
1 Potent Efficacy of Combined PI3K/mTOR and JAK or ABL ...
35
1 Potent Efficacy of Combined PI3K/mTOR and JAK or ABL Inhibition in Murine Xenograft Models of Ph-like Acute Lymphoblastic Leukemia Sarah K Tasian MD, 1,2 David T Teachey MD, 1,2 Yong Li, 1 Feng Shen, 1 Richard C Harvey PhD, 4 I-Ming Chen DVM MS, 4 Theresa Ryan, 1 Tiffaney L Vincent, 1 Cheryl L Willman MD, 4 Alexander E Perl MD MS, 2,3 Stephen P Hunger MD, 1,2 Mignon L Loh MD, 5,6 Martin Carroll MD, 2,3 Stephan A Grupp MD PhD 1,2 1 Department of Pediatrics, Division of Oncology, Center for Childhood Cancer Research, Children’s Hospital of Philadelphia; Philadelphia, PA 2 University of Pennsylvania Perelman School of Medicine 3 Department of Medicine, Division of Hematology, Abramson Cancer Center, University of Pennsylvania; Philadelphia, PA 4 Department of Pathology, University of New Mexico: Albuquerque, NM 5 Department of Pediatrics, Division of Hematology/Oncology, University of California, San Francisco Benioff Children’s Hospital and School of Medicine; San Francisco, CA 6 Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, CA. Corresponding author: Sarah K Tasian, MD 3501 Civic Center Boulevard, CTRB 3010 Philadelphia, Pennsylvania 19104 Phone: 215.590.5476 Fax: 215.590.3770 Email: [email protected] Short title: PI3K inhibition in Ph-like ALL Abstract word count: 232 Text word count: 3988 Number of figures/tables: 5 Number of supplemental figures: 5 Number of references: 60 Scientific category: Lymphoid Neoplasia Presented in part at the American Society of Hematology 2015 annual meeting (New Orleans, LA) and the International Society of Paediatric Oncology 2014 annual meeting (Toronto, ON) Blood First Edition Paper, prepublished online October 24, 2016; DOI 10.1182/blood-2016-05-707653 Copyright © 2016 American Society of Hematology For personal use only. on February 5, 2018. by guest www.bloodjournal.org From
Transcript of 1 Potent Efficacy of Combined PI3K/mTOR and JAK or ABL ...

1
Potent Efficacy of Combined PI3K/mTOR and JAK or ABL Inhibition in Murine Xenograft Models of Ph-like Acute Lymphoblastic Leukemia Sarah K Tasian MD,1,2 David T Teachey MD,1,2 Yong Li,1 Feng Shen,1 Richard C Harvey PhD,4 I-Ming Chen DVM MS,4 Theresa Ryan,1 Tiffaney L Vincent,1 Cheryl L Willman MD,4 Alexander E Perl MD MS,2,3 Stephen P Hunger MD,1,2 Mignon L Loh MD,5,6 Martin Carroll MD,2,3 Stephan A Grupp MD PhD1,2
1 Department of Pediatrics, Division of Oncology, Center for Childhood Cancer Research, Children’s Hospital of Philadelphia; Philadelphia, PA 2 University of Pennsylvania Perelman School of Medicine 3 Department of Medicine, Division of Hematology, Abramson Cancer Center, University of Pennsylvania; Philadelphia, PA 4 Department of Pathology, University of New Mexico: Albuquerque, NM 5 Department of Pediatrics, Division of Hematology/Oncology, University of California, San Francisco Benioff Children’s Hospital and School of Medicine; San Francisco, CA 6 Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, CA. Corresponding author: Sarah K Tasian, MD 3501 Civic Center Boulevard, CTRB 3010 Philadelphia, Pennsylvania 19104 Phone: 215.590.5476 Fax: 215.590.3770 Email: [email protected] Short title: PI3K inhibition in Ph-like ALL Abstract word count: 232 Text word count: 3988 Number of figures/tables: 5 Number of supplemental figures: 5 Number of references: 60 Scientific category: Lymphoid Neoplasia Presented in part at the American Society of Hematology 2015 annual meeting (New Orleans, LA) and the International Society of Paediatric Oncology 2014 annual meeting (Toronto, ON)
Blood First Edition Paper, prepublished online October 24, 2016; DOI 10.1182/blood-2016-05-707653
Copyright © 2016 American Society of Hematology
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

2
KEY POINTS
1. PI3K/mTOR inhibition potently inhibited leukemia proliferation and signal
transduction in vivo in human Ph-like ALL xenograft models.
2. Combined PI3K/mTOR and JAK or ABL inhibition was superior to monotherapy in
CRLF2/JAK-mutant and ABL/PDGFR-mutant Ph-like ALL models.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

3
ABSTRACT
Philadelphia chromosome-like B-cell lymphoblastic leukemia (BCR-ABL1-like or
Ph-like ALL) is associated with activated JAK/STAT, SRC/ABL, and/or PI3K/Akt/mTOR
signaling and poor clinical outcomes. Inhibitors of PI3K pathway signaling (PI3Ki) have
been minimally investigated in Ph-like ALL to date. We hypothesized that targeted
inhibition of PI3Kα, PI3Kδ, PI3K/mTOR, or TORC1/TORC2 would decrease leukemia
proliferation and abrogate aberrant kinase signaling. We further hypothesized that
combined PI3K pathway and JAK inhibition or PI3K pathway and SRC/ABL inhibition
would have superior efficacy compared to inhibitor monotherapy. We treated ten
childhood ALL patient-derived xenograft models harboring various Ph-like genomic
alterations with four discrete PI3K pathway protein inhibitors and observed marked
leukemia reduction and in vivo signaling inhibition in all models. Treatment with the dual
PI3K/mTOR inhibitor gedatolisib resulted in near-eradication of ALL in CRLF2/JAK-
mutant models (n=7) with mean 92.2% (range 86.0-99.4%) reduction versus vehicle
controls (p<0.0001) and in prolonged animal survival. Gedatolisib also inhibited ALL
proliferation in ABL/PDGFR-mutant models (n=3) with mean 66.9% (range 42.0-87.6%)
reduction versus vehicle (p<0.0001). Combined gedatolisib and ruxolitinib treatment of
CRLF2/JAK-mutant models more effectively inhibited ALL proliferation than either
inhibitor alone (p<0.001) and further enhanced survival. Similarly, superior efficacy of
combined gedatolisib and dasatinib was observed in ABL/PDGFR-mutant models
(p<0.001). Overall, PI3K/mTOR inhibition potently decreased ALL burden in vivo, and
anti-leukemia activity was further enhanced with combination inhibitor therapy. Clinical
trials testing combinations of kinase inhibitors in patients with Ph-like ALL are indicated.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

4
INTRODUCTION
B-cell acute lymphoblastic leukemia (B-ALL), the most common childhood
cancer, is caused by somatic genetic mutations that result in aberrant arrest of normal
lymphoid maturation, dysregulated cellular proliferation, and evasion of programmed cell
death.1-3 Increased understanding of the biologic heterogeneity of childhood ALL has led
to modern risk stratification, which incorporates the critical contributions of genetic
subgroups and induction chemotherapy responses to deliver appropriately intensive
therapy to achieve cure.4-6 Unfortunately, approximately 15% of children with ALL have
recurrent disease, and relapsed ALL remains a leading cause of pediatric cancer
mortality.7 Adults with ALL fare even more poorly with >50% relapse rates and 20-40%
overall survival.8,9
Genomic profiling of high-risk (HR) ALL cases has identified the Philadelphia
chromosome-like subtype of B-ALL (Ph-like ALL), which comprises 10-20% of HR B-ALL
in children and adolescents and nearly 30% in young adults.10-15 Ph-like ALL is defined
by lack of BCR-ABL1 fusion (as in Ph+ ALL), but has a kinase-activated gene
expression signature similar to that of Ph+ ALL, frequent deletion of IKZF1, and
mutations in other kinase and cytokine receptor signaling pathway genes.12-14,16-18
Rearrangements of cytokine receptor-like factor 2 (CRLF2) occur in approximately 50%
of Ph-like ALL cases and result in CRLF2 overexpression.19-21 Half of CRLF2-rearranged
ALLs have concomitant alterations in Janus kinase (JAK) pathway-associated genes,
including JAK1 and JAK2, and a small number of these cases also harbor interleukin-7
receptor alpha (IL7RA) mutations.22-24 JAK2 point mutations are the most frequent co-
existing genetic abnormality in CRLF2-rearranged ALL and also occur in non-Ph-like
ALL, including trisomy 21-associated B-ALL and T-ALL.25,26 Genomic rearrangements of
Abelson kinase 1 and Abelson kinase 2 (ABL1 and ABL2), colony stimulating factor-1
receptor (CSF1R), platelet-derived growth factor receptor-beta (PDGFRB), JAK2,
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

5
erythropoietin receptor (EPOR), and other genes account for an additional 30-40% of
Ph-like ALL and also drive constitutive kinase signaling.12-14,26,27 Patients with Ph-like
ALL often have high rates of minimal residual disease (MRD) at the end of induction
chemotherapy with some patients experiencing overt induction failure.14,21,28
Retrospective analyses of high-risk children and adolescents and young adults (AYAs)
with Ph-like ALL demonstrate greatly increased risk of treatment failure and poor overall
survival (OS) regardless of underlying Ph-like genomic alterations.10,13,14 New therapies
are indicated for these patients.
All Ph-like ALL-associated mutations identified to date activate kinase signaling,
particularly of CRLF2-, JAK-, ABL-, and PDGFRB-associated pathways. Biochemical
sequelae of these mutations have been primarily studied to date in the most common
CRLF2-rearranged subset of Ph-like ALL with the goal of identifying potential signal
transduction inhibitor (i) therapies, but perturbed intracellular signaling mechanisms of
these genomic alterations remain incompletely characterized. Others and we previously
reported constitutive and/or cytokine-inducible JAK/signal transduction activator of
transcription (JAK/STAT) and phosphatidylinositol 3-kinase/protein kinase B/mammalian
target of rapamycin (PI3K/Akt/mTOR) signaling in CRLF2-rearranged ALL.29,30 We
further observed robust inhibition of constitutively activated signal transduction with JAK
or mTOR inhibition in vitro and in vivo.24,31-33 More recent studies focused upon non-
CRLF2-rearranged Ph-like ALL cases have demonstrated in vitro and in vivo efficacy of
other kinase inhibitors, including anecdotal clinical responses in children and adults with
Ph-like ALL harboring ABL1, ABL2, or PDGFRB rearrangements and fusion proteins
(“ABL class” rearrangements) treated with imatinib or dasatinib.14,34,35
While preclinical32,36 and early clinical studies of JAK inhibition in CRLF2/JAK-
mutant and SRC/ABL inhibition in ABL/PDGFR-mutant Ph-like ALL are ongoing
(NCT02723994, NCT02883049), therapeutic disruption of aberrant PI3K pathway
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

6
signaling has been minimally investigated. Clinical efficacy of the mTORi rapamycin and
its analogues has proven suboptimal in various cancers, at least in part due to
upregulation of Akt signaling, a known sequela of mTORi monotherapy and a common
resistance mechanism.37 Newer-generation kinase inhibitors that target multiple PI3K
pathway signaling proteins or that selectively inhibit PI3K isoforms may have superior
anti-leukemia cytotoxicity, as well as avoid compensatory upregulation of salvage
signaling pathways.38,39 Such “next generation” PI3K pathway inhibitors (PI3Ki) have
been minimally evaluated in ALL to date.39 Furthermore, the efficacy of simultaneously
targeting multiple oncogenic signaling networks in Ph-like ALL has not been
investigated, such as combination therapy with PI3Ki and JAKi.
Using patient-derived xenograft (PDX) models of childhood Ph-like ALL, we
demonstrate the in vivo therapeutic efficacy of and pharmacodynamic signaling inhibition
by four clinically promising PI3K pathway inhibitors with particularly potent efficacy of the
dual PI3K/mTORi gedatolisib. We further demonstrate augmented leukemia cytotoxicity
in vivo with combined gedatolisib and ruxolitinib (JAK1/2i) treatment of CRLF2/JAK-
mutant Ph-like ALL and with gedatolisib and dasatinib (SRC/ABLi) treatment of
ABL/PDGFR-mutant Ph-like ALL. These data provide compelling rationale for testing
combinations of kinase inhibitors without or with multi-agent cytotoxic chemotherapy in
children and adults with Ph-like ALL.
METHODS
Ph-like ALL specimens
Viably cryopreserved leukemia cells from children and AYAs with de novo Ph-like
ALL (n=8) were obtained from the Children’s Oncology Group (COG) for
xenotransplantation studies as described.12,14,32 Additional specimens from patients with
multiply relapsed Ph-like ALL (n=2) were obtained from the Children’s Hospital of
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

7
Philadelphia (CHOP) and University of California, San Francisco leukemia
biorepositories under approved institutional research protocols after obtainment of
written informed consent in accordance with the Declaration of Helsinki (Table 1). Ph-like
genomic alterations were identified by polymerase chain reaction and Sanger
sequencing and/or fluorescence in situ hybridization (FISH) assays as described.21,40,41
RNA from primary and corresponding xenografted leukemia specimens were also
assessed for an activated kinase Ph-like ALL gene expression signature using a 15-
gene low density microarray (LDA) classifier as described.40
Patient-derived xenograft models
Studies to date have largely focused upon studying the most common Ph-like
ALL subset harboring CRLF2 rearrangements. However, Ph-like ALL is now known to be
highly genetically diverse with a variety of mutations that induce kinase
hyperactivation.14 In these studies, we thus assessed the efficacy of PI3K pathway
inhibition in patient-derived xenograft (PDX) models of CRLF2-rearranged, JAK1-mutant,
JAK2-rearranged, ABL1-rearranged, and PDGFRB-rearranged Ph-like ALL (Table 1).
Individual PDX models were established in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ
(NSG) mice as described.32,42 To assess initial ALL engraftment and during therapeutic
efficacy analyses, peripheral venous blood or splenocyte suspensions from vehicle or
PI3Ki-treated mice were erythrocyte-lysed, Fc-blocked, and stained with mouse anti-
human CD10-PE-Cy7, CD19-PE, and CD45-APC antibodies (eBioscience). Specimens
were analyzed on an Accuri C6 flow cytometer (BD Biosciences) with CountBright
absolute counting beads (ThermoFisher) for quantification of human leukemia cells as
described.32
Experimental randomization to treatment with vehicle or PI3Ki commenced when
all mice demonstrated ≥5% ALL in peripheral blood. Animals were sacrificed humanely
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

8
at planned study endpoint or if ill-appearing in accordance with the Panel on Euthanasia
of the American Veterinary Medical Association’s guidelines. Animal studies were
conducted under a CHOP Institutional Animal Use and Care Committee (IACUC)-
approved protocol.
In vivo therapeutic efficacy studies
For each of the PDX models, cohorts of 5 engrafted mice were randomized to
daily treatment with vehicle (0.5% hydroxypropyl methylcellulose/0.2% Tween-80 in
water via oral gavage), vehicle (0.3% lactic acid in 5% dextrose in water [D5W]
intraperitoneally), PI3Kαi BYL719 (50 mg/kg/dose via oral gavage), PI3Kδi idelalisib
(formerly CAL-101; 50 mg/kg/dose via oral gavage), PI3K/mTORi gedatolisib (formerly
PKI-587 or PF-05212384; 10 mg/kg intraperitoneally), or TORC1/2i AZD2014 (10 mg/kg
intraperitoneally).
For combination studies in CRLF2/JAK-mutant ALL models, animals were
treated as above with vehicle, gedatolisib, ruxolitinib (2 g/kg ruxolitinib-infused rodent
chow continuously provided), or simultaneous gedatolisib and ruxolitinib. For
combination studies in ABL/PDGFR-mutant ALL models, animals were treated with
vehicle, gedatolisib, dasatinib (10 mg/kg/dose twice daily via oral gavage), or
simultaneous gedatolisib and dasatinib. Studies were terminated and all mice sacrificed
after 28 days of treatment or sooner if ill-appearing. Spleens and bone marrow were
harvested as described for quantification of human ALL and for cryopreservation for
future studies.32
Survival analyses were conducted in some PDX models with vehicle, gedatolisib,
ruxolitinib, dasatinib, combined gedatolisib and ruxolitinib, or combined gedatolisib and
dasatinib treatment (n = 5 mice/cohort) as above for up to 120 days to assess potential
longer-term therapeutic efficacy of inhibitor therapies.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

9
Vehicle reagents were obtained from Sigma-Aldrich. BYL719, idelalisib, and
AZD2014 were purchased from Active Biochem. Gedatolisib and ruxolitinib were
provided by Pfizer and Incyte, respectively. Dasatinib was purchased from LC Labs.
Pharmacodynamic assessment of signal transduction inhibition
For measurements of in vivo signaling inhibition, additional PDX mice (n=3-4
animals/treatment) were administered vehicle or inhibitors as above for 72 hours, then
sacrificed at one hour following final inhibitor dose for leukemia cell-specific phosphoflow
cytometry analysis as described.32 Fixed and permeabilized splenocytes from vehicle-
and PI3Ki-treated animals were Fc-blocked and stained with mouse anti-human CD10-
PE-Cy7, CD19-APC-Cy7, and/or TSLPR-PE antibodies (BD Biosciences or
eBioscience). Cells were also stained with rabbit anti-human antibodies against
phosphorylated (p) 4EBP1T37/46 or pAktS473 (Cell Signaling Technologies; CST) with
secondary Pacific Blue-conjugated IgG H+L (Invitrogen/Life Technologies) and with
pS6S240/244-Ax488 (CST), pERKT202/Y204-Ax647 (BD Biosciences), or pSTAT5Y694-Ax647
(BD Biosciences). Data were compensated, acquired, and analyzed on an LSRII flow
cytometer (BD Biosciences) as described.29,32 Phosphosignaling data were gated and
measured using fluorescence-minus-one (FMO) controls for each phosphoprotein
fluorophore.43 Immunoblotting of splenic lysates from end-study animals (after 3 or 4
weeks of treatment) for phosphorylated pSTAT5Y694, pS6S240/244, 4EBP1T37/46,
corresponding total proteins, and β-actin (all antibodies from CST) was performed as
described for the combination inhibitor experiments.32 For experiments with dasatinib
treatment, immunoblotting was also performed for total and pCrkLY207 (CST), a
downstream effector of ABL-mediated signaling.44,45
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

10
Statistical analyses
For therapeutic efficacy studies, mean human ALL cell numbers in peripheral
blood of inhibitor- and vehicle-treated animals for each xenograft model were calculated
at each measured time point. Kaplan-Meier curves for survival analyses were compared
using the log-rank test. Human ALL cell number in murine spleens (efficacy studies) and
phosphoflow cytometry data for each phosphoprotein (pharmacodynamic studies) were
compared via ANOVA with Dunnett’s or Tukey’s post-test for multiple comparisons. All
statistical analyses were conducted using Prism version 6.0h for Mac (GraphPad).
RESULTS
Genetic Characterization of Ph-like ALL PDX models
LDA data confirmed the Ph-like signature in relevant PDX models (n=9 Ph-like,
n=1 non-Ph-like), which were concordant with gene expression signature patterns of
primary ALL specimens from which PDX models were derived (Supplemental Figure 1).
Known Ph-like driver lesions (e.g., CRLF2 rearrangements, JAK1 and JAK2 point
mutations, ABL1 or PDGFRB fusions) were also identified in all xenografted leukemias
by FISH and RT-PCR or PCR with Sanger sequencing, confirming genetic fidelity of
studied models (not shown).
PI3K pathway inhibitors robustly inhibit leukemia proliferation in Ph-like ALL
Based upon our prior studies demonstrating aberrant PI3K pathway signaling in
CRLF2-rearranged ALL and in vitro signaling inhibition with kinase inhibitors,29,32 we
hypothesized that “next generation” PI3Ki could robustly inhibit in vivo leukemia
proliferation and oncogenic signal transduction. We tested four clinically-available,
selective inhibitors of PI3K pathway proteins to identify the most potent therapeutic
approach(es) of targeting activated PI3K signaling in Ph-like ALL. In therapeutic efficacy
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

11
analyses, engrafted mice were treated daily with vehicle or PI3Ki for 2-4 weeks
depending upon the rapidity of leukemia progression in control animals. We assessed
tumor burden in peripheral blood during treatment (line graphs) and in spleens at the
conclusion of treatment (column graphs; Figure 1). In total, ten PDX models representing
genetically diverse subsets of Ph-like ALL were studied. Importantly, all PI3Ki-treated
models demonstrated inhibition of ALL proliferation in blood and spleens in comparison
to vehicle-treated control animals (Figure 1 and representative flow cytometry data in
Supplemental Figure 2). Inhibitor treatment appeared well-tolerated with normal physical
appearance and stability of weight in animals during studies (Supplemental Figure 3).
Therapeutic responses varied modestly among PDX models for each PI3Ki tested
depending upon the underlying genomic alteration(s) and the disease status of the
primary ALL specimen engrafted (de novo versus relapse).
Treatment with the PI3K/mTORi gedatolisib was remarkably efficacious in all
tested models with mean reduction in splenic ALL cell counts of 91.8% (range 82.5-
99.4%) in comparison to vehicle-treated mice (p<0.0001 for all models). Treatment with
the PI3Kαi BYL719, PI3Kδi idelalisib, or TORC1/TORC2i AZD2014 inhibited ALL
proliferation with mean leukemia reductions of 55.0% (range 27.5-72.6%), 38.1% (range
13.9-53.1%), and 52.8% (range 20.1-88.7%), respectively, versus vehicle controls.
Leukemia inhibition by the two isoform-selective PI3K inhibitors, BYL719 and idelalisib,
were generally similar with greater responses to BYL719 in some models (Figure 1).
Therapeutic responses to AZD2014 were greater in the two tested relapsed ALL models
(ALL121, ALL4364) in which high basal p4EBP1 activation was observed in the
pharmacodynamic studies reported below. Overall, these data suggest that Ph-like ALL
requires PI3K pathway signaling activation for survival in vivo and demonstrate potent
efficacy of the PI3K/mTORi gedatolisib in suppressing ALL cell growth.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

12
Pharmacodynamic inhibition of signaling proteins
We next addressed the question of whether PI3Ki could abrogate constitutive
PI3K pathway signaling in vivo and whether compensatory upregulation of alternative
pathways occurs after drug administration (Figure 2A). Long-term treatment of human
cancer cells with kinase inhibitors can lead to selection for alternative signaling
activation.39,46 Engrafted PDX mice were treated with vehicle or PI3Ki for 72 hours, then
sacrificed for immediate quantification of target protein phosphorylation. We first defined
basal activation of PI3K pathway phosphoproteins, pERK, and pSTAT5 by leukemia cell-
specific phosphoflow cytometry measurements and displayed data as the percentage of
cells in each positive FMO phosphoprotein gate (Figure 2B). Overall, constitutive
activation of pSTAT5, pS6, and pERK was observed in most models regardless of
underlying Ph-like genetic alteration(s). Basal activation of pAktS473 and p4EBP1 was
more heterogeneous across models (Figure 2C).
We then compared leukemia signaling in PI3Ki-treated animals to vehicle-treated
controls to assess potential in vivo phosphoprotein inhibition (Figure 3 and Supplemental
Figure 5). Treatment with the PI3Kα and PI3Kδ isoform-selective inhibitors BYL719 and
idelalisib resulted in significant inhibition of pS6 in 6/10 and 7/10 PDX models,
respectively (p<0.05 for each). Both PI3Ki also decreased p4EBP1 (1/10 models for
BYL719, 3/10 for idelalisib), pAkt (2/10 models for each inhibitor), and pERK (3/10
models for each inhibitor), suggesting potent inhibition of downstream mTORC1
signaling and potential crosstalk with or direct inhibition of the Ras/MAPK/ERK pathway
(Figure 3). The PI3K/mTORi gedatolisib also potently inhibited pS6 and p4EBP1 in vivo
in 8 of 10 models versus vehicle controls, as well as decreased pAktS473 (3/10 models)
and pERK (1 model) (p<0.05 for each; Figure 3). Importantly, minimal upregulation of
pAktS473 was observed in BYL719-, idelalisib-, or gedatolisib-treated animals (Figure 3),
suggesting generally effective blockade of potential TORC2-mediated feedback
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

13
signaling loops that may reactivate proximal PI3K pathway signaling. Similarly, treatment
with the TORC1/TORC2i AZD2014 significantly inhibited pS6 and p4EBP1 in 6 of 10
models (p<0.05). Surprisingly, minimal inhibition of the TORC2 phosphoprotein pAktS473
was observed in AZD2014-treated animals (1/10 models), although many PDX models
did not have basal pAktS473 activation (Figure 2C).
Consistent with our earlier studies,32 we observed marked constitutive pSTAT5
activation in all tested CRLF2/JAK-mutant or ABL/PDGFR-mutant Ph-like ALL PDX
models (Figure 2C). Perhaps surprisingly, in vivo inhibition of pSTAT5 occurred with
BYL719 (1 model), idelalisib (2 models), gedatolisib (2 models), and AZD2014 (1 model)
treatment (Figure 3). These data suggest potential off-target effects of PI3K pathway
inhibitors in these models and/or possible interconnection of PI3K/Akt/mTOR and
JAK/STAT signaling networks previously described in Ph-like ALL.29,30,32
Three conclusions can be drawn from these experiments. First, the remarkable
efficacy of gedatolisib treatment correlates with inhibition of pS6 and p4EBP1 in Ph-like
ALL models. Second, minimal upregulation of AktS473 occurs after PI3K pathway
inhibition with tested agents. Third, a complex interplay of signaling pathway regulation
apparently occurs after in vivo administration of PI3K pathway inhibitors, which may be
difficult to assess fully at a single optimized pharmacodynamic time point in these
preclinical models.
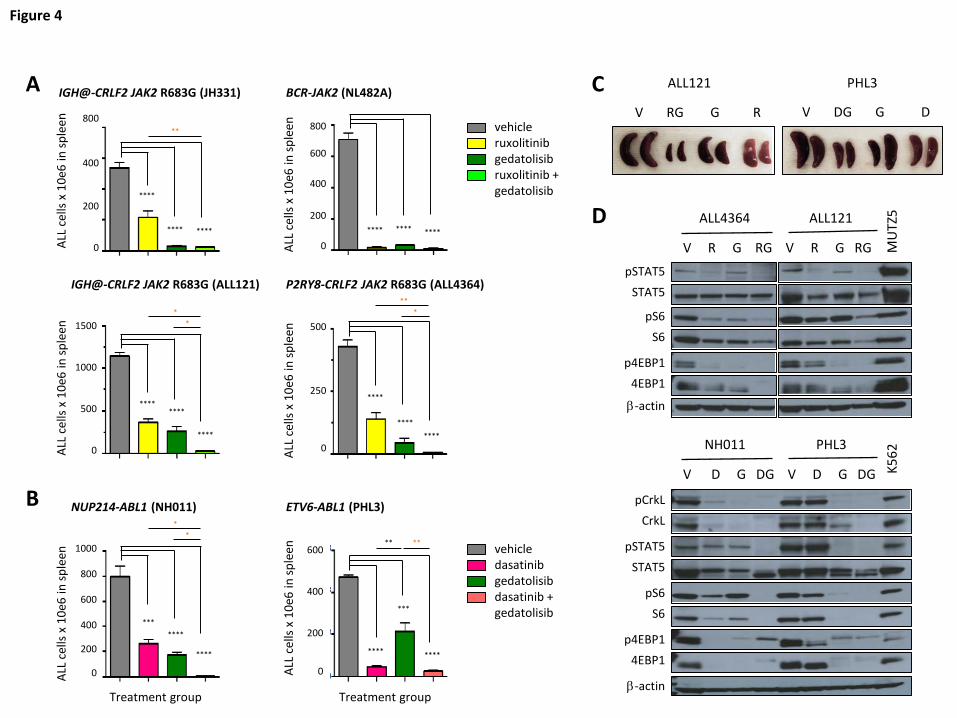
Superior anti-leukemia efficacy of combination signal transduction inhibitor treatment
Given the potent, but incomplete, therapeutic efficacy and pharmacodynamic
effects of PI3K/mTORi monotherapy in Ph-like ALL models, we further assessed the
potential for enhanced efficacy of gedatolisib when co-administered with ruxolitinib
(CRLF2/JAK-mutant models; Figure 4A) or dasatinib (ABL1/PDGFRB-mutant models;
Figure 4B). Indeed, combined inhibitor therapy resulted in superior inhibition of ALL
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

14
proliferation in murine spleens than was observed with inhibitors alone with near-
complete eradication of leukemia in most models (orange asterisks in Figures 4A-4B;
Figure 4C). Furthermore, greater in vivo inhibition of JAK/STAT, PI3K, and/or ABL
pathway signaling phosphoproteins was also observed with combination inhibitor
treatment, as shown by immunoblotting of splenic lysates from vehicle- and inhibitor-
treated animals after 3-4 weeks of treatment (Figure 4D). Combination inhibitor therapy
also appeared well-tolerated with stability of animal weights in most PDX models during
treatment (Supplemental Figure 4). Taken together, these data demonstrate enhanced
anti-leukemia efficacy of simultaneous inhibition of multiple signaling nodes in Ph-like
ALL and provide strong preclinical rationale to advance such treatment approaches to
clinical testing.
Survival benefit of inhibitor treatment
Given the robust potency of gedatolisib monotherapy and enhanced signaling
inhibition when combined with ruxolitinib or dasatinib, we next queried whether inhibitor
treatment of established leukemia could confer a survival benefit. To address this
question, we conducted longer-term survival analyses of vehicle, gedatolisib
monotherapy, and combination inhibitor treatment in four PDX ALL models (2
CRLF2/JAK, 2 ABL/PDGFR). Mice were first injected with human ALL cells and
monitored for engraftment. Once ≥5% leukemia was detected in peripheral blood, daily
treatment with vehicle or inhibitor(s) was commenced, and animals were followed by
weekly peripheral blood analysis as above.
As anticipated, gedatolisib monotherapy significantly prolonged survival versus
vehicle controls in all tested models (Figure 5; p<0.05 for each). Similar survival benefit
was also observed with ruxolitinib monotherapy treatment (Figure 5A; ALL121 and
JH331 models) and with dasatinib monotherapy treatment (Figure 5B; NH011 and PHL3
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

15
models). Enhanced survival was further observed with combined gedatolisib and
ruxolitinib or gedatolisib and dasatinib treatment for up to 120 days. When possible,
spleens from expiring animals were analyzed by flow cytometry, which confirmed
presence of residual or recurrent human leukemia. These studies thus further
demonstrate superior efficacy of combination kinase inhibitor approaches in Ph-like ALL.
DISCUSSION PI3K/Akt/mTOR Signal Transduction in Human Leukemias
PI3K pathway proteins play a central, highly complex role in the metabolic
regulation and survival of cancer cells. Constitutive activation of PI3K/Akt/mTOR
signaling occurs frequently in a variety of human cancers and likely contributes to
avoidance of normal apoptotic death mechanisms following cellular injury.47,48
Dysregulated PI3K/Akt/mTOR signaling has also been associated with resistance to
cytotoxic chemotherapy.49,50 However, the specific role and necessity of PI3K pathway
signaling in B-precursor ALL remains inadequately characterized.39
We previously observed constitutive PI3K/Akt/mTOR activation in childhood Ph-
like ALL and signaling inhibition with in vitro treatment of primary leukemia cells with
PI3K pathway inhibitors.29 We also previously demonstrated in vivo efficacy of
rapamycin treatment in some Ph-like ALL xenograft models also used in the current
study (JH331, JH612, NH362, NL482A, JL491), although therapeutic responses to
rapamycin were incomplete and did not appear to correlate with pharmacodynamic
inhibition of TORC1 phosphoproteins.32 In the current studies, we demonstrate that
treatment with the PI3K/mTORi gedatolisib profoundly inhibited in vivo Ph-like ALL
proliferation. Furthermore, extended gedatolisib treatment was well-tolerated in PDX
models and significantly prolonged animal survival. Treatment with BYL719 and
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

16
idelalisib reduced ALL burden by approximately half of that in vehicle-treated animals in
most models. The comparable efficacy of both PI3Kαi and PI3Kδi is perhaps surprising,
as B-cell lymphoid malignancies are not generally PI3KCA mutation-driven and have
higher expression of PI3Kδ.38,51 However, PI3Kα dependence of Ras-driven myeloid
leukemias and therapeutic activity of BYL719 in AML cell line xenograft models was
recently reported, providing additional rationale for further study of PI3Kαi in human
leukemias.52
Although development of PI3Ki resistance or upregulation of alternate signaling
pathways are plausible potential explanations for incomplete therapeutic efficacy, the
partial treatment responses we observed with BYL719 and idelalisib during relatively
short-duration animal experiments likely reflect at least in part known solubility issues for
oral gavage and poor pharmacokinetic properties of PI3K isoform-selective inhibitors in
mice.38,51,53 Similarly, the TORC1/TORC2i AZD2014 moderately inhibited leukemia
proliferation in CRLF2/JAK-mutant ALL PDX models, particularly in two aggressive
relapsed ALL models (ALL121, ALL4364), which may correlate with basal activation of
pAktS473. Interestingly, no appreciable reduction in leukemia burden was observed with
AZD2014 treatment in the tested BCR-JAK2 fusion model (NL482A) or ABL/PDGFR-
mutant models (NL432, NH011, PHL3) despite constitutive mTORC1 activation (pS6
and p4EBP1). However, minimal basal TORC2 signaling activation (pAktS473) was
detected in these four models. These data suggest biologic differences and signaling
dependencies among the genetically heterogeneous Ph-like ALL subgroups, as well as
highlight potential limitations of our pharmacodynamic assays with respect to
measurement of target inhibition of multiple phosphoproteins at a single time point in
animals treated with various inhibitors. Additional studies are indicated to assess such
response differences.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

17
As anticipated, we observed augmented anti-leukemic efficacy with simultaneous
targeting of two discrete oncogenic pathways in Ph-like ALL. Firstly, gedatolisib
monotherapy demonstrated markedly greater anti-leukemia efficacy in these studies in
comparison to prior studies testing single-agent ruxolitinib or dasatinib in Ph-like ALL
models.12,14,27,32 Interestingly, while highly effective in all tested models, gedatolisib more
potently inhibited ALL proliferation in CRLF2/JAK models than in ABL/PDFGR models,
highlighting potential differential signaling dependencies driven by underlying Ph-like
genetic alterations. In addition, combined gedatolisib and ruxolitinib or gedatolisib and
dasatinib treatment of CRLF2/JAK or ABL/PDGFR models, respectively, resulted in
superior therapeutic efficacy, pharmacodynamic phosphoprotein inhibition, and survival
benefit. Such dual pathway-targeting approaches may also minimize potential for
compensatory signaling upregulation that is a common mechanism of treatment
resistance.38,39
In pharmacodynamic studies, we observed few increased signaling sequelae in
animals treated with PI3Ki. It is plausible that dynamic signaling changes occur over a
longer treatment time period than 4 weeks or in other phosphoproteins than were
evaluated in our studies. However, use of next generation inhibitors that target proximal
PI3K pathway proteins or “vertically” inhibit multiple PI3K pathway nodes may more
potently shut down aberrant PI3K signaling in Ph-like ALL and perhaps ultimately evoke
superior therapeutic efficacy.
Our results are consistent with studies by Badura and colleagues, who assessed
effects of various PI3K pathway inhibitors incubated in vitro with primary BCR-ABL1-
rearranged and non-rearranged B-ALL cells.54 They also observed greatest anti-
proliferative and pro-apoptotic activity of dual PI3K/mTOR inhibition (BGT-226, BEZ-235)
in ALL cells versus pan-PI3K (BKM-120), mTOR (everolimus), or TORC1/TORC2 (Torin
1, PP242, KU-0063794) inhibition. Cytotoxic effects of the various inhibitors were similar
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

18
among Ph+ and non-Ph+ ALL specimens in their study, suggesting that PI3Ki efficacy
may not be restricted to specific ALL subsets. Similarly, Kharas et al. and Wong et al.
demonstrated potent in vitro cytotoxicity of PI3K/mTORi incubated with murine or human
ALL cell lines, as well as reduction in leukemia burden and prolonged survival in murine
and human ALL cell line-engrafted mice treated with PI3K/mTORi.55,56 We also observed
greatest phosphosignaling inhibition with PI3K/mTORi (gedatolisib) without upregulation
of pAktS473, suggesting potent blockade of the PI3K pathway and consistent with
preclinical data from Mallon et al.57
Unlike the Badura study, we did not observe appreciably increased TORC2-
mediated pAktS473 with PI3K isoform-selective or TORC1/TORC2 inhibitor treatment,
possibly due to differences in specific PI3Ki drugs tested and in vivo versus in vitro
systems analyzed. The lack of observed pS6 and p4EBP1 inhibition in a few PDX
models may also reflect characteristics of utilized phosphoantibody reagents,
mechanism(s) of action of each PI3Ki, and dynamic signaling networks evaluated in vivo
at a single time point. Although we did not specifically investigate selective Akt inhibition
given mixed preclinical and clinical data to date,50 our observed basal activation of pAkt
in some Ph-like ALL models and recent data demonstrating reversal of steroid
resistance in T-ALL with newer Akt inhibitors may warrant future study of these agents.58
We thus demonstrate that activated PI3K pathway signaling is prevalent in Ph-
like ALL and can be effectively targeted in vivo using clinically available kinase inhibitors.
We report particularly potent efficacy of the PI3K/mTORi gedatolisib as monotherapy
and enhanced efficacy in combination with ruxolitinib or dasatinib in specific genetic
subsets of Ph-like ALL. Our pharmacodynamic assays also identified constitutive basal
activation of PI3K and JAK/STAT phosphoproteins in Ph-like ALL, which may be
informative biomarkers to guide precision medicine-focused clinical selection of specific
kinase inhibitors for individual leukemias. However, significant and/or dose-limiting
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

19
toxicities of various PI3K pathway inhibitors in adults with cancer have been reported, as
well as modest clinical activity of some inhibitor monotherapies.38,59,60 Careful evaluation
of the biochemical mechanisms and anti-leukemia efficacy of PI3K/mTORi and their
toxicity profiles, particularly in combination with JAKi or ABLi, is warranted to facilitate
rapid clinical translation of novel kinase inhibitor-based therapies for children and adults
with Ph-like ALL.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

20
ACKNOWLEDGMENTS We thank Dr Miles Pufall (University of Iowa) and Dr David Fruman (University of
California, Irvine) for thoughtful discussions about this work and Dr Shannon Maude
(Children’s Hospital of Philadelphia) and Dr Elliot Stieglitz (University of California, San
Francisco) for assistance with experimental reagents. Support for these studies was
from a National Institutes of Health/National Cancer Institute (NIH/NCI) Mentored Career
Development Award K08CA184418 (SKT), an NIH/NCI Paul Calabresi Career
Development Award for Clinical Oncology award to the University of Pennsylvania
K12CA076931 (SKT), an NIH/National Center for Research Resources award to the
University of Pennsylvania Institute for Translational Medicine and Therapeutics
UL1RR024134 (SKT), Alex’s Lemonade Stand Foundation (ALSF) Young Investigator
and Center of Excellence awards (SKT), the Rally Foundation for Childhood Cancer
Research (SKT), and the Leukemia and Lymphoma Society Specialized Center of
Research Program (DTT and SAG). This work was also supported by grants to the
Children’s Oncology Group, specifically U10 CA98543 (Chair’s grant and supplement to
support the COG High Risk ALL TARGET Project), U10 CA98413 (Statistical Center),
and U24 CA114766 (specimen banking). SKT was an ALSF Scholar in Developmental
Therapeutics. DTT is supported by a Research Scholar Grant (RSG-14-022-01-CDD)
from the American Cancer Society. CLW is the Maurice and Marguerite Liberman
Distinguished Chair in Cancer Research� Professor of Pathology and Medicine. SPH is
the Jeffrey E. Perelman Distinguished Chair in the Department of Pediatrics. MLL is the
Deborah and Arthur Ablin Chair in Molecular Pediatric Oncology. MC is supported by
Veterans Administration Merit Award 1I01BX000918. SAG is the Yetta Deitch Novotny
Endowed Chair in Pediatric Oncology.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

21
AUTHORSHIP CONTRIBUTIONS
SKT designed and directed research, performed research, analyzed data, and wrote the
manuscript. DTT and CLW provided key reagents and interpreted data. YL, FS, RCH,
TR, TLV, and I-MC performed research and analyzed and interpreted data. AEP, SPH,
MLL, MC, and SAG contributed to research design and data interpretation. SPH and MC
edited the manuscript. All authors critically reviewed the manuscript prior to submission.
CONFLICT OF INTEREST DISCLOSURES
SKT and MC have a research collaboration with Incyte Corporation without financial
support. RCH, I-MC, CLW, and SPH are co-inventors on US Patent No. 8,568,974 B2
“Identification of novel subgroups of high-risk pediatric precursor-B acute lymphoblastic
leukemia, outcome correlations and diagnostic and therapeutic methods related to
same.” This technology has not been licensed, and there is no income. The remaining
authors declare no relevant competing financial interests for this work.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom
22
TABLES
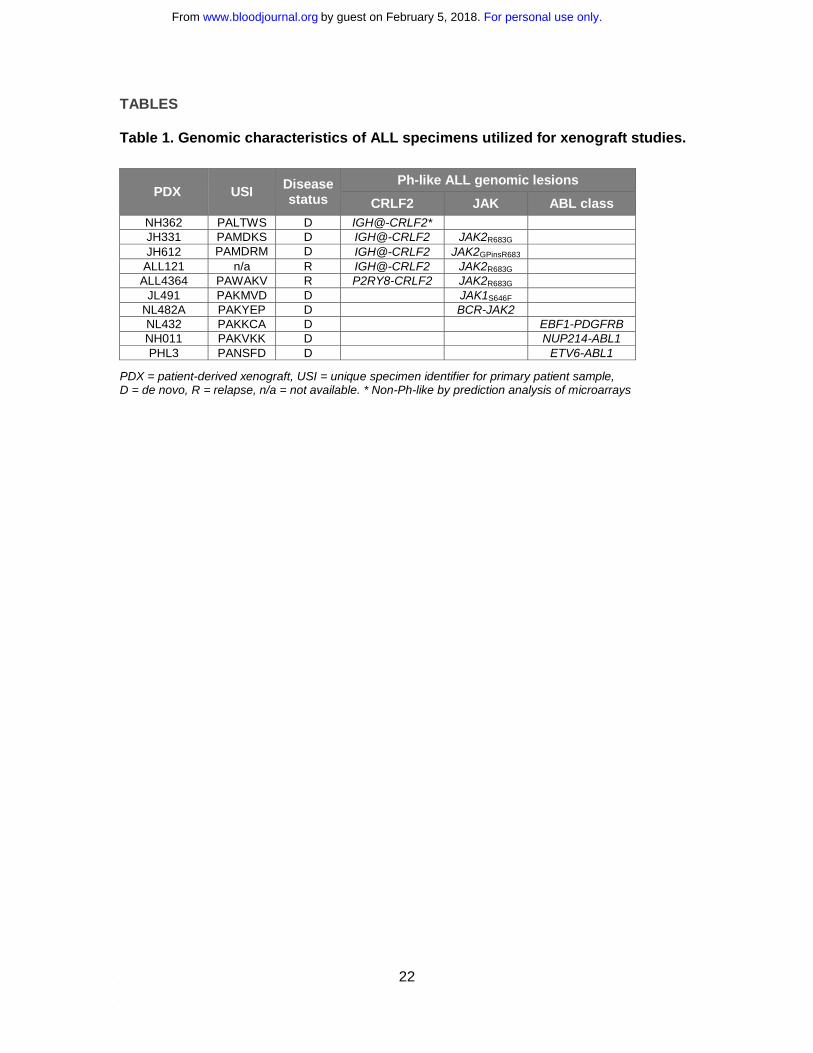
Table 1. Genomic characteristics of ALL specimens utilized for xenograft studies.
PDX = patient-derived xenograft, USI = unique specimen identifier for primary patient sample, D = de novo, R = relapse, n/a = not available. * Non-Ph-like by prediction analysis of microarrays
PDX USI Disease status
Ph-like ALL genomic lesions
CRLF2 JAK ABL class NH362 PALTWS D IGH@-CRLF2* JH331 PAMDKS D IGH@-CRLF2 JAK2R683G JH612 PAMDRM D IGH@-CRLF2 JAK2GPinsR683
ALL121 n/a R IGH@-CRLF2 JAK2R683G ALL4364 PAWAKV R P2RY8-CRLF2 JAK2R683G
JL491 PAKMVD D JAK1S646F NL482A PAKYEP D BCR-JAK2 NL432 PAKKCA D EBF1-PDGFRB NH011 PAKVKK D NUP214-ABL1 PHL3 PANSFD D ETV6-ABL1
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

23
FIGURE LEGENDS
Figure 1. In vivo efficacy of PI3K pathway inhibition in Ph-like ALL. Patient-derived
xenograft (PDX) models were treated with vehicle, PI3Kα inhibitor BYL719,
PI3Kδ inhibitor idelalisib, PI3K/mTOR inhibitor gedatolisib, or TORC1/TORC2 inhibitor
AZD2014 (n=5 mice/treatment arm) daily for 2-4 weeks depending on rapidity of
leukemia progression in vehicle-treated control animals. PI3K isoform or TORC1/TORC2
inhibition resulted in significant suppression of leukemia proliferation in peripheral blood
and end-of-study spleens of most PDX models of (A) CRLF2-rearranged, (B) JAK1-
mutant, (C) JAK2 fusion, and (D) ABL/PDGFR-mutant ALL versus vehicle controls.
Gedatolisib most effectively inhibited ALL proliferation with near-eradication of leukemia
in CRLF2/JAK-mutant models and marked suppression in ABL/PDGFR-mutant models.
Treatment groups were compared to the vehicle control for each PDX model via one-
way ANOVA with Dunnett’s post-test for multiple comparisons with α=0.05. *p<0.05,
**p<0.01, *** p<0.001, **** p<0.0001.
Figure 2. Constitutive activation of signaling phosphoproteins in Ph-like ALL PDX
models. (A) Schema of signaling nodes assessed and targets of kinase inhibitors. (B)
Gating strategy for phosphoflow cytometry analyses: live singlet CD10+ CD19+ (and
TSLPR+ if CRLF2-rearranged) human ALL cells in murine spleens were gated.
Fluorescence-minus-one (FMO) controls were used for each fluorophore-conjugated
phosphoprotein antibody to set negative (FMO-) and positive (FMO+) gates. The
percentage of cells in the FMO+ gate was calculated for all phosphoproteins for each
STI treatment and displayed graphically. All studied ALL samples were uniformly CD10+.
(C) Basal levels of each measured phosphoprotein in vehicle-treated mice from each
PDX model are displayed as percentages of human ALL cells in FMO+ gates. Data are
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

24
depicted for each PDX model (colored columns) as mean percentage of cells in FMO+
gate on the y-axis with standard error of the mean (black bars).
Figure 3. PI3K pathway inhibitors inhibit signaling phosphoproteins and induce
minimal compensatory signaling upregulation. Ph-like ALL PDX models were treated
with vehicle, BYL719, idelalisib, gedatolisib, or AZD2014 (n=3-4 mice/treatment) for 72
hours, then sacrificed at 1 hour after final dose for pharmacodynamic measurement of in
vivo target inhibition via phosphoflow cytometry. Heatmap data depict significant
changes in phosphoprotein levels in inhibitor- versus vehicle-treated controls using
%FMO+ cells (described in Figure 2) for each PDX model. Colorimetric scale depicts
normalization of each model to vehicle controls (white squares; left-most columns) with
statistically significant decreased phosphorylation (blue colors) or increased
phosphorylation (green colors) with inhibitor treatment as calculated by ANOVA with
Dunnett’s post-test for multiple comparisons. FMO+ data for all individual mice are
displayed in greater detail in Supplemental Figure 5.
Figure 4. Superior efficacy of combination STI treatment. Mice engrafted with (A)
CRLF2/JAK-mutant Ph-like ALL were treated with vehicle, ruxolitinib, gedatolisib, or both
ruxolitinib and gedatolisib for 3 or 4 weeks. Human ALL in murine spleens after
treatment completion was measured by quantitative flow cytometry as in Figure 1.
Significantly greater inhibition of ALL proliferation was observed with combined
ruxolitinib and gedatolisib treatment (orange asterisks), as measured by one-way
ANOVA with Tukey’s post-test for multiple comparisons with α=0.05. (B) ABL1-
rearranged Ph-like ALL PDX models were treated with vehicle, dasatinib, gedatolisib, or
both dasatinib and gedatolisib for 3 weeks. Enhanced anti-leukemia efficacy was also
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

25
observed in these models with combined dasatinib and gedatolisib treatment (orange
asterisks) versus dasatinib alone or gedatolisib alone. (C) Combination inhibitor
treatment markedly reduced splenomegaly in ALL-engrafted PDX mice treated as in (A)
or (B). (D) Immunoblotting of total and phosphorylated signal transduction proteins from
murine splenic lysates (obtained after 3 or 4 weeks of treatment) demonstrate greatest
inhibition of target phosphoproteins with combination inhibitor treatment of PDX models.
Total protein loss is observed with inhibitor treatment in some models. MUTZ5 (a
CRLF2/JAK2-mutant ALL cell line) and K562 (a BCR-ABL1-rearranged chronic myeloid
leukemia cell line) lysates were used as positive signaling controls, and β-actin
immunoblotting was used as a protein loading control. V=vehicle, R=ruxolitinib,
G=gedatolisib, RG= ruxolitinib + gedatolisib combination, D=dasatinib, DG=dasatinib +
gedatolisib combination. *p<0.05, **p<0.01, *** p<0.001, **** p<0.0001.
Figure 5. Survival advantage with inhibitor monotherapy and combination
inhibitor treatment. Animals engrafted with (A) relapsed (ALL121) or de novo (JH331)
CRLF2/JAK-mutant Ph-like ALL were treated with vehicle, ruxolitinib chow, gedatolisib
10 mg/kg intraperitoneally daily, or both ruxolitinib and gedatolisib until moribund (n=5
mice/treatment cohort) for up to 120 days. Similarly, animals engrafted with (B) de novo
ABL1-mutant ALL (NH011, PHL3) were treated with vehicle, dasatinib 10 mg/kg twice
daily via oral gavage, gedatolisib, or both dasatinib and gedatolisib for up to 120 days. X-
axes depict duration of inhibitor treatment (week 0 = treatment initiation) in mice after
documentation of human leukemia engraftment (≥5% ALL in peripheral blood). Kaplan-
Meier survival curves for each xenograft model were compared statistically using the
log-rank test (p-values indicated on graphs for each model). Dotted lines delineate
median survival in each study with corresponding values for listed each treatment (black
numbers).
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

26
REFERENCES
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70. 2. Teitell MA, Pandolfi PP. Molecular genetics of acute lymphoblastic leukemia.
Annu Rev Pathol. 2009;4:175-198. 3. Tasian SK, Loh ML, Hunger SP. Childhood acute lymphoblastic leukemia:
Integrating genomics into therapy. Cancer. 2015;121(20):3577-3590. 4. Pui C, Robison L, Look A. Acute lymphoblastic leukaemia. Lancet.
2008;371(9617):1030-1043. 5. Jeha S, Pui C. Risk-adapted treatment of pediatric acute lymphoblastic leukemia.
Hematol Oncol Clin North Am. 2009;23(5):973-990. 6. Schultz KR, Pullen DJ, Sather HN, et al. Risk- and response-based classification
of childhood B-precursor acute lymphoblastic leukemia: a combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children's Cancer Group (CCG). Blood. 2007;109(3):926-935.
7. Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children's Oncology Group study. Leukemia. 2008;22(12):2142-2150.
8. Fielding AK, Richards SM, Chopra R, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109(3):944-950.
9. Sive JI, Buck G, Fielding A, et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG2993 trial. Br J Haematol. 2012;157(4):463-471.
10. Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. The Lancet Oncology. 2009;10(2):125-134.
11. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. The New England Journal of Medicine. 2009;360(5):470-480.
12. Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153-166.
13. Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood. 2013;121(3):485-488.
14. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. The New England Journal of Medicine. 2014;371(11):1005-1015.
15. Izraeli S. Beyond Philadelphia: 'Ph-like' B cell precursor acute lymphoblastic leukemias - diagnostic challenges and therapeutic promises. Current Opinion in Hematology. 2014;21(4):289-296.
16. Roberts KG, Pei D, Campana D, et al. Outcomes of Children With BCR-ABL1-Like Acute Lymphoblastic Leukemia Treated With Risk-Directed Therapy Based on the Levels of Minimal Residual Disease. J Clin Oncol. 2014.
17. van der Veer A, Waanders E, Pieters R, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122(15):2622-2629.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

27
18. Boer JM, Koenders JE, van der Holt B, et al. Expression profiling of adult acute lymphoblastic leukemia identifies a BCR-ABL1-like subgroup characterized by high non-response and relapse rates. Haematologica. 2015;100(7):e261-264.
19. Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243-1246.
20. Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688-2698.
21. Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010.
22. Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. The Journal of Experimental Medicine. 2011;208(5):901-908.
23. Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008;372(9648):1484-1492.
24. Mullighan CG, Zhang J, Harvey RC, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106(23):9414-9418.
25. Gaikwad A, Rye CL, Devidas M, et al. Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol. 2009;144(6):930-932.
26. Hertzberg L, Vendramini E, Ganmore I, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115(5):1006-1017.
27. Iacobucci I, Li Y, Roberts KG, et al. Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell. 2016;29(2):186-200.
28. Schwab C, Ryan SL, Chilton L, et al. EBF1-PDGFRB fusion in paediatric B-cell precursor acute lymphoblastic leukaemia (BCP-ALL): genetic profile and clinical implications. Blood. 2016.
29. Tasian SK, Doral MY, Borowitz MJ, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120(4):833-842.
30. Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010;107(1):252-257.
31. Tasian SK, Loh ML. Understanding the biology of CRLF2-overexpressing acute lymphoblastic leukemia. Crit Rev Oncog. 2011;16(1-2):13-24.
32. Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012;120(17):3510-3518.
33. Suryani S, Bracken LS, Harvey RC, et al. Evaluation of the In Vitro and In Vivo Efficacy of the JAK Inhibitor AZD1480 against JAK-Mutated Acute Lymphoblastic Leukemia. Molecular Cancer Therapeutics. 2015;14(2):364-374.
34. Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

28
lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013;98(11):e146-148.
35. Weston BW, Hayden MA, Roberts KG, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol. 2013;31(25):e413-416.
36. Tasian SK, Loh ML, Rabin KR, et al. A Phase 1 Study of Ruxolitinib in Children with Relapsed/Refractory Solid Tumors, Leukemias, or Myeloproliferative Neoplasms: A Children's Oncology Group Phase 1 Consortium Study (ADVL1011). J Clin Oncol. 2014;32(5s):American Society of Clinical Oncology Annual Meeting 2014 abtract 10019.
37. O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Research. 2006;66(3):1500-1508.
38. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews Drug discovery. 2014;13(2):140-156.
39. Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015;3:2.
40. Harvey RC, Kang H, Roberts KG, et al. Development and Validation Of a Highly Sensitive and Specific Gene Expression Classifier To Prospectively Screen and Identify B-Precursor Acute Lymphoblastic Leukemia (ALL) Patients With a Philadelphia Chromosome-Like (“Ph-like” or “BCR-ABL1-Like”) Signature For Therapeutic Targeting and Clinical Intervention. Blood. 2013;122(21):ASH Annual Meeting 2013 abstract #2826.
41. Reshmi SC, Harvey RC, Smith A, et al. Abstract 4729: Frequency of actionable gene fusions in patients with Philadelphia chromosome-like (Ph-like) B-acute lymphoblastic leukemia (ALL): A retrospective study from the Children's Oncology Group (COG). Cancer research. 2015;75(15 Supplement):4729.
42. Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759-1767.
43. Perl AE, Kasner MT, Shank D, Luger SM, Carroll M. Single-cell pharmacodynamic monitoring of S6 ribosomal protein phosphorylation in AML blasts during a clinical trial combining the mTOR inhibitor sirolimus and intensive chemotherapy. Clin Cancer Res. 2012;18(6):1716-1725.
44. ten Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J. Tyrosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood. 1994;84(6):1731-1736.
45. La Rosee P, Holm-Eriksen S, Konig H, et al. Phospho-CRKL monitoring for the assessment of BCR-ABL activity in imatinib-resistant chronic myeloid leukemia or Ph+ acute lymphoblastic leukemia patients treated with nilotinib. Haematologica. 2008;93(5):765-769.
46. Gioeli De. The dynamics of the cell signaling network; implications for targeted therapies. Targeted Therapies: Mechanisms of Resistance: Springer 2011:33-53.
47. Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17(3):590-603.
48. Martelli AM, Evangelisti C, Follo MY, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cells. Curr Med Chem. 2011;18(18):2715-2726.
49. Martelli AM, Tazzari PL, Evangelisti C, et al. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

29
leukemia therapy: from bench to bedside. Curr Med Chem. 2007;14(19):2009-2023.
50. Kim D, Dan HC, Park S, et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci. 2005;10:975-987.
51. Fruman DA, Cantley LC. Idelalisib--a PI3Kdelta inhibitor for B-cell cancers. The New England journal of medicine. 2014;370(11):1061-1062.
52. Gritsman K, Yuzugullu H, Von T, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. The Journal of Clinical Investigation. 2014;124(4):1794-1809.
53. Fritsch C, Huang A, Chatenay-Rivauday C, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Molecular Cancer Therapeutics. 2014;13(5):1117-1129.
54. Badura S, Tesanovic T, Pfeifer H, et al. Differential effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway in acute lymphoblastic leukemia. PloS one. 2013;8(11):e80070.
55. Kharas MG, Janes MR, Scarfone VM, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. The Journal of Clinical Investigation. 2008;118(9):3038-3050.
56. Wong J, Welschinger R, Hewson J, Bradstock KF, Bendall LJ. Efficacy of dual PI-3K and mTOR inhibitors in vitro and in vivo in acute lymphoblastic leukemia. Oncotarget. 2014.
57. Mallon R, Feldberg LR, Lucas J, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011;17(10):3193-3203.
58. Piovan E, Yu J, Tosello V, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24(6):766-776.
59. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550-562.
60. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143-153.
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

A
vehicle
B
C
D
Figure 1
BYL719 idelalisib gedatolisib AZD2014
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

Figure 2
A
C
Basa
l pho
spho
prot
ein
activ
atio
n (%
FMO
+)B
NH3
62JH
331
JH61
2AL
L121
ALL4
364
JL49
1N
L482
AN
L432
NH0
11PH
L3
pAkt S473p4EBP1pS6 pERK pSTAT5
NH3
62JH
331
JH61
2AL
L121
ALL4
364
JL49
1N
L482
AN
L432
NH0
11PH
L3
NH3
62JH
331
JH61
2AL
L121
ALL4
364
JL49
1N
L482
AN
L432
NH0
11PH
L3
NH3
62JH
331
JH61
2AL
L121
ALL4
364
JL49
1N
L482
AN
L432
NH0
11PH
L3
NH3
62JH
331
JH61
2AL
L121
ALL4
364
JL49
1N
L482
AN
L432
NH0
11PH
L3
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

Figure 3
pS6
vehi
cle
BYL7
19id
elal
isib
geda
tolis
ibAZ
D201
4
p4EBP1
vehi
cle
BYL7
19id
elal
isib
geda
tolis
ibAZ
D201
4
NH362JH331JH612
ALL121ALL4364
JL491NL482A
NL432NH011
PHL3
pAkt S473
vehi
cle
BYL7
19id
elal
isib
geda
tolis
ibAZ
D201
4
pERK
vehi
cle
BYL7
19id
elal
isib
geda
tolis
ibAZ
D201
4
pSTAT5
vehi
cle
BYL7
19id
elal
isib
geda
tolis
ibAZ
D201
4
p <0.0001p <0.001p <0.01p <0.05no changep <0.05p <0.01p <0.001p <0.0001
Decreasedphosphorylation
Increasedphosphorylation
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom
IGH@-CRLF2 JAK2 R683G (JH331)
800
400
200
0ALL
cells
x 1
0e6
in sp
leen
****
**** ****
BCR-JAK2 (NL482A)
800
600
400
200
0ALL
cells
x 1
0e6
in sp
leen
**** **** ****
IGH@-CRLF2 JAK2 R683G (ALL121)
ALL
cells
x 1
0e6
in sp
leen
********
****
1500
1000
500
0
P2RY8-CRLF2 JAK2 R683G (ALL4364)AL
L ce
lls x
10e
6 in
sple
en
500
250
0
****
****
****
NUP214-ABL1 (NH011)
Treatment group
ALL
cells
x 1
0e6
in sp
leen
1000
800
600
400
200
0
*******
****
ETV6-ABL1 (PHL3)
Treatment group
600
400
200
0ALL
cells
x 1
0e6
in sp
leen
****
***
****
vehicleruxolitinibgedatolisibruxolitinib + gedatolisib
vehicledasatinibgedatolisibdasatinib + gedatolisib
A C
B
D
pSTAT5STAT5
V R G RG V R G RG
ALL4364 ALL121
MU
TZ5
pS6S6
p4EBP14EBP1
β-actin
V RG G R V DG G D
ALL121 PHL3
V D G DG V D G DG
NH011 PHL3
K562
pSTAT5STAT5
pS6S6
p4EBP14EBP1
β-actin
pCrkLCrkL
Figure 4
***
**
**
**
****
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom
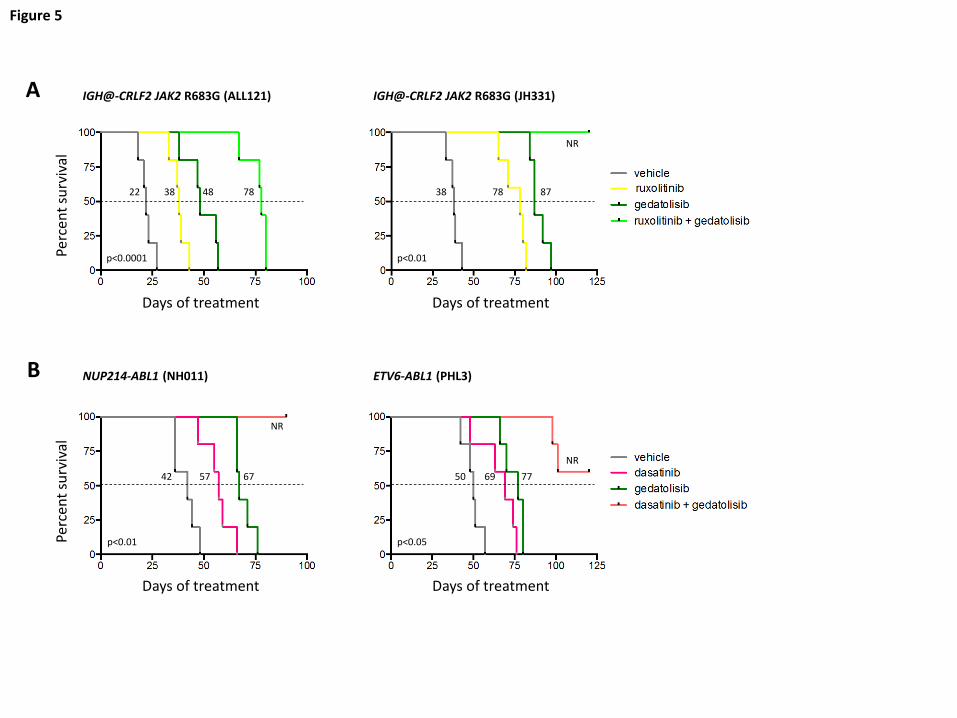
Figure 5
Days of treatment
Perc
ent s
urvi
val
A
B
Days of treatment
Perc
ent s
urvi
val
IGH@-CRLF2 JAK2 R683G (JH331)IGH@-CRLF2 JAK2 R683G (ALL121)
NUP214-ABL1 (NH011) ETV6-ABL1 (PHL3)
Days of treatment Days of treatment
p<0.01
p<0.01
p<0.0001
22 38 48 78 38 78 87
42 57 67 50 69 77
NR
NR
p<0.05
NR
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

doi:10.1182/blood-2016-05-707653Prepublished online October 24, 2016;
Carroll and Stephan A. GruppTiffaney L. Vincent, Cheryl L. Willman, Alexander E. Perl, Stephen P. Hunger, Mignon L. Loh, Martin Sarah K. Tasian, David T. Teachey, Yong Li, Feng Shen, Richard C. Harvey, I-Ming Chen, Theresa Ryan, murine xenograft models of Ph-like acute lymphoblastic leukemiaPotent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. indexed by PubMed from initial publication. Citations to Advance online articles must include final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of
For personal use only.on February 5, 2018. by guest www.bloodjournal.orgFrom

![The Dual PI3K/mTOR Inhibitor NVP-BEZ235 Is a Potent ... · actively developed for tumor therapy (reviewed in Liu et al. [1] and Garcia-Echeverria and Sellers [2]). Dual PI3K-mTOR](https://static.fdocuments.net/doc/165x107/60220782d84f464be711e04e/the-dual-pi3kmtor-inhibitor-nvp-bez235-is-a-potent-actively-developed-for-tumor.jpg)
















