







Languages
Pages
Legal



ISBN DE LA OBRAISBN-13: 978-84-611-2176-2
REUMATOLOGÍA (3ª edición)ISBN-13: 978-84-612-6347-9
DEPÓSITO LEGALM-39965-2006
ACADEMIA DE ESTUDIOS MIR, S.L. (AMIR)[email protected]
MAQUETACIÓN E ILUSTRACIONESIceberg Visual
IMPRESIÓNGrafinter, S.L.
La protección de los derechos de autor se extiende tanto al contenido redaccionalde la publicación como al diseño, ilustraciones y fotografías de la misma, por loque queda prohibida su reproducción total o parcial sin el permiso del propietariode los derechos de autor.

R e u m a t o l o g í a
3] AUTORES [
REUMATOLOGÍA
Dirección editorialBORJA RUIZ MATEOS
JAIME CAMPOS PAVÓNJAVIER ALONSO GARCÍA-POZUELO
AIDA SUAREZ BARRIENTOSÓSCAR CANO VALDERRAMA
Autores principalesMIRIAM ESTÉBANEZ MUÑOZ
BORJA RUIZ MATEOSPAULA MARTÍNEZ SANTOS
MARÍA DE LAS MERCEDES SIGÜENZA SANZDAVID BERNAL BELLO
MARÍA MOLINA VILLARJAVIER ALONSO GARCÍA-POZUELO
Relación de autores
A U T O R E S
Hospital Universitario Clínico San Carlos. MadridHospital Universitario 12 de Octubre. MadridHospital Universitario de La Princesa. MadridHospital Universitario Clínico San Carlos. MadridHospital Universitario Clínico San Carlos. Madrid
Hospital Universitario La Paz. MadridHospital Universitario Clínico San Carlos. MadridHospital Universitario Fundación de AlcorcónHospital Universitario Puerta de Hierro. MadridHospital Universitario de GuadalajaraHospital Severo Ochoa de Leganés. MadridHospital Universitario de La Princesa. Madrid
AIDA SUÁREZ BARRIENTOSALBERTO TOUZA FERNÁNDEZ
ALFONSO JURADO ROMÁNALICIA JULVE SAN MARTÍN
ALONSO BAU GONZÁLEZÁLVARO GONZÁLEZ ROCAFORT
ANA DELGADO LAGUNAANA GÓMEZ ZAMORA
ANA MARÍA VALVERDE VILLARBORJA VARGAS ROJOCARMEN VERA BELLA
CLARA MARCUELLO FONCILLASCRISTIAN IBORRA CUEVAS
CRISTINA IGUALADA BLÁZQUEZCRISTINA VIRGINIA TORRES DÍAZ
DAVID BUENO SÁNCHEZEDUARDO FORCADA MELERO
ELISEO VAÑÓ GALVÁNENRIQUE JOSÉ BALBACID DOMINGO
ESTELA LORENZO HERNANDOFERNANDO CARCELLER LECHÓN
FERNANDO MORA MÍNGUEZFRANCISCO ARNALICH MONTIELGONZALO BARTOLOMÉ GARCÍA
GUILLERMO SCHOENDORFF RODRÍGUEZINMACULADA GARCÍA CANO
JAIME CAMPOS PAVÓNJORGE ADEVA ALFONSO
JORGE ASO VIZÁNJOSÉ MANUEL GONZÁLEZ LEITEJOSÉ MANUEL MARTÍNEZ DIEZJUAN JOSÉ GONZÁLEZ FERRERJUAN MIGUEL ANTÓN SANTOS
JUAN PEDRO ABAD MONTES
(11)(14)(10)(10)(14)(11)(10)(19)(19)(5)(19)(11)(19)(18)(2)(19)(10)(11)(11)(10)(7)(6)(22)(18)(1)(10)(10)(18)(10)(18)(19)(11)(14)(18)
KAZUHIRO TAJIMA POZOLAIA CANAL DE LA IGLESIA
LUIS BUZÓN MARTÍNLUIS MANUEL MANSO SÁNCHEZ
MANUEL GÓMEZ SERRANOMANUEL GONZÁLEZ LEYTE
MANUEL LEOPOLDO RODADOMARCO SALES SANZ
MARÍA ASENJO MARTÍNEZMARÍA DEL PILAR ANTÓN MARTINMARÍA LUISA GANDÍA GONZÁLEZ
MARÍA TERESA RIVES FERREIROMARÍA TERESA TRUCHUELO DÍEZ
MARTA MORADO ARIASMERCEDES SERRANO GIMARE
MONCEF BELAOUCHIOLGA NIETO VELASCO
ÓSCAR CANO VALDERRAMAPABLO DÁVILA GONZÁLEZ
PABLO SOLÍS MUÑOZPALOMA IGLESIAS BOLAÑOS
PATRICIO GONZÁLEZ PIZARRORICARDO SALGADO ARANDA
ROBERTO MOLINA ESCUDEROROCÍO CASADO PICÓN
RODRIGO FERNÁNDEZ JIMÉNEZRUTH LÓPEZ GONZÁLEZSARA BORDES GALVÁN
SARA ELENA GARCÍA VIDALSILVIA PÉREZ TRIGO
SUSANA GARCÍA MUÑOZGURENSUSANA PERUCHO MARTÍNEZTERESA BASTANTE VALIENTE
VERÓNICA SANZ SANTIAGO
(11)(12)(5)(10)(11)(18)(23)(22)(2)(14)(19)(19)(22)(19)(8)(4)(10)(11)(11)(10)(14)(19)(10)(18)(10)(11)(11)(11)(11)(11)(3)(13)(10)(7)
(1)(2)(3)(4)(5)(6)(7)(8)(9)(10)(11)(12)
(13)(14)(15)(16)(17)(18)(19)(20)(21)(22)(23)
Clínica Universitaria de Navarra. NavarraFundación Jiménez Díaz. Madrid
Hospital de Ciudad RealHospital de la Santa Creu i San Pau. Barcelona
Hospital General de Móstoles. MadridHospital Infanta Leonor. Madrid
Hospital Niño Jesús. MadridHospital Sant Joan de Déu. Barcelona
Hospital Severo Ochoa de Leganés. MadridHospital Universitario 12 de Octubre. Madrid
Hospital Universitario Clínico San Carlos. MadridHospital Universitario de Bellvitge. Barcelona
Hospital Universitario de Fuenlabrada. MadridHospital Universitario de Getafe. Madrid
Hospital Universitario de GuadalajaraHospital Universitario de La Princesa. MadridHospital Universitario Fundación de Alcorcón
Hospital Universitario Gregorio Marañón. MadridHospital Universitario La Paz. Madrid
Hospital Universitario Príncipe de Asturias. MadridHospital Universitario Puerta de Hierro. Madrid
Hospital Universitario Ramón y Cajal. MadridHospital Universitario Virgen del Rocío. Sevilla


5] ORIENTACIÓN MIR [


R e u m a t o l o g í a
7
• TEMA 1 INTRODUCCIÓN ..........................................................................................91.1. DIAGNÓSTICO DIFERENCIAL DE TRASTORNOS MÚSCULOESQUELÉTICOS ..................11
• TEMA 2 ARTRITIS POR MICROCRISTALES ...............................................................122.1. HIPERURICEMIA Y GOTA............................................................................................122.2. CLASIFICACIÓN DE LAS HIPERURICEMIAS ..................................................................122.3. RIÑÓN Y GOTA ..........................................................................................................132.4. DIAGNÓSTICO ...........................................................................................................132.5. TRATAMIENTO...........................................................................................................142.6. ARTRITIS DEBIDA A DEPÓSITO DE CRISTALES DE CALCIO...........................................14
• TEMA 3 VASCULITIS .................................................................................................163.1. PANARTERITIS NODOSA.............................................................................................173.2. POLIANGEITIS MICROSCÓPICA ..................................................................................193.3. VASCULITIS GRANULOMATOSA DE CHURG-STRAUSS
(ANGEITIS Y GRANULOMATOSIS ALÉRGICA)..............................................................193.4. GRANULOMATOSIS DE WEGENER .............................................................................203.5. ARTERITIS DE LA TEMPORAL O DE CÉLULAS GIGANTES .............................................213.6. ARTERITIS DE TAKAYASU...........................................................................................223.7. VASCULITIS POR HIPERSENSIBILIDAD..........................................................................233.8. ENFERMEDAD DE KAWASAKI ....................................................................................243.9. SÍNDROME DE BEHÇET ..............................................................................................253.10. ENFERMEDAD DE BUERGER O TROMBOANGEITIS OBLITERANTE ................................263.11. OTRAS VASCULITIS ....................................................................................................27
• TEMA 4 ARTRITIS REUMATOIDE..............................................................................27• TEMA 5 ARTRITIS CRÓNICA JUVENIL Y ENFERMEDAD DE STILL DEL ADULTO ....32
5.1. ARTRITIS CRÓNICA JUVENIL .......................................................................................325.2. ENFERMEDAD DE STILL DEL ADULTO .........................................................................33
• TEMA 6 LUPUS ERITEMATOSO SISTÉMICO.............................................................34• TEMA 7 ESPONDILOARTROPATÍAS SERONEGATIVAS ...........................................39
7.1. ESPONDILITIS ANQUILOSANTE ...................................................................................397.2. ARTRITIS REACTIVA (SÍNDROME DE REITER) ...............................................................427.3. ARTROPATÍA PSORIÁSICA ..........................................................................................447.4. ARTROPATÍAS ENTEROPÁTICAS (EII) ...........................................................................44
• TEMA 8 ENFERMEDADES METABÓLICAS ÓSEAS ...................................................458.1. OSTEOPOROSIS..........................................................................................................458.2. OSTEOMALACIA - RAQUITISMO ................................................................................478.3. ENFERMEDAD DE PAGET ...........................................................................................48
• TEMA 9 ESCLEROSIS SISTÉMICA PROGRESIVA.......................................................499.1. SÍNDROMES ESCLERODERMIFORMES.........................................................................52
• TEMA 10 ENFERMEDAD MIXTA DEL TEJIDO CONECTIVO .......................................53• TEMA 11 ARTRITIS SÉPTICAS .....................................................................................53
11.1. ARTRITIS NO GONOCÓCICAS ....................................................................................5311.2. ARTRITIS GONOCÓCICA ............................................................................................5411.3. ARTRITIS TUBERCULOSA ............................................................................................5511.4. ARTRITIS BRUCELÓSICA .............................................................................................5511.5. ARTRITIS POR ESPIROQUETAS ....................................................................................5611.6. ARTRITIS VÍRICAS .......................................................................................................5611.7. ARTRITIS MICÓTICAS .................................................................................................56
• TEMA 12 OTRAS ARTROPATÍAS ................................................................................5612.1. POLICONDRITIS RECIDIVANTE ....................................................................................5612.2. ARTROPATÍA NEUROPÁTICA DE CHARCOT................................................................5712.3. OSTEOARTROPATÍA HIPERTRÓFICA............................................................................5712.4. FIBROMIALGIA ...........................................................................................................5812.5. POLIMIALGIA REUMÁTICA .........................................................................................58
• TEMA 13 AMILOIDOSIS..............................................................................................5813.1. FIEBRE MEDITERRÁNEA FAMILIAR ..............................................................................59
• TEMA 14 SÍNDROME DE SJÖGREN............................................................................59• TEMA 15 ARTROSIS ....................................................................................................60• TEMA 16 POLIMIOSITIS Y DERMATOMIOSITIS.........................................................62
ANEXO ........................................................................................................62
] ÍNDICE [
Í N D I C E


R e u m a t o l o g í a
9] INTRODUCCIÓN [
En reumatología la anamnesis y la exploración física articularson fundamentales.
Anamnesis reumatológica- Antecedentes familiares.- Antecedentes personales.- Clínica actual: el síntoma principal es el dolor. El cartílago ar-ticular es avascular y no tiene inervación de manera que eldolor se produce cuando se distiende la cápsula articular. Ellohace que los pacientes mantengan la articulación en la posi-ción en la que la cápsula adopte un mayor volumen para dis-minuir la presión y con ello el dolor (suele ser en flexiónparcial). Se debe interrogar sobre: el tiempo de evolución y lo-calización del dolor, impotencia funcional y limitación de lamovilidad asociada; deformidades, manifestaciones no articu-lares, tratamientos previos,...
Exploración físicaSe explorarán todas las articulaciones y se completará con unaexploración sistemática de las estructuras no articulares. La ins-pección, la palpación y el examen de la movilidad articular seefectuarán comparando siempre las articulaciones deambos lados del cuerpo para detectar asimetrías. Se debe in-vestigar la presencia de crepitación, tumefacción, rubor, calor yderrame articular; existencia de deformidades congénitas o ad-quiridas (la deformidad aparece en enfermedades de larga du-ración); palpación de crujidos articulares; limitación de lamovilidad, que puede ser parcial o total (anquilosis) o, por elcontrario, movilidad excesiva (laxitud articular). La afectación ar-ticular puede ser monoarticular, oligoarticular (dos o tres articu-laciones afectas) o poliarticular (más de tres articulacionesafectas).
Pruebas complementariasMétodos de imagen
- Exploración radiográfica. Es preciso tener en cuenta queen muchos procesos el período de latencia radiológica eslargo, de meses e incluso años (sacroileítis de la espondilitisanquilosante, espondilitis tuberculosa, erosiones de la artritisreumatoide). Lo habitual es que una imagen sitúe el procesodentro de un grupo de enfermedades, pero sin especificar unaentidad. - Ecografía. Es útil en el diagnóstico de afecciones tendinosas,musculares y del tejido subcutáneo. Técnica de elección en lapatología del manguito de los rotadores, quistes sinoviales y enla displasia de cadera en niños menores de 3 meses. En gene-ral, por su accesibilidad y bajo coste, suele ser la primeraprueba a realizar ante lesiones de partes blandas (la RNMaporta más información de partes blandas pero es menos dis-ponible y más costosa). La ECO supera incluso a la RNM en lasepitrocleítis, bursitis y la evaluación del manguito de los rota-dores.- Tomografía computerizada (TC). Es muy útil en el estudiodel raquis, tanto de las estructuras óseas como de los tejidosradiotransparentes (para evaluar hernias lumbares tiene unrendimiento similar a la RNM pero no en el caso de herniascervicales donde la RNM es claramente superior). Indicada, enespecial, para el estudio de las articulaciones occipito-a tloaxoideas, temporomaxilares, esternoclaviculares y costo-
vertebrales. Supera a la RNM en la evaluación de las estenosisdel canal medular.- Resonancia magnética. Permite delimitar los tejidos blan-dos y el hueso (sobre todo la médula ósea), poniendo de ma-nifiesto las alteraciones no visibles con las técnicas de imagencitadas anteriormente. Es una excelente técnica de exploraciónpara el raquis, la cadera (en especial para el diagnóstico tem-prano de la necrosis avascular de la cabeza femoral), para losproblemas mecánicos de la rodilla y para el diagnóstico de laosteomielitis.- Examen gammagráfico. El isótopo que se emplea es el po-lifosfato de tecnecio, (permite identificar cualquier tipo deafección osteoarticular); el galio es más específico de infecciónosteoarticular. En los trastornos óseos, la gammagrafía es su-mamente útil en la detección de metástasis, por ejemplo, quese manifiestan por este medio antes de que se evidencie laimagen radiográfica; lo mismo sucede en las osteomielitis.
Pruebas de laboratorioHemograma completo, VSG y proteína C reactiva (PCR), pará-metros bioquímicos incluyendo complemento, sedimento deorina y proteinuria. En los casos seleccionados se realizaránpruebas más específicas para el diagnóstico, como la determi-nación de la tasa de antiestreptolisina (ASLO), anticuerpos anti-nucleares (ANA) y anticuerpos anti-DNA, estudio de antígenosHLA, etc.
Biopsia sinovialSólo son patognomónicos los granulomas de la tuberculosis ysarcoidosis y la observación de cristales de ácido úrico y de pi-rofosfato.
Examen del líquido sinovialEl líquido sinovial es un dializado del plasma más ácido hialuró-nico. Es la prueba diagnóstica más rentable en el estudio de lasenfermedades reumatológicas y la primera prueba a realizarante una monoartritis aguda (MIR 08, 83; MIR 01,76; MIR00,117; MIR 00, 121; MIR 97, 105; MIR 97F, 90). No debe re-alizarse si existe celulitis periarticular (por el riesgo de contami-nar e infectar la articulación).Indicaciones:
- Monoartritis (aguda, crónica).- Traumatismo con derrame articular.- Sospecha de infección articular (tinción Gram y cultivo), ar-tritis por cristales (visualización con microscopio de luz polari-zada) o hemartrosis.
Su análisis proporciona datos fundamentales para el diagnósticodiferencial de muchas enfermedades y es la clave diagnósticade las artritis sépticas y microcristalinas. Desde el punto de vistaclínico, los derrames se clasifican en tres grupos (ver tabla 1).
Pruebas analíticas- Reactantes de fase aguda: son útiles para el seguimientode enfermedades reumáticas, dándonos una idea de su activi-dad, sin embargo rara vez tienen valor diagnóstico.- VSG: es la prueba más utilizada como indicador de respuestade fase aguda. Es una medida indirecta del aumento enplasma de las proteínas de fase aguda.- Proteína C reactiva (PCR): es el mejor parámetro en la AR,por su mejor correspondencia con el tiempo de evolución. Mu-chos pacientes con LES tienen PCR normal.- Otros: proteína sérica amiloide A, haptoglobina, fibrinógeno,ceruloplasmina, fracción C3 del complemento…- Factor reumatoide (FR): Es un anticuerpo dirigido contra lafracción Fc de la inmunoglobulina G (IgM). Existen tambiénanticuerpos IgG e IgA. Las dos técnicas utilizadas clásicamentepara su determinación (pruebas de látex y Waaler-Rose) sólodetectan los anticuerpos IgM, mientras que los métodos de
TEMA 1 INTRODUCCIÓN
Hay que tener muy claras las características del líquido sinovialy sacar una idea básica sobre los anticuerpos que te ayude enlos temas siguientes. Por lo demás, éste es sólo un tema de in-troducción.
ENFOQUE MIR

Manual A Mir
10 ] INTRODUCCIÓN [
www.academiamir.com
RIA y ELISA, permiten detectar otros factores reumatoides dis-tintos del IgM. La presencia de FR es uno de los criterios diag-nósticos de la artritis reumatoide (AR) (MIR), pero no esespecífico de la AR pudiendo aparecer en otras circunstancias:
• Personas sanas: 5-10% (10-20% en mayores de 65 años,su frecuencia aumenta con la edad).• Familiares de pacientes con AR.• Enfermedades reumáticas:
- AR: 70-90%.- Síndrome de Sjögren (SS): 75-95% (a títulos altos).- Crioglobulinemia mixta: 40-100% (a títulos altos).- Enfermedad mixta del tejido conectivo: 50-60%.- Lupus eritematoso sistémico: 15-35%.- Dermatopolimiositis: 5-10%.
• Enfermedades infecciosas (bacterianas, víricas y parasita-rias).• Enfermedades inflamatorias crónicas: hepatopatía crónica,sarcoidosis, bronquitis crónica...(MIR).• Neoplasias.
- Autoanticuerpos antinucleares: son inmunoglobulinas diri-gidas contra antígenos autólogos intracelulares (la mayoría lo-calizados en el núcleo). Su presencia indica la existencia de unareacción inmunológica pero no necesariamente una enferme-dad. Suelen detectarse mediante inmunofluorescencia indi-recta. La positividad a títulos altos es específica dedeterminados procesos: LES (95%), lupus por fármacos(100%), enfermedad mixta del tejido conectivo (EMTC 100 %)y esclerodermia (60-90%). También están presentes, aunquea títulos bajos, en un pequeño tanto por ciento en la poblaciónnormal y en procesos inflamatorios o infecciosos. El título deanticuerpos se expresa como una proporción (1:40; 1:1.600,etc). Cuanto mayor es el denominador, mayor es el título deanticuerpos.En la tabla 2 se presentan los principales autoanticuerpos y sufrecuencia en algunas enfermedades autoinmunes (MIR 00,119).- Otros autoanticuerpos:
• Anticuerpos frente al citoplasma de neutrófilos(ANCA):
- c-ANCA (antiproteinasa 3): patrón citoplasmático diri-gido contra la a proteinasa 3; aparece en Granulomatosisde Wegener (90%), Poliangeitis microscópica (15%).- p-ANCA (antimieloperoxidasa): patrón perinuclear di-rigido contra la mieloperoxidasa; PAN (10-50%), Poliangei-tis microscópica (con la que más se asocia), enfermedad deChurg-Strauss, Granulomatosis de Wegener…
• Antifosfolípidos: como el anticoagulante lúpico (provocael alargamiento del TTPA) o los anticuerpos anticardiolipina(dan lugar a un falso positivo en test de sífilis VRDL). Relacio-nados con el síndrome antifosfolípido, aumentando el riesgode trombosis, abortos de repetición, trombopenia, valvulopa-tía de Libman-Sachs. No están siempre presentes en el LES nirelacionados con su actividad (MIR 00,119).• Anti-neuronales: LES (60%) en títulos altos guardan rela-ción con alteraciones en SNC.• Anti-proteínas P ribosomales: asociados a psicosis lú-pica.• Sistema HLA: sistema de genes localizados en al cromo-soma 6 que regulan la expresión de unas glicoproteínas cuyafunción principal es la presentación del antígeno a las célulasresponsables de la respuesta inmune. Tienen herencia codo-minante. Pueden ser de clase I (A, B, C), clase II (D), clase III.Existen ciertas asociaciones con enfermedades reumatológi-cas:
- Artritis reumatoide: DR4 (DR1).- Artropatía psoriásica con predominio de afectación peri-férica: B38.- Beçhet: B5.- LES: B8 / DR3 / DQW2 / C4AQO.- Sjögren primario: B8 / DR3 / DRW52.- PM / DM: B8 / DRW52.
Su valor clínico para el diagnóstico de enfermedades se limita alHLA-B27 y la espondilitis anquilosante.
GRUPO III
No inflamatorio(mecánico)
TIPO DE LÍQUIDO ASPECTO LEUCOCITOSVISCOSIDAD
Claro(hemorrágicoen traumas)
Similaral plasma
Mononucleares(<3.000)Alta
Normal(1,5-2,5)
GRUPO II
GRUPO I
GLUCOSA ACIDO LÁCTICO PATOLOGÍASPROTEÍNAS(gr/dl)
N
- Artrosis- Traumatismos- Artropatía neu-ropática
- Osteonecrosis- Osteocondritis- Amiloidosis
- Artritis inflama-toria
- AR, LES, EA- Gota y pseudo-gota
- Algunas sépti-cas (fúngicas)
Inflamatorio TurbioAmarillo N/↓Polimorfonucleares
(3.000 - 50.000)
Baja
Altas(>2,5)
↑
- Artritis sépticas- Algunas infla-
matorias (Rei-ter, a/v AR ygota)
Infeccioso(MIR 07, 83)
Turbioopaco ↓
Polimorfonucleares(>50.000)
Altas(>3)
Tabla 1. Características del líquido sinovial.
Regla de las 4 Ps:p-ANCA
perinuclearAntimieloperoxidasa
PAN
REGLA NEMOTÉCNICA
La artrocentesis y análisis del líquido articular esobligado ante toda monoartritis aguda (aunque el
diagnóstico parezca evidente).
RECUERDA

R e u m a t o l o g í a
11] INTRODUCCIÓN [
1.1.- Diagnóstico diferencial de los trastornos mús-culo-esqueléticos
Antinucleares (ANA)
Anti-ADNss (cadena simple)
Anti- ADNds (cadena doble o nativo)
Anti-Sm
Anti-RNP
Anti-histona
Anti-Ro (SS-A) MIR
Anti-La (SS-B)
Anti-Jo 1
Anti-topoisomerasa I (Anti-Scl 70)
Anti-centrómero
Antinucleolo
LES (98%). Lupus inducido por fármacos (100%)
LES (90%; no específicos ), aparecen en otras enfermedades del tejido conectivo
LES (70%; muy específicos), títulos altos se relacionan con nefritis y actividad de la enfermedad
LES (30%, el más específico ). En el LES se asocia a vasculitis, leucopenia y afectación del SNC
EMTC (en casi todos los pacientes, los marcadores U1-RNP son los más característicos)
LES (bajo riesgo de nefritis)
Esclerodermia
Polimiositis
Lupus inducido por fármacos (90%)
LES (60%)
AR (20%)
Síndrome de Sjögren primario (60%)
LES (30%, neonatal, del anciano, asociado a déficit hereditario de complemento, nefritis si no se asocia a anti-La)
LES con AAN negativos
Lupus cutáneo subagudo, bloqueo cardíaco congénito
Síndrome de Sjögren primario (50%)
LES (asociado a anti-Ro), protector de la presencia de nefritis
Polimiositis (30%, con enfermedad intersticial pulmonar)
AR
Esclerosis sistémica (20%, el más específico, la mayoría en forma de esclerosis difusa, se asocia a neumonitis intersticial)
Esclerosis sistémica (50%, la mayoría en la forma limitada-CREST) también en paciente sólo con Raynaud puede indicar
aparición posterior de afección cutánea
Esclerosis sistémica (20%)
Tabla 2. Principales anticuerpos.
Trastornos musculoesqueléticos
Oligoarticular
TraumatismoArtrosis
OsteonecrosisArtropatías
neuropáticas
No Articular Articular
InflamatorioNoInflamatorioSistémico
FibromialgiaPolimialgiareumática
PolimiositisOsteomalacia
LocalizadoBursitis
TendinitisOsteomielitis
TúnelCarpiano
Monoarticular
Artritis sépticaGota
PseudogotaReumatismopalindrómico
SimétricaAsimétrica
CrónicaAguda CrónicaAguda
Artritispsoriásica
EAACJ
oligoarticular(ANA+, B27)
Artritisreactiva
Sdr. BeçhetFiebre
reumáticaEnf. de LymeArtropatías
enteropáticasVasculitissistémicas
ARLES
EsclerosissistémicaDM y PMArtritis
psoriásicaACJ (FR+)
Artritis gotosacrónica
Artritis crónicapor pirofosfato
Hepatitis BRubeola
Artritis de EII
Figura 1. Diagnóstico diferencial de los trastornos músculo-esqueléticos.

Manual A Mir
12 ] ARTRITIS POR MICROCRISTALES [
www.academiamir.com
2.1.- Hiperuricemia y Gota
Con el término gota se designan las manifestaciones clínicasproducidas por el depósito de cristales de urato monosódico(UMS) sobre todo en la cavidad articular, pero también en otrostejidos. El ácido úrico es el resultado final del catabolismo de las purinas,que se realiza en tejidos que contienen xantina oxidasa, fun-damentalmente en el hígado y en el intestino delgado. La mayorparte del urato se elimina por los riñones (60-70%); el resto porel intestino. La hiperuricemia se define como la concentración plasmática deurato mayor de 7 mg/dl. A pesar de que la precipitación de cris-tales de urato monosódico requiere niveles de ácido úrico porencima del de saturación, sólo un porcentaje reducido de indi-viduos con hiperuricemia padecen gota, porcentaje que au-menta a medida que los niveles de ácido úrico sérico suben,acercándose al 50% en aquéllos cuyo ácido úrico sérico es su-perior a 9 mg/dL. De ello se infiere que, aunque los niveles ele-vados de ácido úrico sérico son necesarios para la formación decristales, se requieren de otros factores por ahora desconocidos. Los niveles de ácido úrico se mantienen muy bajos antes de lapubertad, aumentando progresivamente con la edad. En lasmujeres ascienden después de la menopausia (MIR 99, 88).El UMS se produce a partir de la síntesis endógena de purinas,de la dieta y de la degradación de los ácidos nucleicos. Existendos puntos clave en esta vía de síntesis:Los niveles de fosforribosil-pirofosfato (PRPP): se relacionan di-rectamente con los niveles de UMS.La enzima hipoxantin-guanin-fosforribosil transferasa (HGPRt):es una vía de eliminación de UMS.
2.2.- Clasificación de las hiperuricemias
Los niveles séricos de ácido úrico aumentan por dos posiblesmecanismos: aumento de la síntesis y disminución de la excre-ción renal, que es el mecanismo más común.
Hiperuricemia por aumento de síntesis de ácido úricoDefectos enzimáticos hereditariosLos dos defectos enzimáticos relacionados con el aumento acu-sado de la síntesis de ácido úrico se trasmiten ligados al cromo-soma X: el aumento de la actividad de la5-fosforribosil-1-pirofosfato-sintetasa (PRPPs) y la ausencia total oparcial de hipoxantina-guanina-fosforribosiltransferasa (HGPRt).
- Aumento de la PRPP sintetasa: se caracteriza por la sobre-producción de purinas e hiperuricemia, con aparición de cál-culos de ácido úrico y gota antes de los 20 años de edad.- Déficit de HGPRt: existiendo variantes en función de si el dé-ficit es completo o parcial:
• Completo: síndrome de Lesch-Nyhan, niños con gota y li-tiasis renal, que presentan también retraso mental, tendenciaa la automutilación, coreoatetosis y espasticidad.• Parcial: síndrome de Kelley-Seegmiller, presentan gota ycálculos renales.
Hiperuricemia por aumento del catabolismo de purinas(MIR 01, 64)En la mayoría de los pacientes con hiperuricemia por aumentode síntesis del ácido úrico, la anomalía subyacente es un au-mento del catabolismo de purinas. Éste se produce en enferme-dades mieloproliferativas y linfoproliferativas, mieloma múltipleu otros tumores, anemias hemolíticas, anemia perniciosa, he-moglobinopatías y policitemia vera, así como la destrucción degran cantidad de células durante el tratamiento de neoplasiasmediante quimioterápicos. Otras situaciones como la psoriasisextensa, la enfermedad de Paget e incluso un ejercicio físico in-tenso, también resultan en un aumento de producción y excre-ción de ácido úrico.
Hiperuricemia por defecto de excreción renal de ácidoúricoEste mecanismo causa el 90% de las hiperuricemias. La excre-ción renal de ácido úrico es compleja, ya que, tras filtrarse en elglomérulo, el ácido úrico es en gran parte reabsorbido por eltúbulo, secretándose de nuevo distalmente; el ácido úrico quellega a las vías excretoras resulta por tanto del equilibrio entrela filtración glomerular, la reabsorción tubular y la secreciónpostreabsortiva. No está bien definido en cuál o cuáles de estospasos se encuentra el defecto que ocasiona la disminución de laexcreción renal y, con ella, la hiperuricemia. Se plantean tres po-sibilidades:
- Disminución de la filtración glomerular de urato: contri-buye a la hiperuricemia de la insuficiencia renal (30%), es cu-rioso que, salvo en la poliquistosis renal, la insuficiencia renalraras veces se acompaña de gota o tofos; sin embargo los pa-cientes con insuficiencia renal crónica en hemodiálisis sí pue-den sufrir ataques recurrentes de artritis o periartritis agudatanto por cristales de urato como de calcio u oxalato cálcico.- Aumento de la absorción de uratos: se produce en situa-ciones en las que hay disminución del volumen extracelular:diabetes insípida, diuréticos sobre todo tiazídicos, etc. (MIR02, 76; MIR 01F, 83). El uso de diuréticos constituye en la ac-tualidad la causa identificable más común de hiperuricemia.- Disminución de la secreción de urato: la competición conel ácido úrico para la excreción renal de otros ácidos orgánicosexplica la hiperuricemia asociada a la cetoacidosis diabética,alcohólica, láctica, acidosis de los estados de malnutrición ytambién a la toma de fármacos como el ácido acetilsalicílico endosis bajas (en dosis altas 2 gr/día es uricosúrico) (MIR). La pi-razinamida disminuye la excreción renal al inhibir la secrecióntubular de ácido úrico.- Otras: el ácido nicotínico, el etambutol y la ciclosporina sontambién causa de hiperuricemia por disminución de excreciónrenal a través de mecanismos mal definidos. La hiperuricemiaque acompaña al hipotiroidismo, hiperparatiroidismo, hipopa-ratiroidismo y al pseudohipoparatiroidismo probablementetiene también origen renal. La intoxicación crónica por plomoreduce el aclaramiento renal de ácido úrico y es causa de hi-peruricemia y de la denominada “gota saturnina”.
Hiperuricemia de causa mixta- Alcohol: la ingesta de alcohol, independientemente de ladisminución de la excreción renal de ácido úrico que provoca(por hiperlactacidemia), incrementa la uricemia al acelerar elcatabolismo del ATP.- Otros: déficit de fructosa-1-fosfato aldolasa (causa intole-rancia a la fructosa) o el déficit de glucosa-6-fosfatasa (cursacon hiperuricemia e hiperlactacidemia).
Manifestaciones clínicasLas manifestaciones clínicas son de dos tipos:
- Inflamación, por lo general articular, pero que puede locali-zarse en otras estructuras sinoviales, como tendones o bolsas
TEMA 2 ARTRITIS PORMICROCRISTALES
Debes diferenciar correctamente la artritis gotosa y la condro-calcinosis. Son las más importantes (ver tabla 3); en la gota fíjateen el tratamiento y en la condrocalcinosis en las enfermedadessistémicas asociadas. El depósito de cristales en la articulaciónpuede dar clínica similar a la artritis reumatoide, espondilitis an-quilosante, sin ser específico de cada cristal (MIR 08, 82).
ENFOQUE MIR

R e u m a t o l o g í a
13] ARTRITIS POR MICROCRISTALES [
de deslizamiento tendinoso.- Aparición de agregados clínicamente detectables de estoscristales formando los tofos. En relación directa con la gota ocon el aumento de excreción de ácido úrico, puede tambiénafectarse el riñón.
Podemos hablar de 4 fases:1. Hiperuricemia asintomática. Se caracteriza por niveles deuratos elevados sin síntomas artríticos, ni tofos o cálculos deácido úrico. La posibilidad de sufrir artritis gotosa y nefrolitiasisaumenta con el nivel de hiperuricemia y su duración. Casitodos los pacientes con gota están hiperuricémicos, pero sóloel 5% de hiperuricémicos desarrollan gota. Esta fase concluyecuando aparece el primer ataque de gota o nefrolitiasis. Lagota aparece después de 20 ó 30 años de hiperuricemia sos-tenida.2. Artritis gotosa aguda. Ante determinados factores preci-pitantes como cambios bruscos de uricemia (sobre todo des-censos bruscos, aunque puede ocurrir con ácido úriconormal), uso de diuréticos, alcohol, fármacos, traumatis-mos, situaciones de estrés (hospitalización, infecciones,ayuno o disminución de peso...) se desencadena el ataqueagudo de gota. Los primeros ataques tienen un comienzoagudo, en ocasiones nocturno y, dejados a su evolución natu-ral, ceden en días o escasas semanas. La inflamación articularsuele ser intensa y muy dolorosa, no soportando en ocasionesni la más ligera presión sobre la articulación; sin embargo, éstano es una regla estricta y la artritis puede tener inicio solapado,duración prolongada e intensidad moderada. Es una monoartritis, localizada en la mitad de los casos en laarticulación metatarsofalángica del dedo gordo del pie –queorigina la clásica podagra (suele ser la forma de inicio de la en-fermedad)– o bien en tarso, tobillo, bolsa preaquílea, rodilla,muñeca o alguna articulación metacarpofalángica o interfa-lángica de la mano o en la bolsa olecraniana. En un pequeñoporcentaje de pacientes, sobre todo en mujeres, la gota co-mienza con inflamación simultánea de más de una articula-ción. Pueden aparecer además síntomas sistémicosacompañantes como: fiebre, leucocitosis y aumento de la VSG.En los niños no se suelen afectar las rodillas.3. Gota intercrítica. Se refiere a los períodos asintomáticosentre los episodios agudos de gota. Aproximadamente el 75%de pacientes sufren un segundo ataque de gota en los dosaños siguientes.4. Tofos y artritis gotosa crónica. Con el tiempo, si no haytratamiento, se puede desarrollar una poliartritis de grandes ypequeñas articulaciones con tendencia a la simetría y con laaparición de nódulos o tofos (agregados de cristales de uratomonosódico rodeados por una reacción granulomatosa) quetienen importante capacidad de erosión. Los tofos puedenapreciarse en la superficie de extensión de los codos y en laproximidad de diversas articulaciones o a lo largo de algunos
tendones, como el aquíleo. A menudo el color blanco de lostofos se aprecia a través de la piel. Una localización frecuentees el borde externo del pabellón auricular como pequeñosagregados blanquecinos y opacos a la transiluminación, lo quepermite diferenciarlos de otras formaciones locales. Los tofosno llegan a formarse cuando el diagnóstico y el tratamientode la gota son adecuados y precoces. Pueden fistulizar al ex-terior, dejando salir un material blanco compuesto casi exclu-sivamente por cristales de urato monosódico. Mediante eltratamiento normouricemiante los tofos se disuelven con len-titud y van disminuyendo su tamaño hasta desaparecer.
2.3.- Riñón y gota
La hiperuricemia puede ocasionar diversos trastornos renales:- Nefrolitiasis: los pacientes gotosos padecen mayor frecuen-cia de litiasis renal por cálculos de ácido úrico. El aumento deexcreción de ácido úrico en la orina –en pacientes que presen-tan esta característica– es el factor que influye más directa-mente en la aparición de litiasis y tiene correlación con el nivelsérico de ácido úrico. Los individuos gotosos tienden a padeceren exceso litiasis cálcicas pues parece que la precipitación ini-cial de cristales de ácido úrico podría servir de centro de nucle-ación para otros tipos de cristales (MIR).- Nefropatía gotosa: hace referencia a la nefropatía inters-ticial resultante del depósito de cristales de ácido úrico en elparénquima renal desencadenando una reacción inflamatoriaque, en casos avanzados, desencadena una fibrosis medular.Se considera un síntoma tardío de gota grave. En la actualidad,gracias al uso de fármacos es mucho menos frecuente. Se pos-tula que detrás de una nefropatía gotosa existe una intoxica-ción subrepticia por plomo.- Nefropatía aguda por ácido úrico: por lo general sin rela-ción con la gota, es consecuencia del depósito masivo de cris-tales de ácido úrico en los túbulos excretores renales,produciendo una insuficiencia renal aguda reversible. Sueledeberse a un aporte masivo de ácido úrico al riñón y se asociaa leucemias o tumores, sobre todo a causa de la destrucciónmedular masiva provocada por el tratamiento, y probable-mente tras ejercicio excesivo, convulsiones o rabdomiólisis.También puede ocurrir en pacientes gotosos, con hiperpro-ducción acusada de ácido úrico.
2.4.- Diagnóstico
- Clínica: monoartritis característica.- Analítica: aumento de reactantes de fase aguda y posibleleucocitosis. Hay hiperuricemia en el 95% de gota aguda y en100% de gota tofácea crónica no tratada. Líquido articular in-flamatorio con abundantes PMN, con cristales de UMS intra yextracelulares, con forma de aguja y birrefringencia negativa almicroscopio de luz polarizada (MIR 03, 227).
Figura 1. Tofo gotoso.
Figura 2. Cristales de urato monosódico con forma de aguja y birrefringencia ne-gativa.

Manual A Mir
14 ] ARTRITIS POR MICROCRISTALES [
www.academiamir.com
- Radiología: aguda: inespecífico; crónica: aumento de partesblandas, calcificaciones punteadas y condrocalcinosis, erosio-nes óseas en sacabocados, en los márgenes articulares oste-ólisis y geodas (quistes intraóseos yuxtaarticulares).
2.5.- Tratamiento
- Hiperuricemia asintomática. En la actualidad, no esta in-dicado el tratamiento de la hiperuricemia asintomática (MIR00, 123). Sólo está indicado para prevenir el desarrollo de lanefropatía aguda por ácido úrico, mediante una profilaxis conalopurinol antes del tratamiento citotóxico y, en caso de queya haya hiperuricemia, impedir que se deposite mediante diu-réticos, alcalinizando la orina (bicarbonato / inhibidores de laanhidrasa carbónica) y con alopurinol. Se deben corregir losproblemas asociados como la HTA, hipercolesterolemia, la dia-betes mellitus o la obesidad (MIR).- Artritis gotosa aguda: Colchicina + AINEs (MIR 06, 83;MIR 02, 84).
• Colchicina: el fármaco más empleado (la respuesta al fár-maco es diagnóstica de gota). Será el fármaco de elecciónen pacientes en los que no se dispone de diagnóstico de cer-teza. Inhibe la liberación del factor quimiotáctico leucocitarioinducido por los cristales. Los efectos secundarios más fre-cuentes son gastrointestinales (sobre todo diarrea). En algu-nos casos también puede presentarse toxicidadhematológica, renal o hepática. También puede adminis-trarse vía intravenosa (MIR 07, 82).• Antiinflamatorios no esteroideos (AINEs): fundamental-mente, la indometacina. Son mejor tolerados que la colchi-cina y constituyen probablemente el tratamiento de elecciónen pacientes con el diagnóstico establecido de artritis gotosaaguda. El tratamiento es más eficaz cuanto más precoz seinicie y se mantiene 3-4 días después de la desaparición delos signos de inflamación.• Glucocorticoides: la asociación de un ataque de gota conuna hemorragia digestiva o con cualquier afección que im-pida el uso de AINEs o colchicina orales y en casos resistentes,constituyen una situación especial. En estos casos, cuandose tiene certeza diagnóstica y no existe un proceso sépticoconcomitante, es útil la inyección intraarticular de una pe-queña dosis de glucocorticoides (MIR 97, 109; MIR 97F, 92).
- Gota intercrítica, debe tratarse la hiperuricemia de todoslos pacientes con artritis aguda recidivante, los que han pre-sentado artritis gotosa y nefrolitiasis, y los que presentan artro-patía crónica tofácea. El papel de la dieta es limitado siendonecesario recurrir a medidas farmacológicas. Disponemos dedos grupos de fármacos:
• Antihiperuricémicos: alopurinol. Inhibe la síntesis deácido úrico (inhibe la xantina oxidasa). Los efectos secunda-rios son: alteraciones digestivas, erupción cutánea, fiebre, ne-crólisis epidérmica, alopecia y depresión. Es importanteademás tener en cuenta las posibles interacciones farmaco-lógicas, pues puede potenciar la acción de la azatioprina, la6-mercaptopurina y la ciclofosfamida.
• Uricosúricos: sulfinpirazolona, probenecid, benzbroma-
nona (el disponible en España) y la benziodarona. Aumentanla excreción de ácido úrico perdiendo eficacia a medida quese reduce el aclaramiento de creatinina, siendo ineficacescuando la filtración glomerular cae por debajo de 30 ml/min.No tienen propiedades antinflamatorias. El AAS puede inhibirel efecto uricosúrico de esto fármacos. Pueden desencadenarla aparición de nefrolitiasis. Para evitarlo, se inicia el trata-miento con dosis bajas forzando la hidratación y alcalini-zando la orina. Los efectos secundarios son erupcionescutáneas y molestias digestivas; es rara la toxicidad gravepero pueden desencadenar necrosis hepática y síndrome ne-frótico.
El descenso brusco del urato plasmático como consecuenciadel inicio del tratamiento con alopurinol o agentes uricosúri-cos puede prolongar o precipitar un ataque agudo, de formaque, antes de iniciar el tratamiento, el paciente no debe tenersigno alguno de inflamación y debe haber comenzado atomar colchicina como profilaxis de 6 a 12 meses antes (sesolapan ambos tratamientos y posteriormente se suspende lacolchicina) (MIR). En pacientes con respuesta insuficiente ala monoterapia y en pacientes normo o hiposecretores congota tofácea grave se ha planteado la posibilidad de un tra-tamiento combinado (MIR).
- Nefrolitiasis: se recomienda la ingesta de agua suficientepara crear un volumen de orina >2 l/día, alcalinizar la orinacon bicarbonato sódico o acetazolamida, y administrar alopu-rinol.- Nefropatía por ácido úrico: se necesita hidratación intra-venosa intensa y asociación con furosemida (aumenta al flujoen los túbulos y diluye el ácido úrico), aumento de la alcalini-zación de la orina y alopurinol.
2.6.- Artritis debida a depósito de cristales de calcio
Cristales de pirofosfato cálcico dihidratado (MIR)El hallazgo de cristales de pirofosfato cálcico dihidratado (PPCD)permite establecer el diagnóstico de esta enfermedad y diferen-ciarla de otras artropatías. La prevalencia de condrocalcinosisaumenta claramente con la edad y constituye una de las prime-ras causas de artropatía en el anciano.
- Normosecretores o uricosuria elevada(>700 mg/día)- Intolerancia o contraindicación a uricosúricos:
• Alteración de la función renal (depuración plasmática de creatinina <80 ml/min)
• Nefrolitiasis (MIR)- Gota con tofos sea cual sea la función renal- Pacientes con nefropatía por ácido úrico o riesgo de padecerla (MIR)
INDICACIONES
Tabla 1. Indicaciones de alupurinol.
- Función renal normal- Ausencia de antecedentes de nefrolitiasis (MIR 98F, 208)- Eliminación renal de ácido úrico <700 mg/día con dieta normal
INDICACIONES
Tabla 2. Indicaciones de uricosúricos.
Aunque el diagnóstico parezca evidente, si debuta comomonoartritis aguda, se debería realizar artrocentesis.
Los cristales tienen forma de aguja y birrefringencia negativa.
Las bajadas agudas de uricemia precipitan ataques de gota.Por ello una uricemia normal no descarta la artritis gotosa.
Recuerda los diuréticos, los traumatismos y la hospitalizacióncomo precipitantes típicos.
El AAS a dosis altas de 2 gr/día es uricosúrico.
Para emplear fármacos antihiperuricemiantes el pacientedebe estar asintomático y llevar varios meses con colchicina
profiláctica.
RECUERDA

R e u m a t o l o g í a
15] ARTRITIS POR MICROCRISTALES [
Se conocen tres tipos de condrocalcinosis:
Esporádica o idiopáticaEs, con diferencia, la forma más frecuente de condrocalcinosis.La edad, la artrosis y la lesión articular por traumatismo previoson factores predisponentes para esta forma clínica.
FamiliarComo una artropatía de inicio temprano, entre la tercera y laquinta décadas de la vida, con afectación poliarticular grave e in-capacitante. El mecanismo de transmisión se cree que es de tipoautosómico dominante.
Asociada a otras enfermedadesRepresenta sólo el 10% de todas las condrocalcinosis, pero sedebe descartar siempre ante cualquier caso nuevo de condrocal-cinosis. Entre los procesos metabólicos que estimulan el depósitoencontramos algunas enfermedades metabólicas o endocrinas,como la hipofosfatasia, la hipomagnesemia, la hemocromatosisy el hiperparatiroidismo o la gota tofácea crónica (MIR 00F,97). Su importancia radica en que puede ser la primera manifes-tación de dichas enfermedades. En menor grado, también existeasociación con el hipotiroidismo (MIR), la hipercalcemia hipocal-ciúrica familiar, el síndrome de Bartter, gota, amiloidosis, acro-megalia, enfermedad de Wilson y ocronosis.La presencia de artritis por PPCD en sujetos menores de 50 añosdebe hacer pensar en las dos últimas situaciones y deberemosrealizar estudios de agregación familiar y solicitar niveles séricosde calcio, fósforo, magnesio, ferritina, fosfatasa alcalina y hor-monas tiroideas (MIR 98, 231; MIR 97, 106).
Manifestaciones clínicasCuando existen manifestaciones clínicas, consisten en una ar-tritis aguda, una artropatía crónica de tipo degenerativo-des-tructivo o una combinación de ambas. La articulación que másse afecta es la rodilla.Artritis aguda (pseudogota)Se caracteriza por episodios autolimitados de artritis de iniciobrusco, con intensos signos inflamatorios, seguidos de períodosasintomáticos intercríticos, a veces muy prolongados. Es pare-cida a la gota pero con crisis menos dolorosas. La mayoría de losepisodios son monoarticulares, siendo la rodilla la localizaciónpreferente (50%), seguida de la muñeca, el hombro, el tobillo,el codo y las articulaciones de las manos y los pies (MIR 99F,99), incluyendo la primera metatarsofalángica (pseudopodagra).En el 10% de los casos, se afecta más de una articulación. Enocasiones, la artritis se acompaña de febrícula e incluso fiebreelevada (hasta 40°C). Puede aparecer de forma espontánea otras los mismos desencadenantes que describimos en la gota(hospitalización, cirugía, embarazo,..) (MIR 00F, 94).Artropatía crónica (artropatía por pirofosfato)Constituye el 50% de las formas con manifestaciones, sobretodo, en las mujeres mayores de 65 años. Su semiología es si-milar a la de la artrosis primaria (diagnóstico diferencial), perocon una distribución simétrica y progresiva, mayor gravedad ymoderada inflamación articular. La localización articular, salvopara las articulaciones de carga (que se afectan en los dos casos)es diferente, de forma que la artrosis casi nunca afecta a las ar-ticulaciones metacarpofalángicas, muñecas, hombros, codos, ytobillos. Son frecuentes las contracturas en flexión, las deformi-dades articulares (genu varum o valgum) y la inestabilidad arti-cular. La afectación de las muñecas puede provocar unsíndrome del túnel carpiano por calcificación del ligamentotriangular del carpo.
Diagnóstico- Analítica: leucocitosis y elevación de reactantes de faseaguda. Presencia de cristales de PPCD en líquido sinovial (rom-
boidales con birrefringencia positiva débil) (MIR 98, 234).- Radiografía: en la mayoría de los pacientes se puede apre-ciar, mediante radiografías convencionales, una calcificaciónlineal muy característica del cartílago fibroso o hialino (con-drocalcinosis), secundaria al depósito de cristales de pirofos-fato cálcico, (aunque también se ha encontrado en otrasenfermedades) junto a calcificaciones en la sínfisis del pubis,ligamento triangular del carpo, o en discos intervertebrales.Suele ser bilateral y simétrica (MIR 04, 19; MIR 01F, 78). La ar-tropatía crónica tiene una radiografía similar a la de la artrosis.
TratamientoAspiración de la articulación y AINE o inyección intraarticular decorticoides. La colchicina puede ser útil como profilaxis en pa-cientes con episodios recurrentes. En la forma crónica el trata-miento es similar al de la artrosis.
Artropatía por hidroxiapatita cálcicaLa mayoría de las calcificaciones de partes blandas del orga-nismo son producidas por la hidroxiapatita (HA), que constituyeel principal mineral del hueso y de los dientes. La mayoría de lascalcificaciones de partes blandas son idiopáticas, pero existenademás diversas asociaciones:
- Lesión tisular local.- Enfermedades del colágeno (esclerodermia, dermatomiositisinfantil y LES).- Enfermedades metabólicas (hipercalcemia, hiperfosforemia,intoxicación por vitamina D, insuficiencia renal crónica, hemo-diálisis y diabetes mellitus).- Trastornos neurológicos.
Manifestaciones clínicasLa mayoría de las calcificaciones de partes blandas son asinto-máticas. Se han descrito:
- Periartritis calcificante. El hombro (manguito de los ro-tadores, bolsa subacromial o tendón del bíceps) es la localiza-ción preferente. Se caracteriza por la presencia decalcificaciones en partes blandas, zonas de inserción tendinosao bolsas sinoviales. La periartritis calcificante es asintomática ose acompaña de dolor crónico localizado que aumenta al con-traer contra resistencia el tendón afecto. En la radiografía seobserva una calcificación oval, redondeada o algodonosasobre la estructura clínicamente implicada. - Artritis. Episodio agudo de artritis similar a los provocadospor cristales de urato monosódico o pirofosfato cálcico. - Artrosis. Los cristales de hidroxiapatita se han identificado enel líquido sinovial de las articulaciones con artrosis, con unafrecuencia que varía entre el 20 y el 60% de los pacientes es-
1ª MTF
Tardías:Erosionesóseas +reborde
resaltadoDestrucciónintraarticular
↓Excreciónidiopática
↑Síntesis por:Recambio
celular↑PRPPs
Lesch-Nyhan
TRATAMIENTO
Solosintomática:
AINEsColchicinaAlopurinol
Uricosúricos
GOTA
ARTIC. TÍPICA MANIF. RX ETIOLOGÍA
Rodilla
Calcificacióncartílago,
ligamento ytendón
AspiraciónAINEs
Corticoidesintra-
articularesColchicina
CONDRO-CALCINOSIS
IdiopáticaOtras:
HemocromatosisHiper PTH 1º
HipomagnesemiaHipofofatasia
HipotiroidismoGota Tofácea
OcronosisFormas fami-
liares
Tabla 3. Diagnóstico diferencial entre gota y condrocalcinosis.

Manual A Mir
16 ] VASCULITIS [
www.academiamir.com
tudiados. Su observación se relaciona con la gravedad radio-lógica de la artrosis. Se desconoce su origen y sus implicacio-nes patogénicas, pero se cree que aparecen secundariamentea la degeneración del cartílago articular.- Artropatía destructiva. Forma rara de artropatía rápida-mente destructiva relacionada con el depósito de cristales dehidroxiapatita, que predomina en mujeres mayores de 60años. Se localiza en un hombro (hombro de Milwaukee) o unarodilla, aunque se pueden afectar varias articulaciones, en cuyocaso es bilateral y simétrica. Clínicamente se acompaña dedolor leve o moderado, rigidez, inestabilidad articular y granincapacidad funcional.
Diagnóstico- Analítica. Los cristales son muy pequeños y sólo puedenverse con el microscopio electrónico. Los acúmulos de cristalesse tiñen con la tinción de Wright y con alzarina roja. Comoparticularidad diferencial con el resto de artritis microcristali-nas, el análisis del líquido sinovial suele tener características noinflamatorias, siendo característico el hallazgo de un derramesinovial copioso de aspecto hemático, con escasos leucocitosy gran cantidad de cristales de hidroxiapatita. - Radiografía. Puede ser normal o encontrar calcificacionesintra o periarticulares, con cambios destructivos o hipertróficos.
TratamientoInespecífico, aspiración de la articulación y AINE o la inyecciónintraarticular de corticoides, que parece acortar la duración eintensidad de los síntomas.
Cristales de oxalato cálcicoEl oxalato cálcico (OXCA) es el producto final del metabolismodel ácido ascórbico y de algunos aminoácidos. Aunque existeuna forma de oxalosis primaria, enfermedad hereditaria rara queasocia nefrocalcinosis, insuficiencia renal y muerte antes de los20 años (artritis y periartritis durante últimos años de la enfer-medad), la mayoría de los casos se incluyen dentro del grupo
de la oxalosis secundaria en situaciones de excreción renal dis-minuida (insuficiencia renal crónica, hemodiálisis o diálisis peri-toneal) y otros factores como la ingesta elevada de vitamina C.
Manifestaciones clínicasSe presenta como una poliartritis aguda simétrica de curso cró-nico, con afectación de manos y rodillas, con tenosinovitis o sinella. Otras manifestaciones incluyen artritis de grandes articula-ciones, bursitis, episodios de podagra y condrocalcinosis en me-tacarpofalángicas y rodillas. El líquido sinovial es claro ohemático, con escasos leucocitos y abundantes cristales de oxa-lato cálcico, principalmente extracelulares, alguno de los cualesposee una forma bipiramidal muy característica, con intensa bi-rrefringencia. El curso suele ser crónico y rebelde al tratamientocon AINE, colchicina e infiltraciones de glucocorticoides.
TratamientoEn la oxalosis primaria, el transplante hepático reduce el depó-sito de cristales. La artropatía por cristales de OXCA se trata conAINEs, colchicina y corticoides intraarticulares. Actualmente seevitan los suplementos de vitamina C en pacientes con insufi-ciencia renal.
Bajo el término de vasculitis se engloban un grupo heterogéneode procesos que reconocen como sustrato patológico la presen-cia de inflamación en los vasos sanguíneos, pudiendo asociarnecrosis de la pared vascular.La afectación inflamatoria difusa vascular determina la apariciónde sintomatología general (fiebre, astenia, afectación delestado general, etc.) y el desarrollo de manifestaciones orgá-nicas locales (síntomas neurológicos, dolor abdominal, compro-miso renal, etc.) como consecuencia de la isquemia o el infartovisceral por oclusión de los vasos. La localización de los vasos, sudiferente tamaño y la distinta histopatología, en la que predo-minará la lesión necrosante o la granulomatosa, constituyen lascaracterísticas que definen los diferentes síndromes vasculíticosy permiten su individualización. Pueden ser la única manifestación de enfermedad y constituir elgrupo de vasculitis primarias (poliarteritis nodosa o granulo-matosis de Wegener) o asociarse a otra entidad nosológica yconsiderarse vasculitis secundarias (artritis reumatoide, lupuseritematoso sistémico…).
ClasificaciónLa heterogeneidad de los síndromes vasculíticos, su solapa-miento clínico-patológico y la ausencia de datos patognomóni-cos y de agente etiológico reconocido para la mayoría de elloshan dificultado sobremanera su clasificación. A continuación in-cluimos dos clasificaciones (FAUCI 1978):
Tabla 4. Resumen de la artritis microcristalina (MIR 05, 85).
Romboidalpequeño
Muypequeños BipiramidalFORMA DEL
CRISTAL
UMS OXCAHAPPCD
Aguja
Debil + No Muy+++BIRREFRIN-GENCIA Muy---
Inflamatorio(Pred.
Neutrófilos)
MecánicoMononucleares
<1000 Cel
MecánicoMononucleares
Neutros<2000 Cels
LÍQUIDOSINOVIAL
Inflamatorio24000 Cels.
(Pred.Neutrófilos)
Condro-calcinosissimétrica
Calcificacionesdistróficas ymetastásicas
Condro-calcinosisRADIOLOGÍA Erosiones
Geodas
RodillaMuñecaTobillo
RodillaHombro Cualquiera
LOCALIZACIÓNMÁS
FRECUENTE
1ª Metatarso-falángica(Podagra)
Mic. Luzpolarizada
M.E.Alzarina Roja
Wright
Mic. LuzpolarizadaDIAGNÓSTICO Mic. Luz
polarizada
Ancianos conArtrosis
(Si <50 añospensar en alt.Metabólica
o Hereditaria)
Ancianos
Oxalosis 1ª(<20 años)Oxalosis 2ª
(I RenalTerminal)
EDAD MÁSFRECUENTE
Varón 50años
Asintomático(a veces
“pseudo-gota”aguda)
AsintomáticoArtritis aguda
Artp.Destructiva
Sinovitis eninsuficiencia
renal terminal
PRESENTACIÓNCLÍNICA Gota aguda
TEMA 3 VASCULITIS
Constituye el capítulo al que debéis dedicar más tiempo puesaglutina el mayor porcentaje de preguntas de la asignatura. Laspreguntas suelen ser tipo caso clínico; una buena tabla con lascaracterísticas particulares de cada una de ellas simplifica el es-tudio. Los criterios diagnósticos y el tratamiento de las más im-portantes no deben dejar de estudiarse.
ENFOQUE MIR

R e u m a t o l o g í a
17] VASCULITIS [
EtiopatogeniaAunque hay casos aislados con agregación familiar no hay rela-ción con ningún patrón HLA, excepto en el Behçet (B5), arteritisde la temporal (DR4 y DRB1) y arteritis de Takayasu (DR2 y 4/MB1 y 3).
- Para la mayoría de los síndromes vasculíticos la etiología esdesconocida, aunque parecen mediadas por mecanismos in-munológicos.- El depósito de inmunocomplejos con activación del comple-mento parece ser el mecanismo fundamental.- La presencia de granulomas en algunos tipos de vasculitis(granulomatosis de Wegener, vasculitis granulomatosa y alér-gica de Churg-Strauss, etc.) confiere protagonismo a la lesióninmunológica mediada por células, aunque este patrón histo-lógico puede ser inducido también por inmunocomplejos.- La citotoxicidad celular dependiente de anticuerpos se ha de-mostrado en la enfermedad de Kawasaki, en la que se descri-bieron anticuerpos dirigidos contra antígenos de las célulasendoteliales durante la fase activa de la enfermedad.- Se ha concedido importancia patogénica a otros anticuerposdirigidos contra constituyentes enzimáticos (proteinasa 3, elas-tasa, mieloperoxidasa, etc.) del citoplasma de los neutrófilos(ANCA).
DiagnósticoLas vasculitis se presentan como cuadros de difícil diagnósticocon síntomas inespecíficos referidos a diferentes órganos.
Sospecha diagnóstica- Síndrome constitucional: fiebre, astenia, malestar, artro-mialgias…- Cutáneas: púrpura palpable - es la manifestación más ca-
racterística -, que suele comenzar en zonas declives, nódulossubcutáneos, urticaria crónica, livedo reticularis, úlceras, telan-giectasias del lecho ungueal, infarto o gangrena digital…- Renales: la afectación renal es lo más característico y fre-cuente de la PAN. Puede cursar con glomerulonefritis, protei-nuria, hematuria, insuficiencia renal, HTA…- Pulmonares: hemoptisis, tos, disnea, crisis asmáticas, altera-ciones radiográficas (nódulos, infiltrados).- Digestivas: melenas, dolor abdominal, náuseas, vómitos, in-farto y perforación intestinal…- Nerviosas: mononeuritis múltiple (manifestación más suge-rente de vasculitis, dolor y parestesias en la distribución de unnervio periférico y déficit motor del mismo nervio en las horaso días siguientes), cefalea, ACV, convulsiones…- Oculares: iritis, uveítis, conjuntivitis…- ORL: congestión nasal, epistaxis, sinusitis de repetición…- Cardíacas: pericarditis, miocarditis, infarto, insuficiencia car-díaca congestiva…
Pruebas de laboratorio- Biopsia: diagnóstico de confirmación.- Estudio de sangre: reactantes de fase aguda, ANCA, factorreumatoide.- Angiografía: útil cuando la biopsia no es fácil, especial-mente en la PAN y en la arteritis de Takayasu, también enChurg-Strauss.- Pruebas dirigidas a encontrar la enfermedad subyacente (au-toanticuerpos, serología VHB, VHC, crioglobulinas…) y a de-tectar la afectación orgánica (análisis de orina, sangre ocultaen heces, Rx tórax…).
3.1.- Panarteritis nodosa
La PAN es el prototipo de las vasculitis necrosantes sistémicas,en la que el proceso inflamatorio afecta arterias musculares demediano y pequeño tamaño y, secundariamente, arteriolas y vé-nulas adyacentes.
EpidemiologíaLa PAN es una enfermedad infrecuente, cuya incidencia se es-tima entre 4 y 10 casos por millón de habitantes. Puede apare-cer a cualquier edad, pero es excepcional en la infancia(MIR), y los casos descritos como PAN infantil probablementecorrespondan a enfermedad de Kawasaki. Predomina en los va-rones entre los 40 y los 60 años, con una proporción de 2-2,5:1en relación con las mujeres. Existe una asociación con el VHB(20-30% presentan antígenos) (MIR 97, 114), el 5% tienen in-fección por VHC. Se ha encontrado también asociación con latricoleucemia y con el consumo de anfetaminas.
Anatomía patológicaLa lesión fundamental consiste en un proceso inflamatorio ne-crosante que afecta primariamente a arterias musculares de pe-queño y mediano tamaño, aunque las arteriolas y las vénulassubyacentes pueden afectarse de forma secundaria. La lesión esde distribución segmentaria y se localiza sobre todo en zonasde bifurcación de los vasos. Existe necrosis fibrinoide e infil-tración con predominio de polimorfonucleares neutrófilos en lafase aguda y de células mononucleares cuando la lesión se cro-nifica. Se pueden visualizar dilataciones aneurismáticas (seproducen por la rotura de la elástica interna), hiperplasia de laíntima con trombosis y oclusión de la luz vascular; un hallazgocaracterístico es la observación de lesiones en diversos esta-dios evolutivos, en las que se combinan alteraciones inflama-torias agudas con otras residuales cicatriciales. No es típico de laenfermedad la presencia de infiltrados eosinofílicos o la forma-ción de granulomas.Aunque cualquier órgano puede afectarse por la enfermedad,
Vasculitis Necrotizantes Sistémicas- Poliarteritis nodosa clásica (PAN)- Vasculitis granulomatosa de Churg-Strauss- Síndrome poliangítico de superposición
Granulomatosis de WegenerArteritis de Células Gigantes- Arteritis de la temporal- Enfermedad de Takayasu
Vasculitis de Hipersensibilidad- Antígenos exógenos:
• Púrpura de Schönlein-Henoch• Enfermedad del suero• Vasculitis por fármacos• Vasculitis asociadas a infecciones
- Antígenos endógenos:• Vasculitis asociadas a neoplasias• Vasculitis asociadas a enfermedades del colágeno• Vasculitis asociadas a deficiencias del complemento• Vasculitis asociadas a otras entidades
- Otras Entidades; entre ellas:• Enfermedad de Kawasaki• Vasculitis aislada del SNC• Enfermedad de Buerger
Vasculitis de Grandes Vasos- Arteritis de células gigantes o de la temporal- Arteritis de Takayasu
Vasculitis de Mediano Calibre- Poliarteritis nodosa clásica (PAN)- Enfermedad de Kawasaki- Enfermedad de Buerger
Vasculitis de Pequeño Vaso- Granulomatosis de Wegener- Poliarteritis microscópica- Vasculitis granulomatosa de Churg-Strauss- Púrpura de Schönlein-Henoch- Vasculitis de la crioglobulinemia mixta esencial- Vasculitis leucocitoclástica cutánea- Enfermedad de Behçet
Tabla 1. Clasificación de la vasculitis.

Manual A Mir
18 ] VASCULITIS [
www.academiamir.com
el riñón y el corazón, con más del 70% en las series de autop-sia, son los más involucrados. Por el contrario, el pulmón y elbazo suelen estar respetados.
Cuadro clínicoComo enfermedad multisistémica, la PAN presenta una sinto-matología variada, expresión de los diversos territorios vascula-res afectados. Las manifestaciones generales (fiebre, afectacióndel estado general, adelgazamiento, etc.) están presentes en lamayoría de los enfermos (siendo la fiebre su manifestación fun-damental). La gravedad de la enfermedad depende de la exten-sión y la localización de las lesiones; así, la afectación del SNC,cardíaca, digestiva y renal entraña un peor pronóstico.
- La afectación renal es la más característica y de mayor tras-cendencia, presente en el 60%. Se traduce en proteinuria, mi-crohematuria y cilindros celulares, aunque en ocasiones puedeexistir un síndrome nefrótico completo o fracaso renal agudo.Es habitual que se desarrolle una insuficiencia renal moderada.La hipertensión arterial, presente en más del 50% de losenfermos, es consecuencia de la afectación vascular y de lanefropatía - La afectación articular es frecuente (50-60%), como artral-gias y mialgias. En menor proporción puede aparecer tambiénuna poliartritis no deformante asimétrica, localizada sobretodo en las grandes articulaciones de las extremidades.- La neuropatía periférica puede aparecer hasta en el 70%de los pacientes y, aunque su forma más característica es lamononeuritis múltiple, puede exteriorizarse como polineuro-patía sensitivomotora. Con frecuencia es la manifestación ini-cial y afecta de forma similar las extremidades superiores einferiores. El comienzo brusco, con síntomas sensitivos inten-sos seguidos en corto tiempo, a veces horas, de déficit motoresgraves es característico de esta entidad (MIR 97, 117). La afec-tación del SNC es mucho menos frecuente (cuadros de disfun-ción cerebral o cerebelosa -vasculitis difusa- o como defectosmotores focales -isquemia, hemorragia o infarto-).- Dentro de la afectación gastrointestinal (44%) el dolorabdominal difuso es su síntoma más frecuente, también po-demos encontrar náuseas, vómitos, estreñimiento, hemateme-sis o melenas, como consecuencia de cuadros suboclusivos,perforativos o ulcerativos (MIR). La afectación hepática es
variable y más frecuente en los casos de PAN asociada a losvirus de la hepatitis B o C, con aumento de fosfatasas alcalinas.También puede ocasionar aneurismas de la arteria hepática.Otras manifestaciones digestivas menos frecuentes de la PANson la colecistitis, el infarto hepático masivo o el infarto pan-creático por afectación vascular. - Las manifestaciones cutáneas (43%) están presentes casien la mitad de los pacientes. La más característica es la púrpurapalpable. También pueden aparecer livedo reticularis, ulcera-ciones o acrocianosis. En ocasiones, la PAN puede quedar limi-tada a la piel como única expresión de la enfermedad.
- La afectación coronaria (34%) se traduce por arritmias ymanifestaciones de cardiopatía isquémica, con insuficienciacardíaca o sin ella, o pericarditis. La afectación genital sepuede traducir en orquiepididimitis o prostatitis. La vasculitisde los vasos retinianos se traduce por la presencia de hemo-rragias o exudados que, si afectan la mácula, pueden compro-meter la visión.
Se acepta que el pulmón está respetado en esta entidad(diagnóstico diferencial con PAN microscópica), y la presenciade infiltrados pulmonares descritos en ocasiones en la PAN apo-yaría la existencia de un síndrome de solapamiento u overlapcon la vasculitis de Churg-Strauss o con la granulomatosis deWegener. Debemos replantear el diagnóstico si existe afectaciónpulmonar. Es frecuente la asociación de manifestaciones propiasde diferentes vasculitis en el mismo paciente (MIR 04, 12).
Pruebas de laboratorioNo existen parámetros de laboratorio específicos de la enferme-dad (MIR 99, 85). Se observa un aumento de la VSG y de reac-tantes de fase aguda (plaquetas, fibrinógeno, haptoglobina,
Figura 1. Vasculitis con oclusión vascular.
Figura 2. Evolución de la afectación cutánea de la PAN con úlceras cutáneas.

R e u m a t o l o g í a
19] VASCULITIS [
ferritina, etc.), leucocitosis con neutrofilia sin eosinofilia. Puededetectarse hipergammaglobulinemia, FR, crioglobulinas e inmu-nocomplejos circulantes. Los valores de complemento (C3 y C4)pueden estar disminuidos por consumo en fase aguda de la en-fermedad. En un 20-30% de los pacientes existen marcadores del virus dela hepatitis B. Los anticuerpos antinucleares (ANA) rara vez sonpositivos y los ANCA son + en el 30%, siendo la variedad p-ANCA la más común (MIR 00F, 96).
DiagnósticoDebe tenerse en cuenta ante las siguientes situaciones:
El diagnóstico definitivo se establece mediante la confirmaciónhistológica (biopsias superficiales de piel, músculo, nervio peri-férico…o profunda- de riñón o hígado). Dado el carácter seg-mentario de las lesiones, una biopsia negativa no excluye eldiagnóstico, estando indicada la realización de una arteriografíaselectiva abdominal (tronco celíaco y arterias renales) apare-ciendo microaneurismas en el 70% de casos aunque no sonespecíficos, su presencia en el contexto de un cuadro clínicocompatible permite establecer el diagnóstico (en los casos enlos que la biopsia no sea posible) (MIR 08, 81).
TratamientoLos corticoides son la base del tratamiento (1mg/Kg/día). La ad-ministración de inmunosupresores (fundamentalmente la ciclo-fosfamida) está indicada: inicialmente en caso de afectaciónsevera, en caso de ausencia de respuesta a corticoides y comofármacos “ahorradores de corticoides”. El pronóstico es malosin tratamiento (supervivencia del 15% a los 5 años sin trata-miento frente al 90% con tratamiento).
3.2.- Poliangeitis microscópica (MIR 07, 97)
Vasculitis necrotizante sistémica, que afecta a pequeños vasossin formación de aneurismas. Las lesiones anatomopatológi-cas se encuentran en el mismo estadio y cursa con leucocitocla-
sia (restos nucleares de neutrófilos). Presenta manifestacionescomunes a la PAN clásica, salvo que la glomerulonefritis es muyfrecuente y que a menudo aparece una capilaritis pulmonar.Las manifestaciones más frecuentes son consecuencia de laafectación cutánea y pulmonar, siendo la complicación másgrave la hemorragia alveolar. La HTA, al contrario que en la PANclásica, es rara.Existe una débil asociación con VHB, y los ANCA son + en másdel 50% de pacientes con patrón p-ANCA fundamentalmentey c-ANCA (MIR 04, 12).El tratamiento es similar al anterior y el pronóstico malo sintratamiento (supervivencia del 75% a los 5 años con trata-miento).
3.3.- Vasculitis granulomatosa de Churg-Strauss (an-geítis y granulomatosis alérgica)
Vasculitis necrotizante sistémica granulomatosa caracterizadapor afectación pulmonar, historia de asma grave y atopia,infiltración eosinófila tisular e intensa eosinofilia periférica.Su etiología es desconocida y se acepta que su patogenia es in-munoalérgica por reacción antígeno-anticuerpo mediada porIgE. Es una forma rara de vasculitis, más frecuente en varones,(1,5:1) y con predominio en la cuarta década de la vida, aunquepuede aparecer desde la juventud hasta edades avanzadas.
Anatomía patológicaVasculitis necrotizante sistémica granulomatosa que afecta avasos de pequeño y mediano calibre, sobre todo pulmonares. Secaracteriza por la tríada clásica:
- Vasculitis necrotizante de arterias musculares de pequeño ymediano calibre, capilares, vénulas y venas.- Infiltración tisular por eosinófilos.- Granulomas vasculares y extravasculares.
Cuadro clínicoLas manifestaciones generales del proceso (fiebre, adelgaza-miento, malestar general) son similares a las de la PAN clásica,
- Proteinuria (síndrome nefrótico)- Hematuria- I. renal- HTA
RENAL (60-70)MÁS FRECUENTE
MÚSCULOESQUELÉTICA(50-60)
Artralgias-mialgias, poliartritis nodeformante
CUTÁNEO (50) Púrpura palpable, lívedo reticularisinfartos piel, acrocianosis, gangrena digital
SNP (50) Mononeuritis múltiple, PN sensitivo-motora
ÓRGANO (%) MANIFESTACIÓN CLÍNICA
GI (40) Dolor abdominal, ↑ F. alcalinas, colecistitis.
CORAZÓN (30) I. cardíaca, IAM, pericarditis
GENITOURINARIO (25) Dolor testicular
OJOS Hemorragias / exudados retinianos
Tabla 2. Manifestaciones clínicas de la PAN.
- Fiebre de origen desconocido- Cuadro multisistémico- Síntomas isquémicos en cualquier localización, sobre todo en jóvenes- Púrpura palpable y/o nódulos cutáneos con necrosis- Mononeuritis múltiple- Glomerulonefritis
La gran simuladora
Tabla 3. Datos clínicos que deben hacer sospechar PAN.
Figura 3. Arteriografía en un enfermo con PAN. Nótese la presencia de aneuris-mas en las arterias de mediano calibre. Hallazgos similares pueden apreciarse enla vasculatura mesentérica, sobre todo en la arteria hepática.

Manual A Mir
20 ] VASCULITIS [
www.academiamir.com
de la que se diferencia fundamentalmente por la afectación res-piratoria que domina el cuadro. Aparece en personas con ante-cedentes atópicos, historia de alergia estacional, rinitis, sinusitisrecidivante o poliposis nasal y precede, en ocasiones hasta en 30años, a la aparición de las manifestaciones sistémicas. Puede en-contrarse afectación crónica de los senos paranasales (a diferen-cia de la PAN y al igual que puede producirse en la enfermedadde Wegener) (MIR 04, 12; MIR 99F, 95; MIR 97, 227).La afectación pulmonar se caracteriza por infiltrados pul-monares migratorios, no cavitados y bilaterales. La afecta-ción renal es menos frecuente y menos grave que en la PAN.El corazón es uno de los órganos afectados con mayor frecuen-cia. La afectación cardíaca es responsable de más del 50% de lasmuertes en estos pacientes. La lesión característica es una mio-cardiopatía restrictiva con obliteración de las cavidades porsustitución del tejido miocárdico normal por infiltración granu-lomatosa eosinófila y fibrosis, que determina la aparición de in-suficiencia cardíaca progresiva y rebelde al tratamiento. Elinfarto de miocardio por vasculitis coronaria es una complica-ción frecuente y la afectación del pericardio puede conducir auna pericarditis constrictiva.
Pruebas de laboratorioEl hallazgo más típico de la enfermedad es la existencia de unaimportante eosinofilia periférica (>1000 eos/ml) durante la faseaguda de la enfermedad, que es un buen marcador de la res-puesta al tratamiento (MIR). El aumento de los reactantes defase aguda es constante durante las fases de actividad de la en-fermedad; podemos encontrar elevados los niveles de IgE; pre-sentan p-ANCA + en 65% de los casos.
DiagnósticoSospecha clínica y de laboratorio, y confirmación mediante biop-sia cutánea, de pulmón, nervio o cualquier órgano potencial-mente afectado y sugerido por la clínica.
TratamientoEl tratamiento es similar al de la PAN, generalmente con buenarespuesta a corticoides. La enfermedad se comporta de formasimilar a la PAN, con afectación sistémica (piel, SNC,...) y tieneun mejor pronóstico que la PAN clásica.
3.4.- Granulomatosis de Wegener
La granulomatosis de Wegener es una vasculitis granulomatosanecrosante que afecta a las vías respiratorias superiores e infe-riores, glomérulos renales y en grado variable a otros órganospor vasculitis de pequeños vasos. La etiología es desconocida,se acepta que el sistema inmune participa en su patogenia.
Anatomía patológicaLas lesiones histológicas de las vías respiratorias y del parén-quima pulmonar se caracterizan por la presencia de granulomasnecrosantes intra y extravasculares, con abundantes células gi-gantes multinucleadas y por una vasculitis necrosante queafecta a pequeñas arterias y venas.
Cuadro clínicoLa granulomatosis de Wegener puede ocurrir en personas decualquier edad, pero es más frecuente hacia los 40 años. Lasmanifestaciones clínicas iniciales de la enfermedad suelen serlas correspondientes a la afección de las vías respiratorias supe-riores, que se pueden asociar a un cuadro general de astenia,anorexia y pérdida de peso. Desde el inicio o bien a lo largo dela evolución, suelen aparecer alteraciones en otras localizacionesanatómicas, entre las que destacan las renales, articulares, cu-táneas, oculares y neurológicas.
- Las vías respiratorias superiores se afectan en más del90% de los pacientes, con desarrollo de sinusitis crónica(forma más frecuente de presentación), rinitis, otitis media,estenosis traqueal, se han descrito ulceraciones nasales, per-foraciones del tabique nasal y deformidades de la nariz ensilla de montar (granuloma de la línea media).- En los pulmones se observan alteraciones radiológicas en lamayoría de los pacientes (más del 85% de los casos) en formade infiltrados pulmonares múltiples y/o nódulos con ele-vada tendencia a la cavitación, no migratorios (MIR). Susmanifestaciones clínicas suelen ser tos, disnea, dolor torácicoy hemoptisis que puede llegar a ser masiva. Es la vasculitisque más frecuentemente afecta al pulmón.- La afectación renal no es precoz, pero ocurre en más del80% de los pacientes durante los primeros 2 años de evolu-ción. Las manifestaciones iniciales incluyen proteinuria y alte-raciones en el sedimento, con hematuria y cilindros hemáticos.Es posible observar distintos grados de afectación glomerular,que pueden evolucionar hacia una glomerulonefritis necroti-zante proliferativa extracapilar (glomerulonefritis rápida-mente progresiva con formación de semilunas), condeterioro rápido de la función renal (MIR 05, 86). La HTA esrara.- Clínica general, con artralgias, artritis, fiebre, astenia, afec-tación cutánea (40%), afectación ocular (posibilidad de prop-tosis del globo ocular). En el 20% de los pacientes, seproducen manifestaciones neurológicas que incluyen, en elSNC, la lesión de pares craneales y de la hipófisis y, en el SNP,la aparición de mononeuritis múltiple o una polineuritis simé-trica. Las manifestaciones cardíacas son la pericarditis, miocar-diopatía o vasculitis coronaria (8%) (MIR 98, 232).
Pruebas de laboratorioSuele existir anemia normocítica y normocrómica, leucocitosisneutrofílica sin eosinofilia y trombocitosis con elevación de laVSG. Se puede observar hipergammaglobulinemia (IgA) y posi-tividad del factor reumatoide con negatividad de los ANA. Se pueden encontrar p-ANCA en vasculitis diferentes al Wege-ner, varias glomerulonefritis o en el síndrome de Goodpasture;sin embargo, la presencia de c-ANCA es muy específica (95%)y sensible (88%) en el Wegener (MIR 08, 78; MIR 05, 86). Enmás del 95% de los pacientes con enfermedad activa generali-zada y en el 70% de los que la presentan limitada, se detectanc-ANCA, con especificidad para la proteinasa-3. Los títulos de c-ANCA se correlacionan con la actividad de la enfermedad,(aunque debemos tener presente que pueden aparecer en otrassituaciones, como la PAN, y la policondritis recidivante…).
DiagnósticoSospecha clínica y de laboratorio (c-ANCA), y confirmación porbiopsia pulmonar (es la de mayor rentabilidad).
TratamientoEn la actualidad el tratamiento de elección es la ciclofosfamida(inicialmente, 2 mg/Kg/día). En el comienzo del tratamiento esimportante añadir prednisona (1 mg/Kg/día durante el primermes, que se irá disminuyendo paulatinamente). La ciclofosfa-mida debe mantenerse el menos durante 1 año después de
Criterios diagnósticos:- Asma- Eosinofilia >10% del recuento leucocitario- Mononeuropatías, mononeuritis múltiple o polineuropatía- Infiltrados pulmonares fugaces no cavitados.- Historia de sinusitis aguda o crónica o veladura radiológica de los senos pa-
ranasales- Presencia de infiltración eosinófila extravascularLa presencia de 4 criterios o más da una sensibilidad del 85% y especificidaddel 99,7%
Tabla 4. Criterios diagnósticos de la enfermedad de Churg-Strauss.

R e u m a t o l o g í a
21] VASCULITIS [
haber obtenido la mejoría clínica, comenzando entonces unadisminución de la dosis de manera progresiva. Más del 90% delos pacientes experimentan una mejoría clínica (75% remisióncompleta), e incluso las recaídas responden también a la reintro-ducción del tratamiento (MIR 06, 86; MIR 05, 86; MIR 04, 12;MIR 00F, 229; MIR 98, 225).Si el paciente no tolera la ciclofosfamida puede ser útil el uso demetotrexate (hasta 20-25 mg/semana).El pronóstico es malo sin tratamiento (parecido al del Churg-Strauss) (MIR 05, 86). La mortalidad (<15%) se relaciona fun-damentalmente con la afectación renal.
3.5.- Arteritis de la temporal o de células gigantes oenfermedad de Horton
Vasculitis granulomatosa que afecta a arterias de mediano ygran calibre, especialmente a ramas de la arteria carótida (sobretodo a la arteria temporal); en cualquier caso se trata de un pro-ceso sistémico que puede afectar a arterias de múltiples locali-zaciones.
La etiología es desconocida, parece existir cierta predisposicióngenética (casos familiares y mayor prevalencia de HLA DR4 yDRB1), y participación autoinmune. Afecta fundamentalmentea individuos mayores de 50 años. Su máxima incidencia ocurreen la octava década de la vida y es unas 2 veces más frecuenteen la mujer que en el varón. Aproximadamente el 50% de lospacientes presenta polimialgia reumática asociada, síndromeclínico caracterizado por la existencia de dolor y rigidez en lascinturas escapular y pelviana.
Anatomía patológicaLa pared arterial se halla ocupada por un infiltrado inflamatoriocompuesto por linfocitos y macrófagos que adoptan una orga-nización granulomatosa. Es frecuente la presencia de células gi-gantes multinucleadas. Se produce una proliferación de laíntima con fragmentación de la lámina elástica. Aunque laafectación de las ramas de la carótida es la que tiene mayor ex-presividad clínica, en estudios necrópsicos se ha comprobadoque, prácticamente en todos los casos, hay compromiso de laaorta y de sus ramas principales.
Cuadro clínicoCon frecuencia la enfermedad lleva varias semanas o meses deevolución en el momento del diagnóstico, debido a que, a me-nudo, las manifestaciones iniciales son inespecíficas, con unaimportante repercusión sobre el estado general, con astenia,anorexia, pérdida de peso y depresión.La cefalea hemicraneal es uno de los síntomas más caracterís-ticos (presente en 60-98%). Suele ser de instauración recienteo de características distintas a las habituales. Con frecuencia seacompaña de dolor en el cuero cabelludo, algias faciales atípicasen las zonas occipital o cervical. La claudicación mandibular,aunque se ha observado también en otros procesos (amiloidosis,PAN), sugiere fuertemente el diagnóstico (MIR).Pueden hallarse anomalías en la exploración física de las arteriastemporales: el endurecimiento y la disminución o ausencia depulso a la palpación de las arterias temporales superficiales uotras arterias craneales constituyen datos clínicos importantes. Pueden existir complicaciones oculares; así, la pérdida de visiónunilateral o bilateral es una de las complicaciones más gravesde la arteritis temporal. Se produce como consecuencia de la
ANATOMÍAPATOLÓGICA
PAN
Necrosis fibrinoide (vasculitis necrotizante)Afect. segmentaria
Afect. típica del riñonFormación de aneurismas20% infección por VHB
No afecta pulmón (PAN clásica)
DIAGNÓSTICO
CLÍNICA
TRATAMIENTO
CHURG-STRAUSS WEGENER
PNX
Parecida a PAN pero también se afectancapilares y vénulas
Granulomas intra y extravascularesEosinofilia tisular y sanguínea
Afectación predominio de Pulmón
Vasc. necrotizante(A. mediano y pequeño calibre,
capilares y vénulas)Granulomas (intra y extravasculares)
Muy polimorfa(sínt. generales y afecta locomotor, renal, piel, etc,)
Mononeuritis múltiple (muy típica)
Asma grave, rinitis, pólipos nasalesInfiltrados pulmonares
fugaces (no cavitados) bilateralesRiñón (afectación menos frec y grave
que en PAN)80% Eosinofilia intensa
(>1000 cels/mm3)Los demás órganos similar a PAN
Vías respiratorias superiores:Sinusitis crónica
Vías respiratorias inferiores:Nódulos pulmonares cavitados,
múltiples y bilateralesRiñon:
La GNF suele dominar la clínica
1º. Biopsia de órganos afectos2º. Angiografía (aneurismas):
cuando la biopsia no sea posible
BiopsiaP-ANCA
Biopsia:Pulmón (la de >rendimiento)
Riñon: se ve GNF (raro ver granulomas)↑ VSG, IgA y FR
c-ANCA
Corticoides+
CiclofosfamidaCorticoides +/- Ciclofosfamida
Ciclofosfamida(de elección)
+Corticoides
Si HBsAg +:Vidarabina +
Plasmaféresis +Corticoides
Si ef. secundariosgraves: metotrexate o
azatioprina +corticoides
Depende del tratamiento Muerte por alt. cardíaca Muerte por alt. renal
Tabla 5. Diagnóstico diferencial de la vasculitis de pequeño y mediano vaso.
La PAN no afecta al pulmón.
La PAN microscópica sí afecta al pulmón.
En la PAN microscópica es rara la HTA (que es frecuente en laPAN clásica) y no forma aneurismas.
La principal causa de muerte en el Churg-Strauss es la afecta-ción cardíaca (miocardiopatía restrictiva) mientras que en el
Wegener es la afectación renal.
Las vasculitis, en general, se tratan con esteroides, recurrién-dose a inmunosupresores si no hay respuesta. En el caso del
Wegener, se emplea la ciclofosfamida de entrada.
En los casos clínicos del MIR, es muy típico que se hable de eleva-ción de la VSG en la arteritis de la temporal y en el mieloma.
RECUERDA

Manual A Mir
22 ] VASCULITIS [
www.academiamir.com
neuritis óptica isquémica anterior. La pérdida de visión puedeser total (amaurosis) o consistir en amputaciones del campo vi-sual (hemianopsias o cuadrantanopsias). Suele ser de apariciónbrusca, y en ocasiones se halla precedida por episodios de pér-dida de visión transitoria (amaurosis fugaz) o diplopía (MIR).Aproximadamente el 15% de los pacientes sufre defectos vi-suales permanentes.Entre la afectación de otros territorios vasculares destacanla claudicación intermitente de extremidades, con aparición es-porádica de fenómenos isquémicos y el desarrollo de aneurismaaórtico. Con menor frecuencia se produce un accidente vascularcerebral por compromiso carotídeo o vertebral, o una cardiopa-tía isquémica por lesión coronaria.Polimialgia reumática (50%). Dolor y debilidad localizado enlas cinturas escapular o pelviana que se exacerba con la movili-zación y se acompaña de una notable rigidez, especialmentematutina. Puede preceder en varios meses, coincidir o aparecerposteriormente a la sintomatología craneal típica de la arteritistemporal. La polimialgia reumática puede existir como entidad propia enausencia de arteritis temporal.
Pruebas de laboratorioEs muy característica la elevación de la VSG, con frecuenciasuperior a 100 mm/h (dato de laboratorio más útil para la mo-nitorización), también elevación de reactantes de fase aguda.Es común la presencia de anemia y trombocitosis (MIR 08, 80).Con frecuencia existe un aumento de GGT y fosfatasa alca-lina hepática (por granulomatosis hepática). Los niveles de CPKson normales. El sedimento urinario también es normal (MIR01, 81).
DiagnósticoSospecha clínica (cefalea, fiebre, anemia) y analítica (hemo-grama, VSG y bioquímica sérica). La confirmación se realiza me-diante biopsia de la arteria temporal superficial (MIR 98, 229).Ésta debe realizarse lo antes posible, ya que el tratamiento cor-ticoideo, al cabo de unos días, puede negativizar los hallazgoshistológicos. La ausencia de lesiones en la biopsia no excluye eldiagnóstico debido a la naturaleza segmentaria de las lesiones.
TratamientoEl tratamiento de elección lo constituyen los glucocorticoides.Ante la sospecha clínica, para prevenir la ceguera, se debe iniciartratamiento inmediatamente (¡¡es lo primero a realizar!!), antesde proseguir el estudio (MIR 06, 79; MIR 04,11). Además deproducir una remisión rápida y completa de la sintomatología,se ha demostrado su eficacia en la prevención de complicacio-nes isquémicas. La dosis inicial es de 1 mg/Kg/día de prednisona.A partir de las 3-4 semanas, o cuando se ha obtenido la remi-sión clínica y la mejoría de los parámetros biológicos alterados,
se inicia una reducción gradual. Aproximadamente a los 3meses debe alcanzarse una dosis de mantenimiento de 8-12mg/día. Suele ser preciso mantener el tratamiento durante pe-ríodos prolongados, generalmente superiores a un año. Los pacientes con polimialgia aislada, en los que se ha excluidorazonablemente la existencia de arteritis temporal, presentanuna evolución favorable con una dosis inicial de 8-15 mg/día deprednisona; también se pueden dar AINES.
3.6.- Arteritis de Takayasu
Es una vasculitis granulomatosa que afecta sobre todo a la aortay a sus ramas principales (los troncos supraaórticos son los máscomúnmente afectados). Su etiopatogenia es desconocida, pre-domina en mujeres asiáticas durante la segunda y la terceradécadas. Parece existir una asociación con DR2 y 4/MB1 y 3.
Anatomía patológicaLas lesiones se distribuyen fundamentalmente en la aorta y lostroncos supraórticos. También se afectan con frecuencia las ar-terias pulmonares, femorales, renales, mesentéricas, coronariasy vertebrales. Se caracteriza por un infiltrado linfomonocitariocon formación de granulomas y la presencia esporádica de cé-lulas gigantes. En estadios más avanzados la luz se halla redu-cida de manera segmentaria por una fibrosis de la media y dela íntima, que se halla engrosada.
Cuadro clínicoLa enfermedad se instaura de manera insidiosa, de forma que eshabitual un retraso diagnóstico de meses e incluso años tras laaparición de los primeros síntomas (fiebre, malestar, dolor arte-rial). Posteriormente, en la llamada “fase oclusiva” se producensíntomas derivados de la hipoperfusión en los territorios distales.El 70-80% de los pacientes presenta soplos vasculares, disminu-ción o ausencia de pulsos y/o claudicación intermitente de lasextremidades superiores fundamentalmente (es la llamada “co-artación invertida”). En el 30-60% de los casos hay hiper-tensión arterial, que puede ser debida a estenosis de arteriarenal, pérdida de elasticidad aórtica o estenosis de la aorta to-rácica. Es también frecuente la aparición de cefalea y vértigo.Otras complicaciones más graves son: accidente vascular cere-bral, amaurosis fugaz o permanente, hipertensión pulmonar,cardiopatía isquémica y disfunción valvular aórtica. La arteriaafectada con más frecuencia es la subclavia seguida de la ca-rótida común. Sin embargo los síntomas más frecuentes sonlos neurológicos derivados de la afectación de las arterias delSNC. Aproximadamente el 50% de los pacientes presenta ma-nifestaciones sistémicas en forma de artralgias, mialgias, febrí-cula y pérdida de peso. En algunos casos puede aparecer uneritema nodoso (MIR 02, 50).
DiagnósticoSospecha clínica acompañada de elevación de VSG y de reactan-tes de fase aguda. La confirmación se establece por arteriografíaconvencional, en la que se aprecian estenosis vasculares, circu-
90 %
% MANIFESTACIONES CLÍNICAS
Cefalea
Arteria temporal anormal (endurecimiento, disminución oausencia de pulso y signos inflamatorios)75 %
50 % Síntomas constitucionales, importante repercusión sobre elestado general, astenia anorexia, febrícula ( FOD?)
50 % Polimialgia reumática
30 % Claudicación mandibular
10 % Ceguera o síntomas de neuropatía óptica isquémica
7 % Accidente cerebrovascular, disección de aortaVSG, FA y GGT. Anemia
Tabla 6. Manifestaciones clínicas de la arteritis de la temporal.
Arteria subclavia (93%)
AFECTACIÓN DE ARTERIA
Claudicación m. sup., Raynaud
Alt. visión, síncope, AIT/ACVArteria carótida común (60%)
Aorta abdominal (50%) Dolor abdominal, nauseas, vómitos
Arteria Renal (40%) HTA, I. renal
MANIFESTACIÓN
Raíz aórtica (35%) I. aórtica, ICC, C. isquémica
Arteria Vertebrales (35%) Alt. visión, acúfenos
Tabla 7. Arteritis de Takayasu.

R e u m a t o l o g í a
23] VASCULITIS [
lación colateral o aneurismas en las zonas afectadas. La histolo-gía es similar a la encontrada en la arteritis de la temporal.
TratamientoEl tratamiento se basa en la angioplastia y en la cirugía vascular.El 60% de los pacientes responde inicialmente al tratamientocon prednisona (1 mg/kg/día, con reducción paulatina). Algunospacientes que no responden al tratamiento, alcanzan la remisióncon la adición de fármacos inmunodepresores (metotrexato, ci-clofosfamida). El 20% de los enfermos nunca alcanza la remi-sión completa, y sus lesiones progresan a pesar del tratamiento.Hasta el 50% de los casos requiere cirugía derivativa por hiper-tensión vasculorrenal, grave compromiso de las arterias caróti-das o vertebrales, isquemia de extremidades o lesión coronaria.La angioplastia transluminal es eficaz en el 50% de los casos,pero las reestenosis son frecuentes y precoces. La mortalidad (hasta 20%) suele ser debida a la insuficienciacardíaca, al infarto de miocardio y a los ACV.
3.7.- Vasculitis por hipersensibilidad
El término vasculitis por hipersensibilidad engloba un amplio yheterogéneo grupo de síndromes que se caracterizan por la in-flamación, mediada por inmunocomplejos, de pequeños vasos(vénulas postcapilares). La afectación predominante de la pielhace que también se las llame vasculitis predominantementecutáneas. Tienen menor afectación visceral lo que les confiereun mejor pronóstico. Es una de las formas más frecuentesde vasculitis, puede afectar a todas las edades y muestra undiscreto predominio en las mujeres.
EtiologíaLos agentes causantes de este síndrome son diversos, algunosexógenos como productos químicos, antígenos alimentarios omicrobianos y otros endógenos como el DNA, inmunoglobulinaso antígenos tumorales. En muchas ocasiones no es posible iden-tificar la causa de este proceso.
ClasificaciónDe las vasculitis por hipersensibilidad, según sus causas:
Anatomía patológicaSu denominador común es la afectación de los pequeños vasosde la piel, sobre todo de las vénulas postcapilares. Es característicala leucocitoclasia o “polvo celular” (restos nucleares de neutró-filos que han invadido los vasos y sus alrededores durante la faseaguda). En los estadios subagudo y crónico predominan las célu-las mononucleares. Es frecuente la extravasación de hematíes,dando lugar a púrpura palpable (MIR 03, 220; MIR 97F, 239).
Cuadro clínico:El signo distintivo de la vasculitis por hipersensibilidad es la pre-sencia de púrpura palpable en la piel (MIR). Se trata de pá-pulas eritematosas cuyo color no desaparece al ejercervitropresión. Estas lesiones pueden encontrarse en combina-ción con otras como: urticaria (denominándose vasculitis urtica-riforme), pústulas, ampollas e incluso úlceras necróticas (MIR).Pueden localizarse en cualquier zona del cuerpo, de forma simé-trica, pero asientan con preferencia en áreas declives (extremi-dades inferiores y nalgas), y aparecen en forma de brotes,únicos o recurrentes. Cada lesión individual persiste durante 1-4 semanas y, al curar, deja un área de hiperpigmentación.La erupción cutánea puede acompañarse de fiebre y diversossignos de afectación sistémica, sobre todo articular (artralgias,raras veces, artritis), digestiva (dolor abdominal y signos de he-morragia digestiva) y renal (el signo más frecuente de afectaciónrenal es una hematuria asintomática). En general, las manifes-taciones extracutáneas son menos graves que en las vasculitissistémicas.
Granulomas cels. gigantesInflamación de ramas de caró-
tida (s/t temporal; a vecessegmentaria)
cels mononuclearesFragmentación de
elástica interna
TRATAMIENTO
ARTERITIS DE LA TEMPORAL
ANATOMÍAPATOLÓGICA
CLÍNICA
DIAGNÓSTICO
ARTERITIS DE TAKAYASU(MIR 07, 80)
Cayado aórtico ytroncos
supraaórticosGranulomas: cel.
gigantes y mononucleares
CefaleaNeuritis
isquémica anteriorClaudicación mandibular
(muy específica)Polimialgia
reumática (asociado frec.)
Sínt. generalesSínt. de isquemia de zonasafectadas; mujeres jóvenes
(orientales)
Clínica + biopsia (confirma)en cortes seriados
↑ VSG y FA (afectación hepática)Anemia
CPK normal
Sospecha clínicaArteriografía (confirma)
↑ VSG y IgsAnemia
Corticoides precozDosis bajas o aines en
polimialgia aislada
Angioplastia (cirugía) +corticoides +/- inmuno
supresores
Tabla 8. Diagnóstico diferencial de las vasculitis de gran vaso.
- Primarias o idiopáticas- Secundarias a antígenos exógenos:
• Fármacos (MIR 03, 220), proteínas heterólogas, productos químicos- Secundarias a otras enfermedades:
• Neoplasias: sobre todo linfoproliferativas• Enfermedades del tejido conjuntivo: lupus eritematoso, artritis reumatoide,
síndrome de Sjögren• Otras vasculitis: panarteritis nodosa• Enfermedades infecciosas: estreptococos, estafilococos, micobacterias, VEB,
VHB, HIV, E. coli• Deficiencias congénitas del sistema del complemento• Otras: cirrosis biliar primaria, colitis ulcerosa, endocarditis, síndrome de
derivación intestinal, déficit de α1-antitripsina, síndrome de Goodpasture, policondritis recidivante, crioglobulinemia mixta, púrpura de Waldenström
- Formas con características propias:• Enfermedad del suero• Síndrome de Schönlein-Henoch• Vasculitis-urticaria• Eritema elevatum diutinum
Tabla 9. Clasificación de las vasculitis por hipersensibilidad.
Figura 4. Púrpura “palpable” de las vasculitis.

Manual A Mir
24 ] VASCULITIS [
www.academiamir.com
DiagnósticoPúrpura palpable y biopsia del órgano afectado. No hay datosespecíficos de laboratorio. Generalmente encontramos leucoci-tosis y aumento de la VSG, en algunos casos hay crioglobulinasy factor reumatoide positivo. Puede asociarse a deficiencias defactores del complemento.
TratamientoEn caso de identificar un posible agente causal, la primera me-dida es el tratamiento etiológico. En caso de sospecha de causafarmacológica, se deberá retirar el tratamiento. En casos leves,bastará con tratamiento sintomático, reservándose los gluco-corticoides para los casos en los que existan síntomas sistémicos,afectación cutánea diseminada o afectación de otros órganos.
Crioglobulinemia mixta esencialLas crioglobulinas son inmunoglobulinas capaces de precipitarcon la exposición al frío. Su presencia en concentraciones supe-riores a la normalidad (crioglobulimenia), puede causar daño sise produce su precipitación en los tejidos:Encontramos dos tipos:
- Crioglobulinemia mixta esencial: no se acompaña de tras-tornos sistémicos conocidos (30%)- Crioglobulinemia secundaria (MIR 07, 96): asociada atrastornos linfoproliferativos y mieloma, vasculitis sistémicas,LES, AR y otras conectivopatías, infección crónica, cirrosis….- También pueden dividirse en formas monoclonales (tipo I) ypoliclonales (tipo II y III). Las formas policlonales cursan condescenso del complemento.
Anatomía patológicaVasculitis leucocitoclástica de pequeños vasos.
ClínicaPredominantemente cutánea (púrpura, Raynaud, livedo reticu-laris…), artralgias, fiebre, adenopatías, hepatoesplenomegalia,afectación renal…
AnalíticaInmunocomplejos circulantes, hipocomplementemia, presenciade anticuerpos anti-VHC en un alto porcentaje de pacientes,también, pero en menor porcentaje, VHB. En algunos tipos, seencuentra Factor Reumatoide.
TratamientoMala respuesta al tratamiento, se usa plasmaféresis junto concorticoides y ciclofosfamida. Si existe infección VHC asociada,se usa interferón alfa.
Púrpura de Schönlein-HenochEs más frecuente en la infancia, en varones y suele aparecer trasinfecciones de las vías respiratorias. También se han involu-crado fármacos, ciertos alimentos, picaduras de insectos y vacu-naciones. La inmunoglobulina mediadora de esta reacción es laIgA.
Anatomía patológicaVasculitis leucocitoclástica, se ha podido demostrar el depósitode inmunocomplejos (formados por IgA) (MIR 98F, 144) en laslesiones de la piel y en el mesangio renal por inmunofluores-cencia directa (IFD).
Clínica (MIR 2007, 255)Más frecuente en primavera y con frecuencia sigue a una infec-ción respiratoria de vías altas. La púrpura palpable no trom-bopénica (en las nalgas sobre todo aunque también enmiembros inferiores) es la manifestación más frecuente; la ma-nifestación sistémica más común es la digestiva (dolor abdomi-
nal, hemorragia…), la afectación renal puede manifestarsecomo una hematuria o proteinuria (en una minoría puede pro-ducir síndrome nefrótico que evolucione a insuficiencia renalcrónica). El compromiso articular incluye artralgias e inclusopoliartritis no erosiva (afecta a tobillos, rodillas, carpos y pe-queñas articulaciones de las manos) que no deja secuelas (MIR05, 186; MIR 00,127; MIR 00, 213).
AnalíticaLeucocitosis y elevación de IgA (MIR).
TratamientoEn general sólo requiere tratamiento sintomático. En casos gra-ves, dar glucocorticoides a dosis altas durante un tiempo limi-tado (ej. síndrome nefrótico) (MIR 99, 183). Es un cuadroautolimitado en la mayoría de los casos, con tendencia a recidi-var en un período de semanas o meses, y sólo en un pequeñoporcentaje, evoluciona a la cronicidad.
Enfermedad del sueroEstá mediada por IgE y se observa tras la administración de de-terminados fármacos. El proceso es autolimitado y cursa con fie-bre, artralgias, poliadenopatías y una erupción cutáneaurticariforme.
Vasculitis-urticariaSe manifiesta en forma de habones que persisten 24-48 h ydejan pigmentación residual. La erupción tiene un carácter re-currente y, en ocasiones, se acompaña de una alteración mode-rada del estado general y disminución del complemento sérico.El proceso puede ser idiopático o acompañar a otros procesos,como el lupus eritematoso sistémico o el síndrome de Sjögren.
Eritema elevatum diutinumEs la expresión de una reacción de hipersensibilidad persistentey mantenida, en pacientes con infecciones estreptocócicas defaringe y senos. Se expresa en forma de placas o nódulos dér-micos, localizados en la superficie de extensión de las extremi-dades, que persisten meses o años. En ocasiones apareceasociado a gammapatías monoclonales IgA. Puede tratarsecon sulfona.
3.8.- Enfermedad de Kawasaki
Es una vasculitis necrosante difusa que afecta siempre a arte-riolas, capilares y vénulas, de manera casi constante las arteriascoronarias y, a menudo, grandes arterias no intraparenquima-tosas. Se presenta de manera esporádica o en forma de peque-ños brotes epidémicos. Es más frecuente en Japón. El 85% delos pacientes tienen menos de 5 años. Predomina en los varones(1,5:1). En casi todos los casos mortales, los estudios histopato-lógicos han revelado lesiones de vasculitis coronaria, con aneu-rismas múltiples (en “cuentas de rosario”) y trombosiscoronaria, indiferenciables de la PAN infantil.
EtiopatogeniaEs desconocida, están implicados agentes que actuarían comosuperantígenos
Cuadro clínicoSe trata de una enfermedad aguda, febril, autolimitada (9-60días). También denominada síndrome linfomucocutáneo, cuyosrasgos clínicos fundamentales están descritos en la tabla 10.
DiagnósticoEl diagnóstico se acepta en presencia de fiebre de más de 5 días,junto con cuatro o más de los otros criterios y siempre que elproceso no pueda explicarse por otra causa.

R e u m a t o l o g í a
25] VASCULITIS [
Debido al carácter multisistémico de la enfermedad, puedehaber artralgias, artritis, dolor abdominal, meningitis y especial-mente alteraciones cardiovasculares, como miocarditis, insufi-ciencia cardíaca y arritmias, aneurismas coronarios (15-25%) ypericarditis (la gravedad viene marcada por el grado de afecta-ción cardíaca). Para su monitorización se recomiendan la ecocar-diografía cardíaca.
Pruebas de laboratorioLos datos de laboratorio son inespecíficos. En el 75% de loscasos, hay una elevación de la IgE entre la tercera y la quintasemana, momento en el que se desarrollan aneurismas corona-rios y aparece la trombocitosis. A veces se detectan ANCA y an-ticuerpos anticélula endotelial.
Evolución y pronósticoLa mayoría de los pacientes se recuperan por completo y sólo el3,9% recidiva. Algunos fallecen (0,5-2,8%), y la muerte, confrecuencia súbita, ocurre en las fases iniciales por miocarditisaguda o arritmias, y, en las tardías, por ruptura de aneurismascoronarios o por trombosis con infarto de miocardio.
TratamientoSe basa en la administración precoz y combinada de salicilatos(80-100 mg/kg/día durante los primeros 14 días y luego 3-5mg/kg/día) y altas dosis de gammaglobulina intravenosa (400mg/kg y día, 4 días consecutivos, o en dosis única de 2 g/kg). Enla fase aguda de la enfermedad pueden requerirse digital y diu-réticos para tratar la insuficiencia cardíaca.El tratamiento trombolítico, la derivación coronaria y el tras-plante cardíaco son opciones terapéuticas adicionales que sehan de valorar en cada caso concreto.
3.9.- Síndrome de Behçet
Es una enfermedad multisistémica, crónica y recidivante. El sus-trato patológico es una vasculitis leucocitoclástica o linfocítica,preferentemente de capilares y vénulas. Aunque infrecuente, esposible también la afectación de vasos de mayor calibre.Es más frecuente en los países mediterráneos orientales y enJapón, pero su distribución es universal. Su prevalencia es difícilde estimar porque muchos casos no son diagnosticados. Predo-mina en los varones (2-4:1) y puede aparecer a cualquier edad,con una frecuencia máxima en la tercera década.
EtiopatogeniaLa etiopatogenia es desconocida. El antígeno HLA-B5 (B51 yB52) es 3-4 veces más frecuente entre los enfermos que en loscontroles.
Cuadro clínicoEs característica la aparición de aftas orales (100%, imprescin-dible para el diagnóstico) recidivantes, dolorosas, que curan en1-3 semanas, generalmente sin dejar cicatriz. En la mayoría delos casos son la primera manifestación (MIR 98, 233). En elvarón, las aftas genitales (60-80%) suelen ser dolorosas, aveces de gran tamaño y curan lentamente dejando cicatriz. Enla mujer, pueden pasar inadvertidas.
- La afectación ocular (60-70%), casi siempre bilateral, es lacomplicación más grave. La participación del segmento ante-rior en forma de uveítis es la más característica, pero la afec-tación del segmento posterior, sola o junto con la del anterior(panuveítis), es más frecuente.- La afectación cutánea es frecuente (70-80%) y variada (le-siones papulopustulosas, pioderma, eritema nodoso, vas-culitis cutánea, nódulos acneiformes, foliculitis, etc). Existe el fenómeno de patergia, que consiste en el desarrollode una reacción inflamatoria tras la administración de suerosalino intradérmico.
- La afectación articular. Las artralgias son más comunes que
- Fiebre de 38-40°C- Inyección conjuntival no exudativa bilateral- Alteraciones orofaríngeas (una o varias), consistentes en lengua de fram-
buesa, eritema difuso, labios enrojecidos, secos y agrietados, con fisurasangulares
- Alteraciones en las manos y los pies (una o varias), como eritema palmar yplantar, edema indurado, descamación, en general limitada a los dedos(fase de convalecencia)
- Exantema polimorfo en las superficies extensoras de los miembros y tronco;uno de los signos iniciales es una erupción perianal
- Poliadenopatías cervicales agudas no supuradas (al menos una adenopatía>1,5 cm de diámetro)
Tabla 10. Criterios diagnósticos de la Enfermedad de Kawasaki.
E. KAWASAKI(SDR. LINFO-
MUCOCUTÁNEO)
Aneurismascoronarios (25%)
Proliferacióntípica de íntima
(histologíasimilar a PAN)
Infiltración mono-nuclear de vasos
VASCULITISHIPERSENS
SCHÖNLEIN-HENOCH
ANATOMÍAPATOLÓGICA
FiebreAdenitis cervical
no supurativaAlt. en piel y muco-sas (eritema bucal,
descamaciónyemas dedos,..)
No responde a ATBAneurismas
coronarios "encuentas de rosa-rio" (3ª-4ª sem)
Vasculitisleucocitoclástica
CLÍNICA
TRATAMIENTO
DIAGNÓSTICO
Vasculitisleucocitoclástica
Afectación +frec: vénulaspostcapilares
Púrpura palpable(nalgas)
ArtralgiasDolor abdominalGNF, hematuria
Piel:púrpura palpableen zonas declives
Clínica (Criterios Dx)Útil Ecocardio
BiopsiaICs (IgA)
Biopsia pielBuscar etiología
Tratamiento precoz:Globulinas iv.
1º- 4º d+ Salicilatos:Fase febril:dosis altas
(antiinflamatorias)Fase
convalecencia:dosis bajas
(antiagregante)
No precisaTratamientoCorticoides
Tratamientoetiológico
+/-Corticoides
Tabla 11. Resumen de vasculitis cutáneas y Kawasaki.
BeChetHLA B Cinco
REGLA MNEMOTÉCNICA Figura 5. Behçet: A. Aftas orales B. Aftas genitales C. Uveitis posterior.
A CB

Manual A Mir
26 ] VASCULITIS [
www.academiamir.com
las artritis (30-60%); la artritis suele ser monoarticular u oligo-articular, no deformante ni erosiva y de curso subagudo auto-limitado recidivante. Las rodillas, los tobillos, los codos y lasmuñecas son las más afectadas. ¡No es un criterio diagnóstico!(MIR).- La afectación de vasos distintos de los de la microcirculaciónse halla presente en un tercio de los casos y es fundamental-mente venosa. Se trata de tromboflebitis superficiales(25%) y, con menos frecuencia, de trombosis del sistema ve-noso profundo; el compromiso arterial es raro (MIR).- Las manifestaciones neurológicas (4-48%), en ocasiones,(5%) constituyen la primera manifestación, con preferenciapor el tronco cerebral y los núcleos de la base. Destacan por sufrecuencia, las cefaleas difusas, la meningitis aséptica reci-divante, la hipertensión intracraneal benigna (pseudotu-mor cerebrii) y síndromes de tronco cerebral. En el LCR es casiconstante una hiperproteinorraquia moderada, con pleiocitosisdiscreta, a veces de predominio polimorfonuclear y otras, lin-focitario o mixto. La tomografía computarizada (TC) puede sernormal o mostrar alteraciones diversas e inespecíficas. Los po-tenciales evocados y la resonancia magnética (RM) craneal tie-nen un alto rendimiento diagnóstico y son de utilidad paradetectar lesiones subclínicas. - Las manifestaciones digestivas (5-40%) consisten por lo ge-neral en episodios de diarrea, dolor abdominal, distensión,náuseas y vómitos. Pueden aparecer lesiones ulcerosas en es-pecial en la región ileal o ileocecal y causar complicacionesagudas (perforación, hemorragia masiva). - Otras manifestaciones menos frecuentes son fiebre, orquie-pididimitis y miositis.
Pruebas de laboratorioLos hallazgos son inespecíficos. En el 50% de los casos se puedeencontrar anticuerpos contra la mucosa oral.
DiagnósticoEl diagnóstico es clínico. Los criterios diagnósticos de la enferme-dad son (MIR 05, 148; MIR 98F, 209):
Evolución y pronósticoPor lo general es una enfermedad benigna, pero en ocasionesse asocia a complicaciones graves. Muchos de los pacientes conafectación del segmento posterior del ojo sufren una pérdidagrave de la visión. La mayoría de los casos mortales presentanafectación neurológica, ulcerativa intestinal o vascular. Algunosdesarrollan una amiloidosis sistémica de tipo AA.
TratamientoLa elección del tratamiento depende de las manifestaciones clí-nicas y de su gravedad. Los glucocorticoides son efectivos parala mayoría de las manifestaciones, pero no evitan las recidivas.Las dosis elevadas (1 mg/kg/día) se reservan para las formas gra-ves de la enfermedad. Rara vez hay que utilizar citotóxicos (clo-rambucilo, azatioprina). La ciclosporina A (3-5 mg/kg/día) es útilpara el tratamiento de la afección ocular. Para las aftas oralespueden emplearse la colchicina o los glucocorticoides tópicos.Un porcentaje importante de enfermos requiere sólo cuidadosmínimos.
3.10.- Enfermedad de Buerger o tromboangeítisobliterante
Es una vasculopatía inflamatoria, oclusiva, segmentaria y recidi-vante de etiología desconocida que afecta arterias y venas demediano y pequeño calibre de las extremidades y, sólo rarasveces, a arterias viscerales y cerebrales. Predomina en varonesjóvenes, orientales, de 20 a 40 años, en su mayoría (95%) fuma-dores importantes, pero se ignora el mecanismo mediante elcual el tabaco influye en su aparición. Es dudosa la intervenciónde factores inmunológicos. No produce afectación visceral.
Anatomía patológicaLa especificidad histológica de las lesiones vasculares es muy ele-vada en los estadios agudos y muy baja en los crónicos. En el es-tadio agudo, hay proliferación de la íntima y formación detrombos inflamatorios oclusivos con microabscesos rodeadospor una reacción granulomatosa con células gigantes, hallazgoque no se observa en ningún otro tipo de enfermedad vascular.Es importante destacar que las lesiones agudas pueden versetanto en arterias como en venas. El estadio crónico se carac-teriza por fibrosis de la pared vascular y recanalización deltrombo; asimismo pueden existir lesiones en distintos estadiosevolutivos.
Cuadro clínicoLas manifestaciones clínicas dependen de la isquemia periféricapor oclusión arterial, que empieza distalmente y, con el tiempo,progresa en dirección proximal. La claudicación (fundamental-mente del pie) es el motivo de consulta en el 30-60% de loscasos. A menudo se comprueba la ausencia de pulsos dista-les. Cuando la isquemia es importante aparece dolor en reposo.El 40-50% de los pacientes presentan tromboflebitis migransen algún momento de la enfermedad. Los trastornos tróficosconstituyen la manifestación inicial en el 30-50% de los casos,se localizan sobre todo en los dedos de los pies y manos ypronto se ulceran y sobreinfectan. Son muy dolorosos y en elmejor de los casos cicatrizan, aunque dejan un defecto estéticoy/o funcional, pero, en muchos otros evolucionan hacia la gan-grena y obligan a la amputación.La angiografía muestra estenosis u oclusiones segmentarias, bi-laterales, múltiples, con normalidad de los vasos proximales. Elaspecto “en sacacorchos” que ofrecen las colaterales es tam-bién típico de la enfermedad.
DiagnósticoSe basa en criterios clínicos, angiográficos e histológicos Debesospecharse automáticamente en varones menores de 40 añoscon claudicación o isquemia digital. Los estudios de laboratorioen general no aportan datos orientativos. A diferencia de la arteriosclerosis, los pacientes con tromboan-gitis son más jóvenes, presentan fenómeno de Raynaud, trom-boflebitis migratoria y afectación de las extremidades superiorescon mayor frecuencia y las úlceras periféricas y gangrena sonmás precoces.
Evolución y pronósticoEl pronóstico funcional es bastante malo, ya que con frecuenciaes necesaria la amputación, pero el pronóstico vital es excelente.En los pacientes que siguen fumando o que reanudan el hábito,el proceso es progresivamente mutilante, mientras que la mayo-ría de los que abandonan el tabaco mejoran.
TratamientoLa supresión total y definitiva del tabaco es en realidad la únicamedida terapéutica efectiva, además de tratamiento analgésicopara el control del dolor y la administración de antibióticos porvía general en el caso de lesiones infectadas. La cirugía de revas-
- Ulceras orales (sin cicatriz) recurrentes (3 episodios/año) asociadas a 2 crite-rios de los siguientes:• Ulceras genitales recurrentes (con cicatriz)• Lesión ocular (posterior o anterior): es lo más grave!• Lesiones cutáneas (eritema nodoso, foliculitis, acné, pioderma...)• Fenómeno de patergia positivo
Tabla 12. Criterios diagnósticos de la Enfermedad de Behçet.

R e u m a t o l o g í a
27] ARTRITIS REUMATOIDE [
cularización no suele ser, en general, practicable debido al ta-maño de los vasos afectos. Las indicaciones de la simpatectomíaquedan limitadas a los casos evolutivos, hiperalgésicos y con is-quemia grave, que no mejoran con otras medidas.
3.11.- Otras vasculitis
Vasculitis aislada del SNCEsta entidad se ha definido como una enfermedad con signosclínicos, angiográficos e histopatológicos de vasculitis confinadaal SNC, en ausencia de vasculitis sistémica. Su etiología es des-conocida. Muestra ligero predominio en los varones y en laquinta década. Clínicamente se caracteriza por la apariciónaguda o subaguda de cefalea intensa, alteraciones de la memo-ria y personalidad, confusión y defectos neurológicos multifoca-les. El LCR es casi siempre patológico, con discreta pleocitosislinfocítica e hiperproteinorraquia. En la arteriografía cerebral, laimagen presenta un aspecto arrosariado. Ante la sospecha,debe practicarse una biopsia cerebral (corteza y leptomeninges).El tratamiento se realiza con glucocorticoides (1 mg/kg y día) yciclofosfamida (2 mg/kg y día).
Síndrome de CoganEsta rara enfermedad de etiología y patogenia desconocidas,afecta a adultos jóvenes de ambos sexos y cursa con episodiosagudos de queratitis intersticial y de disfunción cocleovestibu-lar. La insuficiencia aórtica por valvulitis o aortitis está presenteen el 10% de casos (siendo causa de muerte). La hipoacusiaresponde mal al tratamiento con glucocorticoides y, a menudo,es permanente. En ocasiones, es necesario el empleo de agentescitotóxicos para el control de la vasculitis.
Enfermedad de EalesSe trata de una vasculitis retiniana aislada que afecta a adul-tos jóvenes y causa hemorragias recidivantes en la retina y el ví-treo, con pérdida de la visión.
La artritis reumatoide es una enfermedad crónica sistémica, pre-dominantemente articular, cuyo signo característico es la sino-vitis persistente, generalmente en articulaciones periféricas yde forma simétrica, capaz de producir la destrucción del car-tílago articular y deformidades óseas, aunque su evoluciónpuede ser muy variable.
EpidemiologíaLa artritis reumatoide afecta alrededor del 1% de la población.Puede presentarse a cualquier edad, aunque su máxima inci-dencia se sitúa entre los 40 y 60 años. Predomina en la mujer enuna proporción de 3:1 en relación con el varón.
EtiologíaSe considera que la enfermedad reumatoide es el resultado dela acción de un antígeno en un individuo que tiene una base
genética predisponente. La naturaleza del factor desencade-nante es desconocida; podría tratarse de un antígeno exógenoo de un autoantígeno.
- Base genética. Existe una predisposición genética a padecerla enfermedad. Así lo indica la tendencia a la agregación fami-liar y la asociación significativa con el HLA-DR4 (70%) (MIR02, 79). Los subtipos DRB1*0401 (DW4) y DRB1*0404(DW14) parecen asociarse a una enfermedad más agresiva,mientras que con el DRB1*0101 (DR1) se relaciona una pro-gresión más lenta. Otros genes del sistema HLA aparecen, sinembargo, con menor frecuencia como el DR5, DR2, DR3 (quese asocia a una mayor toxicidad renal por sales de oro y D-pe-nicilamina; y a toxicidad cutánea y hematológica por sales deoro) y DR7.- Respuesta inmunológica. El antígeno provoca una res-puesta inmune en el huésped, de la cual se deriva una reaccióninflamatoria. La sinovitis reumatoidea se caracteriza por unaactividad inmunológica persistente. La célula infiltrante predo-minante es el linfocito T, (con marcadores de actividad en lasuperficie CD4 y CD8); también infiltran linfocitos B que se di-ferencian en células plasmáticas productoras de anticuerpos(policlonales y FR) y macrófagos activados (MIR 04, 25; MIR03, 225). Las manifestaciones sistémicas de la AR se explicanpor la liberación de moléculas efectoras inflamatorias del tejidosinovial (interleucina 2, IL-6, TNF-alfa, el interferón gamma, elfactor inhibidor de la migración de los macrófagos, el factorquimiotáctico de los monocitos y el factor inhibidor de la mi-gración de los leucocitos) (MIR 04, 25; MIR 02, 79).- Reacción inflamatoria. Los polimorfonucleares atraviesan elendotelio y migran hacia el tejido sinovial, donde fagocitan loscomplejos inmunes y liberan enzimas lisosómicas que perpe-túan la respuesta inflamatoria (MIR). En la inflamación desen-cadenada por la respuesta inmune en el medio sinovial, seactivan numerosos procesos que perpetúan la inflamación: sis-temas del complemento, de cininas, de coagulación y de fibri-nólisis. La mayor parte de la destrucción del cartílago seproduce en yuxtaposición a la sinovial inflamada o pannus (te-jido de granulación vascular que produce gran cantidad de en-zimas de degradación).
Anatomía patológicaLa lesión microvascular y el aumento de las células del revesti-miento sinovial parecen ser las lesiones más precoces. La sinovialreumatoide, va engrosándose y forma vellosidades que hacenrelieve en la cavidad articular. El tejido sinovial se adhiere a losbordes del cartílago hialino y se transforma en un tejido de gra-nulación o pannus, que progresivamente destruye y reemplazaal cartílago. Los cambios anatómicos de destrucción más pre-coces empiezan, por tanto, en las intersecciones capsulares. Lassuperficies opuestas quedan conectadas por masas de fibrinaque puede organizarse y provocar anquilosis fibrosa u ósea. Sihay grandes destrucciones epifisarias, los segmentos óseos pier-den su alineación normal y se producen desviaciones y luxacio-nes; estas formas destructivas y deformantes pueden afectar acualquier articulación, pero son más frecuentes en las pequeñasarticulaciones de manos y pies.Otras veces el cartílago se destruye, el hueso subcondral se es-clerosa y se desarrollan osteofitos en sus bordes (artrosis secun-daria). La sinovial de las vainas tendinosas se comporta demanera similar a la de las articulaciones (tenosinovitis). Los ten-dones se deterioran por la propagación de la sinovitis, y esta al-teración puede causar necrosis y rotura.La alteración histológica del nódulo reumatoide se componede tres zonas: una central, necrótica, con material fibrinoide(MIR 08, 231); una intermedia constituida por histiocitos dis-puestos en empalizada y, a su alrededor, una zona de límitesimprecisos, con gran infiltración de células redondas, tejido fi-broso y vasos sanguíneos.
TEMA 4 ARTRITISREUMATOIDE
La clínica y localización articular nos dan los criterios diagnós-ticos junto a los signos radiográficos, esto junto a la clínica sis-témica ayudará a resolver los casos clínicos. Recordad tambiénlas complicaciones y los criterios de uso de uno u otro fármaco.
ENFOQUE MIR

Manual A Mir
28 ] ARTRITIS REUMATOIDE [
www.academiamir.com
Cuadro clínicoLa clínica típica es de poliartritis inflamatoria simétrica y bilateralcon afectación de pequeñas y grandes articulaciones.
Formas de comienzoSon habituales manifestaciones inespecíficas como astenia, ano-rexia, pérdida de peso y febrícula. Los síntomas específicos apa-recen generalmente de forma gradual en varias articulaciones(sobre todo muñecas, manos, rodillas y pies) de forma simétrica(MIR 98F, 201). Alrededor del 25% tienen un comienzo dife-rente (poliarticular aguda, monoarticular o tenosinovitis).
ArtropatíaSe trata de una poliartritis crónica, simétrica, aditiva, erosiva,deformante y anquilosante. Las muñecas se afectan en casitodos los casos, también es casi constante la afectación de laarticulación MCF (la más frecentemente afectada), IFP (segun-das en frecuencia), MTF y rodillas, mientras que es rara la afec-tación de IFD y del esqueleto axial (excepto la región cervical)(MIR). También puede afectarse la articulación cricoaritenoidea,la temporomandibular, esternoclavicular y hombros.
Período de comienzoEn la forma de comienzo más frecuente, la artritis afecta variasarticulaciones de manera simultánea o aditiva, preferentementelas de las muñecas, las manos, los pies y las rodillas, con tenden-cia a la simetría y evolución lentamente progresiva.En otros casos, la enfermedad empieza siendo biarticular y simé-trica o monoarticular, durante semanas, meses o más de un añoantes de que se generalice. Otras posibles formas de comienzoson la tenosinovitis, en especial de los flexores de los dedos –quepuede causar un síndrome del túnel carpiano–, la localizaciónde la artritis en articulaciones de una sola extremidad o en lasgrandes articulaciones proximales.
Período de estadoClínicamente, la sinovitis se manifiesta por dolor, calor, tumefac-ción y disminución de la movilidad articular. La rigidez articulardespués de la inactividad es otro síntoma muy frecuente. Es ca-racterística la rigidez matutina, sensación de entumecimientode las manos que se nota al despertarse por la mañana y quepuede durar más de una hora. Al cabo de algunos meses de evolución se aprecia atrofia en losmúsculos próximos a las articulaciones afectas, como los mús-culos interóseos en la artritis de la mano.
Período de secuelasLas deformidades articulares son consecuencia de la destruccióndel cartílago y del hueso, de la hiperlaxitud o de la retracciónde las formaciones cápsulo-ligamentosas, de alteraciones tendi-nosas y de la contractura o atrofia muscular. Las deformidadesmás características en la mano son:
- Desviación cubital de los dedos, a menudo con subluxaciónpalmar de las falanges proximales.- Hiperextensión de las interfalángicas proximales, conflexión compensadora de las interfalángicas distales (deforma-ción en cuello de cisne).- Flexión de la interfalángica proximal y extensión de la dis-tal (deformidad en boutonnière o en ojal).- En el primer dedo, hiperextensión de la interfalángica y fle-xión de la metacarpofalángica.
Las muñecas tienden a colocarse en flexión, los codos en semi-flexión y los hombros en aducción. En el pie la deformación máscaracterística consiste en el hundimiento del antepié y ensan-chamiento del metatarso, además de hallux valgus, subluxaciónplantar de la cabeza de los metatarsianos, dedos en martillo ycon desviación lateral (MIR 98, 228), de manera que a veces elprimer dedo se sitúa por encima o por debajo del segundo. En
la rodilla, la deformación más frecuente es la actitud en flexión.El quiste de Baker, es una prominencia que aparece en la caraposterior de la rodilla (precisa que la cavidad articular de la ro-dilla comunique con una bolsa serosa del hueco poplíteo). No esespecífico de la enfermedad y puede darse en otros procesosque aumenten el líquido sinovial (artrosis, por ejemplo). En oca-siones, se rompe y provoca un dolor brusco y tumefacción en lapantorrilla que hacen pensar en una tromboflebitis.
La columna cervical es el único segmento vertebral que se afecta
Figura 1. Desviación cubital de los dedos con subluxación palmar de las falanges.
Figura 2. Deformidad en cuello de cisne.
Figura 3. Quiste de Baker.

R e u m a t o l o g í a
29] ARTRITIS REUMATOIDE [
en la artritis reumatoide. La evidencia radiográfica de subluxa-ción atloaxoidea anterior, provocada por la rotura o laxitud delligamento transverso del atlas, es frecuente en casos de larga du-ración; también pueden producirse subluxaciones en los planosvertical, lateral y rotacional. También son frecuentes las subluxa-ciones a otros niveles, la artritis interapofisaria y la espondilitis. Esexcepcional la alteración de sacroilíacas (MIR 98, 236).
Además de afectación articular, se produce tenosinovitis (en losflexores de la mano en el túnel carpiano puede originar un sín-drome del mediano).
Manifestaciones extraarticularesParecen más frecuentes en pacientes con títulos altos de FR (+)y en varones.
- Nódulos reumatoides (MIR 07, 78). Se hallan en alrededordel 20% de los enfermos, pueden localizarse en cualquier ór-gano, pero habitualmente se localizan en tejido celular subcu-táneo en estructuras periarticulares, superficies extensoras yáreas sometidas a presión mecánica, siendo los codos la loca-lización más frecuente.Su consistencia es firme; pueden ser móviles sobre planos pro-fundos o estar adheridos al periostio o a los tendones. No sue-len ser dolorosos. El fenómeno inicial parece ser una vasculitislocal (MIR).
- Vasculitis reumatoide. En la artritis reumatoide puedenaparecer diversos tipos de vasculitis. La variedad de mayor tras-cendencia es la vasculitis necrosante. Se asocia a títulos altosde factor reumatoide, IgM e IgG, y disminución del comple-mento sérico (crioglobulinemia). Las manifestaciones isquémi-
cas pueden localizarse en órganos muy diversos: piel (necrosisy ulceración), tejido nervioso periférico (polineuropatía o mo-noneuritis múltiple), mesenterio (infarto visceral) u otras es-tructuras (es infrecuente la vasculitis renal). La arteritis digitalproduce infartos hemorrágicos en el lecho ungueal y en el pul-pejo de los dedos. - Manifestaciones pleuropulmonares. Se observan sobretodo en los varones. Pueden aparecer antes que la afectaciónarticular. Incluyen (MIR 02, 26):
• La pleuritis es la manifestación pulmonar más frecuente ypuede ser unilateral o bilateral. En el líquido pleural el datomás significativo es la tasa baja de glucosa y complemento;la deshidrogenasa (LDH) y la adenosindesaminasa (ADA)están elevadas. • La fibrosis intersticial difusa es una alteración de tiporestrictivo. • La bronquiolitis obliterante, obstrucción de pequeñosbronquios y bronquiolos. La evolución es mortal en el cursode algunos meses.• Los nódulos pulmonares tienen un tamaño variable y selocalizan preferentemente en la periferia de los campos pul-monares. En los mineros de carbón con artritis reumatoide seha descrito una silicosis nodular con opacidades redondeadasque se denomina síndrome de Caplan; anatómicamente seidentifica porque existen partículas de carbón en el centrode los nódulos reumatoides. Los nódulos pulmonares tienenlas mismas características histológicas que los subcutáneos.Pueden cavitarse y producir neumotórax o fístulas bronco-pleurales (MIR).• La hipertensión pulmonar es rara pero grave; puede seridiopática, pero se ha asociado a hiperviscosidad sérica, fi-brosis intersticial y vasculitis pulmonar.
- Alteraciones cardíacas. Son raramente sintomáticas (50%de necropsias); lo más frecuente es la pericarditis (líquido bajoen glucosa), puede llegar a evolucionar a una pericarditis cons-trictiva crónica; pueden existir bloqueos por granulomas en elsistema de conducción,... - Manifestaciones neurológicas. Pueden tener tres orígenes:a) polineuropatía o mononeuritis múltiple relacionada con lavasculitis; b) compresión de nervios periféricos que están si-tuados cerca de una sinovial engrosada, y c) manifestacionesderivadas de las alteraciones cervicales. - Manifestaciones oculares. La queratoconjutivitis seca re-lacionada con un síndrome de Sjögren es la manifestaciónmás frecuente (20%), la epiescleritis (transitoria, benigna) yla escleritis (dolorosa y más grave, puede adelgazar las capasmás profundas del ojo y evolucionar a escleromalacia perfo-rante), son menos frecuentes (MIR 01, 83; MIR 00F, 92).
Figura 4. Subluxación atloaxoidea.
Figura 5. Nódulos reumatoides.
Figura 6. Escleromalacia perforante.

Manual A Mir
30 ] ARTRITIS REUMATOIDE [
www.academiamir.com
- Osteoporosis. Es una complicación frecuente de la enfer-medad debida a la inmovilización, esteroides...- Síndrome de Felty. Consiste en la asociación de artritis reu-matoide, esplenomegalia y neutropenia (MIR). Se asocia a tí-tulos elevados de FR y a HLA-DR4. Es una artritis reumatoidenodular, seropositiva, a menudo con anticuerpos antinuclearesy, a veces, con disminución del complemento sérico y crioglo-bulinemia. En la mayoría de los enfermos esta tríada sintomá-tica se acompaña de otras manifestaciones: adelgazamiento,pigmentación cutánea, fiebre, úlceras cutáneas, vasculitis, neu-ropatía, adenopatías, pleuritis y pericarditis, anemia y trombo-citopenia. El riesgo de infecciones graves es muy elevado. - Nefropatía. La afectación renal en el curso de la artritis reu-matoide es frecuente. Por lo común, se relaciona con amiloi-dosis, vasculitis o toxicidad farmacológica (penicilamina, salesde oro, AINEs).- Hematológica. La anemia es multifactorial. Es la manifestaciónhematológica y la manifestación extraarticular más frecuente.- Amiloidosis. Es una complicación de la artritis reumatoidemuy avanzada. Habitualmente se manifiesta con signos deafección renal (proteinuria de rango nefrótico y aumento deltamaño renal) (MIR 98F, 203).
Diagnóstico- Pruebas de laboratorio. La mayoría de los sueros de enfer-mos con artritis reumatoide contienen anticuerpos dirigidoscontra determinantes antigénicos Fc de las moléculas de IgG;son los denominados factores reumatoides (MIR 04, 25). Hayfactores reumatoides de clase IgM, IgG e IgA. El factor reu-matoide que más se detecta en los laboratorios es IgM. En lapoblación sana el factor reumatoide es positivo en el 5% delos individuos, y por encima de los 65 años en alrededor del20%. En la artritis reumatoide, es positivo entre el 60 y 70%de los enfermos. El factor reumatoide no es específico de la ar-tritis reumatoide, pero sí que se ha relacionado con el pronós-tico, de forma que los pacientes seropositivos parecen teneruna afectación más grave, con afectación extraarticular. Nomonitoriza la actividad de la enfermedad (MIR 99, 84).Los anticuerpos anticitrulinados (antiCCP) tienen una sensibi-lidad similar al FR para el diagnóstico de AR, pero la especifi-cidad de los anti-CCP es mayor, por lo que puede ser útil enel diagnóstico diferencial de la AR temprana. Los anticuerposantiCCP son útiles en la clínica para el diagnóstico de exclusiónde AR en pacientes con poliartritis.En el 15-40% de los casos, se hallan anticuerpos antinucleares,generalmente a títulos bajos, con patrón homogéneo. No sehallan anticuerpos anti-DNA. La anemia, la VSG, PCR y otrosreactantes de fase aguda suelen estar elevados y se correla-cionan con la actividad (MIR).
El líquido sinovial es de tipo inflamatorio, la actividad del com-plemento hemolítico total, del C3, del C2 y del C4 está dismi-nuida.- Signos radiológicos. En las fases iniciales, la imagen radio-lógica de la articulación puede ser normal, o mostrar un dis-creto aumento de partes blandas periarticulares. Con eltiempo, aparece una desmineralización epifisaria que se tra-duce en una hipertransparencia de los extremos óseos (osteo-penia “en banda”o descalcificaciones localizadas), reducciónde la interlínea articular, imágenes radiológicas osteolíticas,deformidades, actitudes viciosas, luxaciones y subluxaciones,muy visibles en las radiografías; en fases avanzadas se puedeañadir ligera esclerosis subcondral y osteofitosis en las articu-laciones que soportan peso, o bien anquilosis ósea.
TratamientoEl tratamiento no es curativo. Buscamos aliviar el dolor, mejorarla función y prevenir las secuelas. Las medidas generales alter-nan reposo (disminuye la intensidad de la artritis) con ejercicio(evita la rigidez, previene las deformidades, mantiene el tonomuscular).El reposo absoluto en cama, que no debe ser prolon-gado, sólo está indicado en las fases agudas de la enfermedad,y cuando hay una gran alteración del estado general. El tratamiento médico (analgésicos-antiinflamatorios, fármacosantirreumáticos de acción lenta o FARAL e inmunosupresores) setienden a utilizar precozmente y se mantienen por tiempo inde-finido, ya que suelen recaer cuando se suspende el medicamento.La cirugía se reserva para articulaciones gravemente lesionadas(artroplastia de rodilla y cadera). De forma más precoz puederealizarse una sinovectomía para alivio sintomático y reducir eldaño óseo que ocasiona el pannus sinovial.
Fármacos de primera líneaAntiinflamatorios no esteroideos (AINEs) Parte de la acción que ejercen los AINEs en la AR se debe a la in-hibición de la ciclooxigenasa y, con ello, de la síntesis de pros-Figura 7. Erosiones articulares en la AR. Nótese la presencia de zonas con des-
calcificacines localizadas o de “osteopenia en banda” (punta de flecha).
A. Rigidez matutina articular de al menos una hora de duración antes desu mejoría máxima.
B. Artritis de tres o más áreas articulares. Al menos tres de ellas tienenque presentar simultáneamente hinchazón de tejidos blandos o líquidosinovial (no sólo crecimiento óseo) observados por un médico. Las 14posibles áreas articulares son las interfalángicas proximales, las meta-carpofalángicas, las muñecas, los codos, las rodillas, los tobillos y lasmetatarsofalángicas.
C. Artritis de las articulaciones de las manos, manifestada por hinchazónen al menos una de las siguientes áreas articulares: muñeca, metacar-pofalángicas o interfalángicas proximales.
D. Artritis simétrica, con afectación simultánea de las mismas áreas articu-lares (como se exige en b) en ambos lados del cuerpo (se acepta laafectación bilateral de interfalángicas proximales, metacarpofalángicaso metatarsofalángicas aunque la simetría no sea absoluta).
E. Nódulos reumatoides, subcutáneos, sobre prominencias óseas o en su-perficies extensoras o en regiones yuxtarticulares, observados por unmédico.
F. Factor reumatoide sérico, demostrado por cualquier método, que seapositivo en menos del 5% de los controles normales.
G. Alteraciones radiológicas típicas de artritis reumatoide en las proyeccio-nes posteroanteriores de las manos y de las muñecas, que pueden in-cluir erosiones o descalcificación ósea indiscutible localizada o másintensa junto a las articulaciones afectas (la presencia única de altera-ciones artrósicas no sirve como criterio).
CRITERIOS DE AMERICAN COLLEGE OF RHEUMATOLOGY (MIR 00F, 89)
Se afirma que un enfermo tiene una artritis reumatoide si satisface al menoscuatro de los siete criterios. Los cuatro primeros criterios deben estar presentesal menos durante 6 semanas (MIR 08, 77). No obstante, el hecho de no cumplirestos criterios, sobre todo en las primeras fases de la enfermedad, no excluye eldiagnóstico.
Tabla 1. Criterios diagnósticos de AR.

R e u m a t o l o g í a
31] ARTRITIS REUMATOIDE [
taglandinas. El efecto antiinflamatorio es rápido y puede apre-ciarse ya en el primer día, pero alcanza su máximo en el cursode los 7-14 días que siguen al inicio de su administración y des-aparece rápidamente al suspender el tratamiento. Los AINEs nointerfieren en la evolución de la enfermedad a largo plazo. Esuna medida necesaria en casi todos los casos para conseguir unalivio rápido del dolor, disminuir la inflamación y mejorar la ca-lidad de vida. La doctrina clásica según la cual el ácido acetilsa-licílico es el AINE de preferencia debe revisarse. La dosisnecesaria para que el ácido acetilsalicílico actúe como antiinfla-matorio es de 4-5 g/día, con lo cual los efectos secundarios sonfrecuentes y pueden ser graves. Existen dudas sobre si la indo-metacina sería el AINE de preferencia en la artritis reumatoide.El desarrollo de fármacos inhibidores selectivos de la ciclooxige-nasa-2 (meloxicam) podría evitar la aparición de los efectos ad-versos relacionados con la inhibición de la ciclooxigenasa-1(irritación gástrica, hiperazoemia, disfunción plaquetaria, exa-cerbación de rinitis alérgica y asma). Otros inhibidores selectivosde la ciclooxigenasa-2 como el rofecoxib o el colecoxib poseenmenor toxicidad gástrica que los AINEs clásicos, aunque se les harelacionado con un aumento relativo de infarto agudo de mio-cardio, lo que ha limitado su uso. Otros efectos como la erup-ción cutánea, alteraciones hepáticas y depresión medular noestán relacionados con la inhibición de la ciclooxigenasa-1 (MIR97, 116).
GlucocorticoidesDosis bajas de corticoides orales ayudan al control de los sínto-mas. La inyección intrarticular de glucocorticoides es un recursoque permite lograr una mejoría local, que dura desde unospocos días hasta varios meses. No es recomendable repetir lasinfiltraciones con intervalos inferiores a 3 meses.
Fármacos de segunda línea, modificadores de la enferme-dad ó FARALDenominados también antirreumáticos de acción lenta, permi-ten una mejoría clínica y serológica (reducción de FR, VSG, PCR)y frenan la progresión de la enfermedad. Se dispone de las sales de oro, la D-penicilamina, la cloro-quina, la sulfasalazina y el metotrexato. Sus efectos bene-ficiosos no suelen apreciarse hasta pasados, por lo menos, unos3 meses, excepto con el metotrexato, que tiene una acción másrápida. Deben administrarse junto a los AINEs dado que suefecto antiinflamatorio o analgésico es mínimo. Presentan engeneral alta toxicidad, siendo los antipalúdicos, la sulfasalazinay la auranofina, los menos tóxicos, recomendándose en pacien-tes con menor actividad. En los casos más activos se utilizan lassales de oro, D-penicilamina, y metotrexate que es en la actua-lidad el fármaco de elección (se pauta una vez a la semana)(MIR 06, 81).
Inhibidores del TNF-alfaSon el Infliximab (anticuerpo monoclonal quimérico frente alFactor de Necrosis Tumoral alfa -TNF-alfa) y el Etanercept (re-ceptor TNF-alfa tipo II unido a IgG1). Son eficaces desde la pri-mera semana y han demostrado controlar sintomáticamente laenfermedad en aquellos pacientes sin respuesta favorable a losFARAL (tienen la mejor tasa de respuesta); parecen reducir sig-nificativamente la progresión radiológica asociados aMTX. Sus efectos adversos aunque son infrecuentes requierenun control estrecho, consistiendo en infecciones oportunistas yaparición de anticuerpos anti-DNA sin desarrollo concomitantede LES, ni síntomas relacionados. Se administran por vía paren-teral una vez al mes. Su coste es elevado.Antes de iniciar el tratamiento con un fármaco anti-TNF es ne-cesario hacer un screening de tuberculosis latente (Mantoux yradiografía de tórax).
InmunosupresoresNo son más efectivos que los FARAL. Se usan en caso de escasarespuesta a éstos o ante manifestaciones severas extraarticula-res, como vasculitis. Incluye este grupo: azatioprina, ciclofosfa-mida, ciclosporina y leflunomida.
Enfoque del paciente1. AINEs.2. Fármacos modificadores de la enfermedad o FARAL(MTX). Actualmente hay una tendencia a darlos de forma pre-coz tras el diagnóstico (si la evolución es agresiva a los 1-3meses del comienzo del proceso) y también se utilizan combi-naciones de varios de ellos para aumentar su eficacia (MIR 05,79; MIR 03, 226; MIR 01, 79; MIR 98F, 205; MIR 97, 256).3. ANTI-TNF alfa. Se usan de rescate una vez al mes cuandono hay respuesta a los FARAL (se añade al tratamiento conAINEs y FARAL).4. Inmunosupresores. Si hay inflamación persistente o mani-festaciones extraarticulares graves.
Retinopatía (maculopatía en "ojo de buey")Requiere controles oftalmológicos periódicos
(MIR 00, 109)La hidroxicloroquina es menos tóxica
ANTIPALÚDICOS(CLOROQUINA,
HIDROXICLOROQUINA)
SALES DE ORO(AURANOFINA, AURO-TIOMALATO SÓDICO)
Alteraciones mucocutáneas, trombocitopenia,alt. GI, afectación renal
D-PENICILAMINA
Alteraciones gastrointestinales(las más frecuentes)
La leucopenia es la complicación más graveSíndrome nefrótico
(más frecuente que con sales de oro)Ageusia, inducción de enfermedades
autoinmunes (LES, pénfigo, miastenia gravis…)
EFECTOS SECUNDARIOS:ALTERACIONES MUCOCUTÁNEAS,
HEMATOLÓGICAS (DEPRESIÓN DE MÉDULAÓSEA) Y GASTROINTESTINALES
SULFASALAZINA Gastrointestinales ( las más frecuentes)
METOTREXATO(ANTAGONISTA DEL
AC. FÓLICO)
Aplasia medular(revierte con ácido folínico o fólico)
Fibrosis hepática:No dar en hepatitis crónica activaNeumonitis por hipersensibilidad
Otras: gastrointestinales, neurológicos…
CICLOFOSFAMIDA Aplasia medular, disfunción gonadal,cistitis hemorrágica, carcinogénesis
AZATIOPRINA Leucopenia, trombocitopenia,hepatotoxicidad
Tabla 2. Principales efectos adversos de los fármacos emplados en la AR.
- AINES: efecto rápido, sintomático.- FAME + AINES: tratamiento de elección
• MTX: de elección. Retrasa erosión ósea. Bien tolerado• Otros: FME solos o en asociación con Mtx: cloroquinas, salazopirina, oroim, azatipioprina, etc.
- Glucocorticoides a bajas dosis (asocia para trat síntomas)- Anti-TNF solos o asociados a MTX
• Eficacia clínica relevante• Intervienen de forma significativa en la progresión de la enfermedad• Efectos adversos: inmunodepresión - infecciones
- Otros (paliativos): Qx, artroplastia, fijación de articulaciones
Tabla 3. Actitud terapéutica a seguir en el paciente diagnosticado de AR (MIR07, 77).

Manual A Mir
32 ] ARTRITIS CRÓNICA JUVENIL Y ENFERMEDAD DE STILL DEL ADULTO [
www.academiamir.com
Evolución y pronósticoLa evolución es muy variable y difícil de predecir, la mayoríamantiene una actividad de carácter fluctuante con grado varia-ble de deformidad. Mejoran durante el embarazo, aunque em-peoran tras el parto.Las causas más frecuentes de mortalidad son: infecciones, he-morragia digestiva y efectos secundarios de fármacos.
5.1.- Artritis crónica juvenil
Enfermedad poco frecuente de etiología desconocida y base po-siblemente autoinmune, en la que se produce una sinovitis simi-lar a la de la AR, responsable a largo plazo de erosiones, fibrosis,luxaciones y anquilosis.Las articulaciones que se afectan con más frecuencia son las mu-ñecas y las rodillas. Es frecuente la artritis de IFD. Los nódulos
subcutáneos son poco frecuentes. No se acompaña necesa-riamente de FR. Es relativamente frecuente la aparición de sín-tomas generales (fiebre, adenopatías, hepatoesplenomegalia,exantema…). Es más frecuente en mujeres.Se establecieron tres formas de comienzo según su evoluciónlos primeros 6 meses:
- Sistémica (artritis con fiebre intermitente, rash o afectaciónvisceral).- Poliarticular (5 o más articulaciones).- Oligoarticular (4 o menos articulaciones). Causa más fre-cuente de monoartritis crónica en la infancia.
EtiologíaComo todas las enfermedades con un trasfondo autoinmune, sesospecha una etiología multifactorial, en la que estarían impli-cados factores de predisposición genética junto con desenca-denantes ambientales, posiblemente infecciosos. El subgrupomás claramente ligado a un determinante genético es el oligo-articular HLA-B27 positivo.
Cuadro clínicoLa enfermedad presenta diferentes manifestaciones clínicas quese hallan en relación con su forma de comienzo.
- Forma sistémica: es más frecuente antes de los 10 años. Lafiebre es el síntoma primordial, con uno o dos picos. Hay unexantema maculopapuloso, no pruriginoso, más evidentecuando la fiebre se eleva, lo mismo ocurre con las artralgias,mialgias y artritis. El número de articulaciones que se afecta esvariable; en algunos pacientes, el componente articular per-siste tras la remisión febril. Puede acompañarse de hepatoes-plenomegalia, adenopatías, pleuritis y pericarditis y, más rarasveces, de miocarditis. Se detecta una elevación de los reactan-tes de fase aguda, en especial al inicio de la enfermedad y,según su actividad, se elevan la VSG, la proteína C y algunasinmunoglobulinas. La leucocitosis puede ser intensa, y asociaruna anemia normocítica o ligeramente microcítica. Pueden de-tectarse anticuerpos antinucleares positivos en un número bajode pacientes. El factor reumatoide es negativo. Cursa en formade brotes de meses o años de duración.Una vez que ha remitido el brote, el enfermo puede permane-cer asintomático o reaparecer otro brote meses o años des-pués, incluso en la edad adulta. En los pacientes de evoluciónprolongada, en quienes persiste una alteración de la concen-tración sérica de las distintas inmunoglobulinas y de la proteínaC, el riesgo de amiloidosis es mayor. El tratamiento inicial escon AAS. Si no se controla añadiremos glucocorticoides y, sipersiste, inmunosupresores (metotrexato, azatioprina o ciclos-porina).- Forma poliarticular:
• Seropositiva. Es más frecuente en niñas, a partir de los10 años de edad. Representa el 5% de todas las formas decomienzo de la enfermedad. Desde el inicio suelen afectarselas pequeñas articulaciones de las manos y, progresivamente,rodillas, tobillos, metatarsofalángicas de forma simétrica.Pueden aparecer otras manifestaciones extrarticulares: nó-dulos, pleuritis, pericarditis, vasculitis. Su evolución es similara la de la artritis reumatoide y, habitualmente, la enfermedadpersiste en la edad adulta. Las lesiones radiológicas articula-res pueden ser tempranas, implicando peor pronóstico. Lasformas más graves pueden evolucionar hacia la amiloidosis,aunque en menor proporción que las formas sistémicas. Cur-san con una VSG más o menos elevada según el grado de ac-tividad; anemia normocítica o ligeramente microcítica ymoderada trombocitosis. El factor reumatoide es positivo yen un porcentaje elevado de enfermos, los anticuerpos anti-nucleares son positivos con títulos moderados y patrón ho-mogéneo. • Seronegativa. Aunque puede comenzar a cualquier edad,
El HLA DR4 se asocia a AR (los HLA de “los lados”, es decir elHLA DR3 y DR5, protegen del desarrollo de AR).
En la AR es rara la afectación de IFD, que es frecuente en laartropatía psoriásica o en la artritis crónica juvenil.
Síndrome nefrótico en paciente con AR no tratada, sospecharamiloidosis. En caso de que reciba tratamiento, sospechar D-
penicilamina o sales de oro como causa.
El FR no se relaciona con la actividad de la enfermedad.
RECUERDA
TEMA 5 ARTRITISCRÓNICA JUVENILY ENFERMEDAD DE STILL DEL ADULTO
Es un tema de mínima importancia en el MIR. En lo referente ala ACJ con la tabla de criterios diagnósticos, la tabla resumen yel tratamiento, es más que suficiente (nunca ha sido pregun-tada). Sobre la enfermedad de Still del adulto sólo ha aparecidouna pregunta.
ENFOQUE MIR
FR -Varones
<40 añosPródromos escasos y pocos
síntomas generalesUnilateral o asimétrica
Aguda
BUENOS
FR ++Reactantes de fase aguda++,
anemia, trombocitosisMujeres de raza blancaNódulos subcutáneosErosiones en RX (MIR)
Actividad sostenida >1 añoNivel socioeconómico bajo>20 articulaciones afectas
MALOS
Tabla 4. Factores pronósticos en la AR.
- Exclusión de otras causas (incluyendo las espondiloartropatías)- Inicio antes de los 16 años- Artritis persistente durante más de 6 semanas
Tabla 1. Criterios diagnósticos de la ACJ.
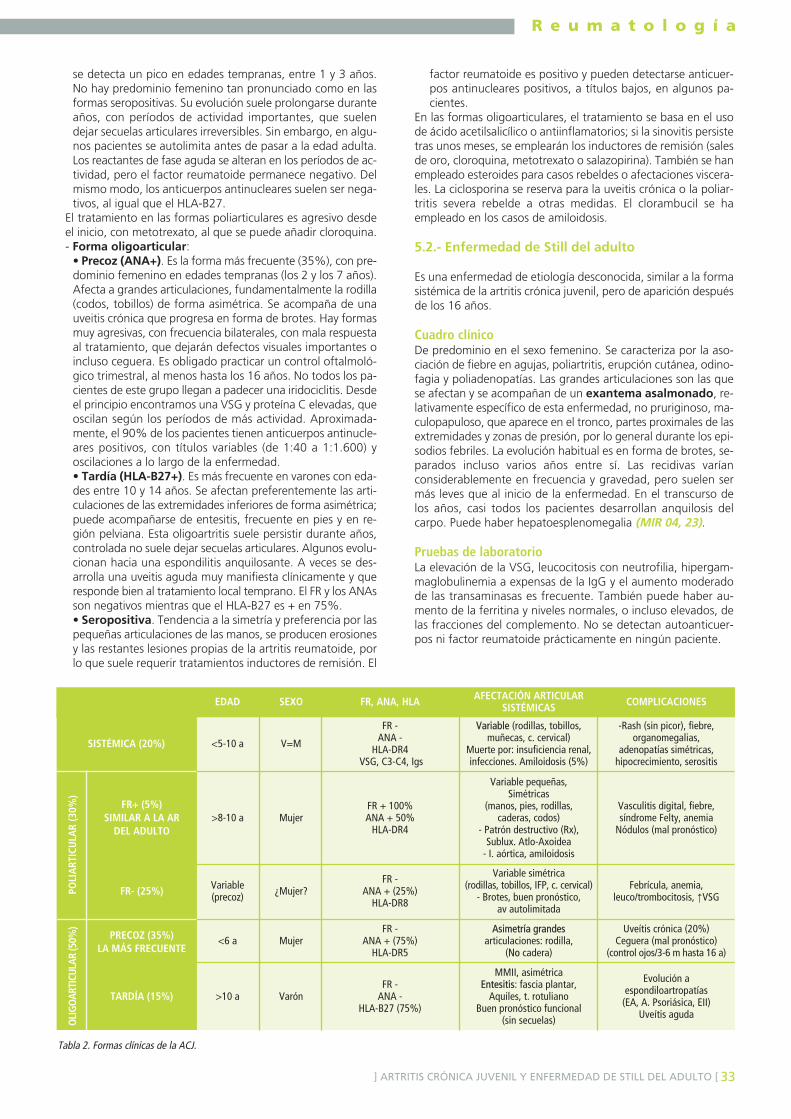
R e u m a t o l o g í a
33] ARTRITIS CRÓNICA JUVENIL Y ENFERMEDAD DE STILL DEL ADULTO [
se detecta un pico en edades tempranas, entre 1 y 3 años.No hay predominio femenino tan pronunciado como en lasformas seropositivas. Su evolución suele prolongarse duranteaños, con períodos de actividad importantes, que suelendejar secuelas articulares irreversibles. Sin embargo, en algu-nos pacientes se autolimita antes de pasar a la edad adulta.Los reactantes de fase aguda se alteran en los períodos de ac-tividad, pero el factor reumatoide permanece negativo. Delmismo modo, los anticuerpos antinucleares suelen ser nega-tivos, al igual que el HLA-B27.
El tratamiento en las formas poliarticulares es agresivo desdeel inicio, con metotrexato, al que se puede añadir cloroquina.- Forma oligoarticular:
• Precoz (ANA+). Es la forma más frecuente (35%), con pre-dominio femenino en edades tempranas (los 2 y los 7 años).Afecta a grandes articulaciones, fundamentalmente la rodilla(codos, tobillos) de forma asimétrica. Se acompaña de unauveitis crónica que progresa en forma de brotes. Hay formasmuy agresivas, con frecuencia bilaterales, con mala respuestaal tratamiento, que dejarán defectos visuales importantes oincluso ceguera. Es obligado practicar un control oftalmoló-gico trimestral, al menos hasta los 16 años. No todos los pa-cientes de este grupo llegan a padecer una iridociclitis. Desdeel principio encontramos una VSG y proteína C elevadas, queoscilan según los períodos de más actividad. Aproximada-mente, el 90% de los pacientes tienen anticuerpos antinucle-ares positivos, con títulos variables (de 1:40 a 1:1.600) yoscilaciones a lo largo de la enfermedad.• Tardía (HLA-B27+). Es más frecuente en varones con eda-des entre 10 y 14 años. Se afectan preferentemente las arti-culaciones de las extremidades inferiores de forma asimétrica;puede acompañarse de entesitis, frecuente en pies y en re-gión pelviana. Esta oligoartritis suele persistir durante años,controlada no suele dejar secuelas articulares. Algunos evolu-cionan hacia una espondilitis anquilosante. A veces se des-arrolla una uveitis aguda muy manifiesta clínicamente y queresponde bien al tratamiento local temprano. El FR y los ANAsson negativos mientras que el HLA-B27 es + en 75%.• Seropositiva. Tendencia a la simetría y preferencia por laspequeñas articulaciones de las manos, se producen erosionesy las restantes lesiones propias de la artritis reumatoide, porlo que suele requerir tratamientos inductores de remisión. El
factor reumatoide es positivo y pueden detectarse anticuer-pos antinucleares positivos, a títulos bajos, en algunos pa-cientes.
En las formas oligoarticulares, el tratamiento se basa en el usode ácido acetilsalicílico o antiinflamatorios; si la sinovitis persistetras unos meses, se emplearán los inductores de remisión (salesde oro, cloroquina, metotrexato o salazopirina). También se hanempleado esteroides para casos rebeldes o afectaciones viscera-les. La ciclosporina se reserva para la uveitis crónica o la poliar-tritis severa rebelde a otras medidas. El clorambucil se haempleado en los casos de amiloidosis.
5.2.- Enfermedad de Still del adulto
Es una enfermedad de etiología desconocida, similar a la formasistémica de la artritis crónica juvenil, pero de aparición despuésde los 16 años.
Cuadro clínicoDe predominio en el sexo femenino. Se caracteriza por la aso-ciación de fiebre en agujas, poliartritis, erupción cutánea, odino-fagia y poliadenopatías. Las grandes articulaciones son las quese afectan y se acompañan de un exantema asalmonado, re-lativamente específico de esta enfermedad, no pruriginoso, ma-culopapuloso, que aparece en el tronco, partes proximales de lasextremidades y zonas de presión, por lo general durante los epi-sodios febriles. La evolución habitual es en forma de brotes, se-parados incluso varios años entre sí. Las recidivas varíanconsiderablemente en frecuencia y gravedad, pero suelen sermás leves que al inicio de la enfermedad. En el transcurso delos años, casi todos los pacientes desarrollan anquilosis delcarpo. Puede haber hepatoesplenomegalia (MIR 04, 23).
Pruebas de laboratorioLa elevación de la VSG, leucocitosis con neutrofilia, hipergam-maglobulinemia a expensas de la IgG y el aumento moderadode las transaminasas es frecuente. También puede haber au-mento de la ferritina y niveles normales, o incluso elevados, delas fracciones del complemento. No se detectan autoanticuer-pos ni factor reumatoide prácticamente en ningún paciente.
SISTÉMICA (20%)
EDAD
<5-10 a
FR- (25%)
FR+ (5%)SIMILAR A LA AR
DEL ADULTO
Variable (rodillas, tobillos,muñecas, c. cervical)
Muerte por: insuficiencia renal,infecciones. Amiloidosis (5%)
FR -ANA -
HLA-DR4 VSG, C3-C4, Igs
V=M
SEXO FR, ANA, HLA AFECTACIÓN ARTICULARSISTÉMICAS COMPLICACIONES
-Rash (sin picor), fiebre,organomegalias,
adenopatías simétricas,hipocrecimiento, serositis
<6 aAsimetría grandes
articulaciones: rodilla,(No cadera)
FR -ANA + (75%)
HLA-DR5Mujer
Uveítis crónica (20%)Ceguera (mal pronóstico)
(control ojos/3-6 m hasta 16 a)
>8-10 a
Variable pequeñas,Simétricas
(manos, pies, rodillas,caderas, codos)
- Patrón destructivo (Rx),Sublux. Atlo-Axoidea
- I. aórtica, amiloidosis
FR + 100%ANA + 50%
HLA-DR4Mujer
Vasculitis digital, fiebre,síndrome Felty, anemia
Nódulos (mal pronóstico)
Variable(precoz)
Variable simétrica(rodillas, tobillos, IFP, c. cervical)
- Brotes, buen pronóstico,av autolimitada
FR -ANA + (25%)
HLA-DR8¿Mujer? Febrícula, anemia,
leuco/trombocitosis, ↑VSGPOLI
ART
ICU
LAR
(30%
)
TARDÍA (15%)
PRECOZ (35%)LA MÁS FRECUENTE
OLI
GO
ARTI
CULA
R (5
0%)
>10 a
MMII, asimétricaEntesitis: fascia plantar,
Aquiles, t. rotulianoBuen pronóstico funcional
(sin secuelas)
FR -ANA -
HLA-B27 (75%)Varón
Evolución aespondiloartropatías
(EA, A. Psoriásica, EII)Uveítis aguda
Tabla 2. Formas clínicas de la ACJ.

Manual A Mir
34 ] LUPUS ERITEMATOSO SISTÉMICO [
www.academiamir.com
TratamientoEl ácido acetilsalicílico y la indometacina son efectivos en mu-chos pacientes. El 40-60% de los casos requieren glucocorticoi-des para tratar las manifestaciones sistémicas. En ocasiones esnecesario asociar azatioprina, ciclofosfamida o metotrexato,aunque sus resultados son variables según los pacientes.
El lupus eritematoso sistémico (LES) es una enfermedad infla-matoria multisistémica, de carácter crónico, de causa descono-cida en la que se produce una lesión tisular citológica pordepósito de autoanticuerpos e inmunocomplejos. Tiene predilección por el sexo femenino (aunque en la infanciay en pacientes mayores de 65 años la diferencia de sexos se re-duce) y por la raza negra (es 3 veces más frecuente y más agre-sivo). Los hispanos y asiáticos también son más susceptibles. Laprevalencia varía entre 15-50 casos/10.0000 hab (MIR 98, 239).Puede afectar la piel, las articulaciones, los riñones, los pulmo-nes, el sistema nervioso, las serosas, el tubo digestivo y el apa-rato cardiocirculatorio. Es, posiblemente, el paradigma de lasenfermedades autoinmunes: los enfermos lúpicos desarrollanun sinnúmero de alteraciones inmunológicas, entre las que des-taca la formación de anticuerpos antinucleares (ANA). Aunqueel pronóstico ha mejorado, no se dispone de tratamiento reso-lutivo.
Etiología y patogeniaLa propuesta etiopatogénica más aceptada es la que consideraque un estímulo o varios estímulos etiológicos (agente infec-cioso, radiación u otros) actuarían sobre una serie de variablesde un huésped genéticamente susceptible, como la inmunidadcelular, la inmunidad humoral, el sistema mononuclear fagocí-tico y/o el sistema del complemento. La interacción de éstos ori-ginaría la aparición de mediadores (inmunocomplejoscirculantes, anticuerpos citotóxicos, células citotóxicas y/o me-diadores químicos) que, como sistemas efectores, serían respon-sables de las distintas manifestaciones de la enfermedad.
- Factores genéticos: el LES tiene mayor prevalencia (0,4-5%)entre los familiares de pacientes que en la población normal.Parece existir una asociación con el HLA-B8, HLADR3 y HLA-DR2. El factor genético más relacionado con el LES, es un alelodefectuoso de clase III, el C4AQO. En la raza negra, el LES es3 veces más frecuente, y algunos déficits de factores delcomplemento (C2 y C4) se pueden relacionar con él. - Influencia hormonal: mayor frecuencia del LES en mujeresen edad fértil y en individuos con síndrome de Klinefelter.- Factores ambientales: como luz UVB, virus, fármacos (pro-cainamida, hidralazina…)- Trastorno en la regulación de la inmunidad. Se pueden
detectar alteraciones de casi todos los componentes del sis-tema inmune (humoral y celular). Existe una hiperactividad delos linfocitos B, que se traduce en gran producción de anti-cuerpos, y una supresión de algunas funciones reguladoras delos linfocitos T y de los macrófagos. Algunos de estos anticuer-pos tienen una acción directa antigenoespecífica, como pue-den ser los antihematíe, antiplaqueta o antifosfolípido; otros,como los antilinfocito, podrían representar un papel en el pro-pio trastorno de la inmunorregulación; la mayoría de ellos for-marían complejos inmunes, cuyo depósito tisular se seguiríade una respuesta inflamatoria.
Cuadro clínicoEn el 65% de los casos, la enfermedad comienza entre los 20 y40 años.
- Manifestaciones generales (95%). Astenia, anorexia, fie-bre, pérdida de peso y malestar general. Las artromialgias sonprácticamente constantes.- Manifestaciones musculoesqueléticas (95%). Son la ma-nifestación clínica más frecuente. Se trata fundamentalmentede artralgias y mialgias (MIR 98, 224; MIR 97F, 94); tambiénpuede aparecer una poliartritis no erosiva, no deformantey simétrica (diagnóstico diferencial con AR), más frecuente enlas manos. En el 10% de los enfermos se observan deformida-des reducibles en flexión, desviación cubital, laxitud articular ydedos en cuello de cisne (artropatía de Jaccoud), debidas ala inestabilidad articular por la laxitud de los tendones, liga-mentos y cápsula articular. La necrosis avascular de la ca-beza femoral y humeral puede estar presente en el 30% delos casos, relacionada, en la mayoría de las ocasiones, con laadministración de glucocorticoides.La afectación muscular se manifiesta en el 20-30% de los en-fermos en forma de mialgias y debilidad muscular y, rarasveces, como una verdadera miositis. Los pacientes pueden pre-sentar una miopatía medicamentosa (por glucocorticoides oantipalúdicos). - Manifestaciones cutáneas (70-80%). Casi el 60% de losenfermos con LES tienen fotosensibilidad, y su expresión clínicaes en forma de lesiones agudas, subagudas, discoides y, enocasiones, ampollares y urticariformes. La alopecia se observaen el 40-60% de los enfermos. La afectación cutánea se divideen tres formas clínicas:
• Lupus cutáneo agudo (50%), cuya manifestación más ca-racterística es el eritema malar, en alas de mariposa, relacio-nado con la exposición solar y con las exacerbaciones de laenfermedad; no deja cicatrices. Puede aparecer también unrash eritematoso en otras áreas, fundamentalmente cara,cuero cabelludo, cuello, región del escote, hombros, super-ficies de extensión de los brazos y dorso de las manos.
• Lupus cutáneo subagudo (10%) (MIR 05, 142), lesiones
TEMA 6 LUPUS ERITEMA-TOSO SISTÉMICO
Volvemos a insistir en los criterios diagnósticos (hay que saber-los al detalle); fijaos también en las manifestaciones cutáneasy renales. El subtipo de anticuerpo que se asocia a una enferme-dad concreta puede orientar el diagnóstico. Las manifestacionesrenales del LES están completadas con lo que es suficiente paracontestar las preguntas del LES que aparecen dentro del bloquede Nefrología y las preguntas de Anatomía Patológica. Así quehaciendo un esfuerzo añadido ahora se rentabilizará mucho elestudio.
ENFOQUE MIR
Figura 1. Eritema malar.

R e u m a t o l o g í a
35] LUPUS ERITEMATOSO SISTÉMICO [
en forma de pápulas eritematosas, de distribución simétrica,con tendencia a confluir, con regresión central, que no dejancicatriz pero pueden dejar una zona de hipo o hiperpigmen-tación; afectan los hombros y las superficies de extensión delos brazos, la región del escote y la región dorsal del tórax. Sedistinguen dos tipos morfológicos: el psoriasiforme y elanular policíclico.A menudo tienen manifestaciones articulares y fotosensibi-lidad (es la manifestación cutánea más frecuente), aunqueno hay afectación renal ni del SNC. Algunos son ANA nega-tivos, la mayoría son antiRo (SS-A) (MIR 05, 82; MIR 01F,150), o antiDNA (ss) positivos. También se asocia con la pre-sencia de HLA DR3, DQW1, DQW2. • Lupus cutáneo crónico, incluye:
- Lupus eritematoso discoide: es la forma más frecuentede lupus cutáneo. Son placas eritematosas elevadas esca-mosas en folículos pilosos. Dejan una cicatriz y alopecia ci-catricial permanente. Las localizaciones más frecuentesson áreas fotoexpuestas como cara, cuero cabelludo, cuelloy región auricular. Es rara la evolución hacia la formasistémica de lupus y, si se produce, las manifestacionesson menos graves. El 15% de los enfermos con LES presen-tan lesiones discoides al comienzo de la enfermedad y, casiel 25%, las desarrolla durante su curso clínico. - Lupus profundo: consiste en nódulos subcutáneos, in-durados, que respetan la epidermis y se localizan, principal-mente, en las extremidades, dejando atrofia. En el 70% delos casos se acompaña de lesiones de lupus discoide.- Lupus discoide hipertrófico, cuyas lesiones tienen un as-pecto verrugoso hiperqueratósico (herpes cretáceo de De-vergie).
La inmunofluorescencia directa (IFD), demostrará depósitos deIgM o IgG en la membrana basal de la piel lesional; sobre lapiel sana lo hará en casos agudos, algunos subagudos y raravez en el discoide. Otras manifestaciones cutáneas son: exan-tema maculopapular, telangiectasias, livedo reticularis, úlcerasisquémicas de tipo crónico, urticaria, púrpura vasculítica, vas-culitis de pequeñas arterias con infartos en la punta de losdedos o sin ellos, eritema periungueal…El 40% de los enfermos presentan afección de la mucosa oraly en las fosas nasales, en forma de pequeñas úlceras super-ficiales no dolorosas, que se consideran criterio diagnóstico.- Manifestaciones hematológicas (85%). Lo más frecuentees una anemia de trastornos crónicos (70%), también puedeaparecer una anemia hemolítica (que es menos frecuenteque la anterior, pero que es la que constituye un criterio diag-nóstico). La asociación anemia hemolítica con trombocitopeniao neutropenia autoinmune, se denomina síndrome deEvans. Existe además leucopenia (65%) y linfopenia (50%),que en general, no favorece las infecciones; la trombopeniano suele tener repercusión clínica.Se han demostrado anticuerpos frente a diversos factores dela coagulación (II, VIII, IX, XI, XII y XIII), pero la presencia dediátesis hemorrágica es rara. Son más frecuentes los fenóme-nos tromboembólicos. El hallazgo más común, dentro de lasalteraciones de la coagulación, es la detección de anticuerposantifosfolípido (30-50%). - Manifestaciones neurológicas (65%). El LES puede afectara cualquier zona del encéfalo, meninges, médula espinal y ner-vios craneales y periféricos. Con frecuencia hay alteracionesen el EEG y en el LCR. Clínicamente se traduce en disfunciónpsíquica o cognitiva leve (es la manifestación neurológica másfrecuente), cefaleas, depresión y ansiedad, epilepsia (granmal), neuropatía, ACV, mielitis transversa, ataxia cerebelosa,meningitis (infecciosa y aséptica)… Se consideran criteriosdiagnósticos las convulsiones y la psicosis lúpica. Parece quelas manifestaciones neurológicas se deben a un síndrome an-tifosfolípido.
- Manifestaciones pleuropulmonares (50%). Pleuritis, amenudo bilateral, que se manifiesta con dolor pleurítico o de-rrame pleural moderado (exudado, linfocitario o neutrofílico,con complemento bajo, glucosa normal, adenosindesaminasa(ADA) normal, anticuerpos anti-DNA y células LE). Se conside-ran criterios diagnósticos las serositis (pleuritis o pericardi-tis). La infección pulmonar es la causa más frecuente deinfiltrados pulmonares en el LES (MIR 00, 54; MIR 00F, 31).La neumonitis aguda lúpica es una manifestación grave peropoco frecuente (5-12%) con infiltrados pulmonares difusos depredominio basal en la radiografía de tórax. Otros enfermosdesarrollan una neumonitis crónica, también rara (0-9%) quepresenta un patrón restrictivo. Puede haber ausencia de clínicarespiratoria. La manifestación pulmonar que más mortalidadasocia es la hemorragia alveolar masiva. No existen anti-cuerpos específicos.- Manifestaciones cardíacas (50%). La más frecuente es lapericarditis que raras veces provoca taponamiento y constric-ción; son menos frecuentes la miocarditis, endocarditis ve-rrugosa de Libman-Sacks… Entre las manifestacionesvasculares destacan: el fenómeno de Raynaud (20%), laHTA secundaria a tratamiento con glucocorticoides y/o nefro-patía, la trombosis venosa y arterial.- Manifestaciones renales (50% tienen afectación clínica -anivel histológico la mayoría tiene lesiones-). Es la más trascen-dente de todas las manifestaciones clínicas por ser la que con-diciona el pronóstico (MIR 01, 80). Su presencia constituyeun signo de mal pronóstico. El sedimento urinario puede mos-tra todo tipo de cilindros (“sedimento telescopado”) consi-derándose criterios diagnósticos la proteinuria y los cilindroscelulares. El depósito, o la formación “in situ”, de inmuno-complejos DNA-anti-DNA sobre la membrana basal glomerulary la consiguiente activación del complemento componen la se-cuencia patogénica, cuyo resultado final es la lesión lúpica delglomérulo, en forma de glomerulonefritis. En todas las formasse detecta, en mayor o menor medida, depósito de inmuno-globulinas y de complemento con patrón granular. Es fre-cuente pasar de una forma de glomerulonefritis a otra:
• Riñón normal o cambios mínimos (I).• Forma mesangial (II): las alteraciones del sedimento y laproteinuria son mínimas y el pronóstico es bueno. No sueleevolucionar a insuficiencia renal. Es la afectación renal másfrecuentemente encontrada en individuos asintomáticos.• Forma proliferativa focal y segmentaria (III) (alteraciónde menos del 50% de glomérulos): la proteinuria está casisiempre presente, aunque el síndrome nefrótico es raro; tam-bién se detecta hematuria, pero la función renal se halla pre-servada durante mucho tiempo (sin alteración del filtradoglomerular). • Forma proliferativa difusa (IV) (alteración de más del50% de glomérulos): es la forma más frecuente y graveen pacientes sintomáticos. El síndrome nefrótico es comúny se acompaña de hematuria y/o cilindros hemáticos, hiper-tensión y un filtrado glomerular (FG) que se halla ya reducidoen el 50% de los casos en el momento del diagnóstico (alte-ración de la función renal). Son típicos los depósitos en “asade alambre”. Se detecta también necrosis fibrinoide y lostípicos cuerpos hematoxinofílicos (MIR).La forma proliferativa focal y segmentaria y la proliferativadifusa pueden evolucionar hacia glomerulonefritis rápida-mente progresiva (GNRP o extracapilares). Se detecta histo-lógicamente, por la proliferación del epitelio de la cápsula deBowman con la consiguiente formación de semilunas(MIR).• Forma membranosa (V) puede producir proteinuria enrango nefrótico con una buena función renal, que con elpaso de los años sufre un deterioro progresivo. Se suele en-contrar en LES incipientes (incluso que todavía no han des-

Manual A Mir
36 ] LUPUS ERITEMATOSO SISTÉMICO [
www.academiamir.com
arrollado ANAs).• Esclerosis renal (VI). Es inútil dar tratamiento agresivo porla fibrosis renal que presenta. En las formas III y IV está indicado el tratamiento agresivopara detener la progresión y evitar la irreversibilidad de laslesiones. La biopsia renal es el mejor método para diagnosticar la ne-fritis lúpica, pero su correcta indicación es un asunto contro-vertido; es útil en los casos sospechosos de nefritisproliferativa difusa (sospechar ante ausencia de respuesta altratamiento) y para determinar el grado de actividad y selec-cionar el tratamiento más adecuado. Los pacientes con LESque presentan mayor riesgo de desarrollar una nefritis graveson aquellos con: altos niveles de anti-DNA, hipocomplemen-temia y anomalías persistentes del sedimento. En estos trescasos está indicada la biopsia renal.
La microangiopatía trombótica es otra complicación temi-ble que puede abocar a insuficiencia renal en pacientes conLES. Es una anemia hemolítica microangiopática que cursacon HTA y esquistocitosis en sangre periférica (hematíes frag-mentados).
- Manifestaciones gastrointestinales (45%). Lo más fre-cuente son síntomas inespecíficos (anorexia, náuseas, dolorleve, diarrea…). La más grave es la vasculitis intestinal. Otras:ascitis, alteraciones de enzimas hepáticos…- Otras. Abortos (30% de embarazos), queratoconjuntivitisseca, vasculitis retiniana, esplenomegalia (20%), adenopatías(50%), SIADH o hipotiroidismo subclínico… Rara vez evolu-ciona a amiloidosis.
Pruebas de laboratorioVamos a encontrar anemia, leucopenia, trombocitopenia, eleva-ción de la VSG (en algunos pacientes se correlaciona con la en-fermedad), aumento moderado de proteína C reactiva (siaumenta mucho, se debe pensar en un proceso infeccioso aso-ciado). El FR aparece aumentado en un 20-30% de casos (casiexclusivo de pacientes sin nefritis). Es frecuente la hipergamma-globulinemia (sobre todo IgG), puede asociarse a un déficit deIgA, al igual que ocurre en la AR.Se produce un consumo del complemento (C3, C4, CH50),siendo el C4 el más útil en la valoración de la evolución y del tra-tamiento al ser la fracción más sensible a modificarse (MIR).Autoanticuerpos en el LES: la alteración más característica(no específica) del LES es la presencia de autoanticuerpos, fun-damentalmente ANA (MIR 08, 97).Los pacientes ANA negativos presentan mayor fotosensibilidady fenómeno de Raynaud. No tienen manifestaciones renales nineurológicas.Otros anticuerpos que pueden estar presentes son:
- Anticardiolipina: relacionado con síndrome antifosfolípido(MIR 02, 51).- Antieritrocito: una pequeña proporción de pacientes presen-tan una hemólisis franca.- Antiplaquetas: trombocitopenia.- Antilinfocito: puede tener relación con leucopenia y alteracio-nes en la función de linfocitos T.- Antineurona: relacionado con afectación difusa del SNC.
Formas clínicas especialesLupus medicamentoso (pseudolupus)Existen numerosos fármacos que inducen un cuadro clínico einmunológico prácticamente idéntico al LES, pero sin afecta-ción renal o del SNC y con negatividad para anti-DNA na-tivo y anti-Sm. El 100% de los pacientes tienen ANAs, siendolos anticuerpos antihistona + en el 90% de los casos. Los fár-macos más relacionados son la procainamida (produce ANA +en el 75% de pacientes con tratamiento y de ellos 20% desa -rrollan LES clínico) y la hidralazina (MIR 97F, 87). Tambiénpueden producirlo la D-penicilamina, isoniazida, fenitoína, in-terferón alfa, etc. Tienen mayor probabilidad de desarrollar unlupus los individuos acetiladores lentos. Hay una estrecha aso-ciación con el HLA-DR4. La primera medida terapéutica es la su-presión del fármaco con resolución de los síntomas, pudiendoen algunas ocasiones requerir tratamiento con corticoides. Encualquier caso, estos fármacos no están contraindicados en pa-cientes con LES idiopático.
Necrosis glomerularSemilunas epiteliales
Infiltrados inflamatorios intersticialesVasculitis necrotizante
Proliferación endocapilar"Asa de alambre"
LESIONES REVERSIBLES
EsclerosisSemilunas fibrosasFibrosis intersticial
Atrofia tubular
LESIONES IRREVERSIBLES CRÓNICAS
Tabla 1. Lesiones de la nefritis lúpica.
No específico
CARACTERÍSTICAS
90
Característico del LES inducidopor fármacos, dónde aparece en el 90%40-60
Muy específico de LESTítulos altos se relacionan con
nefritis y actividad clínica (MIR 07, 81)40-60
El más específico de LES (MIR 00F, 204).Asociado a vasculitis,
leucopenia y afec. del SNC30
ANTI-DNA SS(CADENA SIMPLE)
ANTI-DNA DS(NATIVO,
CADENA DOBLEO BICATENARIO)
ANTIHISTONA
ANTI-SM
TIPO DE Ac %
Títulos altos en pacientes con rasgosde esclerosis sistémica, EMTC,
fenómeno de Raynaud, edema manos…40ANTI-RNP
LES ANA negativoLES con déficit de complemento (MIR)
Lupus neonatal y del ancianoLupus cutáneo subagudo
Mayor riesgo de nefritis (sin SSB)Además aparece en el Sdr. de Sjögren
30ANTI RO (SSA)
Siempre asociado al SSA,disminuye riesgo de nefritis
También aparece en Sdr. Sjögren10ANTI LA (SSB)
Asociado a manifestacionesneuropsiquiátricas20ANTI-RIBOSOMALES
(ANTI-P)
Tabla 2. Autoanticuerpos en el LES.
- Eritema malar- Lupus discoide- Fotosensibilidad- Úlceras orales o nasofaríngeas- Artritis (no erosiva que afecta dos o más articulaciones periféricas)- Serositis (pleuritis o pericarditis)- Afección renal (proteinuria o cilindros celulares)- Afección neurológica (convulsiones o psicosis)- Alteración hematológica:
• Anemia hemolítica• Leucopenia: <4000/mm3
• Linfopenia: <1500/mm3
• Trombocitopenia: <100000 plaquetas/ mm3
- Alteración inmunológica(célula LE positiva, anti-DNA, anti-Sm o VDRL falso positivo )
- Anticuerpos antinucleares
Para el diagnóstico de LES se requiere la presencia secuencial o simultáneade, como mínimo, 4 de ellos.
Tabla 3. Criterios diagnósticos del LES (MIR 07, 144; MIR 03, 230; MIR99F, 102; MIR 97F, 99).

R e u m a t o l o g í a
37] LUPUS ERITEMATOSO SISTÉMICO [
Lupus cutáneo subagudoEl 10% de los enfermos con LES presentan lesiones en la pielcompatibles con la forma subaguda descrita, mientras que en lamitad de casos de lupus cutáneo subagudo se observan mani-festaciones sistémicas, como artralgias, astenia, fiebre y, muyraras veces, afectación renal o neurológica. A menudo se de-muestra la presencia de anticuerpos anti-Ro (SS-A) (MIR 05, 82;MIR 01F, 150) y anti-La (SS-B), así como asociación con el ha-plotipo HLA-B8, DR3.
Lupus y embarazoLa tasa de fertilidad es normal, aunque existe elevada frecuenciade amenorrea, aborto espontáneo y de muerte intraútero (10-30%), especialmente en mujeres con anticoagulante lúpico y/oanticardiolipina. Sin enfermedad renal ni cardíaca grave y con laactividad controlada, la mayoría terminan el embarazo (el LES noes contraindicación absoluta de embarazo). No se aconseja lagestación en fases de actividad de la enfermedad. Durante elembarazo el fármaco de elección es la prednisona o hidrocorti-sona, que pueden ser degradados por los enzimas placentarios.Evitar AINEs e inmunosupresores.El embarazo tiene un efecto variable sobre la actividad del LES.En algunos casos se desarrollan brotes de la enfermedad o in-cluso el debut, en el período postparto (MIR 00, 42).
Lupus neonatalSe produce por la acción de los anticuerpos anti-Ro (SS-A) y/oanti-La (SS-B) transferidos de la madre al feto. Aunque las ma-dres no siempre tienen LES, pueden hallarse afectas de otras en-fermedades del tejido conjuntivo o estar asintomáticas. Secaracteriza por lesiones cutáneas de tipo cutáneo subagudo,que desaparecen a los 6 meses (al igual que el Ac anti-Ro), he-patoesplenomegalia, anemia hemolítica y trombocitopenia tran-
sitorias y/o bloqueo cardíaco congénito permanente. Es raroque posteriormente desarrollen un LES.
Evolución y pronósticoEl LES tiene una evolución crónica y cursa con períodos de acti-vidad y de remisión. Se han comunicado diversas causas quedesencadenan su comienzo o exacerbación, como las infeccio-nes, las intervenciones quirúrgicas, la exposición solar, el emba-razo, la toma de anticonceptivos estrogénicos o el aborto. Lasupervivencia ha mejorado, siendo >75% a los 10 años del diag-nóstico. Los principales factores que influyen en el pronóstico yla mortalidad son el grado de proteinuria y de uremia, la anemiade cualquier tipo y la afectación del SNC. También indican malpronóstico la raza negra, hipoalbuminemia, hipocomplemente-mia, HTA y la trombopenia. Las causas de muerte más frecuen-tes son las infecciones, la nefropatía y las lesiones neurológicas.
Tratamiento- Medidas generales: evitar la exposición a los rayos ultra-violeta en los enfermos con fotosensibilidad; prestar una ade-cuada atención a las situaciones que pueden reactivar laenfermedad (embarazo, infecciones, aborto, intervencionesquirúrgicas, toma de anticonceptivos).- Tratamiento farmacológico:
• AINES: en fiebre, artralgias, artritis y serositis (pleuroperi-carditis) moderada.• Antipalúdicos (hidroxicloroquina): indicada en el manejode las manifestaciones cutáneas (las lesiones cutáneas agudasresponden mal), astenia y artritis (formas leves). Requierencontroles oftalmológicos periódicos. En general, para los sín-tomas leves como la fatiga, artralgias y lesiones cutáneas, losantimaláricos son la primera elección. Se prefiere la hidroxiclo-roquina a la cloroquina por su menor toxicidad ocular.
Figura 2. Criterios diagnósticos del LES.
Alteraciones Hematológicas:anemia hemolítica, leucopenia,linfopenia o trombocitopenia
Úlceras orales Afectación Neurológica:psicosis o convulsiones
Serositis: Artritis Fotosensibilidad y ANAs Afectación Renal: Alteraciones Inmunológicaspleuritis o pericarditis lesiones cutáneas: proteinuria o
eritema malar y lupus discoide cilindros celulares
Regla mnemotécnica
"Hoy UN SAFARI"

Manual A Mir
38 ] LUPUS ERITEMATOSO SISTÉMICO [
www.academiamir.com
• Glucocorticoides: su uso debe restringirse, si es posible, alperíodo de brote y, después, reducir la dosis de forma pau-latina. A bajas dosis (<20 mg/día) se usan para el manejo si-tuaciones leves (sin riesgo vital) pero que no se controlan contratamientos menos agresivos (antipalúdicos, AINEs) comoson la artritis y serositis (MIR 99, 86). Las dosis altas se re-servan para afectación del SNC, glomerulonefritis prolifera-tiva, neumonitis, anemia hemolítica autoinmune ytrombocitopenia grave.• Inmunosupresores: ciclofosfamida (el más usado, en pul-sos mensuales en la glomerulonefritis proliferativa difusa, queproduce menos toxicidad vesical que las dosis diarias), azatio-prina (el menos tóxico), o el metotrexato (especialmente útilen la afectación articular persistente). Se emplean en pacien-tes con afección grave que no responden al tratamiento conglucocorticoides a dosis altas, o como fármacos ahorradores
de los mismos. Es posible que el micofenolato mofetilo y elmetotrexato sean fármacos útiles en los casos que no res-pondan a la ciclofosfamida+corticoides (MIR).
Además: en el manejo del paciente con LES, en función de lasmanifestaciones clínicas, podemos requerir utilizar antipsicóti-cos, anticonvulsivantes, tratamiento anticoagulante o antiagre-gante (en fenómenos trombóticos o abortos de repetición,asociados a la presencia de Ac antifosfolípido, que no respon-den al tratamiento inmunosupresor) (MIR). Algunas manifesta-ciones neuropsiquiátricas no son sensibles al tratamientocorticoideo, y de hecho, las de tipo psicótico pueden empeorartras el mismo (los esteroides son causa de psicosis farmacoló-gica). Ante la presencia de insuficiencia renal terminal, se tratarácon diálisis y transplante. La supervivencia de los pacientes LEStratados con estos procedimientos es similar a la de los pacientescon insuficiencia renal derivada de otras glomerulonefritis (MIR00F, 257). Las tasas de recidiva en el riñón transplantado sonbajas.
Se caracteriza por la aparición de trombosis (sobre todo venosas profundasen las extremidades inferiores y también en arterias cerebrales) (MIR 98,227), abortos de repetición (sobre todo en 2º y 3er trimestre) y trombocito-penia. Se acompaña de la presencia de anticuerpos antifosfolípidos (anticar-diolipina IgG e IgM). Los anticuerpos antifosfolípidos se pueden detectardirectamente por pruebas inmunológicas o bien por pruebas analíticas, yaque los anticuerpos antifosfolípidos son los responsables del resultados depruebas reagínicas (VDRL) falsamente positivas (MIR) y de la prolongaciónde TTPA que no corrige con plasma fresco (es en relación a esta última pro-piedad por lo que se le conoce como anticoagulante lúpico).
Más frecuente en mujeres (80%) entre los 0 y 40 años y en pacientes diag-nosticados de LES. También se han descrito casos en otras enfermedades eincluso en individuos sanos (en este último caso sería un síndrome antifos-folípidos primario). En ocasiones puede complicarse con anemia microangio-pática.
Criterios (MIR 02, 51; MIR 99F, 97; MIR 98F, 213):- Clínicos (1 ó +):
• Trombosis arterial / venosa• Abortos de repetición
- Analíticos (1 ó +):• Ac. anticardiolipina (IgG o IgM)• Anticoagulante lúpico
Gastropatía(por alt. barrera mucosa)
↑Enz. hepáticosMeningitis asépticaInsuficiencia renal
EFECTOS SECUNDARIOS
Formas leves: artralgias,mialgias, astenia, artritis,
fiebre, serositis leve,..
Aspecto cushingoide,HTA, hiperglucemia, infec-ciones, osteoporosis, ne-crosis ósea isquémica,...
Ojo! Psicosis!
Mielosupresión,infecciones, hemorragias,..
AZA: hepatotóxica,pancreatitis,..
Ciclofosfamida:cistitis hemorrágica
Formas graves, st GNF PD:Control de actividad y
↓dosis corticoides
Toxicidad retiniana(maculopatía
"en ojo de buey")Exantema, miopatía,
neuropatía
DermatitisAsteniaArtritis
AINEs
INMUNOSUPRESORES
ANTIPALÚDICOS
CORTICOIDES
INDICACIONES
Formas graves:GNF proliferativa,
vasculitis, alt. SNC,..
Tabla 4. Tratamiento del LES.
Tabla 7. Síndrome antifosfolípidos.
La asociación del LES con el HLA-DR2 HLADR-TRES(la AR lo hacía con el HLA-DR4)
En el lupus inducido por fármacos y en el lupus cutáneo sub-agudo, la afectación renal o del SNC es rara.
RECUERDA
Artralgias / ArtritisLesiones cutáneas
AsteniaSerositis leve
¿Afectación de órgano interno?- Riñón
- SN- Pulmón
No Sí
- Hidroxicloraquina- AINES
- Prednisona <10 mg
Esteroides i.v. (pulsos)Ciclofosfamida i.v. (pulsos)
Esteroides oralesAzatioprina
Micofenolato mofetilRespuesta
Mantener el tratamientohasta remisión y tratar
de mantener sólo con hi-droxicloraquina
No
MTX
Tabla 5. Tratamiento del LES.
- Infección- Nefritis lúpica
- Eventos car-diovasculares
Temprana Tardía
Tabla 6. Mortalidad del lupus.

R e u m a t o l o g í a
39] ESPONDILOARTROPATÍAS SERONEGATIVAS [
En este grupo se incluyen artropatías de etiología desconocidaque tienen en común:
7.1.- Espondilitis anquilosante
Es una enfermedad inflamatoria crónica del raquis, que afectaen todos los casos a las articulaciones sacroilíacas y, con menorfrecuencia, a las articulaciones periféricas, y que evoluciona conuna acusada tendencia a la anquilosis.
EtiologíaSe desconoce su etiología. Se presuponen factores genéticos(HLA B27 y agregación familiar) y ambientales (reactividad cru-zada con Klebsiella pneumoniae, bacterias entéricas…). Los por-tadores de HLA DR4, parecen tener mayor frecuencia deafectación periférica. ¿Relación con HLA-B27?: Más del 90% delos pacientes con espondilitis anquilosante presentan el antí-geno de histocompatibilidad HLA-B27, mientras que en la po-blación general los individuos B27 positivos constituyen el 6-8%(MIR 98, 237). El número de individuos HLA-B27-positivos quedesarrolla una espondilitis anquilosante es del 2%. La prevalen-cia estimada de la enfermedad es del 0,2% en poblaciones ge-neral y del 20% en los familiares de primer grado de lospacientes afectos. Teniendo en cuenta que la enfermedad semanifiesta también en individuos HLA-B27 negativos, es evi-dente que han de intervenir otros factores genéticos todavía nodeterminados.El HLA B27 no se incluye en los criterios diagnósticos deespondilitis y es independiente de la gravedad del proceso.En las restantes enfermedades del grupo, existe también unaclara correlación entre la afección del raquis y positividad delHLA-B27, aunque dicha correlación no es tan alta como en la es-pondilitis anquilosante.
Anatomía patológicaLa espondilitis anquilosante es una afección de las entesis, queson las zonas de inserción ligamentaria en los huesos, acompa-ñada de lesiones reactivas en el hueso adyacente. El infiltrado in-flamatorio está integrado por linfocitos y macrófagos, lo quesugiere que la alteración está mediada inmunológicamente. Lalesión inflamatoria inicial va seguida de una reacción fibroblás-tica inmediata, la cual reemplaza progresivamente al infiltradoinflamatorio; el tejido fibroblástico se organiza y origina cicatri-ces fibrosas densas, con gran tendencia a calcificarse y a osifi-carse. En las articulaciones periféricas hay una sinovitis y, si laevolución es crónica, se forma un pannus parecido al de la ar-tritis reumatoide, aunque no idéntico: hay menos necrosis deltejido sinovial y raras veces se observan folículos linfoides.
Cuadro clínico (MIR 06, 84)La enfermedad es más frecuente en varones. Suele comenzar
- Artritis periférica mono u oligoarticular, asimétrica (salvo en EII), depredominio en miembros inferiores
- Seronegativa (factor reumatoide negativo), ausencia de nódulosreumatoides
- Sacroileitis radiológica con espondilitis anquilosante (afectación axial) o sin ella
- Manifestaciones mucocutáneas, oculares, intestinales o genitourinarias(MIR 08, 79)- Tendencia a la agregación familiar- Elevada prevalencia del antígeno HLA-B27- Presencia de entesitis (inflamación de inserciones ligamentosas)
- Espondilitis anquilosante (MIR 00, 116)- Artritis reactiva (enfermedad o síndrome de Reiter)- Artritis psoriásica- Artritis de la colitis ulcerosa y de la enteritis regional
(enfermedad de Crohn)- Artritis de la enfermedad de Whipple- Espondiloartropatía indiferenciada- Síndrome SAPHO: Sinovitis, Acné, Pustulosis, Hiperostosis, Osteítis- Espondilitis juvenil
Tabla 1. Características de las espondiloartropatías seronegativas.
Tabla 2. Entidades clínicas. Figura 1. Entesitis típica en la inserción del tendón de Aquiles en el calcáneo.
TEMA 7 ESPONDILO-ARTROPATÍASSERONEGATIVAS
Tercer tema en importancia de Reumatología para el MIR, sobretodo centrado en la espondilitis anquilosante (criterios diagnós-ticos), comparándola con el resto de artritis. Mira la tabla com-parativa, haciendo énfasis en los aspectos diferenciadores entreellas.
ENFOQUE MIR
Embarazada
Embarazada
No embarazo
No embarazo
No embarazo
No embarazo
ANTI-FOSFOLÍPIDO
POSITIVO
* En embarazada con antecedente de abortos recurrentes o muerte fetal, la heparinasc. se administra a dosis profiláctica asociado a la antiagregación.
Tabla 8. Tratamiento de síndrome antifosfolípido.
No
Si
Venosos
Arteriales
Recurrentes
No
EVENTOSTROMBÓTICOS
PREVIOSACTITUD
Adiro 100 mg/día
HBPM sc. (a dosis terapéuticas*) +Adiro 100 mg/día
Sintrom INR 2-3
Sintrom INR 3-4
Sintrom INR 2-3 (± AAS 100)
- ¿AAS 100 mg/día?- Heparina BPM a dosis profiláctica en situaciones de riesgo

Manual A Mir
40 ] ESPONDILOARTROPATÍAS SERONEGATIVAS [
www.academiamir.com
entre los 15 y 30 años. En más del 80% de los pacientes, el co-mienzo es insidioso, con síntomas discretos, de manera quetranscurren de 1 a 3 años hasta que se establece el diagnósticocorrecto.
Manifestaciones articulares (MIR 07, 79)- El dolor lumbar es el síntoma inicial en 3/4 partes de pa-cientes, acompañado de sensación de rigidez en nalgas y caraposterior de los muslos, que corresponden a la inflamación dela región lumbar y de las articulaciones sacroilíacas (MIR 98,223). Es un dolor de carácter inflamatorio que empeora con elreposo. Con frecuencia, las algias se manifiestan con mayorintensidad entre las 3 y las 5 de la madrugada, de modo queel enfermo tiene que levantarse de la cama y caminar por lahabitación unos minutos; el dolor puede agravarse con los es-fuerzos. La exploración neurológica generalmente es normal.Este cuadro se halla presente durante meses y va seguido, engeneral, de períodos de remisión de duración variable, hastaque aparece un nuevo brote sintomático. Con la evolución dis-minuye el dolor, aumenta la rigidez, la cifosis y la fusión.- Artritis periférica (es oligoarticular asimétrica no erosiva),el 30% tienen artritis de caderas, (articulación no axial másfrecuentemente afecta) y hombros. La afectación de otras ar-ticulaciones periféricas es mucho más rara, con síntomas tran-sitorios. La artritis de las caderas, es crónica, generalmente
bilateral, y determina una importante incapacidad. También haylocalizaciones en las articulaciones temporomandibular, manu-brioesternal, condrosternales y condrocostales. La afectación deotras articulaciones periféricas es más rara, como la de rodillas,tobillos, carpos y metacarpofalángicas, que tienen un patrónoligoarticular y asimétrico, suele producir síntomas leves y tran-sitorios y es excepcional que sea erosiva (MIR 00F, 90).- Osteoporosis. Es temprana y frecuente en el raquis y puedefavorecer la aparición de fracturas.
Manifestaciones extraarticulares- Uveítis anterior aguda, no granulomatosa, es la manifes-tación extraarticular más frecuente (25-30%) (MIR) aveces cómo síntoma inicial (2%). No guarda relación con la in-tensidad de la espondilitis anquilosante, aunque es más fre-cuente en los enfermos con artritis periférica. En la mayoría delos enfermos se produce durante los primeros 10 años de evo-lución. En general es unilateral, con tendencia a recidivar.Cursa con dolor, fotofobia y lagrimeo. No suele dejar secuelas. - Afectación cardiovascular: la más característica es la insu-ficiencia aórtica por inflamación de la aorta y de la válvula aór-tica (MIR). Es más frecuente en la espondilitis anquilosante delarga duración, especialmente en las que cursan con artritis pe-riférica importante y con manifestaciones generales (fiebre,adelgazamiento y anemia). Otras manifestaciones son la insufi-ciencia cardíaca, la cardiomegalia y los defectos de conducción.- Lesiones del SNC y de las raíces. La rigidez vertebral hacesusceptibles a estos enfermos a los traumatismos y fracturasvertebrales (C5-C6; C7-C8). Por otra parte existe cierta inesta-bilidad en C1-C2 que puede determinar una subluxaciónatloidoaxoidea y lesiones de la médula cervical. En raras oca-siones puede establecerse un síndrome de la cola de caballopor la aparición de divertículos o quistes aracnoideos. La causade estos quistes es desconocida y se atribuye a una aracnoiditiscrónica.- Alteraciones pulmonares. Se producen tardíamente. Lomás característico es una fibrosis apical bilateral, con patrónquístico que puede ser colonizado por Aspergillus o Myco-bacterium tuberculosis. La afectación de la mecánica respira-toria puede motivar que las pruebas funcionales respiratoriasestén alteradas en algunos enfermos; asimismo es posible de-tectar una hipoxemia en algunos de ellos. - Amiloidosis. Es poco frecuente. Se produce en las formas delarga evolución (< 6% de los casos). - Alteraciones genitourinarias. Fundamentalmente, pros-tatitis y la nefropatía IgA.- Alteración inflamatoria intestinal. Es una alteración histo-lógica que no suele dar síntomas.
Exploración físicaDebemos explorar la movilidad articular, para determinar elgrado de limitación de la movilidad de la columna lumbar y to-rácica y la afectación sacroilíaca:
- Test de Schöber: valora la limitación de la movilidad lumbar.Se realiza midiendo 10 cm por encima y 5 cm por debajo de launión lumbosacra en posición firme; con la flexión del troncose determina la distancia existente entre las dos marcas (en con-diciones normales aumenta más de 5 cm) (MIR 99F, 96).- Maniobra de Volkmann: con el paciente en decúbito supinosobre un plano duro, al efectuar la separación forzada deambas espinas ilíacas anterosuperiores aparece dolor en una oambas sacroilíacas.- Maniobra de Erichsen: con el paciente en decúbito supino,aparece dolor en sacroilíacas al hacer una aproximación for-zada de las crestas ilíacas.- Maniobra de Fabere (= f: flexión; ab: abducción; ere: rotaciónexterna): con el paciente en decúbito supino, aparece doloren la sacroilíaca al colocar el muslo homolateral en flexión, ab-Figura 2. Afectación articular en la EA.

R e u m a t o l o g í a
41] ESPONDILOARTROPATÍAS SERONEGATIVAS [
ducción y rotación externa máximas.La cifosis dorsal fisiológica se acentúa, de modo que los hom-bros y la cabeza se proyectan hacia delante (el signo de la flechade Forestier, que consiste en la distancia entre el occipital y lapared, con el paciente en bipedestación y la espalda apoyada enla pared, es útil para controlar la evolución de la cifosis). La ex-pansión respiratoria está limitada por la alteración de las articu-laciones costovertebrales y costotransversas. La amplitudrespiratoria normal (a la altura del 4º espacio intercostal en va-rones o submamario en mujeres) es superior a 25 mm, pero enestos casos está disminuida. La región cervical pierde movilidaden todos los sentidos y puede quedar totalmente anquilosadaen actitudes variables. Los grados de deformidad varían muchode un paciente a otro, según la intensidad de la enfermedad ytambién en relación con la correcta terapéutica realizada encuanto al tratamiento antiinflamatorio y medidas rehabilitado-ras. El paciente espondilítico mal tratado adquiere la clásica ac-titud cifósica con la cabeza proyectada hacia delante, enrotación lateral.
RadiologíaLas alteraciones que aparecen en las articulaciones sacroilíacasson fundamentales para el diagnóstico; se trata de una afectaciónbilateral y simétrica. Los cambios aparecen inicialmente en lamitad inferior de la articulación, primero afectando al borde ilíaco,y siguen distintos grados en función del grado de afectación.
Además aparecen:- Sindesmofitos y osificación ligamento vertebral anterior y ani-llos fibrosos (imagen en caña de bambú).- Signo de Romanus: erosión en el ángulo anterior de 2 cuer-pos vertebrales contiguos- Cuadratura de cuerpos vertebrales (más común en vérte-bras torácicas): “squaring”- Pérdida de lordosis lumbar fisiológica y aumento de cifosistorácica y cervical.- Osteoporosis.
La gammagrafía ósea permite detectar la sacroilitis temprana-mente, así como los focos de osteítis. No obstante, no es unatécnica que se deba utilizar en la clínica habitual. La tomografíacomputarizada (TC) no ha desplazado a la radiología conven-cional. Tiene sus indicaciones en el estudio de imágenes dudosasde alteración sacroiliaca o vertebral. La resonancia magnética(RM) es una excelente técnica para el estudio de la sacroileitis yde otras alteraciones raquídeas (como el síndrome de cola decaballo), motivadas por la enfermedad.
AnalíticaLa VSG está acelerada, pero carece de valor como parámetrode la actividad de la enfermedad (sí se relaciona con la afecta-ción periférica); también la proteína C reactiva se eleva y poseeuna discreta correlación con la actividad global de la enferme-dad. Aparece hipergammaglobulinemia a expensas de laIgA. No es propio de la enfermedad la presencia de ANA ni deFR. El antígeno de histocompatibilidad HLA-B27 se halla pre-sente en más del 90% de los pacientes; su positividad no es perse diagnóstica de enfermedad (MIR).
Diagnóstico
La primera exploración a realizar es la Rx pelvis (MIR 05,81; MIR 02, 80; MIR 99, 89; MIR 97F, 95). Se diagnosticaráuna espondilitis anquilosante cuando se cumpla un criterio ra-diológico y al menos uno clínico. Se debe realizar el diagnósticodiferencial con la hiperostosis anquilosante vertebral idiopática(enfermedad de Forestier). De etiología desconocida es una pa-tología más frecuente en varones de edad más avanzada. Cursacon una osificación anterolateral del raquis con formación depuentes óseos gruesos produciendo gran rigidez, generalmenteindolora.
EvoluciónEs muy variable, el dolor suele ser persistente en las primerasfases y luego cursa con períodos de remisión. No suele acortarla supervivencia, a pesar de la cronicidad de la enfermedad. Lamayoría de los pacientes puede realizar un trabajo normal. Seconsideran factores de mal pronóstico el comienzo precoz,antes de los 16 años, y la afectación persistente de articulacio-nes periféricas. El problema más grave e invalidante es la artritisde cadera, que, en la actualidad, puede solventarse en muchoscasos con una artroplastia, la cual permite que el enfermo rea-nude una vida con mayores posibilidades que anteriormente.
TratamientoNo hay un tratamiento definitivo, los objetivos son controlar lainflamación y evitar la anquilosis y la deformidad.
- Tratamiento postural y ejercicio físico. El reposo absolutoestá contraindicado por la gran tendencia a la anquilosis(MIR). Entre las actividades más beneficiosas se encuentra lanatación.
Sacroilíacas normales0
4 Anquilosis completa de la articulación
AFECTACIÓN
1 Borramiento del hueso subcondral =pseudoensanchamiento del espacio articular
2 Estrechamiento del espacio articular, esclerosispor la osteítis reactiva y erosiones
3 Formación de puentes óseos (fusión parcial)
GRADO
Tabla 3. Clasificación radiológica de la sacroileitis.
Figura 3. Sacroileitis en la EA.
(1984). Los criterios de diagnóstico que se utilizan son:
Criterios clínicos (≥1)- Limitación de movimientos de la región lumbar en los tres planos(flexión anterior, flexiones laterales y extensión)
- Historia de dolor, o presencia de dolor, en la unión dorsolumbaro en la región lumbar
- Expansión respiratoria igual o inferior a 25 mm, medida en elcuarto espacio intercostal
Criterios radiológicos (criterio mayor) (MIR 05, 81; MIR 02, 80)- Sacroileítis bilateral grado 2 o superior- Sacroileítis unilateral grado 3 o 4
Tabla 4. Criterios diagnósticos de la EA.
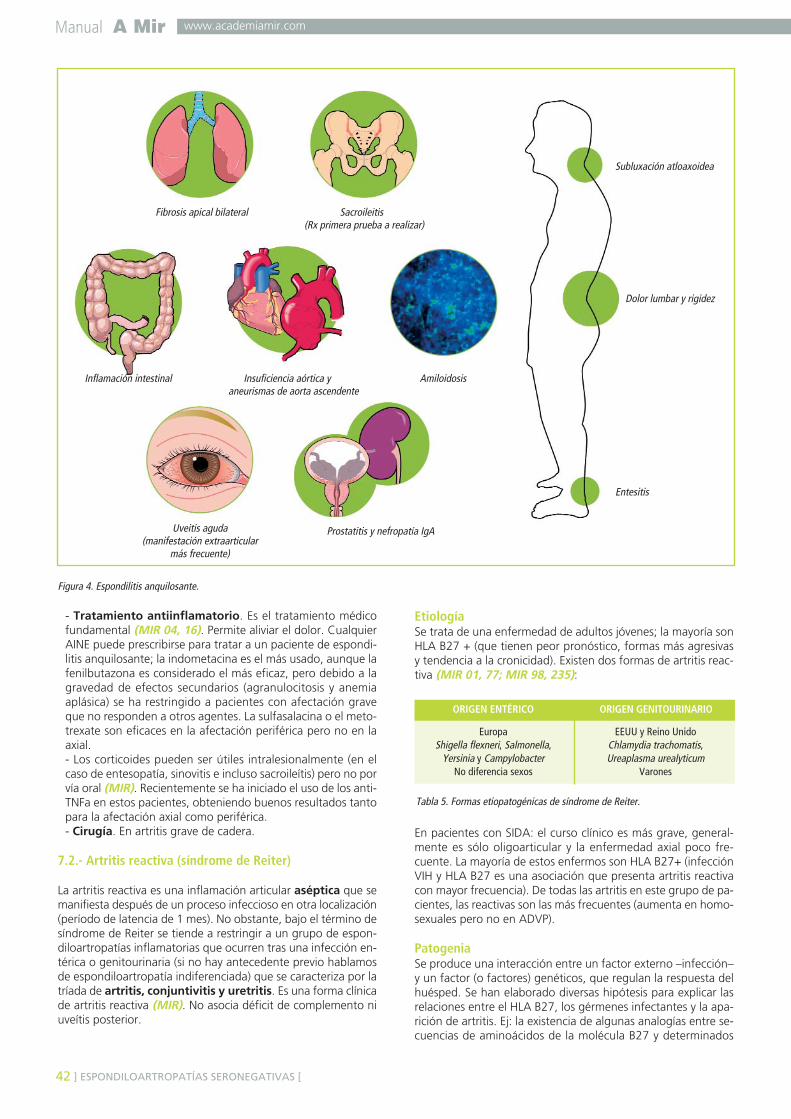
Manual A Mir
42 ] ESPONDILOARTROPATÍAS SERONEGATIVAS [
www.academiamir.com
- Tratamiento antiinflamatorio. Es el tratamiento médicofundamental (MIR 04, 16). Permite aliviar el dolor. CualquierAINE puede prescribirse para tratar a un paciente de espondi-litis anquilosante; la indometacina es el más usado, aunque lafenilbutazona es considerado el más eficaz, pero debido a lagravedad de efectos secundarios (agranulocitosis y anemiaaplásica) se ha restringido a pacientes con afectación graveque no responden a otros agentes. La sulfasalacina o el meto-trexate son eficaces en la afectación periférica pero no en laaxial.- Los corticoides pueden ser útiles intralesionalmente (en elcaso de entesopatía, sinovitis e incluso sacroileítis) pero no porvía oral (MIR). Recientemente se ha iniciado el uso de los anti-TNFa en estos pacientes, obteniendo buenos resultados tantopara la afectación axial como periférica.- Cirugía. En artritis grave de cadera.
7.2.- Artritis reactiva (síndrome de Reiter)
La artritis reactiva es una inflamación articular aséptica que semanifiesta después de un proceso infeccioso en otra localización(período de latencia de 1 mes). No obstante, bajo el término desíndrome de Reiter se tiende a restringir a un grupo de espon-diloartropatías inflamatorias que ocurren tras una infección en-térica o genitourinaria (si no hay antecedente previo hablamosde espondiloartropatía indiferenciada) que se caracteriza por latríada de artritis, conjuntivitis y uretritis. Es una forma clínicade artritis reactiva (MIR). No asocia déficit de complemento niuveítis posterior.
EtiologíaSe trata de una enfermedad de adultos jóvenes; la mayoría sonHLA B27 + (que tienen peor pronóstico, formas más agresivasy tendencia a la cronicidad). Existen dos formas de artritis reac-tiva (MIR 01, 77; MIR 98, 235):
En pacientes con SIDA: el curso clínico es más grave, general-mente es sólo oligoarticular y la enfermedad axial poco fre-cuente. La mayoría de estos enfermos son HLA B27+ (infecciónVIH y HLA B27 es una asociación que presenta artritis reactivacon mayor frecuencia). De todas las artritis en este grupo de pa-cientes, las reactivas son las más frecuentes (aumenta en homo-sexuales pero no en ADVP).
PatogeniaSe produce una interacción entre un factor externo –infección–y un factor (o factores) genéticos, que regulan la respuesta delhuésped. Se han elaborado diversas hipótesis para explicar lasrelaciones entre el HLA B27, los gérmenes infectantes y la apa-rición de artritis. Ej: la existencia de algunas analogías entre se-cuencias de aminoácidos de la molécula B27 y determinados
EuropaShigella flexneri, Salmonella,
Yersinia y CampylobacterNo diferencia sexos
EEUU y Reino UnidoChlamydia trachomatis,Ureaplasma urealyticum
Varones
ORIGEN ENTÉRICO ORIGEN GENITOURINARIO
Tabla 5. Formas etiopatogénicas de síndrome de Reiter.
Figura 4. Espondilitis anquilosante.
Uveitis aguda(manifestación extraarticular
más frecuente)
Prostatitis y nefropatía IgA
Inflamación intestinal Insuficiencia aórtica y Amiloidosisaneurismas de aorta ascendente
Fibrosis apical bilateral Sacroileitis(Rx primera prueba a realizar)
Subluxación atloaxoidea
Dolor lumbar y rigidez
Entesitis

R e u m a t o l o g í a
43] ESPONDILOARTROPATÍAS SERONEGATIVAS [
proteínas bacterianas, permitiría la aparición de reacciones cru-zadas con producción de anticuerpos.
Cuadro clínicoLa intensidad de la infección causal no se correlaciona con lagravedad de la artritis. La artritis es aditiva, asimétrica, con sig-nos inflamatorios muy acusados (dolor, tumefacción), y afectacon mayor frecuencia las extremidades inferiores (90-95% delos enfermos), rodillas, tobillos, articulaciones metatarsofalán-gicas e interfalángicas de los dedos de los pies y de las manos.Al igual que ocurre en la artropatía psoriásica la afectación si-multánea de IFD e IFP produce la dactilitis o dedo en salchicha.Aparece una entesitis responsable del dolor en áreas de inser-ción de tendones (talalgia, dolor en tendón de Aquiles), asícomo la bursitis. Puede aparecer también dolor lumbar (por sa-croileítis, entesitis…).Entre las manifestaciones extraarticulares destacamos (MIR03, 222):
- Urogenitales: uretritis, prostatitis, cervicitis, salpingitis… de-bidas al agente infeccioso desencadenante o al proceso reac-tivo estéril. (Pueden aparecer aunque el origen sea entérico).- Diarrea no infecciosa, (aunque el origen sea genitourinario).- Alteraciones mucocutáneas: úlceras orales (asintomáticastransitorias), distrofia ungueal (hiperqueratosis subungueal).- Ocular: conjuntivitis, uveítis anterior (menos frecuente, nuncaes posterior).
Si el origen es genitourinario podemos encontrar:- Queratodermia blenorrágica: tardía (1-6 meses), vesículas con hi-perqueratosis que, antes de desaparecer dejan costra localizadasen palmas y plantas, con una histología de psoriasis pustular.- Balanitis circinada: erosión superficial indolora en el glande.- Alteraciones oculares: la conjuntivitis suele ser la primera ma-
nifestación oftalmológica. Generalmente bilateral y leve. Lauveítis anterior es menos frecuente.- Otras: puede aparecer afectación del SNC y SNP, alteracionesde la conducción, infiltrados pulmonares…
Pruebas de laboratorioLa VSG está muy elevada y en algunos enfermos se descubreuna leucocitosis que guarda relación con la actividad de la en-fermedad. El líquido articular es inflamatorio.
RadiologíaLas alteraciones radiográficas típicas son la afectación de lospies, con relativa indemnidad de manos. Destaca la especial afi-nidad por el calcáneo y articulaciones interfalángicas del dedogordo y metatarsofalángicas, así como, la aposición de huesoperióstico cerca de las articulaciones afectadas (espolón) conerosiones marginales.
EvoluciónLa artritis reactiva no suele limitarse a un único ataque. En el 61%de casos de artritis reactiva, se producen recurrencias o un cursocrónico. Los pacientes HLA B27 (60-80%) presentan peor evolu-ción (MIR). Al cabo de 15 años de seguimiento se observa sacroi-leítis en el 36% de los pacientes y sindesmofitos en el 10% (puedellegar a ser indistinguible de la espondilitis anquilosante). Se haplanteado la posibilidad de que los casos relacionados con la Yer-sinia enterocolítica, tengan menor tendencia a la cronicidad.
DiagnósticoSe diagnosticará la enfermedad de Reiter cuando un enfermopresente una artritis periférica de más de un mes de duración(asimétrica y de predominio en miembros inferiores), acompa-ñada de conjuntivitis, uretritis y/o cervicitis. En el momento enel que el cuadro clínico queda establecido, los cultivos (exudadouretral, coprocultivo y hemocultivo) son negativos. Para el diag-nóstico etiológico será de utilidad la determinación serológica,posible en las infecciones por Shigella, Salmonella, Yersinia,Campylobacter y Chlamydia, sin olvidar la determinación de latasa de antiestreptolisina O (ASLO).
Diagnóstico diferencialEl diagnóstico diferencial se debe establecer fundamentalmentecon las siguientes entidades:Figura 5. Úlcera oral en el Reiter.
Figura 6. Balanitis circinada.
REITER A. GONOCÓCICA A.PSORIÁSICA
SíCONJUNTIVITIS SíNo
SíUVEÍTIS Sí Sí
SíÚLCERASORALES No
SíBALANITIS No
SíQUERATODERMIABLENORRÁGICA No +/-
+/-SACROILEÍTIS No +/-
MMIIARTRITIS MMSSMMSS MMII
+/- no excluyeartritis reactiva
CULTIVO,GONOCOCO No
En sangre, lesionescutáneas, o líquidosinovial: enf. gono-cócica diseminada
80%HLA B 27 10% 50%
RecurrenteCURSO Agudo Crónico
NoRESPUESTAA PENICILINA Sí No
Tabla 6. Diagnóstico diferencial.

Manual A Mir
44 ] ESPONDILOARTROPATÍAS SERONEGATIVAS [
www.academiamir.com
TratamientoNo hay tratamiento curativo. El tratamiento de la infección des-encadenante no influye sobre la evolución de la artritis (sólo pa-rece ser eficaz en caso de la Chlamydia con tetraciclinas). En lasfases de brote, es necesario el reposo absoluto en cama, ya quelos pacientes se hallan seriamente incapacitados. El tratamientosintomático se basa en la administración de AINEs a dosis ple-nas. Los glucocorticoides son menos eficaces que los antiinfla-matorios no esteroideos y, en general, no son recomendables.Pueden ser utilizados intralesionalmente en las entesitis. Encasos con sintomatología persistente, se han empleado el me-totrexato, sulfasalazina o azatioprina.
7.3.- Artropatía psoriásica
Es una artropatía inflamatoria y seronegativa que afecta al 5-10% de pacientes afectos de psoriasis. La psoriasis se registra enel 1,5-2% de la población general; en contraste, su prevalenciaen pacientes con artritis seronegativa es del 20%.
EtiologíaDe causa desconocida, se plantea la relación con factores am-bientales y genéticos (agregación familiar, antígenos de histo-compatibilidad B17, B37, A26, B38, Cw6 y DR7a; el antígenoDR4 puede ser un marcador de la gravedad de la artritis; el HLA-B27 se encuentra en el 30-60% de los enfermos con sacroileítiso espondilitis anquilosante).
Manifestaciones clínicasExiste un ligero predominio masculino. El comienzo ocurre a unaedad media de la vida. La psoriasis precede cronológicamente ala artritis en más del 60% de los casos y, a menudo, durantemuchos años. Encontramos varios patrones artropáticos. Entodos ellos es muy frecuente la afectación de IFD -las articulacio-nes más afectadas- y dactilitis de manos y pies. En las formasde larga evolución, puede desarrollarse amiloidosis.
- Oligoartritis asimétrica. Tradicionalmente era el patrónmás frecuente, aunque en las últimas series ocupa el segundolugar (30%). Es de evolución episódica, con brotes que puedendurar semanas o meses y que dejan lesiones residuales o nin-guna secuela. Afecta a los dos sexos por igual. Es frecuente laasociación con onicopatía (pitting). Es la forma con compli-caciones oculares más frecuente (MIR 02, 78; MIR 99F,100).- Poliartritis simétrica. En la actualidad es el patrón más fre-cuente (40%). Parecida a la artritis reumatoide (sin nódulos),aunque menos invalidante y con carácter evolutivo, en general,más leve. Parece predominar en mujeres y se asocia a HLADR4. Puede ser FR positivo.- Espondilitis anquilosante (7-10%). La espondilitis psoriá-
sica puede comenzar después de los 40 años y, a veces, seacompaña de afectaciones radiográficas importantes. Los pa-cientes son HLA B27 positivos en el 30-60% de los casos. El20-30% de los pacientes con artritis psoriásica tienen una sa-croileítis no acompañada de espondilitis. - Artritis exclusiva de IFD (5%). Asociada a onicopatía delas uñas correspondientes. Generalmente, es una forma de co-mienzo que evoluciona luego a los otros patrones.- Forma mutilante (5%). Se caracteriza por una lisis y reab-sorción de los huesos y articulaciones de los dedos de lasmanos y de los pies, así como de las metacarpofalángicas ymetatarsofalángicas. Puede ser una forma aislada o bien laevolución de las otras (MIR 00F, 91; MIR 97F, 89).
DiagnósticoEs un diagnóstico fundamentalmente clínico, ante la coexisten-cia de psoriasis y de artritis (MIR 01F, 84).
Datos de laboratorioSe produce un aumento de reactantes de fase aguda y puededisminuir el complemento. Podemos encontrar una anemia nor-mocítico-normocrómica, propia de procesos crónicos. El FR sueleser negativo (en la forma poliarticular simétrica es positivo en el25% de los casos). Podemos encontrar una hipergammaglobu-linemia IgA e hiperuricemia (MIR).
RadiologíaComparte rasgos con la artritis reumatoide (tumefacción de par-tes blandas, disminución del espacio articular, erosiones, quistessubcondrales…) pero a diferencia de ésta, no es propia la oste-oporosis, es generalmente asimétrica, se afectan las IFD y existemayor frecuencia de reabsorciones óseas (imagen en lápiz-copay lápiz-lápiz).
TratamientoEl tratamiento de la artritis se fundamenta en la administracióncontinuada de AINEs. Se han observado resultados favorablescon antirreumáticos de acción lenta como las sales de oro, conla misma pauta terapéutica de la artritis reumatoide (aunque noparecen eficaces en la afectación axial). Así mismo, se han ob-tenido resultados favorables con salazopirina, etretinato y me-totrexato (útil frente a la afectación articular y cutánea); losantipalúdicos no se emplean por el riesgo de empeorar lasmanifestaciones cutáneas. Recientemente se ha iniciado el usode los anti-TNFa en aquellos pacientes con una gran afectacióncutánea.
7.4.- Artropatías enteropáticas (EII)
Las manifestaciones articulares de la enfermedad inflamatoria in-Figura 7. Dactilitis o”dedos en salchicha”. Típicos de la artritis psoriásica y delReiter, aunque también pueden verse en la esclerodermia.
Figura 8. Artropatía psoriásica con afectación de IFD (a diferencia de la AR).

R e u m a t o l o g í a
45] ENFERMEDADES METABÓLICAS ÓSEAS [
testinal (colitis ulcerosa, enfermedad de Crohn) son muy semejan-tes, siendo la manifestación extraintestinal más frecuente.En la mayoría de los casos, la afección intestinal se manifiestaantes que la artritis. La artritis se inicia entre los 25 y 45 años,existiendo sincronismo entre los brotes de artritis y las exacer-baciones de la enteritis. Se asocia con el HLA BW62. No hayasociación con HLA-B27, salvo en un grupo reducido con afec-tación axial espondilitis/sacroileítis, de curso independiente alas manifestaciones gastrointestinales, que pueden aparecer conposterioridad a la afectación axial.En el 10-20% de los casos de enfermedad inflamatoria intesti-nal, aparece una artritis periférica, que se localiza básicamenteen las rodillas y los tobillos, con carácter simétrico. Se trata deuna sinovitis episódica, que se acompaña de remisiones queno dejan secuelas (no es erosiva). En algunos casos, por el con-trario, puede observarse una artritis crónica con deformidades.En la colitis ulcerosa, la artritis se da con mayor frecuencia en losenfermos que tienen pseudopólipos, enfermedad perianal, eri-tema nodoso, úlceras orales o uveítis.La artritis de las enfermedades intestinales inflamatorias crónicaspuede tratarse con AINEs, fármacos que entrañan el riesgo deagravar la enfermedad intestinal; en tal circunstancia, es mejor
administrar glucocorticoides a dosis bajas durante los brotes deartritis. Se han observado remisiones persistentes con la colec-tomía.
Enfermedad de Whipple (MIR 99F, 98)Artritis (90% en rodillas y tobillos), generalmente de formaaguda, migratoria, transitoria (sin erosiones), y con una evolu-ción independiente al cuadro intestinal. Aparece espondilitis an-quilosante en un 20% de casos y HLA B27 (30%) en pacientescon artritis axial.Otras enfermedades intestinales que asocian síntomas articula-res son las derivaciones intestinales, enfermedad pancreática,colitis colágena, esteatorrea idiopática…
8.1.- Osteoporosis
Enfermedad caracterizada por pérdida de masa ósea (tanto mi-neral como matriz colágena) debido a una tasa de resorciónósea superior a la de síntesis. Da lugar al desarrollo de fracturasespontáneas y ante traumatismos desproporcionadamente pe-queños, como consecuencia de una disminución de la masaósea.
E. ANQUILOPOYÉTICAVARÓN 15-30 A
90% +
ARTRITISPERIFÉRICA
M. EXTRA-ARTICULARES
TRATAMIENTO
ESPONDILITIS/
SACROILEITIS
HLA B27
A. REACTIVA ARTRITIS PSORIÁSICAENF. INFLAMATORIA
INTESTINAL (EII)
50-75% + (solo espondilitis)88%: bilateral22%: unilateral60-80% +
- Manif predominante- Precoz, simétrica, ascendente- Final: columna "caña de bambú"
y anquilosis sacroilíacas
- 20%- Curso no paralelo a E. intestinal- Varón
- 25% (2ª + frec)- Similar a E. anquilosante
- 25%- Más tardía (no predomina en el
cuadro clínico)
- Uveítis anterior- Fibrosis L. sup. pulmón + infección
Aspergillus- I. aórtica- M. intestinales ( Crohn)- Nefropatía IgA- Prostatitis crónica- Subluxación atlo-axoidea- Sdr. cola caballo- Fracturas vertebrales
- 4 formas (oligoarticular asimétrica)- 1º alt. cutánea- MMSS- Dactilitis (IFD)
- Recidivas frecs. conjuntivitis yuveítis ant
- Ulceras orales- Onicopatía- Balanitis circinada y queratoder-
mia blenorrágica (sólo en etiolo-gía urogenital)
- Uretritis, cervicitis- Síndrome de Reiter = artritis +
uretritis + conjuntivitis
- Fisioterapia- AINES- Cirugía de cadera
- Espondilitis: AINEs- A. periférica: Tto de pat. intestinal
- + frec si hay alt piel extensa- Onicopatía- IRITIS (7%)- Imagen "lápiz-copa"
- Indometacina- F. refractarias: sulfasalacina o MTX
- Cadera (30%), hombro- Distales (poco frec)- HLA DR4
- Mono/oligoarticular, asimétrica,aditiva
- MMII- Dactilitis (IFP + IFD)- Entesitis
- 25% casos EII- Curso paralelo a EII- + frec si otras M. extraintestinales- Inicio agudo- No asociado a HLA B27
- Acropaquias- Amiloidosis- Osteomalacia (malabsorción)- Osteoporosis
- Similar a AR- MTX: muy eficaz (en piel y artics)
Tabla 7. Resumen de las espondiloartropatías seronegativas.
HLA B27 no se incluye en los criterios diagnósticos de EA y esindependiente de la gravedad (en el Reiter sin embargo la
asociación con HLA B27 se asocia a peor pronóstico).
La afectación axial de la EA es erosiva, pero no lo es laafectación periférica.
La dactilitis o “dedo en salchicha” se produce por afectación dela IFP e IFD. Es típico del Reiter (más en pies) y de la psoriasis(más en manos). Recuerda que en la AR no se afecta la IFD.
La artritis periférica de la EII se asocia a la actividad de la en-fermedad por lo que mejora con el mismo tratamiento. La
afectación axial es, sin embargo, independiente del curso dela enfermedad intestinal. El uso de AINES puede empeorar la
clínica digestiva e incluso precipitar un brote.
RECUERDA
TEMA 8 ENFERMEDADESMETABÓLICASÓSEAS
Destacar la osteoporosis (tipo I y II) y la enfermedad de Paget.El raquitismo está completado con los aspectos pediátricos dela enfermedad. Con lo que aquí se expone es más que suficienteen lo referente al raquitismo de cara al MIR (para ambas asig-naturas).
ENFOQUE MIR

Manual A Mir
46 ] ENFERMEDADES METABÓLICAS ÓSEAS [
www.academiamir.com
EpidemiologíaSe trata de la enfermedad metabólica ósea más frecuente (30-40% de todas las mujeres posmenopáusicas y casi la mitad delos mayores de 75 años). La osteoporosis es un trastorno gene-ralizado del esqueleto, pero sus secuelas clínicas principales de-penden de las fracturas de vértebras, muñecas, cadera, húmeroy tibia.
ClasificaciónDiferenciamos dos grandes grupos etiológicos:
Se consideran factores de riesgo para el desarrollo de osteopo-rosis: edad avanzada, sexo femenino, raza blanca, antecedentesfamiliares de osteoporosis, déficit de estrógenos (anovulaciónen la anorexia, menopausia precoz, ejercicio excesivo…), pesocorporal bajo para su estatura (pero no obesidad), alcohol, ta-baco (disminuye los estrógenos) o cafeína, consumo de sal…(MIR 98F, 64; MIR 97, 110).
Cuadro clínicoEs asintomática hasta que aparecen las fracturas. Se consideracaracterístico del hueso osteoporótico una densidad metabólicaósea <2,5 desviaciones estándar respecto a la de un adultojoven del mismo sexo (t-score).
- Fractura vertebral: produce un dolor de inicio agudo irra-diado a abdomen, tras flexiones óseas o sin factor desencade-nante claro. Se acompaña de cifosis progresiva conacuñamiento vertebral y disminución de talla. La localizaciónmás frecuente es en vértebras dorsales (medias y bajas) y encolumna lumbar (MIR 08, 84). La presencia de fracturas ver-tebrales por encima de D4 debe hacer pensar en otros facto-res etiológicos (ej: fracturas patológicas por metástasis) (MIR).- Fractura de huesos largos: las de cadera son las que cau-san complicaciones más graves. Habitualmente, son conse-cuencia de una caída y se acompañan de dolor agudo ydeformidad importante.
Diagnóstico- Clínica: cuando los signos clínicos despiertan la sospecha deosteoporosis, ésta suele estar muy evolucionada.- Analítica: no existe ningún parámetro analítico patognomó-nico. Los marcadores de recambio (turn-over) son normales.Aunque en un 20% de los casos de la tipo I se encuentra hi-percalciuria (MIR).- Radiografía: se aprecian cambios cuando la pérdida ósea es
>30%. A nivel vertebral, reproduce una pérdida de la estria-ción horizontal, resalte de los platillos vertebrales y aplasta-miento vertebral. En los huesos largos se produce unensanchamiento medular y un adelgazamiento cortical.- Densitometría: es la técnica de elección para el estudio dela osteopenia. Indicada en: mujeres con déficit estrogénico,pacientes con osteopenia radiológica o anormalidades verte-brales, pacientes en tratamiento corticoideo, pacientes con hi-perparatiroidismo primario asintomático… Es estos caso deberealizarse una densitometría de control cada año y medio.Sirve para confirmar que la densidad mineral ósea (DMO) esbaja (lo que sugiere que la fractura es realmente osteoporó-tica) y para valorar la eficacia del tratamiento (MIR 01F, 77;MIR 97F, 209).
• >-1 DE: normal.• -1/-2.5 DE: osteopenia.• <-2.5 DE: osteoporosis.
TratamientoEl objetivo será:
- Alcanzar el máximo pico de masa ósea en la edad adultajoven: dieta rica en calcio, realización de ejercicio físico y supri-mir factores de riesgo.- Prevenir la pérdida de masa ósea, en especial en los períodosen los que ésta acelerada - Actuación sobre la osteoporosis ya establecida para impedirque aparezcan fracturas y evitar caídas o traumatismos.
Fármacos- Estrógenos. El tratamiento con estrógenos ha descendidosignificativamente por las contraindicaciones y efectos adver-sos que ahora se conocen. El tratamiento con estrógenos enla mujer postmenopáusica posiblemente esté indicado cuandoexistan síntomas climatéricos persistentes y cuando no se to-leran otros fármacos.
• Absolutas: antecedente de cáncer de útero o de mama,melanoma, TEP reciente, hepatopatía severa, obesidad mór-bida, HTA severa.• Relativas: pancreatitis, tabaquismo, endometriosis, TVP,mastopatía fibroquística severa.
Incrementan el riesgo de padecer cáncer de mama a partir delos 5 años de tratamiento; su uso obliga a la realización decontroles ginecológicos y mamografías anuales- Modificadores selectivos de los receptores estrogénicos oSERM (Raloxifeno y Tamoxifeno) (MIR 07, 260). Reducenel recambio y la pérdida de masa ósea. También reducen elriesgo de cáncer de mama y mejoran el perfil lipídico, aunqueesto no se correlaciona con un efecto protector de la enferme-dad cardiovascular. El raloxifeno no aumenta ni disminuye elriesgo de cáncer de endometrio en comparación con placebo,mientras que el tamoxifeno sí que aumenta el riesgo. Ambosfármacos aumentan el riesgo de tromboembolismos, aunqueel riesgo es mayor con tamoxifeno. El raloxifeno no disminuyela aparición de sofocos. La indicación de estos fármacos esmuy estricta.- Calcitonina. Hormona polipeptídica sintetizada en paratiroi-des. Actúa sobre los osteoclastos, inhibiendo la resorciónósea. Útil en el tratamiento de la osteoporosis con alto recam-bio óseo, como en ciertas osteoporosis postmenopáusicas (demás de 5 años de evolución y donde los estrógenos están con-traindicados), y en las relacionadas con el uso de glucocorticoi-des. También tiene efecto analgésico, empleándose ante doloróseo.- Bifosfonatos (alendronato, risedronato). Son potentes inhi-bidores de la resorción ósea, indicados en las formas con au-mento de la resorción o de alto remodelado. Son de elecciónen el tratamiento de las formas severas. Tiene especial utili-dad en las formas inducidas por esteroides. Sus reacciones
Primaria- Involutiva o senil:
• Tipo I o postmenopáusica: mujeres (51-75 años). Se caracteriza por una pérdida acelerada de hueso trabecular; clínicamente son másfrecuentes las fracturas de cuerpos vertebrales y distal de brazo(fractura de Colles)
• Tipo II o senil: mujeres y varones > 70 años. Es debida a la pérdida no tan acelerada de hueso trabecular y cortical proximal de húmero,cuello femoral, tibia y pelvis (MIR)
- Idiopática juvenil o del adulto joven
Secundaria- Alimentaria: Ingesta baja de calcio, malabsorción, exceso de proteínas,
déficit de vitamina D…- Endocrinopatías. Hiperparatiroidismo, hipertiroidismo, hipercortisolismo,
hipogonadismo, déficit de GH, hiperprolactinemia…- Metabólicas. Diabetes, acidosis, hemocromatosis, intolerancia a la
lactosa…- Genéticas. Osteogénesis imperfecta, homocistinuria, síndrome de Ehlers-
Danlos, síndrome de Marfan- Farmacológicas. Corticoides, heparina, antiestrógenos...- Tumores. Tumores primarios o metastáticos, mieloma…- Otros. Inmovilización, artritis reumatoide, ingravidez, alcoholismo…
Tabla 1. Clasificación de la osteosporosis.

R e u m a t o l o g í a
47] ENFERMEDADES METABÓLICAS ÓSEAS [
adversas son, sobre todo, gastrointestinales, tipo esofagitis,especialmente el alendronato. Se recomiendan como trata-miento de primera línea en la osteoporosis de la mujer pos-tmenopáusica. El raloxifeno se reserva en casos de notolerancia a los bifosfonatos.- Calcio: la ingesta mínima adecuada para los adultos es de 1gr/día y 1.5 gr/día en mujeres embarazadas. Durante la lactan-cia y desde la menopausia, cuando no se aporta a través de ladieta, se añaden suplementos de calcio en las comidas.- PTH sc (recombinante humana): estimula la formación ósea.Es efectiva en reducir el riesgo de fractura en mujeres con os-teoporosis. También es efectiva en hombres.- Vitamina D (800 UI/día): se recomienda en osteoporosis se-niles junto a los suplementos de calcio, en especial en pacien-tes limitados con escasa exposición solar.- Fluoruro sódico: estimula los osteoblastos y produce au-mento de las trabéculas óseas sin modificar el hueso cortical.Indicado en osteoporosis seniles con fracturas vertebrales ybaja densidad de masa ósea.- Tiacidas: diuréticos que reducen la excreción renal de calcio.- Anabolizantes: en la osteoporosis del varón con hipogona-dismo.- Ralenolato de estroncio: fármaco recien comercializado,parece ser un tratamiento eficaz y bien tolerado para mujerescon osteoporosis establecida.
8.2.- Osteomalacia - Raquitismo
Trastorno caracterizado por la disminución de la mineralizacióndel hueso, con matriz ósea normal. El raquitismo es el mismoproceso, pero en niños.
EtiologíaDéficit de Vitamina DEs la causa más frecuente de osteomalacia. Debido a:
- Factores extrínsecos. Déficit de aporte (causa más fre-cuente), exposición inadecuada al sol, malabsorción intestinal,incluyendo la enfermedad hepatobiliar y la insuficiencia pan-creática crónica.- Alteraciones metabólicas. Alteraciones hepáticas (déficithidroxilación C25), insuficiencia renal (déficit hidroxilación C1),consumo de antiepilépticos (que trasforman el colecalciferolen un metabolito inactivo), y dos trastornos hereditarios:
• Raquitismo hereditario dependiente de vitamina D tipo I,por déficit de 25-hidroxilasa.• Raquitismo hereditario dependiente de vitamina D tipo II,por alteración del receptor de la vitamina D.
HipofosforemiaTanto por déficit de aporte (abuso de antiácido con aluminio),como por la pérdida tubular (raquitismo resistente a vitamina Dligado al cromosoma X, acidosis tubular renal tipo I, sín-drome de Fanconi).
Cuadro clínicoEn el raquitismo encontraremos deformidades óseas, fracturaspatológicas, debilidad muscular y trastornos importantes en elcrecimiento. En algunos casos incluso con tetania debida a lahipocalcemia severa. A nivel radiológico destaca: ensancha-miento de suturas (craneotabes o cráneo “en pelota de ping-pong”), rosario raquítico (prominencia de las uniones
condrocostales), deformidades y arqueamientos óseos. De ini-cio, aumenta el varo fisiológico de los niños causando poste-riormente deformaciones en valgo. Se suele realizarradiografía de muñeca donde se aprecian alteraciones en la me-táfisis, con cartílago de crecimiento aumentado con la típicaforma de copa.En la osteomalacia aparece un dolor esquelético difuso (sín-toma más frecuente) e hiperestesia ósea, acompañado de debi-lidad muscular de predominio proximal. Las deformidades óseassuelen ser menos llamativas que en el caso anterior. A nivel ra-diográfico destaca la disminución de densidad ósea, estructuratrabecular borrosa (imagen en vidrio esmerilado); son caracterís-ticas las líneas de pseudofractura de Looser-Milkman (ban-das radiotranparentes perpendiculares a la cortical) (MIR).
El diagnóstico definitivo se realiza con la biopsia ósea (tejidoosteoide aumentado de grosor, con un tiempo de desfase en lamineralización de más de 100 días, detectado mediante marcajecon tetraciclinas).
Es la desnutrición la que se asocia a osteoporosis. La obesi-dad, al estar aumentada la conversión periférica de andróge-
nos a estrógenos, previene el desarrollo de osteoporosis.
RECUERDA
Figura 1. Osteomalacia: líneas de pseudofractura de Looser-Milkman.
Normalo bajo
CALCIOSÉRICO
FOSFATOSÉRICO
FOSFATASAALCALINA
OTROS
Bajo Alta
- 25(OH) Ddisminuida
- 1,25(OH) 2Dnormal o
aumentados- PTH elevada(hiperparati-
roidismosecundario)
DÉFICIT DEVIT D.
(MIR 00, 124)
Normal Bajo Alta -PTH normalTRASTORNO
TUBULARRENAL
Bajo Alto Alta
- 25(OH) Dnormal
- 1,25(OH) 2Ddisminuida
- PTH elevada(hiperparati-
roidismosecundario)
IRC
Bajo Bajo Alta - 25(OH)Dbaja
HEPATOPATÍA UOBSTRUCCIÓNBILIAR EXTRA-
HEPÁTICA
Tabla 1. Hallazgos de laboratorio.

Manual A Mir
48 ] ENFERMEDADES METABÓLICAS ÓSEAS [
www.academiamir.com
Diagnóstico diferencial- Debilidad muscular: miopatías.- Cuadros con dolor óseo: metástasis óseas.- Hipocalcemia: hipoparatiroidismo.- Fosfatasa alcalina elevada: hepatopatías a expensas de laforma hepática, otras osteopatías (enfermedad de Paget).
TratamientoOrientado a suplir la carencia; en general suele existir una buenarespuesta, fundamentalmente en la forma carencial (lo primeroque mejoran son las alteraciones musculares, lo último lasóseas).
- Déficit de vitamina D (formas carenciales): vitamina D2 (er-gocalciferol) o vit D3 (colecalciferol) oral, añadiendo calcio.- Malabsorción intestinal. vitamina D (50000-100000 UI) ygran cantidad de calcio (4 gr de carbonato cálcico)- Insuficiencia renal crónica: calcitriol (0.25 microgr/día)- Hipofosfatemia (osteomalacia resistente a la vitaminaD): fósforo (1-4 gr/d) más calcitriol (0.2 microgr/día)- Tratamiento anticonvulsivante: vitamina D (1000 UI/día).
8.3.- Enfermedad de Paget
Se trata de una enfermedad del tejido óseo, al que afecta deforma focal, caracterizada por una anomalía de la remodelaciónósea que es excesiva y anárquica, con estructura patológica (mé-dula fibrosa, trabéculas toscas y hueso laminar desorganizadocon imagen en mosaico).Afecta con más frecuencia a varones, aumentando su prevalen-cia con la edad. Se desconoce la etiología: la presencia de inclu-siones virales (paramixovirus como el sarampión, VRS o delmoquillo canino), en el núcleo de osteoclastos sugiere la posibi-lidad de un origen viral en individuos predispuestos genética-mente (15% tienen historia familiar) (MIR).
Cuadro clínicoLa mayoría de los pacientes permanecen asintomáticos, siendoel diagnóstico muchas veces un hallazgo casual. El dolor óseoprimario, no relacionado con el movimiento, es el síntoma clí-nico más frecuente. Otras manifestaciones clínicas son el au-mento de tamaño del cráneo (cráneo pagético), la cefalea, elaumento de la temperatura local, por aumento de la red vascu-lar, y la aparición de deformidades típicas, como la tibia ensable y el fémur en cayado. Podemos encontrar estrías angioi-des en la retina.
Destacamos como complicaciones:- Hipoacusia (por afectación de huesos del oído o compresióndel VIII par).- Compresión nerviosa, sobre todo intercostal.- Elevación del gasto cardíaco por aumento de la vasculariza-ción y comunicaciones arteriovenosas en el hueso. Puede llegara producir insuficiencia cardíaca de alto gasto, estenosisaórtica.- Complicaciones neurológicas, por crecimiento de la base delcráneo.- Mayor frecuencia de litiasis urinaria, por aumento de la cal-ciuria (rara vez hipercalcemia), hiperuricemia y gota.- Fracturas patológicas.- Anemia por compresión medular.- Sarcoma (1%): es la complicación más grave. Se trata de laprincipal causa de sarcoma óseo en el adulto. Clínicamente,se debe considerar ante aumento del dolor y de la tumefacciónjunto con elevación exagerada de los niveles de fosfatasa alca-lina (MIR 97F, 101).
Diagnóstico- Alteraciones radiográficas. La pelvis es la estructura másafectada seguida de fémur, cráneo, tibia, columna lumbosa-cra, clavículas, costillas… Encontramos dos fases:
• Lítica: osteoporosis circunscrita.• Esclerótica: aumento de la densidad ósea, frecuente enhuesos faciales y vértebras (en marco o en marfil), con au-mento del tamaño óseo (por incremento del hueso corticalsubperióstico).
- Analítica. No se altera ni el hemograma ni la VSG. Se puedeacompañar de calciuria pero raras veces de hipercalcemia. Losparámetros bioquímicos de formación (fosfatas alcalina, oste-ocalcina, procolágeno) y de resorción ósea suelen estar eleva-dos (fosfatasa ácida, piridolina, desoxipiridolina ehidroxiprolina) (MIR 98F, 215).- TAC.- Biopsia ósea (confirmación).- Gammagrafía con bifosfonatos marcados con Tc-99, paradeterminar la extensión de la enfermedad, incluso en el estu-dio inicial de la enfermedad (MIR 06, 89; MIR 02, 81).
Figura 2. Cráneo pagético con zonas de osteoporosis circunscrita.
Figura 3. Tibia en sable. La imagen de la derecha muestra una gammagrafía quevalora la extensión de la enfermedad.

R e u m a t o l o g í a
49] ESCLEROSIS SISTÉMICA PROGRESIVA [
TratamientoLa mayoría no requiere tratamiento, ya que la enfermedadsuele ser localizada y asintomática (MIR 05, 80). Las indicacio-
nes para el tratamiento serán: dolor óseo persistente, compre-sión nerviosa, deformidad rápidamente progresiva con trastor-nos de la postura y de la marcha, insuficiencia cardíaca,hipercalciuria intensa, fracturas repetidas y preparación para lacirugía ortopédica. En estos casos, se utilizan calcitonina y bifos-fonatos, estos últimos destacan como tratamiento de elección(MIR 00, 118).
Trastorno multisistémico de etiología desconocida caracterizadopor una fibrosis tisular y alteraciones estructurales del lecho vas-
Figura 5. Patrón vertebral en marco típico del Paget.
ETIOPATOGENIA
OSTEOPOROSIS
Reducción de masa ósea (pérdida paralela dematriz y mineral)- Primaria:
• Tipo I: Mujeres de 50-75 años, fracturas vertebrales y Colles
• Tipo II: Hombres y mujeres>70 años,fracturas de fémur y vertebrales.
- Secundarias
DIAGNÓSTICO
RAQUITISMO Y OSTEOMALACIA ENF. DE PAGET
Defecto de acción de la Vit D por diversas causas:- Reducción en la mineralización de la matriz
ósea- Niño: raquitismo. Afecta al cartílago de creci-
miento- Adulto: osteomalacia
- Infección viral (paramixovirus) en pacientegenéticamente predispuesto
- ↑ de reabsorción ósea (por hiperactividad delos osteoclastos) y ↑compensatorio de la sín-tesis de hueso
- Hipervascularización ósea- Varón (65 a)
CLÍNICA
- Raquitismo: deformidades óseas (cráneo pro-minente, rosario costal, extremidades ar-queadas,…), fracturas patológicas, hipotoníay retraso del crecimiento
- Osteomalacia: dolor óseo difuso, debilidadproximal y fracturas patológicas
Fracturas, s/t vertebrales(dorsal-lumbar) → dolor de espalda
y cifosis dorsal progresiva (acuñamiento)
- Frecuentemente asintomática- Dolor, deformidad, y tumefacción en las ex-
tremidades ( Tª local)- Fracturas- Complicaciones: sordera, compresión medu-
lar, ICC de alto gasto, litiasis, degeneraciónen sarcoma
- Ca normal o ↓, P↓ y FA↑: 25(OH)2D↓
- 1,25(OH)2D es N o incluso ↑ en fases iniciales- Rx: - Raquitismo: Epífisis en copa, rosario ra-
quítico, craneotabesOsteomalacia:Pseudofracturas de Looser - Milkman, vérte-bras:"vidrio esmerilado"Dx definitivo: biopsia ósea
- Ca++, P y F. alcalinas: normales- Rx: (si ↓DMO > 30%)
•Vértebra bicóncava•Colapso vertebral (ANT)
- Densitometría ósea
- Ca++ normal, FA e hidroxiprolina ↑- Rx:
• Fase osteoporosis "circunscrita" Y• Fase esclerótica• Huesos más afectados: pelvis, fémur y
cráneo, tibia• Vértebra en "marco" o "marfil"
- G-grafía y biopsia ósea
- Vit D2 o D3- Suplementos de calcio- Tratamiento etiológico
- Estrógenos, raloxifeno: menopausia- Calcitonina- Bifosfonatos- Suplementos de Ca++ más Vit D- Fluoruro sódico- Otros: tiazidas, anabolizantes (varón)
- Sólo es necesario en determinadas ocasiones- Bifosfonatos (alendronato o risedronato v.o.
o pamidronato iv.)- Calcitonina + Ca++ más Vit D
TRATAMIENTO
Tabla 2. Resumen de las enfermedades metabólicas óseas.
La enfermedad de Paget suele cursar de manera asintomática.Es la causa más frecuente de elevación de la fosfatasa alca-lina en el anciano. Ello hace que muchos pacientes se diag-
nostiquen estando asintomáticos por encontrar una elevaciónde la fosfatasa alcalina en revisiones rutinarias.
La pelvis es el hueso que más se afecta en el Paget.
La enfermedad de Paget puede ocasionar estenosis aórtica(lo habitual en las enfermedades reumatológicas en que
condicionen insuficiencia aórtica).
RECUERDA
Figura 4. Osteoporosis circunscrita en la rótula.
TEMA 9 ESCLEROSISSISTÉMICAPROGRESIVA
Debemos tener clara la clasificación y las complicaciones, sobretodo renales. No olvidéis prestar atención a cuál es la principalcausa de muerte. Suelen preguntarla en forma de caso clínico.
ENFOQUE MIR

Manual A Mir
50 ] ESCLEROSIS SISTÉMICA PROGRESIVA [
www.academiamir.com
cular. Afecta, fundamentalmente, la piel y ciertos órganos in-ternos, como tubo digestivo, pulmón, corazón y riñón.
EtiopatogeniaDesconocida. Se plantean tres alteraciones básicas:
- Alteraciones vasculares (daño en el endotelio).- Trastorno en la síntesis de colágeno.- Anomalías inmunológicas (alteración fundamentalmente ce-lular).
Podrían intervenir factores genéticos (asociación familiar, HLADR1, DR2, DR3 y DR5), y ambientales. La sospecha más fuerteha podido establecerse entre el contacto con diversas sustanciasy la aparición de manifestaciones clínicas parecidas a la esclero-dermia: cloruro de polivinilo (polimerización), sílice (mineros),algunos disolventes orgánicos (tricloroetileno, hidrocarburosaromáticos), silicona (prótesis mamarias), tratamientos con ble-omicina y pentazocina o ingestión de aceite tóxico.
Clasificación- Esclerosis sistémica difusa (ver tabla 1).- Esclerosis sistémica limitada o síndrome de Crest (Calci-nosis, Raynaud, alteración de la motilidad Esofágica, eSclero-dactilia y Telangiectasias) (MIR).- Esclerosis sistémica sin afectación cutánea. La afectaciónvisceral sin esclerodermia es un cuadro raro.- Esclerodermia exclusivamente cutánea.
• Morfea en placas: placas nacaradas con un halo violáceo(lilac ring).• Esclerodermia lineal: en cuero cabelludo y frente (coup desabre) o en extremidades. Más frecuente en niños.• Morfea en gotas.• Morfea generalizada.
Cuadro clínico (MIR 01F, 82)La esclerodermia predomina en mujeres, con una edad mediade comienzo a los 40 años. El comienzo suele ser insidioso condolores generalizados, rigidez, fatiga y/o pérdida de peso.
Fenómeno de Raynaud (90-100%)Aparece en la práctica totalidad de los pacientes (90-100%)(MIR). Es la manifestación inicial en la mayoría de los casos, pre-cediendo en años a las manifestaciones de las formas localiza-das, mientras que en las formas difusas los demás síntomas se
manifiestan en menos de 1 año. Es el resultado de la vasocons-tricción episódica de arteriolas y arterias de pequeño calibre delos dedos, desencadenada por el frío, emociones, estímulos vi-bratorios…, y cursa con tres fases clínicas: palidez (vasoes-pasmo), cianosis (vasodilatación) y rubor (hiperemia reactiva);suele acompañarse de dolor.
Alteraciones cutáneas (98-100%)Son la manifestación más característica. Se produce en unaserie de fases: inicialmente, es una fase edematosa que afectafundamentalmente a los dedos de las manos (dedos en salchi-cha), progresivamente se pasa a una fase indurativa en la queencontramos una piel engrosada y tirante difícil de pellizcar, queen la cara da lugar a una falta de expresividad, microstomía yaumento de pliegues peribucales, y en los dedos a una esclero-dactilia. Con el tiempo la piel se adelgaza, -fase atrófica-, quelleva en la mano a la aparición de contracturas en flexión, úlce-ras en las yemas de los dedos y en prominencias óseas, pérdidade pelo, alteraciones de la pigmentación (hiper e hipo), calcino-sis, telangiectasias.
Alteraciones musculoesqueléticas (40-70%)Fundamentalmente en las manos y en las rodillas, que se mani-fiestan en forma de artralgias, en ocasiones incluso como unapoliartritis simétrica parecida a la AR. Encontramos engrosa-mientos tendinosos y atrofias musculares por inmovilización. Enla radiografía aparece, de forma característica, calcificación departes blandas y reabsorción de falanges terminales (inclusoen clavículas y ángulo mandibular). Puede aparecer síndrome
Progresión gradual,dedos región distal
de extremidades o cara
LIMITADA
Generalizadoen extremidades,
cara y tronco
- Tardía(es rara la afectación
cardíaca o renal <1%)- Esófago, HTP
(afección pulmonarmás frecuente), (MIR 04, 15)
- Cirrosis biliar primaria
Anticentrómero:70-80%
Antitopoisomerasa I(anti Scl-70: 40%)
Anticentrómero: 10%
De años de evolución,previo a clínica
Reciente(<1 a o inicio con clínica)
ENGROSAMIENTOCUTÁNEO
SIMÉTRICO
ANA
FENÓMENO DERAYNAUD
AFECTACIÓNVISCERAL
DIFUSA
Temprana y extensa:- Fibrosis pulmonar
intersticial- Crisis HTA
renovasculares- Gastrointestinales
- Cardíaca: ICC
Asas dilatadas, sinpérdida capilar
Megacapilares ypérdida capilarCAPILAROSCOPIA
Mejor(muerte por HTP o cirrosis
biliar de años de evolución)
Malo, peor en varones.Muerte por afectación
renal, cardíaca o pulmonarPRONÓSTICO
Tabla 1. Diferencia entre la ESP difusa y localizada.
Figura 1. Fenómeno de Raynaud.
Figura 2. Fases evolutivas de la esclerodermia.

R e u m a t o l o g í a
51] ESCLEROSIS SISTÉMICA PROGRESIVA [
del túnel carpiano. La radiología puede mostrar reabsorción delas falanges distales (acroosteólisis) siendo típicas las calcifi-caciones de partes blandas. La miositis no es frecuente.
GastrointestinalesSon las manifestaciones viscerales más frecuentes. El esófagoes el órgano más frecuentemente afectado (80% presenta alte-raciones manométricas, 50% presenta clínica), sobre todo ensu porción distal y en el esfínter esofágico, lo que se traduceen pirosis y reflujo, disfagia… Puede complicarse con esofagitispor reflujo, estenosis esofágica y esófago de Barret con elconsecuente riesgo de degeneración maligna. También puedeafectarse el estómago y el intestino en todo su trayecto (dilata-ción, atonía, dolores abdominales, íleo paralítico, malabsor-ción, divertículos..). La forma localizada puede complicarse concirrosis biliar primaria.
PulmonarEs la segunda manifestación visceral más frecuente. Implica malpronóstico (en la actualidad es la primera causa de muerte).Clínicamente, la disnea de esfuerzo es el síntoma más frecuente.La fibrosis pulmonar de lóbulos inferiores es la alteracióncaracterística y aumenta la frecuencia de carcinoma broncogé-nico y de células alveolares (típico el carcinoma bronquialve-olar). Pueden producirse neumonías por aspiración debido alreflujo gástrico. La HTP puede ser primaria (sin fibrosis pulmo-nar; ocurre en el 10% de los pacientes con afectación cutánealimitada) o secundaria a la fibrosis pulmonar, y desencadenarun empeoramiento progresivo de la disnea con aparición de in-suficiencia cardíaca derecha (MIR 97F, 93).
CardíacaSuele ser asintomática. Se trata de pericarditis, insuficienciacardíaca, miocardiopatía, arritmias o bloqueos. La necrosis enbanda es una alteración patológica característica, resultado delvasoespasmo.
RenalImplica mal pronóstico, se manifiesta desde proteinuria hastauna crisis renal esclerodérmica (más frecuente en las formasdifusas). La crisis renal cursa con insuficiencia renal rápidamenteprogresiva, HTA maligna por activación del eje renina-angioten-sina, precedida a veces de anemia microangiopática y de-rrame pericárdico. Responde a IECAS, lo que ha hecho que laafectación renal ya no sea la principal causa de muerte (MIR00F, 98; MIR 99, 87; MIR 98, 209).
OtrasSíndrome seco, neuropatía periférica y neuralgia del trigémino,endocrinopatías como hipogonadismo, tiroiditis de Hashimoto,hipotiroidismo e hipertiroidismo.
DiagnósticoCriterios diagnósticosSe establece cuando se da 1 criterio mayor y 2 menores.
Datos de laboratorioEncontramos datos inespecíficos como VSG acelerada, anemiamultifactorial (de enfermedad crónica, hemorragia digestiva, dé-ficit de vitamina B12 y/o ácido fólico), factor reumatoide positivo(25%), anticuerpos antipolimiositis cuando asocian poliomiosi-tis, hipergammaglobulinemia y/o inmunocomplejos circulantes.Los anticuerpos antinucleares (ANA) positivos en el 90%, cons-tituyen el hallazgo inmunológico de mayor interés. El patrón deinmunofluorescencia más habitual es el moteado, aunque esmás específico el nucleolar.
- Anti-topoisomerasa I o anti-Scl 70 (20-25%); la mayoría delos pacientes con este anticuerpo positivo, pertenecen a laforma difusa; se asocia con neumonitis intersticial (MIR 06, 88).- Anticuerpos anticentrómero (50% del total de formas),son característicos de la forma limitada, donde se hallan pre-sentes hasta en el 70-80% de los casos. No aparecen en otrasconectivopatías (MIR).- Anticuerpos antinucleolares (20%). Anti-RNA polimerasaI, II y III en la forma difusa y antifibrilarina (muy específica) delas formas con afectación intestinal, HTP y alteraciones muscu-loesqueléticas.
EvoluciónLa historia natural de la enfermedad es variable. En la mayoríade los enfermos, tiene un curso prolongado y no es infrecuenteuna supervivencia superior a 20 años, después del diagnóstico.Un porcentaje reducido desarrolla un curso rápidamente pro-gresivo hacia el deterioro orgánico y la muerte. Las formas difu-sas tienen peor pronóstico que las limitadas, porque presentancon mayor frecuencia afectación pulmonar, renal y/o cardíaca(MIR 05, 84).
Figura 3. Calcificaciones de partes blandas.
Esclerodermia proximal a MCF(suele ser bilateral y proximal)
CRITERIOS MAYORES
EsclerodactiliaÚlceras digitales puntiformes
Resorción digital distalFibrosis pulmonar bibasal
CRITERIOS MENORES
Tabla 2. Criterios diagnósticos de la esclerodermia.
Figura 4. Criterios diagnósticos menores de la esclerodermia.

Manual A Mir
52 ] ESCLEROSIS SISTÉMICA PROGRESIVA [
www.academiamir.com
TratamientoNo hay un tratamiento satisfactorio. El tratamiento de los órga-nos afectos puede aliviar los síntomas y mejorar su función. De-bemos controlar la tensión arterial y realizar recuentossanguíneos, con control de función renal y pulmonar.
- Medidas generales. Se ha ensayado un sinfín de medica-mentos, entre ellos:
• Glucocorticoides: en casos de miopatía grave, pericarditis,artritis refractarias a AINEs y para reducir el edema en la pri-mera fase de afectación cutánea.• D-penicilamina: es un inmunosupresor que interfiere en lasíntesis del colágeno. Se trata del fármaco más utilizado conrespuestas desiguales. Su indicación se concretaría en lasfases iniciales de la forma difusa con afectación orgánica mí-nima, mientras que en etapas de la enfermedad ya estable-cida o en las formas limitadas, su uso no parece tanadecuado.• Otros: colchicina, interferón alfa, fotoquimioterapia.
- Tratamiento específico. Orientado en función de las mani-festaciones clínicas.
9.1.- Síndromes esclerodermiformes
Fascitis eosinofílicaConsiste en la inflamación y el engrosamiento de la fascia quepueden extenderse hacia el músculo, el tejido celular subcutá-neo y la dermis. Al comienzo, la fascia y el tejido subcutáneoestán edematosos, con la progresión de la enfermedad, dichostejidos aparecen engrosados y fibróticos.Con frecuencia, el comienzo de la enfermedad se puede relacio-nar con un ejercicio físico intenso o con un traumatismo. El pa-ciente presenta dolor y edema de las extremidades, seguidosrápidamente de induración de la piel y del tejido subcutáneo. Lapiel adquiere un aspecto eritematoso, brillante, que se ha com-parado a la piel de naranja. La presencia de un síndrome deltúnel carpiano es frecuente. Puede afectar el tronco y la cara; elfenómeno de Raynaud y la afectación visceral están, de formacaracterística, ausentes. La VSG está acelerada y se detecta unaeosinofilia en sangre periférica y en los tejidos afectados, sobretodo al comienzo, así como hipergammaglobulinemia e inmu-nocomplejos circulantes. Muchos enfermos mejoran espontá-neamente o con pequeñas dosis de glucocorticoides en el plazode 3 a 5 años.
Cuadros clínicos esclerodérmicos inducidos por sustanciasquímicasSíndrome mialgia-eosinofiliaDe carácter epidémico relacionado con la ingesta de L-triptó-fano adulterado. Se caracterizaba inicialmente por la apariciónde mialgias generalizadas, neumonitis, miocarditis, encefalopa-tía… Posteriormente se producían los cambios cutáneos escle-rodermiformes, polineuropatía ascendente, que podía inclusoproducir parálisis de la musculatura respiratoria, alteración cog-
La esclerodermia difuSA se asocia conanticuerpos AntitopoisomeraSA
RECUERDA
- Warfarina (?)- Protector rígido, vendaje
oclusivo, limpieza mecá-nica, ATB (si se infecta)
- D-Penicilamina, colchicina,ác p-aminobenzoico y Vit E
TRATAMIENTO
- Piel dura y rígida- Calcinosis periarticular- Úlceras en yemas de
dedos y sobre prominen-cias óseas
- Acroosteolisis, pérdida deanejo cutáneos
-Microstomía y pérdida deexpresión facial
- AINEs o corticoides (10 mg/d)- Fisioterapia
- Medidas dietéticas y pos-turales, Anti-H2 y antise-cretores, procinéticos
- Antibióticos (ciclos inter-mitentes) + supl. nutricio-nales
- Ablandadores de heces,laxantes suaves
- Esofagitis por reflujo- Malabsorción (hipomotili-
dad duodenal, sobrecreci-miento bacteriano)
- Estreñimiento (hipomotili-dad del colon)
- Antag. del Ca++, Ketanse-rina, Iloprost + proteccióndel frío, evitar estrés y nofumar
- Simpatectomía, bloqueoganglio estrellado
- Manifestación habitual(95%), que suele ser inicial
- Palidez o cianosis o rubor(PCR)
CUTÁNEAS
CARDÍACAS
FENÓMENO DERAYNAUD
RENALES
CLÍNICA
- Poliartritis simétrica- Sdr. del túnel carpiano
- IECAs o diálisis (IR progresiva)- Hipotensores: propranolol,
clonidina y minoxidilo
- Insuf. renal- HTA (crisis renal)
MÚSCULO-ESQUELÉTICAS
- Corticoides (no indicadosa largo plazo)
- Digital, diuréticos (vigilan-cia estrecha)
- Pericarditis (derrame peri-cárdico) y miositis
- I cardíaca- Otros: arritmias, bloqueos…
DIGESTIVAS
PULMONARES
- Fase inicial (alveolitis):corticoides e inmunosu-presores (AZA), D-penici-lamina
- Iloprost, oxigenoterapia,nifedipino
- Fibrosis pulmonar
- Hipertensión pulmonar
Tabla 4. Resumen de las manifestaciones clínicas del la esclerodermia y su tratamiento.
Figura 5.Fascitis eosinofílica.
1. Protección frente a factores desencadenantes (frío)2. Uso de vasodilatadores: calcioantagonistas
(nifedipino y diltiacem)3. Perfusión de prostaglandinasen caso de úlceras necróticas
FENÓMENO DERAYNAUD
Esofágicas: antiácidos y anti H2Malabsorción: ATBs y suplementos nutricionales
HTA MODERADA YEN CRISIS RENAL
ESCLERODÉRMICA
MANIFESTACIONESGASTROINTESTINALES
ARTRALGIAS YRIGIDEZ
AFECTACIÓNPULMONAR
AINES
1. Etapas iniciales: D-penicilamina, glucocorticoides2. HTP: vasodilatadores como hidralazina o nifedipino
IECAS de elección(captopril y enalapril)
(MIR)
Figura 4. Tratamiento dirigido en la esclerodermia.

R e u m a t o l o g í a
53] ENFERMEDAD MIXTA DEL TEJIDO CONECTIVO / ARTRITIS SÉPTICAS [
nitiva…. Cursa en la mayoría de casos con eosinofilia periféricay ANA+ (50%). Las fases iniciales se controlaban con glucocor-ticoides sin encontrar un tratamiento eficaz en la fase crónica.
Síndrome del aceite tóxicoEn relación con el consumo de aceite de colza (1981 en Es-paña). Cuadro clínico parecido al anterior, la fase crónica se pro-ducía a partir del 6º mes, con neuromiopatía, lesiones cutáneasesclerodérmicas, disfagia, síndrome seco, hipertensión arterial y,a diferencia del anterior, con frecuencia, fenómeno de Raynaudy alteraciones tromboembólicas. También se acompaña de eosi-nofilia y ANA + (50%). El tratamiento es similar al anterior.
OtrosPor consumo de cloruro de polivinilo, bleomicina, pentazocina.
Síndromes pseudosclerodermiformesExisten múltiples enfermedades que producen lesiones cutáneasque pueden parecerse a las de la esclerodermia, como el sín-drome carcinoide, la porfiria cutánea tarda o la amiloidosis. Otra entidad de este apartado sería el escleredema de Bus-chke, que se caracteriza por una induración cutánea de co-mienzo súbito, simétrica, localizada en las zonas posterior ylateral del cuello con extensión posterior a hombros, brazos ycara. En muchos casos, existe una infección, generalmente es-treptocócica o vírica y, además, se ha comprobado que el 50%de los pacientes son diabéticos. La evolución es hacia la resolu-ción en un plazo de 3-5 años, excepto en las formas asociadasa diabetes.Por último, el escleromixedema es una entidad muy rara en laque hay una induración cutánea con erupción liquenoide gene-ralizada, acompañada de una gammapatía monoclonal (IgM).
Combinación de signos clínico semejantes a AR, lupus, esclero-dermia (con manifestaciones cutáneas similares a la forma loca-lizada) y polimiositis junto con títulos elevados de anti RNP (antiribonucleoproteína U1). Afecta sobre todo a las mujeres (80%),por lo general al final de la cuarta década de la vida, y se handescrito casos de asociación familiar.
Cuadro clínicoEl fenómeno de Raynaud es el síntoma más precoz. Puede sergrave, con gangrena de las puntas de los dedos. Otras manifes-taciones precoces son artralgias, esclerodactilia, con tumefac-ción de los dedos, en forma de salchicha, e hinchazón del dorsode las manos, fiebre y neuralgia del trigémino. También es muyfrecuente la artritis erosiva y deformante (80-90%), que sueleser simétrica y afecta sobre todo las articulaciones interfalángi-cas proximales y las metacarpofalángicas. Los pulmones se afec-tan, aproximadamente, en el 80% de los pacientes (alteraciónde la capacidad de difusión del CO, pleuritis, fibrosis pulmonar
difusa, o hipertensión pulmonar, que es la manifestación másgrave). En un 20% de los pacientes se desarrolla pericarditis y al-teraciones de la conducción cardíaca, miocarditis e insuficienciacardíaca congestiva. Puede aparecer afectación gastrointestinaly renal en el transcurso de la enfermedad. La neuropatía perifé-rica y la meningitis aséptica son las manifestaciones neurológicasmás frecuentes.
Pruebas de laboratorioLos marcadores serológicos característicos son los anticuerposanti RNP, que se detectan a títulos elevados. Se observan tam-bién anticuerpos antinucleares (ANA) con patrón moteado. Elfactor reumatoide está presente en el 25% de los casos.
DiagnósticoSe establece con el criterio serológico y 3 criterios clínicos.
EvoluciónEn muchos pacientes, evoluciona hacia el desarrollo de esclero-sis sistémica o, más raras veces, a lupus eritematoso sistémico(LES), con reducción gradual de la hipergammaglobulinemia, asícomo la desaparición de los anticuerpos anti-U1-RNP. El pro-nóstico depende de la naturaleza y la gravedad de la afectaciónde los órganos principales.
TratamientoNo existe un tratamiento específico. El control de cada síntomase realiza igual que en el resto de conectivopatías.
La artritis séptica (AS) es una reacción inflamatoria de la sinovialsecundaria a la colonización de la cavidad articular por un ger-men, con tendencia a la supuración y a la destrucción articular.La vía de contaminación más frecuente es la hematógena a par-tir de un foco primario. Menos frecuentes son la inoculación di-recta y la extensión local a partir de un foco vecino (osteomielitiso bursitis) (MIR 98F, 204). La respuesta inflamatoria que se pro-duce depende tanto de factores del huésped (estado de los me-canismos de defensa), como del microorganismo (virulencia ytamaño del inóculo).
11.1.- Artritis no gonocócicas
La mayoría de las artritis bacterianas se manifiestan en formade monoartritis aguda que, por orden de frecuencia, afectan arodilla, cadera, hombro, muñeca, tobillo y otras articulaciones;en el grupo de adictos a drogas por vía parenteral, se afectan
TEMA 10 ENFERMEDADMIXTA DELTEJIDOCONECTIVO
Enfermedad a tener en cuenta en el diagnóstico diferencial deotros procesos. Memoriza cuál es el anticuerpo típico e intentarelacionar los síntomas con enfermedades que ya conoces.
ENFOQUE MIR
Anticuerpos anti RNPimpresindible
CRITERIOS SEROLÓGICOS
Edema de manosMiositis
Fenómeno de RaynaudEsclerodactilia
Sinovitis
CRITERIOS CLÍNICOS
Tabla 1. Criterios diagnósticos de la EMTC.
TEMA 11 ARTRITISSÉPTICAS
Con las dos tablas que te ofrecemos queda muy bien resumidode cara al MIR.
ENFOQUE MIR

Manual A Mir
54 ] ARTRITIS SÉPTICAS [
www.academiamir.com
sobre todo las articulaciones axiales: sacroilíacas, esternoclavicu-lares y condrosternales. En el 10% de los enfermos, puedenafectarse dos o más articulaciones. La vía principal de infecciónes la hematógena y, menos frecuentemente, por extensióndesde tejidos vecinos infectados o inoculación directa.
Etiología
ClínicaSe caracteriza por la presencia de síntomas generales (fiebre,malestar…) y clínica articular. Se trata, en la mayoría de loscasos, de una sinovitis monoarticular con predilección por arti-culaciones de carga (rodilla y cadera) (MIR 99F, 119). La articu-lación aparece caliente, eritematosa, dolorosa y edematosa. Lasprincipales complicaciones asociadas a una artritis séptica serán:
- Degeneración del cartílago, que comienza en las 48 horas si-guientes a la infección.- Distensión capsular, puede producir una luxación patológica.- Empiema articular.- Flemón pericapsular.
A grandes rasgos, podemos afirmar que:- Cocos Gram +: provocan un proceso agudo asociado a sín-tomas generales, con hinchazón, dolor, aumento de la tempe-ratura y limitación de la movilidad.- Bacilos Gram - : curso más indolente, con moderados sín-tomas generales y molestias menos importantes, con lo cual,el diagnóstico se demora y existe mayor frecuencia de osteo -mielitis simultáneas al hacer el diagnóstico.
Analítica y diagnóstico- Laboratorio: la artrocentesis permite obtener un líquido ar-ticular (dato clave) (MIR 00, 115), de características inflama-torias/sépticas y permite también el diagnóstico definitivo conidentificación del agente etiológico:
• Frotis del líquido sinovial: positivo en 75% de infeccionespor S. aureus y estreptococos (con menor porcentaje si sonotros gérmenes).• Cultivo del líquido sinovial: positivo en 90% de los casos.
También se puede identificar al germen en hemocultivos (po-sitivos en el 50% para S. aureus); otras alteraciones analíticasincluyen elevación de VSG, leucocitosis con desviación (aunquepuede faltar hasta en el 50%). - Radiografía: inicialmente se aprecia distensión de la cápsulaarticular y aumento de partes blandas periarticulares; poste-riormente, se acompaña de osteoporosis, disminución del es-pacio interarticular y erosiones óseas (indicador de malpronóstico). Una radiografía normal no la descarta (MIR).- Otras: la ECO es útil en derrames de caderas; la Gammagra-fía con Tc-99 sólo es de utilidad pasados 6 meses de la colo-cación de la prótesis de cadera infectada; TAC/RM, si existeafectación del esqueleto axial.
TratamientoDrenaje de la articulaciónSe realiza mediante artrocentesis con aguja, diariamente, mien-tras la articulación se halle tumefacta.
- Los antibióticos se han de utilizar por vía parenteral durantelas primeras 2 semanas, por lo menos, y a dosis plenas. Se hande iniciar inmediatamente después de haber extraído las mues-tras necesarias para cultivos. El tratamiento de elección inicialdepende del tipo de paciente, del foco infeccioso sospechadoy, sobre todo, de la tinción de Gram del líquido articular (vertabla 2); posteriormente se adaptará a los resultados de loscultivos y al antibiograma correspondiente. En las artritis bac-terianas no gonocócicas, la duración global del tratamientoserá de 6 semanas.
• Cefalosporina de 3ª generación + Cloxacilina u Oxacilina,dan cobertura a la mayoría de las infecciones en el adulto.• Vancomicina: fármaco de elección en enfermos con alergiaa penicilina y en infecciones de articulación protésica debidaa S. epidermidis resistente a meticilina.
- Inmovilización. Se recomienda colocar la articulación en re-poso, mediante una inmovilización en la posición de tensiónmínima de la cápsula, durante los primeros días, mientras eldolor sea intenso. Si la evolución es buena, a las 48-72 h de ini-ciado el tratamiento pueden comenzarse los movimientos pa-sivos y, si la mejoría persiste, suspenderse la inmovilización einiciar movimientos activos y contra resistencia. El apoyo sepermitirá cuando no exista ningún signo inflamatorio.- Indicaciones del drenaje quirúrgico:
• Mala respuesta clínica a los 4-7 días de iniciado el trata-miento.• Persistencia de cultivos positivos de líquido articular.• Presencia de tabiques intrarticulares que dificultan el va-ciado, en cuyo caso puede ser útil la artroscopia.• Artritis infecciosas no tratadas, de varias semanas de evo-lución. La principal indicación quirúrgica en las espondilodis-citis es la presencia de complicaciones neurológicas.
11.2.- Artritis gonocócica
Es la forma principal de artritis bacteriana en adultos jóvenes yadolescentes (< 40 años) (MIR 99, 103), con un ligero predomi-nio en las mujeres. Se sospecha ante la existencia de promiscui-dad sexual o antecedentes de infección venérea en el últimomes (menos del 25% de los pacientes con infección gonocócicadiseminada presentan síntomas genitourinarios) (MIR 01F, 79).
SEGÚN EDADESSEGÚN SITUACIÓN
DE BASEEN GENERAL
- < 2 meses:S. aureus
S. aureus(75% de pioartrosis
no gonocócicas)
- Artritis reumatoide: S. aureus
- ADVP: S. Aureus (MIR98, 167) y estreptoco-cos de su propia flora,Pseudomonas y otrosGram negativos de losinstrumentos de inyec-ción.
- Inmunodeprimidos:bacilos gram negativos
- Prótesis postquirúrgicas:S. epidermidis (es elmás frecuente), S. au-reus. Pueden cursardeforma aguda o condolor crónico sin sínto-mas generales (trata-miento: erradicar laprótesis + ATB).
- Inoculación directa: S.aureus, S. epidermidis
- Arañazo o mordedurade gato: Pasteurellamultocida.
- Alcohólicos, hemoglobi-nopatías: neumococo
- Fracturas abiertas:polimicrobiana
- Ulceras, abscesos abdo-men: anaerobios
- 2-24 meses:H. influenzae (desdela vacuna contra enHaemophilus han
predominado S.aureusy S. pyogenes)
2-15 años:S. aureus
Adulto joven yadolescente: gonococo
(MIR)
> 40 años: S. aureus
Nota: En ADVP y en pacientes con SIDA, el germen más frecuente causal deespondilodiscitis piógena y de la afectación esternoclavicular, es la Pseudo-mona aeruginosa, seguida de S. aureus. La afectación del cartílago esterno-costal es típica de infección por Candida.
GÉRMENES MÁS FRECUENTES
Tabla 1. Etiología de las artritis.

R e u m a t o l o g í a
55] ARTRITIS SÉPTICAS [
Forma parte del cuadro de la infección gonocócica diseminada,con artritis, tenosinovitis, dermatitis, meningitis, miopericaditisy sepsis clínica; los síntomas predominantes son la artritis y latenosinovitis, con exantema o sin él. Se consideran factores predisponentes la menstruación, el em-barazo y el déficit de factor final del complemento (C5 - C6 - C7- C8) (MIR). La bacteriemia proviene de una infección gonocó-cica, o, más frecuentemente, de la colonización asintomática dela mucosa de uretra, cuello uterino o faringe.
Clínica- Fase inicial: la forma de presentación más frecuente es lapresencia de fiebre, escalofríos junto a un exantema y sínto-mas articulares. Las alteraciones cutáneas aparecen en 1/3 decasos como lesiones vesículo-pustulosas (en tronco y enporción distal de extremidades); los síntomas articulares sontenosinovitis y la presencia de monoartritis (40%), oligoartri-tis (35%) o poliartritis (25%), precedida durante 1 ó 2 días depoliartralgias, generalmente migratorias (rodillas, tobillos,muñecas, pies y manos). Los hemocultivos en esta fase puedenser positivos, el gram y cultivo del líquido sinovial suelen ser ne-gativos (se piensa que las lesiones cutáneas y articulares sonconsecuencia de una reacción inmunitaria y depósito de inmu-nocomplejos).- Fase final: los síntomas generales y la dermatitis ceden, laafectación articular empeora, produciendo una artritis puru-lenta mono o pauciarticular. Los hemocultivos son negativosen la mayoría de casos, los cultivos del líquido sinovial puedenser positivos.
DiagnósticoRequiere además de lo expuesto para las artritis no gonocócicas,la realizaciónde cultivo en medio de Thayer-Martin de un frotisuretral, cervical, rectal o faríngeo para determinar la coloniza-ción del gonococo.
TratamientoAntibioterapia con Ceftriaxona 1 gr iv. o im. cada 24 horas du-
rante 6-12 días, además del drenaje de la articulación infectada.
11.3.- Artritis tuberculosa (MIR 07, 192)
Se trata de una artritis de carácter insidioso cuyo diagnóstico seretrasa semanas o meses, debido a que no se acompaña de sín-tomas sistémicos y, sólo en algunos casos, se acompaña de TBCpulmonar activa. La infección articular se produce tras la activa-ción de focos linfohematógenos latentes, siendo la forma depresentación más frecuente una monoartritis granulomatosacrónica, que afecta fundamentalmente a articulaciones decarga (cadera en niños y rodilla en adultos). La osteomielitis porTBC es más frecuente a nivel vertebral y se inicia en el hueso es-ponjoso vertebral, alcanzando luego el espacio discal vecino(mal de Pott).
La enfermedad de Poncet, es una poliartritis simétrica y reac-tiva que afecta a pacientes con TBC visceral o diseminada (nohay bacilos en las articulaciones)El liquido sinovial es de características inflamatorias (20000cels/mm3), se observan BAAR en un 20% de casos, el cultivo espositivo en un mayor porcentaje (80%) aumentando la sensibi-lidad si se hace biopsia sinovial (lesiones granulomatosas).El tratamiento se basa en el uso de antituberculosos durante6-12 meses.
11.4.- Artritis brucelósica
La brucelosis es una zoonosis que cursa con manifestaciones ar-ticulares en el 85% de los casos. La artritis puede cursar deforma aguda, crónica o recurrente. Lo más característico es unamonoartritis, siendo lo más frecuente la afectación axial (espon-dilitis, sacroileítis), pero también son posibles las artralgias y ar-tritis leves e incluso una artritis destructiva, siendo esto últimomás frecuente en la cadera (pseudocoxalgia mediterránea).La artritis y la sacroileítis son complicaciones del aparato locomo-tor que aparecen en fases agudas de la brucelosis, afectandocon más frecuencia a jóvenes, mientras que la espondilitis lohace en cuadros más cronificados generalmente en mayores de40 años.En nuestro medio, se debe pensar en una brucelosis ante cual-quier coxitis, espondilitis o sacroileítis aguda o cuadro articularcon cuadro de fiebre ondulante o sudores, astenia, estreñi-miento, esplenomegalia y orquitis.Se puede diagnosticar mediante hemocultivo en fase aguda(medio de Ruiz Castañeda) y muchas veces mediante pruebasserológicas. En un 50% de casos, el cultivo de líquido sinovial espositivo, en el resto se considera una artritis reactiva mediada
Figura 1. Mal de Pott (espondilitis). Puede ocasionar aplastamiento vertebral ycifosis.
ETIOLOGÍATRATAMIENTO DE
ELECCIÓNDIAGNÓSTICO
S. aureusNiños <2 meses Cefalosporina 3ª gen.+ Cloxacilina
H. influenzaeNiños 2- 24 meses Cefalosporina 3ª gen.+ Cloxacilina
S. aureus2- 14 años Cefalosporina 3ª gen.+ Cloxacilina
N. gonorrhoeae15-40(artritis gonococica) Ceftriaxona 1 gr/día iv.
S. aureus>40 años
Terapia basadaen el Gram:
Cefalosporina 3ª+Cloxacilina: gram +
Ciprofloxacino +Cloxacilina: gram -
S. epidermidisInfección postquirúrgica,protésica, tras inyección
intraarticular
Vancomicina +Ciprofloxacino
(o Aztreonam, Cefepima)
CGPaerobios/anaerobios,
ClostridiumFracturas abiertas Amoxicilina/clavulánico
S. aureus
UDVP
Cloxacilina +Gentamicina
P. aeruginosa Ceftazidima +Amikazina
Tabla 2. Tratamiento de la artritis séptica por grupos de edad y por agente (MIR00, 140; MIR 98F, 206).

Manual A Mir
56 ] OTRAS ARTROPATÍAS [
www.academiamir.com
por inmunocomplejos.El tratamiento se realiza mediante tetraciclinas y estreptomicinao la combinación de doxiciclina, rifampicina y cotrimoxazol.
11.5.- Artritis por espiroquetas
Artritis sifilíticaPodemos encontrar afectación articular en la sífilis congénita(osteocondritis, pseudoparálisis de Parrot y sinovitis crónica - ar-ticulación de Clutton), en la sífilis secundaria (artralgias, artritis,sacroileítis…), en la sífilis terciaria (afectación de la membranasinovial por goma sifilítico en grandes articulaciones) y el la sífiliscuaternaria (con tabes dorsal y articulación neuropática de Char-cot).
Enfermedad de LymeInfección producida por la Borrelia burgdorferi, que cursa conclínica cutánea, articular, neurológica y cardíaca. La afectaciónarticular puede ser:
- Artralgias y mialgias en los días-semanas siguientes a la pica-dura.- Artritis en el 70% de los no tratados: episodios intermitentesde mono u oligoartritis (rodilla+++), sinovitis crónica erosiva ydestructiva…
El tratamiento se realiza mediante doxiciclina o amoxicilina du-rante 20-30 días. En caso de no respuesta o en fases avanzadas,se utiliza ceftriaxona o penicilina benzatina 2,4 millones UI se-manal durante 2-3 semanas.
11.6.- Artritis víricas
Se producen por infección del tejido sinovial directamente du-rante la infección sistémica, o mediante una reacción inmuno-lógica que afecte a las articulaciones. Las más frecuentes sonlas debidas al VHB y a la rubéola. Otras: parvovirus B19, paroti-ditis, enterovirus, arbovirus… Suelen ser autolimitadas (no haylesión articular permanente) y de predominio poliarticular (MIR).El virus de Epstein-Barr (mononucleosis infecciosa) no produceartritis.
Hepatitis BPoliartritis de comienzo rápido, con afectación simétrica degrandes y pequeñas articulaciones (también puede ser asimé-trica o migratoria), por orden de frecuencia se afectan: dedos delas manos, muñecas, rodilla, hombro. Se cree que está mediadapor mecanismo inmunológico. Suele acompañarse de exantemay aparece antes que la ictericia. Cuando la hepatitis ya es evi-dente por la clínica, la artritis suele desaparecer. No hay artritispor VHA (MIR 97, 112).
RubéolaMás frecuente en mujeres jóvenes, es una poliartritis de muñe-cas y pequeñas articulaciones de las manos, simétrica. Coincidecon el exantema o aparece poco después.
11.7.- Artritis micóticas
En general tienden a cursar lentamente y el diagnóstico suele sertardío. La articulación más frecuentemente afectada es la rodilla.La infección por Cándida se produce por intervenciones quirúr-gicas, inyecciones intraarticulares o diseminación hematógena(pacientes críticos, diabetes mellitus, insuficiencia renal, trata-miento inmunosupresor…). En ADVP es característica la infec-ción por Candida en la columna dorsal, sacroilíacas y en otrasarticulaciones fibrocartilaginosas, siendo típica la artritis con-drocostal o esternocostal.El tratamiento se realiza con anfotericina B, fluconazol o itraco-nazol, requiriendo en ocasiones un desbridamiento quirúrgico.
12.1.- Policondritis recidivante
Se trata de una enfermedad poco frecuente y de carácter infla-matorio, que a afecta las estructuras cartilaginosas de todo el or-ganismo, así como los órganos auditivo y visual, el riñón y elsistema cardiovascular.
EtiologíaDesconocida. La detección en algunos pacientes de anticuerposcontra el colágeno tipo II sugiere una base inmunológica. En un30%, se asocia a otra enfermedad reumatológica autoinmune(AR, EA, síndrome de Reiter, LES y síndrome de Sjögren).
Anatomía patológicaEl cartílago es invadido por un infiltrado inflamatorio en el queprimero predominan los neutrófilos y, más tarde, las células re-dondas. Como consecuencia de ello, el cartílago pierde su ba-sofilia, degenera y finalmente es sustituido por tejido fibroso,que puede calcificarse.
Cuadro clínicoPredomina entre los 40 y los 60 años, cursando de forma epi-sódica y recidivante (MIR).La condritis de los pabellones auriculares es la manifestaciónclínica más frecuente y la forma de inicio de la mitad de loscasos. El comienzo de la condritis es súbito, con dolor intenso yun enrojecimiento violáceo de la parte cartilaginosa del pabellónauricular, respetando el lóbulo. La afectación del oído internoproduce un síndrome vestibular. La artritis es la segunda manifestación más frecuente de inicio,con oligoartritis o poliartritis asimétrica no erosiva que afecta acualquier articulación, pero, en especial, las paraesternales. La condritis nasal se traduce en episodios súbitos de dolor y tu-mefacción con sensación de obstrucción nasal, rinorrea e inclusoepistaxis. La inflamación reiterada produce una deformidad tí-pica de la nariz en silla de montar. En los ojos la episcleritis, la conjuntivitis y la uveítis son las le-siones más frecuentes.La afectación del cartílago del tracto respiratorio produce ron-quera, tos, disnea, estridor y dolor a la palpación del cartílago ti-roides y de la tráquea. La cuarta parte de los pacientes presentan lesiones cutáneas,sobre todo vasculíticas.En el sistema cardiovascular, puede producir insuficiencia aór-tica por dilatación del anillo valvular, insuficiencia mitral y aneu-risma aórtico. En ocasiones puede presentarse como fiebre de origen desco-nocido.
DiagnósticoEs clínico y sólo en los casos en que éste es dudoso está indicadala biopsia del cartílago. La VSG suele aumentar durante los bro-tes. En algunos casos se detecta positividad del factor reuma-toide, de anticuerpos antirribonucleoproteína y de losanticitoplasma de neutrófilo (ANCA) (MIR).
TratamientoSe basa en el uso de corticoides a dosis altas. En caso de nohaber respuesta, se emplean inmunosupresores.
TEMA 12 OTRASARTROPATÍAS
Idea general de todas.
ENFOQUE MIR

R e u m a t o l o g í a
57] OTRAS ARTROPATÍAS [
12.2.- Artropatía neuropática de Charcot
Es una forma grave de osteoartritis que se asocia a una pérdidade sensibilidad al dolor, propioceptiva o ambas, con dismi-nución de los reflejos musculares. Sin estos mecanismos las ar-ticulaciones están sometidas a traumatismos repetidos quecausan una lesión progresiva del cartílago.
EtiologíaEn la actualidad la polineuropatía diabética es la causa másfrecuente a nivel del tarso y MTF (MIR). En niños la causa másfrecuente es el mielomeningocele. Otras causas son: tabesdorsal, siringomielia (con afectación glenohumeral, del codo ydel carpo) (MIR), la amiloidosis, la lepra o las inyecciones in-traarticulares repetidas de glucocorticoides. No aparece en el hi-potiroidismo (MIR 00F, 95).
ClínicaLa articulación aumenta de tamaño, como consecuencia del cre-cimiento óseo y del derrrame sinovial. Conforme progresa la en-fermedad aparece inestabilidad, subluxación y crepitación: lo máscaracterístico es que el dolor es inferior al que cabría esperar.
TratamientoSe basa en estabilizar la articulación (soportes externos, férulas).Está contraindicado el reemplazamiento articular por prótesis. Eltratamiento de la enfermedad causal NO suele alterar la enfer-medad articular.
12.3.- Osteoartropatía hipertrófica
Esta afección se manifiesta generalmente en la edad media dela vida y se caracteriza por una hipertrofia de manos y pies. Di-ferenciamos una forma primaria, que puede ser idiopática omás frecuentemente, familiar (enfermedad de Touraine-Solente-Calvé de herencia AD) y una forma secundaria asociada a neo-
plasias de pulmón (carcinoma broncogénico y pleural), infec-ciones pulmonares, fibrosis quística, sarcoidosis, neumonitis in-tersticial crónica, cardiopatía congénita con shunt de derecha aizquierda, cor pulmonale, neoplasias gastrointestinales, enfer-medad inflamatoria intestinal, endocarditis bacteriana subaguda(S. viridans sobre todo), etc. (MIR 97, 107; MIR 97F, 96).
- Síndrome articular. Cursa con dolor, tumefacción y de-rrame en las grandes articulaciones distales.- Síndrome morfológico. Hay una deformación de los dedosde las manos y de los pies, con una incurvación de la uña, enforma de vidrio de reloj, junto con un ensanchamiento de lafalange terminal (acropaquia o dedos en palillo de tambor).
- Síndrome radiológico. La primera exploración a realizar esla Rx tórax. A nivel óseo, las diáfisis afectas aparecen recubier-
Figura 1. Policondritis recidivante.
Epiescleritis,conjuntivitis y
uveitis
Afectación decartílago respiratorio(ronquera, estridor)
Artritis dearticulacionesparaesternales
Condritis auricularLo más característico y frecuente
Condritis nasal
Insuficiencia aórtica yaneurismas de aorta ascendente
Dx: clínico
Tratamiento: esteroides ± inmunosupresores
Figura 2. Acropaquias.
Vasculitiscutánea

Manual A Mir
58 ] AMILOIDOSIS [
www.academiamir.com
tas por una o varias capas óseas estratificadas, en forma decorteza de árbol, lo cual se traduce por un doble o triple con-torno óseo.
La instauración del proceso es rapidísima y bastan a veces sólo15 días para que se forme la acropaquia, especialmente en loscasos que son debidos a un tumor maligno. La evolución pos-terior depende de la enfermedad de base y, si ésta cura, puedendesaparecer.
TratamientoDepende de la enfermedad de base. El tratamiento sintomáticode la dolencia se realiza con glucocorticoides, analgésicos y an-tiinflamatorios.
12.4.- Fibromialgia
Proceso frecuente caracterizado por dolores musculoesqueléti-cos, rigidez, sueño no reparador y tendencia a cansarse con fa-cilidad. Afecta con más frecuencia a mujeres de entre 25-45años. La característica fundamental es la palpación de zonasmucho más dolorosas a la palpación (“puntos gatillo” agrupa-das en 18 puntos). La palpación de 11 de estos 18 puntos gatilloestablece el diagnóstico. La exploración articular y las pruebasde laboratorio son normales dado que no se trata de una enfer-medad inflamatoria.No es degenerativa. El tratamiento se basa en salicilatos y AINES,incluso se emplean en su manejo antidepresivos tricíclicos.
12.5.- Polimialgia reumática
Enfermedad caracterizada por dolor, rigidez e impotencia fun-cional en la cintura escapular y pelviana, acompañada en oca-siones de síntomas generales como astenia, anorexia, fiebre…Al contrario que la anterior, puede ser muy invalidante. Puedeaparecer asociada a la arteritis de la temporal. Afecta con másfrecuencia a mujeres por encima de los 50 años.Se caracteriza por elevación de VSG (marcador de actividad dela enfermedad al igual que ocurría en la arteritis de la temporal),aumento de fosfatasa alcalinas y anemia de trastornos crónicos.No cursa con elevación de enzimas musculares (MIR 98F,217). El tratamiento se basa en el uso de AINE y corticoides adosis bajas (la mejoría clínica apoya el diagnóstico, ya que sueleser muy marcada) (MIR 03, 229).
La amiloidosis engloba un grupo de entidades clínicas de etio-logía desconocida producidas por el depósito extracelular deproteínas de estructura fibrilar (proteína principal), que provocaalteraciones diversas, según la cuantía del depósito y el órganoafecto. Podemos clasificarlas en:
- Amiloidosis primaria, o asociada a mieloma múltiple (tipoAL, cadenas ligeras).- Amiloidosis secundaria (tipo AA), asociadas a enfermedadesinfecciosas crónicas (TBC, lepra…) o inflamatorias crónicas(AR) (MIR 98F, 17).- Amiloidosis heredofamiliar, donde se incluye la fiebre me-
diterránea familiar (tipo AA), síndromes polineuropáticos comola polineuropatía amiloide familiar. La mutación en el gen dela transtirretina es la forma más frecuente de polineuropatíaamiloidótica (MIR 02, 60).- Amiloidosis focal (lesión ocupante de espacio) en órganosaislados, sobre todo endocrinos (carcinoma medular de tiroi-des con depósitos intracitoplasmáticos) (tipo AE).- Amiloidosis asociada a envejecimiento.- Amiloidosis secundaria a hemodiálisis crónica y diálisis pe-ritoneal: depósito de β2-microglobulina (MIR 01F, 80; MIR01F, 215).
En todos los casos el riñón es el órgano más frecuentementeafectado, siendo la primera causa de muerte en las formas se-cundarias (AA), mientras que en las primarias (AL) lo es la afec-tación cardíaca (MIR). La clínica es, en cualquier caso, multisistémica y se deberá sos-pechar especialmente en caso de hepatoesplenomegalia (conpoca afectación de la función hepática) y proteinuria (síndromenefrótico, insuficiencia renal), así como elevación moderada dela fosfatasa alcalina y la GGT (MIR 97, 113). A veces puede pro-ducir un síndrome de Fanconi.Síntomas característicos de la amiloidosis primaria (AL) son lamacroglosia, la aparición de pápulas cutáneas de aspectocéreo muy pruriginosas (liquen amiloideo), el síndrome deojo negro (lesiones purpúricas periorbitarias “en mapa-che”), la infiltración cardíaca pudiendo cursar con miocardio-patía restrictiva (ecorrefringencia aumentada del miocardiocon el típico centelleo o moteado granular) y atrapamientos denervios periféricos (síndrome del túnel carpiano) (MIR 02, 85).La afectación del sistema nervioso autónomo provoca hipoten-sión ortostática. A nivel pulmonar puede ocasionar enferme-dad intersticial y nódulos pulmonares.Ante la sospecha de amiloidosis sistémica se debe obtener unamuestra tisular, generalmente de grasa subcutánea abdominalo de mucosa gingival, biopsia renal o rectal. La presencia desustancia amiloide, en muestras teñidas con rojo Congo (MIR07, 232) tiene una birrefringencia verde al microscopio deluz polarizada. Si tratamos el material con permanganato potá-sico, las formas primarias son resistentes, mientras que la se-cundarias son sensibles al mismo.
TratamientoEn el caso de amiloidosis AL se han utilizado citostáticos. El usode melfalán y prednisona junto con colchicina puede mejorar lasupervivencia. En las formas secundarias, se basa en el trata-miento de la enfermedad de base.El pronóstico de las formas generalizadas es malo.
TEMA 13 AMILOIDOSIS
Tema en auge en los últimos años del MIR. Recuerda la formaprimaria y las formas secundarias. Fíjate en las manifestacionescutáneas típicas para no tener que estudiarlas en el capítulo deDermatología.
ENFOQUE MIR
Figura 1. Rojo congo y luz polarizada en la amiloidosis.

R e u m a t o l o g í a
59] SÍNDROME DE SJÖGREN [
13.1.- Fiebre mediterránea familiar
También llamada poliserositis paroxística, es una enfermedadhereditaria (Autosómica Recesiva) de etiología desconocida, ca-racterizada por episodios recurrentes de fiebre, peritonitis y/opleuritis (MIR).Aparece entre los 5-15 años, como episodios recurrentes de fie-bre elevada (24-48 horas) acompañada de dolor abdominal(95%), dolor torácico, dolor articular (en grandes articulaciones)o manifestaciones cutáneas (áreas eritematosas dolorosas en laporción inferior de la pierna, maleolo interno…). El dolor abdo-minal puede llegar a ser tan intenso, que algunos de estos pa-cientes son sometidos a laparotomías exploradoras antes dellegar al diagnóstico.La amiloidosis sistémica (tipo AA) es la complicación más gravey se manifiesta en forma de nefropatía de aparición precoz yevolución rápida a la insuficiencia renal.En su diagnóstico se han utilizado pruebas de provocación conmetaraminol. El tratamiento se basa en el uso de colchicina paraprevenir la amiloidosis y disminuir el número de brotes (MIR 06,258; MIR 00, 122; MIR 99F, 101).
Trastorno inmunitario crónico, de etiología desconocida que sedefine por la asociación de xeroftalmía (queratoconjuntivitisseca) y xerostomía (manifestación más frecuente). Afecta conmás frecuencia a mujeres de edad media y puede ser primarioo asociado a otras enfermedades autoinmunes, sobre todo reu-matológicas (el 30% de pacientes con patología reumática pre-sentan un síndrome seco, fundamentalmente la AR que es lacausa más frecuente de síndrome de Sjögren secundario, perotambién el LES, ESP, PM, DM…) y hepáticas (cirrosis biliar pri-maria y hepatitis crónica activa).
EtiopatogeniaA partir de estímulos antigénicos internos o externos, se produceun infiltrado de glándulas exocrinas por linfocitos T con fenotipoCD4+. Según evoluciona el proceso inflamatorio se produce unadestrucción acinar junto a una hipofunción glandular.
Cuadro clínico (MIR)Los síntomas más frecuentes son la sequedad ocular y bucal (casien 100%). Las manifestaciones iniciales pueden no ser específi-cas y pasan años hasta el desarrollo total de la enfermedad.
- Afectación ocular: se caracteriza por una queratoconjunti-vitis seca (sensación de arenilla y cuerpo extraño, enrojeci-miento ocular), úlceras corneales, hipertrofia lagrimal…- Oral: xerostomía, aumento de parótidas o de otras glándulassalivares mayores.- Respiratorio: tos, disfonía.- Digestivo: atrofia de mucosa esofágica, gastritis atrófica,pancreatitis subclínica.- Genital: dispareunia, prurito.- Sequedad de piel y mucosas.
Las manifestaciones extraglandulares son propias del Sjögrenprimario y no suelen aparecer en las formas secundarias. Sonlas que marcarán el pronóstico del paciente. Por orden de fre-cuencia, aparecen: artralgias/ artritis (60%), Raynaud (40%),adenopatías, afectación pulmonar (neumonitis intersticial), renal(nefritis intersticial), vasculitis y polineuropatías.En los pacientes con síndrome de Sjögren, hay una mayor inciden-cia de linfomas no Hodgkin de células B y macroglobulinemiade Waldenström. El linfoma se sospecha ante tumefacción pa-rotídea prolongada, reducción de FR, linfadenopatías y nódulospulmonares (MIR), aumento de la B2 microglobulina, ↑ LDH,banda monoclonal, negativización de los anticuerpos, negativi-zación del FR, presencia de crioglobulinemia.
DiagnósticoAnalítica: VSG elevada, FR en 80% de pacientes. La mayoríatienen ANA pero no anti-DNA. Los anticuerpos característicos,sobre todo de las formas primarias, son anti- Ro (SS-A) y anti-La (SS-B), siendo el último más específico. Su presencia se asociaa comienzo más precoz y mayor duración de la enfermedad.
Criterios diagnósticos (MIR 02, 83)- Síntomas oculares.
El amiloide AL se asocia a Mieloma y a la forma PRImaria(el ALMa es lo PRImero).
La amioloidosis hereditaria hay que sospecharla en pacientescon neuropatía, disautonomía, afectación renal, cardíaca o
gastrointestinal, es decir, síntomas parecidos a la formaprimaria, pero con mayor protagonismo de la afectación
neurológica y sin afectación mucocutánea (por ello la macro-glosia no es característica de esta enfermedad).
La amiloidosis secundaria habrá que sospecharla en pacientecon enfermedades inflamatorias o infecciosas crónicas quedesarrollan proteinuria o clínica digestiva (la amiloidosissecundaria afecta más frecuentemente al riñón, hígado,
bazo y ganglios).
En la FMF es útil la colchicina (esto ocurre también en otrasserositis como la pericarditis).
RECUERDA
TEMA 14 SÍNDROME DESJÖGREN
No muy importante. Recuerda qué autoanticuerpos se relacio-nan con este síndrome. Con la tabla es más que suficiente parael MIR. Recuerda el linfoma de parótida.
ENFOQUE MIR
Figura 1. Xerostomía, que es la manifestación más frecuente.

Manual A Mir
60 ] ARTROSIS [
www.academiamir.com
- Síntomas bucales.- Signos oculares (test de Schirmer < 5 mm en 5 min o rosa deBengala +).- Biopsia de glándulas salivares menores (es el lugar más ren-table).- Afectación de glándulas salivares visto por gammagrafía, sia-lometría o flujo salival.- Presencia de uno de los siguientes FR, ANA ,anti- Ro (SSA) yanti-La (SS-B).
Diagnostico diferencial:Cuadros que afectan las glándulas salivares y lagrimales: amiloi-dosis, linfoma, sarcoidosis, VIH
TratamientoNo existe tratamiento eficaz contra la destrucción glandular.Empleamos tratamiento sintomático con lágrimas artificiales,nebulizaciones nasales e ingesta abundante de líquidos. Es im-portante evitar fármacos diuréticos, antihipertensivos y antide-presivos (disminuyen la secreción glandular). La pilocarpina oralmejora las manifestaciones de sequedad. Se utilizan glucocorti-coides e inmunosupresores para el manejo de la afectación ex-traglandular.
La artrosis es la segunda causa de incapacidad permanente, des-pués de las enfermedades cardiovasculares. Se trata de un sín-drome que engloba un grupo heterogéneo de procesos convariados mecanismos etiopatogénicos, que terminan condicio-nando el fracaso de la articulación debilitando el cartílago, queno puede soportar las fuerzas normales o claudica ante fuerzasanormalmente intensas. También llamada osteoartritis o artro-patía degenerativa (progresiva pérdida de cartílago articular,junto con proliferación de hueso nuevo y de tejidos blandosintra y periarticular).
EpidemiologíaSe trata de la forma más frecuente de enfermedad articular y laprincipal causa de incapacidad en el anciano. La incidencia au-menta con la edad (es el factor de riesgo más importante). Hastalos 55 años, es igual de frecuente en ambos sexos pero, por en-cima de los 55 años, predomina en mujeres, donde ademássuele ser más sintomática (MIR 99F, 210). La artrosis de manosy rodillas es más frecuente en mujeres, la artrosis de cadera esmás frecuente en el hombre (MIR 07, 87).
EtiologíaSegún el American Collage of Reumatology (1986), se clasificanlas artrosis en:
IdiopáticaEs aquella en la que no se conoce la causa, aunque se han iden-tificado una serie de factores que pueden influir en la génesis dela enfermedad (edad, factores genéticos, sexo, obesidad, estró-genos, microtraumatismos) Localizada
- Manos: nódulos de Heberden y Bouchard, rizartrosis del pul-gar, artrosis erosiva interfalángica.- Pies: hallus valgus, hallus rigidus, artrosis talonavicular.- Rodilla: compartimento femorotibial y femoropatelar.- Cadera: excéntrica (superior), concéntrica (medial), difusa.- Raquis: interapofisaria, discovertebral, ligamentaria (hipe-rostosis anquilosante vertebral), espondilosis.- Otras localizaciones: hombro, acromioclavicular…
GeneralizadaPequeñas articulaciones periféricas y del raquis; grandes articu-laciones centrales y raquis; mixta.
SecundariaSe incluyen en este grupo las relacionadas con:
- Traumatismos.- Patología congénita o del desarrollo (enfermedad de Perthesy otras displasias, dismetrías…).- Alteraciones metabólicas: hemocromatosis, Wilson, enferme-dad de Gaucher.- Alteraciones endocrinas: acromegalia, hiperparatiroidismo,DM, obesidad, hipotiroidismo…- Alteraciones neuropáticas: artropatía de Charcot.- Enfermedad articular inflamatoria: por depósito de microcris-tales, infecciones, artritis reumatoide, artropatías seronegativas.- Otras: necrosis avascular, enfermedad de Paget…
Anatomía patológicaInicialmente se produce un reblandecimiento focal en un áreade la superficie cartilaginosa sometida a cargas; aumenta el con-
- Ingesta de líquidos- Bromhexina y pilocarpina
TRATAMIENTO
- Xerostomía (sequedadbucal)
- Disfagia (sensación urente)- Hipertrofia parotídea en el
Sjögren 1º
- Sequedad vaginal: gelesde ácido propiónico
- AINEs- Glucocorticoides e inmu-
nosupresores- Hidroxicloroquina:
artralgias(↓VSG, Ac antiLa/SS-B ehipergammaglobulinemia)
-Artritis no erosiva-Enfermedad pulmonarintersticial
-Nefritis intersticial,Sdr. de Fanconi, GNF
-Vasculitis necrotizanteasociado a enf. neurológica
-Linfoma no Hodgkin;Waldenström
- Lágrimas artificiales- Si ulceración: pomada de
ácido bórico y oclusión- Evitar: diuréticos, antide-
presivos, hipotensores
- Xeroftalmia (sequedadocular)
- Queratoconjuntivitis seca:eritema, picor, cansancioocular
MANIFESTACIONESBUCALES
MANIFESTACIONESOCULARES
CLÍNICA
- Sequedad traqueobronquial- Atrofia de mucosa esofágica- Gastritis atrófica- Dispareunia- Sequedad cutánea
OTRASGLÁNDULASEXOCRINAS
MANIFESTACIONESSISTÉMICAS
Tabla 1. Síndome de Sjögren.
TEMA 15 ARTROSIS
Recordad cuadros específicos por artrosis según cada articula-ción, la sucesión de cambios radiológicos y las indicaciones detratamiento médico o quirúrgico.
ENFOQUE MIR
Figura 2. Sjögren primario. Linfoma de parótida.

R e u m a t o l o g í a
61] ARTROSIS [
tenido en agua y disminuye el de proteoglicanos, los condrocitosproliferan y forman grupos localizados de gran actividad. Segui-damente, aparecen fisuras superficiales, tangenciales o perpen-diculares, que dan un aspecto fibrilar al cartílago. El grado dedestrucción del cartílago es variable y puede progresar desde le-siones superficiales y moderadas, a francas ulceraciones que ex-ponen el hueso subcondral; ello depende de las fuerzas queactúen sobre la articulación y de la eficacia del proceso repara-dor del condrocito.El hueso subcondral responde activamente a la agresión con au-mento de la remodelación y una neta ganancia de hueso, au-mentando el grosor y la densidad de la placa ósea(osteosclerosis) y formando excrecencias óseas en los márgenesde la articulación (osteofitos), en las inserciones cápsulo-liga-mentarias.En la cadera, y más raras veces en otras articulaciones, se for-man quistes (geodas) intraóseos, yuxtarticulares que se originancomo consecuencia de la hiperpresión articular que escapa através de fallas de la cortical.
ClínicaSe caracteriza por un comienzo insidioso. En los primeros esta-dios es indolora, posteriormente comienza a aparecer un dolorde características mecánicas (se agrava con el ejercicio y mejoraen reposo), que suele ser la primera y principal manifestación; larigidez de la articulación después del reposo es de breve dura-ción (matutina, de <15-30 minutos). Hay limitación de la movi-lidad articular, crepitación ósea, tumefacción de consistenciaósea, deformidad en estadios avanzados. No aparecen signosclínicos ni analíticos de afectación sistémica. Destacan como for-mas clínicas:
- IFD (nódulos de Heberden). Forma más frecuente de oste-oartritis idiopática. Más frecuente en mujeres y >45 años. Setrata de un problema estético más que funcional. Se habla deuna tendencia familiar heredada de forma AD en la mujer y ARen el varón.- IFP (nódulos de Bouchard) (MIR).
- Base del pulgar (articulación trapeciometacarpiana). Es lasegunda localización más frecuente de la artrosis. También esmás frecuente en mujeres.- Coxartrosis. Es una de las formas de artrosis más incapaci-tante, junto con la rodilla. En la mayoría de los casos, es secun-daria a anomalías congénitas o del desarrollo. Es másfrecuente en varones y unilateral (20% bilateral). La impoten-cia funcional se traduce en cojera. Se produce dolor en regióninguinal, también en nalgas, en región proximal del muslo e in-cluso en la rodilla. Inicialmente se afecta la rotación internade la cadera.
- Gonartrosis. Es más frecuente en mujeres. De inicio unila-teral, con tendencia a hacerse bilateral. Es menos frecuenteque la artrosis de manos y pies y más que la de la cadera. Laafectación más frecuente es la degeneración artrósica del com-partimento femorotibial medial o interno, provocando un varode la rodilla (MIR).- Artrosis vertebral. Dolor y rigidez, localizados más frecuen-temente en la columna cervical (C5-C6) y lumbar (L4-L5 y L5-S1) Diferenciamos entre espondilosis para referirnos a laenfermedad degenerativa de los discos y artrosis vertebralcuando se afectan las articulaciones interapofisarias.- Esternoclavicular. Causa frecuente de consulta por motivosestéticos, pero casi siempre asintomática.- Artrosis generalizada idiopática. Forma clínica con fuertecomponente genético, más frecuente en mujeres perimeno-páusicas. Se caracteriza por brotes de inflamación con afecta-ción de 3 o más articulaciones. Remite paulatinamente en lamayoría de los casos y aparecen deformidades de los dedosde poca repercusión funcional (sólo una pequeña proporciónrequiere tratamiento médico prolongado o cirugía).- Osteoartritis erosiva. Es la forma más agresiva de artrosisque cursa con brotes de inflamación aguda articular y destruc-ción progresiva de articulaciones (más frecuentemente IFD eIFP), produciendo deformidad y alteración funcional. En RX estípico el colapso de la placa ósea subcondral.
DiagnósticoEstá basado en la clínica y en los hallazgos radiológicos. En la ra-diografía, lo más precoz es el pinzamiento de la línea articu-lar, otros hallazgos son el aumento de la remodelación(esclerosis subcondral y osteofitos) quistes o geodas sub-condrales y deformidad articular (MIR 97, 118). No hay corre-lación entre la intensidad de los síntomas y el grado dealteración radiográfica.Encontramos un líquido sinovial de características mecánicas(grupo I o no inflamatorio). A nivel analítico: VSG normal y FRnegativo (MIR 01F, 81).
Tratamiento (MIR 07, 84; MIR 07, 87)Dirigido a aliviar el dolor y mantener la función, lo más impor-tante es la reducción de la sobrecarga articular. La reducción dela carga articular puede hacerse mediante el control del sobre-peso y el uso de bastón.
- Fisioterapia: calor, hielo, ejercicios (mejor isométricos queisotónicos, ya que reducen la sobrecarga articular). - Tratamiento farmacológico: analgésicos como primergrupo de fármacos a utilizar (paracetamol), AINEs si no res-ponden a analgésicos (alivian el dolor más por su efecto anal-gésico que por el antiinflamatorio). Los opiáceos menores
Figura 1. Nódulos de Bouchard.
Figura 2. Coxartrosis.

Manual A Mir
62 ] POLIOMIOSITIS Y DERMATOMIOSITIS [
www.academiamir.com
(Tramadol, codeína) son la segunda alternativa cuando no hayrespuesta al parecetamol solo. Se ha aprobado la inyección in-traarticular de ácido hialurónico en la artrosis de rodilla resis-tente a otros tratamientos, pues parece tener un efecto máslento pero más duradero que el de los corticoides intraarticu-lares. No están indicados los glucocorticoides vía sistémica. Lainyección intra o periarticular de glucocorticoides provoca unamejoría sintomática, (no realizar más de 1 vez cada 1-4 mesesen la misma articulación). - Cirugía: artroplastia total (artrosis avanzada y fracaso del tra-tamiento médico intenso), osteotomías, artroscopia (eliminarfragmentos de cartílago desprendidos).
Trastornos de etiología desconocida, en los que el sistema mus-culoesquelético resulta dañado por un proceso inflamatorio depredominio linfocítico. La polimiositis respeta la piel, mientrasque la dermatomiositis presentará alteraciones cutáneas carac-terísticas acompañando a la afectación muscular. Ambos proce-sos pueden aparecer en el 20% de los casos asociados adistintas enfermedades (AR, LES, conectivopatías…) y, en un10%, a neoplasias.
EtiologíaSe han planteado diferentes hipótesis:
- Infecciosa: en relación con virus (no el VHB).- Inmune: por la presencia de autoanticuerpos circulantes (anti-Jo, anti-Mi, anti-PM1, anti-PM/Scl) y de linfocitos CD8 + y ma-crófagos que invaden fibras musculares.- Genética: predisposición genética, con HLA DR3 y DRW52.
Cuadro clínico- Alteraciones musculares. Vienen marcadas por la presenciade debilidad muscular aguda o subaguda (generalmente deinicio insidioso), simétrica y difusa, con preferencia por muscu-latura proximal de extremidades (cintura pélvica y escapular),tronco y cuello. En la mayoría de los casos es indoloro. Con eltiempo, desarrollan atrofia, contracturas y disminución de losreflejos.- Alteraciones cutáneas. La más frecuente en la DM es unaerupción cutánea eritematoviolácea que afecta a cuello, caray tórax. Es característico también, el eritema heliotropo (enpárpados), que puede extenderse a otras zonas fotoexpues-tas), las pápulas de Gottron (localizadas en los nudillos), te-langiectasias periungueales, a veces ulceración dérmica ycalcinosis (fundamentalmente en la DM infantil).- Articulares. Artralgias, artritis transitorias, no erosivas, contendencia a la simetría.- Otras. Afectación cardíaca variable (alteración ECG, arritmia,miocarditis), pulmonar (fibrosis intersticial asociada con antiJo-1), renal (muy rara), fenómeno de Raynaud.
Figura 1. Eritema heliotropo en su localización típica.
Figura 2. Pápulas de Gottron en el dorso de los nudillos (esta ubicación no es tí-pica del LES).
Sin clínica cutánea30%
- Mayor frecuencia de calcinosis,contracturas musculares y vasculitis en piel,
músculos y aparato gastrointestinal- Mortalidad de 1/3
10%
- Erupción cutánea típica +miosistis sin demostración de neoplasia,
vasculitis o conectivopatía- Alteraciones cutáneas pueden
preceder o aparecer con posterioridadal síndrome muscular
30%
- Lesiones musculares ycutáneas indistinguibles de otros tipos
- Más frecuente en > 60 años- Las neoplasias pueden preceder o
aparecer con posterioridad a la miositis(hasta 2 años)
- Neoplasias más frecuentes: pulmón, ovario,mama, gastrointestinales y mieloproliferativas
10%
I.PM IDIOPÁTICA
PRIMARIA
II.DM IDIOPÁTICA
PRIMARIA
IV.DM Y PM INFANTIL
ASOCIADA AVASCULITIS
III.DM O PMASOCIADA
A NEOPLASIA
- ES, AR, EMTC, LES- La respuesta a corticoides es peor
que en las formas puras20%
V.PM O DM
ASOCIADA CONENFERMEDAD DELTEJIDO CONECTIVO
Tabla 1. Clasificación de la PM y DM.
TEMA 16 POLIMIOSITISDERMATO-MIOSITIS
A tener en cuenta en relación al diagnóstico diferencial, y enpreguntas tipo caso clínico. Recuerda el eritema heliotropo ylas pápulas de Gottron.
ENFOQUE MIR

R e u m a t o l o g í a
63] ANEXO [
Diagnóstico- Analítica: aumento de VSG y de enzimas musculares, (CPK,aldolasa, GOT, GPT, LDH). La CPK es la más sensible y la queguarda una mejor correlación clínica con la actividad de la en-fermedad y la valoración de recaídas. El FR es + en 20% y ANAes + en 10-30%. Si la destrucción muscular es intensa, puedeproducir mioglobinuria.- Destacan anticuerpos:• Anti-Jo1: en casos de PM asociado a neumonitis intersticial(síndrome antisintetasa-miosistis, fibrosis pulmonar, artritis noerosiva y fenómeno de Raynaud).• Anti-PM1 o PM-Scl: asociación con esclerodermia.• Anti-Mi, en DM.• Antimioglobina.- EMG: es fundamental. Muestra cambios miopáticos, con es-casa amplitud, polifásicos y reclutamiento anormalmente pre-coz de potenciales de acción de unidad motora (MIR 97F, 97).- Biopsia: la afectación muscular es parcheada, caracterizadapor una infiltración inflamatoria perivenular y necrosis muscular.
Tratamiento- Glucocorticoides. Son el tratamiento de elección a altas dosis.- Inmunosupresores. En caso de no respuesta a gucocorticoi-des tras 3 meses de tratamiento o ante recidivas frecuentes. Laazatioprina es el más usado.
En los pacientes en los que reaparece la sintomatología y que seencuentran en tratamiento esteroideo, se debe hacer el diagnós-tico diferencial con una recidiva franca o una miopatía esteroi-dea. Se debe disminuir la dosis y ver la respuesta, que será haciala mejoría en caso de miopatía esteroidea, o hacia el empeora-miento, en caso de que fuera una recidiva de la enfermedad.
LES - queratoconjuntivitis secaAR - S. Sjöegren (20%) secundario. Episcleritis. Escleritis, escleromalacia per-forante.EA - uveítis anterior aguda no granulomatosa unilateralArtritis psoriásica - conjuntivitis > uveítis anteriorEnfermedad de Behçet - uveítis bilateral anterior o posteriorArteritis de la temporal - NOIAEnfermedad de Kawasaki - conjuntivitis no exudativa bilateral
Afectación ocular de las enfermedades reumáticas.
ANEXO

Manual A Mir
64
www.academiamir.com
NOTAS

R e u m a t o l o g í a
65

Manual A Mir
66
www.academiamir.com


Top Related