






![, VT[5] LIVE! and VT[5] LIVE SDI! VT[5] VT[5]LIVE] VT[5 ... · Virtual Studios SDI switcher HD/SD Editing VT[5] ... FEATURES Live video mixer ... Dual-channel upstream Effects bus](https://static.fdocuments.net/doc/165x107/5b0b5ac27f8b9ae61b8da9b2/-vt5-live-and-vt5-live-sdi-vt5-vt5live-vt5-studios-sdi-switcher.jpg)
Languages
Pages
Legal


www.design.che.vt.edu/VT-2005.html
VTVT--2005 Sigma Profile Database2005 Sigma Profile DatabaseA detailed tutorial for generating sigma profiles using A detailed tutorial for generating sigma profiles using
Accelrys’ Materials Studio v3.2 software packageAccelrys’ Materials Studio v3.2 software package
Eric Mullins, Y.A. Liu, Richard Oldland, Shu Wang, Stanley I. Sandler, Chau-Chyun Chen, Michael Zwolak, and Kevin Seavey
Honeywell Center of Excellence in Computer-Aided DesignSINOPEC/FPCC/AspenTech Center of Excellence in Process Systems Engineering
Department of Chemical Engineering, Virginia TechBlacksburg, VA 24060

www.design.che.vt.edu/VT-2005.html
ObjectivesObjectives
• An Introduction to Sigma Profiles, P(σ)• Using Accelrys’ Materials Studio (MS)
– Detailed procedure for creating a molecule– Detailed procedures for optimizing molecular geometry and
performing COSMO calculations
• Overview of the Sigma Profile averaging program

www.design.che.vt.edu/VT-2005.html
Sigma Profiles at a GlanceSigma Profiles at a Glance
• Sigma Profiles, P(σ), depict the surface charge density distribution over the entire molecule.
• Graphically: Area vs. Charge/Area
• Profiles are available with our database.
• Each molecule’s profile is unique.
• Profiles are sensitiveto conformation 0
2
4
6
8
10
12
14
16
18
20
-0.025 -0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020 0.025
Sigma (e/A^2)
P(s
igm
a)*A
rea(
sigm
a) (A
^2)
1,4-dioxanewatermethyl acetate

www.design.che.vt.edu/VT-2005.html
COSMOCOSMO--based models used sigma profiles to based models used sigma profiles to predict physical properties.predict physical properties.
• Solubility (SLE)• Vapor-Liquid (VLE)• Partition
coefficients (Kow)• pKa
T =333.15 K
0
1
2
3
4
5
6
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Phenol Mole Fraction
Pre
ssur
e (k
Pa)
COSMO-SAC
COSMO-SACExperimental Data
Experimental DataCOSMO-RS
COSMO-RS

www.design.che.vt.edu/VT-2005.html
The VTThe VT--2005 sigma profile database currently 2005 sigma profile database currently contains 1266 compounds.contains 1266 compounds.
• Compounds include alkanes, alkenes, alkynes, alcohols, amines, acids, aldehydes, aromatics, epoxides, esters, ethers, fluorocarbons, chlorocarbons, ketones and others.
• Compounds consist of the following atoms:N, C, O, H, F, Cl, P, S, I, and Br

www.design.che.vt.edu/VT-2005.html
Using Accelrys’ Materials Studio (MS), we will Using Accelrys’ Materials Studio (MS), we will construct our model molecule.construct our model molecule.
• We will use propane for our example.• Molecules can be drawn manually or downloaded from
other online sources as *.mol files and imported into MS.– NIST database
http://webbook.nist.gov/chemistry/
– Scifinder™http://www.cas.org/SCIFINDER/SCHOLAR/index.html
– National Library of Medicinehttp://chem.sis.nlm.nih.gov/chemidplus/

www.design.che.vt.edu/VT-2005.html
We begin by creating a new project in MS, and We begin by creating a new project in MS, and then opening a new molecule window.then opening a new molecule window.• Select a new “3D
Atomistic Document.”
• Right click on the “3D Atomistic Document” in the project window and select “Rename.”
• Type “Propane” and hit Enter.

www.design.che.vt.edu/VT-2005.html
We display the molecule with the “Ball and We display the molecule with the “Ball and Stick” option.Stick” option.• Right click in the
molecular window and select “Display Style.”
• Select “Ball and Stick” in the display style window and then close the window.

www.design.che.vt.edu/VT-2005.html
Use the sketch tool to draw propane (CUse the sketch tool to draw propane (C33HH88))
• Select the “Sketch Atom” tool from the toolbar.
• With the drop-down menu, select the Carbon atom.

www.design.che.vt.edu/VT-2005.html
Draw the three carbon atoms, but do not draw Draw the three carbon atoms, but do not draw the hydrogen atoms for now.the hydrogen atoms for now.• Left click once to place the first atom. Move your cursor
and click again; it will automatically create a second atom connected with a single bond.
• Draw a three carbon backbone.
• Hitting “Esc” will stopadding atoms.
• In the instance of double bonds, simply clicking on the singlebond will change itsbond type.

www.design.che.vt.edu/VT-2005.html
The program automatically adds the required The program automatically adds the required hydrogen for each molecule with the “Adjust hydrogen for each molecule with the “Adjust Hydrogen” tool.Hydrogen” tool.
• Click the “Adjust Hydrogen” icon from the toolbar to add the hydrogen atoms.
www.design.che.vt.edu/VT-2005.html
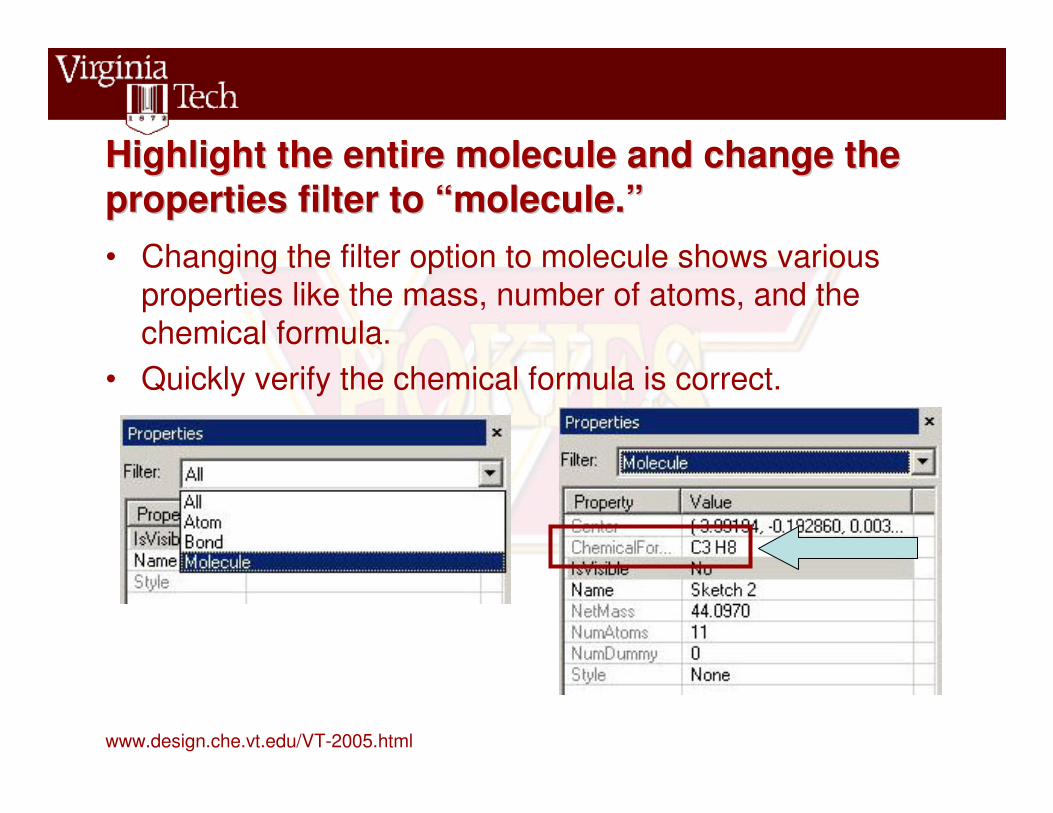
Highlight the entire molecule and change the Highlight the entire molecule and change the properties filter to “molecule.”properties filter to “molecule.”• Changing the filter option to molecule shows various
properties like the mass, number of atoms, and the chemical formula.
• Quickly verify the chemical formula is correct.

www.design.che.vt.edu/VT-2005.html
Now we use the “Clean” tool to correct the Now we use the “Clean” tool to correct the bond lengths and bond angles.bond lengths and bond angles.• Left click on the “Clean” tool icon on the toolbar.• The “Clean” tool does a rough geometry optimization
and adjusts the bond lengths and angles to the correct values for each atom and bond type.
• We are now finished constructing our example molecule, Propane.

www.design.che.vt.edu/VT-2005.html
Now we move onto setting up the Geometry Now we move onto setting up the Geometry Optimization calculation.Optimization calculation.• Select the “DMol3 calculation” from the “Modules”
Menu or use the icon on the toolbar.• The calculation dialog box will appear.

www.design.che.vt.edu/VT-2005.html
Select the task option in the DMol3 calculation Select the task option in the DMol3 calculation window.window.• Geometry optimization fixed the atomic coordinates for
the molecule by minimizing the total energy of the molecule.

www.design.che.vt.edu/VT-2005.html
Adjust the Geometry Optimization tolerance to Adjust the Geometry Optimization tolerance to “Fine.”“Fine.”• This can be done in the Setup or
Electronic tab.• This is the accuracy of the
Hamiltonian matrix element convergence.
• This specifies the accuracy to which the SCF (Self-Consistent Field) equations are converged. The “Fine” setting is recommended by the software documentation for highly accurate geometry optimizations. It represents a convergence of 10-6.

www.design.che.vt.edu/VT-2005.html
Select DNP as the Basis Set in the Electronic Select DNP as the Basis Set in the Electronic tab.tab.• DNP = Double Numerical basis with
Polarization functions, i.e., functions with angular momentum one higher than the highest occupied orbital in the free atom.
• According to the software documentation, “minimal basis sets are generally inadequate for anything but qualitative results, while DNP sets are the most reliable.” The DNP option instructs the program to ignore extraneous functions that “eliminate” certain atomic orbitals.
• When Quality it set to “Fine,” DNP is the default basis set.

www.design.che.vt.edu/VT-2005.html
Select GGA (local correlation) and VWNSelect GGA (local correlation) and VWN--BP BP (gradient(gradient--corrected functional) for the corrected functional) for the Functional option in the Setup tab.Functional option in the Setup tab.
• GGA = Generalized Gradient Approximation.• VWN-BP = BP functional (Becke, 1986; Pedrew, 1986)
with local correlation replaced by the VWN functional (Volsko, Wilk, and Nusair, 1980). Koch and Holthausen(2001) give excellent guidance about the selection of appropriate functionals.

www.design.che.vt.edu/VT-2005.html
Select the “More” button in the Job Control Select the “More” button in the Job Control tab.tab.• Check the “Retain server files” box. This will save
the files to the hard drive in the “jobs” directory.

www.design.che.vt.edu/VT-2005.html
Click the “Files” button at the bottom of the Click the “Files” button at the bottom of the DMol3 calculation dialog box.DMol3 calculation dialog box.• Click the “Save Files” button. This will create the input
files necessary for the calculation. They are visible in the Project window.

www.design.che.vt.edu/VT-2005.html
Open “propane.input” from the Project Open “propane.input” from the Project window.window.• Insert the code “Basis_version v4.0.0” into the input file. • This statement instructs the program to use version 4.0.0 DNP basis
set instead of the default v3.5 DNP basis set. The software documentation recommends v4.0.0 for COSMO calculations.

www.design.che.vt.edu/VT-2005.html
Leave the “Spin Unrestricted” box unchecked Leave the “Spin Unrestricted” box unchecked in the Setup tab.in the Setup tab.• Checking the “Spin Unrestricted” is necessary when
working with radicals, charged molecules, and organo-metallics.
• Although it is possible to run an organic molecule with the “Spin Unrestricted” setting, it will be much slower computationally.
• Checking the “Symmetry” box in the setup tab can also apply to certain molecules, but not all.

www.design.che.vt.edu/VT-2005.html
Click the “Run Files” button to begin the DMol3 Click the “Run Files” button to begin the DMol3 Geometry Optimization.Geometry Optimization.• The geometry optimization predicts the energy
level of the molecule in the ideal gas phase. • This calculation is the longest step of the
procedure and will require ~75% of the time to produce a sigma profile.

www.design.che.vt.edu/VT-2005.html
The Project window will show the files created The Project window will show the files created and tracks the progress of the calculation.and tracks the progress of the calculation.• The *.xsd file is the optimized
geometry. All further calculations should use this file.
• The *.outmol file will show the ideal gas phase energy. There are additional files that do not show up in the Project window but are stored in the “Jobs” folder. (C:\Program Files\Accelrys\MS Modeling 3.2\Gateway\root_default\dsd\jobs)

www.design.che.vt.edu/VT-2005.html
We have now completed the Geometry We have now completed the Geometry Optimization and will proceed to the COSMO Optimization and will proceed to the COSMO calculation.calculation.• Begin by opening the
geometry optimization output, vt-0003.xsd, and open another DMol3 calculation dialog box.
• Select the task, “Energy.”• Leave all other options the
same as the geometry optimization.

www.design.che.vt.edu/VT-2005.html
Click the “Files” button in the calculation Click the “Files” button in the calculation window.window.• Click the “Save Files” button to create the input
files.

www.design.che.vt.edu/VT-2005.html
Open the propane.input file and add the Open the propane.input file and add the COSMO calculation keywords.COSMO calculation keywords.• The COSMO keywords tell the program which parameters to use in
the COSMO calculation including atomic radii, basis set version, etc.

www.design.che.vt.edu/VT-2005.html
Now we review the parameters and settings in Now we review the parameters and settings in the COSMO keywords.the COSMO keywords.• This statement turns the
COSMO calculation on.• Again, we will use basis set
version 4.0.0 instead of the default v3.5.
• The first number is the atomic number and the second term is the fitted atomic radii. Klamt (1998) fitted these parameters to several sets of data.

www.design.che.vt.edu/VT-2005.html
Now we will run the COSMO calculation.Now we will run the COSMO calculation.
• Click on the “Run Files” button from the calculation window to begin the energy calculation.
• Upon completion, the Project window will contain a *.cosmo and *.outmol file. There are additional files stored in the “Jobs” folder.

www.design.che.vt.edu/VT-2005.html
The *.The *.cosmocosmo file contains much useful file contains much useful information.information.• This file contains the
volume of the cavityaround the molecule in the theoretical conducting medium, and it is used in the COSMO-RS/COSMO-SAC models.
• It also contains the condensed phase energy, the number of surface segments, and their charge.

www.design.che.vt.edu/VT-2005.html
We edit the *.We edit the *.cosmocosmo file to make it compatible file to make it compatible with the sigma averaging FORTRAN program.with the sigma averaging FORTRAN program.• Record the number of surface segments.• Delete all information above the last table, which
contains the segment charges and coordinates. • Then save as a *.txt text file.
Delete alltext abovethis line.

www.design.che.vt.edu/VT-2005.html
Our sigma averaging program uses the Our sigma averaging program uses the modified *.modified *.cosmocosmo file to calculate the sigma file to calculate the sigma profile. profile. • We average the surface charge densities from the
*.cosmo file to find an effective surface charge density on a standard surface segment.
2 2 2*
2 2 2 2
2 2 2
2 2 2 2
exp
exp
n av mnn
n n av n avm
n av mn
n n av n av
r r dr r r r
r r dr r r r
σσ
� �−� �+ +� �=
� �−� �+ +� �
�
�

www.design.che.vt.edu/VT-2005.html
The averaging program creates a new text file, The averaging program creates a new text file, the sigma profile.the sigma profile.• The output file is “ ‘Chemical name’ Sigma-profile.txt” is
located in the C:\Profiles\ directory. This directory must be created manually before running the averaging program.
• The output can be input directly into the COSMO-SAC program or can be plotted graphically.

www.design.che.vt.edu/VT-2005.html
We run the FORTRAN program to average the We run the FORTRAN program to average the surface charges over a standardized bonding surface charges over a standardized bonding site.site.• Run sigmaprofile.exe.• Follow the prompts and enter:
– New COSMO output (c:/propane.txt)
– Chemical Name (Propane)– The Number of Surface Segments

www.design.che.vt.edu/VT-2005.html
The sigma averaging program output is text, but The sigma averaging program output is text, but can be easily converted into graphical form.can be easily converted into graphical form.
Sigma values, (e/A2) [X-axis]
P(σσσσ) values, (A2) [Y-axis]

www.design.che.vt.edu/VT-2005.html
Using Microsoft™ Excel, we display the sigma Using Microsoft™ Excel, we display the sigma profile below.profile below.
Propane Sigma Profile
0
5
10
15
20
25
-0.025 -0.020 -0.015 -0.010 -0.005 0.000 0.005 0.010 0.015 0.020 0.025
Sigma, σσσσ e/A2
P( σσ σσ
)*A
rea(
σσ σσ),
A2
VT-0003 Propane

www.design.che.vt.edu/VT-2005.html
Acknowledgments Acknowledgments
• We want to thank Alliant Techsystems (particularly Anthony Miano, Vice President), AspenTech(particularly Dustin MacNeil, Director of Worldwide University Programs, Larry Evans, Board Chairman, and Chau-Chyun Chen), China Petroleum and Chemical Corporation (particularly Wilfred Wang, President) and Honeywell Specialty Materials and Honeywell International Foundation for supporting our educational programs in computer-aided design and process systems engineering at Virginia Tech. Michael Zwolak gratefully appreciates support from an NSF Graduate Fellowship.

www.design.che.vt.edu/VT-2005.html
ReferencesReferences
Andzelm, Jan; Klamt, Andreas; and Kölmel, Christoph, “Incorporation of solvent effects into density functional calculations of molecular energies and geometries,” J. Chem. Phys., 103, 9312-9321 (1995)
Bustamante, P.; Pena, M.A.; Barra, J., The Modified Extended Hansen Method to Determine Partial Solubility Parameters of Drugs Containing a Single Hydrogen Bonding Group and Their Sodium Derivatives: Benzoic Acid/Na and Ibuprofen/Na,Int. J. of Pharmaceutics, 2000, 194, 117.
Giles, Neil F. and Wilson, Grant M., “Phase Equilibria on Seven Binary Mixtures,” J. Chem. Eng. Data., 45, 146-153 (2000).
Klamt, Andreas, G. Shurmann, “COSMO-A New Approach

www.design.che.vt.edu/VT-2005.html
ReferencesReferences
Lin, Shiang-Tai; Chang, Jaeeon; Wang, Shu; Goddard III, William A.; Sandler, Stanley I., “Prediction of Vapor Pressures and Enthalpies of Vaporization Using COSMO Solvation Model,” J. Phys. Chem., 108, 7429-7439, (2004).
Lin, Shiang-Tai and Stanley I. Sandler, “A Priori Phase Equilibrium Prediction from a Segment Contribution Solvation Model”, Ind. Eng. Chem. Res, 41, 899(2002a).
Lin, Shiang-Tai and Stanley I. Sandler, “Reply to Comments on “A Priori Phase Equilibrium Prediction from a Segment Contribution Solvation Model,” Ind. Eng. Chem. Res., 41, 2332 (2002b).
Panayiotou, Costas, “Equation-of-State Models and Quantum Mechanics Calculations,” Ind. Eng. Chem. Res, 42, 1495 (2003).
Top Related