







Languages
Pages
Legal


Surya Saha [email protected]
BTI PGRP Summer Internship Program 2014
Slides: https://bitly.com/BioinfoInternEx2014
Quality Control of NGS Data

1. Evaluation
2. Preprocessing
Quality Control of NGS Data
7/8/2014 BTI PGRP Summer Internship Program 2014 2 Slide credit: Aureliano Bombarely

Goal:
Learn the use of read evaluation programs keeping
attention in relevant parameters such as quality score and
length distributions and reads duplications.
Data: (Illumina data for two tomato ripening stages)
/home/bioinfo/Data/ch4_demo_dataset.tar.gz
Tools: tar -zxvf (command line, untar and unzip the files)
head (command line, take a quick look of the files)
mv (command line, change the name of the files)
grep (command line, find/count patterns in files)
FASTX toolkit (command line, process fasta/fastq)
FastQC (gui, to calculate several stats for each file)
Evaluation
7/8/2014 BTI PGRP Summer Internship Program 2014 3 Slide credit: Aureliano Bombarely

Exercise 1:
1. Untar and Unzip the file:
/home/bioinfo/Data/ch4_demo_dataset.tar.gz
2. Raw data will be found in two dirs: breaker and
immature_fruit. Print the first 10 lines for the files:
SRR404331_ch4.fq, SRR404333_ch4.fq,
SRR404334_ch4.fq and SRR404336_ch4.fq.
Question 1.1: Do these files have fastq format?
3. Change the extension of the .fq files to .fastq
Evaluation
7/8/2014 BTI PGRP Summer Internship Program 2014 4 Slide credit: Aureliano Bombarely

Exercise 1:
4. Count number of sequences in each fastq file using
commands you learnt earlier.
5. Convert the fastq files to fasta.
6. Explore other tools in the FASTX toolkit.
7. Now count the number of sequences in fasta file and see
if the number of sequences has changed.
Evaluation
Tip: Use ‘grep’
Tip: Use ‘fastq_to_fasta -h’ to see help Use Google if you are stuck
7/8/2014 BTI PGRP Summer Internship Program 2014 5 Slide credit: Aureliano Bombarely
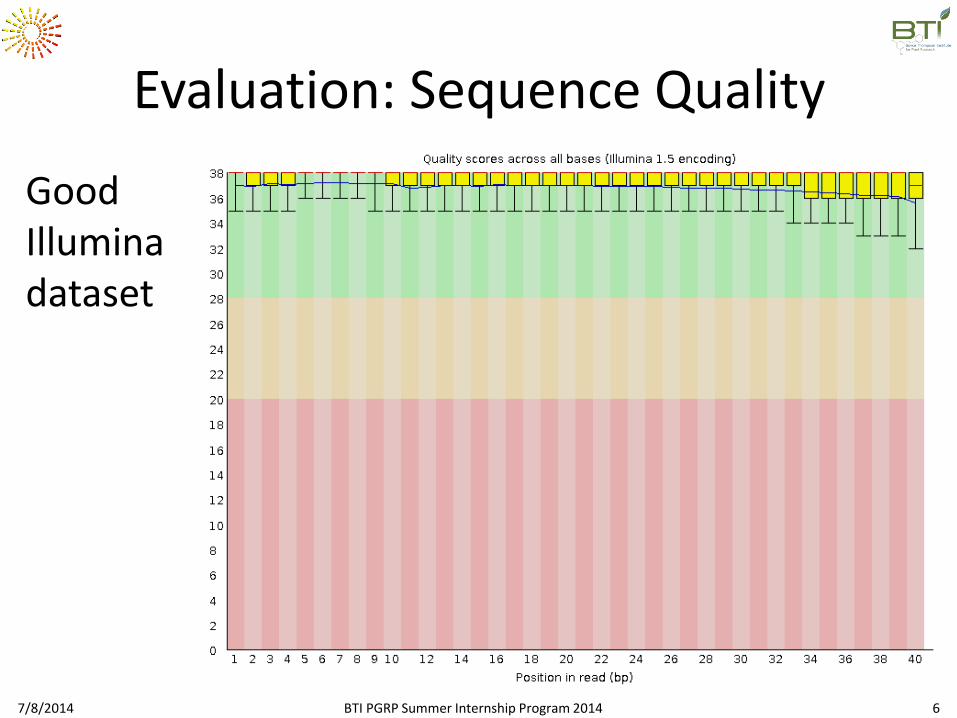
Evaluation: Sequence Quality
Good Illumina dataset
7/8/2014 BTI PGRP Summer Internship Program 2014 6
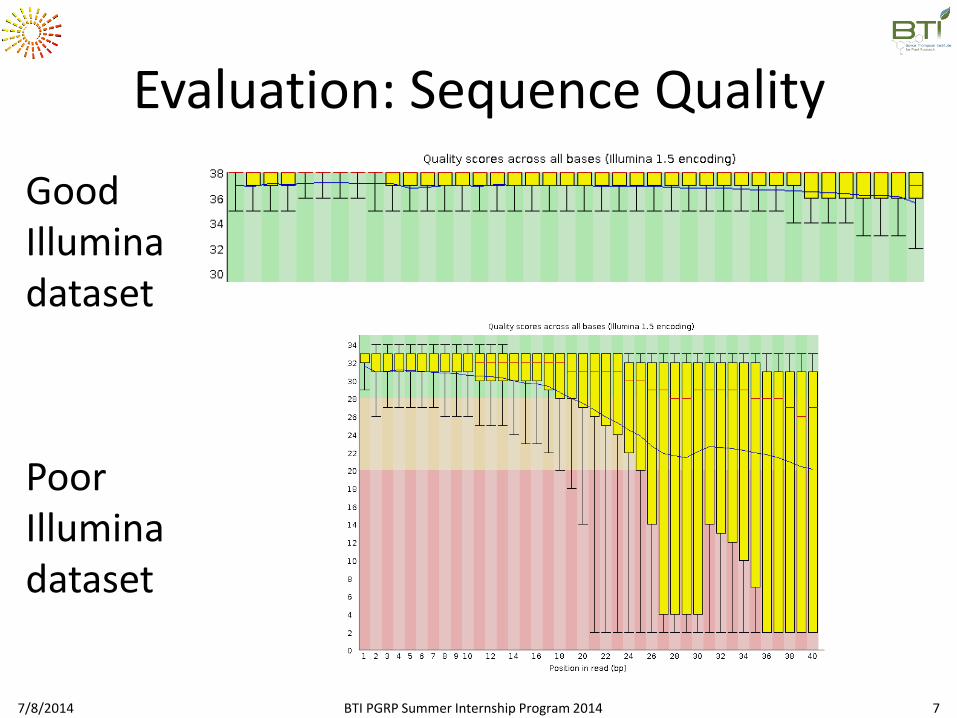
Evaluation: Sequence Quality
7/8/2014 BTI PGRP Summer Internship Program 2014 7
Good Illumina dataset
Poor Illumina dataset
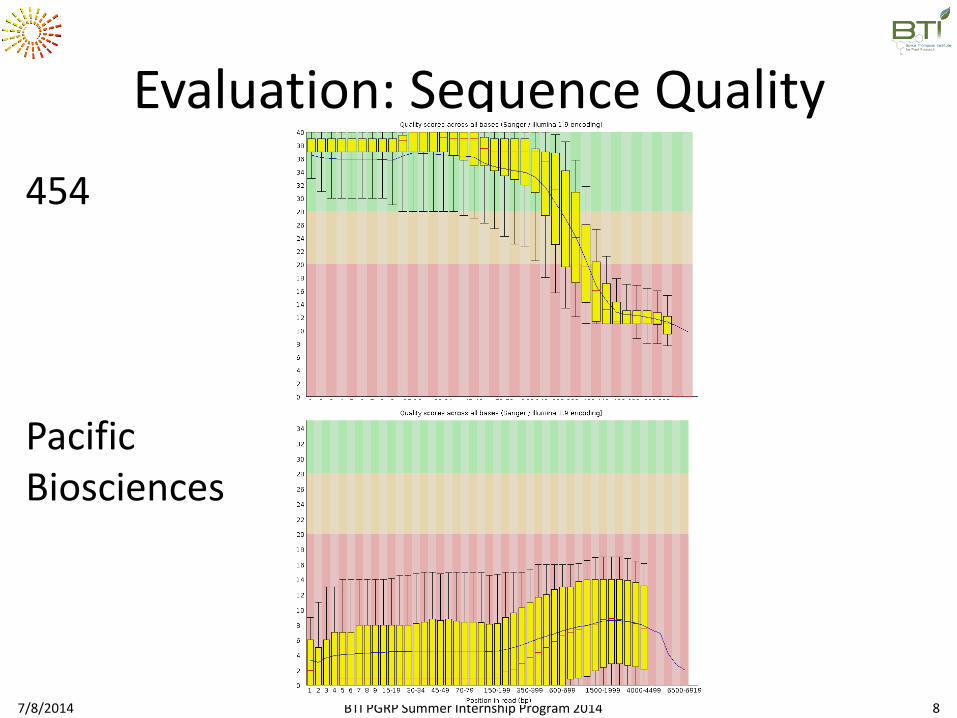
Evaluation: Sequence Quality
7/8/2014 BTI PGRP Summer Internship Program 2014 8
454
Pacific Biosciences
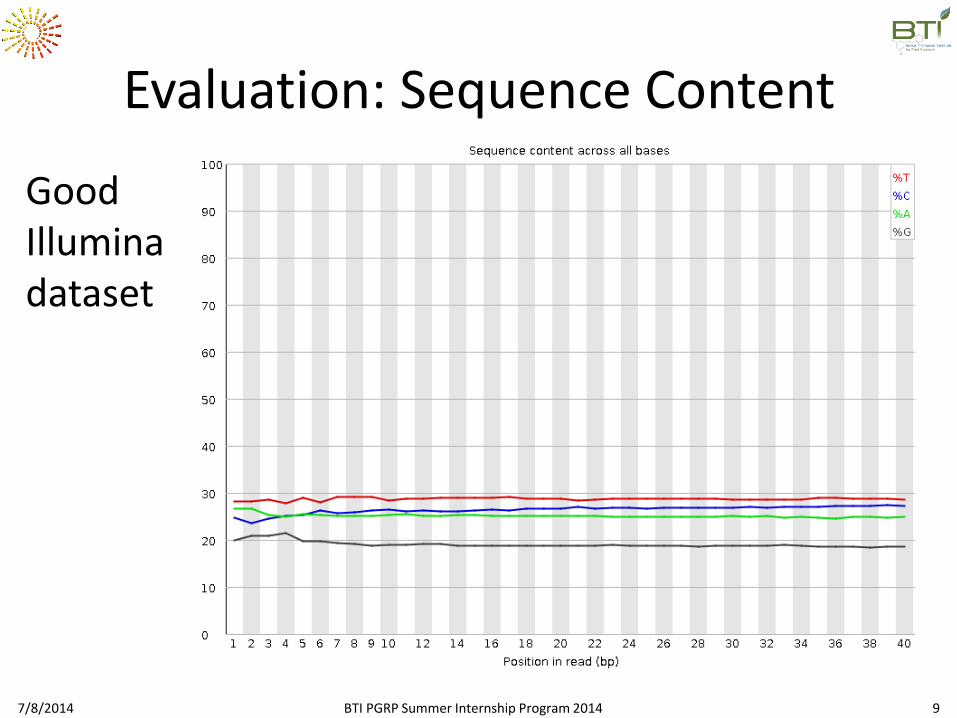
Evaluation: Sequence Content
Good Illumina dataset
7/8/2014 BTI PGRP Summer Internship Program 2014 9
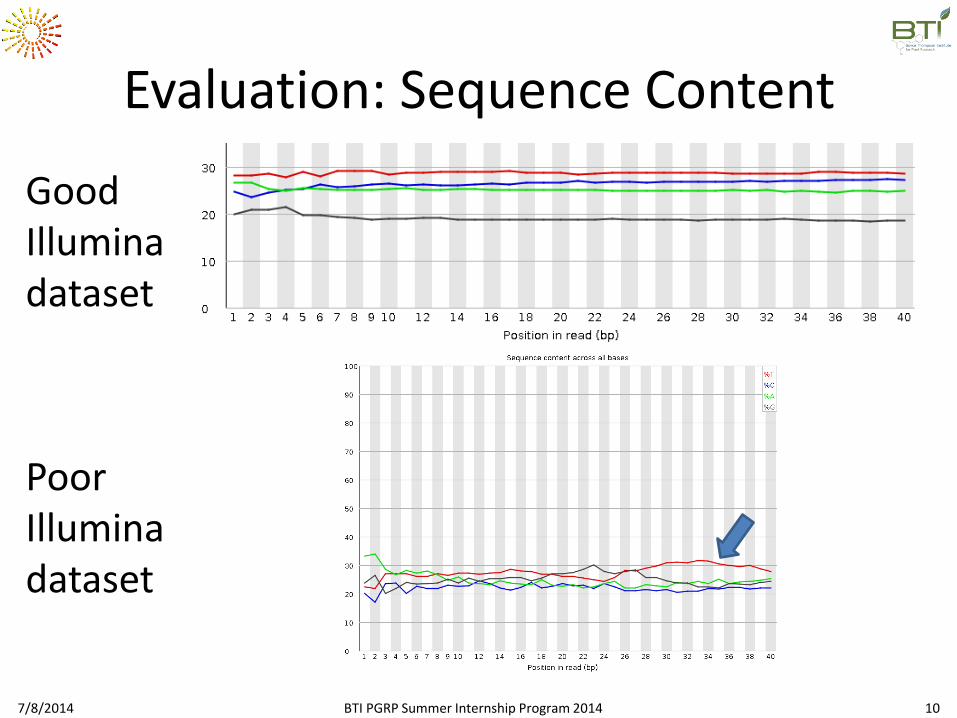
Evaluation: Sequence Content
7/8/2014 BTI PGRP Summer Internship Program 2014 10
Good Illumina dataset
Poor Illumina dataset

Evaluation: Duplication
Good Illumina dataset
7/8/2014 BTI PGRP Summer Internship Program 2014 11

Evaluation: Duplication
7/8/2014 BTI PGRP Summer Internship Program 2014 12
Good Illumina dataset
Poor Illumina dataset

Evaluation: Overrepresented Sequences
Good Illumina dataset
7/8/2014 BTI PGRP Summer Internship Program 2014 13

Evaluation: Overrepresented Sequences
7/8/2014 BTI PGRP Summer Internship Program 2014 14
Good Illumina dataset
Poor Illumina dataset
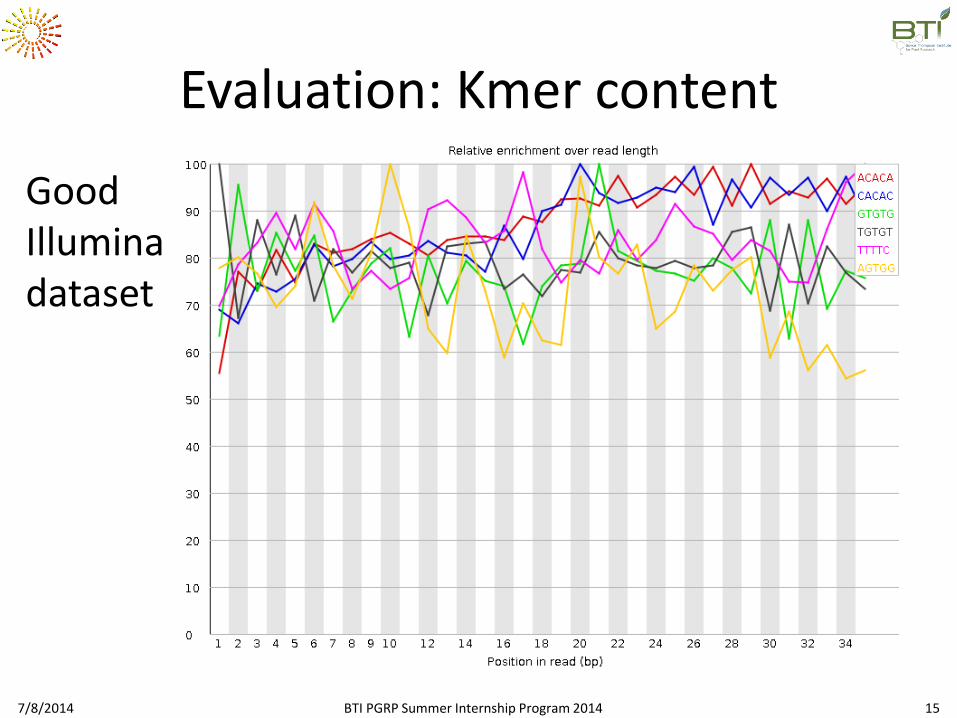
Evaluation: Kmer content
Good Illumina dataset
7/8/2014 BTI PGRP Summer Internship Program 2014 15

Evaluation: Kmer content
7/8/2014 BTI PGRP Summer Internship Program 2014 16
Good Illumina dataset
Poor Illumina dataset
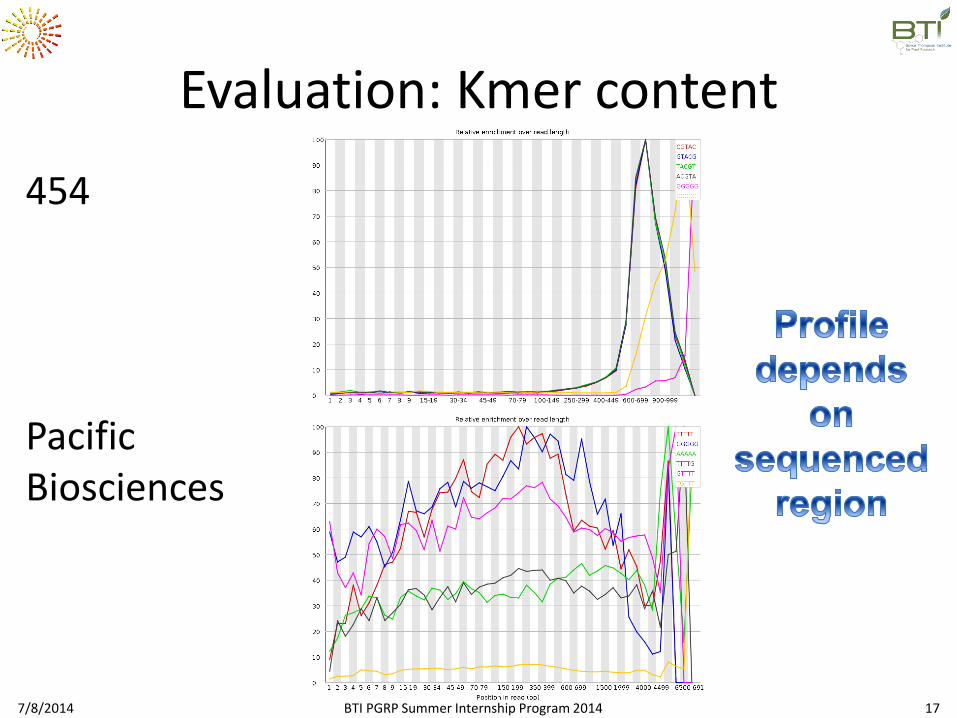
Evaluation: Kmer content
7/8/2014 BTI PGRP Summer Internship Program 2014 17
454
Pacific Biosciences

Question 2.2: How many sequences there are per file in FastQC?
Question 2.3: Which is the length range for these reads?
Question 2.4: Which is the quality score range for these reads? Which
one looks best quality-wise?
Question 2.5: Do these datasets have read overrepresentation?
Question 2.6: Looking into the kmer content, do you think that the samples
have an adaptor?
Evaluation Exercise 2:
1.Type ‘fastqc’ to start the FastQC program. Load the four
fastq sequence files in the program.
7/8/2014 BTI PGRP Summer Internship Program 2014 18

Goal:
Trim the low quality ends of the reads and remove
the short reads.
Data: (Illumina data for two tomato ripening stages)
ch4_demo_dataset.tar.gz
Tools: fastq-mcf (command line tool to process reads)
FastQC (gui, to calculate several stats for each file)
Preprocessing
7/8/2014 BTI PGRP Summer Internship Program 2014 19

Exercise 3:
• Download the file: adapters1.fa from ftp://ftp.solgenomics.net/user_requests/aubombarely/courses/RNAseqCorpoica/a
dapters1.fa
• Run the read processing program over each of the datasets
using
• Min. qscore of 30
• Min. length of 40 bp
• Type ‘fastqc’ to start the FastQC program. Load the four
new fastq sequence files. Compare the results with the
previous datasets.
Preprocessing
Tip: Use ‘fastqc -h’ to see help
7/8/2014 BTI PGRP Summer Internship Program 2014 20

Need Help??
7/8/2014 BTI PGRP Summer Internship Program 2014 21
Solutions: https://bitly.com/BioinfoInternExSol2014
Top Related