







Languages
Pages
Legal


LA MALATTIA DI MELE
Pierangelo Veggiotti
Fondazione Istituto neurologico nazionale casimiro mondino
Pavia

Il termine Malattie Mitocondriali comprende numerose patologie caratterizzate da anomalie nel
metabolismo energetico con un grado variabile di disfunzione della catena respiratoria mitocondriale.
Le malattie mitocondriali
Il mitocondrio-organello
subcellulare- è in grado di svolgere
molteplici funzioni. La più
importante tra esse consiste
nell’estrarre energia dai substrati
organici che gli arrivano per
produrre un gradiente ionico che
viene sfruttato per produrre
adenosintrifosfato (ATP).
Gli altri processi in cui il
mitocondrio interviene
sono:
• l’apoptosi,
• regolazione del ciclo
cellulare,
• regolazione dello stato
redox della cellula,
• sintesi dell’eme,
• sintesi del colesterolo,
• produzione di calore. La produzione di energia è la funzione principale
del mitocondrio e viene svolta utilizzando i
principali prodotti della glicolisi: il piruvato ed il
NADH. Essi vengono sfruttati in due processi: il
ciclo di Krebs e la fosforilazione ossidativa.

L’origine del mitocondrio: la teoria endosimbiotica
Il mitocondrio presenta alcune caratteristiche tipiche dei
batteri: presenza di molecole di cardiolipina ed assenza di
colesterolo nella membrana interna, la presenza di un DNA
circolare a doppia eliche e la presenza di ribosomi propri e
di una doppia membrana.
Come i batteri, i mitocondri non hanno istoni ed i loro
ribosomi sono sensibili ad alcuni antibiotici (come il
cloramfenicolo).
In più i mitocondri sono organelli semiautonomi in quanto
replicano, per scissione binaria, autonomamente rispetto
alla cellula.
Stante queste similitudini, la teoria endosimbiotica afferma
che i mitocondri deriverebbero da ancestrali batteri, dotati di
metabolismo ossidativo, che sarebbero stati inglobati dalle
cellule eucariote con conseguente mutuo beneficio.
Successivamente i batteri avrebbero trasferito gran parte del
loro materiale genetico a quello cellulare, divenendo così,
mitocondri
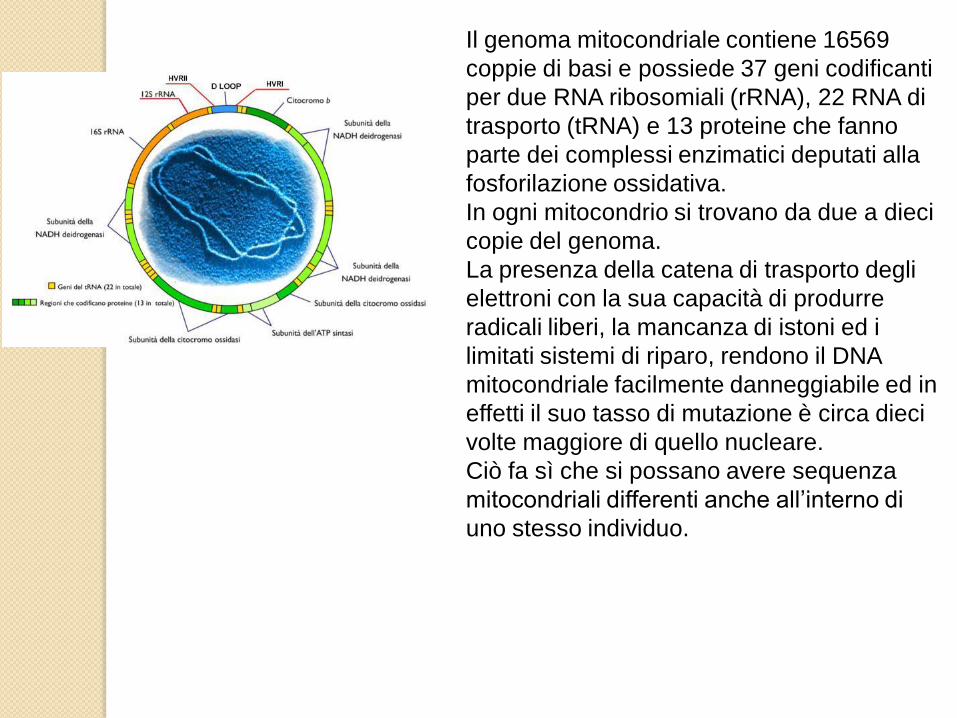
Il genoma mitocondriale contiene 16569
coppie di basi e possiede 37 geni codificanti
per due RNA ribosomiali (rRNA), 22 RNA di
trasporto (tRNA) e 13 proteine che fanno
parte dei complessi enzimatici deputati alla
fosforilazione ossidativa.
In ogni mitocondrio si trovano da due a dieci
copie del genoma.
La presenza della catena di trasporto degli
elettroni con la sua capacità di produrre
radicali liberi, la mancanza di istoni ed i
limitati sistemi di riparo, rendono il DNA
mitocondriale facilmente danneggiabile ed in
effetti il suo tasso di mutazione è circa dieci
volte maggiore di quello nucleare.
Ciò fa sì che si possano avere sequenza
mitocondriali differenti anche all’interno di
uno stesso individuo.
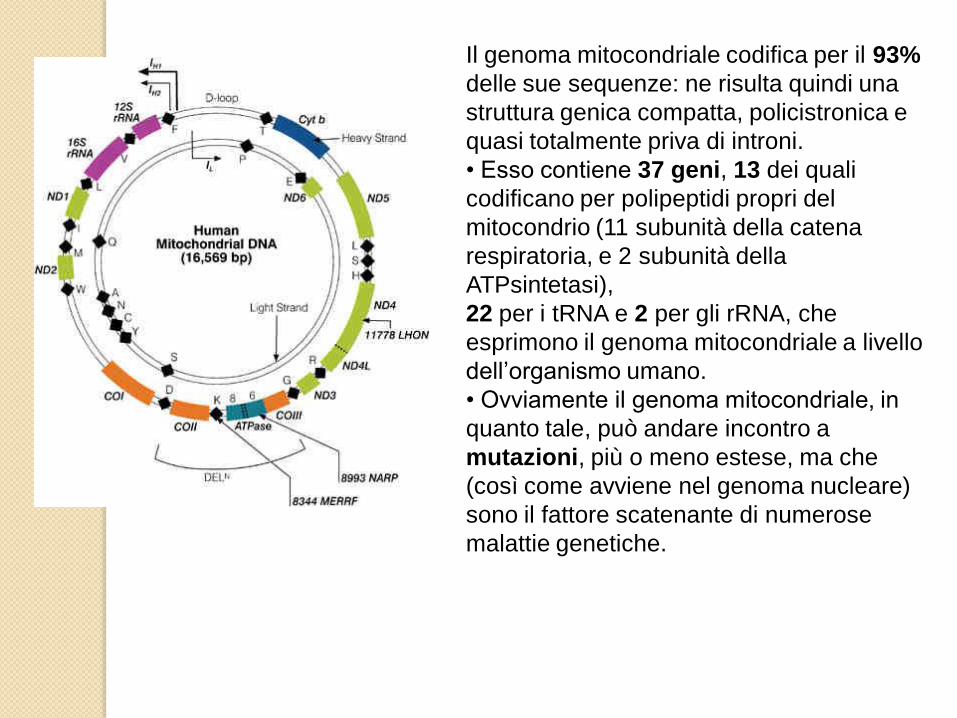
Il genoma mitocondriale codifica per il 93%
delle sue sequenze: ne risulta quindi una
struttura genica compatta, policistronica e
quasi totalmente priva di introni.
• Esso contiene 37 geni, 13 dei quali
codificano per polipeptidi propri del
mitocondrio (11 subunità della catena
respiratoria, e 2 subunità della
ATPsintetasi),
22 per i tRNA e 2 per gli rRNA, che
esprimono il genoma mitocondriale a livello
dell’organismo umano.
• Ovviamente il genoma mitocondriale, in
quanto tale, può andare incontro a
mutazioni, più o meno estese, ma che
(così come avviene nel genoma nucleare)
sono il fattore scatenante di numerose
malattie genetiche.

Fosforilazione ossidativa: la catena di trasporto
degli elettroni
Attraverso un complesso multienzimatico avente le
funzioni di catena di trasporto, gli elettroni dopo una
serie di passaggi intermedi, vengono ceduti all’ossigeno
molecolare (O2) che viene ridotto ad acqua. Nel
mitocondrio si possono isolare ben quattro complessi
poliproteici responsabili del trasporto degli elettroni:
• Complesso I (NADH deidrogenasi) che contiene
almeno 30 diversi polipeptidi, una flavoproteina e 9
centri ferro-zolfo e per ogni coppia di elettroni fatta
passare vengono trasferiti tre o quattro protoni,
• Complesso II (Succinato deidrogenasi) che, oltre a
catalizzare una reazione del ciclo di Krebs, consente il
trasferimento di elettroni al FAD ed all’ubichinone ma
non permette il passaggio di protoni,
• Complesso III (Citocromo c riduttasi) che contiene
circa 10 polipeptidi e gruppi eme ed un centro ferro-
zolfo, permette il passaggio di elettroni dall’ubichinone
ridotto al citocromo c e per ogni coppia di elettroni
trasferisce quattro protoni,
• Complesso IV (Citocromo c ossidasi) che contiene
almeno 13 polipeptidi permette il trasferimento di
elettroni dal citocromo c all’ossigeno ed anche lo
spostamento dei protoni anche se non ne è ben chiaro
il numero (forse quattro per ossigeno ridotto).
Successivamente i protoni vengono rifatti passare
attraverso la membrana interna, in un processo di
diffusione facilitata, tramite l’enzima ATP sintetasi che
ottiene così l’energia sufficiente per produrre molecole
di ATP, trasferendo un gruppo fosfato a dell’ADP.

Il mitocondrio funziona da centrale d’integrazione degli stimoli
apoptotici. Essi possono essere di molteplice natura (caspasi,
ceramide, vari tipi di chinasi, ganglioside GD3, ecc…) e sono
in grado di determinare l’apertura di un complesso
poliproteico chiamato poro di transizione mitocondriale. Come
risultato finale, il mitocondrio si riempe di liquido e la
membrana esterna scoppia liberando nel citoplasma fattori
stimolanti l’apoptosi come AIF, (Apoptosis Inducing Factor)
che è in grado di raggiungere il nucleo ed attiva una via
indipendente dalle caspasi in grado di degradare il DNA, ed il
citocromo c che si lega alle proteine Apaf-1 (apoptotic
protease activating factor) e caspasi 9 ed una molecola di ATP
formando un complesso definito apoptosoma. La caspasi 9
presente diviene in grado di attivare altre caspasi che danno il
via ad una cascata molecolare che si conclude con la
degradazione del DNA ad opera di fattori nucleari
Il mitocondrio e l’apoptosi

Dal punto di vista clinico, le malattie mitocondriali possono essere tessuto-specifiche oppure avere
carattere multisistemico.
L’elevata richiesta energetica da parte del Sistema Nervoso Centrale e dei muscoli scheletrici
conferisce a questi tessuti una maggiore vulnerabilità alle disfunzioni mitocondriali. Per questo
motivo, ci si riferisce alle malattie mitocondriali spesso con l’espressione “Encefalomiopatie
mitocondriali”. Questo rende anche ragione dell’interesse preminentemente neurologico del loro
studio.
Le patologie mitocondriali possono essere distinte, su base genetica in 2 categorie:
1) Malattie causate da mutazioni del DNA mitocondriale (mtDNA)
2) Malattie mitocondriali causate da mutazioni in geni nucleari che codificano per proteine
residenti nel mitocondrio
Malattie Mitocondriali: clinica

Le normali funzioni
mitocondriali dipendono
dalla relazione simbiotica
di DNA nucleare (frecce blu
e nere) e DNA
mitocondriale (frecce
rosse).
L’omeostasi cellulare è sotto
il doppio controllo di questi
due genomi. Le
patologie mitocondriali
possono sorgere da
disfunzioni che scaturiscono
dall’uno o dall’altro genoma
(testo in rosso: malattie del
DNA
mitocondriale; testo in blu:
malattie del DNA nucleare).

Ereditarietà malattie da alterazioni del DNA
mitocondriale:
• La malattia si trasmette con ereditarietà
materna.
Il padre di un probando non è a rischio di
avere la malattia.
La madre invece,che possiede
sicuramente la mutazione,può presentare
o meno i sintomi.
La diagnosi prenatale è possibile solo se la
malattia è già stata rilevata nella madre.
Tuttavia non si può prevedere il fenotipo.
Le malattie mitocondriali conseguenti ad alterazioni del
DNA nucleare seguono le leggi della genetica
mendeliana
Al momento della fecondazione il DNA
mitocondriale di provenienza paterna è
espulso
Ereditarietà

Classificazione genetica delle malattie mitocondriali da disordini del
genoma mitocondriale
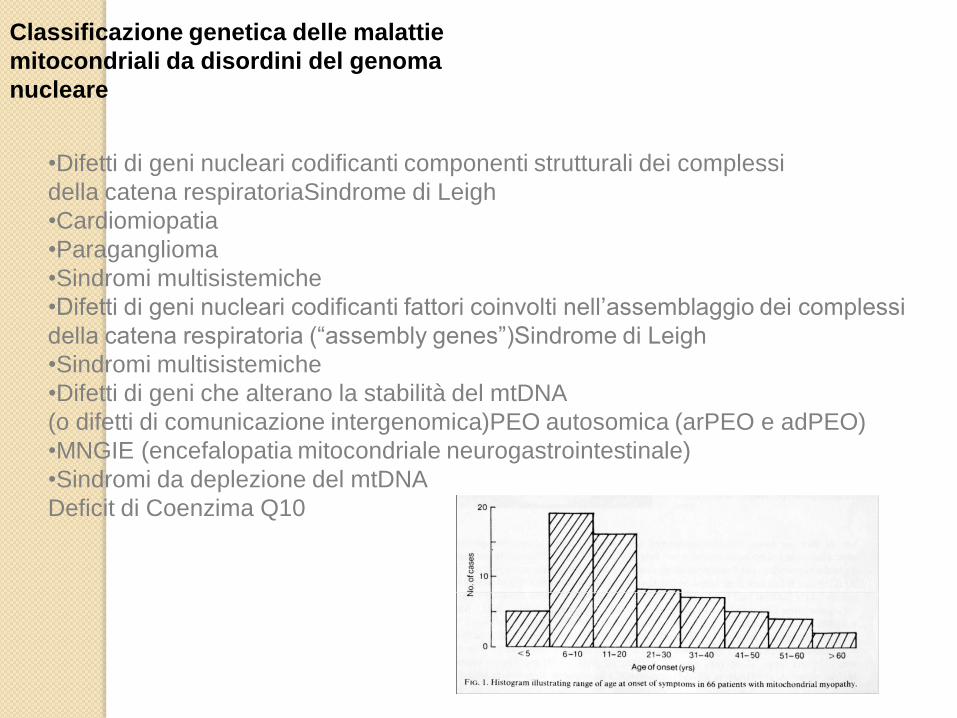
Classificazione genetica delle malattie
mitocondriali da disordini del genoma
nucleare
•Difetti di geni nucleari codificanti componenti strutturali dei complessi
della catena respiratoriaSindrome di Leigh
•Cardiomiopatia
•Paraganglioma
•Sindromi multisistemiche
•Difetti di geni nucleari codificanti fattori coinvolti nell’assemblaggio dei complessi
della catena respiratoria (“assembly genes”)Sindrome di Leigh
•Sindromi multisistemiche
•Difetti di geni che alterano la stabilità del mtDNA
(o difetti di comunicazione intergenomica)PEO autosomica (arPEO e adPEO)
•MNGIE (encefalopatia mitocondriale neurogastrointestinale)
•Sindromi da deplezione del mtDNA
Deficit di Coenzima Q10
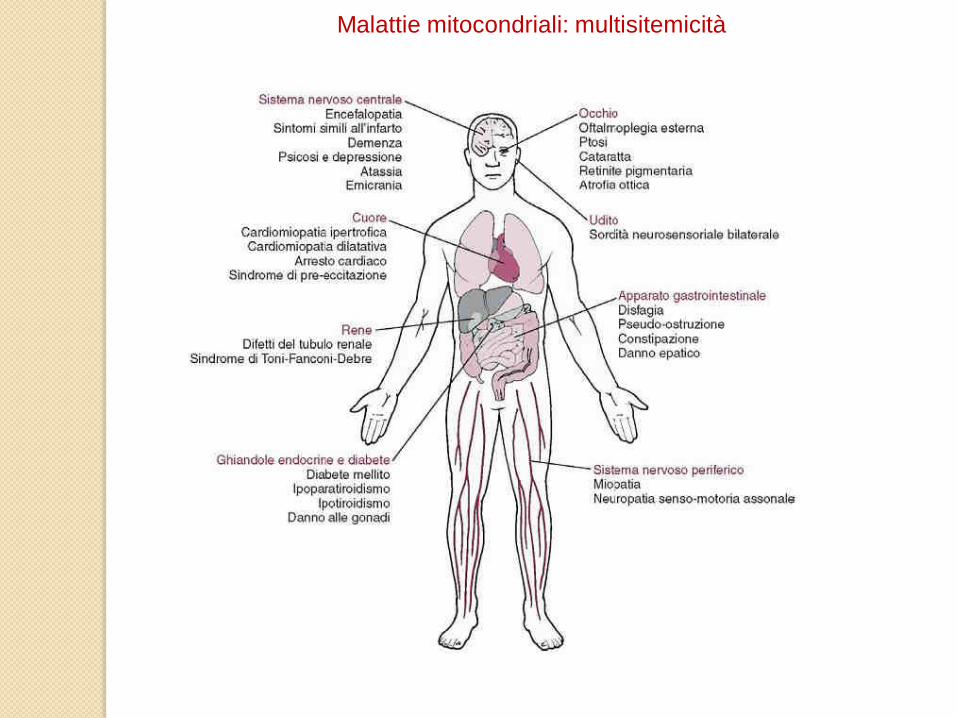
Malattie mitocondriali: multisitemicità

Epilepsy definition
Epilepsy affect 65 million of people worldwide and entails major burden in seizures related disability, mortality,comorbidities stigma and cost



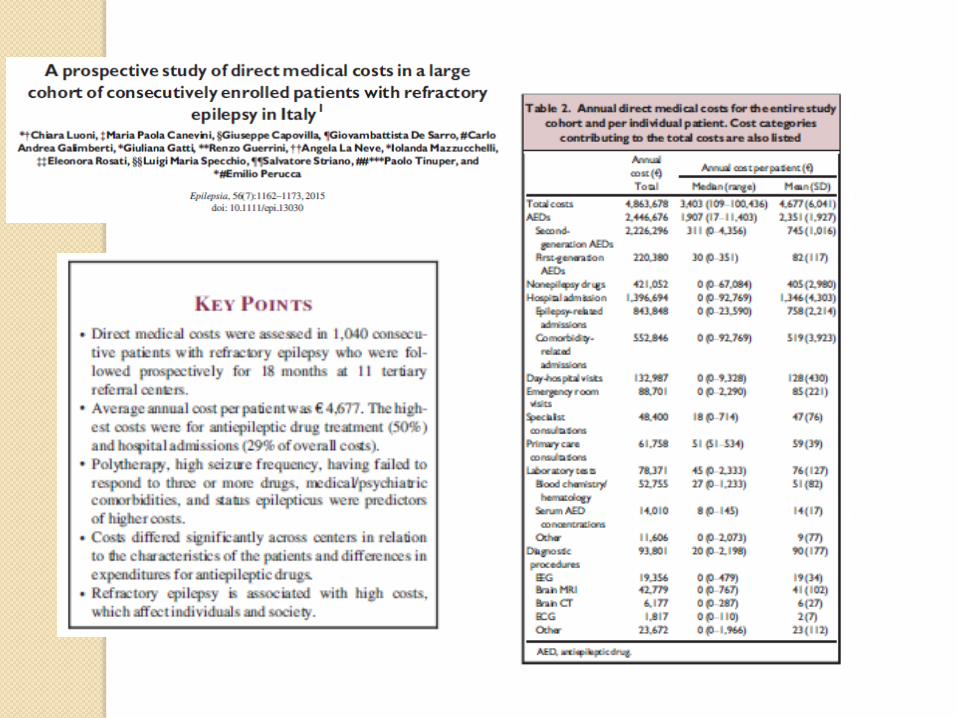

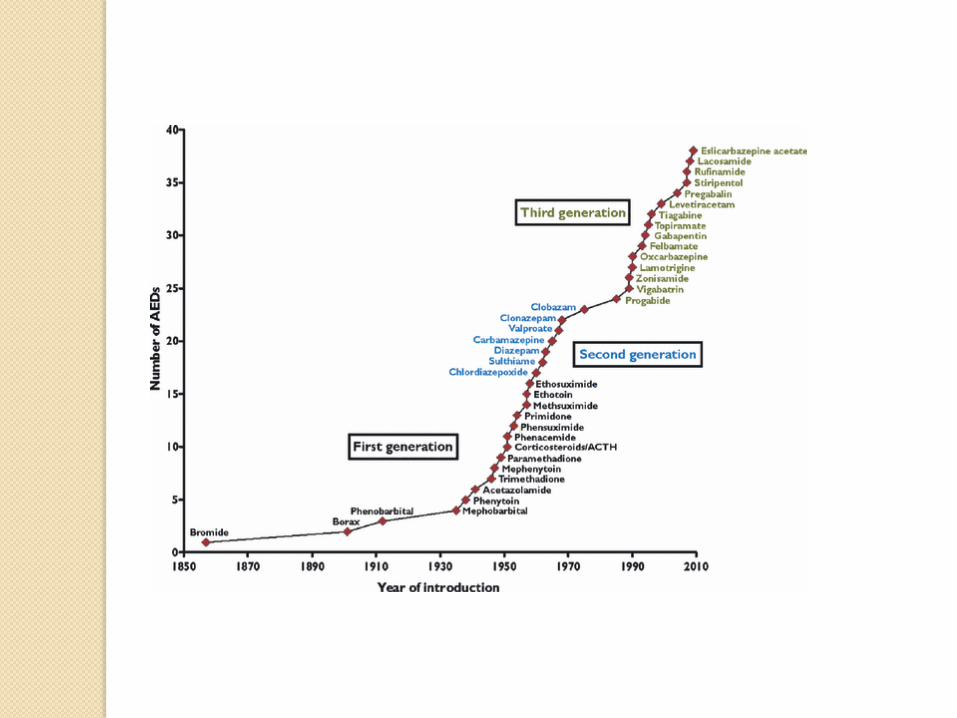

KETOGENIC DIET
1920 1970 1990 2000 2015
Fasting ?
MAD


Grazie a tutti quelli che mi sopportano/supportano ogni giorno
Top Related