







Languages
Pages
Legal


GUSTAVO MONTEIRO VIANA
INVESTIGAÇÃO MOLECULAR E PERFIL BIOQUÍMICO DE
PACIENTES COM MUCOPOLISSACARIDOSES
BELÉM/PA
2010

Livros Grátis
http://www.livrosgratis.com.br
Milhares de livros grátis para download.

GUSTAVO MONTEIRO VIANA
INVESTIGAÇÃO MOLECULAR E PERFIL BIOQUÍMICO DE PACIENTES COM MUCOPOLISSACARIDOSES
Dissertação de Mestrado submetida ao Programa de Pós-graduação em Neurociências e Biologia Celular da Universidade Federal do Pará como requisito parcial para obtenção do grau de Mestre em Neurociências e Biologia Celular. Orientador: Prof. Dr. Luiz Carlos Santana da Silva, ICB – UFPA.
BELÉM/PA
2010

GUSTAVO MONTEIRO VIANA
INVESTIGAÇÃO MOLECULAR E PERFIL BIOQUÍMICO DE PACIENTES COM MUCOPOLISSACARIDOSES
Dissertação de mestrado submetida ao Programa de Pós-graduação em Neurociências e Biologia Celular da Universidade Federal do Pará como requisito parcial para obtenção do grau de Mestre em Neurociências e Biologia Celular. Orientador: Prof. Dr. Luiz Carlos Santana da Silva, ICB – UFPA.
Orientador: Prof. Dr. Luiz Carlos Santana da Silva (ICB – UFPA) Banca examinadora Avaliador1: Prof. Dr. João Farias Guerreiro (ICB / UFPA) Avaliador2: Prof. Dr. José Ricardo dos Santos Vieira (ICB/UFPA) Avaliador3: Profa. Dra. Rita Mousinho Ribeiro (ICB/UFPA) Avaliador4 (suplente): Prof. Dr. André Salim Khayat (ICB / UFPA) Avaliador5 (suplente): Prof. Dr. José Eduardo Melo dos Santos (ICB/UFPA)
BELÉM/PA 2010

Dedico esse trabalho aos meus pais, verdadeiros alicerces da minha vida.

AGRADECIMENTOS
À Deus, por me proporcionar serenidade e paciência durante a realização
deste trabalho.
Aos meus pais, que sempre estiveram ao meu lado, me apoiaram em todas as
minhas decisões e me incentivaram em todos os momentos;
Ao meu orientador Prof. Dr. Luiz Carlos Santana da Silva, pela orientação, por
todo empenho, sabedoria, exigência e, acima de tudo, pelo companheirismo e
amizade que demonstrou ao longo da realização deste trabalho.
Ao Prof. Dr. Anderson Manoel Herculano de Oliveira pelo apoio e envolvimento
dados no momento da minha qualificação.
Aos meus colegas do LEIM, pelo companheirismo e amizade.
Aos meus amigos Erik Alves e Cléber Cruz, por terem sido essenciais na
elaboração deste trabalho e pelo apoio fornecido nos momentos mais difíceis.
À Dra. Isabel Cristina Neves de Souza e à Dra. Raimunda Helena Feio, pela
importante participação no projeto ao fornecer os dados clínicos dos pacientes.
Ao grupo do Serviço de Genética Médica do Hospital de Clínicas de Porto
Alegre, pela hospitalidade e pelos ensinamentos a mim dedicados.
Aos pacientes com MPS e seus familiares, pela paciência e compreensão
durante a realização do estudo.
Ao CNPq, pelo apoio financeiro.
E a todas as pessoas que, direta ou indiretamente, contribuíram para a
realização deste trabalho.

“A verdadeira ignorância não reside na falta de conhecimentos, mas na falta de vontade de aceitá-los”
Karl Popper

RESUMO
As Mucopolissacaridoses (MPS) consistem em um raro grupo de doenças
metabólicas hereditárias e são causadas pela deficiência de enzimas
lisossomais específicas responsáveis pela degradação de glicosaminoglicanos
(GAG). Uma vez não degradados, os GAG acumulam-se em vários tecidos e
órgãos, levando ao desenvolvimento de uma doença crônica de caráter
progressivo e multisistêmico. A Terapia de Reposição Enzimática (TRE) tem
demonstrado eficácia na redução da acumulação tecidual e excreção urinária
de GAG. Além disso, a TRE também permitiu uma melhora dos sintomas
clínicos apresentados pelos pacientes com MPS. O presente estudo investigou
as características bioquímicas e moleculares de 10 pacientes com MPS
provenientes da região Norte do Brasil (2 com MPS I, 4 com MPS II, 1 com
MPS IIB e 3 com MPS VI), sendo alguns submetidos à TRE. Para avaliar a
eficácia da TRE durante o tratamento, a excreção urinária de GAG foi
mensurada através do ensaio colorimétrico utilizando o reagente Azul de
Dimetileno (DMB). A análise molecular revelou a presença da mutação P533R
nos pacientes com MPS I. Nos pacientes com MPS II, foi detectada a mutação
R468W. Em pacientes com MPS VI, dois polimorfismos anteriormente descritos
(V358M e V376M) também foram identificados. Após 48 semanas de
tratamento, observou-se uma redução significativa da excreção urinária de
GAG (p<0,01) em todos os pacientes com MPS submetidos à TRE. Os
resultados obtidos no presente estudo estão de acordo com a literatura,
confirmando a eficácia da TRE na redução da excreção urinária de GAG em
pacientes com MPS e a elevada heterogeneidade molecular da doença. Este é
o primeiro estudo bioquímico e molecular realizado em pacientes com MPS no
Estado do Pará.

ABSTRACT
Mucopolysaccharidoses (MPS) are rare lysosomal disorders caused by the
deficiency of specific lysosomal enzymes responsible for glycosaminoglycan
(GAG) degradation. When not degraded, GAG accumulates in various tissues
and organs, leading to a chronic, progressive and multisystem disease. Enzyme
Replacement Therapy (ERT) has been shown to reduce accumulation and
urinary excretion of GAG. It has also been demonstrated that ERT is able to
improve some clinical signs of MPS patients. We studied biochemical and
molecular characteristics of 10 MPS patients (2 MPS I, 4 MPS II, 1 MPS IIB and
3 MPS VI) undergoing ERT in Northern Brazil. To evaluate the efficacy of the
ERT during the treatment, urinary GAG excretion was measured by colorimetric
assay using Dimethylene Blue (DMB). Patients were screened for 8 known
MPS related mutations using PCR, RFLP, SSCP and direct sequencing.
Molecular analysis of two MPS I patients revealed the previously reported
mutation P533R. In MPS II patients, mutation analysis identified one previously
reported mutation (R468W) and in MPS VI patients, two previously detected
polymorphisms (V358M and V376M) were also found. In the biochemical
analysis, total urinary GAG excretion significantly decreased in patients with
MPS I (p<0,01) and MPS VI (p<0,01) after 48 weeks of ERT. Our findings are in
agreement with literature, confirming the efficacy of ERT on the reduction of
urinary GAG excretion in MPS patients and the high molecular heterogeneity of
this disease. This is the first biochemical and molecular study of MPS patients
in the State of Pará.

SUMÁRIO
1. INTRODUÇÃO 01
1.1 Mucopolissacaridoses (MPS) 01
1.1.1 Generalidades 01
1.1.2 Classificação 02
1.1.3 Diagnóstico clínico 05
1.1.4 Diagnóstico laboratorial 08
1.1.5 Tratamento 09
1.1.6 Aspectos epidemiológicos 12
1.1.7 Bases Celulares das MPS 13
1.1.8 Bases Moleculares das MPS 15
a) Mucopolissacaridose I (MPS I) 15
b) Mucopolissacaridose II (MPS II) 18
c) Mucopolissacaridose IIIB (MPS IIIB) 20
d) Mucopolissacaridose VI (MPS VI) 21
2. OBJETIVOS 23
2.1 Objetivo Geral 23
2.2 Objetivos Específicos 23
3. MATERIAL E MÉTODOS 24
3.1 MATERIAL 24
3.1.1 Caracterização da amostra 24
3.2 Critérios de inclusão 24
3.3 Aspectos éticos 27
3.4 Coleta e armazenamento das amostras 27
3.2 MÉTODOS 27
3.2.1. Análise bioquímica 27
a) TRE 27
b) Dosagem de cratinina urinária 28
c) Dosagem dos GAG urinários 28
3.2.2 Análise molecular 29
a) Extração do DNA 29
b) Reação em cadeia da polimerase (PCR) 29

c) Análise por digestão com endonucleases de restrição 32
d) Análise de Polimorfismos Conformacionais de Cadeia Simples (SSCP) 34
e) Seqüenciamento direto 34
f) Análise estatística 35
4. RESULTADOS 36
4.1 MPS I 36
4.1.1 Perfil bioquímico 36
4.1.2 Perfil molecular (gene IDUA) 37
4.2 MPS II 40
4.2.1 Perfil molecular (gene IDS) 40
4.3 MPS IIIB 43
4.3.1 Perfil molecular (gene NAGLU) 43
4.4 MPS VI 43
4.4.1 Perfil bioquímico 43
4.4.2 Perfil molecular (gene ARSB) 44
5. DISCUSSÃO 48
5.1 Investigação molecular 48
5.2 Perfil bioquímico 54
5.3 Considerações finais 58
6. CONCLUSÕES 60
7. REFERÊNCIAS BIBLIOGRÁFICAS 61
8. ANEXOS 74
8.1 Carta de aceite do Comitê de Ética em Pesquisa com Seres Humanos 74
8.2 Termo de Consentimento Livre e Esclarecido 75

LISTA DE QUADROS E FIGURAS
QUADRO 01: Classificação das mucopolissacaridoses 04 QUADRO 02: Perfil bioquímico inicial dos pacientes com MPS antes da TRE.
25
QUADRO 03: Resumo das características clínicas dos pacientes com MPS
26
QUADRO 04: Oligonucleotídeos iniciadores (primers) utilizados para a amplificação de fragmentos específicos dos genes IDUA (MPS I), IDS (MPS II), NAGLU (MPS IIIB) e ARSB (MPS VI).
30
QUADRO 05: Protocolo básico para as reações de amplificação por PCR.
31
QUADRO 06: Condições usadas nas reações de PCR. 31 QUADRO 07: Protocolo empregado nas reações com enzimas de restrição.
33
QUADRO 08: Protocolo básico de digestão com endonucleases de restrição
33
QUADRO 09: Protocolo básico utilizado nas reações de seqüenciamento
35
FIGURA 01: Rota metabólica da degradação do dermatan sulfato. Nac (N-acetil-galactosamina)
01
FIGURA 02: Esquema celular das Mucopolissacaridoses 14

LISTA DE TABELAS
TABELA 01: Mutações e polimorfismos encontrados nos pacientes com MPS e seus parentes.
47
TABELA 02: Características moleculares, bioquímicas e clínicas dos pacientes com MPS.
58

LISTA DE ABREVIA TURAS E SIGLAS
ARSB: Arilsulfatase B BCTMA: Brometo de cetil-trimetil-amônio DLD: Doença de depósito lisossômico DNA: Ácido desoxirribonucléico
EIM: Erros inatos do metabolismo GAG: glicosaminoglicanos IDS: iduronato-sulfatase IDUA: α-L-iduronidase MPS: mucopolissacaridoses NAGLU: α-N-acetil-glicosaminidase PCR: polimerase chain reaction (reação em cadeia da polimerase) RFLP: restriction fragment lenght polymorphism (digestão por endonucleases de restrição)
SSCP: single stranded conformational polymorphism (polimorfismo conformacional de cadeia simples)

1
1. INTRODUÇÃO
1.1. Mucopolissacaridoses
1.1.1. Generalidades
As mucopolissacaridoses (MPS) correspondem a um grupo de doenças
lisossômicas de depósito (DLD), causadas pela deficiência de enzimas lisossomais
(Figura 01) essenciais para o metabolismo de componentes da matriz extracelular
como os glicosaminoglicanos (GAG). Uma vez não degradados, os GAG são
estocados nos compartimentos lisossômicos celulares, promovendo uma série de
complicações patológicas que iniciam desde o período fetal, até a intensa
progressão dos sintomas na fase infantil (Nelson & Cox, 2000; Scriver et al., 2001).
Figura 01 . Rota metabólica da degradação do dermatan sulfato. Nac (N-acetil-galactosamina). Fonte: Scriver, 2001.

2
1.1.2. Classificação
A Mucopolissacaridose do tipo I corresponde a um tipo de DLD
caracterizada pela deficiência da enzima alfa-L-iduronidase (IDUA; E.C.: 3.2.1.76),
responsável pela quebra dos GAG dermatan sulfato e heparan sulfato. A MPS I é
dividida em três subformas, de acordo com a apresentação clínica dos pacientes:
Síndrome de Hurler (forma grave, OMIM: 607014), Síndrome de Hurler-Scheie
(forma intermediária, OMIM: 607015) e Síndrome de Scheie (forma atenuada, OMIM:
607016). Os sintomas apresentados estão inseridos em um amplo espectro clínico,
de caráter progressivo, que geralmente resultam na morte do indivíduo ainda na fase
infantil, em função de diagnósticos tardios e/ou da ausência de um tratamento
específico (Neufeld & Muenzer, 2001).
A Síndrome de Hunter ou MPS II (OMIM: +309900), corresponde a um
tipo de DLD caracterizada pela deficiência da enzima iduronato-sulfatase (IDS; E.C.:
3.1.6.13), responsável pela quebra dos GAG dermatan sulfato e heparan sulfato.
Sua herança, diferentemente dos outros tipos de MPS, é ligada ao cromossomo X.
Pacientes com essa doença apresentam praticamente os mesmos quadros clínicos
característicos da MPS I, com exceção do opacificação de córnea e do
comprometimento progressivo do sistema nervoso central (Neufeld & Muenzer,
2001).
As MPS do tipo III (Síndrome de Sanfilippo) correspondem, também, a um
grupo de doenças autossômicas recessivas que afetam o metabolismo dos GAG em
função da deficiência ou ausência de hidrolases lisossomais específicas. De acordo
com a deficiência enzimática, as MPS III podem ser dividas em 4 diferentes subtipos,
de acordo com a enzima deficiente: MPS IIIA (heparan-N-sulfatase, EC.: 3.10.1.1;
OMIM:252900); MPS IIIB (α-N-acetilglicosaminidase, E.C.: 3.2.1.50 OMIM: 252920);
MPS IIIC (acetil-CoA:alfa-glicosaminídeo acetiltransferase, E.C.: 2.3.13; OMIM:
252930) e MPS IIID (N-acetilglicosamina-6-sulfatase, E.C.: 3.1.6.14 OMIM: 252930).
A MPS IIIA parece ser a forma mais grave, com rápida progressão dos sintomas e
baixa expectativa de vida (van de Kamp et al., 1981; Yogalingam et al., 2000).

3
A Mucopolissacaridose do tipo IV (Síndrome de Morquio) é classificada em
dos subtipos (conforme a deficiência da enzima lisossomal específica): MPS IVA (N-
acetilgalactosamina-6-sulfatase, OMIM: 253000) e MPS IVB (β-D-galactosidase,
OMIM: 253010). A doença é causada por uma alteração no gene GALNS,
responsável pela expressão da enzima lisossomal inativa, o que leva a uma falha na
degradação do GAG keratan sulfato (Oshima et al., 1991; Neufeld & Muenzer,
2001).
A Síndrome de Maroteaux-Lamy, ou MPS VI (OMIM: 253200), corresponde
a uma DLD cuja principal característica é a ausência total ou parcial da enzima
arilsulfatase B (ARSB; EC. 3.1.6.12), responsável pelo catabolismo do GAG
dermatan sufato. Inicialmente, descrita em 1963 como uma síndrome semelhante à
Síndrome de Hurler (MPS I), a MPS VI é, também caracterizada, pelo baixo índice
de alterações do desenvolvimento intelectual (Scriver et al., 2001, Siddharth et al.,
2004).
A Síndrome de Sly (MPS VI, OMIM: 253220) é ocasionada pela deficiência
da enzima β-glicuronidase (gene GUSB), responsável pela degradação dos GAG
dermatan sulfato, heparan sulfato e condroitin sulfato (Neufeld & Muenzer, 2001).
A Mucopolissacaridose do tipo IX (Deficiência da enzima hialuronidase,
OMIM: 610492), é a MPS descoberta mais recentemente. Apesar da existência de
apenas 1 caso na literatura, os sintomas apresentados são semelhantes àqueles
evidentes em pacientes com MPS (baixa estatura, acúmulo intracelular de
substratos em função da degradação ineficaz do ácido hialurônico (Triggs-Raine et
al., 1999; Neufeld & Muenzer, 2001).
Todos os tipos de MPS, com exceção da MPS II (herança ligada ao
cromossomo X) são doenças de herança autossômica recessiva. O resumo da
classificação das MPS quanto à deficiência enzimática, excreção urinária de GAG e
o tipo de herança está descrito no quadro 01:

4
QUADRO 01: Classificação das mucopolissacaridoses
Tipo de MPS Sinonímia Herança GAGs na
urina
Deficiência
enzimática
MPS I H/HS/S
Hurler/ Hurler-
Scheie/
Scheie
HAR DS + HS α-L-
Iduronidase
MPS II
(grave/leve) Hunter HLX DS + HS
Iduronato
sulfatase
MPS III A Sanfilippo A HAR HS Heparan-N-
sulfatase
MPS III B Sanfilippo B HAR HS
α-N-Acetil-
glicosaminidas
e
MPS III C Sanfilippo C HAR HS
Acetil-CoA:α-
glicosamina
acetiltransfera
se
MPS III D Sanfilippo D HAR HS
N-
acetilglicosami
na 6-sulfatase
MPS IV A Morquio A HAR QS Galactose-6-
sulfatase
MPS IV B Morquio B HAR QS β-
galactosidase
MPS VI Maroteaux-
Lamy HAR DS Arilsulfatase B
MPS VII Sly HAR DS + HS β-
glicuronidase
MPS IX ------ HAR CS Hialuronidase
DS: dermatan sulfato; HS: heparan sulfato; QS: quer atan sulfato; CS: condroitin sulfato; HAR:
herança autossômica recessiva; HLX: Herança ligada ao X; H: Hurler; S: Sheie; HS: Hurler-
Sheie. Adaptada de Neufeld & Muenzer, 2001.

5
1.1.3. Diagnóstico clínico
Apesar de apresentarem, em comum, características crônicas e
progressivas, os sintomas apresentados por pacientes com MPS variam de acordo
com o tipo. Dentre os sintomas descritos estão organomegalia, disostose múltipla,
hepatoesplenomegalia, mão em garra e fácies grosseiras, todos relacionados ao
acúmulo de GAG nos tecidos (Neufeld & Muenzer, 2001; Muenzer, 2004).
Os pacientes com MPS I classificados como portadores da Síndrome de
Hurler apresentam uma evolução clínica progressiva, sistêmica (envolvimento
múltiplo de órgãos e tecidos) e com baixa expectativa de vida, levando à morte ainda
na infância. O diagnóstico dessa síndrome é realizado entre o 4º e o 18º mês de
vida, período no qual os sintomas se tornam mais evidentes. Deformidades ósseas,
infecções nasais e auriculares recorrentes, hérnias umbilicais e inguinais,
descaracterização facial, opacificação, córnea, hepatoesplenomegalia e
macroglossia são identificados nos estágios iniciais. Além disso, alguns pacientes
morrem em função de uma cardiomiopatia fatal antes de completarem 1 ano de
idade, sem receber o diagnóstico clínico de MPS. A desaceleração no crescimento
também ocorre precocemente (entre o 6º e o 18º mês), alcançando uma estatura
máxima de 110 cm em média. (Neufeld & Muenzer, 2001; Muenzer, 2004).
A Síndrome de Hurler-Scheie (MPS I HS) corresponde a uma forma clínica
intermediária na qual os sintomas mais evidentes, resultantes do envolvimento
somático progressivo (disostose múltipla, opacificação córnea, comprometimento
articular e cardíaco) ocorrem entre a infância e a juventude. Esses pacientes
geralmente apresentam micrognastia, que causa uma descaraterização facial
característica dessa síndrome. Apesar do envolvimento cardíaco e da obstrução de
vias áreas serem responsáveis pela maior parte dos eventos que causam a
mortalidade clínica desses pacientes, o aparecimento dos sintomas geralmente
ocorre entre o 3º e o 8° ano de vida e a sobrevivên cia até a vida adulta é comum
(Neufeld & Muenzer, 2001; Muenzer, 2004).

6
Os pacientes com a forma atenuada da MPS I (Síndrome de Scheie, MPS I
S) apresentam, em geral, comprometimento articular, doença aórtica valvar,
opacificação da córnea e algumas outras alterações somáticas. A inteligência e a
estatura são normais, entretanto a má formação facial ainda é presente. Mãos em
garra, síndrome do túnel do carpo e deformidades nos pés são sintomas que podem
levar os pacientes a limitações funcionais. Alguns pacientes podem necessitar,
eventualmente, de traqueostomia para reverter a apnéia do sono causada pela
doença obstrutiva das vias aéreas. A surdez também foi descrita em pacientes com
MPS I S, porém a etiologia dessa alteração é desconhecida. O aparecimento dos
sintomas geralmente começa após os 5 anos de idade, e o diagnóstico, realizado
entre o 10º e o 20º ano de vida (Neufeld & Muenzer, 2001; Muenzer, 2004).
A Síndrome de Hunter (MPS II), apesar de apresentar duas classificações
específicas (moderada e grave), possui um amplo espectro de apresentação clínica.
A forma grave da síndrome apresenta características semelhantes à MPS I H, com
exceção da opacificação córnea e da lenta progressão do comprometimento
somático e do sistema nervoso central. Descaracterização facial, baixa estatura,
deformidades ósseas, comprometimento articular e retardo mental são
características dessa forma, assim como degeneração retiniana grave, convulsões,
obstrução das vias aéreas e comprometimento cardiopulmonar, que constituem as
principais causas de mortes entre 10 e 15 anos de idade. A forma moderada da
Síndrome de Hurler é análoga à MPS I HS ou MPS I S, possuindo expectativa de
vida prolongada e lenta progressão da deteriorização somática, com preservação
intelectual (Neufeld & Muenzer, 2001; Muenzer, 2004).
A Síndrome de Sanfilippo (MPS III), apesar de ter 4 subtipos
bioquimicamente distintos conforme a enzima lisossomal deficiente (MPS IIIA, IIIB,
IIIC e IIIB), possui um espectro clínico semelhante em todas as suas formas. A
síndrome é caracterizada pela grave degeneração do sistema nervoso central e pelo
moderado comprometimento somático. Hiperatividade com comportamento
agressivo, atraso no desenvolvimento, hipertricose, hirsutismo, desordens do sono,
e hepatoesplenomegalia também são evidentes nesses pacientes. Apesar do
relativo comprometimento ósseo, pacientes com MPS III apresentam grave
degeneração neurológica entre os 6 e os 10 anos de idade, acompanhado com

7
rápida deteriorização das interações sociais e capacidades adaptativas (van de
Kamp et al., 1981; Yogalingam et al., 2000; Neufeld & Muenzer, 2001; Muenzer,
2004).
A Mucopolissacaridose do tipo IV ou Síndrome de Morquio (MPS IV)
apresenta um amplo espectro de manifestações clínicas, das quais as
predominantes são as alterações ósseas e comprometimento do sistema nervoso
central. Como na maioria das MPS, pacientes com MPS IV são aparentemente
normais após o nascimento. Os sintomas iniciais, que ocorrem entre o 1º e o 4º ano
de idade, são cifose, retardo no crescimento com tronco e pescoço curtos e o “andar
de galope” com tendência à queda. As alterações ósseas típicas em pacientes com
MPS IV são o nanismo (com tronco curto), platispondilia, hipoplasia odontóide,
hiperlordose, escoliose e osteoporose. Outras manifestações como
comprometimento da audição, opacificação córnea moderada, hepatomegalia,
obstrução das vias aéreas superiores, lesões em válvulas cardíacas e
descaracterização facial (ex.: prognatismo) também já foram descritas em pacientes
com essa síndrome (Neufeld & Muenzer, 2001; Muenzer, 2004).
O desenvolvimento neurológico em pacientes com Síndrome de Maroteuax-
Lamy (MPS VI) é geralmente normal, apesar do comprometimento físico e visual
impedir o desempenho psicomotor. Alterações somáticas também são evidentes na
forma grave da doença, tais como hérnias umbilicais e/ou inguinais, atraso no
crescimento e hepatoesplenomegalia após o 6º ano de vida, opacificação córnea,
comprometimento articular, mãos em forma de garra e síndrome do túnel do carpo.
As alterações ósseas encontradas em pacientes com MPS VI são semelhantes
àquelas descritas na MPS I H, todas relacionadas a uma disostose múltipla.
Disfunção valvar (aórtica e mitral) são as mais comuns alterações cardíacas
encontradas em pacientes com a forma intermediária. Entretanto, a maioria dos
pacientes com a forma grava da doença morreram entre a segunda e a terceira
década de vida em função da falência das funções cardíacas (Neufeld & Muenzer,
2001; Muenzer, 2004).
A Mucopolissacaridose do tipo VII ou Síndrome de Sly é caracterizada pela
presença de sintomatologias semelhantes às MPS I H ou II. Dentre as

8
apresentações clínicas mais proeminentes, estão a descaracterização facial,
hepatoesplenomegalia, hérnia umbilical, deformidades vertebrais e deficiência
mental moderada. A opacificação córnea aparece, geralmente, após os 8 anos de
idade.Já a deficiência mental é evidente a partir dos 3 anos de idade, porém,
aparentemente, de caráter não-progressivo. Disostose múltipla, baixa estatura e
episódio de pneumonia também podem ser apresentados por esses pacientes
(Neufeld & Muenzer, 2001; Muenzer, 2004).
A Mucopolissacaridose IX, causada pela deficiência da enzima
hialuronidase, possui apenas 1 caso relatado na literatura. Dentre os sintomas
apresentados, todos estão relacionados ao acúmulo de massas periarticulares e
intra-articulares,, causados pela incapacidade da enzima em degradar o ácido
hialurônico que é normalmente encontrado em altas concentrações em cartilagens e
no líquido sinovial (Neufeld & Muenzer, 2001; Muenzer, 2004).
1.1.4. Diagnóstico laboratorial
A investigação bioquímica em pacientes com suspeita de MPS começa a
partir de testes de triagem urinária, com teste do Brometo de Cetil-Trimetil-Amônio
(BCTMA), Azul de Toluidina, dosagem de GAG na urina (quantitativo) e
cromatografia de GAG em camada delgada (qualitativo).
O BCTMA corresponde a um teste preliminar, no qual pode-se verificar a
presença de GAG na urina através da reação destes metabólitos com o reagente
(BCTMA), resultando na formação de um líquido turvo e com precipitado. O teste do
azul de toluidina permite avaliar qualitativamente uma possível excreção de GAG na
urina, através da impregnação de pequenas quantidades urina em papel filtro. Caso
os testes de BCTMA e Azul de Toluidina forem positivos, parte-se para a dosagem
de GAG urinários e para a cromatografia em camada delgada, na qual pode-se
avaliar os tipos de GAG excretados e, assim, verificar qual a MPS possivelmente
relacionada.

9
O diagnóstico definitivo das MPS é realizado através da mensuração da
atividade das enzimas lisossomais deficientes em cada subtipo de MPS (Wajner et
al., 2001).
1.1.5. Tratamento
A partir da década de 70, ocorreram tentativas para correção do
metabolismo dos GAG em culturas de células de pacientes com MPS, entretanto, as
primeiras tentativas de reposição enzimática in vivo falharam, provavelmente, devido
à administração da enzima inadequada. Até 1980, apenas terapias paliativas e não
específicas foram aplicadas para pacientes com MPS. Estas condutas terapêuticas
falharam em virtude de não produzir nenhuma mudança no quadro clínico,
provavelmente, devido esses procedimentos produzirem somente uma quantidade
insignificante de enzima (Neufeld & Muenzer, 2001; Muenzer, 2004).
Hobbs et al. (1981) relataram que um transplante alogênico de medula
óssea em um menino de 9 anos de idade portador da síndrome de Hurler (MPS I)
proporcionou uma considerável mudança no aspecto da doença. Desde então, o
entendimento dos benefícios e das limitações do transplante de medula óssea vêm
caminhando junto com as extensivas experiências clínicas com pacientes portadores
de MPS e com numerosos estudos experimentais em modelos animais. O advento
do transplante de medula óssea beneficiou alguns casos, principalmente, as formas
graves da MPS I e MPS VI (Muenzer, 2004).
Outra alternativa terapêutica ainda em período de testes é a terapia
gênica, que consiste, basicamente, na utilização de vetores para inserção de células
modificadas que expressam a enzima deficiente em um tecido deficiente. Apesar de
ter demonstrado diversos resultados positivos na melhoria da sintomatologia da
MPS em modelos animais, sua implantação em estudos clínicos com humanos
ainda é improvável. Entretanto, a terapia gência ainda é considerada promissora
para o tratamento de pacientes com MPS durante os estágios inicias do
desenvolvimento infantil (Ponder et al., 2007; Tolar et al., 2008).

10
A Terapia de Reposição Enzimática (TRE), uma outra alternativa recente
de tratamento, repõe a enzima ausente através de infusões intravenosas. Com o
advento das técnicas de engenharia genética, a enzima humana recombinante pôde
ser gerada, eliminando a necessidade de purificação da enzima de humanos ou de
tecidos animais. A maior vantagem da TRE está no fato que ela pode distribuir mais
enzimas do que as produzidas pelo transplante de células tronco hematopoiéticas. A
TRE está associada a um significante baixo risco e é apropriada para um número
maior de pacientes (Muenzer, 2004).
Os protocolos clínicos utilizando a TRE têm mostrado resultados
promissores no tratamento de pacientes com MPS I, II e VI. Recentemente, a TRE
para MPS I e VI tornou-se comercialmente disponível (Rohrbach & Clarke, 2007;
Harmatz et al., 2005; Wraith et al., 2008).
De forma geral, as enzimas recombinantes utilizadas nas TRE mimetizam
os efeitos biológicos da enzima nativa por serem molecularmente desenhadas para
que possam ser facilmente captadas pelas células dos pacientes. Resíduos de
manose-6-fosfato são inseridos nas cadeias glicoprotéicas da enzima recombinante,
o que permite que a mesma se ligue em receptores específicos de manose-6-fosfato
da superfície celular, seja captada pela célula e direcionada para organelas
específicas (lisossomos), onde irão atuar no catabolismo dos GAG acumulados
(Wraith et al., 2007).
Estudos recentes mostram uma diminuição da excreção de GAG urniários
em pacientes que encontram-se em TRE. Em relação aos pacientes com MPS I, os
quais recebiam a enzima recombinante (laronidase®), Miebach et al., (2005),
Thomas et al. (2006), Arora et al. (2007) e El Dib et al. (2007) descreveram uma
redução significativa da quantidade de GAG excretados na urina, assim como uma
redução da organomegalia, apesar de outros aspectos clínicos (comprometimento
músculo-esquelético, respiratório e medular) não apresentarem melhora significativa.

11
Em pacientes com MPS VI, que recebiam a enzima recombinante (rhASB),
foram verificadas melhorias em alguns aspectos clínicos, tais como capacidade
respiratória e articular, além de uma redução dose-dependente de GAG urinários,
principalmente naqueles pacientes que apresentavam formas mais graves da
doença e que receberam maiores doses da enzima (Harmatz et al., 2004; Harmatz
et al., 2005; Harmatz et al., 2008; Feio et al., 2008).
Karageorgeos et al. (2007) descreveram os padrões genotípicos e
fenotípicos de pacientes com o mesmo subtipo de MPS que já encontravam-se em
TRE, indicando que, juntamente à anáise de GAG urinários, outros parâmetros
bioquímicos (mensuração das atividades enzimáticas residuais em fibroblastos dos
pacientes) são necessários para a determinação do espectro clínico em cada
paciente.
A maior desvantagem é que a administração intravenosa da enzima não
ultrapassa a barreira hemato-encefálica. Dessa forma, nos casos de MPS onde há
sintomas neurológicos, a TRE não poderá reverter esse aspecto clínico (Muenzer,
2004).
A TRE não interfere significativamente na progressão óssea e neurológica
da doença, devido ao elevado peso molecular das enzimas recombinantes. Porém,
estudos com diferentes formas de administração como a aplicação intratecal e intra-
articular da enzima vêm sendo realizados com o objetivo de superar este obstáculo
(Munoz-Rojas, 2008).
Além disso, o uso da enzima recombinante demonstrou, em modelos
felinos e em humanos, que quanto mais precoce o início da terapia menor o
comprometimento das articulações, ossos, válvulas cardíacas e cartilagem traqueal
(Harmatz et al., 2008; Clarke 2008).

12
1.1.6. Aspectos epidemiológicos:
As MPS estão entre as DLD mais freqüentes, com uma incidência estimada
em 1:10000 e 1:25000 recém-nascidos. As MPS I e II são consideradas as mais
freqüentes, enquanto a MPS VII parece ser o tipo mais raro. Todas as MPS, com
exceção da MPS II são herdadas de forma autossômica recessiva. A MPS II possui
herança ligada ao X (Solyon, 1996; Meikle et al., 1999).
Em função da escassez de centros de referência para a investigação de
EIM e à falta de informação dos médicos, as MPS ainda são pouco diagnosticadas.
Entretanto, a partir de 1990, com a implantação de novos centros de referência,
esse retrospecto vem mudando gradativamente. Atualmente, ainda não existem
trabalhos que descrevam a epidemiologia das MPS no Brasil, porém, Coelho et al.
(1997) verificaram que dentre as MPS estão entre os EIM mais freqüentemente
diagnosticados em pacientes de alto risco no Brasil.
O Laboratório de Erros Inatos do Metabolismo da Universidade Federal do
Pará (LEIM/UFPa) e o Hospital Universitário Bettina Ferro de Souza (HUBFS) são,
atualmente, os principais centros de referência regional no diagnóstico bioquímico e
molecular, assim como no tratamento de pacientes com MPS, respectivamente. Em
colaboração com esses centros regionais, está o Laboratório de Referência de Erros
Inatos do Metabolismo do Serviço de Genética Médica do Hospital de Clínicas de
Porto Alegre (LREIM/SGM/HCPA), o qual oferece suporte auxiliar na identificação de
várias doenças hereditárias metabólicas.
A rede MPS Brasil foi uma iniciativa criada em 2004 para aprimorar o
diagnóstico e o manejo de pacientes com MPS por todo o Brasil (Vieira et al., 2008).
Entre 249 pacientes diagnosticados com MPS, 60 apresentaram MPS I, 82 MPS II,
31 MPS III (7 MPS IIIA, 14 MPS IIIB e 10 MPS IIIC), 15 MPS IV (11 MPS IVA e 4
MPS IVB), 57 MPS VI e 4 com MPS VII (Vieira et al., 2008).
Embora não consista um estudo epidemiológico (devido ao número
pequeno de casos identificados), Castro et al. (2007) descreveram a freqüência de

13
MPS no Estado do Pará. Dentre as MPS mais freqüentemente diagnosticadas estão
a MPS II e VI (1:960.005 por nascidos vivos), seguida da MPS I (1:1.140.000 por
nascidos vivos). A freqüência de 1:411.430 encontrada para nascidos vivos com
MPS não classificada reflete a necessidade da implantação de protocolos de
investigação dessa doença no Estado do Pará.
1.1.7. Bases celulares das MPS
Os GAG são moléculas não-ramificadas de heteropolissacarídeos formadas
por unidades repetitivas de dissacarídeos. A natureza do monossacarídeo que
compõe os dissacarídeos define, especificamente, o subtipo de GAG: o heparan
sulfato é formado por glicosamina e iduronatos de ácidos hexurônicos ou
glicuronatos; o dermatan sulfato é composto por galactosamina e iduronato de ácido
hexurônico; o condroitin sulfato é formado por galactosamina e glicuronato de ácido
hexurônico; o queratan sulfato, por galactose e glicosamina e o ácido hialurônico por
glicuronato e glicosamina (Clarke, 2008).
Uma vez produzidos, os proteoglicanos serão encaminhados tanto para a
superfície quanto para matriz celular, onde sofrerão mudanças na composição de
seus açúcares. Na superfície celular (ex.: proteoglicanos do heparan sulfato), as
cadeias laterias de heparan sulfato podem ser clivadas e liberadas para o meio
extracelular através da enzima heparanase (Prydz & Dalen, 2000).
A degradação dos GAG ocorre a partir da clivagem de moléculas de
proteoglicanos por proteases e pela remoção de suas porções endoglicosídicas. A
catálise desses compostos pode, portanto, ocorrer tanto na matriz extracelular
quanto nos compartimentos lisossomais celulares. Uma vez no meio extracelular, os
GAG podem, também, seqüestrar fatores humorais (figura 02), modulando seus
efeitos biológicos (Clarke, 2008).

14
Figura 02: Esquema celular das Mucopolissacaridoses . A figura ilustra os diversos processos
fisiológicos que podem ser afetados pela deficiência na degradação de GAG, com conseqüente
aumento do depósito desses compostos na matrix extracelular. (a) Macrófago disfuncional em função
do acúmulo de GAG nos lisossomos; (b) padrão alterado de receptores da membrana plasmática; (c)
Alteração na captação de citocinas e fatores de crescimento; (d) alteração no recrutamento de
citocinas circulantes; (e) Alteração na apresentação de citocinas aos receptores; (f) interferência no
tráfico celular; (g) Depósito lisossômico adicional pela interferência de hidrolases lisossomais; (h)
interações anormais da matriz extracelular; (i) adesão celular alterada. Adaptado de Clarke, 2008.
A ausência ou deficiência de uma ou mais enzimas relacionadas a esse
processo de regulação da catálise de GAG ocasiona a deposição destes compostos
em diversos tecidos orgânicos. Modelos animais têm demonstrado um excessivo
acúmulo de GAG em tecidos hepáticos, renais, pulmonares, cardíacos e esplênicos,
com leve comprometimento de tecidos nervosos (Li et al., 1999; Vogler et al., 2001;
Vogler et al., 2005).

15
Além disso, outras implicações sistêmicas estão relacionadas com a
deficiência enzimática característica das MPS, tais como alteração na atividade de
outras enzimas lisossomais (ex.: β-hexoaminidase, α-glicosidase, β-galactosidase e
β-glicuronidase), alterações no metabolismo de gangliosídeos (GM2 e GM3), o que
ocasionalmente representa um dos principais fatores para o comprometimento
neural progressivo, evidente em alguns pacientes com MPS (Canstantopoulos &
Dekaban, 1978; Jones et al., 1997; Walkley, 2004).
Como conseqüência da catálise deficiente, os GAG não degradados são,
portanto, excretados na urina, o que consiste em um importante parâmetro para a
investigação de MPS. Dependendo do tipo de MPS, o catabolismo de diferentes
GAG (ex.: heparan sulfato, dermatan sulfato, keratan sulfato) pode ser bloqueado
isoladamente ou em conjunto (Clarke, 2008).
1.1.8. Bases moleculares das MPS
Após a clonagem dos genes que codificam a maioria das enzimas
relacionadas à catálise de GAG na década de 90, vários estudos têm sido realizados
com o objetivo de elucidar quais as principais mutações relacionadas às deficiências
enzimáticas presentes em cada tipo de MPS. Apesar da grande variabilidade
apresentada pelos pacientes de diferentes tipos de MPS, algumas mutações
relatadas em pacientes com essa patologia são bastante freqüentes, embora
algumas possuam um predomínio em determinadas regiões geográficas e/ou em
populações específicas (Isogai et al., 1998; Venturi et al., 2002; Lin et al, 2008).
a) Mucopolissacaridose I (MPS I)
A partir de critérios clínicos, é possível distinguir diferentes padrões
fenotípicos relacionados a MPS I, os quais são didaticamente divididos em três
subtipos: Hurler (forma leve), Hurler-Scheie (forma moderada) e Scheie (grave).

16
Cada subtipo possui um padrão fenotípico distinto, com variações, por exemplo, na
taxa de excreção urinária de GAG, no nível de comprometimento ósseo e mental e
na funcionalidade de alguns órgãos (Hopwood & Morris, 1990).
O gene que expressa a enzima IDUA está localizado na região p16.3 do
cromossomo 4, possui 19 kb, incluindo 14 éxons, os quais codificam um proteína
com 627 resíduos de aminoácidos. A enzima IDUA apresenta peso molecular de
70.029 kDa (Scott et al., 1990).
Em relação às mutações patogênicas descritas no gene IDUA, Scott et al.,
(1992) mostraram a existência de algumas mutações freqüentes em populações
caucasianas como, por exemplo, W402X e Q70X e outra menos freqüente como
P533R. Essas mutações são responsáveis pela ausência da produção da enzima
funcional e, consequentemente, pelo aparecimento de formas graves e/ou
intermediárias. Outras mutações também foram descritas em pacientes com MPS I
(mutações sem sentido, destruição do sítio de splicing e mudança na matriz de
leitura), correlacionadas a diferentes quadros clínicos.
Yamagishi et al. (1996) verificaram que a maioria dos pacientes japoneses
com MPS I apresentava a mutação R89Q, a qual também está presente em
caucasianos e está relacionada a um fenótipo intermediário.
Matte et al. (2003) descreveram 13 novas mutações em pacientes
brasileiros, além das anteriormente descritas (W402X e Q70X). As mutações mais
comuns encontradas em pacientes brasileiros foram a W402X (fenótipo grave) e a
P533R (fenótipo intermediário), sendo que a primeira foi encontrada com a mesma
freqüência em populações ibero-americanas (60%) (Gort et al., 1998), e a segunda
foi, também, encontrada com a mesma freqüência (10%) em populações italianas
(Gatti et al., 1997).
Estudos in vitro têm sido amplamente utilizados como ferramenta na tentativa
de estabelecer padrões entre uma determinada alteração genética e suas
conseqüências nos processos de expressão, transcrição, tradução e tráfego
intracelular, além das alterações estruturais na molécula protéica que podem levar a

17
depleção da atividade enzimática. Ainda segundo esses estudos, uma vez
conhecidas as mutações e suas implicações intracelulares, pode-se estimar o grau
de comprometimento clínico do paciente e, assim, identificar o fenótipo apresentado.
Sugawara et al. (2008) descreveram as alterações estruturais causadas na
enzima α-L-iduronidase por diversas mutações previamente descritas. Com base na
correlação genótipo/fenótipo feita por estudos anteriores, foram utilizados modelos
estruturais dessa enzima, cada um resultante de 33 alterações de aminoácidos
(apenas mutações missense). Tais alterações eram responsáveis pelo aparecimento
de diversas formas clínicas de MPS I (17 graves, 8 intermediárias e 8 atenuadas).
Segundo o estudo, as mutações descritas como responsáveis pelo
aparecimento de fenótipos graves (ex.: A75T, L218P, I270S e A327P) causam
alterações em um número maior de átomos da molécula do que as que são
responsáveis por fenótipos atenuados. Em pacientes com fenótipo grave, as
mutações encontravam-se, em sua maioria, na parte interna da proteína, o que
poderia acarretar em sérias alterações conformacionais da proteína, gerando uma
falha no processo de empacotamento e/ou tráfego intracelular e, posteriormente, a
degradação da enzima pelo sistema de controle de qualidade do retículo
endoplasmático antes da sua chegada ao lisossomo.
Já as mutações apresentadas por pacientes com fenótipos atenuados (ex.:
R89Q) ocorriam mais frequentemente na superfície da estrutura protéica
tridimensional. Isso sugere que essas alterações permitem que uma pequena parte
da enzima mutante possa ser processada pelo retículo endoplasmático e
transportada para o lisossomo sem perder, necessariamente, sua atividade
enzimática total. Assim, a atividade enzimática, ainda que residual, permite certa
degradação dos substratos uma vez acumulados, levando ao desenvolvimento
tardio da doença e, dessa forma, ao desenvolvimento de um fenótipo atenuado.
O estudo realizado por Vazna et al. (2009) também verificou que mutações do
tipo missense, quando presentes na parte interior da proteína, são responsáveis
pelo aparecimento de fenótipos graves, assim como mutações na superfície
molecular acarretam fenótipos atenuados. Além disso, os autores também sugeriram

18
que outras mutações, que ocorrem na porção C-terminal da proteína, também levam
a manifestações clínicas da doença, indicando a importância dessa região proteica
na investigação molecular de pacientes com MPS I.
b) Mucopolissacaridose II (MPS II)
O gene IDS que codifica a enzima iduronato-sulfatase, deficiente na MPS II,
está localizado na região q28 do cromossomo X, sendo composto por
aproximadamente 24 kb distribuídos por 9 éxons. O gene também possui um
pseudogene que compreende cópias dos éxons 2 e 3 e do íntron 7, localizado 20kb
distante da região codificante do gene (Timms et al., 1995; Froissart et al., 2007
apud Wilson et al., 1993).
A molécula de cDNA produzida pelo gene IDS contém, aproximadamente,
2,3 kb. Ela codifica um polipeptídeo de 550 aminoácidos, incluindo um peptídeo sinal
de 25 aminoácidos. Os precursores de 76 kDa e 90 kDa da enzima IDS, após vários
processos pós-transcricionais como glicosilação, fosforilação e proteólise são
convertidos a várias formas intermediárias que são, por fim, transformados em
formas maduras de 55 kDa e 45 kDa (Froissart et al., 1995).
Mais de 300 mutações já foram identificadas no gene IDS (Lin et al., 2006).
Dentre as mutações apresentadas no gene IDS de pacientes com MPS II, estão as
alterações estruturais, tais como deleções parciais e totais, inserções e mutações no
sítio de splicing, representando aproximadamente 20% do total de alterações
encontradas.
Essas alterações, geralmente associadas ao aparecimento de fenótipos
graves (com elevada freqüência de comprometimento neural), estão relacionadas a
processos de falha no emparelhamento cromossômico, o que leva a recombinações
homólogas entre seqüências do gene IDS e de seu pseudogene (IDS-2) (Lagerstedt
et al., 1997; Karsten et al., 1997). As alterações mais freqüentes são as mutações do

19
tipo missense, nonsense e pequenas inserções e deleções (Li et al., 1999;
Sukegawa-Hayasaka et al., 2006; Froissart et al., 2007; Martin et al., 2008).
Outros estudos, no entanto, relatam que mutações que afetam o processo de
splicing representaram cerca de 50% das mutações apresentadas em pacientes com
MPS II (Alves et al., 2006). Outras alterações como mutações pontuais e pequenas
alterações estruturais do gene aparecem em, aproximadamente, 80% dos pacientes
(Hopwood et al., 1993).
Apesar da ampla heterogeneidade molecular relacionada ao gene IDS
algumas mutações foram descritas como mais freqüentes em algumas populações.
Isogai et al. (1998) verificaram em pacientes japoneses uma elevada prevalência de
algumas mutações, concentradas nos éxons 5 e 9, além de mutações no sítio de
splicing do éxon 3 e mutações intrônicas, as quais estão relacionadas a um fenótipo
grave da doença.
Alves et al. (2006) estudaram a freqüência de mutações do gene IDS em
pacientes portugueses e verificaram que a maioria das mutações corresponde a
mutações no sítio de splicing, ocasionadas tanto por substituições intrônicas quanto
exônicas, principalmente aquelas que ocorrem no éxon 3.
A análise do perfil mutacional em populações italianas foi realizada por
Filocamo et al. (2001), que descreveram uma elevada freqüência de mutações do
tipo missense (55,2%), nonsense (20,7%), e pequenas deleções ou mutações em
sítios de splicing em pacientes italianos. A maioria das pequenas deleções
encontradas (c.472delTCC, c.533delTT e c.1131-1142del) ocorreu em regiões do
gene muito susceptível à ocorrência de alterações gênicas (éxons 3, 8 e 9). O
estabelecimento de uma correlação do genótipo com o fenótipo em pacientes com
MPS II é muito difícil, haja vista a ampla heterogeneidade de mutações no gene IDS
e a falta de uma classificação clínica dos pacientes em relação às diferentes
apresentações clínicas.

20
c) Mucopolissacaridose IIIB (MPS IIIB)
O gene NAGLU que codifica a enzima N-acetil-galactosaminidase,
deficiente na MPS IIB, está localizado na região q21 do cromossomo 17, contendo,
aproximadamente, 8,5 kb, dividido em 6 éxons. A molécula de cDNA do gene
contém 2575 pb, a qual codifica uma proteína de 743 resíduos de aminoácidos, que
contem um peptídeo sinal de 20 a 23 aminoácidos imediatamente antes da porção
aminoterminal e seis possíveis pontos de glicosilação. Mais de 100 diferentes
mutações foram associadas ao gene NAGLU, as quais podem estar relacionadas
com a ampla heterogeneidade do fenótipo clínico apresentado pelos pacientes
(Weber et al., 1996; Zhao et al., 1996).
Beesley et al. (1998) amplificaram as 10 regiões codificantes do gene
NAGLU com o objetivo de encontrar alterações genéticas em pacientes europeus
com MPS III. Para isso, foram feitos testes de triagem molecular (SSCP) e análise
de sequenciamento para investigação de possíveis mutações. Das 16 mutações
encontradas, 3 estavam presentes em mais de um paciente (Y140C, R297X e
R626X) e, juntas, corresponderam a 25% dos alelos mutantes analisados.
Tessitore et al. (2000) demonstraram uma ampla heterogeneidade de
mutações entre 20 pacientes italianos. Das 28 mutações identificadas (todas
relacionadas a um fenótipo grave, com exceção da S534Y), 14 eram novas (L35F,
204delC, 221insGCGCG, G82D, W156C, 507delC, IVS3+1G→A, E336X, V501G,
R520W, S534Y, W649C, 1953insGCCA, 2185delAGA), além das descritas
anteriormente (Y140C, R234C, G292R, e901delAA). Algumas mutações, tais como a
L35F, G292R e Y140C foram descritas como alterações comuns em populações
italianas.
Tanaka et al. (2002) realizaram uma triagem molecular em pacientes
japoneses para investigar mutações no gene NAGLU. De acordo com o estudo,
foram encontradas duas novas mutações (F314L e V241M), as quais foram
relacionadas a fenótipos moderados da doença, enquanto que a mutação R565P

21
correspondia a um fenótipo grave, como comprovado também por Chinen et al.
(2005).
Mais recentemente, Mangas et al. (2008) realizaram um levantamento
acerca do padrão de mutações do gene NAGLU em pacientes ibero-americanos. A
partir da análise de 11 pacientes com MPS IIIB, 9 mutações foram encontradas,
sendo 5 novas (M1K, W147X, G304V, S522P e R533X) e 4 anteriormente descritas
(W168X, R234C, R565W e R643C), sendo que a mutação R234C, relacionada a um
fenótipo grave da doença, foi encontrada com relativa freqüência (32% dos alelos)
nessa população.
d) Mucopolissacaridose VI (MPS VI)
O gene ARSB, codificador da enzima arilsulfatase B (deficiente na MPS VI)
está localizado na região q13-q14 do cromossomo 5 e possui 8 exons. Mutações em
homozigose desse gene promovem as formas mais graves da doença. As mutações
mais freqüentes costumam ocorrer nas regiões correspondentes aos éxons 4 e 5
(Karageorgos et al., 2007).
Em relação ao padrão de mutações desse gene, Litjens et al. (1996)
encontraram alterações freqüentes em pacientes australianos (R95Q, Y210C e
H393), as quais estavam relacionadas a padrões fenotípicos distintos.
Villani et al. (1998) descreveram uma ampla heterogeneidade de mutações
em pacientes italianos, sendo que as alterações genéticas encontradas nesses
pacientes (S65F, P116H, R315Q, Q503X, P531R) eram responsáveis por falhas no
rearranjo tridimensional da enzima ARSB e, conseqüentemente, pelo
desenvolvimento de diferentes formas da doença (leve, moderada e grave).
Petry et al. (2005) também descreveram a variabilidade de mutações nesse
gene (a maioria no éxon 1), através da análise de 12 pacientes sul-americanos (11
brasileiros e 1 chileno). A mutação 1533del23 (éxon 8), uma mutação comum entre

22
pacientes brasileiros e que está associada com fenótipo grave e intermediários, foi
encontrada em 23,1% dos alelos analisados neste estudo.
Em relação à correlação genótipo/fenótipo, Wicker et al. (1991) analisaram o
perfil molecular de um paciente com a forma leve de MPS VI. Foi possível detectar a
presença de 3 alterações, sendo duas polimorfismos e uma mutação (G137V). De
acordo com os autores, essa mutação não afetaria a síntese da enzima, entretanto
reduziria significativamente a estabilidade do precursor enzimático, o que levaria a
uma degradação prematura desse precursor em estágios iniciais no tráfego
intracelular e, portanto, apenas uma pequena parte desse conteúdo seria
encaminhada para os lisossomos. A apresentação do fenótipo leve seria relacionada
à atividade residual dessa pequena porção de enzima restante no lisossomo.
Outro estudo, realizado por Bradford et al. (2002) também verificou as
implicações intracelulares da mutação Y210C (fenótipo atenuado) através da
expressão de células de ovário de hamster (CHO-K1) contendo essa mutação.
Segundo o estudo, a mutação Y210C não influencia a expressão da enzima, porém
causa falhas no processamento, tráfego e estabilidade da molécula. Apesar disso, a
enzima ainda consegue ser direcionada para o lisossomo, portando uma atividade
enzimática residual, o que permite uma degradação parcial dos substratos e, dessa
forma, características clínicas atenuadas.
A heterogeneidade do padrão de mutações do gene ARSB foi, também,
comprovada por outros trabalhos como o de Garrido et al. (2007), o qual descreveu
19 diferentes mutações em 16 pacientes hispânicos (espanhóis e argentinos).
Dentre as mutações, apenas duas (c.1143-1G>C e c.1143-8T>G) estavam
presentes em 30% dos pacientes analisados. A análise de haplótipos identificou uma
origem comum das mutações mais freqüentes encontradas nessas populações.

23
2. OBJETIVOS
2.1. Objetivo Geral
• Analisar o perfil molecular e bioquímico de pacientes e familiares com
Mucopolissacaridoses no Estado do Pará.
2.2. Objetivos Específicos
• Investigar mutações no gene IDUA (éxons 2, 9 e 11) de pacientes com MPS I;
• Investigar mutações no gene IDS (éxons 3 e 9) de pacientes com MPS II;
• Investigar mutações no gene NAGLU (éxons 2, 4 e 6) de paciente com MPS
IIIB;
• Investigar mutações no gene ARSB (éxons 5 e 8) de pacientes com MPS VI.
• Avaliar o padrão de excreção de GAG dos pacientes submetidos à TRE antes
e durante a terapia (MPS I e VI);
• Avaliar a eficácia da TRE nos pacientes submetidos à terapia por meio da
dosagem urinária de GAG (monitoramento bioquímico);
• Descrever possíveis correlações entre os perfis clínicos, bioquímicos e
moleculares apresentados pelos pacientes com MPS I e VI;

24
3. MATERIAL E MÉTODOS
3.1. MATERIAL
3.1.1. Caracterização da Amostra:
O estudo contou com 10 pacientes com MPS (02 com MPS I, 04 com MPS
II, 01 com MPS IIIB e 03 com MPS VI) e 9 familiares desse pacientes, resultando em
um total de 19 indivíduos. Os pacientes foram encaminhados pelo o Hospital
Universitário Bettina Ferro de Souza.
A pré-caracterização da amostra consistiu na determinação do perfil clínico
e na realização de testes bioquímicos iniciais (testes de triagem) para cada paciente,
ambos realizados no Hospital Universitário Bettina Ferro de Souza da Universidade
Federal do Pará – HUBFS/UFPA, no Laboratório de Erros Inatos do Metabolismo do
Instituto de Ciências Biológicas da Universidade Federal do Pará – LEIM/ICB/UFPa.
Já o diagnóstico bioquímico (dosagem inicial de GAG urinários e a mensuração de
cada atividade enzimática específica) foram realizados no Laboratório de Referência
em Erros Inatos do Metabolismo do Serviço de Genética Médica do Hospital de
Clínicas de Porto Alegre – LREIM/SGM/HCPA.
3.2 Critérios de inclusão
Os critérios de inclusão do estudo adotados pelo presente estudo foram o
diagnóstico bioquímico confirmado para MPS (pacientes com níveis urinários de
GAG elevados e atividade enzimática abaixo dos valores normais) e o diagnóstico
clínico previamente realizado pelo corpo médico responsável (HUBFS/UFPa) a partir
da avaliação de determinados parâmetros clínicos, que possibilitaram a
determinação fenotípica para cada paciente (Quadro 03).
Apenas os pacientes com MPS do tipo I e VI foram submetidos à terapia de
reposição enzimática. Dos 4 pacientes com MPS II, apenas 1 estava sob TRE, o que
levou a exclusão da análise bioquímica em MPS II. Em relação aos pacientes com

25
MPS IIIB, como ainda não há tratamento baseado na reposição enzimática, só foi
realizada, portanto, a investigação molecular.
Quadro 02 : Perfil bioquímico inicial dos pacientes com MPS antes da TRE.
Pacientes GAG urinários
(mg/mmol CREA)
Valores de referência (mg/mmol
CREA)*
Atividade enzimática em
leucócitos (nmoles/h/mg prot)
Valores de referência
(nmoles/h/mg prot)
P1 (MPS I)
76,4 1,5 – 7,0
(18-20 anos) 0,03**
6,8-13,7 (IDUA) §
P2 (MPS I)
18,2 3,4 – 11,0 (< 14 anos)
0,25 32-52 (IDUA)
P3 (MPS II)
50,0 5,2 – 12,0 (< 9 anos)
8,9 31-110 (IDS)
P4 (MPS II)
79,0 5,2 – 12,0 (< 9 anos)
ND** 122-463 (IDS) §
P5 (MPS II)
12,4 5,2 – 12,0 (< 9 anos)
ND 31-110 (IDS)
P6 (MPS II)
4,7 1,5 – 7,0 (18-20 anos)
ND 31-110 (IDS)
P7 (MPSIIIB)
43,5 3,4 – 11,0
(< 14 anos) 0,13**
11-37 (NAGLU) §
P8 (MPS VI)
31,9 3,4 – 11,0
(< 14 anos) 8,0
72-176 (ARSB)
P9 (MPS VI)
27,0 3,4 – 11,0
(< 14 anos) 10,0
72-176 (ARSB)
P10 (MPS VI)
19,0 3,4 – 11,0 (< 14 anos)
11,0 72-176 (ARSB)
CREA: Creatinina; GAG: glicosaminoglicanos; prot: proteína; ARSB: arilsulfatase b; NAGLU: N-acetil-glicosaminidase; IDUA: α-L-iduronidase; ND: não detectável. *Os valores de referência de GAG urinários correspondem à faixa etária de cada paciente no momento da realização da primeira dosagem. **Valor de atividade enzimática dosada em plasma (nmoles/h/mL). §Valores de referência em plasma humano (nmoles/h/mL). Fonte: LEIM/ICB/UFPa e LREIM/SGM/HCPA.

26
Quadro 03 : Resumo das características clínicas dos pacientes com MPS
CS: consangüinidade; OC: opacificação córnea; DC: dano cardíaco; IIS: Idade do início dos sintomas; IUA: Idade da última avaliação; ND: Não disponível. G/I: fenótipo grave a
intermediário; I: fenótipo intermediário; A: fenótipo atenuado. # Paciente faleceu durante a realização do estudo.
Pacientes Origem Gênero Idade (anos) CS IIS (meses) IUA
(anos) Altura (cm) OC DC FC
P1 (MPS I)
Belém (PA) M 20 Sim < 6 20 138 ++++ Insuf. Mitral/ Aórtica G/I
P2 (MPS I)
Belém (PA) M 15 Não 60 15 152 ++ Insuf. Mitral A
P3 (MPS II)
Belém (PA) M 10 Não 12 10 133 - - G/I
P4 (MPS II)
Anajás
(PA) M # Não 48 08 100 - Insuf. mitral I
P5 (MPS II)
Portel (PA) M 08 Não 36 08 104 - Insuf. aórtica G/I
P6 (MPS II)
Breu
Branco
(PA)
M 10 Não 24 09 110 - - G/I
P7 (MPS IIIB)
Abaetetuba
(PA) F 10 Não ND ND ND ND ND ND
P8 (MPS VI)
Cametá
(PA) F 16 Não 14 16 121,5 +++ Insuf. Mitral/ aórtica G/I
P9 (MPS VI)
Cametá
(PA) F 14 Não 24 14 121,5 ++
Insuf. Mitral /
Aórtica G/I
P10 (MPS VI)
Colares
(PA) M # Não 24 10 97 ++ - G/I

27
3.3 Aspectos éticos
O presente estudo levou em consideração os princípios éticos básicos das
diretrizes e normas regulamentadoras de pesquisa envolvendo seres humanos. As
famílias dos pacientes que aceitaram participar deste estudo receberam um termo
de consentimento livre e esclarecido, além de informações sobre os objetivos da
pesquisa conforme protocolo de pesquisa (número 172/08) aprovado no Comitê de
Ética em Pesquisa do Instituto de Ciências da Saúde, na reunião do dia 11 de
novembro de 2008 (anexo 01).
Embora alguns pacientes apresentarem idade superior a 18 anos, eles não
foram considerados seus próprios responsáveis legais, em função das limitações
impostas pela doença, cabendo aos familiares tal responsabilidade.
3.4 Coleta e armazenamento das amostras
As amostras de sangue total (5 a 10 mL) foram obtidas através de punção
venosa e colhidas em frascos de vidro de 5 mL (tipo vacutainer), contendo
anticoagulante EDTA. Em seguida, as mesmas foram adequadamente identificadas
com o nome do paciente e a data da coleta e armazenadas a -20ºC até o momento
da extração de DNA. As amostras de urina (30mL) dos pacientes com MPS I e MPS
VI foram coletadas semanalmente, antes de cada infusão de reposição enzimática.
3.2. MÉTODOS
3.2.1. Análise bioquímica
a) TRE
As enzimas recombinantes Laronidase (Aldurazyme®, Biomarin
Pharmaceutical Inc., Novato, CA, USA; Genzyme Corporation , Cambridge, MA,

28
USA) e Galsulfase (Naglazyme®, BioMarin Pharmaceutical Inc., Novato, CA, USA)
foram administradas por via endovenosa, semanalmente, durante 48 semanas, nas
doses de 1 mg/kg e 0,58 mg/kg nos pacientes com MPS I e VI, respectivamente. As
infusões foram realizadas no Hospital Universitário Bettina Ferro de Souza da
Universidade Federal do Pará.
b) Dosagem de creatinina urinária:
A dosagem de creatinina, descrita por Dick (1970), na urina é um
procedimento inicial prévio à separação e à quantificação de GAG’s que visa estimar
a quantidade de soluto da amostra em função da excreção do metabólito creatinina.
A creatinina, determinada pela mensuração espectofotométrica da coloração obtida
a partir da reação da amostra da urina com os reagentes da técnica (ácido pícrico e
hidróxido de sódio), serviu como parâmetro para a correção do valor de excreção
urinária de GAG.
c) Dosagem dos GAG urinários:
A dosagem dos GAG urinários consiste em um método simples e de fácil
emprego, comumente utilizado por laboratórios que realizam a triagem urinária para
investigação de MPS. Esse método produz resultados mais confiáveis em relação a
falsos-negativos e indica a quantidade de GAG excretados na urina do paciente
através do método espectrofotométrico descrito por De Jong et al. (1989), o qual
mede a absorbância da reação obtida entre uma solução tamponada de azul de
dimetileno (DMB) e os GAG presentes na urina. Os resultados obtidos foram
expressos em mg de GAG / mmol de creatinina ou ug de GAG / mg de creatinina.

29
3.2.2. Análise molecular
a) Extração do DNA:
As amostras de sangue de cada pacientes com MPS e seus familiares (pais
e/ou irmãos) foram coletadas para a extração de DNA, segundo o método do Fenol-
Clorofórmio (Sambrook et al., 2001).
b) Reação em cadeia da polimerase (PCR):
Para a amplificação dos fragmentos de interesse (Quadro 04), foi utilizada
a técnica de PCR. Os oligonucleotídeos iniciadores (primers) utilizados neste estudo,
com exceção dos utilizados na amplificação do éxon 6 do gene NAGLU (Yogalingam
et al., 2000) foram desenhados com o auxílio do programa Primer3
(http://www.genome.wi.mit.edi/cgi-bin/primer3_www.cgi):

30
Quadro 04: Oligonucleotídeos iniciadores (primers) utilizados para a amplificação de fragmentos específicos dos genes IDUA (MPS I), IDS (MPS II), NAGLU (MPS IIIB) e ARSB (MPS VI):
Tipo de MPS Éxon Primer (5’ ���� 3’) Tamanho do fragmento
(pb)
MPS I
2* GGCTTGAACGTGTGTGTCAG(F) TCCCATCTGTGCCTCTGTAA(R)
274
9* CTGGGGACTCCTTCACCAAG(F) CAGAGACCTCCCTGGAACC (R)
354
11* GTGTGGGTGGGAGGTGGA (F) TTAGGGGACTGCCACTTGC (R)
250
MPS II
3* AAGCATCTGCTGGTTTCAGG(F) CAGACTCTGGACATGGAGCA(R)
423
9I* CATATGGAGCCCAGACAGGT(F) ATGCTGCGTATGGAATAGCC(R)
399
9II* CCCGTGAACTGATTGCCTAT (F) ACTAGCCCTCAGGCTGCTTC(R)
400
MPS IIIB
2* GATGGGGGATTTGTTCCAG(F) GTGGGGAAGGGACAGTGG(R)
246
6a** GGCCCTCTGTTTCATCACTC(F) AAATCTGGCACTGGGTCCTT(R)
444
6b** GCATCAGCCAGAACGAAGTG(F) CCAGCTCCTTGCTCAGGTAG(R) 426
6c** CAACCGATCTGATGTGTTTG(F) TTGGCATAGTCCAGGATGTT(R)
386
6d** GCTGGCTAGTGACAGCCGCTT(F) CTGGTGCTGTTGGAAAGGGAT(R)
261
6e** GCCGAGGCCGATTTCTAC(F)
GCGAATCTATCACCAAGAGC(R) 345
6f** CGTTCTCAGCAAGCAGAGGTA(F) CAAGCGTGGCAGCAGTGACC(R)
373
4* CTGCGTGTATCCTGGGAGAT(F) AGCTAAGTGGAGGGGGTGAG(R)
197
MPS VI 5*
TCATCCTCATGCCAAGACCT (F) GAAAAAGGGCAGGGTGTAGG(R)
300
8* CCTCTGTGCTTCTCCCTCAG (F) CTTCCAATTGAAAGGTTTTC (R)
347
(F) forward; (R) reverse; (pb) pares de bases *LEIM/ICB/UFPA **Yogalingam et al., 2000
Os primers foram diluídos com água Mili-Q até uma concentração de 1000
pmoles/µL (solução estoque). A solução de uso foi preparada para uma
concentração de 10 pmoles/µL. O protocolo de preparo e as condições das reações
encontram-se resumidos nos quadros 05 e 06, respectivamente.

31
Quadro 05 : Protocolo básico para as reações de amplificação por PCR.
Soluções 1X
DNTPs (2 mM) 2,5 µL
Tampão de PCR 10x (sem MgCl2) 2,5 µL
Primer 1,5 ou 2,0 µL
Primer 1,5 ou 2,0 µL
MgCl2 (50 mM) 2,0 ou 1,5 µL
Taq polimerase 1,0; 0,5 ou 0,25 µL
DNA (100 ng/µL) 2,0 µL
Água Mili-Q estéril q.s.p 25 µL
Quadro 06: Condições usadas nas reações de PCR.
Estágios Número de ciclos Temperatura Tempo (minuto s)
Desnaturação inicial 1 94ºC 5
Desnaturação
30
94ºC 1
Anelamento 55ºC a 66ºC* 1
Extensão 72ºC 1
Extensão final 1 72ºC 5
* Intervalo que compreende as temperaturas de anelamento de todos os éxons investigados.
Após as reações de amplificação, os produtos de PCR (5,0 µL) serão
submetidos à análise por eletroforese horizontal em gel de agarose 1,5%, 2,0% ou
3,0% (p/v) (com brometo de etídio), de acordo com o fragmento analisado. Após a
eletroforese, o gel de agarose será transferido para um transiluminador com luz
ultravioleta, para visualização dos fragmentos de DNA. O tamanho do fragmento
amplificado será comparado ao marcador de peso molecular (50 pb ou 100pb), com
o objetivo de confirmar a amplificação desejada.

32
c) Análise por digestão com endonucleases de restri ção:
Com base em alguns estudos moleculares realizados em pacientes com
MPS (Beesley et al., 1998; Beesley et al., 2001; Garrido et al., 2008), algumas
mutações específicas e comuns nos genes correspondentes a cada tipo da doença
foram analisadas por digestão com enzimas de restrição (Quadros 07 e -08). As
mutações estudadas foram: Q70X, W402X e P533R (MPS I); Y140C e R234C (MPS
IIIB); e R315Q (MPS VI). A mutação P533R
O protocolo empregado nesta metodologia está representado no Quadro 07.
Após as reações, os produtos da digestão enzimática (10,0 µL) foram submetidos à
análise por eletroforese horizontal em gel de agarose 1,5%, 2,0% ou 3,0% (p/v) (com
brometo de etídio), de acordo com o tamanho do fragmento analisado. Após a
eletroforese, o gel de agarose foi transferido para um transiluminador com luz
ultravioleta, para visualização dos fragmentos digeridos. O tamanho do fragmento
digerido foi comparado ao marcador de peso molecular (50 pb ou 100 pb), com o
objetivo de verificar o tamanho dos fragmentos clivados e/ou não clivados.

33
Quadro 07: Protocolo empregado nas reações com enzimas de restrição.
Tipo de
MPS
Éxon Mutação Enzima
de restrição
Produto de PCR
(pb)
Fragmentos esperados
Normal (pb) Mutante
(pb)
MPS I
2 Q70X –Sau96I 274 122+95+57 179+95
MPS I
9 W402X +BfaI 354 354 127 + 227
MPS II
9I R468W -MspI 399 316 + 83 399
MPS II
9I R468Q -TaqI 399 241 + 158 399
MPS II
9I P467L -NciI 399 316 + 83 399
MPS IIIB
2 Y140C -Fnu4HI 246 196 + 31 + 19 76 + 170
MPS IIIB
4 R234C -HhaI 197 103 + 94 197
MPS VI
5 R315Q -TaqI 300 77 + 179 + 44 256 + 44
(-) perda do sítio de restrição; (+) criação de nov o sítio de restrição; (pb) pares de bases
Quadro 08 : Protocolo básico de digestão com endonucleases de restrição
Soluções 1X
Tampão da enzima de restrição (10X) 2,0 µL
Enzima de restrição (500, 1000 ou 5000 U/µL)
1,0 µL
Produto da PCR 10,0 ou 20,0 µL
Água Mili-Q estéril q.s.p

34
d) Análise de Polimorfismos Conformacionais de Cade ia Simples (SSCP):
A análise por SSCP (Labrune et al., 1991) é um método de triagem
molecular, baseado na desnaturação do DNA em alta temperatura (acima de 90ºC),
originando 2 fitas simples. Possíveis alterações na seqüência gênica dos produtos
da desnaturação podem ser visualizadas em gel de poliacrilamida não-desnaturante,
corado com nitrato de prata. A presença de uma alteração na seqüência normal
provoca um padrão de migração eletroforético alterado e diferente do indivíduo
controle (normal), o qual pode ser visualizado ao final da análise.
Os produtos de PCR das seqüências gênicas que não apresentaram
mutações ou não foram submetidos à análise por digestão com endonucleases de
restrição foram submetidos à análise por SSCP, a fim de identificar regiões dos
genes com alterações na seqüência gênica.
e) Seqüenciamento direto:
Todos os fragmentos gênicos analisados foram, posteriormente, submetidos
à análise por seqüenciamento direto automatizado, com o intuito de identificar
alterações na seqüência gênica.
A análise por seqüenciamento direto foi realizada em um Seqüenciador
Automático ABI-PRISM 377 (Applied Biosystems), com o auxílio do kit ABI PRISM
BigDye Terminator Cycle Sequencing (Applied Biosystems, USA). Este
procedimento foi realizado no Laboratório de Genética Humana e Médica da
Universidade Federal do Pará (LGHM/UFPa). O Quadro 09 mostra o protocolo
básico utilizado nas reações de seqüenciamento.
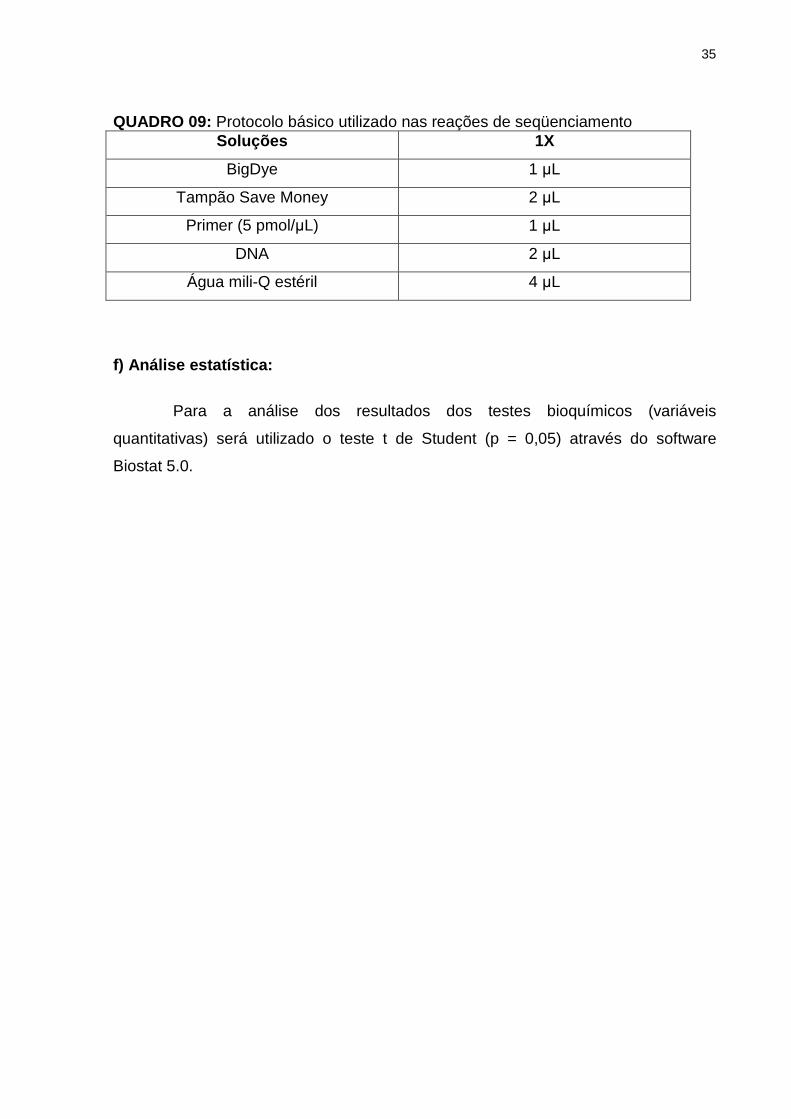
35
QUADRO 09: Protocolo básico utilizado nas reações de seqüenciamento Soluções 1X
BigDye 1 µL
Tampão Save Money 2 µL
Primer (5 pmol/µL) 1 µL
DNA 2 µL
Água mili-Q estéril 4 µL
f) Análise estatística:
Para a análise dos resultados dos testes bioquímicos (variáveis
quantitativas) será utilizado o teste t de Student (p = 0,05) através do software
Biostat 5.0.

36
4. RESULTADOS
4.1 MPS I
4.1.1 Perfil Bioquímico
A análise da concentração de GAG urinários revelou uma diminuição
significativa da excreção de GAG em todos os pacientes com MPS I submetidos à
TRE em relação aos níveis anteriores a terapia. A concentração inicial de GAG
inicial de 76,38 mg/mmol CREA diminuiu, significativamente, para 12,34±9,35
mg/mmol CREA, correspondendo a uma redução de aproximadamente 79%
(p<0,001) na taxa de excreção do paciente P1 durante as 6 primeiras semanas de
TRE. Essa redução se tornou mais evidente nas semanas posteriores, chegando a
10,20±6,89 mg/mmol CREA (redução de 91%) a partir da 12a semana e se manteve
em níveis relativamente constantes até o final do período analisado (Figura 03).
Já em relação ao paciente P2, a concentração inicial de GAG de 17,27
mg/mmol CREA reduziu para 9,17±4,1 mg/mmol CREA, uma diminuição de
aproximadamente 47% (p<0,05). Entretanto, esses valores oscilaram
acentuadamente, chegando a valores bem próximos ao valor inicial durante a 12a
semana (16,4±7,6 mg/mmol CREA; p>0,05) e retornando a valores menores durante
a 18a e 24a (semana (11,1±7,2 mg/mmol CREA e 4,3±0,53 mg/mmol CREA,
respectivamente). Não foi possível realizar as dosagens de GAG urinários deste
paciente após a 24ª semana em função da interrupção do tratamento do mesmo
durante tal período (Figura 03).

37
Figura 03: Concentração de GAG urinários de pacientes com MPS I antes e durante a TRE. Os
valores representam as médias das concentrações de GAG urinários encontradas em um período de
seis semanas, ao longo de 48 semanas. As barras de erros representam o desvio padrão de cada
amostra
4.1.2 Perfil Molecular (gene IDUA)
Em relação ao SSCP, os dois pacientes com MPS I apresentaram alterações
no padrão de migração de bandas quando comparados a um controle negativo. A
Figura 04 ilustra um exemplo de padrões de migração (linhas 2 e 3) diferentes dos
padrão apresentado pelo indivíduo saudável (linha 1). Para confirmar se essas
alterações correspondiam necessariamente a mutações, os mesmos fragmentos
foram, posteriormente, submetidos ao seqüenciamento direto.

38
Figura 04: Análise de SSCP do éxon 11 do gene IDUA (MPS I). Indivíduo controle (linha 1), pacientes com padrões de migração alterados (linhas 2 e 3). A técnica de seqüenciamento direto das amostras amplificadas permitiu
confirmar que os padrões de migração alterados no SSCP correspondiam a uma
mutação patogênica (P533R). Essa mutação é resultante da troca de um resíduo de
aminoácido prolina por um de arginina na posição 533 da cadeia polipeptídica e é
responsável pela alteração estrutural da conformação tridimensional da enzima, o
que leva a uma perda significativa de sua função enzimática e, consequentemente, à
falha na degradação do substrato (GAG).
Além disso, por meio da técnica de seqüenciamento, foi possível identificar o
paciente homozigoto e o heterozigoto para a mutação. A Figura 05 ilustra a
seqüência de DNA do indivíduo saudável (Fig. 05A), seguida da seqüência do
paciente homozigoto (Fig. 05B) e do heterozigoto (Fig. 05C).
1 2 3

39
Figura 05 : Sequenciamento do éxon 11 do gene IDUA (MPS I). 01: Alelo selvagem; 02: Homozigoto
para P533R (paciente P1); 03: Heterozigoto para P533R (paciente P2).
C>G (P533R)
homozigoto
B
A
C
C>G (P533R)
heterozigoto

40
4.2 MPS II
4.2.1 Perfil Molecular (gene IDS)
A investigação molecular dos pacientes com MPS II começou a partir da
amplificação por PCR e análise do padrão de migração dos fragmentos amplificados
pelo SSCP. Dentre todos os pacientes analisados, apenas o paciente P5
demonstrou alteração no éxon 9, através de um padrão de migração diferente do
padrão de um indivíduo normal, o que demonstra uma possível alteração no
fragmento analisado (Figura 06).
Figura 06: Análise de SSCP do éxon 9 do gene IDS (MPS II). Indivíduos controles (linhas 1, 2, 3, 4, 5, 7 e 8) e padrão de migração alterado (linha 6).
A análise da digestão por endonucleases de restrição revelou que a
alteração encontrada no éxon 9 do paciente P5 consistia na mutação R468W
(enzima MspI) como mostra a Figura 07. A mutação R468W promove a destruição
do sítio de restrição da enzima em função de uma troca C>T (Arg � Trp) no éxon 9
1 2 3 4 5 6 7 8

41
do gene IDS, causando a não clivagem do produto de PCR de 399 pb. Em
indivíduos normais, esse fragmento é clivado em dois subprodutos: um com 316 pb
e outro 83 pb.
Figura 07 : Digestão enzimática do exon 9 do gene IDS para a detecção da mutação R468W (MPS II).
M: Marcador de 100 pb; ND: Produto não-digerido; 01: Paciente apresentando padrão de digestão
correspondente à mutação R468W; 02: Indivíduo controle.
O seqüenciamento direto (Figura 08) confirmou a presença da mutação
R468W (em homozigose) no paciente P5. A alteração, como dito anteriormente,
corresponde a uma troca C>T no nucleotídeo 1526 do gene IDS, a qual resulta na
mudança de um resíduo de aminoácido Arginina (R) para um de Triptofano (W) no
precursor polipeptídico da enzima. Embora não existam estudos in vitro que avaliam
o efeito dessa mutação na proteína, acredita-se que essa alteração cause uma
desestruturação da conformação tridimensional da enzima iduronato-sulfatase,
levando a um déficit na atividade enzimática.
M100 ND 01 02

42
Figura 08 : Sequenciamento do éxon 9 do gene IDS (MPS II). A: Alelo selvagem; B: Paciente com a
mutação R468W (paciente P3).
A
B
C>T (R468W)

43
4.3 MPS IIIB
4.3.1 Perfil Molecular (gene NAGLU)
A paciente com MPS IIIB não apresentou alterações em nenhum éxons
investigado por meio das técnicas moleculares utilizadas (PCR, digestão por
endonucleases de restrição, SSCP e sequenciamento direto). Entretanto, é
necessária a investigação de mutações em outros éxons do gene para a efetiva
identificação de mutações e/ou polimorfismos.
4.4 MPS VI
4.4.1 Perfil Bioquímico
A concentração de GAG nas urinas das pacientes com MPS VI também foi
significativamente reduzida após o início da TRE. Em relação à paciente P8, a
dosagem inicial de 31,9 mg/mmol CREA reduziu para 6,17 mg/mmol CREA durante
as 6 semanas inicias, representando uma redução de, aproximadamente, 80%
(p<0,01). Esses valores permaneceram relativamente constantes até a 36ª semana,
quando se verificou um discreto aumento na concentração, porém não-significativo
em relação à dosagem inicial. Já em relação à paciente P9, verificou-se, também,
uma redução significativa de, aproximadamente, 94% nas 6 semanas iniciais (6,1
mg/mmol CREA) em relação à dosagem inicial (31,90mg/mmol CREA) anterior ao
início da TRE (Figura 09). O paciente P10 não foi submetido à TRE em função da
indisponibilidade da mesma durante o período em que o paciente estava vivo.

44
Figura 09: Concentração de GAG urinários de pacientes com MPS I antes e durante a TRE. Os
valores representam as médias das concentrações de GAG urinários encontradas durante um
período de 6 semanas, ao longo de 48 semanas.. As barras de erros representam o desvio padrão de
cada amostra,
4.4.2 Perfil Molecular (gene ARSB)
Após amplificação, os fragmentos obtidos foram submetidos à digestão por
endonucleases de restrição, análise de SSCP e seqüenciamento direto. Embora a
análise por digestão enzimática não tenha indicado a presença de mutação, a
análise de SSCP demonstrou uma alteração no padrão de migração de bandas
entre os pacientes e o controle negativo no éxon 5 (Figura 10). Além disso, o padrão
de migração apresentado pelas pacientes P8 e P9 (linhas 3 e 4, respectivamente) foi
diferente daquele observado pelo paciente P10 (linha 2) no mesmo éxon, o que
indica a presença de diferentes alterações entre esses pacientes.

45
Figura 10: Análise de SSCP do éxon 5 do gene ARSB (MPS VI). Indivíduo controle (linha 1), pacientes com padrões de migrações alterados (linhas 2, 3 e 4).
Após o seqüenciamento direto das amostras (Figura 11), os polimorfismos
V358M e V376M (éxon 5), descritos pela literatura como freqüentes em pacientes
com MPS VI, foram detectados. O polimorfismo V358M foi identificado em
homozigose nas pacientes P8 e P9. Já o polimorfismo V376M foi identificado em
homozigose no paciente P10.
1 2 3 4

46
Figura 11 : Sequenciamento do éxon 5 do gene ARSB (MPS VI). A e C: Alelos selvagens; B:
Homozigoto para polimorfismo V358M; D: Homozigoto para polimorfismo V376M.
A
B
C
D
G>A (V376M)
A>G (V358M)

47
Dos 10 éxons investigados em pacientes com diferentes tipos de MPS,
apenas 3 apresentaram alterações: MPS I (éxon 11), MPS II (éxon 9) e MPS VI
(éxon 5). A Tabela 01 mostra as mutações e polimorfismos encontrados nos
pacientes e familiares (mães) de acordo com o protocolo de investigação molecular
adotado pelo presente estudo.
TABELA 01: Mutações e polimorfismos encontrados nos pacientes com MPS e seus parentes.
Paciente Mutação Polimorfismo Mãe
MPS I P1 P533R/P533R - P533R
P2 P533R/? - P533R
MPS II
P3 - - -
P4 - - -
P5 R468W - R468W
P6 - - -
MPS IIIB
P7 - - -
MPS VI
P8 - V358M/V358M V358M
P9 - V358M/V358M V358M
P10 - V376M/V376M V358M/V376M

48
5. DISCUSSÃO
A região Norte do Brasil, mais especificamente o Estado do Pará é
caracterizado pela longa extensão territorial e pela dispersão habitacional. A
população residente em regiões distantes dos grandes centros urbanos, por não
possuir um adequado sistema de apoio de saúde local, busca na capital uma
alternativa para um atendimento adequado.
Em função disso, vários pacientes com MPS residentes nessa região têm o
seu diagnóstico realizado tardiamente. Em alguns casos, o paciente passa por
diversas avaliações clínicas até que haja, por fim, a suspeita de MPS. Quando isso
ocorre, o paciente, geralmente, já possui uma idade elevada e as intervenções
terapêuticas podem surtir pouco ou nenhum efeito significativo.
Outro fator que implica no manejo desses pacientes é o fato de que a maioria
das análises bioquímicas e moleculares (essenciais para a adoção de medidas
terapêuticas adequadas) seja realizada fora do Estado. O período compreendido
entre o envio da amostra e o recebimento do exame pode durar alguns meses e,
durante esse período, os sintomas das MPS continuam a progredir.
5.1 Investigação molecular
A investigação molecular em pacientes com MPS no Norte do Brasil consistiu
na busca por mutações freqüentes em pacientes brasileiros com MPS, por meio da
amplificação de fragmentos gênicos específicos descritos como regiões propensas à
ocorrência dessas mutações. Dos 10 pacientes analisados, apenas 3 apresentaram
mutações (2 com MPS I e 1 com MPS II) e 3 apresentaram apenas polimorfismos (3
com MPS VI). O restante dos pacientes não apresentou alterações em nenhum dos
éxons investigados.

49
O presente estudo demonstrou uma heterogeneidade da mutação P533R em
relação ao desenvolvimento de fenótipos clínicos em pacientes com MPS I. O
paciente P1, homozigoto para a mutação P533R, apresentou características clínicas
e bioquímicas condizentes com um fenótipo clínico grave a intermediário, enquanto
o heterozigoto (P2) apresentou características clínicas e bioquímicas relacionadas a
um fenótipo atenuado.
A ampla heterogeneidade da doença também é evidenciada por outros
estudos que tentaram correlacionar a presença de uma determinada mutação com
um fenótipo clínico específico. Gatti et al. (1997) avaliou em um grupo de 27
pacientes italianos com MPS I, a presença de mutações no gene IDUA. As
mutações mais frequentemente encontradas foram a W402X e a Q70X,
correspondendo a 11% e 13% dos alelos, respectivamente. A mutação P533R,
também encontrada em uma freqüência elevada (11%), foi encontrada em
homozigose em 2 pacientes com diferentes fenótipos clínicos (intermediário e leve).
Venturi et al. (2002) identificou 23 mutações diferentes, sendo 10 novas e 13
já conhecidas. Dentre as mutações encontradas, apenas a P533R, Q70X e G51D
foram encontradas em mais de 1 paciente, indicando uma elevada freqüência nessa
população. Os pacientes portadores da mutação P533R (tanto em homozigose
quanto em heterozigose) também possuíam fenótipo clínico variado.
Em relação à população brasileira, Matte et al. (2003) descreveu como
freqüente a mutação P533R (11,6% dos alelos). Os pacientes homozigotos para a
mutação apresentavam um fenótipo classificado como intermediário enquanto os
heterozigotos, como grave a intermediário. O fenótipo intermediário apresentado
pelos homozigotos corresponde à atividade residual da enzima mutante. Uma
segunda alteração paralela a essa pode trazer intensas modificações na estrutura
tridimensional da proteína, levando a uma menor capacidade catalítica da enzima e,
assim, ocasionar fenótipos mais graves.

50
Ainda não há estudos que avaliem a interferência da mutação P533R na
estrutura tridimensional da enzima α-L-iduronidase. Entretanto, é provável que essa
mutação, quando em homozigose, seja responsável por uma alteração significativa
na conformação tridimensional protéica, conforme fenótipo apresentado pelo
paciente P1 (grave a intermediário). A variação fenotípica apresentada por pacientes
com essa mesma mutação, tal como em pacientes heterozigotos (ex.: paciente P2)
pode estar relacionada a diversos fatores, dentre eles, a interação da mutação com
polimorfismos e/ou haplótipos, background genético e fatores ambientais e sociais.
Para a identificação da mutação presente no outro alelo do paciente P2
(heterozigoto composto), uma análise molecular mais detalhada do gene deve ser
realizada, através da amplificação de outros fragmentos gênicos e de estudos de
expressão in vitro, a fim de verificar o impacto das mutações na formação da
proteína e, assim, determinar quais implicações as alterações na enzima terão na
caracterização clínica do paciente.
A ampla heterogeneidade clínica e molecular da MPS II também pôde ser
comprovada pelos achados do presente estudo. O paciente P3, portador da mutação
R468W, foi diagnosticado clinicamente como portador de um fenótipo grave a
intermediário, resultado que difere da maioria dos estudos que tentaram traçar tal
correlação. Essa dificuldade em determinar correlações genótipo-fenótipo pode ser
considerada resultado de alguns fatores, como a raridade da doença e o fato de que
a maioria das mutações apresentadas pelos pacientes corresponde a mutações
privadas, ou seja, mutações que ocorrem apenas em uma determinada população.
Alguns estudos sugerem que certas mutações que envolvam transições C�T
ou G�A em dinucleotideos CpG do gene IDS são causados por processos de
metilação-deaminação in situ e que, por isso, essas regiões são consideradas como
“hot spots” ou “pontos-chave” para a identificação de mutações. A mutação R468W
ocorre devido a uma troca de C�T em regiões CpG e, portanto, é considerada como
uma “mutação-chave”. Entretanto, sua caracterização enquanto ao seu papel no
desenvolvimento do fenótipo do paciente ainda não é totalmente definida devido à
elevada heterogeneidade molecular do gene (Rathmann et al., 1996; Tomatsu et al.,
2004)

51
A mutação R468W corresponde a uma troca de um resíduo de arginina por
um de triptofano na posição 468 do polipetídeo precursor da enzima iduronato-
sulfatase. Apesar da ausência de estudos in vitro que possam avaliar o impacto
dessa mutação na estrutura protéica, é provável que essa alteração promova uma
diminuição significativa da atividade enzimática. Entretanto, essa depleção da
atividade enzimática aparentemente é variável, pois diferentes fenótipos clínicos são
associados a mesma alteração genética, como mostram alguns estudos.
Segundo Froissart et al. (1998), dos 8 pacientes com fenótipo atenuado, 7
carreavam uma mutação do tipo missense, incluindo a R468W. Além disso, Jonsson
et al. (1995) também descreveram 3 pacientes com essa mutação, todos
apresentando um fenótipo leve. Em contraste, Isogai et al. (1998), Lin et al. (2006) e
Froissart et al., (2007) relataram fenótipos graves associados a essa mesma
mutação em pacientes diferentes. Já Li et al. (1999) não relatou a classificação
fenotípica em função da falta de dados clínicos do paciente.
A posterior investigação de outras mutações que possam estar presentes em
outros éxons do gene IDS nos pacientes com MPS II pertencentes a este estudo é
imprescindível, pois com a recente implantação da TRE para esse tipo de MPS no
Estado no Pará, o diagnóstico molecular, juntamente com o bioquímico e clínico
servirá como base para a elaboração de abordagens terapêuticas mais eficazes
para esses pacientes.
A única paciente com MPS IIIB participante deste estudo (P8) não apresentou
alterações nos éxons investigados (éxons 2, 4 e 6). Além disso, em função da
ausência de dados clínicos e bioquímicos, não foi possível determinar o seu fenótipo
e, assim, correlacionar o perfil mutacional com o perfil bioquímico e clínico. Para a
identificação da mutação e/ou polimorfismos causadores da doença, uma análise
molecular completa deve ser realizada, por meio da amplificação dos fragmentos
que ainda não foram amplificados. Além disso, a análise bioquímica (mensuração da
atividade enzimática e da concentração de GAG urinários) também deve ser
realizada para que, junto com o diagnóstico molecular, o caracterização clínica
completa seja realizada.

52
Os polimorfismos encontrados nos pacientes com MPS VI no presente estudo
(V358M e V376M) não podem ser estritamente relacionados a um padrão fenotípico
específico, pois podem estar associados a diversas mutações causadoras de várias
apresentações clínicas. Entretanto, esses polimorfismos podem servir como base
para uma investigação molecular mais específica, uma vez que a presença desses
polimorfismos, em muitos casos, está ligada, também, a presença de determinadas
mutações.
Assim como nos outros demais tipos de MPS, a heterogeneidade molecular
também é evidenciada na MPS VI. Vários estudos demonstram que o padrão
genotípico apresentado não está, na maioria dos casos, diretamente relacionada
com o perfil bioquímico e clínico dos pacientes.
O estudo realizado por Villani et al. (1999) identificaram, em pacientes com
MPS VI, algumas mutações (S65F, P116H, R315Q, Q503X, P531R) e polimorfismos
(I114I, L124L, G324G e V358M) e, também, avaliaram o impacto das mesmas
através da análise da estrutura tridimensional da proteína mutante. De acordo com
os autores, a mutação R315Q, presente em um dos pacientes e responsável pelo
aparecimento do fenótipo intermediário, consiste em uma torça de G>A no
nucleotídeo 944 do éxon 5 do gene ARSB, o que resulta em uma troca de Arg para
Gln no resíduo 315. Essa alteração promove sérias mudanças na estrutura da
proteína, levando a falhas no processo de empacotamento e, consequentemente, de
estabilidade da forma madura da proteína.
Karageorgeos et al. (2007) descreveram um raro caso no qual um paciente
com MPS VI apresentava, ao mesmo tempo, 3 mutações no gene ARSB (L72R,
R315Q e S384N) além do polimorfismo V358M. A mutação R315Q foi descrita
anteriormente como causadora de um fenótipo intermediário, enquanto a S384N era
responsável por um fenótipo de rápida progressão dos sintomas. A outra mutação
(L72R) é possivelmente relacionada com uma significante alteração na estrutura
tridimensional da proteína e também é responsável pelo aparecimento rápido de
sintomas (fenótipo grave). Além disso, através da clonagem de produtos de RT-PCR
e posterior seqüenciamento, foi possível detectar que as mutações R315Q e S384N

53
estão ligadas no mesmo alelo, enquanto a mutação L72R e o polimorfismo V358M
estão localizados em outro.
Outro trabalho realizado por Petry et al. (2005) também relatou a presença
das mutações R315Q e 1533del23 e dos polimorfismos V358M, V376M e P397P.
Segundo os autores, a mutação R315Q, relacionada anteriormente fenótipos
intermediários, foi identificada em 2 pacientes gêmeos com fenótipos diferentes (um
grave e o outro grave a intermediário). Além disso, outros pacientes, que possuiam
classificação clínica tipicamente grave, também apresentaram essa mutação. Em
relação a mutação 1533del23, encontrada tanto em homozigose quanto em
heterozigose, foi relacionada a um fenótipo grave.
Segundo Karageorgeos et al. (2004), os polimorfismos V358M, V376M e
P397P, causados por transições em dinucleotideos CpG, quando ligados a
determinadas mutações causadoras de MPS VI, podem contribuir para diminuir a
atividade residual da enzima e, assim, influenciar no desenvolvimento clínico do
paciente.
Todas as mutações e/ou polimorfismos encontrados nos pacientes com MPS
e em seus familiares já foram descritas anteriormente, entretanto a correlação entre
essas alterações genéticas e o fenótipo clínico apresentado pelos pacientes difere
significativamente entre os estudos publicados. Assim, o presente estudo buscou
verificar correlações entre os fenótipos bioquímicos e clínicos apresentado por cada
paciente e comparar os resultados com aqueles obtidos na literatura.
Embora estudos sofisticados como a análise de expressão in vitro e a
cristalografia de proteínas permitam verificar o impacto de mutações na estrutura e
atividade das enzimas, as implicações celulares que essas mutações causam
diferem de paciente para paciente, conforme outras variáveis como fatores
ambientais, sociais e genéticos e, dessa forma, caracterísiticas clínicas diferentes
são apresentadas por pacientes com a mesma mutação, impedindo, portanto,
possívies correlações genótipo/fenótipo.

54
6.2 Perfil bioquímico
O tratamento das MPS era, antigamente, realizado através do controle dos
sintomas apresentados pelos pacientes. Entretanto, com o advento de novas
tecnologias terapêuticas, esse tratamento foi tornando-se mais específico e eficaz.
Dentre as terapias atualmente disponíveis, a TRE ainda constitui a mais eficaz para
pacientes com MPS (Arora et al., 2007; Harmatz et al., 2008; Muenzer et al., 2009).
Embora estudos recentes tenham demonstrado que outras formas de tratamento
como a terapia gênica (em modelos animais) (Fu et al., 2007; Wang et al, 2008;
McIntyre et al., 2008), injeção enzimática intratecal para casos com
comprometimento neurológico (Kakkis et al., 2004; Dickson et al., 2007) e
transplante de medula óssea (Zheng et al., 2004; Ellinwood et al., 2007; Sprangers
et al., 2009) apresentam resultados positivos na melhora dos quadros clínicos, a
TRE ainda é a abordagem que possui menor proporção entre efeitos adversos e a
melhora clínica dos paciente.
Com isso, alguns grupos de pesquisa vêm desenvolvendo uma série de
ensaios clínicos a fim de determinar qual a melhor forma intervenção em pacientes
submetidos à TRE (dosagem, intervalo entre as infusões e tempo total de
tratamento). Alguns parâmetros bioquímicos, como a dosagem de GAG urinários,
são utilizados como ferramentas para determinação da eficácia do tratamento, uma
vez que esse teste permite avaliar a variação da degradação desses compostos
após a infusão da enzima recombinante.
O estudo realizado por Harmatz et al. (2004) verificou que a dosagem da
excreção total de GAG na urina fornece forte evidência bioquímica de um efeito
estável, dependendo da dose de TRE administrada nesses pacientes. A dose mais
elevada de 1.0 mg/kg, como esperado, é mais eficaz do que doses mais baixas na
diminuição da excreção urinária de GAG em pacientes com MPS.
Posteriormente, para confirmar a eficácia e a segurança da TRE em pacientes
com MPS, Harmatz et al. (2006) avaliaram, por meio de um estudo duplo-cego,
randomizado, controlado por placebo, multicêntrico e multinacional em um período

55
de 24 semanas, 39 pacientes com MPS. Os pacientes foram divididos em 2 grupos
distintos (intervenção e placebo) e monitorados de acordo com parâmetros clínicos
(mobilidade articular e capacidade cardiopulmonar) e bioquímicos (excreção urinária
de GAG).apresentados ao longo da terapia. Ao final do período, foi possível verificar
que além de melhorias significativas nos aspectos clínicos, houve, também, uma
redução drástica da excreção de GAG urinários no grupo intervenção quando
comparado ao placebo.
Em relação ao presente estudo, é importante ressaltar que apesar de
todos os pacientes terem mais de 5 anos e apresentarem a doença em estágio
avançado, a TRE foi capaz de trazer benefícios à qualidade de vida dos mesmos.
Embora alguns dados sobre melhoras clínicas pós-TRE não tenham sido incluídos
nos resultados do presente estudo, foi possível observar uma diminuição
significativa da concentração de GAG urinários em todos os pacientes submetidos à
TRE. E de acordo com a literatura, a redução de GAG está intimamente ligada ao
aperfeiçoamento da condição clínica dos pacientes.
Os resultados obtidos com a TRE em pacientes com MPS em estágio
avançado indicam a capacidade de estabilização dos sintomas já desenvolvidos,
entretanto a administração precoce da enzima recombinante parece evitar a
progressão da doença e, conseqüentemente, de sintomas mais graves. Estudos
utilizando modelos animais (ex.: modelo felino) demonstram que quanto mais cedo
for o iniciado o tratamento, maiores serão as melhorias nos quadros esqueléticos
(Crawley et al., 1997), uma vez que a implantação do tratamento prévio antes do
aparecimento dos sintomas irreversíveis da doença pode maximizar os benefícios da
terapia.
A terapia precoce depende da triagem eficiente em recém–nascidos e
identificação de pacientes com risco de ter a doença. Programas para triagem de
recém-nascidos tem sido propostos (Whitley et al., 1989; Meikle et al., 1999; Whitley
et al., 2002), mas ainda estão em desenvolvimento e não estão disponíveis para
uso.

56
Em relação aos pacientes com MPS I, a redução de GAGs encontrada em
P1 foi de 79% na 6ª semana e de 91% nas semanas posteriores. O paciente P2
apresentou uma redução de 47% na 6ª semana e, após um período de oscilação
decorrente da interrupção do tratamento, a redução observada foi de 75% após a
25ª semana. Esta redução é superior à descrita na literatura, a qual varia entre
54,1% e 59,7% em 26 semanas, mantêm-se 59,1% em 52 semanas e alcança
patamares entre 69,1% e 88,4% em um estudo que durou seis anos (Wraith et al.,
2004; Sifuentes et al., 2007; Wraith et al., 2007).
Em tais estudos, por outro lado, a quantidade de enzima por kg corpóreo
aplicada por infusão foi menor – 0,58mg/kg, enquanto neste estudo aplicou-se
1mg/kg. Em pacientes que receberam 1,06mg/kg corpóreo, Wraith et al. (2007)
relataram em 52 semanas uma redução média de 67,7% - valor ainda inferior ao
encontrado no presente estudo, mas superior ao encontrados no mesmo estudo com
uma dosagem inferior da enzima.
Por outro lado, diferentemente do relatado na literatura (et al., 2004;
Sifuentes et al., 2006; Wraith et al., 2007), a quantidade de GAG excretada na urina
dos pacientes continuou acima do patamar normal por um período maior do que o
esperado. Para a faixa etária de P1 (19 anos), os valores de referência são entre 3,5
e 7 mg / mmol de creatinina; já para P2, entre 3,4 e 11 mg / mmol de creatinina (13
anos) (Fonte: Hospital de Clínicas de Porto Alegre).
Houve uma interrupção da TRE para P1 (MPS I) durante dois meses.
Neste período foi observada uma pequena elevação dos níveis de GAG na 12ª
semana após o início da TRE. No entanto, este incremento não foi significativo
(p>0,01) quando comparado com a última dosagem realizada na 4º semana de
tratamento. Foi também observada uma interrupção da TRE para P2 (MPS I)
durante 10 meses, porém, também os valores encontrados de GAG não diferiram
estatisticamente da última infusão (p>0,01).
Tais resultados sugerem que a enzima recombinante apresenta atividade
residual por um tempo ainda não conhecido. Na literatura há poucos artigos
relatando efeitos da descontinuidade do tratamento. Anbu et al. (2006) relataram

57
uma grande regressão clínica em uma paciente que teve a TRE suspensa após 80
semanas de tratamento. Neste caso, a interrupção foi por um período muito mais
longo – 2 anos.
As pacientes com MPS VI (P8 e P9) fizeram parte do estudo realizado por
Harmatz et al. (2004) no início do tratamento, onde foi relatada uma diminuição
progressiva na excreção de GAGs (71% na 24º semana e 76% na 48º) em relação
ao valores encontrados antes do início da terapia. Nossos resultados demonstram
que esses valores reduzidos mantiveram-se relativamente constantes como uma
média de 73% em P8 e 77% em P9 até a 48º semana. A ausência de estudos dos
efeitos da galsulfase na excreção de GAG em longo prazo torna os resultados aqui
encontrados particularmente relevantes.
Byers et al. (1998) relataram em estudos preliminares que a injeção de
rhASB nas junções ósseas de gatos auxilia na preservação da cartilagem. O dado
mais relevante desse estudo foi o achado de que essas injeções poderiam ser
administradas em um curto intervalo de tempo, dando a esta terapia potencial de
uso em humanos. Simonaro et al. (2001) descreveram a diminuição da liberação de
óxido nítrico e de fatores de necrose tumoral em cultura de condrócitos das
articulações de humanos com MPS VI, sugerindo que a TRE deve atuar reduzindo a
inflamação ao redor das junções. No estudo de Harmartz et al. (2008) foi observada
regressão nos sintomas referentes às junções com diminuição da dor e melhora na
capacidade de movimento.
A Tabela 02 ilustra os dados obtidos pela caracterização molecular,
bioquímica e clínica dos pacientes com MPS analisados pelo presente estudo. Foi
possível observar uma heterogeneidade entre subtipo da doença, uma vez que em
alguns casos, a elevada excreção inicial de GAG correspondia a uma dosagem
enzimática baixa, juntamente com uma apresentação clínica de grave a
intermediária (ex.: P1). Em outros casos, porém, a mesma apresentação clínica foi
identificada em pacientes que apresentavam uma excreção urinária inicial de GAG
bem menor e níveis maiores de atividade enzimática (ex.: P8, P9 e P10). Esses
resultados, assim como demonstrado pela literatura, indicam a ampla

58
heterogeneidade das MPS e a dificuldade em estabelecer correlações entre o
genótipo e o fenótipo apresentado pelos pacientes.
TABELA 02: Características moleculares, bioquímicas e clínicas dos pacientes com MPS.
Paciente Mutação Polimorfismo
[ ]
inicial
GAG
%
redução
GAG*
A.E. Fenótipo
clínico
MPS I P1 P533R/P533R - 76,4 94% 0,03** G/I
P2 P533R/? - 18,2 41% 0,25 A
MPS II
P3 - - 50,0 - 8,9 G/I
P4 - - 79,0 - ND I
P5 R468W - 12,4 - ND G/I
P6 - - 4,7 - ND G/I
MPS
IIIB P7
- - 43,5 - 0,13** -
MPS VI
P8 - V358M/V358M 31,9 77% 8,0 G/I
P9 - V358M/V358M 27,0 73% 10,0 G/I
P10 - V376M/V376M 19,0 - 11,0 G/I
A.E.: Atividade enzimática; ND: não detectável; G/I: Grave a intermediário; I: Intermediário; A:
Atenuado. (*) A redução da excreção de GAG (%) corresponde à diferença entre a dosagem inicial e
a média de redução durante o período analisado apenas em pacientes submetidos à TRE (MPS I e
VI). (**) Atividade enzimática dosada em plasma.
5.3 Considerações finais
A identificação de mutações causadoras de MPS é uma importante
ferramenta para o desenvolvimento de uma intervenção terapêutica mais eficaz, pois
permite a elucidação de possíveis alterações em mecanismos intracelulares e de
seus efeitos na sintomatologia que os pacientes possam apresentar no futuro. Além
disso, a identificação de mutações e/ou polimorfismos em familiares dos pacientes
permite um maior esclarecimento sobre os riscos inerentes a uma nova gestação
(aconselhamento genético).

59
A estratégia de investigação molecular adotada pelo presente estudo, embora
tenha permitido a identificação das mutações e polimorfismos em alguns pacientes,
não foi suficiente para a detecção das alterações presentes nos demais pacientes.
Novos protocolos de investigação molecular, tais como o seqüenciamento completo
de cada gene responsável pelos subtipos da doença, devem ser utilizados, a fim de
determinar o padrão genotípico de todos os indivíduos.
Paralela ao diagnóstico molecular, a dosagem de GAG urinários consiste em
uma importante ferramenta tanto para o diagnóstico de MPS quanto para o
acompanhamento da TRE, uma vez que esse monitoramento permite avaliar
possíveis flutuações desses metabólitos urinários em eventuais interrupções do
tratamento e, dessa forma, auxiliar no melhor entendimento sobre a
farmacodinâmica dessas substâncias no organismo dos pacientes.

60
6. CONCLUSÕES
• Dentre as 8 mutações investigadas, foi possível identificar apenas 2:
P533R (MPS I) e R468W (MPS II). Além disso, 2 polimorfismos foram
encontrados em 3 pacientes com MPS VI (2 com V358M e 1 com V376M);
• Houve uma redução significativa da taxa de excreção urinária de GAG
dos pacientes com MPS I e VI após o início da TER, indicando a eficácia
da terapia.
• As correlações entre os fenótipos clínico, bioquímico e molecular
diferiram da maioria dos estudos encontrados na literatura, o que
comprova a elevada heterogeneidade das MPS e a necessidade de mais
estudos envolvendo análises mutacionais in vitro paralelamente a
determinação fenotípica.

61
7. REFERÊNCIAS BIBLIOGRÁFICAS
ALVES, S.; MANGAS, M.; PRATA, M.J.; RIBEIRO, G.; LOPES, L.; RIBEIRO, H.;
PINTO-BASTO, J.; REIS LIMA, M.; LACERDA, L. Molecular characterization of
Portuguese patients with mucopolysaccharidosis type II shows evidence that the
IDS gene is prone to splicing mutations. J Inherit Metab Dis ., 29:743–754, 2006.
ANBU, A.T.; MERCER, J.; WRAITH, J.E. Effect of discontinuing of laronidase in a
patient with mucopolysaccharidosis type I. J Inherit Metab Dis., 29(1):230-1,
2006.
ARORA, R. S.; MERCER, J.; THORNLEY, M.; TYLEE, K.; WRAITH, J. E. Enzyme
replacement therapy in 12 patients with MPS I–H/S with homozygous
p.Leu490Pro mutation. J. Inherit. Metab. Dis. , 30:281., 2006.
BEESLEY, C.L.; YOUNG, E.P.; VELLODI, A.; WINCHESTER, B.G. Identification of
12 novel mutations in the a-N-acetylglucosaminidase gene in 14 patients with
Sanfilippo syndrome type B (mucopolysaccharidosis type IIIB). J. Med. Genet .,
35:910-914, 1998.
BEESLEY, C.E.; MEANEY, C.A.; GREENLAND, G.; ADAMS, V.; VELLODI, A.;
YOUNG, E.P.; WINCHESTER, B.G. Mutational analysis of 85
mucopolysaccharidosis type I families: frequency of known mutations,
identification of 17 novel mutations and in vitro expression of missense mutations.
Human Genetics. 109(5):503-11, 2001.
BYERS, S; ROZAKLIS, T; BRUMFIELD, LK; RANIERI, E; HOPWOOD, JJ.
Glycosaminoglican accumulation and excretion in mucopolyssacharidosis:
characterization and basis of the diagnostic test for MPS. The Journal of
Pediatrics 65: 222-290. 1998.
BRADFORD, T.M.; LITJENS, T.; PARKINSON, E.J.; HOPWOOD, J.J.; BROOKS,
D.A. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): a Y210C
mutation causes either altered protein handling or altered protein function of N-
acetylgalactosamine 4-sulfatase at multiple points in the vacuolar network.
Biochemistry; 41(15):4962-71, 2002

62
BUIST, N.R. Set of simple side-room urine tests for detection of inborn errors of
metabolism.. British Medical Journal , 2:745-749. 1968.
BYERS, S; ROZAKLIS, T; BRUMFIELD, LK; RANIERI, E; HOPWOOD, JJ.
Glycosaminoglican accumulation and excretion in mucopolyssacharidosis:
characterization and basis of the diagnostic test for MPS. The Journal of
Pediatrics 65: 222-290. 1998.
CANSTANTOPOULOUS, G. & DEKABAN, A.S. Neurochemistry of the
mucopolysaccharidoses: brain lipids and lysosomal enzymes in patients with four
type of mucopolysaccharidosis and in normal controls. J. Neurochem. 30:965-
973, 1978.
CASTRO, N.S.S.; BENTES, E.S.; SOUZA, I.C.N.; GIUGLIANI, R.; SANTANA-DA-
SILVA, L.C. Estimativa do perfil epidemiológico de mucopolissacaridoses no
Estado do Pará. Pediatria Moderna , XLIII: 23-28, 2007.
CHINEN, Y.; TOHMA, T.; IZUMIKAWA, Y.; UEHARA, H.; OHTA, T. Sanfilippo type B
syndrome: five patients with an R565P homozygous mutation in the alpha-N-
acetylglucosaminidase gene from the Okinawa islands in Japan. J. Hum. Genet.
50(7):357-359, 2005.
CLARKE, L.A. The mucopolysaccharidosis: a success of molecular medicine. Expert
Reviews in Molecular Medicine, 10(1), 2008.
CLEARY, M.A. & WRAITH, J.E. Management of mucopolysaccharidosis type III.
69(3):403-406, 1993.
COELHO, J.C.; WAJNER, M.; BURIN, M.G.; VARGAS, C.R.; GIUGLIANI, R.
Selective screening of 10,000 high-risk Brazilian patients for the detection of
inborn errors of metabolism. Eur J Pediatr; 156:650–654, 1997. CRAWLEY JN, BELKNAP JK, COLLINS A, CRABBE JC, FRANKEL W,
HENDERSON N, HITZEMANN RJ, MAXSON SC, MINER LL, SILVA AJ,
WEHNER JM, WYNSHAW- BORIS A, PAYLOR R. Behavioral phenotypes of
inbred mousestrains: Implications and recommendations for molecular studies.
Psychopharmacology 132:107-24. 1997.
DE JONG, J.G.; WEVERS, R.A.; LAARAKKERS, C.; POORTHIUS, B.J.
Dimethylmethylene blue-based spectrophotometry of glycosaminoglicans in

63
untreated urine: a rapid screening procedure for mucopolysaccharidoses. Clin.
Chem. 35(7):1472-1477, 1989.
DICK, T. Praticas de bioquímica . Porto Alegre, Instituto de Pesquisas Bioquímicas.
1970.
DICKSON, P.; MCENTEE, M.; VOGLER, C.; LE, S.; LEVY, B.; PEINOVICH, M.;
HANSON, S.; PASSAGE, M.; KAKKIS, E. Intrathecal enzyme replacement
therapy: successful treatment of brain disease via the cerebrospinal fluid. Mol.
Genet. Metab.; 91(1):61-8, 2007.
DIFERRANTE, N.M. The measurement of urinary mucopolysaccharides. Analytical
Biochemistry , 21: 98-106. 1967.
EL DIB, R.P. & PASTORES, G.M. Laronidase for treating mucopolysaccharidosis
type I. Genetics and Molecular Research., 6(3):667-674, 2007.
ELLINWOOD, N.M.; COLLE, M.A.; WEIL, M.A.; CASAL, M.L.; VITE, C.H.; WIEMELT,
S.; HASSON, C.W.; O'MALLEY, T.M.; HE, X.; PROCIUK, U.; VEROT, L.;
MELNICZEK, J.R.; LANNON, A.; AGUIRRE, G.D.; KNOX, V.W.; EVANS, S.M.;
VANIER, M.T.; SCHUCHMAN, E.H.; WALKLEY, S.U.; HASKINS, M.E. Bone
marrow transplantation for feline mucopolysaccharidosis I. Mol. Genet. Metabol.;
91(3):239-50, 2007.
FEIO, R.H.; SOUZA, I.C.N.; SIQUEIRA, M.L.; SILVA, M.; SILVA, R.; SANTANA-DA-
SILVA, L.C. Mucopolissacaridose Tipo VI e I: Três casos em terapia de reposição
enzimática no HUBFS. In: Congresso Brasileiro de Neurologia, 23, Belém
(Brasil), p-669, 2008.
FILOCAMO, M.; BONUCCELLI, G.; CORSOLINI, F.; MAZZOTTI, R.; CUSANO, R.;
GATTI, R. Molecular analysis of 40 patients with mucopolysaccharidosis type II:
New mutations in the iduronate-2-sulfatase (IDS) gene. Human Mutation ,
18(2):164-165, 2001.
FROISSART, R.; MILLAT, G.; MATHIEU, M.; BOZON, D.; MAIRE, I. Processing of
iduronate-2-sulphatase in human fibroblasts. Biochem. J. 309(Pt 2):425-430,
1995.

64
FROISSART, R.; MAIRE, I.; MILLAT, G.; CUDRY, S.; BIROT, A.M.; BONNET, V.;
BOUTON, O.; BOZON D. Identification of iduronate sulfatase gene alterations in
70 unrelated Hunter patients. Clin. Genet.; 53(5):362-8, 1998.
FROISSART, R.; MOREIRA-DA-SILVA, I.; MAIRE, I. Mucopolysaccharidosis type II:
na update on mutation spectrum. Acta Paediatr , 96:71-77, 2007.
FU. H,; KANG, L.; JENNINGS, J.S.; MOY, S.S.; PEREZ, A.; DIROSARIO, J.;
MCCARTY, D.M.; MUENZER, J. Significantly increased lifespan and improved
behavioral performances by rAAV gene delivery in adult mucopolysaccharidosis
IIIB mice. Gene Ther.; 14(14):1065-77, 2007.
GARRIDO, E.; CHABAS, A.; COLL, M.J.; BLANCO, M.; DOMÍNGUEZ, C.;
GRINBERG, D.; VILAGELIU, L.; CORMAND, B. Identification of themolecular
defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux–Lamy
syndrome) patients, including 9 novel mutations. Mol Genet Metab, 92:122–30.,
2007.
GARRIDO, E.; CORMAND, B.; HOPWOOD, J.J.; CHABÁS, A.; GRINBERG, D.;
VILAGELIU, L. Maroteaux-Lamy syndrome: functional characterization of
pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol Genet
Metab.; 94(3):305-12, 2008.
GATTI., R.; DINATALE, P.; VILLANI, G.R.D.;FILOCAMO, M.; MULL, V.; GUO, X.-H.;
NELSON, P.V.; SCOTT, H.S.; HOPWOOD, J.J. Mutations among Italian
mucopolysaccharidosis type I patients. J. Inher. Metab. Dis, 20:803–806, 1997.
GORT, L.; CHABÁS, A.; COLL, M.J. Analysis of five mutations in 20
mucopolyssacharidosis type I patients: high prevalence of the W402X mutation,
Human Mutation . 11:332–333, 1998.
HARMATZ, P.; WHITLEY, C.D.; WABER, l.; PAIS, R.; STEINER, R.; PLECKO, B.
KAPLAN, P.; SIMON, J.; BUTENSKY, E.; HOPWOOD, J.J. Enzyme replacement
therapy in Mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome). The Journal
of Pediatrics, 144:574-580, 2004.
HARMATZ, P.; GIUGLIANI, R.; SCHWARTZ, I.V.; GUFFON, N.; TELES, E.L.;
MIRANDA, M.C.; WRAITH, J.E.; BECK, M.; ARASH, L.; SCARPA, M.;
KETTERIDGE, D.; HOPWOOD, J.J.; PLECKO, B.; STEINER, R.; WHITLEY,

65
C.B.; KAPLAN, P.; YU, Z.F.; SWIEDLER, S.J.; DECKER, C.; MPS VI STUDY
GROUP. Long-term follow-up of endurance and safety outcomes during enzyme
replacement therapy for mucopolysaccharidosis VI: Final results of three clinical
studies of recombinant human N-acetylgalactosamine 4-sulfatase. Molecular
Genetics and Metabolism, 94:469-475, 2005.
HARMATZ, P.; KETTERIDGE, D.; GIUGLIANI, R.; GUFFON, N.; TELES, E.L.;
MIRANDA, M.C.S.; YU, Z.; SWIEDLER, S.J.; HOPWOOD, J.J. Direct comparison
of measures of endurance, mobility, and joint function during enzyme-
replacement therapy of Mucopolysaccharidosis VI (Maroteaux-Lamy Syndrome):
results after 48 weeks in a phase 2 open-label clinical study of recombinant
human N-acetylgalactosamine-4-sulfatase. Pediatrics, 115:681-689, 2008.
HOBBS, J.R. Bone marrow transplantation for inborn errors. Lancet. 2(8249):735-
739, 1981.
HOPWOOD, J.J. & MORRIS, C.P. The mucopolysaccharidosis: Diagnosis, molecular
genetics and treatment. Mol. Biol. Med., 7(5):381-404, 1990.
HOPWOOD, J.J.; BUNGE, S.; MORRIS, C.P.; WILSON, P.J.; STEGLICH, C.; BECK,
M.; SCHWINGER, E.; GAL, A. Molecular basis of mucopolysaccharidosis type II:
mutations in the iduronate-2-sulphatase gene. Hum. Mutat.; 2(6):435-42, 1993.
HUMBEL , R & CHAMOLES, N.A. Sequential thin layer chromatography of urinary
acidic glycosaminglycans. Clinica Chimica Acta , 40: 290-293, 1972.
ISOGAI, K.; SUKEGAWA, K.; TOMATSU, S.; FUKAO, T.; SONG, X-Q.; YAMADA,
Y.; FUKUDA, S.; ORII, T.; KONDO, N. Mutation analysis in the iduronate-2-
sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II
(Hunter disease). J. Inher. Metab . Dis. 21:60-70, 1998.
JONES, M.Z.; ALROY, J.; RUTLEDGE, J.C.; TAYLOR, J.W.; ALVORD, E.C. Jr.;
TOONE, J.; APPLEGARTH, D.; HOPWOOD, J.J.; SKUTELSKY, E.; IANELLI, C.;
THORLEY-LAWSON, D.; MITCHELL-HERPOLSHEIMER, C.; ARIAS, A.;
SHARP, P.; EVANS, W.; SILLENCE, D.; CAVANAGH, K.T. Human
mucopolysaccharidosis IIID: clinical, biochemical, morphological and
immunohistochemical characteristics. J. Neuropathol. Exp. Neurol. 56(10):1158-
1167, 1997.

66
JONSSON, J.J.; ARANOVICH, E.L.; BRAUN, S.E.; WHITLEY, C.B. Molecular
diagnosis of Mucopolysaccharidosis type II (Hunter syndrome) by automated
sequencing and computer-assisted interpretation: toward mutation of the
iduronate-2-sulfatase gene. Am. J. Hum. Genet.; 56:597-607, 1995.
KAKKIS, E.; MCENTEE, M.; VOGLER, C.; LE, S.; LEVY, B.; BELICHENKO, P.;
MOBLEY, W.; DICKSON, P.; HANSON, S.; PASSAGE M. Intrathecal enzyme
replacement therapy reduces lysosomal storage in the brain and meninges of the
canine model of MPS I. Mol. Genet. Metab.; 83(1-2):163-74, 2004.
KARAGEORGOS, L.; HARMATZ, P.; SIMON, J.; POLLARD, A.; CLEMENTS, P.R.;
BROOKS, D.A.; HOPWOOD, J.J. Mutational analysis of mucopolysaccharidosis
type VI patients undergoing a trial of enzyme replacement therapy. Hum. Mutat.;
23(3):229-33, 2004.
KARAGEORGOS L.; BROOKS D.A.; HARMATZ P.; KETTERIDGE, D.; POLLARD,
A.; MELVILLE, E.L.; PARKINSON-LAWRENCE, E.; CLEMENTS, P.R.;
HOPWOOD, J.J. Mutational analysis of mucopolysaccharidosis type VI patients
undergoing a phase II trial of enzyme replacement therapy. Mol Genet Metab ,
90:164–70, 2007.
KARSTEN, S.L.; LAGERSTEDT, K.; CARLBERG, B.M.; KLEIJER, W.J.; ZAREMBA,
J.; VAN DIGGELEN, O.P.; CZARTORYSKA, B.; PETTERSSON, U.;
BONDESON, M.L. Two distinct deletions in the IDS gene and the gene W: a
novel type of mutation associated with the Hunter syndrome. Genomics;
43(2):123-9, 1997.
LABRUNE, P.; MELLE, D.; REY, F.; BERTHELON, M.; CAILLAUD, C.; REY, J.;
MUNNICH, A.; LYONNET, S. Single-strand conformational polymorphism for
detection of mutations and base substitutions in phenylketonuria. Am. J. Hum.
Genet. 48(6):1115-1120, 1991.
LAGERSTEDT, K.; KARSTEN, S.L.; CARLBERG, B.M.; KLEIJER, W.J.;
TÖNNESEN, T.; PETTERSSON, U.; BONDESON, M.L. Double-strand breaks
may initiate the inversion mutation causing the Hunter syndrome. Hum Mol
Genet.; 6(4):627-33, 1997.
LI, H.H.; YU, W.H.; ROZENGURT, N.; ZHAO, H.Z.; LYONS, K.M.;
ANAGNOSTARAS, S.; FANSELOW, M.S.; SUZUKI, K.; VANIER, M.T.;

67
NEUFELD, E.F. Mouse model of Sanfilippo syndrome type B produced by
targeted disruption of the gene encoding a-N-acetylglucosaminidase. Proc Natl
Acad Sci U S A, 96(25):14505-14510, 1999.
LIN, S.P.; CHANG, J.H.; LEE-CHEN, G.J.; LIN, H.Y.; CHUANG, C.K. Detection of
Hunter syndrome (mucopolyssacharidosis type II) in Taiwanese: biochemical and
linkage studies of the iduronate-2-sulphatase gene defects in MPS II patients and
carriers. Clin. Chim. Acta, 15;369(1):29-34, 2006.
LIN, W.; LIN, S.; WANG, C.; HWU, W.; CHUANG, C.; LIN, S.; TSAI, Y.; CHEN, C.;
TSAI, F. Genetic analysis of mucopolysaccharidosis type VI in Taiwanese
patients. Clinica Chimica Acta 394:89–93, 2008.
LIPIELLO,L & MANKIN,H.J. Thin-layer chromatographic separation of the isomeric
chondroitin sulfates, dermatan sulfate, and keratan sulfate. Analytical
Biochemistry, 39: 54-58. 1971.
LITJENS, T.; BROOKS, D.A.; PETERS, C.; GIBSON, G.J.; HOPWOOD, J.J
Identification, expression, and biochemical characterization of N-
acetylgalactosamine-4-sulfatase mutations and relationship with clinical
phenotype in MPS-VI patients. Am. J. Hum. Genet . 58:1127-1134, 1996.
MANGAS, M.; NOGUEIRA, C.; PRATA, M.J.; LACERDA, L.; COLL, M.J.; SOARES,
G.; RIBEIRO, G.; AMARAL, O.; FERREIRA, C.; ALVES, C. COUTINHO, M.F.;
ALVES, S. Molecular analysis of mucopolysaccharidosis type IIIB in Portugal:
evidence of a single origin for a common mutation (R234C) in the Iberian
Peninsula. Clin Genet ., 73:251–256, 2008.
MARTIN, R.; BECK, M.; ENG. C.; GIUGLIANI, R.; HARMATZ, P.; MUÑOZ, V.;
MUENZER, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter
Syndrome). Pediatrics , 121(2):e377-86, 2008.
MATTE, U., YOGALINGAM, G.; BROOKS, D.; LEISTNER, S.; SCHWARTZ, I.; LIMA,
L.; NORATO, D.Y.; BRUM, J.M.;BEESLEY, C.; WINCHESTER, B.; GIUGLIANI,
R.; HOPWOOD, J.J. Molecular Identification and characterization of 13 new
mutations in mucopolysaccharidosis type I patients. Genetics and Metabolism
78:37–43, 2003.

68
MCINTYRE, C,; DERRICK ROBERTS, AL,; RANIERI, E,; CLEMENTS, P.R,;
BYERS, S,; ANSON DS. Lentiviral-mediated gene therapy for murine
mucopolysaccharidosis type IIIA. Mol. Genet. Metab.; 93(4):411-8, 2008.
MEIKLE, P.J.; HOPWOOD, J.J.; CLAGUE, A.E.; et al. Prevalence of lysossomal
storage diseases. JAMA , 28:249-254, 1999.
MIEBACH, E. Enzyme replacement therapy in Mucopolysaccharidosis type I. Acta
Paediatr Suppl . 94(447):58-60, 2005.
MUENZER, J. The mucopolysaccharidoses: a heterogeneous group of disorders with
variable pediatric presentations. The Journal of Pediatrics, 144: 27-34. 2004.
MUENZER, J.; WRAITH, J.E.; CLARKE, L.A.; INTERNATIONAL CONSENSUS
PANEL ON MANAGEMENT AND TREATMENT OF
MUCOPOLYSACCHARIDOSIS I. Mucopolysaccharidosis I: management and
treatment guidelines. Pediatrics; 123(1):19-29, 2009.
MUNOZ-ROJAS, M.V.; VIEIRA, T.; COSTA, R.; FAGONDES, S.; JOHN, A.; JARDIM,
L.B.; VEDOLIN, L.M.; RAYMUNDO, M.; DICKSON, P.I.; KAKKIS E.; GIUGLIANI,
R. Intrathecal enzyme replacement therapy in a patient with
mucopolysaccharidosis type I and symptomatic spinal cord compression.
American Journal of Medical Genetics , 146A(19):2538-44, 2008.
NELSON, D.L. & COX, M.M. Lehninger Principles of Biochemistry , 3ª ed. Ed.
Worth Publishers. New York, 2000.
NEUFELD, EF. & MUENZER J. In: Metabolic and Molecular bases of iherited
disease . SCRIVER, C.R.; BEAUDET, A.L.; SLY, W.S.; et al, eds. New York:
McGraw Hill; 2001.
OSHIMA, A.; YOSHIDA, K.; SHIMMOTO, M.; FUKUHARA, Y.; SAKURABA, H.;
SUZUKI, Y. Human beta-galactosidase gene mutations in Morquio B disease.
Am. J. Hum. Genet. 49:1091, 1991.
PETRY, M.F.; NONEMACHER, K.; SEBBEN, J.C.; SCHWARTZ, I.V.; AZEVEDO,
A.C.; BURIN, M.G.; DE REZENDE, A.R.; KIM, C.A.; GIUGLIANI, R.; LEISTNER-
SEGAL, S. Mucopolysaccharidosis type VI: identification of novel mutations on
the arylsulphatase B gene in South American patients. J. Inherit. Metab. Dis .
286:1027–1034, 2005.

69
PONDER, K.P. & HASKINS, M.E. Gene therapy for mucopolysaccharidosis. Expert
Opin Biol Ther. 7(9)1333-45, 2007.
PRYDZ, K. & DALEN, K.T. Synthesis and sorting of proteoglycans. J. Cell. Sci.
113:193-205, 2000.
RATHMANN, M.; BUNGE, S.; BECK, M.; KRESSE, H.; TYLKI-SZYMANSKA, A.;
GAL, A. Mucopolysaccharidosis type II (Hunter syndrome): mutation "hot spots" in
the iduronate-2-sulfatase gene. Am. J. Hum. Genet.; 59(6):1202-9, 1996.
ROHRBACH & CLARKE. Treatment of lysosomal storage disorders : progress with
enzyme replacement therapy. Drugs . 67(18):2697-716, 2007.
SAMBROOK, J. & RUSSEL D.W. Molecular cloning – A Laboratory Manual . 3a.
edição, New York: Cold Spring Harbor Laboratory Press, 2001.
SCHWARTZ, I.V.D.; MATTE, U.S.; ARTIGALAS, O.; BROILLO, F.; BURIN, G.M.;
GIUGLIANI, R. MPS no Brasil: Estudos clínicos e Dados Epidemiológicos
[online]. Disponível em http://www.hcpa.ufrgs.br/genetica,
gené[email protected], 2002.
SCOTT, H.S., GUO, X-H, HOPWOOD, J.J,; MORRIS, C.P. Structure and sequence
of the human a-L-iduronidase gene. Genomics 13:1311-1313, 1990.
SCOTT, H.S.; LITJENS, T.; HOPWOOD, J.J.; MORRIS, C.P. A common mutation for
mucopolysaccharidosis type I associated with a severe Hurler syndrome
phenotype. Human Mutation , 1:103-108, 1992.
SCRIVER C.R..; BEAUDET A.L..; SLY, W.S. & VALLE, D. The metabolic and
Molecular Basis of Inherited Disease , 8th ed. New York: McGraw-Hill, Inc; 2001.
SIDDHARTH, L.; SHARMA, A.; SHRIVASTAVA, G.P.; JAIN, S.K. Maroteaux-Lamy
Syndrome. Indian Journal of Pediatrics . 71:933-936, 2004.
SIFUENTES, M; DOROSHOW, R; HOFT, R; MASON, G; WALOT, I; DIAMENT, M;
OKASAKI, S; HUFF, K; COX, GF; SWIDLER, SJ; KAKKIS E. A follow-up study of
MPS I patients treated with laronidase enzyme replacement therapy for 6 years.
Mol Gen. Metabolic , 2006.
SIMONARO CM, HASKINS ME, SCHUCHMAN EH. Articular chondrocytes from
animals with a dermatan sulfate storage disease undergo a high rate of apoptosis

70
and release nitric oxide and inflammatory cytokines: a possible mechanism
underlying degenerative joint disease in the mucopolysaccharidoses. Lab Invest
81:1319-28. 2001.
SOLYON, E. Incidence data for Mucopolysaccharidoses in Hungary. In: 4th
International Symposium on Mucopolysaccharide and r elated diseases
program. Wollongong (Austrália), p.75, 1996.
SPRANGERS, B.; VAN WIJMEERSCH, B.; LUYCKX, A.; SAGAERT, X.; DE
SOMER, L.; RUTGEERTS, O.; LENAERTS, C.; LANDUYT, W.; BOECKX, N.;
DUBOIS, B.; DE WOLF-PEETERS, C.; WAER, M.; BILLIAU, A.D. Allogeneic
bone marrow transplantation and donor lymphocyte infusion in a mouse model of
irradiation-induced myelodysplastic/myeloproliferation syndrome (MD/MPS):
evidence for a graft-versus-MD/MPS effect. Leukemia; 23(2):340-9, 2008.
SUKEGAWA-HAYASAKA, K.; KATO, Z.; NAKAMURA, H.; TOMATSU, S.; FUKAO,
T.; ORII, T.; KONDO, N. Effect of Hunter disease (mucopolysaccharidosis on
molecular phenotypes of iduronate-2-sulphatase: Enzimatic activity, protein
processing and structural analysis. J. Inherited Metab. Dis , 29:755-761, 2006.
TANAKA, A.; KIMURA, M.; LAN, H.T.N.; TAKAURA, N.; YAMANO, T. Molecular
analysis of the α-N-acetylglucosaminidase gene in seven Japanese patients from
six unrelated families with mucopolysaccharidosis IIIB (Sanfilippo type B),
including two novel mutations. J. Hum. Genet. 47:484-487, 2002.
TESSITORE, A.; VILLANI, G.R.D.; DOMENICO C.D.; FILOCAMO, M.; GATTI, R.; DI
NATALE, P. Molecular defects in the α-N-acetylglucosaminidase gene in Italian
Sanfilippo type B patients. Hum Genet , 107 :568–576, 2000.
THOMAS, G.H. & HOWELL,R.R. Selected screening tests fot genetic metabolic
diseases. Chicago, Year Book Medical Publishers Inc,pag 36, 1973.
THOMAS, J. A.; JACOBS, S.; KIERSTEIN, J.; VAN HOVE, J. Outcome after three
years of laronidase enzyme replacement therapy in a patient with Hurler
syndrome. J. Inherit. Metab. Dis. , 29:762, 2006.
TIMMS, K.M.; LU, F.; SHEN, Y.; PIERSON, C.A.; MUZNY, D.M.; GU, Y.; et al. 130
kb of DNA sequence reveals two new genes and a regional duplication distal to
the human iduronate-2-sulfate sulfatase locus. Genome Res ; 5: 71–8, 1995.

71
TOLAR, J.; ORCHARD, P.J. Alpha-L-iduronidase therapy for mucopolysaccharidosis
type I. Biologics; 2(4):743-51, 2008.
TOMATSU, S.; ORII, K.O.; BI, Y.; GUTIERREZ, M.A.; NISHIOKA, T.; YAMAGUCHI,
S.; KONDO, N.; ORII, T.; NOGUCHI, A.; SLY, W.S. General implications for CpG
hot spot mutations: methylation patterns of the human iduronate-2-sulfatase gene
locus. Hum. Mutat.; 23(6):590-8, 2004.
TRIGGS-RAINE, B.; SALO, T.J.; ZHANG, H.; WICKLOW, B.A.; NATOWICZ, M.R.
Mutations in HYAL1, a member of a tandemly distributed multigene family
encoding disparate hyaluronidase activities, cause a newly described lysosomal
disorder, mucopolysaccharidosis IX. Proc. Nat.l Acad. Sci. USA ; 96(11):6296-
300, 1999.
VAN DE KAMP, J.J.; NIERMEIJER, M.F.; VON FIGURA, K.; GIESBERTS, M.A.
Genetic heterogeneity and clinical variability in the Sanfilippo syndrome (types A,
B, and C). Clin. Genet. 20(2):152-160, 1981.
VENTURI, N.; ROVELLI, A.; PARINI, R.; MENNI, F.; BRAMBILLASCA, F.;
BERTAGNOLIO, F.; UZIEL, G.; GATTI, R.; FILOCAMO, M.; DONATI, M.A.;
BIONDI, A.; GOLDWURM, S. Molecular Analysis of 30 Mucopolysaccharidosis
Type I Patients: Evaluation of the Mutational Spectrum in Italian Population and
Identification of 13 Novel Mutations. Human Mutation, Mutation in Brief #522,
2002.
VIEIRA, T.; SCHWARTZ, I.; MUÑOZ, V.; PINTO, L.; STEINER, C.; RIBEIRO, M.;
BOY, R.; FERRAZ, V.; DE PAULA, A.; KIM, C.; ACOSTA, A.; GIUGLIANI, R.
Mucopolysaccharidoses in Brazil: what happens from birth to biochemical
diagnosis? American Journal of Medical Pediatrics . 146A(13):1741-7,
2008.
VILLANI, G.R.D.; BALZANO, N.; DI NATALE, P. Two novel mutations of the
arylsulfatase B gene in two Italian patients with severe form of
mucopolysaccharidosis, Hum. Mutat . 11:410, 1998.
VOGLER, C.; BARKER, J.; SANDS, M.S.; LEVY, B.; GALVIN, N.; SLY, W.S. Murine
mucopolysaccharidosis VII: impact of therapies on the phenotype, clinical course,

72
and pathology in a model of a lysosomal storage disease. Pediatr. Dev. Pathol. 4,
421–33, 2001.
VOGLER, C.; LEVY, B.; GALVIN, N.; LESSARD, M.; SOPER, B.; BARKER, J. Early
onset of lysosomal storage disease in murine model of mucopolysaccharidosis
type VII: undegraded substrate accumulates in many tissues in the fetus and very
young MPS VII mouse. Pediatr. Devel. Pathol. , 8(4), 453-462, 2005.
WAJNER, M.; VARGAS, R.C.; BURN, G.M.; GIUGLIANI, R. & COELHO, J.C.
Investigação de Erros Inatos do Metabolismo. Revista do Hospital de Clínicas
de Porto Alegre 21 (3): 343-360, 2001.
WALKLEY, S.U. Pathologic cascades and brain dysfunction. In: Lysosomal
Disorders of the Brain. (Platt, F.M. and Walkley, S.U., eds), pp 290-324, Ed.:
Oxford, 2004.
WANG, D.; WORSHAM, D.N.; PAN, D. Co-expression of MGMT(P140K) and alpha-
L-iduronidase in primary hepatocytes from mucopolysaccharidosis type I mice
enables efficient selection with metabolic correction. J. Gene. Med.; 10(3):249-
59, 2008.
WEBER, B.; BLANCH, L.; CLEMENTS, P.R.; SCOTT, H.S.; HOPWOOD, J.J.
Cloning and expression of the gene involved in Sanfilippo B syndrome
(mucopolysaccharidosis III B). Hum. Mol. Genet . 5:771-777, 1996.
WICKER, G.; PRILL, V.; BROOKS, D.; GIBSON, G.; HOPWOOD, J.; VON FIGURA,
K.; PETERS, C. Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). An
intermediate clinical phenotype caused by substitution of valine for glycine at
position 137 of arylsulfatase B. J. Biol. Chem.; 266(32):21386-91, 1991.
WILSON, P.J.; MEANEY, C.A.; HOPWOOD, J.J.; MORRIS, C.P.; Sequence of the
human iduronate 2-sulfatase (IDS) gene. Genomics , 17: 773–5, 1993.
WHITLEY CB, DRAPER KA, DUTTON CM, BROWN PA, SEVERSON SL, FRANCE
LA. Diagnostic test for mucopolysaccharidosis II. Rapid quantification of
glycosaminoglycan in urine samples collected on a paper matrix. Clin Chem
35:2074-81. 1989.
WHITLEY CB, SPIELMAN RC, HERRO G, SEVERSON ST. Urinary
glycosaminoglycan excretion quantified by an automated semimicro method in

73
specimens conveniently transported from around the globe. Mol Genet Metabol
75:56-64. 2002.
WRAITH, J.E.; BECK, M.; LANE, R.; VAN DER PLOEG, A.; SHAPIRO, E.; XUE, Y.;
KAKKIS, E.D.; GUFFON, N. Enzyme replacement therapy in patients who have
mucopolysaccharidosis I and are younger than 5 years: results of a multinational
study of recombinant human a-L-iduronidase (Laronidase). Pediatrics
120(1):e37-e46, 2007.
WRAITH, J.E.; BECK, M.; GIUGLIANI, R.; CLARKE, J.; MARTIN, R.; HOS
INVESTIGATORS. Initial report from the Hunter Outcome Survey. Genet. Med.
10(7):508-516, 2008.
YAMAGISHI. A.; TOMATSU, S.; FUKUDA, S.; UCHIYAMA, A.; SHIMOZAWA, N.;
SUZUKI, Y.; KONDO, N.; SUKEGAWA, K.; ORII, T. Mucopolysaccharidosis type
I: identification of common mutations that cause Hurler and Scheie syndromes in
Japanese populations. Human Mutation , 7(1):23-9, 1996.
YOGALINGAM, G.; WEBER, B.; MEEHAN, J.; ROGERS, J.; HOPWOOD, J.J.
Mucopolysaccharidosis type IIIB: characterization and expression of wild-type and
mutant recombinant alpha-N-acetylglucosaminidase and relationship with
Sanfilippo phenotype in an attenuated patient. Biochim. Biophys. Acta.
1502:415-425, 2000.
ZHAO, H.G.; LI, H.H.; BACH, G.; SCHMIDTCHEN, A.; NEUFELD, E.F. The
molecular basis of Sanfilippo syndrome type B, Proc. Natl. Acad. Sci. USA 93
6101-6105, 1996.
ZHENG, Y.; RYAZANTSEV ,S.; OHMI, K.; ZHAO, H.Z.; ROZENGURT, N.; KOHN,
D.B.; NEUFELD, E.F. Retrovirally transduced bone marrow has a therapeutic effect
on brain in the mouse model of mucopolysaccharidosis IIIB. Mol. Genet. Metabol.;
82(4):286-95, 2004.

74
8. ANEXOS 8.1 Carta de aceite do Comitê de Ética em Pesquisa com Seres Humanos

75
8.2 Termo de Consentimento Livre e Esclarecido
INFORMAÇÕES PARA OS PAIS OU RESPONSÁVEIS LEGAIS
TÍTULO DO ESTUDO: “CARACTERIZAÇÃO MOLECULAR E BIOQU ÍMICA DE
PACIENTES COM MUCOPOLISSACARIDOSES”
O QUE SÃO MUCOPOLISSACARIDOSES: Mucopolissacaridoses são doenças genéticas
raras, causadas pela ausência ou insuficiência de enzimas responsáveis pela quebra dos
mucopolissacarídeos. Também são conhecidas por: MPS, Síndrome de Hurler (MPS-IH),
Síndrome de Scheie (MPS-IS), Síndrome de Hurler-Scheie (MPS-IHS), Síndrome de Hunter
(MPS-II), Síndrome de Sanfilippo (MPS-III), Síndrome de Morquio (MPS-IV), Síndrome de
Maroteaux-Lamy (MPS-VI) e Síndrome de Sly (MPS-VII). Os mucopolissacarídeos -
atualmente denominados GAGs ou glicosaminoglicanos - não são corretamente processados
nem eliminados pelo organismo. O acúmulo dessas substâncias causa distúrbios, incluindo o
progressivo mau funcionamento físico e/ou mental. Os sintomas podem aparecer nos
primeiros meses de vida ou demorar alguns anos, a medida em que mais e mais células são
danificadas.
QUAIS AS CONSEQUÊNCIAS DESSA DOENÇA?: Uma das características de muitas
dessas síndromes pode ser observada nas mãos que ficam com dedos "encurvados" para
dentro e "grossos". São freqüentes algumas alterações no rosto, enrijecimento das
articulações, dificuldades respiratórias e cardíacas, alterações no crescimento, deformações
ósseas, aumento do fígado e baço e das mucosas em geral. Deficiência visual, auditiva e
retardamento mental também podem ocorrer.
COMO O MÉDICO FAZ O DIAGNÓSTICO?: O diagnóstico para MPS é realizado
através de uma triagem inicial na urina do paciente. Caso a triagem detecte alterações
indicativas de uma excreção excessiva de GAG (característico em pacientes com MPS), testes
mais específicos (ex.: dosagem enzimática em leucócitos do sangue periférico) são realizados
para confirmação do diagnóstico.

76
COMO SE TRATA?: A Terapia de Reposição Enzimática (TER) consiste no principal
tratamento para pacientes com MPS. O tratamento, entretanto, só está disponível atualmente
apenas para alguns tipos de MPS (MPS I, II e VI). O procedimento terapêutico consiste na
administração semanal de uma enzima recombinante intravenosamente. Esta enzima irá atuar
de forma semelhante à enzima deficiente, causando um melhora significativa do quadro
clínico geral do paciente, apesar da enzima recombinante não possuir efeito na reversão de
quadros com comprometimento ósseo e/ou neurológico. O controle do tratamento consiste na
medida semanal dos níveis de GAG na urina do paciente em tratamento.
POR QUE ESTE ESTUDO ESTÁ SENDO REALIZADO?: O objetivo desse estudo é
identificar a causa genética (mutações – alterações no DNA) e sua possível correlação com os
sintomas clínicos apresentados pelos pacientes com MPS.
COMO E ONDE ESTE ESTUDO SERÁ REALIZADO?: Deverão participar deste estudo
pacientes do Estado do Pará. Os pacientes selecionados serão solicitados a fornecer
informações sobre sua história médica. Se você permitir, estas informações poderão ser
obtidas através do seu médico ou de registros médicos hospitalares. O seguinte procedimento
será realizado: Coleta de sangue (5 a 10mL) e Coleta de urinas (30 mL): O material
coletado será processado e analisado no Laboratório de Erros Inatos do Metabolismo da
UFPA.
QUAIS OS RISCOS DESTE ESTUDO? A coleta de sangue poderá causar um desconforto
temporário por causa da picada de agulha, hematoma e raramente, infecção. Às vezes, uma
pessoa pode ficar tonta ou desmaiar quando o sangue for coletado.
QUAIS SÃO OS BENEFÍCIOS DESTE ESTUDO?: Como esse diagnóstico, poderá ser
possível estabelecer correlação com a gravidade da doença. Além disso, para as famílias
interessadas, este estudo pode ser oferecido para encontrar pessoas portadoras da mutação, ou
seja, identificar pessoas da mesma família que têm o risco de ter um filho com MPS.
POSSO RECUSAR A PARTICIPAÇÃO? Seu filho não é obrigado a participar deste
estudo. A sua participação neste estudo é voluntária. A recusa em participar não terá
conseqüências para os seus cuidados presentes e futuros. Seu filho poderá se retirar do estudo
a qualquer momento.
TENHO QUE PAGAR PARA PARTICIPAR? Não há nenhum custo para participar dessa
pesquisa. Seu filho não será pago para participar deste estudo.
E SE EU / MEU FILHO FOR PREJUDICADO?: O Dr. Luiz Carlos Santana da Silva
deverá ser notificado se você suspeitar que seu filho foi prejudicado por estar no estudo.

77
AS INFORMAÇÕES SOBRE MEU FILHO SE TORNARÃO PÚBLICAS ? A identidade
de seu filho e outras informações pessoais obtidas neste estudo serão confidenciais.
Informações científicas e médicas obtidas neste estudo, das quais a identidade de seu filho não
poderá ser revelada, deverão ser apresentadas em encontros e publicadas a fim de tornar as
informações obtidas neste estudo de benefício para os outros.
COM QUEM POSSO TIRAR DÚVIDAS?: Você está livre para fazer perguntas sobre este
estudo. Quando você tiver dúvidas relacionadas a este estudo, poderá falar com: Dr. Luiz
Carlos Santana da Silva e/ou Mestrando Gustavo Monteiro Viana.
Lab. Erros Inatos do Metabolismo, Instituto de Ciências Biológicas. UFPA. Av. Augusto
Correa, S/N, Guamá, CEP. 666075900 Belém-Pa. Fone: 32018030; e-mail: [email protected]

78
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO (PACIENTE)
Eu manifesto meu
consentimento com envolvimento do meu(minha) filho(a) no projeto de pesquisa intitulado:
“CARACTERIZAÇÃO MOLECULAR E BIOQUÍMICA DE PACIENTES COM
MUCOPOLISSACARIDOSES”.
1. A natureza e objetivo do projeto de pesquisa, descritos na folha de informação em anexo,
foram explicadas a mim. Eu compreendo e concordo em participar.
2. Eu compreendo que meu (minha) filho (a) poderá não ter benefício direto por participar
do estudo
3. Eu entendo que os possíveis riscos e/ou efeitos adversos, desconforto e inconveniências,
como foi destacado na folha de informações, foram explicadas a mim.
4. Eu compreendo que, apesar das informações obtidas no estudo poderem ser publicadas,
elas serão confidenciais e meu (minha) filho(a) não será identificado(a) a partir delas.
5. Eu compreendo que posso retirar meu (minha) filho(a) do estudo em qualquer etapa e que
isto não irá afetar os cuidados médicos ou quaisquer outros aspectos da relação recebidos
a ele(a).
6. Eu compreendo que não haverá pagamento para meu(minha) filho(a) por participar deste
estudo.
7. Eu tive a oportunidade de discutir a participação de meu(minha) filho(a) neste projeto de
pesquisa com um membro da família ou amigo e/ou tive a oportunidade de ter um
membro da família ou amigo presente enquanto o projeto de pesquisa estava sendo
explicado pelo pesquisador.
8. Eu estou ciente de que devo guardar uma cópia do Termo de Consentimento, quando
completo, e da folha de Informações.
9. Eu concordo que o material (sangue e urina) coletado de meu(minha) filho(a) seja
utilizado no projeto acima.
Assinatura do Responsável:
Relação de parentesco com o paciente:
Nome completo do paciente:
Data: __ / __ / __

79
TERMO DE CONSENTIMENTO LIVRE E ESCLARECIDO
(FAMILIAR DO PACIENTE)
Eu manifesto meu
consentimento em participar do projeto de pesquisa intitulado: “CARACTERIZAÇÃO
MOLECULAR E BIOQUÍMICA DE PACIENTES COM MUCOPOLISSACARIDOSES”.
1. A natureza e objetivo do projeto de pesquisa, descritos na folha de informação em anexo,
foram explicadas a mim. Eu compreendo e concordo em participar.
2. Eu compreendo que poderei não ter benefício direto por participar do estudo.
3. Eu entendo que os possíveis riscos e/ou efeitos adversos, desconforto e inconveniências,
como foi destacado na folha de informações, foram explicadas a mim.
4. Eu compreendo que, apesar das informações obtidas no estudo poderem ser publicadas,
elas serão confidenciais e eu não serei identificado(a) a partir delas.
5. Eu compreendo que posso me retirar do estudo em qualquer etapa e que isto não irá afetar
os cuidados médicos ou quaisquer outros aspectos da relação recebidos.
6. Eu compreendo que não haverá pagamento em nenhuma espécie por participar deste
estudo.
7. Eu tive a oportunidade de discutir a minha participação neste projeto de pesquisa com um
membro da família ou amigo e/ou tive a oportunidade de ter um membro da família ou amigo
presente enquanto o projeto de pesquisa estava sendo explicado pelo pesquisador.
8. Eu estou ciente de que devo guardar uma cópia do Termo de Consentimento, quando
completo, e da folha de Informações.
9. Eu concordo com a utilização do meu material biológico (sangue) como amostra do projeto
acima citado.
Assinatura do Participante:
Nome do paciente do qual possui parentesco:
Grau de parentesco:
Data: __ / __ / __

Livros Grátis( http://www.livrosgratis.com.br )
Milhares de Livros para Download: Baixar livros de AdministraçãoBaixar livros de AgronomiaBaixar livros de ArquiteturaBaixar livros de ArtesBaixar livros de AstronomiaBaixar livros de Biologia GeralBaixar livros de Ciência da ComputaçãoBaixar livros de Ciência da InformaçãoBaixar livros de Ciência PolíticaBaixar livros de Ciências da SaúdeBaixar livros de ComunicaçãoBaixar livros do Conselho Nacional de Educação - CNEBaixar livros de Defesa civilBaixar livros de DireitoBaixar livros de Direitos humanosBaixar livros de EconomiaBaixar livros de Economia DomésticaBaixar livros de EducaçãoBaixar livros de Educação - TrânsitoBaixar livros de Educação FísicaBaixar livros de Engenharia AeroespacialBaixar livros de FarmáciaBaixar livros de FilosofiaBaixar livros de FísicaBaixar livros de GeociênciasBaixar livros de GeografiaBaixar livros de HistóriaBaixar livros de Línguas

Baixar livros de LiteraturaBaixar livros de Literatura de CordelBaixar livros de Literatura InfantilBaixar livros de MatemáticaBaixar livros de MedicinaBaixar livros de Medicina VeterináriaBaixar livros de Meio AmbienteBaixar livros de MeteorologiaBaixar Monografias e TCCBaixar livros MultidisciplinarBaixar livros de MúsicaBaixar livros de PsicologiaBaixar livros de QuímicaBaixar livros de Saúde ColetivaBaixar livros de Serviço SocialBaixar livros de SociologiaBaixar livros de TeologiaBaixar livros de TrabalhoBaixar livros de Turismo
Top Related