![Large scale computational motif finding · ENCODE project: GENCODE consensus human gene set GENCODE [ENCODE] Transcription Tom Gingeras/ENCODE Structural Biology EU Biosapiens Nomenclature](https://static.fdocuments.net/doc/165x107/5faa836c4070c305dd409a12/large-scale-computational-motif-finding-encode-project-gencode-consensus-human.jpg)







Languages
Pages
Legal


Biological Motif Discovery
ConceptsMotif Modeling and Motif InformationEM and Gibbs SamplingComparative Motif Prediction
ApplicationsTranscription Factor Binding Site PredictionEpitope Prediction
Lab PracticalDNA Motif Discovery with MEME and AlignAceCo-regulated genes from TB Boshoff data set

Regulatory Motifs
Find promoter motifs associated with co-regulated or functionally related genes

Transcription Factor Binding Sites
• Very Small
• Highly Variable
• ~Constant Size
• Often repeated
• Low-complexity-ish
Slide Credit: S. Batzoglou

Other “Motifs”
• Splicing Signals– Splice junctions– Exonic Splicing Enhancers (ESE)– Exonic Splicing Surpressors (ESS)
• Protein Domains– Glycosylation sites– Kinase targets– Targetting signals
• Protein Epitopes– MHC binding specificities

• Modeling Motifs– How to computationally represent motifs
• Visualizing Motifs– Motif “Information”
• Predicting Motif Instances– Using the model to classify new sequences
• Learning Motif Structure– Finding new motifs, assessing their quality
Essential Tasks

Modeling Motifs

Consensus Sequences
Useful for publication
IUPAC symbols for degenerate
sites
Not very amenable to computation
Nature Biotechnology 24, 423 - 425 (2006)

Probabilistic Model
.2
.2
.5
.1
.7.2.2.1.3
.1.2.4.5.4
.1.2.2.2.2
.1.4.1.2.1ACGT
M1 MKM1
Pk(S|M)
Position FrequencyMatrix (PFM)
1 K Count frequenciesAdd pseudocounts

Scoring A Sequence
11
( | ) ( | )( | )log log log
( | ) ( | ) ( | )
N Ni i i i
ii i i
P S PFM P S PFMP S PFMScore
P S B P S B P S B
To score a sequence, we compare to a null model
A: 0.25
T: 0.25
G: 0.25
C: 0.25
A: 0.25
T: 0.25
G: 0.25
C: 0.25
Background DNA (B)
.2
.2
.5
.1
.7.2.2.1.3
.1.2.4.5.4
.1.2.2.2.2
.1.4.1.2.1ACGT
Log likelihoodratio
-0.3
-0.3
1
-1.3
1.4-0.3-0.3-1.30.3
-1.3-0.30.610.6
-1.3-0.30.3-0.3-0.3
-1.30.6-1.3-0.3-1.3ACGT
Position WeightMatrix (PWM)PFM

Scoring a Sequence
MacIsaac & Fraenkel (2006) PLoS Comp Bio
Common threshold = 60% of maximum score

Visualizing Motifs – Motif Logos
Represent both base frequency and conservation at each position
Height of letter proportionalto frequency of base at that position
Height of stack proportionalto conservation at that position

Motif InformationThe height of a stack is often called the motif information at
that position measured in bits
{ , , , }
Motif Position Information 2 logb bb A T G C
p p
Info
rmat
ion
Why is this a measure of information?

1Uncertainty = log
eventpUncertainty = - log eventp
1Uncertainty
eventp
Uncertainty and probability
“The sun will rise tomorrow”
“The sun will not rise tomorrow”
Uncertainty is inversely related to probability of event
Not surprising (p~1)
Very surprising (p<<1)
Uncertainty is related to our surprise at an event

Average Uncertainty
A “The sun will rise tomorrow”
B “The sun will not rise tomorrow”
P(A)=p1
P(B)=p2
Two possible outcomes for sun rising
1 1 2 2
( )Uncertainty(A) ( )Uncertainty(B)
log log
logi i
P A P B
p p p p
p p
= Entropy
What is our average uncertainty about the sun rising

Entropy
Entropy measures average uncertainty
Entropy measures randomness
If log is base 2, then the units are called bits
2( ) logi ii
H X p p

Entropy versus randomness
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 10
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
Entropy is maximum at maximum randomness
P(heads)
Example: Coin Toss
En
tro
py
P(heads)=0.1 Not very randomH(X)=0.47 bits
P(heads)=0.5 Completely randomH(X)=1 bits

Entropy Examples
( ) [0.25log(0.25) 0.25log(0.25)
0.25log(0.25) 0.25log(0.25)]
2 bits
H X
1 2 3 40
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
P(x
)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1 2 3 4
P(x
) ( ) [0.1log(0.1) 0.1log(0.1)
0.1log(0.1) 0.75log(0.75)]
0.63 bits
H X
A T G C
A T G C

Information Content
Information is a decrease in uncertainty
Once I tell you the sun will rise, your uncertainty aboutthe event decreases
Hbefore(X) Hafter(X)-Information =
Information is difference in entropy after receiving information

Motif Information
2 -Motif Position Information ={ , , , }
logb bb A T G C
p p
Hbackground(X) Hmotif_i(X)
Prior uncertainty aboutnucleotide
Uncertainty after learning it isposition i in a motif
H(X)=2 bits
A T G C0
0.10.20.30.40.50.60.70.80.9
1
P(x
)
H(X)=0.63 bits
A T G C0
0.10.20.30.40.50.60.70.80.9
1
P(x
)
Uncertainty at this position has been reduced by 0.37 bits

Motif Logo
Conserved ResidueReduction of uncertainty
of 2 bits
Little ConservationMinimal reduction of
uncertainty

{ , , , }
logb bb A T G C
p p
Background DNA Frequency
2 -Motif Position Information =
Hmotif_i(X)
H(X)=1.9 bitsA T G C
00.10.20.30.40.50.60.70.80.9
1
P(x
)
Some motifs could have negative information!
= -0.2 bits
The definition of information assumes a uniform background DNA nucleotide frequency
What if the background frequency is not uniform?
Hbackground(X)
H(X)=1.7 bitsA T G C
00.10.20.30.40.50.60.70.80.9
1
P(x
)
(e.g. Plasmodium)
1.7

A Different MeasureRelative entropy or Kullback-Leibler (KL) divergence
Divergence between a “true” distribution and another
{ , , , }
( )( || ) ( ) log
( )motif
KL motif background motifi A T G C background
P iD P P P i
P i
“True” Distribution Other Distribution
DKL is larger the more differentPmotif is from Pbackground
Same as Information ifPbackground is uniform
Properties
motif backgroundif and only if P =P
0
0
( || ) ( || )
KL
KL
KL KL
D
D
D P Q D Q P

Comparing Both Methods
Information assuminguniform background
DNA
KL Distance assuming20% GC content
(e.g. Plasmodium)

Online Logo Generation
http://weblogo.berkeley.edu/ http://biodev.hgen.pitt.edu/cgi-bin/enologos/enologos.cgi

Finding New Motifs
Learning Motif Models

A Promoter ModelLength K
Motif
The same motif model in all promoters
.2
.2
.5
.1
.1.2.2.1.3
.2.2.4.5.4
.4.2.2.2.2
.3.4.1.2.1ACGT
M1 MKM1
Background DNA
A: 0.25
T: 0.25
G: 0.25
C: 0.25
A: 0.25
T: 0.25
G: 0.25
C: 0.25
P(S|B)Pk(S|M)

Probability of a Sequence
1 60 65 100
Given a sequence(s), motif model and motif location
59 6
631
100
1 66
|( | 10, ) ( | ) ( | )i ii
ki
kk
P Seq Mstart Model P S B PP SS M B
Si = nucleotide at position i in the sequence
A T A T G C
.2
.2
.5
.1
.1.2.2.1.3
.2.2.4.5.4
.4.2.2.2.2
.3.4.1.2.1ACGT
M1 MKM1

Parameterizing the Motif ModelGiven multiple sequences and motif locations but no motif model
A
C
G
T
M1 M6M1
3/4
3/4
ETC…
AATGCGATATGGATATCGGATGCA
Count Frequencies
Add pseudocounts

Finding Known MotifsGiven multiple sequences and motif model but no motif locations
x
x
x
xP(Seqwindow|Motif)
window
Calculate P(Seqwindow|Motif) for every starting location
Choose best starting location in each sequence

Discovering Motifs
Given a set of co-regulated genes, we need to discoverwith only sequences
We have neither a motif model nor motif locationsNeed to discover both
How can we approach this problem?
(Hint: start with a random motif model)

Expectation Maximization (EM)
Remember the basic idea!
1.Use model to estimate (distribution of) missing data2.Use estimate to update model
3.Repeat until convergence
Model is the motif model
Missing data are the motif locations

EM for Motif Discovery
1. Start with random motif model
2. E Step: estimate probability of motif positions for each sequence
3. M Step: use estimate to update motif model
4. Iterate (to convergence)
.2
.2
.5
.1
.1.2.2.1.3
.2.2.4.5.4
.4.2.2.2.2
.3.4.1.2.1ACGT
.2
.2
.5
.1
.1.2.2.1.3
.1.5.4.5.4
.1.2.2.3.2
.3.1.1.1.1ACGT
ETC…
At each iteration, P(Sequences|Model) guaranteed to increase

MEME
http://meme.sdsc.edu/meme/
• MEME - implements EM for motif discovery in DNA and proteins
• MAST – search sequences for motifs given a model

P(Seq|Model) Landscape
P(S
eq
ue
nc
es
|pa
ram
s1
,pa
ram
s2
)
EM searches for parameters to increase P(seqs|parameters)
Useful to think of P(seqs|parameters)
as a function of parameters
Parameter1 Parameter2
EM starts at an initial set ofparameters
And then “climbs uphill” until it reaches a local maximum
Where EM starts can make a big difference

Search from Many Different Starts
P(S
eq
ue
nc
es
|pa
ram
s1
,pa
ram
s2
)
To minimize the effects of local maxima, you should searchmultiple times from different starting points
Parameter1 Parameter2
MEME uses this idea
Start at many points
Run for one iteration
Choose starting point that gotthe “highest” and continue

Gibbs Sampling
A stochastic version of EM that differs from deterministic EM in two key ways
1. At each iteration, we only update the motif positionof a single sequence
2. We may update a motif position to a “suboptimal” new position

1. Start with random motif locations and calculate a motif model
2. Randomly select a sequence, remove its motif and recalculate tempory model
3. With temporary model, calculate probability of motif at each position on sequence
4. Select new position based on this distribution
5. Update model and Iterate
Gibbs Sampling
.2
.2
.5
.1
.1.2.2.1.3
.2.2.4.5.4
.4.2.2.2.2
.3.4.1.2.1ACGT
.2
.2
.5
.1
.1.2.2.1.3
.1.5.4.5.4
.1.2.2.3.2
.3.1.1.1.1ACGT
ETC…
“Best”Location New
Location

Gibbs Sampling and Climbing
P(S
eq
ue
nc
es
|pa
ram
s1
,pa
ram
s2
)
Because gibbs sampling does always choose the best new locationit can move to another place not directly uphill
Parameter1 Parameter2
In theory, Gibbs Sampling less likely to get stuck a local maxima

AlignACE
http://atlas.med.harvard.edu/cgi-bin/alignace.pl
• Implements Gibbs sampling for motif discovery– Several enhancements
• ScanAce – look for motifs in a sequence given a model
• CompareAce – calculate “similarity” between two motifs (i.e. for clustering motifs)

Assessing Motif QualityScan the genome for all instances and associate
with nearest genes
• Category Enrichment – look for association between motif and sets of genes. Score using Hypergeometric distribution– Functional Category
– Gene Families
– Protein Complexes
• Group Specificity – how restricted are motif instances to the promoter sequences used to find the motif?
• Positional Bias – do motif instances cluster at a certain distance from ATG?
• Orientation Bias – do motifs appear preferentially upstream or downstream of genes?

Comparative Motif Prediction

Kellis et al. (2003) Nature

Scer TTATATTGAATTTTCAAAAATTCTTACTTTTTTTTTGGATGGACGCAAAGAAGTTTAATAATCATATTACATGGCATTACCACCATATACA Spar CTATGTTGATCTTTTCAGAATTTTT-CACTATATTAAGATGGGTGCAAAGAAGTGTGATTATTATATTACATCGCTTTCCTATCATACACA Smik GTATATTGAATTTTTCAGTTTTTTTTCACTATCTTCAAGGTTATGTAAAAAA-TGTCAAGATAATATTACATTTCGTTACTATCATACACA Sbay TTTTTTTGATTTCTTTAGTTTTCTTTCTTTAACTTCAAAATTATAAAAGAAAGTGTAGTCACATCATGCTATCT-GTCACTATCACATATA * * **** * * * ** ** * * ** ** ** * * * ** ** * * * ** * * *
Scer TATCCATATCTAATCTTACTTATATGTTGT-GGAAAT-GTAAAGAGCCCCATTATCTTAGCCTAAAAAAACC--TTCTCTTTGGAACTTTCAGTAATACGSpar TATCCATATCTAGTCTTACTTATATGTTGT-GAGAGT-GTTGATAACCCCAGTATCTTAACCCAAGAAAGCC--TT-TCTATGAAACTTGAACTG-TACGSmik TACCGATGTCTAGTCTTACTTATATGTTAC-GGGAATTGTTGGTAATCCCAGTCTCCCAGATCAAAAAAGGT--CTTTCTATGGAGCTTTG-CTA-TATGSbay TAGATATTTCTGATCTTTCTTATATATTATAGAGAGATGCCAATAAACGTGCTACCTCGAACAAAAGAAGGGGATTTTCTGTAGGGCTTTCCCTATTTTG ** ** *** **** ******* ** * * * * * * * ** ** * *** * *** * * *
Scer CTTAACTGCTCATTGC-----TATATTGAAGTACGGATTAGAAGCCGCCGAGCGGGCGACAGCCCTCCGACGGAAGACTCTCCTCCGTGCGTCCTCGTCTSpar CTAAACTGCTCATTGC-----AATATTGAAGTACGGATCAGAAGCCGCCGAGCGGACGACAGCCCTCCGACGGAATATTCCCCTCCGTGCGTCGCCGTCTSmik TTTAGCTGTTCAAG--------ATATTGAAATACGGATGAGAAGCCGCCGAACGGACGACAATTCCCCGACGGAACATTCTCCTCCGCGCGGCGTCCTCTSbay TCTTATTGTCCATTACTTCGCAATGTTGAAATACGGATCAGAAGCTGCCGACCGGATGACAGTACTCCGGCGGAAAACTGTCCTCCGTGCGAAGTCGTCT ** ** ** ***** ******* ****** ***** *** **** * *** ***** * * ****** *** * ***
Scer TCACCGG-TCGCGTTCCTGAAACGCAGATGTGCCTCGCGCCGCACTGCTCCGAACAATAAAGATTCTACAA-----TACTAGCTTTT--ATGGTTATGAASpar TCGTCGGGTTGTGTCCCTTAA-CATCGATGTACCTCGCGCCGCCCTGCTCCGAACAATAAGGATTCTACAAGAAA-TACTTGTTTTTTTATGGTTATGACSmik ACGTTGG-TCGCGTCCCTGAA-CATAGGTACGGCTCGCACCACCGTGGTCCGAACTATAATACTGGCATAAAGAGGTACTAATTTCT--ACGGTGATGCCSbay GTG-CGGATCACGTCCCTGAT-TACTGAAGCGTCTCGCCCCGCCATACCCCGAACAATGCAAATGCAAGAACAAA-TGCCTGTAGTG--GCAGTTATGGT ** * ** *** * * ***** ** * * ****** ** * * ** * * ** ***
Scer GAGGA-AAAATTGGCAGTAA----CCTGGCCCCACAAACCTT-CAAATTAACGAATCAAATTAACAACCATA-GGATGATAATGCGA------TTAG--TSpar AGGAACAAAATAAGCAGCCC----ACTGACCCCATATACCTTTCAAACTATTGAATCAAATTGGCCAGCATA-TGGTAATAGTACAG------TTAG--GSmik CAACGCAAAATAAACAGTCC----CCCGGCCCCACATACCTT-CAAATCGATGCGTAAAACTGGCTAGCATA-GAATTTTGGTAGCAA-AATATTAG--GSbay GAACGTGAAATGACAATTCCTTGCCCCT-CCCCAATATACTTTGTTCCGTGTACAGCACACTGGATAGAACAATGATGGGGTTGCGGTCAAGCCTACTCG **** * * ***** *** * * * * * * * * **
Scer TTTTTAGCCTTATTTCTGGGGTAATTAATCAGCGAAGCG--ATGATTTTT-GATCTATTAACAGATATATAAATGGAAAAGCTGCATAACCAC-----TTSpar GTTTT--TCTTATTCCTGAGACAATTCATCCGCAAAAAATAATGGTTTTT-GGTCTATTAGCAAACATATAAATGCAAAAGTTGCATAGCCAC-----TTSmik TTCTCA--CCTTTCTCTGTGATAATTCATCACCGAAATG--ATGGTTTA--GGACTATTAGCAAACATATAAATGCAAAAGTCGCAGAGATCA-----ATSbay TTTTCCGTTTTACTTCTGTAGTGGCTCAT--GCAGAAAGTAATGGTTTTCTGTTCCTTTTGCAAACATATAAATATGAAAGTAAGATCGCCTCAATTGTA * * * *** * ** * * *** *** * * ** ** * ******** **** *
Scer TAACTAATACTTTCAACATTTTCAGT--TTGTATTACTT-CTTATTCAAAT----GTCATAAAAGTATCAACA-AAAAATTGTTAATATACCTCTATACTSpar TAAATAC-ATTTGCTCCTCCAAGATT--TTTAATTTCGT-TTTGTTTTATT----GTCATGGAAATATTAACA-ACAAGTAGTTAATATACATCTATACTSmik TCATTCC-ATTCGAACCTTTGAGACTAATTATATTTAGTACTAGTTTTCTTTGGAGTTATAGAAATACCAAAA-AAAAATAGTCAGTATCTATACATACASbay TAGTTTTTCTTTATTCCGTTTGTACTTCTTAGATTTGTTATTTCCGGTTTTACTTTGTCTCCAATTATCAAAACATCAATAACAAGTATTCAACATTTGT * * * * * * ** *** * * * * ** ** ** * * * * * *** *
Scer TTAA-CGTCAAGGA---GAAAAAACTATASpar TTAT-CGTCAAGGAAA-GAACAAACTATASmik TCGTTCATCAAGAA----AAAAAACTA..Sbay TTATCCCAAAAAAACAACAACAACATATA * * ** * ** ** **
GAL10
GAL1
TBP
GAL4 GAL4 GAL4
GAL4
MIG1
TBPMIG1
Factor footprint
Conservation islandslide credits: M. Kellis
Conservation of Motifs

Spar
Smik
Sbay
Scer
Evaluate conservation within: Gal4 Controls
4:1 1:3(2) Intergenic : coding
12:0 1:1(3) Upstream : downstream
A signature for regulatory motifs
22% 5%(1) All intergenic regions
Genome-wide conservation

1. Enumerate all “mini-motifs”
2. Apply three tests
1. Look for motifs conserved in intergenic regions
2. Look for motifs more conserved intergenically than in genes
3. Look for motifs preferentially conserved upstream or downstream of genes
CT A C GAN
slide credits: M. Kellis
Finding Motifs in Yeast GenomesM. Kellis PhD Thesis

Test 1: Intergenic conservation
Total count
Con
serv
ed c
ount
CGG-11-CCG
slide credits: M. Kellis

Test 2: Intergenic vs. Coding
Coding Conservation
Inte
rgen
ic C
onse
rvat
ion
CGG-11-CCG
Higher Conservation in Genes
slide credits: M. Kellis

Test 3: Upstream vs. Downstream
CGG-11-CCG
MostPatterns
Downstream Conservation
Ups
trea
m C
onse
rvat
ion
slide credits: M. Kellis
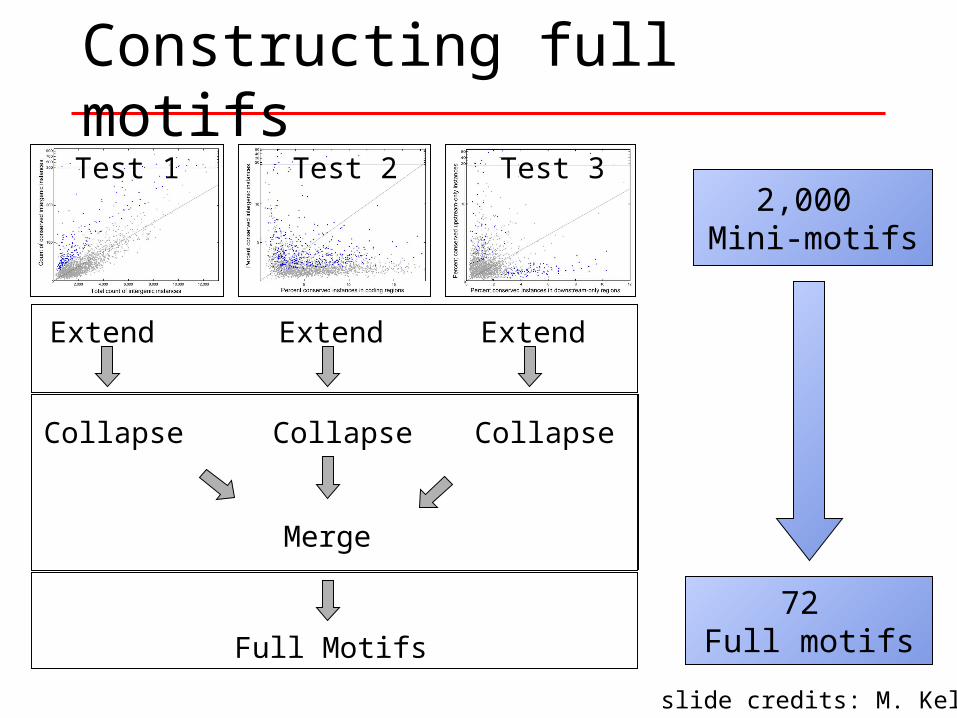
Extend
Collapse
Full Motifs
Constructing full motifs
2,000 Mini-motifs
72 Full motifs
6CT A C GAR R
CT GR C C GA AA CCTG C GA A
CT GR C C GA ACT RA Y C GA A
Y 5Extend Extend Extend
Collapse Collapse Collapse
Merge
Test 1 Test 2 Test 3
slide credits: M. Kellis

Rank Discovered MotifKnown
TF motifTissue
EnrichmentDistance
bias
1 RCGCAnGCGY NRF-1 Yes Yes
2 CACGTG MYC Yes Yes
3 SCGGAAGY ELK-1 Yes Yes
4 ACTAYRnnnCCCR Yes Yes
5 GATTGGY NF-Y Yes Yes
6 GGGCGGR SP1 Yes Yes
7 TGAnTCA AP-1 Yes
8 TMTCGCGAnR Yes Yes
9 TGAYRTCA ATF3 Yes Yes
10 GCCATnTTG YY1 Yes
11 MGGAAGTG GABP Yes Yes
12 CAGGTG E12 Yes
13 CTTTGT LEF1 Yes
14 TGACGTCA ATF3 Yes Yes
15 CAGCTG AP-4 Yes
16 RYTTCCTG C-ETS-2 Yes Yes
17 AACTTT IRF1(*) Yes
18 TCAnnTGAY SREBP-1
Yes Yes
19 GKCGCn(7)TGAYG
Yes Yes
• 174 promoter motifs 70 match known TF motifs 60 show positional bias
75% have evidence
• Control sequences< 2% match known TF motifs
< 3% show positional bias
< 7% false positives
slide credits: M. Kellis
Results

Antigen Epitope Prediction

Genome to “Immunome”
• Looking for a needle…– Only a small number of epitopes are typically antigenic
• …in a very big haystack– Vaccinia virus (258 ORFs): 175,716 potential epitopes (8-, 9-,
and 10-mers)– M. tuberculosis (~4K genes): 433,206 potential epitopes – A. nidulans (~9K genes): 1,579,000 potential epitopes
Can computational approaches predict all antigenic epitopes from a genome?
Pathogen genome sequences provide define all proteins that could illicit an immune response

Antigen Processing & Presentation

Modeling MHC Epitopes
• Have a set of peptides that have been associate with a particular MHC allele
• Want to discover motif within the peptide bound by MHC allele
• Use motif to predict other potential epitopes

Motifs Bound by MHCs
• MHC 1– Closed ends of grove– Peptides 8-10 AAs in length– Motif is the peptide
• MHC 2– Grove has open ends– Peptides have broad length
distribution: 10-30 AAs– Need to find binding motif
within peptides

MHC 2 Motif Discovery
Nielsen et al (2004) Bioinf
Use Gibbs Sampling!
462 peptides known to bind to MHC II
HLA-DR4(B1*0401)
9-30 residues in length
Goal: identify a common length 9 binding motif

Vaccinia Epitope PredictionMutaftsi et al (2006)
Nat. Biotech.
• Predict MHC1 binding peptides
• Using 4 matrices for H-2 Kb and Db
• Top 1% predictions experimentally validated
49 validated epitopes accounting for 95% of
immune response

Top Related