数 平均値 標準偏差 最大値 中央値 最小値
0週 33 -2.42 1.85 0.78 -2.47 -8.35
4週 33 -0.58 2.13 4.32 -0.47 -7.04 8週 32 0.40 2.37 5.19 0.51 -6.16
12週 32 -0.09 2.43 5.32 -0.23 -6.36 LY群
24週 31 0.82 2.28 6.81 0.98 -4.25 28週 31 -0.74 1.95 3.13 -0.93 -6.96
32週 31 -0.73 2.04 3.90 -0.84 -6.32
36週 31 -0.34 2.00 2.36 -0.13 -7.82 40週 30 0.11 1.31 2.26 0.38 -3.45
44週 30 0.18 1.56 2.98 0.24 -3.06
48週 29 0.65 1.11 2.30 0.44 -1.20 60週 30 0.48 1.29 3.23 0.63 -3.00
LY/LY48w群
72週 30 0.51 1.25 2.82 0.58 -1.81
0週 28 -2.33 1.42 -0.30 -2.41 -4.92 4週 28 -0.81 2.02 2.34 -1.21 -5.21
8週 28 -0.59 1.81 3.65 -0.68 -3.96
12週 28 -0.12 1.66 2.77 -0.16 -2.78 16週 28 0.05 1.56 2.75 0.16 -2.71
20週 28 0.07 1.46 2.04 0.55 -3.53
24週 27 0.32 1.70 2.51 0.76 -3.81 36週 28 0.67 1.65 2.68 0.98 -3.69
プラセボ/LY48w群
48週 27 0.38 1.67 4.23 0.60 -3.69
また、K01A試験では、登録された 64例中 26例で投与開始前の IGF-Iが基準値範囲内であっ
た。特に成人においては、IGF-I は GH のみならず栄養状態や肥満度などにより影響を受け、
AGHD患者であっても必ずしも血清 IGF-I低値を示さないことが知られている。しかしながら
GH欠乏下では、たとえ血清 IGF-I濃度が正常を示す患者であったとしても、IGF-Iを介さない
GH の直接作用は欠如したままであり、それに起因する臨床症候が生じると考えられる。この
ような患者で GH補充療法を実施する際には、血清 IGF-I濃度が基準範囲を超えないように GH
投与量の調整を行い、IGF-I 異常高値を誘導するような GH の補充は避けなければならない。
一方では、上記に述べたとおり、GH 補充治療実施時に血清 IGF-I 濃度が増加することは本剤
の有効性の発現につながるものであるため、有効性及び安全性の両面から血清 IGF-I濃度を参
照しつつ用量調整を行うことが重要である。
(b) 有効性に関する臨床情報の解析
LY72w群及びプラセボ/LY48w群の LBM平均変化率を「2.7.3.3.2全有効性試験の結果の比
較検討」の表 19及び表 20に示した。LY72w群におけるベースラインから投与 24週時、投与
72週時までの LBMの平均変化率はそれぞれ 4.7%、5.8%であり、ともに投与前後で統計学的有
意差が認められた(p<0.001)。用量調整法を用いた投与がなされた LY72w群の投与 24週以降
についても、LBMの改善が維持されていた。
用量調整法を用いたプラセボ/LY48w群におけるベースラインから投与 48週時までの LBM
平均変化率は 4.5%であり、投与前後で統計学的有意差が認められた(p<0.001)。

ヒューマトロープ CTD(2.7 臨床概要) Page 69
69
以上の結果から、臨床症候及び血清 IGF-I濃度を指標とした用量調整法は有効であることが
確認された。
(c) 安全性に関する臨床情報の解析
K02A試験において認められた有害事象の要約を表 74に示した。また、K01A試験及び K02A
試験における有害事象による投与中止例又は減量例を表 75に示した。
表 74 有害事象の要約(K02A試験)
区分*1 LY群 (N=33)
LY/LY48w群 (N=31)
プラセボ/LY48w群 (N=28)
死亡 0 0.0% 0 0.0% 0 0.0%
重篤な有害事象 1 1 3.0% 2 2 6.5% 1 1 3.6%
有害事象による中止*2 1 1 3.0% 0 0 0.0% 0 0 0.0%
有害事象による減量*2 6 6 18. 2% 4 4 12. 9% 3 3 10.7%
有害事象 173 28 84.8% 266 30 96.8% 214 26 92.9%
副作用 38 14 42.4% 31 15 48.4% 27 15 53.6%
*1 件数、例数、発現率%、 *2 重篤な有害事象例を除く
用量漸増法を用いた LY群において、重篤な有害事象が 1例認められた。用量調整法を用い
た LY/LY48w群及びプラセボ/LY48w群において、重篤な有害事象はそれぞれ 2例、1例認
められた。LY/LY48w群で認められた頭蓋咽頭腫の 1例(因果関係:不明)を除き、治験責
任医師によって本剤との因果関係は「なし」と判断された。
有害事象による投与中止例は、LY 群で関節痛を発現した 1 例のみであった。一方、用量調
整法を行ったLY/LY48w群及びプラセボ/LY48w群では有害事象による投与中止例は認めら
れなかった。LY群で認められた関節痛の 1例は、本剤との因果関係は「あり」と判断された。
LY群において、投与 24週間に認められた有害事象による減量例は 6例であった。そのなか
で有害事象と本剤との因果関係が「あり」と判断された例は、関節痛 2例、浮腫 1例、筋骨格
硬直 1例の 4例であった。用量調整法を行った LY/LY48w群の投与 48週間において、4例の
減量例が認められた。そのなかで有害事象と本剤との因果関係が「あり」と判断された例は、
関節痛 2例であった。また、K02A試験より本剤投与を開始し、用量調整法を行ったプラセボ
/LY48w 群の投与 48 週間において、3例の減量例が認められた。そのなかで有害事象と本剤
との因果関係が「あり」と判断された例は、末梢性浮腫 1例、浮腫 1例の合計 2例であった。
これらのことより、強制的に投与量を増量する用量漸増法より、臨床症候及び血清 IGF-I濃
度を指標とした用量調整法が適切であることが示唆された。
以上、安全性を考慮し、0.021 mg/kg/週から 0.084 mg/kg/週の用量範囲の臨床症候及び血清
IGF-I濃度を指標とした用量調整法は適切であると考えられた。

ヒューマトロープ CTD(2.7 臨床概要) Page 70
70
表75 投与中止例又は減量例(
K01A試験及び
K02A試験)
試験番号
投与群
症例番号
発症時期
*4年齢
(歳)
性別
有害事象
*1
重症度
因果関係
発現までの投与期間
有害事象の持続時間
(日)
中止
/減量
転帰
治験薬の処置
処置の有無
A
O
3
女性
蕁麻疹NOS
軽度
関連なし
15
7
減量
回復
休薬
併用薬剤あり
A
O
5
女性
関節痛
中等度
関連あり
36
継続中
中止及び減量
未回復
*2
投与中止
なし
A
O
4
女性
関節痛
中等度
関連あり
29
継続中
減量
未回復
減量
なし
C
O
2
男性
胃腸炎NOS
中等度
関連なし
74
7
減量
回復
休薬
併用薬剤あり
A
O
4
女性
浮腫NOS
中等度
関連あり
14
5 21
減量
回復
休薬
併用薬剤あり
LY群
*3
AO
5
男性
筋骨格硬直
中等度
関連あり
92
継続中
減量
未回復
減量
なし
C
O
3
男性
インフルエンザ
軽度
関連なし
10
0 10
減量
回復
休薬
併用薬剤あり
K01
A試験
プラセボ群
A
O
5
女性
良性下垂体腫瘍NOS
中等度
不明
16
2 継続中
中止
未回復
投与中止
なし
*1 M
edD
RA
(J) 5
.1 P
T、
*2 追跡調査により、消失を確認した。
*3 本症例は
K02
A試験期間中、同一事象により再度減量された。
*4
AO;成人期発症型、
CO;小児期発症型

ヒューマトロープ CTD(2.7 臨床概要) Page 71
71
表 7
5 投与中止例又は減量例(
K01A試験及び
K02A試験)(続き)
試験番号
投与群
症例番号
発症時期
*3年齢
(歳)
性別
有害事象
*1
重症度
因果関係
発現までの投与期間
有害事象の持続時間
(日)
中止
/減量
転帰
治験薬の処置
処置の有無
C
O
4
女性
関節痛
中等度
関連あり
33
6 継続中
減量
軽快
減量
なし
C
O
3
女性
鼻咽頭炎
中等度
関連なし
44
1 5 減量
回復
休薬
併用薬剤あり
A
O
4
女性
関節痛
中等度
関連あり
45
8 29
減量
回復
減量
なし
LY
/LY
48w群
A
O
3
男性
高血圧
NO
S 軽度
不明
19
7 継続中
減量
*2
未回復
*4
減量
なし
A
O
4
女性
末梢性浮腫
中等度
関連あり
10
6 22
減量
回復
減量
なし
A
O
4
男性
浮腫
NO
S 軽度
関連あり
20
9 12
0 減量
回復
減量
なし
K02
A試験
プラセボ
/LY
48w群
A
O
5
女性
関節痛
中等度
不明
59
15
0 減量
回復
減量
なし
*1 M
edD
RA
(J) 5
.1 P
T *2
K02
A試験投与
24週以前に発現した有害事象であるが、指示投与量の減量は投与
24週以降に行われた
*3 A
O;成人期発症型、
CO;小児期発症型
*4
本症例は
K02
A試験を完了した。
K02
A試験完了時の収縮期血圧は
152m
mH
g、拡張期血圧は
104m
mH
gであった。

ヒューマトロープ CTD(2.7 臨床概要) Page 72
72
3) 血清 IGF-I濃度を指標とした用量調整法に関する結論
有効性及び安全性の観点から、AGHD患者に対する GH補充療法として、0.021 mg/kg/週から
投与開始し、最大 0.084 mg/kg/週まで増量できる用量範囲で、副作用の発現及び血清 IGF-I濃度
を指標とした用量調整法を本効能・効果に対する用法・用量(案)として推奨した。
(3) 1日投与量の上限
GRSコンセンサス・ガイドラインでは、1日投与量の上限に関する以下の指針が示されてい
る。
• 維持用量は個々で異なるが、1日 1 mg以上になることは稀である。そのため、1日投与量
が 1 mgを超えないこと。
この指針を踏まえ、国内臨床試験では 1日投与量が 1 mgを超えないこととした。
国内臨床試験成績において 1日 1 mg以上投与された症例はいなかった。本療法は補充療法
であり、1日 1 mgを超えて投与することは、ほとんどの患者にとって基準範囲を超える過量投
与となり、安全性の観点から問題があると判断される。したがって、本効能・効果の用法・用
量として「1日量として 1 mgを超えないこと」を規定した。
(4) 投与量の性差に関する考察
K02A試験における本剤投与量を男女別に比較した(表 76)。また、性別ごとの投与量を表
77に示した。
LY/LY48w群+プラセボ/LY48w群の投与 4週時以外に男女間で統計学的有意差は認めら
れなかった。LY/LY48w群+プラセボ/LY48w群の投与 4週時における男女間の差は、体重
換算によって算出される投与量の幅(許容範囲)によるものであり、平均投与量は男女とも
0.021 mg/kg/週であったことから臨床的意義はないと考えた。なお、K02A試験における投与 4
週時の指示投与量は固定用量である 0.021 mg/kg/週であった。
また、LY/LY48w群+プラセボ/LY48w群の投与48週時における男性の平均投与量は 0.047
mg/kg/週、女性は 0.052 mg/kg/週であり、統計学的有意差はないものの、全般的に女性で投与
量が高くなる傾向がみられた。女性ホルモンが肝での IGF-I産生を抑制するため、血清 IGF-I濃
度を指標とした用量調整法を行う場合、女性の投与量は男性の投与量と比較し高くなることが
知られている 14。今回みられた投与量の性差による違いは、これまで得られた知見と一致して
いた。一方、血清 IGF-I濃度を指標とした用量調整法を採用したプラセボ/LY48w群における
性別ごとの本剤投与 48週間の LBM平均変化率は、男性 3.0%、女性 5.9%であり、性別にかか
わらず改善がみられた。なお、投与 48週時における IGF-I SDSはそれぞれ 0.71、0.10であり、
男女とも正常範囲に含まれていた。
以上のことから、投与量に性差は認められたが、血清 IGF-I濃度を指標とした用量調整法を
実施することで安全に効果を示すことができると判断した。また、性ホルモン補充の影響に関
しても、性ホルモン補充の有無にかかわらず、性別及び年齢別の IGF-I標準値を参照し、血清
IGF-I濃度を指標とした用量調整法を用いるため、GH補充療法の有効性及び安全性に影響があ
るとは考えられなかった。

ヒューマトロープ CTD(2.7 臨床概要) Page 73
73
表 76 投与量の男女間比較(K02A試験)
男性 女性 投与群 来院
例数 平均値±標準偏差 例数 平均値±標準偏差
検定*1
p値
4週 15 0.021 ± 0.0004 16 0.021 ± 0.0005 0.060
8週 15 0.021 ± 0.0005 16 0.021 ± 0.0005 0.621
12週 15 0.032 ± 0.0108 15 0.032 ± 0.0096 0.820
16週 15 0.040 ± 0.0192 15 0.038 ± 0.0166 0.885
20週 15 0.046 ± 0.0234 15 0.047 ± 0.0244 0.950
24週 15 0.049 ± 0.0248 14 0.055 ± 0.0229 0.585
36週 15 0.050 ± 0.0263 15 0.049 ± 0.0231 0.901
LY/LY48w群
48週 15 0.051 ± 0.0260 15 0.048 ± 0.0233 0.836
4週 14 0.021 ± 0.0005 14 0.021 ± 0.0007 0.408
8週 14 0.021 ± 0.0006 14 0.021 ± 0.0005 0.748
12週 14 0.035 ± 0.0176 14 0.034 ± 0.0104 0.927
16週 14 0.035 ± 0.0174 14 0.042 ± 0.0219 0.662
20週 14 0.039 ± 0.0199 14 0.049 ± 0.0229 0.280
24週 13 0.045 ± 0.0215 14 0.053 ± 0.0244 0.254
36週 14 0.043 ± 0.0223 14 0.055 ± 0.0258 0.206
プラセボ/LY48w群
48週 13 0.041 ± 0.0235 14 0.056 ± 0.0262 0.198
4週 29 0.021 ± 0.0004 30 0.021 ± 0.0006 0.041
8週 29 0.021 ± 0.0005 30 0.021 ± 0.0005 0.873
12週 29 0.034 ± 0.0142 29 0.033 ± 0.0098 0.988
16週 29 0.038 ± 0.0182 29 0.040 ± 0.0191 0.630
20週 29 0.042 ± 0.0217 29 0.048 ± 0.0233 0.451
24週 28 0.047 ± 0.0230 28 0.054 ± 0.0232 0.204
36週 29 0.047 ± 0.0242 29 0.052 ± 0.0242 0.401
LY/LY48w群+プラセボ/LY48w群
48週 28 0.047 ± 0.0249 29 0.052 ± 0.0247 0.518
投与量(mg/kg/週)を示す *1 2標本のWilcoxon検定

ヒューマトロープ CTD(2.7 臨床概要) Page 74
74
表 77 性別ごとの投与量の要約統計量(K02A試験)
投与群 性別 来院 例数 平均値 標準偏差 最大値 中央値 最小値
4週 15 0.021 0.0004 0.022 0.021 0.020
8週 15 0.021 0.0005 0.022 0.021 0.020
12週 15 0.032 0.0108 0.043 0.039 0.010
16週 15 0.040 0.0192 0.081 0.041 0.010
20週 15 0.046 0.0234 0.084 0.041 0.011
24週 15 0.049 0.0248 0.084 0.042 0.011
36週 15 0.050 0.0263 0.084 0.042 0.010
男性
48週 15 0.051 0.0260 0.085 0.042 0.010
4週 16 0.021 0.0005 0.022 0.021 0.020
8週 16 0.021 0.0005 0.022 0.021 0.020
12週 15 0.032 0.0096 0.043 0.034 0.020
16週 15 0.038 0.0166 0.084 0.042 0.020
20週 15 0.047 0.0244 0.084 0.041 0.020
24週 14 0.055 0.0229 0.084 0.051 0.020
36週 15 0.049 0.0231 0.084 0.043 0.021
LY/LY48w群
女性
48週 15 0.048 0.0233 0.085 0.042 0.020
4週 14 0.021 0.0005 0.022 0.021 0.020
8週 14 0.021 0.0006 0.022 0.021 0.020
12週 14 0.035 0.0176 0.084 0.031 0.021
16週 14 0.035 0.0174 0.085 0.035 0.020
20週 14 0.039 0.0199 0.085 0.036 0.020
24週 13 0.045 0.0215 0.084 0.042 0.021
36週 14 0.043 0.0223 0.084 0.039 0.021
男性
48週 13 0.041 0.0235 0.083 0.030 0.021
4週 14 0.021 0.0007 0.022 0.021 0.020
8週 14 0.021 0.0005 0.022 0.021 0.020
12週 14 0.034 0.0104 0.043 0.041 0.020
16週 14 0.042 0.0219 0.084 0.041 0.020
20週 14 0.049 0.0229 0.084 0.043 0.020
24週 14 0.053 0.0244 0.084 0.045 0.020
36週 14 0.055 0.0258 0.085 0.052 0.020
プラセボ/LY48w群
女性
48週 14 0.056 0.0262 0.084 0.052 0.020
投与量(mg/kg/週)を示す

ヒューマトロープ CTD(2.7 臨床概要) Page 75
75
表 77 性別ごとの投与量の要約統計量(K02A試験)(続き)
投与群 性別 来院 例数 平均値 標準偏差 最大値 中央値 最小値
4週 29 0.021 0.0004 0.022 0.021 0.020
8週 29 0.021 0.0005 0.022 0.021 0.020
12週 29 0.034 0.0142 0.084 0.039 0.010
16週 29 0.038 0.0182 0.085 0.041 0.010
20週 29 0.042 0.0217 0.085 0.041 0.011
24週 28 0.047 0.0230 0.084 0.042 0.011
36週 29 0.047 0.0242 0.084 0.042 0.010
男性
48週 28 0.047 0.0249 0.085 0.042 0.010
4週 30 0.021 0.0006 0.022 0.021 0.020
8週 30 0.021 0.0005 0.022 0.021 0.020
12週 29 0.033 0.0098 0.043 0.040 0.020
16週 29 0.040 0.0191 0.084 0.041 0.020
20週 29 0.048 0.0233 0.084 0.042 0.020
24週 28 0.054 0.0232 0.084 0.045 0.020
36週 29 0.052 0.0242 0.085 0.043 0.020
LY/LY48w群+ プラセボ/LY48w群
女性
48週 29 0.052 0.0247 0.085 0.043 0.020
投与量(mg/kg/週)を示す
2.7.3.5 効果の持続、耐薬性
2.7.3.3.2 項で述べたように、国内臨床試験において、LY72w 群の投与後 24 週時に認められ
た LBMの変化率 4.7%は、投与後 48週時でも 4.7%に維持されていた。更に、投与後 72週時で
も 5.8%であり、本剤を 48週間以上投与した場合でも効果は維持されていた。また、本剤投与
開始時より用量調整法を行ったプラセボ/LY48w群の投与後24週時に認められたLBMの変化
率 3.4%は、投与後 48週時でも 4.5%に維持されていた。
また、2.7.3.4.2項で述べたように、用量調整法を用いた LY/LY48w群の投与量は投与後 48
週以降大きく変更されることはなく、また、プラセボ/LY48w群でも投与後 24週以降大きく
変更されることはなかった。
外国臨床試験の E003/4 試験及び E005/6 試験においても、投与後 24 週時の LBM の変化量
(E003/4試験:2.59 kg、E005/6試験:3.68 kg)は、投与後 48週時及び投与後 72週時において
も維持されていた(投与後 48週時:E003/4試験;1.61 kg、E005/6試験;3.20 kg、投与後 72週
時:E003/4試験;2.07 kg、E005/6試験;3.58 kg、表 23)。
また、上記外国試験では抗 GH抗体も測定したが、抗 GH抗体は E003/4試験の 98例中 3例、
E005/6試験の 67例中 1例に認められたのみであった。抗体産生が認められた 4例のうち、1例
では GH補充療法に対する効果の減弱傾向が認められたものの、他の 3例では効果は減弱しな
かった。抗体産生と効果の減弱の関係については、該当症例数が少ないことから明確な結論は
出せないものの、抗 GH抗体が産生されたすべての患者で効果が減弱する可能性は低いと考え
られた。
以上のことから、GHの長期投与による効果の減弱及び耐薬性は認められず、長期投与によっ
ても効果は持続するものと考えられた。
ヒューマトロープ CTD(2.7 臨床概要) Page 76
76
表78 国内臨床試験
試験
番号
試験施設数
場所
試験開始日
登録状況
日付
総登録数
/ 登録目標数
デザイン
対照の種類
試験薬、比較対照薬
投与量、投与経路
投与方法
試験の目的
登録時
/完了時
群別被験者数
試験期間
男性
/女性
年齢の中央値
(範囲)
診断
選択基準
主たる
エンド
ポイント
K01
A試験
*
25施設
科
等
20
年 月
~
20
年
月
65/6
8
無作為化
二重盲検
並行群間比較
プラセボ
LY13
7998群:
6 m
g 皮下投与:
1週間に
6~7回
0~
4週:
0.02
1 m
g/kg
/週
4~12週:
0.04
2 m
g/kg
/週
12~
24週:
0.08
4 m
g/kg
/週
プラセボ群
有効性と
安全性
LY群
33
/31
プラセボ群
32
/29
24週間
31/3
3 35
(18~
63)
AG
HD
成長ホルモン分
泌刺激試験にお
いて
GH頂値が
3.0
ng/m
L未満の
者
24週後に
おける全
身LB
Mの
変化率
K02
A試験
*
25施設
科
等
20
年 月
~
20
年
月
非盲検
無対照
LY13
7998群:
6 m
g 皮下投与
:1週間に
6~7回
0~
8週:
0.02
1 m
g/kg
/週
8~48週:
0.02
1~0.
084
mg/
kg/
週
安全性と
有効性
LY/
LY48
w群
31/3
0 プラセボ/
LY48
w群
29
/27
48週間
29/3
0 35
(18~
64)
同上
48週間に
おける治
療の安全
性
*:K
01A試験で対象とされた
AG
HD患者のうち、治験責任医師又は治験分担医師によって安全性の観点から長期投与への移行が問題ないと判断され、かつ長期投与移行の同
意が得られた患者が対象となるため、症例数設定は行わない。
* 新薬承認情報提供時に置き換えた

ヒューマトロープ CTD(2.7 臨床概要) Page 77
77
表79 外国臨床試験
試験
番号
試験施設数
場所
登録開始日
登録状況
日付
総登録数
/ 登録目標数
デザイン
対照の種類
試験薬、比較対照薬
投与量、投与経路
投与方法
試験の目的登録時
/完了時
群別被験者数
試験期間
男性
/女性
年齢の中央
値(範囲)
診断
選択基準
主たる
エンド
ポイント
無作為化
二重盲検
並行群間比較
プラセボ
LY13
7998群:
5.92
mg
皮下投与:
1日
1回
0~
1ヵ月:
6.25
µg/
kg/日
1~
6ヵ月:
12.5
µg/
kg/日
プラセボ群
LY群
52
/49
プラセボ群
46
/44
E0
03/4
試験
*
7施設
フランス
ドイツ
スペイン
スウェーデン
オランダ
イギリス
19
年 月
~
19
年
月
10
2/10
0 (二重盲検期を
終えた後
) 非盲検
長期
LY13
7998群:
5.92
mg
6~7ヵ月:
6.25
µg/
kg/日
7~
18ヵ月:
12.5
µg/
kg/日
有効性と
安全性
LY群
49
/44
プラセボ群
44
/40
1ヵ月間の
プラセボ
投与観察
期の後、
6ヵ月間二
重盲検
続いて
12ヵ月間
非盲検
62/3
6 43
.5
(21.
3~60
.9)
成人期発症型
AG
HD
GH分泌刺激試験にお
いて
GH頂値が
5.0
ng/m
L未満
・体組成
・脂質パラ
メータ
・
QO
L
無作為化
二重盲検
並行群間比較
プラセボ
LY13
7998群:
5.92
mg
皮下投与:
1日
1回
0~
1ヵ月:
6.25
µg/
kg/日
1~
6ヵ月:
12.5
µg/
kg/日
プラセボ群
LY群
32
/28
プラセボ群
35
/30
E0
05/6
試験
*
8施設
フランス
イタリア
スペイン
ドイツ
ノルウェー
デンマーク
19
年 月
~
19
年
月
71
/100
(二重盲検期を
終えた後
) 非盲検
長期
LY13
7998群:
5.92
mg
皮下投与:
1日
1回
6~
7ヵ月:
6.25
µg/
kg/日
7~
18ヵ月:
12.5
µg/
kg/日
有効性と
安全性
LY群
28
/22
プラセボ群
30
/26
1ヵ月間の
プラセボ
投与観察
期の後、
6ヵ月間
二重盲検
続いて
12ヵ月間
非盲検
49/1
8 28
.4
(18.
0~48
.2)
小児期発症型
AG
HD
GH分泌刺激試験にお
いて
GH頂値が
5.0
ng/m
L未満
・体組成
・脂質パラ
メータ
・
QO
L
* 新薬承認情報提供時に置き換えた
ヒューマトロープ CTD(2.7 臨床概要) Page 78
78
表79 外国臨床試験(続き)
試験
番号
試験施設数
場所
試験開始日
登録状況
日付
総登録数
/ 登録目標数
デザイン
対照の種類
試験薬、比較対照薬
投与量、投与経路
投与方法
試験の目的
登録時
/完了時
群別被験者数試験期間
男性
/女性
年齢
診断
選択基準
主たる
エンド
ポイント
G
DED
試験
*
56施設
オーストリア
チェコ
ハンガリー
ポーランド
スイス
スロバキア
カナダ
イタリア
イギリス
オーストラリ
ア
ロシア
クロアチア
スロベニア
19
年 月
~
19
年
月
59
5/60
0
無作為化
非盲検
並行群間
比較試験
LY13
7998:
6.66
mg又は
13.3
3 m
g皮下投与:
1日
1回
低用量群:
0~3ヵ月:
3 µg
/kg/日
3~6ヵ月:
6 µg
/kg/日
高用量群:
0~3 ヵ月:
6 µg
/kg/日
3~6 ヵ月:
12 µ
g/kg
/日
安全性と
有効性
低用量群
30
2/27
4
高用量群
293/
246
24週間
低用量群:
174/
128
中央値
43
.40
範囲
17.9~
78.4
高用量群:
185/
108
中央値
43.7
0 範囲
18.0~
78.1
成人期発症型
AG
HD
及び
小児期発症型
AG
HD
GH分泌刺激試験にお
いて
GH頂値が
3.0
µg/L未満
体組成の
改善
T0
02
試験
*
61施設
フランス
ドイツ
イタリア
イギリス
アメリカ
プエルトリコ
19
年 月
~
20
年
月
38
7/37
0
無作為化
非盲検
並行群間
比較試験
LY13
7998:
6.0
mg又は
12.0
mg
皮下投与:
1日
1回
FD群:
0~16週:
4 µg
/kg/日
16~
24週:
8 µg
/kg/日
24~
32週:
12 µ
g/kg
/日
ID群:
0~
8週:
200
µg/日
8~16週:
400
µg/日
16~
24週:
600
µg/日
24~
32週:
800
µg/日
安全性と
有効性
FD群
20
0/17
4
ID群
18
7/15
8
32週間
FD群:
11
5/85
平均値
± 標準偏差
48.1
±13.
2
ID群:
10
9/78
平均値
± 標準偏差
45
.1±1
4.1
成人期発症型
AG
HD
及び
小児期発症型
AG
HD
GH分泌刺激試験にお
いて
GH頂値が
3.0
µg/L未満
体組成の
改善
* 新薬承認情報提供時に置き換えた


















