







Languages
Pages
Legal


Principales canalopathies à l'origine d'arythmies
ventriculaires graves chez l'adulte
Camilleri Elise DESC de réanimation médicale
2ème annéeJuin 2010

Quel contexte en réanimation?
• Patient admis pour MORT SUBITE RECUPEREE• QUE FAIRE?
– Âge du patient– Facteurs de risque cardiovasculaires– Prises médicamenteuses– Interrogatoire familial sur antécédents du patients/famille
• Antécédents de mort subite familiale– Examen clinique:
• Anomalies associées, diagnostic syndromique• Manifestations cliniques variées:
– Douleur thoracique– Antécédent de syncope, – palpitations
• Insuffisance cardiaque– Analyse précise de l’électrocardiogramme– Radiographie du thorax– Enzymes cardiaques– Question posée: ischémie myocardique, canalopathie?

Le tableau est-il compatible avec une canalopathie?
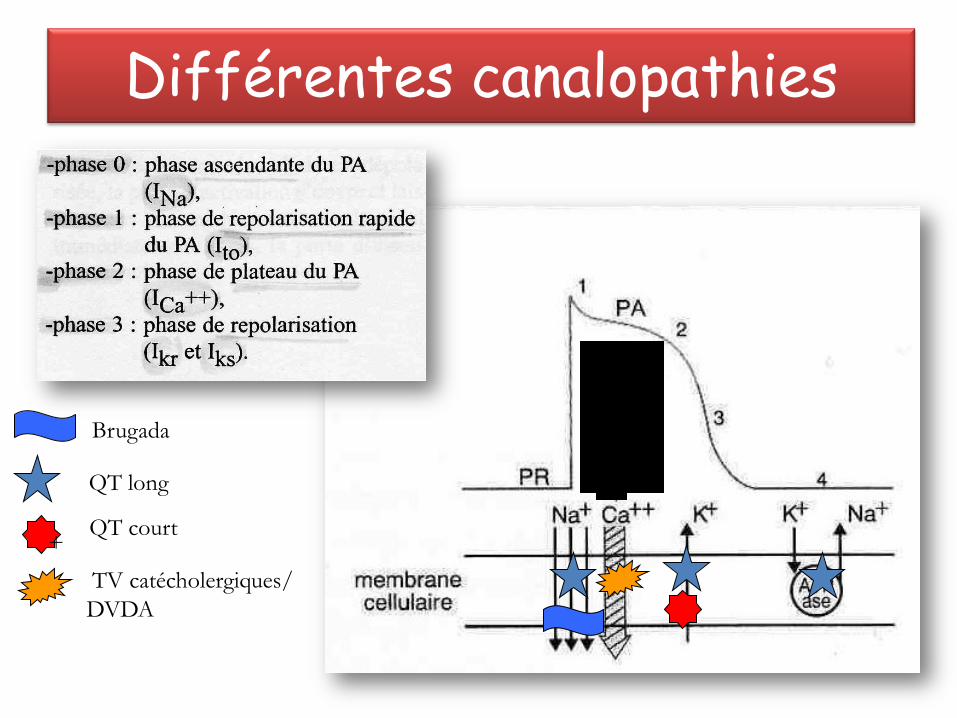
Différentes canalopathies
QT long
TV catécholergiques/
DVDA
QT court
Brugada
+

Canaux sodiques
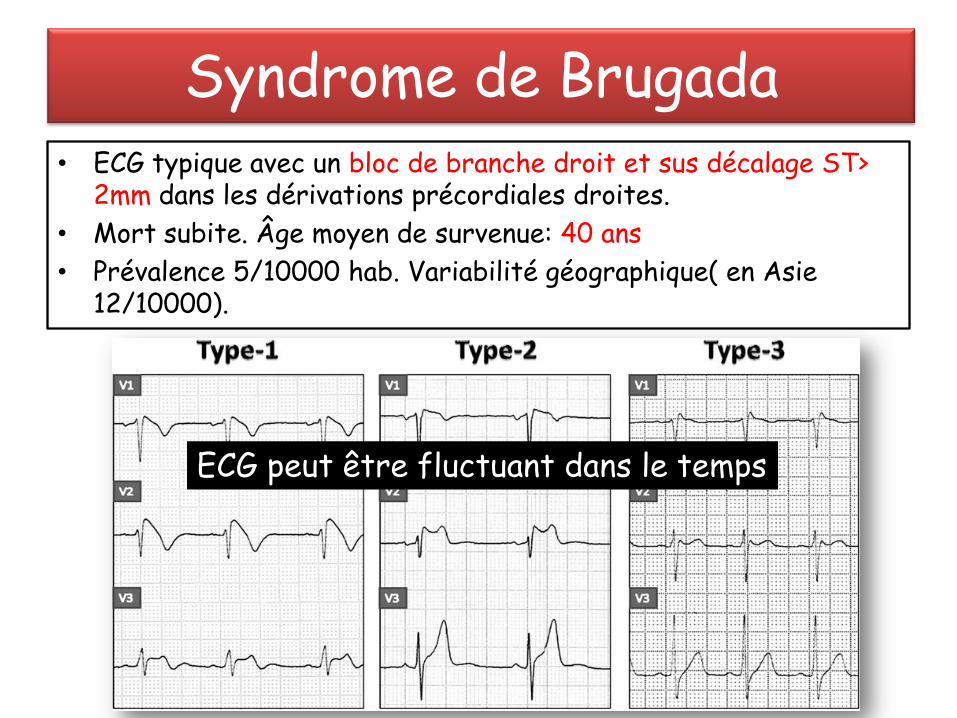
Syndrome de Brugada• ECG typique avec un bloc de branche droit et sus décalage ST>
2mm dans les dérivations précordiales droites.
• Mort subite. Âge moyen de survenue: 40 ans
• Prévalence 5/10000 hab. Variabilité géographique( en Asie .(12/10000
ECG peut être fluctuant dans le temps

Syndrome de Brugada
• Critères diagnostiques
Circulation 2002;106:2514-9.

Syndrome de Brugada
– Aspect génétique du syndrome• Transmission autosomique dominante
• Mutation du gène SCN5A codant pour la sous unité α du canal sodique.
• Mutation présente chez 30 % des syndromes de Brugada.
• Perte de fonction qualitative et quantitative du canal sodique.
• Le test génétique n’est pas une obligation pour le diagnostic
Circulation Cardiovascular Genetics 2009
– Physiopathologie • Perte de la balance des courants ioniques entrants et sortants
durant la phase 1 du potentiel d'action.
From M Alings Circulation 1999

Tests de provocation
• Ajmaline (1mg/kg IV en 5 mn) ou flécaïnide (2mg/kg IV en 10mn)
• Critères de positivité et d’arrêt du test:– ECG type 1
– ESV
– Elargissement du QRS de plus de 130%
Stimulation ventriculaire programmée
• Positivité : induction de TV polymorphe ou FV
• Facteur pronostic
Eckardt European heart journal 2002
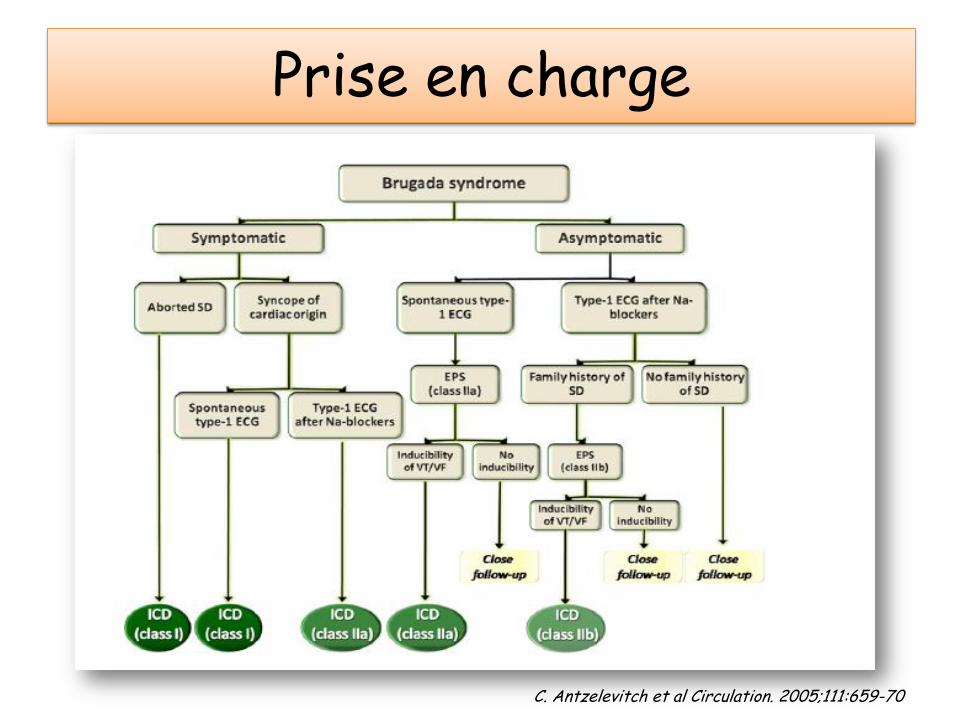
Prise en charge
C. Antzelevitch et al Circulation. 2005;111:659-70

Prise en charge
• SEUL TRAITEMENT EPROUVE: le DAI
• Quinidiniques en cours d'essai thérapeutique– Belhassen Circulation 2004– Ikr et Ito bloqueur– Adjuvent du DAI pour prévenir de nombreux CEI appropriés
• Possibilité de proposer l’ablation par radiofréquence de la zone d’émergence d’extrasystoles ventriculaires déclenchant les accès de FV ou TV polymorphes
• Exclusivement réservée dans les cas d’accès ventriculaires fréquents et responsables de défibrillations internes fréquentes
Haissaguerre et al. Circulation 2003

Diagnostic differentiel

Canaux calciques

Dysplasie arythmogène du ventricule droit
– Définition • Maladie génétique à transmission autosomique dominante, à
pénétrance partielle et expression variable
• Gènes codant les protéines du desmosome impliqués dans la communication intercellulaire
• Les cellules musculaires cardiaques sont progressivement remplacées par de la graisse et du tissu fibreux
• Entre 1/5000 et 1/10 000 dans la pop générale
Circulation 2003

Dysplasie arythmogène du ventricule droit
• ECG: inversion T en précordial droit(le plus fréquent), bloc de branche, ESV avec aspect BBG, onde epsilon(1/3 cas)
• UN ECG NORMAL N’ELIMINE PAS LE DIAGNOSTIC
• Holter ECG: troubles du rythme intermittent
• Epreuve d’effort: à visée diagnostique ou pronostique
• ECG haute amplification: peut mettre en évidence des zones de conduction électrique ralentie dans le myocarde
• ETT: diagnostic différentiel/ dilatation VD
• IRM myocardique: mettre en évidence les zones fibroadipeuses. Anévrisme localisé de la paroi VD
• Angiographie de contraste: examen progressivement remplacé par l’IRM
• SVP: inductibilité des arythmies (par réentrée)
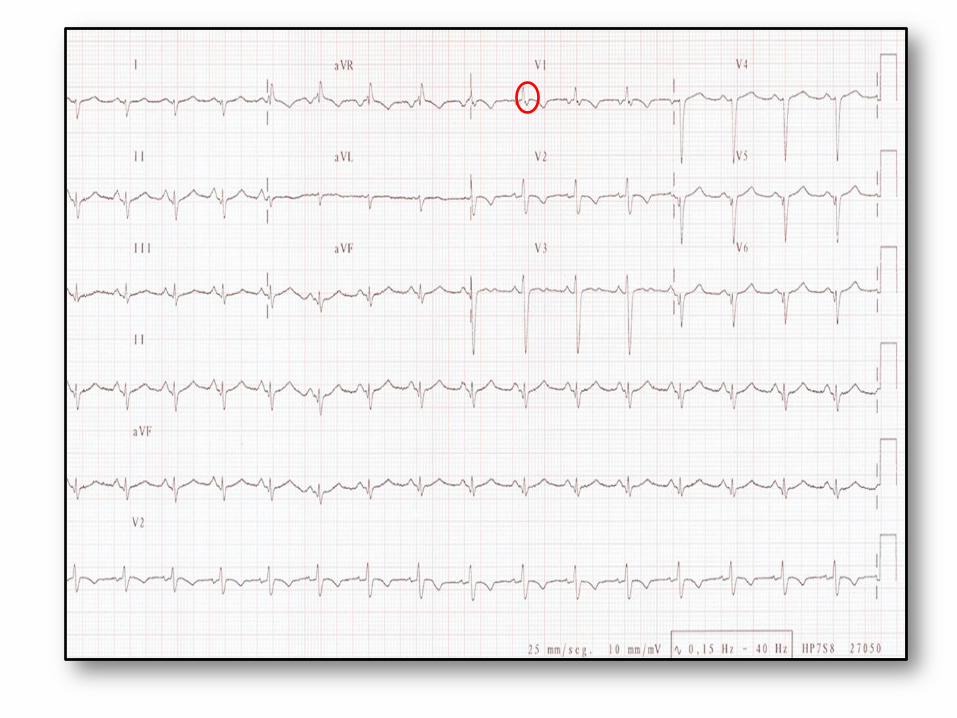

Dysplasie arythmogène du ventricule droit
• Le traitement des troubles du rythme peut faire appel selon les cas:
- aux médicaments antiarythmiques : βbloquants, Sotalol, Amiodarone
- à des interventions d’ablation par radiofréquence
- à l’implantation d’un défibrillateur automatique.

TV polymorphes cathécolergiques
• Autosomique dominant => mutation RyRr (ryanodine) 50%
• Autosomique récessive => mutation CASQ2 (calsequestrine)
• Canal Ca++ => surcharge de Ca++ dans la cellule• Enfants (3,5- - 16,5 ans). Ne touche pas les nourrissons à la
différence du QT long• Homme = femme• Clinique : Syncope à effort/stress
« comitialité » d’effortRetard diagnostic++ (épilepsie, spasme, vagal..): enfants longtemps traités à tortHistoire familiale de MS (30%)

TV polymorphes cathécolergiques
• ECG : FC 60/mn (bradycardie/enfant)QTC = NormalFibrillation atrialeTV bidirectionnelles +++
• Traitement : BBloquant = Nadolol 50mg/m²/j ( 2x/j)
DAI ?
• Risque : lié au moment de survenue des symptômes
• Suivi : Holter (FC < 130/mn effort; plus d’ESV répétitive/effort, non polymorphes.)
Leenhart , Circulation 1995

Canaux potassiques
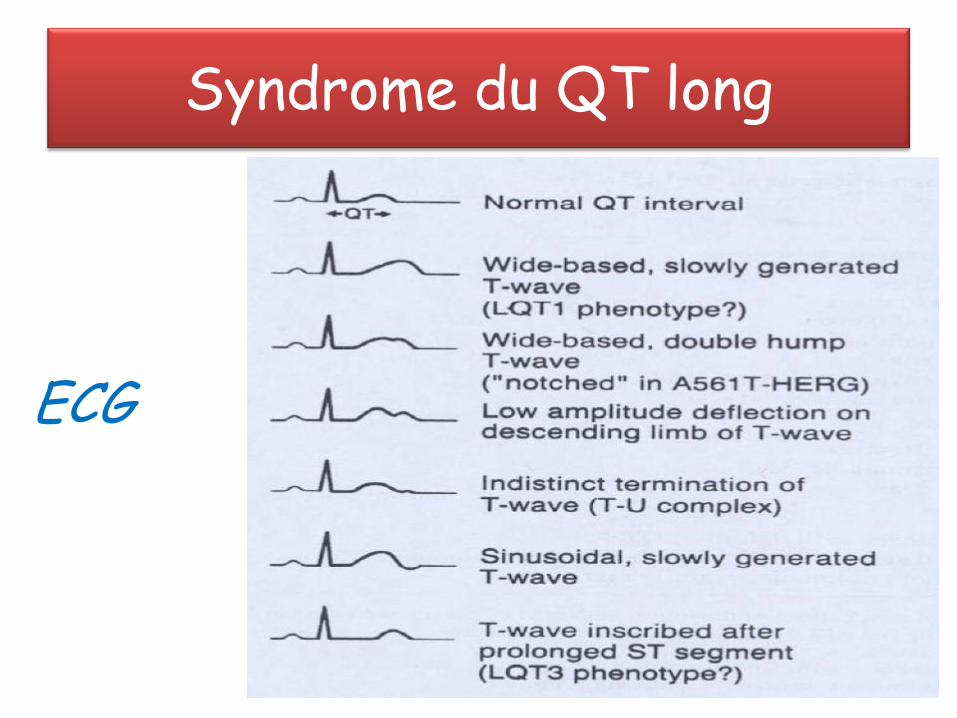
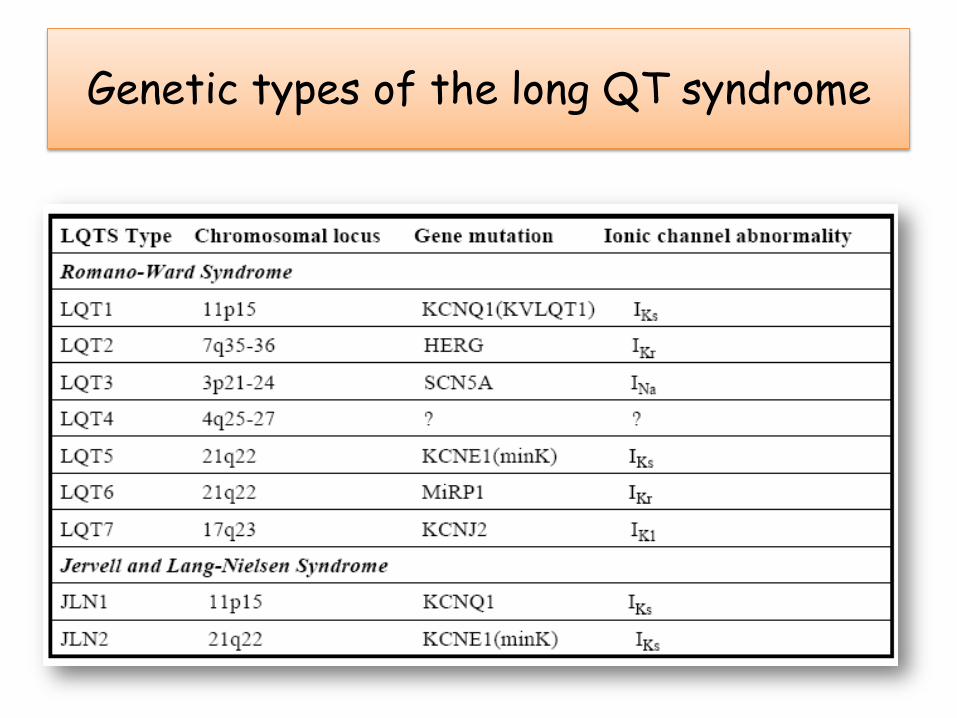
Syndrome du QT long
ECG

Syndrome du QT long
• QT> 440 ms; QTc = QTm/ √RR
• Associé à des syncopes ou mort subite dues à des arythmies ventriculaires: torsade de pointe, fibrillation ventriculaire
• Formes acquises plus fréquentes que les formes familiales.
• 2 formes majeures familiales:– Syndrome de Jervell et Lange-Nielson
• Transmission autosomique récessive
• Canal Iks
– Syndrome de Romano-Ward
• Transmission autosomique dominante

Syndrome du QT long
• Clinique : Syncope inexpliquée sur cœur normal (1er symptôme)Arrêt cardiaque ou mort subite chez enfant ou jeune
• Facteur : adrénergie (stress, émotions, effort, noyade) LQT3 : la nuit (et les efforts aussi) Médicaments
• Contexte : histoire de syncope ou MS familialehistoire de « comitialité »
de QT chez un autre / famille
• Ergométrie et Holter (jour/nuit) le ST ne diminue pas à l’effort
• Risques évolutifs: liés à la longueur du QT > 500mstype de mutation LQT3,1,2Première syncope: risque de mort subite de 70%
Priori et al. N Engl. J Med 2003;348:1866-74
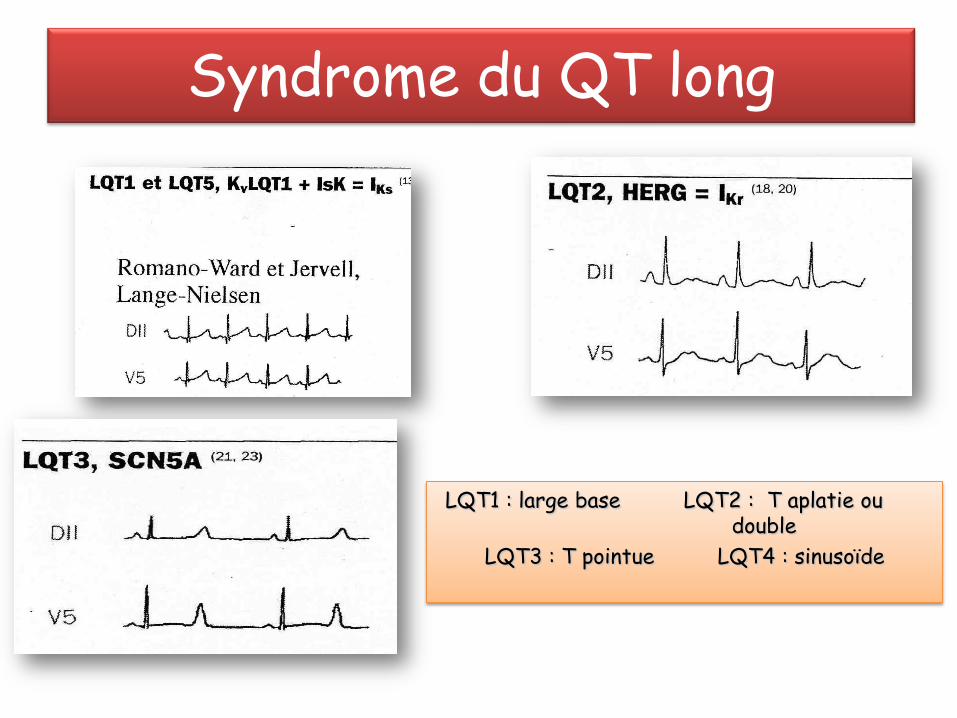
Syndrome du QT long
LQT1 : large base LQT2 : T aplatie ou double
LQT3 : T pointue LQT4 : sinusoïde

Prise en charge
• Traitement :1 - BBloquant ++
Nadolol (Corgard*) 50mg/m² /j (adulte1x/j et enfant en 2x/j)
=> Eduquer les patients (compliance++)2- Liste des médicaments interdits 3 – Pas de sport de compétition (loisir ok si BB)4 – Enquête familiale (50% atteinte)
5 - DAI :TDP/syncope sous BB (IIa,B)
LQT1, LQT2 > 500ms (IIb, B)LQT3 homme (peu de syncope mais très léthal) (IIb, B)
• Suivi : 1x/an => holter (en activité) =>

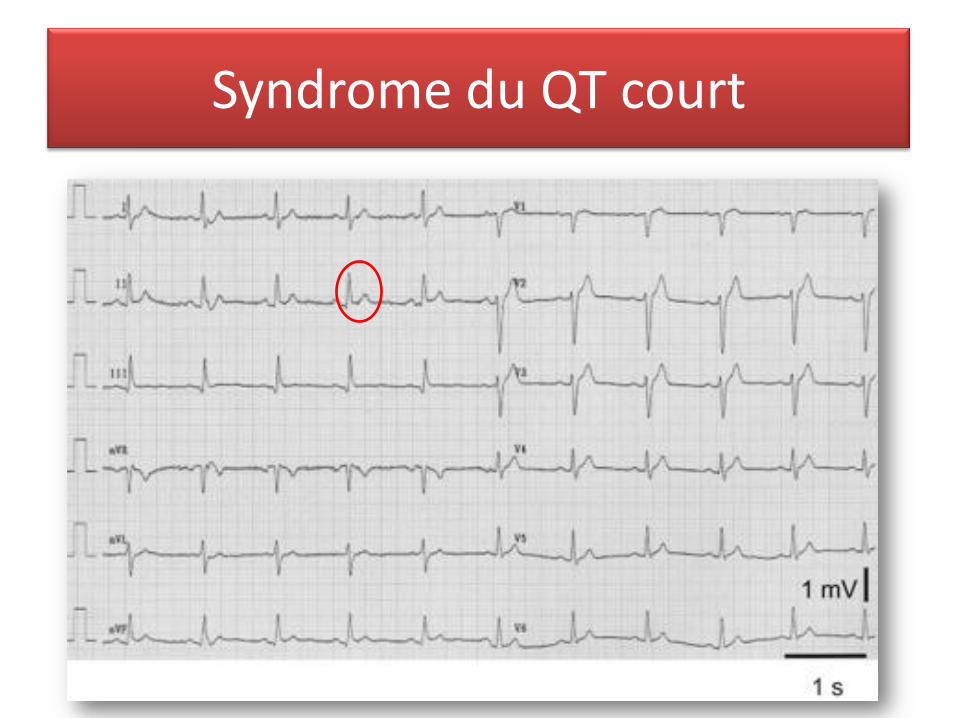
Syndrome du QT court
• Autosomique dominant
• Très rare: 10 cas publiés depuis 2000
• Homme = Femme
• Surtout jeunes enfants (qqmois) et adultes jeunes
• Définition : QT < 320 ms QTc < 340 ms
T pointue qui commence dès la fin de l’onde S
Absence d’adaptation du QT à la fréquence cardiaque parfois
• Génétique: AD Mutation Canal K
SQT1 KCNH2 IKr
SQT2 KCNQ1 IKs
SQT3 KCNJ2 IK1
• = gain de fonction (diminution Pot. Action)

Syndrome du QT court
• Clinique : – Asymptomatique, syncope, palpitations, Mort Subite, FV, FA
– Mort subite familiale, FA familiale idiopathique
• 1er symptômes: 25% Mort Subite 24% FA
• ECG : ESV infundibulaire
• SVP : PREV basse ++ (1ESV => FV)
PREOD basse ++ (ESA=> FA)

Prise en charge
• Prise en charge familiale: analyse ECG et clinique des patients au sein d’une famille à risque
• Diagnostic des patients pré-symptomatiques: test diagnostique après information claire sur les propositions thérapeutiques
• Diagnostic prénatal possible à la dixième semaine de grossesse
• Traitements en cours de développement:– Sotalol
– Quinidine
– Propafénone
– DAI !!!!!!!

CONCLUSION
• AU TOTAL– Y penser devant toute mort subite récupérée en réanimation
lorsqu’une cause ischémique est écartée– Réitérer les ECG régulièrement – Mesurer le QT– Quels sont les trts favorisants éventuels?– Pas de thérapeutique vraie– DAI

ANNEXES
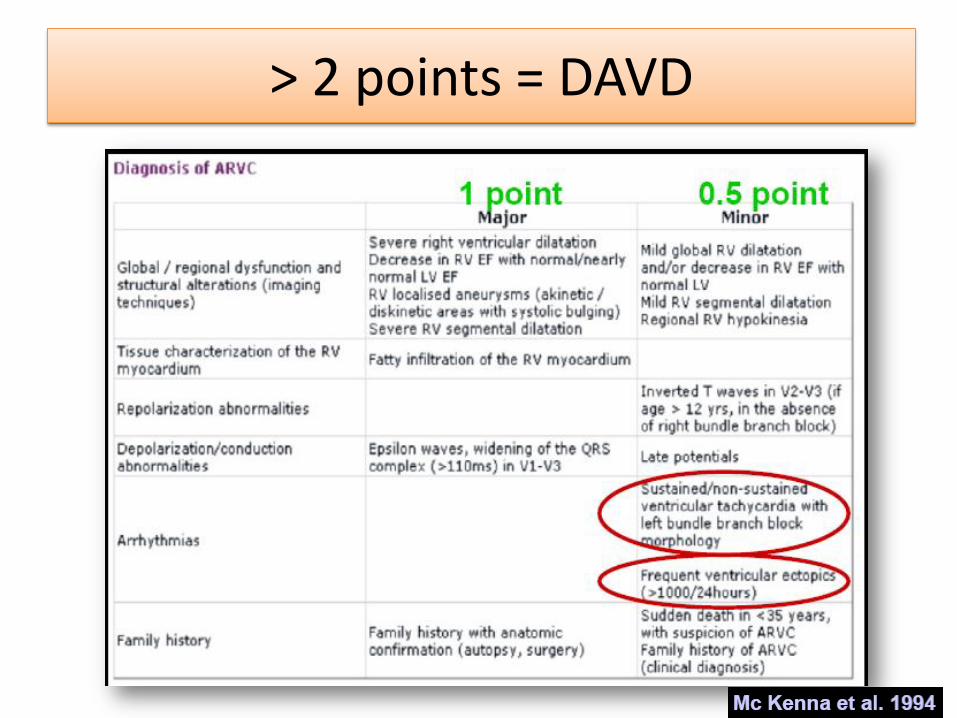
> 2 points = DAVD

Diagnostic criteria of LQTS(2-3 intermediate probability; >4 high)
Genetic types of the long QT syndrome
Top Related