
Current Topics in Medicinal Chemistry 949-961 9 4 9 Macrolide … · 2016-06-15 · Macrolide...
12
Current Topics in Medicinal Chemistry 2003, 3, 949-961 949 1568-0266/03 $41.00+.00 © 2003 Bentham Science Publishers Ltd. Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance Marne Gaynor and Alexander S. Mankin* Center for Pharmaceutical Biotechnology – M/C 870, University of Illinois, 900 S. Ashland Ave., Chicago, IL 60607, USA Abstract: Macrolides are among the most clinically important antibiotics. However, many aspects of macrolide action and resistance remain obscure. In this review we summarize the current knowledge, as well as unsolved questions, regarding the principles of macrolide binding to the large ribosomal subunit and the mechanism of drug action. Two mechanisms of macrolide resistance, inducible expression of Erm methyltransferase and peptide-mediated resistance, appear to depend on specific interactions between the ribosome-bound macrolide molecule and the nascent peptide. The similarity between these mechanisms and their relation to the general mode of macrolide action is discussed and the discrepancies between currently available data are highlighted. Key words: macrolides, erythromycin, ketolides, azithromycin, clarithromycin., tylosin, carbomycin, spiramycin, ribosome, resistance. INTRODUCTION Macrolides belong to one of the most commonly used families of clinically important antibiotics used to treat infections caused by Gram-positive bacteria such as Staphylococcus aureus, Streptococcus pneumoniae and Streptococcus pyogenes . Chemically, macrolides are represented by a 14-, 15- or 16-membered lactone ring carrying one or more sugar moieties and additional substitutions linked to various atoms of the lactone ring, Fig (1). Erythromycin A, a 14-membered ring drug, was the first clinically used macrolide. Drug delivery problems resulting from acid instability, prompted the design of newer macrolides. Increased acid stability as well as an increase in the range of antimicrobial activity characterized the second generation, which includes 14-membered ring drugs, such as clarithromycin and roxithromycin as well as the 15- membered ring azithromycin. In addition, the 16-membered ring macrolides such as tylosin, carbomycin A and spiramycin and others also exhibited significant antimicrobial activity, and were originally thought to be the answer to the growing occurrence of erythromycin resistant infections. Unfortunately, all of these drugs became prone to the selection of resistant strains. The newest generation of macrolides, the ketolides, whose clinical use is in its early stage, are characterized by improved activity against some of the resistant strains. It has been well documented that macrolides bind to the large ribosomal subunit in the vicinity of the peptidyl *Address correspondence to this author at the Center for Pharmaceutical Biotechnology – M/C 870, University of Illinois, 900 S. Ashland Ave., Chicago, IL 60607, USA; Tel: 312-413-1406; Fax: 312-413-9303; E-mail: [email protected] transferase center and cause cell growth arrest due to inhibition of protein synthesis [1,2]. However, in spite of more than 50 years of research, the mode of macrolide inhibition of ribosome activity is understood only in the most general terms. The extensive use of these antibiotics has led inevitably to the spread of resistant strains. Expression of some of the resistance determinants is inducible by macrolides. Of particular interest are Erm methyltransferases, which specifically methylate a unique nucleotide within the macrolide binding site. The mechanism of Erm induction depends on ribosome stall within the translated regulatory open reading frame preceding the Erm cistron, and is apparently closely related to the general mode of macrolide action on protein synthesis. However, the details of the mechanism of Erm induction are not known. In addition, a novel mechanism of macrolide resistance has recently been described which is mediated by the expression of specific short peptides in the cell. Although it is not entirely clear how expression of short peptides can render cells resistant to macrolides, the underlying principles maybe related to the mechanisms of inducible resistance and the mode of drug-induced inhibition of translation. In this brief review we try to summarize the current understanding of the mechanism of macrolide action, and the molecular mechanisms of inducible erm expression and peptide-mediated macrolide resistance. Several reviews dealing with these subjects appeared in the past and we refer the reader interested in specific details to those publications [1-4]. Instead, we will try to focus on the apparent discrepancies and less clearly defined details in the existing models. We strongly believe that understanding these discrepancies exist and finding ways to resolve them will
Transcript of Current Topics in Medicinal Chemistry 949-961 9 4 9 Macrolide … · 2016-06-15 · Macrolide...

Current Topics in Medicinal Chemistry 2003, 3, 949-961 949
1568-0266/03 $41.00+.00 © 2003 Bentham Science Publishers Ltd.
Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance
Marne Gaynor and Alexander S. Mankin*
Center for Pharmaceutical Biotechnology – M/C 870, University of Illinois, 900 S. AshlandAve., Chicago, IL 60607, USA
Abstract: Macrolides are among the most clinically important antibiotics. However,many aspects of macrolide action and resistance remain obscure. In this review wesummarize the current knowledge, as well as unsolved questions, regarding theprinciples of macrolide binding to the large ribosomal subunit and the mechanism ofdrug action. Two mechanisms of macrolide resistance, inducible expression of Ermmethyltransferase and peptide-mediated resistance, appear to depend on specificinteractions between the ribosome-bound macrolide molecule and the nascent peptide.The similarity between these mechanisms and their relation to the general mode ofmacrolide action is discussed and the discrepancies between currently available data arehighlighted.
Key words: macrolides, erythromycin, ketolides, azithromycin, clarithromycin., tylosin, carbomycin, spiramycin, ribosome,resistance.
INTRODUCTION
Macrolides belong to one of the most commonly usedfamilies of clinically important antibiotics used to treatinfections caused by Gram-positive bacteria such asStaphylococcus aureus, Streptococcus pneumoniae andStreptococcus pyogenes. Chemically, macrolides arerepresented by a 14-, 15- or 16-membered lactone ringcarrying one or more sugar moieties and additionalsubstitutions linked to various atoms of the lactone ring,Fig (1).
Erythromycin A, a 14-membered ring drug, was the firstclinically used macrolide. Drug delivery problems resultingfrom acid instability, prompted the design of newermacrolides. Increased acid stability as well as an increase inthe range of antimicrobial activity characterized the secondgeneration, which includes 14-membered ring drugs, such asclarithromycin and roxithromycin as well as the 15-membered ring azithromycin. In addition, the 16-memberedring macrolides such as tylosin, carbomycin A andspiramycin and others also exhibited significantantimicrobial activity, and were originally thought to be theanswer to the growing occurrence of erythromycin resistantinfections. Unfortunately, all of these drugs became prone tothe selection of resistant strains. The newest generation ofmacrolides, the ketolides, whose clinical use is in its earlystage, are characterized by improved activity against some ofthe resistant strains.
It has been well documented that macrolides bind to thelarge ribosomal subunit in the vicinity of the peptidyl
*Address correspondence to this author at the Center for PharmaceuticalBiotechnology – M/C 870, University of Illinois, 900 S. Ashland Ave.,Chicago, IL 60607, USA; Tel: 312-413-1406; Fax: 312-413-9303; E-mail:[email protected]
transferase center and cause cell growth arrest due toinhibition of protein synthesis [1,2]. However, in spite ofmore than 50 years of research, the mode of macrolideinhibition of ribosome activity is understood only in themost general terms.
The extensive use of these antibiotics has led inevitablyto the spread of resistant strains. Expression of some of theresistance determinants is inducible by macrolides. Ofparticular interest are Erm methyltransferases, whichspecifically methylate a unique nucleotide within themacrolide binding site. The mechanism of Erm inductiondepends on ribosome stall within the translated regulatoryopen reading frame preceding the Erm cistron, and isapparently closely related to the general mode of macrolideaction on protein synthesis. However, the details of themechanism of Erm induction are not known.
In addition, a novel mechanism of macrolide resistancehas recently been described which is mediated by theexpression of specific short peptides in the cell. Although itis not entirely clear how expression of short peptides canrender cells resistant to macrolides, the underlying principlesmaybe related to the mechanisms of inducible resistance andthe mode of drug-induced inhibition of translation.
In this brief review we try to summarize the currentunderstanding of the mechanism of macrolide action, and themolecular mechanisms of inducible erm expression andpeptide-mediated macrolide resistance. Several reviewsdealing with these subjects appeared in the past and we referthe reader interested in specific details to those publications[1-4]. Instead, we will try to focus on the apparentdiscrepancies and less clearly defined details in the existingmodels. We strongly believe that understanding thesediscrepancies exist and finding ways to resolve them will

2 Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 Gaynor and Mankin
O
O
O
OHOH
HO
O
O
O
OMe
OH
OHO
N
O
O
O
OMeOH
HO
O
O
O
OMe
OH
OHO
N
O
O
OHOH
HO
O
O
O
OMe
OH
OHO
N
N
O
O
O
ON
O
O
OOHO
N
O
N
N
N
O
O
O
ON
O
O
OOHO
N
OH
N
O
O
O
OOHO
N
O
OH
O
O
OH
OH
OO
HO
OMeOMe
O
O
O
OO
OHO
N
O
OAc
O
O
OH
O
O
O
Erythromycin A Clarithromycin Azithromycin
Tel ithromycin (HMR-3647) Cethromycin (ABT-773)
Tylosin
Carbomycin A
1 2 3
45
67
89
1011
1213
14
Fig. (1). Chemical structures of the macrolide antibiotics. First row: drugs of the first generation (erythromycin) and secondgeneration (14-membered ring clarithromycin and 15-membered ring azithromycin). Second row: ketolides, the macrolides of thethird generation. Third row: examples of 16-membered ring macrolides.
help to direct further research focused on the molecularmechanisms of macrolide action and drug resistance.
THE MACROLIDE BINDING SITE
The general location of the macrolide binding site on thelarge ribosomal subunit has been initially mapped using acombination of biochemical and genetic methods [2,5-9].Nonetheless, the details of the molecular interactions of thevarious classes of macrolides with the ribosome have juststarted to emerge with the release of several crystallographicstructures of the archaeal and bacterial large ribosomalsubunits and their complexes with antibiotics [10-13].
The X-ray structures confirm conclusions drawn frombiochemical data, which, indicated that RNA constitutes theprimary component of the macrolide binding site, Fig. (2A).A number of nucleotide residues in domain V of 23S rRNAinteract with the macrolide molecule. Important contacts,which contribute markedly to the strength of interaction ofthe macrolide molecule with the ribosome, are formedbetween the C5 mono- or disaccharide side chains of 14- 15-and 16-membered ring macrolides and rRNA. Thedesosamine sugar of erythromycin and other related 14-membered ring macrolides form hydrogen bond interactionswith the nitrogen bases of the nucleotide residues A2058,A2059 (here and throughout the entire paper we will use E.coli nucleotide numbering to facilitate the discussion). The

Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 3
base pair 2611-2057, more specifically, the nucleotideoccupying position 2057, may also be involved in hydrogenbonding with the C5 desosamine of 14-membered ringmacrolides (as seen in the bacterial, D e i n o c o c c u srad iodurans structures) [12], or it may establishhydrophobic interactions with the lactone ring (as seen in thecomplexes of 15- and 16-membered ring macrolides with thearchaeal 50S ribosomal subunit) [13]. In addition, thedesosamine sugar can potentially interact with the backbonephosphate oxygen of G2505. The mycaminose O2 of themycaminose-mycarose disaccharide of 16-membered ringmacrolides also forms a hydrogen bond with adenine at theposition 2058 revealing this nucleotide as one of the mainbinding determinants of all macrolides. The C5mycaminose-mycarose disaccharides of tylosin, carbomycinA and spiramycin form a number of additional interactions,mainly of a hydrophobic nature. The interactions of the C5sugar residues with positions 2057-2059 explain whymutations at these nucleotides or dimethylation of A2058can render macrolide resistance [12].
Interaction of the lactone ring with the ribosome mayaccount for more then 25% of the free binding energy of thedrug [14]. Hydrophobic interactions appear to contributesignificantly to this interaction. In particular, the lactonering interacts hydrophobically with a crevice formed byrRNA bases 2057-2059 [14]. The crystallographic structuresof complexes of the 16-membered ring macrolides, tylosin,carbomycin A and spiramycin, with the large ribosomalsubunit of Haloarcula marismortui [13] suggest that theacetaldehyde group at C6 position of the lactone ring formsa reversible covalent bond with the N6 of A2062, whichmay contribute to the binding energy of these drugs. Thisinteraction is not possible for the 14-membered ringmacrolides of the erythromycin group or the 15-member ringazithromycin, which carry either a hydroxyl or an ester at theC6 position of the lactone ring. This explains why 2062mutation confers resistance specifically to 16-member ring,but not 14- or 15-membered ring macrolides [15].
Ketolides are the most recently introduced generation ofmacrolides. They are characterized by the replacement of theC3 cladinose sugar with a keto group. In addition, mostclinically relevant ketolides also contain extended alkyl-arylside chain appendages as well as an 11, 12-carbamate cycle.Ketolides exhibit increased binding to the ribosomecompared to macrolides of previous generations [16,17]. Incontrast to expectations, the recently solved structure of acomplex of a ketolide, ABT-773, with the D. radioduransribosome (Schlünzen and Yonath, personal communication),did not provide clear clues into the molecular determinantsof increased ketolide affinity. The 11-OH and 12-OH lactonehydroxyls of erythromycin and related drugs could formhydrogen bonds with O4 of U2609. The carbamate cycle thatreplaces these hydroxyls in ABT-773 (and other ketolides) isseen interacting with O4 of U2609 in D. radiodurans 50Ssubunits. Biochemical data show that this interaction ismore important for ketolides than for the drugs of theerythromycin group because the mutation of U2609 to Cconfers resistance specifically to ketolides but not othermacrolides [14]. However, crystal structures did not providea clear explanation of the significance of this mutation.
Biochemical and genetic data revealed interaction ofvarious macrolides, including erythromycin, ketolides andtylosin, with the loop of helix 35 in domain II of 23S rRNA[7,9]. Mutations or changes in posttranscriptionalmodification in this rRNA region confer weak resistance tomacrolides [7,18]. In addition, a bound macrolide eitherenhanced or decreased accessibility of nucleotides in thehelix 35 loop to chemical modification in the RNA probingexperiments [5,7,9,14]. Protection or enhancement ofaccessibility of A752 to dimethyl-sulfate modificationappeared to correlate with the chemical structure of the alkyl-aryl side chain of ketolides, suggesting that the side chainmight contact the loop of helix 35 [16], (Xiong and Mankin,unpublished). Although crystallographic structures show thatthe mucinose side chain of 16-member ring tylosin canapparently reach the loop of helix 35 [14], no interactions of14-membered ring macrolides of the erythromycin type,ABT-773 or azithromycin with this rRNA region wererevealed [12], (Schlünzen and Yonath, unpublished). Thequinolylallyl side chain of ABT-773 forms hydrogen bondswith C1782 in domain IV of 23S rRNA and U790 indomain II. However, the latter interaction it is not closeenough to the loop of helix 35 to explain the protection ofA752 by ABT-773 observed in the RNA footprintingexperiments [14]. Thus, it remains unclear whether theketolide side chain and/or the loop of helix 35 in the crystalstructure are positioned exactly as in the “live and breathing”ribosome. Altogether, crystallographic studies did not showany strong interactions of the ketolide side chain with theribosome, leaving the 11, 12-carbamate and its interactionwith U2609 as potentially the major factor enhancingketolide binding.
A macrolide molecule is coordinated in its binding siteby multiple hydrophobic and hydrogen bonds (and possibly,a covalent bond in case of some 16-membered ringmacrolides) between its functional groups and 23S rRNA.These interactions with RNA account for most of the freeenergy of drug binding. In addition, some macrolides canreach ribosomal proteins L4 and/or L22. The mucinose sugarof tylosin interacts with L22 and the furosamine residue ofspiramycin interacts with L4 [14]. The proximity of theseproteins to the macrolide binding site explains whymutations in L22 and L4 protein genes can render cellsresistant to macrolides [19-22].
When analyzing crystallographic structures of theribosome-macrolide complexes, it is important to keep inmind that although the general features of the drug-ribosomeinteractions observed with the 50S subunit of a halophilicarchaeon (H. marismortui) and a bacterium (D. radiodurans)are in general agreement, some details vary. For example,the precise position and conformation of the lactone ring ofseveral macrolides seen in structures solved by the Yonathgroup [12] differ from those seen by the Steitz laboratory[14]. At this point it is unclear whether the discrepancies aredue to uncertainty in the crystallographic structures,differences in the ribosomes used, or the difference incrystallization procedures (co-crystallization versus crystalsoaking). It is important to note, that in the 23S rRNA ofH. marismortui several positions involved in macrolidebinding are different from those in the bacterial 23S rRNA.

4 Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 Gaynor and Mankin
These include such important positions as 2058 (A inbacteria, G in archaea) and 2609 (U in bacteria, C in H.marismortui). The nucleotide sequence differences mightpotentially affect the affinity of the macrolide molecule tothe ribosome and precise molecular interactions in thebinding site. Indeed, the mutation of G2058 to A in theclosely related archaeon, Halobacterium halobium, signifi-cantly increases sensitivity of the mutant to erythromycinand other macrolides (Mankin, Xiong, Tait-Kamradt,unpublished). However, even the drug complexes with thebacterial ribosome of D. radiodurans should be consideredcautiously: having only one bacterial structure is notsufficient to distinguish between the general and species-specific determinants of macrolide binding. For example,two molecules of azithromycin were found bound to the D.radiodurans ribosome in the co-crystallization experiments.One of the azithromycin molecules occupied the “conven-tional” site while the other is seen bound immediately nextto it, making direct contact with the proteins L4 and L22 aswell as domain II of 23S rRNA. The significance of thissecond site with regard to azithromycin inhibitory actionremains unclear. It could possibly be a species-specificfeature because the amino acid Gly60 in protein L4 that iscontacted by the drug is unique in D. radiodurans comparedto other bacteria (Franceschi, personal communication).
LOCATION OF THE MACROLIDE BINDING SITEIN THE RIBOSOME AND THE MECHANISM OFACTION
The precise mechanism of protein synthesis inhibition bymacrolides depends on the specific chemical structure of thedrug molecule. This affects its interaction with the ribosomeas well as the mode of the inhibitory action. Four modes ofinhibition of protein synthesis have been ascribed tomacrolides: 1) Inhibition of the progression of the nascentpeptide chain during early rounds of translation [23,24]; 2)Promotion of peptidyl tRNA dissociation from the ribosome[25]; 3) Inhibition of peptide bond formation [23]; and 4)Interference with 50S subunit assembly [26]. All of thesemechanisms have some correlation with the location of themacrolide binding site on the ribosome.
The macrolide binding site is located on the largeribosomal subunit inside the nascent peptide exit tunnel nearthe peptidyl transferase center, Fig. (2B and 2C ). Itsproximity to the peptidyl transferase center explains theinhibitory effect of some macrolides on peptide bondformation. The sugar residues attached at the C5 position ofthe lactone ring protrude towards the peptidyl transferasecenter. The long disaccharide mycaminose-mycarose sidechains of the 16-membered ring drugs tylosin, spiramycinand carbomycin A stretch far enough towards the active siteof the peptidyl transferase to directly interfere with thecatalysis of peptide bond formation [13,27]. The shorterdesosamine monosaccharide residues of the 14-memberedring macrolides do not reach the peptidyl transferase, whichexplains the lack of inhibitory effects of these drugs on thereaction of transpeptidation [24,28,29]. In contrast, somemild stimulation of peptidyl transfer by erythromycin isobserved in the puromycin reaction [23].
The main mechanism of inhibition of protein synthesisby macrolides is related to their binding in the nascentpeptide exit tunnel. The exit tunnel is formed primarily by23S rRNA. It starts at the peptidyl transferase center andspans the entire body of the subunit finally opening at its“back” [10,30-32]. Although originally viewed as an inertconduit for nascent peptides of any sequence synthesized bythe ribosome, the tunnel is now considered an active anddynamic functional entity [33]. Several studies showed thatinteractions between the ribosome and the nascent peptidethat take place inside the exit tunnel affect the progression ofprotein synthesis as well as the reactions catalyzed by theribosomal peptidyl transferase [34,35], (see [36] for review).The tunnel is relatively wide (15 Å average). However, itcontains a constriction, (ca. 10 Å wide) formed by proteinsL4 and L22, which is located a short distance from thepeptidyl transferase center. Macrolides bind close to thisconstriction and pose a molecular road block for the growingpolypeptide chain [12,13,33]. In the presence of bound 14-and 15-membered ring macrolides, polymerization of thefirst several amino acids continues unperturbed andinhibition of polypeptide growth occurs only after thenascent peptide becomes large enough to reach the boundmacrolide near the tunnel constriction. The inhibition ofpeptide progression eventually results in the dissociation ofpeptidyl tRNA from the ribosome. The latter apparentlytakes place during an attempted act of translocation duringwhich the contacts of tRNA with the ribosome must be“loosened”.
This mechanistic model explains biochemicalobservations regarding macrolide activity and is generallyconsistent with the macrolide location in the ribosome[24,25,37]. However, the main question that remainsunresolved is the length of the nascent peptide that themacrolide-bound ribosome can synthesize. In poly (A)-dependent cell-free translation systems, macrolides werereported to cause accumulation of di-lysine but inhibitedsynthesis of longer peptides. In poly (U)-dependent proteinsynthesis, accumulation of Phe2 and Phe3 was observed inthe presence of erythromycin, while formation of longerpeptides was inhibited [28,29]. Also noteworthy is theobservation that the exact point of erythromycin inhibitionof elongation appears to be dependent on the amino acidsequence of the nascent peptide. While the drug does notaffect transfer of Ac-Gly-Gly to puromycin, the transfer ofAc-Pro-Gly is efficiently inhibited [23]. The effect oferythromycin on elongation of very short peptides appears tocontradict more recent observations using an in vitrotranslation system driven by synthetic mRNAs, in whicherythromycin caused the accumulation of peptidyl-tRNAscarrying significantly longer - 6 to 8 amino acid residue-longpeptides (Tenson, personal communication). Furthermore,the ketolide telithromycin allowed the ribosome tosynthesize nascent peptides that were even longer – 9 to 12amino acids long. Modeling studies suggest that 6 to 8amino acids can potentially fit between the peptidyltransferase active site and the macrolide roadblock [12]. Ifso, then how can erythromycin affect elongation of a di-peptide? And how can a 12-amino-acid-long nascent peptidesynthesized in the presence of ketolides fit? Is it folded in aspecific way within the ribosome? Another question that
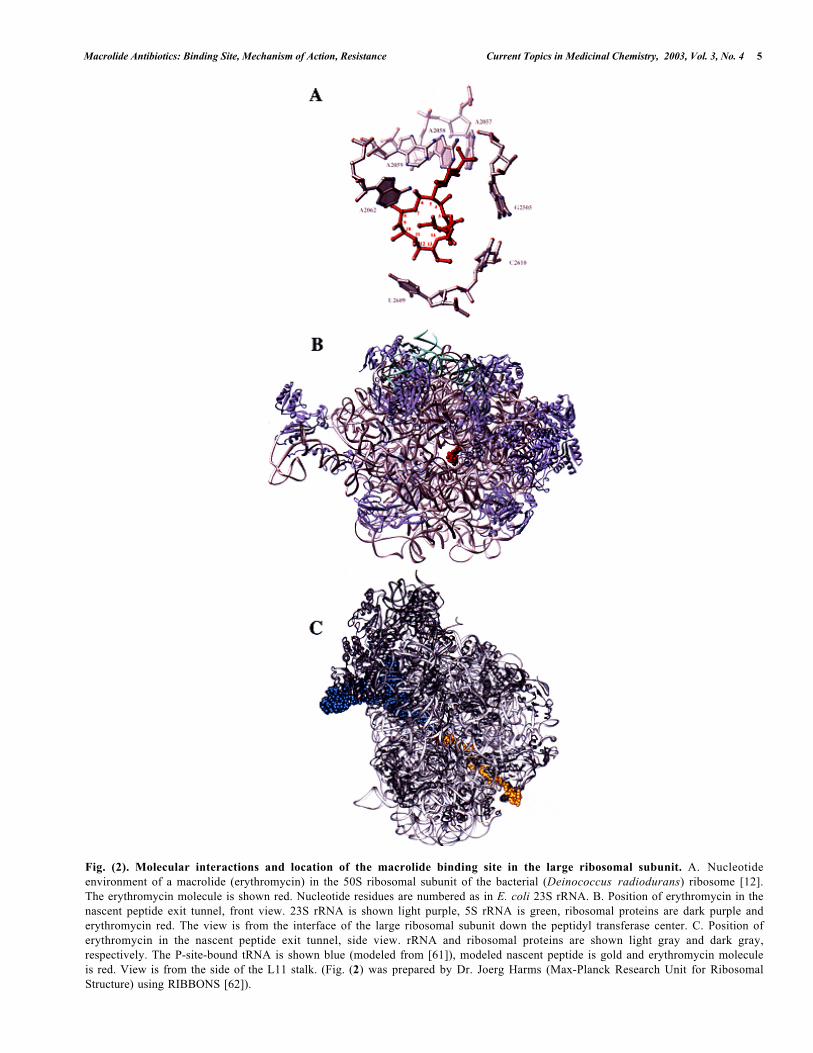
Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 5
Fig. (2). Molecular interactions and location of the macrolide binding site in the large ribosomal subunit. A. Nucleotideenvironment of a macrolide (erythromycin) in the 50S ribosomal subunit of the bacterial (Deinococcus radiodurans) ribosome [12].The erythromycin molecule is shown red. Nucleotide residues are numbered as in E. coli 23S rRNA. B. Position of erythromycin in thenascent peptide exit tunnel, front view. 23S rRNA is shown light purple, 5S rRNA is green, ribosomal proteins are dark purple anderythromycin red. The view is from the interface of the large ribosomal subunit down the peptidyl transferase center. C. Position oferythromycin in the nascent peptide exit tunnel, side view. rRNA and ribosomal proteins are shown light gray and dark gray,respectively. The P-site-bound tRNA is shown blue (modeled from [61]), modeled nascent peptide is gold and erythromycin moleculeis red. View is from the side of the L11 stalk. (Fig. (2) was prepared by Dr. Joerg Harms (Max-Planck Research Unit for RibosomalStructure) using RIBBONS [62]).

6 Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 Gaynor and Mankin
remains unclear is whether the length of the nascent peptideproduced in the presence of erythromycin or relatedmacrolides depends on the peptide amino acid compositionand/or sequence. One would expect that the growth of anascent peptide composed of bulky amino acids would bearrested earlier than that of a peptide composed of smallerresidues. Furthermore, since the macrolide molecule and thenascent peptide appear to be competing for the same space inthe exit tunnel, it is possible that certain peptides mightexhibit affinity for the components of the tunnel wall thatinteract with a macrolide molecule, and could competitivelyremove macrolide from its binding site. In this scenario,synthesis of different proteins can be inhibited to a differentextent by macrolides and such specificity may depend bothon the structure of the drug and the structure of the nascentpeptide and their interactions with the ribosome.
Interference with ribosome assembly may contributefurther to the overall inhibitory activity of macrolides[38,39]. In the presence of macrolides, precursors of 50Sribosomal subunits accumulate. These precursors do notcontain a full protein complement of the 50S subunit andsediment in a sucrose gradient as 30S particles [40]. The23S rRNA in 50S precursor particles is more accessible forchemical modification than in the fully assembled 50Ssubunits (Xiong, Champney and Mankin, unpublished).Although the location of the macrolide binding site in theassembly intermediates remains to be determined; it isconceivable that binding of the macrolide to the subunitprecursor (in its “conventional” site or a different site) mayprevent specific conformational rearrangements and/orprotein binding required for the completion of assembly[41]. A correlation between the inhibition of proteinsynthesis and inhibition of ribosome assembly suggests thatnot only drug binding to the precursor, but also eithergeneral reduction of protein synthesis or inhibition ofproduction of a specific protein(s) (for example, chaperonswhich may contribute to ribosome biogenesis [42,43]) isrequired for inhibiting assembly of the 50S subunit bymacrolides. The importance of assembly inhibition for theoverall inhibitory action of macrolides remains to bedetermined. Since inhibition of the assembly takes placewithin the background of the arrested protein synthesis, it isunclear whether reduced production of new ribosomesamplifies the cell’s “suffering” when protein synthesis isalready being inhibited.
MACROLIDE RESISTANCE DUE TO THE TARGETSITE MODIFICATION
High efficacy and safety of macrolides as well as theiruse as an alternate therapy for penicillin-intolerant patientsmade them popular drugs. The wide use of these antibioticsled inevitably to the spread of the resistance strains. The twomost common mechanisms of resistance are excretion of thedrug from the cell and modification of the drug target site[2]. The latter mechanism comes in the form of a site-specific posttranscriptional modification of 23S rRNA ormutations in 23S rRNA or ribosomal proteins.
Mutations in protein L4 directly or allosterically affectmacrolide binding and cause resistance by preventing drug
binding to the ribosome [21,22]. Mutations in anotherribosomal protein, L22, do not significantly perturb drugaffinity, but appear to act through an indirect mechanism.These mutants exhibit a wider opening of the tunnel so thatthe nascent peptide can apparently “slip by” the macrolidemolecule bound in the tunnel (and/or possibly displace thedrug) [21,33]. Mutations in ribosomal protein genes are afrequent cause of drug resistance because a single mutationalevent is sufficient to render cells resistant to a macrolide. Incontrast, resistance due to rRNA mutations is less common[44]. Because of the multiplicity of rRNA genes in themajority of bacteria, the beneficiary (for the pathogen) effectof the mutation in one rRNA gene copy is usually maskedby the abundance of wild type rRNA transcribed fromunmutated gene copies. Therefore, this mechanism ofresistance is more often found in the organisms with a lowcopy number of rRNA cistrons (for example, Helicobaterpylori that has two rRNA operons) (see [2,3] for review).
The most frequently found mechanism of macrolidebinding site modification is dimethylation of a single 23SrRNA nucleotide, A2058, by Erm-type methyltransferases.The erm genes are found in macrolide producers from whichthey were apparently disseminated to the clinical pathogensthrough horizontal gene transfer [45]. Dimethylation ofA2058, which is located within the macrolide binding site,drastically decreases drug affinity due to steric hindrancethus, rendering bacteria resistant to high concentrations ofmacrolide antibiotics [12,16,46]. A2058 is also intimatelyinvolved in the binding of lincosamides and streptograminB [12]. Hence, Erm-based methylation (termed MLSBresistance) renders the cell resistant to at least three unrelatedclasses of antibiotics. Importantly, the fully assembledribosome is not a substrate for erm methylation becauseA2058 is buried deep inside the large ribosomal subunit andapparently is not accessible to Erm methyltransferase [47].Methylation of A2058 can take place only during ribosomeassembly, which leaves a very narrow time window for theerm enzyme to methylate its rRNA target.
Expression of erm genes can either be constitutive orinducible. Inducible erm-based resistance, where productionof Erm methyltransferase is activated only when cells areexposed to the drug, is found in some antibiotic producersas well as in many clinical pathogens. The frequentoccurrence of inducibly expressed erm [48,49] suggests thatA2058 dimethylation is apparently “unhealthy” forunperturbed protein synthesis. Bacteria appear to avoidunnecessary methylation of this nucleotide unless it becomesessential for survival in the presence of the drug.
The molecular mechanisms of Erm induction are relatedto the general mode of macrolide action, but they are largelyenigmatic. The inducible erm cassette consists of twofunctional parts – the Erm gene whose expression isnormally repressed due to sequestration the erm ribosomebinding site in the mRNA secondary structure and aconstitutively translated leader peptide cistron that precedeserm [50,51], Fig. (3A). In the presence of erythromycin, theribosome is believed to stall around the 8th –9th codon of theleader peptide open reading frame (ORF). The stall of theribosome triggers a conformational rearrangement in mRNAresulting in the opening of the erm ribosome binding site

Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 7
Met Gly Ile Phe Ser Ile Phe Val Ile Ser Thr Val His Tyr GLn Pro Asn Lys Lys
Met Gly Ile Phe Ser Ile Phe Val Ile Ser Thr
Pro
Gln
Tyr
His
Val
Asn
Met
Lys
Lys
ErmErm
1 2 3 4
MetS.D.E
2 3
ErmErm
A
B
S.D.L
S.D.L
S.D.E
Fig. (3). Inducible Erm resistance. The secondary structure of the leader region of the bi-cistronic mRNA coding for the leaderpeptide and Erm methyltransferase in the non-induced (A) and induced (B) configuration [51]. The sequence of the short peptideencoded in the leader open reading frame and its Shine-Dalgarno region (S.D.L) are shown. Shine-Dalgrano region (S.D.E) of the Ermcistron and the location of Met initiator codon are indicated. In the presence of an inducing macrolide antibiotic, the ribosome stallsaround 8th-9th codon of the leader cistron. Stalling causes rearrangement of the mRNA secondary structure which renders thetranslation initiation region of the Erm cistron accessible.
and thus, activation of erm translation, Fig. (3B) [52,53].Since stall on the leader ORF occurs only in the presence ofthe drug, the ribosome translating the leader peptide mustbind a macrolide molecule. On the other hand, the ribosomethat will translate the erm cistron, should be drug free atleast during early steps of erm translation in order to be ableto synthesize the entire polypeptide. This implies thatefficiency of erm induction should depend critically onmacrolide concentration. At very low drug concentrations,too few ribosomes will carry the antibiotic and thus stallingon the leader ORF will not occur. On the other hand, if thedrug concentration abruptly becomes very high, translationof the Erm cistron maybe inhibited and expression of the
resistance may be delayed. Moreover, only newly assembledribosomes can carry methylated 23S rRNA, as assembledribosomes are not erm methlyase targets (see above). Thus,the ability of a macrolide to induce erm resistance maydepend on the kinetic interplay of drug binding anddissociation from the ribosome and the rate of translation. Itis conceivable that macrolides with a high off-rate and amoderate-affinity binding will induce erm expression at abroader range of drug concentrations, while the tightlybinding macrolides with a slow off-rate will be less-efficientinducers. Indeed, the ketolides, which bind much tighter tothe ribosome than macrolides appear to be poor inducers oferm [16,17,54].

8 Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 Gaynor and Mankin
E-peptidesE-peptides
ATG CTT TTA CGT ATC TAA M L L R IM L L R IATG ACA TTA AAA GTC TAA M T L K VM T L K VATG ATT CTA AAG TTA TAA M I L K LM I L K LATG ATG CTA AAA TTG TAA M M L K LM M L K LATG CTG CTT ACG GTA TAA M L L T VM L L T VATG CTG TTG TTG GTG TAA M L L L VM L L L VATG CTG TTG CTG GTG TAA M L L L VM L L L VATG CTG CTA TTG GTT TAA M L L L VM L L L VATG GTT TTG TTT GTT TAA M V L F VM V L F VATG CGT TTA TTT GTT TAA M R L F VM R L F VATG TTA CTT TGG GTT TAA M L L W VM L L W VATG GTA ATT TTG GTA TAA M V I L VM V I L VATG GTA ATT TTG GTA TAA M V I L VM V I L VATG TTA ATC ACA GTA TAA M L I T VM L I T VATG GCT TTA AAA TAC TAA M A L Y TM A L Y TATG GTA CAA ACA GTA TAA M V Q T VM V Q T VATG GTA TAC ACA ATC TAA M V Y T IM V Y T I
K-peptidesK-peptides
ATG AAA TTA AAA CTC TAA M K L K LM K L K LATG AAA CTG AAG CTC TAA M K L K LM K L K LATG AAA ATG AAA GTT TAA M K M K VM K M K VATG AAA ATG AAA CTC TAA M K M K LM K M K LATG CGC TTT TTT GTC TAA M R F F V M R F F VATG CGG TTC TTT GTT TAA M R F F VM R F F VATG CGG TTC TTT GCT TAA M R F F AM R F F AATG AAG TTC TTT GTT TAA M K F F VM K F F VATG CGA GTA TAC CGA TAA M R V Y RM R V Y RATG AGG CGT TTT ATT TAA M R R F IM R R F IATG CTT CGT TGG TGG TAA M L R W WM L R W W
Fig. (4). Short peptides conferring resistance to erythromycin (E-peptides) or ketolide ABT-773 (K-peptides) selected from arandom five-codon-long mini-gene library [57]. The nucleotide sequences of the mini-genes and the amino acid sequences of theencoded peptides are shown.
The critical and the least understood component of themechanism of erm induction is the dependence of ribosomestall on both the sequence of the leader nascent peptide andthe chemical nature of the inducing macrolide. This suggeststhat a specific interaction between the peptide and the boundmacrolide molecule is required for ribosome stall [51]. Sincethe latest experiments show that macrolides (erythromycin)normally inhibit ribosome progression when the nascentpeptide is between 2 and 8 amino acids long (Tenson,personal communication), it appears that the ribosomeshould stall at approximately the correct place in the leaderpeptide ORF to elicit induction, irrespective of the leaderpeptide sequence. However, this is not true. The identity ofleader peptide amino acids 5-9 (S-I-F-V-I) is critical forinduction [55]. Since these C-terminus-proximal residues,but not the N-terminus of the nascent peptide appear todetermine the efficiency of ribosome stall, the nascentpeptide in the stalled complex might be folded in a specificway to allow direct contact between the critical amino acidsand the drug Fig. (5A). The immediate functional conse-quences of such interactions are not clear. One possibility isthat stall occurs because dissociation of peptidyl-tRNA isinhibited due to a specific drug-nascent peptide interaction.However, other mechanisms are possible.
PEPTIDE-MEDIATED MACROLIDE RESISTANCE.
The potential structure-specific interaction between thenascent peptide and the macrolide on the ribosome, whichmay be the key component of the erm induction mechanism,
is reminiscent of interactions that were proposed to takeplace in a completely different mechanism of macrolideresistance. In this mechanism, resistance to macrolides ismediated by expression of specific peptides, 4-6 amino acid-long [56,57] (see [4] for review). Biochemical evidenceindicates that the peptides act in cis, rendering the ribosomeon which they were translated refractory to inhibition bymacrolides. Screening of random 5-codon-long mini-genelibraries showed that only mini-genes coding for peptideswith a specific amino acid sequence could confer resistance,Fig. (4). Thus, peptides conferring the highest resistance toerythromycin (E-peptides) are characterized by the consensussequence M-(L)-L/I-(F)-V where the third position is almostalways occupied by Leu or Ile, while the second and fourthpositions show strong preference for hydrophobic aminoacids, more frequently Leu/Ile and Phe, respectively [57].Although cells expressing E-peptides also exhibit someresistance to ketolides, the peptides conferring the highestketolide resistance (K-peptides) conform to a differentconsensus: M-K/R-(F/L/V)-X-X, where X indicate positionswhich do not show an obvious amino acid preference [58].Peptides with yet different consensus sequences render cellsresistant to the 14-membered ring drugs oleandomycin andtroleandomycin, 15-membered ring azithromycin, 16-membered ring josamycin and other macrolides [4] and(Gaynor, Mankin and Tenson, unpublished). The strongcorrelation between the sequence of the resistance peptideand the structure of the drug suggests that a direct interactionbetween the drug and the peptide takes place on theribosome. The current model proposes that the newlysynthesized resistance peptide interacts with the ribosome-

Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 9
Met
Gly
Ile
Phe
SerIle
Phe
Val Ile
Met
ValLeu
Phe
Val
Rel
ease
fac
tor
A B
Fig. (5). Putative similarity of the mechanisms of Erm induction and peptide-mediated macrolide resistance. A. Schematicrepresentation of the ribosome stalled on the Erm leader cistron. The ribosome is shown with the 8th (Val codon) in the P-site and the9th (Ile) codon in the A-site. Erythromycin (black octagonal) is shown bound near the constriction of the exit tunnel. The amino acidresidues whose identities are important for ribosome stall are shown gray. The model reflects our proposal that the nascent peptidemay fold back in order to allow direct contact between the critical amino acid residues and the macrolide. B. The pre-terminationcomplex formed by the ribosome which has finished translation of a resistance peptide. The sequence of one of the E-peptides (seeFig. (4)) is shown. According to the “bottle-brush” model [58], the release of the E-peptide will promote dissociation of the boundmacrolide (in this case, erythromycin) from the ribosome. The amino acid residues whose identities are important to confererythromycin resistance are shown in gray.
bound macrolide molecule and evicts the drug moleculefrom its binding site on the ribosome therefore allowing theribosome to get re-engaged, at least temporarily, intranslation of cellular proteins [58]. Since the size of thepeptide appears to be important [56], the act of translationtermination (release of the nascent peptide from peptidyl-tRNA) and/or subsequent events may contribute to the drugdisplacement [59].
The parallel between the mechanism of erm inductionand peptide-mediated macrolide resistance is obvious. Inboth cases, the mode of ribosome function appears to dependdirectly on interaction between the short nascent peptide andthe drug molecule bound in the exit tunnel. Strikingly, theconsensus sequence of the pentapeptides that render cellsresistant to erythromycin, M-(L)-L/I-(F)-V, has a clearresemblance to the C-terminal amino acids of the erm leaderpeptide in the stalled complex, (… I-F-V). It is tempting tothink that this is not a mere fortuitous coincidence and thatthe two mechanisms exploit similar interactions between thedrug and the nascent peptide on the ribosome, Fig. (5).However, the consequences of such interactions may bedifferent – ribosome stalling during erm induction and drugdissociation (probably accompanied by the release of theribosome from the mini-gene transcript) in the case of E-peptide expression. How either of these events may occurremains unclear.
FUTURE DIRECTIONS
We mentioned some main achievements and problems inunderstanding the mechanism of action of macrolideantibiotics and mechanisms of macrolide resistance.However, it is clear that comprehensive knowledge of theprinciples of drug interaction with the ribosome and themolecular mechanisms of drug resistance are essentialprerequisites for the development of better drugs. The atomicstructures of the ribosome and the ribosome-drug complexeswill definitely aid the rational design of new macrolidederivatives, which might exhibit tighter binding to thetarget. It is unclear, however, whether it is possible todesign a molecular platform that would strengthen theinteraction of the lactone ring and its side chains with theribosome while avoiding steric clash with the A2058dimethyl moiety of an erm-methylated ribosome.
The mechanism of macrolide action is only starting toemerge. Again, the precise knowledge of the mode ofmacrolide binding to the ribosome will be instrumental inunderstanding how various macrolides affect different aspectsof protein synthesis. The length of the nascent peptidesynthesized in the presence of macrolides, and the relationbetween inhibition of elongation and the nascent peptidesequence need to be investigated. Kinetics and mechanismsof peptidyl-tRNA dissociation induced by macrolides alsorequire more detailed biochemical experimentation. The

10 Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 Gaynor and Mankin
release of the ribosome from mRNA, resulting frompeptidyl-tRNA dissociation, should potentially allow thisribosome to re-engage in protein synthesis. One wouldwonder, therefore, if blocking the exit tunnel farther awayfrom the peptidyl transferase center would produce a moreefficient inhibition of protein synthesis. The longer nascentpeptide stuck in the exit tunnel may prevent peptidyl-tRNAfrom dissociating and thus, could irreversibly incapacitatethe large ribosomal subunit.
Though the general inhibition of protein synthesis bymacrolides is obviously an important factor in cell growtharrest, it remains to be elucidated whether production of allproteins is inhibited to the same extent or if translation ofsome cistrons is more sensitive to macrolide inhibition thanthe others. The importance of the inhibition of ribosomeassembly is yet another parameter which must be studied inmore detail.
The mechanism of ribosome stall during induction oferm resistance and its relation to peptide-mediated resistanceis unclear and thus, will require attention. Modeling ordirect crystallographic studies of short nascent peptides onthe ribosome may provide a useful structural background toapproach these problems. It is obvious that detailedknowledge of the correlation between the structures ofinducing macrolides and the sequences of the nascentpeptides mediating ribosome stall will be essential.
Although no clinical cases of peptide-mediated macrolideresistance have been reported to date, this type of resistancecan be easily selected in the laboratory setting [56,60], andthus may potentially contribute to the overall resistance of aclinical isolate to macrolide antibiotics. Bacterial genomescontain multiple short open reading frames, some encodingpeptide sequences, which can potentially confer macrolideresistance. Expression of such ORFs may easily avoiddetection. Thus a more focused investigation is required toevaluate the clinical importance of this resistancemechanism.
ACKNOWLEDGEMENTS
We thank Joyce Sutcliffe (Rib-X), Francois Franceschi(Rib-X) and Frank Schlünzen (Max-Planck Research Unit forRibosomal Structure) for valuable advice and discussion,Tanel Tenson (Tartu University) and Ada Yonath (WeizmannInstitute) for communicating results prior to publication,Joerg Harms (Max-Planck Research Unit for RibosomalStructure) for preparing Fig. (2). The work was supported bya grant from the National Institutes of Health to A.S.M.(GM 53762).
REFERENCES
[1] Vazquez,D. Inhibitors of protein biosynthesis. Springer-Verlag: Berlin, Heidelberg, New York, 1979.
[2] Weisblum, B. Erythromycin resistance by ribosomemodification. Antimicrob. Agents Chemother. 1995, 39,577-585.
[3] Vester, B.; Douthwaite, S. Macrolide resistanceconferrred by base substitutions in 23S ribosomal RNA.Antimicrob. Agents Chemother. 2001, 45, 1-12.
[4] Tenson, T.; Mankin, A.S. Short peptides conferringresistance to macrolide antibiotics. Peptides 2002, 22,1661-1668.
[5] Moazed, D.; Noller, H.F. Chloramphenicol, erythromycin,carbomycin and vernamycin B protect overlapping sitesin the peptidyl transferase region of 23S ribosomal RNA.Biochimie 1987, 69, 879-884.
[6] Ettayebi, M.; Prasad, S.M.; Morgan, E.A.Chloramphenicol-erythromycin resistance mutations ina 23S rRNA gene of Escherichia coli. J. Bacteriol. 1985,162, 551-557.
[7] Xiong, L.; Shah, S.; Mauvais, P.; Mankin, A.S. A ketolideresistance mutation in domain II of 23S rRNA revealsproximity of hairpin 35 to the peptidyl transferasecentre. Mol. Microbiol. 1999, 31, 633-639.
[8] Vester, B.; Garrett, R.A. A plasmid-coded and site-directed mutation in Escherichia coli 23S RNA thatconfers resistance to erythromycin: implications for themechanism of action of erythromycin. Biochimie 1987,69, 891-900.
[9] Hansen, L.H.; Mauvais, P.; Douthwaite, S. The macrolide-ketolide antibiotic binding site is formed by structuresin domains II and V of 23S ribosomal RNA. M o l .Microbiol. 1999, 31, 623-632.
[10] Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A.The complete atomic structure of the large ribosomalsubunit at 2.4 A resolution. Science 2000, 289, 905-920.
[11] Harms, J.; Schluenzen, F.; Zarivach, R.; Bashan, A.; Gat,S.; Agmon, I.; Bartels, H.; Franceschi, F.; Yonath, A. Highresolution structure of the large ribosomal subunit froma mesophilic eubacterium. Cell 2001, 107, 679-688.
[12] Schlunzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj,A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structuralbasis for the interaction of antibiotics with the peptidyltransferase centre in eubacteria. Nature 2001, 413, 814-821.
[13] Hansen, J.; Ippolito, J.A.; Ban, N.; Nissen, P.; Moore,P.B.; Steitz, T.A. The structures of four macrolideantibiotics bound to the large ribosomal subunit. Mol.Cell 2002, 10, 117-128.
[14] Garza-Ramos, G.; Xiong, L.; Zhong, P.; Mankin, A.Binding site of macrolide antibiotics on the ribosome:new resistance mutation identifies a specific interactionof ketolides with rRNA. J. Bacteriol. 2002, 183, 6898-6907.
[15] Depardieu, F.; Courvalin, P. Mutation in 23S rRNAresponsible for resistance to 16-membered macrolidesand streptogramins in Streptococcus pneumoniae.Antimicrob. Agents Chemother. 2001, 45, 319-323.
[16] Douthwaite, S.; Hansen, L.H.; Mauvais, P. Macrolide-ketolide inhibition of MLS-resistant ribosomes isimproved by alternative drug interaction with domain IIof 23S rRNA. Mol. Microbiol. 2000, 36, 183-193.
[17] Capobianco, J.O.; Cao, Z.S.; Shortridge, V.D.; Ma, Z.K.;Flamm, R.K.; Zhong, P. Studies of the novel ketolide

Macrolide Antibiotics: Binding Site, Mechanism of Action, Resistance Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 11
ABT-773: Transport, binding to ribosomes, andinhibition of protein synthesis in S t r e p t o c o c c u spneumoniae. Antimicrob. Agents Chemother. 2000, 44,1562-1567.
[18] Liu, M.; Kirpekar, F.; Van Wezel, G.P.; Douthwaite, S. Thetylosin resistance gene tlrB of Streptomyces fradiaeencodes a methyltransferase that targets G748 in 23SrRNA. Mol. Microbiol. 2000, 37, 811-820.
[19] Chittum, H.S.; Champney, W.S. Ribosomal protein genesequence changes in erythromycin-resistant mutants ofEscherichia coli. J. Bacteriol. 1994, 176, 6192-6198.
[20] Pardo, D.; Rosset, R. Properties of ribosomes fromerythromycin resistant mutants of Escherichia coli. Mol.Gen. Genet. 1977, 156, 267-271.
[21] Spahn, C.M.T.; Prescott, C.D. Throwing a spanner in theworks: Antibiotics and the translation apparatus. J. Mol.Med. 1996, 74, 423-439.
[22] Gregory, S.T.; Dahlberg, A.E. Erythromycin resistancemutations in ribosomal proteins L22 and L4 perturb thehigher order structure of 23 S ribosomal RNA. J. Mol.Biol. 1999, 289, 827-834.
[23] Mao, J.C.-H.; Robishaw, E.E. Erythromycin, apeptidyltransferase effector. Biochem. 1972, 11, 4864-4872.
[24] Vazquez, D. The Macrolide Antibiotics. In Antibiotics III.Mechanism of action of antimicrobial and antitumoragents; Corcoran,J.W.; Hahn, F.E. Eds.; Springer-Verlag:New York, Heidelberg, Berlin, 1975, pp. 459-479.
[25] Menninger, J.R.; Otto, D.P. Erythromycin, carbomycin,and spiramycin inhibit protein synthesis by stimulatingthe dissociation of peptidyl-tRNA from ribosomes.Antimicrob. Agents Chemother. 1982, 21, 810-818.
[26] Champney, W.S.; Burdine, R. Macrolide antibioticsinhibit 50S ribosomal subunit assembly in Bacillussubtilis and Staphylococcus aureus. Antimicrob. AgentsChemother. 1995, 39, 2141-2144.
[27] Poulsen, S.M.; Kofoed, C.; Vester, B. Inhibition of theribosomal peptidyl transferase reaction by the mycarosemoiety of the antibiotics carbomycin, spiramycin andtylosin. J. Mol. Biol. 2000, 304, 471-481.
[28] Mao, J.C.-H.; Robishaw, E.E. Effects of macrolides onpeptide-bond formation and translocation. Biochem.1971, 10, 2054-2061.
[29] Tanaka, S.; Otaka, T.; Kaji, A. Furhter studies on themechanism of erythromycin action. Biochim. Biophys.Acta 1973, 331, 128-140.
[30] Bernabeu, C.; Lake, J.A. Nascent polypeptide chainsemerge from the exit domain of the large ribosomalsubunit: immune mapping of the nascent chain. Proc.Natl. Acad. Sci. USA 1982, 79, 3111-3115.
[31] Yonath, A.; Leonard, K.R.; Wittmann, H.G. A tunnel inthe large ribosomal subunit revealed by three-dimensional image reconstruction. Science 1987, 236,813-816.
[32] Frank, J.; Zhu, J.; Penczek, P.; Li, Y.; Srivastava, S.;Verschoor, A.; Radermacher, M.; Grassucci, R.; Lata, R.K.;Agrawal, R.K. A model of protein synthesis based on
cryo-electron microscopy of the E. coli ribosome. Nature1995, 376, 441-444.
[33] Gabashvili, I.S.; Gregory, S.T.; Valle, M.; Grassucci, R.;Worbs, M.; Wahl, M.C.; Dahlberg, A.E.; Frank, J. Thepolypeptide tunel system in the ribosome and its gatingin erythromycin resistance mutants of L4 and L22. EMBOJ. 2001, 8, 181-188.
[34] Nakatogawa, H.; Ito, K. The ribosomal exit tunnelfunctions as a discriminating gate. Cell 2002, 108, 629-636.
[35] Gong, F.; Yanofsky, C. Instruction of translatingribosome by nascent peptide. Science 2002, 297, 1864-1867.
[36] Tenson, T.; Ehrenberg, M. Regulatory nascent peptidesin the ribosomal tunnel. Cell 2002, 108, 591-594.
[37] Menninger, J.R. Mechanism of inhibition of proteinsynthesis by macrolide and lincosamide antibiotics. J.Basic Clin. Phys. Pharmacol. 1995, 6, 229-250.
[38] Chittum, H.S.; Champney, W.S. Erythromycin inhibitsthe assembly of the large ribosomal subunit in growingEscherichia coli cells. Curr. Microbiol. 1995, 30, 273-279.
[39] Champney, W.S.; Tober, C.L.; Burdine, R. A comparisonof the inhibition of translation and 50S ribosomalsubunit formation in Staphylococcus aureus cells bynine different macrolide antibiotics. Curr. Microbiol.1998, 37, 412-417.
[40] Usary, J.; Champney, W.S. Erythromycin inhibition of50S ribosomal subunit formation in Escherichia colicells. Mol. Microbiol. 2001, 40, 951-962.
[41] Champney, W.S.; Tober, C.L. Inhibition of translationand 50S ribosomal subunit formation in Staphylococcusaureus cells by 11 different ketolide antibiotics. Curr.Microbiol. 1998, 37, 418-425.
[42] Alix, J.H.; Guerin, M.F. Mutant DnaK chaperones causeribosome assembly defects in Escherichia coli. Proc.Natl. Acad. Sci. USA 1993, 90, 9725-9729.
[43] Maki, J.A.; Schnobrich, D.J.; Culver, G.M. The DnaKchaperone system facilitates 30S ribosomal subunitassembly. Mol. Cell 2002, 10, 129-138.
[44] Tait-Kamradt, A.; Davies, T.; Appelbaum, P.C.;Depardieu, F.; Courvalin, P.; Petitpas, J.; Wondrack, L.;Walker, A.; Jacobs, M.R.; Sutcliffe, J. Two newmechanisms of macrolide resistance in clinical strains ofStreptococcus pneumoniae from Eastern Europe andNorth America. Antimicrob. Agents Chemother. 2002, 44,3395-3401.
[45] Cundliffe, E. How antibiotic-producing organisms avoidsuicide. Annu. Rev. Microbiol. 1989, 43, 207-233.
[46] Hansen, J.L.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A.Structural insights into peptide bond formation. Proc.Natl. Acad. Sci. USA 2002, 99, 11670-11675.
[47] Skinner, R.; Cundliffe, E.; Schmidt, F.J. Site of action ofa ribosomal RNA methylase responsible for resistance toerythromycin and other antibiotics. J. Biol. Chem. 1983,258, 12702-12706.

12 Current Topics in Medicinal Chemistry, 2003, Vol. 3, No. 4 Gaynor and Mankin
[48] Rosato, A.; Vicarini, H.; Leclercq, R. Inducible orconstitutive expression of resistance in clinical isolatesof streptococci and enterococci cross-resistant toerythromycin and lincomycin. J. Antimicrob. Chemother.1999, 43, 559-562.
[49] Giovanetti, E.; Montanari, M.P.; Mingoia, M.; Varaldo,P.E. Phenotypes and genotypes of erythromycin-resistant Streptococcus pyogenes strains in Italy andheterogeneity of inducibly resistant strains. Antimicrob.Agents Chemother. 1999, 43, 1935-1940.
[50] Dubnau, D. Induction of ermC requires translation of theleader peptide. EMBO J. 1985, 4, 533-537.
[51] Weisblum, B. Macrolide resistance. Drug Resist. Updates1998, 1, 29-41.
[52] Gryczan, T.J.; Grandi, G.; Hahn, J.; Grandi, R.; Dubnau, D.Conformational alteration of mRNA structure and theposttranscriptional regulation of erythromycin-induceddrug resistance. Nucleic Acids Res. 1980, 8, 6081-6097.
[53] Mayford, M.; Weisblum, B. Conformational alterationsin the ermC transcript in vivo during induction. EMBO J.1989, 8, 4307-4314.
[54] Zhong, P.; Cao, Z.; Hammond, R.; Chen, Y.; Beyer, J.;Shortridge, V.D.; Phan, L.Y.; Pratt, S.; Capobianco, J.;Reich, K.A.; Flamm, R.K.; Or, Y.S.; Katz, L. Induction ofribosome methylation in MLS-resistant Streptococcuspneumoniae by macrolides and ketolides. Microb. DrugResist. 1999, 5, 183-188.
[55] Mayford, M.; Weisblum, B. The ermC leader peptide:amino acid alterations leading to differential efficiency
of induction by macrolide-lincosamide-streptogramin Bantibiotics. J. Bacteriol. 1990, 172, 3772-3779.
[56] Tenson, T.; DeBlasio, A.; Mankin, A. A functionalpeptide encoded in the Escherichia coli 23S rRNA. Proc.Natl. Acad. Sci. USA 1996, 93, 5641-5646.
[57] Tenson, T.; Xiong, L.; Kloss, P.; Mankin, A.S.Erythromycin resistance peptides selected from randompeptide libraries. J. Biol. Chem. 1997 , 272 , 17425-17430.
[58] Tripathi, S.; Kloss, P.S.; Mankin, A.S. Ketolide resistanceconferred by short peptides. J. Biol. Chem. 1998, 273,20073-20077.
[59] Heurgué-Hamard, V.; Karimi, R.; Mora, L.; MacDougall,J.; Leboeuf, C.; Grentzmann, G.; Ehrenberg, M.;Buckingham, R.H. Ribosome release factor RF4 andtermination factor RF3 are involved in dissociation ofpeptidyl-tRNA from the ribosome. EMBO J. 1998, 17,808-816.
[60] Douthwaite, S.; Prince, J.B.; Noller, H.F. Evidence forfunctional interaction between domains II and V of 23Sribosomal RNA from erythromycin resistant mutant.Proc. Natl. Acad. Sci. USA 1985, 82, 8330-8334.
[61] Yusupov, M.M.; Yusupova, G.Z.; Baucom, A.; Lieberman,K.; Earnest, T.N.; Cate, J.H.; Noller, H.F. Crystal structureof the ribosome at 5.5 A resolution. Science 2001, 292,883-896.
[62] Carson, M. Ribbons. Methods Enzymol. 1997, 277, 493-505.


















