







Languages
Pages
Legal

Once again I want to thank Larry Moran over at Sandwalk for these posts. I've collected them in one place so they aren't lost and that I can find them again when I would like to use them [non-profit, teaching only].
Part 1:
The Cori Cycle
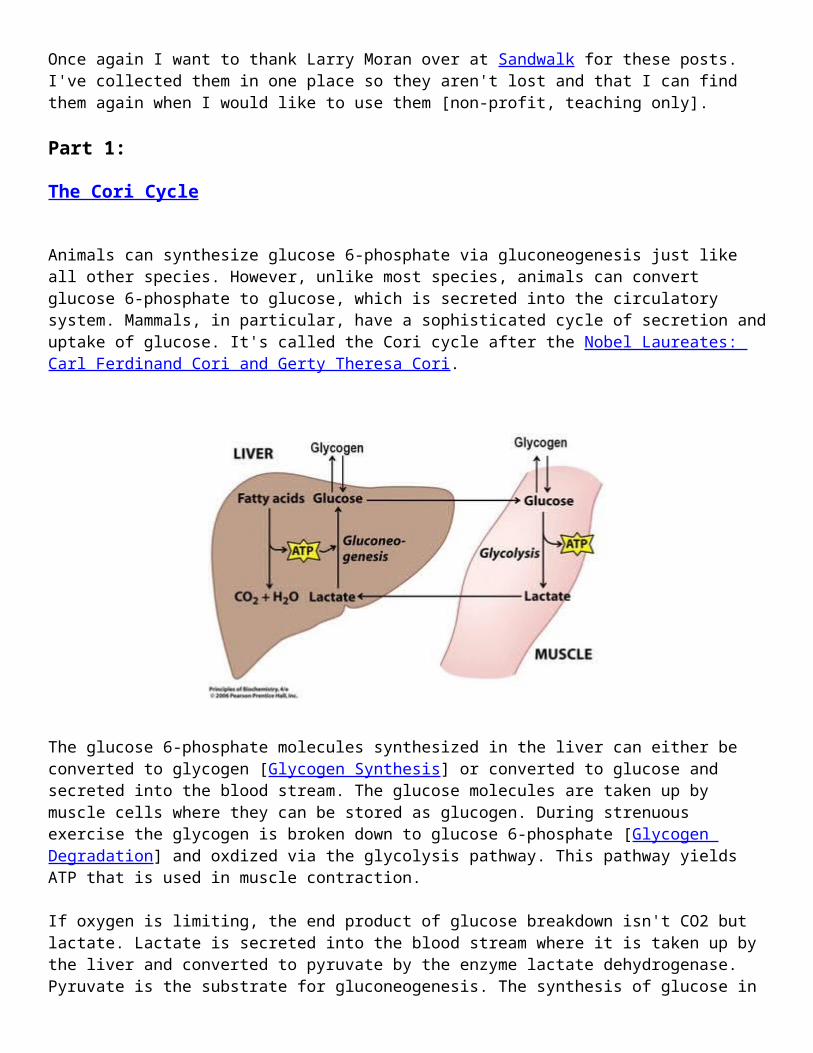
Animals can synthesize glucose 6-phosphate via gluconeogenesis just like all other species. However, unlike most species, animals can convert glucose 6-phosphate to glucose, which is secreted into the circulatory system. Mammals, in particular, have a sophisticated cycle of secretion and uptake of glucose. It's called the Cori cycle after the Nobel Laureates: Carl Ferdinand Cori and Gerty Theresa Cori.
The glucose 6-phosphate molecules synthesized in the liver can either be converted to glycogen [Glycogen Synthesis] or converted to glucose and secreted into the blood stream. The glucose molecules are taken up by muscle cells where they can be stored as glucogen. During strenuous exercise the glycogen is broken down to glucose 6-phosphate [Glycogen Degradation] and oxdized via the glycolysis pathway. This pathway yields ATP that is used in muscle contraction.
If oxygen is limiting, the end product of glucose breakdown isn't CO2 but lactate. Lactate is secreted into the blood stream where it is taken up by the liver and converted to pyruvate by the enzyme lactate dehydrogenase. Pyruvate is the substrate for gluconeogenesis. The synthesis of glucose in the liver requires energy in the form of ATP and this energy is supplied by a variety of sources. The breakdown of fatty acids is the source shown in the figure.
The Cori cycle preserves carbon atoms. The six carbon molecule, glucose, is split into two 3-carbon molecules (lactate) that are then converted to another 3-carbon molecule (pyruvate). Two pyruvates are joined to make glucose.
Part 2:

Glycogen Synthesis
All cells are capable of making glucose. The pathways is called gluconeogenesis and the end product is not actually glucose but a phosphorylated intermediate called glucose-6-phosphate.
Glucose-6-phosphate (G6P) serves as the precursor for synthesis of many other compounds such as the ribose sugars needed in making DNA and RNA. The only organisms that make free glucose are multicellular organisms, such as animals, that secrete it into the circulatory system so it can be taken up and used by other cells (Glucose-6-phosphate cannot diffuse across the membrane so it's retained within cells.)
In times of plenty, G6P may not be needed in further biosynthesis reactions so cells have evolved a way of storing, or banking, excess glucose. The stored glucose molecules can then be retrieved when times get tough. Think of bacteria growing in the ocean, for example. There may be times when an abundant supply of CO2 combined with a surplus of inorganic energy sources (e.g., H2S) allows for synthesis of lots of G6P. These cells can store the excess G6P by making glycogen—a polymer of glucose residues.
Glycogen consists of long chains of glucose molecules joined end-to-end through their carbon atoms at the 1 and 4 positions. The chains can have many branches. Completed chains can have up to 6000 glucose residues making glycogen one of the largest molecules in living cells.
The advantage of converting G6P to glycogen is that it avoids the concentration effects of having too many small molecules floating around inside the cell. By compacting all these molecules into a single large polymer the cell is able to form large granules of stored sugar (see photo above).
The first step in the synthesis of glycogen is the conversion of glucose-6-phosphate to glucose-1-phosphate by the action of an enzyme called phosphoglucomutase. (Mutases are enzymes that rearrange functional groups, in this case moving a phosphate from the 6 position of glucose to the 1 position.) The glycogen synthesis reaction requires adding new molecules that will be connected to the chain through their #1 carbon atoms so this preliminary reaction is required in order to "activate" the right end of the glucose residue.

Glucose-1-phosphate is the "Cori ester" [Monday's Molecule #25] that was discovered by Carl Cori and Gerty Cori while they were working out this pathway [Nobel Laureates: Carl Cori and Gerty Cori].
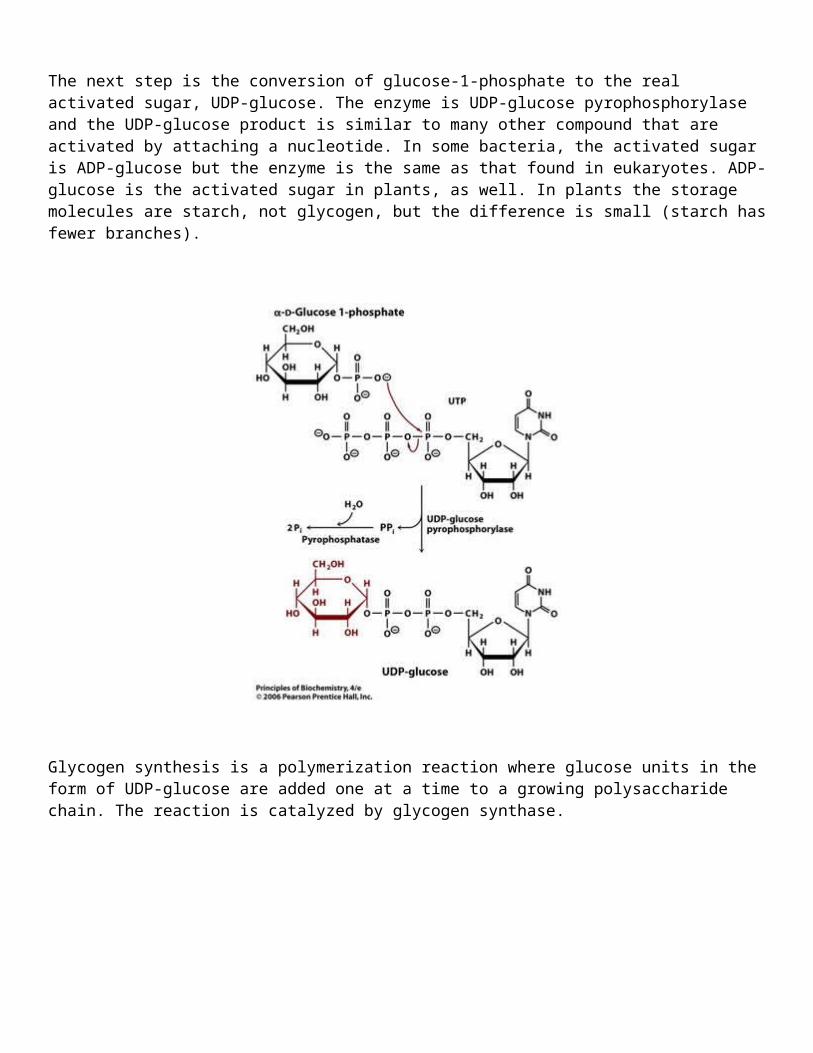
The next step is the conversion of glucose-1-phosphate to the real activated sugar, UDP-glucose. The enzyme is UDP-glucose pyrophosphorylase and the UDP-glucose product is similar to many other compound that are activated by attaching a nucleotide. In some bacteria, the activated sugar is ADP-glucose but the enzyme is the same as that found in eukaryotes. ADP-glucose is the activated sugar in plants, as well. In plants the storage molecules are starch, not glycogen, but the difference is small (starch has fewer branches).
Glycogen synthesis is a polymerization reaction where glucose units in the form of UDP-glucose are added one at a time to a growing polysaccharide chain. The reaction is catalyzed by glycogen synthase.
[©Laurence A. Moran. Some of the text is from Principles of Biochemistry 4th ed. ©Pearson/Prentice Hall]
Part 3:
Glycogen Degradation/Utilization
Glucose is stored as the intracellular polysaccharides starch and glycogen. Starch occurs mostly in plants. Glycogen is an important storage polysaccharide in bacteria, protists, fungi and animals. Glycogen is stored in large granules. In mammals, these granules are found in muscle and liver cells. In electron micrographs, liver glycogen appears as clusters of cytosolic granules with a diameter of 100 nm—much larger than ribosomes. The enzymes required for synthesis of glycogen are found in muscle and liver cells [Glycogen Synthesis]. Those same cells contain the enzymes for glycogen degradation.
The glucose residues of starch and glycogen are released from storage polymers through the action of enzymes called polysaccharide phosphorylases: starch phosphorylase (in plants) and glycogen phosphorylase (in many other organisms). These enzymes catalyze the removal of glucose residues from the ends of starch or glycogen. As the name implies, the enzymes catalyze phosphorolysis—cleavage of a bond by group transfer to an oxygen atom of phosphate. In contrast to hydrolysis (group transfer to water), phosphorolysis produces phosphate esters. Thus, the first product of polysaccharide breakdown is α-D-glucose 1-phosphate, not free glucose.

Glucose 1-phosphate is one of the precursors required for glycogen synthesis. It is the "Cori ester" [Monday's Molecule] discovered by Carl Cori and Gerty Cori [Nobel Laureates: Carl Cori and Gerty Cori]. The Cori's also discovered and characterized glycogen phosphorylase.
In order for glucose 1-phosphate to be used in other pathways it has to be converted to glucose 6-phosphate by the enzyme phosphoglucomutase. This is the same enzyme that's used in the synthesis of glycogen from glucose 6-phosphate. Glucose 6-phosphate can be oxidzed by the glycolysis pathway to produce ATP. This is what happens in muscle cells. Glucose is stored as glycogen during times of rest but during exercise the glycogen is broken down to glucose 6-phosphate and glycolysis is activated. The resulting ATP is used in muscle activity.Obviously, there has to be a balance between the synthesis and degradation of glycogen and this balance is maintained by regulating the activities of the biosynthesis and degradation enzymes. This regulation occurs at many levels. Regulation by hormones is one of the classic examples of a signal transduction pathway in mammals.
[©Laurence A. Moran. Some of the text is from Principles of Biochemistry 4th ed. ©Pearson/Prentice Hall]
Part 4:

Regulating Glycogen Metabolism
Mammalian glycogen stores glucose in times of plenty (after feeding, a time of high glucose levels) and supplies glucose in times of need (during fasting or in “fight-or-flight” situations). In muscle, glycogen provides fuel for muscle contraction. In contrast, liver glycogen is largely converted to glucose that exits liver cells and enters the bloodstream for transport to other tissues that require it [The Cori Cycle]. Both the mobilization and synthesis of glycogen are regulated by hormones.
The regulation of glycogen metabolism is a good way to introduce the idea of signal transduction. This is a very popular part of modern biochemistry. It's basically a way in which signals from outside the cell are transduced through a chain of molecules to affect a particular biochemical reaction. In this case, we'll examine how the hormones glucagon, epinephrine, and insulin regulate glycogen synthesis and glycogen degradation.
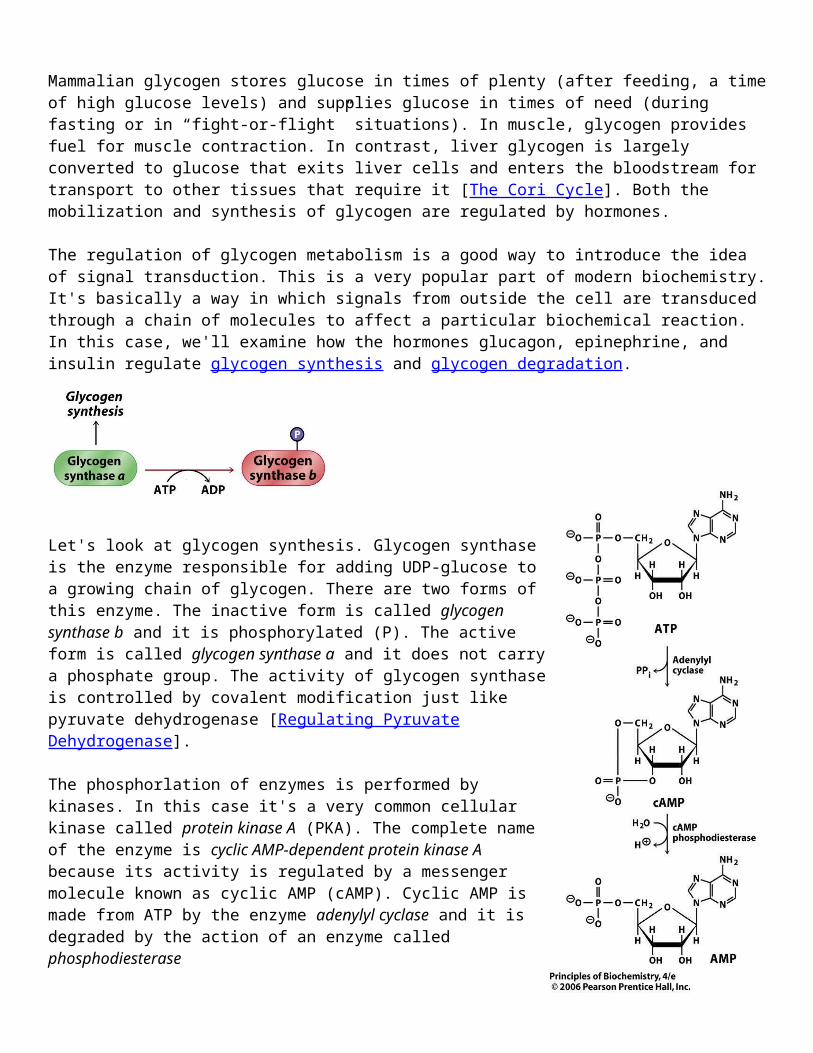
Let's look at glycogen synthesis. Glycogen synthase is the enzyme responsible for adding UDP-glucose to a growing chain of glycogen. There are two forms of this enzyme. The inactive form is called glycogen synthase b and it is phosphorylated (P). The active form is called glycogen synthase a and it does not carry a phosphate group. The activity of glycogen synthase is controlled by covalent modification just like pyruvate dehydrogenase [Regulating Pyruvate Dehydrogenase].
The phosphorlation of enzymes is performed by kinases. In this case it's a very common cellular kinase called protein kinase A (PKA). The complete name of the enzyme is cyclic AMP-dependent protein kinase A because its activity is regulated by a messenger molecule known as cyclic AMP (cAMP). Cyclic AMP is made from ATP by the enzyme adenylyl cyclase and it is degraded by the action of an enzyme called phosphodiesterase
When cAMP is present inside the cell it binds to protein kinase A and activates it so that it can phosphorylate glycogen synthase. This shuts down glycogen synthesis by deactivating the enzyme. The key to hormonal regulation is the effect of the hormones on the production of cAMP. This takes place on the cell surface when the hormone binds to a cell surface receptor molecule.
Insulin, glucagon, and epinephrine are the principal hormones that

control glycogen metabolism in mammals. Insulin, a 51-residue protein is synthesized by the cells of the pancreas. It is secreted when the concentration of glucose in the blood increases. Thus, high levels of insulin are associated with the fed state of an animal. Insulin stimulates glycogen synthesis in the liver. This makes sense since high concentrations of glucose indicate that it's time to store it as glycogen.
Glucagon, a peptide hormone containing 29 amino acid residues, is secreted by the cells of the pancreas in response to a low blood glucose concentration. Glucagon restores the blood glucose concentration to a steady-state level by stimulating glycogen degradation. Only liver cells are rich in glucagon receptors, so glucagon is extremely selective in its target. The effect of glucagon is opposite that of insulin, and an elevated glucagon concentration is associated with the fasted state.
The adrenal glands release the catecholamine epinephrine (also known as adrenaline) in response to neural signals that trigger the fight-or-flight response. Epinephrine stimulates the breakdown of glycogen to glucose 1-phosphate, which is converted to glucose 6-phosphate. The increase in intracellular glucose 6-phosphate increases both the rate of glycolysis in muscle and the amount of glucose released into the bloodstream from the liver. Note that epinephrine triggers a response to a sudden energy requirement; glucagon and insulin act over longer periods to maintain a relatively constant concentration of glucose in the blood.
Epinephrine binds to β-adrenergic receptors of liver and muscle cells and to α1-adrenergic receptors of liver cells. The binding of epinephrine to β-adrenergic receptors or of glucagon to its receptors activates the adenylyl cyclase signaling pathway. The second messenger, cyclic AMP (cAMP), then activates protein kinase A.
For now let's just take it as a given that glucagon and epinephrine trigger cAMP synthesis and this leads to shutting down of glycogen synthesis.
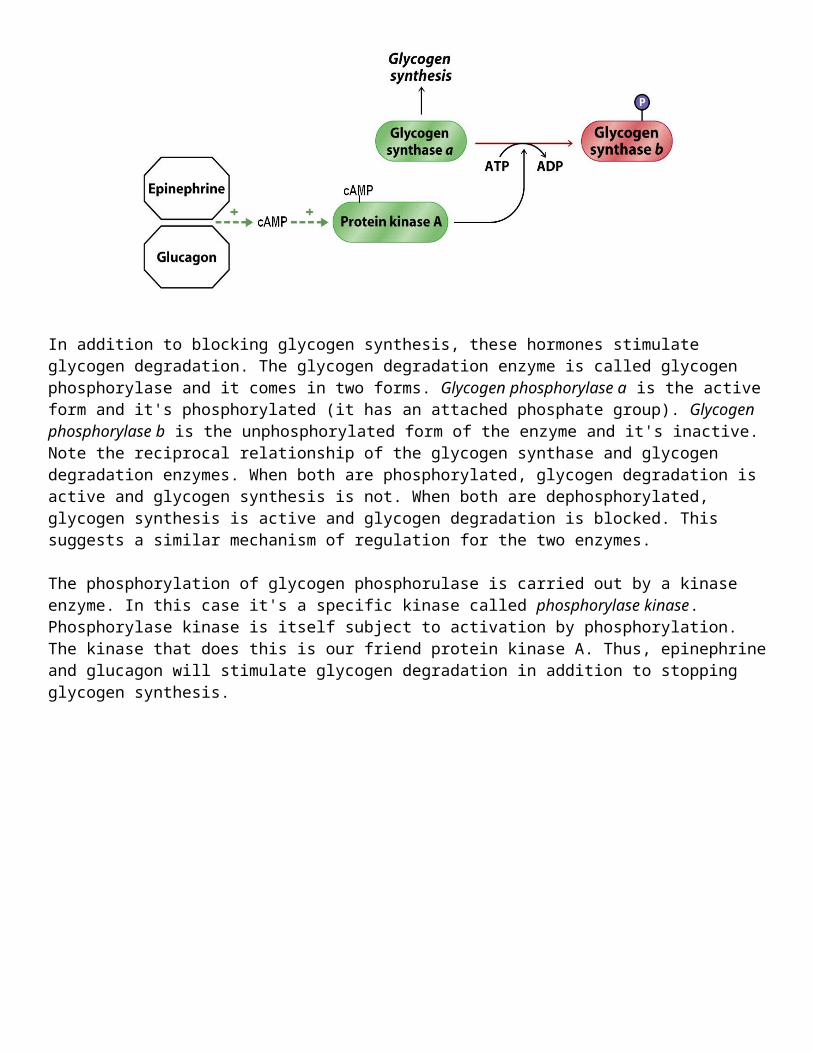
In addition to blocking glycogen synthesis, these hormones stimulate glycogen degradation. The glycogen degradation enzyme is called glycogen phosphorylase and it comes in two forms. Glycogen phosphorylase a is the active form and it's phosphorylated (it has an attached phosphate group). Glycogen phosphorylase b is the unphosphorylated form of the enzyme and it's inactive. Note the reciprocal relationship of the glycogen synthase and glycogen degradation enzymes. When both are phosphorylated, glycogen degradation is active and glycogen synthesis is not. When both are
dephosphorylated, glycogen synthesis is active and glycogen degradation is blocked. This suggests a similar mechanism of regulation for the two enzymes.
The phosphorylation of glycogen phosphorulase is carried out by a kinase enzyme. In this case it's a specific kinase called phosphorylase kinase. Phosphorylase kinase is itself subject to activation by phosphorylation. The kinase that does this is our friend protein kinase A. Thus, epinephrine and glucagon will stimulate glycogen degradation in addition to stopping glycogen synthesis.
For every kinase there's a phosphatase that removes phosphate groups from proteins. Recall that insulin is released when glucose levels in the blood are high. The effect of insulin is the exact opposite of the effect of glucagon and epinephrine. Insulin binds to a cell surface receptor and triggers a pathway that leads to activation of protein phosphatase-1. This enzyme dephosphorylates the three enzymes shown above leading to activation of glycogen synthesis and deactivation of glycogen degradation. Insulin causes glucose to be stored as glycogen.
These kinds of kinase/phosphatase cascades are very common in eukaryotes. Believe it or not, this is one of the simpler examples.
Now, let's return to the effect of the hormone on cAMP synthesis. This is the key part of any signaling pathway and it's best illustrated by using a general model based on cAMP production. (There are other types of signaling pathways.)

The details aren't important unless you're seriously into signaling—like 50% of all biochemistry graduate students these days. Hormone binds to a cell surface receptor. The signal is transferred through the cell membrane to the inside part of the receptor molecule. This interacts with a G protein so that when hormone binds, the G protein is activated.
G protein then diffuses to the membrane bound adenylyl cyclase molecule and, when the two proteins connect, the activity of adenylyl cyclase is stimulated and cAMP is produced. This leads to activation of protein kinase A. The stimulatory effect of the signal transduction pathway is transient because cAMP is rapidly degraded by phosphodiesterase. Thus, hormone must usually be continuously present in order to get stimulation.
There are other hormones that inhibit cAMP production by activating different G proteins (Gi) that block adenylyl cyclase.
[©Laurence A. Moran. Some of the text is from Principles of Biochemistry 4th ed. ©Pearson/Prentice Hall]
Part 4? Glycogen Storage Diseases
Type 0: Hypoglycemia due to lack of glycogen synthase [OMIM 240600, OMIM 138571]Glycogen synthase is the enzyme required for glycogen synthesis [Glycogen Synthesis]. There are two forms of the enzyme; liver and muscle. The muscle form is found in many different tissues but the liver version of the enzyme is only found in liver cells. Mutations in the gene for the liver enzyme (GTS2) cause glycogen storage disease type 0.
The disease is usually recognized in infants who have very low blood sugar (hypoglycemia) after a short fast. The low sugar is due to the fact that there's no store of glycogen in the liver. In normal cases, the liver stores glucose as glycogen right after a meal then breaks it down as blood glucose is depleted. In the absence of liver glycogen synthase the maintenance of blood sugar levels is impaired.Here's what OMIM has to say about typical cases.Gitzelmann et al. (1996) described 3 children with liver glycogen synthase deficiency from 2 German families and compared the observations with the previously published 3 families comprising 8 patients. The 2 index cases presented with morning fatigue, had ketotic hypoglycemia when fasting which rapidly disappeared after eating, and hepatic glycogen deficiency with absent or very low hepatic glycogen synthase activity. Metabolic profiles comprising glucose, lactate, alanine, and ketones in blood were typical for hepatic glycogen synthase deficiency. Symptoms were rapidly relieved and chemical signs corrected by introducing frequent protein-rich meals and nighttime feedings of suspensions of uncooked corn starch. The discovery of oligosymptomatic and asymptomatic sibs suggested that there are persons with undiagnosed hepatic glycogen synthase deficiency. Gitzelmann et al. (1996) stated that the disorder should be sought in children who, before the first meal of the day, present with drowsiness, lack of attention, pallor, uncoordinated eye movements, disorientation, or convulsions, and who have hypoglycemia and acetone in the urine.
Type I: Von Gierke's Disease: Deficiency in glucose 6-phosphatase [Ia OMIM 232200, Ib OMIM 602671, Ib OMIM 232220, Ic OMIM #232240]
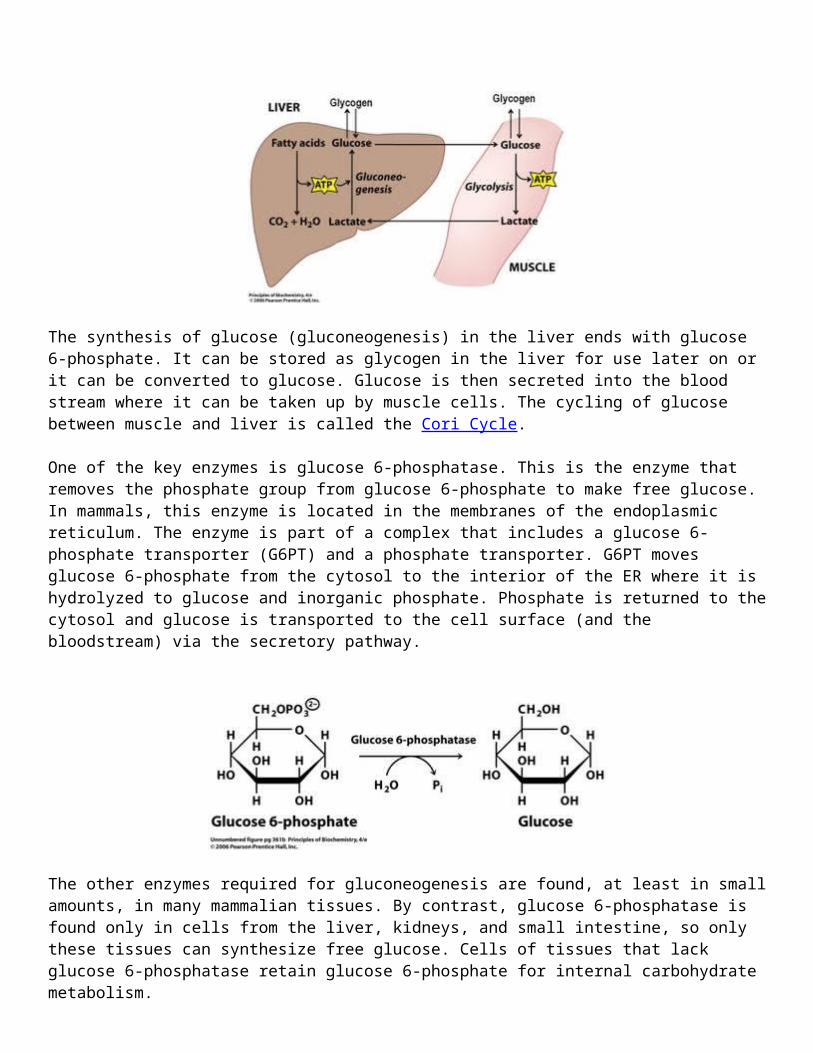
The synthesis of glucose (gluconeogenesis) in the liver ends with glucose 6-phosphate. It can be stored as glycogen in the liver for use later on or it can be converted to glucose. Glucose is then secreted into the blood stream where it can be taken up by muscle cells. The cycling of glucose between muscle and liver is called the Cori Cycle.
One of the key enzymes is glucose 6-phosphatase. This is the enzyme that removes the phosphate

group from glucose 6-phosphate to make free glucose. In mammals, this enzyme is located in the membranes of the endoplasmic reticulum. The enzyme is part of a complex that includes a glucose 6-phosphate transporter (G6PT) and a phosphate transporter. G6PT moves glucose 6-phosphate from the cytosol to the interior of the ER where it is hydrolyzed to glucose and inorganic phosphate. Phosphate is returned to the cytosol and glucose is transported to the cell surface (and the bloodstream) via the secretory pathway.
The other enzymes required for gluconeogenesis are found, at least in small amounts, in many mammalian tissues. By contrast, glucose 6-phosphatase is found only in cells from the liver, kidneys, and small intestine, so only these tissues can synthesize free glucose. Cells of tissues that lack glucose 6-phosphatase retain glucose 6-phosphate for internal carbohydrate metabolism.
Defects in glucose 6-phosphatase affect mostly liver and kidneys where stored glycogen can accumulate to high levels due to the fact that it can't be broken down to free glucose for secretion into the blood stream. Glycogen storage disease Ia results from mutations in the catalytic subunit of glucose 6-phosphatase (G6PC gene) while Ib and Ic are caused by mutations in the transporter subunits.
The major problem is hypoglycemia (low glucose) and lactic acidemia due to inefficient conversion of lactic acid to free glucose. These can be fatal but nowadays the symptoms are treated by feeding carbohydrates at regular intervals throughout the day and though a gut tube at night. This is an autosomal recessive disease.
Type II (Pompe Disease): Deficiency of α:-glucosidase [OMIM #232300, OMIM 606800]Glycogen granules are taken up by lysosomes where they are broken down by a pathway that's different from the normal glycogen degradation pathway. One of the key lysosomal enzymes is α1,4-glucosidase. Mutations in the gene for this enzyme cause glycogen storage disease type II.
This is a very severe form of the disease. Although glycogen breakdown in lysosomes is relatively minor in terms of overall glycogen metabolism, the inability to process glycogen granules leads to their accumulation in lysosomes and consequent disruption of many important lysosomal functions. This disruption takes place in all cells and all tissues.
In the classic cases, infants are inactive and hypertonic with enlarged hearts. Death usually occurs before the first year, usually from heart failure. An adult onset version is known. It usually begins with respiratory difficulties and often ends in death from ruptures of the arteries or respiratory failure.

Type III: (Cori Disease): Defects in glycogen debranching enzyme [OMIM 232400, OMIM 610860]
The glycogen debranching enzyme is required for the complete mobilization of glucose from glycogen. The standard glycogen phosphorylase enzyme will lop off glucose residues until it come to within for residues of a branch point in the glycogen chain. This produces a truncated glycogen molecule known as limit dextrin.
Further degradation of glycogen requires the activity of the debranching enzyme which actually has two separate activities: a glucanotransferase activity that transfers glucose residues from the end of one branch to the end of another, and a glucosidase activity that chops off the last glucose residue on a branch. The gene is the AGL gene (amylo-1,6-glucosidase, 4-α -glucanotransferase) in humans and the GDE gene (glycogen debranching enzyme) in many other species.
There are several subtypes of type III glycogen storage disease. They all result from mutations in the AGL gene. The most common type is IIIa where debranching activity is missing in both liver and muscle cells [see The Cori Cycle]. Patients have muscle weakness and liver problems similar to those in von Gierke's Disease (type I) but the symptoms are milder and not usually life threatening.
In type IIIb the deficiency in debranching enzyme is only detectable in liver. This is probably due to lower production of functional enzyme that only affects liver cells where more debranching enzyme is needed than in muscle cells.
Types IIIc and IIId are quite rare. They only affect the glucanotransferase activity (IIIc) or the glucosidase activity (IIId).
Type IV (Anderson Disease): Deficiency in glycogen branching enzyme [OMIM #232500, OMIM 607839]Glycogen storage disease IV is caused by a deficiency of glycogen branching enzyme (amylo-(1,4 → 1,6)-transglycosylase). This is the enzyme that adds new branches for glycogen during synthesis. A deficiency in this enzyme results in reduced ability to store glucose residues in glycogen.(This is the enzyme responsible for the wrinkled pea phenotype that Gregor Mendel studied. [ Biochemist Gregor Mendel Studied Starch Synthesis.)
The disease is severe according to OMIM.Glycogen storage disease type IV is a clinically heterogeneous disorder. The typical 'classic' hepatic presentation is liver disease of childhood, progressing to lethal cirrhosis. The neuromuscular presentation of GSD IV is distinguished by age at onset into 4 groups: perinatal, presenting as fetal akinesia deformation sequence (FADS) and perinatal death; congenital, with hypotonia, neuronal involvement, and death in early infancy; childhood, with myopathy or cardiomyopathy; and adult, with isolated myopathy or adult polyglucosan body disease (Bruno et al., 2004). The enzyme deficiency

results in tissue accumulation of abnormal glycogen with fewer branching points and longer outer branches, resembling an amylopectin-like structure, also known as polyglucosan (Tay et al., 2004).
Type V (McArdle Disease): Deficiency of muscle glycogen phosphorylase [OMIM 23600, OMIM 608455]Glycogen phosphorylase is the enzyme that degrades glycogen [Glycogen Degadation]. Deficiencies in the muscle form of the enzyme lead to severe muscle cramps. Patients are not able to perfom strenuous exercise. The lack of muscle glycogen phosphorylase prevents breakdown of glycogen in muscle and consequent lack of glucose to fuel ATP production via glycolysis. One of the characteristic symptoms is an absence of blood lactate since muscle cells are unable to convert glycogen to glucose and then to lactate.
Muscle tissue breaks down due to lack of ATP leading to general weakness, especially in adults. The disease is not fatal; in fact, it is relatively harmless as long as patients avoid exercise.
Type VI (Hers Disease): Deficiency in liver phosphorylase [OMIM 23700]Deficiencies of the liver form of glycogen phosphorylase are not as harmful as deficiencies of the muscle version (type V). The disease is inherited as an autosomal recessive and it's due to mutations in the gene for the liver form of glycogen phosphorylase.
The symptoms are mild compared to other forms of glycogen storage disease, giving rise to enlarged liver with mild hypoglycemia, mild ketosis, and retarded growth.
Type VII (Tarui Disease): Muscle phosphofructokinase deficiency [OMIM #232800, OMIM 610681]According to OMIM,Glycogen storage disease VII is an autosomal recessive metabolic disorder characterized clinically by exercise intolerance, muscle cramping, exertional myopathy, and compensated hemolysis. Myoglobinuria may also occur. The deficiency of the muscle isoform of PFK results in a total and partial loss of muscle and red cell PFK activity, respectively. Raben and Sherman (1995) noted that not all patients with GSD VII seek medical care because in some cases it is a relatively mild disorder.Muscle phosphofructokinase (PFKM) is an enzyme required for glycolysis. When glycolysis is blocked in muscle cells glycogen cannot be broken down and there is no abundant supply of ATP available for muscle activity.
Type IXa (X-linked liver glycogenosis): Deficiency of liver phosphorylase kinase [OMIM 306000Phosphorylase kinase is the enzyme that phosphorylates glycogen phosphorylase in order to regulate its activity [Regulating Glycogen Metabolism]. Defects in the phosphorylase kinase gene (PHK) cause glycogenstorage disease type IXa— a very mild form of the disease according to OMIM.
Deficiency of liver phosphorylase kinase (PHK; ATP:phosphotransferase; EC 2.7.1.38) produces one of the mildest of the glycogenoses of man. The clinical symptoms include hepatomegaly, growth retardation, elevation of glutamate-pyruvate transaminase and glutamate-oxaloacetate transaminase, hypercholesterolemia, hypertriglyceridemia, and fasting hyperketosis (Schimke et al., 1973; Willems et al., 1990). With age, these clinical and biochemical abnormalities gradually disappear and most adult patients are asymptomatic.Phosphorylase kinase consists of α, β, γ, and δ sunbuits each of which is encoded by specific genes. Defects in the α subunit gene (PHKA) are what causes glycogen storage disease type IXa. There are two different genes for α subunits on the X chromosome: one for the liver specific version of the enzyme (PHA2) and one for the muscle specific version of the enzyme (PHA1). Mutations in either one cause the disease, which is why it is called an X-linked glycogen storage disease.
Top Related