


![Unique Glycemic and Cardio-Renal Protective Effects of ... · beneficial changes in blood rheology, serum lipid profile, and putative anti-ischemic effects [11,12]. Although exact](https://static.fdocuments.net/doc/165x107/5af0a06c7f8b9a8b4c8db581/unique-glycemic-and-cardio-renal-protective-effects-of-changes-in-blood-rheology.jpg)




Languages
Pages
Legal


Synthesis of Novel Dual Acting Cardio-
protective Agents
VIRAJ H MANKAR
[Masters of Science (Drug Chemistry)]
A Thesis Submitted in Fulfilment of the Requirements for the Degree of
Doctor of Philosophy.
School of Chemistry, Physics and Mechanical Engineering
Science and Engineering Faculty
Queensland University of Technology
Brisbane, Australia
[2016]

II
Keywords
Adenosine, adenosine receptors, allosteric modulator, antioxidants, chemistry,
nitroxides, free radicals, synthesis, cardio-protection, ischaemia, oxidative
stress, dual-acting drug.

III
Abstract
Adenosine and adenosine derivatives are recognised as cardioprotective
agents due to their ability to activate myocardial adenosine receptors.
Antioxidants such as nitroxide radicals have also been demonstrated to provide
cardioprotective effects by scavenging oxygen based damaging free radicals.
In this thesis, three series of C2-linked adenosine 5-N-ethylcarboxamide
derivatives bearing antioxidant nitroxide and phenolic moieties were designed
and synthesised. It was envisaged that linking of the antioxidant moiety to the
adenosine skeleton would produce adenosine analogues that bind to and
activate adenosine receptors and, in addition, scavenge harmful oxygen-
centered free radicals generated in the body.
Evaluation of the prepared compounds as A2AAR agonists using the cAMP
accumulation assay identified a number of potent and selective agonists. Of the
compounds examined, the adenosine 5–N-ethylcarboxamide compounds,
possessing various nitroxides linked through para-(2-aminoethyl)aniline
linkages (namely compounds designated as 162, 164, 166, 168 and 170 in this
Thesis) displayed high selectivity, with more than 200-fold selectivity for the
adenosine A2A receptors over adenosine A1 receptors, whilst retaining
reasonable selectivity over adenosine A3 receptors.
Of the adenosine compounds examined, PROXYL nitroxide (166) and tetraethyl
isoindoline nitroxide (170) possessing a para-(2-aminoethyl)aniline linked
adenosine 5–N-ethylcarboxamide unit approached high selectivity for the A2A
receptors over the other adenosine receptor subtypes. Four of the

IV
highly-selective adenosine compounds (162, 164, 166, and 170) were further
investigated in the simulated ischaemia model using rat atrial cardiomyoblast
cells. All four of these compounds showed strong protective effects in this
assay. There were some additional cardioprotective effects observed at the
highest agonist concentration in the presence of the antagonist. This result
indicates that the antioxidant moiety may contribute to the compounds
cardioprotective effects at high levels of agonist concentration.
Ethynyl- and ethyl-linked structural classes of adenosine 5–N-ethylcarboxamide
compounds were also synthesised bearing tetraalkylisoindoline nitroxides. All
compounds from these series are less-potent A2A agonists than the previously-
mentioned candidates. However, the ethynyl linked tetramethylisoindoline (178)
and tetraethylisoindoline nitroxide (182) bearing adenosine 5-N-
ethylcarboxamide and ethyl linked tetraethylisoindoline nitroxide bearing
adenosine 5-N-ethylcarboxamide (184) have a higher selectivity for A2B
receptors than other adenosine receptor subtypes.
This project has resulted in the synthesis and characterization of twelve novel
C2-substituted adenosine analogues with three different linker groups. Most of
the target compounds were tested for their binding ability to adenosine receptor
subtypes, and some analogues were found to be potent and selective
adenosine receptor subtype agonists.

V
Table of Contents
Keywords II
Abstract III
Table of Contents V
List of Figures XIV
List of Schemes XVI
List of Tables XIX
List of Abbreviations XX
Statement of Original Authorship XXVI
Acknowledgements XXVII
Chapter 1: Introduction & Literature Review 1
1.1 Cardiovascular diseases 2
1.1.1 Ischaemic heart disease 3
1.1.2 Reperfusion injury 5
1.1.3 Free radicals 6
1.1.4 Oxidative stress 7
1.2 Antioxidants 9
1.2.1 Nitroxide radical 13
1.2.2 Application of nitroxides 18
1.2.2.1 Radical trapping and spin labelling 18
1.2.2.2 Nitroxide antioxidants 20
1.2.3 Phenolic antioxidants 21
1.3 Adenosine 25
1.3.1 Structural features of adenosine 25

VI
1.3.2 Adenosine receptors 26
1.3.2.1 Adenosine A1 receptors 31
1.3.2.2 Adenosine A2A receptors 32
1.3.2.2 Adenosine A2B receptors 34
1.3.2.2 Adenosine A3 receptors 35
1.3.3 The concept of agonist and antagonists 36
1.3.3.1 Adenosine agonists 37
1.3.3.2 Adenosine antagonists 38
1.3.4 Active adenosine A2AAR agonists and antagonists in clinical
trials
40
1.3.4.1 Adenosine A2AAR agonists 40
1.3.4.2 Adenosine A2AAR antagonists 42
1.3.4.3 Adenosine A2BAR agonists 45
1.3.4.4 Adenosine A2BAR antagonists 46
1.4 Dual action adenosine agonists: -Rationale for the research 48
Chapter 2: Result and Discussion: Part A: Adenosine analogue with
para-aminophenylethylamine linker possessing antioxidant moieties
57
2.1 Preliminary considerations 58
2.2 Synthesis of C2-substituted TMIO adenosine analogues
possessing substituent attached to ‘aminoethylaniline
linker’ moiety (162)
64
2.2.1 Synthesis of antioxidant carboxy TMIO nitroxide
intermediates possessing substituent attached to
‘aminoethylaniline linker’ moiety (127)
65
2.2.1.1 Synthesis of antioxidant carboxy TMIO intermediate (106) 65
2.2.1.2 Synthesis of antioxidant carboxy TMIO linked
aminoethylaniline chain linker intermediate (127)
67
2.2.2 Synthesis of C2-substituted adenosine intermediates (146) 72

VII
2.2.2.1 Convergent approach to synthesis of adenosine
intermediate
73
2.2.2.2 Adenosine intermediate for C2-functionalization with a
Linear approach
75
2.2.3 Synthesis of TMIO adenosine analogue with
aminoethylaniline linker (161) (Approach 1).
90
2.2.4 Synthesis of TMIO adenosine analogue with
aminoethylaniline linker (161) (Approach 2).
94
2.3 Synthesis of C2-substituted CTEMPO adenosine analogues
possessing substituent attached to ‘aminoethylaniline linker’
moiety (164)
101
2.4 Synthesis of C2-substituted CPROXYL adenosine
analogues possessing substituent attached to
‘aminoethylaniline linker’ moiety (166).
104
2.5 Synthesis of C2-substituted MCTMIO adenosine analogues
possessing substituent attached to ‘aminoethylaniline linker’
moiety (168).
106
2.6 Synthesis of C2-substituted CTEIO adenosine analogues
possessing substituent attached to ‘aminoethylaniline linker’
moiety (170)
110
2.6.1 Synthesis of antioxidant carboxy TEIO intermediate (114) 110
2.6.2 Coupling of carboxy TEIO intermediate with
aminoethylaniline linked adenosine moiety (169)
111
2.7 Synthesis of C2-substituted di-tert-butylhydroxycinnamic
acid adenosine analogues possessing substituent attached
to ‘aminoethylaniline linker’ moiety (172)
114
2.7.1 Synthesis of antioxidant di-tert-butylhydroxycinnamic acid
linked ‘aminoethylaniline linker’ intermediate (133)
115
2.7.2 Synthesis of C2-substituted di-tert-butylhydroxycinnamic
acid adenosine analogue possessing substituent attached
by ‘aminoethylaniline linker’ moiety (171) (Approach 1).
118
2.7.3 Synthesis of C2-substituted di-tert-butylhydroxycinnamic
acid adenosine analogue possessing substituent attached
by ‘aminoethylaniline linker’ moiety (172) (Approach 2).
120

VIII
2.8 Result of Biological testing of adenosine analogue 123
2.8.1 Adenosine receptor binding assays 123
2.8.2 Simulated ischemia assay. 129
Chapter 3: Result and Discussion: Part B: Adenosine analogue
with ethynyl linker possessing antioxidant moieties.
132
3.1 Synthesis of C2-substituted TMIO adenosine analogues
possessing substituent attached to ‘ethynyl linker’ moiety
(178)
134
3.1.1 Synthesis of the 2-iodo-2,3-O-isopropylideneadenosine-5-
N-ethylcarboxamide intermediate (149).
134
3.1.2 Synthesis of ethynyl TMIO (ETMIO) intermediate (120). 137
3.1.3 Coupling of ETMIO with iodo-adenosine intermediate(178). 139
3.2 Synthesis of C2-substituted TEIO adenosine analogues
possessing substituent attached to ‘ethynyl linker’ moiety
(182)
143
3.3 Synthesis of C2-substituted phenyl adenosine analogues
possessing substituent attached to ‘ethynyl linker’ moiety
(185)
146
3.4 Result of biological testing of adenosine analogue 147
Chapter 4: Result and Discussion: Part C: Adenosine analogue
with ethyl linker possessing antioxidant moieties
151
4.1 Synthesis of C2-substituted TMIO adenosine analogues
possessing substituent attached to ‘ethyl linker’ moiety (180)
153
4.2 Synthesis of C2-substituted TEIO adenosine analogues
possessing substituent attached to ‘ethyl linker’ moiety (184)
156
4.3 Synthesis of C2-substituted non-nitroxide phenyl adenosine
analogues possessing substituent attached to ‘ethyl linker’
moiety (188)
158
4.4 Result of Biological testing of adenosine analogue 161
Chapter 5: Conclusion 165
5.1 Synthetic chemistry 166

IX
5.1.1 Adenosine based antioxidant compounds with para-
aminoethylaniline linker
167
5.1.2 Adenosine based antioxidant compounds with ethynyl linker 168
5.1.3 Adenosine based antioxidant compounds with ethyl linker 169
5.2 Biological analysis 170
5.2.1 Adenosine based antioxidant compounds with para-
aminoethylaniline linker
170
5.2.2 Adenosine based antioxidant compounds with ethynyl linker 172
5.2.3 Adenosine based antioxidant compounds with ethyl linker 172
Chapter 6: Future work 174
Chapter 7: Experimental 177
7.1 General experimental 178
7.2 Synthetic procedures 180
7.2.1 Synthesis of 2-benzyl phthalimide (100) 180
7.2.2 Synthesis of 2-benzyl-1, 1, 3, 3- tetramethylisoindoline (101) 181
7.2.3 Synthesis of 5-bromo-1,1,3,3-tetramethylisoindoline (102) 182
7.2.4 Synthesis of 5-bromo-1, 1, 3, 3- tetramethylisoindoline-2-
yloxyl (103)
183
7.2.5 Synthesis of 5-iodo -1, 1, 3, 3- tetramethylisoindoline (104) 185
7.2.6 Synthesis of 5-iodo-1, 1, 3, 3- tetramethylisoindoline-2-yloxyl
(105)
186
7.2.7 Synthesis of 5-carboxy-1, 1, 3, 3- tetramethylisoindoline-2-
yloxyl (106)
187
7.2.8 Synthesis of 2-benzyl-5-methylphthalimide (108) 189
7.2.9 Synthesis of 2-benzyl-5-methyl-1,1,3,3-tetraethylisoindoline
(109)
190
7.2.10 Synthesis of 5-methyl-1,1,3,3-tetraethylisoindoline (110) 191

X
7.2.11 Synthesis of 5-methyl-1,1,3,3-tetraethylisoindoline-2-yloxyl
(111)
192
7.2.12 Synthesis of 2-acetoxy-5-methyl-1,1,3,3-tetraethylisoindoline
(112)
193
7.2.13 Synthesis of 2-acetoxy-5-carboxy-1,1,3,3-
tetraethylisoindoline (113)
194
7.2.14 Synthesis of 5-carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl
(114)
195
7.2.15 Synthesis of 5-(3-hydroxy-3-methyl)butynyl-1,1,3,3-
tetramethylisoindoline-2-yloxyl) (119)
196
7.2.16 Synthesis of 5-ethynyl-1,1,3,3-tetramethylisoindoline-2-yloxyl
(120)
197
7.2.17 Synthesis of N-tert-butoxycarbonyl-2-(4-
aminophenyl)ethylamine (125)
198
7.2.18 Synthesis of N-tert-butoxycarbonyl-2-(4-N-(5-carboxy-
1,1,3,3-tetramethylisoindoline-2-
yloxyl)aminophenyl)ethylamine (126)
199
7.2.19 Synthesis of 2-(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl)aminophenyl)ethylamine
(127)
201
7.2.20 Synthesis of (E)-tert-4-(3-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamide)phenethylcarbamate (132)
202
7.2.21 Synthesis of (E)-N-(4-(2-aminoethyl)phenyl)-3-(3,5-di-tert-
butyl-4-hydroxyphenyl)acrylamide (133)
204
7.2.22 Synthesis of 2,3-O-isopropylideneguanosine (139) 205
7.2.23 Synthesis of 2,3-O-isopropylideneguanosine-5-carboxylic
acid (140)
206
7.2.24 Synthesis of 2,3-O-isopropylideneguanosine-5-
ethylcarboxylate (142)
207
7.2.25 Synthesis of 2,3-O-isopropylideneguanosine-5-N-
ethylcarboxamide (143)
209
7.2.26 Synthesis of O6-(benzotriazol-1-yl)-2′,3′-O-
isopropylideneguanosine-5′-N-ethylcarboxamide (144)
210

XI
7.2.27 Synthesis of 2-fluoro-O6-benzotriazole-1-yl-2,3-O-
isopropylideneguanosine-5-N-ethylcarboxamide (145)
211
7.2.28 Synthesis of 2-fluoro-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (146)
213
7.2.29 Synthesis of 2-iodo-O6-benzotriazole-1-yl-2,3-O-
isopropylideneguanosine-5-N-ethylcarboxamide (148)
214
7.2.30 Synthesis of 2-iodo-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (149)
215
7.2.31 Synthesis of 2-(2-(4-aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (159)
217
7.2.32 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-
2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide
(161).
219
7.2.33 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (162)
221
7.2.34 Synthesis of 2-(2-(4-N-(4-carboxy-2,2,6,6-
tetramethylpiperidine-1-yloxyl)aminophenyl))ethylamino-
2,3,-O-isopropylidineadenosine-5-N-ethylcarboxamide
(163)
223
7.2.35 Synthesis of 2-(2-(4-N-(4-carboxy-2,2,6,6-
tetramethylpiperidine-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (164)
224
7.2.36 Synthesis of 2-(2-(4-N-(3-carboxy-2,2,5,5-
tetramethylpyrrolidine-1-yloxyl)aminophenyl))ethylamino-2,
3,-O-isopropylidineadenosine-5-N-ethylcarboxamide (165)
226
7.2.37 Synthesis of 2-(2-(4-N-(3-carboxy-2,2,5,5-
tetramethylpyrrolidine-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide(166)
227

XII
7.2.38 Synthesis of 2-(2-(4-N-(5-methylcarboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-
2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide
(167)
229
7.2.39 Synthesis of 2-(2-(4-N-(methylcarboxy-1,1,3,3-
tetramethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (168)
230
7.2.40 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-
tetraethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (169)
232
7.2.41 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-
tetraethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (170)
234
7.2.42 Synthesis of 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido) phenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (171)
236
7.2.43 Synthesis of 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido) phenyl))ethylaminoadenosine-5-
N-ethylcarboxamide (172)
238
7.2.44 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethynyl)-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (177)
240
7.2.45 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethynyl)adenosine-5-N-ethylcarboxamide (178)
241
7.2.46 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethyl)-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (179)
243
7.2.47 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethyl)adenosine-5-N-ethylcarboxamide (180)
244
7.2.48 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-
ethynyl)-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (181)
246

XIII
7.2.49 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-
ethynyl)adenosine-5-N-ethylcarboxamide (182)
247
7.2.50 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-
ethyl)-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (183)
249
7.2.51 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-
ethyl)adenosine-5-N-ethylcarboxamide (184)
250
7.2.52 Synthesis of 2-(2-phenylethynyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (185)
251
7.2.53 Synthesis of 2-(2-phenylethyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (187)
253
7.2.54 Synthesis of 2-(2-phenylethyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (188)
254
Chapter 8: Bibliography 256

XIV
List of Figures
Figure 1.1 The chemical structure of triphenylmethyl radical. 6
Figure 1.2 Dietary and synthetic antioxidants. 14
Figure 1.3 General chemical structure of a nitroxide. 14
Figure 1.4 Structures of stable nitroxide radicals. 16
Figure 1.5 The chemical structure of nitroxide, TEMPOL. 19
Figure 1.6 Phenolic and radical antioxidants 23
Figure 1.7 Examples of derivatives of hydroxycinnamic acids. 24
Figure 1.8 Structural features of adenosine. 26
Figure 1.9 Three-dimensional crystal structure of the G-protein coupled
receptor (GPCR) embedded in the cell membrane attached to
heterotrimeric G protein.
28
Figure 1.10 Intracellular adenosine synthesis. 29
Figure 1.11 Adenosine receptor activation. 30
Figure 1.12 Mode of action of agonists. 38
Figure 1.13 Mode of action of competitive and non-competitive antagonists. 39
Figure 1.14 Adenosine A2AAR agonists. 40
Figure 1.15 Pharmacologically active A2AAR agonists. 42
Figure 1.16 Adenosine A2AAR antagonists. 43
Figure 1.17 Comparison of receptor-ligand interaction binding for the A2AR
with the agonists adenosine (39) and NECA (62) along with
antagonists ZM 241385 (51).
44
Figure 1.18 Adenosine A2BAR agonists. 46
Figure 1.19 Adenosine A2BAR antagonists. 47
Figure 1.20 Dual action compounds effective for ischaemia. 48
Figure 1.21 Bifunctional adenosine receptor-binding analogues and binary
conjugates.
49
Figure 1.22 Pharmacologically active A2AAR and A3AR selective analogues. 51
Figure 1.23 Pharmacologically dual-action antioxidant coupled A2AAR and
A3 AR-selective analogues.
52

XV
Figure 1.24 Adenosine receptor crystal structure in complex with A) UK432,
097 (42) B) compound VCP728 (70).
53
Figure 1.25 Antioxidants coupled p-aminoethylaniline linked adenosine
target compounds.
55
Figure 1.26 Antioxidants coupled with ethynyl and ethyl linked adenosine
target compounds.
56
Figure 2.1 Adenosine A2AAR selective compound CGS21680 (46). 60
Figure 2.2 Candidate moieties and their actions in the first structural class
of target adenosine analogues.
63
Figure 2.3 Dual acting A2AAR agonists - antioxidants decrease cell death
within an in vitro model of simulated ischemia. A)
Concentration-dependent, decrease in cell death and B). The
decrease in cell death was inhibited in the presence of the
A2AAR antagonist, SCH442415.
130
Figure 3.1 Candidate moieties and their actions in the second structural
class of target adenosine analogues.
133
Figure 3.2 Adenosine A3AR agonists PENECA (186). 134
Figure 4.1 Candidate moieties and their actions in the third structural class
of target adenosine analogues
152

XVI
List of Schemes
Scheme 1.1 DPPH radical scavenging assay principle. 13
Scheme 1.2 Resonance stabilisation of a nitroxide radical. 14
Scheme 1.3 Bimolecular degradation of a phenyl substituted nitroxide. 15
Scheme 1.4 β-Hydrogen induced disproportion of nitroxide. 16
Scheme 1.5 Redox reaction of nitroxide. 18
Scheme 2.1 Overview of first approach synthesis of 161. 64
Scheme 2.2 Overview of second approach synthesis of 161. 65
Scheme 2.3 Synthesis of 5-carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl
(C-TMIO) 106.
66
Scheme 2.4 Overview of N-Boc protection of linker amine. 67
Scheme 2.5 Mechanism of N-Boc protection of linker amine. 68
Scheme 2.6 Overview of EDCI amide coupling of nitroxide to para-
(aminoethyl)aniline linker moiety (127).
70
Scheme 2.7 Mechanism of EDCI promoted amide formation. 71
Scheme 2.8 Convergent approach to adenosine A2AAR agonists. 74
Scheme 2.9 Overview of synthesis of the key adenosine intermediate. 76
Scheme 2.10 Overview of 2, 3-alcohol protection (139). 77
Scheme 2.11 Overview of the 5-hydroxyl oxidation of (140). 79
Scheme 2.12 Mechanism of primary-hydroxyl oxidation . 80
Scheme 2.13 Overview of 5-ethyl ester formation (142). 82
Scheme 2.14 Overview of 5-amide formation (143). 83
Scheme 2.15 Overview of leaving group formation (144). 85
Scheme 2.16 Mechanism of leaving group formation. 86
Scheme 2.17 Overview of fluoro substitution at the C2 position (145). 88
Scheme 2.18 Overview of N6 amino substitution (146). 89
Scheme 2.19 Overview of coupling of nitroxide with aminoethylaniline linker
to adenosine intermediate (Approach 1) (161).
91

XVII
Scheme 2.20 SNAr Mechanism for the synthesis of C2-substituted target
compound (161)
92
Scheme 2.21 Overview of coupling of carboxy nitroxides with
aminoethylaniline linked adenosine intermediate (161)
(Approach 2)..
95
Scheme 2.22 Overview of coupling of aminoethylaniline linker to adenosine
intermediate (159)..
95
Scheme 2.23 Synthesis of TMIO-substituted adenosine analogue
possessing a substituted aniline linker moiety (161)
(Approach 2).
98
Scheme 2.24 Synthesis of 3,5-dinitrobenzoic acid-coupled adenosine
analogue possessing a substituted aniline linker.
100
Scheme 2.25 Reaction of coupling reagents with adenosine analogue
possessing a substituted aniline linker.
101
Scheme 2.26 Synthesis of TEMPO-substituted adenosine analogue
possessing a substituted aniline linker moiety (164).
102
Scheme 2.27 Synthesis of proxyl-substituted adenosine analogue
possessing a substituted aniline linker moiety (166).
104
Scheme 2.28 Synthesis of MCTEIO-substituted adenosine analogue
possessing a substituted aniline linker moiety (168).
107
Scheme 2.29 Synthesis of 5-Carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl
(CTEIO) 114.
110
Scheme 2.30 Synthesis of CTEIO-substituted adenosine analogue
possessing a substituted aniline linker moiety (170).
112
Scheme 2.31 Overview of synthesis of di-tert-butylhydroxycinnamic acid
coupled aminoethylaniline linker moiety (133).
115
Scheme 2.32 Overview of coupling of di-tert-butylhydroxycinnamic acid with
linker to adenosine (Approach 1) (171).
118
Scheme 2.33 Synthesis of di-tert-butylhydroxycinnamic acid adenosine
analogue possessing a substituted aniline linker moiety
(Approach 2).
120
Scheme 3.1 Overview of synthesis of C2 iodo adenosine intermediate
(149).
135
Scheme 3.2 Synthesis of 5-ethyne-1,1,3,3-tetraethylisoindoline-2-yloxyl
(E-TMIO) (120).
138

XVIII
Scheme 3.3 Sonogshira coupling reaction mechanism. 140
Scheme 3.4 Synthesis of ETMIO-substituted adenosine analogue (178). 141
Scheme 3.5. Synthesis of ETEIO-substituted adenosine analogue (182). 144
Scheme 3.6. Synthesis of the phenylacetylene-substituted adenosine
analogue (185).
146
Scheme 4.1. Overview the synthesis of the TMIO-nitroxide ethyl linked
adenosine compound (180).
154
Scheme 4.2. Overview synthesis of TEIO-nitroxide ethyl linked adenosine
compound (184).
156
Scheme 4.3. Overview synthesis of the non-nitroxide phenyl ethyl linked
adenosine compound (188).
159

XIX
List of Tables
Table 2.1 cAMP analysis of aminoethylaniline linked adenosine
compounds
126
Table 3.1 cAMP analysis of ethynyl linked adenosine compounds. 148
Table 4.1 cAMP analysis of ethyl linked adenosine compounds. 163

XX
LIST OF ABBREVIATIONS
3 h — 3 hours
1 M — One molar
2 M — Two molar
4 M — Four molar
7TM — 7-Trans membrane
AR — Adenosine receptors
A1R — Adenosine A1 receptors
A2AR — Adenosine A2A receptors
A2BR — Adenosine A2B receptors
A3R — Adenosine A3 receptors
AMP — Adenosine monophosphate
ADP — Adenosine diphosphate
ATP — Adenosine triphosphate
ADORA2A — Adenosine A2A receptors
BAIB — Bis(acetoxy)iodobenzene
BOP—(Bezotriazole-1-yloxy)tris(dimethylamino)phosphonium
hexafluorophospohate

XXI
BHT — Butylhydroxytoluene
BOC — Butyloxycarboxyl
CDCl3 — Deuterated chloroform
13CNMR — 13C nuclear magnetic resonance
CHO — Chinese hamster ovary
CO2 — Carbon dioxide
cAMP — Cyclic adenosine monophosphate
CAD — Coronary Artery Disease
CHD — Chronic heart Disease
CRP — C-reactive protein
CSC — (E)-8-(3-chlorostyryl)caffine
CTMIO — 5-Carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl
CTEIO — 5-Carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl
CTEMPO — 4-Carboxy-2,2,6,6-tetramethylpieridine-1-yloxyl
CPROXYL — 3-Carboxy- 2,2,5,5-tetramethylpyrrolidine-1-yloxyl
CuI — Copper iodide
DABCO — 1,4-Diazabicyclo(2.2.2)octane
DBU — 1,8-Diazabicycloundec-7-ene
DCM — Dichloromethane

XXII
DIPEA — N,N-diisopropylethylamine
DMF — N,N-dimethylformamide
DMPX — 3,7-Dimethyl-1-propargylxanthine
DMAP — 4-Dimethylaminopyridine
DMSO — Dimethyl sulfoxide
DNA — Deoxyribonucleic acid
DPPH — 2,2-Diphenyl-1-picrylhydrazyl
DTBN — Di-tert-butylnitroxide
ET — Electron transfer
EtOAc — Ethyl acetate
EI — Electron ionisation
E-TMIO — 5-Ethynyl-1,1,3,3-tetramethylisoindoline-2-yloxyl
E-TEIO — 5-Ethynyl-1,1,3,3-tetraethylisoindoline-2-yloxyl
EDCI — 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
Et2O — Diethyl ether
EtOH — Ethanol
EIMS — Electron ionisation mass spectroscopy
GDP — Guanosine diphosphate
GTP — Guanosine triphosphate

XXIII
GSH-Px — Glutathione peroxidase
GPCR — G-coupled protein receptor
H2O2 — Hydrogen peroxide
HCl — Hydrochloric acid
HCA — Hydroxycinnamic acid
HF — Hydrofluoric acid
HOBt — Hydroxy benzotriazole
HPLC — High-pressure liquid chromatography
HRMS — High-resolution mass spectroscopy
H9c2 — Rat cardiomyoblast cell line
HENECA — 2-Hexynyl-5-N-ethylcarboxamide
IL-4 — Interleukin-4
I2 — Iodine
IOP — Internal ocular pressure
Ki — Inhibitory constant
KMnO4 — Potassium permanganate
KOH — Potassium hydroxide
LC/MS — Liquid chromatography-mass spectroscopy
LDL — Low-density lipoprotein

XXIV
MeCN — Acetonitrile
MeOH — Methanol
MCTMIO — 5-Methylcarboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl
MS — Mass spectroscopy
MPLC — Medium pressure liquid chromatography
MPI — Myocardial perfusion imaging
mRNA — Messenger ribonucleic acid
NADPH — Nicotinamide adenine dinucleotide phosphate
NaOH — Sodium hydroxide
Na2WO4.2H2O — Sodium Tungstate dihydrate
nM — Nanomolar
NECA — N-ethylcarboxamide
NMR — Nuclear magnetic resonance
NO — Nitric oxide
n-BuLi — n-Butyl Lithium
O2 — Oxygen
PbO2 — Lead dioxide
Pd — Palladium
Pd/C — Palladium on carbon

XXV
Pd(PPh3)2Cl2 — Palladium bis(triphenylphosphene) dichloride
PS — Petroleum spirit
PTSA — Para-toluene sulphonic acid
PROXYL — 2,2,5,5-Tetramethylpyrrolidine-1-yloxyl
pEC50 — Logarithimic 50% effective concentration
ROS — Reactive oxygen species
RNS — Reactive nitrogen species
SAR — Structure-activity relationship
SNAr — Nucleophilic aromatic substitution
SI — Simulated Iscaemia
SOD — Superoxide dismutase
TEA — Triethylamine
THF — Tetrahydrofuran
TEMPO — 2,2,6,6-Tetramethylpiperidin-1-yloxyl
TEMPOL — 2,2,6,6-Tetramethylpiperidin-1-yloxyl
TEIO — 1,1,3,3-Tetraethylisoindoline-2-yloxyl
TMIO — 1,1,3,3-Tetramethylisoindoline-2-yloxyl
TLC — Thin layer chromatography

XXVI
Statement of Original Authorship
The work contained in this thesis has not been previously submitted for a
degree or diploma from this or any other higher education institution. To the
best of my knowledge and belief, the thesis contains no material previously
published or written by another person except where due reference is made.
Signature
Date: ------------------------------------------
QUT Verified Signature

XXVII
Acknowledgements
Thanks to Professor Steven Bottle for his guidance and continuous support for
allowing me to move thousands of kilometres away from my home country and
further develop my skills and expertise.
Thanks to Professor Peter Scammells and co-workers from Monash Institute of
Pharmaceutical Sciences Melbourne for the biological testing of my synthesised
chemical compounds and providing guidance and their assistance throughout
the project.
Thanks to Dr Kathryn Fairfull-Smith for her valuable guidance and support, and
help in understanding the policies and procedures in QUT.
Thanks to Dr James Blinco and Dr Kye Masters for their support inside and
outside the laboratory.
Thanks to my parents and siblings for their love and support and
encouragement throughout the Ph.D. tenure.
Thanks to all friends and colleagues Mr Komba Thomas, Mr Liam Walsh, Mr
Jason Morries, Ms Vanessa Luccini, Mr Paul Lederhose, Mr. Chong, Mr Viraj
Jayawardhana and Mr Kai Anders-Hanson for their support and friendly
environment in and outside the laboratory.
Thanks to all other inhabitants of M6 laboratory who directly or indirectly helped
me.
Thanks all the QUT chemistry technicians, Dr Chris Carvalho, Dr Mark Wellard
and all technical staff for their support in learning the instrument handling.

XXVIII
Thanks to QUT administrative staff and all research centre staff for their direct
or indirect support throughout my QUT life.
Thanks to QUT counselling and medical centre services for their support.
Thanks to professional editor Laurel Mackinnon, provided copyediting and
proofreading services, according to the guidelines laid out in the university-
endorsed guidelines and the Australian standards for editing research theses’.
Thanks to the Australian Research Council and Centre of Excellence for Free
Radical Chemistry and Biotechnology for a living allowance scholarship.

1
Introduction & Literature Review 1

2
1.1 Cardiovascular diseases
Cardiovascular diseases are a leading public health concern throughout the
world. Globally, more than 17 million people die from cardiovascular diseases
every year.1 Of these, 80% of deaths occur in low-income and middle-income
people or developing countries. Heart disease, a type of cardiovascular
disease, is the most common cause of death in developed countries.1
Cardiovascular disease is classified further as peripheral vascular diseases and
cerebrovascular diseases, and includes coronary heart disease (CHD),
rheumatic heart disease, stroke, chronic heart failure, heart attack (myocardial
infarction), and diabetes.2
The pathophysiology of cardiovascular diseases is very complex because of the
involvement of several biological pathways, which all originate in the vascular
endothelium.3 Interference in these pathways is considered to be the main
factor in cardiovascular risk. Activation of the vascular endothelium and
oxidative stress play important roles in atherosclerosis4 and hypertension,5, 6
which eventually result in the progression of cardiovascular damage. High
cholesterol levels,7 impaired glucose tolerance8 and inflammation also
contribute to cardiovascular damage.
During the past several decads, extensive research has focused on treating
cardiovascular diseases from a different perspective such as the use of
precautionary therapies and curative medications. Several factors have been
found to contribute to cardiovascular diseases. These factors are classified into

3
two types: unavoidable and avoidable factors. Unavoidable factors include age,
sex, menopausal status and genetic composition. Avoidable factors can include
environmental pollution, diet, smoking, some diseases (e.g., diabetes,
hypertension), lack of physical activity9 and ethanol consumption.10
Cardiovascular diseases result from an inadequate blood supply to the brain,
heart and peripheral vasculature.2 The term ischaemia describes the deficiency
or shortage of blood supply (to hold back blood).11 These ischaemia-related
heart diseases are the most common cause of death in the world,12 and
research has focused on finding new ways to design and synthesise drugs that
will achieve cardioprotection.
1.1.1 Ischaemic heart diseases
Coronary artery disease (CAD) is also called ischaemic heart disease.12
According to the World Health Organization, in 2008 ischaemic heart disease
accounted for 7.25 million deaths worldwide and, by 2015, 25% of the global
adult population will be affected by the major risk of hypertension.13 Ischaemic
heart disease involves a narrowing of the coronary arteries, the vessels that
supply blood to the mayocardium, mainly because of deposition of plaque in the
arterial walls, a process called atherosclerosis.14 Plaques are composed of
cholesterol-rich fatty deposits, collagen, other proteins, and excess smooth
muscle cells.14 Thickening and narrowing of the arterial walls, as well as
impeding the flow of blood and starving the heart of oxygen, are the symptoms
of this disease (also called “ischaemia”).15 This disease can cause a muscle
cramp-like chest pain called angina pectoris.16

4
Ischaemia occurs where there is an interruption or occlusion of the blood supply
to an organ or tissues, which deprives the cells of oxygen and nutrients, and the
ability to generate energy, a condition called hypoxia.11 This hypoxia can
initiate a series of chemical events that result in cell dysfunction, cellular chaos,
interstitial oedema, and necrosis, and eventually lead to anaerobic cell death.11
Hypoxia can affect various parts of the body, resulting in cardiac ischaemia,
brain ischaemia,17 cutaneous ischaemia, and ischaemia of the bowel and
limb.11 Hypoxia can cause serious disorders by damaging metabolically active
tissues and eventually various organs linked by the circulatory system.11 A
deficiency of oxygen supply (hypoxia) increases the intercellular concentration
of lactic acid through a process called acidosis, which alters normal enzyme
kinetics within cells.11
Ischaemia often occurs during surgical procedures, especially vascular
procedures and transplantations.18 Abnormalities can be detected in the
electrocardiogram (ECG), which may show the first indications of ischaemic
heart disease.18 These abnormalities may occur even with an absence of
symptoms of angina or myocardial infarction and may indicate the possible
presence of ischaemic heart disease.18 In hypoxic muscle cells, glucose
consumption can increase by 12-fold.15 The depletion of cellular energy stores,
particularly of adenosine 5-triphosphate (ATP) is considered to reflect the
failure of cellular homoeostasis and is diagnosed by the loss of the ion gradient
across the cell membrane.15

5
Hypoxia develops within 10 seconds of the coronary blockage.11 Different
tissues can remain hypoxic for different periods. Skeletal muscles can recover
after some period of ischaemia; however, irreversible neuronal injury typically
occurs within minutes.19 Cardiac cells can survive for about 20 minutes under
ischaemic conditions and, if blood flow is restored within this time, the normal
aerobic mechanism resumes.15 In aerobic condition, the toxic metabolites are
removed from the site of the ischaemic event. However, this restoration of the
blood supply may also lead to further local tissue injury.11 Ischaemia and
reperfusion injuries are the root causes of the pathophysiology of cerebral
ischaemia, stroke, myocardial infarction and some of the problems with organ
transplantation.20
1.1.2 Reperfusion injury
Reperfusion is the restoration of blood supply to ischaemic tissues, which
facilitates the removal of toxic metabolites from these tissues.11 These toxic
metabolites flow into and throughout the systemic circulation and can lead to
serious injury including ischaemic tissue injury, called ischaemia–reperfusion
injury.11 This restoration of blood initiates a series of events that can lead to
further tissue injury.21 Once the blood supply is restored, a cascade of
biochemical and molecular changes occur that lead to free radical-mediated
damage.21 Oxygen-centred free radicals, including superoxide radicals, are
mainly involved in ischaemia–reperfusion injury.21 These free radicals are
produced intracellularly within the mitochondria in the post-ischaemic
endothelium and may be responsible for major changes in the endothelium.15
Superoxide radicals may be responsible for the production of highly reactive

6
and damaging hydroxyl radicals.22 Superoxide radical production is controlled
naturally by the endogenous enzyme superoxide dismutase in normal and non-
stressed physiological conditions.23-26 This natural defence mechanism is
overwhelmed in ischaemia and reperfusion conditions, and the hydroxyl radicals
produced can cause damage to a variety of biological components including
amino acids, proteins, enzymes and nucleic acids.22, 27-30
1.1.3 Free radicals
Free radicals are ions, atoms or molecules with one or more unpaired electrons
in their outer shell. Radicals are formed by homolytic bond cleavage. Free
radicals can react readily with other radicals to form stable species and can be
oxidised to cations or reduced to anions. In 1900, the first organic free radical
triphenylmethyl radical (Figure 1.1) was identified by Moses Gomberg.31
Figure 1.1: Chemical structure of a triphenylmethyl radical.
Free radicals are generated in biological systems by a wide variety of processes
such as ionising radiation, inflammation or exposure to toxic xenobiotics, and as
metabolites of membrane lipid transformation.32 These radical species also can
be generated by external sources including cigarette smoke33 and other forms
of pollution, by exposure to radiation and by exposure to chemical processes

7
such as those involving photosensitized decomposition and the decomposition
of non-endogeneous diacetyl peroxide, peresters and azo compounds.34
However, not all radicals are highly reactive or damaging. Free radicals can
also play an important role as mediators of cell signalling processes that
regulate vascular function.35, 36
1.1.4 Oxidative stress
Oxidation and reduction reactions represent the basis for several biochemical
mechanisms that generate metabolic changes in biological systems.37
Oxidative stress is defined as the imbalance between the increased production
of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and the
lowered activity of radical-scavenging mechanisms in biological systems.38
Oxidative stress is involved in the various mechanisms of injury in many types
of disease processes. The free radical species ROS and RNS are produced in
low amount in all layers of the vascular membrane, such as the endothelium
and smooth muscle and in several metabolic processes via the actions of
various enzymes.39 These enzymes include nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase, xanthine oxidase, nitric oxide (NO) synthases,
myeloperoxidase (MPO) and some mitochondrial enzymes.40
The involvement of ROS and RNS is widely recognised as being important in
several aspects of pathophysiological processes.39, 41 ROS are known to play
an important role in oxidative stress. Increased oxidative stress can cause
damage to cellular structures. The overproduction of radical species (the active

8
form of oxygen or nitrogen) can damage biological molecules including proteins,
amino acids, nucleic acids and DNA molecules by various mechanisms.42
The toxic effects of oxygen were investigated in several animal species. In
1954, Gerschman et. al. reported that the observed oxygen toxicity could be
attributed to the partially reduced form of oxygen.43 These ROS and RNS
encompass hydrogen peroxide (H2O2), superoxide anion radicals (O2–),
hydroxyl radicals (OH), nitric oxide (NO), and peroxynitrite (ONOO–) radicals.44,
45 An excess of the active forms of these radicals (R) or other ROS, leads to
the oxidation of other cellular components and forms the peroxyl radical
(HOO), which can initiate the chain oxidation reaction. This radical chain
process leads to more damage and generates oxidative stress conditions.
ROS are responsible for the increased oxidative stress and decreased lifespan
in various living species.46 The free radical theory of ageing states that the
organism’s age depends on the accumulation of damage over time, attributed to
oxidative stress induced by ROS.47 This theory is supported by the imbalance
between the ROS produced by mitochondria, and the activity of internal radical-
scavenging defence mechanisms.48
Physical exercise, can also increase oxidative stress. Intense physical activity
increases mitochondrial activity in skeletal muscles and the subsequent
production and leakage of ROS from mitochondria.49 Ischaemia–reperfusion
injury and activation of neutrophils, may occur during intense physical exercise,
and also further increase in the production of ROS.50 Cigarette smoke can also

9
increase oxidative stress because it contains a large number of free radicals
that can cause endothelial cell dysfunction and mitochondrial damage.33, 51
In living organisms, free radicals may cause more damage to the immature
brain than the adult's brain because of underdeveloped free radical-scavenging
mechanisms and the presence of a higher concentration of iron that can
promote radical formation.17 As a result, free radical damage in infants can
cause neurological complications such as birth asphyxia, perinatal brain injury
and ischaemic injury etc.17 However, antioxidant analogues are recognised to
provide protective effects for this type of free radical damage.
1.2 Antioxidants
Antioxidants are compounds that prevent or defend against the cellular damage
caused by free radicals via inhibition of oxidation of atoms or molecules.32
Oxidation is a chemical reaction in which loss of a hydrogen atom or an electron
or addition of oxygen to a molecule.52 Oxidation can generate free radicals,
which then initiate a chain radical reaction that can damage the cellular
components of biological systems.53 Antioxidants terminate this chain reaction
by scavenging the free radical intermediates and preventing further oxidation of
other species.53 The antioxidant activity of species such as thiols, ascorbic acid
and polyphenols is due to their ability to act as reducing agents (i.e. they
undergo oxidation themselves).53 The antioxidant mechanism of
cardioprotection typically involves either the generation of lynchpin radicals in
ischaemia–reperfusion injury or the direct scavenging of the superoxide anion.54
Clinical studies have shown that anti-inflammatory agents act as antioxidants

10
significantly reduce the levels of C-reactive protein (CRP) and membrane attack
complex (MAC) protein in infarcted tissues.54 These CRP and MAC protein
levels increase during inflammatory and cardiovascular conditions.55
In biological systems, the potential damaging effects of free radicals are
regulated through intracellular body fluids by the actions of several enzymes
including superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and
glutathione-S-transferase.56 The family of glutathione-S-transferase enzymes
catalyses the transformation of thiols, glutathione and some reactive
electrophiles to thioethers called S-conjugates.56 These reactive intermediates
are formed by several mechanisms including NADPH-dependent enzymatic
dethiolation and thiol exchange or radical-mediated processes.56
Non-enzymatic defence mechanisms act by preventing the excessive
production of free radicals. The body employs several means to prevent the
overproduction of free radicals. The first method is metal chelation, which
involves binding metal ions, particularly iron and copper ions, at the site of
initiation of the chain reaction. This method is a potent technique for preventing
the radical production and it is used as a major strategy for preventing lipid
peroxidation and DNA fragmentation.57 Metal binding to the proteins transferrin,
ceruloplasmin and metallothionein is broadly effective to counteract radical
generation. A second process involves the modification of the target site to
make it resistant to the metal ion-dependent oxidation.53 This process can
protect cells from radiation-derived oxidation; for example, to protect melanin

11
from ultraviolet radiation and carotenoid from electronically excited species such
as singlet oxygen.37
A third biological process that prevents the overproduction of free radicals is the
introduction of non-enzymatic antioxidants to protect the tissues. The main
issue is that once the radical species are formed, they initiate a radical chain
reaction and produce more radical compounds. This radical generation can be
prevented by the production of the non-radical and unreactive end products.
This process of deactivation of the radical chain reaction by inserting
antioxidants or other radical species prevents further damage.
Another strategy is to move radical species away from potential target species
to avoid cellular damage. High-efficiency breakup mechanisms are used in
cellular systems, where only a few antioxidant molecules are required to
deactivate thousands of potential target molecules. Such chain-breaking
mechanisms often involve polyphenolic antioxidants including -tocopherol,
which reduces peroxyl radicals and is itself converted to the tocopheroxyl
radical. This radical is then scavenged by external dietary antioxidants
including ascorbate vitamin C (2) and thiols.58 Several other naturally occurring
dietary antioxidants include retinol vitamin A (8), kaempferol (4) and quercetin
(5) (Figure 1.2).
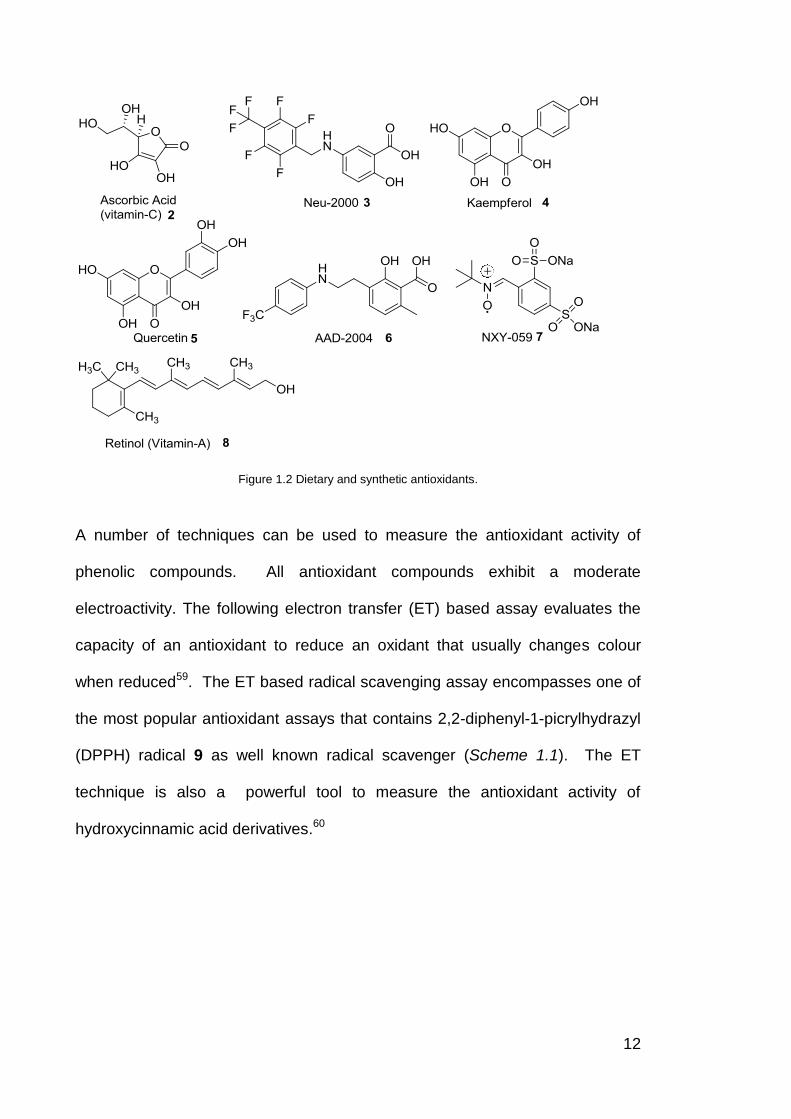
12
Figure 1.2 Dietary and synthetic antioxidants.
A number of techniques can be used to measure the antioxidant activity of
phenolic compounds. All antioxidant compounds exhibit a moderate
electroactivity. The following electron transfer (ET) based assay evaluates the
capacity of an antioxidant to reduce an oxidant that usually changes colour
when reduced59. The ET based radical scavenging assay encompasses one of
the most popular antioxidant assays that contains 2,2-diphenyl-1-picrylhydrazyl
(DPPH) radical 9 as well known radical scavenger (Scheme 1.1). The ET
technique is also a powerful tool to measure the antioxidant activity of
hydroxycinnamic acid derivatives.60

13
Scheme 1.1: DPPH radical scavenging assay principle.
Several synthetic compounds have also been reported as radical scavengers
that have different mechanisms of action. After the discovery of superoxide
dismutase (SOD) activity in 1969, there has been a significant development in
the understanding of radical chemistry61 and, since 1988, in the arginine-
dependant synthesis of NO radicals.62 It has also been demonstrated that
nitroxides are very efficient radical scavengers that exhibit antioxidant actions.63
For example, several synthetic compounds reported as radical scavengers
including AAD-2004 (6)64, Neu-2000 (3)65 and NXY-059 (7)66, 67 (Figure 1.2)
exhibit anti-inflammatory and antioxidant properties by spin trapping in cortical
neuron culture.
1.2.1 Nitroxide radicals
Nitroxides (also known as aminoxyls or nitroxyls) are synthetically stable free
radicals. In 1965, Griffith and McConnell demonstrated that nitroxides are free
radical species with paramagnetic properties.68 Nitroxides exhibit paramagnetic
behaviour because of an unpaired electron. The general molecular formula is
R2NO (Figure 1.3).

14
Figure 1.3: General chemical structure of a nitroxide.
The majority of stable nitroxides are secondary amine N-oxide radical species
with quaternary α–carbon atoms. The stability of a nitroxide radical depends on
several factors. The delocalization of an unpaired electron (resonance) over the
N–O bond and hetero atoms provides remarkable kinetic and thermodynamic
stability to the nitroxide moiety (Scheme 1.2). Therefore, it shows resistance to
dimerization.69
Scheme 1.2: Resonance stabilisation of a nitroxide radical.
The groups attached to the radical centre can also strongly influence the
stability of the nitroxide radical. Nitroxides become more stable with bulky
groups around the nitroxyl moiety. This steric effect resists the bimolecular
combination reaction that occurs between a nitroxide and a more reactive
radical.
The stability of nitroxides is also maintained by their resistance towards the
formation of stable adducts with other oxygen centred radical species. Although
nitroxides can undergo reaction with a variety of reactive oxygen species, this is
not through direct interaction with the oxygen centred radicals. Dimerization
does not occur due to the loss in delocalisation stabilisation of two nitroxide

15
species. The dimerization of two nitroxide molecules is also energetically
unfavourable (2 x ~130 kJ/mol) because of the weak nature of the resultant N–
O–O–N bond (~140 kJ/mol) that would form.70, 71 This nature maintains the
stability of nitroxides and is also useful for protecting the nitroxide radical from
dimerization.72, 73
The stability of nitroxides is dependent on the unpaired electron remaining
delocalised over the two hetero atoms. The nitroxide stability is, therefore,
dependent on the absence of degradation pathways, which mainly arise through
the substituents present on the carbon α to the nitroxide moiety.74 The
delocalisation of unpaired electron spin density in the aromatic ring of tert-butyl
phenyl nitroxide increases the thermodynamic stability of the nitroxide system.
However, the reactive carbon centred radical thereby formed promotes the
bimolecular degradation of the nitroxide moiety to give the corresponding amine
and nitrone species74 (Scheme 1.3).
Scheme 1.3: Bimolecular degradation of a phenyl substituted nitroxide.
The degradation pathways that affect the stability of nitroxides arise mainly due
to the presence of methyl (CH3-), methylene (RCH2-) and methine (R2CH-)
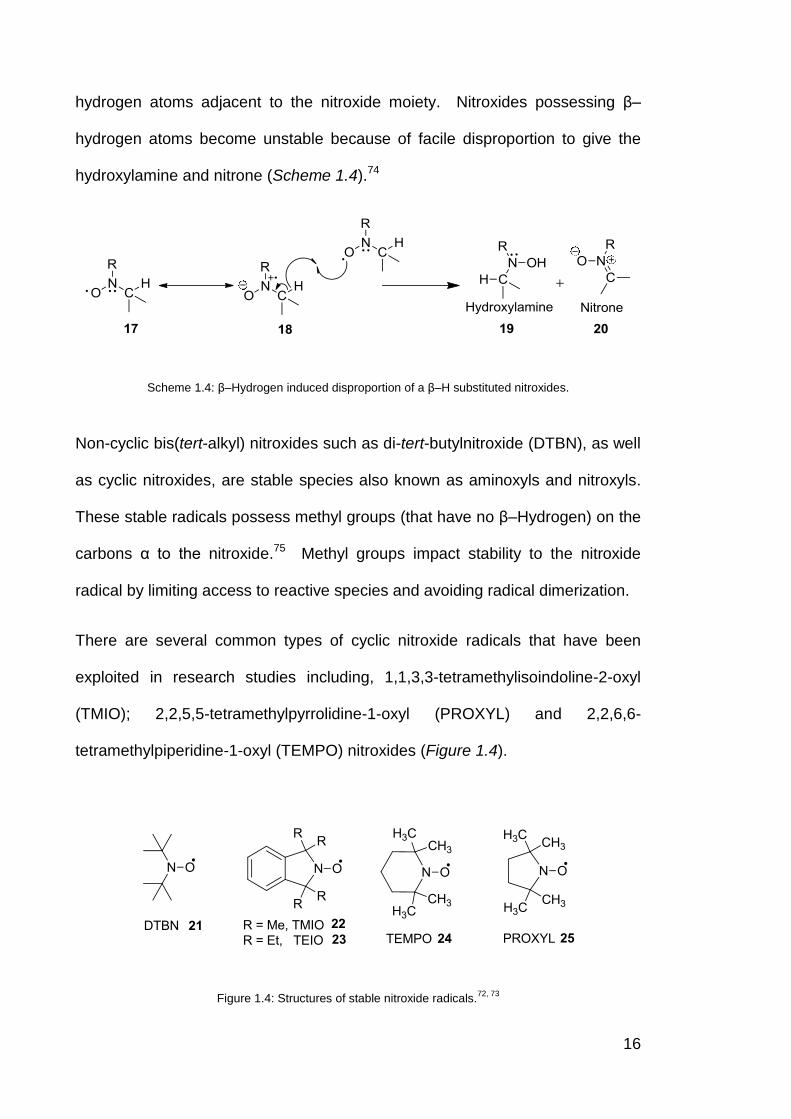
16
hydrogen atoms adjacent to the nitroxide moiety. Nitroxides possessing β–
hydrogen atoms become unstable because of facile disproportion to give the
hydroxylamine and nitrone (Scheme 1.4).74
Scheme 1.4: β–Hydrogen induced disproportion of a β–H substituted nitroxides.
Non-cyclic bis(tert-alkyl) nitroxides such as di-tert-butylnitroxide (DTBN), as well
as cyclic nitroxides, are stable species also known as aminoxyls and nitroxyls.
These stable radicals possess methyl groups (that have no β–Hydrogen) on the
carbons α to the nitroxide.75 Methyl groups impact stability to the nitroxide
radical by limiting access to reactive species and avoiding radical dimerization.
There are several common types of cyclic nitroxide radicals that have been
exploited in research studies including, 1,1,3,3-tetramethylisoindoline-2-oxyl
(TMIO); 2,2,5,5-tetramethylpyrrolidine-1-oxyl (PROXYL) and 2,2,6,6-
tetramethylpiperidine-1-oxyl (TEMPO) nitroxides (Figure 1.4).
Figure 1.4: Structures of stable nitroxide radicals.72, 73

17
Nitroxides have been reported as effective ROS scavengers with some
advantages over other reactive radical trapping agents. Specifically, the
isoindoline-based aminoxyls, namely 1,1,3,3-tetramethylisoindoline-2-oxyl
(TMIO)76 (22) and its analogues, are known to have several advantages77
compared to other nitroxides including the PROXYL (25) and TEMPO (24)
containing species. The fused aromatic ring of the isoindoline skeleton provides
resistance to ring-opening reactions over the decomposition pathways of other
aminoxyls and demonstrates excellent electron paramagnetic resonance (EPR)
properties with narrower line-widths. In addition, these nitroxides demonstrated
excellent thermal and chemical stability in several chemical reactions.
Substitution on the aromatic ring of the isoindoline allows the creation of more
complex structures suitable for several applications with little impact on the
stability and reactivity of nitroxide radical species.78, 79 These modifications of
isoindoline nitroxide can be beneficial in increasing the water solubility of
nitroxides, and this has been used to expand the potential scope of the
application of nitroxides in various therapeutic contexts.80 Isoindoline nitroxides
are implicated in radical trapping and as potent antioxidants in biological
systems.81, 82
As nitroxides are more stable than other reactive radical species, their use has
been exploited in various applications. Nitroxides demonstrate their effect
through electron transfer mechanisms, including redox metabolism via one or
two electron-reduction reactions involving either an interconvertible
hydroxylamine, an oxoammonium cation or a radical ion. These three-stage

18
oxidative electron-transfer processes also explain the ROS-metabolizing action
of nitroxides (Scheme 1.5).81, 83
Biological studies have found that some nitroxides can accumulate in cells at
high concentration with lifetimes of over 100 minutes.84 This property of
nitroxides has been exploited in EPR imaging and biomarkers.75 These findings
have initiated further research into nitroxides and their applications in the
therapeutic field.85
Scheme 1.5: Redox reactions of a nitroxide.81, 83
1.2.2 Applications of nitroxides:
Radical trapping and spin labelling: 1.2.2.1
The biological activity of nitroxides was first investigated in 1964 by Emmerson
and Howard-Flanders.86 Further studies on nitroxides described the protective
antitumor activity of nitroxides,87 and also their roles as powerful bioregulatory
molecules in the nervous system, immune system and cardiovascular system.88
Nitroxides are kinetically stable systems compared with other radical species,

19
and their intracellular lifespan has been found to be long enough to be detected
by electron paramagnetic resonance (EPR) spectroscopy. Therefore, these
stable nitroxide radicals have been used in several animal models of disease
and also in human diseases to try to find ways to avoid the production of ROS,
which can generate oxidative stress.
The nitroxide TEMPOL (4-hydroxy-2, 2, 6, 6-tetramethylpiperidine-N-oxyl) 28
(Figure 1.5) is the most widely studied nitroxide, and it has been shown to be
permeable through cell membranes and to reduce the formation of toxic
hydroxyl radicals.89
Figure 1.5: The chemical structure of nitroxide, TEMPOL.
In 1990, Schnackenberg, Welch, and Wilcox90 reported that the intravenous
administration of TEMPOL decreased blood pressure and lipid peroxidation in a
hypertensive rat model. TEMPOL also reduced glutathione levels in human
blood cells,91 and was found to protect cells and tissues from the damaging
effects of ROS. In vivo, nitroxides rapidly undergo reduction to the
corresponding hydroxylamine, which demonstrates their involvement in redox
biochemistry.92

20
Nitroxide antioxidants 1.2.2.2
Nitroxides act as antioxidants against free radical-induced oxidative damage in
biological systems and provide chemo-protection and radioprotection.93
Extensive research has focused on the action of nitroxides as antioxidants in
several biological systems ranging from isolated molecules to membranes, cells
to organs and in whole animals.94, 95 These nitroxides can operate at different
levels in the cell, for example, preventing radical formation, intercepting formed
radicals, repairing oxidative damage, and increasing the elimination of damaged
molecules as well as promoting the death of cells with excessively damaged
DNA96. Antioxidant mechanisms are studied in cells using isolated
biomolecules created by microsomal preparation methods.97 Cell protection is
observed by preserving the colony-formation ability of the cells and by avoiding
DNA degradation. The mechanism proposed to explain this protection is that
nitroxides oxidise reduced metals and limit the production of H2O2 and,
consequently, the production of ROS.
Nitroxide radicals react with free radicals and intercept them before they can
damage lipids, proteins and DNA molecules. These radicals also act as
cytotoxic mediators in the immune system and as neurotransmitters in the
central nervous system.98 Nitroxide analogues and their derivatives have been
investigated as potential protective agents in rat models of myocardial
ischaemia–reperfusion injury.99 Experiments have confirmed that, under the
ischaemic condition, nitroxides are completely reduced in the heart, and this is
decreased by ~33% in non-ischaemic conditions.100

21
Several stable nitroxides have been reported to exhibit intracellular radical-
scavenging actions with recycling capacities.101, 102 These nitroxides were
developed and tested for their free radical-scavenging and antioxidant
abilities.63, 75, 76 TEMPO and its analogues have been shown to protect against
cerebral and cardiovascular ischaemia103 and reperfusion injury89 by decreasing
the infract size. This discovery of the actions of nitroxide radicals has led to an
interest in the role of dietary antioxidants and evidence suggesting that
antioxidants may protect against many neurological and cardiovascular
diseases.104-107 Several hindered phenolic and polyphenolic compounds also
exhibit antioxidant and anticancer activity.108-111
1.2.3 Phenolic antioxidants
Phenolic compounds also exhibit antioxidant properties similar to those of
nitroxides. In particular, hindered phenolic compounds have been reported to
demonstrate excellent antioxidant activity and are known as “natural”
antioxidants. These sterically hindered phenolic compounds have improved
reactivity towards small free radicals and are more biologically stable than non-
hindered phenolic species when oxidised. Phenolic antioxidants including
butylated hydroxytoluene (BHT) generate stable free radicals by donating a
hydrogen atom to diffuse the more reactive radical species. This phenoxy
radical is more stable, but it can still react with other radicals or can be
polymerised, although these reactions are slower than those that are initiated by
more reactive species.

22
Substituents on the ring play an important role in determining the antioxidant
activity of phenolic antioxidants. Electron-donating substituents on the phenyl
ring increase the radical-scavenging potency of phenolic compounds by
weakening the phenolic O–H bond.112 Substitutions at the ortho position
determine the stability of the phenoxy radical. A bulkier substituent produces a
more hindered phenoxy radical, which helps increase the radical-scavenging
activity. Substitution at the para position avoids the dimerization of the phenolic
radical compounds.
A variety of compounds incorporating the phenolic structures have been
reported. Some if these phenolic compounds have been claimed to be
responsible for the trapping and stabilising of reactive free radicals, including
single oxygen, hydroxyl, superoxide and peroxyl radicals and also lipid soluble
radicals. These properties of phenolic compounds (some examples of which
are shown in Figure 1.6) provide desirable effects in various biological
functions.
In particular, the hindered phenolic species have been reported to reduce low-
density lipoprotein (LDL) oxidation and morbidity in the lipoprotein and have
reactivity towards radicals.112 Many novel phenolic antioxidant compounds
capable of oxidative modification of LDL have been reported. LDL oxidation
has been found to be a major contributor to atherosclerosis, and logically its
inhibition by antioxidants should be beneficial in preventing and treating
atherosclerosis. The well-known phenolic compound -tocopherol (vitamin E)

23
exhibits antioxidant activity by trapping and stabilising reactive free radicals and
efficiently inhibiting lipid peroxidation.113
The antioxidant capacity of a compound is measured by its reactivity towards
the reactive radical species. Likewise vitamin E (29), Galvinoxyl (34) is also a
free radical inhibitor that is often used to investigate the antioxidant activity.
The rates of reaction of other phenolic compounds have been compared with
that of Galvinoxyl. BO-653 (31) and vitamin E (29) were found to have similar
reactivity, which is higher than that of another commonly used hindered
phenolic compound Probucol (32)114(Figure 1.6).
Figure 1.6: Phenolic and radical antioxidants.
The hydroxycinnamic acids (HCA) are also a class of phenolic antioxidants,
which are highly abundant in food and may account for one-third of the phenolic
compounds in our diet. Hydroxycinnamic acid compounds are of increasing
interest in health research because they are recognised as potent
antioxidants.115, 116 These compounds have been reported as radical chain

24
breaking antioxidants that achieve this action through radical scavenging
activity. This activity has been attributed to hydrogen or electron-donating
capacity and their ability to delocalise or stabilise the resulting radical within the
structure. The additional unsaturation incorporated in hindered
hydroxycinnamic acid compounds gives extra stabilisation to the phenoxy
radical species accordingly increasing their antioxidant activity. These active
phenolic compounds are employed in various biological applications.117, 118
HCA derivatives display their effects on cell membrane stabilisation through
their anti-hypoxic, hepatoprotective and anti-inflammatory mode of action. It
has been reported that there is a direct relationship between the antioxidant
activity and the membrane stabilisation activity of these compounds. HCA
derivatives (p-caumaric (35), ferulic (36), and caffeic (37) acids) also
demonstrate high antibacterial, antifungal activity and also antiviral activity119.
Some of the alkyl and cycloalkylamino derivatives of cinnamic acids also exhibit
hypolipidemic and antiatherosclerotic effects120 (Figure 1.7). Several adenosine
combining hindered phenolic antioxidant species are found to be biologically
active and useful in various therapeutic applications.121
Figure 1.7: Examples of derivatives of hydroxycinnamic acids.

25
1.3 Adenosine
Antioxidants are not only radical scavenging and H-transfer molecules, but they
can also generate an effect through switching on endogenous cellular
antioxidant mechanisms. Many clinical and experimental studies have
confirmed that stimulation of the adenosine receptor activates the antioxidant
mechanism of enzymes.122, 123 By stimulating neutrophils, adenosine inhibits
the superoxide anion generation. This adenosine-induced inhibition acts via
activation of cell surface receptors without cellular uptake of adenosine.124
Neuronal cells are highly sensitive to free radical induced oxidative stress.
Adenosine is used as a neuro-protective agent in the condition of free radical-
induced ischaemia-reperfusion injury and ischaemic stroke.125
1.3.1 Structural features of adenosine
Adenosine (39) is a naturally occurring nucleoside, formed during physiological
processes by degradation of the adenosine triphosphate (ATP) nucleoside (a
cell energy source). Its structure is composed of an adenine-based purine
nucleoside attached to a ribose sugar at the N9-position. The ribose sugar ring
is numbered independently in the chemical structure (Figure 1.8). Adenosine
derivatives are usually synthesised by modification at the exocyclic amine (N6),
the heteroaromatic ring carbon (C2) or the exocyclic (C5) ribose ring methylene
group. Adenosine is the only endogenous agonist for adenosine receptors and
regulates a number of physiological processes through the activation of
adenosine receptors.

26
Figure 1.8: Chemical structure of adenosine (39)
1.3.2 Adenosine receptors
A receptor is a protein macromolecular complex often found on the surfaces of
the cell membrane that receives or transmits chemical signals through the cell
wall. The chemical signals are generated through the binding of a particular
agent to these receptors which is extremely structurally selective. A ligand
binds to the receptor through various interactions including covalent, ionic,
dipole–dipole, and ion–dipole, hydrogen bonding and the charge-transfer
complex. The binding of a ligand generates a chemical signal, which is further
converted to a specific cellular and/or tissue response. Several ‘drug-receptor
interaction theories’ have been proposed to explain how a drug can generate or
stimulate a specific biological response.126
Several types of receptors are involved in various physiological processes. One
of these types of receptors is called a purinergic receptor. They are involved in
signalling actions mediated by purine nucleosides or nucleotides. The term
purinergic receptor is used to describe classes of plasma membrane receptors
that are also called purinoceptors. Purinergic receptors are involved in a

27
number of cellular functions including the migration of neuronal stem cells,
proliferation, vascular action, apoptosis and cytokine secretion.127, 128 The
purinergic receptors are categorised into three types: P1 (adenosine) receptors,
P2Y (all nucleotides) and P2X (ATP), where P1 and P2Y are G-protein-coupled
receptors (GPCR) and P2X are ligand-gated ion channels.129
GPCRs are receptors with a peptide sequence that pass through the cell
membrane seven times, and therefore, are known as seven-transmembrane
domain receptors or 7TM receptors. GPCRs are the most heavily targeted
receptors in the pharmaceutical industry, because of their significant role in
many diseases. Resulting in 40% of modern drugs being based on targeting
these receptors.130 GPCRs interact with different types of proteins and
participate in several important biological functions. They interact with hetero-
trimeric G-proteins (Figure 1.9) and with the accessory proteins called GPCR-
interacting proteins (GIP).131
The GPCR couples with the membrane-associated heterotrimeric G-protein
made up of G, G and G subunits.132 These subunits are in complex form
when in the resting state. The G subunit is the flexible signalling subunit. In
the inactive complex form, the G subunit binds to guanosine diphosphate
(GDP). After the activation of this receptors, a nucleotide-exchange reaction
occurs on the G subunit and the GDP is replaced by guanosine triphosphate
(GTP).133

28
These G-proteins exist in three different families of proteins including Gi, Gs and
Gq, which express selectivity for particular receptors. Gs and Gi proteins
stimulate and inhibit the adenylate cyclase enzyme, and Gq protein interacts
with the enzyme phospholipase-C respectively.134 The G subunits of these G-
proteins differ in structure (Figure 1.9).
Figure 1.9: Three-dimensional crystal structure of the G-protein coupled receptor (GPCR) embedded in the cell membrane attached to heterotrimeric G protein.
135
Adenosine receptors are the P1-type G-protein coupled receptor, which are
activated by the adenosine nucleotide released in cells. The adenosine
nucleotide is found in living cells and is involved in several metabolic functions
through both intracellular and extracellular mechanisms. In cells, adenosine
participates in all metabolic processes, and its concentration is modified by
pathophysiological conditions (i.e. during disease or disorder).136, 137

29
Adenosine is produced intracellularly by two metabolic pathways, which both
involve the hydrolysis of AMP to adenosine by 5-nucleotidase followed by the
catabolism of S-adenosylhomocysteine137 (Figure 1.10). The adenosine
concentration increases within the cell membrane during pathophysiological
conditions and is released from the cell. The released adenosine can then bind
to adenosine receptors and acts as a local metabolic regulator or, what has
been described as a “retaliatory metabolite”.138 Adenosine is also involved in
the regulation of several types of cells including endothelial cells, smooth
muscle cells and macrophages.139
Figure 1.10: Intracellular adenosine synthesis.140
Several studies have demonstrated the strong effect of adenosine purine
nucleoside on the regulation of various physiological and biochemical

30
functions.141 The local tissue concentration of adenosine increases when
exposed to oxidative stress. This ischaemic event protects the heart from the
further ischaemic injury, which has focused attention on understanding the
protective effects of adenosine.142-144 Adenosine exerts its effect through
several receptors called adenosine receptors. Adenosine receptors are
classified into four different receptor subtypes, namely A1, A2A, A2B, and A3
subtypes20 (Figure 1.11). These receptor subtypes are identified according to
their pharmacological properties and cloning studies. These adenosine
receptor subtypes are members of the GPCR family, which are classified
according to the receptor coupling to adenylate cyclase through Gi (A1, A3) and
Gs (A2A, A2B) protein. The receptor subtypes are involved in the inhibition (A1,
A3) and stimulation (A2A, A2B) of adenylate cyclase activity and subsequent
cyclic adenosine monophosphate (cAMP) production.145, 146
Figure 1.11: Adenosine receptor activation.147

31
Adenosine receptors were first investigated as potential drug targets in 1981.148,
149 Adenosine A1 receptor (A1AR) agonists are involved in cardiac arrhythmias
and neuropathic pain. Adenosine A2A receptor (A2AAR) agonists are useful for
myocardial perfusion imaging and as anti-inflammatory agents. Adenosine A2B
receptor (A2BAR) agonists are used in the treatment of cardiac ischaemia. The
Adenosine A3 receptor (A3AR) subtype is involved in inflammation, cancer and
cardioprotection. These cardioprotective effects are generated through binding
interactions of the four extracellular adenosine receptor subtypes. The
protective effect of adenosine in reperfusion injury has also been reported.150 A
study of acute myocardial infarction showed that administration of adenosine
reduced the infarct size.151, 152 In particular, adenosine plays a role in ischaemic
preconditioning to protect the heart from ischaemia153 and also protects against
acute myocardial infarction.
Adenosine A1 receptors (A1AR) 1.3.2.1
The A1 receptor is the first subtype of the G-protein coupled receptors that was
discovered. It is involved in a wide range of biological functions through
different signalling pathways, which are based on the coupling of the A1AR to
the different G-proteins.154, 155 In this mechanism, the A1AR agonist binds to the
Gi1/2/3 and G0 proteins and inhibits the enzyme adenylate cyclase by decreasing
the concentration of the secondary messenger cAMP.156, 157 The binding of the
A1AR activates K+ ion channels and inhibits Ca2+ ion channels that activate the
enzyme phospholipase C158-160 throughout the body.

32
The A1AR are distributed widely throughout the body and mediate different
biological functions. This receptor is present in the brain, spinal cord, heart,
liver, testis and white adipose tissues in higher concentrations.161 The A1AR is
found in lower concentrations in the lungs, kidney and small intestine162, 163 and
is present in the smooth muscles throughout the vascular system.164
Activation of the A1AR is involved in several myocardial pathological conditions
including ischaemia, reperfusion injury in arrhythmogenesis, coronary and
ventricular dysfunction, and chronic heart failure.165-168 In the heart, stimulation
by the A1AR acts as a myocardial depressant by decreasing the conduction of
electrical impulses and suppressing the pacemaker cell function, resulting in a
decrease of the heart rate. A1AR have also been detected in several types of
cancer, with suspicion that they may be involved in the stimulation of
chemotaxis of tumour cells.169
Adenosine A2A receptors (A2AAR) 1.3.2.2
The adenosine A2AR was first suggested in 1979 by Calker et al. 170 and in
1980 by Londos and co-workers.157 A2AR are classified further according to
their ligand binding affinity as A2AAR (high affinity for adenosine 0.1-1.0 µM) and
A2BAR (low affinity for adenosine ≥ 10 µM) by Bruns171 on the basis of initial
work by Daly and co-workers.172 It is a type of GPCR, whose activity is
mediated by G-proteins and stimulates adenylate cyclase and the synthesis of
cAMP.

33
A2AAR are present throughout the body and are found in low abundance in the
heart, lungs, blood vessels, and other areas of the brain.160. These receptors
are highly expressed in blood platelets, leukocytes, thymus, spleen and
olfactory bulb. These receptors have been involved in several
pathophysiological conditions. Current research has focused on developing
new A2AAR agonists in order to determine the structure of the A2AAR receptor.
This development has been achieved by cloning the A2AAR from several
species including rat,173 human brain,174 mouse175 and guinea pig brain.176 The
A2AAR agonists have also been found to increase blood flow during a cardiac
nuclear stress test.177
The A2AAR agonists play a key role in several diseases including
inflammation,178 ischaemia-reperfusion injury179 and cancer180 and have
cardioprotective effects.141 The A2AAR provides cardioprotection via
vasodilatation by increasing myocardial blood flow, and by supporting the
synthesis of new blood vessels as well as protecting tissues from collateral
inflammatory tissue damage.181 The A2AAR is beneficial in controlling
inflammatory leukocytes by reducing oxidative metabolites such as superoxide
and H2O2 in activated neutrophils.124, 182, 183
The activation of A2AAR may contribute in the pathophysiology of glaucoma due
to the increase in ocular perfusion pressure.184 Current evidence suggests that
the increased internal ocular pressure (IOP) leads to blindness via induction of
ischaemia and lowering IOP via blood reperfusion to cause ischemia-

34
reperfusion injury and retinal inflammation.185 Intravenous infusion of
adenosine in healthy volunteers led to the reduction of IOP and increasing
blood flow to the optic nerves. Experimental evidence has also shown that
application of adenosine and selective A2AAR agonists induced dilation of retinal
blood vessels via activation of A2AAR.186 Therefore, A2AAR agonists might be
an ideal agent in the treatment of glaucoma.
In addition, activation of the A2AAR on cells of the immune system plays a role
in controlling the activity of inflammatory cells including neutrophils,
macrophages and T lymphocytes. Therefore, A2AAR agonists may be
applicable as endogenous regulators of inflammation and cardioprotection.187,
188 However, A2AAR also negatively regulates over-reactive immune cells. The
A2AAR influence the activity of the indirect pathway of the basic ganglia in the
brain. In the brain, the A2AAR mediate the release of glutamate and dopamine,
making them a potential therapeutic target for neurological diseases.
Adenosine A2B receptors (A2BAR) 1.3.2.3
The A2BAR are low-affinity GPCRs that are involved in the activation of
adenylate cyclase.160 These receptors are small in number, and a high
concentration of adenosine is needed to generate a response. The adenosine
A2BAR has been implicated in the proliferation and differentiation of mast cell-
mediated angiogenesis.189-191 Additionally, adenosine stimulates the production
of interleukin 4 (IL-4) and IL-13 in mast cells via A2BAR activation.192

35
A2BAR have been suggested to be present in a relatively high density in, the
gastrointestinal tract, caecum, large intestine and urinary bladder. They are
thought to be present at a lower concentration in the lung, blood vessels, eye
and mast cells.193 Adipose tissue and the adrenal gland, brain, kidney, liver,
ovary and pituitary gland are thought to have a very low concentration of the
A2BAR.160
A2BAR are involved in several pathophysiological conditions including coronary
artery disorders,194 atherosclerosis,195 pulmonary hypertension associated with
lung disease,196 liver ischaemia–reperfusion injury,197 colitis198 and type II
diabetes.193
Adenosine A3 receptors (A3AR) 1.3.2.4
A3AR are GPCRs that bind specifically to Gi/ Gq proteins and are involved in
many pathophysiological functions. Activation of the A3AR inhibits adenylate
cyclase activity, which decreases the cAMP level and stimulates phospholipase
C and D, resulting in the elevation of intracellular inositol 1,4,5-triphosphate and
Ca2+ levels.199 The A3AR is involved in neutrophil degranulation in neutrophil-
induced tissue injury and the regulation of other immune cells of the innate and
adaptive immune systems. The A3AR also participates in intracellular signalling
and stimulates mitogen-activated protein kinases including extracellular signal-
regulated kinase1/2 and p38 by upstream activation of protein PI3K.200
A3AR are distributed widely in the body. The A3AR can be detected using radio-
ligand binding and functional assays in organs from various species. The

36
highest levels of these receptors are present in the testis, liver and brain. They
are present at low levels in the central nervous system, placenta, heart, bladder,
uterus, spleen, and eye of sheep, rats and humans.201-203 Some of the
functional A3AR are detected on the cell surface.204 Expression of the A3AR
can be detected at the mRNA level in both rodent and human organs. The
A3AR is overexpressed in cancer and inflammatory cells, rheumatoid arthritis,
psoriasis and Crohn's disease and exhibits low expression in normal cells.205-207
Adenosine expresses itself in all the receptor-binding and biological functions
via agonist and antagonist mechanisms.
1.3.3 The concept of agonism and antagonism
Endogeneous natural ligands, including hormones and neurotransmitters bind
to their receptors, which induces a further biological response. The ligand of a
adenosine receptors induces a conformational change in the receptor (a
transmembrane protein) which initiates the intracellular response. The agonism
and antagonism concept explains the different types of binding of a drug
molecule to its receptor.
Adenosine receptor selective ligands are synthesised by first studying their
binding interaction with GPCRs and are usually categorised as (partial)
agonists, neutral antagonists, (partial) inverse agonists, and positive and
negative allosteric modulators.

37
Over the past two decades, it has become clear that the available GPCR
ligands preferentially stimulate one signal-transduction pathway over another
signalling pathway. This phenomenon is referred to as functional selectivity or
biased signalling.208-210 Biased signalling implies that an agonist for one
pathway may act as an antagonist or inverse agonist for another signal-
transduction cascade. Exploiting the functional selectivity of compounds is an
important research field that will help develop drugs with fewer side
effects.209,211 Selective activation of the beneficial signalling pathways without
simultaneous activation of side effect-inducing pathways can be used to
improve the therapeutic window of a drug.
An agonist is a compound that binds to a receptor, reorients receptor and
protein conformation, and causes similar biological actions as the endogenous
species (such as hormones and neurotransmitters) which triggers the natural
response. An antagonist is a chemical or biological entity that inhibits the
effects of a natural agonist ligands (hormone, neurotransmitter) and is also
called a blocker of the receptor binding site or competitive inhibitors.
Adenosine agonists 1.3.3.1
An agonist can cause complete or partial effects when it binds to its receptor
under different physiological conditions. Agonists can be exogenous ligands
(such as drugs) or endogenous ligands for the receptor. Agonists are
categorised according to the binding ability of the ligand as full agonists, partial
agonists and inverse agonists (Figure 1.12). Full agonists activate all sites of

38
adenosine receptors, and side effects are associated with receptor activation.
Partial agonists bind to their receptor and show a partial response, and that can
provide the solution to avoiding side effects by activating only the specific site of
the receptor.212 The inverse agonists show a negative intrinsic effect or partially
or fully block the receptor, resulting in low or no biological response and are
also called antagonists.
Figure 1.12: Mode of action of a agonists.213
Adenosine antagonists 1.3.3.2
Antagonists that bind at the same site as the natural ligands are called
competitive antagonists, and agents that bind to mixed-receptor sites are known
as nonselective or non-competitive antagonists (Figure 1.13). Caffeine,

39
theophylline and other methylxanthines are non-selective antagonists for
adenosine receptors. Several antagonists are used in therapeutic
applications.214 It has been proposed recently that antagonists of distinct
adenosine receptor subtypes may be beneficial in the treatment of asthma215, 216
or certain neurological diseases, such as Parkinson’s disease.217
Figure 1.13: Mode of action of a competitive and noncompetitive antagonists.213
Adenosine mediates various physiological and pathophysiological functions via
agonist and antagonist mechanisms. All four adenosine receptors can be
influenced differently via these mechanisms. Structural modifications to the
parent adenosine molecule can significantly change activity towards these
receptors, which are essential in maintaining various pathophysiological
conditions. Specifically, cardiovascular diseases are a major health concern in

40
the world and therefore research has been focused towards the adenosine A2
receptor subtypes, which are believed to be mainly involved in the
pathophysiology of cardiovascular diseases.
1.3.4 Active adenosine A2AR agonists and antagonists in clinical trials
Adenosine A2AAR agonists 1.3.4.1
In the past two decades, a number of biologically important agonists and
antagonists that that can selectively exhibit A2AAR affinity, have been
synthesised. The study of these compounds has progressed further with the
discovery of the importance of the A2AAR agonist–ligand interactions. The C5-
modification of adenosine produces the potent and nonselective adenosine
receptor agonist 5-N-ethylcarboxamide (NECA) (Figure 1.14).
Figure 1.14: Adenosine A2AAR agonists.
Substitution at the C2-position of adenosine has been determined to be
essential for the A2AAR activity and selectivity. Most of the A2AAR agonists are
synthesised by C2-modification of NECA (40). In this regard, the C2-modified
ammonium salt of 4-[2-[(6-amino-9-b-D-ribofuranosyl-9H-purin-2-yl) thio] ethyl]
benzenesulfonic acid (PSB 0777, 41) is a potent full A2AAR agonist that has

41
been reported to stimulate acetylcholine-induced contractions in rat intestine
segments.218 This compound is expected to be potentially useful for the
treatment of inflammatory bowel disease (Figure 1.14).
C2-modified alkynyl linked adenosine compounds are also known to possess
A2AAR agonist activity and selectivity. In this regard, Apadenoson (48) is a
highly selective A2AAR agonist’s which has potential utility as a pharmacological
stress agent in myocardial perfusion imaging (MPI).219 The selective A2AAR
agonist YT 146 (46) acts as a vasodilator that also inhibits the synthesis of
inflammatory mediators to reduce ischemia/reperfusion injury.220 Furthermore,
this alkynyl analogue HENECA (YT146, 46), is being studied for
cardioprotective effects.221
A synthetic analogue A2AAR-selective agonist UK-432097 (42) was designed for
the treatment of chronic obstructive pulmonary disease, but it failed phase II
clinical trials and still being used as an internal probe to examine the active site
structure. The protein-binding capacity of C2-terminal amino acid conjugates,
the A2AAR agonist CGS 21680222 has been studied and it has shown 140 times
greater selectivity for A2AAR compared with the A1AR and it has effects on
neuronal transmission.223-225
The highly selective A2AAR agonists Apadenoson (48) (StedivazeTM) and
Binodenoson (CorVueTM) are in phase III trials for myocardial perfusion

42
imaging.226 Regadenoson (Lexican, 43) is used as a coronary vasodilator and
Sonedenoson is being evaluated as a new therapy for wound healing and
human diabetic foot ulcers.227 ATL313 (47), a A2AAR selective agonist
profoundly protect the mouse liver from reperfusion injury (Figure 1.15).
Figure 1.15: Pharmacologically active A2AAR agonists.
Adenosine A2AAR antagonists 1.3.4.2
A2AAR antagonists also play important roles in elucidating structure activity
relationships (SARs) and are synthesised primarily by modification of the
xanthine ring with caffeine and theophylline (from tea) as well-known examples.

43
The xanthine synthetic analogue DMPX (49)228 and the caffeine analogue (E)-8-
(3-chlorostyryl)caffine (CSC)229 were reported to be selective antagonists of the
A2AAR. In addition, the A2AAR antagonist Istradefylline (KW 6002, 51) has been
used in the treatment of Parkinson’s disease to reduce the movement disorder
(dyskinesia) caused by conventional anti-Parkinson’s drug therapy (Figure
1.16).230, 231
Further insights into the allowable structural diversity of receptor antagonists for
these receptor subtypes were discovered through the X-Ray crystal analysis of
a non-xanthine-based analogue ZM 241385 (51) bound to the A2AAR. This
compound has a 90-fold selectivity for the A2AAR over the A2BAR.232
Preladenant (SCH420814, 52) and several other similar analogues were
developed with longer hydrophobic chains at the C2-position. These
compounds were found to be highly potent and selective adenosine A2AAR
antagonists and were intended for use in the treatment of Parkinson’s
disease233 (Figure 1.16).
Figure 1.16: Pharmacologically active A2AAR antagonists.

44
The hypotensive and bradycardic activity of adenosine was first investigated in
1929 by Drury and Sezent Gyorgyi.234 Adenosine is currently used in the
treatment of several cardiovascular diseases under the brand name Adenocard
and Adenoscan. The recent determination of the crystal structure of some
adenosine receptors confirms that the 7TM topology is common between
various families of GPCR receptors. Adenosine receptor agonists have more
beneficial effects than do antagonists, and the crystal structure provides
information about the binding interaction of agonists to the protein receptors, as
shown in Figure 1.17.
Figure 1.17: Comparison of receptor-ligand interaction binding for the A2AR with the agonists adenosine
(39) and NECA (62) along with antagonists ZM 241385 (51).235
Structure of human A2AR in cartoon
representation are bound to the ligands (a) ZM 241385; (b) NECA; and (c) adenosine; (d,e) polar and non-polar interactions involved in agonist binding to human A2AR are shown for NECA and adenosine. Figure b and c the hydrogen bond interactions (H3 and H7) activates the A2A receptor. However, these bonding is abscent in antagonists Figure a. Figure d and e van der waals interactions with amino acid residues.

45
There is a great deal of interest in studying GPCR–drug interactions, particularly
those concerning the A2AARs. A2AAR agonists that act in the periphery to cause
vasodilation and to decrease blood pressure, can also induce cardiovascular
side effects such as hypotension.188 These effects can trigger a baroreflex
(body’s homeostatic mechanism to maintain blood pressure) in the heart,
leading to increases in heart rate and cardiac output, and therefore the demand
for oxygen. These conditions would be seriously detrimental under prolonged
ischaemic conditions and would work against any cardioprotective effects of
A2AAR activation in cardiomyocytes. However, it has been demonstrated that
adenosine itself has cardioprotective effects during myocardial ischemia236 and
reperfusion,237 and is expected that an A2AAR subtype-specific agent would
cause fewer side effects.
Adenosine A2BAR agonists 1.3.4.3
Several ligands for the A2BAR have been identified in recent years193, 238 and
have been studied as pharmacological targets and to evaluate their therapeutic
potential. However, the important advances in identifying A2BAR agonists
through improved in vitro pharmacological profiling have been recently
published.239 Previous SAR studies of NECA and adenosine derivatives have
indicated that N6 is a useful position for A2B receptor binding-site recognition.
Some examples of N6-substituted adenosine derivatives endowed with
satisfactory levels of A2BAR potency have been reported. In particular, the
introduction of (substituted) phenyl rings at the N6-position of adenosine led to
the A2BAR selective compounds.238

46
Based on some patent claims, a series of substituted 2-amino-4-phenyl-6-
phenylsulfanylpyridine-3,5-dicarbonitriles were synthesised as agonists for
adenosine receptors.240 A series of five 2-amino-6-(1H-imidazol-2-
ylmethylsulfanyl)-4-(substituted) phenyl pyridine-3,5-dicarbonitrile derivatives
has been also reported to display high-potency agonistic activity for the A2BAR
over the A3AR subtype.241 The A2BAR agonist 2-phenylaminoadenosine
(CV1808, 53) has been shown to be effective as a coronary vasodilator and as
an antihypertensive and antipsychotic agent.242, 243 The potent and highly
selective non-adenosine compound A2BAR agonist 2-((6-amino-3,5-dicyano-4-
(4-(cyclopropylmethoxy)phenyl)-2-pyridinyl)thio)-acetamide (BAY60-6583, 54)
has been shown to have similar potency to NECA 244, 245 (Figure 1.18).
Figure 1.18: Adenosine A2BAR agonists
Adenosine A2BAR antagonists 1.3.4.4
Several A2BAR antagonists with high affinity and good selectivity have been
identified among structures based upon a xanthine core suitably substituted at
the 1-, 3- and 8- positions.246 Kim et al.247 reported that a substituted
phenylcarbamoyl-methoxy-phenyl chain at the 8 position of a series of 1,3-
dipropylxanthines could specifically direct the antagonist activity to the A2BAR.

47
C8-modifications of adenosine analogues without a ribose ring are found to give
the selective A2BAR antagonists that are also involved in cardiac actions. In this
regard, 3-ethyl-3,9-dihydro-1-propyl-8-[1-[[3-(trifluoromethyl)phenyl]methyl]-1H-
pyrazol-4-yl]-1H-purine-2,6-dione (GS6201, 55) attenuates an inflammatory
response during acute myocardial infarction in mice and reduces caspase-1
activity in the heart.195 The highly potent human A2BAR inverse antagonist N-(4-
acetylphenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-
yl)phenoxy]acetamide (MRS 1706, 56) with extended chain was reported to be
a selective A2BAR antagonist with Ki value of 1.93 nM. (Figure 1.19).
Figure 1.19: Adenosine A2BAR antagonists
Despite the number of selective A2AAR agonists and antagonists that have been
reported, only a few adenosine derivatives have succeeded in clinical trials for
cardiovascular diseases. This failure is due to widespread signalling effects of
adenosine and unwanted side effects. It was believed that dual acting
adenosine molecule would be beneficial to overcome these complications.

48
1.4 Dual action adenosine agonists-Rationale for the research
The focus of current research is shifting towards the development of lead
compounds that can act on more than one target. This modern approach to
drug design combines two or more active parts of different molecules within one
molecule to generate a multitargeted drug.
The novel dual acting antioxidant 5-(3-methyl-pyrazole-5-ol-1-yl)-1,1,3,3-
tetramethylisoindoline-2-yloxyl (57) with an isoindoline nitroxide within the
Edaravone structure is act as a potential active drug for cardiovascular101 and
cerebrovascular ischemia in the brain-protection.248 The dual acting compound
UA-8 ( 13-(3-propylureido)tridec-8-enoic acid, 58) provides the cardioprotective
effect against the ischaemia-reperfusion injury249 (Figure 1.20).
Figure 1.20: Dual action compounds in Ischaemia
The majority of research on multi-targeted adenosine ligands has been carried
out by Ken Jacobson and co-workers. In 1985, Jacobson et. al. first published
their investigation into adenosine “binary conjugates” molecules. These
compounds are coupled to organic molecules such as peptides and amines and
act as ligands at adenosine receptors (Figure 1.21). This work initiated
research that resulted in the coupling of both adenosine agonists and
antagonists to biotin (a compound that forms a complex with glycoprotein
avidin), substance P-receptor binding entities as well as lipids. At the time,
49
some of the work was patented and many additional publications detailed the
synthesis of bifunctional adenosine analogues.250
Figure 1.21: Bifunctional adenosine receptor-binding analogues and binary conjugates.
The broader interest in the field of binary conjugates of adenosine A1AR and
A3AR agonists commenced with a publication in 2000, 251 and a continuance of
the original patent was granted in 2003.252 Further work in the field of
bifunctional adenosine drugs rose again in 2003 when Liang and Jacobson filed
a patent for the use of adenosine receptor agonists and antagonists together as
a novel approach for treating ischaemic injury to the heart.252 In the patent, the
activity of the binary conjugate MRS1528 (62) was explained, i.e. it acts as an
agonist for the A3AR and as an antagonist at the A2AAR.

50
In 1986, Burns et al characterised the A2 adenosine receptors and termed them
A2AR. During the 1989s, Hutchison and co-workers found that C2-substituted
adenosine compounds were selective A2AR agonist ligands and possessed
hypotensive activity.253 In 1992, Cristalli and colleagues published further
research that demonstrated that C2-substituted alkynyl linked with 5-
ethylcarboxamide adenosine compounds increased A2AAR selectivity.254 In
1997 255 and 2001,183 Martin and Sullivan et al further developed C2-substituted
adenosine compounds by preparing compounds containing a 2-cyclohexyl
linked with hydrazine chain (63) and an alkynyl chain (64). These compounds
enhanced A2AAR selectivity and were effective in coronary vasodilation and
inhibiting human neutrophil oxidation. Pfizer patented several C2-, N6- and C5-
modified adenosine compounds (65 and 66) as anti-inflammatory agents with
inhibition of neutrophil function at a concentration of < 40 nM 256 (Figure 1.22).
51
Figure 1.22: Pharmacologically active A2AAR and A3AR selective analogues.
Interest in A2AAR has been rekindled by a finding that suggests both agonisms
of this receptor and its inhibition by antagonists may have therapeutic
applications. In 2001, Ohta and Sitkovsky reported that A2AARs are
imperatively involved in the limitation and termination of the prolonged
inflammation.257
In 2007, Gregg and co-workers published new research on dual acting A1AR
agonists that contained antioxidant species and had the potential to produce
cardioprotective effects.258 Furthermore, in 2009 and 2012, Foitzik and Hausler

52
reported the cardioprotective effects of A2AAR selective adenosine
compounds259 attached by an aminoethylaniline linker possessing antioxidant
functionality, which were shown to reduce ROS activity. The C2- and C5-
modified adenosine compounds VCP874 (69) and VCP728 (70) attached by an
aminoethylaniline linker were shown to have an excellent A2AAR potency and
selectivity. They demonstrated good EC50 values of 45 nM and 6.6 nM,
respectively121 (Figure 1.23).
Figure 1.23: Pharmacologically dual-action antioxidant coupled A2AAR and A3AR-selective analogues.
Adenosine compounds that act via A2AAR on leucocytes significantly inhibit the
inflammatory process. Adenosine compounds with the para-(2-
aminoethyl)aniline linker at the C2-position are known to have A2AAR selectivity.
Attachment of the antioxidant structure by a flexible linker is predicted to
contribute to the A2AAR pharmacophore, improving A2AAR selectivity and thus

53
localising antioxidant function to the desired site of action. Once positioned, this
can move in the vicinity of its intended site of action, scavenging cytotoxic ROS
before they can cause further injury (the antioxidant species will be active from
the moment of administration and the ROS scavenging properties are therefore
not strictly limited to A2AAR orthosteric site, but apply more generally) (Figure
1.24).
The value of this approach has been given support by binding studies
undertaken using molecular modelling that have analysed the crystal structure
of bifunctional compound 70. Here the attached antioxidant moiety would
extend beyond the orthosteric binding site of the adenosine derivative. The 2.7
Ǻ-resolution crystal structure of the human A2AAR agonist in complex with a
selective A2AAR agonist UK-432097 has shed light on the ligand-induced GPCR
binding and activation260 (Figure 1.24).121
A B
Figure 1.24: Adenosine receptor crystal structure in complex with A)UK-432, 097 (42), B) compound VCP728 (70).

54
Currently, there is a need for potent A2AAR agonists with combined antioxidant
species to test this theory. In addition, cardiovascular and other inflammatory
diseases have complex pathogenesis, and currently available adenosine and
antioxidant compounds are still not able to do enough on their own, treating only
a limited number of these complications. Treatment involving adenosine
analogues have selectivity problems and hence forming unwanted side effects
and low efficacy. Antioxidant therapy is more effective in a combination of other
related treatments.
These days, it is common practice to administer a cocktail of drugs with
complementary effects to patients. However, this combined drug therapy
introduces complications associated with pharmacokinetics, distribution, toxicity,
and patient's compliance and elevate the risk of side effects. To circumvent
some of these issues, the focus of current research is shifting towards the
generation of multi-targeted lead compounds. This new generation of designed
drug compounds combines two or more active parts in a single molecule that
each initiates independent but complementary action when administered. A
single administration of multi-targeted compounds can have benefit in the
predictability of their pharmacokinetics (PK) and pharmacodynamics (PD)
properties and also in improved patient compliance. It is anticipated that, this
bifunctional approach could have beneficial effects from the combined entities
which would not only be additive but synergistic in nature, resulting in increased
efficacy.

55
It has been demonstrated that both the stimulation of adenosine receptors and
presence of antioxidants such as nitroxide radicals have independently shown
beneficial effects in reducing the duration of ischemia. These effects also limit
ischemia-reperfusion injury in models of acute myocardial infraction. It was
proposed that compounds that possess both adenosine-receptor binding and
antioxidant capabilities would demonstrate potent cardioprotective actions and
be more effective than the two independent moieties.
The goal of this project therefore was to explore multi-targeted compounds that
can act as selective A2AAR agonists. These compounds (some initial target
structures are shown in Figure 1.25 and 1.26) would also possess a moiety that
is able to react with ROS (and other free radical species). Thus, these
compounds could potentially act as effective preventive treatments for
inflammatory diseases and also provide cardiovascular protection.
Figure 1.25: Antioxidant coupled p-aminoethylaniline linked adenosine target compounds (Series 1).
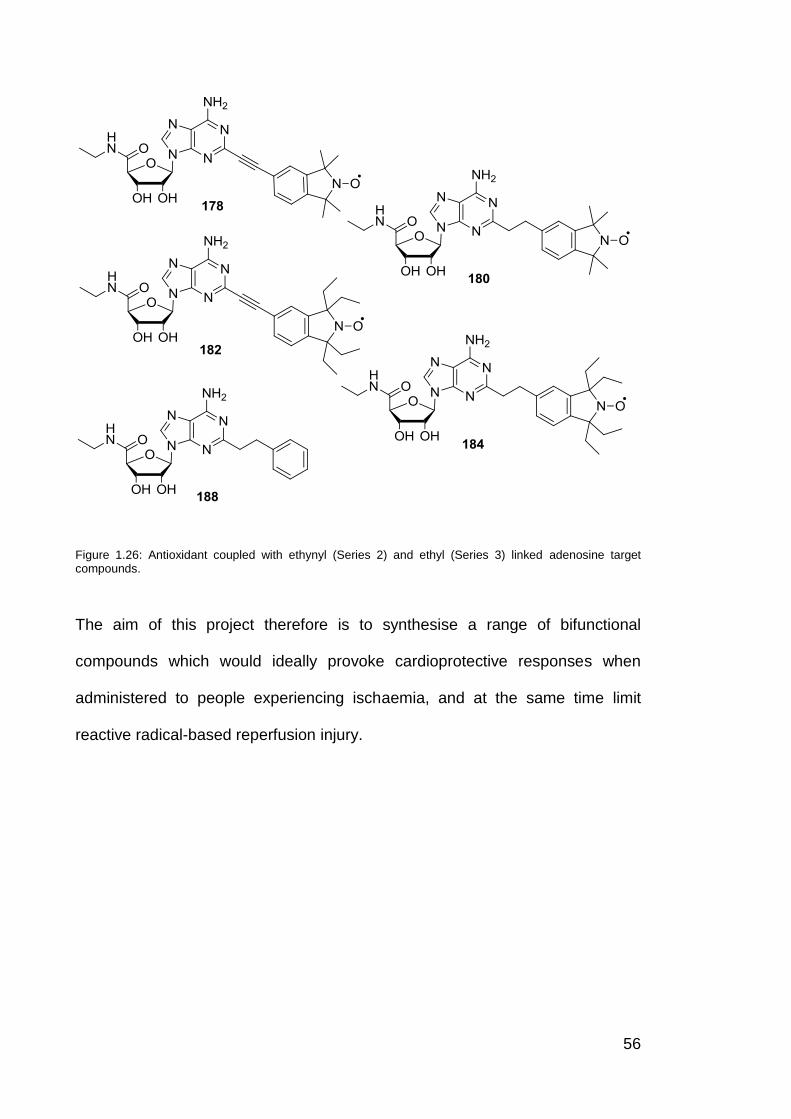
56
Figure 1.26: Antioxidant coupled with ethynyl (Series 2) and ethyl (Series 3) linked adenosine target compounds.
The aim of this project therefore is to synthesise a range of bifunctional
compounds which would ideally provoke cardioprotective responses when
administered to people experiencing ischaemia, and at the same time limit
reactive radical-based reperfusion injury.

57
Results and Discussion Part A: Adenosine 2analogues with para-aminoethylaniline linker
possessing antioxidant moieties

58
2.1 Preliminary Considerations
There have been many efforts to attempt to elucidate the various SARs for the
adenosine receptor subtypes. These efforts are continuing, and the
understanding of key structural features has improved, which is essential to
enhance subtype affinity and selectivity.256 Most of the current adenosine
receptor ligands are based on the structure of endogenous adenosine, and
therefore tend to be polar in nature.261 The nitrogen atoms at position 3 and 7
on the adenosine structure are particularly essential for high affinity for the
adenosine receptors (Figure 2.1).256, 262
The ribose moiety attached to the N9-position of the adenosine molecule is the
β-D-ribofuranose stereoisomer. The experimental evidence clearly indicates
that the ribose group binds to the receptor pocket via its hydroxyl groups and,
therefore, is essential for binding affinity and adenosine receptor activation.263
Further evidence shows that the adenosine molecule is arranged in an anti-
conformation within the receptor binding pocket, and thus the essential
pharmacophore must possess this particular ribose stereoisomer.264 This
ribose stereoisomer with at least one free 2- or 3-hydroxyl group is necessary
for the agonist activity at adenosine receptors.
Although all available ribose hydroxyl groups are hydrogen-bonded to the
receptor pocket, modifications at the C5- and even C3-position are known to
be tolerated.256 Even the furanose oxygen can be replaced by a carbon with
retention of activity.261 In addition, C2-256 and N6-positions on the purine ring

59
are most tolerant towards modifications with other groups. Indeed, structural
modification at C2 and N6 is thought to be responsible for the increased
receptor subtype selectivity and affinity.265 Most of the C6-modifications on the
purine ring undertaken to date lead to more potent and selective A1AR agonists.
However, co-substitution at the C2- and N6-positions does not necessarily lead
to additional affinity for the A2AAR over the A1AR.265, 266
The C6–amino proton appears to be necessary for high receptor affinity.263
This property may indicate either some unfavourable steric interaction for
N,N-disubstituted adenosine analogues or a hydrogen bond forming the role of
the C6-amino proton with the N6 binding domain of the orthosteric binding site.
However, certain bulky N6-substituents are known to be A2AAR tolerated such
as the hydrophobic 6-diphenylethylamino group. Almost all other N6
modifications considered favour A1AR affinity. Non-bulky modification at the
endogenous C6–amino223, 262, 267-269 group is also known to be tolerated by
A2AAR.270 Consequently, the additions of non-bulky side chains at the
C6-amino group were employed for all of the synthetic targets used in this
project.
The 5-modification most essential for an increase in agonist receptor affinity
such as the N-ethylcarboxamide moiety (5-NECA). The NECA itself is a
prototypic A2A adenosine receptor agonist.223, 266, 268, 271 This modification is
also known to improve pharmacokinetic stability to adenosine derivatives
because of the lack of interaction with adenosine deaminase.270 An ethyl amide
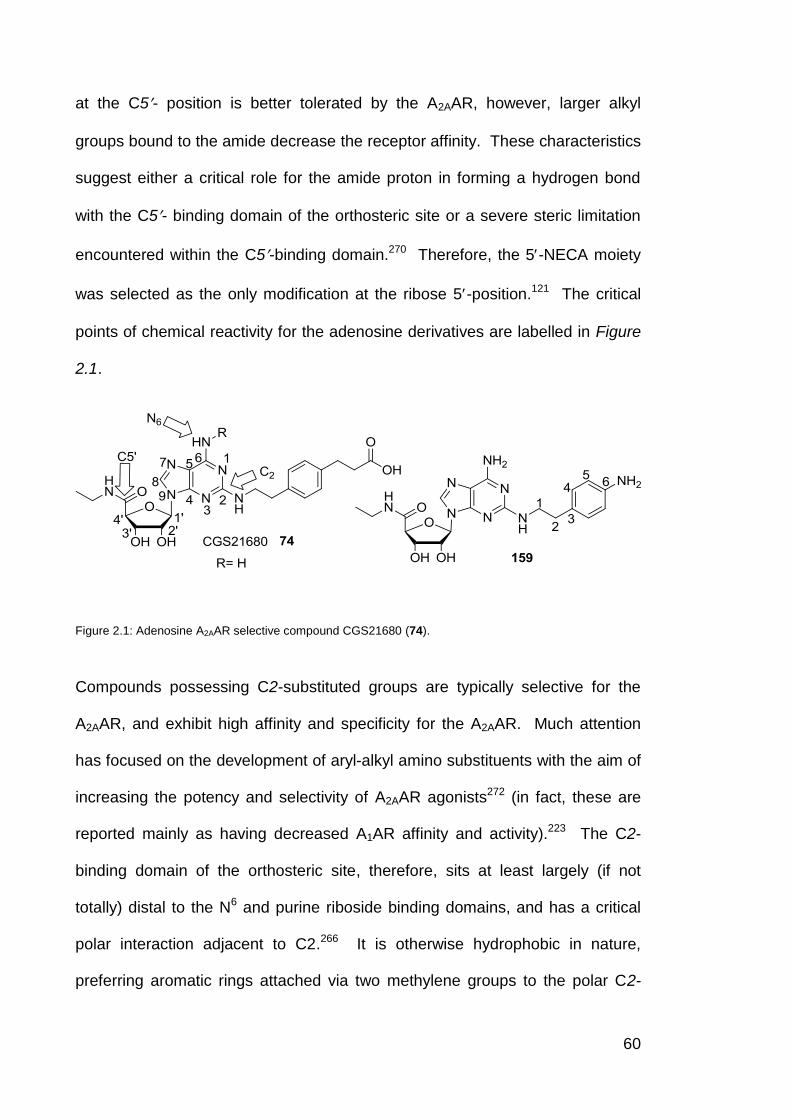
60
at the C5- position is better tolerated by the A2AAR, however, larger alkyl
groups bound to the amide decrease the receptor affinity. These characteristics
suggest either a critical role for the amide proton in forming a hydrogen bond
with the C5- binding domain of the orthosteric site or a severe steric limitation
encountered within the C5-binding domain.270 Therefore, the 5-NECA moiety
was selected as the only modification at the ribose 5-position.121 The critical
points of chemical reactivity for the adenosine derivatives are labelled in Figure
2.1.
Figure 2.1: Adenosine A2AAR selective compound CGS21680 (74).
Compounds possessing C2-substituted groups are typically selective for the
A2AAR, and exhibit high affinity and specificity for the A2AAR. Much attention
has focused on the development of aryl-alkyl amino substituents with the aim of
increasing the potency and selectivity of A2AAR agonists272 (in fact, these are
reported mainly as having decreased A1AR affinity and activity).223 The C2-
binding domain of the orthosteric site, therefore, sits at least largely (if not
totally) distal to the N6 and purine riboside binding domains, and has a critical
polar interaction adjacent to C2.266 It is otherwise hydrophobic in nature,
preferring aromatic rings attached via two methylene groups to the polar C2-

61
linking atom (Figure 2.1). The size of the hydrophobic C2-domain appears to
be better suited to an attached aromatic ring because the affinity of the
saturated cyclohexyl-bearing compound is significantly less than that of C2-
arylalkyl amino compounds.265
It is proposed that a linker involving a ‘bulky’ substituent linked with a two
carbon chain at the C2-position of adenosine could endow the generated
analogue with some subtype selectivity at adenosine receptors. C2-
substituents are well tolerated, and a range of different substituents display
variations in subtype selectivity. In particular, A2AAR selectivity appears to be
maximised by an electronegative bonding atom,261-263, 265, 267, 270, 272 a two or
three-atom spacer and a bulky, nonpolar group such as a cyclohexyl or phenyl
ring,259, 263 which can be itself further substituted.
It has been further proposed that the introduction of radical species attached to
the bulky linker substitution at C2-position of adenosine may improve the
receptor subtype selectivity and also be able to scavenge free radicals present
at the site. To test this theory, it was speculated that the antioxidant moieties
be tethered to the receptor binding adenosine using a ‘linker side chain’ that
would extend outside the binding pocket of the receptor. In such compounds,
the antioxidant functionality would still be joined to the receptor binding entity,
but, the ability of such dual action compounds to interact with the receptor
would not be compromised.

62
Another advantage of the ‘linker’ approach with an aniline-based unit such as
the aryl alkylamino chain linker would be that the antioxidant portion of the
molecule would be located outside the hydrophobic part of receptor pocket.
The antioxidant fuctionality would consequently interact more easily with
reactive free radical species.121
In the literature of adenosine analogues, a number of different linker groups
have been used. This linker groups were used to bridge from the receptor-
binding adenosine functionality to another moiety, including a fluorescent group,
a radiolabelled group, or another receptor binding entity. The compound
CGS21680 (46) has been shown to possess high affinity and specificity at the
A2AAR. By incorporating the commercially available para-(2-aminoethyl)aniline
(124), which has an aniline ring attached through an ethylamino group at the
C2-position in the target compounds. This type of compound preserves the 6-
carbon distance between two amino groups and also maintains high A2AAR
selectivity (Figure 2.1 and 2.2).

63
Figure 2.2: Candidate moieties and their actions in the initial structural class of target adenosine analogues.
In this study, all three series of compounds were synthesised with C2-
substitution on the adenosine NECA molecule. Further design and different
extensions to this structural C2-modification on adenosine was achieved with
appropriate linker groups and the desired antioxidant function. This approach
confers a bifunctional mode of action upon the target molecules without
compromising binding efficiency.
Nucleophilic C2-substitutions on adenosine can be accessed via an SNAr
(nucleophilic aromatic substitution) process. The literature provides an example
of the most nucleophilic atoms (i.e. S265, 273, 274, O261, 266 and N,121, 223, 259, 274),
which are primary choices of nucleophilic atoms that can be used to attack at
the C2-position of adenosine. This position is activated for SNAr by the
installation of a lower (highly electronegative) halogen atom (F, Cl, I). These
synthetic variables are discussed below in further detail.
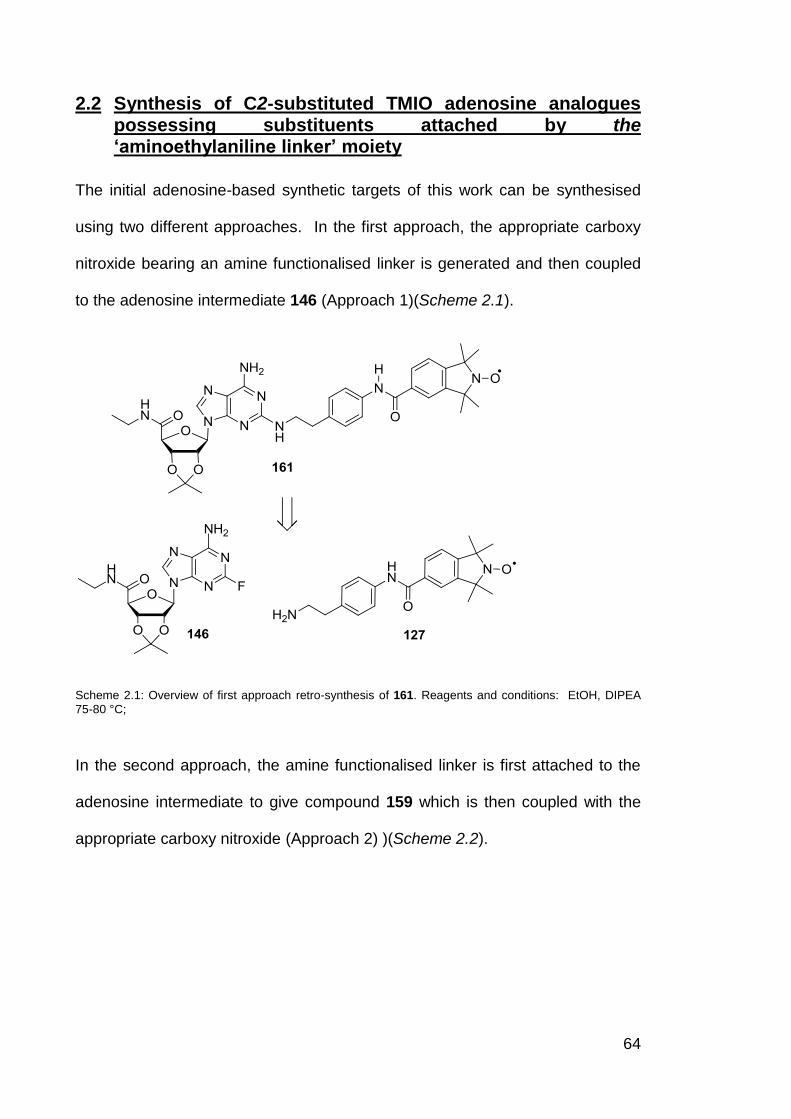
64
2.2 Synthesis of C2-substituted TMIO adenosine analogues possessing substituents attached by the ‘aminoethylaniline linker’ moiety
The initial adenosine-based synthetic targets of this work can be synthesised
using two different approaches. In the first approach, the appropriate carboxy
nitroxide bearing an amine functionalised linker is generated and then coupled
to the adenosine intermediate 146 (Approach 1)(Scheme 2.1).
Scheme 2.1: Overview of first approach retro-synthesis of 161. Reagents and conditions: EtOH, DIPEA
75-80 °C;
In the second approach, the amine functionalised linker is first attached to the
adenosine intermediate to give compound 159 which is then coupled with the
appropriate carboxy nitroxide (Approach 2) )(Scheme 2.2).
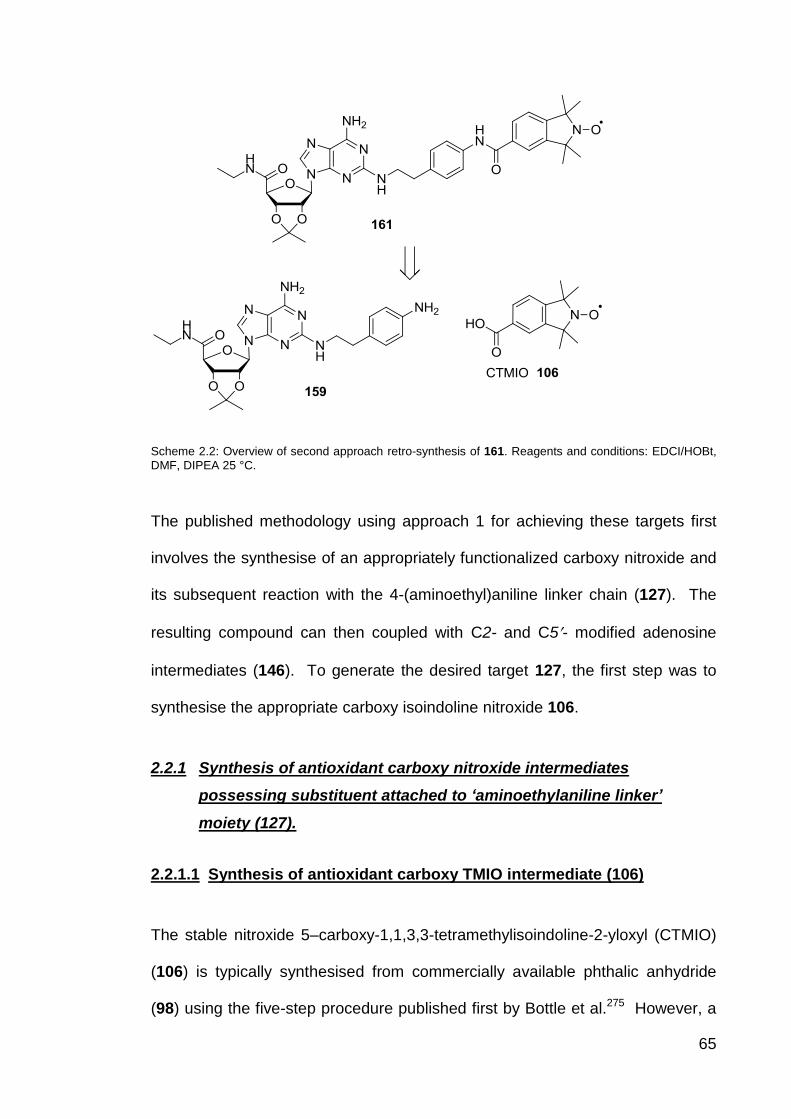
65
Scheme 2.2: Overview of second approach retro-synthesis of 161. Reagents and conditions: EDCI/HOBt,
DMF, DIPEA 25 °C.
The published methodology using approach 1 for achieving these targets first
involves the synthesise of an appropriately functionalized carboxy nitroxide and
its subsequent reaction with the 4-(aminoethyl)aniline linker chain (127). The
resulting compound can then coupled with C2- and C5- modified adenosine
intermediates (146). To generate the desired target 127, the first step was to
synthesise the appropriate carboxy isoindoline nitroxide 106.
2.2.1 Synthesis of antioxidant carboxy nitroxide intermediates
possessing substituent attached to ‘aminoethylaniline linker’
moiety (127).
Synthesis of antioxidant carboxy TMIO intermediate (106) 2.2.1.1
The stable nitroxide 5–carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl (CTMIO)
(106) is typically synthesised from commercially available phthalic anhydride
(98) using the five-step procedure published first by Bottle et al.275 However, a
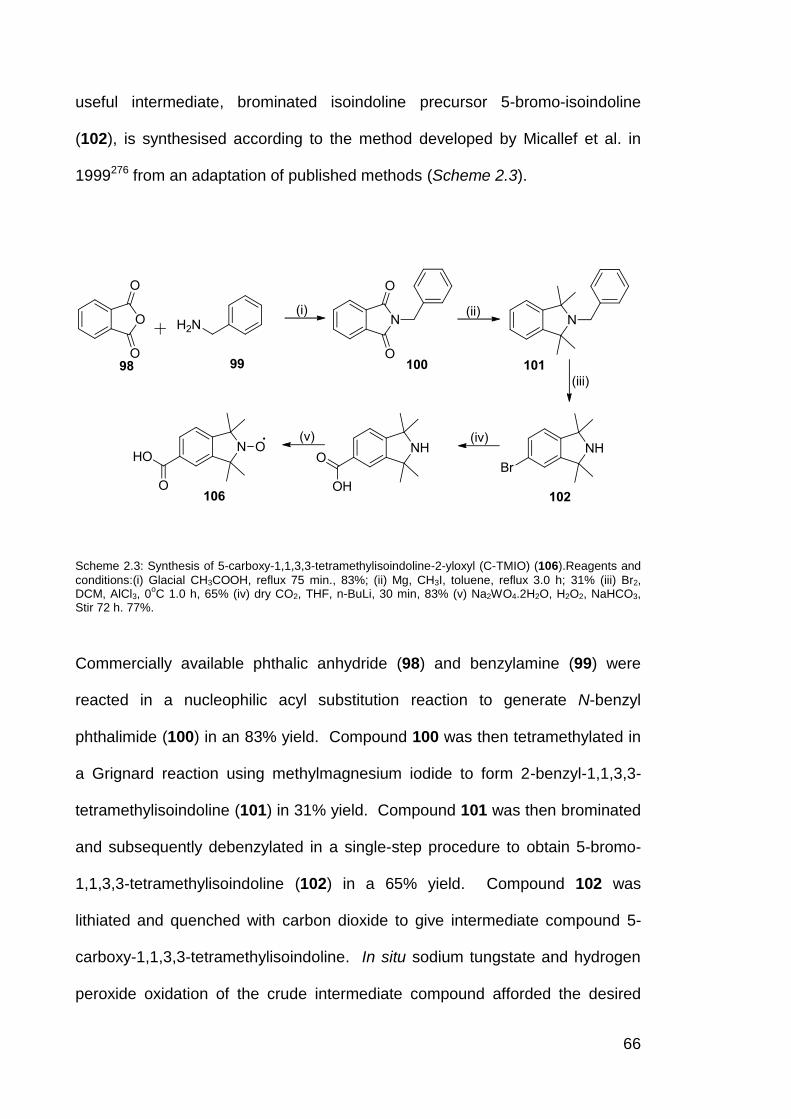
66
useful intermediate, brominated isoindoline precursor 5-bromo-isoindoline
(102), is synthesised according to the method developed by Micallef et al. in
1999276 from an adaptation of published methods (Scheme 2.3).
Scheme 2.3: Synthesis of 5-carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl (C-TMIO) (106).Reagents and
conditions:(i) Glacial CH3COOH, reflux 75 min., 83%; (ii) Mg, CH3I, toluene, reflux 3.0 h; 31% (iii) Br2, DCM, AlCl3, 0
oC 1.0 h, 65% (iv) dry CO2, THF, n-BuLi, 30 min, 83% (v) Na2WO4.2H2O, H2O2, NaHCO3,
Stir 72 h. 77%.
Commercially available phthalic anhydride (98) and benzylamine (99) were
reacted in a nucleophilic acyl substitution reaction to generate N-benzyl
phthalimide (100) in an 83% yield. Compound 100 was then tetramethylated in
a Grignard reaction using methylmagnesium iodide to form 2-benzyl-1,1,3,3-
tetramethylisoindoline (101) in 31% yield. Compound 101 was then brominated
and subsequently debenzylated in a single-step procedure to obtain 5-bromo-
1,1,3,3-tetramethylisoindoline (102) in a 65% yield. Compound 102 was
lithiated and quenched with carbon dioxide to give intermediate compound 5-
carboxy-1,1,3,3-tetramethylisoindoline. In situ sodium tungstate and hydrogen
peroxide oxidation of the crude intermediate compound afforded the desired

67
nitroxide 5-carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl (106) in a yield of
77% over two steps.(Scheme 2.3).
The antioxidant carboxy TMIO 106 was then in hand to react with linker 4-(2-
aminoethyl)aniline moiety (124) to generate the appropriate antioxidant bearing
side chain to synthesise the C2-modified adenosine target compound (161).
Synthesis of antioxidant carboxy TMIO coupled with para-2.2.1.2
(aminoethyl)aniline chain linker (127).
The antioxidant-bearing side chain 127 was initially synthesised separately in a
three-step reaction sequence involving coupling of the core compound 124 with
the antioxidant carboxy isoindoline nitroxide 106 to form a stable amide (125).
In the first-step, the more reactive primary aliphatic amine of the commercially
available compound 124 was protected with di-tert-butyl dicarbonate to give the
mono-Boc-protected compound N–tert-butoxycarbonyl-2-(4-aminophenyl)
ethylamine (125) (Scheme 2.4).
Scheme 2.4: Overview of N-Boc protection of linker amine. Reagents and conditions: (i) (Boc)2O, DCM, 100 min., 89%.
This was accomplished using a standard amine N-boc-protection method using
di-tert-butyl dicarbonate.277, 278 Dichloromethane (DCM) was substituted for
chloroform as the solvent for convenience, and the reaction was maintained at 0
°C to minimise undesirable reactivity at the 4–aminophenyl group. After 100

68
min, the starting material was consumed and < 5% of the undesired di-boc-
protected species was recovered. The reaction was conducted on a 1–2 g
scale.
The primary aliphatic amine is a stronger nucleophile than the aniline and
attacks the anhydride carbonyl to form the sp3 hybridised intermediate.
Reformation of the carbonyl eliminates a tert-butyloxycarboxylate anion, which
quickly releases CO2 and the tert-butoxide anion. This anion abstracts the extra
proton from the Boc-protected amine to form the product 125 and tert-butyl
alcohol (Scheme 2.5). The addition of the boc group to the primary amine
lowers the overall polarity of the compound, giving an Rf value of 0.67 in (9:1)
DCM/MeOH.
Scheme 2.5: Mechanism of N-boc protection of linker amine

69
The structure of 125 was supported by 1H NMR and 13C NMR spectroscopy and
mass spectrometry (MS) with the obtained spectra in agreement with the
published values. The Boc-protected amine was characterised using electron
ionisation MS (EIMS), which showed the expected molecular ion of M+Na at
259.1433 m/z (EI+ (HRMS) showed a deviation of 0.0011 from a calc. mass of
C13H20NaN2O2) and by a melting point of 56–58°C which supported the
successful synthesis of 125.121 In the 1H NMR spectrum, a new peak appeared
as a singlet at 1.45 ppm for the nine protons of the tert-butyl group. In the 13C
NMR spectrum, three additional peaks appeared. A strong peak corresponding
to the equivalent Boc carbons appeared at 28.4 ppm, the quaternary Boc
carbon appeared at 79.1 ppm, and the new Boc-amide carbonyl appeared at
155.9 ppm.
The Boc-protected amine linker (125) was coupled with the carboxylic acid
functionality of isoindoline nitroxide antioxidant 106 to form an amide. The
standard peptide coupling agent EDCI and reaction conditions were employed
in the formation of the amide.
The novel compound N-tert-butoxycarbonyl-2(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline)aminophenyl)ethylamine (126) was synthesized from
compound 125 following the published procedure for amide coupling by
employing the carboxy isoindoline nitroxide compound 106 and EDCI in the
presence of HOBt to give N-tert-butoxycarbonyl-2(4-N-(5-carboxy-1,1,3,3-

70
tetramethylisoindoline)aminophenyl)ethylamine (126) in 77% yield (Scheme
2.6).
Scheme 2.6: Overview of EDCI amide coupling of CTMIO nitroxide with para-aminoethylaniline linker moiety (127), Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C, 77%; (ii) 4 M HCl solution, 1,4-
dioxane:water(1:1), 55–60 °C, 5 h.,89%.
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) acts as an
acid activating agent, forming O-acylisourea a reactive ester intermediate in situ
with the carboxylic acid of the antioxidant moiety. This O-acylisourea formed
has an activated leaving group and is attacked by the 1-Hydroxybenzotriazole
hydrate (HOBt) to form HOBt ester. A thermodynamically stable urea by-
product is released. The nucleophilic aniline is reacted with HOBt ester to give
the novel amide target (Scheme 2.7).
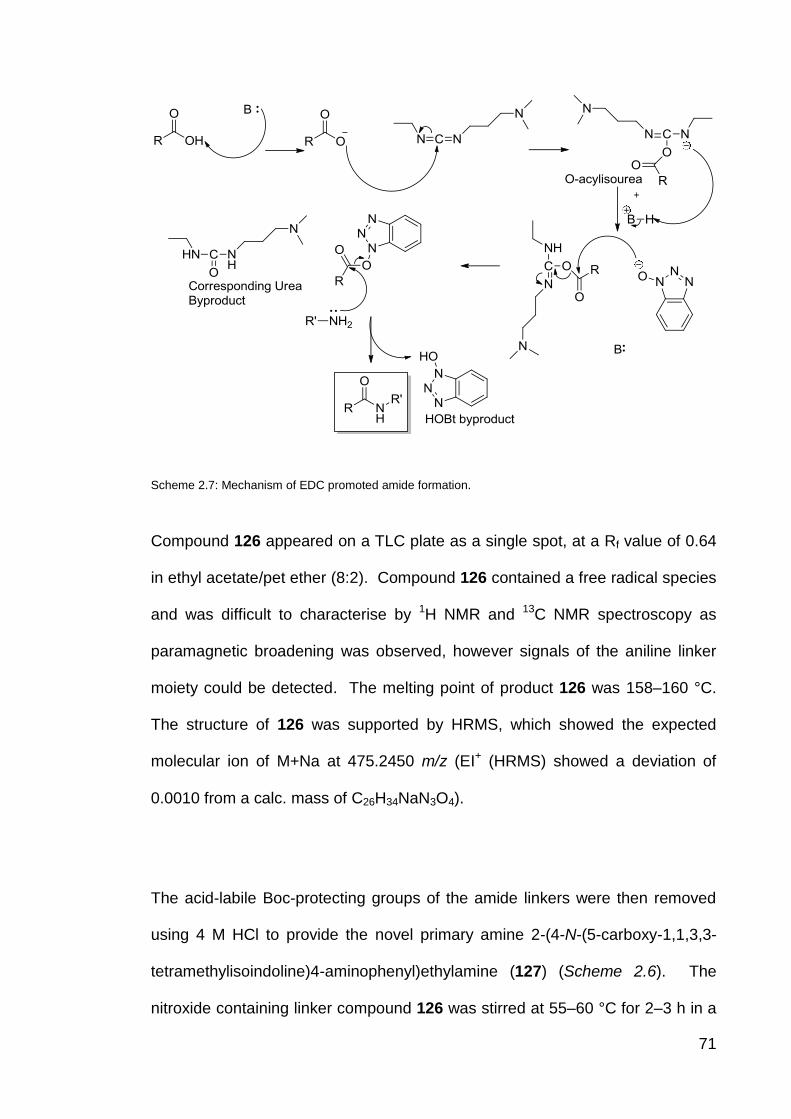
71
Scheme 2.7: Mechanism of EDC promoted amide formation.
Compound 126 appeared on a TLC plate as a single spot, at a Rf value of 0.64
in ethyl acetate/pet ether (8:2). Compound 126 contained a free radical species
and was difficult to characterise by 1H NMR and 13C NMR spectroscopy as
paramagnetic broadening was observed, however signals of the aniline linker
moiety could be detected. The melting point of product 126 was 158–160 °C.
The structure of 126 was supported by HRMS, which showed the expected
molecular ion of M+Na at 475.2450 m/z (EI+ (HRMS) showed a deviation of
0.0010 from a calc. mass of C26H34NaN3O4).
The acid-labile Boc-protecting groups of the amide linkers were then removed
using 4 M HCl to provide the novel primary amine 2-(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline)4-aminophenyl)ethylamine (127) (Scheme 2.6). The
nitroxide containing linker compound 126 was stirred at 55–60 °C for 2–3 h in a

72
mixture of 1,4-dioxane and water with a dropwise addition of 4 M HCl over 10
minute. Analysis by TLC indicated a loss of starting material, and ninhydrin
staining highlighted the generation of a product expected to be the reformed
primary amine 127 on the TLC plate with an intense purple colour with an Rf of
0.46 in chloroform/methanol (9:1). The compound was purified by column
chromatography to give an 89% yield of the deprotected primary amine product
127 with the sharp melting point of 176–178 °C.
The structure of 127 was supported HRMS, which showed the expected
molecular ion of M+H at 353 m/z (EI+ (HRMS) showed a deviation of 0.0011
ppm from a calc. mass of C21H27N3O2). In the 1H NMR spectrum, paramagnetic
broadening was observed due to the nitroxide radical that resulted in some
signals not being detected or giving poor integration. However, signals
corresponding to the aniline and the ethylene linker moiety could be detected,
the aromatic signals at 7.21 and 7.61ppm.
2.2.2 Synthesis of C2-substituted adenosine intermediates (146)
Adenosine receptors were first investigated as a potential drug target in 1981148,
149 and the compound NECA was quickly identified as the prototypic A2
adenosine receptor agonist.279 Adenosine analogues with amide functionality at
the C5-position have been the focus of research since that time. Further
investigation of the A2A adenosine receptor in 1990280 and the discovery that the
C5 amide adenosine analogues are active at this receptor has subsequently
prompted more work in this area.

73
Convergent approach to synthesis of adenosine intermediate 2.2.2.1
A general synthesis of C2-modified N-alkyl-5-N-carboxamide was published by
Jacobson and Kim et al. in 1994 and has been subsequently adopted as the
standard procedure for generating such analogues. The synthetic route most
commonly reported in the literature 223, 259, 263, 272, 274, 281-283 has several
drawbacks for the targets of this research project. Convergent synthesis has
been performed on numerous occasions with varying degrees of success, and
this approach was initially explained here. Such synthetic methods involve
separate modification of the appropriate –D-ribose sugar, followed by a
Vorbrüggen coupling of the anomeric N9 of the purine ring compound. This
compound has been modified at C6 and which can then be substituted at C2 as
required259, 262, 270, 272, 274, 281, 282 (Scheme 2.8).

74
Scheme 2.8: Convergent approach to adenosine A2AAR agonists: Reagents and conditions: (i) Candida rugosa lipase, 1,4-dioxane, 0.1 M sodium phosphate buffer (pH = 7); (ii) TEMPO/BAIB, MeCN/H2O (1:1);
(iii) EDCI, DMAP, MeOH; (iv) R-NH2, t-BuOH, 80 °C; (v) HMDS, MeCN, (NH4)2SO4, TMSOTf; (vi) EtNH2, THF; (vii) R-NH2, EtOH, 120 °C.
The literature shows that the convergent approach gives poorer yields than a
linear approach. Modification of the ribose ring has proved challenging, time-
consuming, and only produced moderate yields. The Vorbrüggen reaction was
difficult to scale up, and substitution at the 2-chloro requires sufficiently harsh
conditions, which threaten the stability of the ribose and antioxidant moieties.
Instead, a linear approach proved more reliable than the convergent method
and afforded acceptable yields.259

75
A linear synthetic approach to this class of compounds tends to have many
steps and, therefore, provides reduced overall yields. The literature proves that
the potential starting materials, including inosine284, 285 and xanthosine286, 287
derivatives, are often difficult to work with. This difficulty is because of their
poor solubility, which requires the use of harsh reaction conditions including the
use of DMSO, DMF, and strongly acidic media. In addition, the similarity in the
reactive groups, which make the regioselectivity modification difficult. This
linear synthetic approach towards C2-substituted adenosine compounds was
based on the work of Hutchison and was improved upon here by modifying the
methods of Foitzik121, 259 and Scammells.121 This work began from guanosine
as the starting material and progressed to selective N6-, C2- and C5-modified
adenosine derivatives. The single change of 5-ethylcarboxylate was
introduced instead of 5-methylcarboxylate in this scheme because of
convenience and because it produced a better yield and improved the quality of
the product synthesised. The synthetic methodology was published121, 259 and
is presented here in detail as it pertains to this project.
Adenosine intermediate for C2-functionalization with a linear 2.2.2.2
approach121
The planned series of adenosine target compounds was obtained successfully
via the 2-fluoro-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (146)
intermediate, which was synthesised from guanosine in seven steps of efficient
synthesis (Scheme 2.9).

76
Scheme 2.9: Overview of synthesis of the key adenosine intermediate (146).
Because all of the target compounds bear the 5–NECA modification (Figure
1.15 and 2.1), this was installed first. Then C6-modifications were achieved
with regioselective substitution by the installation of an active, leaving group at
the C6-position (144). The nucleophilic aromatic substitution (SNAr)-reactive
fluorine was achieved at C2 (145). The C6-leaving group was quickly replaced
by the amino group (146). The detailed reaction steps are discussed below.
The secondary 2,3-alcoholic groups of guanosine (138) are more nucleophilic
than the primary 5-alcoholic group, which is the required target for our initial
part of the synthesis. To avoid any side reactions from these secondary
alcoholic groups throughout the process, they were protected by forming a
cyclic acetal with an isopropylidene-protecting group, according to the
procedure originally outlined by Hampton288 (Scheme 2.10).

77
Scheme 2.10: Overview of 2, 3-alcohol protection (139)288
Reagents and conditions: (i) Acetone; 2, 2-
dimethoxypropane, 19 h. 75%.
Another benefit of the formation of protected guanosine is the increasing
solubility of the substrate in a variety of solvents other than DMSO. Solubility of
guanosine is one of the obstacles in the development of the efficient synthesis,
and guanosine is a highly polar species and is less soluble in solvents other
than DMSO.
The reaction was carried out using the acid catalyst para-toluene sulfonic acid
under anhydrous conditions with acetone as the solvent, however, acetal
formation was hindered by the presence of water with 2, 2-dimethoxypropane.
The workup involved using water and sodium bicarbonate to neutralise the
acidic environment and to precipitate the desired product 2,3-O-
isopropylideneguanosine (139) in a yield of 75%. Complete drying of product
139 was very difficult because of the aqueous work-up, so a quick methanol
wash was introduced after filtration of the product from the aqueous medium.
The use of para-toluenesulfonic (tosic) acid in 10-fold excess as the acid
catalyst aided the solubility of the substrate in this reaction because it formed
acetone-soluble salts with the purine nucleoside.288 The isopropylidene

78
protecting group is very labile under acidic conditions, and all further reactions
were carried out using neutral or basic conditions.
Because of the overall length of the synthetic sequence, the first reaction was
undertaken on a scale of 10 g of starting material. Yields were typically
excellent (75-90%). It remains a highly effective reaction in terms of its ease
and general applicability to protect nucleoside 2,3–hydroxyl groups, and it
continues to feature prominently in the recent literature.289,290,291,271, 292,293
The reaction gives the single reaction product (as identified on TLC) with a Rf of
the product of 0.72 in EtOAc/MeOH (7:3). In the 1H NMR (d6-DMSO) spectrum
of compound 139, two singlets appeared at 1.5 ppm and 1.3 ppm with an
integral corresponding to three protons each. In the 13C NMR spectrum, two
additional signals corresponding to the two methyl groups of the acetal moiety
appeared at 25.6 ppm and 27.5 ppm. The melting point of product 139 is 260–
262 °C and is consistant with literature value. The HRMS of the isolated
compound supported the synthesis of 139, which showed the expected
molecular ion of M+H at 324.1286, (EI+ HRMS showed a deviation of 0.0014
ppm from the calc. mass of C13H18N5O5).
The next step in the synthesis of 146 was the oxidation of the 5-hydroxy group
of 139. The traditional oxidising agents used to oxidise primary alcohols involve
harsh conditions and toxic reagents (e.g., Cr(VI) oxides; periodate/ruthenium;294

79
KMnO4; Pd/O2).295 The standard methods of oxidation of the 5-hydroxy group
gave poor yields and were carried out in strong acidic or alkaline conditions,
which resulted in degradation of the substrate.223 These methods can also
create a problem with regioselectivity for compounds with more than one
oxidisable group. The 5-hydroxy group of compound 139 was then oxidised to
the corresponding 2,3-O-isopropylideneguanosine-5-carboxylic acid (140).
The Epp and Widlanski296 oxidation implemented using the mild oxidising agent
system including, the free radical species TEMPO and a stoichiometric amount
of the organic oxidant bis(acetoxy)iodobenzene (BAIB) (Scheme 2.11).295
Scheme 2.11: Overview of the 5-hydroxyl oxidation (140). Reagents and conditions: (i) [Bis (acetoxy)
iodo] benzene (BAIB), TEMPO, MeCN, H2O, 18 h,79%.
This oxidation progressed under aqueous conditions (1:1 solution of
acetonitrile/water) with the two-step mechanism that led to the formation of the
aldehyde which was oxidised further to give the 5-carboxylic acid (Scheme
2.11). The aqueous conditions were used to allow the reaction to stir overnight
(Epp & Widlanski296 (3 h) and Middleton292 (4 h). In this process, the
hydroxylamine formed was reoxidised to TEMPO in the presence of BAIB,
which contains hypervalent (+3) iodine 279. The nitroxide radical is very stable
because of the absence of -protons on the ring, which can cause
disproportionate reactions and form a cyclic hydroxylamine and nitrone.297

80
Although it is not completely proven, the following mechanism was proposed by
de Mico (Scheme 2.12).293
Scheme 2.12: Mechanism of the primary hydroxyl oxidation.
The reaction has several additional benefits including the use of relatively non-
toxic reagents that can be used safely in the presence of a wide range of
functional groups and protecting groups such as acetal and ethers296 and
secondary alcohols.279 These reagents are highly selective for the oxidation of
primary alcohols.279 The by-products of oxidation were iodobenzene and acetic
acid, which could be easily removed via precipitation of the product from
acetone and diethyl ether.298,289 Often, in oxidation reactions, the solution is
homogeneous and opaque; however; in these reaction conditions, the solution
was heterogeneous and biphasic before the workup.

81
The workup, which involved adding some acetone and then adding the solution
to a large volume of Et2O, produced a precipitate. This precipitate was filtered
and gave reliable crude yields in the range of 70–80%. The Rf of compound
140 in 4:1 EtOAc/MeOH returned to 0 (as identified on TLC) because of the
increase in polarity around the 5-carbon from the introduction of the
electronegative oxygen to form the carbonyl group. The purification of the acid
was not successful using recrystallisation and column chromatography.
There are two main characteristics of the 1H NMR (d6-DMSO) spectrum of
crude compound 140. The signal at 3.54 ppm for the 5-methylene protons was
absent because of their removal by the successful oxidation of the 5-carbon.
Conversion to the carboxylic acid also removed the signal observed at 5.06
ppm for the remaining hydroxyl proton. In the 13C NMR spectrum, the signal for
the oxidised 5-carbon moved 112 ppm downfield from the corresponding signal
for 139 because of the powerful deshielding effect of the electron-withdrawing
carboxyl group and reappeared at 171.37 ppm. The melting point of the desired
acid 140 was 210–212 °C259 and is consistent with literature value. The
synthesis of compound 140 supported by HRMS, which showed the expected
molecular ion of M+H at 338.1134 m/z, (EI+ HRMS showed a deviation of
0.0033 ppm from the calc. mass of C13H16N5O6).
Despite evidence that the compound produced was desired target 140, purity of
the carboxylic acid was not satisfactory. Forming a novel intermediate ethyl
ester 142 at the 5–carboxyl group was a more reliable and higher-yielding route

82
to the desired 2,3-O-isopropylideneguanosine-5-N-ethylcarboxamide (143).299
Novel 5-ethylester 142 was synthesised under standard esterification
conditions (Scheme 2.13).
Scheme 2.13: Overview of 5–ethyl ester formation (142). Reagents and conditions: (iii) SOCl2, ethanol, 15
h, 64%.
Compound 140 was reacted at 0 °C in absolute ethanol with a fourfold excess
of thionyl chloride to give the reactive acid chloride in situ. The mixture was
stirred at room temperature overnight, which allowed the ethyl ester to form
through nucleophilic acyl substitution.300 The alcohol was used in large excess
(as solvent) optimising the yield of 2,3-isopropylideneguanosine-5-
ethylcarboxylate (142) (Scheme 2.13). The reaction solution was then basified
with an excess saturated NaHCO3 solution to quench any remaining thionyl
chloride and HCl. The acidity of the conditions did not appear to affect the
purine riboside linkage adversely.
The workup involved removal of ethanol by distillation after basification, and the
residue was stirred with the water to remove excess NaHCO3. The desired
product 142 was obtained in 65% yield. The Rf of the product on TLC in 7:3

83
EtOAc/MeOH increased to 0.71 because of the decrease in polarity around the
5-carbon from the introduction of the ethyl group to form the ester group.
In the 1H NMR (d6-DMSO) spectrum of 142, a multiplet appeared at 3.7–3.9
ppm with an integral corresponding to two protons and a triplet appeared at 0.9
ppm for three protons of the ethyl group of the new ester substituent. An extra
two peaks corresponding to the carbon of the ethyl group also appeared in the
13C NMR spectrum at 61.1 ppm and 13.8 ppm, respectively. The melting point
(272–274 °C) of the isolated compound 142 was consistant and sharp. The
high-resolution mass spectrum of the isolated compound supported the
synthesis of 142, which showed the expected molecular ion of M+H at 366.1449
m/z, (EI+ HRMS showed a deviation of 0.0035 ppm from the calc. mass of
C15H20N5O6).
Compound 142 was converted to the desired 2,3-O-isopropylideneguanosine-
5-N-ethylcarboxamide (NECA) (143) by using liquid ethylamine (Scheme 2.14).
Scheme 2.14: Overview of 5-amide formation (143). Reagents and conditions: (iv) Ethylamine, EtOH, -
20°C, 3 h. 88%.

84
Formation of the NECA 143 was achieved following a novel procedure by
combining the 5-ethyl ester 142 directly in an excess of dry ethylamine that was
used both as a reagent and as a solvent at a temperature of –20 °C for 3.0 h.
The reaction mixture was allowed to room temperature and stirred overnight at
room temperature. This overnight stirring was used for to react the unreacted
starting material and to remove the excess of ethylamine. The reaction mixture
was then diluted with absolute ethanol. The product mixture was then adsorbed
on silica and separated by column chromatography to obtain pure compound
143 in an 82 % yield.
The conditions were sufficient to enable nucleophilic acyl substitution of the
ethoxy group by the nucleophilic amine to form the amide in excellent yield.
The reaction was conducted no more than a 3 g scale. Formation of the amide
bond increased the polarity of the molecule, and the Rf value of compound 143
declined to 0.43 (9:1 DCM/MeOH).
The successful reaction to produce 143 was analysed by the 1H NMR (d6-
DMSO) spectroscopy. A multiplet appeared at 2.7–2.9 ppm with an integral
corresponding to the two protons and a triplet appeared at 0.6 ppm for three
protons of the ethyl group of the new amide substituent. An extra two peaks
corresponding to the carbons of the ethyl group also appeared in the 13C NMR
spectrum at 61.1 ppm and 13.8 ppm, respectively. The results of analysis using
HRMS and the melting point (278–280 °C) of the isolated compound were
consistent with the expected results for desired compound 143, which showed
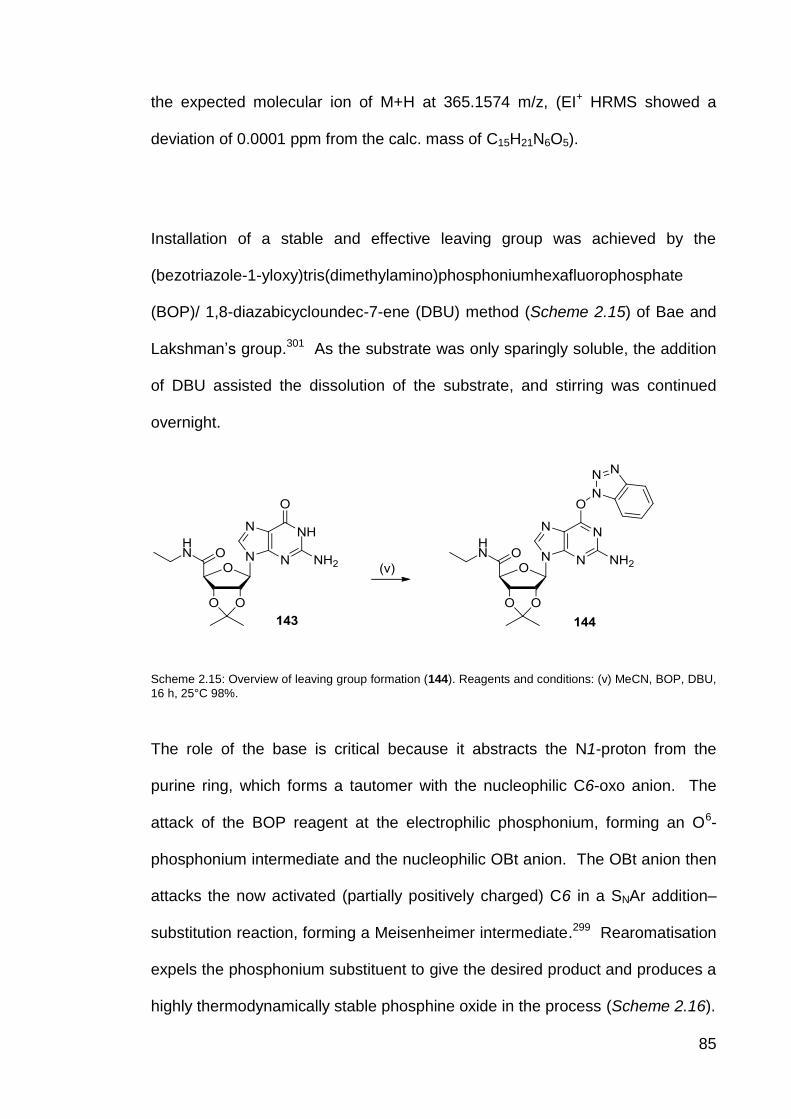
85
the expected molecular ion of M+H at 365.1574 m/z, (EI+ HRMS showed a
deviation of 0.0001 ppm from the calc. mass of C15H21N6O5).
Installation of a stable and effective leaving group was achieved by the
(bezotriazole-1-yloxy)tris(dimethylamino)phosphoniumhexafluorophosphate
(BOP)/ 1,8-diazabicycloundec-7-ene (DBU) method (Scheme 2.15) of Bae and
Lakshman’s group.301 As the substrate was only sparingly soluble, the addition
of DBU assisted the dissolution of the substrate, and stirring was continued
overnight.
Scheme 2.15: Overview of leaving group formation (144). Reagents and conditions: (v) MeCN, BOP, DBU,
16 h, 25°C 98%.
The role of the base is critical because it abstracts the N1-proton from the
purine ring, which forms a tautomer with the nucleophilic C6-oxo anion. The
attack of the BOP reagent at the electrophilic phosphonium, forming an O6-
phosphonium intermediate and the nucleophilic OBt anion. The OBt anion then
attacks the now activated (partially positively charged) C6 in a SNAr addition–
substitution reaction, forming a Meisenheimer intermediate.299 Rearomatisation
expels the phosphonium substituent to give the desired product and produces a
highly thermodynamically stable phosphine oxide in the process (Scheme 2.16).
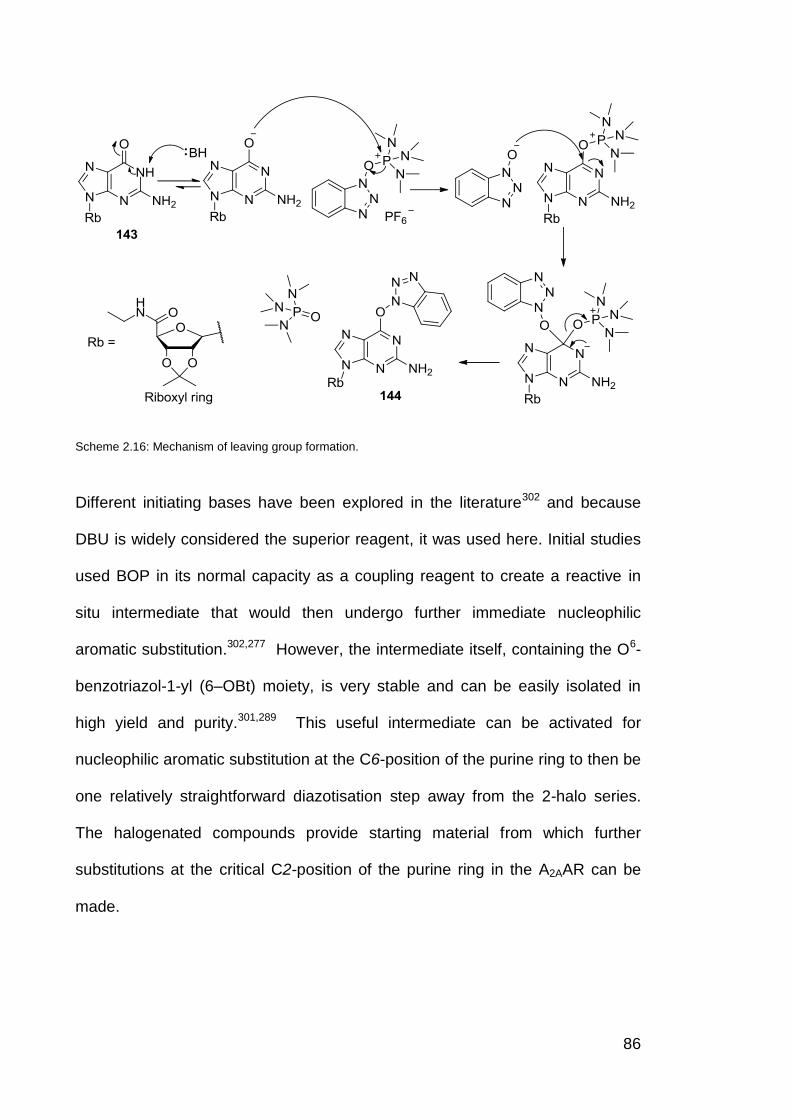
86
Scheme 2.16: Mechanism of leaving group formation.
Different initiating bases have been explored in the literature302 and because
DBU is widely considered the superior reagent, it was used here. Initial studies
used BOP in its normal capacity as a coupling reagent to create a reactive in
situ intermediate that would then undergo further immediate nucleophilic
aromatic substitution.302,277 However, the intermediate itself, containing the O6-
benzotriazol-1-yl (6–OBt) moiety, is very stable and can be easily isolated in
high yield and purity.301,289 This useful intermediate can be activated for
nucleophilic aromatic substitution at the C6-position of the purine ring to then be
one relatively straightforward diazotisation step away from the 2-halo series.
The halogenated compounds provide starting material from which further
substitutions at the critical C2-position of the purine ring in the A2AAR can be
made.

87
The introduction of the benzotriazole group decreased the polarity around the
C6-position of guanosine molecule to form the better leaving group. The Rf
value of the product O6-(benzotriazol-1-yl)-2,3-O-isopropylideneadenosine-5-
N-ethylcarboxamide (144) decreased to 0.38 in 8:2 DCM/MeOH. In the 1H
NMR spectrum (d6-DMSO) of 144, four additional peaks from the phenyl
protons of the heteroaromatic OBt substituent were located between 7.5 ppm
and 8.3 ppm, with a multiplet at 7.55 ppm with an integral corresponding to one
proton, a multiplet at 7.66 ppm with an integral corresponding to two protons,
and a doublet at 8.19 ppm with an integral corresponding to one proton. The
multiplet at 7.66 ppm corresponds to the two aromatic protons furthest from the
triazole heterocyclic and the doublet to the aromatic proton most proximal to the
nitrogen–oxygen bond. Six new peaks corresponding to the two quaternary and
four proton-bearing OBt phenyl carbons appeared in the 13C NMR spectrum
between 110 ppm and 130 ppm. The melting point of the isolated compound
144 was 140–142 °C. The HRMS supported the synthesis of 144, which
showed the expected molecular ion of M+H at 482.1898 m/z, (EI+ HRMS
showed a deviation of 0.0002 ppm from the calc. mass of C21H24N9O5).
Functional group interconversion of the C2-amine of compound 144 to the
nucleophilic aromatic substitution-promoting fluorine was accomplished using
the method of Kim et al.273 (Scheme 2.17). This method proceeded smoothly
with good yields (60–75%) of 2-fluoro-O6-(benzotriazol-1-yl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (145) obtained on scales
ranging from 50 mg to 2.0 g.

88
Scheme 2.17: Overview of fluoro substitution at the C2-position (145). Reagents and conditions: (vi) t-
BuONO, 70% HF in pyridine, 68%.
The reaction conditions employed were reasonably harsh with HF (70% in
pyridine) used as the fluorinating agent and pyridine as the solvent. This
reaction required the temperature to be kept between –50 °C and –30 °C.
Although distinguishable by TLC, the Rf value for the product 145 was 0.45 in
1:4 P.S./EtOAc (Rf of 0.37 for the C2-amino compound 144).This change in Rf
value indicates that the polarity of the molecule was decreased only slightly by
the introduction of the highly electronegative fluorine atom.
In the 1H NMR spectrum (d6-DMSO), the broad singlet at 6.68 ppm
corresponding to the heteroaromatic aniline at the C2-position in 144 was not
present in the product 145. In the 13C NMR spectrum of the product 145, the
peak for the C2-carbon of the purine ring was split by coupling with the fluorine
to produce a doublet at 157.1 ppm with a coupling constant of 221.8 Hz.
Further, less pronounced splitting occurred at C4 and C6 (J = 16.4 Hz). No
splitting was observed at sites more remote from C2. The melting point of the
isolated compound 145 was 145–147 °C. The synthesis of isolated compound

89
145 was also supported using HRMS, which showed the expected molecular
ion of M+H at 485.1699 m/z, (EI+ HRMS showed a deviation of 0.0007 ppm
from the calc. mass of C21H22FN8O5).
The labile benzotriazolyl leaving group present in compound 145 is readily
displaced by nucleophilic amines.299, 302 Ammonia solution was used to form a
6-amino product 2-fluoro-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (146) (Scheme 2.18). This SNAr reaction occurred smoothly
overnight in acetonitrile at room temperature.
Scheme 2.18: Overview of N6-amino substitution (146) Reagents and conditions: (vii) 28% ammonia sol,
MeCN, 2 h, 85%.
The Rf of the product 146 decreased as the benzotriazolyl group was lost, and
the polarity increased slightly. The Rf of compound 146 was determined to be
0.62 in ethyl acetate/MeOH (7:3). Purification via column chromatography gave
good yields of a white solid for 250 mg to 1 g scale reactions. A second
component was separated during the column chromatography was identified as
a small amount of amino-substituted product at the C2-position. The diamino
substitution was observed to be present in < 5% most typically when longer

90
reaction times led to some of the fluorine group being replaced by the amino
group.
In d6-DMSO, the major features of the 1H NMR spectrum of the fluoro
adenosine intermediate 146 are the absence of the signals from the
benzotriazole aromatic protons, and the appearance at 7.85 ppm of a broad
singlet with an integral corresponding to the two N6 protons. The signals from
the 2’- and 3’-protons completely coalesce from two resolved doublets in the
starting material to a singlet peak at 5.35 ppm with an integral corresponding to
two protons in the product. Likewise, in the 13C NMR spectrum, six peaks for
the benzotriazolyl aromatic carbons are no longer present. Two identifiable
signals, separated by 0.03 ppm were observed for the 2’- and 3’-carbons. The
melting point of the product 146 was 194–196 °C. The synthesis of compound
146 was supported by HRMS spectrum of 146, which showed the expected
molecular ion of M+ at 366.1451 m/z, (EI+ HRMS showed a deviation of 0.0049
ppm from the calc. mass of C15H19FN6O4).
2.2.3 Synthesis of TMIO adenosine analogues with aminoethylaniline
linker (161)(Approach 1)
Coupling of the antioxidant linker with functionalised adenosine was the next
step in the process towards target 161. In this work, the novel C2-substituted
target compound 2-(2-(4-N-(5-carboxy-1,1,3,3-tetramethylisoindoline-1-yloxyl)
aminophenyl)) ethylamino-2,3,-O-isopropylideneadenosine-5-N-
ethylcarboxamide (161) was synthesised through the reaction of the 2-fluoro
adenosine analogue (146) with an appropriate antioxidant-bearing
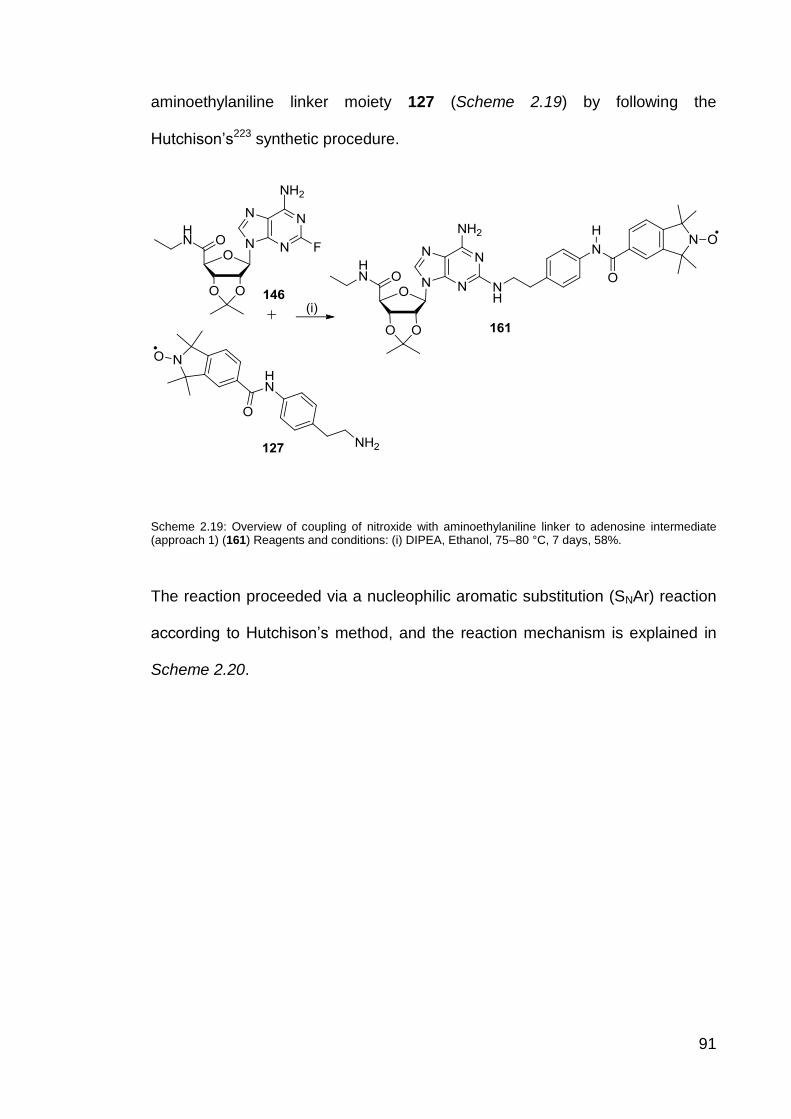
91
aminoethylaniline linker moiety 127 (Scheme 2.19) by following the
Hutchison’s223 synthetic procedure.
Scheme 2.19: Overview of coupling of nitroxide with aminoethylaniline linker to adenosine intermediate (approach 1) (161) Reagents and conditions: (i) DIPEA, Ethanol, 75–80 °C, 7 days, 58%.
The reaction proceeded via a nucleophilic aromatic substitution (SNAr) reaction
according to Hutchison’s method, and the reaction mechanism is explained in
Scheme 2.20.
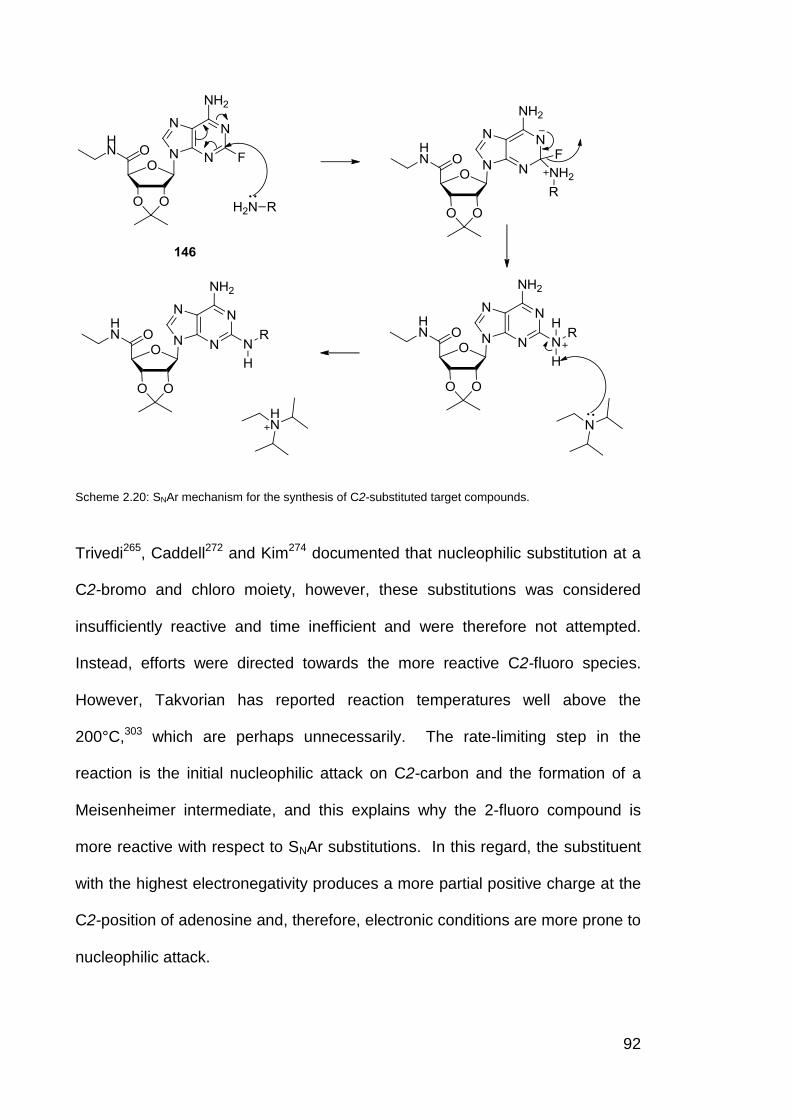
92
Scheme 2.20: SNAr mechanism for the synthesis of C2-substituted target compounds.
Trivedi265, Caddell272 and Kim274 documented that nucleophilic substitution at a
C2-bromo and chloro moiety, however, these substitutions was considered
insufficiently reactive and time inefficient and were therefore not attempted.
Instead, efforts were directed towards the more reactive C2-fluoro species.
However, Takvorian has reported reaction temperatures well above the
200°C,303 which are perhaps unnecessarily. The rate-limiting step in the
reaction is the initial nucleophilic attack on C2-carbon and the formation of a
Meisenheimer intermediate, and this explains why the 2-fluoro compound is
more reactive with respect to SNAr substitutions. In this regard, the substituent
with the highest electronegativity produces a more partial positive charge at the
C2-position of adenosine and, therefore, electronic conditions are more prone to
nucleophilic attack.

93
The synthesis of product 161 was achieved in ethanol using Hunig’s base (N,
N-diisopropylethylamine)(DIPEA) by stirring at 75–80 °C for 7 days. The Rf
value of the product on TLC in EtOAc/MeOH (9:1) was 0.51 and the compound
was purified by column chromatography using this solvent mixture to generate
product 161 in 58% yield.
The structure of the nitroxide-coupled adenosine product 161 was supported by
the HRMS, which showed the expected molecular ion of M+H at 699.3506 m/z,
(EI+ HRMS showed a deviation of 0.0017 ppm from the calc. mass of
C36H45N9O6). In the 1H NMR spectrum, paramagnetic broadening was
observed as expected due to the nitroxide radical that resulted in some peaks
not being detected and giving poor integration. However, the signal of the
ethylene group from aniline linker could be detected at 2.91 and 2.98 ppm and
the adenosine ribose ethylamide proton signals could be detected at 0.71 ppm
and acetal two methyl protons appeared at 1.24 and 1.42 ppm. The melting
point of the isolated compound 161 was 66–68°C.
In this approach, the reaction conditions used were very harsh, and the product
was only generated in low yields. This low yield was attributed to
decomposition of the starting reactants or product formed. In addition, this
approach required extended reaction times (7 days). Accordingly, an
alternative approach was explored. This approach first involved the attachment
of the aminoethylaniline linker to the adenosine intermediate, followed by
subsequent coupling with the carboxy nitroxide. As the amine linkers could be

94
sourced in large scale, greater efficiency for the synthesis was thought to be
possible through coupling the adenosine intermediate 146 with excess amine
124 and then coupling this system to the nitroxide.
2.2.4 Synthesis of TMIO adenosine analogues with aminoethylaniline
linker (161) (Approach 2)
As outlined in previous section, the reaction of 2-fluoro adenosine precursor 146
with an excess of amine 124 was undertaken in order to give the maximum
possible levels of the preliminary target 2-(2-(4-aminophenyl)) ethylamino-2,3-
O-isopropylideneadenosine-5-N-ethylcarboxamide (159).
In this approach, the para-(aminophenyl)ethylamine-linked adenosine
intermediate 159 was synthesised first and then the appropriate carboxy-
nitroxide was coupled directly to the adenosine intermediate 159. The
advantage of this approach is that various carboxy-nitroxides such as 5-
carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl (CTMIO, 106), 4-carboxy-
2,2,6,6-tetramethylpieridine-1-yloxyl (CTEMPO, 116), 3-carboxy- 2,2,5,5-
tetramethylpyrrolidine-1-yloxyl (CPROXYL, 117), 5-methylcarboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl (MCTMIO, 115) and 5-carboxy-1,1,3,3-
tetraethylisoindoline-2-yloxyl (CTEIO, 114) could also be coupled to the para-
aminoethylaniline linked adenosine intermediate 159 using very mild amide
coupling conditions (Scheme 2.21).

95
Scheme 2.21: Overview of coupling of carboxy nitroxides with aminoethylaniline linked adenosine intermediate (Approach 2)
Firstly, the adenosine intermediate 159 was synthesised from the reaction of
compound 146 with amine 124 by using Hunig’s base DIPEA in 97% yields
(Scheme 2.22). The reaction was proceeded by an SNAr reaction mechanism,
which is explained in Scheme 2.20.
Scheme 2.22: Overview of coupling of aminoethylaniline linker to adenosine intermediate (159), Reagents
and conditions: (i) DIPEA, MeCN, MW 180 °C;7 h.

96
The adenosine intermediate 159 was synthesised by using the conventional
method of heating in oil bath. This synthetic method takes 7 days to complete
the reaction and only gives moderate yields (50%). This methodology is time-
consuming and failed to achieve desired yield of product 159. This low yield
(50%) was attributed to the longer heating time which may decompose the
starting reagents, or the product formed. Accordingly, an alternative approach
was implemented which successfully prepared the desired C2-
aminoethylaniline adenosine analogue 159 through a substitution reaction using
microwave assisted conditions in the reduced time span of 7 h with an excellent
yield of 97%. This approach provided a convenient synthetic route to the
synthesis of the desired adenosine analogues.
The reaction was carried out successfully either in ethanol or acetonitrile with oil
bath heating (at 75–80 °C). For complete conversion of the starting material,
the reaction still took 7 days to complete. This longer reaction time, is avoided
by undertaken using microwave irradiation in which compound 146, compound
124 and DIPEA were combined in acetonitrile, sealed in a microwave tube and
heated at 180 °C. Multiple experiments at various reaction times were
conducted under microwave conditions and after 7 h, the complete conversion
of the starting material was observed. After which time, the reaction mixture
was purified using column chromatography.
The structure of 159 was supported by 1H NMR and 13C NMR spectroscopy and
MS spectrometry. In the 1H NMR spectrum, a new peak corresponding to the

97
ethylene protons of the linker appeared in the aliphatic region as two multiplets
at 2.82 and 2.86 ppm integrated for four protons. Two additional doublets
corresponding to the aromatic ring of the linker appeared at 6.67 and 7.06 ppm
which integrated for four protons. In the 13C NMR spectrum, three additional
signals appeared as strong peaks corresponding to the two methyl carbons at
33.7 and 34.3 ppm. The additional aromatic ring signals appeared at 79.1 ppm.
The C2-substituted linked adenosine compound 159 was supported by HRMS,
which showed the expected molecular ion of M+H at 483.2482 m/z, (EI+ HRMS
showed a deviation of 0.022 ppm from the calc. mass of C23H31N8O4). The
melting point of 150-152 °C of isolated product 159 is consistent. The obtained
data were in agreement with the published values.121
Compound 159 was then coupled with nitroxide compound (CTMIO) 106 using
EDCI/HOBt coupling reagent and Hunig’s base in dry DMF to give 2-(2-(4-N-(5-
carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-2,3-
O-isopropylideneadenosine-5-N-ethylcarboxamide (161) (Scheme 2.23). HOBt
helps in the formation of active esters in the reaction mixture by reacting with
the carboxylic acid at ambient temperatures.
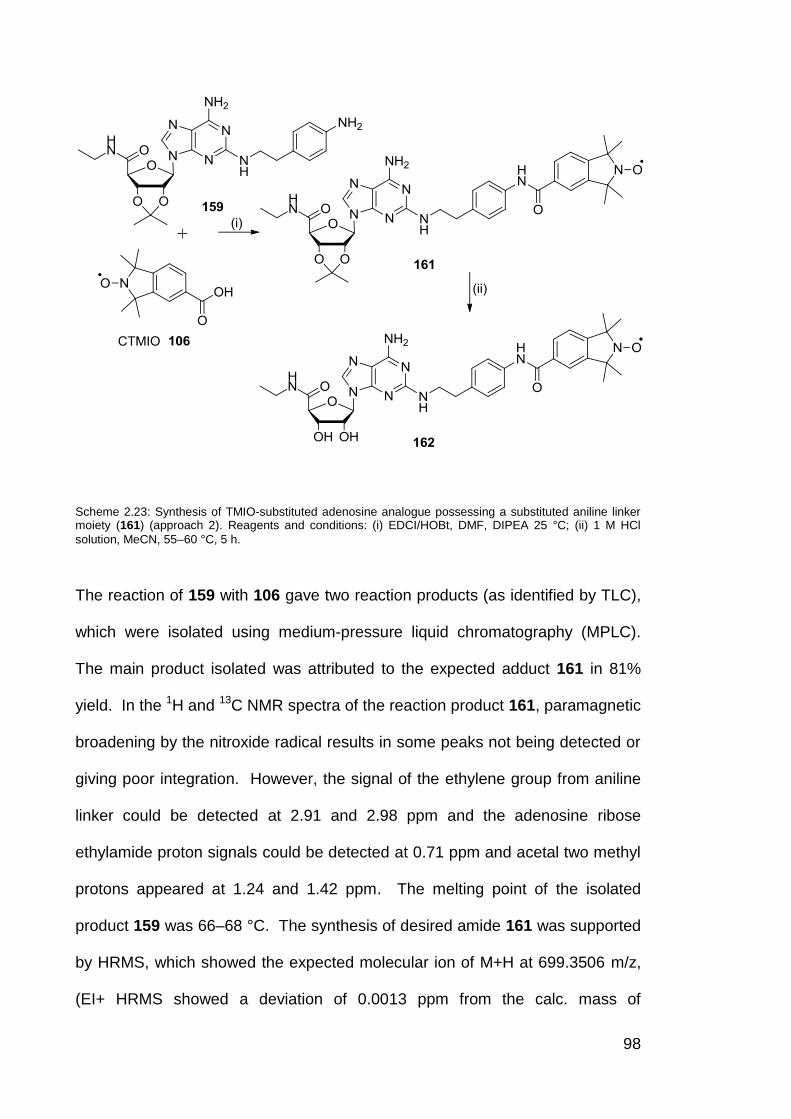
98
Scheme 2.23: Synthesis of TMIO-substituted adenosine analogue possessing a substituted aniline linker moiety (161) (approach 2). Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C; (ii) 1 M HCl
solution, MeCN, 55–60 °C, 5 h.
The reaction of 159 with 106 gave two reaction products (as identified by TLC),
which were isolated using medium-pressure liquid chromatography (MPLC).
The main product isolated was attributed to the expected adduct 161 in 81%
yield. In the 1H and 13C NMR spectra of the reaction product 161, paramagnetic
broadening by the nitroxide radical results in some peaks not being detected or
giving poor integration. However, the signal of the ethylene group from aniline
linker could be detected at 2.91 and 2.98 ppm and the adenosine ribose
ethylamide proton signals could be detected at 0.71 ppm and acetal two methyl
protons appeared at 1.24 and 1.42 ppm. The melting point of the isolated
product 159 was 66–68 °C. The synthesis of desired amide 161 was supported
by HRMS, which showed the expected molecular ion of M+H at 699.3506 m/z,
(EI+ HRMS showed a deviation of 0.0013 ppm from the calc. mass of

99
C36H45N9O6). The purity was found to be 97.6% by analytical HPLC in 80%
methanol/20% water system.
Deprotection of the isopropylidene group from compound 161 was achieved
using 1 M HCl at at 55–60 °C in 4.0 h to give 2-(2-(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl) aminophenyl)) ethylaminoadenosine-5-N-
ethylcarboxamide (162) (Scheme 2.23).
The deprotection reaction gave a single reaction product 162 (as identified on
TLC), which was isolated using MPLC and partial crystallisation in a mixture of
methanol/ water solvent system with a yield of 70%. The structure of
deprotected product 162 was supported by HRMS, which showed the expected
molecular ion of M+H at 659.3197 m/z, (EI+ HRMS showed a deviation of
0.0010 ppm from the calc. mass of C33H41N9O6). A melting point of 241–243 °C
(decomp) was obtained for the product 162. The purity was found to be 99.9%
using analytical HPLC in an 80% methanol/20% water system. In the proton
NMR spectrum, paramagnetic broadening was observed which resulted in
some peaks not being detected and giving poor integration of compound 162.
However, signals of the aniline linker and adenosine moieties could be
detected.
The coupling reaction also gave a small amount of unknown impurity at the
nonpolar region (as identified on TLC) in each reaction. To confirm the
structure of the by-product, which was suspected to be the dimer, several

100
experiments were undertaken. Firstly, the compound 159 was coupled with 3,5-
dinitrobenzoic acid (175) using EDCI/HOBT-coupling reagents (Scheme 2.24).
The same reaction conditions gave the product 176 with a similar impurity to
those of the coupling reactions with radical species (a identified by TLC). The
coupled compound was not isolated further.
Scheme 2.24: Synthesis of 3,5-dinitrobenzoic acid-coupled adenosine analogue possessing a substituted aniline linker moiety (176) Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C.
We next performed another experiment by assuming that the impurity may have
formed because of a side reaction or decomposition of the starting material 159
(Scheme 2.25). This assumption is confirmed by stirring the aniline linked
adenosine compound 159 under the same reaction conditions and without any
carboxy acid starting material. However, no change was observed on TLC,
after three days of continued stirring at room temperature and starting
compound 159 was recovered.

101
Scheme 2.25: Reaction of coupling reagents with adenosine analogue possessing a substituted aniline linker, Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C.
From this experiment, it was confirmed that the unknown compound impurity
was arising from the other starting material the carboxy nitroxide. After analysis,
it was found that the impurity interferring in this reaction was the dimer
compound (anhydried) formed from two CTMIO molecules.
2.3 Synthesis of C2-substituted CTEMPO adenosine analogues possessing substituent attached to ‘aminoethylaniline linker’ moiety (164)
Compound 159 was coupled with commercially available 4-carboxy- 2,2,6,6 -
tetramethylpiperidine-1-yloxyl (CTEMPO) (116) using the EDCI/HOBT amide
coupling reagent to give the novel compound 2-(-2-(4-N-(4-carboxy-2,2,6,6
tetramethylpieridine-2-yloxyl)aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (163) in 60% yield (Scheme
2.26).

102
Scheme 2.26: Synthesis of TEMPO-substituted adenosine analogue possessing a substituted aniline linker moiety (164), Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C; (ii) 1 M HCl solution,
MeCN, 55–60 °C, 5 h.
The reaction was carried out in freshly distilled DMF under an argon
atmosphere at room temperature. The reaction went to completion in 3 days
(as identified by TLC).
The reaction of 116 with 159 produced two products (as identified by TLC)
which were isolated and purified twice by MPLC, as the two components ran
very close to each other. In the 1H spectrum of the reaction product 163,
paramagnetic broadening by the nitroxide radical resulted in some peaks not
being detected or giving poor integration. However, the signal of the aniline
linker and adenosine moieties could be detected. The results of HRMS of the
isolated compound were consistent and supported the structure of 163, which
showed the expected molecular ion of M+H at 665.3658 m/z, (EI+ HRMS
showed a deviation of 0.0018 ppm from the calc. mass of C33H47N9O6). A purity

103
of 99.4% was found using analytical HPLC in an 80% methanol/20% water
system. The melting point of compound 163 was measured as 148–150 °C.
The second spot isolated was found to be the mass corresponding to the
anhydride of the CTEMPO (116), which was found according to the mass
analysis.
Deprotection of the isopropylidene group from compound 163 was achieved
using similar procedure as used for synthesis of compound 162 by employing 1
M HCl at elevated temperatures to give the novel target compound 2-(2-(4-N-(4-
carboxy-2,2,6,6-tetramethylpieridine-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide (164)
(Scheme 2.26). The reaction went to completion in 5.0 h at 55–60 °C.
The reaction gave a single reaction product (as identified on TLC) with an Rf
value of 0.43 in (8:2) IPA: MeOH, and which was purified using MPLC and
partial crystallisation using a 75% methanol/25% water system to give 164 in
70% yield. The melting point of 211-213 °C (decomp) was obtained for the
isolated product 164. The structure of 164 was supported by HRMS, which
showed the expected molecular ion of [M+H]: 625.3341 m/z (EI+ HRMS showed
a deviation of 0.0011 from the calc. mass of C30H43N9O6). In the 1H spectrum of
the reaction product 164, paramagnetic broadening by the nitroxide radical
resulted in some peaks not being detected or giving poor integration, however
the signal of the aniline linker and adenosine moieties could be detected.
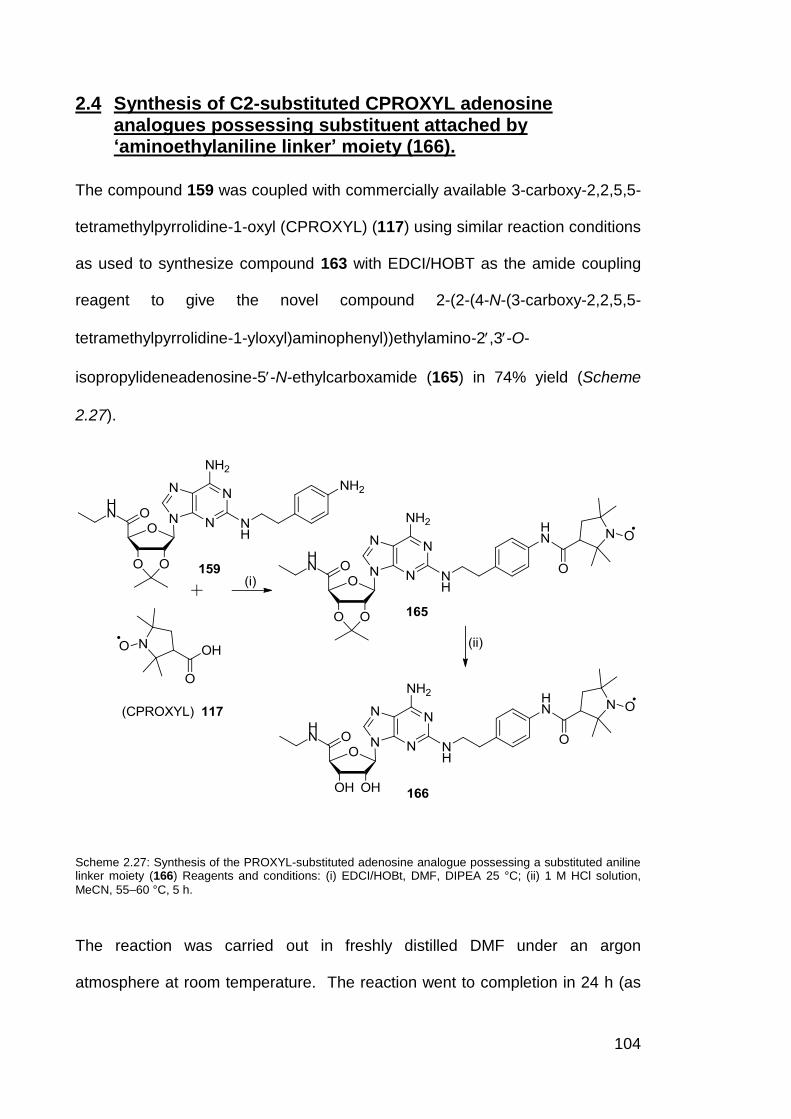
104
2.4 Synthesis of C2-substituted CPROXYL adenosine analogues possessing substituent attached by ‘aminoethylaniline linker’ moiety (166).
The compound 159 was coupled with commercially available 3-carboxy-2,2,5,5-
tetramethylpyrrolidine-1-oxyl (CPROXYL) (117) using similar reaction conditions
as used to synthesize compound 163 with EDCI/HOBT as the amide coupling
reagent to give the novel compound 2-(2-(4-N-(3-carboxy-2,2,5,5-
tetramethylpyrrolidine-1-yloxyl)aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (165) in 74% yield (Scheme
2.27).
Scheme 2.27: Synthesis of the PROXYL-substituted adenosine analogue possessing a substituted aniline linker moiety (166) Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C; (ii) 1 M HCl solution,
MeCN, 55–60 °C, 5 h.
The reaction was carried out in freshly distilled DMF under an argon
atmosphere at room temperature. The reaction went to completion in 24 h (as

105
identified by TLC). The reaction of 117 with 159 gives two products (as
identified by TLC), which were subsequently isolated by MPLC in a 75%
methanol/25% water system. The results of HRMS of the main component
isolated were consistent and supported structure of 165, which showed the
expected molecular ion of M+H at 651.3493 m/z, (EI+ HRMS showed a
deviation of 0.0003 ppm from the calc. mass of C32H45N9O6). The melting point
of the isolated product 165 was found to be 140–142 °C. The purity was found
to be 98% by analytical HPLC in a 75% methanol/25% water system. The
second component isolated from the reaction mixture was found to be the
unreacted substrate compound 159 by MS analysis.
The novel compound 2-(2-(4-N-(3-carboxy-2,2,5,5-tetramethylpyrrolidine-1-
yloxyl) aminophenyl)) ethylaminoadenosine-5-N-ethylcarboxamide (166) was
synthesised by deprotection of the isopropylidene group from compound 165
using 1 M HCl at 50–60 °C for 5.0 h in 62.5% yield. (Scheme 2.27).
The reaction gave a single reaction product 166 (as identified on TLC), which
was isolated using MPLC and partial crystallisation using 75% methanol/25%
water system. The results of HRMS of the isolated compound were consistent
with the expected results for 166, which showed the expected molecular ion of
M+H at 611.3183 m/z, (EI+ HRMS showed a deviation of 0.0001 ppm from the
calc. mass of C29H41N9O6). In the 1H spectrum of the compound 166,
paramagnetic broadening by the nitroxide radical resulted in some peaks not
being detected or giving poor integration of compound 166, however signals of

106
the aniline linker and adenosine moieties could be detected. A melting point of
176–178 °C (decomp) was measured for the isolated product 166. The purity
was found to be >99.9% using analytical HPLC in a 75% methanol/25% water
system.
2.5 Synthesis of C2-substituted MCTMIO adenosine analogues possessing substituent attached to “aminoethylaniline linker” moiety (168)
A novel adenosine compound was synthesised using a mixture of the one
carbon extended methylene carboxy isoindoline nitroxide 115, and compound
106 which was synthesised from compound 102 and obtained from advanced
stage synthesis in the laboratory. The compound 159 was coupled with a
mixture of nitroxide 5-methylenecarboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl
(MCTMIO, 115) and 106 using EDCI/HOBT as the amide coupling reagent to
give a mixture of 2-(2-(4-N-(5-methylenecarboxy-1,1,3,3-tetramethylisoindoline-
2-yloxyl)aminophenyl))ethylamino-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (167) and compound 161 (Scheme 2.28).

107
Scheme 2.28: Synthesis of MCTEIO-substituted adenosine analogue possessing a substituted aniline linker moiety (168). Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C; (ii) 1 M HCl solution,
MeCN, 55–60 °C, 5 h.
The reaction was carried out in freshly distilled DMF under an argon
atmosphere at room temperature. Addition of the mixture of nitroxides after the
coupling reagents, improves the reaction completion time from 48 h to 24 h and
product 167 was obtained in a high yield. Moreover, complete consumption of
the starting material was observed. The addition of EDCI and HOBt first to
DMF under an argon atmosphere followed by the addition of carboxy nitroxide
115 and DIPEA, and later the portion-wise addition of the linker amine 159 led
to completion of the reaction in 24 h instead of 3 days at room temperature (as
identified by TLC). The work-up involved filtration of the coupled solid product
formed at 0 °C after the 30 min reaction, quenching and washing many times

108
with chilled water. As per TLC analysis, the similar Rf values were observed for
the mixture of compounds 167 and 161.
The synthesis of the one carbon extended isoindoline nitroxide derivative of
adenosine compound 167 was attempted by following a similar amide coupling
procedure used for the synthesis of compound 165. The reaction of mixture of
115 and 106 with 159 produced three components (as identified by MPLC),
however, a TLC analysis shows only two components. These components was
firstly purified by using MPLC. The first fraction was found to be unreacted
starting amine 159, which was isolated from the product. The second fraction
isolated from the 300mg reaction by MPLC was 150 mg of a pale yellow
component. The second component on TLC, what appeared to be a single new
product has two close components (as identified by HPLC). However, it was
found difficult to isolate these two compounds present in the sample by MPLC
and therefore, this mixture was used as it is for the next step of deprotection
reaction.
The mixture was analysed using analytical HPLC in a 75% methanol/25% water
system and was found to be a 60:40 mixture of compound 167 with compound
161 respectively. The results of HRMS of the isolated product were consistent
with the expected results for 167 and 161, which showed the expected
molecular ions of M+H at 713.5496 and 699.9465 m/z respectively. The NMR,
MS and HPLC analysis revealed that two structures-related isoindoline
compounds were present and determined that a component had a molecular

109
mass corresponding to the desired MCTMIO adenosine derivative 167 with the
CTMIO adenosine derivative 161 in a 60:40 mixture.
A mixture of compound 167 and 161 was used as starting material for the
deprotection reaction. Deprotection of the isopropylidene group from a mixture
of compound 167 and compound 161 was achieved using 1 M HCl at elevated
temperatures (Scheme 2.28). The reaction went to completion in 5.0 h at 50–
55 °C and produced a mixture of the final target compound 2-(2-(4-N-(5-
methylcarboxy-1,1,3,3-tetramethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide (168) and
162 as an off-white solid compound. (Note: compound 162 is present because
the starting material contained two components). The product was filtered after
quenching of the reaction using saturated sodium bicarbonate solution at 0 °C
and was washed with plenty of chilled water.
The reaction gave a mixture of compounds 168 with 162 (single spot as
identified on TLC) that was not isolated using MPLC but partially crystallised in
75% methanol/25% water system. The results of HRMS of the isolated
compound were consistent with the expected results for 168 and 162, which
showed the expected molecular ion of M+2H at 674.6586, and 661.0205 m/z.
The isolated off-white solid was found to be a 60:40 mixture of compound 168
with 162 using analytical HPLC in a 75% methanol/25% water mobile-phase
system. This compound was analysed by 1H NMR spectroscopy, which
indicated clearly the absence of the two methyl signals of isopropylidene
protection.

110
2.6 Synthesis of C2-substituted adenosine analogue possessing TEIO nitroxide substituent attached to the ‘aminoethylaniline linker’ moiety (170).
2.6.1 Synthesis of carboxy TEIO nitroxide antioxidant
The stable nitroxide 5-carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl (CTEIO)
114 was synthesised in six steps as per the procedure first published by our
group 102, 275 (Scheme 2.29).
Scheme 2.29: Synthesis of 5-Carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl (CTEIO) 114.102
Reagents and
conditions: (i) Glacial CH3COOH, reflux 75 min, 80%; (ii) Mg, C2H5I, toluene, reflux 3.0 h, 32%; (iii) 60 psi H2, 10% Pd/C catalyst, glacial CH3COOH, 3 h, 92%; (iv) Na2WO4. 2H2O, H2O2, NaHCO3, stirred 72 h, 47%; (v) AcCl, Pd/C,Et3N, 99%; (vi) KMnO4 , MgSO4, 78%; (vii) LiOH, 89%.
Commercially available 5-methyl phthalic anhydride (107) and benzylamine
were reacted in a nucleophilic acyl substitution to generate 5-methyl-N-benzyl
phthalimide (108) in 80% yield. Compound 108 was tetraethylated in a
Grignard reaction using ethylmagnesium iodide to form 5-methyl-N-benzyl-
1,1,3,3-tetraethylisoindoline (109) in 32% yield. The 2-benzyl group of 109 was

111
cleaved through hydrogenation of the protected isoindoline in a Parr-apparatus
using 50 psi H2 gas pressure to generate 5-methyl-1,1,3,3-tetraethylisoindoline
amine (110) in 92% yield. The debenzylated amine compound 110 was then
oxidized304 with 30% H2O2 in the presence of a Na2WO4.2H2O catalyst to form
the desired nitroxide 5-methyl-1, 1, 3, 3-tetraethylisoindoline-2-yloxyl (111) in
47% yield. Compound 111 was subsequently reduced with hydrogen using a
Pd/C catalyst and then acylated305 with acetyl chloride to form 2-acetoxy-5-
methyl-1,1,3,3-tetraethylisoindoline (112) in 99% yield.102 The methyl group of
compound 112 was then oxidized using 0.4 M solution of potassium
permanganate in the presence of magnesium sulfate306 at 70 °C to generate the
desired carboxylic acid 2-acetoxy-5-carboxy-1,1,3,3-tetraethylisoindoline (113)
in 78% yield.102 Compound 113 was finally hydrolysed by lithium hydroxide and
reoxidised by lead oxide to generate the desired nitroxide 5-carboxy-1,1,3,3-
tetraethylisoindoline-2-yloxyl (114) in 89% yield (Scheme 2.29). The
characterization of all compounds in this scheme is in agreement with the
published data.102
2.6.2 Coupling carboxy TEIO adenosine compound with
aminoethylaniline linker (170)
The compound 159 was coupled with a 5-carboxy-1,1,3,3-tetraethylisoindoline-
2-yloxyl (CTEIO) (114) using the EDCI/HOBT as the amide coupling reagent to
give the novel compound 2-(2-(4-N-(5-carboxy-1,1,3,3-tetraethylisoindoline-2-
yloxyl)aminophenyl))ethylamino-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (169) in 70% yield. (Scheme 2.30)
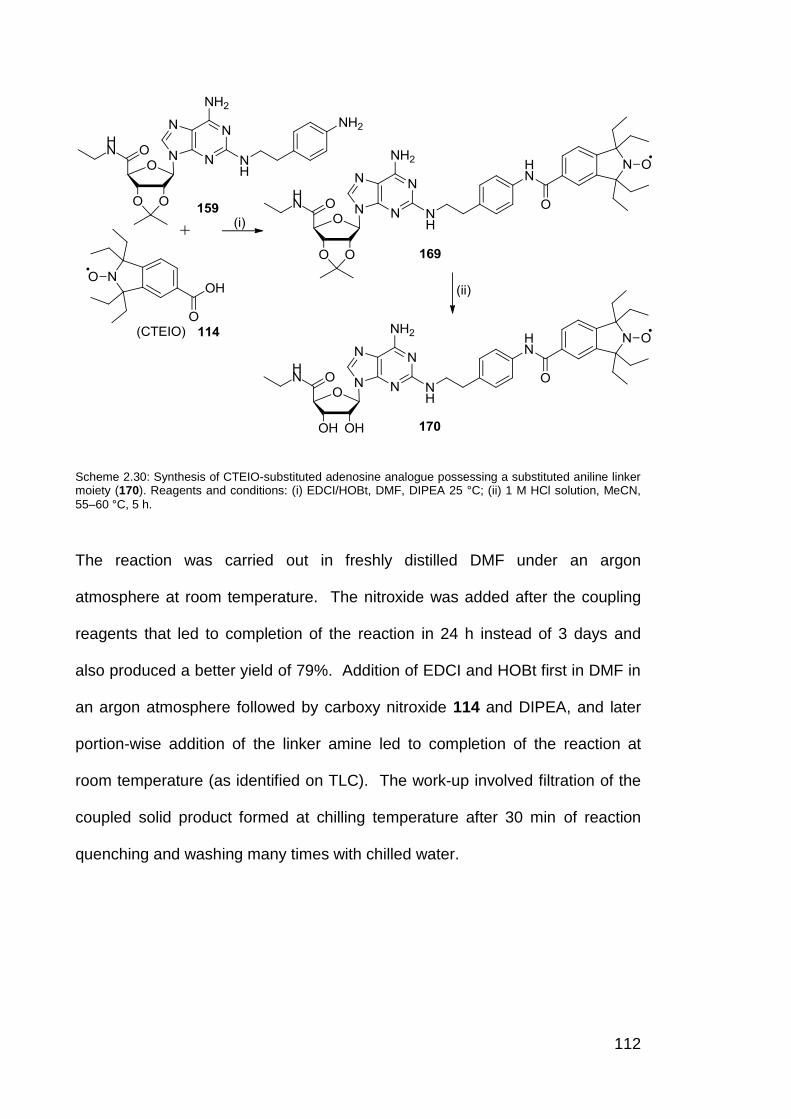
112
Scheme 2.30: Synthesis of CTEIO-substituted adenosine analogue possessing a substituted aniline linker moiety (170). Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C; (ii) 1 M HCl solution, MeCN,
55–60 °C, 5 h.
The reaction was carried out in freshly distilled DMF under an argon
atmosphere at room temperature. The nitroxide was added after the coupling
reagents that led to completion of the reaction in 24 h instead of 3 days and
also produced a better yield of 79%. Addition of EDCI and HOBt first in DMF in
an argon atmosphere followed by carboxy nitroxide 114 and DIPEA, and later
portion-wise addition of the linker amine led to completion of the reaction at
room temperature (as identified on TLC). The work-up involved filtration of the
coupled solid product formed at chilling temperature after 30 min of reaction
quenching and washing many times with chilled water.

113
The reaction of 114 with 159 produced two products (as identified by TLC),
which were isolated by MPLC. HRMS of one isolated product was consistent
with the expected results for 169, which showed the expected molecular ion of
M+H at 755.4118 m/z, (EI+ HRMS showed a deviation of 0.0001 ppm from the
calc. mass of C40H53N9O6). The purity was found to be 95% by HPLC in a 75%
methanol/25% water system. The second component isolated was found to be
starting material. In the1H NMR spectroscopic analysis of compound 169,
signals of the aniline linker and the adenosine moieties could be detected.
Deprotection of the isopropylidene group from compound 169 was achieved
using 1 M HCl at elevated temperatures. The reaction went to completion in 5.0
h at 50–55 °C, to give the novel target compound 2-(2-(4-N-(5-carboxy-1,1,3,3-
tetraethylisoindoline-2-yloxyl) aminophenyl)) ethylaminoadenosine-5-N-
ethylcarboxamide (170) as a pale yellow solid compound (Scheme 2.30). The
product was filtered after quenching of the reaction using saturated sodium
bicarbonate solution at 0 °C and washed with plenty of chilled water.
The reaction gave a single reaction product 170 (as identified on TLC), which
was isolated using MPLC and partial crystallisation using 75% methanol/25%
water system, to give product in 71% yields. The results of the HRMS of the
isolated compound were consistent with the expected results for 170, which
showed the expected molecular ion of M+Na at 737.3506 m/z, (EI+ HRMS
showed a deviation of 0.0021 ppm from the calc. mass of C37H48NaN9O6). The
purity of product 170 was found to be 98.5% using analytical HPLC in a 75%

114
methanol/25% water mobile-phase system. This compound was analysed by
1H NMR spectroscopy, which indicated paramagnetic broadening by the
nitroxide radical resulting in some peaks not being detected or giving poor
integration, however the signals of the aniline linker and adenosine moieties
and the disappearance of the two acetal methyl signals at 1.17 and 1.19 ppm
could be detected.
2.7 Synthesis of C2-substituted di-tert-butylhydroxycinnamic acid adenosine analogues possessing substituent attached to ‘aminoethylaniline linker’ moiety (172).
Phenolic compounds also exhibit antioxidant properties similar to those of
nitroxides. In particular, hindered phenolic compounds, including BHT with
adenosine molecule demonstrated excellent results in the biological analysis.
Similarly, the phenolic compound hydroxycinnamic acid is also a potent
antioxidant. Additionally, this may deliver an excellent antioxidant activity and
A2AAR selectivity results when attached to the adenosine molecule.
It was speculated that the nitroxide compounds were degrading through out the
reaction conditions and having low yield using first approach synthesis,
therefore to confirm this behavoiur the novel hydroxy cinnamic acid based
adenosine compound 171 was synthesised using two different synthetic
approaches in a manner similar to synthesise compound 161. Firstly coupling
of the antioxidant moiety 118 with the linker amine is performed to form
compound 133 and then 133 is coupled to the adenosine intermediate 146.

115
2.7.1 Synthesis of di-tert-butylhydroxycinnamic acid possessing
substituent attached by ‘aminoethylaniline linker’ intermediate (133)
The novel compound N-tert-butoxycarbonyl-2(4-N-(3,5-ditert-butyl-4-
hydroxycinnamic acid)aminophenyl)ethylamine (132) was synthesized from
compound 125 by amide coupling with 3,5-ditert-butyl-4-hydroxycinnamic acid
(118) using EDCI in the presence of a catalytic amount of HOBt and DIPEA
along with using DMF as the basic solvent in 99% yield (Scheme 2.31).
Scheme 2.31: Overview of synthesis of di-tert-butyl-4-Hydroxycinnamic acid coupled aminophenylethylamine linker moiety (133). Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA 25 °C,
99%; (ii) 1 M HCl solution, MeCN, 55–60 °C, 5 h., 65% .
Compound 132 was synthesized by following the same amide coupling
procedure as that used to generate the nitroxide linker compounds. EDCI was
used as an acid activating agent, forming a reactive ester intermediate in situ
with the carboxylic acid of the hydroxy cinnamic acid antioxidant moiety. This
carboxylic acid ester has an activated leaving group that is attacked by the

116
nucleophilic amine group from the linker aniline. A thermodynamically stable
urea is released to give the novel desired amide product 132 with the amide
coupling mechanism explained in Scheme 2.7. HOBt was used to drive the
reaction in the desired direction, which limits side reactions and improves yields.
The reaction was run overnight at room temperature. A workup procedure
involving pouring the reaction solution into excess water and extracting the
resulting precipitate with DCM and washing several times with water afforded
high yields. The compound 132 was purified by column chromatography using
a mixture of CHCl3: MeOH: 28% aqueous ammonia solution (7.9:2:0.1), to give
product 132 in 99% yields on a 50–100 mg scale. Compound 132 appeared on
a TLC plate as a single spot. In the 1H NMR (CDCl3) spectrum of 132, a
multiplet appeared at 1.4 ppm with an integral corresponding to the eighteen
protons from the two tert-butyl groups and two triplet appeared at 2.73 and 3.36
ppm for the four protons of the ethyl linker group from the new aminoethylaniline
substituent. An extra two peaks corresponding to the carbons of the ethene
group also appeared in the 13C NMR spectrum at 117.3 ppm and 136.3 ppm,
respectively. The melting point of 126–128 °C of the isolated compound 132
was consistent and sharp. The structure of 132 was supported by MS analysis,
which showed the expected molecular ion of M+Na at 517.3006 m/z (EI+
(HRMS) showed a deviation of 0.0034 from the calc. mass of C30H42NaN2O4).

117
The novel target compound primary amine (E)-N-(4-(2-aminoethyl)phenyl)-3-
(3,5-di-tert-butyl-4-hydroxyphenyl)acrylamide (133) was obtained in 65% yield
from the Boc-protected amine 132 (Scheme 2.31). Removal of the acid-labile
Boc-protecting groups of amine 132 can be effected by reaction with 4 M HCl.
By following the same procedure as that used for the deprotection of 169, the
desired target 133 was obtained from the protected precursor 132. The
hydroxycinnamic acid containing linker compound 132 was stirred at 55–60 °C
for 2–3 h in a mixture of 1,4-dioxane and water with dropwise addition of 4 M
HCl over ten minutes. Analysis by TLC indicated consumption of starting
material, and ninhydrin staining highlighted the reformed primary amine 133 on
the TLC plate with a strong purple colour with an Rf of 0.13 in 9:1
chloroform/methanol. The compound was purified by column chromatography
to give 65% yield of the deprotected primary amine product 133 with a sharp
melting point of 130–132 °C.
In the 1H NMR (CDCl3) spectrum of 133, a multiplet appeared at 1.43 ppm with
an integral corresponding to eighteen protons from two tert-butyl groups and a
broad singlet appeared at 2.95 ppm for the two primary amine protons. An
extra two peaks corresponding to the carbons of the ethene group also
appeared in the 13C NMR spectrum at 125.0 ppm and 136.6 ppm, respectively.
The structure of 133 was supported by MS, which showed the expected
molecular ion of M+H at 395.2651 m/z (EI+ (HRMS) showed a deviation of
0.0048 from the calc. mass of C25H35N2O2).
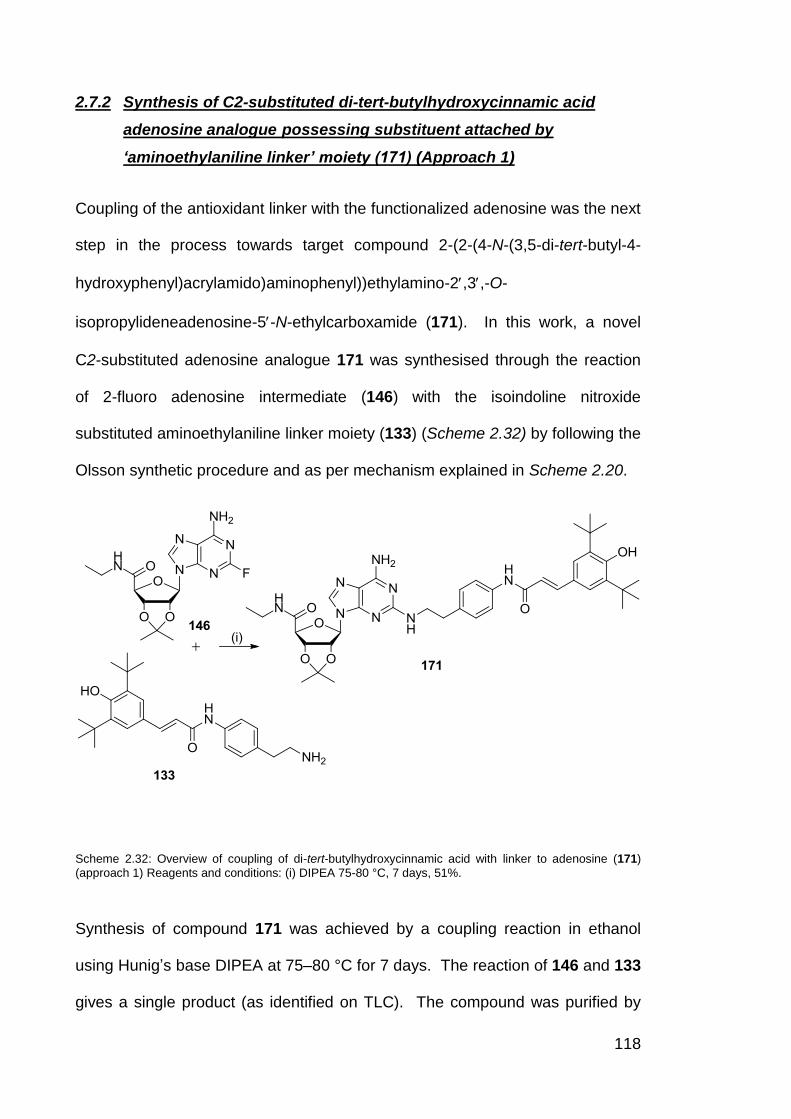
118
2.7.2 Synthesis of C2-substituted di-tert-butylhydroxycinnamic acid
adenosine analogue possessing substituent attached by
‘aminoethylaniline linker’ moiety (171) (Approach 1)
Coupling of the antioxidant linker with the functionalized adenosine was the next
step in the process towards target compound 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido)aminophenyl))ethylamino-2,3,-O-
isopropylideneadenosine-5-N-ethylcarboxamide (171). In this work, a novel
C2-substituted adenosine analogue 171 was synthesised through the reaction
of 2-fluoro adenosine intermediate (146) with the isoindoline nitroxide
substituted aminoethylaniline linker moiety (133) (Scheme 2.32) by following the
Olsson synthetic procedure and as per mechanism explained in Scheme 2.20.
Scheme 2.32: Overview of coupling of di-tert-butylhydroxycinnamic acid with linker to adenosine (171)
(approach 1) Reagents and conditions: (i) DIPEA 75-80 °C, 7 days, 51%.
Synthesis of compound 171 was achieved by a coupling reaction in ethanol
using Hunig’s base DIPEA at 75–80 °C for 7 days. The reaction of 146 and 133
gives a single product (as identified on TLC). The compound was purified by

119
column chromatography using ethyl acetate and ammonia solvent system in
50% yield.
The structure of the hydroxycinnamic acid-coupled adenosine compound 171
was supported by MS, which showed the expected molecular ion of M+H at
741.4490 m/z by the HRMS (EI+ HRMS showed a deviation of 0.040 ppm from
the calc. mass of C40H53N8O6). The consistent sharp melting point of 138–140
°C supported the synthesis of the isolated compound 171.
Similarly, to the nitroxide possessing adenosine analogues the hydroxycinnamic
acid coupling reaction with approach 1 only gives moderate yields (50%). The
reaction conditions were very harsh. As this approach required harsh reaction
conditions and extended reaction times (7 days) an alternative approach
(approach 2) was explored. As the amine linkers could be sourced in large
scale, greater efficiency for the synthesis was thought to be possible through
first coupling the adenosine intermediate 146 with excess amine 124 and then
coupling this system to the hydroxycinnamic acid 118. Moreover, the number of
steps are also reduced by using adenosine intermediate 159 as the starting
material.
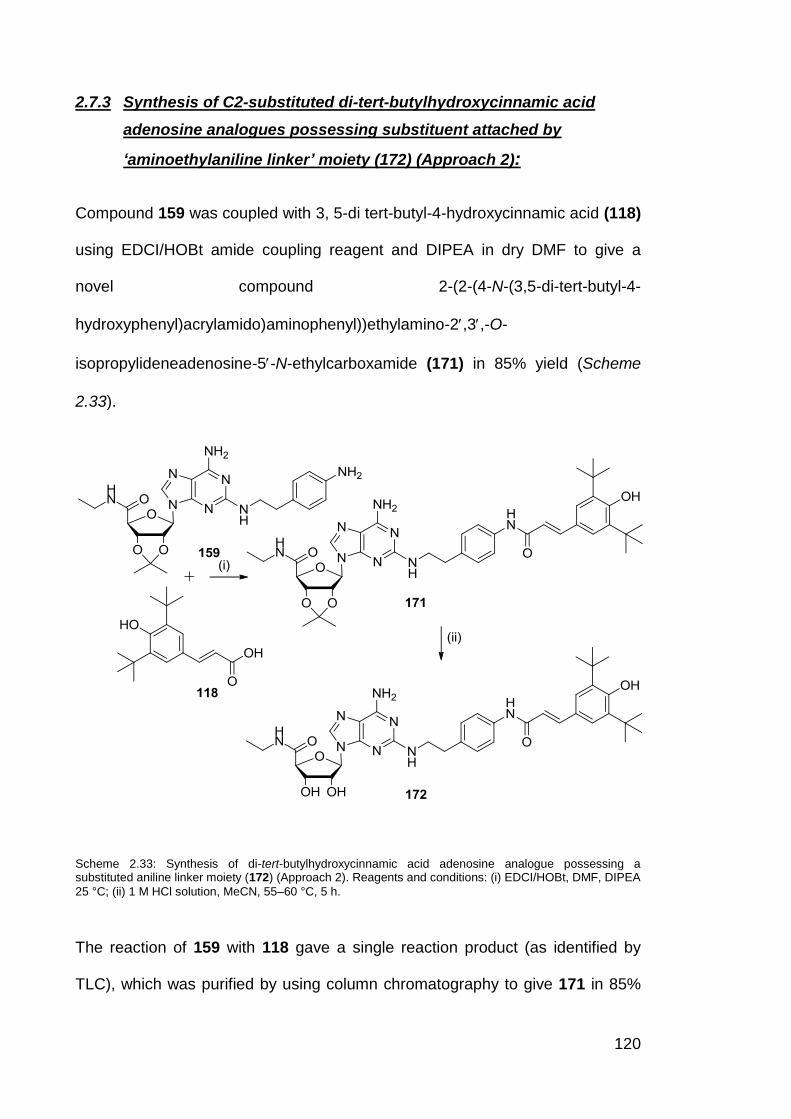
120
2.7.3 Synthesis of C2-substituted di-tert-butylhydroxycinnamic acid
adenosine analogues possessing substituent attached by
‘aminoethylaniline linker’ moiety (172) (Approach 2):
Compound 159 was coupled with 3, 5-di tert-butyl-4-hydroxycinnamic acid (118)
using EDCI/HOBt amide coupling reagent and DIPEA in dry DMF to give a
novel compound 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido)aminophenyl))ethylamino-2,3,-O-
isopropylideneadenosine-5-N-ethylcarboxamide (171) in 85% yield (Scheme
2.33).
Scheme 2.33: Synthesis of di-tert-butylhydroxycinnamic acid adenosine analogue possessing a substituted aniline linker moiety (172) (Approach 2). Reagents and conditions: (i) EDCI/HOBt, DMF, DIPEA
25 °C; (ii) 1 M HCl solution, MeCN, 55–60 °C, 5 h.
The reaction of 159 with 118 gave a single reaction product (as identified by
TLC), which was purified by using column chromatography to give 171 in 85%

121
yield. In the 1H NMR (CD3OD) spectrum, of the product 171, two alkene
protons appeared at 7.27 and 6.65 ppm as doublets and the phenolic hydroxy
proton appeared as a broad singlet at 5.57 ppm. Two multiplets at 2.77 ppm
with an integral corresponding to two protons and a triplet appeared at 0.59
ppm with an integral corresponding to three ethyl group protons from the ribose
amide moiety. An extra two peaks corresponding to the carbon of the ethene
group also appeared in the 13C NMR spectrum at 117 ppm and 138 ppm,
respectively.
The structure of 171 was supported by high-resolution mass spectrometry,
which showed the expected molecular ion of M+H at 741.4490 m/z (EI+ HRMS
showed a deviation of 0.040 ppm from the calc. mass of C40H53N8O6). The
melting point of 138–140 °C of the isolated compound 171 was sharp. The
purity of compound 171 was found to be >99.9% by analytical high-pressure
liquid chromatography (HPLC).
Deprotection of the isopropylidene group from compound 171 was achieved
using 1 M HCl at at 55–60 °C. in 4.0 h to give 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido)phenyl))ethylamino)adenosine-5-N-
ethylcarboxamide (172) in 70% yield (Scheme 2.33).
The deprotection reaction gave a single reaction product 172 (as identified on
TLC), which was isolated and purified using column chromatography and partial

122
crystallisation with 75% methanol/ 25% water system. In the 1H NMR
(CD3COCD3) spectrum of 172, a doublet of two alkene protons appeared at
7.24 and 5.92 ppm and the phenolic hydroxy proton appeared as a broad
singlet at 5.59 ppm. Two multiplets at 3.07 and 3.36 ppm with an integral
corresponding to two protons and a triplet appeared at 1.03 ppm with an
integral corresponding to three protons for the ethyl group from the ribose
amide substituent. An extra two peaks corresponding to the carbon of the ethyl
group linker also appeared in the 13C NMR spectrum (CD3OD) at 34.1 ppm and
42.5 ppm, respectively and two ethene group carbons appears at 117.2 and
142.6 ppm. The structure of the deprotected product 172 was supported by
MS, which showed the expected molecular ion of M+H at 701.4096 m/z, (EI+
HRMS showed a deviation of 0.032 ppm from the calc. mass of C37H49N8O6).
The melting point of 241–243 °C (decomposed) of isolated compound 172 was
sharp. The purity of compound 172 was found to be >99.9% using analytical
HPLC.
All the novel C2-substituted para-aminoethylaniline linked adenosine analogues
joined with antioxidant moieties were found to be difficult to purify and isolate by
column chromatography. The main products generated have close Rf values
similar to starting materials and a byproduct. The use of medium pressure
chromatography (MPLC) was found to be a better technique to purify those
components.

123
2.8 Result of biological testing of adenosine analogues
2.8.1 Adenosine receptor-binding assay
A number of C2-modified nitroxide adenosine compounds with various linker
groups were successfully synthesised. The potency and agonist activity (rather
than antagonists or inverse agonists) of each adenosine analogues was
assessed with a functional cAMP assay using transfected Chinese Hamster
Ovary (CHO) cell lines, in which cells overexpress one of the relevant human
A1, A2A, A2B, and A3 adenosine receptor subtypes. This analysis was undertaken
at the Monash Pharmaceutical Institute, Melbourne.
These functional assays provide an indication of the physiologically-relevant
mode of interaction between test compounds and the target receptor
subtypes307 and determination of the agonist activity of the compound in this
case at human adenosine receptor subtypes (hAXAR, X=1, 2A, 3). Functional
assays that measure a cAMP production were used to assess the (A1, A2A, A2B,
and A3) receptor binding affinity for each of the novel compounds.
This assay is based on a competition between cAMP attached to special
fluorescence material present in the test well with the test cells, and the
stimulation or inhibition of intracellular cAMP production by receptor agonism.
The generation of fluorescent material is relative to the total amount of cAMP at
the end of the experiment, and is measured by photo-stimulating the fluorescent
material and measuring the degree of fluorescence.

124
The potency for the A2AAR, and A2BAR is measured by pEC50, which is the
logarithmic value of concentration at which exactly half of the receptor
population is in a fully-activated state (i.e. the concentration of an agonists that
produces a 50% of physiological response). The A1AR and A3AR data is
reported as a pIC50 value, which is also a measure of potency, but more
commonly used for antagonists (this is because A1AR and A3AR act
biochemically in opposition to the A2AAR) and therefore the assay reports the
inverse of the pEC50 for the A1AR and A3AR.121 In this case, both assays are
equivalent measures of relative agonist activity at the adenosine receptor
subtype in question, and are expressed here in nanomoles (nM).
Next, the activity of a compound can be compared to that of other compounds
for each of the adenosine receptor subtypes assayed. The values are an
indication of relative functional selectivity and are expressed as a unitless ratio.
Functional selectivity ratios are calculated simply by dividing the pEC50 of the
compound at A2AAR by the pIC50 of the compound at the A1AR (or A3AR)
subtype. The value is an indication of how much more functionally active the
compound is at A2AAR than the other receptor subtypes (i.e. a value of A2A/ A1
of 100 means the compound is 100 times more potent in terms of functional
selectivity at A2AAR than A1AR). Desirable figures of this measure tend towards
more than a thousand fold in favour of the target receptor subtype relative to all
other receptor subtypes. The assays were conducted upon all three structural
classes of compounds at A1AR, A2AAR, A2BAR and A3AR receptor subtypes,
and the results are presented in Table 1.

125
cAMP Accumulation Assay. Adenosine is found naturally in nanomolar
concentrations in human interstitial fluid, and this level is essential for any
potential drug acting at the adenosine receptor. However, in this work,
compounds that exhibited nanomolar pEC50 and pIC50 values were described
here, which could act as desirable targets for further development as
cardioprotective agents.
The receptor binding results generated by the adenosine compounds from this
structural class are listed in Table 1.
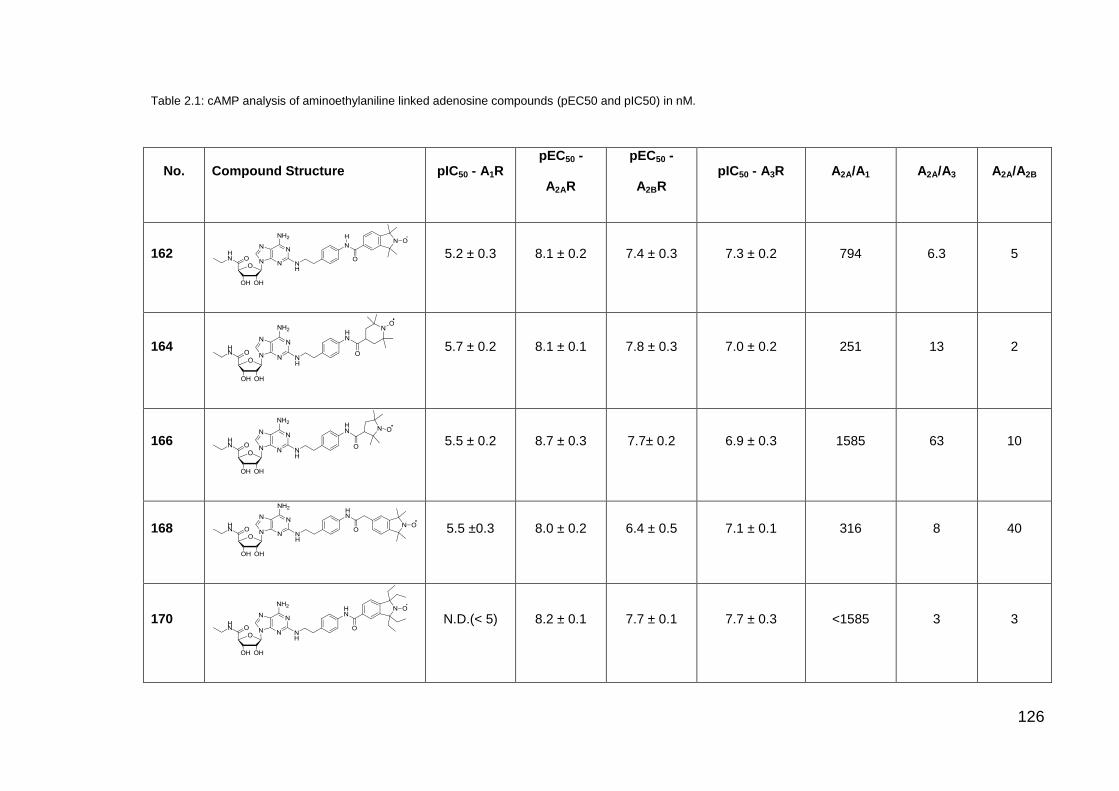
126
Table 2.1: cAMP analysis of aminoethylaniline linked adenosine compounds (pEC50 and pIC50) in nM.
No. Compound Structure pIC50 - A1R pEC50 -
A2AR
pEC50 -
A2BR pIC50 - A3R A2A/A1 A2A/A3 A2A/A2B
162
5.2 ± 0.3 8.1 ± 0.2 7.4 ± 0.3 7.3 ± 0.2 794 6.3 5
164
5.7 ± 0.2 8.1 ± 0.1 7.8 ± 0.3 7.0 ± 0.2 251 13 2
166
5.5 ± 0.2 8.7 ± 0.3 7.7± 0.2 6.9 ± 0.3 1585 63 10
168
5.5 ±0.3 8.0 ± 0.2 6.4 ± 0.5 7.1 ± 0.1 316 8 40
170
N.D.(< 5) 8.2 ± 0.1 7.7 ± 0.1 7.7 ± 0.3 <1585 3 3

127
The dual action of adenosine agonist compounds possessing effective radical
scavenger nitroxide moieties and the C2-binding domain of A2AAR selective
compounds has not been explored to date. To investigate such selectivity, a
number of C2-modified nitroxide adenosine compounds were synthesised with
various linker groups. The potency and agonist activity (rather than antagonists
or inverse agonists) of the adenosine analogues was assessed with a this type
of downstream cAMP assay using transfected CHO lines. These cells
overexpress one of the human A1, A2A, A2B, and A3 adenosine receptor
subtypes. In general the C2-substituted nitroxides with 4-(2-aminoethyl)aniline
linked N6-aminoadenosine-5-N-ethylcarboxamide compounds (162-172) have
shown a good A2AAR affinity.
It is well known that the C2-binding domain with particular linkers can
accommodate a wide range of aryl and cycloalkyl groups. The incorporation of
very bulky tetramethylisoindoline-2-yloxyl, tetraethylisoindoline-2-yloxyl,
tetramethylpiperidine-1-yloxyl, and tetramethylpyrrolidine-1-yloxyl nitroxide
groups in this position afforded agonists with excellent affinity for the A2AAR. In
order to improve receptor affinity, the antioxidant nitroxide moiety was attached
via an aminoethylaniline linker. Linkers of this type have previously proven to
be effective in connecting adenosine receptor agonists with antioxidant moieties
(compounds 48 and 49).121

128
In some cases, large selectivity increases were seen with C2-substituted
nitroxides possessing (4-aminophenyl) ethylaminoadenosine-5-N-
ethylcarboxamide (namely compounds 162, 164, 166, 168 and 170) showing
more than 200-fold selectivity for the A2AAR over A1AR and reasonable
selectivity over A3AR.
All adenosine compounds in the first structural class with aminoethylaniline
linked series are potent A2AAR agonists with pEC50 values ranging from 8.0-8.7.
Compound 166 is marginally the most potent adenosine analogue with pEC50
value of 8.7. These compounds also showed good selectivity versus A1AR (251
to 1585) and modest selectivity versus A3AR (up to 63 fold). Similarly, these
compounds also have modest selectivity versus A2BAR (2 to 40 fold).
In a comparison of structurally related antioxidant groups in the first structural
class with aminoethylaniline linked series series, structural modification has
limited effect on the A2AAR selectivity. For example the tetramethylisoindoline
nitroxide 162 has a pEC50 of 8.1 at the A2AAR. The incorporation of a
methylene spacer in compound 168 has no (statistically significant) effect on
A2AAR activity (168; pEC50 of 8.0 ± 0.2). Likewise, tetraethyl substitution had no
statistical effect on A2A potency (cf. 162 v 170 with pEC50 values of 8.1 ± 0.2
and 8.2 ± 0.1, respectively). The above facts support the concept that the
antioxidant group is protruding into the extracellular space when the agonist is
bound to the A2AAR.

129
2.8.2 Simulated Ischemia Assay121.
A number of potent and selective C2-modified aminoethylaniline linked nitroxide
adenosine agonists were successfully assessed with this type of simulated
ischaemia assay. This assay is one measure of investigating cardioprotective
action of agonists. This ischaemia assay was conducted using a previously
reported cell culture hypoxia model to determine cell protection in the ischemic
induced rat arterial cardiomyoblast cell line H9c2.258, 308-310 This cell line cells
are incubating in hypoxic simulated ischaemia. This analysis was undertaken at
the Monash Pharmaceutical Institute, Melbourne.
The assay was conducted using the most potent and selective dual functionality
adenosine compounds 162, 164, 166, and 170 along with known compound
VCP874 (49) for further analysis. All compounds possess antioxidant moieties
combined with aminoethylaniline linked adenosine analogues. These
antioxidants possessing aminoethylaniline linked adenosine compounds have
good A2AAR selectivity (with pEC50 values of 8.0-8.7 nM) and have
demonstrated positive control in the study of ischemic cell protection as shown
in Figure 2.3 (panel A).
Specifically, the adenosine compounds 162 and 170 were selected for further
examination in an ischemic cell culture hypoxia assay. These compounds were
analysed by combining with a known A2AAR antagonist SCH442415, to observe
the effect of the antioxidants. This study shows that these dual acting ligands
gave significant cardioprotective effects, however, these effects were largely

130
inhibited by A2AAR antagonist SCH442415. This means that these effects were
primarily mediated by A2AAR activation and a limited effect by the nitroxide
antioxidant species was evident. There were some additional cardioprotective
effects observed at the highest concentration in the presence of antagonist.
These results may indicate that the antioxidant activity makes some contribution
in cardioprotection at that point of high concentration as shown in Figure 2.3
(panel B).
A B
Figure 2.3: Dual acting A2AAR agonists - antioxidants decrease cell death within an in vitro model of simulated ischemia. A) 162 (), 164 (), 166 () and 170 () stimulate an equivalent, concentration-
dependent, decrease in cell death to the reference ligand VCP874 (). B) The decrease in cell death mediated by 162 () and 170 () was inhibited in the presence of the A2AAR antagonist, SCH442415 ( 162 + 100 nM SCH442415; 170 + 100 nM SCH442415). Cell death was assessed by determining the
proportion of non-viable, propidium iodide positive, cells; N=2-4.
The smaller ring nitroxide adenosine compounds linked with the
aminoethylaniline linker group were found to be more potent and selective to
the adenosine receptors than other larger nitroxides containing adenosine
analogues. Furthermore, tetraethyl modified nitroxide adenosine compounds
also have better adenosine agonist activity. However, these compounds have
no major differences in the selectivities to the particular adenosine receptors. In
addition, side chains do not limit binding and are not detrimental to bioassays.
-8 -7 -6 -5SI
60
80
100
120
[Agonist] Log M
Ce
ll D
ea
th (%
of S
I)
VCP874
1VHM157C
1VHM168C2
1VHM170B
1VHM187
-8 -7 -6 -5SI
60
80
100
120
[Agonist] Log M
Ce
ll D
ea
th (%
of S
I)
1VHM157C
1VHM157C + 100 nM SCH442416
1VHM187
1VHM187 + 100 nM SCH442416

131
This means that even small or large group modifications are also tolerated in
the adenosine receptor selectivity; therefore, a small spacer linker group such
as alkynyl-linked nitroxide possessing adenosine compounds were synthesised.
In addition, it has been previously reported that the alkyne containing
compounds at adenosine C2-position are selective to the A2AAR. In order to
expand the possible side chain variations all of which still possess a nitroxide or
phenolic antioxidant, a further target of ethynyl-linked extended groups were
developed.

132
Result and Discussion Part B: Adenosine 3analogues with ethynyl linker possessing
antioxidant moieties.

133
Compounds having groups linked by an alkyne joined at the C2-position of the
adenosine are reported to be selective adenosine receptor agonists. A series of
2-alkynyl-5-N-ethylcarboxamide compounds were previously synthesised.311
These compounds were studied for the binding and functional assays to assess
their potencies for the A2AR compared to the A1AR. These compounds are
known to have desirable effects on vasorelaxation without impacting on heart
rate. C2-alkynyl-5-N-ethylcarboxamide compounds were found to be selective
A2AR agonists243 (Figure 3.1). Moreover, some of these alkynyl-linked
compounds were also found to possess good A1AR/A2AR affinity. Some of the
hydroxy alkynyl adenosine derivatives are potent inhibitors of platelet
aggregation and could be beneficial in the treatment of cardiovascular
disorders.311
Figure 3.1: Candidate moieties and their actions in the second structural class of target adenosine analogues.
The alkynyl linker was intended to promote specific receptor recognition.
Binding studies312 have shown that the presence of a phenylethynyl group in the

134
C2-position of adenosine 65 (Figure 3.2) favoured the interaction with A3AR,
with the resulting compounds displaying high affinity and selectivity for the
adenosine A3AR. Additional substitution at the N6- and C4-positions interact
with the hydrophilic pocket on the adenosine receptor to increase both the A3AR
agonists affinity and also the H-bonding at the 3- and 5-positions which are
required for A3AR agonism.313-315
Figure 3.2: Adenosine A3AR agonists (PENECA) 65
The initial adenosine synthetic targets of this work can be synthesised from the
adenosine intermediate 2-iodo-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (149) by coupling with the appropriate alkyne isoindoline
nitroxide (120, and 122) or non-nitroxide moiety. The published methodology
for achieving these targets involves the Sonogashira coupling reaction.
3.1 Synthesis of C2-substituted TMIO adenosine analogues possessing substituents attached by an ‘ethynyl linker’moiety (178)
3.1.1 Synthesis of the 2-iodo-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide intermediate (149)
Functional group interconversion of the C2-amine of O6-(benzotriazol-1-yl)-2,3-
O-isopropylideneadenosine-5-N-ethylcarboxamide (144) to the nucleophilic

135
aromatic substitution-promoting iodine was accomplished using the method of
Kim273 (Scheme 3.1) The reaction proceeded smoothly achieving 2-iodo-O6-
(benzotriazol-1-yl)-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide
(148) in 86% yield on a scale ranging from 50-200 mg.
Scheme 3.1: Overview of synthesis of C2-iodo adenosine intermediate (149) Reagents and conditions: (i)
CH2I2, t-BuONO, in MeCN, 68 %. (ii) 28% aq. ammonia MeCN.
The reaction conditions were reasonably harsh because diiodomethane was
used as an iodination agent and MeCN as the solvent, and the reaction was
carried out at a temperature of 65–70 °C. The solvent and excess of
diiodomethane was removed by evaporating the reaction mixture under vacuum
for a long time. The impurity was successfully removed by excessive washing
with ethyl acetate. The Rf value for the product 148 was 0.42 in 1:4
DCM/MeOH and was distinguishable from 144 by TLC, (Rf. 0.37 for the C2-
amino compound 144). These values indicates that the polarity of the molecule
slightly decreased with the introduction of the electronegative iodine atom and
the removal of the amine.

136
In the 1H NMR spectrum (d6-DMSO), the broad singlet at 6.67 ppm
corresponding to the heteroaromatic amino at C2-position of compound 144
was not present. The C8-proton appeared to be a sharp singlet. This singlet
was shifted to the more deshielded region from 8.04 to the 8.19 ppm for the
iodo substituted adenosine compound 148. In the 13C NMR spectrum, a peak
for the C2-carbon of the purine ring appeared at 144.0 ppm. Additionally, there
was a signal at 168 ppm which was assigned to the amide carbonyl carbon.
The melting point of 194–196 °C for the product 148 was sharp. The structure
of deprotected product 148 was supported by HRMS, which showed the
expected molecular ion of M+Na at 615.0808 m/z, (EI+ HRMS showed a
deviation of 0.0371 ppm from the calc. mass of C21H21INaN8O5)
Ammonia solution was used to displace the more labile benzotriazolyl leaving
group from compound 148 with nucleophilic amines262, 282 and form a 6-amino
product 2-iodo-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (149)
(Scheme 3.1). This SNAr reaction proceeded smoothly in acetonitrile at room
temperature overnight. Purification via column chromatography gave good
yields between 70–80% of an off-white solid on a 250 mg to 1 g scale. A
second component separated by column chromatography was identified as a
small amount of amino-substituted product at the C2-position.
As expected, the Rf value of 149 on TLC decreased as the benzotriazolyl group
was lost and the polarity increased slightly. The Rf value was measured at 0.62
using 7:3 ethyl acetate/MeOH. In d6-DMSO, the major features of the 1H NMR

137
spectrum of 149 were the absence of signals from the benzotriazolyl aromatic
protons and the broad singlet signal which appeared at 7.85 ppm with an
integral corresponding to two N6-protons. Signals from the 2- and 3-protons
coalesced completely from two resolved doublets to a singlet peak at 5.35 ppm
with an integral corresponding to two protons. Similarly, in the 13C NMR
spectrum, signals from the benzotriazolyl aromatic carbons disappeared.
Furthermore, the signals for the 2- and 3-carbons coalesced to give two
identifiable signals separated by 0.03 ppm. The melting point of 196–198 °C of
the product 149 was sharp. The structure of the product 149 was supported by
MS, which showed the expected molecular ion of M+H at 497.0390 m/z, (EI+
HRMS showed a deviation of 0.0030 ppm from the calc. mass of
C15H19INaN6O4). The purity of product 149 was found to be 99.99% by
analytical HPLC in a 80% methanol/20% water system.
A number of adenosine molecules were generated with either tetramethyl and
tetraethyl nitroxide or a phenyl side chain joined by with an ethynyl linker by
modification of adenosine at the C2-position and with 5-N-ethylcarboxamide
modification of the ribose ring.
3.1.2 Synthesis of ethynyl TMIO (ETMIO) intermediate (120)
In this synthesis, firstly the nitroxide moiety 5-ethynyl-1,1,3,3-
tetramethylisoindoline-2-yloxyl (ETMIO) nitroxide 120 was synthesised from 5-
bromo-tetramethylisoindoline (102) according to the procedure reported in the
literature316 (Scheme 3.2).

138
Scheme 3.2: Synthesis of 5-ethynyl-1,1,3,3-tetraethylisoindoline-2-yloxyl (ETMIO) (120) 316
. Reagents and conditions: (i) I2, n-BuLi, H2O2, Stir 30 min, 69% (ii) Na2WO4.2H2O,H2O2,NaHCO3, Stir 72 h. 79%. (iii)
DABCO, Pd(OAc)2, 2-methyl-3-butyn-2-ol, MeCN, 80 %; (iv) KOH, Toluene, 90 %.
5-Bromo-1,1,3,3-tetramethylisoindoline (102) was reacted with n-BuLi in dry
THF and then quenched with iodine to obtain a di-iodo intermediate, in which
iodination occurred on the ring as well as on the nitrogen atom. This product
was subsequently treated with H2O2 to generate a mono-iodo compound 5-iodo-
1,1,3,3-tetramethylisoindoline (104) in 69% yield. Compound 104 was then
oxidised by 30% H2O2 in the presence of the Na2WO4.2H2O catalyst in MeCN to
give 5-iodo-1,1,3,3-tetramethylisoindoline-2-yloxyl (105) in 79% yield. The
compound 105 was then coupled with 2-methyl-3-butyne-2-ol (160) using
palladium diacetate (Pd(OAc)2) in the presence of 1,4-diazabicyclo(2.2.2)
octane (DABCO) as a base using triethylamine as a solvent to obtain 5-(2-
methyl-3-butyne-2-ol)-1,1,3,3-tetramethylisoindoline-2-yloxyl (119)316 in 80%
yield.
The reaction mechanism of alkyne coupling with the palladium reagent is
detailed in Scheme 3.3. Deprotection of the dimethylpropanol group from
compound 119 with solid KOH in refluxing MeCN resulted in the formation of E-

139
TMIO 120 in 90% yield (Scheme 3.2). The melting point of 96–98 °C of the
recrystallised product was consistent and in agreement with the published
values316 for the desired product 120.
3.1.3 Coupling of E-TMIO with iodo-adenosine intermediate (178)
The ethynyl isoindoline nitroxides and phenyl acetylene were then coupled in
separate reactions with the adenosine iodo intermediate under Sonogashira
coupling conditions to achieve coupling products 177, 181 and 185. The details
of the reaction mechanisms are explained as follows.
The palladium-catalysed alkynylation called the Sonogashira reaction is a
convenient method to synthesise aryl and other acetylene-substituted
compounds. This methodology was first discovered in 1975 by Cassar,317
Dieck, Heck318 and Sonogashira et al.319 The method used was an
amalgamation of the Castro–Stephens reaction320, 321, involving Cu-promoted
alkynylation and alkyne implementation of the Heck reaction. Sonogashira et
al. used Cu (I) in the presence of the Pd (0) catalyst, which favours the alkynyl
transformation at room temperature. The reaction was achieved under much
milder conditions and is now commonly referred to as the Sonogshira coupling
reaction.
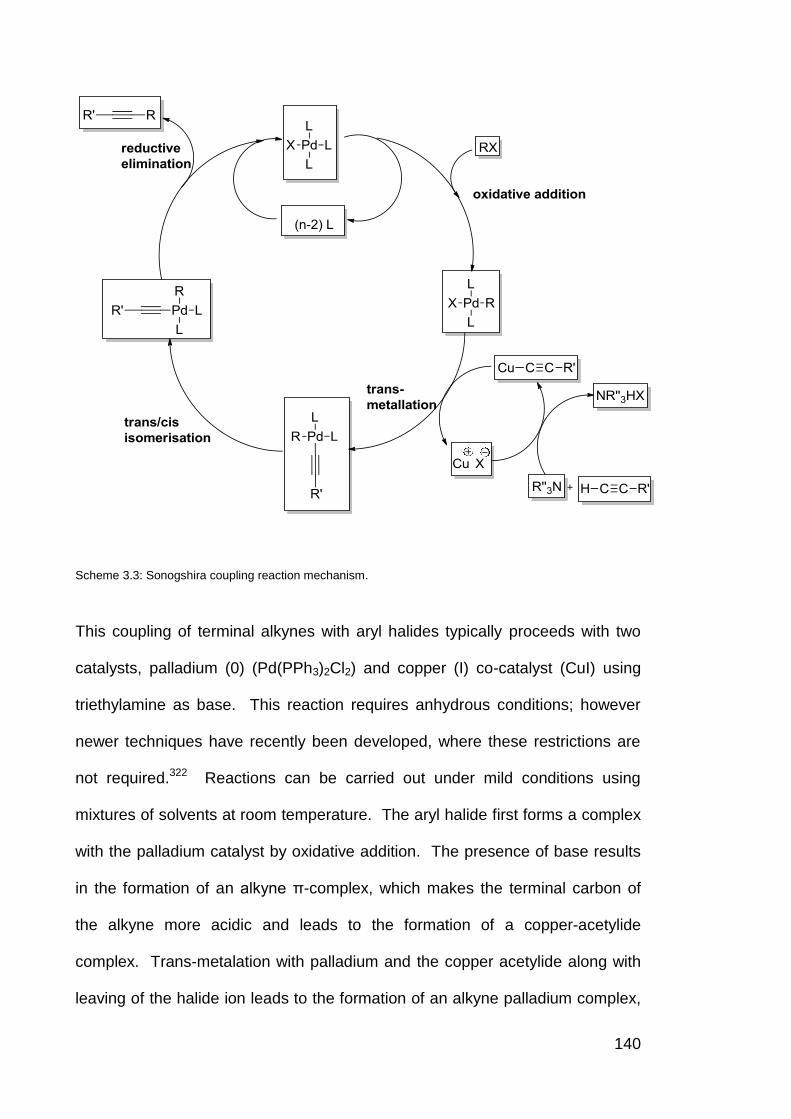
140
Scheme 3.3: Sonogshira coupling reaction mechanism.
This coupling of terminal alkynes with aryl halides typically proceeds with two
catalysts, palladium (0) (Pd(PPh3)2Cl2) and copper (I) co-catalyst (CuI) using
triethylamine as base. This reaction requires anhydrous conditions; however
newer techniques have recently been developed, where these restrictions are
not required.322 Reactions can be carried out under mild conditions using
mixtures of solvents at room temperature. The aryl halide first forms a complex
with the palladium catalyst by oxidative addition. The presence of base results
in the formation of an alkyne π-complex, which makes the terminal carbon of
the alkyne more acidic and leads to the formation of a copper-acetylide
complex. Trans-metalation with palladium and the copper acetylide along with
leaving of the halide ion leads to the formation of an alkyne palladium complex,
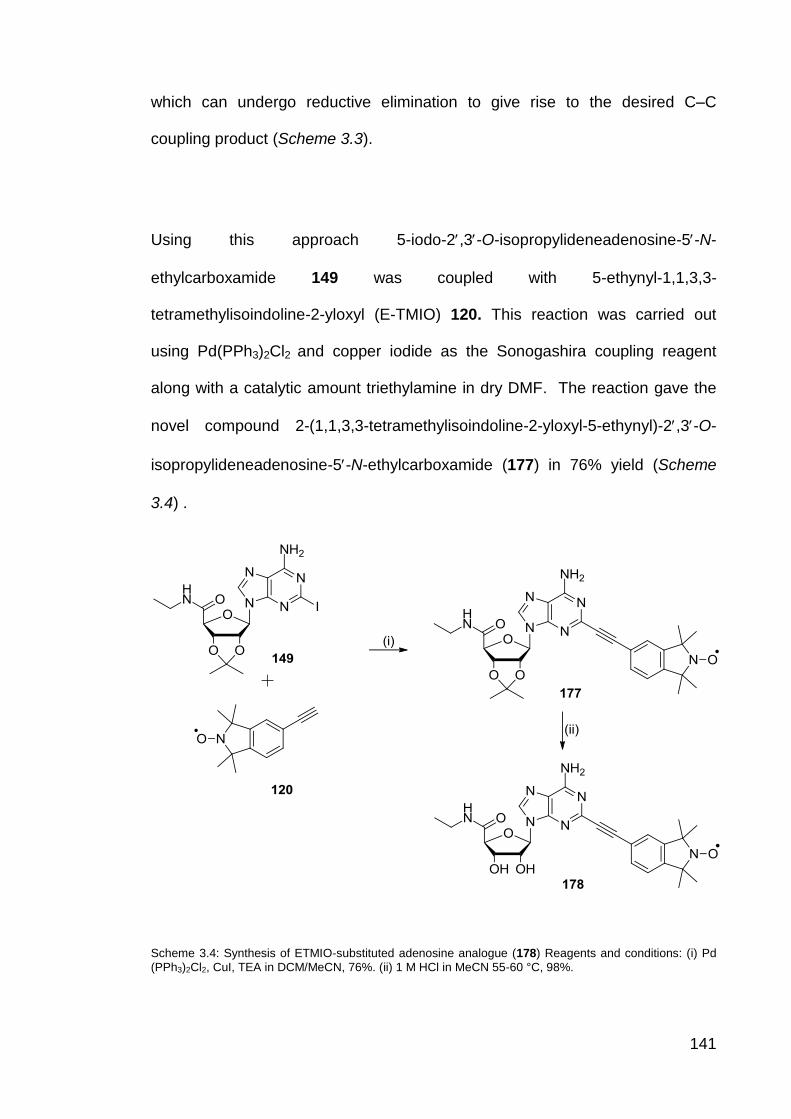
141
which can undergo reductive elimination to give rise to the desired C–C
coupling product (Scheme 3.3).
Using this approach 5-iodo-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide 149 was coupled with 5-ethynyl-1,1,3,3-
tetramethylisoindoline-2-yloxyl (E-TMIO) 120. This reaction was carried out
using Pd(PPh3)2Cl2 and copper iodide as the Sonogashira coupling reagent
along with a catalytic amount triethylamine in dry DMF. The reaction gave the
novel compound 2-(1,1,3,3-tetramethylisoindoline-2-yloxyl-5-ethynyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (177) in 76% yield (Scheme
3.4) .
Scheme 3.4: Synthesis of ETMIO-substituted adenosine analogue (178) Reagents and conditions: (i) Pd
(PPh3)2Cl2, CuI, TEA in DCM/MeCN, 76%. (ii) 1 M HCl in MeCN 55-60 °C, 98%.

142
The reaction was carried out in a mixture of freshly distilled DCM and dry MeCN
under an argon atmosphere at room temperature. The reaction was followed by
TLC and went to completion in 20 h with a product Rf value of 0.64 in
chloroform: ammonia solution (9.8:0.2) as the mobile phase solvent system.
The reaction of 120 with 149 produced a single adduct 177 along with some
alkyne nitroxide starting material (<5%) (as identified by TLC). The product was
isolated and purified by column chromatography using ethyl acetate and
aqueous ammonia solution (9.8:0.2) as the mobile phase. In the 1H NMR
spectrum, paramagnetic broadening was observed as expected due to the
nitroxide radical. This broadening resulted in some peaks not being detected
and giving poor integration. However, the signal of the adenosine moieties
could be detected. The synthesis of product 177 was supported by HRMS,
which showed the expected molecular ion of M+H at 561.2682 m/z, (EI+ HRMS
showed a deviation of 0.0018 ppm from the calc. mass of C29H35N7O5). The
melting point of 146–148 °C of the isolated compound 177 was sharp. The
purity of product 177 was found to be 97.4% by analytical HPLC in an 80%
methanol/20% water system.
Deprotection of the isopropylidene group from compound 177 was achieved
using 1 M HCl at 55–60 °C in 8.0 h. The reaction gave the novel target
compound 2-(1,1,3,3-tetramethylisoindoline-2-yloxyl-5-ethynyl)-adenosine-5-N-
ethylcarboxamide 178 in 98% yield (Scheme 3.4).

143
The deprotection reaction gave a single product 178 (as identified on TLC),
which was isolated by column chromatography using (9.8:0.2) chloroform and
aqueous ammonia solution as the mobile phase. In the 1H NMR spectrum,
paramagnetic broadening was observed as expected due to the nitroxide
radical. This broadening resulted in some peaks not being detected and giving
poor integration. However, the signal of the adenosine moieties could be
detected. The synthesis of product 178 was supported by HRMS, which
showed the expected molecular ion of M+Na at 543.2281 m/z, (EI+ HRMS
showed a deviation of 0.0075 ppm from the calc. mass of C26H30NaN7O5). The
melting point of 261 °C (decomp) for the isolated compound 178 was sharp.
The purity of product 178 was found to be 99.9% using analytical HPLC in an
80% methanol/20% water system.
3.2 Synthesis of C2-substituted TEIO adenosine analogues possessing substituents attached by an ‘ethynyl linker’ moiety (182)
A novel compound 5-ethynyl-1,1,3,3-tetraethylisoindoline-2-yloxyl (E-TEIO) 122,
which was synthesized from compound 105 and obtained from advanced stage
synthesis in the laboratory. Compound 149 was coupled with compound 122
using Pd(PPh3)2Cl2 and copper iodide as the coupling reagents. The reaction is
employed a catalytic amount of triethylamine in dry DMF to produce a novel
compound 2-(1,1,3,3-tetraethylisoindoline-2-yloxyl-5-ethynyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (181) in 83% yield (Scheme
3.5).

144
Scheme 3.5: Synthesis of ETEIO-substituted adenosine analogue (182) ,Reagents and conditions: (i) Pd
(PPh3)2Cl2, CuI, TEA in DCM/MeCN, 83%. (ii) 1 M HCl in MeCN 55-60 °C, 88%.
The reaction was carried out with a mixture of freshly distilled DCM and dry
MeCN under an argon atmosphere at room temperature. The reaction went to
completion in 20 h (as identified by TLC) with Rf value of product 0.47 in (DCM /
MeOH, 9:1).
The reaction of 122 with 149 produced a single product 181 along with some
alkynyl nitroxide starting material (<5%) (as identified by TLC). The product
was isolated and purified by column chromatography in 83% yield. In the 1H
NMR spectrum, paramagnetic broadening was observed as expected due to the
nitroxide radical. This broadening resulted in some peaks not being detected
and giving poor integration. However, the signal of the adenosine moieties

145
could be detected. The synthesis of product 181 was supported by HRMS,
which showed the expected molecular ion of M+H at 617.3296 m/z, (EI+ HRMS
showed a deviation of 0.0030 ppm from the calc. mass of C33H43N7O5). The
melting point of 188–190 °C of the isolated compound 181 was sharp. The
purity of the product 181 was found to be 97.4% using analytical HPLC in an
80% methanol/20% water system.
The deprotection of isopropylidene group from compound 181 was achieved
using 1 M HCl at 55–60 °C in 8.0 h. The reaction generated the novel target
compound 2-(1,1,3,3-tetraethylisoindoline-2-yloxyl-5-ethynyl)-adenosine-5-N-
ethylcarboxamide (182) (Scheme 3.5).
The deprotection reaction of 181 gave a single product 182 (as identified on
TLC), which was purified by column chromatography to give product 182 in 88%
yield. In the 1H NMR spectra, paramagnetic broadening was observed as
expected due to the nitroxide radical that resulted in some peaks not being
detected and giving poor integration. However, signals of the adenosine moiety
could be detected. The synthesis of product 182 was supported by HRMS,
which showed the expected molecular ion of M+Na at 577.3012 m/z, (EI+
HRMS showed a deviation of 0.0001 ppm from the calc. mass of C30H39N7O5).
The melting point of 196–198 °C (decomp) of the isolated compound 182 was
sharp. The purity of the product 182 was found to be 99.9% using analytical
HPLC in an 80% methanol/20% water system.

146
3.3 Synthesis of C2-substituted non-nitroxide phenyl adenosine analogues possessing substituents attached by an ‘ethynyl linker’ moiety (185)
The compound 149 was coupled with phenylacetylene (123) using Pd(PPh3)2Cl2
and copper iodide as the coupling reagent as well as a catalytic amount of
triethylamine in dry DMF. The coupling reaction produced the expected product
2-(2-phenylethynyl)-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide
(185) in 97% yield (Scheme 3.6).
Scheme 3.6: Synthesis of the phenylacetylene-substituted adenosine analogue (185). Reagents and
conditions: (i) Pd (PPh3)2Cl2, CuI, TEA in DCM/MeCN, 97%.
The reaction was carried out with a mixture of freshly distilled DCM and dry
MeCN under an argon atmosphere. The reaction went to completion in 15 h (as
identified by TLC) at room temperature.
The reaction of 149 with 123 produced a single adduct 185 along with some
detectable alkyne nitroxide starting material (< 5%) (as identified by TLC). The
product was isolated and purified by column chromatography using100%
chloroform. In the 1H NMR spectrum, a new peak appeared in the aromatic
region as a multiplet for three protons from the phenyl group at 7.35–7.40 ppm
along with one multiplet for two aromatic protons which appeared at 7.58 ppm.

147
In the 13C NMR spectrum, three additional signals appeared as strong peaks
corresponding to the aromatic phenyl carbons at 128.4, 129.5 and 132.3 ppm.
The other signals appeared at 84.9 and 114.9 ppm for the alkyne carbons. The
synthesis of product 185 was supported by HRMS, which showed the expected
molecular ion of M+H at 449.1881 m/z, (EI+ HRMS showed a deviation of
0.0057 ppm from the calc. mass of C23H25N6O4). The melting point of 174–176
°C of the isolated compound 185 was sharp. The purity of product 185 was
found to be 99.9% using analytical HPLC in a 80% methanol/20% water
system.
3.4 Results of biological testing of adenosine analogues
3.4.1 Adenosine receptor binding assays
A number of C2-modified nitroxide adenosine compounds were synthesised
with ethynyl linker groups. The potency and agonist activity (rather than
antagonists or inverse agonists) of the adenosine analogues were assessed
with this type of downstream cAMP assay. This test was implemented using
transfected CHO lines in which cells overexpress one of the human A1, A2A, A2B,
and A3 adenosine receptor subtypes. This assay was carried out by our
collaborators at the Monash University Melbourne.
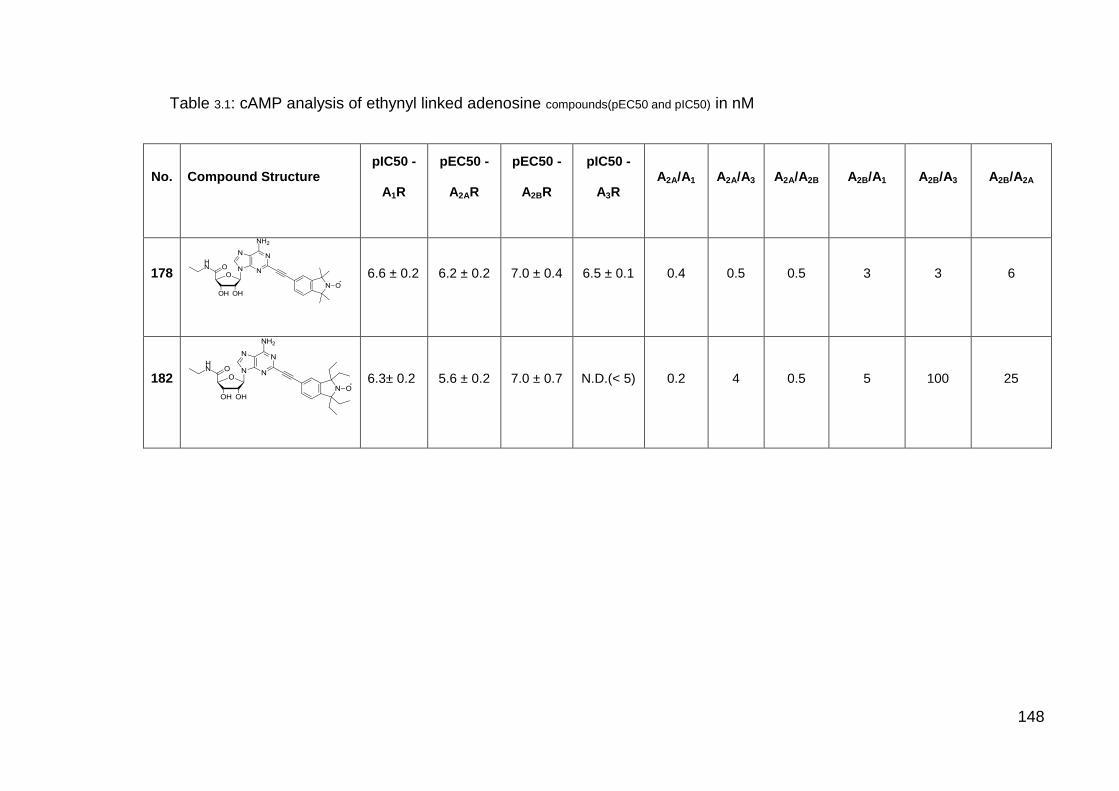
148
Table 3.1: cAMP analysis of ethynyl linked adenosine compounds(pEC50 and pIC50) in nM
No. Compound Structure pIC50 -
A1R
pEC50 -
A2AR
pEC50 -
A2BR
pIC50 -
A3R A2A/A1 A2A/A3 A2A/A2B A2B/A1 A2B/A3 A2B/A2A
178
6.6 ± 0.2 6.2 ± 0.2 7.0 ± 0.4 6.5 ± 0.1 0.4 0.5 0.5 3 3 6
182
6.3± 0.2 5.6 ± 0.2 7.0 ± 0.7 N.D.(< 5) 0.2 4 0.5 5 100 25

149
The molecules from this series of adenosine linked nitroxide were found to have
low A2AAR selectivity with pEC50 values ranging from 5.6-6.2 nM. The alkyne
(ethynyl) linked tetramethylisoindoline-2-yloxyl nitroxide adenosine compounds
(178; pEC50 of 6.2 ± 0.2) and tetraethylisoindoline-2-yloxyl nitroxide adenosine
(182; pEC50 of 5.6 ± 0.2) were significantly less selective for A2AAR over the
other receptor subtypes.
However, this structural class of nitroxide linked with ethynyl moieties to
adenosine displayed a slightly higher selectivity for A2BAR with pEC50 values
(178; pEC50 of 7.0 ± 0.4) and (182; pEC50 of 7.0 ± 0.7) respectively. The above
results indicate that the nitroxide adenosine compounds with two carbon
(ethynyl) linker are better in A2BAR potency and selectivity than the non-
nitroxide adenosine compounds.
These analogues demonstrate slightly higher selectivity for the adenosine
A2BAR over the other adenosine receptor subtypes. Specifically, the adenosine
analogue 178 shows 6 fold better selectivity for A2BAR over A2AAR and 3 fold
better selectivity over other receptor subtypes. Adenosine analogue 182 shows
25 fold selectivity for adenosine A2BAR over A2AAR and 5 and 100 fold
selectivity over adenosine A1AR and A3AR subtypes respectively ( Table 3.1).
These ethynyl linked nitroxide adenosine compounds synthesised have no
significant impact on the affinity of the A2AAR. However, these modified

150
adenosine analogues were found to be selective for the A2BAR. This
modification shows the promising characteristics to develop the new A2BAR
selective compounds. It was postulated that the flexible chain linker might
increase the selectivity for the A2AAR agonists. To find out the effect of
flexibility on the adenosine receptor selectivity, it was decided to reduce the
ethynyl triple bond between the adenosine and antioxidant nitroxide moiety in
order to generate a new class of compounds.

151
Results and Discussion Part C: Adenosine 4analogues with ethyl linker possessing
antioxidant moieties

152
The adenosine synthetic targets of this work have been synthesised starting
from adenosine-5-N-ethylcarboxamide coupled with nitroxides and non-
nitroxide compounds joined by an ethynyl linker chain to generate nitroxides
and non-nitroxides possessing ethyl linked adenosine compounds. These
ethylene-linked adenosine targets possessing nitroxide antioxidant moieties
were expected to be highly selective towards the A2AR. This selectivity is
expected to arise based on previous reports showing that A2AR selectivity is
provided through the incorporation of 2 (or 3) atom chain spacers connected to
the adenosine C2-position. In this project, a number of nitroxide and non-
nitroxide compounds possessing a 5-amide group (the 5-NECA modification)
were synthesised by modification of the C2-position using a two methylene
carbon linkage, with no polar atoms attached. It was expected that this would
provide good A2AR specificity whilst allowing a range of structurally-diverse
antioxidant groups to be incorporated (Figure 4.1).
Figure 4.1: Candidate moieties and their actions in the third structural class of target adenosine analogues.

153
A number of adenosine molecules were generated with tetramethyl and
tetraethylisoindoline nitroxides as well as a phenyl group as the non-radical,
non-antioxidant analogue and each was joined to adenosine at the adenosine
C2-position by an ethyl linker were achieved by the reaction of the appropriate
alkyne at the adenosine C2-position. These compounds also possesse the 5-
N-ethylcarboxamide modification to enhance receptor subtype specificity.
In this approach, the alkynyl linked nitroxides and non-nitroxide adenosine
compounds were reacted further with palladium on carbon under hydrogen gas
at room temperature in methanol to reduce the triple bond and afford the
‘saturated-linker’ adenosine compounds.
4.1 Synthesis of C2-substituted TMIO adenosine analogues possessing substituents attached by an ‘ethyl linker’ moiety (180)
2-(1,1,3,3-Tetramethylisoindoline-2-yloxyl-5-ethynyl)-2,3-
isopropylideneadenosine-5-N-ethylcarboxamide (177) was reacted with
palladium on carbon under a hydrogen atmosphere at room temperature to
afford the novel reduced compound 2-(1,1,3,3-tetramethylisoindoline-2-yloxyl-5-
ethyl)-2,3-isopropylideneadenosine-5-N-ethylcarboxamide (179) in 84% yield
(Scheme 4.1).
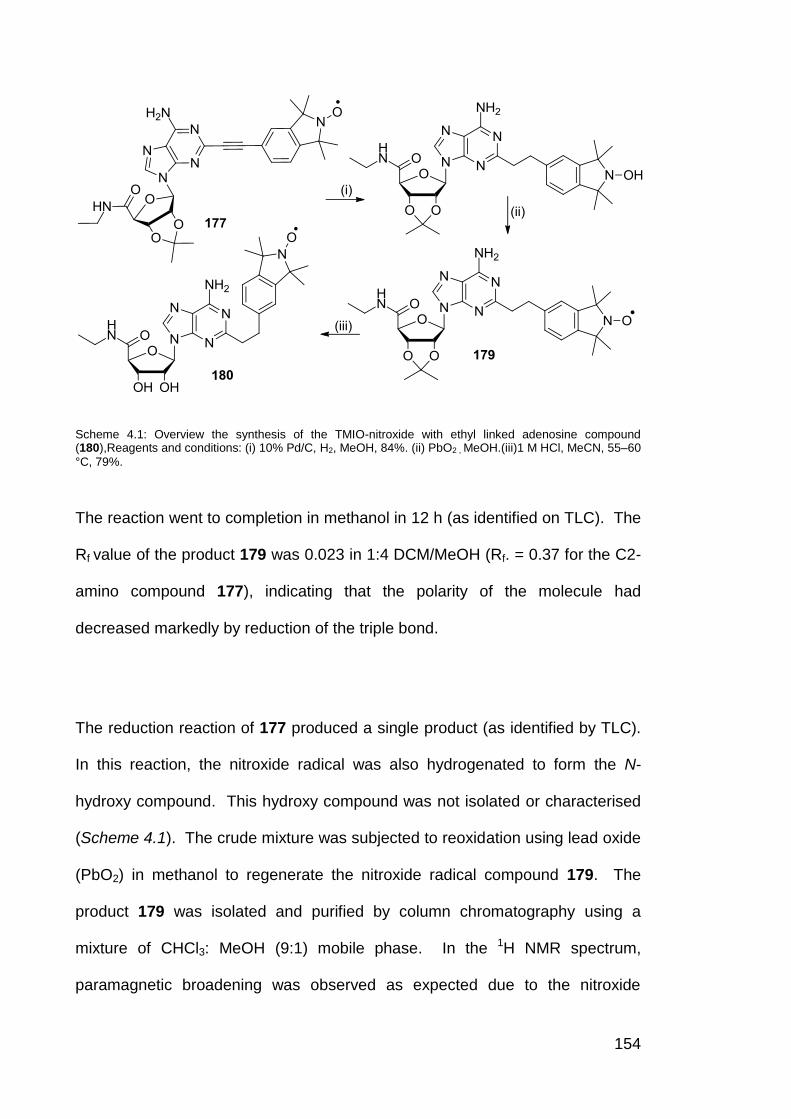
154
Scheme 4.1: Overview the synthesis of the TMIO-nitroxide with ethyl linked adenosine compound (180),Reagents and conditions: (i) 10% Pd/C, H2, MeOH, 84%. (ii) PbO2 , MeOH.(iii)1 M HCl, MeCN, 55–60
°C, 79%.
The reaction went to completion in methanol in 12 h (as identified on TLC). The
Rf value of the product 179 was 0.023 in 1:4 DCM/MeOH (Rf. = 0.37 for the C2-
amino compound 177), indicating that the polarity of the molecule had
decreased markedly by reduction of the triple bond.
The reduction reaction of 177 produced a single product (as identified by TLC).
In this reaction, the nitroxide radical was also hydrogenated to form the N-
hydroxy compound. This hydroxy compound was not isolated or characterised
(Scheme 4.1). The crude mixture was subjected to reoxidation using lead oxide
(PbO2) in methanol to regenerate the nitroxide radical compound 179. The
product 179 was isolated and purified by column chromatography using a
mixture of CHCl3: MeOH (9:1) mobile phase. In the 1H NMR spectrum,
paramagnetic broadening was observed as expected due to the nitroxide

155
radical which resulted in some peaks not being detected and giving poor
integration, however the signal of the adenosine moieties could be detected.
The synthesis of product 179 was supported by HRMS, which showed the
expected molecular ion of M+H at 564.3276 m/z, (EI+ HRMS showed a
deviation of 0.033 ppm from the calc. mass of C29H38N7O5). The melting point
of 108–110 °C of the isolated compound 179 was sharp. The purity of the
product 179 was found to be 96.7% using analytical HPLC in an 80%
methanol/20% water system.
Deprotection of the isopropylidene group from compound 179 was achieved
using 1 M HCl at 55–60 °C in 8.0 h and generated the novel target compound 2-
(1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-ethyl)-adenosine-5-N-
ethylcarboxamide (180) in 79% yield (Scheme 4.1).
The deprotection of compound 179 produced a single product 180 (as identified
by TLC). The Rf value of the product 180 was 0.16 in CHCl3/MeOH (8:2). The
product was isolated and purified by column chromatography using a mixture of
CHCl3: MeOH (9:1) mobile phase. In the 1H NMR spectra, paramagnetic
broadening was observed as expected due to the nitroxide radical which
resulted in some peaks not being detected and giving poor integration, however
the signal of the adenosine moieties could be detected. The synthesis of
product 180 was supported by HRMS, which showed the expected molecular
ion of M+H at 525.2833 m/z, (EI+ HRMS showed a deviation of 0.013 ppm from
the calc. mass of C26H35N7O5). The melting point of 122–124 °C of the isolated

156
compound 180 was sharp. The purity of product 180 was found to be 99.9%
using analytical HPLC in a 80% methanol/20% water system.
4.2 Synthesis of C2-substituted TEIO adenosine analogues possessing substituents attached by ‘ethyl linker’ moiety (184).
2-(2-(1,1,3,3-Tetraethylisoindoline-2-yloxyl)-5-ethynyl)-2,3-
isopropylideneadenosine-5-N-ethylcarboxamide (181) was reacted with
palladium on carbon under hydrogen atmosphere using a hydrogen gas balloon
at room temperature afford the novel reduced compound 2-(1,1,3,3-
tetraethylisoindoline-2-yloxyl-5-ethyl)-2,3-isopropylideneadenosine-5-N-
ethylcarboxamide (183) in 97% yield (Scheme 4.2).
Scheme 4.2: Overview synthesis of TEIO-nitroxide with ethyl linked adenosine compound (184) Reagents
and conditions: (i) 10% Pd/C, H2, MeOH, 97%. (ii) PbO2 , MeOH. (iii) 1 M HCl, MeCN, 55–60 °C, 70%.

157
The reaction was carried out in a mixture of methanol with 10% palladium on
carbon as the solvent under a hydrogen atmosphere at room temperature. The
reaction went to completion in 12 h (as identified on TLC). The Rf value of
product 183 was 0.023 in a mixture of DCM/MeOH (1:4), indicating that the
polarity of the molecule was decreased markedly by reduction of the triple bond.
The reduction of compound 181 produced a single component (as identified by
TLC). In this reaction, the nitroxide radical also hydrogenated to form N-
hydroxy compound. This hydroxy compound was not isolated or characterised
(Scheme 4.2). The crude mixture was subjected to reoxidation using lead oxide
(PbO2) in methanol to regenerate the nitroxide radical product 183. The product
183 was isolated and purified by column chromatography using DCM/MeOH
(9:1) mobile phase. In the 1H NMR spectrum, paramagnetic broadening was
observed as expected due to the nitroxide radical which resulted in some peaks
not being detected and giving poor integration, however the signal of the
adenosine moieties could be detected. The synthesis of product 183 was
supported by HRMS, which showed the expected molecular ion of M+H at
621.3630 m/z, (EI+ HRMS showed a deviation of 0.0009 ppm from the calc.
mass of C33H47N7O5). The melting point of 88–90 °C of the isolated compound
183 was sharp. The purity of product 183 was found to be 89% using analytical
HPLC in an 80% methanol/20% water system.
Deprotection of the isopropylidene group from compound 183 was achieved
using 1 M HCl at 55–60 °C in 8.0 h and generated a novel target compound 2-

158
(1,1,3,3-tetraethylisoindoline-2-yloxyl-5-ethyl)adenosine-5-N-ethylcarboxamide
(184) in 70 % yield (Scheme 4.2).
The deprotection of compound 183 produced a single product 184 (as identified
by TLC). The Rf value of the product 184 was 0.69 in DCM/MeOH (9:1). The
product was isolated and purified by column chromatography using (100%)
chloroform as mobile phase. In the 1H NMR spectra, paramagnetic broadening
was observed as expected due to the nitroxide radical which resulted in some
peaks not being detected and giving poor integration, however the signal of the
adenosine moieties could be detected. The synthesis of product 184 was
supported by HRMS, which showed the expected molecular ion of M+H at
581.3277 m/z, (EI+ HRMS showed a deviation of 0.0049 ppm from the calc.
mass of C30H43N7O5). The melting point of 138–140 °C of the isolated
compound 184 was consistent. The purity of product 184 was found to be
>99.9% using analytical HPLC in 80% methanol/20% water system.
4.3 Synthesis of C2-substituted non-nitroxide phenyl adenosine analogues possessing substituents attached by an ‘ethyl linker’ moiety (188).
2-(2-Phenylethynyl)-2,3-isopropylideneadenosine-5-N-ethylcarboxamide (185)
was reacted with palladium on carbon under hydrogen atmosphere at room
temperature to afford the novel reduced compound 2-(2-phenylethyl)-2,3-
isopropylideneadenosine-5-N-ethylcarboxamide (187) in 93% yield (Scheme
4.3).
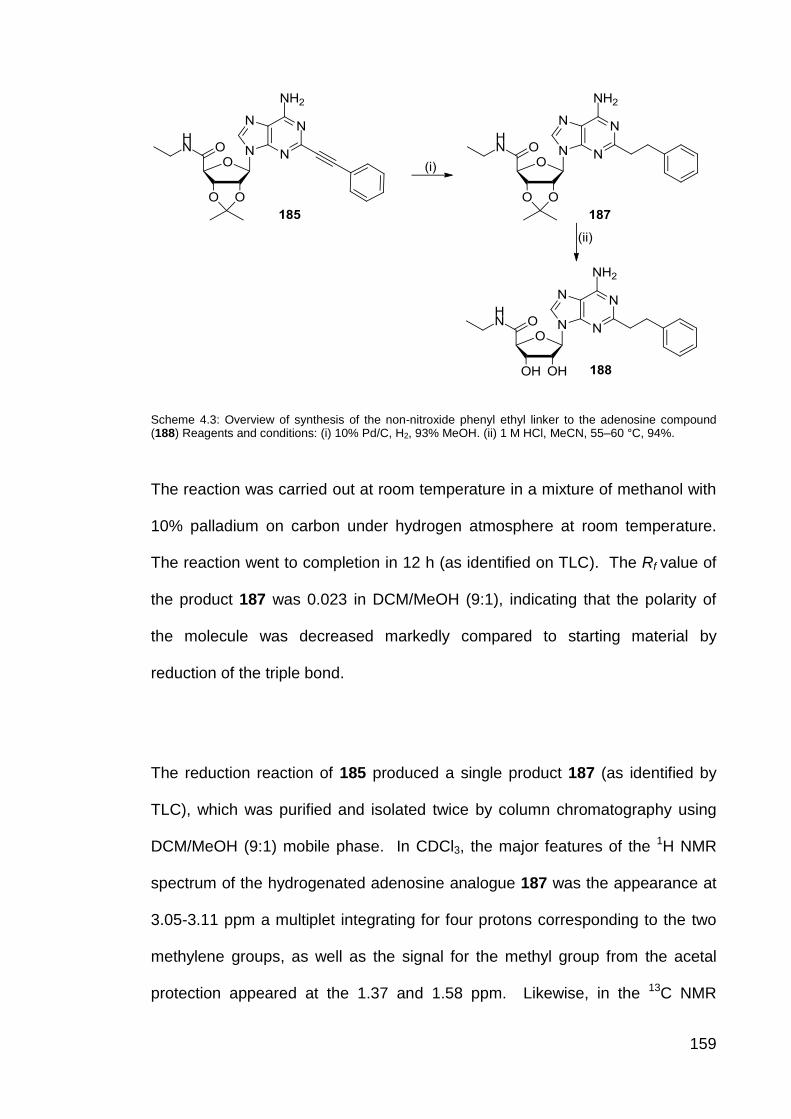
159
Scheme 4.3: Overview of synthesis of the non-nitroxide phenyl ethyl linker to the adenosine compound (188) Reagents and conditions: (i) 10% Pd/C, H2, 93% MeOH. (ii) 1 M HCl, MeCN, 55–60 °C, 94%.
The reaction was carried out at room temperature in a mixture of methanol with
10% palladium on carbon under hydrogen atmosphere at room temperature.
The reaction went to completion in 12 h (as identified on TLC). The Rf value of
the product 187 was 0.023 in DCM/MeOH (9:1), indicating that the polarity of
the molecule was decreased markedly compared to starting material by
reduction of the triple bond.
The reduction reaction of 185 produced a single product 187 (as identified by
TLC), which was purified and isolated twice by column chromatography using
DCM/MeOH (9:1) mobile phase. In CDCl3, the major features of the 1H NMR
spectrum of the hydrogenated adenosine analogue 187 was the appearance at
3.05-3.11 ppm a multiplet integrating for four protons corresponding to the two
methylene groups, as well as the signal for the methyl group from the acetal
protection appeared at the 1.37 and 1.58 ppm. Likewise, in the 13C NMR

160
spectrum, two methylene carbon signals appeared at 33.7 and 40.5 ppm. The
synthesis of the product 187 was supported by HRMS, which showed the
expected molecular ion of M+H at 453.2243 m/z, (EI+ HRMS showed a
deviation of 0.0007 ppm from the calc. mass of C23H29N6O4). The melting point
of 70–72 °C of the isolated compound 187 was sharp. The purity of the product
187 was found to be 98% using analytical HPLC in an 80% methanol/20%
water system.
Deprotection of the isopropylidene group from compound 187 was achieved
using 1 M HCl at 55–60 °C in 8 h and generated the novel target compound 2-
(2-phenylethyl)adenosine-5-N-ethylcarboxamide (188) in 94% yield (Scheme
4.3). The deprotection of compound 187 produced a single product 188 (as
identified by TLC). The Rf value of the product 188 was 0.69 in DCM/MeOH
(1:4), The product was purified and isolated by column chromatography using
100% chloroform as the mobile phase. In CDCl3, the major features of the 1H
NMR spectrum of deprotected adenosine analogue 188 were the
disappearance of the methyl signals from the acetal protection at the 1.37 and
1.58 ppm. Likewise, in the 13C NMR spectrum, two methylene carbon signals
disappeared at 25.2 and 26.9 ppm. The synthesis of the product 188 was
supported by HRMS, which showed the expected molecular ion of M+H at
413.2019 m/z, (EI+ HRMS showed a deviation of 0.0082 ppm from the calc.
mass of C20H25N6O4). The melting point of 216–217 °C (decomp). of the
isolated compound 188 was sharp. The purity of the product 188 was found to
be >99.9% using analytical HPLC in an 80% methanol/ 20% water system.

161
4.4 Results of Biological testing of adenosine analogues
A number of C2-modified nitroxide adenosine analogues were synthesised with
ethyl linker groups. The potency and agonist activity (rather than antagonists or
inverse agonists) of the adenosine analogues was assessed with a downstream
cAMP assay using transfected CHO lines. These cells overexpress one of the
human A1, A2A, A2B, and A3 adenosine receptor subtypes. The details about the
cAMP Accumulation Assay have recently been described.121 This assay was
carried out at Monash University in Melbourne.
The tetraethylisoindoline-2-yloxyl nitroxide adenosine analogue 184 possessing
a two carbon chain (ethyl) linker moiety showed less A2AAR affinity (184; pEC50
of 5.2 ± 0.2) than non-nitroxide species (188; pEC50 of 6.0 ± 0.1) when
compared to all other receptors subtypes. The compound 188 with ethylene
linked phenyl was prepared in this structural class of comppounds is to compare
the effect of radical and non-radical species on adenosine receptors. The
compound 180 with tetramethyl isoindoline nitroxide linker was not tested for
cAMP assay because it was synthesised after the last batch of compounds was
tested.
All compounds in the final series are less-potent A2AAR agonists with pEC50
values ranging from 5.2–6.0. However, compound 184 is a slightly higher
potent adenosine analogue for A2BAR with pEC50 of 7.0 ± 0.5) compared to
other receptor subtypes. The non-nitroxide adenosine analogue also showed

162
somewhat good selectivity for A1AR and A3AR (pEC50 of 7.1± 0.2 and 7.0± 0.2)
respectively, compared to other adenosine receptor subtypes.

163
Table 4.1: cAMP analysis of ethyl linked adenosine compounds (pEC50 and pIC50 values in nM.
No. Compound Structure pIC50 -
A1R
pEC50 -
A2AR
pEC50 -
A2BR
pIC50 -
A3R A2A/A1 A2A/A3
A2A/A2
B A2B/A1 A2B/A3
A2B/
A2A
184
6.0± 0.2 5.2± 0.2 7.0± 0.5 N.D.(< 5) 0.2 <2 <2 10 100 63
188
7.1± 0.2 6.0± 0.1 6.3± 0.6 7.0 ± 0.2 0.08 0.1 0.1
A1/A2B
13
A3/A2B
10
-

164
Specifically, radical species (TEIO nitroxide) possessing adenosine analogue
184 with an ethyl linker moiety has been demonstrated to have 63 fold better
selectivity at the adenosine A2BAR over the adenosine A2AAR subtype.
However, it was 10 and 100 fold selective for the A1AR and A3AR subtype
respectively. Non-radical, non-nitroxide species (phenyl group) possessing
ethyl linked adenosine derivative 188, exhibited slightly increased selectivity
towards the adenosine A1AR and A3AR subtypes (10–13 fold), over the
adenosine A2AAR and A2BAR subtypes.
However, it was confirmed that the nitroxide possessing adenosine compounds
with ethyl linkers also have the similar receptor subtype affinity and selectivity at
A2BAR as the ethyne linked nitroxide adenosine compounds from the previous
chapter. In addition, it was also confirmed that flexibility of the linker chain has
no significant impact on receptor-binding .

165
Conclusion 5

166
5.1 Synthetic chemistry
This project focused on the design, synthesis and biological evaluation of novel
adenosine compounds that incorporate radical antioxidant species. These
hybrid analogues are designed to act potentially as selective adenosine
receptor agonists. They are specifically designed to target the A2A adenosine
receptors. The A2A adenosine receptors are one of the primary drug targets in
the management of ischaemia reperfusion injury. It has previously been
reported that having a potent antioxidant molecule within a molecular framework
of A2AAR agonists significantly increases their therapeutic efficacy. Therefore,
this work was based on linking/combining antioxidant molecules with known
adenosine receptor agonists. The main classes of antioxidant molecules
explored are nitroxides and phenolic compounds. These compounds were
linked via enzymatically cleavable (esters, amides) and non-clevable
(aminoethylaniline, ethynyl, ethyl) bonds.
Facilitating this approach, three structural classes of adenosine analogues were
designed and synthesised using a linear methodology. In total, twelve novel
hybrid adenosine compounds were synthesised in nine to twelve synthetic
steps. In eleven of those new adenosine analogues, the antioxidant moieties
were incorporated at the C2-position of the adenosine skeleton. These
analogues were designed to act as bifunctional ligands at the adenosine A2AAR
subtype. In the twelveth adenosine compound, the C2-position was modified
with a phenyl group instead of an antioxidant. This was intended to act as a
control to compare the effect of the presence of an antioxidant moiety.

167
5.1.1 Adenosine based antioxidant compounds with para-
aminoethylaniline linkers
In the first set of antioxidant-linked adenosine analogues, the C2-position of the
adenosine intermediate 146 was functionalised with a series of sterically
hindered antioxidant nitroxides via a para-aminoethylaniline linker. Different
ring classes of nitroxides, having tetramethyl substitution predominantly at the
α-carbon (some with tetraethyl substituent), were used. Adenosine intermediate
146 was synthesised in high yield from commercially available guanosine 138 in
seven steps. The key step in the synthesis of adenosine N-ethylcarboxamide
(NECA) 146 involves the generation of 143 which was achieved through the
synthesis of a novel guanosine-5-ethyl ester analogue 142. The synthesis of
143 was accomplished successfully in high yield by the esterification of
appropriately substituted guanosine-5-carboxylic acid 140.
Addition rate and addition sequence of the reagents and reactants make a
significant difference in the time span in completion of the amide coupling
reaction. The amide coupling reaction was complete in 21 h compared to 48 h,
when the carboxy nitroxide reactant was added after the addition of the other
coupling reagents to give the C2-substituted adenosine analogue in reduced
time.
The deprotection of the isopropylidene group was carried out using dilute
hydrochloric acid (HCl). Using this easier and cheaper deprotection reagent
gave better yields and compared to the reported resin mediated isopropylidene

168
deprotection.323 However, the isopropylidene deprotection of adenosine
derivatives with dilute HCl gave the deprotected adenosine analogues in (50–
71%) moderate yields. These moderate yields were attributed to the low
solubility of the deprotected adenosine analogues. The synthesis of the target
adenosine analogues was achieved in a with ten step reaction scheme starting
from guanosine and achieved in an overall yield of 9–12%.
Purification and isolation of the novel antioxidant possessing aminoethylaniline
linked adenosine analogues was found to be difficult using silica gel column
chromatography. The product generated has an Rf value close to the starting
materials and a byproduct. The use of medium pressure chromatography
(MPLC) with a reverse phase column was found to be a better technique to
purify those components. All the adenosine were analogues generated in >
98% pure and all the novel target adenosine analogues were isolated with >
99.9% purity (as analysed by HPLC).
5.1.2 Adenosine based antioxidant compounds with ethynyl linkers
In a second structural class of adenosine compounds, two novel alkynyl
(ethynyl) linked adenosine analogues were successfully synthesised
possessing sterically bulky isoindoline nitroxide substituents attached at the C2-
position of the adenosine skeleton. In this synthesis, the key step was the
formation of the ethynyl coupled product between the 2-iodo adenosine 149 and
the appropriate terminal alkyne functionality of 120 and 122 and 123. This
synthesis was achieved using the Sonogshira coupling procedure under mild
reaction conditions. The ethynyl linked adenosine product was successfully

169
synthesised in good overall yields of 53% and by 58% respectively. All the
novel target adenosine analogues were isolated in this class were > 99.9%
purity (as analysed by HPLC).
5.1.3 Adenosine based antioxidant compounds with ethyl linkers
In a third structural class of adenosine compound, three novel adenosine
analogues were successfully synthesised possessing ethyl linked sterically
bulky isoindoline nitroxides and a phenyl group substituents at the C2-position
of the adenosine skeleton. In this project, a four-stage reaction scheme was
employed to synthesise the target adenosine analogues. The isoindoline
nitroxide possessing ethynyl linked adenosine analogues 177 and 181 and 185
(from the chapter 3) were the key materials used as starting compounds. The
target adenosine analogues were achieved in excellent overall yields of 62%
and 64% respectively. In addition, a single non-nitroxide ethyl linked adenosine
derivative was synthesised using a three-step reaction scheme from the iodo
adenosine intermediate 149 in a high overall yield of 74%. All the novel target
adenosine analogues isolated in this class were > 99.9% purity (as analysed by
HPLC).

170
5.2 Biological Testing
In this project, three structural classes comprising of twelve novel hybrid
adenosine compounds were synthesised. In eleven of those novel compounds,
an antioxidant functionality was incorporated into the adenosine framework to
generate the bifunctional compounds. Each structural class of adenosine
analogue was tested for the adenosine agonist activity and selectivity at A1AR,
A2AAR, A2BAR, and A3AR. All three structural classes of adenosine analogues
were found to act as adenosine agonists to all adenosine receptor subtypes.
5.2.1 Adenosine based antioxidant compounds with para-
aminoethylaniline linkers
In this work, seven novel antioxidants possessing aminoethylaniline linked
adenosine analogues were found to be selective as the human adenosine
receptor agonists. Specifically, these adenosine analogues are acted as
selective bifunctional ligands at the adenosine A2AAR subtype. The adenosine
A2AARs are one of the primary drug targets in the pathophysiology of
cardiovascular diseases. The activation of adenosine A2AAR is required to
achieve higher cardioprotection via ischemic preconditioning. In addition, these
receptor subtype are strongly involved in anti-ischemic action to avoid damage
induced by reperfusion injury. In this regard, all these synthesised adenosine
analogues were shown to have very promising characteristics and may be
implicated in potential cardiac treatments.
The nitroxide radical functionality and the ring size of the cyclic nitroxide moiety
have no significant difference in the receptor-binding ability at any of the four

171
adenosine receptor subtypes. From this, it can be postulated that the nitroxide
functionality of the five compounds did not interact in any great extent within the
binding pocket of the adenosine receptors.
However, all these adenosine derivatives retained more than 200-fold selectivity
to adenosine A2AAR over the A1AR subtype. Specifically, the adenosine
analogue possessing PROXYL nitroxide (166) and TEIO nitroxide (170)
substituents being displayed more than 800 fold selectivity for the A2AAR over
the A1AR. These compounds have low selectivity versus A2BAR (3 and 10 fold).
In addition, these analogues demonstrate modest selectivity for A2AAR over the
A3AR (63 and three fold). Therefore, it is beneficial to implicate some of these
adenosine A2AAR selective analogues in further ischemic evaluations.
All the adenosine analogues from this first structural class were analysed in the
further cardioprotection testing by using cell-based simulated ischemia assay.
All compounds have demonstrated positive control in the study of an ischaemia
cell protection assay. The test indicates that, the positive cardioprotective effect
is primarily mediated by A2AAR activation. Furthermore, additional
cardioprotective effects were observed at the highest concentration is may be
due to the contribution of antioxidant activity. However, it can be said with
confidence that the incorporation of antioxidant functionality does not have a
measurable adverse effect on selectivity as adenosine A2AAR agonists. It is
reemphasized here that selectivity at the receptor subtype is an equally
important characteristic than compounds potency. This feature justifies the

172
original hypothesis that the C2-binding domain can accommodate sufficient
structural variations. This property makes feasible the design and application of
bifunctional compounds of this nature in the cardioprotection.
5.2.2 Adenosine based antioxidant compounds with ethynyl linkers
Compounds from a second structural class include antioxidant linked adenosine
analogues with ethynyl linkers. These adenosine analogues demonstrated no
significant impact on selectivity at adenosine A2AAR. However, these
analogues show slightly higher selectivity for the adenosine A2BAR over the
other adenosine receptor subtypes. This characteristic activity of these
analogues as a selective adenosine A2BAR agonists may be implicated in their
therapeutic applications.
5.2.3 Adenosine based antioxidant compounds with ethyl linkers
Compounds from a third structural class include two adenosine analogues
possessing various antioxidant isoindoline nitroxides linked with two carbon
chain (ethyl) linker moieties. Similarly, to the second class nitroxide adenosine
compounds, these nitroxide compounds exhibit high selectivity at A2BAR over
the other receptor subtypes. However, no significant selectivity at A2AAR was
observed.
However, non-nitroxide phenyl group possessing ethyl linked adenosine
derivative 188, exhibited slightly increased selectivity towards the adenosine
A1AR and A3AR subtypes over the adenosine A2AAR and A2BAR subtypes.
There is no significant difference in selectivities of non-nitroxide adenosine

173
analogues with an ethynyl or ethyl linkers, all are selective at adenosine A1AR
and A3AR subtypes.
In this structural class of compounds, the comparison between adenosine
compounds possessing antioxidant groups with non-antioxidant groups was
found to have significant effect on the selectivity of particular adenosine
receptor subtypes. The adenosine compound possessing antioxidant groups
has higher selectivity at the adenosine A2BAR subtype. However, the
adenosine compound possessing non-antioxidant groups has greater selectivity
at the A1AR and A3AR subtypes compared to other receptor subtypes. In this
regard, the characteristics of this adenosine analogues possessing nitroxide
antioxidant moiety as A2BAR selective analogues may be implicated in the
further testing in the potential treatment as coronary vasodilator or
antihypertensive agents.

174
Future work 6

175
No further pharmacological testing of the adenosine compounds was able to be
conducted within the time frame of the project and this remains an missing
piece of this work. While the functional data has confirmed that the compounds
possess agonists function, testing for compound antioxidant efficacy remains to
be performed.
The functional efficacy of the antioxidant combined adenosine analogues would
be more challenging to effectively measure. The work described in this thesis
has been an initial exploration into the concept of ligands for selective
adenosine receptors that also possess an antioxidant species. No further
pharmacological testing was conducted within the time frame of this project.
Although this research has resulted in the synthesis of compounds that are
potent adenosine agonists ligands, there are three main areas that can be
identified where there is opportunity for further development of the concept of
dual action adenosine receptor ligands. These areas are i) Synthesis of novel
adenosine compounds that are designed to be selective adenosine A2AAR
agonists with propynyl linkers linked to nitroxide antioxidant capabilities. This
joining of three carbon distance with a triple bond (alkyne) compounds
increases the selectivity for A2AAR and antioxidant function will also have
additional effects. ii) Synthesis of tetraethyl modified smaller ring nitroxides with
aminoethylaniline linked adenosine compounds will increase the selectivity for
A2AAR. The nitroxide functionality will be blocked at some extent by more
hindered tetraethyl group. This phenomenon will achieve better
cardioprotection. iii) More extensive biological evaluation of the novel
compounds synthesised in this work remained to be performed, as the

176
functional data have confirmed that the compounds possess agonist function,
testing for compound antioxidant efficacy remains to be performed.
More extensive biological testing of the novel adenosine derivatives described
in this work is required in order to measure their potential as cardioprotective
treatments. Although the antioxidant activity of both BHT and TMIO and other
nitroxide functional groups are mentioned in the literature, the novel compounds
synthesised in this work have not yet been tested for the comprehensive
assessment of antioxidant activity. In vitro, evaluation of the antioxidant
capabilities of these antioxidant coupled adenosine analogues should be
performed. In addition, the further biological assay should be performed in a
more complex biological system such as isolated perfused rat hearts
(Langendorff heart assay) and live animal models which stimulate heart attack
and ischaemia-reperfusion injury. However, only after the biological evaluation
of these compounds in a more complex system, their viability can be assessed
as a potential cardioprotective treatment.

177
Experimental work 7

178
7.1 General Experimental
All starting materials and required chemicals were purchased from Sigma-
Aldrich with purities of more than 95% and all solvents used were of analytical
reagent (AR) grade except for HPLC. Solvents were dried according the
appropriate drying procedure under an argon atmosphere. Solvents used such
as acetone, tetrahydrofuran (THF), Dichloromethane (DCM), Chloroform
(CHCl3), and methanol (MeOH), N, N-dimethylformamide (DMF) for air and
moisture sensitive reactions were distilled under an argon atmosphere. The
solvents such as diethyl ether (Et2O), and toluene were dried under sodium wire
and acetonitrile (MeCN) was dried under predried molecular sieves and used
absolute ethanol (EtOH) for moisture sensitive reactions. Demineralised (DM)
water was used for all reaction work-ups. Thin layer chromatography was
performed on aluminium backed silica F254 plates with UV light (254 nm) for
chemical compound detection. Column chromatography was performed using
kieselgel 60 silica gel (200- 300 mesh) with eluents.
Nuclear magnetic resonance spectra were recorded on a Bruker Advance 300
WB (1H at 400 MHz and 13C at 75 MHz) or a Varian (1H NMR at 400 MHz and
13C NMR at 101 MHz) FT- NMR spectrometer instrument. NMR spectra are
reported based on integration, chemical shift (δ ppm) and multiplicity (s =
singlet, d = doublet, dd = doublets of doublet, t = triplet, q = quartet, m =
multiplet, br = broad, coupling constant and assignment. 13C NMR spectra are
reported according to the chemical shift, multiplicity, coupling constant (if
appropriate) and assignment. Deuterated solvents acetone (1H NMR 2.05/13C

179
NMR 29.84 ppm), chloroform (1H NMR 7.26/13C NMR 77.16 ppm); DMSO (1H
NMR 2.50/13C NMR 39.53 ppm) and methanol (1H NMR 3.31/13C NMR 49.00
ppm) was used for recording nuclear magnetic resonance spectra with their
respective solvent residual peaks.
High resolution mass analysis were performed on the Micromass platform II
single quadrupole mass spectrometer equipped with the dual electron spray ion
source. Melting points were measured using a Gallenkamp melting point
apparatus with a calibrated thermometer. All the target final compounds (161-
174 and 177-188) were purified using a medium pressure liquid
chromatography (MPLC) instrument using filtered HPLC grade methanol and
HPLC grade water using C18 reverse phase column with 1 mL/ minute flow rate
of mobile phase and 264 nM UV detector. The purities of novel compounds
(161-174 and 177-188) were analysed using analytical high performance liquid
chromatography (HPLC) using a C18 reverse phase column with a 1 mL/minute
flow rate with various solvent (methanol/water) ratios of mobile phase and 264
nM UV detector.

180
7.2 Synthetic procedure
7.2.1 Synthesis of 2-benzyl phthalimide (100).
A suspension of phthalic anhydride (98) (20 g, 135 mmol, 1 equiv) in acetic acid
(93.5 mL) was treated with benzylamine (99) (22.42 mL, 205 mmol, 1.52 equiv).
The reaction mixture was heated at 120 °C and stirred for 1 h. The resulting
solution was poured onto an ice/ water mixture (150 mL) and collect the white
precipitate by filteration. The solid was recrystallised from ethanol to give 2-
benzyl phthalimide (100) as a white solid (26.5 g, 83%). Rf = 0.83 (EtOAc /
Hexane, 1:1).
1H NMR (400 MHz, CDCl3) δ ppm: 4.85 (s, 2 H) 7.29 - 7.35 (m, 3 H) 7.43 (d,
J=6.99 Hz, 2 H) 7.67 - 7.72 (m, 2 H) 7.84 (dd, J=5.40, 3.06 Hz, 2 H).
13C NMR (101MHz, CDCl3) δ ppm: 41.6, 123.3, 127.8, 128.6, 128.7, 132.1,
134.0, 136.3, 168.1. The obtained spectroscopic data was consistent with that
previously reported.324
Melting point: 116–118 °C (lit.325 116 °C).
HRMS (ESI): m/z calcd for C15H11NNaO2+ [M+Na]: 260.0687, found: 260.0813

181
7.2.2 Synthesis of 2-benzyl-1,1,3,3-tetramethylisoindoline (101).
A solution of compound 100 (40 g,168 mmol,1.0 equiv) in anhydrous toluene
(320 mL) was treated with methyl magnesium iodide [freshly prepared from
methyl iodide (62.75 mL,1008 mmol, 6.0 equiv) and pre-dried magnesium
turnings (33 g, 1348 mmol, 8.0 equiv) in anhydrous Et2O (400 mL)]. The Et2O
was distilled off via a Dean-stark. The reaction mixture was heated to reflux,
stirred for 3 h and then concentrated to half its volume. Hexane (600 mL) was
added, and the resulting mixture was filtered through celite and washed
thoroughly with extra hexane (600 mL). The filtrate was passed through a
column of basic alumina and evaporated under reduced pressure to give a
colourless oil that solidified at ambient temperature. This solid was
recrystallised in MeOH to give 2-benzyl-1,1,3,3-tetramethylisoindoline (101) as
a white crystalline solid (14 g, 31%), Rf = 0.9 (EtOAc / Hexane, 1:1).
1H NMR (400 MHz, CDCl3) δ ppm: 1.33 (s, 12 H) 4.02 (s, 2 H) 7.16 (dd, J=5.52,
3.17 Hz, 2 H) 7.26 (br. s., 2 H) 7.27 - 7.34 (m, 3 H) 7.49 (d, J=7.23 Hz, 2 H).
13C NMR (101 MHz, CDCl3) δ ppm: 28.4, 46.3, 65.2, 120.6, 122.1, 126.0, 127.2,
127.5, 128.6, 129.0, 143.5 and 147.9. The obtained spectroscopic data was
consistent with that previously reported.76
Melting point: 63–65 °C (lit.76 63–64 °C).

182
HRMS (ESI): m/z calcd for C19H24N+ [M+H]: 266.1909, found: 266.1936.
7.2.3 Synthesis of 5-bromo-1,1,3,3-tetramethylisoindoline (102)275
A solution of compound 101 (5 g, 18.8 mmol, 1 equiv) in DCM (30 mL) under an
atmosphere of argon was cooled to 0 °C in an ice bath. A solution of liquid Br2
(2.15 mL, 41.47 mmol, 2.2 equiv) in DCM (42 mL) was cautiously added
followed by the immediate addition of anhydrous AlCl3 (9.5 g, 65.8 mmol, 3.5
equiv). The reaction was stirred at 0 °C for 1 h and then poured onto ice (200
mL) and stirred vigorously for 30 minutes after which time the solution was
basified with 10 M NaOH solution up to pH 14 and extracted with DCM (3 × 50
mL). The combined DCM layers were washed with brine (3 × 50 mL) and
evaporated under reduced pressure to give 2, 5-dibromoisoindoline as a yellow
oily residue.
The residue was dissolved in methanol (30 mL), and NaHCO3 (280 mg) was
added to the solution. Aqueous H2O2 (30% aqueous solution, 15 mL) was then
added slowly until no further effervescence could be detected (ensuring that
some NaHCO3 remained). The mixture was acidified with 2 M H2SO4 aqueous
solution (up to pH 1) and extracted with DCM (3 × 50 mL) to remove the
benzaldehyde by-product. The combined organic layers were back extracted
with 2 M H2SO4 to recover any removed product. The combined aqueous
phases were washed with DCM (3 × 50 mL), after which time they were cooled

183
in ice bath, basified with 10 M NaOH aqueous solution (up to pH 14) and
extracted with DCM (3 × 50 mL). The combined organic phases were washed
with brine (3 × 50 mL) and dried (over anhydrous Na2SO4). The solvent was
removed under reduced pressure to give a golden oil which immediately
crystallised to give yellowish-white solid, which was purified by silica gel column
chromatography using a mixture of DCM: MeOH (8:2) to give 5-bromo-1,1,3,3-
tetramethylisoindoline (102) as a pale yellow solid (3.15 g, 64.5%). Rf = 0.65
(EtOAc: Hexane, 1:1).
1H NMR (400 MHz, CDCl3): δ ppm 1.45 (s, 6 H) 1.47 (s, 6 H) 1.80 (br s, 1 H)
7.00 (d, J=8.03 Hz, 2 H) 7.25 (d, J=1.58 Hz, 2 H) 7.35 - 7.39 (m, 3 H).
13C NMR (101 MHz, CDCl3): δ ppm 31.8, 62.7, 120.8, 123.1, 123.2, 124.7,
147.9, 151.3. (The obtained spectroscopic data was consistent with that
previously reported)276
Melting point: 58–60 °C (lit.276 58–60 °C).
HRMS (ESI): m/z calcd for C12H17NBr+ [M+H]: 254.0544 and 256.0524, found:
254.0568 and 256.0549.
7.2.4 Synthesis of 5-Bromo-1,1,3,3-tetramethylisoindoline-2-yloxyl
(103)276.

184
To a solution of compound 102 (1.0 g, 3.92 mmol, 1 equiv), NaHCO3 (400 mg,
4.312 mmol, 1.1 equiv) and Na2WO4.2 H2O (169 mg, 0.78 mmol, 0.2 equiv) in
methanol (15 mL) was added 30% H2O2 solution (2.7 mL, 28.6 mmol, 7.3
equiv). The reaction mixture was stirred for 24 h, after which time added a
second portion of NaHCO3 (400 mg, 4.312 mmol, 1.1 equiv), Na2WO4.2 H2O
(169 mg, 0.78 mmol, 0.2 equiv) and 30% H2O2 (2.7 mL, 28.6 mmol, 7.3 equiv)
were added and stirring was continued for 48 hours. Water (40 mL) was then
added into the reaction solution and the resulting mixture was extracted with
DCM (3× 40 mL). The combined organic phases were washed with 2 M HCl
solution (40 mL), brine solution (3 × 40 mL) and dried over anhydrous Na2SO4.
The solvent was concentrated under reduced pressure. The residue was
recrystallised (MeCN) to give 5-bromo-1, 1, 3, 3- tetramethylisoindoline-2-yloxyl
(103) as a yellowish crystalline solid (1.0 g, 95%), Rf = 0.01 (EtOAc / MeOH,
8:2).
Melting point: 108–110 °C (lit.276 109 °C).
HRMS (ESI): m/z calcd for C12H16NBrO+ [M+2H]: 270.0494, found: 270.0487.

185
7.2.5 Synthesis of 5-iodo-1,1,3,3- tetramethylisoindoline (104)276.
Compound 102 (300 mg, 1.176 mmol, 1 equiv) was dissolved in dry THF (3.4
mL) under an atmosphere of argon. The solution was cooled in a dry ice and
acetone bath at –78 °C. A solution of n-BuLi (1.6 M in hexanes, 1.95 mL, 3.22
mmol, 2.74 equiv) was added dropwise to the stirring mixture, followed by the
addition of a solution of iodine (0.9 g, 65.8 mmol, 6 equiv) in dry THF (7.5 mL).
The reaction mixture was cooled to room temperature and then quenched by
pouring onto an ice water mixture (150 mL). The mixture was basified with 5 M
NaOH solution (up to pH 14). The resulting mixture was extracted with DCM (3
× 25 mL), the combined organic phases were washed with brine (3 × 25 mL)
and dried over anhydrous Na2SO4 and the solvent was concentrated under
reduced pressure to give a yellow oily residue.
The residue was taken up in MeOH (7.5 mL) and NaHCO3 (37.5 mg) was
added, followed by the addition of 30% H2O2 solution, until no further
effervescences could be detected. The mixture was treated with 2 M H2SO4 (50
mL) and washed with DCM (3 × 25 mL). The combined organic layers were
back extracted with 2 M H2SO4 to recover any removed product. The combined
aqueous phases were washed with DCM (25 mL) after which time they were
cooled in ice bath and basified with 5 M NaOH solution (up to pH 14). The
resulting solution was extracted with DCM (3 × 25 mL). The combined organic
phases were washed with brine (3 × 25 mL), dried over anhydrous Na2SO4 and

186
the solvent was evaporated under reduced pressure to give a golden oil which
immediately crystallised to give 5-iodo -1, 1, 3, 3- tetramethylisoindoline (104)
as a pale yellow solid (243 mg, 69%). Rf = 0.67 (EtOAc / Hexane, 3:7).
1H NMR (400 MHz, CDCl3) δ ppm: 1.43 (s, 6 H) 1.44 (s, 6 H) 1.93 (br. s., 1 H)
6.88 (d, J=7.92 Hz, 1 H) 7.44 (d, J=1.03 Hz, 1 H) 7.56 (dd, J=7.92, 1.22 Hz, 1
H).
13C NMR (101 MHz, CDCl3) δ ppm: 31.7, 62.7, 92.2, 123.5, 130.8, 136.1, 148.6,
and 151.4. (The obtained spectroscopic data was consistent with that previously
reported)79
ESMS calcd for C12H17IN+ [M+H]: 302.0406, found: 302.0400.
7.2.6 Synthesis of 5-iodo-1,1,3,3-tetramethylisoindoline-2-yloxyl (105)79
To a stirring solution of compound 104 (180 mg, 0.59 mmol, 1 equiv) in MeOH
(2.7 mL), NaHCO3 (60 mg, 0.705 mmol, 1.195 equiv), Na2WO4.2H2O (25.4 mg,
0.705 mmol, 0.135 equiv) and 30% H2O2 (0.5 mL, 4.307 mmol, 7.3 equiv) was
added and the reaction mixture was stirred at room temperature for 24 h. A
second portion of NaHCO3 (60 mg, 0.705 mmol, 1.195 equiv), Na2WO4.2H2O
(25.4 mg, 0.705 mmol, 0.135 equiv) and 30% H2O2 solution (0.5 mL, 4.307
mmol, 7.3 equiv) was then added. After 48 h, water (30 mL) was added and the
resulting mixture was extracted with DCM (3 × 20 mL). The combined organic
phases were washed with 2 M HCl solution (25 mL) and brine solution (3 × 25

187
mL) and dried over anhydrous Na2SO4. The solvent was concentrated under
reduced pressure to give a yellow crystalline solid and which was recrystallised
(MeCN) to give 5-iodo-1,1,3,3-tetramethylisoindoline-2-yloxyl (105) as an
orange crystalline solid (150 mg, 79%). Rf = 0.71 (EtOAc / Hexane, 3:7).
Melting point: 133–135 °C (lit.79 133–135 °C).
ESMS calcd for C12H16INNaO+ [M+Na]: 339.0096, found: 339.0090.
7.2.7 Synthesis of 5-carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl
(106)275.
A solution of n-BuLi (1.6 M in hexanes, 1.95 mL, 3.22 mmol, 2.74 equiv ) was
added dropwise to a stirring solution of compound 102 (300 mg, 1.176 mmol, 1
equiv) in a freshly distilled dry THF (3.4 mL) at –78 °C under an atmosphere of
argon. After stirring for 10 min, the solution was added slowly to a mixture of
crushed dry ice (0.9 g, 65.8 mmol, 6 equiv) in freshly distilled dry THF (7.5 mL).
The reaction mixture was warmed to room temperature under an atmosphere of
argon and then concentrated under reduced pressure. 2 M HCl (15 mL) was
added to the resulting residue and the solution was washed with Et2O (3 × 10
mL). The combined organic phases were washed with 2 M HCl (15 mL). The
combined aqueous phases were neutralised with saturated Na2CO3 solution (15
mL) and extracted with Et2O (3 × 20 mL). The ether layers were washed with

188
water (3 × 20 mL) and brine (3 × 20 mL) and dried over anhydrous Na2SO4.
The solvent was evaporated under reduced pressure to give yellow oily residue.
The yellow oily residue was dissolved in MeOH (2 mL) at room temperature.
NaHCO3 (40 mg, 3.92 mmol, 1.1 equiv), Na2WO4.2H2O (24 mg, 0.78 mmol, 0.2
equiv) and 30% H2O2 solution (0.56 mL, 28.6 mmol, 7.3 equiv) were added and
the reaction mixture was stirred for 24 h. A second portion of NaHCO3 (40 mg,
3.92 mmol, 1.1 equiv), Na2WO4.2 H2O (24 mg, 0.78 mmol, 0.2 equiv) and 30%
H2O2 (0.56 mL, 28.6 mmol, 7.3 equiv) was added. The reaction mixture was
stirred again for 48 h, after which time water (30 mL) was added to the reaction
mixture and the resulting mixture was extracted with DCM (3× 40 mL). The
combined organic extracts were washed with 2 M HCl solution (40 mL) and
brine solution (40 mL) and dried over Na2SO4. The solvent was concentrated
under reduced pressure to give a yellow crystalline solid, which was
recrystallised (MeCN) to give 5-carboxy-1, 1, 3, 3- tetramethylisoindoline-2-
yloxyl (106) as a yellowish crystalline solid (140 mg, 76.5%). Rf = 0.01 (EtOAc /
MeOH, 8:2).
IR: 2977 (alkylCH3)), 1680 (C=O), 1423, 1357 (N-O), 1256, 1160, 908, 840,
774, 744.
Melting point: 216–216 °C (lit.275 214–218 °C).
ESMS calcd for C13H17 NO3+ [M+H+]: 235.1208, found: 235.1198.
EPR (methanol): typical 3-line nitroxide signal, g 1.9823, aN 1.5007 mT.

189
7.2.8 Synthesis of 2-benzyl-5-methylphthalimide(108)102.
Benzylamine (20.5 mL, 92.6 mmol, 1.52 equiv) was added to a stirring
suspension of 5-methylphthalic anhydride (107) (20.0 g, 123 mmol, 1 equiv) in
acetic acid (94 mL). The reaction mixture was refluxed at 120 °C for 1 h and
then poured onto ice/ water mixture (500 mL) and stirred vigorously for 30
minutes and the obtained white precipitate was filtered. The residue was
recrystallised from EtOH to give 2-benzyl-5-methylphthalimide (108) as a white
solid (24.8g, 80%) Rf = 0.83 (100% EtOAc),
1H NMR (400 MHz, CDCl3) δ ppm: 2.45 - 2.54 (m, 3 H) 4.83 (s, 2 H) 7.22 - 7.35
(m, 3 H) 7.39 - 7.51 (m, 3 H) 7.60 - 7.67 (m, 1 H) 7.67 - 7.78 (m, 1 H).
13C NMR (101 MHz, DMSO- d6) δ ppm: 22.0, 41.5, 123.3, 123.9, 127.7, 128.5,
128.6, 129.5, 132.9, 134.5, 136.8, 145.3, 168.1 and 168.2. (The obtained
spectroscopic data were consistent with that previously reported).102
Melting point:128–130 °C .(lit 102 128–130 °C)
HRMS (ESI): m/z calcd for C16H14NO2+ [M+H]: 252.1025, found: 252.1067.

190
7.2.9 Synthesis of 2-benzyl-5-methyl-1,1,3,3-tetraethylisoindoline(109)102.
A solution of compound 108 (10.0 g, 39.8 mmol, 1 equiv) in anhydrous toluene
(80 mL) was treated with ethyl magnesium iodide [freshly prepared from ethyl
iodide (19.2 mL, 239 mmol, 6.0 equiv) and pre-dried magnesium turnings (7.78
g, 318.4 mmol, 8.0 equiv.) in anhydrous Et2O (100 mL)]. The Et2O was distilled
off via dean-stark. The reaction mixture was heated to reflux, stirred for 3 h and
then concentrated to half its volume. Hexane (4 x 200 mL) was added to the
reaction mixture. The resulting mixture was filtered through celite and washed
thoroughly with extra hexane (600 mL). The extracted hexane was bubbled
overnight to dryness. The residue was dissolved in hexane (600 mL) and
passed through a column of basic alumina. The resulting filtrate was
evaporated under reduced pressure to give 2-benzyl-5-methyl-1,1,3,3-
tetraethylisoindoline (109) as a colourless oil (4.25 g, 32%). Rf = 0.92 (100%
EtOAc).
1H NMR (400 MHz, CDCl3) δ ppm: 0.78 (td, J=7.34, 2.93 Hz, 12 H) 1.53 (dd,
J=13.90, 7.30, 3.30 Hz, 4 H) 1.84 - 1.97 (m, 4 H) 2.38 (s, 3 H) 4.01 (s, 2 H) 6.87
(s, 1 H) 6.95 (d, J=7.78 Hz, 1 H) 7.00 - 7.06 (m, 1 H) 7.24 (br. s., 1 H) 7.27 -
7.34 (m, 2 H) 7.46 (d, J=7.04 Hz, 2 H).
13C NMR (75 MHz, CDCl3) δ: 9.6, 21.5, 30.3, 46.8, 71.0, 71.2, 123.2, 124.0,
125.3, 126.5, 127.7, 128.2, 129.1, 129.3, 135.0, 141.7, 142.5, and 144.8; (The

191
obtained spectroscopic data was consistent with that previously reported for this
compound).102
HRMS (ESI): m/z calcd for C24H34N+ [M+H]: 336.2691, found: 336.2677.
7.2.10 Synthesis of 5-methyl-1,1,3,3-tetraethylisoindoline (110).
To a solution of compound 109 (4.2 g, 12.5 mmol, 1.0 equiv) in acetic acid (100
mL) was added palladium (10% charcoal = 1.0 g). The reaction mixture was
shaken under hydrogen pressure (50 psi in Parr apparatus) at room
temperature for 3 h. The mixture was filtered through celite and concentrated
under reduced pressure. The resulting residue was dissolved in DCM (150 mL)
and the resulting solution was washed with saturated sodium bicarbonate
(Na2HCO3) solution (3 x 50 mL). The organic phase was dried over anhydrous
Na2SO4 and concentrated under reduced pressure to give 5-methyl-1,1,3,3-
tetraethylisoindoline (110) as a yellow orange oil (2.82 g, 92%). Rf = 0.92
(EtOAc: MeOH, 8:2).
1H NMR (400 MHz, CDCl3) δ ppm: 0.80 - 0.91 (m, 12 H) 1.59 - 1.78 (m, 8 H)
2.35 (s, 3 H) 3.49 (s, 1 H) 6.86 (s, 1 H) 6.91 - 6.97 (m, 1 H) 7.01 (s, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 9.1, 21.5, 33.9, 47.0, 68.1, 68.2, 122.3,
123.2, 127.5, 136.2, and 147.9. (The obtained spectroscopic data was
consistent with that previously reported for this compound)102.

192
HRMS (ESI): m/z calcd for C17H28N+ [M+H]: 246.2222, found: 246.2226.
7.2.11 Synthesis of 5-methyl-1,1,3,3-tetraethylisoindoline-2-yloxyl (111).
A solution of compound 110 (2.8 g,11.4 mmol, 1 equiv), NaHCO3 (1.04 g,125
mmol, 1.1 equiv) and Na2WO4.2 H2O (0.42 g,15.5 mmol, 1.15 equiv) in MeOH
(80 mL), was added dropwise 30% H2O2 solution (9.2 mL,105 mmol, 7.3 equiv
). The reaction mixture was stirred at room temperature for 24 h after which
time a second portion of NaHCO3 (1.04 g,125 mmol, 1.1 equiv) and Na2WO4.2
H2O (0.42 g,15.5 mmol, 1.15 equiv) and then 30% H2O2 solution (9.2 mL,105
mmol, 7.3 equiv) were added and the stirring was continued for an additional 48
h. Water (160 mL) was then added to a reaction mixture and the resulting
mixture was extracted in DCM (3 x 50 mL). The combined organic phases were
washed with 2 M H2SO4 solution (3 x 50 mL), brine (3 x 50 mL) solution and
dried over anhydrous Na2SO4, filtered. The solvent was evaporated under
reduced pressure. The resulting residue was purified by silica gel column
chromatography (eluent 100 % DCM) to give 5-methyl-1,1,3,3-
tetraethylisoindoline-2-yloxyl (111) as a yellowish orange oil (1.35 g, 47%). Rf =
0.53 (1:1 DCM/ Hexane).
HRMS (ESI): m/z calcd for C17H27NaN+ [M+Na]: 283.191, found: 283.1934. (The
obtained spectroscopic data was consistent with that previously reported for this
compound).102

193
7.2.12 Synthesis of 2-acetoxy-5-methyl-1,1,3,3-tetraethylisoindoline (112).
To a stirring solution of compound 111 (1.35 g, 5.17 mmol, 1 equiv) in a dry
THF (70 mL) was added palladium (10% charcoal, 137 mg). The reaction
mixture was stirred under balloon of hydrogen for 1 h. The reaction mixture was
cooled at 0 °C in ice bath and, triethylamine (1.44 mL, 10.34 mmol, 2 equiv) and
acetyl chloride (0.92 mL) were added. A mixture was allowed to warm up to 25
°C and stirred for 1 h. Argon gas bubbled over the mixture for 10 minutes and
the reaction mixture was filtered through celite and evaporated under reduced
pressure. Water was poured onto the residue and the resulting mixture was
extracted with EtOAc (3 x 50 mL). The organic extracts were dried over
anhydrous Na2SO4 and evaporated by distillation under reduced pressure. The
resulting residue was purified by silica gel column chromatography (eluent 1:1
DCM/ Hexane) to give 2-acetoxy-5-methyl-1,1,3,3-tetraethylisoindoline (112) as
a colourless oil which solidifies upon standing (1.55 g, 99%). Rf = 0.78 (DCM/
Hexane, 1:1).
1H NMR (400 MHz, CDCl3) δ ppm: 0.79 (br. s., 6 H) 0.95 (d, J=4.70 Hz, 6 H)
1.60 - 1.80 (m, 4 H) 1.83 - 2.02 (m, 4 H) 2.10 (s, 3 H) 2.36 (s, 3 H) 6.86 (s, 1 H)
6.94 (d, J=7.63 Hz, 1 H) 7.06 (d, J=7.48 Hz, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 8.7, 9.6, 19.6, 21.7, 29.0, 30.4, 73.7, 73.8,
123.5, 124.2, 127.6, 136.3, 138.8, 141.9, and 170.7. (The obtained

194
spectroscopic data was consistent with that previously reported for this
compound).102
Melting point: 78–80 °C (Lit102 76–78 °C).
HRMS (ESI): m/z calcd for C19H30NO2+ [M+H]: 304.2277, found: 304.2293.
7.2.13 Synthesis of 2-acetoxy-5-carboxy-1,1,3,3-tetraethylisoindoline(113).
A solution of compound 112 (1.55 g, 5.12 mmol, 1 equiv) in tert-butyl alcohol
(35 mL) was warmed to 40 °C. Anhydrous MgSO4 (0.62 g, 5.12 mmol, 1 equiv)
and potassium permanganate (0.4 M solution in water, 60 mL, 20.68 mmol, and
4.04 equiv) were added to the solution and heated to 70 °C for 24 h and the
reaction progress was followed by TLC analysis DCM: EtOAc (6:4). The
solution was cooled to 40 °C, was treated with 2-propanol (50 mL) and the
reaction mixture was stirred at room temperature for 16 h, after which time it
was filtered through celite. The filtrate was then evaporated to half its volume
and acidified with 2 M HCl solution (50 mL) up to pH 2. Water was added to the
mixture (50 mL) and extracted with Et2O (4 x 50 mL). The organic layers were
dried over anhydrous Na2SO4. The combined organic phases were evaporated
under reduced pressure. The resulting residue was purified by using silica gel
column chromatography (eluent DCM/ EtOAc, 6:4) to give 2-acetoxy-5-carboxy-
1,1,3,3-tetraethylisoindoline (113) as a white solid (1.32 g, 78%).

195
1H NMR (400 MHz, CDCl3) δ ppm: 0.80 (br. s., 6 H) 0.97 (br. s., 6 H) 1.57 - 1.85
(m, 4 H) 1.85 - 2.06 (m, 4 H) 2.12 (s, 3 H) 7.17 (d, J=8.07 Hz, 1 H) 7.80 (s, 1 H)
8.03 (d, J=7.92 Hz, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 8.5, 9.4, 19.3, 28.9, 30.2, 73.7, 74.0, 123.7,
125.4, 128.0, 128.9, 142.3, 148.1 , 170.3., 170.8 (The obtained spectroscopic
data was consistent with that previously reported for this compound).102
Melting point: 168–170 °C. (Lit102 168–170 °C).
HRMS (ESI): m/z calcd for C19H28NO4+ [M+H]: 334.2018, found: 334.2042.
7.2.14 Synthesis of 5-carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl (114).
Compound 113 (1.3 g, 3.91 mmol, 1 equiv) was suspended in water (26 mL)
and the mixture was cooled using ice/salt mixture up to 0–5 °C. Lithium
hydroxide (LiOH) (0.462 g, 19.47 mmol, 4.98 equiv) was added to the reaction
mixture and ice bath was removed. The mixture was stirred at room
temperature for 20 h and the reaction progress was followed by TLC analysis
EtOAc: MeOH (8:2) The resulting yellow solution was again cooled in ice bath
and acidified with 2 M HCl (50 mL) solution and extracted with Et2O (5 x 50 mL).
The combined ether layers were treated with lead oxide (PbO2) (0.462 g, 1.95
mmol, 0.5 equiv) and stirred for 30 minutes. The ether extract was dried using
anhydrous Na2SO4, filtered. The filtrate was evaporated under reduced

196
pressure to give yellow oil that solidifies upon standing, which was recrystallised
(MeCN) to give 5-carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl (114) as yellow
crystals. (1.02 g, 89%).
Melting point: 102–104 °C. (lit.102 97–99 °C)
HRMS (ESI): m/z calcd for C17H24NNaO3+ [M+Na]: 313.1654, found: 313.1680.
EPR (methanol): typical 3-line nitroxide signal, g 1.9928, aN 1.4418 mT.
7.2.15 Synthesis of 5-(3-hydroxy-3-methyl)butynyl-1,1,3,3-
tetramethylisoindoline-2-yloxyl) (119).
To a solution of compound 105 (770 mg, 2.435 mmol, 1 equiv) in an anhydrous
triethylamine (15 mL) was added in a stirring solution of Pd(OAc)2 (13 mg,
0.021 mmol, 0.1 equiv), and DABCO (820 mg, 7.31 mmol, 3 equiv) and 2-
methyl-3-butyne-2-ol (204). The argon was bubbled for 10 minutes in a mixture.
The reaction mixture was stirred at 80–85 °C for 3 days and after which time,
CHCl3 (50 mL) was added to the reaction mixture. The organic phase was
washed with water (3 x 50 mL) and brine solution (3 x 50 mL) and dried over
anhydrous Na2SO4. The solvent was evaporated under reduced pressure and
the resulting residue was purified by silica gel column chromatography using a
mixture of (1:9) EtOAc: Hexanes to give 5-(3-hydroxy-3-methyl)butynyl-1,1,3,3-

197
tetramethylisoindoline-2-yloxyl) (119) as a brown-oil (530 mg, 80 %), Rf = 0.71
(DCM/ Hexanes, 2:8).
HRMS (ESI): m/z calcd for C17H22NO2+ [M+]: 272.1651, found: 272.1654. (The
obtained spectroscopic data was consistent with that previously reported for this
compound).316
7.2.16 Synthesis of 5-ethynyl-1,1,3,3-tetramethylisoindoline-2-yloxyl (120).
To a solution of compound 119 (200 mg, 0.734 mmol, 1 equiv), in an anhydrous
MeCN (15 mL) added solid KOH (330 mg, 5.87 mmol, 8.0 equiv). The reaction
mixture was stirred at 80–85 °C for 4 h and after which time water (30 mL) was
added to the reaction mixture. The resulting mixture was extracted with EtOAc
(3 x 50 mL). The combined organic phases were washed with water (15 mL)
and brine solution (15 mL) and dried over an anhydrous Na2SO4, filtered. The
solvent was evaporated under reduced pressure and the resulting residue was
purified on silica gel column chromatography using a mixture of DCM: Hexanes
(9:1) to give 5-ethynyl-1,1,3,3-tetramethylisoindoline-2-yloxyl (120) as a
yellowish-purple oil (141 mg, 90 %). Rf = 0.71 (DCM/ Hexanes, 2:8).
Ʋmax (ATR-FTIR) cm-1: 3726 (aryl C-H), 3192 (ΞC-H), 3044, 2978 (alkyl CH3),
2166 (CΞC), 1487 (aryl C-C), and 1428 (N-O).
Melting point: 96–98 °C (lit).

198
HRMS (ESI): m/z calcd for C14H17NO+ [M+Na]: 237.1130, found: 237.1126.
(The obtained spectroscopic data was consistent with that previously reported
for this compound).316
EPR (methanol): typical 3-line nitroxide signal, g 1.9908, aN 1.1266 mT.
7.2.17 Synthesis of N-tert-butoxycarbonyl-2-(4-aminophenyl)ethylamine
(125)277, 326
A solution of 2-(4-aminophenyl)ethylamine (124) (550 mg, 4.04 mmol, 1 equiv)
in DCM (50 mL) was cooled to –5 °C and tert-butoxycarbonyl anhydride (882
mg, 4.04 mmol, 1 equiv) was slowly added. The reaction mixture was stirred for
100 minutes at –5 to –10 °C, and the reaction progress was followed by TLC
analysis using DCM / MeOH (9:1) and poured onto an ice/ water mixture. After
stirring vigorously for 30 minutes, the organic phase was separated and washed
with water (3 × 25 mL). The combined aqueous layers were then washed with
DCM (3 × 25 mL). The combined organic layers were washed with brine
solution (3 × 25 mL) and dried, over anhydrous MgSO4, filtered. The solvent
was evaporated under reduced pressure and the resulting residue purified by
silica gel column chromatography to give N-tert-butoxycarbonyl-2-(4-
aminophenyl)ethylamine (125) as an orange solid (847 mg, 88.7%). Rf = 0.71
(DCM / MeOH, 9:1).
Melting point: 56–58 °C (lit.121 55–60 °C).

199
1H NMR (400 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.95 - 3.07 (m, 2 H) 3.17 (d,
J=4.36 Hz, 2 H) 4.12 (d, J=5.06 Hz, 1 H) 4.85 (s, 2 H) 6.47 (d, J=8.33 Hz, 2 H)
6.82 (d, J=8.25 Hz, 2 H).
13C NMR (101 MHz, CDCl3) δ ppm: 28.4, 35.2, 41.9, 79.1, 115.3, 128.8, 129.6,
144.7, and 155.9.
Ʋmax (ATR-FTIR) cm-1: 3369 (NH2), 3188 (aryl C-H), 2976 (alkyl CH3), 1678
(NC=OO), 1511 (aryl C-C), 1362 (O-C(CH3)3), 1244 (H-NCOO),
ESMS calcd for C13H20NaN2O2+ [M + Na]: 259.1422, found: 259.1433.
7.2.18 Synthesis of N-tert-butoxycarbonyl-2-(4-N-(5-carboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl)aminophenyl)ethylamine (126).
Compound 125 (244 mg, 1.03 mmol, 1.21 equiv) was added to a solution
containing 5-carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl (106) (200 mg,
0.853 mmol, 1 equiv), EDCI (174 mg, 0.904 mmol, 1.06 equiv), HOBt (126 mg,
0.929 mmol, 1.09 equiv), and DIPEA (160 µL, 0.895 mmol, 1.05 equiv) in dry
DMF (10 mL). The reaction mixture was stirred at room temperature for 16 h
and the reaction progress was followed by TLC analysis using (DCM / EtOAc,
7:3). The solution was quenched using water (30 mL) at 0–5 °C and the oily
reaction mixture was extracted in EtOAc (3 × 25 mL). The combined extracts
were washed with water (30 mL) and brine solution (30 mL) and dried over

200
anhydrous MgSO4. The solvent was evaporated under reduced pressure and
the resulting residue purified by silica gel column chromatography using a (8:2)
mixture of EtOAc and petroleum ether to give N-tert-butoxycarbonyl-2-(4-N-(5-
carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl)aminophenyl)ethylamine (126)
as a brownish-white solid (220 mg, 57%). Rf = 0.64 (DCM / EtOAc, 7:3).
1H NMR (400 MHz, CDCl3) δ ppm 1.47 (br. s., 9 H) 1.57 (br. s., 12 H) 2.82 (br.
s., 3 H) 3.42 (br. s., 4 H) 4.55 (br. s., 1 H) 7.62 (br. s., 3 H) 7.79 (br. s., 1 H)
Note that paramagnetic broadening by the nitroxide radical results in some
peaks not being detected or giving poor integration in the proton NMR
spectrum.
Melting point: 158–160 °C.
ESMS calcd for C26H34NaN3O4+ [M+Na]: 475.2440, found: 475.2450.
Elemental analysis: found C 68.58, H 7.51, N 8.88, expected C 69.00, H 7.57,
and N 9.28.
HPLC: 91%
Ʋmax (ATR-FTIR) cm-1: 3360 (NH), 3289 (NH), 2969 (alkylCH3); 2929 (aryl CH),
1689 (NC=O), 1662, 1599 (amide C=O), 1514 (H-NCOO), 1359 (O-C(CH3)3),
1319 and 1356 (NO), 1276-1248 (N-CO-O), 1168 (H-NCOO).
EPR (methanol): typical 3-line nitroxide signal g 1.9922, aN 1.4875 mT.

201
7.2.19 Synthesis of 2-(4-N-(5-carboxy-1,1,3,3-tetramethylisoindoline-2-
yloxyl) aminophenyl) ethylamine (127).
4 M HCl (0.75 mL) was added to a solution of compound 126 (100 mg, 0.94
mmol, 1.0 equiv) in a (1:1) mixture of 1, 4–dioxane (4.3 mL) and water (4.3 mL).
The reaction mixture was stirred for 2 h at 55–60 °C and the reaction progress
was followed by TLC analysis using DCM / EtOAc (7:3). Upon completion, the
solution was evaporated to half its volume under reduced pressure. Water (30
mL) was added and the resulting solution was basified with 28% aqueous
ammonia solution and extracted with EtOAc (3 x 25 mL). The combined
organic layers were washed with water (30 mL) and brine solution (30 mL), and
dried over anhydrous MgSO4, filtered. The filtrate was concentrated under
reduced pressure and the resulting residue purified by silica gel column
chromatography using a (8.9:1:0.1) mixture of CHCl3: methanol: 28% aqueous
ammonia solution to give (127) as a yellowish solid (95 mg, 91%), Rf = 0.26
(DCM / EtOAc, 7:3).
1H NMR (400 MHz, CDCl3) δ ppm 0.79 - 0.97 (m, 2 H) 1.28 (br. s., 13 H) 1.38 -
1.63 (m, 12 H) 2.06 (s, 2 H) 2.62 (s, 1 H) 2.68 - 2.86 (m, 3 H) 3.17 (br. s., 1 H)
3.81 (br. s., 2 H) 4.14 (d, J=6.90 Hz, 1 H) 7.21 (br. s., 3 H) 7.61 (br. s., 4 H).
Note that paramagnetic broadening by the nitroxide radical results in some

202
peaks not being detected or giving poor integration in the proton NMR
spectrum.
Ʋmax (ATR-FTIR) cm-1: 3285 (NH2), 2972 (alkylCH3); 2921(aryl CH), 1654
(C=O), 1598 and 1574 (H-NCO), 1514 (aryl C-C), 1356, and 1316 and (NO).
Melting point: 176–178 °C.
ESMS calcd for C21H27N3O2+ [M + H]: 353.2100, found: 353.2110.
EPR (methanol): typical 3-line nitroxide signal, g 1.9924, aN 1.4738 mT..
HPLC: 97%
7.2.20 Synthesis of (E)-tert-4-(3-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamide)phenethylcarbamate (132).
3, 5-di tert-butyl-4-hydroxycinnamic acid (118) (135 mg, 0.486 mmol, 1.15
equiv) was added to a mixture of EDCI (93.5 mg, 0.486 mmol, 1.15 equiv),
HOBt (65.6 mg, 0.486 mmol, 1.15 equiv), DIPEA (85 µL, 0.486 mmol, 1.15
equiv), and compound 125 (100 mg, 0.423 mmol, 1 equiv) in anhydrous DMF (2
mL). The reaction mixture was stirred to room temperature for 21 h and the
reaction progress was followed by TLC analysis in IPA/ MeOH (8:2). DCM was
added in to the reaction mixture and the solution was quenched by using water
(30 mL) at 0–5 °C. The resulting mixture was extracted in DCM (3 × 25 mL).

203
The combined extracts were washed with water (3 × 25 mL) and brine solution
(2 × 25 mL) and dried over anhydrous MgSO4, filtered. The solvent was
evaporated under reduced pressure and the oily residue was purified by silica
gel column chromatography using a (8:2) mixture of isopropanol: methanol to
give (E)-tert-4-(3-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamide)phenethylcarbamate (132) as a brownish-oil (208
mg, 99.5 %), Rf = 0.78 (CHCl3/ MeOH, 8:2),
1H NMR (400 MHz, CDCl3) δ ppm: 1.40 - 1.50 (m, 18 H) 1.60 (br. s., 9H) 2.73 -
2.81 (m, 2 H) 3.36 (d, J=5.53 Hz, 2 H) 5.30 (s, 1 H) 5.49 (s, 1 H) 6.39 (d,
J=15.41 Hz, 1 H) 7.17 (d, J=8.27 Hz, 2 H) 7.29 (s, 1 H) 7.39 (s, 2 H) 7.55 (d,
J=8.17 Hz, 1 H) 7.70 (d, J=15.41 Hz, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 28.4, 30.2, 34.3, 35.6, 41.8, 117.3, 120.1,
125.3, 125.9, 129.4, 136.3, 143.4, and 155.9.
Ʋmax (ATR-FTIR) cm-1: 3624 (NH2), 3282 (OH), 2956 (alkyl CH3), 1690 (NC=O),
1619 (C=C), 1511, 1425 (aryl C-C), 1390, 1365 (tBu-O), 1238 (H-NCO), 1168,
978, 849.
Melting Point: 126–128°C.
HRMS (ESI): m/z calcd for C30H42NaN2O4+ [M+Na]: 517.3042, found: 517.3006.

204
7.2.21 Synthesis of (E)-N-(4-(2-aminoethyl)phenyl)-3-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamide (133).
4 M HCl (2 mL) was slowly added to a stirring solution of compound 33 (205
mg, 0.414 mmol, 1 equiv) in a (1:1) mixture of 1, 4-dioxane (9 mL) and water (9
mL). The solution was stirred for 2 h at 55–60 °C and the reaction progress
was followed by TLC analysis in CHCl3/ MeOH (8:2). Upon completion, the
solution was treated with water (25 mL) and basified up to pH 14 by using 28%
aqueous ammonia solution. The resulting mixture was extracted with EtOAc (3
x 25 mL). The combined organic phases were washed with water (3 x 25 mL)
and brine solution (2 x 25 mL) and dried over anhydrous Na2SO4, filtered. The
filtrate was evaporated under reduced pressure and the resulting residue was
purified by silica gel column chromatography using a mixture of CHCl3: MeOH:
28% aqueous ammonia (7.9:2:0.1) solution to give (E)-N-(4-(2-
aminoethyl)phenyl)-3-(3,5-di-tert-butyl-4-hydroxyphenyl)acrylamide (133) as a
yellowish-green solid (105 mg, 65%), Rf = 0.13 (CHCl3/ MeOH, 8:2).
1H NMR (400 MHz, CDCl3) δ ppm: 1.43 (s, 18 H) 2.70 - 2.74 (m, 2 H) 2.95 (br.
s., 2 H) 5.30 (s, 1 H) 6.48 (d, J=15.41 Hz, 1 H) 7.11 (d, J=7.24 Hz, 2 H) 7.37 (s,
2 H) 7.49 (d, J=6.94 Hz, 2 H) 7.70 (d, J=15.36 Hz, 1 H).

205
13C NMR (101 MHz, CDCl3): δ ppm: 30.1, 34.3, 55.9, 63.9, 125.0, 125.3, 126.0,
129.2, 136.2, 136.6 137.9, 143.2 146.2, 153.5 and 155.8.
Ʋmax (ATR-FTIR) cm-1: 3624 (NH2), 2955 (alkyl CH3), 1660 (NC=O), 1617
(C=C), 1513, 1421 (aryl C-C), 1237 (H-NCO), 1174, 979, 848, 771, 565, 508.
Melting point: 130–132 °C.
HRMS (ESI): m/z calcd for C25H35N2O2+ [M+H]: 395.2699, found: 395.2651.
7.2.22 Synthesis of 2,3-O-isopropylideneguanosine (139).288
Guanosine 138 (10.00 g, 35.3 mmol, 1.0 equiv) and para-toluenesulfonic acid
monohydrate (6.72 g, 35.3 mmol, 1.0 equiv) were added to a stirring solution of
acetone (350 mL) and 2, 2-dimethoxypropane (88.7 mL, 723 mmol, 20.5 equiv).
The reaction was stirred overnight at 25 °C. The reaction mixture was
evaporated to dryness and water (250 mL) and saturated NaHCO3 solution (70
mL) was added. The mixture was stirred for a further 1 h. The resulting
suspension was filtered and washed with cold water (250 mL) to give 2,3-O-
isopropylideneguanosine (139) as a white solid (8.6 g, 75%), Rf = 0.72 (EtOAc /
MeOH, 7:3).
1H NMR (400 MHz, DMSO-d6) δ ppm:1.34 (s, 3 H) 1.49 (s, 3 H) 3.49- 3.55 (m,
2H) 4.10- 4.15 (m, 1 H) 4.97 (dd, J=6.31, 3.08, 1 H) 5.05 (br. s., 1 H) 5.19(dd,

206
J=6.24, 2.86 Hz, 1 H) 5.93 (d, J=2.79, 1 H) 6.52 (s, 2 H) 7.92 (s, 1 H) 10.70 (s,
1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 25.2, 27.0, 62.1, 81.2, 83.6, 86.6, 88.4,
113.0, 116.7, 135.8, 150.7, 153.7, 156.7.
Melting point: 260–262 °C (lit.259 260–262 °C)
HRMS (ESI): m/z calcd for C13H18N5O5+ [M + H]: 324.1300, found: 324.1286.
(The obtained spectroscopic data was consistent with that previously reported
for this compound).259
7.2.23 Synthesis of 2,3-O-isopropylideneguanosine-5-carboxylic acid
(140).259
Compound 139 (6.7 g, 21 mmol, 1 equiv), TEMPO (809 mg, 5.25 mmol, 0.25
equiv) and bis (acetoxy) iodobenzene (BAIB) (13.5 g, 42 mmol, 2.25 equiv)
were combined in MeCN/ water (1:1, 88 mL). The reaction mixture was stirred
overnight at 25 °C and the reaction progress was followed by TLC analysis
using a EtOAc / MeOH (4:1). After which time, acetone (160 mL) was added
and the solution was stirred for 1 h. The solution was poured into a well-stirred
volume of Et2O (930 mL) and the mixture was stirred for a further 1 h. The
resulting precipitate was filtered to give 2,3-O-isopropylideneguanosine-5-
carboxylic acid (140) as a pale orange solid (5.5 g, 78.8%). Rf = 0.16 (4:1
EtOAc / MeOH).

207
1H NMR (400 MHz, DMSO- d6) δ ppm: 1.34 (s, 3 H) 1.49 (s, 3 H) 4.61 (s, 1 H)
5.31 (d, J=6.02 Hz, 1 H) 5.56 (d, J=6.02 Hz, 1 H) 6.10 (s, 1 H) 6.46 (br. s., 2 H)
7.80 (s, 1 H) 10.68 (s, 1 H).
13C NMR (101 MHz, DMSO- d6) δ ppm: 25.4, 26.9, 84.0, 84.2, 85.9, 89.5,
112.9, 116.9, 137.1, 151.3, 153.8, 157.2, and 171.4.
Melting point: 210–212 °C (lit.259 210–212 °C).
HRMS (ESI): m/z calcd for C13H16N5O6+ [M + H]: 338.1101, found: 338.1134.
(The obtained spectroscopic data was consistent with that previously reported
for this compound).259
7.2.24 Synthesis of 2,3-O-isopropylideneguanosine-5-ethylcarboxylate
(142).
A suspension of 140 (5 g, 15.7 mmol, 1 equiv) in EtOH (500 mL) was stirred at
0 °C for 30 minutes. SOCl2 (4.5 mL, 67.6 mmol, 4.3 equiv) was slowly added.
The reaction mixture was warmed to room temperature and stirred overnight
and the reaction progress was followed by TLC analysis using (7:3) EtOAc /
MeOH. The reaction mixture was basified up to pH 8 by adding saturated
NaHCO3 solution and the resulting mixture was filtered. The filtrate was
evaporated under reduced pressure. The resulting residue was purified by
silica gel column chromatography using a mixture of EtOAc / MeOH (7:3) to

208
give 2,3-O-isopropylideneguanosine-5-ethylcarboxylate (142) as an off-white
solid (4.5 g, 84%). Rf = 0.71 (7:3 EtOAc / MeOH).
1H NMR (400 MHz, DMSO-d6) δ ppm: 0.98 (t, J=7.19 Hz, 3 H) 1.35 (s, 3 H) 1.49
(s, 3 H) 3.71 - 3.81 (m, 1 H) 3.82 - 3.91 (m, 1 H) 4.74 (s, 1 H) 5.34 (d, J=6.02
Hz, 1 H) 5.72 (d, J=5.87 Hz, 1 H) 6.18 (s, 1 H) 6.43 (br. s., 2 H) 7.78 (s, 1 H)
10.69 (br. s., 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 13.8, 25.3, 26.7, 61.1, 83.9, 84.8, 86.3,
89.9, 112.7, 117.1, 137.7, 151.1, 153.7, 157.2 and 169.8.
Ʋmax (ATR-FTIR) cm-1: 3413 (NH2), 3162 (NH), 2731 (alkyl CH3), 1696 (OC=O),
1628, 1596 (aryl C-N), 1540, 1488 (aryl C-C), 1382 (Et-O), 1271, 1205, 1157,
1057 (C-O-C), 866, 782.
Melting point: 272–274 °C.
HRMS (ESI): m/z calcd for C15H20N5O6+ [M + H]: 366.1414, found: 366.1449.
HPLC purity: - 99.99 %.
Elemental analysis: found C 49.30, H 5.17, and N 19.06. Expected C 49.31, H
5.24, and N 19.17

209
7.2.25 Synthesis of 2,3-O-isopropylideneguanosine-5-N-
ethylcarboxamide (143)259.
Compound 142 (774 mg, 2.12 mmol, 1 equiv) and ethylamine (22 mL) were
stirred at 0 to –20 °C for 3 h. The reaction progress was followed by TLC
analysis using (8:2 DCM: MeOH) and after which time the solution was allowed
to warm to room temperature and stirred overnight and the reaction progress
was followed by TLC analysis using a DCM / MeOH (7:3). The reaction mixture
was added to a stirring Et2O (50 mL) and the resulting mixture was stirred for 1
h, after which time the obtained precipitate was filtered and washed with Et2O
(25 mL). The resulting residue was purified by silica gel column
chromatography using a mixture of Pet Spirits/ EtOAc (2:8) to give 2,3-O-
isopropylideneguanosine-5-N-ethylcarboxamide (143) as a bluish-white solid
(725 mg, 94%), Rf = 0.85 (DCM / MeOH, 8:2).
1H NMR (400 MHz, DMSO-d6) δ ppm: 0.75 (t, J=7.19 Hz, 3 H) 1.33 (s, 3 H) 1.51
(s, 3 H) 2.78 - 2.90 (m, 1 H) 2.90 - 3.01 (m, 1 H) 4.47 (d, J=2.20 Hz, 1 H) 5.27
(dd, J=6.09, 1.25 Hz, 3 H) 5.44 (dd, J=6.09, 2.27 Hz, 3 H) 6.13 (s, 1 H) 6.42 (br.
s., 2 H) 7.37 (t, J=5.80 Hz, 1 H) 7.81 (s, 3 H) 10.65 (br s, 2 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.7, 15.8, 17.5, 25.4, 75.9, 76.0, 80.1,
82.9, 105.1, 108.6, 130.3, 142.8, 145.6, 149.9, and 162.5.
Melting point: 278–280 °C (lit.259 199–200 °C).

210
HRMS (ESI): m/z calcd for C15H21N6O5+ [M + H]: 365.1573, found: 365.1574.
(The obtained spectroscopic data was consistent with that previously reported
for this compound).259
7.2.26 Synthesis of O6-(benzotriazol-1-yl)-2′,3′-O-isopropylideneguanosine-
5′-N-ethylcarboxamide (144).259
Compound 143 (285 mg, 0.78 mmol, 1 equiv) was suspended in MeCN (27
mL).and BOP (554 mg, 1.25 mmol, 1.6 equiv) and DBU (187 µL, 1.25 mmol, 1.6
equiv) were added. The reaction mixture was stirred at room temperature for
20 h and the reaction progress was followed by TLC analysis using (Pet Spirits:
EtOAc, 1:4). The reaction mixture was diluted with EtOAc (90 mL) and the
resulting solution was washed with water (3 x 90 mL) and brine solution (90
mL). The EtOAc layer was filtered and dried over anhydrous MgSO4. The
solvent was evaporated under reduced pressure and the resulting residue was
purified via silica gel column chromatography using a gradient from (1:1)
petroleum spirits/ EtOAc to (1:4) petroleum spirits/ EtOAc to give O6-
(benzotriazol-1-yl)-2′,3′-O-isopropylideneguanosine-5′-N-ethylcarboxamide
(144) as a pale yellow solid (500 mg, 98%), Rf = 0.95 (Pet Spirits / EtOAc, 1:4).

211
1H NMR (400 MHz, DMSO-d6): δ ppm 0.67 (t, J=7.19 Hz, 3 H) 1.35 (s, 3 H) 1.52
(s, 3 H) 2.71 - 2.98 (m, 2 H) 4.53 (s, 1 H) 5.40 (d, J=6.16 Hz, 1 H) 5.52 (d,
J=6.02 Hz, 1 H) 6.29 (s, 1 H) 6.68 (s, 2 H) 7.48 (t, J=5.72 Hz, 1 H) 7.51 - 7.58
(m, 1 H) 7.66 (d, J=3.81 Hz, 2 H) 8.18 (d, J=8.51 Hz, 1 H) 8.21 (s, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.4, 25.4, 27.0, 33.5, 83.6, 87.0, 90.5,
109.5, 113.4, 120.6, 126.1, 128.6, 130.2, 143.1, 147.4, 155.0, 156.1, 156.2,
159.8, 160.0 and 168.4.
Melting point: 140–142 °C (lit.259 140–141 °C).
HRMS (ESI): m/z calcd for C21H24N9O5+ [M+H]: 482.1900, found: 482.1898.
(The obtained spectroscopic data was consistent with that previously reported
for this compound)259.
7.2.27 Synthesis of 2-fluoro-O6-benzotriazole-1-yl-2,3-O-
isopropylideneguanosine-5-N-ethylcarboxamide (145).259
Pyridine (2.0 mL) was cooled to –50 °C and 70% HF in pyridine (6.5 mL) was
added slowly followed by compound 144 (1 g, 2.08 mmol, 1 equiv) with vigorous
stirring. The temperature was allowed to rise up to –30 °C and the reaction
mixture was stirred to dissolve all solid. Tert-butylnitrite (2.1 mL, 17.68 mmol,
8.5 equiv) was added dropwise into the reaction mixture and the mixture was

212
stirred at –30 °C for a further 15 minutes. The reaction progress was followed
by TLC analysis using (2:8 Pet Spirits: EtOAc). The reaction mixture was
poured onto iced water (200 mL) and stirred for 30 minutes. The resulting
suspension was extracted in DCM (3 x 50 mL) and the combined DCM extracts
were washed with water (3 x 50 mL) and brine solution (3 x 50 mL). The DCM
extract was dried over anhydrous MgSO4, filtered. The filtrate was evaporated
under reduced pressure and the resulting residue was purified by silica gel
column chromatography using a mixture of Pet Spirits / EtOAc (2:8) to give 2-
fluoro-O6-benzotriazole-1-yl-2,3-O-isopropylideneguanosine-5-N-
ethylcarboxamide (145) as an off-white solid (0.68 g, 68%). Rf = 0.52 (2:8 Pet
Spirits / EtOAc).
1H NMR (400 MHz, DMSO-d6): δ ppm 0.65 (t, J=7.12 Hz, 3 H) 1.36 (s, 3 H) 1.54
(s, 3 H) 2.66 - 2.98 (m, 2 H) 4.65 (s, 1 H) 5.38 (d, J=4.40 Hz, 1 H) 5.45 (d,
J=5.87 Hz, 1 H) 6.46 (s, 1 H) 7.59 (t, J=6.0 Hz, 1 H) 7.66 - 7.82 (m, 3 H) 8.24
(d, J=8.36 Hz, 1 H) 8.78 (s, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.4, 25.5, 27.0, 33.5, 83.7, 87.1, 90.6,
109.6, 113.5, 118.2, 120.6, 126.1, 128.7, 130.2, 143.2, 147.5, 155.0, 157.1,
159.9, 168.4.
Melting point: 145–147 °C (decomp) (lit.259 140–141 °C).
HRMS (ESI): m/z calcd for C21H22FN8O5+ [M+H]: 485.1692, found: 485.1699.
(The obtained spectroscopic data was consistent with that previously reported
for this compound).259

213
7.2.28 Synthesis of 2-fluoro-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (146).259
A solution of compound 145 (700 mg, 1.24 mmol. 1 equiv)) in MeCN (9 mL) was
cooled at 0 °C in ice bath and 28% ammonia solution (500 µL) was cautiously
added to the stirring solution. The reaction mixture was allowed to rise to room
temperature and the stirring was continued for 2 h and the reaction progress
was followed by TLC analysis using EtOAc: MeOH (7:3). The reaction mass
was partitioned between water (50 mL) and EtOAc (50 mL). The aqueous
phase was extracted with EtOAc (3 x 30 mL) and the combined organic phases
were washed with water (3 x 50 mL) and brine solution (3 x 50 mL) and dried
over anhydrous MgSO4, filtered. The filtrate was evaporated under reduced
pressure. The resulting residue was purified by silica gel column
chromatography using EtOAc as the eluent to give 2-fluoro-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (146) as a pale yellow solid
(450 mg, 85%). Rf = 0.63 (EtOAc / MeOH, 7:3).
1H NMR (400 MHz, DMSO-d6): δ ppm 0.66 (t, J=7.19 Hz, 3 H) 1.34 (s, 3 H) 1.53
(s, 3 H) 2.72 - 2.97 (m, 2 H) 4.54 (s, 1 H) 5.36 (br s, 2 H) 6.27 (s, 1 H) 7.55 (t,
J=5.58 Hz, 1 H) 7.75 (br s, 2 H) 8.23 (s, 1 H).

214
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.4, 25.5, 27.1, 33.5, 83.5, 83.7, 86.5,
89.8, 113.3, 117.9, 141.0, and 150.6 (d, J = 20.1 Hz), 158.0 (d, J = 21.1 Hz),
158.2, 168.5.
Melting point: 194–196 °C (lit 259 194–196 °C).
HRMS (ESI): m/z calcd for C15H19FN6O4+ [M+]: 366.1500, found: 366.1451. (The
obtained spectroscopic data was consistent with that previously reported for this
compound).259
7.2.29 Synthesis of 2-iodo-O6-benzotriazole-1-yl-2,3-O-
isopropylideneguanosine-5-N-ethylcarboxamide (148)259.
Tert-butylnitrite (0.2 mL, 10.39 mmol, 4 equiv) was added to a mixture of
compound 144 (200 mg, 0.415 mmol, 1 equiv) and diiodomethane (1 mL, 49.92
mmol, 30 equiv) in MeCN (3 mL). The reaction mixture was vigorously stirred at
65–70 °C for 4 h under argon atmosphere and the reaction progress was
followed by TLC analysis using DCM: MeOH (9:1). The reaction solution was
diluted with EtOAc (50 mL), and washed with water (3 x 50 mL), and brine
solution (3 x 50 mL) and dried over anhydrous MgSO4. The filtrate solvent was
evaporated under reduced pressure and the resulting residue was purified by
silica gel column chromatography using a mixture of DCM and MeOH (9:1) to

215
give 2-iodo-O6-benzotriazole-1-yl-2,3-O-isopropylideneguanosine-5-N-
ethylcarboxamide (148) as a brown solid (212 mg, 86%). Rf = 0.42 (DCM /
MeOH, 9:1).
1H NMR (400 MHz, DMSO-d6) δ ppm: 0.64 (t, J=7.19 Hz, 3 H) 1.33 (s, 3 H) 1.50
(s, 3 H) 2.54 - 2.59 (m, 2 H) 4.58 (d, J=2.20 Hz, 1 H) 5.38 (m, 2 H) 6.44 (br. s., 1
H) 7.53 - 7.57 (m, 2 H) 7.64 - 7.67 (m, 2 H) 8.17- 8.20 (m, 1 H) 8.63 (s, 1 H).
13C NMR (101 MHz, DMSO-d6) δ ppm: 14.7, 25.1, 27.0, 30.9, 34.2, 82.9, 83.2,
86.3, 91.6, 108.3, 114.9, 116.3, 120.0, 120.7, 125.0, 128.6, 129.0, 143.4, 143.9,
154.2, 157.9 and 167.8.
Melting point: 194–196 °C (lit 259 195–196 °C).
HRMS (ESI): m/z calcd for C21H21INaN8O5+ [M+Na]: 615.0577, found:
615.0808. (The obtained spectroscopic data was consistent with that previously
reported for this compound).259
7.2.30 Synthesis of 2-iodo-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (149).259
An aqueous ammonia (28%) solution (1.1 mL) was added to the solution of
compound 148 (652 mg, 1.1 mmol, 1 equiv) in MeCN (52 mL) at 25 °C. The
solution was stirred for three days at room temperature, and the reaction

216
progress was followed by TLC analysis using EtOAc: MeOH (7:3). After which
time, the reaction mass was partitioned between water (50 mL) and EtOAc (50
mL). The aqueous phase was extracted with EtOAc (4 x 70 mL). The
combined organic phases were washed with water (3 x 50 mL), and brine
solution (4 x 50 mL) and dried over anhydrous MgSO4. The organic phase was
filtered, and the solvent was evaporated under reduced pressure. The resulting
residue was purified by silica gel column chromatography using 100% EtOAc to
give 2-iodo-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (149) as a
pale yellow solid (400 mg, 77%), Rf = 0.63 (EtOAc / MeOH, 7:3).
1H NMR (400 MHz, CDCl3) δ ppm 0.92 (t, J=7.24 Hz, 3 H) 1.37 (s, 3 H) 1.59 (s,
3 H) 3.11 - 3.35 (m, 2 H) 4.66 (d, J=1.81 Hz, 2 H) 5.27 (d, J=2.74 Hz, 1 H) 5.28
(d, J=2.64 Hz, 1 H) 5.32 (d, J=1.96 Hz, 1 H) 5.33 (d, J=2.01 Hz, 1 H) 6.00 (d,
J=2.69 Hz, 2 H) 6.02 (br. s., 2 H) 6.48 (br. s., 1 H) 7.74 (s, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 14.7, 25.2, 27.1, 30.9, 34.3, 82.6, 83.4,
85.9, 91.5, 114.7, 120.1, 139.8, 149.7, 155.2, and 168.2.
Ʋmax (ATR-FTIR) cm-1: 3390 (NH2), 3308 (NH), 3168, 3079 (aryl C-H), 2982
(alkyl C-H), 1647 (NC=O), 1586 (aryl C-N), 1520 (aryl C-C), 1456 (N-CO), 1256
(H-NCO) 1183, 1091 (C-O-C), 1052.
Melting point: 196–198 °C(lit 259 196–198 °C).
HRMS (ESI): m/z calcd for C15H19INaN6O4+ [M+Na]: 497.0410, found:
497.0390.

217
7.2.31 Synthesis of 2-(2-(4-aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine -5-N-ethylcarboxamide (159).
Approach 1
Compound 146 (150 mg, 0.41 mmol, 1 equiv) was combined with compound
124 (93 mg, 0.389 mmol, 1.25 equiv), and DIPEA (1.1 mL) in absolute EtOH
(3.0 mL). The reaction mixture was stirred at 70–75 °C for 7 days, after which
time the reaction mixture was treated with DM water at 0–5 °C. The resulting
oily reaction mixture was extracted in DCM (4 x 30 mL), and the combined DCM
extract was washed with water (50 mL), saturated NaHCO3 solution (50 mL)
and brine solution (50 mL). The solvent was dried over anhydrous MgSO4 and
evaporated under reduced pressure. The resulting residue was purified by
silica gel column chromatography using a mixture of 75% MeOH and 25%
water, to give 2-(2-(4-aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (159) as a yellowish solid.
Approach 2
Compound 146 (400 mg, 0.33 mmol, 1 equiv), 4-(2-aminoethyl)anilne 124 (83
mg, 0.69 mmol, 2 equiv), and DIPEA (1.5 mL) in MeCN (36 mL) was stirred in a

218
sealed reaction vessel at 180 °C in microwave conditions for 7 h. The reaction
progress was followed by TLC analysis in a mixture of (9.9:0.1) EtOAc and 28%
aqueous ammonia solution. After which time, the reaction mixture was
evaporated under reduced pressure. The resulting residue was purified by
silica gel column chromatography using a mixture of (9.9:0.1) EtOAc and 28%
aqueous ammonia solution to give 2-(2-(4-aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (159) as a yellowish solid (510
mg, 97%), Rf = 0.32 (EtOAc/ 28% NH3 solution, 9.5:0.5).
1H NMR (400 MHz, CDCl3) :δ ppm 0.71 (t, J=7.26 Hz, 3 H) 1.40 (s, 3 H) 1.62 (
s., 3 H) 2.82 (t, J=6.82 Hz, 2 H) 2.86 - 3.08 (m, 2 H) 3.45 - 3.61 (m, 1 H) 3.62 -
3.72 (m, 1 H) 4.68 (s, 1 H) 4.85 (br. s., 1 H) 5.26 (br. s., 2 H) 5.54 (d, J=6 Hz, 1
H) 5.63 (d, J=4.84 Hz, 1 H) 6.04 (br. s., 2 H) 6.67 (d, J=8.22 Hz, 2 H) 7.06 (d,
J=8.07 Hz, 2 H) 7.53 (s, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 13.9, 20.1, 25.0, 26.7, 33.8, 34.8, 83.3,
84.0, 87.7, 91.4, 105.7, 113.7, 115.4, 129.3, 129.6, 137.0, 144.7, 151.3, 155.6,
159.5 and 168.9.
Ʋmax (ATR-FTIR) cm-1: 3350 (NH2), 3292 (NH), 2968 (alkyl CH3), 1689 (NC=O),
1662, 1602 (aryl C-N), 1513 (aryl C-C), 1437 (N-CO), 1364, 1253 (H-NCO),
1170 (C-O-C).
Melting point: 150–152 °C (lit 121 95–104 °C).
HRMS (ESI): m/z calcd for C23H31N8O4+ [M+H]: 483.2460, found: 483.2482.
(The obtained spectroscopic data was consistent with that previously reported
for this compound).121
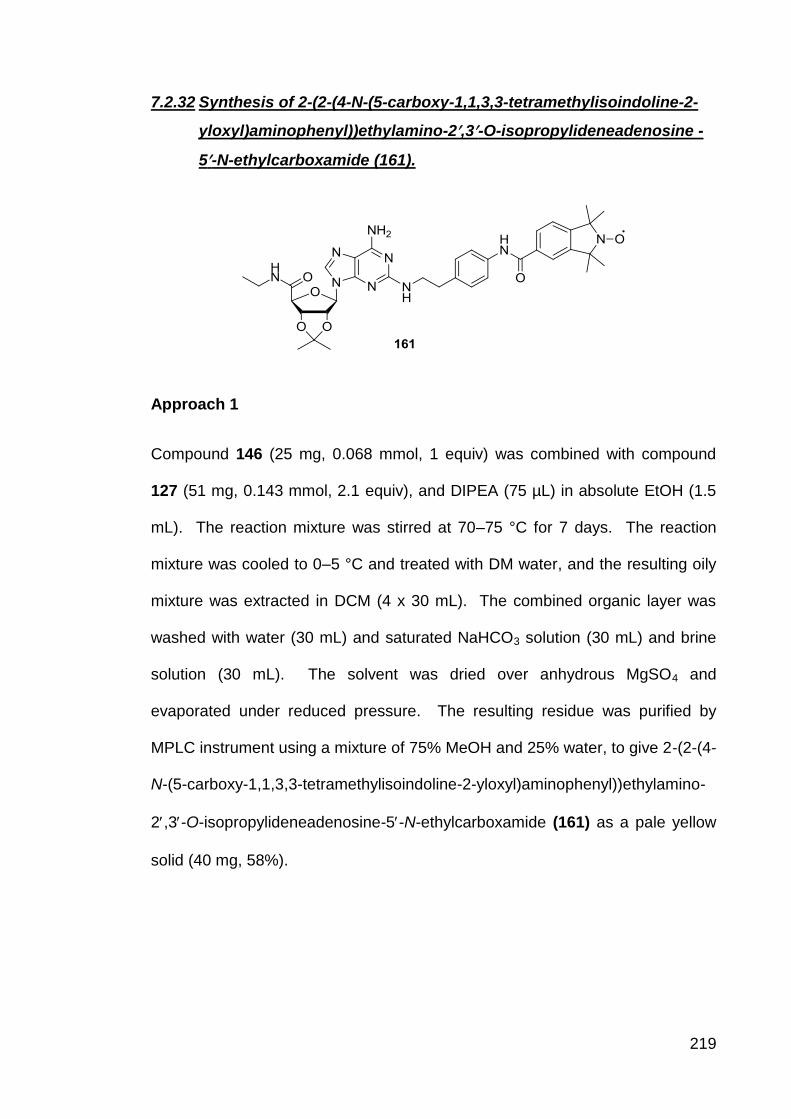
219
7.2.32 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-tetramethylisoindoline-2-
yloxyl)aminophenyl))ethylamino-2,3-O-isopropylideneadenosine -
5-N-ethylcarboxamide (161).
Approach 1
Compound 146 (25 mg, 0.068 mmol, 1 equiv) was combined with compound
127 (51 mg, 0.143 mmol, 2.1 equiv), and DIPEA (75 µL) in absolute EtOH (1.5
mL). The reaction mixture was stirred at 70–75 °C for 7 days. The reaction
mixture was cooled to 0–5 °C and treated with DM water, and the resulting oily
mixture was extracted in DCM (4 x 30 mL). The combined organic layer was
washed with water (30 mL) and saturated NaHCO3 solution (30 mL) and brine
solution (30 mL). The solvent was dried over anhydrous MgSO4 and
evaporated under reduced pressure. The resulting residue was purified by
MPLC instrument using a mixture of 75% MeOH and 25% water, to give 2-(2-(4-
N-(5-carboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-
2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (161) as a pale yellow
solid (40 mg, 58%).

220
Approach 2
Compound 159 (143 mg, 0.311mmol, 1 equiv) was added to a mixture
containing 5-carboxy-1,1,3,3-tetramethylisoindoline-1-yloxyl 106 (88 mg, 0.37
mmol, 1.25 equiv), EDCI (96 mg, 0.356 mmol, 1.2 equiv), HOBt (100 mg, 0.356
mmol, 1.2 equiv), and DIPEA (1 mL) were combined in dry DMF (3 mL) under
argon atmosphere. The reaction mixture was stirred at room temperature for 2
days. The solution was quenched by using water (30 mL) at 0–5 °C and the
resulting oily reaction mixture was extracted in DCM (4 x 30 mL). The
combined DCM extract was washed with water (30 mL), saturated NaHCO3
solution (30 mL) and brine solution (30 mL). The solvent was evaporated under
reduced pressure and the resulting residue was purified by MPLC instrument
using a mixture of 75% MeOH and 25% water to give 2-(2-(4-N-(5-carboxy-
1,1,3,3-tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (161) as a pale yellow solid
(168 mg, 81%), Rf = 0.48 (DCM / IPA/ MeOH, 4.5:4.5:1).
1H NMR (400 MHz, CDCl3) δ ppm: 0.71 (t, 3 H) 1.23 (d, J=6.13 Hz, 12 H) 1.42
(s, 3 H) 1.62 (s, 3 H) 2.91 (s, 2 H) 2.98 (s, 2 H) 4.69 - 4.70 (m, 1 H) 5.32 (s, 1 H)
6.05 - 6.07 (m, 1 H) 6.24 - 6.26 (m, 1 H) 7.00 - 7.03 (m, 1 H) 7.55 (s, 1 H) 8.03 -
8.05 (m, 1 H) Note that paramagnetic broadening by the nitroxide radical results
in some peaks not being detected or giving poor integration in the proton NMR
spectrum.
Melting point: 66–68 °C (decomp.)
HRMS (ESI): m/z calcd for C36H45N9O6+ [M + H]: 699.3493, found: 699.3506.
EPR (methanol): typical 3-line nitroxide signal, g 1.9823, aN 1.4871 mT.

221
HPLC purity- 97.64%.
7.2.33 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-tetramethylisoindoline-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide
(162).
1 M HCl (3.3 mL) was added to a solution of compound 161 (101 mg, 0.94
mmol) in MeCN (8.0 mL). The solution was stirred for 4 h at 55–60 °C, and the
reaction progress was followed by TLC. Upon completion, the reaction mixture
was basified with saturated NaHCO3 solution up to pH 8 at 0–5 °C, the resulting
precipitate was extracted with EtOAc (3 x 25 mL). The combined organic phase
was washed with water (3 x 25 mL), brine solution (3 x 25 mL) and dried over
anhydrous MgSO4. The solvent was filtered and evaporated under reduced
pressure. The resulting residue was purified by MPLC instrument using a
mixture of 80% MeOH and 20% water with partial crystallisation to give 2-(2-(4-
N-(5-carboxy-1,1,3,3-tetramethylisoindoline-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide (162) as a
pale yellow solid (66 mg, 69.5%), Rf = 0.65 (EtOAc: MeOH, 8:2).
1H NMR (400 MHz, MeOH-d4) δ ppm: 1.00 - 1.12 (m, 3 H) 1.27 (s, 2 H) 2.12 -
2.25 (m, 1 H) 2.61 - 2.69 (m, 1 H) 2.86 - 3.00 (m, 1 H) 3.09 - 3.22 (m, 1 H) 3.33
(br. s., 20 H) 3.46 - 3.61 (m, 1 H) 3.63 - 3.77 (m, 1 H) 4.36 - 4.47 (m, 1 H) 4.49 -
4.55 (m, 1 H) 4.58 - 4.72 (m, 1 H) 4.87 (s, 74 H) 5.01 - 5.09 (m, 1 H) 5.90 - 6.02

222
(m, 1 H) 7.54 - 7.75 (m, 1 H) 7.93 - 8.04 (m, 1 H) 8.05 - 8.12 (m, 1 H) Note that
paramagnetic broadening by the nitroxide radical results in some peaks not
being detected or giving poor integration in the proton NMR spectra.
Ʋmax (ATR-FTIR) cm-1: 3400-3000 (NH2, NH, OH), 2926 (alkyl CH3), 1691
(NC=O), 1597 (aryl C-N), 1529 (aryl C-C), 1408 (OC-N), 1258 (H-NCO), 1044
(C-O-C).
Melting point: 241-243 °C (decomp).
HRMS (ESI): m/z calcd for C33H41N9O6+ [M+H]: 659.318, found: 659.3197.
EPR (methanol): typical 3-line nitroxide signal, g 1.9950, aN 1.4928 mT.
HPLC purity: >99.9 %.
Elemental analysis: found C 57.64, H 6.10, N 17.91. Expected C 60.17, H 6.12,
N 19.14

223
7.2.34 Synthesis of 2-(2-(4-N-(4-carboxy-2,2,6,6-tetramethylpiperidine-1-
yloxyl)aminophenyl))ethylamino-2,3,-O-isopropylidineadenosine-
5-N-ethylcarboxamide (163).
Compound 159 (300 mg, 0.622 mmol, 1 equiv), was added to a mixture
containing 4-carboxy-TEMPO-1-yloxyl 116(156 mg, 0.7789 mmol, 1.25 equiv),
EDCI (179 mg, 0.933 mmol, 1.5 equiv), and HOBt (101 mg, 0.746 mmol, 1.2
equiv), and DIPEA (2 mL) in dry DMF (10 mL) under argon atmosphere. The
resulting reaction mixture was stirred at room temperature for two days. The
solution was quenched by adding water (30 mL) at 0–5 °C and the resulting oily
mixture was extracted in DCM (3 x 30 mL). The combined extracts were
washed with water (2 x 30 mL), and saturated NaHCO3 solution (2 x 30 mL) and
brine solution (2 x 30 mL) and dried over anhydrous MgSO4. The solvent was
evaporated under reduced pressure. The resulting brownish residue was
purified by MPLC instrument using a mixture of 75% MeOH and 25% water to
give 2-(2-(4-N-(4-carboxy-2,2,6,6-tetramethylpiperidine-1-
yloxyl)aminophenyl))ethylamino-2,3,-O-isopropylidineadenosine-5-N-
ethylcarboxamide (163) as a yellowish solid (346 mg, 82%). Rf = 0.43 (IPA:
MeOH, 8:2).

224
Ʋmax (ATR-FTIR) cm-1: 3400-3000 (NH, NH2), 2976 (alkyl CH3), 1655 (NC=O),
1596 (aryl C-N), 1530 (aryl C-C), 1411 (N-CO), 1326 (N-O), 1239 (H-NCO)
1087 (C-O-C), 789.
Melting point: 148–150 °C decomp.
HRMS (ESI): m/z calcd for C33H47N9O6+ [M + H]: 665.364, found: 665.3658.
EPR (methanol): typical 3-line nitroxide signal, g 1.9823, aN 1.4871 mT.
HPLC purity: 99.38%.
Elemental analysis: found: C 59.58, H 7.35, and N 18.43. Expected: C 59.62, H
6.97, and N 18.96.
7.2.35 Synthesis of 2-(2-(4-N -(4- carboxy-2,2,6,6-tetramethylpiperidine-1-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide
(164).
1 M HCl (11 mL) was slowly added to a solution of compound 163 (177 mg,
0.267 mmol, 1 equiv) in MeCN (6.0 mL). The reaction mixture was stirred for 5
h at 55–60 °C, and the reaction progress was followed by TLC in IPA: MeOH
(8:2). Upon completion, the solution was basified up to pH 8 by adding
saturated NaHCO3 solution at 0–5 °C, and the resulting precipitate was
extracted with EtOAc (3 x 20 mL). The combined organic phase was washed

225
with water (3 x 20 mL), brine solution (3 x 20 mL) and dried over anhydrous
MgSO4, filtered. The solvent was evaporated under reduced pressure and the
resulting residue was purified by MPLC instrument using a mixture of 70%
MeOH and 30% water with a partial crystallisation to give 2-(2-(4-N-(4-carboxy-
2,2,6,6-tetramethylpiperidine-1-yloxyl)aminophenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (164) as a pale yellow solid (100 mg, 60 %). Rf = 0.43 (IPA:
MeOH, 8:2).
Ʋmax (ATR-FTIR) cm-1: 3400-3100 (NH2, NH, OH), 2926 (alkyl CH3), 1691
(NC=O), 1650, 1600 (aryl C-N), 1532 (aryl C-C), 1411 (OC-N), 1240 (H-NCO),
1324 (N-O), 1044 (C-O-C).
Melting point: 211-213°C (decomp).
HRMS (ESI): m/z calcd for C30H43N9O6+ [M+H]: 625.333, found: 625.3341.
EPR (methanol): typical 3-line nitroxide signal, g 1.9893, aN 1.6011 mT.
HPLC Purity – <99.9 %.
Elemental analysis: found C 59.79, H 7.25, and N 19.32. Expected C 57.68, H
6.78, and N 20.18.

226
7.2.36 Synthesis of 2-(2-(4-N-(3-carboxy-2,2,5,5-tetramethylpyrrolidine-1-
yloxyl) aminophenyl)) ethylamino-2,3,-O-isopropylidineadenosine-
5-N-ethylcarboxamide (165).
3-Carboxy-PROXYL 117(145 mg, 0.778 mmol, 1.25 equiv) was added to a
stirring solution containing compound 159 (300 mg, 0.622 mmol, 1 equiv), EDCI
(179 mg, 0.933 mmol, 1.5 equiv), HOBt (101 mg, 0.746 mmol, 1.2 equiv), and
DIPEA (2 mL) were combined in dry DMF (10.5 mL) under argon atmosphere.
The reaction mixture was stirred at room temperature for two days. The
resulting solution was quenched by adding water (20 mL) at 0–5 °C and the
resulting oily reaction mixture was extracted in DCM (3 x 20 mL). The
combined extracts were washed with water (3 x 25 mL) and saturated NaHCO3
solution (3 x 25 mL) and brine solution (3 x 25 mL) and dried over anhydrous
MgSO4. The solvent was evaporated under reduced pressure and the resulting
residue was purified by MPLC instrument using a mixture of 75% MeOH and
25% water to give 2-(2-(4-N-(3-carboxy-2,2,5,5-tetramethylpyrrolidine-1-
yloxyl)aminophenyl))ethylamino-2,3,-O-isopropylidineadenosine-5-N-
ethylcarboxamide (165) as a yellowish-white solid (300 mg, 74 %), Rf = 0.29
(IPA / MeOH, 9:1).
Ʋmax (ATR-FTIR) cm-1: 3500-3000 (OH, NH, NH2), 2923 (alkyl CH3), 1670
(NC=O), 1600 (aryl C-N), 1532 (aryl C-C), 1411 (N-CO), 1356-1320 (N-O),
1195, 1087 (C-O-C), 789.

227
Melting point: 140–142 °C.
HRMS (ESI): m/z calcd for C32H45N9O6+ [M + H]: 651.349, found: 651.3493,
EPR (methanol): typical 3-line nitroxide signal, g 1.9808, aN 1.5239 mT.
HPLC purity- 97.96 %.
Elemental analysis: found C 59.57, H 7.30, and N 19.08. Expected C 59.06, H
6.82, and N 19.37.
7.2.37 Synthesis of 2-(2-(4-N -(3-carboxy-2,2,5,5-tetramethylpyrrolidine-1-
yloxyl) aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide
(166).
1 M HCl (3.3 mL) was added to a stirring solution of compound 165 (101 mg,
0.94 mmol, 1 equiv) in MeCN (8.0 mL). The reaction mixture was stirred for 4 h
at 55–60 °C, and the reaction progress was followed by TLC analysis using
(DCM: IPA: MeOH, 4:5:1). Upon completion, the solution was basified by using
saturated NaHCO3 solution up to pH 8 at 0–5 °C and the resulting precipitate
was extracted with EtOAc (4 x 20 mL). The organic phases were washed with
water (3 x 20 mL) and brine solution (3 x 20 mL) and dried over anhydrous
MgSO4, filtered. The solvent was evaporated under reduced pressure and the
resulting residue was purified by MPLC instrument using a mixture of 70%
methanol and 30% water with a partial crystallisation of solid to give 2-(2-(4-N-
(3-carboxy-2,2,5,5-tetramethylpyrrolidine-1-

228
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide(166) as a
brownish white solid (65 mg, 62.5 %), Rf = 0.10 (DCM: IPA: MeOH, 4:5:1).
1H NMR (400 MHz, CD3OD) δ ppm: 0.97 - 1.11 (m, 3 H) 2.17 (s, 1 H) 2.82 -
3.01 (m, 1 H) 3.05 - 3.19 (m, 1 H) 3.33 (d, J=1.47 Hz, 18 H) 3.46 - 3.59 (m, 1 H)
3.61 - 3.74 (m, 1 H) 4.37 - 4.54 (m, 1 H) 4.97 - 5.12 (m, 1 H) 5.87 - 5.99 (m, 1
H) 7.14 - 7.33 (m, 1 H) 7.38 - 7.61 (m, 1 H) 7.91 - 8.04 (s, 1 H). Note that
paramagnetic broadening by the nitroxide radical results in some peaks not
being detected or giving poor integration in the proton NMR spectrum.
Ʋmax (ATR-FTIR) cm-1: 3322-3000 (OH, NH, NH2), 2926 (alkyl CH3), 1634
(NC=O), 1597 (aryl C-N), 1538 (aryl C-C), 1411 (N-CO), 1354 (N-O), 1255 (H-
NCO), 1048 (C-O-C).
Melting point: 176–178 °C (decomp).
HRMS (ESI): m/z calcd for C29H41N9O6+ [M+H]: 611.3184, found: 611.3183.
EPR (methanol): typical 3-line nitroxide signal, g 1.9939, aN 1.5006 mT.
HPLC purity- >99.9%
Elemental analysis: found C 56.40, H 7.19, and N 19.22. Expected C 57.04, H
6.60, and N 20.64.

229
.
7.2.38 .Synthesis of 2-(2-(4-N-(5-methylcarboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine -5-N-ethylcarboxamide (167).
Compound 159 (100 mg, 0.207 mmol, 1 equiv), was added in a stirring mixture
of (60:40) 5-methylcarboxy-1,1,3,3-tetramethylisoindoline-2-yloxyl (115) and
compound 8 (65 mg, 0.259 mmol, 1.25 equiv), EDCI (64 mg, 0.33 mmol,
1.6equiv), HOBt (35 mg, 0.259 mmol, 1.25 equiv), and DIPEA (1.4 mL) in
anhydrous DMF (8 mL). The reaction mixture was stirred at room temperature
for 4 days and the reaction progress was followed by TLC analysis (EtOAc:
MeOH, 8:2). The solution was quenched by adding of water (50 mL) at 0–5 °C
and the oily reaction mixture was extracted in DCM (3 x 50 mL). The combined
extracts were washed with water (3 x 50 mL) and brine solution (3 x 50 mL).
The solvent was evaporated under reduced pressure. The resulting residue
was purified by MPLC instrument using a mixture of 75% MeOH and 25% water
to give a (60:40) mixture of 2-(2-(4-N-(5-methylcarboxy-1,1,3,3-
tetramethylisoindoline-2-yloxyl)aminophenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (167) and compound (161) as
a pale yellow solid (150 mg, 71% w/w)

230
Melting point: 175–180 °C (decomp).
HRMS (ESI): m/z calcd for C37H47N9O6+ [M + H] 713.3649 and C36H45N9O6+:
699.3493, found: 713.5496 and 699.9465.
HPLC purity: 60:40 mixtures of compound 167 and 161
EPR (methanol): typical 3-line nitroxide signal, g 1.9912, aN 1.5084 mT.
(This mixture was hard to separate so taken as it is for further for next reaction)
7.2.39 Synthesis of 2-(2-(4-N-(methylcarboxy-1,1,3,3-
tetramethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide
(168).
1 M HCl (3.3 mL) was added slowly in to a mixture of compound 167 and 161
(86 mg, 0.94 mmol, 1 equiv)) in MeCN (8.0 ml). The solution was stirred for 9 h
at 55–60 °C, and the reaction progress was followed by TLC analysis in (DCM:
IPA: MeOH, 4:5:1). Upon completion, the solution was basified up to pH 8 by
slow adding saturated NaHCO3 solution and, the resulting mixture was
extracted with EtOAc (5 x 50 mL). The EtOAc layers were washed with water
(3 x 25 mL) and brine solution (3 x 25 mL), and dried over anhydrous MgSO4,
filtered. The filtrate was evaporated under reduced pressure and the resulting

231
residue was purified by MPLC instrument using 70% MeOH and 30% water to
give a mixture of 2-(2-(4-N-(methylcarboxy-1,1,3,3-tetramethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide (168) and
compound (162) as an off-white solid (74 mg,). Rf = 0.37 (DCM: MeOH, 9:1).
Ʋmax (ATR-FTIR) cm-1: 3400-3117 (OH, NH, NH2), 2973 (alkyl CH3), 2927, 1656
(NC=O), 1599 (aryl C-N), 1409 (N-CO), 1355 (N-O), 1256 (H-NCO), 1045 (C-O-
C).
Melting point: 175–180 °C (decomp).
HRMS (ESI): m/z calcd for C34H43N9O6+ and C33H41N9O6
+ [M+2H]: 673.3336
and 659 56 found: 674.6586.and 661.0205.
HPLC purity: 60:40 mixture.
EPR (methanol): typical 3-line nitroxide signal, g 1.9928, aN 1.4952 mT.

232
7.2.40 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-tetraethylisoindoline-2-
yloxyl) aminophenyl)) ethylaminoadenosine-5-N-ethylcarboxamide
(169).
Compound 159 (150 mg, 0.311 mmol, 1 equiv) was added in a mixture
containing 5-carboxy-1, 1, 3, 3-tetraethylisoindoline-2-yloxyl (114) (113 mg,
0.389 mmol, 1.25 equiv), EDCI (95.4 mg, 0.498 mmol, 1.6 equiv), HOBt (53 mg,
0.389 mmol, 1.25 equiv), and DIPEA (1.2 mL) were combined in anhydrous
DMF (10 mL). The reaction mixture was stirred at room temperature for 19 h
and the reaction progress was followed by TLC analysis (DCM: MeOH, 9:1).
The solution was quenched by adding water at 0–5 °C and the resulting oily
reaction mixture was extracted in DCM (5 x 50 mL). The combined DCM
extracts were washed with water (3 x 25 mL) and saturated NaHCO3 solution (3
x 25 mL) and brine solution (2 x 25 mL) and dried over anhydrous MgSO4,
filtered. The solvent was evaporated under reduced pressure. The resulting
residue was purified by MPLC instrument using a mixture of 75% MeOH and
25% water to give 2-(2-(4-N-(5-carboxy-1,1,3,3-tetraethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide (169) as a
pale yellow solid (101 mg, 65%).
Rf = 0.80 (DCM / MeOH, 9:1).

233
1H NMR (400 MHz, METHANOL-d4) δ ppm 0.54 - 0.68 (t, 3 H) 1.17 - 1.19 (d, 6
H) 1.53 - 1.63 (m, 1 H) 2.71 - 2.86 (m, 1 H) 2.86 - 3.03 (m, 1 H) 3.44 - 3.58 (m,
1 H) 3.67 - 3.82 (m, 1 H) 5.56 - 5.64 (m, 1 H) 5.73 - 5.81 (m, 1 H) 6.20 - 6.27
(m, 1 H) 7.29 - 7.39 (m, 1 H) 7.59 - 7.73 (m, 1 H) 7.83 - 7.90 (m, 1 H) Note that
paramagnetic broadening by the nitroxide radical results in some peaks not
being detected or giving poor integration in the proton NMR spectrum.
Ʋmax (ATR-FTIR) cm-1:3400-3000 (NH2 and NH), 2924 (alkyl CH3), 1645
(NC=O), 1593 (C-N), 1528 (aryl C-C), 1409 (OC-N), 1256 (H-NCO), 1088 (C-O-
C).
Melting point: 138–140 °C dec;
HRMS (ESI): m/z calcd for C40H53N9O6+ [M + H]: 755.4119, found: 755.4118.
HPLC purity – <95%.
EPR (methanol): typical 3-line nitroxide signal, g 2.0288, aN 1.4456 mT.
Elemental analysis: found C 66.59, H 8.03, and N 15.97. Expected C 63.64, H
6.94, and N 16.70.

234
7.2.41 Synthesis of 2-(2-(4-N-(5-carboxy-1,1,3,3-tetraethylisoindoline-2-
yloxyl)aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide
(170).
1M HCl (6.5 mL) added slowly in to a stirring solution of compound 169 (100
mg, 0.132 mmol, 1 equiv) in MeCN (3.0 mL). The solution was stirred for 5 h at
55–60 °C and the reaction progress was followed by TLC analysis (EtOAc:
MeOH, 8:2) indicated that all starting material had been consumed. Upon
completion, the solution was basified up to pH 8 with saturated NaHCO3
solution, and the resulting mixture was extracted with EtOAc (3 x 50 mL). The
combined organic phases were washed with water (3 x 25 mL) and brine
solution (3 x 25 mL) and dried over anhydrous MgSO4, filtered. The filtrate was
evaporated under reduced pressure. The resulting residue was purified by
MPLC instrument using a mixture of 75% MeOH and 25% water as the eluent to
give 2-(2-(4-N-(5-carboxy-1,1,3,3-tetraethylisoindoline-2-yloxyl)
aminophenyl))ethylaminoadenosine-5-N-ethylcarboxamide (170) as a pale
yellow solid (67 mg, 71%), Rf = 0.48 (EtOAc: MeOH, 8:2).
1H NMR (400 MHz, METHANOL-d4) δ ppm 1.00 - 1.20 (m, 12 H) 2.95 (br. s., 8
H) 3.22 (br. s., 4 H) 3.56 - 3.84 (m, 7 H) 4.47 (br. s., 9 H) 5.99 (br. s., 4 H) 7.31
(br. s., 8 H) 7.67 (br. s., 7 H) 8.11 (br. s., 3 H) Note that paramagnetic

235
broadening by the nitroxide radical results in some peaks not being detected or
giving poor integration in the proton NMR spectrum.
Melting point: 175–180 °C (decomp).
Ʋmax (ATR-FTIR) cm-1: 3286-3000 (OH, NH, NH2), 2962 and 2926 (alkyl CH3),
1622 (NC=O), 1591 (aryl C-N), 1515 (aryl C-C), 1407 (OC-N), 1320 (N-O), 1050
(C-O-C).
HRMS (ESI): m/z calcd for C37H48NaN9O6+ [M+ Na]: 737.3625, found:
737.3883.
HPLC purity- 98.5%.
EPR (methanol): typical 3-line nitroxide signal, g 1.9923, aN 1.4380 mT.
Elemental analysis: found C 59.58, H 7.35, and N 18.43. Expected C 59.62, H
6.97, and N 18.96.
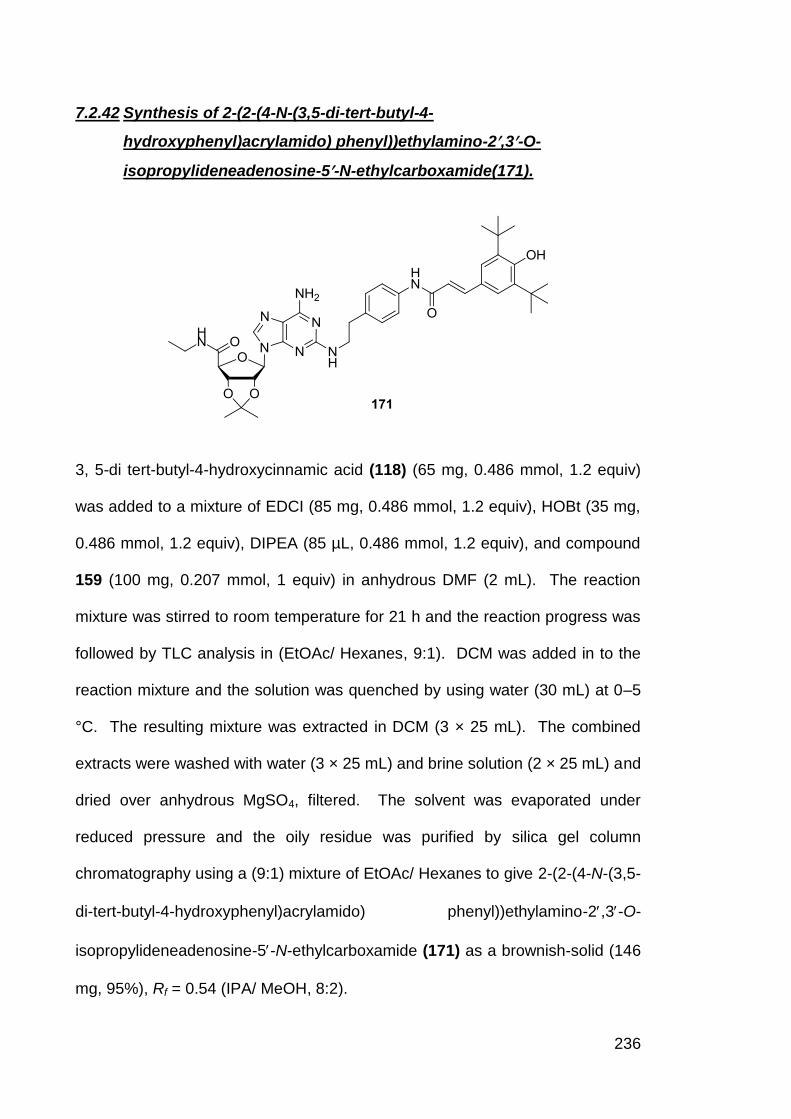
236
7.2.42 Synthesis of 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido) phenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide(171).
3, 5-di tert-butyl-4-hydroxycinnamic acid (118) (65 mg, 0.486 mmol, 1.2 equiv)
was added to a mixture of EDCI (85 mg, 0.486 mmol, 1.2 equiv), HOBt (35 mg,
0.486 mmol, 1.2 equiv), DIPEA (85 µL, 0.486 mmol, 1.2 equiv), and compound
159 (100 mg, 0.207 mmol, 1 equiv) in anhydrous DMF (2 mL). The reaction
mixture was stirred to room temperature for 21 h and the reaction progress was
followed by TLC analysis in (EtOAc/ Hexanes, 9:1). DCM was added in to the
reaction mixture and the solution was quenched by using water (30 mL) at 0–5
°C. The resulting mixture was extracted in DCM (3 × 25 mL). The combined
extracts were washed with water (3 × 25 mL) and brine solution (2 × 25 mL) and
dried over anhydrous MgSO4, filtered. The solvent was evaporated under
reduced pressure and the oily residue was purified by silica gel column
chromatography using a (9:1) mixture of EtOAc/ Hexanes to give 2-(2-(4-N-(3,5-
di-tert-butyl-4-hydroxyphenyl)acrylamido) phenyl))ethylamino-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (171) as a brownish-solid (146
mg, 95%), Rf = 0.54 (IPA/ MeOH, 8:2).

237
1H NMR (400 MHz, CD3OD) δ ppm: 0.59 (t, J=7.21 Hz, 3 H) 0.84 - 0.92 (t,
J=7.21 Hz, 3 H) 1.28 (s, 6 H) 1.45 (s, 9 H) 1.51 - 1.58 (m, 2 H) 1.97 (s, 1 H)
2.01 (s, 1 H) 2.13 - 2.20 (m, 1 H) 2.30 (d, J=6.80 Hz, 1 H) 2.62 (s, 1 H) 2.76 (dd,
J=13.74, 7.14 Hz, 1 H) 2.85 (s, 1 H) 2.87 - 2.95 (m, 1 H) 2.99 (s, 1 H) 3.48 (d,
J=6.50 Hz, 1 H) 3.66 - 3.76 (m, 1 H) 4.55 - 4.62 (m, 1 H) 5.58 (d, J=5.09 Hz, 1
H) 5.73 (d, J=5.87 Hz, 1 H) 6.21 (s, 1 H) 6.50 - 6.55 (m, 1 H) 6.63 (d, J=15.80
Hz, 1 H) 6.95 (s, 1 H) 7.13 (s, 1 H) 7.26 (d, J=7.87 Hz, 1 H) 7.42 (s, 1 H) 7.53 -
7.63 (m, 1 H) 7.84 (br. s., 1 H) 7.97 (s, 1 H)
13C NMR (101 MHz, CD3OD) δ ppm 12.5, 23.9, 25.5, 29.2, 29.3, 29.5, 33.4,
34.1, 83.6, 84.1, 87.9, 91.1, 113.0, 117.2, 119.8, 122.0, 124.6, 126.0, 128.9,
135.6, 136.9, 138.0, 142.5, 156.2, 159.4, 163.4, 165.9, 170.2 .
Ʋmax (ATR-FTIR) cm-1: 3621(NH2), 3500-3200 (NH, OH), 2922 and 2852 (alkyl
CH3), 1655 (NC=O), 1601 (C=C), 1515 (aryl C-N), 1467 (aryl C-C), 1425 (N-
CO), 1381, 1357, 1207 (H-NCO), 1184, 1118, 1087 (C-O-C).
Melting point: 138–140 °C.
HRMS (ESI): m/z calcd for C40H53N8O6+ [M+H]: 741.4088, found: 741.4490.
HPLC purity: >99.9%.

238
7.2.43 Synthesis of 2-(2-(4-N-(3,5-di-tert-butyl-4-
hydroxyphenyl)acrylamido) phenyl))ethylaminoadenosine-5-N-
ethylcarboxamide (172) (2VHM162)
1 M HCl (3.3 mL) was added to a solution of compound 171 (121 mg, 0.94
mmol) in MeCN (8.0 mL). The solution was stirred for 4 h at 55–60 °C, and the
reaction progress was followed by TLC. Upon completion, the reaction mixture
was basified with saturated NaHCO3 solution up to pH 8 at 0–5 °C, the resulting
precipitate was extracted with EtOAc (3 x 25 mL). The combined organic phase
was washed with water (3 x 25 mL), brine solution (3 x 25 mL) and dried over
anhydrous MgSO4. The solvent was filtered and evaporated under reduced
pressure. The resulting residue was purified by MPLC instrument using a
mixture of 80% MeOH and 20% water with partial crystallisation to give 2-(2-(4-
N-(3,5-di-tert-butyl-4-hydroxyphenyl)acrylamido) phenyl))ethylaminoadenosine-
5-N-ethylcarboxamide (172) as a pale yellow solid (46 mg, 50%), Rf = 0.65
(EtOAc: MeOH, 8:2).
1H NMR (400 MHz, (CD3)2CO) δ ppm 1.02 (t, J=7.09 Hz, 3 H) 1.23 - 1.34 (m, 9
H) 1.44 - 1.52 (m, 9 H) 2.05 (d, J=1.03 Hz, 2 H) 3.07 - 3.17 (m, 1 H) 3.26 - 3.36
(m, 1 H) 3.51 - 3.58 (m, 1 H) 3.59 - 3.69 (m, 1 H) 4.36 (s, 1 H) 4.46 (br, s, 1 H)
5.02 - 5.09 (m, 2 H) 5.59 - 5.63 (s, 1 H) 5.91 (d, J=6.55 Hz, 1 H) 6.33 (br, s, 1 H)

239
6.69 (d, J=15 Hz, 2 H)) 7.23 (d, J=8.12 Hz, 2 H) 7.38 (s, 1H) 7.45 (s, 2 H) 7.64
(s, 1 H) 7.69 (s, 1 H) 7.70 (d, J=7.87 Hz, 2 H) 7.87 (s, 1 H) 9.26 (s, 1 H).
13C NMR (101 MHz, CD3OD) δ ppm 21.4, 29.1, 33.4 , 34.9, 35.5, 42.5, 83.6,
84.1, 87.9, 91.1, 113.9, 117.2, 119.8, 122.0, 126.0, 128.9, 135.7, 136.8, 137.0,
138.1, 142.6, 152.4 156.1, 156.2, 159.3, 159.5, 163.5, 165.9, 170.2.
Ʋmax (ATR-FTIR) cm-1: 3700-3000 (NH2, NH, OH), 2955 and 2852 (alkyl CH3),
1620 (C=C), 1596 (NC=O), 1514 (aryl C-N), 1467 (aryl C-C), 1424 (N-CO),
1356, 1236 (H-NCO), 1206, 1184, 1116 (C-O-C).
Melting point: 241-243 °C (decomp).
HRMS (ESI): m/z calcd for C37H49N8O6+ [M+H]: 701.3775, found: 701.4096.
HPLC purity: >99.9%.
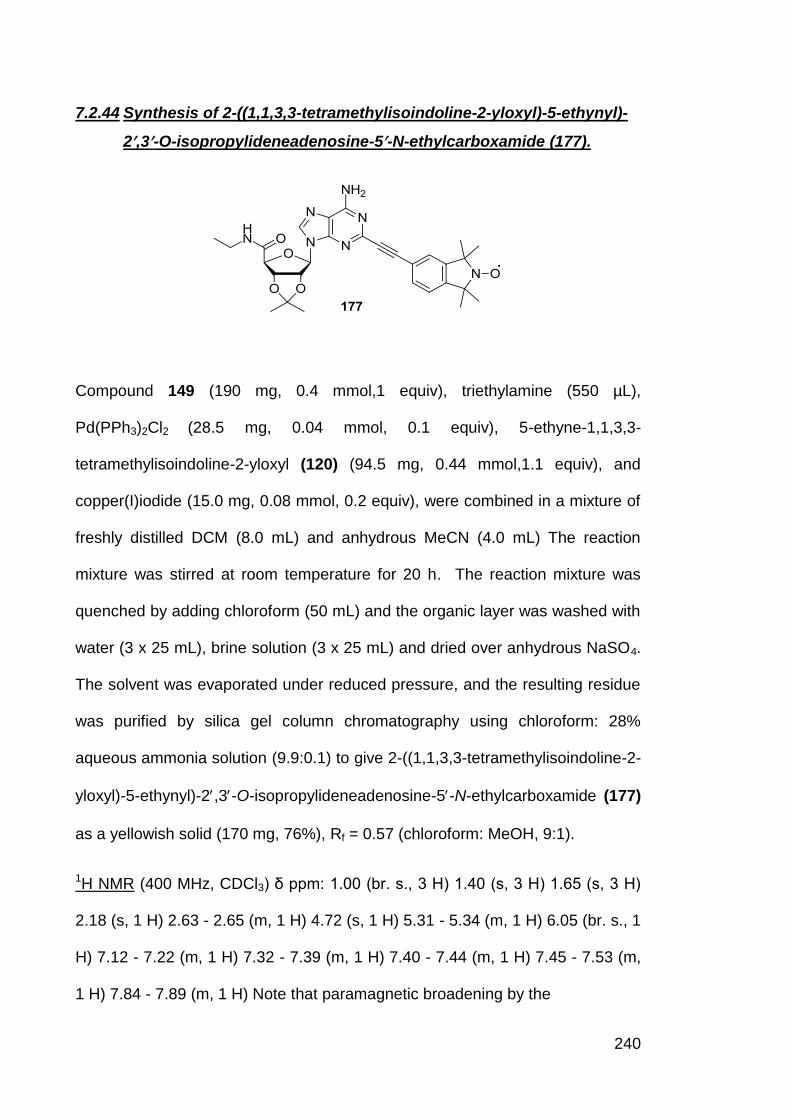
240
7.2.44 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-ethynyl)-
2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (177).
Compound 149 (190 mg, 0.4 mmol,1 equiv), triethylamine (550 µL),
Pd(PPh3)2Cl2 (28.5 mg, 0.04 mmol, 0.1 equiv), 5-ethyne-1,1,3,3-
tetramethylisoindoline-2-yloxyl (120) (94.5 mg, 0.44 mmol,1.1 equiv), and
copper(I)iodide (15.0 mg, 0.08 mmol, 0.2 equiv), were combined in a mixture of
freshly distilled DCM (8.0 mL) and anhydrous MeCN (4.0 mL) The reaction
mixture was stirred at room temperature for 20 h. The reaction mixture was
quenched by adding chloroform (50 mL) and the organic layer was washed with
water (3 x 25 mL), brine solution (3 x 25 mL) and dried over anhydrous NaSO4.
The solvent was evaporated under reduced pressure, and the resulting residue
was purified by silica gel column chromatography using chloroform: 28%
aqueous ammonia solution (9.9:0.1) to give 2-((1,1,3,3-tetramethylisoindoline-2-
yloxyl)-5-ethynyl)-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (177)
as a yellowish solid (170 mg, 76%), Rf = 0.57 (chloroform: MeOH, 9:1).
1H NMR (400 MHz, CDCl3) δ ppm: 1.00 (br. s., 3 H) 1.40 (s, 3 H) 1.65 (s, 3 H)
2.18 (s, 1 H) 2.63 - 2.65 (m, 1 H) 4.72 (s, 1 H) 5.31 - 5.34 (m, 1 H) 6.05 (br. s., 1
H) 7.12 - 7.22 (m, 1 H) 7.32 - 7.39 (m, 1 H) 7.40 - 7.44 (m, 1 H) 7.45 - 7.53 (m,
1 H) 7.84 - 7.89 (m, 1 H) Note that paramagnetic broadening by the

241
nitroxide radical results in some peaks not being detected or giving poor
integration in the proton NMR spectrum.
Ʋmax (ATR-FTIR) cm-1: 3634 (NH2), 3050 (alkyl CH3), 2220 (CΞC), 1668
(NC=O), 1572 (aryl C-N), 1490 (aryl C-C), 1454 (N-CO), 1377 (N-O), 1206 (H-
NCO), 1084 (C-O-C).
Melting point: 146–148 °C.
HRMS (ESI): m/z calcd for C29H35N7O5+ [M+H]: 561.2700, found: 561.2682.
HPLC purity: 97.4%.
EPR (methanol): typical 3-line nitroxide signal, g 1.9919, aN 1.4972 mT.
7.2.45 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethynyl)adenosine-5-N-ethylcarboxamide (178).
1 M HCl solution (3.5 mL) was added slowly in a stirring solution of compound
177 (55 mg, 0.98 mmol, 1 equiv) in MeCN (3 mL). The reaction mixture was
stirred for 8 h at 55–60 °C and the reaction was followed by TLC analysis.in
(EtOAc: MeOH, 8:2). Upon completion, the pH of the solution was adjusted up
to 8 via the slow addition of saturated NaHCO3 solution (25 mL). The resulting
mixture was extracted with EtOAc (5 x 50 mL). The organic phases were
washed with water (3 x 25 mL), brine solution (3 x 25 mL), and dried over

242
anhydrous Na2SO4. The solvent was filtered and the filtrate was evaporated
under reduced pressure to give 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethynyl)adenosine-5-N-ethylcarboxamide (178) as a pale yellow solid (50 mg,
98%), Rf = 0.57 (EtOAc: MeOH, 8:2).
1H NMR (400 MHz, acetone-d6) δ ppm: 1.10 (br. s., 12 H) 4.37 (br. s., 2 H) 4.43
(s, 3 H) 4.68 - 4.81 (m, 2 H) 4.91 (br. s., 6 H) 6.04 (d, J=6.55 Hz, 3 H) 7.96 -
7.98 (m, 1 H) 8.31 (s, 3 H) 8.58 - 8.61 (m, 1 H). Note that paramagnetic
broadening by the nitroxide radical results in some peaks not being detected or
giving poor integration in the proton NMR spectrum.
Ʋmax (ATR-FTIR) cm-1: 3634 (NH2), 3050 (alkyl CH3), 2220 (CΞC), 1668
(NC=O), 1572 (aryl C-N), 1490 (aryl C-C), 1454 (N-CO), 1377 (N-O), 1206 (H-
NCO), 1084 (C-O-C).
Melting point: 261 °C (decomp).
HRMS (ESI): m/z calcd for C26H30NaN7O5+ [M+Na]: 543.2206, found: 543.2281.
EPR (methanol): typical 3-line nitroxide signal, g 1.9928, aN 1.4876 mT.
HPLC purity: 99.99%.

243
7.2.46 Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-ethyl)-
2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (179).
Palladium (10% charcoal = 190 mg) was added to a stirring solution of
compound 177 (190 mg, 0.178 mmol, 1.equiv) in MeOH (50 mL). The reaction
mixture was stirred at room temperature using a balloon of H2 gas for 12 h and
the reaction progress was followed by TLC analysis in (CHCl3: MeOH, 9:1).
The reaction mixture was filtered through celite and the solvent was evaporated
under reduced pressure. The resulting residue was dissolved in MeOH and
added lead oxide (100 mg). The mixture was stirred for 1 h at room
temperature after which time filtered and dried through anhydrous Na2SO4 and
the solvent was evaporated under reduced pressure. The resulting residue was
purified by silica gel column chromatography using CHCl3: MeOH (9:1) to give
2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-ethyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (179) as an off-white solid (161
mg, 84%). Rf = 0.023 (CHCl3: MeOH, 9:1),
1H NMR (400 MHz, CDCl3) δ ppm 0.67 (t, J=7.29 Hz, 3 H) 1.40 (s, 3 H) 1.60 (s,
3 H) 1.75 - 1.87 (m, 12 H) 2.75 - 2.87 (m, 1 H) 2.95 - 3.04 (m, 1 H) 3.04 - 3.11
(m, 2 H) 3.11 - 3.18 (m, 2 H) 3.48 (s, 1 H) 4.69 (d, J=1.37 Hz, 1 H) 5.42 (d,

244
J=6.80 Hz, 1 H) 5.59 - 5.67 (m, 3 H) 6.04 (t, J=5.40 Hz, 1 H) 6.13 (s, 1 H) 7.00
(s, 1 H) 7.05 (d, J=7.82 Hz, 1 H) 7.29 (s, 1 H) 7.81 (s, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm 14.1, 25.2, 26.8, 33.7, 34.3, 40.6, 83.4, 83.8,
87.4, 91.4, 114.0, 121.3 (d, J=5.34 Hz, 1 C) 139.8, 155.2, 168.5. Note that
paramagnetic broadening by the nitroxide radical results in some peaks not
being detected or giving poor integration in the NMR spectrum.
Melting point: 108–110 °C. (Decomp.)
HRMS (ESI): m/z calcd for C29H38N7O5+ [M+]: 564.2934, found: 564.3276.
EPR (methanol): typical 3-line nitroxide signal, g 1.9928, aN 1.5066 mT.
HPLC purity- 96.72%.
7.2.47 .Synthesis of 2-((1,1,3,3-tetramethylisoindoline-2-yloxyl)-5-
ethyl)adenosine-5-N-ethylcarboxamide (180).
1 M HCl (10 mL) was slowly added in a stirring solution of compound 179 (150
mg, 0.155 mmol, 1 equiv) in MeCN (12.0 mL). The reaction mixture was stirred
for 5 h at 55–60 °C, and the reaction progress was followed by TLC analysis in
(CHCl3: MeOH, 8:2). Upon completion, the solution was basified up to pH 8 by
slow addition of saturated NaHCO3 solution (50 mL). The resulting suspension

245
was extracted with EtOAc (5 x 50 mL). The combined organic layers were
washed with water (3 x 50 mL) and brine solution (3 x 50 mL) and dried over
anhydrous Na2SO4, filtered. The solvent was evaporated under reduced
pressure to give a tan solid which was recrystallised (MeCN) to give 2-((1,1,3,3-
tetramethylisoindoline-2-yloxyl)-5-ethyl)adenosine-5-N-ethylcarboxamide (180)
as a pale yellow solid (110 mg, 79%). Rf = 0.16 (CHCl3: MeOH, 8:2).
1H NMR (400 MHz, Acetone-d6) δ ppm: 1.08 (t, J=7.16 Hz, 3 H) 1.31 - 1.31 (m,
1 H) 1.31 - 1.42 (m, 16 H) 2.98 - 3.08 (m, 2 H) 3.08 - 3.16 (m, 2 H) 3.26 (d,
J=7.09 Hz, 1 H) 3.31 - 3.43 (m, 1 H) 3.55 - 3.61 (m, 1 H) 4.38 - 4.44 (m, 2 H)
4.88 (dd, J=7.24, 4.60 Hz, 1 H) 6.02 (d, J=7.43 Hz, 1 H) 6.68 (br. s., 1 H) 7.00
(s, 1 H) 7.04 - 7.09 (m, 1 H) 7.09 - 7.15 (m, 1 H) 8.20 (s, 1 H) 8.23 (br. s., 1 H).
13C NMR (101 MHz, Acetone-d6) δ ppm: 14.7, 31.5, 31.6, 33.5, 34.7, 41.3, 62.0
(d, J=13.73 Hz, 1 C) 72.0, 73.7, 84.9, 88.7, 121.2 (d, J=3.05 Hz, 1 C) 127.1,
140.5, 140.7, 146.6, 149.1, 150.3, 156.2, 164.5, 169.2. Note that paramagnetic
broadening by the nitroxide radical results in some peaks not being detected or
giving poor integration in the NMR spectra
Ʋmax (ATR-FTIR) cm-1: 3198(OH), 2924 (alkyl CH3), 1636 (NC=O), 1581, 1388
(N-O), 1120, 1046, 797.
Melting point: 122-124 °C
HRMS (ESI): m/z calcd for C26H35N7O5+ [M+H]: 525.2700, found: 525.2833.
EPR (methanol): typical 3-line nitroxide signal, g 1.9939, aN 1.5006 mT.
HPLC purity- >99.9%.

246
7.2.48 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-ethynyl)-
2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (181).
Compound 149 (100 mg, 0.211 mmol, 1 equiv), 5-ethyne-(1,1,3,3-
tetraethylisoindoline-2-yloxyl (122) (57 mg, 0.211 mmol, 1 equiv), Pd(PPh3)2Cl2
(15 mg, 0.021 mmol, 0.1 equiv), copper iodide (8 mg, 0.042 mmol, 0.2 equiv),
and triethylamine (0.25 mL) were combined in a mixture of freshly distilled DCM
(6 mL) and anhydrous MeCN (3 mL). Argon was bubbled for 10 min, and the
reaction mixture was stirred at room temperature for 3 days and the reaction
progress was followed by TLC analysis using (DCM / MeOH, 9:1). The
chloroform (25 mL) was added to the reaction mixture and the organic layer was
washed with water (3 x 25 mL), and brine solution (3 x 25 mL) and dried over
anhydrous Na2SO4. The solvent was evaporated under reduced pressure and
the resulting residue was purified by silica gel column chromatography using a
mixture of (9:1) EtOAc: Hexane to give 2-((1,1,3,3-tetraethylisoindoline-2-
yloxyl)-5-ethynyl)-2,3-O-isopropylideneadenosine-5-N-ethylcarboxamide (181)
as a pale yellow solid (107 mg, 83%), Rf = 0.47 (DCM / MeOH, 9:1).
1H NMR (400 MHz, CDCl3)(Me trap) δ ppm: 0.58 - 1.05 (m, 5 H) 1.23 (br. s., 8
H) 1.57 (br. s., 13 H) 1.85 - 2.18 (m, 3 H) 2.97 (s, 5 H) 3.47 (br. s., 1 H) 3.66 (br.
s., 1 H) 4.63 (br. s., 1 H) 4.89 (br. s., 1 H) 5.60 - 5.92 (m, 1 H) 7.02 (d, J=6.85
Hz, 1 H) 7.36 - 7.57 (m, 2 H) 7.58 - 7.69 (m, 1 H) 7.89 (br. s., 1 H). Note that

247
paramagnetic broadening by the nitroxide radical results in some peaks not
being detected or giving poor integration in the proton NMR spectrum.
Ʋmax (ATR-FTIR) cm-1: 3400-3100 (OH, NH2), 2971 and 2960 (alkyl CH3 and
CH2), 2214 (CΞC), 1668 (NC=O), 1593 (aryl C-N), 1521 (aryl C-C), 1487 (N-
CO), 1374 (N-O), 1259, 1206 (H-NCO), 1156, 1083 (C-O-C).
Melting point: 188–190 °C.
HRMS (ESI): m/z calcd for C33H43N7O5+ [M + H]: 617.3326, found: 617.3296.
EPR (methanol): typical 3-line nitroxide signal, g 1.9913, aN 1.4200 mT.
HPLC purity- >99.9%.
7.2.49 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-
ethynyl)adenosine-5-N-ethylcarboxamide (182).
1 M HCl solution (5 mL) was added slowly.in a stirring solution of compound
181 (70 mg, 0.94 mmol, 1 equiv) was dissolved in MeCN (5.0 mL). The
reaction mixture was stirred for 3 h at 55–60 °C, and the reaction progress was
followed by TLC analysis in 100% EtOAc. Upon completion, the solution was
basified up to pH 8 by adding saturated NaHCO3 solution (25 mL). The
resulting suspension was extracted with EtOAc (5 x 50 mL). The organic
extracts were washed with water (3 x 25 mL) and brine solution (3 x 25 mL) and
dried over anhydrous Na2SO4, filtrated. The solvent was evaporated under

248
reduced pressure and the resulting residue was purified by silica gel column
chromatography using EtOAc to give 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-
5-ethynyl)adenosine-5-N-ethylcarboxamide (182) as a tan solid (57 mg, 88%),
Rf = 0.53 (100% EtOAc).
1H NMR (400 MHz, CDCl3) δ ppm: 0.73 (br. s., 3 H) 0.81 - 1.00 (m, 8 H) 1.11
(s., 2H)1.23 (br. s., 15 H) 2.02 (br. s., 5 H) 2.97 (s, 3 H) 3.47 (br. s., 2 H) 3.54 -
3.74 (m, 4 H) 4.49 - 4.69 (m, 1 H) 4.89 (br. s., 1 H) 5.68 - 5.88 (m, 1 H) 7.00 (d,
J=5.18 Hz, 1 H) 7.44 (d, J=1.52 Hz, 1 H) 7.52 (d, J=6.46 Hz, 1 H) 7.58 - 7.69
(m, 1 H) 7.90 (br. s., 1 H) Note that paramagnetic broadening by the nitroxide
radical results in some peaks not being detected or giving poor integration in the
proton NMR spectrum.
Ʋmax (ATR-FTIR) cm-1: 3400-3100 (OH, NH2), 2971 and 2960 (alkyl CH3 and
CH2), 2214 (CΞC), 1668 (NC=O), 1593, 1521, 1487, 1374 (N-O), 1259, 1206,
1156, 1083, 861,796.
Melting Point: 196–198 °C (decomp).
HRMS (ESI): m/z calcd for C30H39N7O5+ [M+H]: 577.3013, found: 577.3012.
EPR (methanol): typical 3-line nitroxide signal, g 1.9928, aN 1.4113 mT.
HPLC purity- >99.9%.

249
7.2.50 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-ethyl)-2,3-
O-isopropylideneadenosine-5-N-ethylcarboxamide (183).
Palladium (10 % charcoal = 50 mg) was added in a stirring solution of
compound 181 (68 mg, 0.11 mmol, 1.0 equiv) in MeOH (4 mL). The reaction
mixture was stirred at room temperature under a balloon full of H2 gas for 12 h
and the reaction progress was followed by TLC analysis in (DCM: MeOH, 9:1).
The mixture was filtered through celite and the solvent was evaporated under
reduced pressure. The resulting residue was dissolved in MeOH and added
lead oxide (100 mg). The mixture was stirred for 1 h at room temperature after
which time filtered and dried through anhydrous Na2SO4 and the solvent was
evaporated under reduced pressure. The resulting residue was purified by
silica gel column chromatography using CHCl3 to give 2-((1,1,3,3-
tetraethylisoindoline-2-yloxyl)-5-ethyl)-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (183) as a yellow solid (66 mg, 96.5%), Rf = 0.76 (DCM:
MeOH, 9:1).
1H NMR (400 MHz, CDCl3) δ ppm 0.68 (t, J=6.90 Hz, 3 H) 1.38 (s, 12 H) 1.52 -
1.65 (m, 11 H) 2.84 - 2.86 (m, 2 H) 3.07 - 3.12 (m, 4 H) 4.68 (s., 1 H) 5.52 (br s,
2 H) 6.09-6.12 (s, 1 H) 6.18 (br s, 2 H) 6.89 (s, 1 H) 6.94 - 6.99 (m, 1 H) 7.12 -
7.14 (d, 1 H) 7.79 (br. s., 1 H).

250
13C NMR (101 MHz, CDCl3) δ ppm: 9.0, 25.3, 26. 9, 33.8, 34.4, 67.9, 82.1,
83.1, 83.3, 83.8, 87.3, 90.0, 91.4, 103.7, 110.0, 114.0, 118.3, 139.7, 168.5,
185.2, 217.3.
Melting point: 88–90 °C.
HRMS (ESI): m/z calcd for C33H47N7O5+ [M+H]: 621.3639, found: 621.3630.
HPLC purity: 88.72%.
EPR (methanol): typical 3-line nitroxide signal, g 1.9892, aN 1.4462 mT.
7.2.51 Synthesis of 2-((1,1,3,3-tetraethylisoindoline-2-yloxyl)-5-
ethyl)adenosine-5-N-ethylcarboxamide (184).
1 M HCl solution (5 mL) was added slowly in a stirred solution of compound 183
(66 mg, 0.106 mmol, 1 equiv) in MeCN (5.0 mL). The solution was stirred for 5
h at 55–60 °C and the reaction progress was followed by TLC analysis in DCM:
MeOH (9:1). Upon completion, the solution was basified up to pH 8 by adding
saturated NaHCO3 solution (25 mL). The resulting suspension was extracted
with EtOAc (5 x 50 mL). The combined organic phases were washed with
water (3 x 25 mL) and brine solution (3 x 25 mL) and filtered and dried over
anhydrous Na2SO4. The solvent was evaporated under reduced pressure

251
to give a tan solid, which was recrystallised (MeCN) to give 2-((1,1,3,3-
tetraethylisoindoline-2-yloxyl)-5-ethyl)adenosine-5-N-ethylcarboxamide (184)
as a pale yellow solid (43 mg, 70%). Rf = 0.69 (DCM: MeOH, 9:1).
Ʋmax (ATR-FTIR) cm-1: 3400-3100 (NH2, OH,), 2925 (alkyl CH3), 2214 (CΞC),
1636 (NC=O), 1581(aryl C-N), 1459 (N-CO), 1389 (N-O), 1119, 1046 (C-O-C).
Melting point: 138–140 °C
HRMS (ESI): m/z calcd for C30H43N7O5+ [M+H]: 581.3326, found: 581.3277.
HPLC purity:- 99.99%.
EPR (methanol): typical 3-line nitroxide signal, g 1.9921, aN 1.4609 mT.
7.2.52 Synthesis of 2-(2-Phenylethynyl)-2,3-O-isopropylideneadenosine -
5-N-ethylcarboxamide (185).
Compound 149 (100 mg, 0.211 mmol, 1 equiv), phenylacetylene 123 (57 mg,
0.232 mmol, 1.1 equiv), Pd(PPh3)2Cl2 (15 mg, 0.021 mmol, 0.1 equiv), copper
iodide (8 mg, 0.042 mmol, 0.2 equiv), and triethylamine (0.3 mL) were
combined in a mixture of freshly distilled DCM (6 mL) and anhydrous MeCN (3
mL). Argon was bubbled through a mixture for 10 minutes. The reaction
mixture was stirred at room temperature for 15 h and the reaction progress was

252
followed by TLC analysis in (DCM: MeOH, 9:1). The CHCl3 was added to a
mixture and resulting solution was washed with water (3 x 25 mL) and brine
solution (3 x 25 mL). The solvent was evaporated under reduced pressure and
the resulting residue was purified by silica gel column chromatography using
CHCl3 to give 2-(2-Phenylethynyl)-2,3-O-isopropylideneadenosine-5-N-
ethylcarboxamide (185) as a brownish solid (91 mg, 97%).
1H NMR (400 MHz, CDCl3) δ ppm: 0.94 - 1.00 (m, 3 H) 1.36 (s, 3 H) 1.62 (s, 3
H) 3.16 - 3.30 (m, 1 H) 3.37 (dt, J=13.60, 6.94 Hz, 1 H) 4.69 (s, 1 H) 5.24 (d,
J=5.18 Hz, 1 H) 5.30 (br. s., 1 H) 6.00 (d, J=1.37 Hz, 1 H) 7.35 (br. s., 1 H) 7.35
- 7.40 (m, 3 H) 7.58 – 7.61(m, 2H).
13C NMR (101 MHz, CDCl3) δ ppm: 14.7 , 25.3 , 27.3 , 34.1 , 76.7 , 77.0 , 77.2 ,
77.3 , 82.0 , 83.4 , 84.9 , 114.9 , 128.4 , 129.5 , 132.3 , 168.5 .
Ʋmax (ATR-FTIR) cm-1: 3356 (NH2), 3156 (OH), 2214 (CΞC), 1654 (NC=O),
1588 (aryl C-N), 1522 (aryl C-C), 1492, 1457 (N-CO), 1380, 1203 (H-NCO),
1155, 1082 (C-O-C).
Melting point: 174–176 °C ;(lit )
HRMS (ESI): m/z calcd for C23H25N6O4+ [M + H]: 449.1937, found: 449.1881.
HPLC purity- 99.99%.

253
7.2.53 Synthesis of 2-(2-phenylethyl)-2,3-O-isopropylideneadenosine-5-
N-ethylcarboxamide (187).
Palladium (10% charcoal = 80 mg) was added to a stirred solution of compound
185 (80 mg, 0.178 mmol, 1.0 eq) in MeOH (10 mL). The reaction mixture was
stirred at room temperature using a balloon of H2 gas atmosphere for 12 h and
the reaction progress was followed by TLC analysis in (DCM: MeOH, 9:1). The
mixture was filtered through celite and the filtrate was evaporated under
reduced pressure. The resulting residue was purified by silica gel column
chromatography using a DCM: MeOH (9:1) to give 2-(2-phenylethyl)-2,3-O-
isopropylideneadenosine-5-N-ethylcarboxamide (187) as a yellow solid (75 mg,
93 %). Rf = 0.56 (DCM: Methanol, 9:1).
1H NMR (400 MHz, CDCl3): δ ppm 0.67 (t, J=7.24 Hz, 4 H) 1.37 (s, 3 H) 1.58 (s,
3H) 2.77 - 2.86 (m, 1 H) 2.99 - 3.05 (m, 1 H) 3.05 - 3.14 (m, 4 H) 4.65 (d, J=1.66
Hz, 1 H) 5.25 (d, J=4.65 Hz, 1 H) 5.47 - 5.55 (m, 3 H) 6.05 (d, J=1.37 Hz, 1 H)
7.15 (d, J=6.60 Hz, 1 H) 7.19 - 7.23 (m, 3 H) 7.76 (s, 1 H).
13C NMR (75 MHz, DMSO- d6): δ ppm: 14.1, 25.2, 26.9, 33.7, 34.5, 40.5, 83.2,
83.8, 86.2, 91.4, 114.0, 118.3, 125.8, 128.3, 128.4, 139.7, 141.5, 155.5, 165.4
and 168.5.

254
Melting point: 70–72 °C.
HRMS (ESI): m/z calcd for C23H29N6O4+ [M+H]: 453.2250, found: 453.2243.
HPLC purity- 98.32%.
7.2.54 Synthesis of 2-(2-phenylethyl)adenosine-5-N-ethylcarboxamide
(188).
1 M HCl solution (6 mL) was added slowly in a stirring solution of compound
187 (70 mg, 0.155 mmol, 1 equiv) in MeCN (6.0 mL). The reaction mixture was
stirred for 5 h at 55–60 °C and the reaction progress was followed by TLC
analysis in (DCM: MeOH, 9:1). Upon completion, the solution was basified up
to the pH 8 by adding saturated NaHCO3 solution (25 mL). The resulting
suspension was extracted with EtOAc (5 x 25 mL) and the combined organic
layers were washed with water (3 x 25 mL) and brine solution (3 x 25 mL) and
dried over anhydrous Na2SO4. The solvent was evaporated under reduced
pressure to give a brownish solid which was recrystallised (MeCN) to give 2-(2-
phenylethyl)adenosine-5-N-ethylcarboxamide (188) as a pale yellow solid (60
mg, 94%). Rf = 0.38 (DCM: MeOH, 9:1).
1H NMR (400 MHz, CDCl3) δ ppm: 1.06 (t, J=7.26 Hz, 3 H) 2.03 (s, 2 H) 3.07 (d,
J=4.55 Hz, 4 H) 3.16 - 3.21 (m, 1 H) 3.37 - 3.43 (m, 1 H) 4.57 (s, 1 H) 4.63 -

255
4.68 (m, 2 H) 5.63 (br. s, 2 H) 5.83 (d, J=5.48 Hz, 1 H) 7.15 - 7.19 (m, 3 H) 7.23
(br. s., 1 H) 7.24(br. s, 1 H) 7.84 (s, 1 H).
13C NMR (101 MHz, CDCl3) δ ppm: 14.7 , 25.3 , 27.3 , 34.1 , 76.7 , 77.0 , 77.2 ,
77.3 , 82.0 , 83.4 , 84.9 , 114.9 , 128.4 , 129.5 , 132.3 and 168.5.
Ʋmax (ATR-FTIR) cm-1: 3330 (NH2), 3225 (OH), 2921 (aryl C-H), 1643 (NC=O),
1588 (aryl C-N), 1522 (aryl C-C), 1467 (N-CO), 1396, 1315, 1203 (H-NCO),
1196, 1143, 1115, 1080 (C-O-C), 1043, 968, 870.
Melting point: 216-217 °C.(decomp.)
HRMS (ESI): m/z calcd for C20H25N6O4+ [M+H]: 413.1937, found: 413.2019.
HPLC purity- 100%.

256
BIBLIOGRAPHY 8

257
1. Ogunseitan, Oladele ed., I. Cardiovascular Diseases. Green Health: An A-to-Z Guide. SAGE Publications, Inc. SAGE Publications, Inc.: Thousand Oaks, CA, 2011, 9.
2. Guttmacher, A. E. M. D.; Collins, F. S. M. D. P.; Nabel, E. G. M. D. Cardiovascular disease. The New England Journal of Medicine 2003, 349, 60-72.
3. Cai, H.; Harrison, D. G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation Research 2000, 87, 840-844.
4. Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafi, M. Oxidative stress in atherosclerosis development: the central role of LDL and oxidative burst. Endocrine, Metabolic & Immune Disorders-Drug Targets (Formerly Current Drug Targets-Immune, Endocrine & Metabolic Disorders) 2012, 12, 351-360.
5. Touyz, R. M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension what is the clinical significance? Hypertension 2004, 44, 248-252.
6. Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertension Research 2011, 34, 431-440.
7. Anderson, K. M.; Odell, P. M.; Wilson, P. W.; Kannel, W. B. Cardiovascular disease risk profiles. American Heart Journal 1991, 121, 293-298.
8. Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arteriosclerosis, Thrombosis, and Vascular Biology 2004, 24, 816-823.
9. Ganzit, G. P.; Stefanini, L. Cardiovascular diseases and physical activity. In 1 ed.; SEEd: Torino, 2012.
10. Abegunde, D. O.; Mathers, C. D.; Adam, T.; Ortegon, M.; Strong, K. The burden and costs of chronic diseases in low-income and middle-income countries. The Lancet 2007, 370, 1929-1938.
11. Grace, P. A. Ischaemia-reperfusion injury. British Journal of Surgery 1994, 81, 637-647.
12. Bhatia, S. K. Biomaterials for clinical applications. Springer Science & Business Media: 2010.
13. Mittal, B. V.; Singh, A. K. Hypertension in the developing world: challenges and opportunities. American Journal of Kidney Diseases 2010, 55, 590-598.
14. Selzer, A., M.D. Understanding heart disease. . In University of California Press: Berkeley, 1992.

258
15. McCord, J. M. Oxygen-derived free radicals in postischemic tissue injury. New England Journal of Medicine 1985, 312, 159-163.
16. Gutterman, D. D. Silent myocardial ischemia. Circulation Journal 2009, 73, 785-797.
17. Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia–ischemia in the developing brain. Free Radical Biology and Medicine 2006, 40, 388-397.
18. Randall, O. S. R., Deborah S. Segerson, Nathan M. . The encyclopedia of the heart and heart disease. In 2nd ed.; Facts On File New York, NY, USA 2010.
19. Chervu, A.; Moore, W. S.; Homsher, E.; Quinones-Baldrich, W. J. Differential recovery of skeletal muscle and peripheral nerve function after ischemia and reperfusion. Journal of Surgical Research 1989, 47, 12-9.
20. Kerrigan, C. L., Stotland, Mitchell A. Ischemia reperfusion injury: A review. Microsurgery 1993, 14, 165-175.
21. Anaya-Prado, R.; Toledo-Pereyra, L. H.; Lentsch, A. B.; Ward, P. A. Ischemia/reperfusion injury. Journal of Surgical Research 2002, 105, 248-258.
22. Halliwell, B.; Gutteridge, J.; Cross, C. Free radicals, antioxidants, and human disease: where are we now? The Journal of Laboratory and Clinical Medicine 1992, 119, 598.
23. Wink, D. A.; Mitchell, J. B. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radical Biology and Medicine 1998, 25, 434-456.
24. Granger, D. N. Role of xanthine oxidase and neutrophils in ischemia-reperfusion injury in rabbit lung. 1990; Vol. 69, p 2012-2018.
25. Tyler, D. D. Polarographic assay and intracellular distribution of superoxide dismutase in rat liver. Biochem. J 1975, 147, 493-504.
26. Granger, D. N. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. American Journal of Physiology-Heart and Circulatory Physiology 1988, 255, H1269-H1275.
27. Halliwell, B.; Gutteridge, J. Biologically relevant metal ion-dependent hydroxyl radical generation An update. Febs Letters 1992, 307, 108-112.
28. Wink, D. A.; Wink, C. B.; Nims, R. W.; Ford, P. C. Oxidizing intermediates generated in the Fenton reagent: kinetic arguments against the intermediacy of the hydroxyl radical. Environmental Health Perspectives 1994, 102, 11.
29. Halliwell, B.; Clement, M. V.; Ramalingam, J.; Long, L. H. Hydrogen peroxide. Ubiquitous in cell culture and in vivo? IUBMB life 2000, 50, 251-257.

259
30. Halliwell, B.; Clement, M. V.; Long, L. H. Hydrogen peroxide in the human body. Febs Letters 2000, 486, 10-13.
31. Gomberg, M. An instance of trivalent carbon: Triphenylmethyl. Journal of the American Chemical Society 1900, 22, 757-771.
32. Fang, Y.-Z.; Yang, S.; Wu, G. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872-879.
33. Kalra, J.; Chaudhary, A.; Prasad, K. In Cigarette-smoking and oxygen free-radicals producing activity of polymorphonuclear leukocytes, Clinical Pharmacology & Therapeutics, 1989; Mosby-year book Inc 11830 Westline Industrial Dr, St Louis, Mo 63146-3318: 1989; pp 178-178.
34. Nonhebel, D. a. J. W. Free radical chemistry: structure and mechanism
1974.
35. Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V. B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: the new wave? Trends in Plant Science 2011, 16, 300-309.
36. Zhang, M.; Shah, A. M. ROS signalling between endothelial cells and cardiac cells. Cardiovascular Research 2014, 102, 249-257.
37. Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions 2006, 160, 1-40.
38. Maddika, S.; Elimban, V.; Chapman, D.; Dhalla, N. S. Role of oxidative stress in ischemia-reperfusion-induced alterations in myofibrillar ATPase activities and gene expression in the heart. Canadian Journal of Physiology & Pharmacology 2009, 87, 120-129.
39. Westerblad, H.; Allen, D. G. Emerging roles of ROS/RNS in muscle function and fatigue. Antioxidants & Redox Signaling 2011, 15, 2487-2499.
40. Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Disease Markers 2013, 35, 773-790.
41. Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radical Biology and Medicine 2003, 34, 1507-1516.
42. Binger, C. A.; Faulkner, J. M.; Moore, R. L. Oxygen poisoning in mammals. The Journal of Experimental Medicine 1927, 45, 849.
43. Gerschman, R.; Gilbert, D. L.; Nye, S. W.; Dwyer, P.; Fenn, W. O. Oxygen poisoning and x-irradiation: a mechanism in common. Science of Aging Knowledge Environment 2005, 2005, cp1.

260
44. Michaelson, L. P.; Iler, C.; Ward, C. W. ROS and RNS signaling in skeletal muscle: Critical signals and therapeutic targets. Annual Review of Nursing Research 2013, 31, 367-X.
45. Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nature Reviews Drug Discovery 2007, 6, 662-680.
46. Finkel, T.; Holbrook, N. J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239-247.
47. Harman, D. Aging: a theory based on free radical and radiation chemistry. University of California Radiation Laboratory: 1955.
48. Harman, D. The biologic clock: the mitochondria? Journal of the American Geriatrics Society 1972, 20, 145-147.
49. Leeuwenburgh, C.; Heinecke, J. W. Oxidative stress and antioxidants in exercise. Current Medicinal Chemistry 2001, 8, 829-38.
50. Preiser, J.-C. Oxidative stress. Journal of Parenteral and Enteral Nutrition 2012, 36, 147-154.
51. Morrow, J. D.; Frei, B.; Longmire, A. W.; Gaziano, J. M.; Lynch, S. M.; Shyr, Y.; Strauss, W. E.; Oates, J. A.; Roberts, L. J. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers—smoking as a cause of oxidative damage. New England Journal of Medicine 1995, 332, 1198-1203.
52. Wikipedia. Oxidation http://simple.wikipedia.org/wiki/Oxidation.
53. Sies, H. Oxidative stress: oxidants and antioxidants. Experimental Physiology 1997, 82, 291-295.
54. Lockwood, S. F.; Gross, G. J. Disodium disuccinate astaxanthin (CardaxTM): antioxidant and antiinflammatory cardioprotection. Cardiovascular Drug Reviews 2005, 23, 199-216.
55. Pepys, M. B.; Hirschfield, G. M.; Tennent, G. A.; Gallimore, J. R.; Kahan, M. C.; Bellotti, V.; Hawkins, P. N.; Myers, R. M.; Smith, M. D.; Polara, A. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 2006, 440, 1217-1221.
56. Dafré, A. L.; Sies, H.; Akerboom, T. Protein S-thiolation and regulation of microsomal glutathione transferase activity by the glutathione redox couple. Archives of Biochemistry and Biophysics 1996, 332, 288-294.
57. Aronovitch, Y.; Godinger, D.; Israeli, A.; Krishna, M. C.; Samuni, A.; Goldstein, S. Dual activity of nitroxides as pro- and antioxidants: Catalysis of copper-mediated DNA breakage and H2O2 dismutation. Free Radical Biology & Medicine 2007, 42, 1317-1325.

261
58. Brivba K., S. H. Non enzymatic antioxidant defense systems, in Natural Antioxidants in Human Health and Disease. San Diego: Academic Press: Orlando, FL, USA., 1994. ; p pp. 107–128.
59. Huang, D.; Ou, B.; Prior, R. L. The chemistry behind antioxidant capacity assays. Journal of Agricultural and Food Chemistry 2005, 53, 1841-1856.
60. Teixeira, J.; Gaspar, A.; Garrido, E. M.; Garrido, J.; Borges, F. Hydroxycinnamic acid antioxidants: an electrochemical overview. BioMed Research International 2013, 2013.
61. McCord, J. M.; Fridovich, I. SOD enzyme function for erythrocuprein. J. Biol. Chem. 1969, 224, 6049-6055.
62. Palmer, R. M. J.; Ashton, D. S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664-666.
63. Lam, M. A.; Pattison, D. I.; Bottle, S. E.; Keddie, D. J.; Davies, M. J. Nitric oxide and nitroxides can act as efficient scavengers of protein-derived free radicals. Chemical Research in Toxicology 2008, 21, 2111-2119.
64. Cho, J.; Son, S.; Ko, S.; Lee, M.; Byun, S.; Lee, Y.; Park, S.; Kim, J.; Gwag, B. In Nonclinical safety and pharmacokinetic profile of AAD-2004 as a putative disease-modifying drug for neurodegenerative diseases, The 9th international conference AD/PD, 2009, pp 11-15.
65. Shin, J. H.; Lee, Y. A.; Lee, J. K.; Lee, Y. B.; Cho, W.; Im, D. S.; Lee, J. H.; Yun, B. S.; Springer, J. E.; Gwag, B. J. Concurrent blockade of free radical and microsomal prostaglandin E synthase-1-mediated PGE2 production improves safety and efficacy in a mouse model of amyotrophic lateral sclerosis. Journal of Neurochemistry 2012, 122, 952-961.
66. Jackson, D.; Sammut, I. Oxygen free radical traps for the treatment of ischemia-associated organ injury. Current Opinion in Investigational Drugs (London, England: 2000) 2004, 5, 50-54.
67. Lees, K. R.; Zivin, J. A.; Ashwood, T.; Davalos, A.; Davis, S. M.; Diener, H.-C.; Grotta, J.; Lyden, P.; Shuaib, A.; Hårdemark, H.-G. NXY-059 for acute ischemic stroke. New England Journal of Medicine 2006, 354, 588-600.
68. Griffith, O. H.; McConnell, H. M. A nitroxide-maleimide spin label. Proceedings of the National Academy of Sciences of the United States of America 1966, 55, 8.
69. Griffith, O. H.; Cornell, D. W.; McConnell, H. M. Nitrogen hyperfine tensor and g tensor of nitroxide radicals. The Journal of Chemical Physics 2004, 43, 2909-2910.
70. Forrester, A. R.; Hepburn, S. P. Nitroxide radicals. Part XV. p-Methoxy-and p-phenoxy-phenyl t-butyl nitroxides. Journal of the Chemical Society, Perkin Transactions 1 1974, 2208-2213.

262
71. Mukherjee, S. Syntheses of nitroxide diradicals and tetraradicals. Ph.D., The University of Nebraska - Lincoln, Ann Arbor, 2006.
72. Nonhebel, D. C.; Walton, J. C. Free-radical chemistry; structure and mechanism. CUP Archive: 1974.
73. Hicks, R. Stable radicals: fundamentals and applied aspects of odd-electron compounds. John Wiley & Sons: 2011.
74. Volodarsky, L. B.; Reznikov, V. A.; Ovcharenko, V. I. Synthetic chemistry of stable nitroxides. CRC Press Boca Raton, FL: 1994.
75. Soule, B. P.; Hyodo, F.; Matsumoto, K.-i.; Simone, N. L.; Cook, J. A.; Krishna, M. C.; Mitchell, J. B. The chemistry and biology of nitroxide compounds. Free Radical Biology and Medicine 2007, 42, 1632-1650.
76. Griffiths, P.; Moad, G.; Rizzardo, E. Synthesis of the radical scavenger 1,1,3,3-Tetramethylisoindolin-2-yloxyl. Australian Journal of Chemistry 1983, 36, 397-401.
77. Bottle, S.; Busfield, W.; Grice, I.; Heiland, K.; Jenkins, I.; Meutermans, W.; Monteiro, M. Some recent developments in the aminoxyl radical trapping technique. In Progress in Pacific Polymer Science 3, Springer: 1994; pp 85-97.
78. Keddie, D. J.; Johnson, T. E.; Arnold, D. P.; Bottle, S. E. Synthesis of profluorescent isoindoline nitroxides via palladium-catalysed Heck alkenylation. Organic and Biomolecular Chemistry 2005, 3, 2593-2598.
79. Fairfull-Smith, K. E.; Blinco, J. P.; Keddie, D. J.; George, G. A.; Bottle, S. E. A novel profluorescent dinitroxide for imaging polypropylene degradation. Macromolecules 2008, 41, 1577-1580.
80. Hosokawa, K.; Chen, P.; Lavin, F. M.; Bottle, E. S. The impact of carboxy nitroxide antioxidants on irradiated ataxia telangiectasia cells. Free Radical Biology and Medicine 2004, 37, 946-952.
81. Krishna, M. C.; Halevy, R. F.; Zhang, R. L.; Gutierrez, P. L.; Samuni, A. Modulation of streptonigrin cytotoxicity by nitroxide sod mimics. Free Radical Biology and Medicine 1994, 17, 379-388.
82. Mitchell, J. B.; Xavier, S.; DeLuca, A. M.; Sowers, A. L.; Cook, J. A.; Krishna, M. C.; Hahn, S. M.; Russo, A. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Radical Biology and Medicine 2003, 34, 93-102.
83. Kuppusamy, P.; Chzhan, M.; Vij, K.; Shteynbuk, M.; Lefer, D. J.; Giannella, E.; Zweier, J. L. Three-dimensional spectral-spatial EPR imaging of free radicals in the heart: a technique for imaging tissue metabolism and oxygenation. Proceedings of the National Academy of Sciences 1994, 91, 3388-3392.

263
84. Kao, J. P.; Barth, E. D.; Burks, S. R.; Smithback, P.; Mailer, C.; Ahn, K.
H.; Halpern, H. J.; Rosen, G. M. Very‐low‐frequency electron paramagnetic resonance (EPR) imaging of nitroxide‐loaded cells. Magnetic Resonance in Medicine 2007, 58, 850-854.
85. Brustad, T.; Bugge, H.; Jones, W.; Wold, E. Reactions between organic nitroxyl free radicals and radiation-induced transients in the DNA bases. International Journal of Radiation Biology 1972, 22, 115-129.
86. Emmerson, P. T.; Howard-Flanders, P. Sensitization of anoxic bacteria to x-rays by di-t-butyl nitroxide and analogues. Nature 1964, 204, 1005-1006.
87. Gubskaya, V. P.; Berezhnaya, L. S.; Gubaidullin, A. T.; Faingold, I. I.; Kotelnikova, R. A.; Konovalova, N. P.; Morozov, V. I.; Litvinov, I. A.; Nuretdinov, I. A. Synthesis, structure and biological activity of nitroxide malonate methanofullerenes. Organic & Biomolecular Chemistry 2007, 5, 976-981.
88. Likhtenshtein, G. I. Novel fluorescent methods for biotechnological and biomedical sensoring: assessing antioxidants, reactive radicals, NO dynamics, immunoassay, and biomembranes fluidity. Applied Biochemistry and Biotechnology 2009, 152, 135-155.
89. Kato, N.; Yanaka, K.; Hyodo, K.; Homma, K.; Nagase, S.; Nose, T. Stable nitroxide Tempol ameliorates brain injury by inhibiting lipid peroxidation in a rat model of transient focal cerebral ischemia. Brain Research 2003, 979, 188-193.
90. Wilcox, C. S.; Pearlman, A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacological Reviews 2008, 60, 418-469.
91. Quinu, J.; Delbridge, L.; Proietto, J.; Ritchie, R. In Cardioprotective actions of the antioxidant tempol in GLUT4-deficient mice, Clinical and Experimental Pharmacology and Physiology, 2004; Blackwell Publishing Asia 54 University St, Po Box 378, Carlton, Victoria 3053, Australia: 2004; pp A25-A25.
92. Goldstein, S.; Samuni, A.; Russo, A. Reaction of cyclic nitroxides with nitrogen dioxide: the intermediacy of the oxoammonium cations. Journal of the American Chemical Society 2003, 125, 8364-8370.
93. Tabaczar, S.; Talar, M.; Gwoździński, K. [Nitroxides as antioxidants-possibilities of their application in chemoprevention and radioprotection]. Postepy Higieny i Medycyny Doswiadczalnej (Online) 2010, 65, 46-54.
94. Weil, J. T.; Van der Veen, J.; Olcott, H. Stable nitroxides as lipid antioxidants. 1968.
95. Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? British Journal of Pharmacology 2004, 142, 231-255.

264
96. Gutteridge, J. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clinical Chemistry 1995, 41, 1819-1828.
97. Chen, X.; Sun, F.; Ma, L.; Wang, J.; Qin, H.; Du, G. In vitro evaluation on the antioxidant capacity of triethylchebulate, an aglycone from Terminalia chebula Retz fruit. Indian Journal of Pharmacology 2011, 43, 320.
98. Vincent, S. R. Nitric oxide: A radical neurotransmitter in the central nervous system. Progress in Neurobiology 1994, 42, 129-160.
99. Chatterjee, P. K.; Cuzzocrea, S.; Brown, P. A.; Zacharowski, K.; Stewart, K. N.; Mota-Filipe, H.; Thiemermann, C. Tempol, a membrane-permeable radical scavenger, reduces oxidant stress-mediated renal dysfunction and injury in the rat. Kidney International 2000, 58, 658-673.
100. Zhu, X.; Zuo, L.; Cardounel, A. J.; Zweier, J. L.; He, G. Characterization of in vivo tissue redox status, oxygenation, and formation of reactive oxygen species in postischemic myocardium. Antioxidants and Redox Signaling 2007, 9, 447-455.
101. Walker, J. R.; Fairfull-Smith, K. E.; Anzai, K.; Lau, S.; White, P. J.; Scammells, P. J.; Bottle, S. E. Edaravone containing isoindoline nitroxides for the potential treatment of cardiovascular ischaemia. MedChemComm 2011, 2, 436-441.
102. Fairfull-Smith, K. E.; Brackmann, F.; Bottle, S. E. The synthesis of novel isoindoline nitroxides bearing water-solubilising functionality. European Journal of Organic Chemistry 2009, 2009, 1902-1915.
103. Samuni, A. M.; Degraff, W.; Krishna, M. C.; Mitchell, J. B. Nitroxides as antioxidants: Tempol protects against EO9 cytotoxicity. Molecular and Cellular Biochemistry 2002, 234-235, 327-33.
104. Wu, G.; Meininger, C. J. Regulation of nitric oxide synthesis by dietary factors. Annual Review of Nutrition 2002, 22, 61-86.
105. Cong, J.-H.; Sun, C.-P.; Tian, X.-H.; Fang, Y.-Z. Effect of Lu-Duo-Wei on Scavenging Superoxide and Hydroxyl Radicals in vitro. The American Journal of Chinese Medicine 1998, 26, 153-158.
106. Jackson, M. J. An overview of methods for assessment of free radical activity in biology. Proceedings of the Nutrition Society 1999, 58, 1001-1006.
107. Gilbert, D. L. Fifty years of radical ideas. Annals of the New York Academy of Sciences 2000, 899, 1-14.
108. Rice-Evans, C.; Miller, N.; Paganga, G. Antioxidant properties of phenolic compounds. Trends in Plant Science 1997, 2, 152-159.
109. Cai, Y.; Luo, Q.; Sun, M.; Corke, H. Antioxidant activity and phenolic compounds of 112 traditional Chinese medicinal plants associated with anticancer. Life Sciences 2004, 74, 2157-2184.

265
110. Rice-Evans, C. A.; Miller, N. J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Biology and Medicine 1996, 20, 933-956.
111. Wright, J. S.; Johnson, E. R.; DiLabio, G. A. Predicting the activity of phenolic antioxidants: theoretical method, analysis of substituent effects, and application to major families of antioxidants. Journal of the American Chemical Society 2001, 123, 1173-1183.
112. Noguchi, N.; Niki, E. Phenolic antioxidants:: A rationale for design and evaluation of novel antioxidant drug for atherosclerosis. Free Radical Biology and Medicine 2000, 28, 1538-1546.
113. Pryor, W. A. Vitamin E and heart disease:: basic science to clinical intervention trials. Free Radical Biology and Medicine 2000, 28, 141-164.
114. Noguchi, N.; Iwaki, Y.; Takahashi, M.; Komuro, E.; Kato, Y.; Tamura, K.; Cynshi, O.; Kodama, T.; Niki, E. 2,3-Dihydro-5-hydroxy-2,2-dipentyl-4,6-di-tert-butylbenzofuran: Design and evaluation as a novel radical-scavenging antioxidant against lipid peroxidation. Archives of Biochemistry and Biophysics 1997, 342, 236-243.
115. Agadzhanyan, V.; Oganesyan, E.; Abaev, V. Targeted search for a lead compound in a series of cinnamic acid derivatives possessing antiradical activity. Pharmaceutical Chemistry Journal 2010, 44, 360-365.
116. Durand, E.; Bayrasy, C.; Laguerre, M.; Barouh, N.; Lecomte, J.; Durand,
T.; Balas, L.; Wrutniak‐Cabello, C.; Cabello, G.; Villeneuve, P. Regioselective synthesis of diacylglycerol rosmarinates and evaluation of their antioxidant activity in fibroblasts. European Journal of Lipid Science and Technology 2015.
117. Kwak, S.-Y.; Yang, J.-K.; Choi, H.-R.; Park, K.-C.; Kim, Y.-B.; Lee, Y.-S. Synthesis and dual biological effects of hydroxycinnamoyl phenylalanyl/prolyl hydroxamic acid derivatives as tyrosinase inhibitor and antioxidant. Bioorganic & Medicinal Chemistry Letters 2013, 23, 1136-1142.
118. Fresco, P.; Borges, F.; Diniz, C.; Marques, M. New insights on the anticancer properties of dietary polyphenols. Medicinal Research Reviews 2006, 26, 747-766.
119. Simonyan, A. Activity of cinnamic acid derivatives and new methods for their synthesis (review). Pharmaceutical Chemistry Journal 1993, 27, 92-100.
120. Shepherd, R. G. Certain cinnamic acid or propiolic acid derivatives. 1983.
121. Hausler, N. E.; Devine, S. M.; McRobb, F. M.; Warfe, L.; Pouton, C. W.; Haynes, J. M.; Bottle, S. E.; White, P. J.; Scammells, P. J. Synthesis and pharmacological evaluation of dual acting antioxidant A2A adenosine receptor agonists. Journal of Medicinal Chemistry 2012, 55, 3521-3534.

266
122. Arch, J. R.; Newsholme, E. A. The control of the metabolism and the hormonal role of adenosine. Essays in Biochemistry 1978, 14, 82-123.
123. Maggirwar, S. B.; Dhanraj, D. N.; Somani, S. M.; Ramkumar, V. Adenosine acts as an endogenous activator of the cellular antioxidant defense system. Biochemical and Biophysical Research Communications 1994, 201, 508-515.
124. Cronstein, B. N.; Kramer, S. B.; Weissmann, G.; Hirschhorn, R. Adenosine: a physiological modulator of superoxide anion generation by human neutrophils. The Journal of Experimental Medicine 1983, 158, 1160-1177.
125. Williams-Karnesky, R. L.; Stenzel-Poore, M. P. Adenosine and stroke: Maximizing the therapeutic potential of adenosine as a prophylactic and acute neuroprotectant. Current Neuropharmacology 2009, 7, 217.
126. Silverman, R. B. The organic chemistry of drug design and drug action. Academic press: 2004.
127. Ulrich, H.; Abbracchio, M.; Burnstock, G. Extrinsic purinergic regulation of neural stem/progenitor cells: Implications for CNS development and repair. Stem Cell Reviews and Reports 2012, 8, 755-767.
128. Burnstock, G. Introduction to purinergic signalling in the brain. In Glioma Signaling, Barańska, J., Ed. Springer Netherlands: 2013; Vol. 986, pp 1-12.
129. King, B. F.; Burnstock, G. Purinergic receptors. Understanding G Protein-coupled Receptors and their Role in the CNS 2002, 422-438.
130. Overington, J. P.; Al-Lazikani, B.; Hopkins, A. L. How many drug targets are there? Nature Reviews Drug Discovery 2006, 5, 993-996.
131. Bockaert, J.; Fagni, L.; Dumuis, A.; Marin, P. GPCR interacting proteins (GIP). Pharmacology & Therapeutics 2004, 103, 203-221.
132. Hurowitz, E. H.; Melnyk, J. M.; Chen, Y.-J.; Kouros-Mehr, H.; Simon, M. I.; Shizuya, H. Genomic characterization of the human heterotrimeric G protein α, β, and γ subunit genes. DNA Research 2000, 7, 111-120.
133. Hamm, H. E. The many faces of G protein signaling. Journal of Biological Chemistry 1998, 273, 669-672.
134. Dickenson, J. M.; Hill, S. J. Involvement of G-protein βγ subunits in coupling the adenosine A1 receptor to phospholipase C in transfected CHO cells. European Journal of Pharmacology 1998, 355, 85-93.
135. Snyder, B. Where are the new drugs. The push to improve the pipeline.(Article) Picture by William Oldham 2005.
136. Berne, R. Cardiac nucleotides in hypoxia: possible role in regulation of coronary blood flow. 1963; Vol. 204, p 317-322.

267
137. Olsson, R.; Pearson, J. Cardiovascular purinoceptors. Physiological Reviews 1990, 70, 761-845.
138. Newby, A. C. Adenosine and the concept of ‘retaliatory metabolites’. Trends in Biochemical Sciences 1984, 9, 42-44.
139. Müller, C. E. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Edited by Gyorgy Hasko, Bruce N. Cronstein and Csaba Szabo. ChemMedChem 2008, 3, 1789-1789.
140. Best, B. Is Caffeine a health hazard. The American Journal of Psychiatry 1999, 156, 223-228.
141. Headrick, J. P.; Peart, J. A3 adenosine receptor-mediated protection of the ischemic heart. Vascular Pharmacology 2005, 42, 271-279.
142. Miura, T.; Ogawa, T.; Iwamoto, T.; Shimamoto, K.; Iimura, O. Dipyridamole potentiates the myocardial infarct size-limiting effect of ischemic preconditioning. Circulation 1992, 86, 979-985.
143. Tsuchida, A.; Miura, T.; Miki, T.; Shimamoto, K.; Iimura, O. Role of adenosine receptor activation in myocardial infarct size limitation by ischaemic preconditioning. Cardiovascular Research 1992, 26, 456-461.
144. Miura, T.; Iimura, O. Infarct size limitation by preconditioning: its phenomenological features and the key role of adenosine. Cardiovascular Research 1993, 27, 36-42.
145. Poulsen, S. A.; Quinn, R. J. Adenosine receptors: New opportunities for future drugs. Bioorganic & Medicinal Chemistry 1998, 6, 619-641.
146. Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacological Reviews 1998, 50, 413-492.
147. Ham, J.; Evans, B. A. An emerging role for adenosine and its receptors in bone homeostasis. Frontiers in endocrinology 2012, 3.
148. Daly, J. W.; Padgett, W.; Thompson, R. D.; Kusachi, S.; Bugni, W. J.; Olsson, R. A. Structure-activity-relationships for N6-substituted adenosines at a brain adenosine-A1-receptor with a comparison to an adenosine-A2 receptor regulating coronary blood-flow. Biochemical Pharmacology 1986, 35, 2467-2481.
149. Daly, J. W. Adenosine receptors: targets for future drugs. Journal of Medicinal Chemistry 1982, 25, 197-207.
150. Yang, Z.; Day, Y. J.; Toufektsian, M. C.; Xu, Y.; Ramos, S. I.; Marshall, M. A.; French, B. A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056-2064.

268
151. Mahaffey, K. W.; Puma, J. A.; Barbagelata, N. A.; Dicarli, M. F.; Leesar, M. A.; Browne, K. F.; Eisenberg, P. R.; Bolli, R.; Casas, A. C.; Molina-Viamonte, V.; Orlandi, C.; Blevins, R.; Gibbons, R. J.; Califf, R. M.; Granger, C. B. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction - Results of a multicenter, randomized, placebo-controlled trial: the acute myocardial infarction study of adenosine (AMISTAD) trial. Journal of the American College of Cardiology 1999, 34, 1711-1720.
152. Ross, A. M.; Gibbons, R. J.; Stone, G. W.; Kloner, R. A.; Alexander, R. W. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). Journal of the American College of Cardiology 2005, 45, 1775-1780.
153. Ninomiya, H.; Otani, H.; Lu, K.; Uchiyama, T.; Kido, M.; Imamura, H. Complementary role of extracellular ATP and adenosine in ischemic preconditioning in the rat heart. American Journal of Physiology-Heart and Circulatory Physiology 2002, 282, H1810-H1820.
154. Freissmuth, M.; Schütz, W.; Linder, M. E. Interactions of the bovine brain A1-adenosine receptor with recombinant G protein alpha-subunits. Selectivity for rGi alpha-3. Journal of Biological Chemistry 1991, 266, 17778-17783.
155. Munshi, R.; Pang, I. H.; Sternweis, P. C.; Linden, J. A1 adenosine receptors of bovine brain couple to guanine nucleotide-binding proteins Gi1, Gi2, and Go. Journal of Biological Chemistry 1991, 266, 22285-9.
156. Calker, D. V.; Muller, M.; Hamprecht, B. Adenosine inhibits the accumulation of cyclic AMP in cultured brain cells. Nature 1978, 276, 839-841.
157. Londos, C.; Cooper, D. M.; Wolff, J. Subclasses of external adenosine receptors. Proceedings of the National Academy of Sciences 1980, 77, 2551-2554.
158. Iredale, P. A.; Alexander, S. P. H.; Hill, S. J. Coupling of a transfected human brain A1 adenosine receptor in CHO-K1 cells to calcium mobilisation via a pertussis toxin-sensitive mechanism. British Journal of Pharmacology 1994, 111, 1252-1256.
159. Megson, A. C.; Dickenson, J. M.; Townsend-Nicholson, A.; Hill, S. J. Synergy between the inositol phosphate responses to transfected human adenosine A1-receptors and constitutive P2-purinoceptors in CHO-K1 cells. British Journal of Pharmacology 1995, 115, 1415-1424.
160. Fredholm, B. B.; IJzerman, A. P.; Jacobson, K. A.; Klotz, K.-N.; Linden, J. International union of pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacological Reviews 2001, 53, 527-552.
161. Reppert, S. M.; Weaver, D. R.; Stehle, J. H.; Rivkees, S. A. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Molecular Endocrinology 1991, 5, 1037-1048.

269
162. Stehle, J. H.; Rivkees, S. A.; Lee, J. J.; Weaver, D. R.; Deeds, J. D.; Reppert, S. M. Molecular cloning and expression of the cDNA for a novel A2-adenosine receptor subtype. Molecular Endocrinology 1992, 6, 384-393.
163. Dixon, A. K.; Gubitz, A. K.; Sirinathsinghji, D. J. S.; Richardson, P. J.; Freeman, T. C. Tissue distribution of adenosine receptor mRNAs in the rat. British Journal of Pharmacology 1996, 118, 1461-1468.
164. Tawfik, H. E.; Schnermann, J.; Oldenburg, P. J.; Mustafa, S. J. Role of A1 adenosine receptors in regulation of vascular tone. American Journal of Physiology-Heart and Circulatory Physiology 2005, 288, H1411-H1416.
165. Reichelt, M. E.; Willems, L.; Molina, J. G.; Sun, C.-X.; Noble, J. C.; Ashton, K. J.; Schnermann, J.; Blackburn, M. R.; Headrick, J. P. Genetic deletion of the A1 adenosine receptor limits myocardial ischemic tolerance. Circulation Research 2005, 96, 363-367.
166. Liao, Y.; Takashima, S.; Asano, Y.; Asakura, M.; Ogai, A.; Shintani, Y.; Minamino, T.; Asanuma, H.; Sanada, S.; Kim, J.; Ogita, H.; Tomoike, H.; Hori, M.; Kitakaze, M. Activation of adenosine A1 receptor attenuates cardiac hypertrophy and prevents heart failure in murine left ventricular pressure-overload model. Circulation Research 2003, 93, 759-766.
167. Peart, J.; Headrick, J. P. Intrinsic A1 adenosine receptor activation during ischemia or reperfusion improves recovery in mouse hearts. American Journal of Physiology-Heart and Circulatory Physiology 2000, 279, H2166-H2175.
168. Finegan, B. A.; Lopaschuk, G. D.; Gandhi, M.; Clanachan, A. S. Inhibition of glycolysis and enhanced mechanical function of working rat hearts as a result of adenosine A1 receptor stimulation during reperfusion following ischaemia. British Journal of Pharmacology 1996, 118, 355-363.
169. Merighi, S.; Mirandola, P.; Varani, K.; Gessi, S.; Leung, E.; Baraldi, P. G.; Tabrizi, M. A.; Borea, P. A. A glance at adenosine receptors: novel target for antitumor therapy. Pharmacology & Therapeutics 2003, 100, 31-48.
170. Calker, D. v.; Müller, M.; Hamprecht, B. Adenosine regulates via two different types of receptors, the accumulation of cyclic AMP in cultured brain cells. Journal of Neurochemistry 1979, 33, 999-1005.
171. Bruns, R. F.; Lu, G. H.; Pugsley, T. A. Characterization of the A2 adenosine receptor labeled by [3H] NECA in rat striatal membranes. Molecular Pharmacology 1986, 29, 331-346.
172. Daly, J. W.; Butts-Lamb, P.; Padgett, W. Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cellular and Molecular Neurobiology 1983, 3, 69-80.
173. Chern, Y.; King, K.; Lai, H.-L.; Lai, H.-T. Molecular cloning of a novel adenosine receptor gene from rat brain. Biochemical and Biophysical Research Communications 1992, 185, 304-309.

270
174. Furlong, T. J.; Pierce, K. D.; Selbie, L. A.; Shine, J. Molecular characterization of a human brain adenosine A2 receptor. Molecular Brain Research 1992, 15, 62-66.
175. Ledent, C.; Vaugeois, J.-M.; Schiffmann, S. N.; Pedrazzini, T.; El Yacoubi, M.; Vanderhaeghen, J.-J.; Costentin, J.; Heath, J. K.; Vassart, G.; Parmentier, M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 1997, 388, 674-678.
176. Meng, F.; Xie, G.-x.; Chalmers, D.; Morgan, C.; Watson Jr, S. J.; Akil, H. Cloning and expression of the A2A adenosine receptor from guinea pig brain. Neurochemical Research 1994, 19, 613-621.
177. Botvinick, E. H. Current methods of pharmacologic stress testing and the potential advantages of new agents. Journal of Nuclear Medicine Technology 2009, 37, 14-25.
178. Lappas, C. M.; Sullivan, G. W.; Linden, J. Adenosine A2A agonists in development for the treatment of inflammation. Expert Opinion on Investigational Drugs 2005, 14, 797-806.
179. Laubach, V. E.; French, B. A.; Okusa, M. D. Targeting of adenosine receptors in ischemia–reperfusion injury. Expert Opinion on Therapeutic Targets 2011, 15, 103-118.
180. Fishman, P. J., K. A.; Ochaion, A.; Cohen, S.; Bar-Yehuda, S. The anti-cancer effect of A3 adenosine receptor agonists: a novel, targeted therapy. Immun., Endocr. Metab. Agents in Med. Chem 2007, 7.
181. de Lera Ruiz, M.; Lim, Y.-H.; Zheng, J. Adenosine A2A receptor as a drug discovery target. Journal of Medicinal Chemistry 2013, 57(9), 3623-3650.
182. Roberts, P.; Newby, A.; Hallett, M.; Campbell, A. Inhibition by adenosine of reactive oxygen metabolite production by human polymorphonuclear leucocytes. Biochem. J 1985, 227, 669-674.
183. Sullivan, G. W.; Rieger, J. M.; Michael Scheld, W.; Macdonald, T. L.;
Linden, J. Cyclic AMP‐dependent inhibition of human neutrophil oxidative activity by substituted 2‐propynylcyclohexyl adenosine A2A receptor agonists. British Journal of Pharmacology 2001, 132, 1017-1026.
184. Grieshaber, M. C.; Flammer, J. Blood flow in glaucoma. Current Opinion in Ophthalmology 2005, 16, 79-83.
185. Leske, M. C. Ocular perfusion pressure and glaucoma: clinical trial and epidemiologic findings. Current Opinion in Ophthalmology 2009, 20, 73.
186. Konno, T.; Uchibori, T.; Nagai, A.; Kogi, K.; Nakahata, N. Effect of 2-(6-cyano-1-hexyn-1-yl) adenosine on ocular blood flow in rabbits. Life sciences 2007, 80, 1115-1122.

271
187. Sitkovsky, M. V.; Lukashev, D.; Apasov, S.; Kojima, H.; Koshiba, M.; Caldwell, C.; Ohta, A.; Thiel, M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine a2a receptors*. Annual Review Immunology 2004, 22, 657-682.
188. Cronstein, B. N. Adenosine, an endogenous anti-inflammatory agent. Journal of Applied Physiology 1994, 76, 5-13.
189. Feoktistov, I.; Goldstein, A. E.; Biaggioni, I. Role of p38 mitogen-activated protein kinase and extracellular signal-regulated protein kinase kinase in adenosine a2breceptor-mediated interleukin-8 production in human mast cells. Molecular Pharmacology 1999, 55, 726-734.
190. Feoktistov, I.; Ryzhov, S.; Goldstein, A. E.; Biaggioni, I. Mast cell–mediated stimulation of angiogenesis cooperative interaction between A2B and A3 adenosine receptors. Circulation Research 2003, 92, 485-492.
191. Auchampach, J. A.; Jin, X.; Wan, T. C.; Caughey, G. H.; Linden, J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Molecular Pharmacology 1997, 52, 846-860.
192. Ryzhov, S.; Goldstein, A. E.; Matafonov, A.; Zeng, D.; Biaggioni, I.; Feoktistov, I. Adenosine-activated mast cells induce IgE synthesis by B lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. The Journal of Immunology 2004, 172, 7726-7733.
193. Volpini, R.; Costanzi, S.; Vittori, S.; Cristalli, G.; Klotz, K.-N. Medicinal chemistry and pharmacology of A2B adenosine receptors. Current Topics in Medicinal Chemistry 2003, 3, 427-443.
194. Eckle, T.; Krahn, T.; Grenz, A.; Köhler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M. A.; Osswald, H.; Thompson, L. F.; Unertl, K. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581-1590.
195. Toldo, S.; Zhong, H.; Mezzaroma, E.; Van Tassell, B. W.; Kannan, H.; Zeng, D.; Belardinelli, L.; Voelkel, N. F.; Abbate, A. GS-6201, a selective blocker of the A2B adenosine receptor, attenuates cardiac remodeling after acute myocardial infarction in the mouse. Journal of Pharmacology and Experimental Therapeutics 2012, 343, 587-595.
196. Karmouty-Quintana, H.; Zhong, H.; Acero, L.; Weng, T.; Melicoff, E.; West, J. D.; Hemnes, A.; Grenz, A.; Eltzschig, H. K.; Blackwell, T. S. The A2B
adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. The FASEB Journal 2012, 26, 2546-2557.
197. Zimmerman, M. A.; Grenz, A.; Tak, E.; Kaplan, M.; Ridyard, D.; Brodsky, K. S.; Mandell, M. S.; Kam, I.; Eltzschig, H. K. Signaling through hepatocellular A2B adenosine receptors dampens ischemia and reperfusion injury of the liver. Proceedings of the National Academy of Sciences 2013, 110, 12012-12017.

272
198. Frick, J.-S.; MacManus, C. F.; Scully, M.; Glover, L. E.; Eltzschig, H. K.; Colgan, S. P. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. The Journal of Immunology 2009, 182, 4957-4964.
199. Gessi, S.; Merighi, S.; Varani, K.; Leung, E.; Mac Lennan, S.; Borea, P. A. The A3 adenosine receptor: An enigmatic player in cell biology. Pharmacology & Therapeutics 2008, 117, 123-140.
200. Schulte, G.; Fredholm, B. B. Signalling from adenosine receptors to mitogen-activated protein kinases. Cellular Signalling 2003, 15, 813-827.
201. Zhou, Q.-Y.; Li, C.; Olah, M. E.; Johnson, R. A.; Stiles, G. L.; Civelli, O. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proceedings of the National Academy of Sciences 1992, 89, 7432-7436.
202. Linden, J. Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends in Pharmacological Sciences 1994, 15, 298-306.
203. Rivkees, S. A.; Thevananther, S.; Hao, H. Are A3 adenosine receptors expressed in the brain? Neuroreport 2000, 11, 1025-1030.
204. Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nature Reviews Drug Discovery 2008, 7, 759-770.
205. Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A.; Gullini, S.; Leung, E.; Mac-Lennan, S.; Borea, P. A. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clinical Cancer Research 2004, 10, 5895-5901.
206. Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clinical Cancer Research 2004, 10, 4472-4479.
207. Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Barer, F.; Patoka, R.; Amital, H.; Reitblat, T.; Reitblat, A.; Ophir, J.; Konfino, I.; Chowers, Y.; Ben-Horin, S.; Fishman, P. The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn's disease. Cellular Immunology 2009, 258, 115-122.
208. Rajagopal, S.; Rajagopal, K.; Lefkowitz, R. J. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nature Reviews Drug Discovery 2010, 9, 373-386.
209. Patel, C. B.; Noor, N.; Rockman, H. A. Functional Selectivity in Adrenergic and Angiotensin Signaling Systems. Molecular Pharmacology 2010, 78, 983-992.

273
210. Kenakin, T. Functional Selectivity and Biased Receptor Signaling. Journal of Pharmacology and Experimental Therapeutics 2011, 336, 296-302.
211. Whalen, E. J.; Rajagopal, S.; Lefkowitz, R. J. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends in Molecular Medicine 2011, 17, 126-139.
212. Albrecht-Küpper, B. E.; Leineweber, K.; Nell, P. G. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signalling 2012, 8, 91-99.
213. Jakubowski, H. Transport and kinetics, biochemistry online: An approach based on chemical logic. In Agonist and antagonist of ligand binding, biowiki ucdavis.edu
214. Moro, S.; Gao, Z. G.; Jacobson, K. A.; Spalluto, G. Progress in the pursuit of therapeutic adenosine receptor antagonists. Medicinal Research Reviews 2006, 26, 131-159.
215. Feoktistov, I.; Biaggioni, I.; Polosa, R.; Holgate, S. Adenosine A2B receptors: a novel therapeutic target in asthma? Trends in pharmacological sciences 1998, 19, 148-153.
216. Fozard, J.; McCarthy, C. Adenosine receptor ligands as potential therapeutics in asthma. Current opinion in investigational drugs (London, England: 2000) 2002, 3, 69-77.
217. Richardson, P. J.; Kase, H.; Jenner, P. G. Adenosine A2A receptor antagonists as new agents for the treatment of Parkinson's disease. Trends in Pharmacological Sciences 1997, 18, 338-344.
218. El-Tayeb, A.; Michael, S.; Abdelrahman, A.; Behrenswerth, A.; Gollos, S.; Nieber, K.; Muller, C. E. Development of polar adenosine A2A receptor agonists for inflammatory bowel disease: synergism with A2B antagonists. ACS Medicinal Chemistry Letters 2011, 2, 890-895.
219. Hendel, R.; Stilley, W. B.; Williams, S. P. Unit Dosage of Apadenoson. U. S. Patent application No 14/103,130, 2013.
220. Harada, N.; Okajima, K.; Murakami, K.; Usune, S.; Sato, C.; Ohshima, K.; Katsuragi, T. Adenosine and selective A2A receptor agonists reduce ischemia/reperfusion injury of rat liver mainly by inhibiting leukocyte activation. Journal of Pharmacology and Experimental Therapeutics 2000, 294, 1034-1042.
221. Yoneyama, F.; Yamada, H.; Satoh, K.; Taira, N. Vasodepressor mechanisms of 2-(1-octynyl)-adenosine (YT-146), a selective adenosine A2 receptor agonist, involve the opening of glibenclamide-sensitive K+ channels. European Journal of Pharmacology 1992, 213, 199-204.
222. Deflorian, F.; Kumar, T. S.; Phan, K.; Gao, Z.-G.; Xu, F.; Wu, H.; Katritch, V.; Stevens, R. C.; Jacobson, K. A. Evaluation of molecular modeling of agonist

274
binding in light of the crystallographic structure of an agonist-bound A2A adenosine receptor. Journal of Medicinal Chemistry 2011, 55, 538-552.
223. Hutchison, A. J.; Williams, M.; De Jesus, R.; Yokoyama, R.; Oei, H. H.; Ghai, G. R.; Webb, R. L.; Zoganas, H. C.; Stone, G. A.; Jarvis, M. F. 2-(Arylalkylamino)adenosin-5'-uronamides: a new class of highly selective adenosine A2 receptor ligands. Journal of Medicinal Chemistry 1990, 33, 1919-1924.
224. Mayer, C. A. Adenosine A2A receptors mediate GABAergic inhibition of respiration in immature rats. 2006; Vol. 100, p 91-97.
225. Diógenes, M. J.; Fernandes, C. C.; Sebastião, A. M.; Ribeiro, J. A. Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. The Journal of Neuroscience 2004, 24, 2905-2913.
226. Zoghbi, G. J. M. D.; Iskandrian, A. E. M. D. Selective adenosine agonists and myocardial perfusion imaging. Journal of Nuclear Cardiology 2012, 19, 126-41.
227. Müller, C. E.; Jacobson, K. A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochimica et Biophysica Acta (BBA) - Biomembranes 2011, 1808, 1290-1308.
228. Brackett, L. E.; Daly, J. W. Relaxant effects of adenosine analogs on guinea pig trachea in vitro: xanthine-sensitive and xanthine-insensitive mechanisms. Journal of Pharmacology and Experimental Therapeutics 1991, 257, 205-213.
229. Mathôt, R. A.; Gubbens-Stibbe, J. M.; Soudijn, W.; Jacobson, K. A.; Ijzerman, A. P.; Danhof, M. Quantification of the in vivo potency of the adenosine A2 receptor antagonist 8-(3-chlorostyryl)caffeine. Journal of Pharmacology and Experimental Therapeutics 1995, 275, 245-53.
230. LeWitt, P. A.; Guttman, M.; Tetrud, J. W.; Tuite, P. J.; Mori, A.; Chaikin,
P.; Sussman, N. M. Adenosine A2A receptor antagonist istradefylline (KW‐6002) reduces “off” time in Parkinson's disease: A double‐blind, randomized, multicenter clinical trial (6002‐US‐005). Annals of Neurology 2008, 63, 295-302.
231. Mizuno, Y.; Hasegawa, K.; Kondo, T.; Kuno, S.; Yamamoto, M. Clinical efficacy of istradefylline (KW-6002) in Parkinson's disease: A randomized, controlled study. Movement Disorders 2010, 25, 1437-1443.
232. Jaakola, V.-P.; Griffith, M. T.; Hanson, M. A.; Cherezov, V.; Chien, E. Y. T.; Lane, J. R.; Ijzerman, A. P.; Stevens, R. C. The 2.6 angstrom crystal structure of a human [image] adenosine receptor bound to an antagonist. Science 2008, 322, 1211-1217.
233. Salamone, J. D. Preladenant, a novel adenosine A (2A) receptor antagonist for the potential treatment of parkinsonism and other disorders. IDrugs: The Investigational Drugs Journal 2010, 13, 723-731.

275
234. Drury, A.; Szent-Györgyi, A. v. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. The Journal of Physiology 1929, 68, 213-237.
235. Lebon, G.; Warne, T.; Edwards, P. C.; Bennett, K.; Langmead, C. J.; Leslie, A. G. W.; Tate, C. G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521-5.
236. Ely, S.; Berne, R. Protective effects of adenosine in myocardial ischemia. Circulation 1992, 85, 893-904.
237. Peart, J.; Paul Matherne, G.; Cerniway, R. J.; Headrick, J. P. Cardioprotection with adenosine metabolism inhibitors in ischemic–reperfused mouse heart. Cardiovascular Research 2001, 52, 120-129.
238. Baraldi, P. G.; Tabrizi, M. A.; Fruttarolo, F.; Romagnoli, R.; Preti, D. Recent improvements in the development of A2B adenosine receptor agonists. Purinergic Signalling 2008, 4, 287-303.
239. Auchampach, J. A.; Kreckler, L. M.; Wan, T. C.; Maas, J. E.; van der Hoeven, D.; Gizewski, E.; Narayanan, J.; Maas, G. E. Characterization of the A2B adenosine receptor from mouse, rabbit, and dog. Journal of Pharmacology and Experimental Therapeutics 2009, 329, 2-13.
240. Rosentreter, U.; Henning, R.; Bauser, M.; Krämer, T.; Vaupel, A.; Hübsch, W.; Dembowsky, K.; Salcher-Schraufstätter, O.; Stasch, J.-P.; Krahn, T. Substituted 2-thio-3, 5-dicyano-4-aryl-6-aminopyridines and the use thereof. U.S. Patent No. 7,135,486, 2006.
241. Beukers, M. W.; Chang, L. C.; von Frijtag Drabbe Künzel, J. K.; Mulder-Krieger, T.; Spanjersberg, R. F.; Brussee, J.; IJzerman, A. P. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. Journal of Medicinal Chemistry 2004, 47, 3707-3709.
242. Kawazoe, K.; Matsumoto, N.; Tanabe, M.; Fujiwara, S.; Yanagimoto, M.; Hirata, M.; Kikuchi, K. Coronary and cardiohemodynamic effects of 2-phenylamino-adenosine (CV-1808) in anesthetized dogs and cats. Arzneimittelforschung Drug Research 1980.
243. Abiru, T.; Miyashita, T.; Watanabe, Y.; Yamaguchi, T.; Machida, H.; Matsuda, A. Nucleosides and nucleotides. 107. 2-(cycloalkylalkynyl) adenosines: adenosine A2 receptor agonists with potent antihypertensive effects. Journal of Medicinal Chemistry 1992, 35, 2253-2260.
244. Thimm, D.; Schiedel, A. C.; Sherbiny, F. F.; Hinz, S.; Hochheiser, K.; Bertarelli, D. C.; Maaß, A.; Muller, C. E. Ligand-specific binding and activation of the human adenosine A2B receptor. Biochemistry 2013, 52, 726-740.

276
245. Hinz, S.; Lacher, S. K.; Seibt, B. F.; Müller, C. E. BAY60-6583 acts as a partial agonist at adenosine A2B receptors. Journal of Pharmacology and Experimental Therapeutics 2014, 349, 427-436.
246. Kim, S.-A.; Marshall, M. A.; Melman, N.; Kim, H. S.; Müller, C. E.; Linden, J.; Jacobson, K. A. Structure-activity relationships at human and rat A2B adenosine receptors of xanthine derivatives substituted at the 1-, 3-, 7-, and 8-positions. Journal of Medicinal Chemistry 2002, 45, 2131-2138.
247. Kim, Y.-C.; Ji, X.-d.; Melman, N.; Linden, J.; Jacobson, K. A. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. Journal of Medicinal Chemistry 2000, 43, 1165-1172.
248. Watanabe, T.; Tanaka, M.; Watanabe, K.; Takamatsu, Y.; Tobe, A. [Research and development of the free radical scavenger edaravone as a neuroprotectant]. Yakugaku Zasshi Journal of the Pharmaceutical Society of Japan 2004, 99-111.
249. Batchu, S.; Lee, S.; Qadhi, R.; Chaudhary, K.; El‐Sikhry, H.; Kodela, R.; Falck, J.; Seubert, J. Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. British Journal of Pharmacology 2011, 162, 897-907.
250. Jacobson, K. A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nature Reviews Drug discovery 2006, 5, 247-264.
251. Jacobson, K. A.; Xie, R.; Young, L.; Chang, L.; Liang, B. T. A novel pharmacological approach to treating cardiac ischemia binary conjugates of A1 and A3 adenosine receptor agonists. Journal of Biological Chemistry 2000, 275, 30272-30279.
252. Liang, B. T.; Jacobson, K. A. Methods and compositions for reducing ischemic injury of the heart by administering adenosine receptor agonists and antagonists. U. S. Patents 6586413, 2003.
253. Hutchison, A.; Oei, H.; Ghai, G.; Williams, M. CGS21680, an A2 selective adenosine (ADO) receptor agonist with preferential hypotensive activity. FASEB J 1989, 3, A281.
254. Cristalli, G.; Eleuteri, A.; Vittori, S.; Volpini, R.; Lohse, M. J.; Klotz, K. N. 2-Alkynyl derivatives of adenosine and adenosine-5'-N-ethyluronamide as selective agonists at A2 adenosine receptors. Journal of Medicinal Chemistry 1992, 35, 2363-2368.
255. Martin, P. L.; Barrett, R. J.; Linden, J.; Abraham, W. M. Pharmacology of
2‐cyclohexylmethylidenehydrazinoadenosine (WRC‐0470), a novel, short‐acting adenosine A2A receptor agonist that produces selective coronary vasodilation. Drug Development Research 1997, 40, 313-324.

277
256. Yan, L.; Burbiel, J. C.; Maaß, A.; Müller, C. E. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opinion on Emerging Drugs 2003, 8, 537-576.
257. Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916-920.
258. Gregg, A.; Bottle, S. E.; Devine, S. M.; Figler, H.; Linden, J.; White, P.; Pouton, C. W.; Urmaliya, V.; Scammells, P. J. Dual acting antioxidant A1 adenosine receptor agonists. Bioorganic & Medicinal Chemistry Letters 2007, 17, 5437-5441.
259. Foitzik, R. C.; Devine, S. M.; Hausler, N. E.; Scammells, P. J. Linear and convergent approaches to 2-substituted adenosine-5′-N-alkylcarboxamides. Tetrahedron 2009, 65, 8851-8857.
260. Xu, F.; Wu, H.; Katritch, V.; Han, G. W.; Jacobson, K. A.; Gao, Z.-G.; Cherezov, V.; Stevens, R. C. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322-327.
261. Mantell, S.; Jones, R.; Trevethick, M. Design and application of locally delivered agonists of the adenosine A2A receptor. Expert Review of Clinical Pharmacology 2010, 3, 55-72.
262. Bosch, M. P.; Campos, F.; Niubó, I.; Rosell, G.; Díaz, J. L.; Brea, J.; Loza, M. I.; Guerrero, A. Synthesis and biological activity of new potential agonists for the human adenosine A2A receptor. Journal of Medicinal Chemistry 2004, 47, 4041-4053.
263. Francis, J. E.; Webb, R. L.; Ghai, G. R.; Hutchison, A. J.; Moskal, M. A.; DeJesus, R.; Yokoyama, R.; Rovinski, S. L.; Contardo, N. Highly selective adenosine A2 receptor agonists in a series of N-alkylated 2-aminoadenosines. Journal of Medicinal Chemistry 1991, 34, 2570-2579.
264. Jacobson, M. A. Adenosine receptor agonists. Expert Opinion on Therapeutic Patents 2002, 12, 489-501.
265. Trivedi, B. K.; Bruns, R. F. C2, N6-Disubstituted adenosines: synthesis and structure-activity relationships. Journal of Medicinal Chemistry 1989, 32, 1667-1673.
266. Cristalli, G.; Lambertucci, C.; Taffi, S.; Vittori, S.; Volpini, R. Medicinal chemistry of adenosine A2A receptor agonists. Current Topics in Medicinal Chemistry 2003, 3, 387-401.
267. Niiya, K.; Thompson, R. D.; Silvia, S. K.; Olsson, R. A. 2-(N'-aralkylidenehydrazino) adenosines: potent and selective coronary vasodilators. Journal of Medicinal Chemistry 1992, 35, 4562-4566.

278
268. Rieger, J. M.; Brown, M. L.; Sullivan, G. W.; Linden, J.; Macdonald, T. L. Design, synthesis, and evaluation of novel A2A adenosine receptor agonists. Journal of Medicinal Chemistry 2001, 44, 531-539.
269. Manera, C.; Saccomanni, G. A2A receptor ligands: past, present and future trends. Current Topics in Medicinal Chemistry 2010, 10, 902-922.
270. Hutchinson, S.; Scammells, P. A1 adenosine receptor agonists: medicinal chemistry and therapeutic potential. Current Pharmaceutical Design 2004, 10, 2021-2039.
271. Adachi, H.; Palaniappan, K. K.; Ivanov, A. A.; Bergman, N.; Gao, Z.-G.; Jacobson, K. A. Structure−activity relationships of 2,N6,5‘-substituted adenosine derivatives with potent activity at the A2B adenosine receptor. Journal of Medicinal Chemistry 2007, 50, 1810-1827.
272. Caddell, J. M.; Chapman, A. M.; Cooley, B. E.; Downey, B. P.; LeBlanc, M. P.; Jackson, M. M.; O'Connell, T. M.; Phung, H.-M.; Roper, T. D.; Xie, S. Efficient synthesis of an adenosine A2A agonist: Glycosylation of 2-haloadenines and an N 2-alkyl-6-chloroguanine. The Journal of Organic Chemistry 2004, 69, 3212-3215.
273. Kim, H. S.; Ohno, M.; Xu, B.; Kim, H. O.; Choi, Y.; Ji, X. D.; Maddileti, S.; Marquez, V. E.; Harden, T. K.; Jacobson, K. A. 2-Substitution of adenine nucleotide analogues containing a bicyclo[3.1.0]hexane ring system locked in a northern conformation: Enhanced potency as P2Y1 receptor antagonists. Journal of Medicinal Chemistry 2003, 46, 4974-4987.
274. Kim, H. O.; Ji, X.-d.; Siddiqi, S. M.; Olah, M. E.; Stiles, G. L.; Jacobson, K. A. 2-Substitution of N6-benzyladenosine-5'-uronamides enhances selectivity for A3 adenosine receptors. Journal of Medicinal Chemistry 1994, 37, 3614-3621.
275. Bottle, S. E.; Gillies, D. G.; Hughes, D. L.; Micallef, A. S.; Smirnov, A. I.; Sutcliffe, L. H. Synthesis, single crystal X-ray structure and W-band (95 GHz) EPR spectroscopy of a new anionic isoindoline aminoxyl: synthesis and characterisation of some derivatives. Journal of the Chemical Society, Perkin Transactions 2 2000, 1285-1291.
276. S. Micallef, A.; C. Bott, R.; E. Bottle, S.; Smith, G.; M. White, J.; Matsuda, K.; Iwamura, H. Brominated isoindolines: precursors to functionalised nitroxides. Journal of the Chemical Society, Perkin Transactions 2 1999, 65-72.
277. Howell, S. J.; Spencer, N.; Philp, D. Recognition-mediated regiocontrol of a dipolar cycloaddition reaction. Tetrahedron 2001, 57, 4945-4954.
278. Wuts, P. G.; Greene, T. W. Greene's protective groups in organic synthesis. John Wiley & Sons: 2006.
279. Olsson, R. A.; Kusachi, S.; Thompson, R. D.; Ukena, D.; Padgett, W.; Daly, J. W. N6-Substituted N-alkyladenosine-5'-uronamides: bifunctional ligands

279
having recognition groups for A1 and A2 adenosine receptors. Journal of Medicinal Chemistry 1986, 29, 1683-1689.
280. Maenhaut, C.; Van Sande, J.; Libert, F.; Abramowicz, M.; Parmentier, M.; Vanderhaegen, J.-J.; Dumont, J. E.; Vassart, G.; Schiffmann, S. RDC8 codes for an adenosine A2 receptor with physiological constitutive activity. Biochemical and Biophysical Research Communications 1990, 173, 1169-1178.
281. Devine, S. M.; Scammells, P. J. An efficient convergent synthesis of adenosine-5′-N-alkyluronamides. Tetrahedron 2008, 64, 1772-1777.
282. Bookser, B. C.; Raffaele, N. B. High-throughput five minute microwave accelerated glycosylation approach to the synthesis of nucleoside libraries. The Journal of Organic Chemistry 2007, 72, 173-179.
283. Hutchinson, S. A.; Baker, S. P.; Linden, J.; Scammells, P. J. New potent and selective A1 adenosine receptor agonists. Bioorganic & Medicinal Chemistry 2004, 12, 4877-4884.
284. Ahmadibeni, Y.; Dash, C.; Le Grice, S. F.; Parang, K. Solid-phase synthesis of 5′-O-β, γ-methylenetriphosphate derivatives of nucleosides and evaluation of their inhibitory activity against HIV-1 reverse transcriptase. Tetrahedron Letters 2010, 51, 3010-3013.
285. El-Farargy, A.; Ghoneim, A. A. Synthesis of Some Purine Nucleoside Derivatives with Expected Biological Activity. Current Organic Chemistry 2009, 13, 1842-1847.
286. Chen, G. S.; Chen, C. S.; Chien, T. C.; Yeh, J. Y.; Kuo, C. C.; Talekar, R.
S.; Chern, J. W. Nucleosides. IX. Synthesis of purine N 3, 5′‐cyclonucleosides and N 3, 5′‐Cyclo‐2′, 3′‐seconucleosides via Mitsunobu reaction as TIBO‐like derivatives. Nucleosides, Nucleotides and Nucleic Acids 2004, 23, 347-359.
287. Aerschot, A. V.; Mag, M.; Herdewijn, P.; Vanderhaeghe, H. Double protection of the heterocyclic base of xanthosine and 2′-deoxyxanthosine. Nucleosides & Nucleotides 1989, 8, 159-178.
288. Hampton, A. Nucleotides. II.1 A new procedure for the conversion of ribonucleosides to 2',3'-O-isopropylidene derivatives. Journal of the American Chemical Society 1961, 83, 3640-3645.
289. Ashton, T. D.; Scammells, P. J. An improved synthesis of 5′-fluoro-5′-deoxyadenosines. Bioorganic & Medicinal Chemistry Letters 2005, 15, 3361-3363.
290. Blum, T.; Ermert, J.; Wutz, W.; Bier, D.; Coenen, H. H. First no-carrier-added radioselenation of an adenosine-A1 receptor ligand. Journal of Labelled Compounds and Radiopharmaceuticals 2004, 47, 415-427.
291. Ciuffreda, P.; Loseto, A.; Santaniello, E. Deamination of 5′-substituted-2′,3′-isopropylidene adenosine derivatives catalyzed by adenosine deaminase (ADA, EC 3.5.4.4) and complementary enzymatic biotransformations catalyzed

280
by adenylate deaminase (AMPDA, EC 3.5.4.6): a viable route for the preparation of 5′-substituted inosine derivatives. Tetrahedron 2002, 58, 5767-5771.
292. Middleton, R. J.; Briddon, S. J.; Cordeaux, Y.; Yates, A. S.; Dale, C. L.; George, M. W.; Baker, J. G.; Hill, S. J.; Kellam, B. New fluorescent adenosine A1-receptor agonists that allow quantification of ligand−receptor interactions in microdomains of single living cells. Journal of Medicinal Chemistry 2007, 50, 782-793.
293. De Mico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. A versatile and highly selective hypervalent iodine (iii)/2,2,6,6-tetramethyl-1-piperidinyloxyl-mediated oxidation of alcohols to carbonyl compounds. The Journal of Organic Chemistry 1997, 62, 6974-6977.
294. Singh, A. K.; Varma, R. S. Ruthenium tetraoxide: a mild reagent for the oxidation of 2′,3′-O-isopropylidene purine nucleosides. Tetrahedron Letters 1992, 33, 2307-2310.
295. Marcus M. Sá, G. P. S., Marcelo S. Castilho, Fernando Pavão and Glaucius Oliva. Synthesis of acylated nucleosides and ribonic-1,4-lactones as inhibitors of trypanosomal glyceraldehyde-3-phosphate dehydrogenase (gGAPDH) (AP-507HP) ARKIVOC: (Gainesville, FL United States) 2002, 8, 112-124.
296. Epp, J. B.; Widlanski, T. S. Facile preparation of nucleoside-5'-carboxylic acids. The Journal of Organic Chemistry 1999, 64, 293-295.
297. Nooy, A. E. J. d.; Besemer, A. C.; Bekkum, H. v. On the use of stable organic nitroxyl radicals for the oxidation of primary and secondary alcohols. Synthesis 1996, 1996, 1153-1176.
298. Smith, M. B., March, J. . March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure.; John Wiley & Sons, Inc. Hoboken, N.J.: 6 ed. 2007, 1254-1255.
299. Wan, Z.-K.; Wacharasindhu, S.; Binnun, E.; Mansour, T. An efficient direct amination of cyclic amides and cyclic ureas. Organic Letters 2006, 8, 2425-2428.
300. Bae, S.; Lakshman, M. K. Synthetic utility of an isolable nucleoside phosphonium salt. Organic Letters 2008, 10, 2203-2206.
301. Smith, M. B., March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. John Wiley & Sons, Inc.: Hoboken, N.J 6 th ed, 2007, 854-857.
302. Kang, F.-A.; Sui, Z.; Murray, W. V. Phosphonium Coupling in the Direct Bond Formations of Tautomerizable Heterocycles via C–OH Bond Activation. European Journal of Organic Chemistry 2009, 2009, 461-479.

281
303. Takvorian, A. G.; Combs, A. P. Microwave-assisted organic synthesis using minivials to optimize and expedite the synthesis of diverse purine libraries. Journal of Combinatorial Chemistry 2004, 6, 171-174.
304. Sholle, V. D. Akad. Nauk. SSSR, Ser, Khim. 1969, 1, 149-151.
305. Keana, J. F. W.; Heo, G. S.; Gaughan, G. T. Stereospecific synthesis of difunctionalized 2,5-disubstituted cis-2,5-dimethylpyrrolidine (azethoxyl) nitroxides by oxidative cleavage of protected 8-azabicyclo[3.2.1]octane precursors. The Journal of Organic Chemistry 1985, 50, 2346-2351.
306. Rogister, F.; Laeckmann, D.; Plasman, P.-O.; Van Eylen, F.; Ghyoot, M.; Maggetto, C.; Liégeois, J.-F.; Géczy, J.; Herchuelz, A.; Delarge, J.; Masereel, B. Novel inhibitors of the sodium–calcium exchanger: benzene ring analogues of N-guanidino substituted amiloride derivatives. European Journal of Medicinal Chemistry 2001, 36, 597-614.
307. Ferguson, G. N. Synthesis and pharmacological evaluation of novel heteroaromatic ligands of the A1 adenosine receptor. . PhD, Monash University, Melbourne 2010.
308. Urmaliya, V. B.; Church, J. E.; Coupar, I. M.; Rose'Meyer, R. B.; Pouton, C. W.; White, P. J. Cardioprotection induced by adenosine A1 receptor agonists in a cardiac cell ischemia model involves cooperative activation of adenosine A2A and A2B receptors by endogenous adenosine. Journal of Cardiovascular Pharmacology 2009, 53, 424-433.
309. Urmaliya, V. B.; Pouton, C. W.; Devine, S. M.; Haynes, J. M.; Warfe, L.; Scammells, P. J.; White, P. J. A novel highly selective adenosine A1 receptor agonist VCP28 reduces ischemia injury in a cardiac cell line and ischemia–reperfusion injury in isolated rat hearts at concentrations that do not affect heart rate. Journal of Cardiovascular Pharmacology 2010, 56, 282-292.
310. Kolamunne, R. T.; Clare, M.; Griffiths, H. R. Mitochondrial superoxide anion radicals mediate induction of apoptosis in cardiac myoblasts exposed to chronic hypoxia. Archives of biochemistry and biophysics 2011, 505, 256-265.
311. Cristalli, G.; Volpini, R.; Vittori, S.; Camaioni, E.; Monopoli, A.; Conti, A.; Dionisotti, S.; Zocchi, C.; Ongini, E. 2-Alkynyl derivatives of adenosine-5'-N-ethyluronamide: selective A2 adenosine receptor agonists with potent inhibitory activity on platelet aggregation. Journal of Medicinal Chemistry 1994, 37, 1720-1726.
312. Dal Ben, D.; Lambertucci, C.; Taffi, S.; Vittori, S.; Volpini, R.; Cristalli, G.; Klotz, K.-N. Molecular modelling study of 2-phenylethynyladenosine (PEAdo) derivatives as highly selective A3 adenosine receptor ligands. Purinergic Signalling 2006, 2, 589-594.
313. Kim, Y.-C.; Ji, X.-d.; Jacobson, K. A. Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. Journal of Medicinal Chemistry 1996, 39, 4142-4148.

282
314. Kim, S.-K.; Gao, Z.-G.; Jeong, L. S.; Jacobson, K. A. Docking studies of agonists and antagonists suggest an activation pathway of the A3 adenosine receptor. Journal of Molecular Graphics and Modelling 2006, 25, 562-577.
315. Jacobson, K. A.; Klutz, A. M.; Tosh, D. K.; Ivanov, A. A.; Preti, D.; Baraldi, P. G. Medicinal chemistry of the A3 adenosine receptor: agonists, antagonists, and receptor engineering. In Adenosine Receptors in Health and Disease, Springer: 2009; pp 123-159.
316. Keddie, D. J.; Fairfull-Smith, K. E.; Bottle, S. E. The palladium-catalysed copper-free Sonogashira coupling of isoindoline nitroxides: a convenient route to robust profluorescent carbon-carbon frameworks. Organic & Biomolecular Chemistry 2008, 6, 3135-3143.
317. Cassar, L. Synthesis of aryl-and vinyl-substituted acetylene derivatives by the use of nickel and palladium complexes. Journal of Organometallic Chemistry 1975, 93, 253-257.
318. Dieck, H.; Heck, F. Palladium catalyzed synthesis of aryl, heterocyclic and vinylic acetylene derivatives. Journal of Organometallic Chemistry 1975, 93, 259-263.
319. Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron letters 1975, 16, 4467-4470.
320. Stephens, R.; Castro, C. The Substitution of Aryl Iodides with Cuprous Acetylides. A Synthesis of Tolanes and Heterocyclics1. The Journal of organic chemistry 1963, 28, 3313-3315.
321. Castro, C.; Stephens, R. Substitutions by ligands of low valent transition metals. A preparation of tolanes and heterocyclics from aryl iodides and cuprous acetylides. Journal of Organic Chemistry 1963, 28, 3313-3315.
322. Liang, B.; Dai, M.; Chen, J.; Yang, Z. Copper-free Sonogashira coupling reaction with PdCl2 in water under aerobic conditions. The Journal of Organic Chemistry 2005, 70, 391-393.
323. Park, K., Yun, YJ., Lee, SG. Efficient clevage of terminal acetonide group:Chirospecific synthesis of 2, 5-dideoxy-2, 5-imino-D-mannitol. Tetrahedran Letters 1994, 9737.
324. Hsieh, J.-C.; Cheng, C.-H. Nickel-catalyzed coupling of isocyanates with 1,3-iodoesters and halobenzenes: a novel method for the synthesis of imide and amide derivatives. Chemical Communications 2005, 4554-4556.
325. Mankske, R. H. F. Organic Synthesis 1932, XII, 10-11.
326. Greene, T. W., Wuts, P.G.M. Protective Groups in Organic Synthesis. John Wiley & Sons, Inc.: Brisbane, 1999, p 518–525.

283
Top Related